Embed Size (px)
Citation preview

Cellular Signalling 23 (2011) 152–160
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event ofp73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblasticleukemia cell apoptosis
Abdurazzag Abusnina a,1, Mahmoud Alhosin a,1, Thérèse Keravis a, Christian D. Muller b, Guy Fuhrmann a,Christian Bronner a,2, Claire Lugnier a,⁎,2
a CNRS UMR 7213 Laboratoire de Biophotonique et Pharmacologie, Université de Strasbourg, Faculté de Pharmacie, 74 route du Rhin, 67401 Illkirch, Franceb CNRS UMR 7200 Laboratoire d'Innovation Thérapeutique, Université de Strasbourg, Faculté de Pharmacie, 74 route du Rhin, 67401 Illkirch, France
⁎ Corresponding author. Université de Strasbourg, CNBiophotonique et Pharmacologie, Faculté de Pharmacie67401 Illkirch, France. Tel.: +33 3 68 85 42 64; fax: +3
E-mail address: [email protected] (C. Lugnier)1 Equal first authors.2 Equal senior contributors.
0898-6568/$ – see front matter © 2010 Elsevier Inc. Aldoi:10.1016/j.cellsig.2010.08.015
a b s t r a c t
a r t i c l e i n f oArticle history:Received 4 May 2010Received in revised form 24 August 2010Accepted 24 August 2010Available online 31 August 2010
Keywords:Acute lymphoblastic leukemiaJurkat cellCyclic nucleotide phosphodiesterasePDE1Tumor suppressor p73Epigenetic integrator UHRF1
Thymoquinone (TQ), the active principle of Nigella sativa black seeds, has anti-proliferative properties onnumerous cancer cell types. Others and we have previously reported that TQ acts as agent that triggers cellcycle arrest and apoptosis through either a p53- or p73-dependent pathway. However, the immediatetargets recruited upon TQ-induced cytotoxicity have not yet been clearly identified. We therefore askedwhether cyclic nucleotide phosphodiesterases (PDEs) could be involved in TQ-triggered pro-apoptoticreactivity; PDEs are regulators of intracellular levels of cyclic nucleotides and therefore can modulate cAMPand cGMP-dependent cell death pathways. Our results showed that TQ specifically repressed PDE1Aexpression in the acute lymphoblastic leukemia Jurkat cell line. This effect is concomitant with the previouslydescribed sequential deregulation of the expression of the tumor suppressor protein p73 and the epigeneticintegrator UHRF1 (Ubiquitin-like, PHD Ring Finger 1). Interestingly, RNA-interference knock-down of PDE1Aexpression as well as decreased PDE1A expression induced growth inhibition of Jurkat cells, cell cycle arrestand apoptosis through an activation of p73 and a repression of UHRF1. Conversely, PDE1A re-expressioncounteracted the cellular pro-apoptotic effects of TQ in association with a p73 repression and UHRF1 re-expression. Altogether, our results show that TQ induced an initial down-regulation of PDE1A with asubsequent down-regulation of UHRF1 via a p73-dependent mechanism. This study further proposes thatPDE1A might be involved in the epigenetic code inheritance by regulating, via p73, the epigenetic integratorUHRF1. Our findings also suggest that a forced inhibition of PDE1A expression might be a new therapeuticstrategy for the management of acute lymphoblastic leukemia.
RS UMR 7213, Laboratoire de, 74 route du Rhin, B.P. 60024,3 3 68 85 43 13..
l rights reserved.
© 2010 Elsevier Inc. All rights reserved.
1. Introduction
Thymoquinone (TQ), the most abundant constituent of the seed oilextract of Nigella sativa, has been shown to have cytoprotective effectsthat aremainlymediated through its anti-oxidant and anti-inflammatoryproperties [1]. Conversely, TQ has also an anti-neoplastic activity ondifferent types of cancer [2–4]. It has been recurrently reported that TQacts as a DNA damage agent and exerts its anti-cancer effects byinhibiting cell growth, arresting cell cycle progression fromG1 to S phasesand triggering apoptosis [3,5–9]. By using the acute lymphoblasticleukemia Jurkat cell model, we have recently demonstrated that TQ-
induced cell cycle arrest and apoptosis are mediated through a p73activation, which targets UHRF1 (Ubiquitin-like, PHD Ring Finger 1) [10].
UHRF1 is a multi-domain protein that serves as a fidelity factor forthe maintenance of the DNA methylation pattern throughout cellduplication [11–16]. The Set and Ring Associated domain (SRAdomain) of UHRF1 has the unique feature to recognize a particularstate of DNA, i.e. hemi-methylated. The SRA–DNA interaction mayserve as an anchor to keep UHRF1 at a hemi-methylated CpG sitewhere it recruits the DNA methyltransferase 1 (DNMT1) for DNAmethylation maintenance [13–16]. Thus, UHRF1 plays a fundamentalrole in the inheritance of the DNA epigenetic marks from the mothercell to the daughter cells. It also appears that preventing thetransmission of these marks via knock-down of UHRF1, leads to anactivation of pro-apoptotic pathways [16,17].
The initial molecular mechanisms which induce the previouslyreported up-regulation of the tumor suppressor p73 and subsequentdown-regulation of the epigenetic integrator UHRF1 in TQ-treatedJurkat cells [10], have so far not yet been elucidated. The involved

153A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
pathway could likely be common to various pro-oxidant DNA-damaging agents including polyphenolic compounds [18,19]. Inter-estingly, polyphenolic compounds are able to inhibit PDEs [20–22].One potential early target of TQ could be one of the eleven members(PDE1 to PDE11) of PDE super-family. PDEs are ubiquitouslydistributed in the different mammalian cell types and their specificcontribution to tissue function in physiological or physiopathologicalconditions designate them as new therapeutic targets [23]. PDEs playa major role in cell signaling by hydrolyzing cAMP and cGMP. Thesecyclic nucleotides are pivotal molecules for achieving crosstalkadaptation between different signaling pathways, i.e. those involvedin cell death processes [24,25]. Cyclic AMP has been shown to regulatecellular proliferation in that it produces a cytostatic effect in early G0to G1 transition and mid-G1 phase in many cell types [26,27]. Forseveral years, it has also been shown that PDE1 inhibition inducesapoptosis in human leukemic cells [28]. Moreover, PDE4 inhibitordoes not allow cells to progress to S phase, suggesting thatphosphodiesterases play a key role in the G1/S transition [29]. Morerecently, it has been observed that IC86340, a PDE1A inhibitor, andPDE1A shRNA caused a dose-dependent increase in the amount ofvascular smooth muscle cells in G1 and a decrease in cells in the S andG2 phases [30]. Furthermore, inhibition of the activity of PDE1A byIC86340 or reducing nuclear PDE1A expression by shRNA significantlyincreased the number of apoptotic cells and upregulated tumorsuppressor [30]. These results suggest that PDEs, notably PDE1A orPDE4 have anti-apoptotic activity by promoting the entry into the Sphase. Strikingly, UHRF1 exhibits similar behavior [11], supportingthe idea that searching for a putative relationship between phospho-diesterases and p73/UHRF1 would be highly interesting.
This study was therefore aimed to: i) characterize PDE isozymeactivity and expression in Jurkat cells that are considered as an in vitroacute lymphoblastic leukemia model; ii) investigate the modulatoreffects of TQ on PDE isozyme activity and expression; and iii) evaluatethe incidence of their modifications on p73/UHRF1 expression status.Altogether, our findings demonstrate that the pro-apoptotic activityof TQ in Jurkat cells is linked to a repression of PDE1 activity andexpression that trigger a sequential deregulation of p73 and UHRF1.
2. Material and methods
2.1. Drugs and chemicals
Nimodipine was a generous gift from Bayer (Berlin, Germany).Rolipram was synthesized by Dr. J.J. Bourguignon as described [31].Cilostamide was synthesized as described elsewhere [32]. 1, 3dimethyl-6-(2-propoxy-5-methane sulphonylamidophenyl)-pyrazolo[3,4-d]pyrimidin-4-(5H)-one (DMPPO) was a kind gift of Dr P. Grondin[33]. Propidium iodide, RNase solution and thymoquinone (TQ) werefrom Sigma-Aldrich (St. Louis, MO, USA). The cocktail, proteaseinhibitor cocktail (set III # 539134), was from Calbiochem. Antibodiesused in this study were: polyclonal PDE1A, PDE1B, PDE1C, PDE4A,PDE4B, PDE4C and PDE4D antibodies (FabGennix Inc., Frisco, TX);polyclonal PDE5 antibody (Cell Signaling), monoclonal UHRF1 antibody[34];monoclonal p73 antibody (BD Biosciences Pharmingen, San Diego,CA); monoclonal β-actin and β-tubulin antibodies (Sigma-Aldrich);monoclonal GAPDH antibody (Chemicon International, Temecula, CA).Polyclonal PDE3A (424–460 aa as immunogen) and PDE3B (2–20 aa asimmunogen) antibodies were a generous gift of Dr. V.C. Manganiello(NIH, Bethesda, MA).
2.2. Cell culture and treatment
Human acute lymphoblastic leukemia (ALL) p53-mutated cells,Jurkat cells (clone E6-1) were cultured as previously described [10].TQ was prepared as a 100 mM stock solution in 100% dimethylsulf-oxide (DMSO). Appropriate working concentrations were prepared
with the cell culture medium. The final concentration of DMSO was0.1% in both control and treatment conditions.
2.3. Cell apoptosis and proliferation assays
Cells were seeded in 96-well plates at a density of 1.5×105cells/mland grown for 24 h. Cells were then treated with PDE inhibitors and orTQ at different concentrations and allowed to proliferate for a further24 h period. PDE inhibitors were dissolved in DMSO and the finalconcentration of DMSOwas 0.1% throughout the assays. Cell apoptosisrate was determined by capillary cytometry (Guava EasyCyte plus HPsystem) using the Annexin V Binding Guava Nexin® Assay, followingthe manufacturer's recommendations (Guava Technologies Inc,Hayward, CA, USA). Cell proliferation rate was determined by acolorimetric assay using the “CellTiter 96 Aqueous One Solution CellProliferation Assay” (Promega, Charbonnières-les-bains, France).
2.4. Cell cycle analysis
Jurkat cells were prepared as described elsewhere [10]. Briefly,cells were seeded in 6-well plates at a density of 1.5×105cells/ml andgrown for 24 h, then exposed to different drugs for a further 24 hperiod. Cells were then harvested by centrifugation (200×g for 5 minat room temperature), washed with phosphate-buffered saline (PBS)solution and resuspended in 100% methanol. After 1 h incubation at4 °C, cells were centrifuged at 200×g for 10 min at 4 °C and washedoncewith PBS buffer. Cells were then resuspended in 500 μl PBS buffercontaining 50 μg/ml propidium iodide and 50 μg/ml RNase solutionand were kept in dark for 30 min at 37 °C. Cellular DNA content wasassessed by capillary cytometry (Guava EasyCyte Plus HP system,Guava Technologies Inc.).
2.5. Transient transfection
Cells were seeded in 24-well plates at a density of 4×105 cells perwell. For plasmid transfections, cells were transfected using theQiagen Expression Kit/Mammalia (Qiagen # 39003, Courtaboeuf,France) with either 1 μg of HS_PDE1A_IM_1 plasmid or 1 μg ofHS_CDC2_IM_1 plasmid (control vector). Untransfected cells werealso used as additional control. Cells were allowed to grow for 24 hand were then treated for 24 h with TQ (20 μM) or vehicle (0.1%DMSO). siRNA transfections were performed using lipofectamine(Invitrogen, Carlsbad, CA); cells were transfected with either 100 nMPDE1A siRNA (Qiagen #SI03057187) or 100 nM scramble siRNA(Sigma-Aldrich) and allowed to grow for 48 h. The target sequence ofthe siRNA for PDE1A was 5′-CACCACGTGAGTGCAGCTTAT-3′. Thesequence of the scramble siRNA was 5′-GGACUCUCGGAUUGUAA-GAdTdT-3′.
2.6. cAMP and cGMP assays
Cells were seeded in 75 cm2 culture flasks at a density of1.5×105cells/ml. Cells were treated for 24 h with TQ (10 and20 μM) or vehicle (0.1% DMSO). Cells were then collected, washed 3times with PBS and intracellular cAMP and cGMP levels weredetermined by a competitive enzyme immunoassay (EIA) systemfollowing manufacturer's recommendations (Assay Designs Inc., AnnArbor, MI).
2.7. Cell extracts
Cells were seeded in 75 cm2 culture flasks at a density of1.5×105cells/ml, then treated for 24 h. Cells were centrifuged at200×g for 5 min at room temperature. Cell pellets were then washedwith PBS and resuspended in the homogenizing buffer (20 mM Tris,5 mM EGTA, 150 mM NaCl, 20 mM Na glycerophosphate, 1 μM H-89,
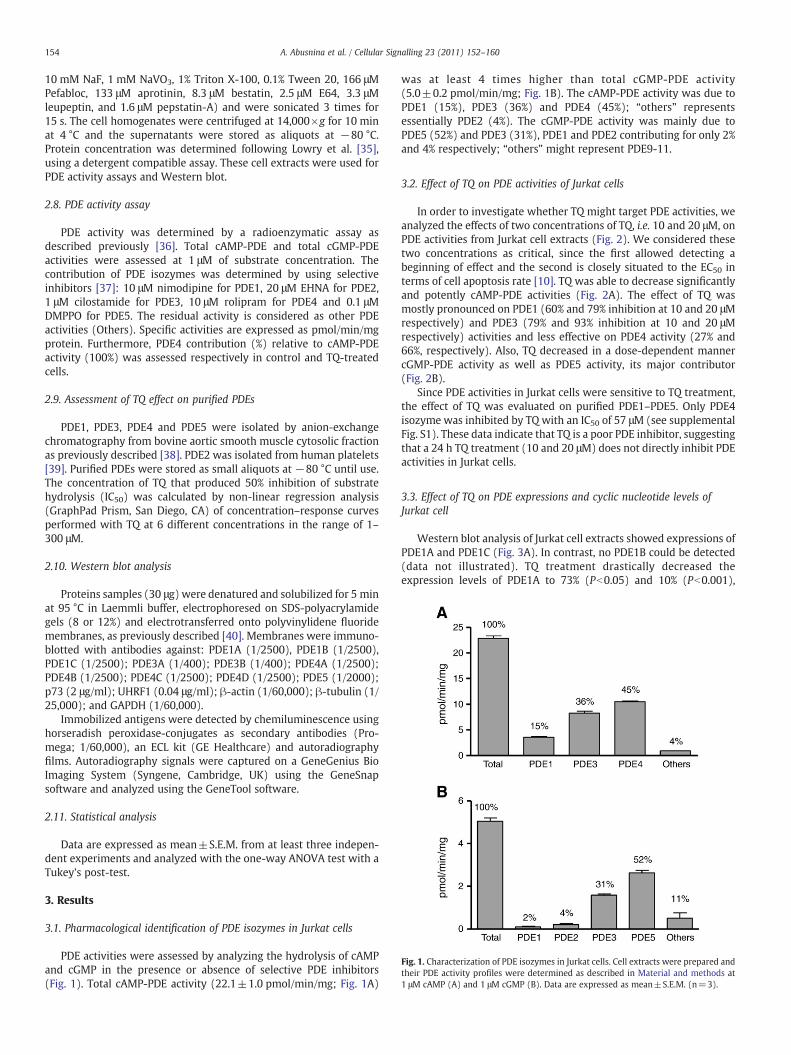
Fig. 1. Characterization of PDE isozymes in Jurkat cells. Cell extracts were prepared andtheir PDE activity profiles were determined as described in Material and methods at1 μM cAMP (A) and 1 μM cGMP (B). Data are expressed as mean±S.E.M. (n=3).
154 A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
10 mM NaF, 1 mM NaVO3, 1% Triton X-100, 0.1% Tween 20, 166 μMPefabloc, 133 μM aprotinin, 8.3 μM bestatin, 2.5 μM E64, 3.3 μMleupeptin, and 1.6 μM pepstatin-A) and were sonicated 3 times for15 s. The cell homogenates were centrifuged at 14,000×g for 10 minat 4 °C and the supernatants were stored as aliquots at −80 °C.Protein concentration was determined following Lowry et al. [35],using a detergent compatible assay. These cell extracts were used forPDE activity assays and Western blot.
2.8. PDE activity assay
PDE activity was determined by a radioenzymatic assay asdescribed previously [36]. Total cAMP-PDE and total cGMP-PDEactivities were assessed at 1 μM of substrate concentration. Thecontribution of PDE isozymes was determined by using selectiveinhibitors [37]: 10 μM nimodipine for PDE1, 20 μM EHNA for PDE2,1 μM cilostamide for PDE3, 10 μM rolipram for PDE4 and 0.1 μMDMPPO for PDE5. The residual activity is considered as other PDEactivities (Others). Specific activities are expressed as pmol/min/mgprotein. Furthermore, PDE4 contribution (%) relative to cAMP-PDEactivity (100%) was assessed respectively in control and TQ-treatedcells.
2.9. Assessment of TQ effect on purified PDEs
PDE1, PDE3, PDE4 and PDE5 were isolated by anion-exchangechromatography from bovine aortic smooth muscle cytosolic fractionas previously described [38]. PDE2 was isolated from human platelets[39]. Purified PDEs were stored as small aliquots at −80 °C until use.The concentration of TQ that produced 50% inhibition of substratehydrolysis (IC50) was calculated by non-linear regression analysis(GraphPad Prism, San Diego, CA) of concentration–response curvesperformed with TQ at 6 different concentrations in the range of 1–300 μM.
2.10. Western blot analysis
Proteins samples (30 μg) were denatured and solubilized for 5 minat 95 °C in Laemmli buffer, electrophoresed on SDS-polyacrylamidegels (8 or 12%) and electrotransferred onto polyvinylidene fluoridemembranes, as previously described [40]. Membranes were immuno-blotted with antibodies against: PDE1A (1/2500), PDE1B (1/2500),PDE1C (1/2500); PDE3A (1/400); PDE3B (1/400); PDE4A (1/2500);PDE4B (1/2500); PDE4C (1/2500); PDE4D (1/2500); PDE5 (1/2000);p73 (2 μg/ml); UHRF1 (0.04 μg/ml); β-actin (1/60,000); β-tubulin (1/25,000); and GAPDH (1/60,000).
Immobilized antigens were detected by chemiluminescence usinghorseradish peroxidase-conjugates as secondary antibodies (Pro-mega; 1/60,000), an ECL kit (GE Healthcare) and autoradiographyfilms. Autoradiography signals were captured on a GeneGenius BioImaging System (Syngene, Cambridge, UK) using the GeneSnapsoftware and analyzed using the GeneTool software.
2.11. Statistical analysis
Data are expressed as mean±S.E.M. from at least three indepen-dent experiments and analyzed with the one-way ANOVA test with aTukey's post-test.
3. Results
3.1. Pharmacological identification of PDE isozymes in Jurkat cells
PDE activities were assessed by analyzing the hydrolysis of cAMPand cGMP in the presence or absence of selective PDE inhibitors(Fig. 1). Total cAMP-PDE activity (22.1±1.0 pmol/min/mg; Fig. 1A)
was at least 4 times higher than total cGMP-PDE activity(5.0±0.2 pmol/min/mg; Fig. 1B). The cAMP-PDE activity was due toPDE1 (15%), PDE3 (36%) and PDE4 (45%); “others” representsessentially PDE2 (4%). The cGMP-PDE activity was mainly due toPDE5 (52%) and PDE3 (31%), PDE1 and PDE2 contributing for only 2%and 4% respectively; “others” might represent PDE9-11.
3.2. Effect of TQ on PDE activities of Jurkat cells
In order to investigate whether TQ might target PDE activities, weanalyzed the effects of two concentrations of TQ, i.e. 10 and 20 μM, onPDE activities from Jurkat cell extracts (Fig. 2). We considered thesetwo concentrations as critical, since the first allowed detecting abeginning of effect and the second is closely situated to the EC50 interms of cell apoptosis rate [10]. TQ was able to decrease significantlyand potently cAMP-PDE activities (Fig. 2A). The effect of TQ wasmostly pronounced on PDE1 (60% and 79% inhibition at 10 and 20 μMrespectively) and PDE3 (79% and 93% inhibition at 10 and 20 μMrespectively) activities and less effective on PDE4 activity (27% and66%, respectively). Also, TQ decreased in a dose-dependent mannercGMP-PDE activity as well as PDE5 activity, its major contributor(Fig. 2B).
Since PDE activities in Jurkat cells were sensitive to TQ treatment,the effect of TQ was evaluated on purified PDE1–PDE5. Only PDE4isozyme was inhibited by TQ with an IC50 of 57 μM (see supplementalFig. S1). These data indicate that TQ is a poor PDE inhibitor, suggestingthat a 24 h TQ treatment (10 and 20 μM) does not directly inhibit PDEactivities in Jurkat cells.
3.3. Effect of TQ on PDE expressions and cyclic nucleotide levels ofJurkat cell
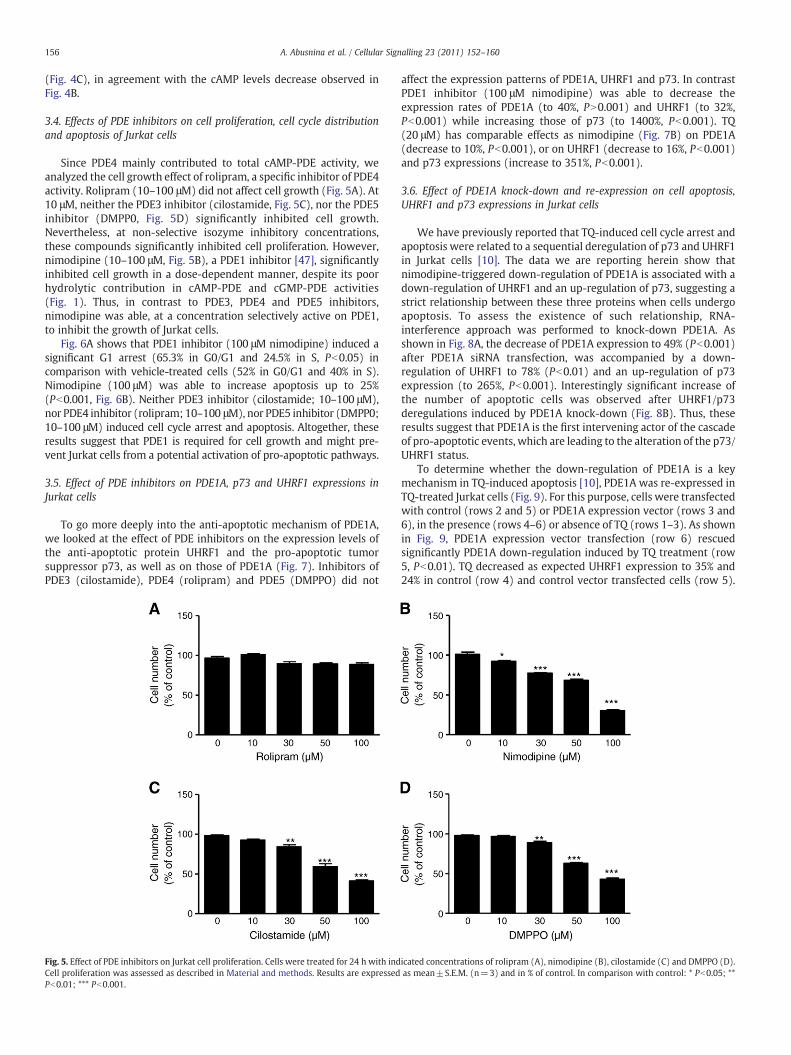
Western blot analysis of Jurkat cell extracts showed expressions ofPDE1A and PDE1C (Fig. 3A). In contrast, no PDE1B could be detected(data not illustrated). TQ treatment drastically decreased theexpression levels of PDE1A to 73% (Pb0.05) and 10% (Pb0.001),
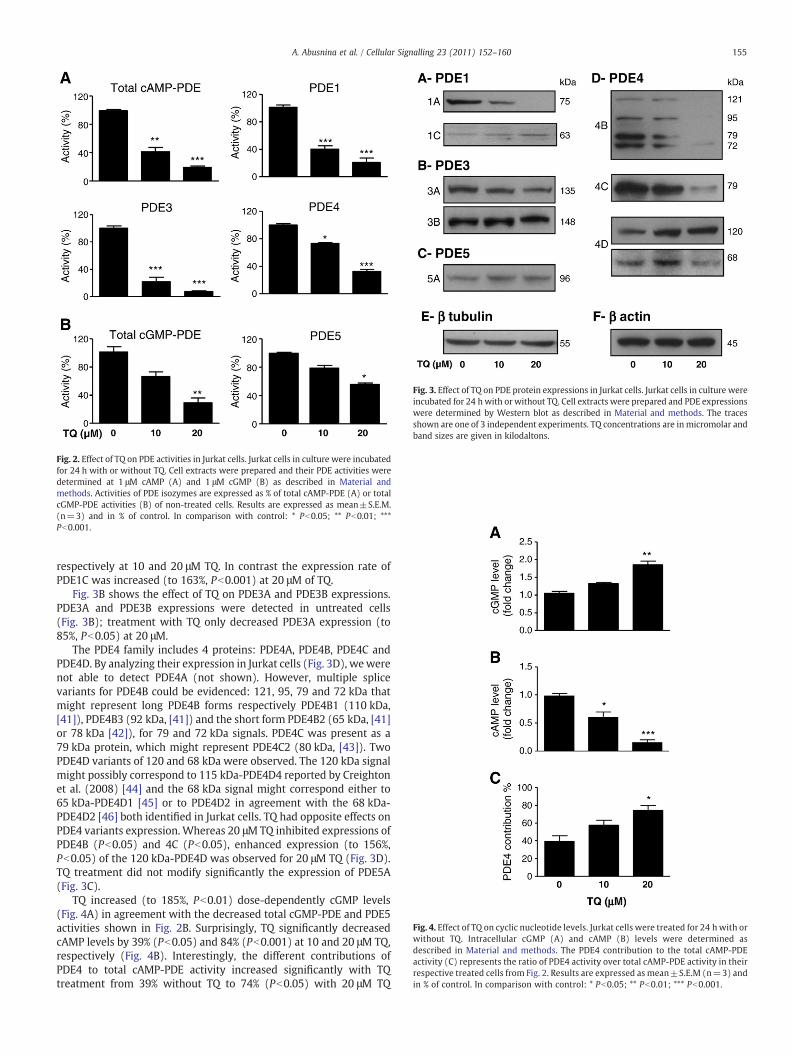
Fig. 2. Effect of TQ on PDE activities in Jurkat cells. Jurkat cells in culture were incubatedfor 24 h with or without TQ. Cell extracts were prepared and their PDE activities weredetermined at 1 μM cAMP (A) and 1 μM cGMP (B) as described in Material andmethods. Activities of PDE isozymes are expressed as % of total cAMP-PDE (A) or totalcGMP-PDE activities (B) of non-treated cells. Results are expressed as mean±S.E.M.(n=3) and in % of control. In comparison with control: * Pb0.05; ** Pb0.01; ***Pb0.001.
Fig. 3. Effect of TQ on PDE protein expressions in Jurkat cells. Jurkat cells in culture wereincubated for 24 hwith or without TQ. Cell extracts were prepared and PDE expressionswere determined by Western blot as described in Material and methods. The tracesshown are one of 3 independent experiments. TQ concentrations are in micromolar andband sizes are given in kilodaltons.
Fig. 4. Effect of TQ on cyclic nucleotide levels. Jurkat cells were treated for 24 hwith orwithout TQ. Intracellular cGMP (A) and cAMP (B) levels were determined asdescribed in Material and methods. The PDE4 contribution to the total cAMP-PDEactivity (C) represents the ratio of PDE4 activity over total cAMP-PDE activity in theirrespective treated cells from Fig. 2. Results are expressed as mean±S.E.M (n=3) andin % of control. In comparison with control: * Pb0.05; ** Pb0.01; *** Pb0.001.
155A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
respectively at 10 and 20 μM TQ. In contrast the expression rate ofPDE1C was increased (to 163%, Pb0.001) at 20 μM of TQ.
Fig. 3B shows the effect of TQ on PDE3A and PDE3B expressions.PDE3A and PDE3B expressions were detected in untreated cells(Fig. 3B); treatment with TQ only decreased PDE3A expression (to85%, Pb0.05) at 20 μM.
The PDE4 family includes 4 proteins: PDE4A, PDE4B, PDE4C andPDE4D. By analyzing their expression in Jurkat cells (Fig. 3D), wewerenot able to detect PDE4A (not shown). However, multiple splicevariants for PDE4B could be evidenced: 121, 95, 79 and 72 kDa thatmight represent long PDE4B forms respectively PDE4B1 (110 kDa,[41]), PDE4B3 (92 kDa, [41]) and the short form PDE4B2 (65 kDa, [41]or 78 kDa [42]), for 79 and 72 kDa signals. PDE4C was present as a79 kDa protein, which might represent PDE4C2 (80 kDa, [43]). TwoPDE4D variants of 120 and 68 kDa were observed. The 120 kDa signalmight possibly correspond to 115 kDa-PDE4D4 reported by Creightonet al. (2008) [44] and the 68 kDa signal might correspond either to65 kDa-PDE4D1 [45] or to PDE4D2 in agreement with the 68 kDa-PDE4D2 [46] both identified in Jurkat cells. TQ had opposite effects onPDE4 variants expression. Whereas 20 μM TQ inhibited expressions ofPDE4B (Pb0.05) and 4C (Pb0.05), enhanced expression (to 156%,Pb0.05) of the 120 kDa-PDE4D was observed for 20 μM TQ (Fig. 3D).TQ treatment did not modify significantly the expression of PDE5A(Fig. 3C).
TQ increased (to 185%, Pb0.01) dose-dependently cGMP levels(Fig. 4A) in agreement with the decreased total cGMP-PDE and PDE5activities shown in Fig. 2B. Surprisingly, TQ significantly decreasedcAMP levels by 39% (Pb0.05) and 84% (Pb0.001) at 10 and 20 μM TQ,respectively (Fig. 4B). Interestingly, the different contributions ofPDE4 to total cAMP-PDE activity increased significantly with TQtreatment from 39% without TQ to 74% (Pb0.05) with 20 μM TQ
156 A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
(Fig. 4C), in agreement with the cAMP levels decrease observed inFig. 4B.
3.4. Effects of PDE inhibitors on cell proliferation, cell cycle distributionand apoptosis of Jurkat cells
Since PDE4 mainly contributed to total cAMP-PDE activity, weanalyzed the cell growth effect of rolipram, a specific inhibitor of PDE4activity. Rolipram (10–100 μM) did not affect cell growth (Fig. 5A). At10 μM, neither the PDE3 inhibitor (cilostamide, Fig. 5C), nor the PDE5inhibitor (DMPP0, Fig. 5D) significantly inhibited cell growth.Nevertheless, at non-selective isozyme inhibitory concentrations,these compounds significantly inhibited cell proliferation. However,nimodipine (10–100 μM, Fig. 5B), a PDE1 inhibitor [47], significantlyinhibited cell growth in a dose-dependent manner, despite its poorhydrolytic contribution in cAMP-PDE and cGMP-PDE activities(Fig. 1). Thus, in contrast to PDE3, PDE4 and PDE5 inhibitors,nimodipine was able, at a concentration selectively active on PDE1,to inhibit the growth of Jurkat cells.
Fig. 6A shows that PDE1 inhibitor (100 μM nimodipine) induced asignificant G1 arrest (65.3% in G0/G1 and 24.5% in S, Pb0.05) incomparison with vehicle-treated cells (52% in G0/G1 and 40% in S).Nimodipine (100 μM) was able to increase apoptosis up to 25%(Pb0.001, Fig. 6B). Neither PDE3 inhibitor (cilostamide; 10–100 μM),nor PDE4 inhibitor (rolipram; 10–100 μM), nor PDE5 inhibitor (DMPP0;10–100 μM) induced cell cycle arrest and apoptosis. Altogether, theseresults suggest that PDE1 is required for cell growth and might pre-vent Jurkat cells from a potential activation of pro-apoptotic pathways.
3.5. Effect of PDE inhibitors on PDE1A, p73 and UHRF1 expressions inJurkat cells
To go more deeply into the anti-apoptotic mechanism of PDE1A,we looked at the effect of PDE inhibitors on the expression levels ofthe anti-apoptotic protein UHRF1 and the pro-apoptotic tumorsuppressor p73, as well as on those of PDE1A (Fig. 7). Inhibitors ofPDE3 (cilostamide), PDE4 (rolipram) and PDE5 (DMPPO) did not
Fig. 5. Effect of PDE inhibitors on Jurkat cell proliferation. Cells were treated for 24 h with indCell proliferation was assessed as described in Material and methods. Results are expressedPb0.01; *** Pb0.001.
affect the expression patterns of PDE1A, UHRF1 and p73. In contrastPDE1 inhibitor (100 μM nimodipine) was able to decrease theexpression rates of PDE1A (to 40%, PN0.001) and UHRF1 (to 32%,Pb0.001) while increasing those of p73 (to 1400%, Pb0.001). TQ(20 μM) has comparable effects as nimodipine (Fig. 7B) on PDE1A(decrease to 10%, Pb0.001), or on UHRF1 (decrease to 16%, Pb0.001)and p73 expressions (increase to 351%, Pb0.001).
3.6. Effect of PDE1A knock-down and re-expression on cell apoptosis,UHRF1 and p73 expressions in Jurkat cells
We have previously reported that TQ-induced cell cycle arrest andapoptosis were related to a sequential deregulation of p73 and UHRF1in Jurkat cells [10]. The data we are reporting herein show thatnimodipine-triggered down-regulation of PDE1A is associated with adown-regulation of UHRF1 and an up-regulation of p73, suggesting astrict relationship between these three proteins when cells undergoapoptosis. To assess the existence of such relationship, RNA-interference approach was performed to knock-down PDE1A. Asshown in Fig. 8A, the decrease of PDE1A expression to 49% (Pb0.001)after PDE1A siRNA transfection, was accompanied by a down-regulation of UHRF1 to 78% (Pb0.01) and an up-regulation of p73expression (to 265%, Pb0.001). Interestingly significant increase ofthe number of apoptotic cells was observed after UHRF1/p73deregulations induced by PDE1A knock-down (Fig. 8B). Thus, theseresults suggest that PDE1A is the first intervening actor of the cascadeof pro-apoptotic events, which are leading to the alteration of the p73/UHRF1 status.
To determine whether the down-regulation of PDE1A is a keymechanism in TQ-induced apoptosis [10], PDE1A was re-expressed inTQ-treated Jurkat cells (Fig. 9). For this purpose, cells were transfectedwith control (rows 2 and 5) or PDE1A expression vector (rows 3 and6), in the presence (rows 4–6) or absence of TQ (rows 1–3). As shownin Fig. 9, PDE1A expression vector transfection (row 6) rescuedsignificantly PDE1A down-regulation induced by TQ treatment (row5, Pb0.01). TQ decreased as expected UHRF1 expression to 35% and24% in control (row 4) and control vector transfected cells (row 5).
icated concentrations of rolipram (A), nimodipine (B), cilostamide (C) and DMPPO (D).as mean±S.E.M. (n=3) and in % of control. In comparison with control: * Pb0.05; **
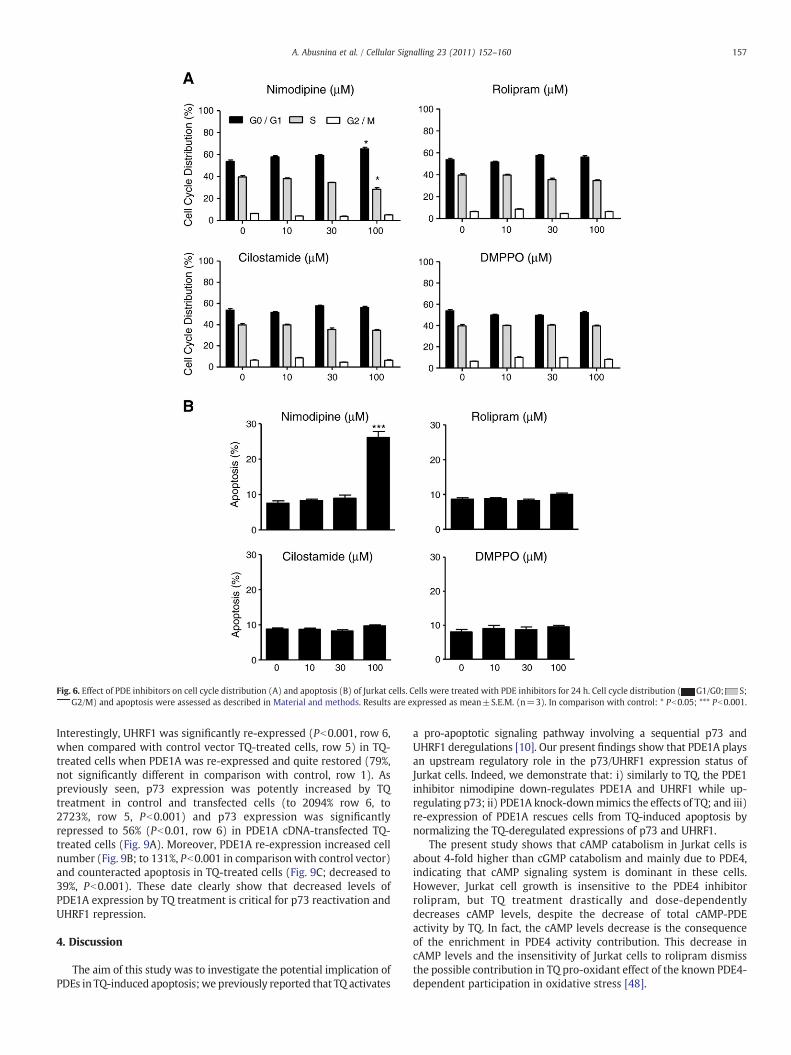
Fig. 6. Effect of PDE inhibitors on cell cycle distribution (A) and apoptosis (B) of Jurkat cells. Cells were treated with PDE inhibitors for 24 h. Cell cycle distribution ( G1/G0; S;G2/M) and apoptosis were assessed as described in Material and methods. Results are expressed as mean±S.E.M. (n=3). In comparison with control: * Pb0.05; *** Pb0.001.
157A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
Interestingly, UHRF1 was significantly re-expressed (Pb0.001, row 6,when compared with control vector TQ-treated cells, row 5) in TQ-treated cells when PDE1A was re-expressed and quite restored (79%,not significantly different in comparison with control, row 1). Aspreviously seen, p73 expression was potently increased by TQtreatment in control and transfected cells (to 2094% row 6, to2723%, row 5, Pb0.001) and p73 expression was significantlyrepressed to 56% (Pb0.01, row 6) in PDE1A cDNA-transfected TQ-treated cells (Fig. 9A). Moreover, PDE1A re-expression increased cellnumber (Fig. 9B; to 131%, Pb0.001 in comparison with control vector)and counteracted apoptosis in TQ-treated cells (Fig. 9C; decreased to39%, Pb0.001). These date clearly show that decreased levels ofPDE1A expression by TQ treatment is critical for p73 reactivation andUHRF1 repression.
4. Discussion
The aim of this study was to investigate the potential implication ofPDEs in TQ-induced apoptosis;we previously reported that TQ activates
a pro-apoptotic signaling pathway involving a sequential p73 andUHRF1 deregulations [10]. Our present findings show that PDE1A playsan upstream regulatory role in the p73/UHRF1 expression status ofJurkat cells. Indeed, we demonstrate that: i) similarly to TQ, the PDE1inhibitor nimodipine down-regulates PDE1A and UHRF1 while up-regulating p73; ii) PDE1A knock-downmimics the effects of TQ; and iii)re-expression of PDE1A rescues cells from TQ-induced apoptosis bynormalizing the TQ-deregulated expressions of p73 and UHRF1.
The present study shows that cAMP catabolism in Jurkat cells isabout 4-fold higher than cGMP catabolism and mainly due to PDE4,indicating that cAMP signaling system is dominant in these cells.However, Jurkat cell growth is insensitive to the PDE4 inhibitorrolipram, but TQ treatment drastically and dose-dependentlydecreases cAMP levels, despite the decrease of total cAMP-PDEactivity by TQ. In fact, the cAMP levels decrease is the consequenceof the enrichment in PDE4 activity contribution. This decrease incAMP levels and the insensitivity of Jurkat cells to rolipram dismissthe possible contribution in TQ pro-oxidant effect of the known PDE4-dependent participation in oxidative stress [48].
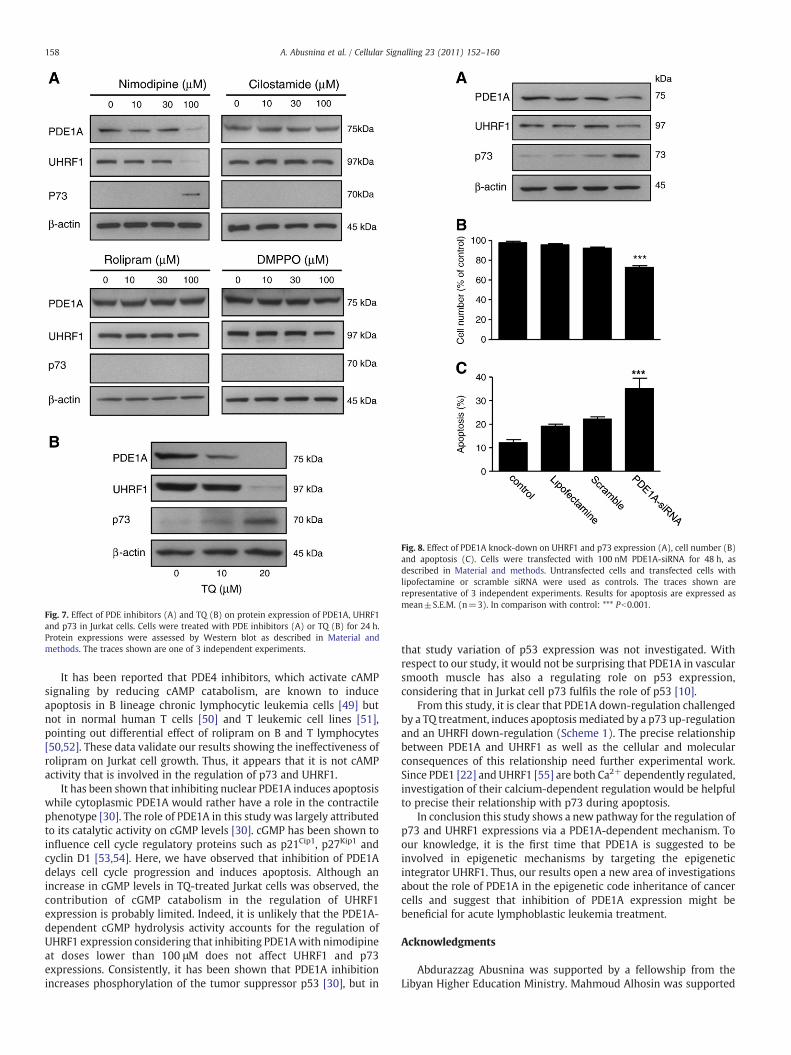
Fig. 7. Effect of PDE inhibitors (A) and TQ (B) on protein expression of PDE1A, UHRF1and p73 in Jurkat cells. Cells were treated with PDE inhibitors (A) or TQ (B) for 24 h.Protein expressions were assessed by Western blot as described in Material andmethods. The traces shown are one of 3 independent experiments.
Fig. 8. Effect of PDE1A knock-down on UHRF1 and p73 expression (A), cell number (B)and apoptosis (C). Cells were transfected with 100 nM PDE1A-siRNA for 48 h, asdescribed in Material and methods. Untransfected cells and transfected cells withlipofectamine or scramble siRNA were used as controls. The traces shown arerepresentative of 3 independent experiments. Results for apoptosis are expressed asmean±S.E.M. (n=3). In comparison with control: *** Pb0.001.
158 A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
It has been reported that PDE4 inhibitors, which activate cAMPsignaling by reducing cAMP catabolism, are known to induceapoptosis in B lineage chronic lymphocytic leukemia cells [49] butnot in normal human T cells [50] and T leukemic cell lines [51],pointing out differential effect of rolipram on B and T lymphocytes[50,52]. These data validate our results showing the ineffectiveness ofrolipram on Jurkat cell growth. Thus, it appears that it is not cAMPactivity that is involved in the regulation of p73 and UHRF1.
It has been shown that inhibiting nuclear PDE1A induces apoptosiswhile cytoplasmic PDE1A would rather have a role in the contractilephenotype [30]. The role of PDE1A in this study was largely attributedto its catalytic activity on cGMP levels [30]. cGMP has been shown toinfluence cell cycle regulatory proteins such as p21Cip1, p27Kip1 andcyclin D1 [53,54]. Here, we have observed that inhibition of PDE1Adelays cell cycle progression and induces apoptosis. Although anincrease in cGMP levels in TQ-treated Jurkat cells was observed, thecontribution of cGMP catabolism in the regulation of UHRF1expression is probably limited. Indeed, it is unlikely that the PDE1A-dependent cGMP hydrolysis activity accounts for the regulation ofUHRF1 expression considering that inhibiting PDE1Awith nimodipineat doses lower than 100 μM does not affect UHRF1 and p73expressions. Consistently, it has been shown that PDE1A inhibitionincreases phosphorylation of the tumor suppressor p53 [30], but in
that study variation of p53 expression was not investigated. Withrespect to our study, it would not be surprising that PDE1A in vascularsmooth muscle has also a regulating role on p53 expression,considering that in Jurkat cell p73 fulfils the role of p53 [10].
From this study, it is clear that PDE1A down-regulation challengedby a TQ treatment, induces apoptosis mediated by a p73 up-regulationand an UHRFI down-regulation (Scheme 1). The precise relationshipbetween PDE1A and UHRF1 as well as the cellular and molecularconsequences of this relationship need further experimental work.Since PDE1 [22] and UHRF1 [55] are both Ca2+ dependently regulated,investigation of their calcium-dependent regulation would be helpfulto precise their relationship with p73 during apoptosis.
In conclusion this study shows a new pathway for the regulation ofp73 and UHRF1 expressions via a PDE1A-dependent mechanism. Toour knowledge, it is the first time that PDE1A is suggested to beinvolved in epigenetic mechanisms by targeting the epigeneticintegrator UHRF1. Thus, our results open a new area of investigationsabout the role of PDE1A in the epigenetic code inheritance of cancercells and suggest that inhibition of PDE1A expression might bebeneficial for acute lymphoblastic leukemia treatment.
Acknowledgments
Abdurazzag Abusnina was supported by a fellowship from theLibyan Higher Education Ministry. Mahmoud Alhosin was supported
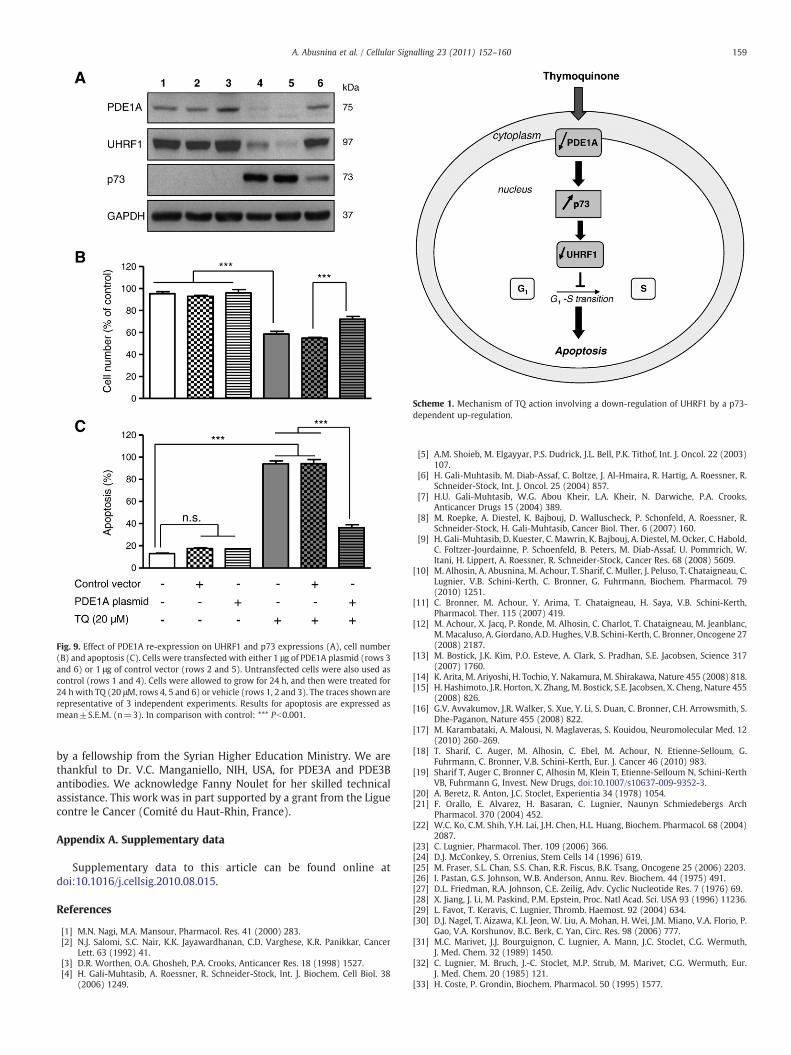
Fig. 9. Effect of PDE1A re-expression on UHRF1 and p73 expressions (A), cell number(B) and apoptosis (C). Cells were transfected with either 1 μg of PDE1A plasmid (rows 3and 6) or 1 μg of control vector (rows 2 and 5). Untransfected cells were also used ascontrol (rows 1 and 4). Cells were allowed to grow for 24 h, and then were treated for24 hwith TQ (20 μM, rows 4, 5 and 6) or vehicle (rows 1, 2 and 3). The traces shown arerepresentative of 3 independent experiments. Results for apoptosis are expressed asmean±S.E.M. (n=3). In comparison with control: *** Pb0.001.
Scheme 1. Mechanism of TQ action involving a down-regulation of UHRF1 by a p73-dependent up-regulation.
159A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
by a fellowship from the Syrian Higher Education Ministry. We arethankful to Dr. V.C. Manganiello, NIH, USA, for PDE3A and PDE3Bantibodies. We acknowledge Fanny Noulet for her skilled technicalassistance. This work was in part supported by a grant from the Liguecontre le Cancer (Comité du Haut-Rhin, France).
Appendix A. Supplementary data
Supplementary data to this article can be found online atdoi:10.1016/j.cellsig.2010.08.015.
References
[1] M.N. Nagi, M.A. Mansour, Pharmacol. Res. 41 (2000) 283.[2] N.J. Salomi, S.C. Nair, K.K. Jayawardhanan, C.D. Varghese, K.R. Panikkar, Cancer
Lett. 63 (1992) 41.[3] D.R. Worthen, O.A. Ghosheh, P.A. Crooks, Anticancer Res. 18 (1998) 1527.[4] H. Gali-Muhtasib, A. Roessner, R. Schneider-Stock, Int. J. Biochem. Cell Biol. 38
(2006) 1249.
[5] A.M. Shoieb, M. Elgayyar, P.S. Dudrick, J.L. Bell, P.K. Tithof, Int. J. Oncol. 22 (2003)107.
[6] H. Gali-Muhtasib, M. Diab-Assaf, C. Boltze, J. Al-Hmaira, R. Hartig, A. Roessner, R.Schneider-Stock, Int. J. Oncol. 25 (2004) 857.
[7] H.U. Gali-Muhtasib, W.G. Abou Kheir, L.A. Kheir, N. Darwiche, P.A. Crooks,Anticancer Drugs 15 (2004) 389.
[8] M. Roepke, A. Diestel, K. Bajbouj, D. Walluscheck, P. Schonfeld, A. Roessner, R.Schneider-Stock, H. Gali-Muhtasib, Cancer Biol. Ther. 6 (2007) 160.
[9] H. Gali-Muhtasib, D. Kuester, C. Mawrin, K. Bajbouj, A. Diestel, M. Ocker, C. Habold,C. Foltzer-Jourdainne, P. Schoenfeld, B. Peters, M. Diab-Assaf, U. Pommrich, W.Itani, H. Lippert, A. Roessner, R. Schneider-Stock, Cancer Res. 68 (2008) 5609.
[10] M. Alhosin, A. Abusnina, M. Achour, T. Sharif, C. Muller, J. Peluso, T. Chataigneau, C.Lugnier, V.B. Schini-Kerth, C. Bronner, G. Fuhrmann, Biochem. Pharmacol. 79(2010) 1251.
[11] C. Bronner, M. Achour, Y. Arima, T. Chataigneau, H. Saya, V.B. Schini-Kerth,Pharmacol. Ther. 115 (2007) 419.
[12] M. Achour, X. Jacq, P. Ronde, M. Alhosin, C. Charlot, T. Chataigneau, M. Jeanblanc,M.Macaluso, A. Giordano, A.D. Hughes, V.B. Schini-Kerth, C. Bronner, Oncogene 27(2008) 2187.
[13] M. Bostick, J.K. Kim, P.O. Esteve, A. Clark, S. Pradhan, S.E. Jacobsen, Science 317(2007) 1760.
[14] K. Arita, M. Ariyoshi, H. Tochio, Y. Nakamura, M. Shirakawa, Nature 455 (2008) 818.[15] H. Hashimoto, J.R. Horton, X. Zhang, M. Bostick, S.E. Jacobsen, X. Cheng, Nature 455
(2008) 826.[16] G.V. Avvakumov, J.R. Walker, S. Xue, Y. Li, S. Duan, C. Bronner, C.H. Arrowsmith, S.
Dhe-Paganon, Nature 455 (2008) 822.[17] M. Karambataki, A. Malousi, N. Maglaveras, S. Kouidou, Neuromolecular Med. 12
(2010) 260–269.[18] T. Sharif, C. Auger, M. Alhosin, C. Ebel, M. Achour, N. Etienne-Selloum, G.
Fuhrmann, C. Bronner, V.B. Schini-Kerth, Eur. J. Cancer 46 (2010) 983.[19] Sharif T, Auger C, Bronner C, Alhosin M, Klein T, Etienne-Selloum N, Schini-Kerth
VB, Fuhrmann G, Invest. New Drugs, doi:10.1007/s10637-009-9352-3.[20] A. Beretz, R. Anton, J.C. Stoclet, Experientia 34 (1978) 1054.[21] F. Orallo, E. Alvarez, H. Basaran, C. Lugnier, Naunyn Schmiedebergs Arch
Pharmacol. 370 (2004) 452.[22] W.C. Ko, C.M. Shih, Y.H. Lai, J.H. Chen, H.L. Huang, Biochem. Pharmacol. 68 (2004)
2087.[23] C. Lugnier, Pharmacol. Ther. 109 (2006) 366.[24] D.J. McConkey, S. Orrenius, Stem Cells 14 (1996) 619.[25] M. Fraser, S.L. Chan, S.S. Chan, R.R. Fiscus, B.K. Tsang, Oncogene 25 (2006) 2203.[26] I. Pastan, G.S. Johnson, W.B. Anderson, Annu. Rev. Biochem. 44 (1975) 491.[27] D.L. Friedman, R.A. Johnson, C.E. Zeilig, Adv. Cyclic Nucleotide Res. 7 (1976) 69.[28] X. Jiang, J. Li, M. Paskind, P.M. Epstein, Proc. Natl Acad. Sci. USA 93 (1996) 11236.[29] L. Favot, T. Keravis, C. Lugnier, Thromb. Haemost. 92 (2004) 634.[30] D.J. Nagel, T. Aizawa, K.I. Jeon, W. Liu, A. Mohan, H. Wei, J.M. Miano, V.A. Florio, P.
Gao, V.A. Korshunov, B.C. Berk, C. Yan, Circ. Res. 98 (2006) 777.[31] M.C. Marivet, J.J. Bourguignon, C. Lugnier, A. Mann, J.C. Stoclet, C.G. Wermuth,
J. Med. Chem. 32 (1989) 1450.[32] C. Lugnier, M. Bruch, J.-C. Stoclet, M.P. Strub, M. Marivet, C.G. Wermuth, Eur.
J. Med. Chem. 20 (1985) 121.[33] H. Coste, P. Grondin, Biochem. Pharmacol. 50 (1995) 1577.

160 A. Abusnina et al. / Cellular Signalling 23 (2011) 152–160
[34] R. Hopfner, M. Mousli, J.M. Jeltsch, A. Voulgaris, Y. Lutz, C. Marin, J.P. Bellocq, P.Oudet, C. Bronner, Cancer Res. 60 (2000) 121.
[35] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, J. Biol. Chem. 193 (1951) 265.[36] T. Keravis, R. Thaseldar-Roumie, C. Lugnier, Meth. Mol. Biol. 307 (2005) 63.[37] J.C. Stoclet, T. Keravis, N. Komas, C. Lugnier, Exp. Opin. Investig. Drugs 4 (1995)
1081.[38] C. Lugnier, P. Schoeffter, A. Le Bec, E. Strouthou, J.C. Stoclet, Biochem. Pharmacol.
35 (1986) 1743.[39] J.F. Kameni Tcheudji, L. Lebeau, N. Virmaux, C.G. Maftei, R.H. Cote, C. Lugnier, P.
Schultz, J. Mol. Biol. 310 (2001) 781.[40] M. Campos-Toimil, T. Keravis, F. Orallo, K. Takeda, C. Lugnier, Br. J. Pharmacol. 154
(2008) 82.[41] C. D'Sa, L.M. Tolbert, M. Conti, R.S. Duman, J. Neurochem. 81 (2002) 745.[42] H. Abrahamsen, G. Baillie, J. Ngai, T. Vang, K. Nika, A. Ruppelt, T. Mustelin, M.
Zaccolo, M. Houslay, K. Taskén, J. Immunol. 173 (2004) 4847.[43] G.S. Baillie, S.J. MacKenzie, I. McPhee, M.D. Houslay, Br. J. Pharmacol. 131 (2000)
811.[44] J. Creighton, B. Zhu, M. Alexeyev, T. Stevens, J. Cell Sci. 121 (2008) 110.
[45] S. Erdogan, M.D. Houslay, Biochem. J. 321 (1997) 165.[46] J. Seybold, R. Newton, L.Wright, P.A. Finney, N. Suttorp, P.J. Barnes, I.M. Adcock, M.A.
Giembycz, J. Biol. Chem. 173 (1998) 20575.[47] C. Lugnier, A. Follenius, D. Gerard, J.C. Stoclet, Eur. J. Pharmacol. 98 (1984) 157.[48] G. Dent, M.A. Giembycz, K.F. Rabe, P.J. Barnes, Br. J. Pharmacol. 103 (1991) 1339.[49] A. Lerner, D.H. Kim, R. Lee, Leuk. Lymphoma 37 (2000) 39.[50] J.A. Meyers, D.W. Su, A. Lerner, J. Immunol. 182 (2009) 5400.[51] S. Tiwari, H. Dong, E.J. Kim, L. Weintraub, P.M. Epstein, A. Lerner, Biochem.
Pharmacol. 69 (2005) 473.[52] L. Zhang, F. Murray, A. Zahno, J.R. Kanter, D. Chou, R. Suda, M. Fenlon, L. Rassenti,
H. Cottam, T.J. Kipps, P.A. Insel, Proc. Natl Acad. Sci. USA 105 (2008) 19532.[53] J. Sato, K. Nair, J. Hiddinga, N.L. Eberhardt, L.A. Fitzpatrick, Z.S. Katusic, T. O'Brien,
Cardiovasc. Res. 47 (2000) 697.[54] S. Fukumoto, H. Koyama, M. Hosoi, K. Yamakawa, S. Tanaka, H.Morii, Y. Nishizawa,
Circ. Res. 85 (1999) 985.[55] A.Q. Abbady, C. Bronner, K. Bathami, C.D. Muller, M. Jeanblanc, E. Mathieu, J.P.
Klein, E. Candolfi, M. Mousli, Biochem. Pharmacol. (2005) 570.





























