Embed Size (px)
Citation preview

1
De novo germline disorders of the Ras-MAPK pathway:
clinical delineation, molecular diagnosis and pathogenesis
A thesis submitted to the University of Manchester for the
degree of Doctor of Philosophy in the Faculty of Medical and Human Sciences
2013
Emma Mary Milborough Burkitt Wright
Institute of Human Development
School of Medicine

2
LIST OF CONTENTS
1 INTRODUCTION………………………………………………………...20
1.1 Introduction to cardio-facio-cutaneous syndrome (CFC) and other disorders
of the Ras-MAPK pathway ...................................................................................................... 21
1.2 Aims of project ......................................................................................................... 24
1.3 The neuro-cardio-facio-cutaneous syndromes (NCFCs) .................................... 25
1.3.1 Cardiac features of the NCFCs .............................................................................. 26
1.3.2 Cancer risk across the NCFCs ................................................................................ 26
1.3.3 Cardio-facio-cutaneous (CFC) and Costello syndromes .................................... 26
1.4 Characteristic clinical aspects of cardio-facio-cutaneous syndrome (CFC) ..... 31
1.5 Disorders demonstrating clinical overlap with CFC syndrome ......................... 33
1.5.1 Noonan syndrome (NS) and Noonan syndrome with multiple lentigines
(formerly LEOPARD) syndrome (NSML) ........................................................................... 33
1.5.2 Costello syndrome .................................................................................................... 34
1.5.3 Clinical overlap and distinction between the NCFCs ......................................... 35
1.6 The Ras-MAPK pathway and its role in cancer ................................................... 36
1.7 The molecular basis of the NCFCs ........................................................................ 40
1.7.1 Comparison of the molecular basis of the NCFCs with the mutational
spectrum observed in cancers ................................................................................................. 45
1.7.2 Genomic factors that may affect Ras-MAPK pathway activity ......................... 50
1.7.3 Genetic testing in the NCFCs and implications of a molecular diagnosis ....... 51
1.7.4 New genomic and genetic technologies for investigation of the NCFCs ........ 53

3
1.7.5 Genotype phenotype correlations across the NCFCs ......................................... 57
1.8 Molecular pathogenesis of the NCFCs ................................................................. 60
1.8.1 Functional effects of CFC-associated mutations ................................................. 61
1.9 Animal and other models of CFC and related conditions .................................. 64
1.9.1 Mouse models of the NCFCs ................................................................................. 66
1.9.2 Zebrafish models of the NCFCs ............................................................................ 67
1.9.3 Human-derived cellular models of the NCFCs ................................................... 68
1.10 Avenues for therapy of Ras-MAPK pathway disorders ..................................... 68
1.11 Summary of conclusions from the literature ........................................................ 71
2 MATERIALS AND METHODS ......................................................................... 73
2.1 Reagents and supplies .............................................................................................. 74
2.2 Clinical and molecular diagnosis of patients with Ras-MAPK disorders ......... 74
2.2.1 Identification of patient cohort .............................................................................. 74
2.2.2 Clinical phenotyping of patient cohort .................................................................. 75
2.2.3 Molecular analysis of exon 2 of SHOC2 in patients previously tested for
Costello or cardio-facio-cutaneous syndromes ..................................................................... 75
2.3 Massively parallel sequencing approaches for molecular diagnosis .................. 76
2.3.1 Target enrichment sequencing of selected patients ............................................. 76
2.3.2 Whole exome sequencing of patient-parent trios ................................................ 78
2.4 Cell culture work ....................................................................................................... 79
2.4.2 Site-directed mutagenesis ........................................................................................ 80

4
2.4.3 Transfection using jet PEI reagent ........................................................................ 80
2.4.4 Western blotting ....................................................................................................... 81
2.4.5 Dual luciferase assay ................................................................................................. 82
2.4.6 In vitro kinase assays ................................................................................................ 82
2.4.7 Transient transfection in the H9C2 cell line ......................................................... 83
2.4.8 Stable transfection of the H9C2 cell line .............................................................. 83
2.5 Characterisation of mouse models of the NCFCS .............................................. 84
2.5.1 The B-Raf LSLV600E/+ mouse model of CFC syndrome .............................. 84
2.5.2 Cardiac phenotyping in the B-Raf LSLV600E/+ mouse ................................... 85
2.5.3 Cardiac expression microarrays in mouse models of the NCFCs ..................... 86
3 CLINICAL AND MOLECULAR DIAGNOSIS OF PATIENTS WITH
GERMLINE RAS-MAPK PATHWAY DISORDERS ..................................................... 89
3.1 Chapter overview ...................................................................................................... 90
3.2 Mutational spectrum observed in patients with CS/CFC phenotypes ............. 90
3.3 Clinical features of patients with mutation-proven CFC .................................... 95
3.3.1 Patients with a BRAF mutation ............................................................................. 95
3.3.2 Patients with a MAP2K1 mutation ...................................................................... 104
3.3.3 Patients with other mutations causing CFC ....................................................... 108
3.3.4 Findings across the group of patients with CFC-associated mutations ......... 109
3.4 Extending the molecular basis of CFC: SHOC2 is an important disease gene in
this patient group .................................................................................................................... 110

5
3.4.1 Clinical presentations of patients diagnosed with SHOC2 p.(Ser2Gly)
mutations………. ................................................................................................................... 117
3.5 Discussion of chapter results ................................................................................ 128
3.5.1 Genotype-phenotype correlation in CFC ........................................................... 128
3.5.2 Clinical features in patients with p.(Ser2Gly) mutation in SHOC2 ................. 129
4 MASSIVELY PARALLEL SEQUENCING APPROACHES FOR
MOLECULAR DIAGNOSIS .............................................................................................. 132
4.1 Chapter overview .................................................................................................... 133
4.2 Target enrichment approach ................................................................................. 134
4.2.1 Development of gene list ...................................................................................... 134
4.2.2 Selection of patient samples .................................................................................. 142
4.2.3 Results of target enrichment experiment ............................................................ 145
4.2.4 Results of testing in each of the samples ............................................................ 150
4.2.5 Use of the data set for further filtering of candidate variants .......................... 161
4.2.6 PTK2 as a novel candidate disease gene .............................................................. 166
4.2.7 Sequencing of PTK2 as a candidate gene for germline human disease ........... 168
4.2.8 Iterative review of target enrichment results as further genes for NCFCs
identified……… ..................................................................................................................... 169
4.2.9 RIT1 : a gene with significant structural and functional similarities to RAS
genes………….. ...................................................................................................................... 170
4.2.10 Confirmation of diagnosis by RIT1 sequencing and clinical implications ..... 171
4.3 Whole exome sequencing for gene identification in CFC ................................ 172

6
4.3.1 Selection of patient samples .................................................................................. 172
4.3.2 Whole exome sequencing ...................................................................................... 172
4.3.3 Bioinformatic analysis ............................................................................................ 172
4.3.4 Resequencing affected patients’ exomes using Illumina HiSeq2000............... 175
4.3.5 Mutation in NF1 in a patient with a clinical diagnosis of CFC syndrome ..... 176
4.4 Discussion of chapter results ................................................................................ 177
5 FUNCTIONAL CONSEQUENCES OF DE NOVO GERMLINE
MUTATIONS CAUSING RAS-MAPK PATHWAY DISORDERS ........................... 181
5.1 Chapter overview .................................................................................................... 182
5.2 Characterisation of the effect of mutations in BRAF on ERK pathway activity
in the HEK293T and HEK293 cell lines ............................................................................ 183
5.2.1 Verification of plasmids for expression of BRAF in cell culture .................... 183
5.2.2 Site-directed mutagenesis to generate CFC-associated mutations in BRAF .. 186
5.2.3 Western blotting for phospho-ERK1/2 ............................................................. 186
5.2.4 In vitro kinase assay of BRAF activity using myelin basic protein ................. 188
5.2.5 Site-directed mutagenesis to assess novel variants in BRAF identified in
patients with CFC syndrome ................................................................................................. 189
5.2.6 Work in the HEK293 cell line .............................................................................. 190
5.2.7 Western blotting to assess ERK1/2 phosphorylation by novel variants in
BRAF identified in patients with CFC syndrome .............................................................. 190
5.2.8 In vitro kinase assay to assess novel variants in BRAF identified in patients
with CFC syndrome ................................................................................................................ 190

7
5.2.9 Dual luciferase assay to measure ELK1 transcriptional activity ...................... 193
5.3 Effects of mutated BRAF in the H9C2 cardiomyoblast cell line .................... 195
5.3.1 Stable transfection of the H9C2 cell line ............................................................ 197
5.4 Discussion of results .............................................................................................. 198
6 INVESTIGATION OF THE CARDIAC PHENOTYPES OF MOUSE
MODELS OF THE NCFCS……………………………………………………….202
6.1 Characterisation of the cardiac phenotype of the B-Raf LSLV600E/+
mouse………………………………………………………………………………203
6.1.1 Embryonic development of the heart of the B-Raf LSLV600E/+
mouse………….……………………………………………………………...…….206
6.1.2 Pathway analysis of microarray data from the B-Raf LSLV600E/+mouse
model…………..…………………………………………………………..………..215
6.1.3 Validation of findings of microarray by quantitative fluorescent PCR (qPCR)
………………..……………………………………………………………….……216
6.1.4 Western blotting to assess Myh7 protein concentration in the B-Raf LSLV600E/+
mouse model…. ...................................................................................................................... 221
6.2 Investigation of the cardiac phenotype of the H-Ras G12V/G12V mouse model of
Costello syndrome ................................................................................................................... 223
6.2.1 Affymetrix Mouse Genome 430A arrays in the H-Ras G12V/G12Vmouse
model……………………………………………………………………………….223
6.2.2 Quantitative fluorescent PCR (qPCR) to investigate Hras transcript
abundance…………..………………………………………………………………226
6.3 Investigation of the cardiac phenotype of the K-Ras V14I/+ mouse model of
CFC/NS by expression microarray ...................................................................................... 231

8
6.3.1 Affymetrix Mouse Genome 430A arrays in the K-Ras V14I/+ mouse
model……………………………………………………………………………….231
6.3.2 Pathway analysis in the K-Ras V14I/+ mouse model ........................................... 236
6.3.3 Comparative analysis of the cardiac phenotype across the mouse models of
the NCFCs…….. .................................................................................................................... 236
6.3.4 Cluster analysis ........................................................................................................ 242
6.4 Discussion of chapter results ................................................................................ 246
7 DISCUSSION......................................................................................................... 251
7.1 Overview .................................................................................................................. 252
7.2 Clinical phenotypes of the NCFCs ...................................................................... 253
7.2.1 CFC syndrome ........................................................................................................ 253
7.2.2 SHOC2- related phenotypes ................................................................................. 254
7.3 Molecular diagnosis of the NCFCs by massively parallel sequencing ............ 257
7.3.1 Target enrichment approaches ............................................................................. 257
7.3.2 Exome sequencing approaches ............................................................................ 259
7.4 Cellular and organism level effects of NCFC-associated mutations ............... 260
7.4.1 Cell culture ............................................................................................................... 260
7.4.2 Mouse models of the NCFCs ............................................................................... 261
7.4.3 Expression microarray ........................................................................................... 262
7.4.4 Novel means of modelling the NCFCs ............................................................... 263
7.5 Review of techniques used and possible alternatives ........................................ 264

9
Genetic / Genomic Medicine and the NCFCs .................................................. 265 1.1
7.6 Conclusions ............................................................................................................. 267
8 REFERENCES ...................................................................................................... 269
9 APPENDICES ....................................................................................................... 305
9.1 Appendix 1: Germline mutations described in the genes causing CFC
syndrome…… ......................................................................................................................... 306
9.2 Appendix 2 .............................................................................................................. 313
9.2.1 Appendix 3a ............................................................................................................ 345
9.2.2 Appendix 3B............................................................................................................ 346
9.2.3 Appendix 3c: ........................................................................................................... 355
9.3 Appendix 4: Primers and PCR conditions .......................................................... 372
9.4 Appendix 5: Histograms of coverage of diagnostically relevant genes in
patients TE1-TE10 ................................................................................................................. 380
9.5 Appendix 6: Transcripts with greatest differential expression in
interventricular septum of mouse models of the NCFCs ................................................. 385
9.6 References for appendices ..................................................................................... 438
9.7 Appendix 7: Reprints of articles relating to the work undertaken (in
chronological order): ............................................................................................................... 442
Word Count: 76657

10
LIST OF TABLES
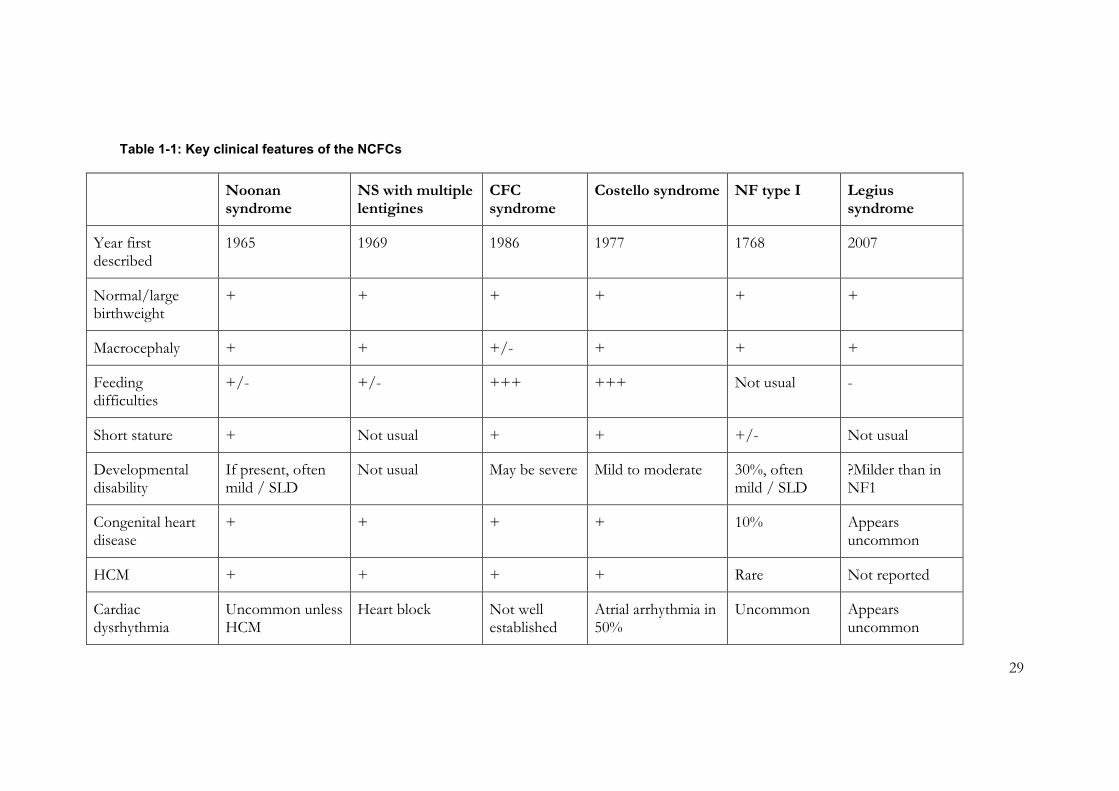
Table 1-1: Key clinical features of the NCFCs ..................................................................... 29
Table 1-2: Somatic mutations in genes of the RAS-MAPK pathway in human tumours
...................................................................................................................................................... 39
Table 1-3: Numbers of patients with mutations in genes causing NS, CFC and
genotypically overlapping conditions, as represented in the NS Euronet database ........ 43
Table 1-4: Animal models of CFC, NS and CS .................................................................... 64
Table 2-1: Antibodies used for Western blotting ................................................................. 81
Table 3-1: Samples tested for mutations in CFC/CS genes ............................................... 91
Table 3-2 Clinical features of patients with BRAF mutations ............................................ 96
Table 3-3 Clinical features of patients M1-M5 ..................................................................... 99
Table 3-4: Clinical features of patients S1-7 ........................................................................ 111
Table 3-5: Clinical features of patients S8 – 14 ................................................................... 114
Table 4-1: Genes included in target enrichment experiment ............................................ 134
Table 4-2: Samples included in target enrichment experiment......................................... 143
Table 4-3: Variants identified in target enrichment experiment ....................................... 151
Table 4-4: Variants called in whole exome sequencing ..................................................... 175
Table 5-1: Effects of CFC-associated mutations in BRAF assessed in cell culture ...... 199
Table 6-1: The 20 transcripts most highly expressed in B-Raf LSLV600E/+ mouse heart. . 211
Table 6-2: The 20 transcripts with most reduced expression in B-Raf LSLV600E/+ mouse
heart. .......................................................................................................................................... 213
Table 6-3: The 20 transcripts with most increased expression in the B-Raf LSLV600E/+
mouse heart. ............................................................................................................................. 213
Table 6-4: Pathways identified by KEGG analysis of transcripts with altered expression
in the B-Raf LSLV600E/+ mouse model. ..................................................................................... 215
Table 6-5: Transcripts selected for validation by qPCR: ................................................... 218
Table 6-6: The 20 most highly expressed transcripts in the IVS of the H-Ras G12V/G12V
mouse model. ........................................................................................................................... 225
Table 6-7: 20 transcripts with most decreased expression in the H-Ras G12V/G12V mouse
model......................................................................................................................................... 227
Table 6-8: 20 transcripts with most increased expression in the H-Ras G12V/G12V mouse
model......................................................................................................................................... 228

11
Table 6-9: 20 transcripts with highest expression in the K-Ras V14I/+ mouse model 233
Table 6-10: 20 transcripts with greatest fold decrease in expression in the K-Ras mouse
model......................................................................................................................................... 234
Table 6-11: 20 transcripts with greatest fold increase in expression in the K-Ras mouse
model......................................................................................................................................... 234
Table 6-12: Pathways identified by differentially expressed transcripts in the K-Ras V14I/+
expression microarray ............................................................................................................. 236
Table 6-13: Number of transcripts with ‘q’ value below thresholds 0.05, 0.1 and 0.2 in
the three sets of microarrays. ................................................................................................. 239
Table 6-14: Genes with ‘q’ value <0.1 in B-Raf LSLV600E/+ and K-Ras V14I/+ expression
microarrays ............................................................................................................................... 241

12
LIST OF FIGURES
Figure 1-1: The Ras-MAPK pathway and disorders due to mutations in its genes ........ 23
Figure 1-2: Features of CS and CFC in early life: ................................................................. 28
Figure 1-3: The severity of effects of a mutation can influence the context in which it is
observed ...................................................................................................................................... 41
Figure 1-4: Molecular basis of CFC syndrome in patients on the NSEuronet database. 43
Figure 1-5: Genes in which mutations have been found in patients with NCFCs .......... 44
Figure 3-1: Molecular diagnosis of Costello and CFC syndromes (Manchester Regional
Genetics Laboratory) 2006-2012. ........................................................................................... 92
Figure 3-2: Mutations identified in samples referred for CS and CFC gene testing. ...... 93
Figure 3-3: Patients with BRAF mutations ........................................................................ 103
Figure 3-4: Patients with MAP2K1 p.(Tyr130Cys) mutations .......................................... 106
Figure 3-5: Serial photographs of patients with MAP2K1 p.(Tyr130Cys) mutations ... 107
Figure 3-6: Adults with SHOC2 p.(Ser2Gly) mutations .................................................... 125
Figure 3-7: Children with SHOC2 p.(Ser2Gly) mutations ................................................ 125
Figure 3-8: Serial photographs of patients with SHOC2 p.(Ser2 Gly) mutations .......... 127
Figure 4-1: Coverage across the exons of the three RAS genes KRAS, HRAS and
NRAS. ...................................................................................................................................... 146
Figure 4-2 Coverage of genes of diagnostic relevance ....................................................... 148
Figure 4-3 Coverage across genes known to be mutated in human disease in samples
TE1-TE10. ............................................................................................................................... 150
Figure 4-4: Bidirectional Sanger sequencing of chr2:39262581. ...................................... 162
Figure 4-5: Algorithm for hierarchical filtering of candidate variants. ............................ 163
Figure 4-6: Bidirectional Sanger sequencing of chr8: 141900836 in patient TE4 and her
mother. ...................................................................................................................................... 165
Figure 4-7: Bidirectional Sanger sequencing of chr6: 166845926 in patient TE7. ........ 166
Figure 4-8: The N-terminal portion of human FAK (product of PTK2 NM_153831.3)
aligned against the protein sequence of other species. ...................................................... 167
Figure 4-9: Amino acid sequence encoded by exon 5 of RIT1. ....................................... 171
Figure 4-10: Algorithm for filtering of candidate variants identified through whole
exome sequencing of trios. .................................................................................................... 174

13
Figure 4-11: Massively parallel sequencing has the potential to transform the diagnostic
process. ..................................................................................................................................... 180
Figure 5-1: Mutations and uncharacterised variants in BRAF in patients with CFC
syndrome. ................................................................................................................................. 183
Figure 5-2: Verification of pEF-BRAF wild-type and p.(Val600Glu) plasmids. ........... 185
Figure 5-3: Western blotting in HEK293T cell lysates after transient transfection of
pEF-BRAF plasmids demonstrated effects upon ERK1/2 phosphorylation. .............. 187
Figure 5-4: In-vitro kinase assay to assess previously characterised mutations in BRAF.
.................................................................................................................................................... 188
Figure 5-5: Western blotting (A) and in vitro kinase assay (B) of HEK293 cells transiently
transfected with CFC-associated BRAF alleles................................................................... 192
Figure 5-6: Dual luciferase assay results in HEK293 cells transfected with CFC-
associated mutations in BRAF. ............................................................................................. 194
Figure 5-7 Transient transfection of H9C2 cells with pEF-BRAF plasmids by
electroporation. ........................................................................................................................ 196
Figure 5-8 Abnormal morphology of H9C2 cells in the attempt to generate stable cell
lines. ........................................................................................................................................... 198
Figure 6-1 A: Schematic of exons 14-16 of the B-Raf LSLV600E allele. ................................ 205
Figure 6-2: Histological appearance of embryonic heart of the B-Raf LSLV600E/+ mouse
and wild-type counterparts. ................................................................................................... 207
Figure 6-3: High power magnification of interventricular septum in the B-Raf LSLV600E/+
mouse model and wild-type counterpart. ............................................................................ 208
Figure 6-4: Principal component analysis of raw data from comparative B-Raf LSLV600E/+
vs wild-type expression microarray. ...................................................................................... 210
Figure 6-5: Expression of Myh6 and Myh7 transcripts in the heart of the B-Raf LSLV600E/+
mouse model. ........................................................................................................................... 220
Figure 6-6 Expression of further targets suggested by microarray results in the B-Raf
LSLV600E/+ mouse model. .......................................................................................................... 221
Figure 6-7: Western blot for Myh7 in B-Raf LSLV600E/+ and B-Raf +/+ hearts. .................. 222
Figure 6-8: Principal component analysis of raw data from H-Ras G12V/G12V expression
microarrays. .............................................................................................................................. 224
Figure 6-9: Hras expression in heart, muscle, brain and liver of the H-Ras G12V mouse
model......................................................................................................................................... 229

14
Figure 6-10: Principal component analysis of raw data from K-Ras V14I/+ expression
microarrays. .............................................................................................................................. 232
Figure 6-11 Myh7 expression in K-Ras V14I/+ and H-RasG12V/G12V mouse models............ 237
Figure 6-12: Similarity of the 50 most highly expressed transcripts identified across the
three mouse models. ............................................................................................................... 238
Figure 6-13: Cluster analysis of transcripts altered in the B-Raf LSLV600E/+ microarray. .. 243
Figure 6-14: Close-up representation of cluster ‘B’ (of Figure 6-13)............................... 245

15
ABSTRACT
This work sought to investigate the clinical phenotypes and molecular basis of cardio-facio-cutaneous syndrome (CFC), a germline disorder of the Ras-MAPK pathway, like Noonan syndrome (NS) and neurofibromatosis type I, caused by mutations in genes encoding proteins that act within this signal transduction pathway. CFC is most commonly due to mutation in BRAF, and less commonly MAP2K1, MAP2K2 or KRAS. A proportion of patients currently have no mutation identified.
Mutations and clinical features of patients with a molecular diagnosis of CFC were investigated, which demonstrated a wide range of causative mutations, and some unclassified variants. Both known and novel clinical features of CFC were identified. A strong association between severe contractures and the p.(Tyr130Cys) mutation in MAP2K1 was found, which has not previously been reported.
In contrast to the large number of patients with a confirmed molecular diagnosis, several with a highly suggestive clinical phenotype have been found to have no mutation in any of the known CFC genes. The molecular basis of these presentations was investigated by conventional Sanger sequencing of candidate genes. Fourteen patients with the p.(Ser2Gly) mutation in SHOC2 were identified, with clinical presentations consistent with CFC, NS or CS. Target enrichment and massively parallel sequencing of selected genes was undertaken in ten patients. Mutations in known genes were identified in four patients (including the positive control). Candidate causative variants in novel genes were suggested in two further patients, one of which was confirmed on Sanger sequencing. Whole exome sequencing of patient-parent trios was also undertaken to identify de novo variants. Three trios were analysed, and in one patient with a clinical diagnosis of CFC, a frameshift mutation in NF1 was identified, which was confirmed by Sanger sequencing to be present and de novo.
The molecular effects of CFC-associated mutations in BRAF on Ras-MAPK pathway signalling were studied in cell culture systems, using Western blotting for ERK1/2 phosphorylation, in vitro kinase assays and luciferase assays, to assess activity of downstream targets of the Ras-MAPK pathway. Altered pathway activity was demonstrated for novel variants that had not previously been characterised at the molecular level, which was in keeping with the findings of the effects of previously studied mutations.
The cardiac phenotype in animal models of CFC, CS and NS/CFC was explored using expression microarrays to identify potentially important genes and pathways in the pathogenesis of hypertrophic cardiomyopathy (a progressive but potentially treatable disease feature) in these conditions. A signature of increased expression of Myh7, the embryonic form of myosin, was identified in the heart of the mouse model of CFC due to a B-Raf mutation at four weeks postnatal age, but comparative analysis suggested significant differences in either the mechanisms causing cardiac phenotypes, or the timescales over which these may exert their effects, in the three models.
In summary, the most significant findings of this work were that SHOC2 mutation is a frequent cause of a severe NCFC presentation, and massively parallel sequencing can be an effective means of molecular investigation of this group of disorders. Novel features of CFC syndrome that were identified include severe contractures in association with p.(Tyr130Cys) mutations in MAP2K1. The analysis of mouse models of the NCFCs was hampered by heterogeneity within the expression microarray results, and low levels of expression of the H-Ras mutant allele in the mouse model of Costello syndrome.

16
DECLARATION
No portion of the work referred to in the thesis has been submitted in support of
an application for another degree or qualification of this or any other university or other
institute of learning.

17
COPYRIGHT STATEMENT
i. The author of this thesis (including any appendices and/or schedules to this
thesis) owns certain copyright or related rights in it (the “Copyright”) and she has given
The University of Manchester certain rights to use such Copyright, including for
administrative purposes.
ii. Copies of this thesis, either in full or in extracts and whether in hard or electronic
copy, may be made only in accordance with the Copyright, Designs and Patents Act
1988 (as amended) and regulations issued under it or, where appropriate, in accordance
with licensing agreements which the University has from time to time. This page must
form part of any such copies made.
iii. The ownership of certain Copyright, patents, designs, trade marks and other
intellectual property (the “Intellectual Property”) and any reproductions of
copyright works in the thesis, for example graphs and tables (“Reproductions”), which
may be described in this thesis, may not be owned by the author and may be owned by
third parties. Such Intellectual Property and Reproductions cannot and must not be
made available for use without the prior written permission of the owner(s) of the
relevant Intellectual Property and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property and/or
Reproductions described in it may take place is available in the University IP Policy (see
http://documents.manchester.ac.uk/DocuInfo.aspx?DocID=487), in any relevant
Thesis restriction declarations deposited in the University Library, The University
Library’s regulations (see http://www.manchester.ac.uk/library/aboutus/regulations)
and in the University’s policy on Presentation of Theses.

18
ACKNOWLEDGEMENTS AND DEDICATION
The help and support of supervisors Professor Graeme Black, Dr Bronwyn Kerr
and Dr Alan Whitmarsh, clinical and laboratory colleagues in MCGM, and laboratory
colleagues in the groups of Professor Graeme Black, Dr Alan Whitmarsh and Professor
Mariano Barbacid is acknowledged with gratitude. Particular thanks are due to the
following individuals for their specific roles in generating the data for this work.
Information regarding the molecular diagnoses made in the MCGM’s diagnostic
laboratory was provided by Dr Jenny Shorto. Referring clinicians of patients with
SHOC2 mutations kindly provided further clinical information about these individuals.
Target enrichment and exome sequencing experiments were co-ordinated by Dr James
O’Sullivan and other members of the MCGM next generation sequencing team. Initial
bioinformatic analysis was performed by Dr Sanjeev Bhaskhar and Dr Simon Williams.
The mouse model work described in chapter 6 was performed collaboratively with Dr
Mari Carmen Guerra and Drs Jelena Urosevic and Isabel Hernandez at CNIO.
Histological slides were prepared by the CNIO histopathology unit. Expression
microarrays were performed in the University of Manchester’s Faculty of Life Sciences
Genomics Core Facility by Dr Mike Smiga, and initial bioinformatic analysis by Dr Leo
Zeef.
Outside the work arena, the support of my husband Mike and my parents has been
essential, especially since the arrival of our daughter Iris in March 2013. This thesis is
therefore dedicated to them.

19
PREFACE
The author undertook this thesis whilst out of programme from specialist training
in clinical genetics. Her research training fellowship was initially funded by the
Manchester Biomedical Research Centre (October 2009 – April 2010), and subsequently
by the award of a Wellcome Trust clinical research training fellowship (May 2010 –
October 2013, with maternity leave from March 2013-September 2013).
A note on nomenclature
For human genetic variations, HGVS approved nomenclature has been used,
including the notation for predicted (rather than proven) changes in the protein level in
parentheses, e.g. p.(Gly12Val). With reference to animal models, however, for ease of
reference to, and consistency with, the published literature, the description of these
models has been to use the notation of the original publication, e.g. H-Ras G12V/G12V.
Where the term mutation is used, this is intended to indicate a pathogenic variant.
Where pathogenicity is unproven, or the variant is considered non-pathogenic, the term
variant has been used (with qualifiers where necessary).

20
1 INTRODUCTION

21
1.1 Introduction to cardio-facio-cutaneous syndrome (CFC) and
other disorders of the Ras-MAPK pathway
Cardio-facio-cutaneous syndrome (CFC) is one of the germline disorders of the
Ras-MAPK pathway, a group of conditions that includes Noonan syndrome (NS) and
neurofibromatosis type I (NF1). These are united by shared clinical features and a
common strand to their molecular pathogenesis, namely dysregulated Ras-MAPK
pathway signal transduction, as shown in Figure 1-1. These conditions are also termed
the neuro-cardio-facio-cutaneous syndromes (NCFCs), or Rasopathies. The Ras-MAPK
pathway has long been known to be dysregulated in cancer, with HRAS being the first
identified human oncogene (1, 2). This pathway has therefore been more fully
characterised than many other cellular pathways, and agents to modulate its activity
have been developed in the context of cancer chemotherapeutics.
The spectrum of clinical presentations of the NCFCs, as the name suggests,
involves many organ systems of the human body. CFC frequently has a severe
presentation, with large proportions of patients experiencing significant morbidity and
mortality due to neurological features, including learning disability and epilepsy,
cardiovascular features, particularly congenital heart disease and hypertrophic
cardiomyopathy (HCM), and also skin and musculoskeletal problems. Feeding
difficulties and growth failure are further features that are particularly common in CFC,
and these, like the features listed above, also occur in Costello syndrome (CS) and to a
variable extent in other NCFCs.
Classification of these disorders has been assisted by the development of robust
clinical criteria for disorders such as NF1 (3), and the identification of causative
mutations in many patients. Confirmation of the diagnosis of other NCFCs, however,
particularly NS and CFC, can be difficult. These two conditions demonstrate both
strong clinical overlap with one another, and extensive genetic heterogeneity. In
addition, up to one third of patients with a clinical diagnosis of one of these conditions
have no mutation currently identifiable (4, 5), suggesting that further causative genes
may exist. Assessment of genotype-phenotype correlation is hindered by this extensive
genetic and allelic heterogeneity: most individual causative mutations are rare.
International efforts to address this challenge are now underway, for example through

22
the development of the NSEuronet database (6). Clinical and molecular heterogeneity,
and the high proportion of causative mutations that occur de novo, have hampered the
search for novel NS- and CFC-associated genes by traditional gene identification
methods. New massively parallel sequencing techniques, however, hold promise in
identifying the molecular basis for the clinical presentations of this group of patients.
Identifying mutations in new genes for the NCFCs, and pursuing extended genotype-
phenotype correlation studies will permit definitive diagnosis in more affected
individuals and improve medical and scientific understanding of these disorders, hence
allowing for better clinical management. Such work, together with the extensive
previous study of the Ras-MAPK pathway in the context of cancer, can also provide the
basis for future drug treatments for these disorders. Therapeutic trials using agents to
modulate Ras-MAPK pathway activity, have already been initiated in NF1 (7). However,
varied effects of NCFC-associated mutations upon Ras-MAPK signalling have been
observed, depending upon both the gene and the specific mutation involved. For many
of the genes, such as NF1, and SPRED1, known mutations have been demonstrated to
result in similar downstream effects (8, 9), but for some, such as BRAF, mutations
which result in similar clinical phenotypes appear to have divergent effects on pathway
activity and activation in experimental systems (10). This emphasises the need for
caution in any such approach and the particular need for better understanding of the
molecular basis of the clinical features of these conditions. The accurate identification
of which of these phenotypic elements develop through the lifespan, rather than being
congenital (and irrevocable by the time of diagnosis) will be important to know in order
to guide what aspects may be amenable to future treatments.

23
Figure 1-1: The Ras-MAPK pathway and disorders due to mutations in its genes
This simplified schematic shows the proteins with known roles in signal transduction. Arrows indicate the recognised direction of signalling: Ras proteins activate Raf proteins, which are the first kinases in a triple cascade. Proteins are colour coded for the disorders that are known to arise when each of the relevant genes are mutated. The overlap between NS and CFC is demonstrated by the several bicoloured ellipses.

24
1.2 Aims of project
This study sought to investigate the clinical phenotypes and molecular basis of CFC
syndrome, in the following areas:
1. To study the spectrum of mutations and clinical features of patients with a
molecular diagnosis of CFC syndrome.
The Manchester Centre for Genomic Medicine (MCGM) diagnostic laboratory has
offered genetic testing for CS and CFC since the publication of the genes mutated in
these conditions, meaning that a significant proportion of the UK’s patients with this
condition have been tested in this centre, along with many overseas patients,
representing a large cohort of patients with CS/CFC with molecular data available.
Clinical phenotypic data were sought to explore the clinical phenotypes of CFC and
assess for genotype-phenotype correlation.
2. To investigate the molecular basis of NCFC presentations in patients who had
been tested for CFC, in whom no mutation had been found.
For this, three approaches were used: firstly, sequencing of genes identified in
NCFC disorders by conventional sequencing, secondly, target enrichment and massively
parallel sequencing of a selected panel of genes, and thirdly, whole exome sequencing of
patient-parent trios.
3. To investigate the effects of CFC-associated mutations in BRAF on Ras-MAPK
pathway signalling in cell culture systems.
A variety of assays of downstream effects were used for this: Western blotting for
ERK1/2 phosphorylation, in vitro kinase assays and luciferase assays were each
performed to assess activity of downstream targets of the Ras-MAPK pathway.
4. To explore the cardiac phenotype in animal models of the NCFCs, using
expression microarrays.
These were used as a means to identify genes and pathways that may be important
to the pathogenesis of hypertrophic cardiomyopathy (a progressive but potentially
treatable disease feature) in patients with these conditions.

25
1.3 The neuro-cardio-facio-cutaneous syndromes (NCFCs)
The NCFCs have an estimated collective prevalence of between 1 in 700 and 1 in
1250 of the population (11, 12), this large range reflecting the uncertain prevalence of
NS (13). Because many of the causative genes have only recently been identified, and
genetic testing has only recently become available, substantial numbers of affected
individuals have clinical diagnoses that are yet to be confirmed by molecular testing.
The incidence of NS has never been determined, but has been suggested that between 1
in 1000 and 1 in 2500 people may be affected (13). CFC is known to be a much rarer
presentation than NS, associated with more severe sequelae (14), as discussed in 1.4, but
its incidence, like that of NS, has not been determined. NF1 is a disorder for which
effective clinical diagnostic criteria exist (3), for which the birth incidence has been
estimated at 1 in 2000 in the population (11). The birth incidence of Costello syndrome
(CS) has been estimated at 1 in 381000 (15). Incidence and prevalence figures for
mutated genes and specific mutations, however, depend upon the presence of an
identifiable phenotype to prompt testing, and to what extent survival is affected by the
mutation: presentations may be too mild to be recognised, or so severe as to be lethal
prior to recognition (16).
Key clinical features of this group of disorders are summarised in Table 1-1.
Features that are common across the pathway disorders include similarities of physique
including short stature, relative or absolute macrocephaly, facial characteristics including
downslanting palpebral fissures, ptosis and hypertelorism, congenital heart disease,
hypertrophic cardiomyopathy, feeding difficulties, developmental delay and
predisposition to a range of early-onset tumours (see reference (17), appendix 7 for an
overview).

26
1.3.1 Cardiac features of the NCFCs
Cardiac anomalies are one of the hallmarks of the NCFCs. The commonest
structural cardiac anomalies found in CFC are pulmonary valve stenosis and atrial septal
defect (18), whilst neonatal supraventricular arrhythmias are common in CS (19, 20).
Hypertrophic cardiomyopathy, which may be of very early onset, can occur in both CS
(1/3 of patients (21)) and CFC (1/4 of patients (14)) as well as being found in a similar
proportion of NS (22). Patients with Noonan syndrome with multiple lentigines
(NSML) show particularly high rates of HCM (23), which may be present in 80% of
these individuals (24). NS is frequently associated with pulmonary stenosis (particularly
with a dysplastic valve), but a wide range of other congenital heart disease is also seen
(25). Cardiac anomalies are not common in the NF1 population as a whole, but
pulmonary stenosis is a recognised feature in a subgroup with missense NF1 mutations
(26).
1.3.2 Cancer risk across the NCFCs
The risk of childhood cancer has been estimated at 4% for NS, predominantly of
juvenile myelomonocytic leukaemia (JMML) (27), and a similar risk of malignancy has
been suggested for CFC, though this has not yet been established due to the small
numbers of patients identified (14). In contrast, due to the well-defined patient group
and high incidence of specific tumours, the childhood cancer risk in CS has been able to
be accurately estimated, at 17% (28). A wide range of childhood tumours is recognised
in association with NF1, with greatly increased relative risks compared to the general
population, but low absolute risks (29).
1.3.3 Cardio-facio-cutaneous (CFC) and Costello syndromes
Cardio-facio-cutaneous (CFC) and Costello syndromes (CS) are the NCFCs with
the most predictably severe presentation, with individuals often demonstrating prenatal
features such as fetal oedema and polyhydramnios, with a consequent high risk of
premature birth and neonatal problems, including significant mortality (30). Severe
feeding problems and failure to thrive are both present in the majority of cases of CFC
and CS (19, 31). Intestinal malrotation or pyloric stenosis may occur, contributing to
nutritional difficulties (18). Nearly all patients with CS and CFC have significant
developmental delay. Abnormal scalp hair is very common, which may be unusually

27
sparse, thick or very curly (31). Facial features may be unremarkable in infancy, but
coarsen with age, and are often similar in the two conditions, though distinguishable in
classical cases, see Figure 1-2. Strabismus and nystagmus are common in both patient
groups (31).
28
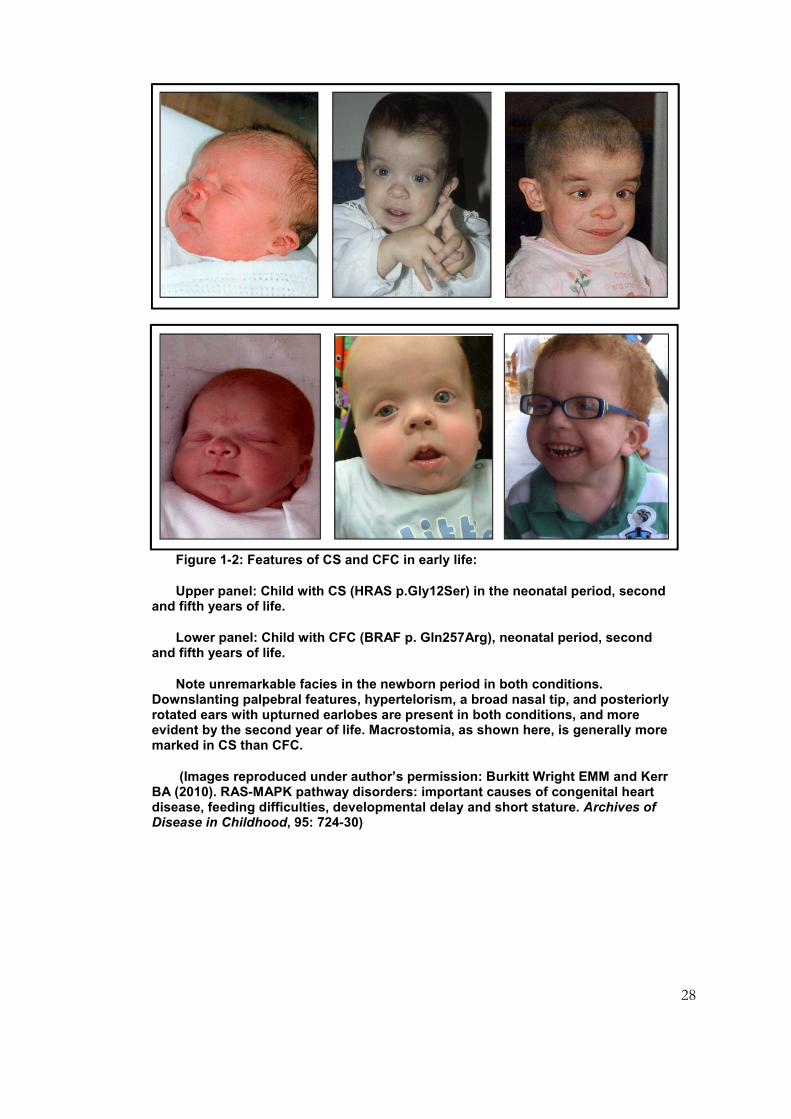
Figure 1-2: Features of CS and CFC in early life:
Upper panel: Child with CS (HRAS p.Gly12Ser) in the neonatal period, second and fifth years of life.
Lower panel: Child with CFC (BRAF p. Gln257Arg), neonatal period, second and fifth years of life.
Note unremarkable facies in the newborn period in both conditions. Downslanting palpebral features, hypertelorism, a broad nasal tip, and posteriorly rotated ears with upturned earlobes are present in both conditions, and more evident by the second year of life. Macrostomia, as shown here, is generally more marked in CS than CFC.
(Images reproduced under author’s permission: Burkitt Wright EMM and Kerr BA (2010). RAS-MAPK pathway disorders: important causes of congenital heart disease, feeding difficulties, developmental delay and short stature. Archives of Disease in Childhood, 95: 724-30)
29
Table 1-1: Key clinical features of the NCFCs
Noonan syndrome
NS with multiple lentigines
CFC syndrome
Costello syndrome NF type I Legius syndrome
Year first described
1965 1969 1986 1977 1768 2007
Normal/large birthweight
+ + + + + +
Macrocephaly + + +/- + + +
Feeding difficulties
+/- +/- +++ +++ Not usual -
Short stature + Not usual + + +/- Not usual
Developmental disability
If present, often mild / SLD
Not usual May be severe Mild to moderate 30%, often mild / SLD
?Milder than in NF1
Congenital heart disease
+ + + + 10% Appears uncommon
HCM + + + + Rare Not reported
Cardiac dysrhythmia
Uncommon unless HCM
Heart block Not well established
Atrial arrhythmia in 50%
Uncommon Appears uncommon

30
Noonan syndrome
NS with multiple lentigines
CFC syndrome
Costello syndrome NF type I Legius syndrome
Cutaneous features
Occasional CAL Lentigines Ulerythema ophrhyogenes;
Keratosis pilaris
Excess skin;
papillomata;
hyperkeratosis
CAL;
Cutaneous neurofibromas
CAL; depigmented macules; lipomas
Sensorineural deafness
Rare Common Rare Rare Rare Not reported
Tumour risk Leukaemias (JMML, AML); giant cell tumours, ?modest risk for solid tumours
Not established;
single reports [13]
Not established; individual reports [25, 26]
High: 17% for childhood cancer, rhabdomyosarcoma; bladder cancer
Increased risk for a wide range of tumours
Not established; single report of Wilms tumour [29]
Variant phenotypes/ genotype-phenotype correlation
CRAF: HCM [21]
SOS1: skin, hair [22]
SHOC2: skin, hair, GHD [23]
CBL: leukaemia [24]
Not established Not established
Severe lethal phenotype with certain mutations;
Mild phenotype with others
Certain hypomorphic mutations: CAL only [28]
Gene deletion: high NF burden, tall stature
Not established

31
1.4 Characteristic clinical aspects of cardio-facio-cutaneous
syndrome (CFC)
CFC was first described as a clinical entity in 1986 by Reynolds et al (32), who
studied a group of 8 patients with similar cardiac, facial and cutaneous features, in the
setting of significant developmental disability and growth failure. Prior to the
identification of the genes mutated in CFC, publications on this condition were few, and
hence the natural history was poorly defined.
Patients with CFC typically have more severe developmental delay and worse long-
term neurological outcomes than those with other NCFCs: ventriculomegaly,
hydrocephalus, structural cerebral abnormalities, and epilepsy are all common in CFC
(31). 50% of patients have seizures, which may present as infantile spasms, and may also
be hard to control (33). EEG abnormalities are present in many patients with CFC,
including some who do not have seizures (14). A large variety of structural congenital
brain abnormalities have been reported, but often only in a very few patients (14), but
optic nerve hypoplasia is a recurrently identified feature (4).
Ectodermal abnormalities are also typically present in patients with CFC, particularly
absent eyebrows (ulerythema ophryogenes) and keratosis pilaris (34). Palmoplantar
keratoderma and ichthyosis are both well-recognised sequelae, and a significant
proportion of patients develop significant numbers of naevi (34). Sparse and/or very
curly hair are both reported in the very large majority of patients with CFC ascertained to
date, but the true frequency of these characteristics in CFC has potential to be
overestimated as they are considered so typical that the diagnosis may be less likely to be
considered in a patient without such findings.
Gastrointestinal dysfunction is common in patients with CFC (14), but whilst
structural features such as malrotation have been found in some (31), the underlying
basis for the severe feeding difficulties that are frequently observed is not clear.
Disproportionately severe feeding difficulties are also a feature of CS (21), and it seems
likely that a common basis may exist for these problems (as for other shared features)
that occur in both CFC and CS. Similarly, facial morphology in patients with CFC, NS
and CS is often similar, with macrocephaly, broad forehead, downslanting palpebral

32
fissures and hypoplasia of the supraorbital ridges, together with a short nose with
depressed nasal bridge and posteriorly rotated ears with large upturned lobes, as shown
in Figure 1-2. 3D analysis of facial morphometry has demonstrated the significant
overlap recognised clinically across NS, CFC and CS, with CFC showing greater
similarities with the other two conditions than these do with one another (Dr P.
Hammond, unpublished data). Aside from this frequently characteristic appearance,
however, the extension of genetic testing to patients with less classical presentation has
revealed increasing numbers of individuals with atypical features (including
microcephaly) who have CFC-associated mutations.
Cardiac abnormalities in CFC show considerable overlap with those observed in
other NCFCs, particularly NS and CS, with pulmonary stenosis, atrial septal defect and
HCM all commonly reported (31). Again, the very high estimates of around 80% of
patients having a cardiac anomaly (14) have potential to be inflated by the lower
likelihood of consideration of the diagnosis in a patient without an identified cardiac
phenotype.
It is thought that the musculoskeletal phenotype in CFC is usually less severe than
that seen in CS (21), but again due to the young age at which many patients have been
assessed and the lack of large series, it is hard to draw conclusions on the published data
(14). In the largest published group of CFC patients, pectus deformity was present in
around half of patients, and scoliosis in around one third (4), suggesting that many
patients do have musculoskeletal elements to their phenotype.
Whilst no increased risk of cancer has been confirmed in CFC, a case of
hepatoblastoma, in a patient immunosuppressed after cardiac transplantation (35), and
two cases of acute lymphoblastoid leukaemia (36) have been reported. As the
denominator total number of patients diagnosed to date with this condition worldwide is
thought to be around 400 (37), these data are compatible with a modestly increased risk
of childhood cancer: the background population risk for any individual developing such a
cancer is around 1 in 600 (and in any case, the number of patients with CFC is
sufficiently small that a lack of association could never be proven). This appears broadly
similar to the situation for NS (with the exception of the specific risk for leukaemia that

33
is well documented in NS) (27). High early mortality from a severe neurological
phenotype or other sequelae of CFC (14), may also be an important contributor to the
absence of any identifiable cancer predisposition; the variable clinical presentation and
current lack of a definitive diagnostic test may also confound comprehensive assessment
of the true risk.
Overall, the natural history of CFC remains poorly defined in the literature, and
requires further clarification. Prior to the identification of its genetic basis, Kavamura et
al proposed an inventory of 82 items on which to calculate the ‘CFC index’ (38). This
was designed to be a means of distinguishing patients with CFC from those with other
conditions with which it shows the greatest clinical and genetic overlaps, particularly NS
and its variants, and CS. The emerging diversity of phenotypes associated with mutations
in the various CFC genes demonstrates the extreme limitations of any such classification
(see Sarkozy et al, 2009 (39), for the example of BRAF).
1.5 Disorders demonstrating clinical overlap with CFC syndrome
1.5.1 Noonan syndrome (NS) and Noonan syndrome with multiple
lentigines (formerly LEOPARD) syndrome (NSML)
As discussed above, the prevalence of NS has never definitively been ascertained
(13), and it is thought that many individuals remain undiagnosed, as adults especially may
be asymptomatic. NS shows extreme phenotypic variability, from mildly short stature
with normal intelligence to severe congenital heart disease or hypertrophic
cardiomyopathy (HCM), or (rarely) severe learning disability (40). Typical features on
examination include ptosis, downslanting palpebral fissures and pterygium colli.
Cryptorchidism is common, and contributes to the reduced fertility observed in NS
males (12). Childhood leukaemia is an occasional finding in NS, but there is a better
prognosis for myeloproliferative disorders occurring in NS than in other situations, with
instances of spontaneous remission reported (41). Work to assess the psychological and
psychiatric features of NS has identified high levels of anxiety and attentional difficulties
in this patient group, such that underperformance at school and in other social situations

34
may be a problem, even for those patients whose general intelligence is measured within
the normal range (42).
Various subtypes of NS are now recognised, including Noonan syndrome with
multiple lentigines (NSML, previously termed LEOPARD syndrome, for lentigines,
electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal
genitalia, retardation of growth, and deafness). Sensorineural deafness, HCM and cardiac
conduction abnormalities are common presenting features (23), whilst short stature and
learning disability are both less common than in the classical NS population. NSML is
therefore usually readily distinguishable from CFC and CS due to this frequently mild
and rather specific phenotype, particularly when numerous lentigines develop from early
childhood, predominantly over the face and trunk. The tumour risk in NSML is not
thought to be high, but, analogous to observations in NS, individual cases of
neuroblastoma, myelodysplasia, acute leukaemia and other forms of neoplasia have been
described (23). Like NS in general, the majority of patients with NSML have a mutation
in PTPN11, but with considerable genetic heterogeneity demonstrable in the remainder,
including some patients with no currently identifiable mutation, as discussed further in
1.7.3. Whilst HCM is a common feature in patients with NSML, affecting 75% (24), it is
also seen in 20% of patients with classical NS (22).
Further groups of patients with distinctive NS-like presentations have also been
described. ‘Noonan-like syndrome with loose anagen hair’ was described by Mazzanti et
al in 2003, with various features of NS such as short stature and shared facial
characteristics, but also prominent ectodermal features, including easily plucked hair with
characteristic histology, and a hypernasal voice (43). This phenotype appears to have
been described previously in single patients, for example the child described by Baraitser
and Patton (44). The existence of such variants, which may show considerable overlap
with patients with CFC, emphasises the difficulty in assigning diagnostic terms across this
group of related conditions with such variable clinical severity and genetic cause.
1.5.2 Costello syndrome
The phenotype of Costello syndrome (CS) is frequently characteristic, and, even prior
to the advent of a definitive genetic test for CS, the existence of a distinct patient group

35
to study meant that, despite its rarity, the clinical aspects of this condition could start to
be successfully delineated (45-47). A mutation in HRAS is present in all patients with CS,
meaning that, firstly, genetic testing is easily accomplished, and secondly, this represents a
definitive inclusion criterion. As discussed in section 1.7.1.1, classical CS arises most
frequently in association with a mutation of codon 12 of HRAS, frequently p.(Gly12Ser),
and milder phenotypes may arise in association with mutations of other codons, for
example the p.(Gly13Cys) substitution.
Especially in early life, patients with CS may have many features in common with
those with CFC, including severe and frequently prolonged feeding difficulties, cardiac
hypertrophy, and macrocephaly (21). Whilst the initial presentation of CS may be similar
or even more severe than that of CFC, with substantial mortality in the first year of life
(20), patients with CS often achieve higher levels of function than those with CFC in the
long term (20). As in CFC, short stature and developmental delay in childhood are
usually present, but so too are additional features more characteristic of CS. These
include neonatal atrial arrhythmia, ulnar deviation, excess skin which darkens with age,
papillomata (usually developing after age 2 years) especially at the interfaces of mucous
membranes and skin, and childhood cancers, particularly embryonic rhabdomyosarcoma
and bladder carcinoma (the latter typically from teenage years onwards) (20). The facial
appearance of individuals with CS is often coarser than that seen in other NCFCs,
though this develops over the first few years and the facies in infancy may not be
remarkable, as seen in Figure 1-2. It is particularly important to identify children with
HRAS mutations at the earliest opportunity because of the associated high childhood
cancer risk (48).
1.5.3 Clinical overlap and distinction between the NCFCs
NF1, whilst a very variable condition in terms of severity and variety of possible
complications, is a further example of a genetically homogeneous and usually clinically
recognisable disorder, which again has been extensively characterised (11). In contrast,
NS is a very heterogeneous condition both genetically and phenotypically, and it is likely
that some affected individuals with sufficiently mild or nebulous phenotypes remain
undiagnosed. This heterogeneity has presented challenges for both molecular and clinical
characterisation, and in particular the overlap of NS and CFC in certain patients has also

36
led to challenges in the classification and nomenclature of these disorders. This has more
than semantic significance: for families, the name assigned as the diagnosis may provide
the key to their search for appropriate information, tailored clinical care and appropriate
support from similarly affected peers.
1.6 The Ras-MAPK pathway and its role in cancer
In the 3 decades since HRAS was identified as the first human oncogene (1), the
genes and proteins of the Ras-MAPK pathway have been the subject of intense scrutiny.
Collectively, mutations in KRAS, NRAS or HRAS have been reported in up to 40% of
human cancers (49). KRAS mutations are most common, occurring particularly in lung
and many gastrointestinal cancers (50, 51). NRAS mutations are seen in melanoma and
haematological malignancies (51, 52), whilst HRAS mutations, less common in cancers
than mutations in KRAS or NRAS, are found in some bladder tumours (53). The
mutational spectrum of these genes in cancer is discussed below and in section 1.7.1.
Mutations in genes for other proteins acting in the pathway, particularly BRAF, are
also frequent, being identified in around 25% of tumours (49). Malignant melanomas
(and the naevi from which they frequently develop), thyroid, lung and colorectal cancers
are particularly commonly identified to have such mutations (51, 54), Similarly, gain-of-
function PTPN11 mutations are not infrequently found in leukaemias and other
haematological malignancies (55), and loss-of-function mutations in NF1 are seen in a
broad range of tumours, in keeping with the known tumour predisposition of patients
with germline NF1 mutations (56), which appears highest in those with whole gene
deletions (57).
There is good evidence that each of these mutations are ‘driver’ rather than
‘passenger’ mutations, that is, they are key players in the development or progression of
tumours, rather than being acquired coincidentally in cells in which normal regulatory
mechanisms have failed. They cluster in distinct functional domains, are highly recurrent,
and are usually mutually exclusive: presence of a KRAS mutation, for example, predicts
that a BRAF mutation is very unlikely to be present (50, 55). Which mutation is present
in a tumour can have important therapeutic consequences, for example EGFR inhibitors
such as cetuximab may be effective in treating tumours with mutations in the gene for

37
this receptor, whilst this agent may be ineffective for tumours with mutations in genes
further down the pathway, for example in KRAS (58). Similarly, the potential for
dramatic treatment responses in advanced melanoma by use of selective inhibitors of
mutant BRAF such as vemurafenib (59) is a compelling indication for determining
tumour genotype, as discussed further in 1.10. Testing for selected somatic mutations in
cancers is therefore entering routine clinical practice as a means of optimising therapy,
both for provision of specific treatments and the avoidance of toxicity and other
substantial costs of agents that would be ineffective (60). Cancer-associated mutations,
where gains of function are implicated, show a marked hotspot effect, clustering in
specific functional domains of each gene. For KRAS, NRAS and HRAS, the majority of
mutations found in cancer are in codons 12, 13 or 61 (49). Substitutions at these residues
(and also at residues 59 and 63), close to the position of the gamma-phosphate of GTP,
have long been known to have transforming potential (61), rendering the protein
insensitive to GAP-mediated GTP hydrolysis, and hence constitutively active (62).
The mutation in BRAF that accounts for over 80% of those identified in cancer is
p.(Val600Glu) (49). This, like the majority of other BRAF mutations reported in cancer,
results in increased activation and downstream pathway activity (54). In contrast,
mutations in CRAF and ARAF (the latter being an X-linked gene) both appear to be
very rare in cancer, with mutations found in less than 1% tumour samples analysed, as
recorded on the COSMIC database (49). The mechanisms by which BRAF mutations
drive oncogenesis are not yet fully understood, but recent work investigating previously
observed paradoxical responses to inhibitors of the pathway has shed light on this (63-
66). Homodimerisation of BRAF and BRAF-CRAF heterodimerisation appear to be key
processes. At low concentrations, RAF inhibitors have been observed to activate
pathway activity. Poulikakos et al (63) present the model that this is due to
transactivation, that is, that at low concentrations, it is common for just one of the two
protomers to be drug-bound, and this binding serves to activate the other non-drug-
bound protomer. At higher concentrations, both protomers become drug bound, causing
a reduction in downstream pathway activity.
Mutations in other genes of the Ras-MAPK pathway in cancers are less commonly
seen than those in the Ras genes and BRAF, and are summarised in Table 1-2. In

38
keeping with their respective roles in the pathway, kinase mutations that are recurrently
found in cancer are usually missense substitutions, altering the activity of the kinase,
analogous to the effect of BRAF p.(Val600Glu), whereas mutations in genes encoding
regulatory factors show a wider variety of loss of function mutations (49, 55).

39
Table 1-2: Somatic mutations in genes of the RAS-MAPK pathway in human
tumours
Gene % of tumours in which mutated
Commonest hotspot mutations
Domain(s) Tumour types in which most commonly found
Reported in germline?
PTPN11 6% p.(Glu76Lys) N-SH2 Haematological malignancy
No; p.(Glu76Asp) reported in NS
HRAS 3% p.(Gly12Val)
p.(Gln61Arg)
p.(Gln61Leu)
G-motifs, assemble close to γ-phosphate of GTP
Bladder carcinoma
p.(Gly12Val): reported in severe CS; other codon 12 mutations
KRAS 22% p.(Gly12Asp)
p.(Gly12Val)
G-motifs, assemble close to γ-phosphate of GTP
Lung, colorectal, many other cancers
No; p.(Gly12Ser) reported in CFC
NRAS 8% p.(Gly61Arg) G-motifs, assemble close to γ-phosphate of GTP
Haematological malignancy, malignant melanoma
No
BRAF 20% p.(Val600Glu) Activation segment
Malignant melanoma, thyroid, colorectal
No; p.(Val600Gly) found in one CFC patient
CRAF <1% No strongly recurrent mutations identified
NF1 10% Wide variety of loss-of function alleles observed
SPRED1 <1% No strongly recurrent mutations identified
MAP2K1MAP2K2
Not yet determined
SOS1 <1% No recurrent mutations identified
SHOC2 Not found

40
The advent of massively parallel sequencing has enabled more comprehensive
assessment of tumour genomes. Whilst two single mutations in MAP2K1, p.(Phe129Leu)
and p.(Asp67Asn), had previously been identified in individual cell lines derived from
colorectal and ovarian cancers respectively (67, 68), recurrent mutations in MAP2K1 and
MAP2K2, unlike most known genes for NCFCs, were not identified in cancers until
2012. Recurrent substitutions of both of these genes have, however, now been found in
melanoma cell lines by exome sequencing techniques (69).
1.7 The molecular basis of the NCFCs
The NCFCs result from germline mutations in genes encoding kinases and other
proteins that interact in the Ras-MAPK pathway, as shown in Figure 1-1. Long before
the discovery of the related molecular basis for the NCFCs, phenotypic overlaps between
these conditions were recognised: clinical recognition of the overlap between Noonan
syndrome (NS) and cardio-facio-cutaneous syndrome (CFC) is a prime example (70), as
is the existence of a Noonan-like phenotype in some patients with neurofibromatosis
type I (NF1) (71, 72). In the years since the first genes for these conditions were
identified (NF1 in NF1 in 1990 (73) and PTPN11 in NS in 2001 (74)), there has been
much work undertaken to characterise the resultant phenotypes and how mutations
cause them, but this has been more straightforward for some conditions than others. For
example, Costello syndrome (CS) is a genetically homogeneous condition, with all
patients having a mutation in HRAS (19, 62), meaning that this condition can be
distinguished confidently from other NCFCs on the basis of a single genetic test. This
test is important for clinical prognostication, most specifically the uniquely high risk of
childhood cancer in CS for which specific screening strategies are recommended (75).
In keeping with other autosomal dominant disorders with severe phenotypes,
germline disorders of the Ras-MAPK pathway frequently arise due to de novo mutation in
gametes, meaning that a substantial proportion of affected individuals have no family
history of such a condition. This is especially the case for severe presentations (where
increased mortality is seen, and affected patients are unlikely to reproduce due to multiple
comorbidities including intellectual disability), such as CFC and CS (20, 31). Whilst the
milder disorders may be inherited through the generations, the more severe conditions

41
nearly always arise from new dominant mutations, as shown in Figure 1-3 For NF1,
around a half of cases represent new mutations (11), and in NS, two thirds of cases
appear de novo (though this is harder to confirm, because not all patients have a
causative mutation identified at present, and clinical manifestations may be extremely
mild or absent) (25).
Figure 1-3: The severity of effects of a mutation can influence the context in which it is observed
Examples of Ras-MAPK pathway gene mutations are shown, from those at the top that are sufficiently mild to allow transmission from parent to child, to those at the bottom that are so severe as to be likely to be lethal to the developing embryo, and hence not observed in the germline.

42
NS most commonly arises due to mutations in PTPN11 (which encodes the SHP2
protein), but SOS1, CRAF (RAF1), RIT1, SHOC2, KRAS, NRAS, and CBL have each
been found to be mutated in smaller proportions of patients (5, 76), as discussed below.
Similarly, CFC may arise due to mutations in genes that include BRAF, KRAS, MAP2K1
and MAP2K2 (10, 77). CFC and NS are allelic disorders (25); both severe and mild
presentations may be caused by mutations in the same gene. Despite this overlap, certain
genotype-phenotype correlations are demonstrable across the spectrum, and are
discussed further in section 1.7.3. BRAF mutations, for example, are most commonly
associated with a severe phenotype classical for CFC (39), whilst PTPN11 mutations are
most frequently associated with milder, classical NS presentations (78, 79).
The genes identified to date to be mutated in patients with CFC are shown in Figure
1-4 and Table 1-3, which also shows the other genes responsible for NS and its variants.
The numbers of mutations in these genes that are included on the NSEuronet database
(6) are shown in this table. These cannot be claimed to be representative of the
worldwide prevalence of each these mutations, however, because at present the database
entries are heavily influenced by publication bias, with most of these being either those
retrieved from the published literature, or those known to the team creating the database
(C. Lissewski, in the group of Prof. M. Zenker). As such, noteworthy mutations or
clinical presentations are therefore over-represented (witnessed by the inclusion of 407
HRAS mutations on this database, despite the rarity of CS), and recently described genes
are likely to be under-represented, as testing in the widest possible cohorts of patients
may not yet have been reported. It nonetheless demonstrates the relative frequency of
mutations in these genes in the patients currently represented in the literature (and for
more common disorders, greater numbers of patients are likely to have been identified
overall). RIT1 mutations, too recently described to be in the NSEuronet database at
present, have been described to account for approximately 10% of NS (76), suggesting
that these may be about as common as mutations in CRAF. A list of mutations identified
to date in patients with CFC syndrome is given in appendix 1.

43
Figure 1-4: Molecular basis of CFC syndrome in patients on the NSEuronet database.
Note the high proportion of individuals with mutations in BRAF, with smaller proportions of presentations due to mutations in KRAS, MAP2K1 and MAP2K2. Patients with mutations in SHOC2 are discussed further in chapter 3. The very small numbers of patients with mutations in PTPN11, SOS1 and CRAF illustrate the overlap with NS.
Table 1-3: Numbers of patients with mutations in genes causing NS, CFC and genotypically overlapping conditions, as represented in the NS Euronet database
Gene NS NSML NFNS CFC NSLAH undetermined
PTPN11 897 163 3 3 - 1
SOS1 234 - - 2 - 3
CRAF 81 4 - 2 - -
BRAF 14 3 - 209 - 2
KRAS 39 - - 16 - 2
MAP2K1 3 - - 47 - 1
MAP2K2 - - - 34 - -
CBL 27 - - - - -
NRAS 7 - - - - -
SHOC2 9 - - 10 93 5

44
For many dominant de novo disorders, mutations have been identified to originate
In keeping with other autosomal dominant disorders with severe phenotypes, CFC-
and CS-associated mutations, along with many NS-associated mutations, arise de novo in
gametes. These gain-of-function mutations occur almost exclusively in spermatogenesis
(80). Selective clonal advantage of CS-associated mutated HRAS alleles has been
demonstrated: the mutation confers a proliferative advantage to spermatogonia, causing
an enrichment for mutated cells in the course of spermatogenesis (81). This helps to
explain the very high rate of mutation observed at codon 12 of HRAS in the germline,
compared to the baseline mean rate of mutation at any given nucleotide per generation
(81). In keeping with these findings, an increasing proportion of sperm have been
identified to carry such mutations with increasing age of the donor (81). The spectrum of
causative mutations for the NCFCs overlaps with those observed in human tumours, but
alleles that are compatible with germline development usually display less extreme effects
upon signal transduction than those seen in cancer and other instances of somatic
Figure 1-5: Genes in which mutations have been found in patients with NCFCs
The left ellipse includes NS and NSML- associated genes, the right ellipse, CFC-associated genes. A significant overlap is seen: there are many genes that, when mutated, can cause presentations of either type. Whilst NF1 and Legius syndrome are usually clinically distinct from NS, the small number of patients with mutations in NF1 or SPRED1 and an NS phenotype is indicated by these being
shown overlapping the border of the NS ellipse.

45
mutation (16, 55). Factors promoting survival or clonal advantage of any given mutant
allele may differ in gametic, embryonic and fetal stages and across the postnatal lifespan.
1.7.1 Comparison of the molecular basis of the NCFCs with the
mutational spectrum observed in cancers
1.7.1.1 HRAS
HRAS mutations cause Costello syndrome (CS). p.(Gly12Ser) is much the
commonest mutation found in patients with CS (20), accounting for 80% of patients
diagnosed. Other mutations of codon 12 also occur, accounting for the majority of the
remainder of identified patients. Mutations of codon 13 have been identified as a rare
cause of CS, with a milder presentation (82). Severe neonatal phenotypes have been
reported in association with the common p.(Gly12Ser) mutation (83), but are usually due
to mutations that are less common in the germline (83), and more commonly observed in
cancers (16, 49). Mutations at codons 12 and 13 affect the kinase domain of the protein,
resulting in it being constitutively active (19) The degree of activation of the protein may
dictate the severity of the phenotype, and mutations resulting in milder CS phenotypes
(84) are much less frequently observed as somatic events in cancers (49).
1.7.1.2 KRAS
KRAS mutations are a rare cause of CFC, and a rare cause of NS (85). The mutations
described are predominantly de novo, and at least mild learning difficulties have been
present in the large majority of patients, whether their clinical diagnosis was NS or CFC.
Familial transmission of KRAS mutations has, however, now been documented in two
families (86), where, as would be expected, the phenotype in the parent carrying the
mutation was relatively mild. From the small number ascertained to date, it appears that
ectodermal features may be relatively mild in this patient group, emphasising the
frequently intermediate clinical presentation between NS and CFC of germline KRAS
mutations. Patients’ phenotypes may also show significant overlap with those of
individuals with Costello syndrome (87).
One of the commonest KRAS mutations in cancer, like that seen in HRAS, is
p.(Gly12Val) (49). Whilst HRAS p.(Gly12Val) and p.(Gly12Asp) have, rarely, been

46
recorded in the germline of individuals with severe, lethal, Costello syndrome (62, 83),
neither KRAS p.(Gly12Val) nor p.(Gly12Asp) have ever been reported in the germline.
KRAS p.(Gly12Ser) has been reported as a rare cause of CFC (88), but no other
mutations of this codon of KRAS have been reported in the germline. The spectrum of
mutations described in CFC/NS is wide, and the number of patients quite small, but
p.(Val14Ile) and p.(Asp147Val) appear to be the most commonly identified to date (6).
This wide spectrum is in contrast to the high preponderance of mutations at codons 12
and 13 in cancers, accounting for over 90% of entries on the COSMIC database (49).
The mutations for which familial transmission has been observed have not yet been
found recurrently in further patients with NS/CFC (6).
1.7.1.3 NRAS
NRAS mutations have only been identified in a very few patients with NS, with only
seven such patients identified (6) since the gene discovery in 2010 (89). This is in contrast
to the high number of tumours in which this gene has been seen to be mutated: 3447 of
60233 samples analysed (5.7%). Four of seven known patients with germline mutations
had the p.(Gly60Glu) mutation. Mutation at this codon is a very infrequent finding in
tumours (11/60233), in contrast to mutation at codon 61, at which two-thirds of all
known somatic mutations in this gene have been identified (2224/60233 samples).
Nearly all of the remainder of tumour-associated mutations have been identified at
codons 12 and 13. Mutation at codon 12 has been reported in blood and mouth swab
DNA from a patient with autoimmune lymphoproliferative syndrome (90), and an
apparently germline mutation in codon 13 in a patient with JMML (91).

47
1.7.1.4 BRAF
Mutation in BRAF is the commonest identified molecular cause of a CFC
phenotype, with around two-thirds of identified causative mutations being in this gene
(37). BRAF mutations associated with CFC cluster in exon 6, encoding the cysteine-rich
domain, and exons 11-16, encoding the protein kinase domain. Within exon 6, a
mutational hotspot is observed at codon 257, with p.(Gln257Arg) accounting for up to
50% of all molecularly diagnosed CFC (92). In cancer, as discussed above, a wide variety
of mutations in BRAF have been described (49). The degree of overlap between cancer-
associated and CFC-causing BRAF mutations is considerably less than is the case for
HRAS mutations in cancer and CS, and more in keeping with the comparison between
KRAS somatic and germline mutations. BRAF p.(Val600Glu) has been shown to be
embryonic lethal in mouse (93). This observation, in conjunction with the fact that this
mutation has not been observed in the germline, suggests that it would also be lethal in
human development. In contrast, many of the rarer cancer-associated mutations
clustered in exons 11, 14 and 15 of this gene have also been described in the germline in
CFC patients (37). Where these substitutions have been functionally assessed, they
appear to have variable effects on downstream pathway signalling, as discussed in section
1.8. A germline p.(Val600Gly) mutation in BRAF has been described in a single patient
with CFC; transient transfection experiments demonstrated that this mutation generates
higher levels of ERK1/2 phosphorylation and ELK-1 transcription than wild-type
BRAF, but not as high as those observed with p.(Val600Glu) (94).
1.7.1.5 CRAF
CRAF (RAF1) mutations are an uncommon cause of NS, and similarly, CRAF
mutations appear to be rare in cancer. Codon 257 is observed as the most commonly
mutated residue in both cancer and NS, with p.(Ser257Leu) mutations accounting for
over half of all NS-associated mutations recorded (6).

48
1.7.1.6 MAP2K1 and MAP2K2
MAP2K1 and MAP2K2 mutations collectively account for approximately 20% of
molecularly diagnosed CFC (37). Until recently, all germline MAP2K1 and MAP2K2
mutations had been found to be de novo, where assessed, but two families, one with
affected members across four generations, have now been published with vertical
transmission of MAP2K2 mutations, the first descriptions of familial CFC, with relatively
mild presentations, in which skin manifestations were predominant (95, 96). As discussed
in 1.6, recurrent mutations in these genes have only recently been described in cancers
(69), and are much rarer than those observed in other CFC-associated genes BRAF and
KRAS.
1.7.1.7 PTPN11
PTPN11 mutations are identifiable in half of all patients with NS. These are nearly all
missense substitutions, clustered in distinct regions of various domains of the SHP2
protein. Codon 308, in the protein tyrosine phosphatase domain, is the site most
commonly mutated (6). As discussed in 1.7.5, specific mutations are associated with
Noonan syndrome with multiple lentigines (NSML), and distinct functional effects can
be observed for such substitutions (as discussed in section 1.8). Somatic mutations in
PTPN11 have been identified in approximately 3% of tumours studied (49), particularly
myeloid malignancies, characteristically juvenile myelononocytic leukaemia (JMML) (97),
as is also seen in NS (78). PTPN11 mutations appear rare in solid tumours (49), in
keeping with the low prevalence of such tumours in NS (22). As observed for KRAS,
some degree of overlap between somatic and germline substitutions is observed, but, as
for other genes discussed above, mutations compatible with germline survival appear to
have less dramatic effects upon downstream signal transduction (25). Similarly, a
favourable prognosis for NS-associated JMML has been observed compared to that
which is due to somatic, more highly activating, mutations in PTPN11(98).

49
1.7.1.8 SOS1
SOS1 mutations are thought to account for around 10% of all instances of NS (98).
These mutations affect residues with key roles in autoinhibition of the protein, resulting
in increased baseline activity of the Ras-MAPK pathway (99). In contrast to PTPN11,
somatic mutations in this gene have rarely been identified (49), suggesting that these are
not a major contributor to carcinogenesis. However, rhabdomyosarcomas have now
been recorded in multiple patients with SOS1-associated NS (100), suggesting that their
germline mutations may be significant contributors to the development of this tumour,
which is also characteristically seen in CS (101).
1.7.1.9 CBL
CBL mutations appear to be a rare cause of NS, reported in only a few families so far
(102, 103). The characteristic feature of these families was a high risk of JMML. Other
features of NS appeared to be very mild, such that in several families mutations had been
transmitted by a parent without features of NS (102, 103). In keeping with this mild
phenotype, somatic mutations in CBL in cancer also appear to be rare (49), but are
clustered in the same region of the gene as the germline mutations, displaying an almost
identical relative frequency, with p.(Tyr371His) being the most commonly observed
mutation in the germline (6) and in malignancies (49).
1.7.1.10 SHOC2
In the published literature, germline SHOC2 mutations have been exclusively
p.(Ser2Gly) (104, 105). This mutation was identified to cause the protein to relocate to
the cell membrane, and to hence recruit RAS proteins to this location, where they could
be activated (104), hence resulting in increased downstream Ras-MAPK signal
transduction. SHOC2 mutations have not been recurrently identified in cancers, despite
testing of over 8000 samples (49).

50
1.7.1.11 RIT1
RIT1 mutations are a very recently described cause of NS (76). As for the other genes
described above, missense substitutions conferring a gain of function have been
described, that are clustered at specific residues of this small Ras-like protein. Whilst
somatic mutations in this gene have rarely been described (49), the p.(Met90Ile)
substitution that has been observed in the germline in patients with NS (76) is the most
commonly observed RIT1 substitution in cancers (49).
1.7.1.12 NF1 and SPRED1
NF1 and SPRED1 are both negative regulators of the Ras-MAPK pathway (9). In
keeping with these roles, the mutations identified in patients with neurofibromatosis type
I and Legius syndrome are a wide spectrum, representing loss-of-function alleles (9, 106).
10% of tumours harbour alterations in NF1, and, like in germline samples from patients
with NF1, frameshift or nonsense changes constitute the majority of mutations
identified. In neurofibromas and other NF1-associated tumours, the wild-type NF1 allele
is lost, that is, these lesions contain cells with no functional copy of NF1 (107, 108). In
contrast to these observations, the effects of NF1 upon the brain appear, in mouse, to be
mediated by haploinsufficiency, the loss of one allele being sufficient to place individuals
at risk of the learning and attentional difficulties known to be frequent in this condition
(109). Sequencing of SPRED1 in tumours has demonstrated mutations in less than 1%
of samples (49), and these have been largely missense changes of uncertain pathogenicity
and non-recurrent, suggesting that this gene has, in contrast to NF1, no major role in
tumorigenesis.
1.7.2 Genomic factors that may affect Ras-MAPK pathway activity
Increased activity due to increased copy number of PTPN11 has been reported as a
rare cause of NS (110), and one patient has been reported with duplication including
CRAF, in association with an NS-like phenotype (111), but there are as yet very few
other published reports of duplication of other pathway genes in patients with an NCFC
phenotype. Tandem duplication of 7q34 including BRAF was reported as a means of
Ras-MAPK pathway activation in low-grade astrocytomas (112), but the precise
mechanism of activation may instead be via the creation of a novel fusion gene encoding

51
the kinase domain of BRAF (113), which has now been found in many different tumour
samples analysed (49).
A further mechanism by which severe NCFC phenotypes have been described is the
presence of two separate and functionally important Ras-MAPK pathway associated
mutations in the same patient. Examples of this include an individual with biallelic
inherited mutations in PTPN11 (114), where both parents were affected with NS and the
baby had a lethal outcome; coexistent inherited PTPN11 and de novo SHOC2 mutations
(115); and patients with both a PTPN11 and a SOS1 mutation (116). Similarly, in rare
patients with neurofibromatosis type 1-Noonan syndrome, mutations in both NF1 and
PTPN11 have occasionally been found (117, 118) (although missense mutations in NF1
are a much more common cause of such presentations (72)). The frequency with which
more than one mutation is an important contributor to an affected individual’s NCFC
phenotype is currently unclear, but the small possibility of multiple contributory
mutations is important information for those counselling affected families. The
recognition of such patients may increase with the increasing use of broader spectrum
genetic testing, for example by massively parallel sequencing, as discussed in section
1.7.4.1.
1.7.3 Genetic testing in the NCFCs and implications of a molecular
diagnosis
Molecular confirmation of a genetic diagnosis is important for many reasons.
Neurodevelopmental outlook or cancer risk may depend upon the result, accurate genetic
counselling can be provided for families regarding recurrence risk, confirmatory testing
may be indicated in further family members, and prenatal diagnosis can be offered. The
need to differentiate between CS and CFC is particularly apparent, as there is a direct
impact upon management when a molecular diagnosis of CS is confirmed: screening for
rhabdomyosarcoma in early years and bladder neoplasia from the second decade of life
are recommended (75). Whilst no such immediate management implications are currently
recognised following a molecular diagnosis of CFC, such a confirmation is important for
advising families as to the likely complications and developmental outcomes (14).

52
Low recurrence risks for offspring of the parents of children with molecularly
diagnosed NCFCs can be confirmed if neither parent carries the mutation in lymphocyte
DNA. Recurrences have been reported however, due to either demonstrated somatic or
presumed gonadal mosaicism. A father with a mosaic HRAS p.(Gly12Ser) mutation, and
some features of CS, was reported in the literature after his child was born with non-
mosaic CS (119). Sibling recurrence of CS due to parental gonadal mosaicism has also
been reported (120), and the first instance of sibling recurrence of CFC syndrome, due to
a mutation in BRAF with presumed parental gonadal mosaicism, has also been observed
recently in the MCGM diagnostic laboratory (Prof. A. Green, unpublished data). An
unusual further report of two siblings with severe NCFC phenotypes, one having a
KRAS mutation and the other an HRAS mutation (121), cannot be adequately explained
currently other than as a coincidence. Prenatal diagnosis of fetuses with CFC and CS has
now been reported (122, 123) when amniocentesis was performed for suggestive features
on antenatal scans, including characteristic hand posture and polyhydramnios. Whilst
such tests can allow informed decisions regarding the pregnancy to be made, they are
currently only rarely available, that is, in the fetus with both a highly suggestive prenatal
presentation and readily identifiable mutation. The majority of individuals with these
conditions are therefore likely to continue to be ascertained postnatally in the foreseeable
future. Local experience demonstrates a high take-up of prenatal testing by chorionic
villus sampling for parents with a previous child with CFC or CS: to date, 8 such requests
have been actioned in the MCGM diagnostic laboratory, all with normal results (as would
be expected for the low recurrence risk). In contrast, whilst prenatal testing for a 50%
risk of NF1 and NS is available where the mutation has been identified in the affected
parent, to date it has not frequently taken up by families, presumably in view of the
highly variable and often mild phenotypes involved.

53
1.7.4 New genomic and genetic technologies for investigation of the
NCFCs
Patients with NCFC presentations are a worthwhile group to study by new genomic
and genetic technologies, because of the frequent difficulty of achieving a specific
diagnosis on clinical grounds (48) and the large number of genes that may be involved
(17). Additionally, some patients’ presentations may be due to genomic copy number
imbalance, rather than a single gene disorder. Single nucleotide polymorphism based
array comparative genomic hybridisation has already demonstrated 17q21.31
microdeletions in several patients with a CFC phenotype, for example (124), appendix 7,
and individual instances of other novel chromosomal microdeletions, such as deletion of
6q25-q27, have also been described in patients where previously CFC syndrome was the
likely clinical diagnosis (Dr B. Kerr, personal communication). Of note, this latter
deletion included ARID1B, a gene subsequently implicated in patients with Coffin Siris
syndrome, a description which may include individuals with phenotypic features in
common with those of certain CFC patients (125, 126). For genetic disorders, massively
parallel sequencing is transforming the prospects for molecular diagnosis. The ability to
sequence many genes at once may be of particularly obvious value both for the diagnosis
of genetically heterogeneous disorders and for the identification of further causative
genes, as discussed below in sections 1.7.4.1 and 1.7.4.2.
1.7.4.1 Massively parallel sequencing for molecular diagnosis
Ongoing innovation in all phases of the processes involved in massively parallel
sequencing of human DNA has brought enormous reductions in cost, and reciprocal
increases in efficiency and effectiveness of these techniques. As a result, these
technologies are being applied to increasingly diverse aspects of molecular diagnosis.
Molecular diagnostic testing by conventional Sanger sequencing for genetically
heterogeneous Mendelian disorders, such as NS and CFC, has historically been extremely
laborious, and hence prohibitive for many patients assessed by clinical genetic services
across the world, including the United Kingdom. A recent audit in the MCGM clinical
department demonstrated this (unpublished data), with only 1 of a group of 30
individuals with a confident clinical diagnosis of NS but no known mutation having had

54
testing of all available genes for the condition. Targeted resequencing of all known genes
for disorders demonstrating extreme genetic heterogeneity has now successfully entered
clinical diagnostics, such as for retinal dystrophies, where over 100 genes are known to
cause disease, and sequencing of all of these genes can now be integrated into a single
investigation (127). Accurate phenotyping will be a prerequisite to success of any such
targeted approach, just as has always been the case for molecular testing of single genes.
For de novo disorders, diagnostic exome sequencing of affected individuals and their
unaffected parents may be an elegant means of identifying the mutation responsible for
the phenotype (128). Whilst this is dependent upon samples from both parents being
available for analysis, and the generation of high quality coverage across each of three
samples, such a technique, when accurate phenotypic data are available, can be an
effective means of achieving a diagnosis. Whilst decisions about which genes to sequence
may not need to be made, phenotypic data may be crucial in guiding data analysis in the
event that multiple possibly causal, but unclassified, variants are identified. Whole exome
sequencing approaches may initially be most appropriate when more selective genetic
testing has failed to demonstrate the cause for their condition, but will increasingly be
employed at an earlier stage in the diagnostic process, as technologies and strategies for
performing such analyses become more established and streamlined. Additionally, it
represents a means of investigating phenotypes such as autism or intellectual disability
that may be due to mutation in any one of a very large number of genes (128, 129), and
hence not previously amenable to diagnostic genetic testing.
Multiple techniques and platforms have been developed for massively parallel
sequencing (130, 131). As algorithms for base calling and alignment improve (132), in
addition to increased confidence of nucleotide sequence results, accurate assessment of
copy number variation (CNV) can now be achieved through analysis of the short
sequencing reads (133). A significant proportion of patients with undiagnosed syndromic
presentations have causative genomic imbalances (estimated at 15% of those tested by
array CGH for developmental delay and congenital anomalies (134)), and hence the
ability to assess CNV increases the potential utility of massively parallel sequencing for
clinical molecular diagnosis. For the NCFCs, where a small number of patients may have
increased copy number of a relevant gene (110, 111), this additional possibility may

55
increase the utility of the diagnostic test. Similarly, the presence of multiple relevant
variants in different genes, as is present in a small proportion of patients with NCFCs
(discussed in section 1.7.2), is a further reason in favour of comprehensive testing for
these individuals.
Just as massively parallel sequencing is applicable to germline disorders, it can also be
harnessed for somatic mutation detection in a clinical context. In tumour tissue, where a
wide variety of mutations may exist, this can be of great value, characterising
malignancies in a timely manner to guide treatment. For malignancies where sequential
samples are readily obtainable, such as haematological malignancies, repeated massively
parallel sequencing tests can be used to diagnose the molecular progression of the
malignancy, and be a guide to prognosis and treatment (135).
1.7.4.2 Massively parallel sequencing for novel gene discovery
Several strategies for discovery of novel causal variants using massively parallel
sequencing have been developed. Comparison of massively parallel sequencing results
with data from other genome-wide analysis methods such as linkage (136), autozygosity
mapping (137), or loss of heterozygosity/copy number variation (138) has been
successful in this. Use of such collateral information to narrow the genomic interval of
interest increases the chance of successful gene identification by greatly reducing the
number of candidate variants that need to be assessed. To identify the causative gene for
de novo germline disorders, pedigree-based analyses such as linkage or autozygosity
mapping are not generally relevant, but copy number information (138) may be of
relevance, for example when disorders are caused by loss of function mutations.
Alternatively, where a gain-of-function mutation (as are responsible for NS/CFC/CS) is
present, subsequent loss of the wild-type allele may give rise to additional dysregulation at
the cellular level, such as occurs with loss of the wild-type HRAS allele in CS-associated
tumour tissue (139). In this situation, loss of heterozygosity data from tissue likely to
have sustained a second hit may suggest a genomic region for which the sequencing data
should be particularly closely scrutinised.
Successful gene identification has been achieved for conditions where similar, or
indistinguishable, phenotypes result from gross genomic deletion. An example is the

56
17q21.31 microdeletion syndrome (138). Previous molecular characterisation of this
syndrome by array comparative genomic hybridisation (aCGH) had demonstrated a small
number of deleted genes as candidates for major contributors to the phenotype (140).
This finding allowed focused analysis of these genes, by massively parallel sequencing
methods, to identify loss of function mutations in KANSL1 in patients with phenotypes
suggestive of 17q21.31 microdeletion but no abnormality on aCGH (138).
A further successful strategy has relied upon meticulous phenotyping to identify
groups of patients with very similar phenotypes, in order to maximise the chance of
genetic homogeneity within the study population, and hence the chance of identifying a
single gene (or functionally related set of genes, such as those for subunits of a protein
complex) in which affected individuals each have a mutation. This has proven an elegant
solution for many de novo dominant disorders, particularly those which are genetically
homogeneous and where mutations are exquisitely clustered within a single functional
domain, such as Schinzel Giedion syndrome (141), Shprintzen Goldberg syndrome (142)
and Myhre syndrome (143, 144). It has also been used with success for disorders with a
degree of genetic heterogeneity but a characteristic phenotype, for example Kabuki
syndrome: Ng et al (102) identified mutations in 7 of 10 exomes sequenced from patients
with a clinical diagnosis of this condition.
Comparative massively parallel sequencing has also been successfully used to
determine the basis of developmental mosaic phenotypes, including those caused by
mutation in genes of the Ras-MAPK and other signal transduction pathways. Mosaic
mutations in HRAS and KRAS have been identified to cause Schimmelpenning
syndrome (145), which describes a severe phenotype of skin and brain abnormality.
Similarly, mutations at codon 61 of NRAS have recently been identified to underly
neurocutaneous melanosis (146). The effects of such mutations will be exquisitely
influenced by the point in development at which the mutation is sustained and the
resulting cell lineages and tissue distribution affected. Milder or more limited phenotypes
may be found to be due to similar or identical mutations with a yet more limited tissue
distribution.

57
A further context in which exome or genome sequencing for somatic mutations has
been highly successful is in analysis of cancer genomes. Comparative sequencing of
thousands of tumour samples is now possible, identifying many novel recurrently
mutated genes, including MAP2K1 and MAP2K2 (69), as discussed in section 1.6, and
hence highlighting further pathways that are involved in tumour pathogenesis.
For each of the situations described above in which new generation sequencing
(NGS) can be used for research, there is potential for translation of the findings into
clinical practice. Novel genes that are identified to have clinical significance in the
research setting can then be tested in patients, either by conventional or NGS techniques.
1.7.5 Genotype phenotype correlations across the NCFCs
As the molecular basis for increasing numbers of patients’ individual conditions is
found, the spectrum of disease associated with each causative gene is being expanded
(39, 147). The scope for genotype-phenotype correlation also increases as larger numbers
of patients’ mutations are known.
1.7.5.1 Noonan syndrome
In NS, the best example of genotype-correlation is the strong association between
certain mutations in PTPN11 and Noonan syndrome with multiple lentigines (NSML)
(148), but it has also been noted that apparently identical mutations may generate either
an NS or a NSML phenotype (6, 149). Further identified correlations in NS include a
higher prevalence of cardiac defects, particularly pulmonary stenosis, in patients with
PTPN11 mutations than in those with mutations in other genes (150), whilst patients
with SOS1-associated NS often have prominent ectodermal features and are less likely to
be of short stature (150).
A further robust association is the very high frequency of HCM observed in patients
with CRAF mutations (151, 152). Of note, due to the relative rarity of such mutations,
and the large proportion of total NS due to PTPN11 mutations (12), for any given
patient with NS and HCM, PTPN11 remains the most likely causative gene prior to
molecular investigation (25). With many early genotype-phenotype assessments in NS,
the only comparisons that could be made were between PTPN11 and non-PTPN11-

58
associated NS, and the limitations of such an approach have been emphasised as the
extent of genetic and phenotypic heterogeneity of this latter group has emerged.
There remain many NS-associated genotypes for which the clinical spectrum is yet to
be fully explored, due to only very small numbers of affected patients having been
identified to date. Examples include NRAS mutations, which appear to be a rare cause of
a classical NS phenotype (89), and CBL mutations, which have a variable presentation
with often very mild NS features (which have also been identified retrospectively in
several clinically unaffected parents with mutations) and a high risk of juvenile
myelomonocytic leukaemia (102, 103). Similarly, the phenotypic spectrum associated with
SHOC2 and RIT1 mutations will be clarified by the study of larger numbers of patients
with mutations in these genes.
Assessments of facial morphology across the different NS-associated genes have
suggested that, whilst classical facial features of NS are most common in patients with
PTPN11 mutations, significant variability is seen in facial appearance for patients with
mutations in both this and other causative genes (153). Similarly, no clear genotype-
phenotype correlations with respect to facial morphology have yet emerged within CFC
cohorts (4), and the only identified correlation in a large series of patients with CFC has
been a higher prevalence of pulmonary stenosis in patients with mutations in BRAF (4),
illustrating the need for systematic clinical and molecular characterisation of larger
numbers of affected individuals.
Mutations in SHOC2 have been described as the cause for ‘Noonan-like syndrome
with loose anagen hair’ (104), the clinical presentation of which is discussed above, but
also, as discussed in chapter 3, in patients with phenotypes clinically described as CFC or
NS. Affected patients in the literature, aside from the distinctive ectodermal phenotype,
are known to have a particularly high rate of growth hormone deficiency, and the large
majority have had at least some degree of learning disability. Whilst this has been
reported to be a relatively homogeneous phenotype (104), the spectrum of
manifestations is likely to expand with the testing of wider cohorts of patients.

59
1.7.5.2 Costello syndrome
In CS, genotype-phenotype correlation is skewed by the fact that 80% of affected
patients have the same mutation, p.(Gly12Ser). However, severe presentations have been
identified to be associated with more dramatically activating mutations, particularly
p.(Gly12Val), the HRAS mutation most commonly observed in cancer (16). Milder
mutations, producing less classical CS phenotypes, have also been described, such as
p.(Gly13Cys) (82).
1.7.5.3 CFC syndrome
Few genotype-phenotype correlations have yet to be identified in CFC, due to the
small number of patients and relatively large number of different genes implicated.
However, a higher frequency of pulmonary stenosis has been identified in patients with a
mutation in BRAF, compared to those with mutations in other genes (4). Whilst only a
small number of patients have been identified with MAP2K2 mutations, vertical
transmission of such mutations has been identified in at least two families (95, 96),
suggesting that the neurodevelopmental phenotype in this patient group may be milder
than that seen in patients with BRAF or MAP2K1 mutations.
1.7.5.4 NF1 and Legius syndrome
NF1 and Legius syndrome, caused by loss-of-function mutations in NF1 and
SPRED1, which encode negative regulators of the Ras-MAPK pathway, have had
relatively few specific genotype-phenotype correlations identified to date, but as greater
numbers of patients have molecular diagnoses made, this situation may change. Existing
genotype-phenotype associations known for NF1 include the high tumour burden and
greater degree of learning disability seen with whole gene deletions (where co-deleted
genes or altered genomic architecture may also play a role), and the isolated café-au-lait
patches phenotype seen with a specific recurrent 3 basepair deletion,
c.2970_2972delAAT [NM_00267.3] (154). Neurofibromatosis-Noonan syndrome
(NFNS) and NF1-associated pulmonary stenosis have each been identified in association
with missense changes in NF1 (26, 71).

60
1.7.5.5 Germline mutations in Ras-MAPK pathway genes resulting in non-
NCFC phenotypes
A number of phenotypes that do not correspond to NCFC disorders have now been
described in association with germline mutations in genes that are also involved in NCFC
syndromes. The phenotypes do not appear directly relevant to those of the NCFCs, and
as such, the finding of mutations in this different context can provide insights into the
pathogenesis of the NCFCs.
Exome sequencing demonstrated loss-of-function mutations in PTPN11 to cause of
metachondromatosis (155), an incompletely penetrant, rare, autosomal dominant
disorder characterised by development of multiple exostoses and enchondromatoses.
Loss of the wild-type PTPN11 allele was demonstrated in these benign tumours, and,
together with the lack of a phenotype in other tissues, this suggests that complete loss of
SHP2 function, specifically in chondrocytes of the cartilage core, is required to generate
the features of this disorder, and that haploinsufficiency for PTPN11 alone does not
cause a cellular phenotype. This finding emphasises that ‘loss-of-function’ per se may not
be the most appropriate way to describe NSML-associated mutations for which impaired
phosphatase activity has been demonstrated (156). The alteration of PI3K pathway
signalling caused by such mutations (157) is a further illustration of this.
A frameshift mutation in SOS1, likely to result in a protein with an altered C-
terminus, has been reported as the cause of isolated gingival fibromatosis in a single large
family (158), and increased proliferation was shown in fibroblasts transfected with the
mutant allele (159). However, the very circumscribed nature of this phenotype and the
unique nature of the mutation, private to a single family, mean that it is unclear to what
extent the molecular pathophysiology of this disorder may be relevant to the multisystem
phenotype of the NCFCs.
1.8 Molecular pathogenesis of the NCFCs
Irrespective of the gene involved, the mutations that cause NCFC presentations
appear to exert their effect by altering signalling activity through the Ras-MAPK
pathway, usually increasing this (55), and potentially also affecting other signal
transduction pathways (157). NF1 and SPRED1 encode negative regulators of the Ras-

61
MAPK pathway, and the loss-of-function alleles identified in patients with NF1 and
Legius syndrome (9, 11) therefore result in increased Ras-MAPK pathway activity. NF1
is a RasGTPase-activating protein (GAP), acting to convert Ras proteins into their
inactive, GDP-bound form, and its loss (by haploinsufficiency or mutation involving a
critical residue) therefore causes Ras proteins to spend longer in their active GTP bound
form (160). The roles of SPRED1 in Ras-MAPK signal transduction are not yet as well
characterised as those of NF1, but SPRED1 has recently been shown to interact with
NF1 and appears to recruit it to the plasma membrane where it can inactive Ras proteins
(161). For most NCFC-associated mutations, evidence of increased Ras-MAPK pathway
activity has been demonstrated, including for many that cause CFC, but as discussed
below in 1.8.1 and explored in chapter 5, this is not the case for all variants identified
(162) . Whilst the HRAS mutations described in classical CS have all been noted to
increase Ras-MAPK signal transduction, a much wider variety of effects have been
observed for the BRAF and KRAS mutations described in NS/CFC. Some of these
germline variants appear to have no major effects upon downstream pathway signal
transduction (87), by currently available methodologies.
The situation for PTPN11 mutations in NS and NSML is also complex. PTPN11
encodes SHP2, a tyrosine phosphatase (74). Classical NS is most commonly associated
with mutations that demonstrate increased downstream pathway activity, whilst NSML-
associated mutations have more commonly been identified to result in impaired catalytic
activity of SHP2 in vitro , and reduced signalling through ERK1/2 (163). This
classification is challenged, however, by the variable presentation of certain mutations in
different individuals (149), and suggests that other molecular factors may be important in
determining phenotype. The recent work animal models that has demonstrated
involvement of the PI3kinase signalling pathway in the pathogenesis of myocardial
hypertrophy due to PTPN11 mutations (157) opens up a potential new route for therapy
of this disease feature.
1.8.1 Functional effects of CFC-associated mutations
At present, the evidence is inconclusive regarding the mechanisms by which CFC-
associated point mutations, of BRAF in particular, exert their effect. The majority of
such mutations have been noted to increase downstream ERK1/ERK2 pathway

62
signalling, as assessed by effects on phosphorylation of these targets, or by in vitro kinase
assay (162), after transient transfection in cell lines. In such assays, some mutations
appear to have contradictory effects, demonstrating decreased kinase activity and
decreased ERK1/2 phosphorylation, when compared to wild-type alleles (162).
In their initial report of BRAF and KRAS mutations in CFC, Niihori and colleagues
transiently cotransfected unstimulated NIH 3T3 cells with BRAF or KRAS constructs
and the Elk1-GAL4/GAL4-luciferase reporter system (77). This demonstrated increased
relative luciferase activity (as a proxy for downstream pathway activity) for some mutants,
such as KRAS p.(Asp153Val), and BRAF p.(Leu485Phe) and p.(Lys499Glu) but not
others, such as KRAS p.(Gly60Arg), or BRAF p.(Gly469Glu) and p.(Glu501Gly).
Rodriguez-Viciana et al (10, 162) achieved a similar spectrum of results when they
transfected HEK293T cells with constructs bearing BRAF, MAP2K1 and MAP2K2
mutations described in CFC, and then assessed downstream pathway activation by
ERK1/2 phosphorylation, using Western blotting with phospho-specific antibodies.
BRAF activity was also assessed by immune complex kinase assay (using myelin basic
protein as the substrate), and four of the six CFC-associated BRAF mutations,
p.(Gln257Arg), p.(Ser467Ala), p.(Leu485Phe) and p.(Lys499Glu), showed increased
activity, whilst two, (p.(Glu501Gly) and p.(Gly596Val), showed decreased activity,
compared to the wild-type protein. Sarkozy et al (39) transfected NIH3T3 cells with
BRAF p.(Thr241Pro), p.(Glu275Gly), p.(Tyr531Cys), p.(Leu597Val), p.(Thr599Arg),
p.(Lys601Gln) and p.(Val600Glu), and identified that the kinase activity of each of the
CFC-associated alleles was increased, though to a lesser extent than was the case for
p.(Val600Glu).
Schubbert et al (85) demonstrated the effect of KRAS mutations found in patients
with NS and CFC, and compared these to those observed with the three KRAS
mutations commonly described in cancer, using GTP hydrolysis assays and Western blots
for the phosphorylated kinases MEK (MAP2K), ERK, AKT and S6 kinase. Different
functional effects for the different mutations were suggested, with p.(Pro34Arg) and
p.(Asp153Val) having normal intrinsic GTPase activity, and p.(Val14Ile), p.(Thr58Ile)
and p.(Phe156Leu) having reduced intrinsic activity (as is the case for p.(Gly12Asp), the

63
common KRAS mutation in cancer). p.(Pro34Arg) demonstrated reduced responsiveness
to both neurofibromin and p120GAP. The lack of any observable difference between
p.(Asp153Val) and wild-type KRAS in this regard is in contrast to the increased
downstream transcriptional activity induced by this mutant, demonstrated by Niihori et al
by luciferase assay (77). The complexity of the effects of germline KRAS mutations was
further explored by Gremer et al (87), with at least four classes of mechanism suggested
by which such mutations could exert their influence. Many of the mutations of KRAS
studied have decreased binding efficiency to RAF (which in itself would be considered
likely to reduce signal propagation), but also decreased GAP-stimulated GTPase activity,
which would be likely to increase downstream signalling (87). The balance of the
magnitude of each of these effects may be distinct for different alleles. The complexity of
this situation (even before further genetic or other factors are considered) may be a
factor in the variable phenotypic effects of germline mutations in KRAS (147), but the
numbers of patients known with such mutations at present are not sufficient for
genotype-phenotype correlations to have been established to assess this hypothesis
further.
Senawong et al (164) transfected HEK293T and COS-7 cells with constructs bearing
MAP2K1 and MAP2K2 mutations, and demonstrated that, as previously found by
Rodriguez Viciana et al (162), these all appear to increase downstream pathway activity as
measured by ERK1/2 phosphorylation on Western blot. This appeared to occur via
phosphorylation of the two key serine residues (218 and 222 of MAP2K1, 222 and 226
of MAP2K2), was dependent on the presence of BRAF, and amenable to MAP2K
inhibition with the MAP2K inhibitor UO126. The increased activity of MAP2K1
mutants was also modulated by RAF inhibition, but this was not found for the MAP2K2
mutant p.(Phe53Ser), suggesting a difference in the way that these two MAP2K proteins
may function.
Further work is necessary to identify how the mutations described in association with
human disease exert their phenotypic effects. The work that has been undertaken so far
to characterise CFC-associated mutations has predominantly relied upon transient
transfection of constructs bearing the altered genes. The situation in these cells, with
dramatic overexpression of mutant protein, is therefore far from that occurring in vivo in
64
patients with heterozygous mutations, where it would be expected that there would be
equal expression of wild-type and mutant alleles, and native stoichometry with key
interactors such as CRAF (165). This is a powerful reason for moving towards more
physiologically and developmentally relevant systems, such as germline expression of
mutant alleles in animal models, to assess the effects of mutations found in CFC patients.
1.9 Animal and other models of CFC and related conditions
There are now several animal models of the various NCFCs. Animal models for NF1
(109) and Legius syndrome (166) are well-established, but as these conditions are
substantially distinct from NS, CFC and CS, they are not further considered here.
Similarly, knockout mouse models for many of the genes known to cause NS and CFC
have been reported, but as the mutations implicated to date in these conditions are
thought to represent gain-of-function alleles, and loss-of-function of these genes either
causes a phenotype unrelated to the NCFCs (such as metachondromatosis, discussed in
1.7.5.5), or no known human phenotype, these are not considered further. Animal
models that have been characterised with mutations in genes that are known to cause
CFC, NS or CS in humans are listed in Table 1-4. Vertebrate model organisms, mouse
and zebrafish, that have been developed, are discussed in more detail in 1.9.1 and 1.9.2.
Table 1-4: Animal models of CFC, NS and CS
Gene Organism Mutation, how introduced, target tissue
Phenotype Ref.
BRAF Mouse
LSLp.(Val600Glu) germline knockin
Small size, cardiomyopathy, seizures, abnormal behaviour, premature ageing
(167)
p.(Val600Glu) conditional melanocyte-specific (tyrosinase-driven Cre recombinase)
Postnatal induction: benign melanocytic hyperplasia**, increased pigmentation, melanoma in older age
(168)
Germline induction: lethality around birth, hydrocephalus, cardiac and eye defects
(169)

65
p.(Val600Glu) germline knockin
Embryonic lethal at E7.5 (93)
p.(Leu597Val) germline knockin
NS/CFC features (170)
Zebrafish Overexpression of a range of mutant BRAF alleles by injection into embryos
Severe developmental defects, ameliorated by BRAF or MEK inhibition
(171, 172)
KRAS Mouse
p.(Val14Ile) germline knockin
Enlarged heart, myeloproliferative disorder
*
Zebrafish p.(Gly12Val) Cre/loxP neuronal progenitor specific
Lethal: massive apotosis and brain oedema
(173)
SHOC2 C.elegans p.(Ser2Gly) Abnormal vulva (104)
PTPN11 Mouse p.(Gln79Arg) conditional cardiomyocyte expression
Postnatal: no phenotype
Prenatal: abnormal cell cycling, ventricular noncompaction, VSD
(174)
p.(Tyr279Cys) germline Short stature, craniofacial dysmorphism, hypertrophic cardiomyopathy
(157)
p.(Asp61Gly) Conditional in endocardium
Cardiac defects due to endocardial-mesenchymal transformation defects
(175)
p.(Asp61Gly) germline Embryonic lethal in homozygosity
Reduced viability in heterozygotes: cardiac defects, short stature, myeloproliferative disease
(176)
Cardiomyocyte-specific overexpression of
Left ventricular dysfunction and dilatation, increased apoptosis
(177)

66
p.(Gln510Glu)
Drosophila
(csw gene)
p.(Tyr279Cys)
p.(Thr468Met)
Abnormal wing veins (both alleles); abnormal eyes (Y279C)
(178)
p.(Ala72Ser) p.(Ile282Val) p.(Asn308Asp) p.(Asp61Tyr) p.(Glu76Lys) p.( Thr73Ile) expressed in mushroom body of central nervous system
Impaired long term memory formation
(179)
HRAS Mouse p.(Gly12Val) germline knockin
Facial dysmorphism, cardiomyopathy, hypertension
(180)
Zebrafish p.(Gly12Val) transgene in germline
Scoliosis, hypertelorism, shortened body, tumour predisposition
(181)
* Hernandez et al, manuscript in preparation
1.9.1 Mouse models of the NCFCs
The development of mouse models of the NCFCs broadly mirrors the sequence of
identification and clinical impact of the causative mutations, such that Ptpn11 mouse
models of NS have been the most extensively studied to date (176). Homozygosity for
the Ptpn11 p.(Asp61Gly) mutation was noted to be embryonic lethal, whilst presence of
this mutation in the heterozygous state generated reduced viability and phenotypic
features of NS: severe cardiac defects in 50%, alongside short stature, craniofacial
dysmorphism and myeloproliferative abnormalities. Further characterisation using tissue-
specific inducible Ptpn11 p.(Asp61Gly) alleles showed evidence that increased duration of
endocardial-mesenchymal transformation in the endocardium may underly the cardiac
defects observed in association with this mutation (175).
The first mouse model of CFC to be generated was the B-Raf LSLV600E/+ mouse, which
was made and initially characterised in Professor Mariano Barbacid’s laboratory at the

67
Spanish National Cancer Centre (CNIO) (167), and which is further described in chapter
6. Mice bearing this mutation, which is only partially expressed due to a leaky stop
cassette (167), have a multisystem phenotype strongly reminiscent of human CFC, with
excess mortality, enlarged heart size, growth failure, abnormal behaviour and epilepsy.
Two further mouse models of the NCFCs, of Costello syndrome: H-Ras G12V/G12V
(180), and Noonan/CFC syndrome: K-Ras V14I/+(I. Hernandez, PhD thesis, Autonomous
University of Madrid) have also been generated at CNIO, and these are described further
in chapter 6. A further mouse model of the NCFCs has recently been published (170),
with conditional expression of the intermediate activity BRAF p.(Leu597Val) mutation.
This allele has been identified both in the germline of CFC/NS patients (39, 182) and in
tumours (49), though in this latter situation it is much less common than BRAF
p.(Val600Glu) (49). Mice bearing this mutation were noted to have cardiac abnormalities
and craniofacial dysmorphism, analogous to the phenotype of CFC. Significantly, as has
been observed for humans with CFC/NS, no strong tumour predisposition was
identifiable when this allele was expressed in the germline as the sole mutation (unlike
when it was coexpressed in tissue with oncogenic KRAS p.(Gly12Asp), when a wide
range of tumours developed). Further mouse and other animal models with NCFC-
associated mutations, including in B-Raf, Map2k1/2 and Shoc2 are known to be in
preparation around the world (as discussed in a round table workshop forum at the 2012
Rare Disorders of the Ras-MAPK Pathway meeting, Nuremberg).
1.9.2 Zebrafish models of the NCFCs
The zebrafish (Danio rerio) is a model organism with significant potential in the study
of tissue-specific and developmental phenotypes, and for the early evaluation of potential
treatments. Several mutations associated with NCFC disorders have successfully been
assessed in this species. CFC-associated BRAF alleles have been assessed by
microinjection of capped human mRNAs into single cell embryos (171, 172),
demonstrating a range of early developmental defects reminiscent of features of the
human syndrome. This model was used to demonstrate both time- and dosage-sensitive
responses to MEK inhibition: when applied either within a very specific developmental
window (for 1 hour, 4.5-5.5 hours post fertilisation), or continuously at a much lower
concentration, treatment with MEK inhibitors appeared to abolish evidence of these

68
early defects (171, 172). Zebrafish with germline expression of a transgene, comprising
human HRAS p.(Gly12Val) tagged with green fluorescent protein, exhibited phenotypic
features analogous to those of human CS, including scoliosis, hypertelorism, heart wall
thickening, shorter body length and tumour predisposition (181). Oncogene-induced
senescence at the cellular level, as assessed by decreased BrdU incorporation and
increased β-galactosidase staining, was also found in certain tissues including brain and
heart (but not others, such as skin or gut).
1.9.3 Human-derived cellular models of the NCFCs
An important recent addition to the means of assessing functional effects of NCFC-
associated mutations in a live cell system is the successful reprogramming of skin-derived
fibroblasts from patients into pluripotent stem cells (iPSCs). The initial published
examples were from two patients with NSML caused by p.(Thr468Met) mutations in
PTPN11 (183). Cardiomyocytes derived from these two patients’ iPSCs (generated from
skin-derived fibroblasts) had a greater median surface area than those without the
mutation, though this was variable between different lines derived from the same patient.
Nuclear localisation of NFATC4, a calcineurin-regulated transcription factor known to
play a role in hypertrophy, was also increased in cells bearing the mutation compared to
those without. The possibility of generating patient-specific lines of many different
genres of cells makes this technique promising for yielding further important discoveries
about this group of disorders, especially regarding the possibility of mimicking the
behaviour of cells from tissues for which human biopsy material is very seldom available,
such as heart, or central or peripheral nervous system tissue.
1.10 Avenues for therapy of Ras-MAPK pathway disorders
At present, there are few specific recommendations regarding the treatment of the
complications of the NCFCs, as compared to these disease features occurring in a non-
syndromic context. HCM and epilepsy are examples for which the current management
recommendations are the same as for these disorders in the general population (184).
However, the example of optic pathway glioma in NF1 (where radiotherapy treatment
should be avoided, due to the high risk of second tumours (185)) serves as a warning that
specific situations may call for adjustments to management of NCFC-associated

69
complications, and vigilance regarding potential sequelae of treatment is required. There
is no current evidence of progression of HCM or increased cancer incidence after
administration of growth hormone in patients with NCFCs, but the data are currently
sparse (184). The occurrence of hepatoblastoma in an immunosuppressed patient with
CFC (after cardiac transplantation) may be incidental to the immunosuppression, but the
possibility of a connection cannot be excluded. Conversely, the risk of exacerbation of
cardiac compromise by certain cancer chemotherapeutic agents may be an important
consideration for individual patients.
When considering the possibility of treatments directed at normalising Ras-MAPK
pathway activity, the relatively well-characterised nature of this pathway, and in particular
its importance in the pathogenesis of cancer, has led to the development of targeted
agents to modulate signal transduction activity. The discovery of germline mutations in
the same genes as are mutated in cancers has made the potential extension of such
therapeutic agents into developmental disorders a natural consideration, as if the
dysregulation of pathway activity could be treated, then potentially there may be effects
upon many of the different phenotypes observed in these disorders. Clinical trials in NF1
have already been reported (7), and many more are recruiting or being planned
(www.nfconsortium.org). These may identify agents to be trialled in other NCFCs, if a
positive therapeutic effect were shown in the NF1 population.
As mentioned in 1.6, specific inhibitors to various kinases known to be important in
cancer have been developed, from receptor tyrosine kinases such as cetuximab to block
EGFR (186) and Herceptin to block ERBB2 (187), to inhibitors of BRAF such as
vemurafenib (59), and MAP2K1/2 such as trametinib (188). Whilst the clinical response
of BRAF-mutated tumours to specific inhibitors of this kinase, namely vemurafenib and
dabrafenib, may be dramatic (189), it is often short-lived, with the cancer evolving
mechanisms to circumvent this inhibition. These mechanisms are the subject of intense
ongoing investigation. Combined therapies, using cocktails of inhibitors to inhibit
multiple kinases simultaneously, may be of clinical use in prolonging treatment response
and survival (190). Better understanding of the role of cross-talk between MAPK
signalling pathways is needed to better predict how modulation of one route may impact
upon signal transduction along other routes. The recent demonstration of major effects

70
of PI3K-Akt pathway inhibition in mouse models of cardiac hypertrophy driven by
mutant Shp2 (157) is one such example. Ras proteins (and hence those upstream from
them, such as SHP2) are known to signal through the AKT pathway, but whether cardiac
hypertrophy driven by mutations further down the Ras-Raf-MAP2K-ERK cascade might
also be amenable to such agents remains unclear. Such cross-talk may also be very
different in different tissues and at different stages of development and within a disease
course, meaning that caution is required in extrapolating results across these boundaries.
The skin phenotypes such as keratoacanthoma and squamous cell carcinoma noted in
many patients with advanced melanoma following therapeutic inhibition of mutant
BRAF have been identified to be due frequently to Ras gene mutations, most commonly
in HRAS (191). A recent report of leukaemia with a p.(Gly12Arg) mutation in NRAS in
a patient receiving vemurafenib for advanced BRAF-mutated melanoma (192) further
emphasises the potential for initiation, unmasking or promotion of, for example, Ras-
driven neoplastic processes in this treatment setting.
The observations of side effects of pathway modulation such as the cutaneous
eruptions seen in nearly all responders to vemurafenib therapy (191) suggest extreme
caution will be necessary when considering the use of agents to modulate signal
transduction pathway activity in patients with germline Ras-MAPK pathway disorders.
The potential for generating severe or enduring side effects (including the promotion of
tumours as discussed above) illustrates the very different risk-benefit analysis for patients
with developmental disorders, whose condition may be largely stable and for whom
treatment may need to be considered over many years, compared to that for patients with
advanced, multiply resistant (and frequently rapidly lethal) tumours. Suitable models for
assessing the long-term effects of these agents are therefore required, and again the long
term administration of such potential treatments to suitable animal models could be an
important first step. The advantage of harnessing existing therapies developed for
cancers or other diseases is that the ‘first in human’ trials have already been achieved, and
hence there are individual patients with life-threatening NCFC-associated phenotypes
who have already been treated for these, for example a patient with severe neonatal
HCM (who was on a heart transplant list) has received rapamycin therapy (Prof. M.

71
Zenker, personal communication) on the strength of the good response to this drug
observed in the mouse model with the relevant NSML-associated mutation (157).
1.11 Summary of conclusions from the literature
The discovery of the molecular basis for many cases of CFC and the other NCFCs
has started to explain the overlapping clinical phenotypes frequently observed in patients
with these conditions, as well as providing genotyped cohorts for focused research. To
date, the clinical phenotype of CFC has not been particularly successfully elucidated (4),
due to the small numbers of patients available for assessment, and their wide
geographical distribution.
The proportion of patients with CFC or NS and no current molecular diagnosis (4,
22) suggests the involvement of further loci in causing the features of these conditions.
The great variation in severity of phenotype observed with apparently similar mutations,
for example causing mild NS in one patient and severe CFC syndrome in another (6),
suggests that genetic and other modifiers may play roles in determining the phenotypic
effects of any given mutation. The apparent divergence in the resultant degree of
downstream pathway activation and activity, as assessed by ERK phosphorylation and in
vitro kinase assay, for example, of BRAF mutations identified in CFC syndrome (162),
requires further investigation. Further outstanding issues in this group of disorders
include their classification and nomenclature: whilst some disease entities such as NF1
and CS are readily distinguishable (by clinical criteria and definitive molecular testing),
NS, NSML and CFC syndrome have sufficient clinical and genetic overlaps to suggest
that some alternative form of classification, taking into account both genotypic and
phenotypic information might be necessary in future, particularly regarding potential
targeted treatment strategies.
In patients with NCFC phenotypes and no identifiable mutation by current
diagnostic means (either due to the involvement of hitherto unknown genes, or the
previously prohibitive cost of achieving effective testing across all candidate exons),
novel sequencing technologies hold considerable promise for achieving molecular
diagnoses in the near future. Conversely, whole exome (and potentially whole genome)
sequencing of far greater numbers of patients with developmental phenotypes will

72
demonstrate the true clinical spectra of the NCFCs, as for other genetic disorders. In
this, it appears likely that further groups of patients not previously clinically diagnosed
with a particular disorder involving dysregulation of the Ras-MAPK pathway will be
ascertained.

73
2 MATERIALS AND METHODS

74
2.1 Reagents and supplies
All laboratory chemicals and reagents used were sourced from Sigma-Aldrich or
Fisher Scientific, unless otherwise specified. Custom primers were designed using the
online Primer3 program: http://frodo.wi.mit.edu/primer3/ and sourced from
Invitrogen.
2.2 Clinical and molecular diagnosis of patients with Ras-MAPK
disorders
2.2.1 Identification of patient cohort
The Manchester Centre for Genomic Medicine (MCGM)’s diagnostic laboratory
offers comprehensive molecular diagnostic testing for Costello and cardio-facio-
cutaneous syndromes, the only laboratory in the United Kingdom’s National Health
Service to have done so since the initial publication of genes responsible for these
conditions in 2005 and 2006. This means that a high proportion of UK patients clinically
identified to have these conditions have had samples submitted to the laboratory. Many
overseas patients with these conditions have also been tested in this laboratory.
The amplicons analysed in this diagnostic service cover all coding exons (2 - 6) and
exon-intron boundaries of HRAS (NM_005343.2 & NM_176795.3) and KRAS
(NM_004985.3 & NM_033360.2); exons 6, 11, 12, 13, 14, 15 and 16 of BRAF
(NM_004333.4), exons 2, 3, 6 and 7 of MAP2K1 (NM_002755.3), and exons 2, 3, 5, 6
and 7 of MAP2K2 (NM_030662.3). These have been selected as the exons in which
recurrent mutations have been described in the published literature.
Clinicians who had referred patients for testing were contacted to ask whether they
felt it appropriate for the family to be invited to participate in a clinical study of CFC
syndrome and related disorders. If so, information was sent via this clinician to the
family. Similarly, patients attending a UK family conference of the Costello and CFC
support group were invited. Both children and adults with these conditions were eligible
to take part in the study, necessitating the development of several different information
sheets, consent and consultee declaration forms (appendix 2)

75
Application for ethics committee approval was made through the North West 6
Research Ethics Committee (Greater Manchester South), reference number
10/H1003/77, see appendices for the study flow chart and protocol.
2.2.2 Clinical phenotyping of patient cohort
A proforma for clinical data collection was developed (appendix 3c), with reference
to the published literature on these conditions. Patients were seen in person (in clinic, in
their own home, or at the conference venue) wherever possible, but where this was not
the case, data were sought from the referring geneticist. Linked anonymised patient data
were entered onto a Microsoft Access database. In addition to comparisons across the
groups of patients enrolled in the study, comparisons to the phenotypes of patients with
clinical and molecular diagnoses of CFC published in the literature were also made.
2.2.3 Molecular analysis of exon 2 of SHOC2 in patients previously
tested for Costello or cardio-facio-cutaneous syndromes
Samples for inclusion in this work (undertaken as a pilot for service development in
the MCGM diagnostic laboratory) were identified with reference to records in this NHS
laboratory’s database. Genomic DNA referred for testing of HRAS, KRAS, BRAF,
MAP2K1 or MAP2K2 was eligible for inclusion, if sufficient sample remained. DNA
extraction had already been undertaken in the accredited referring molecular diagnostic
laboratory from which the sample originated, according to local protocols.
2.2.3.1 PCR, DNA sequencing and analysis
Primers were designed (appendix 4) to amplify the second exon of SHOC2
(NM_007373.3). This exon includes the translation start site and second codon, in which
disease-associated germline mutations have been described. PCR reactions using
ReddyMix Custom Master Mix (Abgene; CM102) were set up and products checked by
agarose gel electrophoresis using ethidium bromide or SafeView nucleic acid stain as per
standard protocols. PCR products were purified using the Ampure magnetic bead system
(Agencourt), on a Beckman Coulter Biomek Multichannel robot in accordance with the
standard CPA accredited laboratory protocol.

76
DNA sequencing was performed using the BigDye 3.1 system (reagents and reaction
conditions as shown in appendix 4). These were run on a Veriti 96 well thermal cycler
(Applied Biosystems). Purification of sequencing reactions was done either through
Sephadex columns in Millipore multiscreen plates or using the CleanSeq magnetic bead
system on a Beckman Coulter Biomek Multichannel robot, in accordance with the
standard CPA accredited laboratory protocol. Samples were analysed on an ABI3730
analyser (Applied Biosystems) as per standard protocol. Sequence traces were compared
using Staden chromatogram analysis programs (Pregap version 1.4b1, Gap4 version 4,
8b1; MRC Laboratory of Molecular Biology, Cambridge) and the Chromas Lite program
(www.technelysium.com.au/chromas_lite.html).
For samples in which a mutation was found, this was confirmed by repeat sequencing
in the NHS laboratory, so that a diagnostic report could be issued to the referring
clinician. Further clinical data regarding patients found to have a SHOC2 mutation was
requested where possible.
2.3 Massively parallel sequencing approaches for molecular
diagnosis
2.3.1 Target enrichment sequencing of selected patients
DNA samples from a further cohort of patients were selected as a panel in which to
test the utility of a massively parallel sequencing approach for molecular diagnosis of the
NCFCs. Patient samples for inclusion in the target enrichment experiment were selected
as described below.
As it would be expected that many further genes would be involved in the
pathogenesis of NCFCs, a list of approximately 200 genes was drawn up for inclusion in
the target enrichment, to reflect both the capacity of the experimental technique and the
potential size of a future comprehensive testing strategy. This list was designed to include
all previously implicated genes for NCFC disorders and selected others representing top
candidates for additional novel causative genes. These genes are shown in chapter 4.
Targets for inclusion were selected by literature searching to identify genes whose
products had most robust published evidence for relevant interactions with known Ras-

77
MAPK proteins, with cross-referencing to available online pathway analysis and gene
ontology tools.
A sample from a patient with a known pathogenic mutation in a NS-associated gene
was used as a positive control. This was the extent of information known at the time of
analysing this sample, as the diagnosis had been confirmed in another diagnostic
laboratory (South West Thames Regional Genetics Laboratory). The result had been
communicated to the patient’s geneticist, but was not known to anyone else involved in
the target enrichment experiment. Unblinding regarding this diagnostic test result was
performed after running the target enrichment analysis.
The majority of other samples included were those which had been subject to
extensive previous analysis by Sanger sequencing of CFC and CS-associated genes, for
two principal reasons. This group of patients had well-defined phenotypes that were
strongly suggestive of CFC syndrome or a related NCFC, and the previous diagnostic
testing meant that SNPs in relevant genes had been identified previously in these
samples, providing data that could be used to verify the effectiveness of the target
enrichment method to identify such variants. As a group of patients without a molecular
diagnosis, they also had the greatest chance of potentially having a mutation identifiable
in a previously unsequenced exon of a known gene, or in a novel gene. A third category
of sample included was from patients with a convincing NCFC clinical phenotype, but
who (due to recent presentation or other circumstances) had not had previous testing.
These were included on the basis of a clear NCFC phenotype, aiming to assess the
feasibility of mutation detection in previously untested samples.
Coverage across all genes included in the test was calculated, and a further detailed
analysis of genes with known clinical importance was performed, to assess the potential
for direct translation of this experimental approach into the diagnostic setting.
2.3.1.1 Filtering of variants identified in target enrichment experiment
As for variants identified through exome sequencing, basic filtering at a bioinformatic
level was carried out to exclude variants most likely to be due to artefact or sequencing
error. The algorithm designed to identify the assumed single causative mutation is shown
in chapter 4. Variants with the highest likelihood of pathogenicity were highest priority

78
for this validation, following the hierarchy described in chapter 4 if necessary to select
which candidate substitutions should be followed up by Sanger sequencing.
Primers were designed to verify candidate nucleotide changes that had been
identified in the target enrichment, with 100-200 basepairs of flanking sequence. PCR,
purification and sequencing analysis of these amplicons was carried out as described
above in 2.2.3.1.
Where the candidate variant was a previously identified pathogenic mutation, and
confirmed on Sanger sequencing, this was sent directly for diagnostic confirmation so
that a report from an accredited laboratory could be sent to the referring clinician. Where
the candidate was in a novel gene, and was confirmed on Sanger sequencing, primers for
all coding exons of the relevant gene were generated (appendix 4) to permit sequencing
of all of these exons in the patient’s sample. Sequencing of all exons of the gene was also
undertaken in a panel of samples from patients with no identified pathogenic mutation.
Given the likelihood of de novo mutations being responsible for the phenotypes under
study, parental samples were also sought for analysis of the identified variant where
possible.
2.3.2 Whole exome sequencing of patient-parent trios
Patients with a clinical diagnosis of CFC in whom exhaustive molecular testing of
known genes for CFC syndrome had shown no abnormalities were selected for exome
trio analysis. For this, DNA samples from both parents needed to be available.
Targeted enrichment and sequencing were performed by the next generation
sequencing team in the MCGM laboratory. 3 µg of DNA extracted from peripheral
blood from the affected individual and his or her unaffected parent was used.
Enrichment was performed with the SureSelect Human All Exon 50 MB Kit v3 (Agilent,
Santa Clara, CA, USA) for the ABI SOLiD system following the manufacturer's
protocols. Samples were indexed and sequenced on a SOLiD4 sequencer (Life
Technologies, Carlsbad, CA, USA) in accordance with the manufacturer's protocols.
Sequence data were mapped with SOLiD Lifescope software (Life Technologies)
against version hg19 of the reference human genome by Dr Sanjeev Bhaskar. Unique

79
mapping of an average of 5.00 gigabases of sequence was achieved, with an aim that
more than 65% of the targeted exome should be covered in at least 20-fold depth.
Variants were called with a combination of the diBayes tool in the Lifescope software
suite with medium stringency and Samtools, and then filtered for those SNPs with 5-fold
or greater coverage. SNPs were annotated using Ensembl v61 and Ensembl's defined
consequence hierarchical system, with retention of the variant of greatest predicted
consequence in any given gene. Variants were excluded from analysis if they were
annotated as non-functional in dbSNP134 (unless identified in the Human Gene
Mutation Database) or previously seen in the in-house variant database (consisting of
data from over 100 exomes at the time of analysis). To ensure only high quality calls were
included in the list of candidate variants, sequence data were further filtered. A variety of
mean quality values for novel allele calls were used as a cut-off, with a minimum novel
allele count of 10, this strategy aiming to maximise data quality and generate a
manageable number of variants for further analysis.
2.4 Cell culture work
2.4.1.1 Restriction digest
Plasmids pEF-BRAF and pEF-BRAFV600E that express the BRAF proteins fused to a
myc epitope-tag (gift of Claudia Wellbrock, Faculty of Life Sciences, University of
Manchester), were verified by restriction digest with two restriction endonucleases,
BamHI and Xba1, each of these enzymes having a single target site for cleavage within
the construct. Plasmid DNA was incubated at 37C for 3 hours in NEB buffer number 4
(New England Biolabs), and run on a 1% agarose gel, alongside Hyperladder I (Bioline)
for measurement, and the two expected fragment sizes of approximately 8000 and 4000
basepairs were observed.
2.4.1.2 Transformation of E. coli and plasmid DNA purification
E. coli DH5α were used to amplify purified plasmid DNA. After incubating on ice
with plasmid DNA for 30 minutes, these were heat shocked (42°C for 2 minutes), and
then chilled on ice (2 minutes). 1 ml of LB broth was added, for a 1 hour incubation at
37°C with shaking. Cells were then cultured on ampicillin-containing agar plates
overnight at 37°C. Colonies were selected and cultured overnight in LB broth containing

80
1:1000 ampicillin. Purification of plasmids from the resultant saturated cultures was
performed using either using the QIAprep Spin Miniprep or Plasmid Maxi Kits (Qiagen,
cat. 27104 or 12163), according to the manufacturer’s instructions.
2.4.2 Site-directed mutagenesis
Primers for BRAF mutagenesis are shown in appendix 4. Mutations were selected to
represent a range of those found in patients with CFC syndrome, namely p.(Thr241Pro),
p.(Gln257Arg), p.(Gln262Pro), p.(Gly469Glu), p.(Thr470Pro), p.(Lys499Glu),
p.(Glu501Gly) and p.(Leu525Gln). These were selected as either commonly identified
mutations, or those that had been identified in the diagnostic laboratory but not
published elsewhere. As such, no functional data regarding effects of these mutations
was previously available (and the diagnostic reports issued had had to be inconclusive).
Mutations were made using pEF-BRAF as the template. pEF-BRAFV600E plasmid, which
expresses the common cancer-associated mutation p.Val600Glu was used as a
comparator to the CFC-associated mutations. The QuikChange Lightning Mutagenesis
kit was used as per the manufacturer’s instructions (Agilent, cat.210518). Plasmid
sequence was checked using dideoxy DNA sequencing according to standard protocols.
Precipitation of DNA prior to sequencing in the University of Manchester’s DNA
sequencing facility was carried out using ethanol and sodium acetate.
2.4.3 Transfection using jet PEI reagent
Cells were cultured in high glucose Dulbecco’s modified Eagle Medium, (Invitrogen,
cat.21969-035), supplemented with 10% bovine serum albumin, 1% penicillin /
streptomycin (Invitrogen, cat.15140-122) and 2 mM glutamine solution (Glutamax ™,
Invitrogen, cat.35050-038). Cells were plated the day before transfection into 6 well
plates, aiming for 70% confluency (50-80%). 1 µg DNA* was transfected using the
reagent volumes specified in the jetPEI protocol (Polyplus, cat.101-40N). Due to rapid
proliferation, cells were harvested as described below 30 hours after transfection.
(* for all BRAF plasmids except p.Gly469Glu, which required 2.5µg DNA to be
added for equivalent expression of the c-myc tagged protein to be obtained).

81
2.4.4 Western blotting
Plates were placed on ice and the media aspirated. Cells were washed with phosphate
buffered saline (PBS), scraped into 0.5ml PBS, pelleted by spinning at 2000 rpm for 5
minutes (4 °C), and resuspended in 50 - 100 µl Triton lysis buffer. Lysis occurred on ice
for 15 minutes, then the lysates were clarified by centrifugation (4 °C) at 14000 rpm for
15 minutes. Supernatants were transferred to chilled fresh tubes. These cell lysates were
boiled with SDS loading buffer for 5 minutes and run on 12% polyacrylamide gels, with a
protein marker for comparison (All Blue, BioRad). Gels were rinsed in Western transfer
buffer and transferred to polyvinylidene fluoride membranes (Immobilon FL, Millipore)
using a semi-dry technique (Semiphor, Hoefer; 15V for 3 hours). Membranes were
blocked by incubation in 5% non-fat milk in Western blotting buffer (WBB; 1 hour at
room temperature), then incubated overnight in primary antibody (diluted as shown in
Table 2-1below) at 4 °C. After washing in 0.1% Tween-20 in WBB, incubation with
fluorescent secondary antibody (Li-cor), diluted in 5% milk in WBB was performed for
all blots except those for phosphoERK1/2, which were in 1% milk in WBB, each with
addition of 1:10000 sodium dodecylsulphate. After 40 - 60 minutes at room temperature,
membranes were washed 4 times in 0.1% Tween-20 in WBB, rinsed in WBB and imaged
on the Odyssey machine (Li-cor).
Table 2-1: Antibodies used for Western blotting
Target Origin Dilution storage Source/manufacturer
c-myc Mouse
monoclonal
1:5000 -20°C Millipore (cat. 05-724)
ERK1 Rabbit 1:2000 4°C Santa Cruz (cat. sc-94)
ERK2 Rabbit 1:2000 4°C Santa Cruz (cat. sc-154)
p-ERK Mouse 1:1000 -20°C Cell Signaling (cat.9101)
Secondary:
Odyssey 800
and 680 goat
anti-mouse
and goat
anti-rabbit
Goat 1:10000 4°C
(stock
-20°C)
Li-cor (cat. 926-32210,
926-32211, 926-68020,
926-68021)

82
2.4.5 Dual luciferase assay
This was undertaken in HEK293 cells for the BRAF mutations described above,
using a dual luciferase assay kit (Promega, Madison, WI, USA; cat E1910). Cells were
transfected using jetPEI with pEF BRAF, p-SG424Elk/C, p-GSTK-luc and p-RL-TK
plasmids. Cells transfected with p-SG424 instead of p-SG424Elk/-C acted as a negative
control. These were harvested in passive lysis buffer, and 20µl lysate transferred into
luminometer tubes containing 50 µl LARII reagent. Firefly luciferase activity was then
measured in a single chamber TD20/20 luminometer (Turner systems), before the
reaction was quenched using 50 µl Stop&Glo reagent for measurement of background
Renilla luciferase activity. Aliquots of lysate were reserved for a Western blot to cross-
reference for the level of expression of the constructs. Assays were performed in
triplicate. Luminometer readings for firefly luciferase activity for each construct were
averaged, and represented as a fold-change of background Renilla luciferase activity.
2.4.6 In vitro kinase assays
Cells were transfected in 6 well plates with wild-type and mutant BRAF plasmids and
harvested as described above to generate cell lysates. A small volume of each lysate was
reserved for a Western blot as a cross reference for protein expression. For each reaction,
antibody against the myc epitope tag that is fused to the BRAF proteins was pre-bound
to protein G-Sepharose: 1 µg was incubated with 400 µl TLB and 20µl of 50% slurry of
protein G-Sepharose (30 minutes, 4 °C, with rotation). Antibody-bound protein G-
Sepharose was washed twice with TLB, cell lysates were added, and the volume made up
to 400 µl with TLB. Immunoprecipitation was undertaken for 3 hours at 4°C with
rotation. Beads were washed three times in TLB and twice in kinase buffer, which was
then removed and kinase reaction mix added. For the myelin basic protein experiment,
this comprised 27 µl kinase buffer, 2 µl of 2 mg/ml myelin basic protein (a MAPK
substrate) (Sigma cat.M1891), 1 µl 1 mM ATP and 0.5 µl 10 mCi/ml [γ32-P]ATP (6000
Ci/mmol). For the GST-MEK experiment, this comprised 27 µl kinase buffer, 1.6 µl of
GST-MEK1 (Millipore) as a specific substrate for BRAF, 1 µl 1 mM ATP and 0.5 µl 10
mCi/ml [γ32-P]ATP (6000Ci/mmol). Reactions were incubated at 30 °C for 30 minutes
and terminated by boiling with 10 µl SDS loading buffer. Samples were run on 14%
SDS-PAGE gels at 150 V, before being stained with Coomassie blue (to check equal

83
quantities of substrate present) and destained overnight. Gels were dried onto Whatman
3 mm filter paper on a gel dryer under vacuum at 80 °C for 1 hour and exposed to X-ray
film overnight to demonstrate the degree of incorporation of radioactive ATP.
2.4.7 Transient transfection in the H9C2 cell line
In order to investigate the functional consequences of mutations in BRAF in a more
physiologically relevant cell line, attempts were made to transfect H9C2 cells (a cell line
of BD1X rat cardiomyoblast origin; sourced from ATCC, gift of Joy Wang, Faculty of
Life Sciences, University of Manchester). These were maintained in supplemented
growth media as for HEK293 above. Cells were passaged at 50-70% confluency to
prevent differentiation into myotubes. As the proliferation of H9C2 is much slower, and
the cells significantly larger, than HEK293, passaging was undertaken by slow
centrifugation (600 rpm) of trypsinised cells to permit resuspension of 20% of the seed
flask in each new flask. Transfections were attempted in 6 well plates, as above, with the
same pEF BRAF wild-type and mutant plasmids, using jetPEI or Lipofectamine 2000
(Invitrogen, cat.11668-027), according to the manufacturers’ protocols. Using either of
these, BRAF expression by Western blot was barely detectable, and so transfection using
the Amaxa system (LonzaBio) was also attempted, as per the manufacturer’s instructions.
Test transfection of a green fluorescent protein (GFP) plasmid, pmaxGFP, resulted in
very low levels of GFP expression, with approximately 80% of surviving cells
demonstrating no fluorescence. Whilst BRAF expression was just detectable by Western
blot in cell lysates 48 hours after transfection, there was no observable difference in the
degree of ERK1/2 phosphorylation between untransfected H9C2 cells and those in
which transfection with BRAF p.Val600Glu had been attempted. In the light of
insufficient transfection efficiency to cause demonstrable effects on pathway activity, it
was decided to attempt to generate stably transfected H9C2 cell lines as described below.
2.4.8 Stable transfection of the H9C2 cell line
The Flp-in system (Invitrogen) was used with an aim of generating stably transfected
H9C2 cell lines that could be regarded as congenic, in that the various different alleles of
BRAF should be integrated into the same locus in each line. This involved two rounds of
transfection, firstly to transfect pFRT/lacZeo plasmids into cells, using Zeocin resistance

84
for selection of a clone with a successfully integrated copy of this plasmid. The BRAF
alleles of interest were to be cloned into the pcDNA5/FRT expression vector and this
plasmid transfected into pFRT/lacZeo positive cells, along with pOG44, which expresses
Flp recombinase. The recombinase acts to cause integration of the BRAF expression
vector into the same locus (at the site of integration of pFRT/lacZeo) in each line of cells.
For the first phase, a kill curve of untransfected H9C2 was generated. This was
complicated by the slow division rate of this cell line, but was undertaken as per the
manufacturer’s suggested protocol, using concentrations of 50, 100, 200, 300, 400, 600,
800 and 1000 µg/ml of Zeocin. The lethal dose of Zeocin for untransfected H9C2 cells
was 400 µg/ml; at lower concentrations, cells were able to divide and survive longer than
3 weeks.
However, cells transfected with pFRT/lacZeo and exposed to this concentration
repeatedly died within 2 weeks. The kill curve was repeated with cells exposed to mock
transfection (electroporation using the same protocol but without plasmid), and a much
lower Zeocin concentration, 100 µg/ml, was found to inhibit proliferation. 100 µg/ml
was therefore used for further attempts at selecting successfully transfected pFRT/lacZeo
positive cells. Following transfection, cells were cultured for 24 hours to recover from
the stress of electroporation. Individual clones were detached by local application of 10
µl of trypsin/EDTA solution and very gentle aspiration using a soft tipped plastic pipette
for transfer to 12 well plates. These clones were cultured in media containing 100 µg/ml
Zeocin, and did proliferate, but extremely slowly, with significant numbers of clones
demonstrating differentiation into myotubes and rounded senescent colonies before
adequate numbers could be obtained. The attempt to generate stably transfected cells was
therefore abandoned.
2.5 Characterisation of mouse models of the NCFCS
2.5.1 The B-Raf LSLV600E/+ mouse model of CFC syndrome
The B-RafLSLV600E/+ mouse had been generated as previously described (193). Its
phenotype was analysed collaboratively in Professor Mariano Barbacid’s laboratory at the
Spanish National Cancer Centre (CNIO). Due to the inability of B-RafLSLV600E/+ mice to

85
breed spontaneously, in vitro fertilisation had to be used to generate mutant animals and
littermate controls. To maximise genetic similarity between individuals in the colony, the
mutant allele had previously been backcrossed onto the inbred C57Black6 background
for six generations.
2.5.2 Cardiac phenotyping in the B-Raf LSLV600E/+ mouse
2.5.2.1 Cell proliferation in embryonic heart of the B-Raf LSLV600E/+mouse
Two wild-type female mice fertilised with sperm from mutant B-RafLSLV600E/+ males
were intraperitoneally injected with bromodeoxyuridine (BrdU) dissolved in PBS 12.5
days after conception. 24 hours after this, they were sacrificed by carbon dioxide
asphyxiation, in accordance with the animal care regulations of the CNIO, and their
embryos dissected out. Embryos were fixed and sectioned by staff of the histology
department of CNIO. Sections were taken in the coronal plane, stained with anti-BrdU
antibody and counterstained with haematoxylin, with an aim of providing four-chamber
views of the heart. 11 embryos yielded sections suitable for analysis. The interventricular
septum was viewed at 630 x magnification. The midpoint of the IVS was positioned in
the centre of the image, such that the whole field of view was covered with septal tissue,
and photographs were taken. Cells with nuclei staining positive for BrdU were then
counted within this field of view. Counting was repeated for three non-overlapping fields
of view for each slide, with blinding to the genotype on each occasion.
2.5.2.2 Cardiac expression microarray in B-Raf LSLV600E/+ mouse
Sufficient animals were born from IVF of ova from 4 wild-type females with sperm
of B-RafLSLV600E/+ males to allow for analysis of tissue from trios of male heterozygous B-
RafLSLV600E/+ animals and a sex-matched littermate control. Four week old mice were
sacrificed and tissues harvested immediately. Hearts were dissected out and the atria
removed. The heart was divided into right ventricular wall, interventricular septum, left
ventricular wall and apex (the latter constituting the remainder of the ventricular mass).
These samples were immersed in RNAlater solution (Life Technologies) for ease of
storage, transport and preparation.

86
Interventricular septum (IVS) was chosen for microarray analysis, as a readily
identifiable region that could be sampled in a repeatable manner, and which is known in
humans to be frequently affected by hypertrophic cardiomyopathy, including early in the
disease course in many cases. Total RNA was prepared from 30 µg samples of IVS using
a proprietary microcentrifugation kit (RNEasy Tissue RNA extraction kit, Qiagen,
Hilden, Germany) and Matrix D lysis tubes (MP Biomedicals, Santa Ana, CA, US). An
automated tissue lyser (Hybaid, Cambridge, UK) was used for initial disruption of these
samples, with 2 pulses of 40 seconds’ duration at maximum speed. RNA samples were
subjected to analysis including automated electrophoresis on a 2100 Bioanalyzer using
2100 Expert software (Agilent), to ensure suitable quality for expression microarrays.
RIN indices of >8.0 were obtained for all samples, indicating sufficiently high quality
RNA was present. These samples were submitted to the University of Manchester
Genomic Technologies facility for Mouse Genome 430A arrays, run on the Affymetrix
Genechip system (Affymetrix, Santa Clara, CA).
2.5.3 Cardiac expression microarrays in mouse models of the NCFCs
Crosses were set to generate pups of the CNIO’s two further mouse models, of
Costello syndrome: H-Ras G12V/G12V; and Noonan/CFC syndrome: K-RasV14I/+. Identical
procedures to those described in 2.5.2.2 were then used to generate samples from four
week old male mice of these other two models, so that three way comparisons would be
possible.
2.5.3.1 Microarray analysis
Analysis was undertaken by Dr Leo Zeef (Faculty of Life Sciences, University of
Manchester) using a Robust Multi-array Average (RMA) technique in Bioconductor. The
integrity of the data obtained was assessed by principal component analysis.
Targets identified in the microarray analysis were selected for validation by
quantitative PCR and Western blotting as described below. A cluster analysis was
performed on targets that met the threshold q value of <0.05, and had demonstrated a
fold change of greater than 1.5 in either direction between mutant and control mice in
the B-Raf array. Sets of transcripts demonstrating increased or decreased expression over
this threshold were also subjected to DAVID pathway analysis.

87
2.5.3.2 Quantitative PCR analysis
This was undertaken to validate findings from the microarray analysis. Genes that
were differentially expressed between mutant and control animals were identified. In
combination with the pathway analysis, manual interrogation of the data sets was
undertaken to select transcripts with particular potential significance to the pathogenesis
of cardiomyopathy, other features of the NCFCs or the observed murine phenotypes.
Extracted total RNA (obtained as described in 2.5.2.2) was reverse-transcribed into
single-stranded cDNA using a High Capacity RNA-to-cDNA Kit (Applied Biosystems).
RT-PCR was performed using first-strand cDNA with TaqMan Fast Universal PCR
Master Mix (Applied Biosystems). Assay numbers for mRNA target genes and
endogenous control (Gapdh) were as follows:
Quantitative PCR was performed on an Applied Biosystems 6900 Real-Time PCR
system (Applied Biosystems), using the following conditions: 50 °C incubation for 2 min,
95 °C for 10min, 40 cycles of PCR at 95 °C for 15 s, and 60°C for 1 min. All reactions
were performed in a 10 µl reaction volume in triplicate, and mRNA expression levels
were determined by the 2−∆Ct method.
2.5.3.3 Western blot analysis of Myh7 expression:
A rabbit monoclonal Myh7 antibody was acquired for Western blot analysis (Abcam,
Cambridge, UK; cat. ab172967). Tissues were lysed in RIPA buffer with addition of a
protease inhibitor cocktail (cOmplete Mini tablets, Roche, Mannheim). Protein
Target Assay number
Acta1, alpha-1 actin Mm00808218_g1
Myh6, myosin heavy chain 6 Mm00440359_m1
Myh7, myosin heavy chain 7 Mm01319006_g1
Nppb, natriuretic precursor protein B Mm01255770_g1
Hras, H-Ras Mm01275932_g1
Gapdh, (endogenous control) Mm99999915_g1

88
concentration was verified using a Bradford assay kit (Biorad), according to the
manufacturer’s instructions. Lysates of normalised concentration were run on
polyacrylamide gels, as described above, which contained 7.5% acrylamide (due to the
relatively high molecular mass of Myh7 (220kDa)).

89
3 CLINICAL AND MOLECULAR DIAGNOSIS OF PATIENTS
WITH GERMLINE RAS-MAPK PATHWAY DISORDERS

90
3.1 Chapter overview
Collectively, the Ras-MAPK pathway disorders are commonly encountered
conditions in clinical genetic practice. Individual presentations may be suggestive of a
specific disorder of this group, or it may be difficult to define at presentation which of
these conditions a patient may have. This emphasises the value of molecular diagnosis,
which is also a prerequisite to assessment of genotype-phenotype correlation, providing
insights into the biology of these conditions and in turn leading to tailored anticipatory
management of specific features of these. The MCGM diagnostic laboratory has
undertaken genetic testing for Costello syndrome (CS) and CFC since causative genes for
these conditions were first published (in 2005 and 2006 respectively (10, 62, 77)), and as
such has served a large proportion of the UK affected population, as well as many
patients from overseas.
Referrals to this laboratory were therefore examined to investigate the prevalence of
different gene mutations in this patient population, and to identify patients for invitation
into a clinical study. For the group of patients with a known molecular diagnosis,
genotype-phenotype correlations could be assessed. Patients in whom no molecular
diagnosis had been made, who might have mutations in exons of known genes that had
not previously been tested, or alternatively might have mutations in novel genes, were
also identified as potential candidates for involvement in the work described in chapter 4.
3.2 Mutational spectrum observed in patients with CS/CFC
phenotypes
In order to ascertain the frequency of mutations identified in patients with clinical
diagnoses of CS and CFC, data regarding the samples received for testing of the genes
for these conditions was obtained. By the end of 2012, 418 samples had been referred for
sequencing of one or more of HRAS, BRAF, KRAS, MAP2K1, MAP2K2 and SHOC2.
Sequencing of exon 2 of SHOC2 (NM_007373.3) was added to the diagnostic battery
following the work described below, in 2010. The number of mutations identified to date
in this patient cohort is shown in Table 3-1 and Figure 3-1.

91
Table 3-1: Samples tested for mutations in CFC/CS genes
Gene HRAS BRAF KRAS MAP2K1 MAP2K2
Samples tested 241 212 212 217 215
Mutations found 43 48 7 19 7
Non-pathogenic/unclassified
variants found 3 17 20 4 7
Normal results 195 192 185 194 201
Samples not tested for this gene 177 161 206 201 203
Not all samples had been tested for mutations in all genes, according to each
referring clinician’s request, as shown in Table 3-1, but for patients where the diagnosis
of a Ras-MAPK pathway disorder was strongly suspected clinically, diagnostic testing of
each of the genes described above had usually been undertaken (often sequentially, until
a mutation was identified). Similarly, many of these patients had also had some testing of
genes associated with Noonan syndrome (NS) in a different laboratory service, but data
regarding this was not routinely available for patients who had been seen outside the
MCGM clinical genetics department.
The data demonstrate a large number of molecular diagnoses of CS. This reflects the
enrichment of this cohort for patients with a firm clinical diagnosis of this condition who
had been gathered over many years (as a result of Dr Kerr’s clinical expertise in CS). A
high proportion of these mutations were identified as soon as the test was available: 25 of
43 CS molecular diagnoses were confirmed before the end of 2006. The numbers of
molecular diagnoses made over time are shown in Figure 3-1, demonstrating the
diagnosis of an increasingly high proportion of CFC-associated mutations with time. The
figures observed in this more recent patient cohort, whilst less enriched for definite CS
phenotypes, could nonetheless still be skewed by the indications for genetic testing that
prompt referral of samples to the laboratory. This is because CS is
• Frequently readily identifiable clinically, meaning that a high proportion of
affected patients will be recognised to have the disorder
• Genetically homogeneous, and testing of a single amplicon will detect ~90% of
mutations, meaning that molecular testing is effective and inexpensive

92
• Associated with cancer risk, and definitive diagnosis allows for appropriate
screening, meaning that molecular confirmation and exclusion alter clinical
management.
In contrast, CFC is frequently less characteristic in its presentation, with greater
overlaps with NS, genetically very heterogeneous, and not associated with a high cancer
risk, and hence no specific proactive management is currently indicated on confirmation
of the diagnosis. Each of these factors can make genetic testing less effective, less
pressingly indicated, and more costly. Whilst it has been possible to determine an
approximate birth prevalence of 1 in 381000 for CS in the UK (15), such data are not
possible to obtain for CFC. What can be determined is that CFC is more common than
CS, as much higher numbers of affected patients are being identified prospectively in the
CS/CFC cohort undergoing testing at MCGM: by the end of 2012, 46 diagnoses of CFC
(not including patients with SHOC2 mutations) had been confirmed, in contrast to 15
diagnoses of CS, since the beginning of 2009. The higher numbers of patients receiving a
molecular diagnosis of CS up to 2008 reflects the existence of a group of patients in
whom the clinical diagnosis had been established prior to the availability of genetic
testing.
Figure 3-1: Molecular diagnosis of Costello and CFC syndromes (Manchester Regional Genetics Laboratory) 2006-2012.
The major feature is that an increasing proportion of the diagnoses confirmed are of CFC. The large number of CS mutations identified before 2009 reflects the presence of a large series of patients known with a clinical diagnosis of this condition prior to the availability of genetic testing.

93
The uncertainty regarding CFC incidence or prevalence is mirrored by the situation
for NS, where even for this common condition, there are no definitive data. Published
estimates appear to have relied heavily upon the early empirical statement of Nora and
Nora in 1974 (13) that the prevalence of NS was unknown, but suggested to be between
1 in 1000 and 1 in 2500. However, whilst the phenotypic overlap between CFC and NS
hinders complete ascertainment of each of these conditions, the significant numbers of
patients with NS phenotypes that are sufficiently mild not to come to medical attention
are likely to represent the largest ‘iceberg’ of currently undiagnosed individuals with the
latter condition.
A further factor that may complicate accurate ascertainment of the total patient
cohort for any condition is the possibility that presentations may be sufficiently severe to
cause death before a diagnosis has been made. Severe, neonatal lethal, CS is one such
example, as shown in (16), appendix 7. Of the cohort of four patients with lethal
presentations due to p.(Gly12Val) in this series, two of the diagnoses were made some
time after the baby’s death, and it is possible that such presentations may continue to
occur without being recognised.
Figure 3-2 (overleaf): Mutations identified in samples referred for CS and CFC gene testing.
The x axis shows the number of individuals identified with each mutation. Note the prevalence of BRAF mutations as the most commonly identified cause of CFC syndrome, with 21/48 mutations being seen in only one or two patients to date. The high number of patients identified with the p.(Tyr130Cys) mutation in MAP2K1 is also evident. Note too that all mutations yet identified for CS in this laboratory have been at codon 12 of HRAS (codon 13 mutations being associated with a recognisable, but usually milder, phenotype).
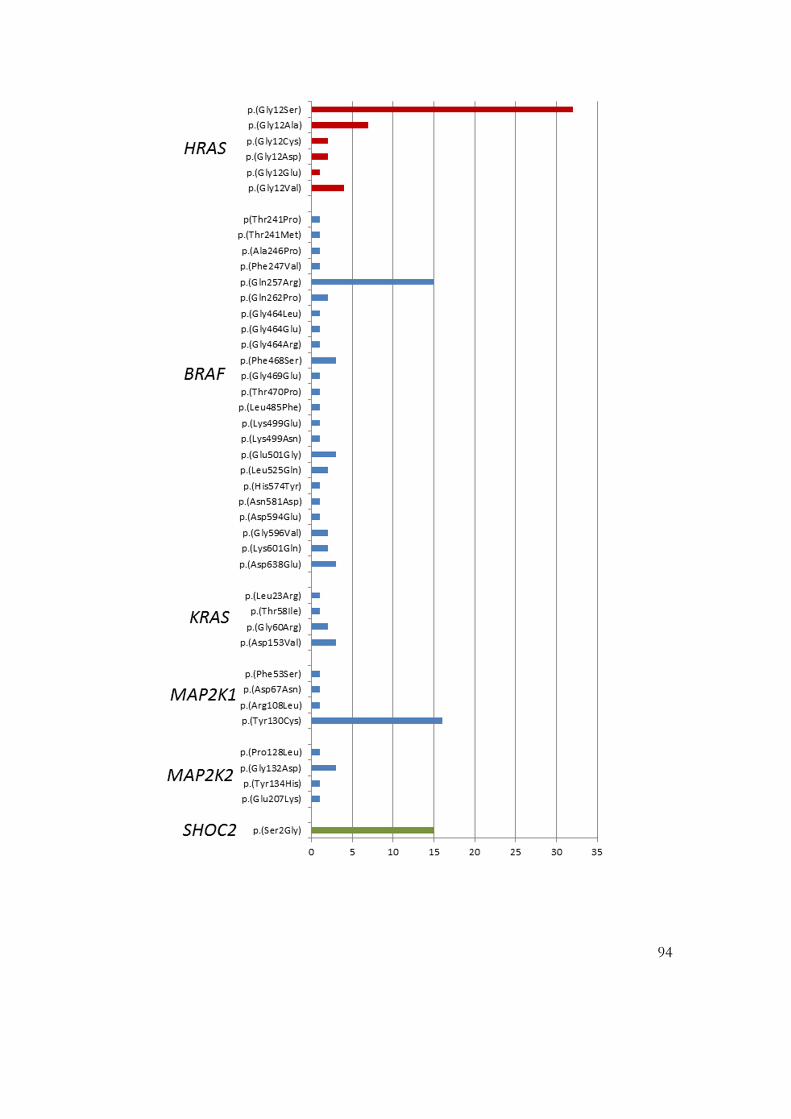
94

95
3.3 Clinical features of patients with mutation-proven CFC
78 molecular diagnoses of CFC (not including the patients with a mutation in
SHOC2, who are discussed in section 3.4) had been made in the laboratory by the end of
2012, as shown in Figure 3-2. Summary details of these patients are described below,
together with more detailed information about individuals whose families had consented
to involvement in the clinical study.
3.3.1 Patients with a BRAF mutation
48 patients had been diagnosed with a mutation in BRAF. Of these, 15 had the
mutation p.(Gln257Arg), which has been identified in all series as the commonest CFC-
associated substitution (37). Whilst mortality had been observed in the cohort of patients
with BRAF mutations, this appeared to be lower than has been noted to be the case for
patients with CS (i.e. HRAS mutations (16)): only 1 patient with a BRAF p.(Gln257Arg)
mutation was known to have died, at age 6 years from a presumed lower respiratory tract
infection, but further details were not available. Phenotypic data of 8 patients with a
mutation in BRAF were available, 4 of whom carried the common mutation,
p.(Gln257Arg), and 4 of whom were patients with other mutations in BRAF.
Photographs were available for 7 patients, Figure 3-3. Features of patients reviewed in
person are shown in Table 3-2 and Table 3-3.
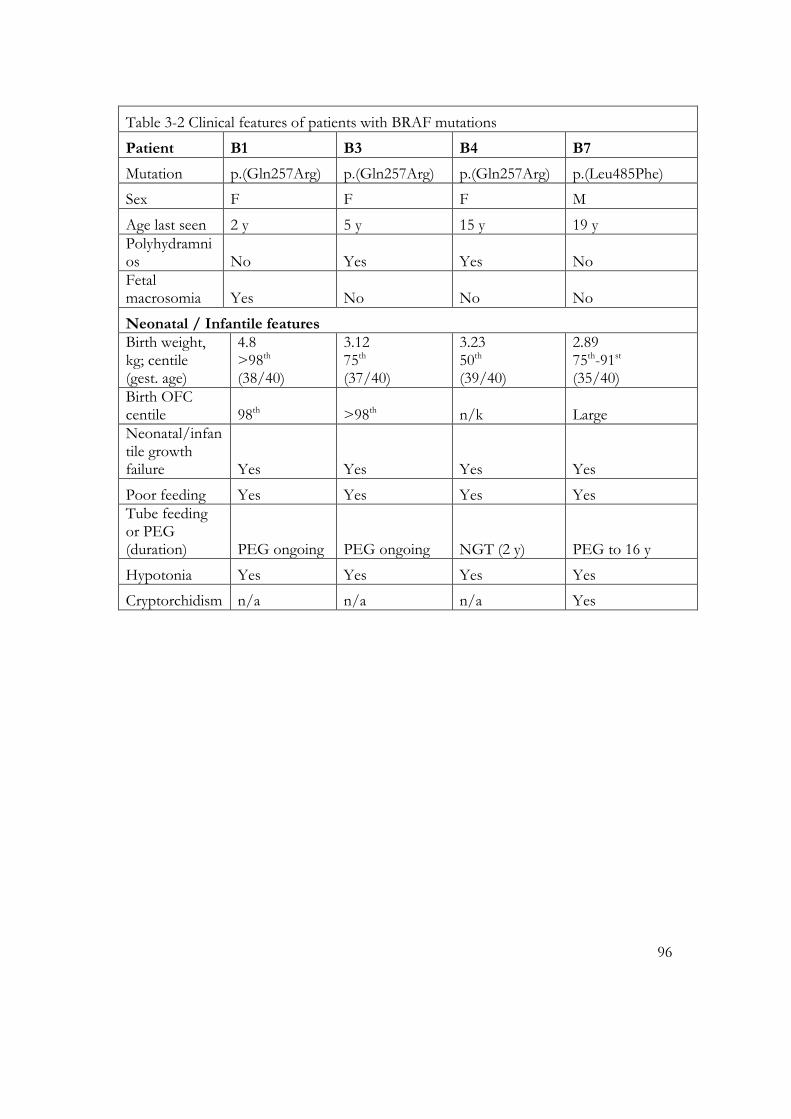
96
Table 3-2 Clinical features of patients with BRAF mutations
Patient B1 B3 B4 B7
Mutation p.(Gln257Arg) p.(Gln257Arg) p.(Gln257Arg) p.(Leu485Phe)
Sex F F F M
Age last seen 2 y 5 y 15 y 19 y Polyhydramnios No Yes Yes No Fetal macrosomia Yes No No No
Neonatal / Infantile features Birth weight, kg; centile (gest. age)
4.8 >98th (38/40)
3.12 75th (37/40)
3.23 50th (39/40)
2.89 75th-91st (35/40)
Birth OFC centile 98th >98th n/k Large Neonatal/infantile growth failure Yes Yes Yes Yes
Poor feeding Yes Yes Yes Yes Tube feeding or PEG (duration) PEG ongoing PEG ongoing NGT (2 y) PEG to 16 y
Hypotonia Yes Yes Yes Yes
Cryptorchidism n/a n/a n/a Yes

97
Growth
Height, cm; centile (age)
Weight 10kg (9th 21m) <0.4th
134.0 0.4th after GH (12 y) Short stature
Bone age n/k n/k n/k n/k
GH deficiency n/k n/k yes No; received GH Delayed puberty n/a n/a Yes Yes Developmental delay? Yes Yes, severe Yes, moderate
Yes, moderate – severe
Age at walking n/a 4 y 2 y 6 m 3 y
Craniofacial features
OFC centile 99.6th >98th 98th >98th
Downslanting palpebral fissures Yes Yes Yes Yes
Hypertelorism Yes Yes Yes Yes
Strabismus Yes Yes Yes No Epicanthic folds Yes Yes No No Palpebral ptosis Yes Yes Yes No Flat nasal bridge Yes Yes Yes Yes Broad nasal root Yes Yes No Yes Prominent philtrum Yes No No No
Thick lips No Yes No No
Macrostomia No Yes Yes No
Low-set ears Yes Yes Yes Yes Thickened helix Yes Yes Yes Yes Large, thick ear lobe yes Yes yes Yes

98
Cardiac features Pulmonary valve stenosis Yes No Yes Yes Atrial septal defect No Yes No Yes
HCM Yes Yes No No
Other Coarctation, VSD
Cutaneous features
Dark skin No (yes) No No Keratosis pilaris faciei No Yes Yes No
Hyperkeratosis No Yes, mild Yes No Sparse/absent scalp hair Yes Yes No Yes
Curly hair Yes Yes Yes Yes Thin dystrophic nails No No No No Eczema or ichthyosis No No Yes No
Pruritus No Yes Yes No
Redundant skin No No No No
Other Skin picking
Haematological / immunological
Hepatospleno-megaly No No Yes, neonatal No Musculo-skeletal symptoms No
Arthralgia Myalgia No
Behavioural
Anxiety or irritability Yes Yes Yes Yes Hypersensitivity to light, sound, touch Yes Yes Yes Yes Short attention span Yes Yes Yes Yes
Poor sleep Yes Yes Yes Yes Repetitive behaviour Yes Yes No Yes

99
Table 3-3 Clinical features of patients M1-M5
Patient M1 M2 M3 M4 M5
Sex M M F M F
Age last seen 2 y 13y 13 y 19 y 25 y Polyhydramnios yes no n/k Yes Yes Fetal macrosomia no no No No Yes
Neonatal / Infantile features Birth weight centile (gest. age)
75th (37/40)
50th (40/40)
25th (40/40)
50th (39/40)
91st (27/40)
Neonatal/infantile growth failure yes yes Yes Yes Yes
Poor feeding yes yes Yes Yes Yes Tube feeding or PEG (duration)
PEG (ongoing) PEG to 3y No
PEG (ongoing) no
Hypotonia yes yes Yes Yes Yes
Cryptorchidism no no n/a No n/a
Growth
Height centile (age)
Weight 2nd centile (2 y)
<0.4th
(12y)
<0.4th (8 y 8 m)
<0.4th (19 y)
<0.4th (adult)
Delayed puberty n/a
Pre-pubertal at 13 y
Pre-pubertal at 13 y
Yes, early puberty at 19 y
Yes, primary ameno-rrhoea

100
Craniofacial features
OFC centile 91st 91st 50th 98th 91st Downslanting palpebral fissures Yes yes no No Yes
Hypertelorism Yes yes yes No Yes
Strabismus yes no yes No Yes Epicanthic folds no no no No No Palpebral ptosis no no yes Yes Nes Flat nasal bridge yes no yes Yes Yes Broad nasal root yes no yes Yes Yes Prominent philtrum no no no, short no, short No
Thick lips yes no no Yes Yes
Macrostomia yes yes yes Yes yes
Low-set ears yes yes yes Yes yes Thickened helix yes yes yes Yes yes Large, thick ear lobe yes yes yes Yes yes
Cardiac features
Pulmonary valve stenosis no yes yes
peripheral pulmonary stenosis yes
Atrial septal defect no no no No no
HCM no no no No no
Other Coarctation

101
Cutaneous features
Dark skin No yes No Yes no Keratosis pilaris faciei No yes Yes Yes yes
Hyperkeratosis No no No No yes Sparse/absent scalp hair Yes no No No yes
Curly hair Yes yes Yes Yes yes Thin dystrophic nails No no No No no Eczema or ichthyosis No no No Yes no
Pruritus No no Yes Yes no
Redundant skin No no No No no
Other Hyper-hidrosis
Pressure sores
Musculoskeletal symptoms
contractures
Severe contractures
Severe contractures contractures
Behavioural
Anxiety or irritability Yes yes Yes Yes yes Hypersensitivity to light, sound, touch Yes yes Yes Yes yes Short attention span Yes yes Yes Yes yes
Poor sleep Yes yes Yes Yes no Repetitive behaviour No yes Yes Yes no
3.3.1.1 p.(Gln257Arg): the archetypical CFC-associated mutation
Referral data regarding the 15 patients with this mutation demonstrated a variable
severity of presentation. Initial referral reasons provided were a clinical suspicion of CFC
in 9, balanced between CFC and CS in two, and possible CS in 4 (For comparison, all
samples in which a mutation in HRAS was found were from patients with a clinical
suspicion of CS, though this observation is likely to be influenced by the more pressing
need to confirm/refute a CS diagnosis as discussed above, and the fact that many of
these latter patients had been assessed by Dr Kerr).

102
The 4 patients with p.(Gln257Arg) mutations for whom further phenotypic data were
available demonstrated significant similarities to one another, and all had presentations in
keeping with a classical CFC phenotype. As would be expected (4), facial characteristics
in this group included a prominent forehead, sparse eyebrows and ptosis (Figure 3-3).
Sparse hair was common until the age of around 5 years, at which point hair growth
tended to improve. Very curly hair was present in all 4 patients, and was either red or of
gingery colour in the 3 of Caucasian origin, whilst the patient of mixed Caucasian and
black ethnic origin (B3) had black hair.
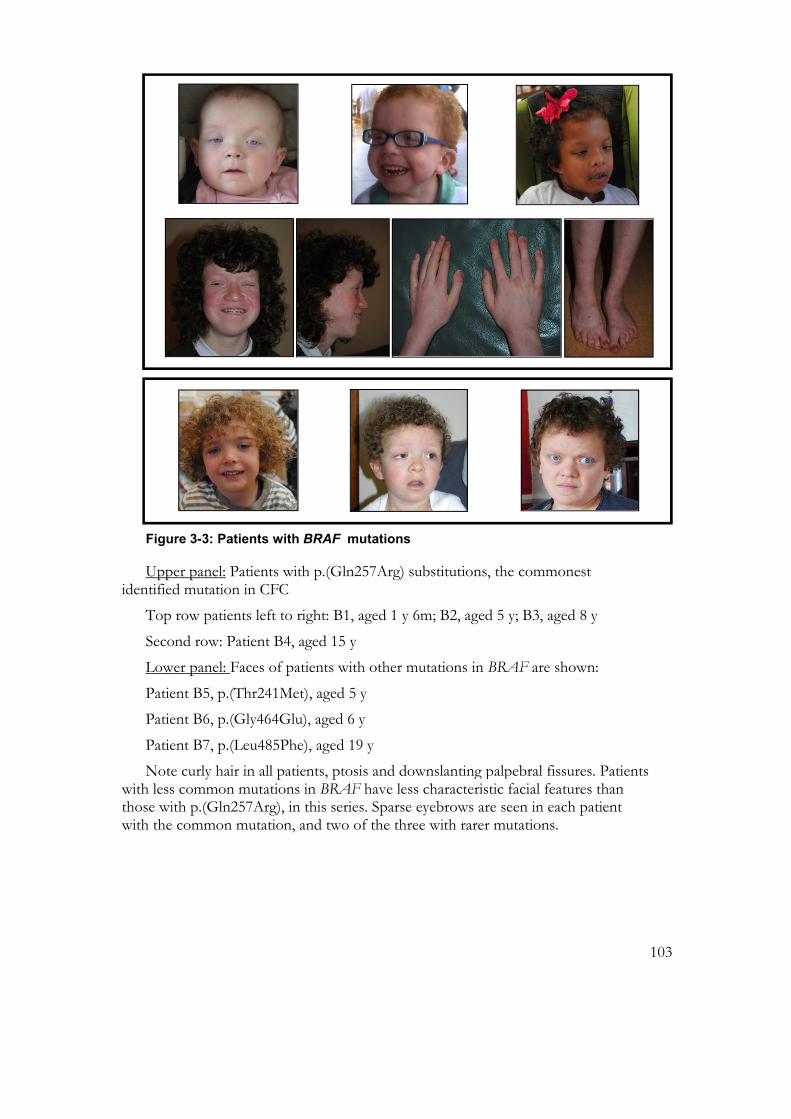
103
Figure 3-3: Patients with BRAF mutations
Upper panel: Patients with p.(Gln257Arg) substitutions, the commonest identified mutation in CFC
Top row patients left to right: B1, aged 1 y 6m; B2, aged 5 y; B3, aged 8 y
Second row: Patient B4, aged 15 y
Lower panel: Faces of patients with other mutations in BRAF are shown:
Patient B5, p.(Thr241Met), aged 5 y
Patient B6, p.(Gly464Glu), aged 6 y
Patient B7, p.(Leu485Phe), aged 19 y
Note curly hair in all patients, ptosis and downslanting palpebral fissures. Patients with less common mutations in BRAF have less characteristic facial features than those with p.(Gln257Arg), in this series. Sparse eyebrows are seen in each patient with the common mutation, and two of the three with rarer mutations.

104
3.3.1.2 Other mutations in BRAF
Alongside allelic heterogeneity, the group of patients identified to have one of the
less common mutations in BRAF also appeared to demonstrate more phenotypic
variability than those with the p.(Gln257Arg) substitution. Some of the presentations
observed were more severe than was seen in association with p.(Gln257Arg), and some
less severe. As the large majority of mutations have been identified in only one or two
patients in this cohort, and some are unique to the individual, it is not possible to draw
many inferences on genotype-phenotype correlation.
The spectrum of ability in this patient group varied from children who were
managing with extra assistance in mainstream school, to a patient in her teenage years
with a p.(Leu525Gln) mutation who had never walked, had no speech, and was affected
with intractable seizures, respiratory failure due to probable pulmonary lymphangiectasia
and multiple other medical problems. She had been born with multiple structural
anomalies, namely an atrial septal defect and absent right kidney. This constellation of
difficulties is reminiscent of reports of other patients with CFC (4), but more severe than
for many patients. A novel association included the presence of a plexiform unicystic
ameloblastoma of the right mandible aged 3 y, in a patient with a p.(Gly464Arg)
mutation. A further patient, patient B6, had severe cyclical vomiting syndrome, in
association with a p.(Gly464Glu) mutation.
3.3.2 Patients with a MAP2K1 mutation
19 patients have been diagnosed with a mutation in this gene in the MCGM
laboratory. p.(Tyr130Cys) appears particularly over-represented in this cohort (16 of the
19 patients), and all 5 patients for whom extensive data were obtained had this same
mutation. Importantly, three of these patients were in their second decade, and a fourth
was a young adult (25 y), and hence significantly older than the majority of patients
known to have CFC. Features of the five patients reviewed in person are shown in Table
3-3.
The most striking feature in these patients, especially M3 and M4, but which was also
present in M2 and M5, was the presence of multiple joint contractures. These appeared
disproportionate to the patients’ level of movement (as none had had prolonged periods

105
of immobility), and were severe enough in M3 and M4 to make these patients non-
ambulant and unable to stand up: M4 mobilised by means of a custom-built chair on
wheels, and M3 by bottom shuffling around her home. The other two patients, M2 and
M5, were also wheelchair users, but were able to stand with assistance, and M2 was able
to mobilise short distances. The only patient with this mutation in this series without
contractures was very young when assessed.
On reviewing the faces of these patients, there are significant similarities amongst
them, and also possible differences from those with BRAF mutations. In these
individuals, a more triangular face shape appeared to be present. Significant coarsening of
the face over time was seen in older patients for whom serial photographs were available
(Figure 3-5), with similarities to the evolution of facial characteristics that is well
recognised in CS (21).

106
Figure 3-4: Patients with MAP2K1 p.(Tyr130Cys) mutations
Top row: patient M1, aged 2 y
Middle row: patient M2, aged 13 y
Bottom row: patient M3, aged 10 y, patient M4, aged 18 y, patient M5, aged 23 y.
Note sparse eyebrows, curly hair and long, slender fingers. Facial characteristics appeared to coarsen with time (see Figure 3-5).

107
Figure 3-5: Serial photographs of patients with MAP2K1 p.(Tyr130Cys) mutations
Upper panel: patient M5 from 1 y to 25 y
Lower panel: patient M4 from 1 y to 18 y
Note coarsening of facial features with increasing age.

108
3.3.3 Patients with other mutations causing CFC
Very few patients have been diagnosed with mutations in KRAS or MAP2K2 in the
MCGM laboratory, mirroring the rarity of such mutations in the published literature (6).
Of the 7 patients diagnosed with KRAS mutations, 4 were referred with a clinical
suspicion of CS, and two are deceased. One patient was reported previously with a large
congenital ulcerating haemangioma (194).
7 patients have also been diagnosed with mutations in MAP2K2. Of note, the three
patients with the p.(Gly132Asp) mutation are the mother and sons reported by Linden
and Price (96), who each had severe ectodermal manifestations. Limited information was
available on one other patient with a MAP2K2 mutation, who also had a dramatic
ectodermal phenotype, with exuberantly curly hair and the development of very large
numbers of naevi in the second decade of life. For this patient, like each of the familial
cases reported in the literature, she had a degree of intellectual disability, but this was
relatively mild, as compared to other patients known to have BRAF p.(Gln257Arg) or
MAP2K1 p.(Tyr130Cys) mutations.
All patients in whom a mutation in MAP2K2 was identified were clinically considered
to have CFC, rather than CS or NS. These limited observations are consistent with the
hypothesis that MAP2K2 mutations are strongly associated with both significant
ectodermal features and intellectual disability, each of which may be considered hallmarks
of CFC. The degree of the latter may, however, be milder than is frequently observed
with mutations in other CFC-associated genes. In support of this hypothesis, multiple
families with dominant transmission, including over four generations, have now been
identified worldwide (95, 96), but data sets including larger numbers of patients could
assist in confirming this.

109
3.3.4 Findings across the group of patients with CFC-associated
mutations
The wide variety of manifestations observed in this patient group mirrors
observations in other studies of patients with CFC. Many had arrived at the correct
diagnosis following an initial diagnosis of NS (or sometimes CS). Other genetic
conditions, such as Williams syndrome, had also sometimes been suggested by clinicians
in individual patients, as is commonly the case for children with developmental delay,
dysmorphic features and/or congenital abnormalities.
The behavioural phenotype of CFC requires further assessment. Hypersensitivity to a
variety of stimuli, particularly sound, bright light and tactile stimuli, appears very
common in this patient group, and, along with irritability (sometimes with self-injurious
behaviour) was a significant source of difficulty for these patients and their families.
Tactile defensiveness is a feature that has been apparent in many patients in this cohort,
which has significant impact on day-to-day abilities and activities. High levels of anxiety
in children with CFC syndrome were also reported by parents and caregivers. All of these
factors were reported as having impacts upon daily living such as responses to being in
public places or a schoolroom.
The question of autistic features across the disorders of the Ras-MAPK pathway has
been the source of much debate in recent years, but the information gathered in this
study is not strongly in support of this hypothesis in the CFC patients studied. Significant
intellectual disability and high levels of anxiety and tactile defensiveness appeared to
combine in some individuals, and these could be sufficient to result in behaviour that
may resemble that seen in children with autistic spectrum disorders. However, high rates
of autistic traits have been identified in patients with NF1, and further assessment of this
is warranted. This is now in progress through an MCGM collaboration with the
University of Manchester’s Department of Child Psychiatry (‘Cognitive profiling in the
genetic disorders of the Ras-mitogen-activated protein kinase (Ras-MAPK) pathway:
towards pathogenesis-based treatment’; Newlife foundation for disabled children).

110
3.4 Extending the molecular basis of CFC: SHOC2 is an important
disease gene in this patient group
Of patients with CS or CFC referred to the MCGM diagnostic laboratory prior to
2010 for molecular testing of these conditions, there were 162 without a mutation
identified. 140 had sufficient DNA remaining for further testing. Testing for the
p.(Ser2Gly) mutation in SHOC2 was undertaken to test the hypothesis (104) that this
gene could cause CFC or CS-like phenotypes. Of these patients, 10 were shown to have
heterozygous c.4A>G mutations in SHOC2, p.(Ser2Gly), previously described as the
cause for ‘Noonan-like syndrome with loose anagen hair’ (104), demonstrating that this
mutation is indeed a common, and hence important, cause of a severe NCFC phenotype.
Detailed clinical information was available on these 10 patients, and 4 further patients
in whom mutations were subsequently identified. These individuals are described below
in detail and pictured in Figure 3-7, Figure 3-6 and Figure 3-8. A summary of growth
parameters and other clinical features is shown in Table 3-4 and Table 3-5. Where DNA
samples were available from both parents (6 families), the mutation was demonstrated to
have arisen de novo in all cases, rather than being inherited from a parent. This is in
keeping with all previously published cases of SHOC2 mutation to date (104, 105).
Detailed clinical information about the phenotypes of each of these patients was
sought, where possible, by meeting the patient in person (patients 5,6,11,12 and 14), and
where this was not possible, from the clinician who had referred the patient’s sample for
testing (patients 1-4,7-10 and 13). This data was of particular significance in view of the
newly described nature of the p.(Ser2Gly) mutation in SHOC2 , and the possibility that it
would confer a unique and recognisable phenotype. The clinical presentations of these
patients demonstrate a broader spectrum of phenotypic features than have previously
been reported in the literature, as discussed below. Two patients had been referred with
clinical diagnoses of CS, six with CFC, and one with NS. Two patients had a clinical
diagnosis poised between NS and CFC, and three patients (S12-14) presented recently
enough to be identified prospectively as being likely to have p.(Ser2Gly) mutations.
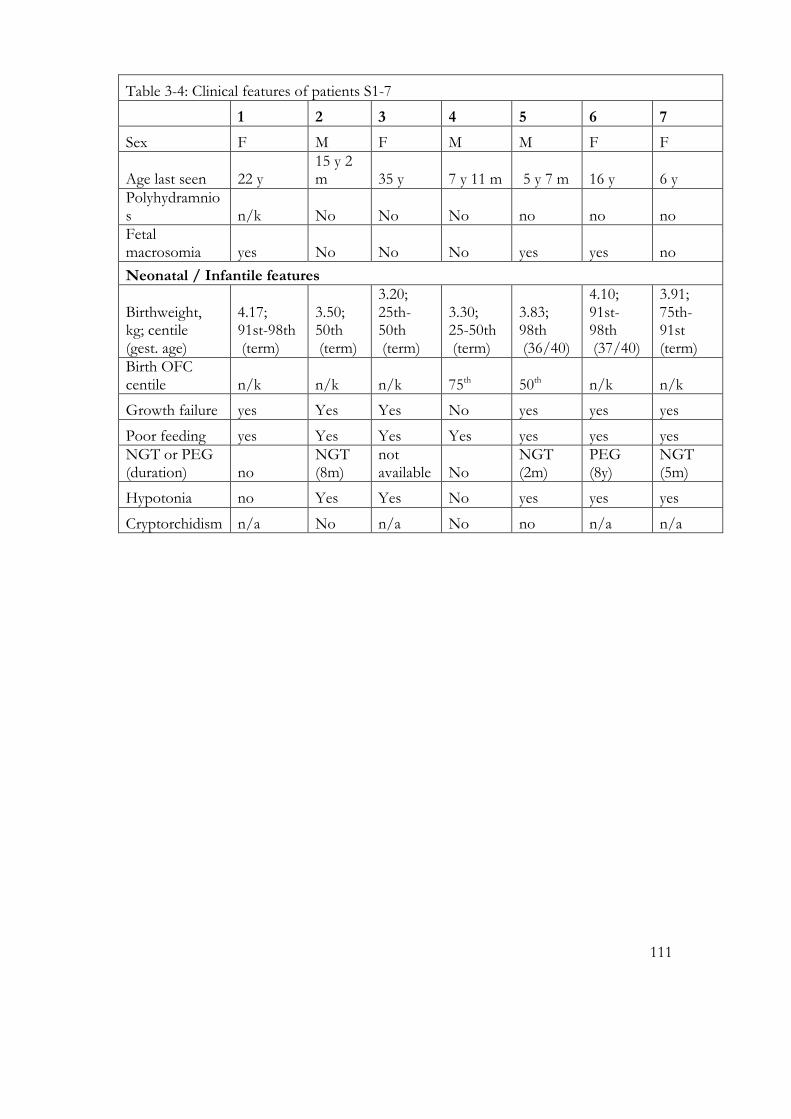
111
Table 3-4: Clinical features of patients S1-7
1 2 3 4 5 6 7
Sex F M F M M F F
Age last seen 22 y 15 y 2 m 35 y 7 y 11 m 5 y 7 m 16 y 6 y
Polyhydramnios n/k No No No no no no Fetal macrosomia yes No No No yes yes no
Neonatal / Infantile features
Birthweight, kg; centile (gest. age)
4.17; 91st-98th (term)
3.50; 50th (term)
3.20; 25th-50th (term)
3.30; 25-50th (term)
3.83; 98th (36/40)
4.10; 91st-98th (37/40)
3.91; 75th-91st (term)
Birth OFC centile n/k n/k n/k 75th 50th n/k n/k
Growth failure yes Yes Yes No yes yes yes
Poor feeding yes Yes Yes Yes yes yes yes NGT or PEG (duration) no
NGT (8m)
not available No
NGT (2m)
PEG (8y)
NGT (5m)
Hypotonia no Yes Yes No yes yes yes
Cryptorchidism n/a No n/a No no n/a n/a

112
Growth
Height, cm; centile (age)
156.9; 9th (adult)
141; <0.4th (15y2m)
149; 0.4th (adult)
102.7; <0.4th (7y5m)
101; 0.4th 5y7m)
146.5; 0.4th (16y)
104.8; 0.4th (6y)
Bone age 2½y at 4½y n/k n/k n/k n/k
11y at 14y n/k
GH deficiency resistant Yes Yes borderline
not tested resistant pending
Delayed puberty yes Yes Yes n/a n/a yes n/a
Craniofacial characteristics OFC, cm; centile (age)
56.9 (adult)
58.5 (15y)
56.5 (adult)
52.5 (3y0m)
53.5 (5y7m)
57.6; (16y)
>97th (6y)
Downslanting palpebral fissures yes Yes no Yes yes yes yes
Hypertelorism yes Yes no Yes yes yes yes
Strabismus no Yes yes No yes no yes Epicanthic folds no No no No no no no Palpebral ptosis yes Yes no Yes yes yes yes Flat nasal bridge no No no Yes yes no no Broad nasal root no No no No no no no Prominent philtrum yes No no No no no no
Thick lips yes Yes yes No yes no no
Macrostomia no No yes No no yes no
Low-set ears yes Yes yes Yes yes yes yes Thickened helix yes Yes yes Yes yes yes yes Fleshy ear lobes yes Yes yes Yes yes yes no

113
Cardiac features Pulmonary valve stenosis yes No no No no yes no Atrial septal defect no No no No no no no
HCM no No no No yes no no
Other
coarctation, SVAS
Cutaneous features
Dark skin no Yes yes No no yes no Keratosis pilaris no No no No yes yes no
Hyperkeratosis no Yes no No yes no no Sparse/absent scalp hair yes Yes no Yes yes yes yes
Curly hair no No no No no no no Thin dystrophic nails no No yes No no no no Eczema or ichthyosis no Yes no Yes severe infancy infancy
Pruritus no No yes Yes yes yes no
Redundant skin yes Yes yes Yes yes yes no
1 CAL
papillomas 1 CAL
hyper-hidrosis
strawberry naevus
Haematological / immunological
Hepatospleno-megaly yes Possible autoimmune disease Yes ?MCTD
Juvenile arthritis
Myeloproliferative abnormalities

114
Table 3-5: Clinical features of patients S8 – 14
Patient 8 9 10 11 12 13 14
Sex F F F F M M M
Age last seen 3 y 6 m 25 y 11 y 11 y 5 m 1 y 6 m 9 y 18 y
Polyhydramnios no No no Yes no no no Fetal macrosomia yes Yes no Yes no no no
Neonatal / Infantile features Birth weight, kg; centile (gest. age)
3.60; 91st (38/40)
3.80; 75th-91st (term)
3.90; >99.6th (36/40)
4.35 98th (term)
2.99; 9th-25th (term)
2.41; 75th-91st (33/40)
1.87; 2nd (36/40)
Birth OFC centile 90-97th n/k 98th >99.6th 75th n/k n/k Neonatal/infantile growth failure yes Yes yes Yes yes n/k yes
Poor feeding yes Yes yes Yes yes (yes) yes
Tube feeding or PEG (duration)
PEG (ongoing) No
NGT (1 m) No no
NGT (4 m)
NGT (6 y)
Hypotonia no No yes No yes yes yes
Cryptorchidism n/a n/a n/a n/a no n/k yes
Growth
Height, cm; centile (age)
94; 9th (3y6m)
141; <0.4th (15y)
124; 0.4th (10y6m)
86; 0.4th (3y6m)
72; <0.4th (1y6m)
106.5; <0.4th (7y5m)
158.2; 0.4th (18y)
Bone age n/k n/k 9y at 11y 4y at 6y6m no delayed n/k
GH deficiency not at 3y6m Yes Resistant
GH resistant
not yet tested resistant n/k
Delayed puberty n/a n/k n/a n/a n/a n/a no, slow
Craniofacial features OFC, cm (centile) (90-97th) Relative (75th) 53 (98th) 49 (50th) 53.5 (50th)
57.1 (50th)
Downslanting palpebral fissures yes n/k No Yes yes no
upslanting
Hypertelorism yes n/k Yes Yes yes yes yes
Strabismus no n/k No Yes no no no
Epicanthic folds no n/k Yes No no no no
Palpebral ptosis yes n/k Yes Yes yes yes yes

115
Flat nasal bridge no n/k No No yes no no Broad nasal root no n/k No No no no no Prominent philtrum no n/k No No yes no no
Thick lips no n/k Yes No no no no
Macrostomia no n/k No No no no yes
Low-set ears no n/k Yes Yes yes yes yes
Thickened helix yes n/k No Yes yes yes no Large, thick ear lobe yes n/k No Yes yes yes yes
Cardiac features Pulmonary valve stenosis no No No Yes yes no yes Atrial septal defect no No No Yes no no yes
HCM no No No Yes no yes no
Other MVP small VSD

116
Cutaneous features
Dark skin no No Yes No no no yes Keratosis pilaris faciei no No No Yes no no yes
Hyperkeratosis no No No Yes no no yes Sparse/absent scalp hair yes No Yes Yes yes yes yes
Curly hair yes No Yes Yes no no no Thin dystrophic nails no No No Yes yes no no Eczema or ichthyosis yes Yes No Yes yes yes yes
Pruritus no Yes No Yes no no no
Redundant skin yes Yes Yes Yes no yes yes
Other 2 CAL Haem-angioma
haemangioma 1 CAL papillomas poor teeth
6 CAL,
hyper-hidrosis
Capillary mal-formation, 1 CAL
Haematological / immunological
Hepatospleno-megaly Yes Yes yes Possible autoimmune disease
Crohn's disease Joint pain
Joint pain
Myeloproliferative abnormalities
Transient neonatal
MVP: mitral valve prolapse; VSD: ventricular septal defect

117
3.4.1 Clinical presentations of patients diagnosed with SHOC2
p.(Ser2Gly) mutations
Patient S1 was the youngest child of non consanguineous parents of Vietnamese and
Chinese origins. Family history was non-contributory, but her father was 37 when she
was born. The pregnancy had been complicated by vomiting and glycosuria, but a
glucose tolerance test was normal. Dysmorphism was noted at birth, with a very wide
neck. Cardiology assessment for dusky episodes revealed mild pulmonary stenosis and a
slightly enlarged left atrium.
At 3 months she remained below her birth weight, following severe congenital
oedema and poor feeding. Excess nuchal skin, a short chest with widely spaced nipples
and hypertelorism with downslanting palpebral fissures were present, but no ptosis.
Posteriorly rotated ears, one café au lait patch and abnormal palmar creases were also
noted. Her karyotype was 46,XX, including extended counts for mosaicism, and the
diagnosis of NS was considered clinically. Significant developmental delay was evident:
she sat unsupported at 14 months, and at 18 months bottom-shuffled and had 2-3 words
with meaning. She walked at 2½ y.
She received special schooling for learning difficulties, but developed English and
Cantonese language with adenoidal speech. Aged 5 y, she had very sparse wispy hair
which had never been cut, eczema, poor teeth and a squint. Aged 9 y, endocrine
investigations showed normal cortisol response to glucagon stimulation, but GH
response was blunted at only 16.3mU/l. Gonadotropins were present and appropriately
prepubertal. She had a low IGF1 and IGFBP3. Her growth velocity was only
3.5cm/year, and she received growth hormone (GH) therapy until age 17, when
menarche occurred. She was attending college for life skills and had friends but was very
shy.
Patient S2 was a 15 y old boy with a clinical diagnosis of CFC. He received extra
help in mainstream school for mild to moderate learning difficulties. There were no
problems in the pregnancy, but he lost weight due to poor neonatal feeding, requiring

118
tube feeding from 2 to 8 months. Hypotonia and global developmental delay were
apparent. GH treatment was administered from age 13 for short stature, without GH
deficiency. His onset of puberty was delayed, but he was otherwise medically well until he
developed an acute severe idiopathic pericardial effusion aged 14, which showed an
excellent response to steroid treatment. He had sparse, thin hair which improved with
age, and darker skin than the rest of his family. His dentition was disorganised, but he
had no other ectodermal features.
Patient S3 was the first child of second cousin Iranian parents. Her family history
was non-contributory, and the pregnancy uneventful. She was an unsettled, crying baby
with marked feeding difficulties, hypotonia and a very poor suck. Her family had no
access to medical intervention in Iran so constantly poured milk into her mouth and then
puréed food: if tube feeding had been available, this would have been indicated. She had
marked failure to thrive: at 1 y, she was said to be 6 months’ size, and she continued to
be small for her age.
She had marked motor delay: head control was achieved at one year of age, sitting at
3 y and walking at 4 y. She started using single words at 18-24 months, and sentences
from 3 y. Aged 6 y, she required special education for learning difficulties and had
bilateral alternating esotropia and hypermetropia. As an adult, she had a friendly
disposition and good self help skills, helping with house work, but did not understand
arithmetic or money, and could not tell the time using analogue or digital clocks. She had
very limited reading skills, and answered questions verbally at a similar level to a 7-8 y old
child. Her hearing was normal. She required orthodontic braces but dentition was
otherwise normal.
She had an anxious personality, and was taking sertraline 50 mg daily. She previously
had normal mobility, but age 35 y, she presented to medical attention as her walking had
deteriorated due to unsteadiness. Her upper limbs were neurologically normal but
increased tone was present in her lower limbs with brisk lower limb reflexes and a wide
based gait (genetic testing for spinocerebellar ataxia types 1, 2, 3 and 7 and Friedreich
ataxia showed normal results). She had mild small joint laxity, and excess palmar and
plantar creases, thick ankles but no pitting oedema and no joint contractures. Menarche

119
was at age 17, and menses were very irregular. Other secondary sexual characteristics
were also affected, with scant pubic and absent axillary hair, and very little breast tissue.
Her scalp hair was dry, and soft not wiry, but grew slowly, fell out easily and looked over-
permed when it had never been chemically treated. Similarly, her nails grew slowly and
split easily. She had posteriorly rotated and mildly low set ears, and two small papillomas
on her left ala nasi (not visible on the photographs in Figure 3-6). Her skin was dry and
itchy, darker than other family members’ skin, and was noted to be loose and soft,
especially on the dorsum of her hands. Her face had an aged appearance, with
prematurely wrinkled skin. She had patchy hyperpigmentation on back of both her legs,
varicose veins on one leg. Extensive genetic and other laboratory investigations were
normal, including karyotype 46, XX, subtelomere MLPA screen, urine amino and organic
acids and glycosaminoglycan profiles, and transferrin isoelectric focussing. Vitamin E and
B and folate levels and thyroid function tests were also normal, but she had iron
deficiency anaemia. An MRI scan of brain, cervical and thoracic spines showed
generalised mild cerebral atrophy only.
Patient S4 was an 8 y old boy with a clinical diagnosis of CFC. His mother and
father were 31 and 46 years old when he was born. He was admitted to the neonatal unit
with a suspected chest infection. He had feeding problems but managed to gain weight
without artificial feeding. Hepatosplenomegaly was noted in the neonatal period, and he
also had ichthyosis and pruritus since that time. His skin was otherwise unremarkable,
but his hair was fine and sparse and fell out easily (though this improved with age), and
his tooth enamel was dysplastic. He had short stature, below the 0.4th centile for height,
with borderline pituitary function tests: peak GH following a stimulation test was
12.8ng/ml (normal peak: >10ng/ml). Relative macrocephaly was present, and prominent
ventricles and cortical sulci were noted on MRI scan. He attended special school, with
integration into mainstream classes half a day per week. He had a short temper, and
showed hyperactivity and poor concentration.
Patient S5 was a 6 y old boy with a clinical diagnosis of CFC. Antenatally, enlarged
cerebral ventricles were noted, and delivery was induced at 36 weeks’ gestation because
of a poor biophysical profile, and maternal gestational diabetes and cholestasis. His
birthweight was 3.83kg, but he was oedematous and he weighed 3.50kg 4 weeks

120
postnatally. Whilst on the neonatal intensive care unit with polycythaemia requiring
partial exchange transfusion, and poor feeding, hypertrophic cardiomyopathy was
diagnosed, but this did not require treatment. Severe atopy and food allergies, including
to cow’s milk and soya were present. Eczema required extensive treatment, including
steroid and tacrolimus ointments and daily wraps. He had allergic conjunctivitis of
sufficient severity for a mixed connective tissue disorder to have been considered. An
extensive metabolic workup showed normal results including urine organic and amino
acids, mucopolysaccharide screen, and very long chain fatty acids. His physique and facial
appearance were in keeping with NS or CFC, as alongside relative macrocephaly, he had
ptosis, downslanting palpebral fissures, low set posteriorly rotated ears and pectus
excavatum. He presented to hospital at 8 y of age with a hypertensive encephalopathy.
Bilateral phaeochromocytomas, not previously reported in CFC or patients with SHOC2
mutations, were identified on abdominal ultrasound.
Patient S6 had a longstanding clinical diagnosis of NS, but with particularly
prolonged and severe feeding difficulties, remaining partially gastrostomy fed until the
age 8. Pulmonary stenosis and duplex kidneys were diagnosed in the neonatal period. The
latter required a heminephrectomy and removal of the duplex system. She also had
severe infantile eczema. Her skin was darker than other family members, with keratosis
pilaris. She was diagnosed with mild von Willebrand disease aged 15, following blood
tests prior to dental work (orthodontics and fillings to carious teeth). She received GH
from the age of 10 y for short stature. Menarche was delayed, at 15 ½ y, following
oestrogen therapy. Idiopathic juvenile arthritis developed at 13 y, requiring intra-articular
steroids, but was then quiescent. She had mild generalized joint laxity. She received 1:1
support and small group teaching in a mainstream school, but continued to be unable to
do sums or tell the time. She had a short attention span, and was impulsive with a poor
sense of danger, and also some obsessive traits. Melatonin therapy helped her sleep
problems, but constant hunger and frequent snacking (including at night) was a feature.
Patient S7 presented at 20 weeks’ gestation with a pleural effusion requiring
drainage. No other scan abnormalities were detected prenatally. Amniocentesis
demonstrated a normal karyotype. Emergency Caesarian delivery was necessary for fetal
distress and presence of meconium. Coarctation of the aorta and mild supravalvular

121
aortic stenosis were diagnosed postnatally. Repiratory distress due to heart failure
developed, and the coarctation was operated on at 6 weeks of age. She had significant
feeding difficulties, and was fed by nasogastric (NG) tube until 5 months. Severe eczema
was present from one week to 7 months of age. She had an egg allergy. Gross motor
delay was apparent, as was fluctuation conductive hearing loss, probably related to glue
ear. Her developmental milestones were mildly delayed. Her hair was sparse, and aged 6 y
had never needed cutting.
Patient S8 was clinically suspected to have CFC, due to typical facial features,
including dry, eczematous skin requiring daily emollients and mild steroid
(hydrocortisone) treatment, and sparse hair. She required a gastrostomy for nutrition
until age 4 y. Global developmental delay was present, with independent walking from 22
months and delayed speech (using short phrases at 3½ y). MRI brain in infancy showed
mild cortical atrophy but was otherwise normal.
Patient S9 was clinically suspected to have CFC, having presented with poor feeding,
ichthyosis, eczema and mild joint hyperextensibility. She received GH treatment for short
stature and attended a special school. As an adult, she developed a depressive disorder,
for which she received pharmacological treatment, and had also been diagnosed with
Crohn’s disease and required surgery for intestinal malrotation.
Patient S10 was clinically suspected to have Costello syndrome. She was born at 36
weeks’ gestation weighing 3.9 kg (>97th centile), and spent 2 days in NICU with
respiratory distress. She was then discharged home but readmitted at 3 weeks with failure
of feeding, treated with a NG tube. Early milestones were delayed, sitting at 14 months
and walking at 22 months. Speech delay was also reported. Hepatosplenomegaly was
noted, and in early childhood, she developed multiple ulcerating capillary haemangiomas
(Figure 3-8), which required surgical management, but she was otherwise well. Her
exercise tolerance, reportedly due to pain in her legs, was reduced, limiting activities of
daily living such as shopping trips. Echocardiography at 10y demonstrated myxomatous
mitral and tricuspid valves with billowing leaflets and trivial incompetence. She also had
delayed eruption and enamel dysplasia of her primary dentition, cutting her first tooth at
14 months, but secondary dentition was normal. Her skin was darker than that of other

122
family members, with excess skin of the palms and soles (Figure 3-7) and easy bruising.
Aged 15 y, she had still not entered puberty. Special educational needs were identified,
and addressed in mainstream school and then by home schooling. Overfriendliness with
strangers was a source of considerable anxiety to her parents, and other aspects of her
behavioural phenotype included hypersensitivity to sounds such as those of a vacuum
cleaner. Her facial appearance and overall body habitus was consistent with a Ras-MAPK
pathway disorder, and in retrospect classical for patients with SHOC2 mutations in
particular.
Following the identification of such a high proportion of patients in the CFC/CS-like
cohort having this mutation, testing for this was incorporated into the Manchester
Regional Genetics Laboratory’s diagnostic testing service. In the subsequent two years,
five further patients with this same c.4A>G mutation in SHOC2 were identified. Further
clinical data was available on four of these (patients S11 to 14).
Patient S11 presented an intermediate phenotype, being considered to have CFC by
some clinicians, and NS by others. She had pulmonary stenosis, and atrial and ventricular
septal defects, with easy bruising and short stature. Very curly hair was present, but this
grew very slowly in early childhood. She developed papillomas just inside her alae nasi at
the age of 8 y. She had extensive genetic testing for CFC and NS, with no mutation
confirmed until SHOC2 testing was performed. She had poor sleep, with night terrors,
hypersensitivity to noise and high levels of stranger anxiety. Irritability and possible
autistic tendencies were also features.
Patient S12 presented with a spiral fracture of the femur at the age of 18 months,
with no apparent precipitating cause. For investigation of this, a bone biopsy was
performed, which showed abnormal trabeculation (195). He was already known to
paediatrics with poor feeding, failure to thrive and eczema, and had been noted to have
sparse hair. He was an irritable baby, and had been noted to be developmentally delayed.
The possibility of a mutation in SHOC2 was considered the most likely diagnosis when
he was seen by geneticists prior to any genetic testing.
Patient S13, a 9 y old boy was noted to have had a transient, spontaneously resolving
myeloproliferative disorder in early childhood. He also had very poor vision, being

123
registered partially sighted due to severe myopia. Mild nystagmus was also present. He
developed short stature but had a good response to GH therapy, growing 6 cm in the
first 6 months of treatment aged 8 y. His intellect was normal on IQ tests, but he had
some obsessive features, with particular enthusiasm for computers and lottery numbers,
for example.
Patient S14 had presented with low birth weight (1.87 kg at 36 weeks’ gestation,
emergency Caesarian section delivery for fetal distress) and poor feeding from birth,
which required admission to the neonatal intensive care unit and supplementary feeding
by NG tube until age 6. He was diagnosed with atrial septal defect and pulmonary
stenosis, and cryptorchidism and inguinal hernias were present. Dysmorphic features
were noted: consistent with a diagnosis of NS were pectus excavatum, low posterior
hairline, low-set, posteriorly rotated ears, a broad mouth and long eyelashes, but his
palpebral fissures were upslanting (Figure 3-6). He required eye patching for strabismus,
and wore hearing aids for a short period in childhood, but his hearing improved after
grommets, adenoidectomy and myringoplasty for a perforated eardrum. Short stature was
noted, and he received GH from the age of 13 y. Age of entry into puberty was normal,
and progress was accelerated on GH therapy. He experienced nocturnal epileptic seizures
around puberty, but these did not require treatment and resolved spontaneously.
Moderate developmental delay was noted, with independent walking at 3 y and
significant speech delay. He received special schooling, but developed computer skills on
a college course. Genetic testing for NS revealed no mutations in PTPN11, SOS1, CRAF
or KRAS. His body habitus was very thin, and remained so even as an adult when eating
normal portions of food. In early childhood, his hair did not grow, and he did not need
his first haircut until the age of approximately 8 y, though his hair as an adult was
unremarkable. His skin was dry and sensitive, requiring regular emollients, especially to
the palms and soles, and appeared prematurely aged. One café-au-lait mark was present
on his right thigh, and a capillary malformation of approximately 8 cm in length extended
down the right side of his neck (Figure 3-6).

124

125
Figure 3-7 (Previous page): Children with SHOC2 p.(Ser2Gly) mutations
Upper panel: patients S11 and S4 in second year of life
Central panel: patient S4 at 6 y
Lower left panel: patient S10 at 10 y
Lower right panel: patient S2 at 8 y and 14 y
Figure 3-6 (overleaf): Adults with SHOC2 p.(Ser2Gly) mutations
Upper panel: patient S14 at 18 y
Centre panel: patient S1 at 19 y
Lower panel: patient S3 at 32 y

126

127
Figure 3-8: Serial photographs of patients with SHOC2 p.(Ser2 Gly) mutations
Upper panel: patient S10 aged 2 days, in second and third years of life and at 14 y.
Lower panel: patient S14 as an infant, in second and fourth years of life, and at 18 y.

128
The 14 patients identified with SHOC2 mutations bear significant phenotypic similarities
to one another, with severe feeding difficulties, including the need for gastrostomy
feeding, present in many of them. Sparse hair has also been extremely common in
younger years, with improvement over the age of about 6 years, and short stature (with
or without growth hormone deficiency) was observed in the large majority: two patients
had height on the 9th centile, but for 12/14 it was at or below the 0.4th. In contrast to
classical CFC, eyebrow growth appeared to be normal in many patients, but a variety of
skin problems, some of which have been severe, such as eczema requiring occlusive
bandaging, have been observed. In the light of the many ectodermal features present in
this group, it is unsurprising that a clinical suspicion of CFC had been present in the
majority. A variety of cardiac manifestations are observed across the patient group, with
no clear pattern identifiable in this modest number of individuals, aside from the
observation that pulmonary stenosis may be less common in these individuals (5/14)
than in the overall population with NS, for example. The observation of a variety of
autoimmune phenomena in these patients is discussed further below.
3.5 Discussion of chapter results
3.5.1 Genotype-phenotype correlation in CFC
With the exception of sparse or very curly hair, and prevalence of pulmonary stenosis
(4), each of which have been suggested to be more common in patients with BRAF
mutations, very few genotype-phenotype correlations have previously been identified in
patients with CFC (4). As discussed in the introduction, the means of ascertainment of
affected patients is a crucial consideration in any subsequent assessment. Whilst
extremely high proportions of patients are reported to have, for example, curly hair, it is
possible that affected patients without this characteristic are less likely to be successfully
ascertained, leading to a bias in the known patient cohort. Curly hair is known to be a
prominent feature in patients with BRAF mutations (31), as seen in this study, and whilst
this was also present in patients with mutations in other genes, notably thick and
exuberant curls appear to be particularly common in the BRAF patient group.
Facial characteristics have not previously been extensively considered across the
genotypes in CFC. From the photographs available for this study, there do appear to be

129
potential differences between the facial appearance of patients with BRAF (most
classically p.(Gln257Arg)) mutations and those with MAP2K1 p.(Tyr130Cys)
substitutions, but this observation requires further extension across a larger data set.
Other genotypes have been insufficiently commonly observed for data to be available for
comparison.
Whilst a variety of musculoskeletal features are known to occur in CFC (14), the
presence of severe and progressive joint contractures in multiple patients with the
MAP2K1 p.(Tyr130Cys) mutation appears to be a novel and important finding.
Contractures have been sufficiently advanced in two patients to date to preclude
independent standing or ambulation, and require specialist equipment or unusual
techniques for mobilising.
Irrespective of the causative gene, a feature that appears common across patients
with CFC is a slim body habitus despite good appetite (or high energy artificial nutrition).
This suggests that further work to investigate whether differences in aspects of
metabolism might exist between these patients and other individuals to account for their
low body mass index. That a similar habitus may be seen in many individuals with CS
(20) and NS (22) suggests that a common factor due to Ras-MAPK pathway
dysregulation may be implicated.
3.5.2 Clinical features in patients with p.(Ser2Gly) mutation in SHOC2
Patients with mutations in SHOC2 have been reported to have a characteristic
phenotype, which in many can be clinically recognisable (104). This is to an extent
recapitulated in the cohort studied here, but with notable exceptions. Features that are
common in this cohort are
• Short stature, GH deficiency and delayed puberty (seen in the majority of
patients of this cohort who have attained the appropriate age).
• Attentional difficulties have not been remarked upon ab initio in all, but when
patients have been specifically assessed, features of attention deficit disorder have
been present in the majority. Other behavioural features such as anxiety and
hypersensitivity to noise or tactile stimuli are also reported in this patient group,

130
in keeping with the fact that these are known to be common in other NCFCs
(CFC, NS and CS).
• Severe and prolonged feeding difficulties have been present in several of
these patients, including the need for gastrostomy or NG tube feeding for several
years (up to age 8). It should be noted however that such severe feeding
difficulties can be a strong clinical indicator towards a diagnosis of both CS and
CFC, and as such our cohort is biased towards this group regarding
ascertainment.
• Eczema, particularly in the first year of life, has been present in nearly all
patients, and severe in some. Like the hair phenotype, this has usually improved
or normalised with time. Darker skin than other family members has also been a
consistent feature.
• Autoimmune disorders (or those with a possible autoimmune component),
namely idiopathic juvenile arthritis, Crohn’s disease and pericardial effusion have
each been present in individual patients in this series. Significant allergies have
also been a problem for two further patients. Unexplained joint pain in further
patients in this cohort could reflect effects of hypermobility, but a possible risk of
immunologically-mediated disease processes in this patient group suggests that
such symptoms may warrant investigation in the individual and consideration for
further exploration in the scientific arena.
• Coarsening of the facial features, reminiscent of that which occurs in Costello
syndrome, was seen in several patients, particularly patients S1 and S3 (Figure
3-6). They also had markedly deep creases in their palmar and plantar skin. This
had also been reported in several other patients in the group (for whom
photographs were not available). A prematurely aged appearance has been noted
in patients as they have matured, and is particularly striking in older patients in
this cohort, such as patients S3, S6 and S14.
The older median age of the cohort of patients identified in this series, compared to
other patients reported in the literature (104, 105), may be one reason why late-onset
sequelae (such as autoimmune disorders) might appear to be more common in this
group. A further possibility is that the patients in this cohort are, globally, more severely

131
affected than those of other cohorts, especially as they were ascertained by being
clinically suspected to have CFC or CS, as opposed to the, often milder, NS. Such
hypotheses can only be addressed by studying as large as possible a series of patients (by
international collaboration) and attempting to obtain long-term follow up clinical data on
as large a number of affected individuals as possible. Notably, an increased prevalence of
autoimmune disorders has recently been suggested across the NCFCs (196), particularly
systemic lupus erythematosus in patients with SHOC2 mutations (196, 197), as discussed
further in chapter 7.

132
4 MASSIVELY PARALLEL SEQUENCING APPROACHES FOR
MOLECULAR DIAGNOSIS

133
4.1 Chapter overview
Many patients with presentations suggestive of Ras-MAPK pathway disorders have
had a molecular diagnosis made by conventional means, that is, Sanger sequencing of the
gene or genes judged most likely to be responsible on the basis of clinical assessment, as
discussed in Chapter 3. Other patients with similar phenotypes have not been
diagnosable in this way, either because of an insufficiently characteristic presentation, or
a lack of resources for the genetic tests required (which may have been too numerous,
with too low a diagnostic yield to warrant funding). Massively parallel sequencing
approaches can overcome the need for multiple rounds of molecular testing, and offer
unprecedented power to diagnose genetic disorders. Target enrichment refers to a set of
genes being selected for sequencing and a custom experiment being designed to achieve
this. A set of probes are designed to capture the sequence containing the exons of these
genes from genomic DNA. These fragments are then amplified by PCR, so that they can
then be sequenced on a new generation sequencer such as an ABI SOLiD or Illumina
HiSeq machine, which can provide data on hundreds of millions (108 -109) or more short
sequence reads per run (198).
Massively parallel sequencing is a very recent development, with the first articles
reporting its use in identification of mutations responsible for human genetic disorders
being published in 2009 (199, 200), and the first SOLiD3+ sequencer being installed in
the MCGM laboratory in January 2010. These technologies have therefore become
available within the time period of this study, allowing novel approaches to the
investigation of genetic disorders to be developed. In order to better understand the
molecular basis of the Ras-MAPK disorders, target enrichment of a panel of selected
genes with known or potential relevance to the Ras-MAPK pathway disorders was
developed and tested in a panel of patients with clinical presentations suggestive of these
conditions but no known molecular diagnosis. Whole exome sequencing was also
performed on DNA samples from three affected patients, with comparison to samples
from both of their parents, with an aim of identifying de novo variants that may be
responsible for presentations consistent with a germline disorder of the Ras-MAPK
pathway.

134
4.2 Target enrichment approach
4.2.1 Development of gene list
Genes for inclusion in the initial SureSelect target enrichment panel were selected by
literature searching for those genes already known (in June 2010) to be responsible for
germline Ras-MAPK pathway disorders (shown in black) and disorders with phenotypic
overlap (shown in green in Table 4-1). Published literature and online resources such as
the UCSC genome browser and Decipher database were consulted to determine the
genes with altered copy number in known genomic disorders with phenotypic overlap
(shown in purple; Table 4-1). Genes included on the basis of Ras-MAPK pathway
involvement are shown in blue; the strategy to select these is described below the table.
Table 4-1: Genes included in target enrichment experiment
Gene name OMIM Condition Reference
PTPN11 176876 NS (74)
HRAS 190020 CS (62)
KRAS 190070 NS/CFC (77)
NRAS 164790 NS (89)
SOS1 182530 NS (201)
CBL 165360 NS with JMML (102)
NF1 613113 NF1 (202)
SHOC2 602775 NSLAH (104)
BRAF 164757 CFC (77)
CRAF 164760 NS (151)
SPRED1 609291 Legius syndrome (9)
MAP2K1 176872 CFC (10)
MAP2K2 601263 CFC (11)
RPS6KA3 303600 Coffin Lowry syndrome (203)
FGD1 305400 Aarskog syndrome (204)
TBX1 188400 22q11 deletion syndrome (205)
JAG1 118450 Alagille syndrome (206)
135
NOTCH2 610205 Alagille syndrome (207)
PRKAR1A 160980 Carney complex (208)
ROR2 268310 Robinow syndrome (209)
PTEN 158350 Cowden syndrome (210)
PTCH1 109400 Gorlin syndrome (211)
PTCH2 603673 Gorlin syndrome (212)
CRHR1 122561 del17q2.31 (140)
IMP5 608284 del17q2.31 (140)
MAPT 157140 del17q2.31 (140)
STH 607067 del17q2.31 (140)
CCDC91 - dup12p11.22 (MCGM arrayCGH)
PTHLH 168470 dup12p11.22 (MCGM arrayCGH)
ARID1B 614556 del6q25.3 (125)
ZDHHC14 - del6q25.3 (MCGM arrayCGH)
SNX9 605952 del6q25.3 (MCGM arrayCGH)
SYNJ2 609410 del6q25.3 (MCGM arrayCGH)
SERAC1 614725 del6q25.3 (MCGM arrayCGH)
GTF2H5 608780 del6q25.3 (MCGM arrayCGH)
TULP4 - del6q25.3 (MCGM arrayCGH)
TMEM181 613209 del6q25.3 (MCGM arrayCGH)
DYNLT1 601554 del6q25.3 (MCGM arrayCGH)
SYTL3 - del6q25.3 (MCGM arrayCGH)
EZR - del6q25.3 (MCGM arrayCGH)
LOC202459 - del6q25.3 (MCGM arrayCGH)
ACK1 606994 (213)
ADRBK1 109635 (214)
AGER 600214 (215)
ARAF 311010 (216)
ARRB1 107940 (217)
ARRB2 107941 (218)
BCL2 151430 (219)

136
BRAP 604986 (220)
C6ORF21 611404 (221)
CALM1 114180 (222)
CAV1 601047 (223)
CDC37 605065 (224)
CDC42 116952 (225)
CDK1 116940 (226)
CDKN2A 600160 (227)
CNKSR1 603272 (228)
CNKSR2 300724 (228)
CRKL 602007 (229)
DAB2 601236 (230)
DAB2IP 609205 (231)
DOK3 611435 (232)
DUSP1 600714 (233)
DUSP2 603068 (234)
DUSP4 602747 (235)
DUSP5 603069 (236)
DUSP6 602748 (237)
DUSP7 602749 (238)
DUSP9 300134 (238)
EGFR 131550 (239)
ERBB2 164870 (240)
ERBB2IP 606944 (241)
ETS1 164720 (242)
ETS2 164740 (242)
FGD3 - (243)
FRS2 607743 (244)
FYN 137025 (245)
GAB1 604439 (246)
GAB2 606203 (247)

137
GPS1 601934 (248)
GPS2 601935 (248)
GRAF 605370 (249)
GRB2 108355 (250)
GSK3B 605004 (251)
HRASLS 606487 (252)
HSP90 140571 (253)
ILI7RD 606807 (254)
INSR 147670 (255)
IQGAP1 603379 (256)
IRS1 147545 (255)
JAK1 147795 (257)
JAK2 147796 (258)
KSR1 601132 (224)
KSR2 610737 (224)
LGALS1 150570 (259)
LGALS3 153619 (260)
MAPK7 602521 (261)
MAPKAP1 610558 (262)
MAPKSP1 603296 (263)
MARK3 602678 (264)
MKNK1 606724 (265)
MLLT4 159559 (266)
MOS 190060 (267)
MRAS 608435 (268)
NCL 164035 (269)
NF2 607379 (270)
NGFR 162010 (271)
NOTCH1 190198 (272)
NPM1 164040 (273)
NPM2 608073 (269)

138
NPM3 606456 (269)
NUP153 603948 (274)
NUP214 114350 (275)
PAK1 602590 (276)
PAK2 605022 (277)
PAK3 300142 (278)
PEA15 603434 (279)
PEBP1 604591 (280)
PHB 176705 (281)
PHLPP1 609396 (282)
PIK3CA 171834 (283)
PLCG1 172420 (284)
PPP2CA 176915 (285)
PPP2R1A 605983 (285)
PPP2R2B 604235 (285)
PPP3R1 601302 (285)
PRKCA 176960 (214)
PTK2 600758 (286)
PTK2B 601212 (287)
PTPN23 606584 (288)
PTPN5 176879 (289)
PTPN7 176889 (289)
PTPRR 602853 (289)
PXN 602505 (290)
RALGDS 601619 (291)
RAP1A 179520 (292)
RAP1B 179530 (292)
RAP1GA1 600278 (293)
RAPGEF1 600303 (294)
RASA1 139150 (295)
RASA2 601589 (296)

139
RASA3 605182 (297)
RASA4 607943 (298)
RASAL1 604118 (299)
RASGRF2 606614 (300)
RASGRP1 603962 (301)
RASGRP3 609531 (302)
RASGRP4 607320 (303)
RASIP1 609623 (304)
RASSF2 609492 (305)
RASSF4 610059 (306)
RGS12 602512 (307)
RGS14 602513 (307)
RHEB 601293 (308)
RIN1 605965 (309)
RIN2 610222 (310)
RIT1 609591 (311)
RIT2 609592 (311)
RPS6KA1 601684 (312)
RPS6KA2 601685 (313)
RRAS 165090 (314)
RSU1 179555 (315)
SFN 601290 (316)
SH2B2 605300 (317)
SH2B3 605093 (318)
SH3BP2 602104 (319)
SH3KBP1 300374 (320)
SHC1 600560 (321)
SIGLEC7 604410 (322)
SIT1 604964 (323)
SMEK1 610351 (324)
SMEK2 610352 (324)

140
SOS2 601247 (325)
SPRED2 609292 (326)
SPRED3 609293 (327)
SPRY1 602465 (328)
SPRY2 602466 (328)
SPRY3 300531 (328)
SPRY4 607984 (328)
STAT5A 601511 (329)
STAT5B 604260 (329)
STK3 605030 (330)
SYNGAP1 603384 (331)
TBC1D10C 610831 (332)
TET2 612839 (333)
TRIB1 609461 (334)
TRIB2 609462 (334)
TRIB3 607898 (335)
VAV1 164875 (336)
WDR83 - (337)
YWHAB 601289 (338)
YWHAE 605066 (338)
YWHAH 113508 (338)
YWHAQ 609009 (338)
YWHAZ 601288 (338)
ZDHHC9 300646 (339)
Table 4-1 shows the genes included in the target enrichment experiment. Genes
known to cause NCFC disorders are shown in black, those mutated in disorders with
phenotypic overlap are shown in green, those with copy number variation in patients
with suggestive phenotypes are shown in purple, and those identified to have potentially
relevant Ras-MAPK pathway interaction are shown in blue.

141
A literature review was performed using PubMed (http://www.pubmed.gov) and
OMIM (http://www.omim.org) to define a list of genes encoding proteins with known
significance to Ras-MAPK pathway function that might represent further top candidates
for genes which when mutated might cause similar phenotypes to the NCFCs (shown in
blue; Table 4-1). The PubMed search focussed on reviews of Ras-MAPK pathway
activity, aiming to identify proteins with well characterised interactions with proteins
known to be implicated in germline Ras-MAPK disorders. The search terms used in
OMIM were the names of genes already known to be responsible for these disorders.
This abbreviated strategy was used in place of a formal systematic literature review, as
this latter approach was precluded by the very large number of potentially relevant
articles (340). A bias in the resultant list of genes according to the content of the
published literature was inevitable, and a feature common to any targeted investigative
approach.
The numbers of pages cross-referenced within OMIM was variable for the different
genes, from 3 for SHOC2 to 172 for HRAS (reflecting the quantity of relevant literature,
heavily influenced by the time elapsed since identification of the significance of each gene
to human disease). Titles and abstracts of the articles retrieved were scanned to identify
those that described direct interactions with other proteins or genes. The relevant articles
were then consulted to determine whether the subject represented an appropriate
candidate: those falling within the search due to less relevant factors (such as the use of
Ras-transformed cells in a largely unrelated field), or genes whose proteins had very
circumscribed tissue expression were not included (the latter condition was as the
disorders under investigation involve many body systems, so a generalised expression
pattern of the protein would be expected).
The draft list was discussed with Dr Kerr regarding information on the patients with
suggestive phenotypes and copy number variants identified on array CGH within
MCGM, and Dr Whitmarsh with respect to the candidates with best molecular evidence
for Ras-MAPK pathway interaction. As a result of these searches and discussions, 196
genes were selected for inclusion (Table 4-1). In order to ensure coverage of alternatively
spliced exons, transcripts were selected inclusively, so that all exons would be included,

142
to allow for sequencing of as much relevant genetic material as possible. The total size of
this enrichment was 1.52Mb, comprising 376 RefSeq transcripts covering the 196 genes.
4.2.2 Selection of patient samples
The size of the enrichment meant that samples from 10 individuals could be included
in this experiment (a single run on the SOLiD 5500 sequencer). The 10 samples selected
for inclusion are shown in Table 4-2. They were from six female and four male patients
with good clinical phenotypic data, strongly suggestive of a Ras-MAPK pathway
disorder. Within this group, a mixture of clinical diagnoses were represented, from NS,
through intermediate NS/CFC phenotypes, to more classical CFC syndrome. These
patients had had variable degrees of previous molecular testing. As shown in Table 4-2,
the majority had had previous diagnostic testing of all or most clinically available exons,
but other individuals were selected without having had such testing, analogous to the
potential future situation of samples being subject to diagnostic testing without previous
molecular investigations having been performed. One patient’s sample was included as a
positive control: this patient had a clinical diagnosis of NS that was known to have been
confirmed in a diagnostic laboratory (South West Thames Regional Genetics Laboratory
(SWTRGL)), but the mutation was not known to anyone involved in this experiment.
This blinding was to prevent bias in interpretation of the raw data from the experiment.

143
Table 4-2: Samples included in target enrichment experiment
Patient Clinical diagnosis
Previous molecular testing
BRAF exons 6,11,12,1314,15,16
KRAS all coding exons
MAP2K1 exons 2,3,6,7
MAP2K2 exons 2,3,5,6,7
PTPN11 exons 2,3,4,7,8, 12,13
SOS1 exons 3,6,10
SOS1 exons 7,8,11, 13,14,16
CRAF exons 6,13,16
CBL exons 7,8,9
HRAS all coding exons
Other genetic testing
TE1 NS yes yes no no ?* ?* ?* ?* no no none
TE2 CFC yes yes yes yes yes yes yes yes no no none
TE3 NS yes yes yes yes yes yes yes yes yes no none
TE4 CFC yes yes yes yes yes yes yes yes no no none
TE5 NS; two childhood tumours
yes yes yes yes yes no no no no yes TP53 all coding exons

144
Patient Clinical diagnosis
Previous molecular testing
BRAF exons 6,11,12,1314,15,16
KRAS all coding exons
MAP2K1 exons 2,3,6,7
MAP2K2 exons 2,3,5,6,7
PTPN11 exons 2,3,4,7,8,12,13
SOS1 exons 3,6,10
SOS1 exons7,8,11,13,14,16
CRAF exons 6,13,16
CBL exons 7,8,9
HRAS all coding exons
Other genetic testing
TE6 NSML yes yes yes yes yes yes yes yes no no none
TE7 CFC yes yes yes yes yes no no no no yes none
TE8 CFC/NS yes yes yes yes yes yes no yes no no none
TE9 ?NS (newly referred)
no no no no no no no no no no none
TE10 CFC (newly referred)
no no no no no no no no no no none
Table 4-2: shows the samples included in the target enrichment experiment, with clinical diagnosis for each patient and the extent of previous molecular
testing. *For patient TE1, ? refers to the previous testing known to have taken place to confirm her diagnosis, the results of which were blinded to all
involved in the experiment.

145
4.2.3 Results of target enrichment experiment
4.2.3.1 Coverage across the panel of genes and the panel of patients
DNA sequencing reads were mapped to human genome reference hg19. Whilst the
overall mean coverage of the targeted material at a depth of at least 20 reads was 69.8%,
and at least 30 reads, 68.4%, considerable variability was observed at the gene and exon
levels. For each patient, the variants identified were filtered to remove synonymous
substitutions and nucleotides covered at less than 5 reads’ depth.
The coverage of genes already known to be clinically relevant to the NCFCs was
examined specifically. The percentage of bases successfully (greater than 20x) covered in
certain of these genes, particularly HRAS, was low (Figure 4-1). This is likely to have
been due to a combination of factors: high homology to other RAS genes and
pseudogenes, and a higher GC content (Figure 4-1) are possible contributors.

146
Figure 4-1: Coverage across the exons of the three RAS genes KRAS, HRAS and NRAS.
Upper panel: Coding regions of these genes are indicated between the dotted lines. Near complete (99-100%) coverage was seen for all coding exons of NRAS, in contrast to HRAS, which had a low percentage of bases covered (at 20x), whilst KRAS demonstrated good coverage for all but one probed region.
Lower panel: The GC content of the coding exons of the three genes is tabulated, showing the higher GC content of HRAS.
Summary statistics for each of these clinically implicated genes at the gene level, that
is, taking the average coverage across all bases of this gene, are shown for sample TE1 in
the upper panel of Figure 4-2, and the lower panel depicts exon-by exon coverage of
SHOC2 in this sample. This shows that whilst the overall level of coverage recorded
across this gene was high, at 91%, there were several individual exons that were poorly

147
covered. Regions that are known to be challenging to cover successfully using NGS
assays bear similarities to those that can also be problematic for Sanger sequencing, due
to the common necessity of PCR amplification, and can include GC rich regions (341),
highly repetitive sequences and others with extremely high degrees of homology to other
genomic loci (342). However, the precise reasons why an individual region might not be
successfully amplified or aligned may be multifactorial or poorly understood. A close
correlation was observed between samples: genes that were well covered for one sample
were well covered for each of the others, Figure 4-3.

148
Figure 4-2 Coverage of genes of diagnostic relevance
Upper panel: The overall
coverage of genes listed (those
known to be mutated in human
DNA) is shown for sample TE1,
as the percentage of bases of the
gene covered at 20x depth. This
spans from 29% for MAP2K2 to
99% for NRAS.
Lower panel: Exon-by-exon
coverage of SHOC2 is shown. In
both panels, horizontal axes
represent the % of bases covered
at 20x depth.
% coverage 0% 100%
% coverage
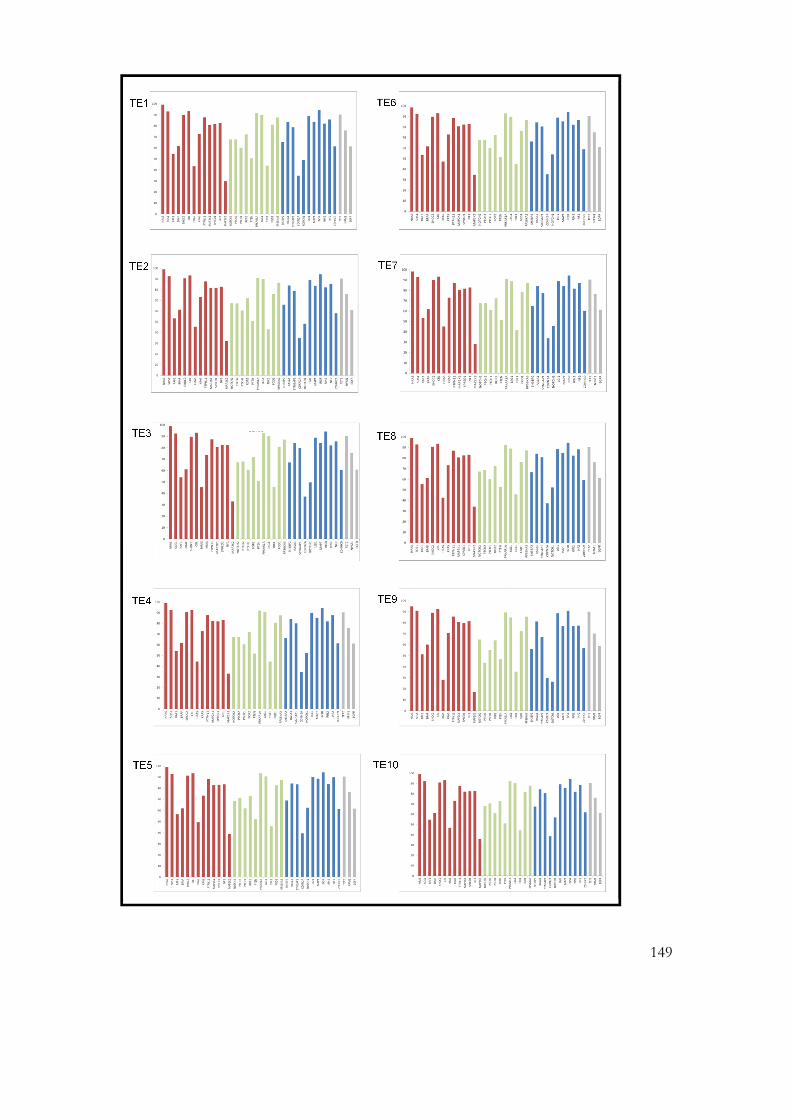
149
TE1
TE2
TE3
TE4
TE5
TE6
TE7
TE8
TE9
TE10

150
Figure 4-3 (previous page) Coverage across genes known to be mutated in human disease in samples TE1-TE10.
The same genes as shown in Figure 1-2 are depicted, and the pattern of percentage of bases successfully covered for each gene is seen to be very similar in each of the samples. Small images are shown here for the purposes of comparison; the full size histograms are shown in the appendix.
4.2.4 Results of testing in each of the samples
Initial bioinformatic filters for data quality used cutoff quality scores of MQV>18 for
novel and reference alleles. A filter to exclude non-coding and synonymous substitutions
was then applied, denoted as the ‘A’ filter in Figure 4-5. This resulted in a list of variants
predicting non-synonymous substitutions, premature stop codons or variation at essential
splice sites. Between 6 and 24 such variants were present in each sample, as shown in
Table 4-3.The genotypes of the heterozygous variants identified are shown, with the
number of reads in support of the reference allele denoted by RAC (reference allele
count) and in support of the novel allele by NAC (novel allele count). The quality scores
attributed to each of these calls are denoted by RMQV (reference MQV) and NMQV
(novel MQV). This value represents the mean quality of the reads at this nucleotide,
giving an indication of the confidence with which this variant has been called.
Conservation at the nucleotide, as assessed by GERP (genomic evolutionary rate
profiling) score, which aims to compare the nucleotide across 30 placental mammals
(343).

151
Table 4-3: (10 pages) Variants identified in target enrichment experiment
TE1
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein DepthRAC/NAC
RMQV/NMQV SIFT Polyphen Cons.
X 54496517 FGD1 ENST00000375135 NSC G/C c.1033G>C p.Glu345Gln 240 154/31 31/19 tol(0.17) unk(0) 4.271 26888093 RPS6KA1 ENST00000403732 NSC A/G c.377G>A p.Arg126Gln 257 134/122 30/34 tol(0.16) poss(0.917) 2.841 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 403 316/59 28/24 unk(0) unk(0) 1.461 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 172 121/33 23/18 tol(0.53) poss(0.686) 3.312 227660544 IRS1 ENST00000305123 NSC G/A c.2911G>A p.Gly971Arg 84 58/21 31/31 tol(0.47) prob(0.996) 4.524 124323077 SPRY1 ENST00000394339 NSC G/T c.331T>G p.Leu111Val 951 576/165 23/18 tol(0.12) prob(0.998) -5.425 80511755 RASGRF2 ENST00000265080 NSC C/T c.3415C>T p.Leu1139Phe 262 189/50 28/23 del(0.01) prob(1) 4.625 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 159 84/40 22/24 4.256 32150320 AGER ENST00000375067 NSC G/A c.805G>A p.Gly269Arg 450 265/181 32/35 del(0) unk(0) 3.36 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 280 124/63 24/19 del(0) prob(1) 4.66 159029716 TMEM181 ENST00000367090 NSC A/T c.1241A>T p.Tyr414Phe 202 135/33 24/19 del(0.02) prob(0.999) 5.667 55249095 EGFR ENST00000454757 NSC C/T c.2234T>C p.Leu745Pro 264 188/38 29/18 del(0) prob(1) 4.549 98209594 PTCH1 ENST00000375274 NSC T/T c.3941C>T p.Pro1314Leu 50 0/48 0/34 del(0.01) prob(0.955) 3.989 135983523 RALGDS ENST00000372062 NSC G/A c.962A>G p.Glu321Gly 140 68/32 27/21 del(0.01) poss(0.664) 2.8712 112926270 PTPN11 ENST00000351677 NSC C/T c.1403C>T p.Thr468Met 1202 647/551 36/38 del(0) prob(1) 3.6917 37879588 ERBB2 ENST00000540147 NSC A/G c.1873A>G p.Ile625Val 229 126/96 34/36 tol(0.86) poss(0.637) -1.117 61712068 MAP3K3 ENST00000361357 ESS A/G c.127-1G>A - 422 242/83 24/27 3.9517 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 379 214/104 26/23 del(0.03) benign(0.014) 0.8519 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 141 72/52 30/21 del(0.04) benign(0.333) 3.6219 7168103 INSR ENST00000341500 NSC:SS G/A c.1486G>A p.Glu496Lys 280 196/44 27/28 tol(0.09) benign(0.06) 3.619 38901933 RASGRP4 ENST00000405332 NSC T/G c.1769G>T p.Arg590Ile 91 66/19 29/29 unk(0) 3.0720 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 939 558/131 26/24 tol(0.16) benign(0.039) 4.4422 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 328 243/51 36/21 del(0) prob(0.997) 1.5822 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 575 440/98 36/18 3.95
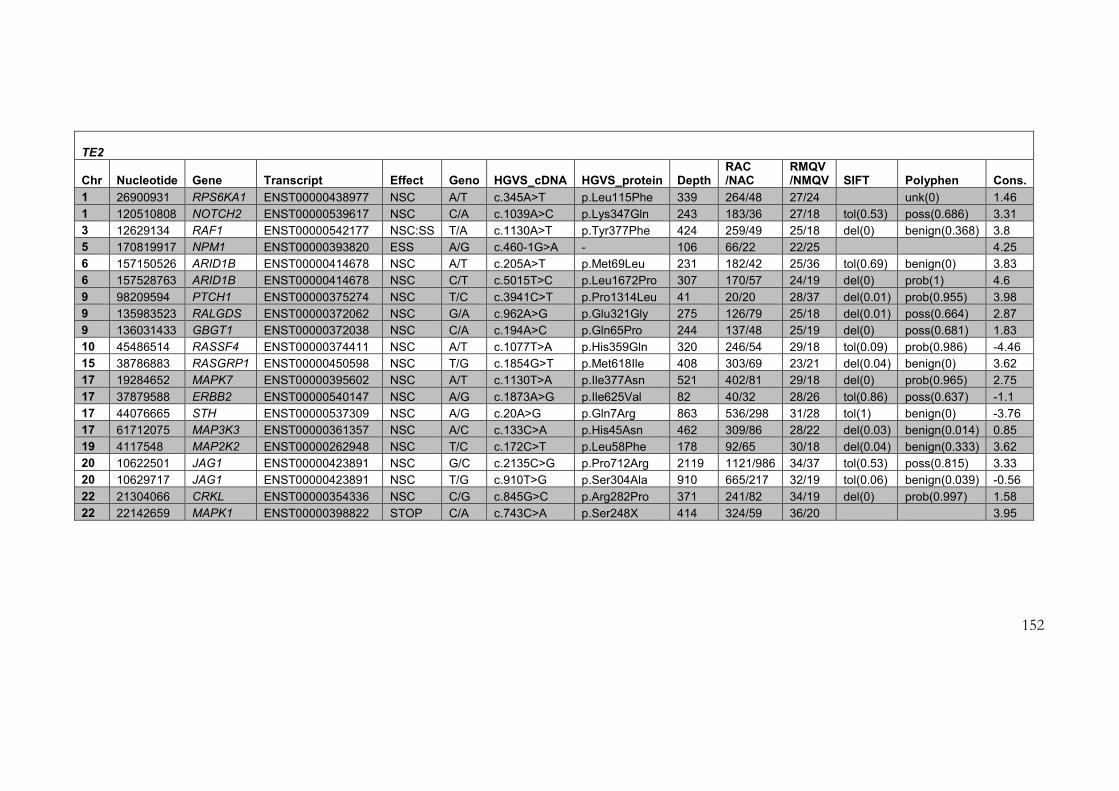
152
TE2
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
1 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 339 264/48 27/24 unk(0) 1.46
1 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 243 183/36 27/18 tol(0.53) poss(0.686) 3.31
3 12629134 RAF1 ENST00000542177 NSC:SS T/A c.1130A>T p.Tyr377Phe 424 259/49 25/18 del(0) benign(0.368) 3.8
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 106 66/22 22/25 4.25
6 157150526 ARID1B ENST00000414678 NSC A/T c.205A>T p.Met69Leu 231 182/42 25/36 tol(0.69) benign(0) 3.83
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 307 170/57 24/19 del(0) prob(1) 4.6
9 98209594 PTCH1 ENST00000375274 NSC T/C c.3941C>T p.Pro1314Leu 41 20/20 28/37 del(0.01) prob(0.955) 3.98
9 135983523 RALGDS ENST00000372062 NSC G/A c.962A>G p.Glu321Gly 275 126/79 25/18 del(0.01) poss(0.664) 2.87
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 244 137/48 25/19 del(0) poss(0.681) 1.83
10 45486514 RASSF4 ENST00000374411 NSC A/T c.1077T>A p.His359Gln 320 246/54 29/18 tol(0.09) prob(0.986) -4.46
15 38786883 RASGRP1 ENST00000450598 NSC T/G c.1854G>T p.Met618Ile 408 303/69 23/21 del(0.04) benign(0) 3.62
17 19284652 MAPK7 ENST00000395602 NSC A/T c.1130T>A p.Ile377Asn 521 402/81 29/18 del(0) prob(0.965) 2.75
17 37879588 ERBB2 ENST00000540147 NSC A/G c.1873A>G p.Ile625Val 82 40/32 28/26 tol(0.86) poss(0.637) -1.1
17 44076665 STH ENST00000537309 NSC A/G c.20A>G p.Gln7Arg 863 536/298 31/28 tol(1) benign(0) -3.76
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 462 309/86 28/22 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 178 92/65 30/18 del(0.04) benign(0.333) 3.62
20 10622501 JAG1 ENST00000423891 NSC G/C c.2135C>G p.Pro712Arg 2119 1121/986 34/37 tol(0.53) poss(0.815) 3.33
20 10629717 JAG1 ENST00000423891 NSC T/G c.910T>G p.Ser304Ala 910 665/217 32/19 tol(0.06) benign(0.039) -0.56
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 371 241/82 34/19 del(0) prob(0.997) 1.58
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 414 324/59 36/20 3.95

153
TE3
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
1 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 531 402/88 27/23 unk(0) unk(0) 1.46
1 155874285 RIT1 ENST00000368322 NSC T/G c.297T>G p.Phe99Leu 1466 798/665 39/38 tol(0.06) prob(0.999) 0.06
5 80511755 RASGRF2 ENST00000265080 NSC C/T c.3415C>T p.Leu1139Phe 232 167/47 30/20 del(0.01) prob(1) 4.62
6 157405907 ARID1B ENST00000319584 NSC A/C c.376C>A p.Gln126Lys 897 546/340 33/35 del(0) poss(0.93) 4.54
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 239 101/57 23/18 del(0) prob(1) 4.6
9 98209594 PTCH1 ENST00000375274 NSC T/C c.3941C>T p.Pro1314Leu 63 26/36 35/29 del(0.01) prob(0.955) 3.98
9 135983523 RALGDS ENST00000372062 NSC G/A c.962A>G p.Glu321Gly 221 118/45 26/19 del(0.01) poss(0.664) 2.87
15 66727444 MAP2K1 ENST00000307102 NSC C/T c.160C>T p.Leu54Phe 290 168/52 28/19 del(0.04) benign(0.333) 3.45
17 4623513 ARRB2 ENST00000412477 NSC:SS G/T c.923T>G p.Val308Gly 141 97/42 34/33 del(0.01) poss(0.629) -
17 61712068 MAP3K3 ENST00000361357 ESS A/G c.127-1G>A - 467 324/68 27/27 3.95
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 436 254/121 29/23 del(0.03) benign(0.014) 0.85
20 19955510 RIN2 ENST00000255006 NSC A/C c.988A>C p.Ser330Arg 395 203/167 30/32 tol(0.24) prob(0.999) 4.43
20 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 687 436/88 27/24 tol(0.16) benign(0.039) 4.44
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 557 432/85 37/19 3.95
22 30090772 NF2 ENST00000397822 NSC C/G c.1780C>G p.Pro594Ala 256 187/60 34/36 unk(0) 3.94
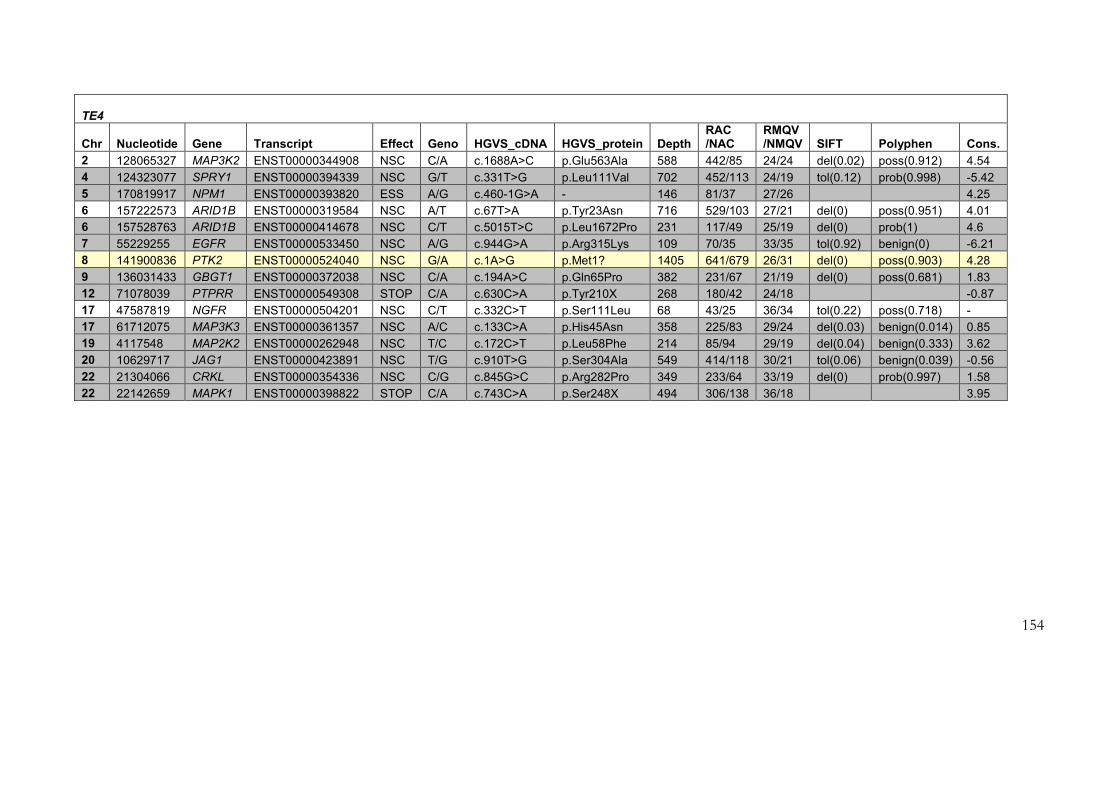
154
TE4
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
2 128065327 MAP3K2 ENST00000344908 NSC C/A c.1688A>C p.Glu563Ala 588 442/85 24/24 del(0.02) poss(0.912) 4.54
4 124323077 SPRY1 ENST00000394339 NSC G/T c.331T>G p.Leu111Val 702 452/113 24/19 tol(0.12) prob(0.998) -5.42
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 146 81/37 27/26 4.25
6 157222573 ARID1B ENST00000319584 NSC A/T c.67T>A p.Tyr23Asn 716 529/103 27/21 del(0) poss(0.951) 4.01
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 231 117/49 25/19 del(0) prob(1) 4.6
7 55229255 EGFR ENST00000533450 NSC A/G c.944G>A p.Arg315Lys 109 70/35 33/35 tol(0.92) benign(0) -6.21
8 141900836 PTK2 ENST00000524040 NSC G/A c.1A>G p.Met1? 1405 641/679 26/31 del(0) poss(0.903) 4.28
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 382 231/67 21/19 del(0) poss(0.681) 1.83
12 71078039 PTPRR ENST00000549308 STOP C/A c.630C>A p.Tyr210X 268 180/42 24/18 -0.87
17 47587819 NGFR ENST00000504201 NSC C/T c.332C>T p.Ser111Leu 68 43/25 36/34 tol(0.22) poss(0.718) -
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 358 225/83 29/24 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 214 85/94 29/19 del(0.04) benign(0.333) 3.62
20 10629717 JAG1 ENST00000423891 NSC T/G c.910T>G p.Ser304Ala 549 414/118 30/21 tol(0.06) benign(0.039) -0.56
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 349 233/64 33/19 del(0) prob(0.997) 1.58
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 494 306/138 36/18 3.95
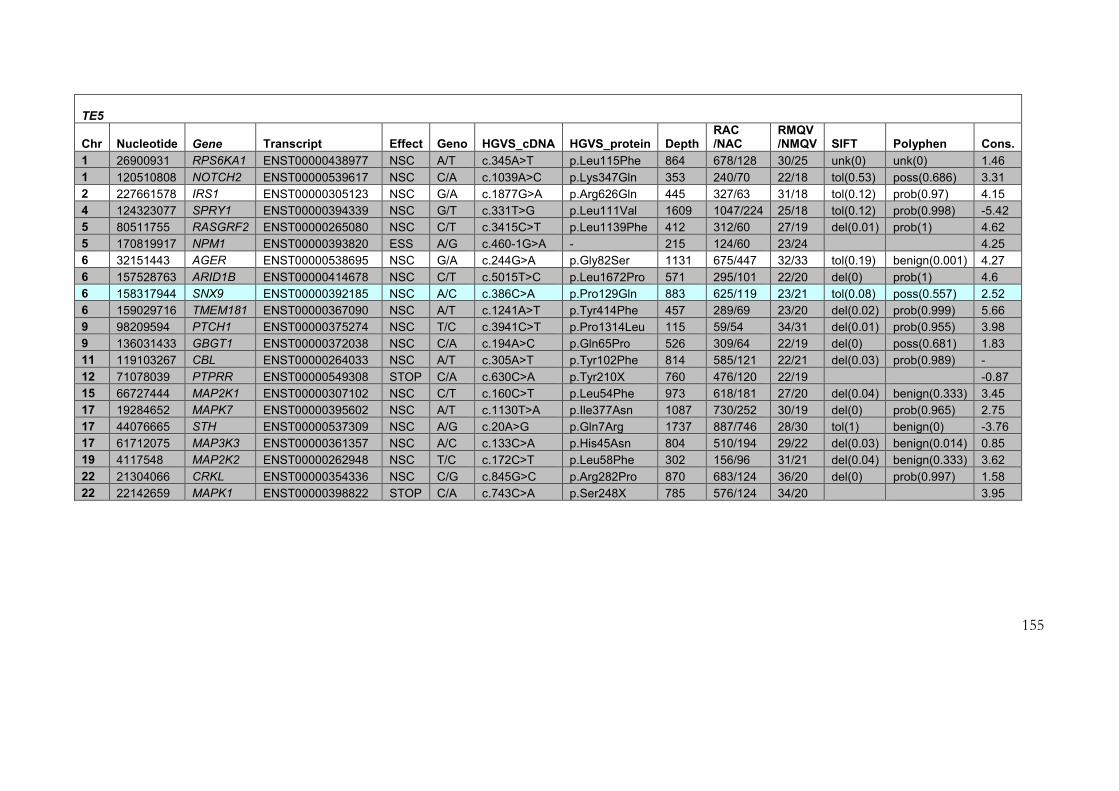
155
TE5
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
1 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 864 678/128 30/25 unk(0) unk(0) 1.46
1 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 353 240/70 22/18 tol(0.53) poss(0.686) 3.31
2 227661578 IRS1 ENST00000305123 NSC G/A c.1877G>A p.Arg626Gln 445 327/63 31/18 tol(0.12) prob(0.97) 4.15
4 124323077 SPRY1 ENST00000394339 NSC G/T c.331T>G p.Leu111Val 1609 1047/224 25/18 tol(0.12) prob(0.998) -5.42
5 80511755 RASGRF2 ENST00000265080 NSC C/T c.3415C>T p.Leu1139Phe 412 312/60 27/19 del(0.01) prob(1) 4.62
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 215 124/60 23/24 4.25
6 32151443 AGER ENST00000538695 NSC G/A c.244G>A p.Gly82Ser 1131 675/447 32/33 tol(0.19) benign(0.001) 4.27
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 571 295/101 22/20 del(0) prob(1) 4.6
6 158317944 SNX9 ENST00000392185 NSC A/C c.386C>A p.Pro129Gln 883 625/119 23/21 tol(0.08) poss(0.557) 2.52
6 159029716 TMEM181 ENST00000367090 NSC A/T c.1241A>T p.Tyr414Phe 457 289/69 23/20 del(0.02) prob(0.999) 5.66
9 98209594 PTCH1 ENST00000375274 NSC T/C c.3941C>T p.Pro1314Leu 115 59/54 34/31 del(0.01) prob(0.955) 3.98
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 526 309/64 22/19 del(0) poss(0.681) 1.83
11 119103267 CBL ENST00000264033 NSC A/T c.305A>T p.Tyr102Phe 814 585/121 22/21 del(0.03) prob(0.989) -
12 71078039 PTPRR ENST00000549308 STOP C/A c.630C>A p.Tyr210X 760 476/120 22/19 -0.87
15 66727444 MAP2K1 ENST00000307102 NSC C/T c.160C>T p.Leu54Phe 973 618/181 27/20 del(0.04) benign(0.333) 3.45
17 19284652 MAPK7 ENST00000395602 NSC A/T c.1130T>A p.Ile377Asn 1087 730/252 30/19 del(0) prob(0.965) 2.75
17 44076665 STH ENST00000537309 NSC A/G c.20A>G p.Gln7Arg 1737 887/746 28/30 tol(1) benign(0) -3.76
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 804 510/194 29/22 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 302 156/96 31/21 del(0.04) benign(0.333) 3.62
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 870 683/124 36/20 del(0) prob(0.997) 1.58
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 785 576/124 34/20 3.95
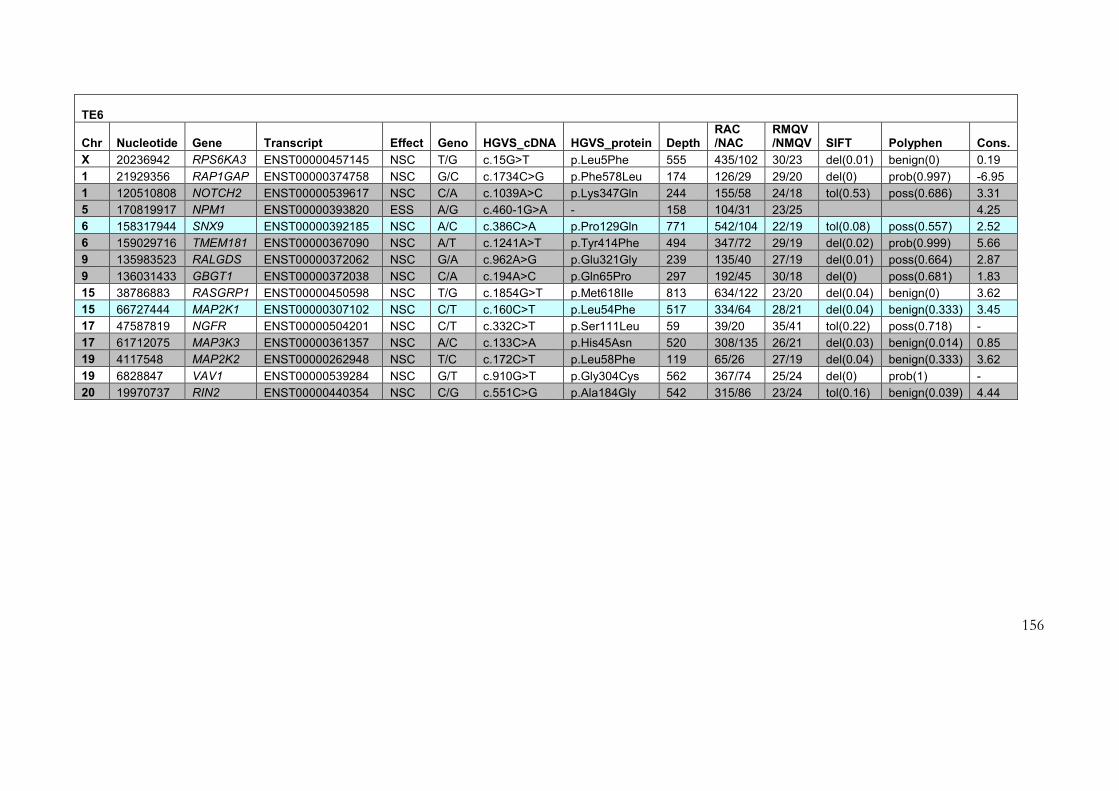
156
TE6
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
X 20236942 RPS6KA3 ENST00000457145 NSC T/G c.15G>T p.Leu5Phe 555 435/102 30/23 del(0.01) benign(0) 0.19
1 21929356 RAP1GAP ENST00000374758 NSC G/C c.1734C>G p.Phe578Leu 174 126/29 29/20 del(0) prob(0.997) -6.95
1 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 244 155/58 24/18 tol(0.53) poss(0.686) 3.31
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 158 104/31 23/25 4.25
6 158317944 SNX9 ENST00000392185 NSC A/C c.386C>A p.Pro129Gln 771 542/104 22/19 tol(0.08) poss(0.557) 2.52
6 159029716 TMEM181 ENST00000367090 NSC A/T c.1241A>T p.Tyr414Phe 494 347/72 29/19 del(0.02) prob(0.999) 5.66
9 135983523 RALGDS ENST00000372062 NSC G/A c.962A>G p.Glu321Gly 239 135/40 27/19 del(0.01) poss(0.664) 2.87
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 297 192/45 30/18 del(0) poss(0.681) 1.83
15 38786883 RASGRP1 ENST00000450598 NSC T/G c.1854G>T p.Met618Ile 813 634/122 23/20 del(0.04) benign(0) 3.62
15 66727444 MAP2K1 ENST00000307102 NSC C/T c.160C>T p.Leu54Phe 517 334/64 28/21 del(0.04) benign(0.333) 3.45
17 47587819 NGFR ENST00000504201 NSC C/T c.332C>T p.Ser111Leu 59 39/20 35/41 tol(0.22) poss(0.718) -
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 520 308/135 26/21 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 119 65/26 27/19 del(0.04) benign(0.333) 3.62
19 6828847 VAV1 ENST00000539284 NSC G/T c.910G>T p.Gly304Cys 562 367/74 25/24 del(0) prob(1) -
20 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 542 315/86 23/24 tol(0.16) benign(0.039) 4.44

157
TE7
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT PolyPhen Cons.
1 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 166 117/34 25/22 tol(0.53) poss(0.686) 3.31
1 154940250 SHC1 ENST00000412170 NSC G/A c.496G>A p.Glu166Lys 177 116/25 22/20 tol(0.08) benign(0.059) 4.42
4 124323077 SPRY1 ENST00000394339 NSC G/T c.331T>G p.Leu111Val 709 434/96 22/18 tol(0.12) prob(0.998) -5.42
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 100 59/23 20/23 4.25
6 32151443 AGER ENST00000538695 NSC G/A c.244G>A p.Gly82Ser 485 257/223 36/33 tol(0.19) benign(0.001) 4.27
6 157222573 ARID1B ENST00000319584 NSC A/T c.67T>A p.Tyr23Asn 754 510/138 26/22 del(0) poss(0.951) 4.01
6 166845926 RPS6KA2 ENST00000405189 NSC T/G c.1118G>T p.Arg373Leu 795 390/370 29/34 tol(0.19) benign(0.018) 4.47
9 134039498 NUP214 ENST00000438605 NSC A/G c.1147A>G p.Arg383Gly 378 230/75 23/18 tol(0.07) prob(0.999) 3.46
9 135983523 RALGDS ENST00000372062 NSC G/A c.962A>G p.Glu321Gly 295 140/66 25/18 del(0.01) poss(0.664) 2.87
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 264 173/45 25/18 del(0) poss(0.681) 1.83
11 119103267 CBL ENST00000264033 NSC A/T c.305A>T p.Tyr102Phe 526 393/72 21/19 del(0.03) prob(0.989) -
12 71078039 PTPRR ENST00000549308 STOP C/A c.630C>A p.Tyr210X 253 171/38 22/19 -0.87
17 19284652 MAPK7 ENST00000395602 NSC A/T c.1130T>A p.Ile377Asn 447 332/68 32/21 del(0) prob(0.965) 2.75
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 415 242/115 27/22 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 117 46/52 27/19 del(0.04) benign(0.333) 3.62
19 51656450 SIGLEC7 ENST00000305628 STOP A/C c.1073C>A p.Ser358X 238 170/41 28/18 -6.04
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 379 299/55 36/18 del(0) prob(0.997) 1.58
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 379 266/80 37/18 3.95

158
TE8
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
1 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 453 336/89 31/22 unk(0) unk(0) 1.46
1 120510808 NOTCH2 ENST00000539617 NSC C/A c.1039A>C p.Lys347Gln 229 169/37 24/24 tol(0.53) poss(0.686) 3.31
2 39262581 SOS1 ENST00000428721 NSC T/G c.754G>T p.Asp252Tyr 809 575/231 35/36 del(0.01) poss(0.487) 4.09
2 227659816 IRS1 ENST00000305123 NSC G/C c.3639C>G p.Ser1213Arg 170 105/24 22/19 tol(0.06) benign(0.101) 2.62
4 3432268 RGS12 ENST00000338806 NSC C/G c.1756C>G p.Pro586Ala 605 464/115 29/18 del(0.02) benign(0.001) -4.72
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 164 110/25 24/22 4.25
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 332 166/73 23/19 del(0) prob(1) 4.6
9 136031433 GBGT1 ENST00000372038 NSC C/A c.194A>C p.Gln65Pro 201 111/35 23/22 del(0) poss(0.681) 1.83
11 119103267 CBL ENST00000264033 NSC A/T c.305A>T p.Tyr102Phe 980 725/134 21/21 del(0.03) prob(0.989) -
15 66727444 MAP2K1 ENST00000307102 NSC C/T c.160C>T p.Leu54Phe 721 413/137 24/19 del(0.04) benign(0.333) 3.45
17 19284652 MAPK7 ENST00000395602 NSC A/T c.1130T>A p.Ile377Asn 703 518/131 31/19 del(0) prob(0.965) 2.75
17 44076665 STH ENST00000537309 NSC A/G c.20A>G p.Gln7Arg 1066 593/404 31/33 tol(1) benign(0) -3.76
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 508 335/69 31/23 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 230 153/58 31/20 del(0.04) benign(0.333) 3.62
20 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 549 322/64 28/25 tol(0.16) benign(0.039) 4.44
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 459 330/86 34/20 del(0) prob(0.997) 1.58

159
TE9
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
4 124323077 SPRY1 ENST00000394339 NSC G/T c.331T>G p.Leu111Val 211 125/25 22/23 tol(0.12) prob(0.998) -5.42
6 32151443 AGER ENST00000538695 NSC G/A c.244G>A p.Gly82Ser 114 60/52 34/35 tol(0.19) benign(0.001) 4.27
15 66727444 MAP2K1 ENST00000307102 NSC C/T c.160C>T p.Leu54Phe 123 69/30 29/20 del(0.04) benign(0.333) 3.45
17 44076665 STH ENST00000537309 NSC A/G c.20A>G p.Gln7Arg 199 128/62 28/35 tol(1) benign(0) -3.76
20 10622501 JAG1 ENST00000423891 NSC G/C c.2135C>G p.Pro712Arg 566 240/318 33/38 tol(0.53) poss(0.815) 3.33
20 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 169 101/22 24/26 tol(0.16) benign(0.039) 4.44

160
Table 1-3 shows the variants identified in each patient’s sample. Geno: observed genotype at this nucleotide; RAC/NAC: reference allele count/novel allele count; RMQV/NMQV: reference MQV/novel MQV; Cons: conservation as judged by GERP score. Pathogenic variants are shown in yellow. Variants identified in a sample with a known mutation are shaded grey (indicating the low likelihood of relevance to pathogenesis). Variants identified in more than one sample without a mutation are shown in turquoise. Variants meeting criteria for validation as potentially pathogenic are shaded pale yellow.
TE10
Chr Nucleotide Gene Transcript Effect Geno HGVS_cDNA HGVS_protein Depth RAC /NAC
RMQV /NMQV SIFT Polyphen Cons.
X 21581461 CNKSR2 ENST00000379510 NSC G/T c.1499G>T p.Gly500Val 1374 954/408 36/38 del(0.02) poss(0.943) 3.7
X 54496517 FGD1 ENST00000375135 NSC G/C c.1033G>C p.Glu345Gln 470 324/60 28/19 tol(0.17) unk(0) 4.27
1 26900931 RPS6KA1 ENST00000438977 NSC A/T c.345A>T p.Leu115Phe 503 374/79 29/26 unk(0) unk(0) 1.46
2 227659816 IRS1 ENST00000305123 NSC G/C c.3639C>G p.Ser1213Arg 300 175/37 20/18 tol(0.06) benign(0.101) 2.62
5 170819917 NPM1 ENST00000393820 ESS A/G c.460-1G>A - 138 90/26 32/27 4.25
6 157488212 ARID1B ENST00000400790 NSC C/T c.80T>C p.Met27Thr 1679 890/740 35/35 del(0) benign(0.214) 4.35
6 157528763 ARID1B ENST00000414678 NSC C/T c.5015T>C p.Leu1672Pro 294 146/56 26/18 del(0) prob(1) 4.6
6 159029716 TMEM181 ENST00000367090 NSC A/T c.1241A>T p.Tyr414Phe 347 237/51 23/20 del(0.02) prob(0.999) 5.66
7 55229255 EGFR ENST00000533450 NSC A/G c.944G>A p.Arg315Lys 133 81/49 33/37 tol(0.92) benign(0) -6.21
9 95792228 FGD3 ENST00000538555 NSC A/G c.439A>G p.Thr147Ala 402 205/192 37/36 tol(0.31) benign(0.018) -5.98
9 98209594 PTCH1 ENST00000375274 NSC T/C c.3941C>T p.Pro1314Leu 111 61/49 38/33 del(0.01) prob(0.955) 3.98
12 71078039 PTPRR ENST00000549308 STOP C/A c.630C>A p.Tyr210X 561 385/72 21/20 -0.87
15 66727483 MAP2K1 ENST00000425818 NSC A/G c.17G>A p.Asp7Asn 665 384/278 37/40 del(0.03) poss(0.932) 3.45
17 19284652 MAPK7 ENST00000395602 NSC A/T c.1130T>A p.Ile377Asn 657 437/124 28/21 del(0) prob(0.965) 2.75
17 44076665 STH ENST00000537309 NSC A/G c.20A>G p.Gln7Arg 1261 703/469 27/29 tol(1) benign(0) -3.76
17 61712075 MAP3K3 ENST00000361357 NSC A/C c.133C>A p.His45Asn 434 282/82 28/20 del(0.03) benign(0.014) 0.85
19 4117548 MAP2K2 ENST00000262948 NSC T/C c.172C>T p.Leu58Phe 340 224/82 28/20 del(0.04) benign(0.333) 3.62
20 10629717 JAG1 ENST00000423891 NSC T/G c.910T>G p.Ser304Ala 1005 636/339 31/22 tol(0.06) benign(0.039) -0.56
20 19970737 RIN2 ENST00000440354 NSC C/G c.551C>G p.Ala184Gly 619 375/80 26/25 tol(0.16) benign(0.039) 4.44
22 21304066 CRKL ENST00000354336 NSC C/G c.845G>C p.Arg282Pro 512 397/78 34/19 del(0) prob(0.997) 1.58
22 22142659 MAPK1 ENST00000398822 STOP C/A c.743C>A p.Ser248X 597 461/96 37/19 3.95

161
4.2.5 Use of the data set for further filtering of candidate variants
An algorithm was developed for hierarchical filtering of the putative variants identified, as
shown in Figure 4-5. Firstly, the variant list was examined for mutations that had been
previously described in patients with Ras-MAPK pathway disorders. Of the 10 samples, three
(TE1, TE8 and TE10) had variants identified within them that were identical to mutations
previously found in patients with NS and CFC.
In patient TE1 (the positive control), this was the mutation PTPN11 c.1403C>T,
p.(Thr468Met). Unblinding revealed this to be the variant previously diagnosed in her, and
accounting for her diagnosis of classical NS. This demonstrated a proof of principle of this
technique to identify a mutation in the genes tested.
In patient TE8, who had a phenotype intermediate between NS and CFC, the mutation
SOS1 c.925G>T, p.(Asp309Tyr) was identified. This is a recurrently described, but
uncommon, cause of NS, representing 6 of 239 mutations in this gene included in the NS
Euronet database (6). Exon 7, in which this substitution is present, had not previously been
analysed in this patient (who had had SOS1 exons 3,6, and 10 tested by dHPLC in stage 2 of
the South West Thames Regional Genetics Laboratory NS testing protocol). This variant was
confirmed by bidirectional Sanger sequencing (Figure 4-4), prior to export for confirmatory
diagnostic testing in that laboratory.

162
Figure 4-4: Bidirectional Sanger sequencing of chr2:39262581.
The presence of the heterozygous variant in SOS1 (NM_005633.3) c.925G>T p.(Asp309Tyr) is demonstrated in patient TE8.
In patient TE10, a recurrent but uncommon mutation in MAP2K1, c. 199G>A,
p.(Asp67Asn) was identified, again consistent with her classical CFC phenotype. This
mutation has been identified in 4 of 51 patients in the NSEuronet database with a mutation in
this gene (6). Again, this was confirmed in the MCGM diagnostic laboratory so that a
diagnostic report could be issued to the referring clinician. For the samples in which no such
obvious mutation was identified, the algorithm shown in Figure 4-5 was applied.
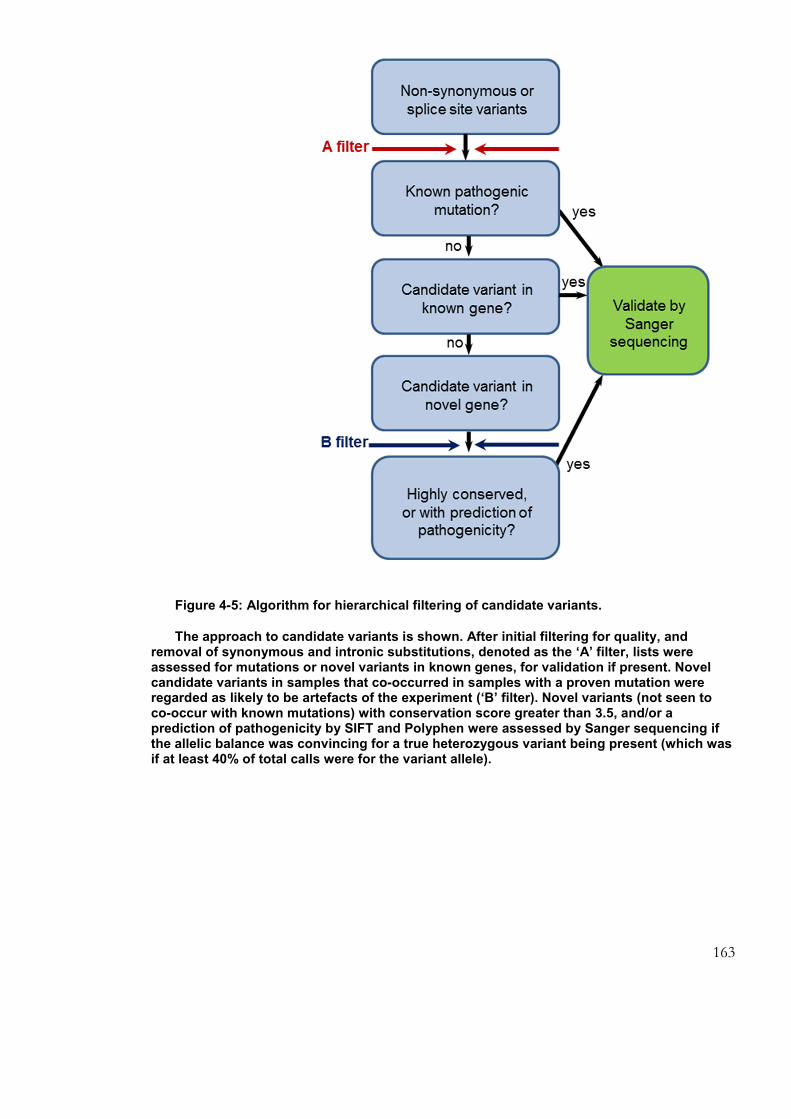
163
Figure 4-5: Algorithm for hierarchical filtering of candidate variants.
The approach to candidate variants is shown. After initial filtering for quality, and removal of synonymous and intronic substitutions, denoted as the ‘A’ filter, lists were assessed for mutations or novel variants in known genes, for validation if present. Novel candidate variants in samples that co-occurred in samples with a proven mutation were regarded as likely to be artefacts of the experiment (‘B’ filter). Novel variants (not seen to co-occur with known mutations) with conservation score greater than 3.5, and/or a prediction of pathogenicity by SIFT and Polyphen were assessed by Sanger sequencing if the allelic balance was convincing for a true heterozygous variant being present (which was if at least 40% of total calls were for the variant allele).

164
Datasets were examined for novel variants in known genes, as these could be obvious
candidates for the causative mutation. If present, any such variant would have been the next
highest priority for validation by Sanger sequencing. The blue arrow ‘B filter’ denotes the
point at which putative variants were compared to those identified in other patient samples.
Where a variant had also been called in a data set in which a known pathogenic mutation was
identified, this information was used as a means of excluding the variant, as it was much more
likely to represent an artefactual finding in this situation. These bases are shaded in grey in
Table 4-3. Putative variants with less than 40% of calls for the variant nucleotide were also
excluded at this stage, as such a skewed allelic balance was not likely to represent a true
heterozygous variant being present. Variants called in multiple samples that did not have a
known mutation identified are shaded in blue in Table 4-3. These could not be excluded from
being pathogenic, but those in genes within which multiple variants were called across the data
sets were judged less likely to represent disease-associated alleles, as the chance of an
artefactual finding was higher.
Candidate mutations in novel genes for validation by Sanger sequencing required, in
addition to passing the quality filter above, to have either a high level of evolutionary
conservation at that nucleotide, or in silico predictions of pathogenicity by SIFT and Polyphen.
In addition to this filtering, the unfiltered data sets were also scanned for variant calls in
known genes that nearly met the quality criteria, as the biological odds of such a variant being
pathogenic could be higher than for other variants, and hence a higher index of suspicion of
pathogenicity might be warranted (no such borderline variants were present in this
experiment).
Verification of novel variants by Sanger sequencing

165
Two novel variants meeting criteria for confirmation by Sanger sequencing were
identified, PTK2 c.1A>G [NM_151831.3] in patient TE4 (chr8:141900836) and RPS6KA2
c.1118G>T [NM_001006932.1] in patient TE7 (chr6:166845926). These were sequenced, with
approximately 150 bp of 5’ and 3’ flanking sequence. The start codon variant called in PTK2
was confirmed to be correct, being present in lymphocyte DNA of patient TE4 by
bidirectional sequencing (Figure 4-6). The variant in RPS6KA2 was not present in sample TE7
(Figure 4-7), indicating this finding to have been an artefact of the target enrichment
experiment.
Figure 4-6: Bidirectional Sanger sequencing of chr8: 141900836 in patient TE4 and her mother.
The heterozygous variant c.1A>G in PTK2 [NM_151831.3] is demonstrated in the patient’s lymphocyte and fibroblast DNA, and its absence from her mother’s lymphocyte DNA.

166
4.2.6 PTK2 as a novel candidate disease gene
The start codon mutation in PTK2 identified in patient TE4 represented a potentially
significant finding. Whilst it is difficult to be sure of what effect this might have at the
molecular level, the most likely effect would be that an alternative translation initiation some
75 bases 3’ would become active, resulting in the production of an abnormal N-terminally
truncated protein (missing the first 25 amino acid residues).
PTK2 encodes focal adhesion kinase (FAK), a highly conserved protein with known roles
in many cell types, with, as the name suggests, a key role in cell-cell adhesion. FAK contains
tyrosine residues that are phosphorylated when focal adhesions are present, and
dephosphorylated when cells are detached (344). This phosphorylation has been suggested to
be an important early step in intracellular signal transduction (345), and ERK1 has been
shown to be required for downstream signalling from FAK in dermal fibroblasts (346). The
N-terminus of FAK has been demonstrated to interact with neurofibromin, the NF1 gene
product (347). A role of FAK in regulation of cardiomyocyte hypertrophy has also been
established (348). Whilst the crystal structure of the N-terminal FERM (four-point-one, ezrin,
radixin, moesin) domain of FAK has been elucidated (345), the model does not extend to the
extreme N-terminus, hence it remains unclear whether and how this domain would be
impacted by the loss of the initial 25 amino acid residues. However, the N-terminus of this
protein does show a high degree of evolutionary conservation, as shown in Figure 4-8. The
FERM domain’s role in autoinhibition, as well as its interactions with other proteins, suggests
the possibility the N-terminal truncation predicted by the c.1A>G mutation might result in
constitutive activity, as has been shown to be the case for FAK lacking amino acid residues 1-
Figure 4-7: Bidirectional Sanger sequencing of chr6: 166845926 in patient TE7.
The candidate variant at this nucleotide is shown not to be present, indicating it to have been an artefact of the target
enrichment experiment.

167
384 (344) and other N-terminally truncated kinases such as MAP2K1 proteins engineered with
internal deletions (349).
Figure 4-8: The N-terminal portion of human FAK (product of PTK2 NM_153831.3) aligned against the protein sequence of other species.
Alamut version 2.2 (using data from Ensembl) was used to retrieve the alignment shown. The extensive sequence homology shown indicates a high level of evolutionary conservation of this region of the protein, part of which lies within the FERM domain, as shown in the schematic A. FERM indicates the FERM domain, which is involved in autoinhibition and protein-protein interactions, P, the proline-rich regions, and L, the linker domain. The kinase domain is indicated in red, and, at the C-terminus, the focal adhesion targeting domain (FAT) is indicated in green. Schematic B shows the likely effect of the c.1A>G mutation, which is production of an N-terminally truncated protein. Whilst the residues absent from this do not participate in the solved crystal structure of the FERM domain, their very close proximity to this means that this series of residues at the extreme N-terminus may nonetheless be involved in this domain’s function.

168
Patient TE4 was a 20 year old woman with a clinical diagnosis of CFC, made on the basis
of significant learning disability, epilepsy, coarsening of her facial features and suggestive
ectodermal features, namely curly hair and palmoplantar keratoderma. None of these features
were present in any other family member, although phenotypic data on paternal family
members were scarce.
Following the identification of a single potentially pathogenic mutation in this gene in
patient TE4, sequencing of all coding exons of PTK2 was undertaken in her sample. No
further mutations were identified in this gene, all variants identified being previously recorded
in databases of genetic variation. Exon 3, in which the start codon is found, was sequenced in
the patient’s mother, and the c.1A>G variant was not present (Figure 4-6). Unfortunately, the
possibility of paternal inheritance could not be assessed, as the father was deceased and there
was no contact with any other member of the paternal family. Sequencing of DNA extracted
from cultured skin-derived fibroblasts from patient TE4 also demonstrated the mutation to be
present, with no evidence in the chromatographic traces for mosaicism in either of the tissues
examined (Figure 4-6).
4.2.7 Sequencing of PTK2 as a candidate gene for germline human disease
In view of the potential finding that PTK2 could represent a novel gene for a phenotype
with significant similarities to the Ras-MAPK pathway disorders, Sanger sequencing of all 31
coding exons of this gene was undertaken in further patient samples. A panel of DNA
samples from 70 patients with CFC and related phenotypes and no known mutation was
available for testing. These patients had clinical diagnoses of possible CS, CFC or NS and had
therefore had relevant exons of BRAF, MAP2K1, MAP2K2, KRAS, SHOC2 and/or HRAS
sequenced previously in the MCGM diagnostic laboratory.
As would be expected, several known common SNPs were recurrently identified in these
samples, but no novel substitutions or indels were present, meaning that no further candidate
pathogenic mutations were identified in this cohort. Sequencing for the start codon mutation
identified in patient TE4 was also extended to a further set of 30 patients on whom only very
scant DNA was available, again with no further mutations identified. Collaborations were
developed to extend the spectrum of patients tested for this mutation in PTK2: large cohorts
known to Professors Martin Zenker (University of Magdeburg) and Marco Tartaglia (Istituto
Superiore di Sanità, Rome) with a clinical diagnosis of NS were tested by melt curve analysis

169
and sequencing of exon 3 respectively, but with no positive results to date in approximately
200 patients sequenced (personal communications). Whilst this suggests that this mutation is
not a common cause of a CFC/NS phenotype, it remains possible that it is the underlying
cause for some or all aspects of the phenotype observed in this single patient, and could also
be responsible for Mendelian disease in further as yet unidentified individuals. With the
patient’s unusual combination of phenotypic features, of intellectual disability, epilepsy, facial
features suggestive of NS/CFC and palmoplantar keratoderma, it is possible that the variant
identified in PTK2 is the major cause for some but not all of these features. Discussions with
other geneticists with an interest in NS and skin disorders did not reveal further patients
known to them with similar phenotypic combinations. If identified, such individuals would be
good candidates in whom to test for the presence of further mutations in PTK2.
4.2.8 Iterative review of target enrichment results as further genes for
NCFCs identified
Since this target enrichment experiment was performed, further genes mutated in patients
with NCFC presentations have been identified: the discovery by Aoki et al (76) of RIT1
mutations in a substantial number of patients with NS, suggests that this gene is the cause of a
significant proportion of previously molecularly undiagnosed NS. Where it could be
ascertained, the mutations were shown to have arisen de novo, again suggesting the possibility
that mutations in this gene could potentially confer a severe phenotype.

170
4.2.9 RIT1 : a gene with significant structural and functional similarities
to RAS genes
RIT1 had been selected for inclusion in the list of target enrichment genes on the basis of
being a strong candidate for harbouring mutations to cause an NCFC phenotype. This was
due to its homology to RAS genes, and evidence of GTPase activity (311). When its
involvement in human germline NCFC disorders was confirmed, the dataset from the target
enrichment experiment was reviewed for evidence of mutations in this gene in the six patients
without a proven molecular diagnosis. One patient, TE3, had a candidate variant, c.246T>G,
encoding p.(Phe82Leu), identified in this gene [NM_006912.4]. This same substitution had
been identified by Aoki et al, in two of the 17 patients in their series, and hence it was
extremely likely to represent the causative mutation in this patient. Retrospectively, the
likelihood of this variant being a true finding of the target enrichment experiment (rather than
artefact) was high, as coverage was very good, with a read depth of 1466, 798 for the reference
and 665 for the novel allele, with very high quality scores for both alleles (MQV of 39 and 38
respectively). The reason that this had not been validated in the first instance was the
prediction by SIFT that this variant would be tolerated, and that this nucleotide had a low
GERP score, suggesting that it was not highly conserved. The reason for the low score for
evolutionary conservation is not clear, from available alignments (Figure 4-9), but it could be
that the region was not correctly aligned to that of other placental mammals used to calculate
the GERP score when the dataset was produced.

171
4.2.10 Confirmation of diagnosis by RIT1 sequencing and clinical
implications
All coding exons of RIT1 were sequenced in patient TE3, confirming the presence of this
mutation, and the absence of any other variants in this gene. No further mutations were found
when the DNA of ten further patients with a CFC/NS presentation (and no molecular
diagnosis) was sequenced. The patient in whom the mutation was identified had some features
characteristic of a Ras-MAPK pathway disorder, specifically NS, as she had curly hair, some
suggestive facial characteristics, lymphoedema, and mild developmental delay. However, this
diagnosis had not been considered until after, importantly, she had also been diagnosed with
myelodysplastic syndrome (MDS) at the age of 4 years, requiring allogenous bone marrow
transplant. Myeloid malignancies are well-recognised in NS, but no patient with a RIT1
mutation has yet been reported with such (one case of acute lymphoblastic leukaemia was
present in Aoki et al’s series (76)). Monosomy 7 was identified in the myelodysplastic bone
marrow of patient TE3. This cytogenetic finding is characteristic of a group of patients with
paediatric MDS and juvenile myelomonocytic leukaemia, occurring in up to half of cases,
frequently as the sole cytogenetic lesion (350). The coexistence of this somatic lesion with a
presumed germline NS-associated mutation in patient TE3 has potential implications for the
investigation and care of this genre of patients.
Figure 4-9: Amino acid sequence encoded by exon 5 of RIT1.
Note a very high degree of conservation across the species included in this alignment (Alamut version 2.2, data from Ensembl). The site of the mutation p.(Phe82Leu) is indicated by the red box.

172
4.3 Whole exome sequencing for gene identification in CFC
4.3.1 Selection of patient samples
DNA samples from three patients with a CFC phenotype in whom no mutation was
identifiable in the clinically tested genes were selected for whole exome sequencing, when
samples from both parents were also available. All three patients selected had had sequencing
of BRAF, MAP2K1, MAP2K2 and KRAS (as described above) and comparative genomic
hybridisation to exclude large deletions or duplications as the cause for their phenotype. In
addition, patients WE1 and WE3 had also had all clinically available genetic tests for Noonan
syndrome, again with normal results.
4.3.2 Whole exome sequencing
As shown in Table 4.8, trios 1 and 2 were run using a SOLiD4 sequencer, and trio 3 an
Illumina HiSeq machine, with coverage statistics as shown.
Table 4.8: Samples on which whole exome sequencing was performed
Sample Platform % at 1x depth
% at 10x depth
% at 20x depth
% at 30x depth
WE1 SureSelect 50Mb exome enrichment, SOLiD4 sequencer
87.3 68.4 57.4 48.5
WE1 father
SureSelect 50Mb exome enrichment, SOLiD4 sequencer
93.0 81.8 75 69.6
WE1 mother
SureSelect 50Mb exome enrichment, SOLiD4 sequencer
93.0 81.6 74.8 69.5
WE2 SureSelect 50Mb exome enrichment, SOLiD4 sequencer
82.8 70.8 63.8 55.0
WE2 father
SureSelect 50Mb exome enrichment, SOLiD4 sequencer
81.2 67.9 61.1 51.7
WE2 mother
SureSelect 50Mb exome enrichment, SOLiD4 sequencer
80.7 67.9 60.4 50.9
WE3 SureSelect 38Mb exome enrichment, Illumina HiSeq (BGI)
95.3 72.7 57.8 46.6
WE3 father
SureSelect 38Mb exome enrichment, Illumina HiSeq (BGI)
96.0 76.2 63.0 53.1
WE3 mother
SureSelect 38Mb exome enrichment, Illumina HiSeq (BGI)
96.4 78.0 65.4 56.0
4.3.3 Bioinformatic analysis

173
The trios of samples were run in parallel and the primary analysis was designed (by Dr
Sanjeev Bhaskar, of the MCGM next generation sequencing group) to filter out variants that
were also present in the parents, as the expectation would be that it was a de novo mutation
causing the patient’s phenotype. Analysis of each of the parental exomes was therefore limited
to nucleotides at which candidate variants had been identified in the affected child. As would
be expected, this approach presented considerable bioinformatic challenges, and several
iterative adjustments to the analysis were required, informed by the results of Sanger
sequencing of candidate variants selected on the basis of initial bionformatic analyses. The
algorithm for assessment of variants is shown in Figure 4-10. The initial quality filter applied
was a depth of at least 20 reads, with an MQV score of at least 18 for both novel and
reference alleles.
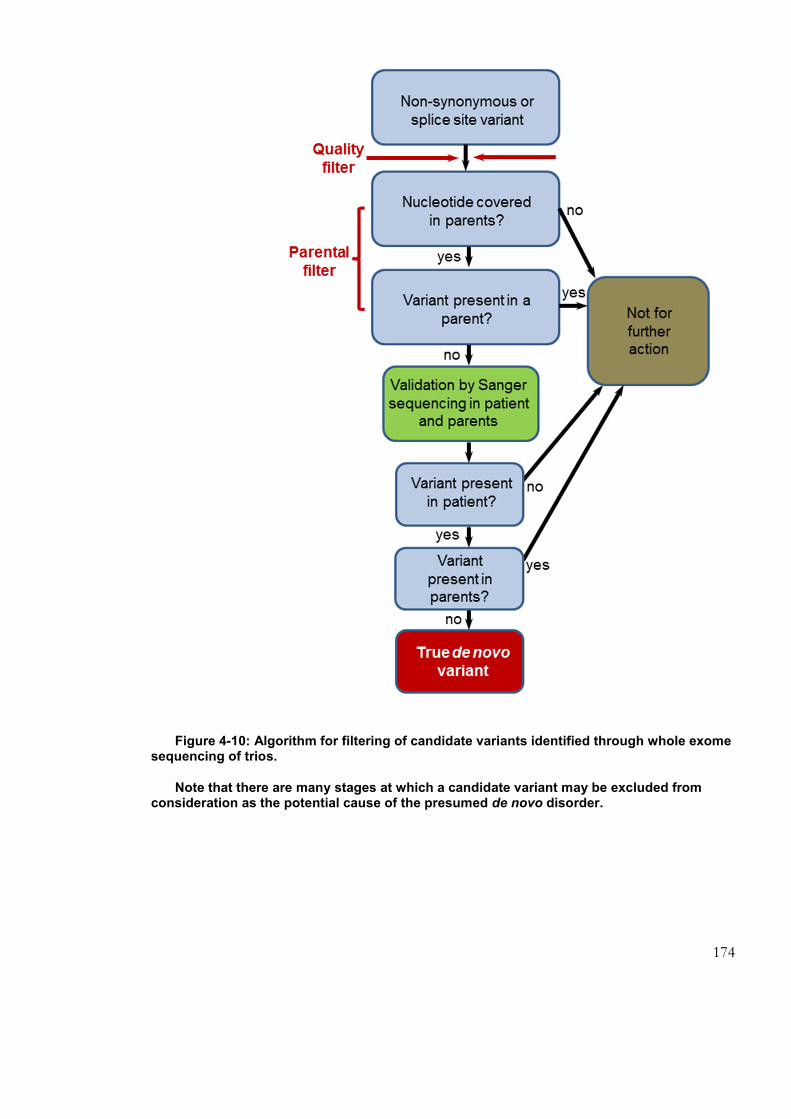
174
Figure 4-10: Algorithm for filtering of candidate variants identified through whole exome sequencing of trios.
Note that there are many stages at which a candidate variant may be excluded from consideration as the potential cause of the presumed de novo disorder.

175
Table 4-4: Variants called in whole exome sequencing Patient WE1 Patient WE2 Patient WE3 Number of variants in exome that met initial quality criteria
3008 3147 664
Number of above variants not seen in parental samples
140 67 106
The initial analysis designed to identify de novo variants yielded the numbers of variants
shown in Table 4-4, which were too large in number to validate individually. 32 candidate
variants were therefore selected that appeared from the bioinformatic data to have arisen de
novo in the affected individual for validation by Sanger sequencing. No bona fide de novo
variants were identified by this approach. 17 variants that had been called by the next
generation sequencing in the affected individuals’ samples were not present, and 15 had been
inherited from a parent (seven from the mother, and eight from the father).
4.3.4 Resequencing affected patients’ exomes using Illumina HiSeq2000
Due to the relatively low coverage across the exome sequencing described above, affected
patients’ samples were re-sequenced on an Illumina HiSeq, with the aim of both improving
coverage across all genes of the exome, and providing greater depth. This provided a further
bioinformatics challenge, to integrate and compare the data generated by this platform with
that obtained from the previously run parental samples on the SOLiD sequencer. This
reannotation and other formatting to ensure compatibility were successfully achieved by Dr
Simon Williams, using version 1.8 of the MCGM next generation sequencing group’s
analytical pipeline.
Using this platform, improvements in coverage were evident, and, compared to the initial
analyses, the lists of candidate variants were enriched for genes appearing to be potentially
plausible candidates for the phenotypes of the patients. However, as (due to resource
limitations) it was only the patients’ samples that had been re-run, the data from parental
exomes were still limited as previously. For trios 1 and 2, no novel candidate variants were
identified by this re-analysis.
In trio 3, one of the variants identified in the child was a heterozygous 4 base pair deletion
in the NF1 gene [ NM_00142492.2], c.499_502delTGTT ; p.(Cys167Glnfs*10), resulting in a
frameshift and premature termination codon. This was verified by Sanger sequencing in his

176
and his parents’ DNA, and confirmed to be present as a de novo mutation in this individual.
Data on coverage at this locus in this patient’s original exome sequencing run was not
available, due to the sequencing having been performed at BGI. Possible reasons for non-
identification of this variant in that experiment could be poor coverage across this locus, or
the fact that as a frameshift mutation, it may not have been possible to align the sequence
reads across the mutated allele correctly, resulting in the deletion not being identified.
4.3.5 Mutation in NF1 in a patient with a clinical diagnosis of CFC
syndrome
Patient WE3 was a 12 year old boy who had been included in the exome sequencing
project on the basis of a severe phenotype strongly suggestive of a Ras-MAPK pathway
disorder. His initial presentation was with severe hypotonia with bulbar muscle involvement
necessitating tracheostomy, feeding difficulties and severe gastrooesophageal reflux
necessitating gastrostomy, with facial features (relative macrocephaly, downslanting palpebral
fissures, and thickened ear helices) and body habitus consistent with NS/CFC. This
combination, with his severe neurological manifestations, was most suggestive of CFC
syndrome, particularly as he later also developed marked pigmentary changes. The possibility
of CS was also raised as his facial features coarsened, and gingival hypertrophy, macroglossia
and excessive skin of the hands and feet were present, with significant joint laxity. The
development of a hepatoblastoma at 7 months of age was an unusual feature. Such tumours
have been reported previously in one individual with CFC syndrome (35) and one with NF1
(351), but not in CS (though the much greater rarity of this disorder should be borne in mind).
Aged 10 y, he developed a mass in the left forearm, thought to be a plexiform neurofibroma.
By this time, he was also developing multiple café-au-lait patches on his trunk and an
increasing number of melanocytic naevi. Skinfold freckling and acanthosis nigricans were also
present. The profound feeding difficulties and otherwise stormy early course of this patient
was very reminiscent of the presentation of many patients with CFC and CS, and would not
normally be considered a feature of NF1. The patient had had all known CFC and NS genes
analysed on the basis of his clinical phenotype, with normal results. Whilst the presence of an
additional mutation in a gene causing CFC or a similar phenotype (that was not identified by
this exome sequencing experiment) cannot be excluded as a possibility, the NF1 mutation that
was identified is highly likely to represent the cause for much of this patient’s clinical
presentation.

177
4.4 Discussion of chapter results
With the pace at which massively parallel sequencing techniques have proven effective in
the investigation and diagnosis of genetic disease, it is inevitable that increasing numbers of
patients with Ras-MAPK pathway disorders will be identified through whole exome (or
genome) (WES/WGS) sequencing techniques. This may be of particular significance for the
group of patients with non-characteristic presentations, who may not have been recognised
and hence could not be diagnosed prior to the availability of comprehensive unbiased genetic
investigations. The precise timescale for adoption of clinical WES/WGS may vary around the
world according to local factors such as access to the necessary resources and the effectiveness
of current alternative means of genetic testing, but this change is likely to be enacted in the
next five years. Currently, the clinical utility of such techniques may be limited by the
substantial resources involved and potential difficulties in analysis, and in this interim, the
testing of targeted panels of genes for this purpose may be useful in providing molecular
diagnosis for this group of patients. Such panels have been a major advance in molecular
testing for genetically heterogeneous disorders (127). The experiments described in section 0
demonstrate that this approach can be effective for Ras-MAPK pathway disorders. Further
work will be necessary to refine the panel of genes to be included in a unified Ras-MAPK
pathway disorder test. Inclusion of the growing number of known genes for these disorders
may be complemented by including genes for phenotypically similar disorders, such as that for
Aarskog syndrome, which has many overlaps with NS, but if this were extended to include all
disorders with short stature, developmental delay and congenital heart disease, then a large
number of disorders would need to be considered for inclusion.
Other factors that could improve the utility of this approach would be modifications to
the bioinformatic filtering and processing of the data. The limitations of reliance upon the
GERP score for assessing conservation are well demonstrated by the low observed score for
the RIT1 nucleotide that was actually very highly conserved (Figure 4-9) across evolution.
Similarly, the prediction of pathogenicity by SIFT was also incorrect for this variant. Such
observations also open the possibility that other variants identified in this experiment might
also be pathogenic, but have not been identified as good candidates due to erroneous
bioinformatic predictions.

178
The finding of a RIT1 mutation in a patient with NS and previous MDS (section 4.2.10) is
important information to haematologists managing patients with MDS, as it is possible that
other patients with myelodysplasia may have similar predisposing germline mutations, even if
monosomy 7 is identified in their affected bone marrow. The RIT1 mutation, present in
lymphocyte DNA in this patient, should be confirmed in a further tissue such as buccal
epithelial cells, hair root follicle or skin fibroblasts, to investigate the possibility of somatic
mosaicism for this mutation. Knowledge of such a germline mutation in a patient would
influence genetic counselling regarding recurrence risk for future siblings and offspring, and
the possible coexistence of other features of a Ras-MAPK pathway disorder. Many of the
features of NS that might be present in such patients, such as short stature, gonadal
dysfunction, unusual hair texture for the family, developmental delay or cardiac pathology may
also recapitulate (and hence be potentially confused with) sequelae of bone marrow
transplantation in childhood (352, 353). Clinical management may also need to be tailored in
response to finding such a mutation, for example, the high risk of HCM identified in the initial
cohort of patients with RIT1 mutations (76), may warrant particular consideration of the risk
of worsening cardiac function in such patients if cardiotoxic chemotherapeutics are being
considered.
In contrast to the ease of identifying mutations in data from the targeted gene sequencing
panel, exome trio sequencing, using the then currently available technology, was more difficult
to interpret and, despite the much broader remit of the experiment, effective only in one of
the three trios assessed (and then only after repeat sequencing of the patient’s sample). There
are several contributors to this observation. As discussed in the introduction, the molecular
basis of genetically heterogeneous disorders is more difficult to investigate and diagnose, as
the causal variants in each affected individual may each be in a different gene, and whilst the
sequencing of trios with parental samples can overcome this, additional considerations are
encountered. For each patient exome investigated, high quality data is required from each of
three exomes. The need for a depth coverage of 20 reads (as a bare minimum) in each sample
for confident calling of heterozygous variants is a further challenge, alongside the relatively
high proportion of genes that displayed poor coverage in this exome sequencing project, the
initial runs of which were amongst the first performed in the MCGM laboratory. The
improvement of the available technology in the course of this study is evident by the
identification of a causative mutation in repeat sequencing of the child of the third trio, which

179
was not identified in the original sequencing. Of note, such insertion/deletion mutations may
be more challenging to identify accurately, and had this not been in a clearly clinically relevant
gene, then it might have been a lower priority for validation (due to the higher chance of it
representing a false-positive finding). The success of exome sequencing in one of three
instances in this study is in keeping with observations from other studies, such as the
identification of the causative mutation in one quarter of patients referred for clinical exome
sequencing in one centre (Baylor College of Medicine (354)). Such figures will improve as
experimental technologies make coverage of the genome more complete, bioinformatic
analyses are streamlined, and knowledge of greater numbers of causative genes for disorders
allows for more focussed stratified analyses.
The finding of a frameshift mutation in NF1 in a patient with a severe developmental
delay phenotype, not characteristic for NF1 (section 4.3.5), has implications for the
understanding of the NCFCs. It emphasises the overlapping clinical nature of these disorders,
and the value of considering testing of extended panels of disease-associated genes in patients
with atypical presentations. The true spectrum of disease associated with mutations in specific
genes will only be fully appreciated once whole exome sequencing has been applied to large
numbers of patients with a range of phenotypes in a relatively unbiased way (as is now in
progress through initiatives such as the Deciphering Developmental Disorders project (355)).
Hepatoblastoma was diagnosed in patient WE3 at eight months of age, and has been
reported in only one previous individual with NF1, but it may be that this is an underestimate:
further rare patients may have been similarly affected, but have succumbed to the tumour
before they could develop other features of NF1 and be diagnosed. The NF1 mutation is
highly likely to be a significant contributor to the moderate to severe learning disability present
in this patient, but the possibility of further genetic or genomic contributors to this phenotype
cannot be excluded.
Whether individual patients have targeted or whole exome sequencing performed, it is
highly likely that such investigations will be performed earlier in the diagnostic and
management process, as depicted in Figure 4-11. The cost of sequencing is falling, and its
clinical utility for diagnosis in patients with congenital disorders is rising. Even currently, the
cost of exome sequencing is comparable to many other diagnostic tests, for example magnetic

180
resonance imaging investigations, or, especially for children or other individuals with
additional needs, any of the many investigations that may require a general anaesthetic.
Figure 4-11: Massively parallel sequencing has the potential to transform the diagnostic
process.
This is particularly the case for genetically heterogeneous disorders, and leads to better
care for patients. The left hand panel indicates the often difficult process of reaching a
diagnosis when genetic testing was limited to single genes at a time. Frequently, multiple forms
of investigation would be required to guide choice of gene for testing. If a mutation was not
identified in the first iteration of testing, then further rounds of investigations and genetic tests
might be indicated, with potential implications for health, psychosocial wellbeing and health
economics.

181
5 FUNCTIONAL CONSEQUENCES OF DE NOVO GERMLINE
MUTATIONS CAUSING RAS-MAPK PATHWAY DISORDERS

182
5.1 Chapter overview
CFC syndrome arises due to heterozygous mutations in genes encoding various proteins
involved in the Ras-MAPK pathway, and around three quarters of mutations identified in
affected patients are in BRAF. As discussed in the introduction and in chapter 3, mutations in
BRAF are most commonly associated with a classical CFC syndrome presentation, but a
variety of phenotypes including occasional clinical diagnoses of NS or NSML have also been
identified in association with such variants (39).
Whilst a wide range of mutations, judged to be causative by their de novo occurrence, and
presence in multiple patients, have been identified for CFC syndrome, the functional
consequences of these have not been so extensively studied. Additionally, several mutations
had been identified in individual patients in the MCGM diagnostic laboratory that had not
been reported in other cohorts, and hence it was not possible to confirm their pathogenicity.
A list of mutations described to date in CFC syndrome was compiled using the data from the
MCGM laboratory (chapter 3) and the published literature, and is shown in appendix 1. This
was cross-checked by reference to the NSEuronet database (6) once this was in operation.
The previously characterised variants selected for inclusion in the experiments were those
that had been recurrently described in association with CFC syndrome, and for which a variety
of downstream effects had been demonstrated in the published literature. These are shown in
normal type in Figure 5-1, with the novel, previously uncharacterised, substitutions shown in
bold type. Each of the novel mutations had been described in a single patient with CFC
syndrome, with one, p.(Gln262Pro) being described subsequently in a second patient whose
sample was referred to the MCGM laboratory, and another, p.(Thr470Pro), subsequently
being described in one further published patient in 2012 (356).
The effects of these variants on downstream pathway signalling were investigated in cell
culture using three methods: Western blotting with specific antibodies against phosphorylated
ERK1 and ERK2, in vitro kinase assays and dual luciferase assays, as described below.

183
Figure 5-1: Mutations and uncharacterised variants in BRAF in patients with CFC syndrome.
This schematic shows the BRAF substitutions selected for molecular analysis in this study. Those shown in bold are novel variants identified in diagnostic testing in the MCGM, and not previously characterised at the molecular level. Other CFC-associated mutations for which the functional effects have been assessed are shown in normal black type. These were selected for their recurrent identification in CFC, and to represent a range of effects on downstream signalling, according to the results of previously published work. p.(Val600Glu), the cancer-associated mutation which has not been seen in the germline, is shown in red. RBD indicates the Ras-binding domain, and CRD the cysteine-rich domain (which contains a high proportion of the mutations known to cause CFC syndrome, including the most common, p.(Gln257Arg)).
5.2 Characterisation of the effect of mutations in BRAF on ERK
pathway activity in the HEK293T and HEK293 cell lines
5.2.1 Verification of plasmids for expression of BRAF in cell culture
pEF-BRAF wild-type and p.(Val600Glu) plasmids (the gift of Dr. Claudia Wellbrock,
Faculty of Life Sciences, University of Manchester), containing full length human BRAF
cDNA with a c-myc tag, were amplified in DH5α cells and verified by restriction digest using
BamHI and Xba1 restriction enzymes, which yielded the expected fragment sizes of 8 kb and
4 kb (Figure 5-2A).
For further verification, 2 µg of DNA of these two plasmids was transfected into
HEK293T cells, as this line is straightforward to culture, retaining its characteristics over many
passages, readily transfectable and high expression levels of transfected products can be
achieved. Expression of the transfected BRAF alleles was assessed by Western blotting for the

184
c-myc tag (Figure 5-2B). This demonstrated that the BRAF alleles were expressed at similar
levels from the two plasmids.
Phospho-ERK1/2 activity as a result of these transfections was assessed by Western
blotting using an antibody which detects phosphorylation of threonine at position 202 and
tyrosine at position 204 of these proteins. Total ERK1/2 was also probed for comparison,
and as a loading control. No significant differences in ERK1/2 expression were seen between
untransfected cells and those transfected with the pEF-BRAF plasmids, indicating appropriate
loading of the gel. ERK1/2 phosphorylation was seen to be markedly increased in the cells
transfected with pEF-BRAF p.(Val600Glu), and modestly increased in cells transfected with
the wild-type pEF-BRAF plasmid (Figure 5-2B).

185
Figure 5-2: Verification of pEF-BRAF wild-type and p.(Val600Glu) plasmids.
A: Restriction digest of plasmids using BamHI and Xba1 restriction endonucleases demonstrates two fragments of 8kb and 4kb, consistent with the expected presence of single sites for these enzymes to act within these plasmids. The digest was performed in duplicate; WT indicates the wild-type plasmid, and the p.(Val600Glu) plasmid is labelled. B: Western blotting of HEK293T cell lysates transfected with these plasmids. Expression of the c-myc tagged BRAF is seen at equivalent levels following transfection of pEF-BRAF wild-type and pEF-BRAF p.(Val600Glu). ERK1/2 is shown as a loading control, and for comparison to phospho-ERK1/2 (pERK1/2). Note increased ERK1/2 phosphorylation in cells transfected with pEF-BRAF p.(Val600Glu), and modestly increased ERK1/2 phosphorylation in cells overexpressing wild-type BRAF, compared to untransfected cells.

186
5.2.2 Site-directed mutagenesis to generate CFC-associated mutations in
BRAF
Site-directed mutagenesis of the pEF- BRAF wild-type plasmid was performed using
custom designed primers (appendix 4) to generate alleles p.(Thr241Pro), p.(Gln257Arg),
p.(Gly469Glu), p.(Lys499Glu) and p.(Glu501Gly), each of which were mutations recurrently
identified in CFC syndrome, that had been subject to previous molecular investigation (10, 77,
162). These alleles were chosen so that the results of the assays could be compared to those in
the published literature; their positions within the protein are shown in the schematic in Figure
5-1. The BRAF cDNA within each of these plasmids was sequenced, demonstrating
successful generation of each mutation, and that no additional sequence changes had been
introduced.
5.2.3 Western blotting for phospho-ERK1/2
Western blotting was performed to assess the level of phosphorylation of ERK1 and
ERK2, downstream targets of the RAF-MEK-ERK kinase cascade, as described above, in
lysates of cells transfected with pEF-BRAF wild-type and mutated alleles. In order to ensure
validity of the observed effects, the level of expression of the c-myc tagged BRAF first had to
be as equal as possible for each of these transfected alleles. With equal amounts of DNA of
each plasmid, it was evident that the p.(Gly469Glu) allele showed a lower level of expression,
an observation which could be due to intrinsic properties of this plasmid, reduced stability of
the BRAF p.Gly469Glu protein, or a combination of factors. When the amount of the
p.(Gly469Glu) plasmid DNA was increased to 2.5 µg, compared to 1 µg for all other alleles in
the transfection experiment, approximately equal expression of each of the constructs was
obtained (Figure 5-3; lane 4), hence all subsequent experiments used this ratio. In this
experiment, BRAF mutations p.(Thr241Pro), p.(Gln257Arg) and p.(Lys499Glu) (lanes 2,3 and
5) each showed increased ERK1/2 phosphorylation compared to the wild-type protein (lane
1), but less than that which was observed for p.(Val600Glu) (lane 7). p.(Gly469Glu) and
p.(Glu501Gly) (lanes 4 and 6) did not show evidence of increased ERK1/2 phosphorylation
compared to the wild-type protein.

187
Figure 5-3: Western blotting in HEK293T cell lysates after transient transfection of pEF-BRAF plasmids demonstrated effects upon ERK1/2 phosphorylation.
Overexpression of the panel of mutations in BRAF was achieved by transfection of 1 µg of plasmid DNA for each allele, except for p(Gly469Glu), which required 2.5 µg to obtain the equivalent level of expression, demonstrated here by blotting for the c-myc tag attached to the BRAF protein. A variety of effects upon ERK1/2 phosphorylation are seen. Overexpression of wild-type BRAF causes increased ERK1/2 phosphorylation, compared to the level observed in untransfected cells. Mutated alleles p.(Thr241Pro) and p.(Gln257Arg), p.(Lys499Glu) and p.(Val600Glu) show greater increases in ERK phosphorylation than is the case for the wild-type protein, whilst p.(Gly469Glu) and p.(Glu501Gly) appear to produce similar levels of ERK1/2 phosphorylation to the wild-type protein. In keeping with published work, the effects of p(Val600Glu) are more extreme than those observed with other mutations.
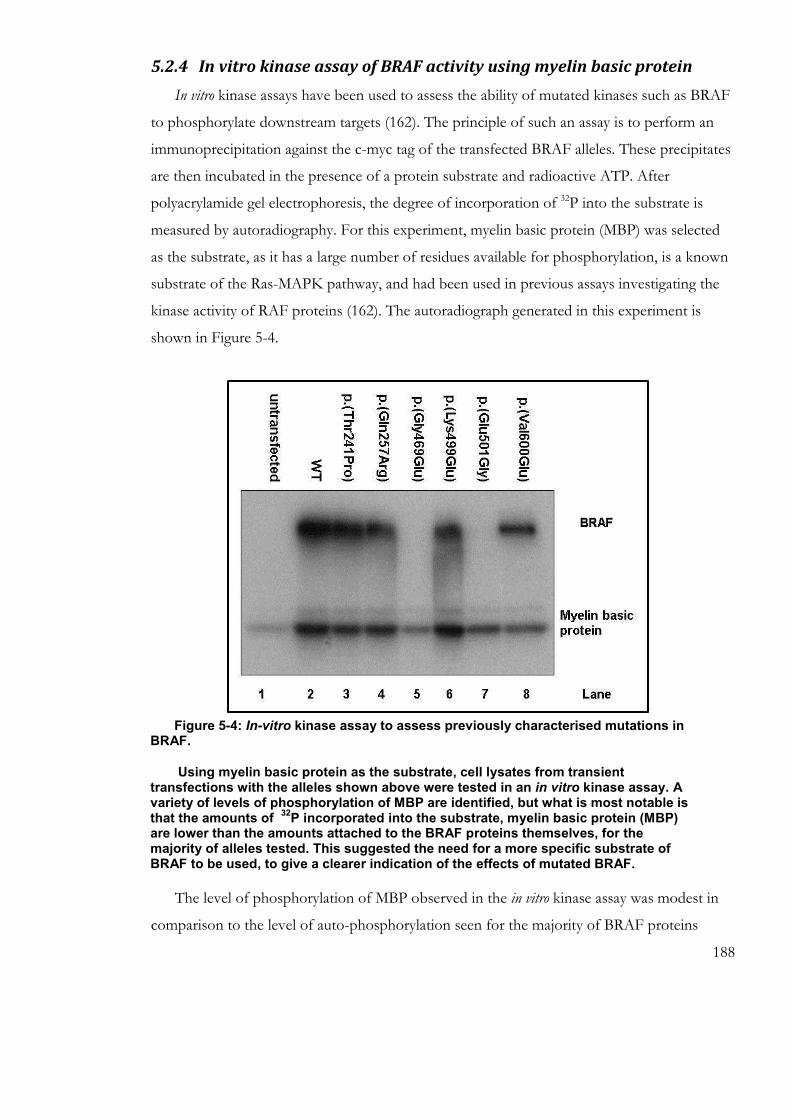
188
Figure 5-4: In-vitro kinase assay to assess previously characterised mutations in BRAF.
Using myelin basic protein as the substrate, cell lysates from transient transfections with the alleles shown above were tested in an in vitro kinase assay. A variety of levels of phosphorylation of MBP are identified, but what is most notable is that the amounts of
32P incorporated into the substrate, myelin basic protein (MBP)
are lower than the amounts attached to the BRAF proteins themselves, for the majority of alleles tested. This suggested the need for a more specific substrate of BRAF to be used, to give a clearer indication of the effects of mutated BRAF.
5.2.4 In vitro kinase assay of BRAF activity using myelin basic protein
In vitro kinase assays have been used to assess the ability of mutated kinases such as BRAF
to phosphorylate downstream targets (162). The principle of such an assay is to perform an
immunoprecipitation against the c-myc tag of the transfected BRAF alleles. These precipitates
are then incubated in the presence of a protein substrate and radioactive ATP. After
polyacrylamide gel electrophoresis, the degree of incorporation of 32P into the substrate is
measured by autoradiography. For this experiment, myelin basic protein (MBP) was selected
as the substrate, as it has a large number of residues available for phosphorylation, is a known
substrate of the Ras-MAPK pathway, and had been used in previous assays investigating the
kinase activity of RAF proteins (162). The autoradiograph generated in this experiment is
shown in Figure 5-4.
The level of phosphorylation of MBP observed in the in vitro kinase assay was modest in
comparison to the level of auto-phosphorylation seen for the majority of BRAF proteins

189
tested. This is likely to be due to BRAF auto-phosphorylation being a significantly more
efficient process than its phosphorylation of MBP, as the latter is a non-physiological target of
BRAF. Due to this, differences between the mutated alleles could have been masked, with
potential overestimation of the ability of the less active mutants to phosphorylate downstream
targets represented by MBP. Overall, the results were difficult to interpret, but suggested the
need to use a more specific and physiologically relevant substrate of BRAF to assess the
effects of the mutant alleles. For this reason, a direct target of BRAF, inactive MEK, was
chosen for a further in vitro kinase assay, in which the effects of the wider panel of mutated
alleles described below could also be assessed.
5.2.5 Site-directed mutagenesis to assess novel variants in BRAF identified
in patients with CFC syndrome
Novel variants that had been identified in patients but which were uncharacterised at the
molecular level were identified from the results of testing in the MCGM diagnostic service
laboratory. p.(Gln262Pro), p.(Gly464Glu) and p.(Leu525Gln) were each substitutions that had
(at that time) been observed only in single patient, and p.(Thr470Pro) had been observed in
two patients. Whilst de novo status of each of these variants had been demonstrated, strongly
suggesting that they represented the cause of the patients’ clinical presentations, no further
patients with these variants had been described, and hence no previous molecular work had
been performed. Each of these patients had a clinical diagnosis of CFC syndrome, and no
other variants in BRAF, MAP2K1, MAP2K2 or KRAS had been identified in diagnostic
sequencing of these genes in the MCGM laboratory. Limited information on the phenotypes
of the patients with p.(Gln262Pro) and p.(Thr470Pro) was available, the former being males
of 11 and 18 years old and the latter being a male 36 years of age. The patient with the
p.(Gly464Glu) mutation was an 8 year old boy, patient B6 (chapter 3), whose presentation was
milder than for many patients with CFC, except for the fact that he had severe cyclical
vomiting syndrome. The patient with the p.(Leu525Gln) mutation had a very severe
phenotype, being non-ambulant and without speech, as described in chapter 3.
Site-directed mutagenesis reactions were set up, using pEF-BRAF and the primers listed in
appendix 4, to generate the mutated alleles. The BRAF cDNA from each of the mutagenesis
reactions was sequenced, as described previously, confirming the presence of the desired
substitution and the absence of any further alterations.

190
5.2.6 Work in the HEK293 cell line
In view of emerging evidence obtained by the Whitmarsh laboratory that the HEK293T
cell line exhibited high levels of basal MAPK pathway activity (Dr R. Monaghan, personal
communication), it was decided to base further assays in the HEK293 cell line, for which such
high basal activity was not demonstrated, but which nonetheless had similar culture properties
to the HEK293T cell line, being rapidly growing and readily transfectable, and hence
particularly suitable for assays in which many different alleles were tested (and which hence
required large numbers of cells).
5.2.7 Western blotting to assess ERK1/2 phosphorylation by novel
variants in BRAF identified in patients with CFC syndrome
HEK293 cells were transfected with BRAF alleles p.(Thr241Pro), p.(Gln257Arg),
p.(Gln262Pro), p.(Gly464Glu), p.(Gly469Glu), p.(Thr470Pro), p.(Lys499Glu), p.(Glu501Gly),
p.(Leu525Gln) and p.(Val600Glu) as described above, with 1 µg DNA of each plasmid except
p.(Gly469Glu), which required 2.5 µg to generate equivalent expression (as shown by the c-
myc tag, Figure 5-5). Western blotting for phospho-ERK1/2 was performed, with total
ERK1/2 probed for comparison and as a loading control (Figure 5-5).
Previously characterised mutations showed similar results to those previously observed in
HEK293T cells (Figure 5-3) and in the published literature (162): p.(Thr241Pro),
p.(Gln257Arg), p.(Lys499Glu) and p.(Val600Glu) (lanes 3, 4, 9 and 12) showed increased
levels of ERK1/2 phosphorylation compared to wild-type, whilst p.(Gly469Glu) and
p.(Glu501Gly) (lanes 7 and 10) showed unaltered or possibly reduced levels. As also observed
previously, overexpression of wild-type BRAF protein (lane 2) caused increased ERK1/2
phosphorylation, when compared to untransfected cells. Overexpression of the novel BRAF
alleles was shown to have effects on ERK1/2 phosphorylation, as shown in Figure 5-5.
p.(Gln262Pro), p.(Gly464Glu), p.(Thr470Pro) and p.(Leu525Gln) (lanes 5, 6, 8 and 11) each
showed increased ERK1/2 phosphorylation as compared to wild-type BRAF, but less than
that which was observed with p.(Val600Glu) (lane 12).
5.2.8 In vitro kinase assay to assess novel variants in BRAF identified in
patients with CFC syndrome

191
Building on the observations made around the in vitro kinase assay shown in Figure 5-4, a
modified assay was designed to assess the effects of novel and previously characterised
mutations in BRAF. A synthesised inactive MEK protein, tagged with GST, was selected as
the substrate for this in vitro kinase assay, as this protein has a direct interaction with BRAF,
and has previously been used in this kinase assays (357). Using a form of MEK as the
substrate meant that the degree of phosphorylation of this, the molecule directly
phosphorylated by BRAF in the RAS-RAF-MEK-ERK signal transduction cascade, could be
assessed. As for MBP in the assay described above, the level of phosphorylation of the GST-
MEK was assessed by incorporation of radiolabelled ATP demonstrated on an
autoradiograph. Analogous to the results of the Western blotting, a range of effects of the
CFC-associated mutations was once again observed (Figure 5-5). In keeping with previous
work, the majority of variants showed increased MEK phosphorylation, compared to wild-
type BRAF. Of the novel mutations, p.(Gln262Pro) (lane 5), like other mutations affecting the
cysteine-rich domain, such as p.(Thr241Pro) and p.(Gln257Arg) (lanes 3 and 4), showed
increased activity. Increased activity was also observed for p.(Leu525Gln) (lane 11), which is
located within the kinase domain. p.(Gly464Glu) and p.(Thr470Pro) (lanes 6 and 8), variants
within a region of the kinase domain that may also be involved in RAF dimerisation (358),
showed kinase activity that was similar to, or lower than, wild-type BRAF (lane 2). This was
not as severely impaired, however, as with the adjacent p.(Gly469Glu) mutation (lane 7), a
substitution that had been identified in patients with CFC in the MCGM laboratory and in the
published literature, and was known to have reduced kinase activity (10).

192
Figure 5-5: Western blotting (A) and in vitro kinase assay (B) of HEK293 cells transiently transfected with CFC-associated BRAF alleles.
Panel A shows increased ERK1/2 phosphorylation for the majority of CFC-associated alleles, but less extreme than is observed with BRAF p.(Val600Glu). The degree of ERK1/2 phosphorylation for newly assessed variants shows a similar range to that demonstrated by previously characterised mutations, with all demonstrating increased activity compared to wild-type. Panel B: In vitro kinase assay demonstrates analogous findings with respect to MEK phosphorylation. The majority of mutated alleles result in increased MEK phosphorylation, including newly characterised mutations p.(Gln262Pro) and p.(Leu525Gln). The other newly characterised substitutions, p.(Gly464Glu) and p.(Thr470Pro), show reduced catalytic activity (similar to or less than the wild-type protein) compared to substitutions elsewhere in the protein, seen in their effect of GST-MEK phosphorylation and autophosphorylation. This effect is less extreme than is observed with the previously characterised p.(Gly469Glu). The Coomassie stained gel is shown on the right to demonstrate presence of equal amounts of GST-MEK in each of the reactions.

193
5.2.9 Dual luciferase assay to measure ELK1 transcriptional activity
As a third means of assessing the downstream effects of mutated BRAF, dual luciferase
assays were performed to assess the panel of mutant alleles. This technique uses cells
transiently transfected with plasmids containing the variant alleles of interest, cotransfected
with a plasmid containing the sequence of transcriptional activation domain of ELK1 (a
downstream target of Ras-MAPK pathway signalling) fused to GAL4 (a heterologous DNA
binding domain), and another containing a firefly luciferase reporter which contains GAL4
binding sites. A further plasmid containing Renilla luciferase provides a control measurement
(as its expression is unaffected by the activity of the allele under test). When the ELK1 domain
is activated by MAPK, this leads to production of firefly luciferase, and hence emission of
light. The light emitted is in direct proportion to the level of ELK1 activation, and is
quantified using a luminometer. The advantages of this approach are that measurements can
be compared rapidly in a large number of samples and there is an internal control (Renilla
luciferase activity) for each measurement. Analogous to the results of the Western blotting, a
range of effects on downstream ELK1 transcriptional activation were observed, as shown in
Figure 5-6. The pattern of activity appeared to mirror the degrees of effect of the different
mutants observed above by Western blotting, with the majority demonstrating increased
activity, compared to wild-type BRAF. However, this observed difference only reached
statistical significance for p.(Gln262Pro), p.(Lys499Glu), p.(Leu525Gln) and p.(Val600Glu)
(judged by one way ANOVA and Dunnett’s post hoc analysis). The similarity between the
effects of p.(Leu525Gln) and p.(Val600Glu) in this experiment could be due to the extreme
activity of the p.(Val600Glu) allele lying outside the dynamic range of this particular assay, but
it is notable that p.(Leu525Gln) also appears to have similar effects to p.(Val600Glu) with
respect to ERK1/2 phosphorylation (Figure 5-5). Clinical details available on the single patient
with this mutation indicated that she had a very severe phenotype, raising the possibility that
this mutation might, like p.(Val600Glu), have more extreme effects upon cellular and tissue
function than other CFC-associated BRAF mutations. On the basis of a single patient,
however, it is not likely to be possible to draw definitive conclusions. Information regarding
any further unpublished patients with this mutation was sought from other investigators with
an interest in CFC syndrome, and none were identified, highlighting that this is likely to be a
very unusual mutation (an observation that could be consistent with it having an extreme
effect if present in the germline of an individual).

194
Figure 5-6: Dual luciferase assay results in HEK293 cells transfected with CFC-associated mutations in BRAF.
The difference between the variant and wild-type alleles is shown on the y axis. Data points represent the mean of triplicate repeats, and error bars show 95% confidence intervals (calculated using one way ANOVA and Dunnett’s post hoc analysis). pLink indicates the empty vector, C1, control cells transfected with luciferase assay plasmids but no BRAF allele, and C2 , cells transfected with BRAF p.(Val600Glu) but not ELK1. Each of these controls are seen to result in significantly less luciferase activity than transfection with wild-type BRAF. The majority of identified substitutions show a tendency to increased downstream pathway activity (as assessed by ELK1 transcription), but this only reached statistical significance (95% confidence interval) for p.(Gln262Pro), p.(Lys499Glu), p.(Leu525Gln) and p.(Val600Glu). Mutations for which no increase in downstream pathway activity is shown are p.(Gly464Glu), p.(Gly469Glu), p.(Thr470Pro) and p.(Glu501Gly), similar observations to the results seen in the Western blot and in vitro kinase assay.

195
5.3 Effects of mutated BRAF in the H9C2 cardiomyoblast cell line
In order to assess the effects of CFC-associated mutations in a more physiologically
relevant context, cells of the H9C2 cell line, an untransformed line derived from neonatal rat
cardiomyoblasts, were obtained (gift of Dr Joy Wang, Faculty of Life Sciences, University of
Manchester). Transient transfections of these were attempted using chemical reagents jetPEI
and Lipofectamine 2000, but without successful demonstration of myc-tagged BRAF
expression by Western blotting. Transfection by electroporation, using the Amaxa system with
a protocol optimised for this cell type, demonstrated only modest expression of the myc-
tagged BRAF by Western blotting (Figure 5-7). Transfection efficiency for this technique was
estimated at approximately 20% by counting of cells on microscopy 48 hours after a test
transfection with green fluorescent protein.
Whilst the expression of BRAF was detectable after electroporation, unfortunately, no
observable difference was present between the level of ERK1/2 phosphorylation in the
lysates from wild-type pEF-BRAF transfection and the levels observed in the lysates from
wells transfected with CFC-associated mutations p.(Gln257Arg) and p.(Glu501Gly) (Figure
5-7). As would be expected, p.(Val600Glu) did generate a higher level of ERK1/2
phosphorylation, but overall the results of this experiment demonstrated that transient
transfection of H9C2 cells was unlikely to represent a tractable means of investigating the
functional consequences of CFC-associated mutations of BRAF, as the resolution of the p-
ERK1/2 assay was not sufficient to show a difference in ERK1/2 phosphorylation between
these and the wild-type protein. A major reason for this could have been the low transfection
efficiency. The benefit of being able to assess the effect of such mutations comparatively and
in a cell line with potential physiological relevance to the human phenotypes of CFC
syndrome suggested that a modified approach, of generating stable transfections of H9C2
cells, would be worthwhile.

196
Figure 5-7 Transient transfection of H9C2 cells with pEF-BRAF plasmids by electroporation.
This shows low levels of expression of BRAF after transfection, as assessed by Western blotting for the c-myc tag. Note approximately equal expression of BRAF with each of the transfected alleles shown, with total ERK1/2 expression shown as a loading control. However, whilst the level of ERK1/2 phosphorylation produced by transfection of pEF-BRAF p(Val600Glu) can be seen to be greater than that observed for wild-type pEF-BRAF, there is no observable difference between wild-type and CFC-associated mutations p.(Gln257Arg) and p.(Glu501Gly). This suggested that this approach would not be suitable for assessing the effects of such mutations in the H9C2 cell line.

197
5.3.1 Stable transfection of the H9C2 cell line
The Flp-in system (Invitrogen) was used with an aim of generating a set of congenic stably
transfected H9C2 cell lines containing a range of alleles of BRAF at the same locus in each
line. Cells first needed to be stably transfected with a pFRT/lacZeo plasmid, then the BRAF
expression vectors cloned into the site of integration of this plasmid, using cotransfected Flp
recombinase.
The initial step of identifying the lethal concentration of Zeocin for untransfected H9C2
cells was complicated by their slow proliferation, but this was eventually demonstrated to be
400 µg/ml: at lower concentrations, H9C2 cells were able to divide and survive longer than 3
weeks.
However, cells transfected with pFRT/lacZeo and subsequently exposed to this
concentration repeatedly died within 2 weeks. When the experiment was repeated with cells
exposed to mock transfection (electroporation using the same protocol but without plasmid),
a much lower Zeocin concentration, 100 µg/ml, was found to inhibit proliferation, and was
hence used for further attempts to select successfully transfected pFRT/lacZeo positive cells.
Individual clones were cultured in media containing 100 µg/ml Zeocin, and did proliferate,
but extremely slowly. Before any clonal colony was of sufficient size to be able to be passaged
and a sample taken for analysis, it was evident that cellular morphology had become altered:
some cells within clonal colonies demonstrated differentiation into myotubes, i.e. with a
multinucleated morphology distinct from the desired cardiomyoblasts (Figure 5-8C), but much
greater numbers of cells had become senescent, evidenced by the presence of rounded
colonies, that did not increase in size (Figure 5-8A,B). The attempt to generate stably
transfected cells was therefore abandoned. In retrospect, the finite number of passages
through which H9C2 cells, as an untransformed cell line, can be cultured made the chances of
successfully generating stable lines from these cells very low. Stable cell lines from this line
have been generated in the past (359-361), but the additional number of passages required for
the two rounds of selection involved in the Flp-in system would be more than has been
required for each of the experiments published in the literature.

198
Figure 5-8 Abnormal morphology of H9C2 cells in the attempt to generate stable cell lines. After electroporation with pFRT/lacZeo and 3 passages in the course of selection by Zeocin resistance, low power microscopy (original magnification 100x) demonstrated rounded senescent colonies (A), as outlined in red. At higher magnification (200x), abnormal cell morphology is evident within these colonies (B). In panel C, a different clonal culture to panels A&B is shown, where cellular morphology is less grossly abnormal, but multinucleated myotubes are present, as indicated by the black arrow (original magnification 200x).
5.4 Discussion of results
Collectively, the results of these assays demonstrate that, in keeping with previous work,
the effects of CFC-associated mutations in BRAF appear to be variable in these experimental
systems. The majority of mutations have identifiable effects on downstream signal
transduction, be it at the next step along the pathway with MEK phosphorylation (as assessed
in the in vitro kinase assay), with ERK1/2 phosphorylation (as assessed in the Western
blotting), or with respect to transcriptional targets downstream of ERK1/2 such as ELK1
activity (as demonstrated in the dual luciferase assay).

199
Table 5-1: Effects of CFC-associated mutations in BRAF assessed in cell culture
ERK1/2 phosphorylation by WB
Kinase activity by in vitro kinase assay
ELK1 transcription by luciferase assay
published (10, 39, 162)
this study published (162)
this study
published (77)
this study
p.(Thr241Pro) ↑ ↑ ↑ ↔ p.(Gln257Arg) ↑ ↑ ↑ ↑ ↑ ↔ p.(Gln262Pro) ↑ ↑ ↑ p.(Gly464Glu) ↑ ↔ ↔ p.(Gly469Glu) ↓ ↔ ↔ ↓ ↓ ↔ p.(Thr470Pro) ↑ ↓ ↔ p.(Lys499Glu) ↑↑ ↑ ↑↑ ↑↑ ↑↑ ↑ p.(Glu501Gly) ↔ ↔ ↔ ↔ ↓ ↔ p.(Leu525Gln) ↑ ↑ ↑↑ p.(Val600Glu) ↑↑↑ ↑↑ ↑↑↑ ↑↑ ↑↑↑ ↑↑
The direction and magnitude of effects observed for each of the variants in the assays
described here broadly concur with those in the literature. Substitutions in the cysteine-rich
domain, affecting residues 241-262, resulted in increased ERK1/2 phosphorylation and
increased activity in the in vitro kinase assay, when overexpressed in HEK293 cells. The effect
of p.(Gln262Pro), newly assessed here, appears similar to the effects of the other two
mutations in this domain, p.(Thr241Pro) and p.(Gln257Arg). These two mutations have also
been shown to increase ELK1 transcription in luciferase assays (77). Whilst the results
obtained for these mutations in the dual luciferase assay in this study did not reach statistical
significance, the values obtained were higher than those observed for wild-type BRAF, and
hence are not inconsistent with those in the literature. The smaller difference observed may be
due to lower expression of the BRAF alleles in HEK293 cells in these experiments than in the
NIH3T3 cells used by Niihori et al. Mutations of residues 464-470, again in keeping with
those in the literature, showed similar effects upon downstream pathway signaling to one
another, with decreased kinase activity as previously observed for p.(Gly469Glu). The final
newly-assessed mutation, p.(Leu525Gln), showed increased activity on each of the assays, and
this was particularly marked in the dual luciferase assay, where the result was nearly as elevated
as that observed with p.(Val600Glu).
There are several reasons why the range of mutations of BRAF identified in patients with
CFC syndrome (with clinically similar features) could appear to have divergent effects at the
molecular level. The shortcomings common to all experiments involving overexpression of

200
proteins by transient transfection are important considerations. Firstly, there may be technical
difficulties with obtaining even expression of a variety of constructs, witnessed by the need to
transfect significantly greater quantities of DNA for the p.(Gly469Glu) construct in order to
have as close to equal expression to the other BRAF mutants as possible (confirmed by
Western blotting for the c-myc tag). Secondly, the physiological relevance of heavily
overexpressed proteins from transient transfections may be limited. This may be of particular
significance for proteins with a chaperoning or scaffolding role, as appears to be the case for
BRAF (63, 64). Overexpression of a mutated protein that at a physiological level increases
signal transduction along a pathway might have a paradoxical effect, if in excess it sequesters
its target rather than promoting excessive signal transduction. Thirdly, it is clear that the
HEK293T and HEK293 cell lines are very far from any known tissue in vivo, and hence the
results of experiments done in such systems cannot take into account the potentially crucial
differences that may be present in different tissues and organs.
The factors described above each relate to the practicalities of the experimental
approaches used, but there are also genuine biological factors that may lead to divergent
results.
In considering signal transduction as a whole, it is inevitable that important factors will
exist that cannot be accounted for in a laboratory assay. The complexities of such cascades
include the possibility of multiple levels of feedback loops, cross-talk between other pathways
and temporal effects. Divergent effects upon downstream pathway function have been
observed not only for CFC-associated mutations in BRAF, but also in KRAS (87, 362).
Additionally, the crucial but complex nature of Raf protein dimerisation and interactions with
other proteins should be considered: heterodimerisation between wild-type and mutant BRAF
and heterodimerisation between BRAF and CRAF have been shown to be important (63).
Modelling these circumstances would be very difficult in any in vitro assay, emphasising the
need for model organisms or cell culture systems with germline mutations in order to explore
these complexities in more biologically relevant experimental conditions.
Cell culture techniques can be rapidly tractable in the experimental setting, with site-
directed mutagenesis being an effective means of generating a range of mutated alleles to
assess comparatively in transfection experiments. However, the limitations of this approach, as
discussed above, are considerable. Such assays may be sufficient to demonstrate an effect of

201
the mutation as evidence for pathogenicity, particularly if a result is obtained that is similar to
those recorded for variants for which the pathogenicity is well-established, but absence of an
effect upon a given assay may not exclude pathogenicity, for the reasons cited above, and such
assays, whilst achievable over a short time scale, are nonetheless too resource-intensive to be
used, for example, routinely in support of diagnostic confirmation.

202
6 INVESTIGATION OF THE CARDIAC PHENOTYPES OF MOUSE
MODELS OF THE NCFCS

203
6.1 6.1 Characterisation of the cardiac phenotype of the B-Raf
LSLV600E/+ mouse
Mouse models have been a crucial means to understanding the biology of many human
diseases. Mouse is the organism in which transgenesis is most well established, and hence is
the species in which the study of the effects of mutations in the mammalian germline have
most commonly been investigated, as the genes responsible for Mendelian disorders have
been identified (363). Additionally, mice have a short life cycle, meaning that many generations
can be bred within a relatively short time span, and animals are small enough that the costs
involved in maintenance of colonies of the appropriate size (both financial and in terms of
animal welfare) are less prohibitive than would be the case for larger animals.
In order to explore the mechanisms involved in the development of a specific feature of
CFC and other NCFCs, hypertrophic cardiomyopathy (HCM), a mouse model of CFC
syndrome, the first to be developed (180, 193), was studied. This B-Raf LSLV600E/+ mouse was
generated in the Barbacid laboratory at the Spanish National Cancer Centre (CNIO), with an
aim of studying melanoma and other BRAF-driven tumours. Animals of this model’s
genotype were generated (with an aim of future crosses to generate cell type-specific
expression of B-Raf LSLV600E, and these were not expected to exhibit a phenotype. This was due
to the presence of the ‘stop’ cassette that had been inserted 5’ to exon 15 of B-Raf , the exon
which contains the p.(Val600Glu) mutation (Figure 6-1). The presence of a stop codon within
the inserted portion of coding DNA, which also contains the hygromycin resistance cassette
(used for selection of recombinant embryonic stem cells), means that this allele should not be
expressed, and that nonsense-mediated decay of this allele’s transcript may be expected (364).
No effects of haploinsufficiency for BRAF have been reported in vivo in humans (365) or
other organisms, and therefore, in heterozygosity, this mutation would not be expected to
generate a phenotype. However, B-Raf LSLV600E/+ animals had a multisystem phenotype
including small size, unusual behaviour, seizures, and increased heart size (167). A low level of
expression of the B-Raf LSLV600E protein was confirmed by Western blotting (167) of embryos
at E13.5, and adult brain and heart samples. This was quantified at 5-10% of the level of
expression of the wild-type allele (167).
Due to the inability of these mutant mice to breed spontaneously, in vitro fertilisation had
to be used to generate mutant animals and littermate controls. To maximise genetic similarity

204
between individuals in the colony, the mutant allele had previously been backcrossed onto the
inbred C57Black6 background for six generations.
Initial characterisation of the heart of this mouse model had demonstrated increased
cardiac mass (167, 193), which was due to hyperplasia of the tissue, rather than hypertrophy of
the cells within it. This cardiomegaly was associated with functional effects, with impaired
filling of the ventricles and increased ejection fraction (analogous to a situation that may be
seen in human hypertrophic cardiomyopathy) (167). Testing of blood pressure in this mouse
model had been performed (Dr. V. Sauzeau, then in the laboratory group of Prof. X. Bustelo,
University of Salamanca), and no evidence of hypertension had been identified, but mutant
animals were too small and unstable when confronted by any stressor to be suitable for
further dynamic testing. Histological analysis revealed differences in gross morphology of the
hearts of mutant compared to wild-type animals, with increased heart mass and numbers of
cardiac myocytes per field of vision, but no increase in cell size was evident (167).

205
Figure 6-1 A: Schematic of exons 14-16 of the B-Raf LSLV600E
allele.
The exons are indicated by the numbered rectangles, and two FRT sites (that would be used to excise the stop cassette if full expression of B-Raf
V600E was desired) are shown by
the slim black isosceles triangles. The stop cassette and adjacent Hygromycin resistance cassette (used for selection of recombinant embryonic fibroblasts) are shown by STOP and Hyg respectively.
Figure 6-1 B,C: Macroscopic appearance of the B-Raf LSLV600E/+
mouse, face and profile.
Mutant and wild-type littermates are shown here at 2 months of age. Note smaller size of the mutant animals, with an altered head shape that is particularly evident in profile.

206
6.1.1 Embryonic development of the heart of the B-Raf LSLV600E/+ mouse
Bromodeoxyuridine (BrdU) incorporation is a means of studying cell proliferation. In
vitro fertilisation of a wild-type mouse of the B-Raf colony with B-Raf LSLV600E/+ sperm was used
to generate B-Raf LSLV600E/+ and B-Raf+/+ embryos. The pregnant mouse was injected with BrdU
at 12.5 days post fertilisation, to allow for this to permeate and stain the nuclei of dividing
cells. Embryos were dissected out at E13.5, genotyped and sectioned to allow for examination
of overall cardiac structure (Figure 6-2) and for evidence of hyperplasia (Figure 6-3). No
differences in cardiac size or structure were evident at this embryonic stage. Comparisons
were made of the number of BrdU positive nuclei in the interventricular septum of mutant
and wild-type hearts per field of vision (Figure 6-3), with no differences observed (n=6
embryos in each group, with 3 non-overlapping fields of vision counted for each animal)
(Figure 6-3). Given the very small amount of material available from mouse hearts at this
gestation, and the lack of a demonstrable histological phenotype, it was decided to base
further investigations of the cardiac phenotype in postnatal animals, whose larger size could
also yield more material for study.

207
Figure 6-2: Histological appearance of embryonic heart of the B-Raf LSLV600E/+
mouse and wild-type counterparts.
x 50 original magnification of interventricular septum of two B-Raf LSLV600E/+
and paired wild-type littermate control mice at E13.5, 24 hours after BrdU administration. Note similar appearances in cardiac morphology between wild-type and mutant mice, and no demonstrable structural cardiac defects. The bracket in each of the two upper images indicates the interventricular septum.

208
Figure 6-3: High power magnification of interventricular septum in the B-Raf LSLV600E/+
mouse model and wild-type counterpart.
Upper panel: x 630 original magnifications of interventricular septum of two B-Raf LSLV600E/+
and paired wild-type control mice at E13.5 are shown, 24 hours after BrdU administration. Note similar appearances in the two groups. Lower panel: quantification of number of nuclei with positive BrdU staining per field of vision in mutant and wild-type mice, confirming no observable difference in proliferation at this stage of development. Error bars represent +/- 2 standard deviations.

209
6.1.1.1 Affymetrix Mouse Genome 430A arrays in the B-Raf LSLV600E/+mouse
In order to investigate the cardiac phenotype of this mouse further, and to identify
pathways that were differentially regulated in the mutant mouse heart, that could be
contributory to the observed cardiomegaly, expression microarrays were performed on RNA
extracted from 30 µg of interventricular septal (IVS) cardiac tissue from three B-Raf LSLV600E/+
animals taken at 4 weeks of age. These were compared to a single wild-type littermate control
sample. The IVS was chosen as the site for sample acquisition as it is readily identifiable on
dissection, and the IVS is frequently seen on echocardiography to be involved in the course of
human HCM, including early in the disease course. Four week old mice were selected for this,
as this was a time point at which no overt increase in cardiac size was present: the aim was to
identify genes that might be differentially regulated early in the disease course. At later time
points, observed differences might have been more likely to reflect responses to established
cardiac pathology, rather than primary disease processes.
These arrays demonstrated high expression of transcripts of genes known to be
important in cardiac structure and function (Table 6-1), in keeping with the cardiac origin of
the tissue studied. Multiple myosins, tropomyosin, cardiac actin and troponin T were all
represented within these 20 most abundant transcripts.
Alteration in expression of many transcripts, including several with known roles in
cardiac structure and function, was also identified. A principal component analysis (PCA),
designed to be a two-dimensional representation of a distillation of the covariance of the
most altered variables between the individual samples (366), was performed by Dr Leo Zeef
(Faculty of Life Sciences, University of Manchester). The smallest number of variables which
can explain as much of the variance as possible are selected, and their variances represented as
vectors. The sum of these vectors generates a point on the graph for each sample’s results.
Distinct separation of mutant and each of the wild-type samples in this experiment was
demonstrated (Figure 6-4). PCA includes the components demonstrating the greatest variance
sequentially, and cannot therefore take into account the potential biological relevance of each
of the observations. This means that, whilst the variance between the data points is used to
separate the samples, this division may not be along the lines of greatest biological
significance. Whilst this is unavoidable, the inclusion of many different variables is designed to
reflect as much of the diversity of the samples as possible.

210
Figure 6-4: Principal component analysis of raw data from comparative B-Raf LSLV600E/+
vs wild-type expression microarray.
This plot is designed to show separation between samples with respect to gene expression, being a two-dimensional representation of variability at a large number of loci. Separation along the ‘principal component 1’ x-axis is particularly clearly demonstrated between wild-type and mutant samples.

211
Table 6-1: The 20 transcripts most highly expressed in B-Raf LSLV600E/+
mouse heart.
The most highly expressed transcripts in these samples are shown. Consistent with the
cardiac origin of the tissue, myosins, troponins, tropomyosin and titin are all seen to be
abundant in the samples tested.

212
6.1.1.2 Differential expression of transcripts in B-Raf LSLV600E/+ microarray
The microarray results were analysed to identify transcripts that were over- or under-
expressed in the mutant animals’ hearts. A total of 691 probes were identified for which the
expression of the transcript was decreased by more than 4 fold, and 716 for which it was
increased by more than 4 fold. A significant number of these probes, especially those over-
expressed in the mutant hearts, corresponded to poorly characterised loci. The 20 transcripts
with greatest negative and positive differential expression are shown in Tables 6-2 and 6-3,
and the 100 most altered in either direction are shown in appendix 7.

213
Table 6-2: The 20 transcripts with most reduced expression in B-Raf LSLV600E/+
mouse heart.
The 20 transcripts with greatest negative differential expression between the mean of the mutant and control heart are shown. The 100 most underexpressed transcripts are shown in appendix 7.

214
Table 6-3: The 20 transcripts with most increased expression in the B-Raf LSLV600E/+
mouse heart.
The 20 transcripts with greatest positive differential expression between the mean of the mutant and control heart are shown. The 100 most overexpressed transcripts are shown in appendix 7.

215
6.1.2 Pathway analysis of microarray data from the B-Raf LSLV600E/+mouse
model
In order to explore the findings of the microarrays, in silico analysis using the DAVID
package (367) was used. Differentially expressed transcripts were identified, using a fold
change of 1.5 in either direction and ‘q’ value of <0.05 to attempt to select for robust findings
and to minimise the false discovery rate. This yielded 874 transcripts for inclusion, 677 with
reduced expression and 197 with increased expression in the mutant hearts. KEGG pathway
analysis (Table 6-4) identified many pathways for which relevant transcripts had shown altered
expression in the B-Raf LSLV600E/+ mouse model. This identified plausible candidates for
biological involvement in the cardiac phenotype of this mouse model. Whether the number of
transcripts identified by the analysis was greater than would be expected on the basis of the
number of total genes that were included in the analysis, is assessed by Benjamini correction,
as shown in Table 6-4.
Table 6-4: Pathways identified by KEGG analysis of transcripts with altered expression in
the B-Raf LSLV600E/+ mouse model.
Pathway identified by KEGG analysis Number of transcripts
Benjamini correction
Circadian rhythm 5 0.14
Glycolysis / Gluconeogenesis 9 0.22
Gap junction 10 0.19
Tyrosine metabolism 6 0.40
Galactose metabolism 5 0.39
Dilated cardiomyopathy 9 0.39
Purine metabolism 12 0.47
Pathways in cancer 19 0.61
Complement and coagulation cascades 7 0.63
Focal adhesion 13 0.61
Pyruvate metabolism 5 0.59
ECM-receptor interaction 7 0.67
Glioma 6 0.65
Hypertrophic cardiomyopathy (HCM) 7 0.63

216
These multiple pathways were identified by the inclusion of the number of relevant
transcripts shown in the right hand column. For domains such as ‘pathways in cancer’, the
number of associated genes is large (n=19), suggesting multiple levels at which such a network
may be impacted at the transcriptional level. However, the denominator of total genes that fall
into the description of ‘pathways in cancer’ is also very large (323 in total), hence the
observation of a high value for the Benjamini correction, as shown in the right hand column.
Due to effects of and correction for multiple testing, a specific cut-off for significance by
Benjamini value is not defined, but the lower the value in this column, the more weight can
potentially be placed on the item identified. The large values for each of the pathways
identified in this analysis suggest that, whilst each pathway identified may be implicated in
cellular processes occurring in the tissue studied, the strongest evidence exists for differential
regulation of the circadian rhythm, glycolysis and gap junction pathways. However, the high
Benjamini values for all of the pathways identified suggested that caution would be required.
The classification and nomenclature of these pathways, as with all such in silico analysis, is
based upon, and hence reflects any bias of, the published scientific literature. However, the
finding of differential expression of elements of multiple pathways controlling processes such
as dilated cardiomyopathy, focal adhesion, gap junction and hypertrophic cardiomyopathy, all
of which may be relevant to cardiac hypertrophy pathogenesis, indicates that these and the
other pathways identified may be of true biological significance.
6.1.3 Validation of findings of microarray by quantitative fluorescent PCR
(qPCR)
The microarray data was mined to identify targets for which expression was altered,
and hence might have potential significance to the pathogenesis of myocardial disease in this
model. Quantitative fluorescent PCR (qPCR) was the technique selected to validate
differentially expressed genes identified by the microarray. Given the very high number of
transcripts with significant fold changes in the microarray, a set of criteria had to be applied to
identify those transcripts for which validation of the findings might be of most value.
Selection of targets for validation was performed according to the following criteria:
• Altered expression demonstrated between the mean of the test samples and the
control sample. Whilst small degrees of difference may be biologically significant, the

217
chance of successfully confirming any such difference was greater for transcripts that
had shown a greater magnitude of fold change.
• Appropriate ‘q’ value of <0.05 (an indicator of readout quality, analogous to the ‘p’
value but corrected for multiple testing).
• The gene should needed to have a high enough level of expression suggested by the
microarray that qPCR was likely to be feasible (transcripts with extremely low
expression levels being less likely to be successfully detected).
Transcripts meeting these criteria were considered on the basis of known or potential
biological significance. As no one pathway was particularly strongly suggested by the pathway
analysis, genes were considered on an individual basis, with those for which the greatest fold
changes had been observed being considered with the highest priority. Literature searching to
decide upon the most appropriate targets focussed on the OMIM database (www.omim.org),
as this is highly enriched for data regarding gene function in human disease processes, and the
primary literature is both well represented and easily accessible through the pages of this
database. The genes selected are shown in Table 6-5. Alongside those selected on the basis of
differential expression, Gapdh was included as the endogenous control, and Myh6 was selected
for purposes of comparison with Myh7, as the ratio between these two proteins has been
observed to be altered in hypertrophic cardiomyopathy (368). The rationale for selection of
each of the genes is described below the table.

218
Table 6-5: Transcripts selected for validation by qPCR:
Gene Symbol
Gene Title Pathway Observation
Acta1 actin, alpha 1, skeletal muscle
Smooth muscle contraction
reduced expression observed in microarray
Tuba8 tubulin, alpha 8 --- reduced expression observed in microarray
Nppb natriuretic peptide precursor type B
--- reduced expression observed in microarray
Myh7 myosin, heavy polypeptide 7, cardiac muscle, beta
Striated muscle contraction
increased expression observed in microarray
Myh6 myosin, heavy polypeptide 6, cardiac muscle, alpha
Striated muscle contraction
for comparison to Myh7
Gapdh Glyceraldehyde-3-phosphate dehydrogenase
Glycolysis and gluconeogenesis
endogenous control for all other test probes
Myosin heavy chain 7 (Myh7) is the embryonically expressed form of myosin, which
is also expressed in human HCM. Germline mutations in this gene are a common cause for
genetic HCM (369). This gene was noted to be significantly over-expressed in heart tissue
from B-Raf LSLV600E/+ mice, with a fold change of +7 (on the single available probe set). The
ratio between Myh7 and myosin heavy chain 6 (Myh6; the adult form) has been used as a
marker of hypertrophy in animal experiments into HCM (370). Myh6 was therefore also
included for comparison, as a relevant gene, the transcript of which did not appear to have
shown alteration between mutant and wild-type mice in the expression microarray. As shown
in Figure 6-5, Myh7 expression was markedly increased in all eight samples tested, with the
increase ranging from 3 to 70-fold, validating the finding of the microarray and suggesting that
this finding might be significant.
Tubulin alpha 8 (Tuba8) was seen to be significantly under-expressed in the mutant
mouse hearts, with a fold change of -6 for one probe set and -3 for the other. A homozygous
loss of function founder mutation in this gene has previously been shown in individuals with a
severe neurological developmental disorder with both structural and functional brain
anomalies (371). This protein is more highly expressed in mammalian skeletal and heart

219
muscle than in brain (372), and given the degree of down-regulation of this transcript, and its
likely relevance to human development, this transcript was therefore selected for validation by
qPCR. The consistent down-regulation observed in the samples included in the microarray
was, however, not replicated by qPCR of samples from the same region of the heart (Figure
6-6).
Nppb, encoding the B-type natriuretic peptide (BNP) hormone, was also noted to be
markedly under-expressed in the mutant mouse hearts, with a fold change of -4 (this gene
being covered by a single probe set in this array). This molecule has long been known to have
altered expression in heart failure (373), and its loss in the Nppb-/- mouse has been shown to
result in cardiac fibrosis (374), and this finding was therefore selected for validation. Again,
the findings from the array were not corroborated by this testing, with the results of qPCR
showing, if anything, a possible modest increase across the samples tested by this technique
(Figure 6-6).
Actin alpha 1 was a further transcript noted to be under-expressed in the mutant
mouse hearts, with a fold change of -9 (this gene being covered by a single probe set in this
array). This is a major component of total cellular protein, and forms the thin filaments of the
sarcomere (375). When mutated in the human germline, ACTA1 causes skeletal (nemaline)
myopathy, often of early onset (376). Whilst predominantly skeletal in expression, ACTA1 is
also expressed to a significant degree in cardiac tissue, and given the skeletal and cardiac
muscle phenotypes present in patients with CFC, this gene therefore appeared a plausible and
potentially important target to validate by qPCR. The results of this testing did not, however,
replicate the findings of the array, with no consistent pattern of reduced expression in the
samples tested (Figure 6-6).

220
Figure 6-5: Expression of Myh6 and Myh7 transcripts in the heart of the B-Raf LSLV600E/+
mouse model.
These two transcripts were measured by qPCR. Myh6, identified to have similar levels of transcript present in wild-type and mutant mice, is confirmed (blue bars) to have similar expression by qPCR, with the modest exception of mutant sample 8. Myh7, by contrast, is confirmed to have greatly increased expression at the RNA level (red bars) in all mutant samples tested, ranging from a 3- to 70- fold increase. These results suggest that this gene is indeed differentially expressed between B-Raf
LSLV600E/+ and wild-type mouse hearts at this
stage of development.

221
Figure 6-6 Expression of further targets suggested by microarray results in the B-Raf LSLV600E/+
mouse model.
Three targets, Nppb (green), Acta1 (turquoise) and Tuba8 (purple) were selected for validation by qPCR, as each had been noted to have reduced expression in the microarray. However, these findings from the microarray were not replicated for each of these transcripts in this experiment.
6.1.4 Western blotting to assess Myh7 protein concentration in the B-Raf
LSLV600E/+ mouse model
Following identification of the increased level of Myh7 transcript in the microarray,
which was successfully validated by qPCR, Western blotting was undertaken on lysed IVS
tissue to try to confirm whether an alteration could be identified at the protein level. However,
this failed to demonstrate a difference between mutant and wild-type hearts, with very similar
amounts of this protein appearing to be present in these two groups (Figure 6-7). This
apparent finding could be for several reasons. It is possible that the level of transcript was
upregulated in the mutant animals in response to a greater need for Myh7 protein, due to
increased protein turnover in the mutant than wild-type heart. A more likely explanation,

222
however, may be related to potential limitations of the reagents available for this experiment.
It was not possible to check the specificity of the Myh7 antibody, due to not having any
Myh7-/- material with which to verify this. The possibility therefore remains that the observed
result of this Western blot could be due to cross-reactivity of the antibody with other proteins
of a similar size, most likely other myosin species, specifically Myh6. Indicators in favour of
this hypothesis would be that Myh6 shares 94% sequence homology (at the nucleic acid level,
by BLAST analysis at http://genome.ucsc.edu) with Myh7, and is much more abundant in
postnatal muscle than it. Additional evidence in support of this explanation is that unaltered
expression of Myh6 at the RNA level had already been ascertained in this tissue (Figure 6-5).
Figure 6-7: Western blot for Myh7 in B-Raf LSLV600E/+
and B-Raf +/+
hearts.
ERK1/2 is shown as a control to indicate total protein content of the lysates. Note no apparent difference between samples from wild-type and B-Raf
LSLV600E/+ animals in this
experiment, which could either be due to a genuine lack of difference of Myh7 protein abundance in these samples, or a lack of specificity of the Myh7 antibody.

223
6.2 Investigation of the cardiac phenotype of the H-Ras G12V/G12V
mouse model of Costello syndrome
In order to gain further information about the pathways involved in cardiac
dysfunction in the NCFCs, tissues from further mouse models of NCFC disorders, also
generated at CNIO, were available for study. The first of these was the H-Ras G12V/G12V mouse
model of Costello syndrome (CS).
The initial characterisation of this model has previously been published (180). Whilst these
H-Ras G12V/G12V animals are of normal size, they have craniofacial differences when compared
to wild-type littermates, and develop papillomas and mammary hyperplasia. A behavioural
phenotype has also been demonstrated (377). H-Ras G12V/G12V mice develop HCM and
hypertension, in association with cardiac hypertrophy (180). As for the B-Raf LSLV600E/+ mouse
model, in the H-Ras G12V/G12V animals studied, mutant alleles had been backcrossed onto the
inbred C57Bl6 background for at least six generations, to maximise the similarity of genetic
background between wild-type and mutant littermates.
6.2.1 Affymetrix Mouse Genome 430A arrays in the H-Ras G12V/G12Vmouse
model
Microarrays were performed on RNA extracted from the IVS of three animals with the H-
Ras G12V/G12V mutation, under identical conditions to those performed on the samples from the
B-Raf LSLV600E/+ mouse model.
PCA performed for the four samples involved in this experiment showed wide
separation of the three mutant H-Ras G12V/G12V samples, one of which almost exactly overlay
the control sample. The genotype of this mutant animal was reconfirmed, to exclude mis-
genotyping as the reason for this, and its homozygous H-Ras G12V/G12V status was confirmed.

224
Figure 6-8: Principal component analysis of raw data from H-Ras G12V/G12V
expression microarrays.
Mutant samples are shown as triangles, the control as a circle. This analysis demonstrates that, for the data included in this analysis (to account for >95% of the variance between the samples, the three mutant samples are separated far apart, and one mutant animal’s sample directly overlies the control sample.
The 20 transcripts with the highest overall expression are shown in Table 6-6. As for the
B-Raf LSLV600E/+ mouse model, these were consistent with the cardiac origin of the tissue.

225
Table 6-6: The 20 most highly expressed transcripts in the IVS of
the H-Ras G12V/G12V
mouse model.

226
6.2.1.1 Differential expression of transcripts in the H-RasG12V/G12V microarray
The 20 transcripts with most reduced expression are shown in Table 6-7, and the 20 with
most increased expression are shown in Table 6-8. In the comparative microarray between H-
Ras G12V/G12V animals and a wild-type animal, there was only one dramatic difference observed.
This was that Hras itself was expressed to a much lower level in the H-Ras G12V/G12V animals.
This was therefore the key finding to validate from this experiment.
6.2.2 Quantitative fluorescent PCR (qPCR) to investigate Hras transcript
abundance
For the H-Ras G12V/G12V mouse model, the low expression of Hras in mutant animals
was the major finding from the array, and hence validation of this result was sought by qPCR.
Given the germline nature of the mutation, and that it would hence be expected to exert
effects in all body tissues, qPCR was undertaken in multiple tissues: heart, skeletal muscle,
brain and liver (Figure 6-9). H-Ras G12V/G12V mice were compared to heterozygous (H-Ras
G12V/+) and wild-type animals, to assess for dosage effects of the mutant allele. Reduced levels
of transcript were present in all tissues examined, with a dose-response effect demonstrated by
the intermediate level of transcript present in heterozygotes compared to homozygotes.
Expression of Hras in the brain appeared less dramatically reduced than was the case for other
tissues, but this finding may have been influenced by the small numbers of animals (especially
wild-type individuals) from which samples were available.

227
Table 6-7: 20 transcripts with most decreased expression in the H-Ras G12V/G12V
mouse
model

228
Table 6-8: 20 transcripts with most increased expression in the H-Ras G12V/G12V
mouse model

229
WT
H-R
as
G12V/+
H-R
as
G12V/G
12V
WT
H-R
asG
12V/+
H-R
asG12V/G
12V
WT
H-R
asG12V/+
H-R
asG12V/G
12V
WT
H-R
asG
12V/+
H-R
asG12V/G
12V
Figure 6-9: Hras expression in heart, muscle, brain and liver of the H-Ras G12V mouse model.
Bars for the three genotypes represent the mean of samples from three animals, with the error bars representing the standard error of the mean. These are compared to a single wild-type animal’s samples (WT; the single wild-type littermate available). The level of Hras transcript present is clearly reduced, with a dose-dependent effect evident for the H-Ras
G12V
allele. This effect was particularly well demonstrated in heart, skeletal muscle and liver, and less marked in brain.

230
The finding of reduced expression of the H-Ras G12V allele could help to explain why, in
this mouse model, the mutation generates, even in homozygosity, an only modestly severe
phenotype in which the mice survive a normal lifespan and achieve normal growth. This is in
contrast to this mutation in heterozygosity in the human germline, which has to date been
associated with a severe, lethal, phenotype (16). Another H-Ras mouse model with a
p.(Gly12Val) mutation demonstrates a much more severe phenotype (378). The low
expression observed in the model under investigation here is most likely to be due to an effect
intrinsic to the mutated allele’s insertion into the mouse germline, a phenomenon, whilst
poorly represented in the scientific literature, thought to be common to many knock-in mouse
models. This could be due to altered genomic architecture when the mutant allele is inserted
through misaligned homologous recombination, disruption of medium- or long-range
regulatory sequences, or an effect of, for example, the neomycin resistance cassette residually
present in the genome of this particular mouse model.
A reduced level of Hras protein, with a dose-response relationship to the H-Ras G12V
allele, in brain of this mouse model has been demonstrated in the laboratory of Prof. Y.
Elgersma (T. van der Vaart, PhD thesis in preparation, Erasmus University, Rotterdam).
Protein expression in the hippocampus of mutant homozygous mice has been measured at
40% of the level of the wild-type. This, like the results of the expression microarray and
qPCR, is consistent with a genomic reason for this model’s reduced Hras expression, though
the possibility of a negative feedback loop whereby increased Ras-MAPK pathway activity
might be affecting the level of Hras transcript (and hence also the level of the protein) cannot
currently be excluded.
Due to the very small number of transcripts showing marked differences in expression
between mutant and wild-type animals, and the much greater observed differences of Hras
expression than any other transcript, pathway analysis was not performed on the microarray
results from this individual model.

231
6.3 Investigation of the cardiac phenotype of the K-Ras V14I/+ mouse
model of CFC/NS by expression microarray
The K-Ras V14I/+ mouse recapitulates features of NS/CFC, with a particularly marked
haematological phenotype with splenomegaly and bone marrow abnormalities, reminiscent of
those seen in NS. KRAS p.(Val14Ile) has been rarely but recurrently described in the germline,
in association with clinical diagnoses of both CFC and NS (6).
This mouse model also develops cardiac hypertrophy (I. Hernandez, PhD thesis in
preparation, Autonomous University of Madrid; unpublished data). In this model, the K-Ras
V14I mutant allele was successfully backcrossed onto this background for four generations.
Further generations of backcrossing than this are associated with significant excess lethality, a
phenomenon seen in other NS models such as the PTPN11 D61G mouse (176).
6.3.1 Affymetrix Mouse Genome 430A arrays in the K-Ras V14I/+ mouse
model
Microarrays were performed on RNA extracted from the IVS of three animals with the K-
Ras V14I/+ mutation and a wild-type littermate, as described above, under identical conditions to
the those performed on the samples described above.
Principal component analysis (PCA) plots demonstrated moderately good separation
for the four samples in this experiment, with the three K-Ras V14I/+ mutant animals each
showing a distinction from the wild-type (Figure 6-10).

232
Figure 6-10: Principal component analysis of raw data from K-Ras V14I/+
expression microarrays.
Mutant samples are shown as triangles, the control as a circle. Note less clear separation than was the case for the B-Raf
LSLV600E/+ model, but better separation than was
seen for the H-Ras G12V/G12V
model.
The cardiac origin of the tissue was apparent, as for the other models tested, by the high
expression of genes encoding myosins, tropomyosin, and troponin T (Table 6-9).
6.3.1.1 Differential expression of transcripts in K-Ras V14I/+ microarray
There were many fewer marked differences in expression between K-Ras V14I/+mutant
and wild-type mouse hearts than were observed in the B-Raf LSLV600E/+ experiment, with only
one transcript reduced by more than twofold as a mean (encoding sarcolipin; but with extreme
variability observed between the three mutant samples), and only nine increased by more than
twofold. However, there were further transcripts that were altered to more subtle extents, with
appropriate ‘q’ values to suggest that the observed differences may be of significance. Tables
6-10 and 6-11 show the 20 transcripts with most decreased and most increased expression
respectively.

233
Table 6-9: 20 transcripts with highest expression in the K-Ras V14I/+ mouse model

234
Table 6-10: 20 transcripts with greatest fold decrease in expression in the K-Ras mouse
model
Table 6-11: 20 transcripts with greatest fold increase in expression in the K-Ras mouse model

235

236
6.3.2 Pathway analysis in the K-Ras V14I/+ mouse model
As the number of differentially expressed transcripts identified in the K-Ras V14I/+ mouse
model was much smaller than that observed in the B-Raf LSLV600E/+ mouse model, pathway
analysis was attempted using less stringent inclusion criteria, to maximise the number of genes
that could be included. All 386 transcripts with ‘q’ value <0.2, irrespective of observed fold
change, were therefore analysed, and the pathways identified in this data set are shown in
Table 6-12. Note, however, the high Benjamini values, suggesting that for all except
‘endocytosis’, there is little evidence of overall enrichment of these categories (but this does
not exclude the possibility of enrichment for certain subsets within the pathways listed, which
would lie beyond the scope of the DAVID analysis tool).
Table 6-12: Pathways identified by differentially expressed transcripts in the K-Ras V14I/+
expression microarray
Pathway identified by KEGG analysis Number of transcripts
Benjamini correction
Endocytosis 10 0.21
Gap junction 5 0.83
Dilated cardiomyopathy 5 0.77
GnRH signalling pathway 5 0.73
Melanogenesis 5 0.69
Wnt signalling pathway 6 0.67
MAPK signalling pathway 8 0.73
Adherens junction 4 0.72
6.3.3 Comparative analysis of the cardiac phenotype across the mouse
models of the NCFCs
6.3.3.1 Myh7 expression in the K-Ras V14I/+ and H-Ras G12V/G12V mouse models
Following the finding of significantly altered Myh7 expression in the B-Raf LSLV600E/+
microarrays, the hypothesis that this might be a feature common to these other mouse models
of the NCFCs was tested. RNA from heart tissue of K-Ras V14I/+, K-Ras+/+, H-Ras G12V/G12V and
H-Ras+/+ animals was tested by qPCR (Figure 6-11). This demonstrated no consistent pattern
of increased expression, suggesting that at the four week postnatal time point, the K-Ras V14I/+
and H-Ras G12V/G12V mouse models did not share this characteristic with the B-Raf LSLV600E/+

237
mouse model. This finding was in keeping with the results of microarrays, in which no
alteration of Myh7 expression had been identified in K-Ras V14I/+or H-Ras G12V/G12V animals.
Figure 6-11 Myh7 expression in K-Ras V14I/+
and H-RasG12V/G12V
mouse models.
Results from (left graph) K-Ras V14I/+
and (right graph) H-Ras G12V/G12V
animals are shown. Note that there is no pattern of increased Myh7 expression observed across these models. No consistent alterations are seen in the K-Ras
V14I/+ samples, and the levels of expression
seen in the H-Ras G12V/G12V
model are very consistent with that seen in the wild-type control. These results recapitulate the findings of the microarrays, that, in contrast to findings in the B-Raf
LSLV600E/+ model, Myh7 is not differentially expressed in these two models at this stage
of development.
6.3.3.2 Comparative analysis of microarray findings across the mouse models of
the NCFCs
As shown in Figure 6-12, the most highly expressed transcripts in the heart samples
from each of the models studied demonstrated an extremely similar profile. This level of
similarity was also apparent when the 50 most highly expressed transcripts were analysed, as
shown in Figure 6-12. This was as would be expected, as samples had been selected to be as
anatomically similar to one another as was possible to ascertain macroscopically. 41 of the 50
most highly expressed transcripts were common to all models, and 49 of the 50 in the B-Raf
LSLV600E/+ and K-Ras V14I/+ animals fell within the 100 most abundant transcripts identified in the
other models (Figure 6-12). Similarly, all 50 of the most highly expressed transcripts in the H-
Ras G12V/G12V model were within the most abundant 100 transcripts in the other models (the
two ‘lone’ transcripts in the B-Raf LSLV600E/+ and K-Ras V14I/+ model’s top 50’s also being found
within the top 200 of the other models).

238
Figure 6-12: Similarity of the 50 most highly expressed transcripts identified across the three mouse models.
A very high level of correlation across the models is demonstrated in this Venn diagram, which shows that 41 of the 50 most abundant transcripts in each of the mouse models were within the top 50 most abundant transcripts in the other two models. Transcripts not in the top 50 in common were each identified in the top 200 for other each other model, further emphasising the similarities within the samples analysed.
In seeking to identify common pathways that could be important to the generation of
the cardiac phenotypes of the three mouse models, differentially expressed transcripts in each
set of microarrays were examined. The differences in numbers of differentially expressed
transcripts in the different experiments were marked. The number of transcripts with low ‘q’
values, suggestive of data points of high quality, in each set of experiments is shown in Table
6-13.

239
Table 6-13: Number of transcripts with ‘q’ value below thresholds 0.05, 0.1 and 0.2 in the three sets of microarrays.
Model ‘q’ value <0.05 ‘q’ value <0.1 ‘q’ value <0.2
B-Raf LSLV600E/+ 2839 4950 8743
K-Ras V14I/+ 9 114 386
H-Ras G12V/G12V 2 2 2
The fact that Hras was, as discussed above, the only gene dramatically differently
expressed in the H-Ras G12V/G12V mouse limited the extent to which three-way analysis was
possible. No transcripts in common to all 3 models were present with a ‘q’ value of less than
0.2, (as the only one with a low ‘q’ value in the H-Ras G12V/G12V array was Hras itself). The B-Raf
LSLV600E/+ and K-Ras V14I/+ experiments were therefore examined pairwise. 24 probe sets were
identified for which the ‘q’ value was less than 0.1 in both models, which are listed in

240
Table 6-14. These transcripts were too few in number to undertake pathway analysis, but
could represent targets for which further exploration of their roles in myocardial development
and growth may be warranted.

241
Table 6-14: Genes with ‘q’ value <0.1 in B-Raf LSLV600E/+
and K-Ras V14I/+
expression microarrays
Gene Symbol Gene Title Pathway
Xrn2 5'-3' exoribonuclease 2 mRNA processing binding Reactome
--- ---
Bdh1 3-hydroxybutyrate dehydrogenase, type 1
2310045N14Rik RIKEN cDNA 2310045N14 gene
--- ---
Plxdc2 plexin domain containing 2
Kcnd2 potassium voltage-gated channel, Shal-related family, member 2
Cldnd1 claudin domain containing 1
--- ---
4931406P16Rik RIKEN cDNA 4931406P16 gene
Senp8 SUMO/sentrin specific peptidase 8
Trim24 tripartite motif-containing 24
Arglu1 arginine and glutamate rich 1
B230208H17Rik RIKEN cDNA B230208H17 gene
Nfia nuclear factor I/A
Slc35f1 solute carrier family 35, member F1
Atp6v1h ATPase, H+ transporting, lysosomal V1 subunit H
Nsd1 Nuclear receptor-binding SET-domain protein 1
--- ---
Itgb6 integrin beta 6 Integrin-medicated cell adhesion / TGF Beta Signalling Pathway
Dep1 diabetic embryopathy 1
Ggnbp2 gametogenetin binding protein 2
--- ---
Bdh1 3-hydroxybutyrate dehydrogenase, type 1

242
The number of transcripts with ‘q’ values <0.2 in common between the B-Raf LSLV600E/+
and K-Ras V14I/+ mouse models was quite small (144), but was large enough to be subjected to
KEGG pathway analysis, to try to identify common elements across the two models. This
identified only two pathways, that of the ‘Gap junction’ (with 3 genes represented) and
‘Endocytosis’ (4 genes represented). These two pathways are well known to have involvement
in cardiac muscle structure and function. Altered function of gap junction proteins is known
to be important to the pathogenesis of cardiac phenotypes including arrhythmogenic right
ventricular cardiomyopathy (379). The significance of endocytosis is widespread, due to it
being the mechanism by which many cellular processes are achieved, including the cycling of
cell surface receptors (such as receptor tyrosine kinases (380)), and further investigation of the
specific elements demonstrated to be altered in these arrays may be worthwhile.
As an alternative means to interrogate the data set, the B-Raf LSLV600E/+ data set were
selected as the primary focus for a cluster analysis, which aimed to investigate whether there
were a set of genes that were differentially expressed in the B-Raf LSLV600E/+ model that were
also more subtly altered in the other two models. Such a pattern could be suggestive of the
presence of similar pathological processes, for example, occurring at a lower intensity in the K-
Ras V14I/+ and H-Ras G12V/G12V models. Whilst very few transcripts were strikingly differentially
expressed in the H-Ras G12V/G12V mouse model, this observation did not preclude the possibility
that more subtle alterations might be present within the data set.
6.3.4 Cluster analysis
The transcripts for which significantly differential expression had been suggested in
the B-Raf LSLV600E/+ microarray experiment, by ‘q’ value <0.05, and fold change greater than 1.5
(in either direction) were identified. A cluster analysis was performed (381) to generate a heat
map of these transcripts. This resolved these genes into three principal groupings (labelled A,
B and C; Figure 6-13). A was those genes over-expressed in the B-Raf LSLV600E/+ model and
over-expressed to a lesser extent in the K-Ras V14I/+ model, but not in the H-Ras G12V/G12V
model; B was those over-expressed in the B-Raf LSLV600E/+ model, but under-expressed in the
K-Ras V14I/+ and H-Ras G12V/G12V models, and C was those under-expressed in the B-Raf
LSLV600E/+ and H-Ras G12V/G12Vmodels, but not in the K-Ras V14I/+ model. None of these
groupings were particularly indicative of a set of processes that were common to all the
models, serving more to highlight potential differences between them. In this regard, cluster B

243
was of interest, as this group of genes were exclusively overexpressed in the B-Raf LSLV600E/+
mouse, and not in the other two models. This set of transcripts could be of significance to the
cardiomyocyte hyperplasia observed solely in this mouse model. As shown in Figure 6-14, the
genes altered only in the B-Raf LSLV600E/+ mouse model included Tnnt2, encoding troponin T,
and Tpm1, encoding tropomyosin, both genes that, when mutated, cause human hypertrophic
cardiomyopathy. Of note, however, was also the presence of multiple genes with roles in
circadian rhythm management (Per1, Per2 and Per3), which may, despite all efforts to
standardise experimental conditions across the three sets of experiments, be suggestive of
altered environmental factors between wild-type and mutant mice in the B-Raf LSLV600E/+
experiment. If this were the case, then such an environmental factor could also be a
contributor to the much larger number of differentially regulated transcripts in this experiment
than those observed in the other two sets of arrays.
Figure 6-13: Cluster analysis of transcripts altered in the B-Raf LSLV600E/+
microarray.
Three groupings of transcripts (clusters) are demonstrated. A: those transcripts over-expressed in the B-Raf
LSLV600E/+ model, modestly over-expressed in the K-Ras
V14I/+ model,
and unaltered/reduced in expression in the H-Ras G12V/G12V
model; B: those transcripts over-

244
expressed in the B-Raf LSLV600E/+
model, and not in the other two models; C: the largest group, transcripts under-expressed in the B-Raf
LSLV600E/+ and H-Ras
G12V/G12V models, and
over-expressed in the K-Ras V14I/+
model.

245
Figure 6-14: Close-up representation of cluster ‘B’ (of Figure 6-13).
This shows the genes over-expressed in the B-Raf LSLV600E/+
model, and not in the K-Ras V14I/+
or H-Ras G12V/G12V
models. Red indicates higher expression in the mutant than the wild-type control, blue lower expression, with yellow indicating no difference between the two. The left hand stack refers to the B-Raf
LSLV600E/+ results, the centre to K-Ras
V14I/+ and the right
to H-Ras G12V/G12V
. Note Tnnt2, encoding troponin-T2, and Tpm1, encoding tropomyosin, are both indicated as over-expressed in the B-Raf
LSLV600E/+ model (but not the other two
models). These two genes both have key roles in cardiac sarcomere organisation, and their differential expression at this stage of development may be important to the pathogenesis of HCM in this mouse model.

246
6.4 Discussion of chapter results
In this chapter, the cardiac phenotype of three mouse models of the NCFCs has been
assessed, with an aim of identifying pathways influenced by dysregulated Ras-MAPK signal
transduction, that might be contributory to the cardiac phenotypes observed in these models.
Whilst each of the models studied has abnormalities of the heart, and increased heart size, the
results obtained suggest that significant differences may exist at the molecular level between
the samples studied from these three models. The relative lack of molecular overlaps identified
in the cardiac phenotypes of the three models could have several contributory factors. Such
inter-model variability may reflect that different molecular pathways are the key players in
pathogenesis in the different models, or that, if the same disease processes are involved, that
they may take place over a different timescale. Both the specific mutations involved and
differences in genetic background of each model may contribute to the divergent results.
6.4.1.1 Limitations of expression microarrays
The large inter-sample variability that was observed when comparing different
individual animals of the same model illustrates some of the limitations of expression
microarrays. This variability could have arisen due to a combination of reasons. The
differences may reflect genuine molecular heterogeneity between the hearts, which could
reflect that some individual animals were at different pathological stages of the same disease
course. It appears feasible that there may be significant microscopic regional variation within
individual hearts, which could have led to differences in the samples extracted. An alternative
approach to counter this could have been to use laser capture microdissection, to ensure
higher accuracy that each sample was from the same anatomical position, but the time and
heat involved in this procedure make it unlikely that a sufficient quantity and quality of RNA
could be obtained in this way. One further possibility is that differences in individual sample
storage or preparation could have affected certain parameters, though all precautions were
taken to ensure identical treatment of all samples. As mentioned above, environmental factors
in the hours or days prior to sacrifice of the animals for this experiment could also influence
the results obtained.
The strategy used, of performing arrays on three mutant animals and comparing the
mean of these results to the results of a single wild-type is well-established (ref), but can also
generate potential problems. Firstly, if the wild-type animal is atypical for that genotype, then

247
comparisons with this will be of limited value. Secondly, if some mutant animals’ hearts are
differently affected to others, then this may be masked by taking the mean of the three
samples (for this reason, individual results were examined before selecting targets for
validation, in case of any outlying results). For Acta1, where significantly reduced expression
was noted in the B-Raf LSLV600E/+ mouse model, when the expression was compared to that
observed in the two other mouse models, it appeared that it was in fact the B-Raf +/+ wild-
type control that was an outlier, with very high expression, and the mutant animals in this
experiment demonstrated similar levels to both wild-type and mutant animals in the other two
models.
A further complication when analysing disease processes through expression
microarrays is the uncertainty as to whether differentially expressed transcripts represent
drivers of primary pathological changes, or molecular responses to a primary disease process.
One means of addressing this, though the resources required would be likely to be prohibitive,
would be to perform serial arrays at different stages of mouse development, and correlate
these with contemporary histological findings. One further consideration is the possibility of
significant physiological inter-individual variability in Gapdh expression: in human
myocardium, GAPDH expression may vary sufficiently to reduce its utility as a reference
transcript in this tissue (382), and it is possible that a similar situation could also hold true for
murine heart tissue, and account for some of the inconsistencies observed between array and
qPCR findings.
6.4.1.2 Limitations of qPCR and Western blotting for validation of microarray
results
The failure to validate several of the targets identified in the microarray by qPCR limits the
conclusions that can be drawn from this data. The fold changes observed in the array were
relatively modest for many transcripts, and several of the targets were only covered by a single
probe set. Both of these factors could have made it less likely that the observed change would
be successfully validated by qPCR. As the RNA samples tested in the array were used up by
the array experiment itself, the RNA tested by qPCR was extracted from immediately
adjoining tissue, and this, as in the situation described above, could have exacerbated any
anatomical variation present in the sampling.

248
Whilst the increase in Myh7 expression that was confirmed at the RNA level could not be
confirmed at the protein level, the results of qPCR in eight samples were strongly in support
of an increased level of this transcript. This could be worth pursuing by further techniques,
but many methods of detection at the protein level rely on the existence of reliable antibodies,
which as discussed above, can be problematic for many proteins, especially where much more
abundant closely related molecules may be present in the sample. Techniques such as mass
spectrometry may be effective for the individual detection of such proteins and comparison
across different samples.
6.4.1.3 Advantages and disadvantages of in silico pathway analysis
The benefit of both the cluster analysis and pathway analysis tools used is their power
to integrate information about a large number of targets, some of which may be altered in a
very subtle manner. In this case, the cluster analysis’ utility was limited by the very uneven
number of transcripts demonstrating changes in the three models, and hence the necessity to
base the analysis upon transcripts altered in the B-Raf LSLV600E/+ model, in order to generate a
heat map. Had this analysis identified a cluster of transcripts with a similar expression profile
across the three models, this would have been a useful means of interrogating the molecular
pathology further. The lack of a consistent signature is in itself useful information, as it adds
weight to the hypothesis that different processes may be occurring in the hearts of each of
these animal models at the time point studied.
The disadvantages of pathway analysis include that, as discussed above, it cannot
necessarily identify if sub-sets of genes in a particular pathway are differentially expressed, and
hence such a pattern in the data may not be identified. In more general terms, the pathway
analysis depends upon the current understanding of biochemical pathways, which is
incomplete. There is therefore potential for misidentification of signatures of pathway
involvement that may be present.
6.4.1.4 Advantages and disadvantages of the mouse models studied
The possibility of comparing three mouse models of the NCFCs, each with a cardiac
phenotype, was a significant opportunity to seek common pathways important to cardiac
function that were affected by altered Ras-MAPK pathway signal transduction. However, the
results obtained suggest that different pathways, or different time frames, may dictate the
phenotypes observed.

249
The presence of hypertension in the H-Ras G12V/G12V mouse (and to a lesser extent, its
heterozygous H-Ras G12V/+ counterpart (180)) may be important to the pathogenesis of cardiac
hypertrophy in this model. Hypertension has not been identified in either of the other two
models to date, and treatment of the H-Ras G12V/G12V mouse with antihypertensive agents led to
improvement of the cardiac hypertrophy (180), further evidence to suggest that this might be
the case. The highlighting of differential troponin T and tropomyosin expression in the B-Raf
LSLV600E/+ model by cluster analysis, alongside the increased Myh7 expression, all three of which
are genes involved in human hypertrophic cardiomyopathy (369, 383) suggests that further
investigation of these molecules in this tissue may be worthwhile. In particular,
immunohistochemistry that could help to both quantify protein and determine its localisation
and arrangement within the cardiac tissue could provide valuable information towards the
mechanism of myocardial hypertrophy in this model.
Mouse models with knock-in mutations identified in human disease are a crucial
means of advancing scientific understanding of conditions such as cancer and Mendelian
disorders, but they are expensive both to create and to maintain. The models available in the
course of this study were those whose generation had been initiated as part of a major cancer
research programme, prior to the identification of these genes’ involvement in human
germline disorders. The potential benefit of appropriating such models is considerable,
particularly when considering very rare disorders like CFC and CS (where resources available
for research may be particularly scarce), but compromises are inevitable if the mutations
engineered into the models differ from those observed in the germline. The organism level of
phenotype of the B-Raf LSLV600E/+ mouse model (167) closely recapitulates the features of
human patients with CFC syndrome due to BRAF mutations, and the common mechanism
for generation of these features appears to be solely through the altered functioning of B-Raf,
but the stochastic relationships involved are likely to be distinct between the human situation,
where a heterozygous mutation is, as far as has yet been established, expressed in a 1:1 ratio
with a wild-type allele, and the mouse, where it is a low level of expression, through a ‘leaky’
stop cassette (167), that permits germline survival of the extremely activating B-RafV600E allele.
Similarly, the lack of a severe phenotype with presence of a heterozygous H-Ras G12V mutation
in the CS mouse model (180), a situation analogous to many mouse models, where
homozygosity for a mutation is often necessary to generate a measurable phenotype (384),
means that altered stochastic relationships of proteins within the cells of this organism will be

250
a further consideration over and above the inter-species differences that also need to be
considered. With cancer-associated mutations often having a more extreme effect upon
cellular function than mutations occurring in the germline (25), it is, however, entirely possible
that model organisms with the ‘correct’ mutations for these disorders will not demonstrate
such robust phenotypes, due to both inter-species differences and the potential complications
that may be encountered in genetic engineering of mutant alleles as discussed above. Whilst
presence of mutations in the germline of animal models that are the same as those described
in human disease is desirable from the perspective of studying intermolecular relationships,
phenotypes at the organism level for animals to be of most use as models of the human
disorders in which to assess the suitability of potential therapies.

251
7 DISCUSSION

252
7.1 Overview
The aim of this work was to delineate the clinical phenotype of individuals with CFC
syndrome, to seek the molecular basis of NCFC phenotypes in patients without a molecular
diagnosis, and to investigate how the mutations that cause these conditions might exert their
effect at the cellular level. Each of these strands has demonstrated some of the complexities
encountered in studying this group of disorders, factors which each need to be considered
when managing patients with these conditions and considering possible treatments for them
in the future. The implications of the findings of the work are discussed, with consideration of
the strengths and weaknesses of the techniques and resources used and potential further
avenues for investigation.
A large variety of mutations in BRAF and other genes have been identified in patients
with CFC syndrome (section 3.2), and variable effects of these have been observed at the
cellular level, as shown in chapter 5 and the published literature (4, 10, 77). These observations
highlight the need for consideration of mutation-specific features, both at the clinical level and
with respect to molecular interactions, and suggest that caution will be required in designing
treatments for CFC and related disorders, as the effects of any therapy may depend upon
genotype. This also underlines the need for model organisms and systems in which the effects
of mutations can be assessed comparatively prior to trials in patients, as discussed in 7.4.4.
The phenotypic variability observed in groups of patients with the same mutation,
exemplified by the p.(Ser2Gly) mutation in SHOC2 (section 3.4), emphasises the need for
study of the largest possible numbers of affected patients, and suggests the importance of
genetic and non-genetic modifiers to this phenotype. Conversely, the identification of
mutations in genes not previously clinically suspected to be responsible for individual patients’
phenotypes (as discussed in section 4.3.5) highlights the need to consider the NCFCs as a
spectrum of disorders over and above a set of individual disorders, and demonstrates the need
for comprehensive molecular testing in these patients.

253
7.2 Clinical phenotypes of the NCFCs
The work described in chapter 3 has reinforced known and drawn out novel features of
patients with CFC syndrome and those with SHOC2 p.(Ser2Gly) mutations, as discussed
below. The difficulty in making accurate clinical distinctions between each of the NCFCs is
illustrated by a number of findings of this study. When molecular testing is performed,
mutations may be identified in genes that were not clinically suspected to be the cause of a
patient’s disorder. The significant number of patients with BRAF mutations whose samples
were referred with the clinical suspicion of CS (section 3.3.1) demonstrates this. A further
example, of the finding of a pathogenic mutation in NF1 in a patient with a severe NCFC
presentation not characteristic of NF1 (section 4.3.5) is discussed below in 7.3. The existence
of a group of patients with clinical presentations convincing of a Ras-MAPK pathway disorder
who do not have a mutation identified in any of the known genes suggests that further loci
responsible for these conditions remain to be discovered.
7.2.1 CFC syndrome
Since the identification of the genes for CFC syndrome, the near-unified approach that
has prevailed for genetic testing for CFC syndrome in the UK has meant that a high
proportion of individuals identified to have this condition in this country have been tested in
the MCGM laboratory (along with many from overseas). These circumstances make this
cohort of great value in investigating this disorder. Whilst biases will exist, for example,
patients with classical presentations may be more likely to have had samples referred for
testing, and there may also be enrichment for severe phenotypes (as the need for confirmation
of a diagnosis may be more pressing in this situation), this cohort is proportionally larger and
hence more likely to be representative than those known to centres in countries where genetic
testing systems are more fragmented.
The large proportion of patients identified to have mutations in BRAF confirms this
gene’s status as the most common cause of a classical CFC phenotype. In keeping with
previously published series (10, 31, 77), extensive allelic heterogeneity was observed, with 21
of the total 48 variants being identified in only one or two patients to date (section 3.2). This
variability is sufficient to generate a significant number of situations where the conclusion is
that the variant identified is of uncertain pathogenicity. In this circumstance, parental samples

254
to assess the de novo nature of such variants are required, and if unavailable, the variant is likely
to remain unclassifiable (unless further patients with the same variant are identified).
The variability in phenotype, particularly with respect to the degree of developmental
delay, present in patients with CFC is in keeping with the published literature (4, 31), and may
range from the need for educational support in mainstream school, to special schooling for
those with severe learning disability. It is nonetheless important to bear in mind that the
cohort studied here may be enriched for severe phenotypes, compared to those ascertained
within the context of a clinical suspicion of NS.
The identification of severe and progressive contractures in multiple patients with the
MAP2K1 p.(Tyr130Cys) mutation (section 3.3.2) suggests that this is a recurrent, if not
necessarily universal, feature of the presentation in certain patients. Whether other patients
previously identified with this mutation have gone on to develop such contractures is currently
unknown. Clinicians caring for patients with this mutation should nonetheless become alert to
this possibility, ensuring that all such individuals receive appropriate input such as targeted
physiotherapy to maximise their level of function and minimise disability. Evidence for the
efficacy of specific interventions may be lacking (385), but early assessment is generally
recommended (386). Additionally, it suggests a potential avenue for further research to assess
the molecular effects of this mutation, as a tissue-specific effect of this allele may be
implicated that would be worthy of further characterisation. Whether the pathogenetic
mechanism by which these arise is the same as for the contractures seen in CS (frequently
requiring surgical treatment of the Achilles tendons (21), but also commonly observed in
proximal lower limb joints and the upper limbs (21)) could also be an important question to
address.
7.2.2 SHOC2- related phenotypes
The recent recognition of p.(Ser2Gly) mutations in SHOC2 as the cause of a NCFC
phenotype (104), and the young age of the patients reported in the literature, mean that little is
known about the longer term natural history of this disorder. The older age of patients
(median 13 y 4 m) identified with such mutations in this study, compared to those in the
published literature (104, 105), and including a significant proportion of young adult patients,
means that the phenotypes of the group identified here may provide insights into the natural
history of this recently defined disorder. The observation of autoimmune (or potentially

255
autoimmune) phenotypes such as arthritis, Crohn’s disease and idiopathic pericarditis in
individual patients within this group (section 3.4.1) is a potentially important observation. It
remains to be seen whether such phenotypes will be recapitulated in other cohorts with this
mutation, especially as they age. Multiple case reports of patients with SHOC2 mutations and
autoimmune disease, specifically systemic lupus erythematosus (196, 197), are also strongly in
support of the hypothesis that this mutation may play a key role in the development of
autoimmune phenotypes, the mechanism for which requires further molecular investigation.
The existence of reports of SLE in other patients with NS suggests that this may be a
manifestation of dysregulated Ras-MAPK pathway signalling. The patient previously reported
with childhood NS by Alanay et al (197) was found, like the patient reported by Bader-
Meunier et al (196) to have a SHOC2 mutation. Another patient with NS and SLE had no
mutation identified on testing of PTPN11 (387), and further testing was not performed (Dr S.
Lewis, personal communication), but the clinical presentation of the patient was not strikingly
reminiscent of those seen with SHOC2 mutations, suggesting that other genes might be more
likely to be responsible in this individual. It appears likely from the current available evidence
that SHOC2 p.(Ser2Gly) mutations are particularly likely to be associated with autoimmune
phenomena, and as such, this mutation could represent a monogenic model for autoimmunity,
the processes of which underlie a large group of debilitating common disorders (388). The
emerging spectrum of such late-onset complications in patients with SHOC2 mutations
emphasises the need for continued follow up of patients with this group of disorders for their
own clinical care, especially given the non-specific nature of many presenting symptoms of
autoimmune disorders, such as fatigue (389). Such follow-up would also permit the
identification of any further associated long-term sequelae across the patient group. In order
to gain the most broadly applicable data regarding the phenotypic spectrum due to SHOC2
mutation, the patients in this series have been combined with series collated by other groups
around Europe (M. Zenker, manuscript in preparation). These include over 70 patients
ascertained through testing of many hundreds of patients with a possible NS diagnosis. On
the basis of available data, SHOC2 mutations appear to produce a distinctive syndromic
presentation in some patients (‘Noonan-like syndrome with loose anagen hair’ (43, 104), as
described in chapter 1), or alternatively, a constellation of features that could be described as
intermediate between classical CFC, NS and CS presentations, as described in section 3.5.2.

256
The occurrence of bilateral phaeochromocytomas in a patient (S5) with a SHOC2
p.(Ser2Gly) mutation, as discussed in section 3.4.1, is a further illustration of the need to
consider possible pathway-wide manifestations when assessing patients with Ras-MAPK
pathway disorders. These are a known association of NF1 (390), but have not been reported
previously in other NCFCs. The need to remain alert for such features may be particularly
great in those disorders for which the molecular basis, and hence the definitive patient groups,
have only recently been clarified, and hence risks are not yet well defined. This is an
illustration of the possibility that phenotypes identified in particular NCFCs may in fact be
manifestations of Ras-MAPK pathway disorders in general. Numerous examples of this now
exist, for example the identification of rhabdomyosarcoma, common in CS, in multiple
patients with SOS1-associated NS (100), and the identification of nasolabial papillomas,
considered almost pathognomic of CS (21) in multiple patients with SHOC2 mutations
(section 3.4.1).
In characterising the clinical effects of mutations in specific genes for the NCFCs, it is
important to consider the route via which affected individuals have been ascertained. A
clinical suspicion of CS or CFC was present for the large majority of patients identified in this
study (section 3.2), and this could reflect a more severe overall phenotype being present,
which in itself could potentially place such individuals at higher risk of further complications
than those with milder presentations presenting with a clinical diagnosis of possible NS.
Nonetheless, the high prevalence of SHOC2 mutations in the cohort referred for testing of CS
and CFC genes demonstrates p.(Ser2Gly) to be a relatively common cause of a severe NCFC
phenotype. Patients with this mutation had frequently been clinically described as having CFC,
due to the presence of prominent ectodermal features. The current observation that all such
mutations, where assessed, have been de novo ((104) and section 3.4) is also good evidence of
the likelihood that SHOC2-associated phenotypes may be severe enough to have significant
impact upon reproductive success. Whether SHOC2 mutations might demonstrate positive
selection within the spermatogonial lineage, as has been demonstrated for, amongst other
mutations, HRAS substitutions of codon 12 (391), remains to be seen. If this were to be
shown, this might be one reason for the relatively high observed incidence of this mutation in
the human germline, witnessed by the substantial number of patients with this single
mutation, analogous to the situation for HRAS p.(Gly12Ser) or BRAF p.(Gln257Arg).

257
7.3 Molecular diagnosis of the NCFCs by massively parallel
sequencing
Chapter 4 demonstrates some of the potential benefits and limitations that may be
encountered in the application of massively parallel sequencing techniques to the investigation
of germline Ras-MAPK pathway disorders. These are discussed in sections 7.3.1 and 7.3.2.
The identification of further genes involved in these conditions in will improve understanding
of this pathway’s role in human health and disease, as well as providing improved diagnosis
for patients. In the medium term, whole genome sequencing is likely to become the routine
method for investigation and diagnosis of genetic disorders (392), with the same
considerations for analysis as currently apply for the use of exome sequencing in this context,
but on a grander scale. The distinction of normal human variation from pathological or high
risk alleles will be achieved with the sequencing of large numbers of individual genomes, and
further development of algorithms to assess this (393). Significant ethical and cultural issues
are posed by the application of these technologies, for example regarding the possible
identification of highly penetrant risk alleles for late-onset diseases. Algorithms for return of
results, including unexpected results, to the individuals sequenced have been developed (394).
If genome sequencing early in life were to become routine, then patients with NCFCs and
other germline disorders could be diagnosed at a very early stage of life, which could be of
particular significance if treatments were to become available. If such testing were undertaken
prenatally, as has now been reported on fetal DNA obtained from maternal plasma (395), then
prospective parents could also be in a position to decide whether or not to continue a
pregnancy in which the fetus were shown to be affected with one of these disorders.
7.3.1 Target enrichment approaches
Target enrichment sequencing of selected genes may compare favourably to traditional
sequencing approaches for genetically heterogeneous disorders, such as the NCFCs, and is
likely to remain of value until whole exome sequencing is routinely adopted for genetic testing
(354). The clinical utility of this approach was demonstrated in section 4.2.4. For patients
where there is a strong clinical suspicion of such a disorder, a comprehensive one-step
investigation may represent a significant improvement upon the previous need to embark
upon multiple rounds of genetic testing, each with a lower diagnostic yield than the last, a

258
potentially stressful process for the patient, that also may not be feasible in resource-limited
healthcare systems.
When designing panels of loci for target enrichment, the inclusion of genes for disorders
that fall within the differential diagnosis may be worth considering, as patients with clinically
overlapping phenotypes may be referred for such testing, but the true utility or otherwise of
this approach will only emerge with the testing of large numbers of patients, especially those
with less clinically characteristic phenotypes. It is also self-evident that such panels will require
frequent revision as new causative genes are identified. The strategy of including good
potential candidate genes, as tested in the target enrichment strategy described in section 4.2.1,
could also potentially be considered (witnessed by the identification of a mutation in RIT1 in a
patient in this series, before this gene’s identification as a true NS-associated gene (76)), but
this would complicate the diagnostic process, as possible mechanisms for revisiting the
primary data would need to be considered, as is the case for whole exome or genome
sequencing approaches (394). Databases to facilitate such activities across different sequencing
centres internationally could be worthwhile, but the resources involved in the generation and
maintenance of any curated database are considerable, and may not be worth the necessary
effort, unless detailed clinical phenotypic data were also included and interrogable.
The identification of a variant considered likely to be pathogenic, the c.1A>G variant in
PTK2, in a single patient in the target enrichment cohort (section 4.2.6), is an example of a
situation frequently encountered at present in massively parallel sequencing experiments,
where the biological significance of a finding in a novel gene is very difficult to confirm when
it has only been observed in a single patient, particularly if de novo status of the variant cannot
be ascertained, as was the case for this patient.
Increased knowledge of the molecular basis of a wide spectrum of genetic diseases will
add to the utility of targeted testing, for example when the target enrichment panel (section
4.2.1) was designed, the gene responsible for 17q21.1 microdeletion syndrome had not been
identified, and in fact the causative gene, KANSL1 (138), was not included in the list of genes
from this locus. A redesign of the panel would therefore include this gene, given the
phenotypic overlaps for certain patients with this syndrome with those of the Ras-MAPK
pathway disorders, specifically CFC syndrome (124), and the other genes co-deleted in the
microdeletion syndrome (that are unlikely to be causative of relevant phenotypes) need not be

259
included. Similarly, in the target enrichment panel (section 4.2.1), a set of contiguous genes
deleted in a single patient with a CFC-like phenotype were included, which included ARID1B.
Subsequently, heterozygous loss-of-function mutations in, or haploinsufficiency for, this gene
have been identified to cause Coffin-Siris syndrome (125), which may show significant
overlaps with CFC syndrome. This finding makes it likely that in this patient, the loss of
ARID1B could be the major contributor to the CFC-like phenotype, and hence the other
contiguous genes are less likely to be significant when seeking the cause for similar conditions
in other patients (though they may, of course, be major contributors to the severe phenotype
of that particular individual).
A further possibility to increase the utility of a target enrichment panel would be to
include genes which, when mutated, cause conditions that may enter the differential diagnosis
for the NCFCs. Such conditions might include Aarskog syndrome, which may frequently bear
similarities to NS (396), or other conditions characterised by combinations of short stature,
congenital heart disease or developmental delay. This approach could have particular value for
patients with intermediate phenotypes, and the confirmation of any disorder, such as Aarskog
syndrome, that shows an inheritance pattern other than autosomal dominant transmission
may have specific and distinct implications for other family members.
7.3.2 Exome sequencing approaches
Conceptually, exome trio sequencing offers an extremely powerful and elegant method for
the identification of de novo mutations. Where no family history or other factor to suggest a
specific disorder or inheritance pattern is in evidence, this may soon become a first line test
for children with developmental disorders (355). However, the very high quality of sequencing
data required in all three tested individuals is such that it represents a significant challenge at
present, and a large burden on bioinformatic and other laboratory resources, as discussed in
section 4.4. As greater numbers of individuals have their exomes sequenced, and the chemistry
and informatics pipelines for these processes improve, the yield of such testing is likely to
increase greatly. The sequencing of trio WE3 described in chapter 4 is a successful example of
this. An individual with a phenotype characteristic for a Ras-MAPK pathway disorder, but not
typical of any individual disorder within this group, was identified to have a mutation in NF1,
a well-characterised gene impacting upon the Ras-MAPK pathway. As for other patients with
severe or atypical phenotypes, the possibility of other genetic or genomic factors being

260
significant contributors to his presentation cannot be excluded. In future, greater knowledge
of such factors, coupled with the possibility of more complete exome (or genome) data for
such individuals, will allow for further investigation of genetic or genomic modifiers. In
contrast to the successful diagnosis made in patient WE3, the failure of the technology to
identify the cause for the clinical presentation of patients WE1 or WE2 is in keeping with
observations from other series, that exome sequencing may deliver the answer in around one
third of individuals assessed (354).
Whilst in the medium term, whole exome or genome sequencing may become routine in
the investigation of all genetic disease, with the examples of Baylor College of Medicine and
others’ current projects towards this (354, 397), significant barriers exist to effective adoption
of this technology in the context of routine molecular diagnosis. As costs reduce further, and
utility increases with improved understanding of more genes’ roles in pathogenesis, these
barriers will disappear.
7.4 Cellular and organism level effects of NCFC-associated mutations
There is a demonstrable need for further investigation of the cellular effects of the
mutations identified in patients with Ras-MAPK pathway disorders, to improve understanding
of the molecular basis for the phenotypes observed, and to assess pathogenicity of unclassified
variants. This may be particularly valuable for genes such as BRAF, KRAS and PTPN11,
where mutations with divergent effects in cell culture assays appear to result in similar effects
at the organism level. Revealing the mechanisms by which these may occur could enhance the
understanding of Ras-MAPK pathway function, both in these disorders and in other
situations, such as normal human development and cancer pathogenesis.
In order to investigate the cellular effects of mutations in genes responsible for CFC
syndrome, as described in chapter 5, cell culture experiments were used to compare the effects
of a panel of such mutations, and heart tissue from mouse models was used to study the
development of cardiomyopathy in the NCFCs.
7.4.1 Cell culture
The results of the work undertaken in HEK293 cells using Western blotting with
phospho-specific antibodies, in-vitro kinase and dual luciferase assays show good correlation
with one another, and with similar experiments in the published literature (10, 77, 162).

261
However (as discussed in chapter 5), there are several shortcomings to overexpression of
proteins in transient transfection experiments, and the issues of temporal and tissue specificity
remain major challenges for investigation of the molecular basis of many human disorders,
whether due to germline or somatic mutations.
For this reason, the development of a set of cell lines in which stable expression of the
different BRAF mutations could be obtained would have been a valuable tool to assess the
effects of such substitutions in a context closer to the physiological situation, as discussed in
chapter 5. Stable expression of CFC-associated mutations in a cell line of cardiomyoblast
origin (such as the H9C2 rat cardiomyoblast cell line) could have provided further insights
into the apparently divergent roles and effects of these mutations at the cellular level, but
nonetheless with major limitations, including that the mutant alleles would have been
integrated randomly into a locus within the genome of the cell line, and that the two
endogenous copies of BRAF would be presumed to both still be present. A factor in
common to many forms of cell culture would have been the very limited number of passages
for which any such cell line could be maintained, which would also limit the scope of the
experiments that could be performed in such a system.
7.4.2 Mouse models of the NCFCs
Mouse models are a key resource for furthering understanding of the effects of mutations
in vivo. Techniques to knock-in mutant alleles into the endogenous genomic locus give the
possibility of expression of such alleles under the control of the native promoter, and, in
heterozygous animals, potentially in proportion with the expression of the wild-type allele.
This is well-demonstrated by the K-Ras V14I/+ mouse, where a significant phenotype is
observed in heterozygous animals (section 6.3; I. Hernandez, thesis in preparation) due to a
mutation observed in patients with a NS/CFC phenotype (147, 398). Some complications of
this approach, however, are demonstrated by the other two mouse models compared in the
microarrays described in section 6.2.1 and 6.3.1. The B-Raf LSLV600E/+ mouse model’s mutation
would be expected not to be expressed until excision of the ‘stop’ cassette by Flp-recombinase
mediated recombination, but this allele has been shown to cause expression of the
p.(Val600Glu) mutation at low levels (167). This is distinct from the situation in human
patients with CFC, where heterozygous mutations in BRAF are, as far as is known, expressed
in a 1:1 ratio with the wild-type allele. Additionally, the p.(Val600Glu) allele has been

262
demonstrated to be embryonic lethal in heterozygosity in the mouse germline (93), and would
be expected to be so too in humans. Despite these differences, the B-Raf LSLV600E/+ mouse
demonstrates a gross phenotype very reminiscent of human CFC. It is difficult to know to
what extent histological findings in the mouse, for example, cardiomyocyte hyperplasia (167),
might have implications for human disease, principally due to the lack of samples available
from patients with CFC. This finding does appear distinct from the histology of HCM in
patients with NS, however, where the pattern appears indistinguishable from that of non-
syndromic HCM (22). The more recent generation of a mouse with the p.(Asp597Val)
mutation in BRAF (170) gives potential for study of a bona fide CFC/NS mutation, that , due
to its less extreme effects than p.(Val600Glu), has been observed in the germline in humans
(147, 398). This will be valuable in assessing the response of individuals with this mutation to
potential therapies, with direct implications for the very small patient group with this precise
substitution. This may also have relevance to other patients with CFC, including those with
other mutations in BRAF, particularly those that have been shown to have similar, modestly
activating effects in vitro to those of p.(Asp597Val) (162, 170).
As discussed in section 6.2.2, the Barbacid group’s H-Ras G12V/G12V mouse model’s utility
for modelling CS is limited by the low expression of the mutant allele. The resultant mild
phenotype is also not as convincingly reminiscent of the human condition. The H-Ras G12V/G12V
mouse generated by the Chen group has a more severe phenotype (378), more in keeping with
the severe phenotype attributable to p.(Gly12Val) mutations in the human germline (16), and
this could be further investigated. However, the extreme nature of this mutation raises a
caveat to generalisation of such findings to CS in general, where p.(Gly12Ser) is so much more
prevalent. The generation of a mouse model with this latter mutation may shortly become
available (Prof. D. Lacombe, personal communication), and the characterisation of this model
is awaited with interest.
7.4.3 Expression microarray
Cardiac molecular phenotyping by expression microarray is a technique that has previously
been used to good effect in mouse models of heart disease (370). The lack of robust candidate
targets identified in the arrays carried out across the mouse models of the NCFCs, described
in section 6.3.3, reflects some of the limitations of these techniques. The high sensitivity of an
expression microarray renders it at risk of identifying variation that is exquisitely time and

263
location specific, possibly beyond the resolution of the technique used for tissue sampling.
The expense of running a microarray experiment means that it is necessary to consider the
samples for inclusion very carefully. Very high RNA quality is required, and hence retrieval of
samples and placement in safe storage needs to be undertaken as fast as possible. Expression
microarrays quantitate RNA transcripts in cell lysates, and hence cannot take into account the
many physiologically important differences between samples that arise at levels other than that
of overall concentrations of mRNAs within a sample. Myh7 transcript was seen to be
significantly elevated in the B-Raf LSLV600E/+ mouse model, but no increase at the protein level
was identified by Western blotting of this tissue. As discussed in chapter 6, the lack of
observed differential expression at the protein level may relate to failure of the available
antibody to detect Myh7 specifically, or the probe set that detected altered expression may
relate to a minor transcript rather than the predominant product of this gene. At present, there
is insufficient data regarding this to be able to assess this further (399). Alternatively, one or
more processes of post-transcriptional regulation may be implicated in this observed disparity.
Cellular levels of mRNA and protein have frequently, when compared, been identified not to
be closely correlated. The higher level of Myh7 mRNA may not be correctly localised for
translation to occur, for example if processes necessary to generate mature mRNA, such as
RNA editing, or nuclear export, were affected. Antisense transcripts have recently been
described to influence gene expression at the Myh6/Myh7 locus (368), and these, or other as
yet unidentified factors, may play a role in the observed differences between the hearts of
wild-type and mutant animals.
7.4.4 Novel means of modelling the NCFCs
New, more elegant and potentially less costly means of generating knock-in animal models
such as nuclease-mediated genome editing, for example using TALENs (400), may permit
creation of multiple animal models at once, and therefore suggest realistic possibilities for in
vivo or ex vivo comparative analysis of genetically and allelically heterogeneous conditions such
as CFC and NS. Such comprehensive assessment of these disorders has previously been
hampered by the often prohibitively large resources required for individual model organism
generation. Similarly, greater opportunities for mutagenesis at intrinsic loci mean that the
phenotypes of the resultant models may more closely reflect the effects of the spontaneous
mutations that cause human disease. However, whilst such techniques may render the
generation of better models in shorter time-frames, the high costs associated with

264
maintenance of any animal model, particularly any mammalian species, will still be a significant
barrier to such projects in many settings. One further technique that could be worthy of
consideration in such projects is the generation of primary cultures of tissues from animals
bearing mutations relevant to human disease. Such techniques have been developed for
several organ systems, the resulting cultures being valuable to study live cells in a tissue-
relevant context, such as from heart (401) or brain (402).
A promising avenue already being successfully used by other groups is the creation of
patient-derived induced pluripotent stem cells (iPSCs), allowing various cell types to be
generated. Whilst there may also be limitations to this technique, ongoing refinements to
reprogramming technologies have further potential to increase the similarities between iPSCs
and native tissues, and yield further insights into pathogenesis at the molecular and cellular
levels (for example, as has been demonstrated in cardiomyocytes derived from patients with
NSML (183)). For disorders where many different organ systems may be involved (and rare
disorders where biological specimens may be particularly rarely available), the possibility of
generating cells to model many aspects of an individual’s phenotype is an exciting prospect.
Techniques for culturing cell types derived from iPSCs in conditions that are closer to the in
vivo context are also being developed (403), giving the potential to create more accurate
models of human disease.
7.5 Review of techniques used and possible alternatives
The group of patients with NCFC disorders who were studied in this work (chapters 3
and 4) has provided much valuable information, but also has potential to provide further
insights into these conditions. Since the inception of the study, increased numbers of patients
have been referred for diagnostic testing, and hence much could be gained by further clinical
and molecular assessment of this cohort. The presence of patients with a longstanding
diagnosis who have been ascertained retrospectively is of value for investigating the natural
history of the NCFCs, whilst the inclusion of patients who are presenting newly, often in
infancy, gives the potential for a prospective study of these disorders.
Molecular diagnosis of these disorders by Sanger sequencing still represents the gold
standard for confirmation of mutations (404). However, the possibility of a unified diagnostic
test across the NCFCs by target enrichment of the genes for these disorders would be of great
value, and is now possible, as shown in 4.2.4. The next step would be to evaluate the

265
effectiveness of this approach in clinical molecular diagnostics. If it were shown to be
effective, this would mean that a single stage test could be offered that covered the NCFCs,
and as discussed in 4.4, other genetic conditions within the differential diagnosis of these
disorders. Such a test would represent a significant advance, particularly as such platforms can
be updated iteratively as new causative genes are identified. In terms of a future research
strategy, to test patients without a clinical NCFC diagnosis by target enrichment would be a
valuable first step, with the possibility of proceeding to exome sequencing of patient and
parents (if unaffected) if no causative mutation was identified in the target enrichment
experiment. For those patients whose samples were the subject of testing described in chapter
4, but in whom no diagnosis was made, repeating the testing now that much higher percentage
coverage across the exons of interest can be achieved (over 95% or 98% of bases in a targeted
enrichment can be expected to be covered at 30x or greater depth in runs now performed in
the MCGM laboratory (Dr J. O’Sullivan, personal communication), as compared to the 70%
achieved in the experiment reported in section 4.2.3).
Whilst molecular diagnosis across the NCFCs is now much more readily achievable if the
techniques above can be used, methods for assessment of functional effects of mutations
remain more laborious. Transient transfections of cell lines to over express candidate variants
(section 5.2) can provide limited data, but, as discussed in section 5.4, further development of
such techniques appears unlikely to yield significant information of relevance to patients’
phenotypes. Mouse models with mutations in Ras-MAPK pathway genes can provide valuable
information about the biology of the NCFCs. When considering the use of these for trials of
potential treatments prior to first testing of these in humans, it would appear important to use,
if possible, animals with the same mutations that have been described in the germline of
patients, as discussed in section 6.4.1.4, provided that this mutation causes a murine
phenotype that is measurable and hence appropriate for the assessment of treatment effects.
For further evaluation of the biology of the NCFCs at the cellular and organ level, the newly
available mouse models discussed above may be of value. Those with the same mutations
known to cause germline human disease may be of particular relevance for assessing the
potential effects of therapies modulating Ras-MAPK pathway activity, such as MEK inhibitors
(405).

266
7.6 Genetic / Genomic Medicine and the NCFCsThe role of clinical genetic services is changing, and the NCFCs provide a case in point for
how this may occur, as molecular diagnostic testing becomes available to non-genetic medical
specialties (‘mainstreaming’ (406)). Molecular testing at an earlier stage, as discussed in section
4.4, may greatly reduce the number of steps necessary to achieve a definitive diagnosis,
improving patient care as a result. The skills of traditional dysmorphological assessment will
therefore be of less significance for diagnosis, and the specific indications for genetic
consultation will alter as the proportion of patients attending with a confirmed diagnosis
increases. Clinical dysmorphology skills may still be of particular relevance, however, in the
setting of unclassified genetic or genomic variants being identified. In this situation, any
features of the conditions associated with the gene or genes in which a change has been
identified need to be sought and evaluated, as a complementary tool to the data mining and
bioinformatic prediction of variants’ pathogenicity. Genetic counselling remains a core skill
which is distinct from those practised in other areas of medicine, and this will still be required
by families in which genetic diagnoses are made. The high take-up of prenatal diagnosis for
the low recurrence risk of CFC or CS in families with an affected child (chapter 3) is an
example of this.
Detailed, current and accurate information about rare disorders is increasingly available on
the internet (407) and hence may be accessible both to patients and all doctors caring for
affected individuals and families. However, the value which patients and families place on a
face-to-face meeting with an expert (408) is such that such consultations will, irrespective of
the quality and accessibility of the information available online, also continue to be a major
part of the practice of clinical specialists, including those in genetic / genomic medicine.
Management of genetic disorders affecting one or a few body systems may be achieved
effectively by specialists in the most relevant discipline, for example, nephrologists for genetic
renal cystic disease, or neurologists for genetic neuropathies. The situation for disorders such
as the NCFCs, however, is more complex as most patients will have manifestations in multiple
organ systems (17), and no one specialist will necessarily be in a good position to manage each
of these. For affected children, paediatric services may be well-configured to manage such
complex presentations, but for adults, frequently no co-ordinating specialist is available, and
this may be especially problematic when the condition is associated with intellectual disability,
as is the case for the NCFCs. Specialists in genetic / genomic medicine may have an

267
important role to play in ensuring optimal management of such multisystem genetic disorders,
and where a group of such conditions are recognised to have a common set of clinical features
and shared pathological mechanisms, a particularly strong case may be made for a dedicated
clinic for these patients (409).
As discussed in chapter 1, and illustrated in chapters 3 and 4, the NCFCs represent a
spectrum of clinical presentations, and a proportion of affected patients have phenotypes that
do not fit neatly into one of the named disorders, or their phenotype is atypical for what has
been identified in association with their genotype. For these patients in particular, the use of
an overarching nomenclature may be helpful. ‘The neuro-cardio-facio-cutaneous syndromes’
or ‘NCFCs’ are helpful descriptions from a clinical perspective, but are terms that are hard to
articulate, and hence are not readily useable, especially by patients and families. The term
‘rasopathies’ has also been coined for this group of disorders, and this is a memorable
description that effectively indicates the dysregulation of Ras-MAPK pathway signal
transduction in many of these patients. However, there is no current evidence for
dysregulation of Ras per se in patients whose mutation affects a gene encoding a protein
downstream of Ras, (such as MAP2K1, for example) and so it is not an ideal description for
this group of disorders. Effective descriptions for individual patients may include specific
genotypic information, and whilst this may not be automatically thought useful for families, it
is notable that in correspondence amongst the Costello syndrome family community, parents
frequently identify themselves, for example, as ‘mother/father of [child’s name] HRAS G12S’.
As trials start to recruit patients, as is the case for the clinical trial of rapamycin for NS-
associated HCM in the USA (410), genotypic information will become crucial to families and
doctors managing this patient group, and this may drive patients’ self-identification and how
their diagnoses are described. An appropriate description of a phenotype may permit
appropriate health, educational and social service input to be obtained, and as such is a key
issue. The description of ‘Noonan-like syndrome with loose anagen hair’ is a case in point: this
label may be entirely inappropriate for the child with significant learning disability, congenital
heart disease, severe eczema and food allergies in whom a p.(Ser2Gly) mutation in SHOC2 is
identified. Patients with a clinical diagnosis of CFC in whom such a mutation is identified are
likely to be best served by the retention of this clinical description for themselves, as
presenting the best fit for their phenotype.

268
7.7 ConclusionsIn summary, for effective investigation and care of patients with NCFCs, several factors
are necessary. Firstly, good clinical phenotyping is a prerequisite to define the natural history
of these disorders and to provide the focus for, and identify any caveats regarding, future
treatments. Suitable endpoints for clinical trials also require definition, and will draw on
information from detailed phenotyping exercises. Comprehensive molecular diagnosis is
necessary to define patient cohorts, identify all responsible genes for these disorders and
permit assessments of genotype-phenotype correlation. Clinical and molecular definition are
both hence prerequisites to the identification of appropriate goals for future treatments. Good
cellular and organism-level models are necessary to dissect the molecular basis of these
conditions and provide initial evidence for effects of any treatments under consideration. In all
of these domains, collaboration between research groups will be important: every known
patient’s phenotype can be important to the understanding of the condition, and hence
ascertainment and co-operation on an international scale are required (6). The emergence of
mutation-specific phenotypes in the patients studied underlines the need for such
collaborative approaches, to ensure that each such association may be identified and explored.
This study has highlighted some of the challenges that this group of disorders present, not
least in classification and nomenclature (19). The clarification of the molecular bases and
clinical phenotypic spectra of this group of disorders will assist in this effort.

269
8 REFERENCES

270
1. Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene
is homologue of Harvey sarcoma virus ras gene. Nature, 1982;297:474-8.
2. Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder
carcinoma oncogene is an activated form of the normal human homologue of BALB- and
Harvey- MSV transforming genes. Nature, 1982;298:343-7.
3. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines
for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet,
2007;44:81-8.
4. Allanson JE, Anneren G, Aoki Y, Armour CM, Bondeson ML, Cave H, et al. Cardio-
facio-cutaneous syndrome: does genotype predict phenotype? Am J Med Genet C Semin Med
Genet, 2011;157:129-35.
5. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders.
Best Pract Res Clin Endocrinol Metab, 2011;25:161-79.
6. NSEuronet. Available from: www.nseuronet.com.
7. van der Vaart T, Plasschaert E, Rietman AB, Renard M, Oostenbrink R, Vogels A, et
al. Simvastatin for cognitive deficits and behavioural problems in patients with
neurofibromatosis type 1 (NF1-SIMCODA): a randomised, placebo-controlled trial. Lancet
Neurol, 2013;12:1076-83.
8. Messiaen L, Yao S, Brems H, Callens T, Sathienkijkanchai A, Denayer E, et al. Clinical
and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA, 2009;302:2111-8.
9. Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, et al. Germline
loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat
Genet, 2007;39:1120-6.
10. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al.
Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous
syndrome. Science, 2006;311:1287-90.
11. Huson S. Neurofibromatosis: emerging phenotypes, mechanisms and management.
Clin Med, 2008;8:611-7.
12. van der Burgt I. Noonan syndrome. Orphanet J Rare Dis, 2007;2:4.
13. Nora JJ, Nora AH, Sinha AK, Spangler RD, Lubs HA. The Ullrich-Noonan syndrome
(Turner phenotype). Am J Dis Child, 1974;127:48-55.
14. Roberts AE. The clinical phenotype of cardiofaciocutaneous syndrome (CFC).
Noonan syndrome and related disorders. Basel: Karger; 2009. p. 66-72.

271
15. Giannoulatou E, McVean G, Taylor IB, McGowan SJ, Maher GJ, Iqbal Z, et al.
Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS
mutations in the paternal germline. PNAS, 2013;in press.
16. Burkitt-Wright EM, Bradley L, Shorto J, McConnell VP, Gannon C, Firth HV, et al.
Neonatal lethal Costello syndrome and unusual dinucleotide deletion/insertion mutations in
HRAS predicting p.Gly12Val. Am J Med Genet A, 2012;158A:1102-10.
17. Wright EM, Kerr B. RAS-MAPK pathway disorders: important causes of congenital
heart disease, feeding difficulties, developmental delay and short stature. Arch Dis Child,
2010;95:724-30.
18. Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, et al. The
cardiofaciocutaneous syndrome. J Med Genet, 2006;43:833-42.
19. Kerr B, Allanson J, Delrue MA, Gripp KW, Lacombe D, Lin AE, et al. The diagnosis
of Costello syndrome: nomenclature in Ras/MAPK pathway disorders. Am J Med Genet A,
2008;146A:1218-20.
20. Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, et al. Genotype-
phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med
Genet, 2006;43:401-5.
21. Kerr B. The clinical phenotype of Costello syndrome. In: Zenker M, editor. Noonan
syndrome and related disorders. Basel: Karger; 2009. p. 83-93.
22. Allanson J. The Clinical Phenotype of Noonan Syndrome. In: Zenker M, editor.
Noonan Syndrome and Related Disorders. 1st ed. Basel: Karger; 2009.
23. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis,
2008;3:13.
24. Digilio MC. The Heart in Ras-MAPK Pathway Disorders. In: Zenker M, editor.
Noonan Syndrome and Related Disorders. 1st ed. Basel: Karger; 2009. p. 109-18.
25. Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular
pathogenesis. Mol Syndromol, 2010;1:2-26.
26. Ben-Shachar S, Constantini S, Hallevi H, Sach EK, Upadhyaya M, Evans GD, et al.
Increased rate of missense/in-frame mutations in individuals with NF1-related pulmonary
stenosis: a novel genotype-phenotype correlation. Eur J Hum Genet,
2012;10.1038/ejhg.2012.221.
27. Tartaglia M, Niemeyer CM, Shannon KM, Loh ML. SHP-2 and myeloid malignancies.
Curr Opin Hematol, 2004;11:44-50.

272
28. Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan,
Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med
Genet, 2011;157:83-9.
29. Narod SA, Stiller C, Lenoir GM. An estimate of the heritable fraction of childhood
cancer. Br J Cancer, 1991;63:993-9.
30. Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach G, et al.
Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello
syndrome. Clin Genet, 2008;73:62-70.
31. Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome:
clinical features of 38 individuals with proven mutations. J Med Genet, 2008;45:249-54.
32. Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, et al. New
multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous
involvement--the CFC syndrome. Am J Med Genet, 1986;25:413-27.
33. Aizaki K, Sugai K, Saito Y, Nakagawa E, Sasaki M, Aoki Y, et al. Cardio-facio-
cutaneous syndrome with infantile spasms and delayed myelination. Brain Dev, 2011;33:166-9.
34. Siegel DH, McKenzie J, Frieden IJ, Rauen KA. Dermatological findings in 61
mutation-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol,
2011;164:521-9.
35. Al-Rahawan MM, Chute DJ, Sol-Church K, Gripp KW, Stabley DL, McDaniel NL, et
al. Hepatoblastoma and heart transplantation in a patient with cardio-facio-cutaneous
syndrome. Am J Med Genet A, 2007;143A:1481-8.
36. Makita Y, Narumi Y, Yoshida M, Niihori T, Kure S, Fujieda K, et al. Leukemia in
Cardio-facio-cutaneous (CFC) syndrome: a patient with a germline mutation in BRAF proto-
oncogene. J Pediatr Hematol Oncol, 2007;29:287-90.
37. Tidyman WER, K.A. Molecular Causes of the Cardio-Facio-Cutaneous Syndrome. In:
Zenker M, editor. Noonan Syndrome and Related Disorders. Basel: Karger; 2009. p. 73-82.
38. Kavamura MI, Peres CA, Alchorne MM, Brunoni D. CFC index for the diagnosis of
cardiofaciocutaneous syndrome. Am J Med Genet, 2002;112:12-6.
39. Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, et al. Germline
BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular
diversity and associated phenotypic spectrum. Hum Mutat, 2009;30:695-702.
40. Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of Noonan
syndrome. Arch Dis Child, 1992;67:178-83.

273
41. Fukuda M, Horibe K, Miyajima Y, Matsumoto K, Nagashima M. Spontaneous
remission of juvenile chronic myelomonocytic leukemia in an infant with Noonan syndrome. J
Pediatr Hematol Oncol, 1997;19:177-9.
42. Lee DA, Portnoy S, Hill P, Gillberg C, Patton MA. Psychological profile of children
with Noonan syndrome. Dev Med Child Neurol, 2005;47:35-8.
43. Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, Forabosco A.
Noonan-like syndrome with loose anagen hair: a new syndrome? Am J Med Genet A,
2003;118A:279-86.
44. Baraitser M, Patton MA. A Noonan-like short stature syndrome with sparse hair. J
Med Genet, 1986;23:161-4.
45. White SM, Graham JM, Jr., Kerr B, Gripp K, Weksberg R, Cytrynbaum C, et al. The
adult phenotype in Costello syndrome. Am J Med Genet A, 2005;136:128-35.
46. Delrue MA, Chateil JF, Arveiler B, Lacombe D. Costello syndrome and neurological
abnormalities. Am J Med Genet A, 2003;123A:301-5.
47. Lin AE, Grossfeld PD, Hamilton RM, Smoot L, Gripp KW, Proud V, et al. Further
delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet, 2002;111:115-29.
48. Gripp KW, Lin AE, Nicholson L, Allen W, Cramer A, Jones KL, et al. Further
delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps
differentiate cardio-facio-cutaneous syndrome from Costello syndrome. Am J Med Genet A,
2007;143A:1472-80.
49. COSMIC database [database on the Internet].
50. Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, et al. Similarity of the
phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia.
Cancer Res, 2002;62:6451-5.
51. Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS
mutations in human lung cancer and melanoma. Cancer Res, 2002;62:6997-7000.
52. Janssen JW, Steenvoorden AC, Lyons J, Anger B, Bohlke JU, Bos JL, et al. RAS gene
mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders,
and myelodysplastic syndromes. Proc Natl Acad Sci U S A, 1987;84:9228-32.
53. Visvanathan KV, Pocock RD, Summerhayes IC. Preferential and novel activation of
H-ras in human bladder carcinomas. Oncogene Res, 1988;3:77-86.
54. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the
BRAF gene in human cancer. Nature, 2002;417:949-54.

274
55. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and
cancer. Nat Rev Cancer, 2007;7:295-308.
56. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of
malignant neoplasms, in people with neurofibromatosis: population-based record-linkage
study. Br J Cancer, 2012;10.1038/bjc.2012.535.
57. Mautner VF, Kluwe L, Friedrich RE, Roehl AC, Bammert S, Hogel J, et al. Clinical
characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb
type-1 NF1 deletions. J Med Genet, 2010;47:623-30.
58. Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation
status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res,
2006;66:3992-5.
59. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al.
Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J
Med, 2011;364:2507-16.
60. Lievre A, Blons H, Laurent-Puig P. Oncogenic mutations as predictive factors in
colorectal cancer. Oncogene, 2010;29:3033-43.
61. Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, Wigler M. Analysis of the
transforming potential of the human H-ras gene by random mutagenesis. Proc Natl Acad Sci
U S A, 1984;81:4008-12.
62. Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline
mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet, 2005;37:1038-40.
63. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate
RAF dimers and ERK signalling in cells with wild-type BRAF. Nature, 2010;464:427-30.
64. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al.
Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF.
Cell, 2010;140:209-21.
65. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al.
RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth.
Nature, 2010;464:431-5.
66. Cichowski K, Janne PA. Drug discovery: inhibitors that activate. Nature,
2010;464:358-9.
67. Estep AL, Palmer C, McCormick F, Rauen KA. Mutation analysis of BRAF, MEK1
and MEK2 in 15 ovarian cancer cell lines: implications for therapy. PLoS One, 2007;2:e1279.

275
68. Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, et al. Identification of the
MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK
inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res, 2011;71:5535-45.
69. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome
sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat
Genet, 2012;44:133-9.
70. Fryer AE, Holt PJ, Hughes HE. The cardio-facio-cutaneous (CFC) syndrome and
Noonan syndrome: are they the same? Am J Med Genet, 1991;38:548-51.
71. Baralle D, Mattocks C, Kalidas K, Elmslie F, Whittaker J, Lees M, et al. Different
mutations in the NF1 gene are associated with Neurofibromatosis-Noonan syndrome
(NFNS). Am J Med Genet A, 2003;119A:1-8.
72. De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, et al. NF1 gene
mutations represent the major molecular event underlying neurofibromatosis-Noonan
syndrome. Am J Hum Genet, 2005;77:1092-101.
73. Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The
neurofibromatosis type 1 gene encodes a protein related to GAP. Cell, 1990;62:599-608.
74. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al.
Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan
syndrome. Nat Genet, 2001;29:465-8.
75. Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase
pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med,
2012;14:285-92.
76. Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, et al. Gain-of-
Function Mutations in RIT1 Cause Noonan Syndrome, a RAS/MAPK Pathway Syndrome.
Am J Hum Genet, 2013;93:173-80.
77. Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline KRAS and
BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet, 2006;38:294-6.
78. Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, et al. PTPN11
mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and
phenotypic heterogeneity. Am J Hum Genet, 2002;70:1555-63.
79. Ion A, Tartaglia M, Song X, Kalidas K, van der Burgt I, Shaw AC, et al. Absence of
PTPN11 mutations in 28 cases of cardiofaciocutaneous (CFC) syndrome. Hum Genet,
2002;111:421-7.

276
80. Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW. Paternal bias in
parental origin of HRAS mutations in Costello syndrome. Hum Mutat, 2006;27:736-41.
81. Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial
selection: causes and consequences for human disease. Am J Hum Genet, 2012;90:175-200.
82. Gripp KW, Hopkins E, Sol-Church K, Stabley DL, Axelrad ME, Doyle D, et al.
Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am J Med
Genet A, 2011;155A:706-16.
83. Lo IF, Brewer C, Shannon N, Shorto J, Tang B, Black G, et al. Severe neonatal
manifestations of Costello syndrome. J Med Genet, 2008;45:167-71.
84. Gripp KW, Innes AM, Axelrad ME, Gillan TL, Parboosingh JS, Davies C, et al.
Costello syndrome associated with novel germline HRAS mutations: an attenuated
phenotype? Am J Med Genet A, 2008;146A:683-90.
85. Schubbert S, Bollag G, Lyubynska N, Nguyen H, Kratz CP, Zenker M, et al.
Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol,
2007;27:7765-70.
86. Brasil AS, Malaquias AC, Kim CA, Krieger JE, Jorge AA, Pereira AC, et al. KRAS
gene mutations in Noonan syndrome familial cases cluster in the vicinity of the switch II
region of the G-domain: report of another family with metopic craniosynostosis. Am J Med
Genet A, 2012;158A:1178-84.
87. Gremer L, Merbitz-Zahradnik T, Dvorsky R, Cirstea IC, Kratz CP, Zenker M, et al.
Germline KRAS mutations cause aberrant biochemical and physical properties leading to
developmental disorders. Hum Mutat, 2011;32:33-43.
88. Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, et al. Cardio-facio-
cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway:
genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet,
2007;44:763-71.
89. Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, et al. A restricted
spectrum of NRAS mutations causes Noonan syndrome. Nat Genet, 2010;42:27-9.
90. Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP, et al. NRAS mutation
causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A,
2007;104:8953-8.

277
91. De Filippi P, Zecca M, Lisini D, Rosti V, Cagioni C, Carlo-Stella C, et al. Germ-line
mutation of the NRAS gene may be responsible for the development of juvenile
myelomonocytic leukaemia. Br J Haematol, 2009;147:706-9.
92. Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of
Ras/MAPK pathway dysregulation. Curr Opin Genet Dev, 2009;19:230-6.
93. Mercer K, Giblett S, Green S, Lloyd D, DaRocha Dias S, Plumb M, et al. Expression
of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in
mice and transformation of primary fibroblasts. Cancer Res, 2005;65:11493-500.
94. Champion KJ, Bunag C, Estep AL, Jones JR, Bolt CH, Rogers RC, et al. Germline
mutation in BRAF codon 600 is compatible with human development: de novo p.V600G
mutation identified in a patient with CFC syndrome. Clin Genet, 2011;79:468-74.
95. Rauen KA, Tidyman WE, Estep AL, Sampath S, Peltier HM, Bale SJ, et al. Molecular
and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome:
transmission through four generations. Am J Med Genet A, 2010;152A:807-14.
96. Linden HC, Price SM. Cardiofaciocutaneous syndrome in a mother and two sons with
a MEK2 mutation. Clin Dysmorphol, 2011;20:86-8.
97. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic
mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and
acute myeloid leukemia. Nat Genet, 2003;34:148-50.
98. Tartaglia M, Gelb BD. Molecular Genetics of Noonan Syndrome. In: Zenker M,
editor. Noonan syndrome and related disorders. first ed. Basel: Karger; 2009. p. 20-39.
99. Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, et al. Gain-of-
function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet,
2007;39:75-9.
100. Hastings R, Newbury-Ecob R, Ng A, Taylor R. A further patient with Noonan
syndrome due to a SOS1 mutation and rhabdomyosarcoma. Genes Chromosomes Cancer,
2010;49:967-8.
101. Kerr B, Eden OB, Dandamudi R, Shannon N, Quarrell O, Emmerson A, et al.
Costello syndrome: two cases with embryonal rhabdomyosarcoma. J Med Genet,
1998;35:1036-9.
102. Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F, et al.
Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan
syndrome-like phenotype. Am J Hum Genet, 2010;87:250-7.

278
103. Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, et al.
Germline CBL mutations cause developmental abnormalities and predispose to juvenile
myelomonocytic leukemia. Nat Genet, 2010;42:794-800.
104. Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V,
et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-
like syndrome with loose anagen hair. Nat Genet, 2009;41:1022-6.
105. Komatsuzaki S, Aoki Y, Niihori T, Okamoto N, Hennekam RC, Hopman S, et
al. Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic
malignancies. J Hum Genet, 2010;55:801-9.
106. Upadhyaya M, Maynard J, Osborn M, Huson SM, Ponder M, Ponder BA, et al.
Characterisation of germline mutations in the neurofibromatosis type 1 (NF1) gene. J Med
Genet, 1995;32:706-10.
107. Sawada S, Florell S, Purandare SM, Ota M, Stephens K, Viskochil D.
Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat Genet,
1996;14:110-2.
108. Xu W, Mulligan LM, Ponder MA, Liu L, Smith BA, Mathew CG, et al. Loss of
NF1 alleles in phaeochromocytomas from patients with type I neurofibromatosis. Genes
Chromosomes Cancer, 1992;4:337-42.
109. Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, et al.
A mouse model for the learning and memory deficits associated with neurofibromatosis type
I. Nat Genet, 1997;15:281-4.
110. Graham JM, Jr., Kramer N, Bejjani BA, Thiel CT, Carta C, Neri G, et al.
Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med
Genet A, 2009;149A:2122-8.
111. Luo C, Yang YF, Yin BL, Chen JL, Huang C, Zhang WZ, et al.
Microduplication of 3p25.2 encompassing RAF1 associated with congenital heart disease
suggestive of Noonan syndrome. Am J Med Genet A, 2012;158A:1918-23.
112. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF
gene duplication constitutes a mechanism of MAPK pathway activation in low-grade
astrocytomas. J Clin Invest, 2008;118:1739-49.
113. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et
al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority
of pilocytic astrocytomas. Cancer Res, 2008;68:8673-7.

279
114. Becker K, Hughes H, Howard K, Armstrong M, Roberts D, Lazda EJ, et al.
Early fetal death associated with compound heterozygosity for Noonan syndrome-causative
PTPN11 mutations. Am J Med Genet A, 2007;143A:1249-52.
115. Ekvall S, Hagenas L, Allanson J, Anneren G, Bondeson ML. Co-occurring
SHOC2 and PTPN11 mutations in a patient with severe/complex Noonan syndrome-like
phenotype. Am J Med Genet A, 2011;155A:1217-24.
116. Fahrner JA, Frazier A, Bachir S, Walsh MF, Applegate CD, Thompson R, et
al. A rasopathy phenotype with severe congenital hypertrophic obstructive cardiomyopathy
associated with a PTPN11 mutation and a novel variant in SOS1. Am J Med Genet A,
2012;158A:1414-21.
117. Pasmant E, Amiel J, Rodriguez D, Vidaud M, Vidaud D, Parfait B. Two
independent de novo mutations as a cause for neurofibromatosis type 1 and Noonan
syndrome in a single family. Am J Med Genet A, 2012;158A:2290-1.
118. Bertola DR, Pereira AC, Passetti F, de Oliveira PS, Messiaen L, Gelb BD, et al.
Neurofibromatosis-Noonan syndrome: molecular evidence of the concurrence of both
disorders in a patient. Am J Med Genet A, 2005;136:242-5.
119. Sol-Church K, Stabley DL, Demmer LA, Agbulos A, Lin AE, Smoot L, et al.
Male-to-male transmission of Costello syndrome: G12S HRAS germline mutation inherited
from a father with somatic mosaicism. Am J Med Genet A, 2009;149A:315-21.
120. Gripp KW, Stabley DL, Geller PL, Hopkins E, Stevenson DA, Carey JC, et al.
Molecular confirmation of HRAS p.G12S in siblings with Costello syndrome. Am J Med
Genet A, 2011;155A:2263-8.
121. Sovik O, Schubbert S, Houge G, Steine SJ, Norgard G, Engelsen B, et al. De
novo HRAS and KRAS mutations in two siblings with short stature and neuro-cardio-facio-
cutaneous features. J Med Genet, 2007;44:e84.
122. Kuniba H, Pooh RK, Sasaki K, Shimokawa O, Harada N, Kondoh T, et al.
Prenatal diagnosis of Costello syndrome using 3D ultrasonography amniocentesis
confirmation of the rare HRAS mutation G12D. Am J Med Genet A, 2009;149A:785-7.
123. Witters I, Denayer E, Brems H, Fryns JP, Legius E. The cardiofaciocutaneous
syndrome: prenatal findings in two patients. Prenat Diagn, 2008;28:53-5.
124. Wright EB, Donnai D, Johnson D, Clayton-Smith J. Cutaneous features in
17q21.31 deletion syndrome: a differential diagnosis for cardio-facio-cutaneous syndrome.
Clin Dysmorphol, 2011;20:15-20.

280
125. Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, et al.
Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat
Genet, 2012;44:376-8.
126. Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, et al.
Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris
syndrome. Nat Genet, 2012;44:379-80.
127. O'Sullivan J, Mullaney BG, Bhaskar SS, Dickerson JE, Hall G, O'Grady A, et
al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med
Genet, 2012;49:322-6.
128. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et
al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med,
2012;367:1921-9.
129. O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, et al. Exome
sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat
Genet, 2011;43:585-9.
130. Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet,
2010;11:31-46.
131. Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A
tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific
Biosciences and Illumina MiSeq sequencers. BMC Genomics, 2012;13:341.
132. Kosugi S, Natsume S, Yoshida K, Maclean D, Cano L, Kamoun S, et al. Coval:
improving alignment quality and variant calling accuracy for next-generation sequencing data.
PLoS One, 2013;8:e75402.
133. Duan J, Zhang JG, Deng HW, Wang YP. CNV-TV: a robust method to
discover copy number variation from short sequencing reads. BMC Bioinformatics,
2013;14:150.
134. Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A
copy number variation morbidity map of developmental delay. Nat Genet, 2011;43:838-46.
135. Kohlmann A, Grossmann V, Nadarajah N, Haferlach T. Next-generation
sequencing - feasibility and practicality in haematology. Br J Haematol, 2013;160:736-53.
136. Morris JA, Barrett JC. Olorin: combining gene flow with exome sequencing in
large family studies of complex disease. Bioinformatics, 2012;28:3320-1.

281
137. Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, et
al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial
ATP-dependent chambered protease. Am J Hum Genet, 2013;92:605-13.
138. Koolen DA, Kramer JM, Neveling K, Nillesen WM, Moore-Barton HL,
Elmslie FV, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31
microdeletion syndrome. Nat Genet, 2012;44:639-41.
139. Kerr B, Mucchielli ML, Sigaudy S, Fabre M, Saunier P, Voelckel MA, et al. Is
the locus for Costello syndrome on 11p? J Med Genet, 2003;40:469-71.
140. Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, et al. A
new chromosome 17q21.31 microdeletion syndrome associated with a common inversion
polymorphism. Nat Genet, 2006;38:999-1001.
141. Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, et al.
De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet, 2010;42:483-5.
142. Carmignac V, Thevenon J, Ades L, Callewaert B, Julia S, Thauvin-Robinet C,
et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome.
Am J Hum Genet, 2012;91:950-7.
143. Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, et al.
Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome.
Nat Genet, 2012;44:85-8.
144. Caputo V, Cianetti L, Niceta M, Carta C, Ciolfi A, Bocchinfuso G, et al. A
restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre
syndrome. Am J Hum Genet, 2012;90:161-9.
145. Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M,
et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning
syndrome. Nat Genet, 2012;44:783-7.
146. Kinsler V. Multiple congenital melanocytic naevi and neurocutaneous
melanosis are caused by mosaicism for NRAS codon 61 mutations, leading to an increased
risk of melanoma in affected tissues 23rd Mammalian Genetics and Development Workshop;
22nd November 2012; UCL Institute of Child Health, London2012.
147. Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R, et al.
Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline
mutations. J Med Genet, 2007;44:131-5.

282
148. Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig
M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet, 2002;39:571-4.
149. Sarkozy A, Obregon MG, Conti E, Esposito G, Mingarelli R, Pizzuti A, et al.
A novel PTPN11 gene mutation bridges Noonan syndrome, multiple lentigines/LEOPARD
syndrome and Noonan-like/multiple giant cell lesion syndrome. Eur J Hum Genet,
2004;12:1069-72.
150. Sarkozy A, Digilio M, Marino B, Dallapiccola B. Genotype-phenotype
Correlations in Noonan Syndrome. In: Zenker M, editor. Noonan Syndrome and Related
Disorders. Basel: Karger; 2009. p. 40-54.
151. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al.
Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with
hypertrophic cardiomyopathy. Nat Genet, 2007;39:1007-12.
152. Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, et al.
Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet,
2007;39:1013-7.
153. Allanson JE, Bohring A, Dorr HG, Dufke A, Gillessen-Kaesbach G, Horn D,
et al. The face of Noonan syndrome: Does phenotype predict genotype. Am J Med Genet A,
2010;152A:1960-6.
154. Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini
S, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in
exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1
genotype-phenotype correlation. Am J Hum Genet, 2007;80:140-51.
155. Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafe L, Superti-
Furga A, et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not
Ollier disease or Maffucci syndrome. PLoS Genet, 2011;7:e1002050.
156. Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine
phosphatase Shp2 in development and cancer. Adv Cancer Res, 2010;106:53-89.
157. Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, et al.
Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD
syndrome-associated PTPN11 mutation. J Clin Invest, 2011;121:1026-43.
158. Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, et al. A
mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet,
2002;70:943-54.

283
159. Jang SI, Lee EJ, Hart PS, Ramaswami M, Pallos D, Hart TC. Germ line gain of
function with SOS1 mutation in hereditary gingival fibromatosis. J Biol Chem,
2007;282:20245-55.
160. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et
al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras
p21. Cell, 1990;63:843-9.
161. Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernandez H, et
al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and
Neurofibromatosis-1. Genes Dev, 2012;26:1421-6.
162. Rodriguez-Viciana P, Rauen KA. Biochemical characterization of novel
germline BRAF and MEK mutations in cardio-facio-cutaneous syndrome. Methods Enzymol,
2008;438:277-89.
163. Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, et al.
Diversity and functional consequences of germline and somatic PTPN11 mutations in human
disease. Am J Hum Genet, 2006;78:279-90.
164. Senawong T, Phuchareon J, Ohara O, McCormick F, Rauen KA, Tetsu O.
Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and
RAF inhibition: implications for therapeutic options. Hum Mol Genet, 2008;17:419-30.
165. Rushworth LK, Hindley AD, O'Neill E, Kolch W. Regulation and role of Raf-
1/B-Raf heterodimerization. Mol Cell Biol, 2006;26:2262-72.
166. Inoue H, Kato R, Fukuyama S, Nonami A, Taniguchi K, Matsumoto K, et al.
Spred-1 negatively regulates allergen-induced airway eosinophilia and hyperresponsiveness. J
Exp Med, 2005;201:73-82.
167. Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, Wright EM, et
al. Constitutive activation of B-Raf in the mouse germ line provides a model for human
cardio-facio-cutaneous syndrome. Proc Natl Acad Sci U S A, 2011;108:5015-20.
168. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE,
Jr., et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet,
2009;41:544-52.
169. Dhomen N, Da Rocha Dias S, Hayward R, Ogilvie L, Hedley D, Delmas V, et
al. Inducible expression of (V600E) Braf using tyrosinase-driven Cre recombinase results in
embryonic lethality. Pigment Cell Melanoma Res, 2010;23:112-20.

284
170. Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, et al. The
intermediate-activity L597VBRAF mutant acts as an epistatic modifier of oncogenic RAS by
enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev, 2012;26:1945-58.
171. Anastasaki C, Estep AL, Marais R, Rauen KA, Patton EE. Kinase-activating
and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish
development and are sensitive to small molecule inhibitors. Hum Mol Genet, 2009;18:2543-
54.
172. Anastasaki C, Rauen KA, Patton EE. Continual low-level MEK inhibition
ameliorates cardio-facio-cutaneous phenotypes in zebrafish. Dis Model Mech, 2012;5:546-52.
173. Seok SH, Na YR, Han JH, Kim TH, Jung H, Lee BH, et al. Cre/loxP-
regulated transgenic zebrafish model for neural progenitor-specific oncogenic Kras
expression. Cancer Sci, 2010;101:149-54.
174. Nakamura T, Colbert M, Krenz M, Molkentin JD, Hahn HS, Dorn GW, 2nd,
et al. Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of
Noonan syndrome. J Clin Invest, 2007;117:2123-32.
175. Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan
syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance
endocardial-mesenchymal transformation. Proc Natl Acad Sci U S A, 2009;106:4736-41.
176. Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al.
Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of
Ptpn11 mutation. Nat Med, 2004;10:849-57.
177. Schramm C, Fine DM, Edwards MA, Reeb AN, Krenz M. The PTPN11 loss-
of-function mutation Q510E-Shp2 causes hypertrophic cardiomyopathy by dysregulating
mTOR signaling. Am J Physiol Heart Circ Physiol, 2012;302:H231-43.
178. Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, Kim IK, et al. Phosphatase-
defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects
during Drosophila development. Hum Mol Genet, 2009;18:193-201.
179. Pagani MR, Oishi K, Gelb BD, Zhong Y. The phosphatase SHP2 regulates the
spacing effect for long-term memory induction. Cell, 2009;139:186-98.
180. Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid
M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition.
J Clin Invest, 2008;118:2169-79.

285
181. Santoriello C, Deflorian G, Pezzimenti F, Kawakami K, Lanfrancone L,
d'Adda di Fagagna F, et al. Expression of H-RASV12 in a zebrafish model of Costello
syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech, 2009;2:56-
67.
182. Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E,
Rauen KA, et al. Effects of germline mutations in the Ras/MAPK signaling pathway on
adaptive behavior: cardiofaciocutaneous syndrome and Noonan syndrome. Am J Med Genet
A, 2010;152A:591-600.
183. Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, et al.
Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome.
Nature, 2010;465:808-12.
184. Group DNSGD. Management of Noonan Syndrome: A Clinical Guideline. In:
Manchester Uo, editor. 1 ed. Manchester: University of Manchester; 2010. p. 1-30.
185. Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, et al.
Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial
risks after radiotherapy. J Clin Oncol, 2006;24:2570-5.
186. Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di
Nicolantonio F, et al. Gene copy number for epidermal growth factor receptor (EGFR) and
clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol,
2005;6:279-86.
187. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al.
Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer
that overexpresses HER2. N Engl J Med, 2001;344:783-92.
188. Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase
II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients With Metastatic BRAF-
Mutant Cutaneous Melanoma Previously Treated With or Without a BRAF Inhibitor. J Clin
Oncol, 2012;10.1200/JCO.2012.43.5966.
189. Ponti G, Tomasi A, Pellacani G. Overwhelming response to Dabrafenib in a
patient with double BRAF mutation (V600E; V600M) metastatic malignant melanoma. J
Hematol Oncol, 2012;5:60.
190. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al.
Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J
Med, 2012;10.1056/NEJMoa1210093.

286
191. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations
in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J
Med, 2012;366:207-15.
192. Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et
al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med,
2012;367:2316-21.
193. Urosevic J. Study of the role of B-RAF protein in cancer using animal tumour
model systems: University of Belgrade; 2009.
194. Tang B, Reardon W, Black GC, Kerr BA. Congenital ulcerating hemangioma
in a baby with KRAS mutation and cardio-facio-cutaneous syndrome. Clin Dysmorphol,
2007;16:203-6.
195. Avatapalle B, Padidela R, Clayton-Smith J, Freemont A, Burkitt-Wright E,
Mughal Z. A case of Noonan syndrome with a SHOC2 mutation associated with cortical and
trabecular osteopenia and early onset fragility fractures. Endocrine Abstracts, 2012;30:P10.
196. Bader-Meunier B, Cave H, Jeremiah N, Magerus A, Lanzarotti N, Rieux-
Laucat F, et al. Are RASopathies new monogenic predisposing conditions to the development
of systemic lupus erythematosus? Case report and systematic review of the literature. Semin
Arthritis Rheum, 2013;43:217-9.
197. Alanay Y, Balci S, Ozen S. Noonan syndrome and systemic lupus
erythematosus: presentation in childhood. Clin Dysmorphol, 2004;13:161-3.
198. ten Bosch JR, Grody WW. Keeping up with the next generation: massively
parallel sequencing in clinical diagnostics. J Mol Diagn, 2008;10:484-92.
199. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al.
Exome sequencing identifies the cause of a mendelian disorder. Nat Genet, 2010;42:30-5.
200. Volpi L, Roversi G, Colombo EA, Leijsten N, Concolino D, Calabria A, et al.
Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with
neutropenia gene. Am J Hum Genet, 2010;86:72-6.
201. Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA,
et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet,
2007;39:70-4.
202. Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, et al.
Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus.
Cell, 1990;62:187-92.

287
203. Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, et al.
Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature, 1996;384:567-
70.
204. Pasteris NG, Cadle A, Logie LJ, Porteous ME, Schwartz CE, Stevenson RE, et
al. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene:
a putative Rho/Rac guanine nucleotide exchange factor. Cell, 1994;79:669-78.
205. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role
of TBX1 in human del22q11.2 syndrome. Lancet, 2003;362:1366-73.
206. Oda T, Elkahloun AG, Meltzer PS, Chandrasekharappa SC. Identification and
cloning of the human homolog (JAG1) of the rat Jagged1 gene from the Alagille syndrome
critical region at 20p12. Genomics, 1997;43:376-9.
207. McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA,
et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch
signaling pathway. Am J Hum Genet, 2006;79:169-73.
208. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al.
Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in
patients with the Carney complex. Nat Genet, 2000;26:89-92.
209. Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, et
al. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by
mutation of ROR2. Nat Genet, 2000;25:419-22.
210. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline
mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer
syndrome. Nat Genet, 1997;16:64-7.
211. Levanat S, Gorlin RJ, Fallet S, Johnson DR, Fantasia JE, Bale AE. A two-hit
model for developmental defects in Gorlin syndrome. Nat Genet, 1996;12:85-7.
212. Fan Z, Li J, Du J, Zhang H, Shen Y, Wang CY, et al. A missense mutation in
PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet,
2008;45:303-8.
213. Manser E, Leung T, Salihuddin H, Tan L, Lim L. A non-receptor tyrosine
kinase that inhibits the GTPase activity of p21cdc42. Nature, 1993;363:364-7.
214. Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase
inhibitor from Raf-1 to GRK-2. Nature, 2003;426:574-9.

288
215. Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, et al.
Advanced glycation end products induce tubular epithelial-myofibroblast transition through
the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol, 2004;164:1389-97.
216. Yuryev A, Ono M, Goff SA, Macaluso F, Wennogle LP. Isoform-specific
localization of A-RAF in mitochondria. Mol Cell Biol, 2000;20:4870-8.
217. Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ,
et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase
complexes. Science, 1999;283:655-61.
218. Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, et al.
Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem,
1992;267:17882-90.
219. Wang HG, Takayama S, Rapp UR, Reed JC. Bcl-2 interacting protein, BAG-1,
binds to and activates the kinase Raf-1. Proc Natl Acad Sci U S A, 1996;93:7063-8.
220. Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA. Ras
regulates assembly of mitogenic signalling complexes through the effector protein IMP.
Nature, 2004;427:256-60.
221. De Vet EC, Aguado B, Campbell RD. Adaptor signalling proteins Grb2 and
Grb7 are recruited by human G6f, a novel member of the immunoglobulin superfamily
encoded in the MHC. Biochem J, 2003;375:207-13.
222. Moreto J, Vidal-Quadras M, Pol A, Santos E, Grewal T, Enrich C, et al.
Differential involvement of H- and K-Ras in Raf-1 activation determines the role of
calmodulin in MAPK signaling. Cell Signal, 2009;21:1827-36.
223. Hunter I, Nixon GF. Spatial compartmentalization of tumor necrosis factor
(TNF) receptor 1-dependent signaling pathways in human airway smooth muscle cells. Lipid
rafts are essential for TNF-alpha-mediated activation of RhoA but dispensable for the
activation of the NF-kappaB and MAPK pathways. J Biol Chem, 2006;281:34705-15.
224. Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan KL. Kinase suppressor
of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell
Biol, 1999;19:5523-34.
225. Wu G, Li H, Yang Z. Arabidopsis RopGAPs are a novel family of rho
GTPase-activating proteins that require the Cdc42/Rac-interactive binding motif for rop-
specific GTPase stimulation. Plant Physiol, 2000;124:1625-36.

289
226. Borysov SI, Guadagno TM. A novel role for Cdk1/cyclin B in regulating B-raf
activation at mitosis. Mol Biol Cell, 2008;19:2907-15.
227. Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al.
Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev,
1997;11:2822-34.
228. Ziogas A, Moelling K, Radziwill G. CNK1 is a scaffold protein that regulates
Src-mediated Raf-1 activation. J Biol Chem, 2005;280:24205-11.
229. de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Crkl is complexed with
tyrosine-phosphorylated Cbl in Ph-positive leukemia. J Biol Chem, 1995;270:21468-71.
230. Diwakar R, Pearson AL, Colville-Nash P, Baines DL, Dockrell ME. Role
played by disabled-2 in albumin induced MAP Kinase signalling. Biochem Biophys Res
Commun, 2008;366:675-80.
231. Xie D, Gore C, Zhou J, Pong RC, Zhang H, Yu L, et al. DAB2IP coordinates
both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc Natl Acad Sci U S A,
2009;106:19878-83.
232. Honma M, Higuchi O, Shirakata M, Yasuda T, Shibuya H, Iemura S, et al.
Dok-3 sequesters Grb2 and inhibits the Ras-Erk pathway downstream of protein-tyrosine
kinases. Genes Cells, 2006;11:143-51.
233. Calvisi DF, Pinna F, Meloni F, Ladu S, Pellegrino R, Sini M, et al. Dual-
specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated
control of growth in human hepatocellular carcinoma. Cancer Res, 2008;68:4192-200.
234. Grumont RJ, Rasko JE, Strasser A, Gerondakis S. Activation of the mitogen-
activated protein kinase pathway induces transcription of the PAC-1 phosphatase gene. Mol
Cell Biol, 1996;16:2913-21.
235. Gaedcke J, Grade M, Jung K, Camps J, Jo P, Emons G, et al. Mutated KRAS
results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone
methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer, 2010;49:1024-34.
236. Ouyang B, Knauf JA, Smith EP, Zhang L, Ramsey T, Yusuff N, et al.
Inhibitors of Raf kinase activity block growth of thyroid cancer cells with RET/PTC or BRAF
mutations in vitro and in vivo. Clin Cancer Res, 2006;12:1785-93.
237. Tsang M, Maegawa S, Kiang A, Habas R, Weinberg E, Dawid IB. A role for
MKP3 in axial patterning of the zebrafish embryo. Development, 2004;131:2769-79.

290
238. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer.
Cancer Metastasis Rev, 2008;27:253-61.
239. Kim S, Salibi N, Hardie AD, Xu J, Lim RP, Lee VS, et al. Characterization of
adrenal pheochromocytoma using respiratory-triggered proton MR spectroscopy: initial
experience. AJR Am J Roentgenol, 2009;192:450-4.
240. Carraway CA, Carvajal ME, Carraway KL. Association of the Ras to mitogen-
activated protein kinase signal transduction pathway with microfilaments. Evidence for a
p185(neu)-containing cell surface signal transduction particle linking the mitogenic pathway to
a membrane-microfilament association site. J Biol Chem, 1999;274:25659-67.
241. Dai P, Xiong WC, Mei L. Erbin inhibits RAF activation by disrupting the sur-
8-Ras-Raf complex. J Biol Chem, 2006;281:927-33.
242. Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, et al.
Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence.
Nature, 2001;409:1067-70.
243. Hayakawa M, Matsushima M, Hagiwara H, Oshima T, Fujino T, Ando K, et al.
Novel insights into FGD3, a putative GEF for Cdc42, that undergoes SCF(FWD1/beta-
TrCP)-mediated proteasomal degradation analogous to that of its homologue FGD1 but
regulates cell morphology and motility differently from FGD1. Genes Cells, 2008;13:329-42.
244. Easton JB, Royer AR, Middlemas DS. The protein tyrosine phosphatase, Shp2,
is required for the complete activation of the RAS/MAPK pathway by brain-derived
neurotrophic factor. J Neurochem, 2006;97:834-45.
245. Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired
long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice.
Science, 1992;258:1903-10.
246. Nakaoka Y, Nishida K, Fujio Y, Izumi M, Terai K, Oshima Y, et al. Activation
of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein
Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ Res, 2003;93:221-9.
247. Zhao C, Yu DH, Shen R, Feng GS. Gab2, a new pleckstrin homology domain-
containing adapter protein, acts to uncouple signaling from ERK kinase to Elk-1. J Biol
Chem, 1999;274:19649-54.
248. Spain BH, Bowdish KS, Pacal AR, Staub SF, Koo D, Chang CY, et al. Two
human cDNAs, including a homolog of Arabidopsis FUS6 (COP11), suppress G-protein- and

291
mitogen-activated protein kinase-mediated signal transduction in yeast and mammalian cells.
Mol Cell Biol, 1996;16:6698-706.
249. Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-
activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol,
1996;16:3169-78.
250. Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, et al. The
SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras
signaling. Cell, 1992;70:431-42.
251. Schrage YM, Briaire-de Bruijn IH, de Miranda NF, van Oosterwijk J, Taminiau
AH, van Wezel T, et al. Kinome profiling of chondrosarcoma reveals SRC-pathway activity
and dasatinib as option for treatment. Cancer Res, 2009;69:6216-22.
252. Ito H, Akiyama H, Shigeno C, Nakamura T. Isolation, characterization, and
chromosome mapping of a human A-C1 Ha-Ras suppressor gene (HRASLS). Cytogenet Cell
Genet, 2001;93:36-9.
253. Grammatikakis N, Lin JH, Grammatikakis A, Tsichlis PN, Cochran BH.
p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol Cell Biol,
1999;19:1661-72.
254. Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal,
2009;21:462-9.
255. Werner H, Le Roith D. The insulin-like growth factor-I receptor signaling
pathways are important for tumorigenesis and inhibition of apoptosis. Crit Rev Oncog,
1997;8:71-92.
256. Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding
protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO J,
1996;15:2997-3005.
257. So EY, Oh J, Jang JY, Kim JH, Lee CE. Ras/Erk pathway positively regulates
Jak1/STAT6 activity and IL-4 gene expression in Jurkat T cells. Mol Immunol, 2007;44:3416-
26.
258. Tefferi A, Gilliland DG. JAK2 in myeloproliferative disorders is not just
another kinase. Cell Cycle, 2005;4:1053-6.
259. Cimmino F, Schulte JH, Zollo M, Koster J, Versteeg R, Iolascon A, et al.
Galectin-1 is a major effector of TrkB-mediated neuroblastoma aggressiveness. Oncogene,
2009;28:2015-23.

292
260. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale
JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci
U S A, 2006;103:5060-5.
261. Hayashi D, Kudoh S, Shiojima I, Zou Y, Harada K, Shimoyama M, et al. Atrial
natriuretic peptide inhibits cardiomyocyte hypertrophy through mitogen-activated protein
kinase phosphatase-1. Biochem Biophys Res Commun, 2004;322:310-9.
262. Schroder WA, Buck M, Cloonan N, Hancock JF, Suhrbier A, Sculley T, et al.
Human Sin1 contains Ras-binding and pleckstrin homology domains and suppresses Ras
signalling. Cell Signal, 2007;19:1279-89.
263. Mouchel-Vielh E, Bloyer S, Salvaing J, Randsholt NB, Peronnet F.
Involvement of the MP1 scaffold protein in ERK signaling regulation during Drosophila wing
development. Genes Cells, 2008;13:1099-111.
264. Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1
regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell, 2001;8:983-
93.
265. Kostenko S, Shiryaev A, Dumitriu G, Gerits N, Moens U. Cross-talk between
protein kinase A and the MAPK-activated protein kinases RSK1 and MK5. J Recept Signal
Transduct Res, 2011;31:1-9.
266. Kuriyama M, Harada N, Kuroda S, Yamamoto T, Nakafuku M, Iwamatsu A,
et al. Identification of AF-6 and canoe as putative targets for Ras. J Biol Chem, 1996;271:607-
10.
267. Lenormand JL, Benayoun B, Guillier M, Vandromme M, Leibovitch MP,
Leibovitch SA. Mos activates myogenic differentiation by promoting heterodimerization of
MyoD and E12 proteins. Mol Cell Biol, 1997;17:584-93.
268. Louahed J, Grasso L, De Smet C, Van Roost E, Wildmann C, Nicolaides NC,
et al. Interleukin-9-induced expression of M-Ras/R-Ras3 oncogene in T-helper clones. Blood,
1999;94:1701-10.
269. Inder KL, Lau C, Loo D, Chaudhary N, Goodall A, Martin S, et al.
Nucleophosmin and nucleolin regulate K-Ras plasma membrane interactions and MAPK
signal transduction. J Biol Chem, 2009;284:28410-9.
270. Jung JR, Kim H, Jeun SS, Lee JY, Koh EJ, Ji C. The Phosphorylation status of
merlin is important for regulating the Ras-ERK pathway. Mol Cells, 2005;20:196-200.

293
271. Blochl A, Blumenstein L, Ahmadian MR. Inactivation and activation of Ras by
the neurotrophin receptor p75. Eur J Neurosci, 2004;20:2321-35.
272. Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, et al.
Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal
adenocarcinoma. Cancer Res, 2010;70:4280-6.
273. Braoudaki M, Papathanassiou C, Katsibardi K, Tourkadoni N, Karamolegou
K, Tzortzatou-Stathopoulou F. The frequency of NPM1 mutations in childhood acute
myeloid leukemia. J Hematol Oncol, 2010;3:41.
274. Vomastek T, Iwanicki MP, Burack WR, Tiwari D, Kumar D, Parsons JT, et al.
Extracellular signal-regulated kinase 2 (ERK2) phosphorylation sites and docking domain on
the nuclear pore complex protein Tpr cooperatively regulate ERK2-Tpr interaction. Mol Cell
Biol, 2008;28:6954-66.
275. Kraemer D, Wozniak RW, Blobel G, Radu A. The human CAN protein, a
putative oncogene product associated with myeloid leukemogenesis, is a nuclear pore complex
protein that faces the cytoplasm. Proc Natl Acad Sci U S A, 1994;91:1519-23.
276. Smith SD, Jaffer ZM, Chernoff J, Ridley AJ. PAK1-mediated activation of
ERK1/2 regulates lamellipodial dynamics. J Cell Sci, 2008;121:3729-36.
277. Martin GA, Bollag G, McCormick F, Abo A. A novel serine kinase activated
by rac1/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. EMBO
J, 1995;14:1970-8.
278. Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione
RA, et al. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet,
1998;20:25-30.
279. Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B,
et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell, 2001;1:239-
50.
280. Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of
Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature, 1999;401:173-7.
281. Rajalingam K, Rudel T. Ras-Raf signaling needs prohibitin. Cell Cycle,
2005;4:1503-5.
282. Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian
oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively
regulates MAPK pathway. J Biol Chem, 2003;278:14920-5.

294
283. Meier F, Schittek B, Busch S, Garbe C, Smalley K, Satyamoorthy K, et al. The
RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the
effective treatment of advanced melanoma. Front Biosci, 2005;10:2986-3001.
284. Guan KL. The mitogen activated protein kinase signal transduction pathway:
from the cell surface to the nucleus. Cell Signal, 1994;6:581-9.
285. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory
subunits and cancer. Biochim Biophys Acta, 2009;1795:1-15.
286. Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal
transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature,
1994;372:786-91.
287. Della Rocca GJ, Maudsley S, Daaka Y, Lefkowitz RJ, Luttrell LM. Pleiotropic
coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role
of focal adhesions and receptor tyrosine kinases. J Biol Chem, 1999;274:13978-84.
288. Cao L, Zhang L, Ruiz-Lozano P, Yang Q, Chien KR, Graham RM, et al. A
novel putative protein-tyrosine phosphatase contains a BRO1-like domain and suppresses Ha-
ras-mediated transformation. J Biol Chem, 1998;273:21077-83.
289. Barr AJ, Knapp S. MAPK-specific tyrosine phosphatases: new targets for drug
discovery? Trends Pharmacol Sci, 2006;27:525-30.
290. Short SM, Talbott GA, Juliano RL. Integrin-mediated signaling events in
human endothelial cells. Mol Biol Cell, 1998;9:1969-80.
291. Rodriguez-Viciana P, McCormick F. RalGDS comes of age. Cancer Cell,
2005;7:205-6.
292. Rousseau-Merck MF, Pizon V, Tavitian A, Berger R. Chromosome mapping
of the human RAS-related RAP1A, RAP1B, and RAP2 genes to chromosomes 1p12----p13,
12q14, and 13q34, respectively. Cytogenet Cell Genet, 1990;53:2-4.
293. Daumke O, Weyand M, Chakrabarti PP, Vetter IR, Wittinghofer A. The
GTPase-activating protein Rap1GAP uses a catalytic asparagine. Nature, 2004;429:197-201.
294. Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, et al.
C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src
homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci U S A,
1994;91:3443-7.

295
295. Trahey M, Wong G, Halenbeck R, Rubinfeld B, Martin GA, Ladner M, et al.
Molecular cloning of two types of GAP complementary DNA from human placenta. Science,
1988;242:1697-700.
296. Maekawa M, Li S, Iwamatsu A, Morishita T, Yokota K, Imai Y, et al. A novel
mammalian Ras GTPase-activating protein which has phospholipid-binding and Btk
homology regions. Mol Cell Biol, 1994;14:6879-85.
297. Cullen PJ, Hsuan JJ, Truong O, Letcher AJ, Jackson TR, Dawson AP, et al.
Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family.
Nature, 1995;376:527-30.
298. Lockyer PJ, Kupzig S, Cullen PJ. CAPRI regulates Ca(2+)-dependent
inactivation of the Ras-MAPK pathway. Curr Biol, 2001;11:981-6.
299. Jin H, Wang X, Ying J, Wong AH, Cui Y, Srivastava G, et al. Epigenetic
silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new
mechanism of Ras activation in human cancers. Proc Natl Acad Sci U S A, 2007;104:12353-8.
300. Fan WT, Koch CA, de Hoog CL, Fam NP, Moran MF. The exchange factor
Ras-GRF2 activates Ras-dependent and Rac-dependent mitogen-activated protein kinase
pathways. Curr Biol, 1998;8:935-8.
301. Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ,
et al. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1.
Nature, 2003;424:694-8.
302. Rebhun JF, Castro AF, Quilliam LA. Identification of guanine nucleotide
exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP
interaction. J Biol Chem, 2000;275:34901-8.
303. Reuther GW, Lambert QT, Rebhun JF, Caligiuri MA, Quilliam LA, Der CJ.
RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J Biol Chem,
2002;277:30508-14.
304. Mitin NY, Ramocki MB, Zullo AJ, Der CJ, Konieczny SF, Taparowsky EJ.
Identification and characterization of rain, a novel Ras-interacting protein with a unique
subcellular localization. J Biol Chem, 2004;279:22353-61.
305. Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel
K-Ras-specific effector and potential tumor suppressor. J Biol Chem, 2003;278:28045-51.

296
306. Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037
is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res, 2004;64:8688-
93.
307. Sierra DA, Gilbert DJ, Householder D, Grishin NV, Yu K, Ukidwe P, et al.
Evolution of the regulators of G-protein signaling multigene family in mouse and human.
Genomics, 2002;79:177-85.
308. Mizuki N, Kimura M, Ohno S, Miyata S, Sato M, Ando H, et al. Isolation of
cDNA and genomic clones of a human Ras-related GTP-binding protein gene and its
chromosomal localization to the long arm of chromosome 7, 7q36. Genomics, 1996;34:114-8.
309. Han L, Wong D, Dhaka A, Afar D, White M, Xie W, et al. Protein binding and
signaling properties of RIN1 suggest a unique effector function. Proc Natl Acad Sci U S A,
1997;94:4954-9.
310. Basel-Vanagaite L, Sarig O, Hershkovitz D, Fuchs-Telem D, Rapaport D, Gat
A, et al. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS
syndrome. Am J Hum Genet, 2009;85:254-63.
311. Shao H, Kadono-Okuda K, Finlin BS, Andres DA. Biochemical
characterization of the Ras-related GTPases Rit and Rin. Arch Biochem Biophys,
1999;371:207-19.
312. Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell
survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -
independent mechanisms. Science, 1999;286:1358-62.
313. Zhao Y, Bjorbaek C, Weremowicz S, Morton CC, Moller DE. RSK3 encodes a
novel pp90rsk isoform with a unique N-terminal sequence: growth factor-stimulated kinase
function and nuclear translocation. Mol Cell Biol, 1995;15:4353-63.
314. Lowe DG, Capon DJ, Delwart E, Sakaguchi AY, Naylor SL, Goeddel DV.
Structure of the human and murine R-ras genes, novel genes closely related to ras proto-
oncogenes. Cell, 1987;48:137-46.
315. Cutler ML, Bassin RH, Zanoni L, Talbot N. Isolation of rsp-1, a novel cDNA
capable of suppressing v-Ras transformation. Mol Cell Biol, 1992;12:3750-6.
316. Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, et al.
Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade.
Recent Prog Horm Res, 2001;56:127-55.

297
317. Yokouchi M, Suzuki R, Masuhara M, Komiya S, Inoue A, Yoshimura A.
Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains
that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene, 1997;15:7-15.
318. Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, et al. Lnk
constrains myeloproliferative diseases in mice. J Clin Invest, 2010;120:2058-69.
319. Bell SM, Shaw M, Jou YS, Myers RM, Knowles MA. Identification and
characterization of the human homologue of SH3BP2, an SH3 binding domain protein within
a common region of deletion at 4p16.3 involved in bladder cancer. Genomics, 1997;44:163-70.
320. Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S. Cloning and
characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem
Biophys Res Commun, 2000;268:321-8.
321. Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, et al. A
novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal
transduction. Cell, 1992;70:93-104.
322. Falco M, Biassoni R, Bottino C, Vitale M, Sivori S, Augugliaro R, et al.
Identification and molecular cloning of p75/AIRM1, a novel member of the sialoadhesin
family that functions as an inhibitory receptor in human natural killer cells. J Exp Med,
1999;190:793-802.
323. Marie-Cardine A, Kirchgessner H, Bruyns E, Shevchenko A, Mann M,
Autschbach F, et al. SHP2-interacting transmembrane adaptor protein (SIT), a novel disulfide-
linked dimer regulating human T cell activation. J Exp Med, 1999;189:1181-94.
324. Mendoza MC, Du F, Iranfar N, Tang N, Ma H, Loomis WF, et al. Loss of
SMEK, a novel, conserved protein, suppresses MEK1 null cell polarity, chemotaxis, and gene
expression defects. Mol Cell Biol, 2005;25:7839-53.
325. Webb GC, Jenkins NA, Largaespada DA, Copeland NG, Fernandez CS,
Bowtell DD. Mammalian homologues of the Drosophila Son of sevenless gene map to
murine chromosomes 17 and 12 and to human chromosomes 2 and 14, respectively.
Genomics, 1993;18:14-9.
326. Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K, et al.
Spred is a Sprouty-related suppressor of Ras signalling. Nature, 2001;412:647-51.
327. Kato R, Nonami A, Taketomi T, Wakioka T, Kuroiwa A, Matsuda Y, et al.
Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk
activation. Biochem Biophys Res Commun, 2003;302:767-72.

298
328. Edwin F, Anderson K, Ying C, Patel TB. Intermolecular interactions of
Sprouty proteins and their implications in development and disease. Mol Pharmacol,
2009;76:679-91.
329. Nyga R, Pecquet C, Harir N, Gu H, Dhennin-Duthille I, Regnier A, et al.
Activated STAT5 proteins induce activation of the PI 3-kinase/Akt and Ras/MAPK pathways
via the Gab2 scaffolding adapter. Biochem J, 2005;390:359-66.
330. O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in
suppression of apoptosis by the proto-oncogene product Raf-1. Science, 2004;306:2267-70.
331. Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding
partners in CNS axon outgrowth. Genes Dev, 2004;18:541-58.
332. Pan F, Sun L, Kardian DB, Whartenby KA, Pardoll DM, Liu JO. Feedback
inhibition of calcineurin and Ras by a dual inhibitory protein Carabin. Nature, 2007;445:433-6.
333. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al.
Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature,
2010;468:839-43.
334. Gilby DC, Sung HY, Winship PR, Goodeve AC, Reilly JT, Kiss-Toth E.
Tribbles-1 and -2 are tumour suppressors, down-regulated in human acute myeloid leukaemia.
Immunol Lett, 2010;130:115-24.
335. Tang M, Zhong M, Shang Y, Lin H, Deng J, Jiang H, et al. Differential
regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through
TRB3/MAPK signaling pathway. Cell Mol Life Sci, 2008;65:2924-32.
336. Bustelo XR, Ledbetter JA, Barbacid M. Product of vav proto-oncogene
defines a new class of tyrosine protein kinase substrates. Nature, 1992;356:68-71.
337. Vomastek T, Schaeffer HJ, Tarcsafalvi A, Smolkin ME, Bissonette EA, Weber
MJ. Modular construction of a signaling scaffold: MORG1 interacts with components of the
ERK cascade and links ERK signaling to specific agonists. Proc Natl Acad Sci U S A,
2004;101:6981-6.
338. Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, et al. The
structural basis for 14-3-3:phosphopeptide binding specificity. Cell, 1997;91:961-71.
339. Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, et al.
DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H-
and N-Ras. J Biol Chem, 2005;280:31141-8.

299
340. Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and
transcriptional regulation. Gene, 2013;513:1-13.
341. Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-
short read data sets from high-throughput DNA sequencing. Nucleic Acids Res, 2008;36:e105.
342. Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing:
computational challenges and solutions. Nat Rev Genet, 2012;13:36-46.
343. Cooper G, Stone E, Asimenos G, Program NCS, Green E, Batzoglou S, et al.
Distribution and intensity of constraint in mammalian genomic sequence. Genome Res,
2005;15:13.
344. Jacamo RO, Rozengurt E. A truncated FAK lacking the FERM domain
displays high catalytic activity but retains responsiveness to adhesion-mediated signals.
Biochem Biophys Res Commun, 2005;334:1299-304.
345. Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain:
organizing the structure and function of FAK. Nat Rev Mol Cell Biol, 2010;11:802-14.
346. Wong VW, Rustad KC, Akaishi S, Sorkin M, Glotzbach JP, Januszyk M, et al.
Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat
Med, 2012;18:148-52.
347. Kweh F, Zheng M, Kurenova E, Wallace M, Golubovskaya V, Cance WG.
Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol
Carcinog, 2009;48:1005-17.
348. Santos AM, Schechtman D, Cardoso AC, Clemente CF, Silva JC, Fioramonte
M, et al. FERM domain interaction with myosin negatively regulates FAK in cardiomyocyte
hypertrophy. Nat Chem Biol, 2012;8:102-10.
349. Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, et al.
Transformation of mammalian cells by constitutively active MAP kinase kinase. Science,
1994;265:966-70.
350. Silva AG, Maschietto M, Vidal DO, Pelicario LM, Velloso ED, Lopes LF, et
al. Array-CGH as an adjuvant tool in cytogenetic diagnosis of pediatric MDS and JMML. Med
Oncol, 2013;30:734.
351. Ucar C, Caliskan U, Toy H, Gunel E. Hepatoblastoma in a child with
neurofibromatosis type I. Pediatr Blood Cancer, 2007;49:357-9.
352. Dvorak CC, Gracia CR, Sanders JE, Cheng EY, Baker KS, Pulsipher MA, et
al. NCI, NHLBI/PBMTC first international conference on late effects after pediatric

300
hematopoietic cell transplantation: endocrine challenges-thyroid dysfunction, growth
impairment, bone health, & reproductive risks. Biol Blood Marrow Transplant, 2011;17:1725-
38.
353. Gurney JG, Ness KK, Rosenthal J, Forman SJ, Bhatia S, Baker KS. Visual,
auditory, sensory, and motor impairments in long-term survivors of hematopoietic stem cell
transplantation performed in childhood: results from the Bone Marrow Transplant Survivor
study. Cancer, 2006;106:1402-8.
354. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al.
Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N Engl J Med,
2013;10.1056/NEJMoa1306555.
355. Firth H, Wright CF, Study D. The Deciphering Developmental Disorders
(DDD) study. Dev Med Child Neurol, 2011;53:2.
356. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, et al.
Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med
Genet A, 2012;158A:1077-82.
357. Ziogas A, Lorenz IC, Moelling K, Radziwill G. Mitotic Raf-1 is stimulated
independently of Ras and is active in the cytoplasm. J Biol Chem, 1998;273:24108-14.
358. Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its
inhibition on normal and disease-associated Raf signaling. Mol Cell, 2013;49:751-8.
359. Bienegraeber M, Ozcan C, Terzic A. Stable transfection of UCP1 confers
resistance to hypoxia/reoxygenation in a heart-derived cell line. J Molec Cell Cardiol
2003;35:861-5.
360. Van Nieuwenhoeven FA, Luiken JJ, De Jong YF, Grimaldi PA, Van der Vusse
GJ, Glatz JF. Stable transfection of fatty acid translocase (CD36) in a rat heart muscle cell line
(H9c2). J Lipid Res, 1998;39:2039-47.
361. Chong KY, Lai CC, Lille S, Chang C, Su CY. Stable overexpression of the
constitutive form of heat shock protein 70 confers oxidative protection. J Molec Cell Cardiol,
1998;30:599-608.
362. Stark Z, Gillessen-Kaesbach G, Ryan MM, Cirstea IC, Gremer L, Ahmadian
MR, et al. Two novel germline KRAS mutations: expanding the molecular and clinical
phenotype. Clin Genet, 2012;81:590-4.
363. Searle AG, Edwards JH, Hall JG. Mouse homologues of human hereditary
disease. J Med Genet, 1994;31:1-19.

301
364. Maquat LE. Nonsense-mediated mRNA decay in mammals. J Cell Sci
2005;118:1773-6.
365. Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al.
Decipher: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl
Resources. Am J Hum Genet, 2009;84:524-33.
366. Sherlock G. Analysis of large-scale gene expression data. Brief Bioinform,
2001;2:350-62.
367. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis
of large gene lists using DAVID bioinformatics resources. Nat Protoc, 2009;4:44-57.
368. Scheuermann JC, Boyer LA. Getting to the heart of the matter: long non-
coding RNAs in cardiac development and disease. EMBO J, 2013;32:1805-16.
369. Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, et
al. Characteristics and prognostic implications of myosin missense mutations in familial
hypertrophic cardiomyopathy. N Engl J Med, 1992;326:1108-14.
370. Schoenfeld JR, Vasser M, Jhurani P, Ng P, Hunter JJ, Ross J, Jr., et al. Distinct
molecular phenotypes in murine cardiac muscle development, growth, and hypertrophy. J Mol
Cell Cardiol, 1998;30:2269-80.
371. Abdollahi MR, Morrison E, Sirey T, Molnar Z, Hayward BE, Carr IM, et al.
Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve
hypoplasia. Am J Hum Genet, 2009;85:737-44.
372. Stanchi F, Corso V, Scannapieco P, Ievolella C, Negrisolo E, Tiso N, et al.
TUBA8: A new tissue-specific isoform of alpha-tubulin that is highly conserved in human and
mouse. Biochem Biophys Res Commun, 2000;270:1111-8.
373. Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, et al. Brain
natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual
natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest,
1991;87:1402-12.
374. Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, et al.
Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A,
2000;97:4239-44.
375. Laing NG, Dye DE, Wallgren-Pettersson C, Richard G, Monnier N, Lillis S, et
al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum
Mutat, 2009;30:1267-77.

302
376. Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K,
et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and
nemaline myopathy. Nat Genet, 1999;23:208-12.
377. Viosca J, Schuhmacher AJ, Guerra C, Barco A. Germline expression of H-
Ras(G12V) causes neurological deficits associated to Costello syndrome. Genes Brain Behav,
2009;8:60-71.
378. Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, et al.
Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with
copy number imbalances of the oncogene. Proc Natl Acad Sci U S A, 2009;106:7979-84.
379. Saffitz JE. Arrhythmogenic cardiomyopathy and abnormalities of cell-to-cell
coupling. Heart Rhythm, 2009;6:S62-5.
380. Goh LK, Sorkin A. Endocytosis of receptor tyrosine kinases. Cold Spring
Harb Perspect Biol, 2013;5:a017459.
381. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display
of genome-wide expression patterns PNAS, 1998;95:14863-8.
382. Pilbrow AP, Ellmers LJ, Black MA, Moravec CS, Sweet WE, Troughton RW,
et al. Genomic selection of reference genes for real-time PCR in human myocardium. BMC
Med Genomics, 2008;1:64.
383. Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et
al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic
cardiomyopathy: a disease of the sarcomere. Cell, 1994;77:701-12.
384. Nagy. Manipulating the Mouse Embryo: A Laboratory Manual (third edition).
third ed. Cold Spring Harbour: CSH Press; 2003 2003. 764 p.
385. Katalinic OM, Harvey LA, Herbert RD. Effectiveness of stretch for the
treatment and prevention of contractures in people with neurological conditions: a systematic
review. Phys Ther, 2011;91:11-24.
386. King M, Bewes P, Cairns J, Thornton J. Primary orthopaedics. In: King M,
editor. Primary surgery: volume I: non-trauma. Bonn: University of Bonn; 1999.
387. Lopez-Rangel E, Malleson PN, Lirenman DS, Roa B, Wiszniewska J, Lewis
ME. Systemic lupus erythematosus and other autoimmune disorders in children with Noonan
syndrome. Am J Med Genet A, 2005;139:239-42.
388. Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune
disease. Nature, 2005;435:584-9.

303
389. Ramsey-Goldman R, Rothrock N. Fatigue in systemic lupus erythematosus
and rheumatoid arthritis. PM R, 2010;2:384-92.
390. Zinnamosca L, Petramala L, Cotesta D, Marinelli C, Schina M, Cianci R, et al.
Neurofibromatosis type 1 (NF1) and pheochromocytoma: prevalence, clinical and
cardiovascular aspects. Arch Dermatol Res, 2011;303:317-25.
391. Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, et
al. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital
disorders and testicular tumors. Nat Genet, 2009;41:1247-52.
392. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et
al. Exome sequencing as a tool for Mendelian disease discovery. Nat Rev Genet, 2011;12:745-
55.
393. Korf BR. Integration of genomics into clinical practice. Discov Med,
2013;16:241-8.
394. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG
recommendations for reporting of incidental findings in clinical exome and genome
sequencing. Genet Med, 2013;15:565-74.
395. Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-
invasive prenatal measurement of the fetal genome. Nature, 2012;487:320-4.
396. Orrico A, Galli L, Faivre L, Clayton-Smith J, Azzarello-Burri SM, Hertz JM, et
al. Aarskog-Scott syndrome: clinical update and report of nine novel mutations of the FGD1
gene. Am J Med Genet A, 2010;152A:313-8.
397. Yu Y, Wu BL, Wu J, Shen Y. Exome and whole-genome sequencing as clinical
tests: a transformative practice in molecular diagnostics. Clin Chem, 2012;58:1507-9.
398. Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline
KRAS mutations cause Noonan syndrome. Nat Genet, 2006;38:331-6.
399. Yu H, Wang F, Tu K, Xie L, Li Y-Y, Li Y-X. Transcript-level annotation of
Affymetrix probesets improves the interpretation of gene expression data. BMC
Bioinformatics, 2007;8:194.
400. Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH
assembly of TALENs for high-throughput genome editing. Nat Biotechnol, 2012;30:460-5.
401. Xu X, Colecraft HM. Primary culture of adult rat heart myocytes. J Vis Exp,
2009;10.3791/1308.

304
402. Shimizu S, Abt A, Meucci O. Bilaminar co-culture of primary rat cortical
neurons and glia. J Vis Exp, 2011;10.3791/3257.
403. Hynds RE, Giangreco A. Concise review: the relevance of human stem cell-
derived organoid models for epithelial translational medicine. Stem Cells, 2013;31:417-22.
404. Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van
den Berg MP, et al. Targeted next-generation sequencing can replace Sanger sequencing in
clinical diagnostics. Hum Mutat, 2013;34:1035-42.
405. Stones CJ, Kim JE, Joseph WR, Leung E, Marshall ES, Finlay GJ, et al.
Comparison of responses of human melanoma cell lines to MEK and BRAF inhibitors. Front
Genet, 2013;4:66.
406. Burton H, Alberg C, Stewart A. Mainstreaming genetics: a comparative review
of clinical services for inherited cardiovascular conditions in the UK. Public Health Genomics,
2010;13:235-45.
407. Black AP, Baker M. The impact of parent advocacy groups, the Internet, and
social networking on rare diseases: the IDEA League and IDEA League United Kingdom
example. Epilepsia, 2011;52 Suppl 2:102-4.
408. Wright EB, Holcombe C, Salmon P. Doctors' communication of trust, care,
and respect in breast cancer: qualitative study. BMJ, 2004;328:864.
409. Gripp KW, Lin AE. Costello syndrome: A Ras/mitogen activated protein
kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med,
2011;10.1097/GIM.0b013e31822dd91f.
410. Kontaridis MI. Therapeutic approach to HCM in Noonan syndrome with
multiple lentigines Third International Meeting on Genetic Syndromes of the Ras/MAPK
Pathway: Towards a Therapeutic Approach; August 3rd 2013; Orlando, Florida2013.

305
9 APPENDICES
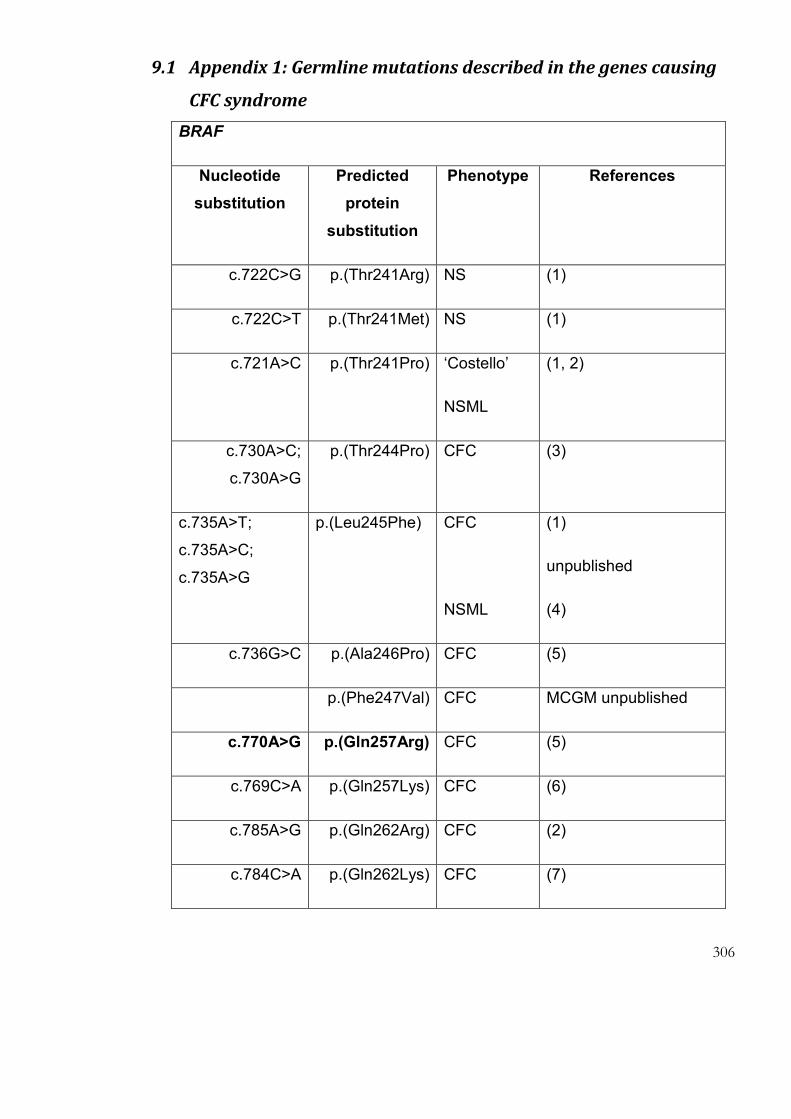
306
9.1 Appendix 1: Germline mutations described in the genes causing
CFC syndrome
BRAF
Nucleotide
substitution
Predicted
protein
substitution
Phenotype References
c.722C>G p.(Thr241Arg) NS (1)
c.722C>T p.(Thr241Met) NS (1)
c.721A>C p.(Thr241Pro) ‘Costello’
NSML
(1, 2)
c.730A>C;
c.730A>G
p.(Thr244Pro) CFC (3)
c.735A>T;
c.735A>C;
c.735A>G
p.(Leu245Phe) CFC
NSML
(1)
unpublished
(4)
c.736G>C p.(Ala246Pro) CFC (5)
p.(Phe247Val) CFC MCGM unpublished
c.770A>G p.(Gln257Arg) CFC (5)
c.769C>A p.(Gln257Lys) CFC (6)
c.785A>G p.(Gln262Arg) CFC (2)
c.784C>A p.(Gln262Lys) CFC (7)

307
p.(Gln262Pro) CFC? MCGM unpublished
c.823G>A p.(Glu275Lys) CFC (1)
p.(Arg384Gly) NS (8)
c.1390G>C p.(Gly464Arg) CFC (2, 9)
c.1391G>T p.(Gly464Val) CFC (10)
c. p.(Gly466Arg) CFC (2)
p.(Gly466Glu) CFC (11)
c.1399T>G p.(Ser467Ala) CFC (12)
c.1403T>C p.(Phe468Ser) CFC (12)
c.1406G>A p.(Gly469Glu) CFC (5)
p.(Thr470Pro) CFC (13); MCGM
c.1472G>T p.(Val471Phe) CFC (14)
c.1455G>C p.(Leu485Phe) CFC (5)
c.1454T>C p.(Leu485Ser) CFC (10)
c.1460T>G p.(Val487Gly) CFC (6)
c.1497A>C p.(Lys499Asn) CFC (7)
c.1495A>G p.(Lys499Glu) CFC/NS (5, 15)
c.1502A>G p.(Glu501Gly) CFC (5)
c.1501G>A p.(Glu501Lys) CFC/NS (5)
c.1502A>T p.(Glu501Val) CFC (2)

308
c.1574T>C p.(Leu525Pro) CFC (3)
c.1574T>A p.(Leu525Gln) CFC MCGM unpublished
c.1593G>C p.(Trp531Cys) NS (1)
c.1600G>C p.(Gly534Arg) CFC (16)
c.1695T>G p.(Asp565Glu) CFC (3)
c.1738A>G p.(Asn580Asp) CFC (6)
c.1741A>G p.(Asn581Asp) CFC (5)
c.1743T>G p.(Asn581Lys) CFC (2)
c.1785T>A;
c.1785T>G
p.(Phe595Leu) CFC (7)
(12)
c.1787G>T p.(Gly596Val) CFC (12)
c.1789C>G p.(Leu597Val) NS (1)
c.1796C>G p.(Thr599Arg) CFC (10)
c.1799T>G p.(Val600Gly) CFC (17)
c.1801A>C p.(Lys601Gln) CFC (1)
c.1802A>C p.(Lys601Thr) CFC (14)
c.1914T>G;
c.1914T>A
p.(Asp638Glu) CFC (1)
16)
c.2126A>G p.(Gln709Arg) CFC (1)
c.1384_1407del del462_469 CFC (10)

309
c.1408_1410del del470Thr CFC (10)
Deletion of
exon 11
CFC (11)
MAP2K1
Nucleotide
substitution
Predicted
protein
substitution
Phenotype References
c.124C>T p.Leu42Phe CFC (18)
c.131A>G p.Glu44Gly NS ? (2)
c.158T>C p.Phe53Ser CFC (12)
c.163A>C p.Thr55Pro ‘Costello’ (2)
c.199G>A p.Asp67Asn CFC (2)
c.371C>A p.Pro124Gln CFC (3)
c.371C>T p.Pro124Leu CFC (6)
c.383G>T p.Gly128Val CFC (7)
c.388T>A p.Tyr130Asn CFC (10)
c.389A>G p.Tyr130Cys CFC (12)
c.388T>C p.Tyr130His CFC (18)
c.607G>C p.Glu203Gln CFC (15)

310
c.175_177del del59 CFC (3)
MAP2K2
Nucleotide
substitution
Predicted
protein
substitution
Phenotype References
c.170T>C p.(Phe57Cys) CFC (12)
c.169T>A p.(Phe57Ile) CFC (19)
c.171T>A p.(Phe57Leu) CFC (15)
c.169T>G p.(Phe57Val) CFC (7)
c.181A>G p.(Lys61Glu) CFC (6)
c.182A>T p.(Lys61Thr) CFC (2)
c.184G>C p.(Ala62Pro) CFC (2)
c.383C>G p.(Pro128Arg) CFC (6)
c.383C>A p.(Pro128Gln) CFC (20)
c.395G>T p.(Gly132Val) CFC (6)
c.395G>A p.(Gly132Asp) CFC (21)
c.401A>G p.(Tyr134Cys) CFC (10)
c.400T>C p.(Tyr134His) CFC (7)
c.818A>G p.(Lys273Arg) CFC (6)

311
c.135_165del del46_55 CFC (2)
c.186_197del del63_66 CFC (18)
KRAS
Nucleotide
substitution
Predicted
protein
substitution
Phenotype Reference
c.15A>T p.(Lys5Asn) ‘Costello’ (22)
c.13A>G p.(Lys5Glu) ‘Costello’
CFC
(23)
(2)
c.34G>A p.(Gly12Ser) CFC (2)
c.40G>A p.(Val14Ile) NS (24)
c.65A>G p.(Gln22Arg) NS (22)
c.64C>G p.(Gln22Glu) CFC (22)
c.77A>T p.(Asn26Ile) NS (25)
c.101C>G p.(Pro34Arg) CFC (24)
c.101C>A p.(Pro34Gln) NS (22)
c.101C>T p.(Pro34Leu) NS (22)
c.108A>G p.(Ile36Met) NS (22)
c.173C>T p.(Thr58Ile) NS (24)
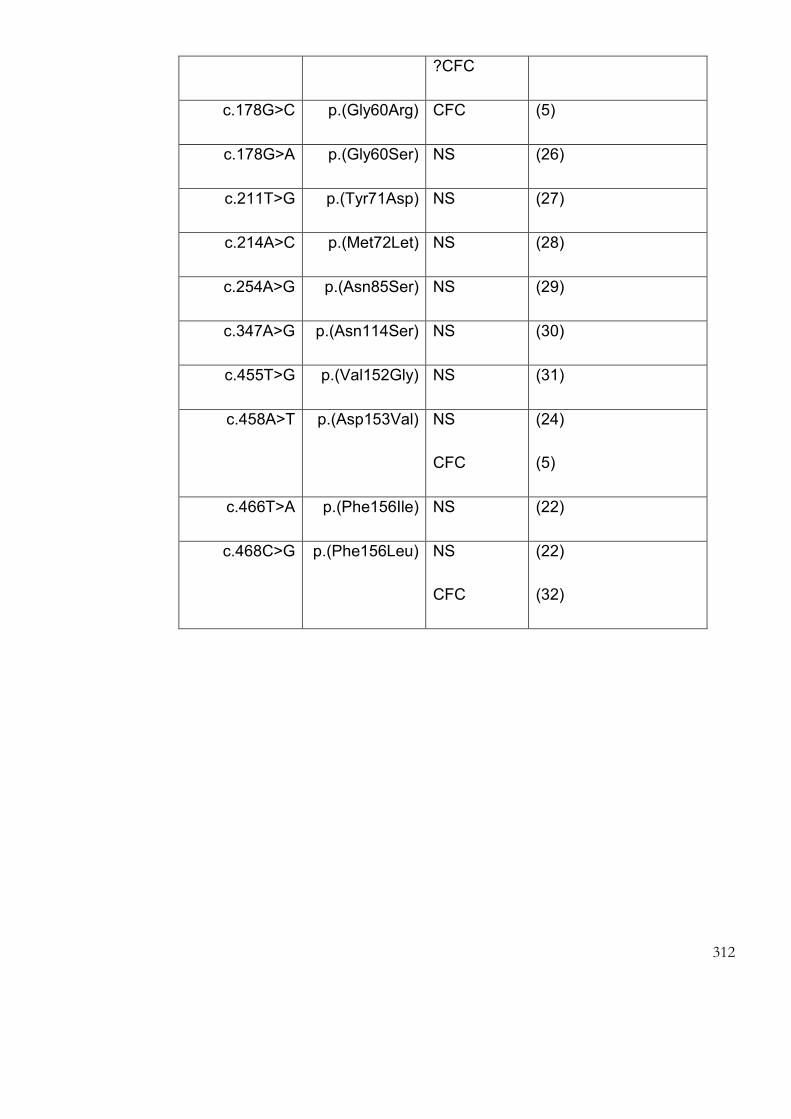
312
?CFC
c.178G>C p.(Gly60Arg) CFC (5)
c.178G>A p.(Gly60Ser) NS (26)
c.211T>G p.(Tyr71Asp) NS (27)
c.214A>C p.(Met72Let) NS (28)
c.254A>G p.(Asn85Ser) NS (29)
c.347A>G p.(Asn114Ser) NS (30)
c.455T>G p.(Val152Gly) NS (31)
c.458A>T p.(Asp153Val) NS
CFC
(24)
(5)
c.466T>A p.(Phe156Ile) NS (22)
c.468C>G p.(Phe156Leu) NS
CFC
(22)
(32)

313
9.2 Appendix 2

314

315
This leaflet gives information about a research study into a group of conditions
called the RAS-MAPK pathway disorders. These disorders arise from changes in
genes, which are the instruction manuals for the body’s cells. RAS-MAPK
disorders include cardio-facio-cutaneous, Noonan and Costello syndromes (CFC,
NS and CS). Please ask if there is anything that is not clear, or if you would like
more information to help you decide whether or not to take part.
Why is the study being done?
We want to know more about the problems that people with these conditions may
have, so that we can look after them better in the future. All of these disorders
involve changes in the way that one particular pathway, called the RAS-MAPK
pathway, works in the cells of the body. Affected people may have a wide variety
of problems including heart disease, poor growth, skin changes and learning
problems. The underlying gene changes that cause these are now clear in many,
but not all, cases. We hope that other genes that cause these conditions can be
identified, as this could also lead to improved understanding of the conditions.
Who is carrying out the research?
The study is being conducted by Dr Emma Burkitt Wright and Dr Bronwyn Kerr
from the Genetic Medicine department at St Mary’s Hospital, Manchester. Dr
Burkitt Wright is doing this study as part of a PhD at the University of Manchester,
and the project will be carried out over the next three years. Dr Kerr is the
Understanding the disorders of the RAS-MAPK pathway:

316
Educational Supervisor for this project. The project has been funded by the
Wellcome Trust, and the North West Ethics Committee has reviewed it (application
number 10/H1003/77).
Why have I been chosen?
We are contacting you because you have Noonan syndrome, cardio-facio-
cutaneous syndrome or Costello syndrome, or a condition with similarities to this
group of disorders.
What will taking part involve?
If you agree to take part in the research project, we will ask your permission to
allow the doctor looking after you to provide us with information about your
medical history and any tests you have had done. We are particularly interested in
growth and development, and any health problems you may have had.
The study assessment itself will involve a clinical history and physical examination
(as happen when you attend other specialist doctors). We would then like to do
some tests that assess learning and memory, which are laptop computer
programs (like simple games). This is because we think that there are some
important differences in how people with conditions like yours think and learn.
Understanding these better could lead to improved ways of teaching and
supporting the learning of children with these conditions in the future. These tests
take a variable amount of time, but overall the time taken by all of these things
should not exceed three hours. If you are tired and need a break at any point then
just ask; this is fine.

317
We will ask you for permission to use your DNA, extracted from a blood or saliva
sample, to check for genetic changes which could be causing your condition. If a
blood sample has been taken previously for genetic testing, this can be used, if
you consent to this. If no stored sample is available, then a new sample can be
taken at the same time as we see you for other parts of the study. If you prefer, a
blood sample could be taken when any other tests are being done by your own
doctors and forwarded to us. If you were having an operation, we would ask if a
tiny piece of tissue could be taken for us to analyse in the study. This is because
we think that there may be important differences in how the RAS-MAPK pathway
acts in different body tissues. No tissue other than what was being operated on
would be taken. There would be no differences to the operation or the getting
better from it as a result.
If a fresh blood sample is being taken, or if a skin biopsy is taken at the time of an
operation, we may ask if you would be prepared for a cell line to be made from this
and gifted to us for use in the research. This means that cells with the DNA of the
affected person could be frozen down and stored securely (like their DNA sample)
and available for further studies.
We might also ask if your parents would be prepared to have blood samples taken
too. This is so that any differences found in your sample could be further
assessed. This is helpful because when a change has arisen newly in a person
with a particular condition, rather than having been inherited from their parents,
this can suggest that the change may cause the condition.
We will ask if we can take photographs of you, or use pictures that have been sent
to us by your clinical geneticist. All information and photographs will be stored
securely on a computer database that is only accessible to members of our

318
research team. Personal, identifying, details will be stored separately, also on a
secure database or in a locked filing cabinet.
We will ask you to sign a consent form for the study and forward a copy to us. If
you decide to take part, and give permission for us to contact your family doctor,
we will let him or her know that you are taking part in the study.
Do I have to take part in the study?
You have a completely free choice about whether to enter the study. If you do not
want to take part, you do not have to give a reason. Deciding not to take part will
not affect the medical care that you receive. You can also choose to stop taking
part in the study at any time. This would not affect your medical care in any way.
Are there any benefits to taking part?
There may be no direct benefits to you personally, but we hope that you find taking
part worthwhile. The findings of this study will help us to understand disorders like
yours better. This could improve the care of people with conditions like yours in the
future. When the work is complete, we will send you a summary of the results.
Are there any disadvantages to taking part in the research?
For anyone having a new blood sample taken, then there is a small risk of bruising
or discomfort at the skin site. This can be minimised by using local anaesthetic
spray. A further possible risk of this study is that testing might reveal a gene
change which is either unexpected or which has implications for the affected
person or other family members. If this situation arises, we will arrange for all
relevant individuals to be seen by a clinical geneticist within their local genetic

319
service, so that this can be explained fully and any important findings acted upon.
Similarly, if anyone becomes distressed in the course of taking part in the study,
we would arrange for an appointment for genetic counselling regarding this.
What if something goes wrong?
It is very unlikely that anyone will come to any harm as a result of taking part in
this research. If taking part causes you any worries, we can arrange for you to
discuss these with a research nurse or with your local clinical geneticist.
Complaints
If you have a concern about any aspect of this study, you should ask to speak to
us, the researchers, and we will do our best to answer your questions. If we are
unable to resolve your concern, or you wish to make a complaint regarding the
study, please contact a University Research Practice and Governance Co-
ordinator on 0161 2757583 or 0161 2758093 or by email to research-
Harm
In the event that something does go wrong and you are harmed during the
research, you may have grounds for a legal action for compensation against The
University of Manchester and Central Manchester Foundation Trust, but you may
have to pay your legal costs. The normal National Health Service complaints
mechanisms will still be available to you.
The University of Manchester has cover for no fault compensation for bodily injury,
mental injury or death where the injury resulted from a trial or procedure you
received as part of the trial. This would be subject to policy terms and conditions.
Any payment would be without legal commitment. (Please ask if you wish more

320
information on this).
The University would not be bound to pay this compensation where the injury
resulted from a drug or procedure outside the trial protocol or the protocol was not
followed.
Will my taking part in the study be kept confidential?
Yes. The names of participants will not be used in any publication or shown to any
person. All information will be kept strictly confidential. Any information will have
names and addresses removed so that no-one can be recognised from it. Any
information that is used in publications or presentations will have all names, dates
of birth and other identifiers removed. Notes and computer files will not be shown
to anyone outside the research team, except for individuals representing the
Research Sponsor or Regulatory authorities (for the purpose of monitoring or
auditing the study).
Will I be paid for participation?
We will not be able to offer any payment for helping with this study, but we will
happily refund any extra costs, such as travel expenses (mileage costs, or public
transport costs, on production of receipts), that you incur as a result of taking part.

321
Further Information
If you require any further information, please contact:
Dr Emma Burkitt Wright Dr Bronwyn Kerr
[email protected] [email protected]
Genetic Medicine, St Mary’s Hospital, Manchester, M13 9WL
Tel: 0161 901 2335 Fax : 0161 276 6145
What happens now?
If you are happy to help with the research, please can you return the consent form
to us in the reply paid envelope provided.
Thank you for taking the time to read about this study

322
This leaflet gives information about a research study into a group of conditions
called the RAS-MAPK pathway disorders. These disorders arise due to changes in
genes, the instruction manuals for the body’s cells. RAS-MAPK pathway disorders
include cardio-facio-cutaneous, Noonan and Costello syndromes (CFC, NS and
CS). We hope that this leaflet will help you to understand why the research is
being done and what it will involve. Please ask if there is anything that is not clear,
or if you would like more information to help you decide whether or not your child
should take part.
What is the purpose of the study?
We want to know more about the problems that people with these conditions may
have, so that we can look after them better in the future. All of these disorders
involve changes in the way that one particular pathway, called the RAS-MAPK
pathway, works in the cells of the body. Affected people may have a wide variety
of problems including heart disease, poor growth, skin changes and learning
problems. The underlying gene changes that cause these are now clear in many,
but not all, cases. We hope that other genes that cause these conditions can be
identified, as this could also lead to improved understanding of the conditions.
Who is carrying out the research?
The study is being conducted by Dr Emma Burkitt Wright and Dr Bronwyn Kerr
from the Genetic Medicine department at St Mary’s Hospital, Manchester. Dr
Burkitt Wright is doing this study as part of a PhD at the University of Manchester,
and the project will be carried out over the next three years. Dr Kerr is the
Understanding the disorders of the RAS-MAPK pathway:

323
Educational Supervisor. The project has been funded by the Wellcome Trust, and
the North West Ethics Committee has reviewed it (application number
10/H1003/77).
Why have I been chosen?
We are contacting you because you have a child with cardio-facio-cutaneous,
Noonan or Costello syndrome, or a condition with similarities to this group of
disorders.
What will taking part involve?
If you agree to involvement in the research project, we will ask your permission to
allow the doctor looking after your child to provide us with information about his or
her medical history and any tests performed to date. This information will focus on
growth, health problems and development.
We will ask permission to use a sample of blood or saliva from your child to check
for genetic changes which may give rise to their condition. If a sample has been
taken previously for genetic testing, this may be used, if you consent to this. If no
stored sample is available, then a new sample can be taken at the same time as
the appointment for other aspects of the study. If you prefer, arrangements could
be made for this to be done locally with any other tests that are being done, and
sent on to us. If your child is having an operation, we will ask if a tiny piece of
tissue could be taken for us to analyse in the study. This is because we think that
there may be important differences in how the RAS-MAPK pathway acts in
different body tissues. No tissue other than that being operated upon would be
taken, and there would be no differences to the operation or recovery from it as a
result. If a fresh blood sample is being taken, or if a skin biopsy is taken at the time

324
of an operation, we may ask if you would be prepared for a cell line to be made
from this and gifted to us for use in the research. This means that cells with the
DNA of the affected person could be frozen down and stored securely (like their
DNA sample) and available for further studies in the University of Manchester.
The study assessment itself involves taking a clinical history and doing a physical
examination of your child (as happens when you attend other specialist doctors).
We would then like to do some tests that assess learning and memory, which are
laptop computer programs (like simple games). This is because we think that there
are some important differences in how children with these disorders think and
learn. Understanding these better could lead to improved ways of teaching and
supporting the learning of your child and other children with these conditions in the
future. The total time taken for these tests is variable, but overall the time taken by
these assessments should not exceed three hours. If you or you child need a
break in this time then just ask, this is fine.
We might also ask if you, as parents, could provide blood samples too, so that any
possible changes found in your child’s sample could be further assessed. This is
helpful because when changes have arisen newly in the individual, rather than
having been inherited from one of their parents, these are more likely to be
important in explaining their pattern of differences.
We will ask you if we can take photographs of your child, or use pictures that have
been sent to us by their clinical geneticist. All information and photographs will be
stored securely on a computer database that is only accessible to members of our
research team. Personal, identifying, details will be stored separately, also on a
secure database or in a locked filing cabinet.

325
We will ask you to sign a consent form for the study and forward a copy to us. If
you decide to take part, and give us permission to contact them, we will let your
family doctor know that your child is taking part in the study.
Do I have to take part in the study?
You have a completely free choice about whether to enter the study. If you do not
want your child to take part, you do not have to give a reason. Deciding not to take
part will not affect the medical care that your family receive. You can also choose
to stop taking part in the study at any time. This would not affect anyone’s medical
care in any way.
Are there any benefits to taking part?
There may be no direct benefits to your family, but we hope that you find taking
part worthwhile. The findings of this study will help us to understand better the
causes and effects of RAS-MAPK pathway disorders. This could improve the care
of people with these conditions in the future. When the work is complete, we will
send you a summary of the results.
Are there any disadvantages to taking part in the research?
For anyone having a new blood sample taken, then there is a small risk of bruising
or discomfort at the skin site. This can be minimised by using local anaesthetic
spray. A further possible risk of this study is that testing might reveal a gene
change which is either unexpected or which has implications for your child or other
family members. If this situation were to arise, we would arrange for all relevant
individuals to be seen by a clinical geneticist within their local genetic service, so
that this can be explained fully and any important findings acted upon. Similarly, if

326
anyone were to become distressed in the course of taking part in the study, we
would arrange for an appointment for genetic counselling regarding this.
What if something goes wrong?
It is very unlikely that any participant will come to any harm as a result of taking
part in this research. If taking part causes you any worries, we can arrange for
you to discuss these with a research nurse or with your local clinical geneticist.
Complaints
If you have a concern about any aspect of this study, you should speak to us, the
researchers, and we will do our best to answer your questions. If we are unable to
resolve your concern, or you wish to make a complaint regarding the study, please
contact a University Research Practice and Governance Co-ordinator on 0161
2757583 or 0161 2758093 or by email to research-
Harm
In the event that something does go wrong and you are harmed during the
research, you may have grounds for a legal action for compensation against The
University of Manchester and Central Manchester Foundation Trust, but you may
have to pay your legal costs. The normal National Health Service complaints
mechanisms will still be available to you.
The University of Manchester has cover for no fault compensation for bodily injury,
mental injury or death where the injury resulted from a trial or procedure you
received as part of the trial. This would be subject to policy terms and conditions.
Any payment would be without legal commitment. (Please ask if you wish more
information on this).

327
The University would not be bound to pay this compensation where the injury
resulted from a drug or procedure outside the trial protocol or the protocol was not
followed.
Will my taking part in the study be kept confidential?
Yes. The names of participants will not be used in any publication or shown to any
person. All information will be kept strictly confidential. Any information will have
names and addresses removed so that no-one can be recognised from it. Any
information that is used in a presentation or publication will have all names, dates
of birth and other identifiers removed. Notes and computer files will not be shown
to anyone outside the research team, except for individuals representing the
Research Sponsor or Regulatory authorities (for the purpose of monitoring or
auditing the study).
Will I be paid for participation?
We will not be able to offer any payment for helping with this study, but we will
happily refund any extra costs, such as travel expenses (mileage costs, or public
transport costs, on production of receipts), that you incur as a result of taking part.

328
Further Information
If you require any further information, please contact:
Dr Emma Burkitt Wright Dr Bronwyn Kerr
Genetic Medicine, St Mary’s Hospital, Manchester, M13 9WL
Tel: 0161 901 2335 Fax : 0161 276 6145
What happens now?
If you are happy to help with the research, please can you return the consent form
to us in the reply paid envelope provided.
Thank you for taking the time to read about this study

329
This leaflet gives information about a research study into a group of conditions
called the RAS-MAPK pathway disorders. These disorders include cardio-facio-
cutaneous, Noonan and Costello syndromes (CFC, NS and CS). You have been
asked to consider whether it could be appropriate for the person you know with
one of these conditions to take part in the study, as they either cannot decide for
themselves or need help with this. Please ask if there is anything that is not clear,
or if you would like more information to help you decide whether or not it may be
for the best for them to take part.
Why is the study being done?
We want to know more about the problems that people with these conditions may
have, so that we can look after them better in the future. All of these disorders
involve changes in the way that one particular pathway, called the RAS-MAPK
pathway, works in the cells of the body. Affected people may have a wide variety
of problems including heart disease, poor growth, skin changes and learning
problems. The underlying gene changes that cause these are now clear in many,
but not all, cases. We hope that other genes that cause these conditions can be
identified, as this could also lead to improved understanding of the conditions.
Who is carrying out the research?
The study is being conducted by Dr Emma Burkitt Wright and Dr Bronwyn Kerr
from the Genetic Medicine department at St Mary’s Hospital, Manchester. Dr
Burkitt Wright is doing this study as part of a PhD at the University of Manchester,
and the project will be carried out over the next three years. Dr Kerr is the
Understanding the disorders of the RAS-MAPK pathway:
330
Educational Supervisor. The project has been funded by the Wellcome Trust, and
the North West Ethics Committee has reviewed it (application number
10/H1003/77).
Why have I been chosen?
We are consulting you because you know a person who has Noonan syndrome,
cardio-facio-cutaneous syndrome or Costello syndrome, or a condition with
similarities to this group of disorders, who would be eligible for this study. Because
of their condition, they are not able to make the decision by themselves as to
whether or not to take part, which is why we would like your opinion.
What will taking part involve?
If you agree that this person could take part in the research project, we will ask the
doctor looking after you to provide us with information about your medical history
and any tests you have had done. We are particularly interested in growth, health
problems and development.
We will use DNA, extracted from a blood or saliva sample, to check for genetic
changes which could be causing the affected person’s condition. If a blood sample
has been taken previously for genetic testing, this can be used. If no stored
sample is available, then a new sample can be taken at the same time as we see
the person for other parts of the study. If it is more suitable, a blood sample could
be taken when any other tests are being done by other doctors, and forwarded to
us. If the person were having an operation, we would ask if a tiny piece of tissue
could be taken for us to analyse in the study. This is because we think that there
may be important differences in how the RAS-MAPK pathway acts in different
body tissues. No tissue other than what was being operated upon would be taken,

331
so there would be no differences to the operation or recovery from it as a result. If
a fresh blood sample is being taken, or if a skin biopsy is taken at the time of an
operation, we may ask if you would be prepared for a cell line to be made from
this, and gifted to us for use in the research. This means that cells with the DNA of
the affected person could be frozen down and stored securely (like their DNA
sample) and available for further studies.
The study assessment itself involves taking a clinical history and doing a physical
examination of the affected person (as happens when attending other specialist
doctors). We would then like to do some tests that assess learning and memory,
which are laptop computer programs (like simple games). This is because we think
that there are some important differences in how people with these conditions
think and learn. Understanding these better could lead to improved ways of
teaching and supporting the learning of children with these conditions in the future.
Where possible, we might also ask if the affected person’s parents would be
prepared to have blood samples taken too. This is so that any differences found
could be further assessed. This is helpful because when a change has arisen
newly in a person with a particular condition, rather than having been inherited
from their parents, this can suggest that the change may be responsible for the
condition.
We will ask you if you think that it would be possible to take photographs of the
affected person, or use pictures that have been sent to us by their clinical
geneticist. All information and photographs will be stored securely on a computer
database that is only accessible to members of our research team. Personal,
identifying, details will be stored separately, also on a secure database or in a
locked filing cabinet.

332
We will ask you to sign a declaration form for the study and forward a copy to us.
If you decide that the person should be able to take part, we will contact his or her
family doctor, to let him or her know that you are taking part in the study.
Does the person have to take part in the study?
If you think that the person would object to taking part in the study, or would be
likely to be adversely affected by doing so, then you should discuss this with us.
Similarly, if in the course of taking part in the study, you think that the person
would be better served by not continuing, you should let us know. This would not
affect their medical care in any way.
Are there any benefits to taking part?
There may be no direct benefits to participants, but the findings of this study will
help us to understand better the causes and effects of RAS-MAPK pathway
disorders. This could improve the care of people with these conditions in the
future. It could be particularly important to include people like the person you know
in order to understand the long-term outcomes for people with these conditions, as
these are not well understood at the moment. When the work is complete, we will
send you a summary of the results if you wish.
Are there any disadvantages to taking part in the research?
For anyone having a new blood sample taken, then there is a small risk of bruising
or discomfort at the skin site. This can be minimised by using local anaesthetic
spray. A further possible risk of this study is that testing might reveal a gene
change which is either unexpected or which has implications for the affected
person or other family members. If this situation were to arise, we would arrange

333
for all relevant individuals to be seen by a clinical geneticist within their local
genetic service, so that this can be explained fully and any important findings
acted upon. Similarly, if any individual were to become distressed in the course of
taking part in the study, we would arrange for an appointment for genetic
counselling regarding this.
What if something goes wrong?
It is very unlikely that any participant will come to any harm as a result of taking
part in this research. If you are worried about any aspect of taking part in the
study, we can arrange for you to discuss this with a research nurse or with your
local clinical geneticist.
Complaints
If you have a concern about any aspect of this study, you should ask to speak to
the researchers who will do their best to answer your questions. If they are unable
to resolve your concern, or you wish to make a complaint regarding the study,
please contact a University Research Practice and Governance Co-ordinator on
0161 2757583 or 0161 2758093 or by email to research-
Harm
In the event that something does go wrong and anyone is harmed during the
research, there may be grounds for a legal action for compensation against The
University of Manchester and Central Manchester Foundation Trust, but you may
have to pay your legal costs. The normal National Health Service complaints
mechanisms will still be available to you.
The University of Manchester has cover for no fault compensation for bodily injury,
mental injury or death where the injury resulted from a trial or procedure you

334
received as part of the trial. This would be subject to policy terms and conditions.
Any payment would be without legal commitment. (Please ask if you wish for more
information on this).
The University would not be bound to pay this compensation where the injury
resulted from a drug or procedure outside the trial protocol or the protocol was not
followed.
Will my taking part in the study be kept confidential?
Yes. The names of participants will not be used in any publication or shown to any
person. All information will be kept strictly confidential. Any information will have
names and addresses removed so that no-one can be recognised from it. We
may use information about participants in publications or presentations about this
research. Any information that is used will have all names, dates of birth and other
identifiers removed. Notes and computer files will not be shown to anyone outside
the research team, except for individuals representing the Research Sponsor or
Regulatory authorities (for the purpose of monitoring or auditing the study).
Will I be paid for participation?
We will not be able to offer any payment for helping with this study, but we will
happily refund any extra costs, such as travel expenses (mileage costs, or public
transport costs, on production of receipts), that might be incurred as a result of
taking part.

335
Further Information
If you require any further information, please contact:
Dr Emma Burkitt Wright Dr Bronwyn Kerr
[email protected] [email protected]
Genetic Medicine, St Mary’s Hospital, Manchester, M13 9WL
Tel: 0161 901 2335 Fax : 0161 276 6145
What happens now?
If you are content that the eligible person could take part in the research, please
can you return the declaration form to us in the reply paid envelope provided.
Thank you for taking the time to read about this study.
Study ID Number:
LLLLLLL

336
Study: Understanding the disorders of the RAS-MAPK pathway
Investigators: Dr Emma Burkitt Wright, Dr Bronwyn Kerr
CONSENT FORM
Participant’s Name (block capitals):LLLLLLLLLLLLLLLLLLL
Age:LLLLLLLLLLLLL
I understand the information that has been given to me about the study. I
am happy with the answers I have had to my questions.
I know that I do not have to take part, and can stop taking part at any time.
This will not have any effect on the care I receive.
I know that the research team and other authorised people may access
relevant parts of my medical records as part of the study.
I know that all information collected in the study will be kept private. Any
information about me that is presented will have my name and other
personal details removed.
PLEASE INITIAL EACH

337
I give consent for my DNA (extracted from a sample of blood or saliva) to be used for research looking at the cause of my condition.
I give consent for a sample of my blood to be used for research looking at the cause of my condition.
I give consent for a cell line to be made from a sample of my blood/skin (delete as appropriate) and gifted to the University of Manchester to be used for research looking at the cause of my condition.
I give consent for a sample of tissue (specify___________________) taken at the time of my operation to be used for research looking at the cause of my condition.
I give consent for each of the samples above to be stored and used for research in future studies into the cause of genetic disorders.
338
I give consent for photographs of me to be taken/reviewed.
These photographs may be:
a. Kept on a computer database accessed only by research staff and in accordance with the data protection act
b. Used to record my physical features as part of the research study
c. Used to teach other medical personnel e.g. in lectures and presentations at medical meetings
The photographs may be kept by the researchers after the end of the study for these purposes.
I am happy for my GP to be informed.
I would like to receive a summary of the research findings at the end of the
study.
I agree to take part in this study
Name of participant Date Signature
Name of person taking consent (if
different from researcher)
Date Signature
Name of Researcher Date Signature

339
Title of Study: Understanding the disorders of the RAS-MAPK pathway
Investigators: Dr Emma Burkitt Wright, Dr Bronwyn Kerr
CONSULTEE DECLARATION FORM
FOR A PERSON SIGNING ON BEHALF OF AN ADULT UNABLE TO CONSENT FOR
HIMSELF OR HERSELF
I have been consulted about LLLL.’s potential participation
in this research project. I have had the opportunity to ask
questions about the study and understand what is involved.
In my opinion he/she would have no objection to taking part in
this study.
I understand that I can request that he/she is withdrawn from
the study at any time, without giving any reason and without
his/her care or legal rights being affected.
I know that the research team and other authorised people may
access relevant parts of his/her medical records as part of the
study.
Study ID Number:
LLLLLLL

340
I know that all information collected in the study will be kept
private. Any information about participants that is presented will
have names and other personal details removed.
I understand that DNA from the participant will be used for research looking at the cause of his/her condition.
I understand that a sample of my blood may be used for research looking at the cause of his/her condition.
I understand that if a cell line is made from a sample of blood or skin from the participant, this would be gifted to the University of Manchester to be used for research looking at the cause of his/her condition.
I understand that a sample of tissue taken at the time of the participant’s operation may be used for research looking at the cause of his/her condition.
I understand that each of the samples above would be stored and used for research in future studies into the cause of genetic disorders.
Name of consultee Date Signature
Name of person undertaking consultation
(if different from researcher)
Date Signature
Name of Researcher Date Signature

341
Parent’s Name (block capitals):LLLLLLLLLLLLLLLLLLL
Participant’s name (block capitals): LLLLLLLLLLLLLLLLLL.
I confirm that I have read and understood the information sheet dated
October 2010, (version 3) for this study. I have been able to consider
the information and ask questions. I am happy with the answers I have
received to my questions.
I have had enough time to think about the study, talk to relatives and
friends about it and to decide without pressure if I want my child to take
part.
I understand that it is my choice whether or not to take part. I am free to
stop taking part in the study at any time, without giving a reason. Doing
this would not affect our medical care or legal rights in any way.
I understand that information collected during this study may be looked
at by the research team and by individuals from regulatory authorities
or from the NHS Trust, where it is relevant to my child taking part in this
research.
I have been assured that all information collected in the study will be
held in confidence. Any information gathered in the study will have
personal details removed before it is presented.
PLEASE INITIAL EACH
Study: Understanding the disorders of the RAS-
MAPK pathway

342
I give consent for a sample of blood/saliva from myself to be used for
research to determine the cause of my child’s condition.
I give consent for my sample to be stored and used for genetic
research in future studies into the cause of genetic disorders.
I agree to take part in this study.
I would like to receive a summary of the research findings at the end of
the study.
Name of person giving sample Date Signature
Name of person taking consent (if
different from researcher)
Date Signature
Name of Researcher Date Signature

343
Study: Understanding the disorders of the RAS-MAPK pathway
Investigators: Dr Emma Burkitt Wright, Dr Bronwyn Kerr
ASSENT FORM FOR CHILDREN
Participant’s Name (block capitals):LLLLLLLLLLLLLLLLLLL
Age:LLLLLLLLLLLLL
(delete any that do not apply)
I understand what the study is about.
I understand that I can choose whether or not I take part.
I know that any information that is collected about me will be kept private.
I understand that a sample from my blood or saliva will be used for research looking at the cause of my condition.
I understand that a sample from my operation may be used for research looking at the cause of my condition.
I understand that the samples will be stored and used for research in future studies.
I understand that photographs of me will be taken and used in the study.
Study ID Number:
LLLLLLL
PLEASE TICK or INITIAL EACH
344
I agree to take part in this study
Name of participant Date Signature
Name of person taking consent (if different
from researcher)
Date Signature
Name of Researcher Date Signature

345
9.2.1 Appendix 3a

346
9.2.2 Appendix 3B
Understanding the disorders of the RAS-MAPK pathway
Investigators: Dr Emma Burkitt Wright, Dr Bronwyn Kerr
Background to the Study
Germline disorders of the RAS-MAPK pathway arise due to dominant mutations which cause
RAS-MAPK pathway dysregulation [1]. Cardio-facio-cutaneous (CFC) and Costello syndrome
(CS) are the most severe conditions in this group of disorders, and have overlapping features,
including congenital heart anomalies, growth failure and learning disability, all of which may
be severe [2]. Such features also occur commonly in other RAS-MAPK disorders including
neurofibromatosis type I and Noonan syndrome, which jointly affect over 1 in 1000 of the
population [2]. Progressive cardiomyopathy, epilepsy, scoliosis and other bone problems can
occur in CFC and related disorders [2].
The RAS-MAPK pathway has been studied for many years in view of its key role in
oncogenesis. Its signalling cascades result in activation of extracellular signal-related kinases,
which, translocating to the nucleus, upregulate transcription of many genes influencing cell
cycling and apoptosis [3]. Somatic mutations of genes of this pathway that are found in cancer,
like those found in the germline disorders, show altered kinase activity [3].
Genetics of germline RAS-MAPK disorders
There are many different genes that are now known to cause RAS-MAPK pathway disorders,
but a proportion of patients with a presentation strongly suggestive of this type of condition
do not have an identifiable mutation at present: as many as 40% of patients with a clinical
diagnosis of CFC or Noonan syndrome have no identifiable mutation currently [2].

347
For the individuals and families we see in clinic, one main reason for trying to find the genetic
basis for their or their child’s condition is so that they can find out accurate information about
the risks of recurrence in further children, any risks for the extended family and the
possibilities of a prenatal test in another pregnancy. A further important reason for
identifying the genetic basis of an individual’s condition is that it may in future predict their
response to potential therapies for these disorders.
Aim of this current study
1. To characterise the clinical presentations of a group of patients with features of
cardio-facio-cutaneous syndrome, a poorly-understood but frequently severe condition
which is due to dysregulated RAS-MAPK pathway signalling. The aim for the number of
patients to be recruited is 30, but this will depend upon how many families wish to take
part in the research.
2. To search for the genetic basis of the clinical presentation in patients with CFC and
related disorders using newer types of genetic analysis including a) high resolution
microarray analysis and b) high throughput sequencing. This will be of benefit to the
individuals and families themselves, and will also be useful in guiding the development of
new diagnostic testing strategies for this group of disorders.
3. To make available any findings from this study so that they can be used to aid patient
management and genetic counselling in the families involved.

348
Plan of investigation
This study will investigate the phenotypes of a group of patients with clinical presentations
suggestive of a germline RAS-MAPK pathway disorder. Some of these will have a previously
confirmed molecular diagnosis, whilst others will not.
a) Recruitment of patients to the study will be via clinical genetics colleagues. Geneticists who
have referred samples to the Manchester Regional Genetics Laboratory for genetic testing of
CFC or CS will be contacted in this regard. The study will also be publicised via the support
groups for these conditions, and potential participants will be invited to contact the
investigators, giving permission for their geneticist to be contacted for further details to assess
their eligibility for inclusion in the study. Interested potential participants will receive (either
via their geneticist or by post) a patient information sheet and consent form with a self
addressed envelope in which to return this.
Capacity to consent to involvement in the research will be made in accordance with the British
Psychological Society guidelines checklist (issued 2008, see reference 4) jointly by the
participant’s geneticist and the research team. The former will assess on the basis of their
knowledge of the patient and his or her level of function, and the latter on the basis of ability
to understand the purpose of the study and retain this information long enough to make a
considered decision whether or not to participate. For potential participants with limited
reading skills, extra help will be offered with verbal explanation of what is involved. For any
participant where there remains a question over capacity to consent, advice and opinion will
be sought from the individual’s next of kin or general practitioner.
Adults who have capacity to give informed consent to participation will be asked to sign a
consent form if they wish to enter the study. For potential participants who are children,
parents/guardians will be asked to give consent to their inclusion. For vulnerable adults, who

349
will be few in number but an important group to include with respect to the long-term natural
history of these conditions, information will be provided in a format that they can understand,
and their capacity to give informed consent will be assessed by the doctor making the initial
approach. Where a potential participant does not have capacity to consent to involvement in
the study, their parent, guardian or advocate will be asked to consider whether or not taking
part in the study is in their best interests. If this person and the doctor making the initial
approach concur that involvement in the study would not be harmful, and could be potentially
beneficial to the individual, then the parent, guardian or advocate will be sent an assent form
to be returned in the same way as the parental consent form for children.
Once consent has been granted, we will contact the patient’s clinical geneticist for clinical
details. Where patients will have had previous DNA studies and DNA has been stored as part
of their clinical care, we will seek to use these samples if possible rather than asking
participants to undergo further venepuncture. Samples from the parents will also be requested,
where this is possible.
Patients will be asked if they consent to their clinical details being entered on a research
database and anonymous data and photographs being reviewed by a panel of
dysmorphologists. They will be asked to consent to the use of their DNA samples and any
tissue samples that may become available for genetic studies of their condition. If consent is
granted, the patient’s GP will be informed about his or her participation in the study.
b) A secure database will be set up, to record the details of the patients, clinical information
and results of molecular testing. This will be a Microsoft Access database, which will be
password protected and accessible only by the investigators and the genetics research co-
ordinator who would only need access to details in an emergency, if the investigators were
absent. Names will be kept on the database as it will be necessary to refer back to individual
families if genetic changes are identified. All other identifying data will be kept on paper
proformas in a locked filing cabinet.

350
c) Participants will be offered an appointment either in clinic or at their home, whichever is
more convenient for them. The content of this visit will consist of clinical history taking and
examination (similar to that which would be undertaken as routine clinical genetic work up),
followed by psychometric assessment using a well-validated set of tests, as listed below:
List of measures
1) The Paired Associated Learning Task from the well-known CANTAB battery, since there is
strong evidence that abnormalities in the Ras-MAPK pathway are associated with deficient
LTP. This test is known to assess hippocampal learning, which is associated with LTP
(Luciana, 2003).
2) Test of Everyday Attention (TEA-Ch), the Continuous Performance Test and the Parent
Version of the Conners ADHD/DSM-IV scale to investigate different aspects of attentional
function, as these are commonly deficient in children with NF1 (Manly et al., 1999; Conners,
2000; Conners, 1997).
3) Stockings of Cambridge, Spatial Working Memory, Stop Signal Task and
Intra/Extradimensional Set Shift from the CANTAB test battery, as well as the Behaviour
Rating Inventory for Executive Function (Gioia et al., 2000). These tests measure executive
function which, again, is commonly impaired in NF1.
4) Benton Judgment of Line Orientation, because this visuospatial test is one of the most
consistent measures on which patients with NF1 are impaired (Benton et al., 1976).

351
5) Wechsler Intelligence Scale for Children (WISC) to assess intellectual ability (Wechsler,
2004).
Test duration
Cantab tests will take approximately 10 minutes each to administer, JLO 10 minutes, the
TEA-Ch test about 25 minutes altogether, WISC about 30 minutes and the CPT-II 14
minutes.
The parental questionnaires should take 5+10 minutes (Conners’ scale) and BRIEF 15
minutes (provided parents don’t have learning problems themselves).
d) DNA samples will be forwarded to the DNA laboratory at St Mary’s Hospital and logged in
on the LIMS information system which is password-protected. Each sample will be given a
number which will be used from then on for identification purposes in the laboratory rather
than using the patient’s name.
Where a new blood sample is being taken, and if consent is granted, part of this sample will be
used to make a cell line. This will enable further studies to be undertaken regarding the effect
of RAS-MAPK pathway signalling in living cells. It will also be a source of further DNA from
the individual, should the original sample be exhausted (thereby preventing the need for repeat
blood sampling).
Where a participant is undergoing a surgical procedure, samples of tissue other than blood
may become available. If this were the case, we would seek consent from the participant or
their parent/guardian to obtain samples of any material made available in this way (for

352
example, skin biopsy). No samples would be taken that would not normally have been
accessible in the course of the procedure.
e) The clinical features of participants will be systematically assessed, in order to better
understand the range of problems encountered by patients with these disorders. Information
will be gathered from the details provided by the referring clinician, and by history taking and
clinical examination in person (by Dr Burkitt Wright). Clinical photographs will also be used
to record facial and other features apparent on examination. These will then be assessed by
the investigators and a consensus reached on the features that are present. Dr Kerr will be
blinded regarding the genotype of these patients, in order to minimise bias in this part of the
data analysis.
f) Microarray studies will be carried out on patient DNA using the Affymetrix SNP6.0
platform to look for small chromosomal imbalances in any patients who have not undergone
molecular cytogenetic studies of this type previously. Data will be analysed using appropriate
computer software in the array laboratory at St Mary’s Hospital. If an imbalance is identified,
FISH or QFPCR analysis (for small imbalances) will be used to verify the abnormality. Parents
will also be screened to check whether the abnormality has occurred de novo, as this would be
of greater significance. The deleted/duplicated region would be checked against Ensembl and
other databases to identify whether any genes lay within or close to the region in question and
these would be considered as candidate genes for future studies. Care will also be taken to
look at genes just outside but close to any region of imbalance which could be implicated due
to a positional effect.
g) A targeted next-generation sequencing approach will be used to screen genes that could be
implicated in germline RAS-MAPK pathway disorders. The genes to be screened will be those
known or predicted to be involved in RAS-MAPK signalling, and will include those in areas of
the genome where chromosomal rearrangements/deletions /duplications have been identified
in patients with suggestive phenotypes. A next generation high throughput sequencer has

353
recently become available through the Manchester Biomedical Research Centre. Where
possible, samples from affected individuals will be run in parallel with those from their
parents, so that possible causal changes identified in the affected person can be verified as
being either de novo or inherited. The results of testing of parental samples will only be
analysed for genes in which possible changes have been identified in the affected individual,
minimising the (already small) risk that results of adverse significance to the asymptomatic
parent would be obtained. In the unlikely event of non-paternity, for example, being suggested
by the results of genetic testing, this information would not be disclosed to participating
individuals. The offer of genetic counselling (through Manchester Regional Genetics Service
or the participant’s local genetic service) will be available for any participant in this study.
Results
Any positive results will be confirmed in a CPA accredited diagnostic laboratory before being
fed back to the families concerned and their clinicians. Findings of importance to the genetic
community will be presented at appropriate meetings and written up for dissemination in
genetic / paediatric journals and as part of the thesis to be submitted by Dr Burkitt Wright to
the University of Manchester for the degree of PhD. No identifying patient data will be used
in presentations/publications. Explicit consent for the use of patient photographs will be
obtained.
At the end of the study we will write to all families to tell them that the study has finished and
inform them of the outcome, irrespective of whether there are positive findings for that family
or not.
Funding

354
Funding for this project has already been obtained: the chief investigator (Dr Emma Burkitt
Wright) has been awarded a research training fellowship by the Wellcome Trust (May 2010-
April 2013).
References
1. Tidyman WE, Rauen KA (2009). The RASopathies: developmental syndromes of
Ras/MAPK pathway dysregulation. Curr Op Genet Dev. 19: 230-6.
2. Burkitt Wright EMM, Kerr BA (2010). RAS-MAPK pathway disorders: Important causes of
congenital heart disease, feeding difficulties, developmental delay and short stature. Arch Dis
Child, epublication ahead of print, April 6th 2010.
3. Yoon S, Seger R (2006). The extracellular signal-regulated kinase: multiple substrates
regulate diverse cellular functions. Growth Factors. 24: 21-44.
4. Conducting Research with people not having the capacity to consent to their participation: a
practical guide for researchers (page 49) Prepared by Catherine Dobson on behalf of the
Mental Capacity Act Working Party (2008). British Psychological Society, London.

355
9.2.3 Appendix 3c:
Proforma for RAS-MAPK study Version 1; May 2010
Geneticist/referring
doctor
completing
questionnaire
Patient surname
Patient forename
Date of birth
Sex male / female
Clinical diagnosis NF1/ Noonan/ CFC / Costello /
other (please specify) / unknown
Genetic tests
undertaken and results
Previously included in a
publication?
yes / no (if yes, please specify)
Family history
Maternal age at delivery
Paternal age at delivery
Number of siblings brothers sisters
356
Ages of siblings
Health or
developmental
problems in other
family members
Pregnancy
Polyhydramnios yes / no / unknown
Fetal macrosomia yes / no / unknown
Fetal oedema / hydrops yes / no / unknown
Altered fetal
movements
yes / no / unknown
Antenatal scans yes / no / unknown (please specify gestations)
Structural heart
anomalies
yes / no / unknown (if present, please specify)
Fetal tachycardia yes / no / unknown (if present, please specify)
Other abnormal
findings on scans
Amniocentesis/ other
invasive test performed?
yes / no / unknown (if done, please specify indication)
Results of any prenatal
testing

357
Birth
Gestation (weeks) weeks
Mode of delivery normal / ventouse / forceps / Caesarian
Apgar at 1 minute
Apgar at 5 minutes
Apgar at 10 minutes
Respiratory distress? yes / no / unknown
Intubation? yes / no / unknown
Birthweight (and
centile)
kg ( centile)
Length (and centile) cm ( centile)
Head circumference
(and centile)
cm ( centile)
Congenital anomalies
noted at birth?
yes / no
358
Neonatal period
Feeding problems yes / no / unknown
Nasogastric tube
feeding
yes / no (if used, for what duration?)
Gastrostomy yes / no (if used, for what duration?)
Gastro-oesophageal
reflux
yes / no (if present, what treatment required?)
Hypertrophic
cardiomyopathy?
yes / no / unknown (please specify whether obstructive
features, if present)
Myocardial thickening? yes / no
Pulmonary stenosis? yes / no / unknown
Other valvular
problems?
yes / no / unknown
Tachycardia? yes / no / unknown
Other cardiac
dysrhythmia?
yes / no / unknown

359
Hypoglygaemic
episodes?
yes / no / unknown
Unusual tone or posture yes / no / unknown (if present, please specify)
Thermoregulatory
disturbance?
yes / no / unknown (if present, please specify)
Chylothorax
yes / no / unknown
Peripheral oedema /
hydrops
yes / no / unknown
Tracheomalacia yes / no / unknown
Other neonatal
problems?
(please specify)
Growth
Growth retardation? yes / no
Height (age at
assessment, centile)
cm ( years months; centile)
Weight (age at
assessment, centile)
kg ( years months; centile)
OFC (age at assessment,
centile)
cm ( years months; centile)

360
Delayed bone age? yes / no / unknown (values if known)
Growth hormone
deficiency?
yes / no / not assessed
Growth hormone
treatment?
yes / no (if received, at what ages?)
Cryptorchidism not applicable / bilateral / unilateral / no (if present, age
at treatment?)
Puberty precocious / normal / delayed (age at menarche for girls)
Menses not applicable / regular / primary amenorrhoea /
secondary amenorrhoea
Sex hormone treatment yes / no (please specify what and dates received)
Neoplasia
Malignant tumour yes / no (specify what, date diagnosed, grade and extent)
Presenting symptoms
Treatment for
malignancy
(specify what, and response to this)

361
Optic nerve glioma Yes / no
Neurofibroma Yes / no (if present, please specify number, size and site)
Type: plexiform / nodular / dermal / other
Other benign tumours Yes / no (please specify what, date diagnosed and
treatment received)
Skin
Hyperkeratosis /
keratosis pilaris
yes / no (if present, in what distribution?)
Excess skin / cutis laxa yes / no (if present, in what distribution?)
Excess palmar skin /
deep skin creases
Yes / no
Eye colour
Skin tone Fair, burns easily / unremarkable / darker than rest of
family

362
Generalised
hyperpigmentation
yes / no
Periorbital
hyperpigmentation
yes / no
Café au lait patches yes / no (if present, please specify number, size and
distribution)
Acanthosis nigricans yes / no (if present, distribution and age at development)
Skin fold freckling yes / no
Papillomata yes / no (if present, distribution and age at development)
Naevus flammeus yes / no (if present, distribution and extent)
Naevi Yes / no (if present, distribution, depth of pigmentation,
size)
Number of naevi
less than 5 / 5-50 / more than 50
Hypomelanotic macules yes / no (if present, number, size and distribution)
363
Scar formation
normal / dystrophic / keloid
Lipoma yes / no (if present, number, site and size)
Juvenile
xanthogranuloma
yes / no (if present, distribution and extent)
Campbell de Morgan
spots
yes / no (if present, number and distribution)
Vascular malformation yes / no (if present, site, size and type)
Hyperhidrosis
yes / no / unknown
Ichthyosis yes / no
Pruritus yes / no
Acne yes / no (if present, age at development)
Hair
Thin, sparse or other
unusual texture to hair
yes / no

364
Alopecia yes / no (if present, in what pattern?)
Ulerythema
ophrhyogenes
yes / no
Slow growing hair yes / no
Hair falls out easily yes / no
Curly hair yes / no
Nails and teeth
Thin, fragile nails yes / no / unknown
Deep-set nails yes / no / unknown
Delayed tooth eruption yes / no / unknown (if delayed, age at first tooth)
Enamel dysplasia yes / no
Disorganized dentition/
malocclusion
yes / no (if present, treatment required)
Tooth agenesis yes / no / unknown

365
Craniofacial features
OFC (age; centile)
Macrocephaly?
cm ( years months; centile)
absolute / relative / proportionate OFC / microcephaly
Coarsened facial
features
yes / no
Macrostomia yes / no
Macroglossia yes / no
High arched palate yes / no
Thick lips yes / no
Gingival hyperplasia yes / no
Hypertelorism yes / no
Downslanting palpebral
fissures
yes / no
Epicanthic folds yes / no
Depressed nasal bridge yes / no
Dysplastic ears yes / no
Low set ears yes / no
Posteriorly rotated ears yes / no
Large or fleshy ear lobes yes / no

366
Short neck yes / no
Ptosis yes / no
Strabismus yes / no
Nystagmus yes / no
Myopia yes / no
Hyperopia yes / no
Optic nerve hypoplasia yes / no
Other ocular anomalies yes / no (specify)
Other features
Hoarse voice yes / no
Hypernasal voice yes / no
Hernias yes / no (if present, where?)
Other features on
examination
(please specify)
Neurological
Hypotonia yes / no / unknown
Seizures? yes / no / unknown (age at onset; precipitants; therapy
received?)

367
Delayed fontanelle
closure?
yes / no / unknown
Cranial imaging: MRI,
CT
yes / no (date performed, result?)
EEG yes / no (date performed; result?)

368
Development
Current age
Age at smiling
Age at sitting
(supported)
Age at sitting
(unsupported)
Age at walking
Age at first words
Age at potty training (or current level of continence)
Current educational/
occupational/ home
provision
Social and behavioural features
Unusual degree of
shyness / stranger
anxiety
yes / no
Irritability yes / no
Hyperactivity or poor
concentration
yes / no
Hypersensitivity to
noise
yes / no

369
Hypersensitivity to
light/sun
yes / no
Sleep problems yes / no (if present, please specify what)
Self-injurious behaviour yes / no (if present, please specify)
Outgoing personality yes / no
Previous diagnosis of
autism or related
condition
yes / no (if present, specify)
Other behavioural
characteristics
Medication? Yes / no (if used, agent, age at commencing therapy,
duration)
Cardiac status
Hypertrophic
cardiomyopathy
yes / no
Pulmonary stenosis yes / no
Other valvular problem yes / no (if present, please specify)
Tachycardia yes / no (if present, please specify)

370
Other dysrhythmia yes / no (if present, please specify)
Echocardiograms yes / no (dates; findings)
Cardiac catheterisations yes / no (dates; findings)
Medication Drug
Dates
administered
Dosage
Musculoskeletal features
Large fingers / broad
distal phalanges
yes / no
Hyperextensible small
joints
yes / no
Ulnar deviation of
hands
yes / no
Joint contractures yes / no (if present, of which joints?)
Developmental
dysplasia of the hip
none / left / right / bilateral
Flat feet yes / no

371
Kyphoscoliosis yes / no (if present, please specify treatment required)
Pectus deformity yes / no (if present, please specify)
Orthopaedic operations
performed
Procedure Date / age performed
Bone densitometry
performed
yes / no (if done, date and result)
osteopenia / osteoporosis
Gait Normal / wide-based / other (please specify)
Mobility Normal exercise tolerance / can walk distances / walks
only short distances / uses a wheelchair outside / inside

372
9.3 Appendix 4: Primers and PCR conditions
Primer
Anneal temp;
ºC
SHOC2exon2F SHOC2exon2R 59.5
TTGCTCTCTTTCCCAAAACC CCATGCTGATTACTTCTTCAAGC
PTK2 exon3F PTK2 exon3R 62
CCTCAGACTCCTTCCGCATA TCTGTAATATGAAAAGTCCCCGATA
PTK2 exon4F PTK2 exon4R 60
TGTTTTGTTTTTGTTTTGTTTTGG GAAATCAAGTGTGCATCACAC
PTK2 exon5F PTK2 exon5R 55
TGGGCCTCTTACTATGCTCTG TTCCTCCAAACGTGAGCTTT
PTK2 exon6F PTK2 exon6R 55
CAGTGTTGTTTTTCCCATTCC TCGCCTAAAATCAGGGAAGA
PTK2 exon7F PTK2 exon7R 55
GCCTCTCCCCCAGTTTTTAT TGGGCCATATACAATGTTAGCTT

373
PTK2 exon8F PTK2 exon8R 60
TGATTTGTCCCATCCTTCCT CCCAAAAGCAATTTACCACTG
PTK2 exon9F PTK2 exon9R 55
TGGCAGTAATTTGAATGTAGGTG TGAAGCTAGGCATGCTGTTTT
PTK2 exon10F PTK2 exon10R 55
TCATCCCAGAGAAACCCTTG CTTACTTGTCCCCCACTCCA
PTK2 exon11F PTK2 exon11R 55
GGCAGCATGGAGAATCTGTT TGTCCTATTCTTGGGGACATTT
PTK2exon12F PTK2 exon12R 55
TCTGGGTTTGCATTTGTCAC CGGATCACCATCCCTAGAAA
PTK2 exon13F PTK2 exon13R 55
CTCCCTGATTCTAGGCACCA GCTCCCCCATAGAACTTAAAGG
PTK2 exon14F PTK2 exon14R 55
TCTTCCTCATCCAAGGCAGT ACAACTTGCTGAGTGATCTGGT

374
PTK2 exon15F PTK2 exon15R 55
CCCTGTTTGCTCTCCGATAA TCTGTTCAGCATCCAAGGAA
PTK2 exon16F PTK2 exon16R 55
TTGATGTGCCACAGATGAAA GACTCATGAAGACAAACAAAAGC
PTK2exon17F PTK2 exon17R 60
TGCCGTTCTGATTCTGTCTG CATGATGGTTTACCTGGAAAAAT
PTK2 exon18F PTK2 exon18R 60
CTCCAGCCTGGTGACAGAAT TCAGAACTCTCCTGAAAATCCAA
PTK2 exon19F PTK2exon19R 55
TTAAAAATGCATTGAAAACAGCA AAGCCATGGCATCTCACTCT
PTK2exon20F PTK2 exon20R 55
TGTCTTTCAAAGTGCCTATTGGT ACCATTGCTGAGAAGGCTGT
PTK2 exon21F PTK2 exon21R 55
GACCTCGAAACACCAAGGAA TACAGAAGCTGTGCCCAGAA
PTK2 exon22F PTK2 exon22R 60

375
TACCTCCCAGCTGGTACTCG TTACTGCAAGGAGGAGAAGCA
PTK2 exon23F PTK2 exon23R 55
TGATCCTTCTTTCCCCTGTG CAACTGCTCATGGCTTCAAA
PTK2 exon24F PTK2 exon24R 55
TTGCCACAGCATCATTTGTT GCTTTCTATCGGCCAAATCA
PTK2 exon25F PTK2 exon25R 60
GGCCATAATTCTGCCTACCA ATGTTGCCCAGTCTGGTTTC
PTK2 exon26F PTK2 exon26R 62
GCACATTAAATTGCCCCCTA ACCTCAGGTGATCTGCTTGC
PTK2 exon27F PTK2 exon27R 55
ACGGGGGAACTTTCAACTCT AGTGCAGAAAACTTCTGTATCTGAAT
PTK2 exon28F PTK2 exon28R 60
GCCCTTTCCACACATGCTAT GCTCACTGCAACCCTAAGGA
PTK2 exon29F PTK2 exon29R 62
GATGGGGTTTCTCCATGTTG ACTAGGGGACACCACAAATCC

376
PTK2 exon30F PTK2 exon30R 60
TATCCCGCACACACAAGAAA TCAACTGGCACCACAGCTTA
PTK2 exon31F PTK2 exon31R 55
TGCCTTGAAACTTGTGGAAA CCACCCCTATGCCCTAGAAT
PTK2 exon32F PTK2 exon32R 60
GAGGCAAGCTTGGACAACAC CTGCTGGTGGAAGGCTAGAG
SOS1exon8F SOS1exon8R 62
GCATAGTCGTGCCCCATAAT TGTGCAGGGTACTCACACAAT
RPS6KA2exon15F RPS6KA2exon15R 62
TGGTCGGGTGTGAGATTGTA CAGCAAGTCAGCTCCAAGTG
RIT1exon2F RIT1exon3R 60
GCATCCCTTTCTTCCCAAA TCCATTAATGTTCAGTAAGAGACA
RIT1exon4F RIT1exon4R 62
TGTAGGTGAAATCTTCAGCTGTG CGCATGTCGATTACCTGCTA

377
RIT1exon5F RIT1exon5R 62
CCTGCCAATCTGGACATTT CGCAAAGTACTGGTGTGAGC
RIT1exon6F RIT1exon6R 62
GCTTGAACACCTCCAGAATTG GTGCAGAGCCAAAAACTTCC
Primers for BRAF mutagenesis
p.(Thr241Pro):
5’ – CTTTGTACGAAAACCGTTTTTCACCTTAGC – 3’
5’ – GCTAAGGTGAAAAACGGTTTTCGTACAAAG - 3’
p.(Gln257Arg):
5’ - GCTGCTTTTCCGGGGTTTCCGCTGTC – 3’
5’ – GACAGCGGAAACCCCGGAAAAGCAGC – 3’
p.(Gln262Pro):

378
5’ – GTTTCCGCTGTCCAACATGTGG – 3’
5’ – CCACATGTTGGACAGCGGAAAC – 3’
p.(Gly464Glu):
5’ – CAAAGAATTGAATCTGGATCATTTG – 3’
5’ – CAAATGATCCAGATTCAATTCTTTG - 3’
p.(Gly469Glu):
5’ – CTGGATCATTTGAAACAGTCTACAAGGG – 3’
5’ – CCCTTGTAGACTGTTTCAAATGATCCAG – 3’
p.(Thr470Pro):
5’ – GGATCATTTGGACCAGTCTACAAG – 3’
5’ – CTTGTAGACTGGTCCAAATGATCC -3’
p.(Lys499Glu):

379
5’ - GCAGTTACAAGCCTTCGAAAATGAAGTAGG – 3’
5’ – CCTACTTCATTTTCGAAGGCTTGTAACTGC – 3’
p.(Glu501Gly):
5’ - CAAGCCTTCAAAAATGGAGTAGGAGTACTC – 3’
5’ – GAGTACTCCTACTCCATTTTTGAAGGCTTG – 3’
p.(Leu525Gln):
5’ – CAAAGCCACAACAGGCTATTGTTAC - 3’
5’ – GTAACAATAGCCTGTTGTGGCTTTG – 3’
Sequencing of plasmids
Sequencing primer for 241/257 residues:
5’ – CCTGGCTTACTGGAGAAG – 3’
Sequencing primer for 469 onwards residues:
5’ – GGAGATTCCTGATGGGCAG – 3’

380
9.4 Appendix 5: Histograms of coverage of diagnostically relevant
genes in patients TE1-TE10

381

382

383

384
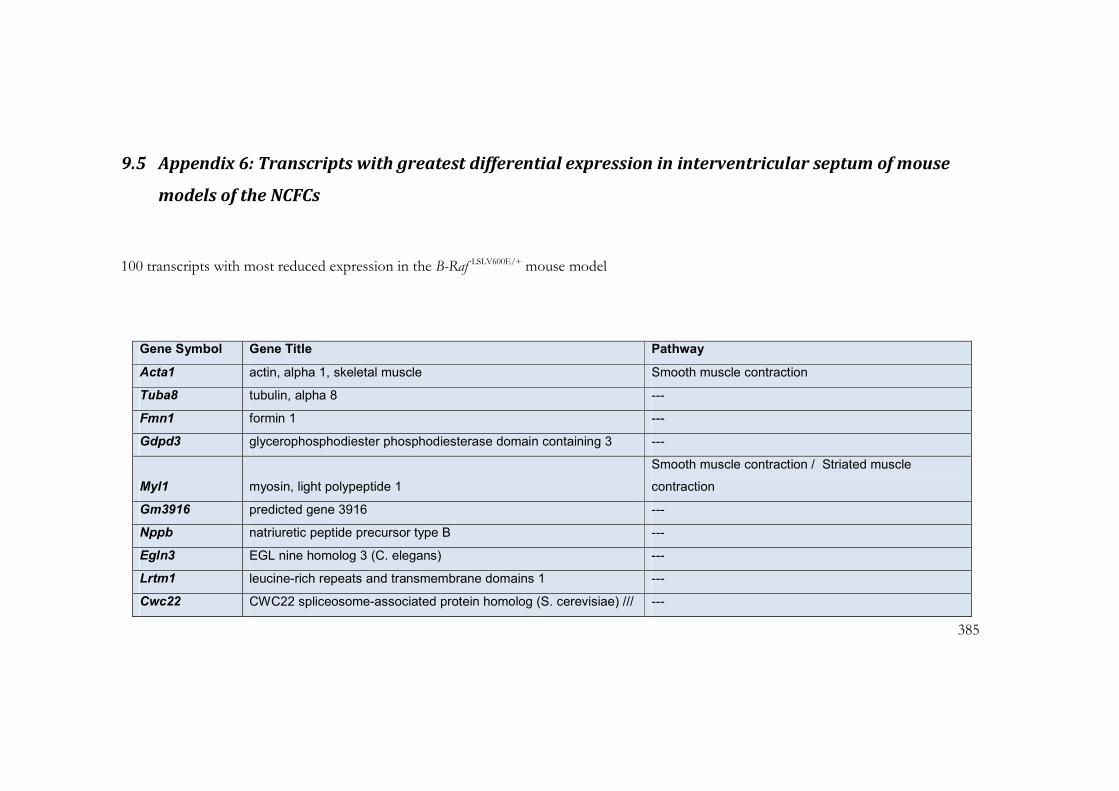
385
9.5 Appendix 6: Transcripts with greatest differential expression in interventricular septum of mouse
models of the NCFCs
100 transcripts with most reduced expression in the B-Raf LSLV600E/+ mouse model
Gene Symbol Gene Title Pathway
Acta1 actin, alpha 1, skeletal muscle Smooth muscle contraction
Tuba8 tubulin, alpha 8 ---
Fmn1 formin 1 ---
Gdpd3 glycerophosphodiester phosphodiesterase domain containing 3 ---
Myl1 myosin, light polypeptide 1
Smooth muscle contraction / Striated muscle
contraction
Gm3916 predicted gene 3916 ---
Nppb natriuretic peptide precursor type B ---
Egln3 EGL nine homolog 3 (C. elegans) ---
Lrtm1 leucine-rich repeats and transmembrane domains 1 ---
Cwc22 CWC22 spliceosome-associated protein homolog (S. cerevisiae) /// ---
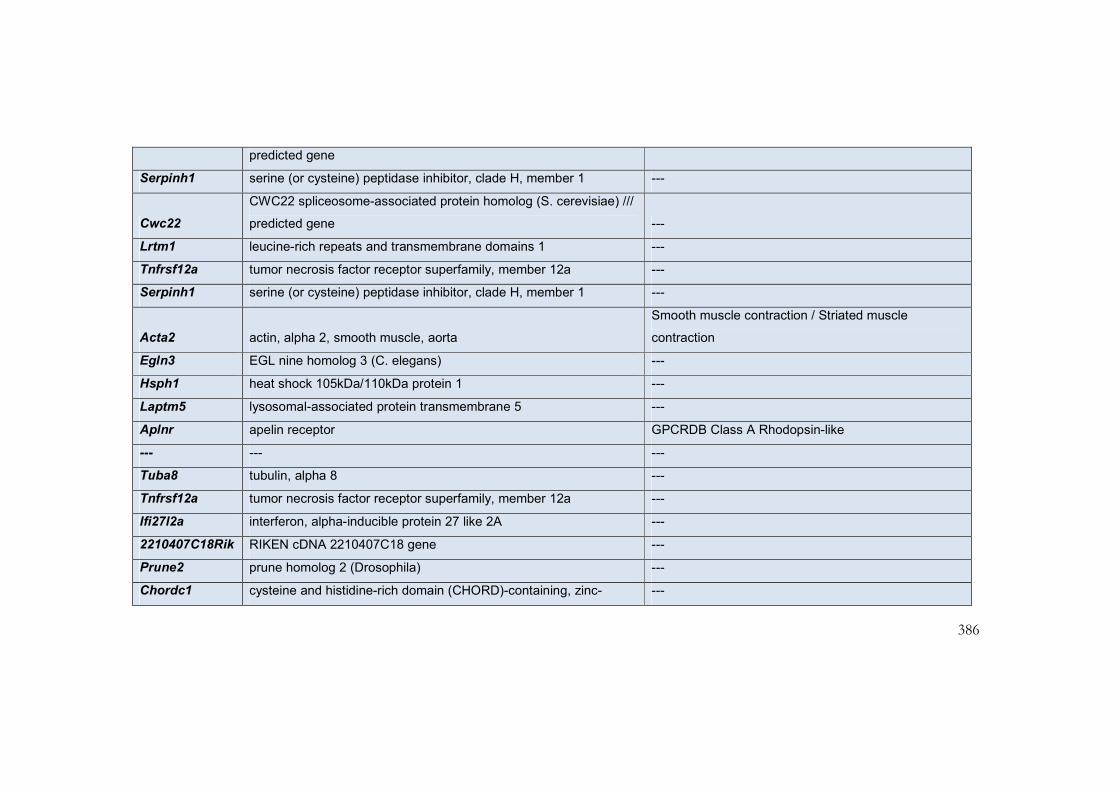
386
predicted gene
Serpinh1 serine (or cysteine) peptidase inhibitor, clade H, member 1 ---
Cwc22
CWC22 spliceosome-associated protein homolog (S. cerevisiae) ///
predicted gene ---
Lrtm1 leucine-rich repeats and transmembrane domains 1 ---
Tnfrsf12a tumor necrosis factor receptor superfamily, member 12a ---
Serpinh1 serine (or cysteine) peptidase inhibitor, clade H, member 1 ---
Acta2 actin, alpha 2, smooth muscle, aorta
Smooth muscle contraction / Striated muscle
contraction
Egln3 EGL nine homolog 3 (C. elegans) ---
Hsph1 heat shock 105kDa/110kDa protein 1 ---
Laptm5 lysosomal-associated protein transmembrane 5 ---
Aplnr apelin receptor GPCRDB Class A Rhodopsin-like
--- --- ---
Tuba8 tubulin, alpha 8 ---
Tnfrsf12a tumor necrosis factor receptor superfamily, member 12a ---
Ifi27l2a interferon, alpha-inducible protein 27 like 2A ---
2210407C18Rik RIKEN cDNA 2210407C18 gene ---
Prune2 prune homolog 2 (Drosophila) ---
Chordc1 cysteine and histidine-rich domain (CHORD)-containing, zinc- ---

387
binding protein 1
Ampd3 adenosine monophosphate deaminase 3 ---
Ccl6 chemokine (C-C motif) ligand 6 ---
Josd2 Josephin domain containing 2 Striated_muscle_contraction
Ccr5 chemokine (C-C motif) receptor 5 GPCRDB Class A Rhodopsin-like
Hsph1 heat shock 105kDa/110kDa protein 1 ---
Manf mesencephalic astrocyte-derived neurotrophic factor ---
Dynll1 dynein light chain LC8-type 1 /// predicted gene 6788 ---
Tpm4 tropomyosin 4 ---
Ccnd1 cyclin D1 G1 to S cell cycle reactome / Wnt signalling
Dynll1 dynein light chain LC8-type 1 /// predicted gene 6788 ---
Dynll1 dynein light chain LC8-type 1 ---
Hspa4 heat shock protein 4 ---
Itga9 integrin alpha 9 Integrin-mediated cell adhesion
C1qa complement component 1, q subcomponent, alpha polypeptide Complement Activation Classical
Col15a1 collagen, type XV, alpha 1 ---
Ccl9 chemokine (C-C motif) ligand 9 ---
Gimap4 GTPase, IMAP family member 4 ---
Myct1 myc target 1 ---
Gm13138 predicted gene 13138 ---

388
Mut methylmalonyl-Coenzyme A mutase ---
Calr calreticulin Calcium regulation in cardiac cells
Hspa4 heat shock protein 4 ---
Tubb2a tubulin, beta 2A ---
Igfbp5 insulin-like growth factor binding protein 5 Smooth muscle contraction
Angptl4 angiopoietin-like 4 ---
Ankrd23 ankyrin repeat domain 23 ---
P4ha1
procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-
hydroxylase), alpha ---
Calr calreticulin Calcium regulation in cardiac cells
Chac2 ChaC, cation transport regulator homolog 2 (E. coli) ---
C1qb complement component 1, q subcomponent, beta polypeptide Complement Activation Classical
Fam101b family with sequence similarity 101, member B ---
Calr calreticulin Calcium regulation in cardiac cells
Alpk2 alpha-kinase 2 ---
Arntl aryl hydrocarbon receptor nuclear translocator-like ---
Ly6e lymphocyte antigen 6 complex, locus E ---
Hist2h3c2 histone cluster 2, H3c2 ---
Gm9844 predicted gene 9844 /// thymosin, beta 10 ---
Itga1 integrin alpha 1 Integrin-mediated cell adhesion
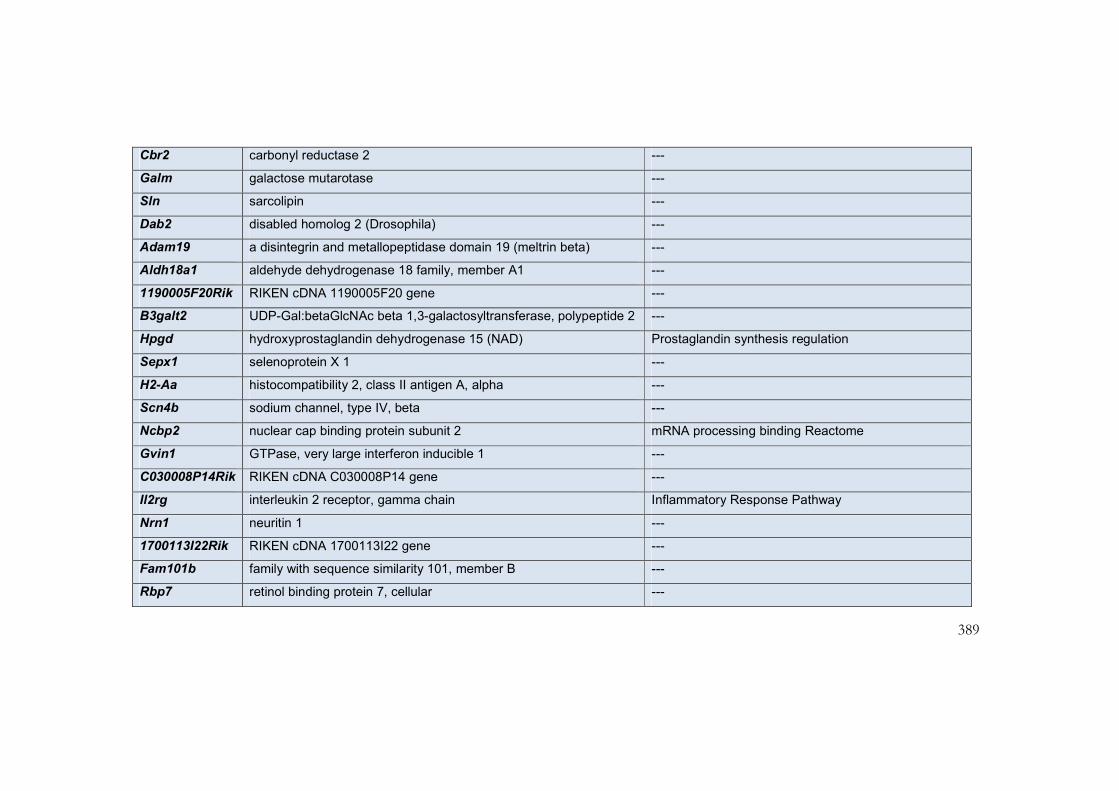
389
Cbr2 carbonyl reductase 2 ---
Galm galactose mutarotase ---
Sln sarcolipin ---
Dab2 disabled homolog 2 (Drosophila) ---
Adam19 a disintegrin and metallopeptidase domain 19 (meltrin beta) ---
Aldh18a1 aldehyde dehydrogenase 18 family, member A1 ---
1190005F20Rik RIKEN cDNA 1190005F20 gene ---
B3galt2 UDP-Gal:betaGlcNAc beta 1,3-galactosyltransferase, polypeptide 2 ---
Hpgd hydroxyprostaglandin dehydrogenase 15 (NAD) Prostaglandin synthesis regulation
Sepx1 selenoprotein X 1 ---
H2-Aa histocompatibility 2, class II antigen A, alpha ---
Scn4b sodium channel, type IV, beta ---
Ncbp2 nuclear cap binding protein subunit 2 mRNA processing binding Reactome
Gvin1 GTPase, very large interferon inducible 1 ---
C030008P14Rik RIKEN cDNA C030008P14 gene ---
Il2rg interleukin 2 receptor, gamma chain Inflammatory Response Pathway
Nrn1 neuritin 1 ---
1700113I22Rik RIKEN cDNA 1700113I22 gene ---
Fam101b family with sequence similarity 101, member B ---
Rbp7 retinol binding protein 7, cellular ---

390
C1qc complement component 1, q subcomponent, C chain Complement Activation Classical
Gm11276 predicted gene 11276 ---
Fam198b family with sequence similarity 198, member B ---
Ppm1a protein phosphatase 1A, magnesium dependent, alpha isoform ---
Bst2 bone marrow stromal cell antigen 2 ---
Nppa natriuretic peptide precursor type A Smooth muscle contraction
Nudt18 nudix (nucleoside diphosphate linked moiety X)-type motif 18 ---
Fscn1
fascin homolog 1, actin bundling protein (Strongylocentrotus
purpuratus) ---
C1qb complement component 1, q subcomponent, beta polypeptide Complement Activation Classical
--- --- ---
Pfkl phosphofructokinase, liver, B-type Glycolysis and Gluconeogenesis
Lyz2 lysozyme 2 ---
Uqcr10 ubiquinol-cytochrome c reductase, complex III subunit ---
Col6a3 collagen, type VI, alpha 3 ---
AW112010 expressed sequence AW112010 ---
100 transcripts with most increased expression in the B-Raf LSLV600E/+ mouse model

391
Gene Symbol Gene Title Pathway
Myh7 myosin, heavy polypeptide 7, cardiac muscle, beta Striated muscle contraction
--- --- ---
Snca synuclein, alpha ---
Cirbp cold inducible RNA binding protein ---
Per1 period homolog 1 (Drosophila) Circadian Exercise
--- --- ---
Per2 period homolog 2 (Drosophila) Circadian Exercise
Dbp D site albumin promoter binding protein ---
Fam107a family with sequence similarity 107, member A ---
Gm129 predicted gene 129 ---
--- --- ---

392
Ddit4 DNA-damage-inducible transcript 4 ---
Sorbs1 sorbin and SH3 domain containing 1 Integrin-mediated cell adhesion
Dbp D site albumin promoter binding protein ---
Hist1h1c histone cluster 1, H1c ---
4833417J20Rik RIKEN cDNA 4833417J20 gene ---
Per2 period homolog 2 (Drosophila) Circadian Exercise
Rprd2 regulation of nuclear pre-mRNA domain containing 2 ---
Nfe2l2 nuclear factor, erythroid derived 2, like 2 ---
Hist1h1c histone cluster 1, H1c ---
6030422H21Rik RIKEN cDNA 6030422H21 gene ---
--- --- ---
Igfbp3 insulin-like growth factor binding protein 3 Smooth muscle contraction

393
--- --- ---
Lamb3 laminin, beta 3 ---
Irx2 ///
LOC100045612 Iroquois related homeobox 2 (Drosophila) ---
Lepr leptin receptor ---
Adh1 alcohol dehydrogenase 1 (class I) ---
Rbm3 RNA binding motif protein 3 mRNA processing binding Reactome
--- --- ---
Hlf hepatic leukemia factor ---
Per3 period homolog 3 (Drosophila) ---
D7Ertd715e DNA segment, Chr 7, ERATO Doi 715, expressed ---
--- --- ---

394
Ppm1k protein phosphatase 1K (PP2C domain containing) ---
--- --- ---
Raph1
Ras association (RalGDS/AF-6) and pleckstrin homology domains
1 ---
Tpm1 tropomyosin 1, alpha Striated muscle contraction
Pcdh7 protocadherin 7 ---
Per3 Period homolog 3 (Drosophila) ---
Ddc dopa decarboxylase Biogenic Amine Synthesis
Sorbs1 sorbin and SH3 domain containing 1 Integrin-mediated cell adhesion
Trpm7 transient receptor potential cation channel, subfamily M, member 7 ---
Nfkbia
nuclear factor of kappa light polypeptide gene enhancer in B-cells
inhibitor, al Apoptosis
Per3 period homolog 3 (Drosophila) ---

395
Kcne1 potassium voltage-gated channel, Isk-related subfamily, member 1 ---
Chic2 cysteine-rich hydrophobic domain 2 ---
2210403K04Rik RIKEN cDNA 2210403K04 gene ---
Rps4y2 ribosomal protein S4, Y-linked 2 ---
Ppil6 peptidylprolyl isomerase (cyclophilin)-like 6 S1P Signaling
4930534B04Rik RIKEN cDNA 4930534B04 gene ---
Nfkbia
nuclear factor of kappa light polypeptide gene enhancer in B-cells
inhibitor, al Apoptosis
Irx3 Iroquois related homeobox 3 (Drosophila) ---
Igfbp3 insulin-like growth factor binding protein 3 Smooth muscle contraction
Tfdp2 Transcription factor Dp 2 G1 to S cell cycle Reactome
Mmrn1 multimerin 1 ---

396
Tsc22d3 TSC22 domain family, member 3 ---
Aqp4 aquaporin 4 ---
Mylip myosin regulatory light chain interacting protein ---
Nfkbia
nuclear factor of kappa light polypeptide gene enhancer in B-cells
inhibitor, al Apoptosis
Arrdc4 arrestin domain containing 4 Statin Pathway PharmGKB
Tspan4 tetraspanin 4 ---
Dusp18 dual specificity phosphatase 18 ---
--- --- ---
Hamp hepcidin antimicrobial peptide ---
Tob2 transducer of ERBB2, 2 ---
Dixdc1 DIX domain containing 1 ---
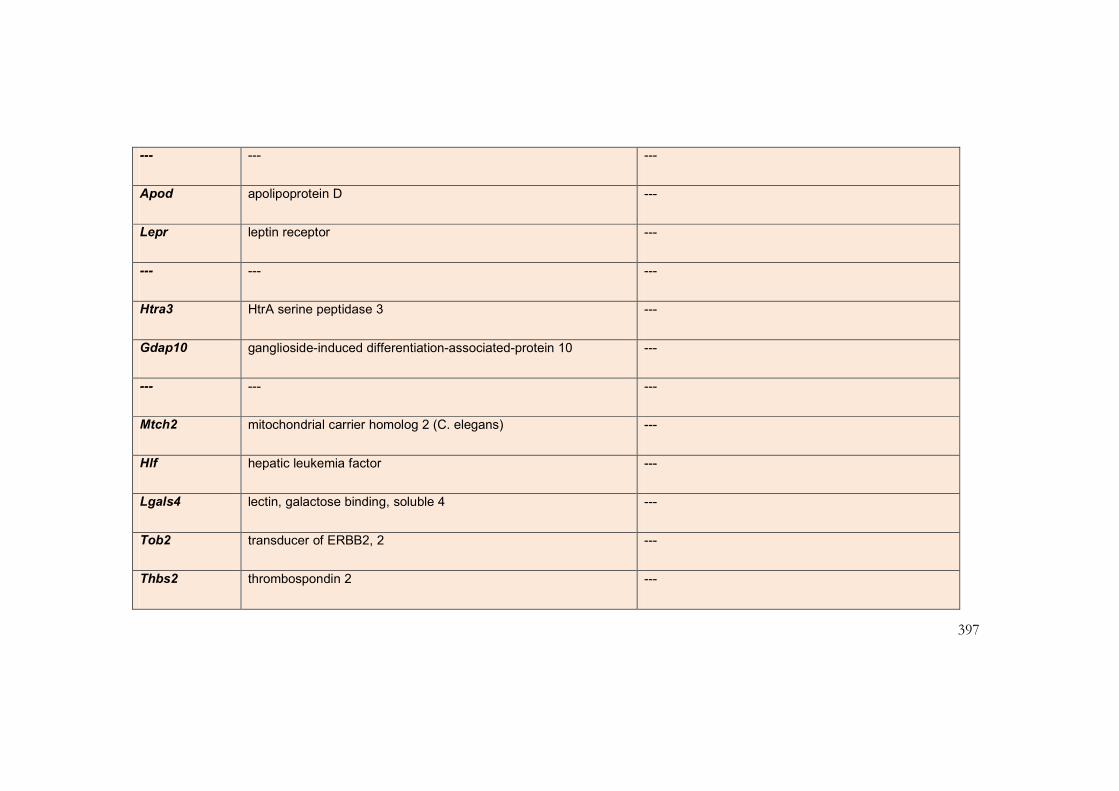
397
--- --- ---
Apod apolipoprotein D ---
Lepr leptin receptor ---
--- --- ---
Htra3 HtrA serine peptidase 3 ---
Gdap10 ganglioside-induced differentiation-associated-protein 10 ---
--- --- ---
Mtch2 mitochondrial carrier homolog 2 (C. elegans) ---
Hlf hepatic leukemia factor ---
Lgals4 lectin, galactose binding, soluble 4 ---
Tob2 transducer of ERBB2, 2 ---
Thbs2 thrombospondin 2 ---

398
--- --- ---
Epm2aip1 EPM2A (laforin) interacting protein 1 ---
Snca synuclein, alpha ---
Cyp2e1 cytochrome P450, family 2, subfamily e, polypeptide 1 ---
Tsc22d3 TSC22 domain family, member 3 ---
Nfkbia
nuclear factor of kappa light polypeptide gene enhancer in B-cells
inhibitor, al Apoptosis
Hist2h2aa1 ///
Hist2h2aa2 histone cluster 2, H2aa1 /// histone cluster 2, H2aa2 ---
1190002H23Rik RIKEN cDNA 1190002H23 gene ---
Fmo2 flavin containing monooxygenase 2 ---
C79242 expressed sequence C79242 ---
BC031353 cDNA sequence BC031353 ---

399
--- --- ---
--- --- ---
Snrnp200 small nuclear ribonucleoprotein 200 (U5) ---
Tnnt2 troponin T2, cardiac Striated muscle contraction
Atp1a2 ATPase, Na+/K+ transporting, alpha 2 polypeptide Calcium regulation in cardiac cells
--- --- ---
--- --- ---
Bclaf1 BCL2-associated transcription factor 1 ---
Dsg2 desmoglein 2 ---
Thbs2 thrombospondin 2 ---

400
100 transcripts with most reduced expression in the K-Ras V14I/+ mouse model
Gene Symbol Gene Title Pathway
Sln Sarcolipin ---
Nr4a1 nuclear receptor subfamily 4, group A, member 1 Nuclear Receptors
E030016H06Rik RIKEN cDNA E030016H06 gene ---
--- --- ---
Kctd12b potassium channel tetramerisation domain containing 12b ---
Sorbs1 sorbin and SH3 domain containing 1 Integrin-mediated cell adhesion
Fus fusion, derived from t(12;16) malignant liposarcoma (human) mRNA processing binding Reactome
Dnmt3a DNA methyltransferase 3A ---
Myl7 myosin, light polypeptide 7, regulatory ---

401
Ablim1 actin-binding LIM protein 1 ---
Irs2 insulin receptor substrate 2 ---
2310040G07Rik RIKEN cDNA 2310040G07 gene ---
Reep3 receptor accessory protein 3 ---
Prox1 prospero-related homeobox 1 ---
Lims1 LIM and senescent cell antigen-like domains 1 ---
--- --- ---
Hist1h2ad histone cluster 1, H2ad ---
Rb1cc1 RB1-inducible coiled-coil 1 ---
Rgs5 regulator of G-protein signaling 5
Calcium regulation in cardiac cells / Smooth muscle
contraction
Zc3h15 zinc finger CCCH-type containing 15 ---

402
Ddx6 DEAD (Asp-Glu-Ala-Asp) box polypeptide 6 mRNA processing binding Reactome
Nipbl Nipped-B homolog (Drosophila) ---
Ppargc1a
peroxisome proliferative activated receptor, gamma, coactivator 1
alpha mRNA processing binding Reactome
Rrm2b ribonucleotide reductase M2 B (TP53 inducible) ---
Nipbl Nipped-B homolog (Drosophila) ---
Pum1 pumilio 1 (Drosophila) mRNA processing binding Reactome
--- --- ---
Dnmt3a DNA methyltransferase 3A ---
Epm2aip1 EPM2A (laforin) interacting protein 1 ---
Ddx6 DEAD (Asp-Glu-Ala-Asp) box polypeptide 6 mRNA processing binding Reactome
Pcdh17 protocadherin 17 ---
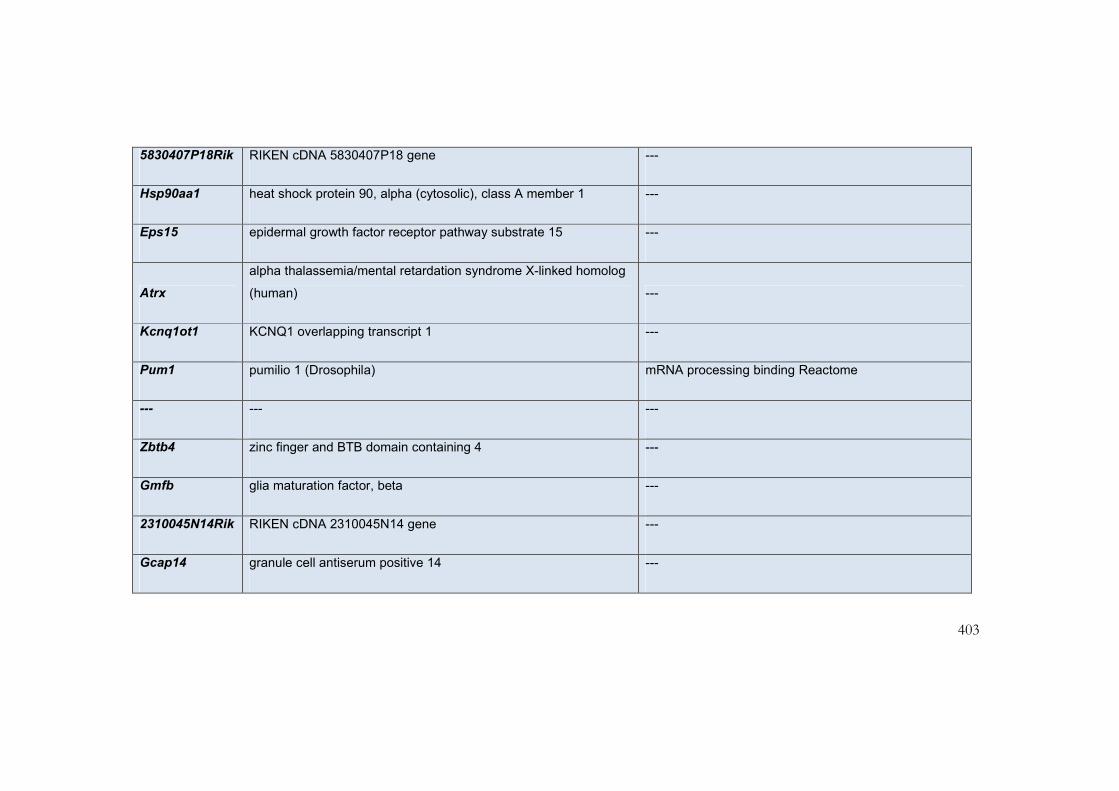
403
5830407P18Rik RIKEN cDNA 5830407P18 gene ---
Hsp90aa1 heat shock protein 90, alpha (cytosolic), class A member 1 ---
Eps15 epidermal growth factor receptor pathway substrate 15 ---
Atrx
alpha thalassemia/mental retardation syndrome X-linked homolog
(human) ---
Kcnq1ot1 KCNQ1 overlapping transcript 1 ---
Pum1 pumilio 1 (Drosophila) mRNA processing binding Reactome
--- --- ---
Zbtb4 zinc finger and BTB domain containing 4 ---
Gmfb glia maturation factor, beta ---
2310045N14Rik RIKEN cDNA 2310045N14 gene ---
Gcap14 granule cell antiserum positive 14 ---

404
Eif5 eukaryotic translation initiation factor 5 Translation Factors
Gbp6 guanylate binding protein 6 ---
Gsk3b glycogen synthase kinase 3 beta Cell Cycle / Glycogen Metabolism / Wnt Signalling
Ppp1r14c Ppp1r14c pseudogene ---
Fbxl7 F-box and leucine-rich repeat protein 7 ---
Ppig peptidyl-prolyl isomerase G (cyclophilin G) ---
Kif5b kinesin family member 5B ---
--- --- ---
Sspn Sarcospan ---
Klf6 Kruppel-like factor 6 ---
1500015O10Rik RIKEN cDNA 1500015O10 gene ---
Ppp2r3a protein phosphatase 2, regulatory subunit B'', alpha ---

405
Nfib nuclear factor I/B ---
Naa15 N(alpha)-acetyltransferase 15, NatA auxiliary subunit ---
Phf20l1 PHD finger protein 20-like 1 ---
Zeb2 zinc finger E-box binding homeobox 2 TGF Beta Signaling Pathway
Dep1 diabetic embryopathy 1 ---
Smc4 structural maintenance of chromosomes 4 ---
2810474O19Rik RIKEN cDNA 2810474O19 gene ---
Igf1r insulin-like growth factor I receptor ---
Eif5 eukaryotic translation initiation factor 5 Translation Factors
Ash1l ash1 (absent, small, or homeotic)-like (Drosophila) ---
Eid1 EP300 interacting inhibitor of differentiation 1 ---
Creb1 cAMP responsive element binding protein 1
G1 to S cell cycle Reactome / Smooth muscle

406
contraction
Klf4 Kruppel-like factor 4 (gut) ---
Mll5 myeloid/lymphoid or mixed-lineage leukemia 5 ---
Sltm SAFB-like, transcription modulator mRNA processing binding Reactome
--- --- ---
Nucks1 nuclear casein kinase and cyclin-dependent kinase substrate 1 ---
4931406P16Rik RIKEN cDNA 4931406P16 gene ---
Evi5 ecotropic viral integration site 5 ---
Nfia nuclear factor I/A ---
Mtap4 microtubule-associated protein 4 ---
Ubxn2a UBX domain protein 2A ---
Mtap1b microtubule-associated protein 1B ---

407
A430081F14Rik RIKEN cDNA A430081F14 gene ---
Thoc2 THO complex 2 ---
Ubn1 ubinuclein 1 ---
Prpf40a PRP40 pre-mRNA processing factor 40 homolog A (yeast) ---
Arid5b AT rich interactive domain 5B (MRF1-like) ---
--- --- ---
Tcf4 transcription factor 4 ---
Tpr
similar to nuclear pore complex-associated intranuclear coiled-coil
protein TPR ---
Prpf38b
PRP38 pre-mRNA processing factor 38 (yeast) domain containing
B ---
Ankrd12 ankyrin repeat domain 12 ---
Ubxn4 UBX domain protein 4 ---

408
Cacna2d1 calcium channel, voltage-dependent, alpha2/delta subunit 1 ---
6720475J19Rik RIKEN cDNA 6720475J19 gene ---
Gm2818 Predicted gene 2818 ---
--- --- ---
Gas2l3 growth arrest-specific 2 like 3 ---
Isoc1 isochorismatase domain containing 1 ---
Mef2c myocyte enhancer factor 2C ---
Crebbp CREB binding protein TGF Beta Signaling Pathway
Smc6 structural maintenance of chromosomes 6 ---
Tpm1 tropomyosin 1, alpha Striated muscle contraction
Itih5 inter-alpha (globulin) inhibitor H5 ---
--- --- ---

409
100 transcripts with most increased expression in the K-Ras V14I/+ mouse model
Gene Symbol Gene Title Pathway
Snca synuclein, alpha ---
Bdh1 3-hydroxybutyrate dehydrogenase, type 1 ---
Fam46c family with sequence similarity 46, member C ---
Laptm5 lysosomal-associated protein transmembrane 5 ---
Txnip thioredoxin interacting protein ---
Cyr61 cysteine rich protein 61 ---
Slc4a1 solute carrier family 4 (anion exchanger), member 1 ---
Alas2 aminolevulinic acid synthase 2, erythroid Haem Biosynthesis

410
C3 complement component 3 Complement Activation Classical
Snca synuclein, alpha ---
Txnip thioredoxin interacting protein ---
Egr1 early growth response 1 Ovarian Infertility Genes
Bpgm 2,3-bisphosphoglycerate mutase ---
Bdh1 3-hydroxybutyrate dehydrogenase, type 1 ---
Aplp2 amyloid beta (A4) precursor-like protein 2 ---
--- --- ---
Bpgm 2,3-bisphosphoglycerate mutase ---
Fmo2 flavin containing monooxygenase 2 ---
Pfn1 profilin 1 G13 Signalling Pathway
Nov nephroblastoma overexpressed gene ---
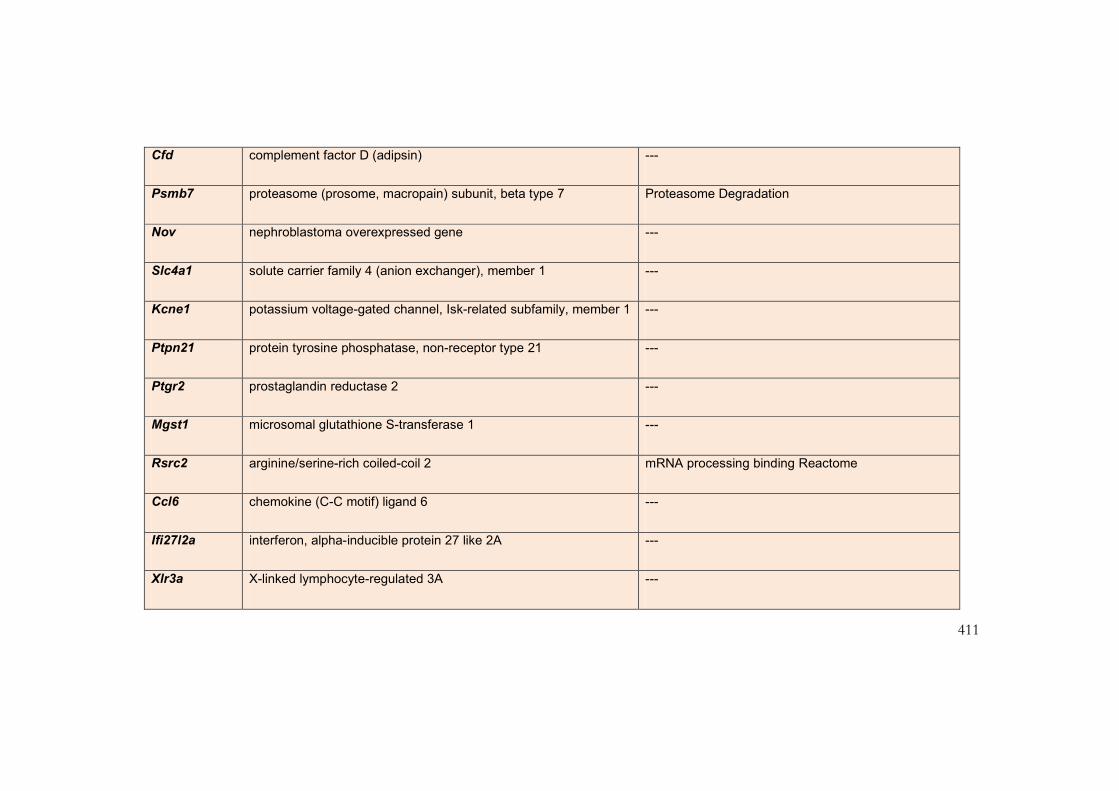
411
Cfd complement factor D (adipsin) ---
Psmb7 proteasome (prosome, macropain) subunit, beta type 7 Proteasome Degradation
Nov nephroblastoma overexpressed gene ---
Slc4a1 solute carrier family 4 (anion exchanger), member 1 ---
Kcne1 potassium voltage-gated channel, Isk-related subfamily, member 1 ---
Ptpn21 protein tyrosine phosphatase, non-receptor type 21 ---
Ptgr2 prostaglandin reductase 2 ---
Mgst1 microsomal glutathione S-transferase 1 ---
Rsrc2 arginine/serine-rich coiled-coil 2 mRNA processing binding Reactome
Ccl6 chemokine (C-C motif) ligand 6 ---
Ifi27l2a interferon, alpha-inducible protein 27 like 2A ---
Xlr3a X-linked lymphocyte-regulated 3A ---
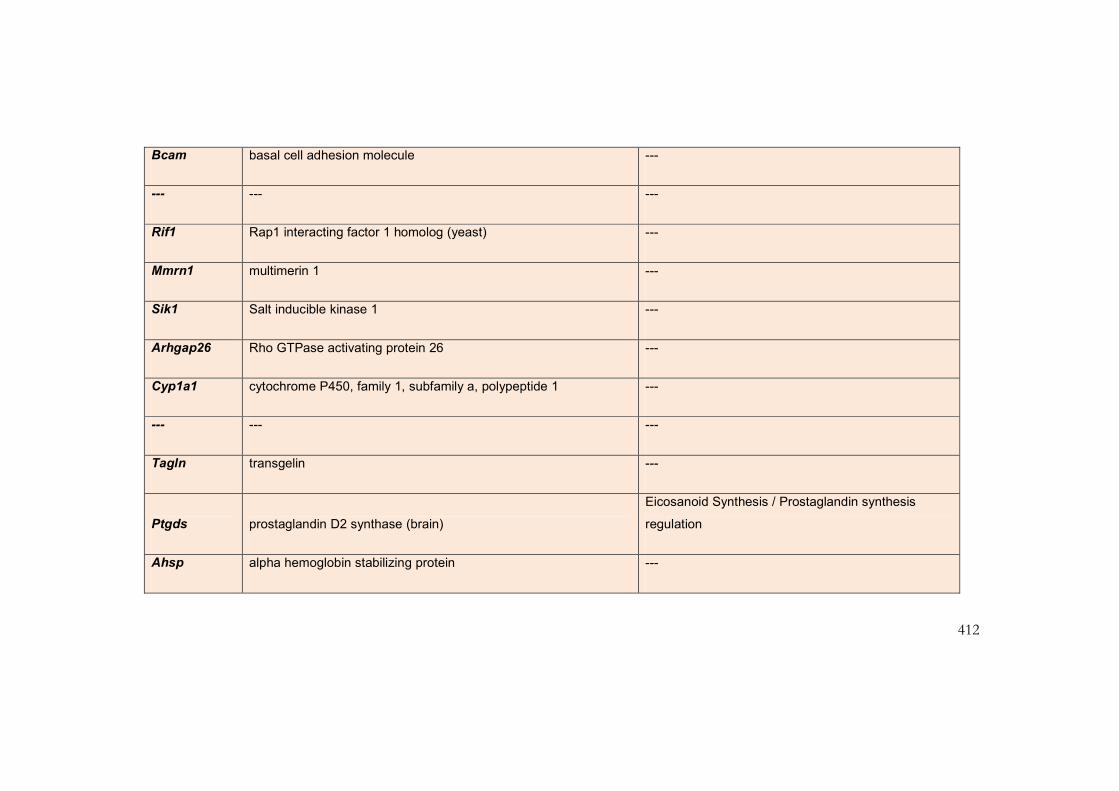
412
Bcam basal cell adhesion molecule ---
--- --- ---
Rif1 Rap1 interacting factor 1 homolog (yeast) ---
Mmrn1 multimerin 1 ---
Sik1 Salt inducible kinase 1 ---
Arhgap26 Rho GTPase activating protein 26 ---
Cyp1a1 cytochrome P450, family 1, subfamily a, polypeptide 1 ---
--- --- ---
Tagln transgelin ---
Ptgds prostaglandin D2 synthase (brain)
Eicosanoid Synthesis / Prostaglandin synthesis
regulation
Ahsp alpha hemoglobin stabilizing protein ---

413
--- --- ---
Cd24a CD24a antigen ---
Anapc11 anaphase promoting complex subunit 11 ---
Cnn1 calponin 1 Smooth muscle contraction
Myh7 myosin, heavy polypeptide 7, cardiac muscle, beta Striated muscle contraction
Hspb7 heat shock protein family, member 7 (cardiovascular) ---
Psme4 Proteasome (prosome, macropain) activator subunit 4 ---
--- --- ---
Pik3cd phosphatidylinositol 3-kinase catalytic delta polypeptide G13 Signaling Pathway
Ifit3 interferon-induced protein with tetratricopeptide repeats 3 ---
Thrsp thyroid hormone responsive SPOT14 homolog (Rattus) ---
Cyr61 cysteine rich protein 61 ---
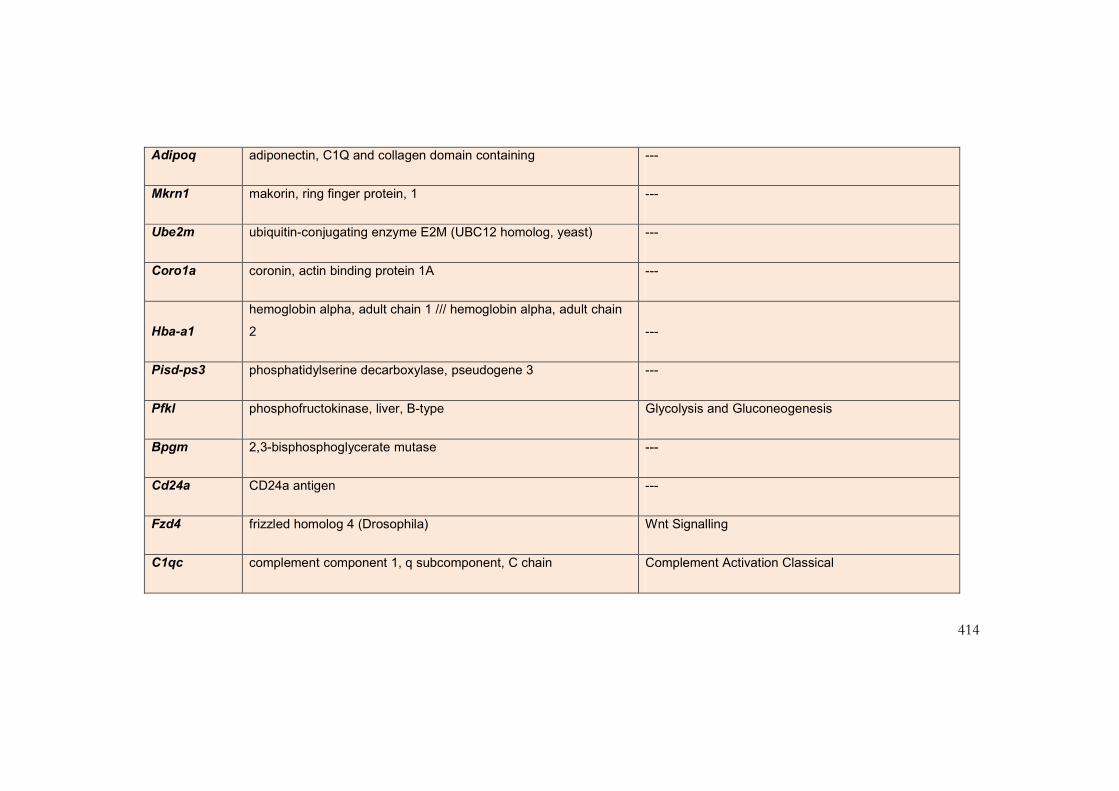
414
Adipoq adiponectin, C1Q and collagen domain containing ---
Mkrn1 makorin, ring finger protein, 1 ---
Ube2m ubiquitin-conjugating enzyme E2M (UBC12 homolog, yeast) ---
Coro1a coronin, actin binding protein 1A ---
Hba-a1
hemoglobin alpha, adult chain 1 /// hemoglobin alpha, adult chain
2 ---
Pisd-ps3 phosphatidylserine decarboxylase, pseudogene 3 ---
Pfkl phosphofructokinase, liver, B-type Glycolysis and Gluconeogenesis
Bpgm 2,3-bisphosphoglycerate mutase ---
Cd24a CD24a antigen ---
Fzd4 frizzled homolog 4 (Drosophila) Wnt Signalling
C1qc complement component 1, q subcomponent, C chain Complement Activation Classical

415
H2-Aa histocompatibility 2, class II antigen A, alpha ---
Cxcl1 chemokine (C-X-C motif) ligand 1 ---
Ccl6 chemokine (C-C motif) ligand 6 ---
Ptgr2 prostaglandin reductase 2 ---
Gm9706 ///
Isg15 predicted gene 9706 /// ISG15 ubiquitin-like modifier ---
Tnnt1 troponin T1, skeletal, slow Striated muscle contraction
Snrpn small nuclear ribonucleoprotein mRNA processing binding Reactome
Xlr4a X-linked lymphocyte-regulated 4A ---
Trp53inp2 Transformation related protein 53 inducible nuclear protein 2 ---
Mmp3 matrix metallopeptidase 3 Matrix Metalloproteinases
Kidins220 kinase D-interacting substrate 220 ---

416
Hadha hydroxyacyl-Coenzyme A dehydrogenase Mitochondrial fatty acid betaoxidation
Apol11b apolipoprotein L 11b ---
Fmo2 flavin containing monooxygenase 2 ---
Fcna ficolin A ---
Zbtb16 zinc finger and BTB domain containing 16 ---
Pisd-ps3 phosphatidylserine decarboxylase, pseudogene 3 ---
Igfbp5 insulin-like growth factor binding protein 5 Smooth muscle contraction
Malat1 metastasis associated lung adenocarcinoma transcript 1 ---
Mid1ip1
Mid1 interacting protein 1 (gastrulation specific G12-like
(zebrafish)) ---
Dpep1 dipeptidase 1 (renal) Eicosanoid Synthesis
Dhx9 DEAH (Asp-Glu-Ala-His) box polypeptide 9 mRNA processing binding Reactome

417
H2-Eb1 histocompatibility 2, class II antigen E beta Inflammatory Response Pathway
Fos FBJ osteosarcoma oncogene
Smooth muscle contraction / TGF Beta Signalling
Pathway
Gypa glycophorin A ---
Cd74
CD74 antigen (invariant polypeptide of major histocompatibility
complex, class I ---
Slc25a37 solute carrier family 25, member 37 ---
Ifitm2 interferon induced transmembrane protein 2 ---
Lgals3 lectin, galactose binding, soluble 3 ---
Etf1 eukaryotic translation termination factor 1 Translation Factors
Cdkn1a cyclin-dependent kinase inhibitor 1A (P21) Cell Cycle / G1 to S cell cycle Reactome
Myh11 myosin, heavy polypeptide 11, smooth muscle ---
Lyrm5 LYR motif containing 5 ---

418
Pisd-ps1 phosphatidylserine decarboxylase, pseudogene 1 ---

419
100 transcripts with most reduced expression in the H-Ras G12V/G12V mouse model
Gene Symbol Gene Title Pathway
Hras1 Harvey rat sarcoma virus oncogene 1
G Protein Signalling / MAPK Cascade / TGF Beta
Signaling Pathway
Ptgds prostaglandin D2 synthase (brain)
Eicosanoid Synthesis / Prostaglandin synthesis
regulation
Ptgds prostaglandin D2 synthase (brain)
Eicosanoid Synthesis / Prostaglandin synthesis
regulation
Hras1 Harvey rat sarcoma virus oncogene 1
G Protein Signalling / MAPK Cascade / TGF Beta
Signaling Pathway
Acta1 actin, alpha 1, skeletal muscle
Smooth muscle contraction / Striated muscle
contraction
H2-Aa histocompatibility 2, class II antigen A, alpha ---

420
Nppa natriuretic peptide precursor type A Smooth muscle contraction
H2-Aa histocompatibility 2, class II antigen A, alpha ---
--- --- ---
Gm10409 predicted gene, 100041874 ---
Sln sarcolipin ---
--- --- ---
Tgtp1 T-cell specific GTPase 1 ---
H2-Q7 histocompatibility 2, Q region locus 7 ---
Gm10406 predicted gene, 100041874 ---
--- --- ---
Gm3916 predicted gene 3916 ---
Mt2 metallothionein 2 ---
421
Adh1 alcohol dehydrogenase 1 (class I) ---
H2-Aa histocompatibility 2, class II antigen A, alpha ---
Prune2 prune homolog 2 (Drosophila) ---
Ifit1 interferon-induced protein with tetratricopeptide repeats 1 ---
Klra3 killer cell lectin-like receptor, subfamily A, member 3 ---
--- --- ---
Xlr4a X-linked lymphocyte-regulated 4A ---
H2-Aa histocompatibility 2, class II antigen A, alpha ---
Mpa2l macrophage activation 2 like ---
Glul glutamate-ammonia ligase (glutamine synthetase) ---
Plac9 placenta specific 9 ---
H2-Ab1 histocompatibility 2, class II antigen A, beta 1 ---

422
Grm1 glutamate receptor, metabotropic 1
GPCRDB Class C Metabotropic glutamate
pheromone
Clec7a C-type lectin domain family 7, member a ---
--- --- ---
Cd74
CD74 antigen (invariant polypeptide of major histocompatibility
complex, class I ---
Tsc22d1 TSC22 domain family, member 1 ---
Myl1 myosin, light polypeptide 1
G13 Signalling Pathway / Smooth muscle
contraction / Striated muscle contraction
Gbp3 guanylate binding protein 3 ---
H2-Eb1 histocompatibility 2, class II antigen E beta Inflammatory Response Pathway
Timp4 tissue inhibitor of metalloproteinase 4 Matrix Metalloproteinases
Thrsp thyroid hormone responsive SPOT14 homolog (Rattus) ---

423
Timp4 tissue inhibitor of metalloproteinase 4 Matrix Metalloproteinases
Gvin1 GTPase, very large interferon inducible 1 ---
Mcf2l mcf.2 transforming sequence-like ---
Ces3 carboxylesterase 3 ---
--- --- ---
Car4 carbonic anhydrase 4 ---
Gbp2 guanylate binding protein 2 ---
Gbp2 guanylate binding protein 2 ---
H2-Q6 histocompatibility 2, Q region locus 6 ---
Adk adenosine kinase ---
Ifi44 interferon-induced protein 44 ---
Colec11 collectin sub-family member 11 ---
424
C1qtnf9 C1q and tumor necrosis factor related protein 9 ---
Mpa2l macrophage activation 2 like ---
Rtp4 receptor transporter protein 4 ---
Xdh xanthine dehydrogenase ---
Fabp4 fatty acid binding protein 4, adipocyte ---
Cd83 CD83 antigen ---
Car4 carbonic anhydrase 4 ---
Hamp hepcidin antimicrobial peptide ---
Trim34 tripartite motif-containing 34 ---
Car7 carbonic anhydrase 7 ---
Ifi203 interferon activated gene 203 ---
C920025E04Rik histocompatibility 2, T region locus 23 ---

425
Ppbp pro-platelet basic protein ---
Sult1a1 sulfotransferase family 1A, phenol-preferring, member 1 ---
Kcna5
potassium voltage-gated channel, shaker-related subfamily,
member 5 ---
Trim54 tripartite motif-containing 54 ---
E030037K03Rik RIKEN cDNA E030037K03 gene ---
Ccl5 chemokine (C-C motif) ligand 5 ---
Mndal myeloid nuclear differentiation antigen like ---
Retsat retinol saturase (all trans retinol 13,14 reductase) ---
Gzma granzyme A ---
Ifi203 interferon activated gene 203 ---
Ankrd1 ankyrin repeat domain 1 (cardiac muscle) ---
426
Cxcl9 chemokine (C-X-C motif) ligand 9 ---
1810011O10Rik RIKEN cDNA 1810011O10 gene ---
Mt1 metallothionein 1 ---
Ces3 carboxylesterase 3 ---
Oasl2 2'-5' oligoadenylate synthetase-like 2 mRNA processing binding Reactome
Ly6e lymphocyte antigen 6 complex, locus E ---
Dpep1 dipeptidase 1 (renal) Eicosanoid Synthesis
Mpeg1 macrophage expressed gene 1 Cell Cycle
Ifit3 interferon-induced protein with tetratricopeptide repeats 3 ---
Adamts9
a disintegrin-like and metallopeptidase (reprolysin type) with
thrombospondin type motif ---
Bdnf brain derived neurotrophic factor ---

427
Gbp6 guanylate binding protein 6 ---
Epha4 Eph receptor A4 ---
H2-K1 histocompatibility 2, K1, K region ---
Lrrc2 leucine rich repeat containing 2 ---
Usp18 ubiquitin specific peptidase 18 ---
Trim30 tripartite motif-containing 30 ---
Kank3 KN motif and ankyrin repeat domains 3 ---
Ifit2 interferon-induced protein with tetratricopeptide repeats 2 ---
H2-Ab1 histocompatibility 2, class II antigen A, beta 1 ---
AW112010 expressed sequence AW112010 ---
Retnla resistin like alpha ---
Slco2b1 solute carrier organic anion transporter family, member 2b1 ---
428
Retsat retinol saturase (all trans retinol 13,14 reductase) ---
Fam107a family with sequence similarity 107, member A ---
100 transcripts with most increased expression in the H-Ras G12V/G12V mouse model
Gene Symbol Gene Title Pathway
Erc1 ELKS/RAB6-interacting/CAST family member 1 ---
Paip1 polyadenylate binding protein-interacting protein 1 Translation Factors
Egr1 early growth response 1 Ovarian Infertility Genes
Car3 carbonic anhydrase 3 ---
Hist1h2ad histone cluster 1, H2ad ---
--- --- ---

429
Il6 interleukin 6
Inflammatory Response Pathway / Smooth muscle
contraction
Paip1 similar to poly(A) binding protein interacting protein 1 Translation Factors
Tubb2c tubulin, beta 2C ---
Cenpf centromere protein F ---
Asns asparagine synthetase ---
Tfrc transferrin receptor ---
Ezr ezrin ---
Uhrf1 ubiquitin-like, containing PHD and RING finger domains, 1 ---
Asns asparagine synthetase ---
A530088H08Rik RIKEN cDNA A530088H08 gene ---
Palld palladin, cytoskeletal associated protein ---

430
--- --- ---
Mid1 midline 1 ---
Tubb6 tubulin, beta 6 ---
Tmem56 transmembrane protein 56 ---
A530088H08Rik RIKEN cDNA A530088H08 gene ---
Kcne1 potassium voltage-gated channel, Isk-related subfamily, member 1 ---
Tuba8 tubulin, alpha 8 ---
2810417H13Rik RIKEN cDNA 2810417H13 gene ---
Ccna2 cyclin A2 Cell Cycle KEGG
Cdt1 chromatin licensing and DNA replication factor 1 DNA replication Reactome
--- --- ---
9430011C21Rik RIKEN cDNA 9430011C21 gene ---

431
Slc4a1 solute carrier family 4 (anion exchanger), member 1 ---
Elavl2
ELAV (embryonic lethal, abnormal vision, Drosophila)-like 2 (Hu
antigen B) mRNA processing binding Reactome
Tubb5 tubulin, beta 5 ---
Ptn pleiotrophin ---
Mcm5
minichromosome maintenance deficient 5, cell division cycle 46 (S.
cerevisiae)
Cell Cycle / DNA replication Reactome / G1 to S cell
cycle Reactome
Fbn2 fibrillin 2 ---
--- --- ---
Ube2c ubiquitin-conjugating enzyme E2C ---
Ckap4 cytoskeleton-associated protein 4 ---
Anln anillin, actin binding protein ---
Tuba4a tubulin, alpha 4A ---

432
Kcne1 potassium voltage-gated channel, Isk-related subfamily, member 1 ---
Slc8a1 solute carrier family 8 (sodium/calcium exchanger), member 1
Calcium regulation in cardiac cells / Smooth muscle
contraction
Birc5 baculoviral IAP repeat-containing 5 Apoptosis
Egr3 early growth response 3 ---
Mki67 antigen identified by monoclonal antibody Ki 67 ---
Tuba8 tubulin, alpha 8 ---
Dpysl3 dihydropyrimidinase-like 3 ---
Nov nephroblastoma overexpressed gene ---
AI605517 expressed sequence AI605517 ---
--- --- ---
2900072G11Rik RIKEN cDNA 2900072G11 gene ---

433
Vcan versican ---
--- --- ---
Nusap1 nucleolar and spindle associated protein 1 ---
Ect2 ect2 oncogene ---
Nr1d2 Nuclear receptor subfamily 1, group D, member 2 Circadian Exercise / Nuclear Receptors
Adipoq adiponectin, C1Q and collagen domain containing ---
Carns1 carnosine synthase 1 ---
Ptprg protein tyrosine phosphatase, receptor type, G ---
Slc4a1 solute carrier family 4 (anion exchanger), member 1 ---
Kif11 kinesin family member 11 ---
Esco2 establishment of cohesion 1 homolog 2 (S. cerevisiae) ---
Cdkn1a cyclin-dependent kinase inhibitor 1A (P21) Cell Cycle / G1 to S cell cycle Reactome

434
Hells helicase, lymphoid specific Apoptosis
Elavl2
ELAV (embryonic lethal, abnormal vision, Drosophila)-like 2 (Hu
antigen B) mRNA processing binding Reactome
Cdc20 cell division cycle 20 homolog (S. cerevisiae) Cell Cycle
Lig1 ligase I, DNA, ATP-dependent ---
Mcm3
similar to DNA replication licensing factor MCM3 (DNA polymerase
alpha holoenzym
Cell Cycle / DNA replication Reactome / G1 to S cell
cycle Reactome
Cdca8 cell division cycle associated 8 ---
2900072G11Rik RIKEN cDNA 2900072G11 gene ---
Fos FBJ osteosarcoma oncogene
Smooth muscle contraction / TGF Beta Signalling
Pathway
--- --- ---
Fam122b family with sequence similarity 122, member B ---

435
Spry4 sprouty homolog 4 (Drosophila) ---
Dclk1 doublecortin-like kinase 1 ---
Top2a topoisomerase (DNA) II alpha ---
--- --- ---
--- --- ---
Fignl1 fidgetin-like 1 ---
Tuba3a tubulin, alpha 3A ---
Mcm6
minichromosome maintenance deficient 6 (MIS5 homolog, S.
pombe) (S. cerevisiae)
Cell Cycle / DNA replication Reactome / G1 to S cell
cycle Reactome
Sqle squalene epoxidase Cholesterol_Biosynthesis
Lpar3 lysophosphatidic acid receptor 3 ---
Aldh1a2 aldehyde dehydrogenase family 1, subfamily A2 ---
436
Ttc27 tetratricopeptide repeat domain 27 ---
Asb4 ankyrin repeat and SOCS box-containing 4 ---
Tnnt1 troponin T1, skeletal, slow Striated muscle contraction
--- --- ---
Mef2a myocyte enhancer factor 2A ---
Ckap4 cytoskeleton-associated protein 4 ---
Neto2 neuropilin (NRP) and tolloid (TLL)-like 2 ---
Nrk Nik related kinase ---
Mapkapk5 MAP kinase-activated protein kinase 5 ---
Serpinb1a serine (or cysteine) peptidase inhibitor, clade B, member 1a ---
Ramp1 receptor (calcitonin) activity modifying protein 1 Smooth muscle contraction
Ccne2 cyclin E2 Cell Cycle / G1 to S cell cycle Reactome

437
Kif2c kinesin family member 2C ---
Fbn2 fibrillin 2 ---
Smc6 structural maintenance of chromosomes 6 ---
Mcm6
minichromosome maintenance deficient 6 (MIS5 homolog, S.
pombe) (S. cerevisiae)
Cell Cycle / DNA replication Reactome / G1 to S cell
cycle Reactome

438
9.6 References for appendices
1. Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, et al.
Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous
syndromes: molecular diversity and associated phenotypic spectrum. Human mutation.
2009;30(4):695-702. Epub 2009/02/12.
2. Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, et al. Cardio-
facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling
pathway: genotype-phenotype relationships and overlap with Costello syndrome.
Journal of medical genetics. 2007;44(12):763-71. Epub 2007/08/21.
3. Gripp KW, Lin AE, Nicholson L, Allen W, Cramer A, Jones KL, et al. Further
delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps
differentiate cardio-facio-cutaneous syndrome from Costello syndrome. American
journal of medical genetics Part A. 2007;143A(13):1472-80. Epub 2007/06/07.
4. Koudova M, Seemanova E, Zenker M. Novel BRAF mutation in a patient with
LEOPARD syndrome and normal intelligence. European journal of medical genetics.
2009;52(5):337-40. Epub 2009/05/07.
5. Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline
KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nature genetics.
2006;38(3):294-6. Epub 2006/02/14.
6. Narumi Y, Aoki Y, Niihori T, Neri G, Cave H, Verloes A, et al. Molecular and
clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical
manifestations with Costello syndrome. American journal of medical genetics Part A.
2007;143A(8):799-807. Epub 2007/03/17.
7. Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach
G, et al. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and
Costello syndrome. Clinical genetics. 2008;73(1):62-70. Epub 2007/11/29.

439
8. Croonen EA, Nillesen WM, Schrander C, Jongmans M, Scheffer H, Noordam
C, et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with
abnormal ultrasound findings. European journal of human genetics : EJHG.
2013;21(9):936-42.
9. Cave H. Gene symbol: BRAF. Disease: Cardio-facio-cutaneous syndrome.
Human genetics. 2008;123(1):108-9. Epub 2008/04/04.
10. Rodriguez-Viciana P, Rauen KA. Biochemical characterization of novel germline
BRAF and MEK mutations in cardio-facio-cutaneous syndrome. Methods in
enzymology. 2008;438:277-89. Epub 2008/04/17.
11. Siegel DH, McKenzie J, Frieden IJ, Rauen KA. Dermatological findings in 61
mutation-positive individuals with cardiofaciocutaneous syndrome. The British journal
of dermatology. 2011;164(3):521-9. Epub 2010/11/11.
12. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS,
et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-
cutaneous syndrome. Science (New York, NY). 2006;311(5765):1287-90. Epub
2006/01/28.
13. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, et al.
Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies.
American journal of medical genetics Part A. 2012;158A(5):1077-82. Epub 2012/04/11.
14. Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurosawa K, et al.
Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous
syndrome in Japan: findings from a nationwide epidemiological survey. American
journal of medical genetics Part A. 2012;158A(5):1083-94. Epub 2012/04/13.
15. Nystrom AM, Ekvall S, Berglund E, Bjorkqvist M, Braathen G, Duchen K, et al.
Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically
overlapping disorders. Journal of medical genetics. 2008;45(8):500-6. Epub 2008/05/06.

440
16. Rauen KA. Distinguishing Costello versus cardio-facio-cutaneous syndrome:
BRAF mutations in patients with a Costello phenotype. American journal of medical
genetics Part A. 2006;140(15):1681-3. Epub 2006/06/29.
17. Champion KJ, Bunag C, Estep AL, Jones JR, Bolt CH, Rogers RC, et al.
Germline mutation in BRAF codon 600 is compatible with human development: de
novo p.V600G mutation identified in a patient with CFC syndrome. Clinical genetics.
2011;79(5):468-74. Epub 2010/08/26.
18. Dentici ML, Sarkozy A, Pantaleoni F, Carta C, Lepri F, Ferese R, et al. Spectrum
of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and
genotype-phenotype correlations. European journal of human genetics : EJHG.
2009;17(6):733-40. Epub 2009/01/22.
19. Kratz CP, Niemeyer CM, Zenker M. An unexpected new role of mutant Ras:
perturbation of human embryonic development. Journal of molecular medicine (Berlin,
Germany). 2007;85(3):227-35. Epub 2007/01/11.
20. Rauen KA, Tidyman WE, Estep AL, Sampath S, Peltier HM, Bale SJ, et al.
Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous
syndrome: transmission through four generations. American journal of medical genetics
Part A. 2010;152A(4):807-14. Epub 2010/04/02.
21. Linden HC, Price SM. Cardiofaciocutaneous syndrome in a mother and two
sons with a MEK2 mutation. Clinical dysmorphology. 2011;20(2):86-8. Epub
2010/12/24.
22. Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R, et al.
Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline
mutations. Journal of medical genetics. 2007;44(2):131-5. Epub 2006/10/24.
23. Bertola DR, Pereira AC, Brasil AS, Albano LM, Kim CA, Krieger JE. Further
evidence of genetic heterogeneity in Costello syndrome: involvement of the KRAS
gene. Journal of human genetics. 2007;52(6):521-6. Epub 2007/05/01.

441
24. Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline
KRAS mutations cause Noonan syndrome. Nature genetics. 2006;38(3):331-6. Epub
2006/02/14.
25. Leventopoulos G, Denayer E, Makrythanasis P, Papapolychroniou C, Fryssira
H. Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS
mutation. Clinical and experimental rheumatology. 2010;28(4):556-7. Epub 2010/09/03.
26. Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, et al.
Craniosynostosis in patients with Noonan syndrome caused by germline KRAS
mutations. American journal of medical genetics Part A. 2009;149A(5):1036-40. Epub
2009/04/28.
27. Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E,
Rauen KA, et al. Effects of germline mutations in the Ras/MAPK signaling pathway on
adaptive behavior: cardiofaciocutaneous syndrome and Noonan syndrome. American
journal of medical genetics Part A. 2010;152A(3):591-600. Epub 2010/02/27.
28. Brasil AS, Malaquias AC, Kim CA, Krieger JE, Jorge AA, Pereira AC, et al.
KRAS gene mutations in Noonan syndrome familial cases cluster in the vicinity of the
switch II region of the G-domain: report of another family with metopic
craniosynostosis. American journal of medical genetics Part A. 2012;158A(5):1178-84.
Epub 2012/04/11.
29. Brasil AS, Pereira AC, Wanderley LT, Kim CA, Malaquias AC, Jorge AA, et al.
PTPN11 and KRAS gene analysis in patients with Noonan and Noonan-like syndromes.
Genetic testing and molecular biomarkers. 2010;14(3):425-32. Epub 2010/06/29.
30. Razzaque MA, Komoike Y, Nishizawa T, Inai K, Furutani M, Higashinakagawa
T, et al. Characterization of a novel KRAS mutation identified in Noonan syndrome.
American journal of medical genetics Part A. 2012;158A(3):524-32. Epub 2012/02/04.
31. Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, et al.
Germline missense mutations affecting KRAS Isoform B are associated with a severe

442
Noonan syndrome phenotype. American journal of human genetics. 2006;79(1):129-35.
Epub 2006/06/15.
32. Sovik O, Schubbert S, Houge G, Steine SJ, Norgard G, Engelsen B, et al. De
novo HRAS and KRAS mutations in two siblings with short stature and neuro-cardio-
facio-cutaneous features. Journal of medical genetics. 2007;44(7):e84. Epub
2007/07/03.
9.7 Appendix 7: Reprints of articles relating to the work
undertaken (in chronological order):
Burkitt Wright EM, Kerr B. RAS-MAPK pathway disorders: important causes of
congenital heart disease, feeding difficulties, developmental delay and short stature.
Arch Dis Child. 2010;95:724-30.
Burkitt Wright E, Donnai D, Johnson D, Clayton-Smith J. Cutaneous features in
17q21.31 deletion syndrome: a differential diagnosis for cardio-facio-cutaneous
syndrome. Clin Dysmorphol. 2011;20:15-20.
Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, Burkitt Wright EM,
Cañamero M, Mulero F, Ortega S, Bustelo XR, Barbacid M. Constitutive activation of
B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous
syndrome. Proc Natl Acad Sci U S A. 2011;108:5015-20.
Burkitt Wright EM, Bradley L, Shorto J, McConnell VP, Gannon C, Firth HV, Park
SM, D'Amore A, Munyard PF, Turnpenny PD, Charlton A, Wilson M, Kerr B.
Neonatal

443
lethal Costello syndrome and unusual dinucleotide deletion/insertion mutations in
HRAS predicting p.Gly12Val. Am J Med Genet A. 2012;158A:1102-10.
Burkitt Wright EM, Sach E, Sharif S, Quarrell O, Carroll T, Whitehouse RW,
Upadhyaya M, Huson SM, Evans DG. Can the diagnosis of NF1 be excluded clinically?
A lack of pigmentary findings in families with spinal neurofibromatosis
demonstrates a limitation of clinical diagnosis. J Med Genet. 2013;50:606-13.
Giannoulatou E, McVean G, Taylor IB, McGowan SJ, Maher GJ, Iqbal Zb, Pfeifer SP,
Turner I, Burkitt-Wright EMM, Shorto J, Itani A, Turner K, Gregory L, Buck D,
Rajpert-De Meyts E, Looijenga LHJ, Kerr B, Wilkie AOM, and Goriely A.
Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS
mutations in the paternal germline. Proc Natl Acad Sci U S A, in press November 2013.

444
This was an invited review for Archives of Disease in Childhood, and hence focused on
the clinical presentation of these disorders, the commonalities and differences between
them.
This was a case series of four patients who had presented with features of CFC
syndrome, with prominent ectodermal features including large numbers of naevi, who
were identified to have 17q21.31 microdeletions.

445
This article described the first mouse model of CFC syndrome, generated by the
Barbacid laboratory. The characterisation of this mouse was a project in which I was
involved over the course of a 4 month collaborative placement in this laboratory in
2008.
This was a case series of four patients with neonatal lethal Costello syndrome, each due
to a p.(Gly12Val) mutation. Two of these patients were only diagnosed some time after
their death, emphasizing the possibility that this phenotype may occasionally go
unrecognized in severely ill neonates.

446
Five families were ascertained through the Manchester service for complex NF1, who
had multiple members affected with the spinal variant of this disorder. These patients
have a high burden of spinal neurofibromas, but few cutaneous or other externally
visible features of this disorder. This is a significant phenotype to ascertain, as it is
difficult to diagnose, requiring specialist imaging, associated with high morbidity, and
the 50% risk of transmission to offspring is important information for families.
This article presents a model for the molecular basis for paternally derived HRAS
mutations in Costello syndrome, and contrasts this to the situation in cancer. Selfish
spermatogonial selection is the term used to describe the process whereby codon 12
mutations in HRAS become enriched in sperm samples as the donor’s age increases.

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069724
1Genetic Medicine, University of Manchester, Manchester Academic Health Science Centre, Central Manchester University Hospitals NHS Foundation Trust, St Mary’s Hospital, Manchester, UK2Genetic and Developmental Medicine, Manchester Biomedical Research Centre, Central Manchester University Hospitals NHS Foundation Trust, Manchester Royal Infi rmary, Manchester, UK
Correspondence toBronwyn Kerr, Genetic Medicine, University of Manchester, Manchester Academic Health Science Centre, Central Manchester University Hospitals NHS Foundation Trust, St Mary’s Hospital, Oxford Road, Manchester M13 9WL, UK; [email protected]
EMMBW and BK contributed equally to this work.
Accepted 20 November 2009
RAS-MAPK pathway disorders: important causes of congenital heart disease, feeding diffi culties, developmental delay and short statureEmma M M Burkitt Wright,1 2 Bronwyn Kerr1
ABSTRACTThe disorders described as the neuro-cardio-facio-
cutaneous conditions (NCFCs) may all present with
symptoms that are common in paediatric practice.
They result from germline mutations in genes encoding
kinases and other proteins interacting in the RAS-MAPK
pathway. This review summarises these disorders,
discussing their presenting features and clinical course,
identifying overarching similarities and, conversely,
features that can help to discriminate one condition from
another. The genetic basis and importance of precise
clinical diagnosis and molecular diagnostic confi rmation
when possible is discussed, given each condition’s
different prognosis, and the need to remain vigilant for
specifi c complications.
INTRODUCTIONNeuro-cardio-facio-cutaneous conditions (NCFCs) have an estimated collective prevalence of between 1 in 700 and 1 in 1250 per head of population,1 2 this large range refl ecting the diversity of esti-mates for the prevalence of Noonan syndrome (NS), the most common of these conditions. As the name implies, each of these conditions may have involvement of similar body systems, par-ticularly the nervous and cardiovascular systems and the skin, with or without striking facial dys-morphism. NS and neurofi bromatosis type 1 (NF1) are the conditions within this group of disorders most familiar to paediatricians, due to both their high prevalence and high incidence of childhood complications. The NCFCs show autosomal dom-inant inheritance, with a high proportion of cases representing de novo mutations. While the milder disorders may be inherited through the genera-tions, the more severe conditions nearly always arise from new dominant mutations.
The genes responsible for these disorders each encode a component of the RAS-MAPK pathway, as shown in fi gure 1. RAS was the fi rst identifi ed human oncogene and mutations in this and other pathway genes are found in around 30% of human malignancies.3 The fact that these same genes are involved in the genesis of congenital abnormali-ties and learning disability in the NCFCs demon-strates their parallel importance in developmental processes. The creation of drugs acting on the pathway, developed to treat cancer, provides pros-pects for therapy of this group of conditions.
CLINICAL APPROACH TO THE NCFCsClinical presentations that are particularly sugges-tive of the NCFCs vary with age. In the newborn
period, congenital heart disease (particularly pulmonary stenosis, atrial septal defect), hyper-trophic cardiomyopathy (HCM) or arrhythmia, with a history of polyhydramnios or high birth weight should raise the index of suspicion. In the fi rst year, severe feeding diffi culties and failure to thrive are common. As childhood progresses, learning disability, short stature with relative macrocephaly and skin abnormalities may be the presenting features.
Key clinical features of RAS-MAPK pathway disorders are summarised in table 1 and typical facial features are shown in fi gure 2, including (relative) macrocephaly, low-set ears, downslant-ing palpebral fi ssures and hypertelorism. These are common across these various disorders and hence it is often not possible to make a specifi c diagno-sis on the basis of facial appearance alone. Once other features are taken into account, a clinical diagnosis of a specifi c NCFC may be possible, but a signifi cant proportion of patients have atypical or overlapping combinations of features, making defi nitive clinical diagnoses diffi cult (and hence also subject to revision over time). In addition, while for some of these conditions the natural his-tory has been well studied, for others the outcome is variable and less certain. Considerable genetic and phenotypic heterogeneity is demonstrated: many different genes may cause the same pheno-type, while identical mutations in the same gene may result in different clinical presentations.
PHENOTYPIC AND GENOTYPIC OVERLAP AMONG NCFCsLong before the discovery of the related molecu-lar basis for the NCFCs, the clinical resemblances and overlaps between these conditions were rec-ognised. A group of patients with NF1 with facial features reminiscent of NS were identifi ed and this is discussed further below. Clinical recognition of an overlap between NS and cardio-facio-cutaneous (CFC) syndrome also predates the molecular era by many years.4 Particularly in early life, clinical differ-entiation between Costello syndrome (CS) and CFC is often diffi cult. Some patients with initial diagno-ses of CS were subsequently diagnosed with CFC and a smaller number with initial CFC diagnoses were later considered to have CS. Quantifi cation of differential facial morphometry in these conditions has demonstrated overlaps between NS and CFC and between CFC and CS, emphasising the diffi -culty in clinical assessment (P Hammond, personal communication). Recognition of the relationship between each of these disorders at the molecular
13_archdischild160069-182717.indd 72413_archdischild160069-182717.indd 724 8/16/2010 5:29:37 PM8/16/2010 5:29:37 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069 725
Table 1 Key clinical features in the neuro-cardio-facio-cutaneous conditions (NCFCs) Noonan syndrome Leopard syndrome CFC syndrome Costello syndrome NF type 1 Legius syndrome
Year of fi rst description 1965 1969 1986 1977 1768 2007Normal or large birth weight
+ + + + + +
Macrocephaly + + ± + + +Feeding diffi culties ± ± +++ +++ Not usual −Short stature + Not usual + + ± Not usualDevelopmental disability
If present, usually specifi c learning disability
Not usual May be severe Mild to moderate If present (30%), often mild or specifi c learning disability
?milder/less frequent than in NF1
Congenital heart disease
+ + + + 10% Appears uncommon
Hypertrophic cardiomyopathy
+ + + + Rare Not reported
Cardiac dysrhythmia – Heart block – Atrial arrhythmia in 50% Uncommon Appears uncommonCutaneous features Occasional café-au-lait
patchesLentigines Many; ulerythema
ophrhyogenes, keratosis pilaris
Excess skin on hands and feet, papillomata, warts, hyperkeratosis
Café-au-lait patches, cutaneous neurofi bromas
Café-au-lait patches, depigmented macules, lipomas
Sensorineural deafness Rare Common Rare Rare Rare Not reportedTumour risk Increased risk of juvenile
myelomonocytic and acute myeloid leukaemias, giant cell tumours
Not established; single reports8
Not established; single reports18 19
High: 17% for a childhood cancer, particularly rhabdomyosarcoma
Increased risk for a wide range of tumours
Not established; single report of Wilms tumour12
Variant phenotypes/genotype–phenotype correlation
cRaf mutations: high rate of HCM SOS1 mutations: more likely to have normal stature and ectodermal abnormalities SHOC2 mutation: loose anagen hair, hypernasal speech
– – Severe neonatal phenotype: frequently lethal, due to respiratory insuffi ciency (central, airway, parenchymal components), rapidly progressive cardiomyopathy or congenital skeletal myopathy
Recurrent three base pair deletion in exon 17: café-au-lait patches only. Microdeletion of whole NF1 gene: high number of neurofi bromas, tall stature. NF
–
CFC, cardio-facio-cutaneous; CS, Costello syndrome; HCM, hypertrophic cardiomyopathy; NF1, neurofi bromatosis type 1.
KRAS
CRAF
MEK1 MEK2
ERK1/2
SPRED1
GRB2SOS1
SHC
Receptor tyrosine kinase
Activation leads to transcription of many genes important in cell proliferation,growth and other processes in both the nucleus and cytosol
SHP2
Neurofibromatosis type I
Costello syndrome
Noonan syndrome and Leopard syndrome
Legius syndrome
Cardio-facio-cutaneous syndrome
Intracellular space
Extracellular space
HRAS
NF1
SHOC2
BRAF
Figure 1 The RAS-MAPK pathway and disorders resulting from mutations in its genes.
13_archdischild160069-182717.indd 72513_archdischild160069-182717.indd 725 8/16/2010 5:29:37 PM8/16/2010 5:29:37 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from
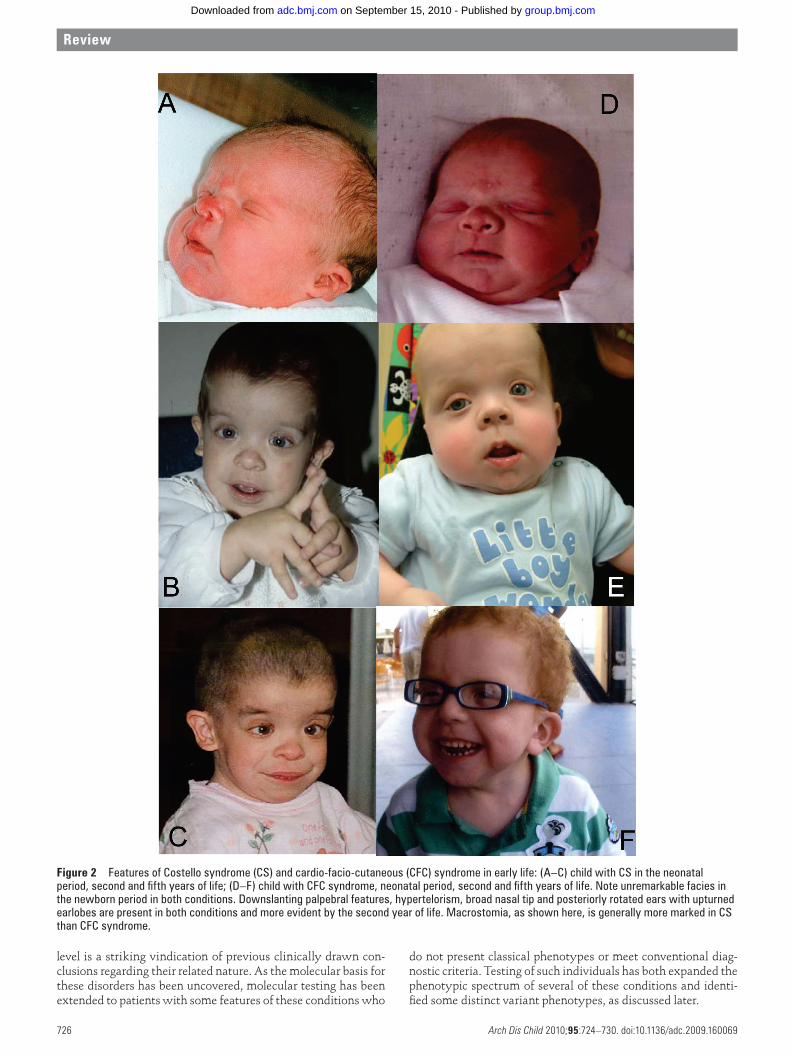
Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069726
Figure 2 Features of Costello syndrome (CS) and cardio-facio-cutaneous (CFC) syndrome in early life: (A–C) child with CS in the neonatal period, second and fi fth years of life; (D–F) child with CFC syndrome, neonatal period, second and fi fth years of life. Note unremarkable facies in the newborn period in both conditions. Downslanting palpebral features, hypertelorism, broad nasal tip and posteriorly rotated ears with upturned earlobes are present in both conditions and more evident by the second year of life. Macrostomia, as shown here, is generally more marked in CS than CFC syndrome.
level is a striking vindication of previous clinically drawn con-clusions regarding their related nature. As the molecular basis for these disorders has been uncovered, molecular testing has been extended to patients with some features of these conditions who
do not present classical phenotypes or meet conventional diag-nostic criteria. Testing of such individuals has both expanded the phenotypic spectrum of several of these conditions and identi-fi ed some distinct variant phenotypes, as discussed later.
13_archdischild160069-182717.indd 72613_archdischild160069-182717.indd 726 8/16/2010 5:29:38 PM8/16/2010 5:29:38 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069 727
THE NCFC SYNDROMESNoonan syndromeThe prevalence of NS is estimated at between 1 in 1000 and 1 in 2500.2 Mutations in several pathway genes are now known to cause NS, as shown in table 2. Irrespective of the causative gene, the effect of mutations is to alter signalling activity through the pathway, usually increasing this.5 It is thought that many individuals with NS remain undiagnosed; adults especially may be asymptomatic. The phenotype of NS shows extreme variability, from mildly short stature with normal intelligence to severe congenital heart disease or HCM, or (rarely) severe learning disability. Typical features on exami-nation include ptosis, downslanting palpebral fi ssures and pterygium colli. Cryptorchidism is common and contributes to the reduced fertility observed in NS males.2 Childhood leukaemia is an occasional fi nding in NS, but there is a better prognosis for the myeloproliferative disorders that occur in NS than in other situations, with spontaneous remission reported, particularly in association with germline c.218C→T (pT73I) mutations in PTPN11.2 Further genotype–phenotype correla-tions for PTPN11 mutations include hotspots in exon 8 for pul-monary stenosis, while patients with SOS1 mutations often have prominent ectodermal features and are less likely to be of short stature.6 Mutations in SHOC2 have recently been found in a group of patients with a Noonan-like syndrome.7 These individuals appear to have prominent ectodermal features, including easily plucked hair with characteristic histology and also a hypernasal voice.
Leopard syndromeLeopard syndrome (LS) has long been recognised to be closely related to NS. Like NS, it most commonly arises due to muta-tions in PTPN11 (85% of cases),8 but mutations in CRAF8 and BRAF9 have also been reported. Individuals with LS typically have a facial gestalt similar to patients with NS, but addition-ally have numerous lentigines from early childhood, predomi-nantly over the face and trunk. These do not darken in the sun, unlike freckles. Sensorineural deafness, HCM and cardiac conduction abnormalities are all commonly found in LS.8 The tumour risk in LS is not thought to be high, but individual cases of neuroblastoma, myelodysplasia, acute leukaemia and other neoplasias have been described.8
Neurofi bromatosis type 1NF1 affects 1 in 2500 people,1 and arises due to a large variety of loss of function mutations in the NF1 gene,1 which result in increased levels of active, GTP-bound RAS. There are well- established diagnostic criteria,1 which enable a clinical diag-nosis to be made. However, its extreme phenotypic variability and the occurrence of café-au-lait patches in several other con-ditions, means that there remains potential for misdiagnosis.
While the large majority of children with NF1 remain healthy, predisposition to many different tumours is now recognised, including optic glioma and a range of childhood cancers.1 While absolute risks for each individual tumour remain low, because of their extreme rarity in the general population, the relative risk of developing an NF1-associated tumour is high.
The spectrum of NF1-associated complications is wide, hence the need for education of care providers and families of affected children, even those with minimal manifestations, that new symptoms should be considered in this context. Communicating small but signifi cant risks to families propor-tionately and managing any resultant anxiety, remain major challenges in NF1 care. Annual review of children with NF1 is an important part of this process, both for informing affected children and their families about the condition, as appropriate to the child’s level of understanding and for clinical assessment, as early recognition of complications can improve outcome.1
Around half of patients with NF1 have new mutations, being the fi rst affected persons in their family.1 Segmental NF1, refl ecting somatic mosaicism for NF1 mutation, occasion-ally occurs in these de novo cases and is associated with lower risks of NF1-associated complications such as learning disabil-ity. These individuals may also be germline mosaics for their mutation, meaning that while they have a risk of passing on non-mosaic NF1 to their children, this risk may be lower than if they themselves had non-mosaic NF1.1
Several other subgroups of patients with NF1 have been rec-ognised, including a group with taller stature, high numbers of neurofi bromas and a high incidence of learning diffi culties. These patients are more likely to have a microdeletion encom-passing the entire NF1 gene.1 Molecular confi rmation enables more accurate counselling that other affected family members would also be at higher risk of this more severe phenotype.
Table 2 Genes known to be mutated in the neuro-cardio-facio-cutaneous conditions (NCFCs)
Gene Alternative gene names Protein name LocusNCFCs in which germline mutations described
De novo or inherited (where assessed)
Mutations present in what proportion
PTPN11 PTP2C SHP2 SHP2 12q24.1 NS Leopard 70% de novo 50% of NS; 85% of LeopardSHOC2 SUR8 SHOC2 10q25 ‘Noonan-like syndrome with
loose anagen hair’All de novo to date Recent description: not yet
knownSOS1 – SOS1 2p22-p21 NS – 10–15% of NSKRAS KRAS2, RASK2, C-KRAS KRAS 12p12.1 NS, CFC de novo 5% of NS. Rare cause of CFCHRAS HRAS1, RASH1, HAMSV HRAS 11p15.5 Costello de novo except rare cases of
parental mosaicismClose to 100% of CS
BRAF BRAF1, RAFB1 BRAF 7q34 CFC, NS, Leopard De novo ?50% of CFC. Rare cause of NS, Leopard
CRAF RAF1 CRAF 3p25 NS, Leopard – RareMEK1 MAP2K1, MKK1, MAPKK1,
PRKMK1MEK1 15q21 CFC De novo ?15% of CFC
MEK2 MAP2K2, MKK2, MAPKK2, PRKMK2
MEK2 19p13.3 CFC De novo ?10% of CFC
NF1 – NF1, neurofi bromin
17q11.2 – 50% de novo Close to 100% of NF1
SPRED1 – SPRED1 15q13.2 Legius syndrome ?usually inherited ?50% of non-NF1 familial CALs
CAL; CFC, cardio-facio-cutaneous; CS, Costello syndrome; HAMSV, Harvey murine sarcoma virus; NF1, neurofi bromatosis type 1; NS, Noonan syndrome.
13_archdischild160069-182717.indd 72713_archdischild160069-182717.indd 727 8/16/2010 5:29:45 PM8/16/2010 5:29:45 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069728
Conversely, a recurrent three base-pair inframe deletion in exon 17 of the NF1 gene causes café-au-lait patches without cutaneous neurofi bromas and preliminary data suggest low risks of other NF1-associated complications.1 The favourable prognosis associated with this mutation provides useful infor-mation for families and clinicians.
Patients with an NF-NS phenotype, intermediate between NF1 and NS, have most commonly been found to have muta-tions in NF1, some of which are also described in classical NF1.10 The co-occurrence of PTPN11 and NF1 mutations in individu-als with the NF-NS phenotype has also been reported.11 Such examples emphasise the complexity and variety of genotypes and phenotypes existing within this group of patients.
Legius syndrome: SPRED1 mutationSince the identifi cation of the NF1 gene, several families with NF1-like features have been described who do not have NF1 mutations. Loss of function mutations in SPRED1 have recently been found in such families, accounting for approxi-mately 3% of the clinical NF1 population.12 These individuals characteristically have café-au-lait patches but not neurofi bro-mas and the incidence of NF1-associated complications such as learning disability or structural malformations appears very low, in the few families ascertained to date. This milder spec-trum of disease makes the NF1-SPRED1 diagnostic distinction important to make. Nearly all reported patients with SPRED1 mutations have a family history of café-au-lait patches, in con-trast to the high rate of new mutations in NF1.12
CS and CFC syndromeThese are the most severe of the NCFCs, with individuals usually presenting with severe feeding problems and failure to thrive. Intestinal malrotation or pyloric stenosis may occur additionally. Both CS and CFC syndrome confer high risks of congenital heart disease and HCM. Nearly all cases are signifi -cantly developmentally delayed. Abnormal scalp hair is very common, which may be unusually sparse, thick or very curly. Facial features may be unremarkable in infancy, but coarsen with age and are often similar in the two conditions, though distinguishable in classical cases (see fi gure 2). Strabismus and nystagmus are common in both patient groups.
Since the identifi cation of genes for CS and CFC syndrome, the breadth of the spectrum of problems encountered within these conditions has been recognised. Importantly, a severe, frequently lethal, CS phenotype can occur.13 Relatively nor-mal facial features at this age (see fi gure 2A) may mean that this diagnosis requires specifi c consideration in this urgent situation. Presenting features of affected individuals are sum-marised in table 1. Importantly, congenital myopathy may occur in CS and hence this should be considered within the differential diagnosis of the congenitally myopathic infant.
Costello syndromeFeatures suggestive of CS include neonatal atrial arrhyth-mias, ulnar deviation, excess skin which darkens with age, papillomata (usually after age 2 years) and childhood cancers, particularly embryonic rhabdomyosarcoma and bladder car-cinoma (the latter typically from teenage years onwards).14 The clinical diagnosis of CS is confi rmed by fi nding a HRAS mutation.15 HRAS mutations frequently affect codons 12 or 13, in the kinase domain of the protein and result in it being constitutively active. Such mutations overlap with those commonly reported in cancers.5 G12S is the most common
mutation found in patients with CS,15 severe neonatal pheno-types are usually due to less common mutations,15 and muta-tions resulting in milder CS phenotypes are also recognised.14 It is particularly important to diagnose children affected with CS because of the associated cancer risk.
CFC syndromePatients with CFC syndrome typically have more severe developmental delay and worse long-term neurological out-comes than those with CS. Ventriculomegaly/hydrocepha-lus, structural abnormalities and epilepsy are all common in CFC syndrome.16 Fifty per cent of patients have seizures,16 which may present as infantile spasms and may also be hard to control. Ectodermal abnormalities are also typically more severe in patients with CFC syndrome: absent eyebrows (ulerythema ophryogenes) and keratosis pilaris are character-istically present.16 While no increased risk of cancer is iden-tifi ed in CFC syndrome, single cases of hepatoblastoma, in a patient immunosuppressed after cardiac transplantation,17 and two cases of acute lymphoblastoid leukaemia18 19 have been reported. BRAF is the most common gene to be mutated in CFC syndrome16; others are shown in fi gure 1 and table 2, but up to 40% of patients may have no molecular diagnosis currently.16 Genotype–phenotype correlation in CFC syn-drome is less well established than for other NCFCs and the molecular effects of mutations also appear variable.
Other conditions due to RAS-MAPK pathway mutationsConditions other than the NCFCs are known to be caused by germline mutations in RAS-MAPK pathway genes, including capillary malformation–arteriovenous malformation syn-drome, due to mutations in RASA1,20 and multiple hereditary gingivomatosis, due to an SOS1 mutation.21 These disorders do not share the cardinal features of the NCFCs, so are not reviewed here.
PROGNOSTIC AND MANAGEMENT IMPLICATIONSThe diagnosis of NCFCs can have important prognostic impli-cations, including for childhood cancer risk. As discussed earlier, HRAS mutations are of particular importance in this context. Diagnosis of the underlying condition predisposing to a cancer can be crucially important in informing management decisions. Management for syndromal complications may be different to the same lesion occurring sporadically, for example patients with NF1 and optic glioma rarely require intervention and if they are treated with radiotherapy, the risk of second tumours is greatly increased.22
Other important aspects of management to be considered once the diagnosis of an NCFC is made are appropriate cardiac monitoring for congenital heart disease, arrhythmia or HCM, assessment of needs for specifi c nutritional or developmen-tal support and monitoring of growth. Short stature is very common and other endocrine problems, including growth hormone defi ciency, may occur.6 Results of extended geno-type–phenotype correlation studies will allow more focused and individualised management plans for affected individuals. This is of particular importance for families with a high bur-den of ongoing follow-up, as is the case for many individuals with these conditions.
Molecular testing and its implicationsMolecular confi rmation is a gold standard for diagno-sis, enabling more accurate counselling about prognosis,
13_archdischild160069-182717.indd 72813_archdischild160069-182717.indd 728 8/16/2010 5:29:45 PM8/16/2010 5:29:45 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069 729
clarifi cation of recurrence risk and confi rmatory testing of other family members. Appropriate health surveillance for particular complications of the condition can also be imple-mented and recognition of further genotype–phenotype cor-relations may guide management. Additionally, a confi rmed diagnosis providing an explanation for why problems have arisen may be of considerable benefi t to families.
Prenatal diagnosis of fetuses with CS and CFC syndrome has now been reported23 24: amniocentesis was performed for suggestive features on antenatal scanning, including charac-teristic hand posture and polyhydramnios. Such techniques make available important information to prospective parents, allowing informed decisions regarding the pregnancy and opti-mising further management as appropriate. Prenatal diagnosis for NF1 or NS is also available for families where the mutation has been identifi ed, though with the frequently milder nature of these conditions, many families currently do not opt for this. Preimplantation genetic diagnosis is now licensed for NF1 in the UK and other countries and the fi rst children excluded from NF1 risk by this arduous process have been born.25
Recurrence riskConfi rming the molecular basis of a child’s disorder optimises assessment of recurrence risk in future siblings. For de novo mutations, low recurrence risks can be confi rmed if neither parent carries the mutation in lymphocyte DNA. However, exceptions to this have been reported, for example, one report of mosaicism in a father with some features of CS and a clas-sically affected non-mosaic son,26 and another of two siblings with severe NCFC phenotypes, one having a KRAS mutation and the other a HRAS mutation.27 Currently, this latter can-not be adequately explained other than as a coincidence.
PROSPECTS FOR TREATMENTChemotherapeutics modulating RAS-MAPK pathway activ-ity are already in limited clinical use: sorafenib is an orally active RAF inhibitor used in various advanced malignancies.3 Animal models for NF128 and NS29 exist and more recently, mouse models for rare NCFCs such as CS have also been gen-erated.30 These can improve understanding of the underlying biology of these conditions and whether RAF or MEK inhibi-tors3 might prove to be effective treatments. Clinical trials of agents affecting pathway signalling, including statins, in NF1, are also ongoing.28
CONCLUSIONThe discovery that the common disorders NS and NF1 and the rare NCFCs, are all due to mutations in genes acting within the same pathway has explained the overlapping clinical phe-notypes observed in these conditions. Genetic testing has per-mitted further defi nition of their phenotypes, both classical and variant. Achieving a molecular diagnosis in the individual allows management to be tailored to the specifi c condition. The long-studied nature of the RAS-MAPK pathway means that, unusually for a group of developmental disorders, there are prospects for treatment on the horizon.
Acknowledgements The authors acknowledge support from the Manchester Biomedical Research Centre. We are also indebted to the support groups for these conditions, the Noonan syndrome support group (http://www.noonansyndrome.org), CFC International (http://www.cfcsyndrome.org), the International Costello Syndrome Support Group (http://www.costellokids.com) and the Association Française des Syndromes de Costello et CardioFacioCutané (http//costello.free.fr) for their instrumental input in allowing the phenotypes of these conditions to be defi ned.
Competing interests None.
Ethics approval This study was conducted with the approval of the Central Manchester Research Ethics Committee.
Provenance and peer review Commissioned; externally peer reviewed.
Detail has been removed from this case description or these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information avail-able and are satisfi ed that the information backs up the case the authors are making.
REFERENCES 1. Huson S. Neurofi bromatosis: emerging phenotypes, mechanisms and
management. Clin Med 2009;8:611–17.
2. van der Burgt I. Noonan syndrome. Orphanet J Rare Dis 2007;2:4.
3. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders
and cancer. Nat Rev Cancer 2007;7:295–308.
4. Fryer AE, Holt PJ, Hughes HE. The cardio-facio-cutaneous (CFC) syndrome and
Noonan syndrome: are they the same? Am J Med Genet 1991;38:548–51.
5. Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes:
dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med 2008;10:e37.
6. Zenker M, Horn D, Wieczorek D, et al. SOS1 is the second most common Noonan
gene but plays no major role in cardio-facio-cutaneous syndrome. J Med Genet
2007;44:651–6.
7. Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes
aberrant protein N-myristoylation and causes Noonan-like syndrome with loose
anagen hair. Nat Genet 2009;41:1022–6.
8. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis
2008;3:13.
9. Sarkozy A, Carta C, Moretti S, et al. Germline BRAF mutations in Noonan,
Leopard, and cardiofaciocutaneous syndromes: molecular diversity and
associated phenotypic spectrum. Hum Mutat 2009;30:695–702.
10. De Luca A, Bottillo I, Sarkozy A, et al. NF1 gene mutations represent the major
molecular event underlying neurofi bromatosis-Noonan syndrome. Am J Hum
Genet 2005;77:1092–101.
11. Thiel C, Wilken M, Zenker M, et al. Independent NF1 and PTPN11 mutations
in a family with neurofi bromatosis-Noonan syndrome. Am J Med Genet A
2009;149A:1263–7.
12. Brems H, Chmara M, Sahbatou M, et al. Germline loss-of-function mutations in
SPRED1 cause a neurofi bromatosis 1-like phenotype. Nat Genet 2007;39:1120–6.
13. Lo IF, Brewer C, Shannon N, et al. Severe neonatal manifestations of Costello
syndrome. J Med Genet 2008;45:167–71.
14. Kerr B, Delrue MA, Sigaudy S, et al. Genotype-phenotype correlation in Costello
syndrome: HRAS mutation analysis in 43 cases. J Med Genet 2006;43:401–5.
15. Kerr B, Allanson J, Delrue MA, et al. The diagnosis of Costello syndrome:
nomenclature in Ras/MAPK pathway disorders. Am J Med Genet A
2008;146A:1218–20.
16. Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous
syndrome: clinical features of 38 individuals with proven mutations. J Med Genet
2008;45:249–54.
17. Al-Rahawan MM, Chute DJ, Sol-Church K, et al. Hepatoblastoma and heart
transplantation in a patient with cardio-facio-cutaneous syndrome. Am J Med
Genet A 2007;143A:1481–8.
18. Makita Y, Narumi Y, Yoshida M, et al. Leukemia in Cardio-facio-cutaneous (CFC)
syndrome: a patient with a germline mutation in BRAF proto-oncogene. J Pediatr
Hematol Oncol 2007;29:287–90.
19. Niihori T, Aoki Y, Narumi Y, et al. Germline KRAS and BRAF mutations in cardio-
facio-cutaneous syndrome. Nat Genet 2006;38:294–6.
20. Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous
malformation, a new clinical and genetic disorder caused by RASA1 mutations.
Am J Hum Genet 2003;73:1240–9.
21. Hart TC, Zhang Y, Gorry MC, et al. A mutation in the SOS1 gene causes
hereditary gingival fi bromatosis type 1. Am J Hum Genet 2002;70:943–54.
22. Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofi bromatosis 1
patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol
2006;24:2570–5.
23. Kuniba H, Pooh RK, Sasaki K, et al. Prenatal diagnosis of Costello syndrome using
3D ultrasonography amniocentesis confi rmation of the rare HRAS mutation G12D.
Am J Med Genet A 2009;149A:785–7.
24. Witters I, Denayer E, Brems H, et al. The cardiofaciocutaneous syndrome:
prenatal fi ndings in two patients. Prenat Diagn 2008;28:53–5.
25. Spits C, De Rycke M, Van Ranst N, et al. Preimplantation genetic diagnosis for
neurofi bromatosis type 1. Mol Hum Reprod 2005;11:381–7.
26. Sol-Church K, Stabley DL, Demmer LA, et al. Male-to-male transmission of
Costello syndrome: G12S HRAS germline mutation inherited from a father with
somatic mosaicism. Am J Med Genet A 2009;149A:315–21.
27. Søvik O, Schubbert S, Houge G, et al. De novo HRAS and KRAS mutations in two
siblings with short stature and neuro-cardio-facio-cutaneous features. J Med
Genet 2007;44:e84.
13_archdischild160069-182717.indd 72913_archdischild160069-182717.indd 729 8/16/2010 5:29:45 PM8/16/2010 5:29:45 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Review
Arch Dis Child 2010;95:724–730. doi:10.1136/adc.2009.160069730
28. Korf BR. Statins, bone, and neurofi bromatosis type 1. BMC Med 2008;6:22.
29. Nakamura T, Colbert M, Krenz M, et al. Mediating ERK 1/2 signaling rescues
congenital heart defects in a mouse model of Noonan syndrome. J Clin Invest
2007;117:2123–32.
30. Schuhmacher AJ, Guerra C, Sauzeau V, et al. A mouse model for Costello
syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest
2008;118:2169–79.
13_archdischild160069-182717.indd 73013_archdischild160069-182717.indd 730 8/16/2010 5:29:46 PM8/16/2010 5:29:46 PM
group.bmj.com on September 15, 2010 - Published by adc.bmj.comDownloaded from

Cutaneous features in 17q21.31 deletion syndrome: adifferential diagnosis for cardio–facio–cutaneous syndromeEmma Burkitt Wrighta, Dian Donnaia, Diana Johnsonb and Jill Clayton-Smitha
Microdeletion of 17q21.31 causes a recurrent recognizable
dysmorphic syndrome. A further four patients with
17q21.31 microdeletions are reported here in whom
an earlier diagnosis of cardio–facio–cutaneous syndrome
was suggested. These patients have significant similarities
of facial gestalt to earlier reported 17q21.31 microdeletion
patients, but a striking feature that has not been
emphasized previously is the large number of naevi and
other pigmentary skin abnormalities that may be present.
These features, together with a coarse facial appearance,
relative macrocephaly and significant learning disabilities,
were what had led to the earlier diagnostic suggestion of
cardio–facio–cutaneous syndrome in each of these four
cases. Clin Dysmorphol 20:15–20 �c 2011 Wolters Kluwer
Health | Lippincott Williams & Wilkins.
Clinical Dysmorphology 2011, 20:15–20
Keywords: cardio–facio–cutaneous syndrome, hyperpigmentation, naevi,17q21.31 microdeletion syndrome
aGenetic Medicine, Manchester Academic Health Science Centre,University of Manchester, Central Manchester University Hospitals NHSFoundation Trust, St Mary’s Hospital, Manchester and bClinical Genetics,Sheffield Children’s NHS Foundation Trust, Sheffield Children’s Hospital,Western Bank, Sheffield, UK
Correspondence to Jill Clayton-Smith, Genetic Medicine, Manchester AcademicHealth Science Centre, University of Manchester, Central Manchester UniversityHospitals NHS Foundation Trust, St Mary’s Hospital, Oxford Road, ManchesterM13 9WL, UKTel: + 44 161 276 6269; fax: + 44 161 276 6145;e-mail: [email protected]
Received 17 May 2010 Accepted 1 July 2010
Introduction17q21.31 microdeletion syndrome is a recently delineated
recognizable dysmorphic syndrome which arises due to
misalignment of low-copy number repeat sequences and
the unusual genomic architecture at this locus: an inver-
sion polymorphism is present in 20% of the Caucasian
population (Koolen et al., 2006; Shaw-Smith et al., 2006).
Currently underdiagnosed because of its only recent re-
cognition, it is thought that 1 in 16 000 individuals may be
affected by this syndrome (Koolen et al., 2008). Haplo-
insufficiency for the microtubule-associated protein tau
(MAPT) gene within the deleted region seems to be the
most likely cause for the learning disabilities and other
anomalies seen in this condition (Koolen et al., 2006;
Shaw-Smith et al., 2006).
Cardio–facio–cutaneous (CFC) syndrome was first de-
scribed in 1986 (Reynolds et al., 1986), and shows con-
siderable overlap with other neuro-cardio–facio–cutaneous
syndromes, particularly Noonan and Costello syndromes.
The reason for these observed overlaps is that the protein
products of all genes known to be mutated in these
conditions interact within the RAS-mitogen-activated
protein kinase (MAPK) pathway (Burkitt Wright and
Kerr, 2010). An earlier published series of 17q21.31
microdeletion patients included several with features
reminiscent of Noonan syndrome, including one in whom
this diagnosis was thought sufficiently likely for Noonan
syndrome genes to have been tested (Tan et al., 2009).
Several individuals in that series also had features of
ectodermal dysplasia, three of 11 individuals had pulmonary
stenosis, and other cardiac lesions were also described
(Tan et al., 2009). Normal birth weight and relative
macrocephaly are features that seem to be present in the
majority of patients both with 17q21.31 microdeletion
(Tan et al., 2009) and RAS pathway disorders (Burkitt
Wright and Kerr, 2010). Variable degrees of develop-
mental delay have been noted in patients with 17q21.31
microdeletion, and this spectrum is still liable to
expansion. Similarly, patients with CFC syndrome may
have variable degrees of intellectual disability (Burkitt
Wright and Kerr, 2010).
PatientsPatient 1
A 21-year-old male patient was born at term, weighing
3.4 kg. He required tube feeding in the first 3 weeks of
life and was hypotonic. Growth has continued between
the 25th and 50th percentile curves, his current height
being 176 cm. He has a predominantly truncal fat distri-
bution. His head circumference has followed the 97th
percentile, and is now 57.6 cm. He has had unusual skin
since birth, with redundant skin folds, coarse hair with
a chaotic growth pattern and keratosis pilaris. He has
always been more highly pigmented than other family
members. A very large number of naevi developed all over
his body through childhood and he also has several cafe
au lait patches. He has slight coarsening of his facial
features, including a broad nose, thick lips and posteriorly
rotated ears with large lobes, as shown in Fig. 1a and e. He
has severe learning difficulties and longstanding epilepsy,
which is well controlled on carbemazepine, but also had
challenging behaviour for which he has been prescribed
thioridazine in addition. He also has strabismus, difficulty
Original article 15
0962-8827 �c 2010 Wolters Kluwer Health | Lippincott Williams & Wilkins DOI: 10.1097/MCD.0b013e32833e8f1e
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Fig. 1
(a) (b) (c) (d)
(h)(g)(f)(e)
(i) (j) (k) (l)
(m) (n)
(a–h) Facial appearances of the three patients, showing coarse features, multiple naevi and the characteristic nose of 17q21.31 deletion. (i–l) Handsof the four patients, showing thickened skin, particularly marked in patient 1. (m) Area of hyperpigmentation on the trunk of patient 3. (n) Soles ofpatient 4, showing deep creases and dry skin.
16 Clinical Dysmorphology 2011, Vol 20 No 1
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

in chewing (from weakness of masseters) and subluxed
patellae. His hands were fleshy with deep skin creases
(Fig. 1i). Endocrinological assessment was made because
of hyperpigmentation and showed normal short synacthen
response and oral glucose tolerance. Chromosome analysis
showed a 46, XY karyotype, with normal fluorescence in-
situ hybridization (FISH) for Prader–Willi and Smith–
Magenis syndromes. Cranial computed tomography scan
and muscle biopsy were normal. Transferrin isoelectric
focusing and steroid sulphatase activity were normal, as was
a urinary mucopolysaccharide screen.
Patient 2
A 22-year-old male patient weighed 2.8 kg at birth
by caesarian delivery at 41 weeks’ gestation for breech
presentation. He was admitted to the special care unit
on day 2 of life with poor feeding and stridor because
of laryngomalacia. Feeding difficulties continued until
weaning. A small secundum atrial septal defect and mild
pulmonary stenosis were shown on echocardiography.
He had undescended testes and hypospadias, which were
surgically corrected, and several urinary tract infections.
A small umbilical hernia resolved spontaneously. He had
joint laxity. Mild delay of motor milestones was present,
but speech delay was marked at the age of 5 years, he was
still using only single words. At the age of 8 years, he
could put three words together. His height (180 cm) and
weight (75 kg) lie between the 50 and 75th percentiles,
whereas his head circumference was on the 91st per-
centile (59.7 cm). Pubertal development was normal. At
the age of 16 years, he sat for several national school
examinations, attaining average grades in some. He cur-
rently lives with his parents, works part time in a super-
market and attends adult education life skills courses. On
review at the age of 20 years, his coarse facial features,
as shown in Fig. 1b and f, were more marked than in
childhood, in particular his broad nose and thick lips.
Naevi were remarkably numerous over his entire body.
His karyotype was normal, 46, XY.
Patient 3
A 16-year-old female patient has short stature and mild-
to-moderate learning difficulties. She had a birth weight
of 2.3 kg at 41 weeks’ gestation, and placental insuffi-
ciency was suspected. She would not breast feed, but had
established bottle feeds by day 10 of life. Joint laxity and
muscular hypotonia were prominent in infancy and early
childhood. She first walked at the age of 20 months. She
had recurrent chest infections in the first 5 years of life,
but has been in good health since, with normal pubertal
development. She has coarse textured, dry hair, relatively
coarse facial features, as shown in Fig. 1c and g, and an
unusual thickened hyperpigmented area of skin on her
upper abdomen, as shown in Fig. 1m. She is developing
naevi, particularly over her face and trunk, and has a mild
pectus deformity. Her karyotype is 46, XX. Her height
remains around the third percentile, and her head cir-
cumference of 55.4 cm is on the 50th percentile.
Patient 4
A 28-year-old female patient was born at term, weighing
2.78 kg. She had global developmental delay; first unsup-
ported sitting was at the age of 2.5 years and walking at
the age of 3 years. She spoke no words until the age of
14 years. She was initially referred to a genetics clinic at
the age of 12 years. She was noted to have generalized
hyperpigmentation for which no cause could be identi-
fied. Porphyria, haemochomatosis and excess of adreno-
corticotropic hormone were excluded. Optic atrophy, left
pelvi–ureteric junction obstruction and delayed bone age
were also present. Her occipito–frontal circumference
measured 51 cm (third percentile) and height 109 cm
({0.4th percentile). She has sensorineural hearing loss
and had chronic serous otitis media. She developed
pneumonia and a pericardial effusion at the age of 11
years, the latter being persistent till the age of 15 years,
but stable. Swollen proximal interphalangeal joints were
noted from the age of 14 years, as were several cafe au
lait patches. A diagnosis within the Noonan/CFC/Watson
spectrum was therefore considered at this stage. Puberty
was delayed, with pelvic ultrasound at the age of 15 years
showing a small uterus and delayed ovarian development.
Menarche was at the age of 17 years. MRI brain scanning
showed left medial temporal sclerosis and a small hip-
pocampus. At the age of 19 years, a thoracic scoliosis
and limitation of elbow extension were noted. At the age
of 28 years, her height is 131.2 cm ({0.4th percentile),
occipito–frontal circumference is 51.5 cm (0.4th percen-
tile) and weight is 35.3 kg (< 0.4th percentile). She has a
prematurely aged skin appearance. She has a broad nasal
tip, upslanting palpebral fissures and a long face with a
thin upper lip (Fig. 1d and h). Her palmar and plantar
creases are deep (see Fig. 1l and n), and she has recently
been diagnosed with cataract. In addition, a small larynx
with a small anterior laryngeal web has been noted.
Her karyotype is 46, XX. Chromosome breakage studies,
creatine kinase, urine amino acids and mucopolysacchar-
ide screen, white cell enzymes, very long-chain fatty
acids, copper levels, sweat test and thyroid function were
also normal, as was FISH testing for deletion at the NF1
locus.
Methods and resultsMutation analysis by sequencing of all exons of BRAF,
KRAS, HRAS, MEK1 and MEK2, in which CFC-
associated mutations have been reported, showed no
such mutation in patient 1. Array comparative genomic
hybridization (aCGH), using the Genechip SNP6.0 array
(Affymetrix, Santa Clara, California, USA), was carried
out on patients 1 and 2, and showed deletions at
17q21.31, of 639 kb in patient 1 and 519 kb in patient
2, as shown in Fig. 2. These deletions included the
Cutaneous features in 17q21.31 deletion syndrome Burkitt Wright et al. 17
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

critical region identified for the condition as indicated,
overlapping as they do with those earlier reported in
17q21.31 microdeletion syndrome (Koolen et al., 2008).
In addition, patient 1 was found to have a 2.4 Mb duplica-
tion of 22q11.21 (data not shown). His 17q21.31 micro-
deletion and 22q11.21 duplication were confirmed by
multiplex ligation probe analysis using the multiplex
ligation-dependent probe amplification P245 kit (MRC
Holland, Amsterdam, the Netherlands). Patient 2 was
noted to have an additional copy number variant, a dupli-
cation of 17q21.31, just distal to his deletion, as shown
in Fig. 2.
In patients 3 and 4, the del 17q21.31 microdeletion pheno-
type was recognized clinically at a review appointment,
and FISH was then used to show deletion of MAPT.
In patient 4, deletion breakpoints of 17q21.31, 41107486
41610161, were indicated by the heterozygous loss of 26
oligonucleotide probes on aCGH, giving an approximate
deletion size of 502 kb, as represented in Fig. 2. The de-
novo nature of the deletions in patients 2 and 3 was con-
firmed by normal FISH results in both parents, whereas
parental samples were not available for patients 1 and 4.
DiscussionThe phenotypic features of 17q21.31 microdeletion and
CFC syndrome are compared in Table 1. A large number
of naevi, in conjunction with other cutaneous features,
coarse facial characteristics, relative macrocephaly, poor
feeding in infancy and significant learning disabilities, led
to the clinical suggestion of CFC syndrome in the four
patients described here. In addition, the striking skin
appearance because of increased pigmentation, sufficient
to warrant endocrine investigation, in patients 1 and 4
in this series is another distinctive feature that is re-
miniscent of RAS–MAPK-pathway disorders. It bears
particular resemblance to the bronzed skin reported in
many patients with Costello syndrome, including those in
Costello’s original report (Costello, 1977).
22q11.2 duplications, as identified in patient 1, have been
associated with a diverse range of features, but overall
phenotypes are most commonly mild, and duplications
are also frequently inherited from phenotypically nor-
mal parents (Firth, 2009). Although it is not possible to
exclude increased copy number at 22q11.2 as a contri-
butor to patient 1’s clinical presentation, his features did
not correlate to those commonly reported in association
with this duplication. This is in contrast to his marked
similarities to other individuals with 17q21.31 microdele-
tion syndrome.
The phenotypic similarity of each of the four individuals
described here to patients with molecularly proven CFC
syndrome raises the question as to whether the 17q21.31
locus could harbour a gene or genes implicated in this
condition. Such genes causing a phenotype by deletion
Fig. 2
Critical region of deletion
11 Patient 1
2 Patient 2
4 (SNP calls not shown)4MAPT ARL17B ARL17AARHGAP27Genes
CRHR1 KIAA1267 NGFPLEKHM1NMT1
17q21.31 17q21.32
41000 kb 42000 kb
Patient 4
Diagram of the 17q21.31 locus, with deletions in patients 1, 2 and 4 is shown. Light grey indicates deleted material, whereas dark grey is adjacentduplicated material in patient 2, in this region, rich in copy number variants. The critical region for the deletion phenotype is shown at the top of thefigures. MAPT, microtubule-associated protein tau; SNP, single nucleotide polymorphism.
18 Clinical Dysmorphology 2011, Vol 20 No 1
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

would suggest haploinsufficiency as a mechanism, with
loss of an allele encoding a protein which inhibits RAS–
MAPK-pathway activity perhaps being sufficient to cause
pathway overactivation, analogous to the mechanism by
which inactivating mutations in NF1 cause neurofibro-
matosis type I (Huson, 2008). The commonly deleted
area on 17q21.31 spans six known genes. At present none
of these are known to interact with the RAS–MAPK
pathway, but two, CRHR1 and MAPT, are each highly
expressed in brain, and therefore represent prime candi-
dates for causing the neurological phenotype seen in
17q21.31 microdeletion (Koolen et al., 2008).
CFC syndrome shows considerable variability in the
severity of learning disability, physical and behavioural
problems shown by affected individuals. A broad spec-
trum of capabilities is also observed in patients with
17q21.31 microdeletion, as shown by the four patients
described here. The variable severity and manifestations
of both CFC syndrome and 17q21.31 microdeletion
means that there may be a significant number of
individuals for whom both conditions should be con-
sidered within the differential diagnosis.
ConclusionThe diagnosis of 17q21.31 microdeletion may be achiev-
able clinically in some patients, but it may not be easy to
recognize in all. Large numbers of naevi and/or pigmen-
tary skin changes, reminiscent of those seen in patients
with CFC syndrome, and like those discussed here,
should prompt consideration of testing for 17q21.31
microdeletion by FISH, multiplex ligation-dependent
probe amplification or aCGH, particularly when a large,
pear-shaped or tubular nose is also present. Patients
in whom a clinical diagnosis of CFC syndrome has been
suggested earlier, or those in whom no mutation has been
found in any of the known genes for CFC syndrome,
should also be reassessed in the light of this possibility.
These findings show the particular value of long-term
clinical review of patients without a molecular diagnosis,
and suggest that aCGH should be considered in all
patients with a clinical diagnosis of CFC syndrome who
do not have a mutation in known CFC genes. The pheno-
typic similarities between several patients with 17q21.31
microdeletions and those with molecularly confirmed
Table 1 Comparison of key features of 17q21.31 microdeletion syndrome, CFC syndrome and patients in this series
17q21.31microdeletion(Koolen et al.,
2008)
CFC syndrome(Armour and
Allanson, 2008) Patient 1 Patient 2 Patient 3 Patient 4
GrowthNormal or high birth weight 16/22 37/38 Yes Yes No YesFeeding difficulties Common 19/30 Yes Yes Mild Not knownFailure to thrive Not common Frequently severe No No Mild Not knownNormal OFC 21/22 Yes Yes Yes YesRelative macrocephaly (OFC morethan + 1SD compared with height)
Notcharacterized
17/33 Yes Yes Yes Yes
Absolute macrocephaly (more than+ 3SD)
— 4/33 No No No No
Short stature (below third percentile) 4/22 27/38 No No Mild SevereNeurological development
Developmental delay 22/22,variabledegree
Present; variable Severe Mild-to-moderate
Mild-to-moderate Severe
Hypotonia 21/22 34/36 Yes Yes Yes Not knownSeizures 11/22 18/37 No No No NoEngaging or amiable personality 16/18 25/32 — Yes Yes —
HeartPulmonary stenosis — 14/33 No Yes No NoASD or VSD 6/27 9/32 No Yes No NoOther cardiac defect Common No No No No
Skin and hairNaevi Present in
some photos28/37 Yes Yes Yes Yes
Other skin pigmentary abnormality Very common, includingcafe au lait patches and
generalizedhyperpigmentation
Yes, generalizedhyperpigmentation
— Yes, discrete area onabdomen (see figure)
Yes, generalizedhyperpigmentation
Keratosis pilaris/hyperkeratosis/ichthyosis/dry skin
A few patients 20/28 Yes Yes Yes Yes
Unusual hair (colour, texture,thickness)
13/22 34/38 Yes Yes Yes Yes
EyesStrabismus 10/22 28/35 Yes No No NoOptic nerve hypoplasia — 11/26 No No No Yes
Numerators are number of patients with the feature, denominators are the number of patients on whom the data was available.ASD, atrial septal defect; CFC, cardio-facio-cutaneous; OFC, occipito–frontal circumference; SD, standard deviation; VSD, ventricular septal defect.
Cutaneous features in 17q21.31 deletion syndrome Burkitt Wright et al. 19
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

CFC syndrome suggest the possibility that one or more
genes at 17q21.31 might influence RAS–MAPK-pathway
activity in some way.
AcknowledgementsE.B.W. holds a Wellcome Trust Clinical Research Training
Fellowship, and is supported by the Manchester Biome-
dical Research Centre. The authors have no other com-
peting interests to declare.
ReferencesArmour CM, Allanson JE (2008). Further delineation of cardio-facio-cutaneous
syndrome: clinical features of 38 individuals with proven mutations. J MedGenet 45:249–254.
Burkitt Wright EMM, Kerr B (2010). RAS pathway disorders: important causes ofcongenital heart disease, feeding difficulties, developmental delay and shortstature. Arch Dis Child . Online first 6th April 2010, doi:10.1136/adc.2009.160069. [Epub ahead]
Costello JM (1977). A new syndrome: mental subnormality and nasal papillomata.Aust Paediatr J 13:114–118.
Firth HV (2009). 22q11.2 duplication. In: Pagon RA, Bird TC, Dolan CR,Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University ofWashington, Seattle.
Huson S (2008). Neurofibromatosis: emerging phenotypes, mechanisms andmanagement. Clin Med 8:611–617.
Koolen DA, Vissers LE, Pfundt R, De Leeuw N, Knight SJ, Regan R, et al. (2006).A new chromosome 17q21.31 microdeletion syndrome associated witha common inversion polymorphism. Nat Genet 38:999–1001.
Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, et al. (2008).Clinical and molecular delineation of the 17q21.31 microdeletion syndrome.J Med Genet 45:710–720.
Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, Opitz JM(1986). New multiple congenital anomalies/mental retardation syndrome withcardio-facio-cutaneous involvement – the CFC syndrome. Am J Med Genet28:413–427.
Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, et al. (2006).Microdeletion encompassing MAPT at chromosome 17q21.3 is associated withdevelopmental delay and learning disability. Nat Genet 38:1032–1037.
Tan TY, Aftimos S, Worgan L, Susman R, Wilson M, Ghedia S, et al. (2009).Phenotypic expansion and further characterisation of the 17q21.31microdeletion syndrome. J Med Genet 46:480–489.
20 Clinical Dysmorphology 2011, Vol 20 No 1
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Constitutive activation of B-Raf in the mousegerm line provides a model for humancardio-facio-cutaneous syndromeJelena Urosevica,1, Vincent Sauzeaub,2, María L. Soto-Montenegroc, Santiago Reigc, Manuel Descoc,d,Emma M. Burkitt Wrighte, Marta Cañamerof, Francisca Mulerof, Sagrario Ortegaf, Xosé R. Bustelob,and Mariano Barbacida,3
aMolecular Oncology Program, Centro Nacional de Investigaciones Oncológicas (CNIO), E-28029 Madrid, Spain; bCentro de Investigación del Cáncer andInstituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Cientificas-University of Salamanca, E-37007 Salamanca, Spain;cUnidad de Medicina y Cirugía Experimental, Hospital General Universitario “Gregorio Marañón,” Centro de Investigación Biomédica en Red de SaludMental (CIBERSAM), E-28007 Madrid, Spain; dDepartamento de Bioingeniería e Ingeniería Aerospacial, Universidad Carlos III, E-28991 Madrid, Spain;eGenetic Medicine, University of Manchester, Manchester Academic Health Science Centre, Central Manchester University Hospitals National Health ServiceFoundation Trust, St. Mary’s Hospital, Manchester M13 9WL, United Kingdom; and fBiotechnology Program, Centro Nacional de Investigaciones Oncológicas(CNIO), E-28029 Madrid, Spain
Edited* by Neal G. Copeland, Institute of Molecular and Cell Biology, Proteos, Singapore, and approved February 11, 2011 (received for review November11, 2010)
RASopathies are a class of developmental syndromes that resultfrom congenital mutations in key elements of the RAS/RAF/MEKsignaling pathway. A well-recognized RASopathy is the cardio-facio-cutaneous (CFC) syndrome characterized by a distinctive fa-cial appearance, heart defects, and mental retardation. Clinicallydiagnosed CFC patients carry germ-line mutations in four differentgenes, B-RAF, MEK1, MEK2, and K-RAS. B-RAF is by far the mostcommonly mutated locus, displaying mutations that most oftenresult in constitutive activation of the B-RAF kinase. Here, we de-scribe a mouse model for CFC generated by germ-line expressionof a B-RafLSLV600E allele. This targeted allele allows low levels ofexpression of B-RafV600E, a constitutively active B-Raf kinase firstidentified in human melanoma. B-Raf+/LSLV600E mice are viable anddisplay several of the characteristic features observed in CFCpatients, including reduced life span, small size, facial dysmor-phism, cardiomegaly, and epileptic seizures. These mice also showup-regulation of specific catecholamines and cataracts, two fea-tures detected in a low percentage of CFC patients. In addition,B-Raf+/LSLV600E mice develop neuroendocrine tumors, a pathologynot observed in CFC patients. These mice may provide a means ofbetter understanding the pathophysiology of at least some of theclinical features present in CFC patients. Moreover, they may serveas a tool to evaluate the potential therapeutic efficacy of B-RAFinhibitors and establish the precise window at which they couldbe effective against this congenital syndrome.
B-Raf signaling | developmental defects | chromaffin-derived tumor
Oncogenic mutations in the B-RAF locus have been found ina variety of human tumors (1, 2), with a single miscoding
mutation, V600E, accounting for more than 80% of the B-RAFmutations identified to date (1, 3). Miscoding mutations in the B-RAF locus have also been observed in the germ line of patients withcardio-facio-cutaneous (CFC) syndrome, a congenital disorderthat shares overlapping defects with other RASopathies such asNoonan, Costello, LEOPARD, and Legius syndromes as well asneurofibromatosis type I (4–8). All of these syndromes result fromconstitutive hyperactivation of the RAS/RAF/MEK/ERK signal-ing cascade, but they display unique characteristic features (8).CFC is characterized by craniofacial defects, short stature, car-diomegaly, ectodermal abnormalities, mental retardation, andneurological defects (9). About 75% of the patients molecularlydiagnosed with CFC carry germ-linemutations in B-RAF (4, 5, 10).The rest display germ-line mutations in other components of thepathway, including K-RAS, MEK1, and MEK2 (4, 11, 12). B-RAFmutations in CFC patients are widely distributed across the codingsequences, and most are predicted to result in hyperactivation ofthe B-RAF kinase. However, these mutations cause more limited
activation of the downstream MEK/ERK kinases than those ob-served in human tumors, particularly those carrying the V600Emutation. In addition, some CFC patients carry B-RAF kinase-impairing mutations (3–5) that may activate the pathway by in-direct mechanisms, possibly activating the related c-RAF kinase(13). Finally, some CFC-associated mutations have previouslybeen identified in tumors (4–6), a feature also observed in patientswith Costello syndrome (14).Here, we report the generation and phenotypic characteriza-
tion of mice expressing a hypomorphic B-RafV600E allele thatphenocopies some of the key developmental defects observed inCFC patients. These mice should provide a suitable model sys-tem to better understand the molecular bases for this congenitaldisease and assay the suitability of potential therapeutic strate-gies (15, 16).
ResultsMouse Model for Human CFC Syndrome. We have taken advantageof a mouse strain, B-Raf+/LSLV600E, that expresses a hypomorphicB-RafV600E allele at 5–10% the levels of the WT counterpart asdetermined by quantitative RT-PCR analyses (Fig. S1). The ki-nase activity of the B-RafV600E oncoprotein has been shown to beabout 10- to 50-fold higher than that of other mutated B-Rafproteins, including those responsible for the CFC syndrome (3–5),and it causes embryonic lethality when expressed during embry-onic development in mice (17). Thus, the low levels of expressionof the B-RafLSLV600E allele are likely to result in constitutive levelsof B-Raf kinase activity similar to those present in CFC patients(Limited Life Span, Size, and Fertility in B-Raf+/LSLV600E Mice).To examine the status of the downstream Mek/Erk pathway in
B-Raf+/LSLV600E mice, we performedWestern blot analysis of cellextracts derived from adult heart and brain, two of the tissuesdisplaying the most defects in B-Raf+/LSLV600E mice (Limited Life
Author contributions: J.U., V.S., X.R.B., and M.B. designed research; J.U., V.S., M.L.S.-M.,S.R., and E.M.B.W. performed research; M.D., M.C., F.M., and S.O. analyzed data; J.U.,E.M.B.W., X.R.B., and M.B. wrote the paper; M.D. and F.M. supervised imaging work; M.C.supervised histopathology work; and S.O. supervised the generation of the recombinantmice.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1Present address: Oncology Programme, Institute for Research in Biomedicine (IRB), ParcCientífic de Barcelona, E-08028 Barcelona, Spain.
2Present address: Institut de Recherche Thérapeutique (IRT-UN), L’institut du thorax-UMR915, 44007 Nantes Cedex 01, France.
3To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1016933108/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1016933108 PNAS | March 22, 2011 | vol. 108 | no. 12 | 5015–5020
MED
ICALSC
IENCE
S

Span, Size, and Fertility in B-Raf+/LSLV600E Mice). As illustrated inFig. 1A, the levels of the B-Raf protein are about one-half of thoseobserved in the corresponding tissues from WT littermates, thusindicating that the mutant B-RafV600E isoform must be expressedat low levels as indicated by RT-PCR analysis. Similar results wereobtained with embryonic day (E)13.5 B-Raf+/LSLV600E embryos(Fig. 1A). Expression of the B-RafV600E isoform did not affecteither the expression or phosphorylation levels of the downstreamMek and Erk proteins (Fig. 1A). Analysis of the overall B-Rafkinase activity in these mutant mice (that is, the kinase activitycontributed by the normal B-Raf kinase and constitutive B-RafV600E hypomorphic isoform) did not increase, at least withinthe sensitivity levels of available in vitro kinase assays (Fig. 1B),thus suggesting that the activation of the Mek/Erk pathway inthese mice must be subtle. These results are reminiscent of thosepreviously obtained with a mouse model of Costello syndromethat expressed an endogenous H-RasG12V oncoprotein (18).
Limited Life Span, Size, and Fertility in B-Raf+/LSLV600E Mice. B-Raf+/LSLV600E mice were born at the expected Mendelian ratio,indicating that limited expression levels of B-RafV600E did notresult in embryonic lethality. However, B-Raf+/LSLV600E miceshowed reduced postnatal fitness, a defect highly dependent ontheir genetic makeup. Mice carrying the B-RafV600E allele ina mixed genetic background derived from C57BL/6J (75%) and129Sv/J (25%) strains (designated as B6/129) displayed a bi-modal survival curve characterized by death of 35% of the miceduring their first 3 wk of life. Moreover, very few of these micesurvived beyond 30 wk (Fig. 2A). This survival rate was furthercompromised when B6/129 B-Raf+/LSLV600E mice were back-crossed into the C57BL/6J background. Most mice carrying theB-RafV600E allele in a genetic background with a 98.5% contri-bution from C57BL/6J (designated as B6) died before theyreached 12 wk of age (Fig. 2A). Conversely, survival increasedwhen the B6 mice were crossed with CD1 females, an outbredstrain (Fig. 2A). About 60% of the resulting progeny (designatedas B6/CD1) survived more than 40 wk (Fig. 2A).Histopathological analysis of tissues obtained from B-
Raf+/LSLV600E mice that became sick during their first 3 wk oflife revealed marked disorganization and atrophy of the thymuswith increased numbers of apoptotic cells in the cortex andmedulla as well as a reduction in the number of immature, ter-minal deoxynucleotidyl transferase (TdT)-positive thymocytes(Fig. S2). In addition, all tissues with lymphoid aggregates, suchas the white pulp in spleen or gastrointestinal Peyer’s patches,showed massive apoptosis (Fig. S3). Most of these animals dis-played a reduction in the cytoplasm/nuclear ratio in most cells,although this phenotype was more evident in the kidney and inpancreatic acinar cells (Fig. S4). Finally, about one-half of themice had reduced numbers and size of sebaceous glands in theskin and decreased white adipose tissue. None of these defects
were observed in healthy animals of similar age. Moreover, theywere present in all genetic backgrounds, albeit that they weremore prevalent in the B6 mice.All B-Raf+/LSLV600E mice, regardless of genetic background,
displayed significant growth defects (Fig. 2B). They had reducedsize and body weight at postnatal day 5 (P5), a phenotype that wasfurther aggravated as the animals aged (Fig. 2B and Fig. S5). B-Raf+/LSLV600E mice also failed to mate. However, B-Raf+/LSLV600E
males were not infertile, because their sperm efficiently fertilizedWT oocytes in vitro. The contribution of the CD1 geneticbackground in B6/CD1 B-Raf+/LSLV600E males amelioratedmating defects, indicating that the breeding problems of B6 and
+/LSLV600E+/+
E13.5 Embryos
+/LSLV600E+/+ +/LSLV600E+/+
Adult Brains Adult Hearts
pMek1/2
Mek1
GAPDH
pErk1pErk2
Erk1Erk2
B-Raf
B-Raf
pMek1IP: B-Raf
Adult Brains Adult Hearts
+/+ +/LSLV600E+/+ +/LSLV600E
A
B
Fig. 1. Activation of the Mek/Erk pathway in B-Raf+/LSLV600E mice. (A) Protein extracts obtained fromB-Raf+/+ and B-Raf+/LSLV600E adult brain and hearttissues as well as E13.5 embryos were submitted toWestern blot analyses using antibodies to B-Raf,pMek1/2, Mek1, pErk1/2, and Erk1/2. GAPDH wasused as loading control. (B) Protein extracts obtainedfrom B-Raf+/+ and B-Raf+/LSLV600E adult brain andheart tissues were incubated with a monoclonal an-tibody against B-Raf, and the resulting immunopre-cipitates were assayed for kinase activity using Mek1as a substrate. The levels of pMek1 were deter-mined by blotting with specific polyclonal antibodiesagainst pMek1. The same membrane was used forWestern blot analyses using B-Raf antibody as load-ing control. Arrowheads indicate the migration ofthe corresponding proteins.
Bod
y w
eigh
t (g)
Age (days)
A
0
20
40
60
80
100
10 20 30 40 50 60 70
Sur
viva
l (%
)
Weeks
B
0
10
20
30
0
10
20
30
40
50
1 5 10 20 30 60 90 1 5 10 20 30 60 90
B6/129 mice B6/CD1 mice
Mal
esF
emal
es
B-Raf+/+
B-Raf+/LSLV600E B6/CD1
B-Raf+/LSLV600E B6/129
B-Raf+/LSLV600E B6
B-Raf+/+
B-Raf+/LSLV600EB-Raf+/+
B-Raf+/LSLV600E
Fig. 2. B-Raf+/LSLV600E mice display decreased survival and growth rates. (A)Survival of B-Raf+/+ mice of all genetic backgrounds (open circles; n = 40) andB-Raf+/LSLV600E mice of various genetic background including B6/CD1 (opentriangles; n = 44), B6/129 (gray triangles; n = 33), and B6 (solid triangles; n =18). (B) Body weights of B-Raf+/+ male and female mice of B6/129 and B6/CD1genetic backgrounds (open circles) compared with those of B-Raf+/LSLV600E
male and female mice of B6/129 (gray triangles) and B6/CD1 (open triangles)genetic backgrounds. Error bars represent SEMs.
5016 | www.pnas.org/cgi/doi/10.1073/pnas.1016933108 Urosevic et al.

B6/129 mice were a consequence of their limited size and/orbehavioral abnormalities.
B-Raf+/LSLV600E Mice Display Craniofacial Dysmorphism and DevelopCataracts. B-Raf+/LSLV600E mice, regardless of their genetic back-ground, displayed more rounded and shorter heads as well asdefects in the shape of their skull vault (Fig. 3). X-ray computedtomography (CT) analysis of 10-wk-old B6/129 B-Raf+/LSLV600E
mice revealed significant differences in 6 of 10 cranial landmarkscompared with WT siblings (Fig. 3C). The most significant devi-ations corresponded to the shape of frontal and parietal bones thatform the skull vault. Milder but significant changes were also ob-served in the base of the cranium (Fig. 3). In addition, 40% of theB-Raf+/LSLV600Emice developed cataracts by 8 wk of age (Fig. S6).This percentage increased with time, with 80%of B-Raf+/LSLV600E
mice being affected by 32 wk of age.
B-Raf+/LSLV600E Mice Develop Defects in Their Central and AutonomousNervous Systems. B-Raf+/LSLV600E mice, regardless of geneticbackground and age, displayed marked hyperactivity character-ized by an increased frequency of repetitive movements and lo-
comotion. Despite these abnormalities, we did not observealterations in locomotor coordination. The most prominent neu-rological alteration of these mice was the development of seizures,highly reminiscent of human tonic-clonic epilepsy (Movie S1).Seizures were first detected at 10–12 wk of age. The percentage ofmice undergoing seizures increased progressively from 15% in 12-wk-old mice up to 50% in 20-wk-old animals (Fig. S7A). Thispercentage remained constant at later ages and did not seem tohave an effect onmortality (Fig. S7A). Seizures normally appearedin response to routine handling and consisted of generalizedconvulsions that lasted 4–8 s (Movie S1). Seizure duration did notincrease significantly with age (Fig. S7B). We also observed that4-mo-old B-Raf+/LSLV600E mice had tachypnea and increased lev-els of noradrenaline, suggesting up-regulation of some sympatheticfunctions (Fig. S8).Histopathological examination of B-Raf+/LSLV600E mice did
not reveal major alterations in brain structure, regardless of thegenetic background analyzed. Moreover, Nissl staining of thehippocampal area did not reveal gross differences in organiza-tion or neuron number. However, immunohistochemical stain-ing for GFAP revealed significant increase in the number of
+/LSLV600E+/+
C
A B6/129 mice
B6/CD1 mice
Merged
OpisthionIntersection of interparietal and parietal bone
Intersection of frontal and parietal bone Base of nasal bone
Tip of nasal bone Center of alveolar ridge over maxillary incisor Spheno-ethmoid synchondrosis
Spheno-occipital synchondrosisIntersphenoidal suture
Basion
0.0081
NS
NS
< 0.00001
< 0.00001
0.0002
0.0066
0.0028
NS
NS
Landmark Anatomical Position
1 2
3 4
5 6 7
8 9
10
E
+/LSLV600E+/+ Merged
B
D
F
Fig. 3. Cranial defects in B-Raf+/LSLV600E mice. (A) Repre-sentative side views of heads (Upper) and sagittal CTimages (Lower) of adult B-Raf+/+ and B-Raf+/LSLV600E mice inB6/129 genetic background illustrating the more roundedskull vault of the mutant mice (arrowheads). (B) Overlayof two representative sagittal CT sections of a B6/129 B-Raf +/+ mouse (white) and a B6/129 B-Raf+/LSLV600E litter-mate (red). (C) Same as A, but images correspond to micein a B6/CD1 genetic background. Note the cataracts in theeye of the two B-Raf+/LSLV600E mice. (D) Overlay of tworepresentative sagittal CT sections of a B6/CD1 B-Raf+/+
mouse (white) and a B6/CD1 B-Raf+/LSLV600E littermate(red). (E) 2D analysis of sagittal CT projections of 10-wk-old adult B-Raf+/+ (n = 6) and B-Raf+/LSLV600E (n = 5) litter-mates (B6/129 genetic background). The anatomical posi-tion of a set of 10 homologous landmarks in a sagittalsection of an adult B-Raf+/+ mouse is indicated by red cir-cles. (F) P values of differences between WT and mutantmice by Hotteling’s T2 test for each landmark coordinate (xand y) depicted in E after generalized procrustes super-imposition. NS, not significant.
Urosevic et al. PNAS | March 22, 2011 | vol. 108 | no. 12 | 5017
MED
ICALSC
IENCE
S
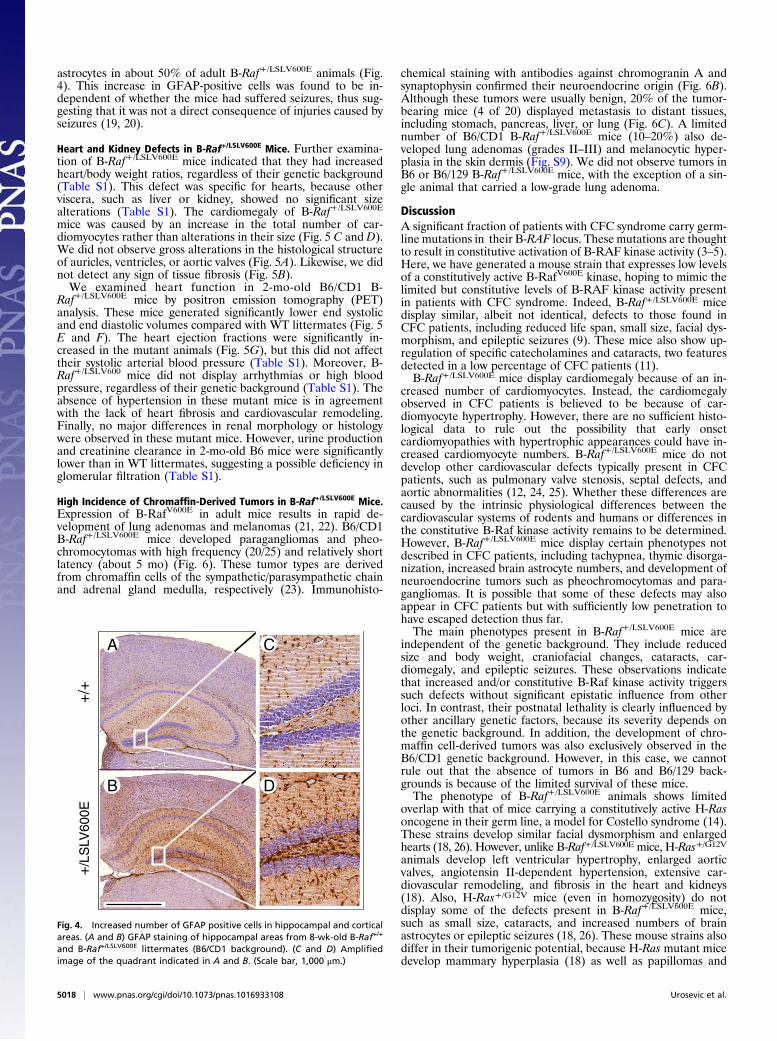
astrocytes in about 50% of adult B-Raf+/LSLV600E animals (Fig.4). This increase in GFAP-positive cells was found to be in-dependent of whether the mice had suffered seizures, thus sug-gesting that it was not a direct consequence of injuries caused byseizures (19, 20).
Heart and Kidney Defects in B-Raf+/LSLV600E Mice. Further examina-tion of B-Raf+/LSLV600E mice indicated that they had increasedheart/body weight ratios, regardless of their genetic background(Table S1). This defect was specific for hearts, because otherviscera, such as liver or kidney, showed no significant sizealterations (Table S1). The cardiomegaly of B-Raf+/LSLV600E
mice was caused by an increase in the total number of car-diomyocytes rather than alterations in their size (Fig. 5 C and D).We did not observe gross alterations in the histological structureof auricles, ventricles, or aortic valves (Fig. 5A). Likewise, we didnot detect any sign of tissue fibrosis (Fig. 5B).We examined heart function in 2-mo-old B6/CD1 B-
Raf+/LSLV600E mice by positron emission tomography (PET)analysis. These mice generated significantly lower end systolicand end diastolic volumes compared with WT littermates (Fig. 5E and F). The heart ejection fractions were significantly in-creased in the mutant animals (Fig. 5G), but this did not affecttheir systolic arterial blood pressure (Table S1). Moreover, B-Raf+/LSLV600 mice did not display arrhythmias or high bloodpressure, regardless of their genetic background (Table S1). Theabsence of hypertension in these mutant mice is in agreementwith the lack of heart fibrosis and cardiovascular remodeling.Finally, no major differences in renal morphology or histologywere observed in these mutant mice. However, urine productionand creatinine clearance in 2-mo-old B6 mice were significantlylower than in WT littermates, suggesting a possible deficiency inglomerular filtration (Table S1).
High Incidence of Chromaffin-Derived Tumors in B-Raf+/LSLV600E Mice.Expression of B-RafV600E in adult mice results in rapid de-velopment of lung adenomas and melanomas (21, 22). B6/CD1B-Raf+/LSLV600E mice developed paragangliomas and pheo-chromocytomas with high frequency (20/25) and relatively shortlatency (about 5 mo) (Fig. 6). These tumor types are derivedfrom chromaffin cells of the sympathetic/parasympathetic chainand adrenal gland medulla, respectively (23). Immunohisto-
chemical staining with antibodies against chromogranin A andsynaptophysin confirmed their neuroendocrine origin (Fig. 6B).Although these tumors were usually benign, 20% of the tumor-bearing mice (4 of 20) displayed metastasis to distant tissues,including stomach, pancreas, liver, or lung (Fig. 6C). A limitednumber of B6/CD1 B-Raf+/LSLV600E mice (10–20%) also de-veloped lung adenomas (grades II–III) and melanocytic hyper-plasia in the skin dermis (Fig. S9). We did not observe tumors inB6 or B6/129 B-Raf+/LSLV600E mice, with the exception of a sin-gle animal that carried a low-grade lung adenoma.
DiscussionA significant fraction of patients with CFC syndrome carry germ-line mutations in their B-RAF locus. These mutations are thoughtto result in constitutive activation of B-RAF kinase activity (3–5).Here, we have generated a mouse strain that expresses low levelsof a constitutively active B-RafV600E kinase, hoping to mimic thelimited but constitutive levels of B-RAF kinase activity presentin patients with CFC syndrome. Indeed, B-Raf+/LSLV600E micedisplay similar, albeit not identical, defects to those found inCFC patients, including reduced life span, small size, facial dys-morphism, and epileptic seizures (9). These mice also show up-regulation of specific catecholamines and cataracts, two featuresdetected in a low percentage of CFC patients (11).B-Raf+/LSLV600E mice display cardiomegaly because of an in-
creased number of cardiomyocytes. Instead, the cardiomegalyobserved in CFC patients is believed to be because of car-diomyocyte hypertrophy. However, there are no sufficient histo-logical data to rule out the possibility that early onsetcardiomyopathies with hypertrophic appearances could have in-creased cardiomyocyte numbers. B-Raf+/LSLV600E mice do notdevelop other cardiovascular defects typically present in CFCpatients, such as pulmonary valve stenosis, septal defects, andaortic abnormalities (12, 24, 25). Whether these differences arecaused by the intrinsic physiological differences between thecardiovascular systems of rodents and humans or differences inthe constitutive B-Raf kinase activity remains to be determined.However, B-Raf+/LSLV600E mice display certain phenotypes notdescribed in CFC patients, including tachypnea, thymic disorga-nization, increased brain astrocyte numbers, and development ofneuroendocrine tumors such as pheochromocytomas and para-gangliomas. It is possible that some of these defects may alsoappear in CFC patients but with sufficiently low penetration tohave escaped detection thus far.The main phenotypes present in B-Raf+/LSLV600E mice are
independent of the genetic background. They include reducedsize and body weight, craniofacial changes, cataracts, car-diomegaly, and epileptic seizures. These observations indicatethat increased and/or constitutive B-Raf kinase activity triggerssuch defects without significant epistatic influence from otherloci. In contrast, their postnatal lethality is clearly influenced byother ancillary genetic factors, because its severity depends onthe genetic background. In addition, the development of chro-maffin cell-derived tumors was also exclusively observed in theB6/CD1 genetic background. However, in this case, we cannotrule out that the absence of tumors in B6 and B6/129 back-grounds is because of the limited survival of these mice.The phenotype of B-Raf+/LSLV600E animals shows limited
overlap with that of mice carrying a constitutively active H-Rasoncogene in their germ line, a model for Costello syndrome (14).These strains develop similar facial dysmorphism and enlargedhearts (18, 26). However, unlike B-Raf+/LSLV600E mice, H-Ras+/G12V
animals develop left ventricular hypertrophy, enlarged aorticvalves, angiotensin II-dependent hypertension, extensive car-diovascular remodeling, and fibrosis in the heart and kidneys(18). Also, H-Ras+/G12V mice (even in homozygosity) do notdisplay some of the defects present in B-Raf+/LSLV600E mice,such as small size, cataracts, and increased numbers of brainastrocytes or epileptic seizures (18, 26). These mouse strains alsodiffer in their tumorigenic potential, because H-Ras mutant micedevelop mammary hyperplasia (18) as well as papillomas and
+/+
+/L
SLV
600E
A C
B D
Fig. 4. Increased number of GFAP positive cells in hippocampal and corticalareas. (A and B) GFAP staining of hippocampal areas from 8-wk-old B-Raf+/+
and B-Raf+/LSLV600E littermates (B6/CD1 background). (C and D) Amplifiedimage of the quadrant indicated in A and B. (Scale bar, 1,000 μm.)
5018 | www.pnas.org/cgi/doi/10.1073/pnas.1016933108 Urosevic et al.
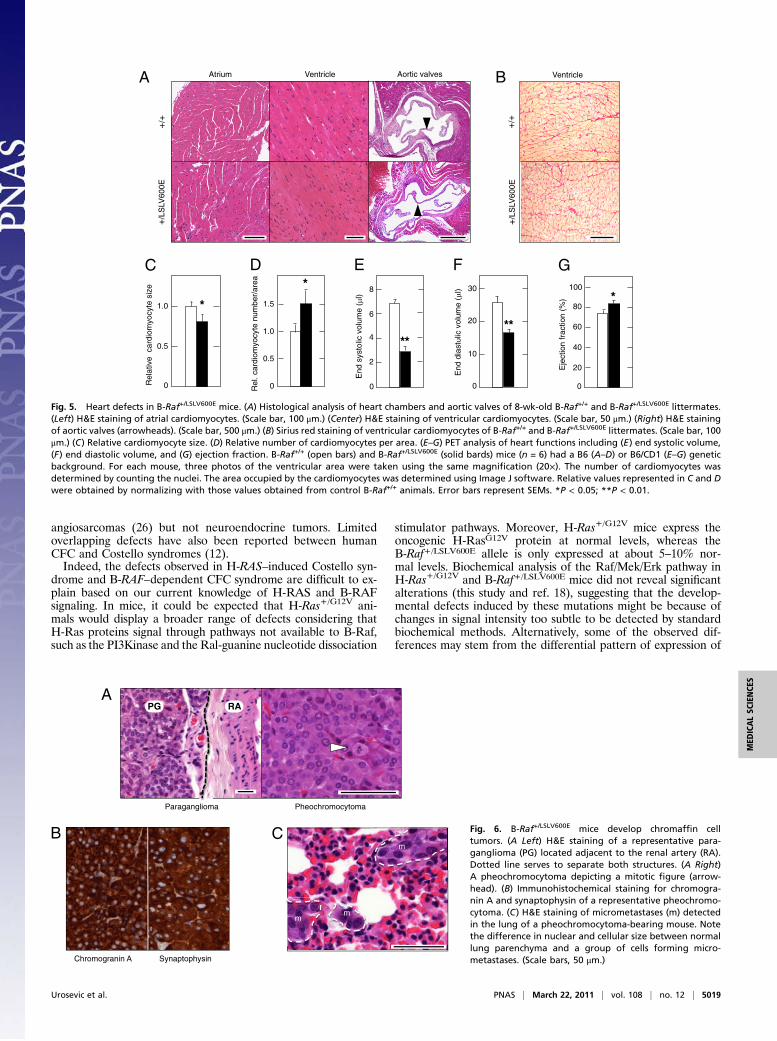
angiosarcomas (26) but not neuroendocrine tumors. Limitedoverlapping defects have also been reported between humanCFC and Costello syndromes (12).Indeed, the defects observed in H-RAS–induced Costello syn-
drome and B-RAF–dependent CFC syndrome are difficult to ex-plain based on our current knowledge of H-RAS and B-RAFsignaling. In mice, it could be expected that H-Ras+/G12V ani-mals would display a broader range of defects considering thatH-Ras proteins signal through pathways not available to B-Raf,such as the PI3Kinase and the Ral-guanine nucleotide dissociation
stimulator pathways. Moreover, H-Ras+/G12V mice express theoncogenic H-RasG12V protein at normal levels, whereas theB-Raf+/LSLV600E allele is only expressed at about 5–10% nor-mal levels. Biochemical analysis of the Raf/Mek/Erk pathway inH-Ras+/G12V and B-Raf+/LSLV600E mice did not reveal significantalterations (this study and ref. 18), suggesting that the develop-mental defects induced by these mutations might be because ofchanges in signal intensity too subtle to be detected by standardbiochemical methods. Alternatively, some of the observed dif-ferences may stem from the differential pattern of expression of
Eje
ctio
nfr
actio
n (%
)
0
20
40
60
80
100
*
End
dias
tolic
volu
me
(µl)
0
10
20
30
**
2
4
6
8
End
syst
olic
volu
me
(µl)
**
0
*
Rel
. car
diom
yocy
tenu
mbe
r/ar
ea
0
1.0
1.5
0.5
Rel
ativ
eca
rdio
myo
cyte
size
0
1.0
0.5
*
+/+
+/L
SLV
600E
Atrium Ventricle Aortic valvesA
C D E F G
+/L
SLV
600E
+/+
B Ventricle
Fig. 5. Heart defects in B-Raf+/LSLV600E mice. (A) Histological analysis of heart chambers and aortic valves of 8-wk-old B-Raf+/+ and B-Raf+/LSLV600E littermates.(Left) H&E staining of atrial cardiomyocytes. (Scale bar, 100 μm.) (Center) H&E staining of ventricular cardiomyocytes. (Scale bar, 50 μm.) (Right) H&E stainingof aortic valves (arrowheads). (Scale bar, 500 μm.) (B) Sirius red staining of ventricular cardiomyocytes of B-Raf+/+ and B-Raf+/LSLV600E littermates. (Scale bar, 100μm.) (C) Relative cardiomyocyte size. (D) Relative number of cardiomyocytes per area. (E–G) PET analysis of heart functions including (E) end systolic volume,(F) end diastolic volume, and (G) ejection fraction. B-Raf+/+ (open bars) and B-Raf+/LSLV600E (solid bards) mice (n = 6) had a B6 (A–D) or B6/CD1 (E–G) geneticbackground. For each mouse, three photos of the ventricular area were taken using the same magnification (20×). The number of cardiomyocytes wasdetermined by counting the nuclei. The area occupied by the cardiomyocytes was determined using Image J software. Relative values represented in C and Dwere obtained by normalizing with those values obtained from control B-Raf+/+ animals. Error bars represent SEMs. *P < 0.05; **P < 0.01.
Chromogranin A Synaptophysin
A
B C
Paraganglioma Pheochromocytoma
mm
m
PG RA
Fig. 6. B-Raf+/LSLV600E mice develop chromaffin celltumors. (A Left) H&E staining of a representative para-ganglioma (PG) located adjacent to the renal artery (RA).Dotted line serves to separate both structures. (A Right)A pheochromocytoma depicting a mitotic figure (arrow-head). (B) Immunohistochemical staining for chromogra-nin A and synaptophysin of a representative pheochromo-cytoma. (C) H&E staining of micrometastases (m) detectedin the lung of a pheochromocytoma-bearing mouse. Notethe difference in nuclear and cellular size between normallung parenchyma and a group of cells forming micro-metastases. (Scale bars, 50 μm.)
Urosevic et al. PNAS | March 22, 2011 | vol. 108 | no. 12 | 5019
MED
ICALSC
IENCE
S

these proteins. For instance, the higher levels of expression ofthe B-Raf locus in the nervous system may account for the sei-zures and neuroendocrine tumors exclusively observed in theB-Raf+/LSLV600E animals. Full understanding of the molecularevents responsible for the developmental defects observed in thevarious RASopathies (8) will require a more profound knowledgeof how these proteins signal in vivo.The B-Raf+/LSLV600E animals described here may help in un-
derstanding the pathophysiology of at least some of the clinicalfeatures present in CFC patients and possibly other RASopathies.Moreover, they could help to identify genetic factors that contrib-ute to the pleiotropic manifestations of the clinical disorders byintroducing the B-RafLSLV600E allele in different genetic back-grounds or combined with other alleles. Recently, MEK and fi-broblast growth factor receptor 1 (FGFR1) inhibitors have beenused to study development of zebra fish embryos carrying CFCmutations (27). Whereas prolonged treatments resulted in axisabnormalities, short exposure during specific developmental win-dows prevented these defects (27).More recently, B-RAF selectiveinhibitors have been developed and shown to have a significantantitumor effect in B-RAFV600E–induced human melanomas (15,16). The B-Raf+/LSLV600E strain could be a tool, not only to de-termine the therapeutic efficacy of these compounds in the treat-ment of CFC but as previously shown in zebra fish (27), to establishthe precise window at which these inhibitors could be effectivelyused to treat this congenital syndrome.
Materials and MethodsGeneration of Mice. The detailed strategy used to generate the B-Raf+/LSLV600E
strain and genotype the corresponding alleles is described in SI Materialsand Methods.
Western Blot and Kinase Assay. Protein extracts obtained from whole-mouseembryos (E13.5; 50 μg), adult heart (70 μg), and adult brain (50 μg) werefractionated in SDS/PAGE gels, transferred onto nitrocellulose membranes,and subjected to immunoblot analysis according to standard procedures (SIMaterials and Methods).
Histopathology and Immunohistochemistry. Tissues were dissected, fixed in10% buffered formalin (Sigma), and embedded in paraffin; 2- to 3-μm-thicksections were stained with H&E. Antibodies used for immunohistochemistry
included those to synaptophysin (1:1; Dako), GFAP (1:25; Dako), chromog-ranin A (1:300; Abcam), active caspase 3 (1:20; R&D Systems), and terminaldeoxynucleotidyl transferase (1:15; Dako). Heart sections were stained withSirius Red (Fluka) to visualize fibrosis (28, 29).
Physiological Parameters. Blood pressure and heart rates were recorded inconscious mice with an automated multichannel system using the tail-cuffmethodandaphotoelectric sensor (Niprem546;CibertecSA) (29,30).Creatinineconcentrations in urine and plasma were determined by a modification ofJaffé’s reaction method (28, 29). Adrenaline and noradrenaline levels weredetermined by using CatCombi ELISA kit (IBL) following the manufacturer’sinstructions. For the determination of breathing activity, animals were lightlyanesthetized with 1 g urethane/kg body weight. Forceps connected to a forcetransducer by a flexible wire were attached to the anesthetized mice at thediaphragm level. Respiratory amplitude and frequency were then collectedusing a digital data recorder (MacLab/4e; AD Instruments), and data were in-tegrated with the Chart v3.4 software (AD Instruments).
CT and PET. Acquisition of CT and PET images was carried out according tostandard protocols using an eXplore Vista PET CT (GE Healthcare). Mor-phometric analysis was carried out as described in SI Materials and Methods.
ACKNOWLEDGMENTS. We thank Isabel Hernandez and Carmen Guerra fortheir comments and Mayte Lamparero and Isabel Aragon for excellenttechnical assistance. We also value the excellent support provided by theComparative Pathology and Transgenic Mice Core Units of the CentroNacional de Investigaciones Oncológicas. Work in the laboratory of M.D.was funded by Consorcios Estratégicos Nacionales en Investigación TécnicaProgram (CDTEAM) Grant TEC2008-06715-C02-01, Centro de InvestigaciónBiomédica en Red Program Grants CB06/01/0079 and PNSD 2007-2010,Fondo de Investigación Sanitaria (FIS) Grant CP08/00017, and Fundación dela Mutua Madrileña del Automovil (FMMA). E.M.B.W. was the holder ofa United Kingdom National Institute for Health Research Academic ClinicalFellowship and was supported by the Manchester Biomedical Research Cen-tre. Work in the laboratory of X.R.B. was funded by National Institutesof Health Grant R01CA073735, Spanish Ministry of Science and Innova-tion (MICINN) Grants SAF2009-07172 and RD06/0020/0001, AutonomousGovernment of Castilla y León (GR97), and Asociación Española contra elCáncer. Work in the laboratory of M.B. was supported by European Union-Framework Programme Grants LSHG-CT-2006-037188 and LSHG-CT-2007-037665 (to M.B.), European Research Council Grant ERC-AG/250297-RASAHEAD (to M.B.), MICINN Grants SAF2006-11773 and CSD2007-00017 (toM.B.), FMMA (to M.B.), FIS Grant PI042124, and Autonomous Community ofMadrid Grant GR/SAL/0349/2004.
1. Davies H, et al. (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954.
2. Wellbrock C, Karasarides M, Marais R (2004) The RAF proteins take centre stage. NatRev Mol Cell Biol 5:875–885.
3. Wan PT, et al. (2004) Mechanism of activation of the RAF-ERK signaling pathway byoncogenic mutations of B-RAF. Cell 116:855–867.
4. Niihori T, et al. (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneoussyndrome. Nat Genet 38:294–296.
5. Rodriguez-Viciana P, et al. (2006) Germline mutations in genes within the MAPKpathway cause cardio-facio-cutaneous syndrome. Science 311:1287–1290.
6. Schubbert S, Shannon K, Bollag G (2007) Hyperactive Ras in developmental disordersand cancer. Nat Rev Cancer 7:295–308.
7. Brems H, et al. (2007) Germline loss-of-function mutations in SPRED1 cause a neu-rofibromatosis 1-like phenotype. Nat Genet 39:1120–1126.
8. Tidyman WE, Rauen KA (2009) The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230–236.
9. Roberts A, et al. (2006) The cardiofaciocutaneous syndrome. J Med Genet 43:833–842.10. Rodriguez-Viciana P, Rauen KA (2008) Biochemical characterization of novel germline
BRAF and MEK mutations in cardio-facio-cutaneous syndrome.Methods Enzymol 438:277–289.
11. Gripp KW, et al. (2007) Further delineation of the phenotype resulting from BRAF orMEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome fromCostello syndrome. Am J Med Genet A 143A:1472–1480.
12. Narumi Y, et al. (2007) Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: Overlapping clinical manifestations with Costello syndrome.Am J Med Genet A 143A:799–807.
13. Heidorn SJ, et al. (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drivetumor progression through CRAF. Cell 140:209–221.
14. Aoki Y, et al. (2005) Germline mutations in HRAS proto-oncogene cause Costellosyndrome. Nat Genet 37:1038–1040.
15. Bollag G, et al. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockadein BRAF-mutant melanoma. Nature 467:596–599.
16. Flaherty KT, et al. (2010) Inhibition of mutated, activated BRAF in metastaticmelanoma. N Engl J Med 363:809–819.
17. Mercer K, et al. (2005) Expression of endogenous oncogenic V600EB-raf inducesproliferation and developmental defects in mice and transformation of primaryfibroblasts. Cancer Res 65:11493–11500.
18. Schuhmacher AJ, et al. (2008) A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest 118:2169–2179.
19. Stringer JL (1996) Repeated seizures increase GFAP and vimentin in the hippocampus.Brain Res 717:147–153.
20. Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434.21. Dankort D, et al. (2007) A new mouse model to explore the initiation, progression,
and therapy of BRAFV600E-induced lung tumors. Genes Dev 21:379–384.22. Dhomen N, et al. (2009) Oncogenic Braf induces melanocyte senescence and
melanoma in mice. Cancer Cell 15:294–303.23. Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL (2003) Pheochromo-
cytoma: The expanding genetic differential diagnosis. J Natl Cancer Inst 95:1196–1204.
24. Armour CM, Allanson JE (2008) Further delineation of cardio-facio-cutaneoussyndrome: Clinical features of 38 individuals with proven mutations. J Med Genet 45:249–254.
25. Nyström AM, et al. (2008) Noonan and cardio-facio-cutaneous syndromes: Twoclinically and genetically overlapping disorders. J Med Genet 45:500–506.
26. Chen X, et al. (2009) Endogenous expression of Hras(G12V) induces developmentaldefects and neoplasms with copy number imbalances of the oncogene. Proc NatlAcad Sci USA 106:7979–7984.
27. Anastasaki C, Estep AL, Marais R, Rauen KA, Patton EE (2009) Kinase-activatingand kinase-impaired cardio-facio-cutaneous syndrome alleles have activity duringzebrafish development and are sensitive to small molecule inhibitors. HumMol Genet18:2543–2554.
28. Sauzeau V, et al. (2006) Vav3 proto-oncogene deficiency leads to sympathetichyperactivity and cardiovascular dysfunction. Nat Med 12:841–845.
29. Sauzeau V, Jerkic M, López-Novoa JM, Bustelo XR (2007) Loss of Vav2 proto-oncogenecauses tachycardia and cardiovascular disease in mice. Mol Biol Cell 18:943–952.
30. Sauzeau V, Sevilla MA, Montero MJ, Bustelo XR (2010) The Rho/Rac exchange factorVav2 controls nitric oxide-dependent responses in mouse vascular smooth musclecells. J Clin Invest 120:315–330.
5020 | www.pnas.org/cgi/doi/10.1073/pnas.1016933108 Urosevic et al.

RESEARCH ARTICLE
Neonatal Lethal Costello Syndrome and UnusualDinucleotide Deletion/Insertion Mutations in HRASPredicting p.Gly12ValEmma M.M. Burkitt-Wright,1 Lisa Bradley,2 Jennifer Shorto,1 Vivienne P.M. McConnell,2
Caroline Gannon,3 Helen V. Firth,4 Soo-Mi Park,4 Angela D’Amore,5 Paul F. Munyard,6
Peter D. Turnpenny,7 Amanda Charlton,8 Meredith Wilson,9 and Bronwyn Kerr1*1Genetic Medicine, Manchester Academic Health Science Centre, University of Manchester and Central Manchester University
Hospitals NHS Foundation Trust, Manchester, UK2Northern Ireland Regional Genetics Service, Belfast, UK3Department of Pathology, Royal Belfast Hospitals, Belfast, UK4Department of Clinical Genetics, East Anglian Medical Genetics Service, Addenbrooke’s Hospital, Cambridge, UK5Department of Neonatology, Addenbrooke’s Hospital, Cambridge, UK6Department of Pediatrics, Royal Cornwall Hospital, Truro, UK7Clinical Genetics Department, Royal Devon & Exeter Hospital, Exeter, UK8Department of Pediatric Pathology, Children’s Hospital at Westmead, Sydney, New South Wales, Australia9Department of Clinical Genetics, Children’s Hospital at Westmead, Sydney, New South Wales, Australia
Received 18 October 2011; Accepted 29 December 2011
De novo heterozygous mutations in HRAS cause Costello syn-
drome (CS), a condition with high mortality and morbidity
in infancy and early childhood due to cardiac, respiratory,
and muscular complications. HRAS mutations predicting
p.Gly12Val, p.Gly12Asp, and p.Gly12Cys substitutions have
been associated with severe, lethal, CS. We report on molecular,
clinical, and pathological findings in patients with mutations
predicting HRAS p.Gly12Val that were identified in our clinical
molecular genetic testing service. Suchmutationswere identified
in four patients. Remarkably, three were deletion/insertion
mutations affecting coding nucleotides 35 and 36. All patients
died within 6 postnatal weeks, providing further evidence that
p.Gly12Val mutations predict a very poor prognosis. High birth
weight, polyhydramnios (and premature birth), cardiac hyper-
trophy, respiratory distress, muscle weakness, and postnatal
growth failure were present. Dysmorphism was subtle or non-
specific, with edema, coarsened facial features, prominent fore-
head, depressed nasal bridge, anteverted nares, and low-set ears.
Proximal upper limb shortening, a small bell-shaped chest,
talipes, and fixed flexion deformities of the wrists were seen.
Neonatal atrial arrhythmia, highly suggestive of CS, was also
present in two patients. One patient had congenital alveolar
dysplasia, and another, born after 36 weeks’ gestation, broncho-
pulmonary dysplasia. A rapidly fatal disease course, and the
difficulty of identifying subtle dysmorphism in neonates requir-
ing intensive care, suggest that this condition remains under-
recognized, and should enter the differential diagnosis for very
sick infants with a range of clinical problems including cardiac
hypertrophy and disordered pulmonary development. Clinical
management should be informed by knowledge of the poor
prognosis of this condition. � 2012 Wiley Periodicals, Inc.
Grant sponsors: Wellcome trust and UK National Institute for Health
Research’s Manchester Biomedical Research Centre.
The authors have no competing interests to declare.
*Correspondence to:
Dr. Bronwyn Kerr, MBBS, FRACP, Genetic Medicine, 6th Floor St Mary’s
Hospital, Central Manchester Foundation Trust, Oxford Road,
Manchester M13 9WL, UK. E-mail: [email protected]
Published online 11 April 2012 in Wiley Online Library
(wileyonlinelibrary.com).
DOI 10.1002/ajmg.a.35296
How to Cite this Article:Burkitt-Wright EMM, Bradley L, Shorto J,
McConnell VPM, Gannon C, Firth HV, Park
S-M, D’Amore A, Munyard PF, Turnpenny
PD, Charlton A, Wilson M, Kerr B. 2012.
Neonatal lethal Costello syndrome and
unusual dinucleotide deletion/insertion
mutations in HRAS predicting p.Gly12Val.
Am J Med Genet Part A 158A:1102–1110.
� 2012 Wiley Periodicals, Inc. 1102

Key words: Costello syndrome; HRAS; neonatal cardiomyop-
athy; congenital alveolar dysplasia; dinucleotide insertion/deletion
mutation
INTRODUCTION
Costello syndrome (CS) is a rare condition which arises due to
heterozygous germline mutations inHRAS, resulting in expression
of constitutively activeHRASproteins [Aoki et al., 2005].Much the
commonest of these is p.Gly12Ser, which accounts for approxi-
mately 80%of diagnosed cases [Kerr et al., 2006]. Common clinical
features include prenatal overgrowth and polyhydramnios, severe
postnatal failure to thrive, short stature, developmental delay,
congenital heart disease, and cardiomyopathy [Kerr, 2009]. Whilst
CS often has a relatively homogenous phenotype, both milder and
more severe phenotypes are now recognized, which often arise due
to less common mutations [Kerr et al., 2006; van der Burgt et al.,
2007; Gripp et al., 2011]. Severe CS and congenital myopathy with
excess of muscle spindles, a variant manifestation of the same
condition, have been described in association with mutations
predicting amino acid substitutions p.Gly12Val [Aoki et al.,
2005; van der Burgt et al., 2007], p.Gly12Asp and p.Gly12Cys
[Lo et al., 2008], but with only a very few patients in the literature.
We present here a series of four further patients with a variety of
HRAS mutations predicting p.Gly12Val. These p.Gly12Val muta-
tions, rare in the germline but much more commonly observed in
cancers, were associatedwith a severe presentation lethal in the first
weeks of life in all four patients.
MATERIALS AND METHODS
Exons 2 to 6 ofHRASwere sequenced and analyzed on an ABI 3730
sequencer, within themolecular diagnostic service offered byMan-
chester Regional Genetics Laboratory. Clinical and histopatholog-
ical details of patients in whom mutations predicting p.Gly12Val
were identified are described below and summarized in Table I.
RESULTS
Patient 1This boy was the second child of healthy unrelated Irish parents, his
mother being 28 and father 33 years of age. His older sister was
stillborn at 41 weeks’ gestation weighing 2,750 g (9th centile), with
no dysmorphic features. No postmortem was performed in her.
Fetal macrosomia was identified in the proband at 34 weeks’
gestation. Maternal HbA1c, glucose tolerance test and a congenital
viral infection screen were normal. He was delivered by cesarean at
36þ6 weeks’ gestation for sudden onset polyhydramnios and
reduced fetal movements, weighing 3,325 g (75th centile), with
an OFC of 38.2 cm (>99.6th centile). Apgar scores were good
(81 and 95), but he quickly became ventilator dependent with
severe central hypotonia. Obstructing vocal cord granulomas
required carbon dioxide laser treatment on day 39. He died on
day 42 of bronchopneumonia.
Dysmorphic features (Fig. 1a,b) included frontal bossing, a box-
shaped face, depressed nasal bridge with anteverted alae nasi, large
dysplastic ears, abundant loose skin around the mandible, a short,
fleshy neck, small thorax with widely spaced nipples, upper limb
rhizomelic shortening, peripheral arthrogryposis with slender
digits, bilateral undescended testes, and talipes equinovarus. Blood
and skin karyotypes showed 46, XY. Subtelomere microdeletion/
duplication multiplex ligation probe amplification (MRC Holland
kit PO36E1), RAPSN analysis, extensive metabolic workup, mag-
netic resonance imaging of brain and abdomen, initial echocardio-
gram, bone age, bone density, and skeletal survey all showednormal
results. Repeat echocardiography on day 36 demonstrated biven-
tricular hypertrophy with a small pericardial effusion.
Postmortem examination revealed length and head circumfer-
ence on the 25th centile, but weight below the 9th, despite apparent
excessive subcutaneous tissue of the limbs, face, and neck. Heart
weight was 34 g (expected: 20 g), biventricular and septal hyper-
trophy (Fig. 2a) with mild interstitial edema were present, but no
fibrosis or myofibrillar disarray. Other muscles were firm and
bulky, especially the diaphragm (Fig. 2b,c). Evidence of bronchop-
neumonia and healing bronchopulmonary dysplasia confirmed
the cause of death. The pancreas showed increased islet cell size
and number, whilst the thymus was small and atrophic. Immature
cryptorchid testes were well above the pelvic rim. The brain
appeared structurally normal, but weighed 602 g (expected:
413 g). Radiographs and the rest of the internal examination
were normal, with no further histological abnormalities evident.
The diagnosis of CS was only established some time after the baby’s
death, when he was presented at an international dysmorphology
meeting.
Patient 2This baby girl was the first child of unrelated Australian parents, a
27-year-oldmother and31-year-old father. Severe polyhydramnios
TABLE I. Summary of Clinical Features of Patients With
p.Gly12Val Mutations
Feature Present inSevere polyhydramnios 5/5Delivery before 37 weeks’ gestation 5/5Birth weight on above 90th centile 4/5Birth OFC on or above 90th centile 5/5Coarsening of facial features 5/6Unusual hand position 5/6Unusual foot position 4/5Short neck 5/6Narrow thorax and protuberant abdomen 3/4High cryptorchidism 2/2Cardiac hypertrophy 6/6Structural findings on echocardiography(abnormal pulmonary outflow tract:1;patent foramen ovale:1)
2/4
Cardiac arrhythmia 2/4Ventilator dependence 5/6Hepatosplenomegaly 2/5Death before 6 weeks postnatal agea 5/6
aThe other previously published patient with a p.Gly12Val mutation also had a lethal course,surviving to 18 months of age [Aoki et al., 2005].
BURKITT-WRIGHT ET AL. 1103

was noted at 25 weeks, requiring amnioreduction of 2 L at 26 weeks
because of threatened premature labor. Ultrasound at 27 weeks
showed persistent mild polyhydramnios, short limbs, prominent
abdomen, and bell-shaped chest. Fetal MRI showed mild
ventricular dilatation. Steroids were administered at 29 weeks,
and emergency cesarean delivery was necessary at 30 weeks for a
non-reassuring CTG. Apgar scores were 41 and 75; she was intu-
bated and ventilated from birth and a single dose of surfactant
given. Her birth weight was 1,926 g (>97th centile), length 25–50thcentile and OFC 90th centile.
Marked generalized edema, a bell-shaped chest, prominent
abdomen, and rhizomelic arm shortening were evident postnatally,
with coarse facies, high prominent forehead, depressed nasal
bridge, pursed lips, microretrognathia, a ‘‘double chin,’’ excess
nuchal skin, edematous fingers, and rocker bottom feet. Her hands
were flexed at the wrists and extended at the metacarpophalangeal
joints, with thumbs adducted and fingers flexed, but without
contractures. There was no organomegaly, ascites, or pleural effu-
sion. Edema resolvedover thefirstweek andherweight decreased to
the 50th centile. Cranial ultrasound showed mild ventricular
dilatation and bilateral grade I intraventricular hemorrhage.
A skeletal dysplasia was initially suspected, but skeletal survey
revealed no additional abnormalities. Abdominal ultrasound, liver
function tests, EEG, transferrin isoforms, very long-chain fatty
acids, white cell enzymes, karyotype, urinary glycosaminoglycans,
amino and organic acids, carnitines and acylcarnitine profile were
also within normal limits. She developed mild hyponatremia and
maximum serum bilirubin was 219 mmol/L.
Requirements for high ventilatory pressures and PiO2 persisted,
with an inability to tolerate ventilatory rates below 40 per min.
Chest X-rays showed non-specific hazy opacities and migrating
lung collapses, and the clinical course suggested pulmonary
hypoplasia/dysplasia. At 3weeks, three episodes of supraventricular
tachycardia occurred in 24 hr, two requiring cardioversion, the
other was brief and resolved spontaneously. Subsequent ECG
showed right axis deviation, borderline left bundle branch block
and T wave inversion in chest leads. Echocardiogram revealed a
patent foramen ovale and mild asymmetric septal hypertrophy.
Clinical genetic assessment revealed deep palmar and plantar
creases and typical hand positioning suggestive of severe neonatal
CS. Given her poor prognosis, persisting high ventilatory require-
ments and features of severe pulmonary dysplasia, the managing
team and parents jointly decided to discontinue mechanical ven-
tilation on day 36, and she died soon afterwards.
At postmortem, body weight was 5th centile, crown-rump and
foot lengths <3rd centile, and OFC 10th centile (for 35 weeks’
FIG. 1. Clinical appearance of patients. a,b: Patient 1, appearance on day 2 of life. Note overall fleshy/edematous appearance to face, trunk and limbs,
rhizomelic shortening with marked circumferential skin folds in the arms and peripheral edema. Dysmorphic facial features including low-set fleshy
ears with overfolded helices and upturned lobes, short nose with depressed nasal bridge and anteverted nares, and micrognathia are present. c–f:Patient 3. c: A deep plantar crease is seen in the foot, which is held in equinovarus. d: Macrosomia, protuberant abdomen, and a small, narrow thorax
with widely spaced nipples are shown. Flexed wrists with hands held in ulnar deviation are also seen. e,f: Coarse facial appearance, a prominent
forehead, depressed nasal bridge with anteverted nares, and low-set, fleshy ears, are shown.
1104 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

gestation). Increased subcutaneous tissue of the face and neck, lax
skin of the trunk and limbs, deep palmar and plantar creases and
perianal papillomata were present. Consent for internal examina-
tion was limited to lung biopsy. The right upper and middle lobes
were removed and appeared heavy, solid, and airless. Histology of
formalin-fixed sections (Fig. 2d–f) showed a diffuse developmental
disorder: development was arrested in the canalicular stage, nor-
mally seen at 17–27 weeks’ gestation [Langston andDishop, 2009].The pulmonary veins were not misaligned, and capillary density
and apposition were normal, thus the features were not of alveolar
capillary dysplasia [Melly et al., 2008]. Delayed maturation was in
keeping with congenital alveolar dysplasia (CAD) [MacMahon,
1948]. The radial alveolar count was normal (3; mean expected
for 32–35 weeks: 3.2� 0.9 [Emery and Mithal, 1960]), and there
was no lymphangiectasia.
Patient 3This baby girl was the product of an IVF/ICSI pregnancy using a
donor ovum from a healthy 29-year-old British Caucasian woman,
due to failureof spontaneous conceptionbyher 48-year-oldmother
and 61-year-old father. The parents had a previous naturally
conceived child with non-disjunctional trisomy 21, born when
the mother was 42 years old.
Fetal macrosomia was identified at 20 weeks’ gestation. By 33þ4
weeks, very marked polyhydramnios was present, with head and
abdominal circumferences well above the 97th centile, but femoral
lengths below the 50th centile, and evidence of gross hepatomegaly.
The kidneys were not visualized, but neither an abdominal wall
defect nor macroglossia were detected. Gestational diabetes was
diagnosed at 24 weeks’ gestation, but was well controlled with
insulin and diet (HbA1c 5.4%), and hence considered unlikely
to be a significant contributor to the abnormalities identified on
ultrasound.
Emergency cesarean was undertaken for fetal distress after
spontaneous onset of labor at 36þ4 weeks. Meconium stained
liquor was present, and there was no respiratory effort at birth.
Apgar scores were 41 and 65.Markedmacrosomia was present, with
birth weight (4,070 g) and OFC (37.3 cm) both greatly above the
99.6th centile. She was blue and hypotonic, with a small thorax,
and required resuscitation, intubation, and ventilation. Marked
respiratory distress necessitated ongoing ventilatory support with
high oxygen requirements and a diaphragmatic breathing pattern
reflecting laryngomalacia and hypotonia. A diagnosis of CS was
considered, given her macrosomia (Fig. 1c,d), disproportionally
large head with prominent forehead, depressed nasal bridge with
anteverted nares, coarse facial appearance and low set, fleshy ears
(Fig. 1e,f). Her protuberant abdomen, narrow thorax with widely
spaced nipples, deep palmar and plantar creases, deep set nails
and bilateral talipes equinovarus, with wrists held in fixed flexion,
were also in keeping with this.
On day 6, she developed a tachycardia of 200 beats/min. ECGs
showed abnormal P wave morphology with multifocal atrial
tachycardia, or sinus tachycardia with intermittent atrial tachycar-
dia. Echocardiography (having been normal on day 2) then showed
ventricular hypertrophy, massive tricuspid regurgitation and an
abnormal pulmonary outflow tract. Subsequent echocardiograms
showedworsening biventricular concentric hypertrophy, particularly
FIG. 2. Postmortem anatomy and histology. a: Macroscopic appearance of coronal section through the heart, showing hypertrophic cardiomyopathy,
with particularly marked septal hypertrophy. b,c: View into chest and abdominal cavity. Note generalized pallor of muscles and thickening of
diaphragm. d: Delayed lung development for 35 weeks’ gestation (adjusted), similar to canalicular phase, is shown. The pulmonary artery and
bronchiole travel together (left), and the pulmonary vein (right) is normally positioned in the interlobular septum. Hematoxylin and eosin stain,
original magnification 200�. e: Cytokeratin (brown chromogen) marks the alveolar lining cells, demonstrating an excess of stroma and too little
airspace. Cytokeratin AE1/3 immunoperoxidase, hematoxylin counterstain, original magnification 200�. f: CD34 (brown chromogen) marks the
capillaries. Capillary apposition and density is not decreased. CD34 immunoperoxidase, hematoxylin counterstain, original magnification 400�.
BURKITT-WRIGHT ET AL. 1105

affecting the right ventricle, and evidence of secondary pulmonary
hypertension. Cardiomegaly and features of pulmonary edema
were also seen on chest X-ray. Skeletal survey confirmed a narrow
ribcage but no other abnormalities, cranial ultrasound scan was
normal, and karyotype was 46, XX. Hepatosplenomegaly persisted
postnatally, with fluctuating conjugated jaundice and raised trans-
aminases after initially normal liver function tests. She also devel-
oped sepsis, anemia, and hyponatremia with high urinary sodium
losses and elevated urinary vanillylmandelic acid. In view of
increasing ventilatory requirements and poor prognosis, the deci-
sionwas reachedwith her parents to discontinue intensivemanage-
ment and she died shortly afterwards on day 39. No postmortem
examination was conducted.
Patient 4This girl was the first child of non-consanguineous British Cau-
casian parents, the father and mother aged 24 and 22 years,
respectively, at the time of conception. Polyhydramnios was noted
on ultrasound at 27 weeks’ gestation, though this was not seen at 28
weeks, when premature labor occurred. Two doses of dexametha-
sone were administered before spontaneous vaginal delivery of the
baby, who weighed 1,377 g (75th–91st centile). Meconium was
present and she made no spontaneous respiratory effort. She was
intubated at 7min and received surfactant (Curosurf). Respiratory
distress syndromewas treatedwithmechanical ventilationuntil day
8. Reintubation and ongoing ventilation became necessary on day
10. Chest X-ray showed persistent right upper lobe collapse.
Echocardiography on day 15 showed a very thick intraventricular
septum and thick ventricular walls, and hypertrophic cardiomy-
opathy was diagnosed. Cranial ultrasonography demonstrated
bilateral periventricular flare. She remained parenterally fed,
became increasingly difficult to ventilate, and died on day 17.
Subtle dysmorphic features had been noted, including a short
neck with possible webbing, widely spaced nipples and rhizomelic
shortening of the limbs. Karyotype was 46, XX and PTPN11
mutation analysis was normal. Eight years later, the family was
referred to the genetics service for investigation of a male sibling
with developmental delay and dysmorphic features (for whom a
diagnosis is still not established). On the strength of a family
photograph of the patient, taken after withdrawal of treatment,
the diagnosis of CSwas considered and her storedDNAwas sent for
HRAS mutation analysis.
MOLECULAR RESULTS
Heterozygous missense mutations in HRAS were identified in all
four patients, each predicting p.Gly12Val at the protein level. A
variety of nucleotide changes were identified, 3 of the 4 arising as
deletion/insertion mutations of nucleotides 35 and 36 (Table II).
Of note, no isolated substitutions of nucleotide 36 (any of which
would be synonymous) have been identified in any other sample of
213 submitted forHRAS sequencing analysis in our laboratory, and
nor is any substitution at this base a recognized polymorphism.
De novo occurrence of the mutations was confirmed by absence
of the changes in parental DNA, where this was available: the
mutation was confirmed to be absent from lymphocyte DNA of
the parents of Patient 1 (Fig. 3) and Patient 2. The latter couple also
opted to have prenatal testing for gonadal mosaic risk in a sub-
sequent pregnancy, with normal results. The parents of other
patients in this series did not pursue genetic testing on their
own accounts in view of low recurrence risks and their personal
circumstances.
The preponderance of dinucleotide deletion/insertion muta-
tions identified in this series is remarkable, as such mutations
are exceedingly rare both in other germline disorders and also in
cases of somatic mutation, such as those that occur in cancer. The
COSMIC database of somatic alterations in cancer (http://www.
sanger.ac.uk/perl/genetics/CGP/cosmic), as of 6th October 2011,
included 749HRASmutations, identified in 21,905 tumor samples
tested. Of these, 453 affected codon 12. These are shown in Figure 4,
and of note none were dinucleotide deletion/insertions. Similar
mutations have, however, been very rarely identified affecting
codon 61 of HRAS (in 6/21905 tumor samples), and also with
extreme rarity in other RAS genes in cancers, for example 54 such
mutations altering codon 12 of KRAS have been included in
COSMIC, in comparison to 17,490 point mutations altering this
codon (in a total of 92,270 tumor samples included at October 6,
2011).
DISCUSSION
Clinical Presentation of p.Gly12Val MutationsThis series demonstrates that a variety of clinical manifestations of
severe CS are present in infants withHRAS p.Gly12Val mutations,
but all of these patients had ultimately similar, lethal, outcomes.
There are only two patients in the literature with similarmutations,
one described as having severe neonatal CS [Aoki et al., 2005],
and one with congenital myopathy with excess of muscle spindles
[van der Burgt et al., 2007, initially reported by de Boode et al.,
1996]. Both died in the first 2 years of life, at 18months and 3weeks,
respectively. Severe CS, lethal in the neonatal period, has also been
described in association with other HRASmutations of codon 12,
both as an unusual manifestation of the commonest CS allele,
p.Gly12Ser, andwith rare p.Gly12Asp, p.Gly12Cys, andp.Gly12Glu
alleles [Kerr et al., 2006; Lo et al., 2008], which similarly are also
identified with greater frequency in tumors (Fig. 4).
Given the subtle or non-specific facial dysmorphism observed
in many of these patients, the variable presentations (such as
congenital skeletal myopathy, cardiomyopathy, or pulmonary
TABLE II. Germline Mutations Predicting p.Gly12Val in Patients
With Severe Costello Syndrome
Patient Nucleotide substitution1 c.35_36delinsTA2 c.35_36delinsTT3 c.35_36delinsTT4 c.35G> TAoki et al. [2005] (Patient COS37) c.35_36delinsTTvan der Burgt et al. [2007] (Patient 1) c.35_36delinsTT
1106 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
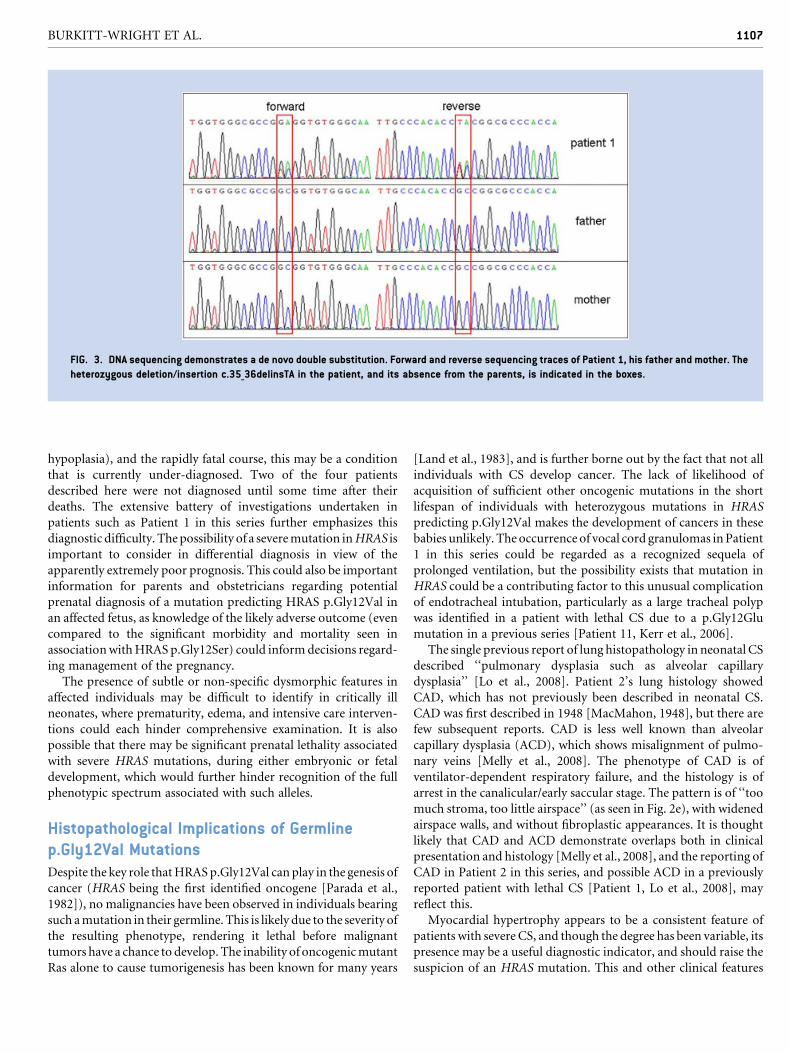
hypoplasia), and the rapidly fatal course, this may be a condition
that is currently under-diagnosed. Two of the four patients
described here were not diagnosed until some time after their
deaths. The extensive battery of investigations undertaken in
patients such as Patient 1 in this series further emphasizes this
diagnostic difficulty. Thepossibility of a severemutation inHRAS is
important to consider in differential diagnosis in view of the
apparently extremely poor prognosis. This could also be important
information for parents and obstetricians regarding potential
prenatal diagnosis of a mutation predicting HRAS p.Gly12Val in
an affected fetus, as knowledge of the likely adverse outcome (even
compared to the significant morbidity and mortality seen in
associationwithHRAS p.Gly12Ser) could inform decisions regard-
ing management of the pregnancy.
The presence of subtle or non-specific dysmorphic features in
affected individuals may be difficult to identify in critically ill
neonates, where prematurity, edema, and intensive care interven-
tions could each hinder comprehensive examination. It is also
possible that there may be significant prenatal lethality associated
with severe HRAS mutations, during either embryonic or fetal
development, which would further hinder recognition of the full
phenotypic spectrum associated with such alleles.
Histopathological Implications of Germlinep.Gly12Val MutationsDespite the key role thatHRASp.Gly12Val can play in the genesis of
cancer (HRAS being the first identified oncogene [Parada et al.,
1982]), no malignancies have been observed in individuals bearing
such amutation in their germline. This is likely due to the severity of
the resulting phenotype, rendering it lethal before malignant
tumors have a chance to develop. The inability of oncogenicmutant
Ras alone to cause tumorigenesis has been known for many years
[Land et al., 1983], and is further borne out by the fact that not all
individuals with CS develop cancer. The lack of likelihood of
acquisition of sufficient other oncogenic mutations in the short
lifespan of individuals with heterozygous mutations in HRAS
predicting p.Gly12Val makes the development of cancers in these
babies unlikely. The occurrence of vocal cord granulomas inPatient
1 in this series could be regarded as a recognized sequela of
prolonged ventilation, but the possibility exists that mutation in
HRAS could be a contributing factor to this unusual complication
of endotracheal intubation, particularly as a large tracheal polyp
was identified in a patient with lethal CS due to a p.Gly12Glu
mutation in a previous series [Patient 11, Kerr et al., 2006].
The single previous report of lung histopathology in neonatal CS
described ‘‘pulmonary dysplasia such as alveolar capillary
dysplasia’’ [Lo et al., 2008]. Patient 2’s lung histology showed
CAD, which has not previously been described in neonatal CS.
CAD was first described in 1948 [MacMahon, 1948], but there are
few subsequent reports. CAD is less well known than alveolar
capillary dysplasia (ACD), which shows misalignment of pulmo-
nary veins [Melly et al., 2008]. The phenotype of CAD is of
ventilator-dependent respiratory failure, and the histology is of
arrest in the canalicular/early saccular stage. The pattern is of ‘‘too
much stroma, too little airspace’’ (as seen in Fig. 2e), with widened
airspace walls, and without fibroplastic appearances. It is thought
likely that CAD and ACD demonstrate overlaps both in clinical
presentation and histology [Melly et al., 2008], and the reporting of
CAD in Patient 2 in this series, and possible ACD in a previously
reported patient with lethal CS [Patient 1, Lo et al., 2008], may
reflect this.
Myocardial hypertrophy appears to be a consistent feature of
patients with severe CS, and though the degree has been variable, its
presence may be a useful diagnostic indicator, and should raise the
suspicion of an HRAS mutation. This and other clinical features
FIG. 3. DNA sequencing demonstrates a de novo double substitution. Forward and reverse sequencing traces of Patient 1, his father and mother. The
heterozygous deletion/insertion c.35_36delinsTA in the patient, and its absence from the parents, is indicated in the boxes.
BURKITT-WRIGHT ET AL. 1107

common to all known patients with mutations predicting
p.Gly12Val are shown in bold in Table I. The finding of atrial
arrhythmia in two of the four patients, as is also commonly
described in other patients with CS, as well as other disorders of
the RAS-MAPK pathway such as Noonan syndrome due to RAF1
mutations [Kobayashi et al., 2010], emphasizes the importance of
such cardiac phenotypes in assisting recognitionof the presenceof a
neurocardiofaciocutaneous disorder in infants with multiple med-
ical problems. The identification of thymic atrophy in Patient 1 is
reminiscent of that reported in a postmortem of an individual
affected with cardio-facio-cutaneous (CFC) syndrome [Manci
et al., 2005], and also of the phenotype of the B-RafLSLV600E mouse
model of CFC syndrome [Urosevic et al., 2011], but at present both
the pathogenesis and effects of these abnormalities are unclear,
and they remain of uncertain significance.
Implications for Mutational Mechanisms: AnUnexplained Preponderance of DinucleotideDeletion/Insertion Mutations in HRAS CausingSevere CSThe very high proportion ofmutations identified to cause severe CS
that arise as dinucleotide deletion/insertions remains unexplained.
FIG. 4. Comparison of HRAS mutations affecting codon 12 identified in Costello syndrome and in cancers. a: HRAS codon 12 mutations identified in
cancers (recorded in the COSMICdatabase) demonstrate ahigh preponderance of substitutions predictingp.Gly12Val. b:HRAS codon12mutations in
patients with CS tested in the Manchester Regional Genetics Laboratory. The p.Gly12Val mutations described in this report are shown hatched. Note
the high preponderance of p.Gly12Sermutations, withmuch lower numbers of other substitutions, in contrast to themutations identified in cancers.
1108 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

Five of the total of six germline mutations predicting p.Gly12Val
identified to date, and also the single identifiedmutation predicting
p.Gly12Glu, are of this nature [Kerr et al., 2006]. This proportion is
in stark contrast to the extreme rarity with which dinucleotide
deletion/insertion mutations in Ras genes have been observed in
cancers, compared to the enormous number of point mutations.
It is also notable that this form of mutation appears extremely rare
in other genetic disorders: very few patients with dinucleotide
deletion/insertion mutations have been described in the literature.
A single patient with a PTPN11 mutation, c.1471_1472delinsTT
[Schuettpelz et al., 2009], of many hundreds known, and a single
patient with a SOS1 mutation, c.1300_1301delinsAA [Lepri et al.,
2011] are examples. Both of these mutations affect codons that are
recurrently mutated in Noonan syndrome, and like the HRAS
mutations that cause CS and occur in cancer, appear to result
in gain-of-function alleles. Similar dinucleotide mutations have
also, rarely, been described in FGFR2, resulting in Apert or Pfeiffer
syndrome [Oldridge et al., 1997; Kan et al., 2002]. A selective
advantage for spermatogonia bearing activatingmutations in genes
including HRAS and FGFR2 has been demonstrated [Goriely
et al., 2009], but the dramatically elevated proportion of germline
mutations predicting HRAS p.Gly12Val that are dinucleotide
deletion/insertions, as opposed to c.35G>T point mutations,
suggests that the mechanisms by which these two classes of muta-
tion are generated, and perhaps their effects upon human develop-
ment, might be distinct, and that this area requires further
investigation.
CONCLUSION
Mutations predicting severely activating HRAS proteins such as
p.Gly12Val are a recognized cause of severe CS, which frequently
has a presentation that is lethal in the neonatal period, though the
common CS p.Gly12Ser mutation has also been described occa-
sionally in similarly severely affected individuals. Recognition of
this condition may be hindered by the extreme illness of these
babies, and their rapid demise. Consideration should be given to
the possibility of severe CS as the cause of a range of presentations
in very sick neonates, especially those born prematurely as a
consequence of extreme polyhydramnios. Identification of this
condition in utero may also be possible in certain cases, and could
be confirmed by prenatal diagnosis.Hypertrophic cardiomyopathy
appears to be a consistent feature, along with hypotonia due
to skeletal muscle weakness. Other features of CS, such as atrial
arrhythmia, or suggestive dysmorphic features,may also bepresent.
The unusual observation of dinucleotide deletion/insertions
as a very high proportion of mutations causing severe CS, and
its contrast with the mutational spectrum observed in cancers,
suggest that differences may exist in the mutational mechanisms
at work in the two contexts.
ACKNOWLEDGMENTS
EBW is a Wellcome Trust clinical research training fellow,
supported by the UK National Institute for Health Research’s
Manchester Biomedical Research Centre.
REFERENCES
Aoki Y, Niihori T, KawameH, KurosawaK,OhashiH, Tanaka Y, FilocamoM, Kato K, Suzuki Y, Kure S, et al. 2005. Germline mutations in HRASproto-oncogene cause Costello syndrome. Nat Genet 37:1038–1040.
de BoodeWP, Semmekrot BA, ter LaakHJ, van der Burgt CJAM,DraaismaJMT, Lommen EJP, Sengers RCA, van Wijk-Hoek JM. 1996. Myo-pathology in patients with a Noonan phenotype. Acta Neuropathol92:597–602.
Emery JL,Mithal A. 1960. Thenumber of alveoli in the terminal respiratoryunit of man during late intrauterine life and childhood. Arch Dis Child35:544–547.
Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ,Pfeifer SP,McVeanGA,Rajpert-DeMeyts E,WilkieAO. 2009. Activatingmutations in FGFR3 and HRAS reveal a shared genetic origin forcongenital disorders and testicular tumors. Nat Genet 41:1247–1252.
Gripp KW, Hopkins E, Sol-Church K, Stabley DL, Axelrad ME, Doyle D,Dobyns WB, Hudson C, Johnson J, Tenconi R, et al. 2011. Phenotypicanalysis of individuals with Costello syndrome due to HRAS p.G13C.Am J Med Genet A 155A:706–716.
Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW,Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, et al. 2002. Genomicscreening of fibroblast growth-factor receptor 2 reveals a wide spectrumof mutations in patients with syndromic craniosynostosis. Am J HumGenet 70:472–486.
Kerr B. 2009. The Clinical phenotype of Costello syndrome. In: ZenkerM,editor. Noonan syndrome and related disorders. Monogr Hum Genet,vol 17. Basel: Karger. pp 83–93.
Kerr B, Delrue M-A, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M,Tang B, Eden OB, O’Sullivan JO, et al. 2006. Genotype-phenotypecorrelation in Costello syndrome: HRAS mutations analysis in 43 cases.J Med Genet 43:401–405.
Kobayashi T, Aoki Y, Niihori T, Cav�e H, Verloes A, Okamoto N, KawameH, Fujiwara I, Takada F, Ohata T, et al. 2010. Molecular and clinicalanalysis of RAF1 in Noonan syndrome and related disorders: Dephos-phorylation of serine 259 as the essential mechanism for mutant acti-vation. Hum Mutat 31:284–294.
Land H, Parada LF, Weinberg RA. 1983. Tumorigenic conversion ofprimary embryo fibroblasts requires at least two co-operating oncogenes.Nature 304:596–602.
Langston C, DishopMK. 2009. Diffuse lung disease in infancy: A proposedclassification applied to 259 diagnostic biopsies. Pediatr Dev Pathol12:421–437.
Lepri F,De LucaA, Stella L, Rossi C, BaldassarreG, Pantaleoni F, CordedduV, Williams BJ, Dentici ML, Caputo V, et al. 2011. SOS1 mutations inNoonan syndrome: Molecular spectrum, structural insights on patho-genic effects, and genotype-phenotype correlations. Hum Mutat 32:760–772.
Lo IF, Brewer C, Shannon N, Shorto J, Tang B, Black G, Soo MT, Ng DK,Lam ST, Kerr B. 2008. Severe neonatal manifestations of Costellosyndrome. J Med Genet 45:167–171.
MacMahon HE. 1948. Congenital alveolar dysplasia of the lungs. Am JPathol 24:919–931.
Manci EA, Martinez JE, Horenstein MG, Gardner TM, Ahmed A, MancaoMC, Gremse DA, Gardner DM, Nimityongskul P, Maertens P, et al.2005. Cardiofaciocutaneous syndrome (CFC)with congenital peripheralneuropathy and nonorganic malnutrition: An autopsy study. Am J MedGenet A 137A:1–8.
Melly L, Sebire NJ, Malone M, Nicholson AG. 2008. Capillary appositionand density in the diagnosis of alveolar capillary dysplasia. Histopathol-ogy 53:450–457.
BURKITT-WRIGHT ET AL. 1109

Oldridge M, Lunt PW, Zackai EH, McDonald-McGinn DM, Muenke M,MoloneyDM,Twigg SR,Heath JK,HowardTD,HogansonG, et al. 1997.Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol Genet 6:137–143.
Parada LF, Tabin CJ, Shih C, Weinberg RA. 1982. Human EJ bladdercarcinoma oncogene is homologue of Harvey sarcoma virus ras gene.Nature 297:474–478.
Schuettpelz LG, McDonald S, Whitesell K, Desruisseau DM, Grange DK,Gurnett CA, Wilson DB. 2009. Pilocytic astrocytoma in a child withNoonan syndrome. Pediatr Blood Cancer 53:1147–1149.
Urosevic J, Sauzeau V, Soto-Montenegro ML, Reig S, Desco M, BurkittWright EM, Ca~namero M, Mulero F, Ortega S, Bustelo XR, et al. 2011.Constitutive activation of B-Raf in themouse germ line provides amodelfor human cardio-facio-cutaneous syndrome. Proc Natl Acad Sci USA108:5015–5020.
van der Burgt I, KupskyW, Stassou S, Nadroo A, Barroso C, DiemA, KratzCP, Dvorsky R, Ahmadian MR, Zenker M. 2007. Myopathy caused byHRAS germline mutations: Implications for disturbed myogenic differ-entiation in the presence of constitutive HRas activation. J Med Genet44:459–462.
1110 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

ORIGINAL ARTICLE
Can the diagnosis of NF1 be excluded clinically?A lack of pigmentary findings in families with spinalneurofibromatosis demonstrates a limitation ofclinical diagnosisEmma MM Burkitt Wright,1,2 Emma Sach,2 Saba Sharif,3 Oliver Quarrell,4
Thomas Carroll,5 Richard W Whitehouse,6 Meena Upadhyaya,7 Susan M Huson,1,2
D Gareth R Evans1,2
1Genetic Medicine ResearchGroup, Faculty of Medical andHuman Sciences, Institute ofHuman Development,University of Manchester,Manchester, UK2Department of GeneticMedicine, St Mary’s Hospital,Central Manchester UniversityHospitals NHS FoundationTrust, Manchester AcademicHealth Science Centre,Manchester, UK3West Midlands RegionalGenetics Service, BirminghamWomen’s Hospital,Birmingham, UK4Sheffield Clinical GeneticsService, Sheffield Children’sHospital, Sheffield, UK5Department of Neurosurgery,Royal Hallamshire Hospital,Sheffield, UK6Department of Radiology,Manchester Royal Infirmary,Central Manchester UniversityHospitals NHS FoundationTrust, Manchester AcademicHealth Science Centre,Manchester, UK7Department of MedicalGenetics, Cardiff UniversitySchool of Medicine, Institute ofMedical Genetics Building,Cardiff, UK
Correspondence toProfessor D Gareth R Evans,Department of GeneticMedicine, St Mary’s Hospital,Oxford Road, Manchester M139WL, UK; [email protected]
Received 6 March 2013Revised 25 May 2013Accepted 28 May 2013Published Online First28 June 2013
To cite: BurkittWright EMM, Sach E,Sharif S, et al. J Med Genet2013;50:606–613.
ABSTRACTBackground Consensus clinical diagnostic criteria forneurofibromatosis type I (NF1) include café-au-laitmacules and skinfold freckling. The former are frequentlythe earliest manifestation of NF1, and as such are ofparticular significance when assessing young children atrisk of the condition. A phenotype of predominantlyspinal neurofibromatosis has been identified in a smallminority of families with NF1, often in association with arelative or absolute lack of cutaneous manifestations.An association with splicing and missense mutations haspreviously been reported for spinal neurofibromatosis,but on the basis of molecular results in only a fewfamilies.Method Patients with spinal NF1 were identifiedthrough the Manchester nationally commissioned servicefor complex NF1.Results Five families with spinal NF1 were identified,with a broad spectrum of NF1 mutations, providingfurther evidence that this phenotype may arise inassociation with any genre of mutation in this gene.Pigmentary manifestations were absent or very mild inaffected individuals. Several further affected individuals,some with extensive spinal root tumours, wereascertained when additional family members wereassessed.Conclusions Clinical NF1 consensus criteria cannot beused to exclude the diagnosis of spinal NF1, especiallyin childhood. This emphasises the importance ofmolecular confirmation in individuals and families withatypical presentations of NF1.
INTRODUCTIONNeurofibromatosis 1 (NF1) is an autosomal domin-ant condition caused by mutations in the NF1 geneon chromosome 17.1 Considerable inter- and intra-familial variability can complicate clinical ascertain-ment of affected individuals.2 Consensus criteria forthe clinical diagnosis of NF1 are well established,3
but these may not be effective in very young chil-dren, especially where there is no family history ofthe disorder. Additional criteria such as T2 hyperin-tensities on MRI of the brain in childhood4 havebeen suggested, but these too are not universallypresent. Notwithstanding these limitations, a defini-tive clinical diagnosis can be made for a large
proportion of patients with NF1. In contrast, pro-cesses for molecular confirmation of NF1 have beencomplicated by the very large size of the gene, itshomology with many pseudogenes, and a lack ofmutational hotspots. Very limited evidence forgenotype–phenotype correlation exists, in keepingwith the likelihood of haploinsufficiency as themolecular mechanism causing pathogenicity for themajority of mutations identified to date. Exceptionsto this include the relatively distinct phenotype ofpatients with whole gene deletions of NF1,5 wherethe altered genomic landscape may play a role, andthe very mild café au lait (CAL) only pattern inpatients with a common three base pair in framedeletion.6 More recent evidence of association ofspecific groups of mutations with optic pathwayglioma7 and pulmonary stenosis8 has also emerged.Situations in which molecular confirmation is
absolutely necessary, such as for prenatal or pre-implantation genetic diagnosis, have to date beenrelatively rarely encountered in NF1. However, therecognition of disorders with overlapping clinicalor radiological phenotypes but differing prognoses,including schwannomatosis9 10 and Legius syn-drome,11 12 emphasise the importance of moleculardiagnosis in an increasing spectrum of patients withNF1 and related disorders.Within the NF1 population, various subtypes of
disease have now emerged, including groups of fam-ilies where several individuals have a high load ofspinal tumours (‘spinal NF1’). As is the case formost patients with NF1, these families have gener-ally been found to have private mutations, but anassociation between the spinal phenotype andsplice and missense variants has previously been sug-gested.13 Five further families are described here inwhich affected individuals have a high spinaltumour load, due to a range of mutations in NF1.Minimal pigmentary manifestations were present inaffected people, complicating clinical diagnosis andmaking it very difficult to reassure at-risk individualsin these pedigrees. This circumstance emphasises thevalue of molecular testing for definitive identifica-tion of individuals at high risk of problematic neuro-fibromas who will require early investigation ofany spinal symptoms in particular, and appropriateMRI surveillance. Clinical presentations, molecular
Open AccessScan to access more
free content
606 Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648
Genotype-phenotype correlations
group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from group.bmj.com on October 30, 2013 - Published by jmg.bmj.comDownloaded from

analysis, and the implications of these findings for families withabsent or non-diagnostic pigmentary features of NF1 arediscussed.
METHODSAffected individuals were ascertained through the Manchesternationally commissioned service for complex NF1 and exam-ined (by DGRE, SMH or OQ) in its specialist clinics (based inManchester). Where indicated, MRI scans of the neuroaxis and/or whole body were carried out, and interpreted along with theclinical phenotypes in the context of a multidisciplinary teamdiscussion. Permission for inclusion of clinical and imagingdetails was sought from the patients reported here.
Molecular analyses were carried out in the clinical pathologyaccredited Manchester Regional Genetics Laboratory and inthe Genetics Research and Development laboratory in Cardiff.RNA and genomic DNA were prepared from peripheral bloodsamples. RNA was reverse transcribed to cDNA using standardprocedures, and direct sequencing performed to demonstratesplicing abnormalities or mutations within the coding sequence.Mutation status was confirmed in genomic DNA. Multiplex liga-tion dependent probe amplification (MLPA) for dosage analysiswas additionally performed in samples without a clearly patho-genic mutation; for example, where a novel sequence variantwith uncertain pathogenicity had been identified.
FAMILY 1This family had individuals affected across at least four genera-tions, as shown in figure 1A. The proband, IV:5, presented aged8 years in New Zealand, when a neurofibroma had been excisedfrom his right arm. At the age of 20 years he developed diffi-culty in walking, pain in his right leg, and occasional paraesthe-sia in his hands. Peripheral subcutaneous neurofibromas, someof which were painful, a thoracolumbar scoliosis, and a neuro-logical deficit in the lower limbs were identified on examination.Serial MRI showed progression of multiple peripheral nerveneurofibromas in his legs and bilaterally in his cervical spine.From the age of 21 he underwent excision of multiple symp-tomatic cervical spine neurofibromas. At 23, multiple subcutane-ous lesions were excised from his right thigh, and wereconfirmed as benign neurofibromas with plexiform elements.Skin examination at age 25 confirmed just one CAL macule, sixsubcutaneous neurofibromas, and one cutaneous neurofibroma.Lisch nodules were noted, but neither axillary nor inguinalfreckling were present.
IV:5’s mother, III:5, was examined in the genetics departmentat the age of 50 years. She had no CAL, and skinfold changeswere also absent apart from a few unilateral axillary freckles.Multiple painful subcutaneous neurofibromas, a plexiformneurofibroma, spinal neurofibromas, scoliosis, and Lischnodules were all present.
III:2 was diagnosed with NF1 in her third decade, when alump excised from her mouth was found to be a neurofibroma.In each of her three pregnancies (at 26–30 years of age), shedeveloped new cutaneous neurofibromas. She was first reviewedin the genetics department at the age of 52, and painful cutane-ous and subcutaneous neurofibromas were noted which weresubsequently histologically confirmed. She had no skin pigmen-tary changes nor any symptomatic spinal tumours, and thereforeMRI imaging was not performed at that time.
III:2’s three sons, IV:2, IV:3, and IV:4, were each examinedon account of their 50% risk of NF1. Two each had one CALdocumented in childhood and one additionally had three sub-cutaneous nodules which subsequently disappeared. No other
pigmentary changes, including Lisch nodules, were identified inany of these individuals. They have remained well with no signsof NF1, but this diagnosis could not be confidently excludedclinically until they were well into adulthood, given the minimalpigmentary findings of the condition in other family members,particularly their mother.
III:1 had a longstanding diagnosis of NF1 on the basis of sub-cutaneous neurofibromas, several of which had been removedand histologically analysed; his daughter (IV:1) also had CALfrom early life. He developed deafness and had a brain scan atthe age of 54 years. No cause for deafness was identified, but anasymptomatic cervical spinal neurofibroma with an intraduralcomponent was seen, with bilateral impingement on theC2 nerve roots. This required surgery, due to the risk of spinalcord compression. On examination at the age of 62, over 100cutaneous and subcutaneous neurofibromas were present, aswere axillary freckles and Lisch nodules. In contrast, only sevenCAL (three of which required ultraviolet light for visualisation)were identified.
III:1’s daughter, IV:1, was diagnosed in early childhood withNF1 on account of her family history and presence of CAL. Atthe age of 7 years, she had no learning difficulties and only fouror five CAL patches, but she also had depigmented areas of skinover her back. By 22 years of age, she had six CAL, minimalaxillary and inguinal freckling, and a small number of histologi-cally proven neurofibromas.
II:2, III:5’s late father, was reported as having a severe disfig-urement due to skin lumps, and had died from what was highlylikely to have been a malignant peripheral nerve sheath tumour(MPNST). His mother (I:1) was also thought by the family tohave been affected with NF1, as her skin was covered in lumps.It was also reported that his late father (I:2, a first cousin of I:1)could have been similarly affected.
For all living affected individuals in this family, a diagnosis ofNF1 could be made by application of the clinical consensus cri-teria. However, this was only possible for many of them wellinto adulthood, due to both the paucity of pigmentary manifes-tations, and the late onset of visible or symptomatic tumours inmost individuals. What was not clinically straightforward wasto exclude NF1 in at-risk individuals. This was a majorsource of anxiety for several family members, who wereworried about risks to their offspring of this disabling condition.Pre-implantation genetic diagnosis was considered by IV:5 andhis wife due to the burdensome nature of his own and hismother’s (III:5) condition, but was not available when he andhis wife were starting a family. After extensive considerationwithin the family, his two daughters were each tested at birthfor the recently identified familial mutation, c.6364+2T>G,which had been found to result in exclusion of exon 33 and asubsequent frameshift resulting in a prematurely terminatedprotein. They were found to be unaffected.
FAMILY 2The proband, I:1 (figure 1B), was seen in the genetics clinic atthe age of 30 years after identification of multiple nerveroot tumours. She had suffered back pain, predominantly in thecervical and lumbar regions, since puberty, which worsened ineach of her three pregnancies and was causing significant disabil-ity. She was under follow-up in the pain clinic which had pre-scribed gabapentin, amitriptyline, and tramadol. MRI scans(figure 2A,B) demonstrated mild thoracic scoliosis and enlarge-ment of multiple segmental nerve roots from C5 to T1 andfrom L2 to S1. Degenerative disc change was also noted at L4/L5. Vestibular schwannomas were absent. One deep
Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648 607
Genotype-phenotype correlations

neurofibroma was present in the left thigh, and one subcutane-ous neurofibroma on the right shoulder. Her height was 173 cm(91st–98th centile) and her head circumference was 56.8 cm(75th–91st centile). No CAL, freckles or plaque skin change(the last as seen in NF2) were present. Three to four Lischnodules were seen in the right eye, and one in the left. The dif-ferential diagnosis on clinical and radiological grounds restedbetween schwannomatosis and NF1. A frameshift NF1 muta-tion, c.5993dupC, resulting in a premature termination codon
10 residues downstream, p.(Thr1999Asnfs*10), was identifiedin her lymphocyte DNA.
I:1’s three children were each assessed in clinic regarding thelikelihood of being affected with NF1. II:1 and II:3 each hadfeatures of attention deficit hyperactivity disorder (ADHD) andlearning delay (at the ages of 8 and 5 years, respectively),with II:3 also having a history of hypotonia and coordinationdifficulties. All head circumferences were around the 50thcentile. II:2 (aged 6) had one CAL, but no other pigmentary
Figure 1 (A–E) Pedigrees offamilies 1–5.
608 Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648
Genotype-phenotype correlations
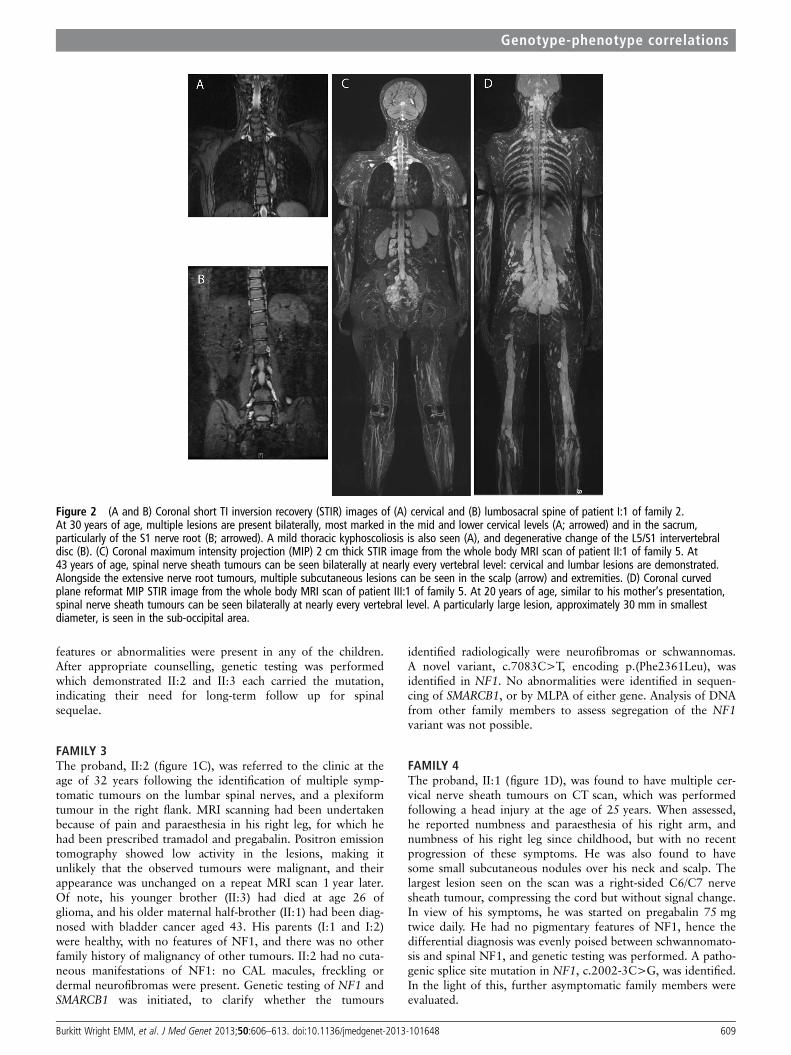
features or abnormalities were present in any of the children.After appropriate counselling, genetic testing was performedwhich demonstrated II:2 and II:3 each carried the mutation,indicating their need for long-term follow up for spinalsequelae.
FAMILY 3The proband, II:2 (figure 1C), was referred to the clinic at theage of 32 years following the identification of multiple symp-tomatic tumours on the lumbar spinal nerves, and a plexiformtumour in the right flank. MRI scanning had been undertakenbecause of pain and paraesthesia in his right leg, for which hehad been prescribed tramadol and pregabalin. Positron emissiontomography showed low activity in the lesions, making itunlikely that the observed tumours were malignant, and theirappearance was unchanged on a repeat MRI scan 1 year later.Of note, his younger brother (II:3) had died at age 26 ofglioma, and his older maternal half-brother (II:1) had been diag-nosed with bladder cancer aged 43. His parents (I:1 and I:2)were healthy, with no features of NF1, and there was no otherfamily history of malignancy of other tumours. II:2 had no cuta-neous manifestations of NF1: no CAL macules, freckling ordermal neurofibromas were present. Genetic testing of NF1 andSMARCB1 was initiated, to clarify whether the tumours
identified radiologically were neurofibromas or schwannomas.A novel variant, c.7083C>T, encoding p.(Phe2361Leu), wasidentified in NF1. No abnormalities were identified in sequen-cing of SMARCB1, or by MLPA of either gene. Analysis of DNAfrom other family members to assess segregation of the NF1variant was not possible.
FAMILY 4The proband, II:1 (figure 1D), was found to have multiple cer-vical nerve sheath tumours on CT scan, which was performedfollowing a head injury at the age of 25 years. When assessed,he reported numbness and paraesthesia of his right arm, andnumbness of his right leg since childhood, but with no recentprogression of these symptoms. He was also found to havesome small subcutaneous nodules over his neck and scalp. Thelargest lesion seen on the scan was a right-sided C6/C7 nervesheath tumour, compressing the cord but without signal change.In view of his symptoms, he was started on pregabalin 75 mgtwice daily. He had no pigmentary features of NF1, hence thedifferential diagnosis was evenly poised between schwannomato-sis and spinal NF1, and genetic testing was performed. A patho-genic splice site mutation in NF1, c.2002-3C>G, was identified.In the light of this, further asymptomatic family members wereevaluated.
Figure 2 (A and B) Coronal short TI inversion recovery (STIR) images of (A) cervical and (B) lumbosacral spine of patient I:1 of family 2.At 30 years of age, multiple lesions are present bilaterally, most marked in the mid and lower cervical levels (A; arrowed) and in the sacrum,particularly of the S1 nerve root (B; arrowed). A mild thoracic kyphoscoliosis is also seen (A), and degenerative change of the L5/S1 intervertebraldisc (B). (C) Coronal maximum intensity projection (MIP) 2 cm thick STIR image from the whole body MRI scan of patient II:1 of family 5. At43 years of age, spinal nerve sheath tumours can be seen bilaterally at nearly every vertebral level: cervical and lumbar lesions are demonstrated.Alongside the extensive nerve root tumours, multiple subcutaneous lesions can be seen in the scalp (arrow) and extremities. (D) Coronal curvedplane reformat MIP STIR image from the whole body MRI scan of patient III:1 of family 5. At 20 years of age, similar to his mother’s presentation,spinal nerve sheath tumours can be seen bilaterally at nearly every vertebral level. A particularly large lesion, approximately 30 mm in smallestdiameter, is seen in the sub-occipital area.
Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648 609
Genotype-phenotype correlations

In II:1’s father, I:1, no CAL marks or other cutaneous stig-mata of NF1 were present, but MRI scanning of his spinedemonstrated multiple lesions of the lumbar nerve roots, mostprominently a mass at the right side of L5 with secondarylumbar canal stenosis at L4/5. Foraminal and extraforaminalportions of other lumbar nerve roots also appeared thickened.
On clinical examination, II:1’s sister, II:2, was found to haveseveral CAL patches on her back and neck, and some axillaryfreckling, but no neurofibromata. Results of MRI scanning ofher spine were awaited. A further sibling was also assessed, butwas not available to ask for permission for publication of clin-ical details. I:1’s sister, I:2, was understandably anxious aboutthe risk of NF1 to herself and her children. She opted for pre-dictive genetic testing, and it was demonstrated that she did notcarry the familial mutation.
FAMILY 5The proband, II:1 (figure 1E), developed multiple subcutaneousnodules from her teenage years. At the age of 26 years, she hada large lesion, reportedly a neurofibroma, excised from the rightposterior aspect of her neck. She subsequently had excisions ofseveral other subcutaneous lesions from her back, but remainedgenerally well until the age of 31, when she presented withincreasing right leg and groin pain. Spinal MRI showed tumourspresent on nerve roots throughout the neuroaxis, particularlyaround the cauda equina, and a large retroperitoneal mass.Surgical intervention was not indicated for any of these, andshe remained under surveillance with annual serial MRI scans.A possible diagnosis of neurofibromatosis type II (NF2) wasraised when a possible lesion at the CPA was identified at theage of 39, which was associated with right-sided vestibularsymptoms and tinnitus. However, pure tone audiometry wasnormal, no CPA abnormality was seen on interval MRI, and hersymptoms did not progress. At the age of 44, she was reviewedin the genetic clinic. She had a very extensive burden of nervesheath tumours, involving the large majority of spinal nerveroots (figure 2C), but no CAL patches or Lisch nodules.Freckling was present across the upper trunk and in the axillae.The absence of CPA abnormality indicated that the diagnosis ofNF2 could be confidently excluded, and the previous clinicalimpression of spinal NF1 was confirmed. Sequencing oflymphocyte DNA demonstrated a previously reported patho-genic mutation in NF1, c.2543G>A, predicting p.(Gly848Glu).Her body habitus was typical for NF1, with short stature of146 cm (<0.4th centile), some facial features suggestive ofNoonan syndrome, and a relatively large head circumference(59 cm, >97th centile).
Her mother, I:1, had a subcutaneous lump, histologicallymost consistent with a plexiform neurofibroma, removed fromher back at the age of 62. She had no pigmentary changes inher skin nor Lisch nodules, but carried her daughter’s mutationin lymphocyte DNA. I:1’s mother had also had a lumpremoved, and died at a young age, but this history could not befurther elucidated.
II:1’s son, III:1, was reviewed at the age of 20, in light of his50% risk of spinal neurofibromatosis. He was asymptomatic andof normal stature (168 cm), with a normal head circumferenceof 57.5 cm, and had no CAL patches, axillary freckling or sub-cutaneous neurofibromas. No Lisch nodules were present, buthe did have some facial features consistent with NF1/Noonansyndrome and he also had pectus carinatum. He had discretelesions of approximately 1 cm in diameter palpable in the leftanterior triangle of the neck. He was tested within a predictivetesting protocol for the NF1 mutation identified in his mother,
and found to carry this. Whole body MRI (figure 2D) showedan extensive tumour burden, including a particularly large lobu-lated lesion in the occipital/posterior neck region (figure 2D).Despite his slim build, this was not palpable. An arachnoid cystof the left middle cranial fossa was also demonstrated.
RESULTSClinical features of affected individuals are summarised intable 1, and the mutations identified in each family are shown intable 2.
DISCUSSIONSpinal tumours are common in NF1, but appear to be onlyrarely symptomatic. However, there is a relative paucity of clin-ical literature regarding this. Thakkar et al16 identified suchtumours in 40% of 30 asymptomatic patients with NF1,but only 2% of people in their large cohort (n=1400, aged5–56 years) had developed symptomatic spinal tumours, andlong term clinical follow-up data of such lesions are lacking.Individuals with symptomatic spinal tumours and typical cutane-ous features of NF1 do not appear more likely than unselectedNF1 patients to have a family history of the disorder.Conversely, multigenerational familial clustering of the spinalNF1 phenotype with few cutaneous features is well recog-nised,17 suggesting that individuals carrying mutations causingthis phenotype may be more likely than the NF1 population ingeneral to have children. Missense mutations have been highlyoverrepresented in the variants reported in association withspinal NF1 (nine of 15 mutations included in the humangenome mutation database18), and a significant further propor-tion have been substitutions affecting splicing.13 19 20 It hastherefore been postulated that the different clinical phenotypeobserved in these families could be a result of milder moleculareffects of these mutations,17 but tissue specific effects may alsobe of importance. In support of the latter hypothesis, a rarefamilial phenotype in which affected individuals had optic nerveglioma, other central nervous system (CNS) tumour, or both,21
has been reported in association with a splice donor site muta-tion in NF1, resulting in skipping of exon 29. At least two indi-viduals with CNS tumours who carried this mutation did nothave typical skin manifestations of NF1, but others did.Differential tissue specific effects for splicing of exon 29 of NF1have also been suggested by the work of Park et al,22 withexpression of certain transcripts including this exon beinglimited to brain, of the tissues examined. This phenomenoncould be a key contributing factor to this family’s unusualphenotype. Such an example further highlights the importanceof mutational analysis, particularly at the RNA level, in familieswith atypical NF1 phenotypes. The data from our series broadlysupport the hypothesis that splice and missense variants are themain causes of a spinal limited NF1 phenotype (as observed infour of five families described here), but that frameshift muta-tions may also, rarely, result in this phenotype, as has previouslybeen reported.23 Similarly, Pizzuti et al24 reported a multi-exondeletion, resulting in nonsense mediated decay of the transcript,in a family with spinal neurofibromatosis where a 73-year-oldman was only found to be affected, with typical spinal MRIfindings, when his daughter was diagnosed.
The late onset of symptomatic manifestations of spinal NF1has several implications for clinical practice. Tumour risk, eitherby malignant transformation of neurofibromas, or due to devel-opment of other neoplasia, has not yet been satisfactorily estab-lished in the small cohort of patients diagnosed with familialspinal NF1, though internal tumour burden is a well recognised
610 Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648
Genotype-phenotype correlations

Table 1 Clinical features in affected individuals
Family Family 1 Family 2 Family 3 Family 4 Family 5
Individual III:1 IV:1 III:2 III:5 IV:5 I:1 II:2 II:3 II:2 II:1 I:1 II:2 I:1 II:1 III:1
Gender; age at most recentreview (year)
Male; 70(deceased)
Female; 23 Female;52
Female; 50 Male; 25 Female; 30 Female;6
Male; 5 Male; 33 Male; 26 Male; 61 Female; 32 Female;66
Female; 44 Male; 20
OFC, cm (centile) 59.0 (90th) 57.0(90th)
55.0(50th)
54.3(25th)
56.4(50th)
56.8(75th–91st)
n/k 52.5(50th–75th)
n/k n/k 60.5 n/k n/k 59.0(>97th)
57.5(50th–75th)
Height, cm (centile) 162 (2nd) n/k 156 (9th) 158 (25th) 167 (9th) 173(91st–98th)
n/k 105 (25th) Normalrange
170(2nd–9th)
173(9th–25th)
n/k n/k 146(<0.4th)
168(2nd–9th)
CAL 7 6 None None 1 None 1 None None None None Yes,multiple
None None None
Axillary or inguinal freckling Yes Few None None None None (None) (None) None None None Yes,axillary
None Yes None
Subcutaneous neurofibroma Yes Few Several 2 Yes Yes (None) (None) None Few None None None Yes NoneCutaneous neurofibroma Yes None Many Several Several No (None) (None) None Yes None None None Yes NonePlexiform neurofibroma None None None Yes, left
clavicleYes, leftthigh
Yes, leftthigh
None None Yes, rightflank
None None None One None None
Histological verification Yes Yes Yes Yes Yes – n/a n/a – – – – Yes – n/aLisch nodules 2, unilateral n/k Bilateral Bilateral Bilateral Yes n/k None n/k no n/k n/k No No Few, bilateralSpinal neurofibroma Yes n/k No Yes Yes Yes n/k n/k Multiple Multiple Multiple n/k Not
scannedMultiple Multiple
Scoliosis No No n/k Yes Yes Yes No No No No n/k n/k No No NoLearning difficulty No Mild Mild No No No No Mild No No Dyslexia n/k No No NoNIH diagnostic criteria met(before spinal investigations)(at age)
Yes (adult) Yes(22 years)
Yes(adult)
Yes (adult) Yes(20 years)
Yes(30 years)
No(6 years)
No (5 years) No(33 years)
No(26 years)
No(61 years)
Yes(32 years)
No(66 years)
Yes(26 years)
No (20 years)
CAL, café au lait macule; n/a, not applicable; n/k, not known; NIH, National Institutes of Health; OFC, occipitofrontal circumference.
BurkittWrightEM
M,etal.J
Med
Genet2013;50:606
–613.doi:10.1136/jmedgenet-2013-101648
611
Genotype-phenotype
correlations

risk factor for development of MPNST.25 Late onset of the NF1phenotype may be an important contributor to the difficulty inestablishing such risks. The large burden of morbidity identifiedto date in this patient group is due to the effects of non-malignant spinal tumours, with high rates of pain, reducedmobility and paraparesis. Patients with NF1 and a heavy internalburden with spinal root involvement represent a considerablemanagement problem. In the future it is likely that they will betreated with pathogenesis based medical therapies that are nowbeing shown to be effective in mouse models.26
Many of the affected patients reported here endured severalyears of such symptoms before the diagnosis was suspected. Asthe first affected individual in a family, this may be unavoidableif no further stigmata of NF1 are evident. Increasing profes-sional awareness of the possibility of the diagnosis in patientswith subtle or few other manifestations of NF1 is important inorder to optimise management, as neurosurgical outcomes maybe improved by presymptomatic intervention in affected indivi-duals. Of note, due to the predominantly extra-axial location ofthe tumours, an extensive tumour burden may be present inasymptomatic patients, and appropriate counselling regardingthis should be undertaken before MRI scanning of these indivi-duals. The frequently late onset of symptoms also means thataffected individuals may have children of their own before theirgenetic risk becomes apparent. Predictive genetic testing proto-cols may therefore be important for asymptomatic at-risk indivi-duals in these families, as the usual clinical assessment inchildhood cannot be relied upon to confirm or refute the diag-nosis. Diagnostic testing of symptomatic individuals is importantfor molecular confirmation, and may obviate the need for surgi-cal biopsy for histological confirmation of the diagnosis, againbenefiting patient care.
CONCLUSIONThe spinal phenotype of NF1, without associated pigmentarymanifestations, can occur in association with a range of muta-tions in NF1. Molecular testing of the gene is warrantedin patients with atypical presentations such as possible spinalNF1, particularly as this diagnosis may not be clinically orradiologically distinguishable from related conditions such asschwannomatosis.
Acknowledgements DGRE is an NIHR senior investigator. This work wasfacilitated by the Manchester Biomedical Research Centre and the GreaterManchester Comprehensive Local Research Network.
Contributors EMMBW, DGRE, SS and SH wrote the body of the paper. DGRE, SH,EMMBW, SS, OQ and TC generated the clinical data. ES and MU were responsiblefor the molecular testing of NF1, and RWW performed and interpreted the imagingstudies. All authors have reviewed and edited this manuscript.
Funding EMMBW is a Wellcome Trust funded Clinical Research Training Fellow(090120).
Competing interests None.
Patient consent Obtained.
Ethics approval This is a retrospective case series. All of the data included wasobtained in the course of clinical care.
Provenance and peer review Not commissioned; externally peer reviewed.
Open Access This is an Open Access article distributed in accordance with theCreative Commons Attribution Non Commercial (CC BY-NC 3.0) license, whichpermits others to distribute, remix, adapt, build upon this work non-commercially,and license their derivative works on different terms, provided the original work isproperly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
REFERENCES1 Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M,
Carey J, Copeland NG, Jenkins NA, White R, O’Connell P. Deletions and atranslocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell1990;62:187–92.
2 Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation inexpression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes.Am J Hum Genet 1993;53:305–13.
3 Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M,Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis andmanagement of individuals with neurofibromatosis 1. J Med Genet 2007;44:81–8.
4 Sabol Z, Resic B, Juraski R Gjergja, Sabol F, Sizgoric M Kovac, Orsolic K, Ozretic D,Sepic-Grahovac D. Clinical sensitivity and specificity of multiple T2-hyperintensitieson brain magnetic resonance imaging in diagnosis of neurofibromatosis type 1 inchildren: diagnostic accuracy study. Croat Med J 2011;52:488–96.
5 Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, Stephens K.Deletions spanning the neurofibromatosis 1 gene: identification and phenotype offive patients. Am J Hum Genet 1994;54:424–36.
6 Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S,Evans DG, Evans DG, Howard E, Kerr B, Griffiths S, Consoli C, Side L, Adams D,Pierpont M, Hachen R, Barnicoat A, Li H, Wallace P, Van Biervliet JP, Stevenson D,Viskochil D, Baralle D, Haan E, Riccardi V, Turnpenny P, Lazaro C, Messiaen L.An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion inexon 17 of the NF1 gene (c.2970–2972 delAAT): evidence of a clinically significantNF1 genotype-phenotype correlation. Am J Hum Genet 2007;80:140–51.
7 Sharif S, Upadhyaya M, Ferner R, Majounie E, Shenton A, Baser M, Thakker N,Evans DG. A molecular analysis of individuals with neurofibromatosis type 1 (NF1)and optic pathway gliomas (OPGs), and an assessment of genotype-phenotypecorrelations. J Med Genet 2011;48:256–60.
8 Ben-Shachar S, Constantini S, Hallevi H, Sach EK, Upadhyaya M, Evans GD,Huson SM. Increased rate of missense/in-frame mutations in individuals withNF1-related pulmonary stenosis: a novel genotype-phenotype correlation. Eur J HumGenet 2013;21:535–9.
9 Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F,Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis.Am J Hum Genet 2007;80:805–10.
10 Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR. Clinicalfeatures of schwannomatosis: a retrospective analysis of 87 patients. Oncologist2012;17:1317–22.
11 Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R,Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A,
Table 2 NF1 mutations identified in the five families
FamilyNF1 genotype(NM_001042492.2)
Exon/intron Effect of mutation
Previously identified in other families withNF1?
1 c.6427+2T>G Intron 42 Splice error: transcript demonstrated exclusion of exon 43 andsubsequent frameshift
No
2 c.6056dup Exon 41 Frameshift mutation resulting in premature termination codon, p.(Thr1999Asnfs*10)
No
3 c.7146C>A Exon 48 p.(Phe2382Leu) missense substitution No; segregation data not available4 c.2002−3C>G Intron 17 Splice error: transcript demonstrated exclusion of exon 18
(r.2002_2251del)No
5 c.2543G>A Exon 21 p.(Gly848Glu) missense substitution Yes14 ; spinal phenotype also reported withp.(Gly848Arg)15
612 Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648
Genotype-phenotype correlations

Legius E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis1-like phenotype. Nat Genet 2007;39:1120–6.
12 Spurlock G, Bennett E, Chuzhanova N, Thomas N, Jim HP, Side L, Davies S,Haan E, Kerr B, Huson SM, Upadhyaya M. SPRED1 mutations (Legius syndrome):another clinically useful genotype for dissecting the neurofibromatosis type 1phenotype. J Med Genet 2009;46:431–7.
13 Upadhyaya M, Spurlock G, Kluwe L, Chuzhanova N, Bennett E, Thomas N, Guha A,Mautner V. The spectrum of somatic and germline NF1 mutations in NF1 patientswith spinal neurofibromas. Neurogenetics 2009;10:251–63.
14 De Luca A, Buccino A, Gianni D, Mangino M, Giustini S, Richetta A, Divona L,Calvieri S, Mingarelli R, Dallapiccola B. NF1 gene analysis based on DHPLC. HumMutat 2003;21:171–2.
15 Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Botella P, Viano J.Familial spinal neurofibromatosis. Neuropediatrics 2007;38:105–8.
16 Thakkar SD, Feigen U, Mautner VF. Spinal tumours in neurofibromatosis type 1: anMRI study of frequency, multiplicity and variety. Neuroradiology 1999;41:625–9.
17 Kluwe L, Friedrich RE, Peiper M, Friedman J, Mautner VF. Constitutional NF1mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheathtumors. Hum Mutat 2003;22:420.
18 Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S,Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update.Hum Mutat 2003;21:577–81.
19 Wimmer K, Muhlbauer M, Eckart M, Callens T, Rehder H, Birkner T, Leroy JG,Fonatsch C, Messiaen L. A patient severely affected by spinal neurofibromascarries a recurrent splice site mutation in the NF1 gene. Eur J Hum Genet2002;10:334–8.
20 Messiaen L, Riccardi V, Peltonen J, Maertens O, Callens T, Karvonen SL, Leisti EL,Koivunen J, Vandenbroucke I, Stephens K, Pöyhönen M. Independent NF1mutations in two large families with spinal neurofibromatosis. J Med Genet2003;40:122–6.
21 Faravelli F, Upadhyaya M, Osborn M, Huson SM, Hayward R, Winter R. Unusualclustering of brain tumours in a family with NF1 and variable expression ofcutaneous features. J Med Genet 1999;36:893–6.
22 Park VM, Pivnick EK. Neurofibromatosis type 1 (NF1): a protein truncation assayyielding identification of mutations in 73% of patients. J Med Genet1998;35:813–20.
23 Ars E, Kruyer H, Gaona A, Casquero P, Rosell J, Volpini V, Serra E, Lázaro C,Estivill X. A clinical variant of neurofibromatosis type 1: familial spinalneurofibromatosis with a frameshift mutation in the NF1 gene. Am J Hum Genet1998;62:834–41.
24 Pizzuti A, Bottillo I, Inzana F, Lanari V, Buttarelli F, Torrente I, Giallonardo AT,De Luca A, Dallapiccola B. Familial spinal neurofibromatosis due to a multiexonicNF1 gene deletion. Neurogenetics 2011;12:233–40.
25 Mautner VF, Asuagbor FA, Dombi E, Funsterer C, Kluwe L, Wenzel R,Widemann BC, Friedman JM. Assessment of benign tumor burden bywhole-body MRI in patients with neurofibromatosis 1. Neuro Oncology2008;10:593–8.
26 Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves DWidemann B, Kim MO, Dombi E, Sabo J, Dudley A Hardiman, Niwa-Kawakita M,Page GP, Giovannini M, Aronow BJ, Cripe TP, Ratner N. MEK inhibition exhibitsefficacy in human and mouse neurofibromatosis tumors. J Clin Invest2013;123:340–7.
Burkitt Wright EMM, et al. J Med Genet 2013;50:606–613. doi:10.1136/jmedgenet-2013-101648 613
Genotype-phenotype correlations

Contributions of intrinsic mutation rate and selfishselection to levels of de novo HRAS mutationsin the paternal germlineEleni Giannoulatoua,1, Gilean McVeanb, Indira B. Taylora, Simon J. McGowana, Geoffrey J. Mahera, Zamin Iqbalb,Susanne P. Pfeiferb,2, Isaac Turnerb, Emma M. M. Burkitt Wrightc, Jennifer Shortoc, Aysha Itanid, Karen Turnerd,Lorna Gregoryb, David Buckb, Ewa Rajpert-De Meytse, Leendert H. J. Looijengaf, Bronwyn Kerrc, Andrew O. M. Wilkiea,3,and Anne Gorielya,1,3
aWeatherall Institute of Molecular Medicine, University of Oxford, Oxford OX3 9DS, United Kingdom; bWellcome Trust Centre for Human Genetics, Universityof Oxford, Oxford OX3 7BN, United Kingdom; cManchester Academic Health Science Centre, University of Manchester, Manchester M13 9WL, UnitedKingdom; dInstitute of Reproductive Sciences, Oxford OX4 2HW, United Kingdom; eDepartment of Growth and Reproduction, Copenhagen UniversityHospital (Rigshospitalet), DK-2100 Copenhagen, Denmark; and fDepartment of Pathology, Erasmus University Medical Centre, 3000 CA Rotterdam,The Netherlands
Edited by Arthur L. Beaudet, Baylor College of Medicine, Houston, TX, and approved October 25, 2013 (received for review June 15, 2013)
The RAS proto-oncogene Harvey rat sarcoma viral oncogene ho-molog (HRAS) encodes a small GTPase that transduces signals fromcell surface receptors to intracellular effectors to control cellularbehavior. Although somatic HRAS mutations have been describedin many cancers, germline mutations cause Costello syndrome(CS), a congenital disorder associated with predisposition to ma-lignancy. Based on the epidemiology of CS and the occurrence ofHRAS mutations in spermatocytic seminoma, we proposed thatactivating HRAS mutations become enriched in sperm througha process akin to tumorigenesis, termed selfish spermatogonialselection. To test this hypothesis, we quantified the levels, inblood and sperm samples, of HRAS mutations at the p.G12 codonand compared the results to changes at the p.A11 codon, at whichactivating mutations do not occur. The data strongly support therole of selection in determining HRAS mutation levels in sperm,and hence the occurrence of CS, but we also found differencesfrom themutation pattern in tumorigenesis. First, the relative prev-alence of mutations in sperm correlates weakly with their in vitroactivating properties and occurrence in cancers. Second, specifictandem base substitutions (predominantly GC>TT/AA) occur insperm but not in cancers; genomewide analysis showed that thissamemutation is also overrepresented in constitutional pathogenicand polymorphic variants, suggesting a heightened vulnerability tothese mutations in the germline. We developed a statistical modelto show how both intrinsic mutation rate and selfish selection con-tribute to the mutational burden borne by the paternal germline.
paternal age effect | male mutation bias | RASopathy | testis
Understanding the factors that influence the apparent rate ofde novo mutations in the genome is central to the study of
genetic diseases and genome diversity. In humans, germlinemutation rates vary by several orders of magnitude, with averagerates of 4–160 × 10−9 per nucleotide for different point muta-tions (1, 2). Mutations also show a parent-of-origin bias that isexplained by differences in the biology of germ cells in males andfemales, with the majority of germline point mutations, smallindels, and nonrecurrent copy number variations showing a strongpaternal bias, believed to originate during the mitotic replicationsof spermatogonial stem cells (SSCs) that continue throughout thereproductive life of the male (3). Direct estimates of germlinemutation rate, based on whole-genome sequencing (WGS) of two-and three-generation families, concur that among the 30–100novel point mutations that are acquired in each generation, ∼80%originate in the paternal germline (4-7). Two recent studies (6, 7)have further suggested that the major determinant of the totalnumber of de novo germline point mutations is the age of thefather at conception, increasing by one to two mutations per year.
However, epidemiologically, this rate of increase would be pre-dicted to result in a modest paternal age effect, with the averagefather of a child with a randomly sampled de novo mutationbeing ∼2.2 y older than the population average (SI Text).We and others have proposed that an additional mechanism
promotes the enrichment of de novo pathogenic mutations in thetestes of aging men (8–11). This process, which we term selfishspermatogonial selection, accounts for the unusual presentationof a group spontaneous dominant diseases that we collectivelycall paternal age effect (PAE) disorders, including Apert syn-drome, achondroplasia, multiple endocrine neoplasia type 2(men2b), Costello syndrome (CS), and Noonan syndrome (11).These disorders occur spontaneously with an apparent birth ratethat is two to three orders of magnitude above the backgroundrate of mutation (up to 1 in 30,000 for achondroplasia; the es-timated birth prevalence of CS in the United Kingdom is∼1:380,000; SI Text), show an extreme paternal bias in origin(male-to-female ratio of mutation >10:1) and are associated with
Significance
Harvey rat sarcoma viral oncogene homolog (HRAS) occupiesan important place in medical history, because it was the firstgene in which acquired mutations that led to activation ofa normal protein were associated with cancer, making it theprototype of the now canonical oncogene mechanism. Here,we explore what happens when similar HRAS mutations occurin male germ cells, an issue of practical importance because themutations cause a serious congenital disorder, Costello syn-drome, if transmitted to offspring. We provide evidence thatthe mutant germ cells are positively selected, leading to anincreased burden of the mutations as men age. Although thereare many parallels between this germline process and classicaloncogenesis, there are interesting differences of detail, whichare explored in this paper.
Author contributions: G.M., A.O.M.W., and A.G. designed research; E.G., I.B.T., G.J.M., andA.G. performed research; A.I., K.T., L.G., D.B., E.R.-D.M., L.H.J.L., and B.K. contributed newreagents/analytic tools; E.G., G.M., S.J.M., Z.I., S.P.P., I.T., E.M.M.B.W., J.S., B.K., A.O.M.W.,and A.G. analyzed data; and E.G., G.M., A.O.M.W., and A.G. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1E.G. and A.G. contributed equally to this work.2Present address: Max F. Perutz Laboratories GmbH, Vienna 1030, Austria.3To whom correspondence may be addressed. E-mail: [email protected] [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311381110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1311381110 PNAS Early Edition | 1 of 6
GEN
ETICS

an increase in average paternal age (∼2.5–8.7 y excess), relativeto the general population. Quantification of specific causativemutations in the FGFR2, FGFR3 (encoding fibroblast growthfactor receptors 2 and 3, respectively), PTPN11, and RET genesin sperm (8, 10, 12) or whole testes (9, 13–16) led to the proposalthat spermatogonial cells that have acquired rare spontaneousPAE mutations are positively selected, leading to their pro-gressive clonal expansion over time (8, 11, 17). Immunohisto-chemical screening of testicular sections from elderly menvisualizes likely clonal expansion events within the seminiferoustubules (18).Supporting the parallels between selfish selection and early
events in tumorigenesis, we reported that strongly activating so-matic mutations in FGFR3 and Harvey rat sarcoma viral oncogenehomolog (HRAS) occur in spermatocytic seminoma (SpS), a raretesticular tumor affecting older men that is thought to representthe extreme outcome of selfish selection. The previous survey (10)identified two tumors with FGFR3 c.1948A>G (p.K650E) muta-tions and five tumors with HRAS mutations [three samples withc.182A>G (p.Q61R) and two with c.181C>T (p.Q61K)]. Thefinding of acquired HRAS mutations was noteworthy becauseheterozygous germline mutations cause CS, which exhibits theepidemiological characteristics of a PAE disorder (11, 19, 20).However, whereas all mutations previously identified in SpS affectthe p.Q61 codon, 88% of published CS mutations localize to the p.G12 codon (Fig. 1) and none has been described at p.Q61 (TableS1). These codons correspond to two of the three hotspots formutation in cancer (p.G12, p.G13, and p.Q61), at which missensesubstitutions act by locking the RAS molecule in a GTP-boundconformation, resulting in a constitutively active state (21).Although HRAS mutations are predicted to be enriched by
selfish selection and have been implicated in SSC growth regu-lation (22), no study has attempted to document their occurrencedirectly in the sperm of healthy males. To explore further thelink between selfish selection and human disease, we quantifiedthe levels in blood and sperm of spontaneous mutations aroundp.G12 of HRAS, the codon most frequently affected both bygermline CS mutations and by somatically acquired oncogenicmutations. Our results illustrate both similarities and differencesbetween selfish selection and classical oncogenic processes.
ResultsOncogenic HRAS p.G12 Codon Mutations Are Elevated in Sperm. Toquantify spontaneous HRAS mutations, we developed a pro-tocol (Fig. S1) combining restriction enzyme digestion, PCR
amplification, massively parallel sequencing, and statistical anal-ysis (SI Text). Observing that every nonsynonymous single nu-cleotide substitution at theHRAS p.G12 codon has been describedin cancer and each is associated with a different transformingactivity (23, 24), we selected for mutations by digesting genomicDNA with the restriction enzyme MspI (cleaves the WT se-quence 5′-CCGG-3′ at c.32_c.35, irrespective of methylationstatus). This strategy allows equal enrichment of all but 1 of 12possible single-nucleotide substitutions at the MspI site (Fig. 1,bold) as well as complex mutations. Additional benefits are thatthe MspI site includes a CpG dinucleotide, enabling comparisonof transition and transversion rates in the context of both CpGand non-CpG nucleotides, and encompasses two adjacentcodons, so that mutation levels at the p.G12 CS/cancer hotspotcan be compared with those at p.A11, at which mutations areanticipated to be selectively neutral. In samples heterozygous forthe SNP rs12628, located 46 bp downstream of c.35G, eachsubstitution within the MspI site can be phased, allowing us toestablish on which of the two HRAS alleles the original con-tributing mutational events took place (Fig. 1; SI Text).To assess the sensitivity and reproducibility of the assay, we
estimated mutation levels in a titration-reconstruction experi-ment using biological replicates containing 10 μg of controlblood DNA (equivalent to ∼3.3 × 106 copies of the haploid ge-nome) supplemented with dilution series of genomic DNA fromfour CS patients heterozygous for HRAS mutations [range ofinput mutant molecules from ∼10 (concentration: 3 × 10−6) to∼1,000 (concentration: 3 × 10−4)]. To quantify mutation levels,samples were spiked with ∼100 mutant copies of genomic DNAfrom a unique CS patient heterozygous for c.35_36GC>AAtandem mutation. We found a good correlation between theamount of input DNA and the mutation levels estimated bymassively parallel sequencing (Fig. 2A). The levels of thec.34G>A transition were overestimated ∼3.6-fold at the lowerdilution (3 × 10−6), but the c.35_36GC>TT tandem mutationexhibited lower mutation levels in blood, and a good correlationbetween estimates and DNA input was observed down to the 3 ×10−6 level.We then used the same strategy to quantify five single-nucle-
otide substitutions at the p.G12 codon and six substitutions atp.A11, in 7 blood and 89 sperm samples from healthy donors(Fig. 2B, Fig. S2A, and Dataset S1). Estimates of mutation levelsfrom blood varied by mutation, with transitions exhibiting higherlevels than transversions, especially within the c.33_34 CpG di-nucleotide (Table S2). These levels are likely to reflect a combi-nation of rare endogenous mutations in blood and artifactsduring PCR, as this technique generates ∼2- to 20-fold moretransition than transversion errors (25). Based on these obser-vations, the results of the titration experiment (Fig. 2A) and theanalysis of skewing with respect to the rs12628 SNP (SI Text andFig. S2C), a sample was considered to carry a given substitutionif the measured levels were >3 × 10−6, except for transitions, forwhich the calling threshold was set at 10−5. We next analyzed thelevels of individual mutations (relevant statistics and correlationwith donor age are summarized in Table S2). The levels for allsubstitutions involving c.32C and c.33C (in p.A11) did not differsignificantly between blood and sperm (Fig. 2B and Fig. S2A).By contrast, the levels at positions c.34G and c.35G (encodingnonsynonymous changes at p.G12) were frequently higher insperm than in blood (Fig. 2B, Middle) and also exhibited positivecorrelation with sperm donor age (Fig. 2C and Fig. S2A): insperm, the c.34G>A (p.G12S) transition was the most widelyoccurring (55/89 samples had levels >10−5) and the most abun-dant mutation, accounting for 62% of total single-nucleotidesubstitutions at codon p.G12. It also showed the strongest posi-tive correlation with donor age (rs = 0.52). The level of c.35G>A(p.G12D) (also a transition but not at a CpG) was on average3.2-fold lower than c.34G>A and was present at >10−5 in 17/89sperm samples. The three other quantifiable single-nucleotidesubstitutions at p.G12 are transversions that exhibited lower
c.34G>A p.G12S (167/58)c.34G>C p.G12R (0/12)c.34G>T p.G12C (6/25)
c.35G>A p.G12D (5/50)c.35G>C p.G12A (22/8)c.35G>T p.G12V (1/253)
c.36C>A p.G12G (0/0)c.36C>G p.G12G (0/0) c.36C>T p.G12G (0/0)
c.32C>A p.A11D (0/0)c.32C>G p.A11G (0/0)c.32C>T p.A11V (0/0)
c.33C>A p.A11A (0/0)c.33C>G p.A11A (0/0)c.33C>T p.A11A (0/2)
c.37G>A p.G13S (0/10)c.37G>C p.G13R (0/69)c.37G>T p.G13C (15/7)
c.38G>A p.G13D (2/11)c.38G>C p.G13A (0/0)c.38G>T p.G13V (0/13)
c.39T>A p.G13G (0/1)c.39T>C p.G13G (0/0)c.39T>G p.G13G (0/0)
c.31G>A p.A11T (0/0)c.31G>C p.A11P (0/0)c.31G>T p.A11S (0/1)
Fig. 1. Genomic context and DNA sequence around the HRAS p.G12 codon.The relative positions of the p.A11, p.G12, and p.G13 codons to the rs12628SNP (dashed box) and the MspI restriction sites used for selection (grayboxes) are indicated. All single-nucleotide substitutions encompassingp.A11-p.G13 codons and corresponding amino acid changes are indicated,with those detected by resistance to MspI digestion in bold. The number ofrecorded instances of mutation as either a germline (Table S1) or somatic(COSMIC) change is indicated as (germline/somatic). Note that c.35G>C,encoding p.G12A, cannot be assayed by MspI digestion as this mutationcreates a new MspI site at position c.35_38.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1311381110 Giannoulatou et al.

background levels and were elevated above 3 × 10−6 in 20/89samples for c.34G>T (p.G12C), in 18/89 samples for c.35G>T(p.G12V) and in only 3/89 samples for c.34G>C (p.G12R).Levels of these transversions were also correlated with donorage, although more weakly so than for the transitions.
Tandem Base Substitutions Are Overrepresented in the Germline.Given that our protocol would select any substitution resistantto MspI digestion, we asked whether multiple nucleotide sub-stitutions could be identified. Unexpectedly, 31 independent eventsinvolving dinucleotide substitutions were observed in sperm sam-ples at levels >3 × 10−6. Aside from c.34G>A;c.36C>T (encodingp.G12S) and c.34_35GG>TT (p.G12F), each observed in singlesperm samples, all other dinucleotide mutations were tandembase substitutions (TBS) involving the last two nucleotides ofcodon p.G12, comprising c.35_36GC>AA (p.G12E) in 1 sample,c.35_36GC>AT (p.G12D) in 4 samples, c.35_36GC>TA(p.G12V) in 3 samples, and c.35_36GC>TT (p.G12V) in 21
samples (Dataset S1, Table S2, and Fig. S2B). Surprisingly, giventhat they both encode the same oncogenic p.G12V change andwould therefore be subject to equivalent selection, the average levelof the most prevalent TBS, c.35_36GC > TT, was 1.7-fold higherthan the level of the c.35G>T single-nucleotide substitution.Levels of this TBS were significantly higher in sperm than blood(P = 0.00002) and correlated strongly (rs = 0.44) with donor age(Fig. 2C and Table S2).To assess the implications of the high prevalence of TBS, par-
ticularly GC>TT, we asked whether they could be identified indifferent human genomic datasets (SI Text). We first interrogatedthe Human Gene Mutation Database (HGMD), in which 441TBS have been cataloged as pathogenic germline mutations. Inagreement with a recently published study (26), themost numerousof all 78 possible TBS involve GC>TT (or its reverse complementGC>AA), representing 14.7%of the total, corresponding to a 10.6-fold enrichment over a uniform distribution of TBS (P< 10−16; Fig.3A and Table S3). Of the 64 coding GC>TT/AA, 26 encode
A B
C
D
Fig. 2. Estimation of HRAS mutation levels within the c.32_35 MspI site (codons p.A11 and p.G12) in sperm and blood samples. (A) Mutation levels estimatedin a titration-reconstruction experiment with serial dilution of four CS samples mixed with blood carrier DNA and spike DNA. (B) Estimation of mutation levelsfor substitutions at p.A11 (Top) and p.G12 (Middle) in 89 sperm (Left) and 7 blood (Right) samples. Color code for each substitution is given on the figure.Samples are organized according to their genotype at the rs12628 SNP (TT to the left, CC in the center, and CT heterozygote to the right). The mutation levelsare plotted independently for the two HRAS alleles with respect to the SNP, so that the total mutation level for CT samples is the sum of the counts on eachallele. The age of the sample donor is given at the Bottom. (C) Average levels for mutations at codon p.G12 in sperm samples binned by 5-y age group. (D)Comparison of levels for different mutations at codon p.G12 in sperm samples (Top) with relative prevalence of mutations reported in CS (Table S1) (Middle)and in cancer (COSMIC) (Bottom). Correlation between sperm data and other measurements are indicated by Spearman (rs) and Pearson (r) correlationcoefficients with statistical significance (NS, not significant; *P = 0.02; **P = 0.009; ***P = 0.000004). The color code used in C and D is identical to B.
Giannoulatou et al. PNAS Early Edition | 3 of 6
GEN
ETICS

changes that (due to the specifics of the genetic code) can onlyarise from a double-nucleotide substitution, including the re-current RET c.2647_2648GC>TT (p.A883F) mutation associatedwith men2B (11, 27).We next examined the distribution of TBS in cancer. Strik-
ingly, although 406 single-nucleotide substitutions are recordedat the HRAS p.G12 codon in the Catalogue Of Somatic Muta-tions in Cancer (COSMIC), there is not a single instance of TBS,suggesting that a different pattern of mutations (either caused bydistinct mutational mechanisms in somatic and germline cellsand/or specific mutagen exposure) is observed in these differentcellular contexts. This interpretation is supported by the profile of3,769 TBS cataloged in COSMIC (Table S3). The most commonTBS in somatic tissues are CC/GG>AA/TT (31.4%) and CC/GG>TT/AA (19.2%), which represent mutagen-specific signa-tures triggered by the action of polycyclic aromatic hydrocarboncomponents found in cigarette smoke or UV exposure (28), re-spectively. By contrast, there were only 107 events (2.8% of TBS)of GC>TT/AA, indicating that there is a much less marked en-richment (2.0-fold over random expectation) for this TBS incancer (Table S3).To explore further the impact of TBS, we analyzed the prev-
alence and distribution of TBS contributing to human variation,based on WGS data (SI Text). We used Cortex, a de novo as-sembly-based variant calling algorithm (29) to assess TBS rep-resentation in 85 human genome sequences from the Luhya inWebuye, Kenya (LWK) dataset of the 1000 Genomes Project(30) and identified 5,425,856 nucleotide variants, among which22,898 (0.42%) involved TBS. Strikingly, the GC>TT/AA changewas the second most common TBS, observed in 1,417 instances(6.2%), representing a 4.5-fold enrichment (Fig. 3B and TableS3). Because the pattern of TBS at the HRAS p.G12 codonsuggested that the CpG dinucleotide at position c.36_37 (Fig. 1)might influence the apparent c.35_36GC>TT mutation rate, wefurther characterized the local sequence context in which the1,417 genomic GC>TT/AA TBS had occurred. Compared withthe relative frequency of single substitutions (G>T or C>A) inthe same sequence context, the GC>TT/AA TBS is three timesas likely to occur as part of a CpG dinucleotide [842 of 103,732events (0.81%) for the single substitutions and 35 of 1,417(2.5%) for TBS; P = 2.2 × 10−8; SI Text]. These genomewideobservations suggest that the sequence context in which the TBS
occurs plays an important, although yet uncharacterized, role,and in particular we propose that hypermutability of the C>Ttransition within the CpG dinucleotide accounts, at least in part,for the high spontaneous GC>TT/AA mutation rate observed inthe germline.
Comparison Between Prevalence of HRAS Mutations in Sperm and inCS, SpS, and Cancer Datasets. To establish the biological relevanceof the measurements of HRAS mutation levels in sperm, wecompared these data to the distribution of published CS alleles,to experimental data generated in our laboratory on mutationsin SpS, and to cancer-associated mutations cataloged in theCOSMIC database.Of the 236 CS cases reported in the literature, 207 (88%) involve
mutations at codon p.G12 (Fig. 1 and Table S1). The c.34G>A(p.G12S) mutation, which is associated with a relatively homog-enous presentation, is by far the most prevalent (81%). Otherp.G12 mutations have also been described, including p.G12A,p.G12C, p.G12D, p.G12V, and p.G12E. These rarer alleles tendto be associated with more severe manifestations, often involvinghypertrophic cardiomyopathy and resulting in neonatal mortality(31–34), consistent with biochemical evidence that p.G12S is lessactivating than any other mutation at this codon (23, 24).We found a strong correlation between the prevalence of
HRAS alleles in sperm and the number of cases reported for eachCS mutation, indicating that the average level of mutation insperm is a major determinant of prevalence of different HRASalleles in the CS population (Fig. 2D). Comparing the sperm datawith observed births of CS, it is apparent that p.G12S is unex-pectedly prevalent in CS compared with other p.G12 substitu-tions, which suggests that these other (more activating) mutationsmay be associated with a higher risk of demise during the preg-nancy (33). In agreement with our finding that TBS are not un-common in sperm, a total of six CS patients carrying similarmutations have been reported (Table S1). Strikingly, amongpatients diagnosed with HRAS mutations encoding p.G12V, fivecases have been associated with TBS (four with c.35_36GC>TTand one with c.35_36GC>TA), whereas only a single patient car-ried the c.35G>T substitution (31–33). The predominance of thec.35_36GC>TT TBS among observed CS alleles supportsthe relevance of our sperm data, as this was themajority (21/30) ofthe TBS observed.We extended our previous survey of SpS by screening (SI Text)
a panel of 33 tumors for hotspot mutations in FGFR3 and HRAS(Table S4). No further FGFR3 mutations were found, but twoadditional tumor samples harbored apparently homozygousHRAS mutations, not observed in matched histologically normaltissue. The mutations were c.37G>C (p.G13R) and c.182A>G(p.Q61R) in tumors from men aged 79 and 81 y, respectively(Fig. S3 A and B). Although these data confirm that HRAS is themost commonly mutated gene in SpS (11%) and mutation pos-itivity is strongly correlated with patient age (Fig. S3C), no mu-tations at codon p.G12 were identified. Currently, all HRASmutations found in SpS are mutually exclusive with CS muta-tions, which may reflect either embryonic or fetal lethality due tothe highly activating nature of the mutations associated with SpS(35) or the inability of mutant SSC to produce differentiatingmeiotic cells and sperm.As illustrated in Fig. 2D, mutations at the p.G12 codon occur
in different relative proportions in sperm compared with cancers.In cancers, HRAS p.G12V, which exhibits the lowest GTPaseactivity (36) and the highest transformation potential (23), accountsfor 64% of mutations at codon p.G12 (Fig. 1), whereas in sperm,HRAS p.G12S (c.34G>A) is most abundant, despite its lowertransforming activity. These observations point to a differentmechanism of mutation and/or selection in spermatogonia thanoccurs in most tumors, which we investigated further by statis-tical modeling.
0
20
40
60
80
0500100015
00200025
003000
CG/CGTA/TAAG/CTGT/ACGG/CCGA/TCCC/GGTC/GATG/CAGC/GCAC/GTAT/ATCT/AGAA/TTTT/AACA/TG
CG
/CG
TA/T
AAT
/AT
CT
/AG
TT
/AA
GT
/AC
GG
/CC
TC
/GA
GC
/GC
TG
/CA
A B
z
x
yCG
/CG
TA/T
AAT
/AT
CT
/AG
TT
/AA
GT
/AC
GG
/CC
TC
/GA
GC
/GC
TG
/CA
Fig. 3. Lego plots representing the prevalence of TBS in the human ge-nome. (A) Data from HGMD. (B) Genomewide variation across 85 LWK wholegenome sequences (Cortex assembler). The x axis represents the originaldinucleotide sequences and their reverse complements, whereas the y axisindicates the mutated sequence and its reverse complement (the y axislegend is the same on both plots and to ease visualization, mutatedsequences are shaded in different colors). Plotted on the z axis is the totalnumber of events for each TBS (Table S3). Owing to the complementarynature of DNA, only 78 different TBS can occur, and gray areas indicatechanges that do not lead to TBS (such as single-nucleotide substitutions) orare identical to their reverse complement.
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1311381110 Giannoulatou et al.

Modeling Mutation Rate and Selective Advantage. Overall, our find-ings suggest that the HRAS mutation levels in sperm are deter-mined by an interplay between the intrinsic genomic mutation rateof a residue and the selective advantage conferred by the resultingmutant protein on the spermatogonial cell (8, 10, 11). To un-derstand the relative impact of these two factors in shaping theoutcome of selfish selection, we developed a statistical model (SIText). We elaborated a simple model (9) in which from the age ofpuberty (13 y), SSC homeostasis is maintained by regular asym-metrical divisions, i.e., each division generates a daughter sper-matogonial cell and a differentiating cell that will ultimatelyproduce sperm. Selfish mutations are predicted to modify the SSCmitotic behavior, allowing occasional symmetric divisions, leadingto an exponential enrichment of mutations in sperm over time. Toaccount for the fact that contributing mutations are anticipated tobe rare, we model their occurrence as a Poisson random variablewith parameter μ (i.e., the mutation rate per cell division). Wethen define a selection coefficient parameter (s) that correspondsto the probability of the occurrence of such symmetric division ateach SSC mitosis. Values of μ and s were then inferred by MonteCarlo simulation for each HRAS substitution at codons p.A11 andp.G12 in the 89 sperm samples, with partitioning of the data forthe 46 individuals heterozygous for the rs12628 SNP across thetwo alleles.The model yields significantly positive values of s for activating
HRAS mutations at p.G12, whereas for the synonymous ornonactivating mutations at p.A11, s is close to zero (Fig. 4 andTable S5). Although s for different mutations at p.G12 reflectstheir documented in vitro activating properties (lowest forp.G12S and highest for p.G12V) (23, 24), the narrow range ofvalues of s, within 1.5-fold, means that the relative abundance ofa given p.G12 mutation is mainly determined by its mutability μwhich varies by a factor sevenfold between c.34G>A (highest;transition at CpG dinucleotide) and c.35G>T (lowest; trans-version at non-CpG). The effect of relative mutability is apparentwhen examining the distribution of mutations on the two HRASalleles in individuals heterozygous for the rs12628 SNP. Whereaslevels of rarer mutations (including TBS) generally exhibit amarked skewing preference on one or other allele, indicatingthat as few as one originating mutation could have contributed tothe final levels, this skewing is much less marked for the c.34G>Atransition (p.G12S) because of its higher intrinsic mutation rate(SI Text and Fig. S2C).
To test further the usefulness of the model, we analyzed threepreviously published datasets of mutation levels quantified insperm for substitutions involving FGFR2 c.755C (8, 12), FGFR2c.758C (12), and FGFR3 c.1948A or c.1949A (10). Althoughthese datasets were obtained using different methodologies,estimates of μ for a given category of substitution broadly agreedboth between the datasets and with previously obtained muta-tion rate estimates (1, 2, 6). Estimates of s and μ for the Apertc.755C>G mutation originating from two independent datasetsare also in good agreement. Notably, selection coefficients forthe most strongly selected mutations in FGFR2 and FGFR3 are1.5- to 2.1-fold higher than for the most strongly selected mu-tation in HRAS, which is likely to account for the lower birthprevalence of CS (SI Text) compared with the disorders associatedwith the specific FGFR2 and FGFR3 mutations (11) (Table S5).
DiscussionThe c.35G>T (p.G12V) substitution in HRAS is of considerablehistorical significance, because in 1982 it was the first missensechange in a proto-oncogene to be implicated in cancer (37, 38).Three decades later, it is known to be the most frequent onco-genic mutation in HRAS (COSMIC), but a rare cause of CSarising through germline mutation (Fig. 1). Here, we have de-termined the distribution of mutation levels at the p.G12 codonin sperm and used these observations to model their occurrencebased on intrinsic mutation rate μ and selection coefficient s. Wefind that although the levels of mutation in sperm are markedlyelevated through a process akin to oncogenesis and are consistentwith a mechanism involving selfish selection, there are also dif-ferences in the outcome between spermatogenesis and classicaloncogenesis. These variations are likely to reflect several underlyingbiological processes, including differences in intrinsic mutation ratesand the specific effects of mutation on proliferation, differentia-tion, and survival of SSC, acting over many years.Regarding the primary mutations that fuel the eventual supply
of mutant sperm, the most surprising observation was of multipleTBS, particularly c.35_36GC>TT, which had an estimated μindistinguishable from the c.35G>T transversion encoding thesame amino acid change, p.G12V (Fig. 4). Considerable confi-dence that these events are real and not experimental artifacts isprovided by the observation of multiple TBS in CS (Table S1).Although changes in two or more nucleotides arising throughindependent mutational events are expected to be extremely rare(∼10−11) (2), several studies suggest that these events are morecommon than expected by chance (26, 39, 40). TBS can eitherresult from a single concomitant mutational event involving ad-jacent nucleotides or have arisen through two hits of increasingselectivity, a mechanism that has been demonstrated in individ-uals heterozygous for rare FGFR2 mutations (17). In the case ofTBS in sperm at the HRAS p.G12 codon, a mechanism involvinga single concomitant mutational event is suggested by threeobservations. First, nearly all complex changes (30/31) involvedbases adjacent to one another within the p.G12 codon (c.34_35or c.35_36). Second, 28/30 TBS encode amino acid changes thatare also observed as single-nucleotide substitutions (encodingp.G12V and p.G12D) and among these, 13/28 occurred in sam-ples that have no detectable levels of the corresponding single-nucleotide substitution (c.35G>A or c.35G>T). Third, most TBS(25/30) involved a C>T transition at position c.36 (encodinga synonymous change as single substitution and not a knownpolymorphism); as c.36C is a part of a CpG dinucleotide(c.36_37), it raises the possibility that hypermutability at thissite could influence the apparent mutation rate of the adjacentnucleotide. This proposal is supported by the threefold enrich-ment of 3′-adjacent guanine nucleotides in the case of GC>TT/AA TBS in humans, compared with GC>TC SNP. Hypermuta-bility associated with methylated CpG sequence context has beendescribed in UV-induced CC>TT dypirimidine changes observedin sun-exposed skin lesions (28). In nucleotide excision repair-deficient cells, methylated CpG sequences frequently undergo
Fig. 4. Contributions of mutation rate (μ) and selection (s) to levels insperm for mutations in HRAS, FGFR2, and FGFR3. Data for HRAS (this work)in black; references for data from previous studies are color-coded as red(8), green (12), and blue (10). Bars and shaded areas represent the 95%confidence intervals (Table S5).
Giannoulatou et al. PNAS Early Edition | 5 of 6
GEN
ETICS

CG>TT tandem mutations in response to oxidative DNA damage(41). Our work highlights a predisposition to specific TBS thatseems largely restricted to germ cells, and should stimulateefforts to investigate its biochemical nature.Our statistical model incorporated a selection parameter s,
defined as the probability of symmetric division at each SSC mi-tosis. Reassuringly, estimates of s for mutations at the neutral p.A11 control codon were close to zero, whereas we found positivevalues of s for all mutations at the p.G12 codon, consistent withclonal expansion and selfish selection. Moreover s was highest forp.G12V (c.35G>T) and lowest for p.G12S (c.34G>A) (Fig. 4 andTable S5), consistent with their relative in vitro transforming po-tential (23, 24). Of note, s may encompass a number of biologicalprocesses other than the balance between symmetric and asym-metric division, including differential survival of cells undergoingstochastic divisions (42) and cell competition (43). In this context,survival simply implies the production of mature sperm, so thiscould be impaired by several pathologies such as spermatogenicarrest, senescence, or apoptosis (44). In any case, the net result ofthe narrow range of s for different mutations at the p.G12 codon ofHRAS is that μ outweighs s in determining that the most prevalentmutation, both in sperm and in CS, is c.34G>A (p.G12S).Finally, it will be of interest to consider the present results
when analyzing de novo mutation load on a genomewide scale.Although direct estimates of mutation rate based on WGS offamily trios have singled out the importance of paternal age asthe major determinant of the total number of de novo mutations
(6, 7), it is apparent that the vast majority of reported mutationsoccur in noncoding parts of the genome and are likely to beneutral. Therefore, characterization of the influence of paternalage, not only on the total mutational load, but specifically fordifferent functional classes of mutations, might provide a meansto estimate what fraction of these newly acquired mutations islikely to be attributable to mechanisms such as selfish selection,and hence the overall role that this process plays in genomediversity and disease.
Materials and MethodsSingle ejaculates from 89 healthy men (aged 22–74 y) were donated anon-ymously, and the age of the donor was recorded. Blood samples wereobtained from seven individuals aged 36–71 y. Written informed consentwas obtained from all donors, and samples were collected with the per-mission of the Oxfordshire Research Ethics Committee (OxREC C03.076). ForSpS analysis, 33 samples were collected from tissue archives. For a detaileddescription of the methods, see SI Text.
ACKNOWLEDGMENTS. We thank Steve Twigg and Oliver Venn for helpfuldiscussions, and the High-Throughput Genomics Group for the generationof the sequencing data. Financial support was provided by Wellcome TrustProgramme Grants 091182 (to A.G., G.M., and A.O.M.W.) and 086084 (toG.M.), Research Training Fellowship 090120 (to E.M.M.B.W.), and Core Award090532 to Wellcome Trust Centre for Human Genetics; Medical ResearchCouncil Hub Grant G0900747; and Danish Cancer Society Grant A2127 (toE.R.-D.M.). I.T. is a recipient of an Engineering and Physical Sciences ResearchCouncil studentship.
1. Nachman MW, Crowell SL (2000) Estimate of the mutation rate per nucleotide inhumans. Genetics 156(1):297–304.
2. Kondrashov AS (2003) Direct estimates of human per nucleotide mutation rates at 20loci causing Mendelian diseases. Hum Mutat 21(1):12–27.
3. Campbell CD, Eichler EE (2013) Properties and rates of germline mutations in humans.Trends Genet 29(10):575–584.
4. Roach JC, et al. (2010) Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328(5978):636–639.
5. Conrad DF, et al.; 1000 Genomes Project (2011) Variation in genome-wide mutationrates within and between human families. Nat Genet 43(7):712–714.
6. Kong A, et al. (2012) Rate of de novo mutations and the importance of father’s age todisease risk. Nature 488(7412):471–475.
7. Michaelson JJ, et al. (2012) Whole-genome sequencing in autism identifies hot spotsfor de novo germline mutation. Cell 151(7):1431–1442.
8. Goriely A, McVean GA, Röjmyr M, Ingemarsson B, Wilkie AOM (2003) Evidence forselective advantage of pathogenic FGFR2 mutations in the male germ line. Science301(5633):643–646.
9. Qin J, et al. (2007) The molecular anatomy of spontaneous germline mutations inhuman testes. PLoS Biol 5(9):e224.
10. Goriely A, et al. (2009) Activatingmutations in FGFR3 andHRAS reveal a shared geneticorigin for congenital disorders and testicular tumors. Nat Genet 41(11):1247–1252.
11. Goriely A, Wilkie AOM (2012) Paternal age effect mutations and selfish spermato-gonial selection: Causes and consequences for human disease. Am J Hum Genet 90(2):175–200.
12. Yoon SR, et al. (2009) The ups and downs of mutation frequencies during aging canaccount for the Apert syndrome paternal age effect. PLoS Genet 5(7):e1000558.
13. Choi SK, Yoon SR, Calabrese P, Arnheim N (2008) A germ-line-selective advantagerather than an increased mutation rate can explain some unexpectedly commonhuman disease mutations. Proc Natl Acad Sci USA 105(29):10143–10148.
14. Choi SK, Yoon SR, Calabrese P, Arnheim N (2012) Positive selection for new diseasemutations in the human germline: Evidence from the heritable cancer syndromemultiple endocrine neoplasia type 2B. PLoS Genet 8(2):e1002420.
15. Shinde DN, et al. (2013) New evidence for positive selection helps explain the paternalage effect observed in achondroplasia. Hum Mol Genet 22(20):4117–4126.
16. Yoon SR, et al. (2013) Age-dependent germline mosaicism of the most commonNoonan syndrome mutation shows the signature of germline selection. Am J HumGenet 92:917–926.
17. Goriely A, et al. (2005) Gain-of-function amino acid substitutions drive positive se-lection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci USA 102(17):6051–6056.
18. Lim J, et al. (2012) Selfish spermatogonial selection: Evidence from an immunohis-tochemical screen in testes of elderly men. PLoS ONE 7(8):e42382.
19. Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW (2006) Paternal bias inparental origin of HRAS mutations in Costello syndrome. Hum Mutat 27(8):736–741.
20. Zampino G, et al. (2007) Diversity, parental germline origin, and phenotypic spectrumof de novo HRAS missense changes in Costello syndrome. Hum Mutat 28(3):265–272.
21. Scheffzek K, et al. (1997) The Ras-RasGAP complex: Structural basis for GTPase acti-vation and its loss in oncogenic Ras mutants. Science 277(5324):333–338.
22. Lee J, et al. (2009) Genetic reconstruction of mouse spermatogonial stem cell self-renewal in vitro by Ras-cyclin D2 activation. Cell Stem Cell 5(1):76–86.
23. Fasano O, et al. (1984) Analysis of the transforming potential of the human H-rasgene by random mutagenesis. Proc Natl Acad Sci USA 81(13):4008–4012.
24. Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD (1984) Biologicalproperties of human c-Ha-ras1 genes mutated at codon 12. Nature 312(5989):71–75.
25. Bracho MA, Moya A, Barrio E (1998) Contribution of Taq polymerase-induced errorsto the estimation of RNA virus diversity. J Gen Virol 79(Pt 12):2921–2928.
26. Chen JM, Férec C, Cooper DN (2013) Patterns and mutational signatures of tandembase substitutions causing human inherited disease. Hum Mutat 34(8):1119–1130.
27. Gimm O, et al. (1997) Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J ClinEndocrinol Metab 82(11):3902–3904.
28. Alexandrov LB, et al.; Australian Pancreatic Cancer Genome Initiative; ICGC BreastCancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain (2013) Signatures ofmutational processes in human cancer. Nature 500(7463):415–421.
29. Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G (2012) De novo assembly andgenotyping of variants using colored de Bruijn graphs. Nat Genet 44(2):226–232.
30. Abecasis GR, et al.; 1000 Genomes Project Consortium (2012) An integrated map ofgenetic variation from 1,092 human genomes. Nature 491(7422):56–65.
31. Aoki Y, et al. (2005) Germline mutations in HRAS proto-oncogene cause Costellosyndrome. Nat Genet 37(10):1038–1040.
32. van der Burgt I, et al. (2007) Myopathy caused by HRAS germline mutations: Im-plications for disturbed myogenic differentiation in the presence of constitutive HRasactivation. J Med Genet 44(7):459–462.
33. Burkitt-Wright EM, et al. (2012) Neonatal lethal Costello syndrome and unusual di-nucleotide deletion/insertion mutations in HRAS predicting p.Gly12Val. Am J MedGenet A 158A(5):1102–1110.
34. Lorenz S, et al. (2012) Two cases with severe lethal course of Costello syndrome as-sociated with HRAS p.G12C and p.G12D. Eur J Med Genet 55(11):615–619.
35. Der CJ, Finkel T, Cooper GM (1986) Biological and biochemical properties of humanrasH genes mutated at codon 61. Cell 44(1):167–176.
36. Colby WW, Hayflick JS, Clark SG, Levinson AD (1986) Biochemical characterization ofpolypeptides encoded by mutated human Ha-ras1 genes. Mol Cell Biol 6(2):730–734.
37. Reddy EP, Reynolds RK, Santos E, Barbacid M (1982) A point mutation is responsiblefor the acquisition of transforming properties by the T24 human bladder carcinomaoncogene. Nature 300(5888):149–152.
38. Tabin CJ, et al. (1982) Mechanism of activation of a human oncogene. Nature300(5888):143–149.
39. Averof M, Rokas A, Wolfe KH, Sharp PM (2000) Evidence for a high frequency of si-multaneous double-nucleotide substitutions. Science 287(5456):1283–1286.
40. Schrider DR, Hourmozdi JN, Hahn MW (2011) Pervasive multinucleotide mutationalevents in eukaryotes. Curr Biol 21(12):1051–1054.
41. Lee DH, O’Connor TR, Pfeifer GP (2002) Oxidative DNA damage induced by copperand hydrogen peroxide promotes CG—>TT tandem mutations at methylated CpGdinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res 30(16):3566–3573.
42. Klein AM, Nakagawa T, Ichikawa R, Yoshida S, Simons BD (2010) Mouse germ linestem cells undergo rapid and stochastic turnover. Cell Stem Cell 7(2):214–224.
43. Moreno E (2008) Is cell competition relevant to cancer? Nat Rev Cancer 8(2):141–147.44. Paul C, Robaire B (2013) Ageing of the male germ line. Nat Rev Urol 10(4):227–234.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1311381110 Giannoulatou et al.

Supporting InformationGiannoulatou et al. 10.1073/pnas.1311381110SI TextEstimation of Epidemiological Paternal Age Effect from Whole GenomeSequencing Data. Birth data, reported for England and Walesduring 2011, were obtained from the UK Office of NationalStatistics. For 2011, the average age of fathers (combining birthswithin and outside marriage/civil partnership) at the time of livebirth was 32.8 y.To model the occurrence of new mutations in this population,
we used the estimation by Kong et al. (1) that the rate of de novopaternal mutations increases by 4.28% per year. Using this rate,we calculated the relative de novo mutation burden anticipatedat each paternal age between 15.5 and 75.5 y (in increments of1 y). Hence, the total number of de novo mutations was obtainedby multiplying the number of births reported for each year offather’s age with the corresponding number of paternal muta-tions, as estimated by the exponential growth model (with rate of4.28%). The resulting distribution of father’s age, given the oc-currence of a de novo mutation, yields a value of 35.0 y for theaverage age at which a mutation would have arisen, corre-sponding to a paternal age effect (PAE) of +2.2 y comparedwith the population average.
Estimation of the Birth Prevalence of Costello Syndrome in the UnitedKingdom. Determining the birth prevalence of rare disorders isdifficult, due to clinical variability and underdiagnosis often as-sociated with lack of professional awareness of uncommon dis-eases. However, through its centralized Regional Clinical Geneticservices as well as a highly specialized Dysmorphology forum thatoperates three times yearly, awareness of the clinical features ofCostello syndrome (CS) is considered to be high in the UnitedKingdom. Therefore, to estimate the birth prevalence of CS, wecollated data on laboratory-ascertained cases diagnosed withinthe United Kingdom.The years 2000–2009 were chosen for estimation of disease
prevalence as this corresponds to a period during which the datawere most likely to be complete; population birth data wereavailable from the relevant Offices of Statistics. During this 10-yperiod, a total of 7,256,368 births were registered across theUnited Kingdom, and 19 CS cases (14 for England and Wales, 4for Scotland, and 1 for Northern Ireland) were molecularly con-firmed, corresponding to a birth prevalence estimate of 1 in381,914. Although this figure is similar to the recently reportedprevalence of CS in Japan of 1 in 290,000 (2), it is likely to rep-resent a minimum estimate. Considering that about 40% of knownCS cases show very poor survival including neonatal mortality (inabout half these cases), usually caused by multiorgan failure, withrespiratory insufficiency and cardiac hypertrophy, it is likely thatthis group of very sick infants will still be unrecognized as CS inmany instances; of note, similar cautions were given by the authorsof the Japanese study who suggested that CS prevalence could be ashigh as 1 in 60,000–1 in 100,000 (2).
Quantification of Harvey Rat Sarcoma Viral Oncogene Homolog MutationLevels in Sperm and Blood Samples. Preparation of sequencing libraries.DNA from sperm and blood samples was extracted followinga protocol previously described (3). DNA quantification wasinitially estimated by fluorometry (Hoefer) and then preciselymeasured at three different dilutions against a dilution series ofhuman genomic DNA (Roche Applied Biosystems) by real-timePCR using a strategy described previously (4).Measurements of mutation levels in blood and sperm samples
within the MspI site located at position c.32–35 of Harvey rat
sarcoma viral oncogene homolog (HRAS) (cDNA RefSeqNM_001130442.1) and encompassing codons p.A11 and p.G12were performed using a strategy similar to that described pre-viously (4), as summarized in Fig. S1A. Triplicate biologicalsamples each containing 10 μg of genomic DNA and either 0.6 ng(two repeats of spiked samples at concentration 3 × 10−5) or 0 ng(unspiked samples) of genomic DNA from an individual hetero-zygous for the HRAS c.35_36GC>AA mutation (referred to asspike DNA) were digested in 1× buffer 4 with 100 U MspI (NewEngland Biolabs) and 20 U MspI (Fermentas; ThermoScientific)for 4 h at 37 °C in a final volume of 120 μL. The digested DNAsamples, flanked by two lanes of pUC19/MspI DNA ladder 23(Fermentas; ThermoScientific), were electrophoresed at 70 Vovernight (4 °C) on a 1.2% (vol/vol) Tris-acetate-EDTA (TAE)gel (without ethidium bromide). This digestion is expected togenerate 410-bp MspI fragments carrying mutant HRAS se-quences at position c.32_35, whereas the MspI WT sequenceyields two fragments of 355 and 55 bp (sizes are based on thegenomic reference sequence NT_009237.18). To select for mu-tant sequences, a gel slice corresponding to a size defined byexclusion of the 489/501-bp band and inclusion of the 404-bpband of the pUC19/MspI DNA marker (the marker lanes werecut out of the gel, stained with ethidium bromide for 20–30 min,rinsed twice, and carefully placed in their original position in thegel) was excized and gel purified using the E.Z.N.A. MicroElutegel purification kit (Omega Bio-Tek).A first PCR amplification (PCR-1) was performed on this
purified material using 1 U Platinum Pfx DNA polymerase(Invitrogen; Life Technologies), 1× Pfx buffer, 1 mM MgSO4,300 μM dNTPs, 0.3 μM primers (primer 1: 5′-CCCTGAGGAG-CGATGACGGAATATAAGCTGGTGG-3′; primer 2: 5′-CCT-ATCCTGGCTGTGTCCTGGGCTCGCCC-3′) each in 40 μLvolume, under the following cycling conditions: 94 °C for 5 minfollowed by 25 cycles of 94 °C for 15 s, 63 °C for 20 s, and 68 °C for30 s, and a final extension at 68 °C for 8 min. A second round ofselection was performed by digesting the PCR products with 70 UMspI (New England Biolabs) for 3 h at 37 °C in 100 μL volume toyield a pool of selected material referred to as PCR-1 product (Fig.S1A). For each biological sample, three aliquots of 5 μL of thePCR-1 product material were used as template for three in-dependent nested PCR amplifications [performed with a uniquelybarcoded forward (BC-Fw) primer and three different reverseprimers (NN-Rev)]. This process generated amplicons referred toas PCR replicates that were pooled to prepare the Illumina li-braries (see below). Titration-reconstruction samples were ampli-fied in parallel following the same protocol (including the threeindependent PCR replicates) but only duplicate samples, eachspiked with 0.6 ng of 35_36GC>AA genomic DNA, were pro-duced during the PCR-1 (i.e., the unspiked DNA sample wasomitted). Finally, the control DNAs (referred to as controls) wereamplified separately following a similar approach, except that theaddition of MspI was omitted at each step, and only 1 μL of PCR-1product was used for the nested PCR replicates. The resultingcontrol PCR products were mixed together, split three ways anddistributed equally between the final PCR pools used to build thethree independent libraries.Titration-reconstruction experiment. DNA samples from three pa-tients heterozygous for the HRAS c.34G>A (p.G12S CS muta-tion), one patient heterozygous for c.35_36GC>TT (p.G12V) (5),and one patient heterozygous for c.35_36GC>AA (p.G12E, re-ferred to as spike DNA) (6) were used in the titration experimentsin which duplicates, each containing 10 μg of blood genomic DNA
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 1 of 17

from an anonymous donor supplemented with 0.6 ng ofc.35_36GC>AA spike genomic DNA, were mixed with a dilutedseries of each HRAS heterozygous CS mutant DNA corre-sponding to final concentrations of 0 (no added mutant DNA),3 × 10−6 (0.06 ng), 10−5 (0.2 ng), 3 × 10−5 (0.6 ng), 10−4 (2 ng),and 3 × 10−4 (6 ng). One dilution sample containing c.34G>Aat a concentration of 10−5 was inadvertently lost during sampleprocessing and is therefore missing from the final analysis (redsample on Fig. 2A). The titration samples were taken throughthe same protocol of selection and amplification as the sperm andblood samples and were analyzed together with the biologicalsamples within two independent libraries.Control DNAs.Themutant control DNAs analyzed were the same asthose used in the titration experiments, i.e., genomic DNA fromperipheral blood of three patients heterozygous for the c.34G>Amutation, one patient heterozygous for c.35_36GC>TT, and onepatient heterozygous for c.35_36GC>AA (spike). In addition,four normal (WT) genomic DNA samples were included in theanalysis. Duplicates of each sample were taken through the sameprotocol of amplification as the sperm, blood, and titrationsamples with the exception that the MspI enzyme was omittedfrom all of the incubation steps; hence, there was no selection forHRAS mutant sequences performed on these samples. We gen-otyped each control sample at the rs12628 SNP, and for theheterozygous patient samples, further established the phase ofthe mutation at p.G12 with respect to the rs12628 SNP.Preparation of the Illumina libraries for massively parallel sequencing.Weconstructed three independent libraries for massively parallelsequencing using a modified version of a previously describedprotocol (4) (Fig. S1A). Each library contained a mixture of 112uniquely barcoded samples (PCR replicates) and was charac-terized by the presence of a given amount of the spike DNA(c.35_36GC>AA), so that library 1O contained only unspikedbiological samples, whereas libraries 2A and 3C contained bloodand sperm samples spiked at a concentration of 3 × 10−5. Am-plicon sequencing of low complexity libraries on the Illuminaplatform is known to be problematic and results in low yields andlower per-base quality scores (7). In our previous study (4), wecircumvented this problem by adding 5–10% (vol/vol) PhiXSpike-In Control DNA to our library to increase complexity, butthis resulted in a decrease of the number of sequences of in-terest. In the present study, we used a different strategy to in-crease library complexity, which is summarized on Fig. S1B. AsIllumina analysis software (RTA) uses images from the first foursequencing cycles to detect the positions of DNA clusters on thesequencing slide, it is essential that a fluorescent signal from eachnucleotide channel (A, C, G, and T) be represented to detect theclusters properly and estimate background noise. Increased li-brary complexity was achieved for Read1 by designing 112 uniquebarcoded forward (BC-Fw) primers consisting of a common 17 bpHRAS sequence preceded by a unique barcode index of 4–8 bp.All primers also contained a 5′ NlaIII (5′-CATG-3′) restrictionsite, preceded by a 2-bp (CG) sequence [BC-Fw: 5′-CG-CATG-(BC)-AAGCTGGTGGTGGTGGG-3′]. When designing the 112BC sequences, we ensured that each of the four nucleotideswould be represented equally at each sequencing cycle [individualbarcode (BC) sequences are available on request; examples ofindividual BC-Fw primer sequences are shown in Fig. S1B]. ForRead2, increased complexity was provided through the use of 15different (five sets of three) nested reverse primers (NN-Rev)such that they all contained a 5′-tail consisting of the genericsequence of the Illumina Gex-adapter2 and 17–20 bp of HRAS-target sequence. However, each reverse primer differed by thepresence of variable sequence stuffer [NN-gg, where NN repre-sents either AA, CC, GG, TT, or no base pair addition and ggrepresents a variable number (0, 1 or 2) of G nucleotides] betweenthese two generic sequences (NN-Rev: 5′-GCATTCCTGCTGA-ACCGCTCTTCCGATCT-NN-gg-GTCGTATTCGTCCACAA-
3′); hence, the amplicons generated with these primers havelengths that vary by ± 1–5 bp, allowing staggering of the nu-cleotides incorporated at the different sequencing cycles (ex-amples of Rev-NN primer sets are shown in Fig. S1B). Each BC-Fwprimer was then assigned to one of the 5 NN-Rev primer sets.This scheme has the added advantage of permitting confirmation,during data analysis, that the BC index of a given BC-Fw primer(obtained at Read1) was correctly matched to their originally as-signed NN-Rev primer sets (obtained at Read2) and that no PCRjumping/template switching had occurred (see below).Five microliters of each PCR-1 sample was used as a template
and submitted to a set of three independent nested PCR (PCRreplicates) performed with Expand High FidelityPLUS PCR sys-tem reagents (Roche Applied Biosystems) in the following con-ditions: 1× HifiPLUS buffer, 2.5 mM MgCl2, 0.75 U HifiPLUS DNApolymerase, 200 μM dNTPs, and 0.1 μM each primer (either oneof three reverse primers from a given NN-Rev set + 1 uniqueBC-Fw primer, as explained above; Fig. S1). The following cy-cling conditions were used: initial 94 °C for 2 min, followed by 22cycles at 94 °C for 10 s, 53 °C for 20 s, and 68 °C for 20 s, followedby a final extension at 68 °C for 8 min.An aliquot of each PCR replicate was electrophoresed on a
3% agarose gel to ensure similar levels of amplification for eachreaction. Following this visual confirmation of amplification, weassumed that by mixing equal volumes of the PCR products, theywould be represented in the library in a near-equimolar ratio. Inparallel, the titration-reconstruction samples were also amplifiedwith unique BC-Fw primers (and specific NN-Rev primer sets)and subsequently added to the biological sample pools. Dupli-cates of control DNAs were amplified separately to avoid cross-contamination. One third of the controls PCR pool was separatelyadded to each library. For each library, the total pooled PCRproducts were then purified on an E.Z.N.A. PCR purificationcolumn (Omega Bio-Tek) before being subjected to digestionwith 100 U of NlaIII (New England Biolabs) for 1.5 h at 37 °C andthen dephosphorylated using 4 U of shrimp alkaline phosphatase(SAP; United States Biochemical) for 1 h at 37 °C followed byheat inactivation for 15 min at 80 °C. Resulting products werepurified on E.Z.N.A. MicroElute PCR purification columns(Omega Bio-Tek) and resuspended in a final volume of 15 μl.Illumina Gex NlaIII Adapter [consisting of the annealed pri-mers: Gex-Adapter1-P (5′Phos-TCGGACTGTAGAACTCTG-AAC-3′) and NlaIII-Gex1 (5′-ACAGGTTCAGAGTTCTACA-GTCCGACATG-3′)] were then ligated to 10 μL of the digested/SAP-treated PCR amplicons using the reagents from the RapidDNA Ligation kit (Fermentas) in a final 20-μL volume reactioncontaining 1× Ligation Buffer, 5 μM annealed Gex NlaIIIAdapter, and 5 U T4 DNA ligase for 20 min at 22 °C, followedby addition of another 5 U of T4 DNA ligase and incubation at22 °C for a further 60 min. The ligation reaction was again pu-rified using an E.Z.N.A. gel purification column (Omega Bio-Tek) and resuspended in 25 μL sterile water. Each library wasthen enriched by a final PCR amplification using 1 μL of 1 in 10dilution of the purified ligated PCR products and 0.8 U PhusionTaq DNA polymerase (New England Biolabs) in 1× HF buffer,0.1 μM each of Gex_PCR_primer1 (5′-AATGATACGGCGA-CCACCGAGATCTACACCGACAGGTTCAGAGTTCTACA-GTCCGA-3′) and Gex_PCR_primer2 (5′-CAAGCAGAAGA-CGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACC-GCTCTTCCGATCT-3′), and 200 μM dNTPs in the followingconditions: 98 °C for 30 s, followed by 9 cycles at 98 °C for 10 s,60 °C for 30 s, and 72 °C for 20 s, and a final extension at 72 °Cfor 10 min (Fig. S1B). The resulting PCR products were run ona 1.2% TAE agarose gel, and a band corresponding to 218 bpwas cut out, extracted with a MicroElute E.Z.N.A. gel purifica-tion column, and resuspended in 20 μL Tris-EDTA buffer (pH8.0). The three libraries were prepared independently to avoid
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 2 of 17

cross-contamination and were quantified using a fluorometer(Qubit) and a Bioanalyzer high sensitivity chip (Agilent).Massively parallel sequencing of the libraries was performed as
paired-end reads on three different lanes of an Illumina HiSeqsequencer for 50 cycles using the Gex-NlaIII sequencing primer(5′-CCGACAGGTTCAGAGTTCTACAGTCCGACATG-3′) forRead1 and the generic Illumina Read2 Seq primer for Read2.Read1 is designed to first capture the unique (4–8 bp) BC indexsequences, followed by a 17-bp HRAS sequence used for amplifica-tion (BC-Fw primers), and read the amplified DNA sequences fromHRAS position c.30 onward, whereas Read2 is designed to capturethe rs12628 C/T SNP phasing information in each read. All Illuminaprimers are proprietary sequences and individual primer combina-tions are available on request.All primers were synthesized by Sigma.Estimation of HRASmutation levels.Each sample was characterized bya unique 4–8 bp BC index within each one of the three libraries (Fig.S1B). Reads with unmatched BC indices (defined at cycles 1–4 tocycles 1–8, depending on the expected length of theBC index) as wellas reads with sequence errors within the HRAS-Fw target sequence(cycles 5–21 to cycles 9–25) were excluded from further analysis.The remainder (85.2%, 88.7%, and 84.5% of all reads in library1O, 2B, and 3C, respectively) was used for each individual sampleto obtain themutation profile around the p.A11 and p.G12 codonsand the phase in relation to the rs12628 C/T SNP for each read.Initial quality control of Read1 and Read2 datasets revealed
a number of reads containing erroneous pairs of forward (BC-Fw)and reverse (NN-Rev set) primers. The percentage of chimericreads (i.e., containing a given BC index in combination withunexpected reverse primers) was 23.17% in library 1O, 23.45% inlibrary 2B and 20.71% in library 3C. This chimera formation wasmost probably the result of annealing of an incompletely extendedprimer and template switching during DNA synthesis (“jumpingPCR”), which is a common feature of low complexity libraries(8). Reads that did not contain matching forward and reverseprimers were discarded from further analysis.To further eliminate errors caused by jumping PCR that might
have occurred at different positions of the amplicon [such as be-tween the BC and the nucleotide positions within the MspI site(c.32_c.35) or between c.32_35 and the rs12628 SNP], the controlDNA samples were used. For these samples, the DNA sequencewithin the MspI site (which is not under MspI selection) and thehaplotypewith respect to the rs12628 SNPare known.We thereforeconstructed confusion matrices for both theMspI site and the SNPby fitting a linearmodel to estimate the rates of jumping at differentpositionsof theampliconsequence,aswellas the ratesofmisreadingat thepositionswithin theMspI site and rs12628 SNP.TheMspI siteconfusion matrix MC = fmijg corresponds to the proportion of er-roneous reads with DNA sequence j when the true sequence is i.Similarly, the SNP confusion matrixMSNP contains the estimatedproportion of reads mapped at the correct position of the MspIsite but with the wrong haplotype at rs12628 SNP. Let r1, r2, and r3denote the respective rates of jumping between the forwardprimer (BC-Fw) and MspI site (c.32_c.35), between the MspI siteand the rs12628 SNP, and between the rs12628 SNPand the assignedreverse primer set (Rev-NN), and e1 and e2 denote the rate ofmisreading at theMspI site and the rate of misreading at the rs12628SNP, respectively. We formulate the following linear model:
1− r1 − r2 − r3 − e1 − e2 =FMSR1− r1 − r2 − e1 − e2 =FMS1− r1 − e1 =FM1− r1 − r2 − r3 − e1 =FM�R1− r1 − r2 − r3 − e2 =F�SR1− r1 − r2 − r3 =F—R;
where FMSR is defined as the percentage of reads that exhibitthe expected assignment of forward primer (F) and reverse
primer set (R), as well as the correct sequence within MspI site(M) and the rs12628 SNP (S), whereas FMS is defined as thepercentage of reads that show the correct assignment of F, M,and S only; FM is defined as the percentage of reads that exhibitthe expected F and M only; FM�R is defined as the percentageof reads with the anticipated F, M and R; F�SR is defined as thepercentage of reads with the correct F, S and R; and F—R isdefined as the percentage of reads with the correct F and R only.We assumed normal distribution of errors and we fitted themodel using linear least squares. Using the control DNA sam-ples, the rates of jumping and misreading at different positionsalong the amplicon were inferred and used to construct the es-timates of the confusion matrices.To estimate the mutation levels of each substitution within
the MspI site (c.32_35), we developed a Bayesian hierarchicalmodel that accounts for sequencing errors, noise of differentsequencing libraries, artifacts introduced during PCR rounds anddigestion.We fitted themodel to the observed read counts at eachpossible substitution in p.A11 and p.G12. Let i index the BC ofa given sample (i= f1; 2; :::;Ng, where N is the total number ofsamples), j index the mutation (j= f1; 2; :::; Jg, where J is thetotal number of mutations), α index the library (α= f1; 2; 3g),and k index the technical (PCR) replicate (k= f1; 2; 3g). Weassume that the counts of the mutations in the sequencing dataCαik = ½Ca
i1k;Cai2k; :::;C
αijk; :::;C
αiJk� are multinomial
Cαik ∼Multinomial
�jCαikj; p
�;
with frequency vector
p= 10Zαik
�XJ
j= 1
10Zαijk ;
where Zαijk is defined as the levels of mutation j in sample i, that
correspond in library α and technical replicate k. To eliminatePCR errors due to the formation of sequence chimeras, theconfusion matrices were used to update this frequency vectoras pcorrected = pMC for the MspI site estimates. Similarly, for eachpossible substitution in p.A11 or p.G12, the SNP confusion ma-trix was used to update the haplotype estimates.To describe the error structure of the data, we used a multilevel
model, where each level was used to construct the priors withina hierarchical structure. For each mutation j in sample i, theselevels were defined as (i) the mutation level of each technicalreplicate k and library α (Zα
ijk), (ii) the mutation level of eachlibrary α (denoted as Y α
ij ) across all technical replicates, and (iii)the uppermost level denoted Xij, which is the final required es-timate. If σiαz denotes the variance within each library and acrosstechnical replicates (of sample i) and σiY denotes the varianceacross libraries (again of sample i), we assume the followingprior structure:
Xij ∼Normalðμ0; σ0ÞY αij ∼Normal
�log10
�10Xij + Ij=spikesα
�; σiY
�Zαijk ∼Normal
�Y αij ; σ
iαz
�σiY ∼ InverseGammaðγ1; γ2Þσiαz ∼ InverseGammaðγ3; γ4Þ:
Xij denotes the inferred mutation levels of mutation j in sample i,and Y α
ij denotes the estimated counts of mutation j in sample i inlibrary α. For calibration purposes, we introduced the spike con-centration information at level (ii) of the hierarchical model; I isan indicator function that takes the value 0 or 1 depending onwhether the mutation in question is the same as the spike(c.35_36GC>AA), and sα is the spike concentration in library
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 3 of 17

α (s0 = 0; s1 = s2 = 3× 10−5). The hyper-parameters for the priorswere chosen to be μ0 = − 2; σ0 = 2; γ1 = γ3 = 3; γ2 = γ4 = 0:05. Theprior on the mutation levels corresponds to concentrated levelsnear zero but with heavy tails to allow for large nonzero values.The choice of the hyper-parameters of the inverse gamma dis-tribution of both variances (across replicates and across libraries)makes the distributions concentrated to values below 0.05 (butwith a long tail).For model inference, we used Metropolis-within-Gibbs sam-
pling, where σiαz and σiY are updated sequentially using Gibbssampling steps, whereas random walk Metropolis was performedfor the sequential updates of Y α
ij and Xij and the vector-wiseupdate of Zα
ik. Multiple runs from different starting points wereconducted to check for convergence. The model was fitted in-dependently on each sample and for all possible single- anddouble-nucleotide substitutions at codons p.A11 and p.G12.As shown in Fig. S2C (top two rows), estimated mutation levels
for biological samples homozygous for the rs12628 SNP exhibitedthe correct relationship in respect to their rs12628 genotype,showing that using this model the errors arising fromPCR jumping/chimera formation have been minimized.
Distribution of HRAS Mutations with Respect to the rs12628 SNP. Weestimated the relative distribution of mutations on the two HRASalleles by establishing the phase of each substitution with respectto the rs12628 SNP in 46 heterozygous sperm samples. Thisanalysis provides both an assessment of background mutationlevels associated with our assay and an estimation of the relativenumber of contributing mutational events to the total mutationcounts for each HRAS substitution within the MspI site. Giventhat each biological sample was processed in triplicate (and eachof these was further subjected to three independent nested PCR;see above and Fig. S1A), we reasoned that in individuals het-erozygous for the rs12628 SNP, low level background mutationsthat had arisen during in vitro processing of a sample (i.e., duringDNA extraction and/or PCR) were more likely to show a ran-dom (and more even) distribution of mutations across the twoHRAS alleles. Consistent with this interpretation, as illustratedon Fig. S2C (vertical dotted lines), preferential skewing on oneor other allele of the rs12628 SNP was only observed at mutationlevels >3 × 10−6, except for transitions where the threshold wasat >10−5.Moreover, in heterozygous sperm samples, we observed that
at mutation levels above background (>3–10 × 10−6), the SNPdistribution for all (single- and double-nucleotide) substitutionsinvolving changes at p.G12 shows a different pattern to thatobserved for the p.A11 codon mutations (Fig. 2B and Fig. S2C).For most sperm samples, mutations resulting in p.G12 activatingmutations exhibit a strong skewing to one or the other HRASallele (i.e., the C allele of the SNP is <20% or >80%), suggestingthat the measured levels are the result of a small number ofmutational events. No preferential skewing to one or the otherallele of the SNP was observed (permutation test P value from0.20 and 0.82 for different mutations). As previously reported inthe case of gain-of-function substitutions within FGFR2 (3, 9),this pattern of skewing at high levels of mutations is incompatiblewith any neutral model of mutation accumulation and stronglysuggests that the elevated levels of mutations observed at codonp.G12 in sperm are the result of ultra-rare mutational eventsfollowed by clonal expansion of the mutant spermatogonial cells.We observed that the c.34G>A mutation (p.G12S) showed amore even distribution across the two HRAS alleles than the otherp.G12 substitutions, consistent with the idea that this transitionoccurs more frequently than the other substitutions (Fig. S2C).However, we note that 7/11 samples with the highest c.34G>Amutation levels (>20 × 10−6) exhibit a significant skewing to one orthe other HRAS allele. This result suggests that, although theoriginating mutations occur more frequently than is the case for
the other substitutions, the high c.34G>A levels are unlikely tohave arisen through many independent mutational events duringspermatogenesis.
Analysis of Tandem Base Substitutions in Genomewide Datasets. Wequeried the Human Gene Mutation Database (HGMD Pro-fessional 2012.3, release date, September 28, 2012), containing130,522 mutations associated with genetic diseases, for tandembase substitutions (TBS) reported as delXXinsXX, where X = [A,G, C, T]. Although 144 different permutations of 2 bp couldtheoretically be anticipated, due to the complementary nature ofDNA, only 78 different TBS can occur (other combinations leadto single-nucleotide substitutions or are identical to their reversecomplements). A total of 441 events of TBS have been reported inHGMD and the GC>TT (and its reverse complement GC>AA)substitution was found in 65 occurrences (Table S3). To determinewhether single-nucleotide substitutions have also been reported atthe positions where GC>TT/AA TBS have been observed, wefurther queried the database for single-nucleotide substitutionspresent either in the first or second genomic position of theGC>TT (or its reverse complement, GC>AA) TBS. A total of 38variants that met either one of these criteria were found inHGMD and 14 mutations were shown to encode an identicalprotein change to that reported as a TBS.We also queried the Catalogue of Somatic Mutations in Cancer
(COSMIC v63) that lists 5,171,967 variants identified in tumors(corresponding to 620,857 unique variants) for TBS listed asXX>XX, where X = [A, G, C, T]. A total of 5,080 TBS havebeen cataloged in COSMIC, among which 3,769 are unique(Table S3). The GC>TT (or its reverse complement GC>AA)TBS has been reported in 107 tumor samples.Genomewide analysis of TBS variants was investigated using
genotype calls generated by a de novo assembly-based variantcaller [Cortex algorithm (10)]. The Cortex algorithm uses coloredde Bruijn graphs that allow the detection and accurate geno-typing of complex variants, such as TBS, that are commonlyfiltered out when mapping-based approaches are used. Theprevalence of TBS was assessed in 85 human genomes from theLWK (Luhya in Webuye, Kenya) population used for phase 1 ofthe 1000 Genomes Project (11). The callset was produced byassembling each sample, pooling them together into one deBruijn graph, error cleaning the graph, calling variants in thegraph, and subsequently genotyping each sample at those sitesusing the per-sample assemblies. The SNP and complex variantcalls were polarized to determine the ancestral allele, using theHuman Ancestral Genome from Ensembl 57. Both the callset(with details of how the calls were made) and the ancestral ge-nome are available at URLs listed at the end of this document.A total of 5,425,856 nucleotide polymorphisms, both single and
complex variants, were identified using this approach, amongwhich 22,898 (0.42%) involved TBS and 4,718 (0.087%) triple orother multinucleotide polymorphisms. The GC>TT/AA changewas the second most common TBS, observed in 1,417 instances(6.2%), including 12 exonic calls. We note that the most commonTBS in the human genome is the quasi-palindromic changeCA>TG (and its reverse complement TG>CA) (11%), althoughthis TBS represents only 0.3% of occurrences in HGMD. Thisfinding is supported by previous studies (12, 13); Schrider et al.(13) have suggested that the genomewide overrepresentation ofthe CA>TGTBSmay be in part explained by the occurrence of anintermediate CpG hypermutable state. Hence this TBS might bethe result of sequential mutational events in the form of CA>CGfollowed by CG>TG. The distribution and prevalence of TBS inthe 85 LWK genome sequences are given in Table S3.To establish whether the local sequence contextmight contribute
to the high mutation rate observed for the HRAS c.35_36GC>TT(p.G12V) TBS, we analyzed the relative distribution of the se-quences adjacent to GC>TT/AA TBS described in the 85 LWK
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 4 of 17

genomes and compared it to the context in which each single-nucleotide polymorphism (G>T or C>T and their reversecomplements C>A or G>A) occurs. To perform this analysis, weextracted G>T SNP Cortex calls occurring 5′ of a cytosine (suchas GC>TC, where the underlined nucleotide is the mutation) orequivalent reverse complement GC>GA). A total of 103,732GC>TC (and GC>GA) SNPs were found across the 85 LWKgenomes. We then conditioned on the nucleotide located 5′ ofthe G>T (or 3′ of C>A) SNP and of the GC>TT (or 3′ ofGC>AA) TBS, in a way that is relevant to the sequence contextfound in HRAS c.34_36. We found that conditioning for thepresence of a guanine 5′ of the SNP or TBS (or a cytosine 3′ tothe reverse complement) did not change the relative frequencyof these polymorphisms (28,335 of 103,732 SNPs [correspondingto 27% of all GG>GT (or CC>AC)], compared with 342 of1,417 TBS [corresponding to 24% of all GGC>GTT (orGCC>AAC)]). However, when we conditioned for the presenceof a guanine located 3′ to the SNP or the TBS (or a cytosine 5′ tothe reverse complement), we found that in this sequence context,TBS occurred almost twice as often as SNPs [2,892 of 103,732(2.8%) CG>TG (or CG>CA) SNPs, compared with 78 of 1,417(5.5%) of GCG>TTG (or CGC>CAA) TBS]. Of note, the smallnumber of observed occurrences of these events is consistentwith the expected under-representation of CpG dinucleotidesequences across the human genome (14). By further condi-tioning for the simultaneous presence of guanines on the 3′ and5′ side of the SNP [GCG>GTG (or CGC>CAC)] or TBS[GGCG>GTTG (or CGCC>CAAC)], we found frequencies of842 of 103,732 (0.81%) and 35 of 1,417 (2.5%), respectively.Hence, in this sequence context, which is identical to the contextin which the HRAS c.35_36GG>TT occurs, TBS are enrichedthreefold (P = 2.2 × 10−8) compared with the corresponding SNPs.Functional annotation of all observed variants was performed
using ANNOVAR 2011Nov01 (15).
Spermatocytic SeminomaMutation Screening.We obtained 33 sper-matocytic seminoma (SpS) collected from tissue archives fromhospitals in Holland, Denmark, and Sweden. DNA was extractedfrom frozen tissues and paraffin-embedded samples as previouslydescribed (4). Mutation hotspots in FGFR3 and HRAS wereanalyzed by PCR amplification followed by Sanger DNA se-quencing and/or restriction enzyme digestion. The primers,PCR, and sequencing conditions used are the same as thosedescribed previously (4), and hotspots analyzed are listed inTable S4. Age information was available for 26 SpS and wascombined with SpS age data from 28 SpS analyzed in a previousstudy (4) (Fig. S3C). The average age at diagnosis of SpS was59.2 y (28–89 y). Mutation-positive samples (n = 9) have sig-nificantly older diagnostic age [average: 72.7 y (67–87 y)] thanmutation-negative samples [average: 55.9 y (28–89 y); Student ttest, P = 0.0002].
Modeling Mutation Rate and Selective Advantage. Human sper-matogenesis is an ongoing process that, from puberty (13 y old)onward, requires regular divisions of spermatogonial stem cellsevery 16 days, corresponding to 23 divisions per year (16). Let Aidenote the age of sample i, the number of spermatogonial celldivisions αi are given by
αi = 23ðAi − 13Þ:
The number of mutations Mi that occur in αi cell divisions aremodeled by a Poisson distribution with parameter μ that denotesthe mutation rate per cell division. Given that for 46 heterozy-gous sperm samples, the HRAS mutation levels in sperm canbe partitioned across the two HRAS alleles (with respect tors12628 SNP), we model the number of mutations occurring ineach allele as
Mheti ∼PoissonðαiμÞ;
whereas for the 43 homozygous samples the number of mutationsis given by
Mhomi ∼Poissonð2αiμÞ:
We assume that the time at which a mutation k happens insample i is uniform on ½0; αi�
dk;i ∼Uniformð0; αiÞ:
To model the selective advantage, we assume a simple scenariowhereat each cell generation, amutant spermatogonial cell dividessymmetrically with probability p to produce two identical mutantcells. The subsequent divisions will lead to exponential growth ofthe mutant cells with rate s producing the total number of mu-tated cells f at time t given by f ðtÞ= f0 expðstÞ, where f0 denotesthe number of mutant cells at time 0. We assume a continuoustime exponential growth approximation, which leads to s≅ p.The total spermmutation level of sample i is the randomsumof all
of the mutations that have accumulated through exponential growthfrom the time of occurrence di;k up to the current cell division αi
Vi =XMi
k= 1
f0 exp�s�αi − dk;i
��:
Our aim was to infer μ and s for each mutation within the MspIsite (HRAS c.32_35) using the estimated mutation levels quan-tified in 89 sperm samples. Because our model does not havea closed-form likelihood, an empirical likelihood had to be ap-proximated via a Monte Carlo approach. Using a grid of valuesfor s in the interval [0, 0.008] with step size of 0.0001, and a gridof values for μ within the interval [1 × 10−12, 1 × 10−8], with stepsize increasing linearly within each order of magnitude, we sim-ulated data under the model for n = 1,000 times. At each sim-ulation we counted the number of occurrences in a discretized2 × 2 grid, which consists of sample age within the interval [20,80] with step size of 6 y, and the log10 total mutation levels (permillion) within the interval [−1, 3] (step size of 0.2). The grid ofoutcomes built by these simulations was then used to calculate fitfor each mutation. For the estimation of 95% confidence inter-vals, we extracted all of the values of s and μ in the grid for whichlnðLmaxÞ− lnðLÞ≤ 1:92 where Lmax is the maximum (empirical)log-likelihood. Table S5 shows the minimum and maximumrange of these values for which the log-likelihood drops off byno more than 1.92 units. Because our values are not continuousbut chosen from a grid, we graphically constructed an area ofvalues within the 95% confidence intervals as shown in Fig. 4.We inferred themutation rate μ and selection coefficient s for each
mutation at HRAS p.A11 and p.G12 codons independently. Muta-tions found in fewer than eight individuals (threshold for callingmutations was 3 × 10−6, except for transitions for which it was 10−5)showed high uncertainty in the inferred parameters and were ex-cluded from downstream analysis. For the remaining mutations, inthe case of substitutions with the same amino acid change [c.35G>T(p.G12V) and c.35_36GC>TT (p.G12V)], we fixed the selectioncoefficient s to be the same to account for the fact that selection actsat the protein level. For thesemutations, bymerging the sample datawe borrow information across both mutations allowing a better es-timate of the mutation rate μ and selection coefficient s.For validation purposes, we applied our model to other datasets
obtained from previous studies of sperm mutation levels occurringin FGFR2 at the p.S252 (3, 17) and p.P253 (17) codons andFGFR3 at the p.K650 codon (4).
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 5 of 17

URLs. UK Office for National Statistics. England & Wales birth sta-tistics (Table S1 was used for birth statistics and Table S2 forcalculation of PAE): http://www.ons.gov.uk/ons/rel/vsob1/birth-summary-tables–england-and-wales/2011–final-/rft-births-summary-tables-2011-final.xls.Scotland birth statistics (Table 3.1b): http://www.gro-scotland.
gov.uk/statistics/theme/vital-events/general/ref-tables/2011/index.html.Northern Ireland birth statistics (Table entitled Live births, 1887–
2011): http://www.nisra.gov.uk/demography/default.asp8.htm.COSMIC. http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/.
README document describing how the Cortex calls were made from1000 Genomes LWK set. ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20120502_phase2_variant_calls/OX/README_20130612_cortex_LWK_calls.1000 Genomes LWK calls. ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20120502_phase2_variant_calls/OX/LWK.wgs.cortex_ox_v3_raw.20111114.snps_indels_and_mnps.low_coverage.genotypes.vcf.gz.Human Ancestral Genome sequence used for allele polarization. ftp://ftp.ebi.ac.uk/pub/databases/ensembl/jherrero/ancestral/homo_sapiens_ancestor_GRCh37_e63.tar.bz2.
1. Kong A, et al. (2012) Rate of de novo mutations and the importance of father’s age todisease risk. Nature 488(7412):471–475.
2. Abe Y, et al.; Costello and CFC syndrome study group in Japan (2012) Prevalence andclinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan:Findings from a nationwide epidemiological survey. Am J Med Genet A 158A(5):1083–1094.
3. Goriely A, McVean GA, Röjmyr M, Ingemarsson B, Wilkie AOM (2003) Evidence forselective advantage of pathogenic FGFR2 mutations in the male germ line. Science301(5633):643–646.
4. Goriely A, et al. (2009) Activating mutations in FGFR3 and HRAS reveal a sharedgenetic origin for congenital disorders and testicular tumors. Nat Genet 41(11):1247–1252.
5. Burkitt-Wright EM, et al. (2012) Neonatal lethal Costello syndrome and unusualdinucleotide deletion/insertion mutations in HRAS predicting p.Gly12Val. Am J MedGenet A 158A(5):1102–1110.
6. Lo IF, et al. (2008) Severe neonatal manifestations of Costello syndrome. J Med Genet45(3):167–171.
7. Krueger F, Andrews SR, Osborne CS (2011) Large scale loss of data in low-diversityillumina sequencing libraries can be recovered by deferred cluster calling. PLoS ONE6(1):e16607.
8. Kanagawa T (2003) Bias and artifacts in multitemplate polymerase chain reactions(PCR). J Biosci Bioeng 96(4):317–323.
9. Goriely A, Wilkie AOM (2012) Paternal age effect mutations and selfish spermatogonialselection: Causes and consequences for human disease. Am J Hum Genet 90(2):175–200.
10. Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G (2012) De novo assembly andgenotyping of variants using colored de Bruijn graphs. Nat Genet 44(2):226–232.
11. Abecasis GR, et al.; 1000 Genomes Project Consortium (2012) An integrated map ofgenetic variation from 1,092 human genomes. Nature 491(7422):56–65.
12. Dawson E, et al. (2001) A SNP resource for human chromosome 22: Extracting denseclusters of SNPs from the genomic sequence. Genome Res 11(1):170–178.
13. Schrider DR, Hourmozdi JN, Hahn MW (2011) Pervasive multinucleotide mutationalevents in eukaryotes. Curr Biol 21(12):1051–1054.
14. Cooper DN, Gerber-Huber S (1985) DNA methylation and CpG suppression. Cell Differ17(3):199–205.
15. Wang K, Li M, Hakonarson H (2010) ANNOVAR: Functional annotation of geneticvariants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164.
16. Crow JF (2000) The origins, patterns and implications of human spontaneousmutation. Nat Rev Genet 1(1):40–47.
17. Yoon SR, et al. (2009) The ups and downs of mutation frequencies during aging canaccount for the Apert syndrome paternal age effect. PLoS Genet 5(7):e1000558.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 6 of 17
Fig. S1. Overview of the experimental design used for quantification of de novo HRAS mutations at codons p.A11 and p.G12. (A) Graphic summary of thedifferent steps involved in processing biological samples, selecting for MspI resistance, PCR amplifying, preparing the Illumina libraries, and quantifying HRASmutations. (B) Overview of the strategy used to prepare the barcode (BC), the PCR replicates and to increase the complexity of the Illumina libraries. Fordetailed description and abbreviations, see SI Text.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 7 of 17

c.35G>C p.G12A vs. age
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
A11 G12GCC GGC
32 33 34 35CC GG
Not assayed
c.32C>T p.A11V vs. ager_s = 0.24
Age(years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.32C>G p.A11G vs. ager_s = 0.0801
Age(years)M
utat
ion
leve
ls (
per
mill
ion)
0.1
110
100
1000
20 30 40 50 60 70
c.32C>A p.A11D vs. ager_s = 0.36
Age(years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.33C>T p.A11A vs. ager_s = 0.248
Age(years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.33C>G p.A11A vs. ager_s = 0.192
Age(years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.33C>A p.A11A vs. ager_s = 0.267
Age(years)M
utat
ion
leve
ls (
per
mill
ion)
0.1
110
100
1000
20 30 40 50 60 70
c.34G>A p.G12S vs. ager_s = 0.521
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.34G>T p.G12C vs. ager_s = 0.192
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35G>A p.G12D vs. ager_s = 0.485
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35G>T p.G12V vs. ager_s = 0.396
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.34G>C p.G12R vs. ager_s = 0.316
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35_36GC>AA p.G12Ec.35_36GC>AG p.G12Ec.35_36GC>AT p.G12Dc.35_36GC>CA p.G12Ac.35_36GC>CG p.G12Ac.35_36GC>CT p.G12Ac.35_36GC>TA p.G12Vc.35_36GC>TG p.G12Vc.35_36GC>TT p.G12V
c.34G>A; c.36C>A p.G12Rc.34G>A; c.36C>G p.G12Rc.34G>A; c.36C>T p.G12Sc.34G>C; c.36C>A p.G12Rc.34G>C; c.36C>G p.G12Rc.34G>C; c.36C>T p.G12Rc.34G>T; c.36C>A p.G12*c.34G>T; c.36C>G p.G12Wc.34G>T; c.36C>T p.G12C
TBS c.34_35GG Non-TBS c.34G; c.36CTBS c.35_36GC
c.34_35GG>AA p.G12Nc.34_35GG>AC p.G12Tc.34_35GG>AT p.G12Ic.34_35GG>CA p.G12Hc.34_35GG>CC p.G12Pc.34_35GG>CT p.G12Lc.34_35GG>TA p.G12Yc.34_35GG>TC p.G12Sc.34_35GG>TT p.G12F
c.34_35GG>TT p.G12F vs. ager_s = 0.162
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35_36GC>AA p.G12E vs. ager_s = 0.127
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35_36GC>TA p.G12V vs. ager_s = 0.195
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35_36GC>AT p.G12D vs. ager_s = 0.443
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.35_36GC>TT p.G12V vs. ager_s = 0.439
Age (years)
Mut
atio
n le
vels
(pe
r m
illio
n)0.
11
1010
010
00
20 30 40 50 60 70
c.34G>A;c.36C>T p.G12S vs. ager_s = 0.0179
Age (years)M
utat
ion
leve
ls (
per
mill
ion)
0.1
110
100
1000
20 30 40 50 60 70
A
B
Cc.32C>T p.A11V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.33C>T p.A11A
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.32C>T p.A11V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.33C>T p.A11A
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.34G>A p.G12S
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.34G>T p.G12C
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35G>A p.G12D
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35G>T p.G12V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35_36GC>AT p.G12D
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35_36GC>TT p.G12V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.34G>A p.G12S
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.34G>T p.G12C
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35G>A p.G12D
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35G>T p.G12V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35_36GC>AT p.G12D
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
c.35_36GC>TT p.G12V
Mutation levels (per million)
Pro
port
ion
on C
alle
le
0.1 1 10 100 1000
0.0
0.2
0.4
0.6
0.8
1.0
Fig. S2. Mutation levels and skewing with respect to C/T rs12628 SNP for individual substitutions selected by digestion with MspI (gray box) and involvingHRAS p.A11 and p.G12 codons. (A) Level of mutation for individual single-nucleotide substitutions plotted against the donor’s age. Note that the panels havebeen organized according to mutation type (with transitions in the top row and transversions in the middle and bottom rows). (B) Sequence context andmutation levels plots for a subset of dinucleotide substitutions observed to be elevated in sperm samples (in bold). Mutation levels are given per million againstthe age of the donor, and r_s represents the Spearman coefficient of correlation between mutation levels and donor’s age. In A and B, red, blood DNA; black,sperm DNA. (C) Individual skewing plots with respect to total mutation levels for samples homozygous (two top rows) and heterozygous (two bottom rows) forrs12628 C/T SNP. Green, CC homozygous samples; blue, TT homozygous samples; black, CT heterozygous sperm samples; red, CT heterozygous blood samples.(Thresholds for mutation calling as defined in text are indicated by a dotted line.)
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 8 of 17

Fig. S3. Analysis of spermatocytic seminoma (SpS) samples. Sequencing traces in the samples [SS24, patient aged 79 y (A); H4T, patient aged 82 y (B)] identifythe homozygous HRAS mutations c.37G>C (p.G13R) and c.182A>G (p.Q61R), respectively, in tumor tissue (Lower, red arrow) but not in the matched histo-logically normal tissue (Upper). The red boxes represent the frame of the codon affected. (C) Age distribution at time of diagnosis for 54 SpS includingmutational status as indicated on the figure. Data are combined with those published in Goriely et al. 2009 (1). Age at diagnosis for SpS with HRAS or FGFR3mutations (average: 72.7 y) was significantly higher than that for the mutation-negative samples (average: 55.9 y) (t test, P = 0.0002).
1. Goriely A, et al. (2009) Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet 41(11):1247–1252.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 9 of 17

Table
S1.
Cases
ofCSasso
ciated
withHRASmutationsreported
intheliterature
p.G12
Sp.G12
Cp.G12
Dp.G12
Ap.G12
Vp.G12
Vp.G12
Vp.G12
Ep.G13
Cp.G13
Dp.Q
22K
p.E37
Dup
p.E37
Dup
p.T58
Ip.E63
KE6
3_D69
dup
p.S89
Cp.K11
7Rp.A14
6Tp.A14
6V
c.34
G>A
c.34
G>T
c.35
G>A
c.35
G>C
c.35
G>T
c.35
_36
GC>TA
c.35
_36
GC>TT
c.35
_36
GC>AA
c.37
G>T
c.38
G>A
c.64
C>A
c.11
0_11
1+1d
upAGG
c.10
8_11
0dupAGA
c.17
3C>T
c.18
7G>A
c.18
7_20
7dup
c.26
6C>G
c.35
0A>G
c.43
6G>A
c.43
7C>T
Refs.
91
(1)
292
31
1(2)
444
1(3–5)
3(6)
11
11
(7)
2(8)
81
(9)
61
23
(10)
11
(11)
233
1(12)
1(13)
1(14)
21
(15)
1(16)
11
(17)
102
1(18)
12(19)
271
15
12
1(20–
22)
1(23)
11
2(24)
11
(25)
1(26)
1(27)
1(28)
1(29)
167
65
221
14
115
21
11
21
11
21
1To
tal
Asthesamepatientsamplesareregularlyusedin
differentstudies,wehav
etake
ncare
tocross-referen
cestudiesto
countpatients
only
once,wherev
erpossible
theoriginal
reference
isgiven
.
1.So
l-ChurchK,Stab
leyDL,
NicholsonL,
Gonza
lezIL,GrippKW
(200
6)Pa
ternal
biasin
paren
talorigin
ofHRASmutationsin
Costello
syndrome.
Hum
Mutat27
(8):73
6–74
1.2.
KerrB,et
al.(200
6)Gen
otype-phen
otypeco
rrelationin
Costello
syndrome:
HRASmutationan
alysisin
43cases.JMed
Gen
et43
(5):40
1–40
5.3.
GrippKW
,et
al.(200
6)HRASmutationan
alysisin
Costello
syndrome:
Gen
otypean
dphen
otypeco
rrelation.Am
JMed
Gen
etA
140(1):1–7.
4.EstepAL,
Tidym
anW
E,Te
itellM
A,C
otter
PD,R
auen
KA(200
6)HRASmutationsin
Costello
syndrome:
Fetectionofco
nstitutional
activa
tingmutationsin
codon12
and13
andloss
ofwild
-typ
eallele
inmalignan
cy.A
mJMed
Gen
etA14
0(1):8–16
.5.
LinAE,
Rau
enKA,GrippKW
,Carey
JC(200
8)Clarificationofpreviouslyreported
Costello
syndromepatients.Am
JMed
Gen
etA
146(7):940
–94
3.6.
vanStee
nselMA,et
al.(200
6)RecurringHRASmutationG12
Sin
Dutchpatients
withCostello
syndrome.
ExpDermatol15
(9):73
1–73
4.7.
vander
BurgtI,et
al.(200
7)Myo
pathycausedbyHRASgermlin
emutations:Im
plicationsfordisturbed
myo
gen
icdifferentiationin
thepresence
ofco
nstitutive
HRas
activa
tion.JMed
Gen
et44
(7):45
9–46
2.8.
ØrstavikKH,Ta
ngeraa
sT,
Molven
A,Presco
ttTE
(200
7)Distalphalan
gea
lcrea
ses—
Adistinctivedysmorphic
feature
indisordersoftheRASsignallin
gpathway
?Eu
rJMed
Gen
et50
(2):15
5–15
8.9.
ZampinoG,et
al.(200
7)Diversity,paren
talgermlin
eorigin,an
dphen
otypic
spectrum
ofdenovo
HRASmissense
chan
ges
inCostello
syndrome.
Hum
Mutat28
(3):26
5–27
2.10
.Lo
IF,et
al.(200
8)Se
vere
neo
natal
man
ifestationsofCostello
syndrome.
JMed
Gen
et45
(3):16
7–17
1.11
.GrippKW
,et
al.(200
8)Costello
syndromeassociated
withnove
lgermlin
eHRASmutations:Anattenuated
phen
otype?
Am
JMed
Gen
etA
146A
(6):68
3–69
0.12
.Schulz
AL,
etal.(200
8)Mutationan
dphen
otypic
spectrum
inpatients
withcardio-facio-cutaneo
usan
dCostello
syndrome.
Clin
Gen
et73
(1):62
–70
.13
.Sk
órkaA,et
al.(201
2)A
girlwithtw
osyndromes:Tu
rner
syndromean
dCostello
syndrome.
Acase
history.Am
JMed
Gen
etA
158A
(6):14
86–14
88.
14.Zh
angH,YeJ,GuX(200
9)RecurringG12
SmutationofHRASin
aChinesech
ildwithCostello
syndromewithhighalka
linephosphataseleve
l.Bioch
emGen
et47
(11-12
):86
8–87
2.15
.LinAE,
etal.(200
9)Pren
atal
featuresofCostello
syndrome:
Ultrasonographic
findingsan
datrial
tach
ycardia.Pren
atDiagn29
(7):68
2–69
0.16
.KunibaH,et
al.(200
9)Pren
atal
diagnosisofCostello
syndromeusing3D
ultrasonographyam
niocentesisco
nfirm
ationoftherare
HRASmutationG12
D.Am
JMed
Gen
etA
149A
(4):78
5–78
7.17
.Gremer
L,et
al.(20
10)DuplicationofGlu37
inthesw
itch
Ireg
ionofHRASim
pairs
effector/GAPbindingan
dunderlie
sCostello
syndromebypromotingen
han
cedgrowth
factor-dep
enden
tMAPK
andAKTactiva
tion.H
um
MolG
enet
19(5):7
90–80
2.18
.GrippKW
,HopkinsE,
DoyleD,D
obyn
sW
B(201
0)Highinciden
ceofprogressivepostnatal
cerebellaren
largem
entin
Costello
syndrome:
Brain
ove
rgrowth
associated
withHRASmutationsas
thelik
elycause
ofstructuralb
rain
andspinal
cord
abnorm
alities.Am
JMed
Gen
etA
152A
(5):11
61–11
68.
19.GrippKW
,et
al.(201
1)Ph
enotypic
analysisofindividualswithCostello
syndromedueto
HRASp.G13
C.Am
JMed
Gen
etA
155A
(4):70
6–71
6.20
.AbeY,e
tal.;Costello
andCFC
syndromestudygroupin
Japan
(201
2)Prev
alen
cean
dclinical
featuresofCostello
syndromean
dcardio-facio-cutaneo
ussyndromein
Japan
:Findingsfrom
anationwideep
idem
iological
survey
.Am
JMed
Gen
etA
158A
(5):10
83–10
94.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 10 of 17

21.Aoki
Y,et
al.(200
5)Germlin
emutationsin
HRASproto-onco
gen
ecause
Costello
syndrome.
Nat
Gen
et37
(10):103
8–10
40.
22.Niih
oriT,
etal.(201
1)HRASmutants
iden
tified
inCostello
syndromepatients
caninduce
cellu
larsenescence:Po
ssible
implicationsforthepathogen
esisofCostello
syndrome.
JHum
Gen
et56
(10):707
–71
5.23
.Simsek-Kiper
PO,et
al.(201
3)Clin
ical
andmolecu
laran
alysisofRASo
pathiesin
agroupofTu
rkishpatients.Clin
Gen
et83
(2):18
1–18
6.24
.Burkitt-W
rightEM
,et
al.(201
2)Neo
natal
lethal
Costello
syndromean
dunusual
dinucleo
tidedeletion/in
sertionmutationsin
HRASpredictingp.Gly12
Val.Am
JMed
Gen
etA
158A
(5):11
02–11
10.
25.Lo
renzS,
etal.(201
2)Tw
ocaseswithseve
relethal
courseofCostello
syndromeassociated
withHRASp.G12
Can
dp.G12
D.Eu
rJMed
Gen
et55
(11):615
–61
9.26
.Ta
jirM,et
al.(201
2)[Costello
syndrome:
report
ofacase].Pa
nAfr
Med
J12
:64.
27.GrippKW
,et
al.(201
2)A
nove
lHRASsubstitution(c.266
C>G;p.S89
C)resultingin
decreased
downstream
signalingsuggests
anew
dim
ensionofRASpathway
dysregulationin
human
dev
elopmen
t.Am
JMed
Gen
etA
158A
(9):21
06–21
18.
28.GrippKW
,et
al.(201
2)Tran
smissionoftherare
HRASmutation(c.17
3C>T;
p.T58
I)further
illustratesitsattenuated
phen
otype.
Am
JMed
Gen
etA
158A
(5):10
95–11
01.
29.Lo
renzS,
etal.(201
3)Fu
nctional
analysisofaduplication(p.E63
_D69
dup)in
thesw
itch
IIregionofHRAS:
new
aspects
ofthemolecu
larpathogen
esisunderlyingCostello
syndrome.
Hum
MolGen
et22
(8):16
43–16
53.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 11 of 17

Table S2. Estimates of levels of single- and double-nucleotide substitutions at HRAS codons p.A11 and p.G12 in sperm (n = 89)and blood (n = 7) samples
HRAS nucleotideand (amino acidsubstitution)
Mean mutationlevel in sperm(per million)
Mutation levelrange (sperm)(per million)
Number ofsperm
samples abovebackground*
Correlation of spermmutation levelswith age (rs)
P value(rs)
†
Mean mutationlevel in blood(per million)
Mutation levelrange (blood)(per million)
P value(sperm vs.blood)‡
c.32C>A (p.A11D) 0.173 0.0126–1.546 0 0.360 0.00052 0.538 0.0359–2.702 0.3542c.32C>G (p.A11G) 0.017 0.0006–0.125 0 0.080 0.4555 0.073 0.007–0.317 0.2347c.32C>T (p.A11V) 2.972 0.237–16.649 3* 0.240 0.0235 2.614 0.328–6.628 0.7463c.33C>A (p.A11A) 1.018 0.137–3.879 4 0.267 0.0114 2.001 0.340–4.907 0.2023c.33C>G (p.A11A) 0.150 0.012–0.788 0 0.192 0.0712 0.314 0.018–1.125 0.3187c.33C>T (p.A11A) 6.214 0.753–41.225 11* 0.248 0.0194 7.882 1.430–19.509 0.5509c.34G>A (p.G12S) 19.954 1.692–115.638 55* 0.521 0.0000002 8.189 2.749–19.682 0.0017c.34G>C (p.G12R) 0.862 0.042–44.039 3 0.316 0.0026 0.227 0.038–0.483 0.2061c.34G>T (p.G12C) 2.998 0.201–34.923 20 0.192 0.0719 1.483 0.409–3.548 0.0611c.35G>A (p.G12D) 6.236 0.503–32.725 17* 0.485 0.000001 1.621 0.448–4.547 0.00002c.35G>T (p.G12V) 2.091 0.045–14.339 18 0.396 0.0001 0.300 0.076–0.570 0.0000c.34_35GG>TT (p.G12F) 0.175 0.0001–13.998 1 0.162 0.1301 0.002 0.0002–0.003 0.2735c.34G>A;c.36C>T (p.G12S) 0.471 0.078–5.204 1 –0.018 0.8680 0.652 0.221–1.392 0.3105c.35_36GC>AA (p.G12E) 0.473 0.049–4.618 1 –0.127 0.2338 0.913 0.235–1.581 0.0527c.35_36GC>AT (p.G12D) 0.512 0.002–11.602 4 0.443 0.00001 0.018 0.005–0.066 0.0114c.35_36GC>TA (p.G12V) 0.135 0.0002–4.169 3 0.195 0.0668 0.003 0.0001–0.008 0.0733c.35_36GC>TT (p.G12V) 3.623 0.039–38.942 21 0.439 0.00002 0.119 0.056–0.214 0.00002
*Samples were considered to be above background for mutation levels >3 × 10−6, except for the indicated single-nucleotide transitions for which the callingthreshold was >10−5.†P values describing the statistical significance of the Spearman coefficient of correlation (rs) between sperm mutation levels and donor age.‡A Student t test was used to assess the statistical significance of the difference between mutation levels observed in sperm and blood samples.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 12 of 17
Table S3. Total number of TBS occurrences observed in HGMD, in the COSMIC database, and across 85 LWK whole genome sequences(Cortex aligner)
TBSTBS (reversecomplement)
Number ofoccurrencesin HGMD
RelativeproportionHGMD
Number ofoccurrencesin COSMIC
RelativeproportionCOSMIC
Number ofvariants in
LWK (Cortex)
Relativeproportion
LWK (Cortex)
GC>AA GC>TT 65 14.7%* 107 2.8%† 1,417 6.2%‡
GA>TT TC>AA 29 6.6% 85 2.3% 898 3.9%CC>AA GG>TT 27 6.1% 1185 31.4% 489 2.1%GC>AG GC>CT 18 4.1% 35 0.9% 359 1.6%CC>TT GG>AA 16 3.6% 725 19.2% 602 2.6%GA>AT TC>AT 16 3.6% 48 1.3% 590 2.6%GC>AT GC>AT 10 2.3% 22 0.6% 519 2.3%AG>CC CT>GG 10 2.3% 12 0.3% 294 1.3%CC>GA GG>TC 10 2.3% 73 1.9% 144 0.6%GA>AG TC>CT 9 2.0% 25 0.7% 621 2.7%AC>GA GT>TC 9 2.0% 7 0.2% 287 1.3%AA>GC TT>GC 8 1.8% 10 0.3% 587 2.6%CA>AG TG>CT 8 1.8% 87 2.3% 497 2.2%AG>CT CT>AG 8 1.8% 26 0.7% 218 1.0%CC>TA GG>TA 8 1.8% 78 2.1% 213 0.9%GC>CA GC>TG 8 1.8% 19 0.5% 152 0.7%CC>GG GG>CC 8 1.8% 16 0.4% 111 0.5%CC>AT GG>AT 7 1.6% 175 4.6% 310 1.4%CC>AG GG>CT 7 1.6% 158 4.2% 300 1.3%CA>TT TG>AA 7 1.6% 38 1.0% 274 1.2%CA>AT TG>AT 7 1.6% 71 1.9% 249 1.1%AC>CT GT>AG 7 1.6% 15 0.4% 195 0.9%GC>TA GC>TA 7 1.6% 16 0.4% 115 0.5%GA>TC TC>GA 6 1.4% 12 0.3% 218 1.0%CG>AA CG>TT 6 1.4% 66 1.8% 203 0.9%GA>CT TC>AG 6 1.4% 19 0.5% 194 0.8%GA>AC TC>GT 5 1.1% 10 0.3% 261 1.1%AG>TT CT>AA 5 1.1% 76 2.0% 244 1.1%AC>CA GT>TG 5 1.1% 14 0.4% 198 0.9%CG>TA CG>TA 5 1.1% 23 0.6% 154 0.7%AA>TT TT>AA 4 0.9% 22 0.6% 489 2.1%AG>GA CT>TC 4 0.9% 17 0.5% 450 2.0%AC>TT GT>AA 4 0.9% 30 0.8% 274 1.2%GA>TG TC>CA 4 0.9% 33 0.9% 223 1.0%AG>TA CT>TA 4 0.9% 40 1.1% 208 0.9%AA>GT TT>AC 4 0.9% 4 0.1% 204 0.9%TA>AT TA>AT 4 0.9% 5 0.1% 189 0.8%AC>TA GT>TA 4 0.9% 14 0.4% 163 0.7%CG>GA CG>TC 4 0.9% 23 0.6% 129 0.6%CA>TG TG>CA 3 0.7% 15 0.4% 2,513 11.0%CC>TG GG>CA 3 0.7% 20 0.5% 401 1.8%AA>GG TT>CC 3 0.7% 8 0.2% 386 1.7%AT>GA AT>TC 3 0.7% 6 0.2% 334 1.5%CA>GG TG>CC 3 0.7% 8 0.2% 331 1.4%CC>GT GG>AC 3 0.7% 27 0.7% 196 0.9%AA>TG TT>CA 3 0.7% 6 0.2% 190 0.8%CA>AC TG>GT 3 0.7% 28 0.7% 172 0.8%CA>GT TG>AC 3 0.7% 9 0.2% 85 0.4%CG>AC CG>GT 3 0.7% 11 0.3% 46 0.2%GC>CG GC>CG 3 0.7% 2 0.1% 21 0.1%AC>GT GT>AC 2 0.5% 9 0.2% 590 2.6%AT>GC AT>GC 2 0.5% 1 0.0% 298 1.3%TA>AG TA>CT 2 0.5% 6 0.2% 273 1.2%AT>CC AT>GG 2 0.5% 4 0.1% 202 0.9%AG>GC CT>GC 2 0.5% 7 0.2% 190 0.8%AT>TA AT>TA 2 0.5% 4 0.1% 172 0.8%AT>CA AT>TG 2 0.5% 5 0.1% 166 0.7%CG>AT CG>AT 2 0.5% 34 0.9% 38 0.2%AG>CA CT>TG 1 0.2% 23 0.6% 494 2.2%AA>CC TT>GG 1 0.2% 4 0.1% 257 1.1%
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 13 of 17
Table S3. Cont.
TBSTBS (reversecomplement)
Number ofoccurrencesin HGMD
RelativeproportionHGMD
Number ofoccurrencesin COSMIC
RelativeproportionCOSMIC
Number ofvariants in
LWK (Cortex)
Relativeproportion
LWK (Cortex)
AG>GT CT>AC 1 0.2% 16 0.4% 185 0.8%TA>CG TA>CG 1 0.2% 2 0.1% 153 0.7%AC>GG GT>CC 1 0.2% 7 0.2% 148 0.6%AA>CG TT>CG 1 0.2% 2 0.1% 123 0.5%CA>GC TG>GC 1 0.2% 3 0.1% 101 0.4%TA>GC TA>GC 1 0.2% 1 0.0% 50 0.2%CG>GC CG>GC 1 0.2% 1 0.0% 48 0.2%AA>TC TT>GA 0 0.0% 7 0.2% 380 1.7%CA>TC TG>GA 0 0.0% 11 0.3% 190 0.8%AA>CT TT>AG 0 0.0% 8 0.2% 166 0.7%AG>TC CT>GA 0 0.0% 11 0.3% 144 0.6%TA>CC TA>GG 0 0.0% 4 0.1% 129 0.6%TA>AC TA>GT 0 0.0% 4 0.1% 120 0.5%AC>TG GT>CA 0 0.0% 8 0.2% 109 0.5%GA>CG TC>CG 0 0.0% 2 0.1% 105 0.5%GA>CC TC>GG 0 0.0% 3 0.1% 89 0.4%AC>CG GT>CG 0 0.0% 0 0.0% 35 0.2%AT>CG AT>CG 0 0.0% 1 0.0% 26 0.1%Total number of TBS 441 3,769 22,898
*This value corresponds to an 10.6-fold enrichment compared with a contribution of 1.4% (2/144 possible TBS) that would be expected from a uniformrepresentation of each TBS (binomial test, P = 2.2 × 10−16).†This value corresponds to a 2.0-fold enrichment (binomial test, P = 2.0 × 10−11).‡This value corresponds to a 4.5-fold enrichment (binomial test P = 2.2 × 10−16).
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 14 of 17

Table
S4.
Sequen
cingresu
ltsofFG
FR3an
dHRAShotspots
in33
spermatocyticseminoma(SpS)
samples
Sample
Mutation
found
Age(y)
FGFR
3ex
on7
FGFR
3ex
on10
FGFR
3ex
on13
FGFR
3ex
on15
FGFR
3ex
on19
HRASex
on2
HRAS
exon3
p.R24
8p.S24
9p.P25
0p.E36
8p.G37
0p.S37
1p.Y37
3p.G37
5p.G38
0p.A39
1p.N54
0p.K65
0p.X80
7p.G12
p.G13
p.Q
61
H1T
44Y
YY
YY
YY
YY
YY
YY
YY
YH2T
48Y
YY
YY
YY
YY
YY
YY
YY
YH3T
48Y
YY
YY
YY
YY
YY
YY
YY
YH4T
HRASc.18
2A>G
82Y
YY
YY
YY
YY
YY
YY
YY
Y(Q
61R)
H5T
70Y
YY
YY
YY
YY
YY
YY
YY
YH6T
66Y
YY
YY
YY
YY
YY
YY
YY
YH7T
28Y
YY
YY
YY
YY
YY
YY
YY
YH8T
55Y
YY
YY
YY
YY
YY
YY
YY
YH9T
48Y
YY
YY
YY
YY
YY
YY
YY
YH10
T36
YY
YY
YY
YY
YY
YY
YY
YY
H11
T49
YY
YY
YY
YY
YY
YY
YY
YY
H12
T60
YY
YY
YY
YY
YY
YY
YY
YY
H13
T53
YY
YY
YY
YY
YY
YY
YY
YY
H14
T72
FF
FY
YY
YY
YY
YY
YY
YY
H15
T30
YY
YF
FF
FF
FF
FY
FY
YY
H16
T43
YY
YY
YY
YY
YY
YY
YY
YY
H17
T47
FY
YF
FF
FF
FF
FY
YY
YY
H18
T51
FF
FF
FF
FF
FF
FF
YY
FY
H19
T62
YY
YY
YY
YY
YY
YY
YY
YY
H20
T70
FF
FY
YY
YY
YY
YY
YY
FY
H21
Tn/a
FF
FF
FF
FF
FF
FY
YY
FY
H22
Tn/a
FF
FY
YY
YY
YY
YY
YF
FF
H23
Tn/a
FF
FY
YY
YY
YY
YY
FY
YY
H24
Tn/a
FF
FY
YY
YY
YY
YY
YF
FF
H25
Tn/a
FF
FF
FF
FF
FF
FY
YY
FY
H26
Tn/a
FF
FY
YY
YY
YY
YY
FY
FY
H27
Tn/a
FY
YY
YY
YY
YY
YY
YY
YY
SS12
67F
FF
FF
FF
FF
FF
FF
YY
YSS17
61F
FF
FF
FF
FF
FF
FF
YY
YSS24
HRASc.37
G>C
79F
FF
FF
FF
FF
FF
FF
YY(G
13R)
YSS25
89F
FF
FF
FF
FF
FF
FF
YY
FSS28
63F
FF
FF
FF
FF
FF
FF
FF
YSS32
44F
FF
FF
FF
FF
FF
FF
YY
YNumber
ofSp
San
alyz
edsuccessfully
1618
1822
2222
2222
2222
2226
2430
2529
Ave
rageag
eofSp
S(y)
57.41
Key
tomutationscreen
ingsymbols:F
,amplifi
cationorsequen
cefaile
d;n
/a,n
otav
ailable;Y
,codonch
ecke
donsequen
cingtrace;
(),m
utationiden
tified
inthetumortissuean
dnotpresentin
thehistologically
norm
almatch
edsample.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 15 of 17

Table
S5.
Values
ofμ(m
utationrate
per
celldivision)an
ds(selectionco
efficien
t)estimated
bymodelingtheprocess
ofselfish
selection
Gen
e
Nucleo
tide
substitution
(cDNA)
Aminoacid
chan
ge
Disea
se-
associated
?Ty
peofnucleo
tide
substitution
μ(×
106)
μlower_C
I(×
106)
μupper_C
I(×
106)
sslower_C
Isupper_C
I
Totalnumber
ofsamplesin
thestudy
Number
of
samples
above
backg
round*
Referen
ces
HRAS
c.32
C>A
p.A11
DNotreported
Tran
sversion_n
onCpG
2.0E
-05
3.0E
-06
8.0E
-05
0.01
%0.01
%0.31
%89
0Th
isstudy
HRAS
c.32
C>G
p.A11
GNotreported
Tran
sversion_n
onCpG
1.0E
-06
1.0E
-06
1.0E
-05
0.09
%0.01
%0.80
%89
0Th
isstudy
HRAS
c.32
C>T
p.A11
VNotreported
Tran
sition_n
onCpG
2.0E
-03
2.0E
-03
3.0E
-03
0.02
%0.01
%0.03
%89
3*Th
isstudy
HRAS
c.33
C>A
p.A11
ANotreported
Tran
sversion_C
pG
1.0E
-03
9.0E
-04
1.0E
-03
0.01
%0.01
%0.05
%89
4Th
isstudy
HRAS
c.33
C>G
p.A11
ANotreported
Tran
sversion_C
pG
1.0E
-06
1.0E
-06
2.0E
-05
0.41
%0.01
%0.80
%89
0Th
isstudy
HRAS
c.33
C>T
p.A11
ANotreported
Tran
sition_C
pG
4.0E
-03
4.0E
-03
4.0E
-03
0.03
%0.03
%0.05
%89
11*
Thisstudy
HRAS
c.34
G>A
p.G12
SCS,
cancer
Tran
sition_C
pG
5.0E
-03
5.0E
-03
5.0E
-03
0.25
%0.24
%0.27
%89
55*
Thisstudy
HRAS
c.34
G>C
p.G12
RCan
cer
Tran
sversion_C
pG
1.0E
-04
5.0E
-05
2.0E
-04
0.50
%0.14
%0.78
%89
3Th
isstudy
HRAS
c.34
G>T
p.G12
CCS,
cancer
Tran
sversion_C
pG
1.0E
-03
9.0E
-04
2.0E
-03
0.36
%0.12
%0.44
%89
20Th
isstudy
HRAS
c.35
G>A
p.G12
DCS,
cancer
Tran
sition_n
onCpG
2.0E
-03
2.0E
-03
2.0E
-03
0.27
%0.22
%0.27
%89
17*
Thisstudy
HRAS
c.35
G>T
p.G12
VCS,
cancer
Tran
sversion_n
onCpG
7.0E
-04
6.0E
-04
8.0E
-04
0.37
%0.35
%0.44
%89
18Th
isstudy
HRAS
c.34
_35G
G>TT
p.G12
FNotreported
TBS
9.0E
-06
1.0E
-06
3.0E
-05
0.40
%0.28
%0.80
%89
1Th
isstudy
HRAS
c.34
G>A;c.36C
>T
p.G12
SNotreported
Other
double
3.0E
-04
2.0E
-04
6.0E
-04
0.14
%0.07
%0.22
%89
1Th
isstudy
HRAS
c.35
_36G
C>AT
p.G12
DNotreported
TBS
7.0E
-05
3.0E
-05
1.0E
-04
0.35
%0.23
%0.44
%89
4Th
isstudy
HRAS
c.35
_36G
C>TA
p.G12
VCS
TBS
4.0E
-05
1.0E
-05
8.0E
-05
0.17
%0.13
%0.26
%89
3Th
isstudy
HRAS
c.35
_36G
C>TT
p.G12
VCS
TBS
7.0E
-04
6.0E
-04
8.0E
-04
0.37
%0.35
%0.44
%89
21Th
isstudy
FGFR
2c.75
5C>A
p.S25
2XNotreported
Tran
sversion_C
pG
2.0E
-03
2.0E
-03
2.0E
-03
0.02
%0.03
%0.03
%10
59
(1)
FGFR
2c.75
5C>G
p.S25
2WApert,cancer
Tran
sversion_C
pG
3.0E
-03
3.0E
-03
3.0E
-03
0.54
%0.51
%0.57
%10
584
(1)
FGFR
2c.75
5C>T
p.S25
2LCrouzo
nTran
sition_C
pG
4.0E
-03
4.0E
-03
4.0E
-03
0.26
%0.24
%0.27
%10
585
(1)
FGFR
2c.75
5C>G
p.S25
2WApert,cancer
Tran
sversion_C
pG
2.0E
-03
2.0E
-03
2.0E
-03
0.55
%0.54
%0.58
%32
523
0(2)
FGFR
2c.75
8C>G
p.P25
3RApert,cancer
Tran
sversion_n
onCpG
9.0E
-04
9.0E
-04
9.0E
-04
0.67
%0.67
%0.67
%32
518
0(2)
FGFR
3c.19
48A>C
p.K65
0QHCH,cancer
Tran
sversion_n
onCpG
8.0E
-06
1.0E
-06
3.0E
-05
0.16
%0.12
%0.80
%88
1(3)
FGFR
3c.19
48A>G
p.K65
0ETD
II,cancer
Tran
sition_n
onCpG
2.0E
-03
2.0E
-03
2.0E
-03
0.67
%0.62
%0.69
%88
80(3)
FGFR
3c.19
48A>T
p.K65
0XNotreported
Tran
sversion_n
onCpG
1.0E
-06
1.0E
-06
1.0E
-05
0.41
%0.01
%0.80
%88
0(3)
FGFR
3c.19
49A>C
p.K65
0TAN,cancer
Tran
sversion_n
onCpG
3.0E
-04
2.0E
-04
3.0E
-04
0.79
%0.75
%0.80
%88
20(3)
FGFR
3c.19
49A>G
p.K65
0RNotreported
Tran
sition_n
onCpG
1.0E
-06
1.0E
-06
1.0E
-05
0.41
%0.01
%0.80
%88
0(3)
FGFR
3c.19
49A>T
p.K65
0MSA
DDAN,cancer
Tran
sversion_n
onCpG
1.0E
-04
5.0E
-05
1.0E
-04
0.53
%0.47
%0.69
%88
8(3)
FGFR
3c.19
50G>A
p.K65
0KNotreported
Tran
sition_n
onCpG
1.0E
-05
1.0E
-06
4.0E
-05
0.02
%0.01
%0.79
%88
0(3)
FGFR
3c.19
50G>C
p.K65
0NHCH
Tran
sversion_n
onCpG
8.0E
-05
6.0E
-05
1.0E
-04
0.37
%0.33
%0.80
%88
8(3)
FGFR
3c.19
50G>T
p.K65
0NHCH
Tran
sversion_n
onCpG
8.0E
-05
6.0E
-05
1.0E
-04
0.37
%0.33
%0.80
%88
5(3)
AN,acan
thosisnigricans;CS,
Costello
syndrome;
HCH,hyp
och
ondroplasia;
TBS,
tandem
basesubstitution;TD
II,than
atophoricdysplasiatype2;
CI,95
%co
nfiden
ceinterval.
*Sam
pleswereco
nsidered
tobeab
ove
backg
roundformutationleve
ls>3×10
−6,e
xcep
tfortheindicated
tran
sitionsforwhichthecallingthreshold
was
10−5.O
nly
mutationswithgreater
than
eightsamples
above
backg
roundhav
ebee
nplotted
inFig.4.
1.GorielyA,McV
eanGA,Röjm
yrM,Ingem
arssonB,W
ilkie
AOM
(200
3)Ev
iden
ceforselectivead
vantageofpathogen
icFG
FR2mutationsin
themalegerm
line.
Science
301(56
33):64
3–64
6.2.
YoonSR
,et
al.(200
9)Th
eupsan
ddownsofmutationfreq
uen
cies
duringag
ingcanacco
untfortheApertsyndromepaternal
ageeffect.PL
oSGen
et5(7):e10
0055
8.3.
GorielyA,et
al.(200
9)Activatingmutationsin
FGFR
3an
dHRASreve
alashared
gen
etic
origin
forco
ngen
ital
disordersan
dtesticulartumors.Nat
Gen
et41
(11):124
7–12
52.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 16 of 17

Dataset S1. Dataset containing estimated mutation levels for nucleotide substitutions at HRAS p.A11 and p.G12 codons, given for eachallele (with respect to the C/T rs12628 SNP) and with 95% ETPI (equal-tailed probability interval)
Dataset S1
Levels are given per million independently for each HRAS allele (estimated with respect to the C/T rs12628 SNP) with their 95% ETPI (equal-tailed probabilityinterval) in separate tables.
Giannoulatou et al. www.pnas.org/cgi/content/short/1311381110 17 of 17





























