Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1770 (2007) 345–359www.elsevier.com/locate/bbagen
Review
Cytochrome P450–redox partner fusion enzymes
Andrew W. Munro ⁎, Hazel M. Girvan, Kirsty J. McLean
Manchester Interdisciplinary Biocentre, School of Chemical Engineering and Analytical Science,University of Manchester, 131 Princess Street, Manchester, M1 7ND, UK
Received 26 May 2006; received in revised form 23 August 2006; accepted 25 August 2006Available online 30 August 2006
Abstract
The cytochromes P450 (P450s) are a broad class of heme b-containing mono-oxygenase enzymes. The vast majority of P450s catalysereductive scission of molecular oxygen using electrons usually derived from coenzymes (NADH and NADPH) and delivered from redox partnerproteins. Evolutionary advantages may be gained by fusion of one or more redox partners to the P450 enzyme in terms of e.g. catalytic efficiency.This route was taken by the well characterized flavocytochrome P450BM3 system (CYP102A1) from Bacillus megaterium, in which soluble P450and cytochrome P450 reductase enzymes are covalently linked to produce a highly efficient electron transport system for oxygenation of fattyacids and related molecules. However, genome analysis and ongoing enzyme characterization has revealed that there are a number of other novelclasses of P450–redox partner fusion enzymes distributed widely in prokaryotes and eukaryotes. This review examines our current state ofknowledge of the diversity of these fusion proteins and explores their structural composition and evolutionary origins.© 2006 Elsevier B.V. All rights reserved.
Keywords: Cytochrome P450; Redox partner; Fusion protein; Flavoprotein; Ferredoxin; Electron transfer; Catalytic cycle; Phthalate dioxygenase reductase;Flavodoxin; Peroxidase; Chimera; Heme binding; Linker; Enzyme mechanism; Domain
1. Introduction
The cytochromes P450 (P450s) are a widely studiedsuperfamily of hemoproteins involved in a vast array ofoxidative reactions [1]. They are distributed through archaea,bacteria and eukaryotes and are involved in numerous cellularfunctions, including xenobiotic oxygenation, synthesis ofsteroids and of lipid mediators of cellular signalling events,manufacture of complex macromolecules including polyketideantibiotics, and the bioconversion of recalcitrant organicmolecules to enable their utilization as carbon sources forgrowth [2–5]. The P450s have a major impact on humanphysiology, and their capacity to perform regio- and stereo-selective oxygenations of numerous organic substrates has alsoattracted interest in the biotechnology sector, since the P450sprovide a cleaner and “greener” route to synthesis of severalhigh value chemicals [6–8].
The classical P450 reaction is the introduction of an atom ofoxygen (derived from molecular oxygen) to facilitate hydrox-
⁎ Corresponding author. Tel.: +44 161 306 5151; fax: +44 161 200 8918.E-mail address: [email protected] (A.W. Munro).
0304-4165/$ - see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.bbagen.2006.08.018
ylation at an unactivated carbon centre on a molecule. Toachieve this reaction, oxygen is bound to ferrous heme at thecentre of a b-type heme bound to the P450. The present modelof P450 activity involves the further reductive activation andprotonation of the oxyferrous species, ultimately leading toformation of a highly reactive ferryl-oxo intermediate and theproduction of a molecule of water (Fig. 1). The ferryl-oxointermediate attacks a substrate molecule bound proximal to theheme iron to perform the oxygenation and to restore the restingstate of the P450. The reaction follows the basic scheme shownbelow (Scheme 1):
In Scheme 1, RH is the substrate and ROH is the oxygenatedproduct. The electrons for catalysis derive (usually) from NAD(P)H and are delivered from one or more redox partnerenzymes. In the most frequently characterized class I and classII P450 redox systems, the electrons are transferred through (1)NAD(P)H ferredoxin reductase and ferredoxin enzymes (class
Scheme 1.

Fig. 1. Catalytic cycle of cytochrome P450. The model cycle of cytochromeP450 involves the binding of substrate (R–H) to the ferric P450 enzyme,followed by delivery of an electron (from the redox partner). This precedesbinding of dioxygen to ferrous heme iron (leading to the ferric superoxyspecies), reduction and protonation (forming ferric hydroperoxy) and dehydra-tion with formation of the ferryl-oxo intermediate considered to be the oxidantthat catalyses oxygenation (frequently hydroxylation) of the substrate. Departureof product from the enzyme restores the resting ferric state. Breakdown ofreactive intermediates is known to occur, producing either superoxide (fromferric superoxy), peroxide (from ferric hydroperoxy) or water (from ferryl-oxo).This uncoupling may occur if electron delivery or protonation is not timely, or ifsubstrate is absent or inappropriately positioned for oxidative attack.
346 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
I), as observed in e.g. the Pseudomonas putida cytochromeP450cam enzyme (CYP101A1, a camphor hydroxylase)—where the redox partners are the FAD-binding NADH-dependent putidaredoxin reductase, and the 2Fe–2S ferredoxinputidaredoxin (Fig. 2A) [9]; or (2) the FAD- and FMN-containing NADPH-dependent cytochrome P450 reductase(CPR, class II), which is an integral membrane protein thatsupports catalytic function of the majority of eukaryotic P450s(Fig. 2B) [10]. It is important to recognize that the cycleintermediate(s) that oxygenate substrate are transient and haveproven difficult to isolate for spectroscopic characterization. AnFe(IV) oxo complex widely referred to as compound I iscommonly accepted as the oxygenating species in most P450reactions, but its lifetime is so short that definitive proof for itsformation remains elusive [11].
It is now well known that there is considerable biologicaldiversity in the systems that provide reducing equivalents forP450 function. For instance, Fusarium oxysporum P450nor(CYP55A1) and its homologues bypass the requirement forprotein partners altogether and NAD(P)H binds directly to theP450 to facilitate reduction of 2 molecules of nitric oxide (NO)to dinitrogen oxide (N2O) [12]. A further class of peroxygenaseP450s interact directly with hydrogen peroxide to catalysehydroxylation of fatty acids [13]. Other P450s have been shownto interact with flavodoxins (e.g. cytochrome P450BioI[CYP107H1] involved in biotin synthesis in Bacillus subtilis),or to be reduced by non-pyridine nucleotide coenzyme
dependent partners (e.g. CYP119 and the 2-oxoacid-ferredoxinoxidoreductase- and ferredoxin-driven system from thearchaeon Sulfolobus tokodaii) [14,15]. However, a furthergeneral class of P450 system is also now recognized whereredox partner enzymes are fused to the P450 to generatecatalytically self-sufficient, multidomain entities, or to reducethe complexity of the P450 system (i.e. decrease the number ofpartner proteins involved). The first example of such a systemwas the P450BM3 fatty acid hydroxylase enzyme, where theP450 is fused to a eukaryotic-like CPR to create a soluble, high-activity oxygenase system [16]. P450BM3 (CYP102A1) is bysome distance the best characterized of the P450–redox partnerfusion systems, but several other distinctive types of P450–redox partner fusions have now been recognized in a largenumber of different organisms, and several attempts have beenmade to artificially engineer P450–redox partner fusions inorder to enhance catalytic efficiency of the P450. This reviewexamines our current knowledge of the diversity of P450–redoxpartner fusion systems, their evolutionary origin, and theircatalytic and biochemical properties.
2. P450 fusion systems—an evolutionary step forward?
As discussed above, the first groups of well characterizedP450 systems were the class I enzymes (typified by the P450camsystem and the eukaryotic mitochondrial adrenodoxin reduc-tase/adrenodoxin systems and their cognate P450s) and the classII enzymes (e.g. mammalian hepatic membrane-bound P450and CPR systems) [10,17]. However, the discovery of the Ba-cillus megaterium P450BM3 enzyme provided the first indica-tion that P450 systems with covalently fused partner enzymesmight exist. Several BM3-type systems have now beenrecognized, as have other forms of redox partner fusions [18].Genome sequences have revealed other novel types of P450enzymes linked to distinct partner proteins. Obvious questionssurround the reasons for the evolution of such systems and thepossible advantages afforded to the host organisms.
The first possible reason is that the fusion arrangementenhances catalytic efficiency, in terms of reaction velocity (i.e.kcat) and efficiency (i.e. less wastage of reducing equivalents innon-specific reduction of dioxygen) [19]. If the properties of theP450BM3 enzyme in any way reflect the general features ofP450–redox partner fusion enzymes, then improved catalyticproperties are certainly associated with this arrangement.P450BM3 catalyses fatty acid hydroxylation at rates of up to∼17,000min−1 (i.e. ∼280s−1), which is at least two orders ofmagnitude faster than observed for eukaryotic (CYP4) fatty acidhydroxylases [20,21]. The Km values of P450BM3 for severalfatty acid substrates are comparable to those for severaleukaryotic fatty acid hydroxylases (CYP4 enzymes), butBM3’s much greater kcat values means that overall catalyticefficiency (i.e. kcat/Km) is dramatically improved. P450BM3 isalso a well-coupled enzyme, producing substantial amounts ofhydroxylated products with rather little wastage of electrons information of superoxide, peroxide or water. A major factorunderlying the high catalytic rate of the P450BM3 enzyme is thefast rate of flavin-to-heme electron transfer afforded by the

347A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
fusion arrangement. Stopped-flow analysis indicates that thisoccurs at 223s−1 for the first electron transfer to the ferric ironin presence of the substrate myristic acid at 25 °C (first re-duction step in Fig. 1), and is thus a likely contributor to ratelimitation [20,22]. Interactions (i.e. productive collisions)between the discrete redox partners in the typical class I andclass II system (whether in biological membranes or in thecytosol) are almost invariably slower and lead to lower flavin-to-heme electron transfer rates. Due to the relative instability ofthe ferrous-oxy P450 complex, there is also the likelihood ofcollapse of this intermediate with release of superoxide ifdelivery of the second electron is not timely (Fig. 1). In the BM3system [23], the proximity of the linked redox partner likelyensures a high frequency of productive interactions betweenFMN and heme domains (see Fig. 2C). In all P450 mono-oxygenases, there is a requirement for temporally distinctdelivery of the two electrons (to enable dioxygen binding priorto further reduction, see Fig. 1), and in at least the prokaryoticclass I systems this likely requires the dissociation of a P450–ferredoxin (Fd)/flavodoxin (Fld) complex to enable the singleelectron carrier Fd/Fld proteins to source the second electronfrom their cognate reductases. This obviously provides one routefor uncoupling, if the reduced Fd/Fld does not re-bind to deliverthe second electron prior to collapse of the ferrous-oxy form.However, it should be noted that P450cam is also a well coupledenzyme and that the putidaredoxin-to-P450 electron transferreaction is sufficiently rapid to support a kcat of ∼4200min−1
(∼70s−1), with either the 1st or 2nd electron transfer to the hemeiron rate-limiting in catalysis according to the putidaredoxinconcentration used [24]. Oxyferrous complexes are oftenstabilized by substrate binding, and may be sufficiently long-lived in many P450s such that collapse of this intermediate priorto second electron delivery does not disable the oxygenase toany considerable degree [25,26]. It should also be noted thatcollapse of oxy intermediates is also linked to the nature of theP450 substrate, with e.g. mammalian hepatic P450s showingmarkedly different degrees of coupling of electron transfer tooxidation of varying substrates (e.g. [27]). Moreover, thephenomenon of heme reduction in the absence of boundsubstrate is commonly observed in eukaryotic P450s (particu-larly in hepatic forms) and this will inevitably lead to oxygenradical formation if a substrate molecule does not bind to theP450 in rapid succession to the reductive event [28]. Thus,factors other than efficient P450 heme iron reduction by theredox partner (and particularly the structural compatibility of thesubstrate to the P450 active site) are important in determining theextent of coupling of electron transfer to the final accumulationof oxygenated product. The tendency of P450s to uncouple with“imperfect” substrates is not restricted to the eukaryotic forms,and has been observed with e.g. P450BM3 and P450cam enzymesin turnover of non-standard substrates such as short chainalkanes, alkenes and alkanoic acids (e.g. [29]). In general,uncoupling in P450s (i.e. collapse of oxy intermediates in thecatalytic cycle, Fig. 1) is associated with such factors as access ofwater to the reactive species, non-productive binding modes ofsubstrate(s) and insufficiently reactive sites on the substrate [30–32]. Thus, for example, mammalian hepatic P450s that have
wide substrate specificity often exhibit significant levels ofuncoupling [33]. However, in the case of P450BM3, at least,redox partner fusion clearly leads to a substantial increase incatalytic rate over that for its isolated P450 and CPR domains,and this may be one major reason for the evolution of this fusionarrangement, as discussed in more detail in Section 3 below.
A second major reason for the evolution of P450–redoxpartner fusions is likely to revolve around advantagesassociated with the regulation of their expression. Coordinationof the transcription and translation of an intact P450 redoxsystem (as in the case of P450BM3) is clearly advantageousover the separate production of either two or three partnerenzymes. In some cases (e.g. the synthesis of P450cam and itspartners on the Pseudomonas putida CAM plasmid, and e.g.for the Mycobacterium tuberculosis CYP51 and cognateferredoxin adjacent on the chromosome), there is evidencefor coordination of transcription of the partners encoded onseparate genes (e.g. [34]). However, this does not occur acrossthe board, and in the case of eukaryotic P450 systems a singleCPR enzyme is not only chromosomally separated from P450partners, but also must interact with several different P450s[10]. In addition, eukaryotic CPR interacts with other cellularpartners, including cytochrome b5, heme oxygenase, squalenemonooxygenase and 7-dehydro-cholesterol reductase (e.g.[35–37]). For P450BM3, a complex regulatory system existsin which expression is controlled by a repressor protein(BM3R1). The repressor is displaced on binding barbiturate-like molecules, fatty acids and other lipophilic molecules—leading to CYP102A1 expression [38]. Further positive andnegative regulatory proteins are also involved in CYP102A1production in Bacillus megaterium [39,40]. Expression of thegene also occurs in Escherichia coli transformants in thestationary phase of growth, suggesting that there is a similarregulatory response in the heterologous host as is seen in theBacillus [41]. While the physiological role of P450BM3 remainsuncertain, it may be important to the Bacillus that expressionof a highly active system occurs at high cell density and/orduring nutrient/oxygen limitation. Thus, coordinated produc-tion of intact P450 redox systems (such as BM3) may offerconsiderable advantages over requirement for regulation over 2or 3 separate systems. In addition to ensuring that a singlecatalytically efficient unit is produced, these systems may alsoensure that the redox partner does not participate in reactionsother than those involving the fused P450, and are thus lesswasteful in terms of non-specific oxidation of NAD(P)H andthe formation of unnecessary products from other P450enzymes. It also means that there can be a concertedproduction of an efficient enzyme system in response toenvironmental changes. In the case of P450BM3 and itshomologues (e.g. the CYP102A2 and CYP102A3 systems),it has been suggested that this may relate to presence ofspecific lipids in the environment (e.g. [42]).
The fusion of redox partner enzymes in systems such asP450BM3 clearly provides some advantages to host cells, as isevident from the fact that this type of P450/CPR fusion has nowbeen found in a wide range of organisms (both prokaryotic andeukaryotic, Fig. 2) and since the CYP102 family continues to
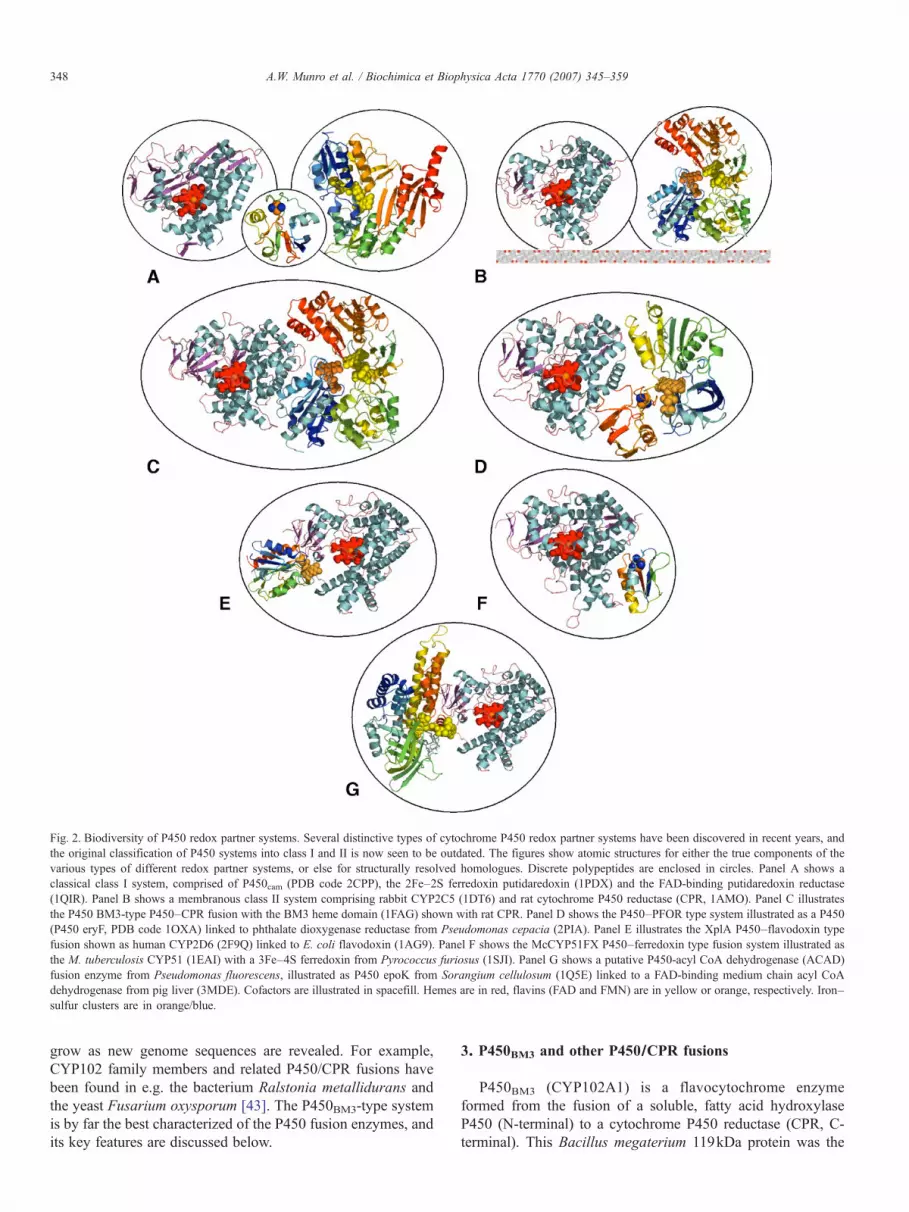
Fig. 2. Biodiversity of P450 redox partner systems. Several distinctive types of cytochrome P450 redox partner systems have been discovered in recent years, andthe original classification of P450 systems into class I and II is now seen to be outdated. The figures show atomic structures for either the true components of thevarious types of different redox partner systems, or else for structurally resolved homologues. Discrete polypeptides are enclosed in circles. Panel A shows aclassical class I system, comprised of P450cam (PDB code 2CPP), the 2Fe–2S ferredoxin putidaredoxin (1PDX) and the FAD-binding putidaredoxin reductase(1QIR). Panel B shows a membranous class II system comprising rabbit CYP2C5 (1DT6) and rat cytochrome P450 reductase (CPR, 1AMO). Panel C illustratesthe P450 BM3-type P450–CPR fusion with the BM3 heme domain (1FAG) shown with rat CPR. Panel D shows the P450–PFOR type system illustrated as a P450(P450 eryF, PDB code 1OXA) linked to phthalate dioxygenase reductase from Pseudomonas cepacia (2PIA). Panel E illustrates the XplA P450–flavodoxin typefusion shown as human CYP2D6 (2F9Q) linked to E. coli flavodoxin (1AG9). Panel F shows the McCYP51FX P450–ferredoxin type fusion system illustrated asthe M. tuberculosis CYP51 (1EAI) with a 3Fe–4S ferredoxin from Pyrococcus furiosus (1SJI). Panel G shows a putative P450-acyl CoA dehydrogenase (ACAD)fusion enzyme from Pseudomonas fluorescens, illustrated as P450 epoK from Sorangium cellulosum (1Q5E) linked to a FAD-binding medium chain acyl CoAdehydrogenase from pig liver (3MDE). Cofactors are illustrated in spacefill. Hemes are in red, flavins (FAD and FMN) are in yellow or orange, respectively. Iron–sulfur clusters are in orange/blue.
348 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
grow as new genome sequences are revealed. For example,CYP102 family members and related P450/CPR fusions havebeen found in e.g. the bacterium Ralstonia metallidurans andthe yeast Fusarium oxysporum [43]. The P450BM3-type systemis by far the best characterized of the P450 fusion enzymes, andits key features are discussed below.
3. P450BM3 and other P450/CPR fusions
P450BM3 (CYP102A1) is a flavocytochrome enzymeformed from the fusion of a soluble, fatty acid hydroxylaseP450 (N-terminal) to a cytochrome P450 reductase (CPR, C-terminal). This Bacillus megaterium 119kDa protein was the

349A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
first characterized example of a P450–CPR fusion enzyme, andthe first time that a bacterial P450 was found to operate a class IIredox system (i.e. use a CPR for electron transfer) [44].Moreover, the enzyme was soluble, lacking the membraneanchors typically found in the P450 and CPR components ofeukaryotic class II systems. P450BM3 was also the first enzymefound to contain FAD, FMN and heme in a single polypeptide,and evolutionary analysis demonstrated clearly that it wasevolved by the fusion of CPR and P450 modules [23] (Fig. 2C).In addition, the CPR component (in both BM3 and eukaryoticCPRs) is evolved from a fusion of ancestral NADP+-ferredoxinoxidoreductase- and flavodoxin-like proteins [45]. Thus, BM3has evolved due to at least two separate gene fusion events.
The P450 domain of the enzyme is related to family 4 of theP450 enzyme superfamily, whose members are mainly fattyacid ω-hydroxylases. While P450BM3 is a fatty acid hydroxy-lase, it generally oxygenates at the ω-1 to ω-3 carbons, withsaturated fatty acids of carbon chain length C12–C18 and variousunsaturated and polyunsaturated fatty acids (e.g. arachidonicacid) being good substrates [20,46]. Structural and catalytic dataindicate that the active site residue Phe 87 interacts with the ω-terminal methyl group of fatty acids and may prevent omegaoxidation by the P450 [47]. However, there are contrastingreports of the catalytic properties of BM3 F87 point mutants—with hydroxylation of substrates reported to occur eitherexclusively at the ω-position, or even further away from thisposition than is observed for wild-type BM3 (e.g. [47,48]). Asstated above, P450BM3 is a very fast enzyme, oxygenatingarachidonic acid at ∼17,000min−1 [20]. This efficient activityclearly has its origins in the proximal fusion of the redoxpartners in P450BM3. The genetically dissected CPR and P450domains of P450BM3 retain cofactor binding and spectroscopicproperties typical of the intact flavocytochrome, but interactonly weakly to reconstitute a low rate of fatty acid hydroxyl-ation [41,49].
A structure of full length flavocytochrome P450BM3 hasproven elusive to date, no doubt due to the complex,multidomain nature of the protein and probably also toconformational and aggregational heterogeneity. For instance,our group has recently demonstrated that the dimeric form ofP450BM3 is active as a fatty acid hydroxylase, with electrontransfer occurring between the CPR of monomer 1, and theheme domain of monomer 2 in the dimer [50]. Previoushydrodynamic studies had indicated that the dimer was apredominant form in solution, and that even higher orderoligomers were present [51]. Notwithstanding the problemsinherent in the crystallization of the full length flavocyto-chrome, genetic dissection of the domains of P450BM3 producedfirst the heme (P450) and CPR domains, and later thecomponent FAD/NADP(H)-binding (NADP+-ferredoxin reduc-tase-like) and FMN-binding (flavodoxin-like) domains of theCPR [42,52,53]. Recently, the FMN-heme domain of theenzyme was also generated and expressed. Atomic structureshave been solved for both this FMN- and heme-bindingpolypeptide, and for the isolated heme domain of P450BM3 [54–56]. Structures for the heme domain in both substrate-free andsubstrate-bound (palmitoleic acid- and N-palmitoylglycine-
bound) forms reveal that a substantial conformational readjust-ment occurs on the binding of substrate, with alterationsobserved in the orientation of the long I helix that runs acrossthe face of the heme, and motions observed for the B/C and F/Ghelices—leading to a closure of the active site and sequestrationof the substrate [55–57]. While it is generally held thatstructural differences between the substrate-free (SF) andsubstrate-bound (SB) forms are a consequence of fatty acidassociation with the P450, it is worthy of note that a pointmutant (A264E, designed for investigations of heme ironligation) adopts the SB conformation in either presence orabsence of substrates, and regardless of whether or not themutated residue (Glu 264) coordinates to the heme iron. Inaddition, the A264E variant binds fatty acid substrates tighter(i.e. with lower Kd value) than does the wild-type P450BM3
[58]. Thus, a likely scenario is that the SB and SF conformationsof P450BM3 heme exist naturally in solution, possibly inequilibrium with other states. The increased affinity forsubstrate observed in A264E may indicate that the SB form isa conformational state which binds substrate more tightly thanthe SF form, rather than a form forced to adopt the SBconformation by substrate association per se [58,59]. In recentstudies, we have solved the atomic structure of the FAD/NADPH-binding domain of P450BM3, meaning that allindividual domains of P450BM3 have now been structurallyresolved [60]. The final prize of the intact flavocytochromeenzyme remains elusive, but the ability to conformationallyrestrain the multidomain enzyme (e.g. by binding of NADP+ toits CPR domain, or by use of the conformationally “locked”A264E variant) may hold the key to crystallizing and obtainingan atomic structure for the intact P450BM3.
P450BM3 shows a strong preference for NADPH overNADH as the reducing coenzyme, as do the eukaryotic CPRenzymes [22,61]. This distinguishes BM3 further from manyother prokaryotic P450 enzymes, which communicate withredox systems that favour NADH. It has been established that amajor determinant of coenzyme selectivity is an aromaticresidue (Trp 1046) whose side chain sits across the face of theisoalloxazine ring of the FAD in P450BM3. Aromatic residuesare conserved at the same position in all known homologues ofP450BM3 (see below) and in the CPRs and other related diflavinreductase enzymes. In both P450BM3 and human CPR (whichhas residue Trp 676 in the conserved position), mutants withnon-aromatic side chains were shown to “switch” pyridinenucleotide coenzyme selectivity considerably from NADPHtowards NADH [62,63]. The effect is most spectacular withP450BM3, in which an almost 6000-fold conversion ofspecificity constant from NADPH to NADH (i.e. the changein ratios of kcat/Km for electron transferase activity with NADHas the donor coenzyme to that with NADPH as donor) wasdetermined [61]. Stopped-flow studies of the human CPRindicate that the aromatic side chain of Trp 676 is mobile and isdisplaced from the face of the FAD by coenzyme binding tofacilitate hydride transfer to the flavin. Removal of this “barrier”effects a dramatic switch in coenzyme specificity in both CPRand P450BM3, and highlights the importance of this aromaticshielding residue in determining coenzyme preference [61]. The

Fig. 3. Active site structure in P450 BM3. Key amino acid residues in the activesite of P450 BM3's heme domain are illustrated. Below the plane of the heme(dark grey at centre of image) Cys 400 provides the thiolate ligand to the hemeiron and Phe 393 interacts with the Fe–S bond system to influenceelectronegativity of the heme iron [68]. Above the heme plane, Phe 87 interactswith the ω-methyl of fatty acid substrates and controls regioselectivity of lipidoxygenation [48]. Thr 260, Ala 264 and Thr 268 are key residues in the I helix,forming important hydrogen bonding interactions (including to the distal wateron the heme iron) and likely defining an oxygen binding pocket. An A264Emutant results in Glu 264 coordination to heme iron [58]. The substratepalmitoleic acid is shown in light grey, with its ω-end stretching towards theheme and Phe 87. The carboxylate group at the other end of the fatty acid makesinteractions with the sidechains of Tyr 51 and Arg 47, and Phe 42 places aphenyl “cap” over the top of the fatty acid binding cavity [20].
350 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
ability to produce NADH-specific enzymes has major con-sequences in relation to the exploitation of P450BM3 forbiotechnological uses. NADPH is a substantially moreexpensive coenzyme than is NADH, and a NADH-specificvariant is of great interest. The substrate specificity profile ofP450BM3, the fastest of the P450 monooxygenases, has beenstudied extensively—and the enzyme and active site mutantsthereof have been shown capable of oxygenating a wide rangeof molecules of biotechnological interest. These include variousalkanes, alkenes, short chain alkanoic acids and aromaticmolecules (e.g. [64,65]). Both rational mutagenesis and forcedevolution methods have been used to achieve alterations insubstrate specificity (e.g. [64,66]). In addition there has been alarge programme of work in several laboratories aimed atdeconvoluting structure and mechanism of this model enzymethrough rational mutagenesis coupled to structural, kinetic andbiophysical analysis. Major breakthroughs in this area includethe discovery of a key role for active site residue Phe 87 in thecontrol of regio-selectivity of substrate oxidation [20,47]; thedemonstration that the phylogenetically conserved residue Phe393 (which stacks against the iron–cysteine bond at theproximal side of the heme) plays a pivotal role in the controlof heme iron reduction potential and in the stability of theferrous-oxy complex [67,68]; establishment of important rolesfor residues Arg 47 and Tyr 51 in the interaction with thecarboxylate group of fatty acid substrates [20]; and thediscovery of key roles for residues Trp 574 and Gly 570 inthe binding of FMN in the reductase domain of the enzyme[22,69] (Fig. 3).
As discussed above, the fatty acid hydroxylation rate ofP450BM3 is extremely high by comparison with other P450monooxygenases, and is expected to have its origins in efficientelectron transport through the flavin and heme cofactors of theflavocytochrome. Stopped-flow kinetic studies indicate that therate of FMN-to-heme electron transfer is substantially more
Fig. 4. Multiple alignment of members of the P450–CPR fusion enzyme family. (A)and related P450–CPR fusion enzymes (residues 1–472 in P450 BM3 are typically efrom Bacillus megaterium P450 BM3 (BM3) heme domain aligned with related enStreptomyces avermitilis, Fusarium oxysporum (CYP505A1, Foxy), Ralstonia metalan absolutely conserved group of four amino acids surrounding Phe 87 (BM3 number96, which contacts the heme macrocycle, is also strictly conserved in this group. Paneand the loop connecting it to the F helix. A heptapeptide section of the F helix (Threncompassing the entire I helix of P450 BM3, and absolute conservation of a twelvsubstrate- and oxygen-binding region. Panel D shows the strongly conserved regionimportant regulator of heme iron reduction potential and stability of the oxy complexBM3 and the same group of P450–CPR fusion enzymes as in (A), including the linkerBM3 are typically expressed as the reductase domain). Sequence similarities are mosconserved region involved in binding the FAD cofactor. The absolutely conserved tyring in these proteins, and BM3 Ser 830 is a component of a catalytic triad of residNADPH to the FAD [98]. Panel B indicates conservation in a region involved in bindthe end of the proteins involved in interactions with the re-face of the FAD cofaphenylalanine; Trp 1046 in BM3), mutants of which in BM3 reductase (W1046A/H) heffect a considerable switch in coenzyme specificity towards NADH [61]. Asp 104involved in catalysis with NADPH [119,120]. Panel D shows the area around the intmuch of the sequence shown. The region indicated starts at the end of the defined hemin the BM3 sequence) are also seen in mammalian CPR enzymes and in related diflavalignment for some of the bacterial enzymes (typically 7–8 residues) suggest that thernotable in this context that the catalytic properties of P450 BM3 and CYP102A3CYP102A2 (where A2 has an extended linker sequence) [76].
rapid than seen in the typical eukaryotic, 2-component class IIP450 systems, and was measured as 223s−1 for the reaction ofNADPH with myristic acid-bound enzyme at 25 °C, usingformation of the carbon monoxy adduct of the ferrous heme iron(at 450nm) as a means of trapping the reduced heme iron.NADPH-dependent reduction of the FAD cofactor and internal
shows selected, highly conserved regions from the heme domains of CYP102A1xpressed as the heme domain). Panel A shows a region in the B′/C helix sectionzymes from Bacillus subtilis (CYP102A2 and 102A3), B. anthracis, B. cereus,lidurans, Neurospora crassa and Aspergillus nidulans. This region encompassesing), a key residue in controlling regioselectivity of substrate oxidation [48]. Trpl B aligns the same group in the protein region corresponding to the BM3 E helix146 to Tr 152) is very strongly conserved. Panel C aligns the group in a regione amino acid stretch is seen. This stretch includes several residues defining thearound the cysteinate ligand to the heme iron (Cys 400), including Phe 393, anin P450 BM3 [68]. B shows selected regions from the reductase domains of P450region connecting the heme and reductase modules (residues 472–1048 in P450t obvious towards the carboxy terminal end of the reductases. Panel A shows therosine (Tyr 829 in BM3) is likely to stack against the si-face of the isoalloxazineues (defined in studies on mammalian CPR) involved in hydride transfer froming the coenzyme (NADPH) pyrophosphate group. Panel C shows the region atctor. This region includes a conserved aromatic residue (either tryptophan orave a profound effect on affinity of the reduced enzyme for NAD(P)+ and which4 is also a further residue from the catalytic triad (with Ser 830 and Cys 999)er-domain linker in these enzymes. The enzymes display quite low similarity fore domain atomic structure for BM3, and conserved regions (starting from∼L482in reductases, indicating that the linker terminates by this point. The gaps in thee is variation in linker length between some of these flavocytochromes P450. It is(both of which have gapped linkers) are more similar than those of BM3 and

351A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
electron transfer between FAD and FMN were also much fasterthan seen for e.g. the human CPR [22,70]. It might be expectedthat the nature of the linker region between the CPR and hemedomains of P450BM3 would be an important regulator of theelectron transfer process between the FMN and heme centres.Precise structural composition of the BM3 linker (in terms ofboth length and any secondary structural content) remainsunknown in the absence of a structure for the full lengthP450BM3, but the linker is probably up to ∼20–30 amino acidsin length on the basis of the expected C-terminus of the hemedomain and the N-terminal of the CPR domain, in turn based onthe amino acid sequences of other P450 and CPR proteins(Fig. 4). Our recent studies indicate that electron transferbetween FMN1 and heme2 occurs in a dimeric P450BM3 enzyme[50], and this is a complicating factor in establishing therelevance of the linker size and structure with respect to control
of inter-domain electron transfer in P450BM3. That is, the linkerregion might be e.g. a segment of the protein whose primary roleis in the stabilization of the dimeric form, as opposed to being ofappropriate conformation to optimise FMN-to-heme electrontransfer within a P450BM3 monomer. In absence of an atomicstructure for the flavocytochrome P450BM3, the relevance oflinker size and composition is clearly best addressed bycomparisons with other (CYP102) family members—andseveral of these have come to light in recent years.
For a short period, P450BM3 stood alone as the only knownP450–CPR fusion enzyme. However, a combination of genomesequence analysis and novel P450 characterization has revealeda growing number of homologues and additions to the CYP102family over the last ∼10–15years. In Fusarium oxysporum, afungal pathogen of rice and other plants, a fatty acid sub-terminal hydroxylase activity (i.e. oxygenation of fatty acids at

352 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
the ω-1, ω-2 and ω-3 positions) was discovered more than20years ago [71], and some time later shown to be associatedwith a BM3-like fatty acid hydroxylase P450–CPR fusionprotein [72]. The enzyme was informally termed P450foxy(CYP505A1) and, like BM3, shown to have a strong preferencefor NADPH over NADH [43,44]. Like P450BM3, an unusualphenomenon is observed in P450foxy whereby binding of fattyacid substrate in the heme domain accelerates reduction ofcytochrome c through the reductase domain of the enzyme[43,73]. This suggests that fatty acid binding induces aconformational change in these flavocytochromes whichfacilitates improved interactions with the electron acceptorprotein. A similar type of phenomenon is observed in steady-state reactions of neuronal nitric oxide synthase, where bindingof the effector protein calmodulin stimulates nitric oxideformation and cytochrome c reduction, possibly by releasing aconformational lock in the protein [74]. P450foxy is purifiedfrom the host organism with the membrane fraction. However, atrue membrane anchor region (as seen at the N-termini ofmammalian P450 enzymes) is not obvious in P450foxy, and thusthe enzyme is likely to be a peripheral membrane protein, ratherthan an integral membrane protein of the membrane-associatedclass. A blast search of the protein sequence database revealshomologues of P450foxy in a number of other eukaryotes,including Neurospora crassa and Aspergillus oryzae. Anothermember of the family (CYP505B1) was found in Fusariumverticilloides, and likely functions as a polyketide hydroxylasein synthesis of the mycotoxin fumonisin [75].
Genome sequencing revealed the presence of two distinctBM3 homologues in Bacillus subtilis. The B. subtilis flavo-cytochromes P450 CYP102A2 and CYP102A3 have beenexpressed and characterized in recent years, revealing interest-ing differences in catalytic properties to those of P450BM3 [76].The BM3 (CYP102A1) and A2/A3 enzymes show 49% overallidentity in amino acid sequence, with 60% identity between A1/A2, 58.5% identity between A1/A3 and 61% identity betweenA2/A3 in pairwise alignments. The A2 and A3 enzymes alsocatalyse hydroxylation of a range of fatty acids at theω-1 toω-3positions, and the phenylalanine residue (Phe 87) found inP450BM3 and shown crystallographically to interact with the ω-terminal methyl group of fatty acids is also conserved in A2/A3(Phe 88 in both cases) and is likely a major determinant of theregioselectivity of fatty acid oxidation in these enzymes (Fig. 4).The substrate selectivity profiles of CYP102A2 and A3 aredifferent from one another, and from those of P450BM3. Whilethe precise physiological roles of P450BM3 and its homologuesremain unclear, it is notable that the B. subtilis enzymes havestrong activity towards various branched chain fatty acids,molecules that are prevalent in the Bacillus membrane. The A2and A3 enzymes also exhibit some unusual features with respectto equilibrium binding and steady-state turnover of various fattyacids. Specifically, spectral titrations of the BM3 enzyme with arange of different fatty acids result in hyperbolic relationshipsbetween induced spectral change and the concentration of thefatty acid ligand (e.g. [77]). The spectral change observed (aprogressive shift in Soret maximum from ∼418nm to∼390nm) reports on the substrate-dependent displacement of
a weakly bound water molecule that is the distal ligand to theheme iron, and the resultant shift in heme iron spin-stateequilibrium from low- towards high-spin. Hyperbolic relation-ships observed for P450BM3 are usually taken to indicate singleoccupancy, reversible binding of fatty acids to the enzymeactive site. Plots of initial rate of BM3-catalysed steady-stateturnover of fatty acids versus fatty acid concentration are alsohyperbolic and display Michaelis–Menten-like behaviour.However, this behaviour is not seen for the A2 and A3 enzymesin their interactions with various lipid substrates (e.g. 12-methylmyristic acid, 13-methyl myristic acid and linoleic acid withCYP102A2; and 13-methyl myristic acid, 14-methyl pentade-canoic acid and linoleic acid with CYP102A3 [76]. In severalcases, the relationships between induced optical change/apparent catalytic rate and substrate concentration are sigmoi-dal. This may reflect e.g. cooperativity of binding of twosubstrate molecules in the active site of the A2/A3, orcooperativity between active sites of the P450 domains in adimeric flavocytochrome [50]. While the structural origins ofthis unusual behaviour remain to be resolved, it is pertinent tonote that there is compelling evidence for the dimeric status ofP450BM3 and the importance of dimerization for fatty acidhydroxylase activity in this enzyme [50,51]. In addition,spectroscopic studies have indicated that BM3 can accommo-date both a fatty acid and a bulky azole derivative simulta-neously in the active site [78]. Thus, relatively small changes inactive site structure between the A2/A3 and BM3 enzymes maybe sufficient to produce the sigmoidal relationships observed forfatty acid binding/turnover seen in the former enzymes.
Further homologues of P450BM3 are also found in otherbacteria, including the heavy metal tolerant bacterium Ralstoniametallidurans, B. anthracis and Bradyrhizobium japonicum.However, these systems have yet to be characterized in detail atthe protein level. Based on similarities between catalyticactivities of the BM3, A2/3 and P450foxy systems, it would beexpected that all these enzymes have fatty acid oxygenaseactivity, and probably similar regioselectivity of substratehydroxylation. Alignments of the protein sequences of theheme and reductase domains of these enzymes reveals thoseregions that are most closely conserved across this class, and arethus most likely to be important for functionality. These includethe heme-binding region that surrounds the cysteinate ligandwhich coordinates the heme iron in the P450s. This region isalso strongly conserved across the P450 superfamily, andincludes the phylogenetically conserved BM3 residue Phe 393(see above). However, there are other very highly conservedregions in the heme domain that are more definitive for theCYP102 family. These include large sections of the I helix andthe region surrounding phenylalanine 87 (Fig. 4A).
In the reductase domain, the most highly conserved regionsare also those that are well maintained in eukaryotic CPRenzymes, and in other members of the diflavin reductase familyof enzymes that includes methionine synthase reductase and thecancer-related enzyme novel reductase 1 (NR1) [79,80]. Mostof these regions define segments essential for binding of theflavin cofactors and pyridine nucleotide coenzyme [45] (Fig.4B, panels A–C). An aromatic residue (Trp or Phe) is conserved

353A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
in all these BM3 homologues close to the C-terminal end ofthese enzymes (usually 0–3 residues from the end). However,related nitric oxide synthase (NOS) enzymes (see below) havea C-terminal peptide extension of up to ∼35 residues. Asdiscussed above, in BM3 the replacement of the relevantaromatic residue (W1046) with either alanine or histidineresulted in a spectacular conversion of coenzyme selectivityfrom NADPH towards NADH. Similar, if less spectacular,switches in specificity were observed for the comparableF1395A/S and W676A mutants of the related neuronal nitricoxide synthase and CPR enzymes, respectively [63,81]. Thus,a single residue, the aromatic side chain of which stacksacross the isoalloxazine ring of the FAD cofactor in thereductase domain, plays a pivotal role in coenzyme discri-mination in both the reductase domain of P450BM3 and inrelated diflavin reductases of higher organisms. Due to theenormous interest in biotechnological exploitation of theP450BM3 enzyme, the specificity conversion to the cheaperNADH coenzyme achieved in W1046A/H variants may be ofconsiderable relevance.
A potentially important feature of P450BM3 and otherCYP102 family enzymes is the composition and length of thelinker region that joins the heme and reductase domains. Withseveral BM3-like enzymes now identified, alignment of theseproteins provides an important means of comparing the linkersections and establishing whether there is a common structuralarrangement observed in this region of the flavocytochromes.As can also be seen in Fig. 4B, there is in fact rather little aminoacid sequence similarity in the peptide region between the hemeand CPR domains of these enzymes. The precise linker length isnot certain, but is unlikely to be more than 30 amino acids, andis probably considerably shorter in most cases. It is clear thatthere is some variation in length, with shorter linkers (by ∼7–8 residues) apparent in P450BM3, CYP102A3 and the enzymesfrom Streptomyces avermitilis and R. metallidurans than in theother enzymes shown (Fig. 4B, panel D). This may have somesignificance with respect to activity, given the rather moresimilar reactivity profile noted for P450BM3 and CYP102A3than for BM3 and CYP102A2 [76]. In view of recent dataindicating that P450BM3 is functional as a fatty acid oxygenasein the dimeric state with electron transfer between monomers,the linker's role may be primarily to stabilize a dimeric form asopposed to facilitating inter-domain interactions between CPRand P450 modules within the monomer [16]. Studies of linkercomposition in P450BM3 indicated that length (rather thanamino acid sequence) was a major determinant of enzymeactivity and FMN-to-heme electron transfer [82].
Although the P450BM3 type structure was the first charac-terized enzyme class in which a heme domain with cysteinate-coordinated iron is linked to a diflavin reductase, it is pertinentto note that the eukaryotic nitric oxide synthase family ofenzymes were discovered soon after P450BM3 and shown tohave similar domain organization [83]. In addition to bindingFAD, FMN and heme, NOS's also bind the redox cofactortetrahydrobiopterin (H4B) in proximity to the heme iron, and theCa2+-binding protein calmodulin as an effector of electrontransport in the enzyme [84,85]. However, despite the fact that
NOS's catalyses P450-like reductive activation of dioxygen, thestructures of the NOS heme domains are considerably differentto that of any P450 enzyme, and the similar reaction chemistryof the NOS and P450 enzymes clearly results from convergentevolution.
While the BM3-type system was the first, and by far the mostintensively studied, P450–redox partner fusion identified, anumber of others have now been recognized. Some have beencharacterized at the protein level. Below, we survey the currentstate of knowledge of other P450 fusion enzymes.
4. Cytochrome P450–PFOR-like fusion enzymes
Analysis of prokaryotic genome sequences first broughtattention to the fact that a novel kind of P450 redox system wasfound in Rhodococcus erythropolis, various species of thepathogen Burkholderia, and in R. metallidurans. These putativeproteins contain a N-terminal P450 domain and a C-terminaldomain with substantial homology (>40% identity) to thereductase subunits of the phthalate family of mono- anddioxygenases (abbreviated as PFOR-representing PhthalateFamily Oxygenase Reductase; [86,87]) (Fig. 2D). The wellcharacterized phthalate dioxygenase reductase (PFOR) enzymecontains both FMN and iron–sulfur (2Fe–2S) centres, andtransfers electrons to phthalate dioxygenase (PDO) [88]. ThePDO system characterized in Burholderia cepacia catalyses thefirst step in the breakdown of the aromatic compound phthalate,forming a cis-dihydrodiol product [89]. A subsequent conver-sion by the next enzymes in the pathway produce protocatech-uate (3,4-dihydroxybenzoate), a common intermediate inaromatic metabolism [90]. PDO is a multimeric proteincontaining one Rieske-type iron–sulfur cluster (2Fe–2S) andone mononuclear Fe (II) centre in each monomer. The PFORtransfers electrons from pyridine nucleotide coenzymes (NADHis preferred) through FMN, then the 2Fe–2S centre and onto theRieske centre in the PDO [91]. In the P450 fusion systemsidentified at the genetic level, the cofactor accepting electronsfrom PFOR should instead be P450 heme iron.
The first such P450–PFOR fusion enzyme characterized atthe protein level was from the Rhodococcus sp. NCIMB 9784[92]. This protein was expressed in E. coli and shown todealkylate 7-ethoxycoumarin, a marker activity for P450enzymes. Through a combination of steady-state and stopped-flow studies, the coenzyme preference was seen to be forNADPH in this P450–PFOR fusion enzyme (by a factor ofmore than 500 according to transient kinetic analysis of flavinreduction rate). Purified enzyme was shown to catalyse 7-ethoxycoumarin dealkylation, although the rate was only∼5min−1, substantially lower than the limiting rate of flavinreduction with NADPH under similar conditions (180s−1). It isunlikely, therefore, that this aromatic substrate is a good mimicof the molecule(s) naturally selected by this enzyme, andprobably by this enzyme class. Such information may beobtained from analysis of the amino acid sequence of the heme(P450) domain of the fusion protein, and its relationships withother P450s of known function. In this respect, the hemedomain of the Rhodococcus sp. NCIMB 9784 P450–PFOR

354 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
fusion protein has substantial similarity (>50% identity) toThcB from Rhodococcus erythropolis, a P450 shown to catalyseoxidative degradation of atrazine and thiocarbamate-basedherbicides, including EPTC (S-ethyl dipropylthiocarbamate)[93]. The P450 is formally named CYP116A1, and is encodedalongside a 2Fe–2S ferredoxin (ThcC, rhodocoxin) and aferredoxin reductase (ThcD, rhodocoxin reductase). Thus, thisP450 appears to be part of a class I redox system. The Rhodo-coccus sp. NCIMB 9784 P450–PFOR fusion is now classifiedas CYP116B2, but capacity to oxidatively degrade herbicideswas not shown for this enzyme. However, in recent work, wehave expressed and characterized the R. metallidurans P450–PFOR fusion (classified as CYP116B1) and have shown thatthis enzyme does possess activity towards thiocarbamateherbicides (Warman, A.J. et al., unpublished data). Thus, itappears possible that a role for these P450–PFOR CYP116family fusion enzymes may be in provision of resistance toenvironmental toxins, although their specific evolution forthiocarbamate degradation may be unlikely.
In the P450BM3-type P450–redox partner fusion enzymes,the arrangement of the redox chain (electron transfer from FAD-to-FMN-to-heme) is in the same linear order as are the bindingsites for the cofactors in the protein chain (heme, FMN, FAD).This arrangement changes in the P450–PFOR CYP116 fusionenzymes, with electron transfer from FMN-to-[2Fe–2S]-to-heme, and the cofactor binding sites arranged heme, FMN,iron–sulfur (Fig. 5). Tertiary and quaternary structure for theCYP116 fusion enzymes has yet to be resolved, but a lineararrangement of domains appears inconsistent with the electrontransfer pathway expected. Thus, the tertiary structure of theseenzymes might result in the “cradling” of the heme domain bythe PFOR domain to bring 2Fe–2S and heme redox centres intoproximity, or enable the mobility of the 2Fe–2S-containingdomain to facilitate communication with both FMN and heme
Fig. 5. Modular construction of P450 redox partner fusion systems. The diagramillustrates the domain organization and order (with block lengths indicating theapproximate relative domain sizes) for various P450–redox partner fusionenzymes. Domains binding heme (i.e. P450 domains) are shown in red, thosebinding FMN in orange, FAD in yellow and iron–sulfur clusters (Fe/S) inbrown. P450 BM3 has a N-terminal fatty acid hydroxylase P450 linked to aFMN- and FAD-containing CPR [23]. The Rhodococcus-type P450-phthalatedioxygenase reductase fusion (PFOR) also has the P450 at the N-terminal [92],as does the Methylococcus capsulatus P450–ferredoxin fusion McCYP51FX[105]. The domain organization is reversed in the flavodoxin–P450 fusion XplA[104], and also in the putative P450-acyl CoA dehydrogenase fusion (P450-ACAD)—with the redox partners at the N-terminal.
centres. Alternatively, a dimeric arrangement (as for P450BM3)may bring about inter-monomer electron transfer.
A recent Blast search indicates that the CYP116 PFORfusion proteins are found in an increasing number of Rhodo-coccus, Burkholderia and Ralstonia species. This implies (asfor P450BM3) that a fundamental role is played in bacterialphysiology and/or survival in the environment. Amazingly, R.metallidurans encodes both CYP102- and CYP116-like fusionenzymes. A major challenge in coming years will be theresolution of these enzymes' roles, and in the case of theCYP116 class to determine whether these enzymes havegenuinely evolved for detoxication purposes. This will also bean issue with the following system discussed.
5. A cytochrome P450–flavodoxin fusion protein
In the typical class II P450 systems (and for the BM3-typeP450–CPR fusion enzyme class), the ultimate electron donor tothe P450 heme iron is a flavodoxin-like module with FMNcofactor bound. The flavodoxins are small (∼14–23kDa)flavoproteins that are found in a wide range of microbes,including both aerobic and anaerobic bacteria and lowereukaryotes (including algae) [94]. In their non-P450 relatedphysiological roles, flavodoxins are generally considered to actas single electron donors with the FMN shuttling between its 2-electron reduced (hydroquinone) and 1-electron reduced(semiquinone) form as the electron in transferred. These rolesinclude participation in nitrogen fixation reactions, reductiveactivation of pyruvate-formate lyase, and reductive activation ofbiotin synthase [95–97]. Studies of the evolution of CPRenzymes indicated that they had emerged from fusion of aferredoxin reductase-like protein to a flavodoxin module. Theseassumptions were borne out by the atomic structure of rat CPR,which clearly defined a flavodoxin-like fold for the FMN-containing domain [98]. Structural studies of the P450BM3 FMNdomain also demonstrated a typical flavodoxin structure.However, this particular domain shows unusual properties interms of destabilizing the semiquinone form in favour of thehydroquinone [99]. Stopped-flow kinetic and EPR studies havealso shown that a transient anionic (red) semiquinone is formedon one electron reduction of the BM3 FMN domain, whereasthe neutral (blue) form is almost invariably seen in otherflavodoxins [99,100].
With the foreknowledge that CPR enzymes communicatewith P450s using a flavodoxin-like domain, it was perhaps nosurprise that bacterial class I P450 redox systems have beenrecognized which can utilize flavodoxin proteins rather than (oras well as) ferredoxins in order to mediate electron transferbetween FAD-containing reductases and the P450s. Examplesinclude the B. subtilis P450BioI protein (CYP107H1) whichplays a key role in the synthesis of biotin (vitamin H). BioIcatalyses fatty acid hydroxylation, but also catalyses theoxidative scission of long chain fatty acyl CoA esters. Theproduct, pimeloyl CoA, is a common intermediate in biotinsynthesis pathways in various bacteria [101]. BioI can interactproductively with either a 4Fe–4S ferreodoxin (Fer) encodedadjacent to the P450 on the genome, or with either of the two

355A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
B. subtilis flavodoxin proteins (YkuN and YkuP) [14,102]. InCitrobacter braakii, P450cin (CYP176A1) catalyses the oxy-genation of cineole to enable its use as an energy source. P450cinreceives electrons from the flavodoxin cindoxin, encodedadjacent to the P450 on the genome [103].
While it is now clear that flavodoxins are viable redoxpartners for microbial P450s, only recently has a P450 beendiscovered in which a flavodoxin is fused directly to the P450(Figs. 2E, 5). In XplA from Rhodococcus rhodochrous strainY-11, a flavodoxin-like protein (N-terminal) is fused to a P450that is able to degrade the explosive RDX (Royal DemolitioneXplosive; hexahydro-1,3,5-trinitro-1,3,5-triazine). The en-zyme was isolated from bacteria cultured in RDX-contami-nated soil. The protein has been purified, although flavincontent appears low from spectral analysis, perhaps indicatinga weak Kd for the FMN. Plant cells expressing the XplAprotein were shown to degrade RDX, raising the possibilitythat degradation of RDX explosive-contaminated soil could bepossible by phytoremediation [104]. A host reductase systemwas not identified as yet, but the enzyme does communicatewith heterologous ferredoxin reductase. An endogenoussubstrate for the P450 has yet to be identified, but one likelyscenario is that RDX is an adventitious substrate for XplA,which has evolved to catabolise other structurally or che-mically related compounds. Detailed studies of structure andmechanism of the enzyme will clearly be critical to revealits catalytic properties and the breadth of its substrateselectivity.
6. A cytochrome P450–ferredoxin fusion protein
One step away from a P450–flavodoxin fusion would be aP450 fused to a ferredoxin protein, a system that would againrequire interaction with a NAD(P)H dependent ferredoxinreductase to complete the part-fused class I-type P450 redoxsystem. One such enzyme has been cloned and expressed.McCYP51FX from Methylococcus capsulatus was clonedand expressed in E. coli and produced a P450 linked at theC-terminal to a ferredoxin module via an alanine rich linker(Figs. 2F, 5) [105]. The fused P450 is a member of the CYP51family of sterol demethylase enzymes, which catalyse the 3-stepoxidation of sterols such as lanosterol (in fungi), 24,25-dihydrolanosterol and lanosterol in animals, and obtusifoliol(in plants), resulting in the cleavage of their 14α-methyl group[106]. M. capsulatus is the only prokaryote for which a role ofCYP51 (in cholesterol synthesis) appears clear. In fungi, theproduct ergosterol is an essential molecule for membraneintegrity. CYP51 inhibition by azole antifungal drugs (e.g.fluconazole, ketoconazole) results in accumulation of methyl-ated sterols and disruption of the membrane, leading to cell death[107]. A CYP51 homologue was discovered recently in thepathogenic bacterium Mycobacterium tuberculosis. The M.tuberculosis CYP51 is the first structurally characterizedmember of the CYP51 family and has high affinity for azoledrugs, although it does not appear to be a drug target enzyme inthis bacterium [108,109]. TheM. capsulatusCYP51–ferredoxinfusion is the first such fusion enzyme identified, and is clearly a
target for structural elucidation. The ferredoxin fusion mayfacilitate a more efficient electron transfer system to expediteP450 activity, in consort with a host ferredoxin reductase.Protein sequence analysis indicated that the ferredoxin islikely to be of the 3Fe–4S cluster-type, although more detailedspectroscopic or structural analysis would be required forsecure assignment. In the M. tuberculosis genome, a 3Fe–4Sferredoxin is encoded alongside the CYP51 on the genome,and was shown to be a viable redox partner [110]. Recentpotentiometric studies suggest that the M. tuberculosis ferre-doxin has a rather more positive redox potential than does itsCYP51 partner, possibly reflecting a strict thermodynamiccontrol over the electron transfer process and product forma-tion [111]. It will be interesting to establish whether the M.capsulatus CYP51–ferredoxin fusion also has similar features.In addition, and as with the above XplA P450–flavodoxinfusion, crystallization and structural elucidation has potentialto provide important information on the nature of P450/redoxpartner interactions.
7. Man-made P450 fusion enzymes
While a detailed discussion of artificial fusion proteins isoutwith the scope of this review, it is pertinent to discuss brieflythe fact that the scientific literature is rich with reports of theproduction and characterization of artificial fusions betweenP450 enzymes and redox partners. The bulk of these reportsfocus on fusions of eukaryotic P450s with cytochrome P450reductase, mimicking the natural P450BM3 system. Sometimes,these systems achieve moderately enhanced turnover ratescompared to those obtained through mixing the individualcomponent proteins. Examples of stable and active P450/CPRfusions include those involving mammalian CYP3A4 andCYP17A P450s [112,113]. Proximity effects (i.e. closejuxtaposition of the redox partners due to their connection viaa short linker peptide) are obviously one major reason whyenhanced catalytic rate constants can be achieved in suchfusions, since rates of productive collisions between the redoxpartners can be enhanced. However, the fact that such P450/CPR fusions have yet to produce chimeric enzymes with kcatvalues even 10% of that of P450BM3 indicates that naturalevolution of the CYP102-type enzymes has resulted in muchmore effective control of e.g. protein dynamics and electrontransfer than has yet been feasible by artificial proteinengineering.
Other attempts to generate novel P450 fusions includeartificial fusions of flavodoxins to P450s (e.g. [114]) and a threeprotein fusion of P450cam to its putidaredoxin and putidaredoxinreductase redox partners [115]. In the latter case, an effectivecamphor hydroxylase system was created and the advantages ofproduction of a single multidomain oxygenase enzyme areobvious. However, there has yet to be exploitation of this modelfor biotechnologically relevant transformations, despite the factthat rational engineering of P450cam has resulted in the creationof variants with abilities to oxygenate molecules such asdiphenylmethane [116]. Although man-made P450–redoxpartner fusions can evidently lead to enhancement of catalytic

356 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
rate, there has been, in general, rather little commercialexploitation of this technology. In fact, after proof of conceptof the catalytic competence of P450/P450 reductase fusionsthrough several studies in the 1980's to 1990's, the productionand characterization of such chimeras now appears lessfashionable and the bulk of biotechnologically orientatedstudies have been done on the natural P450BM3 fusion system(e.g. [64–66]). However, gene shuffling studies and creation ofchimeras between distinct P450 enzymes (e.g. P450cam andCYP2E1) have potential for evolution of novel P450 activities[117]. Fusions of stand-alone P450s to other redox systems (e.g.the PFOR-type redox apparatus) will also be of interest toexplore electron transfer mechanism and possibly to createcatalytically more efficient systems [118].
8. Conclusions and future prospects
The advent of genome sequencing has led to therecognition that several novel types of P450 redox systemexist, and the early classification of P450 oxygenase redoxsystems into class I (prokaryotic: P450, ferredoxin reductaseand ferredoxin) and class II (eukaryotic: P450 and P450reductase) is now clearly proven to be outdated. As shown inFig. 2, a number of new types of P450 redox partner fusionenzymes have been identified in recent years. To thesedistinctive types of P450 systems other unusual members ofthe P450 superfamily can be added, including (i) P450nor fromFusarium oxysporum, which interacts directly with NADH tocatalyse nitric oxide reduction, and bypasses the requirementfor a protein redox partner; and (ii) P450BSβ from B. subtilis(and homologues from other organisms) a peroxygenaseenzyme which uses hydrogen peroxide to provide electrons,protons and oxygen, and again evades requirement for aprotein partner. In addition to these enzymes, further scrutinyof the databases enables identification of other likelycandidates for novel types of P450–redox partner fusions.Among the most interesting is an apparent P450-acyl CoAdehydrogenase fusion is found in Pseudomonas fluorescens(Figs. 2G, 5). This is surely likely to be only the tip of theiceberg with respect to the biodiversity of P450 systems inbiology. It is thus likely that the explosion of genomic data hasushered in a new era of P450 biochemistry in which novelprotein–protein interactions, reaction chemistry and physio-logy are revealed.
Acknowledgements
The author wishes to acknowledge financial support forresearch in his laboratory from the Biotechnology andBiological Sciences Research Council, UK (grant numbersBB/C006879/1, BBS/B/06288 and 91/C19757) and from theEU (grant NM4TB; contract number LSHP-CT-2005-018923).
References
[1] F.P. Guengerich, Cytochrome P450: what have we learned and what arethe future issues? Drug Metab. Rev. 36 (2004) 159–197.
[2] C. Rodriguez-Antona, M. Ingelman-Sundberg, Cytochrome P450pharmacogenetics and cancer, Oncogene 25 (2006) 1671–1679.
[3] I. Pikuleva, M.R. Waterman, Cytochromes P450 in synthesis of steroidhormones, bile acids, vitamin D3 and cholesterol, Mol. Aspects Med. 20(1999) 33–42.
[4] Y. Xue, D.H. Sherman, Biosynthesis and combinatorial biosynthesis ofpikromycin-related macrolides in Streptomyces venezuelae, Metab. Eng.3 (2001) 15–26.
[5] S.G. Sligar, T.M. Makris, I.G. Denisov, Thirty years of microbial P450monooxygenase research: peroxo-heme intermediates—The central busstation in heme oxygenase catalysis, Biochem. Biophys. Res. Commun.338 (2005) 346–354.
[6] V.B. Urlacher, S. Lutz-Wahl, R.D. Schmid, Microbial P450 enzymes inbiotechnology, Appl. Microbiol. Biotechnol. 64 (2004) 317–325.
[7] T. Kubo, M.W. Peters, P. Meinhold, F.H. Arnold, R. Bernhardt,Enantioselective epoxidation of terminal alkenes to (R)- and (S)-epoxidesby engineered cytochromes P450 BM-3, Chemistry 12 (2006)1216–1220.
[8] S.G. Bell, X. Chen, R.J. Sowden, F. Xu, J.N. Williams, L.L. Wong, Z.Rao, Molecular recognition in (+)-alpha-pinene oxidation by cytochromeP450cam, J. Am. Chem. Soc. 125 (2003) 705–714.
[9] I.F. Sevrioukova, H. Li, T.L. Poulos, Crystal structure of putidaredoxinreductase from Pseudomonas putida, the final structural component ofthe cytochrome P450cam monooxygenase, J. Mol. Biol. 336 (2004)889–902.
[10] M.B. Murataliev, R. Feyereisen, F.A. Walker, Electron transfer bydiflavin reductases, Biochim. Biophys. Acta 1698 (2004) 1–26.
[11] T.M. Makris, K. von Koenig, I. Schlichtling, S.G. Sligar, The status ofhigh-valent metal oxo complexes in the P450 cytochromes, J. Inorg.Biochem. 100 (2006) 507–518.
[12] A. Daiber, H. Shoun, V. Ullrich, Nitric oxide reductase (P450nor) fromFusarium oxysporum, J. Inorg. Biochem. 99 (2005) 185–193.
[13] D.S. Lee, A. Yamada, H. Sugimoto, I. Matsunaga, H. Ogura, K.Ichihara, S. Adachi, S.Y. Park, Y. Shiro, Substrate recognition andmolecular mechanism of fatty acid hydroxylation by cytochrome P450from Bacillus subtilis. Crystallographic, spectroscopic, and mutationalstudies, J. Biol. Chem. 278 (2003) 9761–9767.
[14] R.J. Lawson, D. Leys, M.J. Sutcliffe, C.A. Kemp, M.R. Cheesman,S.J. Smith, J. Clarkson, W.E. Smith, I. Haq, J.B. Perkins, A.W.Munro, Thermodynamic and biophysical characterization of cyto-chrome P450 BioI from Bacillus subtilis, Biochemistry 43 (2004)12410–12426.
[15] A.V. Puchkaev, T. Wakagi, P.R. Ortiz de Montellano, CYP119 plus aSulfolobus tokodaii strain 7 ferredoxin and 2-oxoacid:ferredoxinoxidoreductase constitute a high-temperature cytochrome P450 catalyticsystem, J. Am. Chem. Soc. 124 (2002) 12682–12683.
[16] L.O. Narhi, A.J. Fulco, Characterization of a catalytically self-sufficient119,000-dalton cytochrome P-450 monooxygenase induced by barbitu-rates in Bacillus megaterium, J. Biol. Chem. 261 (1986) 7160–7169.
[17] J.J. Muller, A. Lapko, G. Bourenkov, K. Ruckpaul, U. Heinemann,Adrenodoxin reductase–adrenodoxin complex structure suggests elec-tron transfer path in steroid biosynthesis, J. Biol. Chem. 276 (2001)2786–2789.
[18] K.J. McLean, M. Sabri, K.R. Marshall, R.J. Lawson, D.G. Lewis, D.Clift, P.R. Balding, A.J. Dunford, A.J. Warman, J.P. McVey, A.M. Quinn,M.J. Sutcliffe, N.S. Scrutton, A.W. Munro, Biodiversity of cytochromeP450 redox systems, Biochem. Soc. Trans. 33 (2005) 796–801.
[19] P.J. Loida, S.G. Sligar, Molecular recognition in cytochrome P-450:mechanism for the control of uncoupling reactions, Biochemistry 32(1993) 11530–11538.
[20] M.A. Noble, C.S. Miles, S.K. Chapman, D.A. Lysek, A.C. MacKay, G.A.Reid, R.P. Hanzlik, A.W. Munro, Roles of key active-site residues inflavocytochrome P450 BM3, Biochem. J. 339 (1999) 371–379.
[21] P.K. Powell, I. Wolf, R. Jin, J.M. Lasker, Metabolism of arachidonic acidto 20-hydroxy-5,8,11,14-eicosatetraenoic acid by P450 enzymes inhuman liver: involvement of CYP4F2 and CYP4A11, J. Pharmacol.Exp. Ther. 285 (1998) 1327–1336.
[22] A.W. Munro, S. Daff, J.R. Coggins, J.G. Lindsay, S.K. Chapman,

357A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
Probing electron transfer in flavocytochrome P450 BM3 and itscomponent domains, Eur. J. Biochem. 239 (1996) 403–409.
[23] A.W. Munro, D. Leys, K.J. McLean, K.R. Marshall, T.W. Ost, S. Daff,C.S. Miles, S.K. Chapman, D.A. Lysek, C.C. Moser, C.C. Page, P.L.Dutton, P450 BM3: the very model of a modern flavocytochrome,Trends Biochem. Sci. 27 (2002) 250–257.
[24] C.B. Brewer, J.A. Peterson, Single turnover kinetics of the reactionbetween oxycytochrome P-450cam and reduced putidaredoxin, J. Biol.Chem. 263 (1988) 791–798.
[25] R. Perera, M. Sono, G.M. Raner, J.H. Dawson, Subzero-temperaturestabilization and spectroscopic characterization of homogeneous oxyfer-rous complexes of the cytochrome P450 BM3 (CYP102) oxygenasedomain and holoenzyme, Biochem. Biophys. Res. Commun. 338 (2005)365–371.
[26] M. Sono, R. Perera, S. Jin, T.M. Makris, S.G. Sligar, T.A. Bryson, J.H.Dawson, The influence of substrate on the spectral properties ofoxyferrous wild-type and T252A cytochrome P450-CAM, Arch.Biochem. Biophys. 436 (2005) 40–49.
[27] F.P. Guengerich, Human cytochrome P450 enzymes, in: P.R. Ortiz deMontellano (Ed.), Cytochrome P450, Structure, Mechanism andBiochemistry (3rd Edition), Kluwer Academic/Plenum Publishers,New York, 2005, pp. 377–530.
[28] T.M. Makris, I. Denisov, I. Schlichtling, S.G, Sligar, Activation ofmolecular oxygen by cytochrome P450, in: P.R. Ortiz de Montellano(Ed.), Cytochrome P450, Structure, Mechanism and Biochemistry(3rd Edition), Kluwer Academic/Plenum Publishers, New York, 2005,pp. 149–182.
[29] F. Xu, S.G. Bell, J. Lednik, A. Insley, Z. Rao, L.L. Wong, The hememonooxygenase cytochrome P450cam can be engineered to oxidizeethane to ethanol, Angew. Chem. Int. Ed. 44 (2005) 4029–4032.
[30] M. Imai, H. Shimada, Y. Watanabe, Y. Matsushima-Hibiya, R.Makino, H. Koga, T. Horiuchi, Y. Ishimura, Uncoupling of thecytochrome P-450cam monooxygenase reaction by a single mutation,threonine-252 to alanine or valine: possible role of the hydroxyl aminoacid in oxygen activation, Proc. Natl. Acad. Sci. U. S. A. 86 (1989)7823–7827.
[31] P.J. Loida, S.G. Sligar, Engineering cytochrome P-450cam to increase thestereospecificity and coupling of aliphatic hydroxylation, Protein Eng. 6(1993) 207–212.
[32] W.M. Atkins, S.G. Sligar, Metabolic switching in cytochrome P-450cam:deuterium isotope effects on regiospecificity and the monooxygenase/oxidase ratio, J. Am. Chem. Soc. 109 (1987) 3754–3760.
[33] H.G. Shertzer, C.D. Clay, M.B. Genter, M.C. Chames, S.N. Schneider,G.G. Oakley, D.W. Nebert, T.P. Dalton, Uncoupling-mediated generationof reactive oxygen by halogenate aromatic hydrocarbons in mouse livermicrosomes, Free Radic. Biol. Med. 36 (2004) 618–631.
[34] H. Aramaki, Y. Sagara, M. Fujita, In vitro transcriptional analysis of thecytochrome P-450cam hydroxylase operon, Biol. Pharm. Bull. 22 (1999)1110–1112.
[35] J. Wang, P.R. Ortiz de Montellano, The binding sites on human hemeoxygenase-1 for cytochrome P450 reductase and biliverdin reductase,J. Biol. Chem. 278 (2003) 20069–20076.
[36] A. Bridges, L. Gruenke, Y.T Chang, I.A. Vasker, G. Loew, L. Waskell,Identification of the binding site on cytochrome P450 2B4 forcytochrome b5 and cytochrome P450 reductase, J. Biol. Chem. 273(1998) 17036–17049.
[37] H. Nishino, T. Ishibashi, Evidence for requirement of NADPH-cytochrome P450 oxidoreductase in the microsomal NADPH-sterolD7-reductase system, Arch. Biochem. Biophys. 374 (2000) 293–298.
[38] A.J. Fulco, P450BM-3 and other inducible bacterial P450 cytochromes:biochemistry and regulation, Annu. Rev. Pharmacol. Toxicol. 31 (1991)177–203.
[39] G.C. Shaw, A.J. Fulco, Barbiturate-mediated regulation of expression ofthe cytochrome P450BM-3 gene of Bacillus megaterium by Bm3R1protein, J. Biol. Chem. 267 (1992) 5515–5526.
[40] J.S. He, Q. Liang, A.J. Fulco, The molecular cloning and characterizationof BM1P1 and BM1P2 proteins, putative positive transcription factorsinvolved in barbiturate-mediated induction of the genes encoding
cytochrome P450BM-1 of Bacillus megaterium, J. Biol. Chem. 270(1995) 18615–18625.
[41] J.S. Miles, A.W. Munro, B.N. Rospendowski, W.E. Smith, J. McKnight,A.J. Thomson, Domains of the catalytically self-sufficient cytochromeP-450 BM-3. Genetic construction, overexpression, purification andspectroscopic characterization, Biochem. J. 288 (1992) 503–509.
[42] M.C. Gustafsson, C.N. Palmer, C.R. Wolf, C. von Wachenfeldt,Fatty-acid-displaced transcriptional repressor, a conserved regulator ofcytochrome P450 102 transcription in Bacillus species, Arch.Microbiol. 176 (2001) 459–464.
[43] N. Nakayame, A. Takemae, H. Shoun, Cytochrome P450foxy, acatalytically self-sufficient fatty acid hydroxylase of the fungus Fusar-ium oxysporum, J. Biochem. 119 (1996) 435–440.
[44] L.O. Narhi, A.J. Fulco, Identification and characterization of two functionaldomains in cytochrome P-450BM-3, a catalytically self-sufficient mono-oxygenase induced by barbiturates in Bacillus megaterium, J. Biol. Chem.262 (1987) 6683–6690.
[45] T.D. Porter, An unusual yet strongly conserved flavoprotein reductase inbacteria and mammals, Trends Biochem. Sci. 16 (1991) 154–158.
[46] R.T Ruettinger, A.J. Fulco, Epoxidation of unsaturated fatty acids by asoluble cytochrome P-450-dependent system from Bacillus megaterium,J. Biol. Chem. 256 (1981) 5728–5734.
[47] S. Graham-Lorence, G. Truan, J.A. Peterson, J.R. Falck, S. Wei, C.Helvig, J.H. Capdevila, An active site substitution, F87V, convertscytochrome P450 BM-3 into a regio- and stereoselective (14S,15R)-arachidonic acid epoxygenase, J. Biol. Chem. 272 (1997) 1127–1135.
[48] C.F. Oliver, S. Modi, M.J. Sutcliffe, W.U. Primrose, L.Y. Lian, G.C.Roberts, A single mutation in cytochrome P450 BM3 changes substrateorientation in a catalytic intermediate and the regiospecificity ofhydroxylation, Biochemistry 36 (1997) 1567–1572.
[49] A.W. Munro, J.G. Lindsay, J.R. Coggins, S.M. Kelly, N.C. Price,Structural and enzymological analysis of the interaction of isolateddomains of cytochrome P-450 BM3, FEBS Lett. 343 (1994) 70–74.
[50] R. Neeli, H.M. Girvan, A. Lawrence, M.J. Warren, D. Leys, N.S.Scrutton, A.W. Munro, The dimeric form of flavocytochrome P450 BM3is catalytically functional as a fatty acid hydroxylase, FEBS Lett. 579(2005) 5582–5588.
[51] S.D. Black, S.T. Martin, Evidence for conformational dynamics andmolecular aggregation in cytochrome P450 102 (BM-3), Biochemistry 33(1994) 12056–12062.
[52] S.N. Daff, S.K. Chapman, K.L. Turner, R.A. Holt, S. Govindaraj, T.L.Poulos, A.W. Munro, Redox control of the catalytic cycle offlavocytochrome P-450 BM3, Biochemistry 36 (1997) 13816–13823.
[53] S. Govindaraj, T.L. Poulos, The domain architecture of cytochromeP450BM-3, J. Biol. Chem. 272 (1997) 7915–7921.
[54] I.F. Sevrioukova, H. Li, H. Zhang, J.A. Peterson, T.L. Poulos, Structure ofa cytochrome P450–redox partner electron-transfer complex, Proc. Natl.Acad. Sci. U. S. A. 96 (1999) 1863–1888.
[55] H. Li, T.L. Poulos, The structure of the cytochrome P450BM-3 haemdomain complexed with the fatty acid substrate, palmitoleic acid, Nat.Struct. Biol. 4 (1997) 140–146.
[56] K.G. Ravichandran, S.S. Boddupalli, C.A. Hasemann, J.A. Peterson, J.Deisenhofer, Crystal structure of hemoprotein domain of P450BM-3, aprototype for microsomal P450s, Science 261 (1993) 731–736.
[57] D.C. Haines, D.R. Tomchick, M. Machius, J.A. Peterson, Pivotal role ofwater in the mechanism of P450BM-3, Biochemistry 40 (2001)13456–13465.
[58] H.M. Girvan, K.R. Marshall, R.J. Lawson, D. Leys, M.G. Joyce, J.Clarkson, W.E. Smith, M.R. Cheesman, A.W. Munro, FlavocytochromeP450 BM3 mutant A264E undergoes substrate-dependent formationof a novel heme iron ligand set, J. Biol. Chem. 279 (2004)23274–23286.
[59] M.G. Joyce, H.M. Girvan, A.W. Munro, D. Leys, A single mutation incytochrome P450 BM3 induces the conformational rearrangement seenupon substrate binding in the wild-type enzyme, J. Biol. Chem. 279(2004) 23287–23293.
[60] D.S. Sem, C.B. Kasper, Interaction with arginine 597 of NADPH-cytochrome P-450 oxidoreductase is a primary source of the uniform

358 A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
binding energy used to discriminate between NADPH and NADH,Biochemistry 32 (1993) 11548–11558.
[61] R. Neeli, O. Roitel, N.S. Scrutton, A.W. Munro, Switching pyridinenucleotide specificity in P450 BM3: mechanistic analysis of the W1046Hand W1046A enzymes, J. Biol. Chem. 280 (2005) 17634–17644.
[62] O. Dohr, M.J. Paine, T. Friedberg, G.C. Roberts, C.R. Wolf, Engineeringof a functional human NADH-dependent cytochrome P450 system, Proc.Natl. Acad. Sci. U. S. A. 98 (2001) 81–86.
[63] A. Gutierrez, O. Dohr, M.J. Paine, C.R. Wolf, N.S. Scrutton, G.C.Roberts, Trp-676 facilitates nicotinamide coenzyme exchange in thereductive half-reaction of human cytochrome P450 reductase: propertiesof the soluble W676H and W676A mutant reductases, Biochemistry 39(2000) 15990–15999.
[64] T.W. Ost, C.S. Miles, J. Murdoch, Y. Cheung, G.A. Reid, S.K. Chapman,A.W. Munro, Rational re-design of the substrate binding site offlavocytochrome P450 BM3, FEBS Lett. 486 (2000) 173–177.
[65] M.W. Peters, Meinhold A. Glieder, F.H. Arnold, Regio- andenantioselective alkane hydroxylation with engineered cytochromesP450 BM-3, J. Am. Chem. Soc. 125 (2003) 13442–13450.
[66] M. Landwehr, L. Hochrein, C.R. Otey, A. Kasrayan, J.E. Backvall, F.H.Arnold, Enantioselective alpha-hydroxylation of 2-arylacetic acidderivatives and buspirone batalyzed by engineered cytochrome P450BM-3, J. Am. Chem. Soc. 128 (2006) 6058–6059.
[67] T.W. Ost, A.W. Munro, C.G. Mowat, P.R. Taylor, A. Pesseguiero, A.J. Fulco, A.K. Cho, M.R. Cheesman, M.D Walkinshaw, S.K.Chapman, Structural and spectroscopic analysis of the F393H mutantof flavocytochrome P450 BM3, Biochemistry 40 (2001)13430–13438.
[68] T.W. Ost, C.S. Miles, A.W. Munro, J. Murdoch, G.A. Reid, S.K.Chapman, Phenylalanine 393 exerts thermodynamic control over theheme of flavocytochrome P450 BM3, Biochemistry 40 (2001)13421–13429.
[69] M.L. Klein, A.J. Fulco, Critical residues involved in FMN binding andcatalytic activity in cytochrome P450BM-3, J. Biol. Chem. 268 (1993)7553–7561.
[70] A. Gutierrez, L.Y. Lian, C.R.Wolf, N.S. Scrutton, G.C. Roberts, Stopped-flow kinetic studies of flavin reduction in human cytochrome P450reductase and its component domains, Biochemistry 40 (2001)1964–1975.
[71] N. Nakayama, H. Shoun, Subterminal hydroxylation of fatty acids by acytochrome P-450-dependent enzyme system from a fungus, Fusariumoxysporum, J. Biochem. 97 (1985) 755–763.
[72] T. Kitazume, N. Takaya, N. Nakayama, H. Shoun, Fusarium oxysporumfatty-acid subterminal hydroxylase (CYP505) is a membrane-boundeukaryotic counterpart of Bacillus megaterium cytochrome P450BM3,J. Biol. Chem. 275 (2000) 39734–39740.
[73] M.B. Murataliev, R. Feyereisen, Functional interactions in cytochromeP450BM3. Fatty acid substrate binding alters electron-transfer proper-ties of the flavoprotein domain, Biochemistry 35 (1996) 15029–15037.
[74] D.H. Craig, S.K. Chapman, S. Daff, Calmodulin activates electrontransfer through neuronal nitric-oxide synthase reductase domain byreleasing an NADPH-dependent conformational lock, J. Biol. Chem. 277(2002) 33987–33994.
[75] J.A. Seo, R.H. Proctor, R.D. Plattner, Characterization of four clusteredand coregulated genes associated with fumonisin biosynthesis in Fusar-ium verticillioides, Fungal Genet. Biol. 34 (2001) 155–165.
[76] M.C. Gustafsson, O. Roitel, K.R. Marshall, M.A. Noble, S.K. Chapman,A. Pessegueiro, A.J. Fulco, M.R. Cheesman, C. von Wachenfeldt, A.W.Munro, Expression, purification, and characterization of Bacillus subtiliscytochromes P450 CYP102A2 and CYP102A3: flavocytochromehomologues of P450 BM3 from Bacillus megaterium, Biochemistry 43(2004) 5474–5487.
[77] A.W. Munro, M.A. Noble, T.W. Ost, A.J. Green, K.J. McLean, L.Robledo, C.S. Miles, J. Murdoch, S.K. Chapman, Flavocytochrome P450BM3 substrate selectivity and electron transfer in a model cytochromeP450, Sub-cell. Biochem. 35 (2000) 297–315.
[78] A.W. Munro, L. Quaroni, G.D. Chumanov, K.L. Turner, S.K. Chapman,R.P. Hanzlik, A.W. Munro, Imidazolyl carboxylic acids as mechanistic
probes of flavocytochrome P-450 BM3, Biochemistry 37 (1998)15799–15807.
[79] K.R. Wolthers, N.S. Scrutton, Electron transfer in human methioninesynthase reductase studied by stopped-flow spectrophotometry, Bio-chemistry 43 (2004) 490–500.
[80] M.J. Paine, A.P. Garner, D. Powell, J. Sibbald, M. Sales, N. Pratt, T.Smith, D.G. Tew, C.R. Wolf, Cloning and characterization of a novelhuman dual flavin reductase, J. Biol. Chem. 275 (2000) 1471–1478.
[81] A.J. Dunford, K.R. Marshall, A.W. Munro, N.S. Scrutton, Thermody-namic and kinetic analysis of the isolated FAD domain of rat neuronalnitric oxide synthase altered in the region of the FAD shielding residuePhe 1395, Eur. J. Biochem. 271 (2004) 2548–2560.
[82] S. Govidaraj, T.L. Poulos, Probing the structure of the linker connectingthe reductase and heme domains of cytochrome P450BM-3 using site-directed mutagenesis, Protein Sci. 5 (1996) 1389–1393.
[83] D.J. Stuehr, J. Santolini, Z.Q. Wang, C.C. Wei, S. Adak, Update onmechanism and catalytic regulation in the NO synthases, J. Biol. Chem.279 (2004) 36167–36170.
[84] K. Panda, S. Ghosh, D.J. Stuehr, Calmodulin activates intersubunitelectron transfer in the neuronal nitric oxide synthase dimer, J. Biol.Chem. 276 (2001) 23349–23356.
[85] D.J. Stuehr, C.C. Wei, Z. Wang, R. Hille, Exploring the redox reactionsbetween heme and tetrahydrobiopterin in the nitric oxide synthases,Dalton Trans. 2005 (2005) 3427–3435.
[86] R. De Mot, A.H.A. Parret, A novel class of self-sufficient cytochromeP450 mono-oxygenases in prokaryotes, Trends Microbiol. 10 (2002)502–508.
[87] D.T. Gibson, R.E. Parales, Aromatic hydrocarbon dioxygenases inenvironmental biotechnology, Curr. Opin. Biotechnol. 11 (2000)236–243.
[88] G.T. Gassner, M.L. Ludwig, D.L. Gatti, C.C. Correll, D.P. Ballou,Structure and mechanism of the iron–sulfur flavoprotein phthalatedioxygenase reductase, FASEB J. 9 (1995) 1411–1418.
[89] M. Tarasev, D.P. Ballou, Chemistry of the catalytic conversion ofphthalate into its cis-dihydrodiol during the reaction of oxygen with thereduced form of phthalate dioxygenase, Biochemistry 44 (2005)6197–6207.
[90] M. Tarasev, F. Rhames, D.P. Ballou, Rates of the phthalate dioxygenasereaction with oxygen are dramatically increased by interactions withphthalate and phthalate oxygenase reductase, Biochemistry 43 (2004)12799–12808.
[91] C.C. Correll, C.J. Batie, D.P. Ballou, M.L. Ludwig, Phthalatedioxygenase reductase: a modular structure for electron transfer frompyridine nucleotide to [2Fe–2S], Science 258 (1992) 1604–1610.
[92] G.A. Roberts, A. Celik, D.J. Hunter, T.W. Ost, J.H. White, S.K.Chapman, N.J. Turner, S.L. Flitsch, A self-sufficient cytochrome P450with a primary structural organization that includes a flavin domain and a[2Fe–2S] center, J. Biol. Chem. 278 (2003) 48914–48920.
[93] I. Nagy, F. Compernolle, K. Ghys, J. Vanderleyden, R. De Mot, A singlecytochrome P-450 system in involved in degradation of the herbicidesEPTC (S-ethyl dipropylthiocarbamate) and atrazine by Rhodococcus sp.Strain N186/21, Appl. Environ. Microbiol. 61 (1995) 2056–2060.
[94] S.G. Mayhew, G. Tollin, General properties of flavodoxins, in: F. Muller(Ed.), Chemistry and Biochemistry of Flavodoxins, vol. 3, CRC Press,Boca Raton, FL, 1992, pp. 389–426.
[95] K. Fujii, J.H. Galivan, F.M. Hennekens, Activation of methioninesynthase: a further characterization of the flavoprotein system, Arch.Biochem. Biophys. 78 (1977) 662–670.
[96] D. Nieva-Gomez, G.P. Roberts, S. Klevickis, W.J. Brill, Electrontransport to nitrogenase in Klebsiella pneumoniae, Proc. Natl. Acad.Sci. U. S. A. 77 (1980) 2255–2258.
[97] H.P. Blaschkowski, G. Neuer, M. Ludwig-Festl, J. Knappe, Routes offlavodoxin and ferredoxin reduction in Escherichia coli. CoA-acylatingpyruvate: flavodoxin and NADPH flavodoxin oxidoreductases partici-pating in the activation of pyruvate-formation lyase, Eur. J. Biochem. 123(1982) 563–569.
[98] M. Wang, D.L. Roberts, R. Paschke, T.M. Shea, B.S.S. Masters, J.-J.P.Kim, Three dimensional structure of NADPH-cytochrome P450

359A.W. Munro et al. / Biochimica et Biophysica Acta 1770 (2007) 345–359
reductase-prototype for FAD- and FMN-containing enzymes, Proc. Natl.Acad. Sci. U. S. A. 94 (1997) 9411–9416.
[99] S.C. Hanley, S. Daff, The unusual redox properties of flavocytochromeP450 BM3 flavodoxin domain, Biochem. Biophys. Res. Commun. 325(2004) 1418–1423.
[100] M.B. Murataliev, R. Feyereisen, Functional interactions in cytochromeP450 BM3: evidence that NADP(H) binding controls redox potentials ofthe flavin cofactors, Biochemistry 39 (2000) 12699–12707.
[101] M.J. Cryle, J.J. De Voss, Carbon–carbon bond cleavage by cytochromeP450 BioI (CYP107H1), Chem. Commun. 2004 (2004) 86–87.
[102] R.J. Lawson, C. von Wachenfeldt, I. Haq, J. Perkins, A.W. Munro,Expression and characterization of the two flavodoxin proteins of Ba-cillus subtilis, YkuN and YkuP: biophysical properties and interactionswith cytochrome P450 BioI, Biochemistry 43 (2004) 12390–12409.
[103] D.B. Hawkes, G.W. Adams, A.L. Burlingame, P.R. Ortiz de Montellano,J.J. De Voss, Cytochrome P450cin (CYP176A), isolation, expression,and characterization, J. Biol. Chem. 277 (2002) 27725–27732.
[104] E.L. Rylott, R.G. Jackson, J. Edwards, G.L. Womack, H.M.B. Seth-Smith,D.A. Rathbone, S.E. Strand, N.C. Bruce, An explosive-degrading cyto-chrome P450 activity and its targeted application for the phytoremediationof RDX, Nat. Biotechnol. 24 (2006) 216–219.
[105] C.J. Jackson, D.C. Lamb, T.M. Marczylo, A.G.S. Warrilow, N.J. Manning,D.J. Lowe, D.E. Kelly, S.L. Kelly, A novel sterol 14α-demethylase/ferredoxin fusion protein (McCYP51FX) from Methylococcus capsulatusrepresents a new class of the cytochrome P450 superfamily, J. Biol. Chem.277 (2002) 46959–46965.
[106] M.R. Waterman, G.I. Lepesheva, Sterol 14a-demethylase, an abundantand essential mixed-function oxidase, Biochem. Biophys. Res. Commun.338 (2005) 418–422.
[107] F.C. Odds, A.J. Brown, N.A. Gow, Antifungal agent: mechanisms ofaction, Trends Microbiol. 11 (2003) 272–279.
[108] L.M. Podust, T.L. Poulos, M.R.Waterman, Crystal structure of cytochromeP450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosisin complex with azole inhibitors, Proc. Natl. Acad. Sci. U. S. A. 98 (2001)3068–3073.
[109] K.J. McLean, K.R. Marshall, A. Richmond, I.S. Hunter, K. Fowler, T.Kieser, S.S. Gurcha, G.S. Besra, A.W. Munro, Azole antifungals arepotent inhibitors of cytochrome P450 mono-oxygenases and bacterialgrowth in mycobacteria and streptomycetes, Microbiology 148 (2002)2937–2949.
[110] A. Bellamine, A.T. Mangla, W.D. Nes, M.R. Waterman, Characterization
and catalytic properties of the sterol 14α-demethylase fromMycobacteriumtuberculosis, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 8937–8942.
[111] K.J. McLean, A.J. Warman, H.E. Seward, K.R. Marshall, M.R.Cheesman, M.A. Waterman, A.W. Munro, Characterization of the My-cobacterium tuberculosis ferredoxin (Fdx) and interactions with itscognate redox partner: the sterol demethylase P450 CYP51, Biochem-istry 45 (2006) 8427–8443.
[112] C.W. Fisher, M.S. Shet, D.L. Caudle, C.A. Martin-Wixtrom, R.W.Estabrook, High-level expression in Escherichia coli of enzymaticallyactive fusion proteins containing the domains of mammalian cytochromesP450 and NADPH-P450 reductase flavoprotein, Proc. Natl. Acad. Sci.U. S. A. 89 (1992) 10817–10821.
[113] M.S. Shet, C.W. Fisher, P.L. Holmans, R.W. Estabrook, Humancytochrome P450 3A4: enzymatic properties of a purified recombinantfusion protein containing NADPH-P450 reductase, Proc. Natl. Acad. Sci.U. S. A. 90 (1993) 11748–11752.
[114] A. Fantuzzi, Y.T. Meharenna, P.B. Briscoe, C. Sassone, B. Borgia, G.Gilardi, Improving catalytic properties of P450 BM3 haem domainelectrodes by molecular Lego, Chem. Commun. 2006 (2006) 1289–1291.
[115] O. Sibbesen, J.J. de Voss, P.R. Ortiz de Montellano, Putidaredoxinreductase–putidaredoxin–cytochrome P450cam triple fusion protein,J. Biol. Chem. 271 (1996) 22462–22469.
[116] S.M. Fowler, P.A. England, A.C.G. Westlake, D.R. Rough, D.P.Nickerson, C. Blunt, D. Braybrook, S. West, L.L. Wong, S.L. Flitsch,Cytochrome P450cam monooxygenase can be redesigned to catalyze theregioselective aromatic hydroxylation of diphenylmethane, J. Chem.Soc., Chem. Commun. 24 (1994) 2761–2762.
[117] S. Sukumaran, W.M. Atkins, R. Shanker, Engineering cytochrome P-450s: chimeric enzymes, Appl. Biochem. Biotechnol. 102–103 (2002)291–302.
[118] M. Nodate, M. Kubota, N. Misawa, Functional expression system forcytochrome P450 genes using the reductase domain of self-sufficientP450RhF from Rhodococcus sp. NCIMB 9784, Appl. Biochem.Biotechnol. 2005 (2005) 1–8.
[119] P.A. Hubbard, A.L. Shen, R. Paschke, C.B. Kasper, J.J. Kim, NADPH-cytochrome P450 oxidoreductase. Structural basis for hydride andelectron transfer, J. Biol. Chem. 276 (2001) 29163–29170.
[120] O. Roitel, N.S. Scrutton, A.W. Munro, Electron transfer in flavocyto-chrome P450 BM3: kinetics of flavin reduction and oxidation, the role ofcysteine 999, and relationships with mammalian cytochrome P450reductase, Biochemistry 42 (2003) 10809–10821.





























