Embed Size (px)
Citation preview

Journal of Molecular Structure 969 (2010) 55–68
Contents lists available at ScienceDirect
Journal of Molecular Structure
journal homepage: www.elsevier .com/ locate /molst ruc
Conformational stability, r0 structural parameters, barriers to internal rotation,vibrational assignments and ab initio calculations of c-C3H5GeH2CH3
James R. Durig a,*, Savitha S. Panikar a,1, Gamil A. Guirgis b, Todor K. Gounev a, Peter Klæboe c, Anne Horn c,Claus J. Nielsen c, Rebecca A. Peebles d, Sean A. Peebles d, Richard J. Liberatore b
a Department of Chemistry, University of Missouri-Kansas City, Kansas City, MO 64110, USAb Department of Chemistry and Biochemistry, College of Charleston, Charleston, SC 29424, USAc Department of Chemistry, University of Oslo, P.O. Box 1033, 0315 Oslo, Norwayd Department of Chemistry, Eastern Illinois University, Charleston, IL 61920, USA
a r t i c l e i n f o
Article history:Received 30 December 2009Accepted 22 January 2010Available online 1 February 2010
Keywords:Conformational stabilityr0 structural parametersAb initio calculationsVibrational assignmentsMethylgermylcyclopropane
0022-2860/$ - see front matter � 2010 Elsevier B.V. Adoi:10.1016/j.molstruc.2010.01.042
* Corresponding author. Tel.: +1 816 235 6038; faxE-mail address: [email protected] (J.R. Durig).
1 Taken in part from the dissertation of S.S. Panikar wDepartment of Chemistry in partial fulfillment of the P
a b s t r a c t
The infrared spectra (3100–40 cm�1) of gaseous and amorphous solid and Raman spectra (3200–20 cm�1) of the liquid at various temperatures for methylgermylcyclopropane, c-C3H5GeH2CH3, havebeen obtained. Additionally, variable temperature (�55 to �100 �C) studies of the infrared spectra ofthe sample dissolved in xenon have been recorded. From these spectral data, two conformers have beenidentified with one the cis (syn) form where the methyl group is over the three-membered ring and theother the gauche form. By utilizing six conformer pairs of the vibrational bands the enthalpy difference ofthe sample dissolved in xenon has been determined to be 43 ± 11 cm�1 (0.51 ± 0.13 kJ mol�1) with thegauche form the more stable conformer. It is estimated that there is approximately 30 ± 3% of the cis formpresent at ambient temperature. It was not possible to achieve crystallization of the compound byannealing it, and both conformers were present at all temperatures. The Ge–H distances of 1.531 and1.533 Å for the gauche conformer have been determined from their stretching frequencies. By utilizingthe microwave rotational constants of the gauche conformer for five isotopomers (70Ge, 72Ge, 73Ge,74Ge, 76Ge) combined with the structural parameters predicted from the MP2(full)/6-311+G(d,p) calcula-tions, the adjusted r0 structural parameters have been obtained. The heavy atom distances (Å) are:(GeC2) = 1.925(5); (C2C4) = 1.517(3); (C2C5) = 1.519(3); (C4C5) = 1.502(3); (GeC6) = 1.947(5) and the angles(�) are: \CGeC = 110.6(5); \GeC2C4 = 120.1(5); \GeC2C5 = 119.2(5). For the cis form very small differ-ences of 0.003 and 0.001 Å from those of the gauche form are predicted for the GeC bonds whereas theother distances are predicted to be the same or differ at the most by 0.002 Å. A complete vibrationalassignment is given for both the conformers. To support the vibrational assignments, normal coordinatecalculations with scaled force constants from MP2(full)/6-31G(d) calculations were carried out to predictthe fundamental vibrational frequencies, infrared intensities, Raman activities, depolarization values andinfrared band contours. Barriers to internal rotation have been predicted. The results are discussed andcompared to the corresponding properties of some similar molecules.
� 2010 Elsevier B.V. All rights reserved.
1. Introduction
Mono-substitution on the methyl group of methylcyclopropanehas a significant effect on the relative amounts of the two conform-ers usually present in the gas phase at ambient temperature. Ini-tially, it was proposed [1] that the electronegativity of thesubstituent has the most pronounced effect on the relativeamounts of the conformational isomers present since the substitu-tion of the ethynyl and cyano-groups had a relatively larger
ll rights reserved.
: +1 816 235 2290.
hich will be submitted to theh.D. degree.
amount of the cis (syn) form present compared to the amount ofthe gauche conformers. These groups have considerably larger elec-tronegativities [2] i.e. 3.1 and 3.2 than the other substituents suchas CH3 (2.9) and Cl (3.0). However, the determination [3] of the en-thalpy difference of the two conformers of fluoromethylcyclopro-pane clearly showed that electronegativity was not the mostsignificant factor since fluorine has the largest electronegativity(4.0) yet there was a relatively small amount of the cis form pres-ent (12 ± 1%) at ambient temperature. The factor which seems tohave the most pronounced effect on the amount of cis rotamerpresent is the distance of the carbon atom of the methyl group tothe carbon atom of the ring [3].
As a continuation of our structural and spectroscopic studies ofmono-substituted cyclopropanes we were interested in meth-

56 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
ylgermylcyclopropane, c-C3H5GeH2CH3 for a comparison to theconformational stability results of our recently investigated [4]methylsilylcyclopropane, c-C3H5SiH2CH3, as well as to the corre-sponding carbon analogue which we investigated a few years ago[5]. Also of interest were the structural parameters and the enthal-py difference between the two conformers which could possiblyprovide some information on the major factors contributing tothe stability of the more stable form. Therefore, we have recordedthe infrared (Fig. 1) and/or Raman spectra (Fig. 2) of the gas, liquidand solid phases of methylgermylcyclopropane and propose acomplete vibrational assignment (Tables 1 and 2). Additionally,we have recorded variable temperature studies of the infraredspectra of the sample dissolved in liquid xenon. Since we haveobtained the rotational constants of all five isotopomers of germa-nium, we determined the r0 structural parameters with reasonablysmall uncertainties. We have also carried out ab initio calculationsemploying a variety of basis sets up to 6-311++G(3df,3pd) at theMøller–Plesset (MP) level to second order with full electron corre-lation to obtain predicted conformational stabilities and completeequilibrium geometries. Similar studies have also been carried outby density functional theory calculations by using the B3LYP meth-od. The force constants, vibrational frequencies, infrared and Ra-man intensities, and conformational stabilities have also been
Fig. 1. Comparison of experimental and calculated infrared spectra of methylgermylcycloof a mixture of gauche and cis conformers at �100 �C with DH = 43 cm�1; (C) simulated
obtained from the ab initio calculations. The results of these spec-troscopic and theoretical studies are reported herein and compar-isons of these results to those of related compounds are made.
2. Experimental
The sample of c-C3H5GeH2CH3 was prepared in two steps.Firstly, cyclopropylbromide was reacted with magnesium in dryether by using the Grignard method and subsequently coupledwith methyltrichlorogermane in dry ether under dry nitrogengas. Secondly, the product methyldichlorogermylcyclopropanewas separated from the ether under reduced pressure and then re-duced with lithium aluminum hydride in dry dibutyl ether underdry nitrogen. The sample was first purified by trap-to-trap distilla-tion to isolate the highly volatile methylgermane and further puri-fied by using a low-temperature, low-pressure fractionationcolumn. The purity of the sample was verified by comparing theinfrared and Raman spectra with the predicted ones along withthe 1H and 13C NMR data.
The mid-infrared spectrum of the gas was obtained from 3500to 300 cm�1 on a Perkin-Elmer model 2000 Fourier transformspectrometer equipped with a Ge/CsI beamsplitter and a DTGSdetector. Atmospheric water vapor was removed from the spec-
propane: (A) observed infrared spectrum in xenon; (B) simulated infrared spectruminfrared spectrum of cis (Cs); (D) simulated infrared spectrum of gauche (C1).
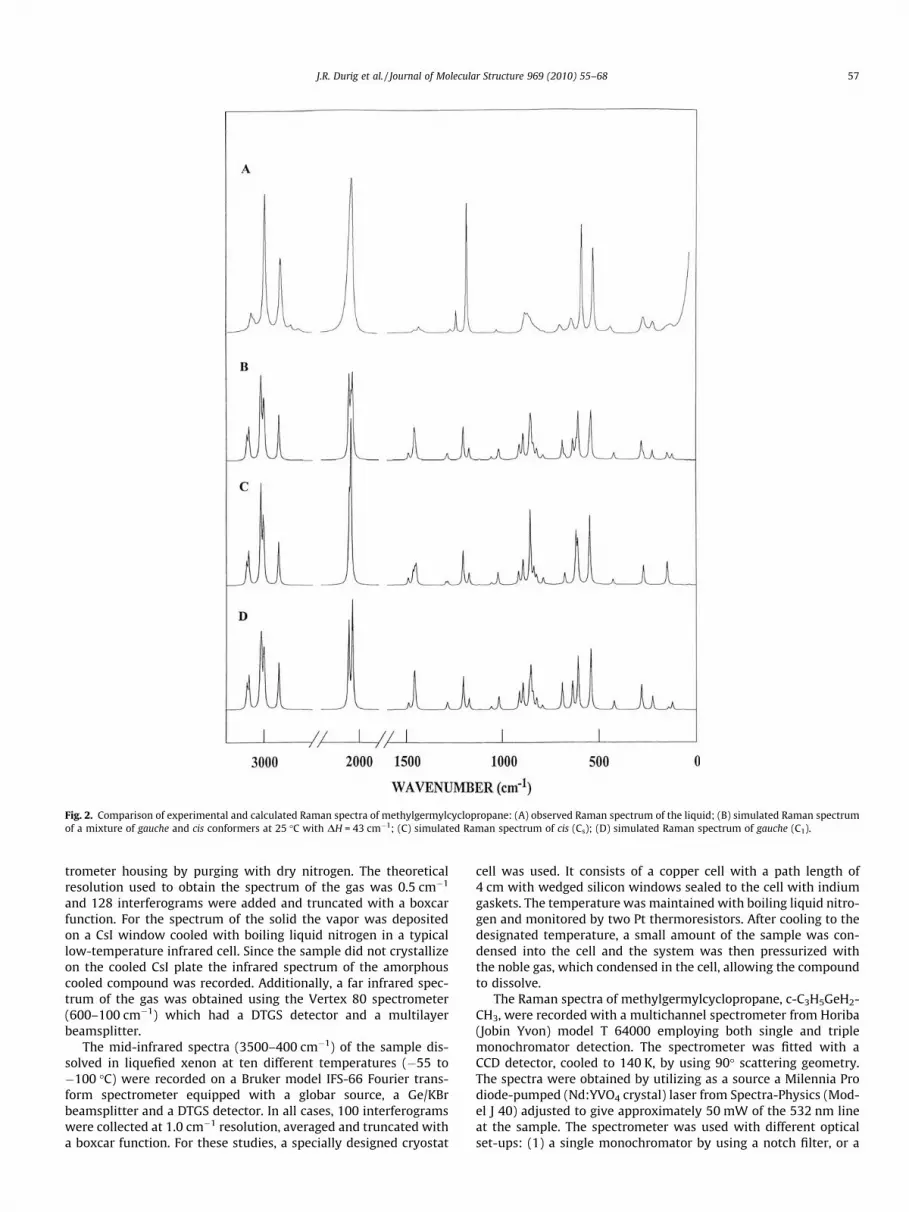
Fig. 2. Comparison of experimental and calculated Raman spectra of methylgermylcyclopropane: (A) observed Raman spectrum of the liquid; (B) simulated Raman spectrumof a mixture of gauche and cis conformers at 25 �C with DH = 43 cm�1; (C) simulated Raman spectrum of cis (Cs); (D) simulated Raman spectrum of gauche (C1).
J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68 57
trometer housing by purging with dry nitrogen. The theoreticalresolution used to obtain the spectrum of the gas was 0.5 cm�1
and 128 interferograms were added and truncated with a boxcarfunction. For the spectrum of the solid the vapor was depositedon a CsI window cooled with boiling liquid nitrogen in a typicallow-temperature infrared cell. Since the sample did not crystallizeon the cooled CsI plate the infrared spectrum of the amorphouscooled compound was recorded. Additionally, a far infrared spec-trum of the gas was obtained using the Vertex 80 spectrometer(600–100 cm�1) which had a DTGS detector and a multilayerbeamsplitter.
The mid-infrared spectra (3500–400 cm�1) of the sample dis-solved in liquefied xenon at ten different temperatures (�55 to�100 �C) were recorded on a Bruker model IFS-66 Fourier trans-form spectrometer equipped with a globar source, a Ge/KBrbeamsplitter and a DTGS detector. In all cases, 100 interferogramswere collected at 1.0 cm�1 resolution, averaged and truncated witha boxcar function. For these studies, a specially designed cryostat
cell was used. It consists of a copper cell with a path length of4 cm with wedged silicon windows sealed to the cell with indiumgaskets. The temperature was maintained with boiling liquid nitro-gen and monitored by two Pt thermoresistors. After cooling to thedesignated temperature, a small amount of the sample was con-densed into the cell and the system was then pressurized withthe noble gas, which condensed in the cell, allowing the compoundto dissolve.
The Raman spectra of methylgermylcyclopropane, c-C3H5GeH2-CH3, were recorded with a multichannel spectrometer from Horiba(Jobin Yvon) model T 64000 employing both single and triplemonochromator detection. The spectrometer was fitted with aCCD detector, cooled to 140 K, by using 90� scattering geometry.The spectra were obtained by utilizing as a source a Milennia Prodiode-pumped (Nd:YVO4 crystal) laser from Spectra-Physics (Mod-el J 40) adjusted to give approximately 50 mW of the 532 nm lineat the sample. The spectrometer was used with different opticalset-ups: (1) a single monochromator by using a notch filter, or a

Table 1Calculateda and observed frequencies (cm�1) for gauche methylgermylcyclopropane, c-C3H5GeH2CH3.
Vib.no.
Approx. description MP2 6-31G(d)
MP2 6-31G(d)scaledb
IR int. Raman act. dp B3LYPcc-PVDZ
B3LYPcc-PVQZ
IR int. IR Ramanliquid
IR solid PEDc Bandcontour
Gas Xenonsolution
A B C
m1 CH2 antisymmetric stretch 3294 3090 13.4 53.3 0.51 3207 3201 20.1 3080 3071 – 3066 97% S1 39 9 52m2 CH2 antisymmetric stretch 3282 3079 0.5 72.2 0.74 3193 3186 0.2 – – 3063 3054 97% S2 18 69 13m3 CH3 antisymmetric stretch 3220 3021 6.1 78.8 0.73 3133 3126 5.6 – 3021 – 2993 100% S3 57 25 18m4 CH3 antisymmetric stretch 3217 3017 6.5 59.0 0.74 3130 3118 13.7 3009 – – 2882 100% S4 8 8 83m5 CH stretch 3213 3014 2.5 94.2 0.22 3128 3118 15.1 3001 – 3003 – 95% S5 13 4 83m6 CH2 symmetric stretch 3200 3002 10.4 104.5 0.06 3116 3116 9.1 2994 2998 2990 – 81% S6, 15% S7 70 14 16m7 CH2 symmetric stretch 3195 2997 11.4 40.3 0.34 3111 3115 21.7 2991 2986 – – 85% S7, 15% S6 – 80 20m8 CH3 symmetric stretch 3117 2924 4.4 97.3 0.00 3040 3039 10.8 2928 2919 2921 2915 100% S8 42 57 1m9 GeH2 antisymmetric stretch 2055 2055 155.1 100.7 0.43 2094 2103 130.9 2061 – – – 84% S9, 16% S10 15 39 46m10 GeH2 symmetric stretch 2036 2036 100.8 124.4 0.09 2084 2096 165.6 2050 2050 2049 2044 84% S10, 16% S9 1 47 52m11 CH2 deformation 1569 1471 1.1 5.0 0.71 1482 1508 0.5 1465 1459 1461 1461 90% S11, 10% S17 51 12 37m12 CH3 antisymmetric
deformation1538 1443 1.5 11.5 0.73 1444 1481 2.0 1437 1435 – 1454 95% S12 39 43 18
m13 CH3 antisymmetricdeformation
1537 1442 3.3 12.8 0.75 1439 1469 3.0 1437 1435 1437 1434 95% S13 5 15 80
m14 CH2 deformation 1533 1438 1.7 10.3 0.75 1438 1469 1.6 1420 1421 1420 1419 100% S14 9 86 5m15 CH bend (in-plane) 1356 1287 4.9 3.6 0.64 1289 1303 3.4 1276 1272 1273 1270 42% S15, 27% S17, 12% S22 22 1 77m16 CH3 symmetric
deformation1352 1268 7.5 2.2 0.01 1251 1276 4.9 1249 1243 1243 1240 98% S16 49 50 1
m17 Ring breathing 1266 1202 5.9 19.1 0.09 1214 1218 5.0 1191 1189 1190 1187 56% S17, 16% S15, 14% S22 91 1 8m18 CH2 rock 1235 1172 0.7 6.3 0.75 1182 1200 0.5 1174 1170 1174 1169 52% S18, 32% S19, 15% S28 4 83 14m19 CH2 twist 1177 1117 2.3 0.4 0.58 1117 1134 0.8 1100 1099 1099 1101 46% S19, 49% S28 13 81 6m20 CH2 wag 1118 1060 3.5 0.2 0.47 1069 1091 2.3 1059 1053 1052 1053 86% S20 2 55 43m21 CH2 wag 1113 1056 11.2 1.5 0.45 1051 1066 11.4 1039 1035 1034 1034 67% S21, 11% S20 32 5 63m22 CH2 twist 1072 1017 2.5 6.1 0.70 1027 1039 1.2 1017 1013 1013 1013 22% S22, 28% S15,
24% S29, 19% S21
36 3 61
m23 Ring deformation 960 911 25.9 7.0 0.64 912 907 19.6 887 890 888 889 87% S23 78 – 21m24 Ring deformation 939 892 1.7 9.9 0.75 897 896 14.9 883 883 882 – 73% S24 1 95 4m25 CH3 rock 905 859 29.7 8.0 0.75 871 875 39.1 872 874 875 875 68% S25, 17% S31 12 3 85m26 GeH2 deformation 897 851 29.0 14.4 0.74 854 862 23.6 841 843 862 861 71% S26, 12% S24, 12% S27 16 82 2m27 CH3 rock 884 840 130.4 4.6 0.70 838 847 139.7 837 833 848 846 53% S27, 20% S26, 17% S30 77 19 4m28 CH bend (out-of-plane) 865 821 33.1 3.7 0.75 819 832 20.9 – 818 818 819 29% S28, 34% S18,
17% S19, 11% S24
44 47 8
m29 CH2 rock 833 791 5.1 1.4 0.74 793 804 3.8 793 792 791 792 60% S29, 33% S22 50 5 45m30 GeH2 wag 725 689 60.7 7.6 0.74 697 706 62.4 705 707 708 706 64% S30, 19% S27 76 23 1m31 GeH2 twist 669 636 25.5 7.0 0.74 638 645 16.0 646 649 650 651 53% S31 45 9 45m32 CGeC antisymmetric stretch 641 608 24.6 12.7 0.22 581 585 28.3 598 597 597 599 54% S32, 42% S20 65 33 1m33 CGeC symmetric stretch 571 542 11.5 12.6 0.32 522 527 13.7 537 538 537 539 35% S33, 19% S31, 18% S32 74 5 21m34 GeH2 rock 445 423 17.9 1.4 0.37 431 436 17.8 446 446 447 450 78% S34 17 19 64m35 Ring-GeH2 bend
(out-of-plane)285 282 2.7 2.1 0.50 274 276 2.0 285 – 279 288 60% S35, 26% S38 3 94 3
m36 Ring-GeH2 bend (in-plane) 229 225 0.4 0.8 0.04 219 220 0.5 233 – 228 237 71% S36 35 1 64m37 CH3 torsion 143 143 0.0 0.1 0.60 133 129 0.0 – – 145 – 93% S37 – – 99m38 CGeC bend 124 123 0.0 0.6 0.73 122 123 0.1 – – 138 – 61% S38, 30% S35 3 82 15m39 Asymmetric torsion 68 68 0.1 0.2 0.66 58 58 0.1 – – – – 98% S39 79 1 20
a MP2(full)/6-31G(d) ab initio calculations, scaled frequencies, infrared intensities (km/mol), Raman activities (Å4/u), depolarization ratios (dp) and potential energy distributions (PEDs).b Scaled ab initio calculations with factors of 0.88 for CH stretches and deformations, 0.9 for all other modes except torsions and heavy atom bends using MP2/6-31G(d) basis set.c Symmetry coordinates with PED contribution less than 10% are omitted.
58J.R
.Durig
etal./Journal
ofM
olecularStructure
969(2010)
55–68
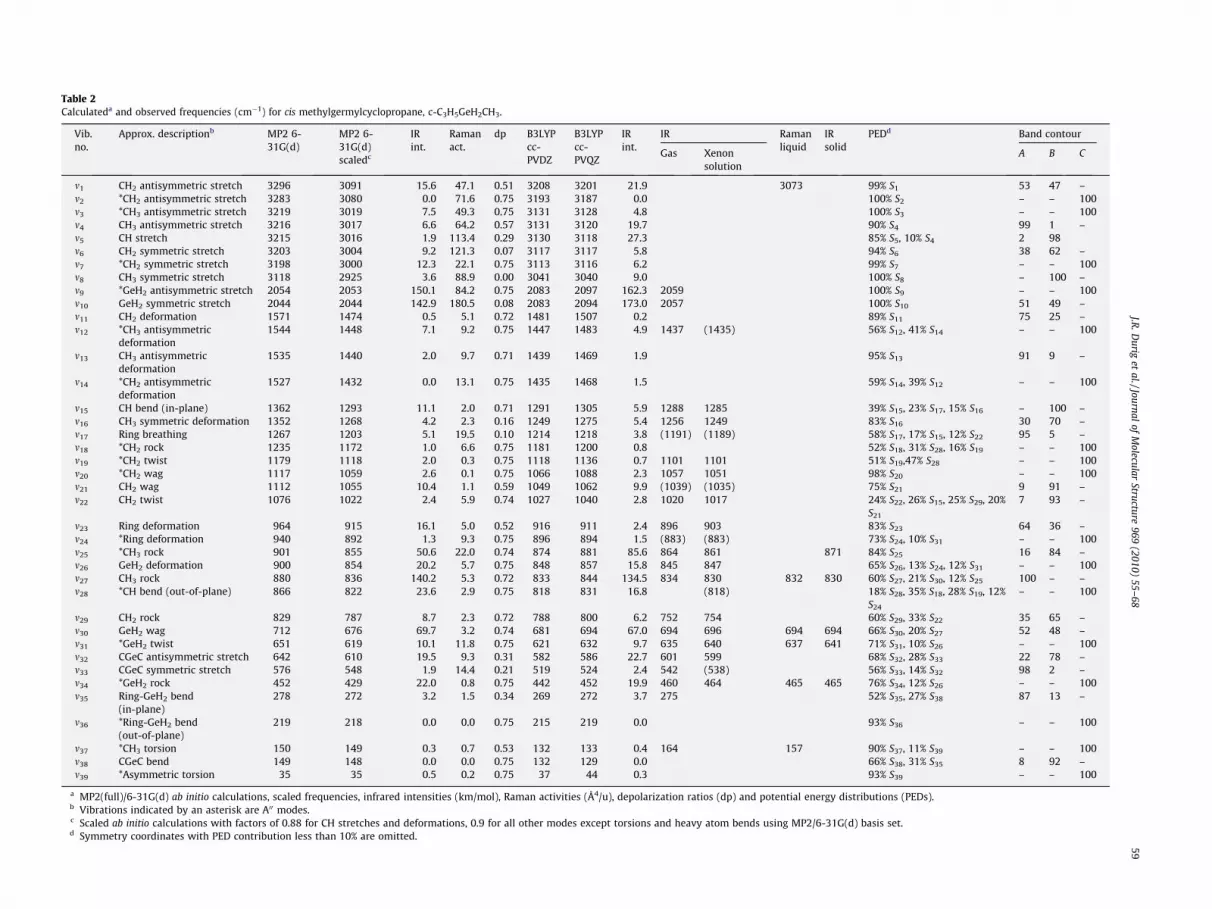
Table 2Calculateda and observed frequencies (cm�1) for cis methylgermylcyclopropane, c-C3H5GeH2CH3.
Vib.no.
Approx. descriptionb MP2 6-31G(d)
MP2 6-31G(d)scaledc
IRint.
Ramanact.
dp B3LYPcc-PVDZ
B3LYPcc-PVQZ
IRint.
IR Ramanliquid
IRsolid
PEDd Band contour
Gas Xenonsolution
A B C
m1 CH2 antisymmetric stretch 3296 3091 15.6 47.1 0.51 3208 3201 21.9 3073 99% S1 53 47 –m2 *CH2 antisymmetric stretch 3283 3080 0.0 71.6 0.75 3193 3187 0.0 100% S2 – – 100m3 *CH3 antisymmetric stretch 3219 3019 7.5 49.3 0.75 3131 3128 4.8 100% S3 – – 100m4 CH3 antisymmetric stretch 3216 3017 6.6 64.2 0.57 3131 3120 19.7 90% S4 99 1 –m5 CH stretch 3215 3016 1.9 113.4 0.29 3130 3118 27.3 85% S5, 10% S4 2 98m6 CH2 symmetric stretch 3203 3004 9.2 121.3 0.07 3117 3117 5.8 94% S6 38 62 –m7 *CH2 symmetric stretch 3198 3000 12.3 22.1 0.75 3113 3116 6.2 99% S7 – – 100m8 CH3 symmetric stretch 3118 2925 3.6 88.9 0.00 3041 3040 9.0 100% S8 – 100 –m9 *GeH2 antisymmetric stretch 2054 2053 150.1 84.2 0.75 2083 2097 162.3 2059 100% S9 – – 100m10 GeH2 symmetric stretch 2044 2044 142.9 180.5 0.08 2083 2094 173.0 2057 100% S10 51 49 –m11 CH2 deformation 1571 1474 0.5 5.1 0.72 1481 1507 0.2 89% S11 75 25 –m12 *CH3 antisymmetric
deformation1544 1448 7.1 9.2 0.75 1447 1483 4.9 1437 (1435) 56% S12, 41% S14 – – 100
m13 CH3 antisymmetricdeformation
1535 1440 2.0 9.7 0.71 1439 1469 1.9 95% S13 91 9 –
m14 *CH2 antisymmetricdeformation
1527 1432 0.0 13.1 0.75 1435 1468 1.5 59% S14, 39% S12 – – 100
m15 CH bend (in-plane) 1362 1293 11.1 2.0 0.71 1291 1305 5.9 1288 1285 39% S15, 23% S17, 15% S16 – 100 –m16 CH3 symmetric deformation 1352 1268 4.2 2.3 0.16 1249 1275 5.4 1256 1249 83% S16 30 70 –m17 Ring breathing 1267 1203 5.1 19.5 0.10 1214 1218 3.8 (1191) (1189) 58% S17, 17% S15, 12% S22 95 5 –m18 *CH2 rock 1235 1172 1.0 6.6 0.75 1181 1200 0.8 52% S18, 31% S28, 16% S19 – – 100m19 *CH2 twist 1179 1118 2.0 0.3 0.75 1118 1136 0.7 1101 1101 51% S19,47% S28 – – 100m20 *CH2 wag 1117 1059 2.6 0.1 0.75 1066 1088 2.3 1057 1051 98% S20 – – 100m21 CH2 wag 1112 1055 10.4 1.1 0.59 1049 1062 9.9 (1039) (1035) 75% S21 9 91 –m22 CH2 twist 1076 1022 2.4 5.9 0.74 1027 1040 2.8 1020 1017 24% S22, 26% S15, 25% S29, 20%
S21
7 93 –
m23 Ring deformation 964 915 16.1 5.0 0.52 916 911 2.4 896 903 83% S23 64 36 –m24 *Ring deformation 940 892 1.3 9.3 0.75 896 894 1.5 (883) (883) 73% S24, 10% S31 – – 100m25 *CH3 rock 901 855 50.6 22.0 0.74 874 881 85.6 864 861 871 84% S25 16 84 –m26 GeH2 deformation 900 854 20.2 5.7 0.75 848 857 15.8 845 847 65% S26, 13% S24, 12% S31 – – 100m27 CH3 rock 880 836 140.2 5.3 0.72 833 844 134.5 834 830 832 830 60% S27, 21% S30, 12% S25 100 – –m28 *CH bend (out-of-plane) 866 822 23.6 2.9 0.75 818 831 16.8 (818) 18% S28, 35% S18, 28% S19, 12%
S24
– – 100
m29 CH2 rock 829 787 8.7 2.3 0.72 788 800 6.2 752 754 60% S29, 33% S22 35 65 –m30 GeH2 wag 712 676 69.7 3.2 0.74 681 694 67.0 694 696 694 694 66% S30, 20% S27 52 48 –m31 *GeH2 twist 651 619 10.1 11.8 0.75 621 632 9.7 635 640 637 641 71% S31, 10% S26 – – 100m32 CGeC antisymmetric stretch 642 610 19.5 9.3 0.31 582 586 22.7 601 599 68% S32, 28% S33 22 78 –m33 CGeC symmetric stretch 576 548 1.9 14.4 0.21 519 524 2.4 542 (538) 56% S33, 14% S32 98 2 –m34 *GeH2 rock 452 429 22.0 0.8 0.75 442 452 19.9 460 464 465 465 76% S34, 12% S26 – – 100m35 Ring-GeH2 bend
(in-plane)278 272 3.2 1.5 0.34 269 272 3.7 275 52% S35, 27% S38 87 13 –
m36 *Ring-GeH2 bend(out-of-plane)
219 218 0.0 0.0 0.75 215 219 0.0 93% S36 – – 100
m37 *CH3 torsion 150 149 0.3 0.7 0.53 132 133 0.4 164 157 90% S37, 11% S39 – – 100m38 CGeC bend 149 148 0.0 0.0 0.75 132 129 0.0 66% S38, 31% S35 8 92 –m39 *Asymmetric torsion 35 35 0.5 0.2 0.75 37 44 0.3 93% S39 – – 100
a MP2(full)/6-31G(d) ab initio calculations, scaled frequencies, infrared intensities (km/mol), Raman activities (Å4/u), depolarization ratios (dp) and potential energy distributions (PEDs).b Vibrations indicated by an asterisk are A00 modes.c Scaled ab initio calculations with factors of 0.88 for CH stretches and deformations, 0.9 for all other modes except torsions and heavy atom bends using MP2/6-31G(d) basis set.d Symmetry coordinates with PED contribution less than 10% are omitted.
J.R.D
uriget
al./Journalof
Molecular
Structure969
(2010)55–
6859

Table 3Calculated electronic energies (hartree) and energy differences (cm�1) for meth-ylgermylcyclopropane, c-C3H5GeH2CH3.
Basis set MP2(full) B3LYP
gauchea cisb gauchea cisb
6-31G(d) 0.166930 248 2.403013 2846-31+G(d) 0.204648 210 2.435976 3206-311G(d,p) 2.796431 54 4.470413 1666-311+G(d,p) 2.800072 36 4.471643 1656-311G(2d,2p) 2.865537 �6 4.476717 1526-311+G(2d,2p) 2.868201 �4 4.477795 1586-311+G(2df,2pd) 2.961535 13 4.484828 1636-311++G(2df,2pd) 2.962060 14 4.484913 1636-311++G(3df,3pd) 3.003872 �9 4.487703 164aug-cc-pVTZ 2.804331 2 4.565841 165
Averagec 13 ± 22 cm�1 162 ± 5 cm�1
a Energy of the gauche conformer is given as �(E + 2231) H.b Energy of the cis conformer is relative to the gauche form.c Average value excluding the two smallest basis sets.
60 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
triple monochromator with (2) additive or (3) subtractive collec-tion. A higher signal to noise ratio was achieved with the singlemonochromator, whereas the triple additive collection gaveslightly higher resolution, and the triple subtractive set-up allowedthe spectra to be recorded at wavenumbers closer to the excitingline. However, the different systems of light collection gave quitesimilar spectra. With the liquid sample the low wavenumbermodes were recorded to 60 cm�1 with the triple subtractive modecompared to 120 cm�1 with the single monochromator and thenotch filter.
The high sensitivity of the CCD detector gave spectra with largesignal/noise ratios, and since narrow slits could be employed, thespectra were recorded at ca. 1 cm�1 resolution. Most spectra wererecorded in the range 3500–100 cm�1 within 5 min. Polarizationmeasurements were carried out in the 90� illumination mode byemploying a polarizer and a scrambler between the sample andthe monochromator. Except for very weak and/or overlappingbands, fairly comprehensive depolarization data were obtained.
Raman spectra of the liquid, including depolarization measure-ments, were primarily recorded at room temperature. Additionalspectra were obtained at 30 temperatures between 283 and143 K in a capillary tube of 2 mm inner diameter. The tube wassurrounded by a Dewar, cooled by gaseous nitrogen evaporatedfrom a reservoir [6]. It was observed that most of the Raman bandswere enhanced after cooling, although some bands more than oth-ers. This effect is undoubtedly a result of the very low barrier forcis–gauche conversion.
The vapor of methylgermylcyclopropane was condensed on acopper finger at 78 K. An amorphous phase was formed, and inspite of numerous attempts at different annealing temperatures,the compound did not crystallize. Thus, the Raman bands of theamorphous solid were similar to those of the liquid. All of the ob-served bands in both the Raman spectra of the liquid and infraredspectra of the gas and solid along with their proposed assignmentsare listed in Tables 1 and 2.
3. Computational methods
The LCAO-MO-SCF restricted Hartree–Fock calculations wereperformed with the Gaussian-03 program [7] using Gaussian-typebasis functions. The energy minima with respect to nuclear coordi-nates were obtained by the simultaneous relaxation of all geomet-ric parameters using the gradient method of Pulay [8]. Severalbasis sets as well as the corresponding ones with diffuse functionswere employed with the Møller–Plesset perturbation method [9]to the second order (MP2(full)) along with the density functionaltheory by the B3LYP method. The predicted conformational energydifferences are listed in Table 3.
In order to obtain a description of the molecular motions in-volved in the fundamental modes of c-C3H5GeH2CH3, a normalcoordinate analysis has been carried out. The force field in Carte-sian coordinates was obtained with the Gaussian-03 program [7]at the MP2(full) level with the 6-31G(d) basis set. The internalcoordinates used to calculate the G and B matrices are given inTable 4 with the atomic numbering shown in Fig. 3. By using theB matrix [10], the force field in Cartesian coordinates was con-verted to a force field in internal coordinates. Subsequently, scalingfactors of 0.88 for CH stretches and CH deformations, and 0.9 forother coordinates except for the heavy atom bends and torsionswere applied, along with the geometric average of the scaling fac-tors for the interaction force constants, to obtain the fixed scaledforce field and resultant wavenumbers. A set of symmetry coordi-nates was used (Table 5) to determine the corresponding potentialenergy distributions (PEDs). A comparison between the observedand calculated wavenumbers, along with the calculated infraredintensities, Raman activities, depolarization ratios and potential
energy distributions for the gauche and the cis conformers arelisted in Tables 1 and 2, respectively.
The vibrational spectra were predicted from the MP2(full)/6-31G(d) calculations. The predicted scaled frequencies were usedtogether with a Lorentzian function to obtain the simulated spec-tra. Infrared intensities were obtained based on the dipole momentderivatives with respect to Cartesian coordinates. The derivativeswere transformed with respect to normal coordinates by olu/oQi) =
Pj(olu/oXj)Lij, where Qi is the ith normal coordinate, Xj is
the jth Cartesian displacement coordinate, and Lij is the transfor-mation matrix between the Cartesian displacement coordinatesand the normal coordinates. The infrared intensities were thencalculated by [(NP)/(3c2)][(olx/oQi)2 + (oly/oQi)2 + (olz/oQi)2]. Acomparison of experimental and simulated infrared spectra ofc-C3H5GeH2CH3 is shown in Fig. 1. Infrared spectra of the gas andthe predicted infrared spectra for the pure gauche and cis conform-ers, as well as the mixture of the two conformers with relativeconcentrations calculated for the equilibrium mixture at 25 �C byusing the experimentally determined enthalpy difference areshown in Fig. 1A–D, respectively. The predicted spectrum is ingood agreement with the experimental spectrum which showsthe utility of the scaled predicted frequencies and predicted inten-sities for supporting the vibrational assignment.
Additional support for the vibrational assignments wasobtained from the simulated Raman spectra. The evaluation ofRaman activity by using the analytical gradient methods has beendeveloped [11–14] and the activity Sj can be expressed as:Sj ¼ gjð45a2
j þ 7b2j Þ, where gj is the degeneracy of the vibrational
mode j, aj is the derivative of the isotropic polarizability, and bj isthe derivative of the anisotropic polarizability. To obtain theRaman scattering cross sections, the polarizabilities are incorpo-rated into Sj by multiplying Sj with (1 � qj)/(1 + qj) where qj isthe depolarization ratio of the jth normal mode. The Raman scat-tering cross sections and calculated wavenumbers obtained fromthe Gaussian-03 program were used together with a Lorentzianfunction to obtain the simulated Raman spectra. Comparison ofexperimental Raman spectra of the liquid and the predicted Ramanspectra for the pure gauche and cis conformers as well as the mix-ture of the two conformers with relative concentrations calculatedfor the equilibrium mixture at 25 �C by using the experimentallydetermined enthalpy difference (43 cm�1) are shown in Fig. 2A–D. The spectrum of the mixture should be compared to that ofthe Raman spectrum of the liquid at room temperature. The pre-dicted spectrum is in reasonable agreement with the experimentalspectrum which indicates the utility of the predicted Raman spec-tra for supporting vibrational assignments.

Table 4Structural parameters (Å and �), rotational constants (MHz) and dipole moments (Debye) for gauche and cis rotamers of methylgermylcyclopropane, c-C3H5GeH2CH3.
Structural parameters Internal coordinates MP2/6-311+G(d,p) B3LYP/6-311+G(d,p) Adjusted r0 gauche Predicted cis
gauche cis gauche cis
r(Ge1C2) R1 1.934 1.937 1.955 1.958 1.925(5) 1.928r(C2C4) R2 1.517 1.517 1.516 1.516 1.517(3) 1.517r(C2C5) R3 1.519 1.517 1.517 1.516 1.519(3) 1.517r(C4C5) R4 1.504 1.504 1.504 1.505 1.502(3) 1.502r(Ge1C6) R5 1.950 1.951 1.971 1.971 1.947(5) 1.948r(C2H3) r1 1.087 1.087 1.086 1.086 1.087(2) 1.087r(C4H7) r2 1.085 1.085 1.085 1.085 1.085(2) 1.085r(C4H8) r3 1.084 1.084 1.084 1.084 1.084(2) 1.084r(C5H9) r4 1.085 1.085 1.085 1.085 1.085(2) 1.085r(C5H10) r5 1.084 1.084 1.084 1.084 1.084(2) 1.084r(Ge1H11) r6 1.529 1.527 1.545 1.544 1.533(2) 1.531r(Ge1H12) r7 1.527 1.527 1.544 1.544 1.531(2) 1.531r(C6H13) r8 1.092 1.093 1.092 1.092 1.092(2) 1.093r(C6H14) r9 1.092 1.092 1.092 1.092 1.092(2) 1.092r(C6H15) r10 1.093 1.092 1.092 1.092 1.093(2) 1.092\Ge1C2H3 c 116.4 115.6 114.4 113.3 116.4(5) 115.6\C2C4C5 A1 60.4 60.3 60.3 60.2 60.4(5) 60.3\C2C5C4 A2 60.2 60.2 60.2 60.2 60.3(5) 60.3\C4C2C5 A3 59.4 59.5 59.5 59.5 59.3(5) 59.4\Ge1C2C4 b1 120.0 120.4 121.1 122.1 120.1(5) 120.5\Ge1C2C5 b2 119.1 120.4 120.9 122.1 119.2(5) 120.5\C2Ge1C6 x 111.1 109.5 111.6 111.4 110.6(5) 109.0\H7C4H8 u1 114.8 114.7 113.9 113.8 114.8(5) 114.7\H9C5H10 u2 114.8 114.7 113.9 113.8 114.8(5) 114.7\C2Ge1H11 l1 106.4 109.9 107.4 109.3 106.4(5) 109.9\C2Ge1H12 l2 110.7 109.9 110.1 109.3 110.7(5) 109.9\Ge1C6H13 h1 110.8 109.8 111.4 109.6 110.8(5) 109.8\Ge1C6H14 h2 110.0 110.9 110.3 111.0 110.0(5) 110.9\Ge1C6H15 h3 110.3 110.9 110.3 111.0 110.3(5) 110.9\H13C6H14 d3 108.7 108.2 108.7 108.3 108.7(5) 108.2\H14C6H15 d1 108.6 108.8 108.6 108.8 108.6(5) 108.8\H13C6H15 d2 108.5 108.2 108.5 108.3 108.5(5) 108.2sC6Ge1C2C4 s2 �152.9 �35.1 �154.5 �35.8 �151.8(5) �35.1sH13C6Ge1C2 s1 177.3 180.0 178.2 180.0 177.3(5) 180.0A 7251 5668 7232 5673 7208 5698B 1911 2136 1857 2053 1926 2147C 1673 1828 1629 1768 1681 1835|la| 0.201 0.530 0.215 0.528|lb| 0.597 0.366 0.634 0.438|lc| 0.322 0.000 0.310 0.000|lt| 0.707 0.644 0.738 0.686
J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68 61
4. Vibrational assignment
To obtain the enthalpy difference between the two stable con-formers it is very important to make confident assignments foras many of the fundamentals for both conformers as possible.There was considerable reliance on the predicted frequencies offundamentals from the ab initio calculations which was supportedby the predicted infrared intensities and band contours as well asthe Raman activities and depolarization values. Additionally, therehave been many studies of mono-substituted cyclopropanes whichresulted in a significant number of group frequencies for the three-membered ring moiety as well as the normal vibrations for themethyl group. Furthermore, the infrared spectrum of the xenonsolution made it possible for several of the fundamentals to be as-signed especially for the cis conformer. Thus, the main emphasiswill be on the assignments for the germyl and ring modes.
The assignments for the eight CH stretches were consistentlyfound in the usual places for the methyl and the CH modes forthe ring. The antisymmetric and symmetric GeH2 stretching modesare assigned for both the gauche and cis conformers as shown in Ta-bles 1 and 2. For the gauche conformer the predicted difference forthese modes is 19 cm�1 whereas the observed difference is11 cm�1. The CH3 and CH2 deformations are observed in the usualregions. The highest frequency ring fundamental is the breathingmode which is expected to be one of the strongest Raman lines
and can be confidently assigned at 1190 cm�1. Its intensity isgrossly under predicted by the ab initio calculations. The othertwo ring modes are predicted with similar frequencies and bothare significantly purer (87% and 73%) than the ring breathing modewhich is only 56% S17 with 16% S15 (CH bend) and 14% S22 (CH2
twist). Only one of these ring modes, m23, could be assigned forthe cis conformer which is a relatively pure (83% S23) mode.
The four GeH2 bending modes were readily assigned at 841,705, 646 and 446 cm�1 with the deformation and rock being rela-tively pure modes whereas the GeH2 twist is only 53% S31 but theremaining contributions are all less than 10%. The two GeCstretches are assigned at 598 and 537 cm�1 with both of themextensively mixed. The CGeC bend is expected to be a very weakband even in the Raman spectrum (0.7 Å4/u) and it is observed at138 cm�1 for the gauche conformer but not for the cis form.
The remaining vibrations to be assigned for the gauche con-former are carbon–hydrogen bends with several of them exten-sively mixed such as m22 (CH2 twist) which has similarcontributions from three other symmetry coordinates, i.e. CH bendip, CH2 wag, and CH2 rock. The m28 (CH bend op) mode also has sig-nificant contributions from four symmetry coordinates but theseare the two worst scenarios with most of the carbon–hydrogenbends having significant contributions from three symmetry coor-dinates with the major one having 50% or more from the approxi-mate described mode.

Fig. 3. cis conformer of methylgermylcyclopropane showing atom numbering and internal coordinates.
62 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
With confident assignments for the fundamentals of the gaucheconformer it was possible to assign many fundamentals for the cisconformer, particularly those arising from the GeH2 modes whichare relatively strong and should be most sensitive to the differentconformers. Of particular help was the comparison of the infraredspectrum of the solution with the spectrum of the gas (Fig. 4). Forseveral of these fundamentals the spectrum of the gas will have ashoulder or an unsymmetrical band shape whereas the band willbe relatively sharp in the low-temperature liquid so it can be read-ily identified. With these data it was possible to assign 11 funda-mentals of the cis conformer in the ‘‘fingerprint” region. Thefrequencies for these fundamentals are very similar to those ofthe corresponding modes of the gauche conformer. Nevertheless,with the significant number of fundamentals identified for the lessstable conformer it was possible to obtain enthalpy values for sev-eral pairs of bands which leads to small uncertainties in the deter-mined values.
5. Conformational stability
To determine the enthalpy difference between the two con-formers of c-C3H5GeH2CH3 the sample was dissolved in liquefied
xenon and the mid-infrared spectra were recorded as a functionof temperature from �55 to �100 �C. Only small interactions areexpected to occur between xenon and the sample and, conse-quently, only small frequency shifts are anticipated when passingfrom the gas to the liquefied noble gas solutions. A significantadvantage of this study is that the conformer bands are better re-solved in comparison with those in the infrared spectrum of thegas (Fig. 4A). From ab initio calculations, the dipole moments ofthe two conformers are predicted to have similar values (Table 4)and the molecular sizes of the two rotamers are nearly the same,so the DH value obtained from the temperature dependent FT-IRstudy is expected to be near to the value for the gas [15–19].
To obtain the best results, one should utilize the low frequencyfundamentals to minimize potential interaction from overtone orcombination bands which could arise from the conformer in nearcoincidence with the fundamental of the other conformer whichis being used for the determination of the enthalpy difference.There are three pairs of bands at 707/696, 649/640, and 446/464 cm�1 which are the GeH2 wags, GeH2 twists and GeH2 rocks,respectively, which meet the criteria that they are not expectedto have adverse interference from the overtone or combinationbands. These fundamentals are expected to be some of the most

Table 5Symmetry coordinates for methylgermylcyclopropane, c-C3H5GeH2CH3.
Description Symmetry coordinatea
CH2 antisymmetric stretch S1 = r2 � r3 + r4 � r5
CH2 antisymmetric stretch S2 = r2 � r3 � r4 + r5
CH3 antisymmetric stretch S3 = 2r10 � r9 � r8
CH3 antisymmetric stretch S4 = r9 � r8
CH stretch S5 = r1
CH2 symmetric stretch S6 = r2 + r3 + r4 + r5
CH2 symmetric stretch S7 = r2 + r3 � r4 � r5
CH3 symmetric stretch S8 = r8 + r9 + r10
GeH2 antisymmetric stretch S9 = r6 � r7
GeH2 symmetric stretch S10 = r6 + r7
CH2 deformation S11 = 4u1 � r1 � g1 � p1 � e1 +4u2 � r2 � g2 � p2 � e2
CH3 antisymmetric deformation S12 = 2d3 � d2 � d1
CH3 antisymmetric deformation S13 = d2 � d1
CH2 deformation S14 = 4u1 � r1 + g1 + p1 � e1
� 4u2 � r2 + g2 + p2 � e2
CH wag (in-plane) S15 = 2c � a2 � a1
CH3 symmetric deformation S16 = d1 + d2 + d3 � h1 � h2 � h3
Ring breathing S17 = R2 + R3 + R4
CH2 rock S18 = r2 � p2 + g2 � e2 � r1 + p1 � g1 + e1
CH2 twist S19 = r2 � p2 � g2 + e2 � r1 + p1 + g1 � e1
CH2 wag S20 = r2 � p2 + g2 � e2 + r1 � p1 + g1 � e1
CH2 wag S21 = r2 + p2 � g2 � e2 + r1 + p1 � g1 � e1
CH2 twist S22 = r2 + p2 + g2 + e2 � r1 � p1 � g1 � e1
Ring deformation S23 = 2R3 � R2 � R4
Ring deformation S24 = R2 � R4
CH3 rock S25 = h2 � h3
GeH2 deformation S26 = /CH3 rock S27 = 2h1 � h2 � h3
CH bend (out-of-plane) S28 = a2 � a1
CH2 rock S29 = r2 + p2 � g2 � e2 � r1 � p1 + g1 + e1
GeH2 wag S30 = l1 + l2 � q1 � q2
GeH2 twist S31 = l1 � l2 � q1 + q2
CGeC antisymmetric stretch S32 = R1 � R5
CGeC symmetric stretch S33 = R1 + R5
GeH2 rock S34 = l1 � l2 + q1 � q2
Ring-GeH2 bend (out-of-plane) S35 = b1 � b2
Ring-GeH2 bend (in-plane) S36 = b1 + b2
CH3 torsion S37 = s1
CGeC bend S38 = xAsymmetric torsion S39 = s2
a Not normalized.
J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68 63
sensitive to the conformational interchange along with the CGeCstretches and ring-GeH2 bends. Unfortunately, the two CGeC anti-symmetric stretches are predicted to be only separated by 2 cm�1
and they appear as a single band at 598 cm�1 in the infrared spec-trum of the xenon solution. Although the GeH2 twists are predictedto be separated by 17 cm�1 they are only separated by 9 cm�1
which makes their intensity measurements a little difficult. Never-theless, we used this pair which made it possible to have nineexperimentally determined enthalpy differences. Finally, we alsoattempted to use the 538 cm�1 band as the intensity of the gaucheconformer while knowing it is due to the CGeC symmetricstretches for both conformers. However, the predicted intensityof this mode for the cis form is only one-sixth of that for the gaucheconformer so the enthalpy difference obtained for the 538 cm�1
band may be a little lower (37 ± 10 cm�1, 0.44 ± 0.12 kJ mol�1)than the values obtained from the other pairs but it could possiblysupport the values obtained from the other pairs.
The intensities of the infrared bands were measured as a func-tion of temperature (Fig. 5) and their ratios were determined. Byapplication of the van’t Hoff equation �ln K = DH/(RT) � DS/R, theenthalpy difference was determined from a plot of �ln K versus1/T, where DH/R is the slope of the line and K is substituted withthe appropriate intensity ratios, i.e. Igauche/Icis. It was assumed thatDH is not a function of temperature in the temperature range stud-ied. By combining the intensities from three of the bands for thegauche conformers with the intensities of three bands for the cis
form nine pairs of bands (Fig. 4) were obtained for the determina-tion of the enthalpy difference. The band pair at 649/696 cm�1 hadan uncertainty larger than the determined value so it was not usedfor the determination of the enthalpy difference. The values ob-tained for each of the eight pairs are listed in Table 6 with eachof the individual uncertainties which, for the most part, are rela-tively small although the range of the values of the enthalpy differ-ence is rather large. Nevertheless, by using all of the data as asingle set, the determined enthalpy difference is 43 ± 11 cm�1
(0.51 ± 0.13 kJ mol�1) where the uncertainty is the statistical stan-dard deviation of one sigma, with the gauche conformer the morestable form. These error limits do not take into account possibleassociations with the liquid xenon which could have different ef-fects on different fundamentals. Also, there is possible interferenceof the combination band of m26 + m37 of the gauche conformer withthe m30 fundamental of the cis conformer or 2m36 with the gaucheform with m34 of the cis form. There is also possible interferenceof the overtone (three possibilities) and combination bands (twopossibilities) of the cis form being in near coincidence with them34 fundamental of the gauche form. The variations are undoubt-edly due in part to these types of interferences as well as the mea-surements of the band intensities. As further support, the relativeintensities of several pairs of conformer bands at a single temper-ature were measured and the predicted ab initio intensities werethen used to obtain the enthalpy differences. The arithmetic aver-age of these values was consistent with the experimentally deter-mined value. The abundance of the less stable cis form present atambient temperature is 30 ± 3%.
We also attempted to obtain the enthalpy difference in the li-quid by using the Raman spectra where a pair of bands at 228and 279 cm�1 were well separated and had relative intensity vari-ations with temperature. The intensities of these two bands weremeasured as a function of temperature first over eight differentvalues from 13 to �130 �C and then over 16 different values inthe same temperature range. Again, by the application of the van’tHoff equation an enthalpy difference, for both of the series, was ob-tained as 52 cm�1 (0.62 kJ mol�1) and 40 cm�1 (0.48 kJ mol�1).These values are in good agreement with the value obtained fromthe xenon solution by utilizing the eight conformer pairs(43 ± 11 cm�1, 0.51 ± 0.13 kJ mol�1). Also, the interferences fromovertones or combination bands is limited to two possibilities sothe uncertainties for this determination in the liquid are mainlydue to the difficulty in obtaining reproducible intensities fromthe Raman lines. Nevertheless, it appears that any interaction inthe liquid which affects the enthalpy difference is small. This deter-mination is similar to the use of the 538 cm�1 transition for the xe-non solution where the 538 cm�1 line was known to include somecontribution from the cis conformer. The average value obtainedutilizing this transition is 37 ± 10 cm�1 (0.44 ± 0.12 kJ mol�1)which is slightly smaller than the value obtained for the liquidfrom the Raman data.
A comparison with the theoretical results shows that the MP2calculations with all but the two smallest basis sets with and with-out diffuse functions give predictions that the enthalpy differenceis near zero, which is consistent with the small experimentallydetermined conformational enthalpy difference. However, the pre-dicted value from the B3LYP density functional theory calculationsof 162 ± 5 cm�1 (1.94 ± 0.06 kJ mol�1) is entirely too large. Itshould be noted that the DFT calculations have little variation withbasis set size.
6. Structural parameters
We have found that good structural parameters for hydrocar-bons and many substituted ones can be determined by adjusting
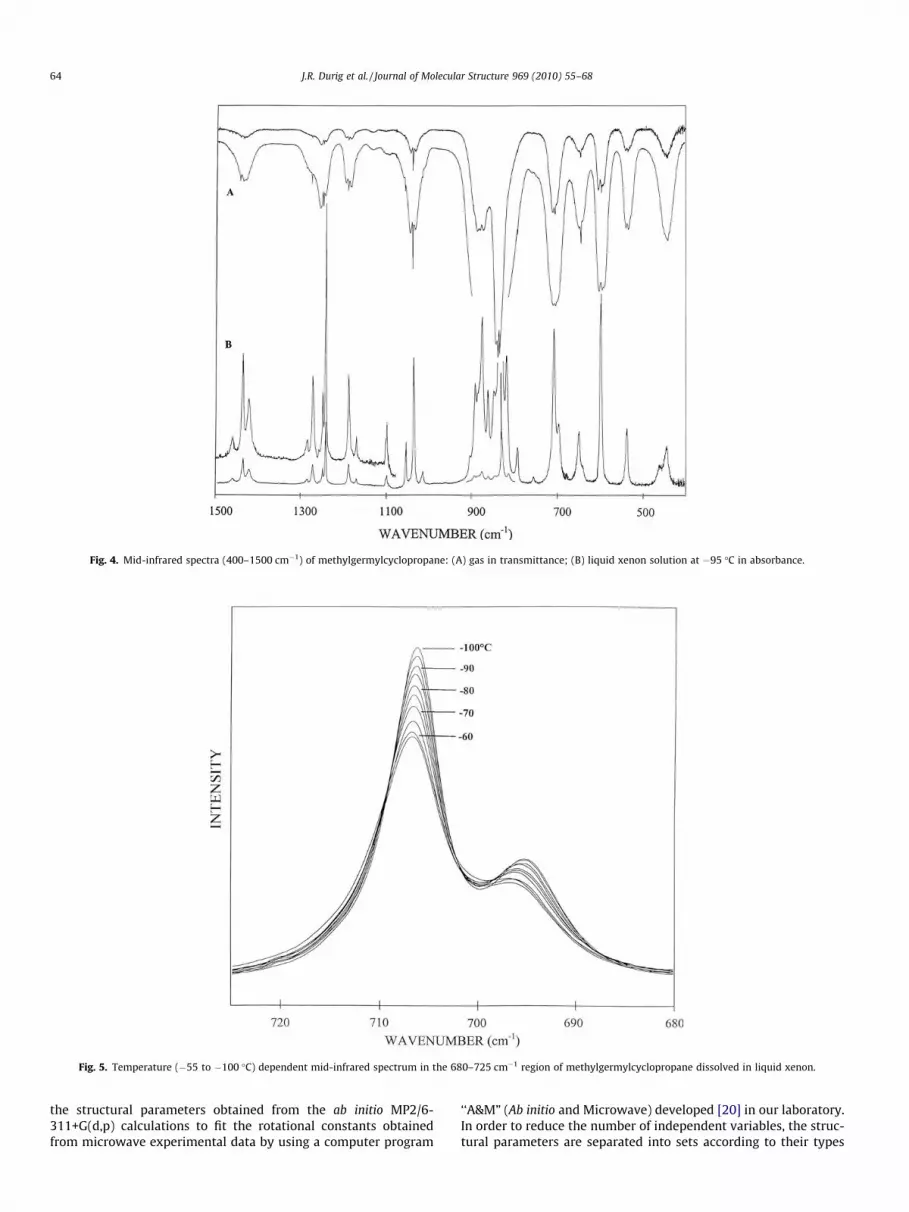
Fig. 4. Mid-infrared spectra (400–1500 cm�1) of methylgermylcyclopropane: (A) gas in transmittance; (B) liquid xenon solution at �95 �C in absorbance.
Fig. 5. Temperature (�55 to �100 �C) dependent mid-infrared spectrum in the 680–725 cm�1 region of methylgermylcyclopropane dissolved in liquid xenon.
64 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
the structural parameters obtained from the ab initio MP2/6-311+G(d,p) calculations to fit the rotational constants obtainedfrom microwave experimental data by using a computer program
‘‘A&M” (Ab initio and Microwave) developed [20] in our laboratory.In order to reduce the number of independent variables, the struc-tural parameters are separated into sets according to their types

Table 6Temperature and intensity ratios of the conformational bands of methylgermylcyclopropane, c-C3H5GeH2CH3 from the infrared spectra of the liquid xenon solution phase.
T (�C) 1/T (�103 K�1) I707/I696 I649/I640 I446/I464 I707/I640 I707/I464 I649/I464 I446 /I696 I446 / I640 I538 / I696 I538 / I464
�55.0 4.584 2.8421 4.7000 – 5.4000 – – 0.9649 – 1.2988 –�60.0 4.692 2.8500 4.5455 – 5.1818 – – 0.8714 4.6654 – –�65.0 4.804 2.8571 4.3333 – 5.0000 8.4233 2.6981 0.8788 – 1.3025 –�70.0 4.923 2.9091 4.2308 2.2336 4.9231 9.0493 2.9276 0.8568 4.9562 1.2757 3.3257�75.0 5.047 2.9130 4.3846 2.2214 5.1538 9.0549 2.9702 0.8888 4.5064 – 3.4600�80.0 5.177 2.9167 4.2143 – 5.0000 9.1279 2.9646 0.8750 4.4255 – 3.4730�85.0 5.315 2.9200 4.0667 2.1517 4.8667 9.4780 3.0713 0.7690 4.2583 1.2836 3.5918�90.0 4.460 3.0400 3.9375 – 4.7500 – – 0.7848 4.1728 1.2637 –�95.0 5.613 3.0000 4.0625 2.1968 4.8750 9.6399 3.1764 0.7894 4.1515 – 3.5948�100.0 5.775 2.9630 3.9412 2.0980 4.7059 – 2.9919 0.7749 4.2129 1.2509 –
DHa (cm�1) 33 ± 7 95 ± 14 40 ± 17 63 ± 13 101 ± 23 71 ± 30 113 ± 23 101 ± 26 22 ± 5 74 ± 22
a Average value: DH = 43 ± 11 cm�1 (0.51 ± 0.13 kJ mol�1) with the gauche conformer the more stable form and the statistical uncertainty (r) obtained by utilizing all of thedata as a single set.
J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68 65
where bond distances in the same set keep their relative ratio, andbond angles and torsional angles in the same set keep their differ-ence in degrees. This assumption is based on the fact that errorsfrom ab initio calculations are systematic. We [21] have also shownthat ab initio MP2/6-311+G(d,p) calculations predict the r0 struc-tural parameters for more than fifty carbon–hydrogen distancesto better than 0.002 Å compared to the experimentally determinedvalues from isolated CH stretching frequencies which were com-pared [22] to previously determined values from earlier micro-wave studies. Therefore, all of the carbon–hydrogen distancescan be taken from the MP2/6-311+G(d,p) predicted values formethylgermylcyclopropane.
It has been shown that GeH bond distances can be obtainedfrom the frequencies of the isolated GeH stretching frequencies[23]. Therefore, we have obtained values of 1.533 and 1.531 Å forthe r6 and r7 distances (Table 4) which are 0.004 Å longer than val-ues for the corresponding distances from the ab initio MP2/6-311+G(d,p) predicted parameters. This longer distance is similarto the difference found for some other GeH distances in otherorganogermanes [24,25]. Thus, with the GeH and CH distancesdetermined within 0.002 Å and the corresponding bond angles toan expected uncertainty of 0.5�, there are only five heavy atom dis-tances and six heavy atom angles to be determined. However,three of the heavy atom angles are CCC angles which can be treatedas a single set and similarly the two GeCC angles also treated as asingle set. Also, two of the CC distances (R2 and R3) can be treatedas a single set which leaves at most seven parameters to bedetermined.
The microwave studies provided rotational constant values forthe five isotopomers 70Ge, 72Ge, 73Ge, 74Ge and 76Ge for the gaucheconformer with the spectral data taken at �10 K from a jet cooledsample. From these data 15 rotational constants for the gaucheconformer are available to be used to determine the seven r0 struc-tural parameters of this conformer. The resulting adjusted param-eters obtained for the gauche form are listed in Table 4, where it isbelieved that the C–C distances should be accurate to ±0.003 Å, theGe–C distances to 0.005 Å, the C–H and Ge–H distances should beaccurate to ±0.002 Å, and the angles should be within ±0.5�. Thepredicted structural parameters for the cis conformer were ob-tained by making the same adjustments to the correspondingparameters to those made for the gauche conformer.
The fit of 15 determined rotational constants (Table 7) by thestructural parameters for the gauche conformer is remarkably goodwith differences of 1.1, 0.9, 0.5, 0.4, 0.2, six of 0.1, and the remain-ing four by 0.0 MHz. Therefore, it is believed that the suggesteduncertainties are realistic values and the determined structuralparameters are probably as accurate as can be obtained for theirvalues for the molecule in the gas phase.
The estimated parameters for the cis conformer are also ex-pected to be quite good since the experimental rotational con-stants for the gauche conformer differed by small values of +43,�15 and �8 MHz for the A, B, and C rotational constants for the70Ge isotopomer with similar differences for the other isotopo-mers. Therefore, the rotational constants predicted from the smalladjustments made to the parameters obtained from the MP2/6-311+G(d,p) calculations are expected to be within a few MHz fromthe experimental ones obtained from the cis conformer whichshould make it relatively easy to assign the microwave spectrumfor this conformer.
7. Barriers to internal and asymmetric rotations
With the assignment of the methyl torsional fundamental forthe gauche conformer from the Raman spectrum of the liquid itis possible to obtain the threefold barrier to internal rotation. Uti-lizing the determined r0 structural parameters listed in Table 4 andthe 145 cm�1 frequency for the CH3 torsional mode, the periodicthreefold barrier (V3) to internal rotation of the CH3 group can becalculated. The equation: V3 = (9/4)Fs is used, where F (cm�1) is re-lated to the reduced moment of inertia of the CH3 top by h/8p2cIr.The dimensionless parameter of the Mathieu equation, s is indi-rectly determined from the observed frequency. The F numberhas a value of 5.47 cm�1 and with the fundamental frequency of145 cm�1 a barrier of 492 cm�1 (5.89 kJ mol�1) is obtained. Three-fold barrier for the methyl rotor has been predicted from the ab ini-tio calculations by using the MP2(full)/6-31G(d) basis set. Thevalue for the methyl barrier obtained from the transition statefrom the rotation of the CH3 moiety is 429 cm�1 (5.13 kJ mol�1).This rather small difference could be due to, in part, to a slightlylarger value for the liquid from the small association in the con-densed phase. For several methyl rotors attached to a carbon atomwe have found that the MP2(full)/6-31G(d) predicted barriers areusually in good agreement with the experimentally determinedvalues [26–30].
The barriers and potential function governing the asymmetricrotor motion, i.e. rotation of the �GeH2CH3 group relative to thethree-membered ring have been predicted from the MP2(full)/6-31G(d) and MP2(full)/6-311+G(d,p) calculations where the energydifferences were predicted to be 242 cm�1 and 36 cm�1, respec-tively, between the gauche and cis conformers. Utilizing the smallerbasis set the gauche to gauche barrier is predicted to be 699 cm�1
and the gauche to cis barrier to be 682 cm�1 whereas from the lar-ger basis set the values of 503 cm�1 and 474 cm�1 (Fig. 6), respec-tively, were obtained. However, the cis to gauche barrier ispredicted to be the same at 440 cm�1 from both basis sets. The dif-ference of �200 cm�1 obtained for the gauche to gauche and the

Table 7Comparison of rotational constants (MHz) obtained from modified ab initio MP2(full)/6-311+G(d,p) predictions, experimental values from microwave spectra, and the adjusted r0
structural parameters for methylgermylcyclopropane, c-C3H5GeH2CH3.
Isotopomer Rotational constant MP2(full)/6-311+G(d,p) Experimental Adjusted r0 |D|
c-C3H570GeH2CH3 A 7561.7 7260.1 7261.0 0.9
B 1928.5 1938.4 1938.4 0.1C 1704.1 1692.9 1693.0 0.1
c-C3H572GeH2CH3 A 7534.7 7233.5 7233.9 0.4
B 1922.8 1932.3 1932.3 0.0C 1698.3 1686.9 1687.0 0.1
c-C3H573GeH2CH3 A 7521.6 7220.6 7220.8 0.2
B 1920.0 1929.3 1929.3 0.0C 1695.5 1684.0 1684.0 0.0
c-C3H574GeH2CH3 A 7508.9 7209.1 7208.0 1.1
B 1917.2 1926.5 1926.4 0.1C 1692.8 1681.1 1681.1 0.0
c-C3H576GeH2CH3 A 7484.2 7183.8 7183.3 0.5
B 1911.8 1920.8 1920.7 0.1C 1687.4 1675.6 1675.5 0.1
66 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
gauche to cis barrier is attributed to the energy difference betweenthe two conformers.
It is possible to obtain values for four of the potential constantsof the function governing the internal rotation (asymmetric rota-tion) of the �GeH2CH3 moiety which has the form:
VðhÞ ¼X4
i¼1
ðVi=2Þð1� cos ihÞ
The series coefficients of the first four Vi terms in the above equa-tion, were determined by a non-linear least-squares fitting of thepredicted energy differences and the torsional dihedral angles fromthe MP2(full)/6-31G(d) calculation for the gauche (121.7�) and cis(0.0�) conformers and the two transition states (62.7� and 180.0�).The potential is nearly a threefold rotation (barrier � 680 cm�1)with the following values of the coefficients: V1 = 145, V2 = �161,V3 = �603, V4 = �16 cm�1. The torsional dihedral angles taken fromthe MP2(full)/6-311+G(d,p) level of calculation for the gauche(121.3�) and cis (0.0�) and the transition states (61.0�, 61.8� and180.0�) gave a smaller threefold barrier of �475 cm�1. The corre-sponding coefficients of the cosine function were: V1 = 4, V2 = �48,V3 = �471, V4 = 5 cm�1. These predicted values are expected to bereasonably near the experimental ones that would be obtained fromthe frequencies of the asymmetric torsional modes from the twoconformers. The resulting potential functions governing conforma-tional interchange with comparison of the values obtained fromthe two basis sets are shown in Fig. 6.
8. Discussion
The determined enthalpy difference of 43 ± 11 cm�1 is consis-tent with the value of 98 ± 13 cm�1 obtained for the DH for the cor-responding silicon molecule where the ab initio MP2(full)predicted a value of 65 ± 8 cm�1 from eight different basis sets.A similar calculation with the same basis sets from density func-tional theory calculations by the B3LYP method gave a value of207 ± 6 cm�1. This difference of about 150 cm�1 higher by densityfunctional theory calculations is similar to what has been observedfor the germanium compound where the predicted value of162 ± 5 cm�1 is about 150 cm�1 higher than the MP2 predicted va-lue of 13 ± 22 cm�1. This larger predicted value from density func-tional theory calculation for the enthalpy difference has beenfound for most of the conformational determinations made whenthe enthalpy difference is relatively small.
The predicted frequencies for the fundamental vibrations withtwo scaling factors compare very favorably with the observed val-ues with an average error of 9 cm�1 which represents a percentage
error of 0.7% for the gauche conformer. This error cannot be ob-tained for the cis conformer since many of the vibrations of thisconformer seem to have nearly the same frequency as the corre-sponding vibrations for the gauche form; thus, only limited numberof fundamentals for this conformer was clearly identified. Never-theless, there is strong evidence from the few normal modes thatwere assigned for the cis form that the error is probably similarto that found for the gauche form for the predicted frequencies.
The predicted frequencies have also been obtained from densityfunctional theory calculations by the B3LYP method and those forthe gauche conformer are also listed in Table 1 for comparison tothose predicted from the MP2 calculations. Predictions were ob-tained with three different basis sets of cc-PVDZ, cc-PVTZ and cc-PVQZ. Very little difference was found for the predictions fromthe cc-PVTZ and cc-PVQZ calculations but the difference was usu-ally 0, 1 or 2 cm�1 with an average difference of 1.3 cm�1. How-ever, there is a significant average difference of 11 cm�1 betweenthe predicted frequencies from the cc-PVDZ calculations and thosefrom the cc-PVTZ calculations but those obtained from the cc-PVDZcalculations are in much better agreement with the observed fre-quencies than those with the larger basis sets (Table 1). Thus, ifthe interest is in making a vibrational assignment of the observedbands the smaller basis set is satisfactory, and the predictions fromthe MP2/6-31G(d) calculations with two scaling factors is evenbetter than those from the predictions from the B3LYP calculationswith the coupled clusters for most substituted hydrocarbons ex-cept for those with carbon–carbon triple bonds. For example, theaverage frequency error for the current molecule studied is42 cm�1 if one includes the predictions for the frequencies of thecarbon–hydrogen stretches, and if they are excluded then the aver-age error is 22 cm�1 which is more than twice the value from theMP2 calculations with the relatively small basis set. There are sixmodes which have significant contributions from three symmetrycoordinates but only two of them, the m15 and m33 modes, have lessthan 52% from the symmetry coordinate listed as the approximatedescription and they have 42% (CH bend ip) and 35% (CGeC sym-metric stretch), respectively.
The mixing of the vibrations is indicated by the potential energydistributions and practically all of those have major contributionsfrom the approximate description given for the vibration. In fact,there are only two vibrations that have significant contributionsfrom four different symmetry coordinates for the gauche con-former. Most of the vibrations have more than 40% of the symme-try coordinate indicated as that vibrational mode. However, m22
which is described as a CH2 twist, has almost equal contributionsfrom four different symmetry coordinates and a similar problemis also found for m28. Nevertheless, for the most part, there are fairly

Fig. 6. The predicted potential function governing the internal rotation of the CH3GeH2-moiety from the gauche to the cis form for methylgermylcyclopropane. The solid lineis from MP2/6-31G(d) calculation; dotted line is from MP2/6-311+G(d,p) calculation.
J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68 67
reasonable potential energy distributions obtained for the individ-ually observed fundamentals as described.
The depolarization values were obtained from ab initio calcula-tions for both the gauche and the cis conformers. The depolariza-tion values from the MP2 basis set calculations and the densityfunctional theory calculations are essentially the same and, ingeneral, the predicted values are in agreement with the experi-mentally determined ones (Fig. 7). In fact, out of the 39 depolariza-tion values predicted for the fundamentals only three of them wereinconsistent with the experimental values. The CH2 wags (m20 andm21) and the ring-GeH2 out-of-plane bend (m35) were predicted tobe significantly polarized (�0.50) but the values from the experi-mental measurements showed these vibrations to be depolarized.This small number of incorrect predictions can be consideredrelatively minor when assessing the reliability of the ab initiopredicted depolarization values which can be very useful whenmaking vibrational assignments.
Fig. 7. Raman spectra (100–3200 cm�1) for the determination of depolarization valuereduced by �25%.
The determined structural parameters are believed to have verylow uncertainties since there are essentially two sets of heavyatom distances along with five heavy atom angles which shouldbe readily determined from the 15 rotational constants. The uncer-tainties of 0.003 Å for the C–C distances and 0.005 Å for the Ge–Cdistances along with 0.002 Å for the C–H and Ge–H parametersare believed to be good estimates for the uncertainties. Uncertain-ties in the angles are a little larger at 0.5� but it is still believed thatthese adjusted r0 parameters are probably as accurate as could bedetermined experimentally for the parameters in the vapor state.
The difference in the Ge–Cr distance to the ring and the Ge–Cm
distance to the methyl moiety is quite close to the difference in thecorresponding silicon analogue for the gauche conformer. Formethylgermylcyclopropane the two distances have a differenceof 0.022 Å whereas for methylsilylcyclopropane the differencewas determined to be 0.019 Å. When comparing the structuralparameters by using the MP2/6-311+G(d,p) basis set the Ge–C as
s for methylgermylcyclopropane. The asterisks indicate that the band intensity is

68 J.R. Durig et al. / Journal of Molecular Structure 969 (2010) 55–68
well as Si–C distances were each 0.016 Å apart whereas in ethylcy-clopropane the corresponding C–C distances had a difference of0.019 Å. The differences obtained from the adjusted r0 structuralparameters are consistent with those obtained from the theoreticalcalculations. We are also able to compare the Ge–Cr and the Ge–Cm
distances with vinylgermane [31] and alkylgermanes [32–34],respectively. In the former molecule the Ge–C adjusted r0 distancewas determined to be 1.930 Å. The closeness of this distance to theGe–Cr distance (1.925 Å) in methylgermylcyclopropane is attrib-uted to the fact that a three-membered ring has an affect that issimilar to a double bond. There have been two previous studies[32,33] carried out on methylgermane with experimentally deter-mined Ge–C distances of 1.945(5) and 1.949(1) Å as well as a studyon ethylgermane [34] also with experimentally determined Ge–Cdistances of 1.949(10) Å. These values are in good agreement withthe Ge–Cm distance (1.947 Å) in our molecule. The adjusted r0
structural parameters have also been calculated [24] for ethylger-mane, although the Ge–Cm distance obtained in this manner isslightly higher at 1.959(5) Å.
The barrier to internal rotation of the methyl group has beendetermined to be 492 cm�1 (5.89 kJ mol�1) from a single vibra-tional frequency of 145 cm�1 in the liquid phase. This barrier willnot be as accurate as that obtained from the splitting of the ob-served microwave transitions (395 cm�1, 4.73 kJ mol�1). The valueof the splitting is directly determined by the potential barrierheight so the barrier value should be much more accurately deter-mined from the microwave data than from the fundamental vibra-tional frequency. The fundamental frequency gives informationnear the bottom of the well and the barrier is then projected fromthis information which may be somewhat different than the actualbarrier. To determine how the presence of a three-membered ringaffected the rotation of a methyl group, the internal rotational bar-rier in methylgermane [32] was considered for comparison pur-poses. An internal barrier of 433 ± 9 cm�1 (5.18 ± 0.1 kJ mol�1)was determined from the transitional splittings. Another micro-wave study [35] on methylgermane gave a much higher barrierof 585 cm�1 (7.00 kJ mol�1) but this was obtained from relativeintensity measurements of a microwave line for the excited stateof the methyl torsion which had a vibrational frequency of195 cm�1. This method is considered to be much less accurate thanthe splitting of the observed microwave transitions or direct obser-vation of the torsional fundamental.
Acknowledgement
J.R.D. acknowledges the University of Missouri-Kansas City for aFaculty Research Grant for partial financial support of thisresearch.
References
[1] W. Caminati, R. Danieli, M. Dakkouri, R. Bitschenauer, J. Phys. Chem. 99 (1995)1867.
[2] N. Inamoto, S. Masuda, Chem. Lett. (1982) 1003.[3] J.R. Durig, Z. Yu, C. Zheng, G.A. Guirgis, J. Phys. Chem. A 108 (2004) 5353.[4] J.R. Durig, S.S. Panikar, G.A. Guirgis, T.K. Gounev, R.M. Ward, R.A. Peebles, S.A.
Peebles, R.J. Liberatore, S. Bell, C.J. Wurrey, J. Mol. Struct. 923 (2009) 1.[5] C.J. Wurrey, S. Shen, T.K. Gounev, J.R. Durig, J. Mol. Struct. 406 (1997) 207.[6] F.A. Miller, B.M. Harney, Appl. Spectrosc. 24 (1970) 291.[7] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson,H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T.Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian,J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O.Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K.Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S.Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J.Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L.Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M.Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A.Pople, Gaussian 03, Revision D.01, Gaussian, Inc., Wallingford, CT, 2004.
[8] P. Pulay, Mol. Phys. 17 (1969) 197.[9] C. Møller, M.S. Plesset, Phys. Rev. 46 (1934) 618.
[10] G.A. Guirgis, X. Zhu, Z. Yu, J.R. Durig, J. Phys. Chem. A 104 (2004) 4383.[11] M.J. Frisch, Y. Yamaguchi, J.F. Gaw, H.F. Schaefer III, J.S. Binkley, J. Chem. Phys.
84 (1986) 531.[12] R.D. Amos, Chem. Phys. Lett. 124 (1986) 376.[13] P.L. Polavarapu, J. Phys. Chem. 94 (1990) 8106.[14] G.W. Chantry, in: A. Anderson (Ed.), The Raman Effect, vol. 1, Marcel Dekker
Inc., New York, NY, 1971 (Chapter 2).[15] M.O. Bulanin, J. Mol. Struct. 19 (1973) 59.[16] B.J. van der Veken, F.R. DeMunck, J. Chem. Phys. 97 (1992) 3060.[17] W.A. Herrebout, B.J. van der Veken, A. Wang, J.R. Durig, J. Phys. Chem. 97
(1995) 578.[18] M.O. Bulanin, J. Mol. Struct. 347 (1995) 73.[19] W.A. Herrebout, B.J. van der Veken, J. Phys. Chem. 100 (1996) 9671.[20] B.J. van der Veken, W.A. Herrebout, D.T. Durig, W. Zhao, J.R. Durig, J. Phys.
Chem. A 103 (1999) 1976.[21] J.R. Durig, K.W. Ng, C. Zheng, S. Shen, Struct. Chem. 15 (2004) 149.[22] D.C. McKean, J. Mol. Struct. 113 (1984) 251.[23] D.C. McKean, M.W. Mackenzie, A.R. Morrison, J. Mol. Struct. 116 (1984) 331.[24] J.R. Durig, C. Pan, G.A. Guirgis, J. Mol. Struct. 607 (2002) 117.[25] J.R. Durig, C. Pan, W. Witkowski, G.A. Guirgis, Can. J. Chem. 82 (2004) 964.[26] J.R. Durig, S. Bell, G.A. Guirgis, Spectrochim. Acta A 52 (1996) 1843.[27] S. Bell, G.A. Guirgis, Y. Li, J.R. Durig, J. Phys. Chem. A 101 (1997) 5987.[28] J.R. Durig, G.A. Guirgis, S. Bell, W.E. Brewer, J. Phys. Chem. A 101 (1997) 9240.[29] S. Bell, G.A. Guirgis, S.W. Hur, J.R. Durig, Spectrochim. Acta A 55 (1999) 2361.[30] S. Bell, B.R. Drew, G.A. Guirgis, J.R. Durig, J. Mol. Struct. 553 (2000) 199.[31] J.R. Durig, K.L. Kizer, Y.S. Li, J. Am. Chem. Soc. 96 (1974) 7400.[32] V.W. Laurie, J. Chem. Phys. 30 (1959) 1210.[33] D.C. McKean, M.W. Mackenzie, A.R. Morrisson, J. Mol. Struct. 116 (1984) 331.[34] J.R. Durig, A.D. Lopata, P. Groner, J. Chem. Phys. 66 (1977) 1888.[35] A.I. Barchukov, A.M. Prokhorov, Opt. Spektrosk. 4 (1958) 799.





























