Embed Size (px)
Citation preview

25
.I. Electraanal. Chem., 344 (1993) 25-44 Elsevier Sequoia S.A., Lausanne
JEC 02276
Computer simulation of large-scale controlled-potential electrolysis. Father-son and grandfather-grandson self-protonation systems
Matthew L. Vincent and Dennis G. Peters l
Department of Chemistry, Indiana University, Bloomington, IN 47405 (USA)
(Received 26 July 1991; in revised form 6 April 1992)
In some electrochemical systems, transfer of an electron from the electrode to the starting material is followed by a reaction in which the electrogenerated intermediate (radical anion) abstracts a proton from unreduced starting material. Products of this self-protonation or father-son proton transfer are a neutral radical, thought to be more easily reduced than the starting material, and the conjugate base of the parent. Linear scan voltammetry fails to discriminate between two mechanistic scenarios, one where transfer of a second electron to the neutral radical occurs at the electrode surface (EC&,) and the other where the second electron is transferred to the neutral radical in solution by the radical anion (DISPl). Reduction of the neutral radical results in an anion that is thought to be more basic than the radical anion; thus this anion should be more reactive toward unreduced starting material than the radical anion. We have utilized computer simulations of controlled-potential electrolyses to investigate systematically the competition between (i) unreduced starting material and the neutral radical for the radical anion in father-son proton-transfer systems, (ii) father-son and grandfather-grandson self-pro- tonation reactions, and (iii) protonation of the son and grandson species by the medium. Under typical experimental conditions for controlled-potential electrolyses, the maximum yield of two-electron reduction product formed via the heterogeneous electron transfer is 64%. In addition, we have found that, when the second-order rate constant for the father-son proton-transfer reaction is less than 2.5 x lo8 M-’ s-l, more than 95% of the two-electron reduction product is formed via the solution electron-transfer reaction. For a mechanistic scheme where father-son and grandfather-grandson proton-transfer reactions both play a role, product distributions vary, as a function of three parameters, from quantitative formation of the two-electron reduction product to a 1:2 mixture of the two-electron reduction product and the conjugate base of the starting material.
l To whom correspondence should be addressed.
0022-0728/93/$06.00 0 1993 - Elsevier Sequoia S.A. All rights reserved

26
INTRODUCTION
Over the last 20 years, several groups of researchers have studied systems where voltammetric and coulometric results indicate that a self-protonation (father-son or grandfather-grandson) reaction is an important step in the mechanism of reduction for the compounds of interest. Electrochemical reduction of phthalimide in nonaqueous solvents has been examined in several laboratories [l-3]. In each investigation, it was observed that the radical anion generated from phthalimide is basic enough to abstract a proton from unreduced starting material to form a neutral radical and the conjugate base of the parent; further reduction of the neutral radical, which is easier to reduce than the starting material, yields an anion that is also protonated by unreduced starting material. These phenomena lead to coulometric and polarographic it values approximately 67% of those determined for N-methylphthalimide. In a separate study, Farnia et al. [4] discovered that, in dimethylformamide containing tetraethylammonium perchlorate, isatin undergoes reduction by the same mechanism found for phthalimide.
Elving and colleagues established that, in dimethyl sulfoxide containing tetra- alkylammonium salts, 2-hydroxypyrimidine [5], uracil [61, thymine [7], uridine [8] and 4-aminopyrimidine [93 all show voltammetric and coulometric evidence that unreduced starting material acts as an acid toward the radical anion derived from the parent molecule. These workers determined that the routes for reduction of these materials are drastically affected by the availability of protons in the medium [lo]. Roffia et al. [ll] employed polarography, cyclic voltammetry and controlled- potential coulometry to demonstrate that the radical anions derived from both 4-nitroimidazole and 2-methyl-5nitroimidazole abstract a proton from unreduced starting material. Other studies have been performed on substrates containing more easily recognized acidic functionalities [12-H]. Farnia and coworkers [12,13] investigated the electrochemistry of some nitrophenols, and Brillas et al. [14] probed the electrochemical behavior of p-nitrobenzoic acid. For each of these systems, the radical anion derived from the starting material plays a key role in the nonelectrochemical consumption of that starting material.
Janata et al. [16] published the first report that a hydrocarbon can act as a proton source for electrogenerated intermediates. These investigators utilized polarography and electron spin resonance spectrometry to show that the electro- generated radical anion derived from 4,5_methylenephenanthrene can very slowly attack unreduced starting material. In a series of papers dealing with the reduction of activated carbon-carbon double bonds, it was found that properly substituted indenes can undergo self-protonation with their electrogenerated intermediates both directly 117-191 and indirectly [20]. Evidence for self-protonation reactions has also been detected for electrochemically induced isomerizations of various acetylenes and allenes [21-251.
Self-protonation reactions are not restricted to the attack of a radical anion on starting material. Vianello and coworkers [26-311 have studied the electrochem- istry of several compounds where the species responsible for nonelectrochemical

21
consumption of starting material results from a rapid cleavage reaction interposed between a pair of heterogeneous electron-transfer steps. These investigators have developed [27] a theoretical analysis of these types of systems where in certain time domains the initial heterogeneous charge-transfer step can become rate limiting; they have applied their findings to the electrochemical reduction of diphenylmeth- ylphenylsulfide as well as several different 2-bromocarboxamides in dimethylform- amide containing tetra-n-butylammonium perchlorate. These self-protonations have been called grandfather-grandson reactions because it is the intermediate gener- ated from a second electron-transfer step that acts as a base toward unreduced starting material.
Amatore et al. [32] have dealt with the theoretical aspects of systems where a father-son proton-transfer reaction is involved in the mechanism of reduction. Their examination of this process involved the use of both linear scan and cyclic voltammetry to extract mechanistic information, particularly the rate constant for the proton-transfer reaction and the nature of the second electron-transfer step (DISP or ECE). Measurements of shifts in peak potentials and changes in peak widths with sweep rate and initial substrate concentration can be used to discrimi- nate between ECE,,, and DISPl on the one hand, and ECE,, and DISP2 on the other hand; however, although it is possible to distinguish the ECE,,, process from the DISP2 process, this approach does not allow for immediate discrimination between the ECE,,, and DISPl processes. These authors reinvestigated earlier work pertaining to the reductions of phthalimide, 2-nitrophenol, 3-nitrophenol, 4-nitrophenol, 4,5methylenephenanthrene and 1,3-diphenyl-2-methylindene; they employed the theory for linear scan and cyclic voltammetry to verify the mecha- nisms by which these materials are reduced and to determine the rate constant for the father-son proton-transfer reaction in each case.
In those situations for Which ECEirr and DISPl processes are indistinguishable, we show in the present paper, through a systematic investigation of product distributions in large-scale controlled-potential electrolysis for a hypothetical fa- ther-son proton-transfer scheme, that the mechanism cannot be purely ECE,,, even when the father-son reaction is diffusion controlled. As the magnitude of the rate constant for the father-son reaction becomes less than the diffusion limit, the mechanism tends rapidly towards DISPl. Moreover, we show how a systematic investigation of product distributions from large-scale controlled-potential electrol- yses can be used to determine the critical parameters that characterize a general- ized self-protonation scheme. Careful attention is paid to the competition between self-protonation reactions and protonation of electrogenerated intermediates by adventitious proton sources in the medium.
EXPERIMENTAL
Principles for the kinetic analysis have already been described by Amatore, Saveant and coworkers [33-401. All homogeneous reactions are assumed to be rapid and all unstable intermediates are confined to a thin layer (reaction layer) of

28
thickness M immediately adjacent to the electrode surface. In the remainder of the diffusion layer, the concentration profiles for all stable reactants and products are linear; outside the diffusion layer, the concentrations of all stable reactants and products are solutions to differential equations that may or may not depend on time. An implicit finite-difference scheme similar to that of Amatore and Saveant was used to solve the simultaneous differential equations which yield the desired concentration profiles in the reaction layer. Algorithms were written in the C programming language and compiled with the Microsoft C optimizing compiler (version 5.1) on a PC’s Ltd. 286 computer operating at a clock rate of 8 MHz. To transform real space and time into their dimensionless counterparts, we introduce the following parameters: s = t(D,Y/W) (time) where D is the diffusion coefficient for the reactant, S is the area of the electrode, V is the volume of the solution and I is the thickness of the diffusion layer, y =x/l (distance), and h = kl*/D and h = kc“l*/D for first- and second-order rate constants respectively. In cases where the distribution of products is dependent on time, a fixed-step-size Runge-Kutta algorithm [41] has been modified and used to integrate the resulting system of time-dependent differential equations.
THEORY
Competition between the neutral radical and unreduced starting material for the electrogenerated radical anion
Consider the reduction mechanism (Scheme 1)
A+e-+B (1)
A+B 2 C+D (2)
Cfe-+EE (3)
B+C 2 A+ED (4)
where the starting material A accepts an electron at the electrode surface to form a radical anion B. In this case the radical anion is basic enough to attack unreduced starting material (or the starting material is acidic enough to protonate the radical anion) to produce a neutral radical C and the conjugate base D. Under these conditions the neutral radical is almost always more easily reduced than the starting material, and so we must consider that the second electron may be transferred to the neutral radical (i) at the electrode surface or (ii) in solution by the radical anion to give the monoanion E. In the majority of instances, species E will be more basic than the radical anion (a case that will be treated later in this paper) and thus should act as a base toward unreduced starting material. However, to investigate the competition between unreduced starting material and the neutral radical for the radical anion in this father-son proton-transfer scheme, we assume here that E is not reactive toward the starting material and we distinguish between

29
E formed at the electrode surface (EE) and E formed via the homogeneous redox process (ED), though they are actually the same species.
We can write the quasi-time-independent diffusion equations (in dimensionless form) as outlined below. In the reaction layer (0 < y < m (m = M/l))
d*a/dy* = h,,ab - h,bc (9
d*b/dy* = h,,ab + h,bc (6)
d*c/dy* = -h,,ab + h,bc (7)
d*d/dy* = -h,,ab (8)
d2eE/dy2 = 0 (9)
d2eD/dy2 = -h,bc (10)
At y=O
(da/dy), + (db/dy), = 0 (11)
(dc/dy), + (deE/dy), = 0 (12)
(dd/dy), = 0 (13)
In the remainder of the diffusion layer (m < y < 1)
d*a/dy* = 0 (14)
d*b/dy* = 0 (15)
d*c/dy* = 0 (16)
d*d/dy* = 0 (17)
d2eE/dy2 = 0 (18)
d2eD/dy2 = 0 (19)
At the boundary between the diffusion layer and the bulk of the solution (y = 1)
(da/dy),_=ab-a,=ab-a, (20)
(db/dy),_ = (dc/dy),_+ 0 (21)
b-+0 (22)
In the bulk of solution
dab/ds = -(da/dy),_ (23)
ddb/ds = -(dd/dy),_ (24)
deETb/ds = - (deE/dy),_ (25)
deDYb/ds = - (deD/dy),_ (26)
At s=O, ab=l and b=c=d=eE=eD=O.

30
For our treatment we shall consider only constant-concentration potentiostatic electrolysis (CCPE) and exhaustive potentiostatic electrolysis (EPE). In each of these techniques the electrode potential is poised at a value on the diffusion-con- trolled portion of the voltammetric wave in question which allows us to set the concentration of depolarizer essentially to zero at the electrode surface (a, = 0). Then
(da/dy),_=ab (27) where ab remains constant for CCPE conditions (ab = 1) and varies with time for EPE conditions (ab = exp( -s)).
We must derive expressions for yields of stable products in terms of consump- tion of starting material. Linear combination of differential equations (5)-(10) gives
d2(a+b+c+d+eE+eD)/dy2=0 (28) Integration over the reaction-layer thickness and then over the remainder of the diffusion layer leads to
(da/dy),_+(dd/dy),_+(deE/dy),_+(deD/dy),_=O (29)
which is a relationship that describes conservation of mass in the system. Now we must relate one or more of the stable products to consumption of starting material. Linear combination of eqns. (5), (6) and (8) gives
d*( a -t b + 2d)/dy2 = 0 (30) Integration of this equation over the thickness of the diffusion layer and considera- tion of the surface boundary conditions (eqn. (11)) give
(da/dy),_ + 2(dd/dy),_ = 0 (31) Combination of eqns. (24), (27) and (31) allows us to relate the appearance of d to the disappearance of a through the following equation:
ddb/ds = +(da/dy),_= +a” (32)
Substitution of eqns. (24)-(27) into eqn. (29) gives
ddb/ds + deE,b/ds + deD,b/ds = ab (33)
Integration of eqns. (32) and (33) gives the following equations:
db = +/‘a” dr (34) 0
db + eE,b + eD,b = ab /
‘dr
0 (35)
where ub may be a function of s depending on the electrolysis technique employed and r is a variable of integration. We can obtain the yield of D if equation (31) is divided by equation (32):
yD = db/( db + eE,b + eDTb) = l/2 (36)

31
Thus, no matter what the electrolysis conditions (EPE or CCPE), the yield of species D is always 50%. However, integration of eqns. (9) and (18), as well as consideration of the surface boundary conditions, gives
(deE/dy),_ = (deE/dy), = - (dc/dy), (37)
Substitution of eqn. (25) into eqn. (37) gives
deEvb/ds = (dc/dy), (38)
Integration of eqn. (38) and combination with eqn. (35) give the expression for the yield of E formed at the electrode surface:
YE E = $,“/( & + $b + $‘.b)
(39)
YED= 1 -YD-YEE (41) We shall now calculate the yields of EE and ED in the context of each type of electrolysis experiment (CCPE and EPE).
Constant-concentration potentiostatic electrolysis (CCPE) We make the following transformations of variables: a* = ahtp/3, b* = bhir,
c* =chtp/” and y * = yhif. A set of three simultaneous differential equations must be solved to determine the slope of the concentration profile for species c for various values of the competition rate parameter w (w = hd/h,,, = k,/k,,). These equations are
d*a*/dy** = a*b* - wb*c*
d*b*/dy ** = a*b* + wb*c*
d*c*/dy ** = -_a*b* + wb*c*
with the following conditions at each boundary. At y* = 0
(da*/dy*), + (db*/dy*),, = 0
a* -0 0- co* = 0
At y*=a
(da*/dy*), = 1
(db*/dy*), + 0 (dc*/dy*), + 0
b*+O
(42)
(43)
(44)
(45)
(46)
(47)
(48)
(49)
32
0.5 -- /__-----
0.4 --
2 0.3--
W F 0.2 --
0.1 --
O.O__~_~_~-~~~
-t I -3.0 -2.0 -1 .o 0.0 1.0 2.0 3.0
LOG(w)
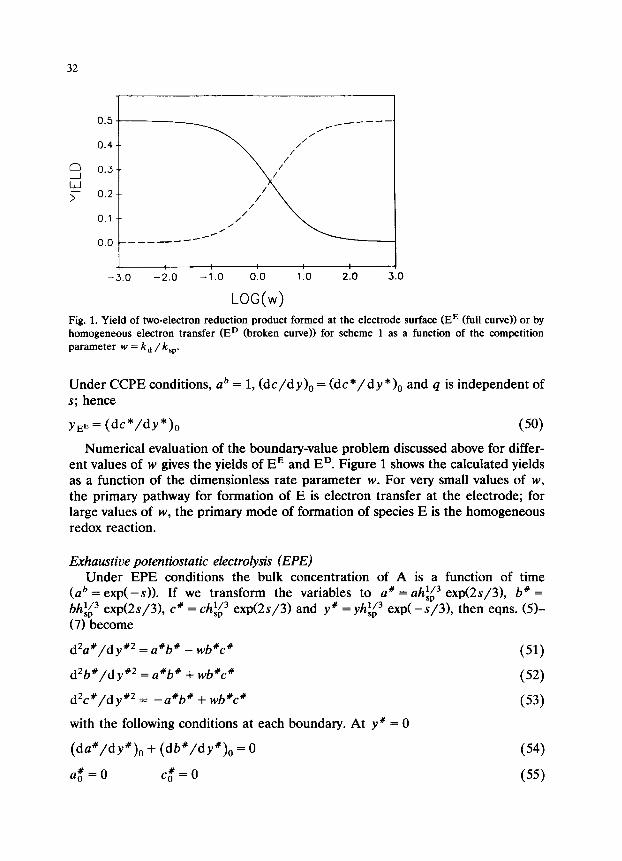
Fig. 1. Yield of two-electron reduction product formed at the electrode surface (EE (full curve)) or by homogeneous electron transfer (ED (broken curve)) for scheme 1 as a function of the competition parameter w = k, /k,,.
Under CCPE conditions, ab = 1, (dc/dy), = (dc*/dy *I,, and 4 is independent of S; hence
yEE = (dc*/dy*),, (50)
Numerical evaluation of the boundary-value problem discussed above for differ- ent values of w gives the yields of EE and ED. Figure 1 shows the calculated yields as a function of the dimensionless rate parameter w. For very small values of w, the primary pathway for formation of E is electron transfer at the electrode; for large values of w, the primary mode of formation of species E is the homogeneous redox reaction.
Exhaustive potentiostatic electrolysis (EPE) Under EPE conditions the bulk concentration of A is a function of time
(ab = exp(-s)). If we transform the variables to an = ah:,/3 exp(2s/3), bX = bhi6 exp(2s/3), cx = ch$” exp(2s/3) and yx = yhip/” exp( -s/3), then eqns. W- (7) become
d*a”/d y #* = a#b# _ wb#c#
d2bX/d y #* = a#b# + wb#c#
d*c#/d y #* = -a’b# + wb#c#
with the following conditions at each boundary. At y” = 0
(da#/dy#), + (db#/dy#), = 0
a: = 0 co” = 0
(51)
(52)
(53)
(54)
(55)

33
At y#==
(da#/dy+‘), = 1 (56)
(db#/dy#), + 0 (dc#/dy#), + 0 (57)
bX + 0 (58)
Under EPE conditions, (dc/dy), = (dc#/dy#), exp(--s). Since the competition parameter derived for this case is the same as that derived for CCPE (w = h&z,,), the slope of the concentration profile for c# is not dependent on s. Thus the yield of EE can be calculated as follows:
/( ’ dc”/dy#) 0 exp( -r) dr
YE 0
E=
/
s (59)
exp( -r) dr 0
YE
(dc’/dy#)oi exp( -r) dr IS=
/
s (60)
exp( -r) dr
yes = (dc#,d;‘), (61)
Thus the yield of E as a function of the competition parameter w is the same for the EPE and CCPE cases (Fig. 1).
Competition between father-son and grandfather-grandson self-protonation reac- tions, and protonation of the electrogenerated intermediates by adventitious proton sources in the medium
Suppose that the parent species is reduced according to the following mecha- nism (scheme 2):
A+e-+B (62)
A+B -% C+D (63) k,
B-+C (64) C+e-+E (65)
B+C 2 A+E (66)
A+E 3 D+F (67) k,
E--,F (68) The starting material A accepts an electron at the electrode surface to form a radical anion B. In this situation, species B is consumed in three ways: 6) B is basic

34
enough to attack unreduced starting material (or the starting material is acidic enough to protonate the radical anion) to produce a neutral radical C and the conjugate base of the parent D; (ii) B is protonated by the solvent-supporting electrolyte via a pseudo-first-order process to yield the same neutral radical; (iii) under the extant conditions the neutral radical is almost always more easily reduced than the starting material, so that the second electron may be transferred in solution by the radical anion as depicted in reaction (66). Since the product of the second electron-transfer reaction is a monoanion E that is generally thought to be more basic than the radical anion, this monoanion should also be able to deprotonate unreduced starting material. Reactions (67) and (68) represent com- petition between protonation of the monoanion by unreduced starting material and by adventitious proton sources in the medium.
In describing scheme 2 we can write the following quasi-time-independent diffusion equations. For 0 < y < m (m = M/I)
d2a/dy2 = h,ab + h,ae - h,bc (69)
d2b/dy2 = h,ab + h,b + h,bc (70)
d2c/dy2 = -h,ab - h,b + h,bc (71)
d2d/dy2 = -h,ab - h,ae (72)
d2e/dy2 = -h,bc + h,ae + h,e (73)
d 2 f/dy2 = -h,ae - h,e (74)
At y=O
(da/dy), + (db/dy), = 0 (75)
(dc/dy)a + (de/dy), = 0 (76)
(dd/dy), = 0 (77)
(df/dyh = 0 (78) In the remainder of the diffusion layer (m < y < 1)
d2a/dy2 = 0 (79)
d2d/dy2 = 0 (80)
d2f/dy2 = 0 (81)
At the boundary between the diffusion layer and the bulk of the solution (y = 1)
(da/dy),_=ab-a,=ab-aa, (82)
(db/dy),_= (dc/dy),_= (de/dy),_+O (83)
b-+0 e+O (84)

35
In the bulk of the solution
da’/ds = (da/dy),_
ddb/ds = (dd/dy),_
df b/ds = (df/dy), -
At s=O, ab=l and b=c=d=e=f=O.
(85)
(86)
(87)
We require to derive expressions for the yields of stable products in terms of consumption of starting material. Linear combination of differential eqns. (69)-(74) yields
d2(a+b+c+d+e+f)/dy2=0 (88)
Integration over the reaction-layer thickness and then over the remainder of the diffusion layer gives
(da/dy),-+(dd/dy),-+(df/dy),-=0 (89)
which is a relationship that describes the conservation of mass in the system. Now we must relate one or more of the stable products to consumption of starting material. Linear combination of eqns. (70), (71), (73) and (74) leads to
d2(b+c+2e+2f)/dy2=0 (90)
Integration of this equation over the thickness of the diffusion layer and considera- tion of the surface boundary conditions give
z(df/dy),- = - (da/dy), - (dc/dy), (91)
Combination of eqns. (87) and (91) allows us to relate the appearance of f to the slopes of the concentration gradients for species a and c:
dfb/ds = ;(da/dy), + +(dc/dy), (92)
Substitution of eqns. (85)-(87) into eqn. (89) gives
ddb/ds + d f b/ds = ah (93)
Integration of eqns. (92) and (93) gives the following equations:
fb = i@da/dy)o + (dc/dy),] dr (94)
db +fb = jSab dr 0
(95)
where ub may be a function of s depending on the electrolysis technique employed and r is a variable of integration. We can obtain the yield of F by dividing eqn. (94) by eqn. (95):
yF =fb/(db +fb) (96)

36
Y,= i, osbWWo + GWWo] dr)
i ‘ab dr
(97) 0
Y,,=l-Y, (98) What remains is to solve the pertinent boundary-value problem to obtain the
slopes of the concentration profiles for species a and c in the context of the electrolysis conditions (CCPE or EPE).
Constant-concentration potentiostatic electrolysis (CCPE) We make the following transformations of variables: a* = hiI* a, b* = hi’* b,
c* =hi/’ c, e* = hi/” e and y* = hi” y. A set of four differential equations must be solved simultaneously to obtain the desired concentration profiles for species a and c (through a* and c*) for various values of the competition parameters:
d*a*/dy** = qa*b* + ua*e* -pb*c* (99
d*b*/dy ** = qa*b* + b* +pb*c* ( 100) &*/dy** = - qa*b* -b* +pb*c* (101)
d*e*/dy** = -pb*c* + ua*e* + ve* ( 104 where q = h,hT3/* = k,k;3/*c”D’/*l-‘, u = h3h;3/* = k,k;3/*coD’/*l-‘, P= hdhT3/* = k,k,3/2c”D “*l-’ and v = h,/h, = k,/k,. The boundary conditions are as follows. At y*=O
(da*/dy*)o + (db*/dy*), = 0 (103) (dc*/dy*)” + (de*/dy*)o = 0 ( 104) a*-0 0- co* =o
( 105)
At y*=m
(da*/dy*), = 1 ( 106) (db*/dy*), = (dc*/dy*), = (de*/dy*), +O ( 107) b*dO ( 108) Under CCPE conditions, ub = 1, (da/dy)o = (da*/dy*), and (dc/dy), = (dc*/ dy *lo. Since q, u, v and p are independent of s, eqn. (97) simplifies to
y, = i(da*/dy*)o + ;(dc*/dy*)o ( 109)
Exhaustive potentiostatic electrolysis (EPE) For EPE conditions the bulk concentration of A is a function of time (ab =
exp(-s)). If the transformations u# = hi” exp(s)a, bX = hi” exp(s)b, cx =

37
hi” exp(sk, ex = h:” exp(s)e and yx = hi’“y are made, eqns. (691, (70), (71) and (73) become
d2ax/dy #2 = q#a”b# + u#a#e’ex _ P #b#cx
d=b#/dy #= = q#a#b# + b# +p#b’c’
d2c#/d y”2 = - q#a”b# - b# + p#b”c’
d2e”/d y #2 = -p”b#c# + u#a#e# + u,x
(110)
(111)
( 112)
(113) where q# = q exd -s>, ux = u exp( -s) and px = p exp( --s), and the following boundary conditions prevail. At y”=O
(da#/dy#), + (db#/dy”), = 0 (114)
(dc#/dy”), + (de#/dy#), = 0 (115)
at = 0 co” = 0 (116)
At y#=m
(da#/dy#), = 1 (117)
(db#/dy#), = (dc#/dyn)oD = (de#/dy”), + 0 (118)
b#+O (119)
Under EPE conditions a6 decays exponentially with time, so that
(da/dy), = (da#/dy#),ab ( 120)
(dc/dy), = (dc#/dy’),ab (121) Thus, for 100% conversion of starting material to products,
y, = $k’[ (da#/dy#), + (dc#/dy#),] exp( -r) dr ( 122)
where the slopes of the concentration profiles for ax and cx are functions of time through q#, u’ and p”. We can consider two limiting cases: (i) the second electron is transferred to the neutral radical by the electrode (ECE process) and (ii) the solution electron-transfer reaction predominates (DISP process).
Heterogeneous electron transfer For this limiting case, p + 0 and the system of equations for the CCPE scenario
is simplified to
d2a*/dye2 = qa*b* + ua*e* ( 123)
d2b*/dy*= =qa*b* +b* ( 124) d2c*/dy”= = vqa*b* _ b*
(125)
d=e*/dy”= = ua*e* + e* ( 126)

38
with all the boundary conditions remaining the same. We can simplify the system further by adding eqns. (124) and (125) to obtain
d’(b*+c*)/dy*‘=O ( 127)
Integration of eqn. (127) results in
-(db*/dy*),, = (dc*/dy*), ( 128)
and substitution of eqn. (103) gives
(da*/dy*), = (dc*/dy*)o ( 129)
Equation (129) coupled with eqn. (109) gives
yr = (da*/dy*)o ( 130)
Now we solve the following boundary-value problem to obtain the slope of the concentration gradient for a (through a*) at the electrode surface:
d2a*/dy*2 = qa*b* + ua*e* (131)
d2b*/dy*2 = qa*b* + b* ( 134
d2e*/dy*2 = ua*e* + ve* (133)
At y*=O
(da*/dy*), + (db*/dy*h,= 0 ( 134)
(da*/dy*)a + (de*/dy*),= 0 (135)
ao* = 0 (136)
At y*=m
(da*/dy*), = 1 ( 137)
(db*/dy*), = (de*/dy*), + 0 ( 138)
b*+O (139)
This scenario is most easily described if one treats the reaction layers for species b* (I,) and e* (I,) separately. In most instances, the monoanion e* that results from reduction of the neutral radical c* will be much more basic than the radical anion b*. If we assume that 1, -=K f,, then h, x=-h, (v + ~1 and species e* is mere reactive than b * toward both the solvent-supporting electrolyte and the unreduced starting material. There is no way to observe how the yields change as a function of q. Attempts were made to set the values of u and v at some arbitrarily large value and then to change the value of q systematically, but the solution of the boundary-value problem always oscillates. However, if u + 0 then h, -=z h;/* which implies that b* is more reactive toward the solvent-supporting electrolyte than e* is toward a*. When these conditions prevail, the solvent-supporting electrolyte solution is too acidic for self-protonation to occur at all. A situation exists where the values of k, and k, are approximately the same and v + 1. This

39
system is dependent upon two parameters 4 and u, and the same problems for systematic investigation of the effects of these parameters exist for this situation as were seen in the previous case.
Homogeneous electron transfer In this limiting case, p + w. Under these conditions the steady-state approxima-
tion can be established for species c and thus
pb*c* =qa*b* + b* ( 140)
For the majority of ECE-ECDISP self-protonation systems, species E is generally thought to be more basic than species B. If we assume that the steady-state approximation is valid for species E, then
pb*c* =ua*e* + ue* (141)
Substituting eqns. (140) and (141) into eqns. (110) and (111) to eliminate e* and c *, we obtain
d*a*/dy**= (uqa** - u)b*/(ua* + LI)
d*b*/dy*‘= 2qa*b* + 2b*
with the following boundary conditions. At y*=O
( 142)
( 143)
(da*/dy*), + (db*/dy*)O = 0
a*-0 0-
At y*=w
( 144)
(145)
(da*/dy*), = 1
b*+O
Production of F is given by
( 146)
( 147)
YF = i(da*/dy*), ( 148)
From results obtained for scheme 1, we predicted that the vast majority of systems, for which father-son proton-transfer is an important step in the reduction mechanism, will proceed via the DISP pathway. In most instances, consumption of the radical anion (species B) will be the rate-determining step in the reaction pathway and so we can consider two limiting situations with respect to the competition parameter u: (i) u + UJ and (ii) u -+ 0. If u -+ ~0, the boundary-value problem becomes
d*a*/dy**= -b*
d*b*/dy** = 2qa*b* + 2b*
(149)
(150)
40
with boundary conditions identical with those just stated, and the yield of F is given by
YF = ;(da*/dy*)o (151) because (dc */d y *I,, = 0 for DISP processes.
Furthermore, the boundary-value problem for the EPE case is exactly the same as that for the CCPE case. For the EPE case, the bulk concentration of starting material is a function of time cab = exp(-8)) and the transformation of variables makes it necessary to account for the time dependence of the boundary-value problem. Using the EPE transformations, we find that the slopes of the concentra- tion gradients for species ax and c’ are dependent on time and that, to compute the yields correctly, we must first calculate the slope of the gradient in question, assume that it will remain constant for a very small step in time and then evaluate the following first-order differential equations:
dab/ds = -ab ( 152) deb/ds = i(da#/dy#),ab (153)
(A)
W F: 0.4.. /--
/ /
0.2 ..
/’
0.0~---’
9 w F
1.0..
(8) 0.8 ..
0.6 ..
0.4.. /I--
/ 0.2 _. /
/
/ 0.0 .+- - - -
4 -3.0 -2.0 -1.0 0.0 1.0 2.0 3.0
LOG(q) 4. 0
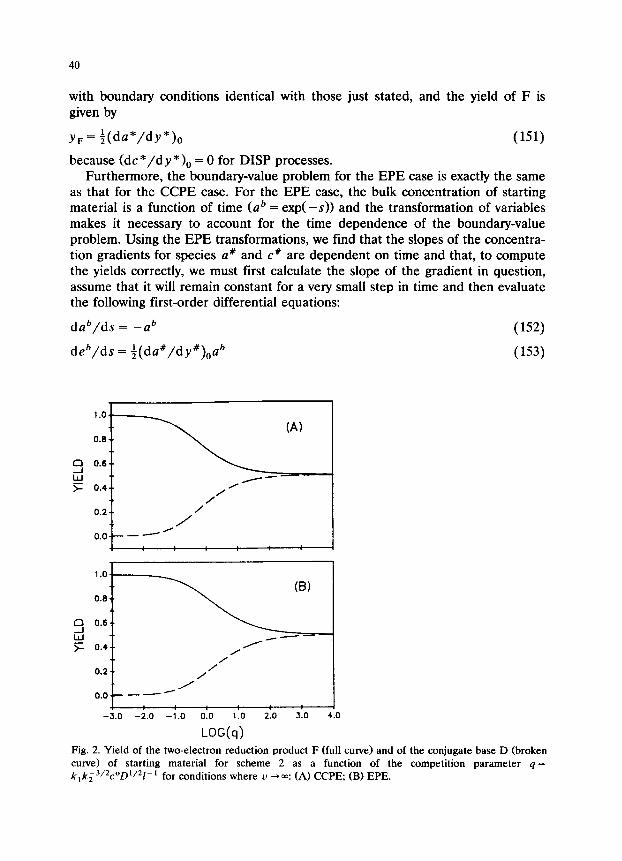
Fig. 2. Yield of the two-electron reduction product F (full curve) and of the conjugate base D (broken curve) of starting material for scheme 2 as a function of the competition parameter q = k,k;3~2coD’~21-’ for conditions where u --tm: (A) CCPE, (B) EPE.
41
0.7.. (B) 0.6.. ,I---
9 ,/
w 0.5.. -.
F
0.4-m
0.3-.
T -i -3.0 -2.0 -1.0 0.0 1.0 2.0 3.0 4.0
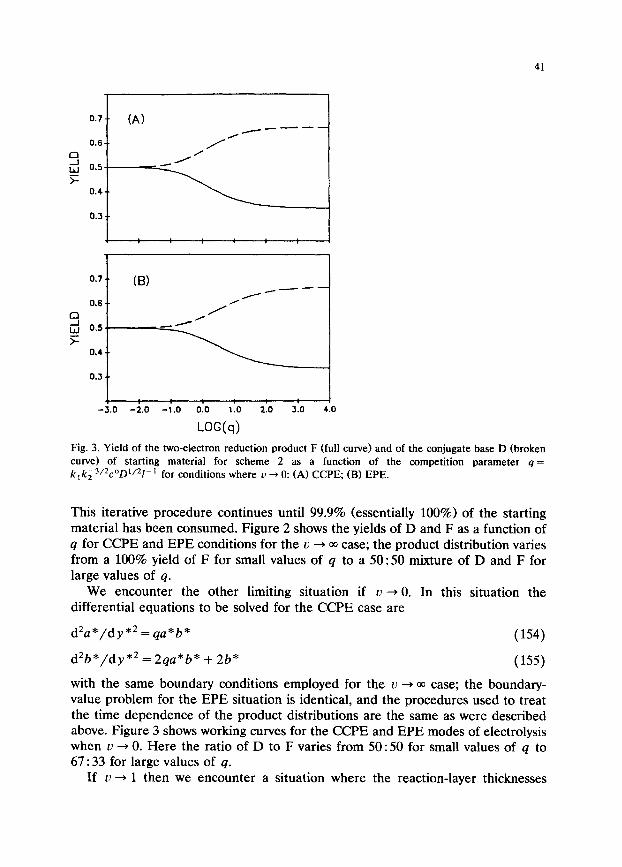
Fig. 3. Yield of the two-electron reduction product F (full curve) and of the conjugate base D (broken curve) of starting material for scheme 2 as a function of the competition parameter q = k,k;3&?‘D’/21-’ for conditions where u + 0: (A) CCPE, (B) EPE.
This iterative procedure continues until 99.9% (essentially 100%) of the starting material has been consumed. Figure 2 shows the yields of D and F as a function of 4 for CCPE and EPE conditions for the u + m case; the product distribution varies from a 100% yield of F for small values of 4 to a 50 : 50 mixture of D and F for large values of 4.
We encounter the other limiting situation if u + 0. In this situation the differential equations to be solved for the CCPE case are
d2a*/dy*2 = qa*b* ( 154)
d2b*/dy*2 = 2qa*b* + 2b* (155)
with the same boundary conditions employed for the u + 00 case; the boundary- value problem for the EPE situation is identical, and the procedures used to treat the time dependence of the product distributions are the same as were described above. Figure 3 shows working curves for the CCPE and EPE modes of electrolysis when u --, 0. Here the ratio of D to F varies from 50 : 50 for small values of q to 67 : 33 for large values of q.
If u --, 1 then we encounter a situation where the reaction-layer thicknesses

42
with respect to b* and e* are approximately equal. In this case the key parameter is u. For u + 0, the boundary-value problem is the same as for the u + 00 case. However, if u -+ oc, then the equations are the same as for the o + 0 case.
DISCUSSION
Product distributions derived for father-son self-protonation (scheme 1) are independent of the experimental parameters (c” and 1 for CCPE and EPE as we have described them), and a coulometric n value equal to unity as well as an equimolar mixture of species D and E will always be obtained. If we consider the E formed via each of the two modes of electron transfer as different species, it can be seen that, for both the EPE and the CCPE cases, the yield of each species is dependent on a single competition parameter which is the ratio w = kd/k,, of rate constants k, and k,, for homogeneous electron transfer and father-son proton transfer respectively. Formation of EE is favored for small values of W, whereas formation of ED is favored for large values of w. As was shown for the equivalent analysis of the classical ECE-DISP scheme [33], the critical parameter governing the competition is the rate constant for the homogeneous reaction that consumes the electrogenerated radical anion. In the present case, as k,, takes on large values the neutral radical C is formed near the electrode surface and thus can be reduced there. As k,, takes on smaller values, C is formed further from the electrode surface which minimizes the chance that it might return to the electrode surface for reduction; thus the solution electron-transfer reaction for consumption of C predominates.
Under conditions where the linear scan voltammetric peak potentials as func- tions of concentration and sweep rate cannot be used to glean definitive informa- tion about the reduction mechanism (ECE,,, vs. DISPl), the rate of the homoge- neous electron-transfer reaction is taken as the diffusion limit (k, = lOlo M-’ s-l) [33]. Thus, from the curves in Fig. 1 the maximum amount of E formed through heterogeneous electron transfer occurs when w = 1 (k, = k,,); this corresponds to 64% of the total amount of E formed. As shown in Fig. 1, the value w = 1 lies on the steepest portion of the full curve so that, as the magnitude of k,, moves further from the diffusion limit, the system rapidly approaches pure DISPl conditions. In fact, if the second-order self-protonation rate constant is less than 2.5 X lo8 M-’ s-l, more than 95% of E is formed via the homogeneous electron- transfer reaction. This is true for electrolyses where the potential of the working electrode is such that the concentration of depolarizer at the electrode surface is zero-a good assumption in most cases. If the depolarizer concentration at the electrode surface is significantly different from zero, these calculations will not be valid [421.
Differential equations that describe father-son self-protonation (scheme 1) are formally the same as those found for DIM2 [43], where the product of the initial heterogeneous electron-transfer reaction couples with unreduced starting material to form a new adduct. Our analysis shows that, in cases where the adduct is more

43
readily reduced than the starting material, transfer of the second electron to the adduct will occur in solution by the radical anion if the rate constant for dimeriza- tion is less than 2.5 x lo8 M-’ s-l. In most cases, species E will be basic enough to attack unreduced starting material. This process will diminish the concentration of A in the reaction layer and, in addition, will make the rate of the father-son proton-transfer reaction slower, thereby forcing the system more toward the DISP pathway.
Scheme 2 allows competition to exist for consumption of B and E by proton donors in the medium. This system is described with the aid of four competition parameters: q = k,k;3/2c”D’/21-1, u = k3k;3~2c”D’~21-‘, p = k,k;3/2coD’/21-1 and u = k,/k,. Because the radical anion is probably the least reactive of all intermediates in the scheme, a process involving its consumption is likely to be the rate-determining step. For the case where heterogeneous transfer of the second electron to the neutral radical is assumed, the system depends on three parame- ters: q, u and U. It is difficult to dissect this boundary-value problem and to investigate systematically the effects of q, u and v independently. It is safe to conclude, however, that q and u must be large in order for maximal self-protona- tion behavior to be observed; if these parameters are small, no self-protonation will be observed.
For the case where homogeneous transfer of the second electron is assumed, it is possible to formulate limiting forms of the problem as a function of a single competition parameter. A scenario in which u -+ CO is illustrative of conditions when reaction (67) exerts a negligible influence on the reaction scheme. This situation can occur when k, and co are small as well as when k, and 1 are large. For the case when u + cQ, the system depends only on q. Substitution of typical experimental conditions (c” = lo-* M, D = lo-’ cm* s-l and I = 10V3 cm) into the expression for q reveals that, when klkT3/* is less than 10, no effects due to self-protonation will be seen whereas, if klkT3/* is larger than 2500, the effects of self-protonation will be maximal.
If u tends toward zero, the yield of F varies from 50% to 33% and the yield of D varies from 50% to 67% as q changes from small to large values. For a typical experiment (co = lo-* M, D = lo-’ cm* s-l and I= lop3 cm), the value of k, ki3/* must be larger than 2000 for CCPE conditions and larger than 6300 for EPE conditions in order to observe coulometric n values corresponding to transfer of two-thirds of an electron per molecule of starting material.
This analysis shows that a hierarchy of rate constants for father-son and grandfather-grandson proton-transfer reactions can be established: h 3 > h 1 > hi/’ and h, > h,. Rate constants for reactions where electrogenerated bases attack unreduced starting material must be much larger than rate constants pertaining to consumption of electrogenerated bases by the solvent-supporting electrolyte. Be- cause the basicities of radical anions derived from organic substrates of similar structure will be comparable, it appears that the kinetic acidity of the starting material is indeed the key parameter in determining whether or not the substrate will be involved in self-protonation reactions when it is reduced electrochemically.

44
ACKNOWLEDGEMENT
Our sincerest thanks go to Dr. Christian Amatore, Ecole Normale SupCrieure, Paris, France, for his helpful suggestions concerning the application of the simula- tion technique.
REFERENCES
1 D.W. Leedy and D.L. Muck, J. Am. Chem. Sot., 93 (1971) 4264. 2 G. Farnia, A. Romanin, G. Capobianco and F. Torzo, J. Electroanal. Chem., 33 (1971) 31. 3 A. Lasia, J. Electroanal. Chem., 52 (1974) 229. 4 G. Farnia, G. Capobianco and A. Romanin, J. Electroanal. Chem., 45 (19731397. 5 T. Wasa and P.J. Elving, J. Electroanal. Chem., 91 (19781 249. 6 T.E. Cummings and P.J. Elving, J. Electroanal. Chem., 94 (19781 123. 7 T.E. Cummings and P.J. Elving, J. Electroanal. Chem., 102 (1979) 237. 8 W.T. Bresnahan, T.E. Cummings and P.J. Elving, Electrochim. Acta, 26 (1981) 691. 9 T. Wasa and P.J. Elving, J. Electroanal. Chem., 142 (1982) 243.
10 P.J. Elving, Can. J. Chem., 5.5 (1977) 3392. 11 S. Roffia, G. Gottardi and E. Vianello, J. Electroanal. Chem., 142 (1982) 263. 12 G. Farnia, G. Mengoli and E. Vianello, J. Electroanal. Chem., 50 (1974) 73. 13 G. Farnia, A. Roque da Silva and E. Vianello, J. Electroanal. Chem., 57 (1974) 191. 14 E. Brillas, G. Famia, M.G. Severin and E. Vianello, Electrochim. Acta, 31 (19861 759. 15 C. Nuntnarumit and M.D. Hawley, J. Electroanal. Chem., 133 (1982) 57. 16 J. Janata, J. Gendell, R.G. Lawton and H.B. Mark Jr., J. Am. Chem. Sot., 90 (1968) 5226. 17 A. Dal Moro, G. Farnia, F. Marcuzzi and G. Melloni, Nouv. J. Chim., 4 (1980) 3. 18 G. Farnia, F. Marcuzzi, G. Melloni and G. Sandonl, J. Am. Chem. Sot., 106 (1984) 6503. 19 G. Farnia, G. Sandona, F. Marcuzzi and G. Melloni, J. Chem. Sot. Perkin Trans. II, (19881 247. 20 G. Farnia, G. Sandona, F. Marcuzzi and G. Melloni, J. Electroanal. Chem., 264 (1989) 297. 21 T.R. Chen, M.R. Anderson and D.G. Peters, J. Electroanal. Chem., 197 (1986) 341. 22 T.R. Chen, M.R. Anderson, S. Grossman and D.G. Peters, J. Org. Chem., 52 (1987) 1231. 23 J.Z. Stemple and D.G. Peters, J. Org. Chem., 54 (1989) 5318. 24 J.Z. Stemple and D.G. Peters, J. Electroanal. Chem., 286 (1990) 89. 25 J.Z. Stemple and D.G. Peters, J. Electroanal. Chem., 286 (1990) 109. 26 F. Maran, E. Vianello, G. Cavicchioni and F. D’Angeli, J. Chem. Sot. Chem. Commun., (1985) 660. 27 M.C. Artvalo, G. Farnia, M.G. Severin and E. Vianello, J. Electroanal. Chem., 220 (1987) 201. 28 F. Maran, E. Vianello, F. D’Angeli, G. Cavicchioni and G. Vecchiati, J. Chem. Sot. Perkin Trans.
II, (1987) 33. 29 F. Maran, M. Fabrizio, F. D’Angeli and E. Vianello, Tetrahedron, 44 (1988) 2351. 30 F. Maran, S. Roffia, M.G. Severin and E. Vianello, Electrochim. Acta, 35 (1990) 81. 31 F. Maran, M.G. Severin and E. Vianello, Tetrahedron Lett., 31 (1990) 7523. 32 C. Amatore, G. Capobianco, G. Farnia, G. Sandona, J.M. Saveant, M.G. Severin and E. Vianello, J.
Am. Chem. Sot., 107 (1985) 1815. 33 C. Amatore and J.M. Saveant, J. Electroanal. Chem., 123 (1981) 189. 34 C. Amatore and J.M. Savtant, J. Electroanal. Chem., 123 (1981) 203. 35 C. Amatore, F. M’Halla and J.M. Saveant, J. Electroanal. Chem., 123 (1981) 219. 36 C. Amatore, J. Pinson and J.M. Saveant, J. Electroanal. Chem., 123 (1981) 231. 37 C. Amatore and J.M. Saveant, J. Electroanal. Chem., 125 (1981) 1. 38 C. Amatore and J.M. Saveant, J. Electroanal. Chem., 125 (1981) 23. 39 C. Amatore and J.M. Saveant, J. Electroanal. Chem., 126 (1981) 1. 40 J.M. Sax&ant, J. Electroanal. Chem., 236 (1987) 31. 41 W.H. Press, B.P. Flannery, S.A. Teukolsky and W.T. Vetterling, Numerical Recipes in C, The Art
of Scientific Computing, Cambridge University Press, New York, 1990, Ch. 15.
42 C. Amatore and J.M. SaGant, J. Electroanal. Chem., 85 (1977) 27. 43 C.P. Andrieux, L. Nadjo and J.M. Savkant, J. Electroanal. Chem., 42 (1973) 223.





























