Embed Size (px)
Citation preview

1
Compensatory Mechanisms Influence Hemostasis in the Setting of eNOS-Deficiency
Mark D. Iafrati, Olga Vitseva, Kahraman Tanriverdi, Price Blair, Sybille Rex,
Subrata Chakrabarti, Sonia Varghese, and Jane E. Freedman
From the From the Department of Surgery, Tufts-New England Medical Center, 750 Washington
Street, Boston, MA 02111(M.D.I.), and the Whitaker Cardiovascular Institute and Evans
Department of Medicine, 700 Albany Street, Boston University School of Medicine, Boston, MA
02118 (O.V., K.T., P.B., S.R., S.C., S.V., J.E.F.).
Address for correspondence: Mark D. Iafrati, Tufts-New England Medical Center, 750
Washington Street, Boston, MA 02111. [email protected]
Hemostasis in eNOS-Deficient Mice
Articles in PresS. Am J Physiol Heart Circ Physiol (November 24, 2004). doi:10.1152/ajpheart.00819.2004
Copyright © 2004 by the American Physiological Society.

2
Abstract
The balance between thrombosis and hemorrhage is carefully regulated. Nitric oxide
(NO) is an important mediator of these processes as it prevents platelet adhesion to the
endothelium and inhibits platelet recruitment. Although endothelial NO synthase (eNOS)-
deficient mice have decreased vascular reactivity and mild hypertension, enhanced thrombosis in
vivo has not been demonstrated. To determine the role of endogenous NO in hemostasis, a
model of carotid arterial injury and thrombosis was performed using eNOS-deficient and wild-
type mice. Paradoxically, the eNOS-deficient animals had a prolongation of time to occlusion as
compared to the wild-type mice (p<0.001). Consistent with this finding, plasma markers
suggesting enhanced fibrinolysis (tissue plasminogen activator (t-PA) activity and antigen and D-
dimer levels) were significantly elevated in eNOS-deficient animals. Vascular tissue expression
of t-PA and platelet activity were not altered. In endothelial cells, t-PA is stored in Weibel-
Palade bodies and exocytosis of these storage granules is inhibited by NO. Thus, in the absence
of NO, release of Weibel-Palade body contents (and t-PA) could be enhanced, an observation
also supported by increased von Willebrand factor (vWF) levels observed in eNOS-deficient
animals. In summary, although eNOS deficiency attenuates vascular reactivity and increases
platelet recruitment, it is also associated with enhanced fibrinolysis due to lack of NO-dependent
inhibition of Weibel-Palade body release. These processes highlight the complexity of NO-
dependent regulation of vascular homeostasis. Such compensatory mechanisms may partially
explain the lack of spontaneous thrombosis, minimally elevated baseline blood pressure, and
normal lifespan seen in animals deficient in a pivotal regulator of vascular patency.
Key words: Platelets, thrombosis, nitric oxide, transgenic mice

3
Introduction
Normal hemostatic balance is maintained by tight regulation of coagulation, fibrinolysis,
and platelet activation. Adhesion of platelets to the endothelium is prevented by several
mechanisms including endothelial cell production of nitric oxide (NO) and prostacyclin (3, 22).
Nitric oxide inhibits platelet adhesion and aggregation (2, 26) and prevents thrombosis (25).
Exogenous NO inhibits the normal activation-dependent increase in the expression of platelet
surface glycoproteins, including P-selectin and the integrin glycoprotein IIb-IIIa complex.
Platelet-derived NO appears to inhibit the primary aggregation response only modestly but NO
release from activated human platelets markedly inhibits platelet recruitment(7) and, thus, may
attenuate the progression of intra-arterial thrombosis. The vascular endothelium, which mediates
vasomotor tone, in part, through NO release, has been extensively characterized. Endothelium-
dependent dilation is impaired in human atherosclerotic coronary arteries as well as patients with
cardiovascular disease (1). Although interventions that are believed to enhanced the systemic
bioavailablity of NO have been clearly shown to enhance peripheral and coronary vascular
relaxation (29), correlative changes in NO production and antithrombotic propensity have not
been directly measured in these studies. In fact, estrogen supplementation, shown in multiple
studies to enhance vascular relaxation (8), was associated with enhanced arterial thrombosis in
prospective clinical studies (11, 18).
Homozygous eNOS-mutant mice are known to have impaired endothelium-derived
relaxing factor activity (9), increased blood pressure, decreased heart rate, and increased plasma
renin concentration (24). In the pulmonary vasculature, eNOS deficiency produces mild
pulmonary hypertension (27). In addition, we previously found that activated platelets from mice
lacking eNOS do not release NO (6) and, although no changes were detected in platelet function,
eNOS-deficiency was associated with shortened bleeding times (6, 19) suggesting enhanced
coagulation. The lack of difference in the platelet activation response in eNOS-deficient animals
suggests that platelet- and endothelial-derived NO deficiency alters the in vivo hemostatic
response by another (non-platelet dependent) mechanism. To further explore this question and
determine if NO alters thrombosis in vivo, these animals were studied using a carotid artery
injury model.

4
Materials and Methods
All studies were approved by the Institutional Animal Care and Use Committee at
Boston University. The generation of mice bearing the NOS III (eNOS) gene deletion has been
previously described in detail (9). Male and female mice weighing 22-24 gm and 10 weeks old
were used for all experiments. Blood was drawn into syringes and anticoagulated with trisodium
citrate by inferior vena caval puncture.
Carotid Thrombosis Model
Mice were subject to a full thickness arterial injury model to determine the rate of
thrombotic vessel occlusion. Briefly, the technique of carotid artery injury(5) was used with
slight modification. After establishing adequate anesthesia, a 1 cm vertical midline incision
was made in the neck and the left common carotid artery was exposed and a disc (0.6mm
diameter) saturated with FeCl3 was applied for 3 minute followed by removal and irrigation.
A mini-Doppler flow probe (model 0.5VB, Transonic Systems) was placed on the distal
carotid artery and monitored on a computerized data acquisition system for time to
thrombotic occlusion. Carotid arteries were then excised and fixed.
Measurements of Fibrinolysis
For determination of plasma t-PA activity, a photometric method using a kit with
chromogenic substrate S-2251 (Coaset® t-PA, Chromogenix AB) was utilized and the
absorbance was measured at 405 nm with a spectrophotometer (UV-160 1PC, Shimadzu).
PAI-1 activity was determined by immunoassays (ELISA; MPAIKTTM, Molecular
Innovations Inc.). Samples were compared with the calibration curve to estimate active PAI-
1 level (ng/ml). The conversion factor for PAI-1 used was PAI-1 IU/ml= 1.34 ng/ml.
D-dimer and fibrinogen were also measured by ELISA. For D-dimer, a 1:5 dilution in
plasma was used (Diagnostica Stago, asserachrom D-Dimer). For fibrinogen, a 1:200
dilution of mouse plasma was used (DiaPharma, Zymutest Fibrinogen).

5
Measurement of Weibel-Palade Body Contents: Plasma Levels
The measurement of t-PA levels has been described above. P-selectin was measured
using sP-selectin immunoassay diluted 1:50 (R&D systems, Inc. Quantikine M). For vWF,
plasma was diluted 1:2 and measured by immunoassay (Corgenix, REAADS von Willebrand
Factor Antigen Test kit). Samples, standards and controls were assayed in triplicate.
Immunofluorescence
Standard immunohistochemical staining procedure was used for frozen sections. Sections
with mouse aorta were fixed in 3.7% formaldehyde for 30 min at RT. The slides were then
washed with TBS and incubated with tPA goat polyclonal antibody at a dilution of 1:50 (Santa
Cruz Biotechnology, Inc.) followed by washing incubation with second mouse anti-goat IgG-
FITC conjugated antibody at a dilution of 1:200 (60 min, 22°C). Sections used for control were
incubated in buffer instead of a primary antibody. The sections were examined using
fluorescence microscopy (Nikon eclipse TE 300) and scanned for intensity.
Expression of t-PA
Aortas were removed from mice and immediately frozen at -80°C until RNA extraction
was performed. Total RNA from aorta samples were isolated by High Pure RNA Tissue Kit
(Roche Applied Sciences). RNA samples were reverse transcribed with 1st Strand cDNA
Synthesis Kit (Roche Applied Science). Real-Time Quantitative PCR was performed for tPA and
G3PD as an internal control by using Assay-on-Demand Gene Expression primers and probes
and TaqMan Universal Master mix (Applied Biosystems) in ICycler Real-Time PCR instrument
(Bio-Rad).
Measurement of Platelet Activation by Flow Cytometry
Blood from wild type and eNOS-deficient mice were isolated from vena cava following
anaesthesia. Murine platelet rich plasma was prepared by centrifugation of whole blood for 20
min at 110 g. The platelets were counted in a Coulter Counter (Coulter Electronics, Miami, FL).
The platelet rich plasma was directly stimulated with thrombin. The samples were immediately

6
fixed with final 1% paraformaldehyde, and immuno-labeled with FITC-conjugated mouse
CD62P, mouse CD41b, or corresponding isotype controls (Pharmingen, BD Biosciences).
Analysis of the FITC-labeled samples was performed with a FACScan flow cytometer
(Becton Dickinson) at an excitation wavelength of 488 nm and a laser power of 15 mW. The
green fluorescence was collected through a 530-nm band-pass filter. The isotypic control was
positioned between fluorescence values of 1 and 10 as reference. For each sample, data from 2 x
105 platelets were recorded in a 1,024-channel distribution showing the logarithmic amount of
green fluorescence. Flow cytometric data were analysed using histogram statistics of the
CellQuest software (Becton Dickinson).
Statistical Analysis
Comparisons between genotypes and genders were performed by unpaired 2-tailed
Student’s t tests. Differences between groups were determined using an unpaired Student’s t test.
The effect of interventions were analyzed using a paired t test. A statistically significant
difference was assumed with a value of P<0.05. All data are expressed as the mean ± SEM.

7
Results
Measurement of Thrombosis in eNOS-Deficient and Wild-Type Mice
Previously, we (6) and others (19) have found that bleeding times are decreased in eNOS-
deficient mice. Bleeding times, however, are primarily dependent on platelet adhesion and
recruitment in the setting of intact endothelium (28). Therefore, to determine the relevance of
vascular NO deficiency on an in vivo model of thrombosis, eNOS-deficient or wild-type mice
underwent a standard characterized murine arterial injury model that is known to induce carotid
artery thrombosis (4). Time to occlusion was measured using a mini-Doppler flow probe in the
distal carotid artery. Using this model, the injury is long relative to the artery diameter. The flow
was monitored and not found to have increased velocities. There was a gradual decrease in
velocity until flow ceased as a result of the thrombosis. Paradoxically, we found that both female
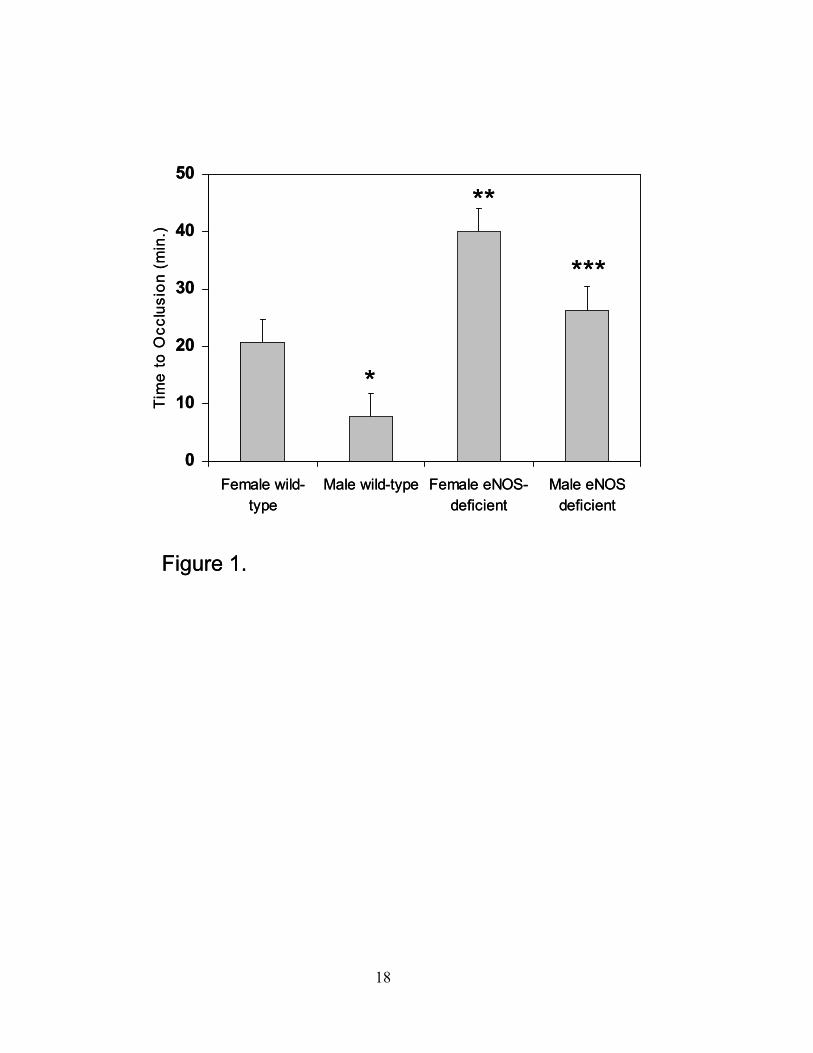
and male eNOS-deficient mice had prolonged measurement of time to occlusion (Figure 1)
suggesting decreased carotid thrombosis. Visual histological examination of cross sectional
analyses of carotid arteries confirmed the limited injury and formation of thrombus as well as the
lysis of thrombus in appropriate specimens.
Measurement of Platelet Activation
We previously found that platelet activation as determined by platelet surface P-selectin
expression in resting and stimulated platelets were not significantly different in the eNOS-
deficient and control animals (6). To confirm and extend these findings, platelet surface
expression of CD41 (GPIIb) and CD62P (P-selectin) was measured in resting and activated
platelets (thrombin 1U/ml) for both male and female animals. For CD41 expression, in resting
platelets, mean fluorescence for eNOS-deficient and wild-type male mice were 58.2 ± 3.4 and
60.8 ± 1.9, respectively (P=ns). For thrombin activated platelets, mean fluorescence for CD41
expression was 71.7 ± 2.7 and 76.9 ± 5.1 for the eNOS-deficient and wild-type male mice,
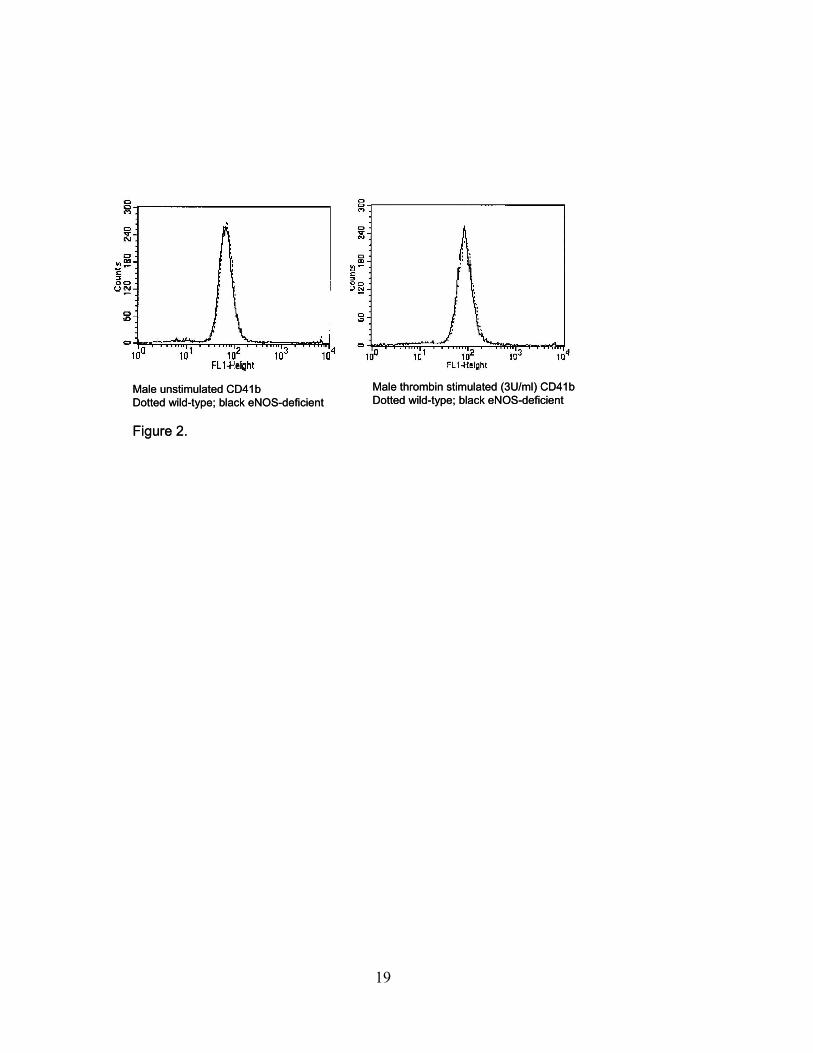
respectively (P=ns). As represented in Figure 2, flow cytometric analysis of glycoprotein IIb
(CD41) demonstrates no significant change in expression between wild-type and eNOS-deficient
platelets. This lack of difference in GPIIb expression between eNOS-deficient and control
platelets was also seen in the female eNOS-deficient and control groups (data not shown).
Consistent with previous results (6), there was no change in P-selectin expression with
stimulation for the male eNOS-deficient mice as compared to the control male mice. Similar

8
findings (P=ns) for surface P-selectin expression were found for the female eNOS-deficient mice
as compared to the wild-type female mice. As measured by change in light transmittance, there
was also no change in ADP (5µM) induced platelet aggregation (n=3, P=ns).
The Effect of NO Deficiency on Fibrinolysis
Importantly, as seen in Figure 1, time to occlusion for eNOS-deficient animals is
prolonged suggesting decreased thrombosis. However, it is also plausible that these findings are
due to enhanced fibrinolysis. To study this question, potential regulators of fibrinolysis were
measured in plasma from NO-deficient and control animals. Activity of t-PA was found to be
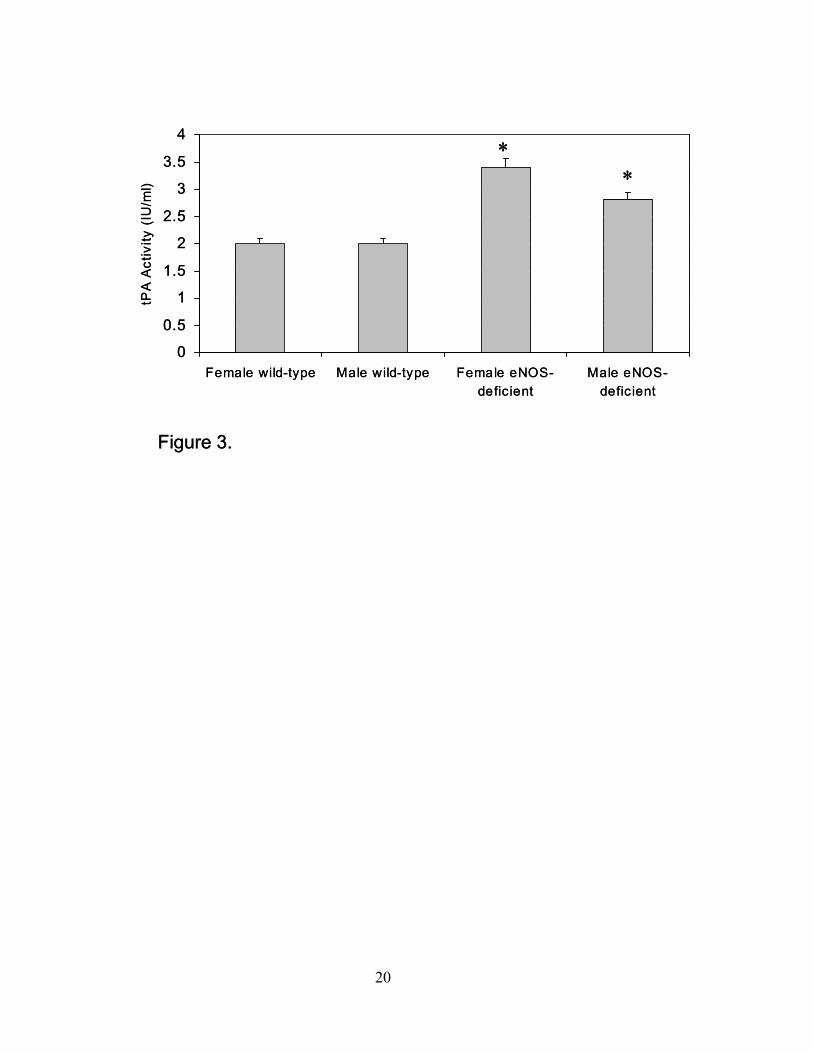
significantly enhanced in female as well as male eNOS-deficient mice as compared to wild-type
controls (Figure 3). Additionally, t-PA activity and antigen were increased in both female and
male eNOS-deficient mice as compared to wild-type mice in the absence of arterial injury (data
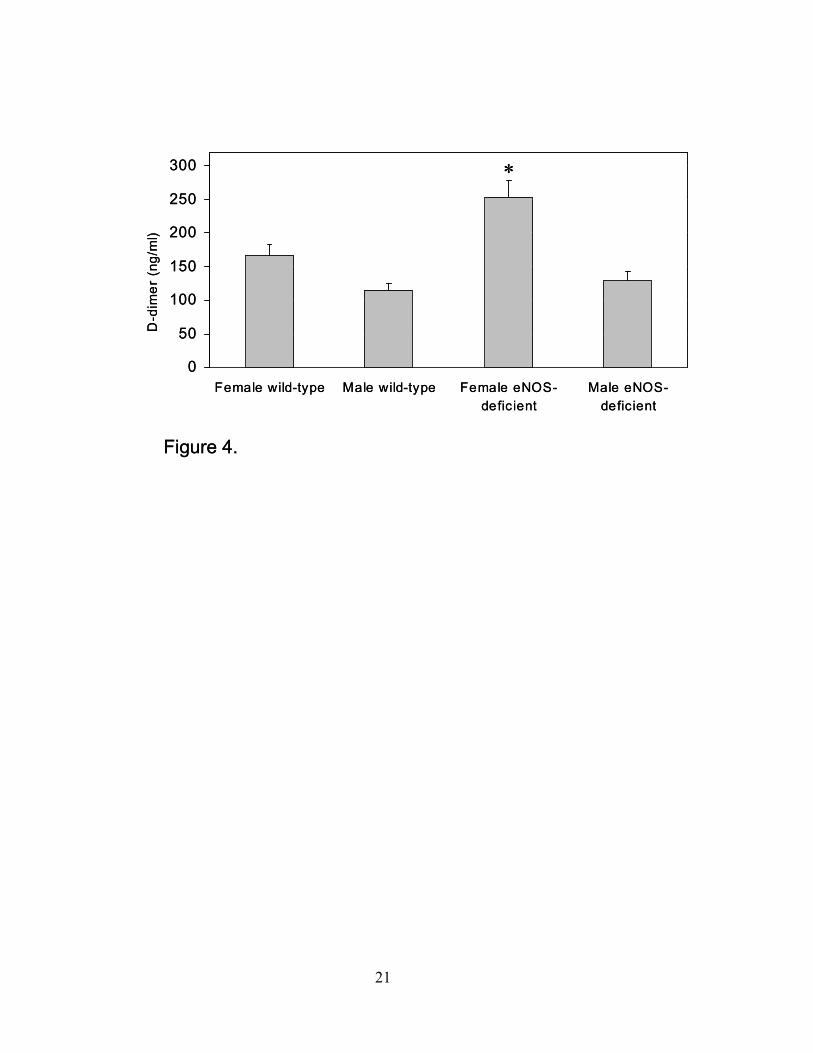
not shown). Consistent with this finding was elevated levels of D-dimer, a specific derivative of
cross-linked fibrin, in the eNOS-deficient animals (Figure 4). The change in levels only reached
statistical significance in the female mice. It should be noted that although fibrinogen was not
different between the groups (22.5 ± 1.0 mg/ml and 19.6 ± 2.6 mg/ml for wild-type and eNOS-
deficient animals, respectively), the levels of D-dimer found in plasma (ng/ml) are considerably
lower than fibrinogen (mg/ml). Additionally, it is unlikely that changes in fibrinogen levels
would be seen in the setting of enhanced lysis. Enhanced fibrinolysis can also be seen in the
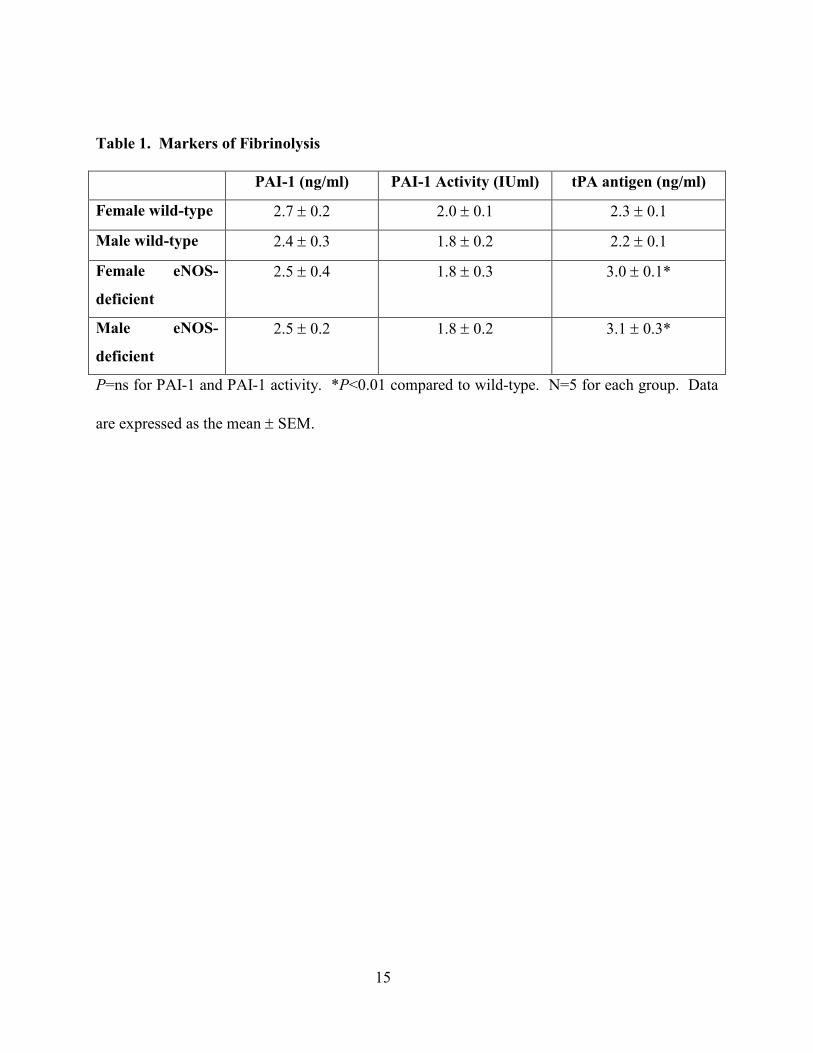
setting of decreased plasminogen activator inhibitor (PAI-1). As shown in Table 1, neither PAI-1
levels nor PAI-1 activity are altered in either gender of eNOS-deficient mouse.
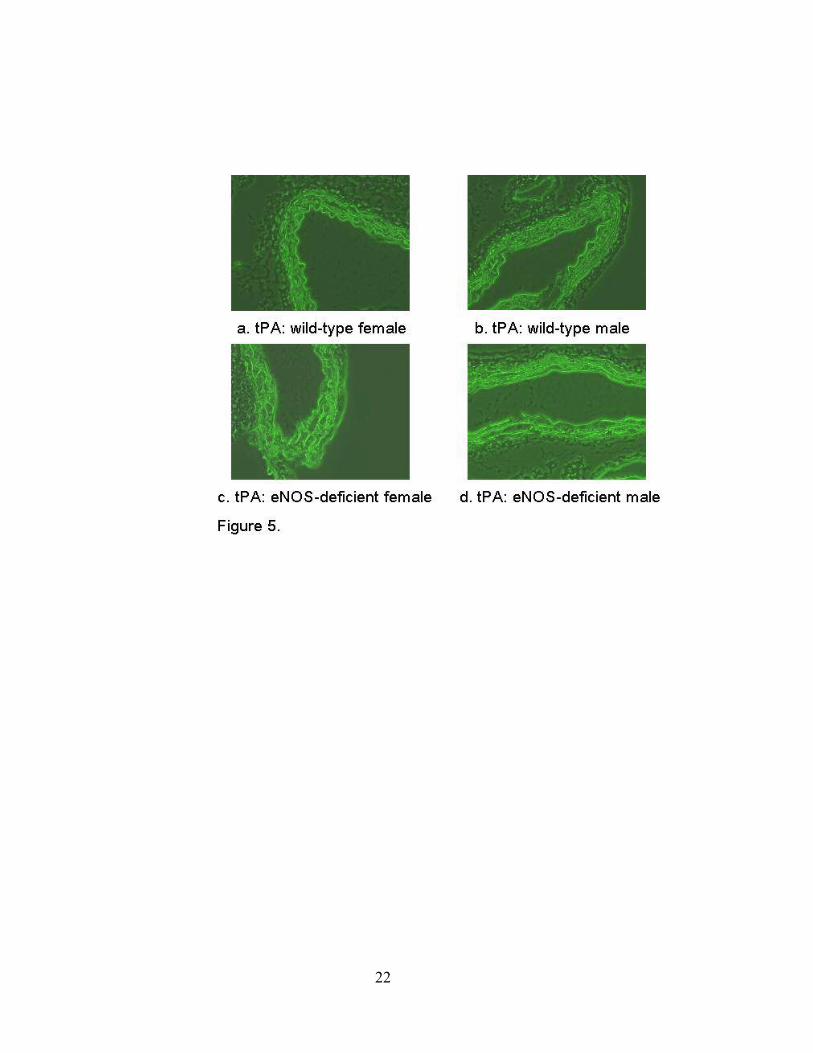
Expression of t-PA in Vascular Tissue
Endothelial cells are the source of t-PA found in the plasma. Enhanced t-PA in the
plasma may be due to enhanced release with or without increased vascular tissue expression. To
determine whether deficiency of eNOS in the vascular tissue is associated with change in t-PA
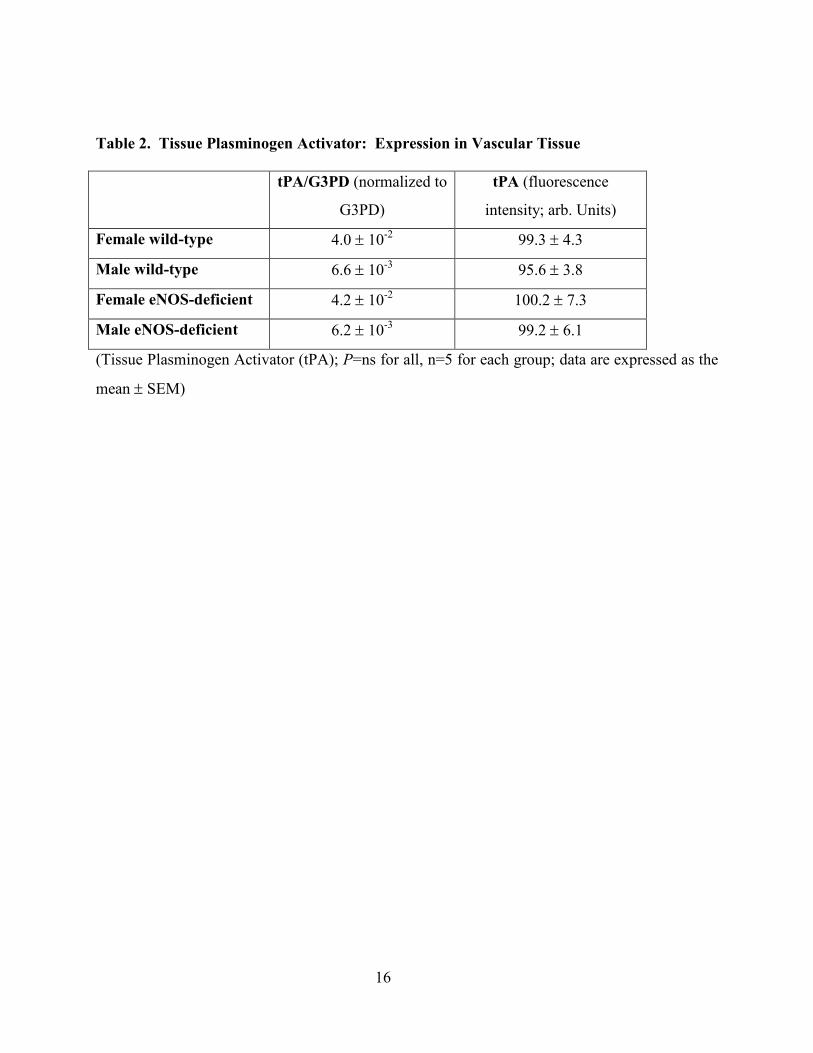
expression, aortic tissue was studied by real-time quantitative PCR for t-PA. As seen in Table 2,
there was no difference between eNOS-deficient and wild-type mice. These findings were
confirmed by immunohistochemistry of aortic tissue (Figure 5).

9
Plasma Levels of Weibel-Palade Body Derived Proteins
Vascular endothelial cells contain vesicles known as Weibel-Palade bodies, which serve
as a storage compartment for vWF, P-selectin, tPA, and other proteins. Exocytosis of Weibel-
Palade bodies has been recently shown to be inhibited by NO(19). Thus, in the absence of NO,
release of Weibel-Palade body contents (and t-PA) could be enhanced. To determine if other
substances stored in Weibel-Palade bodies had increased plasma levels, von Willebrand factor
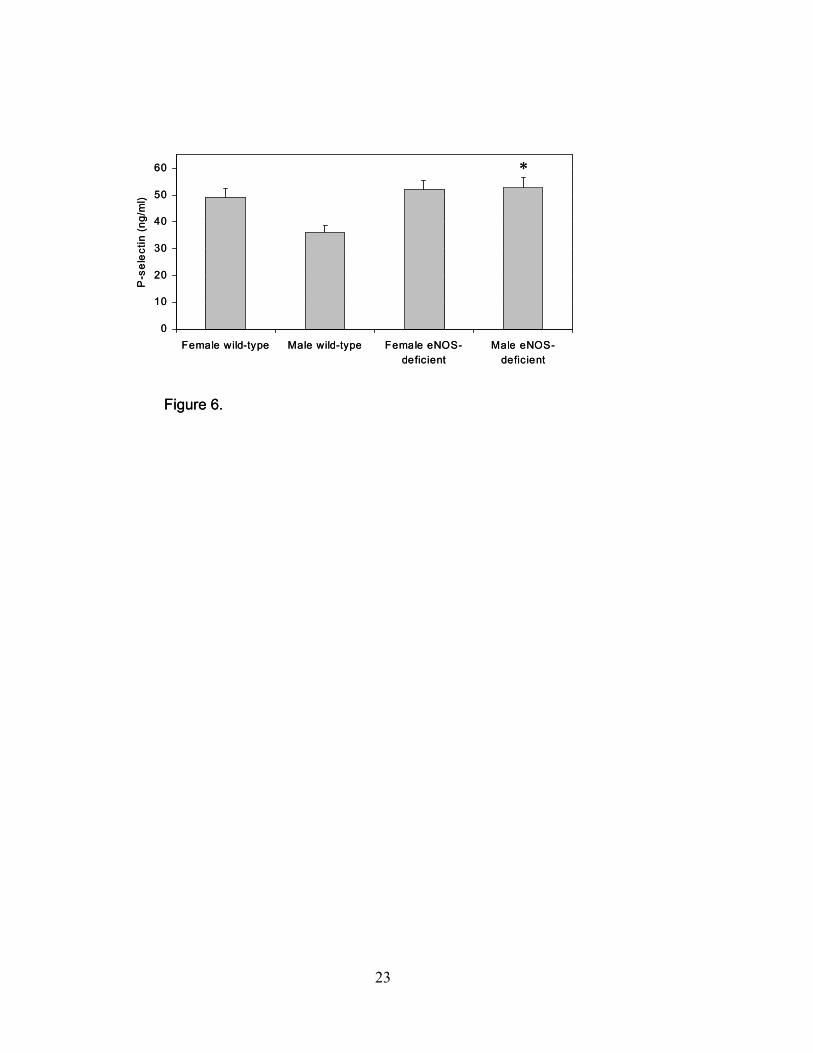
(vWF) and plasma P-selectin levels were measured. P-selectin levels were found to be 51.4 ±
10.3 (ng/ml) and 43.3 ± 9.9 (ng/ml) in eNOS-deficient and control animals, respectively (P<0.05;
Figure 6), although only the male and pooled levels reached statistical significance. This finding
was confirmed with measurement of vWF found to be 57.8 ± 28.1 (%) and 48.3 ± 16.2 (%) in
eNOS-deficient and control animals, respectively (P<0.05).

10
Discussion
In the setting of intimal injury, platelets adhere to the subendothelium leading to platelet
activation. Once activated, platelets facilitate thrombus growth by recruitment of additional
platelets and promoting surface thrombin generation (23). In the intact vessel wall, NO is an
inhibitor of platelet function. While endothelial-derived NO production is known to both
mediate vasorelaxation as well as inhibit platelet adhesion and activation, less is known about the
contribution to thrombosis in vivo. Although deficiency in platelet-NO production is associated
with enhanced platelet activation in a “recruitable” platelet population, this failed to correlate
with extent of vessel-occluding thrombus. These are not the first observations to demonstrate
paradoxical findings in the absence of eNOS. A recent study showed that in the absence of
eNOS, mice with experimentally induced portal hypertension developed a hyperdynamic
circulation (12). Although the mechanism was not directly investigated, it was suggested that
compensatory vasodilator molecules were upregulated (12).
Although the extent of in vivo thrombosis was not correlated with degree of primary
platelet activation, it was correlated with fibrinolysis as demonstrated by enhanced t-PA and D-
dimer levels. Expression of t-PA was not altered (Table 2, Figure 5), suggesting enhanced
release of t-PA from the endothelium into the blood. Tissue plasminogen activator is stored in
vascular endothelial cells and contained in vesicles known as Weibel-Palade bodies (10). Upon
activation of endothelial cells, the Weibel-Palade bodies fuse with the plasma membrane and
release their contents into the blood, allowing endothelial cells an additional mechanism for
participating in hemostasis. Interestingly, as recently shown by Matsushita and colleagues (19),
NO inhibits exocytosis of Weibel-Palade bodies by regulating the activity of N-ethylmaleimide-
sensitive factor (NSF). In this study, NO inhibited NSF disassembly of soluble NSF attachment
protein receptor (SNARE) complexes by nitrosylating critical cysteine residues of NSF.
Consistent with our findings, serum levels of vWF were higher in eNOS-deficient mice as
compared to wild-type mice. Importantly, in the study by Matsushita and colleagues,
pharmacologic modulation of eNOS in human endothelial cell culture also induced these changes
suggesting that our findings are not specific to eNOS-deficient mice.

11
With respect to fibrinolysis, there appears to be some divergence between our findings
and previous studies. Chronic infusion of L-nomega-nitro-L-arginine methyl ester (L-NAME)
has been shown to alter PAI-1 levels (13, 15), however L-NAME can be a nonspecific NOS
inhibitor and chronic L-NAME administration has been shown to induce the expression of iNOS
in the aorta (17). Our findings were specific for eNOS and we have found no change in iNOS
expression in the vascular tissue of the eNOS-deficient animals by RT-PCR or
immunohistochemistry (data not shown). Also potentially conflicting are brachial studies
showing that bradykinin infusion is associated with increased levels of t-PA through a NO
synthase-independent pathway, however, these studies themselves are in opposition to direct
examination of this question (19). In addition, this study examined normal subjects and not those
known to have decreased bioavialable NO. Supportive of our findings is a recent study reporting
increased t-PA antigen D-dimer levels in subjects with increased risk of atherothrombosis, a
group with known impaired NO-dependent vascular function (21). Clearly, the clinical
implications of our findings cannot be assumed.
Von Willebrand factor is a plasma protein that plays an essential role in controlling the
adhesion and aggregation of platelets at sites of vascular injury and may be responsible for the
previously observed decrease in bleeding time in the eNOS-deficient animals (6, 19). The
enhanced release of mediators of both platelet adhesion and fibrinolysis may also explain the
conflicting bleeding time and carotid artery occlusion results. It has been shown in other animal
models of arterial thrombosis that changes in carotid occlusion due to thrombus formation may
not be associated with changes in the bleeding time (28). The release from Weibel-Palade bodies
of both t-PA as well as vWF highlights the complex nature of hemostasis. Although seemingly
paradoxical, such release ensures that while hemorrhage due to vascular trauma is contained, the
unmitigated perpetuation of thrombus is limited to prevent eventual occlusion of the vessel.
These observations are also enhanced by the differences in damage done to the vessel
wall in the setting of injury. Bleeding times reflect normal tissue while the carotid artery
occlusion model studies damaged endothelium. Thus, changes in release of NO from the
endothelium and its contribution to hemostasis would be reflected primarily in the bleeding time.
Previous studies utilizing the carotid injury model have specifically studied thrombolysis at sites
of arterial injury (4) and found a specific contribution to fibrinolysis. This observation is

12
confirmed by a study demonstrating that arterial thrombosis is more influenced by blood
components than by elements within the arterial wall (14). Thus, in this setting of carotid injury,
the secondary effect of NO deficiency, namely the enhanced release of tPA from endothelial
Weibel-Palade bodies is of greater relevance than the release of NO from the endothelium. The
clinical relevance of these findings, while not known, are potentially intriguing.
Although not the specific focus of this study, some differences between the male and
female animals were observed. Gender differences in platelet function in wild-type mice have
been shown recently (16). In this study, female mice had enhanced fibrinogen binding and,
similar to our results, no change in GPIIb expression. These studies found differences only in
washed platelets so the direct relevance to our in vivo observations is not clear. Gender
differences (20) have also been assessed by the response to injury by quantitative morphometry
and measuring the intimal to medial (I/M) volume ratio. In wild-type mice, cuff placement
causes pronounced intimal proliferation. Female mice show less intimal response than do males,
although eNOS mutant female mice still have more response than do wild-type females.
Whether these changes are estrogen-dependent or -independent is not known.
In summary, although eNOS deficiency attenuates vascular reactivity and increases
platelet recruitment, it is also associated with enhanced fibrinolysis due to lack of NO-dependent
inhibition of Weibel-Palade body release. It has been assumed that, for cardiovascular disease, a
relative deficiency of bioactive NO is harmful as it leads to attenuated vascular reactivity and
enhanced thrombosis. However, as suggested by these studies, the processes regulating
thrombosis are more complex. As highlighted by the release of both t-PA and vWF from
Weibel-Palade bodies, hemostasis is a delicate balance between occlusion and patency.
Acknowledgements: This work has been supported in part by NIH grants NIH RO1AG08226
(J.F.), NIH RO1HL62267 (J.F.), and an Established Investigator Award from the American
Heart Association (J.F.).
Conflict-of-Interest Disclosure Statement: The authors (M.D.I., O.V., K.T., P.B., S.R., S.C.,
S.V., J.E.F.) have no conflicts-of-interest to disclose.

13
References
1. Celermajar DS, Sorensen KE, Bull C, Robinson J, and Deanfield JE. Endothelium-dependent dilation in the systemic arteries of symptomatic subjects relates to coronary risk factors and their interaction. J Am Col Card 24: 1468-1474, 1994. 2. Cooke JP, Stamler J, Andon N, Davies PF, McKinley G, and Loscalzo J. Flow stimulates endothelial cells to release a nitrovasodilator that is potentiated by reduced thiol. Am J Physiol 259: H804-H812, 1990. 3. de Graaf JC, Banga JD, Moncada S, Palmer RM, de Groot PG, and Sixma JJ. Nitric oxide functions as an inhibitor of platelet adhesion under flow conditions. Circ 85: 2284-2290, 1992. 4. Farrehi PM, Ozaki CK, Carmeliet P, and Fay WP. Regulation of arterial thrombolysis by plasminogen activator inhibitor-1 in mice. Circ 97: 1002-1008, 1998. 5. Fay WP, Parker AC, Ansari MN, Zheng X, and Ginsburg D. Vitronectin inhibits the thrombotic response to arterial injury in mice. Blood 93: 1825-1830., 1999. 6. Freedman J, Sauter R, Battinelli B, Ault A, Knowles C, Huang P, and Loscalzo J. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOS3 gene. Circ Res 84: 1416-1421, 1999. 7. Freedman JE, Loscalzo J, Barnard MR, Alpert C, Keaney JF, Jr, and Michelson A. Nitric oxide released from activated platelets inhibits platelet recruitment. J Clin Invest 100: 350-356, 1997. 8. Gerhard M, Walsh BW, Tawakol A, Haley EA, Creager SJ, Seely EW, Ganz P, and Creager MA. Estradiol therapy combined with progesterone and endothelium-dependent vasodilation in postmenopausal women. Circ 98: 1158-1163, 1998. 9. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, and Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377:239-242, 1995. 10. Huber D, Cramer EM, Kaufmann JE, Meda P, Masse JM, Kruithof EK, and Vischer UM. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood 99: 3637-3645, 2002. 11. Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, and Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. Jama 280: 605-613., 1998. 12. Iwakiri Y, Cadelina G, Sessa WC, and Groszmann RJ. Mice with targeted deletion of eNOS develop hyperdynamic circulation associated with portal hypertension. Am J Physiol Gastrointest Liver Physiol 283: G1074-1081, 2002. 13. Kaikita K, Fogo AB, Ma L, Schoenhard JA, Brown NJ, and Vaughan DE. Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition. Circ 104: 839-844, 2001. 14. Karnicki K, Owen WG, Miller RS, and McBane RD, 2nd. Factors contributing to individual propensity for arterial thrombosis. Arterioscler Thromb Vasc Biol 22: 1495-1499, 2002. 15. Katoh M, Egashira K, Mitsui T, Chishima S, Takeshita A, and Narita H. Angiotensin-converting enzyme inhibitor prevents plasminogen activator inhibitor-1 expression in a rat model

14
with cardiovascular remodeling induced by chronic inhibition of nitric oxide synthesis. J Mol Cell Cardiol 32: 73-83, 2000. 16. Leng XH, Hong SY, Larrucea S, Zhang W, Li TT, Lopez JA, and Bray PF. Platelets of female mice are intrinsically more sensitive to agonists than are platelets of males. Arterioscler Thromb Vasc Biol 24: 376-381, 2004. 17. Luvara G, Pueyo ME, Philippe M, Mandet C, Savoie F, Henrion D, and Michel JB. Chronic blockade of NO synthase activity induces a proinflammatory phenotype in the arterial wall: prevention by angiotensin II antagonism. Arterioscler Thromb Vasc Biol 18: 1408-1416, 1998. 18. Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, Trevisan M, Black HR, Heckbert SR, Detrano R, Strickland OL, Wong ND, Crouse JR, Stein E, and Cushman M. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med 349: 523-534, 2003. 19. Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, and Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115: 139-150, 2003. 20. Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, and Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest 101: 1225-1232, 1998. 21. Pradhan AD, LaCroix AZ, Langer RD, Trevisan M, Lewis CE, Hsia JA, Oberman A, Kotchen JM, and Ridker PM. Tissue plasminogen activator antigen and D-dimer as markers for atherothrombotic risk among healthy postmenopausal women. Circ 110: 292-300, 2004. 22. Radomski MW, Palmer MJ, and Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Com 148: 1482-1489, 1987. 23. Santos MT, Valles J, Marcus AJ, Safier M, Broekman J, Islam N, Ullman HL, Eiroa AM, and Aznar J. Enhancement of platelet reactivity and modulation of eicosanoid production by intact erythrocytes. A new approach to platelet activation and recruitment. J Clin Invest 87: 571-580, 1991. 24. Shesely E, Maeda N, Kim H, Desai M, Krege J, Laubach V, Sherman P, Sessa W, and Smithies O. Elevated blood pressure in mice lacking endothelial nitric oxide synthase. Procedings of the National Academy of Science, USE 93: 13176-13181, 1996. 25. Shultz PJ and Raij L. Endogenously synthesized nitric oxide prevents endotoxin-induced glomerular thrombosis. J Clin Invest 90: 1718-1725, 1992. 26. Stamler J, Mendelsohn ME, Amarante P, Smick D, Andon N, Davies PF, Cooke JP, and Loscalzo J. N-acetylcysteine potentiates platelet inhibition by endothelium-derived relaxing factor. Circ Res 65: 789-795, 1989. 27. Steudel W, Ichinose F, Huang P, Hurford W, Jones R, Bevan J, Fishman M, and Zapol W. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS3) gene. Circ Res 81: 34-41, 1997. 28. Szalony JA, Taite BB, Girard TJ, Nicholson NS, and LaChance RM. Pharmacological intervention at disparate sites in the coagulation cascade: comparison of anti-thrombotic efficacy vs bleeding propensity in a rat model of acute arterial thrombosis. J Thromb Thrombolysis 14:113-121, 2002. 29. Vita JA and Keaney JF, Jr. Endothelial function: a barometer for cardiovascular risk? Circ 106: 640-642, 2002.
15
Table 1. Markers of Fibrinolysis
PAI-1 (ng/ml) PAI-1 Activity (IUml) tPA antigen (ng/ml)
Female wild-type 2.7 ± 0.2 2.0 ± 0.1 2.3 ± 0.1
Male wild-type 2.4 ± 0.3 1.8 ± 0.2 2.2 ± 0.1
Female eNOS-
deficient
2.5 ± 0.4 1.8 ± 0.3 3.0 ± 0.1*
Male eNOS-
deficient
2.5 ± 0.2 1.8 ± 0.2 3.1 ± 0.3*
P=ns for PAI-1 and PAI-1 activity. *P<0.01 compared to wild-type. N=5 for each group. Data
are expressed as the mean ± SEM.
16
Table 2. Tissue Plasminogen Activator: Expression in Vascular Tissue
tPA/G3PD (normalized to
G3PD)
tPA (fluorescence
intensity; arb. Units)
Female wild-type 4.0 ± 10-2 99.3 ± 4.3
Male wild-type 6.6 ± 10-3 95.6 ± 3.8
Female eNOS-deficient 4.2 ± 10-2 100.2 ± 7.3
Male eNOS-deficient 6.2 ± 10-3 99.2 ± 6.1
(Tissue Plasminogen Activator (tPA); P=ns for all, n=5 for each group; data are expressed as the
mean ± SEM)

17
Figure Legends
Figure 1. Time to occlusion after carotid artery injury. After carotid artery injury, a doppler
flow probe was inserted and time to occlusion was determined in minutes (n=7 in each group;
*P=0.06 compared to female wild-type mice; **P≤0.05 compared to female wild-type mice;
***P≤0.05 compared to male wild-type mice; data are expressed as the mean ± SEM).
Figure 2. Measurement of surface GPIIb (CD41) expression in resting and thrombin activated
platelets from eNOS-deficient and wild-type male mice. Enhanced CD41 expression is seen with
stimulation but no difference is detected between the eNOS-deficient (black line) and wild-type
(dotted line) mice. Similar results were noted for female mice (Results are representative of n=7
for each group, P=ns).
Figure 3. Tissue plasminogen activator (tPA) activity. TPA activity (IU/ml) was measured in
plasma from female and male wild-type and eNOS-deficient mice (*P≤0.05 compared to wild-
type mice, n=5 for each group; data are expressed as the mean ± SEM).
Figure 4. Plasma D-dimer levels. Levels of D-dimer were determined by ELISA in plasma from
female and male wild-type and eNOS-deficient mice (*P≤0.05 compared to female wild-type
mice, n=5 for each group; data are expressed as the mean ± SEM).
Figure 5. Immunohistochemistry or aortic tissue from eNOS deficient male and female mice as
compared to wild-type. No change was detected between the groups as shown in the
representative figures (representative of n=5 animals in each group, P=ns as compared by
intensity of fluorescence).
Figure 6. Plasma P-selectin levels. Levels of P-selectin were determined by ELISA in plasma
from female and male wild-type and eNOS-deficient mice (*P≤0.05 compared to male wild-type
mice, n=5 for each group; data are expressed as the mean ± SEM).
18
0
10
20
30
40
50
Female wild-type
Male wild-type Female eNOS-deficient
Male eNOSdeficient
Tim
eto
Occ
lusi
on(m
in.)
***
**
*
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
0
10
20
30
40
50
Female wild-type
Male wild-type Female eNOS-deficient
Male eNOSdeficient
Tim
eto
Occ
lusi
on(m
in.)
***
**
*
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
19
Male unstimulated CD41b Dotted wild-type; black eNOS-deficient
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
Male thrombin stimulated (3U/ml) CD41bDotted wild-type; black eNOS-deficient
Male unstimulated CD41b Dotted wild-type; black eNOS-deficient
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
Male thrombin stimulated (3U/ml) CD41bDotted wild-type; black eNOS-deficient
20
0
0.5
1
1.5
2
2.5
3
3.5
4
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
tPA
Act
ivity
(IU
/ml)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
**
0
0.5
1
1.5
2
2.5
3
3.5
4
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
tPA
Act
ivity
(IU
/ml)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
**
21
0
50
100
150
200
250
300
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
D-d
imer
(ng/
ml)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
*
0
50
100
150
200
250
300
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
D-d
imer
(ng/
ml)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
*
23
0
10
20
30
40
50
60
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
P-s
elec
tin(n
g/m
l)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
*
0
10
20
30
40
50
60
Female wild-type Male wild-type Female eNOS-deficient
Male eNOS-deficient
P-s
elec
tin(n
g/m
l)
ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�ዊ�
*





























