Embed Size (px)
Citation preview

Combinatorial Chemistry & High Throughput Screening, 2007, 10, 000-000 1
1386-2073/07 $50.00+.00 © 2007 Bentham Science Publishers Ltd.
Microwave-Assisted Reactions in Heterocyclic Compounds with Applica-tions in Medicinal and Supramolecular Chemistry
Antonio de la Hoz*, Ángel Díaz-Ortiz, Andrés Moreno, Ana Sanchéz-Migallón, Pilar Prieto, José Ramón Carrillo, Ester Vázquez, Mª Victoria Gómez and Mª Antonia Herrero
Departamento de Química Orgánica, Facultad de Química, Universidad de Castilla-La Mancha, E-13071 Ciudad Real,
Spain
Abstract: Microwave irradiation has been successfully applied in organic chemistry. Spectacular accelerations, higher yields under milder reaction conditions and higher product purities have all been reported. Indeed, a number of authors have described success in reactions that do not occur under conventional heating and modifications in selectivity (chemo-, regio- and stereoselectivity) have even been reported. Recent advances in microwave-assisted combinatorial chemistry in-clude high-speed solid-phase and polymer-supported organic synthesis, rapid parallel synthesis of compound libraries, and library generation by automated sequential microwave irradiation. In addition, new instrumentation for high-throughput microwave-assisted synthesis continues to be developed at a steady pace. The impressive speed combined with the un-matched control over reaction parameters justifies the growing interest in this application of microwave heating. In this review we highlight our recent advances in this area, with a particular emphasis on cycloaddition reactions of heterocyclic compounds both with and without supports, applications in supramolecular chemistry and the reproducibility and scalabil-ity of organic reactions involving the use of microwave irradiation techniques.
Keywords: Microwave irradiation, heterocycles, DIELS–ALDER, solid supports, supramolecular chemistry.
INTRODUCTION
Microwave heating is very attractive for chemical appli-cations [1] and has become a widely accepted non-conventional energy source for performing organic synthe-sis. This statement is supported by the increasing number of related publications in recent years – particularly since 2003 – that have accompanied the general availability of new and reliable microwave instrumentation [2].
A large number of examples of reactions have been de-scribed in organic synthesis [3]. Several reviews have been published on the application of microwaves to solvent-free reactions [4], cycloaddition reactions [5], the synthesis of radioisotopes [6], fullerene chemistry [7], polymers [8], het-erocyclic chemistry [9], carbohydrates [10], homogeneous [11] and heterogeneous [12] catalysis, medicinal and combi-natorial chemistry [13] and green chemistry [14]. Micro-wave-assisted organic synthesis is characterized by the spec-tacular accelerations produced in many reactions as a conse-quence of a heating profile that cannot be reproduced by classical heating. Higher yields, milder reaction conditions and shorter reaction times can be used and many processes can be improved. Indeed, even reactions that do not occur by conventional heating can be performed using microwaves.
This effect is particularly important in (i) the preparation of isotopically labelled drugs that have a short half-life (11C, t1/2 5 20 min; 122I, t1/2 5 3.6 min and 18F, t1/2 5 100 min) [6], (ii) high throughput chemistry (combinatorial chemistry and parallel synthesis) [13] and (iii) catalysis where the short
*Address correspondence to this author at the Departamento de Química Orgánica, Facultad de Química, Universidad de Castilla-La Mancha, E-13071 Ciudad Real, Spain; Fax: +34 926295318; Tel: +34 926295411; E-mail: [email protected]
reaction times preserve the catalyst from decomposition and increase the catalyst efficiency [15].
The results obtained cannot be explained by the effect of rapid heating alone, and this has led various authors to postu-late the existence of a so-called ‘‘non-thermal microwave effect’’ [16, 17]. Hence, acceleration or changes in reactivity and selectivity could be explained by a specific radiation effect and not merely by a thermal effect. The effect of mi-crowave irradiation in chemical reactions is a combination of the thermal effects, i.e., overheating, hot spots and selective heating, and non-thermal effects arising from the highly po-larizing field, in addition to effects on the mobility and diffu-sion that may increase the probability of effective contacts.
The aim of this review is to show our results on the use of microwave irradiation to improve processes and to obtain better yields in cycloaddition reactions involving heterocyc-lic compounds, both with and without solid supports.
A. CYCLOADDITIONS OF HETEROCYCLIC COM-POUNDS
1. Ketene Acetals
Ketene acetals have been widely employed as dipolaro-philes [18] and dienophiles [19] but their cycloadditions un-der classical thermal conditions are generally performed at high temperatures (>100ºC) with long reaction times (several hours or days) and sometimes under an inert atmosphere. These are typical reaction conditions that are appreciably improved by the use of microwaves. For example, micro-wave irradiation under solvent free conditions induced 1,3-dipolar and hetero-DIELS–ALDER cycloadditions of cyclic ketene acetals (1) under milder reaction conditions [20] than those reported in the literature. Nitrones (2) and nitrile ox-ides (3) were employed as 1,3-dipoles and , -unsaturated

2 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
ketones (4) as hetero-1,3-dienes (Scheme 1). The cycloaddi-tions were performed in a domestic oven in an open vessel at 450 W or 780 W and within 5–15 min they gave excellent yields (69–95%). The reactions were employed and products were isolated directly from the crude reaction mixture with-out polymerization of the ketene acetals having occurred [21].
In these reactions isoxazolidine 5, 6 and dihydropyran 7 derivatives were obtained. Isoxazolidines have found wide-spread application in the synthesis of biotin, aminoglyco-sides, alkaloids, amino acids, -lactams, and herbicide com-pounds [22]. Dihydropyran derivatives are structurally re-lated to many natural products such as carbohydrates, ta-laromycines, avermectins, pheromones, and other natural substances [23].
2. 1,2,3-Triazines
1,2,3-Triazines are useful compounds that have been shown to participate in cycloaddition reactions [24]. These compounds behave as -deficient dienes and undergo in-verse-demand DIELS–ALDER cycloadditions with electron-rich dienophiles. Several reactions of 1,2,3-triazine and its 4- or 5-methyl derivatives with electron-rich dienophiles have been reported to afford pyridines and pyridazines [25]. This approach has been successfully employed with 4-methyl-1,2,3-triazine to prepare alkaloid derivatives [26]. We be-lieve that cycloaddition reactions between dialkyltriazines and enamines derived from cyclic ketones represent useful processes to prepare a wide range of heterocyclic derivatives and natural products (Scheme 2).
However, the presence of alkyl substituents in the triaz-ine ring diminishes the electron deficiency of the ring and increases the steric hindrance experienced in the cycloaddi-tion. For these reasons, cycloaddition reactions of 4,6-dimethyl-1,2,3-triazine (8), especially with enamines [27],
are scarce and reaction conditions are very energetic and yields very poor [28]. Likewise, DIELS–ALDER reactions of 4,5,6-trimethyl-1,2,3-triazine have not been reported [27].
Microwave irradiation in solvent-free conditions is an energy source that is well suited to performing chemical transformations and the utility of this approach has been demonstrated when a reaction requires energetic conditions or unstable reagents and/or products are involved [4, 29].
4,6-Dimethyl-1,2,3-triazine (8) reacts with enamines de-rived from cyclic ketones under microwave irradiation in solvent-free conditions at atmospheric pressure within 20 min to afford the corresponding heterocyclic product 15–21 in 21–71% yield (Table 1). The modest yields obtained in some cases are due to the poor thermal stability of the rea-gents employed (1,2,3-triazines and enamines).
N
NN
NN
8
+
9a-f 15-21
Scheme 2.
Under classical heating, the reported yields for com-pounds 15–21 are in the range 0–27% [27]. Moreover, the microwave procedure and the absence of solvent avoids the necessity of performing reactions in a sealed tube (dry chlo-roform, 100–120ºC, 2 h) or the removal of high boiling sol-vents (dry o-dichlorobenzene, 200–220ºC, 1 h), and it repre-sents a clean and environmentally benign methodology to obtain fused pyridine systems in good or acceptable yields [30].
The reactions described can also be performed with pyr-rolidine and the cyclic ketones used as precursors of the
O
N
Ar
H
Ph
O
OAr N
Ph
O
O
O
C N OAr
MWMW
O R2
R1
MW
OR2
R1 H
O
O
O
N
Ar
O
O
1
23
4
56
7 Scheme 1.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 3
enamines [31]. These cycloadditions were performed under microwave irradiation using the same reaction conditions as described above and they allow the preparation of valuable heterocyclic systems such as compound 21 (Table 1). How-ever, yields are lower than those obtained using enamines. There are two possible reasons for the lower yields. Firstly,
pyrrolidine and the cyclic ketone must form the enamine prior to reaction with the triazine and, secondly, pyrrolidine has a low boiling point (87–88ºC) and could evaporate be-fore reaction with the ketone (in this sense, a small excess, e.g., 2:1 pyrrolidine/ketone molar ratio, is used).
Table 1. Cycloaddition Reactions Between 4,6-Dimethyl-1,2,3-Triazine (8) and Enamines 9 Derived from Cyclic Ketones Under
Microwave Irradiation in Solvent-Free Conditions
Dienophile Reaction Conditions Product Yield (%)
N
9a
40
HN
O
+
10 11
270 W, 20 min 150ºC
N15
34
N
9b
43
HN
+
O
10 12
270 W, 20 min 150ºC
N16 32
N
9c
270 W, 20 min 140ºC
71
HN
+
O
10�
13
270 W, 20 min 150ºC
N17 40
N
9d
270 W, 20 min 130ºC
N18
33
Me
N
9e
270 W, 20 min 130ºC
N
Me
19
30
N
9f
270 W, 20 min 150ºC
N20
21
HN
+ O
10 14
270 W, 3 min 150ºC
N21
18

4 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
3. 1,2,3-Triazoles
1,2,3-Triazole is the essential structural unit in a number of drugs and some of these materials are also potent HIV-1 inhibitors [32], antimicrobial agents [33], or selective 3-adrenergic receptor agonists [34]. The 1,2,3-triazole ring can be easily built by a 1,3-dipolar cycloaddition using an azide derivative [35]. However, the reactivity of this system, par-ticularly in DIELS–ALDER reactions, has barely been stud-ied in comparison with other heterocyclic systems.
1,2,3-Triazoles undergo a DIELS–ALDER cycloaddition with dimethyl acetylenedicarboxylate (DMAD) (23) in sol-vent-free conditions under microwave irradiation within 20 min to afford pyrazole-3,4-dicarboxylates 24 with extrusion of the substituent on position 4 of the triazole as a nitrile (Scheme 3). Yields can be increased significantly by using silica-supported AlCl3 (Si(Al)) as a catalyst [36] (0.1% mol) (Table 2).
The silica-supported Lewis acid catalyst could be re-used five times in the cycloaddition of 22f under microwaves without a decrease in the product yield. This result shows the utility and enhances the environmentally benign applications of this catalyst. In the absence of microwaves and catalyst, and under similar reaction conditions (time and temperature), yields of the products decreased dramatically (4–25%).
4. Pyrazolyl Imines
Pyrazolo[3,4-b]pyridines are very interesting compounds and have received considerable attention as a result of their
biological activity and structural relationship to indoles. A number of pyrazolo[3,4-b]pyridines display interesting anx-iolytic activity [37] (e.g., tracazolate [38]), they are poten-tially biologically active compounds as new inhibitors of xanthine oxidases [39], have proved to be active against Gram positive and Gram negative bacteria [40] and as cho-lesterol formation-inhibiting compounds [41], and are also promising for the treatment of cataracts associated with dia-betes [42].
Pyrazolo[3,4-b]pyridines have generally been prepared by cyclization reactions starting from different heterocyclic reagents [43]. Only two examples have been reported for the preparation of these compounds by cycloaddition reactions: in the first case, the title compounds were obtained by [4+2] cycloaddition of a 1,2,4-triazepine with DMAD and subse-quent 1,3-sigmatropic rearrangement [44]; in the second case, pyrazolo[3,4-b]pyridines were prepared by 1,3-dipolar cycloaddition of cyclic ketene N,O-acetals with diphenylni-trilimine [45].
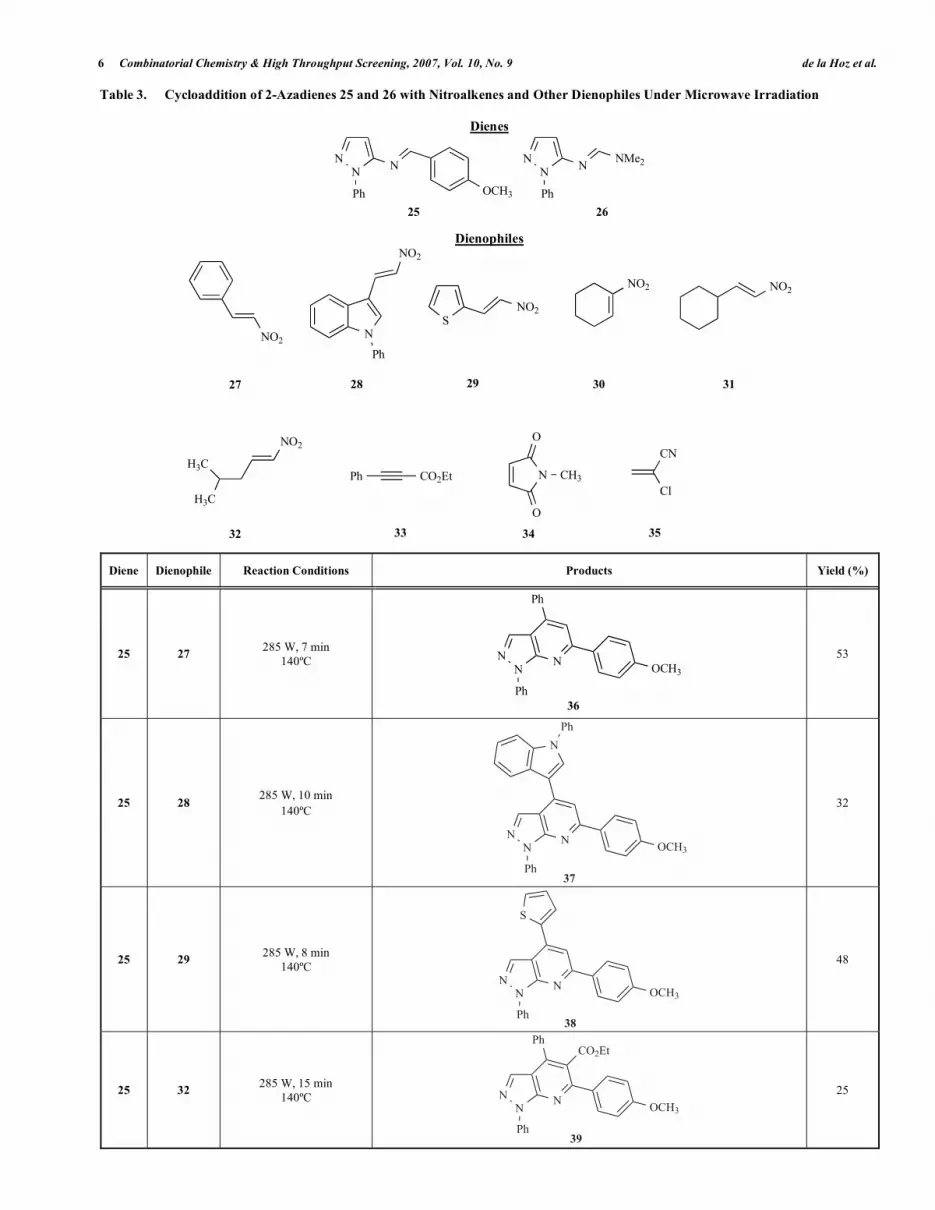
We reported a new, interesting and versatile approach to the preparation of pyrazolo[3,4-b]pyridines through the DI-ELS–ALDER cycloaddition of pyrazolyl imines 25 and 26 with aromatic nitroalkenes under microwave irradiation [46]. This was the first example of a [4+2] cycloaddition involv-ing a 2-azadiene with a pyrazole ring. In order to determine the scope of this reaction and its utility as a new synthetic approach to pyrazolo[3,4-b]pyridines, we studied the cy-cloaddition of 2-azadienes 25 and 26 with aliphatic nitro-alkenes and other dienophiles under microwave irradiation.
N
NN
Ph
R2
R1 CO2Me
CO2Me
N N
NR1
CO2Me
R2
Ph
CO2Me R1 N- N R2
CO2MeMeO2C
N
Ph
Si(Al)+
MW / 80-130 ˚C 20 min
22 23 24
Scheme 3.
Table 2. Product Yield Obtained in the Reaction of 1,2,3-Triazoles 22a–h with DMAD (23) by Classical Heating or Under Micro-
waves
Product Yield (%)a,b
Entry Substrate R1 R2 Product Power (W)c
Temp (ºC) Classical Heating
d Microwaves
1 22a H H 24a 60 120 15 (8) 40 (14)
2 22b C6H5 H 24a 80 120 34 (5) 58 (18)
3 22c CHO H 24a 80 130 7 (4) 35 (15)
4 22d CO2Me H 24a 80 120 12 (5) 38 (12)
5 22e C6H5 CH3 24b 60 120 30 (11) 57 (20)
6 22f CH3CH2 CH3CH2 24c 40 80 46 (25) 89 (56)
7 22g CH3OCH2 CH3OCH2 24d 60 100 62 (12) 94 (20)
8 22h CH3(CH2)3 CH3(CH2)3 24e 80 100 48 (21) 86 (34) aIsolated products. bYields of uncatalyzed reactions are given in brackets. cStarting power in the microwave-assisted reactions. dNo significative increase in the yields is observed at higher reaction times.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 5
All cycloadditions were performed in the absence of solvent at atmospheric pressure in a focused microwave reactor with full control of the incident power and the reaction tempera-ture. The reaction mixture was irradiated until complete con-sumption of the azadiene had been achieved. The pyra-zolopyridines 36–50 were directly isolated from the crude mixture by flash chromatography. Reaction conditions, products and yields are summarized in Table 3.
This is a versatile synthetic procedure that permits the synthesis of several pyrazolopyridines by the introduction of various different substituents in the heterocyclic ring and even allows the preparation of tricyclic derivatives such as 46 or 49.
In order to assess the efficiency of microwave irradiation for [4 + 2] cycloadditions of heterocyclic 2-azadienes of pyrazole we performed some reactions by classical heating under the same reaction conditions: nitrostyrene (27) did not react with 25 and afforded only traces of 41 (3%) in the cy-cloaddition with 26. We studied the reaction of 25 and 26 with 2-chloroacrylonitrile (35) by classical heating in an oil bath under the same reaction conditions (time and tempera-ture). However, in these cases adducts 40 and 50 were not obtained and hydrolysis of the 2-azadiene took place.
5. 4-Pyrazolyl Hydrazones
The first series of bipyrazoles was synthesized in 1893 [47]. Since then many bipyrazolyl derivatives have been re-ported in the literature [48]. Some of these compounds were shown to be useful as potential antiinflamatory agents [49], cytotoxic agents [50], insecticides [51], herbicides [52], fungi-cides [53], and in the photographic and paint industries [54].
In most cases, bipyrazolyl derivatives have been synthe-sized by cyclization reactions [55]. However, 1,3-dipolar cycloaddition constitutes one of the most versatile tools for the construction of five-membered heterocycles [56], and bipyrazoles have also been prepared through these reactions using diarylnitrilimines [57] or diazocompounds [58] as rea-gents.
Hydrazones give a thermal 1,2-hydrogen shift to afford azomethine imine intermediates that can undergo a 1,3-dipolar cycloaddition, a reaction that has been known since 1978 [59]. It has been reported that a protonated substrate can intramolecularly assist, if a carbonyl group is present in an appropriate position, the hydrazone-azomethine imine isomerization under mild conditions [60]. However, in most cases the intermolecular cycloaddition of thermally gener-ated azomethine imines with highly activated dipolarophiles must be performed under reflux in high-boiling solvents (e.g., xylene) with long reaction times (several hours or days) [61]. These requirements reduce the synthetic utility of the reaction.
We report a new approach for the preparation of bipyra-zolyl derivatives by 1,3-dipolar cycloaddition under micro-wave irradiation. The radiation produces the thermal iso-merization of the pyrazolyl hydrazones 51–54 to the corre-sponding azomethine imines (Scheme 4). These intermedi-ates undergo 1,3-dipolar cycloaddition with double or triple bonded dipolarophiles to afford [4,3'] or [5,3'] bipyrazolyl adducts 57–69 in 10–45 min with yields in the range 30–84%. The reaction permits, together with the study of the
periselectivity, the synthesis of useful compounds such as bipyrazoles in good yields by a useful and clean synthetic methodology [62]. The reaction conditions and results are summarized in Table 4.
Cycloadditions were performed at atmospheric pressure in a focused microwave reactor or in hermetically sealed Teflon tubes in a domestic oven. In these cases internal pres-sures were not measured. All cycloadditions were optimized to obtain the best yield and to achieve complete consumption of the starting materials. Reaction products showed a high stability under microwave irradiation. Reactions did not show an equilibrium between reactants and products and the adduct ratio did not change under microwave irradiation.
The thermal hydrazone-azomethine imine isomerization can be easily and efficiently performed under microwave irradiation to give good yields of bipyrazole derivatives within a few minutes. However, the effect of microwave irradiation is not only a reaction acceleration but it also in-duces the cycloaddition of dipolarophiles that do not react on classical heating under comparable reaction conditions.
6. o-Quinodimethanes
Heterocyclic o-quinodimethanes are unstable and reac-tive dienes that must be generated in situ [63]. In solution and in the presence of a dienophile the o-quinodimethanes can be trapped in a DIELS–ALDER reaction [64]. This is the basis for the synthesis of a wide range of polycyclic natural products [65]. Most of the papers in this area describe the generation of o-quinodimethanes in situ and trapping with dienophiles to obtain the DIELS–ALDER adduct. In this regard, the general application of these reactions has not been fully explored.
The generation of o-quinodimethane derivatives gener-ally involves harsh reaction conditions under which the rea-gents are heated to very high temperatures (frequently up to 200ºC). Moreover, o-quinodimethanes rapidly decompose or undergo intramolecular reactions in the absence of an acti-vated dienophile.
We reported a microwave-assisted approach for the gen-eration and DIELS–ALDER reactions of o-quinodimethane derivatives of pyrazine, 1,2,4-triazine, pyrazole and 1,2,3-triazoles [66]. These heterocyclic systems have not been as widely studied as other systems (e.g., indole) and a general synthetic application of o-quinodimethanes derived from these nitrogen-bearing heterocycles is lacking – perhaps ow-ing to the modest results achieved by classical methods.
The 1,2,4-triazine ring system is a key component of commercial dyes, herbicides and insecticides [67]. In addi-tion, some derivatives have shown wide antibiotic and anti-tumor activities [68]. Pyrazoles have long been of pharma-cological interest as antianxiety [69], antipyretic, analgesic and antiinflammatory drugs [70] and have also shown antim-icrobial properties [71]. 1,2,3-Triazole is the essential struc-tural unit in a number of drugs and some of these materials are also potent HIV-1 inhibitors [72], antimicrobial agents [33], or selective 3-adrenergic receptor agonists [73].
Irradiation of bromoderivatives 70–73 under solvent-free conditions in the presence of NaI and a small amount of DMF (0.1 mL, the presence of a small amount of DMF is
6 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
Table 3. Cycloaddition of 2-Azadienes 25 and 26 with Nitroalkenes and Other Dienophiles Under Microwave Irradiation
Dienes
N
N
Ph OCH3
N
25
N
NN
NMe2
Ph
26 Dienophiles
NO2N
Ph
NO2
NO2
S
NO2
NO2
H3C
H3C
NO2
Ph CO2Et N CH3
O
O
CN
Cl
27
32
31302928
353433
Diene Dienophile Reaction Conditions Products Yield (%)
25 27 285 W, 7 min
140ºC N
N
Ph
N
Ph
OCH3
36
53
25 28 285 W, 10 min
140ºC
NN
Ph
N OCH3
NPh
37
32
25 29 285 W, 8 min
140ºC N
N
Ph
N OCH3
S
38
48
25 32 285 W, 15 min
140ºC NN
Ph
N
Ph
OCH3
CO2Et
39
25

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 7
(Table 3) contd…..
Diene Dienophile Reaction Conditions Products Yield (%)
26 35 150 W, 20 min
85ºC NN
Ph
N
NC
OCH3
40
20
26 27 240 W, 5 min
130ºC NN
Ph
N
PhNO2
NN
Ph
N
Ph
41 42
+
64 (41) +
8 (42)
26 28 240 W, 6 min
130ºC
NN
Ph
N
NO2
N
Ph
NN
Ph
N
N
Ph
43 44
+
60 (43) +
9 (44)
26 29 240 W, 5 min
130ºC
NN
Ph
N
NO2S
45
84
26 30 180 W, 10 min
100ºC N
N
Ph
N
46
41
26 31 90 W, 10 min
130ºC
NN
Ph
N
NO2
47
33
26 32 90 W, 10 min
120ºC
NN
Ph
N
NO2
48
55

8 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
necessary to dissolve the sodium salt) gave the correspond-ing o-quinodimethane. Subsequent cycloadditon with elec-tron-deficient dienophiles afforded the corresponding cy-cloadducts 79–93 within 15 min in 51–87% yield. The re-sults are given in Table 5. These findings clearly demon-strate that microwave irradiation provides an excellent way to induce the generation and DIELS–ALDER cycloaddition of heterocyclic o-quinodimethane derivatives [66].
The results represent a general synthetic procedure to prepare within a few minutes a wide range of heterocyclic compounds in good yields. The novelty is not the synthetic approach employed but the application of microwave heating in the development of a general synthetic procedure to pre-pare interesting heteropolycyclic products in good yields.
7. Reactions with Supports: Tandem DIELS–ALDER Aromatization Reaction of Furans
DIELS–ALDER reactions of furan and its derivatives have received a great deal of attention for two main reasons. Firstly, furan and some of its derivatives are inexpensive compounds obtained from agricultural by-products [74].
Secondly, the cycloadducts are versatile intermediates in the preparation of carbohydrates and other biologically active
compounds [75] and these have been further elaborated to substituted arenes [76].
The use of furans as dienes has encountered problems related to their sensitivity to the most common Lewis acid catalysts, the easy reversibility of their reactions, and the formation of by-products. In view of these drawbacks, a range of special reaction conditions has been developed [77].
Heterogeneous catalysts such as ZnCl2, TiCl4 and Et2AlCl supported on silica gel have shown great utility as Lewis acid catalysts. The structures of these systems are not known in great detail but it is believed that silica-supported ZnCl2 consists mainly of a dispersion of small particles of ZnCl2 on the surface of silica gel. On the other hand, treat-ment of silica gel with TiCl4 or Et2AlCl results in the dis-placement of two chloride or ethyl groups, respectively, by the silanol groups of the silica gel (Scheme 5) [78]. These catalysts can be stored and recovered after the reaction with-out loss of catalytic activity.
The reasons outlined above encouraged us to explore the joint use of silica-supported Lewis acids and microwave ac-tivation in DIELS–ALDER reactions between furan deriva-
(Table 3) contd…..
Diene Dienophile Reaction Conditions Products Yield (%)
26 34 210 W, 15 min
170ºC
NN
Ph
N
NOO
CH3
49
50
26 35 150 W, 20 min
85ºC NN
Ph
N
CN
50
39
(
Ph
NHN
R
NN
NN
Ph
NN
RH
NN
Ph
NNH
R
NN
Ph
NN
R
H
51, R = H52, R = NO2
53, R = H54, R = NO2
Scheme 4.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 9
Table 4. Cycloaddition Reactions of 4-Pyrazolyl Hydrazones 51–54 Under Microwave Irradiation
Hydrazone Dipolarophile Reaction Conditions Product Yield (%)
51
CO2Me
MeO2C
55
MW, 255 W, 155ºC, 45 min
NN
NN
Ph
CO2Me
Ph
MeO2C
57
84
51
NO2
Ph
27
MW, 135 W, 135ºC, 10 min
NN
NN
Ph
Ph
Ph
58
67
51 CO2EtPh
33
MW, 780 W, 185ºC, 15 min
NN
NN
Ph
Ph
Ph
EtO2C
59
59
51 CO2EtH
56
MW, 780 W, 170ºC, 15 min
NN
NN
Ph
Ph
EtO2C
NN
NN
Ph
Ph
CO2Et
60
61
+
22 (60) 8 (61)
52
NO2
Ph
27
MW, 135 W, 130ºC, 10 min
NN
NN
Ph
Ph
NO2
NN
NN
Ph
NO2
Ph
62
63
+
70 (62) 5 (63)
53
CO2Me
MeO2C
55
MW, 255 W, 155ºC, 45 min
NN
PhN N
MeO2CCO2Me
Ph64
56

10 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
tives and different dienophiles. The results of these reactions were compared with those obtained under conventional con-ditions.
7.1. DIELS–ALDER Reactions of Furans
Firstly, we compared the behavior of furan (94) and 2,5-dimethylfuran (95) in reactions with methyl acrylate (96) and acrylonitrile (97) catalyzed by silica and by three silica-supported Lewis acids at room temperature (Table 6) [79].
The results obtained were found to depend on the nature of both the diene and the dienophile. As reported previously [80], Zn(Si) is a better catalyst than Al(Si) or Ti(Si) for DI-ELS–ALDER reactions involving , -unsaturated nitriles such as acrylonitrile, and good yields of the corresponding cycloadducts were obtained with this catalyst. The use of this dienophile in conjunction with furan as the diene produced the endo cycloadduct (100n) as the major product, with the situation being reversed on using 2,5-dimethylfuran, which gave the exo cycloadduct (101x) as the major product. Any catalysis due to the support can be ruled out because the re-action was not promoted by silica gel (Table 6).
In the reactions carried out with methyl acrylate as the dienophile, the use of silica-supported Lewis acids resulted in good yields of the corresponding cycloadducts 98n and 98x when furan was used as the diene [81]. However, the yields obtained with this dienophile and 2,5-dimethylfuran were much lower. These results do not indicate that 2,5-dimethylfuran is necessarily less reactive than furan, rather
that the difficulty in obtaining good yields is due to competi-tion from other reactions.
The first of these competing reactions is the retro DI-ELS–ALDER reaction. The equilibrium position changes from one diene/dienophile to another. The second competing reaction, which is not observed in the case of acrylonitrile, is ring-opening of the cycloadducts followed by aromatization to yield methyl 2,5-dimethylbenzoate (102).
In an attempt to improve the results we decided to assess the effect of temperature on the reactions. However, we found that the use of conventional heating gave irreproduci-ble results. It is known that mineral oxides are poor heat conductors and that conventional methods result in inhomo-geneous heating and, in certain cases, to overheating. In or-der to avoid this problem we investigated the use of micro-wave activation in the reaction of 2,5-dimethylfuran (95) with methyl acrylate (96) and acrylonitrile (97) using a fo-cused microwave reactor under pressure and under an argon atmosphere.
In agreement with previous studies, silica gel was found to be fairly inefficient in promoting these reactions. The use of methyl acrylate (96) led to an increase in the amount of aromatic product, probably accompanied by an increase in the retro DIELS–ALDER reaction. In fact, only a small amount of cycloadduct was obtained when a Zn(Si) catalyst was used. In the reactions involving acrylonitrile (97), the corresponding aromatic product was not obtained [79].
(Table 4) contd…..
Hydrazone Dipolarophile Reaction Conditions Product Yield (%)
53
NO2
Ph
27
MW, 90 W, 130ºC, 15 min
NN
PhN N
Ph
Ph
NN
PhN N
Ph
Ph
65
66
+
45 (65) 22 (66)
53 CO2EtPh
33
MW, 240 W, 180ºC, 30 min
NN
PhN N
Ph
PhEtO2C
67
40
54
NO2
Ph
27
MW, 255 W, 130ºC, 12 min
NN
PhN N
Ph
NO2N
N
PhN N
NO2
Ph
68
69
+
33(68) 29(69)

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 11
Table 5. Cycloaddition Reactions of Bromoderivatives 70–73 Under Microwave Irradiation in Solvent-Free Conditions
NN
N CH2Br
CH2BrN
N
Ph
CH2Br
CH2Br N
N
N
Ph
CH2Br
CH2BrN
N CH2Br
CH2Br
70 71 72 73
Substrate Dienophile Temp (ºC) Product Yield (%)
70 Ph
74 90
N
N Ph
79
38
70 Ph Me
75 90
N
N Ph
Me
80
43
70
C CH
76
90 N
N Naph
81
41
70 N
9a
90 N
N
82
41
70
N
9b
90 N
N
83
33
70
NO2
Ph
27
90 N
N Ph
84
69
71 N
O
O
Me
34
90
N
NN
N
O
O
Me
85
68
71 CO2MeMeO2C
23 90
N
NN CO2Me
CO2Me
86
55
72 N
O
O
Me
34
150 NN
N
Ph
Me
O
O87
80

12 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
The differences observed between carbonyl and nitrile compounds were not easy to explain and it was necessary to carry out theoretical studies into the different reaction path-ways [79].
7.2. Synthesis of Benzene Derivatives by Ring-Opening of
DIELS–ALDER Cycloadducts of Substituted Furans
As mentioned previously, one application of the DIELS–ALDER cycloaddition of furan systems stems from the fact that the resultant oxabicycloheptene derivatives are valuable intermediates for the synthesis of arenes. These oxabicycles are easily prepared through cycloaddition reactions and, fur-thermore, the oxygen bridge can be opened by a variety of methods [82]. However, only in a few cases is it possible to obtain benzene derivatives in a single step [83] and a mixture
of compounds is usually obtained [82, 84]. It should be pointed out, however, that this kind of reaction has usually been performed in two steps using homogeneous catalysis and, under these conditions, long reactions times are required.
As described in section 7.1, we observed the formation of the aromatic compound methyl 2,5-dimethylbenzoate in the reaction between 2,5-dimethylfuran and methyl acrylate. When the reaction was carried out in a focused microwave reactor the amount of this product was much larger, showing that its formation could be favored by microwave activation. Our aim was to design an environmentally benign procedure that avoided the use of the polluting homogeneous catalysts commonly employed and, in order to achieve this, reduced reaction times were required [85].
(Table 5) contd…..
Substrate Dienophile Temp (ºC) Product Yield (%)
72
CO2MeMeO2C
23 110
NN CO2Me
CO2Me
Ph 88
51
72 N
N
EtO2C
CO2Et77
110 N
NN
N CO2Et
CO2Et
Ph 89
62
72
O
O78
115 NN
Ph
O
O90
58
73 N
O
O
Me
34
150
NN
NN
Ph
Me
O
O91
87
73
CO2MeMeO2C
23 140
NN
N CO2Me
Ph
CO2Me
92
68
73 N
N
EtO2C
CO2Et77
120 N
NNN
N CO2Et
Ph
CO2Et
93
66
OHOH
OHOH
OO
Al Cl
OO
TiCl
Cl
+ Et2AlCl + 2 C2H6
+ TiCl4+ 2 HCl
Scheme 5.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 13
The process described here is procedurally simple and employs readily available chemicals, thus making it a prom-ising and useful route to a wide variety of polysubstituted benzenes whose synthesis is difficult by other methods. The reactions were performed in hermetically sealed Teflon tubes in a domestic microwave oven in which high pressures are probably reached.
As shown in Scheme 6, furan derivatives undergo DI-ELS–ALDER reactions with dienophiles 55, 97, 34 and 105 to give 7-oxabicyclo[2.2.1]hept-2-enes as intermediates. Desorption of these intermediates would give the oxabicyclic compounds. However, under solvent free conditions this step
is not easy and the coordination of the silica-supported cata-lyst with the oxygen bridge favors ring opening, leading to aromatic products in only one step.
The results of the study into the synthesis of arenes by microwave irradiation using silica-supported catalysts are shown in Table 7. We found that furan derivatives 95, 103 and 104 react with dienophiles 96, 97, 34 and 105 under mi-crowave irradiation within 25–45 min, affording in one pot the aromatic compounds 102 and 106–112 (Table 7). The results obtained depend greatly on the reaction conditions, the nature of the Lewis acid used as a catalyst, and the level of activation of the furan ring.
Table 6. Behavior of Furan (94) and 2,5-Dimethylfuran (95) in Reactions with Methyl Acrylate (96) and Acrylonitrile (97) Cata-
lyzed by Solid Supports
O
R
R
Z
Cat.O R
R Z
R
R
O
Z
COOCH3
CH3
CH3endo exo98-101102
94, R = H95, R = CH3
+ + +
96, Z = COOCH397, Z = CN
98, R = H; Z = COOCH399, R = CH3; Z = COOCH3100, R = H; Z = CN101, R = CH3; Z = CN
Diene Dienophile Catalyst T (h) Yield % (98–101) n/xa Yield of 102 %
SiO2 24 75 (98) 86:14 -
Zn(Si) 24 75 (98) 81:19 - 94 96
Ti(Si) 24 71 (98) 73:27 -
Zn(Si) 3 78 (99) 67:33 - 94 97
Ti(Si) 24 - - -
SiO2 24 24 (100) 66:34 -
Zn(Si) 24 25 (100) 65:37 -
Al(Si) 24 32 (100) 75:25 6
Ti(Si) 2 8 (100) 62:38 -
95 96
- 24 - - 14
SiO2 3 - - -
Zn(Si) 3 79 (101) 37:63 -
Al(Si) 3 - - - 95 97
Ti(Si) 3 15 (101) 38:62 - an = endo, x = exo.
MW
Si(M)O
X
Y
R1
R2
CH
CH
O
Y
X
R2
R1
X
R1
R2
Y96, 97, 34, 105
+
95, X = Y = CH3; 103, X = OCH3, Y = H; 104, X = CH3CH2, Y = H96, R1= COOCH3, R2 = H; 97, R1 = CN, R2 = H 34, R1 = R2 = -CONCH3CO- ; 105, E-isomer R1 = R2 = CN
95, 103, 104 102, 106-112
Scheme 6.

14 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
The nature of the catalyst has, in some cases, a spectacu-lar effect on the yields. The use of Si(Ti) gives the best re-sults and the arene derivatives were obtained in good to ex-cellent yields. It is remarkable that the use of SiO2 (entry 5) does not promote any reaction at all. This result shows the importance of the catalytic activity of the metal. Moreover, microwave irradiation plays an important role in the reaction (Table 7, entry 12 vs entry 13). The use of classical heating in an oil bath under comparable reaction conditions (tem-perature and reaction time) leads to a dramatic decrease in the yield of the aromatic product.
The incorporation of an electron-donating group in posi-tion 2 of the furan has often been employed to enhance the reactivity of the heteroaromatic ring system [83]. In our case, reactions involving 2-methoxyfuran (103) exhibited a very high regioselectivity, with formation of the more crowded compounds when asymmetric dienophiles were used (entries 16 and 20). Moreover, reaction with fumaronitrile (105) af-forded 3-methoxyphthalonitrile (110) in excellent yield (en-try 28). It should be noted that the previously reported syn-thesis of phthalonitriles from furan derivatives required a two-step procedure and ring opening of the cycloadduct is usually performed in basic rather than acidic conditions [86]. On the other hand, the reaction of 2-ethylfuran (104) with N-methylmaleimide (34) demonstrated that this reaction can be successful when less reactive furan derivatives are used, al-beit in moderate yields (entry 32).
Moreover, it should be noted that treatment of the three furan derivatives with N-methylmaleimide (34) gave rise to new N-methylphthalimide derivatives 107, 110 and 112, one of which was obtained in quantitative yield (entry 12).
7.3. One-Pot Synthesis of Phenol Derivatives
Alkynes that possess at least one electron-withdrawing group represent another class of dienophiles that undergo DIELS–ALDER cycloadditions to furans. The resultant 7-oxabicyclo[2.2.1]hept-2,5-dienes are valuable synthetic in-termediates that have been further elaborated to substituted arenes [82b, 86a,c, 87]. These arene 1,4-endoxides are easily prepared through cycloaddition reactions of furans and the oxygen bridge can be removed by a variety of methods [88]. However, only in a few cases is it possible to obtain phenol derivatives in a single step [83] and a mixture of compounds is usually formed [82d, 89].
This kind of reaction is usually performed in two steps using homogeneous catalysis and, under these conditions, long reaction times are required. For this reason we found it of interest to study the reactions of furan derivatives with alkynes using heterogeneous catalysis assisted by microwave irradiation under solvent-free conditions. It was envisaged that this approach could provide a general method to obtain phenol derivatives in a single step. Our aim was to achieve a reduction in the reaction times as well as to design an envi-ronmentally benign procedure that avoids the use of the pol-luting homogeneous catalysts that are commonly employed.
Catalysts used for these reactions were again Si(Zn) [90], Si(Al) [88, 91] and Si(Ti) [88, 91]. A comparative study of the influence of the catalyst on the yield of the phenolic compounds 113–118 was carried out.
As shown in Scheme 7, furan derivatives 95, 103 and 104 undergo DIELS–ALDER reactions with dienophiles 23 and 56 to give 7-oxabicyclo[2.2.1]hept-2,5-dienes as intermedi-ates. Desorption of these intermediates would give the ox-abicyclic compounds. However, under solvent-free condi-tions this step is not easy and the coordination of the silica-supported catalyst with the oxygen bridge favors ring open-ing, leading to phenolic products in only one step [92]. Al-though this conversion can be useful, it can also be complex when other substituents are present. The reaction conditions described give rise to only one product.
This process, owing to its procedural simplicity and the use of readily available chemicals, appears to be a promising and useful route to a wide variety of polysubstituted phenolic compounds 113–118. The reactions were performed in her-metically sealed Teflon tubes in a domestic microwave oven. The internal pressures within the tubes were not measured. Reactions were performed in the absence of solvent and re-action conditions were optimized to obtain the best yield. The results of the study into the synthesis of phenol deriva-tives by microwave irradiation using silica-supported cata-lysts are shown in Table 8.
The results obtained were highly dependent on the reac-tion conditions, the nature of Lewis acid catalyst, and the activation and substitution of the furan ring. The nature of the catalyst has, in some cases, a spectacular effect on the yield. The use of Si(Zn) gives the best results and the pheno-lic derivatives 113–118 were obtained in moderate to good yields (Table 8, entry 1 vs entry 3 and entry 13 vs entry 15). These results can be explained in terms of the different acid characteristics of the catalyst – Si(Zn) is a pure Lewis acid but Si(Al) and Si(Ti) have Lewis/Brønsted acid area ratios [93] of 5.3 and 4.4, respectively. Moreover, in some cases the use of classical heating in an oil bath under comparable reaction conditions (temperature and reaction time) leads to a dramatic decrease in the yield of the phenolic product (Table 8, entry 3 vs entry 4).
A rearrangement reaction does not necessarily take place when monosubstituted furans are used in the aromatization of the adduct, a situation that explains why only in reactions involving 2,5-dimethylfuran (95) was the corresponding ad-duct detected. Thus, reactions involving 2-ethylfuran (104) exhibited a very high regioselectivity, with the formation of 2,4-disubstituted phenol derivatives when methyl propiolate (56) was used as an asymmetric dienophile (entry 15). Moreover, the reaction of 104 with DMAD (23) and methyl propiolate (56) demonstrated that these reactions can be suc-cessful when less reactive furan derivatives, such as 2-ethylfuran (104), are used (entry 11 and 15).
In conclusion, the synthesis of a range of aromatic de-rivatives has been achieved using heterogeneous catalysis under microwave irradiation [79, 85, 92]. The products are easily prepared in a single step by cycloaddition of furans followed by ring opening of the oxabicyclo intermediate through the action of Si(Ti) or Si(Zn). A variety of benzene and phenol derivatives can be synthesised by selecting the appropriate reactants. This method represents a simple, gen-eral and useful alternative for the preparation of some poly-substituted benzene derivatives whose synthesis is difficult by other methods.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 15
Table 7. One-Pot Synthesis of Arenes 102 and 106–112 Catalyzed by Silica-Supported Lewis Acids Using Microwave Irradiation
or Classical Methods
Reaction Conditions
Entry Diene Dienophile Catalyst
M.W.a C.H.
b
Product Yield (%)
1 Si(Zn) 45 min 23
2 Si(Al) 45 min 50
3 Si(Ti) 45 min 50
4
CN 97 Si(Ti) 45 min
CH3
CH3
CN
106
7
5 SiO2 45 min 0
6 Si(Zn) 45 min 25
7 Si(Al) 45 min 65
8 Si(Ti) 45 min 92
9
COOCH3 96
Si(Ti) 45 min
CH3
CH3
COOCH3
102
10
10 Si(Zn) 45 min 50
11 Si(Al) 45 min 83
12 Si(Ti) 45 min 100
13
O
CH3
CH3 95
N
O
CH3
O
34
Si(Ti) 45 min
N
O
O
CH3
CH3
CH3 107
14
14 Si(Zn) 25 min 13
15 Si(Al) 25 min 54
16 Si(Ti) 25 min 60
17
CN 97 Si(Ti) 25 min
OCH3
CN
98
100 29
18 Si(Zn) 25 min <3
19 Si(Al) 25 min 30c
20 Si(Ti) 25 min 62d
21
COOCH3 96 Si(Ti) 25 min
OCH3
COOCH3
109
13c
22 Si(Zn) 25 min 34
23 Si(Al) 25 min 55
24 Si(Ti) 25 min 88
25
N
O
CH3
O
34
Si(Ti) 25 min
N
O
O
CH3
OCH3
110
31
26 Si(Zn) 25 min <3
27 Si(Al) 25 min 9
28 Si(Ti) 25 min 88
29
O
OCH3
103
NC
CN 105 Si(Ti) 25 min
OCH3
CN
CN 111
<3
30 Si(Zn) 45 min 28
31 Si(Al) 45 min 29
32 Si(Ti) 45 min 45
33
O
CH2CH3
104
N
O
CH3
O
34
Si(Ti) 45 min
N
O
O
CH3
CH3CH2
112 19
aMicrowave Irradiation: All reactions were carried out at 780 W. bConventional Heating at 120ºC. The reaction temperature was determined at the end of a blank reaction using microwave irradiation. cIn addition to this product, 10% of methyl 3-methoxybenzoate was obtained. dIn addition to this product, trace amounts of methyl 3-methoxybenzoate were detected.

16 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
O
Y
X R1
R2
MWO X
Y
R1
R2Si(M)
OH
R1X
R2
Y
+
95, 103, 104 23, 56 113-118
95, X = Y = CH3; 103, X = H, Y = OCH3; 104, X = CH2CH3, Y = H23, R1 = R2 = COOCH3; 56, R1 = COOCH3, R2 = H
Scheme 7.
Table 8. One-Pot Synthesis of Phenols 113–118 Catalyzed by Silica-Supported Lewis Acids Using Microwave Irradiation or Clas-
sical Methods
Reaction Conditionsa
Entry Diene Dienophile Catalyst M.W.
b C.H.
c
Product Yield (%)
1 Si(Ti) 30 min 33 (22)d
2 Si(Al) 30 min 25 (32)d
3 Si(Zn) 30 min 65
4
COOCH3
COOCH3
23
Si(Zn) 30 min
90ºC
OH
H3C COOCH3
COOCH3
CH3113
18
5 Si(Ti) 30 min 11
6 Si(Al) 30 min 18
7 Si(Zn) 30 min 22
8
O
CH3
CH395
COOCH3
56
Si(Zn) 30 min 90ºC
OH
CH3
COOCH3H3C
114
21
9 Si(Ti) 30 min 46
10 Si(Al) 30 min 53
11 Si(Zn) 30 min 66
12
COOCH3
COOCH3
23
Si(Zn) 30 min 105ºC
OH
COOCH3
COOCH3
CH2CH3115
68
13 Si(Ti) 30 min 10
14 Si(Al) 30 min 17
15 Si(Zn) 30 min 56
16
O
CH2CH3
104 COOCH3
56
Si(Zn) 30 min 100ºC
OH
COOCH3
CH2CH3
116
28
17 Si(Ti) 30 min 65
18 Si(Al) 30 min 27
19 Si(Zn) 30 min 81
20
COOCH3
COOCH3
23
Si(Zn) 30 min 100ºC
OH
OCH3
COOCH3
COOCH3117
87
Si(Ti) 30 min 35 (15)e
Si(Al) 30 min 34 (24)e
Si(Zn) 30 min 25 (19)e
21
22
23
24
O
OCH3
103 COOCH3
56
Si(Zn) 30 min 100ºC
OH
OCH3
COOCH3
118
26 (10)e
aIn all reactions a molar ratio furan derivative:dienophile of 6:1 and 0.5 g of silica-supported Lewis acid catalyst were used. bMicrowave Irradiation. All reactions were carried out at 450 W. cConventional Heating. Reaction temperature was determined at the end of a blank reaction using microwave irradiation. dIn addition the arene 1,4-endoxide was obtained (yield in parentheses). eIn addition to the main product its corresponding methoxy derivative (methyl 2,5-dimethoxybenzoate) was obtained.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 17
B. SYNTHESIS OF AZOLYLTRIAZINES AND BISA-
ZOLES WITH APPLICATIONS IN SUPRAMOLECU-
LAR CHEMISTRY
Melamine derivatives have usually been synthesized from 2,4,6-trichloro-1,3,5-triazine (119) [94] using a nucleo-philic amine. The substitution of chlorine can be controlled by temperature to run in a stepwise manner. Preparation of trisubstituted derivatives usually required stronger reaction conditions, i.e. higher temperatures for long reaction times, especially with sterically hindered amines. However, the use of microwave irradiation in solvent-free conditions provides a mixture of 2,4-dichloro-6-(2-pyrazol-1-ylphenylamino)-1,3,5-triazine (121) and 2,4,6-tris(2-pyrazol-1-ylphenyl-amino)-1,3,5-triazine (122) in only 10 min [95]. Reactions were performed in a focused microwave reactor with full control of the incident power and reaction temperature (Scheme 8).
On increasing the reaction temperature to 185ºC, deriva-tives 124 with symmetrical substitution were prepared exclu-sively [96] by reaction of cyanuric chloride with the appro-priate amine, using microwave irradiation in solvent-free conditions in only 10 min with moderate yields (42–62%) (Scheme 9). Six equivalents of the amine were required in order to neutralise the hydrogen chloride produced in the reaction.
Similarly, derivatives with asymmetrical substitution patterns [96] were prepared in excellent yield by reaction of
2-chloro-4,6-bis(4-pyrazol-1-ylphenylamino)-1,3,5-triazine (125) with two equivalents of the corresponding amine 123. Reactions were also performed in a focused microwave reac-tor (Scheme 10).
It is remarkable that no reaction occurs by conventional heating in comparable reaction conditions (temperature and time). In order to obtain similar yields by conventional heat-ing, reactions should be performed in THF at reflux for 5 days in the presence of DIPEA as a base. However, even under these conditions the formation of hindered compounds such as the tris-o-pyrazolylphenyl-1,3,5-triazine derivative
(122) and asymmetrically substituted triazines did not take place under conventional heating.
Under these conditions, reaction of 2-chloro-4,6-bis(4-pyrazol-1-ylphenylamino)-1,3,5-triazine (125) [96] with a polymer-supported benzylamine [aminomethylated poly(styrene-co-divinylbenzene) (127)] did not afford the desired trisubstituted triazine 128 because in this case the mixture is not sufficiently polar to be heated to the required temperature under microwave irradiation. However, on addi-tion of a small amount of a polar solvent such as DMSO (1 mL/mmol of triazine) the reaction temperature rose to 130ºC and produced complete conversion within 10 min (Scheme 11). This result opens the possibility of using these polymer supported pyrazolyltriazines in supramolecular polymer sup-ported synthesis, thus taking advantage of the possible inter-actions through hydrogen bonding and/or coordination with transition metals.
N
N
N
Cl
ClCl
NH2
N
NN
N
N
Cl
NCl N
N
HN
N
N
N
NN
N
N
N
H
H
N
N HN
+MW, 90W, 140˚C
10 min
119121
122
+
120
Scheme 8.
N
N
N
Cl
ClCl
N
N
N
N
NN
R
R
R
H
H
H
NN
N
N
NN
NN
NNR:
+ R-NH2
MW, 90 W, 10 min
185˚C
119 124123
57% 62% 50% 51% 42%
Scheme 9.

18 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
2,4-Diamino-1,3,5-triazines 131 [97] have been prepared by reaction of dicyandiamide (130) with nitriles (129) under microwave irradiation, a method that can be considered as a green procedure due to the reduction in the use of solvents during synthesis and purification, the short reaction time and the simplicity of the procedure (Table 9). Even phthalonitrile (132) produced the bistriazine 133 in good yield in only 10 min (Scheme 12).
We also described the preparation of 1,4'-bipyrazolyls 136, 140 and 141 as well as 4-pyrazolylpyrimidines 135, 137, 139 and 142 [98] by reaction of 2-pyrazolyl-3-dimethylamino acrylate (134) and acrylonitrile (138) with double nucleophilic reagents such as hydrazines, urea and guanidine (Schemes 13 and 14). Reactions were performed under microwave irradiation in 5–60 min. This is a useful procedure for the preparation of valuable compounds with applications in medicinal and coordination chemistry.
Moreover, a series of 2-imidazolines 144, 145 and 147 and imidazoles 148 [99] has been synthesized using micro-wave irradiation. Reactions were performed under solvent-
free conditions by cyclization of nitriles 129, 132 and 146 with ethylenediamine (143) at 30 W for 3–30 min. The crude product was washed with cold water and, in most cases, the pure product crystallized. Good to excellent yields (Table 10) of the corresponding imidazolines were obtained using this method.
Aromatization of the imidazolines was performed in toluene in the presence of Magtrieve™, a material designed to moderate the absorption of microwave energy. This is an environmentally friendly oxidant that can be removed mag-netically and recycled (Table 11).
C. REPRODUCIBILITY AND SCALABILITY
One of the reasons for the increased interest in the use of microwave heating was the introduction, at the dawn of the 21st Century, of dedicated monomode and multimode in-struments with appropriate temperature and pressure con-trols, an advance that allows reproducibility of results. How-ever, when the microwave methodology was introduced twenty years ago, most reactions were performed in domes-
N
N
N
Cl
NNH H
N NN N
NN
N
N
N
N
N
NN
R1
H
H
H
N
N
N NN
+ R1-NH2
MW, 90 W, 10 min
125-140˚C
67% 92%
R1 =
125 126
123
Scheme 10.
NH2
N N
N NH
Cl
NN
HN
NN
N N
N NH
NN
HN
NN
NH
+MW, 90 W, 10 min
130˚C, DMSO
95 %
127
125128
Scheme 11.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 19
tic ovens without appropriate temperature control. Even to-day there are several reports that describe the use of these instruments in organic synthesis.
Domestic microwave ovens have also been utilised in combinatorial chemistry, because they offer the possibility of performing multiple reactions in one irradiation experiment. For this reason, different parallel arrays have been performed
in these instruments, allowing faster analog synthesis com-pared with sequential monomode instruments [100].
A plethora of interesting organic transformations have been carried out in domestic ovens. However, reactions per-formed in this kind of instrument, without appropriate tem-perature and pressure control, are generally considered as not
Table 9. Reaction Conditions and Yields for the Preparation of 2,4-Diamino-1,3,5-Triazines 131
R CN H2N
NH
NH
CN MW
KOH, DMSON N
NH2N NH2
R
+
130131
129
R Power/W Time/Min. T/ºC Yield/(%)
60 60
10 10
195 190
85a 91b
OCH3
90 90
15 15
220 220
74a 77b
NO2
90 90
15 15
190 175
74a 85b
Cl
90 90
10 10
190 180
82a 88b
NN
90 10 190 52a
N
N
90 90
10 10
185 185
96a 93b
NN
90 90
10 10
175 175
83a 99b
N
90 90
10 10
205 190
83a 88b
N
90 90
10 10
180 180
71a 88b
N O
90 90
10 10
180 175
71a 73b
aDMSO, 1 mL/15 mmol. b DMSO 3 mL/15 mmol.

20 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
CN
CN
H2N
NH
NH
CN MW
KOH, DMSO
N
N
N
NH2
NH2
N
N N
NH2
NH2
+
80%
132 130
133
Scheme 12.
NN
Br
HN NH
O
O
H2N
H2NO
CO2Et
NN
Br
Me2N
SH2N
H2N
N
Br
O
N
HN NH. HCl
NN
Br
HN NH
O
S
15 W, 60 minMesitylene
15 W, 20 minMesitylene
1) NH2NH2. H2O10 W, 3 minEthanol2) HCl
58% 70%
92%
134
135
136
137
Scheme 13.
NN
Br
N N
NH2
NH2
H2N
H2NNH
NN
Br
CNMe2N
H2N
H2NS
NN
Br
HN N
NH2
N
Br
N
N N
NH2
Ph
.HCl
Br
HN N
NN
NH2
S
15 W, 20 minEthanol
15 W, 20 minMesitylene
NH2NH2. H2O10 W, 4 minEthanol
38%
56%
69%
PhNHNH2.HCl10 W, 15 minEthanol
67%
138
139
140141
142
Scheme 14.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 21
reproducible – a situation that limits the application of such reactions [3f, 14d]. Recently, several reports disclosed the reproducibility of results between monomode and multimode microwave instruments for solution chemistry [101, 102]. However, results have yet to be described for solvent-free reactions, probably because these reactions are much more
sensitive to the reaction conditions and, in particular under microwave irradiation, to the power density. This approach broadens the application of microwave chemistry towards scale up or parallel chemistry and allows reactions to be per-formed in well plates in a fully reproducible manner, com-bining the rate enhancements and higher product yields asso-
Table 10. Reaction Conditions and Yields for the Preparation of Imidazolines 144, 145 and 147
R CN NH2CH2CH2NH2
HN
NR
S, MW
30 W 15-30 min129 143 144
+
CN
CN
NH2CH2CH2NH2
N
HN
N
HN
+ S, MW
30 W 5 min
132143
145
CN
CNN
NH
NH2CH2CH2NH2
N
HN
N
HN
N
NH
+ S, MW
30 W 5 min
146
143
147
R Power/W Time/Min. T/ºC Yield/(%)
N NPh
30 15 110 74
N
NPh
30 30 110 98
NN
30 30 100 98
N
N
30 --
30 30
100 100
82 68a
NN
30 15 110 91
132 30 30
5 3
80 80
42 18b
146
30 30 30
5 15 25
95 95 95
68 89 95
aConventional heating in an oil bath. bTraces of impurities.

22 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
ciated with the use microwave technology with an increase in productivity by utilising parallel synthesis techniques [102]. In this respect, methodologies for the successful trans-lation of conditions from domestic ovens to microwave reac-tors are of great importance in order to update such reactions and make them more reproducible and applicable to combi-natorial chemistry.
We have shown for the first time that solvent-free reac-tions performed in domestic ovens, without appropriate tem-perature and pressure controls, can be reproduced, scaled-up and parallelized in controlled microwave monomode and multimode reactors [103].
In order to achieve this goal, the 1,3-dipolar cycloaddi-tion of nitrile N-oxides with nitriles to give 1,2,4-oxadiazoles was used as a model [104]. Although reproducible results were obtained for this reaction on working with the same domestic oven on the same scale, this was not the case when the same reaction was performed, under the same conditions, in a different oven. This represents a typical example of the lack of reproducibility due to lack of control.
The first step was to study the reproducibility of the reac-tion under non-optimized conditions, because under such conditions reactions are more sensitive to temperature differ-ences. Four identical reactions were performed in parallel in the multimode reactor at 100ºC on a scale five times larger than the one used in the monomode instrument. The results show that there is good reproducibility between the two sys-tems for this solvent-free reaction as the results correlate with the calibration curve obtained in the monomode unit (Fig. 1).
The second step in this study concerned the scalability of the reaction. For this purpose eight different nitriles 129a–h, including those possessing electron-donating or electron-withdrawing groups at different positions as well as com-pounds containing heterocycles, were reacted with 2,4,6-trimethylbenzonitrile oxide 149 at 135ºC for 8 min using conditions optimized for p-nitrobenzonitrile 129a. Reactions were performed in both monomode and multimode micro-wave ovens. Note that the scale used in the latter instrument was again five times larger than that used in the monomode apparatus.
Table 11. Aromatization of Imidazolines 144 and 145 with Magtrieve™ in Toluene Under Reflux
HN
NR
MWHN
NR
MagtrieveTM
144 148
R T/ºC T/Min. Power/W Yield (%)
N NPh
100 105 120 77
105 105 120 64 N
NPh
110 105 a 16
95 75 270 88
NN
110 75 a 56
N
N
95 75 270 70
NN
100 105 270 77
120 105 120 20
110 105 180 2 145
110 24 h a 65 aConventional heating in an oil bath.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 23
N
O
O2N
N
MWN
ON
R
+
149 129a
150a
Monomode reactor Multimode oven
Fig. (1). Reproducibility of the reaction of nitrile N-oxide 149 with nitrile 129a under non-optimized conditions.
It can be seen in all the examples presented in Table 12 that successful scale-up is possible for this class of reaction on using temperature-controlled conditions. On average, the difference in yield between the two instruments is only 2.2%. The maximum difference in yield is 3%. These results are consistent with those previously reported for solution reactions [102].
Table 12. Reactions of Nitrile N-Oxide 149 with Nitriles 129a–
h
NO
R C N
MW N
ON
R
+
135 ˚ C
8 min
149 129a-h 150a-h
Nitrile R Yield (%)
Monomodea
Yield (%)
Multimodeb
129a p-NO2-C6H4 80 78
129b p-F-C6H4 70 67
129c m-F-C6H4 94 97
129d o-F-C6H4 92 91
129e p-CH3O-C6H4 83 81
129f C6H5 81 78
129g 4-pyridyl 79 76
129h 3-pyridyl 83 83 aReactions performed on a 2 mmol scale. b Reactions performed on a 10 mmol scale. In all cases yields were determined by HPLC by integration of peak areas using the pure products as standards.
However, in solvent-free microwave-assisted reactions, the heating profiles of samples differed from one another and, as a consequence, reaction conditions are very different
from one sample to another and the yields differ markedly. For this reason, the parallel scale up approach was not feasi-ble. Products were prepared in separate experiments as the absorption of each reaction needed to be monitored (Table 12). The polarity of the solvent is the most important pa-rameter to consider when microwave reactions are performed in solution. Polar solvents directly absorb the microwave radiation and the polarity of the substrate is relatively unim-portant. In apolar solvents, however, microwave radiation is absorbed by the substrates, so the nature of the substituents has a significant influence on the absorption of microwave radiation. However, in this case the solvent moderates the reaction temperature. In neat reactions, microwave radiation is again absorbed by the substrates but there is no solvent to stabilise the temperature. In this situation the nature of the substituent and the polarity of the substrate influence the absorption of microwave energy and, as a consequence, the yield [4c].
The third step was to study the preparation of 24 different compounds in a multiwell plate under microwave irradiation with the aim of achieving an effective combination of pro-ductivity and speed. In an effort to overcome the problem of the different absorption in each reaction – due to the differ-ent polarity induced by the substituents – we took advantage of the Weflon® well plate described by Nüchter and On-druschka [105]. We speculated that the use of this well plate might minimize the individual absorptions of each reaction as microwave irradiation, within the multimode oven, is mostly absorbed by the Weflon® well plate itself. In this way the temperature throughout the plate would be more uniform and the results more reproducible. The 24-well plate was heated using optimized conditions described previously (135ºC for 8 min). All reactions were performed on a 1 mmol scale.
The results of this experiment are displayed in Table 13 and clearly demonstrate that the microwave parallel ap-proach is also applicable to solvent-free reaction conditions, provided that a uniform distribution of temperature is ob-tained. In all cases good to excellent yields were obtained, with isolated yields ranging from 69% to 94%. There does not appear to be an obvious trend between the yield obtained and the electron density or substitution pattern of the sys-tems, with the temperature being the most important parame-ter. These results open new possibilities in parallel reactions performed under microwave irradiation, as almost half of the reactions described under microwave irradiation are per-formed in solvent-free conditions.
The results show that a solvent-free reaction previously performed in a domestic oven without appropriate tempera-ture control can be reproduced in both monomode and mul-timode microwave instruments. This information allows the reaction to be scaled up or performed in a parallel manner using a multi-well plate. The parallel approach requires the use of a Weflon® multi-well plate to ensure that the absorp-tion in individual vessels across the plate is minimized in order to avoid temperature differences. The results obtained in all applications are comparable. The procedure described here opens up new possibilities to exploit the many useful reactions previously carried out in household microwave ovens and makes them more useful for organic and combina-torial chemists.
50
60
70
80
90
60 80 100 120 140
Temperature (ºC)
Yie
ld (
%)

24 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
ACKNOWLEDGEMENTS
Financial support from the DGICYT of Spain through project CTQ2007-60037/BQU and from the Consejería de Ciencia y Tecnología JCCM through projects PAI-05-019 and PBI-06-0020 is gratefully acknowledged.
REFERENCES
[1] (a) Mingos, D.M.P.; Whittaker, A.G. Microwave Dielectric Heat-
ing Effects in Chemical Synthesis, in Chemistry under Extreme or non Classical Conditions, R. van Eldik and C.D. Hubbard, Eds.; John Wiley & Sons: New York, 1997, pp. 479–545. (b) Gabriel, C.; Gabriel, S.; Grant, E.H.; Halstead B.S.J.; Mingos, D.M.P. Chem.
Soc. Rev., 1998, 27, 213. (c) Loupy, A. Ed.; Microwaves in Or-ganic Synthesis, Wiley-VCH: Weinheim, 2002. 2nd Ed., 2006. (d) Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light, CEM Publishing: Matthews, NC, 2002. (d) Varma, R.S. Microwave
Technology—Chemical Synthesis Applications, in Kirk-Othmer Encyclopedia of Chemical Technology, 5th Ed., John Wiley & Sons, Inc.: New York, 2006.
[2] ISI Web of KnowledgeTM, Thompson Scientific, 2007. [3] (a) Bose, A.K.; Manhas, M.J.; Banik B.K.; Robb, E.W. Res. Chem.
Intermed., 1994, 20, 1. (b) Strauss C.R.; Trainor, R.W. Aust. J.
Chem., 1995, 48, 1665. (c) Caddick, S. Tetrahedron, 1995, 38, 10403. (d) Galema, S.A. Chem. Soc. Rev., 1997, 26, 233. (e) Lid-ström, P.; Tierney, J.; Wathey, B.; Westman, J. Tetrahedron, 2001, 57, 9225. (f) Nüchter, M.; Müller, U.; Ondruschka, B.; Tied A.; Lautenschlager, W. Chem. Eng. Technol., 2003, 26, 1207. (g) Kap-pe, C.O. Angew. Chem., Int. Ed., 2004, 43, 621. (h) Lidström P.; Tierney, J.P. Eds.; Microwave-Assisted Organic Synthesis, Black-well Scientific: Oxford, 2004, 320. (i) Kappe, C.O.; Stadler, A. Mi-
crowaves in Organic and Medicinal Chemistry, Wiley-VCH: Weinheim, 2005. (j) Bogdal, D. Microwave-Assisted Organic Syn-
thesis: One Hundred Reaction Procedures, in Tetrahedron Organic Chemistry Vol 25, Elsevier, 2006.
[4] (a) Loupy, A.; Petit, A.; Hamelin, J.; Texier-Boullet, F.; Jacquault, P.; Mathé, D. Synthesis, 1999, 1213. (b) Varma, R.S. Tetrahedron, 2002, 58, 1235. (c) Loupy, A. Solvent-free Reactions, in Modern
Solvents in Organic Chemistry, Knochel, P. Ed. Springer-Verlag: Heidelberg, 1999, p 155.
[5] Díaz-Ortiz, A.; Langa, F.; de la Hoz, A.; Moreno, A. Eur. J. Org.
Chem., 2000, 4, 3659. [6] Elander, N.; Jones, J.R.; Lu, S. Y.; Stone-Elander, S. Chem. Soc.
Rev., 2000, 29, 239. [7] (a) Langa, F.; de la Cruz, P.; Espíldora, E.; García, J.J.; Pérez,
M.C.; de la Hoz, A. Carbon, 2000, 38, 1641. (b) Langa, F.; de la Cruz, P.; Espíldora E.; de la Hoz, A. Applications of Microwave Ir-
radiation to Fullerene Chemistry, in Fullerenes, The Electrochemi-cal Society: New York, 2000, vol. 9, pp. 168–178.
[8] (a) Zong, L.; Zhou, S.; Sgriccia, N.; Hawley M.C.; Kempel, L.C. J. Microwave Power Electromagn. Energy, 2003, 38, 49. (b) Zhang, C.; Liao, L.; Gong, S. Green Chem., 2007, 9, 303. (c) Bogdal, D.; Prociak, A. Chemistry Today, 2007, 25, 30.
[9] (a) Xu, Y.; Guo, Q.-X. Heterocycles, 2004, 63, 903. (b) Romanova, N.N.; Kudan, P.V.; Gravis, A.G.; Bundel, Y.G. Chem. Heterocycl.
Compd., 2000, 36, 1130. (c) Ashry, E.S.H.E.; Ramadan, E.; Kas-sem, A.A.; Hagar, M. Adv. Heterocyl. Chem., 2005, 88, 1. (d) Mol-teni, V.; Ellis, D.A. Current Org. Synth., 2005, 2, 333. (e) Micro-wave-Assisted Synthesis of Heterocycles, in Topics in Heterocyclic
Chemistry, Van der Eycken, E.; Kappe C. O. Eds.; Springer: Hei-delberg, 2006.
[10] (a) Das, S.K. Synlett, 2004, 915. (b) Corsaro, A.; Chiacchio, U.; Pistara V.; Romeo, G. Curr. Org. Chem., 2004, 8, 511.
[11] Larhed, M.; Moberg, C.; Hallberg, A. Acc. Chem. Res., 2002, 35, 717.
[12] Will, H.; Scholz, P.; Ondruschka, B.; Chem.-Ing.-Tech., 2002, 74, 1057.
[13] (a) Lahred, M.; Hallberg, A. Drug Discov. Today, 2001, 6, 406. (b) Kappe, C.O. Comb. Chem., 2002, 6, 314. (c) Kappe, C.O. Curr.
Opin. Chem. Biol., 2002, 6, 314. (d) Lew, A.; Krutzik, P.O.; Hart M.E.; Chamberlin, A.R. J. Comb. Chem., 2002, 4, 95. (e) Wathey, B.; Tierney, J.; Lidström P.; Westman, J. Drug Discovery Today, 2002, 7, 373. (f) Blackwell, H.E. Org. Biomol. Chem., 2003, 1, 1251. (g) Santagada, V.; Perissutti, E.; Caliendo, G. Current. Med. Chem., 2002, 9, 1251. (h) Santagada, V.; Frencentese, F.; Perissut-ti, E.; Favretto, L.; Caliendo, G. QSAR Comb. Sci., 2004, 239, 919. (i) Martínez-Palou, R. Mol. Div., 2006, 10, 435. (j) Dallinger, D.; Kappe, C.O. Nat. Rev. Drug Discov., 2006, 51.
Table 13. Parallel Reaction of Nitrile N-Oxide 149 with Nitriles 129a–x in a 24-Well Weflon®
Plate
NO
R C N
MW N
ON
R
+
135 ˚ C
8 min149 129a-x 150a-x
Nitrile R Yield (%)a
Nitrile R Yield (%)a
129a p-NO2-C6H4 78 (73) 129m o-Cl-C6H4 79 (75)
129b p-F-C6H4 72 (69) 129n 6-CH3O-2-naphthyl 80 (75)
129c m-F-C6H4 91 (84) 129o m-CH3O-C6H4 84 (79)
129d o-F-C6H4 89 (83) 129p o-CH3-C6H4 92 (88)
129e p-CH3O-C6H4 80 (73) 129q o-CH3O-C6H4 81 (76)
129f C6H5 78 (74) 129r 2-F-5-CF3-C6H3 94 (89)
129g 4-pyridyl 79 (73) 129s 4-F-3-CF3-C6H3 94 (90)
129h 3-pyridyl 78 (76) 129t m-CH3-C6H4 81 (77)
129i 3,4-methylendioxyC6H3 84 (79) 129u 3-F-2-CF3-C6H3 84 (80)
129j 4-phenoxyC6H4 86 (82) 129v p-Cl-C6H4 77 (73)
129k 2,5-F-C6H3 89 (84) 129w 4-Cl-3-NO2-C6H3 74 (69)
129l 2,4-F-C6H3 88 (83) 129x p- CH3-C6H4 88 (84) aYields determined by HPLC measurement of peak areas using the pure products as standards. Isolated yields in brackets.

Microwave-Assisted Reactions in Heterocyclic Compounds Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 25
[14] (a) Varma, R. S. Clean Products and Processes, 1999, 132. (b) Varma, R. S. in Advances in Green Chemistry: Chemical Syntheses Using Microwave Irradiation, AstraZeneca Research Foundation, Kavitha Printers: Bangalore, India, 2002. (c) Bose, A.K.; Manhas, M.S.; Ganguly, S.N.; Sharma A.H.; Banik, B.K. Synthesis, 2002, 1578. (d) Nüchter, M.; Ondruschka, B.; Bonrath, W.; Gum, A. Green Chem., 2004, 6, 128.
[15] Kaiser, N.F.K.; Bremberg, U.; Larhed, M.; Moberg C.; Hallberg, A. Angew. Chem., Int. Ed., 2000, 39, 3595.
[16] (a) Perreux, L.; Loupy, A. Tetrahedron, 2001, 57, 9199. (b) Díaz-Ortiz, A.; de la Hoz, A.; Moreno, A. Chem. Soc. Rev., 2005, 157.
[17] de la Hoz, A.; Díaz-Ortiz, A.; Moreno, A. Curr. Org. Chem., 2004, 8, 903.
[18] (a) Huisgen, R.; Grosley, R.; Seild, H.; Hauck, H. Chem. Ber., 1968, 101, 2559. (b) Keirs, D.; Moffat, D.; Overton, K.; Tomamek, R. J. Chem. Soc., Perkin Trans 1, 1991, 1041.
[19] (a) Bérlanger, A.; Brassard, P. J. Chem. Soc. Chem. Commun., 1972. 863. (b) Posner, G.H.; Harrison, W. J. Chem. Soc., Chem. Commun., 1985, 1785. (c) Gruseck, J.; Heuschmann, M. Chem.
Ber., 1990, 123, 1905. [20] Díaz-Ortiz, A.; Díez-Barra, E.; de la Hoz, A.; Prieto. P.; Moreno,
A. J. Chem. Soc., Perkin Trans 1, 1994, 3595. [21] Díaz Ortíz, A.; Díez-Barra, E.; de la Hoz, A.; Prieto, P.; Moreno,
A.; Langa, F.; Prangé, T.; Neuman, A. J. Org. Chem., 1995, 60, 4160.
[22] (a) Takeuchi, Y.; Furusaki. F. Adv. Heterocycl. Chem., 1977, 21, 207. (b) Confalone, P.N.; Huie, E. M. Org. React., 1988, 36, 1.
[23] Cole, D.C. Tetrahedron, 1994, 32, 9517. [24] Boger, D.L. Tetrahedron, 1983, 39, 2869. [25] (a) Sugita, T.; Koyama, J.; Tagahara, K.; Suzuta, Y. Heterocycles,
1985, 23, 2789. (b) Okatani, T.; Koyama, J.; Suzuta, Y.; Tagahara, K. Heterocycles, 1988, 27, 2213. (c) Itoh, T.; Okada, M.; Nagata, K.; Yamaguchi, K.; Ohsawa, A. Chem. Pharm. Bull., 1990, 38, 2108. (d) Koyama, J.; Ogura, T.; Tagahara, K. Heterocycles, 1994, 38, 1595.
[26] (a) Sugita, T.; Koyama, J.; Tagahara, K.; Suzuta, Y. Heterocycles, 1986, 24, 29. (b) Okatani, T.; Koyama, J.; Tagahara, K.; Suzuta, Y. Heterocycles, 1987, 26, 595.
[27] Okatani, T.; Koyama, J.; Tagahara, K. Heterocycles, 1989, 29, 1809.
[28] Itoh, T.; Ohsawa, A.; Okada, M.; Kaihoh, T.; Igeta, H. Chem.
Pharm. Bull., 1985, 33, 3050. [29] (a) Langa, F.; de la Cruz, P.; de la Hoz, A.; Díaz-Ortiz, A.; Díez-
Barra, E. Contemporary Org. Synth., 1997, 4, 373. (b) Varma, R. S. Green Chem., 1999, 1, 43. (c) Strauss, C. R. Aust. J. Chem., 1999, 52, 83.
[30] (a) Díaz-Ortiz, A.; de la Hoz, A.; Prieto, P.; Carrillo, J.R.; Moreno, A. Synlett, 2001, 236. (b) Prieto, P.; Cossio, F.P.; Carrillo, J.R.; de la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. J. Chem. Soc., Perkin
Trans. 2, 2002, 1257. [31] The formation of enamines by reaction of carbonyl compounds and
secondary amines on montmorillonite K10 under microwave irra-diation has been reported previously: Varma, R.S.; Dahiya, R.; Kumar, S. Tetrahedron Lett., 1997, 38, 2039.
[32] Ölgen, S.; Chung, K. C.Z. Naturforsch., B: Chem. Sci., 2001, 56, 804.
[33] Genin, M.J.; Alwine, D.A.; Anderson, D.J.; Barbachyn, M.R.; Emmert, D.E.; Garmon, S.A.; Graber, D.R.; Grega, K.C.; Hester, J.B.; Hutchison, D.K.; Morris, J.; Reischer, R.J.; Ford, C.W.; Zu-renko, G.E.; Hamel, J.C.; Schaadt, R.D.; Staper, D.; Yagi, B.H. J. Med. Chem., 2000, 43, 953.
[34] Brockunier, L.L.; Parmee, E.R.; Ok, H.O.; Candelore, M.R.; Cas-cieri, M.A.; Colwell, L.F.; Deng, L.; Feeney, W.P.; Forrest, M.J.; Hom, G.J.; MacIntyre, D.E.; Tota, L.; Wyvratt, M.J.; Fisher, M.H.; Weber, A.E. Bioorg. Med. Chem. Lett., 2000, 10, 2111.
[35] (a) Huisgen, R. Proc. Chem. Soc., 1961, 357. (b) Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., 2002, 114, 2708. (c) Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem., 2005, 70, 6526.
[36] Diaz-Ortiz, A.; de Cozar, A.; Prieto, P.; de la Hoz, A.; Moreno, A. Tetrahedron Lett., 2006, 47, 8761.
[37] Hoehn, H.; Denzel, T. U.S. Patent 3755340, 1973. [38] Meiners, B.A.; Salama, A. I. Eur. J. Pharmacol., 1982, 78, 315. [39] Lynck, B.; Khan, M.; Teo, H.; Pedrotti, F. Can. J. Chem., 1988, 66,
420.
[40] El-Dean, A.M.; Aralla, A.A.; Mohamed, T.A.; Geies, A.A.Z.
Naturfosch., Teil B, 1991, 46, 541. [41] Fujikama, Y.; Suzuki, M.; Iwasaki, H.; Sakashita, M.; Kitahara, M.
Eur. Pat. Appl., EP 339,358, 1989. [42] Abdel-Hafed, A.; Awad, I.; Ahmed, R. Collect. Czech. Chem.
Commun., 1993, 58, 1198. [43] (a) Hardy, C.R. Adv. Heterocycl. Chem., 1984, 36, 343. (b) Molina,
P.; Arques, A.; Fresneda, P.M.; Vinader, M.V.; Foces-Foces, M.C.; Hernandez Cano, F. Chem. Ber., 1989, 122, 307. (c) Benoit, R.; Dupas, G.; Bourguignon, J.; Quéguiner, G. Synthesis, 1987, 1124. (d) Bare, T.M.; McLaren, Ch. D.; Campbell, J.B.; Firor, J.W.; Resch, J.F.; Walters, C.P.; Salama, A.I.; Meiners, B.A.; Patel, J.B. J. Med. Chem., 1989, 32, 2561. (e) Sanghvi, Y.S.; Larson, S.B.; Robins, R.K.; Revankar, G.R. J. Chem. Soc., Perkin Trans. 1, 1990, 2943.
[44] Hasnaoui, A.; El Messaoudi, M.; Lavergne, J.P. J. Heterocyclic Chem., 1985, 22, 25.
[45] Möhrle, H.; Dwuletzki, H. Chem. Ber., 1986, 119, 3591. [46] Díaz-Ortiz, A.; Carrillo, J.R.; Gómez-Escalonilla, M.J.; de la Hoz,
A.; Moreno, A.; Prieto, P. Synlett, 1998, 1069. [47] Claisen, L.; Roosen, P. Justus Liebigs Ann. Chem., 1893, 278, 274. [48] Behr, L.C.; Fusco, R.; Jarboe, C.H. in The Chemistry of Heterocyc-
lic Compounds. Vol. 22. Pyrazoles, Pyrazolines, Pyrazolidines, In-
dazoles and Condensed Rings; Wiley, R. H., Ed.; John Wiley & Sons: New York, 1967.
[49] (a) El-Khawass, E.S.M.; Bistawroos, A.E. Alexandria J. Pharm. Chem., 1990, 4, 77. (b) Bruno, O.; Ramise, A.; Bondavalli, F.; Schenone, P.; D’Amico, M.; Filippelli, A.; Filipelli, W.; Rossi, F. Farmaco, 1993, 48, 949.
[50] Cuadro, A.M.; Elguero, J.; Navarro, P. Chem. Pharm. Bull, 1985, 33, 2535.
[51] Tsuboi, S.; Moriie, K.; Hatsutori, Y.; Wada, K.; Sone, S.; Oohigata, T.; Ito, A. Jpn. Kokai Tokkyo Koho JP 06,184,114, 1994 [Chem. Abstr., 1995, 122, 105875].
[52] Hartfiel, U.; Dorfmeister, G.; Franke, H.; Geisler, J.; Johann, G.; Rees, R. Eur. Pat. Appl. EP 542, 388, 1993 [Chem. Abstr., 1993, 119, 180774].
[53] (a) Das, N.B.; Mittra, A.S. J. Indian Chem. Soc., 1978, 55, 829. (b) Nayak, A.; Mittra, A.S. J. Indian Chem. Soc., 1980, 57, 643. (c) Desbordes, P.; Guigues, F.; Peignier, R. PCT Int. Appl. WO 94 29, 300, 1994 [Chem. Abstr., 1995, 123, 143884].
[54] (a) Allen, C.F.H.; Wilson, B.D. U. S. Pat., 3106467, 1963 [Chem. Abstr., 1964, 60, 2945]. (b) Pearson, I. U.S. Pat., 3883549, 1975
[Chem. Abstr., 1975, 84, 81243]. (c) Pragst, F.; Stiefke, B.; Schwertfeger, I. East Ger. Pat.,113229, 1975 [Chem. Abstr., 1976, 84, 137220]. (d) Pfeiffer, W.D.; Bulka, E. Ger. (East) DD 293,347, 1991 [Chem. Abstr., 1992, 116, 41452].
[55] For example see: (a) Khan, M.A.; Cosenza, A.G. Afinidad, 1988, 414, 173. (b) Djerrari, B.; Essassi, E.; Fifani, J. Bull. Soc. Chim.
Fr., 1991, 128, 521. (c) Chary, T.J.M.; Reddy, Ch. V.N.; Murthy, A.K. Indian J. Chem., 1992, 31B, 495. (d) Kuznetsov, M.A.; Doro-feeva, Y.V.; Semenovskii, V.V.; Gindin, V.A.; Studenikov, A N. Tetrahedron, 1992, 48, 1269.
[56] Padwa, A. (Ed.) 1,3-Dipolar Cycloaddition Chemistry, Vol. 1, Wiley: New York, 1984.
[57] (a) Oida, T.; Tanimoto, S.; Ikehira, H.; Okano, M. Bull. Chem. Soc. Jpn., 1983, 56, 1203. (b) Hassaneen, H.M.; Shawali, A.S.; Elwal, N.M. Heterocycles, 1990, 31, 1041. (c) Farag, A.M.; Shawali, A.S.; Abed, N.M.; Dawood, K.M. Gazz. Chim. Ital., 1993, 123, 467.
[58] (a) Franck-Neumann, M.; Geoffroy, P.; Lohmann, J.J. Tetrahedron Lett., 1983, 24, 1775. (b) Padwa, A.; Meske, M.; Rodriguez, A. He-
terocycles, 1995, 40, 191. [59] (a) Grigg, R.; Kemp, J.; Thompson, N. Tetrahedron Lett., 1978,
2827. (b) LeFevre, G.; Hamelin, J. Tetrahedron Lett., 1978, 4503. (c) Grigg, R.; Dowling, M.; Jordan, M.W.; Sridharan, V.; Thian-patanagui, S. Tetrahedron, 1987, 43, 5873.
[60] (a) Sun, B.; Adachi, K.; Noguchi, M. Tetrahedron, 1996, 52, 901. (b) Sun, B.; Adachi, K.; Noguchi, M. Synthesis, 1997, 53.
[61] Grigg, R. Chem. Soc. Rev., 1987, 16, 89. [62] Arrieta, A.; Carrillo, J.R.; Cossio, F.P.; Diaz-Ortiz, A.; Gómez-
Escalonilla, M.J.; de la Hoz, A.; Langa, F.; Moreno, A. Tetrahe-
dron, 1998, 54, 13167. [63] Segura, J.L.; Martín, N. Chem. Rev., 1999, 99, 3199. [64] (a) Chou, T.-S. Rev. Heteroatom Chem., 1993, 8, 65. (b) Collier,
S.J.; Storr, R.C. Prog. Heterocycl. Chem., 1999, 10, 25. (c) Wojci-echowski, K. Eur. J. Org. Chem., 2001, 3587. (d) Ando, K.; Taka-

26 Combinatorial Chemistry & High Throughput Screening, 2007, Vol. 10, No. 9 de la Hoz et al.
yama, H. Heterocycles, 1994, 37, 1417. (e) Van de Water, R.W.; Pettus, T.R.R. Tetrahedron, 2002, 58, 5367.
[65] For instance: (a) Nicolaou, K.C.; Gray, D.L.F. J. Am. Chem. Soc., 2004, 126, 607. (b) Nicolaou, K.C.; Gray, D.L.F.; Tae, J.S. J. Am. Chem. Soc., 2004, 126, 613.
[66] (a) de la Hoz, A.; Díaz-Ortiz, A.; Moreno, A.; Prieto, P.; León, R.; Herrero, M.A. Synlett, 2002, 2037. (b) Díaz-Ortiz, A.; Herrero, M.A.; de la Hoz, A.; Moreno, A.; Carrillo, J.R.A. Synlett, 2006, 579.
[67] (a) Hurst, D.T. Prog. Heterocycl. Chem., 1995, 7, 244. (b) Groger, H.; Sans, J.; Gunther, T. Chim. Oggi 2000, 18, 12. (c) Neunhoffer, H. In Comprehensive Heterocyclic Chemistry, Katrizky, A. R.; Rees, C. W.; Boulton, A.J.; McKillop, A., Eds.; Pergamon Press: Oxford, 1984, Vol. 3.
[68] (a) Hirata, K.; Nakagami, H.; Takashina, J.; Miyamoto, K. Hetero-
cyles, 1996, 43, 1513. (b) Abdel-Rahman, R.M.; Seada, M.; Fawly, M.; El-Baz, I. Farmaco, 1993, 48, 397.
[69] (a) Haufel, J.; Breitmaier, E. Angew. Chem., 1974, 13, 604. (b) Wustrow, D.J.; Capiris, T.; Rubin, R.; Knobelsdorf, J.A.; Akunne, H.; Davis, M.D.; MacKenzie, R.; Pugsley, T.A.; Zoski, K.T.; Heff-ner, T.G.; Wise, D.L. Bioorg. Med. Chem. Lett., 1998, 8, 2067.
[70] (a) Menozzi, G.; Mestri, L.; Fossa, P.; Mattioli, F.; Ghia, M. J. Heterocyclic Chem., 1997, 34, 963. (b) Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; Rogers, R.S.; Ro-gier, D.J.; Yu, S.S.; Anderson, G.D.; Burton, G.D.; Cogburn, J.N.; Gregory, S.A.; Kobodt, C.M.; Perkins, W.E.; Seibert, K.; Veenhui-zen, A.W.; Zhang, Y.Y.; Isakson, P.C. J. Med. Chem., 1997, 40, 1347.
[71] (a) Habib, N.S.; Tawil, G.G. Sci. Pharm., 1981, 49, 42. (b) Devi, S.; Mitro, P.; Mishra, S.B.; Mittra, A.S. J. Indian Chem. Soc., 1983, 60, 679. (c) Daidone, G.; Maggio, B.; Plescia, S.; Raffa, D.; Musiu, C.; Milia, C.; Perra, G.; Marongiu, M.E. Eur. J. Med. Chem., 1998, 33, 375. (d) El-Emary, T.I.; Bakhite, E.A. Pharmazie, 1999, 54, 106.
[72] (a) Alvarez, R.; Velazquez, S.; San-Felix, A.; Aquaro, S.; De Clercq, E.; Perno, C.-F.; Karlsson, A.; Balzarini, J.; Camarasa, M.J. J. Med. Chem., 1994, 37, 4185. (b) Ölgen, S.; Chung, K.C.Z. Na-turforsch, 2001, 56b, 804.
[73] Brockunier, L.L.; Parmee, E.R.; Ok, H.O.; Candelore, M.R.; Cas-cieri, M.A.; Colwell, L.F.; Deng, L.; Feeney, W.P.; Forrest, M.J.; Hom, G.J.; MacIntyre, D.E.; Tota, L.; Wyvratt, M.J.; Fisher, M.H.; Weber, A.E. Bioorg. Med. Chem. Lett., 2000, 10, 2111.
[74] Dean, R.M. Adv. Heterocycl. Chem., 1982, 30, 169. [75] Vogel, P.; Cossy, J.; Plumet, J.; Arjona, O. Tetrahedron, 1999, 55,
13521, and references cited therein. [76] (a) Campbell, M.M.; Kaye, A.D.; Sainsbury, M. Tetrahedron Lett.,
1983, 24, 4745. (b) Campbell, M.M.; Kaye, A.D.; Sainsbury, M.; Yavarzadeh, R. Tetrahedron Lett., 1984, 54, 1629.
[77] (a) Nelson, W.L.; Allen, D.R. J. Heterocycl. Chem., 1972, 9, 561. (b) Dauben, W.G.; Krabbenhoft, H.O. J. Am. Chem. Soc., 1976, 98, 1992. (c) Kotsuki, H.; Nishizawa, H.; Ochi, M.; Matsuoka, K.; Bull. Chem. Soc. Jpn., 1982, 55, 496. (d) Brion, F. Tetrahedron
Lett., 1982, 23, 5299. (e) Moore, J.A.; Partain A. M. J. Org. Chem., 1983, 48, 1105. (f) Laszlo, P.; Luchetti, J. Tetrahedron Lett., 1984, 25, 4367. (g) Narayama Murthi, Y.V.S.; Pillai, C.N. Synth. Com-mun. 1991, 21, 783.
[78] Cativiela, C.; Fraile, J.M.; García, J.I.; Mayoral, J.A.; Pires, E.; Royo, A.J.; Figueras, F.; De Mènorval, L.C. Tetrahedron, 1993, 49, 4073.
[79] Fraile, J.M.; García, J.I.; Gómez, M.A.; de la Hoz, A.; Mayoral, J.A.; Moreno, A.; Prieto, P.; Salvatella, L.; Vázquez, E. Eur. J. Org. Chem., 2001, 2891.
[80] García, J.I.; Mayoral, J.A.; Pires, E.; Brown, D.R; Massam, J. Ca-tal. Lett., 1996, 37, 261.
[81] (a) Fraile, J.M.; García, J.I.; Massam, J.; Mayoral, J.A.; Pires, E. J. Mol. Catal. A, 1997, 123, 43. (b) Fraile, J.M.; García, J.I.; Gracia, D.; Mayoral, J.A.; Pires, E. J. Org. Chem., 1996, 61, 9479.
[82] (a) Hogeveen, H.; Middelkoop, T.B. Tetrahedron Lett., 1973, 3671. (b) Hart, H.; Nwokogu, G. J. Org. Chem. 1981, 46, 1251. (c) Coch-
ran, J.E.; Padwa, A. Tetrahedron Lett., 1995, 36, 3495. (d) Mag-giani, A.; Tubul, A.; Brun, P. Tetrahedron Lett., 1998, 39, 4485.
[83] (a) Padwa, A.; Dimitroff, M.; Waterson, A.G.; Wu, T. J. Org.
Chem., 1997, 62, 4088. (b) Zhu, G.-D.; Staeger, M.A.; Boyd S. A. Org. Lett., 2000, 2, 3345.
[84] Brownbridge, P.; Chan, T.-H. Tetrahedron Lett., 1980, 21, 3423. [85] de la Hoz, A.; Díaz-Ortiz, A.; Fraile, J.M.; Gómez, M.V.; Mayoral,
J.A.; Moreno, A.; Sáiz, A.; Vázquez, E. Synlett, 2001, 753. [86] (a) Wong, H.N.C.; Ng, T.-K.; Wong, T.-Y. Heterocycles, 1983, 20,
1815. (b) Wong, H.N.C. Synthesis, 1984, 787. (c) Wong, H.N.C.; Ng, T.-K.; Wong T.-Y.; Xing, Y.D. Heterocycles, 1984, 22, 875.
[87] (a) Huang, N.Z.; Xing, Y.D.; Xe, D.Y. Synthesis, 1982, 1041. (b) Xing, Y.D.; Huang, N.Z. J. Org. Chem., 1982, 47, 140. (c) Wong, H.N.C.; Xing, Y.D.; Zhou, Y.F.; Gong, Q.Q.; Zhang, C. Synthesis, 1984, 787.
[88] (a) Chambers, R.D.; Roche A.J.; Rock M.H. J. Chem. Soc., Perkin Trans. 1, 1996, 1095. (b) Martin-Matute, B.; Nevado, C.; Cardenas, D.J.; Echavarren, A.M. J. Am. Chem. Soc., 2003, 125, 5757.
[89] Maggiani, A.; Tubul, A.; Brun, P. Synthesis, 1997, 631. [90] Rhodes, C.N.; Brown, D.R. J. Chem. Soc., Faraday Trans., 1993,
89, 1387. [91] Cativiela, C.; Figueras, F.; García, J.I.; Mayoral, J.A.; Pires, E.;
Royo, A.J. Tetrahedron: Asymmetry, 1993, 4, 621. [92] Moreno, A.; Gómez, M.V.; Vázquez, E.; de la Hoz, A.; Díaz-Ortiz,
A.; Prieto, P.; Mayoral, J.A.; Pires, E. Synlett, 2004, 1259. [93] Modified silicas were characterized by IR spectroscopy. Pyridine
adsorption-desorption experiments were carried out in order to de-termine the relative proportion of Lewis and Brønsted acidic sites. Thus, Lewis/Brønsted acid area ratios were determined by the inte-gral areas of Lewis and Brønsted IR bands of pyridine adsorbed onto modified silicas previously described by Fraile, J.M.; García, J.I.; Mayoral, J.A.; Pires, E.; Salvatella, L.; Ten, M. J. Chem. Phys. B, 1999, 103, 1664.
[94] (a) Grzegorz, B. Tetrahedron, 2006, 62, 9507. (b) Giacomelli, G.; Porcheddu, A.; De Luca, L. Current Org. Chem., 2004, 8, 1497.
[95] Díaz-Ortiz, A.; Elguero, J.; Foces-Foces, C.; de la Hoz, A.; Mo-reno, A.; Moreno, S.; Sánchez-Migallón, A.; Valiente G. Org.
Biomol. Chem., 2003, 1, 4451. [96] Díaz-Ortiz, A.; Elguero, J.; de la Hoz, A.; Jiménez, A.; Moreno, A.;
Moreno, S.; Sánchez-Migallón, A. QSAR Comb. Sci., 2005, 24, 649.
[97] Díaz, A; Elguero, J.; Foces-Foces, C.; de la Hoz, A.; Moreno, A.; Mateo, M.C.; Sánchez-Migallón, A.; Valiente, G. New J. Chem., 2004, 28, 952.
[98] Díaz, A; Elguero, J.; de la Hoz, A.; Jiménez, A.; Moreno, A.; Ruiz, A.; Sánchez-Migallón, A. Tetrahedron, 2007, 63, 748.
[99] Díaz, A; Elguero, J.; Foces-Foces, C.; de la Hoz, A.; Mateo, M.C.; Moral, M.; Moreno, A.; Rodriguez, M.L.; Sánchez-Migallón, A. Tetrahedron, 2006, 62, 5868.
[100] (a) Kappe, C.O.; Stadler. A. Microwave-Assisted Combinatorial Chemistry. In Microwaves in Organic Chemistry; Loupy, A. Ed.; Wiley-VCH: Weinheim, 2002. (b) Stadler, A.; Kappe, C.O. J. Comb. Chem., 2001, 3, 624. (c) Lidström, P.; Westman, J.; Lewis, A. Comb. Chem. High Throughput Screen., 2002, 5, 441. (d) Swamy, M.K.; Yeh, W.B.; Lin, M. J.; Sun, C.M. Curr. Med.
Chem., 2003, 10, 2403. [101] (a) Stadler, A.; Yousefi, B.H.; Dallinger, D.; Walla, P.; Van der
Eycken, E.; Kaval, N.; Kappe, C.O. Org. Proc. Res. Develop., 2003, 7, 707. (b) Alcázar, J.; DIELS, G.; Schoentjes, B. QSAR
Comb. Sci., 2004, 906. [102] (a) Alcázar, J. J. Comb. Chem., 2005, 7, 353. (b) Loones, K.T.J.;
Maes, B.U.W.; Rombouts, G.; Hostyna, S.; DIELS, G. Tetrahe-dron, 2005, 61, 10338. (c) Murray, J.K.; Gellman, S.H. J. Comb.
Chem., 2006, 8, 58. [103] Díaz-Ortiz, A.; de la Hoz, A.; Alcazar, J.; Carrillo, J.R.; Herrero
M.A.; Fontana, A.; Muñoz, J. de M. Comb. Chem. High Through-put Screen., 2007, 10, 163.
[104] Díaz-Ortiz, A.; Díez-Barra, E.; de la Hoz, A.; Moreno, A.; Gómez-Escalonilla, M.J.; Loupy, A. Heterocycles, 1996, 43, 1021.
[105] Nüchter, M.; Ondruschka, B. Mol. Div, 2003, 7, 253.
Received: July 15, 2007 Revised: August 18, 2007 Accepted: August 20, 2007

![ChemInform Abstract: ChemInform Abstract: Haloacetylated Enol Ethers: 16[5] Regiospecific Synthesis of 5-Trichloromethyl-pyrazoles](https://img.dokumen.tips/doc/110x75/63196e83e9c87e0c09101e7e/cheminform-abstract-cheminform-abstract-haloacetylated-enol-ethers-165-regiospecific.jpg)



























