Embed Size (px)
Citation preview

1 Anti-Cancer Agents in Medicinal Chemistry, 2008, 8, 000-000
1871-5206/08 $55.00+.00 © 2008 Bentham Science Publishers Ltd.
Cdc25B Phosphatase Inhibitors in Cancer Therapy: Latest Developments, Trends and Medicinal Chemistry Perspective
Antonio Lavecchia1,*, Antonio Coluccia2, Carmen Di Giovanni1 and Ettore Novellino1
1Dipartimento di Chimica Farmaceutica e Tossicologica, Università di Napoli “Federico II”, Facoltà di Farmacia, via D. Monte-
sano 49, I-80131 Napoli, Italy and 2Istituto Pasteur - Fondazione Cenci Bolognetti, Dipartimento di Studi Farmaceutici, Sapienza
Università di Roma, Piazzale Aldo Moro 5,I-00185 Roma, Italy
Abstract: The Cdc25 phosphatases (Cdc25A, Cdc25B, and Cdc25C in humans), which are responsible for dephosphorylating specific ty-rosine/threonine residues on cyclin dependent kinases (CDKs), function as essential regulators of cell cycle control during normal eu-karyotic cell division and as mediators of the checkpoint response in cells with DNA damage. Because overexpression of Cdc25A and Cdc25B has been linked to numerous cancers and often correlates with a poor clinical outcome, both academia and industry have devoted substantial research effort in establishing the basic underlying molecular mechanisms and in identifying novel, specific and potentially useful inhibitors of Cdc25 as potential anticancer drugs. Over the past year, dozens of research papers and patent applications describing new Cdc25 inhibitors belonging to different structural classes have been disclosed. In this review, we give an overview on the current status in the field of medicinal chemistry of Cdc25B inhibitors. In addition, molecular modeling studies aimed to clarify the molecular mechanism of inhibition as well as the pharmacophoric features critical for design of new and selective Cdc25B inhibitors are also dis-cussed.
Key Words: Cancer, phosphatases, Cdc25 inhibitors, cell cycle, antiproliferative agents, docking, molecular modeling.
INTRODUCTION
Tyrosine phosphorylation of proteins is a fundamental mecha-nism of intracellular signal transduction involved in important cel-lular events such as cell growth and differentiation. [1-3]. The phos-phorylation states of proteins are strictly controlled by various protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). PTKs catalyze the covalent attachment of a phosphate group to the amino acid side chain of a protein, whereas PTPs cata-lyze the removal of this group. Eukaryiotic protein phosphatases can be divided into two main subclasses [4-7] based upon their substrate specificity and protein structure: protein serine/threonine phosphatases (PSTPs), which dephosphorylate phosphoserine (pS) and phosphothreonine (pT) in a single-step reaction using a metal-activated water molecule, and PTPs which dephosphorylate phos-photyrosine (pY) a two-step reaction involving a cysteinyl-phosphate enzyme intermediate. Dual-specificity protein phospha-tases (DSPs), which dephosphorylate both pY and pT residues, are considered to be a subfamily of PTPase, because they possess the conserved PTP signature motif HCX5R and employ a similar cata-lytic mechanism [7, 8]. Cdc25 cell cycle regulators are examples of DSPs which dephosphorylate contiguous pY and pT on the cyclin dependent kinases (CdKs) and have been shown to play crucial roles in cell proliferation [9].
Cdc25 PHOSPHATASES IN CELL-CYCLE AND CHECK-
POINTS CONTROL
Cdc25 phosphatases are found in all eukaryotic organisms ex-cept plants [10]. Three Cdc25 homologues are encoded by the hu-man genome [11, 12]: Cdc25A, Cdc25B, and Cdc25C. They share 40-50% amino acid identity and regulate distinct cyclin/CdK com-plexes throughout the cell cycle. Although considerable progress has been made in the characterisation of Cdc25 phosphatases, many aspects of their function and regulation, as well as the identity of their CdK substrate, remain unclear.
CDK complexes are known to be key regulators of cell-cycle progression, and are held inactive by the phosphorylation of two
*Address correspondence to this author at the Dipartimento di Chimica Farmaceutica e Tossicologica, Università di Napoli “Federico II”, Facoltà di Farmacia, D. Montesano 49, I-80131 Napoli, Italy; Tel/Fax: +39-081-678613; E-mail: [email protected]
residues located within the ATP binding loop (T14 and Y15 of CdK1), by the Wee1 and Myt1 kinases [13]. When CdK activity becomes required for progression into the next cell-cycle phase, the dual specificity Cdc25 phosphatases dephosphorylate these two residues, thereby activating the CdK-cyclin complex (Fig. 1a).
In mammalian cells, all three isoforms have been implicated in the control of the G1-S and G2-M transitions by regulating the ac-tivities of CdK1 and CdK2. Cdc25A dephosphorylates cyclin E/CdK2 and cyclin A/CdK2 complexes to regulate the G1 and S phase progression [14] (Fig. 1b,c). It has also a role in the G2-M transition [15, 16] by activating CdK1-cyclin B complexes, which are thought to initiate chromosome condensation [15, 17-19]. Cdc25B and Cdc25C are primarily required for entry into mitosis [20-22]. Cdc25B is proposed to be responsible for the initial activa-tion of CdK1-cyclin B at the centrosome during the G2-M transition [17, 22, 23], which is then followed by a complete activation of CdK1-cyclin B complexes by Cdc25C in the nucleus at the onset of mitosis [24]. It is, therefore, likely that all three CDC25 phospha-tases cooperate in regulating each cell cycle transition and partici-pate in the control of entry into and progression through mitosis [19] (Fig. 1b,c).
The Cdc25 phosphatases also play an important role in the checkpoint response that prevents CdK/cyclin activation following DNA damage [25, 26]. In response to DNA damage by ionizing irradiation, ultraviolet light, replication inhibitors, or other DNA-damaging agents, the Chk1 and Chk2 checkpoint kinases, activated by the ATM (ataxia-telangiectasia mutated) and/or ATR (ataxia-telangiectasia and Rad3-related) kinases, phosphorylate the Cdc25s. This phosphorylation leads to G1/S or G2/M blocks in the cell cycle by subsequent proteasome-mediated degradation of Cdc25A or 14-3-3-mediated sequestration of Cdc25C [19]. The arrest of cell cycle is maintained until the damage has been repaired or the cell under-goes apoptosis. The p38 mitogen-activated protein kinase (MAPK), a stress-activated signalling pathway, may also be involved in the phosphorylation and inactivation of Cdc25B in response to UV-induced DNA damage [26, 27].
Hence, the activities of Cdc25 phosphatases are highly regu-lated, both during the normal cell division cycle and in response to checkpoint activation, in order to ensure that a correct level of CDK-cyclin activity is maintained. This, in turn, controls the pro-gression of the cell through the division cycle in an orderly manner,

2 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
and ensures the maintenance of genomic stability. Therefore, the misregulation of any of these control mechanisms could result in the acquisition of genetic mutations and so contribute to tumori-genesis.
Cdc25 PHOSPHATASES AS TARGETS FOR CANCER THERAPY
Elevated levels of Cd25A and Cdc25B but not Cdc25C have been noted in many human tumor types, such as breast, ovarian, prostate, lung, colorectal, oesophageal, thyroid, laryngeal, hepato-cellular, gastric, pancreatic, endometrial, head and neck cancer, neuroblastoma, glioma and non-Hodgkin lymphoma [19], where there seems to be a remarkable association with high protein levels and either tumor aggressiveness or poor prognosis [28-30]. Cdc25A and Cdc25B have been reported to transform cells in cooperation with the ras oncogene or in the absence of the retinoblastoma tumor suppressor protein [31]. Cdc25B expression is increased after trans-formation of primary fibroblasts with simian virus 40 large T anti-
gen [32] and after treatment with a human pulmonary carcinogen in human lung cells [33]. Cdc25A and B are also transcriptional tar-gets of the c-myc oncogene, and have oncogenic properties in coop-eration with either Ha-ras or Rb1 [34]. Moreover, the transgenic mice with Cdc25B overexpression produces mammary gland hy-perplasia, suggesting that overexpression of Cdc25B is related with the initiation of mammary tumorigenesis [35]. Recently, the in-volvement of Cdc25A in the adhesion-dependent proliferation of acute myeloid leukaemia (AML) cells has also been described [36]. Thus, overexpression of Cdc25 phosphatases in neoplasia might provide a growth advantage through the loss of critical cell cycle checkpoint controlling mechanisms or by loss of normal apoptotic signaling mechanisms [30]. Therefore, there has been considerable interest in identifying selective, cell active inhibitors of Cdc25 phosphatases in both pharmaceutical companies and academic labo-ratories.
This work will review the most recent developments made in the field of the Cdc25B phosphatases inhibitors, with a special em-
a)
b)
c)
Fig. (1). Key cell cycle transitions controlled by Cdc25 phosphatases. a) Cdc25 phosphatases dephosphorylate and activate CdK-cyclin complexes, thus allow-ing catalysis and substrate phosphorylation. Wee1 and Myt1 kinases phosphorylate CdK on Thr14 and Tyr15 of CdK1 (orange P). The CDK-activating kinase (CAK), itself a cyclin-dependent kinase, adds a phosphate (light blue P) to a threonine in the catalytic loop of the CdK-cyclin complexes, thereby stimulating kinase activity. b) The eukaryotic cell cycle consists of four phases, G1, S (DNA synthesis), G2 and M (mitosis). Cdc25A, Cdc25B and Cdc25C phosphatases are all involved in phosphorylating CdK-cyclin complexes, such as CdK2-cyclin E at the G1-S transition or CdK1-cyclin B at the entry into mitosis. c) Cdc25A, Cdc25B and Cdc25C control entry and progression into mitosis. Cdc25B is thought to be responsible for the initial activation of CdK1-cyclin B at the centrosome. Nuclear translocation leads to an autoamplification process of CDC25s that then fire the bulk of CdK1-cyclin B complexes and trigger mito-sis.
Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 3
phasis to molecular modeling studies aimed to clarify the molecular mechanism of inhibition as well as the specific structural features responsible for inhibitory activity.
Cdc25B PHOSPHATASE INHIBITORS
Cdc25 inhibitors so far identified inhibit all three Cdc25 iso-forms and belong to various chemical groups including quinonoids, phosphate surrogates and electrophilic entities. Tables 1-3 report
some significant examples of natural and synthetic molecules dis-playing selectivity toward Cdc25B phosphatases.
Natural Compounds and Their Derivatives
Menadione (Vitamin K3, compound 1, Table 1) was found to inactivate the Cdc25B (IC50 = 3.6 μM) in an irreversible manner by covalently binding to its active site [37, 38]. Moreover, an antipro-liferative activity of menadione, either alone or in combination with
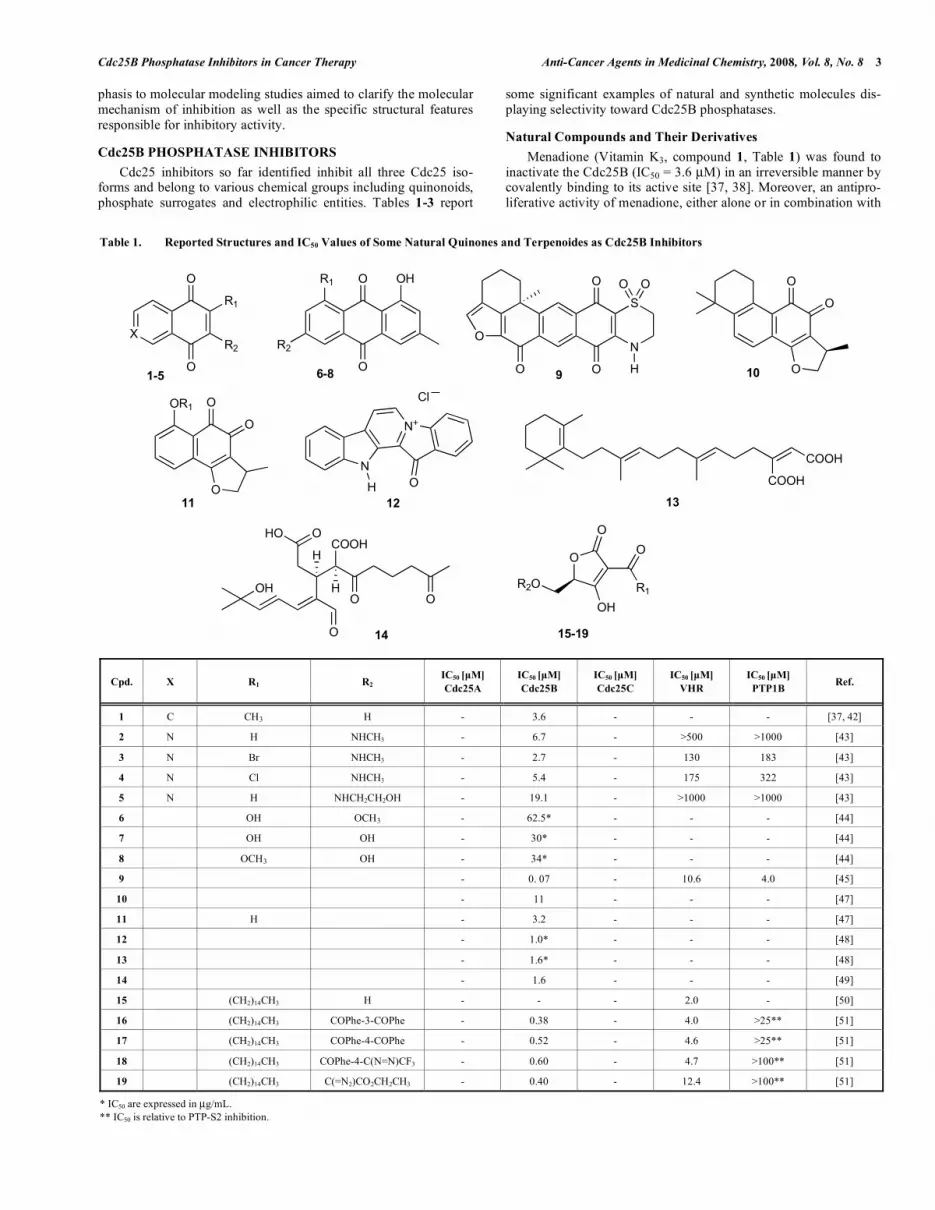
Table 1. Reported Structures and IC50 Values of Some Natural Quinones and Terpenoides as Cdc25B Inhibitors
Cpd. X R1 R2 IC50 [μM]
Cdc25A
IC50 [μM]
Cdc25B
IC50 [μM]
Cdc25C
IC50 [μM]
VHR
IC50 [μM]
PTP1B Ref.
1 C CH3 H - 3.6 - - - [37, 42]
2 N H NHCH3 - 6.7 - >500 >1000 [43]
3 N Br NHCH3 - 2.7 - 130 183 [43]
4 N Cl NHCH3 - 5.4 - 175 322 [43]
5 N H NHCH2CH2OH - 19.1 - >1000 >1000 [43]
6 OH OCH3 - 62.5* - - - [44]
7 OH OH - 30* - - - [44]
8 OCH3 OH - 34* - - - [44]
9 - 0. 07 - 10.6 4.0 [45]
10 - 11 - - - [47]
11 H - 3.2 - - - [47]
12 - 1.0* - - - [48]
13 - 1.6* - - - [48]
14 - 1.6 - - - [49]
15 (CH2)14CH3 H - - - 2.0 - [50]
16 (CH2)14CH3 COPhe-3-COPhe - 0.38 - 4.0 >25** [51]
17 (CH2)14CH3 COPhe-4-COPhe - 0.52 - 4.6 >25** [51]
18 (CH2)14CH3 COPhe-4-C(N=N)CF3 - 0.60 - 4.7 >100** [51]
19 (CH2)14CH3 C(=N2)CO2CH2CH3 - 0.40 - 12.4 >100** [51]
* IC50 are expressed in μg/mL. ** IC50 is relative to PTP-S2 inhibition.
XR2
R1
O
O
O
O
N
S
O
O
O
O
O
O
OR1
H
O O
O
O
1-5 9 10
11
R1
R2
O
O OH
6-8
N
N+
COOH
COOH
OH
O
COOH
O O
H
H
HO
HO O
12 13
14
O
OH
O
R1
O
R2O
15-19
Cl

4 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
other antitumor agents, was shown in parental cancer cell lines and
their multidrug-resistant derivatives. Its lower level of toxicity in vivo compared with other quinone-type chemotherapeutics suggests the potential utility of this agent in cancer therapy [39-42].
Caulibugulones A-D (2-5), selective and potent Cdc25B inhibi-tors bearing a quinonoid moiety, were reported by Lazo's group. These compounds, originally extracted from a marine bryozoan at the NCI, showed to inhibit full-length human Cdc25B in vitro with IC50 values ranging from 2.7 to 19.1 μM (Table 1). All caulibugu-lones exhibited a minimum 30-fold preference as inhibitors against the dual specificity phosphatase Cdc25B compared to DSP Vac-cinia H1-related (VHR) or PTP1B. In addition, a cell growth inhibi-tory activity on human tumor cells was also observed [43]. Very recently, Choi et al. [44] screened more than 1000 plant extracts including 120 herbal medicines against Cdc25B phosphatases. Three Cdc25B anthraquinone inhibitors were isolated from the methanolic extract of the roots of Polygonum multiflorum: physcion (6), emodin (7) and questin (8). These compounds inhibited the enzymatic activity of Cdc25B with IC50 values of 62.5, 30, and 34 μg/mL. Moreover, compounds 7 and 8 strongly inhibited the growth of human colon cancer cell SW620 with GI50 values of 6.1 and 0.9 μg/mL, respectively.
Adociaquinone B (9), a marine sponge derivative extracted from Xestospongia, was described as a potent inhibitor of recombi-nant human Cdc25B with an IC50 value of 0.07 μM, showing a remarkable 19- to 150-fold selectivity against Cdc25B when com-pared to either the closely related VHR or PTP1B. Furthermore, 9 blocked cell cycle progression during mitosis. The authors suggest that selective oxidation of the catalytic cysteine could be one possi-ble mechanism for Cdc25B inhibition [45].
Naphthofuranediones, which have an ortho-quinonoid substruc-ture, were identified as a novel class of competitive and reversible Cdc25 inhibitors. These compounds were shown to block cell cycle progression and to inhibit the growth of human tumor cells in cul-ture [46]. The most interesting compound of the series, the (-)-cryptotanshinone 10, an ortho-quinone diterpene isolated from the rhizome of Salvia miltiorrhiza Bunge, showed to inhibit not only Cdc25B phosphatase with an IC50 of 11 μM, but also the growth of the human lung cancer cell line A549. Its simplified analogue, compound 11, displayed better Cdc25B inhibitory activity and more cytotoxic activity against tumor cells [47].
Fascaplysin (12), an alkaloid extracted from the Thorectandra sp. Sponge, was already known for cytotoxic, antimicrobial and antimalarial properties, along with CdK4 inhibition and DNA bind-ing. No literature report existed about the activity of fascaplysin-type alkaloids against Cdc25B. Nevertheless, compound 12 proved to be the most potent Cdc25B inhibitor of the extracts of Thorec-tandra sp. Sponge (IC50 = 1.0 μg/mL) [48]. The sesterterpenoid 13, isolated from the same extract, whose maleic acid feature could be considered as a phosphate surrogate, also showed good Cdc25B inhibitory properties with an IC50 value of 1.6 μg/mL [48].
Recently, Cao et al. [49] isolated xenicane diterpenoids from a marine anemone of the order Actiniaria and tested them in a Cdc25B assay. Among the four natural products isolated, the diter-penoid 14, which unlike the three other compounds did not cyclized to a hemiacetal, resulted the most active Cdc25B inhibitor, although it did not show any cytotoxicity against the A2780 ovarian cancer cell line.
The hexadecanoyl-5-hydroxymethyl tetronic acid 15 (RK-682) was isolated from the Streptomyces strain 88-682 and was found to be a comparatively potent non-competitive inhibitor of the DSP VHR with an IC50 of 2.0 μM [50]. RK-682 also caused the arrest of cell-cycle progression at the G1 checkpoint in mammalian cells. It has been postulated that the inhibition might be effected by cova-lent modification of the enzyme by a 1,4-Michael-type nucleophilic addition. Further derivatization of RK-682 by Sodeoka’s group,
manipulating the substituents at C-3 and increasing the hydropho-bicity at the C-5 position, resulted in the potent inhibitors 16-19, which showed peculiar selectivity toward Cdc25B [51].
Synthetic Cdc25B Inhibitors
Many synthetic Cdc25 inhibitors have been frequently designed mimicking the phosphate of the natural substrate of the enzyme. The common phosphate surrogates used include sulfate, carboxyl groups, malonate, xanthate, the 3-acyl-tetronic acid core, but also several metal anions [52].
Based on molecular modeling studies, a series of macrocyclic inhibitors synthetically derived from steroids, were prepared and evaluated. The best compound of the series (20 in Table 2) inhib-ited the recombinant human Cdc25B phosphatase (IC50 value of 3.4 μM) with at least 3-fold selectivity versus PTP1B. Computational studies, utilizing both ligand- and receptor-based approaches, sug-gested that the carbonyl moiety rather than the carboxylate group acted as an anchor group in the phosphate binding pocket [53].
Wipf et al. [54] designed a pharmacophore model on the basis of structure-activity relationship (SAR) data available for natural inhibitors of PSTPs. Among the synthesized compounds, the 4-(benzyl - (2[(2,5 - diphenyl - oxaole - 4 - carbonyl) - amino] - ethyl) - carbo-nyl)-2-decanoylamino butyric acid 21 (SC- 9) showed to be the most potent competitive inhibitor of both Cdc25A and B, with an IC50 of 15 M, inhibiting PTP1B non-competitively. SC- 9 in-hibited Cdc25-dependent cell cycle progression in synchronized murine mammary carcinoma cells at both G1 and G2/M phase. It also enhanced tyrosine phosphorylation of both CdK2 and CdK4 and decreased CdK4 kinase activity [55] . A SAR study revealed that the hydrophobicity of the nonyl-residue (R4 in compound 21) and the presence of an aromatic moiety at C-5 of the oxazole were crucial for Cdc25B inhibitory activity. Ducruet et al. [56], using a combinatorial/parallel synthetic approach to rigidify the variable core region and modify the side chains of SC- 9, developed compound 22 (FY21- 09), a partial competitive inhibitor of Cdc25B2 with a Ki value of 7 M. FY21- 09 showed only mod-erate activity against PTP1B (IC50 = 41.4 μM) and inhibited the growth of breast cancer cell lines. It also blocked cell-cycle pro-gression at the G2/M checkpoint, an observation consistent with the intracellular inhibition of Cdc25B2. The SAR observed by the in-fluence of substituents at the oxazole moiety of 21 led Wipf et al. [57] to develop a new heterocyclic scaffold, the sulphonylated aminothiazole 23. A 35-compound library of this compound was prepared and screened for inhibitory activity against Cdc25B, VHR and PTP1B. Among the best inhibitors, compound 24 was found to competitively inhibit Cdc25B with a Ki of 4.6 μM.
A broad screening project allowed Fritzen et al. [58] to identify the isoquinoline 25 (PNU-108937) with weak in vitro inhibitory activity against Cdc25B (IC50 = 70-100 μM). They used a solid phase, combinatorial, synthetic approach to generate several inhibi-tors of Cdc25B including compounds 26 and 27, which exhibited an IC50 of 20 and 15 μM, respectively.
An other screening of the Korea Chemical Bank revealed a novel Cdc25B inhibitor, EK-6136 (28). This pyrazolone derivative inhibited Cdc25B with a IC50 of 6.4 μM and was selective against several phosphatases including PTP1B, CD45, Cdc25A, PP1,VHR and Yop. 28 resulted also to inhibit the proliferation of MCF-7, HT-29 and A549 tumor cell lines in vitro and its antiproliferative activ-ity was associated with the increase in CdK1 phosphorylation and G2/M arrest of the cell cycle. In the inhibition kinetic study, this compound displayed mixed inhibition pattern, suggesting that it bound to both free enzyme and the enzyme-substrate complex or bound to the site adjacent to the active site, the so called anionic binding site. Furthermore, the inhibition was reversible, suggesting that 28 was less likely to induce covalent binding to the active site of Cdc25B [59]. Subsequently, the Korea Research Institute of Technical Technology patented a new series of 5-(1,3-diaryl-1H-
Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 5
pyrazol-4-ylmethylene)-thiazolidine-2,4-diones as selective Cdc25B inhibitors with cytotoxicity toward several cancer cell lines, includ-ing A549, HT-29 and MCF-7. Compound 29 showed activity com-parable to cisplatin in C33A (human uterine cancer cell line) xenografted nude mice but, in contrast to cisplatin, not reduced motility or food intake [60].
Although the naphthyridinone derivative 30 was known to in-hibit Cdc25 phosphatase with IC50 values of 5 M [61] (Table 3), a more potent series of heterocyclic diones was discovered by Lazo et al. [62]. They examined the 1990 compound NCI Diversity Set and then, from their repository of 140 000 compounds, computationally identified eight potent 7-substituted quinolinediones. All submi-cromolar inhibitors showed a strong preference for Cdc25B com-pared with VHR or PTP1B. NSC663284 (31), the most potent com- pound, inhibited Cdc25 in vitro in an irreversible, time-dependent manner with an IC50 of 0.21 μM and exhibited mixed competitive kinetics against Cdc25A, B2 and Cdc25C. 31 showed 20- and 450-fold selectivity for Cdc25B2 compared to VHR or PTP1B phospha-tases, respectively [62]. Computational electrostatic potential map-
ping of the analyzed quinolinequinones suggested the need for an electron-deficient 7-position for maximal inhibitory activity. Sur-prisingly, NSC663284 also covalently modified the active site of Cdc25A through one of the serines (Ser114, corresponding to Ser434 in full-length CDC25A) and not the cysteine [63].
In 2002, Lazo's group evaluated 10,070 compounds in the pub-licly available NCI chemical repository for in vitro inhibitory activ-ity against oncogenic, full-length, recombinant human Cdc25B [64]. The most potent compounds were NSC95397 (32), NSC672121 (33), NSC115447 (34), NSC139049 (35) and NSC135880 (36), which revealed IC50 values for Cdc25B of less than 500 nM. Com-pound 32 displayed mixed inhibition kinetics with in vitro Ki values for Cdc25A, Cdc25B, and Cdc25C of 32, 96, and 40 nM, respec-tively. Moreover, it was 125- to 180-fold more selective for Cdc25A than VH1-related dual-specificity phosphatases or PTP1B, respec-tively. 32 also showed significant growth inhibition against human and murine carcinoma cells and blocked G2/M phase transition. In addition, the di-acid analog of NSC95397, 37, was evaluated for in vitro enzymatic inhibition using recombinant fused MBP-Cdc25B3
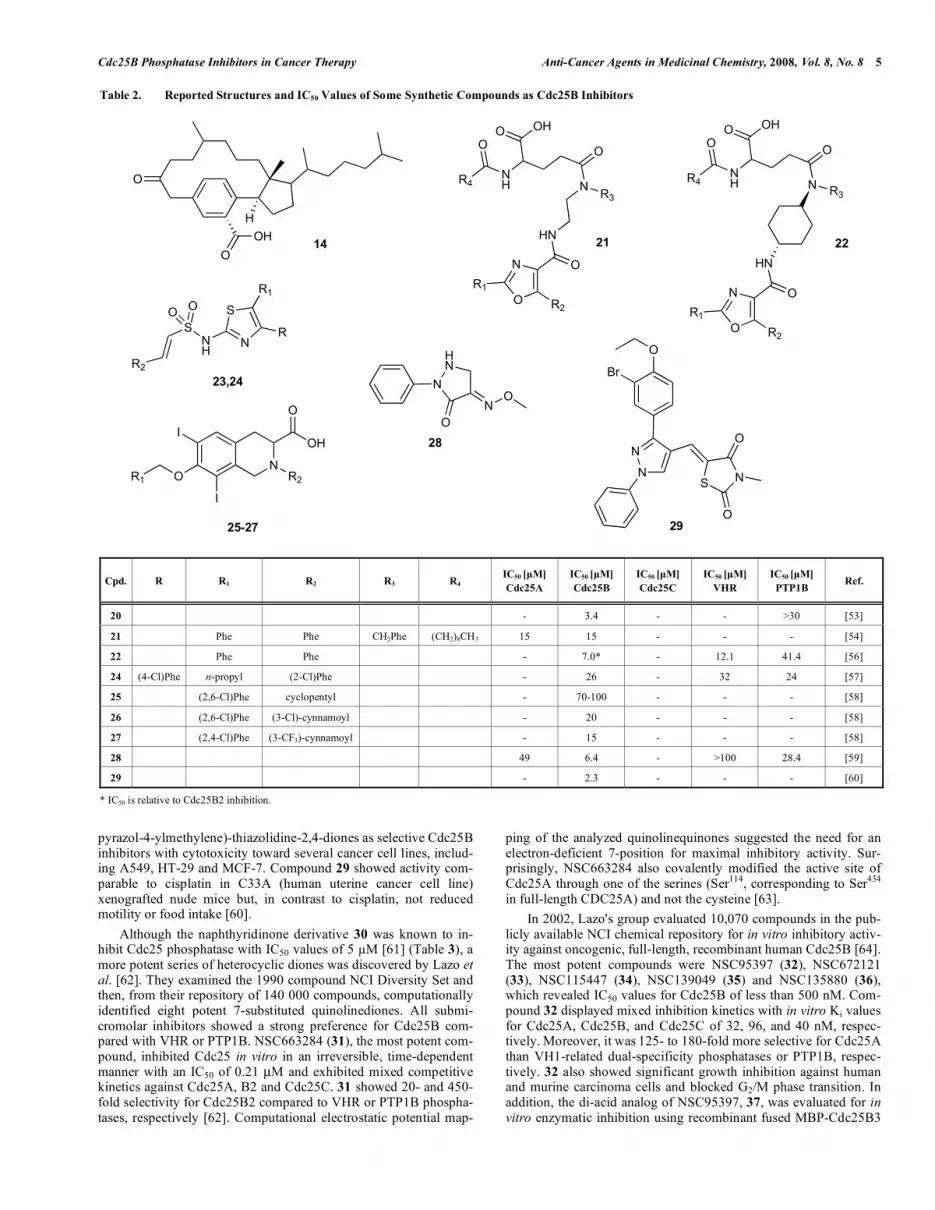
Table 2. Reported Structures and IC50 Values of Some Synthetic Compounds as Cdc25B Inhibitors
Cpd. R R1 R2 R3 R4 IC50 [μM]
Cdc25A
IC50 [μM]
Cdc25B
IC50 [μM]
Cdc25C
IC50 [μM]
VHR
IC50 [μM]
PTP1B Ref.
20 - 3.4 - - >30 [53]
21 Phe Phe CH2Phe (CH2)8CH3 15 15 - - - [54]
22 Phe Phe - 7.0* - 12.1 41.4 [56]
24 (4-Cl)Phe n-propyl (2-Cl)Phe - 26 - 32 24 [57]
25 (2,6-Cl)Phe cyclopentyl - 70-100 - - - [58]
26 (2,6-Cl)Phe (3-Cl)-cynnamoyl - 20 - - - [58]
27 (2,4-Cl)Phe (3-CF3)-cynnamoyl - 15 - - - [58]
28 49 6.4 - >100 28.4 [59]
29 - 2.3 - - - [60]
* IC50 is relative to Cdc25B2 inhibition.
O
H
O
OH14
N
O
R1
R2
O
HN
NR3
O
NH
OHO
R4
O
21
N
O
R1
R2
O
NR3
O
NH
OHO
R4
O
22
HN
NR2
OH
O
I
OR1
I
25-27
N
S
R1
RNH
S
OO
R2
23,24
HN
N
O
NO
28
O
Br
N
N
NS
O
O29
6 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
(one of the revealed splicing variants of CDC25B) with fluorescein diphosphate as the substrate, thus revealing an IC50 value of 1.75 μM. This carboxylic derivative displayed antiproliferative activity on HeLa cells and inhibited cell growth in a clonogenic assay with an effect on cell cycle progression [65].
In 2003, Sohn et al. [66] described a new class of Cdc25 inhibi-tors, the indolyldihydroxyquinones, which bound reversibly to the active site with submicromolar potency. Additionally, these com-pounds exhibited activity in competition with the natural protein substrate in vitro and rapidly led to cell death. The most notable
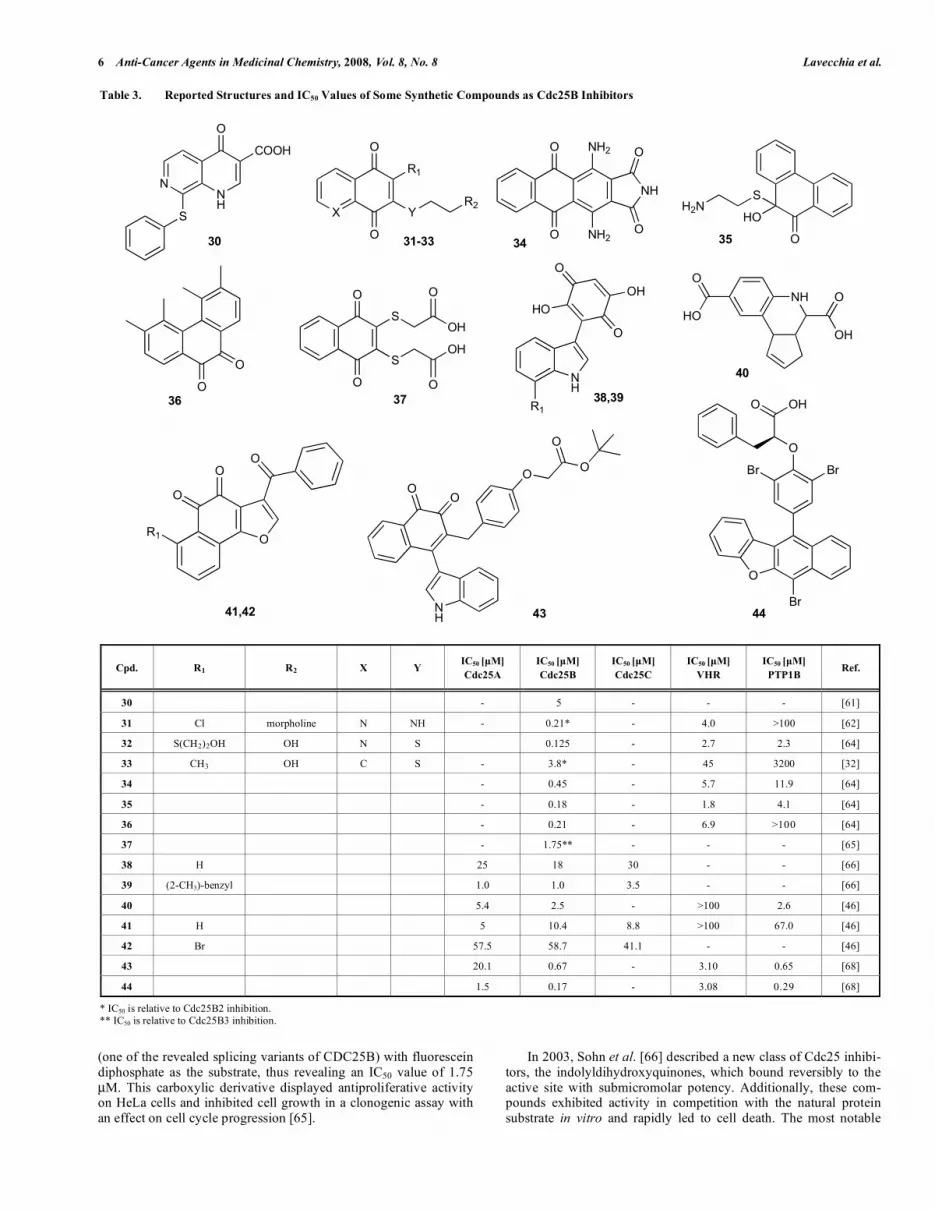
Table 3. Reported Structures and IC50 Values of Some Synthetic Compounds as Cdc25B Inhibitors
Cpd. R1 R2 X Y IC50 [μM]
Cdc25A
IC50 [μM]
Cdc25B
IC50 [μM]
Cdc25C
IC50 [μM]
VHR
IC50 [μM]
PTP1B Ref.
30 - 5 - - - [61]
31 Cl morpholine N NH - 0.21* - 4.0 >100 [62]
32 S(CH2)2OH OH N S 0.125 - 2.7 2.3 [64]
33 CH3 OH C S - 3.8* - 45 3200 [32]
34 - 0.45 - 5.7 11.9 [64]
35 - 0.18 - 1.8 4.1 [64]
36 - 0.21 - 6.9 >100 [64]
37 - 1.75** - - - [65]
38 H 25 18 30 - - [66]
39 (2-CH3)-benzyl 1.0 1.0 3.5 - - [66]
40 5.4 2.5 - >100 2.6 [46]
41 H 5 10.4 8.8 >100 67.0 [46]
42 Br 57.5 58.7 41.1 - - [46]
43 20.1 0.67 - 3.10 0.65 [68]
44 1.5 0.17 - 3.08 0.29 [68]
* IC50 is relative to Cdc25B2 inhibition. ** IC50 is relative to Cdc25B3 inhibition.
S
NNH
O
COOH
X
O
O
R1
YR2
O
HO
SH2N
NH
O
O
NH2
NH2
O
O
O
O
30 31-33 3534
36
NH
O
O
HO
OH
NH
OO
O
OO
O
O
Br Br
Br
O OH38,39
NH
OH
O
40
43 44
O
O
S
SOH
OH
37
O
O
R1
O
O
O
O
R1
41,42
O
HO

Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 7
compounds, 38 and 39, were competitive inhibitors, most likely binding into the pocket adjacent to the active site. Cdc25B inhibi-tion required an intact C-terminal tail, suggesting that this region, essential for binding of protein substrate [67], formed an important part of the binding site for these inhibitors. In addition, treatment of HEK293 embryonic kidney cells with 50 μM of these compounds caused rapid apoptosis by an unknown mechanism.
By screening the PRIME Collection compound library from Chembridge, which consists of 10,000 drug-like small molecules, Brisson et al. [46] identified cyclopentaquinolines (40) and naph-thofuranediones (41 and 42) as a novel class of competitive and reversible Cdc25 inhibitors. The dicarboxylated cyclopentaquino-line 40 (5661118) had IC50 values against Cdc25A, Cdc25B, VHR, and PTP1B of 5.4, 2.5, >100, and 6.6 μM, respectively. This com-pound exhibited mixed partial competitive inhibition with an appar-ent Ki of 9.0 μM and had no noticeable effect on cell cycle progres-sion. Naphthofurandione 41 (5169131), having IC50 values against Cdc25A, Cdc25B, and Cdc25C of 5, 10.4, and 8.8 μM, respec-tively, showed high selectivity toward the DSP VHR and the Ser/Thr phosphatase PP2A2, poor inhibition of PTP1B, and caused G1/S and G2/M arrest. This compound was competitive versus small molecule substrates. Docking studies suggested that it also bound into the pocket adjacent to the active site. The brominated analog of 5169131, compound 42 (5169133), was markedly less effective as phosphatase inhibitor showing IC50 values against Cdc25A, Cdc25B, and Cdc25C of 57.5, 58.7 and 41.1 μM, respectively.
Finally, Cheon et al. [68] screened the Korea Chemical Bank (a chemical library of ~40,000 compounds) in order to find novel scaffolds for PTP1B inhibitor design. Among the five novel tem-plates found, the 1,2-naphthoquinone was chosen for further evaluation and derivatives of this structure were tested for their PTP1B inhibitory activity, but also for selectivity over several phosphatases including Cdc25A and Cdc25B. The best PTP1B inhibitor prepared, KR61639 (43), and its reference compound, KR61170 (44), were found to be active against Cdc25B with IC50 values of 0.67 and 0.17 μM, respectively. 43 exhibited selectivity against several well-known tyrosine or dual specific phosphatases (CD45, LAR, VHR, Yop, Cdc25A and Cdc25C) and serine/threo- nine phosphatases (PP1 and PP2A). In comparison, the reference compound 44 did not show selectivity against CD45, Cdc25B and Yop. A kinetics study revealed that 43 inhibited PTP-1B in a non-competitive manner, indicating that it could bind to the enzyme-substrate complex or interact with the secondary aryl phosphate-binding site near the active site pocket of the enzyme.
Cdc25B CRYSTAL STRUCTURE
The three human Cdc25 isoforms, Cdc25A, Cdc25B and Cdc25C, although sharing functional and sequence homology, are encoded by unique genes that localize to three different chromo-somes: Cdc25A is found on 3p21, Cdc25B on 20p13 and Cdc25C on 5q31. The human Cdc25 family is further complicated by multi-ple splice variants: two for Cdc25A, five for Cdc25B and five for Cdc25C [69, 70]. The Cdc25 proteins have 300-600 residues in length and can be divided into two regions. The N-terminal domain varies in length and contains very little homology between the three isoforms [71]. The amino terminus contains a number of phos-phorylation sites, which regulate the phosphatase activity [72, 73], binding sites for partners, alternatively spliced domains, and func-tionally important domains that control the intracellular localization (nuclear vs cytoplasmic) of the Cdc25 phosphatases [74-76].
The C-terminal domain comprises ~30% of the protein, is mar- ked by a conserved Leu-Ile-Gly-Asp motif and is highly homolo-gous between the three isoforms (~60% pairwise identity over ~200 amino acids). The carboxy-terminal domain houses the catalytic site of the enzyme, which contains the canonical PTPase active site motif HCX5R [77], in which H is a highly conserved histidine resi-
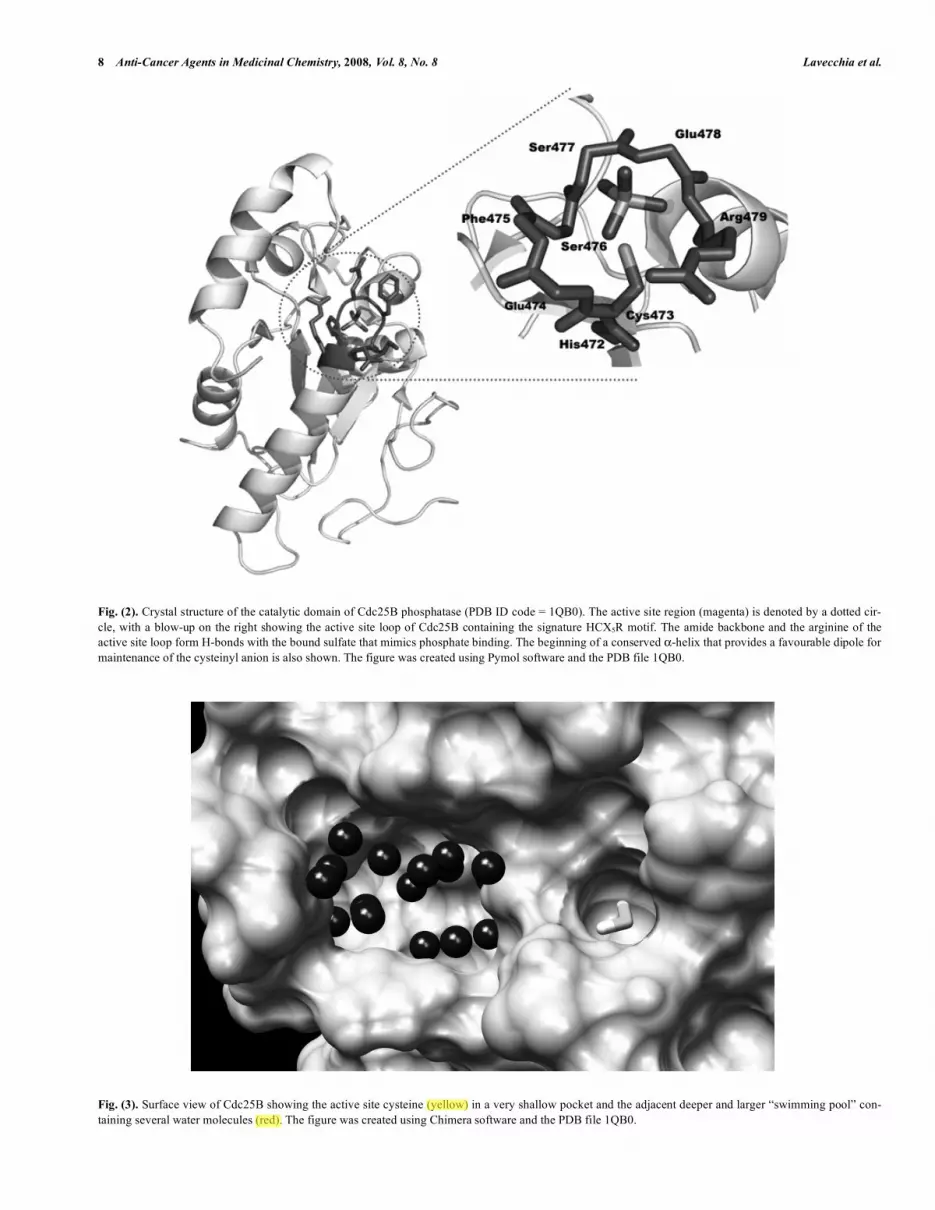
due, C is the catalytic cysteine, Xs are the five residues that form a loop in which all of the amide nitrogens H-bond the phosphate of the substrate, and R is a highly conserved arginine that H-bonds the phosphorylated amino acid of the substrate (Fig. 2).
The crystal structures of the catalytic domains of Cdc25A (PDB ID: 1C25) [78] and Cdc25B (PDB ID: 1QB0) [79] have been re-ported at 2.3 Å and 1.9 Å resolution, respectively. The 31 and 17 C-terminal residues of Cdc25A and Cdc25B, respectively, were not observed in these structures, with an additional -helix seen near the active site of Cdc25B. Both phosphatases contain the canonical HCX5R sequence (Fig. 2), which can be superimposed with essen-tially all other active site loops in PTPs with known structures [80]. Although the overall structure of the catalytic domains of the two Cdc25 phosphatases is similar, key differences in the positions of some residues believed to be essential for catalysis have been ob-served. For example, the carbonyl group of residue 434 in Cdc25A (residue 477 in Ccd25B) points toward the location where phos-phate should bind; the opposite of that observed with Cdc25B and other PTPs [81, 82]. A difference in the positioning of the side chain of Arg436 in Cdc25A has also been observed: this residue appears to be misplaced in Cdc25A when compared to the struc-tures of Cdc25B and other known phosphatases. Cdc25A fails to bind oxyanions in its catalytic site, whereas Cdc25B readily binds tungstate and sulfate in a mode similar to other PTPases and DSPases [79]. The Cdc25A catalytic domain also lacks any loop proximal to the active site that could facilitate substrate binding [78, 79]. Finally, another major difference between Cdc25B and Cdc25A structure lies in the C-terminal region (residues 531-547, Cdc25B numbering). This region, which is well resolved in the Cdc25B crystal structure, contains an -helix that is positioned against the bulk of the protein. Several residues of the helix, such as Met531 and Arg544, point toward the active-site cleft. In contrast, the same region in Cdc25A is undefined beyond Asp492 (Cdc25A numbering scheme), and the few residues that are observed appear to be misplaced. For example, the sequence is directed away from the active site of the protein and toward a symmetrically related molecule in the crystal. Thus, the position of the C-terminus in the Cdc25A structure appears to be determined by packing forces within the crystal.
For the above mentioned reasons, no reliable information with regard to ligand binding can be directly obtained from the Cdc25A protein structure, which means that molecular modeling techniques can not be reliably used to predict ligand binding modes or for ligand design. In contrast, the structure of the catalytic domain of Cdc25B shows a well-ordered active site, which is adjacent to a large cavity, called “swimming pool” by Rudolph [83] for the abundance of well ordered water molecules it contains (Fig. 3). The “swimming pool” is also close to the unstructured C-terminal helix that is involved in protein substrate recognition [67] and 14-3-3 binding [84].
Unlike Cdc25A structure, the crystal structure of Cdc25B could allow the calibration of available SAR data, facilitate the optimiza-tion of lead compounds, and perhaps aid in the design of new classes of Cdc25B inhibitors.
Cdc25B DOCKING STUDIES
Hotspot Mutants and a Docking Model of Cdc25B/CdK2-
pTpY-cyclin A Complex
The binding energy between two proteins is generally governed in large part by just a few (5%–10%) critical residues at the binding interface, the so called hotspots [85-87]. Identification of these hot spots would allow for experiments that probe the details of specific transient protein-protein interactions in vivo. Additionally, such sites could serve as targets for the development of small-molecule compounds that interfere specifically with these protein-protein interactions and serve as leads for drug development.
8 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
Fig. (2). Crystal structure of the catalytic domain of Cdc25B phosphatase (PDB ID code = 1QB0). The active site region (magenta) is denoted by a dotted cir-cle, with a blow-up on the right showing the active site loop of Cdc25B containing the signature HCX5R motif. The amide backbone and the arginine of the active site loop form H-bonds with the bound sulfate that mimics phosphate binding. The beginning of a conserved -helix that provides a favourable dipole for maintenance of the cysteinyl anion is also shown. The figure was created using Pymol software and the PDB file 1QB0.
Fig. (3). Surface view of Cdc25B showing the active site cysteine (yellow) in a very shallow pocket and the adjacent deeper and larger “swimming pool” con-taining several water molecules (red). The figure was created using Chimera software and the PDB file 1QB0.

Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 9
Keeping this in mind, Rudolph et al. [88, 89] studied the tran-sient protein-protein interactions between Cdc25B phosphatase and its protein substrate CdK2-pTpY-cyclin A. By using computational protein docking and molecular dynamics simulations, they pro-posed a docked model of Cdc25B with its substrate CdK2-pTpY-cyclin A. Moreover, the same authors identified the hotspot resi-dues on the protein substrate (Asp206 and Asp210 of the CdK2 subunit) and the corresponding conserved hotspot residues in the enzyme (Arg488, Arg492, Tyr497), located 20-30 Å away from the active site [88, 89]. The docked model made several predictions that were experimentally tested. Most importantly, the hotspot resi-dues at the remote docking site on Cdc25B interacted ionic H-bonds with Asp206 and Asp210 of CdK2 (Fig. 4). As for the origi-nal hotspot mutations on Cdc25B, the mutation of Asp206 in the CdK2-pTpY-cyclin A substrate led to a 200-fold reduction in activ-ity with wild-type Cdc25B, and no change in kinase activity for the mutant CdK-cyclin. The interfacial contacts between the hotspots on Cdc25B and CdK2 were confirmed by double-mutant cycle analysis [89, 90].
The docked model of Cdc25B with its protein substrate cor-rectly predicted the binding of pT14 to the active site, which repre-sents the preferred site of dephosphorylation compared with pY15 [91, 92]. The orientation of the phosphoryl group exactly mimicked the structure of the bound sulfate in the crystal structures of the wild-type enzyme and the substrate-trapping mutant.
The interface between Cdc25B and CdK2-pTpY-cyclin A seems to have many favourable characteristics that should to be disrupted by small-molecule inhibitors. In a recent review [93], Rudolph suggests that there is a pocket on Cdc25B directly adjacent to the hotspots where the binding of suitable ligands could engage one or more of these residues. This pocket is lined primarily by the side chains of 13 residues, which are mostly conserved across the three human Cdc25 isoforms. Thus, core inhibitor structures that inhibit all three Cdc25s and thereby serve as general anticancer agents could be developed.
Docking of Small-Molecule Inhibitors into the Crystal Struc-
ture of CdC25B Catalytic Domain
To predict a potential interaction site of the most active Cdc25B inhibitor 32 (NSC95397 in Table 3, IC50 = 0.125 μM), which exhib-ited mixed inhibition kinetics [63], Lazo et al. [64] scanned the entire surface of the crystal structure of Cdc25B catalytic domain. The most accommodating site found for the active p-naphthoqui- none was the secondary sulfate-binding site located near the cata-lytic site of Cdc25B phosphatases, the so called “swimming pool” [79]. This site, framed by residues Thr547, Phe543, Tyr428, Leu445, Arg479, Met483, Arg482, Glu446, Ser549, Trp550, Arg544, Pro444, and Leu545, is occupied by a network of H-bonded water molecules that were assumed to be displaced during small-molecule occupation. The proposed binding mode of 32, found through a tethered minimization protocol [94, 95], was stabi-lized by the occurrence of H-bonds between the ligand carbonyl oxygens and the guanidinium side chains of both Arg482 and Arg544. The catalytic Cys473 was estimated to be 8.5 Å from the ligand. This complex was further reinforced by hydrophobic inter-actions between the aromatic portion of the inhibitor and the inte-rior of the pocket lined by the lipophilic residues Arg479, Thr547, Tyr428, and Phe543 (Fig. 5).
In a successive study, Lazo et al. [46] discovered two new naphthofurandiones, compounds 41 (5169131 in Table 3, IC50 = 10.4 μM) and 42 (5169133, IC50 = 58.7 μM), displaying competi-tive and reversible Cdc25B inhibition. Docking studies suggested that they also bound in the pocket adjacent to the active site, con-firming the importance of the hydrophobic interactions within the cleft, as well as the weight of Arg482 and Arg544 residues in com-plex stabilization. It is interesting to note how the model also pro-vided a rationale for the less potent brominated inhibitor 42. In fact, the more bulky and electron-negative bromine of the ligand was found to clash sterically with the neighboring Thr547 amino acid on Cdc25B, that is consistent with its decreased potency.
Fig. (4). Docking model of Cdc25B with its substrate CdK2-pTpY-Cyc A highlighting the network of hotspot residues at the remote docking site. Cdc25B (black), CdK2 (gray) and cyclin A (gray) are shown in cartoon. The active site region is denoted by a broken circle and pT14 and pT15 of CdK2 and the active site loop of Cdc25 (Cys473 to Arg478) are shown as stick models. The remote hotspot is denoted by a dotted circle, with a blow-up on the right showing the ionic H-bond interactions that govern the association between Cdc25B and its protein substrate. Residues from CdK2 are underlined. Adapted from Ref. [90].

10 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
Successively, Brun et al. docked into the catalytic domain of Cdc25B the di-acid compound 37 (Table 3, IC50 = 1.75 μM) [65], an analogue of 32, by using the automatic docking programs Surflex [96] and Ligandfit [97]. In contrast with Lazo et al., the ligand bound into the active site with the first carboxylate group involved in a salt-bridge with Arg479, and the second one interact-ing with Arg482 (proposed by Ligandfit) or Arg544 (proposed by Surflex). The authors hypothesized that the strong interaction of the carboxylate group with Arg479 located into the active site most likely brought the naphthoquinone core out of the floor of the sec-ondary sulfate-binding pocket, thus explaining the higher activity of compound 37 in comparison with the remaining synthesized com-pounds of the series.
To explore the ligand-binding mode and clarify the molecular basis of the inhibitory activity of a series of chemically different selective reversible Cdc25B inhibitors, Lavecchia et al. [98] carried out a ligand docking analysis by using the crystal structure of the Cdc25B catalytic domain [79]. All ligand-enzyme complexes were predicted by two automatic docking programs, AutoDock [99, 100] and GOLD, [101] with the aim to achieve a mutual validation of the predicted binding poses.
AutoDock was successful in predicting a single binding posi-tion for compound 31 (Table 3) featuring the quinolinequinone carbonyl oxygens in direct contact with Arg482 and Arg544 within the water pocket, and the pendant chain lying in the enzyme’s cata-lytic site (Fig. 6a). The involvement of Arg482 and Arg544 in bind-ing of this quinone against Cdc25B was in accordance with the binding model postulated by Lazo et al. [64]. On the other hand, GOLD predicted another plausible binding pose for 31 (Fig. 6b), in which the quinolinequinone moiety was lodged near the catalytic Cys473, while the pendant chain occupied the region between Arg482 and Arg544.
These calculations led to the conclusion that two different bind-ing modes were possible for compound 31, thus justifying its partial
or full mixed inhibition kinetics [64]. In short, when the quinone moiety of these ligands fits into the Cdc25B catalytic site (binding pose predicted by GOLD), an oxidation of the crucial Cys473 thio-late might happen [46, 102], thus irreversibly inactivating the en-zyme. On the contrary, when the quinone moiety is placed in the water pocket adjacent to the catalytic site, where it can interact with Arg482 and Arg544 (binding pose predicted by Autodock), a re-versible enzyme binding or, alternatively, a reversible oxidation of the enzyme’s catalytic residues might take place. This latter hy-pothesis is sustained by the evidence that a mild oxidation of pro-tein phosphatases can lead to reversible oxidation of the catalytic cysteine to sulfenic acid (Cys-SO ), which is readily reversible by cellular reductants [102, 103].
A similar binding position was also found for the Cdc25B re-versible indolyldihydroxyquinone 39 (Table 3, IC50 = 1.0 μM) [66], in which the hydroxyquinone ring adapted either in the secondary site between Arg482 and Arg544 (Fig. 7a) or in the Cdc25B cata-lytic site (Fig. 7b).
To map the binding site for the indolyldihydroxyquinone class of inhibitors, Sohn et al. tested their activity toward nine different site-directed mutants located near the active site and in the adjacent water pocket of Cdc25B as well as toward a truncated form of the enzyme lacking the last 17 residues [66]. They found that mutation of Glu474, Phe475 and Arg482, but not Glu478, Arg492, Tyr528, Met531 or Asn532, reduced the amount of inhibition seen for the indolyldihydroxyquinones. Interestingly, these results reinforced the role of Arg482, which is one of the two residues previously suggested by molecular modeling to be required for the binding of the large anionic quinone part of the naphthoquinone [62] and qui-nolinequinone [98] inhibitors. On the other hand, the truncation of the last 17 amino acids of the C-terminal portion of the mutated enzyme caused a loss in activity of 39, while it did not have any effect on the activity of its analogue unsubstituted in position 7 of the indole ring [66]. Taken together, these evidences seem to sup-
Fig. (5). Docking model of compound 32 (NSC95397) with a binding site adjacent to the Cdc25B catalytic domain. The binding of 32 to Cdc25B is denoted by a dotted circle, with a blow-up on the right showing the main interactions between the quinone carbonyls of the inhibitor and the guanidinium side chains of Arg544, Arg482 and the phenolic OH of Tyr428. Adapted from Ref. [64].

Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 11
port the binding pose in which the quinone ring fits into the cata-lytic site, while the substituent in position 7 of indole ring is in close proximity to the C-terminal portion of the enzyme (Fig. 7b). It could be argued that the insertion of this moiety in the catalytic loop might allow reactions with the essential Cys473 thiolate or redox reactions, thus irreversibly inactivating the enzyme. How-ever, the reactivities of the indolyldihydroxyquinones proved to be dissimilar by those reported for quinones 31 and 32 [46, 104]. In fact, as suggested by Finley et al. [105], indolyldihydroxyquinones
are quite electron rich, with two electron-donating hydroxyl groups and an electron-donating indole substituent, which make them much less likely to accept nucleophiles. Moreover, when the redox properties of the indolyldihydroxyquinones were investigated, it was shown that they do not sustain redox cycling (cyclic voltamme-try) in aqueous DMF [106]. These findings might explain why compound 39 and its analogues are not capable of irreversibly in-hibiting the enzyme, even though the quinone ring binds in proxim-ity of the Cdc25B catalytic site.
Fig. (6). Alternative binding modes of compound 31 in the Cdc25B binding cavity as calculated by Autodock (a) and GOLD (b) [98]. For clarity, only inter-acting residues are displayed. The ligand and interacting key residues are represented as stick models, while the protein as a secondary structure cartoon. H-bonds are shown as dashed lines.
Fig. (7). Alternative binding modes of compound 39 in the Cdc25B binding cavity as calculated by Autodock (a) and GOLD (b) [98]. For clarity, only inter-acting residues are displayed. The ligand and interacting key residues are represented as stick models, while the protein as a secondary structure cartoon. H-bonds are shown as dashed lines.

12 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
In conclusion, comparing the binding modes of both reversible and irreversible inhibitors [98], a pharmacophore model (hypothe-sis) for Cdc25B reversible inhibition can be proposed (Fig. 8).
This model could not only to have predictive utility for design-ing new potential Cdc25B inhibitors, but also to provide a poten-tially useful graphical representation of the key structural features suitable for virtual screening. The pharmacophore is defined by four regions:
a) The core structure which, in the majority of cases, consists of an quinone moiety containing the critical oxygen atoms able to accept H-bonds from Arg482 and Arg544.
b) A bulky aromatic system fused to quinone ring (X in Fig. (8)), which affords specific hydrophobic interactions with residues Phe543, Thr547 and Trp550, enhancing the inhibitory activity.
c) An aromatic ring (Y in Fig. (8)), which is usually able to en-gage charge-transfer interactions with the catalytic Phe475. It is worth noting that this group might also be replaced by a H-bond acceptor group able to interact with the backbone NHs of the catalytic HCX5R signature or with Arg479 side chain in close proximity of the catalytic loop.
d) A linker, which connects the quinone system to the aromatic ring Y or the H-bond acceptor group.
Reversible Cdc25B inhibitors bind to the enzyme with the qui-none ring placed into the “swimming pool” between Arg482 and Arg544 and the pendant chain, which frequently terminates with the aromatic ring Y, located within the Cdc25B catalytic site. The bulkiness of the heterocycle fused to quinone ring X seems to be important for reversible inhibition of the enzyme. In fact, the cata-lytic site of Cdc25B is extremely shallow, whereas the “swimming pool” is large and deep, thus representing the only pocket on the surface of Cdc25B capable to accommodate the bulky heterocycle scaffold X. In this way, the enzyme irreversible inhibition mecha-nisms associated with the proximity of the quinone moiety to the
catalytic Cys473 thiolate could be hampered. Moreover, the pres-ence of a chain on the quinone, which terminates alternatively with an aromatic ring or a H-bond acceptor group, seems to be crucial in the competitive inhibition of the enzyme. In fact, this portion of the ligand, occupying the catalytic region of the enzyme, impedes the interaction of Cdc25B with the natural substrate.
CONCLUDING REMARKS AND PERSPECTIVES
The efficient characterization of more selective and specific small molecules that modulate Cdc25 function in vitro and in vivo is at the heart of chemical biology and medicinal chemistry research and the development of new therapies and diagnostics for cancer. In this review we have presented a few structurally different classes of natural products as well as synthetic molecules that have been de-scribed in the literature as Cdc25B inhibitors. Many mechanisms of action have been proposed for these inhibitors. Quinonoid-based compounds may irreversibly oxidize the catalytic cysteine to Cys-SO2
or Cys-SO3 , but can also be considered as Michaël acceptors capable of reacting with the activated thiolate or other electrophilic entities. Phosphate surrogates are thought to interfere with the ar-ginine residues, leading to reversible enzyme inhibition. But some inhibitors can combine in the same molecule several of these mechanisms, thus by fitting into the active site of the enzyme through one part of the molecule and bringing the reactive moiety within the “swimming pool” located in close proximity to the cata-lytic cysteine.
By using the crystal structure of the catalytic domain of Cdc25B, docking models of various Cdc25B inhibitors with the enzyme have been presented [46, 64, 65, 98], shading light onto the molecular mechanism of reversible and irreversible inhibition of ligands. Moreover, a pharmacophoric model for reversible Cdc25B inhibition has been proposed, providing rational directions for fu-ture inhibitor design.
Fig. (8). Pharmacophoric model showing the essential structural components of reversible Cdc25B inhibition.

Cdc25B Phosphatase Inhibitors in Cancer Therapy Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 13
Very recently, Rudolph [80, 93] proposed a new prospect for developing new potential inhibitors against the Cdc25 phospha-tases, which consists in preventing enzyme/substrate (Cdc25B/ CdK2-pTpY-cyclin A) association. The dramatic effect of mutating the critical residues at protein-protein interface (hotspot residues) corroborates the idea that disruption of this single interaction is sufficient for inhibition. An attractive pocket on Cdc25B directly adjacent to the hotspots, where the binding of a suitable ligand could engage one or more of these residues, also has been detected. This potential inhibitor-binding pocket is framed primarily by the side chains of 13 residues, that are mostly conserved across the three human isoforms. Thus, core inhibitor structures that inhibit all three Cdc25s and thereby serve as general anticancer agents could be developed.
ACKNOWLEDGEMENTS
This work was financially supported by the Italian Ministry for University and Research (MIUR), Rome, Italy.
ABBREVIATIONS
PTK = Protein tyrosine kinase
PTP = Protein tyrosine phosphatases
PSTP = Protein serine/threonine phosphatase
pS = Phosphoserine
pT = Phosphothreonine
pY = Phosphotyrosine
DSP = Dual-specificity protein phosphatases
CdK = Cyclin dependent kinase
CAK = CDK-activating kinase
ATM = Ataxia-telangiectasia mutated
ATR = Ataxia-telangiectasia and Rad3-related
MAPK = Mitogen-activated protein kinase
AML = Acute myeloid leukaemia
VHR = Vaccinia H1-related
PP = Protein phosphatases
Yop = Yersinia tyrosine phosphatase
SAR = Structure-activity relationship
REFERENCES
[1] Fischer, E.H.; Charbonneau, H.; Tonks, N.K. Science, 1991, 253, 401.
[2] Stone, R.L.; Dixon, J.E. J. Biol. Chem., 1994, 269, 31323. [3] Sun, H.; Tonks, N.K. Trends Biochem. Sci., 1994, 19, 480 and
references therein. [4] Denu, J.M.; Stuckey, J. A.; Saper, M. A.; Dixon, J. E. Cell, 1996,
87, 361. [5] Fauman, E.B.; Yuvaniyama, C.; Schubert, H.L.; Stuckey, J.A.;
Saper, M.A. J. Biol. Chem., 1996, 271, 18780. [6] Barford, D. Trends Biochem. Sci., 1996, 21, 407. [7] Barford, D.; Das, A. K.; Egloff, M.-P. Annu. Rev. Biophys. Biomol.
Struct., 1998, 27, 133. [8] Zhang, Z.-Y. Crit. Rev. Biochem. Mol. Biol., 1998, 33, 1. [9] Nilsson, I.; Hoffmann, I. Prog. Cell Cycle Res., 2000, 4, 107. [10] Boudolf, V.; Inze, D.; De Veylder, L. Trends Plant Sci., 2006, 11,
474. [11] Strausfeld, U.; Labbe, J.C.; Fesquet, D.; Cavadore, J.C.; Picard, A.;
Sadhu, K.; Russell, P.; and Doree, M. Nature, 1991, 351, 242. [12] Galaktionov, K.; Beach, D. Cell, 1991, 67, 1181. [13] Malumbres, M.; Barbacid, M. Trends Biochem. Sci., 2005, 30, 630. [14] Blomberg, I.; Hoffmann, I. Mol. Cell. Biol., 1999, 19, 6183. [15] Molinari, M.; Mercurio, C.; Dominguez, J.; Goubin, F.; Draetta,
G.F. EMBO Rep., 2000, 1, 71. [16] Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Proc. Natl Acad. Sci.
USA, 2002, 99, 14795.
[17] Lindqvist, A.; Kallstrom, H.; Lundgren, A.; Barsoum, E.; Rosen-thal, C.K. J. Cell. Biol., 2005, 171, 35.
[18] Mailand, N.; Podtelejnikov, A.V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. EMBO J., 2002, 21, 5911.
[19] Boutros, R.; Dozier, C.; Ducommun, B. Curr. Opin. Cell Biol., 2006, 18, 185.
[20] Millar, J.B.; Blevitt, J.; Gerace, L.; Sadhu, K.; Featherstone, C.; Russell, P. Proc. Natl Acad. Sci. USA, 1991, 88, 10500.
[21] Lammer, C.; Wagerer, S.; Saffrich, R.; Mertens, D.; Ansorge, W.; Hoffmann, I. J. Cell Sci., 1998, 111, 2445.
[22] Gabrielli, B.G.; De Souza, C.P.; Tonks, I.D.; Clark, J.M.; Hayward, N.K.; Ellem, K.A. J. Cell Sci., 1996, 109, 1081.
[23] De Souza, C. P.; Ellem, K.A.; Gabrielli, B.G. Exp. Cell Res., 2000, 257, 11.
[24] Gabrielli, B.G.; Clarck, J.M.; McCormack, A.; Ellem, K.A.O. J.
Biol. Chem., 1997, 272, 28607. [25] Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. Oncogene, 2003, 22,
5834. [26] Karlsson-Rosenthal, C.; Millar, J. B. Trends Cell Biol., 2006, 16,
285. [27] Mikhailov, A.; Shinohara, M.; Rieder, C.L. Cell Cycle, 2005, 4, 57. [28] Cangi, M.G.; Cukor, B.; Soung, P.; Signoretti, S.; Moreira, G.;
Ranashinge, M.; Cady, B.; Pagano, M.; Loda, M. J. Clin. Investig., 2000, 106, 753.
[29] Takemasa, I.; Yamamoto, H.; Sekimoto, M.; Ohue, M.; Noura, S.; Miyake, Y.; Matsumoto, T.; Aihara, T.; Tomita, N.; Tamaki, Y.; Sakita, I.; Kikkawa, N.; Matsuura, N.; Shiozaki, H.; Monden, M. Cancer Res., 2000, 60, 3043.
[30] Lyon, M.A.; Ducret, A.P.; Wipf, P.; Lazo, J.S. Nat. Rev. Drug
Discov., 2002, 1, 961. [31] Galaktionov, K.; Lee, A.K.; Eckstein, J.; Draetta, G.; Meckler, J.;
Loda, M.; Beach, D. Science, 1995, 269, 1575. [32] Vogt, A.; Rice, R.L.; Settineri, C.E.; Yokokawa, F.; Yokokawa, S.;
Wipf, P.; Lazo, J.S. J. Pharmacol. Exp. Ther., 1998, 287, 806. [33] Oguri, T.; Singh, S.V.; Nemoto, K.; Lazo, J.S. Cancer Res., 2003,
63, 771. [34] Galaktionov, K.; Chen, X.; Beach, D. Nature, 1996, 382, 511. [35] Ma, Z.Q.; Chua, S.S.; DeMayo, F.J.; Tsai, S.Y. Oncogene, 1999,
18, 4564. [36] Fernandez-Vidal, A.; Ysebaert, L.; Didier, C.; Betous, R.; De Toni,
F.; Prade-Houdellier, N.; Demur, C.; Contour-Galcéra, M.O.; Prévost, G.P.; Ducommun, B.; Payrastre, B.; Racaud-Sultan, C.; Manenti, S. Cancer Res., 2006, 66, 7128.
[37] Ham, S.W.; Park, H.J.; Lim, D.H. Bioorg. Chem., 1997, 25, 33. [38] Tamura, K.; Southwick, E.C.; Kerns, J.; Rosi, K.; Carr, B.I.; Wil-
cox, C.; Lazo, J.S. Cancer Res., 2000, 60, 1317. [39] Nishikawa, Y.; Wang, Z.; Kerns, J.; Wilcox, C.S.; Carr, B.I. J.
Biol. Chem., 1999, 274, 34803. [40] Prasad, K.N.; Edwards-Prasad, J.; Sakamoto, A. Life Sci., 1981, 29,
1387. [41] Noto, V.; Taper, H.S.; Jiang, Y.H.; Janssens, J.; Bonte, J.; De
Loecker, W. Cancer, 1989, 63, 901. [42] Nutter, L.M.; Cheng, A.L.; Hung, H.L.; Hsieh, R.K.; Ngo, E.O.;
Liu, T.W. Biochem. Pharmacol., 1991, 41, 1283. [43] Wipf, P.; Joo, B.; Nguyen, T.; Lazo, J.S. Org. Biomol. Chem.,
2004, 2, 2173. [44] Choi, S.-G.; Kim, J.; Sung, N.-D.; Son, K-H.; Cheon, H.-G.; Kim,
K.-R.; Kwon, B.-M. Nat. Prod. Res., 2007, 21, 487. [45] Cao, S.; Foster, C.; Brisson, M.; Lazo, J.S.; Kingston, D.G. I.
Bioorg. Med. Chem., 2005, 13, 999. [46] Brisson, M.; Nguyen, T.; Vogt, A.; Yalowich, J.; Giorgianni, A.;
Tobi, D.; Bahar, I.; Stephenson, C.R.; Wipf, P.; Lazo, J.S. Mol.
Pharmacol., 2004, 66, 824. [47] Huang, W.G.; Jiang, Y.Y.; Li, Q.; Li, J.; Li, J.Y.; Lu, W.; Cai, J.C.
Tetrahedron, 2005, 61, 1863. [48] Cao, S.; Foster, C.; Lazo, J.S.; Kingston, D.G.I. Bioorg. Med.
Chem., 2005, 13, 5094. [49] Cao, S.; Foster, C.; Lazo, J.S.; Kingston, D.G.I. Bioorg. Med.
Chem., 2005, 13, 5830. [50] Hamaguchi, T.; Sudo, T.; Osada, H. FEBS Lett., 1995, 372, 54. [51] Sodeoka, M.; Sampe, R.; Kojima, S.; Baba, Y.; Usui, T.; Ueda, K.;
Osada, H.J. J. Med. Chem., 2001, 44, 3216. [52] Ham, S.W.; Carr, B.I. Drug Design Rev. Online, 2004, 1, 123.

14 Anti-Cancer Agents in Medicinal Chemistry, 2008, Vol. 8, No. 8 Lavecchia et al.
[53] Bäurle, S.; Blume, T.; Günther, J.; Henschel, D.; Hillig, R.C.; Hu-semann, M.; Mengel, A.; Parchmann, C.; Schmid, E.; Skuballa, W. Bioorg Med. Chem. Lett., 2004, 14, 1673.
[54] Rice, R.L.; Rusnak, J.M.; Yokokawa, F.; Yokokawa, S.; Messner, D.J.; Boynton, A.L.; Wipf, P.; Lazo, J.S. Biochemistry, 1997, 36, 15965.
[55] Tamura, K.; Rice, R. L.; Wipf, P.; Lazo, J.S. Oncogene, 1999, 18, 6989.
[56] Ducruet, A.P.; Rice, R.L.; Tamura, K.; Yokokawa, F.; Yokokawa, S.; Wipf, P.; Lazo, J.S. Bioorg. Med. Chem., 2000, 8, 1451.
[57] Wipf, P.; Aslan, D.C.; Southwick, E.C.; Lazo, J. S. Bioorg. Med.
Chem. Lett., 2001, 11, 313. [58] Fritzen, E.L.; Brightwell, A.S.; Erickson, L.A.; Romero, D.L.
Bioorg. Med. Chem. Lett., 2000, 10, 649. [59] Kim, K.-R.; Kwon, J.-L.; Kim, J.-S.; No, Z.; Kim, H.R.; Cheon,
H.G. Eur. J. Pharmacol., 2005, 528, 37. [60] Kim, H.R.; No, J.; Seo, M.J.; Song, B.G.; Son, B.S.; Kim, J.K.;
Kim, K.-R.; Cheon, H.G.; Lee, G.H. Patent WO 2006/101307, 2006.
[61] El-Subbagh, H.I.; Abadi, A.H.; Al-Khawad, I.E.; Al-Rashood, K.A. Archiv. der Pharmazie, 1999, 332, 19.
[62] Lazo, J.S.; Aslan, D.C.; Southwick, E.C.; Cooley, K.A.; Ducruet, A.P.; Joo, B.; Vogt, A.; Wipf, P. J. Med. Chem., 2001, 44, 4042.
[63] Pu, L.; Amoscato, A.A.; Bier, M.E.; Lazo, J.S. J. Biol. Chem., 2002, 277, 46877.
[64] Lazo, J.S.; Nemoto, K.; Pestell, K.E.; Cooley, K.; Southwick, E.C.; Mitchell, D.A.; Furey, W.; Gussio, R.; Zaharevitz, D.W.; Joo, B.; Wipf, P. Mol. Pharmacol., 2002, 61, 720.
[65] Brun, M.P.; Braud, E.; Angotti, D.; Mondesert, O.; Quaranta, M.; Montes, M.; Miteva, M.; Gresh, N.; Ducommun, B.; Garbay, C. Bioorg. Med. Chem., 2005, 13, 4871.
[66] Sohn, J.; Kiburz, B.; Li, Z.; Deng, L.; Safi, A.; Pirrung, M.C.; Rudolph, J. J. Med. Chem., 2003, 46, 2580.
[67] Wilborn, M.; Free, S.; Ban, A.; Rudolph, J. Biochemistry, 2001, 40, 14200.
[68] Cheon, H.G.; Kim, S.-M.; Yang, S.-D.; Ha, J.D.; Choi, J.-K. Eur. J.
Pharmacol., 2004, 485, 333. [69] Wegener, S.; Hampe, W.; Herrmann, D.; Schaller, H.C. Eur. J. Cell
Biol., 2000, 79, 810. [70] Forrest, A.R.; McCormack, A.K.; DeSouza, C.P.; Sinnamon, J.M.;
Tonks, I.D.; Hayward, N.K.; Ellem, K.A.; Gabrielli, B.G. Biochem.
Biophys. Res. Commun., 1999, 260, 510. [71] Draetta, G.; Eckstein, J. Biochim. Biophys. Acta, 1997, 1332, M53–
M63. [72] Hoffmann, I.; Draetta, G.; Karsenti, E. EMBO J., 1994, 13, 4302. [73] Hoffmann, I.; Clarke, P.R.; Marcote, M.J.; Karsenti, E.; Draetta, G.
EMBO J., 1993, 12, 53. [74] Lopez-Girona, A.; Furnari, B.; Mondesert, O.; Russell, P. Nature,
1999, 397, 172-175. [75] Woo, E.S.; Rice, R.L.; Lazo, J.S. Oncogene, 1999, 18, 2770. [76] Graves, P.R.; Lovly, C.M.; Uy, G.L.; Piwnica-Worms, H. Onco-
gene, 2001, 20, 1839. [77] Gottlin, E.B.; Xu, X.; Epstein, D.M.; Burke, S.P.; Eckstein, J.W.;
Ballou, D.P.; Dixon, J.E. J. Biol. Chem., 1996, 271, 27445.
[78] Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Cell, 1998, 93, 617.
[79] Reynolds, R.A.; Yem, A.W.; Wolfe, C.L.; Deibel, M.R.J.; Chide-ster, C.G.; Watenpaugh, K.D. J. Mol. Biol., 1999, 293, 559.
[80] Rudolph, J. Biochemistry, 2007, 46, 3595. [81] Yuvaniyama, J.; Denu, J., M.; Dixon, J.E.; Saper, M.A. Science,
1996, 272, 1328. [82] Stuckey, J.A.; Schubert, H.L.; Fauman, E.B.; Zhang, Z.Y.; Dixon,
J.E.; Saper, M.A. Nature, 1994, 370, 571. [83] Rudolph, J. Mol. Pharmacol., 2004, 66, 780. [84] Chen, M.S.; Ryan, C.E.; Piwnica-Worms, H. Mol Cell Biol., 2003,
23, 7488. [85] Clackson, T.; Wells, J.A. Science, 1995, 267, 383. [86] Schreiber, S.; Fersht, A.R. J. Mol. Biol., 1995, 248, 478. [87] Bogan, A.A.; Thorn, K.S. J. Mol. Biol., 1998, 280, 1. [88] Sohn, J.; Kristjánsdóttir, K.; Safi, A.; Parker, B.; Kiburz, B.;
Rudolph, J. Proc. Natl Acad. Sci. USA, 2004, 47, 16437. [89] Sohn, J.; Parks, J. M.; Buhrman, G.; Brown, P.; Kristjánsdóttir, K.;
Safi, A.; Edelsbrunner, H.; Yang, W.; Rudolph, J. Biochemistry, 2005, 44, 16563–16573.
[90] Sohn, J.; Rudolph, J. J. Mol. Biol., 2006, 362, 1060. [91] Rudolph, J.; Epstein, D. M.; Parker, L.; Eckstein, J. Anal. Bio-
chem., 2001, 289, 43. [92] Borgne, A.; Meijer, L. J. Biol. Chem., 1996, 271, 27847. [93] Rudolph, J. Nature Rev. Cancer, 2007, 7, 202. [94] Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zahare-
vitz, D.; Bollbuck, B.; Poy, G.; Sackett, D.; Nicolaou, K.C.; Fojo, T. Proc Natl Acad Sci USA, 2000, 97, 2904.
[95] Gussio, R.; Zaharevitz, D.W.; McGrath, C.F.; Pattabiraman, N.; Kellogg, G.E.; Schultz, C.; Link, A.; Kunick, C.; Leost, M.; Meijer, L.; Sausville, E.A. Anticancer Drug Dis., 2000, 15, 53.
[96] Jain, A.N. J. Med. Chem., 2003, 46, 499. [97] Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. J. Mol.
Graphics Modell., 2003, 21, 289. [98] Lavecchia, A.; Cosconati, S.; Limongelli, V.; Novellino, E.
ChemMedChem, 2006, 1, 540. [99] Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart,
W.E.; Belew, R.K.; Olson, A.J. J. Comput. Chem., 1998, 19, 1639. [100] Goodsell, D.S.; Morris, G.M.; Olson, A.J. J. Mol. Recognit., 1996,
9, 1. [101] Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. J. Mol.
Biol., 1997, 267, 727. [102] Buhrman, G.; Parker, B.; Sohn, J.; Rudolph, J.; Mattos, C. Bio-
chemistry, 2005, 44, 5307. [103] Sohn, J.; Rudolph, J. Biochemistry, 2003, 42, 10060. [104] Gant, T.W.; Rao, D.N.; Mason, R.P.; Cohen, G.M. Chem. Biol.
Interact., 1988, 65, 157. [105] Finley, K.T. In The Chemistry of the Quinonoid Compounds; Patai
S, Ed.; Wiley, London, 1974, p. 878. [106] Liu, K.; Xu, L.; Szalkowski, D.; Li, Z.; Ding, V.; Kwei, G.;
Huskey, S.; Moller, D.E.; Heck, J. V.; Zhang, B.B.; Jones, A.B. J.
Med. Chem., 2000, 43, 3487.
Received: March 7, 2008 Revised: April 23, 2008 Accepted: May 5, 2008





























