Embed Size (px)
Citation preview

600
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
Cameo
Nevus depigmentosus and inflammatory linear epidermalnevus – an unusual combination with a note on histology
Adebola O. Ogunbiyi, MB.BS, FMCP(Nig.), Dip.Derm.(London), andJ. Olufemi Ogunbiyi, MB.BS, FWACP(Lab.Med.)
From the Departments of Medicine An 18-year-old woman was seen in the clinic with complaints of a bizarreand Pathology, College of Medicine hypopigmentation of her skin which her mother said had been present from birth and hadand University College Hospital,
been asymptomatic, constant, and permanent. She also complained of a rough eruptionIbadan, Nigeriawhich had developed at one year of age and had progressed slightly in size over the
Correspondence years. It was first noticed in the left axilla, groin, parasternal area and posteriorly on theJ. Olufemi Ogunbiyi neck and was restricted to the left side of the body. It became inflamed and pruritic, wasDepartment of Pathology asymptomatic at times, but never cleared although the patient felt some of the lesion hadCollege of Medicine and University
sloughed off. There were no associated systemic symptoms, no history of seizures, andCollege Hospitalher psychomotor development was normal.Ibadan
Nigeria On examination, the main findings were mucocutaneous; she had hypopigmented
bands, whorls, and streaks following Blaschko’s lines, the lesions affected the front and
the back of her trunk bilaterally, with the arms and thighs also involved. Her face was
spared (Fig. 1a and b). There was no evidence of atrophy or scarring in the
hypopigmented lesions.
The patient also had linear verrucous lesions in left armpit, left hand (dorsal surface),
left groin, left parasternal region (Fig. 2) and posteriorly on the neck (Fig. 1b). The lesions
in some areas were fragile and there was evidence of shedding in some places, leaving
hypopigmented lesions; no vesicles were seen. Her hair, nails, and teeth were normal, as
were the musculo-skeletal, ocular, and central nervous system.
A diagnosis of nevus depigmentosus with inflammatory linear epidermal nevus to rule
out incontinentia pigmenti was made. Skin biopsies were taken from the whorled,
hypopigmented, and verrucous patches.
The biopsy result of the hypopigmented patch showed an essentially normal looking
epidermis and dermis; there was no melanin incontinence. The verrucous patch showed
hyperkeratosis, psoriasiform hyperplasia of the epidermis with regular alternation of
parakeratotic areas with hypergranulosis and orthokeratosis with a reduction in the
granular layer.
Discussion
Nevus depigmentosus (ND) is a congenital pigmentarydisorder characterized by hypopigmented patches, whichcould present in three different ways: (i) isolated (circularor rectangular), (ii) segmental, and (iii) systematized. Thelast variant usually follows Blaschko’s lines.1 There isno known pattern of inheritance2 or sex predominance.The clinical features are constant and permanent through-out life and it is rarely associated with other congenitalabnormalities, although a case of ND with mentalretardation and seizures has been reported.3
ND is believed to result from impaired transfer ofmelanosomes from melanocytes to keratinocytes4 and thenumber of melanocytes are reported to be either normalor slightly reduced.2
The differential diagnoses of ND include hypomelanosisof Ito (incontinentia pigmenti achromians) in which thereare hypopigmented bands and whorls occurring alongBlaschko’s lines, which tend to occur within the firstyear of life, with a variability in clinical presentation.5
A significant number of patients with this disorder haveother associated congenital abnormalities.6 In view ofthe similarity in clinical presentation of hypomelanosisof Ito and ND, some authors have suggested that bothshould be referred to as hypomelanosis of Ito with orwithout systemic abnormalities, respectively7 as the casemay be.
Another differential diagnosis of ND is the fourthstage of incontinentia pigmenti with hypopigmentedatrophic lesions found mainly on the lower limbs.8 Inmost cases there is a previous history of the vesicular,

Ogunbiyi & Ogunbiyi Nevus depigmentosus and inflammatory linear epidermal nevus Cameo 601
Figure 1 Hypopigmented whorls,bands, and streaks on both front(a) and back (b) of patient. Notealso the one-sided distribution ofthe ILVEN (affecting the neck, leftparasternal area, and dorsal surfaceof the hand)
verrucous, and hyperpigmented stages of incontinentiapigmenti and a positive family history. Histology of thehypopigmented lesions in such patients shows an absenceof hair and sweat glands.9
Linear epidermal nevi frequently follow Blaschko’slines and there have been attempts to classify them intovarious clinical types. The presentation of pruritus,recurrent inflammation, and localization to the left sideof the body in this patient suggests inflammatory linearepidermal verrucous nevi (ILVEN) which describes aninflamed variety of the epidermal nevus.10–12 The fragilityof some of the lesions and the sloughed-off areas in someparts are suggestive of the epidermolytic hyperkeratosisvariant,7 although it is generally agreed that the presenceof epidermolytic hyperkeratosis in a nevus is determinedhistologically. The histological appearances in our patientshow a slight variation from the normal description ofILVEN in that the parakeratotic areas are associatedwith hypergranulosis, while the orthokeratotic areas showa reduction in the thickness of the granular layers. Thissuggests that the various clinical entities in linearepidermal nevi may occur as a spectrum and notas distinct variants in themselves. Hypopigmentationsurrounding ILVEN has been previously documented;13
the hypopigmented lesions found in association with the
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
verrucous lesions in our patient were areas where theverrucous lesions had sloughed off.
The occurrence of different clinical types of epidermalnevi in the same individual does exist, but we findthe association of linear epidermal nevi and nevusdepigmentosus in the same patient unusual. In addi-tion, the histological appearances of the lesions in thispatient are definitely uncommon. This combination ofproblems may suggest an underlying gene disorder, butunfortunately we do not have facilities for chromosomalstudies to even start to pursue the basis of thispatient’s problem.
References
1 Ortonne JP, Mosher DB, Fitzpatrick TB. Genetic andcongenital disorders. In: Vitiligo and otherHypomelanoses of Hair and Skin. New York: Plenum,1983: 59, 466.
2 Pinto FJ, Bolognia JH. Disorders of hypopigmentationin children. Paed Clin North Am 1991; 38: 991–1017.
3 Sugerma GI, Read WB. Two unusual neurocutaneousdisorders with focal cutaneous signs. Arch Neurol1969; 21: 242–247.
4 Jimbow K, Fitzpatrick TB, Szabo G, et al. Congenitalcircumscribed hypomelanosis, a characterization based

602 Cameo Wells’ syndrome from centipede bite Friedman et al.
Figure 2 Verrucous lesions of ILVEN affecting the leftarmpit and groin
Cameo
Wells’ syndrome triggered by centipede bite
Ilyse S. Friedman, Robert G. Phelps, MD, Jacob Baral, MD, and Allen N. Sapadin, MD
From the George Washington A 68-year-old African–American woman was awoken from her sleep by a centipede biteUniversity School of Medicine, on the dorsum of her left hand. Several days later, the patient presented to theWashington DC, and the Department
emergency room complaining of fevers, severe itching, swelling, and blistering (Fig. 1)of Dermatology and Division ofof both hands. The past history was unremarkable. There was no history of asthma orDermato Pathology, Mount Sinai
School of Medicine, New York, use of medications. Physical examination at that time was consistent with a bullousNew York cellulitis of the hands. Laboratory investigations revealed: white blood cell count (WBC),Correspondence 10.3 (normal, 4–10.5); differential: 54 neutrophils, 15 lymphocytes, and 31 eosinophils.Allen N. Sapadin, MD The platelet count was 225,000/mm3. A blood chemistry profile was within normal limits.Box 1048
Blood cultures and stool examination for ova and parasites were negative. The patientThe Mount Sinai Medical Center
was treated with oral cephalexin and diphenhydramine.One Gustave L. Levy PlaceNew York, NY 10029–6574
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
on electron microscopic study of tuberous sclerosis,nevus depigmentosus and piebaldism. J InvestDermatol 1975; 64: 50–62.
5 Takematsu H, Sato S, Irgarshi M, et al. Incontinentiapigmenti achromians (Ito). Arch Dermatol 1983; 119:391–395.
6 Ruiz-Moldonado R, Tpissamomt S, Tamayoh ???,et al. Hypomelanosis of Ito. Diagnostic criteria andreport of 41 cases. Pediatr Dermatol 1992; 9: 10.
7 Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko.J Am Acad Dermatol 1994; 31: 157–190.
8 Wiley HE, Frias JL. Depigmented lesions inincontinentia pigmenti – a useful diagnostic sign. Am JDis Child 1974; 128: 546–547.
9 Moss C, Ince P. Anhidrotic and achromiand lesions inincontinentia pigmenti. Br J Dermatol 1987; 116:839–849.
10 Altman J, Mehregan AH. Inflammatory linearverrucose epidermal nevus. Arch Dermatol 1987; 104:385–389.
11 Morag C, Metzker A. Inflammatory linear verrucoseepidermal nevus: a report of seven cases and reviewof the literature. Pediatr Dermatol 1985; 3: 15–18.
12 Rogers M, McCrossin I, Commens C. Epidermal neviand the epidermal nevus syndrome. J Am AcadDermatol 1989; 20: 476–488.
13 Sayag J, Magozzi MB, Adechy H, et al. Nevusepidermique verruquex inflammatoire lineaire aveclesions achromiques. Ann Dermatol Venereol 1978;105: 871–874.

Friedman et al. Well’s syndrome from centipede bite Cameo 603
Two weeks later the patient was seen in the dermatology clinic. She complained of
unabated pruritus and extensive new lesions. Physical examination revealed a moderately
obese woman with normal vital signs. Firm gray plaques with an irregular contour were
present on the left hand. The buttocks (Fig. 2) and extensor aspects of her upper arms
were strikingly indurated and had a blue–gray discoloration. Violaceous, indurated oval
patches were noted on the medial aspect of both knees. Finally, a slate-colored,
irregularly outlined plaque on the heel of the right foot was present. Complete blood count
revealed: WBC, 16.5; differential, 8 neutrophils, 9 lymphocytes, 81 eosinophils, and 2
monocytes. The differential was repeated and confirmed. The platelet count was
568,000/mm3.
A skin biopsy from the patient’s left upper arm revealed a superficial and deep
perivascular dermatitis. In addition, the dermis had a dense inflammatory infiltrate
consisting of numerous interstitial eosinophils, many of which were degranulated.
Collagen bundles coated with eosinophilic granules characteristic of flame figures were
abundant (Fig. 3). The changes were consistent with eosinophilic cellulitis. Chest X-ray
revealed no evidence of infiltrates.
A course of oral corticosteroids was prescribed. One week later, the lesions were
substantially less indurated, but otherwise similar in appearance. Complete blood count
revealed: WBC, 9.9; differential: 21 neutrophils, 19 lymphocytes, 1 basophil, 51
eosinophils, and 8 monocytes. The platelet count was 408,000/mm3. Hematologic
consultation was arranged for bone marrow biopsy and examination. Unfortunately, the
patient did not return for subsequent appointments.
Discussion
Wells’ syndrome is an idiopathic inflammatory dermatosisfirst described by Wells in 19711 as ‘‘recurrent granulo-matous dermatitis with eosinophilia.’’ Spigel andWinkelmann2 named the condition, ‘‘Wells’ syndrome’’ in1979. Wells and Smith3 proposed the ‘‘simpler title’’ ofeosinophilic cellulitis while describing eight additionalcases. The acute or cellulitic stage lasts several days,and is characterized clinically by erythematous, edematousplaques which may form blisters. The lesions are oftenaccompanied by an intense itching or burning sensation.The predilected anatomic sites are the extremities andtrunk. Symptoms, such as arthralgia, malaise, and fever,are occasionally present. The differential diagnosis at thisstage includes urticaria, acute bacterial cellulitis, or ery-sipelas. Some have noted a resemblance to erythemachronicum migrans4 and granuloma annulare.3,5 Allergiccontact dermatitis has also been simulated.6–8
During the subacute or infiltrative stage, the redness andedema gradually subside and the lesions become indurated,obtain a grayish coloration, and may resemble morphea.This stage lasts several weeks to months, after which thereis gradual regression; however, relapse is common withexacerbations and remissions of variable duration.
The characteristic histologic features in the acute stageare dermal edema and diffuse infiltration with eosinophils.Few conditions are characterized by such large numbers of
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
eosinophils in the reticular dermis.9 The subacute stage ischaracterized by scattered ‘‘flame figures,’’ which consist ofa central core of collagen bundles coated with eosinophilicdebris, phagocytic histiocytes, and giant cells. The eosino-philic material contains major basic protein (MBP), anarginine- and lysine-rich protein which comprises morethan 50% of the core of the major granules of theeosinophil.10 MBP has been localized to flame figures byindirect immunofluorescence, and has been implicated intheir formation by binding to and altering collagen.5 The
Figure 1 Left hand reveals a bulla and several vesicles at thesite of the centipede bite

604 Cameo Well’s syndrome from centipede bite Friedman et al.
recurrent nonscarring nature of Wells’ syndrome suggeststhat flame figures do not result from the destruction ofconnective tissue by MBP. This has been confirmed byelectron microscopy.11
The presence of flame figures is the most salient histo-pathologic finding of Wells’ syndrome, but they may beseen in various unrelated disorders, including follicularmucinosis, bullous pemphigoid, herpes gestationis, eczema,urticaria, arthropod bites, and dermatophyte infections.3,4
This has led some to question the existence of Wells’syndrome as a separate entity.4 Others assert that thediagnosis should not be based on the pathologic findingsalone, but should be limited to those patients who also
Figure 2 Blue–gray discoloration and induration of thebuttocks as well as the proximal thighs is seen. Thesechanges became apparent several weeks after the initialblistering reaction of the hands
Figure 3 There is a denseinfiltration of eosinophils in thedermis, many of which havedegranulated. Several collagenbundles coated by anextracellular amorphousmaterial (flame figures, arrow)are evident in the center(hematoxylin and eosin stain;original magnification, 3100)
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
exhibit the characteristic clinical appearance andcourse.7,8,12–14
Our case is unusual in terms of the striking peripheraleosinophilia and the triggering event. Although eosinophiliais not necessary for the diagnosis, it is seen in the majorityof patients and is the most constant blood abnormality. Ina summary of 24 patients, eosinophilia ranged from 2 to48%.6 Nine of the 24 patients had eosinophils greater thanor equal to 30% of the white blood cells, while the highestpercentage reported was 48%. Our patient’ eosinophilsranged from 31 to 81%, and this is the highest rangereported to date.
Although its etiology and pathogenesis are unknown,Wells’ syndrome, like erythema multiforme, erythemanodosum, and leukocytoclastic vasculitis, represents a reac-tion pattern to a number of different stimuli. Reportedtriggers of Wells’ syndrome include fungal3 and parasiticinfections,15 drug eruptions,1–3 underlying hematologicdisorders,1,5,7,9 and insect bites.2–5,7,13,14,16,17 The theorythat insect bites trigger some cases was first proposed byWells and Smith.3 Insect bites are frequently consideredwhen the lesions first appear. In some cases, the patienthas specific knowledge of a bite and may bring the insectto the physician for examination.4 Specific reports haveattributed cases of Wells’ syndrome to mosquitos,3
spiders,4,16,17 ticks,4 fleas,4,5 ‘‘buffalo gnats,’’5 and mites.13
Two patients described being stung by a small bee orwasp.4,14 The present report is the first to describe a casetriggered by a centipede bite.
Centipedes, nocturnal predators of insects and othersmall animals, are commonly found in the USA, India, andAustralia. These 3–250-mm-long arthropods have one pair

Friedman et al. Well’s syndrome from centipede bite Cameo 605
of legs per body segment, which aids in distinguishing themfrom other arthropods. Although they most often hideunder stones, tree bark, and in holes, they commonly setup residence in households. Specifically, Scutigera coleop-trata is found throughout most of eastern North America.The bite of a centipede is seldom fatal to humans, oftencausing only local skin reaction. Several patients haveexperienced burning pain, redness, and edema, sometimesleading to an erysipelas-like state,18,19 while others havereported more severe reactions, such as myonecrosis.20 Wedescribe the first report of eosinophilic cellulitis occurringin association with a centipede bite. The variation ofresponses described is not species specific, and is mostlikely secondary to patients’ individual reactions to thebite.
Treatment of Wells’ syndrome with systemic steroidsleads to resolution in most patients, but this may takeseveral weeks to occur. Dapsone15,21 has been used success-fully on rare occasions. Griseofulvin has also had limitedsuccess.3,21,22 Several cases have demonstrated spontaneousresolution. Response to treatment in our patient could notbe evaluated.
In summary, the present case, precipitated by acentipede bite, displays the characteristic clinical features,course, and histologic findings typical of Wells’ syndrome.This entity, although rare, should be considered in apatient with an atypical ‘‘cellulitis’’ resulting from aninsect bite that does not resolve with oral antibiotics. Insuch cases, eosinophilic cellulitis can be readily confirmedby skin biopsy and remedied with steroid therapy.
References
1 Wells GC. Recurrent granulomatous dermatitis witheosinophils. Trans St Johns Hosp Dermatol 1971; 57:45–56.
2 Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrentgranulomatous dermatitis with eosinophilia. ArchDermatol 1979; 115: 611–613.
3 Wells GC, Smith NP. Eosinophilic cellulitis. BrJ Dermatol 1979; 100: 101–109.
4 Schorr WF, Tauscheck AL, Dickson KB, Melski JW.Eosinophilic cellulitis (Wells’ syndrome): histologic andclinical features in arthropod bite reactions. J Am AcadDermatol 1984; 11: 1043–1049.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
5 Wood C, Miller AC, Jacobs A, et al. Eosinophilicinfiltration with flame figures. Am J Dermatopathol1986; 8: 186–193.
6 Fisher GB, Greer KE, Cooper PH. Eosinophilic cellulitis(Wells’ syndrome). Int J Dermatol 1985; 24: 101–107.
7 Aberer W, Konrad K, Wolff K. Wells’ syndrome is adistinctive disease entity and not a histologic diagnosis.J Am Acad Dermatol 1988; 18: 105–113.
8 Stern JB, Sobel HJ, Rotchford JP. Wells’ syndrome: isthere collagen damage in the flame figures? J CutanPathol 1984; 11: 501–505.
9 Varotti C, Tosti A, Gobbi M, et al. Eosinophiliccellulitis: a new case. Dermatologica 1982; 164:404–406.
10 Ligresti DJ. Rash and eosinophilia. In: Lebwohl M, ed.Difficult Diagnoses in Dermatology. New York:Churchill Livingstone, 1988: 187–201.
11 Peters MS, Schroeter AL, Gleich GJ. Immunofluorescenceidentification of eosinophil granule major basic proteinin the flame figures of Wells’ syndrome. Br J Dermatol1983; 109: 141–148.
12 Melski JW. Wells’ syndrome, insect bites, andeosinophils. Dermatol Clin 1990; 8: 287–293.
13 Clark DP, Anderson PC. Eosinophilic cellulitis caused byarthropod bites. Int J Dermatol 1988; 27: 411.
14 Anderson CR, Jenkins D, Tron V, Prendiville JS. Wells’syndrome in childhood: case report and review of theliterature. J Am Acad Dermatol 1995; 33: 857–864.
15 van den Hoogenband HM. Eosinophilic cellulitis as aresult of onchocerciasis. Clin Exp Dermatol 1983; 8:405.
16 Burket JM. Wells’ syndrome: recurrent granulomatousdermatitis with eosinophilia. Arch Dermatol 1981; 117:759.
17 Poli F, Penso D, Revuz J, et al. Cellulite a eosinophilesapres piqure d’araignee. Ann Dermatol Venereol 1985;112: 753–754.
18 Mohri S, Sugiyama A, Saito K, Nakajima H. Centipedebites in Japan. Cutis 1991; 47: 189–190.
19 Mumcuoglu KY, Leibovici V. Centipede (Scolopendra)bite: a case report. Isr J Med Sci 1989; 25: 47–49.
20 Logan JL, Ogden DA. Rhabdomyolysis and acute renalfailure following the bite of the giant desert centipedeScholopendra heros. West J Med 1985; 142: 549–550.
21 Marks R. Eosinophilic cellulitis: a response to treatmentwith dapsone: case report. Aust J Dermatol 1980; 21:10–12.
22 Dijkstra JEW, Bergfeld WF, Steck WD, TuthillRJ. Eosinophilic cellulitis associated with urticaria. J AmAcad Dermatol 1986; 14: 32–38.

606 Cameo Scrofuloderma Ozkan et al.
Cameo
Scrofuloderma
Sebnem Ozkan, MD, Nalan Gurler, MD, Emel Fetil, MD, Nese Atabey, MD, andAli Tahsin Gunes, MD
From the Departments of A 60-year-old woman presented to the Dermatology Department of Dokuz Eylul UniversityDermatology and Medical Biology, Faculty of Medicine with complaints of hardened reddenings and inflammatory drainageFaculty of Medicine, University of
on her right armpit, groin, and hips. These lesions appeared 2 months after aDokuz Eylul, Izmir, Turkeycholecystectomy operation, which had been performed 4 years ago, and showed
Correspondence unilateral progression. The patient had been treated unnecessarily with prednisoloneSebnem Ozkan, MD (5 mg every other day) for 7 years because of arthralgia.Dokuz Eylul Universitesi Tıp Fakultesi Dermatologic examination revealed erythematous induration, fistulas with central serousDermatoloji Anabilim Dalı
or purulent drainage, and conical-shaped scars on the regio axillaris dextra, regio35340 Inciraltıabdominalis lateralis dextra, and regio lumbalis dextra. A linear operative scar withIzmir
Turkey observable fistula located distally on the regio abdominalis lateralis dextra and another scar
in a lateral position were observed. There were also abscesses, fistulas, and scars on the
regio inguinalis and regio glutealis dextra (Fig. 1). Systemic examination was normal.
A tuberculin test showed erythema (9 cm) and induration in 72 h. Bacterial, mycotic,
and tuberculous cultures of the vacuum and smear samples from the lesions and urine
showed no reproduction.
A histopathologic examination of the biopsy material revealed nonspecific slight
acanthosis, perivascular mononuclear cellular infiltration in the superficial dermis, and a
dense mononuclear cellular infiltration in the subcutis.
The deoxyribonucleic acid obtained from the pus liquid by the phenol–chloroform
method for the detection of mycobacterium tuberculosis with polymerase chain reaction
was amplified using primers specific to microorganisms in the mycobacterium tuberculosis
complex and also specific to the IS 6110 region. At the end of the molecular analysis, a
product of amplification of 123 bp specific to mycobacterium tuberculosis was found.
The erythrocyte sedimentation rate was 92 mm/h. All other laboratory values were
normal, including biochemical analysis, anti-human immunodeficiency virus, venereal
disease research laboratory test, and rheumatoid factor. X-Ray and high-resolution
computerized tomography examination of the chest, rectosigmoidoscopy, esophagus,
stomach, duodenum, and small intestine passage X-ray examinations, and vaginal smear
inspections, performed to investigate other organ tuberculosis, did not show any
pathologic finding. The arthralgic complaints of the patient were assessed as degenerative
arthrosis by considering the bone–joint X-ray examinations.
Isoniazid 300 mg/day and rifampicin 600 mg/day were given to the patient and, since
the sixth week of administration, no drainage from fistulas was observed. At the end of 9
months of treatment, the lesions improved, with scar formation, and no new lesions were
observed (Fig. 2). The improvement has continued in the 4 months following the
completion of therapy.
Discussion
Scrofuloderma, also called tuberculosis cutis colliquativa,is a subacute form of skin tuberculosis, which is character-ized by subcutaneously located, cold abscess formation andsecondary changes of the overlying skin. It can be causedby direct invasion of the bacilli to the skin from the
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
lesions of patients with tuberculosis of the lymph nodes,epididymis, bones, and joints, as well as by exogenousintroduction of the bacilli by trauma or injection.1
Scrofuloderma often originates in children from the necklymph nodes during the primary tuberculosis complex.Despite this, a child case of isolated liver tuberculosis andassociated scrofuloderma development of the overlying

Ozkan et al. Scrofuloderma Cameo 607
Figure 1 Clinical picture before treatment
skin has been reported.2 The elderly often have more thanone lesion, depending on the hematogenous invasion, andthese lesions can be seen on the trunk, inguinal, and glutealregions and on the tongue, in addition to the neck region.3
Occasionally, scrofuloderma can occur by direct exogen-ous inoculation of the bacilli to the skin. Scrofulodermaevolving over the lacrimal duct skin by exogenous inocula-tion and cases of atypical mycobacterium infectiondeveloping after blepharoplasty surgery have beenreported.4,5
In our patient, the initial complaints had begun followinga cholecystectomy operation and showed unilateral pro-gression. Lung and other organ system tuberculosis wasinvestigated and no finding was detected. The administra-tion of low dose corticosteroids for a long time facilitatedthe progression of scrofuloderma in this case.
The sensitivity of tuberculous cultures from skin lesionsis often low, and reproduction may not be establishedevery time.1,6,7 In recent years, it has been reported thatmycobacterial deoxyribonucleic acid analysis by poly-merase chain reaction is the fastest and most importantmethod of diagnosis.8 The advantages of this method arethat it does not necessarily require viable cells and the
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
Figure 2 Clinical picture after treatment
rapid elucidation of the results.6 In developing countries,where tuberculosis can appear frequently and the empiricaladministration of antituberculous drugs is performed dueto the difficulties in establishing the diagnosis by culture,the practice of polymerase chain reaction is primarily ofgreat importance.9 Although the culture taken from theabscess material on the gluteal region in our case showed noreproduction, deoxyribonucleic acid amplification productsunique to mycobacterium tuberculosis were established bypolymerase chain reaction, and this played an importantrole in planning the therapy of the case within a short time.
References
1 Braun-Falco O, Plewig G, Wolff HH, Winkleman RK.Dermatology, 3rd edn. Berlin: Springer-Verlag, 1991:130–146.
2 Gautam A, Singh JP. Isolated hepatic tuberculosis withscrofuloderma. Postgr Med J 1987; 63: 401–402.
3 Farina MDC, Gegundez MI, Pique E, et al. Cutaneoustuberculosis: a clinical, histopathologic and bacteriologicstudy. J Am Acad Dermatol 1995; 33: 433–440.
4 Tur E, Brenner S, Meiron Y. Scrofuloderma (tuberculosiscolliquativa cutis). Br J Dermatol 1996; 134: 350–352.

608 Cameo Linear IgA bullous dermatosis Friedman et al.
5 Moorthy RS, Rao NA. Atypical mycobacterial woundinfection after blepharoplasty. Br J Ophthalmol 1995;79: 93.
6 Narita M, Shibata M, Togashi T, et al. Polymerase chainreaction for detection of mycobacterium tuberculosis.Acta Pediatr 1992; 81: 141–144.
7 Ramesh V, Misra RS, Jain RK. Secondary tuberculosis ofthe skin. Int J Dermatol 1987; 26: 578–581.
Cameo
Captopril-triggered linear IgA bullous dermatosis
Ilyse S. Friedman, Donald Rudikoff, MD, Robert G. Phelps, MD, andAllen N. Sapadin, MD
From the George Washington A 57-year-old African–American woman with a history of long-standing asthma,University School of Medicine, hypertension, and congestive heart failure was referred for an intensely pruritic, blisteringWashington DC, and Department of
eruption of 2 months duration. The blistering began 2 weeks after the initiation of captoprilDermatology and Division offor essential hypertension. It was localized predominantly on the thighs and legs, but hadDermatopathology, Mount Sinai
School of Medicine, New York, recently spread to the upper extremities. Previously, the patient had been treated withNew York albuterol and furosemide. She had been admitted to the hospital with a diagnosis of acute
varicella, which was ruled out by a negative Tzanck preparation. Clarithromycin wasCorrespondence
prescribed upon discharge.Allen N. Sapadin, MD
Physical examination revealed multiple healed and crusted erosions ranging in sizeBox 1048The Mount Sinai Medical Center from 1 to 7 cm on the anteromedial aspect of the thighs and legs (Fig. 1). Discrete andOne Gustave L. Levy Place confluent tense vesicles, some forming arciform or rosette-like patterns, were notedNew York, NY 10029–6574 (Fig. 2). Multiple hypopigmented macules and patches were seen. On the flexural aspect
of the forearms, there were multiple small erosions, some of which appeared excoriated.
There were no urticarial lesions, and the oral mucosa and scalp were spared.
A biopsy specimen of lesional skin from the left thigh revealed epidermal detachment at
the dermal–epidermal junction with prominent festooning of dermal papillae. A dense,
perivascular lymphohistiocytic infiltrate was present in the underlying dermis (Fig. 3). Well-
defined neutrophilic papillary microabscesses were not identified, nor were eosinophils.
Direct immunofluorescence (DIF) staining revealed linear deposition of immunoglobulin A
(IgA) at the dermal–epidermal junction (Fig. 4). Indirect immunofluorescence studies of
the serum were negative for circulating pemphigus and pemphigoid antibodies. On the
basis of these findings, a diagnosis of linear IgA bullous dermatosis was made.
Despite discontinuation of captopril, the patient continued to develop new blisters.
Treatment with dapsone was initiated at a dose of 25 mg t.i.d. Because of continued
blister formation, the dose was increased to 50 mg t.i.d. and prednisone was added to the
regimen. The latter was tapered over a period of 3 months. For the past year, dapsone
has been administered at a dosage of 100 mg b.i.d., and the patient has experienced only
occasional blister formation.
Discussion
Linear immunoglobulin A (IgA) bullous dermatosis (LABD)is a subepidermal vesicobullous eruption occurring in adultsand characterized by a linear pattern of IgA deposition
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
8 Taniguchi S, Chanoki M, Hamada T, et al.Scrofuloderma: the DNA analysis of mycobacteria by thepolymerase chain reaction. Arch Dermatol 1993; 129:1618–1619.
9 Shankar P, Manjunath N, Laksmi R, et al. Identificationof mycobacterium tuberculosis by polymerase chainreaction. Lancet 1990; 335: 423.
along the basement membrane zone. Most characteristic-ally, tense vesicles are arranged in herpetiform, sausage-like, rosette-like, or arciform patterns on erythematousor normal-appearing skin. Histologic examination revealssubepidermal bullae and papillary microabscesses con-

Friedman et al. Linear IgA bullous dermatosis Cameo 609
Figure 1 Multiple healed and crusted erosions on theanteromedial aspect of the left thigh. Hypopigmentedmacules and patches are also evident
taining numerous inflammatory cells—predominantlyneutrophils and occasionally eosinophils. Direct immuno-fluorescence (DIF) is useful in distinguishing LABD fromdermatitis herpetiformis (DH) and bullous pemphigoid(BP). In contrast with the linear pattern seen in LABD,granular deposition of IgA in the tips of the dermal papillaeoccurs in DH. In BP, C3, with or without IgG, is depositedat the dermal–epidermal junction. IgA and IgM may befound in a minority of cases in BP, but not in the absenceof IgG. Indirect immunofluorescence in LABD onlyoccasionally reveals circulating IgA anti-basement-mem-brane-zone antibodies.
Although LABD is usually idiopathic, drug-induced dis-ease has been described. While vancomycin is the agent mostcommonly implicated,1–5 other agents include phenytoin,1
somatostatin,1 amiodarone,6 lithium,7 cefamandole,8 anddiclophenac.9 Captopril, an angiotensin-II-convertingenzyme inhibitor, is a widely used antihypertensive agentthat has been associated with a wide variety of cutaneousreactions, including angioedema,10,11 urticaria,11 lichenoideruptions,12 and pityriasis rosea.13 While its role as anetiologic factor in pemphigus is well established,14,15 itsrole as a causative agent in other bullous dermatoses hasonly rarely been repoted.16–19 It has been implicated in twoprior reports of LABD.1,20
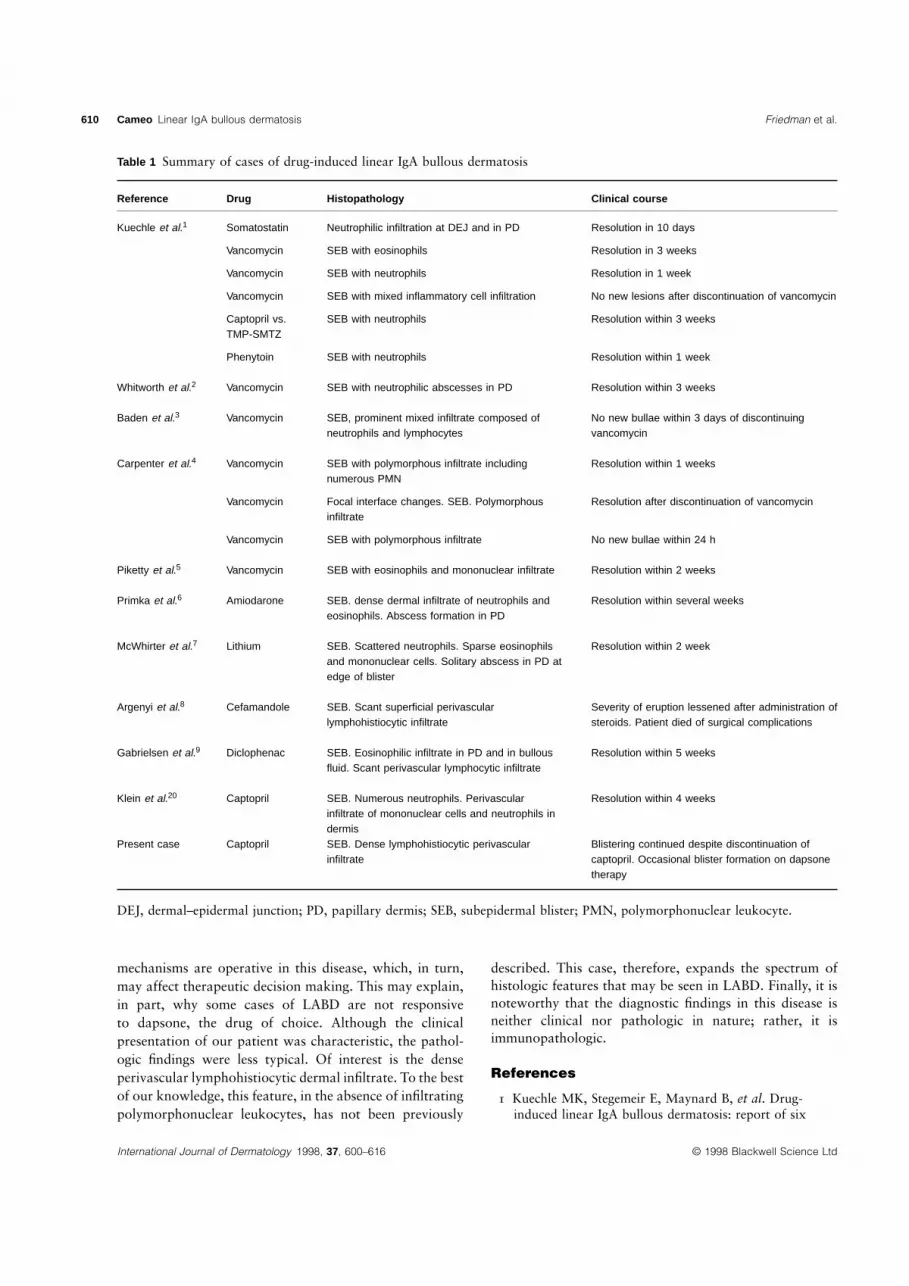
Drug-induced LABD is characterized by the spontaneousremission of the blisters after the removal of the offendingdrug. Idiopathic LABD, in contrast, tends to have a pro-tracted course, frequently requiring treatment with dapsoneand occasionally systemic corticosteroids.3 To date, allreported drug-induced LABD cases have demonstrated theresolution of blisters within days to several weeks ofdiscontinuation of the drug (Table 1). Our patient is uniquein that she continued to blister for 1 year despite the
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
cessation of the implicated drug. In addition, she requiredtreatment with dapsone and corticosteroids for completecontrol of the blistering process. This suggests that thedrug may have induced the expression of a pre-existingbullous dermatosis.
Reports that have unequivocally established a drug asan etiologic factor by virtue of its reintroduction as atherapeutic agent are few in number.7,9 In many cases,multiple drugs were administered simultaneously and onespecific agent could, at best, be labeled the most likelyculprit.2 Although, in the absence of a drug rechallenge,we cannot prove with certainty that captopril induced theeruption in our patient, the temporal relation of hersymptoms to the administration of this drug is highlysuggestive of a drug etiology.
The clinical appearance of LABD is morphologicallyvariable. LABD was originally considered to be a variantof DH, with which it may be confused. Its clinical featuresmay also resemble BP. The diagnosis in the present casewas initially suggested by the characteristic arciformarrangement of blisters in several areas.
The histopathologic features in LABD are equallyvariable. Most typically, histologic examination reveals asubepidermal bulla and papillary microabscesses containingnumerous inflammatory cells—predominantly neutrophilsand occasionally eosinophils; however, the pathologicfeatures often suggest the diagnosis of DH or BP. Rarely,erythema multiforme- or toxic epidermal necrolysis-likeclinical or pathologic changes are identified.8,20
The inconstant clinical and histopathologic manifesta-tions of LABD may indicate that several pathogenic
Figure 2 Confluent tense vesicles in an arciform arrangement
610 Cameo Linear IgA bullous dermatosis Friedman et al.
Table 1 Summary of cases of drug-induced linear IgA bullous dermatosis
Reference Drug Histopathology Clinical course
Kuechle et al.1 Somatostatin Neutrophilic infiltration at DEJ and in PD Resolution in 10 days
Vancomycin SEB with eosinophils Resolution in 3 weeks
Vancomycin SEB with neutrophils Resolution in 1 week
Vancomycin SEB with mixed inflammatory cell infiltration No new lesions after discontinuation of vancomycin
Captopril vs. SEB with neutrophils Resolution within 3 weeksTMP-SMTZ
Phenytoin SEB with neutrophils Resolution within 1 week
Whitworth et al.2 Vancomycin SEB with neutrophilic abscesses in PD Resolution within 3 weeks
Baden et al.3 Vancomycin SEB, prominent mixed infiltrate composed of No new bullae within 3 days of discontinuingneutrophils and lymphocytes vancomycin
Carpenter et al.4 Vancomycin SEB with polymorphous infiltrate including Resolution within 1 weeksnumerous PMN
Vancomycin Focal interface changes. SEB. Polymorphous Resolution after discontinuation of vancomycininfiltrate
Vancomycin SEB with polymorphous infiltrate No new bullae within 24 h
Piketty et al.5 Vancomycin SEB with eosinophils and mononuclear infiltrate Resolution within 2 weeks
Primka et al.6 Amiodarone SEB. dense dermal infiltrate of neutrophils and Resolution within several weekseosinophils. Abscess formation in PD
McWhirter et al.7 Lithium SEB. Scattered neutrophils. Sparse eosinophils Resolution within 2 weekand mononuclear cells. Solitary abscess in PD atedge of blister
Argenyi et al.8 Cefamandole SEB. Scant superficial perivascular Severity of eruption lessened after administration oflymphohistiocytic infiltrate steroids. Patient died of surgical complications
Gabrielsen et al.9 Diclophenac SEB. Eosinophilic infiltrate in PD and in bullous Resolution within 5 weeksfluid. Scant perivascular lymphocytic infiltrate
Klein et al.20 Captopril SEB. Numerous neutrophils. Perivascular Resolution within 4 weeksinfiltrate of mononuclear cells and neutrophils indermis
Present case Captopril SEB. Dense lymphohistiocytic perivascular Blistering continued despite discontinuation ofinfiltrate captopril. Occasional blister formation on dapsone
therapy
DEJ, dermal–epidermal junction; PD, papillary dermis; SEB, subepidermal blister; PMN, polymorphonuclear leukocyte.
mechanisms are operative in this disease, which, in turn,may affect therapeutic decision making. This may explain,in part, why some cases of LABD are not responsiveto dapsone, the drug of choice. Although the clinicalpresentation of our patient was characteristic, the pathol-ogic findings were less typical. Of interest is the denseperivascular lymphohistiocytic dermal infiltrate. To the bestof our knowledge, this feature, in the absence of infiltratingpolymorphonuclear leukocytes, has not been previously
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
described. This case, therefore, expands the spectrum ofhistologic features that may be seen in LABD. Finally, it isnoteworthy that the diagnostic findings in this disease isneither clinical nor pathologic in nature; rather, it isimmunopathologic.
References
1 Kuechle MK, Stegemeir E, Maynard B, et al. Drug-induced linear IgA bullous dermatosis: report of six

Friedman et al. Linear IgA bullous dermatosis Cameo 611
Figure 3 (a) Subepidermalblister with prominentfestooning of dermalpapillae. (b) Dense, perivascularlymphohistiocytic infiltrate inunderlying dermis
cases and review of the literature. J Am Acad Dermatol1994; 30: 187–192.
2 Whitworth JM, Thomas I, Peltz SA, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD). J AmAcad Dermatol 1996; 34: 890–891.
3 Baden LA, Apovian C, Imber MJ, Dover JS.Vancomycin-induced linear IgA bullous dermatosis[letter]. Arch Dermatol 1988; 124: 1186–1188.
4 Carpenter S, Berg D, Sidhu-Malik N, et al. Vancomycinassociated linear IgA dermatosis. J Am Acad Dermatol1992; 26: 45–48.
5 Piketty C, Meeus F, Nochy D, et al. Linear IgAdermatosis related to vancomycin. Br J Dermatol 1994;130: 130–131.
6 Primka II EJ, Liranzo MO, Bergfeld WF, Dijkstra JW.Amiodarone-induced linear IgA disease. J Am AcadDermatol 1994; 31: 809–811.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
7 McWhirter JD, Hashimoto K, Fayne S, Ito K. Linear IgAbullous dermatosis related to lithium carbonate. ArchDermatol 1987; 123: 1120–1122.
8 Argenyi ZB, Bergfeld WF, Valenzuela R, et al. Adultlinear IgA disease associated with an erythemamultiforme-like drug reaction. Cleve Clin J Med 1987;54: 445–450.
9 Gabrielsen TO, Staerfelt F, Thune PO. Drug-inducedbullous dermatosis with linear IgA deposits along thebasement membrane. Acta Derm Venereol (Stock) 1981;61: 439–441.
10 Slater EE, Merrill DD, Guess HA, et al. Clinical profileof angioedema associated with angiotensin converting-enzyme inhibition. JAMA 1988; 260: 967–970.
11 Wood SM, Mann RD, Rawlins MD. Angio-edema andurticaria associated with angiotensin converting enzymeinhibitors. Br Med J 1987; 294: 91–92.

612 Cameo Epidermolysis bullosa acquisita Hayashi et al.
Figure 4 Deposition of IgA in a linear pattern at the dermal–epidermal junction
Cameo
Epidermolysis bullosa acquisita in a 6-year-old Japanese boy
Kazuhiro Hayashi, MD, Masato Ueda, MD, PhD, Makiko Sakai, MD,Yoshiaki Fujikawa, MD, Kenta Tsuru, MD, Masayuki Amagai, MD, PhD, andMasamitsu Ichihashi, MD, PhD
From the Department of Dermatology, A 6-year-old Japanese boy started to develop blisters around the mouth and eyes in MayKobe University School of Medicine, 1996. With provisional diagnoses of herpes simplex or Kaposi’s varicelliform eruption, heKobe and Department of was treated with acyclovir and immune globulin. Blister formation increased, however, andDermatology, Keio University School
he was referred to the Dermatology Clinic of Kobe University School of Medicine inof Medicine, Tokyo, JapanAugust 1996.
Correspondence On physical examination, tense bullae and vesicles were observed on the trunk andMasato Ueda, MD limbs and also on the palms and soles (Fig. 1). Neither mucosal involvement norDepartment of Dermatology cutaneous fragility was observed, and no nail deformity was seen. The blistering on bothKobe University School of Medicine
erythematous bases and normal skin showed a ‘‘string-of-pearls’’ arrangement. Abnormal7–5-1, Kusunoki-cho, Chuo-kulaboratory results were as follows: white blood cell count, 12.43103/mm3 (N 5 4–8.5)Kobe 650
Japan (eosinophils, 25%); platelets, 5143103/mm3 (N 5 130–300); total protein, 4.9 g/dL
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
12 Phillips WG, Vaughan-Jones S, Jenkins R, BreathnachSM. Captopril-induced lichenoid eruption. Clin ExpDermatol 1994; 19: 317–320.
13 Wilken JK, Kirkendall WM. Pityriasis rosea-like rashfrom captopril. Arch Dermatol 1982; 118: 186–187.
14 Korman NJ, Eyre RW, Stanley JR. Drug-inducedpemphigus: autoantibodies directed against thepemphigus antigen complexes are present inpenicillamine and captopril-induced pemphigus. J InvestDermatol 1991; 96: 273–276.
15 Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigusrelated to angiotensin-converting enzyme inhibitors.J Am Acad Dermatol 1992; 26: 364–366.
16 Fitzgerald DA. Subepidermal bullous eruption inducedby captopril. Clin Exp Dermatol 1993; 18: 196–197.
17 Pennel DJ, Nunan TO, O’Doherty MJ, Croft DN. FatalStevens–Johnson syndrome in a patient on captopril andallopurinol. Lancet 1984; 1: 463.
18 Sala F, Crosti C, Monti M, DeBitonto A. Cutaneouspathology due to captopril—observations on 2 clinicalcases. G Ital Dermatol Venereol 1983; 118: 89–93.
19 Flageul B, Foldes C, Wallach D, et al. Captopril-inducedlichen planus pemphigoides with pemphigus-likefeatures. A case report. Dermatologica 1986; 173:284–255.
20 Klein LE, Shmunes E, Carter JB, Walsh MY. Linear IgAbullous dermatosis related to captopril treatment. Cutis1989; 44: 393–396.

Hayashi et al. Epidermolysis bullosa acquisita Cameo 613
(N 5 6.5–7.8); albumin, 2.4 g/dL (N 5 4.1–5.0); glutamate pyruvate transaminase (GPT),
56 IU/L (N 5 7–34); lactate dehydrogenase (LDH), 573 IU/L (N 5 227–416); alkaline
phosphatase (ALP), 384 IU/L (N 5 100–303); C-reactive protein (CRP), 3.08 mg/dL
(N 5 0–0.3). The antinuclear antibody test gave negative results.
Histologic examination of a skin biopsy specimen revealed a subepidermal blister
containing neutrophils and eosinophils, and an infiltrate of lymphocytes with eosinophils
and neutrophils in the superficial dermis. Direct immunofluorescence of perilesional skin
showed a linear band of immunoglobulin G (IgG) and C3 at the dermo–epidermal
junction. Indirect immunofluorescence on 1 mol/L sodium chloride-split normal skin
showed IgG deposits on the dermal side of the blister. Western immunoblot analysis of
the reactivity of the patient’s serum on epidermal and dermal extracts revealed that
autoantibodies were bound to the 290-kDa antigen in the dermal extract (Fig. 2). No
reactivity was observed in the epidermal extract.
The boy was treated with prednisolone (1.3 mg/kg body weight per day) which inhibited
new blistering. Since a reduction of the dose of prednisolone to 1.1 mg/kg body weight per
day induced new blistering,dapsone (1.6 mg/kg body weight per day) and prednisolone
(1.1 mg/kg body weight per day) were given in combination, leading to improvement. A
further decrease in the dose of prednisolone to 0.9 mg/kg body weight per day resulted in
new bulla formation, and the prednisolone was replaced by betamethasone. The disease
activity then markedly subsided and the bullae healed with the formation of milia.
Discussion
Epidermolysis bullosa acquisita (EBA) is an acquiredbullous disease which is characterized by immunoglobulinG (IgG) autoantibodies that react with type VII collagenin the anchoring fibrils, leading to the formation of bullaeat the dermo–epidermal junction.1 EBA is a rare disease inchildhood: we found only 18 cases in the literature.2–12 Inboth children and adults, there are two main clinicalphenotypes of EBA.13 The classic, non-inflammatory,mechanobullous type involves skin fragility, blisters at sitesof trauma, and healing with scarring and milia. Theinflammatory type resembles most chronic bullous disease,including bullous pemphigoid, cicatrical pemphigoid, andlinear IgA bullous disease. During the course of his disease,our patient presented both cutaneous phenotypes of EBA.Although mucosal (oral in particular) involvement is fre-quent and severe, occurring in 13 of 16 cases in whichEBA was described, our patient did not show this sign.Patients of various races have been reported: eight white,five black, one from Iran, and one from the Philippines.Our patient seems to be the first reported Japanese case.
Indirect immunofluorescence on split skin is helpful indifferentiating between EBA and other types of autoimmunebullous diseases, but cannot replace immunoblotting,because, in rare cases, fluorescent binding may also occuron the dermal side in bullous pemphigoid.14
The combination of dapsone and prednisolone appearsto be the most effective treatment for pediatric EBA, andthe prognosis in children seems better than in adults.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
Figure 1 Disseminated tense blisters with an annular patterndeveloping on erythematous plaques on the trunk

614 Cameo Familial mandibuloacral dysplasia Prasad, Padmavathy, and Sethurajan
Figure 2 Western immunoblot analysis on a dermal extract.Lanes 1 and 2, adult EBA serum sample; lane 3, patient’sserum; lane 4, normal control serum
References
1 Woodley DT, Burgeson RE, Lunstrum G, et al.Epidermolysis bullosa acquisita antigen is globularcarboxyl terminus of type VII procollagen. J Clin Invest1988; 81: 683–687.
2 Borok M, Heng MCY, Ahmed AR, et al. Epidermolysisbullosa acquisita in an 8-year-old girl. Pediatr Dermatol1986; 3: 315–322.
3 Rubenstein R, Esterly NB, Fine JD. Childhoodepidermolysis bullosa acquisita. Detection in a 5-year-oldgirl. Arch Dermatol 1987; 123: 772–776.
Cameo
Familial mandibuloacral dysplasi a – a report of four cases
P. V. S. Prasad, MBBS, DV, DD, MD, L. Padmavathy, MBBS, DD, and S. Sethurajan, MBBS,DMRD
From the Departments of Case 1 A 9-year-old girl born to second-degree consanguineous parents was brought toDermatology and Radiology, the outpatient department with a history of pain in the feet and pigmentary changes onRajah Muthiah Medical College and
the skin of 4 years’ duration. Cutaneous examination revealed large symmetrical areas ofHospital, Annamalai University,mottled pigmentation (both hypo and hyper) with atrophy over the face, flexures, hands,Tamil Nadu, India
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
4 McCuaig CC, Chan LS, Woodley DT, et al.Epidermolysis bullosa acquisita in childhood.Differentiation from hereditary epidermolysis bullosa.Arch Dermatol 1989; 125: 944–949.
5 Espagne E, Janssen F, Prost C, et al. Epidermolysebulleuse acquise de l’enfant. A propos d’un cas. AnnDermatol Venereol 1990; 117: 814–816.
6 Arpey CJ, Elewski BE, Moritz DK, et al. Childhoodepidermolysis bullosa acquisita. Report of three casesand review of the literature. J Am Acad Dermatol 1991;24: 706–714.
7 Roger H, Machado P, Nicolas JF, et al. Epidermolysisbullosa acquisita in a 31
2-year-old girl. J Am AcadDermatol 1992; 27: 858–862.
8 Lacour JP, Bernard P, Rostain G, et al. Childhoodacquired epidermolysis bullosa. Pediatr Dermatol 1995;12: 16–20.
9 Inauen P, Hunziker TH, Gerber H, et al. Childhoodepidermolysis bullosa acquisita. Br J Dermatol 1994;131: 898–900.
10 Kirtschig G, Wojnarowska F, Marsden RA, et al.Acquired bullous diseases of childhood: re-evaluation ofdiagnosis by indirect immunofluorescence examinationon 1 M NaCl split skin and immunoblotting.Br J Dermatol 1994; 130: 610–616.
11 Feliciani C, Fasciocco D, Franchi A, et al. Epidermolysisbullosa acquisita in a 10-year-old Albanian boy.Br J Dermatol 1997; 137: 307–308.
12 Callot-Mellot C, Bodemer C, Caux F, et al.Epidermolysis bullosa acquisita in childhood. ArchDermatol 1997; 133: 1122–1126.
13 Gammon WR, Briggaman RA, Woodley DT, et al.Epidermolysis bullosa acquisita—a pemphigoid-likedisease. J Am Acad Dermatol 1984; 11: 820–832.
14 Wakelin SH, Allen J, Wojnarowska F. Childhood bullouspemphigoid—report of a case with dermal fluorescenceon salt-split skin. Br J Dermatol 1995; 133: 615–618.

Prasad, Padmavathy, and Sethurajan Familial mandibuloacral dysplasia Cameo 615
Correspondence and feet. There were multiple, small, keratotic papules (Fig. 1) over the back, joints,P. V. S. Prasad, MBBS, DV, DD, MD palms, and soles. Scalp examination revealed widened suture lines and multiple, soft,B-12, RSA Complex
nodular swellings on the occipital region with sparse and short hair. There was also aAnnamalainagar-608,002small nodule on the scalp which discharged cheesy material and was suggestive ofTamil Nadu
India calcinosis cutis. The right index finger showed a broad terminal phalanx. Gynecologic
examination revealed a small infantile uterus. The patient had normal dentition.
Cases 2 and 3 Two brothers, aged 6 and 3 years, who were brought to the clinic for
screening, also revealed similar skeletal and cutaneous changes, but with less intense
pigmentation. Both had normal development of the genitalia.
Case 4 After 4 years, another girl of 15 years of age was referred to us with complaints
of inability to walk due to pain in the feet. The patient was born to second-degree
consanguineous parents and showed the same clinical features as those of Case 1,
except for short stature (height, 138 cm) and a broad terminal phalanx on the left middle
finger. This female patient also had an infantile uterus.
All four patients were subjected to routine investigations, which were found to be within
normal limits. Examination of the hair under potassium hydroxide mount did not reveal
any abnormality.
Skeletal surveys were performed for all patients. In Case 1, an X-ray of the skull—
anteroposterior (AP) and lateral view (Fig. 2) revealed frontal bossing of the cranial vault.
Coronal, lambdoid, and sagittal sutures were widened, measuring 5, 5, and 7 mm,
respectively. The cranial vault had a ground glass appearance and did not show outer
and inner table differentiation. The metopic and mendosal sutures were seen. The sutures
were seen as straight lines instead of serrated interdigitations. The occipital area showed
only a sea of membranes with islands of ossified bones (Wormian bones). Platybasia was
seen. The mandible was hypoplastic. A chest X-ray revealed a narrow chest cavity with
normal clavicles. An X-ray of the spine was normal. An X-ray of the pelvis showed
increased acetabular angles, collapsed head of femur, and reduced shaft neck angle. An
X-ray of the hand showed popcorn calcification in the soft tissues. There was hypoplasia
of the distal part of the terminal phalanx of the left index finger. An X-ray of the foot also
showed similar soft tissue calcification of the left big and little toes.
Skeletal surveys of the two other siblings revealed Wormian bones and widened suture
lines, but were otherwise normal. A skeletal survey of the fourth case also reflected the
same changes as in Case 1, except that hypoplasia of the terminal phalanx of the left
middle finger was observed.
Histopathology of the scalp nodule of Case 1 and of the keratotic papules of Cases 1
and 4 (Fig. 3) revealed calcified deposits in the mid and lower dermis which stained
positive with Von Kossa.
Discussion
The first three patients from the same family were seen in1992, and the fourth patient was seen 4 years later. Thecombination of poikilodermatous skin lesions with identicalradiologic features led us to suspect one of the prematureaging syndromes.
Familial mandibuloacral dysplasia (MAD) is a very raresyndrome, first described by Danks et al.1 in 1974. Overthe last two decades, very few cases of this syndrome havebeen reported in the world literature.2–5 The conditionappears to be inherited as an autosomal recessive trait andseems to be more common in Italy, according to Tenconi
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 600–616
et al.5 An autosomal recessive mode of inheritance wasfirst suspected by Zina et al.2 in 1981, because two sets ofthird cousins with MAD hailed from a small mountainvillage. Tenconi et al.5 confirmed the mode of inheritancein all their patients. In our series also, the patients wereborn to second-degree consanguineous parents.
The features of MAD syndrome usually start between 3and 5 years of age.5 Schrander et al.3 described a patientwho developed changes at 2 years of age. All of our patientshad normal development until 3–5 years of age, by whichtime they had developed poikilodermatous skin lesions.Although the skin lesions started later, all had defects inthe skull bone at birth.

616 Cameo Familial mandibuloacral dysplasia Prasad, Padmavathy, and Sethurajan
Figure 1 Keratotic papules on the buttocks
Figure 2 Lateral view of X-ray of the skull showing multipleWormian bones in the occipital region
MAD syndrome shows the following features: (i) mandib-ular hypoplasia; (ii) delayed cranial suture closure; (iii)dysplastic clavicles; (iv) abbreviated club-shaped terminalphalanges, associated with acro-osteolysis; (v) atrophy ofthe skin of the extremities; (vi) multiple Wormian bones;(vii) sharp nose with loss of lower teeth; (viii) alopecia;and (ix) autosomal recessive inheritance.
All four patients in our series showed these features,with the exception of dysplastic clavicle and loss of lowerteeth. Interestingly, both of the female patients developedsoft tissue calcification, which was confirmed by histopatho-logy of the nodular and keratotic papules, and by radiologywhich revealed popcorn calcification of the extremities.They also had an infantile uterus. Calcinosis cutis has beenless frequently reported with MAD syndrome, accordingto Tenconi et al.5 Infantile uterus has not been reported.
International Journal of Dermatology 1998, 37, 600–616 © 1998 Blackwell Science Ltd
Figure 3 Histopathological examination from a keratoticpapule showing calcified deposits in the deep dermis (HPE,3400)
MAD syndrome may be mistaken for hereditary scleros-ing poikiloderma. Both conditions show poikilodermatousskin changes, but the presence of keratotic papules andskeletal changes differentiates the two entities. Fryburg andSidhu4 described two siblings of MAD syndrome in whomthe diagnosis of hereditary sclerosing poikiloderma wasmade initially but, on re-evaluation as adults, the clinicalfeatures were perceived to be compatible with MAD syn-drome. This syndrome may also be mistaken for any of thepremature aging syndromes, such as Werner’s syndrome,because of the predominant feature of premature aging,but only MAD syndrome is associated with Wormian bonesin the skull.
References
1 Danks DM, Mayne V, Wettenhall NB, et al.Craniomandibular dermatodysostosis. Birth Defects1974; 10: 99–105.
2 Zina AM, Cravario A, Bundino S. Familial mandibuloacral dysplasia. Br J Dermatol 1981; 105: 719–723.
3 Schrander SC, Spaepen A, Fryns JP, et al. A severe caseof mandibulo acral dysplasia in a girl. Am J Med Genet1992; 43: 877–881.
4 Fryburg JS, Sidhu MN. Long term follow-up ofcutaneous changes in siblings with mandibulo acraldysplasia who were originally considered to havehereditary sclerosing poikiloderma. J Am Acad Dermatol1995; 33: 900–902.
5 Tenconi R, Miotti F, Miotti A, et al. Another Italianfamily with mandibulo-acral dysplasia. Am J Med Genet1986; 24: 357–364.
6 Burton JL. Disorders of connective tissue. In: ChampionRH, Burton JL, Ebling FJG, eds. Textbook ofDermatology. Oxford: Blackwell Scientific Publications,1992: 1816–1818.





























