Embed Size (px)
Citation preview

4Biological Vulnerabilities to theDevelopment of Psychopathology
ROBERT O. PIHL AND AMÉLIE NANTEL-VIVIER
The history of psychopathology isreplete with a fascination withbiological explanations for various
disorders. From the trepanations of earlycave dwellers, in which holes were gougedin the skull; to the humoral theories ofHippocrates, circa 400 BCE; the custodial,animal-like treatment of the mentally ill ofthe Renaissance; the enlightened humanistsperiod; the time of influential theorists suchas Kraeplin, Freud, and Jung; and on to thepresent, the belief in biological mechanismsunderlying pathology has existed. Neverthe-less, today is not yesterday. Generally lackingeven just 30 years ago was credible scientificevidence to support this specific and persis-tent belief. In fact, a sort of antibiologicalmovement grew through the 1950s–1960s,and remnants of this view, which persisttoday, view mental illnesses as sociallylearned disorders treatable solely by behav-ioral interventions. Today, of course, thewell-known vulnerability-stress model pre-dominates as an explanation for the etiologyof most disorders, representing an obviousinteraction of these two historical views.
The relatively recent resurgence of biolog-ical explanations, however, has not beenpredicated on historical influence but ratherhas exploded from a cascading body of evi-dence driven by growth in the neurosciences.This growth, in turn, has benefited directlyfrom powerful new scientific methodologiesable to delineate the vulnerability componentof the vulnerability-stress model. Discriminat-ing biological vulnerabilities is the focus ofthis chapter.
NEW TECHNOLOGIES
The essence of biological studies of mentaldisorders is that there is an ascertainable rela-tionship between a particular disorder andbrain functioning. Measuring brain function-ing is then basic. This, however, is not a simpletask, and for centuries only nonfunctionalmeasures, from the bizarre (phrenology) to theimprecise (X-rays) existed. Advances in phar-macology, neuropsychology, electrophysiol-ogy, and brain imaging are at the forefront oftechnologies that are prime contributors to
C H A P T E R
75
04-Hankin.qxd 2/24/2005 11:20 AM Page 75

new investigations of biological variables.Pharmacology has been the traditional routefor developing biological explanations forpsychopathology. Unfortunately, “miracledrugs,” which implicated specific chemicalsystems in various disorders, seemed over timeto lose their luster, as did the implicated the-ory as well. Recently, the manipulation ofneurotransmitters in both experimental andclinical subjects by regulating amino acid pre-cursors has resulted in the ability, for example,to dramatically decrease brain serotonin(Young, Smith, Pihl, & Ervin, 1985). Similarprocedures exist for dopamine, although likemany pharmacological manipulations, whichare improving, specificity is an issue. Paralleldevelopments in new drugs that are moretargeted in their effects on specific aspects ofa neurotransmitter system have lent supportto biochemical theories, as have drugs cur-rently in pharmaceutical company pipelinesthat will affect the action of genes or targeta single aspect of a neurosystem’s function.Neuropsychology has benefited greatly fromadvances in neuroimaging, because many testshave been refined to assess specific abilities—be they cognitive, motor, or perceptual—andthen validated by concomitant activity in par-ticular brain areas. Neuropsychology has beenat the forefront in determining behavioraldeficits in frontal lobe functioning, which isimplicated in many disorders. This evolution-ary “new brain” seems to have a key role inworking memory, learning, response inhibi-tion, and coordinating sensory input withresponding. Further, perhaps as Luria (1980)suggested, it is this part of the brain that pro-jects the integration of the past, present, andfuture so basic to controlled responding.Many neuropsychological findings have beenvalidated by imaging technology, which incombination can determine the endopheno-types that underlie and are the precursors tovarious clinical disorders.
Relatively recently, there has been a dra-matic growth in noninvasive methods to
monitor the brain and its functioning, all ofwhich are dependent upon the rapid process-ing of complex information, an impossible taskbefore the development of microprocessors.This includes measuring the brain’s electro-physiology and advanced brain imaging tech-nologies that can show structural aspects of thebrain and the level and site of activity when theindividual is presented with various stimuli.Electrophysiology, involving the measurementof brain oscillations, particularly in responseto prescribed tasks, has determined differen-tial patterns of action for various pathologies.More specifically, these patterns have beenshown to represent localized activity and to beheritable and linked to a range of behaviors.Nuclear magnetic resonance imaging (MRI) isa noninvasive technique that involves measur-ing atoms in order to obtain a detailed struc-tural picture of the brain and specific areas.A blood oxygenation signal measured by afunctional MRI (fMRI) allows the assessmentof neural activity without requiring the use ofradioactive substances, which are needed withpositron emission tomography (PET), alsoa valuable, but a more invasive, procedurefor measuring activity to specific brain areas.More powerful technologies are rapidly com-ing online. Diffusion tension imaging, forexample, is an MRI procedure uniquely suitedto study white matter.
Burgeoning findings in other areas alsodirect one to the level of biology. Oneexample is the increasing recognition thatnumerous psychopathological disorders runin families; this implies the existence ofgenetic vulnerabilities and biological mecha-nisms. Questions regarding which genes orgenetic material affect which biochemicalprocesses have drawn our attention first (seeLemery & Doelger, Chapter 7 in this volume,for details on genetic vulnerabilities). Subse-quent concerns about brain development andfunctioning and the interactions of these bio-logical factors with stressful environmentalevents represent the next critical step.
VULNERABILITIES76
04-Hankin.qxd 2/24/2005 11:20 AM Page 76

NORMAL FUNCTIONING OFBRAIN AREAS AND PATHWAYS
Decades of neuropsychological research,especially with the recent technologicaladvances, have elucidated the normal func-tion and purpose of various brain areas andthe pathways that integrate these functionstogether. Here, we briefly review centralbrain areas, pathways, and the normal psy-chological functions that modern neuro-science believes they serve. Understandinghow these brain areas and pathways nor-mally function and what thoughts, behav-iors, and emotions they are associated withis critically important for elucidating howdeficits and dysfunctions in these brain areasmay create biological vulnerabilities for thedevelopment of different psychopathologies.
The frontal lobes have been broadly associ-ated with executive cognitive functioning,which is generally understood to refer to the“ability to plan, initiate, and maintain or altergoal-directed behaviours” (Pihl, Vant, &Assaad, 2003, p. 173). This ability is depen-dent upon specific cognitive functions, such asattention and working memory. The pre-frontal cortex area is believed to subserve therepresentation of goals and the means toachieve them (Miller & Cohen, 2001). Morespecifically, through its connections withother brain areas, the prefrontal cortex is partof an important circuitry that underlies theemergence of appropriate responses and thesimultaneous inhibition of inappropriateactions (Miller & Cohen). Connections to thebasal ganglia, an agglomeration of nucleiwithin the forebrain believed to be importantin motor control, contribute to the organism’sability to show appropriate motor responseand inhibition (Carlson, 2001). Simultaneousconnections to temporal limbic structuresensure the affective appropriateness of theresponse. Davidson and colleagues (Davidson,Pizzagalli, Nitschke, & Putnam, 2002) describethis process as “affect-guided planning and
anticipation” (p. 548), whereby actionsexpected to provide “rewards” will be pur-sued, and actions known to lead to “punish-ment” will be inhibited. Davidson et al.(2002) proposed that the left prefrontal cortexmay be particularly important for the antici-pation of positive outcomes and approachbehaviors, whereas the right prefrontal cortexmay be crucial for appropriate inhibition andwithdrawal. They report that imaging studieshave found the left orbital and ventral regionsto be sensitive to rewards, whereas the sameareas in the right hemisphere were found to beparticularly sensitive to cues of punishment(Davidson et al., 2002).
As mentioned above, the frontal lobesshow strong connections with temporal lim-bic areas such as the amygdala and the hip-pocampus, which are believed to be crucialfor emotional responses (Carlson, 2001).The amygdala has been shown to be impor-tant for promoting vigilance and attention tonovel or affectively salient stimuli, both pos-itive and negative (Davidson et al., 2002).The amygdala is intrinsically connected tothe hippocampus, which has been found tosubserve memory, contextual conditioning,and stress response (Davidson et al., 2002).Upon stress, the hypothalamus, a groupof nuclei located at the base of the brain(Carlson, 2001), secretes corticotropin releas-ing factor (CRF), which in turn triggers syn-thesis and release of adrenocorticotropin(ACTH) by the pituitary. ACTH then stimu-lates synthesis and release of glucocorticoidsby the adrenal cortex (Nestler et al., 2002).The hypothalamic-pituitary-adrenal (HPA)axis has reciprocal feedback connectionswith the hippocampus and the amygdala(Nestler et al.), so that glucocorticoidsrelease triggers inhibition of the HPA axis bythe hippocampus. High, chronic levels of glu-cocorticoids have been suggested to lead tohippocampal damage in the form of reduceddendritic branching and glutamatergic den-dritic spines, as well as reduced genesis of
Biological Vulnerabilities to the Development of Psychopathology 77
04-Hankin.qxd 2/24/2005 11:20 AM Page 77

granule cell neurons (Nestler et al.). A viciouscycle may thus operate, as decreased HPAaxis inhibition due to hippocampal damagewould lead to increasing glucocorticoidlevels, which in turn may lead to furtherhippocampal damage (Nestler et al.).
The prefrontal cortex, basal ganglia, andlimbic structures, together with other brainareas, thus constitute an intrinsic systemunderlying our ability to appropriatelyrespond to our environment. As will bedescribed in sections of this chapter pertain-ing to specific disorders, functional oranatomical anomalies of these regions andtheir interconnections may contribute to dif-ficulties in different spheres of functioning,such as impairments in regulating responses(e.g., motor, affective) to the environment.
CAVEATS
Unfortunately, the road to clarity of under-standing is neither straight nor paved.The powerful methodologies briefly men-tioned above cannot override definitionaland philosophical issues seemingly funda-mental to the study of mental disorders.Psychopathologies are laden with noise,replete with debate, and lacking in speci-ficity. This issue is exemplified by Andreasen(1999), who in proposing a model forschizophrenia wrote,
At present the most important problem inschizophrenia research is not finding thegene or localizing it in the brain and under-standing its neural circuits. Our mostimportant problem is identifying the correcttarget at which to aim our powerful newscientific weapons. Our most pressing prob-lem is at the clinical level: defining whatschizophrenia is. (p. 781)
Similar statements can and shouldbe repeatedly made about most if not all
definitions of mental disorders (see chaptersin Part III of this volume for discussion ofdefinitions and classifications of disorders).It is axiomatic in this area that names arebestowed, not discovered, and names havebeen growing at a prodigious rate. Thenumber of disorders in the different versionsof the American Psychiatric Association’s(APA) Diagnostic and Statistical Manual ofMental Disorders (DSM) has grown fromaround 100 in 1952 to more than 360 intoday’s DSM-IV-TR (APA, 2000). That is athreefold increase, which clearly points torampant inflation in mental disorders. Ofcourse, an increase in definitions is expectedas knowledge is gained and specificitiesrefined. However, the fundamental questionis, “Just how many disorders are there?” Itis almost certain that there are not exactly360 psychological disorders in nature. Afactor analysis of 10 common mental disor-ders from the large, representative NationalComorbidity Survey (NCS; Krueger, 1999)resulted in a three-factor fit, respectivelylabeled Anxious-Misery, Fear, and External-izing. Similar results were obtained in a large,representative sample of children and adoles-cents (Lahey, Applegate, Waldman, Hankin,& Rick, 2004). Further, who gets hospital-ized, both voluntarily and involuntarily, isless a function of diagnostic label than of thedisplay of aggression, be it self- or other-directed (Pihl, 1995). Finally, there is the issueof comorbidity. In the two broad surveys ofmental illness, more than one disorder waspresent in an individual 60% of the time inthe National Institutes of Mental Healthstudy (Robins & Regier, 1991) and 56% ofthe time in the NCS study (Kessler et al.,1994). Comorbidity is the rule with mentaldisorders, not the exception. This fact raisesthe possibility that, instead of many disordersexisting, there may be a relatively smallnumber of underlying biological processesthat, through environmental interactions,result in many diverse behavioral forms.
VULNERABILITIES78
04-Hankin.qxd 2/24/2005 11:20 AM Page 78

A conundrum thus remains. In the DSM-IV-TR, and presumably in the DSM-V whenit arrives, mental disorders are defined on abehavioral level, whereas disorders in generalare seen in the definition of mental disordersas residing within the individual (APA,2000). Thus, the necessary recrafting of cur-rent definitions of mental disorders mustinclude biological factors if the unacceptablevariability in current definitions is to bereduced and terminology become moremeaningful. It is also axiomatic that nosol-ogy necessarily precedes etiology. Thus, it isnecessary to determine which biological vari-ables, as well as those vulnerabilities fromother levels of analyses (see other chapters inPart II of this volume), are relevant to thedefinition of a disorder.
Studying individuals likely to develop adisorder, which should be the basis of riskresearch in psychopathology, provides acontemporaneous recording of events. Thisdesign thus controls for a major error instudies of psychopathologies in which find-ings with patients often reflect having adisorder rather than a causative factor. Putbluntly, by studying already diagnosed andtreated individuals, we may learn more aboutthe explosion than the triggering mecha-nisms. Studying vulnerable individuals alsoallows for the study of “escape” from riskprocesses, which can illustrate the impor-tance of interacting variables, and hetero-geneity of outcomes, where for examplesimilar underlying biological conditions mayhave varying trajectories to divergent disor-ders depending upon interacting factors. Thisresearch also allows for the discrimination ofthe significance of age of onset, which is ofcritical importance, because most disordersare age sensitive. Finally, by assessing thedevelopment of disorders through the studyof vulnerabilities, we are provided with theopportunity to elucidate feedback mecha-nisms, circular processes, and chain effectstypically important in causation.
There are also caveats of import regardingthe “new” methodologies. In the case of neu-roimaging, these range from the general issueof inference to specific concerns regardinghow a region of interest is selected. Regardinginference, mental and physical states are sel-dom measured simultaneously, because alabel is hardly a mental state, and thus anyconnections to a disorder represent specula-tion. Even when testing for specific cognitivestates, converging evidence of that state isrequired; these states are often altered withexperience and context. Even specificparadigms, such as an attentional go-stopprocedure, have multiple neural functioningexplanations (Schall, 2004). Another prob-lem is that the brain is active in general, andPET and fMRI results simply point to one ormore areas being relatively more active thanother areas. The area highlighted is labeledthe region of interest. Because there iswidespread activation in the brain, strongpreexperiment rationale is required for selec-tion of a specific area versus contrastingareas. Further, because the typical design inpsychopathology involves group compar-isons between psychiatric patients and con-trols, resolution is further distorted becauseof imperfect registration, because individualbrains differ both in anatomy and function.Finally, it is important to remember that thebrain is dynamic and ever-changing. Thedeterioration of neuroanatomy with age iswell known, and more recently the negativeramifications of emotional states, pain, anddrugs have been documented. In the case ofstress, the release of emotion-correlated glu-cocorticoid hydrocortisone has been shownto be related to significant hippocampal atro-phy in patients with depression (Sheline,Wang, Gado, Csernansky, & Vannier, 1996)and with posttraumatic stress disorder(Bremner et al., 1995). It thus seems likelythat there are other neuropathological conse-quences of stress, and perhaps other emo-tional states. Longitudinal neuroimaging
Biological Vulnerabilities to the Development of Psychopathology 79
04-Hankin.qxd 2/24/2005 11:20 AM Page 79

studies, for example, show progressivedeterioration in brains of some schizophren-ics over time, yet debate rages as to whetheror not this is a primary feature of the disor-der (Mathalon, Rapoport, Davis, & Krystal,2003). From one perspective, these changesmay reflect the basis of the chronicity of thedisorder, whereas for others it representsconcomitants such as medication histories,comorbid drug use, incidental head injury,and so forth.
The good news is that new technology isbegetting newer technology at an exponentialrate. MRI studies, for example, must continu-ally be reevaluated in light of the developmentof increasingly more powerful scans. Forexample, in the future, temporal and spatialresolution will be at the level of individualneurons. Thus, the following representativereviews of biological vulnerabilities for atten-tion deficit/hyperactivity disorder (ADHD),conduct disorder, depression, and substanceabuse should be seen as current state-of-the artknowledge and theory, yet likely ephemeraland open to change with new developments.What the reviews do underscore, however, isthat currently, “the brain is the game,” andthere exist substantial biological vulnerabili-ties to psychopathologies.
ATTENTION DEFICIT/HYPERACTIVITY DISORDER
ADHD is the most prevalent of the childhood-onset psychiatric disorders. Estimated to affectbetween 3% and 7% of school-age children,with affected boys outnumbering affectedgirls by a ratio of approximately three to one(APA, 2000), it represents 50% of all referralsto child health professionals (Hale, Hariri, &McCracken, 2000; Wicks-Nelson & Israel,2000). ADHD is a heterogeneous syndrome,with the current psychiatric nomenclaturerecognizing three subtypes: inattentive, hyper-active/impulsive, and combined. Comorbiditywith other psychiatric disorders has been
well established, with at least 50% of ADHDchildren receiving an additional diagnosis ofoppositional defiant disorder, conduct disor-der (CD), depression, anxiety, or learningdisability (APA, 2000). Once considered achildhood-limited condition, ADHD isincreasingly perceived as chronic, with symp-toms persisting in as many as 75% of affectedindividuals through adulthood (Wilens,Biederman, & Spencer, 2002).
British pediatrician George Still (1902)first described ADHD symptomatology usingthe label of “defective moral control,” whichhe believed was caused by subtle anomaliesin the structure and activity of neuronsresulting from physical illness or heredity,with minimal environmental contribution.Since then, although inattention, hyperactiv-ity, and impulsivity have been the object ofnumerous categorizations and conceptualiza-tions (Baumeister & Hawkins, 2001), brainanomalies have remained at the core of etio-logical models of the disorder. Researchershave sought to confirm the biological rootsof ADHD using a variety of methods, such asbehavioral and molecular genetics, neuropsy-chology, drug challenges, and investigationof environmental factors likely to affect brainstructure and functioning. Although numer-ous neurobiological theories of ADHD havebeen put forward, most have focused ondysfunctions of the frontal lobes, as well asthe basal ganglia and the cerebellum. Thisemphasis logically follows from the well-established association between frontal lobelesions or deficits with impulsivity and exec-utive function deficits (i.e., problems regulat-ing responses because of impairments inattention, working memory, self-monitoring,and planning) as well as the known impor-tance of the basal ganglia for the regulationof motor activity (Anderson, Polcari, Lowen,Renshaw, & Teicher, 2002). These impair-ments are congruent with the impulsivity,difficulty in planning and concentrating, andcontext-inappropriate hyperactive behaviorsdisplayed by individuals described as having
VULNERABILITIES80
04-Hankin.qxd 2/24/2005 11:20 AM Page 80

ADHD. Together with the cerebellum, thefrontal lobes and the basal ganglia form apathway that is subserved by the mesolimbicand mesocortical dopaminergic systems,neurotransmitter systems directly affectedby stimulant medication (Anderson et al.,2002). Figure 4.1 summarizes the interac-tions between the fronto-striatal-cerebellarpathway, the dopaminergic system, and pre/postnatal moderating influences in leading toADHD symptomatology. However, it is onlywithin the last two decades that the availableneuroimaging technologies have enabledresearchers to directly investigate the specificnature of the brain anomalies putativelyresponsible for the disorder.
Brain Structural andFunctional AbnormalitiesAssociated With ADHD
In the largest structural imaging study ofADHD patients to date, Castellanos and col-leagues (2002) investigated brain volumetricchanges over time, with repetitive MRI scansat 2- to 3-year intervals, in 152 both medicatedand unmedicated patients and 139 matchedcontrols. Results indicated that at initial scan,patients exhibited smaller whole-brain vol-umes (see also Hill et al., 2003; Mostofsky,Cooper, Kates, Denckla, & Kaufmann, 2002,for other supportive evidence, but Filipek et al.,1997; Lyoo et al., 1996, for negative findings).
Biological Vulnerabilities to the Development of Psychopathology 81
Individual (genetic/developmental) vulnerability
Prenatal and postnatal environmentsSubstance exposure during pregnancy
Parenting and schooling environmentStimulant medication use
ADHD symptomatology
Volumetric and/or functional anomaliesFrontal lobes
Striatum and basal ganglia Cerebellum
and other parallel temporal and parietal pathways
Catecholaminergic and other neurotransmitter
Neurochemical Dysregulation
Figure 4.1 Attention Deficit/Hyperactivity Disorder
04-Hankin.qxd 2/24/2005 11:20 AM Page 81

This difference was most significant forunmedicated patients. Unmedicated patientswere found to have smaller total white mattervolumes than medicated patients and controls.They also exhibited smaller cerebellar, tempo-ral gray matter, and total cerebral volumesthan controls. Medicated patients and controlswere not significantly different on white mattervolumes, but they differed for all measuredgray matter regions, with ADHD patientsexhibiting smaller volumes. At follow-up, dif-ferences in total and regional volumes betweenthe groups were found to persist over time,except for the caudate nucleus, which wasoriginally smaller in patients but did not differin size by adolescence. Except for the caudatenucleus, growth curves were found to be lowerin patients but to follow the same shape as incontrols. No significant differences were foundbetween males and females.
The study by Castellanos and colleagues(2002) is very important because it attemptsto provide answers to important questionsleft lingering by previous research. First, thevast majority of studies on ADHD have usedsamples of male participants, so less infor-mation has been available on the possible sexdifferences in the biological underpinningsof the disorder (e.g., Baving, Laucht, &Schmidt, 1999; Ernst et al., 1994). Resultsby Castellanos et al. (2002) suggest that pat-terns of brain volumetric abnormalities inADHD patients are similar in boys and ingirls. Second, because the vast majority ofpatients participating in imaging studies havebeen exposed to stimulant medication, itbecomes difficult to disentangle which brainanomaly may be attributed to the disorderand which may be attributed to medicationuse. Castellanos et al. (2002) suggest thatstimulant medication use is not responsiblefor the reduced total and regional brain vol-umes observed in ADHD children. It may bethat medication contributes to a normaliza-tion of brain volumes in ADHD children.Such findings are consistent with a growing
literature suggesting that stimulant medicationmay help normalize metabolic activityin frontostriatal regions of ADHD patients(Hale et al., 2000). Last, there has beendebate as to whether ADHD constitutes aform of developmental delay whose severitymay lessen over time, or whether the disordershould be characterized as involving a stablebiological vulnerability. Castellanos et al.(2002) showed, using a longitudinal design,that brain volumetric anomalies found inADHD children are present early on and arestable over time, which is consistent with avulnerability perspective and the increasingevidence that significant symptoms may per-sist through adulthood for a great number ofindividuals.
The structural and functional imagingliterature provides support for the presenceof frontal-striatal-cerebellar anomalies inADHD patients. The majority of functionalimaging studies have found evidence forfrontal hypofunction in ADHD patients(Baving et al., 1999; Durston et al., 2003;Lou, Henriksen, & Bruhn, 1984; Sieg,Gaffney, Preston, & Hellings, 1995;Silberstein et al., 1998; Zametkin et al.,1990). Most investigations of the basal gan-glia have focused on the caudate nucleus.Abnormalities of caudate volumes have beenreported, with ADHD patients exhibitingreduced volumes (Castellanos et al., 1996;Filipek et al., 1997; Hynd et al., 1993;Mataro, Garcia-Sanchez, Junque, Estevez-Gonzalez, & Pujol, 1997). PET and fMRIstudies have shown striatal activity to bereduced in ADHD patients (Durston et al.;Lou, Henriksen, Bruhn, Borner, & Nielsen,1989; Lou, Henriksen, & Bruhn, 1990;Vaidya et al., 1998). Volumetric abnormali-ties of the cerebellum have been foundin ADHD patients, but there are inconsisten-cies concerning the specific localization ofthese abnormalities (Berquin et al., 1998;Castellanos et al., 1996; Hill et al., 2003).In addition to the evidence showing that
VULNERABILITIES82
04-Hankin.qxd 2/24/2005 11:20 AM Page 82

ADHD stems from a dysfunction of thefronto-striatal-cerebellar pathway, ADHD isalso influenced by other parallel circuitsinvolving association areas (e.g., temporal,parietal, and occipital lobes) important to theintegration of information (Sowell et al.,2003).
Neurochemistry of ADHD
In light of evidence from neuroimagingstudies of frontal-striatal-cerebellar anatomicand functional anomalies in ADHD patients,as well as the neuropharmacology of stimulantmedications (Kirley et al., 2002), great atten-tion has been given to the contribution ofdopaminergic functioning to the presentationof ADHD. A number of studies using single-photon emission computed tomography tech-nology found evidence of increased striataldopamine transporter density in ADHDpatients (Dougherty et al., 1999; Krause,Dressel, Krause, Lung, & Tatsch, 2000). Theevidence, however, has been somewhat mixed,with some studies finding no differences(van Dyck et al., 2002). PET studies label-ing catecholamine terminals have foundreduced uptake in the left medial prefrontalcortex in adults with ADHD but have alsofound increased uptake in the right midbrainof ADHD adolescents (Ernst, Zametkin,Matochik, Jons, & Cohen, 1998; Ernst et al.,1999). Animal studies have found that bothhypo- and hyperdopaminergic states are posi-tively linked with hyperactive behaviors(Castellanos & Tannock, 2002; Denckla,2003). The pattern of findings thus suggeststhat dysregulation of the catecholaminergicsystem is involved in ADHD, although thenature of the dysregulation is not well defined.It is also possible that other neurotransmittersystems are involved in the etiology of thedisorder. Stimulant medications, as a group,increase synaptic levels of all catecholamines.Methylphenidate, for one, inhibits reuptake ofdopamine and noradrenaline (Denckla). As
well, the noradrenergic system has been shownto be essential to executive functions and maybe especially important for inattention symp-toms (Denckla). Serotonin also may be impli-cated, especially for comorbid aggression. Ithas also been shown to play a role in executivefunctioning (Denckla). However, medicationsthat affect catecholaminergic functioning aregenerally found to be effective, whereas thosethat affect primarily the serotonergic systemhave not (Wilens et al., 2002). In a meta-analysis of 20 genes for dopaminergic, sero-tonergic, and noradrenergic metabolism byComings and colleagues (2000), noradrenergicgenes were found to account for a greater pro-portion of the variance in the ADHD pheno-type than dopamine and serotonin genescombined. It has been suggested that ananomalous balance between levels of differentneurotransmitters, rather than anomalies inone neurotransmitter system per se, may con-tribute to the etiology of ADHD (Oades,2002).
Conduct Disorder
One of the most common comorbidconditions observed in children with a diag-nosis of ADHD is CD (see Hankin, Abela,Auerbach, McWhinnie, & Skitch, Chapter14 of this volume, for greater discussion anddescription), which is defined as a persistentand recurrent disregard for social rules andthe basic rights of others. In contrast toADHD, CD, as a diagnosis, has not receivedas much attention from the field of brainneuroimaging. Instead, research has focusedon the neuroimaging correlates of the mostdisruptive symptoms of CD, aggression andviolence, in both normative adult samplesand in individuals who show the mostextreme forms of antisocial behaviors,namely murderers and psychopaths. Specialattention has also been given to the contribu-tion of serotonergic functioning, testos-terone, cortisol, and sympathetic arousal to
Biological Vulnerabilities to the Development of Psychopathology 83
04-Hankin.qxd 2/24/2005 11:20 AM Page 83

aggression, as well as the potential role ofabnormal prenatal brain development.Current conceptualizations of antisocialbehavior point to dysfunctions in areas andneurochemical systems important to theregulation of impulsivity and aggression(e.g., prefrontal cortex, serotonin), as wellas affect-guided anticipation (e.g., amygdala,hippocampus). Davidson, Putnam, andLarson (2000), for example, suggest thatimpulsive aggression results from anomaliesin the threshold for activating negative affec-tive states and the anticipation of negativeconsequences of aggression. Figure 4.2 illus-trates how these dysfunctions interact withpre- and postnatal moderating factors inleading to symptoms of CD.
THE BRAIN AND AGGRESSION
Lesion studies have provided evidence fora link between damage to specific brainregions and behavioral symptoms similar toconduct disorders and associated antisocialbehaviors. One of the earliest cases—andperhaps the most famous case—is that ofPhineas Gage, who, upon an explosion at hiswork site, had an iron rod blown through hisinferior frontal cortex, more specifically theventromedial region. Previously a conserva-tive and friendly young man, Gage becamehighly irritable and impulsive, showing littleconsideration for the consequences of hisactions (Stuss, Gow, & Hetherington, 1992).This pattern of impulsivity, antisocial
VULNERABILITIES84
Individual (genetic/developmental) vulnerability
Prenatal and postnatal environmentsSubstance exposure during pregnancy
Birth complicationsPsychosocial environment
Conduct disorder symptomatology
Volumetric and/or functional anomaliesPrefrontal cortex
HippocampusAmygdala
Superior parietal cortexCorpus callosum
Neurochemical Dysregulation
Low physiological arousal
Skin conductanceHeart rate
Serotonergic systemTestosterone
Cortisol
Figure 4.2 Conduct Disorder
04-Hankin.qxd 2/24/2005 11:20 AM Page 84

behaviors, and inability to inhibit responsesresulting from damage to the inferior frontaltemporal lobes has been labeled by some as“acquired sociopathy” (Damasio, Tranel, &Damasio, 1990). Damage to the amygdala,in turn, has been found to lead to symptomsreminiscent of psychopathy, such as reducedemotionality and understanding of emo-tions, as well as disturbed fear processing(Hoptman, 2003).
In his recent review of the neuroimagingliterature of antisocial and violent behaviors,Hoptman (2003) notes that anger induction inhealthy adults has been linked with increasedblood flow to the left orbitofrontal cortex, theright ventral anterior cingulate cortex, andbilaterally to the anterior temporal poles, aswell as to the thalamus. In violent patientsand offenders, aggression levels are associatedwith reduced metabolism of the anterior, infe-rior, and medial frontal and temporal lobes,as well as of the thalamus (Hoptman). In aseries of studies by Raine and colleagues(Raine et al., 1994; Raine et al., 1998; Raine,Buchsbaum, & LaCasse, 1997), accused mur-derers were found to have reduced metabolismof the prefrontal and superior parietal cortex,as well as the angular gyrus and corpus callo-sum. Abnormal functional asymmetry of theamygdala, thalamus, and medial temporallobe were also observed. Reductions of pre-frontal activity were more pronounced in indi-viduals whose crime could be described asimpulsive (Raine et al., 1998). It has beensuggested that hypometabolism may be mostpronounced in criminals with a positivepsychosocial background (e.g., from relativelyaffluent socioeconomic conditions, intacthomes, with absence of deprivation or abuse)(Combalbert, Bret-Dibat, & Favard, 2002;Raine, 2002). More recently, Soderstromet al. (2002) observed a negative correlationbetween right frontal and temporal bloodflow and scores on the personality dimensionof a psychopathy interview (Factor 1 of thePsychopathy Checklist-Revised) in violentoffenders. Raine and colleagues (2003)
observed increased interhemispheric connec-tivity in a study of the corpus callosum inpsychopathic, antisocial individuals. The func-tional anomalies observed in violent, antiso-cial individuals appear to be accompaniedby structural anomalies. Raine, Lencz, Bihrle,LaCasse, and Colletti (2000) found that indi-viduals with antisocial personality disorderexhibited an 11% reduction of prefrontal graymatter volumes. As well, hippocampal volu-metric reductions have been negatively associ-ated with psychopathy levels (Laakso et al.,2001). In a recent study of successful andunsuccessful psychopaths, Raine and col-leagues (2004) noted that unsuccessful psy-chopaths exhibit a greater asymmetry of thehippocampus than successful psychopaths andcontrols. This greater asymmetry was causedby decreased left hippocampal and increasedright hippocampal volumes.
Lesion and imaging studies have thuspointed to a number of brain anomalies aspotentially underlying violent and antisocialbehavior. Studies investigating the relation-ship between perinatal complications, pre-natal exposure to toxins, and aggressionsuggest that these brain anomalies may inpart be caused by disrupted prenatal braindevelopment due to very early exposure tostressors. Alcohol and nicotine exposure dur-ing pregnancy have been shown to increasethe risk of later conduct disorders (Raine,2002). Birth complications also increase therisk of CD; delinquency; and impulsive, vio-lent offending, through causing brain dam-age to the frontal lobes and other regions,such as the hippocampus, which have beenfound to be abnormal in certain types ofoffenders (Combalbert et al., 2002; Raine).Exposures to toxins and birth complicationsinteract in predicting conduct outcomes(Combalbert et al.; Raine). Consistent witha vulnerability-stress perspective, Brennan,Grekin, and Mednick (1999) found thatwhereas children whose mothers smoked20 cigarettes per day during pregnancy hada twofold increase in violent offending as
Biological Vulnerabilities to the Development of Psychopathology 85
04-Hankin.qxd 2/24/2005 11:20 AM Page 85

adults, children who were exposed to bothnicotine and delivery complications exhibiteda fivefold increase in violent offending asadults. Thus, it appears that exposure totoxins and obstetric complications interactwith the postnatal psychosocial environmentin predicting later aggression and violence inchildren (Raine).
Aggression and Arousal
Low resting heart rate has been shownto be characteristic of antisocial individuals,especially those who come from privilegedsocial backgrounds (Raine, 2002). A similarpattern is generally found for skin conduc-tance (Raine). Both indices of reactivityappear to have some predictive value, becauselower heart rate and skin conductance at age3 have been shown to predict violence levelsat age 11 (Combalbert et al., 2002). In turn,higher physiological arousal may serve aprotective function, because individuals whodesist from adolescent antisocial behaviorhave been shown to have increased electro-dermal and cardiovascular activity (Raine).Increased heart rate and skin conductancehave also been observed in individuals withcriminal fathers who do not themselves com-mit crimes (Combalbert et al.). Although thespecific mechanism through which low phys-iological arousal may lead to conduct prob-lems is still unclear, it has been hypothesizedthat lowered physiological arousal may resultfrom prefrontal damage and may predisposeindividuals to low levels of fear, high levels ofstimulation seeking, or both, which may inturn cause them to act out (Raine).
The Neurochemistry of Aggression
The contribution of serotonergic function-ing to aggression has been studied in rodents,nonhuman primates, and humans using avariety of methods, such as lesions, measure-ment of serotonin metabolites in CSF and
plasma, tryptophan depletion and supple-mentation, platelet serotonin uptake, andprolactin response to serotonergic agonists(Krakowski, 2003). In rodents, increasedlevels of aggression have been observedfollowing serotonin depletion or neuronaldestruction. In nonhuman primates, lowserotonin levels have been associated withsevere, unrestrained, dysfunctional aggres-sion, whereas no relationship with morepositive, assertive types of aggression hasbeen observed (Krakowski). More specifi-cally, low serotonin early in life has beenlinked with later violence and death, andexperimental manipulations of serotoninlevels have confirmed this inverse relation-ship between aggression and serotonin(Krakowski). In human studies, negative cor-relations have been found between lifetimeaggression in individuals with personalitydisorders and serotonin levels, and low sero-tonin levels are associated with impulsiveviolence and recidivism in offenders(Krakowski). Experimental studies haveshown reduction of irritability and aggres-sion in patients with personality disordersupon treatment with selective serotoninreuptake inhibitors (SSRIs) (e.g., Cherek,Lane, Pietras, & Steinberg, 2002). Further-more, tryptophan depletion and supplemen-tation have been shown to yield changes inaggression levels in the expected directions,although these changes may occur only inindividuals who have a preexisting vulnera-bility (e.g., family history of alcoholism, highhostility) (Krakowski). In a recent meta-analysis (Moore, Scarpa, & Raine, 2002),reduced 5-HIAA levels in antisocial individu-als were found compared with nonantisocialindividuals. However, age was found to be asignificant moderator: Individuals youngerthan 30 years of age exhibited significantlylower 5-HIAA levels. The attenuation withage of decreased serotonergic levels maycontribute to the observed age-related declinein crime (Moore et al.).
VULNERABILITIES86
04-Hankin.qxd 2/24/2005 11:20 AM Page 86

Age differences have also been found withregard to the relationship between testos-terone and aggression. High testosterone lev-els have been associated with increased levelsof aggression in adults. Patterns are less clearin children and adolescents, for whom therelationship between testosterone and aggres-sion has sometimes been found to be reversedor absent (Raine, 2002). Raine has suggestedthat inverse relationships between testos-terone and aggression in younger and olderindividuals may be attributed to the differ-ing social experiences of these age groups.Aggressive children and adolescents usuallyexhibit poor academic and social functioning,and it may be these aversive experiences thatdecrease their testosterone levels. However,as they get older and are perhaps able to usetheir aggression to obtain dominance andsuccess, their testosterone levels rise to theirnaturally high levels (Raine). Activity of theHPA axis has also been proposed as influ-encing aggression and conduct problems.Individuals with a diagnosis of CD who showsignificant levels of anxiety have greater levelsof cortisol, show less aggression, and havefewer police contacts (Pihl et al., 2003). It islikely that testosterone and cortisol interacttogether in contributing to aggression andconduct disorders. Finally, it is also mostlikely that neurochemical functioning inter-acts with psychosocial experiences in predict-ing the presence or absence of antisocialbehaviors. Caspi and colleagues (2002), forexample, found that polymorphism of theMAOA gene, located on the X chromosome,interacts with childhood maltreatment in pre-dicting adult criminal behaviors. The MAOAgene encodes an enzyme that metabolizes anumber of neurotransmitters, including sero-tonin. Although low activity of the MAOAgene was not found to predict adult criminalbehavior by itself, its combination with child-hood maltreatment was significantly relatedto the presence of antisocial behaviors duringadulthood (Caspi et al.).
DEPRESSION
The DSM-IV-TR (APA, 2000) definesmajor depressive episode as being charac-terized by affective (e.g., depressed mood,anhedonia), cognitive (e.g., worthlessnessor guilt, diminished concentration), and veg-etative symptoms (e.g., changes in sleepingor eating) (see Hankin & Abela, Chapter 10in this volume, for greater description anddiscussion about depression). Faced withthe increasing prevalence of depression andits progressively earlier onset, researchersare focusing more and more on the factorsthat render individuals vulnerable todepression. This endeavor, however, is notnovel, because proposals regarding the etio-logical role of both innate and environmen-tal factors have been present since the firstdescriptions of the disorder more than2,000 years ago (Nestler et al., 2002).However, it is only with the use of increas-ingly sophisticated imaging technology thatresearchers have been able to begin describ-ing the specific brain anomalies potentiallyassociated with the disorder. Davidsonand colleagues (2002) proposed a modelwhereby hypoactivation of the left pre-frontal cortex and anterior cingulate cortexand hyperactivation of the right prefrontalcortex would lead to decreased approachbehaviors and increased withdrawal andanxiety, which are often seen in depressedpatients. Decreased activity of the left pre-frontal cortex could in turn lead to adecreased inhibition of the amygdala andthus persistent negative affective states.Abnormalities of the hippocampus werealso suggested to contribute to an absenceof appropriate contextual modulation ofemotions (Davidson et al., 2002). Figure4.3 summarizes brain areas and neuro-chemical systems found to be involved indepressive affective states and their interac-tions with various psychosocial moderatingfactors.
Biological Vulnerabilities to the Development of Psychopathology 87
04-Hankin.qxd 2/24/2005 11:20 AM Page 87

Functional and Structural Anomalies
Little evidence has been provided fordifferences in overall whole-brain volumesin depressed patients when compared withhealthy controls (Strakowski, Adler, &DelBello, 2002). However, consistent with themodel proposed by Davidson et al. (2002), dif-ferences in metabolism and volumes have beenreported for a number of subregions, such asthe prefrontal and cingulate cortex, the hip-pocampus, and the amygdala (Nestler et al.,2002). Total frontal lobe volumes have been
found to be reduced by approximately 7% indepressed patients. The subgenual prefrontalcortex appears to be particularly reduced, withstudies finding reductions of approximately48% (Drevets et al., 1997). As well, reductionof the right-greater-than-left asymmetry of thefrontal lobes usually found in controls has alsobeen observed. The extent of total frontal lobevolumetric reduction and decreased asymme-try has been associated with severity of symp-toms (Beyer & Krishnan, 2002; Strakowskiet al., 2002). Imaging and postmortem studieshave provided support for the presence of
VULNERABILITIES88
Individual (genetic/developmental) vulnerability
Moderating factorsGender
Cognitive styleStress
Social support
Depressive symptomatology
Neurochemical Dysregulation
Volumetric and/or functional anomaliesPrefrontal cortexTemporal cortex
CingulateHippocampus
AmygdalaStriatum
ThalamusCerebellum
Corpus callosum
Family history
Cerebrovasculardisease
Serotonergic systemOther neurotransmitter systems
Cortisol
Figure 4.3 Depression
04-Hankin.qxd 2/24/2005 11:20 AM Page 88

volumetric anomalies in the orbital region,more specifically bilateral volume reductionsand decreased cortical thickness, as well asdecreased neuronal size and density (Lai,Payne, Byrum, Steffens, & Krishnan, 2000;Rajkowska et al., 1999). It is important tonote, however, that a number of studies havenot found evidence for significant volumetricdifferences in these brain regions betweendepressed patients and healthy controls(Nestler et al.).
Decreased bilateral or left prefrontalcortex activation is one of the most oftenreported functional anomalies in the imagingliterature on depression (Davidson et al.,2002). Resting state imaging studies haveshown hypometabolism of the frontal lobes,more specifically of the dorsolateral and dor-somedial prefrontal cortex (Davidson et al.,2002; Drevets, 2000; Liotti & Mayberg,2001). Specificity of reduced activity to theleft hemisphere has been replicated numeroustimes by electroencephalogram (EEG) studies,although negative evidence has also beenfound (Davidson et al., 2002). Partial supporthas been provided for an increase in dorsolat-eral and dorsomedial activity with antide-pressant use, more specifically to the lefthemisphere (Davidson et al., 2002; Drevets).However, although the dorsolateral and dor-somedial prefrontal regions have been foundto be hypoactive in some studies, evidence forhyperactivity of other prefrontal regions indepression has also been provided. Increasedactivity in the subgenual prefrontal cortex, aswell as bilateral posterior orbital cortex, leftventrolateral prefrontal cortex, and anteriorinsula has been found, together with a bodyof findings suggesting that antidepressanttreatment may lead to a reduction of activityin these regions (Drevets).
Evidence for volumetric anomalies of thetemporal lobes and limbic structures has beensomewhat mixed (Beyer & Krishnan, 2002).Patients suffering from major depression havebeen found to have decreased temporal cortical
volumes when compared with healthy controlsin some studies, although a number of investi-gations have reported null findings (Strakowskiet al., 2002). Structural imaging studies havereported reductions of hippocampal volumesin patients with major depression, a findingalso reported in patients with bipolar disorder,posttraumatic stress disorder, and borderlinepersonality disorder (Davidson et al., 2002;Strakowski et al.). Hippocampal volumetricreductions for patients with major depressionhave been estimated to range from 8% to 19%(Davidson et al., 2002). Decreased hippocam-pal activity has also been reported using PETtechnology (Davidson et al., 2002). Findingsfor volumetric anomalies of the amygdala havebeen inconsistent, with different studies report-ing increased, decreased, or no differences involumes in depressed patients compared withcontrols (Davidson et al., 2002). Studies havealso reported a left-greater-than-right asymme-try of the amygdala found in patients but notin controls (Davidson et al., 2002). Functionalstudies have reported increased activity of theamygdala in depressed patients, an increaseevaluated by some to be of a magnitude of44% (Davidson et al., 2002). As well, althoughremission of major depressive symptoms isassociated with a reduction of activity levelsof the amygdala, relapse is associated withincreased amygdala activation (Davidsonet al., 2002). It is interesting to note thatincreased amygdala activity has also beenassociated with bipolar disorder and anxietydisorders (Davidson et al., 2002). Bilateralreductions in anterior cingulate activity havealso been noted and have been found to corre-late with the extent of gray matter reduction,which is consistent with the finding thatantidepressant treatment does not generallylead to a normalization of activity in thisregion (Davidson et al., 2002; Drevets, 2000).
Reduced volumes of the basal ganglia havebeen noted in depressed patients, most specif-ically of the caudate nucleus and the puta-men, in the context, however, of numerous
Biological Vulnerabilities to the Development of Psychopathology 89
04-Hankin.qxd 2/24/2005 11:20 AM Page 89

null findings (Beyer & Krishnan, 2002;Strakowski et al., 2002). Limited research hasbeen done on the presence of cerebellar andcorpus callosum structural anomalies indepression (Beyer & Krishnan). A number ofstudies have found decreased volumes of thecerebellum (Strakowski et al.), whereas someevidence has been provided for increased vol-umes of some subregions of the corpus callo-sum in depressed patients (Beyer & Krishnan).Research on thalamic structural anomalieshas been limited and inconsistent. Volumereductions have been observed in some stud-ies, but it has been suggested that thalamicvolumetric reductions may be more commonin bipolar disorder (Strakowski et al.).Increased, decreased, and no change of thala-mic activity in depressed patients have allbeen reported (Liotti & Mayberg, 2001).
The imaging literature on the brain struc-tural and functional correlates of depressionhas thus produced a number of heteroge-neous findings, with partial empirical sup-port provided for an involvement of thefrontal and temporal lobes, the amygdala,the hippocampus, the basal ganglia and tha-lamus, and the cerebellum and corpus callo-sum. The equivocal nature of evidence forvolumetric and functional anomalies indepression has been attributed by some tothe heterogeneity of the disorder. Morespecifically, it has been suggested that somebrain anomalies may characterize specificsubtypes of patients (Davidson et al., 2002).For example, patients with a family historyof major depression have been shown to bemore likely to have increased activity of theamygdala, orbital cortex, and medial thala-mus and decreased activity in specific subre-gions of the prefrontal cortex, as well asvolumetric anomalies of the basal ganglia(Drevets, 2000). As well, patients with afamily history of major depression do notexhibit the reduced amygdala activity usuallyassociated with remission of symptoms,suggesting that amygdala hyperactivity may
constitute a marker of a depressive traitrather than a depressive state in this subpop-ulation (Davidson et al., 2002). Patients witha late onset of the disorder have also beenshown to exhibit a specific pattern of anoma-lies (Drevets, 2000). They exhibit sulcal andventricular enlargement, as well as reducedvolumes of the frontal lobes and the basalganglia. Infarction to the frontal lobes orstriatum has been associated with increasedrisk of depression, and there has been sug-gestion that late-onset depression maydevelop as a result of cerebrovascular disease(Strakowski et al., 2002). Some brain func-tional and structural anomalies may thus bespecific to familial or late-onset depressioncases (Strakowski et al.). Overlap in some ofthe observed regional anomalies may confera common vulnerability to both subtypes,although different causal mechanisms are atplay (Drevets).
Neurochemical Anomalies
Evidence for a serotonergic contributionto the physiology and treatment of depressioncomes from several lines of evidence. All phar-macological treatments of depression to datehave focused on the monoamines, and the lat-est generation of antidepressants, the SSRIs,has been shown to be effective in reducingsymptoms (Stockmeier, 2003). As well, deple-tion of tryptophan, a serotonergic precursor,has been shown to cause recurrence of symp-toms in unmedicated patients in remission(Young & Leyton, 2002). Reduced levels ofserotonergic metabolites in the cerebrospinalfluids of depressed patients with a history ofsuicide attempts have also been found, as wellas decreased neuroendocrine response to sero-tonin stimuli (Stockmeier). In his recent review,Stockmeier reported a number of anomalies inserotonergic receptor binding sites associatedwith depression. Serotonin-1A receptor bind-ing was found to be reduced in a number ofregions, such as the medial temporal cortex,
VULNERABILITIES90
04-Hankin.qxd 2/24/2005 11:20 AM Page 90

the temporal pole, the orbitofrontal cortex, theanterior cingulate cortex, the insula, and thedorsolateral prefrontal cortex. Alternatively,serotonin-2A receptor binding was found to beincreased in the prefrontal cortex (Stockmeier).However, it is most likely that serotonin is notacting alone in the neurochemistry of depres-sion. Norepinephrine receptor and transporteranomalies have been noted in depressedpatients (Stockmeier). As well, depressedpatients have been shown to have cortical lev-els of the inhibitory neurotransmitter GABAthat are half that of healthy controls, levels thatare restored by antidepressant medications(Stockmeier). Antagonists of substance P, apeptide neurotransmitter, have been shown bysome researchers to be as effective as SSRIs inalleviating symptoms of major depression andanxiety (Stockmeier). In certain brain regions,nearly half of serotonergic neurons are colocal-ized with substance P neurons, and substance Pantagonists have been shown to increase firingactivity of serotonergic neurons in certainbrain areas (Stockmeier). Interactions of anumber of neurochemical systems are thusmost likely involved in the etiology and treat-ment of depression.
Neurochemical explanations of depressionhave also focused on possible hyperactivityof the HPA axis. Research has shown thatabnormally elevated HPA activity may bepresent in approximately 50% of depressedpatients, most importantly those with afamily history of major depression, and thatthese anomalies may be corrected withantidepressant administration (Nestler et al.,2002). Thus, hyperactivity of the HPA axismay contribute to depression through hip-pocampal damage, which is consistent withreduced hippocampal volumes sometimesfound in depressed patients (Nestler et al.).As well, hippocampal volume reductionshave been positively associated with durationof illness, which concurs with exposure tochronically elevated glucocorticoid levels(Davidson et al., 2002). Furthermore, centrally
administered CRF has been shown to triggersymptoms found in depression such as anxi-ety and neurovegetative symptoms. This sug-gests that the contribution of the HPA axishyperactivity to depression may not be lim-ited to the hippocampus, but may alsoextend to other brain regions such as thehypothalamus (Nestler et al.).
The vast majority of studies on the biolog-ical bases of depression have focused on adultdepression, using cross-sectional designs. Thishas made difficult a clear understanding ofthe role biology plays in the emergence of thedisorder and how biological contributions todepression evolve over time. Research withchildren and adolescents has pointed to possi-ble anomalies of the frontal lobes. Steingardand colleagues (2002), for example, havenoted smaller white matter volume but greatergray matter volumes in depressed adolescents.Nolan and colleagues (2002) found thatalthough depressed children and adolescentswith no family history of major depression(MD) had larger left prefrontal volumes thandepressed individuals with a family MD andnormal controls, depressed children and ado-lescents with a family history of MD and con-trols did not significantly differ in terms ofprefrontal volumes. Furthermore, a studyfocusing on depressed preschoolers has pointedto a possible contribution of the stress systemto childhood-onset depression (Luby et al.,2003). Depressed preschoolers exhibitedincreased cortisol levels in response to bothsituations of separation and frustration.Nondepressed preschoolers, however, showeddecreased cortical levels in separation situa-tions and increased cortisol levels when frus-trated. A longitudinal study by Goodyer,Herbert, and Tamplin (2003) has suggestedthat anomalies in steroid levels may precedethe onset of the disorder. Adolescents whodeveloped persistent depressive symptomswithin 2 years initially showed a higher morningcortisol/dehydroepiandrosterone ratio. Morelongitudinal studies focusing on individuals at
Biological Vulnerabilities to the Development of Psychopathology 91
04-Hankin.qxd 2/24/2005 11:20 AM Page 91

risk of developing depression are required inorder to disentangle the biological roots of theemergence and development of the disorder.
SUBSTANCE ABUSE AND DEPENDENCE
Of the many “fat” words in psychopathol-ogy, perhaps the most obese is substanceabuse/dependence (see Kassel, Weinstein,Mermelstein, Skitch, & Veilleux, Chapter 13in this volume, for greater description anddiscussion). The size (the large number of dif-ferential drug diagnoses) and obtuseness (themyriad of paths to the end point) of the diag-nosis make this diagnosis highly prevalentbut often meaningless. Nevertheless, depend-ing on the survey, substance abuse remainsthe most prevalent form of mental disorder,with alcohol abuse and dependence account-ing for a majority of diagnoses (Pihl, 1999).Until recently, there has been a dearth ofknowledge concerning etiology.
It is now clear that some individuals, forvarying reasons, are more likely than others todevelop problems with drugs. To understandreasons for abuse, it is no longer necessary tolook at each drug individually; instead, thefocus is on what a drug or group of drugs doesfor a specific individual. By adopting this per-spective, one can get a glimpse of how genetic,biochemical, physiological, neuropsychologi-cal, and experiential facts interact. Two majordrug groupings are the stimulant drugs and thedepressant drugs, although there are obviousadditional groupings (e.g., pain relief, hallu-cinogenics). Further, this dual, broad classifica-tion also suffers from the fact that some drugshave, for example, both stimulant and depres-sive effects. The most notable substance dis-playing this characteristic is alcohol, becausefor most individuals, on the rising limb of theblood alcohol curve, approximately 30 min-utes postingestion of the drug, one reacts as ifstimulated, whereas later on the blood alcoholcurve, depressant brain effects are normative.
Stimulant Drugs
Research with rats and monkeys hadclearly demonstrated in the 1960s and 1970sthat certain parts of the brain, when stimu-lated electrically or chemically, producedreinforcing effects. That is, animals wouldwork to receive this stimulation and wouldlearn new responses based on pairing somebehavior with the stimulation of the mesolim-bic brain area, particularly the ventraltegmental area and specifically the nucleusaccumbens. Dopamine has been implicatedin this response in that these effects aredirectly related to the density of dopamineneurons. Further, drugs that blocked theseneurons reduced these effects (Fibiger &Phillips, 1988). Stimulant drugs such ascocaine and amphetamines have been shownnot only to directly release dopamine as wellas other neurotransmitters but also to blockthe reuptake process, thus prolongingdopamine’s effects (Koob & Blume, 1988).Some other drugs that increase dopamineflow into the nucleus accumbens are alcohol,marijuana, and opiates. At issue is whetherthese various drugs have a direct or indirecteffect, but it is clear that the dopaminergicsystem, which is related to psychostimula-tion, is affected. Recently, Boileau and col-leagues (2003) used PET technology to showincreased dopamine release in the nucleusaccumbens in humans after consuming analcohol challenge. In addition, the study pro-vided clues to the critical issue of why someindividuals develop drug problems, whereasa large percentage of individuals who try var-ious legal and illegal stimulant drugs do notdevelop abuse or dependency problems (i.e.,a vulnerability to substances). Specifically,individuals who had previously demon-strated an accelerated heart rate responseto alcohol were significantly more likely toshow this dopamine release than individualswho did not previously show an acceleratedheart rate response to alcohol. Althoughmany situations, physical and psychological,
VULNERABILITIES92
04-Hankin.qxd 2/24/2005 11:20 AM Page 92

affect heart rate, this intoxicated heart rateresponse has been shown to distinguish indi-viduals who self-report positive subjectivefeelings to alcohol (Conrod, Peterson, &Pihl, 2001) and those for whom alcohol ispositively reinforcing on a memory task(Bruce, Shestowksy, Mayerovitch, & Pihl,1999). Thus, some individuals at risk fordeveloping problems with alcohol are char-acterized by this elevated heart rate responseon the rising limb of the blood alcohol curve,which has also been shown to be characteris-tic of individuals who reflect externalizingbehaviors such as CD (Pihl et al., 2003) andpathological gambling (Brunelle, Assaad,Pihl, Tremblay, & Vitaro, 2003). The factthat these groups seem to respond with thisresponse as well as having a propensity todevelop stimulant substance abuse problemssuggests particularities in baseline biologicalfunctioning. In rats selectively bred to pre-fer alcohol, increased functioning of thedopamine system has been demonstrated(McBride, Murphy, Lumeng, & Li, 1990).In these particular strains, alcohol facilitatesstimulant-like behaviors, such as increasedmotor activity. In addition, rats selectivelybred to self-administer opiates and cocainedemonstrate heightened place learning whenadministered these drugs, reminiscent of theearly electrical stimulation studies (Guitart,Beitnes-Johnson, & Nestler, 1992). Thesefindings suggest some preexisting genetic vul-nerability. Indeed, there is a large literatureshowing that children of alcoholics, particu-larly sons—who are at four to nine timesincreased risk to develop the disorder—havediscernible neuropsychological, electrophysi-ological, and behavioral responses to alcoholcharacteristics (Pihl, Peterson, & Finn,1990). Further, sensitization (i.e., the factthat the use of one drug makes other similardrugs more reinforcing) has been shown tobe potentiated by environmental stimuli suchas stress (Deminiere, Piazza, Le Moal, &Simon, 1989). However, these effects are notonly the result of biologically determined
vulnerability but also are affected by usingcertain drugs. Some drugs per se do producelong-term potentiation of dopamine-releasingcells in the ventral tegmental area of thebrain. Further, cross-sensitization with stressmeans that stress can reinstate drug taking,perhaps accounting for the difficulties intreatment and the high probability of relapse.This pattern is called neural adaptation andoccurs by raising the excitatory neurotrans-mitter glutamate, which stimulates specificreceptors on dopamine cells (Saal, Dong,Bonci, & Malenka, 2003). Similar effects arelikely to occur with other stimulant drugs,such as nicotine, where withdrawal has beenassociated with a significant decrease indopamine levels. Recent research shows thattreating smokers with a drug designed toincrease dopamine levels is more effectivethan typical nicotine replacement therapy(George & O’Malley, 2004).
The precise nature of the reward whendopamine is released in this particular brainarea is also open to some debate. It appearsnot to be reinforcement in the classical sense,because the behavior affected does not sati-ate. Consequently, we have referred to thissystem as the “cue for reward system” (Pihl& Peterson, 1995) in that it stimulates loco-motor activity and feelings of pleasure as ifsomething is about to happen or is happen-ing. Thus, commonalities are found in behav-ior among individuals who are sensationseekers, conduct disordered, and generallyexternalizing, and the abuse of stimulantdrugs (Pihl & Peterson). These characteristicsare particularly reminiscent of individualsat risk for type 2 alcoholism, which involveshigh heritability, early onset, and associatedantisocial behavior (Pihl et al., 1990).Further, it has been recently demonstratedthat the risk for ADHD and CD is 3.8 and6.6 times greater, respectively, amongchildren whose parents were alcohol depen-dent compared with controls. Twin studieshave shown that shared genetic factorsare largely responsible for this comorbidity
Biological Vulnerabilities to the Development of Psychopathology 93
04-Hankin.qxd 2/24/2005 11:20 AM Page 93
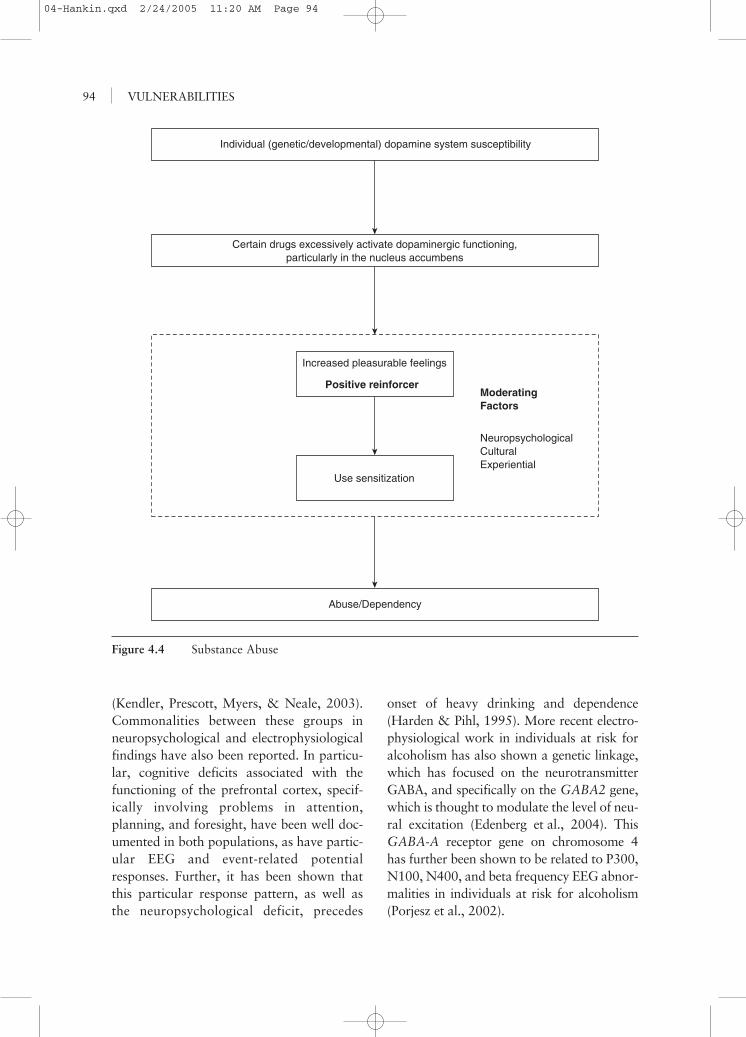
(Kendler, Prescott, Myers, & Neale, 2003).Commonalities between these groups inneuropsychological and electrophysiologicalfindings have also been reported. In particu-lar, cognitive deficits associated with thefunctioning of the prefrontal cortex, specif-ically involving problems in attention,planning, and foresight, have been well doc-umented in both populations, as have partic-ular EEG and event-related potentialresponses. Further, it has been shown thatthis particular response pattern, as well asthe neuropsychological deficit, precedes
onset of heavy drinking and dependence(Harden & Pihl, 1995). More recent electro-physiological work in individuals at risk foralcoholism has also shown a genetic linkage,which has focused on the neurotransmitterGABA, and specifically on the GABA2 gene,which is thought to modulate the level of neu-ral excitation (Edenberg et al., 2004). ThisGABA-A receptor gene on chromosome 4has further been shown to be related to P300,N100, N400, and beta frequency EEG abnor-malities in individuals at risk for alcoholism(Porjesz et al., 2002).
VULNERABILITIES94
Individual (genetic/developmental) dopamine system susceptibility
Certain drugs excessively activate dopaminergic functioning,particularly in the nucleus accumbens
Abuse/Dependency
Increased pleasurable feelings
Positive reinforcer
Use sensitization
ModeratingFactors
NeuropsychologicalCulturalExperiential
Figure 4.4 Substance Abuse
04-Hankin.qxd 2/24/2005 11:20 AM Page 94

Depressant Drugs
Negative reinforcement (i.e., when anaversive situation is reduced) is anotherreward response that can also explain drugabuse. Drugs that reduce anxiety, fear, anddysphoric pain are frequently abused.Individuals with phobias, for example, are2.5 times more likely to abuse alcoholand individuals with panic disorder fourtimes more likely (Weissman, 1988). “Stressresponse dampening” is a phrase used tocharacterize the effect of some drugs on theseand otherwise vulnerable individuals. Theimplied mechanism for abuse is thus seen asa form of self-medication that reduces theintensity of an aversive state. Although someexperimental results in this area have beencontradictory to this explanation (e.g., Steele& Josephs, 1990), like most studies in psy-chopathology the choice of subjects is criti-cal. Vulnerable subjects are required, andone particularly vulnerable group is anxiety-sensitive individuals. In the paradigmaticstudy, these individuals are first shown stim-uli when sober, which results in heightenedphysiological reactivity, which then is greatlydiminished when they are alcohol intoxi-cated (Stewart & Pihl, 1994). Alcohol in thiscase is acting as an antianxiolytic, putativelyby potentiating the action of the inhibitoryneurotransmitter GABA. These drugs andother sedatives operate on selective GABA-Aand benzodiazepine receptor sites. There areactually three subunits of the GABA-A recep-tors and 14 variants of the subunits. What isnotable is that alcohol and benzodiazepinesact on one specific subunit (Suzdak et al.,1986). It is also known that when measuredin plasma, baseline levels of GABA are heri-table and that individuals at risk for develop-ing alcoholism, like alcoholics, have lowerlevels (Song et al., 2003). PET studies withoffspring of alcoholics have also shown areduced GABA response to a drug challenge,supporting the notion of reduced gabanergic
activity. This conclusion is reinforced bynumerous animal studies on selectively bredalcohol-preferring rats; these studies haveshown that such rats have dense gabanergicinnervation in the nucleus accumbens, thatGABA chronically inhibits this system, thatalcohol inhibits the action of the gabanergicinhibitory system, and that this inhibitoryaction accounts for the reward these ratsreceive from alcohol (McBride et al., 1990).Thus, it is suggested that at high dosesneeded to produce intoxication (Stewart,Finn, & Pihl, 1995), alcohol might directlypotentiate inhibition of certain neurons.
Similar autonomic reactivity and dampen-ing in children of alcoholics, particularly sons,have been reported, perhaps as a result of thesame mechanisms just described, but not fromfear or anxiety. This response, however, likelyresults from an overreactivity to novelty, thelatter likely resulting from the aforementionedfrontal neuropsychological deficits (Pihl &Peterson, 1995). Consequently, both highlyanxious and externalizing individuals can dis-play dampening and negative reinforcement,albeit for divergent reasons: those high onanxiety to self-medicate and those with exter-nalizing problems possibly to improve focus.
CONCLUSION
Studies of the biological bases of psy-chopathology have pointed to subtleanatomical, functional, and neurochemicalanomalies being associated with disorderssuch as attention deficit/hyperactivity disor-der, conduct disorder, major depressive dis-order, and substance abuse. Generally,results to date for most disorders have beeninconsistent and of small magnitude, witha great overlap in distributions observedbetween patients and controls. As well, fewbiological findings have been found to bespecific to a single disorder. Therefore, ourunderstanding of biological vulnerabilities
Biological Vulnerabilities to the Development of Psychopathology 95
04-Hankin.qxd 2/24/2005 11:20 AM Page 95

must be refined before we can use suchvulnerabilities as markers or diagnostic toolsfor psychiatry and psychopathology. However,current psychiatry and diagnostic systemsare the problem. The use of broad, heteroge-neous, behavioral definitions as classificationmeasures is undoubtedly the major contribu-tor to the observed lack of consistency. Theassumption that diagnostic categories aredistinct, when in fact psychiatric disordersshow very high comorbidity with each other,is simply spurious. It must be recognized thatpsychiatric nomenclatures represent the waysin which mental health researchers and pro-fessionals have carved out the edges of psy-chopathology in a way that is meaningful tothem and that supposedly allows communi-cation between them, but these are not thetrue frontiers of psychopathology. It may bethat many disorders that we think of as dis-tinct in fact share similar biological etiologi-cal mechanisms and that the same phenotypemay result from a number of etiologicalmechanisms, with multiple pathways.
In keeping with the need to consider themulticausal nature of psychopathology, bio-logical vulnerability must be considered inthe context of the environment in which thehuman organism evolves. The environmentprovides the context in which psychosocialrisk and protective factors interact with bio-logical vulnerability in determining whetherpsychopathology will emerge or not, as wellas the prognosis for each individual. Thescientific literature is replete with examplesof how biology interacts with events in theenvironment in creating or preventing psy-chopathology, as well as in determining itsoutcome. Research on antisocial behaviors,for example, has shown that adolescentboys whose fathers have been convicted ofvarious crimes, but who do not commitcrimes themselves, show higher sympatheticarousal, pointing perhaps to a protectiveeffect of biology (Raine, 2002). Furthermore,low sympathetic arousal is often more
characteristic of criminals from advantagedthan disadvantaged psychosocial back-grounds, suggesting that biological vulnera-bility may add little to the risk associatedwith an upbringing in low socioeconomiccircumstances. In contrast, minor physicalanomalies have been found to predict vio-lence only in individuals growing up inunstable and stressful family environments(Raine). Additionally, the provision of firmstructure, combined with warmth by parentsor teachers, has been found to be particu-larly important for behavioral managementof children with a diagnosis of ADHD(Denckla, 2003; Wicks-Nelson & Israel,2000). Not all individuals exposed to psy-chosocial stress or biological vulnerabilitywill go on to develop signs of psychopathol-ogy, and biological or environmental risksare often not sufficient in themselves to trig-ger onset of psychiatric disorders or todetermine their course. It is the interactionof both types of contributors that deter-mines psychopathology. Biological and psy-chosocial research must thus take intoaccount contributors from each field inorder to create a more complete etiologicalpicture of each psychopathology.
However, it may be in many ways impos-sible to attempt to distinguish the separatecontributions of biological and psychosocialfactors to psychopathology, because bothtypes of factors are in constant interaction,and the boundaries between them are at bestblurry. For example, smoking during preg-nancy could be considered as a stressor thatconfers risk, through biological processes, forthe development of later psychopathologythrough disruption of early brain develop-ment (potentially a biological vulnerability).However, women who smoke during preg-nancy may be likely to create a postnatalenvironment for their child that is very dif-ferent from that created by women whorefrain from smoking (i.e., a more stressfulenvironment). This postnatal environment
VULNERABILITIES96
04-Hankin.qxd 2/24/2005 11:20 AM Page 96

Biological Vulnerabilities to the Development of Psychopathology 97
may then contribute to the emergence ofdifferent psychopathological symptoms, such asdisruptive behaviors observed in children withADHD or CD. Parents with a history of antiso-cial behaviors are more likely to have childrenwho will grow up displaying some forms ofantisocial behaviors as well. The extent towhich this familial aggregation of socially inap-propriate behaviors is due to genetic or biolog-ical transmission, the creation of a disruptive
environment during upbringing, both, or someinteractive combination is, however, unclear.Divisions between different vulnerabilities,especially biological, are thus artificial ones,because all behaviors, thoughts, and emotionsultimately have biological substrates. A multi-causal approach to psychopathology, such asembodied in this volume, is the approach thatwill provide the most complete understandingof psychiatric disorders and their etiology.
REFERENCES
American Psychiatric Association. (2000). Diagnostic and statistical manual ofmental disorders (4th ed., text revision). Washington, DC: Author.
Anderson, C. M., Polcari, A., Lowen, S. B., Renshaw, P. F., & Teicher, M. H.(2002). Effects of methylphenidate on functional magnetic resonance relaxom-etry of the cerebellar vermis in boys with ADHD. American Journal ofPsychiatry, 159, 1322–1328.
Andreasen, N. C. (1999). A unitary model of schizophrenia. Archives of GeneralPsychiatry, 56, 781–787.
Baumeister, A. A., & Hawkins, M. F. (2001). Incoherence of neuroimaging studies ofattention deficit/hyperactivity disorder. Clinical Neuropharmacology, 24(1), 2–10.
Baving, L., Laucht, M., & Schmidt, M. H. (1999). Atypical frontal brain activationin ADHD: Preschool and elementary school boys and girls. Journal of theAmerican Academy of Child and Adolescent Psychiatry, 38, 1363–1371.
Berquin, P. C., Giedd, J. N., Jacobsen, L. K., Hamburger, S. D., Krain, A. L.,Rapoport, J. L., et al. (1998). Cerebellum in attention-deficit hyperactivitydisorder: A morphometric MRI study. Neurology, 50, 1087–1093.
Beyer, J. L., & Krishnan, K. R. R. (2002). Volumetric brain imaging findings inmood disorders. Bipolar Disorders, 4, 89–104.
Boileau, I., Assaad, J. M., Pihl, R. O., Benkelfat, C., Leyton, M., Diksic, M., et al.(2003). Alcohol promotes dopamine release in the human nucleus accumbens.Synapse, 49, 226–231.
Bremner, J., Randall, P., Scott, R., Bronen, A., Seibyl, J., Southwick, G., et al. (1995).MRI-based measurement of hippocampal volume in patients with combat-relatedposttraumatic stress disorder. American Journal of Psychiatry, 152, 973–981.
Brennan, P. A., Grekin, E. R., & Mednick, S. A. (1999). Maternal smoking duringpregnancy and adult male criminal outcome. Archives of General Psychiatry,56, 215–219.
Bruce, K., Shestowksy, J., Mayerovitch, J., & Pihl, R. (1999). Concomitant psy-chomotor stimulation and differentially-enhanced consolidation of emotionallycharged memory following alcohol consumption. Alcoholism: Clinical andExperimental Research, 23, 693–701.
Brunelle, C., Assaad, J. M., Pihl, R. O., Tremblay, R., & Vitaro, F. (2003).Exaggerated ethanol-induced cardiac reactivity as an indicator of increased riskfor gambling. Psychology of Addictive Behaviors, 17, 83–86.
04-Hankin.qxd 2/24/2005 11:20 AM Page 97

VULNERABILITIES98
Carlson, N. R. (2001). Physiology of behavior (7th ed.). Boston: Allyn & Bacon.Caspi, A., McClay, J., Moffitt, T. E., Mill, J., Martin, J., Craig, I. W., et al. (2002). Role
of genotype in the cycle of violence in maltreated children. Science, 297, 851–854.Castellanos, F. X., Giedd, J. N., Marsh, W. L., Hamburger, S. D., Vaituzis, A. C.,
Dickstein, D. P., et al. (1996). Quantitative brain magnetic resonance imagingin attention-deficit hyperactivity disorder. Archives of General Psychiatry, 53,607–616.
Castellanos, F. X., Lee, P. P., Sharp, W., Jeffries, N. O., Greenstein, D. K., Clasen,L. S., et al. (2002). Developmental trajectories of brain volume abnormalitiesin children and adolescents with attention-deficit/hyperactivity disorder.Journal of the American Medical Association, 288, 1740–1748.
Castellanos, F. X., & Tannock, R. (2002). Neuroscience of attention-deficit/hyperac-tivity disorder: The search for endophenotypes. Nature Reviews Neuroscience, 3,617–628.
Cherek, D. R., Lane, S. D., Pietras, C. J., & Steinberg, J. K. (2002). Effects of chronicparoxetine administration on measures of aggressive and impulsive responsesof adult males with a history of conduct disorder. Psychopharmacology, 159,266–274.
Combalbert, N., Bret-Dibat, J. L., & Favard, A. M. (2002). Biological approachfor the study of violent behaviours: Interest and limits. Annales MédicoPsychologiques, 160, 640–648.
Comings, D. E., Gade-Andavolu, R., Gonzalez, N., Wu, S., Muhleman, D., Blake, H.,et al. (2000). Comparison of the role of dopamine, serotonin, and nora-drenaline genes in ADHD, ODD and conduct disorder: Multivariate regressionanalysis of 20 genes. Clinical Genetics, 57, 178–196.
Conrod, P., Peterson, J., & Pihl, R. (2001). Reliability and validity of alcohol-induced heart rate increase as a measure of sensitivity to the stimulant proper-ties of alcohol. Psychopharmacology, 157, 20–30.
Damasio, A. R., Tranel, D., & Damasio, H. (1990). Individuals with sociopathicbehavior caused by frontal damage fail to respond autonomically to social stim-uli. Behavioural Brain Research, 41, 81–94.
Davidson, R. J., Pizzagalli, D., Nitschke, J. B., & Putnam, K. (2002). Depression:Perspectives from affective neuroscience. Annual Review of Psychology, 53,545–574.
Davidson, R. J., Putnam, K. M., & Larson, C. L. (2000). Dysfunction in the neuralcircuitry of emotion regulation: A possible prelude to violence. Science, 289,591–594.
Deminiere, J. M., Piazza, P. V., Le Moal, M., & Simon, H. (1989). Experimentalapproach to individual vulnerability to psychostimulant addiction. Neuroscienceand Biobehavioral Review, 13, 141–147.
Denckla, M. B. (2003). ADHD: Topic update. Brain and Development, 25, 383–389.Dougherty, D. D., Bonah, A. A., Spencer, T. J., Rauch, S. L., Madras, B. K., &
Fischman, A. J. (1999). Dopamine transporter density in patients with atten-tion deficit hyperactivity disorder. Lancet, 354, 2132–2133.
Drevets, W. C. (2000). Neuroimaging studies of mood disorders. BiologicalPsychiatry, 48, 813–829.
Drevets, W. C., Price, J. L., Simpson, J. R., Todd, R. D., Reich, T., Vannier, M.,et al. (1997). Subgenual prefrontal cortex abnormalities in mood disorders.Nature, 386, 824–827.
04-Hankin.qxd 2/24/2005 11:20 AM Page 98

Durston, S., Tottenham, N. T., Thomas, K. M., Davidson, M. C., Eigsti, I. M.,Yang, Y., et al. (2003). Differential patterns of striatal activation in youngchildren with and without ADHD. Biological Psychiatry, 53, 871–878.
Edenberg, H., Dick, D., Xuei, X., Tian, H., Almasy, L., Bauer, L., et al. (2004).Variations in GABRA2 encoding the alpha 2 sub unit of the GABAA receptor,are associated with alcohol dependence and with brain oscillations.Unpublished manuscript.
Ernst, M., Liebenauer, L. L., King, A. C., Fitzgerald, G. A., Cohen, R. M., &Zametkin, A. J. (1994). Reduced brain metabolism in hyperactive girls. Journalof the American Academy of Child and Adolescent Psychiatry, 33, 858–868.
Ernst, M., Zametkin, A. J., Matochik, J. A., Jons, P. H., & Cohen, R. M. (1998).DOPA decarboxylase activity in attention deficit hyperactivity disorder in adults:A FDOPA positron emission tomographic study. Journal of Neuroscience, 18,5901–5907.
Ernst, M., Zametkin, A. J., Matochik, J. A., Pascualvaca, D., Jons, P. H., & Cohen,R. M. (1999). High midbrain [18F]DOPA accumulation in children withADHD. American Journal of Psychiatry, 156, 1209–1215.
Fibiger, H. C., & Phillips, A. G. (1988). Mesocorticolimbic dopamine systems andreward. Annals of the New York Academy of Science, 537, 206–215.
Filipek, P. A., Semrud-Clikeman, M., Steingard, R. J., Renshaw, P. F., Kennedy,D. N., & Biederman, J. (1997). Volumetric MRI analysis comparing subjectshaving attention-deficit hyperactivity disorder with normal controls.Neurology, 48, 589–601.
George, T. P., & O’Malley, S. S. (2004). Current pharmacological treatments fornicotine dependence. Trends in Pharmacological Science, 25, 42–48.
Goodyer, I. M., Herbert, J., & Tamplin, A. (2003). Psychoendocrine antecedents ofpersistent first-episode major depression in adolescents: A community-basedlongitudinal enquiry. Psychological Medicine, 33, 601–610.
Guitart, X., Beitnes-Johnson, D., & Nestler, E. (1992). Fischer and Lewis rat strainsdiffer in basal levels of neurofilament proteins and in their regulation bychronic morphine. Synapse, 12, 242–243.
Hale, T. S., Hariri, A. R., & McCracken, J. T. (2000). Attention-deficit/hyperactiv-ity disorder: Perspectives from neuroimaging. Mental Retardation and Devel-opmental Disabilities Research Reviews, 6, 214–219.
Harden, P., & Pihl, R. (1995). Cognitive deficits and autonomic reactivity in boysat high risk for alcoholism. Journal of Abnormal Psychology, 104, 94–103.
Hill, D. E., Yeo, R. A., Campbell, R. A., Hart, B., Vigil, J., & Brooks, W. (2003).Magnetic resonance imaging correlates of attention-deficit/hyperactivity disor-der in children. Neuropsychology, 17, 496–506.
Hoptman, M. J. (2003). Neuroimaging studies of violence and antisocial behavior.Journal of Psychiatric Practice, 9, 265–278.
Hynd, G. W., Hern, K. L., Novey, E. S., Eliopulos, D., Marshall, R., Gonzalez, J. J.,et al. (1993). Attention deficit-hyperactivity disorder and asymmetry of thecaudate nucleus. Journal of Child Neurology, 8, 339–347.
Kendler, K., Prescott, C., Myers, J., & Neale, M. (2003). The structure of geneticand environmental risk factors for common psychiatric and substance usedisorders in men and women. Archives of General Psychiatry, 60, 929–937.
Kessler, R. C., McGonagle, K. A., Zhao, S., Nelson, C. B., Hughes, M., Eshleman, S.,et al. (1994). Lifetime and 12-month prevalence of DSM-III-R psychiatric
Biological Vulnerabilities to the Development of Psychopathology 99
04-Hankin.qxd 2/24/2005 11:20 AM Page 99

disorders in the United States: Results from the National Comorbidity Survey.Archives of General Psychiatry, 51, 8–19.
Kirley, A., Hawi, Z., Daly, G., McCarron, M., Mullins, C., Millar, N., et al. (2002).Dopaminergic system genes in ADHD: Toward a biological hypothesis. Neu-ropsychopharmacology, 27, 607–619.
Koob, G. F., & Bloom, F. E. (1988). Cellular and molecular mechanisms of drugdependence. Science, 242, 715–723.
Krakowski, M. (2003). Violence and serotonin: Influence of impulse control, affectregulation, and social functioning. Journal of Neuropsychiatry and ClinicalNeurosciences, 15, 294–305.
Krause, K. H., Dressel, S. H., Krause, J., Lung, H. F., & Tatsch, K. (2000). Increasedstriatal dopamine transporter in adult patients with attention deficit hyperactivitydisorder: Effects of methylphenidate as measured by single photon emission com-puted tomography. Neuroscience Letter, 285, 107–110.
Krueger, R. F. (1999). The structure of common mental disorders. Archives ofGeneral Psychiatry, 56, 921–926.
Laakso, M. P., Vaurio, O., Koivisto, E., Savolainen, L., Eronen, M., Aronen, H. J.,et al. (2001). Psychopathy and the posterior hippocampus. Behavioural BrainResearch, 118, 187–193.
Lahey, B. B., Applegate, B., Waldman, I., Hankin, B. L., & Rick, J. (2004). Thestructure of child and adolescent psychopathology: Generating new hypotheses.Journal of Abnormal Psychology, 113, 358–385.
Lai, T.-J., Payne, M. E., Byrum, C. E., Steffens, D. C., & Krishnan, K. R. (2000).Reduction of orbital frontal cortex volume in geriatric depression. BiologicalPsychiatry, 48, 971–975.
Liotti, M., & Mayberg, H. S. (2001). The role of functional neuroimaging in theneuropsychology of depression. Journal of Clinical and Experimental Neuro-psychology, 23, 121–136.
Lou, H. C., Henriksen, L., & Bruhn, P. (1984). Focal cerebral hypoperfusionin children with dysphasia and/or attention deficit disorder. Archives ofNeurology, 41, 825–829.
Lou, H. C., Henriksen, L., & Bruhn, P. (1990). Focal cerebral dysfunction in devel-opmental learning disabilities. Lancet, 335, 8–11.
Lou, H. C., Henriksen, L., Bruhn, P., Borner, H., & Nielsen, J. B. (1989). Striatal dys-function in attention deficit and hyperkinetic disorder. Archives of Neurology,46, 48–52.
Luby, J. L., Heffelfinger, A., Mrakotsky, C., Brown, K., Hessler, M., & Spitznagel,E. (2003). Alterations in stress cortisol reactivity in depressed preschoolersrelative to psychiatric and no-disorder comparison groups. Archives of GeneralPsychiatry, 60, 1248–1255.
Luria, A. R. (1980). Higher cortical functions in man. (B. Haigh, Trans.) (2nd ed.).New York: Basic Books.
Lyoo, I. K., Noam, G. G., Lee, C. K., Lee, H. K., Kennedy, B. P., & Renshaw, P. F.(1996). The corpus callosum and lateral ventricles in children with attention-deficit hyperactivity disorder: A brain magnetic resonance imaging study.Biological Psychiatry, 40, 1060–1063.
Mataro, M., Garcia-Sanchez, C., Junque, C., Estevez-Gonzalez, A., & Pujol, J. (1997).Magnetic resonance imaging measurement of the caudate nucleus in adolescentswith attention deficit hyperactivity disorder and its relationship with neuropsy-chological and behavioral measures. Archives of Neurology, 54, 963–968.
VULNERABILITIES100
04-Hankin.qxd 2/24/2005 11:20 AM Page 100

Mathalon, D., Rapoport, J., Davis, K., & Krystal, J. (2003). Neurotoxicity, neuro-plasticity, and magnetic resonance imaging morphometry. Archives of GeneralPsychiatry, 60, 846–848.
McBride, W. J., Murphy, J. M., Lumeng, L., & Li, T. K. (1990). Serotonin,dopamine and GABA involvement in alcohol drinking of selectively bred rats.Alcohol, 7, 199–205.
Miller, E. K., & Cohen, J. D. (2001). An integrative theory of prefrontal cortexfunction. Annual Review of Neuroscience, 24, 167–202.
Moore, T. M., Scarpa, A., & Raine, A. (2002). A meta-analysis of serotoninmetabolite 5-HIAA and antisocial behavior. Aggressive Behavior, 28, 299–316.
Mostofsky, S. H., Cooper, K. L., Kates, W. R., Denckla, M. B., & Kaufmann,W. E. (2002). Smaller prefrontal and premotor volumes in boys with attention-deficit/hyperactivity disorder. Biological Psychiatry, 52, 785–794.
Nestler, E. J., Barrot, M., Dileone, R. J., Eisch, A. J., Gold, S. J., Monteggia, L. M.(2002). Neurobiology of depression. Neuron, 34, 13–25.
Nolan, C. L., Moore, G. J., Madden, R., Farchione, T., Bartoi, M., Lorch, E., et al.(2002). Prefrontal cortical volume in childhood-onset major depression: Pre-liminary findings. Archives of General Psychiatry, 59, 173–179.
Oades, R. D. (2002). Dopamine may be “hyper” with respect to noradrenalinemetabolism, but “hypo” with respect to serotonin metabolism in children withattention-deficit hyperactivity disorder. Behavioural Brain Research, 130,97–102.
Pihl, R. O. (1995). Violent behavior. Journal of Psychiatry & Neuroscience, 20,101–103.
Pihl, R. O. (1999). Substance abuse. In T. Miller, P. Blaney, & R. D. Davis (Eds.),Oxford textbook of psychopathology (pp. 249–276). New York: OxfordUniversity Press.
Pihl, R. O., & Peterson, J. B. (1995). Alcoholism: The role of different motivationalsystems. Journal of Psychiatry and Neuroscience, 20, 372–396.
Pihl, R. O., Peterson, J. B., & Finn, P. R. (1990). The inherited predisposition toalcoholism: Characteristics of sons of male alcoholics. Journal of AbnormalPsychology, 99, 291–301.
Pihl, R. O., Vant, J., & Assaad, J. M. (2003). Neuropsychological and neuroen-docrine factors. In C. A. Essau (Ed.), Conduct and oppositional defiant disor-ders: Epidemiology, risk factors, and treatment (pp. 163–189). Mahwah, NJ:Lawrence Erlbaum.
Porjesz, B., Almasy, L., Edenberg, H., Wang, K., Charlian, D., Forud, T., et al.(2002). Linkage disequilibrium between the beta frequency of the human EEGand a GABAA receptor gene locus. Proceedings of the National Academy ofScience, 99, 3729–3733.
Raine, A. (2002). Biosocial studies of antisocial and violent behaviour in childrenand adults: A review. Journal of Abnormal Child Psychology, 30, 211–236.
Raine, A., Buchsbaum, M. S., & LaCasse, L. (1997). Brain abnormalities in murderersindicated by positron emission tomography. Biological Psychiatry, 42, 495–508.
Raine, A., Buchsbaum, M. S., Stanley, J., Lottenberg, S., Abel, L., & Stoddard, J.(1994). Selective reductions in prefrontal glucose metabolism in murderers.Biological Psychiatry, 36, 365–373.
Raine, A., Ishikawa, S. S., Arce, E., Lencz, T., Knuth, K. H., Bihrle, S., et al. (2004).Hippocampal structural asymmetry in unsuccessful psychopaths. BiologicalPsychiatry, 55, 185–191.
Biological Vulnerabilities to the Development of Psychopathology 101
04-Hankin.qxd 2/24/2005 11:20 AM Page 101

Raine, A., Lencz, T., Bihrle, S., LaCasse, L., & Colletti, P. (2000). Reducedprefrontal gray matter volume and reduced autonomic activity in antisocialpersonality disorder. Archives of General Psychiatry, 57, 119–127.
Raine, A., Lencz, T., Taylor, K., Hellige, J. B., Bihrle, S., LaCasse, L., et al. (2003).Corpus callosum abnormalities in psychopathic antisocial individuals. Archivesof General Psychiatry, 60, 1134–1142.
Raine, A., Meloy, J. R., Bihrle, S., Stoddard, J., LaCasse, L., & Buchsbaum, M. S.(1998). Reduced prefrontal and increased subcortical brain functioning assessedusing positron emission tomography in predatory and affective murderers.Behavioral Sciences and the Law, 16, 319–332.
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer,H. Y., et al. (1999). Morphometric evidence for neuronal and glial prefrontalcell pathology in major depression. Biological Psychiatry, 45, 1085–1098.
Robins, L. N., & Regier, D. A. (1991). Psychiatric disorders in America: TheEpidemiologic Catchment Area Study. New York: Free Press.
Saal, D., Dong, Y., Bonci, A., & Malenka, R. (2003). Drugs of abuse and stress trig-ger a common synaptic adaptation in dopamine neurons. Neuron, 37, 577–582.
Schall, J. D. (2004). On building a bridge between brain and behavior. AnnualReview of Psychology, 55, 23–50.
Sheline, Y., Wang, P., Gado, M., Csernansky, J., & Vannier, M. (1996). Hippocampalatrophy in recurrent major depression. Proceedings of the National Academy ofScience, 93, 3908–3913.
Sieg, K. G., Gaffney, G. R., Preston, D. F., & Hellings, J. A. (1995). SPECT brainimaging abnormalities in attention deficit hyperactivity disorder. ClinicalNuclear Medicine, 20, 55–59.
Silberstein, R. B., Farrow, M., Levy, F., Pipingas, A., Hay, D. A., & Jarman, F. C.(1998). Functional brain electrical mapping in boys with attention-deficit/hyperactivity disorder. Archives of General Psychiatry, 55, 1105–1112.
Soderstrom, H., Hultin, L., Tullberg, M., Wikkelso, C., Ekholm, S., & Forsman, A.(2002). Reduced frontotemporal perfusion in psychopathic personality.Psychiatry Research Neuroimaging, 114, 81–94.
Song, J., Koller, D. L., Foroud, T., Carr, K., Zhao, J., Rice, J., et al. (2003).Association of GABA(A) receptors and alcohol dependence and the effects ofgenetic imprinting. American Journal of Medical Genetics, 117B(1), 39–45.
Sowell, E. R., Thompson, P. M., Welcome, S. E., Henkenius, A. L., Toga, A. W., &Peterson, B. S. (2003). Cortical abnormalities in children and adolescents withattention-deficit hyperactivity disorder. Lancet, 362, 1699–1707.
Steele, C. M., & Josephs, R. A. (1990). Alcohol myopia: Its prized and dangerouseffects. American Psychologist, 45, 921–933.
Steingard, R. J., Renshaw, P. F., Hennen, J., Lenox, M., Cintron, C. B., Young,A. D., et al. (2002). Smaller frontal lobe white matter volumes in depressedadolescents. Biological Psychiatry, 52, 413–417.
Stewart, S., Finn, P., & Pihl, R. (1995). A dose response study of the effects ofalcohol on the perception of pain and discomfort due to electric shock in menat high risk for alcoholism. Psychopharmacology, 119, 261–267.
Stewart, S., & Pihl, R. (1994). Effects of alcohol administration on psychophysio-logical and subjective-emotional responses to aversive stimulation in anxietysensitive women. Psychology of Addictive Behaviors, 8, 29–42.
VULNERABILITIES102
04-Hankin.qxd 2/24/2005 11:20 AM Page 102

Still, G. F. (1902). Some abnormal conditions in children: Lectures I, II, and III.Lancet, 1, 1008–1012, 1077–1082, 1163–1168.
Stockmeier, C. A. (2003). Involvement of serotonin in depression: Evidence frompostmortem and imaging studies of serotonin receptors and the serotonin trans-porter. Journal of Psychiatric Research, 37, 357–373.
Strakowski, S. M., Adler, C. M., & DelBello, M. P. (2002). Volumetric MRI studiesof mood disorders: Do they distinguish unipolar and bipolar disorder. BipolarDisorders, 4, 80–88.
Stuss, D. T., Gow, C. A., & Hetherington, C. R. (1992). “No longer Gage”: Frontallobe dysfunction and emotional changes. Journal of Consulting and ClinicalPsychology, 60, 349–359.
Suzdak, P., Glewa, J., Crawley, J., Schwartz, R., Skolnick, P., & Paul, S. (1986).A selective imidazobenzodiazepine antagonist of ethanol in the rat. Science,234, 1243–1247.
Vaidya, C. J., Austin, G., Kirkirian, G., Ridlehuber, H. W., Desmond, J. E., Glover,G. H., et al. (1998). Selective effects of methylphenidate in attention deficithyperactivity disorder: A functional magnetic resonance study. Proceedings ofthe National Academy of Science USA, 95, 14494–14499.
van Dyck, C. H., Quinlan, D. M., Cretella, L. M., Staley, J. K., Malison, R. T.,Baldwin, R. M., et al. (2002). Unaltered dopamine transporter availability inadult attention deficit hyperactivity disorder. American Journal of Psychiatry,159, 309–312.
Weissman, M. (1988). Anxiety of alcoholism. Journal of Clinical Psychiatry, 49,17–19.
Wicks-Nelson, R., & Israel, A. C. (2000). Behavior disorders of childhood.Toronto, Ontario, Canada: Prentice Hall Canada.
Wilens, T. E., Biederman, J., & Spencer, T. J. (2002). Attention deficit/hyperactiv-ity disorder across the lifespan. Annual Review of Medicine, 53, 112–131.
Young, S. N., & Leyton, M. (2002). The role of serotonin in human mood andsocial interaction: Insight from altered tryptophan levels. Pharmacology,Biochemistry & Behavior, 71, 857–865.
Young, S., Smith, S., Pihl, R., & Ervin, F. (1985). Tryptophan depletion lowersmood in normal males. Psychopharmacology, 87, 173–177.
Zametkin, A. J., Nordahl, T. E., Gross, M., King, A. C., Semple, W. E., Rumsey, J.,et al. (1990). Cerebral glucose metabolism in adults with hyperactivity of child-hood onset. New England Journal of Medicine, 323, 1361–1366.
Biological Vulnerabilities to the Development of Psychopathology 103
04-Hankin.qxd 2/24/2005 11:20 AM Page 103





























