Embed Size (px)
Citation preview

A
IWa
b
a
ARR2AA
KBNT
1
ttmcmot
apcditohtl
(
0h
Journal of Chromatography A, 1324 (2014) 181– 189
Contents lists available at ScienceDirect
Journal of Chromatography A
jou rn al hom epage: www.elsev ier .com/ locate /chroma
dsorption behavior of proteins on temperature-responsive resins
zabela Poplewskaa, Renata Mucaa, Adam Strachotab,ojciech Piatkowskia, Dorota Antosa,∗
Department of Chemical and Process Engineering, Rzeszów University of Technology, Powstanców Warszawy Ave. 6, 35-959 Rzeszów, PolandInstitute of Macromolecular Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Heyrovskeho nam. 2, CZ-162 00 Praha, Czech Republic
r t i c l e i n f o
rticle history:eceived 11 July 2013eceived in revised form9 September 2013ccepted 20 November 2013vailable online 25 November 2013
a b s t r a c t
The adsorption behavior of proteins on thermo-responsible resins based on poly(N-isopropylacrylamide)and its copolymer containing an anionic co-monomer has been investigated. The influence of the poly-mer composition, i.e., the content of the co-monomer and crosslinker on the thermo-sensitivity of theprotein adsorption has been quantified. The properties of ungrafted polymer as well grafted onto theagarose matrix have been analyzed and compared. Batch and dynamic (column) experiments have beenperformed to measure the adsorption equilibrium of proteins and to quantify the phase transition pro-
eywords:ioseparations-Isopropylacrylamidehermo-responsible resins
cess. As model proteins lysozyme, lactoferrin, �-chymotrypsinogen A and ovalbumin have been used.The adsorption process was found to be governed by ionic interactions between the negatively chargedsurface of resin and the protein, which enabled separation of proteins differing in electrostatic charge. Theinteractions enhanced with increase of temperature. Decrease of temperature facilitated desorption ofproteins and reduced the salt usage in the desorption buffer. Grafted polymers exhibited markedly highermechanical stability and, however, weaker temperature response compared to the ungrafted ones.
. Introduction
Various chromatographic techniques can be used for separa-ion and purification of proteins, which are based on differentypes of interactions between the functional groups of the macro-
olecule and the adsorbent surface. Often, a combination of fewhromatographic stages is integrated; such as ion exchange chro-atography (IEC), hydrophobic interaction chromatography (HIC)
r mixed mode chromatography exploiting both types of interac-ions [1–6].
In both processes the adsorption properties of proteins areltered by the composition of the mobile phase. In IEC the sam-le with the protein mixture is loaded at low concentration of saltontaining counter-ions with respect to the column matrix at pHiffering from the isoelectric point, while elution is imposed by
ncrease of the salt concentration or proper pH change in the direc-ion of pI. In HIC the sample loading is realized at high concentrationf a cosmotropic salt (e.g., ammonium sulfate), which enhancesydrophobic interactions between the adsorbent surface and pro-
eins and, thus, the adsorption strength. The elution is executed byowering the salt concentration.∗ Corresponding author. Tel.: +48 178651853; fax: +48 178543655.E-mail addresses: [email protected], [email protected]
D. Antos).
021-9673/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.chroma.2013.11.040
© 2013 Elsevier B.V. All rights reserved.
A common drawback of both IEC and HIC processes stems fromthe necessity of using highly concentrated salt solutions to pro-mote adsorption or elution of proteins. High viscosity of the mobilephases containing salts results in increase in the pressure drop onchromatographic columns. This reduces flowrates and, in conse-quence, productivity of the process. Moreover, in the final stage ofthe purification process the target protein has to be isolated outof the salt solution, e.g., by use of a membrane process. Becausethe membrane permeability reduces significantly in the presenceof highly concentrated salt solutions the performance of the wholeseparation process is strongly affected by the salt concentration [7].
In case of HIC processes, to overcome this problem the manipu-lation of the salt concentration can be combined with temperatureto alter the adsorption properties of proteins. Typically, increase oftemperature enhances adsorption of proteins, whereas its reduc-tion promotes their elution. Therefore, the effect of temperaturecan partly offset that of the salt concentration [8–10]. However,stability of typical HIC media drops rapidly with increase of tem-perature already above 30 ◦C, at which most of proteins stillpreserve their native properties. Moreover, a number mesophilicand thermophilic proteins exhibit high thermostability and can beseparated at elevated temperatures without denaturation.
Therefore, as an alternative to traditional resins stimuli-
sensitive hydrogels, termed also as smart polymers, have beendeveloped. These polymers exhibit changes in the physico-chemical properties according to external stimuli such as pH, ionicstrength, interaction with the molecules of chemical compounds as
1 matog
wtgrbwtr
it
d(as
u(outwhpNca
mcteramnsap
ecsmaat
be
ovtsotea
apo
a
82 I. Poplewska et al. / J. Chro
ell as temperature [8,11,12]. Among stimuli-sensitive hydrogelshe thermo-responsive polymers gained interest as chromato-raphic media for purification of proteins under mild and envi-onmental friendly conditions. The mechanism of separations isased on temperature-mediated cycles of adsorption–desorption,here a change in temperature is used to manipulate the adsorp-
ion behavior of proteins indirectly, i.e., by altering properties of theesin [13,14].
The most studied temperature-responsive polymer is poly(N-sopropylacrylamide) (PNIPA) which has a lower critical solutionemperature (LCST) of approximately 32 ◦C [15].
At temperatures below the LCST the PNIPA is hydrophilicue to hydrogen bonding with water. At a temperature aboveLCST) water molecules dissociate from polymer leading toggregation of the PNIPA chains, which form a hydrophobicurface.
The LCST of temperature-responsive polymers can be manip-lated by copolymerization of N-isopropylacrylamide monomerNIPAm) with hydrophilic or hydrophobic monomers. Integrationf hydrophilic monomers such as acrylamide into the molec-lar structure of polymer results in an increase in LCST andhe polymer hydrophilicity, whereas copolymerization of NIPAmith hydrophobic monomers, such as butyl acrylamide, impartsydrophobicity and decreases the LCST of the copolymer [12]. Co-olymerization of NIPAm with anionic acrylic acid [16] or cationic,N′-dimethylaminopropylacrylamide [17] produces co-polymersapable of hydrophobic and ionic interactions, the same which arective in HIC, IEC or mixed mode chromatography.
However, despite promising adsorption properties of this poly-er poor mechanical stability often restricts its use in column
hromatography. To tackle this problem different methods ofhe polymer grafting to a rigid matrix have been proposed,.g., in several works [18–27] PNIPA and related temperature-esponsive polymers were grafted onto a silica matrix and useds a stationary phase for HPLC analysis of micro as well asacro-molecules. For the conduction of this analytical tech-
ique salt free mobile phases were used, while the separationelectivity was altered by temperature changes. However, thepplicability of those resins was mostly limited to analyticalrocedures.
Maharjan et al. [28] developed temperature-responsive ion-xchange resins (for IEC) by grafting a PNIPA-based polymer ontoross-linked agarose. The agarose matrix is characterized by largepecific surface area and pore size and it is very often used inanufacturing chromatographic media for protein separations in
preparative and industrial scale. The authors suggested a cyclicdsorption–desorption process for the fractionation of whey pro-eins.
Data presented in the literature listed above have mainly beenased on qualitative description of adsorption properties of differ-nt compounds on the thermo-responsive resins.
The goal of this study was detailed analysis of the patternf adsorption and desorption of proteins on PNIPA-based resinsersus temperature and the polymer composition. Few model pro-eins differing in hydrophobicity and the ionic charge have beentudied such as lysozyme, lactoferrin, �-chymotrypsinogen A andvalbumin. Static and dynamic experiments have been performedo evaluate the adsorption behavior. Moreover, the swelling prop-rties and rate of the phase transition have been determined,nalyzed and compared.
PNIPA-based beads were prepared by polymerization with cross-linking agent. Copolymers were obtained by co-
olymerization of NIPAm, acrylic acid or its sodium salt and,ptionally, with tert-butylacrylamide.The properties of both ungrafted polymer and grafted onto thegarose matrix have been analyzed and compared.
r. A 1324 (2014) 181– 189
2. Materials, instrumentation and methods
2.1. Materials
2.1.1. ProteinsLysozyme from chicken egg white (LYZ) (pI = 11.4, molecular
mass 14.4 kDa), lactoferrin from bovine milk (LTF) (pI = ca. 8.7,molecular mass 87 kDa), �-chymotrypsinogen A from bovine pan-creas (CHTG A) (pI = 9.1, molecular mass 25.7 kDa) and ovalbumin(OVA) (albumin from chicken egg white) (pI = 4.6, molecular mass44.3 kDa) were obtained from Sigma–Aldrich (Poland).
2.1.2. ChemicalsN-Isopropylacrylamide (NIPAm), acrylic acid (AAc),
sodium acrylate (SA), chlorotrimethylsilane (TMS), N,N,N′,N′-tetramethylethylenediamine (TEMED), sodium stearate (SS),1-(ethoxycarbonyl)-2-ethoxy-1,2-dihydro-quinoline (EEDQ), tert-butyl-acrylamide (tBAm), N,N-dimethylformamide (DMF),4,4′-azobis(4-cyanovaleric acid) (ACV) were obtained fromSigma–Aldrich (Poland). Sodium borohydride, N,N′-methy-lenebisacrylamide (BIS), toluene, sodium hydroxide, pyridinewere obtained from Fluka (Poland). Sodium chloride, chloro-form, sodium phosphate dibasic, methanol, ammonia solution,epichlorohydrin were obtained from POCH (Poland). Ethanol,phosphoric acid, ammonium persulfate (APS) were obtained fromChempur (Poland). Sepharose 6Fast Flow (highly cross-linkedagarose 6%), mean particle size 90 �m, was obtained from GEHealthcare (Sweden). Silica gel (Si), particle size 40–63 �m,pore size 60 A, surface area 530 m2/g, was obtained from Merck(Germany).
2.2. Instrumentation
The HPLC instrument Äkta purifier with UV, and conductometricdetectors and a data station (GE Healthcare Life Sciences, Upp-sala, Sweden) were used. The injector was a Rheodyne samplingvalve with 100 �L sample loop. The system was equipped withglass columns (40 mm × 16 mm I.D.). Low temperature thermostatLauda Re110 (Lauda, Lauda-Königshofen, Germany) was used forthe column thermostatting.
2.3. Methods
2.3.1. Preparation of ungrafted polymer PNIPAThe synthesis was performed according to the procedure
described in [29].The following reagents have been used: NIPAm, BIS – cross-
linking agent, SA – ionizing agent, APS – initiator, TEMED – catalyst.Polymers with different chemical composition were synthe-
sized. The quantity of SA was changed within the range 2–10% [g/g](gram SA per gram of NIPAm) and BIS 2–30% [g/g].
1 g of NIPAm, BIS, SA with adequate proportions and water(8 mL) were mixed at ambient temperature under a nitrogen atmo-sphere to dissolve. In the next step, temperature was lowered to5 ◦C. TEMED (0.8 mL) and APS solutions (1.26 g APS in 1.6 mL water)were added to the gel slurry. The gelation process lasted about2 min. The gel was then stored in water at ambient temperature for24 h. PNIPA was chopped into small pieces and rinsed with waterover the next 48 h four times.
The same procedure as described above was performed in asuspension of silica gel to produce the resins meant to be used in
dynamic experiments, i.e., to be packed into the column. Surfactant,sodium stearate (SS), was added to the suspension to reduce thesurface tension between phases. The hydrophobic phase of silica gelwas mixed with a hydrophilic environment of the reaction. NIPAm,
matog
Buwg(ata
2(
bt
i
fti
2(wsltwb
2n1(aaawe
2b(wem3w1t
2
IsdNu
2
5wcbt
I. Poplewska et al. / J. Chro
IS, SA, Si, SS and water (8 mL) were mixed at ambient temperaturender a nitrogen atmosphere to dissolve. Silica gel before the useas endcapped to inactivate adsorption sites. For this purpose silica
el was dried for 2 h in a vacuum oven. Dried silica gel (1 g), TMS0.49 mL), pyridine (0.014 mL) and toluene (12.5 mL) were mixedt boiling temperature for 6 h. The gel was collected by vacuum fil-ration and washed subsequently with chloroform, 100% methanolnd 70% aqueous methanol. The step was repeated twice.
.3.2. Preparation of polymer grafted onto cross-linked agaroseItBA)
Similar procedure of polymerization was used to that describedy Maharjan et al. [28], which was based on polymerization withhe cross-linking agent.
The following reagents were used: NIPAm, BIS, AAc or SA – ion-zing agent, ACV – initiator, EEDQ – catalyst.
ItBA was synthesized using three subsequent procedures: aminounctionalization of agarose bed, immobilization of the polymeriza-ion initiator, development of the polymer matrices with the ACVmmobilized gel.
.3.2.1. Amino functionalization of agarose beads. Sepharose 6FF100 g) and 2 M NaOH solution (100 mL) containing NaBH4 (0.187 g)ere mixed at 28 ◦C for 2 h. Epichlorohydrin (60 mL) and above gel
lurry were mixed at 28 ◦C for 21 h. The epoxy-activated gel was col-ected by vacuum filtration and washed with distilled water. Next,he activated gel (95 g) and 2 M aqueous ammonia solution (95 mL)ere mixed at 28 ◦C for 21 h. The aminated agarose bed was filtrated
y vacuum filtration and washed with distilled water.
.3.2.2. Immobilization of the polymerization initiator. The ami-ated agarose bed was subsequently washed with 50%, 75% and00% aqueous ethanol and filtered by vacuum filtration. ACV5.68 g) and EEDQ (10.02 g) were dissolved in DMF (270 mL). Themino-functionalised gel (90 g) and the DMF solution were mixedt ambient temperature under a nitrogen atmosphere for 45 minnd under an argon atmosphere for 6 h. The ACV immobilized gelas collected by vacuum filtration and washed with DMF and
thanol.
.3.2.3. Development of the polymer matrices with the ACV immo-ilized gel. NIPAm (12.73 g), tBAm (0.795 g), AAc (0.450 g) and BIS0.193 g) were dissolved in ethanol (125 mL). ACV immobilized gelas washed subsequently with 20%, 50%, 75% and 100% aqueous
thanol. Gel (25 g) was added to the solution mixture and wasixed at ambient temperature under a nitrogen atmosphere for
0 min and at 80 ◦C under an argon atmosphere for 16 h. The gelas collected by vacuum filtration and washed subsequently with
00%, 75% 50%, 20% aqueous ethanol and cold water. The gel washen stored in 20% (v/v) aqueous ethanol at 4 ◦C.
.3.3. Chemical characterization of the resinsFor chemical characterization of the synthesized resins the FT-
R spectra were measured using a Thermo Scientific Nicolet 8700pectrometer and KBr pellets. Moreover, the charge density wasetermined by potentiometric titrating the resins with a 0.02 MaOH solution (resins in H+ form) or 0.01 M HCl (resins in Na+ form)sing pH-conductivity meter CPC-501.
.3.4. Adsorption of proteins in batch experimentsSamples of swollen PNIPA and ItBA were conditioned with a
0 mM phosphate buffer (pH 7.0) for 24 h. Then, the swollen gel
as filtered. Solutions of the proteins (lysozyme, lactoferrin, �-hymotrypsinogen A and ovalbumin) were prepared in phosphateuffer (pH 7.0). 2 mL of the protein solution with concentra-ion varying within the range 0.5–8 mg/mL was mixed with 0.2 g
r. A 1324 (2014) 181– 189 183
swollen gel and agitated for 2 h at 5, 25 and 37 ◦C. After 2 h a sam-ple of the solution was analyzed using a UV detector at wavelengthof 280 nm. The equilibrium concentration of proteins was calcu-lated using the detector calibration curves determined for eachprotein. After accomplishing the adsorption measurements the gelwas dried to constant weight. The amount of proteins adsorbed onthe gel, q*, was calculated as follows:
q∗ = V0(cF − c)ma
where ma – mass of dry adsorbent, V0 – volume of the proteinsolution, cF – initial concentration of the sample, c – equilibriumconcentration.
2.3.5. Dynamic experiments2.3.5.1. Chromatographic elution. Ungrafted PNIPA polymerized inthe suspension of silica gel was used for the column experiments.To prevent compression of the gel during dynamic experimentsthe resin was additionally mixed with endcapped silica gel whenpacked into the column. The swollen ItBA was directly packed intothe column as received.
Chromatographic elution of the proteins was studied at 5, 25and 37 ◦C. The concentration profiles were detected at wavelengthof 280 nm. The loading buffer was 50 mM phosphate buffer (pH 7.0).The desorption buffer was 0.1 M or 1 M NaCl solution in phosphatebuffer. The mobile phase flowrate was: 0.5 or 1.0 mL/min. The inletprotein concentration was 1 or 3 g/L in phosphate buffer (pH 7.0).
2.3.5.2. Phase transformation of PNIPA and ItBA. The phase transi-tion of PNIPA and ItBA was studied as a function of the polymercomposition, temperature and time. In the first case polymers con-taining SA and BIS with different content were conditioned withphosphate buffer. The swollen samples of polymers were filtratedusing vacuum filtration, weighed and dried to constant weight. Theswelling degree was determined as a mass ratio of swollen and drypolymer.
To measure the rate of the phase transition the column wasequilibrated in water bath at 25 ◦C. In the next step, the columnwas immersed in another water bath thermostatted at 5 ◦C. Imme-diately after exchange of the water baths several pulses of a tracerwere subsequently injected and their retention time was recordeduntil the column reached the thermal equilibrium, i.e., no furtherchange in the pulse retention (i.e., in the column porosity) wasrecorded. As a tracer 0.1 M NaCl or glucose were used; the resultsin both cases were very similar.
To determine LCST the column was packed with the resin andconditioned in the water bath at different temperature until thethermal equilibrium was achieved. The column porosity was mea-sured at each temperature by recording the retention time of thetracer pulse.
3. Results and discussion
3.1. Chemical characterization of the resins
To analyze the chemical structure of the synthesized PNIPA andItBA resins the FT-IR spectra were measured. The results of mea-surements are depicted in Fig. 1a and b. The repeat units for thechemical structure of polymers are overlaid in the same figures.
The presented results of measurements confirm the expectedstructure of both polymers.
The charge density of ItBA was 158 �mol carboxylic acidgroups/g of dried gel, which was very similar to that measured byMaharjan et al. [28]. For PNIPA the charge density was also similar,i.e., 177 �mol carboxylic acid groups/g of dried gel.

184 I. Poplewska et al. / J. Chromatogr. A 1324 (2014) 181– 189
F hemis 1680–b .
3o
fasoa
pawwbo
ig. 1. FT-IR absorbance spectra of (a) PNIPA and (b) ItBA. The repeat unit for the ctretching bands (3500–3300 cm−1) of secondary amine, amide bands – I band �C O (ands of C(CH3)2, CH2 , C(CH3)3 and C H bending bands (1470–1450 cm−1)
.2. Effect of the polymer composition on the adsorption behaviorf proteins
Adsorption of the proteins was measured on PNIPA resins dif-ering in the polymer composition, i.e., the content of crosslinkernd ionizing agent. The window for the temperature changes waselected within a relatively low temperature range, i.e., 5–37 ◦C,ften preferred in processing of proteins to preserve their biologicalctivity.
The results of the measurements performed for LYZ are com-ared in Fig. 2a and b. As expected, an addition of the ionizinggent enhanced adsorption (see Fig. 2a), however, at simultaneous
idening the adsorption window toward lower temperature,hich indicated possible difficulty in the protein desorptiony temperature reduction. Moreover, increase of the contentf hydrophilic ionizing agent reduced the thermo-sensitivity of
cal structure of polymers is overlaid on the spectra. Prominent IR bands are: N H1630 cm−1) and II band ıN H (1550–1510 cm−1), C H stretching (2900–3000 cm−1)
adsorption within the temperature window investigated, e.g., bydecreasing temperature from 37 to 5 ◦C adsorption of LYZ on PNIPAwith 2% SA [g/g] decreased ca. tenfold, with 4% SA – forth fold,whereas on PNIPA, 10% SA only twice.
It can be observed that the adsorption strength of the proteindeclines with increase of the crosslinker content, BIS (Fig. 2b).Increase of the crosslinking density causes decrease of the gelhydrophilicity and reduces the protein affinity to hydrophilic ioniccenters.
However, the changes of adsorption strength caused by differ-ences in the crosslinking density were much weaker compared tothose involved by the presence of SA. An obvious advantage of
the increase of the crosslinker content was improvement in themechanical stability of the polymer.The results presented above indicate that the adsorption behav-ior of protein can be controlled by tuning the content of ionizing
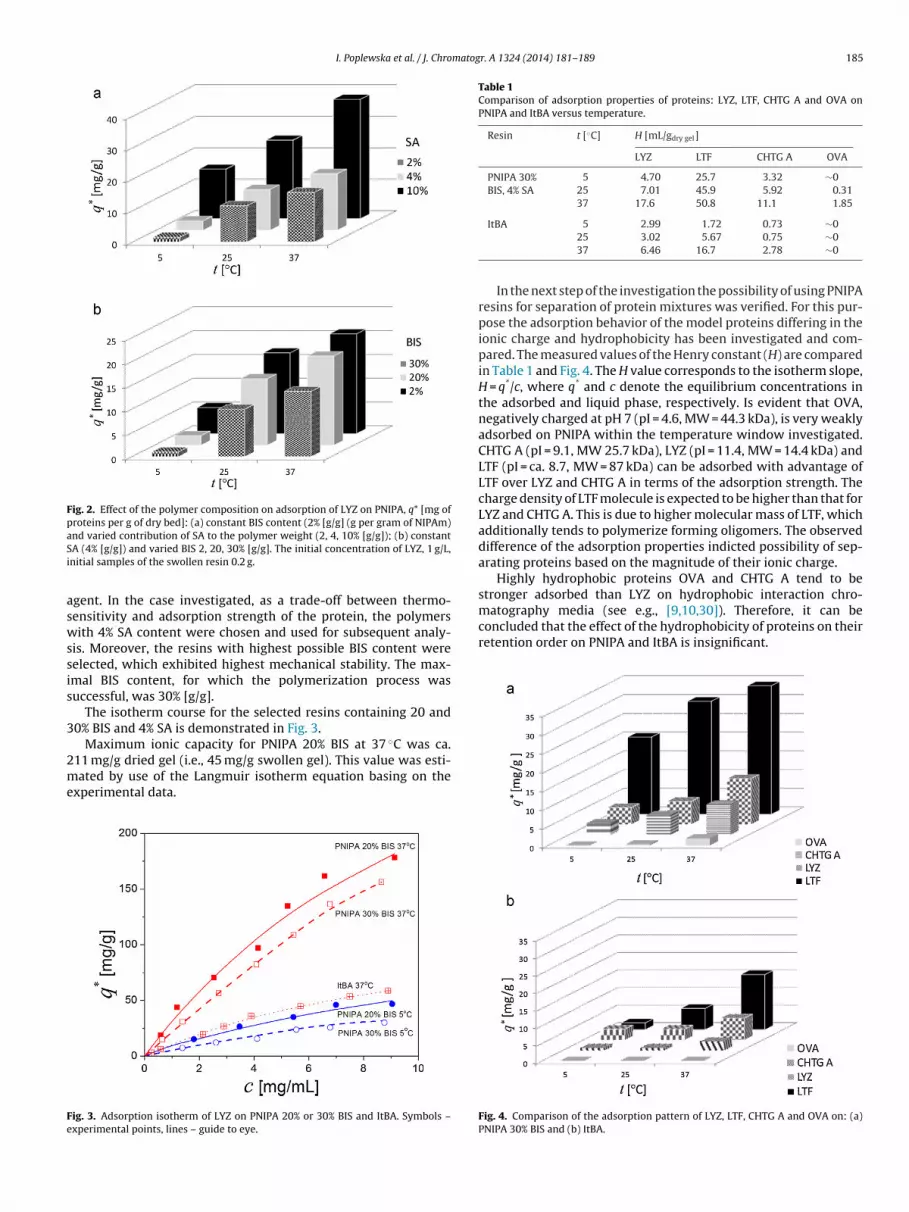
I. Poplewska et al. / J. Chromatogr. A 1324 (2014) 181– 189 185
Fig. 2. Effect of the polymer composition on adsorption of LYZ on PNIPA, q* [mg ofproteins per g of dry bed]: (a) constant BIS content (2% [g/g] (g per gram of NIPAm)aSi
aswssis
3
2me
Fe
Table 1Comparison of adsorption properties of proteins: LYZ, LTF, CHTG A and OVA onPNIPA and ItBA versus temperature.
Resin t [◦C] H [mL/gdry gel]
LYZ LTF CHTG A OVA
PNIPA 30%BIS, 4% SA
5 4.70 25.7 3.32 ∼025 7.01 45.9 5.92 0.3137 17.6 50.8 11.1 1.85
stronger adsorbed than LYZ on hydrophobic interaction chro-matography media (see e.g., [9,10,30]). Therefore, it can beconcluded that the effect of the hydrophobicity of proteins on theirretention order on PNIPA and ItBA is insignificant.
nd varied contribution of SA to the polymer weight (2, 4, 10% [g/g]); (b) constantA (4% [g/g]) and varied BIS 2, 20, 30% [g/g]. The initial concentration of LYZ, 1 g/L,nitial samples of the swollen resin 0.2 g.
gent. In the case investigated, as a trade-off between thermo-ensitivity and adsorption strength of the protein, the polymersith 4% SA content were chosen and used for subsequent analy-
is. Moreover, the resins with highest possible BIS content wereelected, which exhibited highest mechanical stability. The max-mal BIS content, for which the polymerization process wasuccessful, was 30% [g/g].
The isotherm course for the selected resins containing 20 and0% BIS and 4% SA is demonstrated in Fig. 3.
Maximum ionic capacity for PNIPA 20% BIS at 37 ◦C was ca.
11 mg/g dried gel (i.e., 45 mg/g swollen gel). This value was esti-ated by use of the Langmuir isotherm equation basing on thexperimental data.
ig. 3. Adsorption isotherm of LYZ on PNIPA 20% or 30% BIS and ItBA. Symbols –xperimental points, lines – guide to eye.
ItBA 5 2.99 1.72 0.73 ∼025 3.02 5.67 0.75 ∼037 6.46 16.7 2.78 ∼0
In the next step of the investigation the possibility of using PNIPAresins for separation of protein mixtures was verified. For this pur-pose the adsorption behavior of the model proteins differing in theionic charge and hydrophobicity has been investigated and com-pared. The measured values of the Henry constant (H) are comparedin Table 1 and Fig. 4. The H value corresponds to the isotherm slope,H = q*/c, where q* and c denote the equilibrium concentrations inthe adsorbed and liquid phase, respectively. Is evident that OVA,negatively charged at pH 7 (pI = 4.6, MW = 44.3 kDa), is very weaklyadsorbed on PNIPA within the temperature window investigated.CHTG A (pI = 9.1, MW 25.7 kDa), LYZ (pI = 11.4, MW = 14.4 kDa) andLTF (pI = ca. 8.7, MW = 87 kDa) can be adsorbed with advantage ofLTF over LYZ and CHTG A in terms of the adsorption strength. Thecharge density of LTF molecule is expected to be higher than that forLYZ and CHTG A. This is due to higher molecular mass of LTF, whichadditionally tends to polymerize forming oligomers. The observeddifference of the adsorption properties indicted possibility of sep-arating proteins based on the magnitude of their ionic charge.
Highly hydrophobic proteins OVA and CHTG A tend to be
Fig. 4. Comparison of the adsorption pattern of LYZ, LTF, CHTG A and OVA on: (a)PNIPA 30% BIS and (b) ItBA.

186 I. Poplewska et al. / J. Chromatogr. A 1324 (2014) 181– 189
Table 2Influence of the mobile phase flowrate on the adsorption effectiveness of LYZ onPNIPA 30% BIS.
Mobile phase flowrate [mL/min] % mass retained
2 37.71 63.8
(wroaeiattcst(
3
ta
scd
atwawp
iTfi
ifiomtwCti
itm
etid
As it can be noticed by inspecting the contents of Table 3, thedesorbent consumption depends on the adsorption affinity of theprotein to the resin during the loading period. LTF, which wasstronger adsorbed than LYZ, was also more difficult to desorb.
Table 3Comparison of desorption efficiency of LYZ and LTF on PNIPA 30% BIS and ItBA interms of the amount of desorbent (CV) necessary for the protein mass recovery; Des1 – desorbent containing 1 M NaCl, Des 2 – desorbent with 0.1 M NaCl.
Proteins PNIPA ItBA
37 ◦C 25 ◦C, 5 ◦C 37 ◦C 25 ◦C, 5 ◦C
0.5 71.4
The adsorption behavior of proteins on the grafted polymerItBA) followed very similar trend. Similar content of ionizing agentas selected as that for PNIPA. Because the grafted polymer was
igid, BIS content was kept at a low level, i.e., 2% [g/g]. The presencef hydrophobic co-monomer, tert-butylacrylamide (6%), which waslso used in the resin synthesis by Maharjan et al. [28], did not influ-nced noticeably on adsorption of the proteins investigated. As its reported below, it affected only the LCST value. The affinity ofll proteins investigated was however markedly weaker comparedo the ungrafted NIPAm resin. LYZ and CHTG A were almost unre-ained. The maximum of ionic capacity estimated from the isothermourse of LYZ at 37 ◦C (see Fig. 3) was ca. 61 mg/g dried gel (20 mg/gwollen gel). LTF was retained with similar adsorption affinity tohat reported in [28], though weaker than that measured on PNIPAsee Table 1 and Fig. 4a and b).
.3. Dynamic experiments
To investigate the dynamics of separation as well as of the phaseransition, the PNIPA and ItBA resins were packed into the column,s described in Section 2.
Despite high BIS content and the presence of silica gel in theuspension during polymerization, the PNIPA resins were prone toompression. Therefore, they additionally were mixed with silicauring the column packing.
Typical band profiles obtained on PNIPA are depicted in Fig. 5and b. A gradual decrease of the mass of the protein eluted fromhe column was observed with increase of temperature, e.g., at 5 ◦Chole mass of LYZ injected into the column (containing ca. 1 g resin
fter drying) was eluted, at 25 ◦C only 46% – the remaining amountas retained in the column, while at 37 ◦C the whole mass of therotein was retained.
The phenomenon was enhanced for LTF, which elution wasncomplete within the whole temperature window investigated.he adsorption pattern was in agreement with the trend observedor the batch measurements reported in Table 1. CHTG A exhibitedntermediate retention behavior between LYZ and OVA.
The ItBA resin was mechanically stable and at the flowrates usedt did not undergo compression inside the column. The band pro-les of proteins on ItBA are illustrated in Fig. 6a and b. Adsorptionf LYZ was weak regardless the process temperature; e.g., 13% ofass injected was retained in the column at the highest tempera-
ure investigated, 37 ◦C, and no mass was retained (whole amountas eluted with the buffer) at lower temperatures, i.e., 25 and 5 ◦C.HTG A was adsorbed even weaker than LYZ. The affinity of LTF tohe resin was the strongest – at 37 ◦C as much as 73% of the massnjected was retained, at 25 ◦C – 34% and at 5 ◦C – 25%.
The efficiency of adsorption in the chromatographic column wasnfluenced by the residence time. Typical comparison of the adsorp-ion affinity in terms of the mass retained in the column versus the
obile phase flowrate is presented in Table 2.As it can be observed, a long residence time is preferred to
nhance adsorption, which can be attributed to slow kinetics of
he mass transfer as well as possible protein conformation changesn the adsorbed phase. Similar effect, though slightly weaker, wasetected in case of chromatographic elution on the ItBA resins.Fig. 5. Typical chromatograms on the column packed with PNIPA (ca. 1 g of dry bedin the column); (a) LYZ and OVA – the total column loading with the protein 0.3 mg;(b) LTF – the protein loading 0.1 mg.
In the next step of the study, the influence of temperature ondesorption effectiveness was investigated. For this purpose themass retained during the period of the column loading was des-orbed at different temperature within the investigated operatingwindow using solutions of NaCl salt. The desorption effectivenesswas quantified as the volume and concentration of the salt solutionnecessary for the mass recovery of the adsorbed protein.
The data summarized in Table 3 indicate that lowering temper-ature enhances significantly the protein desorption which benefitsin reduction of the desorbent consumption. Moreover, a decrease ofthe salt concentration in the desorption buffer is expected to facil-itate desalting operation following the chromatographic process.
LYZ 10 CV, Des 1 6 CV, Des 1 0.03 CV, Des 2LTF No desorption,
Des 113 CV, Des 1 4 CV, Des 1 0.03 CV, Des 1
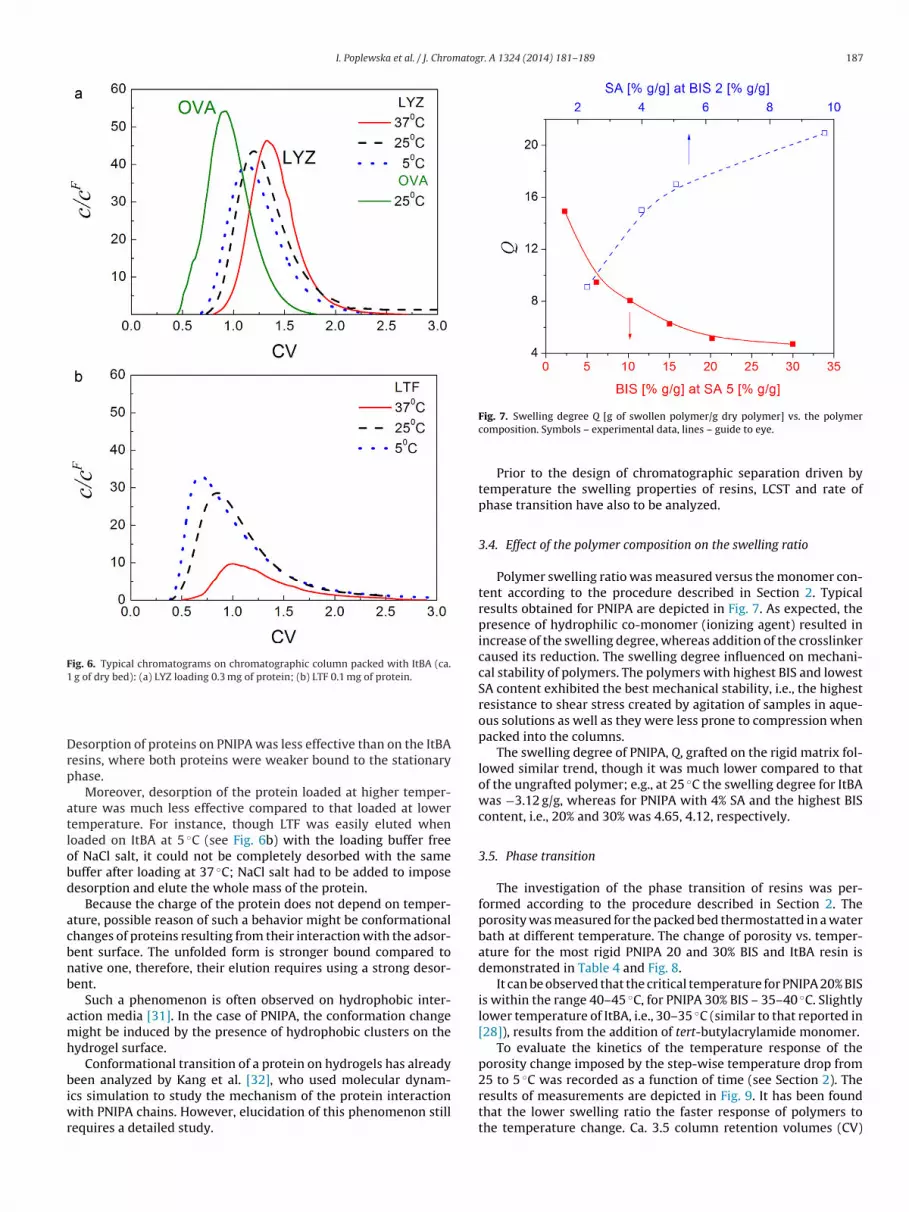
I. Poplewska et al. / J. Chromatogr. A 1324 (2014) 181– 189 187
Fig. 6. Typical chromatograms on chromatographic column packed with ItBA (ca.1
Drp
atlobd
acbnb
amh
biwr
results of measurements are depicted in Fig. 9. It has been found
g of dry bed): (a) LYZ loading 0.3 mg of protein; (b) LTF 0.1 mg of protein.
esorption of proteins on PNIPA was less effective than on the ItBAesins, where both proteins were weaker bound to the stationaryhase.
Moreover, desorption of the protein loaded at higher temper-ture was much less effective compared to that loaded at loweremperature. For instance, though LTF was easily eluted whenoaded on ItBA at 5 ◦C (see Fig. 6b) with the loading buffer freef NaCl salt, it could not be completely desorbed with the sameuffer after loading at 37 ◦C; NaCl salt had to be added to imposeesorption and elute the whole mass of the protein.
Because the charge of the protein does not depend on temper-ture, possible reason of such a behavior might be conformationalhanges of proteins resulting from their interaction with the adsor-ent surface. The unfolded form is stronger bound compared toative one, therefore, their elution requires using a strong desor-ent.
Such a phenomenon is often observed on hydrophobic inter-ction media [31]. In the case of PNIPA, the conformation changeight be induced by the presence of hydrophobic clusters on the
ydrogel surface.Conformational transition of a protein on hydrogels has already
een analyzed by Kang et al. [32], who used molecular dynam-cs simulation to study the mechanism of the protein interaction
ith PNIPA chains. However, elucidation of this phenomenon stillequires a detailed study.
Fig. 7. Swelling degree Q [g of swollen polymer/g dry polymer] vs. the polymercomposition. Symbols – experimental data, lines – guide to eye.
Prior to the design of chromatographic separation driven bytemperature the swelling properties of resins, LCST and rate ofphase transition have also to be analyzed.
3.4. Effect of the polymer composition on the swelling ratio
Polymer swelling ratio was measured versus the monomer con-tent according to the procedure described in Section 2. Typicalresults obtained for PNIPA are depicted in Fig. 7. As expected, thepresence of hydrophilic co-monomer (ionizing agent) resulted inincrease of the swelling degree, whereas addition of the crosslinkercaused its reduction. The swelling degree influenced on mechani-cal stability of polymers. The polymers with highest BIS and lowestSA content exhibited the best mechanical stability, i.e., the highestresistance to shear stress created by agitation of samples in aque-ous solutions as well as they were less prone to compression whenpacked into the columns.
The swelling degree of PNIPA, Q, grafted on the rigid matrix fol-lowed similar trend, though it was much lower compared to thatof the ungrafted polymer; e.g., at 25 ◦C the swelling degree for ItBAwas −3.12 g/g, whereas for PNIPA with 4% SA and the highest BIScontent, i.e., 20% and 30% was 4.65, 4.12, respectively.
3.5. Phase transition
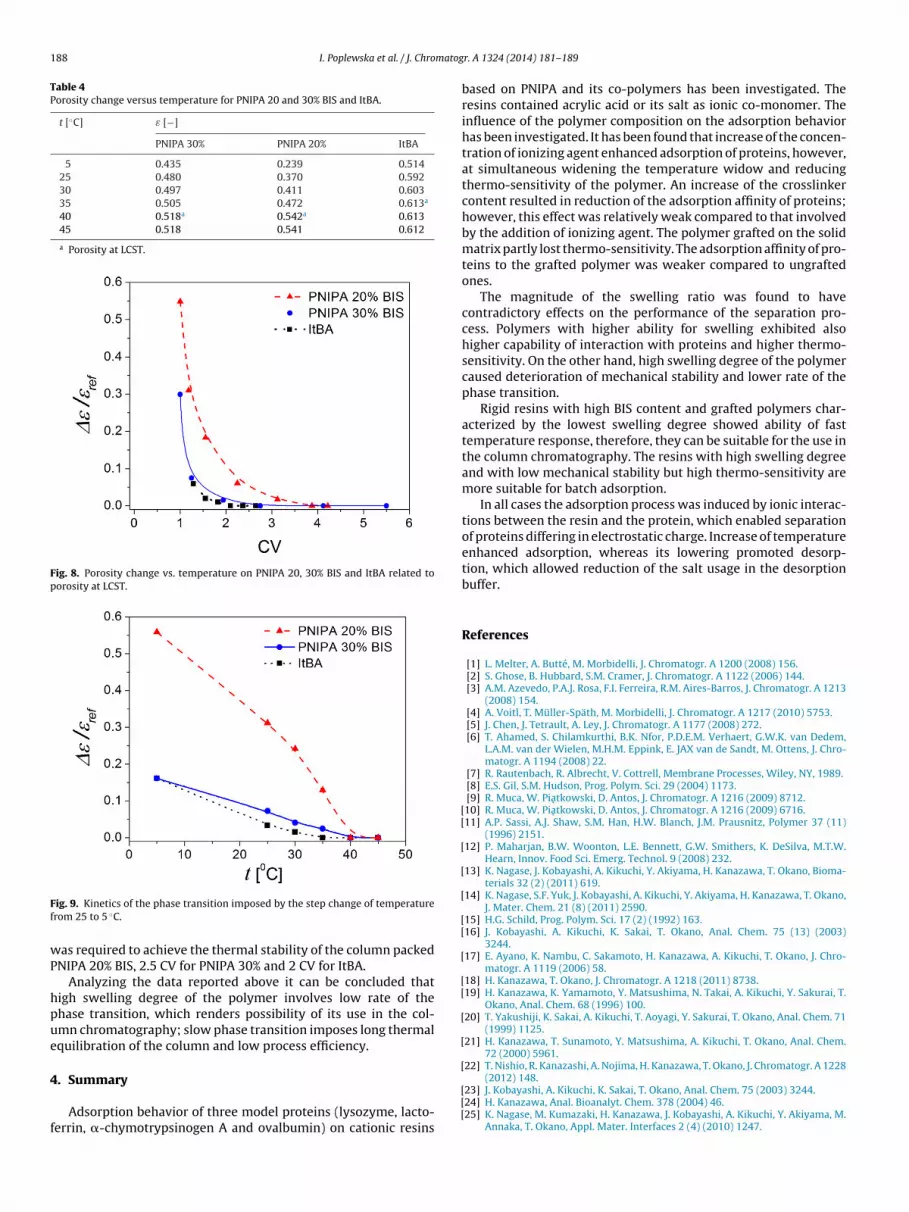
The investigation of the phase transition of resins was per-formed according to the procedure described in Section 2. Theporosity was measured for the packed bed thermostatted in a waterbath at different temperature. The change of porosity vs. temper-ature for the most rigid PNIPA 20 and 30% BIS and ItBA resin isdemonstrated in Table 4 and Fig. 8.
It can be observed that the critical temperature for PNIPA 20% BISis within the range 40–45 ◦C, for PNIPA 30% BIS – 35–40 ◦C. Slightlylower temperature of ItBA, i.e., 30–35 ◦C (similar to that reported in[28]), results from the addition of tert-butylacrylamide monomer.
To evaluate the kinetics of the temperature response of theporosity change imposed by the step-wise temperature drop from25 to 5 ◦C was recorded as a function of time (see Section 2). The
that the lower swelling ratio the faster response of polymers tothe temperature change. Ca. 3.5 column retention volumes (CV)
188 I. Poplewska et al. / J. Chromatog
Table 4Porosity change versus temperature for PNIPA 20 and 30% BIS and ItBA.
t [◦C] ε [−]
PNIPA 30% PNIPA 20% ItBA
5 0.435 0.239 0.51425 0.480 0.370 0.59230 0.497 0.411 0.60335 0.505 0.472 0.613a
40 0.518a 0.542a 0.61345 0.518 0.541 0.612
a Porosity at LCST.
Fig. 8. Porosity change vs. temperature on PNIPA 20, 30% BIS and ItBA related toporosity at LCST.
Ff
wP
hpue
4
f
[[
[
[
[
[[
[
[[
[
[
[
ig. 9. Kinetics of the phase transition imposed by the step change of temperaturerom 25 to 5 ◦C.
as required to achieve the thermal stability of the column packedNIPA 20% BIS, 2.5 CV for PNIPA 30% and 2 CV for ItBA.
Analyzing the data reported above it can be concluded thatigh swelling degree of the polymer involves low rate of thehase transition, which renders possibility of its use in the col-mn chromatography; slow phase transition imposes long thermalquilibration of the column and low process efficiency.
. Summary
Adsorption behavior of three model proteins (lysozyme, lacto-errin, �-chymotrypsinogen A and ovalbumin) on cationic resins
[[[
r. A 1324 (2014) 181– 189
based on PNIPA and its co-polymers has been investigated. Theresins contained acrylic acid or its salt as ionic co-monomer. Theinfluence of the polymer composition on the adsorption behaviorhas been investigated. It has been found that increase of the concen-tration of ionizing agent enhanced adsorption of proteins, however,at simultaneous widening the temperature widow and reducingthermo-sensitivity of the polymer. An increase of the crosslinkercontent resulted in reduction of the adsorption affinity of proteins;however, this effect was relatively weak compared to that involvedby the addition of ionizing agent. The polymer grafted on the solidmatrix partly lost thermo-sensitivity. The adsorption affinity of pro-teins to the grafted polymer was weaker compared to ungraftedones.
The magnitude of the swelling ratio was found to havecontradictory effects on the performance of the separation pro-cess. Polymers with higher ability for swelling exhibited alsohigher capability of interaction with proteins and higher thermo-sensitivity. On the other hand, high swelling degree of the polymercaused deterioration of mechanical stability and lower rate of thephase transition.
Rigid resins with high BIS content and grafted polymers char-acterized by the lowest swelling degree showed ability of fasttemperature response, therefore, they can be suitable for the use inthe column chromatography. The resins with high swelling degreeand with low mechanical stability but high thermo-sensitivity aremore suitable for batch adsorption.
In all cases the adsorption process was induced by ionic interac-tions between the resin and the protein, which enabled separationof proteins differing in electrostatic charge. Increase of temperatureenhanced adsorption, whereas its lowering promoted desorp-tion, which allowed reduction of the salt usage in the desorptionbuffer.
References
[1] L. Melter, A. Butté, M. Morbidelli, J. Chromatogr. A 1200 (2008) 156.[2] S. Ghose, B. Hubbard, S.M. Cramer, J. Chromatogr. A 1122 (2006) 144.[3] A.M. Azevedo, P.A.J. Rosa, F.I. Ferreira, R.M. Aires-Barros, J. Chromatogr. A 1213
(2008) 154.[4] A. Voitl, T. Müller-Späth, M. Morbidelli, J. Chromatogr. A 1217 (2010) 5753.[5] J. Chen, J. Tetrault, A. Ley, J. Chromatogr. A 1177 (2008) 272.[6] T. Ahamed, S. Chilamkurthi, B.K. Nfor, P.D.E.M. Verhaert, G.W.K. van Dedem,
L.A.M. van der Wielen, M.H.M. Eppink, E. JAX van de Sandt, M. Ottens, J. Chro-matogr. A 1194 (2008) 22.
[7] R. Rautenbach, R. Albrecht, V. Cottrell, Membrane Processes, Wiley, NY, 1989.[8] E.S. Gil, S.M. Hudson, Prog. Polym. Sci. 29 (2004) 1173.[9] R. Muca, W. Piatkowski, D. Antos, J. Chromatogr. A 1216 (2009) 8712.10] R. Muca, W. Piatkowski, D. Antos, J. Chromatogr. A 1216 (2009) 6716.11] A.P. Sassi, A.J. Shaw, S.M. Han, H.W. Blanch, J.M. Prausnitz, Polymer 37 (11)
(1996) 2151.12] P. Maharjan, B.W. Woonton, L.E. Bennett, G.W. Smithers, K. DeSilva, M.T.W.
Hearn, Innov. Food Sci. Emerg. Technol. 9 (2008) 232.13] K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa, T. Okano, Bioma-
terials 32 (2) (2011) 619.14] K. Nagase, S.F. Yuk, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa, T. Okano,
J. Mater. Chem. 21 (8) (2011) 2590.15] H.G. Schild, Prog. Polym. Sci. 17 (2) (1992) 163.16] J. Kobayashi, A. Kikuchi, K. Sakai, T. Okano, Anal. Chem. 75 (13) (2003)
3244.17] E. Ayano, K. Nambu, C. Sakamoto, H. Kanazawa, A. Kikuchi, T. Okano, J. Chro-
matogr. A 1119 (2006) 58.18] H. Kanazawa, T. Okano, J. Chromatogr. A 1218 (2011) 8738.19] H. Kanazawa, K. Yamamoto, Y. Matsushima, N. Takai, A. Kikuchi, Y. Sakurai, T.
Okano, Anal. Chem. 68 (1996) 100.20] T. Yakushiji, K. Sakai, A. Kikuchi, T. Aoyagi, Y. Sakurai, T. Okano, Anal. Chem. 71
(1999) 1125.21] H. Kanazawa, T. Sunamoto, Y. Matsushima, A. Kikuchi, T. Okano, Anal. Chem.
72 (2000) 5961.22] T. Nishio, R. Kanazashi, A. Nojima, H. Kanazawa, T. Okano, J. Chromatogr. A 1228
(2012) 148.23] J. Kobayashi, A. Kikuchi, K. Sakai, T. Okano, Anal. Chem. 75 (2003) 3244.24] H. Kanazawa, Anal. Bioanalyt. Chem. 378 (2004) 46.25] K. Nagase, M. Kumazaki, H. Kanazawa, J. Kobayashi, A. Kikuchi, Y. Akiyama, M.
Annaka, T. Okano, Appl. Mater. Interfaces 2 (4) (2010) 1247.

matog
[
[
[
[
I. Poplewska et al. / J. Chro
26] H. Kanazawa, M. Nishikawa, A. Mizutani, C. Sakamoto, Y. Morita-Murase, Y.
Nagata, J. Chromatogr. A 1191 (2008) 157.27] K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa, T. Okano,Biomacromolecules 9 (4) (2008) 1340.
28] P. Maharjan, M.T.W. Hearn, R.W. Jackson, K. De Silva, B.W. Woonton, J. Chro-matogr. A 1216 (2009) 8722.
[[
[
r. A 1324 (2014) 181– 189 189
29] B. Strachotová, A. Strachota, M. Uchman, M. Slouf, J. Brus, J. Plestil, L. Matejka,
Polymer 48 (2007) 1471.30] R. Muca, W. Marek, W. Piatkowski, D. Antos, J. Chromatogr. A 1217 (2010) 2812.31] R. Ueberbacher, E. Haimer, R. Hahn, A. Jungbauer, J. Chromatogr. A 1198/1199
(2008) 154.32] K. Kang, D. Lu, Z. Liu, Chin. J. Chem. Eng. 20 (2) (2012) 284.





























