Embed Size (px)
Citation preview

Adaptation of Protein Surfaces to Subcellular Location
Miguel A. Andrade1, SeaÂn I. O'Donoghue2 and Burkhard Rost2
1European BioinformaticsInstitute, Hinxton, CambridgeCB10 1SD, UK2European Molecular BiologyLaboratory, D-69012Heidelberg, Germany
In vivo, proteins occur in widely different physio-chemical environments,and, from in vitro studies, we know that protein structure can be verysensitive to environment. However, theoretical studies of protein struc-ture have tended to ignore this complexity. In this paper, we haveapproached this problem by grouping proteins by their subcellularlocation and looking at structural properties that are characteristic toeach location. We hypothesize that, throughout evolution, each subcellu-lar location has maintained a characteristic physio-chemical environment,and that proteins in each location have adapted to these environments. Ifso, we would expect that protein structures from different locations willshow characteristic differences, particularly at the surface, which isdirectly exposed to the environment. To test this hypothesis, we haveexamined all eukaryotic proteins with known three-dimensional structureand for which the subcellular location is known to be either nuclear,cytoplasmic, or extracellular. In agreement with previous studies, we ®ndthat the total amino acid composition carries a signal that identi®es thesubcellular location. This signal was due almost entirely to the surfaceresidues. The surface residue signal was often strong enough to accu-rately predict subcellular location, given only a knowledge of which resi-dues are at the protein surface. The results suggest how the accuracy ofprediction of location from sequence can be improved. We concludedthat protein surfaces show adaptation to their subcellular location. Thenature of these adaptations suggests several principles that proteins mayhave used in adapting to particular physio-chemical environments; theseprinciples may be useful for protein design.
# 1998 Academic Press Limited
Keywords: protein evolution; protein three-dimensional-structure; proteinsurface; subcellular location; bioinformatics
Introduction
For over 30 years, researchers have sought todiscover the principles that determine the fold ofglobular proteins in aqueous environments. Thegoals of this research effort are to predict tertiarystructure from sequence, and to facilitate thedesign of proteins with novel structures and func-tions. Several general principles are now wellunderstood: most residues with charged or polarside-chains occur at the proteins surface; most resi-dues with apolar side-chains are buried; many ofthe polar chemical groups that occur in the proteininterior are hydrogen bonded, effectively dampingtheir polarity; this balance of hydrophilic andhydrophobic interactions stabilises the structure.However, these general principles are insuf®cientfor predicting protein fold (e.g. see Rost &O'Donoghue, 1997).
One complexity in studying general propertiesof protein structure is that different proteinsexperience different physio-chemical environments,and that the exact environment in¯uences thestructure. An extreme example is globular versustransmembrane proteins. There are distinct differ-ences in composition between these two groups ofproteins that are well understood; from analysis ofthe sequence, it is possible to predict to whichgroup a protein belongs at more than 97% accuracy(Rost et al., 1996). Theoretical studies have tended toignore the effect of different aqueous environmentson globular proteins, although we know fromin vitro experiments that proteins can be exquisitelysensitive to variations in pH or in the concentrationof various ions. In this paper, we have approachedthis problem by grouping globular proteins by theirsubcellular location; our rationale is that all proteinsin the same subcellular compartment experience a
J. Mol. Biol. (1998) 276, 517±525
0022±2836/98/070517±09 $25.00/0/mb971498 # 1998 Academic Press Limited

similar physio-chemical environment, and each pro-tein usually occurs in only one compartment.Hence, looking for protein structure properties thatare characteristic to each compartment, we mayuncover principles of the in¯uence of speci®cenvironments on protein structures.
The physio-chemical environment of subcellularcompartments varies with different cell types; theextracellular environment varies especially widely.However, certain features are common to almostall cell types. Compared to the extracellularenvironment, the cell interior has a higher overallconcentration of ions, small (usually charged)metabolites, and proteins. The intracellular pH isregulated, typically to be slightly alkaline. All cellsactively export speci®c anions (usually Na� in ani-mal cells and H� in plant cells) to counteract osmo-tic expansion and to facilitate active import ofspeci®c molecules. This transport maintains a vol-tage difference across the plasma membrane ofabout ÿ100 mV (inside more negative) and meansthat certain ions (H�, Na�) are more highly concen-trated in the extracellular environment.
The physio-chemical environments of the cyto-plasm and the nucleus (in eukaryotic cells) aresimilar, since the nuclear pore complexes are per-meable to small (<5 kDa) neutral molecules(Dingwall & Laskey, 1986); however, there aredifferences in ionic strength due to selective ionicpermeability of the nuclear envelope (Dingwall,1991), and the large negative charge arising fromthe DNA phosphate backbone, which leads toaccumulation of anions.
The subcellular location of eukaryotic proteins isdetermined by a traf®cking system, which isreasonably well understood (Pfeffer & Rotteman,1987). The system has two main branches thatdivide at the ®rst stage of protein synthesis on theribosomes: on one branch, proteins are synthesisedin the cytoplasm, and from there can go to thenucleus, mitochondria, or peroxisomes; the secondbranch leads to the endoplasmic reticulum, then tothe Golgi apparatus, and from there to lysosomes,secretory vesicles, or the cell surface (Figure 1). Ateach branch point in the traf®cking system, a``decision'' must be made for each protein; eitherretain the protein in the current compartment ortransport it to the next compartment. These``decisions'' are made by membrane transport com-plexes, which respond to signals on the proteinsthemselves (Verner & Schatz, 1988; Briggs &Gierasch, 1986; SjoÈstroÈm et al., 1987; von Heijne,1985; Nielsen et al., 1997). The best understoodbranch point is the initial division between the twomain branches; proteins destined for the endoplas-mic reticulum/extracellular branch have an N-terminal signal peptide that causes them to betransferred into the endoplasmic reticulum as theyare being synthesised; proteins lacking this signalare synthesised in the cytoplasm. The protein sig-nals used at the other branch points are not alwaysso clear for two reasons: ®rstly, the signals are pre-sented by folded proteins, and hence are not
always contiguous in amino acids sequence; sec-ondly, even where the signals are contiguous insequence, not all of these signal peptides havebeen documented.
Knowing where a protein occurs in the cell is animportant step towards understanding the functionof the protein. Hence, a method for accurately pre-dicting subcellular location from the amino acidsequence alone would be valuable in interpretingthe wealth of data being provided by large scalesequencing projects. Indeed, we believe that pre-diction of physically meaningful and clearlyde®ned quantities, such as location or secondarystructure, may be more fruitful than trying to pre-dict function directly, as ``function'' is necessarilyvery dif®cult to de®ne.
To predict location, the ®rst method would be tosearch for signal peptides in the sequence; unfortu-nately, in many cases no signal peptide can befound. Another approach would be to inferlocation by sequence homology to a protein withknown location; however, such inference can beunreliable (see Results). A third approach wassuggested by the results of Nishikawa et al.(1983a,b) and Nakashima & Nishikawa (1994);they found that the total amino acid compositionof proteins is correlated to the subcellular location.This has recently led to a location prediction meth-od based only on composition (Cedano et al.,
Figure 1. Subcellular locations in eukaryotic cells. In thisstudy, we considered only proteins occurring in thethree major subcellular locations: the nucleus (green),cytoplasm (red), and extracellular space (blue). Proteinsfrom other subcellular compartments (such as the mito-chondria, ribosomes, lysosomes, and the endoplasmicreticulum, all shown in yellow) were not considered.The grey arrows indicate the two major branches of theprotein traf®cking system that determines subcellularlocation: following protein synthesis in the ribosomes,one branch leads into the cytoplasm, and from thereinto the nucleus; the second branch leads into secretoryvesicles and then to the extracellular space.
518 Adaptation of Proteins to Subcellular Location

1997). Probably the most comprehensive locationprediction method to date is the expert systemdeveloped by Nakai et al. (1988) and Nakai &Kanehisa (1991, 1992); this system uses a small num-ber of rules based on composition; it is based mostlyon lists of known signal peptides. No method so farhas combined all three approaches; hence there ismuch scope for developing more accurate and gen-eral solutions to the protein location problem.
Within each subcellular compartment of a givencell type, proteins have co-evolved with the phy-sio-chemical environment so that they are stableand functional in that environment. However, thegeneral features of the nuclear, cytoplasmic, andextracellular environments discussed above havebeen constant factors throughout eukaryotic evol-ution. We hypothesize that these constant factorsimply a set of ``environmental'' constraints on theevolution of protein structure, and that proteinswill have adapted to these different environmentalconstraints. These environmental constraintswould be distinct from the more familiar evol-utionary constraints that conserve residues in theactive site, residues involved in binding othermacromolecules, or residues that anchor the pro-tein within a given structural family (Rost, 1997).Rather than acting on speci®c residues, the effectof environmental constraints would be moreglobal. If the hypothesis is true, we would expectdistinct and measurable differences in structuralproperties of proteins from different compart-ments. The surface residues should be mostaffected, as they are in direct contact with theenvironment, whereas buried residues are largelyshielded from the environment.
To test this hypothesis, we have examined allthree-dimensional structures of eukaryotic proteinsfor which the location is annotated (as eithernuclear, cytoplasmic, or extracellular). In agree-ment with the hypothesis, we found evidence thatprotein surfaces have adapted to the particularenvironment in each compartment.
Results
In compiling the data sets, it rapidly becameclear that subcellular location is not annotated for
most of the proteins in PDB, and hence the Singleand Glycosylated data sets were relatively small,while the Non-located class was large (Table 1). Incompiling the Homology data set, we found tensequence families in which proteins occurred intwo different subcellular locations (two familieshad members in both the nucleus and cytoplasm;eight had members in both the cytoplasm andextracellular space). Thus even at 40% sequenceidentity, it is not safe to infer that two proteinshave the same subcellular location.
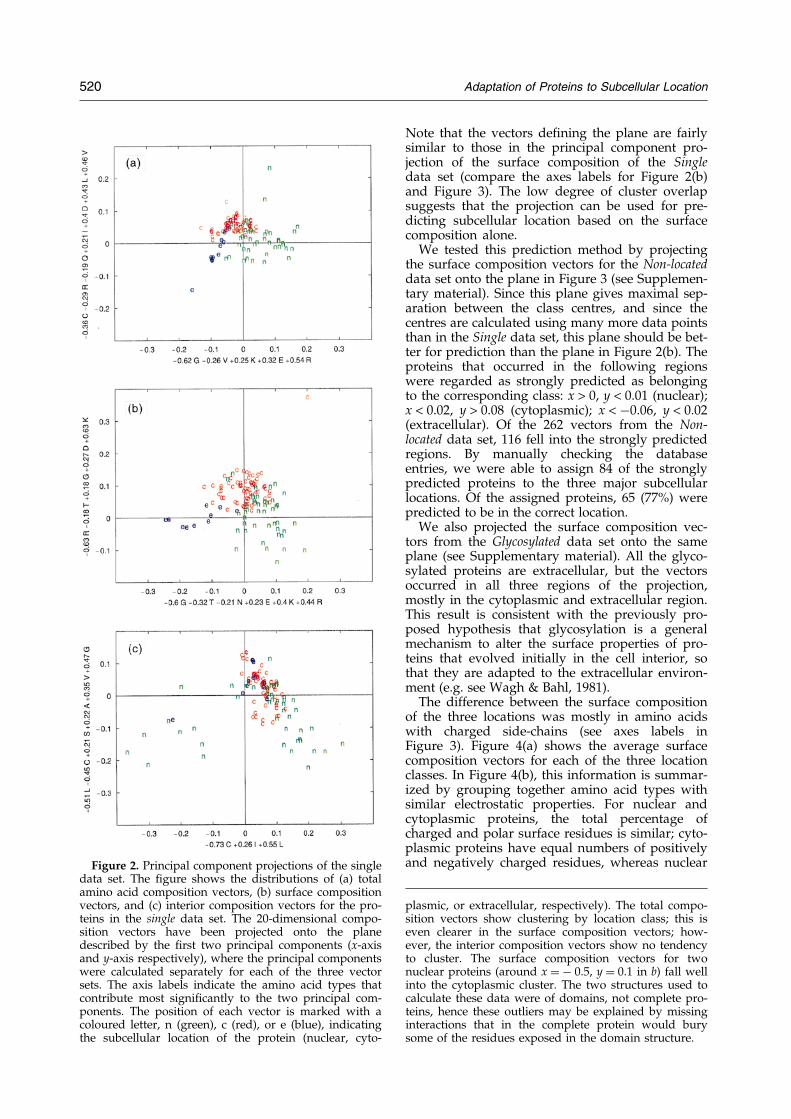
Figure 2(a) shows the total amino acid compo-sition vectors of the Single data set projected ontothe plane de®ned by the ®rst two principal com-ponents. Proteins from the three location classesfall into three clusters; although there is some over-lap between the clusters, the centres are distinct.As discussed in Methods, the occurrence of clustersin the principal component projection indicatesthat the total composition vectors are correlatedwith location, in agreement with the observation ofNishikawa et al. (1983a,b) In Figure 2(b), the planeand projections have been calculated from the sur-face composition vectors of the Single data set.Again, we observe a clustering by location class;the clusters are better de®ned with a larger separ-ation of the cluster centres, and a slight decreasedin overlap. In contrast, the interior compositionvectors of the same data set do not form distinctclusters (Figure 2(c)). Hence we concluded that thesignal observed for the total amino acid compo-sition was due almost entirely to the surface resi-dues.
Figure 3 shows the surface composition vectorsfor the Homology data set projected onto theplane de®ned by the average vectors of each ofthe three location classes. If we accept thehypothesis that the surface composition vectorsare correlated with subcellular location, then thisprojection gives the optimal view to examinehow uniquely de®ned each cluster is. In addition,since the Homology data set contains many moreprotein sequences than the Single data set, it pro-vides a much more telling test of cluster overlap.In fact, proteins from the three location classesare grouped into very clearly de®ned clusterswith little overlap between the classes (Figure 3).
Table 1. Breakdown of the data sets used in this study
Data set Nuclear Cytoplasmic Extracellular Total
Single 44 66 11 121Glycosylated ± ± 16 16Homology 446 885 30 1361Non-located ? ? ? 262
For each of the data sets used in this study, the Table lists the number of proteins in thethree major locations (nuclear, cytoplasm, extracellular) and the total in each data set. TheSingle data set is de®ned as all sequence-distinct, eukaryotic, non-glycosylated proteins inthe PDB, and known to occur in one of the three locations; Glycosylated, as for the Singledata set except glycosylated protein; Homology, all protein sequences with known locationand with high homology to a structure in the Single data set; Non-located, all sequence-distinct eukaryotic structures in the PDB for which the subcellular location has not beenannotated.
Adaptation of Proteins to Subcellular Location 519
Note that the vectors de®ning the plane are fairlysimilar to those in the principal component pro-jection of the surface composition of the Singledata set (compare the axes labels for Figure 2(b)and Figure 3). The low degree of cluster overlapsuggests that the projection can be used for pre-dicting subcellular location based on the surfacecomposition alone.
We tested this prediction method by projectingthe surface composition vectors for the Non-locateddata set onto the plane in Figure 3 (see Supplemen-tary material). Since this plane gives maximal sep-aration between the class centres, and since thecentres are calculated using many more data pointsthan in the Single data set, this plane should be bet-ter for prediction than the plane in Figure 2(b). Theproteins that occurred in the following regionswere regarded as strongly predicted as belongingto the corresponding class: x > 0, y < 0.01 (nuclear);x < 0.02, y > 0.08 (cytoplasmic); x < ÿ0.06, y < 0.02(extracellular). Of the 262 vectors from the Non-located data set, 116 fell into the strongly predictedregions. By manually checking the databaseentries, we were able to assign 84 of the stronglypredicted proteins to the three major subcellularlocations. Of the assigned proteins, 65 (77%) werepredicted to be in the correct location.
We also projected the surface composition vec-tors from the Glycosylated data set onto the sameplane (see Supplementary material). All the glyco-sylated proteins are extracellular, but the vectorsoccurred in all three regions of the projection,mostly in the cytoplasmic and extracellular region.This result is consistent with the previously pro-posed hypothesis that glycosylation is a generalmechanism to alter the surface properties of pro-teins that evolved initially in the cell interior, sothat they are adapted to the extracellular environ-ment (e.g. see Wagh & Bahl, 1981).
The difference between the surface compositionof the three locations was mostly in amino acidswith charged side-chains (see axes labels inFigure 3). Figure 4(a) shows the average surfacecomposition vectors for each of the three locationclasses. In Figure 4(b), this information is summar-ized by grouping together amino acid types withsimilar electrostatic properties. For nuclear andcytoplasmic proteins, the total percentage ofcharged and polar surface residues is similar; cyto-plasmic proteins have equal numbers of positivelyand negatively charged residues, whereas nuclearFigure 2. Principal component projections of the single
data set. The ®gure shows the distributions of (a) totalamino acid composition vectors, (b) surface compositionvectors, and (c) interior composition vectors for the pro-teins in the single data set. The 20-dimensional compo-sition vectors have been projected onto the planedescribed by the ®rst two principal components (x-axisand y-axis respectively), where the principal componentswere calculated separately for each of the three vectorsets. The axis labels indicate the amino acid types thatcontribute most signi®cantly to the two principal com-ponents. The position of each vector is marked with acoloured letter, n (green), c (red), or e (blue), indicatingthe subcellular location of the protein (nuclear, cyto-
plasmic, or extracellular, respectively). The total compo-sition vectors show clustering by location class; this iseven clearer in the surface composition vectors; how-ever, the interior composition vectors show no tendencyto cluster. The surface composition vectors for twonuclear proteins (around x � ÿ 0.5, y � 0.1 in b) fall wellinto the cytoplasmic cluster. The two structures used tocalculate these data were of domains, not complete pro-teins, hence these outliers may be explained by missinginteractions that in the complete protein would burysome of the residues exposed in the domain structure.
520 Adaptation of Proteins to Subcellular Location

proteins have more positively charged residues. Inmarked contrast, extracellular proteins have signi®-cantly fewer charged surface residues, with aboutequal numbers of positively and negativelycharged residues. The lack of charged residues iscompensated by an increase in polar residues. Pro-teins in all three locations have almost the sameproportion of apolar surface residues.
Histograms of solvent accessibility for eachamino acid type differed between the threelocations (see Supplementary material). The differ-ences were correlated with the vectors de®ning theplane separating the clusters (Figure 3). Comparedto proteins from the two other classes, nuclear pro-teins tended to have fewer completely buried resi-dues, suggesting that nuclear proteins may besmaller on average. We con®rmed this trend bycomparing the distribution of protein lengthsbetween the three locations (see Supplementarymaterial). However, the trend was clearly notstrong enough to be used alone for predictinglocation from sequence.
Discussion
While the subcellular locations of almost all theproteins in the PDB would be known, they havenot been entered into either the PDB or SWISS-PROT. This has greatly limited the number ofstructures we could use for this study. For thisreason, we were not able to exclude partial struc-tures (structures of a domain rather than the wholeprotein). Hence some exposed residues may actu-ally be buried in the complete structure, and theseparation between the clusters may actually bebetter than indicated in Figure 3. However, the cur-rent data sets clearly established that the surfacecomposition vectors had a strong signal indicating
the subcellular location. Since we observed this sig-nal in both the Single and Homology data sets, it isvery unlikely to have arisen from bias in the datasets. The total amino acid composition had aweaker location signal, while the interior com-position had little or no location signal.
These results are consistent with our hypothesisthat protein structures have adapted to constraintson the physio-chemical environment of each sub-cellular location. A second class of evolutionaryconstraints may be imposed by the different func-tional roles of proteins in different subcellularlocations. For example, many proteins in thenucleus bind DNA (95% of the nuclear proteins inthe Single data set were DNA-binding), and hencewould be subject to a constant pressure to conservesurface residues favourable for DNA binding. Ineither case, the results suggest that the protein sur-face has been the focus of evolution, in agreementwith the results of Lichtarge et al. (1996).
The difference in electrostatic properties of thesurface of proteins in the three locations(Figure 4(b)) can be summarized as follows. In allthree aqueous environments, about one-third ofthe surface residues were apolar. Of the remainingresidues, the breakdown between polar andcharged residues depended on the total ionicstrength of the environment: the nucleus and cyto-plasm have about the same total ionic strength andthe same proportion of charged surface residues.Outside the cell, where the total ionic strength ismuch lower, the proportion of charged surfaceresidues is also lower; this is compensated by anincrease in polar surface residues. These may begeneral principles by which proteins adapt to theirphysio-chemical environment. In the near future,we intend to test these principles by considering
Figure 3. Surface composition vec-tors for the homology data set. Thesurface composition vectors for thehomology data set are projectedonto the plane de®ned by the threeaverage surface composition vec-tors (one for each location class).Vector positions are marked withcoloured letters to indicate the sub-cellular location (as in Figure 2).The axes are labelled in as Figure 2.
Adaptation of Proteins to Subcellular Location 521

more subcellular location classes, and a broaderrange of organisms (Eubacteria and Archaea).
Cytoplasmic proteins have a balance of acidicand basic surface residues, while extracellular pro-teins have a slight excess of acidic surface residues.However, nuclear proteins have a pronouncedexcess of basic surface residues. This is clearlyrelated to the large negative charge on the DNA,but there are (at least) two explanations. Firstly, themajority of nuclear proteins considered were DNA-binding, and in many the DNA-binding site willhave an excess of basic residues to facilitate bind-ing to the DNA phosphate backbone. Secondly, theexcess positive charge on nuclear proteins may bea result from selective pressure to neutralise theoverall negative charge in the nucleus.
The location signal seen in the surface compo-sition was often strong enough to predict locationclass; this is effectively predicting location from ter-tiary structure. The level of prediction accuracyobtained is impressively high, given that the meth-od used was completely unoptimised; the methodcould clearly be improved by using machine-learn-ing techniques such as neural nets, and by includ-ing additional data. This ability to predictsubcellular location of proteins given the tertiarystructure may be of practical use. Increasingly,new proteins and domains are being discoveredthat are known to be important, either from theircorrelation with some disease state, or from theirassociation with better-characterised proteins, butfor which the exact function or location is unknown.
Figure 4. Average protein surface composition in different subcellular locations. (a) Components of the average sur-face composition vectors for nuclear (top), cytoplasmic (middle), and extracellular (bottom) proteins. The amino acidsare represented by the single-letter code and are coloured by the electrostatic properties of their side-chains: black,apolar; red, acidic; blue, basic; and green, polar. The height of the letters is directly proportional to their contributionto the vector. The numbers in the right-hand column indicate the percentage contributions. The inequalities at thebottom of the Figure indicate which pairwise differences are statistically signi®cant (using the mean difference test,1% con®dence level; e.g. under proline is the inequality N < C, indicating that the average composition of proline inthe nucleus is signi®cantly less than in the cytoplasm). (b) Pie chart representation of the same data as (a), except thatamino acid types have been grouped by the electrostatic properties of their side-chains.
522 Adaptation of Proteins to Subcellular Location

As a result, tertiary structures are being determinedfor proteins of unknown function and location (e.g.the PH domain: Macias et al., 1994). In such cases,the strong correlation between surface compositionand subcellular location may be useful as a steptowards understanding a protein's function.
Our results also suggest that it may be possibleto improve the prediction of location fromsequence alone, using the clusters we observed incombination with a method for accurately predict-ing surface residues (Rost & Sander, 1994). Figure 2suggests that this method would be more accuratethan the existing methods that use total compo-sition (Nishikawa et al., 1983a,b; Cedano et al.,1997). This method would also be complementaryto methods based on signal peptides (Nakai et al.,1988; Nakai & Karehisa, 1991, 1992) or methodsbased on homology. We intend to explore this ideain the future.
In conclusion, we have found a clear signal inthe surface composition of protein structures thatindicates the subcellular location; the signal wasstrong enough to allow accurate prediction oflocation. This supports our hypothesis that proteinstructures have adapted to the different physio-chemical environments in each subcellular location.The results suggest several principles that proteinsuse in adapting to particular physio-chemicalenvironments; if these principles can be establishedby further studies, they may be useful in proteindesign. The results also suggest how the predictionof location from sequence may be improved.
Methods
Definition of the data sets
The protein structure database (PDB: Bernstein,1977), and sequence databases such as SWISS-PROT (Bairoch & Apweiler, 1997) are highly biasedtowards particular protein families (Hobohm et al.,1992). To reduce this bias, we selected our datasets in the following way. We started from the lar-gest sequence-distinct subset of PDB (taken fromthe FSSP database: Holm & Sander, 1996); this sub-set is comprised of 849 protein chains chosen suchthat no pair has more than 25% pairwise sequenceidentity. From this subset we selected all globulareukaryotic proteins that occur in one of the threemain subcellular locations (nucleus, cytoplasm, orextracellular space), based on the annotations inthe SWISS-PROT entries. We excluded proteinsfrom other subcellular locations (ribosome, mito-chondria, chloroplast, vacuole, Golgi apparatus,endoplasmic reticulum, etc.), since our current pur-pose was to establish if a consistent differencecould be observed for proteins from the threemajor location classes. In the remaining set of pro-teins, we distinguished between those that wereand were not glycosylated, as glycosylation greatlyaffects the protein surface properties. Thus wede®ned two data sets: the Glycosylated data set(annotated in SWISS-PROT as glycosylated) and
the Single data set (non-glycosylated, with a singlemember from each sequence family)
We then constructed an extended data set con-sisting of proteins sequences with known locationsfor which a structure could be modelled by hom-ology. This Homology data set was constructed asfollows: for each protein structure in the Singledata set, we searched in SWISS-PROT for alleukaryotic protein sequences with 540% pairwisesequence identity, and with known location (nucle-ar, cytoplasmic, extracellular). At this high level ofsequence homology, it is safe to infer that thesesequences have the same fold as the protein fromthe Single data set (Sander & Schneider, 1991); wechose such a high homology cut-off in order toguarantee conservation of solvent accessibility(Rost & Sander, 1994). Even at this high level ofhomology, it sometimes occurred that two mem-bers within the same sequence family had differentsubcellular locations; in such cases, the entiresequence family was excluded from the Homologydata set. A ®nal data set, the Non-located data set,was constructed with eukaryotic proteins in thesequence-distinct subset of PDB for which the sub-cellular location was not annotated in SWISS-PROT.
For the Single, Glycosylated, and Non-located datasets, the exposure state of each residue was calcu-lated from the solvent-accessible surface area(Connolly, 1983) in the DSSP database (Kabsch &Sander, 1983). The surface area for each residue (inAÊ 2) was normalised by the maximal residue acces-sibility to yield a relative accessibility (as describedby Rost & Sander, 1994). These values were thenused to classify each residue as belonging either tothe surface (relative accessibility 5 25%) or theinterior (relative accessibility < 25%) of the protein(Chothia, 1976; Hubbard & Blundell, 1987). For theHomology data set, the exposure state was calcu-lated similarly except that the surface area valueswere inferred from the corresponding values in thehomologous structure using the sequence align-ments in the HSSP database (Sander & Schneider,1994).
Analysis of composition vector
The total composition vector, ci, for a protein i isde®ned as the row vector ci � {cij}, wherej � 1, . . . , 20 indicates the amino acid type. Thecomposition of the jth amino acid, cij, is de®ned as:
cij � rij
�X20
j�1
rij �1�
where RIJ is the number of residues of amino acidtype j in protein i. We also calculated surface andinterior composition vectors, de®ned as aboveexcept that the RIJ terms were then the number ofresidues of type j at the surface or in the interior,respectively.
Adaptation of Proteins to Subcellular Location 523

For the Single data set, composition vectors werecalculated for all proteins; these were then used tode®ne a sample variance±co-variance matrix, S, asfollows:
S � fsjkg �Xn
i�1
�cij ÿ �cj��cij ÿ �ck�=n( )
�2�
where:
�cj � 1
n
Xn
i�1
cij �3�
is the average composition of the jth amino acidtype over the n proteins in the data set. The princi-pal components of the set of composition vectorsare then the Eigenvectors of S (e.g. see Anderberg,1973). The composition vector for each protein wasthen projected onto the plane de®ned by the ®rsttwo principal components using the standard innerproduct. This provides a two-dimensional view ofhow the component vectors are clustered (Figure 2).Note that in this analysis, the subcellular locationclass of the proteins is not considered. Hence, theresulting view will be unbiased in the sense that ifthe vectors are observed to cluster by locationclass, the clustering would be due to a trend indata, and not due to the analysis. Conversely, how-ever, a cluster in the data may not be observedif the proteins belonging to this class are under-represented compared to the other classes.
For the Homology data set, we calculated a differ-ent projection that speci®cally aims to best separatethe vectors from the three location classes into dis-tinct clusters. This was done by projecting into theplane de®ned by the three average compositionvectors (one for each location class). The plane wascalculated as above except that only the three aver-age vectors were used in de®ning S. Hence onlytwo of the resulting 20 Eigenvectors had non-zeroEigenvalues. Then, the composition vectors foreach protein in the Homology data set where pro-jected into this plane; this plane was also used toproject the composition vectors from the Glycosy-lated and Non-located data sets.
Acknowledgements
For fruitful discussions we are grateful to MichaelNilges, Nigel Brown, and Chris Sander. B.R. thanksChris Sander and Matti Saraste for ®nancial support.M.A.A. was supported from a fellowship of the Euro-pean Union TMR programme. Thanks also to all whodeposit information in public databases, and to thosewho carry the burden of maintaining these valuableevolutionary records. Figure 4(a) was inspired by the``letter-plots'' of Sùren Brunak.
References
Anderberg, M. R. (1973). Cluster Analysis for Applications,Academic Press, New York.
Bairoch, A. & Apweiler, R. (1997). The SWISS-PROTprotein sequence data bank and its new supplementTrEMBL. Nucl. Acids Res. 25, 31±36.
Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer,E. F., Brice, M. D., Rodgers, J. R., Kennard, O.,Shimanouchi, T. & Tasumi, M. (1977). The ProteinData Bank: a computer based archival ®le formacromolecular structures. J. Mol. Biol. 112, 535±542.
Briggs, M. S. & Gierasch, L. M. (1986). Molecular mech-anisms of protein secretion: The role of the signalsequence. Adv. Protein Chem. 38, 109±180.
Cedano, J., Aloy, P., PeÂrez-Pons, J. A. & Querol, E.(1997). Relation between amino acid compositionand celluar location of proteins. J. Mol. Biol. 266,594±600.
Chothia, C. (1976). The nature of the accessible and bur-ied surfaces in proteins. J. Mol. Biol. 105, 1±12.
Connolly, M. L. (1983). Solvent-accessible surfaces ofproteins and nucleic acids. Science, 221, 709±713.
Dingwall, C. (1991). Transport accross the nuclear envel-ope: enigmas and explanations. BioEssays, 13(5),213±218.
Dingwall, C. & Laskey, R. A. (1986). Protein import intothe cell nucleus. Annu. Rev. Cell Biol. 2, 367±390.
Hobohm, U., Scharf, M., Schneider, R. & Sander, C.(1992). Selection of representative protein data sets.Protein Sci. 1, 409±17.
Holm, L. & Sander, C. (1996). The FSSP database: foldclassi®cation based on structure-structure alignmentof proteins. Nucl. Acids Res. 24, 206±210.
Hubbard, T. J. P. & Blundell, T. L. (1987). Comparisonof solvent-inaccessible cores of homologous pro-teins: de®nitions useful for protein modelling. Pro-tein Eng. 1, 159±171.
Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydro-gen bonded and geometrical features. Biopolymers,22, 2577±2637.
Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996). Anevolutionary trace method de®nes binding surfacescommon to protein families. J. Mol. Biol. 257, 342±358.
Macias, M. J., Musacchio, A., Ponstingl, H., Nilges, M.,Saraste, M. & Oschkinat, H. (1994). Structure of thepleckstrin homology domain from b-spectrin.Nature, 369, 675±677.
Nakai, K. & Kanehisa, M. (1991). Expert system for pre-dicting protein localization sites in Gram-negativebacteria. Proteins: Struct. Funct. Genet. 11, 95±110.
Nakai, K. & Kanehisa, M. (1992). A knowledge base forpredicting protein localization sites in eukaryoticcells. Genomics, 14, 897±911.
Nakai, K., Kidera, A. & Kanehisa, M. (1988). Clusteranalysis of amino acid indices for prediction of pro-tein structure and function. Protein Eng. 2, 93±100.
Nakashima, H. & Nishikawa, K. (1994). Discriminationof intracellular and extracellular proteins usingamino acid composition and residue-pairfrequencies. J. Mol. Biol. 238, 54±61.
Nielsen, H., Engelbrecht, J., Brunak, S. & von Heijne, G.(1997). Identi®cation of prokaryotic and eukaryoticsignal peptides and prediction of their cleavagesites. Protein Eng. 10, 1±6.
Nishikawa, K., Kubota, Y. & Ooi, T. (1983a).Classi®cation of proteins into groups based onamino acid composition and other characters. I.Angular distribution. J. Biochem. (Tokyo), 94, 981±995.
524 Adaptation of Proteins to Subcellular Location

Nishikawa, K., Kubota, Y. & Ooi, T. (1983b).Classi®cation of proteins into groups based onamino acid composition and other characters. II.Grouping into four types. J. Biochem. (Tokyo), 94,997±1007.
Pfeffer, S. R. & Rotheman, J. E. (1987). Biosyntheticprotein transport and sorting by the endoplasmicreticulum and Golgi. Annu. Rev. Biochem. 56, 829±852.
Rost, B. (1997). Protein structures sustain evolutionarydrift. Fold. Des. 2, S19±S24.
Rost, B. & O'Donoghue, S. I. (1997). Sisyphus and pro-tein structure prediction. Comput. Appl. Biosci. 13,345±356.
Rost, B. & Sander, C. (1994). Conservation and predic-tion of solvent accessibility in protein families.Proteins: Struct. Funct. Genet. 20(3), 216±226.
Rost, B., Casadio, R. & Fariselli, P. (1996). Topology pre-diction for helical transmembrane proteins at 86%accuracy. Protein Sci. 5, 1704±1718.
Sander, C. & Schneider, R. (1991). Database of hom-ology-derived structures and the structural meaningof sequence alignment. Proteins: Struct. Funct. Genet.9, 56±68.
Sander, C. & Schneider, R. (1994). The HSSP database ofprotein structure ± sequence alignments. Nucl. AcidsRes. 22, 3597±3599.
SjoÈ stroÈm, M., Wold, S., Wieslander, AÊ . & Rilfors, L.(1987). Signal peptide amino acid sequences in
Escherichia coli contain information related to ®nalprotein localization. EMBO J. 6, 823±831.
Verner, K. & Schatz, G. (1988). Protein translocationacross membranes. Science, 241, 1307±1313.
von Heijne, G. (1985). Signal sequences. The limits ofvariation. J. Mol. Biol. 184, 99±105.
Wagh, P. V. & Bahl, O. P. (1981). Sugar residues onproteins. CRC Crit. Rev. Biochem. 10, 303±377.
Edited by F. E. Cohen
(Received 5 September 1997; received in revised form24 October 1997; accepted 24 October 1997)
http://www.hbuk.co.uk/jmb
Supplementary material for this paper is availablefrom JMB Online.
Adaptation of Proteins to Subcellular Location 525





























