Embed Size (px)
Citation preview

ORIGINAL PAPER
A study of the rotational barriers for some organic compoundsusing the G3 and G3CEP theories
Douglas Henrique Pereira & Lucas Colucci Ducati &Roberto Rittner & Rogério Custodio
Received: 20 December 2013 /Accepted: 2 March 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract The G3, G3CEP, MP4, MP4CEP, QCISD(T), andQCISD(T)CEP methods were applied to study 43 internalrotational barriers of different molecules. The calculated G3and G3CEP barriers were accurate with respect to thoseobtained experimentally, typically showing deviations of<0.50 kcal mol−1. The results for the MP4CEP, MP4,QCISD(T), and QCISD(T)CEP calculations were less accu-rate, and larger deviations of approximately ±1 kcal mol−1
were observed. The accuracy of G3CEP was comparable tothat of G3, but a reduction in CPU time of between 5 and 35%was observed when the dependence of the pseudopotentialson the size of the molecule and atom type was taken intoaccount. The behaviors of the energy components show thatthese corrections depend on the molecular environment andwhether the calculations are performed with all electrons orpseudopotentials. Usually, the predominance of a specificeffect follows a distinct pattern when the G3 and G3CEPresults are compared. For the G3 calculations, the most im-portant component of the corrected MP4/6-31G(d) rotationalenergy is ΔE2df,p. Among the 43 molecules, 29 were depen-dent on polarization effects, ΔE2df,p; 19 were dependent on
diffuse functions, ΔE+; and 13 depended on the effects ofmore elaborate basis functions (ΔEG3large). Similar behaviorwas observed for the G3CEP calculations: polarization effectswere more important for 25 molecules, followed closely bythe effect of diffuse functions for 23 molecules, and finally theeffect of large basis sets (19 molecules). ΔEQCI correctionseldom resulted in significant effects on the G3 and G3CEPcalculations.
Keywords Rotational barrier . G3 theory . G3CEP theory .
Electronic effects . Pseudopotential . Compoundmethods
Introduction
Experimental and theoretical studies of internal rotation bar-riers are important for understanding the structural and dy-namic properties of molecules of various types and sizes[1–4]. There is constant interest in the conformational equi-libria of molecules, as demonstrated by the large numbers ofexperimental and theoretical studies performed in this field.Glycine, the simplest amino acid in nature, can be cited as anexample of where a lack of consensus in the literature on themost stable rotamer in solution or the gas phase and the forcesthat govern its stabilization has prompted multiple efforts toelucidate it [5, 6].
Among the various types of theoretical calculations that areapplied to determine thermochemical properties, compositemethods are those that incur a much lower computational costthan high-level ab initio calculations by performing combina-tions of different ab initio calculations, but are still sufficientlyaccurate [7–9]. Such composite methods include the family ofGaussian-n theories, Gn (n=1, 2, 3, 4) [10–19], the completebasis set methods, CBS [20–25], and the Weizmann-n theo-ries, Wn (n=1, 2, 3, 4) [26–28]. The thermochemical valuesobtained by applying any of these methods are usually
Electronic supplementary material The online version of this article(doi:10.1007/s00894-014-2199-3) contains supplementary material,which is available to authorized users.
D. H. Pereira : R. Rittner :R. Custodio (*)Instituto de Química, Universidade Estadual de Campinas, BarãoGeraldo, 13083-970 Campinas, São Paulo, BrazilP. O. Box 6154e-mail: [email protected]
D. H. PereiraDepartamento de Ciências Exatas e Biotecnológicas, UniversidadeFederal do Tocantins, Campus de Gurupi, 77410-530 Gurupi,Tocantins, Brazil
L. C. DucatiInstituto de Química, Universidade de São Paulo, 05508-900 SãoPaulo, São Paulo, BrazilP. O. Box 26077
J Mol Model (2014) 20:2199DOI 10.1007/s00894-014-2199-3

accurate to within 1–2 kcal mol−1. When a large computation-al effort is required to accomplish the calculations, the size ofthe system to be treated is decreased.
A recent alternative that allows an extension of G3 theory[12] to larger molecules or a reduction in the computationaleffort combines pseudopotentials developed by Stevens,Basch, and Krauss (known as compact effectivepseudopotentials, CEPs [29–31]) with the G3 theory, resultingin a method referred to as G3CEP [4, 32]. This method wasused to study 446 chemical species containing atoms of ele-ments from the first and second periods and those of repre-sentative elements from the third period of the periodic table.Five properties were studied: enthalpy of formation, atomiza-tion energy, electron affinity, ionization potential, and protonaffinity, and the results showed a high accuracy, with devia-tions of 1.29 kcal mol−1 for G3CEP and 1.16 kcal mol−1 forG3 from the measured values. The main advantage of G3CEPas compared to G3 is its reduced computational cost—be-tween 7 % and 70 %, depending on the size of the moleculeand types of atoms in it [4, 32].
Unlike the applications described above, only a few reportsin the literature describe the application of these methods toother properties, such as rotational barriers. In this context, wemention the work of Murcko et al. [33], in which the G2 andCBS-Q theories were applied to study internal barriers for therotamers butane, 1-butene, and 1,3-butadiene. The resultsshowed that these theories led to excellent descriptions ofthe systems, except for butane, which showed a greaterdeviation between the calculated and experimental data[33]. In another paper, Ducati et al. [4] used the G3theory to explore the rotational barriers of some simplemolecules (H2O2, H2S2, N2H4, CH3OH, CH3NH2, andC2H6). The results showed deviations of internal rotationalbarriers of less than 0.5 kcal mol−1 from the correspond-ing experimental data. In that paper, different componentsof the G3 energies were used to qualitatively and quanti-tatively evaluate the importance of each specific effectconsidered in the calculation of the rotational barriers, aswell as trends in the patterns of the barriers based on theelectronic neighborhood [4].
The need for accurate calculations of rotational barriers,and the potential use of G3 theory and the recent modificationof it to use pseudopotentials (G3CEP) to calculate them, led usto investigate 43 rotational barriers of molecules—mostlyorganic compounds—to compare the performances of thesetwo methods and the MP4 calculations from which they arederived. That work is reported in the present paper. Once theG3 and G3CEP theories have been used to identify effectsassociated with the basis functions and electron correlationeffects, they can be used to analyze which stereoelectroniceffects are responsible for the most stable conformations, andto determine which of the components contribute most to theheight of the rotational barrier.
Computational procedures: the G3CEP theory
Calculations involving G3 theory are well known and docu-mented in the literature [12], so they are not presented here.The details of the G3CEP theory have also been described inrecent papers [32, 34], so only the main steps will be summa-rized in this manuscript.
The G3CEP energy has the same composition as in the G3theory, and is defined by the equation
EG3CEP ¼ E MP4=CEP−P31G dð Þ½ � þΔEþ þΔE2d;p
þΔEQCI þΔEG3Large þΔESO þΔEHLC
þΔEZPE ð1Þ
The components of Eq. 1 are corrections made to the MP4energy obtained using pseudopotentials along with the CEP-P31G(d) basis function, E[MP4/CEP-P31G(d)].
One of the most important steps in the G3CEP calculationis the selection of the basis set. The G3 theory uses thefollowing basis functions: 6-31G(d), 6-31+G(d), 6-31G(2df,p), and G3large. The basis functions selected forG3CEP are the same valence functions used in the originalG3 theory, namely, P31G(d), P31+G(d), and P31G(2df,p).The letter P emphasizes that Pople’s valence basis set is used.The adjustment of these all-electron basis sets to thepseudopotential environment corresponds to the eliminationof the innermost functions. For the G3large basis functions,only the innermost contracted set is removed; all other Gauss-ian functions are preserved. The contraction coefficients in allthe valence basis functions are the same as the original ones.Therefore, in all of the calculations performed withpseudopotentials, only the valence electrons are considered,and the basis functions are those from the valence all-electronbasis set. These bases are denoted CEP-P31G(d), CEP-P31+G(d), CEP-P31G(2df,p), and G3CEPlarge in the presentwork.
For the G3CEPlarge functions, further adjustment wasnecessary to improve the accuracy of the calculated properties.The s and p basis functions for the N, O, F, P, Cl, and C atomswere scaled using the following parameters: ζC=0.9839, ζN=0.9639, ζO=0.9349, ζF=0.9222, ζP=0.8146, and ζCl=1.0154. As described previously [32, 34], these parametersmust be squared and multiplied by all of the s and p Gaussianexponents from G3CEPlarge.
The other steps involved in the calculation of EG3CEP are asfollows:
Step 1 An initial equilibrium structure is obtained at theHar t ree–Fock (HF) level us ing the CEPpseudopotential and the P31G(d) basis functions,HF/CEP-P31G(d).
2199, Page 2 of 14 J Mol Model (2014) 20:2199

Step 2 The molecular structure obtained in step 1 is used tocalculate the vibrational frequencies and the zeropoint energy (ZPE), EZPE. The ZPE energy is scaledby a factor of 0.8929. The thermal effects are alsocalculated (ΔEZPE).
Step 3 The equilibrium geometries are refined byperforming a new optimization at the MP2/CEP-P31G(d) level. The optimized geometry obtained inthis step is used for all subsequent steps.
Step 4 Corrections to the calculation:
(a) Diffuse functions:
ΔEþ ¼ E MP4=CEP−P31þ G dð Þ½ �−E MP4=CEP−P31G dð Þ½ �
ð2Þ
(b) Polarization functions:
ΔE2df ;p ¼ E MP4=CEP−P31G 2df ; pð Þ½ �−E MP4=CEP−P31G dð Þ½ �
ð3Þ
(c) Higher-order correlation effects:
ΔEQCI ¼ E QCISD Tð Þ=CEP−P31G dð Þ½ �−E MP4=CEP−P31G dð Þ½ �
ð4Þ
(d) Energy due to the effects of using large basis sets,and nonadditivity caused by the difference betweenthe basis sets:
ΔEG3large ¼ E MP2=G3CEPlarge½ �−E½MP2=CEP−P31G 2df ; pð Þ�−E½MP2=CEP−P31þ G dð Þ�þ E½MP2=CEP−P31G dð Þ�
ð5Þ
Step 5 Sum all components according to Eq. 1 to obtainEG3CEP
When calculating rotational barriers, the contributions ofΔEHLC and ΔESO are assumed to be constant and are there-fore not included in the determination of the relative G3 andG3CEP energies. In the G3 theory, the final energy achievedby combining the different ab initio contributions is compara-ble to a QCISD(T,full)/G3large calculation.
All calculations were performed using the Gaussian 09software package [35].
Results and discussion
The G3 and G3CEP theories were applied to calculate therotational barriers of 43 molecules. The molecules, as well as
their rotational angles, are shown in Fig. 1. For all of themolecules, the torsion angle ranged between 0° and 180° inintervals of 10°. The experimental and calculated values of therelative rotation barriers are shown in Table 1. Each theoreticalvalue is presented as the difference between the correspondingexperimental and calculated values, ΔEtheor=Eexp−Etheor. Asshown in Table 1, each molecule was identified by a uniqueinteger (see the first column). The molecular formula of eachmolecule investigated is described in the second column of thetable, the third column contains the experimental barrier, thefourth and fifth columns are the G3CEP and G3 results (notincluding the zero point energy), respectively, the sixth andseventh columns are the MP4/CEP-P31G(d) and MP4/6-31G(d) calculations, the eighth and ninth columns are theG3CEP and G3 data, the tenth and eleventh columns are theQCISD(T)/CEP-P31G(d) and QCISD(T)/6-31G(d) results,and the last column indicates the reference works from whichthe experimental data were obtained. The last two rows ofTable 1 show the values of the mean absolute deviation,
calculated using the equation δi; j ¼ ∑k¼1
N
xik−xjk��
��
!
=N ,
and the Euclidean distance [36], calculated as di; j ¼ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
∑k¼1
N
xik−xjk� �
s
2, where xik is the experimental data and xjk
is the corresponding theoretical result. The mean absolutedeviation and the Euclidean distance demonstrate that theG3 and G3CEP results that do not include the ZPE are moreaccurate than the results of the G3 and G3CEP and the MP4and MP4CEP calculations with respect to the experimentaldata. A comparison of the G3, G3CEP, andMP4 results showsthat they are similar. The application of G3CEP and G3, whenZPE is not included, leads to the smallest mean absolutedeviations (approximately 0.5 kcal mol−1). Including theZPE correction in the original G3 and G3CEP theories yieldsslightly larger mean deviations of approximately0.6 kcal mol−1. The MP4 and MP4CEP calculations presentdeviations of approximately 0.8 kcal mol−1, which are verysimilar to those afforded by the QCISD(T)/CEP-P31G(d) andQCISD(T)/6-31G(d) results.
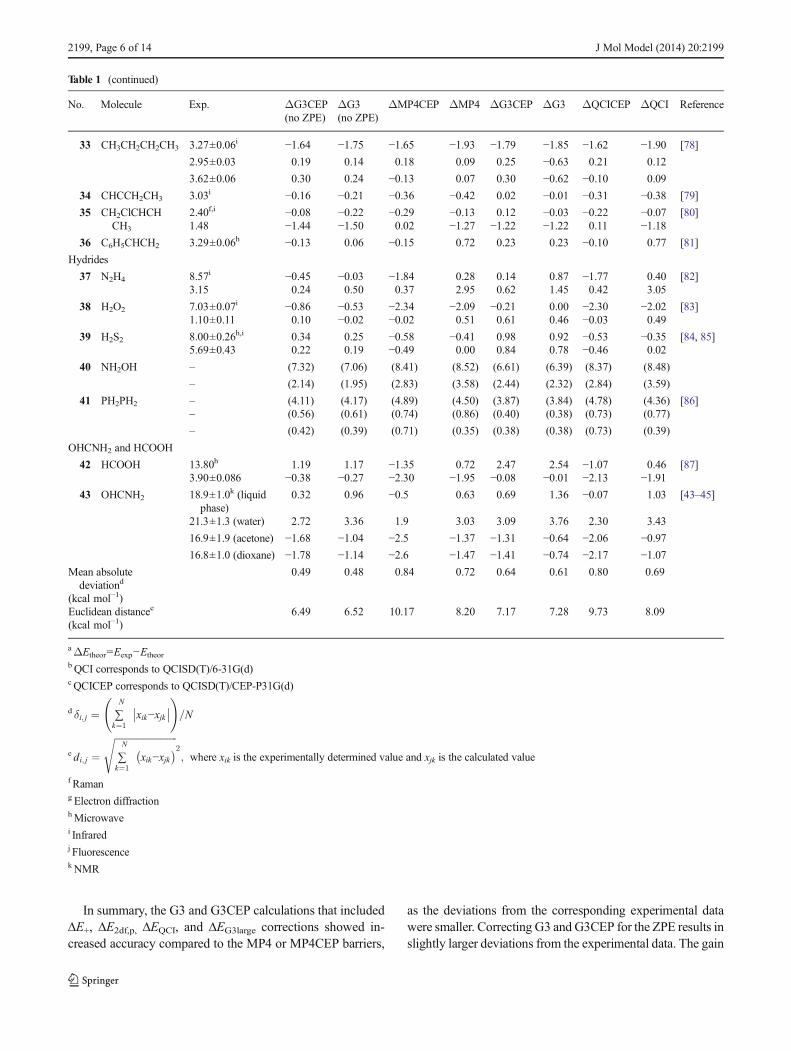
Histograms of the deviations of the calculated resultswith respect to the experimental data for the methodsdiscussed in Table 1 are shown in Fig. 2. Most of theG3 and G3CEP energies (not including ZPE) are scatteredin the range of ±0.5 kcal mol−1 (see Fig. 2a and b),whereas the MP4CEP and MP4 deviations are concentrat-ed in the range of ±1.0 kcal mol−1 (Fig. 2c and d). TheQCISD(T)/CEP-P31G(d) and QCISD(T)/6-31G(d) histo-grams are similar to the MP4CEP and MP4 histograms,respectively. Including the ZPE in the original G3 andG3CEP theories systematically shifts the barriers to devi-ations of between 0 and 1 kcal mol−1 (see Fig. 2e and f).
J Mol Model (2014) 20:2199 Page 3 of 14, 2199

B BCl
Cl
Cl
ClB B
F
F
F
FC OH
H
H
H C C
H
OH
HH
H
H
1) Diboron tetrachloride2) Diboron tetrafluoride
3) methanol 4) ethanol
C CH
H
H
OH
H
C
H
H
H
OH
H3C O CH3C CN O C
H
H
H
H
H
5) propan-2-ol 6) phenol 7) dimethyl ether 8) methoxyacetonitrile
C CH
OO
HH3C C
H
OCH2 C
H
OH3CC
HO
9) oxaldehyde 10) acetaldehyde 11) propanal 12) benzaldehyde
O NO
OCH2CH3 C C
H
H
HH
H
H C C
H
H
HF
H
H C C
H
H
HF
F
H
13) ethyl nitrate 14) ethane 15) fluoroethane 16) 1,1-difluoroethane
C C
H
H
HF
F
F C C
F
F
FF
F
F C C
H
H
HCl
H
H C C
H
H
HCl
Cl
H
17)1,1,1-trifluoroethane
18)hexafluoroethane
19) chloroethane20)
1,1-dichloroethane
C C
H
H
ClCl
H
H C C
H
H
HCl
Cl
Cl H3C COH
OC NH2
H
H
H
21) 1,2-dichloroethane22) 1,1,1-
trichloroethane 23) acetic acid 24) methanamine
C N O
H
H
H H3C PH
HC Si H
H
H
H
H
HN CH3
H
H3C
25)nitrosomethane
26) methylphosphine
27)methyl silane
28)N,N-dimethylamine
C CC
H
H
HH
H
H
C CH3
H
CO
C C HH3C
H
O
H
N CH3
CH3
H3C
29) propene30)
prop-1-en-1-one31)
2-methyloxirane32)
N,N,N-trimethylamine
C CH
H
H
H
H
C
H
H
C
H
H
H
C CH3
H
H
CHC C CH
Cl
H
H
C C
H
H
H
H CCH2H
33) butane 34) but-1-yne35)
(2E)-1-chlorobut-2-ene36) vinylbenzene
N NH
H
H
H
O O
H H
S S
H HHO N
H
H
37) hydrazine38)
hydrogen peroxide39) disulfane 40) hydroxylamine
P PH
H
H
HH C
OH
OC N
H
HO
H
41) diphosphane 42) formic acid 43) formamide
Fig. 1 Molecules studied andtheir respective rotational angles
2199, Page 4 of 14 J Mol Model (2014) 20:2199
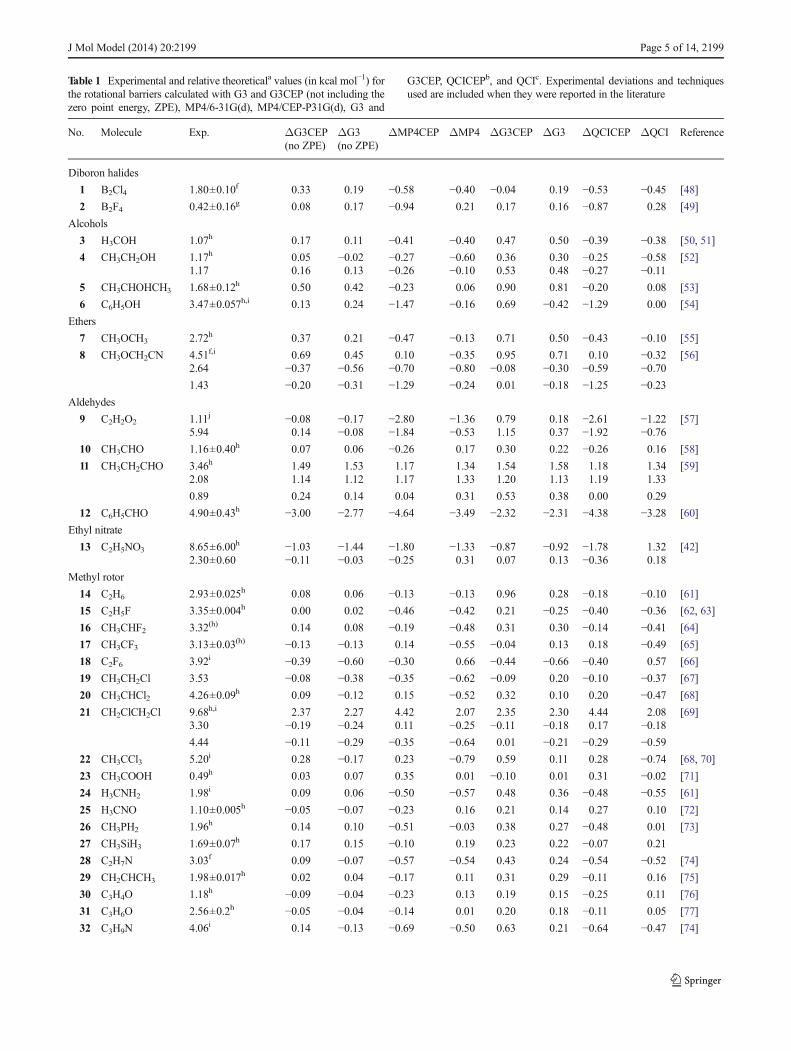
Table 1 Experimental and relative theoreticala values (in kcal mol−1) forthe rotational barriers calculated with G3 and G3CEP (not including thezero point energy, ZPE), MP4/6-31G(d), MP4/CEP-P31G(d), G3 and
G3CEP, QCICEPb, and QCIc. Experimental deviations and techniquesused are included when they were reported in the literature
No. Molecule Exp. ΔG3CEP(no ZPE)
ΔG3(no ZPE)
ΔMP4CEP ΔMP4 ΔG3CEP ΔG3 ΔQCICEP ΔQCI Reference
Diboron halides
1 B2Cl4 1.80±0.10f 0.33 0.19 −0.58 −0.40 −0.04 0.19 −0.53 −0.45 [48]
2 B2F4 0.42±0.16g 0.08 0.17 −0.94 0.21 0.17 0.16 −0.87 0.28 [49]
Alcohols
3 H3COH 1.07h 0.17 0.11 −0.41 −0.40 0.47 0.50 −0.39 −0.38 [50, 51]
4 CH3CH2OH 1.17h 0.05 −0.02 −0.27 −0.60 0.36 0.30 −0.25 −0.58 [52]1.17 0.16 0.13 −0.26 −0.10 0.53 0.48 −0.27 −0.11
5 CH3CHOHCH3 1.68±0.12h 0.50 0.42 −0.23 0.06 0.90 0.81 −0.20 0.08 [53]
6 C6H5OH 3.47±0.057h,i 0.13 0.24 −1.47 −0.16 0.69 −0.42 −1.29 0.00 [54]
Ethers
7 CH3OCH3 2.72h 0.37 0.21 −0.47 −0.13 0.71 0.50 −0.43 −0.10 [55]
8 CH3OCH2CN 4.51f,i 0.69 0.45 0.10 −0.35 0.95 0.71 0.10 −0.32 [56]2.64 −0.37 −0.56 −0.70 −0.80 −0.08 −0.30 −0.59 −0.701.43 −0.20 −0.31 −1.29 −0.24 0.01 −0.18 −1.25 −0.23
Aldehydes
9 C2H2O2 1.11j −0.08 −0.17 −2.80 −1.36 0.79 0.18 −2.61 −1.22 [57]5.94 0.14 −0.08 −1.84 −0.53 1.15 0.37 −1.92 −0.76
10 CH3CHO 1.16±0.40h 0.07 0.06 −0.26 0.17 0.30 0.22 −0.26 0.16 [58]
11 CH3CH2CHO 3.46h 1.49 1.53 1.17 1.34 1.54 1.58 1.18 1.34 [59]2.08 1.14 1.12 1.17 1.33 1.20 1.13 1.19 1.33
0.89 0.24 0.14 0.04 0.31 0.53 0.38 0.00 0.29
12 C6H5CHO 4.90±0.43h −3.00 −2.77 −4.64 −3.49 −2.32 −2.31 −4.38 −3.28 [60]
Ethyl nitrate
13 C2H5NO3 8.65±6.00h −1.03 −1.44 −1.80 −1.33 −0.87 −0.92 −1.78 1.32 [42]2.30±0.60 −0.11 −0.03 −0.25 0.31 0.07 0.13 −0.36 0.18
Methyl rotor
14 C2H6 2.93±0.025h 0.08 0.06 −0.13 −0.13 0.96 0.28 −0.18 −0.10 [61]
15 C2H5F 3.35±0.004h 0.00 0.02 −0.46 −0.42 0.21 −0.25 −0.40 −0.36 [62, 63]
16 CH3CHF2 3.32(h) 0.14 0.08 −0.19 −0.48 0.31 0.30 −0.14 −0.41 [64]
17 CH3CF3 3.13±0.03(h) −0.13 −0.13 0.14 −0.55 −0.04 0.13 0.18 −0.49 [65]
18 C2F6 3.92i −0.39 −0.60 −0.30 0.66 −0.44 −0.66 −0.40 0.57 [66]
19 CH3CH2Cl 3.53 −0.08 −0.38 −0.35 −0.62 −0.09 0.20 −0.10 −0.37 [67]
20 CH3CHCl2 4.26±0.09h 0.09 −0.12 0.15 −0.52 0.32 0.10 0.20 −0.47 [68]
21 CH2ClCH2Cl 9.68h,i 2.37 2.27 4.42 2.07 2.35 2.30 4.44 2.08 [69]3.30 −0.19 −0.24 0.11 −0.25 −0.11 −0.18 0.17 −0.184.44 −0.11 −0.29 −0.35 −0.64 0.01 −0.21 −0.29 −0.59
22 CH3CCl3 5.20i 0.28 −0.17 0.23 −0.79 0.59 0.11 0.28 −0.74 [68, 70]
23 CH3COOH 0.49h 0.03 0.07 0.35 0.01 −0.10 0.01 0.31 −0.02 [71]
24 H3CNH2 1.98i 0.09 0.06 −0.50 −0.57 0.48 0.36 −0.48 −0.55 [61]
25 H3CNO 1.10±0.005h −0.05 −0.07 −0.23 0.16 0.21 0.14 0.27 0.10 [72]
26 CH3PH2 1.96h 0.14 0.10 −0.51 −0.03 0.38 0.27 −0.48 0.01 [73]
27 CH3SiH3 1.69±0.07h 0.17 0.15 −0.10 0.19 0.23 0.22 −0.07 0.21
28 C2H7N 3.03f 0.09 −0.07 −0.57 −0.54 0.43 0.24 −0.54 −0.52 [74]
29 CH2CHCH3 1.98±0.017h 0.02 0.04 −0.17 0.11 0.31 0.29 −0.11 0.16 [75]
30 C3H4O 1.18h −0.09 −0.04 −0.23 0.13 0.19 0.15 −0.25 0.11 [76]
31 C3H6O 2.56±0.2h −0.05 −0.04 −0.14 0.01 0.20 0.18 −0.11 0.05 [77]
32 C3H9N 4.06i 0.14 −0.13 −0.69 −0.50 0.63 0.21 −0.64 −0.47 [74]
J Mol Model (2014) 20:2199 Page 5 of 14, 2199
In summary, the G3 and G3CEP calculations that includedΔE+, ΔE2df,p, ΔEQCI, and ΔEG3large corrections showed in-creased accuracy compared to the MP4 or MP4CEP barriers,
as the deviations from the corresponding experimental datawere smaller. Correcting G3 and G3CEP for the ZPE results inslightly larger deviations from the experimental data. The gain
Table 1 (continued)
No. Molecule Exp. ΔG3CEP(no ZPE)
ΔG3(no ZPE)
ΔMP4CEP ΔMP4 ΔG3CEP ΔG3 ΔQCICEP ΔQCI Reference
33 CH3CH2CH2CH3 3.27±0.06i −1.64 −1.75 −1.65 −1.93 −1.79 −1.85 −1.62 −1.90 [78]
2.95±0.03 0.19 0.14 0.18 0.09 0.25 −0.63 0.21 0.12
3.62±0.06 0.30 0.24 −0.13 0.07 0.30 −0.62 −0.10 0.09
34 CHCCH2CH3 3.03i −0.16 −0.21 −0.36 −0.42 0.02 −0.01 −0.31 −0.38 [79]
35 CH2ClCHCHCH3
2.40f,i −0.08 −0.22 −0.29 −0.13 0.12 −0.03 −0.22 −0.07 [80]1.48 −1.44 −1.50 0.02 −1.27 −1.22 −1.22 0.11 −1.18
36 C6H5CHCH2 3.29±0.06h −0.13 0.06 −0.15 0.72 0.23 0.23 −0.10 0.77 [81]
Hydrides
37 N2H4 8.57i −0.45 −0.03 −1.84 0.28 0.14 0.87 −1.77 0.40 [82]3.15 0.24 0.50 0.37 2.95 0.62 1.45 0.42 3.05
38 H2O2 7.03±0.07i −0.86 −0.53 −2.34 −2.09 −0.21 0.00 −2.30 −2.02 [83]1.10±0.11 0.10 −0.02 −0.02 0.51 0.61 0.46 −0.03 0.49
39 H2S2 8.00±0.26h,i 0.34 0.25 −0.58 −0.41 0.98 0.92 −0.53 −0.35 [84, 85]5.69±0.43 0.22 0.19 −0.49 0.00 0.84 0.78 −0.46 0.02
40 NH2OH – (7.32) (7.06) (8.41) (8.52) (6.61) (6.39) (8.37) (8.48)
– (2.14) (1.95) (2.83) (3.58) (2.44) (2.32) (2.84) (3.59)
41 PH2PH2 – (4.11) (4.17) (4.89) (4.50) (3.87) (3.84) (4.78) (4.36) [86]– (0.56) (0.61) (0.74) (0.86) (0.40) (0.38) (0.73) (0.77)
– (0.42) (0.39) (0.71) (0.35) (0.38) (0.38) (0.73) (0.39)
OHCNH2 and HCOOH
42 HCOOH 13.80h 1.19 1.17 −1.35 0.72 2.47 2.54 −1.07 0.46 [87]3.90±0.086 −0.38 −0.27 −2.30 −1.95 −0.08 −0.01 −2.13 −1.91
43 OHCNH2 18.9±1.0k (liquidphase)
0.32 0.96 −0.5 0.63 0.69 1.36 −0.07 1.03 [43–45]
21.3±1.3 (water) 2.72 3.36 1.9 3.03 3.09 3.76 2.30 3.43
16.9±1.9 (acetone) −1.68 −1.04 −2.5 −1.37 −1.31 −0.64 −2.06 −0.9716.8±1.0 (dioxane) −1.78 −1.14 −2.6 −1.47 −1.41 −0.74 −2.17 −1.07
Mean absolutedeviationd
(kcal mol−1)
0.49 0.48 0.84 0.72 0.64 0.61 0.80 0.69
Euclidean distancee
(kcal mol−1)6.49 6.52 10.17 8.20 7.17 7.28 9.73 8.09
aΔEtheor=Eexp−Etheorb QCI corresponds to QCISD(T)/6-31G(d)c QCICEP corresponds to QCISD(T)/CEP-P31G(d)
d δi; j ¼ ∑k¼1
N
xik−xjk��
��
!
=N
e di; j ¼ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi
∑k¼1
N
xik−xjk� �
s
2; where xik is the experimentally determined value and xjk is the calculated value
f Ramang Electron diffractionhMicrowavei Infraredj FluorescencekNMR
2199, Page 6 of 14 J Mol Model (2014) 20:2199
in accuracy obtained when using G3 or G3CEP is obtained atthe expense of a significant increase in CPU time. This is theprice to be paid in any situation where greater accuracy isrequired. By using pseudopotentials with the G3 theory,G3CEP reduces the computational cost while preserving theaccuracy of the calculations. In this work, the reduction in theCPU time obtained when using G3CEP rather than G3 toperform a full set of calculations with identical initial
conditions, a fixed dihedral angle, and using a single processor(Intel Core2 Quad Q6600) was between 5 % and 35 %. Thelargest relative decreases in CPU time occurred in the stepsinvolving frequency and MP2(full) calculations. The MP4and QCI steps did not present substantial decreases in therelative CPU time when G3CEP was used.
According to Eq. 1, additional information can be obtainedfrom the G3 and G3CEP theories because the final energy
-4 -3 -2 -1 0 1 2 3 40
5
10
15
20
25
30
(f)
Num
ber o
f mol
ecul
es
Deviation / kcal mol-1
0
5
10
15
20
25
30
0
5
10
15
20
25
30
(d)
0
5
10
15
20
25
30
(c)
0
5
10
15
20
25
30
(b)
(a)
(e)
0
5
10
15
20
25
30
G3CEP
G3
MP4CEP
MP4
G3CEP (no ZPE)
G3 (no ZPE)
Fig. 2a–f Histograms of thedeviations (from thecorresponding experimentalvalues) of the internal rotationalbarriers (in kcal mol−1) calculatedusing a G3CEP not includingZPE, b all-electron G3 theory notincluding ZPE, c MP4CEP, dMP4, e G3CEP, and f G3
J Mol Model (2014) 20:2199 Page 7 of 14, 2199

corresponds to the effects of correcting the E[MP4/CEP-P31G(d)] energy. To explore the possible trends associatedwith the G3 andG3CEP energy components, the 43moleculeswere categorized according to their functional groups and/orspecific properties.
Diboron halides
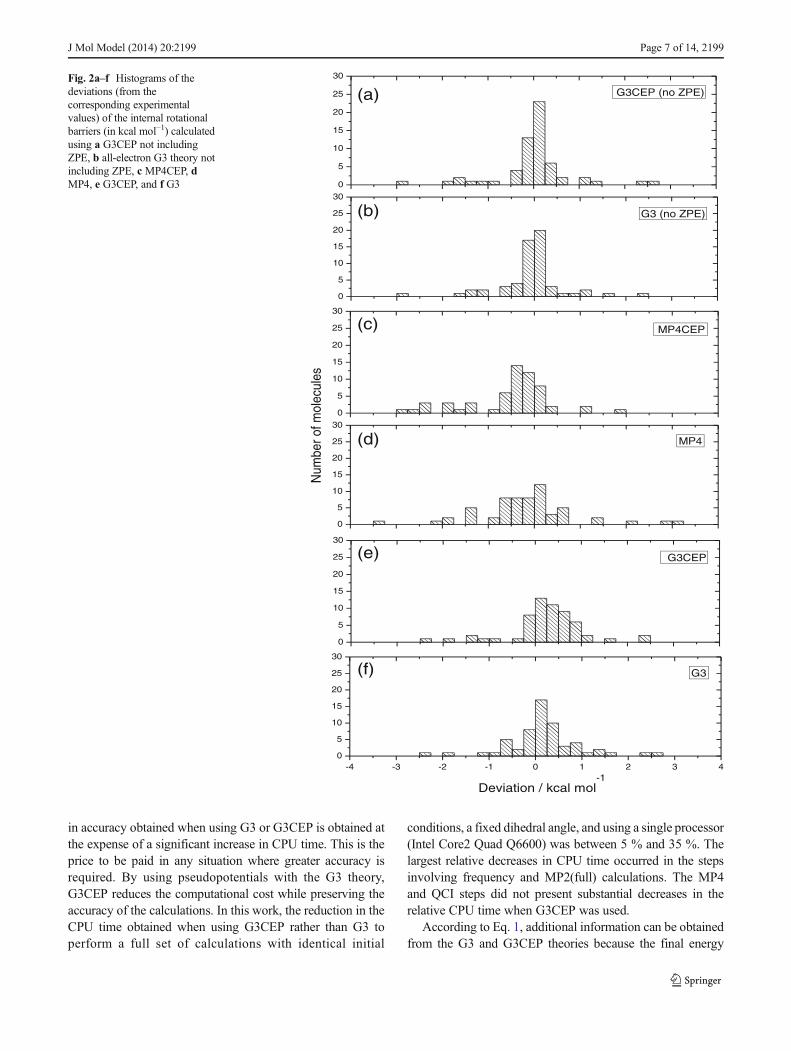
The reference dihedral angle for diboron halides and all of themolecules in this work corresponds to 0°. Figure 3 shows therotational barriers and the relative energy components for bothmolecules. Figures 3a and b show the experimental and the-oretical rotational barriers calculated with G3 and G3CEP forB2Cl2 and B2F2, respectively. Despite the fact that the mole-cules B2F4 and B2Cl4 present similar valence electronic struc-tures, their rotational barriers show contrasting trends. B2F4has energy minima at 0° and 180°, whereas B2Cl4 presentsenergy maxima at these angles, while its minimum is locatedat 90°. A π bond between the boron and fluorine, which doesnot occur between boron and chlorine, is responsible for thedifferent equilibrium conformations of B2F4 and B2Cl4 [37].
Quantitatively, as shown in Table 1, the experimental bar-rier of B2Cl4 (1.80 kcal mol−1) is almost four times higher thanthat of B2F4 (0.42 kcal mol−1). Theoretically, the observeddeviations of the G3 and G3CEP results for B2Cl4 from theexperimental values increased the barriers by 0.19 and0.33 kcal mol−1, respectively, whereas the MP4 and MP4CEPcalculations decreased the barriers by −0.40 and−0.58 kcal mol−1, respectively. For B2F4, a similar trend isobserved, with small increases in the barriers calculated at theG3 and G3CEP levels of theory, 0.17 and 0.08 kcal mol−1,respectively, compared to 0.21 and −0.94 kcal mol−1 for MP4and MP4CEP. As expected, the deviations from the experi-mental data are less for the G3 and G3CEP results. Performingthe ZPE correction on the G3 and G3CEP results tends to leadto similar deviations to those calculated using uncorrected G3and G3CEP.
Because the G3 and G3CEP energies are combinations ofdifferent contributions added to the MP4/6-31G(d) or MP4/CEP-P31G(d) energy, it is possible to evaluate the importanceof each correction to the rotational barrier. Figures 3c and dshow the profiles of the components of the G3 energies ofB2Cl4 and B2F4, respectively. The proximity of theMP4 curverelative to the G3 calculation highlights the good performanceof MP4 in calculating the barriers of these two compounds. Inthe case of B2Cl4, Fig. 3c shows thatΔE+ almost cancels outthe effects of ΔEG3large. The accuracy of the MP4 energy ismainly due to the effect of the polarization function, ΔE2df,p.ΔEQCI is nearly zero. In the case of B2F4, Fig. 3d shows thatthe G3 barrier is significantly smaller than the B2Cl4 barrier,and that all components are equally important to the accuracyof the MP4 energy. Whereas theΔE2df,p andΔE+ corrections
tend to increase the barrier, ΔEG3large and ΔEQCI tend toreduce it.
The behavior of the same components in the G3CEPcalculations for the same molecules can be seen in Figs. 3eand f (B2Cl4 and B2F4, respectively). A comparison betweenFigs. 3c and e shows that the B2Cl4 barrier preserves the sameprofile but that the energy components differ significantly.The trend for the MP4 calculation shows a barrier with oppo-site behavior to that seen for the G3CEP profile. Correctionsdue to theΔEG3large,ΔE2df,p, andΔE+ components determinethe behavior of the barrier. ΔEQCI also appears to have noeffect on the barrier height. For B2F4, a comparison betweenFig. 3d and f allows us to check if the same qualitativebehavior is seen whether pseudopotentials are used or not.ΔEG3large decreases the barrier height whereas ΔE2df,p andΔE+ have the opposite effect. The ΔEQCI component has anegligible effect on the G3CEP barrier (Fig. 3f) with respect tothe all-electron calculations (Fig. 3d).
In general, these two examples show that the barriersobtained from pseudopotential and all-electron calculationscan be similar, but that the components that make up the G3or G3CEP energies can have different effects on the barrierheight.
Alcohols
The alcohols listed in Table 1 are those for which the rotationalbarrier to –OH torsion was studied. Methanol, ethanol,propan-2-ol, and phenol (compounds 3–6) are shown inFig. 1. The experimental internal rotation barriers reported inthe literature for these alcohols [39, 50, 58, 59] increase as thecarbon chain increases in length, illustrating the effect ofneighboring groups on the rotational barrier. The largest bar-rier seen among the alcohols is that of phenol, which can beexplained by the combination of the nonbinding electron pairof the oxygen and the π-aromatic system (nO→π*C–C) [2].The deviations from the experimental data of the G3CEP andG3 calculations for this family of substances are smaller than0.50 kcal mol−1. The deviations for the MP4 and MP4CEPcalculations are usually larger (see Table 1).
The profiles of the G3 and G3CEP rotational barriers showthat, for alcohols, the most stable conformers (synclinal andantiperiplanar) have dihedral angles of 60° and 180° (seeFigs. 4a and S2). Additionally, all of the barriers for this groupoccur between the two energy maxima at 0° and 120° thatseparate the synperiplanar and anticlinal rotamers (Figs. 4aand S2). The profile of the rotational barrier of phenol, asshown in Fig. 4b, is different from those of the other alcohols,as it presents an energy maximum at 90° and a minimumwhen the hydroxyl group is in the plane of the ring.
Figure 4c and e show the barriers and energy componentsfor propan-2-ol, as calculated with G3 and G3CEP, respec-tively. The other alcohols present the same behavior (Fig. S2),
2199, Page 8 of 14 J Mol Model (2014) 20:2199
with the MP4 energy always being greater than the G3 energy,and the most important correction to the MP4 energy is that
provided byΔE+. The relative sizes of the energy componentscan change depending on whether the calculations are
B2Cl4 B2F4
0 20 40 60 80 100 120 140 160 180-2.0
-1.8
-1.6
-1.4
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.0
0.2
E /
kcal
mol
-1
Dihedral Angle Cl-B-B-Cl /degrees
G3G3CEP EXP
(a)
0 20 40 60 80 100 120 140 160 180
0.0
0.1
0.2
0.3
0.4
Dihedral Angle F-B-B-F / degrees
E / k
cal m
ol-1
G3G3CEP EXP
(b)
0 20 40 60 80 100 120 140 160 180-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
Dihedral Angle Cl-B-B-Cl /degrees
E /
kcal
mol
-1
MP4
G3
+2df,pG3LargeQCI
(c)
0 20 40 60 80 100 120 140 160 180-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
E / k
cal m
ol-1
Dihedral Angle F-B-B-F /degrees
MP4G3+2df,pG3LargeQCI
(d)
0 20 40 60 80 100 120 140 160 180
-4.0
-3.0
-2.0
-1.0
0.0
1.0
2.0
3.0
Dihedral Angle Cl-B-B-Cl / degrees
E /
kcal
mol
-1
MP4G3CEP+2df,pG3LargeQCI
(e)
0 20 40 60 80 100 120 140 160 180
-2.0
-1.0
0.0
1.0
E /
kca
l mol
-1
Dihedral Angle F-B-B-F / degrees
MP4G3CEP+2df,pG3LargeQCI
(f)
Fig. 3a–f Theoretical and experimental relative energies as a function of the dihedral angle for a B2Cl4 and b B2F4. G3 components for c B2Cl4 and dB2F4. G3CEP components for e B2Cl4 and f B2F4
J Mol Model (2014) 20:2199 Page 9 of 14, 2199

performed with or without pseudopotentials, as observed forthe diboron halides. However, for the alcohols studied in this
work, the diffuse functions have a similar effect to the othercomponents. These diffuse functions help to describe
CH3CHOHCH3 C6H5OH
0 20 40 60 80 100 120 140 160 180-2.0
-1.5
-1.0
-0.5
0.0
E /
kcal
mol
-1
Dihedral Angle H-C-O-H /degrees
G3 G3CEP EXP
(a)
0 20 40 60 80 100 120 140 160 180-1.0
0.0
1.0
2.0
3.0
4.0
(b)
Dihedral Angle H-O-C-C / degrees
E /
kcal
mol
-1
G3 G3CEP EXP
0 20 40 60 80 100 120 140 160 180-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
MP4
G3
+2df,pG3LargeQCI
Dihedral Angle H-C-O-H / degrees
E /
kcal
mol
-1
(c)
0 20 40 60 80 100 120 140 160 180
-1.0
0.0
1.0
2.0
3.0
4.0MP4
G3
+2df,pG3LargeQCI
(d)
Dihedral Angle H-O-C-C /degrees
E /
kcal
mol
-1
0 20 40 60 80 100 120 140 160 180-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
MP4G3CEP+2df,pG3LargeQCI
Dihedral Angle H-C-O-H / degrees
E /
kcal
mol
-1
(e)
-1.0
0.0
1.0
2.0
3.0
4.0
5.0 MP4G3CEP+2df,pG3LargeQCI
(f)
E /
kcal
mol
-1
Dihedral Angle H-O-C-C /degrees
0 20 40 60 80 100 120 140 160 180
Fig. 4a–f Theoretical and experimental relative energies as a function of the dihedral angle for a CH3CHOHCH3 and b C6H5OH. G3 components for cCH3CHOHCH3 and d C6H5OH. G3CEP components for e CH3CHOHCH3 and f C6H5OH
2199, Page 10 of 14 J Mol Model (2014) 20:2199

hyperconjugation effects of the types nO→σ*C–C and nO→σ*C–H, which stabilize conformers in which these orbitals arein an antiperiplanar arrangement [38].
Ethers
Two ethers were selected for analysis of their rotational bar-riers: dimethyl ether and methoxyacetonitrile ether; com-pounds 7 and 8 in Fig. 1. The rotational torsions were mea-sured around the O–C bond. The barrier profiles for bothcompounds differ considerably because stereoelectronic ef-fects lead to more than one barrier for methoxyacetonitrile(Fig. S4b).
For any of the methods used without ZPE correction, thedeviations of the theoretical results from the correspondingexperimental data for dimethyl ether are less than0.5 kcal mol−1 (see Table 1). The energy minima occur atdihedral angles of 60° and 180° (Figs. S4a and S4b). The moststable conformer of methoxyacetonitrile (synclinal) appears atan angle of 60°, while a less stable antiperiplanar conformeroccurs at 180°.
The G3 and G3CEP components that correct the MP4energies exhibit quantitatively similar effects but show differ-ent qualitative behaviors for the same molecule. For dimethylether, both G3 and G3CEP indicate that the correction due todiffuse functions (ΔE+) is the most important component(Figs. S4c and S4e). For methoxyacetonitrile, the three cor-rectionsΔE+, ΔE2df,p, and ΔEG3large all contribute to the barrierprofile (Figs. S4d and S4f), but the predominant correctiondepends on the dihedral angle, highlighting the difficultyinvolved in suitably adjusting the calculation conditions toobtain accurate results.
Aldehydes
Compounds 9, 10, 11, and 12 in Fig. 1 (oxaldehyde, acetal-dehyde, propanal, and benzaldehyde, respectively) are alde-hydes. For these aldhydes, similar to the alcohols, increasingthe carbon chain length causes the rotational barrier to in-crease, except for oxaldehyde, which has two formyl groups.
Acetaldehyde shows a single rotational barrier, and thedeviations from the experimental data are very small (seeTable 1): 0.07, 0.06, 0.26 and 0.17 kcal mol−1 for G3CEP,G3, MP4CEP, and MP4, respectively, with minima at 0° and120° (Fig. S5b). The behaviors of the G3 and G3CEP com-ponents depend on the dihedral angle and are not identical(Figs. S5d and S5f), but, for both G3 and G3CEP, the ΔE+
component is the main contribution to the correction of theMP4 and MP4CEP energies.
The first two barriers of propanal show deviations of>1 kcal mol−1 for all methods (Table 1). The profiles of theG3 and G3CEP energies for propanal show minima at 0° and
120° (Fig. S6a), with ΔE+ being the most important contri-bution for both energies (Figs. S6c and S6e).
For benzaldehyde and phenol, the barrier profiles present amaximum at 90° when the CHO group is perpendicular to theplane of the ring, which leads to the loss of conjugationbetween πC–O→π*C–C (Fig. S6b) [2]. The barriers presentenergy minima at 0° and 180° when the formyl group is inthe plane of the ring. Similar to propanal, the rotational barrierof benzaldehyde shows significant deviations from the exper-imental value, exceeding 2.30 kcal mol−1 for all methods. Thisdiscrepancy between the calculated and experimental valueshas already been discussed in the literature [39–41]. Table 2compares the results calculated in the present article with theexperimental values obtained from the work of Burcl [39].The experimental values are lower than the rotational barrierof benzaldehyde derived using any of the present calculations.The smallest deviation from the experimental data is on theorder of 1.5 kcal mol−1 for the RASSCF(14)/cc-pVTZ calcu-lations reported in the paper of Burcl [39]. Our results—theG3 and G3CEP data—show behavior similar to that of theCR-CC(2,3)/cc-pVTZ and CASPT2(8)/cc-pVTZ calculationsreported in the literature [39]. Burcl suggests that the discrep-ancy between the experimental and CR-, RASSCF-, andCASPT2-calculated values is caused by approximations thatare used to estimate the barrier height from the experimentaltorsional frequency [39]. Evaluation of the G3 and G3CEPenergy components shows that different corrections dominate(Figs. S6d and S6f). In G3CEP, the ΔEG3large component ispredominant (Figs. S6f), whereas, in the case of G3, thedominant effect is ΔE+ (Fig. S6d).
Oxaldehyde has two formyl groups, which causes a differ-ent energy profile from those seen for the other aldehydes(Fig. S5a). Two conformers, 0° and 180°, are the stablestructures. The antiperiplanar conformer, with a dihedral at
Table 2 Experimental and calculated rotational barriers for benzalde-hyde (in kcal mol−1)
Method Data Reference
Experimental 4.90 [39]
CCSD(T)/cc-pVDZ 8.74 [39]
CCSD(T)/cc-pVTZ 8.22 [39]
CR-CC(2,3)/cc-pVDZ 8.58 [39]
CR-CC(2,3)/cc-pVTZ 7.93 [39]
CASPT2(8)/ cc-pVTZ 7.64 [39]
RASSCF(14)/ cc-pVTZ 6.33 [39]
G3 7.67 This work
G3CEP 7.90 This work
MP4/6-31G(d) 8.39 This work
MP4/CEP-P31G(d) 9.54 This work
QCISD(T)/6-31G(d) 8.18 This work
QCISD(T)/CEP-P31G(d) 9.28 This work
J Mol Model (2014) 20:2199 Page 11 of 14, 2199

180°, presents the highest stability, which is a result ofstereoelectronic effects due to πC–O→π*C–O′ conjugation[2]. The deviations of the theoretical barriers of oxaldehydefrom the corresponding experimental values are small forG3CEP and G3, −0.08 and −0.17 kcal mol−1, respectively(Table 1). Despite the differences in the potential curve ofoxaldehyde from those of other aldehydes, ΔE+ is the compo-nent that contributes most to the G3 energy (Fig. S5c), where-as the largest contributions are associated with three compo-nents: ΔE+, ΔE2df,p, and ΔEG3large, for G3CEP (Fig. S5e).
Ethyl nitrate
Ethyl nitrate is compound number 13 in Fig. 1. The experi-mental values found for the rotational barriers of ethyl nitratewere reported by Scroggin et al. [42]. There is a high uncer-tainty for the first barrier, 8.65±6.00 kcal mol−1, whereas theexperimental error for the second barrier is smaller, 2.30±0.60 kcal mol−1 (Table 1). The deviations from the experi-mental value for the first barrier are −1.03, −1.44, −1.80, and1.33 kcal mol−1, respectively, when G3CEP, G3, MP4CEP,and MP4 are used (Table 1). For the second barrier, thecorresponding deviations are −0.11, −0.33, −0.25, and0.31 kcal mol−1. Corrections for both G3 and G3CEP indicatethat the largest contribution arises from theΔEG3large compo-nent (Figs. S7a and S7b).
Methyl rotor
Several of the molecules studied contain a methyl rotor bond-ed to carbon, silicon, nitrogen, or phosphorus, or substitutionsof hydrogen in the methyl group by halogen atoms. Thesecompounds are shown in Fig. 1 as structures 14–36. This classalso includes twists of the vinyl group of vinyl benzene(structure 33) and the ethyl group in butane (structure 36).
Upon analyzing the rotational barriers, two profiles werefound that describe almost all of the molecules in this class(Figs. S8–S19). In the first set, the molecules present twostable conformers. One is a synclinal conformer at 60°, andthe other is the antiperiplanar structure at 180° separated bytwo barriers at 0° and 120°. In the second set, the moleculespresent energy minima at 0° and 120°. Deviations of theexperimental barriers for these molecules from the data calcu-lated by G3 and G3CEP are very low and usually less than0.5 kcal mol−1 (Table 1), although there are rare exceptionswhere the deviation is greater than 1 kcal mol−1.
The G3 and G3CEP energies differ in terms of whichcomponent is the major contributor to improving the MP4barrier (Figs. S8–S19). Among the 23 molecules containingmethyl groups, G3 calculations indicate that the effect ofpolarization, ΔE2df,p, leads to the greatest improvement inthe MP4 rotational barrier in 18 of them. ΔE+ and ΔEG3largeare important in only 7 and 8 molecules, respectively. In the
case of G3CEP, the distributions are very similar for all threecorrections ΔE+, ΔE2df,p, and ΔEG3large. These correctionswere significant in essentially the same number of molecules:11, 10, and 8, respectively. In almost all cases, the ΔEQCI
correction can be discarded.
H2O2, H2S2, N2H4, P2H4, NH2OH
The set of hydrides H2O2, H2S2 and N2H4 (structures 37–39,Fig. 1) were studied by Ducati et al. [4] using G3, and devia-tions from the available experimental data of <0.5 kcal mol−1
were generally observed. For these hydrides and similar ones,the rotational characteristics of the barriers have been attributedto the lone-pair effect of the heavier atoms. The ΔE2df,p correc-tion was one of the most important contributions to both the G3and G3CEP barriers (Figs. S20–S22).
OHCNH2, HCOOH
Among all of the molecules studied, the rotational barriers ofcompounds 42 and 43 in Fig. 1 (i.e., OHCNH2 and HCOOH)present the highest experimental values, higher than13 kcal mol−1, and were thus studied separately.
The literature contains more than one value for the exper-imental rotation barrier of formamide, and NMR analyseswere performed in the presence of water, acetone, dioxane,or a liquid phase [43–45]. When comparing the calculated andexperimental data, the largest deviations appear when water isused as solvent. Because the present calculations are per-formed for molecules in vacuum, the local hydrogen bondswere the main influence on the large deviations. However, thehigh rotational barrier of this molecule can be attributed to thelack of resonance stabilization for the conformer at the barriermaximum, due to the conjugation of the nonbonded pair ofnitrogen with the p-system through an nN→π*C–O interaction,which occurs for the most stable rotamers [46, 47]. Theprofiles for the corrections from the G3 and G3CEP calcula-tions indicate that no component is more significant than anyother (Figs. S23c and S23e).
Formic acid presents a rotat ional barr ier of13.8 kcal mol−1 and a lower barrier of 3.90 kcal mol−1.The deviations for the highest rotational barrier are between1.0 and 1.5 kcal mol−1 for G3CEP and G3, respectively.For the highest barrier, the MP4 method yields the closestresults to the experimental data, and the deviation wasfound to be 0.72 kcal mol−1 (Table 1). The lower barrierfor formic acid presents low deviations for the G3 andG3CEP calculations, whereas the deviations for MP4 aregreater than 1.95 kcal mol−1. The components of the G3and G3CEP energies indicate that the most important con-tributions are associated with ΔE2df,p and ΔE+ (Figs. S23dand S23f).
2199, Page 12 of 14 J Mol Model (2014) 20:2199

Conclusions
In the present work, the G3 and G3CEP theories, with andwithout ZPE correction, along with the MP4, MP4CEP,QCISD(T)/6-31G(d), and QCISD(T)/CEP-P31G(d) methods,were applied to study the internal rotational barriers of 43different molecules. The results show that G3 and G3CEP areaccurate, general ly yielding values that deviate<0.50 kcalmol−1 from the corresponding experimental values.The results from the MP4CEP, MP4, QCISD(T), andQCISD(T)/CEP calculations are less accurate, with largerdeviations of approximately ±1.0 kcal mol−1 observed. Theaccuracy of the methods is also reflected in the averageabsolute deviations: 0.49, 0.48, 0.84, 0.72, 0.64, 0.61, 0.80,and 0.69 kcal mol−1 for G3CEP (not including ZPE), G3 (notincluding ZPE), MP4CEP, MP4, G3CEP, G3, QCISD(T), andQCISD(T)/CEP, respectively.
The MP4, MP4CEP, QCISD(T), and QCISD(T)/CEPmethods show larger deviations and are less accurate, butthe results for these molecules indicate that the methods arereliable. The G3 and G3CEP energies are calculations thatimprove upon MP4 and MP4CEP and therefore require moreCPU time. Using G3CEP reduced the CPU time by between 5and 35 % as compared with the all-electron G3 theory, de-pending on the size of the molecule and types of atoms presentin the structure.
Because the G3CEP and G3 energies are combinations ofdifferent contributions that are added to the MP4/6-31G(d) andMP4/CEP-P31G(d) energies, the quantitative dependency ofthe final energy on the components of G3CEP and G3 wasassessed, and the contributions of the components were foundto decrease in the following order:ΔEG3large>ΔE2df,p>ΔE+>-ΔEQCI. However, the relative behaviors of the energy compo-nents show that these corrections depend on the molecularenvironment and whether the calculation is performed withall electrons or pseudopotentials. Usually, the predominanceof a specific effect follows a distinct pattern for the G3 andG3CEP calculations. For the G3 results, the most importantcomponent in the correction of the MP4/6-31G(d) rotationalbarrier isΔE2df,p. Among the 43 molecules, 29 were dependenton polarization effects, ΔE2df,p, 19 were dependent on theeffects of diffuse functions, ΔE+, and 13 depended on theeffects of more elaborate basis functions, ΔEG3large. For theG3CEP calculations, polarization effects, ΔE2df,p, were moreimportant for 25 molecules, followed closely by the effects ofdiffuse functions,ΔE+, for 23 molecules, and finally the effectsof large basis sets for 19 molecules, ΔEG3large. The ΔEQCIcorrection seldom proved significant in the correction of eitherG3 or G3CEP calculations.
Some families of molecules were more sensitive to some ofthe energy components. As an example, barriers involvingrotations of bonds containing oxygen atoms were highlyaffected by ΔE+ in the G3 calculations. When the same
calculations were performed with G3CEP, the results rein-forced the importance of correcting ΔE+ but indicated thatthe correction due to ΔE2df,p was also significant. The samepattern was observed for the G3 and G3CEP calculations ofthe methyl rotations. The correction due toΔE2df,p were muchmore important than the other two corrections for G3. ForG3CEP, the three main components ΔEG3large, ΔE2df,p, andΔE+ made important contributions.
In general, employing the G3 and G3CEP theories tocalculate rotation barriers leads to deviations from the exper-imental results of approximately ±0.5 kcal mol−1 when ZPE isnot included and ±0.65 kcal mol−1 when ZPE is included.Using G3CEP rather than G3 reduced the CPU time required,although this reduction in CPU time depended on the numberof electrons in the inner shells and the size of the molecule.
Acknowledgments We acknowledge financial support from FAPESP(Fundação de Amparo à Pesquisa do Estado de São Paulo—Center forComputational Engineering and Sciences: grant 2013/08293-7), CNPq(Conselho Nacional de Desenvolvimento Científico e Tecnológico), andFAEPEX-UNICAMP (Fundo de Apoio ao Ensino, à Pesquisa e àExtensão da UNICAMP). The National Center of High PerformanceComputing in São Paulo (CENAPAD—SP) is acknowledged for makingtheir computational facilities available to us. We also would like to thankDr. Telma Rie Doi Ducati for helpful comments and suggestions.
References
1. Dale J (1978) Stereochemistry and conformational analysis. VerlagChemie, Deerfield
2. Eliel EL, Wilen SH (1994) Stereochemistry of organic compounds.Wiley, New York
3. Veillard A (1974) In: Orville-Thomas WJ (ed) Internal rotation inmolecules. Wiley, London, p 385
4. Ducati LC, Custodio R, Rittner R (2010) Int J Quantum Chem 110:2006–2014
5. Barone V, Biczysko M, Bloino J, Puzzarini C (2013) Phys ChemChem Phys 15:1358–1363
6. Cormanich RA, Ducati LC, Rittner R (2011) Chem Phys 387:85–917. Peterson KA, Feller D, Dixon DA (2012) Theor Chem Acc 131:
1079–10998. Feller D, Peterson KA, Dixon DA (2012) Mol Phys 110:2381–23999. Dixon DA, Feller D, Peterson KA (2012) Annu Rev Comput Chem
8:1–2810. Pople JA, Head-Gordon M, Fox DJ et al (1989) J Chem Phys 90:
5622–562911. Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) J Chem
Phys 94:7221–723012. Curtiss LA, Raghavachari K, Redfern PC et al (1998) J Chem Phys
109:7764–777613. Curtiss LA, Redfern PC, Raghavachari K et al (1999) J Chem Phys
110:4703–470914. Curtiss LA, Redfern PC, Rassolov V et al (2001) J Chem Phys 114:
9287–929515. Curtiss LA, Redfern PC, Raghavachari K (2005) J Chem Phys 123:
124107–12411916. Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 126:
084108–084120
J Mol Model (2014) 20:2199 Page 13 of 14, 2199

17. Curtiss LA, Redfern PC, Raghavachari K (2007) J Chem Phys 127:124105–124113
18. Mayhall NJ, Raghavachari K, Redfern PC et al (2008) J Chem Phys128:144122–144131
19. Mayhall NJ, Raghavachari K, Redfern PC, Curtiss LA (2009) J PhysChem A 113:5170–5175
20. Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (1999) JChem Phys 110:2822–2827
21. Petersson KA, Woon DE, Dunning TH (1994) J Chem Phys 100:7410–7415
22. Ochterski JW, Petersson GA, Montgomery JA (1996) J Chem Phys104:2598–2619
23. Montgomery JA, Ochterski JW, Petersson GA (1994) J Chem Phys101:5900–5909
24. Petersson GA, Bennett A, Tensfeldt TG et al (1988) J Chem Phys 89:2193–2218
25. Petersson GA, Tensfeldt TG, Montgomery JA (1991) J Chem Phys94:6091–6101
26. Martin JML, de Oliveira G (1999) J Chem Phys 111:1843–185627. Boese AD, OrenM, Atasoylu O et al (2004) J Chem Phys 120:4129–
414128. Karton A, Rabinovich E, Martin JML, Ruscic B (2006) J Chem Phys
125:144108–14412529. Stevens WJ, Basch H, Krauss M (1984) J Chem Phys 81:6026–603330. Cundari TR, Stevens WJ (1993) J Chem Phys 98:5555–556531. Stevens WJ, Krauss M, Basch H, Jasien PG (1992) Can J Chem 70:
612–63032. Pereira DH, Ramos AF, Morgon NH, Custodio R (2011) J Chem
Phys 135:034106–03412033. Murcko MA, Castejon H, Wiberg KB (1996) J Phys Chem 100:
16162–1616834. Pereira DH, Ramos AF, Morgon NH, Custodio R (2011) J Chem
Phys 135:219901–21990135. Frisch MJ, Trucks GW, Schlegel HB, et al. (2009) Gaussian 09.
Gaussian, Inc., Wallingford36. Sharaf MA, Illman DL, Kowalski BR (1986) Chemometrics. Wiley-
Interscience, New York37. Clark T, Von Rague Schleyer P (1981) J Comput Chem 2:20–2938. Ducati LC, Freitas MP, Tormena CF, Rittner R (2008) J Mol Struct
THEOCHEM 851:147–15739. Burcl R (2011) J Phys Chem A 115:3605–360640. Speakman LD, Papas BN, Woodcock HL, Schaefer HF (2004) J
Chem Phys 120:4247–425041. Meier RJ (2011) J Phys Chem A 115:3604–360442. Scroggin DG (1974) J Chem Phys 60:1376–138543. Christensen DH (1970) J Chem Phys 53:3912–392244. Sunners B, Piette LH, Schneider WG (1960) Can J Chem 38:681–
68845. Kamei H (1968) Bull Chem Soc Jpn 41:2269–227346. Pedersoli S, Tormena CF, Rittner R (2008) J Mol Struct 875:235–24347. Martins CR, Rittner R, Tormena CF (2005) J Mol Struct
THEOCHEM 728:79–8448. Jones LH (1972) J Chem Phys 57:1012–101349. Danielson DD, Patton JV, Hedberg K (1977) J Am Chem Soc 99:
6484–648750. Venkateswarlu P, Gordy W (1955) J Chem Phys 23:1200–1202
51. Herbst E, Messer JK, De Lucia FC, Helminger P (1984) J MolSpectrosc 108:42–57
52. Kakar RK, Quade CR (1980) J Chem Phys 72:4300–430753. Kondo S, Hirota E (1970) J Mol Spectrosc 34:97–10754. Larsen NW (1986) J Mol Struct 144:83–9955. Pierce L, Hayashi M (1961) J Chem Phys 35:479–48556. Durig JR, Tang Q, Phan HV (1993) J Raman Spectrosc 24:
851–86557. Butz KW, Krajnovich DJ, Parmenter CS (1990) J Chem Phys 93:
1557–156758. Allen LC (1968) Chem Phys Lett 2:597–60159. Randell J, Hardy JA, Cox AP (1988) J Chem Soc Faraday Trans
2(84):1199–121260. Kakar RK, Rinehart EA, Quade CR, Kojima T (1970) J Chem Phys
52:3803–381361. Herzberg G (1967) Electronic spectra and electronic structure of
polyatomic molecules. D. Van Nostrand, New York62. Kraitchman J, Dailey BP (1955) J Chem Phys 23:184–19063. Fliege E, Dreizler H, Demaison J et al (1983) J Chem Phys 78:3541–
354564. Villamanan RM, Chen WD, Wlodarczak G et al (1995) J Mol
Spectrosc 171:223–24765. Meerts WL, Ozier I (1991) Chem Phys 152:241–25966. Mann DE, Plyler EK (1953) J Chem Phys 21:1116–111767. Stahl W, Dreizler H, Hayashi M (1983) Z Naturforsch A 38:1010–
101468. Margulès L, Carvajal M, Demaison J (2008) J Mol Spectrosc 247:
160–16669. Mizushima S, Shimanouchi T, Harada I et al (1975) Can J Phys 53:
2085–209470. Frankiss SG, Harrison DJ (1975) Spectrochim Acta Pt A 31:29–3971. Ilyushin VV, Alekseev EA, Dyubko SF et al (2001) J Mol Spectrosc
205:286–30372. Kollman PA, Allen LC (1970) Chem Phys Lett 5:75–7673. Absar I, van Wazer JR (1971) Chem Commun 611–61274. Nascimento J, Pelegrini M, Ferrão LFA et al (2011) J Braz Chem Soc
22:968–97575. Allen LC, Scarzafava E (1971) J Am Chem Soc 93:311–31476. Bak B (1966) J Chem Phys 45:883–88777. Swalen JD, Herschbach DR (1957) J Chem Phys 27:100–10878. Herrebout WA, van der Veken BJ, Wang A, Durig JR (1995) J Phys
Chem 99:578–58579. Guirgis GA, Durig JR, Bell S (1989) J Mol Struct 196:101–11180. Durig JR, Costner TG, Wang A et al (1993) J Mol Struct 300:257–
27981. Caminati W, Vogelsanger B, Bauder A (1988) J Mol Spectrosc 128:
384–39882. Tsuboi M, Overend J (1974) J Mol Spectrosc 52:256–26883. Hunt RH, Leacock RA, Peters CW, Hecht KT (1965) J Chem Phys
42:1931–194684. Behrend J, Mittler P, Winnewisser G, Yamada KMT (1991) J Mol
Spectrosc 150:99–11985. Herbst E, Winnewisser G (1989) Chem Phys Lett 155:572–57586. Odom JD,Wurrey CJ, Carreira LA, Durig JR (1975) Inorg Chem 14:
2849–285387. Hirao H (2008) Chem Phys 344:213–220
2199, Page 14 of 14 J Mol Model (2014) 20:2199





























