Embed Size (px)
Citation preview

DOI: 10.1126/scitranslmed.3002731, 105ra104 (2011);3 Sci Transl Med
et al.Vivi M. HeineNeonatal Cerebellar InjuryA Small-Molecule Smoothened Agonist Prevents Glucocorticoid-Induced
Editor's Summary
neonatal GC treatment toward more benefit for preterm infants.dose schedule and safety parameters before clinical trials in humans, but SAG may tilt the risk-benefit ratio of are given to preterm infants for life-threatening conditions. Further toxicity studies will need to determine the optimaladjuvant therapy with SAG is safe and might be an effective way to prevent neurotoxic side effects of GCs when they tumors. Most importantly, SAG did not antagonize beneficial effects of GCs for the lung. These findings suggest thatmedulloblastoma or other cancers, a reasonable fear because the Smo pathway is critical for proliferation of some growth-inhibitory effects of GCs. They also showed that this treatment did not induce the cerebellar tumorcrossed the blood-brain barrier to activate Sonic hedgehog targets in the mouse cerebellum and that it prevented the therefore devised a small-molecule mimic of this effect: SAG, an agonist of Smoothened. They showed that SAGGCs. It was known that putting this pathway into genetic overdrive can prevent GC-induced injury, and the authors
bysignaling pathway drives cerebellar cell proliferation during development and that it is this pathway that is disrupted Sonic hedgehog− have built on previous work showing that the Smoothenedet al.address this problem, Heine
the cerebellum, a brain center critical for coordination of movement and higher-order neurological functions. Toglucocorticoids (GCs). However, GC treatment can induce permanent neurological deficits and inhibit the growth of
Preterm babies often develop chronic lung disease, which can be treated by postnatal administration of
Tilting the Risk-Benefit Ratio for Preterm Infants
http://stm.sciencemag.org/content/3/105/105ra104.full.htmlcan be found at:
and other services, including high-resolution figures,A complete electronic version of this article
http://stm.sciencemag.org/content/scitransmed/5/168/168ps2.full.html http://stm.sciencemag.org/content/scitransmed/5/193/193ra90.full.html
http://stm.sciencemag.org/content/scitransmed/5/168/168ra7.full.html http://stm.sciencemag.org/content/scitransmed/5/168/168ra8.full.html
can be found online at:Related Resources for this article
http://www.sciencemag.org/about/permissions.dtl in whole or in part can be found at: article
permission to reproduce this of this article or about obtaining reprintsInformation about obtaining
is a registered trademark of AAAS. Science Translational Medicinerights reserved. The title NW, Washington, DC 20005. Copyright 2011 by the American Association for the Advancement of Science; alllast week in December, by the American Association for the Advancement of Science, 1200 New York Avenue
(print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except theScience Translational Medicine
on
May
8, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
o
n M
ay 8
, 201
4st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om

R E S EARCH ART I C L E
PRETERM B IRTH
A Small-Molecule Smoothened Agonist PreventsGlucocorticoid-Induced Neonatal Cerebellar InjuryVivi M. Heine,1,2*† Amelie Griveau,1,2* Cheryl Chapin,1 Philip L. Ballard,1
James K. Chen,3 David H. Rowitch1,2‡
ay 8
, 201
4
Glucocorticoids are used for treating preterm neonatal infants suffering from life-threatening lung, airway, andcardiovascular conditions. However, several studies have raised concerns about detrimental effects of postnatalglucocorticoid administration on the developing brain leading to cognitive impairment, cerebral palsy, and hy-poplasia of the cerebellum, a brain region critical for coordination of movement and higher-order neurologicalfunctions. Previously, we showed that glucocorticoids inhibit Sonic hedgehog–Smoothened (Shh-Smo) signaling,the major mitogenic pathway for cerebellar granule neuron precursors. Conversely, activation of Shh-Smo in trans-genic mice protects against glucocorticoid-induced neurotoxic effects through induction of the 11b-hydroxysteroiddehydrogenase type 2 (11b-HSD2) pathway. Here, we show that systemic administration of a small-moleculeagonist of the Shh-Smo pathway (SAG) prevented the neurotoxic effects of glucocorticoids. SAG did not interferewith the beneficial effects of glucocorticoids on lung maturation, and despite the known associations of the Shhpathway with neoplasia, we found that transient (1-week-long) SAG treatment of neonatal animals was well toler-ated and did not promote tumor formation. These findings suggest that a small-molecule agonist of Smo haspotential as a neuroprotective agent in neonates at risk for glucocorticoid-induced neonatal cerebellar injury.
n M
ost
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
INTRODUCTION
More than 60,000 very low birth weight (<1.5 kg) preterm neonatesare born yearly in the United States. Such infants suffer from a rangeof neurological impairments such as intracranial hemorrhage, damageto gray and white matter, cerebral palsy, and learning and behavioralproblems (1–3). Cerebellar hypoplasia is common in such infants (2),is a hallmark of severe neurological injury (4–7), and is associated withcognitive and affective disturbances, including autistic syndrome (8).The cerebellum is responsible for coordination of movement andhigher-order relay functions of the brain (7). The most abundant celltype in the cerebellum is the granule neuron precursor (CGNP) (9–11).In humans, CGNP expansion starts at 11 weeks of gestational age andis robust in the third trimester of pregnancy, between 24 and 40 weeksof gestational age, during which deficient growth and foliation can re-sult in cerebellar hypoplasia (12). Human cerebellar growth continuesuntil at least 1 year of age (13). The analogous period of robust CGNPgrowth during mouse development is postnatal days 0 (P0) to 14(P14), and functional studies have demonstrated that Sonic hedgehog(Shh) signaling is the prime driver of this process (11, 14). Shh activatesthe transmembrane receptor protein Smoothened (Smo) to up-regulatethe target genes Gli1 and N-myc, which are essential for CGNP cellcycle progression (15, 16). Mutations that activate the Shh-Smo path-way in CGNPs result in the cerebellar tumor medulloblastoma inmouse and humans (17, 18).
1Division of Neonatology, Department of Pediatrics, University of California, San Francisco(UCSF), San Francisco, CA 94143, USA. 2Howard Hughes Medical Institute and Eli andEdythe Broad Institute for Stem Cell Research and Regeneration Medicine, UCSF, SanFrancisco, CA 94143, USA. 3Department of Chemical and Systems Biology, StanfordUniversity School of Medicine, Stanford, CA 94305, USA.*These authors contributed equally to this work.†Present address: Center for Children with White Matter Disorders, Center forNeurogenomics and Cognitive Research, Department of Pediatrics, VU UniversityMedical Center, 1081 HV Amsterdam, Netherlands.‡To whom correspondence should be addressed. E-mail: [email protected]
www.Science
For several decades, glucocorticoids (GCs) have been used in pre-term infants for the treatment of life-threatening chronic lung disease,as well as for cardiovascular conditions (for example, hypotension) (4, 7).In the immature lung, GCs promote surfactant production and lungepithelial differentiation (19). However, numerous studies have shownthat GCs can have detrimental effects on the development of the brain,leading to impaired cognition, cerebral palsy, and cerebellar hypopla-sia (5–7, 11, 20). 11b-Hydroxysteroid dehydrogenase type 2 (11b-HSD2) catalyzes the interconversion of the endogenous physiologicalGCs cortisol and corticosterone to the inert 11-ketometabolites corti-sone and 11-dehydrocorticosterone, respectively (21, 22). The syntheticGC prednisolone is also inactivated by 11b-HSD2 (23). 11b-HSD2 isexpressed by the cerebellar anlage commencing at embryonic day 12.5(E12.5) in mice (24), and its function is necessary for normal cerebel-lar development (22, 24).
GCs generally regulate gene expression through binding to the in-tracellular mineralocorticoid and/or GC receptor (25) or by directlyaffecting intracellular signaling pathways for protein stability and cellmetabolism (26). Although the molecular basis of GC-induced neuro-toxicity in the developing brain remains incompletely understood, wehave shown that neonatal administration (P0 to P7) of GCs in miceinhibits mitogenic Shh signaling in CGNPs through destabilization ofGli and N-myc proteins (20). Conversely, Shh-Smo signaling can an-tagonize neurotoxic effects of GC signaling in CGNPs. Forced expres-sion of a constitutively active form of Smo (SmoM2) in transgenic miceinduced up-regulation of 11b-HSD2 and prevented neurotoxic effectsof prednisolone in the neonatal cerebellum (20). Nevertheless, such atransgenic approach to protect the cerebellum against GC-induceddamage is not clinically feasible and carries the additional problemthat the animals eventually succumb to medulloblastoma (18).
New and practical strategies are needed to assuage the negative ef-fects of postnatal GC administration on developing brain structures,such as the cerebellum, while preserving their desirable therapeuticprofile in the developing lung. We therefore tested whether systemic
TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 1
R E S EARCH ART I C L E
on
May
8, 2
014
rg
administration of a small molecular agonist of the Shh-Smo pathway (SAG)could antagonize GC-induced cerebellar injury while demonstrating safetyand preserving the beneficial effects of GCs to promote lung maturation.
RESULTS
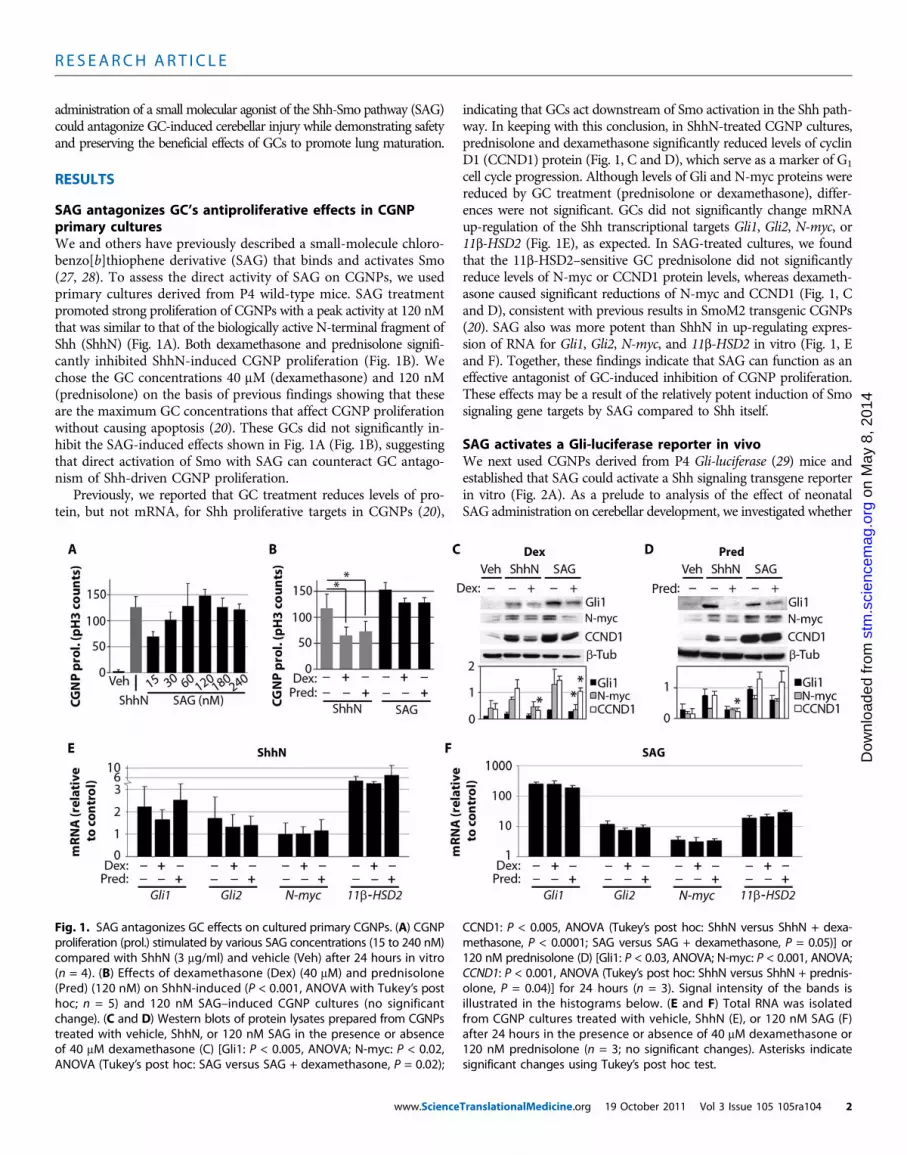
SAG antagonizes GC’s antiproliferative effects in CGNPprimary culturesWe and others have previously described a small-molecule chloro-benzo[b]thiophene derivative (SAG) that binds and activates Smo(27, 28). To assess the direct activity of SAG on CGNPs, we usedprimary cultures derived from P4 wild-type mice. SAG treatmentpromoted strong proliferation of CGNPs with a peak activity at 120 nMthat was similar to that of the biologically active N-terminal fragment ofShh (ShhN) (Fig. 1A). Both dexamethasone and prednisolone signifi-cantly inhibited ShhN-induced CGNP proliferation (Fig. 1B). Wechose the GC concentrations 40 mM (dexamethasone) and 120 nM(prednisolone) on the basis of previous findings showing that theseare the maximum GC concentrations that affect CGNP proliferationwithout causing apoptosis (20). These GCs did not significantly in-hibit the SAG-induced effects shown in Fig. 1A (Fig. 1B), suggestingthat direct activation of Smo with SAG can counteract GC antago-nism of Shh-driven CGNP proliferation.
Previously, we reported that GC treatment reduces levels of pro-tein, but not mRNA, for Shh proliferative targets in CGNPs (20),
www.Science
indicating that GCs act downstream of Smo activation in the Shh path-way. In keeping with this conclusion, in ShhN-treated CGNP cultures,prednisolone and dexamethasone significantly reduced levels of cyclinD1 (CCND1) protein (Fig. 1, C and D), which serve as a marker of G1
cell cycle progression. Although levels of Gli and N-myc proteins werereduced by GC treatment (prednisolone or dexamethasone), differ-ences were not significant. GCs did not significantly change mRNAup-regulation of the Shh transcriptional targets Gli1, Gli2, N-myc, or11b-HSD2 (Fig. 1E), as expected. In SAG-treated cultures, we foundthat the 11b-HSD2–sensitive GC prednisolone did not significantlyreduce levels of N-myc or CCND1 protein levels, whereas dexameth-asone caused significant reductions of N-myc and CCND1 (Fig. 1, Cand D), consistent with previous results in SmoM2 transgenic CGNPs(20). SAG also was more potent than ShhN in up-regulating expres-sion of RNA for Gli1, Gli2, N-myc, and 11b-HSD2 in vitro (Fig. 1, Eand F). Together, these findings indicate that SAG can function as aneffective antagonist of GC-induced inhibition of CGNP proliferation.These effects may be a result of the relatively potent induction of Smosignaling gene targets by SAG compared to Shh itself.
SAG activates a Gli-luciferase reporter in vivoWe next used CGNPs derived from P4 Gli-luciferase (29) mice andestablished that SAG could activate a Shh signaling transgene reporterin vitro (Fig. 2A). As a prelude to analysis of the effect of neonatalSAG administration on cerebellar development, we investigated whether
stm
.sci
ence
mag
.oD
ownl
oade
d fr
om
Fig. 1. SAG antagonizes GC effects on cultured primary CGNPs. (A) CGNPproliferation (prol.) stimulated by various SAG concentrations (15 to 240 nM)compared with ShhN (3 mg/ml) and vehicle (Veh) after 24 hours in vitro(n = 4). (B) Effects of dexamethasone (Dex) (40 mM) and prednisolone(Pred) (120 nM) on ShhN-induced (P < 0.001, ANOVA with Tukey’s posthoc; n = 5) and 120 nM SAG–induced CGNP cultures (no significantchange). (C and D) Western blots of protein lysates prepared from CGNPstreated with vehicle, ShhN, or 120 nM SAG in the presence or absenceof 40 mM dexamethasone (C) [Gli1: P < 0.005, ANOVA; N-myc: P < 0.02,ANOVA (Tukey’s post hoc: SAG versus SAG + dexamethasone, P = 0.02);
CCND1: P < 0.005, ANOVA (Tukey’s post hoc: ShhN versus ShhN + dexa-methasone, P < 0.0001; SAG versus SAG + dexamethasone, P = 0.05)] or120 nM prednisolone (D) [Gli1: P < 0.03, ANOVA; N-myc: P < 0.001, ANOVA;CCND1: P < 0.001, ANOVA (Tukey’s post hoc: ShhN versus ShhN + prednis-olone, P = 0.04)] for 24 hours (n = 3). Signal intensity of the bands isillustrated in the histograms below. (E and F) Total RNA was isolatedfrom CGNP cultures treated with vehicle, ShhN (E), or 120 nM SAG (F)after 24 hours in the presence or absence of 40 mM dexamethasone or120 nM prednisolone (n = 3; no significant changes). Asterisks indicatesignificant changes using Tukey’s post hoc test.
TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 2

R E S EARCH ART I C L E
on
May
8, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
SAG could cross the blood-brain barrierto activate Shh target genes in vivo. WetreatedGli-luciferase pups at P11with var-ious doses of SAG (5.6, 14, and 25.2 mg/g)and, after 4 hours, administered luciferinsubstrate to visualize reporter activity inthe brain and cerebellum. We observeda dose-response profilewithmaximal activa-tion of the reporter at a dosage of 25.2 mg/g(Fig. 2, B and C), and this effect was con-firmed by up-regulation of mRNA tran-scripts for the endogenous Shh targets Gli1and N-myc (Fig. 2D). These findings showthat SAG can cross the blood-brain barrierto activate Shh transcriptional targets in vivo.
Because SAG effects were similar withthe 14 or 25.2 mg/g dose, we chose anintermediate dose of 20 mg/g, which wetermed the “treatment dose.” We alsoused primaryCGNP cultures as a bioassayand determined a second “high dose” byusing a free-base form of SAG that exhibitsgreater potency. The free-base form ofSAG was about seven times more potentthan the treatment dose of SAG, as mea-sured by its ability to stimulate prolifera-tion of CGNPs (Fig. 3C), likely reflectingits higher purity and greater bioavailability.This formwas chosen for high-dose testingto maintain similar volumes of injecteddrug and/or vehicle. The treatment andhigh dosages of SAG were next assessedfor potential toxic effects in mice.
Daily administration of SAG at
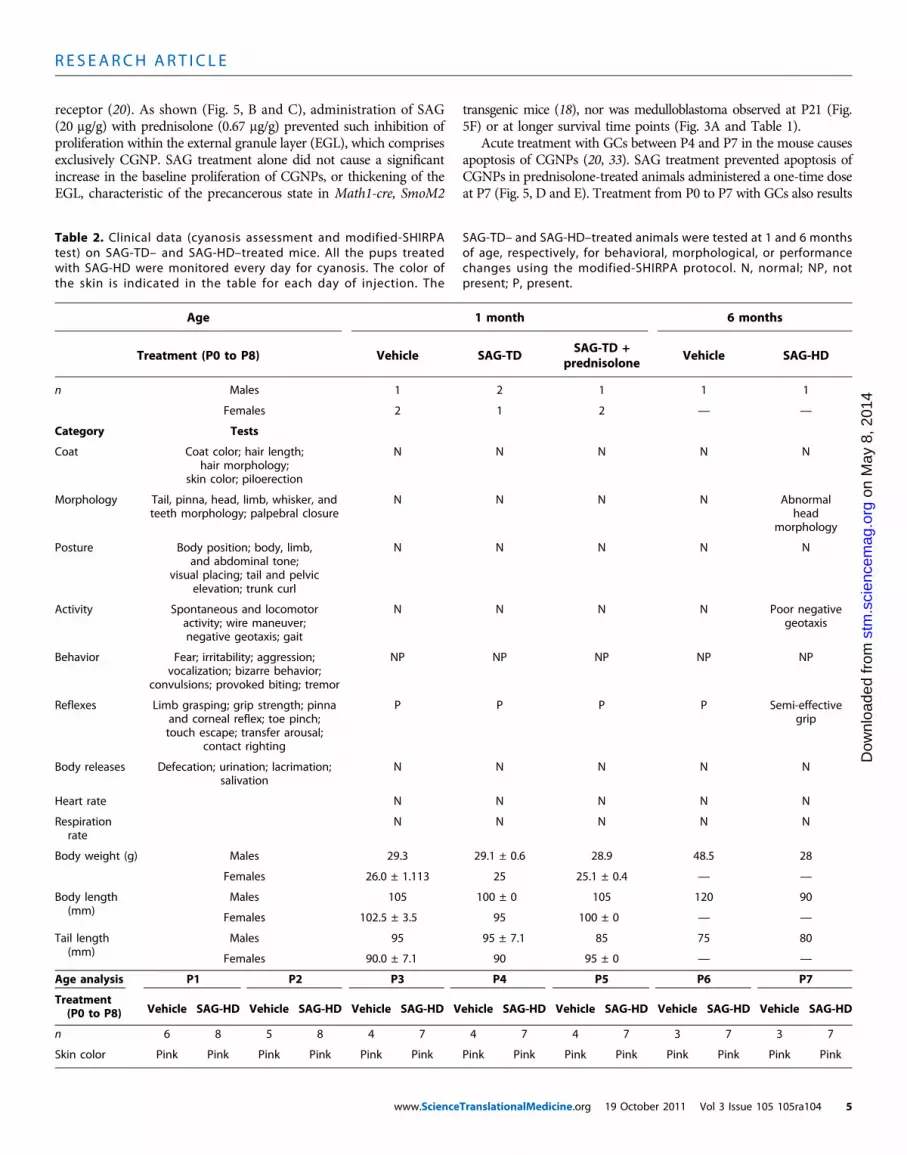
treatment or high dosesis not tumorigenicTheShhpathway is etiologic in several types ofhumancancer (30), includ-ing the cerebellar tumormedulloblastoma (17).Toassesswhether the Smoagonist SAGshowed toxicity and/or tumor formation,we first administeredto pups the treatment dose of SAG (20 mg/g, n = 6) daily from P0 to P7www.Science
(Table 1). At 1 month of age, we performed the modified-SHIRPA (31)protocol to analyze thebehavioral andmorphological effects on the treatedanimals. As shown (Table 2), no motor or behavioral changes were de-tected.All animalswere asymptomatic and showednormalweight gain at2months of age, at which point they were euthanized for inspection of allviscera (heart, lungs, kidneys, pancreas, liver, intestine, and spleen) and
Fig. 2. SAG activates Gli-luciferase reporter transgene in CGNPs in vitroand in vivo. (A) Gli-luciferase (Gli-luc) reporter expression in primaryCGNPs treated with ShhN (3 mg/ml) and SAG (120 nM) for 24 hours in vitro(n = 8). (B) P11 Gli-luciferase mice were injected intraperitoneally with SAG(0, 5.6, 14.0, or 25.2 mg/g) (in saline) and killed after 4 hours. The brains andears were dissected, placed in luciferin (0.4 mg/ml)/phosphate-bufferedsaline for 35 min, and then imaged in the Xenogen IVIS detector for 4 min.A photographic image was taken onto which the pseudocolor imagerepresenting the spatial distribution of photon count was projected. FB,forebrain; CB, cerebellum. (C) Luciferase levels in the cerebellum quantifiedin photons per second at various SAG doses. (D) qRT-PCR analysis of SAGeffects on the Smo targets Gli1 and N-myc in vivo. SAG dosages (14.0 and25.2 mg/g) induced significantly increased N-myc levels over a dosage of5.6 mg/g (P < 0.05, ANOVA with Tukey’s post hoc; n = 2). No significantdifferences were seen between 14.0 and 25.2 mg/g. Asterisks indicatesignificant changes with the Tukey’s post hoc test.
T
Fig. 3. Results of toxicity studieswith SAGat treatment andhighdoses. (A) Histological analysis of the cerebellaof the 6-month-old control and SAG-HD (SAG high dose), and P28 control and SAG-TD (SAG treatment dose)–
treated animals. (B) Immunocytochemistry for the proliferation marker Ki67 in the cerebella of P28 control andSAG-TD–treated animals. DAPI, 4′,6-diamidino-2-phenylindole. (C) Determination of treatment and high dose ofSAG using CGNP proliferation in vitro. Histogram represents the ratio of the number of pH3-positive cellcultures using free-base SAG versus the salt form of SAG at 1 nM (n = 2). (D and E) Growth curves of vehicle,SAG-TD, prednisolone, prednisolone + SAG-TD (D), and SAG-HD–treated (E) mice.ranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 3
R E S EARCH ART I C L E
on
May
8, 2
014
.sci
ence
mag
.org
brain (Table 1 and Fig. 3A). In four treated animals and three controls,complete blood counts were done at 2 months of age; these showed nosigns of leukemia or abnormal hematological counts of leukocytes, ery-throcytes, or thrombocytes. Solid tumors were undetectable in any organsystem including forebrain and cerebellum (Table 1 and Fig. 3, A and B).
We next analyzed animals treated with the high-dose form of SAGdaily fromP1 to P8 at P9 (n = 3, group 1; Table 1), P14 (n = 1, group 2;Table 1), and 4 to 6 months (n = 3, groups 5 and 6; Table 1). No grossormicroscopic evidence of tumor formationwas found in the brain orother viscera of any treated animal. Before medulloblastoma forma-tion in tumor-prone Ptc+/−mice, preneoplastic rests of cells are readilyidentified in the outer layers of the cerebellum as early as 3 weeks of age(32). As shown (Fig. 3A andTable 1), we saw no such precancerous cellsin the cerebellum of animals at P28 or 4 to 6 months after administra-tion of the treatment or high dose of SAG.
In summary, we found no evidence of cancers of blood or solidorgans in any of the 14 animals studied at ages greater than 1 month,nor was there evidence of pretumorigenic lesions of medulloblastoma.We noted that SAG treatment at the high dose resulted in relativelypoor long-term growth (Fig. 3E), which might relate to intestinal hy-perplasia caused by high-dose SAG in some cases (Table 1, groups1 and 2). At the treatment dose, SAG did not cause any such growthor intestinal abnormalities (Fig. 3D and Table 1). Together, these find-ings indicated that transient (1 week) administration of SAG at a treat-ment dose was well tolerated in neonatal mice. Our studies do not ruleout potential side effects of longer-term SAG administration.
SAG administration does not interfere with beneficial GCeffects on pulmonary developmentIn preterm neonates at risk for respiratory distress syndrome, GCs aregiven pre- or postnatally to promote lung maturation and surfactant
www.Science
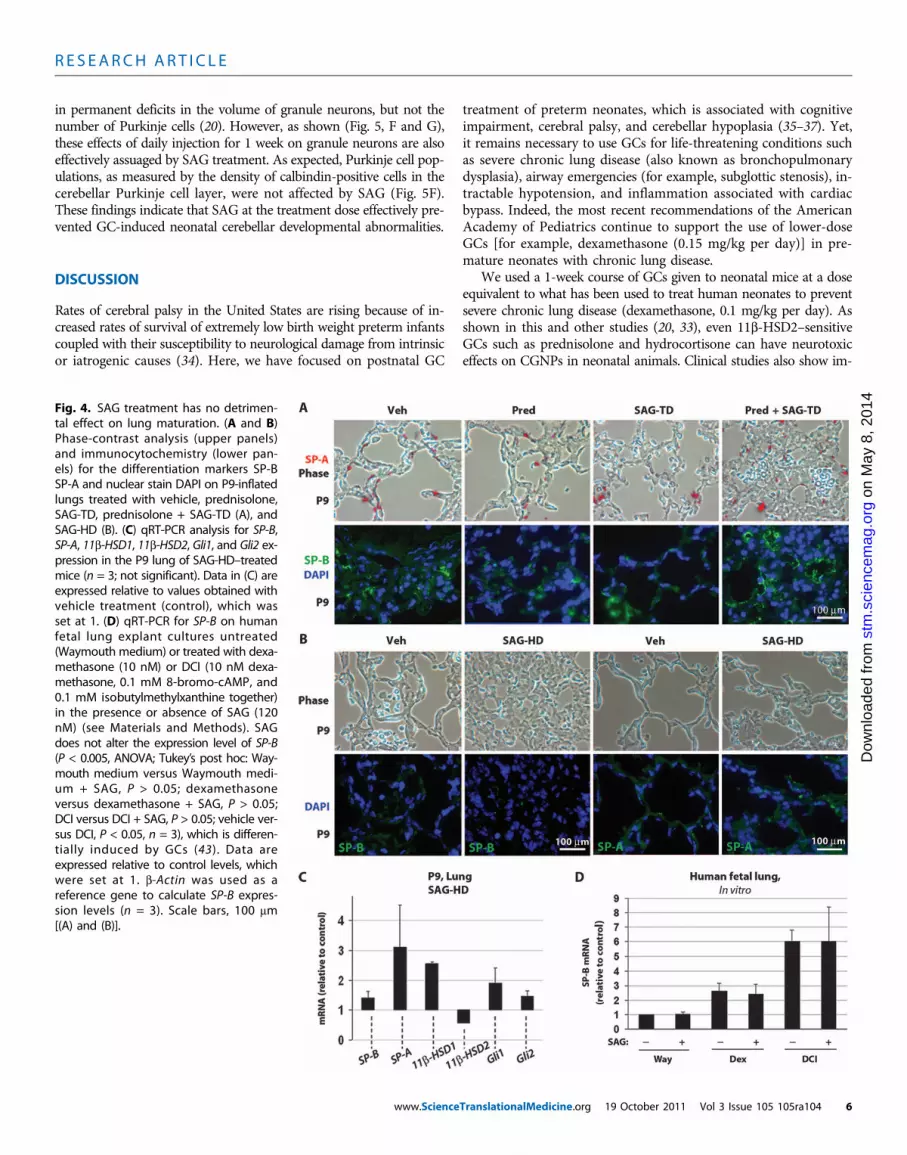
protein production as well as to reduce inflammation. Because SAG isantagonistic to GC activity in the cerebellum, we investigated its effectson neonatal mouse lung to determine whether SAG interfered with GCbeneficial effects in lung. SAG administered at treatment dose alone orin combination with prednisolone did not detectably alter amounts ofthe differentiation markers surfactant proteins A (SP-A) or B (SP-B)(Fig. 4A). When given at high dose, SAG alone appeared to promotelung hyperplasia but did not inhibit expression of the differentiationmarkers SP-A and SP-B (Fig. 4B). Moreover, animals that receivedSAG at treatment dose or high dose did not exhibit respiratory symp-toms such as cyanosis or increased respiratory rate (Table 2).
We next used quantitative polymerase chain reaction (qPCR) toquantitatively assess mRNA levels for the differentiation markersSP-A and SP-B and for genes of the GC and Shh signaling pathwaysin whole lungs of animals treated from P1 to P8 with SAG at the highdose (Fig. 4C). SAG did not significantly change the expression of theShh targets Gli1 and Gli2 and the lung differentiation markers SP-Band SP-A [analysis of variance (ANOVA): not significantly different].In contrast to its effects in the cerebellum, SAG treatment producedno significant changes in 11b-HSD1 or 11b-HSD2 (ANOVA: not sig-nificantly different). Finally, we tested the effects of simultaneous treat-ment of human fetal lung cultures with GC and SAG. As shown (Fig. 4D),SAG did not inhibit GC-induced up-regulation of the differentiationmarker SP-B in human fetal lung cultures.
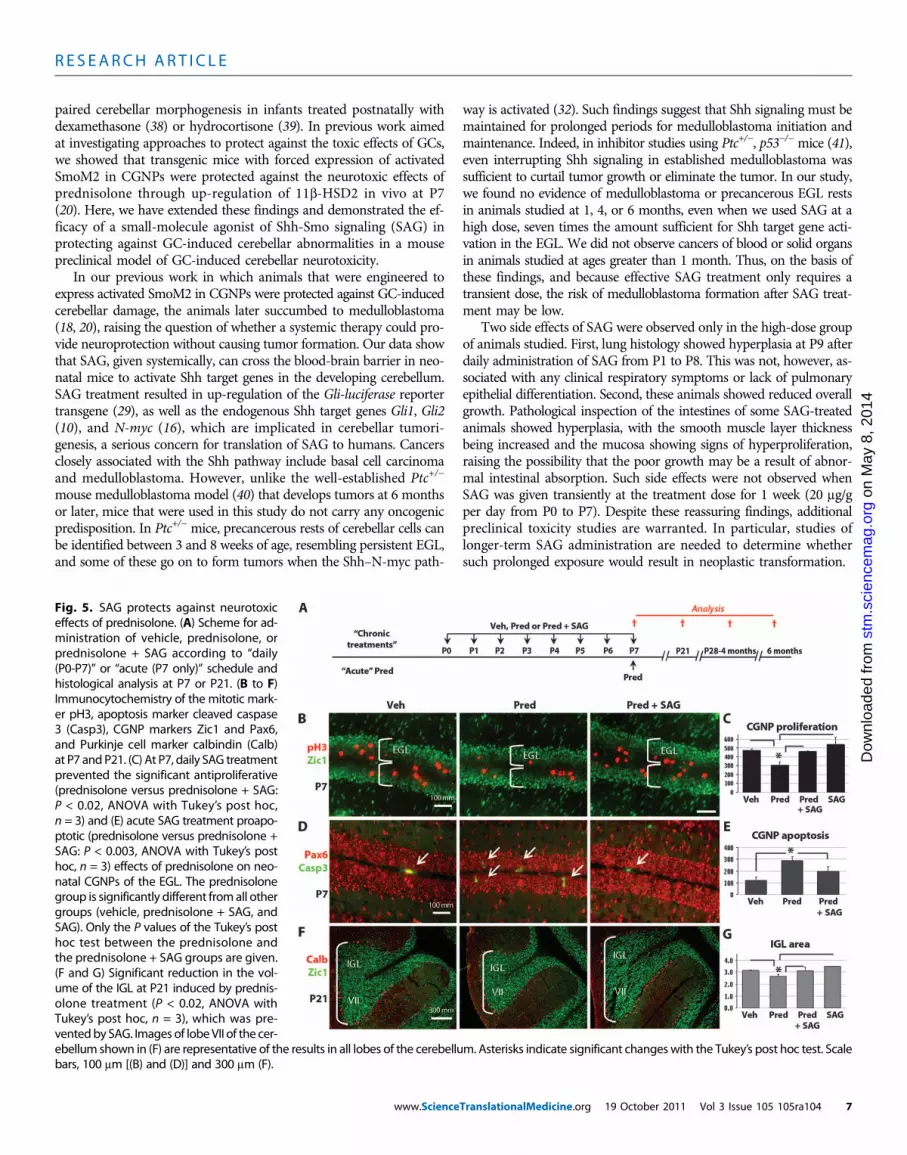
SAG protects against neurotoxic effects of prednisoloneWe next investigated whether SAG can antagonize the neurotoxiceffects of GCs on CGNPs in vivo (Fig. 5A). We have previously shownthat daily treatment with prednisolone from P0 to P7 results in signif-icant inhibition of CGNP proliferation (20), an effect that is mediated bythe GC receptor because mouse CGNPs do not express mineralocorticoid
stm
d fr
om
Table 1. Summary of SAG toxicity studies in neonatal mice. All mice weretreated (intraperitoneally) with SAG treatment dose (SAG-TD) (20 mg/g perday), SAG high dose (SAG-HD) (140 mg/g per day), or vehicle (daily) (P0 to
P7 or P1 to P8). Mice were sacrificed at P9, P14, and P28, and at 2, 4, or6 months of age for necropsy and comprehensive pathological analysis asindicated. Y, yes; N, no; ND, not done.
wnl
oade
Group
Age no
Treatment(P0 to P8)
Gross tumors?(Y/N)
Histological findings
TranslationalMedicine.org
Precancerous lesions“EGL rests” incerebellum?
19 October 2011 Vol 3 Issu
Complete bloodcount
D
1
P9 3 Vehicle N Unremarkable ND NDP9
3 SAG-HD N Intestinal hyperplasia(one of three animals)ND
ND2
P14 1 Vehicle N Unremarkable ND NDP14
1 SAG-HD N Intestinal hyperplasia ND ND3
P28 3 Vehicle N Unremarkable ND NDP28
3 SAG-TD N Unremarkable None NDP28
3 SAG-TD + prednisolone N Unremarkable None ND4
2 months 3 Vehicle N ND ND Within normal limits2 months
4 SAG-TD N ND ND Within normal limits2 months
2 SAG-TD N Unremarkable ND ND5
4 months 2 Vehicle N Unremarkable ND ND4 months
1 SAG-HD N Unremarkable None ND6
6 months 1 Vehicle N Unremarkable None ND6 months
1 SAG-HD N Unremarkable None NDe 105 105ra104 4
R E S EARCH ART I C L E
receptor (20). As shown (Fig. 5, B and C), administration of SAG(20 mg/g) with prednisolone (0.67 mg/g) prevented such inhibition ofproliferation within the external granule layer (EGL), which comprisesexclusively CGNP. SAG treatment alone did not cause a significantincrease in the baseline proliferation of CGNPs, or thickening of theEGL, characteristic of the precancerous state in Math1-cre, SmoM2
Vehicle SAG-HD Vehicle SAG-HD Vehicle SAG-HD V
www.Science
transgenic mice (18), nor was medulloblastoma observed at P21 (Fig.5F) or at longer survival time points (Fig. 3A and Table 1).
Acute treatment with GCs between P4 and P7 in the mouse causesapoptosis of CGNPs (20, 33). SAG treatment prevented apoptosis ofCGNPs in prednisolone-treated animals administered a one-time doseat P7 (Fig. 5, D and E). Treatment from P0 to P7 with GCs also results
Table 2. Clinical data (cyanosis assessment and modified-SHIRPAtest) on SAG-TD– and SAG-HD–treated mice. All the pups treatedwith SAG-HD were monitored every day for cyanosis. The color ofthe skin is indicated in the table for each day of injection. The
SAG-TD– and SAG-HD–treated animals were tested at 1 and 6 monthsof age, respectively, for behavioral, morphological, or performancechanges using the modified-SHIRPA protocol. N, normal; NP, notpresent; P, present.
Age
1 monthehicle SAG-HD Vehicle SAG-HD
TranslationalMedicine.org 19 Octobe
6 months
Treatment (P0 to P8)
Vehicle SAG-TD SAG-TD +prednisolone
VehicleVehicle SAG-HD
r 2011 Vol 3 Issue
SAG-HD
4
n Males 1 2 1 1 101
Females 2 1 2 — —, 2
Category Testsy 8
Coat
n M
a
Coat color; hair length;hair morphology;skin color; piloerection
N
N N N No
Morphologyrg
Tail, pinna, head, limb, whisker, andteeth morphology; palpebral closure
N
N N Ng.o
Abnormalhead
morphology
ma
Postureienc
e
Body position; body, limb,and abdominal tone;
visual placing; tail and pelvicelevation; trunk curl
N
N N N N.sc
Activity
stm
Spontaneous and locomotoractivity; wire maneuver;negative geotaxis; gait
N
N N N Poor negativegeotaxisom
Behaviored fr
Fear; irritability; aggression;vocalization; bizarre behavior;
convulsions; provoked biting; tremor
NP
NP NP NP NPad
Reflexesownl
o
Limb grasping; grip strength; pinnaand corneal reflex; toe pinch;touch escape; transfer arousal;
contact righting
P
P P P Semi-effectivegripD
Body releases
Defecation; urination; lacrimation;salivationN
N N N NHeart rate
N N N N NRespirationrate
N
N N N NBody weight (g)
Males 29.3 29.1 ± 0.6 28.9 48.5 28Females
26.0 ± 1.113 25 25.1 ± 0.4 — —Body length(mm)
Males
105 100 ± 0 105 120 90Females
102.5 ± 3.5 95 100 ± 0 — —Tail length(mm)
Males
95 95 ± 7.1 85 75 80Females
90.0 ± 7.1 90 95 ± 0 — —Age analysis
P1 P2 P3 P4 P5 P6 P7Vehicle SAG-HD
Treatment(P0 to P8)105 105ra1
n
6 8 5 8 4 7 4 7 4 7 3 7 3 7Skin color
Pink Pink Pink Pink Pink Pink Pink Pink Pink Pink Pink Pink Pink Pink04 5
R E S EARCH ART I C L E
in permanent deficits in the volume of granule neurons, but not thenumber of Purkinje cells (20). However, as shown (Fig. 5, F and G),these effects of daily injection for 1 week on granule neurons are alsoeffectively assuaged by SAG treatment. As expected, Purkinje cell pop-ulations, as measured by the density of calbindin-positive cells in thecerebellar Purkinje cell layer, were not affected by SAG (Fig. 5F).These findings indicate that SAG at the treatment dose effectively pre-vented GC-induced neonatal cerebellar developmental abnormalities.
DISCUSSION
Rates of cerebral palsy in the United States are rising because of in-creased rates of survival of extremely low birth weight preterm infantscoupled with their susceptibility to neurological damage from intrinsicor iatrogenic causes (34). Here, we have focused on postnatal GC
www.Science
treatment of preterm neonates, which is associated with cognitiveimpairment, cerebral palsy, and cerebellar hypoplasia (35–37). Yet,it remains necessary to use GCs for life-threatening conditions suchas severe chronic lung disease (also known as bronchopulmonarydysplasia), airway emergencies (for example, subglottic stenosis), in-tractable hypotension, and inflammation associated with cardiacbypass. Indeed, the most recent recommendations of the AmericanAcademy of Pediatrics continue to support the use of lower-doseGCs [for example, dexamethasone (0.15 mg/kg per day)] in pre-mature neonates with chronic lung disease.
We used a 1-week course of GCs given to neonatal mice at a doseequivalent to what has been used to treat human neonates to preventsevere chronic lung disease (dexamethasone, 0.1 mg/kg per day). Asshown in this and other studies (20, 33), even 11b-HSD2–sensitiveGCs such as prednisolone and hydrocortisone can have neurotoxiceffects on CGNPs in neonatal animals. Clinical studies also show im-
on
May
8, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
Fig. 4. SAG treatment has no detrimen-tal effect on lung maturation. (A and B)Phase-contrast analysis (upper panels)and immunocytochemistry (lower pan-els) for the differentiation markers SP-BSP-A and nuclear stain DAPI on P9-inflatedlungs treated with vehicle, prednisolone,SAG-TD, prednisolone + SAG-TD (A), andSAG-HD (B). (C) qRT-PCR analysis for SP-B,SP-A, 11b-HSD1, 11b-HSD2, Gli1, and Gli2 ex-pression in the P9 lung of SAG-HD–treatedmice (n = 3; not significant). Data in (C) areexpressed relative to values obtained withvehicle treatment (control), which wasset at 1. (D) qRT-PCR for SP-B on humanfetal lung explant cultures untreated(Waymouth medium) or treated with dexa-methasone (10 nM) or DCI (10 nM dexa-methasone, 0.1 mM 8-bromo-cAMP, and0.1 mM isobutylmethylxanthine together)in the presence or absence of SAG (120nM) (see Materials and Methods). SAGdoes not alter the expression level of SP-B(P < 0.005, ANOVA; Tukey’s post hoc: Way-mouth medium versus Waymouth medi-um + SAG, P > 0.05; dexamethasoneversus dexamethasone + SAG, P > 0.05;DCI versus DCI + SAG, P > 0.05; vehicle ver-sus DCI, P < 0.05, n = 3), which is differen-tially induced by GCs (43). Data areexpressed relative to control levels, whichwere set at 1. b-Actin was used as areference gene to calculate SP-B expres-sion levels (n = 3). Scale bars, 100 mm[(A) and (B)].
TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 6
R E S EARCH ART I C L E
on
May
8, 2
014
cem
ag.o
rg
paired cerebellar morphogenesis in infants treated postnatally withdexamethasone (38) or hydrocortisone (39). In previous work aimedat investigating approaches to protect against the toxic effects of GCs,we showed that transgenic mice with forced expression of activatedSmoM2 in CGNPs were protected against the neurotoxic effects ofprednisolone through up-regulation of 11b-HSD2 in vivo at P7(20). Here, we have extended these findings and demonstrated the ef-ficacy of a small-molecule agonist of Shh-Smo signaling (SAG) inprotecting against GC-induced cerebellar abnormalities in a mousepreclinical model of GC-induced cerebellar neurotoxicity.
In our previous work in which animals that were engineered toexpress activated SmoM2 in CGNPs were protected against GC-inducedcerebellar damage, the animals later succumbed to medulloblastoma(18, 20), raising the question of whether a systemic therapy could pro-vide neuroprotection without causing tumor formation. Our data showthat SAG, given systemically, can cross the blood-brain barrier in neo-natal mice to activate Shh target genes in the developing cerebellum.SAG treatment resulted in up-regulation of the Gli-luciferase reportertransgene (29), as well as the endogenous Shh target genes Gli1, Gli2(10), and N-myc (16), which are implicated in cerebellar tumori-genesis, a serious concern for translation of SAG to humans. Cancersclosely associated with the Shh pathway include basal cell carcinomaand medulloblastoma. However, unlike the well-established Ptc+/−
mouse medulloblastoma model (40) that develops tumors at 6 monthsor later, mice that were used in this study do not carry any oncogenicpredisposition. In Ptc+/− mice, precancerous rests of cerebellar cells canbe identified between 3 and 8 weeks of age, resembling persistent EGL,and some of these go on to form tumors when the Shh–N-myc path-
www.Science
way is activated (32). Such findings suggest that Shh signaling must bemaintained for prolonged periods for medulloblastoma initiation andmaintenance. Indeed, in inhibitor studies using Ptc+/−, p53−/− mice (41),even interrupting Shh signaling in established medulloblastoma wassufficient to curtail tumor growth or eliminate the tumor. In our study,we found no evidence of medulloblastoma or precancerous EGL restsin animals studied at 1, 4, or 6 months, even when we used SAG at ahigh dose, seven times the amount sufficient for Shh target gene acti-vation in the EGL. We did not observe cancers of blood or solid organsin animals studied at ages greater than 1 month. Thus, on the basis ofthese findings, and because effective SAG treatment only requires atransient dose, the risk of medulloblastoma formation after SAG treat-ment may be low.
Two side effects of SAG were observed only in the high-dose groupof animals studied. First, lung histology showed hyperplasia at P9 afterdaily administration of SAG from P1 to P8. This was not, however, as-sociated with any clinical respiratory symptoms or lack of pulmonaryepithelial differentiation. Second, these animals showed reduced overallgrowth. Pathological inspection of the intestines of some SAG-treatedanimals showed hyperplasia, with the smooth muscle layer thicknessbeing increased and the mucosa showing signs of hyperproliferation,raising the possibility that the poor growth may be a result of abnor-mal intestinal absorption. Such side effects were not observed whenSAG was given transiently at the treatment dose for 1 week (20 mg/gper day from P0 to P7). Despite these reassuring findings, additionalpreclinical toxicity studies are warranted. In particular, studies oflonger-term SAG administration are needed to determine whethersuch prolonged exposure would result in neoplastic transformation.
stm
.sci
enD
ownl
oade
d fr
om
Fig. 5. SAG protects against neurotoxiceffects of prednisolone. (A) Scheme for ad-ministration of vehicle, prednisolone, orprednisolone + SAG according to “daily(P0-P7)” or “acute (P7 only)” schedule andhistological analysis at P7 or P21. (B to F)Immunocytochemistry of the mitotic mark-er pH3, apoptosis marker cleaved caspase3 (Casp3), CGNP markers Zic1 and Pax6,and Purkinje cell marker calbindin (Calb)at P7 andP21. (C) At P7, daily SAG treatmentprevented the significant antiproliferative(prednisolone versus prednisolone + SAG:P < 0.02, ANOVA with Tukey’s post hoc,n = 3) and (E) acute SAG treatment proapo-ptotic (prednisolone versus prednisolone +SAG: P < 0.003, ANOVA with Tukey’s posthoc, n = 3) effects of prednisolone on neo-natal CGNPs of the EGL. The prednisolonegroup is significantly different fromall othergroups (vehicle, prednisolone + SAG, andSAG). Only the P values of the Tukey’s posthoc test between the prednisolone andthe prednisolone + SAG groups are given.(F and G) Significant reduction in the vol-ume of the IGL at P21 induced by prednis-olone treatment (P < 0.02, ANOVA withTukey’s post hoc, n = 3), which was pre-ventedby SAG. Images of lobeVII of the cer-
ebellum shown in (F) are representative of the results in all lobes of the cerebellum. Asterisks indicate significant changeswith the Tukey’s post hoc test. Scalebars, 100 mm [(B) and (D)] and 300 mm (F).TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 7

R E S EARCH ART I C L E
on
May
8, 2
014
stm
.sci
ence
mag
.org
ownl
oade
d fr
om
Our studies with SAG at treatment dose indicate that it is effectivein mice at preventing the neurotoxic effects of prednisolone in the neo-natal cerebellum, confirming and extending previous work with trans-genic mice that show that Shh signaling is antagonistic to GC signalingin CGNPs through an 11b-HSD2–dependent mechanism (20). Specif-ically, SAG co-administration prevented prednisolone-induced CGNPapoptosis and prednisolone inhibition of CGNP proliferation in theneonatal cerebellum, and preserved the normal volume of the cerebel-lar granule neuron populations.
GCs are primarily given to human neonates for their beneficialeffects on lung development and function. Thus, it was importantto determine whether SAG inhibited this beneficial GC activity.Analysis of murine lung in vivo indicated that SAG does not pre-vent normal pulmonary epithelial maturation. Additionally, SAGdid not inhibit GC-induced differentiation of human fetal lungcultures. Therefore, if SAG were to be used in the clinic as an ad-juvant therapy with GCs to prevent neurotoxicity, our data suggestthat it would not interfere with the beneficial effects of GCs in thedeveloping lung.
In summary, our data indicate that SAG crosses the blood-brain barrier to activate Shh-Smo gene targets in the cerebellum,protecting vulnerable neuronal precursors from GC-induced growthinhibition, and that such systemic treatment does not induce medul-loblastoma or other types of cancer. However, it will be important toconfirm these findings with additional toxicity studies to determinethe optimal dose schedule and further establish safety parameters(side effects, tumor development) in rodents and other animalsystems (nonhuman primate) before clinical trials in humans. SAGcan prevent the growth-inhibitory effects of GCs in the cerebellumwithout antagonizing the beneficial effects of GCs in the lung. Thesefindings suggest that adjuvant therapy with SAG could be a practicalway to prevent certain neurotoxic side effects of GCs given to pre-term neonates for life-threatening conditions. Testing of SAG in ad-ditional models of human neonatal brain injury may also bewarranted. Because Shh-Smo signaling is the major driver of cerebel-lar growth, this signaling axis may be affected in cerebellar injurybecause of hemorrhage or hypoxia (2). If so, then small-moleculeagonists of Shh signaling like SAG might help to prevent thesenewborn neurological injuries.
D
MATERIALS AND METHODS
Preparation of SAGSynthesis of SAG has been described (27). The chlorobenzo[b]thiophenederivative SAG (molecular weight, 490.06) was dissolved in dimethylsulfoxide (DMSO) to 5 mM and further diluted with normal saline orculture medium. These experiments used SAG prepared as a trifluoro-acetic acid salt or a free-base form. Vehicle controls comprised salinecontaining an equivalent concentration of DMSO.
AnimalsAll animal procedures were reviewed and approved by the Institution-al Animal Care and Use Committee of University of California, SanFrancisco (UCSF). The SW129/J and C57BL6Jmouse lines were obtainedfrom The Jackson Laboratory. The Gli-luciferase transgenic mouse line,which drives expression of luciferase through Gli-responsive cis-actingDNA regulatory sequences (29), was provided by E. Holland.
www.Science
Systemic administration of prednisolone and SAGOn P0 or P1, C57BL6J or Swiss Webster B pups received daily intra-peritoneal injections of prednisolone (0.67 mg/g, Sigma-Aldrich), SAG(20 mg/g), or prednisolone in combination with SAG, or vehicle, for7 days. For acute treatment, a one-time dose of prednisolone aloneor prednisolone + SAG was given at P7, and the cerebellum was har-vested for analysis 6 hours later.
CGNP primary cell culture, immunohistochemistry, Westernblot, and quantitative reverse transcription–PCRAll the experiments were done as described (20). Quantitative reversetranscription–PCR (qRT-PCR) was performed with SYBR Greenmaster mix (Roche) in LightCycler 480 (Roche). b-Actin was usedas a reference gene to calculate 2−DDCt, and experiments were performedin duplicate for each sample. For all experiments, at least two in-dependent samples were used per genotype and age that we examined(n values are indicated in the figure legends).
Human fetal lung explant cultureHuman fetal lung tissue from a 20-week gestation abortus was ob-tained from Advanced Bioscience Resources under an InstitutionalReview Board–approved protocol of the Children’s Hospital of Phila-delphia. The tissue was minced and placed in culture, as previouslydescribed (42). Explants were cultured in serum-free Waymouth me-dium on a rocking platform with an atmosphere of 95% air/5% CO2
for 24 hours and then cultured for 72 hours in Waymouth medium(control) or in medium containing dexamethasone (10 nM) alone orDCI [10 nM dexamethasone, 0.1 mM 8-bromo-cAMP (8-bromo-3′,5′-adenosine monophosphate), and 0.1 mM isobutylmethylxanthinetogether] in the presence or absence of SAG (120 nM). SP-B inductionin explants was calculated by qRT-PCR using human-specific primers.
Sample size and quantificationSurface area of the entire cerebellar EGL and IGL (internal granulelayer) was calculated as in (20). Results of primary CGNP cultureare representative of experiments repeated with pups from more thanfive independent litters. Total numbers of cells in 10 microscopic fields(magnification, ×20) were counted for the markers pH3 and cleavedcaspase 3 for statistical comparison.
StatisticsStatistical analysis was performed with an ANOVA (single factor). Ifthe ANOVA test gave a significant difference (P < 0.05), a Tukey’spost hoc test was performed.
REFERENCES AND NOTES
1. M. C. Allen, Neurodevelopmental outcomes of preterm infants. Curr. Opin. Neurol. 21, 123–128(2008).
2. L. W. Doyle, P. J. Anderson, Adult outcome of extremely preterm infants. Pediatrics 126,342–351 (2010).
3. M. Yanney, N. Marlow, Paediatric consequences of fetal growth restriction. Semin. FetalNeonatal Med. 9, 411–418 (2004).
4. M. P. Allin, S. Salaria, C. Nosarti, J. Wyatt, L. Rifkin, R. M. Murray, Vermis and lateral lobesof the cerebellum in adolescents born very preterm. Neuroreport 16, 1821–1824(2005).
5. J. B. Bodensteiner, S. D. Johnsen, Cerebellar injury in the extremely premature infant:Newly recognized but relatively common outcome. J. Child Neurol. 20, 139–142(2005).
TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 8

R E S EARCH ART I C L E
on
May
8, 2
014
stm
.sci
ence
mag
.org
Dow
nloa
ded
from
6. C. Limperopoulos, J. S. Soul, K. Gauvreau, P. S. Huppi, S. K. Warfield, H. Bassan, R. L. Robertson,J. J. Volpe, A. J. du Plessis, Late gestation cerebellar growth is rapid and impeded by prematurebirth. Pediatrics 115, 688–695 (2005).
7. J. J. Volpe, Cerebellum of the premature infant: Rapidly developing, vulnerable, clinicallyimportant. J. Child Neurol. 24, 1085–1104 (2009).
8. A. Tavano, R. Grasso, C. Gagliardi, F. Triulzi, N. Bresolin, F. Fabbro, R. Borgatti, Disorders ofcognitive and affective development in cerebellar malformations. Brain 130, 2646–2660(2007).
9. A. M. Kenney, H. R. Widlund, D. H. Rowitch, Hedgehog and PI-3 kinase signaling convergeon Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development131, 217–228 (2004).
10. J. D. Corrales, S. Blaess, E. M. Mahoney, A. L. Joyner, The level of sonic hedgehog signalingregulates the complexity of cerebellar foliation. Development 133, 1811–1821 (2006).
11. R. J. Wechsler-Reya, M. P. Scott, Control of neuronal precursor proliferation in the cerebellumby Sonic hedgehog. Neuron 22, 103–114 (1999).
12. P. Rakic, R. L. Sidman, Histogenesis of cortical layers in human cerebellum, particularly thelamina dissecans. J. Comp. Neurol. 139, 473–500 (1970).
13. R. D. Adams, M. Victor, A. H. Ropper, Principles of Neurology (McGraw-Hill, New York, 1997),p. 1618.
14. P. M. Lewis, A. Gritli-Linde, R. Smeyne, A. Kottmann, A. P. McMahon, Sonic hedgehogsignaling is required for expansion of granule neuron precursors and patterning of themouse cerebellum. Dev. Biol. 270, 393–410 (2004).
15. A. M. Kenney, D. H. Rowitch, Sonic hedgehog promotes G1 cyclin expression and sustained cellcycle progression in mammalian neuronal precursors. Mol. Cell. Biol. 20, 9055–9067 (2000).
16. A. M. Kenney, M. D. Cole, D. H. Rowitch, Nmyc upregulation by sonic hedgehog signalingpromotes proliferation in developing cerebellar granule neuron precursors. Development130, 15–28 (2003).
17. A. M. Dubuc, P. A. Northcott, S. Mack, H. Witt, S. Pfister, M. D. Taylor, The genetics of pediatricbrain tumors. Curr. Neurol. Neurosci. Rep. 10, 215–223 (2010).
18. U. Schüller, V. M. Heine, J. Mao, A. T. Kho, A. K. Dillon, Y. G. Han, E. Huillard, T. Sun, A. H. Ligon,Y. Qian, Q. Ma, A. Alvarez-Buylla, A. P. McMahon, D. H. Rowitch, K. L. Ligon, Acquisition ofgranule neuron precursor identity is a critical determinant of progenitor cell competence toform Shh-induced medulloblastoma. Cancer Cell 14, 123–134 (2008).
19. M. F. Beers, H. Shuman, H. G. Liley, J. Floros, L. W. Gonzales, N. Yue, P. L. Ballard, Surfactantprotein B in human fetal lung: Developmental and glucocorticoid regulation. Pediatr. Res.38, 668–675 (1995).
20. V. M. Heine, D. H. Rowitch, Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11bHSD2-dependent mechanism. J. Clin.Invest. 119, 267–277 (2009).
21. C. R. Edwards, R. Benediktsson, R. S. Lindsay, J. R. Seckl, 11b-Hydroxysteroid dehydro-genases: Key enzymes in determining tissue-specific glucocorticoid effects. Steroids 61,263–269 (1996).
22. M. C. Holmes, M. Sangra, K. L. French, I. R. Whittle, J. Paterson, J. J. Mullins, J. R. Seckl, 11b-Hydroxysteroid dehydrogenase type 2 protects the neonatal cerebellum from deleteriouseffects of glucocorticoids. Neuroscience 137, 865–873 (2006).
23. F. J. Frey, G. Escher, B. M. Frey, Pharmacology of 11b-hydroxysteroid dehydrogenase. Steroids59, 74–79 (1994).
24. R. Diaz, R. W. Brown, J. R. Seckl, Distinct ontogeny of glucocorticoid and mineralocorticoidreceptor and 11b-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brainsuggest a complex control of glucocorticoid actions. J. Neurosci. 18, 2570–2580 (1998).
25. M. Beato, A. Sánchez-Pacheco, Interaction of steroid hormone receptors with thetranscription initiation complex. Endocr. Rev. 17, 587–609 (1996).
26. N. A. Datson, M. C. Morsink, O. C. Meijer, E. R. de Kloet, Central corticosteroid actions:Search for gene targets. Eur. J. Pharmacol. 583, 272–289 (2008).
27. J. K. Chen, J. Taipale, K. E. Young, T. Maiti, P. A. Beachy, Small molecule modulation ofSmoothened activity. Proc. Natl. Acad. Sci. U.S.A. 99, 14071–14076 (2002).
28. M. Frank-Kamenetsky, X. M. Zhang, S. Bottega, O. Guicherit, H. Wichterle, H. Dudek, D. Bumcrot,F. Y. Wang, S. Jones, J. Shulok, L. L. Rubin, J. A. Porter, Small-molecule modulators of Hedgehogsignaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol.1, 10 (2002).
29. O. J. Becher, D. Hambardzumyan, E. I. Fomchenko, H. Momota, L. Mainwaring, A. M. Bleau,A. M. Katz, M. Edgar, A. M. Kenney, C. Cordon-Cardo, R. G. Blasberg, E. C. Holland, Gli activitycorrelates with tumor grade in platelet-derived growth factor–induced gliomas. Cancer Res.68, 2241–2249 (2008).
30. L. Lum, P. A. Beachy, The Hedgehog response network: Sensors, switches, and routers.Science 304, 1755–1759 (2004).
www.Science
31. H. Masuya, M. Inoue, Y. Wada, A. Shimizu, J. Nagano, A. Kawai, A. Inoue, T. Kagami, T. Hirayama,A. Yamaga, H. Kaneda, K. Kobayashi, O. Minowa, I. Miura, Y. Gondo, T. Noda, S. Wakana,T. Shiroishi, Implementation of the modified-SHIRPA protocol for screening of dominantphenotypes in a large-scale ENU mutagenesis program. Mamm. Genome 16, 829–837(2005).
32. J. D. Kessler, H. Hasegawa, S. N. Brun, B. A. Emmenegger, Z. J. Yang, J. W. Dutton, F. Wang,R. J. Wechsler-Reya, N-myc alters the fate of preneoplastic cells in a mouse model ofmedulloblastoma. Genes Dev. 23, 157–170 (2009).
33. K. K. Noguchi, K. C. Walls, D. F. Wozniak, J. W. Olney, K. A. Roth, N. B. Farber, Acute neonatalglucocorticoid exposure produces selective and rapid cerebellar neural progenitor cellapoptotic death. Cell Death Differ. 15, 1582–1592 (2008).
34. S. R. Hintz, D. E. Kendrick, D. E. Wilson-Costello, A. Das, E. F. Bell, B. R. Vohr, R. D. Higgins;NICHD Neonatal Research Network, Early-childhood neurodevelopmental outcomes arenot improving for infants born at <25 weeks’ gestational age. Pediatrics 127, 62–70(2011).
35. S. R. Dalziel, V. K. Lim, A. Lambert, D. McCarthy, V. Parag, A. Rodgers, J. E. Harding, Antenatalexposure to betamethasone: Psychological functioning and health related quality of life31 years after inclusion in randomised controlled trial. BMJ 331, 665 (2005).
36. G. C. Liggins, R. N. Howie, A controlled trial of antepartum glucocorticoid treatment for pre-vention of the respiratory distress syndrome in premature infants. Pediatrics 50, 515–525(1972).
37. D. Wilson-Costello, M. C. Walsh, J. C. Langer, R. Guillet, A. R. Laptook, B. J. Stoll, S. Shankaran,N. N. Finer, K. P. Van Meurs, W. A. Engle, A. Das; Eunice Kennedy Shriver National Institute ofChild Health and Human Development Neonatal Research Network, Impact of postnatalcorticosteroid use on neurodevelopment at 18 to 22 months’ adjusted age: Effects of dose,timing, and risk of bronchopulmonary dysplasia in extremely low birth weight infants.Pediatrics 123, e430–e437 (2009).
38. T. F. Yeh, Y. J. Lin, H. C. Lin, C. C. Huang, W. S. Hsieh, C. H. Lin, C. H. Tsai, Outcomes at schoolage after postnatal dexamethasone therapy for lung disease of prematurity. N. Engl. J. Med.350, 1304–1313 (2004).
39. E. W. Y. Tam, V. Chau, D. M. Ferriero, A. J. Barkovich, K. J. Poskitt, C. Studholme, E. D.-Y. Fok,R. E. Grunau, D. V. Glidden, S. P. Miller, Preterm cerebellar growth impairment after post-natal exposure to glucocorticoids. Sci. Transl. Med. 3, 105ra105 (2011).
40. L. V. Goodrich, L. Milenković, K. M. Higgins, M. P. Scott, Altered neural cell fates andmedulloblastoma in mouse patched mutants. Science 277, 1109–1113 (1997).
41. J. T. Romer, T. Curran, Medulloblastoma and retinoblastoma: Oncology recapitulates ontogeny.Cell Cycle 3, 917–919 (2004).
42. L. W. Gonzales, P. L. Ballard, R. Ertsey, M. C. Williams, Glucocorticoids and thyroid hormonesstimulate biochemical and morphological differentiation of human fetal lung in organculture. J. Clin. Endocrinol. Metab. 62, 678–691 (1986).
43. V. C. Venkatesh, D. M. Iannuzzi, R. Ertsey, P. L. Ballard, Differential glucocorticoid regulationof the pulmonary hydrophobic surfactant proteins SP-B and SP-C. Am. J. Respir. CellMol. Biol. 8, 222–228 (1993).
44. Acknowledgments: We thank A. M. Barette, S. Kaing, and M. Wong for expert technicalassistance; J. Otero, E. Huang, B. Hann, and the UCSF Comprehensive Cancer Center pa-thology core for blood and whole-body solid tumor screening; L. Gonzales for fetal lungcultures; and E. Holland for providing Gli-luciferase mice. Funding: V.M.H. thanks the Nether-lands Organization for Scientific Research for a TALENT stipend. A.G. is grateful for support bya fellowship from the American Brain Tumor Association. This work was supported by grantsfrom the March of Dimes Foundation (to D.H.R.) and NIH (R01 CA136574 to J.K.C.; R01NS047527 to D.H.R.; RO1 HL086323 and PO1 HL024075 to P.L.B.). D.H.R. is a Howard HughesMedical Institute Investigator. Author contributions: V.M.H., C.C., and A.G. acquired and ana-lyzed the data. V.M.H., J.K.C., and D.H.R. conceived the study. All authors designed theexperiments and wrote the manuscript. Competing interests: The authors declare that theyhave no competing interests. The SAG compound was synthesized at Stanford University andis available under a materials transfer agreement.
Submitted 31 May 2011Accepted 29 September 2011Published 19 October 201110.1126/scitranslmed.3002731
Citation: V. M. Heine, A. Griveau, C. Chapin, P. L. Ballard, J. K. Chen, D. H. Rowitch, A small-molecule Smoothened agonist prevents glucocorticoid-induced neonatal cerebellar injury.Sci. Transl. Med. 3, 105ra104 (2011).
TranslationalMedicine.org 19 October 2011 Vol 3 Issue 105 105ra104 9





























