Embed Size (px)
Citation preview

A BEN-domain-containing protein associates withheterochromatin and represses transcription
Kizhakke M. Sathyan, Zhen Shen, Vidisha Tripathi, Kannanganattu V. Prasanth and Supriya G. Prasanth*Department of Cell and Developmental Biology, University of Illinois at Urbana-Champaign, 601S Goodwin Avenue, Urbana, IL 61801, USA
*Author for correspondence ([email protected])
Accepted 2 June 2011Journal of Cell Science 124, 3149–3163� 2011. Published by The Company of Biologists Ltddoi: 10.1242/jcs.086603
SummaryIn eukaryotes, higher order chromatin structure governs crucial cellular processes including DNA replication, transcription and post-transcriptional gene regulation. Specific chromatin-interacting proteins play vital roles in the maintenance of chromatin structure. We
have identified BEND3, a quadruple BEN domain-containing protein that is highly conserved amongst vertebrates. BEND3 colocalizeswith HP1 and H3 trimethylated at K9 at heterochromatic regions in mammalian cells. Using an in vivo gene locus, we have been able todemonstrate that BEND3 associates with the locus only when it is heterochromatic and dissociates upon activation of transcription.
Furthermore, tethering BEND3 inhibits transcription from the locus, indicating that BEND3 is involved in transcriptional repressionthrough its interaction with histone deacetylases and Sall4, a transcription repressor. We further demonstrate that BEND3 isSUMOylated and that such modifications are essential for its role in transcriptional repression. Finally, overexpression of BEND3 causes
premature chromatin condensation and extensive heterochromatinization, resulting in cell cycle arrest. Taken together, our datademonstrate the role of a novel heterochromatin-associated protein in transcriptional repression.
Key words: BEND3, Heterochromatin, Transcriptional repression, SUMO
IntroductionAccurate control of gene expression is crucial for cell survival
and is clearly dependent on the chromatin status (Narlikar et al.,2002). Although gene activation is generally associated witheuchromatic sites with increased histone acetylation, gene
inactivation at heterochromatin is marked with methylation ofhistone H3 at lysine 9 and hypoacetylation of histones (forreviews, see Dillon and Festenstein, 2002; Hubner and Spector,2010; Richards and Elgin, 2002). The heterochromatin protein
HP1 binds to histone H3 trimethylated at lysine 9 (H3me3K9)and contributes to spreading of heterochromatin, resulting insilencing of gene expression at those sites (Bannister et al., 2001;
Jacobs and Khorasanizadeh, 2002; Lachner et al., 2001; Nielsenet al., 2002). Thus, HP1 proteins are implicated in transcriptionalrepression by establishing specialized, higher-order chromatin
structures (Eissenberg and Elgin, 2000; Kellum, 2003a; Kellum,2003b; Kwon and Workman, 2008; Maison and Almouzni, 2004;Nielsen et al., 2001; Stewart et al., 2005). Recent evidence has
also indicated that HP1 proteins participate in transcriptionalrepression both in heterochromatin and euchromatin (Hedigerand Gasser, 2006; Kwon and Workman, 2008). Heterochromaticregions are typically composed of repetitive sequences present at
centromeres and telomeres, are usually late replicating andtranscriptionally silent (Buhler and Gasser, 2009; Fodor et al.,2010; Schoeftner and Blasco, 2009; Vermaak and Malik, 2009).
Our understanding of the molecular network that establishesheterochromatin structure and triggers transcriptional repressionremains far from complete.
Transcriptional repression is mediated through several pathways(Cowell, 1994; Johnson, 1995). One pathway involves therecruitment of chromatin regulators, including chromatin
remodeling complex, that cause localized histone deacetylation
that ultimately results in transcriptional repression of specificgenes (Hassig et al., 1997; Kadosh and Struhl, 1998; Nan et al.,
1998; Rundlett et al., 1998; Xue et al., 1998; Zhang et al., 1997).Another pathway involves the inactivation of transcription
mediated by protein–protein interactions, which prevents theassembly of RNA polymerase or general transcription factors
required to establish the preinitiation complex at transcription startsites (Breiling et al., 2001; Inostroza et al., 1992; Meisterernst and
Roeder, 1991; Um et al., 1995). Furthermore, small ubiquitin-likemodifier (SUMO) protein modification of transcription factors has
been associated with repression (Geiss-Friedlander and Melchior,
2007; Gill, 2005; Hay, 2005; Yang and Sharrocks, 2004).SUMOylation is a covalent post-translational modification,
whereby SUMO is attached by an iso-peptide linkage tolysine(s) residue in the target protein (Bayer et al., 1998;
Johnson, 2004; Mukhopadhyay and Riezman, 2007). Recentstudies have shown that SUMOylation of specific chromatin-
associated factors regulates gene expression by altering chromatinarchitecture, which results in localized heterochromatinization and
inability of the transcription machinery to interact at specificchromatin sites (Stielow et al., 2008a; Stielow et al., 2008b;
Uchimura et al., 2006).
We have identified BEND3 (originally known as KIAA1553),
a protein containing four BEN domains, which are thought to beinvolved in chromatin function and transcription. Localization
studies in mammalian cells showed that BEND3 is aheterochromatin-associated protein in which the BEN domain 4
is crucial for heterochromatin association. Tethering BEND3 to aspecific gene locus results in transcriptional repression, probably
by altering the chromatin structure. Furthermore, BEND3 is
Research Article 3149
Journ
alof
Cell
Scie
nce
SUMOylated and this modification is essential for its repressive
function but not for its association with chromatin. Finally,
overexpression of BEND3 caused premature chromatin
condensation, severe heterochromatinization and cell cycle
arrest. A very recent proteomic study has demonstrated that
BEND3 is a component of Sall4, a repressive NuRD transcription
factor complex implicated in stem cell pluripotency (van den
Berg et al., 2010). We provide evidence that BEND3 associates
with heterochromatin, interacts with histone deacetylases
(HDACs) and Sall4, causes transcriptional repression and can
shut down transcription at an actively transcribing genic region.
We suggest that BEND3, in conjunction with the NuRD complex,
mediates transcriptional repression of specific genes.
ResultsBEND3 is highly conserved amongst vertebrates
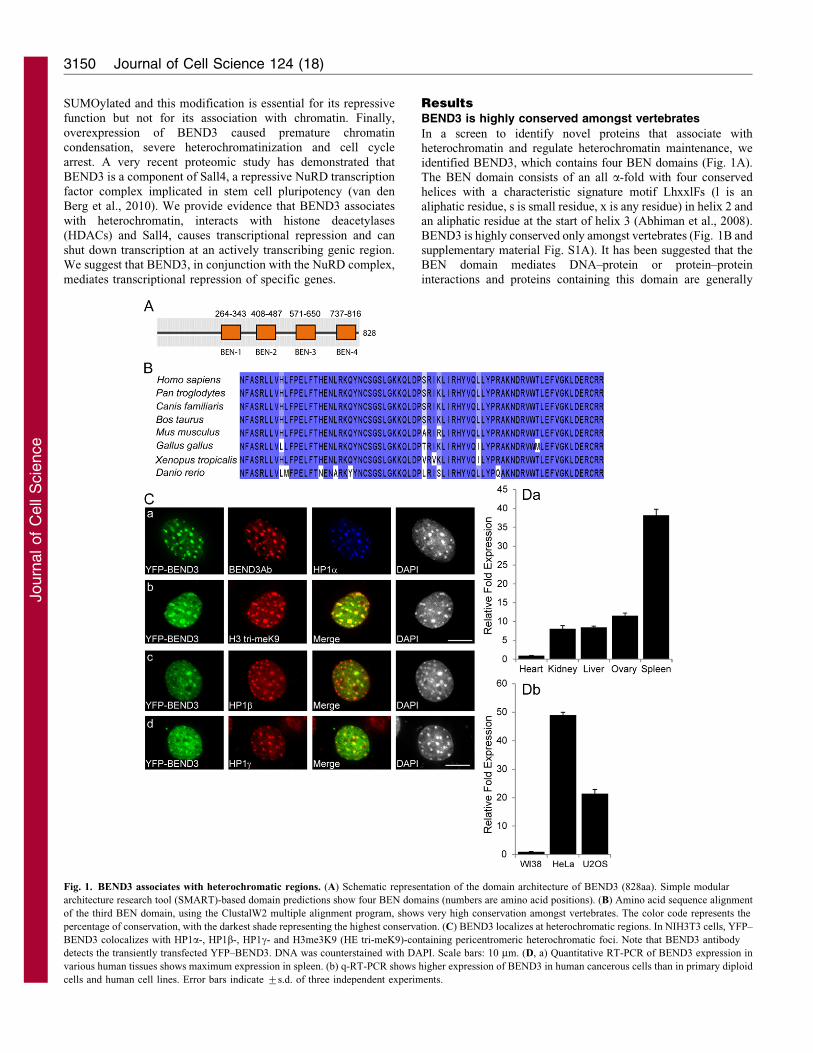
In a screen to identify novel proteins that associate withheterochromatin and regulate heterochromatin maintenance, weidentified BEND3, which contains four BEN domains (Fig. 1A).The BEN domain consists of an all a-fold with four conserved
helices with a characteristic signature motif LhxxlFs (l is analiphatic residue, s is small residue, x is any residue) in helix 2 andan aliphatic residue at the start of helix 3 (Abhiman et al., 2008).
BEND3 is highly conserved only amongst vertebrates (Fig. 1B andsupplementary material Fig. S1A). It has been suggested that theBEN domain mediates DNA–protein or protein–protein
interactions and proteins containing this domain are generally
Fig. 1. BEND3 associates with heterochromatic regions. (A) Schematic representation of the domain architecture of BEND3 (828aa). Simple modular
architecture research tool (SMART)-based domain predictions show four BEN domains (numbers are amino acid positions). (B) Amino acid sequence alignment
of the third BEN domain, using the ClustalW2 multiple alignment program, shows very high conservation amongst vertebrates. The color code represents the
percentage of conservation, with the darkest shade representing the highest conservation. (C) BEND3 localizes at heterochromatic regions. In NIH3T3 cells, YFP–
BEND3 colocalizes with HP1a-, HP1b-, HP1c- and H3me3K9 (HE tri-meK9)-containing pericentromeric heterochromatic foci. Note that BEND3 antibody
detects the transiently transfected YFP–BEND3. DNA was counterstained with DAPI. Scale bars: 10 mm. (D, a) Quantitative RT-PCR of BEND3 expression in
various human tissues shows maximum expression in spleen. (b) q-RT-PCR shows higher expression of BEND3 in human cancerous cells than in primary diploid
cells and human cell lines. Error bars indicate ¡s.d. of three independent experiments.
Journal of Cell Science 124 (18)3150
Journ
alof
Cell
Scie
nce

involved in chromatin organization and transcription (Abhiman etal., 2008).
BEND3 localizes to heterochromatin, which is dependenton the BEN domain 4
To determine the intracellular location of BEND3, we generatedvarious epitope-tagged versions of BEND3, with T7 (an 11 aminoacid peptide encoded in the leader sequence of T7 bacteriophage
gene10), hemagglutinin (HA) or yellow fluorescent protein (YFP)at either the N-terminus or C-terminus of BEND3. YFP–BEND3was found to be primarily nuclear with several punctate foci in
mouse cells (Fig. 1C) as well as in human cells, including U2OS(supplementary material Fig. S1Ba) and HeLa (supplementarymaterial Fig. S1Bb). Similar observations were made with
BEND3–YFP (supplementary material Fig. S1Ca) and T7–BEND3 (supplementary material Fig. S1Cb) demonstrating thatthe protein bearing different tags on either end has similar
localization. Furthermore, in mouse NIH3T3 cells, BEND3clearly associated with HP1a-, HP1b-, HP1c- and H3me3K9-containing heterochromatic foci (Fig. 1Ca–d). We generatedseveral antibodies against BEND3 but none of these detected the
endogenous BEND3 by immunoblots or immunofluorescence (datanot shown). These antibodies, however, could detect exogenouslyexpressed YFP–BEND3 in cells using immunofluorescence,
immunoprecipitation, and also immunoblotting (Fig. 1Ca andsupplementary material Fig. S1D,E). YFP–BEND3 occurs inseveral forms, with residues S379, S489 or S503 phosphorylated
(supplementary material Fig. S1E), as has been reported forendogenous BEND3 in phospho-proteomic screens (Brill et al.,2009; Olsen et al., 2006).
We used a quantitative real-time PCR approach in order toanalyze the expression of endogenous BEND3 in both mouse andhuman cell lines (Fig. 1D). Analysis of BEND3 expression in
human tissues showed maximum levels of expression in spleenand least in heart (Fig. 1Da). Interestingly, BEND3 expressionwas much higher in transformed or cancerous cell-lines (humanU2OS, HeLa; Fig. 1Db and mouse NIH3T3; data not shown)
than in primary diploid fibroblasts (WI38; Fig. 1Db).
To determine the region of BEND3 that is essential for its
localization to heterochromatin, we generated several mutants ofBEND3 spanning each of the four BEN domains (Fig. 2A). YFP-tagged mutants containing the N-terminal fragments, amino acids(aa) 1–357, 1–529 and 1–660 and the C-terminal fragments aa 390–
828, 551–828 and 718–828 were transfected into mouse NIH3T3cells and the localization of these mutants was assessed (Fig. 2B).The mutants expressing YFP–BEND3.718–828 (Fig. 2Be), YFP–
BEND3.551–828 (Fig. 2Bf) and YFP–BEND3.390–828 (Fig. 2Bg)localized to pericentric heterochromatic regions similar to that ofwild-type BEND3 (BEND3.WT; Fig. 2Ba) suggesting that the BEN
domain 4 is vital for heterochromatin localization (Fig. 2A,Be–g).Although the aa 1–357 mutant (Fig. 2Bb) showed few punctate foci,the aa 1–529 and 1–660 mutants showed homogenous nuclear
localization and did not associate with heterochromatic regionsdetected as DAPI-dense foci (Fig. 2Bc,d). Determination of thePearson coefficient of correlation further corroborated theseobservations (Fig. 2B, right side of the panel). These results
indicate that BEN domain 4 is necessary and sufficient for thelocalization of BEND3 to heterochromatic regions.
To examine the role of BEND3 in heterochromatinorganization, we have utilized a modified version of the in vivocell system devised by David Spector’s group (Janicki et al., 2004).
Fig. 2. BEN domain 4 is crucial for heterochromatin localization of
BEND3. (A) Schematic representation of YFP-tagged BEND3 truncation
mutants. The lengths of the bars do not truly reflect the length of the mutants.
(B) NIH3T3 cells were transfected with YFP–BEND3 wild type or truncation
mutants. YFP–BEND3.WT (a) and truncation mutants 718–828 (e), 551–828
(f) and 390–828 (g) localize to the heterochromatic region, indicating that the
C-terminus of the protein is necessary for its localization to heterochromatin.
Mutant 1–357 (b) shows large nuclear foci, whereas 1–529 (c) and 1–660 (d)
are homogenously distributed. DNA was counterstained with DAPI. The
Pearson coefficient of correlation is given on the right-hand side of the panel.
Scale bar: 10 mm.
BEND3 represses transcription 3151
Journ
alof
Cell
Scie
nce
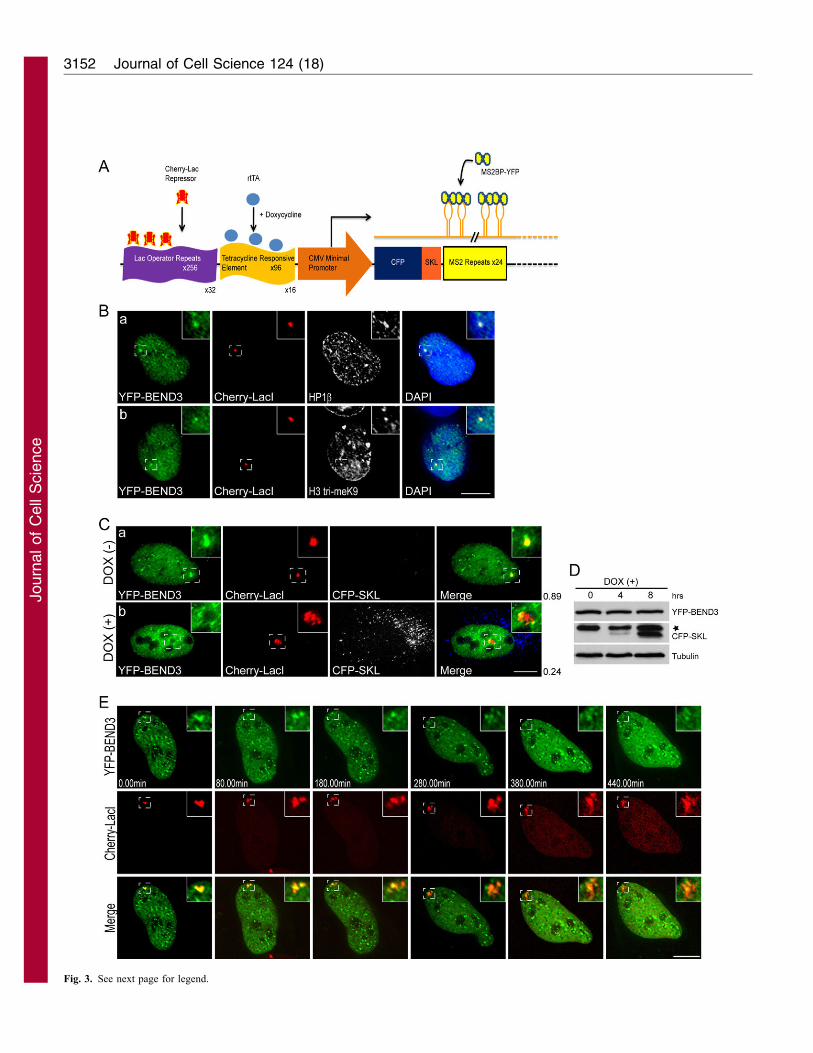
Fig. 3. See next page for legend.
Journal of Cell Science 124 (18)3152
Journ
alof
Cell
Scie
nce

A 200 copy transgene array, with each array having 256 copies of
the Lac operator, had been integrated into human U2OS cell as a
single heterochromatic locus (U2OS-2-6-3) (Janicki et al., 2004).
mCherry–LacI (Lac repressor) and rtTa (reverse tetracycline-
controlled transcriptional activator) were stably integrated in the 2-
6-3 cells, so that the locus containing the transgene array could be
readily visualized in living cells by the presence of mCherry–LacI
and the heterochromatic locus could be decondensed upon
transcriptional activation by the addition of doxycycline (DOX)
in the medium (U2OS-2-6-3 CLTon, mCherry–LacI tetracycline
activator; Fig. 3A) (Bernard et al., 2010; Prasanth et al., 2010;
Shen et al., 2010). YFP–BEND3 localized to the heterochromatic
locus in the U2OS-2-6-3 CLTon cells and showed complete
colocalization with HP1b (Fig. 3Ba), HP1a (supplementary
material Fig. S2B) and H3me3K9 (Fig. 3Bb) at the locus.
Transient transfection of BEND3 mutants in U2OS 2-6-3 CLTon
cells showed that only the constructs containing BEN domain 4
localize to the heterochromatic locus, similar to our observations
of NIH3T3 cells (supplementary material Fig. S2A,C).
We then examined whether association of BEND3 with
chromatin is specified by the state of the chromatin structure, i.e.
whether BEND3 localizes to condensed heterochromatin as well as
decondensed chromatin. Significantly, changing the status of the
gene locus from heterochromatin to euchromatin by activating
DOX-induced transcription as is evident by the decondensation of
the locus (Fig. 3Cb, red) as well as the appearance of the reporter
protein cyan fluorescent protein (CFP)–SKL (serine-lysine-leucine,
peroxisomal targeting signal), resulted in the release of a large
fraction of YFP–BEND3 from the gene locus (Fig. 3C).
Immunoblots corroborated the increase in CFP–SKL reporter
protein in DOX-treated cells (Fig. 3D). Time-lapse imaging of
cells expressing YFP–BEND3 clearly demonstrated the removal of
YFP–BEND3 from the gene locus during chromatin decondensation
(Fig. 3E and supplementary material Fig. S3A, Movies 1 and 2).
BEND3 is SUMOylated at several lysine residues
Recent studies have indicated that SUMOylation modulates the
association of proteins to heterochromatic regions (Reo et al., 2010;
Shin et al., 2005). Similarly, the SUMO-conjugating enzyme Ubc9/Hus5 has been implicated in heterochromatin organization and
transcriptional repression (Shiio and Eisenman, 2003). Using theSILAC (stable isotope labeling with amino acids in cell culture)approach, a previous study compared target protein sets for SUMO1and SUMO2 and found KIAA1553/BEND3 as a potential target
(Vertegaal et al., 2006). Using bioinformatics tools (SUMOplotalgorithm) to predict potential SUMOylation sites on BEND3 weidentified two sites in BEND3 with the SUMO consensus motif
yKx(E/D) (where y is Val, Ile, Leu, Met or Phe, and x is any aminoacid) at residues K20 and K512 (Fig. 4Aa). These two consensussites are highly conserved in mammals (Fig. 4Ab).
To examine the status of SUMOylation of BEND3 and todetermine the lysines that serve as SUMO1 or SUMO2/3acceptors, we transfected YFP–BEND3 with and without HA–SUMO1 or HA–SUMO2 in human U2OS cells. The SUMOylation
status was assessed by immunoblots and immunoprecipitationanalysis. Extracts from cells co-transfected with YFP–BEND3 andHA–SUMO1 clearly showed higher molecular mass forms
representing SUMOylated BEND3 (Fig. 4B). Immunoblots usingGFP antibody showed that YFP–BEND3 yielded an additional,slower migrating form in both SUMO1- and SUMO2-transfected
cells (Fig. 4Ca). This form of YFP–BEND3 was also detected byHA antibody indicating that YFP–BEND3 is SUMOylated bySUMO1 or SUMO2 (Fig. 4Cb, asterisk). In addition, HA alsodetected other higher molecular mass forms that do not correspond
to BEND3 and could represent multiple SUMOylation orSUMOylated proteins that interact with BEND3.
To determine which lysine (K) moiety is SUMOylated, we
generated mutants in which K20, K512 or both sites (henceforthtermed SUMO double mutant, SDM) were mutated to arginines(Fig. 4Aa). Each of the mutants was cotransfected with HA–
SUMO1 (Fig. 4D) or HA–SUMO2 (supplementary material Fig.S3B) and assayed as described above. YFP–BEND3 showed twoslower-migrating forms when co-transfected with HA–SUMO1
(Fig. 4Da). Mutation of K20, abolished one of the slower-migratingforms of BEND3 completely, whereas the second form wassignificantly reduced after transfection with either SUMO1(Fig. 4Da) or SUMO2 (supplementary material Fig. S3Ba). The
mutation in K512 also resulted in only one of the slow-migratingforms, corresponding to SUMOylation at K20 residue (Fig. 4Da andsupplementary material Fig. S3Ba). It is worth noting that in the K20
mutant, a weak, slower-migrating band of YFP–BEND3 waspresent, suggesting that the SUMOylation at K512 predominantlydepends on initial SUMOylation at K20 (Fig. 4Da and
supplementary material Fig. S3Ba). When both K20 and K512were mutated simultaneously (SDM), the YFP–BEND3 mutantshowed reduced mobility in SDS-PAGE and loss of slower-migrating forms of BEND3. It is also possible that SUMO3
contributes to BEND3 SUMOylation. To address this possibility, weco-transfected HA–SUMO3 and YFP–BEND3 and observed thepresence of slower migrating forms of YFP–BEND3 that were
abolished in the YFP–BEND3.K20R, whereas only one of the twoslower migrating bands was abolished in the YFP–BEND3.K512Rmutant (Fig. 4E). The significant reduction of SUMO markers in the
K20R mutant suggests that SUMOylation at K20 may be needed forefficient SUMOylation at the K512 site. Thus, SUMOylation ofBEND3 probably occurs sequentially, with K20 preceding K512.
Quantification of the immunoblots revealed that in YFP–BEND3.K20R mutant, SUMOylation is lost at both the sites,whereas in cells expressing the YFP–BEND3.K512R mutant,
Fig. 3. BEND3 is displaced from decondensed chromatin during
activation of transcription. (A) A schematic representation of the gene locus
in U2OS 2-6-3 CLTon cells [adapted and modified from (Janicki et al.,
2004)]. (B) YFP–BEND3 associates with HP1b- and H3me3K9 (HE tri-
meK9)-containing heterochromatic gene locus in CLTon cells. (C, a) YFP–
BEND3 (green) associates with the artificially generated heterochromatic
gene locus in 2-6-3CLTon cells as shown by its association with mCherry–
LacI-containing (red) gene locus. (b) Activation of transcription (DOX+)
results in the displacement of BEND3 from the de-condensed gene locus.
Note the appearance of CFP–SKL protein in the DOX-treated cells. The
coefficient of correlation (right side of the panel) corroborates the overlap of
YFP–BEND3 with mCherry–LacI at the condensed locus.
(D) Immunoblot analysis of YFP–BEND3-expressing cells, using a GFP
antibody, in untreated (0 hours), DOX-treated (4 hours and 8 hours) cells.
Note the YFP–BEND3 levels remain constant over time, whereas the CFP–
SKL signal increases. (*) denotes non-specific band. Tubulin is shown as a
loading control. (E) Live-cell imaging in 2-6-3 CLTon cells demonstrating
the displacement of YFP–BEND3.WT from the gene locus during
transcriptional activation. The images were taken at every 20 minutes. DOX
(1 mg/ml) was infused into the medium at 0.00 minutes. YFP–BEND3, which
initially localized to the heterochomatic loci was gradually released upon de-
condensation of the gene locus. YFP–BEND3 is still seen localized to the
partially heterochromatic spots remaining at the locus. Scale bars: 10 mm.
BEND3 represses transcription 3153
Journ
alof
Cell
Scie
nce
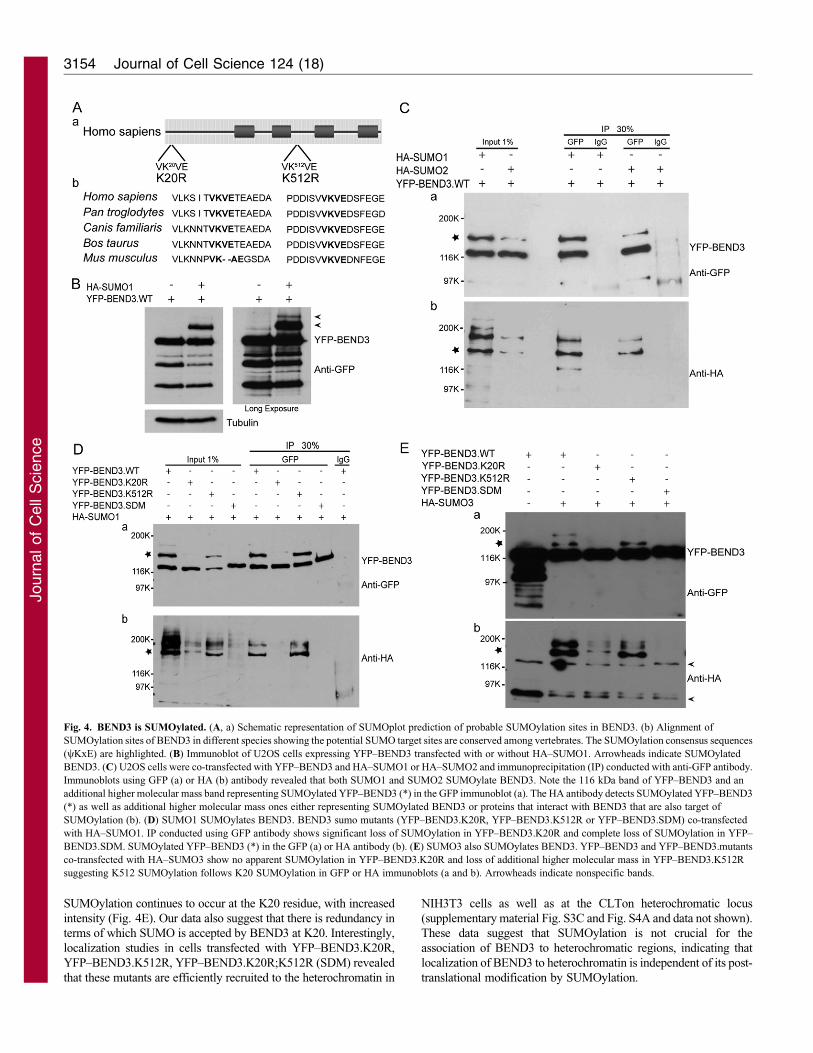
SUMOylation continues to occur at the K20 residue, with increased
intensity (Fig. 4E). Our data also suggest that there is redundancy in
terms of which SUMO is accepted by BEND3 at K20. Interestingly,
localization studies in cells transfected with YFP–BEND3.K20R,
YFP–BEND3.K512R, YFP–BEND3.K20R;K512R (SDM) revealed
that these mutants are efficiently recruited to the heterochromatin in
NIH3T3 cells as well as at the CLTon heterochromatic locus
(supplementary material Fig. S3C and Fig. S4A and data not shown).
These data suggest that SUMOylation is not crucial for the
association of BEND3 to heterochromatic regions, indicating that
localization of BEND3 to heterochromatin is independent of its post-
translational modification by SUMOylation.
Fig. 4. BEND3 is SUMOylated. (A, a) Schematic representation of SUMOplot prediction of probable SUMOylation sites in BEND3. (b) Alignment of
SUMOylation sites of BEND3 in different species showing the potential SUMO target sites are conserved among vertebrates. The SUMOylation consensus sequences
(yKxE) are highlighted. (B) Immunoblot of U2OS cells expressing YFP–BEND3 transfected with or without HA–SUMO1. Arrowheads indicate SUMOylated
BEND3. (C) U2OS cells were co-transfected with YFP–BEND3 and HA–SUMO1 or HA–SUMO2 and immunoprecipitation (IP) conducted with anti-GFP antibody.
Immunoblots using GFP (a) or HA (b) antibody revealed that both SUMO1 and SUMO2 SUMOylate BEND3. Note the 116 kDa band of YFP–BEND3 and an
additional higher molecular mass band representing SUMOylated YFP–BEND3 (*) in the GFP immunoblot (a). The HA antibody detects SUMOylated YFP–BEND3
(*) as well as additional higher molecular mass ones either representing SUMOylated BEND3 or proteins that interact with BEND3 that are also target of
SUMOylation (b). (D) SUMO1 SUMOylates BEND3. BEND3 sumo mutants (YFP–BEND3.K20R, YFP–BEND3.K512R or YFP–BEND3.SDM) co-transfected
with HA–SUMO1. IP conducted using GFP antibody shows significant loss of SUMOylation in YFP–BEND3.K20R and complete loss of SUMOylation in YFP–
BEND3.SDM. SUMOylated YFP–BEND3 (*) in the GFP (a) or HA antibody (b). (E) SUMO3 also SUMOylates BEND3. YFP–BEND3 and YFP–BEND3.mutants
co-transfected with HA–SUMO3 show no apparent SUMOylation in YFP–BEND3.K20R and loss of additional higher molecular mass in YFP–BEND3.K512R
suggesting K512 SUMOylation follows K20 SUMOylation in GFP or HA immunoblots (a and b). Arrowheads indicate nonspecific bands.
Journal of Cell Science 124 (18)3154
Journ
alof
Cell
Scie
nce

BEND3 can efficiently repress transcription
Post-translational modifications, especially SUMO modification,have broad impacts on biological processes. SUMOylation of
transcription factors, chromatin-modifiers and chromatin-bindingproteins has frequently been linked to transcriptional repression(Gill, 2005). To address the functional relevance of BEND3
SUMOylation and its preferential association with heterochromaticregions, we generated a triple fusion protein expressing YFP–LacI–BEND3 so that BEND3 was directly targeted to the stably
integrated gene locus in 2-6-3 CLTon cells through a Lac operator–repressor interaction. As described previously, we can activatetranscription of the reporter gene at the 2-6-3 locus by addition of
doxycycline, which results in decondensation of the chromatinlocus, activation of transcription and finally translation of thereporter protein CFP–SKL that is targeted to the cytoplasmicperoxisomes (Janicki et al., 2004; Prasanth et al., 2010). In cells that
expressed YFP–LacI constructs, addition of DOX resulted inchromatin decondensation and transcriptional activation of areporter gene locus (Fig. 5Aa). These cells also showed the
presence of CFP–SKL in the peroxisomes, indicating efficienttranslation of the DOX-induced reporter RNA. By contrast,tethering of YFP–LacI–BEND3 did not allow DOX-induced
decondensation of the chromatin locus and no visible CFP–SKLprotein product was produced, suggesting that tethering BEND3 tothe gene locus inhibits transcriptional activation (Fig. 5Ab). Notethat a large fraction of YFP–LacI–BEND3 is predominantly
localized at the locus. However, upon overexposure of the image,the punctate nuclear distribution of BEND3 becomes evident(supplementary material Fig. S4D). Similarly, in YFP–LacI–
BEND3-transfected cells treated with DOX, immunoblot analysisrevealed that the CFP–SKL protein levels were reduced to 69% ofthe level in cells transfected with YFP–LacI alone (supplementary
material Fig. S4E). We believe that the CFP–SKL in the YFP–LacI–BEND3-transfected sample, came from the CLTon cells thatwere not transfected with YFP–LacI–BEND3. Tethering YFP–
LacI–BEND3.K20R or –BEND3.K512R to the locus also did notallow activation of transcription in +DOX (DOX treated) cells(Fig. 5Ac,d). Interestingly, targeting YFP–LacI–BEND3.SDM, inthe presence of DOX partially restored chromatin decondensation,
with visible levels of CFP–SKL reporter protein in the cytoplasm,suggesting that SUMOylation of BEND3 is essential for its role inrepression of transcription (Fig. 5Ae). The quantification of the
open (decondensed) and closed (condensed) chromatin locus upontargeting of either YFP–LacI–BEND3 or its mutants to the locus inthe absence or presence of DOX further corroborated that BEND3
prevents decondensation, whereas the double SUMO mutant ofBEND3 restored the DOX-induced chromatin opening of the genelocus (Fig. 5B).
To determine whether BEND3 can repress transcription, we fused
BEND3 or the double SUMO mutant (SDM) of BEND3 to the C-terminus of the GAL4 DNA-binding domain and tested their role intranscriptional activity in human U2OS cells. GAL4–BEND3
expression constructs were transfected together with the GAL4–luciferase reporter, and 48 hours post-transfection, luciferaseactivity was measured and normalized. Although GAL4–BEND3
caused more than a 20-fold repression, the double mutant relievedthe repression significantly (P50.027, t-test; Fig. 5C). Resultsobtained from CLTon cells and from luciferase assays indicate that
BEND3 can efficiently repress transcription, qualitatively andquantitatively and that its SUMOylation is crucial for its repressiveability.
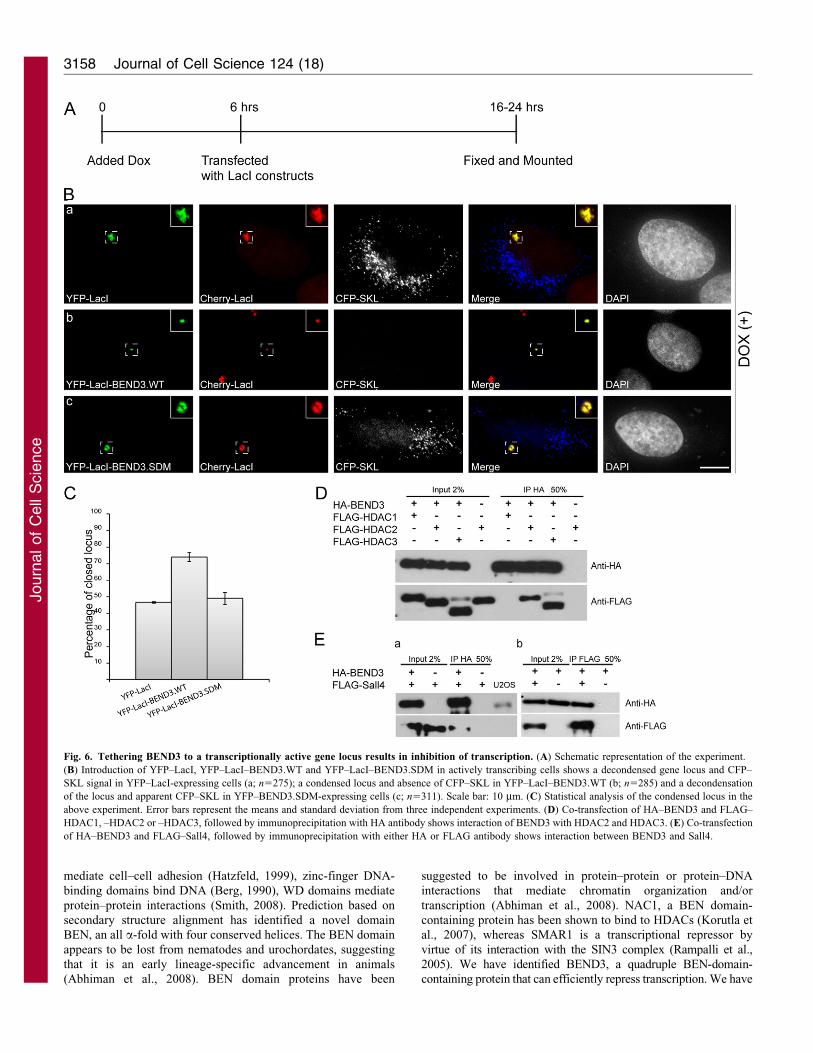
BEND3 can shut down transcription
Using CLTon cells, we demonstrated that BEND3 at the genelocus represses induction of transcription. Next we wanted todetermine whether targeting BEND3 to the transcriptionallyactive gene locus has any effect on continuous transcription. We
activated transcription in the CLTon cells by addition ofdoxycycline. After 6 hours, the cells were transfected withYFP–LacI, YFP–LacI–BEND3 or YFP–LacI–BEND3.SDM and
the decondensation at the locus and the cellular levels of CFP–SKL reporter protein were assessed 24 hours post-transfection(Fig. 6A). In YFP–LacI-expressing control cells the locus
remained decondensed with efficient CFP–SKL reportertranslation (Fig. 6Ba). However, in a large fraction of YFP–LacI–BEND3-expressing cells the locus was condensed (.70%,n5285) and there were reduced levels of CFP–SKL reporter
protein (Fig. 6Bb). In the BEND3.SDM-expressing cells thechromatin was decondensed (similar to that observed for controlYFP–LacI-transfected cells; Fig. 6Bc,C) and CFP–SKL protein
was present. These data indicate that BEND3 can causerecondensation of a highly transcribing gene locus and efficientrepression of transcription.
To determine whether the transcriptional repression induced by
BEND3 is exerted through its interaction with histonedeacetylases, we co-transfected HA–BEND3 with FLAG–HDAC1, 2 or 3 and carried out immunoprecipitation using HA
antibody, and immunoblots with HA and FLAG antibodies. Ourdata showed interaction of BEND3 with HDAC2 and 3 (Fig. 6D).Recent proteomic analysis has identified that BEND3 is a
component of Sall4, a transcription repressor that also associateswith the NuRD complex (van den Berg et al., 2010). We carriedout co-immunoprecipitation of BEND3 and Sall4 in cells co-
transfected with HA–BEND3 and FLAG–Sall4. BEND3 andSall4 could efficiently interact with each other (Fig. 6E),corroborating the previously reported proteomic analysis. Theinteraction of BEND3 with HDAC and Sall4 suggests that the
ability of BEND3 to repress transcription is exerted through itsinteraction with histone deacetylases as well as Sall4.
BEND3-tethered gene locus fails to recruit RNApolymerase II
To determine how BEND3 represses transcription, we examinedthe recruitment of various transcription factors to the gene locus
in the presence of BEND3. In CFP–LacI-expressing control cells,DOX treatment resulted in the production of reporter mRNA, asobserved by the accumulation of MS2BP–YFP (MS2BP–YFP
binds to the bacteriophage MS2-repeat-containing reporter RNA)at the gene locus in ,76% of cells (Fig. 7Aa and supplementarymaterial Fig. S4Ca). CFP–LacI–BEND3-expressing cells did notshow accumulation of MS2BP–YFP at the locus, demonstrating
the absence of active transcription (Fig. 7Ab and supplementarymaterial Fig. S4Ca). However, CFP–LacI–BEND3.SDM-expressing cells continued to respond to DOX and activate
transcription (Fig. 7Ac and supplementary material Fig. S4Ca).We then examined the association of RNA polymerase using anantibody that recognizes the initiation competent form of RNA
polymerase (pol) II [H14 antibody recognizes Ser5 phosphorylatedCTD (C-terminal domain)] (Bregman et al., 1995) (Fig. 7B) as wellas an antibody that recognizes the unphosphorylated form of RNA
pol II (8WG16, supplementary material Fig. S4B). Whereas RNApolymerase was clearly present at the gene locus in 76% of CFP–LacI-expressing cells (Fig. 7Ba and supplementary material Fig.
BEND3 represses transcription 3155
Journ
alof
Cell
Scie
nce
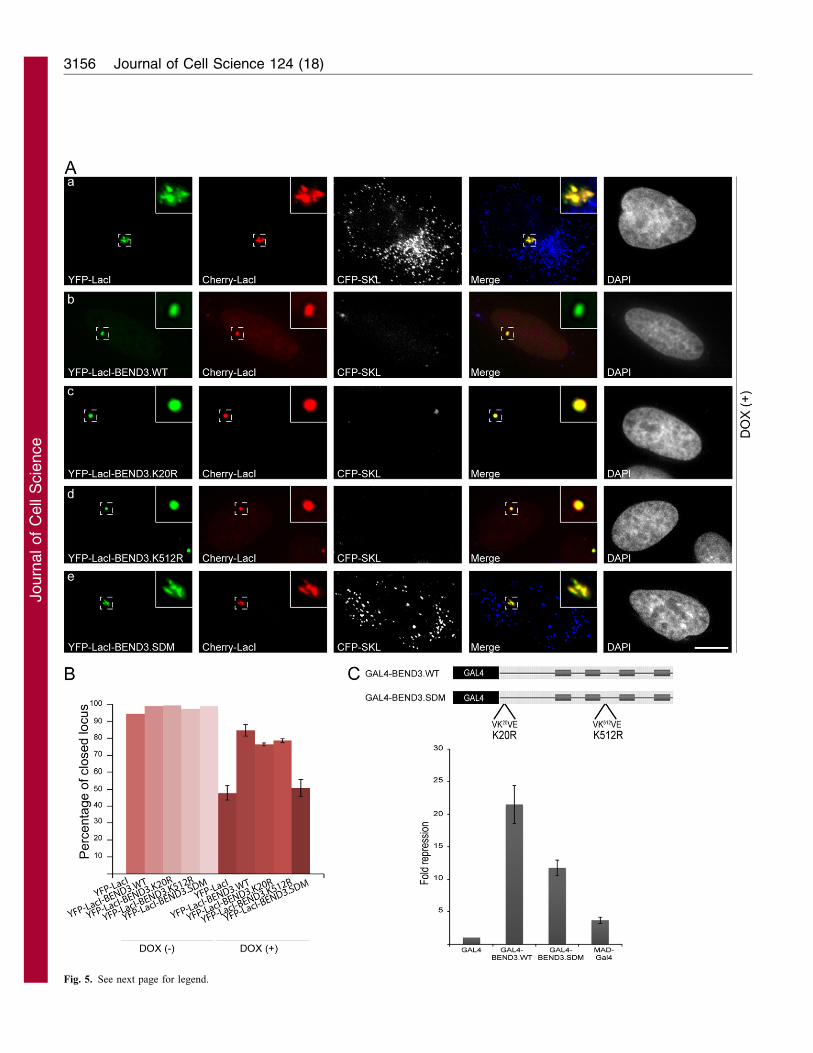
Fig. 5. See next page for legend.
Journal of Cell Science 124 (18)3156
Journ
alof
Cell
Scie
nce

S4Cb), only 40% of CFP–LacI–BEND3-expressing cells showed
recruitment of RNA polymerase II (Fig. 7Bb and supplementary
material Fig. S4Cb). The CFP–LacI–BEND3.SDM-expressing
cells, however, efficiently recruited RNA pol II to the locus
(Fig. 7Bc and supplementary material Fig. S4Cb). Similar results
were obtained with the 8WG16 antibody, with RNA pol II
associated with the CLTon locus in 61% of CFP–LacI-expressing
cells, 25% of CFP–LacI–BEND3-expressing cells and 60% of
CFP–LacI–BEND3.SDM-expressing cells (supplementary material
Fig. S4B). These results were further corroborated by the
association of Cdk9, a component of the phosphorylated TEF-B
kinase complex that phosphorylates the Ser2 of RNA pol II CTD,
which is primarily the elongation-competent form of RNA pol II.
Cdk9 was not enriched in the DOX-induced gene locus in ,74%
of the cells expressing CFP–LacI–BEND3 (Fig. 7Cb and
supplementary material Fig. S4Cc) but continued to associate
with the locus in cells expressing CFP–LacI or CFP–LacI–
BEND3.SDM (Fig. 7Ca,c and supplementary material Fig. S4Cc).
These data indicate that the BEND3-tethered gene locus failed to
assemble the transcription pre-initiation complex possibly by
altering the chromatin structure at that site. Finally, we analyzed
the recruitment of the rtTa transcription activator at the gene locus
in the presence of BEND3. Interestingly, both CFP–LacI- as well
as CFP–LacI–BEND3-expressing cells showed significant
accumulation of YFP–rtTA at the locus (Fig. 8a,b) suggesting
that BEND3 does not influence the recruitment of transcription
activators to the transcription site.
Over-expression of BEND3 induces hyperheterochromatinization
Our data demonstrated that BEND3 localizes to heterochromatin
and is involved in transcriptional repression. To address the
functional relevance of BEND3 association with heterochromatin,
we carried out BEND3 over-expression studies in mouse and human
cells. Strikingly, BEND3-overexpressing cells showed premature
chromatin condensation as evident by a ‘worm-like’ appearance of
DAPI that overlapped with YFP–BEND3 in human U2OS cells
(,11% of cells transfected with 500 ng YFP–BEND3 and ,32%
with 1 mg YFP–BEND3; Fig. 9Ac, supplementary material Fig.
S5A), HeLa (Fig. 9Bb), MCF7 (data not shown) and mouse
embryonic fibroblasts (Fig. 9Cb). The over-heterochromatinized
regions were surprisingly not labeled by H3me3K9-containing
heterochromatin (supplementary material Fig. S5Be) but weredecorated by the polycomb-mediated heterochromatin proteinsdetected by EZH2 and H3K27 staining (compare supplementary
material Fig. S5Ba,c with S5Bd,f). The overall distribution ofH3me3K9 was also significantly altered, suggesting that there waschange in global chromatin organization in BEND3-overexpressedcells (compare, supplementary material Figure S5Bb and e).
Overexpression of YFP–C1 vector alone did not alter chromatinstructure (Fig. 9Aa) indicating that overexpression of BEND3influences chromatin structure. Note that the cells expressing
lower levels of BEND3 showed punctate nuclear staining ofBEND3 (Fig. 9Ab,Ba,Ca and supplementary material Fig. S1B).Interestingly, in generating the YFP–BEND3-expressing stable
cell line, transfection of 1–5 mg (supplementary material Fig. S1D)of YFP–BEND3 into human U2OS cells followed by drugselection resulted in complete cell death within 5 days,
indicating that overexpression of BEND3 is detrimental to cellsurvival. Finally, the stable cell line was generated using 100 ng ofYFP–BEND3 (supplementary material Fig. S5C and Fig. S1E)suggesting that the levels of BEND3 are very carefully maintained
in the cell. Furthermore, exogenous overexpression of BEND3(.1 mg DNA) in human cells resulted in cell cycle arrest, whichwas determined by immunofluorescence analyses using mini-
chromosome maintenance (MCM) and proliferating cell nuclearantigen (PCNA) antibodies (supplementary material Fig. S5D).The wild-type BEND3-expressing cell population consisted of
46.6% MCM-positive (+)/PCNA-negative (2; G1 phase), 49%MCM+/PCNA+ (early S), 1.5% MCM2/PCNA+ (late S) and2.9% MCM2/PCNA2 (G2) in the entire population (Prasanth et
al., 2004a). By contrast, the control cell population consisted of46.4% MCM+/PCNA2, 30% MCM+/PCNA+, 3.6% MCM2/PCNA+ and 20% MCM2/PCNA2. These results suggested thatBEND3 overexpression halts cell cycle progression with cells
arresting in early S phase (supplementary material Fig. S5D).
Our data demonstrate that BEND3 localizes to heterochromaticregions and its overexpression causes extensive heterochromatinization
in mammalian cells. Furthermore, BEND3 is SUMOylated, and thismodification is crucial for its function in repressing transcription.
DiscussionIn eukaryotic cells, repression of transcription is a key mode of
gene regulation and is modulated by several transcriptionalrepressors that have been classified as passive or active (Thiel etal., 2004). Passive repressors compete with activators for DNA
binding, whereas active repressors influence chromatinorganization through histone deacetylation or histone methylationand heterochromatin formation (Thiel et al., 2004). The
methylation on histone H3 at K9 serves as a binding site for theheterochromatin protein HP1 that through homo- andheteromerization results in spreading of heterochromatin status in
the adjoining areas (Brasher et al., 2000; Cowieson et al., 2000; Liet al., 2002; Maison and Almouzni, 2004; Ye et al., 1997). Thus theinterplay of histone methyltransferases and HP1 is crucial in theestablishment and maintenance of heterochromatic sites that ensure
gene silencing at those sites.
Protein domains are independent units within a given proteinthat can function and exist as stable units. The presence of protein
domains is often linked to specific functions; for example, basicleucine zipper domains are found in several DNA-bindingproteins (Busch and Sassone-Corsi, 1990), cadherin repeats
Fig. 5. BEND3 represses transcription and SUMOylation of BEND3
plays crucial roles in transcriptional repression. (A, a) 2-6-3 CLTon cells
show decondensation of the gene locus [YFP–LacI (green) and mCherry–LacI
(red)] upon transcriptional activation (+DOX). CFP–SKL at peroxisomes is a
readout for efficient translation of the reporter mRNA. Transient transfection
of YFP–LacI–BEND3 (b), YFP–LacI–BEND3.K20R (c) and YFP–LacI–
BEND3.K512R (d) prevents the DOX-induced decondensation of the gene
locus. Note that these cells also lack CFP–SKL signal. However, transfection
of YFP–LacI–BEND3.SDM (e) relieved repression. Scale bar: 10 mm.
(B) Statistical analysis of condensed (closed) gene locus of 2-6-3 CLTon cells
(with and without DOX activation). Error bars are ¡ s.d. from three
independent experiments (,150 cells in each experiment). (C) BEND3
represses transcription of the reporter gene in a conventional luciferase assay.
Gal4-BEND3.WT or GAL4-BEND3.SDM co-transfected with LacZ and
luciferase expression vectors into U2OS cells. Relative activations of the
firefly luciferase are presented after normalization against the co-transfected
LacZ. BEND3 represses transcription .20 fold relative to the Gal4 backbone
whereas the BEND3.SDM relieves the repression by ,50%. MAD protein
was used as transcriptional repressor control. The error bars represent the
means and standard deviation from three independent triplicate reactions.
BEND3 represses transcription 3157
Journ
alof
Cell
Scie
nce
mediate cell–cell adhesion (Hatzfeld, 1999), zinc-finger DNA-
binding domains bind DNA (Berg, 1990), WD domains mediate
protein–protein interactions (Smith, 2008). Prediction based on
secondary structure alignment has identified a novel domain
BEN, an all a-fold with four conserved helices. The BEN domain
appears to be lost from nematodes and urochordates, suggesting
that it is an early lineage-specific advancement in animals
(Abhiman et al., 2008). BEN domain proteins have been
suggested to be involved in protein–protein or protein–DNA
interactions that mediate chromatin organization and/or
transcription (Abhiman et al., 2008). NAC1, a BEN domain-
containing protein has been shown to bind to HDACs (Korutla et
al., 2007), whereas SMAR1 is a transcriptional repressor by
virtue of its interaction with the SIN3 complex (Rampalli et al.,
2005). We have identified BEND3, a quadruple BEN-domain-
containing protein that can efficiently repress transcription. We have
Fig. 6. Tethering BEND3 to a transcriptionally active gene locus results in inhibition of transcription. (A) Schematic representation of the experiment.
(B) Introduction of YFP–LacI, YFP–LacI–BEND3.WT and YFP–LacI–BEND3.SDM in actively transcribing cells shows a decondensed gene locus and CFP–
SKL signal in YFP–LacI-expressing cells (a; n5275); a condensed locus and absence of CFP–SKL in YFP–LacI–BEND3.WT (b; n5285) and a decondensation
of the locus and apparent CFP–SKL in YFP–BEND3.SDM-expressing cells (c; n5311). Scale bar: 10 mm. (C) Statistical analysis of the condensed locus in the
above experiment. Error bars represent the means and standard deviation from three independent experiments. (D) Co-transfection of HA–BEND3 and FLAG–
HDAC1, –HDAC2 or –HDAC3, followed by immunoprecipitation with HA antibody shows interaction of BEND3 with HDAC2 and HDAC3. (E) Co-transfection
of HA–BEND3 and FLAG–Sall4, followed by immunoprecipitation with either HA or FLAG antibody shows interaction between BEND3 and Sall4.
Journal of Cell Science 124 (18)3158
Journ
alof
Cell
Scie
nce
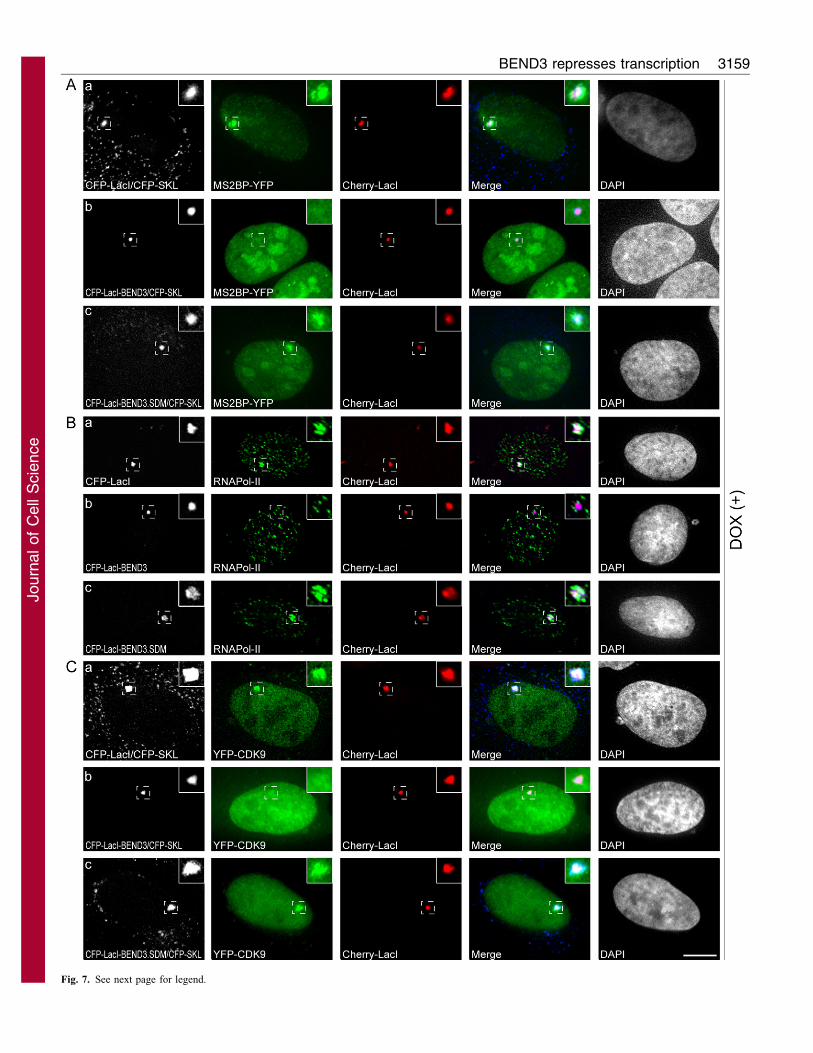
Fig. 7. See next page for legend.
BEND3 represses transcription 3159
Journ
alof
Cell
Scie
nce

also demonstrated that BEND3 associates with heterochromatin and
upon overexpression causes premature chromatin condensation
(PCC), extensive heterochromatinization and cell cycle arrest. This
is similar to overexpression of nimA in fission yeast. Accumulation
of NIMA, a protein kinase required for G2–M transition, has been
shown to induce extensive chromatin condensation (Krien et al.,
1998; O’Connell et al., 1994). In a subset of microcephaly patient
cells PCC has been observed and mutations in MCPH1 or
condensins have been linked to such hyper-condensed DNA
(Griffith et al., 2008; Wood et al., 2008). Previous studies have
demonstrated that distinct heterochromatic structures are formed
upon oncogene-induced cellular senescence, which efficiently result
in silencing of E2F target genes (Narita et al., 2003). Although
several factors can lead to PCC, we suggest that overexpression of
BEND3 results in global repression of transcription by altering
chromatin structure. The heterochromatinized sites are enriched in
the polycomb protein EZH2, a member of the PRC2 complex that is
known to bring about repression through a process of
heterochromatinization (Cao et al., 2002; Margueron et al., 2008).
A recent proteomic study to identify Oct4-interacting proteins
in embryonic stem cells has led to the identification of several
transcription factors, including Sall4, Tcfcp2l1, Esrrb and Dax1
(van den Berg et al., 2010). Sall4, Tcfcp2l1 and Esrrb were found
to associate with BEND3 and Nac1, in addition to their
interaction with the NuRD and SWI–SNF complex (van den
Berg et al., 2010). Nac1, also a BEN-domain-containing protein
has previously been reported to associate with HDACs (Korutla
et al., 2007). Proteomic analysis of Sall4, Tcfcp2lI and Esrrb has
revealed that BEND3 and Nac1 could be part of a complex that
associates with NuRD and mediates stem cell pluripotency (van
den Berg et al., 2010). The NuRD complex is known to have
chromatin remodeling and deacetylase activity, which together
regulate gene expression (Denslow and Wade, 2007). It is also
well established that NuRD, through its interaction with specific
transcription factors, regulates the expression of specific genes.
We provide evidence that BEND3 associates with Sall4 and
HDACs. There is also recent evidence that Sall4 associates with
the NuRD and represses transcription of genes (PTEN and
SALL1) involved in embryonic stem cell leukemogenesis and
kidney development (Lu et al., 2009). Sall4, a pluripotency gene,
is present in adult tissues in the hematopoietic stem cells (HSC)
as well as leukemic stem cells and plays a key role in self-
renewal of stem cells, possibly by recruiting epigenetic
modulators to specific gene targets (Lu et al., 2009). The
interaction of BEND3 with Sall4, its significant enrichment in
spleen, a niche for HSC, tempts us to propose that BEND3 is an
important molecule in stem cell pluripotency. Strikingly, BEND3
also associates with HDAC2, a component of the NuRD complex
that represses genes involved in cell signaling pathways (Wang et
al., 2009).
SUMOylation of BEND3 is required for its ability to repress
transcription. SUMO modification has previously been linked to
transcriptional repression (Gill, 2005), however, the mechanism
by which SUMOylation represses transcription remain to be
elucidated. It has been suggested that SUMOylated proteins are
involved in recruitment of HDACs to chromatin sites, which in
turn results in histone deacetylation. Recent work has also shown
that SUMOylation is crucial for proper heterochromatin
organization in Drosophila through the modification of
SU(VAR)3-7 at K839 (Reo et al., 2010) as well as in MBD1
and MCAF1 (Uchimura et al., 2006). Similarly, SUMO-modified
Sp3 has been shown to repress transcription by promoting locus-
specific heterochromatic gene silencing (Stielow et al., 2008b).
SUMO modification has also been implicated in the maintenance
of heterochromatin stability in fission yeast (Shin et al., 2005).
Interestingly, the SUMO-deficient mutant of BEND3 cannot
repress transcription, but continues to be associated with
heterochromatin in mammalian cells, suggesting that these two
events are independent.
Fig. 8. The rtTa activator is recruited to the locus irrespective of BEND3 localization. Recruitment of YFP–rtTa (activator) at the gene locus (+DOX) in cells
expressing CFP–LacI (a) and CFP–LacI–BEND3 (b).
Fig. 7. BEND3 inhibits pre-initiation complex assembly at the reporter
gene locus in CLTon cells. (A) Recruitment of MS2BP–YFP as an indicator
of active transcription at the gene locus (+DOX) in cells expressing CFP–LacI
(a), CFP–LacI–BEND3 (b) or CFP–LacI–BEND3.SDM (c).
(B) Immunofluorescence localization of the transcription-initiation-competent
form of RNA pol II using H14 antibody in cells expressing CFP–LacI (a)
CFP–LacI–BEND3 (b) or CFP–LacI–BEND3.SDM (c). (C) Recruitment of
YFP–CDK9 at the gene locus (+DOX) in cells expressing CFP–LacI (a),
CFP–LacI–BEND3 (b) and CFP–LacI–BEND3.SDM (c). Note the absence of
RNA pol II and CDK9 at the gene locus in CFP–LacI–BEND3-expressing
cells. Scale bar: 10 mm. Note the loss of CFP–SKL signal in cells where H14
staining has been conducted following a pre-extraction procedure using
detergent.
Journal of Cell Science 124 (18)3160
Journ
alof
Cell
Scie
nce

Utilizing an artificial in vivo gene locus we have demonstrated
that BEND3 inhibits transcription. Similarly, BEND3 can
shutdown transcription of an actively transcribing gene locus
when introduced exogenously. We further provide evidence thata BEND3-tethered gene locus fails to recruit RNA pol II and
associated factors. This might be similar to a group of repressors,
including NC2 and Eve, that inhibit transcription by ‘quenching’
through interaction with activators or general transcriptionfactors thereby inhibiting the interaction between TBP and
TFIIA and TFIIB (Kamada et al., 2001; Kim et al., 2003; Li and
Manley, 1998; Um et al., 1995). Similarly, ZNF76 repressestranscription through its interaction with TBP and further
SUMOylation modulates this repression (Zheng and Yang,2004).
BEND3 is located at 6q21 in humans, a region frequently deletedin leukemias and lymphomas (Zhang et al., 2000), and also
associated with several other human malignancies includingcarcinomas of breast, ovary and prostate (Hyytinen et al., 2002;Morelli et al., 2002; Orphanos et al., 1995a; Orphanos et al.,1995b). A recent study reported a possible tumor suppressor
HACE1 at that locus (Zhang et al., 2007). Interestingly, theoverexpression of BEND3 causes excessive heterochromatinizationand transcriptional shut-down, and therefore in the endogenous
context its fine-tuned balance in the cell is crucial for themaintenance of cellular homeostasis.
Materials and MethodsCell culture and plasmids
HeLa, MCF7, U2OS and wild-type mouse embryonic fibroblast cells were grownin Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose,supplemented with penicillin–streptomycin and 10% fetal bovine serum (FBS).U2OS-2-6-3 CLTon cells were grown in DMEM with 10% Tet system approvedFBS (Clontech). NIH3T3 cells were gown in DMEM supplemented with 10%bovine calf serum (Hyclone).
The coding region of human BEND3 was PCR amplified and cloned intopEYFP-C1 (Clontech), pEYFP-N3, pEYFP-LacI vector and pECFP–LacI vector(modified from pEGFP-LacI; kindly provided by Miroslav Dundr) (Kaiser et al.,2008). BEND3 truncation mutants were created by PCR and inserted into pEYFP-C1 vector. NLS signal sequence was added to the C-terminal mutants. Site-directed mutagenesis was used to make SUMO mutants in pEYFP-BEND3 orpEYFP-LacI–BEND3 as template as per the manufacturer’s instructions(Stratagene). The full-length SUMO1 and SUMO2 genes were PCR amplifiedfrom cDNA and cloned into pCGN vector and HA–SUMO3 was a gift from JieChen (University of Illinois at Urbana-Champaign, Urbana, IL, USA). FLAG–Sall4 was a gift from Hitoshi Niwa and Ryuichi Nishinakamura (Niwa et al., 1991;Yuri et al., 2009). All clones were confirmed by sequencing.
The NCBI accession numbers for BEND3 sequences are: Homo sapiens,NP_001073919.1; Pan troglodytes, XP_527466.2; Bos taurus, XP_603724.2; Mus
musculus, NP_950193.1; Canis familiaris, XP_539070.2; Gallus gallus,XP_419805.2; Danio rerio, XP_001923103.1.
qPCR analysis for BEND3
The expression level of BEND3 was quantified against a standard curve by real-time RT-PCR using the SYBR Green I fluorogenic dye in the StepOnePlus Real-Time PCR System (Applied Biosystems) and data were analyzed using theStepOne system software. The primer sets for human BEND3 were: forward 59-GCAGGACTCCAGCAAACGAAAG-39, reverse 59-GCTGTTCTCACGGTTCC-GCATG-39. Using Q-Gene, a Microsoft Excel script package (Muller et al., 2002),the qPCR values of BEND3 were first normalized against the levels of GAPDH ofthe same samples and then adjusted such that the samples with lowest expressionof BEND3 [heart (see Fig. 1Da) and WI38 cells (see Fig. 1Db)] had a value of 1.The data is presented as mean normalized expression.
Transfection
For transient transfection, Lipofectamine 2000 (Invitrogen) was used according tothe manufacturer’s protocols. For transfection in U2OS 2-6-3 CLTon cells, 100 ngCFP or YFP–LacI constructs were used. For co-transfection, 100 ng LacIconstructs and 500 ng plasmid DNA of the desired proteins were used. Cellswere fixed in 2% (w/v) formaldehyde, 16–24 hours post-transfection, and thenDAPI stained and mounted in p-phenylenediamine (PPD). In order to inducetranscription from the locus in U2OS 2-6-3 CLTon cells, 1 mg/ml DOX was added16–24 hours post-transfection for 4 hours.
Immunofluorescence analysis
Immunofluorescence analysis was carried out as described previously (Prasanth etal., 2004b). Briefly, cells were either fixed in 2% formaldehyde for 15 minutes atroom temperature and then permeabilized in 0.5% Triton X-100 in PBS for7 minutes on ice, or pre-extracted in CSK buffer (10 mM PIPES pH 7.0, 100 mMNaCl, 300 mM sucrose, 3 mM MgCl2) + 0.5% Triton X-100 for 5 minutes on iceand then fixed with 2% formaldehyde. Chilled methanol was applied (for5 minutes) afterwards if needed, according to the specific requirement for theantibodies. The 2-6-3 CLTon cells were pre-extracted in CSK buffer + 0.5% TritonX-100 for 3 minutes and fixed with 1% formaldehyde for 5 minutes. To block the
Fig. 9. Overexpression of BEND3 causes hyper heterochromatinization.
(A–C) Transient overexpression of YFP–BEND3 (but not YFP-C1 vector alone
Aa) (1–2 mg) in U2OS cells (Ac), HeLa (Bb) and MEF (Cb) causes premature
chromosome condensation and extensive heterochromatinization. Note the
worm-like appearance of DNA as visualized by DAPI. Low levels of BEND3
expression with nuclear punctate localization are evident in Ab, Ba and Ca.
DNA was counterstained with DAPI. Scale bar: 10 mm.
BEND3 represses transcription 3161
Journ
alof
Cell
Scie
nce

cells PBS + 1% normal goat serum (NGS) was used. It was incubated with primaryantibodies in a humidified chamber for 1 hour and then secondary antibody wasadded for 45 minutes. All washes between each step were done in PBS + 1% NGS.DNA was stained with DAPI. Cells were mounted in either PPD or Vectashield(Vector Laboratories Inc.). Cells were examined using a Zeiss Axioimager z1fluorescence microscope (Carl Zeiss Inc.) equipped with Chroma filters (ChromaTechnology). Axiovision software (Zeiss) was used to collect digital images from aHamamatsu ORCA-cooled CCD camera. Images were also acquired using a DeltaVision optical sectioning deconvolution instrument (Applied Precision) on anOlympus microscope.
The antibodies used for immunofluorescence were: anti-MCM3 (1:400), anti-PCNA PC10 (1:150), anti-HP1a (1:100; Chemicon), anti-HP1b (1:100;Chemicon), anti-H3–Tri-MeK9 (1:300; Upstate), anti-EZH2 (1:200; BDBiosciences), anti-H3K27 (1:400; Upstate), anti-RNA POLII (H14, 1:50;8WG16, 1:40).
Live-cell microscopy
U2OS 2-6-3 CLTon cells transiently transfected with 100 ng pEYFP-BEND3 wereused for live-cell imaging. 24 hours after transfection, the cells were transferred toa FCS2 live-cell chamber (Bioptechs Inc.) mounted onto the stage of a DeltaVision optical sectioning deconvolution instrument (Applied Precision) on anOlympus microscope and kept at 37 C̊ in L-15 medium (minus Phenol Red)containing 30% FBS. After 20 minutes 1 mg/ml DOX was infused and time-lapseimages acquired with a 633 1.42 NA objective lens were captured using aCoolsnap CCD camera.
Immunoprecipitation and immunoblotting
U2OS cells were co-transfected with 500 ng YFP–BEND3 and 1 mg HA–SUMO1,HA–SUMO2 or HA–SUMO3. Cells were lysed, 24 hours post-transfection, inRIPA buffer containing 25 mM Tris (pH 8), 125 mM NaCl, 0.1% NP-40, 1 mMEDTA, with or without 25 mM N-ethylmaleimide, and protease and phosphataseinhibitors for 30 minutes. Lysate was pre-cleared with Gammabind Sepharosebeads for 1 hour and incubated with appropriate antibodies overnight. The immunecomplex was recovered with Gammabind Sepharose beads by incubating for1.5 hours. The beads were washed three times in RIPA buffer and 60 ml Laemmlibuffer was added. The complex was heat denatured at 95 C̊ for 5 minutes. Forimmunoblots, anti-GFP (1:500; Covance), anti-HA (1:1000), anti-FLAG (M2FLAG 1:500; Sigma) and anti-a-tubulin (1:10,000; Sigma-Aldrich) antibodieswere used.
Luciferase assay
BEND3.WT and BEND3.SDM were PCR amplified and cloned into the pGAL4vector under the SV40 promoter. Gal4–BEND3 (200 ng) or GAL4–BEND3.SDM(200 ng) co-transfected with b-galactosidase expression plasmid CMVb (100 ng)and luciferase expression plasmid (100 ng) into U2OS cells using Lipofectamine2000 reagent. Cells were lysed 48 hours post-transfection using Bright-Glo lysisbuffer (Promega) and luciferase activity was measured using a Bright-Gloluciferase assay kit (Promega). Galactosidase activity was measured usingchlorophenol red–b-D-galactosidase (CPRG) as substrate and used fortransfection normalization. Relative activations of the firefly luciferase arepresented after normalization against the co-transfected LacZ MAD-DBD, whichwas used as a transcriptional repressor control.
We thank members of the Prasanth laboratory for discussions andsuggestions. We thank J. Chen, M. Dundr, B. Freeman, T. Nakamira,R. Nishinakamura, H. Niwa, E. Seto, D. Spector, B. Stillman, E. T.Yeh and R. Zheng for providing reagents and suggestions. We thankP. Newmark and D. Rivier for critical reading of the manuscript. Thiswork was supported by the start-up funds, UIUC and NSF award(0843604) to S.G.P., and ACS award RSG-11-174-01RMC to K.V.P.
Supplementary material available online at
http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.086603/-/DC1
ReferencesAbhiman, S., Iyer, L. M. and Aravind, L. (2008). BEN: a novel domain in chromatin
factors and DNA viral proteins. Bioinformatics 24, 458-461.
Bannister, A. J., Zegerman, P., Partridge, J. F., Miska, E. A., Thomas, J. O.,
Allshire, R. C. and Kouzarides, T. (2001). Selective recognition of methylated
lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120-124.
Bayer, P., Arndt, A., Metzger, S., Mahajan, R., Melchior, F., Jaenicke, R. and
Becker, J. (1998). Structure determination of the small ubiquitin-related modifier
SUMO-1. J. Mol. Biol. 280, 275-286.
Berg, J. M. (1990). Zinc finger domains: hypotheses and current knowledge. Annu. Rev.
Biophys. Biophys. Chem. 19, 405-421.
Bernard, D., Prasanth, K. V., Tripathi, V., Colasse, S., Nakamura, T., Xuan, Z.,
Zhang, M. Q., Sedel, F., Jourdren, L., Coulpier, F. et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression.EMBO J. 29, 3082-3093.
Brasher, S. V., Smith, B. O., Fogh, R. H., Nietlispach, D., Thiru, A., Nielsen, P. R.,
Broadhurst, R. W., Ball, L. J., Murzina, N. V. and Laue, E. D. (2000). Thestructure of mouse HP1 suggests a unique mode of single peptide recognition by theshadow chromo domain dimer. EMBO J. 19, 1587-1597.
Bregman, D. B., Du, L., van der Zee, S. and Warren, S. L. (1995). Transcription-dependent redistribution of the large subunit of RNA polymerase II to discrete nucleardomains. J. Cell Biol. 129, 287-298.
Breiling, A., Turner, B. M., Bianchi, M. E. and Orlando, V. (2001). Generaltranscription factors bind promoters repressed by Polycomb group proteins. Nature
412, 651-655.
Brill, L. M., Xiong, W., Lee, K. B., Ficarro, S. B., Crain, A., Xu, Y., Terskikh, A.,
Snyder, E. Y. and Ding, S. (2009). Phosphoproteomic analysis of human embryonicstem cells. Cell Stem Cell 5, 204-213.
Buhler, M. and Gasser, S. M. (2009). Silent chromatin at the middle and ends: lessonsfrom yeasts. EMBO J. 28, 2149-2161.
Busch, S. J. and Sassone-Corsi, P. (1990). Dimers, leucine zippers and DNA-bindingdomains. Trends Genet. 6, 36-40.
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., Jones,
R. S. and Zhang, Y. (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039-1043.
Cowell, I. G. (1994). Repression versus activation in the control of gene transcription.Trends Biochem. Sci. 19, 38-42.
Cowieson, N. P., Partridge, J. F., Allshire, R. C. and McLaughlin, P. J. (2000).Dimerisation of a chromo shadow domain and distinctions from the chromodomain asrevealed by structural analysis. Curr. Biol. 10, 517-525.
Denslow, S. A. and Wade, P. A. (2007). The human Mi-2/NuRD complex and generegulation. Oncogene 26, 5433-5438.
Dillon, N. and Festenstein, R. (2002). Unravelling heterochromatin: competitionbetween positive and negative factors regulates accessibility. Trends Genet. 18, 252-258.
Eissenberg, J. C. and Elgin, S. C. (2000). The HP1 protein family: getting a grip onchromatin. Curr. Opin. Genet. Dev. 10, 204-210.
Fodor, B. D., Shukeir, N., Reuter, G. and Jenuwein, T. (2010). Mammalian Su(var)genes in chromatin control. Annu. Rev. Cell Dev. Biol. 26, 471-501.
Geiss-Friedlander, R. and Melchior, F. (2007). Concepts in sumoylation: a decade on.Nat. Rev. Mol. Cell Biol. 8, 947-956.
Gill, G. (2005). Something about SUMO inhibits transcription. Curr. Opin. Genet. Dev.
15, 536-541.
Griffith, E., Walker, S., Martin, C. A., Vagnarelli, P., Stiff, T., Vernay, B., Al
Sanna, N., Saggar, A., Hamel, B., Earnshaw, W. C. et al. (2008). Mutations inpericentrin cause Seckel syndrome with defective ATR-dependent DNA damagesignaling. Nat Genet. 40, 232-236.
Hassig, C. A., Fleischer, T. C., Billin, A. N., Schreiber, S. L. and Ayer, D. E. (1997).Histone deacetylase activity is required for full transcriptional repression by mSin3A.Cell 89, 341-347.
Hatzfeld, M. (1999). The armadillo family of structural proteins. Int. Rev. Cytol. 186,179-224.
Hay, R. T. (2005). SUMO: a history of modification. Mol. Cell 18, 1-12.
Hediger, F. and Gasser, S. M. (2006). Heterochromatin protein 1, don’t judge the bookby its cover! Curr. Opin. Genet. Dev. 16, 143-150.
Hubner, M. R. and Spector, D. L. (2010). Chromatin dynamics. Annu. Rev. Biophys.
39, 471-489.
Hyytinen, E. R., Saadut, R., Chen, C., Paull, L., Koivisto, P. A., Vessella, R. L.,
Frierson, H. F., Jr and Dong, J. T. (2002). Defining the region(s) of deletion at6q16-q22 in human prostate cancer. Genes Chromosomes Cancer 34, 306-312.
Inostroza, J. A., Mermelstein, F. H., Ha, I., Lane, W. S. and Reinberg, D. (1992).Dr1, a TATA-binding protein-associated phosphoprotein and inhibitor of class II genetranscription. Cell 70, 477-489.
Jacobs, S. A. and Khorasanizadeh, S. (2002). Structure of HP1 chromodomain boundto a lysine 9-methylated histone H3 tail. Science 295, 2080-2083.
Janicki, S. M., Tsukamoto, T., Salghetti, S. E., Tansey, W. P., Sachidanandam, R.,
Prasanth, K. V., Ried, T., Shav-Tal, Y., Bertrand, E., Singer, R. H. et al. (2004).From silencing to gene expression: real-time analysis in single cells. Cell 116, 683-698.
Johnson, A. D. (1995). The price of repression. Cell 81, 655-658.
Johnson, E. S. (2004). Protein modification by SUMO. Annu. Rev. Biochem. 73, 355-382.
Kadosh, D. and Struhl, K. (1998). Histone deacetylase activity of Rpd3 is important fortranscriptional repression in vivo. Genes Dev. 12, 797-805.
Kaiser, T. E., Intine, R. V. and Dundr, M. (2008). De novo formation of a subnuclearbody. Science 322, 1713-1717.
Kamada, K., Shu, F., Chen, H., Malik, S., Stelzer, G., Roeder, R. G., Meisterernst,
M. and Burley, S. K. (2001). Crystal structure of negative cofactor 2 recognizing theTBP-DNA transcription complex. Cell 106, 71-81.
Kellum, R. (2003a). HP1 complexes and heterochromatin assembly. Curr. Top
Microbiol. Immunol. 274, 53-77.
Kellum, R. (2003b). Is HP1 an RNA detector that functions both in repression andactivation? J. Cell Biol. 161, 671-672.
Journal of Cell Science 124 (18)3162
Journ
alof
Cell
Scie
nce

Kim, M., Park, C. H., Lee, M. S., Carlson, B. A., Hatfield, D. L. and Lee, B. J.(2003). A novel TBP-interacting zinc finger protein represses transcription byinhibiting the recruitment of TFIIA and TFIIB. Biochem. Biophys. Res. Commun. 306,231-238.
Korutla, L., Degnan, R., Wang, P. and Mackler, S. A. (2007). NAC1, a cocaine-regulated POZ/BTB protein interacts with CoREST. J. Neurochem. 101, 611-618.
Krien, M. J., Bugg, S. J., Palatsides, M., Asouline, G., Morimyo, M. and O’Connell,
M. J. (1998). A NIMA homologue promotes chromatin condensation in fission yeast.J. Cell Sci. 111, 967-976.
Kwon, S. H. and Workman, J. L. (2008). The heterochromatin protein 1 (HP1) family:put away a bias toward HP1. Mol. Cells 26, 217-227.
Lachner, M., O’Carroll, D., Rea, S., Mechtler, K. and Jenuwein, T. (2001).Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature
410, 116-120.Li, C. and Manley, J. L. (1998). Even-skipped represses transcription by binding TATA
binding protein and blocking the TFIID-TATA box interaction. Mol. Cell. Biol. 18,3771-3781.
Li, Y., Kirschmann, D. A. and Wallrath, L. L. (2002). Does heterochromatin protein 1always follow code? Proc. Natl. Acad. Sci. USA 99 Suppl. 4, 16462-16469.
Lu, J., Jeong, H. W., Kong, N., Yang, Y., Carroll, J., Luo, H. R., Silberstein, L. E.,Yupoma and Chai, L. (2009). Stem cell factor SALL4 represses the transcriptions ofPTEN and SALL1 through an epigenetic repressor complex. PLoS ONE 4, e5577
Maison, C. and Almouzni, G. (2004). HP1 and the dynamics of heterochromatinmaintenance. Nat. Rev. Mol. Cell Biol. 5, 296-304.
Margueron, R., Li, G., Sarma, K., Blais, A., Zavadil, J., Woodcock, C. L., Dynlacht,B. D. and Reinberg, D. (2008). Ezh1 and Ezh2 maintain repressive chromatinthrough different mechanisms. Mol. Cell 32, 503-518.
Meisterernst, M. and Roeder, R. G. (1991). Family of proteins that interact with TFIIDand regulate promoter activity. Cell 67, 557-567.
Morelli, C., Karayianni, E., Magnanini, C., Mungall, A. J., Thorland, E., Negrini,M., Smith, D. I. and Barbanti-Brodano, G. (2002). Cloning and characterization ofthe common fragile site FRA6F harboring a replicative senescence gene andfrequently deleted in human tumors. Oncogene 21, 7266-7276
Mukhopadhyay, D. and Riezman, H. (2007). Proteasome-independent functions ofubiquitin in endocytosis and signaling. Science 315, 201-205.
Muller, P. Y., Janovjak, H., Miserez, A. R. and Dobbie, Z. (2002). Processing of geneexpression data generated by quantitative real-time RT-PCR. Biotechniques 32, 1372-1374, 1376, 1378-1379.
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N.
and Bird, A. (1998). Transcriptional repression by the methyl-CpG-binding proteinMeCP2 involves a histone deacetylase complex. Nature 393, 386-389.
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., Spector,D. L., Hannon, G. J. and Lowe, S. W. (2003). Rb-mediated heterochromatinformation and silencing of E2F target genes during cellular senescence. Cell 113, 703-716.
Narlikar, G. J., Fan, H. Y. and Kingston, R. E. (2002). Cooperation betweencomplexes that regulate chromatin structure and transcription. Cell 108, 475-487.
Nielsen, A. L., Oulad-Abdelghani, M., Ortiz, J. A., Remboutsika, E., Chambon, P.
and Losson, R. (2001). Heterochromatin formation in mammalian cells: interactionbetween histones and HP1 proteins. Mol. Cell 7, 729-739.
Nielsen, P. R., Nietlispach, D., Mott, H. R., Callaghan, J., Bannister, A., Kouzarides,
T., Murzin, A. G., Murzina, N. V. and Laue, E. D. (2002). Structure of the HP1chromodomain bound to histone H3 methylated at lysine 9. Nature 416, 103-107.
Niwa, H., Yamamura, K. and Miyazaki, J. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193-199.
O’Connell, M. J., Norbury, C. and Nurse, P. (1994). Premature chromatincondensation upon accumulation of NIMA. EMBO J. 13, 4926-4937.
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P. and Mann,
M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signalingnetworks. Cell 127, 635-648.
Orphanos, V., McGown, G., Hey, Y., Boyle, J. M. and Santibanez-Koref, M.(1995a). Proximal 6q, a region showing allele loss in primary breast cancer. Br. J.
Cancer 71, 290-293.Orphanos, V., McGown, G., Hey, Y., Thorncroft, M., Santibanez-Koref, M.,
Russell, S. E., Hickey, I., Atkinson, R. J. and Boyle, J. M. (1995b). Allelicimbalance of chromosome 6q in ovarian tumours. Br. J. Cancer 71, 666-669.
Prasanth, K. V., Camiolo, M., Chan, G., Tripathi, V., Denis, L., Nakamura, T.,
Hubner, M. R. and Spector, D. L. (2010). Nuclear organization and dynamics of7SK RNA in regulating gene expression. Mol. Biol. Cell 21, 4184-4196.
Prasanth, S. G., Mendez, J., Prasanth, K. V. and Stillman, B. (2004a). Dynamics ofpre-replication complex proteins during the cell division cycle. Philos. Trans. R. Soc.
Lond. B 359, 7-16.Prasanth, S. G., Prasanth, K. V., Siddiqui, K., Spector, D. L. and Stillman, B.
(2004b). Human Orc2 localizes to centrosomes, centromeres and heterochromatinduring chromosome inheritance. EMBO J. 23, 2651-2663.
Rampalli, S., Pavithra, L., Bhatt, A., Kundu, T. K. and Chattopadhyay, S. (2005).Tumor suppressor SMAR1 mediates cyclin D1 repression by recruitment of the SIN3/histone deacetylase 1 complex. Mol. Cell. Biol. 25, 8415-8429.
Reo, E., Seum, C., Spierer, P. and Bontron, S. (2010). Sumoylation of Drosophila
SU(VAR)3-7 is required for its heterochromatic function. Nucleic Acids Res. 38,4254-4262.
Richards, E. J. and Elgin, S. C. (2002). Epigenetic codes for heterochromatinformation and silencing: rounding up the usual suspects. Cell 108, 489-500.
Rundlett, S. E., Carmen, A. A., Suka, N., Turner, B. M. and Grunstein, M. (1998).
Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4by RPD3. Nature 392, 831-835.
Schoeftner, S. and Blasco, M. A. (2009). A ’higher order’ of telomere regulation:telomere heterochromatin and telomeric RNAs. EMBO J. 28, 2323-2336.
Shen, Z., Sathyan, K. M., Geng, Y., Zheng, R., Chakraborty, A., Freeman, B.,
Wang, F., Prasanth, K. V. and Prasanth, S. G. (2010). A WD-repeat protein
stabilizes ORC binding to chromatin. Mol. Cell 40, 99-111.
Shiio, Y. and Eisenman, R. N. (2003). Histone sumoylation is associated with
transcriptional repression. Proc. Natl. Acad. Sci. USA 100, 13225-13230.
Shin, J. A., Choi, E. S., Kim, H. S., Ho, J. C., Watts, F. Z., Park, S. D. and Jang,
Y. K. (2005). SUMO modification is involved in the maintenance of heterochromatin
stability in fission yeast. Mol. Cell 19, 817-828.
Smith, T. F. (2008). Diversity of WD-repeat proteins. Subcell. Biochem. 48, 20-30.
Stewart, M. D., Li, J. and Wong, J. (2005). Relationship between histone H3 lysine 9methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol.
Cell. Biol. 25, 2525-2538.
Stielow, B., Sapetschnig, A., Kruger, I., Kunert, N., Brehm, A., Boutros, M. and
Suske, G. (2008a). Identification of SUMO-dependent chromatin-associated
transcriptional repression components by a genome-wide RNAi screen. Mol. Cell
29, 742-754.
Stielow, B., Sapetschnig, A., Wink, C., Kruger, I. and Suske, G. (2008b). SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene
silencing. EMBO Rep. 9, 899-906.
Thiel, G., Lietz, M. and Hohl, M. (2004). How mammalian transcriptional repressors
work. Eur. J. Biochem. 271, 2855-2862.
Uchimura, Y., Ichimura, T., Uwada, J., Tachibana, T., Sugahara, S., Nakao, M. and
Saitoh, H. (2006). Involvement of SUMO modification in MBD1- and MCAF1-
mediated heterochromatin formation. J. Biol. Chem. 281, 23180-23190.
Um, M., Li, C. and Manley, J. L. (1995). The transcriptional repressor even-skipped
interacts directly with TATA-binding protein. Mol. Cell. Biol. 15, 5007-5016.
van den Berg, D. L., Snoek, T., Mullin, N. P., Yates, A., Bezstarosti, K., Demmers,
J., Chambers, I. and Poot, R. A. (2010). An Oct4-centered protein interactionnetwork in embryonic stem cells. Cell Stem Cell 6, 369-381.
Vermaak, D. and Malik, H. S. (2009). Multiple roles for heterochromatin protein 1genes in Drosophila. Annu. Rev. Genet. 43, 467-492.
Vertegaal, A. C., Andersen, J. S., Ogg, S. C., Hay, R. T., Mann, M. and Lamond,
A. I. (2006). Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteinsrevealed by quantitative proteomics. Mol. Cell. Proteomics 5, 2298-2310.
Wang, Y., Zhang, H., Chen, Y., Sun, Y., Yang, F., Yu, W., Liang, J., Sun, L., Yang,
X., Shi, L. et al. (2009). LSD1 is a subunit of the NuRD complex and targets the
metastasis programs in breast cancer. Cell 138, 660-672.
Wood, J. L., Liang, Y., Li, K. and Chen, J. (2008). Microcephalin/MCPH1 associates
with the Condensin II complex to function in homologous recombination repair. J.
Biol. Chem. 283, 29586-29592.
Xue, Y., Wong, J., Moreno, G. T., Young, M. K., Cote, J. and Wang, W. (1998).
NURD, a novel complex with both ATP-dependent chromatin-remodeling andhistone deacetylase activities. Mol. Cell 2, 851-861.
Yang, S. H. and Sharrocks, A. D. (2004). SUMO promotes HDAC-mediatedtranscriptional repression. Mol. Cell 13, 611-617.
Ye, Q., Callebaut, I., Pezhman, A., Courvalin, J. C. and Worman, H. J. (1997).Domain-specific interactions of human HP1-type chromodomain proteins and inner
nuclear membrane protein LBR. J. Biol. Chem. 272, 14983-14989.
Yuri, S., Fujimura, S., Nimura, K., Takeda, N., Toyooka, Y., Fujimura, Y.,
Aburatani, H., Ura, K., Koseki, H., Niwa, H. et al. (2009). Sall4 is essential for
stabilization, but not for pluripotency, of embryonic stem cells by repressing aberranttrophectoderm gene expression. Stem Cells 27, 796-805.
Zhang, L., Anglesio, M. S., O’Sullivan, M., Zhang, F., Yang, G., Sarao, R., Mai,
P. N., Cronin, S., Hara, H., Melnyk, N. et al. (2007). The E3 ligase HACE1 is a
critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med.
13, 1060-1069.
Zhang, Y., Iratni, R., Erdjument-Bromage, H., Tempst, P. and Reinberg, D. (1997).Histone deacetylases and SAP18, a novel polypeptide, are components of a human
Sin3 complex. Cell 89, 357-364.
Zhang, Y., Matthiesen, P., Harder, S., Siebert, R., Castoldi, G., Calasanz, M. J.,
Wong, K. F., Rosenwald, A., Ott, G., Atkin, N. B. et al. (2000). A 3-cM commonly
deleted region in 6q21 in leukemias and lymphomas delineated by fluorescence in situhybridization. Genes Chromosomes Cancer 27, 52-58.
Zheng, G. and Yang, Y. C. (2004). ZNF76, a novel transcriptional repressor targetingTATA-binding protein, is modulated by sumoylation. J. Biol. Chem. 279, 42410-
42421.
BEND3 represses transcription 3163
Journ
alof
Cell
Scie
nce





























