Embed Size (px)
Citation preview

Scleroderma in Children Scleroderma in Children and Young Peopleand Young People
Dr Eileen Baildam
Consultant Paediatric Rheumatologist

Scleroderma in Childhood
• Looking for the evidence
• What direction?

First Incidence Study Funded by RSA
• The majority of children with scleroderma have localised disease (93% in a UK study):– JLS incidence rate 3.4 per million children per
year1
• Systemic sclerosis is extremely uncommon– SSc incidence rate 0.27 per one million
children per year1
Herrick. Arthritis Care & Res 2010

BPSU incidence 2010
87 cases of localised scleroderma
= incidence rate of 3.47 per million
children per year
7 cases of SSc = incidence rate
0.28 per million
children per year
185 notifications
91 excluded
94 verified
84 followed up at 12 months
10 lost to
follow-up
37 out of time-frame20 duplications
34 reporting errors

Scleroderma Diagnosis

Juvenile Scleroderma
• Localised scleroderma (JLS)– Morphea– Linear scleroderma– En coup de sabre– Progressive facial
hemi-atrophy or Parry Romberg syndrome
• Systemic sclerosis (JSSc)– Limited cutaneous– Diffuse cutaneous– Undifferentiated
connective tissue disease
– Overlap syndromes

2 Stages of Disease
• Active inflammatory:– Perivascular infiltration of
mononuclear cells (mainly activated T cells) into skin and blood vessels
– Microvascular change and endothelial cell activation
– Stimulation fibroblasts to increase extracellular matrix components
• Fibrotic stage:– Vascular occlusion
– Interstitial fibrosis

2 stages of disease
• Active inflammatory
• Fibrotic stage ? reversibility
Early diagnosis and treatment during active inflammatory phase with aim to
prevent long term damage and deformity

Delay in accessing care
• 89 cases 8 UK centres• Mean time from first symptom to diagnosis
– 13 m (1-102m) in localised scleroderma– 8 m (0-50m) in systemic sclerosis
Hawley et al. Rheumatology 2011



Assessment of Disease Activity and Damage
Ultrasound gel
Blood vessels
Dermal-epidermal junction
Epidermis

Modified Rodnan Skin Score in Healthy Children
• Mean mRSS 13.9 units (range 4-25 out of possible 54)
• Varied according to age, sex and pubertal status
• No one area scored 3• Scores 1-2 related to
increased subcutaneous fat compared to adults
Foeldvari, Rheum 2006

Epidemiology JLS
UK cohort (n=87)1
International study (n=750)2
Age at onset, mean (S.D.) years
8.3 (3.9) 7.3 (range 0-16 years)
Gender (% female) 63% 76%
Disease duration at diagnosis, median (IQ) months
13.1 (6.9-36.5)
18 (range 0-16.7 months)
ANA positive 43% 42.3%
1Herrick et al. Rheumatology 20112Zulian et al. Rheumatology 2006

Juvenile localised scleroderma: clinical subtypes
BPSU/BAD Incidence Study1
65%
2%7%26%
linear scleroderma
Plaque morphea
GeneralisedmorpheaDeep morphea
Herrick et al. .Rheumatology 2011
Of those with linear subtype: 50% trunk and/or limb only 45% face-head only 5% both

Morphea

JLS not justa skin disease

Extracutaneous manifestations in 22.4% of JLS cases
Zulian et al. Arth & Rheum 2005
In >25% articular, neurological or
ocular involvement was
unrelated to site of skin lesion

Bony/ Bone Marrow Involvement in JLS

• Can co-exist with en coup de sabre• 15/54 patients (28%)
Tollefson, JAAD 2007
Facial Hemi-atrophy/Parry Romberg Syndrome

Neurological involvement• Neurological involvement in craniofacial
scleroderma well reported (18-47%):
– headaches
– epilepsy (10-17%)
– movement disorders
– focal neurological deficits
– neuropsychiatric disorders
– CNS vasculitis
– EEG and/or cerebral MRI abnormalities (including ischaemic, vascular and white matter changes)
Zulian et al. Arth Rheum 2005; Amaral et al, Autoimm Dis 2012; Christen-Zaech et al, JAAD 2008; Marzano et al. Eur J Derm 2003; Herrick et al. Rheum 2011

Intracranial lesions

Natural History
• Previous studies have suggested that localised scleroderma ‘burns itself out’ after 2-7 years
• However:– 27 patients mean onset age 11.5 (3-17)
– followed up for mean 30.6 years
– 89% had continued disease activity into adulthood and 56% had permanent sequelae1
1Saxton-Daniels. Arch Derm 2010

RCT methotrexate in juvenile LSActive disease
MTX po 15mg/m2
(n=46)
Placebo
(n=24)
plus oral prednisolone at 1mg/kg for 3 months
INITIAL RESPONSE SEEN IN ALL
Disease relapse in 15 (32.6%)*
Adverse events 56.5%
Disease relapse in 17 (70.8%)*
Adverse events 45.8%
*p<0.005
Active disease = new or enlarging lesion with erythema and/or positive thermographyResponse = no new lesions; plus SSR<1; plus ↓ lesion temperature by ≤10%
Treatment failure = new lesions; or SSR>1; or increase in lesion temperature. SSR = computerised skin score rate
Zulian et al. Arth & Rheum 2011


Juvenile Systemic Sclerosis
DIFFUSE
LIMITED OVERLAP

Childhood SSc: Have you got the diagnosis right?
Classification Limited cutaneous scleroderma Diffuse cutaneous scleroderma Sine Scleroderma In overlap Undifferentiated/ mixed connective tissue disease
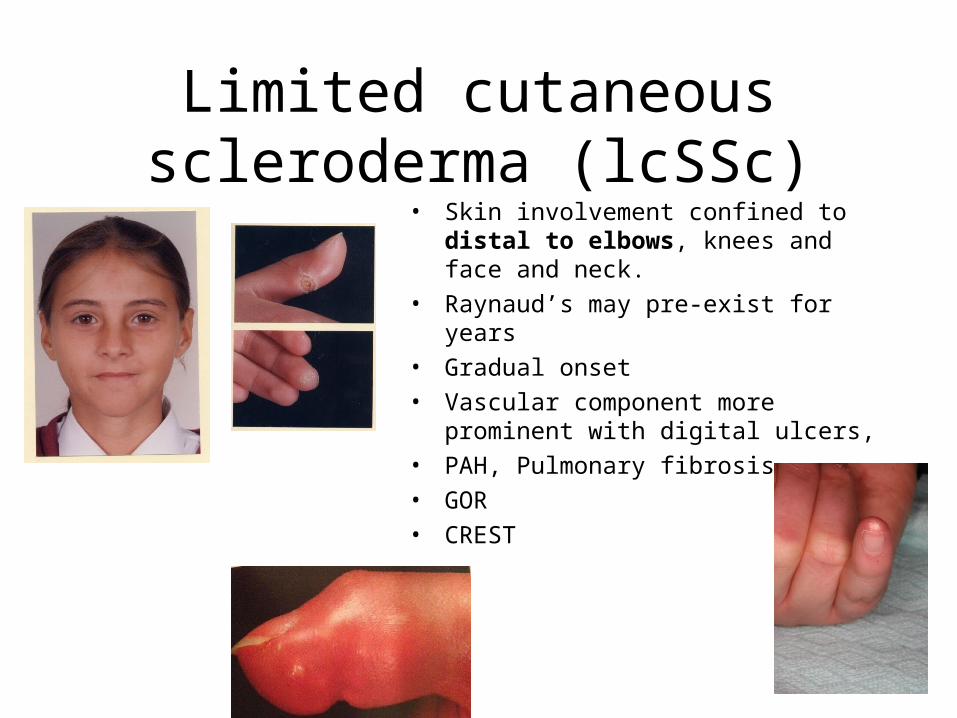
Limited cutaneous scleroderma (lcSSc)
• Skin involvement confined to distal to elbows, knees and face and neck.
• Raynaud’s may pre-exist for years
• Gradual onset
• Vascular component more prominent with digital ulcers,
• PAH, Pulmonary fibrosis
• GOR
• CREST

Diffuse Cutaneous Scleroderma
• Skin involvement extends to proximal limb and to trunk
• More inflammatory• Widespread inflammation in the skin and
musculoskeletal system with oedema• Extensive fibrosis• Raynaud’s usually at onset or even later• Internal organ disease especially kidney

Provisional classification criteria for JSSc (PRES)
Major Criteria required Proximal skin sclerosis/induration
And at least 2 minor criteria Cutaneous Sclerodactly Peripheral vascular Raynauds, nailfold capillary changes, digital
ulcers GIT Dysphagia, GOR Cardiac Arrythmias, heart failure Renal Renal crisis, new onset arterial hypertension Respiratory Pulmonary fibrosis on HRCT, PAH, DLCO reduced Neurological Neuropathy, carpal tunnel syndrome MSK Tendon friction rubs, arthritis, myositis, Serology ANA, SSc specific antibodies
Zulian et al. Arth Rheum 2007

BPSU jSSc No single shared characteristic n=7
0 1 2 3 4 5 6 7
Raynaud's phenomenon
Sclerodactyly
Arthralgia
Digital pitting
Edema
Arthritis
Heliotrope rash
Abnormal muscle strength
Livido reticularis
• 6 (86%) limited cutaneous SSc 2 of these dermatomyositis overlap• 6 ANA +ve • 2 anti-Scl 70 +ve• 1 anti-RNP +ve• 1 anti-centromere +ve

JSSc versus adult onset SSc• 52 pts with JSSc compared with 954 pts
with adult-onset disease:– Overlap cases more common in JSSc
(37% vs 18%, p=0.002)– 10 yr survival significantly better in JSSc
(98% vs 75% p=0.032)– Pulmonary hypertension less (2% vs 14%
p=0.032)– Pulmonary fibrosis equal (22% vs 21%)– Cardiac scleroderma (3% vs 2%)
Foeldvari et al. J Rheum 2010

EUSTAR SSc cohort (n=5000)
• Compared JSSc who are now adults (n=60; 1.2%) with young adult onset SSc (age 20-40) n=910; 18%
• No difference in any organ involvement between groups
• Only difference was rate of ACA 5% JSSc vs 26.9% in aSSc (p<0.005)
• ?related to survival bias Foeldvari, Rheum 2012

JSSc versus young adult onset SSc
• JSSc more overlap particularly with myositis
• Higher frequency of PM-Scl and RNP Ab, less anti-centromere Ab
• Renal disease uncommon in children
• Death (JSSc vs young adult onset SSc vs older):
– Heart involvement 15% vs 9% vs 7% of deaths
– Renal disease 4% vs 7% vs 11% of deathsScalapino, J Rheum 2006

Systemic sclerosis in childhood
• Deaths 15/127 (11.8%)– Cardiac 10 – Renal failure 2– Respiratory failure 2– Septicaemia 2
Cardiac effects•Pericardial effusions•Myocarditis•Diastolic dysfunction•Conduction defects•Restricted or dilated cardiomyopathy•Heart failure
Martini et al. 2006

Predictors of mortality
• Deaths occurred in 16 children all with diffuse disease (4 within 1yr and 10 within 5yrs of onset)
• Significant predictors of mortality:– Fibrosis on CXR (OR 11.2)– Raised creatinine (OR 22.7)– Pericarditis (OR 41.3)
• Short disease duration at diagnosis conferred protection
Martini ; Rheum 2009

JSSc associated with 2 possible courses
• Rapid development of internal organ failure with severe disability leading to death
• Slow course of disease with reduced mortality rates compared to adult onset disease

Systemic sclerosis in childhood (153)
• Mean follow-up 3.9 yrs (0.2-18.8 yrs)
Martini, Arth Rheum 2006
At onset By follow-up
Raynauds 75% 84%
Skin induration 75% 76%
Oedema 35% 46%
Sclerodactyly 6% 66%
Calcinosis 9% 19%

Systemic sclerosis in childhood (n=153)
At onset By FU
Nail fold changes 26% 40%
Finger tip pitting 28% 38%
Digital infarcts 19% 29%
Martini et al. 2006

Systemic sclerosis in childhood (n =153)
• Gastrointestinal involvement
Onset By FU
Dysphagia 10% 24%
GORD 8% 30%
Diarrhoea 2% 10%
Weight loss 18% 27%
Martini et al. 2006

Systemic sclerosis in childhood (n =153)
Renal system
Nervous system
At onset By FU
Raised creatinine/ proteinuria 3% 5%
Renal crisis None 1%
Hypertension 1% 3%
At onset By FU
Seizures 1% 3%
Peripheral neuropathy 1% 1%
Abnormal MRI brain 2% 3%

Scleroderma Topic Specific Group (TSG)
• Aligned to MCRN/Arthritis Research UK Paediatric Rheumatology Clinical Study Group (CSG)
• Open group
• Aim to facilitate collaborative research
• Meet 2-4 monthly via teleconference
• Email [email protected]

International Inception Cohort Study in JSSc
• Prospective cohort study of JSSc (onset <16yrs of age)
• Onset within 18 months of study date
• Skin induration/sclerosis and 1 minor criteria of PRES/ACR/EULAR classification of JSSc
• Standardised protocol of assessmentDr Ivan Foeldvari; UK CI Dr Eileen Baildam

Future direction
• Closer collaborative working of paediatric/adult rheumatology and dermatology, nationally and internationally
• Identification of paediatric disease activity markers
• Prospective studies to describe progression and outcomes
• Clinical trial development

Acknowledgements
• Dr Clare Pain, Professor Ariane Herrick and members of the Scleroderma Topic Specific Group






























