Embed Size (px)
Citation preview

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2013
Consciousness disorders.Comatose conditions
associate professor Ahmed Elmadana

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Classification disorders of CNS
hallucinations
delirium
obtrusive motions
stupor
sopor
coma
Productive disorders Consciousness disorders
Disorders of central neural system
Cognitive dysfunction
Alzheimer's disease dementia
convulsions
induced cognitive dysfunctions
Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
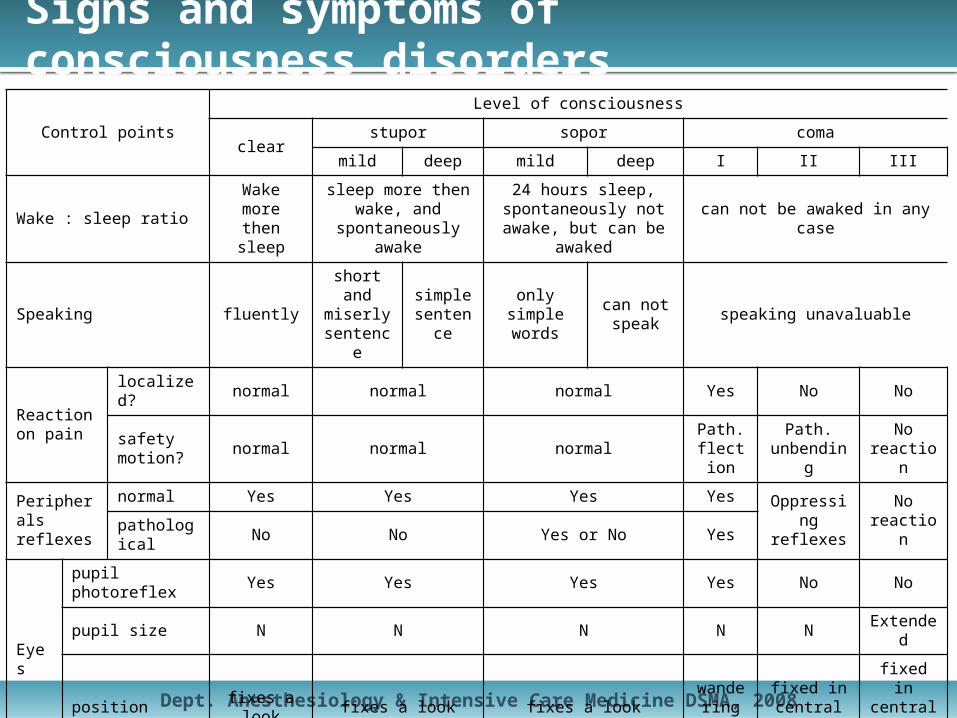
Signs and symptoms of consciousness disorders
Control points
Level of consciousness
clearstupor sopor coma
mild deep mild deep I II III
Wake : sleep ratio Wake more then sleep
sleep more then wake, and spontaneously
awake
24 hours sleep, spontaneously not awake,
but can be awakedcan not be awaked in any case
Speaking fluentlyshort and
miserly sentence
simple sentence
only simple words
can not speak speaking unavaluable
Reaction on pain
localized? normal normal normal Yes No No
safety motion? normal normal normal
Path. flectio
n
Path. unbendin
g
No reaction
Peripherals reflexes
normal Yes Yes Yes Yes Oppressing reflexes
No reactionpathological No No Yes or No Yes
Eyes
pupil photoreflex Yes Yes Yes Yes No No
pupil size N N N N NExtende
d
position fixes a look fixes a look fixes a look wandering sight
fixed in central position
fixed in central position
Function of vital organs
respiration N N N N dyspnoe apnoe
cardiovascular N N N N NCritical hypoten
-sion

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Definition of coma and classification
Coma originally defined as unarousable unresponsiveness in which patient lies with eyes closed (Plum & Posner, 1980)
ComaPrimary cerebralSecondary cerebral
brain injury
hemorrhagic stroke
epilepsy
cerebral infection
intracranial tumor
endocrine disorders
metabolic disorders
hypoxia
poisoning
environment defeat

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Acute brain damage
Causes of acute brain damage
• Trauma • Aneurysmal subarachnoid hemorrhage • Ischemic or hemorrhagic stroke • Infection (encephalitis or meningitis) • Vasculitis (such as systemic lupus erythematosus) • Demyelination (such as acute demyelinating
encephalomyelitis) • Tumor or peritumoral hemorrhage

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Acute brain damage
Indications for intensive carefor patients with head injury
• Not obeying commands after resuscitation and before intubation and ventilation or neurosurgical intervention
• Associated chest injury or multiple injuries that prevent continued assessment of head injury
• Unable to maintain airway or gas exchange • Spontaneous hyperventilation (PaCO2 <3.5 kPa) • Repeated seizures

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Acute brain damage
Indications for intensive carein subarachnoid hemorrhage
• Poor grade aneurysmal subarachnoid hemorrhage • Rapidly decreasing Glasgow coma score or focal neurological deficit
• Complications, including cardiorespiratory dysfunction and particularly neurogenic pulmonary oedema (characterised by acute, severe, but reversible left ventricular dysfunction with associated pulmonary oedema)
• Delayed neurological deficit

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Increased intracranial pressure (IICP)
• Normal intracranial pressure is 5-15 mm Hg• It may be increase by:
– Tumor growth– Edema– Excess cerebrospinal fluid– Hemorrhage

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Increased intracranial pressure (IICP)
If ICP increased and still high, cerebral blood volume is altered:Stage 1 – vasoconstriction and external compression of the
venous system.Compensating, so few symptoms.
If ICP continues to increase, may exceed brain’s ability to adjust.
Stage 2: IICP (gradually rising) causes a decrease of oxygenation of neural tissue
Systemic vasoconstriction occurs to increase blood pressure to get blood to brain. Clinical manifestations transient: episodes of confusion, restlessness, drowsiness, and slight pupillary and breathing changes

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Increased intracranial pressure (IICP)
When ICP begins to = arterial pressure, there is a lack of compensation- beginning decompensationStage 3: Hypoxia and hypercapnia → cytotoxic edema• Decreasing levels of arousal• Widened pulse pressure• May begin Cheynes-Stokes respirations• Bradycardia – due to increased pressure in
carotid arteries• Pupils small and sluggish• Surgical or medical intervention is needed

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Increased intracranial pressure (IICP)
When all compensatory mechanisms have been exhausted:Stage 4:
• Dramatic rise ICP in a short time• Autoregulation is lost, and get vasodilation, further
increasing intracranial volume• ↓ cerebral perfusion = severe hypoxia and acidosis• Brain contents shift (herniate) from area of high
pressure to areas of lower pressure ↓ blood flow• Small hemorrhages develop• Ipsilateral pupil dilation and fixation, progressing to
bilateral fixed and dilated pupils• When mean systolic arterial pressure equal ICP,
cerebral blood flow ceases

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Increased intracranial pressure (IICP)

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Edema in the CNS
• Increase in tissue mass that results from the excess movement of body fluid from the vascular compartment or its abnormal retention in the tissue.
• Why is this a special problem in the brain and spinal cord?
• Enclosed space• Lack of lymphatics• Lack of anastomoses in venous drainage

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Brain edema
Vasogenic edema• Occurs when the blood-brain
barrier is upset– Inflammation due to
infection– Toxic agents that
damage capillary endothelium
– Abnormal capillaries associated with malignant neoplasm
• Leakage of proteins fluid into interstitium → swelling
• Plasma filtrate accumulation alters ionic balance and impairs function
Cytotoxic edema• Intracellular phenomenon• Hypoxia
– Cardiac arrest– Near drowning– Strangulation– Focal edema due to
blockage of an end artery
• Toxic substances that:– Impair sodium/potassium
pump– Impair production of ATP

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Brain edema
• In practice, swelling often caused by both• Treatment is different• If swelling is due to cytotoxicity, can give I.V.
bolus of a hypertonic solution such as mannitol to draw water into the vasculature and out of the brain
• If the cause is vasogenic would this help?• No! would draw fluid into interstitial space and
increase swelling!!

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Standard principlesfor neurological intensive care
• The airway should be protected, generally with an endotracheal tube or tracheostomy
• Normal gas exchange should be maintained using mechanical ventilation if necessary. Especially in conditions of critical cerebral oxygen supplyfor example, acute brain injury the arterial oxygen tension (PaO2) should be kept above 12 kPa and the arterial carbon dioxide tension (PaCO2) at low normal values (4.0-4.5 kPa)
• Maintenance of an adequate cerebral perfusion pressure is essential to maintain cerebral oxygen delivery
• Specialised measurement techniques such as monitoring intracranial pressure assist management
Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
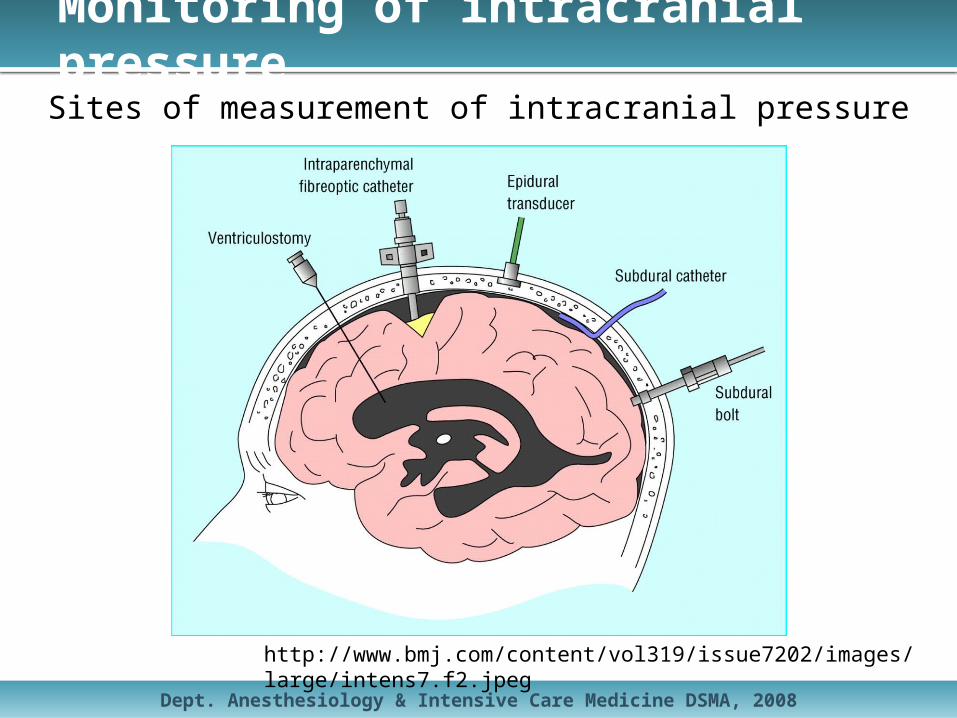
Monitoring of intracranial pressure
http://www.bmj.com/content/vol319/issue7202/images/large/intens7.f2.jpeg
Sites of measurement of intracranial pressure

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
General aspects of neurointensive care
• Noparenteral non-ionic fluid must be given. • Keep plasma sodium concentration >140 mmol/l.
A fall produces an osmotic gradient across the blood-brain barrier and aggravates cerebral oedema.
• Avoid hyperglycaemia and hypoglycaemia.Hyperglycaemia may aggravate ischaemic brain injury by increasing cerebral lactic acidosis.Blood glucose levels >11 mmol/l should be treated.
• Feed through an orogastric tube.Gastric motility drugs can be given as required.
• Anti-thromboembolism stockings; avoid low dose heparin. • 15-30° head up tilt with the head kept in a neutral position
may improve cerebral perfusion pressure.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Compare Glasgow Coma Scale adult and Pediatric
GCSEye Opening
Spontaneous 4Speech 3Pain 2None 1
Verbal ResponseOriented 5Confused 4Inappropriate 3Incomprehensible 2None 1
Motor ResponseObey command 6Localized pain 5Flexor withdrawal 4Flexor posturing 3Extensor posturing 2None 1
Pediatric-GCSEye Opening
Spontaneous 4Speech 3Pain 2None 1
Verbal ResponseCoos, Babbles 5Irritable cries 4Cries to pain 3Moans to pain 2None 1
Motor ResponseNormal spontaneous movement 6Withdrawn to touch 5Withdrawn to pain 4Abnormal flexion 3Abnormal extension 2None 1
Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
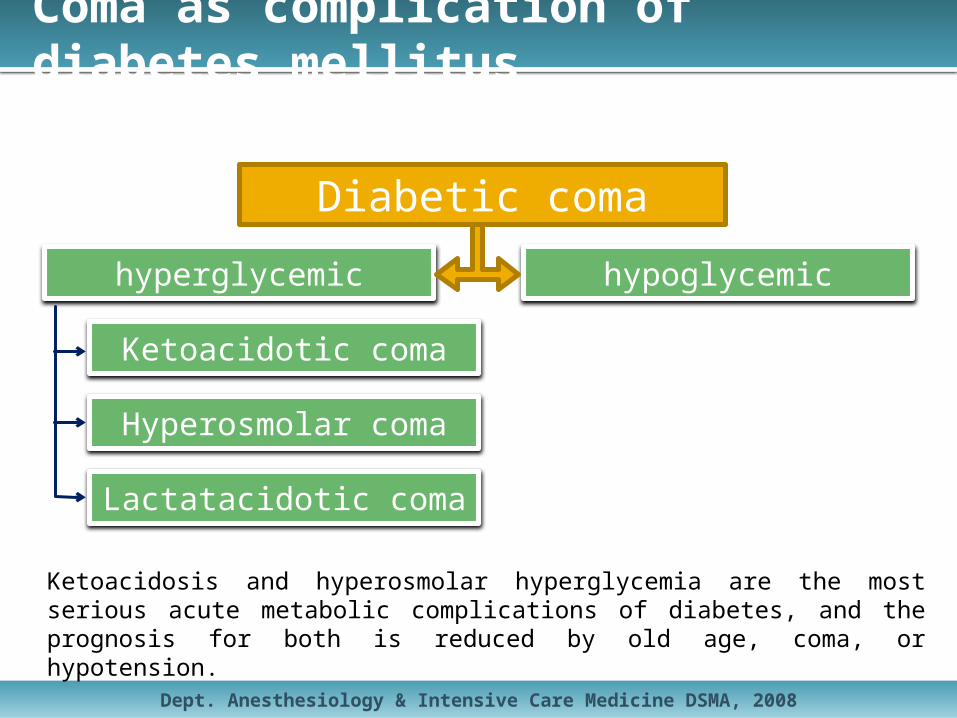
Coma as complication of diabetes mellitus
hyperglycemic hypoglycemic
Ketoacidotic coma
Hyperosmolar coma
Lactatacidotic coma
Diabetic coma
Ketoacidosis and hyperosmolar hyperglycemia are the most serious acute metabolic complications of diabetes, and the prognosis for both is reduced by old age, coma, or hypotension.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Ketoacidosis and hyperosmolar state
PathogenesisThe origin of both diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) is a reduction in the action of circulating insulin along with elevation of counterregulatory hormones.
DKA and HHS are associated with glycosuria and consequent osmotic diuresis with loss of water, sodium, potassium, and other electrolytes.
Precipitating FactorsInfection, cerebrovascular accident, alcohol abuse, pancreatitis, myocardial infarction, trauma, drugs, new onset diabetes, hyperglycemia, and insulin discontinuation or reduction are precipitating factors for DKA or HHS.
Psychological problems and eating disorders may be a contributing factor in 20% of recurrent ketoacidosis in younger type 1 diabetics.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Ketoacidosis and hyperosmolar state
DiagnosisHHS develops over several days to weeks, whereas the evolution of DKA is
shorter.Patient history includes polyuria, polydipsia, polyphagia, weight loss, vomiting,
abdominal pain in DKA, dehydration, weakness, clouding of sensorium, and coma. Physical manifestations may include poor skin turgor, Kussmaul respirations in DKA, tachycardia, hypotension, change in mental status, shock, and ultimately coma.
Hypothermia is a poor prognostic indicator, and abdominal pain can be either a result or a cause of DKA.
Initial lab testing should include plasma glucose, blood urea nitrogen/creatinine, serum ketones, electrolytes, osmolality, urinalysis, urine ketones by dipstick, arterial blood gases, complete blood count with differential, and electrocardiogram. Urine/blood/throat cultures, HbA1c, and chest x-ray should be included if indicated.
Patients with low-normal or low serum potassium concentration need careful cardiac monitoring and more vigorous potassium replacement. Stupor or coma without definitive elevation of effective osmolality should be investigated.
Ketoacidosis is not necessarily an indicator of DKA; it also occurs in starvation ketosis and alcoholic ketoacidosis (AKA).

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Ketoacidosis and hyperosmolar state
TreatmentTreatment requires frequent patient monitoring; dehydration, hyperglycemia,
and electrolyte imbalance correction; and comorbid precipitating event identification.Fluid therapy involves expansion of intravascular/extravascular volume and
restoration of renal perfusion. Effectiveness is evaluated by blood pressure improvement, fluid input/output measurement, and clinical examination.
In pediatric patients, the need for vascular volume expansion must be weighed against the risk of cerebral edema.
Regular insulin by continuous IV infusion is the preferred treatment, with mild DKA patients first receiving a "priming" dose of 0.4-0.6 U/kg body weight, half as an intravenous bolus and half as a subcutaneous or intramuscular injection.
Insulin therapy decreases phosphate concentration.A multidose regimen may be used after DKA resolution and when patients are
able to take fluids orally.Some patients may be discharged on oral medications and dietary therapy.When serum levels fall below 5.5 mEq/L, assuming adequate urine output,
potassium replacement is indicated.Bicarbonate use is controversial.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Ketoacidosis and hyperosmolar state
Complications
DKA and HHS complications include hypoglycemia due to overzealous insulin treatment, hypokalemia due to insulin administration and treatment of acidosis with bicarbonate, hyperglycemia secondary to interruption/discontinuance of insulin therapy, hypoxemia, noncardiogenic pulmonary edema, and rare but frequently fatal cerebral edema.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Ketoacidosis and hyperosmolar state

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
Electrolyte Losses in Hyperosmolar Hyperglycemic StateElectrolytes Losses Electrolytes LossesSodium 7 to 13 mEq/kg Chloride 3 to 7 mEq/kgPotassium 5 to 15 mEq/kg Phosphate 70 to 140 mmol/kgCalcium 50 to 100 mEq/kg Magnesium 50 to 100 mEq/kgWater 100 to 200 mL/kg
The reported sodium level should be corrected when the patient's glucose level is markedly elevated. The types of fluids administered depend on the corrected serum sodium level, which is calculated using the following formula:
Corrected serum sodium =
or
For example, if the reported serum sodium level was 145 mEq/L (145 mmol/L) and the glucose level was 1,100 mg/dL (61.1 mmol/L):
Corrected serum sodium =
100
100)-18(mmol/L) Glucose(65.1Na (mmol/L) Na serumCorrect
5.1615.16145100
100)-181,61(65.1145 (mmol/L) Na serumCorrect

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
Although some formulas for calculating osmolarity include the BUN level, because it is in equal distribution in the intracellular and extracellular spaces, BUN does not contribute to the "effective” serum osmolarity, which is calculated using the following formula:
Effective serum osmolarity =
or mosm/kg = 2[Na+] + Glucose (mmol/L)
Although potassium is included in some formulas, it is not included in the formula recommended by the American Diabetes Association (ADA).
For example, if the sodium level was 150 mEq/L (150 mmol/L), and the glucose level was 1,100 mg/dL:
Effective serum osmolarity =

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
TreatmentThe treatment of hyperosmolar hyperglycemic state involves a five-pronged approach:1) vigorous intravenous rehydration,2) electrolyte replacement,3) administration of intravenous insulin,4) diagnosis and management of precipitating and coexisting problems,5) prevention.
Patients should be managed in the intensive care environment if they have any cardiovascular instability, are unable to maintain an airway, have obtundation or acute abdominal symptoms, or if they cannot be monitored adequately on the general medical ward.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
1) FLUID REPLACEMENTThe first and most important step in the treatment of hyperosmolar hyperglycemic state is aggressive fluid
replacement, which should begin with an estimate of the fluid deficit (usually 100 to 200 mL/kg, or an average total of 9 L). The use of isotonic fluids may cause fluid overload and hypotonic fluids may correct deficits too rapidly with a potential for diffuse myelinolysis and death. Therefore, 1 L of normal saline should be given/hour to start. If the patient is in hypervolemic shock, plasma expanders also may be needed. If the patient is in cardiogenic shock, hemodynamic monitoring is required. Details about the addition of potassium to the intravenous fluids are provided in the next section.
Once there is only mild hypotension, the corrected serum sodium level should be calculated. If the corrected serum sodium level is high (greater than 145 mEq/L) or normal (135 mEq/L [135 mmol/L] to 145 mEq/L), then 0.45 percent sodium chloride may be administered at a rate of 4 to 14 mL/kg/hour depending on the state of dehydration. If the corrected serum sodium level is low (less than 135 mEq/L), 0.9 percent sodium chloride is infused at the same rate. When the serum glucose level is less than 300 mg/dL (16.7 mmol/L), the fluid may be changed to 5 percent dextrose solution with 0.45 percent sodium chloride. One half of the calculated deficit should be given in the first 18 to 24 hours and the remainder over the next 24 hours.
In adults, the risk of cerebral edema is low and the consequences of undertreatment include vascular occlusion and increased rate of mortality. Good clinical judgment should be employed, especially when the patient has comorbid conditions such as acute myocardial infarction, a history of congestive heart failure, or renal failure. In such cases, close hemodynamic monitoring is indicated.
Early in the course of treatment, the plasma glucose level will decrease, even before insulin is started, and this may serve as an index for the adequacy of fluid replacement. If the plasma glucose level fails to decline by 75 to 100 mg/dL (4.2 to 5.6 mmol/L)/hour, this usually implies inadequate fluid volume or renal impairment.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
2) ELECTROLYTE MANAGEMENTElectrolyte replacement is critical. Total body potassium depletion often is unrecognized because the level of
potassium in the blood may be normal or high. The serum potassium level may plummet when insulin is replaced because this forces potassium into the cell. Once urine output is established, potassium replacement should be initiated. Electrolytes should be followed closely (every one to two hours initially) and the patient's cardiac rhythm should be monitored continuously.
If the patient's serum potassium level is less than 3.3 mEq/L (3.3 mmol/L) initially, insulin should be held and potassium given as two thirds potassium chloride and one third potassium phosphate until the potassium level reaches at least 3.3 mEq/L. If the potassium level is greater than 5.0 mEq/L (5.0 mmol/L), potassium should be held until the level is less than 5.0 mEq/L, but the potassium level should be monitored every two hours. If the initial serum potassium level is between 3.3 and 5.0 mEq/L, 20 to 30 mEq of potassium should be given in each liter of intravenous fluid (two thirds as potassium chloride, one third as potassium phosphate) to maintain the serum potassium level between 4.0 mEq/L (4.0 mmol/L) and 5.0 mEq/L.
Despite a lack of evidence that treatment with phosphate, calcium, or magnesium alters outcomes, these electrolytes must be considered. Most studies that have examined the need for phosphate replacement involved patients with diabetic ketoacidosis that developed over hours to days. However, because hyperosmolar hyperglycemic state occurs slowly (over days to weeks), the patient is much more likely to be phosphate depleted.
Although phosphate replacement makes physiologic sense, no controlled data have demonstrated improved outcomes. Phosphate replacement may be considered when the patient's serum phosphate level is below 1.0 mEq/L (1.0 mmol/L) and muscle weakness is a concern, as in patients with respiratory impairment. Because phosphate replacement can cause severe hypocalcemia in this setting, serum calcium levels should be monitored closely.
Hypomagnesemia may manifest as arrhythmias, muscle weakness, convulsions, stupor, and agitation. Hypomagnesemia may be present in up to 90 percent of patients with uncontrolled diabetes. Unless the patient is in renal failure, administration of magnesium is safe and physiologic.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
3) INSULIN THERAPYThe critical point regarding insulin management is to remember that adequate fluids must be
given first. If insulin is administered before fluids, the water will move intracellularly, causing potential worsening of hypotension, vascular collapse, or death. Insulin should be given as an initial bolus of 0.15 U/kg intravenously, followed by a drip of 0.1 U/kg/hour until the blood glucose level falls to between 250 mg/dL (13.9 mmol/L) and 300 mg/dL. If the glucose level does not decrease by 50 to 70 mg/dL/hour, this rate of administration may be doubled. Once the serum glucose concentration is below 300 mg/dL, dextrose should be added to the intravenous fluid and insulin should be titrated by a low-dose sliding scale until the mental obtundation and hyperosmolarity are resolved. When the patient is able to eat, subcutaneous insulin or the previous treatment regimen may be initiated.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Hyperosmolar hyperglycemic state
4) COMPLICATIONS OF TREATMENTComplications from inadequate treatment include vascular occlusions (e.g., mesenteric artery occlusion,
myocardial infarction, low-flow syndrome, and disseminated intravascular coagulopathy) and rhabdomyolysis.
Overhydration may lead to adult respiratory distress syndrome and induced cerebral edema, which is rare but often fatal in children and young adults. Cerebral edema should be treated with intravenous mannitol (Osmitrol) in a dose of 1 to 2 g/kg over 30 minutes and intravenous dexamethasone (Decadron). Slowing the correction of hyperosmolarity in children may prevent cerebral edema.
5) PREVENTION
The patient and another responsible party should be engaged in a significant educational effort that encourages adherence to blood glucose monitoring and compliance with prescribed medications. It is especially important that the patient have access to an adequate water supply. If the patient lives alone, a family member or friend should check in on the patient daily to watch for any changes in mental status and to notify the physician if this occurs. In the nursing home setting, the above recommendations should be followed and the nursing home staff should be educated regarding the signs and symptoms of hyperosmolar hyperglycemic state and the importance of adequate fluid intake and monitoring.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosisBiochemical mechanism diabetic ketoacidosis (DKA)
A key component of DKA is deficit of circulating insulin. DKA occurs mainly, but not exclusively, in Type 1 diabetes because Type 1 diabetes is characterized by a lack of insulin production in the pancreas. It is much less common in Type 2 diabetes because the latter is closely related to cell insensitivity to insulin, not -- at least initially -- to a shortage or absence of insulin. Some Type 2 diabetics have lost their own insulin production and must take external insulin; they have some susceptibility to DKA.
The presence or absence of insulin can by itself regulate most of the pathological effects of DKA; notably, it has a short half-life in the blood of minutes (at least one suggestion is about six), so blood insulin levels decrease rapidly following cessation of insulin release by the pancreas, or by outside sources (eg, injection, though some insulins do not quickly reach the blood, extending their duration of activity).
Most cells in the body are sensitive to one or more of the effects of insulin; the main exceptions are erythrocytes, neurons, some intestinal tissue, and pancreatic beta-cells, none of which require insulin to absorb glucose from the blood. Variation in cell-type sensitivity to insulin is due to the presence of different glucose transporter (GLUT) proteins. Adipocytes and skeletal muscle cells express GLUT-4 proteins, which move to the cell surface membrane when stimulated by a secondary messenger cascade initiated by insulin docking at membrane sensor proteins, thus enabling uptake of glucose. Conversely, when insulin concentrations are low, these transporters dissociate from the cell membrane and so prevent uptake of glucose.
Other effects of insulin include the following: stimulation of the formation of glycogen from glucose and inhibition of glycogenolysis, stimulation of fatty acid (FA) production from stored lipids and inhibition of FA release into the blood, stimulation of FA uptake and storage, inhibition of protein catabolism and of gluconeogenesis, in which glucose is synthesized from non-carbohydrate substrates (such as laktate, alanine and Creb's cycle intermediates). A lack of insulin therefore has many significant effects, several of which contribute to increasing blood glucose levels, to increased fat metabolism and protein degradation. Increased fat metabolism is one of the underlying mechanisms of DKA (ketone bodies are produced during lipid metabolic processing). http://en.wikipedia.org/wiki/Diabetic_ketoacidosis

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis
http://en.wikipedia.org/wiki/Diabetic_ketoacidosis
Ketone body productionUnder low-insulin conditions, regardless of circulating plasma glucose concentration, the
liver acts as though the body is starving and produces another form of fuel, known as ketone bodies. This is an aspect of fat metabolism (beginning with lipolysis) that makes ketone bodies as intermediate products in the fatty acid-processing metabolic sequence. Two of the ketone bodies beta-hydroxybutyrate and acetoacetate enter the blood and can be used as fuel by some organs such as the brain, though the brain still requires a large amount of glucose to function. If large amounts of ketone bodies are produced, the metabolism is in the state termed ketosis; this condition is itself not necessarily harmful, and is normal during times of relatively low carbohydrate availablility (as, for instance, between meals). However, if produced in very large quantities, unprocessed ketone bodies will cause the blood pH to drop, leading to ketoacidosis.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis
P English, G Williams Postgraduate Medical Journal 2004;80:253-261

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis
http://en.wikipedia.org/wiki/Diabetic_ketoacidosis
BrainNormally, ketone bodies are produced in minute quantities, and are used by the heart and
brain as a supplementary energy source. In starvation conditions and DKA, neurons (and therefore the brain) adapt to use ketone bodies as a major energy source since glucose is in short supply.
In non-starvation DKA, a lack of insulin leads to high blood glucose levels. This often significantly increases blood osmolality. At the same time, and also due to low insulin permitted ketosis, large quantities of ketone bodies are produced, two of which -- in addition to increasing the osmolar load of the blood -- are acidic. As a result, the pH of the blood begins to move toward increasing acidity. The normal pH of human blood is 7.35-7.45; in acidosis, the pH dips below 7.35. Very severe acidosis may be as low as 6.9-7.1. (A pH of 6.8 or lower is generally considered to be incompatible with life; i.e., fatal). The acidic shift in the blood is significant because proteins can be significantly denatured by a pH that is either too high or too low, leading to widespread tissue damage, functional deficits, organ failure, and ultimately death.
Glucose begins to spill into the urine as the proteins responsible for reclaiming it from urine reach maximum capacity (the renal threshold for glucose). As glucose is excreted in the urine, it takes a great deal of body water with it, resulting in dehydration. Dehydration further concentrates the blood and worsens the increased serum osmolality. Severe dehydration forces water out of cells and into the bloodstream, with further functional derangement, changing organs behavior. This shift of intracellular water into the bloodstream occurs at a cost as the cells themselves need the water to complete chemical reactions that allow the cells to function as required.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Late signs of diabetic ketoacidosis
DKA is life-threatening and medical attention should be sought immediately.
• Emesis (vomiting), although this is not always a sign of late-stage ketoacidosis, and can occur both in early-stage ketoacidosis and in non-ketoacidic hyperglycaemia.
• Confusion.• Abdominal pain.• Loss of appetite.• Flu-like symptoms.• Lethargy and apathy.• Extreme weakness.• Kussmaul breathing ("air hunger"). A type of hyperventilation where patients
breathe very deeply at a normal or increased rate. This is a sign of severe acidosis.
• Unconsciousness (a variety of diabetic coma) after prolonged DKA. At this stage, speedy medical attention is imperative.

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis EMERGENCY MEASURES
1. Therapeutic flow sheet - One of the most important steps in initiating therapy is to start a flow sheet listing vital signs and the time sequence of diagnostic laboratory values in relation to therapeutic maneuvers. Indices of the metabolic defects include urine glucose and ketones as well as arterial pH, plasma glucose, acetone, bicarbonate, serum urea nitrogen, and electrolytes. Serum osmolality should be measured or estimated and tabulated during the course of therapy. A convenient method of estimating effective serum osmolality is as follows (normal values in humans are 280-300 mosm/kg):
or mosm/kg = 2[Na+] + Glucose (mmol/L)
These calculated estimates are usually 10-20 mosm/kg lower than values measured by standard cryoscopic techniques in patients with diabetic coma. Urea is freely permeable across cell membranes and therefore not included in calculations of effective serum osmolality.
One physician should be responsible for maintaining this therapeutic flow sheet and prescribing therapy. An indwelling urinary catheter is required in all comatose patients but should be avoided if possible in a fully cooperative diabetic because of the risk of introducing bladder infection. Fluid intake and output should be recorded. Gastric intubation is recommended in the comatose patient to correct the commonly associated gastric dilatation that may lead to vomiting and aspiration. The patient should not receive sedatives or narcotics.
http://www.health.am/db/diabetic-ketoacidosis/

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis EMERGENCY MEASURES
2. Insulin replacement• only regular insulin should be used initially in all cases of severe ketoacidosis• insulin should be given immediately after the diagnosis is established.• regular insulin can be given:
• in a loading dose of 0.15 unit/kg as an IV bolus to prime the tissue insulin receptors.• following the initial bolus, doses of insulin as low as 0.1 unit/kg/h are continuously
infused or given hourly as an intramuscular injection;
Replacement of insulin deficiency helps correct the acidosis by reducing the flux of fatty acids to the liver, reducing ketone production by the liver, and also improving removal of ketones from the blood. Insulin treatment reduces the hyperosmolality by reducing the hyperglycemia. It accomplishes this by increasing removal of glucose through peripheral utilization as well as by decreasing production of glucose by the liver. This latter effect is accomplished by direct inhibition of gluconeogenesis and glycogenolysis, as well as by lowered amino acid flux from muscle to liver and reduced hyperglucagonemia. The insulin dose should be "piggy-backed" into the fluid line so the rate of fluid replacement can be changed without altering the insulin delivery rate. If the plasma glucose level fails to fall at least 10% in the first hour, a repeat loading dose is recommended. The availability of bedside glucometers and of laboratory instruments for rapid and accurate glucose analysis has contributed much to achieving optimal insulin replacement. Rarely, a patient with immune insulin resistance is encountered, and this requires doubling the insulin dose every 2-4 hours if hyperglycemia does not improve after the first two doses of insulin.
http://www.health.am/db/diabetic-ketoacidosis/

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic ketoacidosis EMERGENCY MEASURES
3. Fluid replacement - In most patients, the fluid deficit is 4-5 L.• Initially, 0.9% saline solution is the solution of choice to help reexpand the contracted
vascular volume• Fluid replacement should be started in the emergency department as soon as the
diagnosis is established.• The saline should be infused rapidly to provide 1 L/h over the first 1-2 hours.• After the first 2 L of fluid have been given, the intravenous infusion should be at the rate
of 300-400 mL/h.• Use 0.9 % ("normal") saline unless the serum sodium is greater than 150 mEq/L, when
0.45% ("half normal") saline solution should be used.• The volume status should be very carefully monitored. Failure to give enough volume
replacement (at least 3-4 L in 8 hours) to restore normal perfusion is one of the most serious therapeutic shortcomings adversely influencing satisfactory recovery. Excessive fluid replacement (more than 5 L in 8 hours) may contribute to acute respiratory distress syndrome or cerebral edema.
• When blood glucose falls to approximately 250 mg/dL (13,9 mmol/L), the fluids should be changed to a 5% glucose solution to maintain serum glucose in the range of 250-300 mg/dL (13.9-16.7 mmol/L). This will prevent the development of hypoglycemia and will also reduce the likelihood of cerebral edema, which could result from too rapid decline of blood glucose. Intensive insulin therapy should be continued until the ketoacidosis is corrected.
http://www.health.am/db/diabetic-ketoacidosis/

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
4. Sodium bicarbonate - The use of sodium bicarbonate in management of diabetic ketoacidosis has been questioned since clinical benefit was not demonstrated in one prospective randomized trial and because of the following potentially harmful consequences:
1) development of hypokalemia from rapid shift of potassium into cells if the acidosis is overcorrected,
2) tissue anoxia from reduced dissociation of oxygen from hemoglobin when acidosis is rapidly reversed (leftward shift of the oxygen dissociation curve)
3) cerebral acidosis resulting from lowering of cerebrospinal fluid pH.
It must be emphasized, however, that these considerations are less important when severe acidosis exists. It is therefore recommended that bicarbonate be administered to diabetic patients in ketoacidosis if the arterial blood pH is 7.0 or less, with careful monitoring to prevent overcorrection.• One or two ampules of NaHCO3 (one ampule contains 44 mEq/50 mL) should be added
to 1 L of 0.45% saline. (Note: Addition of sodium bicarbonate to 0.9% saline would produce a markedly hypertonic solution that could aggravate the hyperosmolar state already present.) This should be administered rapidly (over the first hour).
• It can be repeated until the arterial pH reaches 7.1, but it should not be given if the pH is 7.1 or greater since additional bicarbonate would increase the risk of rebound metabolic alkalosis as ketones are metabolized. Alkalosis shifts potassium from serum into cells, which could precipitate a fatal cardiac arrhythmia.
Diabetic ketoacidosis EMERGENCY MEASURES

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
5. Potassium - Total body potassium loss from polyuria and vomiting may be as high as 200 mEq. However, because of shifts of potassium from cells into the extracellular space as a consequence of acidosis, serum potassium is usually normal to slightly elevated prior to institution of treatment. As the acidosis is corrected, potassium flows back into the cells, and hypokalemia can develop if potassium replacement is not instituted.
• If the patient is not uremic and has an adequate urinary output, potassium chloride in doses of 10-30 mEq/h should be infused during the second and third hours after beginning therapy as soon as the acidosis starts to resolve.
• Replacement should be started sooner if the initial serum potassium is inappropriately normal or low and should be delayed if serum potassium fails to respond to initial therapy and remains above 5 mEq/L, as in cases of renal insufficiency.
• An ECG can be of help in monitoring the patient's potassium status:− high peaked T waves are a sign of hyperkalemia,− flattened T waves with U waves are a sign of hypokalemia.
• Foods high in potassium content should be prescribed when the patient has recovered sufficiently to take food orally. Tomato juice has 14 mEq of potassium/240 mL, and a medium-sized banana provides about 10 mEq.
Diabetic ketoacidosis EMERGENCY MEASURES

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
6. Phosphate - Phosphate replacement is seldom required in treating diabetic ketoacidosis. However, if severe hypophosphatemia of less than 1 mg/dL (< 0.32 mmol/L) develops during insulin therapy, a small amount of phosphate can be replaced/hour as the potassium salt. Correction of hypophosphatemia helps restore the buffering capacity of the plasma, thereby facilitating renal excretion of hydrogen. It also corrects the impaired oxygen dissociation from hemoglobin by regenerating 2,3-diphosphoglycerate. However, three randomized studies in which phosphate was replaced in only half of a group of patients with diabetic ketoacidosis did not show any apparent clinical benefit from phosphate administration. Moreover, attempts to use the phosphate salt of potassium as the sole means of replacing potassium have led to a number of reported cases of severe hypocalcemia with tetany. To minimize the risk of inducing tetany from too-rapid replacement of phosphate, the average deficit of 40-50 mmol of phosphate should be replaced intravenously at a rate no greater than 3-4 mmol/h in a 60-70-kg person. • A stock solution (Abbott) provides a mixture of 1.12 g KH2PO4 and 1.18 g K2HPO4 in a
5-mL single-dose vial (this equals 22 mmol of potassium and 15 mmol of phosphate).• One-half of this vial (2.5 mL) should be added to 1 L of either 0.45% saline or 5%
dextrose in water.• Two liters of this solution, infused at a rate of 400 mL/h, will correct the phosphate deficit
at the optimal rate of 3 mmol/h while providing 4.4 mEq of potassium/hour. (Additional potassium should be administered as potassium chloride to provide a total of 10-30 mEq of potassium/hour, as noted above.)
• If the serum phosphate remains below 2.5 mg/dL after this infusion, a repeat 5-hour infusion can be given.
Diabetic ketoacidosis EMERGENCY MEASURES

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
7. Hyperchloremic acidosis during therapy - Because of the considerable loss of keto acids in the urine during the initial phase of therapy, substrate for subsequent regeneration of bicarbonate is lost and correction of the total bicarbonate deficit is hampered. A portion of the bicarbonate deficit is replaced with chloride ions infused in large amounts as saline to correct the dehydration. In most patients, as the ketoacidosis clears during insulin replacement, a hyperchloremic, low-bicarbonate pattern emerges with a normal anion gap. This is a relatively benign condition that reverses itself over the subsequent 12-24 hours once intravenous saline is no longer being administered.
8. Treatment of associated infection - Antibiotics are prescribed as indicated. Cholecystitis and pyelonephritis may be particularly severe in these patients.
Diabetic ketoacidosis EMERGENCY MEASURES

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic lactic acidosisDefinition
Severe lactic acidosis is defined as a high anion gap metabolic acidosis with a blood lactate concentration >5.0 mmol/l (normal 0.4–1.2 mmol/l).
Anion gap 10, as calculated using equation:[Na+]–([Cl–]+[HCO3
–]),and should be less than 7–9 mmol/l using current laboratory methods for
chloride concentrations.The pathological elevation of lactate and hydrogen ions may result from
overproduction or delayed clearance of lactate, or a combination of both.
Hyperglycaemia is restricted to the extracellular space so water moves from the intracellular to the extracellular compartment initially, diluting plasma sodium. During the accompanying osmotic diuresis, water is generally lost in excess of sodium until eventually the loss of water is similar for both extracellular and intracellular compartments. Therefore, in DKA, which is usually of relatively brief duration (<24 hours), plasma sodium concentrations may be artificially lowered and can be corrected using the formula:
Corrected [Na+] mmol/l =[plasma Na+] mmol/l+ (1.6x{(plasma glucose–5.6)/5.6})

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic lactic acidosis: causes

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic lactic acidosis: signs
PresentationClinical presentations of lactic acidosis are non-specific, and include:• hyperpnoea (Kussmaul respiration),• nausea,• vomiting,• diarrhoea,• epigastric pain,• anorexia,• lethargy,• thirst,• decreased level of consciousness.
Hypotension, hypothermia, cardiac dysrhythmias, and respiratory failure may also occur in severe metformin-associated lactic acidosis. Blood glucose levels may be low, normal, or high in diabetic subjects and lactic acidosis may also accompany ketoacidosis

Dept. Anesthesiology & Intensive Care Medicine DSMA, 2008
Diabetic lactic acidosis: management
Treatment of lactic acidosis includes appropriate supportive care (usually on an intensive care unit), treatment of any concomitant condition and elimination of any offending drug by renal excretion or dialysis.
Bicarbonate therapy is still one of the principal management modalities for lactic acidosis despite conflicting reports as to its efficacy and even reports of potential adverse consequences including the lowering of mixed venous pH and
intracellular pH when it is used to treat metabolic acidosis associated with concurrent tissue hypoxia.
Metformin is a dialysable drug and the use of bicarbonate in combination with haemodialysis has been successful in the management of metformin associated lactic acidosis.
Other experimental approaches include the use of dichloroacetate, which activates pyruvate dehydrogenase, reducing intracellular lactate formation and increasing lactate disposal. Unfortunately, despite initially promising results, controlled trials have shown no improvements in haemodynamics or survival in acidotic patients treated with this drug.
The use of Carbicarb (sodium bicarbonate and sodium carbonate in equimolar mixture) and THAM (an amino alcohol) is similarly experimental. Despite these management measures the prognosis in lactic acidosis of all causes is poor with
only between 12–17% of patients surviving to discharge in one well conducted study.





























