Embed Size (px)
Citation preview

A stent company struggles with its first Investigational Device Exemption (IDE) application
Moshe Ben Yitzhak, MBA, MSc.CGMP Solutions, Ltd.

Two years ago, Dr. Arthur Harz – a well known cardiologist & surgeon who has developed several innovative surgical procedures and cardiac devices – founded a company (Harz Cardio Ltd.) with several colleagues and contacts from industry.
Dr. Fletcher Kohl, an inventor with several patents to his credit, is the VP of R&D.
Shelly Smith, BSc., is the Regulatory Affairs manager; she nine years experience with Big Pharma and medical device start-up companies.
Bella Carter, BSc, is the QA Manager followed Kohl to Harz Cardio.
Renaldo Cruz, MBA is the Purchasing Manager, he’s been busy sourcing materials, equipment and subcontractors.
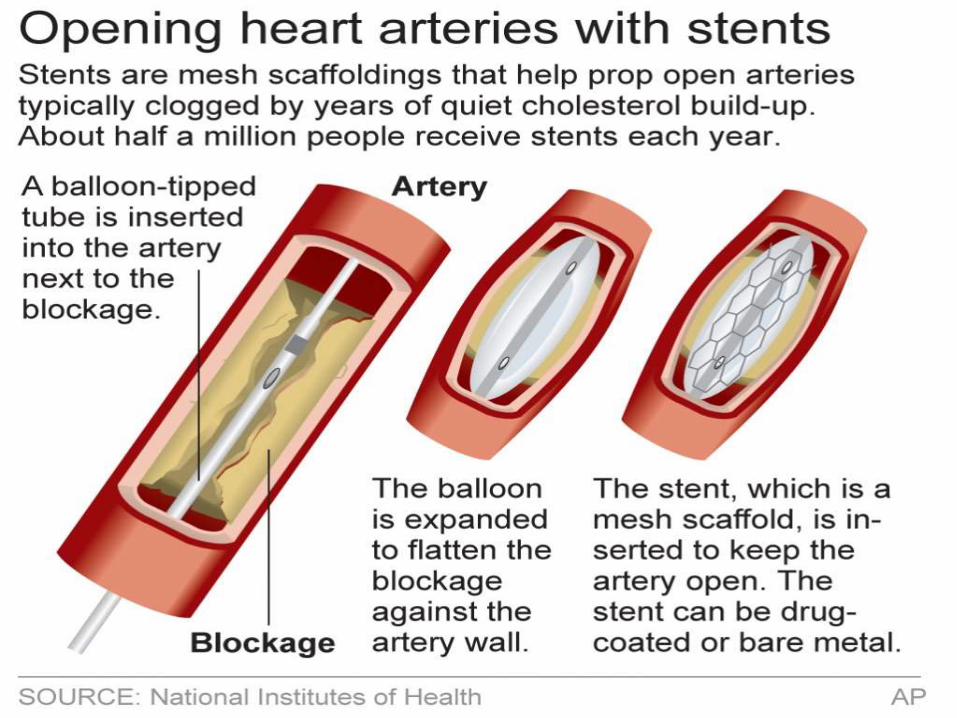

Harz Cardio wants to bring the Novel Bare Metal Coronary Stenting System (NBMCSS) to market. This
special alloy material has the potential to outperform existing stents in flexibility and shape retention,
thereby reducing incidences of thrombosis (blood clotting) and restenosis (narrowing of the affected
artery) in patients after stent implantation.What is their next step?

Arthur, “These are the key facts we should all be aware of,” he began. Clicking on his laptop, he projected a presentation onto the conference room screen.
Nearly 1 million Americans die every year due to cardiovascular diseases.
The market for coronary stents is over $6 billion.
Ideally, a stent procedure will improve blood flow to diseased heart muscles, reviving their normal function.
However, today’s coronary stents have some serious limitations.
There’s a 10 to 40 percent incidence of restenosis within 6 to 12 months after a stent is implanted,
And about 1 to 2 percent of stent patients develop thrombosis at the stent site, which can cause a heart attack, stroke.


“And that’s where we come in,” Dr. Kohl added. “Our early lab experiments have shown that the NBMCSS addresses these issues by means of its superior materials, allowing the stent to sustain pulsating stresses. It can also reduce restenosis and thrombosis. We could be a major player in the market in short order!”
Shelly knew how excited inventors could be, and tried to temper their enthusiasm. “That sounds great, but currently all we have is preliminary data. Before the stent can be marketed, we’ll have to scientifically demonstrate to the FDA that it’s safe, effective, and performs as claimed.
“That’s where you come in,” smiled Arthur.

Shelly connected her laptop to the conference room’s projector and began. “I’m going to give you a roadmap that will take us through the process of getting FDA approval. Keep these three things in mind:”
Does the device work as intended?
Does the device fulfill the claims being made?
Does the device subject patients to undue risk?
“The NBMCSS is a coronary stent (product code MAF) that the Agency will consider a Class III device.”

“So how do we know that we need to submit an IDE?” asked Renaldo. Shelly responded by clicking on the next slide in her presentation and gesturing toward the screen.
Class III devices:
Support or sustain human life, are of substantial importance in preventing impairment of human health, or, can present a potential unreasonable risk of illness or injury when used.
Due to the level of risk, all Class III devices are subject to general controls and special controls.
Require a Pre-Market Approval (PMA), 21 CFR 814.
“Given the facts that implanting a stent is a surgical procedure, and that all surgical procedures entail risk, plus their function in sustaining human life, a stent will be deemed a Class III device, and the IRB will require us to have an IDE.”

“This sounds pretty simple,” Fletcher looked around the table for agreement. He started ticking off points with his fingers. “We know it is a Class III. We know we have to conduct clinical studies. We know that we need FDA approval of the IDE before we conduct those studies. Can’t we submit a form or something?”
Shelly looked at Bella. Bella’s expression told her that she needed to break it down a little bit more for the gifted scientist, who was a bit out-of-touch with Quality Assurance & Regulatory Affairs.
“Actually,” Shelly continued, “there are a couple of steps before that.”

“We’ve started some non-clinical bench testing,” Shelly said nodding towards Fletcher. “R&D is identifying and preparing:
The materials that make up our stenting system;
How the NBMCSS is constructed;
Documenting that all incoming raw materials conform to specifications.”
“Once we’ve gathered this information, we should start putting together our Pre- Submission. A Pre-Submission is a formal written request for feedback from FDA in writing or have a meeting. While it is not required, it is highly recommended.”
“I will take the lead on this by calling CDRH and following-up with an e-mail,” said Shelly.

“I have been through this process a few times with Class II
diagnostic devices, what’s the value of a pre-submission in
this particular case?” asked Arthur.
Shelly made her case. “We can ask FDA for input about our
testing strategy, this will:
Familiarize the Agency with our stent, and
Get input in writing on what our IDE application needs to
include; and
Ultimately, expedite our PMA process while improving our
chances of getting the stent to market.”


“Well, I’m sold,” Arthur grinned. “What’s after that?”
“After we’ve received the Agency’s input,” Shelly continued, “Fletcher will know what the FDA expects to see in our non-clinical testing.”
“Right,” the R&D chief warmed up. “My team will begin in vitro nonclinical engineering tests on the final design of the stent and its delivery system. These tests will ensure that the NBMCSS meets our design specifications, and provide us with reasonable assurance that our device has no functional issues when we start in vivo animal trials.”

“Bench testing should, ideally, help us identify as many of the significant risks associated with the device’s manufacturing and the special controls needed before it ever reaches a patient,” Fletcher concluded.


Shelly began, “Subsection 520(g) of the FDCA encourages the development of medical devices for human use, and allows company’s like ours to conduct scientific investigations of new and predicate devices, to the extent that it is consistent with the protection of the public health, safety and ethical standards.”
The FDA has issued regulations to govern these processes, the most notable of which are:
21 CFR 812 Investigational Device Exemptions
21 CFR 50 Protection for Human Subjects, Informed Consent (IC)
21 CFR 54 Financial Disclosure of Investigators
21 CFR 56 Institutional Review Boards (IRBs)
Section 601 of the FDA Safety and Innovation Act, 2012

“So,” concluded Arthur, “an IDE requires an application.”
“That’s right,” Shelly replied. She continued, “Under §820.1 an IDE permits a device that otherwise would be required to comply with a performance standard or to have a PMA to be shipped lawfully for the purpose of conducting clinical investigations.”
“We don’t have to comply with the QSR?” asked Renaldo.
“An approved IDE exempts the company from complying with certain aspects of the device regulations,” Bella answered.
Shelly clicked on the next slide.

Shelly, “if our IDE submission is approved, here is where we will stand vis-à-vis the device regulations. During clinical trials we will be exempt from:”
Misbranding
Registration
Performance Standards
510(k)
PMA
HDE
Good Manufacturing Practices (GMPs) except Design Controls


“Is all of the data Fletcher and his team generating part of the IDE?” Asked Arthur.
“A large part of it is,” replied Shelly, “plus additional information QA and Regulatory Affairs will provide.” Name and address of sponsor Report of prior investigations and investigational plan Manufacturing, processing, packing, and storage of device Investigator agreement List of the name, address, and chairperson of each IRB Participating institutions Charge for device Environmental assessment Labeling Subject materials including informed consent

“Of course,” she continued, “there will be other information that we will discuss with them in the Pre-submission meeting. At that point, the ball is in the Agency’s court.”
FDA sends acknowledgement with IDE number;
IDE is sent to appropriate review division based on intended use;
The Lead Reviewer assembles team of experts to review the application and make decision within 30 days;
FDA will issue a decision letter, informing us of their decision.

“Some of the tests R&D is performing are quite complex,” Fletcher stated. “What happens if we need to add information after we’ve submitted or if there are minor changes?”
“Do we have any reporting requirements during the time we are conducting clinical trials?” asked Bella.
“Supplements to the submission are covered under CFR 812.35, while reporting requirements are covered under CFR 812.150,” Shelly said, answering both questions.

Arthur had some more concerns. “Can they reject the submission or impose additional requirements on us? That could affect our manpower, skills mix, which investigators & sites we use, quite a few things!”
“Let me address approval first,” Shelly answered. “The Agency will either send a letter stating:
Approving;
Approving with conditions; or
Disapproval.

Arthur shifted in his seat when he saw the word ‘disapproval’ on the screen. “What are the potential reasons for disapproval?”
Shelly clicked on the next slide.
A failure to comply with regulatory requirements;
The application contains an untrue statement of material fact;
The sponsor fails to respond to a request for additional information;

The list continued:
The risks do not outweigh the anticipated benefits to the subjects or the knowledge to be gained;
Informed consent is inadequate;
The proposed investigation is scientifically unsound;
The proposed use of the device is likely to be ineffective; or
The inadequacy of previously reported investigations.

Bella noted the looks of concern on all their faces. “We have to remember, the FDA will not ‘disapprove’ the submission due to concerns over the clinical protocol design.”
“Now I’m totally confused,” exclaimed Fletcher! “She,” he said pointing at Shelly, “just said if there are safety concerns the FDA won’t approve it!”
Shelly quickly jumped in, “Safety, yes. That’s risk versus benefit, and that will be included in the clinical protocol’s inclusion / exclusion criteria for subjects, as well as design controls.”
“However – and I think this is what Bella intended to say – is that the FDA will not disapprove an IDE submission because it thinks the study design won’t support a future PMA.”

Shelly tapped on the keyboard and brought up another slide.
“According to the 2012 revision of the Food, Drug and Cosmetics
Act, the FDA cannot disapprove an IDE application for reasons of:
The investigation may not support a substantial equivalence or de novo
classification determination or approval of a device;
The investigation may not meet a requirement, including a data requirement,
relating to the approval or clearance of a device; or an additional or different
investigation may be necessary to support clearance or approval of the device.
“In other words, the FDA cannot deny an IDE application because
we haven’t definitively proven – yet – that our stent is similar to
other stents or novel, or that more than one study may be
required to prove its effectiveness.”

Arthur stood up and the room became quiet. “It’s like this, the FDA’s goal is to protect patients.”
“It also understands that in order to provide scientific advances to the public, we cannot be expected to know the results in advance. They understand that sometimes we have to move incrementally.”
“As long as we make sure that the benefits outweigh the risks –and that we’ve included all the required data in the IDE – they will give the application a fair review.”
“Fletch, continue your bench testing, while Shelly sets-up the pre-submission meeting. Bella, I want you to start auditing our key component suppliers in support of the bench testing.”

http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/InvestigationalDeviceExemptionIDE/ucm046706.htm
Guidance for Industry - Factors to Consider when making Benefit-Risk Determinations, 2017
Guidance for Industry - Requests for Feedback on Medical Device Submissions, 2014
Guidance for Industry - IDEs for Early Feasibility Studies, 2013
Guidance for Industry - Acceptance and Filing Reviews for Premarket Approval Applications (PMAs), 31 December 2012






























