Embed Size (px)
Citation preview

República Bolivariana de VenezuelaMinisterio del Poder Popular Para la Educación SuperiorUniversidad Nacional Experimental “Rómulo Gallegos“
Área: Ciencias de la Salud Hospital General Dr. Pablo Acosta Ortiz.
Clínica Médica II
San Fernando de Apure, Abril 2015
Síndrome de Guillain-Barré
Bachiller:Felipe Turón
Dra.: Carmen Carreño.
Polineuropatía Aguda Inflamatoria Desmielinizante
(90%)
Variante axonal
Neuropatía Axonal Motora Aguda
Miller Fisher
Neuropatía Aguda Pandisautonómia
Tipos de SGB
Dannia Mohammed

El SGB fue descrito por vez primera por Osler en 1892, pero no fue hasta 1916, en Francia, cuando Guillain, Barré y Strohl; integraron el concepto de esta entidad.
Es la principal causa actual de parálisis fláccida aguda adquirida
El SGB es una polirradiculoneuropatía desmielinizante inflamatoria aguda caracterizada por ser monofásica, frecuentemente grave y de evolución fulminante que tiene origen autoinmunitario.
El riesgo es similar en hombres y mujeres, y en los países occidentales la
enfermedad afecta a los adultos con una frecuencia mayor que a los niños.
Dannia Mohammed

Etiopatogenia
Cursan con el antecedente de una infección respiratoria o gastrointestinal, bacteriana o viral, varias semanas antes del comienzo de los síntomas neurológicos.
Antecedentes infecciosos; mayor frecuencia de infección por:
- Campylobacter jejuni 26-41%
- Cytomegalovirus 10-22%
- Epstein Barr 10%
- M pneumoniae
-Haemophylus influenzae 2-13%
- V Varicela zoster
- Mononucleosis I.
- HIV
Se ha asociado con vacunación (influenza, h1n1 1976), enfermedades
sistémicas (enfermedad de hodgkin, LES, sarcoidosis) y cirugía.
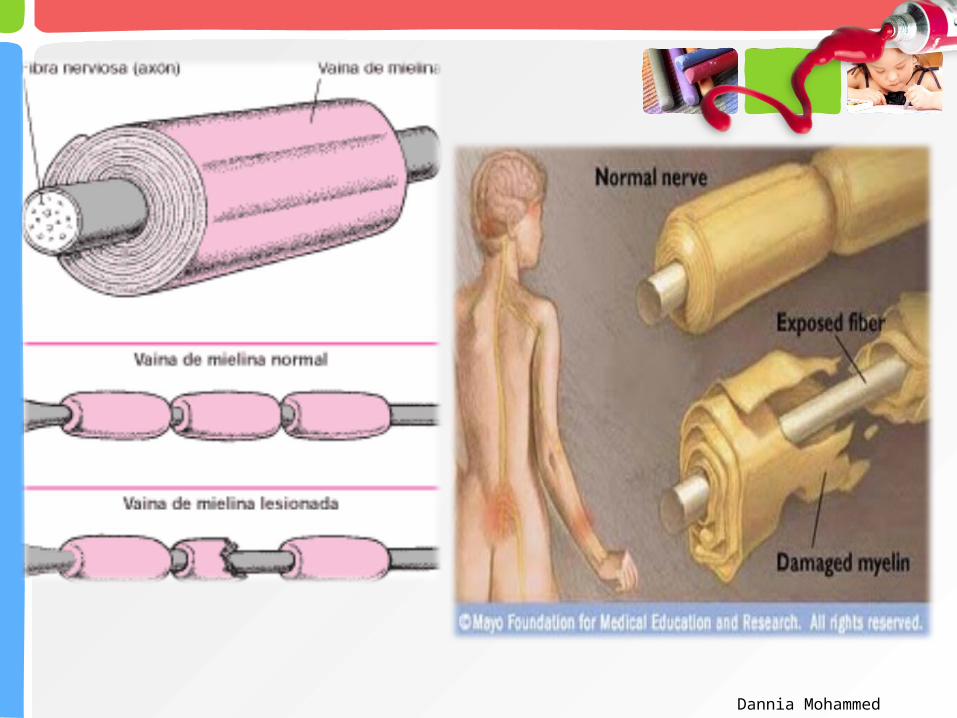
El sistema inmunológico del organismo ataca a sus propios tejidos destruyendo la cobertura de mielina que rodea a los axones de los nervios periféricos, e inclusive a los propios axones.
Dannia Mohammed
Dannia Mohammed
Inmunidad
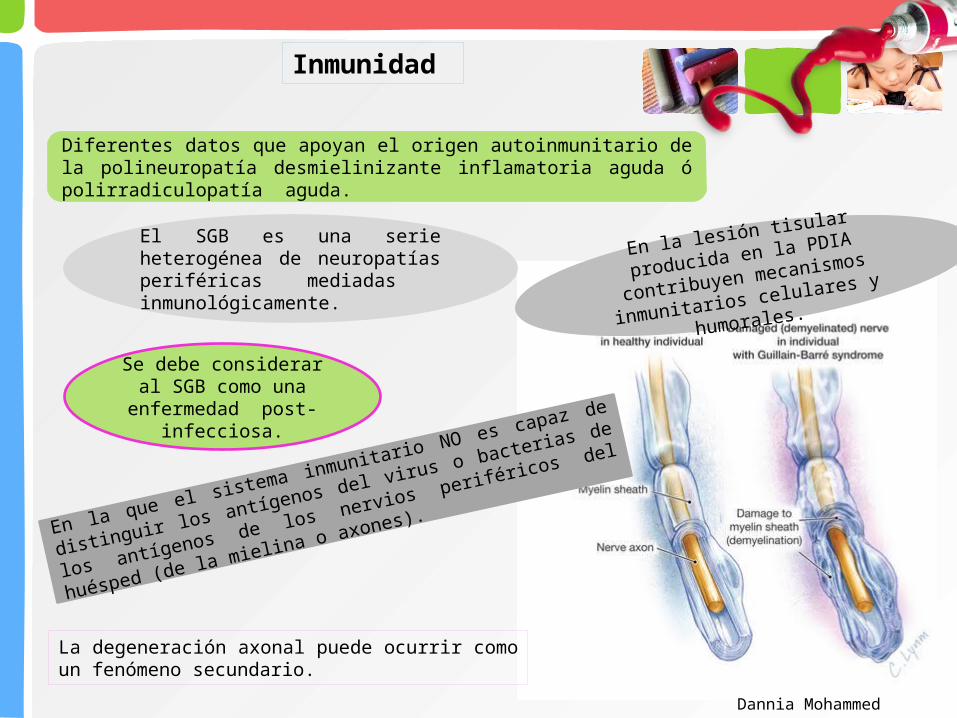
Diferentes datos que apoyan el origen autoinmunitario de la polineuropatía desmielinizante inflamatoria aguda ó polirradiculopatía aguda.
En la lesión tisular producida en la
PDIA contribuyen mecanismos
inmunitarios celulares y humorales.
En la que el sistema inmunitario NO es capaz de distinguir los antígenos
del virus o bacterias de los antígenos de los nervios periféricos del huésped
(de la mielina o axones).
Se debe considerar al SGB como una enfermedad
post-infecciosa.
La degeneración axonal puede ocurrir como un fenómeno secundario.
El SGB es una serie heterogénea de neuropatías periféricas mediadas inmunológicamente.
Dannia Mohammed
Signos y síntomas típicos del SGB son:
Manifestaciones Clínicas
• ´Parestesia .
• Entumecimiento .
• Movimiento descoordinado.
• Defunción autonómica .
• Visión borrosa y visión doble.
• Torpeza y caídas.
• Dificultad para mover los músculos de la cara
• Contracciones musculares.
• Sentir los latidos del corazón (palpitaciones).
Otros síntomas pueden serSíntomas de emergencia
Ausencia temporal de la respiración.
No puede respirar profundamente.
Dificultad respiratoria.
Dificultad para deglutir.
Babeo.
Mareo al pararse.
Desmayo.

CRITERIOS DIAGNOSTICOS
CRITERIOS REQUERIDOS
Debilidad progresiva en más de un miembro.
Arreflexia osteotendinosa universal.
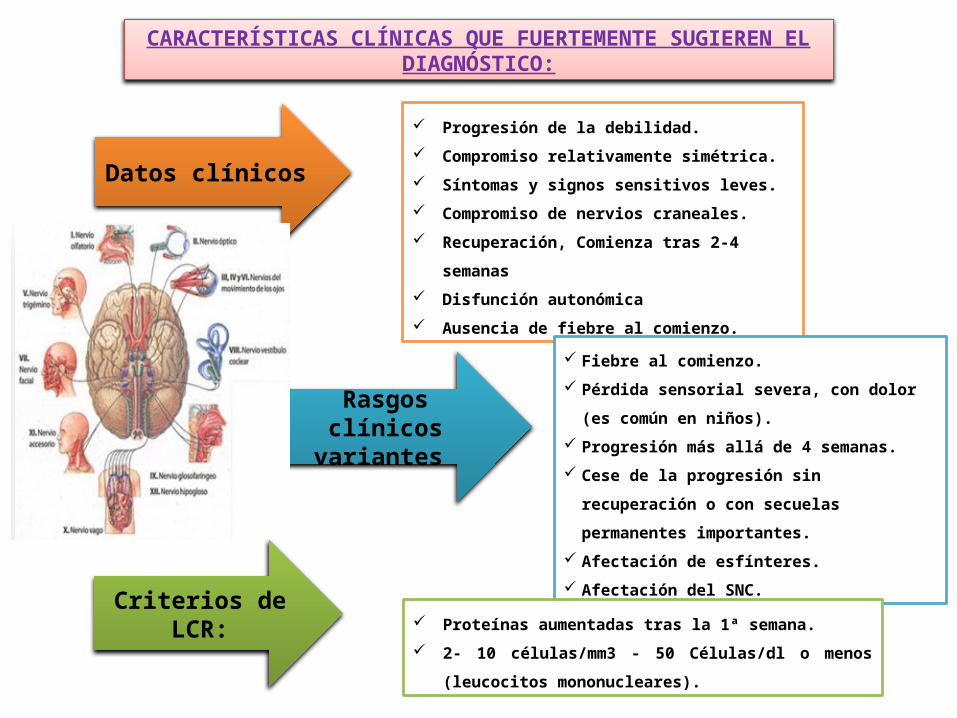
CARACTERÍSTICAS CLÍNICAS QUE FUERTEMENTE SUGIEREN EL DIAGNÓSTICO:
Progresión de la debilidad.
Compromiso relativamente simétrica.
Síntomas y signos sensitivos leves.
Compromiso de nervios craneales.
Recuperación, Comienza tras 2‐4 semanas
Disfunción autonómica
Ausencia de fiebre al comienzo.
Datos clínicos
Rasgos clínicos variantes
Fiebre al comienzo.
Pérdida sensorial severa, con dolor (es común en
niños).
Progresión más allá de 4 semanas.
Cese de la progresión sin recuperación o con
secuelas permanentes importantes.
Afectación de esfínteres.
Afectación del SNC.
Criterios de LCR: Proteínas aumentadas tras la 1ª semana.
2‐ 10 células/mm3 ‐ 50 Células/dl o menos (leucocitos
mononucleares).
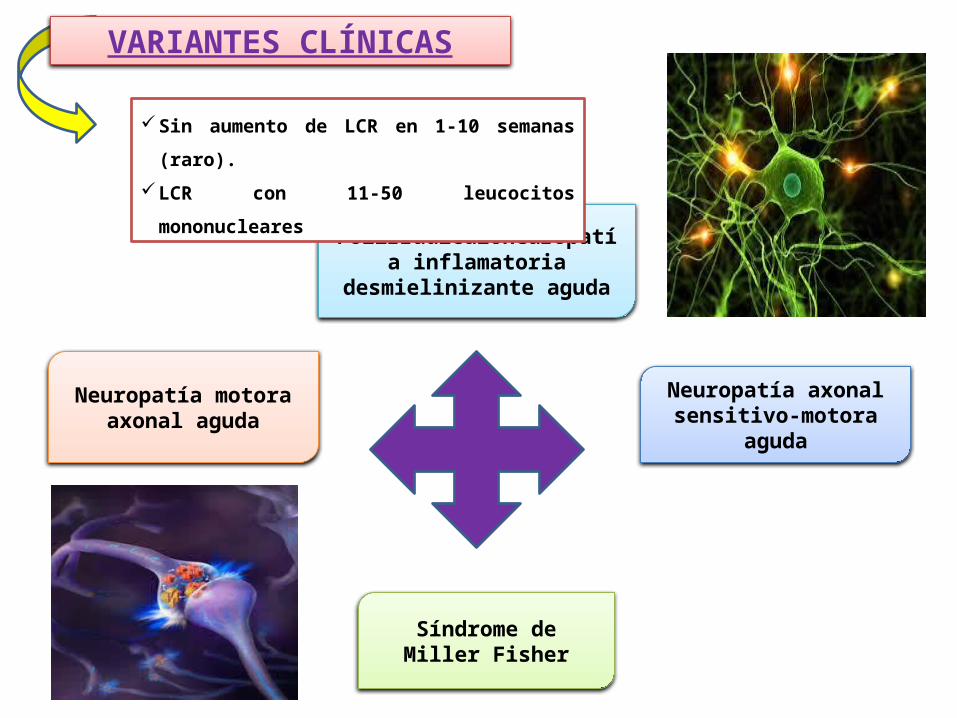
VARIANTES CLÍNICAS
Polirradiculoneuropatía inflamatoria desmielinizante
aguda
Neuropatía axonal sensitivo-motora aguda
Síndrome de Miller Fisher
Neuropatía motora axonal aguda
Sin aumento de LCR en 1‐10 semanas (raro).
LCR con 11‐50 leucocitos mononucleares
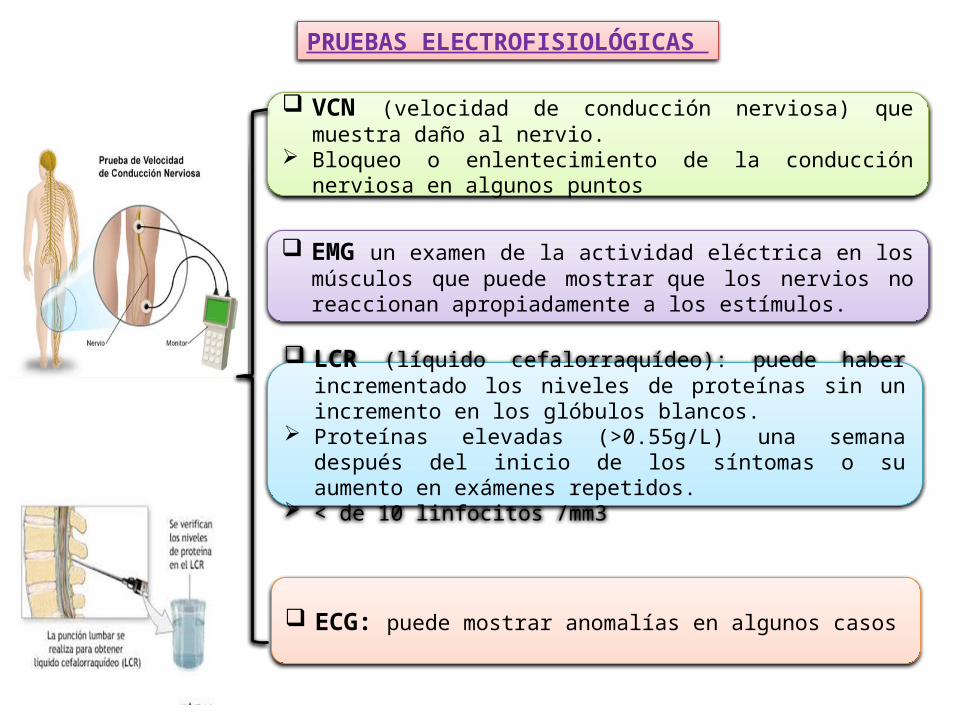
PRUEBAS ELECTROFISIOLÓGICAS
VCN (velocidad de conducción nerviosa) que muestra daño al nervio. Bloqueo o enlentecimiento de la conducción nerviosa en algunos
puntos
EMG un examen de la actividad eléctrica en los músculos que puede mostrar que los nervios no reaccionan apropiadamente a los estímulos.
LCR (líquido cefalorraquídeo): puede haber incrementado los niveles de proteínas sin un incremento en los glóbulos blancos.
Proteínas elevadas (>0.55g/L) una semana después del inicio de los síntomas o su aumento en exámenes repetidos.
< de 10 linfocitos /mm3
ECG: puede mostrar anomalías en algunos casos

HALLAZGOS QUE HACEN EL DIAGNOSTICO DUDOSO
Existencia de un nivel sensorial
Marcada asimetría de signos y síntomas
Disfunción severa y persistente de vejiga y intestino
Más de 50 células/mm3 en LCR
HALLAZGOS QUE EXCLUYEN EL DIAGNÓSTICO
Diagnóstico de botulismo, miastenia,
poliomielitis o neuropatía tóxica
Metabolismo alterado de las porfirinas
Difteria reciente
Síndrome sensorial puro sin fatiga

0 Saludable
1 Síntomas o signos menores de neuropatía pero capaz de trabajo manual/capaz de correr
2 Capaz de caminar sin ayuda de un bastón (5m en un espacio abierto), pero incapaz detrabajo manual/correr
3 Capaz de caminar con un bastón, aparato o soporte (5m en un espacio abierto)
4 Confinados a la cama o silla
5 Requieren ventilación asistida (por cualquier parte del día o de noche)
6 Muerte
Escala de Incapacidad (Hughes)

DIAGNÓSTICO DIFERENCIAL
1.-Neuropatías agudas:• Difteria• Vasculitis• Enfermedad de Lyme• 2.- Enfermedades de la placa
neuromuscular:• Botulismo• Miastenia Gravis

3.- Enfermedades musculares:
Hipokalemia PolimiositisRabdomiolisis

TratamientoEl manejo de la función respiratoria debe incluir la permeabilidad de las vías aéreas.
La capacidad del paciente para toser y expectorar, la evaluación de la mecánica ventilatoria, la habilidad para tragar y la aparición de síntomas y signos de hipoxemia y/o hipercapnia.

Analgesia
El dolor es un síntoma común y se presenta en alrededor de un 50%.El tipo más común es un dolor muscular profundo, que puede ser manejado habitualmente con AINEs . El dolor neuropático es también frecuente y se pude manejar con: Carbamazepina, Ac. Valproico.
Soporte Nutricional Intensivo
Alimentación enteral con 40 a 45 kcal/kg. de calorías no proteicas y 2 a2,5 gr/kg. de proteínas al día.
• En caso de ileo se debería usar nutrición parenteral.

Tratamiento Específico
1.- Plasmaféresis (PF).200 a 250ml de plasma /kg
2.- Inmunoglobulina Endovenosa. 5 infusiones diarias de IG (0,4 g/kg/día) administradas las primeras 2 semanas desde el inicio de la enfermedad.

“...si no fuera por la granvariabilidad entre lospacientes la medicina
podría ser consideradacomo una ciencia y no un
arte”
William Osler, 1892
GRACIAS!!
























