Embed Size (px)
Citation preview

CUTANEOUS LYMPHOMAS
Target Audience: Oncology Fellows, Oncology physicians, Oncologists
Archer Board Review Courseswww.Ccsworkshop.com

CUTANEOUS LYMPHOMAS A variety of T and B-cell neoplasms can
involve skin, either primarily or secondarily.
Primary cutaneous lymphoma : cutaneous T-cell lymphomas (CTCLs) and cutaneous B-cell lymphomas (CBCLs) that present in the skin with no evidence of extracutaneous disease at the time of diagnosis.
Secondary cutaneous lymphomas : systemic lymphomas that secondarily involve the skin.

PRIMARY CUTANEOUS LYMPHOMAS
After the gastrointestinal tract, the skin is the second most common site of extranodal non-Hodgkin lymphoma.
Estimated annual incidence 1:100,000.
Have a completely different clinical behavior and prognosis from histologically similar systemic lymphomas, which may involve the skin secondarily.
Hence, require different types of treatment as opposed to systemic lymphomas.
For that reason, the European Organization for Research and Treatment of Cancer (EORTC) classification for primary cutaneous lymphomas and the World Health Organization (WHO) classification for tumors of hematopoietic and lymphoid tissues included primary cutaneous lymphomas as separate entities. A consensus classification was developed in 2005 referred to as “WHO-EORTC Classification of Cutaneous Lymphomas”.
65% of all Primary Cutaneous Lymphomas are of T-cell type.
CUTANEOUS T-CELL LYMPHOMAS(CTCL)•CLASSIFICATION•CLINICAL FEATURES•TREATMENT
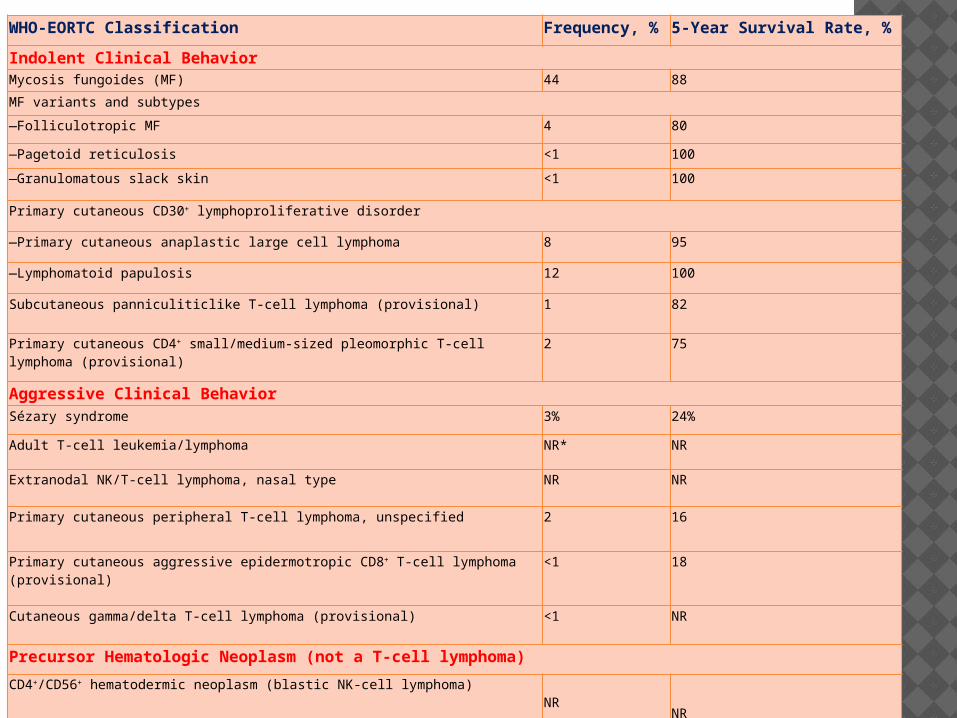
WHO-EORTC Classification Frequency, % 5-Year Survival Rate, %
Indolent Clinical BehaviorMycosis fungoides (MF) 44 88
MF variants and subtypes
—Folliculotropic MF 4 80
—Pagetoid reticulosis <1 100
—Granulomatous slack skin <1 100
Primary cutaneous CD30+ lymphoproliferative disorder
—Primary cutaneous anaplastic large cell lymphoma 8 95
—Lymphomatoid papulosis 12 100
Subcutaneous panniculiticlike T-cell lymphoma (provisional) 1 82
Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma (provisional)
2 75
Aggressive Clinical BehaviorSézary syndrome 3% 24%
Adult T-cell leukemia/lymphoma NR* NR
Extranodal NK/T-cell lymphoma, nasal type NR NR
Primary cutaneous peripheral T-cell lymphoma, unspecified 2 16
Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (provisional)
<1 18
Cutaneous gamma/delta T-cell lymphoma (provisional) <1 NR
Precursor Hematologic Neoplasm (not a T-cell lymphoma)
CD4+/CD56+ hematodermic neoplasm (blastic NK-cell lymphoma)NR
NR

MYCOSIS FUNGOIDES (MF)

MYCOSIS FUNGOIDES Extranodal Non-Hodgkins lymphoma of T-cell
origin, with primary involvement of the skin. First case described in 1806 by Alibert:
“mushroom like tumors” Most common type of CTCL Accounts for almost 50% of all primary
cutaneous lymphomas and 2.2% of all lymphomas.
3 cases/ 1,000,000/ year<1000/year US Peak age 55-60 Male: female 2:1 More common in African-Americans

MF - CLINICAL FEATURES Heterogeneity in presentation. Indolent cutaneous eruption with erythematous
scaly patches or plaques, typically bathing trunk distribution.
Poikiloderma may be seen - presence of mottled pigmentation, epidermal atrophy, and telangiectasia associated with slight infiltration.
3 Phases of progression: Macular erythematous eruption Plaque/Patch phase, resembles eczema/psoriasis Tumor nodules/ generalized erythroderma and associated
adenopathy or visceral involvement ( Often seen in Sezary Syndrome)
Circulating Sezary Cells

CLINICAL PHASES OF MF/SS
Patch & Plaque Tumor
Erythroderma
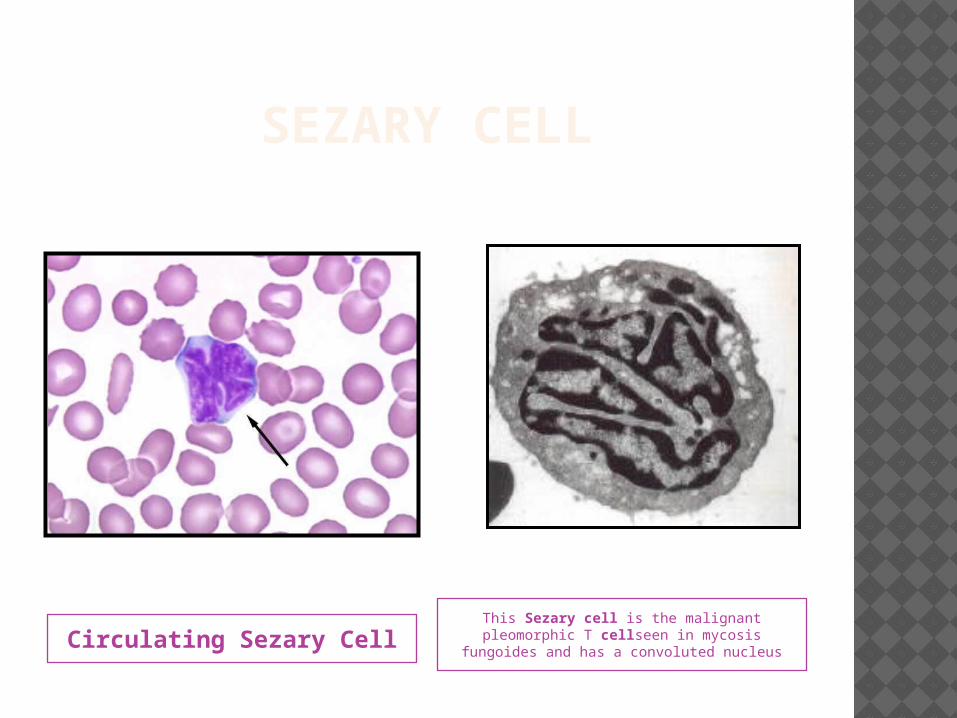
SEZARY CELL
Circulating Sezary CellThis Sezary cell is the malignant pleomorphic T cellseen in mycosis
fungoides and has a convoluted nucleus

SEZARY CELLS Sezary cells are mononuclear cells with
a cerebriform nucleus Small numbers of these cells can be
seen among healthy individuals In MF, an increased number of Sezary
cells seen in the peripheral blood. An absolute count ≥1000 Sezary
cells/cubic mm is a diagnostic criterion for Sezary syndrome.

EXTRACUTANEOUS DISEASE Extracutaneous manifestations :
involvement of regional lymph nodes (approximately 30 percent in MF )
Lungs Spleen Liver Gastrointestinal tract. Bone marrow involvement is rare
Progression to Extracutaneous disease correlates with extent of skin disease Limited patch or plaque very rare Generalized plaque 8 % Tumorous or generalized erythroderma30-40% Hence,
extracutaneous is more commonly seen in Sezary syndrome.
Hoppe RT, Kim YH,Clinical features, staging, and prognosis of mycosis fungoides and Sezary syndrome, Uptodate.com, 11/06
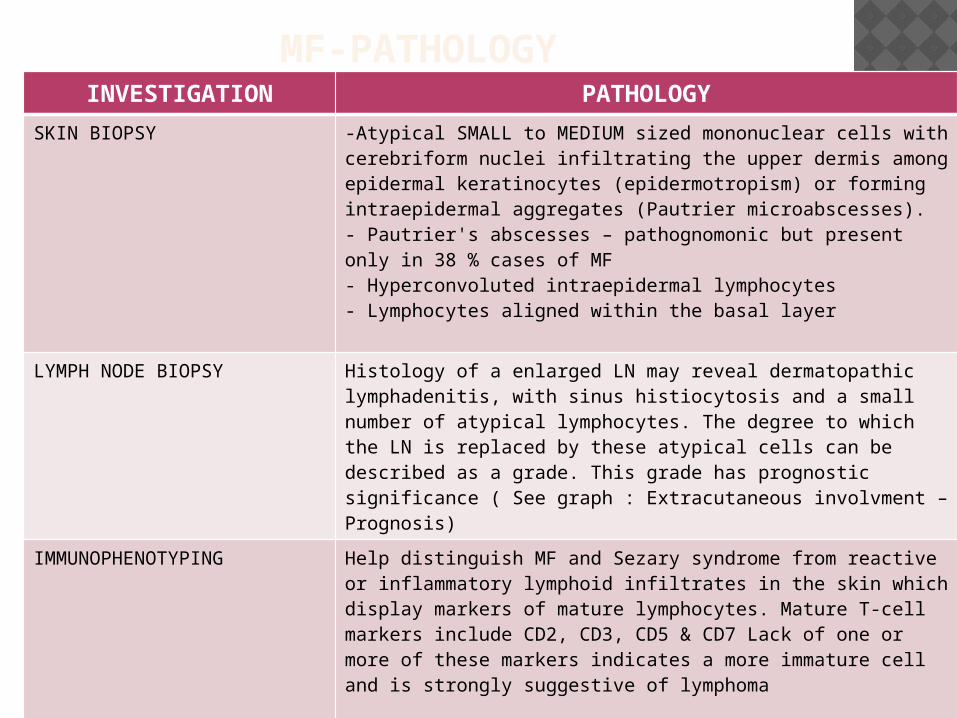
MF-PATHOLOGYINVESTIGATION PATHOLOGY
SKIN BIOPSY -Atypical SMALL to MEDIUM sized mononuclear cells with cerebriform nuclei infiltrating the upper dermis among epidermal keratinocytes (epidermotropism) or forming intraepidermal aggregates (Pautrier microabscesses).- Pautrier's abscesses – pathognomonic but present only in 38 % cases of MF- Hyperconvoluted intraepidermal lymphocytes- Lymphocytes aligned within the basal layer
LYMPH NODE BIOPSY Histology of a enlarged LN may reveal dermatopathic lymphadenitis, with sinus histiocytosis and a small number of atypical lymphocytes. The degree to which the LN is replaced by these atypical cells can be described as a grade. This grade has prognostic significance ( See graph : Extracutaneous involvment – Prognosis)
IMMUNOPHENOTYPING Help distinguish MF and Sezary syndrome from reactive or inflammatory lymphoid infiltrates in the skin which display markers of mature lymphocytes. Mature T-cell markers include CD2, CD3, CD5 & CD7 Lack of one or more of these markers indicates a more immature cell and is strongly suggestive of lymphoma
MOLECULAR ANALYSIS(Southern Blot analysis or PCR amplification method)
TCR gene rearrangements ( to demonstrate clonality neoplastic T cells exhibit clonal TCR gene rearrangements)
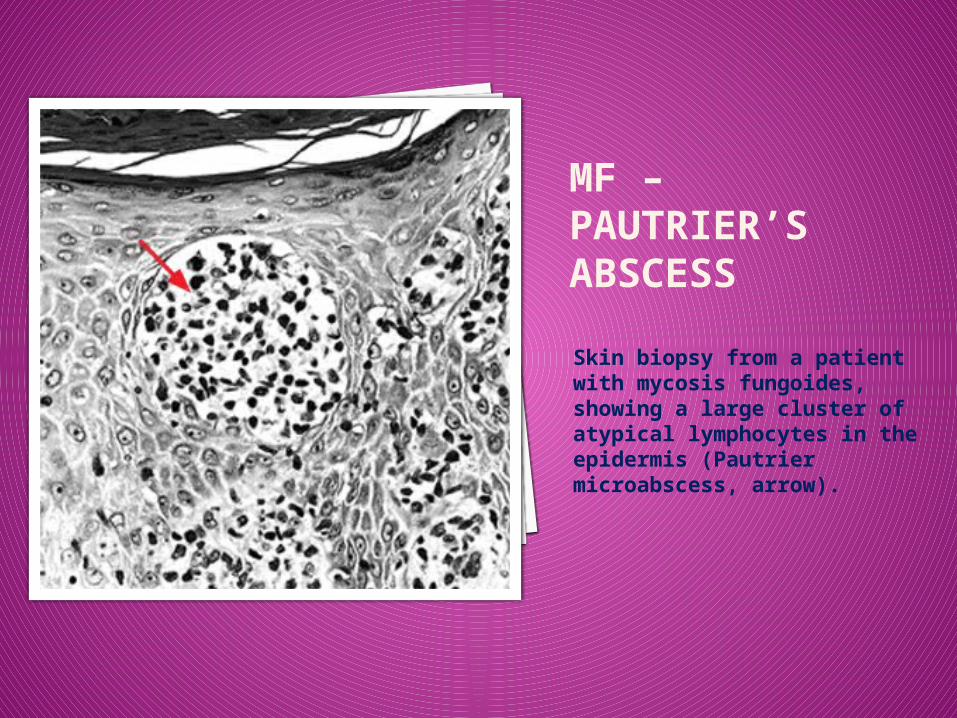
MF – PAUTRIER’S ABSCESS
Skin biopsy from a patient with mycosis fungoides, showing a large cluster of atypical lymphocytes in the epidermis (Pautrier microabscess, arrow).

MF - DIAGNOSIS ISCL/EORTC Diagnostic algorithm :
Point based system A total of 4 points is required for the diagnosis
of MF based on any combination of points from the clinical, histopathologic, molecular biological, and immunopathologic criteria.
Clinical Findings Skin Biopsy ( Histopathology) Molecular criteria Immunophenotyping : CD3+, CD4+, CD8-, CD30-,
CD45RO+, TCR gene rearrangements
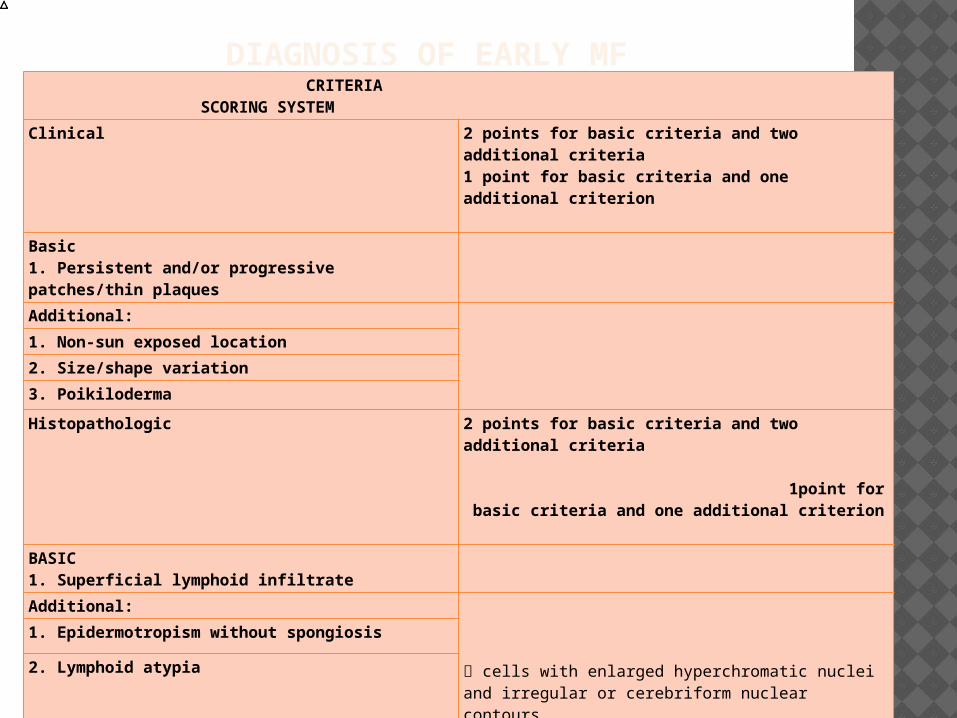
DIAGNOSIS OF EARLY MF CRITERIA SCORING SYSTEM
Clinical 2 points for basic criteria and two additional criteria1 point for basic criteria and one additional criterion
Basic1. Persistent and/or progressive patches/thin plaquesAdditional:
1. Non-sun exposed location
2. Size/shape variation
3. Poikiloderma
Histopathologic 2 points for basic criteria and two additional criteria 1point for basic criteria and one additional criterion
BASIC1. Superficial lymphoid infiltrate
Additional:
cells with enlarged hyperchromatic nuclei and irregular or cerebriform nuclear contours.
1. Epidermotropism without spongiosis
2. Lymphoid atypia
Molecular biological
1. Clonal TCR gene rearrangement 1 point for clonality
Immunopathologic
1. <50 percent CD2+, CD3+, and/or CD5+ T-cells
1 point for one or more criteria
2. <10 percent CD7+ T cells
3. Epidermal/dermal discordance of CD2, CD3, CD5, or CD7

MF – STAGING EVALUATION
Skin evaluation : percentage of involved body surface area must be estimated.
Imaging Studies : CXR, CT Chest/ Abd/ pelvis with or without PET to evaluate the visceral involvement and adenopathy.
Lymph node biopsy : The involved lymphnodes seen clinically or on PET/CT need to be biopsied.
Bone marrow aspirate & Biopsy : Not routinely employed as part of the initial staging procedure for MF. However, indicated in select cases to document visceral disease if marrow involvement is suspected, for eg: as in the setting of B2 blood involvement or in patients with an unexplained hematologic abnormality.
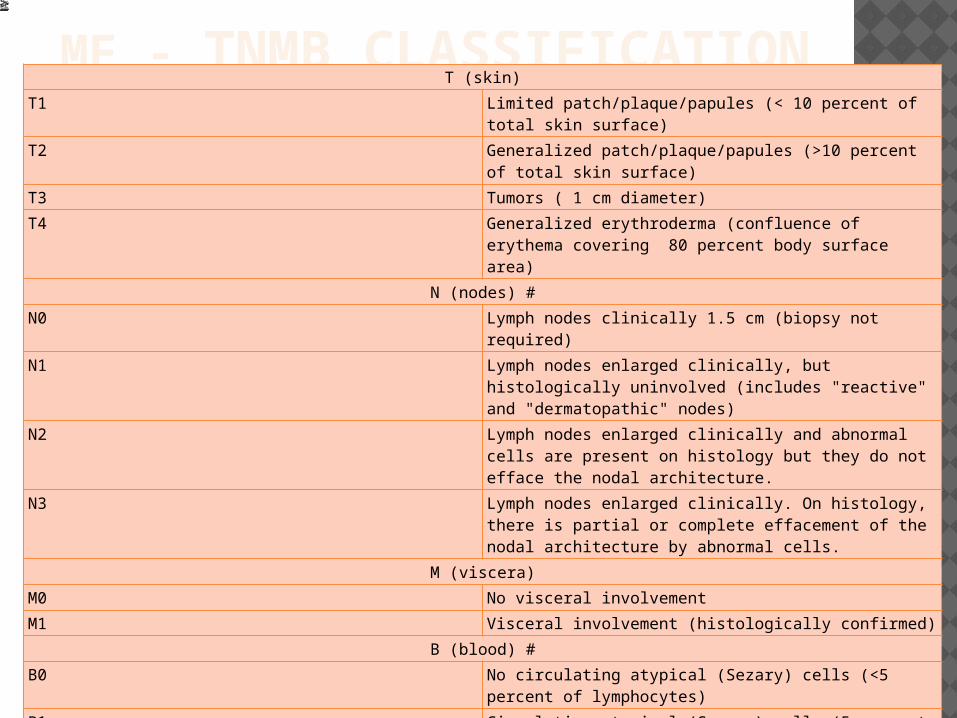
MF - TNMB CLASSIFICATIONT (skin)
T1 Limited patch/plaque/papules (< 10 percent of total skin surface)
T2 Generalized patch/plaque/papules (>10 percent of total skin surface)
T3 Tumors ( 1 cm diameter)
T4 Generalized erythroderma (confluence of erythema covering 80 percent body surface area)
N (nodes) #N0 Lymph nodes clinically 1.5 cm (biopsy not required)
N1 Lymph nodes enlarged clinically, but histologically uninvolved (includes "reactive" and "dermatopathic" nodes)
N2 Lymph nodes enlarged clinically and abnormal cells are present on histology but they do not efface the nodal architecture.
N3 Lymph nodes enlarged clinically. On histology, there is partial or complete effacement of the nodal architecture by abnormal cells.
M (viscera)M0 No visceral involvement
M1 Visceral involvement (histologically confirmed)
B (blood) #B0 No circulating atypical (Sezary) cells (<5 percent of
lymphocytes)
B1 Circulating atypical (Sezary) cells (5 percent of lymphocytes)
B2 High blood tumor burden: 1000/microL Sezary cells with positive clone

MF - STAGINGIA T1 - Patch/PlaqueIB T2 - Patch/PlaqueIIA N1 - Clinical NodesIIB T3 - TumorsIII T4 - ErythrodermaIVA N2-N3 - Path NodesIVB M1 - Visceral Mets
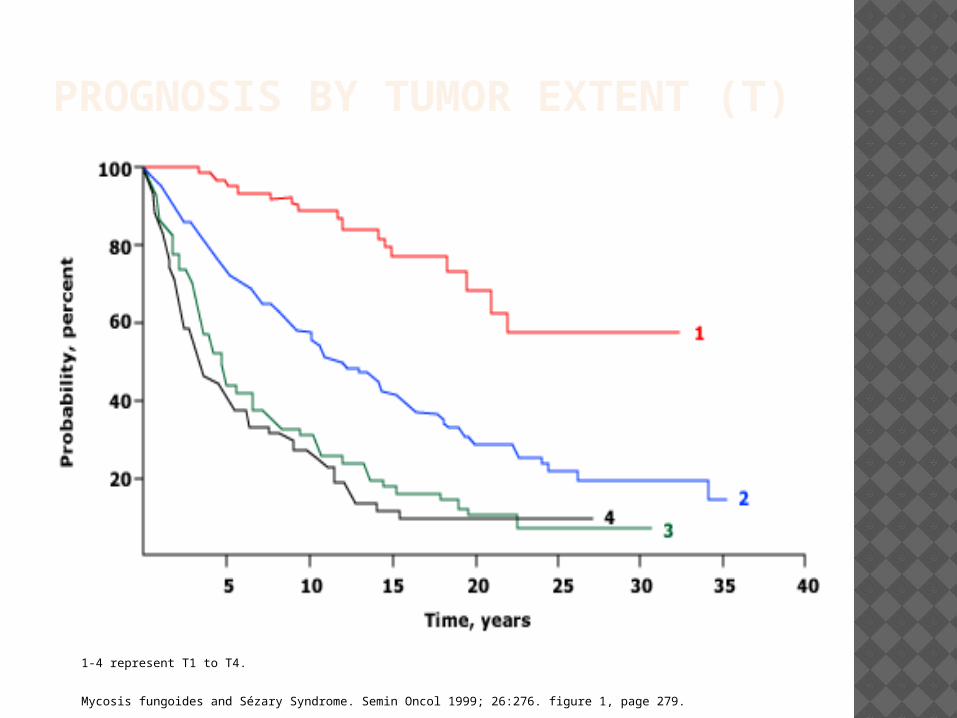
PROGNOSIS BY TUMOR EXTENT (T)
1-4 represent T1 to T4.
Mycosis fungoides and Sézary Syndrome. Semin Oncol 1999; 26:276. figure 1, page 279.
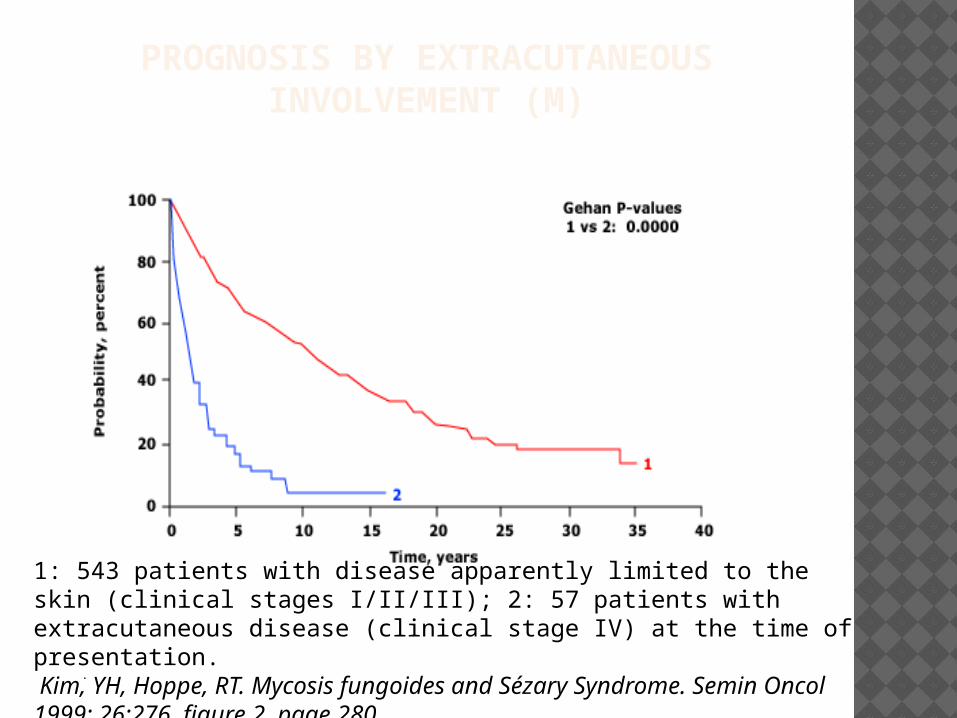
PROGNOSIS BY EXTRACUTANEOUS INVOLVEMENT (M)
.
1: 543 patients with disease apparently limited to the skin (clinical stages I/II/III); 2: 57 patients with extracutaneous disease (clinical stage IV) at the time of presentation. Kim, YH, Hoppe, RT. Mycosis fungoides and Sézary Syndrome. Semin Oncol 1999; 26:276. figure 2, page 280.
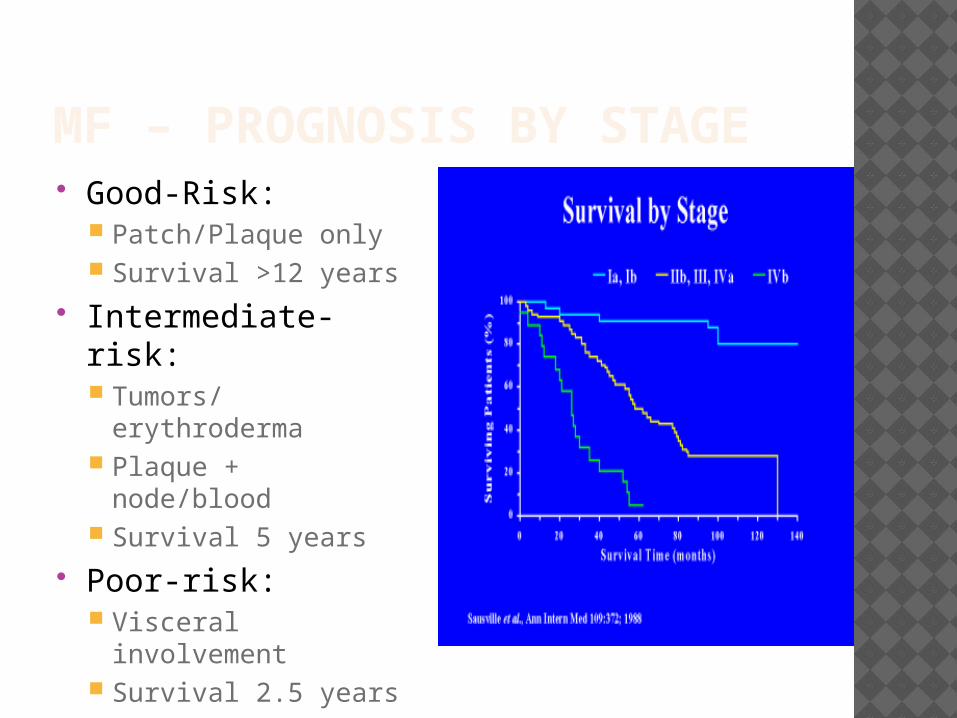
MF – PROGNOSIS BY STAGE Good-Risk:
Patch/Plaque only Survival >12 years
Intermediate-risk: Tumors/erythroderma Plaque + node/blood Survival 5 years
Poor-risk: Visceral involvement Survival 2.5 years

TREATMENT
MYCOSIS FUNGOIDES

MF - TREATMENT Cures generally unattainable Goals of treatment are symptom relief and cosmetic improvement
(palliation) Early aggressive therapy results in high complete remission rates but no
significant difference in DFS or OS.
Patients are susceptible to infections with skin flora; immune suppression is undesirable
Skin Directed Phototherapy : UVB (Ultraviolet B) or PUVA ( Psoralen + UVA photochemotherapy) Topical chemotherapy :- Nitrogen Mustard (HN2) or Carmustine (BCNU) Radiation therapy ( Electron Beam Therapy, TSEBT – Total Skin Electron Beam
Therapy) Topical Retinoids ( Baexarotene) Topical Corticosteroids
Systemic Photopheresis Biologic Therapies : ( IFN alfa, Denileukin diftitox) Retinoids/Rexinoids ( Oral Bexarotene or Isotretinoin) HDAC ( Histone Deacetylase inhibitors) - Vorinostat Chemotherapy ( Single agent chemotherapy – Methotrexate, Doxil, Gemcitabine,
Chlorambucil, Cyclophosphamide)
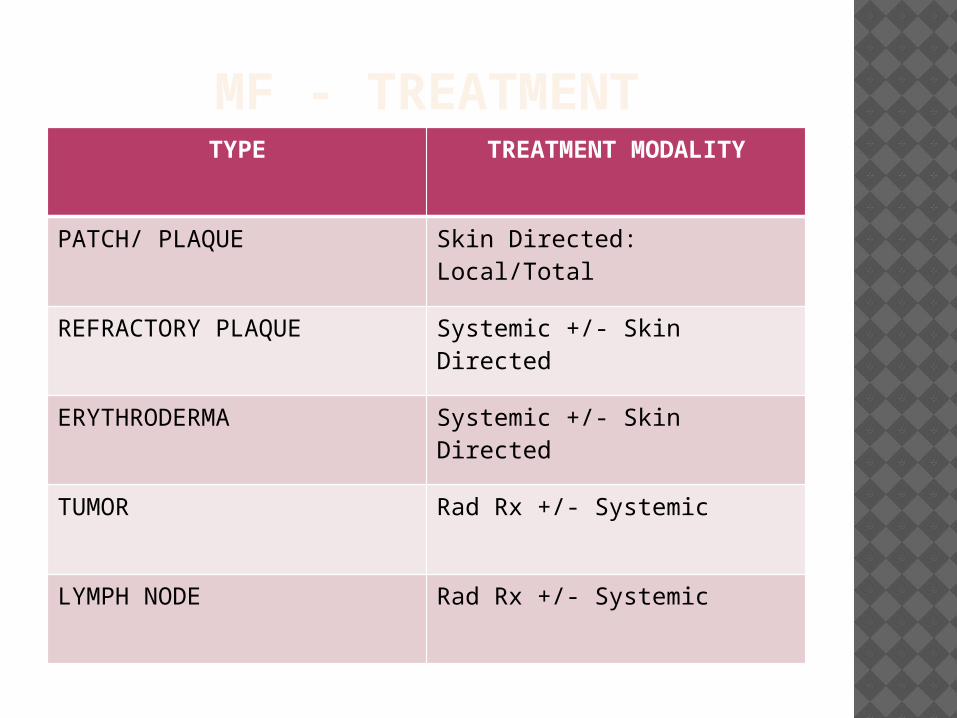
MF - TREATMENTTYPE TREATMENT MODALITY
PATCH/ PLAQUE Skin Directed: Local/Total
REFRACTORY PLAQUE Systemic +/- Skin Directed
ERYTHRODERMA Systemic +/- Skin Directed
TUMOR Rad Rx +/- Systemic
LYMPH NODE Rad Rx +/- Systemic

PUVA Ingestion of 8-methoxypsoralen (0.6mg/kg, 2
hours before UVA exposure) Becomes activated when exposed to UV
light and increases the skin's sensitivity to UV light and hence, improves the effectiveness of UV light therapy
Treatments 3x/wk with subsequent tapering 65% Complete Response, 95% OR, duration
of response 43 months, Mean survival 8.5 years in Stage I
Adverse effects : nausea, erythema, pruritis, dry skin, secondary skin malignancies

TOPICAL CHEMOTHERAPY Nitrogen Mustard (HN2) or Carmustine
(BCNU) Overall Response Rates 70-90% in
Stage I disease Adverse effects: contact dermatitis,
erythema, telangiectasias

RADIATION THERAPY CTCL is very radiosensitive Use of Electron Beam Therapy limits
toxicity, <5% of dose travels beyond 2cm
Standard total dose is 36 Gy CR 56-96% in Stage IA-IIA Given in combination with other
agents to avoid relapse Toxicity: erythema, pain, swelling,
hair and nail loss, secondary skin cancer

PHOTOPHERESIS Reserved for Sezary Syndrome (Stage IVA1)
and Stage IIIB disease Technique: Patient ingests 8-MOP. Leukapheresis,
mononuclear fraction of patient’s WBCs are collected and exposed to UVA, then returned to patient. UVA is toxic to cells and reinfused cells stimulate a selective immune response against malignant cells.
RR (response rate) 73%, median survival 5 years in one study of pt’s with mainly SS

INTERFERON ALFA ORR 79% in pts with all stage disease Maximum dose limited by side effects Started at 3million U and titrated up
to maximum of 15million U 3x/wk In one study combining PUVA with IFN
12 million Units 3x/wk – ORR 88%, CR 62%, response duration 28 months

BEXAROTENE Novel Retinoid – Rexinoid FDA approved for use in advanced MF
i.e; Stage IIB to IVB in patients who have not responded to at least one prior systemic therapy
Selectively activates retinoid X receptors (nuclear hormone receptors)
Acts on retinoid response elements to alter gene expression
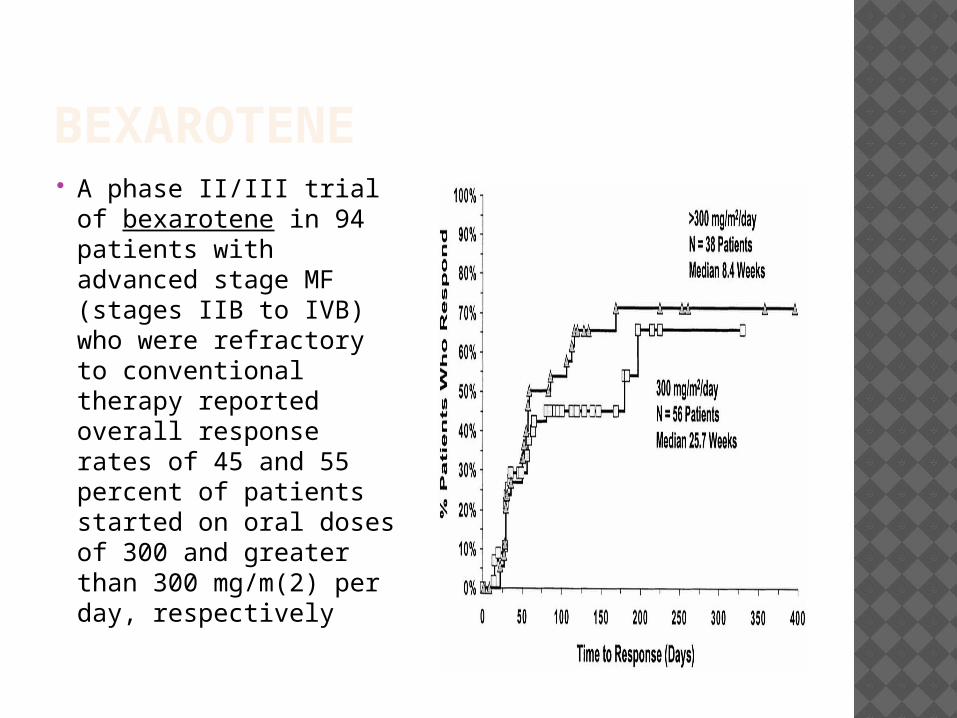
BEXAROTENE A phase II/III trial
of bexarotene in 94 patients with advanced stage MF (stages IIB to IVB) who were refractory to conventional therapy reported overall response rates of 45 and 55 percent of patients started on oral doses of 300 and greater than 300 mg/m(2) per day, respectively

BEXAROTENE Adverse effects :
Hypertriglyceridemia 63% Most patients require drugs to reduce
hypertriglyceridemia such as statin or fibrates. Diet should include Vitamin E and dietary consultation, especially for monotherapy patients.
Hypothyroidism 43% These patients need synthroid supplements.
Leukopenia 7% Dose adjustments control leukopenia
Teratogenic

VORINOSTAT Oral HDAC (Histone Deacetylase) inhibitor Partial response rates in MF of 30 percent Approved by the FDA for the treatment of
cutaneous T-cell lymphoma (CTCL) in patients with progressive, persistent, or recurrent disease on or following two systemic therapies.
Common Side effects : Gastrointestinal symptoms (diarrhea, nausea, anorexia,
weight decrease, vomiting, constipation) Constitutional symptoms (fatigue, chills) Hematologic abnormalities (thrombocytopenia, anemia) Taste disorders (dysgeusia, dry mouth) Abnormal laboratory values include high glucose,
abnormal EKGs
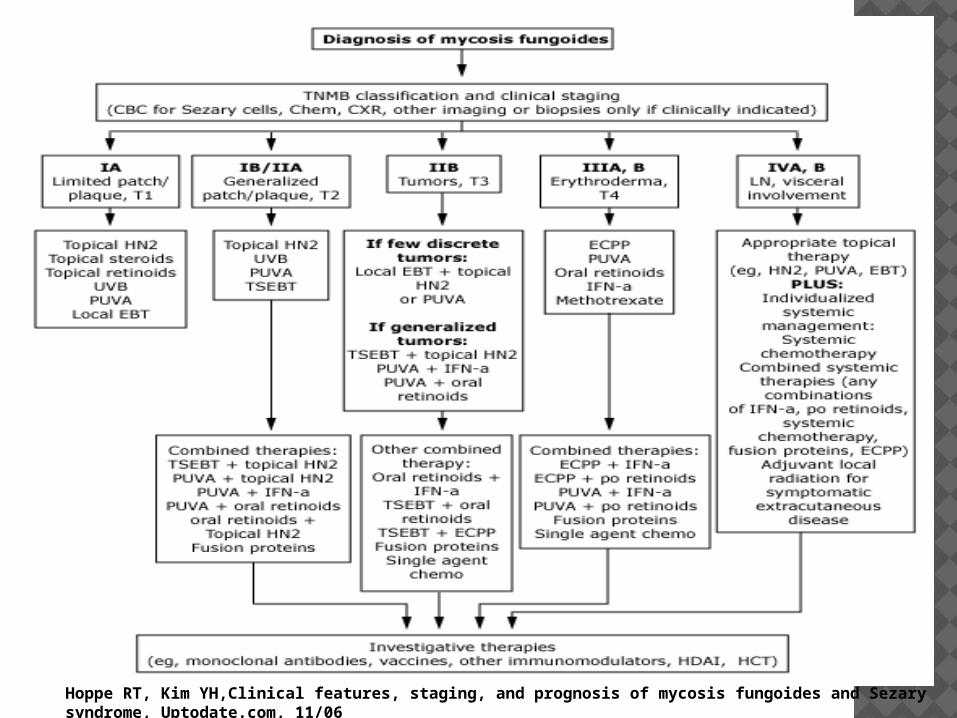
Hoppe RT, Kim YH,Clinical features, staging, and prognosis of mycosis fungoides and Sezary syndrome, Uptodate.com, 11/06

SEZARY SYNDROME

SEZARY SYNDROME Likely represents leukemic phase of mycosis
fungoides. Sezary Syndrome:
Generalized erythroderma + intense Pruritis Lymphadenopathy Atypical T- cells (Sezary cells) in the peripheral blood
( An absolute count ≥1000 Sezary cells/cubic mm is a diagnostic criterion for Sezary syndrome equivalent to the B2 designation in the TNMB classification syndrome).
Low levels of Sezary-like cells can be detected in the peripheral blood of patients with benign skin conditions. Hence, diagnostic criteria of Sezary syndrome uses an absolute Sezary cell count of >1000/microL with positive clones

SEZARY SYNDROME Diagnosis is made when there is a clonal
rearrangement of the T-cell receptor (TCR) in the blood (identified by PCR or southern blot analysis) plus
either an absolute Sezary cell count of at least 1000
cells/microL or one of the following two criteria Increased CD4+ or CD3+ cells with a CD4 to
CD8 ratio of 10 or more. Increased CD4+ cells with an abnormal
phenotype (such as a CD4+ to CD7- ratio ≥40 percent or a CD4+ to CD26- ratio ≥30 percen

SEZARY SYNDROME Treatment includes Extracorporeal Photopheresis
( ECP) alone or in combination with other therapies ( IFNα) OR 30-80% and CR 15-25%.
Recent studies report benefitis with Bexarotene and Alemtuzumab (anti-CD52) therapies – more data needed.
Prognosis - generally poor with a median survival between 2 and 4 years.
Most patients die of opportunistic infections that are due to immunosuppression
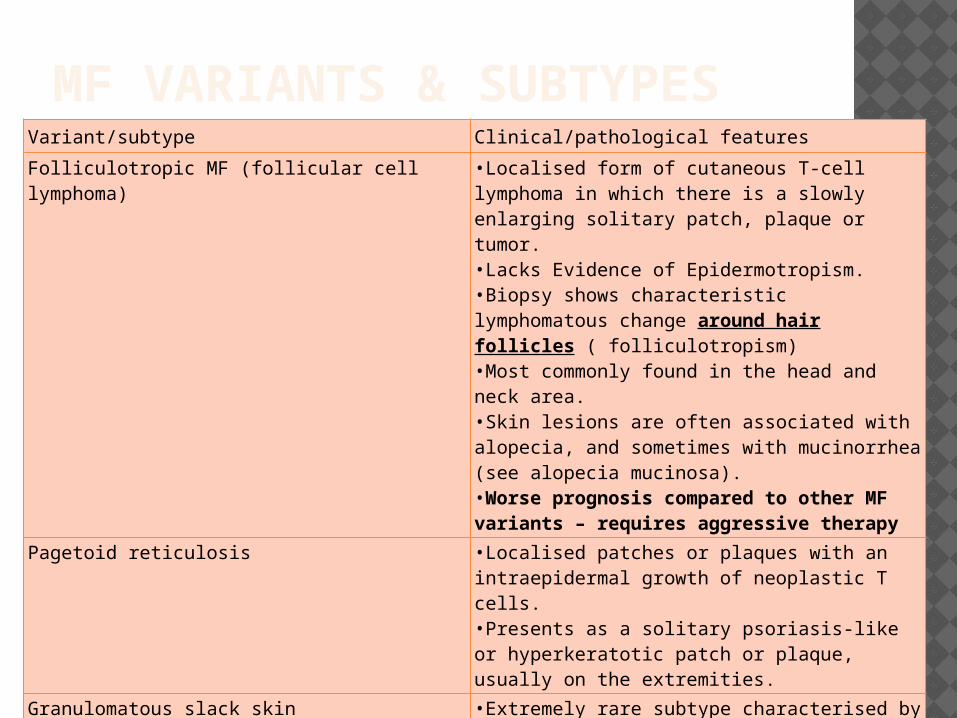
MF VARIANTS & SUBTYPES
Variant/subtype Clinical/pathological features
Folliculotropic MF (follicular cell lymphoma) •Localised form of cutaneous T-cell lymphoma in which there is a slowly enlarging solitary patch, plaque or tumor. •Lacks Evidence of Epidermotropism. •Biopsy shows characteristic lymphomatous change around hair follicles ( folliculotropism) •Most commonly found in the head and neck area.•Skin lesions are often associated with alopecia, and sometimes with mucinorrhea (see alopecia mucinosa).•Worse prognosis compared to other MF variants – requires aggressive therapy
Pagetoid reticulosis •Localised patches or plaques with an intraepidermal growth of neoplastic T cells.•Presents as a solitary psoriasis-like or hyperkeratotic patch or plaque, usually on the extremities.
Granulomatous slack skin •Extremely rare subtype characterised by slow development of folds of lax skin in the major skin folds.•Skin folds show a granulomatous infiltrate with clonal T cells.•Occurs most commonly in the groin and underarm regions.

PRIMARY CUTANEOUS CD30+ LYMPHOPROLIFERATIVE
DISORDERS (LPD)

PRIMARY CUTANEOUS CD30+ LYMPHOPROLIFERATIVE DISORDERS (LPD)
Second most common group of cutaneous T-cell lymphomas (CTCLs)
Accounts for approximately 30% of CTCLs.
C-ALCL and LyP form a spectrum of disease histologic criteria alone are often insufficient to differentiate between these 2 ends of this spectrum.
The clinical appearance and course are used as decisive criteria for the definite diagnosis and choice of treatment.
“Borderline case" refers : refers to cases in which, despite careful clinicopathologic correlation, a definite distinction between C-ALCL and LyP cannot be made. Clinical examination during further follow-up will generally disclose whether the patient has C-ALCL or LyP.
•Primary cutaneous anaplastic large cell lymphoma (C-ALCL)•Lymphomatoid papulosis (LyP)
•Borderline cases
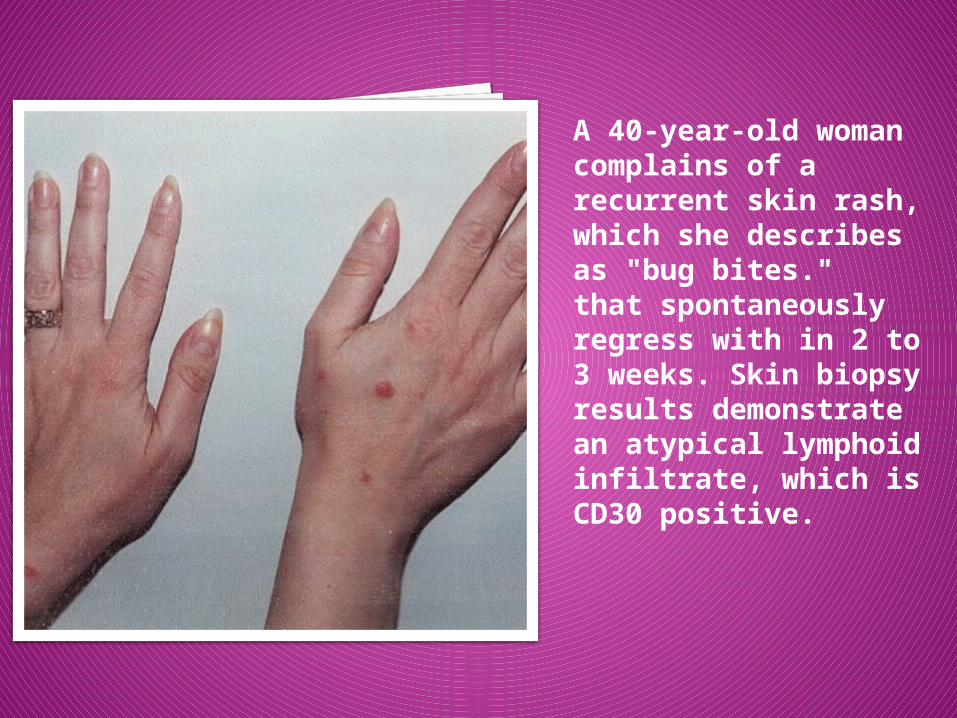
. A 40-year-old woman complains of a recurrent skin rash, which she describes as "bug bites." that spontaneously regress with in 2 to 3 weeks. Skin biopsy results demonstrate an atypical lymphoid infiltrate, which is CD30 positive.

LYMPHOMATOID PAPULOSIS
A chronic, recurrent, self-healing papulonecrotic or papulonodular skin disease with histologic features suggestive
of a (CD30+) malignant lymphoma.

LYMPHOMATOID PAPULOSIS
First described by Macau-ley in 1968 – in his words, he discussed a case in which "repeated biopsies of ... skin lesions consistently reveal an alarming infiltrate of large pleomorphic hyper-chromatic cells which expert histopathologists and hematologists ... variously classified as highest grade malignant lymphoma, malignant reticulosis, metastatic carcinoma, malignant melanoma, undifferentiated malignant tumor."
“A self-healing rhythmical paradoxical eruption, histologically malignant but clinically benign.”
Frequently misdiagnosed.

LYMPHOMATOID PAPULOSIS Despite modern techniques, cannot be definitively
diagnosed by pathologists without pertinent clinical information.
Histologically, the proliferation is malignant and may possess any or all of the features of T-cell malignancy, including aberrant T-cell antigen expression and clonal rearrangement of T-cell receptor genes. Clonal T-cell gene rearrangement can be seen in 60 to 70% cases.
The component cells are highly anaplastic and express the CD30 antigen. Histopathologic features considerably overlap those of anaplastic large-cell lymphoma or in some cases, the histologic features more closely resemble those of mycosis fungoides.

LYMPHOMATOID PAPULOSIS
These difficulties can lead to a mistaken pathologic diagnosis of malignant lymphoma or other types of cancer.
Clinical criteria for the diagnosis :The defining clinical feature of this
disease is spontaneous regression . The patient must be observed without treatment to determine whether spontaneous regression will occur.

LYMPHOMATOID PAPULOSISHistopathology
Histologic picture is extremely variable.
Three histologic subtypes :
Represent a spectrum with overlapping features.
LyP type A : most common ( 75%) - scattered small clusters of large, sometimes multinucleated or Reed-Sternberg-like, CD30+ cells are intermingled with numerous inflammatory cells (histiocytes, small lymphocytes, neutrophils, and/or eosinophils).
LyP type B : uncommon (less than 10%) - characterized by an epidermotropic infiltrate of small atypical cells with cerebriform nuclei similar to that observed in MF. . CD30+ large cells are rare or absent, but epidermotropism is more common in this variant.
LyP type C : demonstrate a monotonous population or large clusters of large CD30+ T cells with relatively few admixed inflammatory cells. Histologically, indistinguishable from Anaplastic Large Cell Lymphoma, with the exception of the minimal subcutaneous invasion.

LYMPHOMATOID PAPULOSIS
Clinical Features
Generally occurs in adults (median age, 45 yrs)
Clinically benign.
Recurrent crops of self-healing, red-brown, centrally hemorrhagic or necrotic papules and nodules on the trunk or extremities, which can evolve in to papulovesicular or pustular lesions.
Lesions are much smaller than anaplastic large cell lymphoma (<2 cm)
Spontaneously resolve in 4-6 weeks, leaving hyperpigmentation or atrophic scars.
Unless accompanied by systemic lymphoma, most patients have no constitutional symptoms

LYMPHOMATOID PAPULOSIS
Clinical Features
No single clinical characteristic at presentation can distinguish Lyp from Lymphoma with absolute certainty. The following features at presentation may indicate the condition is probably malignant :
Presence of a solitary skin lesion > 3cm in diameter.
Persistence without a spontaneous regression.
Presence of significant lymphadenopathy. The involved lymphnode must be biopsied to rule out lymphoma.

LYMPHOMATOID PAPULOSIS
Treatment
Curative therapy is not available.
None of the available treatment modalities affects the natural course of the disease Hence, short-term benefits of active treatment should be balanced carefully against the potential side effects.
Beneficial effects have been reported of PUVA (Oral psoralen plus UVA phototherapy) and topical chemotherapy.
Low-dose oral methotrexate (5-20 mg/wk) - most effective therapy to suppress the development of new skin lesions. However, the disease recurs within 1-2 weeks after discontinuing the therapy.
Therefore, in patients with relatively few and nonscarring lesions, long-term follow-up without active treatment should be considered.

LYMPHOMATOID PAPULOSISPrognosis & Predictive factors
Excellent prognosis.
Waxing and waning course. The disease duration may vary from several months to more than 40 years.
10 to 20% cases may progress to or may have an associated malignancies such as Hodgkin disease, mycosis fungoides, or primary cutaneous anaplastic large cell lymphoma.

LYMPHOMATOID PAPULOSISPrognosis & Predictive factors
No single criterion is available to predict evolution to malignant lymphoma.
Careful long-term follow-up is needed.
Histologically, not possible to definitively diagnose malignant transformation when the disease is confined to skin.
Suggested features indicative of lymphoma are : a high ratio of atypical cells to inflammatory cells, infiltration of atypical cells in to subcutis and a change in the tumor cell immunophenotype with further loss of T-Cell antigens. On the other hand, when the extra-cutaneous dissemination occurs, the diagnosis of lymphoma is straightforward.
In the disease course, the following clinical features may indicate transformation to lymphoma.
A rapidly growing skin lesion that fails to regress spontaneously A lesion that becomes resistant to topical treatment such as PUVA A lesion that exceeds 3 cm in diameter.

PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL
LYMPHOMA (PRIMARY C-ALCL)
A CD30+ anaplastic large cell lymphoma with skin-only involvement without systemic dissemination at presentation.

PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL LYMPHOMA
Accounts for 9% of all cutaneous Lymphomas. No history of prior or concurrent MF or LyP Must be differentiated from Secondary
cutaneous involvement of Systemic Anaplastic Large Cell Lymphoma which requires aggressive chemotherapy.
Primary C-ALCL does not have extracutaneous manifestations at presentation. Patients with widespread systemic and cutaneous disease at first presentation should be considered to have the systemic form with skin involvement.
No t(2;5) translocation in pC-ALCL ( unlike CD30+ systemic ALCL)
No expression of the ALK protein and EMA ( Epithelial Membrane Antigen) in primary cutaneous ALCL.

PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL LYMPHOMA
Differential Diagnosis: Systemic ALCL involving skin Lymphomatoid papulosis Transformed MF
• Biopsy proven history of MF• Infiltrate >25% large T-cells (>x4 small lymphocyte)• In 1/3 cases majority of cells CD30+• Usually correlates with tumour-stage lesions• Very poor outcome: 5-year survival ~20%
Benign lesions with CD30-positive cells• Drug reaction (carbamazepine)• Viral infection (molluscum, herpes simplex)• Arthropod bite reactions (scabies)
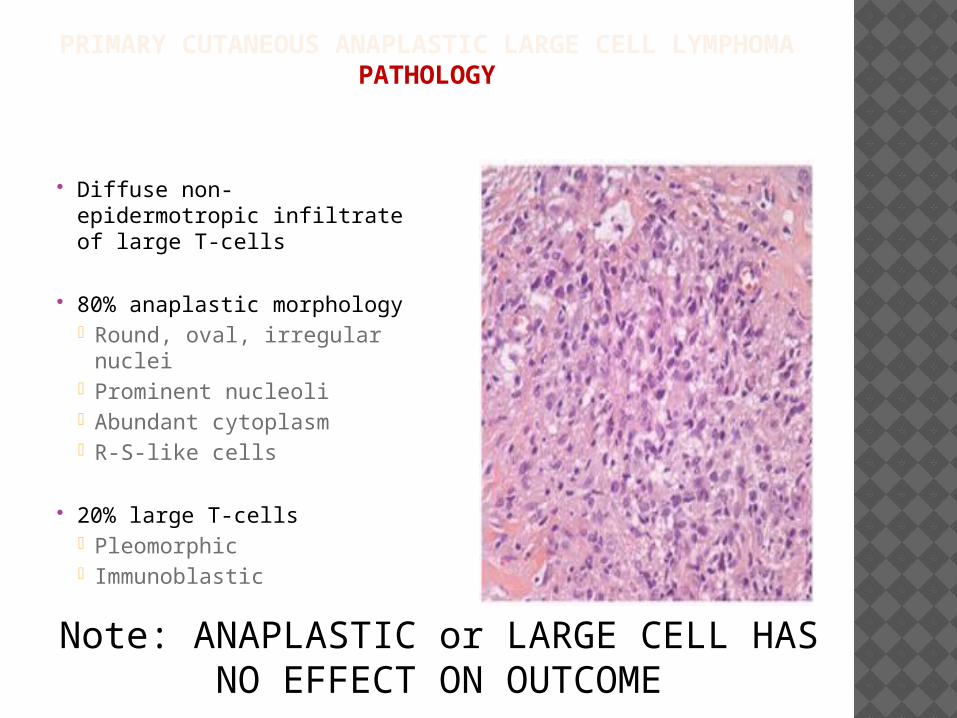
PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL LYMPHOMAPATHOLOGY
Diffuse non-epidermotropic infiltrate of large T-cells
80% anaplastic morphology• Round, oval, irregular
nuclei• Prominent nucleoli• Abundant cytoplasm• R-S-like cells
20% large T-cells• Pleomorphic• Immunoblastic
Note: ANAPLASTIC or LARGE CELL HAS NO EFFECT ON OUTCOME
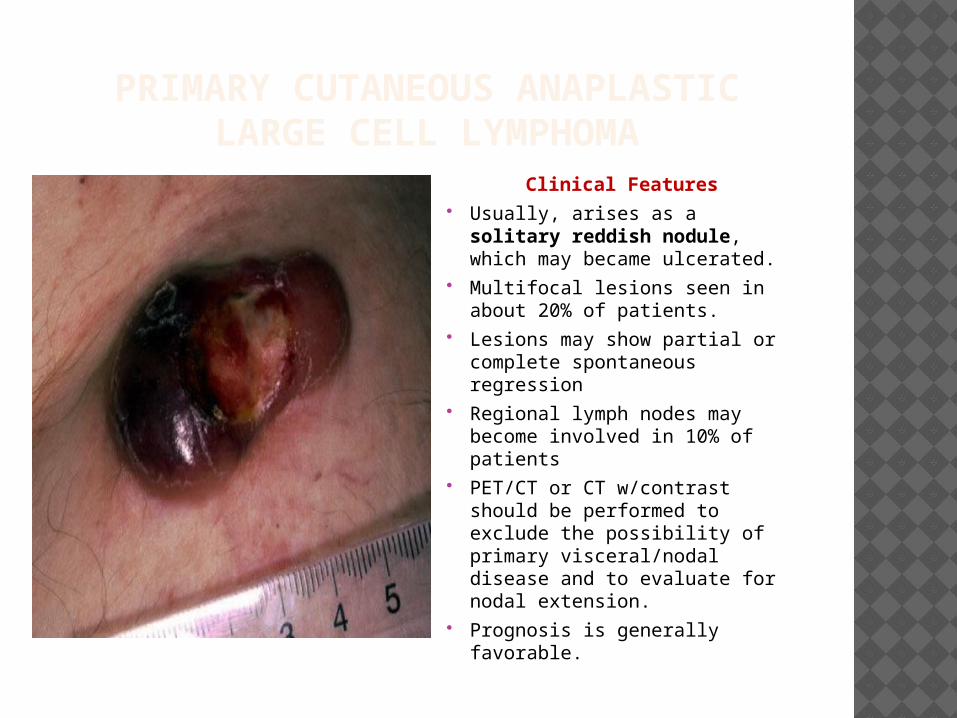
PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL LYMPHOMA
Clinical Features Usually, arises as a solitary
reddish nodule, which may became ulcerated.
Multifocal lesions seen in about 20% of patients.
Lesions may show partial or complete spontaneous regression
Regional lymph nodes may become involved in 10% of patients
PET/CT or CT w/contrast should be performed to exclude the possibility of primary visceral/nodal disease and to evaluate for nodal extension.
Prognosis is generally favorable.

PRIMARY CUTANEOUS ANAPLASTIC LARGE CELL LYMPHOMA
TREATMENT
Patients presenting with a solitary or few localized nodules or tumors Radiotherapy or surgical excision is the first choice of treatment.
Patients presenting with multifocal skin lesions low-dose methotrexate, (as in LyP).
Patients presenting with or developing extracutaneous disease or rare patients with rapidly progressive skin disease doxorubicin-based multiagent chemotherapy.
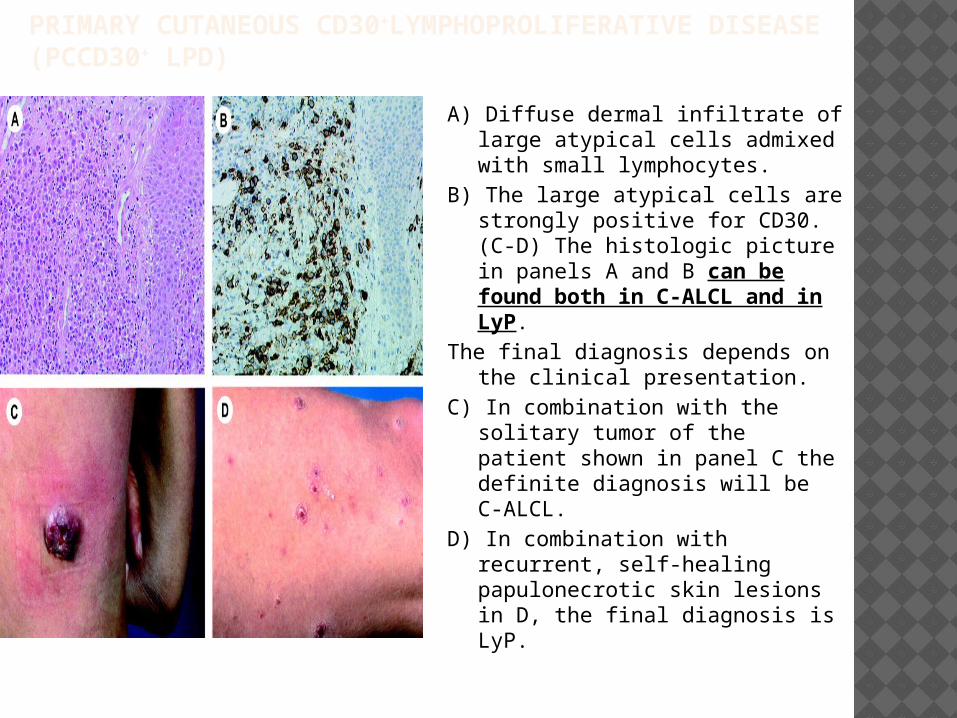
PRIMARY CUTANEOUS CD30+LYMPHOPROLIFERATIVE DISEASE (PCCD30+ LPD)
A) Diffuse dermal infiltrate of large atypical cells admixed with small lymphocytes.
B) The large atypical cells are strongly positive for CD30. (C-D) The histologic picture in panels A and B can be found both in C-ALCL and in LyP.
The final diagnosis depends on the clinical presentation.
C) In combination with the solitary tumor of the patient shown in panel C the definite diagnosis will be C-ALCL.
D) In combination with recurrent, self-healing papulonecrotic skin lesions in D, the final diagnosis is LyP.

SUBCUTANEOUS PANNICULITIS-LIKE T-CELL
LYMPHOMA (SPTCL)
Alpha-beta

SPTCL Very rare Primary Cutaneous T cell Lymphoma ( < 1% of
CTCL) Described in 1991 by Gonzalez et al and is currently
recognized as a distinct lymphoma in WHO classification Malignant alpha/beta+/CD8+, CD4- T cells preferentially
infiltrate the subcutaneous tissue. Two groups of SPTL distinguished with a different histology,
phenotype, and prognosis : SPTCL with α/ β T-cell phenotype
usually CD8+, CD4- restricted to the subcutaneous tissue (no dermal and/or epidermal
involvement) indolent clinical Course.
SPTCL with γ/δ T-cell phenotype CD8-, CD4- and CD56 + neoplastic infiltrates subcutaneous tissue, Epidermis and dermis. very poor prognosis.
WHO-EORTC classification uses the term “SPTL” only for cases with an α/ β T-cell phenotype. Cases with a γ/δ T-cell phenotype are seperately classified as cutaneous γ/δ T-cell lymphomas.

SPTCL Clinical Features :
Multiple subcutaneous nodules involving trunk and extremities, rarely, the face.
Constitutional symptoms : fever, fatigue, and weight loss may be present.
Pancytopenia : usually, due to cytokine mediated BM suppression ( direct BM involvement is seen only in 8% cases)
Hemophagocytic syndrome : less common with SPTCL, more commonly seen with ( in 1/3 cases) cutaneous γ/δ T-cell lymphomas
Extracutaneous dissemination is rare.
Treatment : The clinical course is indolent and 5 year survival 80% for this α/ β, CD8+ T-cell phenotype . Treatment modailities may include Radiotherapy in localized disease and Systemic corticosteroids. Doxorubicin based chemotherapy can be used in extensive or recurrent disease.

CUTANEOUS GAMMA-DELTA
LYMPHOMA

CUTANEOUS GAMMA-DELTA LYMPHOMA A variant of SPTL with clonal T-Cell receptor
gamma-delta chain gene rearrangements. Constitutes 25% of SPTL cases. As per WHO-EORTC classification, this is classified
as an entity distinct from SPTL. Prognosis extremely poor Clinical Features
Multiple subcutaneous nodules with ulceration/necrosis, mostly on the extremities..
Pancytopenia 1/3 cases may present with or complicated by Hemophagocytic
syndrome leading to rapid downhill course { histiocytes engulf RBCs, white cells, platelets infiltrate spleen (splenomegaly), liver(hepatomegaly) and lead to jaundice, liver failure and sometimes, death due to complications from cytopenias}
Rx: Systemic chemotherapy , results disappointing – resistant to multi-agents. Median survival 15 mos.

ADULT T-CELL LEUKEMIA/LYMPHOMA
(ATLL)

ATLL T-cell neoplasm caused by a retrovirus infection with
human T-lymphotropic virus (HTLV I). Endemic in areas with a high prevalence of HTLV-1
eg: southwest Japan,the Caribbean islands, South America, and parts of Central Africa.
ATLL develops in 1% to 5% of seropositive individuals after more than 2 decades of viral persistence.
Characterized large numbers of circulating atypical cells.
Skin lesions resemble MF and histologically, indistinguishable from MF. The neoplastic T cells express a CD3+, CD4+, CD8- phenotype. CD25 is highly expressed
Genetic features : Clonal TCR-Gene rearrangements seen. Clonally integrated HTLV-1 genes found in all cases helpful to differentiate chronic ATLL from classic MF or SS.

ATLL – CLINICAL FEATURES
Can be divided into acute and chronic types. Acute ATLL :
Skin lesions ( nodules, tumors, plaques or papules) similar to those found in mycosis fungoides or Sézary syndrome
Enlarged lymph glands Hypercalcemia Bone lesions. Numerous circulating atypical cells Prognosis is poor for this type with survival ranging from 2
weeks to more than 1 year.
Chronic ATLL skin lesions only ( closely resemble MF) Circulating atypical cells are few or absent Indolent clinical course and better survival, however this may
transform into an acute phase with an aggressive course.
Rx: Skin targeted therapies similar to MF in chronic cases. Acute ATLL requires systemic chemotherapy.

EXTRANODAL NK T-CELL LYMPHOMA, NASAL TYPE

EXTRANODAL NK T-CELL LYMPHOMA, NASAL TYPE
More common in males. Seen in Asia, South America and Central America.
EBV associated Lymphoma. Multiple plaques and tumors on the trunk/
extremities and in the Nose/ Nasopharynx. Systemic symptoms such as fever and weight
loss. An associated hemophagocytic syndrome may be seen
The malignant cells are usually CD2 and CD56 positive (NK cell phenotype) - Epstein-Barr virus (EBV) are commonly positive. Rarely, cells may have a true cytotoxic T-cell phenotype.
Very aggressive disease, Rx with Systemic Chemotherapy, Median survival < 1yr .

PRIMARY CUTANEOUS AGGRESSIVE EPIDERMOTROPIC CD8 CYTOTOXIC
T-CELL LYMPHOMA

characterized by a proliferation of epidermotropic CD8 cytotoxic T-cells and an
aggressive clinical behavior. presents with eruptive papules, nodules,
and tumors with central ulceration. Visceral involvement (CNS, Lung, Testes ) can be seen but LN are usually spared.
Rx: anthracycline-based systemic chemotherapy
Median survival : 32 months

PRIMARY CUTANEOUS PERIPHERAL T-CELL
LYMPHOMA, UNSPECIFIED

PTL, UNSPECIFIED
Heterogeneous group which includes all T-cell neoplasms that do not fit into any of the better defined subtypes of T-cell lymphoma/leukemia.
Out of this group, primary cutaneous aggressive epidermotropic CD8 cytotoxic T-cell lymphoma, cutaneous gamma-delta T-cell lymphoma, and primary cutaneous smallmedium CD4 T-cell lymphoma can be separated out as provisional entities.
Remaining diseases that do not fit into either of these provisional entities must be the designated as PTL, unspecified.
In all these cases a diagnosis of MF must be ruled out by complete clinical examination and an accurate clinical history.
CUTANEOUS B-CELL LYMPHOMAS(C-BCL)•CLASSIFICATION•CLINICAL FEATURES•TREATMENT

CUTANEOUS B-CELL LYMPHOMAS(C-BCL)
Primary cutaneous marginal zone B-cell lymphoma
Primary cutaneous follicle center lymphoma
Primary cutaneous diffuse large B-cell lymphoma, leg type
Primary cutaneous diffuse large B-cell lymphoma, other
Intravascular large B-cell lymphoma
PRIMARY CUTANEOUS MARGINAL ZONE B-
CELL LYMPHOMA
PCMZL

PCMZL An indolent lymphoma composed of small B cells,
including marginal zone (centrocyte-like) cells, lymphoplasmacytoid cells, and plasma cells.
Considered as a part of group of extranodal marginal zone B-cell lymphomas commonly involving mucosal sites, called MALT (mucosa-associated lymphoid tissue) lymphomas.
Variants include primary cutaneous immunocytoma and primary cutaneous plasmacytoma.
Accounts for 10% of all cutaneous lymphomas. In some cases of PCMZL in Europe , an
association with Borrelia burgdorferi infection has been reported in but not in Asian cases or cases from the United States.

PCMZL Clinical Features:
Red to violaceous papules, plaques or nodules on trunk or extremities.
Usually, multifocal lesions. In some cases, spontaneous resolution of lesions may
occur. Anetoderma ( flaccid or herniated-sac like skin due to
loss of dermal elastic tissue) may develop at the site of spontaneous resolution.
Histopathology : Nodular or diffuse skin infiltrates of small
lymphocytes, marginal zone B cells (centrocyte-like cells), and plasma cells with sparing of epidermis.
Immunophenotyping reveals marginal zone B cells expressing CD20, CD79a, and bcl-2, but are negative for CD5, CD10, and bcl-6 (distinction from PCFCL)
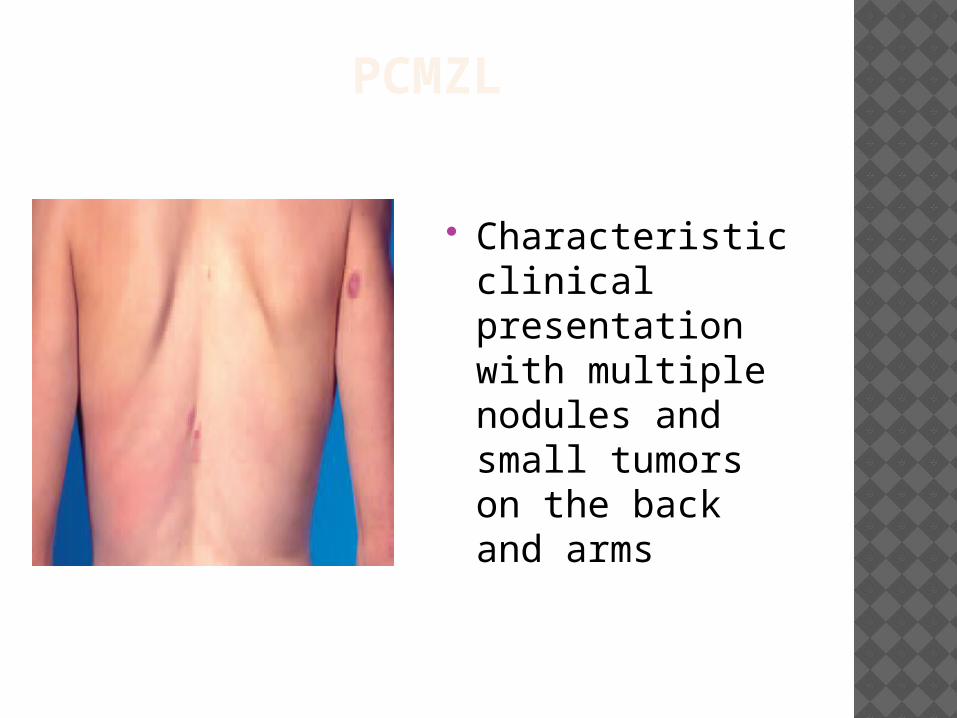
PCMZL
Characteristic clinical presentation with multiple nodules and small tumors on the back and arms

PCMZL - TREATMENT Solitary or a few lesions
Treat with radiotherapy or surgical excision. Patients with associated B.burgdorferi infection
Systemic antibiotics - Doxycycline at 100 mg twice daily for 3 weeks or pulse therapy with cefotaxime
Multifocal skin lesions Chlorambucil or Intralesional or subcutaneous administration of
interferon alpha CR 50% Intralesional or systemic anti-CD20 antibody (Rituximab)
Frequent skin relapses – options topical or intralesional steroids or Observation alone ( as in other indolent B-cell
lymphomas) Prognosis : Excellent, 5 yr survival 100%

PRIMARY CUTANEOUS FOLLICLE CENTER
LYMPHOMA
PCFCL

PCFCL A tumor of neoplastic follicle center cells, usually a
mixture of centrocytes (small and large cleaved follicle center cells) and variable numbers of centroblasts (large noncleaved follicle center cells with prominent nucleoli)
Variants ( according to growth pattern) : follicular, follicular and diffuse, diffuse variants.
Immunophenotype: Neoplastic B cells express CD20, CD79a and bcl-6. bcl-2 protein not expressed by PCFCL( unlike nodal or secondary cutaneous follicular lymphomas). Does not have t(14.18) unlike systemic follicular lymphoma.
C/F : Solitary nodules or grouped plaques/ tumors - found most frequently in the head and neck area. Multifocal lesions are rare
Prognosis : Excellent, 5yr Survival > 90%

PCFCL
(A) Typical presentation with tumors on the chest surrounded by less infiltrated erythematous skin lesions.
(B) Presentation with multiple tumors confined to the scalp.
(C) Diffuse dermal infiltrate mainly consisting of large centrocytes and multilobated cells
(D) Serial sections stained for CD20 (D) and bcl-2 (E). Bcl-2 is expressed by perivascular T cells, but not by the neoplastic B cells.).

PCFCL - TREATMENT Localized or few scattered skin lesions
radiotherapy is the preferred mode of treatment ( even in cases with a predominance of large “cleaved” cells).
Cutaneous relapses can be seen in 20% cases and does not indicate progressive disease. Rx with Radiotherapy.
Extensive cutaneous disease and extracutaneous disease Anthracycline-based chemotherapy .
Systemic or intralesional Rituximab benefits in some studies. Need more data.

PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, LEG TYPE
PLBCL-Leg

PCLBCL - LEG A tumor with predominance or confluent
sheets of centroblasts and immunoblasts ( mostly large B-cells. Small cells are lacking). Characteristically, appears on the lower legs.
Can occur on other parts of the body. Most commonly affects Elderly Women. Neoplastic B-cells express CD-20 and CD79a.
Also, show strong bcl-2 expression and express MUM-1/IRF4 protein ( unlike PCFCL).
t(14,18) is absent.

PCLBCL - LEG Red or bluish-red appear on the lower legs and
frequently grow into large tumors that extend deep into the fat.
Unlike cutaneous follicle center lymphoma, LBCL-L tumors develop quickly over weeks and months, usually becoming open sores and spreading outside the skin ( extracutaneous dissemination).
Prognosis is worse than PCFCL. 5-year survival 55%. PCLBCLs on the leg have an inferior prognosis
compared to PCLBCLs presenting at other sites. The presence of multiple skin lesions at diagnosis is a
significant adverse risk factor. In a recent study, patients presenting with a single skin tumor on one leg had 5-year survival of 100%, whereas patients presenting with multiple skin lesions on one or both legs had a disease-related 5-year survival of 45% and 36%, respectively
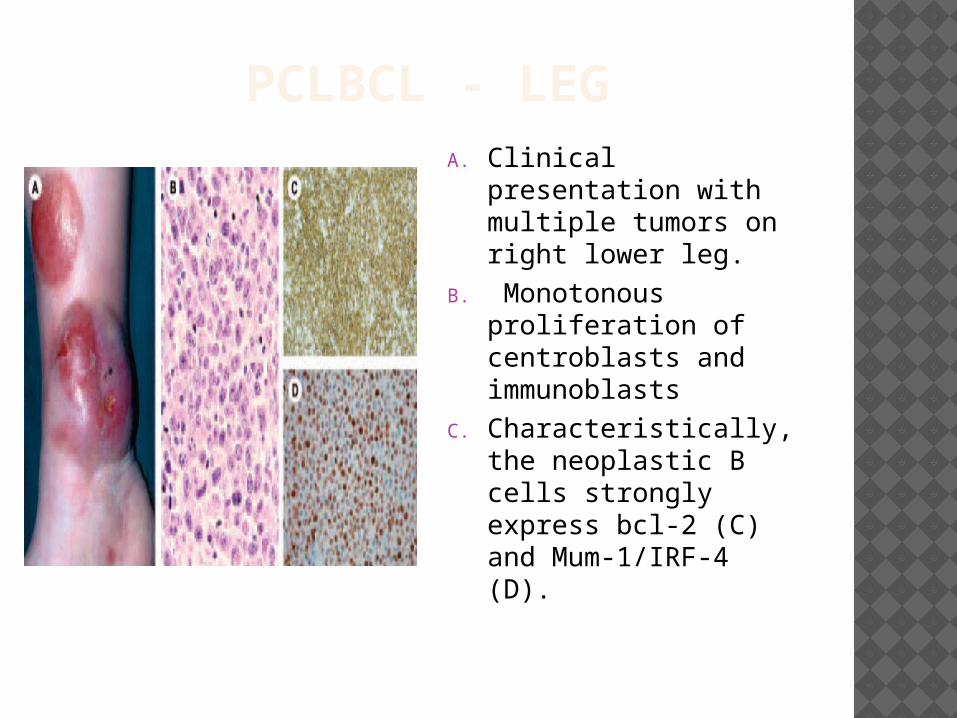
PCLBCL - LEGA. Clinical presentation
with multiple tumors on right lower leg.
B. Monotonous proliferation of centroblasts and immunoblasts
C. Characteristically, the neoplastic B cells strongly express bcl-2 (C) and Mum-1/IRF-4 (D).

PCLBCL - LEG Treated as systemic diffuse large B-cell
lymphomas with anthracycline-based chemotherapy.
In patients presenting with a single small skin tumor radiotherapy may sometimes be considered.
Systemic administration of anti-CD20 antibody (rituximab) has proved effective in some patients, but long-term follow-up data are not available and the place of rituximab in the treatment of PCLBCL, either as single agent therapy or in combination with systemic chemotherapy remains to be established

PRIMARY CUTANEOUS DIFFUSE LARGE B-CELL LYMPHOMA, OTHER
PLBCL-Other

PCLBCL-OTHER “PCLBCL-Other” refers to cases of large B-cell lymphoma
arising in the skin which do not belong to the groups of PCFCL and PCLBCL, leg type.
They include morphologic variants of diffuse large B-cell lymphomas
anaplastic BCL plasmablastic lymphoma T-cell/histiocyte rich large B-cell lymphomas.
The lymphomas usually appear on the head, trunk or extremities. Most often these are cutaneous manifestations of systemic lymphomas and have to be treated the same way.
The prognosis is excellent unlike their nodal counterparts.
Plasmablastic lymphomas are seen almost exclusively in the setting of HIV infection or other immune deficiencies.

INTRAVASCULAR LARGE B-CELL LYMPHOMA
Well-defined subtype of large B-cell lymphoma.
Defined by an accumulation of large neoplastic B cells within blood vessels. Usually, affects the central nervous system, lungs, and skin .
Histologically, dilated blood vessels in the dermis and subcutis are filled and extended by a proliferation of large neoplastic B cells.
Prognosis : poor. Patients often have widely disseminated disease, but cases with only skin involvement may occur. 3 yr Survival 56% vs. 22% for skin only vs. disseminated disease.
CF: Presents as violaceous patches and plaques or teleangiectatic skin lesions usually on the (lower) legs or the trunk.
Rx : Multiagent chemotherapy, both for disseminated and skin-limited disease.
CUTANEOUS PSEUDOLYMPHOMAS•CLASSIFICATION

CUTANEOUS PSEUDOLYMPHOMAS
Group of conditions that simulate a lymphoma , but behave in a harmless manner.
Usually, a reactive process, though a number of these can be difficult to distinguish from a lymphoma
T-cell and B-Cell Pseudolymphomas.

Clinicopathologic condition Simulated malignant lymphoma
•Actinic Reticuloid•Lymphomatoid contact dermatitis•Lymphomatoid drug reaction, T cell type•Solitary T-cell pseudolymphoma (‘unilesional mycosis fungoides’)•Lichenoid (‘lymphomatoid’) keratosis•Lichenoid pigmented purpuric dermatitis•Lichen sclerosus
•Mycosis Fungoides/ Sezary Syndrome
•Atypical lymphoid infiltrates (CD30+) associated with:Orf– Milkers nodule•Herpes simplex &•Molluscum contagiosum•Arthropod (insect) reactions (including nodular scabies)
•Lymphomatoid papulosis / Anaplastic large cell lymphoma – CD30+
•Lupus panniculitis •Subcutaneous T-cell lymphoma
Lymphocytoma cutis •B cell lymphomas Follicle center lymphoma•Marginal zone B-cell lymphoma•Large B-cell lymphoma
•Lymphomatoid drug reaction, B cell type•Pseudolymphoma after vaccination•Pseudolymphoma in tattoos•Pseudolymphoma caused by Hirudo medicinalis therapy
•Follicle center lymphoma•Marginal zone B-cell lymphoma
•Morphoea, inflammatory stage•Syphilis (secondary) • Marginal zone B-cell lymphoma
•Jessner's lymphocytic infiltrate •Chronic lymphocytic leukaemia, B cell type
•Inflammatory pseudotumour •Plasmacytoma•Marginal zone B-cell lymphoma

CUTANEOUS T-CELL PSEUDOLYMPHOMA(DRUG INDUCED)
SYNONYMS:LYMPHOMATOID DRUG REACTION
T-CUTANEOUS LYMPHOID HYPERPLASIA

CUTANEOUS T-CELL PSEUDOLYMPHOMADefinition1. Lymphoid infiltrate highly suggestive
of CTCL
2. Clinical features NOT consistent with CTCL• Identification of causative agent• Uncommon presentation or course

CUTANEOUS T-CELL PSEUDOLYMPHOMA - SUBTYPES1. Well defined clinicopathological entities :
• Drug induced Anticonvulsants; phenytoin, carbamazepine ACE inhibitors Miscellaneous; atenolol, allopurinol, mexilitine, cyclosporine,
antihistamines, griseofulvin
• Insect bite reactions• Lymphomatoid contact dermatitis• Actinic reticuloid;
chronic photosensitive dermatitis Scaly erythema of exposed skin
2. Idiopathic
Clinical course/ HISTOLOGY/ IMMUNOPHENOTYPING/ MOLECULAR ANALYSIS to be used in differentiating them from true CTCL

CUTANEOUS T-CELL PSEUDOLYMPHOMA - HISTOLOGY
TWO PATTERNS:1. Band-like infiltrate (MF-like)
• Subepidermal infiltrate
• Atypical medium sized cerebriform cells +/- blasts, histiocytes
• Few/no eosinophils, plasma cells
Seen in all types of CTCPL except insect bite reactions.
2. Nodular pattern
• Many small round T-cells
• Scattered T-blasts & medium/large cerebriform cells
• Histiocytes usually numerous +/- plasma cells, eosinophils
Seen in
• Drug induced CTCPL
• Persistent arthropod bite reactions
• Idiopathic CTCPL

MF-LIKE CTCPL VS MYCOSIS FUNGOIDES
Features which strongly suggest MF
1. Pautrier’s microabscesses
2. Medium/large cerebriform cells in epidermis
3. Linear epidermotropism
4. Disproportionate epidermotropism
5. ‘Haloed’ lymphocytes in epidermis ACTINIC RETICULOID vs MYCOSIS FUNGOIDES
• CD8+ T-cells, MF usually CD4+• Multinucleate giant cells – fibroblasts,
histiocytes• Vertically orientated collagen in papillary
dermis

USEFUL ADDITIONAL FINDINGS
ABERRANT PHENOTYPE 1.Loss of pan-T-cell antigens
CD2, CD3, CD5, CD7 BUT also lost in some benign conditions (esp CD7)
2. Ratio of CD4:CD8 vast excess dual expression no expression
MONOCLONAL TCR RE-ARRANGEMENT BUT monoclonality found in typically benign lesions
also e.g. lichen planus, pityriasis lichenoides, LSA thus monoclonal but benign infiltrates may arise in the skin

NOTES
Additional Useful Notes


T-CELL RECEPTOR ALPHA CHAIN, BETA CHAIN, GAMMA CHAIN, DELTA CHAIN REARRANGEMENTS
Neoplastic cells show clonal TCR GENE REARRANGEMENTS.
Clonal TCR rearrangements ( dominant T-CELL Clone) does not always indicate malignancy Several benign lymphocyte-mediated dermatoses exist, including lymphomatoid papulosis, Mucha-Habermann disease, lymphomatoid drug eruptions, actinic reticuloid, and lichen planus, all of which demonstrate a dominant T-cell clone in some cases.
Detection of Clonal alpha-beta vs. gamma-delta TCR gene rearrangments is of prognostic significance in SPTL. Gamma-delta carries very poor prognosis.
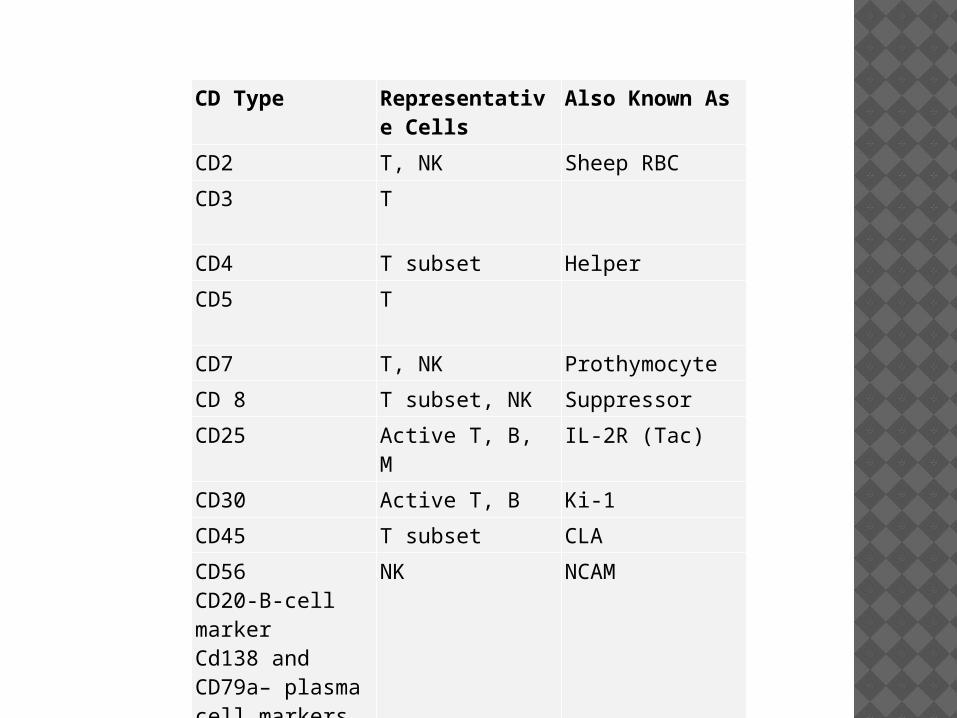
CD Type Representative Cells
Also Known As
CD2 T, NK Sheep RBC
CD3 T
CD4 T subset Helper
CD5 T
CD7 T, NK Prothymocyte
CD 8 T subset, NK Suppressor
CD25 Active T, B, M IL-2R (Tac)
CD30 Active T, B Ki-1
CD45 T subset CLA
CD56CD20-B-cell markerCd138 and CD79a– plasma cell markers 9 usually CD20 –VE)
NK NCAM

ANAPLASTIC LARGE CELL LYMPHOMAS
Immunophenotyping in anaplastic large cell lymphoma (ALCL) exhibits consistently strong CD30 expression in all clinical and pathologic subtypes – Most tumor cells are T Cell Phenotype ( frequent CD3 expression, clonal T-cell receptor gene rearrangements and lack of B-cell – associated markers) or null cell phenotype. In ALCL, B-cell antigenic expression is rare and is commonly observed in the HIV-related clinical form. In fact, these B-cell cases of ALCL are classified separately in the World Health Organization classification under diffuse, large, B-cell lymphoma
Primary systemic anaplastic large cell lymphoma (ALCL) is typically in an advanced stage at patient presentation, and the disease is rapidly progressive. These patients demonstrate an increased frequency of bone marrow involvement (30%) and extranodal involvement, including skin (21%), bone (17%), soft tissues (17%), lung (11%), liver (8%), and, rarely, the gastrointestinal tract and central nervous system. Systemic symptoms are observed in 75% of patients, with fever the most common symptom. The primary systemic form, unlike the primary cutaneous form, generally stains positive for EMA and usually displays the t(2;5) translocation and the chimeric p80 protein ( NPM-ALK fusion protein – NPM on chromosome 5 joined with ALK on chromosome 2) with PCR and antibody studies.
Primary cutaneous anaplastic large cell lymphoma (ALCL) usually manifests as a single or localized cluster of erythematous skin nodules, some of which may demonstrate superficial ulcerations. As many as 25% of patients have some degree of spontaneous regression of these lesions. Although most cases present with local involvement, patients may rarely present with disseminated cutaneous disease and are at higher risk of developing spread to other organs.

ANAPLASTIC LARGE CELL LYMPHOMAS Most cases of HIV-related anaplastic large cell lymphoma
(ALCL) are actually of B-cell origin and seem instead to be related to the anaplastic variant of diffuse large B-cell lymphoma. Many patients demonstrate infection with the Epstein-Barr virus, which is absent in those with the T-cell or null-cell types of anaplastic large cell lymphoma (ALCL).
Secondary anaplastic large cell lymphoma (ALCL) evolves from other lymphomas, most frequently from peripheral T-cell lymphomas, mycosis fungoides, Hodgkin disease, or lymphomatoid papulosis. This form of anaplastic large cell lymphoma (ALCL) tends to arise in older adults, is commonly ALK negative (chr 2, 5 translocation), and carries a poor prognosis.
Patients with anaplastic large cell lymphoma (ALCL) present with either a primary cutaneous form or a systemic form of the illness. Patients may present with isolated lymphadenopathy or with extranodal disease at any site, including the gastrointestinal tract and bone. Patients with infiltration into musculoskeletal tissues (eg, psoas muscle) can present with backache.


















![Primary cutaneous lymphomas: single center experience of … · in tumor stage and approximately 20% in histo-logical lymph node involvement [9-11]. Lymph node,inner organ involvement](https://img.dokumen.tips/doc/110x75/5e5e51adcf8b202fd16e13c3/primary-cutaneous-lymphomas-single-center-experience-of-in-tumor-stage-and-approximately.jpg)










