
1 | P a g e
Relationship of Nitric Oxide and Ascorbic Acid in Coronary Artery Disease
Thesis submitted in partial fulfillment of the requirements for the degree of
Masters of Science
In
Medical Biotechnology
By:
Priya Joshi
Reg. No.: 131702016
M.Sc. Medical Biotechnology
School of Life Sciences
Manipal University
Project Advisor:
Dr Kamna Srivastava
Assistant Professor
B.R. Ambedkar Center for Biomedical Research
University of Delhi
New Delhi
July 2015
Department of Biotechnology
School of Life Sciences
Manipal University, Manipal

2 | P a g e

3 | P a g e
Acknowledgement
I would sincerely and wholeheartedly like to acknowledge many people for their help and co-operation.
Firstly I would like to thank our Director, Professor K. Satyamoorthy for giving me the opportunity to
work on my Masters dissertation.
I would also like to thank my guide, Dr. Kamna Srivastava for guiding me throughout my project,
providing me with constant guidance, patience, support, valuable suggestions and help at every stage of
my project. She gave me constant encouragement and ideas from the beginning of the project till the very
end of it.
I express my sincere gratitude and a warm thank you to Mr. Sudhir Chandra for his help throughout my
project.
I would like to thank my seniors who were very keen to extend a helping hand.
Last but not the least I would like to thank my family and friends for all their support and encouragement.
Ms. Priya Joshi
4 | P a g e
Serial No. Content Page No.
1. Introduction
Coronary artery disease
Nitric oxide
Ascorbic acid
2
2. Literature review
Prevalence of CVD in the world
Prevalence of CVD in India
Genetic evaluation for coronary artery disease
Genetic susceptibility to myocardial infarction and coronary
artery disease
Association of MEF2A gene polymorphism with coronary artery
disease
Lack of MEF2A mutations in coronary artery disease
Gene therapy to treat cardiovascular disease
Genome wide association studies in myocardial infarction and
coronary artery disease
Nitric oxide
Biochemistry of nitric oxide
Chemical structure of nitric oxide
Nitric oxide synthase
Structure of eNOS
Role of HSP in regulation of eNOS is cardiovascular diseases
Vitamin C affects thrombosis/fibrinolysis system and reactive
hyperemia in patients with type II diabetes and coronary artery
disease
11
3. Methods 27
4. Results 32
5. Discussion 40
6. Conclusion 42
7. References 44
8. Synopsis 48
List of figures
1. Figure explaining physiology of atherosclerosis.
2. Figure showing diagnosis, symptoms, and causes of CVD.
3. Inflammation, calcification and scar development in atherosclerosis.
4. Classic risk factors in the formation and progression of atherosclerotic plaque, and possible
pathogenic mechanisms of coronary artery disease risk genes.
5. Physiology of nitric oxide.
6. Conversion of L-arginine to nitric oxide.
7. Canonical structures for the ascorbic anion.
8. Global burden of cardiovascular disease.
9. Restriction fragment length polymorphism for rs325400.
10. Restriction fragment length polymorphism for rs34851361.
11. Nucleotide and amino acid sequences of the repeat region in MEF2A.
12. Pedigree analysis of families affected by CAD.

5 | P a g e
13. A model of interactions of NO with erythrocytes and cell free hemoglobin in an arterial blood
vessel.
14. eNOS protein structure.
15. Reaction between nitric oxide and sulphanilamide to form azo compound.
16. Graph showing concentrations of nitric oxide in control samples.
17. Graph showing concentrations of nitric oxide in case samples.
18. Graph showing mean of control and cases (nitric oxide).
19. Graph showing concentrations of ascorbic acid in control samples.
20. Graph showing concentrations of ascorbic acid in control samples.
21. Graph showing mean of control and cases (Ascorbic acid).
List of tables
1. Gene therapy targets for coronary heart disease.
2. Summary of clinical characteristics
3. Table shows the values of all the samples of nitric oxide as observed after reaction with Griess
reagent. (controls)
4. Table shows the values of all the samples of nitric oxide as observed after reaction with Griess
reagent. (cases)
5. Table shows the values of all the samples of ascorbic acid as observed after reaction with dTCS
reagent. (controls)
6. Table shows the values of all the samples of ascorbic acid as observed after reaction with dTCS
reagent. (cases)
7. Statistical analysis

6 | P a g e
Introduction

7 | P a g e
Coronary artery disease
The worldwide weight of cardiovascular diseases (CVD) is quickly expanding because of a sharp ascent
in the occurrence and predominance of the same in creating nations. In the course of recent years, the
CVD rates have multiplied in India. Expanded hereditary affinity to create CVD and expanding
pervasiveness of cardiovascular danger variables are the reasons proposed for expanded seriousness and
degree of coronary supply route maladies in Indians. Atherosclerotic coronary course malady (CAD)
keeps on being an essential financial issue. A few phone segments, including endothelial, Inflammatory
and smooth muscle cells of the blood vessel divider, take an interest in the era of an atherosclerotic
plaque, the development of which is in charge of a dynamic narrowing of the lumen of the influenced
coronary vessel and the following lessening in oxygen rich blood stream to downstream heart muscle,
which might then be intensely or chronically harmed.
Cardiovascular malady incorporates various diverse infections including coronary illness, stroke and
fringe vascular ailment, hypertension. A normal 17.3 million people kicked the container from CVDs in
2008, identifying with 30% of single overall end. Of these passings, a normal 7.3 million were a result of
coronary ailment and 6.2 million were a direct result of stroke. Low and center pay nations are
disproportionally influenced: more than 80% of CVD passings happen in low and center pay nations and
happen just as in men and ladies. By 2030, just about 25 million individuals will pass on from CVDs,
principally from coronary illness and stroke. 7.5 million Deaths every year, or 13% of all passings can be
ascribed to raised circulatory strain. This incorporates 51% of passings because of stroke and 45%
because of coronary supply route ailment. It may happen through an acquired inclination. In India CVD
was the biggest reason for passings in guys (20.3%) and in addition females (16.9%) and prompted
around 2 million passings every year. (Price et al, 2001)
8 | P a g e
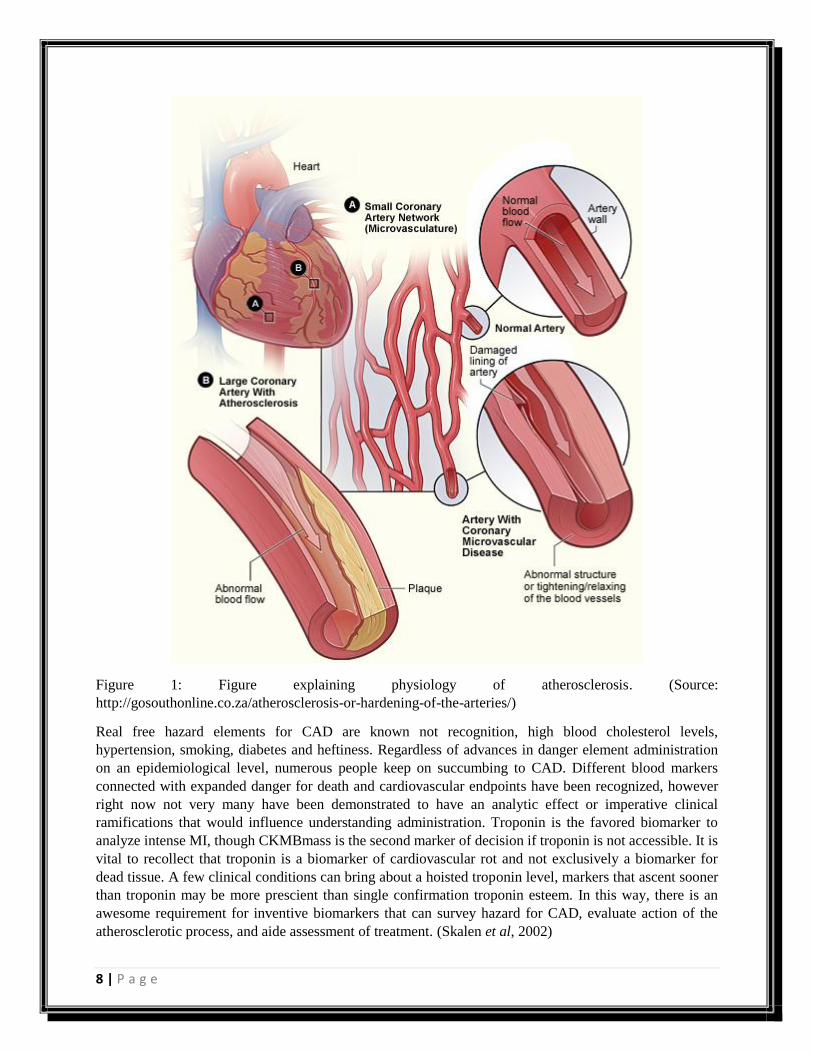
Figure 1: Figure explaining physiology of atherosclerosis. (Source:
http://gosouthonline.co.za/atherosclerosis-or-hardening-of-the-arteries/)
Real free hazard elements for CAD are known not recognition, high blood cholesterol levels,
hypertension, smoking, diabetes and heftiness. Regardless of advances in danger element administration
on an epidemiological level, numerous people keep on succumbing to CAD. Different blood markers
connected with expanded danger for death and cardiovascular endpoints have been recognized, however
right now not very many have been demonstrated to have an analytic effect or imperative clinical
ramifications that would influence understanding administration. Troponin is the favored biomarker to
analyze intense MI, though CKMBmass is the second marker of decision if troponin is not accessible. It is
vital to recollect that troponin is a biomarker of cardiovascular rot and not exclusively a biomarker for
dead tissue. A few clinical conditions can bring about a hoisted troponin level, markers that ascent sooner
than troponin may be more prescient than single confirmation troponin esteem. In this way, there is an
awesome requirement for inventive biomarkers that can survey hazard for CAD, evaluate action of the
atherosclerotic process, and aide assessment of treatment. (Skalen et al, 2002)

9 | P a g e
A few late studies have proposed that flowing microRNAs could be helpful as biomarkers for different
human disappointment and incessant vascular malady. MicroRNAs (miRNAs) are an as of late perceived
class of short (19-25 nts), single stranded, non-coding RNAs that deal with a mixed bag of cell limits
through the debasement and translational requirement of mRNAs that contain correlative groupings. More
than 1000 human miRNAs have been recognized, and, in tissues, miRNAs manage the declaration of
qualities included in discriminating cell procedures, including separation, development, multiplication,
and apoptosis. (Cox et al, 1989) D'Alessandra and associates have shown that flowing muscle inferred
miRNAs may be valuable as biomarkers of intense myocardial localized necrosis (AMI). They further
demonstrated that muscle enhanced miRNAs, for example, miR-1, miR-133a, miR-133b and miR-499-5p
are raised in plasma got from mice after coronary supply route ligation and in addition in people with
AMI. In trial AMI models and in patients, Wang et al demonstrated that the muscle enhanced miR-1,
miR-133a and miR-499 and additionally the cardiovascular particular miR-208 are hoisted in plasma.
Moreover, miR-499, miR-1, miR-133a/b and miR-208 were indicated towards being expanded in little
partners of people after AMI. (Ludmer et al, 1986)
Real free hazard elements for CAD are known not recognition, high blood cholesterol levels,
hypertension, smoking, diabetes and heftiness. Regardless of advances in danger element administration
on an epidemiological level, numerous people keep on succumbing to CAD. Different blood markers
connected with expanded danger for death and cardiovascular endpoints have been recognized, however
right now not very many have been demonstrated to have an analytic effect or imperative clinical
ramifications that would influence understanding administration. Troponin is the favored biomarker to
analyze intense MI, though CKMBmass is the second marker of decision if troponin is not accessible. It is
vital to recollect that troponin is a biomarker of cardiovascular rot and not exclusively a biomarker for
dead tissue. A few clinical conditions can bring about a hoisted troponin level, markers that ascent sooner
than troponin may be more prescient than single troponin levels. (Ohara et al, 1993)

10 | P a g e
Firgure 2: Figure showing diagnosis, symptoms, and causes of CVD (Source:
http://www.imagekb.com/atherosclerosis-and-plaque)
Coronary artery disease (CAD) represents the most important cause of sudden cardiac death. It is a group
of disease that included stable angina, unstable angina, sudden coronary death and myocardial infarction.
Its most common symptom is chest pain or discomfort which might travel to the shoulder, arm, back,
neck or jaw. (Hansson et al, 1989) It might feel like heartburn initially. Symptoms usually occur with
exercise or emotional stress, may last less than a few minutes, and gets better with rest. It is a multi-
factorial disease, risk factors including high blood pressure, diabetes, obesity, smoking, poor diet, high
blood cholesterol, excessive alcohol, environmental factors and a family history of illness among others.
Coronary angiography is the most reliable way to test blockage in arteries. (Shamieh et al, 2012) More
than 80% of sudden cardiac deaths are caused by atherosclerotic CAD; the remaining 20% are caused by
long QT syndrome (LQTS), cardiomyopathies, left ventricular hypertrophy, aortic valve disease, Brugada
syndrome and other cardiac disorders. It is a disease where in a waxy substance called plaque builds up
inside the coronary arteries. These arteries are responsible for supplying oxygen rich blood to the cardiac
muscle. The condition is called atherosclerosis when plaque builds up in the arteries, and the build-up of
this occurs over many years. (Gordon et al, 1989)
Figure 3: Inflammation, calcification and scar development in atherosclerosis. (Source:
http://vpdiagnostics.com/plaque-imaging/)
Atherosclerosis is the thickenings of the endothelial layer of the artery, the intima. It consists of
connective tissue elements, lipids, cells and debris. Immune cells make up a central part of atherosclerotic
lesions (atheromata) along with vascular endothelial and smooth muscle cells. Atheromata are preceded
by a fatty streak, a build-up of lipid laden cells beneath the endothelium. The cells of fatty streak mostly
consist of macrophages along with some T cells. (9) Fatty streaks are even present in young people,
although they may never casus symptoms to show, they might progress to form lesions or disappear
altogether. Foam cells and extracellular lipid droplets form a core region surrounded by a cap of smooth
muscle cells and a collagen rich matrix in the centre of the atheromata. The lesions are infiltrated by T
cells, mast cells and macrophages and are excess in number, especially in the shoulder region where the
atheromata grows. A lot of the immune cells exhibit signs of activation and produce inflammatory
cytokines. (Cox et al, 1989)

11 | P a g e
Figure 4: Classic risk factors in the formation and progression of atherosclerotic plaque, and possible
pathogenic mechanisms of coronary artery disease risk genes. Blue: risk factors associated with
metabolic syndrome (MS) and type 2 diabetes (T2D). Green: risk factor not directly associated with MS
and/or T2D. With respect to diabetes, some mechanisms by which genes modulate the risk of
atherosclerosis are not yet known. (Source: https://www.soc-
bdr.org/orderforms/content/rds/archive/8/2_summer/reviews/genetics_of_diabetes_and_cad/index_en.htm
l?showfulltext=1)
Cytokines produced in the inflamed intima stimulate the monocytes to differentiate into macrophages.
This is crucial for the development of atherosclerosis and is also associated with the up regulation of
pattern recognition receptors for innate immunity. Scavenger receptors engulf a broad range of molecules
and particles presented by pathogen like molecular receptors and destroy apoptotic cell fragments,
bacterial endotoxins and oxidized LDL particles in the process. (Homig et al, 2001) Cholesterol
accumulates as cytosolic droplets, when it cannot be mobilized from the uptake of oxidized LDL particles
which leads to the cell becoming a foam cell, the prototype cell in atherosclerosis. Toll like receptors bind
to pathogen like molecular patterns and initiate a signal cascade that activates the macrophage which in
turn produces inflammatory cytokines, proteases, nitrogen radical molecules and cytotoxic oxygen.
Similar effects are seen in dendritic cells, mast cells and endothelial cells which also express toll like
receptors, heat shock protein 60 and oxidized LDL may also activate these receptors. CD4+ T cell
infiltrates are always present in atherosclerotic lesions; they recognize protein antigens presented to them
bound to major-histocompatibility-complex (MHC) class II molecules. (Gryglewski et al, 1986) Minor T-
cell subpopulation, natural killer T cells are prevalent in early lesions. Natural killer T cells recognize
antigens and their activation increases atherosclerosis in apoE knockout mice. Atherosclerotic lesions also
contain CD8+ T cells, bound to MHC class I antigens. Activation of CD8+ T cells in apoE knockout mice
causes death of arterial cells and accelerates atherosclerosis. The type 1 helper T (Th1) response activates
macrophages, initiates an inflammatory response similar to delayed hypersensitivity, and

12 | P a g e
characteristically functions in the defense against intracellular pathogens. The type 2 helper T (Th2)
response elicits an allergic inflammation. (Hansson et al, 2005)
Myocardial infarction occurs when the atheromatous process prevent blood flow through the coronary
artery. It was earlier thought that the gradual narrowing of the smooth muscle cells by their continued
growth in the plaque was the main cause of infarction. However, upon angiographic studies showed that
lesions did not cause stenosis and it was proven that the activation of plaque, rather than stenosis triggers
ischemia and infarction. (Stary et al, 1994) In most cases infarction is due to the formation of an
obstructing thrombus, however, coronary spasm may also be involved in it to an extent. Coronary
thrombosis can be attributed to two major causes – plaque rupture and endothelial erosion. Plaque rupture
is dangerous because it exposes pro-thrombotic material from the core of the plaque – phospholipids,
platelet aggregating molecules and tissue factors to the blood. Rupture occurs when the fibrous cap is thin
and partly damaged, immune cells are abundant at these sites, producing numerous inflammatory
molecules and proteolytic enzymes that weaken the cap and activate the cells in the core, instigating the
stable plaque to turn into a weak, unstable structure that when ruptures causes acute coronary syndrome.
(Kovanen et al, 1995)
Nitric oxide
The endothelium is a metabolically active organ system which maintains homeostasis by regulation of
solute transport into cell compartment of the vessel wall, extra cellular matrix deposition, local cellular
growth, by modulation vascular tone, regulating inflammatory, homeostatic response to local injury, and
by protecting the vessel from substantial injurious cell circulating in the blood. Growing lists of diseases
(estrogen deficiency, congestive heart failure, aging process, hyperhomocysteinemiaetc) have shown
association to impaired endothelium function. Hence, the vessel walls in these conditions promote
inflammation, smooth muscle proliferation, extracellular matrix deposition, platelet activation, thrombus
formation. Nitric oxide, a soluble gas with a half-life of 6-30 seconds is continually synthesized by the
endothelium and is required for maintaining vascular homeostasis, regulation of local growth, protection
of the vessel from platelets and cells circulating in the blood, and also modulation of vascular dilator tone.
(Ignarro et al, 1987)

13 | P a g e
Figure 5: Physiology of nitric oxide (Source:
http://ethesis.helsinki.fi/julkaisut/laa/biola/vk/lassila/fig4.gif)
Commonly associated risk factors for atherosclerosis such as hypertension and hypercholesterolemia are
associated with low levels of nitric oxide released into the arterial wall. This could be either due to
excessive oxidative degradation or impaired synthesis. (Gordon et al, 1989) Coronary arteries may
constrict during physical exercise or mental stress due to reduced nitric oxide bioactivity and may add to
incitement of myocardial infarction in patients with CAD. Diminished nitric oxide bioactivity may lead to
vascular inflammation which leads to oxidation of lipoproteins and formation of foam cells. A lot of
therapies have been looked into to reverse endothelial dysfunction by increasing the release of nitric oxide
from the endothelium, either by stimulation of nitric oxide synthesis of prevention of its oxidative
inactivation and conversion to toxic molecules such as peroxynitrite. Nitric oxide is synthesized from
amino acid L-arginine in endsothelial cells by calcium calmodulin dependent enzyme nitric oxide
synthase. (Ganz et al, 2003) A five electron oxidation of the basic guanidine nitrogen atoms of L-arginine
in the presence of multiple cofactors and oxygen is catalysed by this heme containing oxygenase.
Furchgott and Zawadzki observed that rabbit aorta with intact endothelium relaxed in response to
acetylcholine but constricted to the same agonist when the endothelium was cleaned off. The substance
responsible for the acetylcholine stimulated relaxation was called endothelium derived relaxant factor and
was later found to be nitric oxide. A variety of agonists including histamine, acetylcholine, ADP,
thrombin, serotonin, isoproterenol, etc can stimulate the synthesis and release of nitric oxide from the
endothelium, but some of the same agonists (histamine, norepinephrine) constrict vascular smooth muscle
in the absence of endothelium. (Homig et al, 2001)
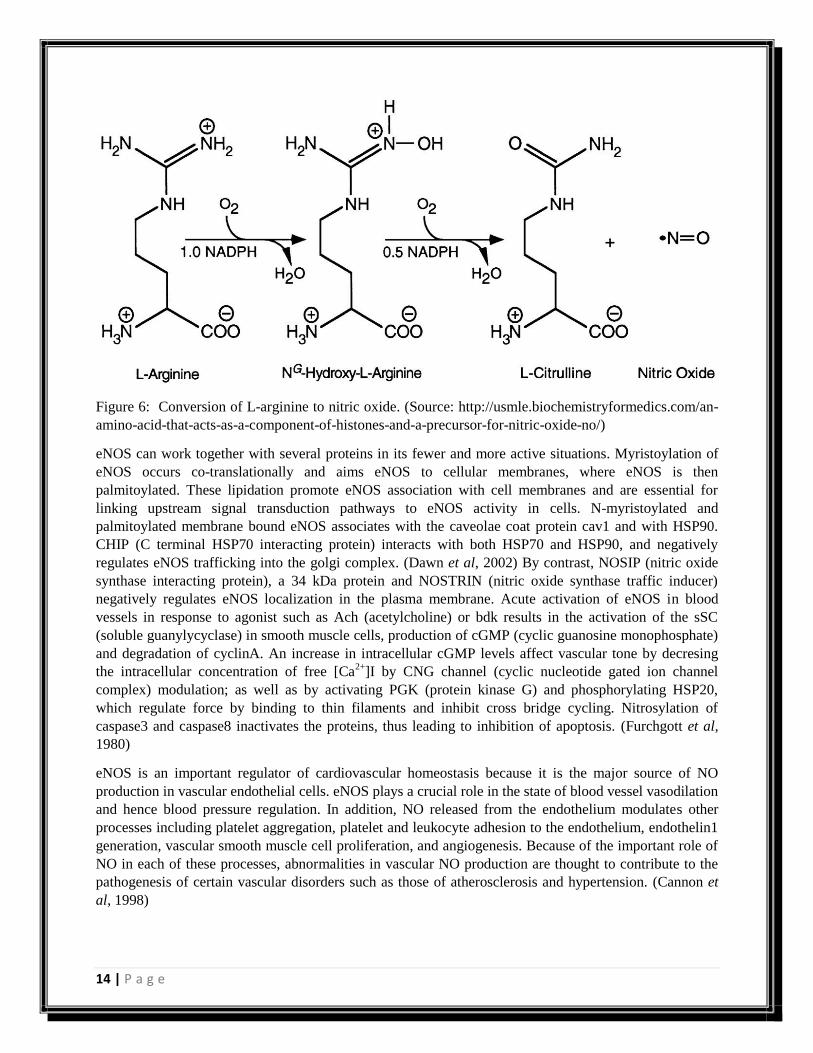
14 | P a g e
Figure 6: Conversion of L-arginine to nitric oxide. (Source: http://usmle.biochemistryformedics.com/an-
amino-acid-that-acts-as-a-component-of-histones-and-a-precursor-for-nitric-oxide-no/)
eNOS can work together with several proteins in its fewer and more active situations. Myristoylation of
eNOS occurs co-translationally and aims eNOS to cellular membranes, where eNOS is then
palmitoylated. These lipidation promote eNOS association with cell membranes and are essential for
linking upstream signal transduction pathways to eNOS activity in cells. N-myristoylated and
palmitoylated membrane bound eNOS associates with the caveolae coat protein cav1 and with HSP90.
CHIP (C terminal HSP70 interacting protein) interacts with both HSP70 and HSP90, and negatively
regulates eNOS trafficking into the golgi complex. (Dawn et al, 2002) By contrast, NOSIP (nitric oxide
synthase interacting protein), a 34 kDa protein and NOSTRIN (nitric oxide synthase traffic inducer)
negatively regulates eNOS localization in the plasma membrane. Acute activation of eNOS in blood
vessels in response to agonist such as Ach (acetylcholine) or bdk results in the activation of the sSC
(soluble guanylycyclase) in smooth muscle cells, production of cGMP (cyclic guanosine monophosphate)
and degradation of cyclinA. An increase in intracellular cGMP levels affect vascular tone by decresing
the intracellular concentration of free [Ca2+
]I by CNG channel (cyclic nucleotide gated ion channel
complex) modulation; as well as by activating PGK (protein kinase G) and phosphorylating HSP20,
which regulate force by binding to thin filaments and inhibit cross bridge cycling. Nitrosylation of
caspase3 and caspase8 inactivates the proteins, thus leading to inhibition of apoptosis. (Furchgott et al,
1980)
eNOS is an important regulator of cardiovascular homeostasis because it is the major source of NO
production in vascular endothelial cells. eNOS plays a crucial role in the state of blood vessel vasodilation
and hence blood pressure regulation. In addition, NO released from the endothelium modulates other
processes including platelet aggregation, platelet and leukocyte adhesion to the endothelium, endothelin1
generation, vascular smooth muscle cell proliferation, and angiogenesis. Because of the important role of
NO in each of these processes, abnormalities in vascular NO production are thought to contribute to the
pathogenesis of certain vascular disorders such as those of atherosclerosis and hypertension. (Cannon et
al, 1998)

15 | P a g e
Ascorbic Acid
Ascorbic acid is antioxidant and mild reducing agent. It gets oxidized by losing an electron to form a
radical cation, when it loses its second electron, it forms a dehydroascorbic acid. It reacts with oxidants of
the reactive oxygen species. It is primarily a white solid, and might appear yellow due to impurities. It is a
form of vitamin C, and is water soluble. It is not produced in the body, hence has to be supplied in the
diet. (Borsook et al, 1997) It is a reductone, and is stabilized by delocalization of its electrons as seen in
the figure. This is why it is more acidic than would be expected if the compound contained only isolated
hydroxyl groups. (Antoniades et al, 2004)
Figure 7: Canonical structures for the ascorbic anion. (Source:
http://wwwchem.csustan.edu/chem1112/1112vitc.htm)
Dietary antioxidants may play a part in the prevention of cardiovascular disease because of modification
of low density lipoproteins enhances their atherogenic potential. Antioxidant vitamins are said to play an
important role in protecting low density lipoproteins from oxidation by free radicals. Vitamin C is very
important in terms of dietary intake and often its low consumption has been linked to increased
cardiovascular disease. Evidence linking CAD to ascorbic acid is very limited. In studies which have
shown a significant correlation between low vitamin C and increased risk of coronary artery disease, the
subjects were younger and their average intake of vitamin C was noticeably higher. (Bordia et al, 1985)

16 | P a g e
Literature Review

17 | P a g e
Prevalence of CVD in the world
Cardiovascular diseases are the number one cause of death globally. More people die annually from CVD
than from any other cause. An estimated 17.3 million people died from CVD in 2008, ie 30% of all global
death. Over 80% of CVD deaths take place in low and middle income countries and occur almost equally
in men and women. And it is estimated by 2020 almost 25 million people will die because of CVD.
CVD is also the leading cause of death in Europe, accounting for over 4 million deaths each year. Nearly
half (49%) of all deaths are from CVD (55% of deaths in women and 43% of deaths in men). In England
CVD is main cause of death. In 2010 there were 147000 deaths in England from CVD nearly one third of
all deaths. Over 2.2 million people living with CHD 1.3 million men, 860000 women. And over 1.2
million people suffered a heart attack CVD related motility rates fell by 5% in Australia, Canada, 60% in
Japan. (Wang et al, 20053)
Figure 8: Global burden of cardiovascular disease (Source:
http://www.nature.com/nrcardio/journal/v11/n11/fig_tab/nrcardio.2014.137_F7.html)
Prevalence of CVD in Chinese population aged between 35-74 years was estimated in 1994 that 40% of
deaths were due to CVD. Furthermore, CVD incidence and mortality in China are projected to increase
substantially during the next 20 years. CVD is common among men than women living in the north as
compared to southern China and in the urban areas than rural. It has been found that the CVD is the
leading cause of motility in China overall 80.5%, 45.9%, 17.2% of adult in China.
While if there should arise an occurrence of Africa more or less 195 individuals for each day because of
CVD and around 20% of every day passings with HIV/AIDS. The United Nations has anticipated that the
rate of populace over 60 years will be more than twofold somewhere around 1999 and 2050 in South
Africa, with the weight because of CVD anticipated to turn into the prime giver to aggregate dismalness
and mortality in the age gathering of individuals more than 50. In any case, CVD is taking its toll even
amongst the more youthful age bunches with passings anticipated to increment by more than 40% in the
35-64 years age gather by 2030. Most cardiovascular sicknesses can be averted by tending to hazard

18 | P a g e
variables, for example, tobacco use, horrible eating regimen and stoutness, physical latency, raised
circulatory strain, diabetes and raised lipids. (Topol et al, 2006)
Prevalence of cardiovascular disease in India
While if there should arise an occurrence of Africa more or less 195 individuals for each day because of
CVD and around 20% of every day passings with HIV/AIDS. The United Nations has anticipated that the
rate of populace over 60 years will be more than twofold somewhere around 1999 and 2050 in South
Africa, with the weight because of CVD anticipated to turn into the prime giver to aggregate dismalness
and mortality in the age gathering of individuals more than 50. In any case, CVD is taking its toll even
amongst the more youthful age bunches with passings anticipated to increment by more than 40% in the
35-64 years age gather by 2030. Most cardiovascular sicknesses can be averted by tending to hazard
variables, for example, tobacco use, horrible eating regimen and stoutness, physical latency, raised
circulatory strain, diabetes and raised lipids.
The overall status on non-transferrable diseases report reported more than 2.5 million passings from CVD
in India in 2008, 66% of which were represented as a result of CHD and 33% as a result of stroke. The
mortality is most lifted in south Indian states, eastern and north eastern states and Punjab in both men and
women, while mortality is the slightest in the central Indian states of Rajasthan, Uttar Pradesh and Bihar.
Sub-examination of mortality examples exhibited that CHD mortality is higher in the south Indian states
while stroke mortality is higher in eastern Indian states. (Stary et al, 1995)
Genetic evaluation for coronary artery disease
Hereditary variations can adjust usefulness of parts in a metabolic pathway, which may bring about
expanded inclination to the improvement and progression of atherosclerosis. A great deal of qualities
have been connected with CAD or MI; in any case, a significant number of these affiliations are
questionable and show shifting results in writing. The absence of predictable discoveries may be because
of positive reports that are because of chance, not coordinating the race/ethnicity of cases and controls or
because of recurrence of polymorphisms in the control populaces. The variable VII locus has been seen to
be included in deciding component VII levels and danger of MI in patients with built up CAD, yet no
relationship of it has been found in the improvement of CAD, recommending that an alternate substrate
for this danger element (atherosclerosis). Essentially, numerous studies have perceived an in number
connection between platelet glycoprotein receptor IIIa (GPIIIa) A2 allele and intense coronary thrombosis
and the level of CAD, be that as it may, different studies have been unsuccessful in demonstrating a
relationship with CAD or MI.
Linkage investigation in factions with untimely CAD has discovered proof for linkage to a locale on
chromosome 2q21.1-q22 and Xq23-q26. The angiotensin receptor 2 quality was recognized when Xq23-
q26 was investigated for hopeful qualities, and it may assume a part in the cardiovascular and focal
sensory system. Chromosome 2q36-q37.3 was discovered to be connected to MI and temperamental
angina before the age 70; it envelops the insulin receptor substrate-1 quality, sort 2 diabetes locus
NIDDM1, and the HDL cholesterol tying protein.
The Framingham Risk Score has been generally used to survey the danger of CAD. It considers the set up
danger elements of sex, smoking, downright cholesterol, diabetes, age, LDL and HDL cholesterol, yet
does not consider family history as a danger component. Consequently, this system thinks little of CAD
dangers for people with a family history of CAD, particularly at younger ages.

19 | P a g e
A few studies have demonstrated that family history reports of CAD are by and large precise. The Health
Family Tree examine, a populace based overview of 122,155 families that CAD in relatives had 67%
positive prescient worth, 96% negative prescient quality, 70% affectability, and 91% specificity. At the
point when the case-control study and the historical backdrop of MI in first degree relatives was
supported, just little contrasts were seen between chances proportion. This test discovered 14% positive
family history of CAD.
The ApoE4 allele has been found to be associated with CAD and individuals with ApoE2/E2
homozygous individuals were seen to be at risk for type III hyperlipoproteinemia, along with increased
risk for atherosclerosis. Carriers of apoE4 allele are more responsive to the LDL lowering effects of low
fat dietary interventions as opposed to non-carriers, and they are also disposed to coronary heart disease
when exposed to diets rich in saturated fats.
Since CAD is a heterogenous issue, it is not down to earth to expect that a solitary way of anticipation
will be compelling against it. The learning of hereditary defenselessness to atherosclerosis can help in the
distinguishing proof of natural contrasts. A few rules for danger appraisal in view of family history are
indicated in the figure beneath. (Scheuner et al, 2003)
Genetic susceptibility to myocardial infarction and coronary artery disease
Atherosclerotic association in coronary supply routes, which brings about heart assaults or unexpected
demise, is an exceptionally normal infection furthermore an intricate human quality. To comprehend the
genomic premise of this ailment, eight linkage investigations of kin sets was performed. Two qualities in
the leukotriene pathway, prior just ensnared in murine atherosclerosis, have indicated to be most likely
connected with the danger of MI. The two quality varieties (ALOX5AP and LTA4) were found to be
feeble variables for myocardial limited rot. GWAS demonstrated proficient flammable cytokine
lymphotoxin-α (LTA) and its key ligand galectin-2 (LGALS2) to be incorporated in the slant for heart
attack. The quality encoding LTA receptor and its central ligand, LGALS2 have been displayed to grow
peril of MI. CFH, the dominating quality for AMD, has additionally demonstrated to assume a part in MI,
in a manner joining two illnesses with a known establishment of irritation and start of the supplement
pathway.Lowered LDL cholesterol that is connected with PCSK9 point changes found in 2-3% of people
has known not security from CAD. Four reliant and boss procedures that have been embroiled in CAD
vulnerability by means of the grouping of qualities are endothelial trustworthiness, thrombosis,
lipoprotein taking care of and blood vessel irritation.
Collectively, important patterns have emerged, that highlight in depth advances in the genomics of CAD
and MI. On the other hand, no single quality has been distinguished inside of an extensive populace
whose danger can be credited to any semblance of TCF7L2 for sort 2 diabetes. One of the principle
purposes behind this may be that CAD and MI are extremely particular and their pathobiology may
additionally be divergent. Except for PCSK9 and Ox40L, the greater part of alternate advancement has
been made in MI. MI happens in a tolerably littler division of populace that convey the atherosclerotic
weight, and it may be said that it has a more prohibitive phenotype. Miscreant needs further study, in light
of the fact that it is fairly common in the created world and has less heritability. Various difficulties have
get to be evident that are expected to make propels in the genomics of atherosclerosis. Since it is a late
onset sickness, a control test may in the end form into a case in a later phase of life. Angiography is an
one-sided system used to focus atherosclerosis, in light of the fact that individuals not influenced by it
don't have to experience the method, which is a really obtrusive one. Late studies with either low LDL or
low HDL recommend that numerous variations in weakness qualities represent a generous rate of
populace to be at a danger for MI and CAD. (Smith et al, 2006)
20 | P a g e
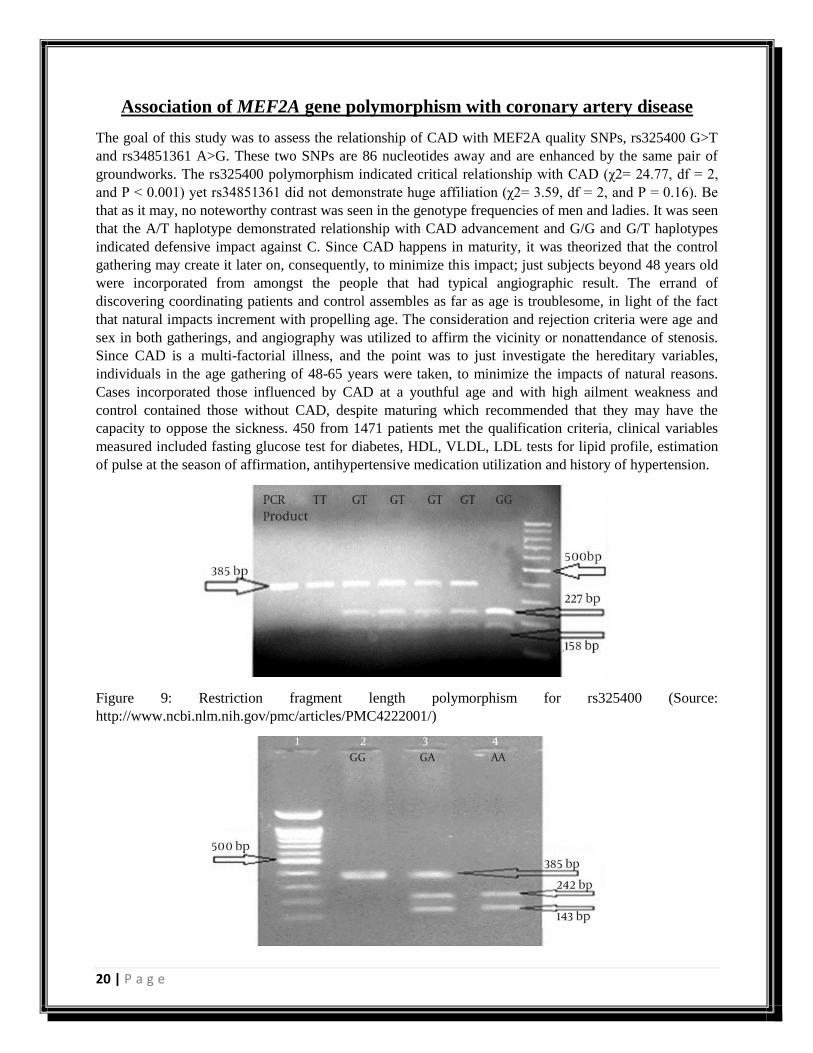
Association of MEF2A gene polymorphism with coronary artery disease
The goal of this study was to assess the relationship of CAD with MEF2A quality SNPs, rs325400 G>T
and rs34851361 A>G. These two SNPs are 86 nucleotides away and are enhanced by the same pair of
groundworks. The rs325400 polymorphism indicated critical relationship with CAD (χ2= 24.77, df = 2,
and P < 0.001) yet rs34851361 did not demonstrate huge affiliation (χ2= 3.59, df = 2, and P = 0.16). Be
that as it may, no noteworthy contrast was seen in the genotype frequencies of men and ladies. It was seen
that the A/T haplotype demonstrated relationship with CAD advancement and G/G and G/T haplotypes
indicated defensive impact against C. Since CAD happens in maturity, it was theorized that the control
gathering may create it later on, consequently, to minimize this impact; just subjects beyond 48 years old
were incorporated from amongst the people that had typical angiographic result. The errand of
discovering coordinating patients and control assembles as far as age is troublesome, in light of the fact
that natural impacts increment with propelling age. The consideration and rejection criteria were age and
sex in both gatherings, and angiography was utilized to affirm the vicinity or nonattendance of stenosis.
Since CAD is a multi-factorial illness, and the point was to just investigate the hereditary variables,
individuals in the age gathering of 48-65 years were taken, to minimize the impacts of natural reasons.
Cases incorporated those influenced by CAD at a youthful age and with high ailment weakness and
control contained those without CAD, despite maturing which recommended that they may have the
capacity to oppose the sickness. 450 from 1471 patients met the qualification criteria, clinical variables
measured included fasting glucose test for diabetes, HDL, VLDL, LDL tests for lipid profile, estimation
of pulse at the season of affirmation, antihypertensive medication utilization and history of hypertension.
Figure 9: Restriction fragment length polymorphism for rs325400 (Source:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4222001/)

21 | P a g e
Figure 10: Restriction fragment length polymorphism for rs34851361 (Source:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4222001/)
Contracting CAD at a younger age hinted at a genetic predisposition and minimal environmental effect.
The goal of this study was to assess the relationship of CAD with MEF2A quality SNPs, rs325400 G>T
and rs34851361 A>G. These two SNPs are 86 nucleotides away and are enhanced by the same pair of
groundworks. The rs325400 polymorphism indicated critical relationship with CAD (χ2= 24.77, df = 2,
and P < 0.001) yet rs34851361 did not demonstrate huge affiliation (χ2= 3.59, df = 2, and P = 0.16). Be
that as it may, no noteworthy contrast was seen in the genotype frequencies of men and ladies. It was seen
that the A/T haplotype demonstrated relationship with CAD advancement and G/G and G/T haplotypes
indicated defensive impact against C. (Foroughmand et al, 2014)
Lack of MEF2A mutations in coronary artery disease
To discover the part of MEF2A transformations in the pathogenesis of nonfamilial instances of CAD,
their exons and flanking intron successions were resequenced in 300 people who procured CAD before
the age of 55 years (men) or 65 years (ladies). The missense change that was found in the CAD gathering
was likewise analyzed in 300 elderly control subjects (men >60 years, ladies >70 years) who did not give
any hints or side effects of CAD. The unlucky deficiency of change in MEF2A in the CAD patients
recommended that it may not be a noteworthy donor to bringing about CAD in white individuals.
Resequencing did however uncover a coding grouping length polymorphism in the last coding exon of
MEF2A that brought about a length variety in a district of poly-glutamine and - proline rehashes. At the
point when the rehash locale was broke down for the situation and control populaces, it demonstrated a 9
length variation alleles covering the poly-glutamine rehash, and all the 9 alleles happened at a
comparative recurrence in both the gatherings. Another 2 length variation was distinguished in the
contiguous poly-proline rehash (5 versus 4 prolines) however more than 98% of people contained 5
deposits of proline, demonstrating no factually critical distinction between the 2 gatherings. This
information demonstrates that there is no relationship between the length polymorphisms in MEF2A and
CAD defenselessness in this arrangement of test of white populace.
Figure 11: Nucleotide and amino acid sequences of the repeat region in MEF2A. The shaded box marks
the polyglutamine and -proline tandem repeats; the 21-bp deletion is boxed with dashed lines; arrows
indicate the primers used for fragment-size analysis. (Source:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1070426/)
A 21-bp cancellation in MEF2A, with a recurrence of more or less 0.15% in the populace likewise does
not indicate relationship with ahead of schedule onset of CAD. On the other hand, the 21-bp cancellation
in MEF2A does show weakness to CAD with variable penetrance, and the way that it demonstrated no
isolation in 3 free families recommends that it doesn't bring about an autosomal overwhelming type of
CAD. (McPherson et al, 2005)
22 | P a g e
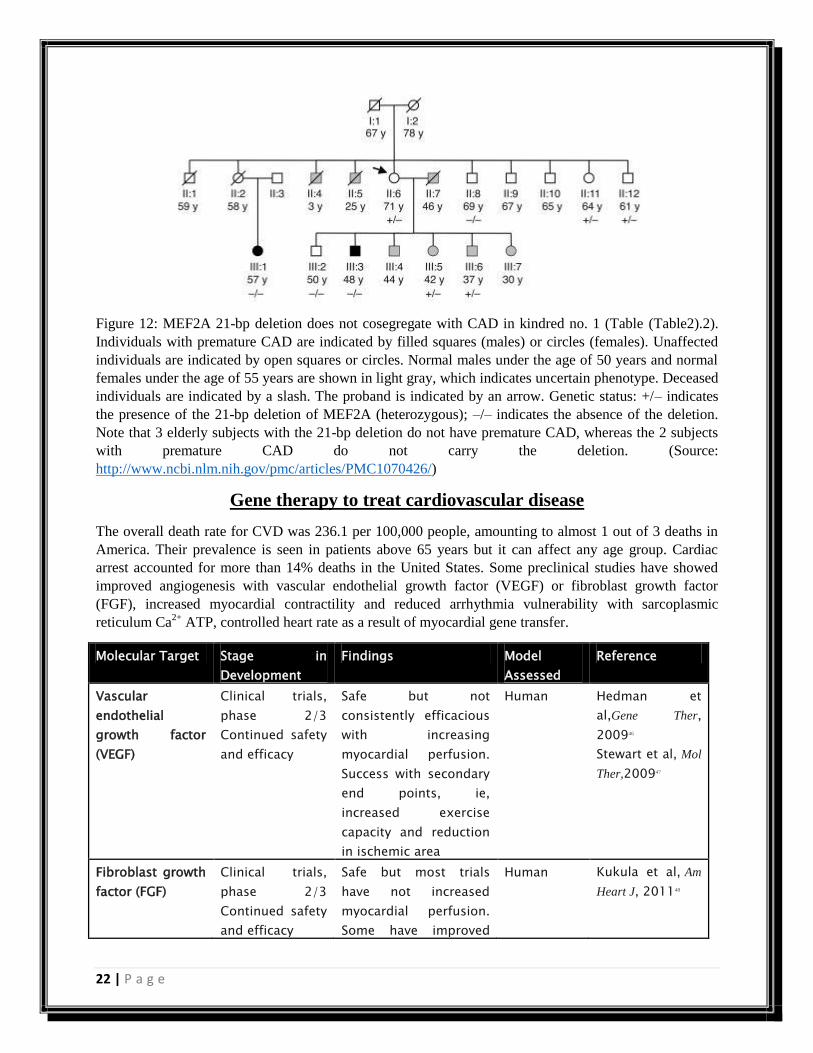
Figure 12: MEF2A 21-bp deletion does not cosegregate with CAD in kindred no. 1 (Table (Table2).2).
Individuals with premature CAD are indicated by filled squares (males) or circles (females). Unaffected
individuals are indicated by open squares or circles. Normal males under the age of 50 years and normal
females under the age of 55 years are shown in light gray, which indicates uncertain phenotype. Deceased
individuals are indicated by a slash. The proband is indicated by an arrow. Genetic status: +/– indicates
the presence of the 21-bp deletion of MEF2A (heterozygous); –/– indicates the absence of the deletion.
Note that 3 elderly subjects with the 21-bp deletion do not have premature CAD, whereas the 2 subjects
with premature CAD do not carry the deletion. (Source:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1070426/)
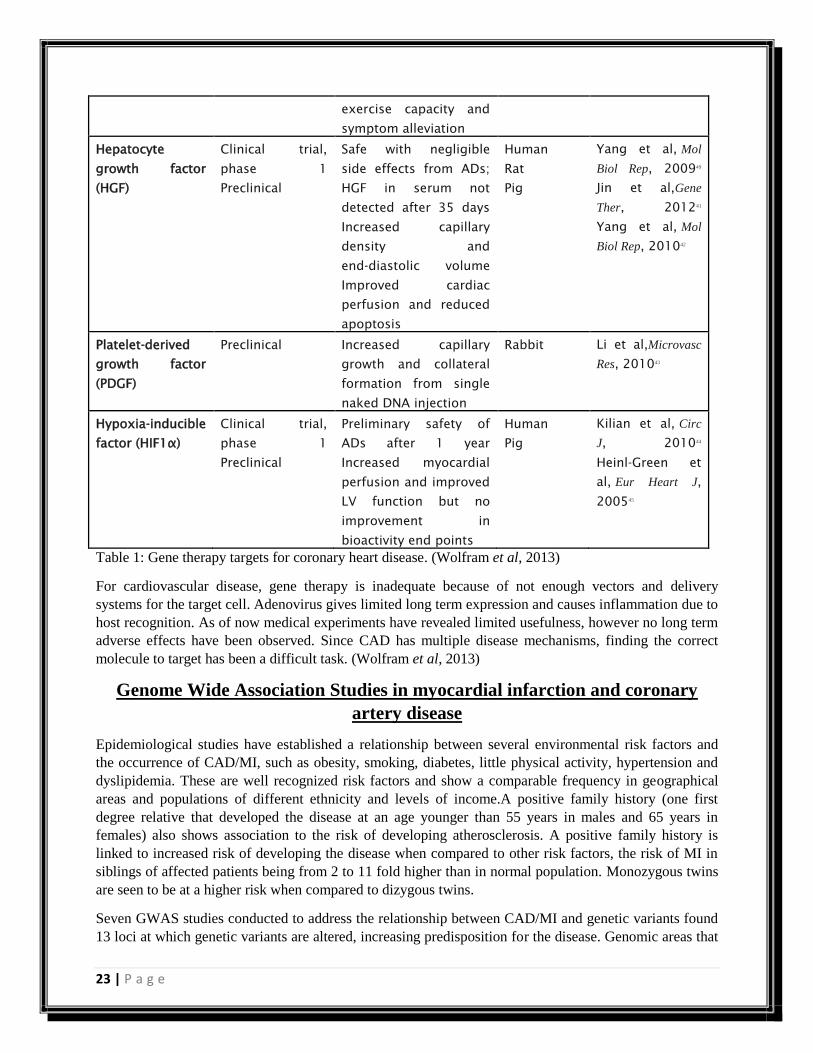
Gene therapy to treat cardiovascular disease
The overall death rate for CVD was 236.1 per 100,000 people, amounting to almost 1 out of 3 deaths in
America. Their prevalence is seen in patients above 65 years but it can affect any age group. Cardiac
arrest accounted for more than 14% deaths in the United States. Some preclinical studies have showed
improved angiogenesis with vascular endothelial growth factor (VEGF) or fibroblast growth factor
(FGF), increased myocardial contractility and reduced arrhythmia vulnerability with sarcoplasmic
reticulum Ca2+
ATP, controlled heart rate as a result of myocardial gene transfer.
Molecular Target Stage in
Development
Findings Model
Assessed
Reference
Vascular
endothelial
growth factor
(VEGF)
Clinical trials,
phase 2/3
Continued safety
and efficacy
Safe but not
consistently efficacious
with increasing
myocardial perfusion.
Success with secondary
end points, ie,
increased exercise
capacity and reduction
in ischemic area
Human Hedman et
al,Gene Ther,
200946
Stewart et al, Mol
Ther,200947
Fibroblast growth
factor (FGF)
Clinical trials,
phase 2/3
Continued safety
and efficacy
Safe but most trials
have not increased
myocardial perfusion.
Some have improved
Human Kukula et al, Am
Heart J, 201148
23 | P a g e
exercise capacity and
symptom alleviation
Hepatocyte
growth factor
(HGF)
Clinical trial,
phase 1
Preclinical
Safe with negligible
side effects from ADs;
HGF in serum not
detected after 35 days
Increased capillary
density and
end‐diastolic volume
Improved cardiac
perfusion and reduced
apoptosis
Human
Rat
Pig
Yang et al, Mol
Biol Rep, 200949
Jin et al,Gene
Ther, 201241
Yang et al, Mol
Biol Rep, 201042
Platelet‐derived
growth factor
(PDGF)
Preclinical Increased capillary
growth and collateral
formation from single
naked DNA injection
Rabbit Li et al,Microvasc
Res, 201043
Hypoxia‐inducible
factor (HIF1α)
Clinical trial,
phase 1
Preclinical
Preliminary safety of
ADs after 1 year
Increased myocardial
perfusion and improved
LV function but no
improvement in
bioactivity end points
Human
Pig
Kilian et al, Circ
J, 201044
Heinl‐Green et
al, Eur Heart J,
200545
Table 1: Gene therapy targets for coronary heart disease. (Wolfram et al, 2013)
For cardiovascular disease, gene therapy is inadequate because of not enough vectors and delivery
systems for the target cell. Adenovirus gives limited long term expression and causes inflammation due to
host recognition. As of now medical experiments have revealed limited usefulness, however no long term
adverse effects have been observed. Since CAD has multiple disease mechanisms, finding the correct
molecule to target has been a difficult task. (Wolfram et al, 2013)
Genome Wide Association Studies in myocardial infarction and coronary
artery disease
Epidemiological studies have established a relationship between several environmental risk factors and
the occurrence of CAD/MI, such as obesity, smoking, diabetes, little physical activity, hypertension and
dyslipidemia. These are well recognized risk factors and show a comparable frequency in geographical
areas and populations of different ethnicity and levels of income.A positive family history (one first
degree relative that developed the disease at an age younger than 55 years in males and 65 years in
females) also shows association to the risk of developing atherosclerosis. A positive family history is
linked to increased risk of developing the disease when compared to other risk factors, the risk of MI in
siblings of affected patients being from 2 to 11 fold higher than in normal population. Monozygous twins
are seen to be at a higher risk when compared to dizygous twins.
Seven GWAS studies conducted to address the relationship between CAD/MI and genetic variants found
13 loci at which genetic variants are altered, increasing predisposition for the disease. Genomic areas that

24 | P a g e
contain genes whose mutation causes familial hypercholesterolemia were found by GWAS. Locus on
chromosome 9p21.3 contains common variants which affect the risk of CAD/MI; this variant has the
largest effect on disease risk (odds ratio – 1.28-1.47). Gene variants at the same locus, genes CDKN2A
and CDKN2B, alter susceptibility to other arterial diseases such as abdominal aortic aneurism and
intracranial aneurism, suggesting that chromosome 9p21.3 may play a role in arterial wall remodelling.
Ye et al found an association with both the occurrence and progression of CAD on chromosome 9p21.
(Mannucci et al, 2010)
Nitric Oxide
Nitric oxide plays an important role in the regulation of cardiovascular diseases by inhibiting vascular
smooth muscle contraction and growth, platelet aggregation and leukocyte adhesion to the endothelium.
Humans with atherosclerosis, diabetes or hypertension often show impaired NO pathways. (Ganz et al,
2003)
Biochemistry of Nitric oxide
Nitric oxide (NO) was first discovered as a colourless and toxic gas. Until 1987, it was not known that it
was produced in the body. Its role in regulating blood pressure and relieving various heart ailments is well
established. It was named ‘molecule of the year’ in 1992, after its biological significance was recognized
by Koshland. Ignerro, Furchgott and Murad were awarded the Nobel Prize for Medicine and Physiology
for identifying NO as a signalling molecule. It plays and important role in the protection against the onset
and progression of cardiovascular diseases. The cardio protective roles of NO include regulation of blood
pressure and vascular tone, inhibition of platelet aggregation and leucocyte adhesion and prevention of
smooth muscle proliferation. Reduced bioavailability of NO is thought to be one of the central factors
common to cardiovascular disease. (Nishikawa et al, 1997)
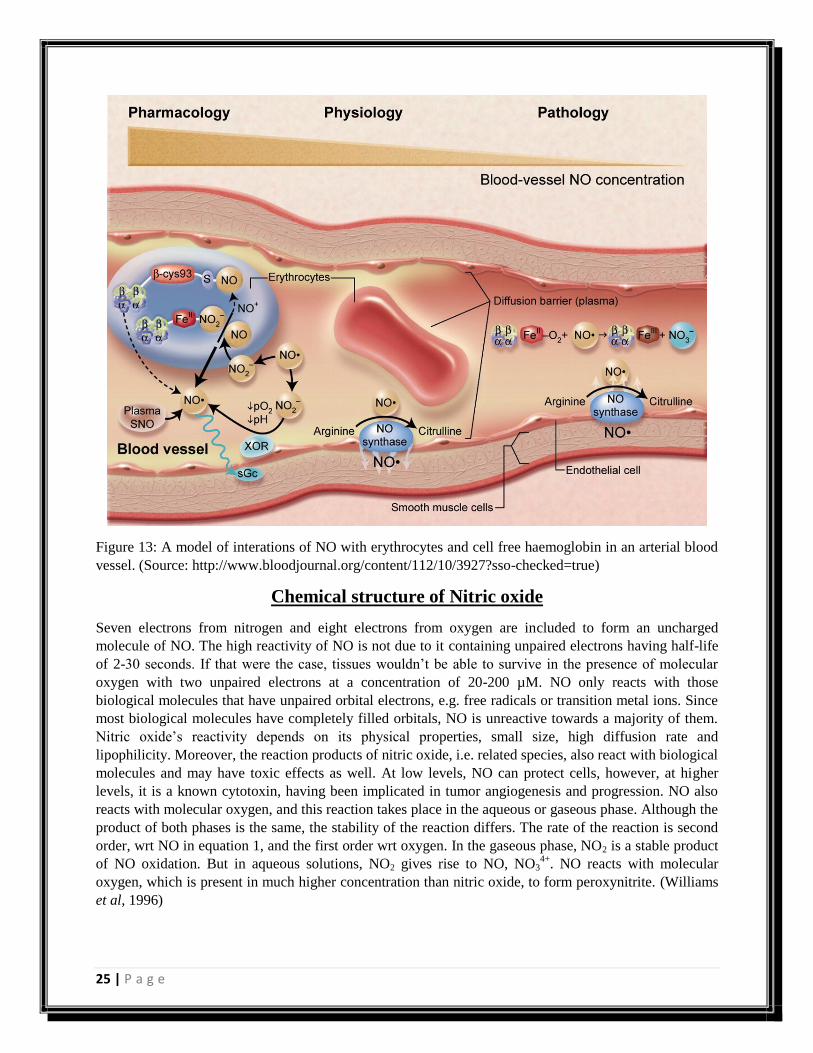
25 | P a g e
Figure 13: A model of interations of NO with erythrocytes and cell free haemoglobin in an arterial blood
vessel. (Source: http://www.bloodjournal.org/content/112/10/3927?sso-checked=true)
Chemical structure of Nitric oxide
Seven electrons from nitrogen and eight electrons from oxygen are included to form an uncharged
molecule of NO. The high reactivity of NO is not due to it containing unpaired electrons having half-life
of 2-30 seconds. If that were the case, tissues wouldn’t be able to survive in the presence of molecular
oxygen with two unpaired electrons at a concentration of 20-200 µM. NO only reacts with those
biological molecules that have unpaired orbital electrons, e.g. free radicals or transition metal ions. Since
most biological molecules have completely filled orbitals, NO is unreactive towards a majority of them.
Nitric oxide’s reactivity depends on its physical properties, small size, high diffusion rate and
lipophilicity. Moreover, the reaction products of nitric oxide, i.e. related species, also react with biological
molecules and may have toxic effects as well. At low levels, NO can protect cells, however, at higher
levels, it is a known cytotoxin, having been implicated in tumor angiogenesis and progression. NO also
reacts with molecular oxygen, and this reaction takes place in the aqueous or gaseous phase. Although the
product of both phases is the same, the stability of the reaction differs. The rate of the reaction is second
order, wrt NO in equation 1, and the first order wrt oxygen. In the gaseous phase, NO2 is a stable product
of NO oxidation. But in aqueous solutions, NO2 gives rise to NO, NO34+
. NO reacts with molecular
oxygen, which is present in much higher concentration than nitric oxide, to form peroxynitrite. (Williams
et al, 1996)

26 | P a g e
Reaction:
NO + NO2 gas NO2 NO + NO3+ O
2 ONOO-
The nitroxyl anion (NO-) is additionally said to be endothelium derived relaxing factor, which is reactive
but a short lived species. It reacts with two nitric oxides to form nitrite and nitrous acid.
Nitric oxide synthase
NO is produced by a various group of enzymes termed as nitric oxide synthase (NOS) which is present in
the body. NO is synthesized when L-arginine is converted to L-citrulline, this reaction is catalysed by
nitric oxide synthases. The two cofactors involved in this reaction are oxygen and NADPH. Moreover,
three isoforms are present whose names are termed on the basis of their activities. These isoforms share
50-60% homology in the amino acid sequence in the oxidase and reductase domains. These isoforms have
distinct characteristics that reflect their specific functions in vivo. Endothelial NO synthase (eNOS or
NOSIII; 7q35-36) and neuronal nitric oxide synthase (nNOS or NOSI; 12q24.2) have a mechanism of
constitutive activation (eNOS). The inducible isoform (iNOS or NOSII; 17cen-q12) is communicated in
strange cell procedures, for example, heart disappointment. After being impelled by cytokines and other
provocative specialists, a high NO stream results. Endothelial NOS is for the most part situated in
endothelial cell compartments named caveolae and is vital for keeping the gauge vascular tone. This tone
is mostly pondered by NO amalgamation, a vascular aggravate that takes part in blood stream regulation
in the diverse vascular quaint little inns in the coronary blood stream. (Homig et al, 2001)
Structure of eNOS
eNOS functions as a dimer consisting of two identical monomers, which in turn can be functionally and
structurally divided into two major domains: a C terminal reductase domain, homologous to cytochrome
P450 which contains binding sites for NADPH, flavine mononucleotide (FMN) and flavine adenine
dinucleotide (FAD); and an N terminal oxidase, which subtracts one electron from the L arginine
substrate and has binding sites for heme iron, for the tetrahydrobiopterin cofactor (BH4) and for L-
arginine. The catalysis reaction of constitutive NOS involves two oxidation stages: L-arginine
hydroxylation into N-g-hydroxy-L-arginine, followed by oxidation of this intermediate compound with
utilization of one NADPH electron, thus forming L-cittruline and NO. This reaction consumes 1.5 mol of
NADPH and 2 moles of oxygen per mol of L-citrulline.

27 | P a g e
Figure 14: eNOS protein structure. (Source:
https://www.caymanchem.com/app/template/Article.vm/article/2185/figure/1%3Bjsessionid=55BADB41
83BD678D2955D650DA29B6F7)
Cofactors such as Heme iron, BH4 and L-arginine have been studied in depth, and their low
bioavailability induces the phenomenon of dysfunctional eNOS. Heme iron is essential for dimerization
of the three isoforms: low concentrations or absence of L-arginine catalyse oxygen reduction in
superoxide (O2-) and decreased levels of BH4 lead to the simultaneous production of NO e O2
-, products
that react with each other forming peroxynitrite (ONOO-). (Vasa et al, 2001)
Role of HSP in regulation of eNOS gene is cardiovascular diseases
Caveolae are specialized invagination of the plasma membrane and are present in most cell types, with
the highest number being present in endothelial cells, adipocytes, fibroblasts and smooth muscle cells.
The main components of caveolae are cholesterol, glycosphingolipids and some structural proteins, such
as caveolin, whereas phospholipids are practically absent. eNOS was reported to be present in caveolae
but not in other parts of plasma membrane. The localization of eNOS within caveolae renders the enzyme
inactive. In caveolae, eNOS activity is inhibited by caveolin-I. NO production from endothelial cells is
stimulated by a variety of mechanical forces such as shear stress and cyclic strain and humoral factors
ranging from growth factors to peptide hormones, including Ach (acetlycholine), VEGF (vascular
endothelial growth factor), Bdk (Bradykinin), Estrogen, S-1P (sphingosine-1 phosphate), H2O2 (hydrogen
peroxide), and angiotensin II. eNOS is a dually acylated peripheral membrane protein that is targeted to
endothelial plasmalemmal caveolae through an interaction with the caveolae structural protein, CavI
(caveolin I).
As we know Cav I inhibition of eNOS is relieved by calm, which causes dissociation of eNOS from
caveolin. This regulatory mechanism is further modified by HSP90 (heat shock protein-90) HSP 70,
which binds to eNOS and facilitates displacement of cav-1 by calm. In addition to these protein
interactions that modulate calm binding, other cellular sigmaling cascades also regulate eNOS activity.
Physiologically, endothelial cells are exposed to the hemodynamic forces of blood including the PI3K
(phosphatidylinositol 3-kinase), PDK (phosphoinositide dependent kinase) and AC (adenylate cyclase)
via cAMP (cyclic adenosine monophosphate), leading to eNOS activation by phosphorylation of serine

28 | P a g e
residues (S617 and S1179 for Akt, and S635 and S1179 for PKA), which promotes eNOS activation.
Additional stimuli such as VEGF, estrogen, S-1P and Bdk, bind to their cognate receptors (RTKs,
VEGFR, ESR, EDG and BdkR) and stimulate PI3K/Akt. However, they also activate PLC-gamma
(phospholipase-C) and PIP2 (phosphatidyl inositol 4, 5-bisphosphate) to increase cytoplasmic calcium
and DAG (diacylglycerol) levels. The increase in cytoplasmic calcium levels activates calm, which binds
to the canonical calm binding domain in eNOS, leading to efficient NO synthesis. In addition, calm
activates calmK-II (calm kinase-II), which phosphorylates eNOS on S1179. Increase in DAG levels
activate PKC (protein kinase C) pathway, which may negatively regulate eNOS or influence its coupling.
Finally, metabolic stress triggers breakdown of ATP, stimulating AMPK (AMP kinase) to phosphorylate
eNOS on S1179. Phosphorylation of this residue by PKA (protein kinase A) is also associated with
increased enzyme activity. Other proteins which are associated with increased eNOS activity or NO
release are dynamin 2 (a GTP binding protein) and porin, which co localize and directly interact with
eNOS. Their interactions with eNOS are stimulated by intracellular Ca2+
and leads to eNOS activation.
Efficient supply with substrate during all this is ensured by localization of the arginine transporter CAT1
(cationic amino acid transporter 1) in caveolae and its direct interaction with eNOS. eNOS can interact
with various proteins in its ‘less active’ and ‘more active’ states. Myristoylation of eNOS occurs co-
translationally and targets eNOS to cellular membranes, where eNOS is then palmitoylated. These
lipidation promote eNOS association with cell membranes and are essential for linking upstream signal
transduction pathways to eNOS activity in cells. N-myristoylated and palmitoylated membrane-bound
eNOS associates with the caveolae coat protein Cav1 and with HSP90. CHIP (C-terminal HSP 70
interacting protein) interacts with both HSP70 and HSP90, and negatively regulates eNOS trafficking into
the golgi complex. By contrast, NOSIP (nitric oxide synthase interacting protein), a 34kDa protein and
NOSTRIN (nitric oxide synthase traffic inducer) negatively regulates eNOS localization in the plasma
membrane. (Cannon et al, 1998)
Acute activation of eNOS in blood vessels in response to agonists such as Acetylcholine or braykinin
leads to the activation of the sGC (soluble guanyly cyclase) in smooth muscle cells, production of cGMP
(cyclic guanosine monophosphate) and degradation of cyclin A. An increase in intracellular cGMP levels
affects vascular tone by decreasing the intracellular concentration of free (Ca2+
) I by CNG channel (cyclic
nucleotide gated ion channel complex) modulation; as well as by activating PKG (protein kinase G) and
phosphorylating HSP20, which regulate force by binding to thin filaments and inhibit cross bridge
cycling. Nitrosylation of caspase 3 and caspase 8 inactivates the proteins, thus leading to inhibition of
apoptosis. eNOS is an important regulator of cardiovascular homeostasis because it is the major source of
NO production in vascular endothelial cells. eNOS plays a crucial role in the state of blood vessel
vasodilation and hence blood pressure regulation. In addition, NO released from the endothelium
modulates other processes including platelet aggregation, platelet and leukocyte adhesion to the
endothelium, endothelin-1 generation, vascular smooth muscle cell proliferation, and angiogenesis.
Because of the important role of NO in each of these processes, abnormalities in vascular NO production
are thought to contribute to the pathogenesis of certain vascular disorders such as those of atherosclerosis
and hypertension. (Gryglewski et al, 1986)
Vitamin C affects thrombosis/fibrinolysis system and reactive hyperemia in
patients with type II diabetes and coronary artery disease
In this study, the impacts of vitamin C were analyzed on patients with sort II diabetes and basic CAD.
Transient treatment with incomprehensible estimations of vitamin C improved RH% and essentially cut
down plasma levels of tPA and vWF in patients with sort II diabetes and atherosclerosis. Hyperglycemia

29 | P a g e
is obstructs the era of NO by blocking endothelial NO synthase order and by growing the making of
responsive oxidation species, superoxide anion (O2-
), in vascular smooth muscles and the endothelium.
Superoxide anion diminishes NO creation by shaping deadly peroxynitrite particle, which causes the
endothelial NO synthase to uncouple, because of oxidation of its cofactor, tetrahydeobiopterin, bringing
about eNOS to deliver O2-
. (Tousoulis et al, 2003)
Vitamin C might improve endothelial function in CAD; however, its effect in T2D along with CAD is
debatable. Increased plasma levels of PAI-1 have been associated with CAD and unstable coronary
syndromes. Evidence proposes that PAI-1 plasma level can be used as a prognosticator for CAD patients
due to the effect it has on the modulation of fibrinolysis and cell migration. (Meister et al, 1994) Due to
its high antigen levels, tPA, an endothelium derived component, has recently been implicated as a
predictor of CAD and MI. Solid confirmation connections expanded oxidative anxiety supplements
impeded fibrinolysis and an increment in PAI-1 and tPA plasma levels, then again, vitamin C and tPA
levels wereinversely associated. The purpose behind this study was to examine the impact of short lived
oral association of vitamin C on lower arm vasodilatory response to responsive hyperemia (RH%),
plasma levels of plasminogen activator inhibitor 1 (PAI-1), tissue plasminogen activator (tPA) in atients
with both T2D and CAD. No critical contrasts were seen in the dietary propensities. RH% was
fundamentally enhanced in vitamin C treatment bunch than in controls. (Levine et al, 1996)

30 | P a g e
Methods

31 | P a g e
Study population
Group 1: 25 CAD patients (age > 25 years, <80 years) were recruited from the Department of
Cardiology, AIIMS. Significant CAD is defined when at least one major epicardial vessel contains more
than 70% stenosis, when assessed by coronary angiography. Hence, angiographically assessed patients
who had 70% or more stenosis in at least one of the major coronary artery were selected for the study.
Patients that were diagnosed with diabetes mellitus and hypertension were also included in the study.
Individuals with less than 50% stenosis, valvular disease or cardiomyopathy were excluded from the
study. Family history of coronary artery disease, diabetes, hypertension, other major illness' in the past,
personal history of smoking, alcohol, dietetic history, drug history was recorded for each subject on a
proforma.
Group 2: Age and sex matched volunteers were selected as controls from a similar population which
includes asymptomatic relatives of the case samples. These were recruited from the staff of AIIMS and
residents of Delhi and surrounding areas. Control subjects had no history of endocrine, metabolic,
ischemic heart disease, renal and cerebrovascular disorders.
Note: Ethical clearance was obtained from the ethics committee of AIIMS, New Delhi in the ICMR
format by the chief investigator of the project.
Informed consent was taken from the patients and controls prior to collection.
Sample collection and processing
Angiographically proven CAD patients with more than 70% stenosis in one or more of their major
arteries were recruited from the Department of Cardiology, All India Institute of Medical Sciences
(AIIMS), New Delhi. The age and sex matched normal healthy individuals, as controls were selected
from similar socio-economic -geographical background. 5 ml blood was withdrawn and collected in a
tube containing EDTA as the anti-coagulant from all subjects after 12 hours of fasting. 3 ml from this was
transferred into a new tube for RNA isolation. The remaining sample was spun down at 3000 rpm for 15
mins at 27°C and the plasma was collected into a 1.5 ml eppendorf tube.
RNA isolation
Total RNA was isolated by Trizol method. (6)
1. 11.5 ml RBC lysis buffer was added to 3 ml of arterial blood in a 15 ml falcon tube. The solutions
were mixed with a pipette. The tube was inverted for proper mixing and was left at room
temperature for 30 mins.
2. It was centrifuged at 3000 g for 10 mins at 4°C.
3. Supernatant was discarded.
4. To this 100-300 µl RBC lysis buffer was added to dissolve the thick layer of red blood cells. The
supernatant was discarded - the whole step was done whilst not disturbing the pellet.
5. The above steps were repeated until a white pellet was obtained.
6. 5 µl of 1x PBS was used to re-suspend the cells. To this another 1000 µl was added, it was mixed
well so as to homogenize the pellet.
7. The solution was transferred to a 1.5 ml eppendorf tube.
8. It was centrifuged at 3000 g for 5 mins at 4°C.
9. The supernatant was discarded. (This is the stopping point)
10. Trizol was added according to the pellet size - 800 µl for small pellet.

32 | P a g e
11. The pellet was dissolved in the solution and kept at room temperature for 15 mins.
12. To this, 200 µl of chloroform was added and the two solutions were mixed by inversion. The tube
was then kept on ice for 15 mins.
13. It was centrifuged at 14000 rpm for 15 mins at 4°C.
14. The aqueous phase containing the RNA was removed carefully into a new tube. It was made sure
that the white interphase layer was not disturbed.
15. 500 µl of ice cold propanol was added to the aqueous phase and was inverted 5-6 times for
thorough mixing. The tube was left on ice for 15 mins.
16. It was inverted again and then centrifuged at 14000 rpm for 15 mins at 4°C.
17. The supernatant was discarded.
18. 1 ml 70% ethanol was added to the pellet and centrifuged at 7500 g for 15 mins at 4°C.
19. The supernatant was discarded and the tubes were left to dry on a tissue paper.
20. 25 µl of RNAase free water was added to dissolve the pellet.
21. The tubes were incubated at 55°C for 5-10 mins.
22. NanoDrop reading was taken to measure the concentration.
Griess reaction assay
Griess reaction was first described in 1879. Because of its simplicity, it has been used extensively in
analysis of numerous biological samples including plasma (serum), urine, CSF, saliva and cell culture
media. In this method, nitrite is first treated with a diazotizing reagent, eg. sulfanilamide (SA), in acidic
media to form a transient diazonium salt. This intermediate is then allowed to react with a coupling
reagent, N-naphthyl-ethylenediamine (NED), to form a stable azo compound. The overall reaction is
described in the scheme below. The intense purple color of the product allows nitrite assay with high
sensitivity and can be used to measure nitrite concentration as low as ~0.5 µM level. The absorbance of
this adduct at 54 nm is linearly proportional to the nitrite concentration in the sample. (34)
Figure 15: Reaction between nitric oxide and sulphanilamide to form azo compound
Preparation of Griess reagent
1% sulphanilamide 2.5% orthophosphoric acid (A)
0.1% napthylenediamine HCL in 2.5% orthophosphoric acid (B)
2.5% Orthophosphoric acid was made in 100ml of milliQ
All three solutions are prepared separately, mixed and stored for a maximum period of 1 month. The
solution was stored in a light sensitive bottle. It was not used if any color was seen.
Estimation of Nitric oxide
A nitrite standard reference curve was prepared for each assay for accurate quantization of NO levels in
each experimental sample.

33 | P a g e
1. NaNO2 standard is prepared by taking 0.69 g of NaNO2 and dissolving in 10 ml water (1 M).
2. A series of serial dilutions were done to obtain different concentrations of NaNO2.
3. 100 µl of plasma is diluted with 100 µl of 1X PBS.
4. In a 96 well plate, the solutions were taken in duplicates.
5. The first set of wells contained the 50 µl standards prepared with 50 µl of Griess reagent.
6. 50 µl of plasma was mixed with 50 µl of Griess reagent.
7. The solutions which were to act as control, contained 50 µl of plasma mixed with 50 µl of 1X
PBS.
8. The plate was covered with aluminum foil and incubated at 37°C for 30-45 mins.
9. The absorbance was measured at 540 nm.
10. Standard curve of absorbance vs concentration was plotted.
Preparation of dTCS reagent
5% Thiourea – 0.2 g in 10ml H2O
0.05% CuSO4 – 0.005 g in 10 ml H2O
3% DNPH – 0.3 g in 10 ml H2O
5 % TCA – 2.5 g in 50 ml H2O
12N H2SO4 – 13.33 ml in 26.67 ml ice cold H2O
Working solution – 800 µl of 5% Thiourea
500 µl of 0.05% CuSO4
900 µl of 3% DNPH
Make up to 10 ml with 9N H2SO4
Estimation of Ascorbic acid
1. 1 mg of ascorbic acid was dissolved in 10 ml of 5% TCA (100mg/dL).
2. This was serially diluted to obtain standards.
3. 40 µl of TCA stabilized samples were taken (50 µl plasma+50 µl 1X PBS+100 µl 5% TCA;
centrifuged at 300 rpm for 20 mins at 4°C; supernatant used as test sample).
4. 2 sets of blanks were taken – 100 µl of 5% TCA and 50 µl of 1X PBS+50 µl of 5% TCA.
5. To this, 8 µl of dTCS reagent was added.
6. They were gently vortexed and covered with aluminum foil.
7. The plate was incubated at 37°C for 2 hours.
8. Samples were vortexed every 30 mins during the incubation period.
9. After the completion of the 2 hour time, the samples were placed on ice and 60 µl of ice cold
H2SO4 was added to the sample.
10. It was vortexed gently, and then incubated in the dark at room temperature for an hour.
11. Samples were transferred to a 96 well plate and the absorbance was read at 512 or 520 nm.
12. Standard curve of absorbance vs concentration was plotted.

34 | P a g e
Results
35 | P a g e
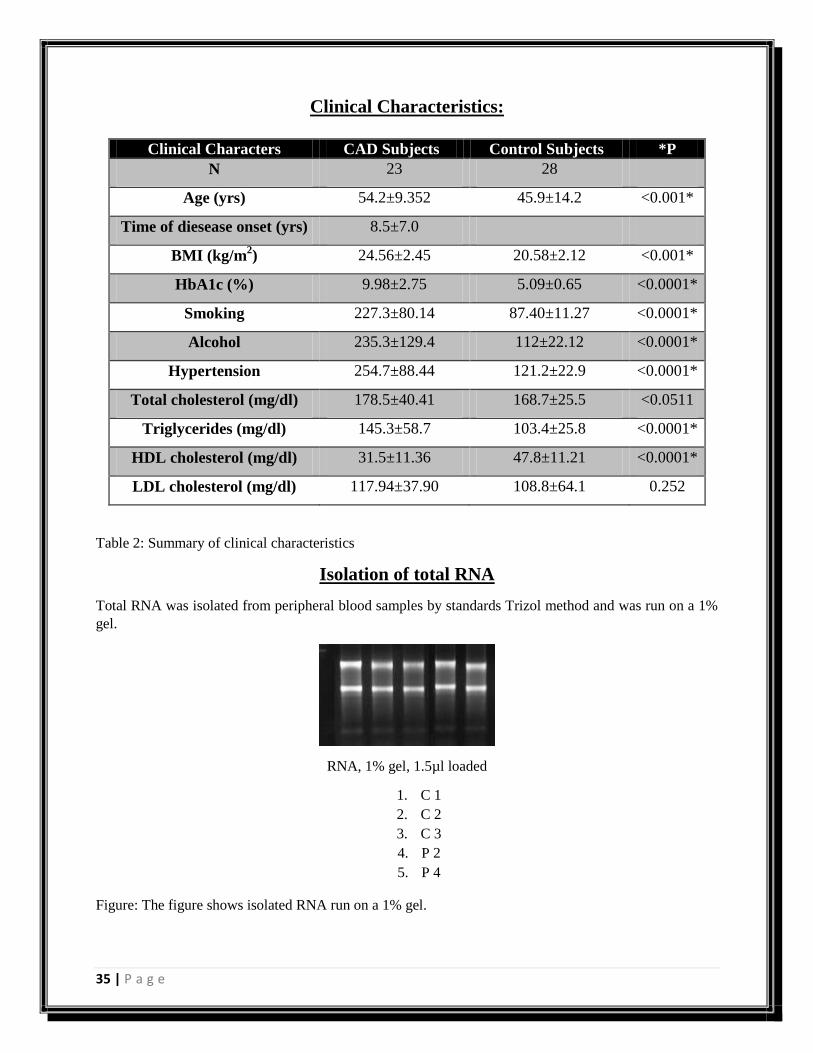
Clinical Characteristics:
Clinical Characters CAD Subjects Control Subjects *P
N 23 28
Age (yrs) 54.2±9.352 45.9±14.2 <0.001*
Time of diesease onset (yrs) 8.5±7.0
BMI (kg/m2) 24.56±2.45 20.58±2.12 <0.001*
HbA1c (%) 9.98±2.75 5.09±0.65 <0.0001*
Smoking 227.3±80.14 87.40±11.27 <0.0001*
Alcohol 235.3±129.4 112±22.12 <0.0001*
Hypertension 254.7±88.44 121.2±22.9 <0.0001*
Total cholesterol (mg/dl) 178.5±40.41 168.7±25.5 <0.0511
Triglycerides (mg/dl) 145.3±58.7 103.4±25.8 <0.0001*
HDL cholesterol (mg/dl) 31.5±11.36 47.8±11.21 <0.0001*
LDL cholesterol (mg/dl) 117.94±37.90 108.8±64.1 0.252
Table 2: Summary of clinical characteristics
Isolation of total RNA
Total RNA was isolated from peripheral blood samples by standards Trizol method and was run on a 1%
gel.
RNA, 1% gel, 1.5µl loaded
1. C 1
2. C 2
3. C 3
4. P 2
5. P 4
Figure: The figure shows isolated RNA run on a 1% gel.
36 | P a g e
Nitric oxide analysis
Griess reagent was used to determine the nitric oxide levels of the case and control samples. The
procedure was standardized using a set of NaNO2 standards of varying concentrations. They were also put
on the testing plate along with the samples. The heating block was set to a temperature of 37°C before the
beginning of the experiment. The absorbance was measured at 540 nm. The concentration obtained from
the standards was used to calculate the concentration of the case and control subjects.
Sample no (Controls)
Nitric oxide
Plasma + GR Plasma + PBS P+GR - P+PBS NO Conc (uM)
C 1 0.095 0.088 0.007 0.84336
C 2 0.076 0.075 0.001 0.12048
C 3 0.069 0.06 0.009 1.08432
C 4 0.064 0.059 0.005 0.6024
C 5 0.06 0.051 0.009 1.08432
C 6 0.072 0.071 0.001 0.12048
C 7 0.064 0.06 0.004 0.48192
C 8 0.066 0.057 0.009 1.08432
C 9 0.103 0.098 0.005 0.6024
C 10 0.088 0.081 0.007 0.84336
C 11 0.1404 0.235 0.0946 11.397
C 12 0.0693 0.9405 0.8712 89.498
C 13 0.1258 0.0675 0.0583 5.989
C 14 0.0794 0.0666 0.0128 1.314
C 15 0.08305 0.074 0.009 0.925
C 16 0.0751 0.0621 0.013 1.34
C 17 0.0847 0.071 0.0137 2.14
C 18 0.0778 0.069 0.0088 1.375
C 19 0.0872 0.084 0.0032 0.5
C 20 0.0818 0.0723 0.0095 1.48
C 21 0.12 0.0906 0.0294 4.59
C 22 0.14365 0.1187 0.0249 3.89
C 23 0.0736 0.0714 0.0022 0.22
C 24 0.0743 0.0724 0.0019 0.19
C 25 0.1053 0.0911 0.0142 1.42
C 26 0.1399 0.0663 0.0736 7.36
C 27 0.2711 0.0744 0.1967 19.67
C 28 0.0857 0.0796 0.0061 0.61
37 | P a g e
Table 3: Table shows the values of all the samples of nitric oxide as observed after reaction with Griess
reagent.
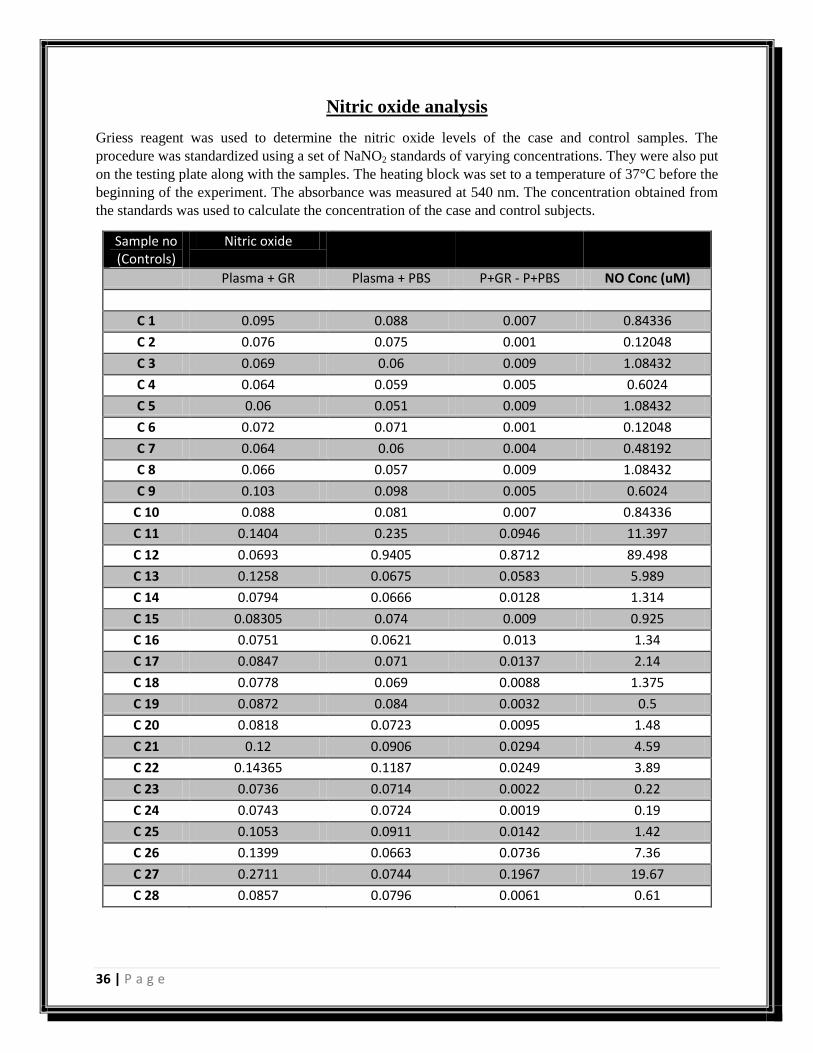
Figure 16: Graph showing concentrations of nitric oxide in control samples
Sample no. (Cases)
Plasma + Griess reagent
Plasma + PBS P+GR – P+PBS Concentration (µg)
P 1 0.199 0.259 0.06 7.25
P 2 0.075 0.124 0.049 5.92
P 3 0.124 0.0723 0.0517 6.24
P 4 0.1 0.096 0.0004 0.048
P 5 0.089 0.1064 0.0174 2.1
P 6 0.076 0.0985 0.0225 2.717
P 7 0.107 0.0794 0.0276 3.34
P 8 0.097 0.0807 0.0163 1.96
P 9 0.069 0.3623 0.2933 35.42
P 10 0.092 0.0788 0.0132 1.59
P 11 0.0845 0.0608 0.0237 2.86
P 12 0.089 0.0608 0.0282 3.405
P 13 0.067 0.0552 0.0118 1.43
P 14 0.069 0.2803 0.2133 25.76
P 15 0.092 0.115 0.023 2.78
P 16 0.087 0.065 0.022 2.65
P 17 0.0628 0.0574 0.0054 0.54
P 18 0.0652 0.079 0.0138 1.417
P 19 0.1156 0.1154 0.00019 0.0195
P 20 0.0973 0.1326 0.0353 3.626
0
20
40
60
80
100
Sample no.
NO conc
NO conc

38 | P a g e
P 21 0.0629 0.0602 0.0027 0.277
P 22 0.0674 0.059 0.0084 0.863
P 23 0.0576 0.0523 0.0053 0.53
Table 4: Table shows the values of all the samples of nitric oxide as observed after reaction with Griess
reagent.
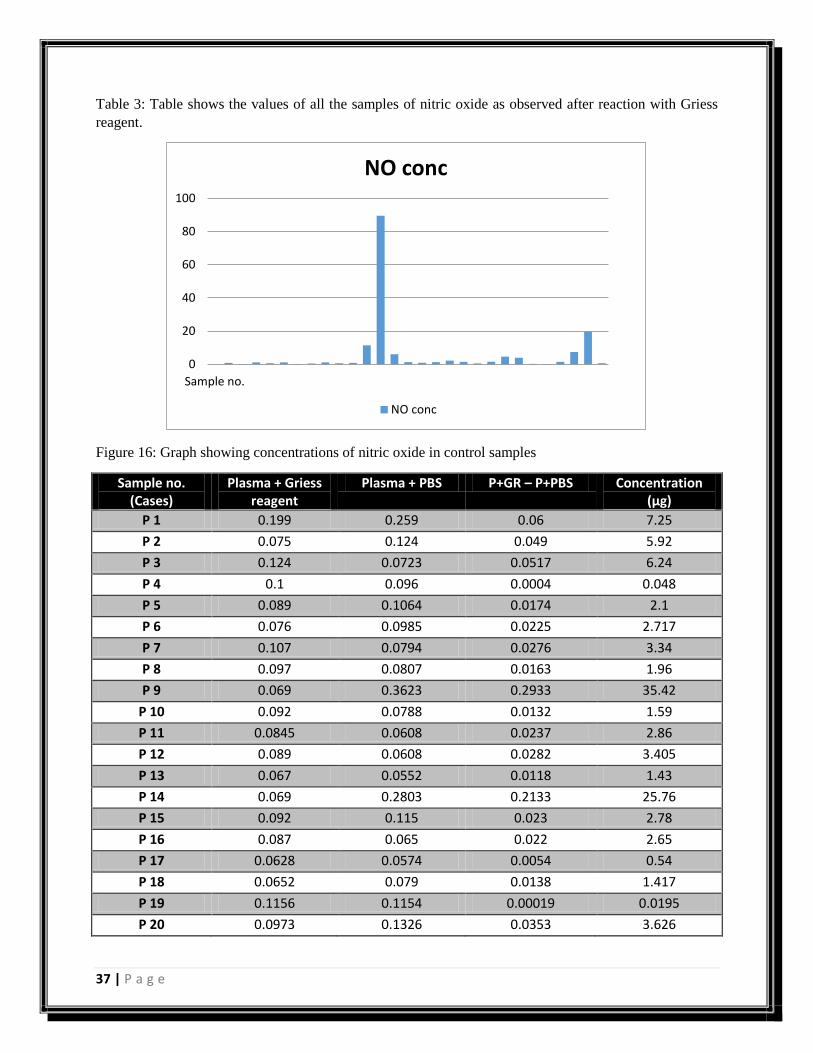
Figure 17: Graph showing concentrations of nitric oxide in case samples.
Figure 18: Graph showing mean of control and cases (nitric oxide).
0
5
10
15
20
25
30
35
40
P 1P 2P 3P 4P 5P 6P 7P 8P 9P 10P 11P 12P 13P 14P 15P 16P 17P 18P 19P 20P 21P 22P 23
Conc NO (ng/ul)
Conc NO (ng/ul)
4.4
4.6
4.8
5
5.2
5.4
5.6
5.8
Controls Cases
Mean (NO)
Mean (NO)
39 | P a g e
Ascorbic acid
dTCS reagent was used to determine the ascorbic acid levels of the case and control samples. The
procedure was standardized using a set of ascorbic acid standards of varying concentrations. They were
also put on the testing plate along with the samples. The heating block was set to a temperature of 37°C
before the beginning of the experiment. The absorbance was measured at 540 nm. The concentration
obtained from the standards was used to calculate the concentration of the case and control subjects.
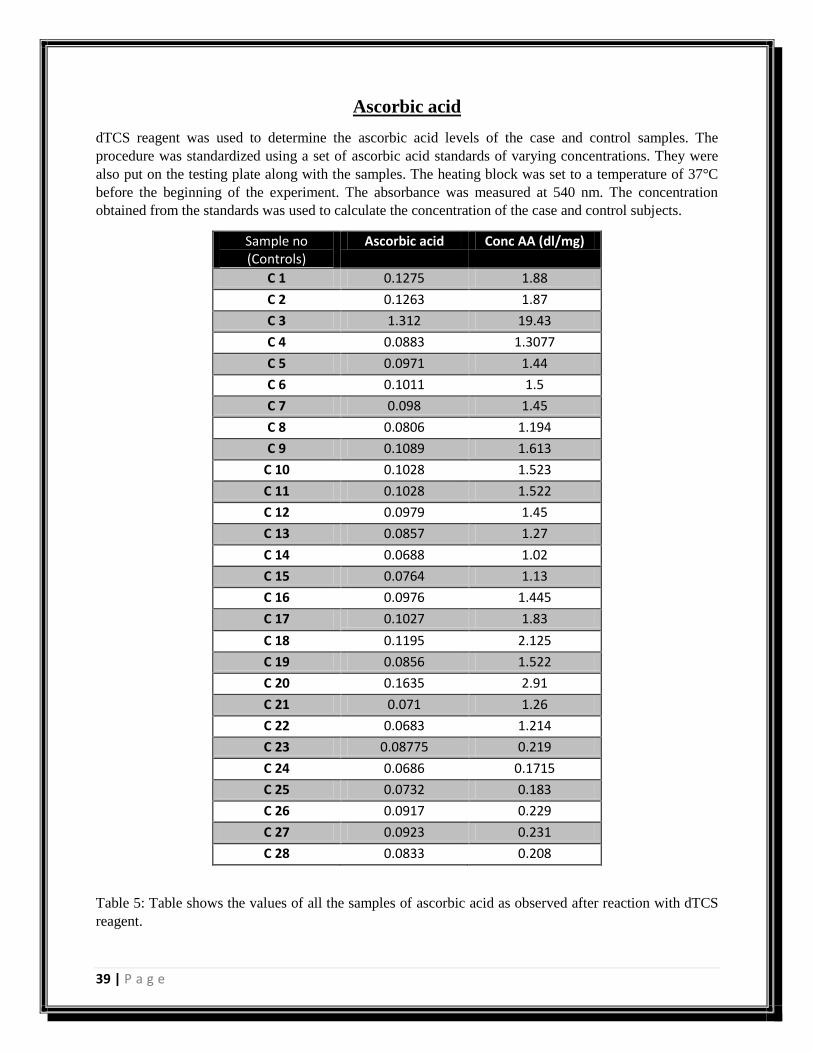
Sample no (Controls)
Ascorbic acid Conc AA (dl/mg)
C 1 0.1275 1.88
C 2 0.1263 1.87
C 3 1.312 19.43
C 4 0.0883 1.3077
C 5 0.0971 1.44
C 6 0.1011 1.5
C 7 0.098 1.45
C 8 0.0806 1.194
C 9 0.1089 1.613
C 10 0.1028 1.523
C 11 0.1028 1.522
C 12 0.0979 1.45
C 13 0.0857 1.27
C 14 0.0688 1.02
C 15 0.0764 1.13
C 16 0.0976 1.445
C 17 0.1027 1.83
C 18 0.1195 2.125
C 19 0.0856 1.522
C 20 0.1635 2.91
C 21 0.071 1.26
C 22 0.0683 1.214
C 23 0.08775 0.219
C 24 0.0686 0.1715
C 25 0.0732 0.183
C 26 0.0917 0.229
C 27 0.0923 0.231
C 28 0.0833 0.208
Table 5: Table shows the values of all the samples of ascorbic acid as observed after reaction with dTCS
reagent.
40 | P a g e
Figure 19: Graph showing concentrations of ascorbic acid in control samples.
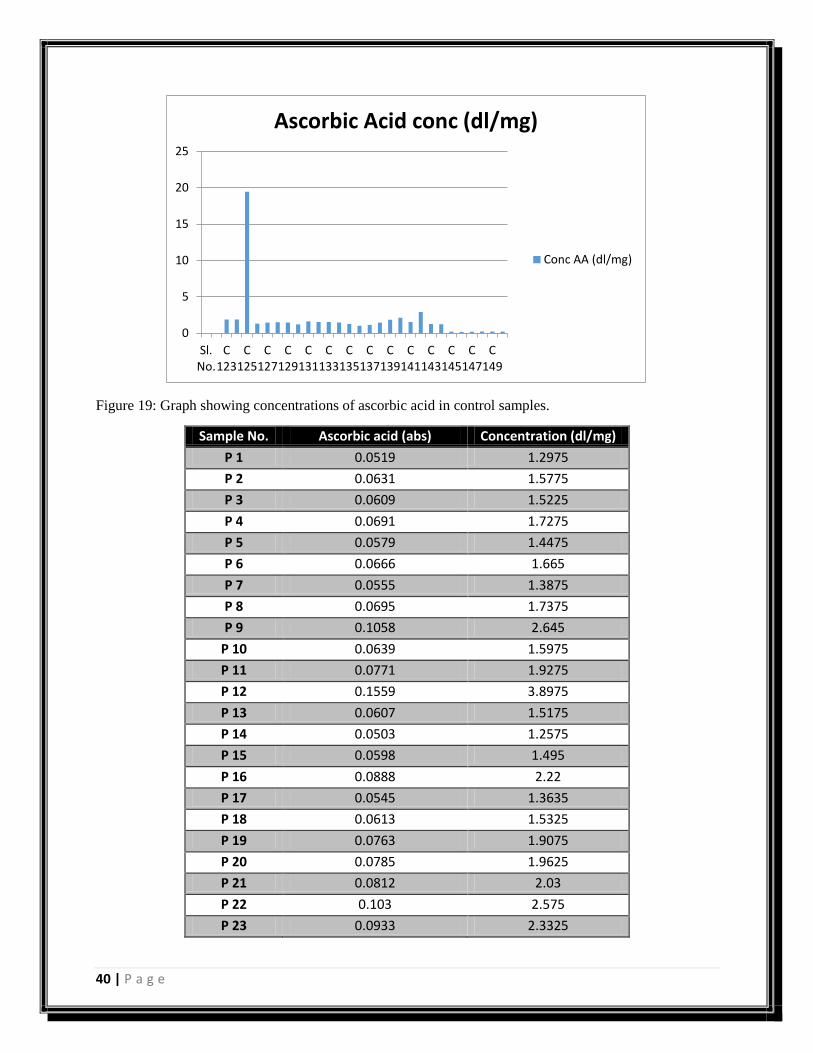
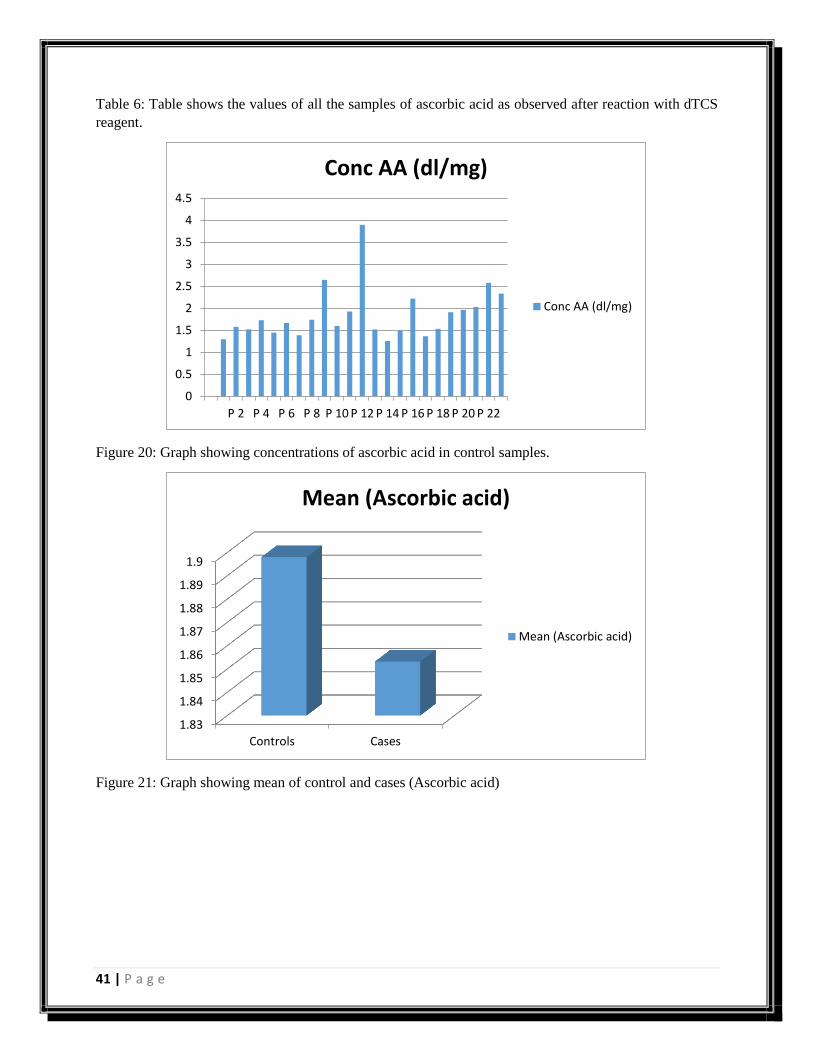
Sample No. Ascorbic acid (abs) Concentration (dl/mg)
P 1 0.0519 1.2975
P 2 0.0631 1.5775
P 3 0.0609 1.5225
P 4 0.0691 1.7275
P 5 0.0579 1.4475
P 6 0.0666 1.665
P 7 0.0555 1.3875
P 8 0.0695 1.7375
P 9 0.1058 2.645
P 10 0.0639 1.5975
P 11 0.0771 1.9275
P 12 0.1559 3.8975
P 13 0.0607 1.5175
P 14 0.0503 1.2575
P 15 0.0598 1.495
P 16 0.0888 2.22
P 17 0.0545 1.3635
P 18 0.0613 1.5325
P 19 0.0763 1.9075
P 20 0.0785 1.9625
P 21 0.0812 2.03
P 22 0.103 2.575
P 23 0.0933 2.3325
0
5
10
15
20
25
Sl.No.
C123
C125
C127
C129
C131
C133
C135
C137
C139
C141
C143
C145
C147
C149
Ascorbic Acid conc (dl/mg)
Conc AA (dl/mg)
41 | P a g e
Table 6: Table shows the values of all the samples of ascorbic acid as observed after reaction with dTCS
reagent.
Figure 20: Graph showing concentrations of ascorbic acid in control samples.
Figure 21: Graph showing mean of control and cases (Ascorbic acid)
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
P 2 P 4 P 6 P 8 P 10 P 12 P 14 P 16 P 18 P 20 P 22
Conc AA (dl/mg)
Conc AA (dl/mg)
1.83
1.84
1.85
1.86
1.87
1.88
1.89
1.9
Controls Cases
Mean (Ascorbic acid)
Mean (Ascorbic acid)
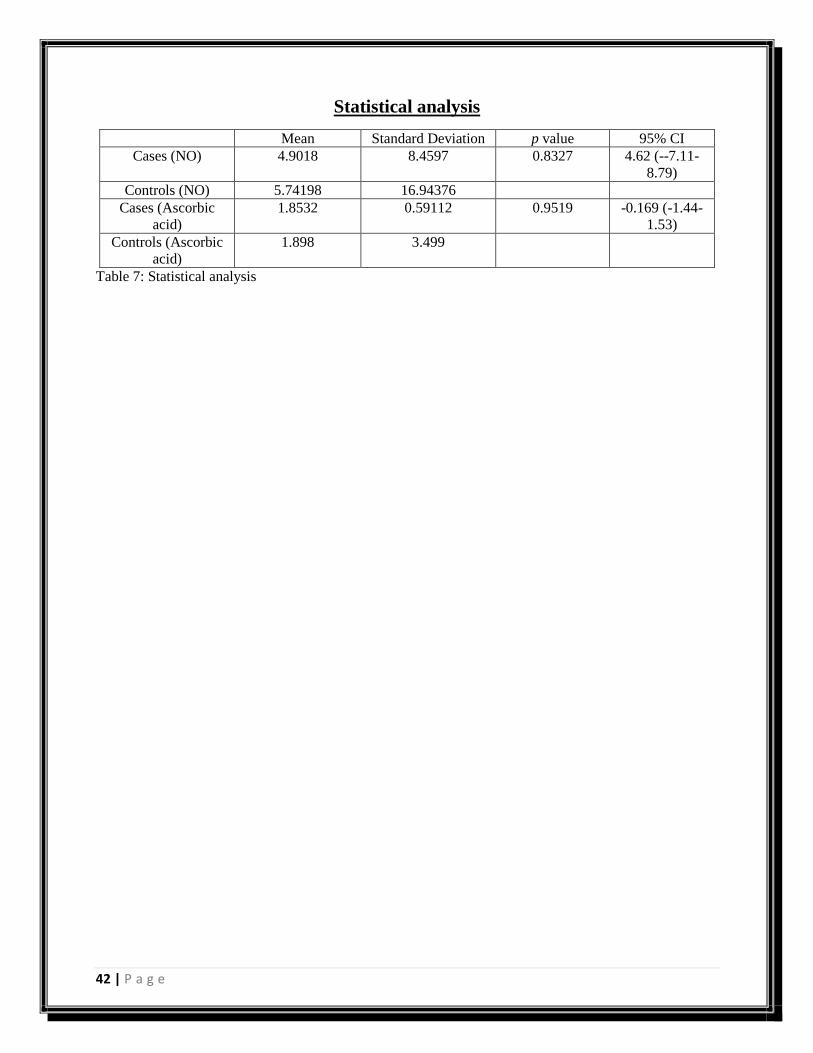
42 | P a g e
Statistical analysis
Mean Standard Deviation p value 95% CI
Cases (NO) 4.9018 8.4597 0.8327 4.62 (--7.11-
8.79)
Controls (NO) 5.74198 16.94376
Cases (Ascorbic
acid)
1.8532 0.59112 0.9519 -0.169 (-1.44-
1.53)
Controls (Ascorbic
acid)
1.898 3.499
Table 7: Statistical analysis

43 | P a g e
Discussion

44 | P a g e
The understandings of disease onset through the variations that occur across the genome are the critical
aspects in developing the emerging branch in personalized medicine, the pharmacogenomics. Studies
reporting the involvement of the gene alterations in various metabolic diseases such as coronary artery
disease are the basis for the current study.
An understanding of the homeostatic function of the endothelium is necessary for modern cardiology.
Nitric oxide’s role in mediating regulatory properties of the endothelium is now well documented, along
with the understanding of diseases and situations can be measured as ‘risk factors’ for atherosclerosis.
The magnitudes of endothelial dysfunction are abundant, including coronary constriction or inadequate
dilation during physical or mental stress, plaques rupture and thrombosis, reperfusion injury after
thrombolysis, production of myocardial ischemia, unstable angina or MI. Several studies have shown that
lowering of lipid levels through therapies; reduce myocardial ischemia in patients with CAD.
A large frame of evidence has shown that accelerated NO inactivation leads to endothelial dysfunction.
Xanthine oxidase, eNOS and NADH/NADPH oxidase are probable sources which add to amplified
production of reactive oxygen species in pathophysiological states. Genetic linkage studies with single
large families have proven to be an effective approach for the identification of disease causing genes for
CAD and MI. MEF2A has been identified as the first disease causing gene for autosomal dominant CAD
and MI and this has exposed a new signaling pathway for the pathogenesis of CAD and MI and has
associated abnormal development of the endothelium as an initial cause for the disease. Further genetic
studies show promise to revolutionize the early diagnosis, treatment and prevention of CAD and MI. One
of the most important benefits of dealing with CAD is that a substantial number of cases are potentially
avertable. Genetic testing will provide with early diagnosis that might help people into making healthier
lifestyle choices.
Nitric oxide is critical for maintaining vasodilation and blood pressure therefore NO plays an important
role coronary artery disease, it has been suggested that there is reduced bioavailability of NO in CAD; this
impairment in bioavailability of NO is due to endothelial dysfunction. NO formation maybe measured via
the determination of its oxidative products, nitrite and nitrate. However, the reported basal nitrite
concentrations in plasma of humans in the literature range from 'non-detectable' over Nano-molar
(450nmol/l) to micro-molars 26µmols.
Kawakatsu et al found that nitrite/nitrate gradually increased with age in healthy male subjects. In
humans’ endothelium dependent vasodilation is reduced in essential hypertensive patients compared to
normal control subjects, suggesting that endothelial function can be impaired in human hypertension.
Panza et al and Koichi et al reported decreased NO products in patients with CAD and some studies show
that the concentration of nitric oxide is low in women with CAD in prehypertensive phases than
normotensive patients but in these studies the role of estrogen and testosterone was ignored. Calver A et
al showed through their study that altered production of NO was proven in knockout mice and humans
with CAD. Vast numbers of studies have shown variation in mean values of nitric oxide. Node K et al in
fact recorded a very similar (43%) decline in patients as compared to controls. The normal range of NO
showed a wide variation in different studies
Prevention of CAD and decrease of its indisposition and death remains one of the chief health challenges
across the world. Atherosclerosis is presently thought to be as a consequence of difference between pro-
and anti-oxidants that produces lesions to grow towards severe thrombotic difficulties and clinical events.
But over the last decade, a lot of evidence has come forward that shows NO plays an important role in the
CAD range, from its origin to the outcome after critical events.

45 | P a g e
Conclusion

46 | P a g e
The main aim of the project was to find correlation between nitric oxide, ascorbic acid and coronary
artery disease. Concluding from the study undertaken, nitric oxide levels are reduced in people affected
with coronary artery disease as compared to the control samples. Nitric oxide is produced in the body
under conditions of stress and it causes vasodilation. This gas has reduced production in CAD patients
which causes obstruction in blood flow, which might lead to formation of clots or myocardial infarction.
Literature surrounding the effect of ascorbic acid in coronary artery disease is very limited. From the
study conducted, the levels of ascorbic acid were decreased in CAD patients. The genetic mechanism of
this disease is poorly understood, because CAD is a multi-factorial disease and is affected by a number of
characteristics, both environmental and genetic. The data obtained from the study however, was found to
be not statistically significant. Further study in extended sample size is warranted.

47 | P a g e
References

48 | P a g e
1. Antoniades, C., D. Tousoulis, C. Tountas, C. Tentolouris, M. Toutouza, C. Vasiliadou, C.
Tsioufis, P. Toutouzas, and C. Stefanadis. "Vascular endothelium and inflammatory process, in
patients with combined Type 2 diabetes mellitus and coronary atherosclerosis: the effects of
vitamin C."Diabetic medicine 21, no. 6 (2004): 552-558.
2. Bordia, A., and S. K. Verma. "Effect of vitamin C on platelet adhesiveness and platelet
aggregation in coronary artery disease patients." Clinical cardiology 8, no. 10 (1985): 552-554.
3. Borsook, Henry, Horace W. Davenport, Cecil EP Jeffreys, and Robert C. Warner. "The oxidation
of ascorbic acid and its reduction in vitro and in vivo." Journal of Biological Chemistry 117, no. 1
(1937): 237-279.
4. Cannon, Richard O. "Role of nitric oxide in cardiovascular disease: focus on the
endothelium." Clinical Chemistry 44, no. 8 (1998): 1809-1819.
5. Celermajer, David S., Keld E. Sorensen, Catherine Bull, Jacqui Robinson, and John E. Deanfield.
"Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to
coronary risk factors and their interaction." Journal of the American College of Cardiology 24,
no. 6 (1994): 1468-1474.
6. Chandra, Sudhir, Rajiv Narang, Vishnubhatla Sreenivas, Jagriti Bhatia, Daman Saluja, and
Kamna Srivastava. "Association of angiotensin II type 1 receptor (A1166C) gene polymorphism
and its increased expression in essential hypertension: a case-control study." (2014): e101502.
7. Cox, D. A., J. A. Vita, C. B. Treasure, R. D. Fish, R. W. Alexander, P. Ganz, and A. P. Selwyn.
"Atherosclerosis impairs flow-mediated dilation of coronary arteries in humans." Circulation 80,
no. 3 (1989): 458-465.
8. Dawn, Buddhadeb, and Roberto Bolli. "Role of nitric oxide in myocardial preconditioning."
Annals of the New York Academy of Sciences 962, no. 1 (2002): 18-41.
9. Das, Sabari, Dinesh Yadav, Rajiv Narang, and Nibhriti Das. "Interrelationship between lipid
peroxidation, ascorbic acid and superoxide dismutase in coronary artery disease." Current
Science-Bangalore- 83, no. 4 (2002): 488-491.
10. El Shamieh, Said, and Sophie Visvikis-Siest. "Genetic biomarkers of hypertension and future
challenges integrating epigenomics." ClinicaChimicaActa 414 (2012): 259-265.
11. Falk, Erling, Prediman K. Shah, and ValentinFuster. "Coronary plaque
disruption." Circulation 92, no. 3 (1995): 657-671.
12. Foroughmand, Ali Mohammad, Zahra Shahbazi, Hamid Galehdari, Mahdi PurmahdiBorujeni,
ParvaneDinarvand, and KhadijeGolabgirkhademi. "Association of MEF2A Gene Polymorphisms
With Coronary Artery Disease." Iranian Red Crescent Medical Journal 16, no. 8 (2014).
13. Furchgott, Robert F., and John V. Zawadzki. "The obligatory role of endothelial cells in the
relaxation of arterial smooth muscle by acetylcholine." Nature 288 (1980): 373-6.
14. Ganz, Peter, and Joseph A. Vita. "Testing endothelial vasomotor function nitric oxide, a
multipotent molecule." Circulation 108, no. 17 (2003): 2049-2053.
15. Gokce, Noyan, John F. Keaney, BalzFrei, Monika Holbrook, MariuszOlesiak, Benoy J.
Zachariah, ChristiaanLeeuwenburgh, Jay W. Heinecke, and Joseph A. Vita. "Long-term ascorbic
acid administration reverses endothelial vasomotor dysfunction in patients with coronary artery
disease." Circulation 99, no. 25 (1999): 3234-3240.
16. Gordon, J. B., P. Ganz, E. G. Nabel, R. D. Fish, J. Zebede, G. H. Mudge, R. W. Alexander, and
A. P. Selwyn. "Atherosclerosis influences the vasomotor response of epicardial coronary arteries
to exercise." Journal of Clinical Investigation 83, no. 6 (1989): 1946.

49 | P a g e
17. Gryglewski, R. J., R. M. J. Palmer, and S. Moncada. "Superoxide anion is involved in the
breakdown of endothelium-derived vascular relaxing factor." (1986): 454-456.
18. Hansson, Goran K., Jan Holm, and L. Jonasson. "Detection of activated T lymphocytes in the
human atherosclerotic plaque." The American journal of pathology 135, no. 1 (1989): 169.
19. Hansson, Göran K. "Inflammation, atherosclerosis, and coronary artery disease." New England
Journal of Medicine 352, no. 16 (2005): 1685-1695.
20. Hornig, Burkhard, Ulf Landmesser, Christoph Kohler, DorotheAhlersmann, Stephan
Spiekermann, Annemarie Christoph, HelmaTatge, and Helmut Drexler. "Comparative effect of
ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in
patients with coronary artery disease role of superoxide dismutase." Circulation 103, no. 6
(2001): 799-805.
21. Ignarro, Louis J., Georgette M. Buga, Keith S. Wood, Russell E. Byrns, and GautamChaudhuri.
"Endothelium-derived relaxing factor produced and released from artery and vein is nitric
oxide." Proceedings of the National Academy of Sciences 84, no. 24 (1987): 9265-9269.
22. Kovanen, Petri T., MaijaKaartinen, and TimoPaavonen. "Infiltrates of activated mast cells at the
site of coronary atheromatous erosion or rupture in myocardial infarction." Circulation 92, no. 5
(1995): 1084-1088.
23. Levine, Glenn N., BalzFrei, Spyridon N. Koulouris, Marie D. Gerhard, John F. Keaney, and
Joseph A. Vita. "Ascorbic acid reverses endothelial vasomotor dysfunction in patients with
coronary artery disease." Circulation 93, no. 6 (1996): 1107-1113.
24. Ludmer, Paul L., Andrew P. Selwyn, Thomas L. Shook, Richard R. Wayne, Gilbert H. Mudge, R.
Wayne Alexander, and Peter Ganz. "Paradoxical vasoconstriction induced by acetylcholine in
atherosclerotic coronary arteries." New England Journal of Medicine 315, no. 17 (1986): 1046-
1051.
25. Mannucci, Pier Mannuccio, Luca A. Lotta, and Flora Peyvandi. "Genome-wide association
studies in myocardial infarction and coronary artery disease." The journal of Tehran Heart
Center 5, no. 3 (2010): 116.
26. Meister, Alton. "Glutathione-ascorbic acid antioxidant system in animals." Journal of Biological
Chemistry-Paper Edition 269, no. 13 (1994): 9397-9400.
27. Nishikawa, Yasuhiro, and Satoshi Ogawa. "Importance of nitric oxide in the coronary artery at
rest and during pacing in humans." Journal of the American College of Cardiology 29, no. 1
(1997): 85-92.
28. Ohara, Yuichi, Timothy E. Peterson, and David G. Harrison. "Hypercholesterolemia increases
endothelial superoxide anion production." Journal of Clinical Investigation 91, no. 6 (1993):
2546.
29. Price, Kendall D., Catherine SC Price, and Robert D. Reynolds. "Hyperglycemia-induced
ascorbic acid deficiency promotes endothelial dysfunction and the development of
atherosclerosis." Atherosclerosis 158, no. 1 (2001): 1-12.
30. Scheuner, Maren T. "Genetic evaluation for coronary artery disease." Genetics in medicine 5, no.
4 (2003): 269-285.
31. Skålén, Kristina, Maria Gustafsson, Ellen Knutsen Rydberg, LillemorMattssonHultén,
OlovWiklund, Thomas L. Innerarity, and Jan Borén. "Subendothelial retention of atherogenic
lipoproteins in early atherosclerosis." Nature 417, no. 6890 (2002): 750-754.

50 | P a g e
32. Stary, Herbert C., A. Bleakley Chandler, Seymour Glagov, John R. Guyton, W. Insull, Michael E.
Rosenfeld, Sheldon A. Schaffer, Colin J. Schwartz, William D. Wagner, and Robert W. Wissler.
"A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the
Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart
Association." Arteriosclerosis, Thrombosis, and Vascular Biology 14, no. 5 (1994): 840-856.
33. Stary, Herbert C., A. Bleakley Chandler, Robert E. Dinsmore, ValentinFuster, Seymour Glagov,
William Insull, Michael E. Rosenfeld, Colin J. Schwartz, William D. Wagner, and Robert W.
Wissler. "A definition of advanced types of atherosclerotic lesions and a histological
classification of atherosclerosis A report from the Committee on Vascular Lesions of the Council
on Arteriosclerosis, American Heart Association." Circulation 92, no. 5 (1995): 1355-1374.
34. Sun, Jie, Xueji Zhang, Mark Broderick, and Harry Fein. "Measurement of nitric oxide production
in biological systems by using Griess reaction assay." Sensors 3, no. 8 (2003): 276-284.
35. Topol, Eric J., Jonathan Smith, Edward F. Plow, and Qing K. Wang. "Genetic susceptibility to
myocardial infarction and coronary artery disease." Human Molecular Genetics 15, no. suppl 2
(2006): R117-R123.
36. Tousoulis, Dimitris, CharalambosAntoniades, CharalambosTountas, EriniBosinakou, Maria
Kotsopoulou, PavlosToutouzas, and ChristodoulosStefanadis. "Vitamin C affects
thrombosis/fibrinolysis system and reactive hyperemia in patients with type 2 diabetes and
coronary artery disease." Diabetes care 26, no. 10 (2003): 2749-2753.
37. Vasa, Mariuca, Stephan Fichtlscherer, Klaudia Adler, Alexandra Aicher, Hans Martin, Andreas
M. Zeiher, and Stefanie Dimmeler. "Increase in circulating endothelial progenitor cells by statin
therapy in patients with stable coronary artery disease." Circulation 103, no. 24 (2001): 2885-
2890.
38. Wang, Qing. "Molecular genetics of coronary artery disease." Current opinion in cardiology 20,
no. 3 (2005): 182.
39. Weng, Li, NihanKavaslar, Anna Ustaszewska, Heather Doelle, Wendy Schackwitz, Sybil Hébert,
Jonathan C. Cohen, Ruth McPherson, and Len A. Pennacchio. "Lack of MEF2A mutations in
coronary artery disease." Journal of Clinical Investigation 115, no. 4 (2005): 1016.
40. Williams, Stephen B., Jorge A. Cusco, Mary-Anne Roddy, Michael T. Johnstone, and Mark A.
Creager. "Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent
diabetes mellitus." Journal of the American College of Cardiology 27, no. 3 (1996): 567-574.
41. Wolfram, Julie A., and J. Kevin Donahue. "Gene therapy to treat cardiovascular disease." Journal
of the American Heart Association 2, no. 4 (2013): e000119.

51 | P a g e
Synopsis

52 | P a g e
Synopsis
Inter-relationship of Nitric Oxide and Ascorbic Acid
at the RNA level in patients with Coronary Artery
Disease
Submitted by
Priya Joshi
131702016
M.Sc. Medical Biotechnology
2nd
Year, 4th
Semester
Manipal University
Under the guidance of
Dr. Kamna Srivastava
Assistant Professor
Dr. B. R. Ambedkar Centre for Biomedical Research
University of Delhi, Delhi
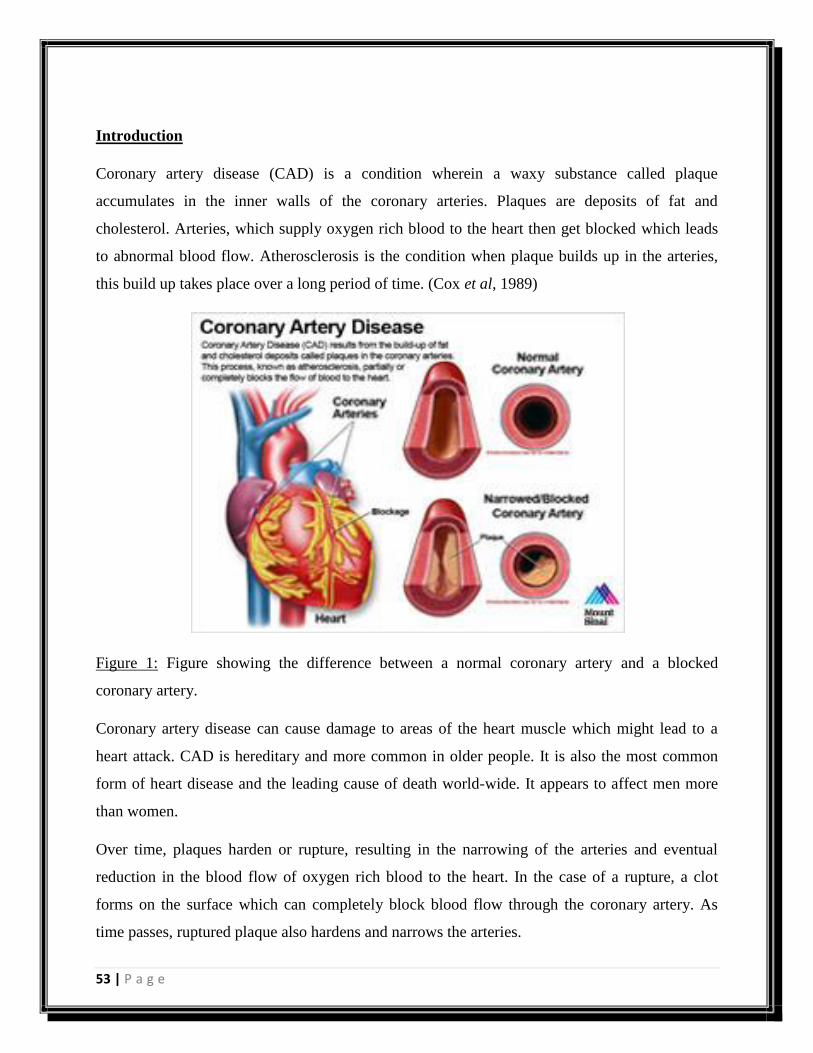
53 | P a g e
Introduction
Coronary artery disease (CAD) is a condition wherein a waxy substance called plaque
accumulates in the inner walls of the coronary arteries. Plaques are deposits of fat and
cholesterol. Arteries, which supply oxygen rich blood to the heart then get blocked which leads
to abnormal blood flow. Atherosclerosis is the condition when plaque builds up in the arteries,
this build up takes place over a long period of time. (Cox et al, 1989)
Figure 1: Figure showing the difference between a normal coronary artery and a blocked
coronary artery.
Coronary artery disease can cause damage to areas of the heart muscle which might lead to a
heart attack. CAD is hereditary and more common in older people. It is also the most common
form of heart disease and the leading cause of death world-wide. It appears to affect men more
than women.
Over time, plaques harden or rupture, resulting in the narrowing of the arteries and eventual
reduction in the blood flow of oxygen rich blood to the heart. In the case of a rupture, a clot
forms on the surface which can completely block blood flow through the coronary artery. As
time passes, ruptured plaque also hardens and narrows the arteries.

54 | P a g e
In the case where flow of oxygen rich blood is reduced or blocked angina or heart attack can
occur. Angina is the feeling of discomfort in the chest, which might give the feeling of immense
pressure or squeezing. The pain may also present in the shoulder, back, arms, jaw as referral
pain. CAD weakens the heart muscle and leads to heart failure and arrhythmias. Heart failure
occurs when the heart cannot pump enough blood to meet the needs of the body, whereas
arrhythmias are problems related to the rhythm of the heartbeat. (Gordon et al, 1989)
Risk factors involved in CAD are:
1. obesity,
2. high blood pressure,
3. high cholesterol
4. diabetes,
5. smoking and
6. physical inactivity etc.
Various molecular factors are also said to be involved which increase the susceptibility
towards the development of coronary artery disease. However the studies carried out on
them is not too extensive.
Disease symptoms and diagnosis
The disease develops without many obvious signs but a few symptoms could develop that could
lead to a heart attack. For example:
1. Angina
2. Shortness of breath
3. Sweating
4. Nausea
5. Weakness
6. Fatigue
7. Cold sweats
8. Pressure in the chest and the middle of the back
Diagnostic tests to detect if a person is experiencing angina or heart attack include blood tests,
ECG, echocardiogram, stress test and coronary angiography.

55 | P a g e
Nitric oxide (NO)
Nitric oxide was detected by Furchgott and Zawadzki when it was seen that the relaxation of
smooth muscle by acetylcholine was dependent on the presence of endothelium. (Ludmeret al,
1986) It gave evidence that relaxation was caused by the release of humoral factors which is also
known as endothelium-derived relaxing factor (EDRF). (Celermajeret al, 1994) Further
investigation helped in concluding that EDRF is actually NO. The benefits of exercise are that it
releases nitric oxide into the circulation through the activation of endothelial NO synthase
(eNOS) due to increase in the stress to the endothelium. NO is produced in the endothelial cells
from the amino acid L-arginine, from where is progresses to locally diffuse to the smooth
muscle. When eNOS is activated, NO is produced in the endothelium. Oxidation of L-arginine
by the action of eNOS leads to the production of NO and L-citrulline, the amino acid is
enzymatically converted to NO when its guanidine nitrogen is oxidized. (Schmidt et al, 1994)
In an alternate hypothesis, it was proposed that NO is made and stored in the vascular endothelial
cells and that the interaction of endothelium-dependent vasodilators with specific endothelial cell
membrane receptors triggers NO exocytosis in a calcium and oxygen dependent manner.
(Ignarroet al, 1989)

56 | P a g e
Figure 2: The mechanism of action of nitric oxide
NO is a powerful vasodilator and has anti-atherosclerotic properties, it acts on the plaques
formed inside the lumen of blood vessels to reduce them. It also reduces the production of tissue
factor which is crucially involved in defining the volume of accumulated atherosclerotic plaques
tha cause intravascular thrombosis. (Ganzet al, 2003) NO also inhibits platelet aggregation,
leukocyte adhesion and smooth muscle proliferation. (Miyazaki et al, 1999) It is also known to
have direct anti-inflammatory properties due to its effect on vascular tone, platelet aggregation
and leucocyte interactions with the endothelium. NO is known to be beneficial in limiting the
effects of ischemia (restriction of blood supply). Its protective effect is due to the dilation of
micro vessels or inhibition of the platelet adherence to the endothelial cells. (Dawn et al, 2002)
Atherosclerosis is much more common in individuals with Diabetes because, Diabetes causes a
reduction in the available NO that is derived from the endothelium. Clear abnormality is
observed in the NO pathway of patients with non-insulin dependent diabetes mellitus. (Williams

57 | P a g e
et al, 1996) The vasodilating capability of NO gives it the ability to increase coronary blood
flow, which is useful when it comes to diseases such as angina pectoris, heart infarct or coronary
vasopasm. (Vasaet al, 2001)
Ascorbic acid
Ascorbic acid (Vitamin C) is the main water soluble antioxidant in the plasma. It controls
intracellular redox state through its interaction with glutathione. (Meister et al, 1994) It is
required to maintain skin, teeth, bone, blood vessels etc. In atherosclerosis, the endothelial
vasomotor function is abnormal. Ascorbic acid reverses this dysfunction in the brachial
circulation of patients with coronary artery disease. (Levineet al, 1996)Vascular superoxide is
produced in large amounts when nitric oxide is inactivated and there is a loss in the endothelium
dilation. (Oharaet al, 1993; Gryglewskiet al, 1986)
Objectives of the study
1. To determine the levels of nitric oxide in patients with coronary artery disease and normal
healthy volunteers.
2. To determine the levels of ascorbic acid in patients with coronary artery disease and normal
healthy volunteers
3. To determine the amount of total RNA in patients with coronary artery disease and normal
healthy volunteers
4. To study the interrelationship of Nitric oxide, Ascorbic acid and total RNA in coronary artery
disease
Materials and Methods
The present study is a hospital based case and control study. The study subjects will be patients
with coronary artery disease (N=25) and Normal healthy volunteers (N= 25) of similar age group
and socioeconomic geographical background.
The present study is a part of project of Principal Investigator, Dr. Kamna Srivastava, approved
by Human ethics committee of ACBR-DU and AIIMS, New Delhi.
The methodology adopted in brief will be:

58 | P a g e
1. Collection of blood samples from the patients and controls and their plasma isolation.
2. Total RNA will be isolated from the whole blood samples using the trizol method. The
concentration of RNA will be measured using nano-drop.
3. The concentration of nitric oxide using Griess reagent
4. The concentration of ascorbic acid will be measured by dTCS reagent.
5. Statistical analysis will be performed to study the interrelationship of the parameters studied.
Expected outcome
The objective of the study is to estimate the levels of nitric oxide and ascorbic acid in patients
with coronary heart disease. The study aims to look into the levels of nitric oxide and ascorbic
acid produced in the body in the case of coronary artery disease, and to measure if it is over
expressed or under expressed. Also to co-relate how the levels of these two components are
associated with the progression of the CAD.The present study will be helpful in the
understanding and management of the coronary artery disease.
References
1. Celermajer, David S., Keld E. Sorensen, Catherine Bull, Jacqui Robinson, and John E.
Deanfield. "Endothelium-dependent dilation in the systemic arteries of asymptomatic
subjects relates to coronary risk factors and their interaction."Journal of the American
College of Cardiology 24, no. 6 (1994): 1468-1474.
2. Cox, D. A., J. A. Vita, C. B. Treasure, R. D. Fish, R. W. Alexander, P. Ganz, and A. P.
Selwyn. "Atherosclerosis impairs flow-mediated dilation of coronary arteries in
humans." Circulation 80, no. 3 (1989): 458-465.
3. Dawn, Buddhadeb, and Roberto Bolli. "Role of nitric oxide in myocardial
preconditioning." Annals of the New York Academy of Sciences 962, no. 1 (2002): 18-41.
4. Ganz, Peter, and Joseph A. Vita. "Testing endothelial vasomotor function nitric oxide, a
multipotent molecule." Circulation 108, no. 17 (2003): 2049-2053.
5. Gordon, J. B., P. Ganz, E. G. Nabel, R. D. Fish, J. Zebede, G. H. Mudge, R. W.
Alexander, and A. P. Selwyn. "Atherosclerosis influences the vasomotor response of

59 | P a g e
epicardial coronary arteries to exercise." Journal of Clinical Investigation 83, no. 6
(1989): 1946.
6. Gryglewski, R. J., R. M. J. Palmer, and S. Moncada. "Superoxide anion is involved in the
breakdown of endothelium-derived vascular relaxing factor." (1986): 454-456.
7. Ignarro, Louis J., Georgette M. Buga, Keith S. Wood, Russell E. Byrns, and
GautamChaudhuri. "Endothelium-derived relaxing factor produced and released from
artery and vein is nitric oxide." Proceedings of the National Academy of Sciences 84, no.
24 (1987): 9265-9269.
8. Levine, Glenn N., BalzFrei, Spyridon N. Koulouris, Marie D. Gerhard, John F. Keaney,
and Joseph A. Vita. "Ascorbic acid reverses endothelial vasomotor dysfunction in
patients with coronary artery disease." Circulation 93, no. 6 (1996): 1107-1113.
9. Ludmer, Paul L., Andrew P. Selwyn, Thomas L. Shook, Richard R. Wayne, Gilbert H.
Mudge, R. Wayne Alexander, and Peter Ganz. "Paradoxical vasoconstriction induced by
acetylcholine in atherosclerotic coronary arteries."New England Journal of Medicine 315,
no. 17 (1986): 1046-1051.
10. Meister, Alton. "Glutathione-ascorbic acid antioxidant system in animals."Journal of
Biological Chemistry-Paper Edition 269, no. 13 (1994): 9397-9400.
11. Miyazaki, Hiroshi, Hidehiro Matsuoka, John P. Cooke, Michiaki Usui, Seiji Ueda, Seiya
Okuda, and Tsutomu Imaizumi. "Endogenous nitric oxide synthase inhibitor a novel
marker of atherosclerosis." Circulation 99, no. 9 (1999): 1141-1146.
12. Ohara, Yuichi, Timothy E. Peterson, and David G. Harrison. "Hypercholesterolemia
increases endothelial superoxide anion production."Journal of Clinical Investigation 91,
no. 6 (1993): 2546.
13. Schmidt, Harald HHW, and Ulrich Walter. "NO at work." cell 78, no. 6 (1994): 919-925.
14. Vasa, Mariuca, Stephan Fichtlscherer, Klaudia Adler, Alexandra Aicher, Hans Martin,
Andreas M. Zeiher, and Stefanie Dimmeler. "Increase in circulating endothelial
progenitor cells by statin therapy in patients with stable coronary artery
disease." Circulation 103, no. 24 (2001): 2885-2890.
15. Williams, Stephen B., Jorge A. Cusco, Mary-Anne Roddy, Michael T. Johnstone, and
Mark A. Creager. "Impaired nitric oxide-mediated vasodilation in patients with non-

60 | P a g e
insulin-dependent diabetes mellitus." Journal of the American College of Cardiology 27,
no. 3 (1996): 567-574.
Recommended











