
Nephrogenic Diabetes Insipidus in Children
Nine V. A. M. Knoersa* and Elena N. LevtchenkobaDepartments of Medical Genetics, University Medical Centre Utrecht, Utrecht, The NetherlandsbDepartment of Pediatric Nephrology, Department of Growth and Regeneration, University Hospitals Leuven,Katholieke Universiteit Leuven, Leuven, Belgium
History
First familial cases with diabetes insipidus were described byMcIlraith in 1892; however, he did notdistinguish between renal and neurohormonal forms of the disorder [1]. The renal type of diabetesinsipidus was appreciated as a separate entity more than 50 years ago, when it was describedindependently by two investigators: Forssman [2] in Sweden and Waring et al. [3] in the UnitedStates. In 1947, Williams and Henry [4] noticed that injection of antidiuretic hormone (ADH) indoses sufficient to induce systemic side effects could not correct the renal concentrating defect. Theycoined the term nephrogenic diabetes insipidus. Subsequent studies revealed active hormone in theserum and urine of affected persons and lent further support to the theory of renal unresponsivenessto vasopressin. Nephrogenic diabetes insipidus is synonymous with the terms vasopressin- or ADH-resistant diabetes insipidus and diabetes insipidus renalis.
Definition and Clinical Manifestations
Congenital nephrogenic diabetes insipidus (NDI) is a rare inherited disorder, characterized byinsensitivity of the distal nephron to the antidiuretic effects of the neurohypophyseal hormonearginine vasopressin (AVP). As a consequence, the kidney loses its ability to concentrate urine,which may lead to severe dehydration and electrolyte imbalance (hypernatremia andhyperchloremia). Patients with NDI have normal birth weight and pregnancies are not complicatedby polyhydramnios. The urine-concentrating defect in NDI is present from birth, and manifestationsof the disorder generally emerge within the first weeks of life. With breast milk feedings, infantsusually thrive and do not develop signs of dehydration. This is because human milk has a low saltand protein content and therefore a low renal osmolar load. With cows’ milk formula feedings, theosmolar load to the kidney increases, resulting in an increased demand for free water. This is usuallynot provided by oral feeding, and therefore hypernatremic dehydration appears. Irritability, poorfeeding, and poor weight gain are usually the initial symptoms [5]. Patients are eager to suck but mayvomit during or shortly after the feeding. Dehydration is evidenced by dryness of the skin, loss ofnormal skin turgor, recessed eyeballs, increased periorbital folding, depression of the anteriorfontanel, and a scaphoid abdomen. Intermittent high fever is a common complication of thedehydrated state, predominantly in very young children. Body temperature can be normalized byrehydration. Seizures can occur but are rare and most often seen during therapy, particularly ifrehydration proceeds too rapidly. Constipation is a common symptom in childrenwith NDI. Nocturiaand nocturnal enuresis are common complaints later in childhood.
*Email: [email protected]
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 1 of 25

Untreated, most patients fail to grow normally. In a retrospective study of 30 male NDI patients,most children grew below the 50th percentile, most of them having standard deviation (SD) scoreslower than�1 [6]. Some well-treated patients, however, may achieve normal adult height. Catch-upgrowth occurs at least in some patients after normalization of water and electrolyte balance,especially in those with adherence to treatment. Bone maturation is generally not delayed [7].Weight-for-height SD scores are initially low, followed by global normalization at schoolage [6]. Initial feeding problems and the ingestion of large amounts of low-caloric fluid resultingin a decreased appetite may play roles in failure to thrive seen in NDI [8, 9]. Furthermore, it ispossible that repeated episodes of dehydration have some as yet undetermined negative effects ongrowth.
Mental retardation has long been considered an important complication of untreated NDI andassumed to be a sequel of recurrent episodes of severe brain dehydration and cerebral edema causedby overzealous attempts at rehydration [10–12]. Additional evidence underscoring the assumptionthat NDI has adverse effects on the cerebrum is provided by several reports describing intracranialcalcifications in NDI patients [13, 14]. Such lesions are generally considered to be the result ofhemorrhage or necrosis. Most of the reported patients with cerebral calcifications were mentallyretarded. Nowadays mental retardation is rare due to earlier recognition and treatment of NDI. Exactestimates of the current frequency of mental retardation under modern treatment are unknown, but inthe largest psychometric study ever reported, only 2 of the 17 male NDI patients (aged 3–30 years)had a total intelligence quotient more than 2 SD below the norm. Fourteen patients had anintelligence score within or above the normal range and one patient had a general index scorebetween �1 and �2 SD [15].
The psychological development of NDI patients is influenced by a persistent desire for drinkingand the need for frequent voiding, which compete with playing and learning. Therefore, many NDIpatients are characterized by hyperactivity, distractibility, short attention span, and restlessness. Inthe psychometric study mentioned earlier, the criteria for attention-deficit/hyperactivity disorderwere met in 8 of 17 tested NDI patients [15].
Persistent polyuria can result in the development of megacystis, trabeculated bladder wall,hydroureter, and hydronephrosis [6, 16, 17]. Urinary tract distension may be seen on ultrasoundexamination even in infants [18] and young children [19]. Potential complications of urinary tractdilatation are rupture of the urinary tract, infection, intractable pain, improper bladder function,and/or kidney failure. These complications may occur as early as the second decade of life. Large-capacity hypotonic bladder dysfunction might require clean intermittent catheterization[17]. Patients should be trained to void regularly in order to assure that maximal urinary bladdercapacity remains within normal range. Both patient groups with AVPR2 and AQP2 mutations candevelop urinary tract dilatation and bladder dysfunction [16, 20].
Diagnostic Procedures
The observation of polyuria in a dehydrated infant, together with the finding of a high serum sodiumconcentration and inappropriately diluted urine (Uosm < Posm), provides presumptive evidencefor a renal concentrating defect. To confirm the concentrating defect and to distinguish the renal formof diabetes insipidus from the central form, a vasopressin test is performed with 1-desamino-8-D-arginine vasopressin (DDAVP), a synthetic analogue of the natural arginine vasopressin that
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 2 of 25

produces a high and prolonged antidiuretic effect. In the test, DDAVP (10 mg for infants<1 year old,20 mg for children>1 year old) is administered intranasally. Urine is collected during the subsequent5.5 h. The first collected portion of the urine should be discarded. The maximal urine osmolality inany collected aliquot is chosen as a measure of the concentrating capacity [21]. After DDAVPadministration, NDI patients are (1) unable to increase urinary osmolality, which remains below200 mOsm/kg H2O (normal values: <1 year old>600, between 1 and 2 years old between 600 and800, >2 years old >800 mOsm/kg H2O), and (2) cannot reduce urine volume or free-waterclearance.
Plasma vasopressin levels are normal or only slightly increased in affected children. Otherlaboratory findings have been described, which mainly result from chronic dehydration. Serumsodium concentration is generally elevated and may be above 170 mmol/L. There is also anincrease in serum chloride concentration and retention of urea and creatinine. All values arenormalized by adequate rehydration. In addition, reduced glomerular filtration rate (GFR) andrenal blood flow can return to normal when a normal hydration state has been achieved.
The primary congenital form of NDI has to be differentiated from central diabetes insipidus (dueto lack of AVP) and from the secondary or acquired forms, which are much more common [22].In our experience, the urinary osmolality obtained after DDAVP administration in secondarydisorders is always higher than in NDI. Several secondary causes, some of which will be discussedlater, are listed in Table 1.
Table 1 Causes of secondary nephrogenic diabetes insipidus
Monogenetic diseases associated with secondary NDI
Renal Fanconi syndromes
Bartter syndrome (type 1 or type 2)
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis
Distal renal tubular acidosis (dRTA)
Apparent mineralocorticoid excess (AME)
Ciliopathies (nephronophthisis, Bardet–Biedl syndrome, etc.)
Other renal diseases
Obstructive uropathy
Renal dysplasia
Postischemic damage
Amyloidosis
Sarcoidosis
Chronic renal failure
Renal impairment in sickle-cell disease or trait
Drug induced
Lithium
Ifosfamide
Amphotericin B
Tetracyclines
Biochemical abnormalities
Hypercalcemia, hypercalciuria, and nephrocalcinosis
Hypokalemia
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 3 of 25

Cellular Physiology of Arginine Vasopressin’s Antidiuretic Action in theDistal Nephron
The physiologic action of vasopressin on the renal collecting duct has been one of the mostintensively studied processes in the kidney. Arginine vasopressin (AVP, ADH) is synthesized onthe ribosomes of the magnocellular neurons of the supraoptic and paraventricular nuclei of thehypothalamus as a large biologically inactive bound form. Within storage granules, the hormone iscleaved into the biologically active form and transported down the neuronal axons to the posteriorpituitary and stored there. Following appropriate stimuli, AVP is secreted from the posterior pituitaryinto the circulation as biologically active hormone. AVP release is regulated by changes in plasmaosmolality (by >2 %) but can also occur in response to nonosmotic stimuli. These nonosmoticstimuli are generally related to changes in either total blood volume or the distribution of extracel-lular fluid. Patients with depleted effective circulating volume may secrete ADH even in thepresence of low plasma osmolality. In addition, physical pain, emotional stress, and certain drugs(e.g., nicotine) influence the release of AVP. In its effector organ, the kidney, AVP binds tovasopressin type-2 (V2) receptors on the basolateral membrane of the principal inner medullarycollecting duct cells and of the arcade cells (Fig. 1, review in Ref. [23]). The arcades are long, highlybranched renal tubule segments that connect distal convoluted tubules of several deep andmidcortical nephrons to the origin of cortical collecting ducts. Upon binding of AVP, the V2 receptoris activated and then stimulates GTP loading of the small GTPase-aGS – subunit of its coupledtrimeric G-protein – eventually leading to dissociation of the G-protein from the receptor. GTP-aGS
can then bind to the membrane-associated adenylate cyclase (AC), activating it, which results in anincrease in intracellular cyclic adenosine monophosphate (cAMP) from adenosine triphosphate(ATP). The elevated cAMP levels stimulate protein kinase A (PKA), leading to phosphorylationof AQP2 which in turn initiates a redistribution of aquaporin-2 (AQP2) water channels fromintracellular vesicles to the apical plasma membrane, rendering this membrane water permeable.The increase in apical membrane permeability allows water to flow from the tubule lumen to thehypertonic medullary interstitium, via AQP2 in the apical membrane and via AQP3 and AQP4,constitutive water channels in the basolateral membrane. This then leads to the formation ofconcentrated urine. Upon fluid intake, AVP release into the blood decreases, AQP2 is redistributedinto intracellular vesicles, and water reabsorption is reduced. Katsura et al. have shown that theAVP-regulated recycling of AQP2 can occur at least six times with the same molecules [24].
In recent years, our knowledge of the AQP2 dynamics in the cell has increased significantly(Fig. 1). For further details the reader is also referred to several excellent reviews on this subject(review in Refs. [23, 25–28]).
AQP2 is 1 of the 13members of the aquaporin family of water channels. After transcription AQP2is folded into its native monomeric conformation in the endoplasmic reticulum, and homotetra-merization takes place [29]. The tetramers are forwarded to the Golgi apparatus, where two out offour monomers are complex N-glycosylated. These functional water channels are then stored inendosomal vesicles to be transported to the apical membrane [30].
Phosphorylation of a PKA-consensus site in AQP2, the serine at position 256 in the cytoplasmiccarboxy-terminus, is absolutely essential for AQP2 delivery to the apical membrane [31, 32]. Inaddition, it has been shown that anchoring of PKA to PKA-anchoring proteins (AKAPs), whichensures targeting of PKA to AQP2-bearing vesicles, is another prerequisite for AVP-mediated AQP2translocation [33]. Studies using oocytes as a model system indicated that for plasma membranelocalization three out of four monomers in an AQP2 tetramer need to be phosphorylated [34].
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 4 of 25

PKA is the main kinase for AQP2 phosphorylation, but other kinases may potentially participatein the regulation of AQP2 trafficking. Besides PKA sites, putative phosphorylation sites for PKG,PKC, and casein kinase II are also present in the AQP2 sequence.
The molecular machinery for the docking and fusion of AQP2-containing vesicles with the apicalmembrane is similar to the process of synaptic vesicle fusion with the presynaptic membrane andinvolves vesicle (v) SNAREs (soluble NSF attachment protein receptors) and target membrane(t) SNAREs. The apical membrane-specific t-SNARE is syntaxin 4, which interacts specifically with
Fig. 1 Intracellular signal transduction pathway initiated by AVP binding to V2R. Via activation of adenylate cyclaseand cAMP-production stimulation, PKA is activated and phosphorylates its target proteins AQP2, Rho-GDI, andCREB-1. The transcription factor CREB-1-p stimulates AQP2 transcription, Rho-GDI-p initiates actin reorganizationrequired for AQP2 transport, and AQP2-p homotetramers are transported to the apical membrane. There they render themembrane permeable for water, which is reabsorbed from the passing pro-urine and transported back into thebloodstream by AQP3 and AQP4. Rab5-mediated AQP2 endocytosis by clathrin-coated vesicles is triggered byshort-chain ubiquitination and leads to termination of the response. Internalized AQP2 vesicles are transported toearly and late endosomes as well as multivesicular bodies (MVBs) for storage. From MVBs they can then either belysosomally degraded or recycled via the Rab11-dependent slow-recycling pathway (From Ref. [23], with kindpermission from Springer Science and Business Media)
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 5 of 25

the v-SNARE protein VAMP2 located on the cytoplasmic side of AQP2-containing endosomalvesicles [35–38]. V- and t-SNAREs are recycled by the AAA-type ATPase NSF. Reorganization ofthe actin cytoskeleton is another important mechanism required for AQP2 transport and accumula-tion at the apical membrane [39, 40]. The actin cytoskeleton most likely provides a network thatanchors the AQP2-bearing vesicles in the unstimulated cell. Vasopressin has been shown todepolymerize apical F-actin in rat inner medullary collecting duct, resulting in the fusion ofAQP2-carrying vesicles with the apical membrane [41], indicating that reorganization of the apicalactin network may be critical in promoting the trafficking of AQP2-bearing vesicles. Rho inhibitionthrough PKA-mediated phosphorylation of Rho-GDP dissociation inhibitor (Rho-GDI) is shown tobe a key event for actin reorganization inducing AQP2 translocation [39, 40].
Counterbalancing increased expression on the plasma membrane, AQP2 is internalized. Duringthis endocytotic process, AQP2 accumulates in clathrin-coated pits and is internalized via a clathrin-mediated process [42]. Endocytosis is regulated by short-chain ubiquitylation at lysine 270 (K270)in the AQP2 terminal tail [43, 44].
To be available for recycling, AQP2-containing endosomes need to be redistributed to theperinuclear region. This process is mediated by dynein-dependent transport along microtubules[45, 46]. Specificity of the endocytotic AQP2 internalization is mediated by Rab5 protein, aneffector-binding factor involved in plasma membrane-to-early-endosome transport [47]. From theendosomal system – early/late endosomes and/or multivesicular bodies (MVBs) – AQP2 is eitherrecycled by the Rab11-dependent slow-recycling pathway or marked for lysosomal degradation[48]. Prolonged K270 ubiquitylation induces MVB trafficking and localization to internal vesiclesof MVBs followed by lysosomal degradation, while deubiquitylation increases localization to earlyendosomes and the limiting membrane of MVBs and enables AQP2 recycling [44].
Long-term adaptation to circulating AVP levels, for instance, in a dehydrated state, is accom-plished by increasing the expression of AQP2 mRNA and protein. PKA-mediated phosphorylationof a cAMP-responsive element-binding protein 1(CREB-1) stimulates synthesis of AQP2 bybinding to the AQP2 gene promotor and activating its transcription, which increases intracellularAQP2 levels [49].
Genetics
Three different inheritance patterns of NDI have been recognized. In most cases (about 90 %), NDIis transmitted as an X-linked recessive trait (MIM304800). In these families, female carriers who areusually unaffected transmit the disease to sons, who display the complete clinical picture [2, 4,50]. In 1988, the major NDI locus was mapped to the distal region of the long arm of theX chromosome (Xq28) [51], and in 1992 mutations in the AVPR2 gene were shown to underlieX-linked NDI [52–54]. In a minority of families (about 10 %), the transmission and phenotypiccharacteristics of NDI are not compatible with an X-linked trait. In these families, females displaythe complete clinical picture of NDI and are clinically undistinguishable from affected male familymembers [55–57]. Family pedigrees suggested the existence of both an autosomal recessive (MIM222000) and an autosomal dominant form (MIM 125800) of NDI. It was subsequently demonstratedthat both autosomal forms of NDI are caused by mutations in the AVP-sensitive aquaporin-2 waterchannel [58, 59]. The prevalence of NDI is not exactly known, but the disease is assumed to be rare.The estimate of the prevalence of NDI in Quebec, Canada, is 8.8:1,000,000 males [60]. In the Dutchpopulation of about 16 million, at least 50 different families are known.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 6 of 25

X-Linked Nephrogenic Diabetes Insipidus: Mutations in the AVPR2 Gene
The X-linked form of NDI is caused by inactivating mutations in the AVPR2 gene (MIM 300538)([52–54], reviews in Refs. [23, 28, 61]). AVPR2 is a relatively small gene, consisting of three exonsseparated by two short intervening sequences (introns); two isoforms are known that are generatedby alternative splicing [62]. AVPR2 is localized on the X chromosome on locus Xq28. The cDNAencodes a receptor protein of 371 amino acids, has a predicted molecular mass of approximately40 kDa, and shares the general structure of a G-protein-coupled receptor consisting of sevenhydrophobic transmembrane helices, connected by extracellular and intracellular loops. The recep-tor contains one unique consensus sequence site for N-linked glycosylation in the extracellularamino-terminus [63] and phosphorylation sites for G-protein-coupled receptor kinases (GRK)represented by a serine cluster in the carboxy-terminus [64, 65]. The N-terminal part of the proteinincluding the first transmembrane domain and the positively charged first intracellular loop areimportant for proper insertion and orientation in the membrane [66]. A conserved glutamate-dileucine motif in the intracellular carboxy-terminal part of the receptor is essential for receptortransport from the endoplasmic reticulum (ER) to the Golgi apparatus [67]. Two conserved adjacentcysteines in the C-terminus are palmitoylated, thereby anchoring the carboxy tail to the plasmamembrane and controlling the tertiary structure of this region of the receptor [68].
At this writing, more than 240 distinct disease-causing mutations in AVPR2 have been identified,and the number is constantly increasing ([69, 70] and review in Refs. [23, 28, 61] and www.hgmd.org). The mutations are not clustered in one domain of the V2 receptor but are scattered throughoutthe protein, except for the part coding for the N- and C-terminal tails of the receptor. More than 50 %of the mutations are missense mutations. Nucleotide deletions and insertions causing frameshifts(26 %), nonsense mutations (13 %), large deletions (7 %), large in-frame insertions/duplications(1 %), splice-site mutations (1 %), and complex rearrangements (2 %) account for the remainder ofmutations ([12, 69], and review in Refs. [23, 28, 61, 71]). In addition to these disease-causingmutations, at least 21 AVPR2 variations that do not lead to disease are known. These non-disease-causing variations are most likely polymorphisms that can be found in more than 1 % of thepopulation. The G12E mutation, for example, has also been found in non-affected individuals,suggesting that it belongs to this class of polymorphisms that do not exert a significant effect onproper functioning of the V2 receptor [72]. Several mutations are recurrent as evidenced by the factthat these mutations were found on different haplotypes in ancestrally independent families. Themost frequent of these recurrent mutations (D85N, R106C, R113W, R137H, S167L, and R337X)occur at potential mutational hot spots.
AVPR2 mutations seem to be present in all ethnical groups tested with no preference of onemutation for any ethnic group over others.
The molecular mechanism underlying the renal insensitivity for AVP differs between mutants. Asupcoming pharmacological treatments for NDI likely depend on the underlying mechanism, GPCRmutations in general and V2 mutations in particular have been divided in different classes accordingto their cellular fate [73, 74].
Class I comprises all mutations that lead to improperly processed or unstable mRNA, likepromoter alterations, exon skipping, or aberrant splicing. This class also holds frameshift andnonsense mutations, which result in truncated proteins like W71X, 458delG (frameshift, 161X),and R337X.
Class II mutations are missense or insertions/deletions of one or more nucleotide triplets, resultingin fully translated proteins. Due to the mutation, however, mutant receptors are misfolded andretained in the endoplasmic reticulum (ER), as the ER is the organelle that has the cellular quality
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 7 of 25

control over proper folding and maturation of synthesized proteins. Misfolded proteins are subse-quently mostly targeted for proteasomal degradation [75]. Intracellular entrapment of missense V2Rmutants and their rapid degradation likely represents the most important cause of NDI, as more than50 % of the mutations in V2R are missense mutations and cellular expression revealed that most ofthese result in ER-retained proteins. The amount of retention and degradation varies within thisclass, since different mutations affect protein folding to a different extent, sometimes allowingpartial transport of at least partially active receptors to the plasma membrane [76].
Class III mutations result in full-length receptors expressed at the cell surface, but interfere withproper interaction with their natural ligands, thereby causing reduced/abolished signaling [77]. ClassIII mutations can be subdivided into two minor groups. IIIa mutations interfere with binding of orsignal transduction to the coupled trimeric G-protein, leading to a reduced activation of adenylatecyclase and thus formation of cAMP. Mutations in this group are missense mutations and in-framedeletions, mostly located in transmembrane and intracellular domains. Examples are the D85N andP322S mutations [78]. IIIb mutations interfere with, or reduce, AVP binding. These mutations,which are also mostly missense and small in-frame deletions or insertions, especially involveresidues thought to be in or close to the AVP-binding pocket, of which delR202 is a clearexample [79].
Finally, class IV is assigned to all mutations that neither interfere with protein synthesis ofmaturation, not with ligand binding, but affect other aspects of protein function. The NDI R137Hmutation, located in the well-conserved DRY/H motif of GPCRs, is the best-characterized exampleof this class. The effect of this mutation is constitutive internalization of V2, leading to reducedexpression of the receptor in the plasma membrane and thereby reduced adenylate cyclase-dependent cAMP signaling upon AVP binding [80, 81].
Sometimes, mutants do not exert a full phenotype of a particular class and then often also showfeatures of another class. For example, some V2R missense mutants are partially ER retained(class II), but are also partially expressed in the plasma membrane, where they might show a reducedG-protein coupling (class IIIa) or AVP binding (class IIIb). As such, it provides an explanation forthe observed small antidiuretic response to high doses of DDAVP in NDI patients harboring suchmutations [82].
Genotype-Phenotype Correlations in X-Linked NDIAlmost all mutations in the V2 receptor gene result in a uniform clinical NDI phenotype withpolyuric manifestations in the first weeks of life and poor growth. There are, however, a fewexceptions to this rule. Several mutations appear to be associated with a milder form of NDI,characterized by a later manifestation, not at birth but later in childhood, and without growthretardation. Examples of mutations causing partial NDI are D85N, V88M, G201D, M311V,N317S, P322S, and S329R [83–86]. Functional studies of some of these mutations by in vitroexpression systems have confirmed the partial phenotype of the NDI. P322S is the most remarkableof these three mutations, since another mutation substituting proline 322, namely, P322H, isassociated with a severe phenotype. By in vitro expression of both P322H and P322S in COS-7cells, Ala et al. [79] have shown that the P322H mutant had totally lost the ability to stimulate theGs/adenylate cyclase system, whereas the P322S mutant was able to stimulate adenylate cyclase,albeit less than the wild-type receptor. Thus, the in vitro experiments closely correspond to theclinical phenotype. On the basis of three-dimensional modeling of the P322H and P322S mutantreceptors, a plausible hypothesis to explain the molecular basis for the mild phenotype of the P322Shas been proposed. Based on this modeling, it is suggested that complete loss of function of the
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 8 of 25

P322H receptor could be due, in part, to hydrogen bond formation between the His322 side chainand the carboxyl group of Asp85, which does not occur in the P322S receptor [79].
Intrafamilial variability of the X-linked NDI phenotype has also been described. A nice exampleis the case described by Kalenga et al., who reported a Belgian family in which the R137H mutationwas associated with severe NDI in the proband but with very mild NDI in his affected brother[87]. Genetic and/or environmental modifying factors are likely to account for this intrafamilialphenotype variability.
The Autosomal Recessive and Autosomal Dominant Forms of NephrogenicDiabetes Insipidus: Mutations in the Aquaporin-2 Water Channel
Both the autosomal recessive and the autosomal dominant types of NDI are caused by mutations inthe AQP2 water channel gene (MIM 107777; GenBank accession number z29491). The humanAQP2 gene is a small gene consisting of 4 exons, comprising 5 kb genomic DNA. The 1,5 kbmRNAencodes a protein of 271 amino acids which has a predicted molecular weight of 29 kDa [88]. AQP2belongs to a family of membrane integral proteins, aquaporins, which function as selective watertransporters throughout the plant and animal kingdom. In mammals, 13 different aquaporins havebeen identified to date, 8 of which (aquaporins 1–4, 6–8, and 11) are highly expressed in the kidney.Like other aquaporins, AQP2 is assembled in the membrane as a homotetramer in which each29 kDa monomer, consisting of six membrane-spanning a-helical domains and intracellular N- andC-termini, is a functional water channel. The six transmembrane domains are connected by fiveloops (A through E). The mechanism of selectivity for water of aquaporins in general has beenrevealed in the homologous AQP1 protein [89, 90] and has further been strengthened by moleculardynamics modeling approaches [91, 92] and was recently also confirmed for AQP2 [93]. The waterpore is formed between the first and sixth transmembrane domains and is lined by the intracellularB-loop and the extracellular E-loop. AQP2 is exclusively localized in the apical membrane and asubapical compartment of collecting duct cells. It is upregulated by dehydration or AVP, indicatingthat it is the AVP-regulated water channel.
To date, 46 putative disease-causing mutations in AQP2 have been identified in families withautosomal recessive NDI ([69, 70, 94–96], reviews in Refs. [23, 61, 71]). These include 38 missensemutations, 2 nonsense mutations, 3 small deletions, and 3 splice-site mutations. Most mutations arefound between the first and last transmembrane domain of AQP2. Expression studies in Xenopuslaevis oocytes have revealed that most AQP2 missense mutations that cause recessive NDI are classII mutations. Thus, these mutations lead to misfolding of the mutant protein, retention in theendoplasmic reticulum (ER), and rapid degradation of AQP2 (review in Refs. [23, 78]). Inagreement with extensive degradation, AQP2 could not be detected in the urine of patients withrecessive NDI [97]. When overexpressed in oocytes and Chinese hamster (CHO) cells, six ofthese AQP2 mutants (A147T, T126M, G64R, L22V, A47V, and T125M) confer water permeability[98, 99]. This indicates that at high expression levels, these AQP2 mutant proteins escape from theER and are routed to the plasma membrane, where they are functional. In terms of possible treatmentstrategies, these results are of high importance, since they suggest that functional channels may bestimulated to reach the plasma membrane by restoring mutant trafficking (discussed later intreatment).
One AQP2 missense mutation, P262L, located in the AQP2 C-terminal tail, a region until thenbelieved to result in dominant NDI, surprisingly was found to be involved in recessive NDI [100].In cell biological experiments, it was shown that the P262L mutant is a functional water channel that
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 9 of 25

forms heterooligomers with wt-AQP2. These wt-AQP2/AQP2-P262L heterotetramers are located inthe apical membrane, indicating that the apical sorting of wild-type AQP2 is dominant over themissorting signal of AQP2-P262L. This is different from dominant NDI, because in this formmutants retain wt-AQP2 in intracellular locations (see below). The recessive inheritance in the twopatients encountered (patients were heterozygous for a R187C or A190T mutation on one allele,combined with a P262Lmutation on the other allele) can be explained as follows: AQP2-R187C andAQP2-A190T are retained in the ER and do not interact with AQP2-P262L. AQP2-P262L foldsproperly and assembles in homotetramers, but will be retained mainly in intracellular vesicles. Theconsequent lack of sufficient AQP2 proteins in the apical membrane of the patients’ collecting ductcells explains their NDI phenotype. In the parents coding for wt-AQP2 and AQP2-R187C or AQP2-A190T, wt-AQP2 will not interact with either mutant but will form homotetrameric complexes, ofwhich the insertion into the apical membrane will be regulated properly by vasopressin and will givea healthy phenotype. In the parents coding for wt-AQP2 and AQP2-P262L, both proteins likelyassemble into heterotetramers. The dominancy of wt-AQP2 sorting on the localization of AQP2-P262Lwill result in proper AVP-regulated trafficking of the heterotetrameric complexes to the apicalmembrane and will also give a healthy phenotype [100].
At present ten families have been described with autosomal dominant NDI, initially uncovereddue to father-to-son transmission of the disease. In these families subsequent sequencing of theAQP2 gene revealed putative disease-causing mutations of one AQP2 allele. The identified muta-tions in AQP2 comprise small deletions, insertions, and missense mutations ([69], review in Refs.[23, 101]). All mutations causing dominant NDI are located in the coding region of the C-terminaltail of AQP2, which is not part of the pore-forming segment but contains important sorting signalsthat govern intracellular transport of the protein [78, 102]. Indeed, all mutants AQP2 proteins foundin dominant NDI appeared to be folded functional water channels that were sorted to othersubcellular locations in the cell than wt-AQP2, e.g., late endosomes/lysosomes and the basolateralmembrane. Because none of these mutants was misfolded, they were, in contrast to AQP2mutants inrecessive NDI, able to interact and form heterotetramers with wt-AQP2. Due to this wt-mutantinteraction and the dominancy of the missorting signals in the mutant protein, the wt-mutantcomplexes are also missorted. Formation of heterotetramers with wt-AQP2 has been shown formost of the dominant AQP2 mutants. For instance, expression studies in polarized cell lines haverevealed that the dominant AQP2-E258K mutant is routed to the Golgi complex or late endosomes/lysosomes [59]. In co-expression studies with wild-type AQP2, a dominant-negative effectwas observed, caused by impaired routing of wild-type AQP2 to the plasma membrane afterhetero-oligomerization with the E258K mutant [103]. Mistargeting to the basolateral membranehas been reported for the AQP2-721delG, AQP2-763-772del, AQP2-812-818del, and AQP2-779-780insA mutants [102, 104]. The AQP2-727delG mutant was shown to interfere with the routing ofwild-type AQP2 to the apical membrane by its mistargeting to the basolateral membrane andlate endosomes/lysosomes [104]. The loss of appropriate AQP2 heterotetramer trafficking indominant NDI is caused by several mechanisms. The phosphorylation site at Ser256, serving asan introducible apical sorting signal, may be inactivated, overruled by basolateral sorting signals, orreprogrammed to induce basolateral sorting, all causing intracellular misrouting ([105–108] andreview in Ref. [23]).
Dominant NDI patients have a milder phenotype when compared to milder recessive forms ofNDI, suggesting that some wt-homotetramers are formed that are able to reach the apicalmembrane [103].
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 10 of 25

Differential Diagnosis Between the X-Linked and the Autosomal Forms of NDIWith a few exceptions, no differences in clinical symptoms between X-linked and autosomalrecessive forms of NDI can be observed, nor in the time of onset of the disease. Only in a minorityof patients with the X-linked form of NDI, namely, those individuals carrying V2R mutations withpartial insensitivity to AVP, the disease onset is not directly after birth but later in childhood. Ingeneral the initial symptoms in most autosomal dominant cases also appear later in childhood.
Male patients with X-linked NDI can be discriminated from patients with autosomal recessiveNDI on the basis of their extrarenal reaction to the intravenous administration of the syntheticV2-vasopressin analogue 1-desamino-8-D-arginine vasopressin (DDAVP). Patients with autosomalrecessive NDI show a decrease in blood pressure, an accelerated heart rate, and an increase in vonWillebrand factor, factor VIII, and tissue-type plasminogen activator levels, whereas in patients withX-linked NDI, these extrarenal responses are absent as a result of an extrarenal mutant V2R [109]. Infemale patients, the interpretation of this intravenous DDAVP test is more complicated. Althoughabsence of the extrarenal responses to intravenous administration of DDAVP in females clearlypoints to the presence of a V2R defect, a normal response cannot be interpreted as indicative of adefect beyond the V2R and thus an AQP2 defect. For instance, a symptomatic female patientdescribed byMoses et al., who was shown to be heterozygous for a V2Rmutation, showed a twofoldincrease in factor VIII activity after administration of DDAVP [110]. The discrepancy between therenal and extrarenal response to DDAVP in these female V2R mutation carriers might be explainedby variability in the pattern of X-inactivation between different tissues.
Nephrogenic Diabetes Insipidus in Females
Several families have been described in which females show classical clinical and laboratoryfeatures of NDI. After the identification of AQP2 mutations as a cause for autosomal recessiveNDI, and in some cases for autosomal dominant NDI, a satisfying explanation for the completemanifestation of the disease in some females had been found. However, several families have beenreported in which symptomatic females do not have an AQP2 defect but are heterozygous for a V2
receptor defect [111–114]. In some of these women, maximal urinary osmolality after DDAVPadministration does not exceed 200 mosmol/L. Of interest, in some of the reported families,asymptomatic female family members shared the same V2 receptor mutation with the manifestingfemales [111, 115]. The most likely explanation for the existence of different phenotypes in carriersof a V2 receptor mutation, varying from no symptoms to complete manifestation of the disorder, isskewed X-inactivation [116]. This hypothesis was underlined by studies investigating theX-inactivation patterns in peripheral blood leukocytes of female carriers via the detection of amethylated trinucleotide repeat in the human androgen receptor gene [117]. In asymptomaticfemales random X-inactivation was found, while in most female carriers who showed clinicalNDI symptoms, skewed X-inactivation patterns occurring preferentially to normal X alleles wererecognized. In a few females with over clinical NDI, however, randomX-inactivation was identified.
In conclusion, clinical NDI phenotypes may correlate with the X-inactivation patterns in femaleswith heterozygote V2R mutations. In some female carriers, however, the clinical phenotype cannotbe predicted by evaluation of X-inactivation patterns in peripheral blood cells, probably due to thefact that X-inactivation ratios within an individual may vary between different tissues.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 11 of 25

Acquired Nephrogenic Diabetes Insipidus
Although the hereditary forms of NDI are relatively rare, a wide range of pathologic conditions anddrug treatments can lead to acquired NDI (Table 1). In our experience, the urine osmolality obtainedafter DDAVP administration in these acquired disorders is always higher than in congenital NDI. Allthese disorders have been shown to coincide with decreased expression of AQP2 or deregulatedAQP2 trafficking to the apical membrane [118–123]. For instance, prolonged treatment withlithium, the drug of choice for treating bipolar disorders and prescribed to 1 in 1,000 of thepopulation, leads to the development of NDI in at least 20% if treated individuals [124]. Thedevelopment of lithium-NDI is believed to occur in two phases. In the first short-time phase, lithiumcauses a decrease in AQP2 expression ([125], review in Ref. [126]). Lithium enters the cells viaepithelial sodium channel (ENaC) and accumulates in principal cells [125, 127]. How lithiumdownregulates AQP2 is not clear but likely involves glycogen synthase kinase type 3b, which isimportant in AVP-regulated antidiuresis and is inhibited by lithium [128–130]. Lithium alsoinfluences AQP2-mediated water reabsorption by increased tubular release of prostaglandin E2[129]. In the second phase, lithium reduces the percentage of principal cells in the collecting duct tothe advantage of intercalated cells, involved in acid-base homeostasis [131]. The exact contributionof this collecting duct remodeling in the lithium-induced resistance to vasopressin remains to beelucidated.
Treatment
Conventional TreatmentSymptomatic treatment of NDI is focused on establishing and maintaining normovolemia byreplacing urinary water losses and reducing urinary volume. Adequate supply of fluid to preventdehydration is the most important component of the therapy. For reducing urine output, a low-solutediet is applied to diminish the renal osmolar load and decrease obligatory water excretion [132]. Ini-tially, a diet low in sodium (1 mmol/kg per day) as well as protein (2 g/kg per day) wasrecommended. However, severe limitations of dietary protein may introduce serious nutritionaldeficiencies. Therefore, it is preferable to prescribe dietary restriction of sodium only.
Diuretics such as hydrochlorothiazide (2–4 mg/kg per 24 h) were the first class of drugs shown tobe effective in lowering the urine volume in NDI [133]. When combined with a reduction of saltintake, hydrochlorothiazide reduces urine volume by 20–50 % of baseline values. However,thiazide-induced hypokalemia may cause further impairment or urine-concentrating ability inpatients with NDI. Another possible risk associated with hypokalemia is cardiac arrhythmia.Simultaneous administration of potassium salt is therefore advised in most cases. Very low dailysodium intake in combination with thiazide diuretics should be avoided to prevent the developmentof hyponatremia.
There is ample evidence that the combined administration of hydrochlorothiazide with either aprostaglandin-synthesis inhibitor, such as indomethacin (2 mg/kg per 24 h), or the potassium-sparing diuretic amiloride, is much more effective in reducing urine volume than the thiazidediuretic alone [134–138]. Prolonged use of prostaglandin-synthesis inhibitors, however, is oftencomplicated by gastrointestinal and hematopoietic side effects. Gastrointestinal complaints andcomplications include anorexia, nausea, vomiting, abdominal pain, ulceration, perforation, andhemorrhage. Hematopoietic reactions include neutropenia, thrombocytopenia, and, rarely, aplastic
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 12 of 25

anemia. In addition, renal dysfunction has been described during indomethacin therapy, most oftenconsisting of a slight reduction in GFR.
In patients, who are not tolerating indomethacin, selective inhibitors of cyclooxygenase-2(COX-2) might be helpful [139]. Caution in using indomethacin and selective COX-2 inhibitorsin NDI is warranted as their administration can potentially lead to the acute deterioration of renalfunction in dehydrated patients.
Amiloride counterbalances the potassium loss from prolonged use of thiazides and thus preventshypokalemia. Since amiloride appears to have only minor long-term side effects, the combination ofhydrochlorothiazide (2–4 mg/kg/24 h) with amiloride (0.3 mg/kg/24 h) is the first choice oftreatment. Our personal experience of more than 20 years with the amiloride-hydrochlorothiazidecombination, however, indicates that amiloride is less well tolerated in young children below the ageof 4–6 years because of persistent nausea. Therefore we advise the temporary use of the combinationof indomethacin-hydrochlorothiazide in these young children.
For a long time the following mechanism for the antidiuretic effect of thiazides in NDI has beenproposed: thiazides reduce sodium reabsorption in the distal tubule by inhibition of the NaClcotransporter (NCC). This subsequently results in increased sodium excretion, extracellular volumecontraction, decreased glomerular filtration rate, and increased proximal sodium and waterreabsorption. Consequently, less water and sodium reach the collecting tubules and less water isexcreted [140, 141]. This long-standing hypothesis has been challenged by Magaldi, who reportednew insights into the possible mechanism of action, based on microperfusion studies in rat innermedullary collecting duct (IMCD) [142, 143]. In these studies it was shown that in the absence ofvasopressin, hydrochlorothiazide, when added to the luminal side, increased osmotic and diffusionalwater permeabilities, thus, decreasing water excretion. When prostaglandins were added, the effectof thiazides decreased. This finding may offer one explanation why indomethacin potentiates theeffect of thiazides in NDI [143]. Antidiuretic effect of thiazides is associated with an increase inAQP2 expression in collecting duct cells [144]. Long-term side effects of chronic thiazide admin-istration such as hyperuricemia, alterations in serum lipid spectrum, and glucose intolerance shouldbe monitored.
Although the drugs mentioned above reduce urine excretion, they are unable to achieve urinevolumes produced in healthy individuals. Therefore the general problem remains, although thesymptoms are relieved. Consequently, current research focuses on methods to treat NDI on a morecausative level than solely try to fight the symptoms (Fig. 2; for detailed reviews [23, 28]).
Therapeutic Strategies for Treatment of X-Linked NDIBecause in vitro expression studies reveal that the majority of AVPR2 mutations in X-linked NDIresult in normal protein that is retained within the endoplasmic reticulum (ER), agents that restoreplasma routing are under investigation as potential treatments. Promising agents are cell-permeableV2R antagonists and agonists that in vitro rescue the intracellular retention of several V2R mutants[145–148]. An important problem with the antagonists is that once the mutant V2R is rescued to thebasolateral membrane, the antagonist needs to be displaced by high concentrations of AVP/DDAVPto induce cAMP signaling. Therefore, low-affinity antagonists are believed to have the highestclinical value. However, their efficiency in rescuing is lower than that of high-affinity ligands, andthe high concentrations required to be administered for sufficient activity by low-affinity antagonistsmight lead to severe complications in patients. The use of non-peptide agonists has somewhatcircumvented this problem since they do not need displacement to activate V2R. All high-affinity
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 13 of 25
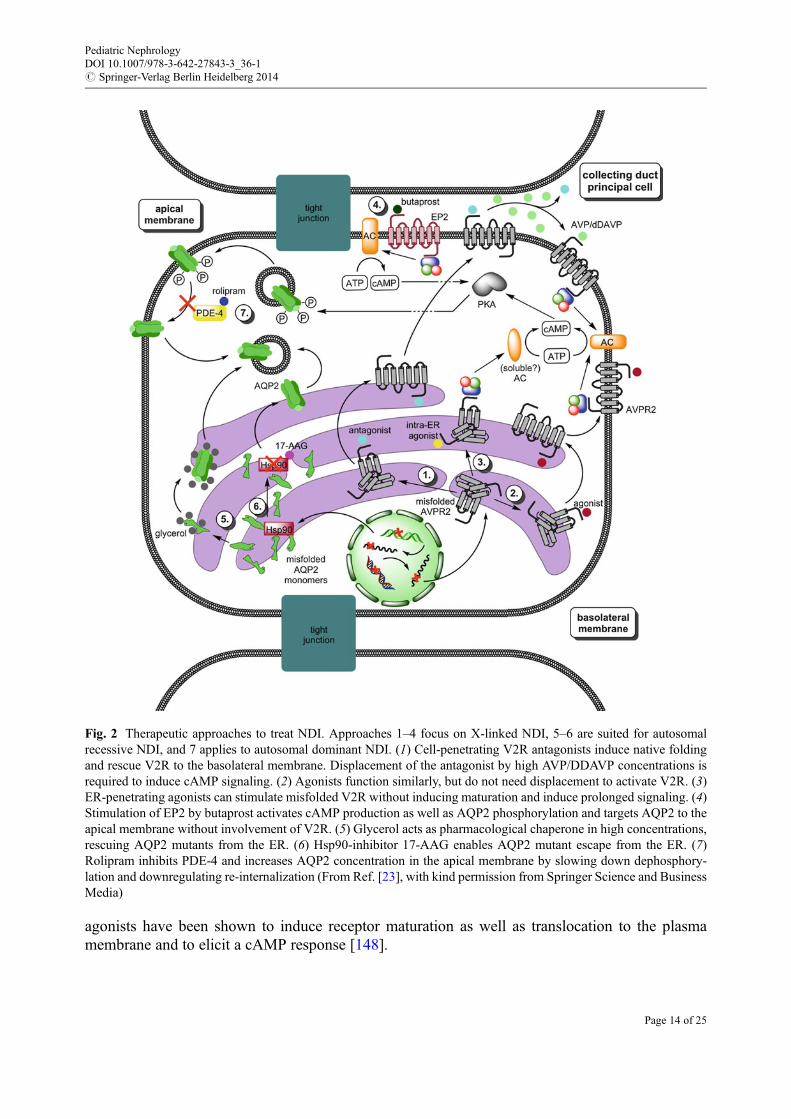
agonists have been shown to induce receptor maturation as well as translocation to the plasmamembrane and to elicit a cAMP response [148].
Fig. 2 Therapeutic approaches to treat NDI. Approaches 1–4 focus on X-linked NDI, 5–6 are suited for autosomalrecessive NDI, and 7 applies to autosomal dominant NDI. (1) Cell-penetrating V2R antagonists induce native foldingand rescue V2R to the basolateral membrane. Displacement of the antagonist by high AVP/DDAVP concentrations isrequired to induce cAMP signaling. (2) Agonists function similarly, but do not need displacement to activate V2R. (3)ER-penetrating agonists can stimulate misfolded V2R without inducing maturation and induce prolonged signaling. (4)Stimulation of EP2 by butaprost activates cAMP production as well as AQP2 phosphorylation and targets AQP2 to theapical membrane without involvement of V2R. (5) Glycerol acts as pharmacological chaperone in high concentrations,rescuing AQP2 mutants from the ER. (6) Hsp90-inhibitor 17-AAG enables AQP2 mutant escape from the ER. (7)Rolipram inhibits PDE-4 and increases AQP2 concentration in the apical membrane by slowing down dephosphory-lation and downregulating re-internalization (From Ref. [23], with kind permission from Springer Science and BusinessMedia)
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 14 of 25

The feasibility of treatment with these so-called pharmacologic “chaperones” has been testedin vivo. In individuals with NDI who have missense AVPR2 mutations, Bernier et al. showed thattreatment with a non-peptide V1a receptor antagonist had beneficial effects on urine volume andosmolality starting a few hours after administration. However, the long-term effect of this drug couldnot be tested because the clinical development of this V1a receptor antagonist was interrupted duringthe course of this study as a result of possible interference with the cytochrome P450 metabolicpathway [147].
Remarkably, certain non-peptide V2R agonists, such as OPC51, VA88, and VA89, were shown tobe able to intracellularly stimulate the V2R and increase cAMP production and AQP2 translocationto the apical membrane [149]. In contrast to pharmacochaperone-assisted folding and rescue of thereceptors, the localization andmaturation state of the V2R did not change upon activation, indicatingthat these compounds do not act as molecular chaperones. The mode of action by which receptorstrapped intracellularly can still activate their coupled G-protein and how this stimulates adenylatecyclase is not yet understood. Future in vivo and clinical testing has to confirm whether thepharmacological chaperones and the intracellularly acting non-peptide agonists have the desiredpositive effects in patients and meet the safety requirements.
In patients with X-linked NDI, bypassing the V2R could be an alternative way to treat the disease.By stimulation of the E-prostanoid receptor EP4, NDI symptoms were greatly reduced in a
conditional AVPR2-deletion mouse model [150]. This was due to raised AQP2 levels, most probablyas a consequence of cAMP production caused by EP4 stimulation. Recently, a similar effect wasseen after stimulation of the EP2 receptor by the agonist butaprost [151]. The EP2 receptor is a moreinteresting candidate for treatment of NDI than the EP4 receptor since EP2 agonists have alreadybeen tested in clinical studies for other diseases and have shown promising results concerning safetyissues. However, clinical trials in NDI are necessary to evaluate the effects and safety of EP2agonists for this disorder.
Another potential therapeutic strategy bypassing the V2R could be an activation of the cGMP-signaling pathway. Several groups have shown that nitric oxide donors and atrial natriuretic factorstimulate the insertion of AQP2 in renal epithelial cells in vitro and in vivo via a cGMP-dependentpathway without increasing the expression of AQP2 [152, 153], and the selective cGMP phospho-diesterase inhibitor sildenafil citrate (Viagra) prevents degradation of cGMP resulting in increasedmembrane expression in AQP2 in vitro and in vivo [154]. In a small number of NDI patientssubjected to clinical trials with sildenafil citrate, no decreases in urine volume or increases in urineosmolality were observed (personal communication in Ref. [28]). Alternative AVP-independentstrategies are the use of calcitonin, which has a vasopressin-like effect on AQP2 trafficking andurine-concentrating ability via cAMP-mediated mechanism [155] and of various statins(simvastatin, fluvastatin) that were reported to increase AQP2 expression and water reabsorptionin the kidney via an as yet unknown mechanism [156, 157]. Very recently, using a systemic high-throughput chemical screening procedure, Nomura et al. identified AG-490 (an EGF receptor andJAK-2 kinase inhibitor) as a compound that stimulates AQP2 exocytosis, induces AQP2 membraneaccumulation, and stimulates urine concentration in an AVP-independent manner [158]. Despitethese promising results in in vitro studies and in animal models, none of these compounds has yetbeen translated into therapy of NDI.
Therapeutic Strategies for Treatment of Autosomal NDISimilarly to V2R mutants, the majority of AQP2 mutants causing autosomal recessive NDI aremissense mutations that lead to aberrant folding of AQP2 in the ER. Hence, finding substances thatare able to reestablish natural AQP2 folding holds comparable promises for treatment of recessive
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 15 of 25

NDI as it has been shown for the X-linked form. In CHO and MDCK cells, glycerol has proven theapplicability of chemical chaperones to AQP2 by restoring ER export in high concentrations [99].
Yang et al. described partial restoration of cellular AQP2 processing upon treatment of conditionalAQP2-T126M knock-in mice with an Hsp90 inhibitor, 17-allylamino-demethoxygeldanamycin(17-AAG), eventually resulting in improved urinary concentrating ability [159]. The preciseexplanation underlying the beneficial effect of this Hsp inhibitor remains to be elucidated. Further-more, it is not unlikely that Hsp90 inhibition may have severe side effects that outweigh theadvantages [160]. Therefore, lengthened studies addressing safety issues of Hsp90 or other chap-erone inhibitors have to be conducted in order to elucidate the applicability of these compounds inNDI therapy.
Based on the improvement of AVP-dependent cAMP signaling of collecting duct cells in ahypercalcemia-induced NDI mouse model, Sohara et al. also tested the phosphodiesterase-4 inhib-itor Rolipram in the knock-in dominant NDI mice [161]. Their data indicated that Rolipram is able toincrease cAMP levels leading to increased AQP2 phosphorylation and translocation to the apicalmembrane. Phosphodiesterase-4 is a common protein that also is involved in immunosuppressiveand anti-inflammatory pathways, and therefore its inhibition may have severe side effects. Rolipramhas been tested in two male patients with X-linked NDI and did not cause any relief of symptoms[162], but the potential for other PDE inhibitors in the treatment of NDI needs to be examinedfurther.
References
1. McIlraith CH. Notes on some cases of diabetes insipidus with marked family and hereditarytendencies. Lancet. 1892;II:767–8.
2. Forssman H. On hereditary diabetes insipidus with special regard to a sex-linked form. ActaMed Scand. 1945;153:3–196.
3. Waring AJ, Kajdi L, Tappan V. A congenital defect of water metabolism. Am J Dis Child.1945;69:323–4.
4. Williams RH, Henry C. Nephrogenic diabetes insipidus: transmitted by females and appearingduring infancy in males. Ann Intern Med. 1947;27:84–95.
5. Kaplan SA. Nephrogenic diabetes insipidus. In: Holliday MA, Barratt TM, Vernier RL,editors. Pediatric nephrology. Baltimore: Williams & Wilkins; 1987. p. 623–5.
6. van Lieburg AF, Knoers NVAM, Monnens LAH. Clinical presentation and follow-up ofthirty patients with congenital nephrogenic diabetes insipidus. J Am Soc Nephrol.1999;10:1958–64.
7. Lejarraga H, Caletti MG, Caino S, et al. Long-term growth of children with nephrogenicdiabetes insipidus. Pediatr Nephrol 2008;23:2007–12.
8. Hillman DA, Neyzi O, Porter P, et al. Renal (vasopressin-resistant) diabetes insipidus:definition of the effects of homeostatic limitation in capacity to conserve water on the physical,intellectual, and emotional development of a child. Pediatrics. 1958;21:430–5.
9. Vest M, Talbot NB, Crawford JD. Hypocaloric dwarfism and hydronephrosis in diabetesinsipidus. Am J Dis Child. 1963;105:175–81.
10. Forssman H. Is hereditary diabetes insipidus of nephrogenic type associated with mentaldeficiency? Acta Psychiatr Neurol Scand. 1955;30:577–87.
11. Macaulay D, WatsonM. Hypernatremia in infants as a cause of brain damage. Arch Dis Child.1967;42:485–91.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 16 of 25

12. Bichet DG. Vasopressin receptor mutations in nephrogenic diabetes insipidus. Semin Nephrol.2008;28:245–51.
13. Kanzaki S, Omura T, Miyake M, et al. Intracranial calcification in nephrogenic diabetesinsipidus. JAMA. 1985;254:3349–50.
14. Schofer O, Beetz R, Kruse K, et al. Nephrogenic diabetes insipidus and intracerebral calcifi-cation. Arch Dis Child. 1990;65:885–7.
15. Hoekstra JA, van Lieburg AF,Monnens LAH, et al. Cognitive and psychosocial functioning ofpatients with nephrogenic diabetes insipidus. Am J Med Genet. 1996;61:81–8.
16. Uribarri J, Kaskas M. Hereditary nephrogenic diabetes insipidus and bilateral nonobstructivehydronephrosis. Nephron. 1993;65:346–9.
17. Shalev H, Romanovsky I, Knoers NV, et al. Bladder function impairment in aquaporin-2defective nephrogenic diabetes insipidus. Nephrol Dial Transplant. 2004;19:608–13.
18. Yoo TH, Ryu DR, Song YS, et al. Congenital nephrogenic diabetes insipidus presented withbilateral hydronephrosis: genetic analysis of V2R gene mutations. Yonsei MedJ. 2006;47:126–30.
19. Hong CR, Kang HG, Choi HJ, et al. X-linked recessive nephrogenic diabetes insipidus: aclinico-genetic study. J Pediatr Endocrinol Metab. 2014;27:93–9.
20. Ulinski T, Grapin C, Forin V, et al. Severe bladder dysfunction in a family with ADH receptorgene mutation responsible for X-linked nephrogenic diabetes insipidus. Nephrol Dial Trans-plant. 2004;19:2928–9.
21. Monnens L, Smulders Y, van Lier H, et al. DDAVP test for assessment of renal concentratingcapacity in infants and children. Nephron. 1991;29:151–4.
22. Bockenhauer D, Bichet DG. Inherited secondary nephrogenic diabetes insipidus: concentrat-ing on humans. Am J Physiol Renal Physiol. 2013;304:F1037–42.
23. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current stateof affairs. Pediatr Nephrol. 2012;27:2183–204.
24. Katsura T, Ausiello DA, Brown D. Direct demonstration of aquaporin-2 water channelrecycling in stably transfected LCC-PK1 epithelial cells. Am J Physiol. 1996;39:F548–53.
25. Noda Y, Sasaki S. Regulation of aquaporin-2 trafficking and its binding protein complex.Biochim Biophys Acta. 2006;1758:1117–25.
26. Sasaki S, Noda Y. Aquaporin-2 protein dynamics within the cell. Curr Opin NephrolHypertens. 2007;16:348–52.
27. Bouley R, Hasler U, Lu HA, et al. Bypassing vasopressin receptor signalling pathways innephrogenic diabetes insipidus. Semin Nephrol. 2008;28:266–78.
28. Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into themolecular background and potential therapies for treatment. Endocr Rev. 2013;34:278–301.
29. Hendriks G, Koudijs M, van Balkom BW, et al. Glycosylation is important for cell surfaceexpression of the water channel aquaporin-2 but is not essential for tetramerization in theendoplasmic reticulum. J Biol Chem. 2004;279:2975–83.
30. Nielsen S, DiGiovanni SR, Christensen EI, et al. Cellular and subcellular immunolocalizationof vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci U S A.1993;90:11663–7.
31. Fushimi K, Sasaki S, Muramo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem.1997;272:14800–4.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 17 of 25

32. Katsura T, Gustafson CE, Ausiello DA. Protein kinase A phosphorylation is involved inregulated exocytosis of aquaporin-2 in transfected LCC-PK1 cells. Am J Physiol. 1997;272:F816–22.
33. Klussmann E, Maric K, Wiesner B, et al. Protein kinase A anchoring proteins are required forvasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principalcells. J Biol Chem. 1999;274:4934–8.
34. Kamsteeg EJ, Heijnen I, van Os CH, et al. The subcellular localization of an aquaporin-2tetramer depends on the stoichiometry of phosphorylated and nonphosphorylated monomers.J Cell Biol. 2000;1:919–30.
35. Jo I, Harris HW, Amendt Raduege AM, Majewski RR, et al. Rat kidney papilla containsabundant synaptobrevin protein that participates in the fusion of antidiuretic hormone-regulated water channel-containing endosomes in vitro. Proc Natl Acad Sci U S A.1995;92:1876–80.
36. Liebenhoff U, Rosenthal W. Identification of Rab3-, Rab5a-, and synaptobrevin II-like pro-teins in a preparation of rat kidney vesicles containing the vasopressin-regulated waterchannel. FEBS Lett. 1995;365:209–13.
37. Nielsen S, Marples D, Birn H, et al. Expression of VAMP2-like protein in kidney collectingduct intracellular vesicles. Colocalization with aquaporin-2 water channels. J Clin Invest.1995;96:1834–44.
38. Mandon B, Chou CL, Nielsen S, Knepper MA. Syntaxin-4 is localized to the apical plasmamembrane of rat renal collecting duct cells: possible role in aquaporin-2 trafficking. J ClinInvest. 1996;98:906–13.
39. Tajika Y, Masuzaki T, Suzuki T, et al. Differential regulation of AQP2 trafficking inendosomes by microtubules and actin filaments. Histochem Cell Biol. 2005;124:1–12.
40. Klussmann E, Tamma G, Lorenz D, et al. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J BiolChem. 2001;276:20451–7.
41. Simon H, Gao Y, Franki N, Hays RH. Vasopressin depolymerizes apical F-actin in rat innermedullary collecting duct. Am J Physiol. 1993;265:C757–62.
42. Sun TX, Van Hoek A, Huang Y, et al. Aquaporin-2 localization in clathrin-coated pits:inhibition of endocytosis by dominant-negative dynamin. Am J Physiol Renal Physiol.2002;282:F998–1011.
43. Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosisand signaling. Science. 2007;315:201–5.
44. Kamsteeg EJ, Hendriks G, BooneM, et al. Short-chain ubiquitination of the aquaporin-2 waterchannel. Proc Natl Acad Sci U S A. 2006;28:18344–9.
45. Vossenkamper A, Nedvetsky PI, Wiesner B, et al. Microtubules are needed for the perinuclearpositioning of aquaporin-2 after its endocytic retrieval in renal principal cells. Am J PhysiolCell Physiol. 2007;293:C1129–38.
46. Marples D, Schroer TA, Ahrens N, et al. Dynein and dynactin colocalize with AQP2 waterchannels in intracellular vesicles from kidney collecting duct. Am J Physiol. 1998;274:F384–94.
47. Palamidessi A, Frittoli E, Garre M, et al. Endocytic trafficking of Rac is required for the spatialrestriction of signaling in cell migration. Cell. 2008;134:135–47.
48. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol.2009;10:513–25.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 18 of 25

49. Matsumura Y, Uchida S, Rai T, et al. Transcription regulation of aquaporin-2 water channelgene by cAMP. J Am Soc Nephrol. 1997;8:861–7.
50. Carter C, Simpkiss M. The “carrier” state in nephrogenic diabetes insipidus. Lancet. 1956;II:1069–73.
51. van den Ouweland AMW, Knoop MT, Knoers NVAM, et al. Colocalization of the gene fornephrogenic diabetes insipidus (DIR) and the vasopressin type-2 receptor (AVPR2) in theXq28 region. Genomics. 1992;13:1350–3.
52. van den Ouweland AMW, Dreesen JCFM, Verdijk M, et al. Mutations in the vasopressin type-2 receptor gene associate with nephrogenic diabetes insipidus. Nat Genet. 1992;2:99–102.
53. Pan Y, Metzenberg A, Das S, et al. Mutations of the V2 receptor are associated with X-linkednephrogenic diabetes insipidus. Nat Genet. 1992;2:103–6.
54. Rosenthal W, Seibold A, Antamarian A, et al. Molecular identification of the gene responsiblefor congenital nephrogenic diabetes insipidus. Nature. 1992;359:233–5.
55. Schreiner RL, Skafish PR, Anand SK, et al. Congenital nephrogenic diabetes insipidus in ababy girl. Arch Dis Child. 1978;53:906–15.
56. Langley JM, Balfe JW, Selander T, et al. Autosomal recessive inheritance of vasopressin-resistant diabetes insipidus. Am J Med Genet. 1991;38:90–4.
57. Brodehl J, Braun L. Familiarer nephrogener diabetes insipidus mit voller auspragung bei einerweiblichen saugling. Klin Wochenschr. 1964;42:563.
58. Deen PMT, Verdijk MAJ, Knoers NVAM, et al. Requirement of human renal water channelaquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–5.
59. Mulders SM, Bichet DG, Rijss JPL, et al. An aquaporin-2 water channel mutant which causesautosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J ClinInvest. 1998;102:57–66.
60. Morello J-P, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607–30.61. Spanakis E, Milord E, Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes
insipidus: review and missense mutation significance. J Cell Physiol. 2008;217:605–17.62. Firsov D, Mandon B, Morel A, et al. Molecular analysis of vasopressin receptors in the rat
nephron. Evidence for alternative splicing of the V2 receptor. Pflugers Arch. 1994;429:79–89.63. Innamorati G, Sadeghi H, Birnbaumer M. A full active nonglycosylated V2 vasopressin
receptor. Mol Pharmacol. 1996;50:467–73.64. Innamorati G, Sadeghi H, Eberle AN, et al. Phosphorylation of the V2 vasopressin receptor.
J Biol Chem. 1997;271:2486–92.65. Innamorati G, Sadeghi HM, Tran NT, et al. A serine cluster prevents recycling of the V2
vasopressin receptor protein. Proc Natl Acad Sci U S A. 1998;95:2222–6.66. Sch€ulein R, Rutz C, Rosenthal W. Membrane targeting and determination of transmembrane
topology of the human vasopressin V2 receptor. J Biol Chem. 1996;271:28844–52.67. Krause G, Hermosilla R, Oksche A, et al. Molecular and conformational features of a
transport-relevant domain in the C-terminal tail of the vasopressin V2 receptor. MolPharmacol. 2000;57:232–42.
68. Sch€ulein R, Liebenhoff U, Muller H, et al. Properties of the human arginine vasopressin V2receptor after site-directed mutagenesis of its putative palmitoylation site. J Biol Chem.1996;313:611–6.
69. Sasaki S, Chiga M, Kikuchi E, Rai T, Uchida S. Hereditary nephrogenic diabetes insipidus inJapanese patients: analysis of 78 families and report of 22 new mutations in AVPR2 andAQP2. Clin Exp Nephrol. 2013;17:338–44.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 19 of 25

70. Duzenli D, Saglar E, Deniz F, Azal O, Erdem B, Mergen H. Mutations in the AVPR2,AVP-NPII, and AQP2 genes in Turkish patients with diabetes insipidus. Endocrine.2012;42:664–9.
71. Knoers NVAM, Deen PMT. Molecular and cellular defects in nephrogenic diabetes insipidus.Pediatr Nephrol. 2001;16:1146–52.
72. Wenkert D, Schoneberg T, Merendino Jr JJ, et al. Functional characterization of five V2vasopressin receptor gene mutations. Mol Cell Endocrinol. 1996;124:43–50.
73. Deen PMT, Brown D. Trafficking of native and mutant mammalian MIP proteins. In:Hohmann S, Agre P, Nielsen S, editors. Aquaporins. San Diego: Academic Press; 2001.p. 235–76.
74. Robben JH, Knoers NV, Deen PM. Characterization of vasopressin V2 receptor mutants innephrogenic diabetes insipidus in a polarized cell model. Am J Physiol Renal Physiol.2005;289:F265–72.
75. Ellgaard L, Helenius A. ER quality control: towards an understanding at the molecular level.Curr Opin Cell Biol. 2001;13:431–7.
76. Hermosilla R, Oueslati M, Donalies U, et al. Disease-causing V(2) vasopressin receptors areretained in different compartments of the early secretory pathway. Traffic. 2004;5:993–1005.
77. Pan Y, Wilson P, Gitschier J. The effect of eight V2 vasopressin receptor mutations onstimulation of adenylyl cyclase and binding to vasopressin. J Biol Chem. 1994;269:31933–7.
78. Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptorand aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol.2006;291:F257–70.
79. Ala Y, Morin D, Sabatier N, et al. Functional studies of twelve mutant V2 vasopressinreceptors related to nephrogenic diabetes insipidus: molecular basis of a mild phenotype.J Am Soc Nephrol. 1998;9:1861–72.
80. Bernier V, Lagace M, Lonergan M, et al. Functional rescue of the constitutively internalizedV2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059.Mol Endocrinol. 2004;18:2074–84.
81. Barak LS, Oakley RH, Laporte SA, et al. Constitutive arrestin-mediated desensitization of ahuman vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc NatlAcad Sci U S A. 2001;98:93–8.
82. Postina R, Ufer E, Pfeiffer R, et al. Misfolded vasopressin V2 receptors caused by extracellularpoint mutations entail congenital nephrogenic diabetes insipidus. Mol Cell Endocrinol.2000;164:31–9.
83. Faerch M, Christensen JH, Corydon TJ, et al. Partial nephrogenic diabetes insipidus caused bya novel mutation in the AVPR2 gene. Clin Endocrinol (Oxf). 2008;68:395–403.
84. Armstrong SP, Seeber RM, Ayoub MA, et al. Characterization of three vasopressin receptor2 variants: an apparent polymorphism (V266A) and two loss-of-function mutations (R181Cand M311V). PLoS One. 2013;8:e65885.
85. Neocleous V, Skordis N, Shammas C, et al. Identification and characterization of a novelX-linked AVPR2 mutation causing partial nephrogenic diabetes insipidus: a case report andreview of the literature. Metabolism. 2012;61:922–30.
86. Bockenhauer D, Carpentier E, Rochdi D, et al. Vasopressin type 2 receptor V88M mutation:molecular basis of partial and complete nephrogenic diabetes insipidus. Nephron Physiol.2010;114:1–10.
87. Kalenga K, Persu A, Goffin E, et al. Intrafamilial phenotype variability in nephrogenicdiabetes insipidus. Am J Kidney Dis. 2002;39:737–43.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 20 of 25

88. Fushimi K, Uchida S, Harra Y, et al. Cloning and expression of apical membrane water channelof rat kidney collecting tubule. Nature. 1993;361:549–52.
89. Jung JS, Preston GM, Smith BL, et al. Molecular structure of the water channel throughaquaporin-CHIP. J Biol Chem. 1994;269:14648–54.
90. Heymann JB, Engel A. Aquaporins: phylogeny, structure, and physiology of water channels.News Physiol Sci. 1999;14:187–93.
91. Hub JS, Grubm€uller H, de Groot BL. Dynamics and energetics of permeation throughaquaporins. What do we learn from molecular dynamics simulations? Handb Exp Pharmacol.2009;190:57–76.
92. de Groot BL, Grubm€uller H. Water permeation across biological membranes: mechanism anddynamics of aquaporin-1 and GlpF. Science. 2001;294:2353–7.
93. Frick A, Eriksson UK, de Mattia F, et al. X-ray structure of human aquaporin 2 and itsimplications for nephrogenic diabetes insipidus and trafficking. Proc Natl Acad Sci U S A.2014;111:6305–10.
94. Park YJ, Baik HW, Cheong HI, et al. Congenital nephrogenic diabetes insipidus with a novelmutation in the aquaporin 2 gene. Biomed Rep. 2014;2:596–8.
95. Rugpolmuang R, Deeb A, Hassan Y, et al. Novel AQP2 mutation causing congenitalnephrogenic diabetes insipidus: challenges in management during infancy. J PediatrEndocrinol Metab. 2014;27:193–7.
96. Leduc-Nadeau A, Lussier Y, Arthus MF, et al. New autosomal recessive mutations inaquaporin-2 causing nephrogenic diabetes insipidus through deficient targeting display normalexpression in Xenopus oocytes. J Physiol. 2010;588:2205–18.
97. Deen PM, van Aubel RA, van Lieburg AF, et al. Urinary content of aquaporin 1 and 2 innephrogenic diabetes insipidus. J Am Soc Nephrol. 1996;7:836–41.
98. Marr N, Kamsteeg EJ, van Raak M, et al. Functionality of aquaporin-2 missense mutants inrecessive nephrogenic diabetes insipidus. Pflugers Arch. 2001;442:73–7.
99. Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetesinsipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–67.
100. De Mattia F, Savelkoul PJ, Bichet DG, et al. A novel mechanism in recessive nephrogenicdiabetes insipidus: wild-type aquaporin-2 rescues the apical membrane expression of intra-cellularly retained AQP2-P262L. Hum Mol Genet. 2004;13:3045–56.
101. Loonen AJM, Knoers NVAM, van Os CH, et al. Aquaporin 2 mutations in nephrogenicdiabetes insipidus. Semin Nephrol. 2008;28:252–65.
102. Kuwahara M, Iwai K, Ooeda T, et al. Three families with autosomal dominant nephrogenicdiabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am J Hum Genet.2001;69:738–48.
103. Kamsteeg E-J, Wormhoudt TAM, Rijss JPL, et al. An impaired routing of wild-typeaquaporin-2 after tetramerization with an aquaporin-2 mutant explains dominant nephrogenicdiabetes insipidus. EMBO J. 1999;18:2394–400.
104. Marr N, Bichet DG, Lonergan M, et al. Heteroligomerization of an aquaporin-2 mutant withwild-type aquaporin-2 and their misrouting to late endosomes/lysosomes explains dominantnephrogenic diabetes insipidus. Hum Mol Genet. 2002;11:779–89.
105. de Mattia F, Savelkoul PJ, Kamsteeg EJ, et al. Lack of arginine vasopressin-induced phos-phorylation of aquaporin-2 mutant AQP2-R254L explains dominant nephrogenic diabetesinsipidus. J Am Soc Nephrol. 2005;16:2872–80.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 21 of 25

106. Savelkoul PJ, De Mattia F, Li Y, et al. p.R254Q mutation in the aquaporin-2 water channelcausing dominant nephrogenic diabetes insipidus is due to a lack of arginine vasopressin-induced phosphorylation. Hum Mutat. 2009;30:E891–903.
107. Kamsteeg EJ, Savelkoul PJ, Hendriks G, et al. Missorting of the aquaporin-2 mutant E258K tomultivesicular bodies/lysosomes in dominant NDI is associated with its monoubiquitinationand increased phosphorylation by PKC but is due to the loss of E258. Pflugers Arch.2008;455:1041–54.
108. Kamsteeg EJ, Stoffels M, Tamma G, et al. Repulsion between Lys258 and upstream argininesexplains the missorting of the AQP2 mutant p.Glu258Lys in nephrogenic diabetes insipidus.Hum Mutat. 2009;30:1387–96.
109. van Lieburg AF, Knoers NVAM, Mallman R, et al. Normal fibrinolytic responses to1-desamino-8-D-arginine vasopressin in patients with nephrogenic diabetes insipidus causedby mutations in the aquaporin-2 gene. Nephron. 1996;72:544–6.
110. Moses AM, Sangai G, Miller JL. Proposed cause of marker vasopressin resistance in a femalewith X-linked recessive V2 receptor abnormality. J Clin Endocrinol Metab. 1995;80:1184–6.
111. van Lieburg AF, Verdijk MAJ, Schoute F, et al. Clinical phenotype of nephrogenic diabetesinsipidus in females heterozygous for a vasopressin type-2 receptor mutation. Hum Genet.1995;96:70–8.
112. Sato K, Fukuno H, Taniguchi T, et al. A novel mutation in the vasopressin V2 receptor gene ina woman with congenital nephrogenic diabetes insipidus. Intern Med. 1999;38:808–12.
113. Chan Seem CP, Dossetor JF, Penney MD. Nephrogenic diabetes insipidus due to a newmutation of the arginine vasopressin V2 receptor gene in a girl presenting withnon-accidental injury. Ann Clin Biochem. 1999;36:779–82.
114. Faerch M, Corydon TJ, Rittig S, et al. Skewed X-chromosome inactivation causing diagnosticmisinterpretation in congenital nephrogenic diabetes insipidus. Scand J Urol Nephrol.2010;44:324–30.
115. Nomura Y, Onigata K, Nagashima T, et al. Detection of skewed X-inactivation on two femalecarriers of vasopressin type 2 receptor gene mutation. J Clin Endocrinol Metab.1997;82:3434–7.
116. Migeon BR. X inactivation, female mosaicism, and sex differences in renal diseases. J Am SocNephrol. 2008;19:2052–9.
117. Satoh M, Ogikubo S, Yoshizawa-Ogasawara A. Correlation between clinical phenotypes andX-inactivation patterns in six female carriers with heterozygote vasopressin type 2 receptormutations. Endocr J. 2008;55:277–84.
118. Marples D, Christensen S, Christensen EI, et al. Lithium-induced down-regulation ofaquaporin-2 water channel expression in rat kidney medulla. J Clin Invest. 1995;95:1838–45.
119. Kwon T-H, Laursen UH, Marples D, et al. Altered expression of renal AQPs and Na+ trans-porters in rats with lithium-induced NDI. Am J Physiol. 2000;279:F552–64.
120. Marples D, Dorup J, Knepper MA, et al. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Am Soc Nephrol. 1996;6:325.
121. Frokiaer J, Marples D, Knepper M, et al. Bilateral ureteral obstruction downregulates expres-sion of the vasopressin-sensitive aquaporin-2 water channel in rat kidney medulla. J Am SocNephrol. 1995;6:1012.
122. Teitelbaum I, Strasheim A, McGuinness S. Decreased aquaporin aquaporin-2 content inchronic renal failure. J Am Soc Nephrol. 1996;7:1273.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 22 of 25

123. Sands JM, Naruse M, Jacobs JD, et al. Changes in aquaporin-2 protein contribute to the urineconcentrating defect in rats fed a low protein diet. J Clin Invest. 1996;97:2807–14.
124. Walker RJ, Weggery S, Bedford JJ, et al. Lithium-induced reduction in urinary concentrationability and aquaporin-2(AQP2) excretion in healthy volunteers. Kidney Int. 2005;67:291–4.
125. Kortenoeven ML, Li Y, Shaw S, et al. Amiloride blocks lithium entry through the sodiumchannel thereby attenuating the resultant nephrogenic diabetes insipidus. Kidney Int.2009;76:44–53.
126. Kishore BK, Ecelbarger CM. Lithium: a versatile tool for understanding renal physiology. AmJ Physiol Renal Physiol. 2013;304:F1139–49.
127. Christensen BM, Zuber AM, Loffing J, et al. alphaENaC-mediated lithium absorption pro-motes nephrogenic diabetes insipidus. J Am Soc Nephrol. 2011;22(2):253–61.
128. Kjaersgaard G, Madsen K, Marcussen N, et al. Tissue injury after lithium treatment in humanand rat postnatal kidney involves glycogen synthase kinase-3b-positive epithelium. AmJ Physiol Renal Physiol. 2012;302:F455–65.
129. Rao R, ZhangMZ, ZhaoM, et al. Lithium treatment inhibits renal GSK-3 activity and promotescyclooxygenase 2-dependent polyuria. Am J Physiol Renal Physiol. 2005;288:F642–9.
130. Rao R, Patel S, Hao C, et al. GSK3beta mediates renal response to vasopressin by modulatingadenylate cyclase activity. J Am Soc Nephrol. 2010;2:428–37.
131. Christensen BM, Marples D, Kim YH, et al. Changes in cellular composition of kidneycollecting duct cells in rats with lithium-induced NDI. Am J Physiol Cell Physiol. 2004;286:C952–64.
132. Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol.2008;19:1076–8.
133. Crawford JD, Kennedy GC. Chlorothiazide in diabetes insipidus renalis. Nature.1959;193:891–2.
134. Monnens L, Jonkman A, Thomas C. Response to indomethacin and hydrochlorothiazide innephrogenic diabetes insipidus. Clin Sci. 1984;66:709–15.
135. Rasher W, Rosendahl W, Henricho IA, et al. Congenital nephrogenic diabetes insipidus:vasopressin and prostaglandins in response to treatment with hydrochlorothiazide and indo-methacin. Pediatr Nephrol. 1987;1:485–90.
136. Jakobsson B, Berg U. Effect of hydrochlorothiazide and indomethacin on renal function innephrogenic diabetes insipidus. Acta Paediatr. 1994;83:522–5.
137. Alon U, Chan JCM. Hydrochlorothiazide-amiloride in the treatment of congenitalnephrogenic diabetes insipidus. Am J Nephrol. 1985;5:9–13.
138. Knoers N, Monnens LAH. Amiloride-hydrochlorothiazide in the treatment of congenitalnephrogenic diabetes insipidus. J Pediatr. 1990;117:499–502.
139. Pattaragarn A, Alon US. Treatment of congenital nephrogenic diabetes insipidus by hydro-chlorothiazide and cyclooxygenase-2 inhibitor. Pediatr Nephrol. 2003;18:1073–6.
140. Early LE, Orloff J. The mechanism of antidiuresis associated with the administration ofhydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest.1962;52:2418–27.
141. Shirley DG, Walter SJ, Laycock JF. The antidiuretic effect of chronic hydrochlorothiazidetreatment in rats with diabetes insipidus. Clin Sci. 1982;63:533–8.
142. Cesar KR, Magaldi AJ. Thiazide induces water reabsorption in the inner medullary collectingduct of normal and Brattleboro rats. Am J Physiol. 1999;277:F750–6.
143. Magaldi AJ. New insights into the paradoxical effect of thiazides in diabetes insipidus therapy.Nephrol Dial Transplant. 2000;15:1903–5.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 23 of 25

144. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-inducednephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Clco-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15:2836–43.
145. Morello J-P, Salahpour A, Laperriere A, et al. Pharmacological chaperones rescue cell-surfaceexpression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest.2000;105:887–95.
146. Robben JH, Sze M, Knoers NV, et al. Functional rescue of vasopressin V2 receptor mutants inMDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus.Am J Physiol Renal Physiol. 2007;292:F253–60.
147. Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment forX-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–43.
148. Jean-Alphonse F, Perkovska S, Frantz MC, et al. Biased agonist pharmacochaperones of theAVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol.2009;20:2190–203.
149. Robben JH, Kortenoeven ML, Sze M, et al. Intracellular activation of vasopressin V2 receptormutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc Natl Acad Sci U S A.2009;106:12195–200.
150. Li JH, Chou CL, Li B, et al. A selective EP4 PGE2 receptor agonist alleviates disease in a newmouse model of X-linked nephrogenic diabetes insipidus. J Clin Invest. 2009;119:3115–26.
151. Olesen ET, Rutzler MR, Moeller HB, et al. Vasopressin-independent targeting of aquaporin-2E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc Natl Acad SciU S A. 2011;108:12949–54.
152. Bouley R, Breton S, Sun TX, et al. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest.2000;106:1115–26.
153. Boone M, Kortenoeven M, Robben JH, et al. Effect of the cGMP pathway on AQP2expression and translocation: potential implications for nephrogenic diabetes insipidus.Nephrol Dial Transplant. 2010;25:48–54.
154. Bouley R, Pastor Soler N, Cohen O, et al. Stimulation of AQP2 insertion in renal epithelialcells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra).Am J Physiol Renal Physiol. 2005;288:F1103–12.
155. Bouley R, Lu HA, Nunes P, et al. Calcitonin has a vasopressin-like effect on aquaporin-2trafficking and urinary concentration. J Am Soc Nephrol. 2011;22:59–72.
156. Procino G, Barbieri C, Carmosino M, et al. Fluvastatin modulates renal water reabsorptionin vivo through increased AQP2 availability at the apical plasma membrane of collecting ductcells. Pflugers Arch. 2011;462:753–66.
157. Li W, Zhang Y, Bouley R, et al. Simvastatin enhances aquaporin-2 surface expression andurinary concentration in vasopressin-deficient Brattleboro rats through modulation of RhoGTPase. Am J Physiol Renal Physiol. 2011;301:F309–18.
158. Nomura N, Nunes P, Bouley R, et al. High-throughput chemical screening identifies AG-490as a stimulator of aquaporin 2 membrane expression and urine concentration. Am J PhysiolCell Physiol. 2014;307:C597–605.
159. Yang B, Zhao D, VerkmanAS. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidusin a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009;23:503–12.
160. Taiyab A, Sreedhar AS, Rao C. Hsp90 inhibitors, GA and 17AAG, lead to ER stress-inducedapoptosis in rat histiocytoma. Biochem Pharmacol. 2009;78:142–52.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 24 of 25

161. Sohara E, Rai T, Yang SS, et al. Pathogenesis and treatment of autosomal-dominant nephrogenicdiabetes insipidus caused by an aquaporin 2 mutation. Proc Natl Acad Sci U S A.2006;103:14217–22.
162. Bichet DG, Ruel N, Arthus MF, et al. Rolipram, a phosphodiesterase inhibitor, in the treatmentof two male patients with congenital nephrogenic diabetes insipidus. Nephron.1990;56:449–50.
Pediatric NephrologyDOI 10.1007/978-3-642-27843-3_36-1# Springer-Verlag Berlin Heidelberg 2014
Page 25 of 25
Recommended

















