HEMOGLOBINOPATIAS

Se ha estimado que aprox. 7% de la población mundial - 400 millones de personas- son portadores heterocigotas de estos desórdenes. Es decir que 300.000- 400.000 RN portadores de alguna hemoglobinopatía severa nace cada año. Se ha estimado que hay unos 100.000 pacientes con homocigosis para la beta talasemia vivos en el mundo.
EPIDEMIOLOGIA
Henri WAJCMAN, MD, Ph.D 2005

• Si bien estas alteraciones se dan con alta frecuencia en regiones tropicales, las migraciones poblacionales han asegurado que en la actualidad se encuentren en todo el mundo.
EPIDEMIOLOGIA

HEMOGLOBINAHEMOGLOBINACLASIFICACIÓN
•A) DEFECTOS ESTRUCTURALES DE LAS GLOBINAS.
•B) DISMINUCIÓN EN LA SÍNTESIS DE GLOBINA.
•C) AMBOS DEFECTOS SIMULTANEAMENTE.

DEFINICIÓN:
ALTERACIONES DE LA GLOBINA, SECUNDARIAS A MUTACIONES GENÉTICAS, CUYA CONSECUENCIA ES UNA MODIFICACIÓN ESTRUCTURAL DE LA MOLÉCULA DE GLOBINA
HEMOGLOBINOPATÍAS ESTRUCTURALES.

• Aunque el sufijo “patía”hace pensar en padecimiento para el enfermo, en su gran mayoría son asintomáticas.

• De las que presentan expresividad clínica, aunque esta es variable, el denominador común es la
anemia hemolítica

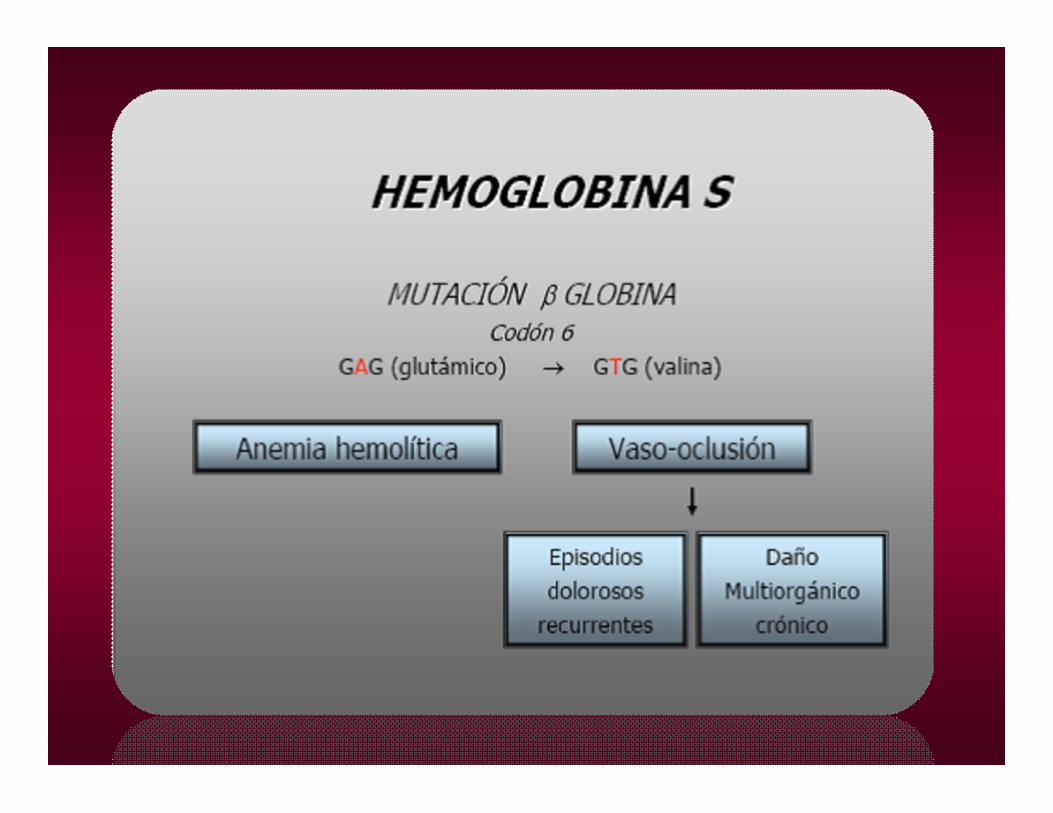
•1910 Herrick describe la anemia falciforme, posteriormente en 1949 Pauling descubre la hemoglobina Scomo causante de dicha anemia.
NOMENCLATURA

• http://globin.bx.psu.edu/hbvar/menu.html• HbVar: A database of Human Hemoglobin
Variants: 983 May, 2008
• NOMENCLATURA CIENTÍFICA:• (A3)6 GLU VAL

HEMOGLOBINOPATIASHEMOGLOBINOPATIAS
•INTERNATIONAL HEMOGLOBIN INFORMATION CENTER DE AUGUSTA, GEORGIA (EEUU), PUBLICA REGULARMENTE EN LA REVISTA HEMOGLOBIN,UN CATÁLOGO ACTUALIZADO DE TODAS LAS HEMOGLOBINOPATÍAS ESTRUCTURALES.

ClasificaciClasificacióónn
• Se pueden clasificar en unos pocos grupos:
• El primer grupo son las Hbs que causan serios problemas de salud: Ejemplo la Hb S en poblaciones de origen africano o la Hb E en poblaciones del sudeste asiático. El propósito es doble: diagnóstico clínico y asesoramiento genético.

ClasificaciClasificacióónn
• El segundo grupo incluye las variantes menos comunes, pero que se encuentran en poblaciones donde hay una variante presente Por ejemplo la Hb C, Hb O Arab or Hb D Punjab se encuentran en las mismas poblaciones que la Hb S. En sí mismas no tienen efecto patológico, pero sí combinadas, conducen a los sickle cell disease syndrome.

ClasificaciClasificacióónn
Otro grupo son las variantes raras que causan desórdenes hematológicos:
• Incluye a las hemoglobinas inestablesque llevan anemia hemolítica crónica
• Variantes con afinidad aumentada por el oxígeno que conducen a eritrocitosis
• Variantes con afinidad disminuída por el oxígeno anemia y cianosis
• HbM (metahemoglobinemia).

DistribuciDistribucióón de la malaria en el n de la malaria en el mundo.mundo.

•PREVALENCIA Y DISTRIBUCIÓN GEOGRÁFICA.
La HbS presenta una incidencia de entre un 40% y 50% en algunas regiones de África, el 25% en Turquía, Arabia Saudí, Israel y sur de la India, y el 32% en algunas zonas muy restringidas del sur de Europa (Sicilia, Chipre, Grecia).
La HbS y la HbC pueden afectar también la raza blanca y su incidencia es variable en diferentes regiones del área mediterránea como, por ejemplo, Grecia, Chipre, sur de Italia, Sicilia, España y Portugal
En Rosario, 5/1500 cordones.( 0,33%)

ClasificaciClasificacióónnHb S también se halla en todos los países donde un importante fracción de la población es de origen africano: América del norte, islas del Caribe. Con las recientes inmigraciones HbS es frecuente en países industrializados, como sucede , por ejemplo, en la región parisina donde aproximadamente un 0.1% de los RN tienen sickle cellsyndrome.
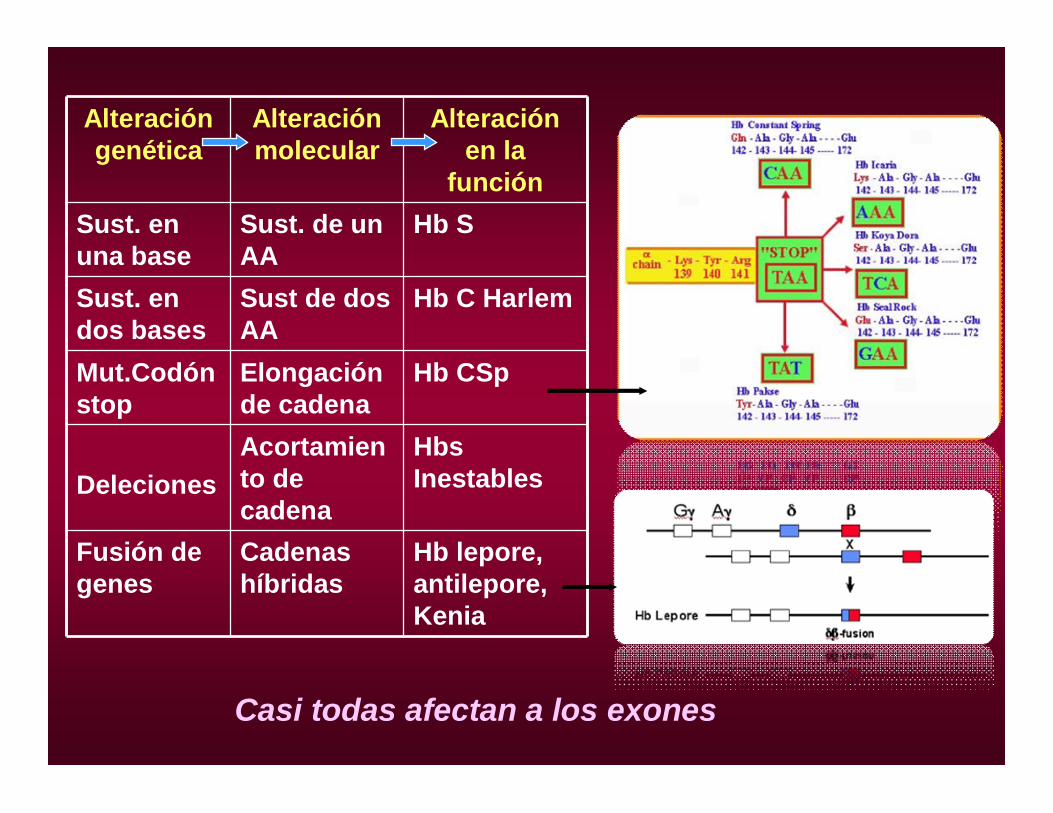
Alteración genética
Alteración molecular
Alteración en la
funciónSust. en una base
Sust. de un AA
Hb S
Sust. en dos bases
Sust de dos AA
Hb C Harlem
Mut.Codónstop
Elongación de cadena
Hb CSp
DelecionesAcortamiento de cadena
HbsInestables
Fusión de genes
Cadenas híbridas
Hb lepore, antilepore, Kenia
Casi todas afectan a los exones
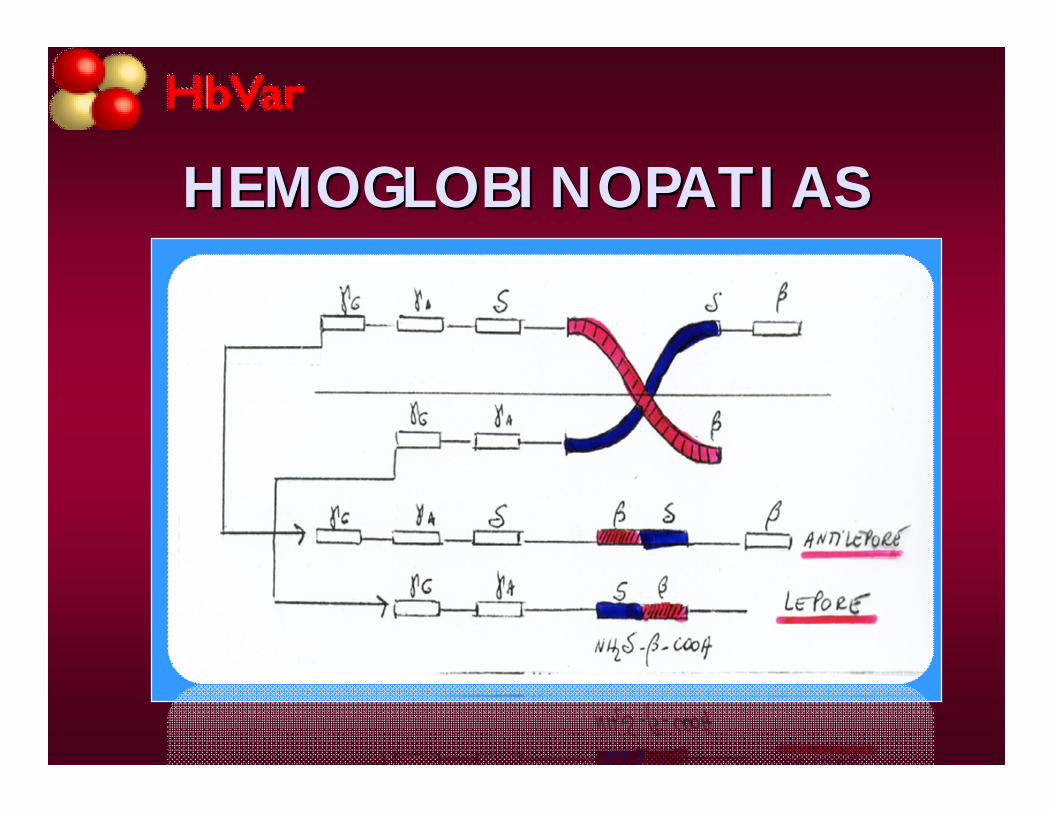
HEMOGLOBINOPATIASHEMOGLOBINOPATIAS

• Las mutaciones puntuales se dan en el 92% de las 982 variantes conocidas, y es el defecto genético responsable más frecuente de hemoglobinas anormales. Las pequeñas deleciones o inserciones de uno a 5 residuos se observan en aproximadamente el 5% de los casos.
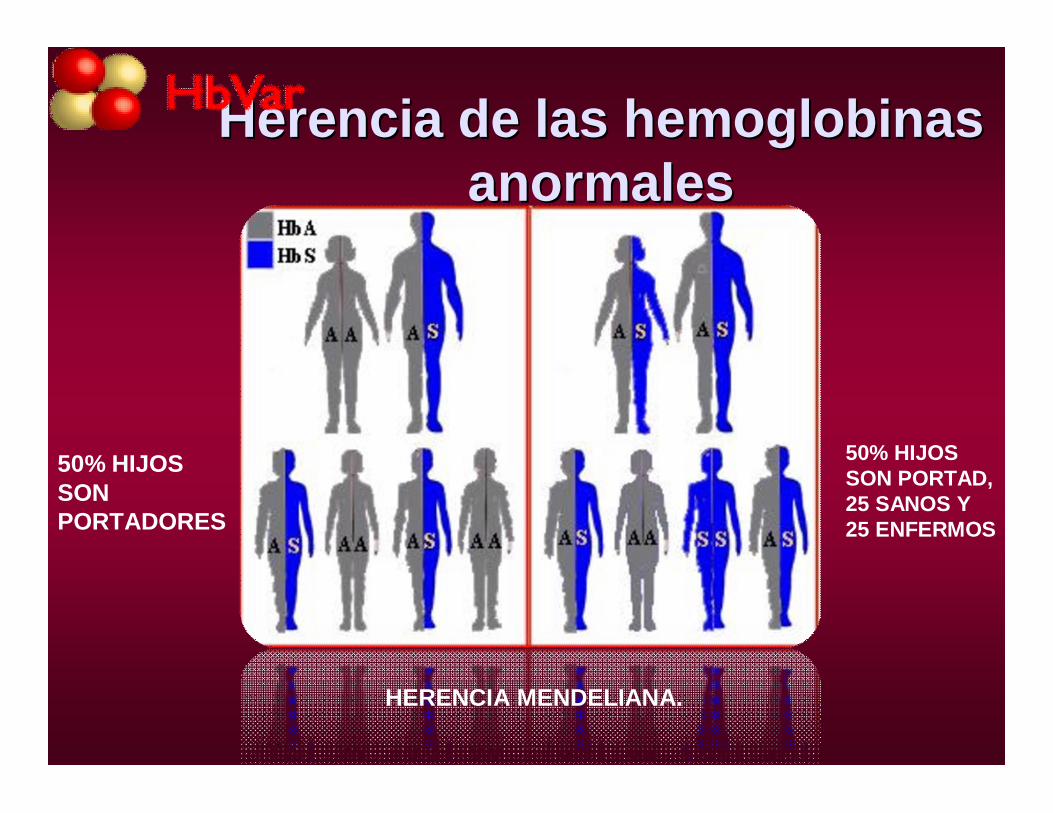
Herencia de las hemoglobinas Herencia de las hemoglobinas anormalesanormales
HERENCIA MENDELIANA.
50% HIJOS SON PORTADORES
50% HIJOS SON PORTAD, 25 SANOS Y 25 ENFERMOS

HEMOGLOBINOPATIASHEMOGLOBINOPATIAS•HERENCIA
•CLÍNICO: AUTOSÓMICO DOMINANTE O RECESIVO.
•LABORATORIO: CODOMINANTES

• Las hemoglobinopatíasestructurales se clasifican en
cuatro grandes grupos:• 1. Hemoglobinas con disminución de la
solubilidad (HbS y HbC).• 2. Hemoglobinas con disminución de la
estabilidad molecular (hemoglobinas inestables).
• 3. Hemoglobinas con alteración de la afinidad por el oxígeno.
• 4. Metahemoglobinas (HbM).

HEMOGLOBINA SHEMOGLOBINA S•INCIDENCIA Y DISTRIBUCIÓN GEOGRÁFICA.
•Corre paralela a las áreas en las que existe o ha existido paludismo endémico.
•La mayor incidencia corresponde al Africa tropical, un 45% de la población es portadora de la mutación.
•En el Caribe 1/100 indiv. es portador y en EEUU la incidencia de anemia drepanocítica es de 1/700 nacimientos

Definiciones.Definiciones.
• “Sickle cell disease”: conjuntos de desórdenes genéticos autosómicos recesivos caracterizados por la presencia de hemoglobina S.
• Sickle cell anemia, tienen dos copias de esta variante S/S, su componente hemoglobínico mayor es la Hb S
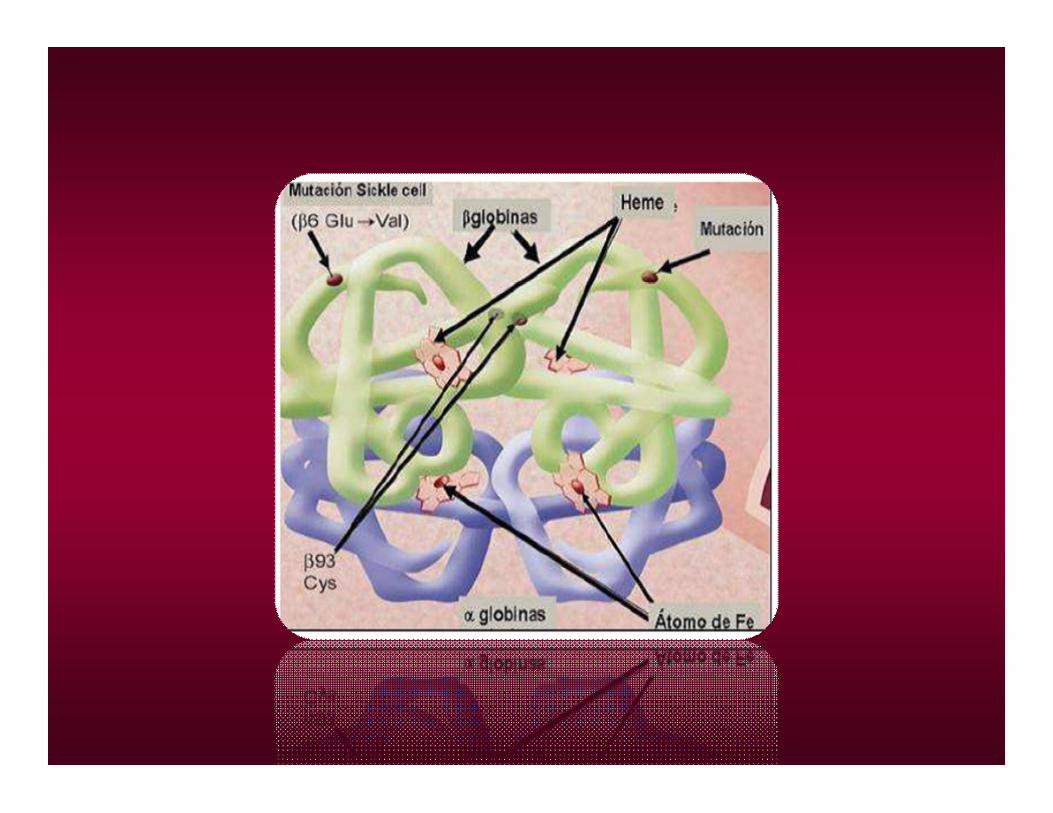

• Los individuos afectados con otros tipos de Sickle cell anemia son dobles heterocigotas, poseen una copia del gen S y otra anormalidad en el otro gen de beta globina. Ej: S/C, S/Beta tal.
• Los individuos portadores, tienen una sola copia del gen beta S y una copia del gen beta normal. A/S. “Sickle cell trait”

Cuando se desoxigena polimeriza
Cada polímero está formado por tetrámeros de desoxihemoglobina S que se disponen formando haces longitudinales que se unen entre sí
HEMOGLOBINA SHEMOGLOBINA SLa polimerización de la Hb S es el disparador de las alteraciones observadas en la enfermedad
Clínica
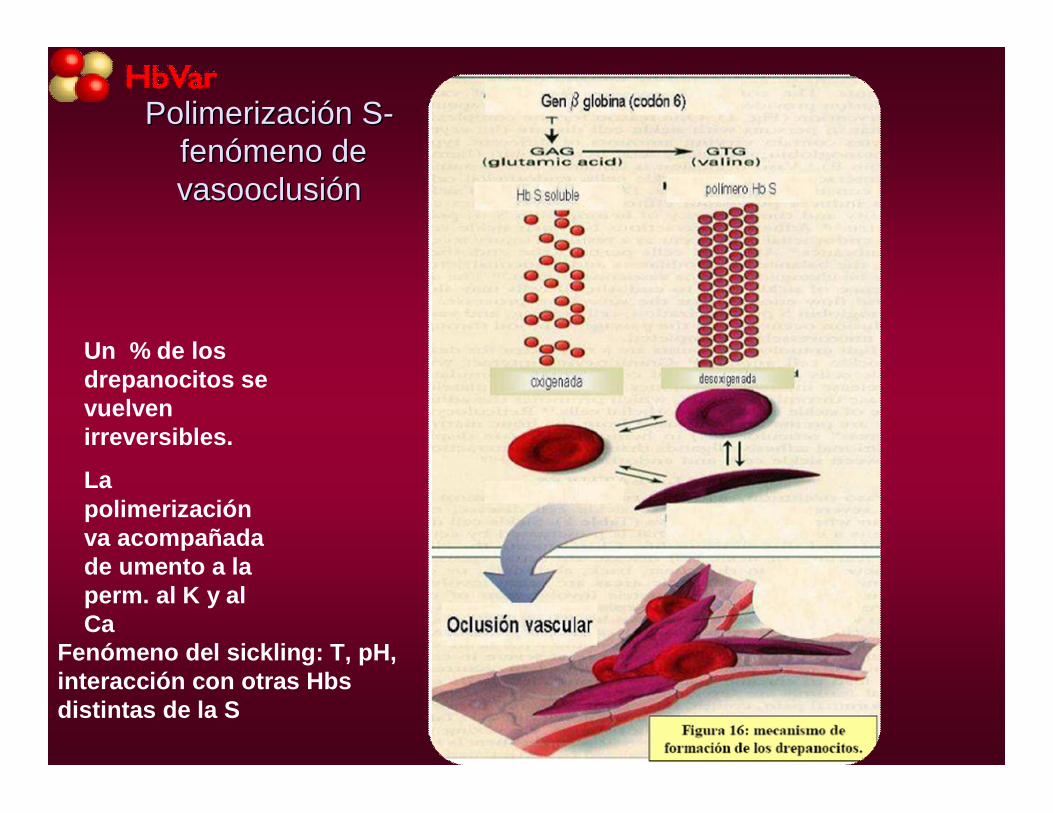
PolimerizaciPolimerizacióón Sn S--fenfenóómeno de meno de vasooclusivasooclusióónn
Fenómeno del sickling: T, pH, interacción con otras Hbsdistintas de la S
Un % de los drepanocitos se vuelven irreversibles.
La polimerización va acompañada de umento a la perm. al K y al Ca
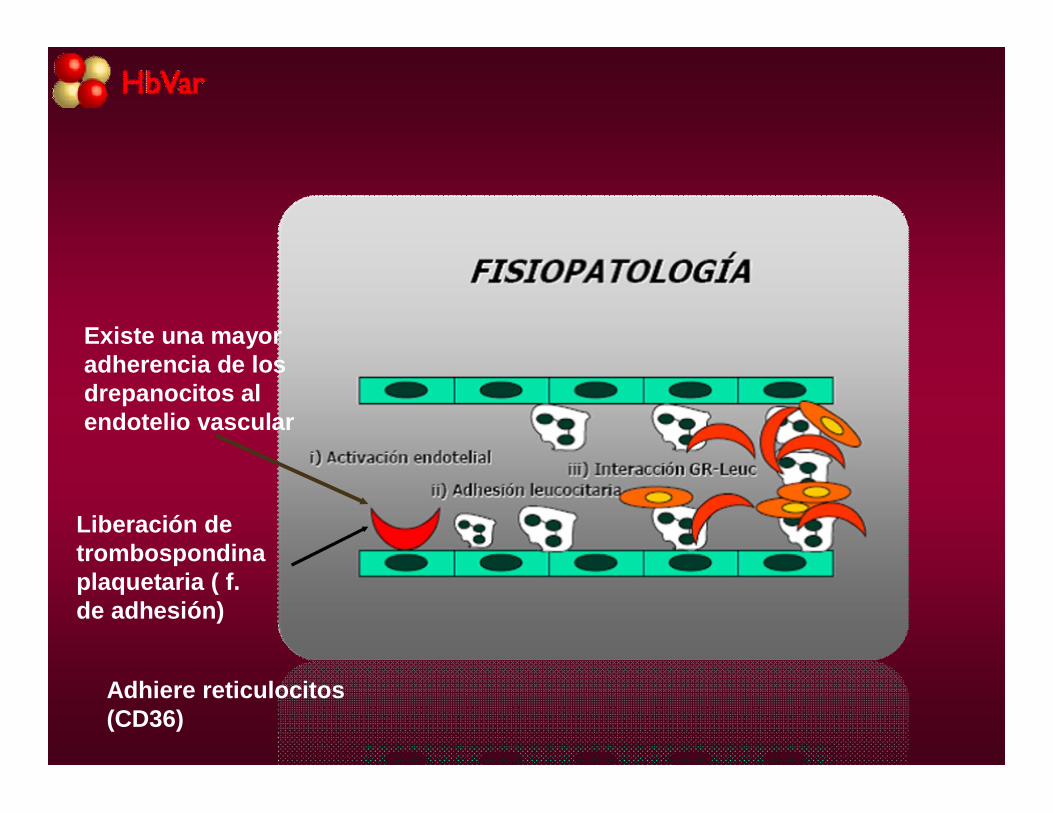
Existe una mayor adherencia de los drepanocitos al endotelio vascular
Liberación de trombospondinaplaquetaria ( f. de adhesión)
Adhiere reticulocitos (CD36)
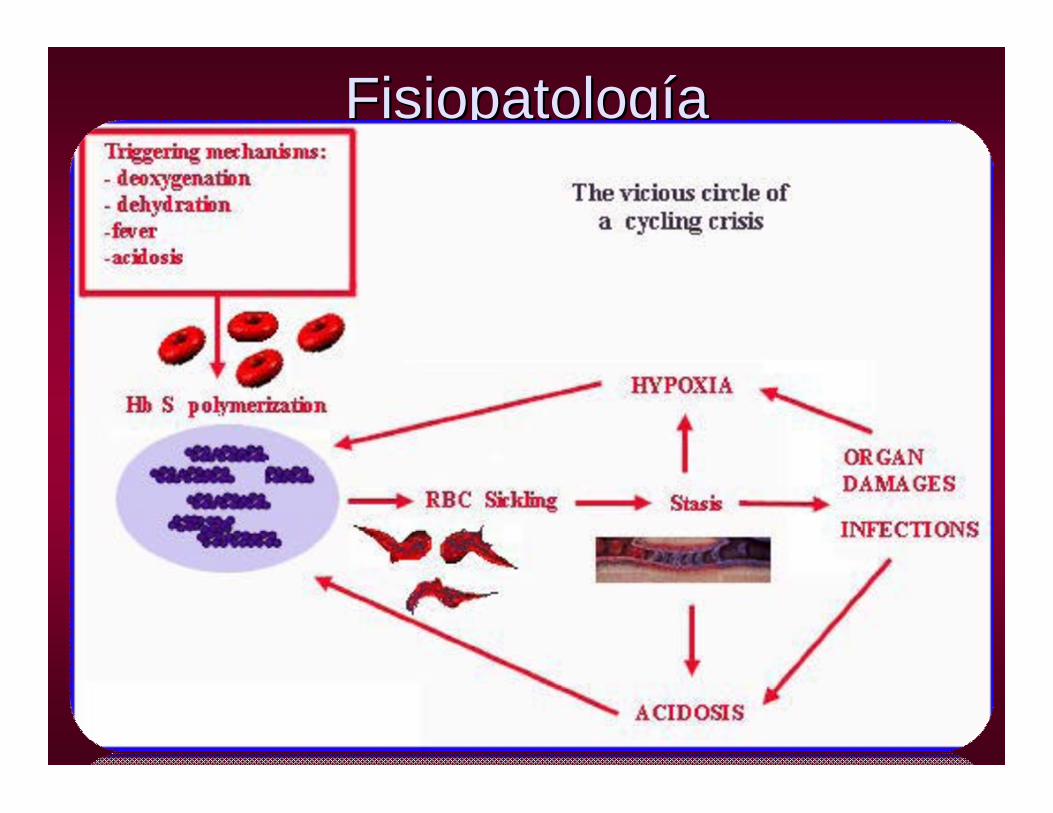
FisiopatologFisiopatologííaa

INTRAVASCULAR : Lisis por mayor sensibilidad al Complemento, y pérdida de la deformabilidad de los drepanocitos
EXTRAVASCULAR: Mayor sensibilidad de los drepanocitos al efecto macrofágico de los monocitos.
• PROCESO DE HEMOLISIS

HEMOGLOBINA SHEMOGLOBINA SMANIFESTACIONES CLÍNICAS.
PORTADORES. ASINTOMÁTICO. A/S
Casi no tienen sintomatología clínica.
Frecuentes: hematurias esporádicas, en condiciones de hipoxia accidentes trombóticoscomo infarto esplénico, altitud, infecciones.

HEMOGLOBINA SHEMOGLOBINA SANEMIA DREPANOCÍTICA: HOMOCIGOTA. S/S
ANEMIA FALCIFORME.
FASE ESTACIONARIA (1- 4 AÑOS)
Anemia hemolítica crónica moderada, hiperesplenismo, con complicaciones vasooclusivas de carácter local que llevan a la autoesplenectomía

HEMOGLOBINA SHEMOGLOBINA SFASE DE EXPRESIVIDAD AGUDA ( a partir de los 4 años)
ANEMIA GRAVE
SÍNDROME VASOOCLUSIVO GENERALIZADO (PULMÓN, RIÑÓN, HUESOS)
CRISIS DE DOLOR AGUDO, MANIFESTACIÓN CARACTERÍSTICA.
INFECCIONES RECIDIVANTES
OSTEOMIELITIS- OCLUSIONES DE VASOS CEREBRALES-SÍNDROMES TORÁCICOS AGUDOS DE ORIGEN VASCULAR.

HEMOGLOBINA SHEMOGLOBINA SFASE DE EXPRESIVIDAD CRÓNICA (pacientes que han logrado sobrevivir la primera infancia) adolescencia y edad adulta.
•RETRASO EN EL CRECIMIENTO
•NECROSIS ÓSEAS
•COMPLICACIONES VISUALES.
•COMPLICACIONES PULMONARES
•COMPLICACIONES CARDÍACAS,RENALES,HEPATOBILIARES

HEMOGLOBINA SHEMOGLOBINA S
•HOMOCIGOTA:
ANEMIA NORMOCÍTICA O LIGERAMENTE MACROCÍTICA
Hb: 6-8 g/dl
reticulocitos altos
SR: macrocitos, megalocitos, drepanocitosreversibles y drepanocitos irreversibles, C de H. Jolly.

HEMOGLOBINA SHEMOGLOBINA S
DIAGNÓSTICO DE LABORATORIO
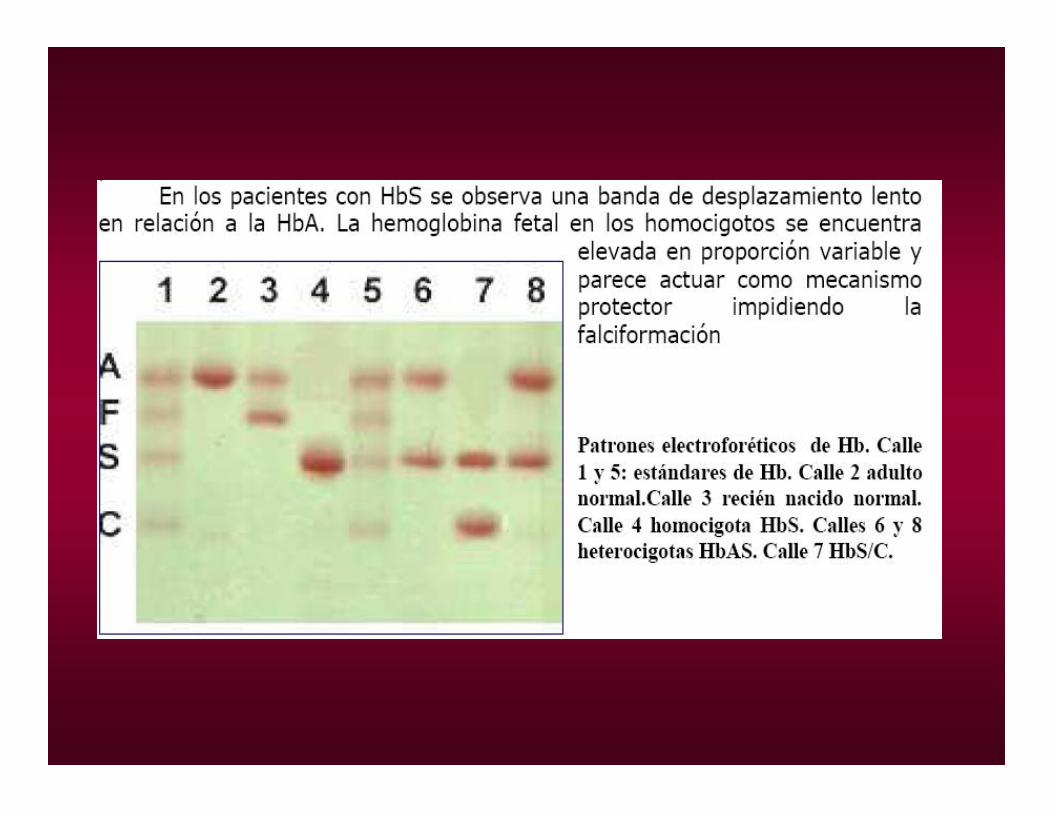
PORTADOR. Hemograma,electroforesis
HOMOCIGOTA a pH alcalino, pbas de solubilidad y de falciformación
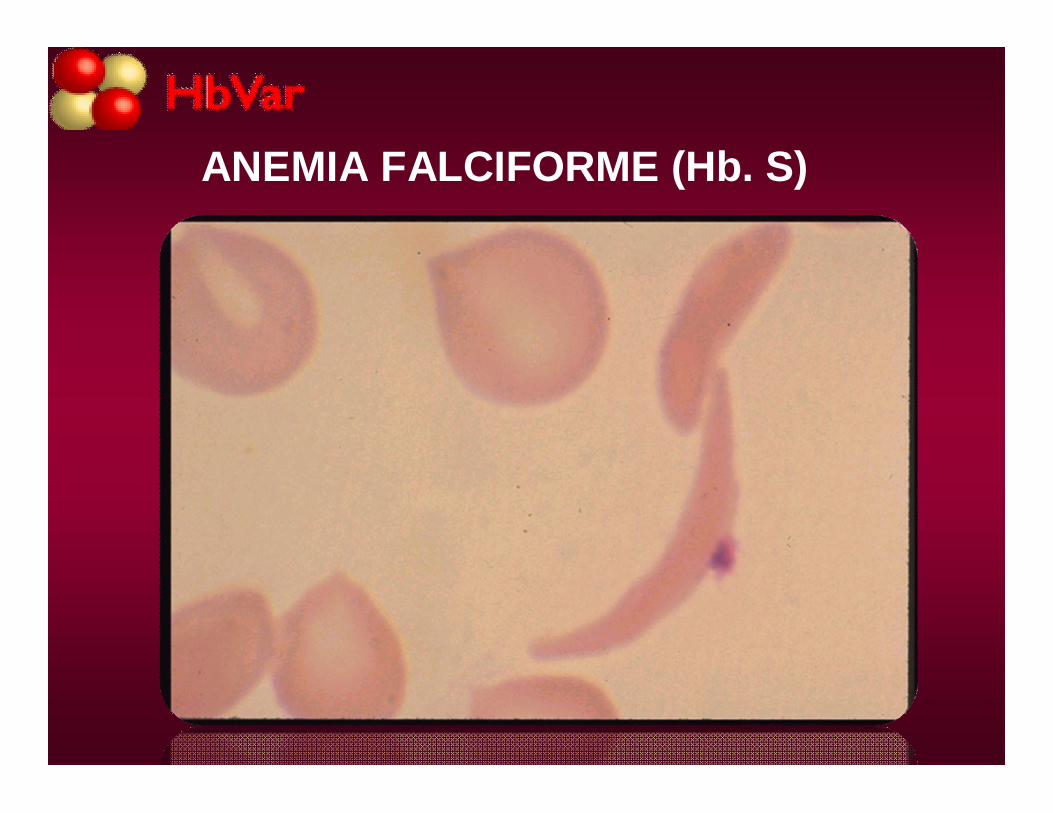
ANEMIA FALCIFORME (Hb. S)
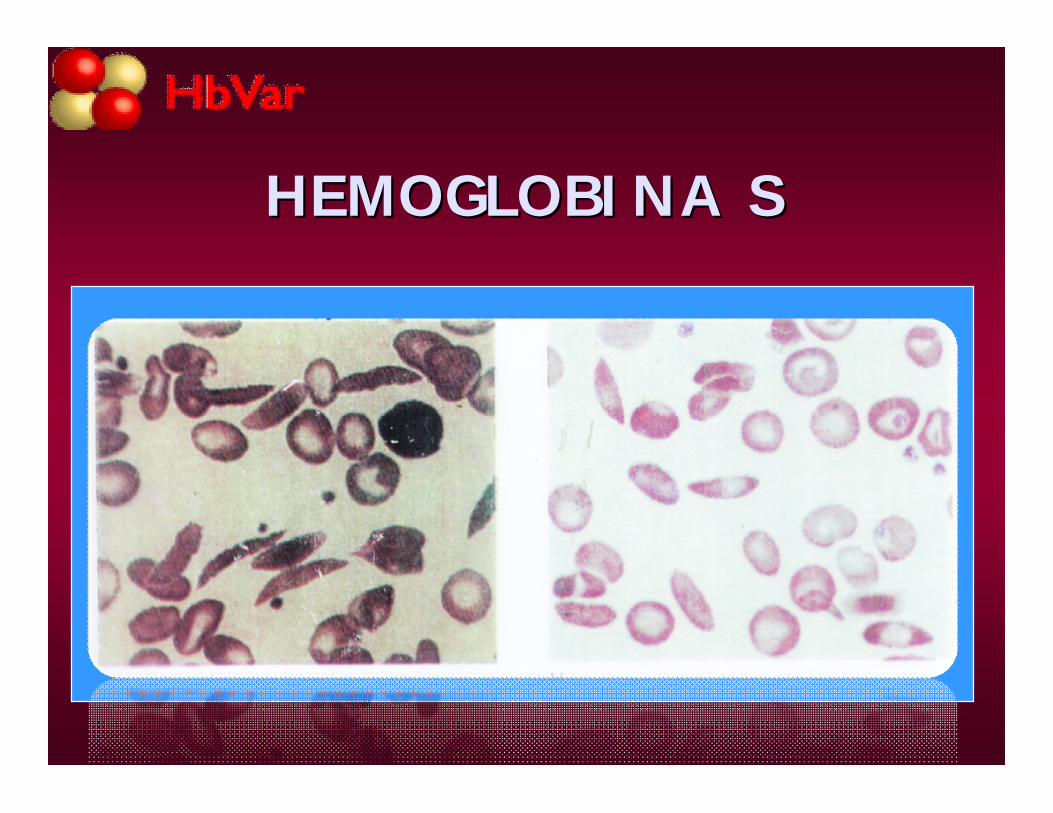
HEMOGLOBINA SHEMOGLOBINA S
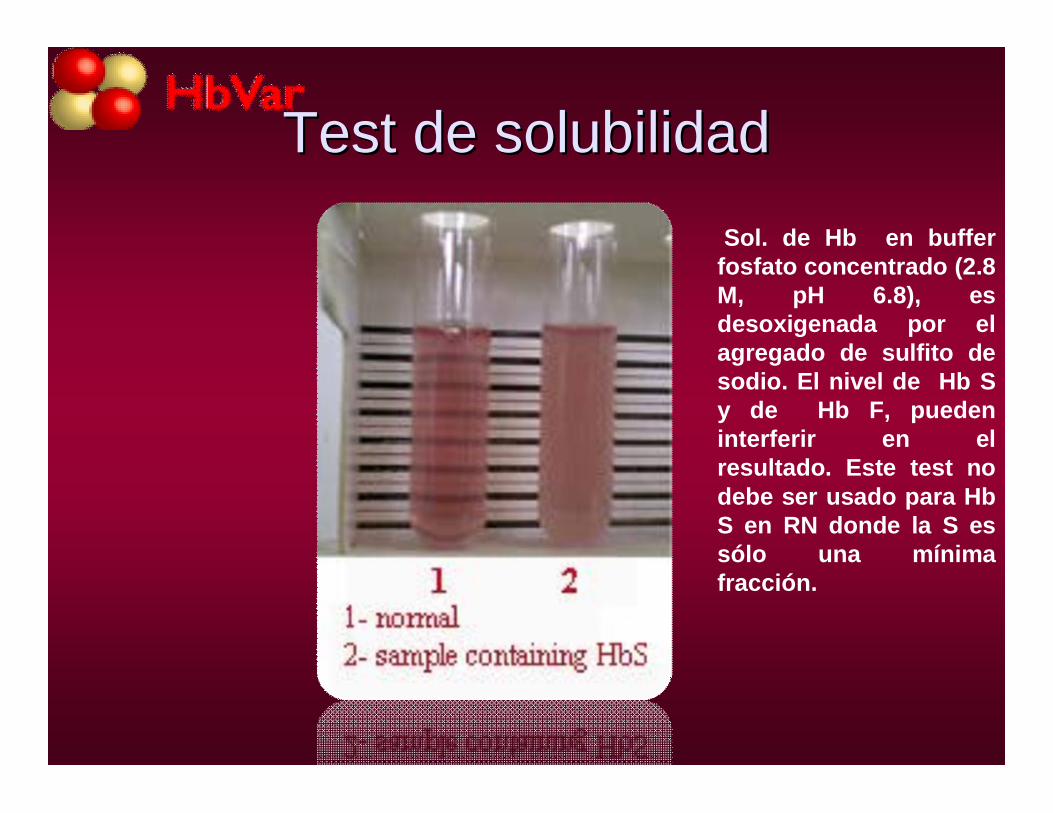
Test de solubilidadTest de solubilidadSol. de Hb en buffer
fosfato concentrado (2.8 M, pH 6.8), es desoxigenada por el agregado de sulfito de sodio. El nivel de Hb S y de Hb F, pueden interferir en el resultado. Este test no debe ser usado para HbS en RN donde la S es sólo una mínima fracción.
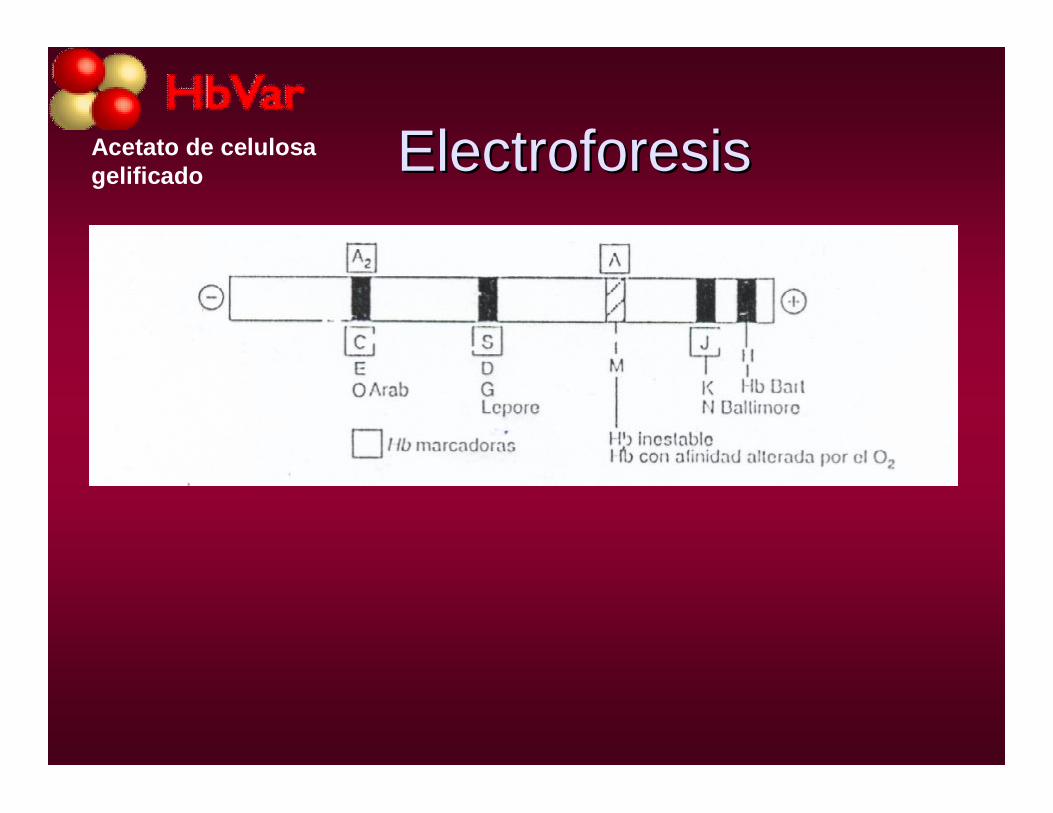
ElectroforesisElectroforesisAcetato de celulosa gelificado
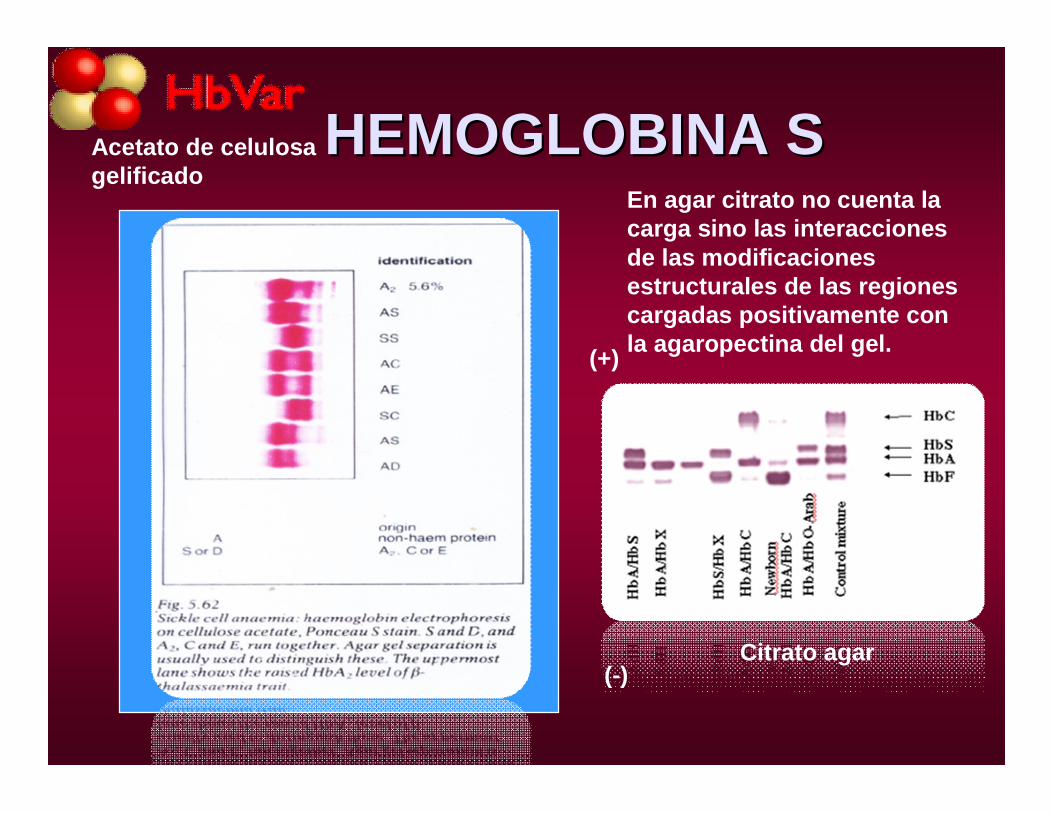
HEMOGLOBINA SHEMOGLOBINA S
Citrato agar
Acetato de celulosa gelificado
En agar citrato no cuenta la carga sino las interacciones de las modificaciones estructurales de las regiones cargadas positivamente conla agaropectina del gel.
(-)
(+)
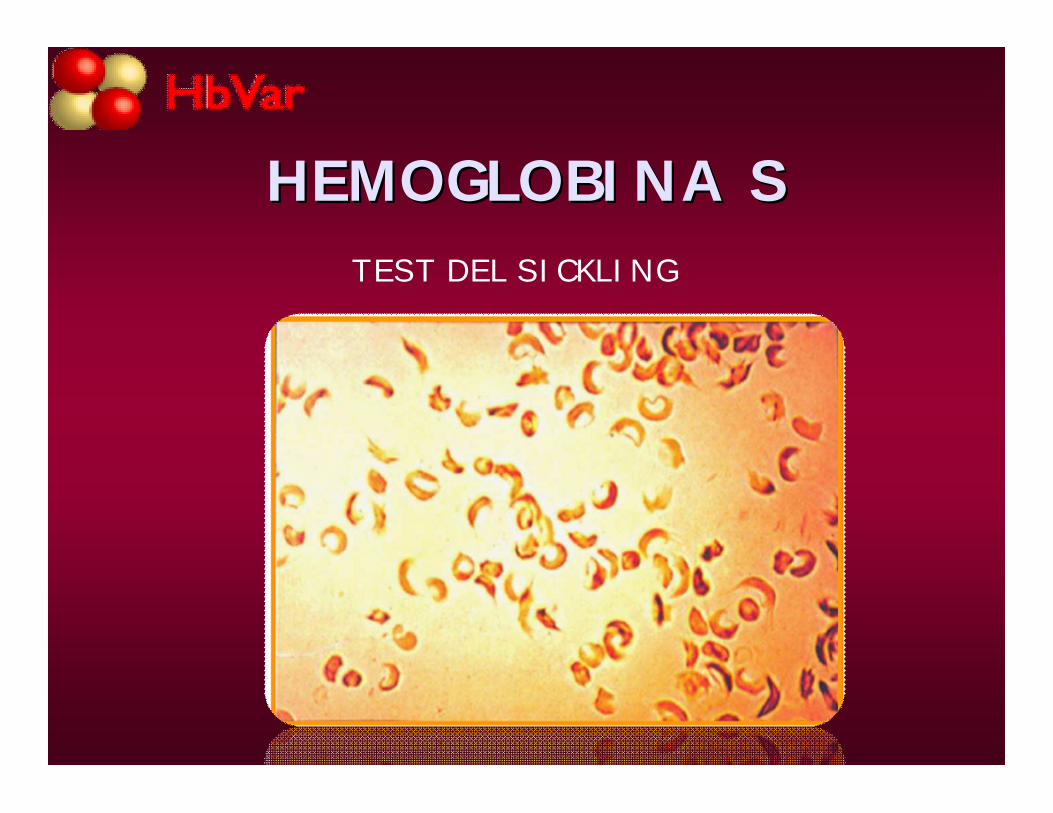
HEMOGLOBINA SHEMOGLOBINA STEST DEL SICKLING
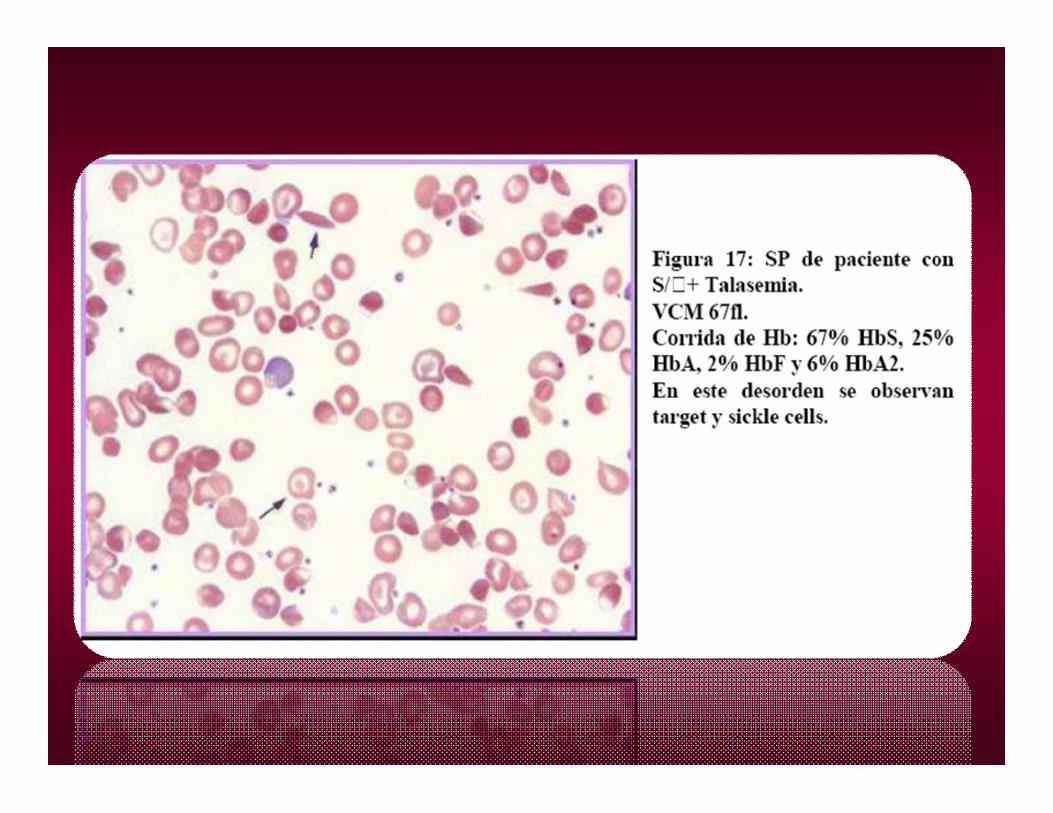

HEMOGLOBINA CHEMOGLOBINA C
•ES LA SEGUNDA CON SOLUBILIDAD ALTERADA MÁS FRECUENTE DESPUÉS DE LA S.
• 6 GLU...LYS, ALTERACIÓN DE LA CARGA ELECTRICA Y DISMINUCIÓN DE LA SOLUBILIDAD.

HEMOGLOBINA CHEMOGLOBINA C•HETEROCIGOTA: ASINTOMÁTICO CLÍNICO Y HEMATOLÓGICO.
•ELECTROFORÉTICAMENTE ES MÁS LENTA QUE LA A,TIENE REDUCIDA SOLUBILIDAD EN CONDICIONES FISIOLÓGICAS, FAVORECECIENDO LA FORMACIÓN DE ESTRUCTURAS PARACRISTALINAS.
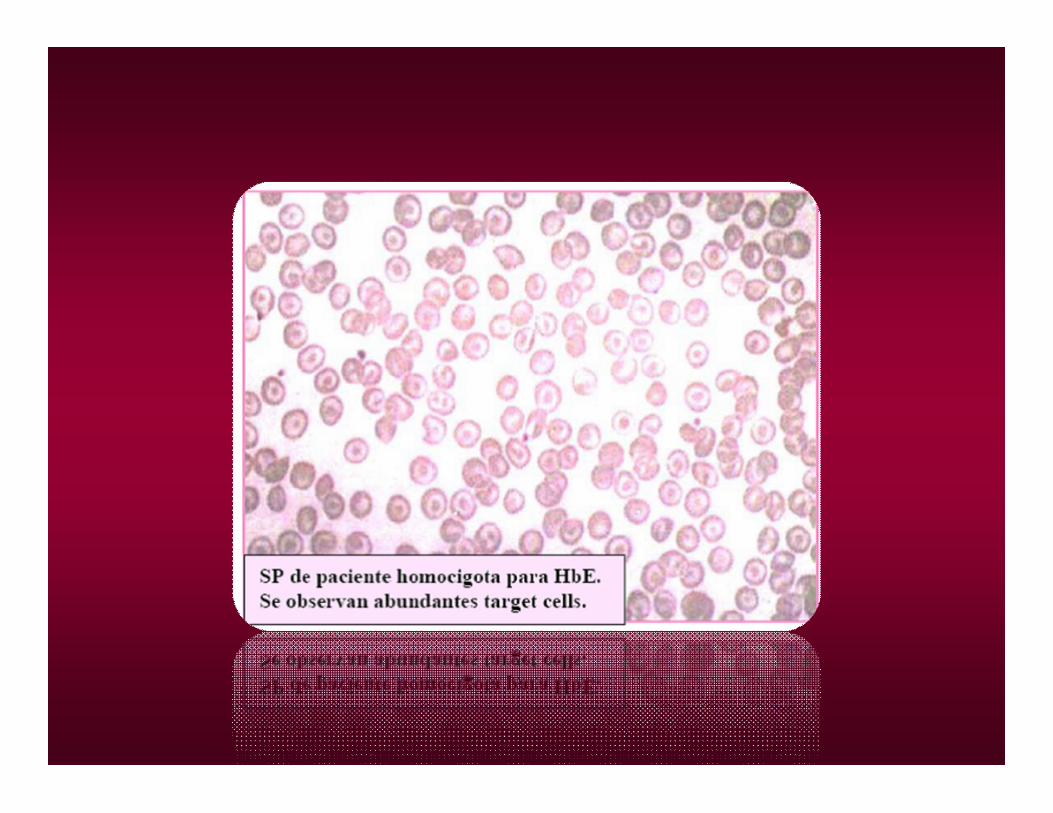
•HOMOCIGOTA (C/C). LIGERA ANEMIA HEMOLÍTICA CRÓNICA Y ESPLENOMEGALIA. TARGET CELLS.

HEMOGLOBINA CHEMOGLOBINA C
•ELECTROFORESIS EN MEDIO ALCALINO
•Hb C=A2=E=OARAB.
•ELECTROFORESIS EN AGAR A pH ÁCIDO.
La asociación SC conduce a una forma de S C disease algo más leve que la homocigosis S pero con frecuentes complicaciones.
36% pacientes desarrollan retinopatía proliferativa, necrosis aséptica de cabeza de fémur u Húmero, hematuria y tendencia a trombosis en embarazo y puerperio.
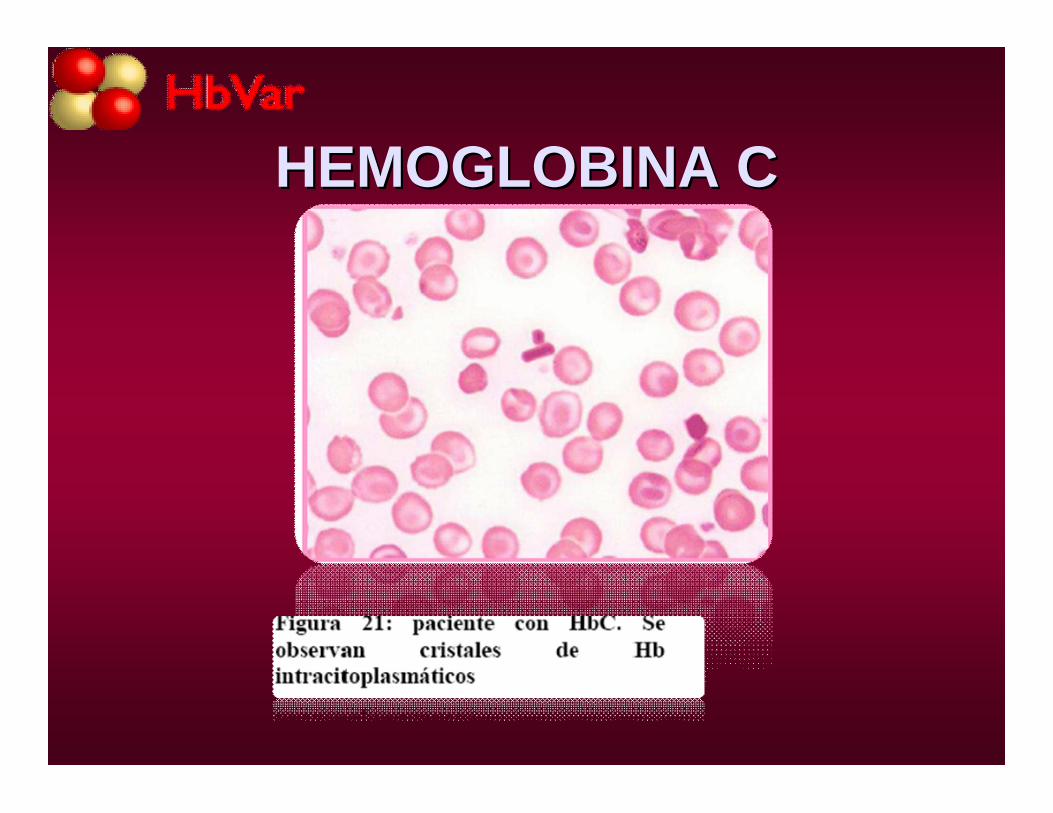
HEMOGLOBINA CHEMOGLOBINA C
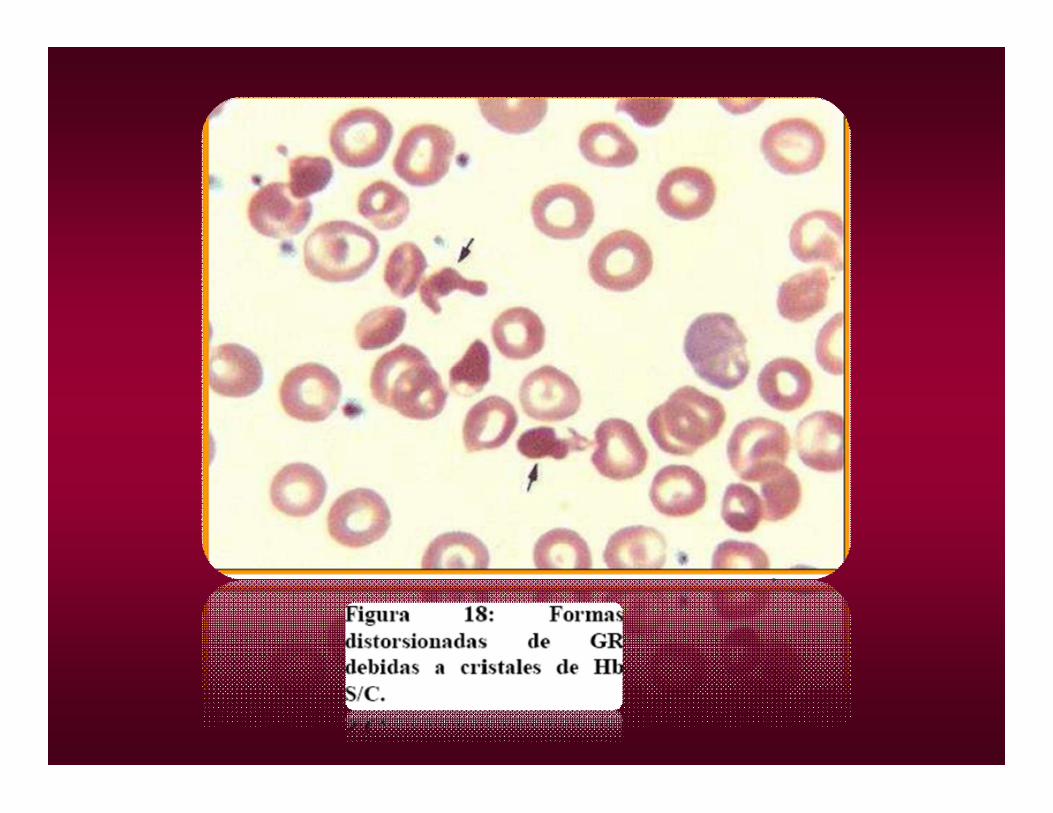

HEMOGLOBINA DHEMOGLOBINA D• HETEROCIGOTA A/D ASINTOMÁTICO.
•HOMOCIGOTA D/D 95 % D, TRAZAS DE A, CON F Y A2 NORMALES, NO DA SICKLING, CORRE COMO LA S. CLÍNICAMENTE ANEMIA HEMOLITICA MODERADA CON ESPLENOMEGALIA.

HEMOGLOBINA EHEMOGLOBINA E • 26 GLU...LYS, MUY FRECUENTE DISTRIBUIDAS POR MUCHAS ÁREAS DE LA GEOGRAFÍA MUNDIAL, ESPECIALMENTE EN EL SUDESTE ASIÁTICO.
•LA MUTACIÓN QUE DA ORIGEN A LA Hb E ACTIVA UNA REGIÓN CRÍPTICA DEL ADN QUE AL COMPETIR CON LA REGIÓN NORMAL DISMINUYE LA SÍNTESIS DE RNA m Y CON ELLO LA SÍNTESIS DE CADENA .
•E/E: INTENSA MICROCITOSIS, HIPOCROMIA, ANEMIA LEVE, INDISTINGUIBLE DE UN SINDROME TALASÉMICO.
Heterocigota asintomático, leve microcitosis e hipocromia.

HEMOGLOBINAS HEMOGLOBINAS INESTABLESINESTABLES
• SON UN GRUPO DE VARIANTES DE Hb QUE EXPERIMENTAN DESNATURALIZACIÓN DENTRO DEL ERITROCITO Y PRECIPITAN DANDO CUERPOS DE HEINZ.
•SE HAN DESCRIPTO MAS DE 200 VARIANTES (sólo la mitad son responsables de trastornos clínicos.)
•GRADO DE HEMOLISIS. DESDE LEVE A MODERADO, DEPENDIENDO DEL TIPO DE SUSTITUCIÓN QUE HAYA TENIDO LUGAR
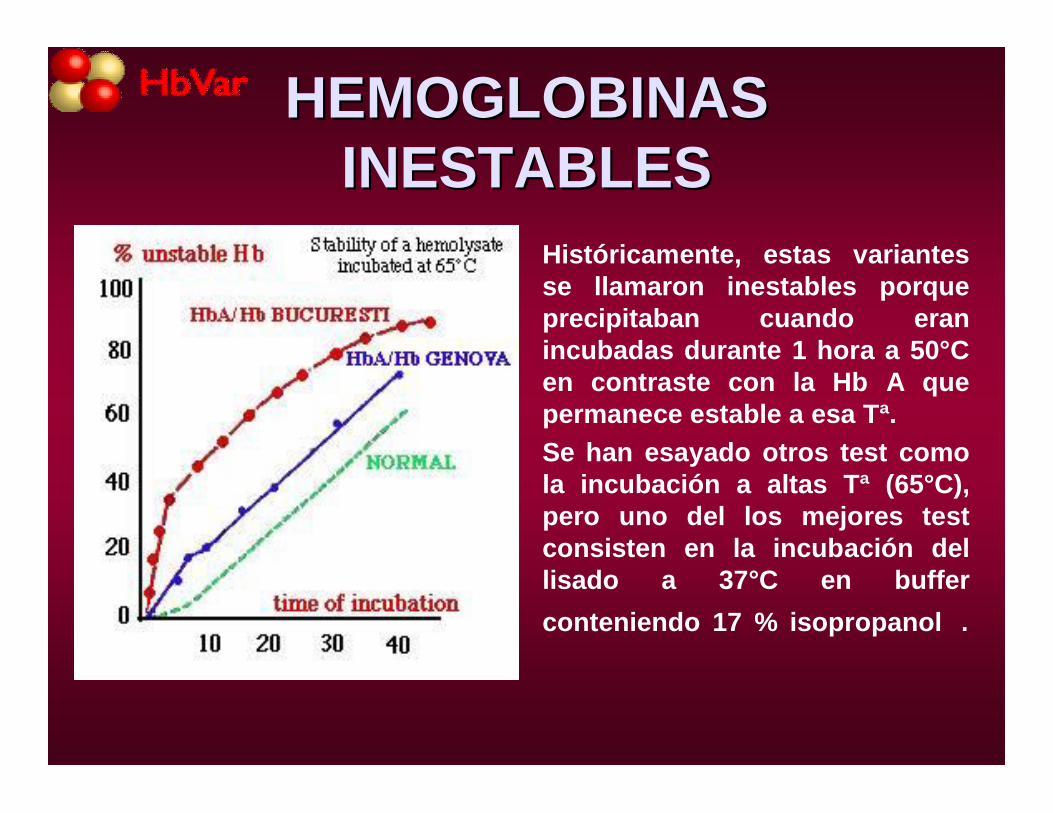
• Históricamente, estas variantes se llamaron inestables porque precipitaban cuando eran incubadas durante 1 hora a 50°C en contraste con la Hb A que permanece estable a esa Tª.
• Se han esayado otros test como la incubación a altas Tª (65°C), pero uno del los mejores test consisten en la incubación del lisado a 37°C en buffer conteniendo 17 % isopropanol .
HEMOGLOBINAS HEMOGLOBINAS INESTABLESINESTABLES

• Desde el punto de vista biológico y clínicolas Hemoglobinas inestables son muy heterogeneas.
• Algunas son sólo inestables in vitro y no hay anormalidades hematológicas.
•En contraste algunas son variantes tan severamentes inestablesson destruídas minutos u horas después de haber sido sintetizadas y conducen biológicamente a eritropoyesis inefectiva y clínicamente thalassemia-like syndromes. Su identificación requiere biología molecular.
HEMOGLOBINAS HEMOGLOBINAS INESTABLESINESTABLES

• La presentación más clásica es intermedia entre estos dos situaciones anteriores: una cantidad significativa de la variante presente en los GR de sangre periférica y precipita dentro de ellos cuando es sometida a stress oxidativo.
• Hay mutaciones de novo con fenotipos dominantes.
HEMOGLOBINAS HEMOGLOBINAS INESTABLESINESTABLES

• HERENCIA:
AUTOSÓMICA DOMINANTE.
EXPRESIÓN COMPLETA EN EL HETEROCIGOTA
•FISIOPATOLOGÍA:
INESTABILIDAD DEPENDE DEL TIPO DE MUTACIÓN QUE HAYA TENIDO LUGAR
HEMOGLOBINAS HEMOGLOBINAS INESTABLESINESTABLES
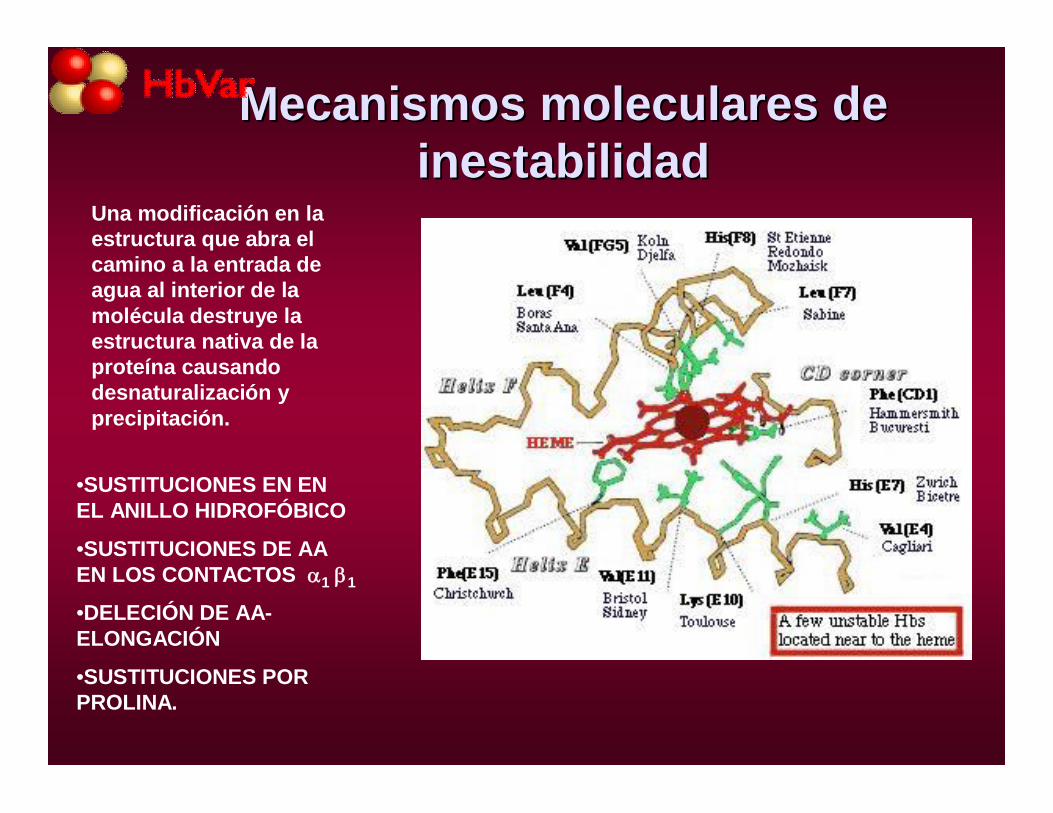
Mecanismos moleculares de Mecanismos moleculares de inestabilidadinestabilidad
Una modificación en la estructura que abra el camino a la entrada de agua al interior de la molécula destruye la estructura nativa de la proteína causando desnaturalización y precipitación.
•SUSTITUCIONES EN EN EL ANILLO HIDROFÓBICO
•SUSTITUCIONES DE AA EN LOS CONTACTOS 1 1
•DELECIÓN DE AA-ELONGACIÓN
•SUSTITUCIONES POR PROLINA.
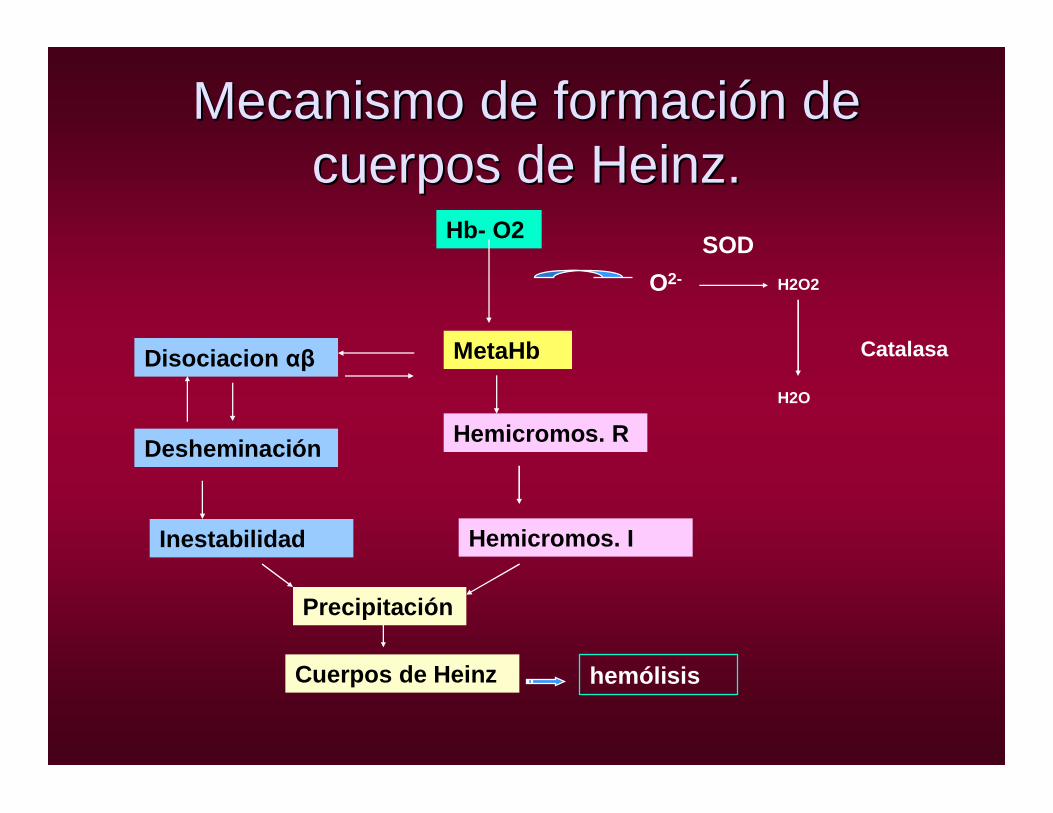
Mecanismo de formaciMecanismo de formacióón de n de cuerpos de Heinz.cuerpos de Heinz.
Hb- O2
MetaHbDisociacion αβ
Desheminación
Inestabilidad
Hemicromos. R
Hemicromos. I
Precipitación
Cuerpos de Heinz hemólisis
O2- H2O2
H2O
Catalasa
SOD

La presentación lo sugiere:
•Anemia hemolítica regenerativa (aumento de reticulocitos)
•Hallazdo de Heinz bodies en los extendidos de sangre periférica,estéril, incubada 24hs, y luego teñida con coloración supravital sugiere el diagnóstico. (Recordar que no son patognomónicos, aparecen en eritroenzimopatías, exposición a varias clases de agentes químicos, etc.)
HEMOGLOBINAS HEMOGLOBINAS INTESTABLESINTESTABLES
DIAGNÓSTICO DE UNA HEMOGLOBINA INESTABLE

HEMOGLOBINAS HEMOGLOBINAS INTESTABLESINTESTABLES
DIAGNÓSTICO DE UNA HEMOGLOBINA INESTABLE
• LABORATORIO: HEMOGLOBINA VARIABLE 7-10 g/dl
•SERIE ROJA: MACROCITOS, FORMAS HEMOLÍTICAS, HEMATIES POLICROMATÓFILOS.•PRUEBA DE ESTABILIDAD HEMOGLOBINICA.
IDENTIFICACIÓN: HPLC, espectroscopia de masa, biologia molecular.
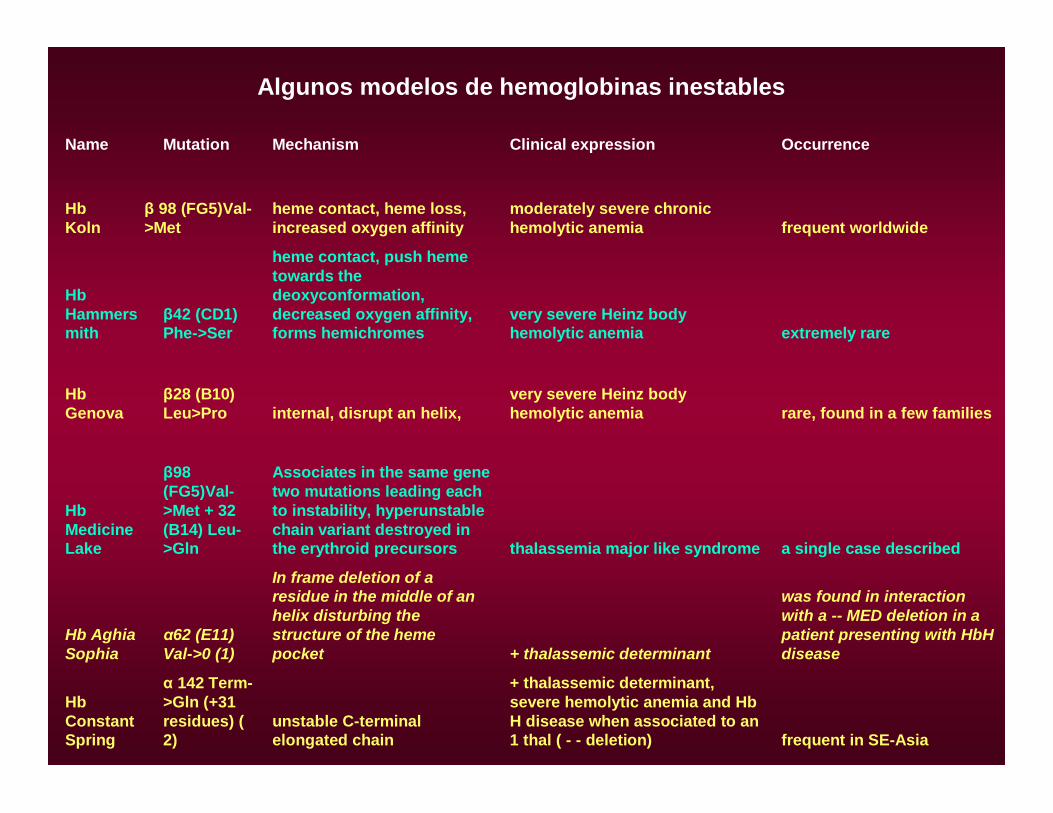
Name Mutation Mechanism Clinical expression Occurrence
Hb Koln
β 98 (FG5)Val->Met
heme contact, heme loss, increased oxygen affinity
moderately severe chronic hemolytic anemia frequent worldwide
Hb Hammersmith
β42 (CD1) Phe->Ser
heme contact, push heme towards the deoxyconformation, decreased oxygen affinity, forms hemichromes
very severe Heinz body hemolytic anemia extremely rare
Hb Genova
β28 (B10) Leu>Pro internal, disrupt an helix,
very severe Heinz body hemolytic anemia rare, found in a few families
Hb Medicine Lake
β98 (FG5)Val->Met + 32 (B14) Leu->Gln
Associates in the same gene two mutations leading each to instability, hyperunstable chain variant destroyed in the erythroid precursors thalassemia major like syndrome a single case described
Hb Aghia Sophia
α62 (E11) Val->0 (1)
In frame deletion of a residue in the middle of an helix disturbing the structure of the heme pocket + thalassemic determinant
was found in interaction with a -- MED deletion in a patient presenting with HbH disease
Hb Constant Spring
α 142 Term->Gln (+31 residues) ( 2)
unstable C-terminal elongated chain
+ thalassemic determinant, severe hemolytic anemia and Hb H disease when associated to an 1 thal ( - - deletion) frequent in SE-Asia
Algunos modelos de hemoglobinas inestables

HEMOGLOBINAS CON AFINIDAD HEMOGLOBINAS CON AFINIDAD ALTERADA POR EL OALTERADA POR EL O22
HERENCIA
AUTOSÓMICA DOMINANTE. AFECTADOS SON HETEROCIGOTAS.
•AUMENTO DE AFINIDAD: ERITROCITOSIS (FAMILIAR) NO SON HEMOLÍTICAS.
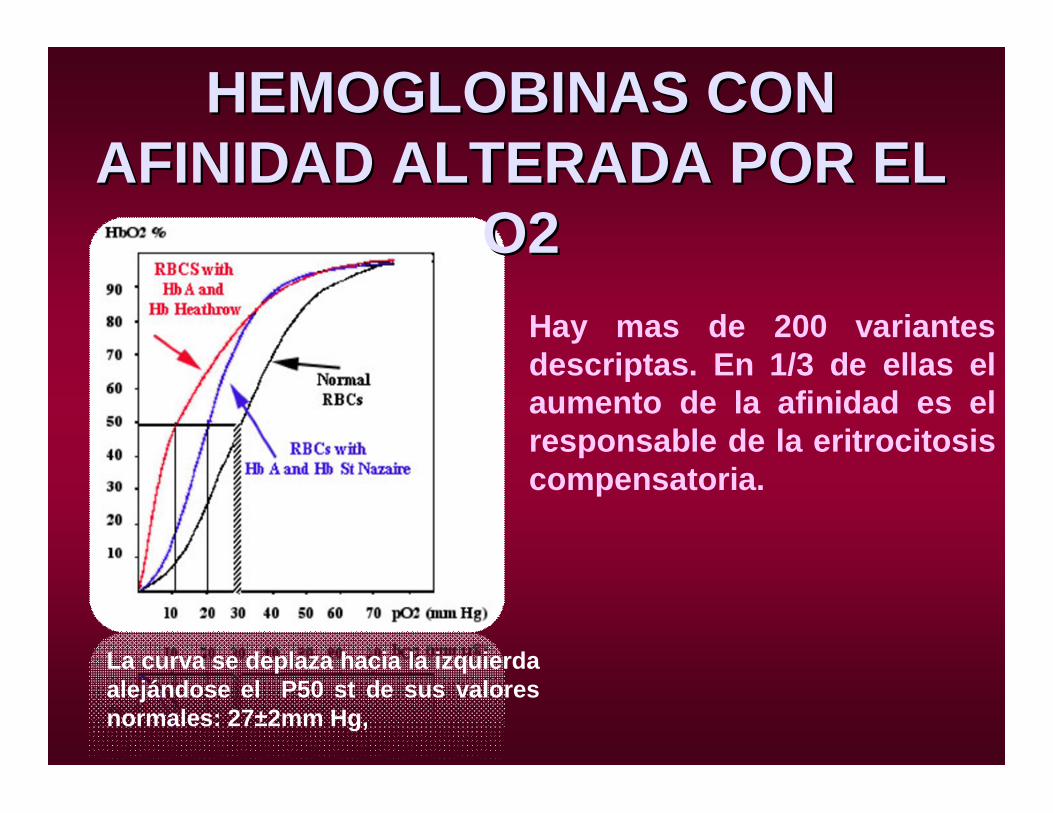
La curva se deplaza hacia la izquierda alejándose el P50 st de sus valores normales: 27±2mm Hg,
Hay mas de 200 variantes descriptas. En 1/3 de ellas el aumento de la afinidad es el responsable de la eritrocitosis compensatoria.
HEMOGLOBINAS CON HEMOGLOBINAS CON AFINIDAD ALTERADA POR EL AFINIDAD ALTERADA POR EL
O2O2

• SUSTITUCIONES:SUPERFICIES DE CONTACTO ALFA1-BETA2, ZONAS DE UNIÓN CON EL 2-3DPG
•DIAGNÓSTICO: ESTUDIO DE LA AFINIDAD. P50 A PARTIR DE SANGRE ENTERA:
•UNA DESVIACIÓN EN +/-3 mmHg DEBE CONSIDERARSE PATOLÓGICA.
HEMOGLOBINAS CON AFINIDAD HEMOGLOBINAS CON AFINIDAD ALTERADA POR EL O2ALTERADA POR EL O2
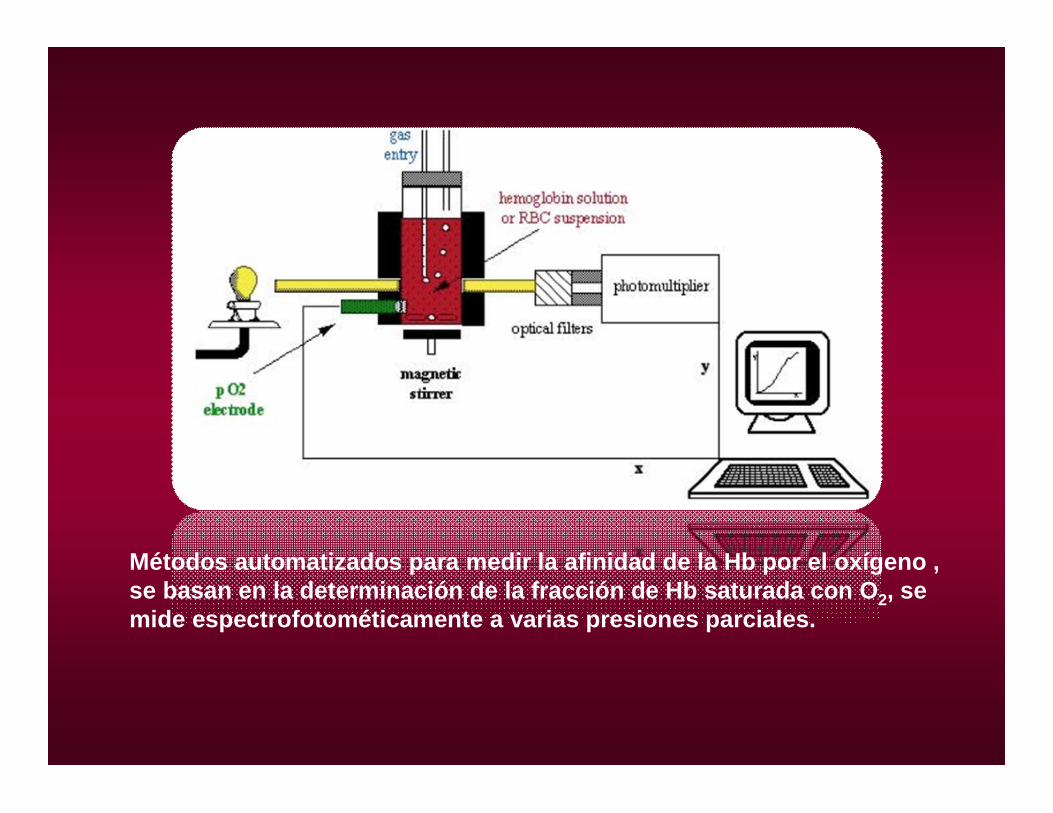
Métodos automatizados para medir la afinidad de la Hb por el oxígeno , se basan en la determinación de la fracción de Hb saturada con O2, se mide espectrofotométicamente a varias presiones parciales.

DISMINUCIÓN DE AFINIDAD: RARAS, ASINTOMÁTICAS, CON LIGERO GRADO DE CIANOSIS POR LA DISMINUCIÓN DE OXIHEMOGLOBINA. HAY SÓLO 16 DESCRIPTAS
Son variantes encontradas en pacientes con cianosis inexplicables, sin enfermedad cardiopulmonar ni meta o sulfohemoglobinemia. Otro caso raro es el diagnóstico diferencial con la deficiencia de piruvato kinasa que acumula 2,3-DPG en el GR
La disminución de afinidad se ve en variantes donde las sustitución crea un enlace adicional que estabilza la forma deoxi de la Hb. El p50 de la sangre está alrededor de 40 mm Hg en el paciente heterocigota-

• La Metamoglobinemia es el estado en el cual el nivel de metamoglobina excede el 1%.
• Normalmente, aproximadamente un 3% del total de hemoglobina es diariamente oxidada, pero es reducida por el sistema de la citocromo b5 reductasa.(NADH-diaforasa)
HEMOGLOBINAS MHEMOGLOBINAS M

HEMOGLOBINAS MHEMOGLOBINAS M
• Se deben distinguir dos tipos de metahemoglobinemias.
1. La hemoglobina normal es oxidada a metahemoglobina.
1. Agentes tóxicos: nitritos. Medicamentos: sulfonas2. Déficit enzimático (diaforasa I)3. Variante con aumento de autooxidación

• 2.-Metahemoglobinemia se debe a la presencia de una variante con propiedades espectrales anormales llamadas Hb M
• Siete anormalidades estructurales conducen a Hb M
HEMOGLOBINAS MHEMOGLOBINAS M

Name Mutation Clinical presentation
Hb M-Boston
α58 (E7) His->Tyr ( 1 or 2)
cyanosis starts at birth and remains at a constant level. Well tolerated
Hb M-Iwate
α87 (F8) His->Tyr ( 1 or 2)
cyanosis starts at birth and remains at a constant level. Well tolerated
Hb M-Saskatoon
β63 (E7) His ->Tyr
cyanosis develops after birth and reach its final level at 6 months. Well tolerated
Hb M-Hyde Park
β92 (F8) His->Tyr
cyanosis develops after birth and reach its final level at 6 months. This Hb is also unstable and leads to some degree of hemolytic anemia
Hb –M Milwaukee
67 (E11) Val -> Glu
cyanosis develops after birth and reach its final level at 6 months. Well tolerated
Hb F-M-Osaka
γG 63 (E7) His->Tyr
Cyanosis is maximum at birth and deceases progressively with the switch from HbF to Hb A
Hb F-M-Fort Ripley
γG 92 (F8) His->Tyr
Cyanosis is maximum at birth and deceases progressively with the switch from HbF to Hb A
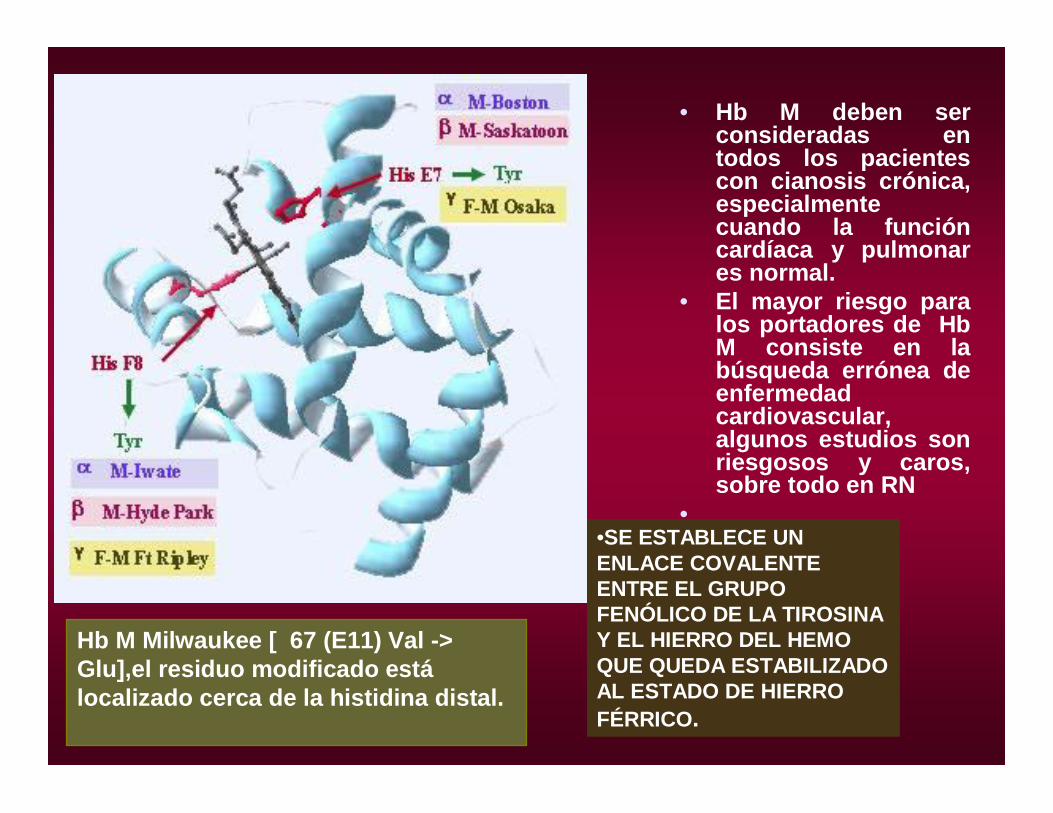
• Hb M deben ser consideradas en todos los pacientes con cianosis crónica, especialmente cuando la función cardíaca y pulmonar es normal.
• El mayor riesgo para los portadores de Hb M consiste en la búsqueda errónea de enfermedad cardiovascular, algunos estudios son riesgosos y caros, sobre todo en RN
••SE ESTABLECE UN ENLACE COVALENTE ENTRE EL GRUPO FENÓLICO DE LA TIROSINA Y EL HIERRO DEL HEMO QUE QUEDA ESTABILIZADO AL ESTADO DE HIERRO FÉRRICO.
Hb M Milwaukee [ 67 (E11) Val -> Glu],el residuo modificado estálocalizado cerca de la histidina distal.

HEMOGLOBINAS MHEMOGLOBINAS M
MANIFESTACIONES CLÍNICAS
•CIANOSIS IRREVERSIBLE, SI HAY INESTABILIDAD MOLECULAR, ANEMIA HEMOLÍTICA, a veces ligera eritrocitosis como consecuencia de la hipoxia hística.
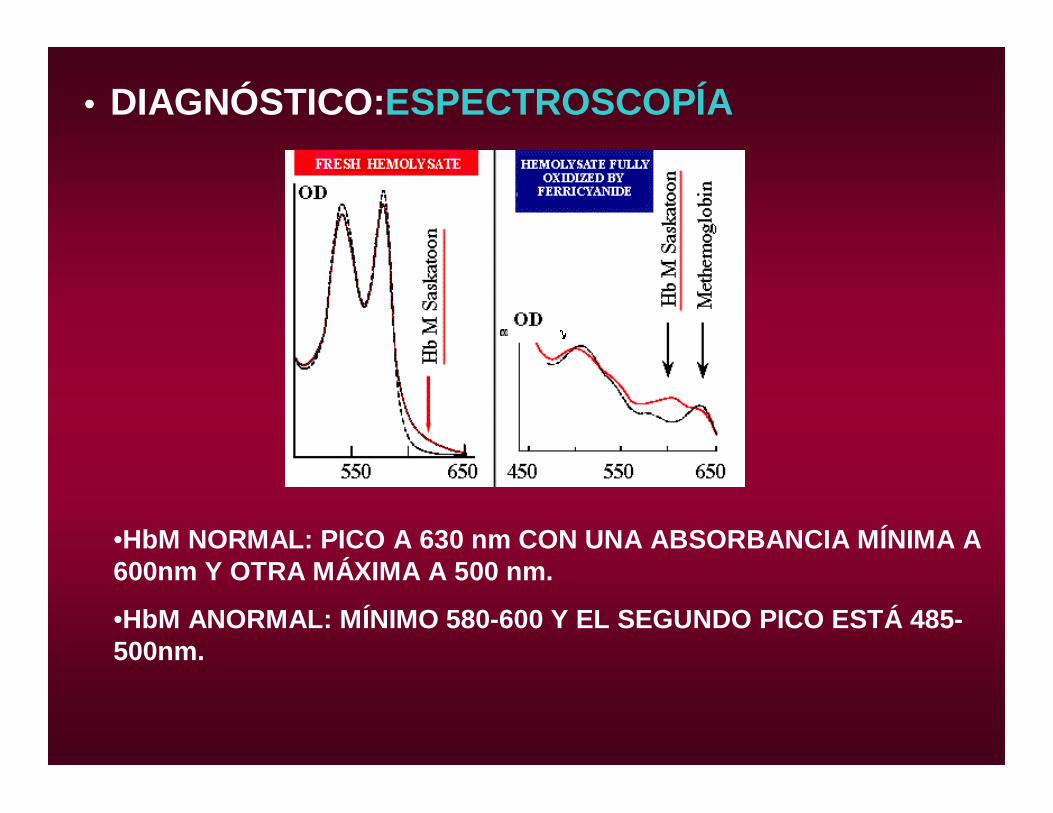
• DIAGNÓSTICO:ESPECTROSCOPÍA
•HbM NORMAL: PICO A 630 nm CON UNA ABSORBANCIA MÍNIMA A 600nm Y OTRA MÁXIMA A 500 nm.
•HbM ANORMAL: MÍNIMO 580-600 Y EL SEGUNDO PICO ESTÁ 485-500nm.

• Para la caracterización de una variante estructural:
–Debemos usar los métodos químicos para proteínas óbiología molecular?

• Respuesta: debería ser lo mismo.
• Algunos casos: Modificaciones post traduccionales, el resultado es diferente-Hb Bristol donde el análisis de la proteína mostro que un aspartato reemplazó a una valina en posicion 67 (E11), y la secuenciación del DNA que el codón mutado codificaba para metionina. La explicación fue la oxidación de la metionina.- Otro ejemplo es la desaminación de la asparagina en aspartato.

• La pregunta es qué hacer?
• Cuando el interés primario es explicar el fenotipo , la respuesta es la química proteica. Si el interés es por ej, el diagnóstico prenatal de una doble heterocigosis severa, la respuesta es que el DNA debe ser investigado.

• La biología molecular es el único enfoque que debe ser considerado seriamente cuando se trata de variantes expresadas a muy bajo nivel.
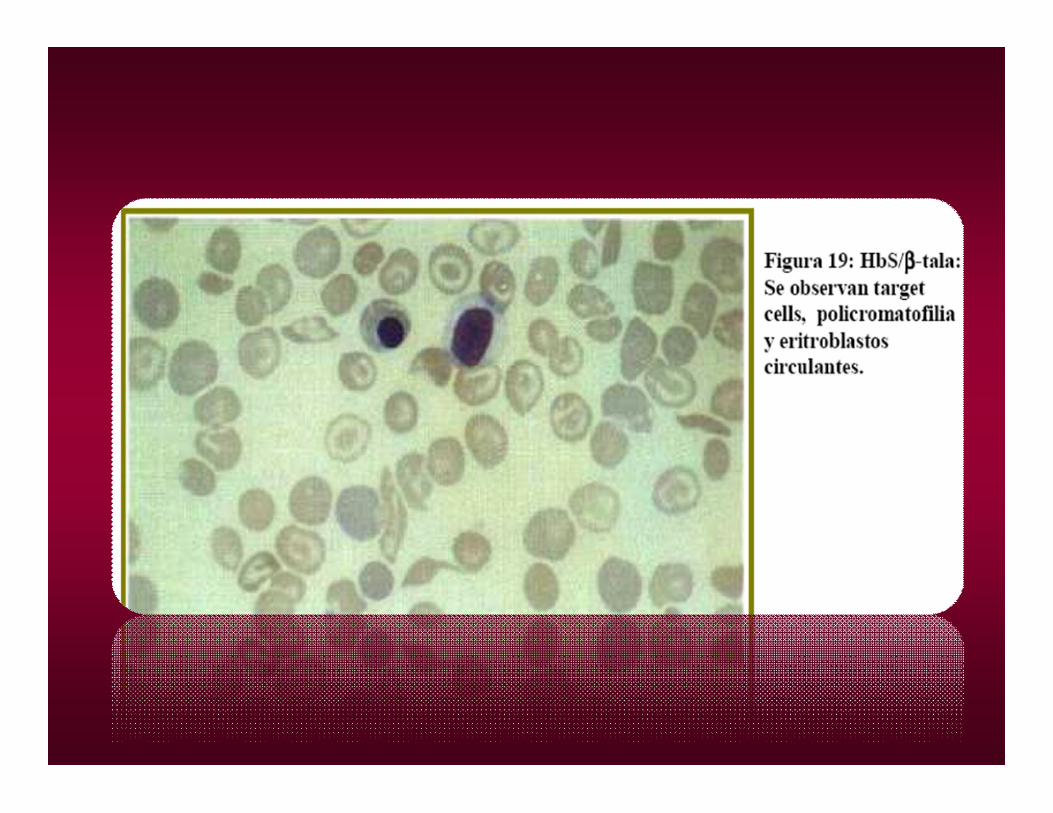
ASOCIACIONESASOCIACIONESS/BETA TALAS.
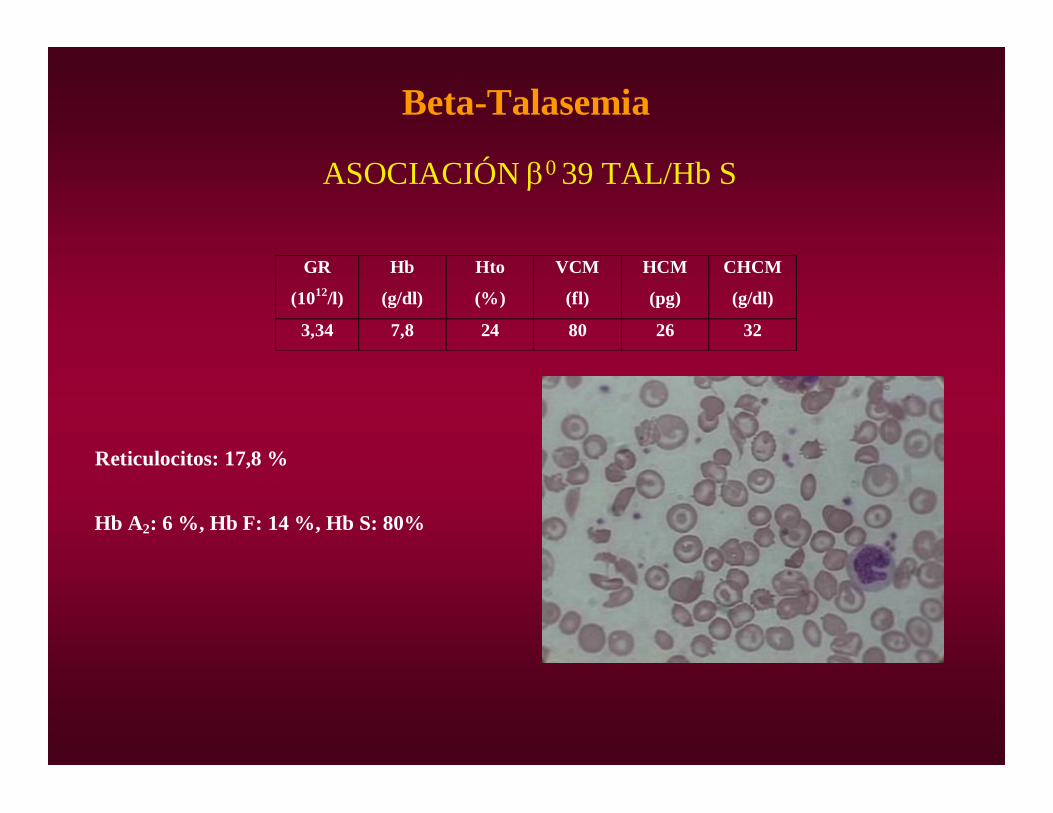
Beta-Talasemia
ASOCIACIÓN 0 39 TAL/Hb S
GR
(1012/l)
Hb
(g/dl)
Hto
(%)
VCM
(fl)
HCM
(pg)
CHCM
(g/dl)
3,34 7,8 24 80 26 32
Reticulocitos: 17,8 %
Hb A2: 6 %, Hb F: 14 %, Hb S: 80%

BIBLIOGRAFIABIBLIOGRAFIA• Aspectos generales:
- Bunn H.F., Forget B.G. (1986) Hemoglobin : Molecular, Genetic and Clinical aspects., W.B.Saunders Company, Philadelphia, London.
-Lehmann H., Huntsman R.G. (1974) Man's Haemoglobins (2nd ed.) North Holland Publ.Comp., Amsterdam
- Stamatoyanopoulos G., Nienhuis A.W., Leder P., Majerus P.W. (1987) The molecular basis of blood diseases, W.B.Saunders Company, Philadelphia, London.
-Steinberg MH, Forget BG, Higgs RD, and Nagel RL. Disorders of Hemoglobin : genetics, pathophysiology, and Clinical Management Cambridge University Press, 2001.

B ) estructura y función:•
-Adair, G. S. (1925) The Hemoglobin System. VI. The Oxygen Dissociation Curve of HemoglobinJ. Biol. Chem. 63, 529-545
- Benesch R.E., Benesch R. (1974) The mechanism of interaction of red cell organic phosphates with hemoglobin. Adv. Protein Chem. 28, 211-237.
-Dickerson R.E., Geis I. (1983) Hemoglobin : structure, function, evolution and pathology. The Benjamin/Cummings Publ.Comp., Menlo Park, California.
- Fermi G., Perutz M.F. Haemoglobin and myoglobin. In Phillips D.C. and Richards F.M. (Eds): Atlas of Molecular Structures in Biology, Oxford, Clarendon Press, 1981
- Imai K.Allosteric effects in haemoglobin. (1981) Cambridge University Press, Cambridge
- Perutz M.F. (1970) Stereochemistry of cooperative effects in haemoglobin. Nature 228, 726-739.
- Perutz M.F. and Lehmann H. (1968) Molecular pathology of human haemoglobin. Nature 219, 902-909.
- Perutz M.F., Muirhead H., Cox J.M. and Coaman L.C.G .(1968) Three dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8Å resolution : the atomic model. Nature, 219, 131-139.

• C ) biosíntesis y aspectos genéticos
- Antonorakis S.E., Kazazian H.H. and Orkin S. DNA polymorphism in the alpha and beta globin gene clusters and molecular etiology of the thalassemia syndromes in man. In Winter W.P. (Edt) Hemoglobin variants in Human Populations, Vol.1, (1986), CRC Press, Boca Raton, Florida
- Benz E.J., Forget B.G. (1974) The biosynthesis of hemoglobin. Semin.Hematol. 11, 463-523.
- Kan Y.W., Dozy A.M. (1978) Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation. Proc. Natl. Acad. Sci. USA 75, 5631-5635.
- Maniatis T., Frich E.F., Lauer J. and Lawn R.M. (1980) The molecular genetics of human hemoglobins. Ann .Rev. Genet., 14, 145-178.
- Weatherall D.J., Clegg J.B. (2001) The thalassaemia syndromes (4th ed.). Oxford, Blackwell Scientific Publications
D) Diagnosis and laboratory
- Hoyer J.D., Kroft S.H. (2003) Color Atlas of Hemoglobin Disorders. College of American Pathologists. Northfield. IL. USA.
- Schneider RG, Barwick RC. (1978) Measuring relative electrophoretic mobilities of mutant hemoglobins and globin chains. Hemoglobin 2, 417-435.
- Nagel RL (2003) Hemoglobin Disorders. Molecular Methods and Protocols. Methods in Molecular Medicine. Humana Press, Totowa, New Jersey, USA

• D) Diagnóstico de laboratorio:
- Hoyer J.D., Kroft S.H. (2003) Color Atlas of Hemoglobin Disorders. College of American Pathologists. Northfield. IL. USA.
- Schneider RG, Barwick RC. (1978) Measuring relative electrophoretic mobilities of mutant hemoglobins and globin chains. Hemoglobin 2, 417-435.
- Nagel RL (2003) Hemoglobin Disorders. Molecular Methods and Protocols. Methods in Molecular Medicine. Humana Press, Totowa, New Jersey, USA
Sans Sabrafen- Hematología Clínica. 2006- Quinta edición. Editorial Elsevier.

Muchas gracias!!!!
Recommended

















