
Funktionelle Interaktionen
von Tau mit anderen Proteinen,
die bei der Alzheimer’schen Krankheit beteiligt sind
Dissertation
zur Erlangung des naturwissenschaftlichen Doktorgrades
des Fachbereiches Biologie/Chemie
der Universität Osnabrück
Julia Leschik
2005

Dunkelgründig, als ob es nichts sei, und doch ist es;
zwanglos aus sich selbst wirkend, gestaltlos und doch
voll zauberischer Kraft. Alle Dinge ernährt es, und
doch wissen diese nichts davon. Dies nennt man den
Ursprung Wurzel. Wer sie erkennt, kennt die Natur.
Dschuang Dsi (365 - etwa 290 v. Chr.),
taoistischer Philosoph

Ich erkläre hiermit, dass ich die vorliegende Dissertation selbständig verfasst und keine
anderen als die angegebenen Hilfsmittel benutzt habe. Die Inhalte anderer wissenschaftlicher
Arbeiten als Referenz sind immer als solche gekennzeichnet. Die Arbeit wurde bisher weder
im In- noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde
vorgelegt. Desweiteren wurden keine früheren Promotionsversuche unternommen.
_______________________________ ___________________________
(Ort, Datum) (Unterschrift)

Verzeichnisse
Inhalt
A Einleitung ........................................................................................................................... 1 A.1 Die Alzheimer-Krankheit ........................................................................................ 1
A.2 Das mikrotubuliassoziierte Protein Tau .................................................................. 2 A.2.1 Tau und seine Funktion im neuronalen Zytoskelett ..............................................................2 A.2.2 Tau und seine potentielle Funktion als axonales „Gerüst“-Protein........................................5 A.2.3 Die Phosphorylierung von Tau ..............................................................................................6 A.2.4 Tau in der Alzheimer-Krankheit ............................................................................................7 A.2.5 PHF-ähnliche Phosphorylierungsmutationen.......................................................................10
A.3 Das Amyloid-Vorläufer-Protein APP und sein Peptidfragment Aβ ..................... 12 A.3.1 Die Prozessierung von APP.................................................................................................13 A.3.2 Aβ in familiären (FAD) und sporadischen Formen der Alzheimer-Krankheit ....................15 A.3.3 Die Neurotoxizität von Aβ...................................................................................................16 A.3.4 Aβ und Tau im funktionellen Zusammenhang ....................................................................17
A.4 Die Preseniline ...................................................................................................... 19 A.4.1 Die biologische und pathologische Rolle der Preseniline....................................................19 A.4.2 Die Rolle der Preseniline im Zelltod....................................................................................22 A.2.3 Presenilin und Tau im funktionellen Zusammenhang .........................................................22
A.5 Ziel der Arbeit ....................................................................................................... 24
B Material und Methoden.................................................................................................. 25
B.1 Material und Geräte............................................................................................... 25 B.1.1 Chemikalien.........................................................................................................................25 B.1.2 Material-Molekularbiologie.................................................................................................25
B.1.2.1 Plasmide....................................................................................................25 B.1.2.2 Primer .......................................................................................................25 B.1.2.3 Bakterienstämme ......................................................................................26 B.1.2.4 Viren .........................................................................................................26 B.1.2.5 Bakterienkultur .........................................................................................26 B.1.2.6 Antibiotika ................................................................................................26 B.1.2.7 Enzyme und Größenmarker ......................................................................26
B.1.3 Material-Zellbiologie ...........................................................................................................27 B.1.3.1 Eukaryotische Zelllinien ...........................................................................27 B.1.3.2 Tiere.........................................................................................................27 B.1.3.3 Zellkulturmedien.......................................................................................27
B.1.4 Material-Biochemie .............................................................................................................28 B.1.5 Antikörper............................................................................................................................29 B.1.6 Puffer und Stammlösungen..................................................................................................30
B.1.6.1 Molekularbiologie.....................................................................................30 B.1.6.2 Zellbiologie...............................................................................................31 B.1.6.3 Protein-Biochemie ............................................................................................32
B.1.7 Geräte und sonstige Materialien ..........................................................................................34 B.1.8 Software...............................................................................................................................38
I

Verzeichnisse
B.2 Methoden............................................................................................................... 38 B.2.1 Molekularbiologische Methoden .........................................................................................38
B.2.1.1 Präparation genomischer DNA aus embryonalem Lebergewebe bzw. adultem Schwanzgewebe der Maus ..........................................................38
B.2.1.2 Polymerase-Kettenreaktion (PCR)................................................................39 B.2.1.3 Analyse des PCR-Produkts bzw. Restriktionsverdaus ..................................41 B.2.1.4 Herstellung von kompetenten Bakterien ........................................................41 B.2.1.5 Transformation...............................................................................................42 B.2.1.6 Präparative Plasmid-DNA-Präparation (Maxipräparation) ............................42 B.2.1.7 Analytischer Restriktionsverdau ....................................................................43 B.2.1.8 Konzentrationsbestimmung von DNA...........................................................44
B.2.2 Zellbiologische Methoden ...................................................................................................44 B.2.2.1 Kultur von Vero 2-2-Zellen ...........................................................................44 B.2.2.2 Kultur von Ratten Pheochromocytoma-Zellen (PC12) ..................................45 B.2.2.3 Kultur von NT2-/NT2-N-Zellen ....................................................................45 B.2.2.4 Einfrieren und Auftauen von Zellen...............................................................46 B.2.2.5 Zellzahlbestimmung.......................................................................................47 B.2.2.6 Präparation und Kultivierung von kortikalen Primärkulturen........................48 B.2.2.7 Herstellung der HSV-1-Amplikons................................................................49 B.2.2.8 Infektion von NT2-N-Zellen und kortikalen Primärkulturen.........................53 B. 2.2.9 Immunfluoreszenzmikroskopie.....................................................................53 B.2.2.9.1 Beschichtung von mikroskopischen Deckgläschen ....................................54 B.2.2.9.2 Fixierung und Färbung von Zellen..............................................................54 B.2.2.9.3 Immunfärbung und Einbettung ...................................................................55 B.2.2.9.4 Zellkernfärbung...........................................................................................55
B.2.3 Protein-Biochemische Methoden.........................................................................................56 B.2.3.1 Präparation von Zelllysat aus kortikalen Primärkulturen...............................56 B.2.3.2 Natriumdodecylsulfat-Polyacrylamidgel-Elektrophorese (SDS-PAGE)........56 B.2.3.3 Proteintransfer durch Elektroblot (Westernblot) ............................................57 B.2.3.4 Immundetektion .............................................................................................58 B.2.3.5 „Strippen“ einer PVDF- Membran.................................................................59 B.2.3.6 Densitometrische Quantifizierung der Immunblotanalyse.............................59 B.2.3.7 ELISA (Enzyme-Linked-Immunosorbant-Assay) .........................................59
C Ergebnisse ........................................................................................................................ 62 C.1 EGFP-PHP-Tau ist neurotoxisch in NT2-N-Zellen .............................................. 62
C.2 Expression der eGFP-Konstrukte in kortikalen Primärkulturen ........................... 66 C.2.1 Protein-biochemische Analyse der Expression der eGFP-Konstrukte in kortikalen
Primärkulturen................................................................................................................67 C.2.2 Immunzytochemische Analyse der Expression der eGFP-Konstrukte in kortikalen
Primärkulturen................................................................................................................69 C.3 PHP-Tau ist neurotoxisch in kortikalen Primärkulturen ....................................... 74
C.3.1 Quantitative Analyse des neuronalen Zelltods in Primärkulturen zu unterschiedlichen Zeitpunkten nach HSV-1-Infektion ...................................................77
C.3.2 Protein-biochemische Analyse des durch PHP-Tau ausgelösten toxischen Effekts in kortikalen Primärkulturen...........................................................................................79
II

Verzeichnisse
C.4 Presenilin 1 wirkt antiapoptotisch auf die durch PHP-Tau ausgelöste Neurotoxizität ........................................................................................................ 81 C.4.1 Quantitative Analyse der PHP-Tau induzierten Neurodegeneration in Presenilin 1
transgenen Primärkulturen .............................................................................................84 C.4.2 Protein-biochemische Untersuchung der durch Presenilin 1 erniedrigten PHP-Tau
induzierten Neurotoxizität ..............................................................................................85 C.4.3 Die Presenilin 1 FAD-Mutation M146L wirkt nicht antiapoptotisch auf die PHP-
Tau induzierte Neurotoxizität.........................................................................................88 C.5 Aβ erhöht die Toxizität wt-Taus möglicherweise durch erhöhte
Phosphorylierung an Serin 396/404 ...................................................................... 89 C.5.1 Quantitative Analyse der Neurodegeneration in Tau-infizierten APPSDL-transgenen
Primärkulturen................................................................................................................92 C.5.2 Protein-biochemische Analyse der Tau-Phosphorylierung in APPSDL-transgenen
Primärkulturen................................................................................................................93
D Diskussion ........................................................................................................................ 97 D.1 Hyperphosphoryliertes Tau ist neurotoxisch......................................................... 97
D.2 Aβ führt zur Neurodegeneration über die Phosphorylierung von Tau (Amyloid-Hypothese)........................................................................................................... 100
D.3 Presenilin 1 wt wirkt antiapoptotisch auf die Toxizität hyperphosphorylierten Taus ..................................................................................................................... 106
D.4 Die Mutation PS1(M146L) vermittelt keinen anti-degenerativen Effekt auf die Toxizität hyperphosphorylierten Taus................................................................. 108
D.5 Eine integrierte Sicht ........................................................................................... 111
E Zusammenfassung......................................................................................................... 113
F Abkürzungen ................................................................................................................. 114
G Literatur......................................................................................................................... 119
H Danksagung ................................................................................................................... 143
III

Verzeichnisse
I Anhang ........................................................................................................................... 144 I.1 Experimente C.1, Fragmentierung von NT2-N-Zellkernen ................................ 144
I.2 Experimente C.2.2, Messungen der Fluoreszenz-Intensität ................................ 145
I.3 Experimente C.3.1, Fragmentierung von Primärneuronen-Zellkernen............... 163
I.4 Experimente C.3.2, Densitometrische Analyse der Caspase-3-Aktivierung in Primärkulturen..................................................................................................... 164
I.5 Experimente C.4.1, Fragmentierung von Zellkernen in PS1-transgenen Primärkulturen..................................................................................................... 165
I.6 Experimente C.4.2, Densitometrische Analyse der Caspase-3-Aktivierung in PS1-transgenen Primärkulturen........................................................................... 166
I.7 Experimente C.4.3, Fragmentierung von Zellkernen in PS1(M146L)-transgenen Primärkulturen..................................................................................................... 167
I.8 Experimente C.5, Aβ40-, Aβ42-ELISA .............................................................. 168
I.9 Experimente C.5.1, Fragmentierung von Zellkernen in APPSDL-transgenen Primärkulturen..................................................................................................... 169
I.10 Experimente C.5.2, Densitometrische Analyse der Immunreaktivität von PHF-1 ........................................................................................................... 170
IV

Verzeichnisse
Abbildungen
Abb. A.1: Schematische Darstellung der 6 ZNS Tau-Isoformen. ....................................... 3
Abb. A.2: Schematische Darstellung der funktionellen Domänen im Tau-Protein. ........... 5
Abb. A.3: Schematische Darstellung eines über ortsgerichtete Mutation hergestellten PHP-Tau-Konstrukts. .................................................................. 11
Abb. A.4: Prozessierung von APP........................................................................................ 14
Abb. A.5: Schematische Darstellung von Presenilin in der Membran...................................... 20
Abb. C.1: Fluoreszenzmikroskopische Analyse der Zellkernmorphologie infizierter NT2-N-Zellen. ................................................................................. 64
Abb. C.2: Quantitative Analyse fragmentierter NT2-N-Zellkerne nach der Infektion mit eGFP-wt-Tau, eGFP-PHP-Tau und eGFP.................................. 65
Abb. C.3: Immunblot Analyse der GFP-/GFP-Tau-Expression in HSV-1 infizierten kortikalen Primärkulturen................................................................................. 68
Abb. C.4: Immunblot-Analyse der Expression von GFP-Tau und GFP an unterschiedlichen Tagen nach HSV-1 Infektion. ............................................. 69
Abb. C.5: Immunfluoreszenzmikroskopische Analyse der Expression von HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP in kortikalen Primärneuronen. ............................................................................................... 71
Abb. C.6: Intensitätsverteilung der GFP-Antikörper-Färbung von eGFP-wt-Tau bzw. eGFP-PHP-Tau exprimierenden Neuronen. ............................................ 72
Abb. C.7: Überblick über HSV-1-eGFP-Tau-infizierte kortikale Primärkulturen............ 75
Abb. C.8: HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP infizierte kortikale Primärkulturen. ................................................................................................. 76
Abb. C.9: Bestimmung des Anteils fragmentierter Zellkerne von Primärneuronen zu unterschiedlichen Zeitpunkten nach HSV-1 Infektion mit eGFP-wt-, eGFP-PHP-Tau und eGFP................................................................................ 78
Abb. C.10: Immunblot-Analyse des durch PHP-Tau ausgelösten Zelltods durch Verwendung eines Antikörpers gegen aktivierte Caspase-3. ........................... 80
Abb. C.11: Densitometrische Analyse der Caspase-3-Aktivierung in HSV-1- infizierten kortikalen Primärkulturen. .............................................................. 81
Abb. C.12: Immunfluoreszenzanfärbungen PS1-transgener und nicht-transgener Primärkulturen. ................................................................................................. 83
Abb. C.13: Quantitative Analyse der Neurodegeneration in PS1-transgenen und nicht-transgenen Primärkulturen........................................................................... 85
Abb. C.14: Immunblot-Analyse der Caspase-3-Aktivierung in PHP-Tau-infizierten PS1-transgenen Primärkulturen. .................................................................................. 86
Abb. C.15: Densitometrische Analyse der Caspase-3-Aktivierung in PHP-Tau-exprimierenden PS1-transgenen Primärkulturen.............................................. 87
V

Verzeichnisse
Abb. C.16: Effekt der PS1(M146L) Mutation auf die Tau-vermittelte Neurodegeneration in Primärkulturen. ............................................................................................... 89
Abb. C.17: Sekretiertes Aβ40 und Aβ42 im konditionierten Medium kortikaler Primärkulturen. ................................................................................................... 91
Abb. C.18: Effekt von APPSDL auf den Anteil von Neuronen mit fragmentierten Zellkernen nach der Infektion mit Tau-Konstrukten................................................................ 93
Abb. C.19: Immunblot-Analyse der Tau-Phosphorylierung in APPSDL-transgenen Primärkulturen an der PHF-1-Seite S396/404.................................................. 95
Abb. C.20: Densitometrische Analyse der PHF-1-Immunoreaktivität in wt-Tau- infizierten APPSDL-transgenen Primärkulturen................................................. 95
Abb. D.1: Vereinfachtes Schaubild der funktionellen Interaktionen von Aβ, Tau und PS1 in der familiären und sporadischen Alzheimer-Pathologie. .................... 112
VI

Einleitung
A Einleitung
Demenzerkrankungen treten überwiegend im höheren Lebensalter auf. Dabei sind die
geistigen Fähigkeiten zunächst gestört, bis sie im Endstadium der Erkrankung gänzlich
verloren gehen. Es werden mehrere Arten von Demenzerkrankungen unterschieden. Etwa 50-
60% aller Betroffenen leiden an der Alzheimer-Demenz (AD). Bei 10-20% der Kranken sind
Durchblutungsstörungen die Ursache fortschreitender geistiger Beeinträchtigungen (vaskuläre
Demenz). Weitere 15-20% aller Kranken leiden sowohl an der Alzheimer-Krankheit als auch
an einer durchblutungsbedingten Demenz. Die übrigen Demenzerkrankungen (ca. 10%)
setzen sich aus vielen zum Teil seltenen Erkrankungsformen und Ursachen zusammen (Jorm,
1991; Katzman & Kawas, 1994).
Demenzerkrankungen sind generell durch ein Absterben von Nervenzellen in bestimmten
Gehirnregionen gekennzeichnet. Aus diesem Grund werden sie zu den neurodegenerativen
Erkrankungen gezählt, die ebenso andere Erkrankungen des Nervensystems wie z.B. Multiple
Sklerose und Amyotrophe Lateralsklerose (ALS) umfassen.
Zur Zeit leiden in Deutschland ca. 1 Million Menschen an einer Demenzerkrankung - mit
steigender Tendenz. Die Alzheimer-Krankheit ist die am weitesten verbreitete Ausprägung
der Demenz, ca. 50% der 85-jährigen zeigen Symptome einer Alzheimer-Krankheit (Bickel,
2000, 2001; Kessler et al., 2000). Auf Grund der Entwicklung der Alterspyramide in den
Industrienationen wird davon ausgegangen, dass die Anzahl der Alzheimer-Patienten in den
nächsten Jahrzehnten drastisch zunehmen wird (Bickel, 2000, 2001).
A.1 Die Alzheimer-Krankheit
Die Alzheimer-Krankheit ist die am häufigsten auftretende neurodegenerative Erkrankung
und ist charakterisiert durch fortschreitenden Gedächtnisverlust, beeinträchtigte Wahr-
nehmung und verändertes Verhalten. Der Rückgang der kognitiven Funktionen ist begleitet
von einem massiven, selektiven Absterben von Neuronen im Hippokampus, der Amygdala
und im Kortex, was schließlich zum Tod des Patienten führt. Die pathologischen Merkmale
im Gehirn wurden bereits 1907 von Alois Alzheimer als Ablagerungen fibrillären Materials
beschrieben (Alzheimer, 1911). Es handelt sich dabei zum einen um Aggregate aus β-
Amyloid-Protein (Aβ), welches als Proteinfragment aus dem Amyloid-Vorläufer-Protein
(APP) entsteht. Anhäufungen von Aβ resultieren in extrazellulären Amyloid-Plaques (Senile
Plaques), während zum anderen intrazellulär das zytoskelettassoziierte Protein Tau,
1

Einleitung
insbesondere im somatodentritischen Kompartiment eine Aggregation in Filamente
(„Neurofibrillary Tangles“, NFTs) aufweist. NFTs entstehen aus Aggregaten von paarigen
helikalen Filamenten (PHFs), welche aus einer Aggregation hyperphosphorylierten Tau-
Proteins bestehen. Immunhistochemische Analysen deuten darauf hin, dass der erhöhte Phos-
phorylierungszustand von Tau ein frühes und wahrscheinlich kritisches Ereignis in der Tau-
Pathologie darstellt (zur Übersicht siehe Brandt et al., 2005). Die Rolle der Tau-Aggregation
während des Krankheitsprozesses ist unklar. Da die Verteilung der NFTs aber eng mit den
Orten der neuronalen Degeneration korreliert (Braak et al., 1993), wird eine ursächliche Rolle
der Tau-Aggregation für das Absterben von Neuronen angenommen.
Zudem lässt die Tatsache, dass verschiedende Amyloid-Plaques auch in kognitiv unbeein-
trächtigten Individuen auftreten, vermuten, dass die neurofibrillären Ablagerungen mit dem
Krankheitsbild der Demenz in einem engen Zusammenhang stehen (Arriagada et al., 1992;
Neve & Robakis, 1998). Interessanterweise führen bei der neurodegenerativen Erkrankung
FTDP-17 (Frontotemporale Demenz mit Parkinsonismus) Mutationen im Tau-Gen zur NFT-
Bildung und zum Absterben von Zellen, die wie im Falle der Alzheimer’schen Erkrankung
aus hyperphosphoryliertem Tau-Protein bestehen (zur Übersicht siehe Goedert et al., 1998).
Dies deutet darauf hin, dass eine Tau-Pathologie, wie sie in AD vorkommt, für eine
Neurodegeneration hinreichend ist.
A.2 Das mikrotubuliassoziierte Protein Tau
A.2.1 Tau und seine Funktion im neuronalen Zytoskelett
Nervenzellen sind polar in ein somatodendritisches (signalempfangendes) und ein axonales
(signalweiterleitendes) Kompartiment organisiert. Diese zelluläre Architektur ist entscheidend
für eine effiziente Informationsverarbeitung und -weiterleitung im Nervensystem und wird
hauptsächlich durch die Ausbildung eines zellulären Zytoskeletts bestimmt. Man
unterscheidet Aktinfilamente, Mikrotubuli und Intermediärfilamente (für eine Übersicht siehe
Alberts et al., 2002). Während Intermediärfilamente überwiegend statische Strukturen sind,
die vor allem der Stabilisierung der Zelle dienen, sind Aktinfilamente und Mikrotubuli dyna-
mische Strukturen, die u.a. für motile Prozesse wie z.B. die Orientierung des Wachstums-
kegels und das Auswachsen der Neuriten zuständig sind (Vale, 1992). Mikrotubuli sind in der
gesamten Nervenzelle ubiquitär verteilt, jedoch in Neuriten konzentriert. Sie haben neben
einer Stabilisierung des Axons vielfältige Funktionen. Eine wichtige Aufgabe ist die
2

Einleitung
Gewährleistung des axonalen Vesikel-Transports, der die Nervenendigung mit essentiellen
Bestandteilen versorgt und einen Rücktransport von endozytiertem Material in den Zellkörper
ermöglicht. Die Integrität des Zytoskeletts ist kritisch für die Funktion und das Überleben der
Nervenzellen. Viele neurodegenerative Erkrankungen sind durch Abnormalitäten im
Zytoskelett charakterisiert. Wie bereits oben erwähnt ist das mikrotubuliassozierte Protein
Tau in der Alzheimer-Krankheit durch Hyperphosphorylierung pathologisch verändert.
Tau-Proteine sind niedermolekulare mikrotubuliassoziierte Proteine (MAPs), die vorwiegend
in Nervenzellen, aber in geringem Maße auch in Astrozyten und Oligodendrozyten vorliegen
(Papazomenos & Binder; 1987, Migheli et al., 1988). In Neuronen ist Tau hauptsächlich im
Axon lokalisiert (Binder et al., 1985). Im humanen zentralen Nervensystem (ZNS) existieren
6 Isoformen, die durch alternatives Spleißen der Exone 2, 3 und 10 einer mRNA generiert
werden (Abb. A.1). Fötal wird nur die kürzeste Isoform exprimiert, wobei im adulten Gehirn
alle 6 Isoformen vorhanden sind (Goedert et al., 1989). Zudem wird in peripheren Nerven-
zellen eine hochmolekulare Isoform von Tau exprimiert (Couchie et al., 1992; Goedert et al.,
1992). Die Expression der spezifischen Tau-Isoformen wird entwicklungsspezifisch reguliert
und ist zudem abhängig von der topographischen Lage der Nervenzellen im ZNS (zur
Übersicht siehe Brandt et al., 1996; Avila et al., 1997).
Abb. A.1: Schematische Darstellung der 6 ZNS Tau-Isoformen.
Die Isoformen unterscheiden sich durch die Präsenz von 29 Aminosäuren (AS) langen Sequenzen im N-terminalen Bereich, kodiert durch Exon 2 oder 3 (hellgraue Kästchen), in der Kombination mit entweder 3 (R1, R3 und R4) oder 4 (R1-R4) „Repeats“ (schwarze Kästchen). Das vierte Mikrotubuli-bindende „Repeat“ (R2) wird von Exon 10 kodiert (Exon 10, dunkelgraues Kästchen). Die kürzeste Isoform besteht nur aus 352 AS (2-3-10-) und wird als einzigste Tau-Isoform im fötalen Gehirn exprimiert.
3

Einleitung
Die Interaktion von Tau und Mikrotubuli wird durch drei oder vier C-terminale sehr
homologe Sequenzen („Repeats“), bestehend aus 31 oder 32 Aminosäuren, mediiert, wobei
das vierte „Repeat“ von Exon 10 kodiert wird. Die Mikrotubuli-Bindestelle wird auch als
„Repeat“-Domäne bezeichnet. Sie ist in vitro für die Tubulin-Polymerisierung und die
Stabilisierung der Mikrotubuli entscheidend (Lee et al., 1989; Butner & Kirschner, 1991).
Desweiteren stärkt ein Sequenzmotiv in der prolinreichen Region (N-terminal von der
„Repeat“-Domäne) sowie eine Region C-terminal von der „Repeat“-Domäne die Bindung an
Mikrotubuli (Matsou et al., 1994; Goedert et al., 1996). Tau fördert außerdem die de novo
Nukleation von Mikrotubuli in vitro. Das hierfür erforderliche Sequenzmotiv liegt ebenfalls in
der prolinreichen Region (Brandt et al., 1993). Tau kann durch seine aminoterminale
Projektionsdomäne mit Komponenten der neuronalen Plasmamembran interagieren (Brandt et
al., 1995). Es wird angenommen, dass Tau eine Vermittlerrolle zwischen der Plasmamembran
und den Mikrotubuli zukommt.
Es ist bekannt, dass Tau auch die Fähigkeit besitzt, an Aktinfilamente zu binden und diese zu
bündeln (Selden & Pollard, 1983; Yamauchi & Purich,1993). Allerdings wird eine
„Crosslinker“-Funktion von Tau zwischen Aktinfilamenten und Mikrotubuli ausgeschlossen,
da die Bindung an Aktin auch durch die Mikrotubuli-bindende-Domäne erfolgt (Correas et
al., 1990). Vielmehr könnte durch eine Bindung von Tau an Aktin die Bindung von Tau an
Miktrotubuli verhindert werden, und umgekehrt (Farias et al., 2002).
Zusätzlich zu der Interaktion von Tau mit Aktinfilamenten und Mikrotubuli wurde auch eine
Bindung von Tau an Neurofilamente beschrieben, zudem kommen Neurofilamente in NFTs
in späteren Stadien von AD vor (Miyata et al., 1986; Schmidt et al., 1989). Die verschiedenen
funktionellen Regionen des Tau-Proteins sind zusammenfassend in Abb. A.2 schematisch
dargestellt.
4

Einleitung
Abb. A.2: Schematische Darstellung der funktionellen Domänen im Tau-Protein.
Bestimmte Funktionen und Eigenschaften von Tau können spezifischen Regionen in der Primärsequenz des Proteins zugeordnet werden. Gezeigt ist die längste adulte Tau-Isoform im ZNS, welche aus 441 AS besteht.
In kultivierten Nervenzellen ist Tau in der distalen Region von auswachsenden Axonen
angereichert. Es handelt sich hierbei um die Region in einer Nervenzelle, die die dyna-
mischste Mikrotubuli-Population enthält (Black et al.,1996). Daneben ist Tau auch im
somatodendritischen Kompartiment vorhanden, wobei sein Phosphorylierungszustand seine
Verteilung zu beeinflussen scheint (Mandell & Banker, 1996). Es ist strittig, welche Rolle
Tau für das Auswachsen von Neuriten hat. Studien mit Anti-sense-Oligonukleotiden in
kultivierten Kleinhirn-Neuronen belegen, dass Tau für das Auswachsen der Neuriten von
Bedeutung ist, insbesondere für die Verlängerung des Axons (Caceres and Kosik,1990;
Caceres et al.,1991). Im Gegensatz dazu hatte eine Inaktivierung von Tau durch mikro-
injizierte Antikörper keinen Effekt auf das Auswachsen von Axonen und der Mikrotubuli-
Dynamik in kultivierten sympathischen Neuronen (Tint et al., 1998). Ebenso zeigten Tau-
„knock-out“ Mäuse keine schwerwiegenden phänotypischen Veränderungen (Harada et al.,
1994). Dies deutet darauf hin, dass Tau während der Entwicklung nicht essentiell ist und
möglicherweise durch die Hochregulierung anderer MAPs ersetzt werden kann. Diese
Annahme konnte kürzlich durch die Herstellung von Tau/MAP-1B-Doppel-„knock-out“
Mäusen bestätigt werden, welche 4 Wochen nach der Geburt starben und schwerwiegendere
phänotypische Veränderungen zeigten als die Einzel-„knock-out“ Tiere (Takei et al., 2000).
A.2.2 Tau und seine potentielle Funktion als axonales „Gerüst“-Protein
Neben seiner Rolle im neuronalen Zytoskelett, kann Tau auch eine Rolle als Verankerungs-
oder Gerüstprotein verschiedener Proteine zugeschrieben werden. Neben der Interaktion mit
5

Einleitung
Komponenten des neuronalen Zytoskeletts, bindet Tau spezifisch eine Vielzahl unter-
schiedlichster Proteine (zur Übersicht siehe Brandt & Leschik, 2004). Ein Großteil dieser
Interaktionspartner sind Komponenten verschiedener Signaltransduktionswege wie z.B. die
Kinasen GSK-3β (Glykogen Synthase Kinase 3 β), NCLK (Cdc2-ähnliche Protein Kinase),
die src-Nicht-Rezeptor-Tyrosin-Kinase Fyn oder die Phosphatase PP2A, die viel stärker
gebunden werden, als man es z.B. von einer Phosphorylierungs-/Dephosphory-lierungs-
reaktion erwartet (Lee et al, 1988; Sontag et al., 1996; Sobue et al., 2000; Sun et al., 2002).
Neben den transienten Interaktionen während einer Phosphorylierung/De-phosphorylierung
könnte über eine direkte Bindung mit Tau oder indirekt z.B. über die Bindung mit 14-3-3-zeta
eine spezifische Lokalisierung im axonalen Kompartiment erfolgen (Agarwal-Mawal, 2003).
Tau könnte so Kinasen und Phosphatasen direkt mit verschiedenen Zielproteinen in
Signalkomplexen in Berührung bringen. Zum Beispiel ist eine relativ stabile Interaktion von
NCLK und Tau in einem Komplex mit hohem Molekulargewicht bekannt, und es wird
vermutet, dass über Tau eine Lokalisation dieses Signalkomplexes an Mikrotubuli erfolgt.
Interessanterweise ist Tau in einem nicht-phosphorylierten Zustand an NCLK gebunden und
die Phosphorylierung von Tau führt zu einer Dissoziation des Komplexes (Sobue et al., 2000).
A.2.3 Die Phosphorylierung von Tau
Tau ist ein Phosphoprotein, und die Phosphorylierung von Tau beeinflusst seine Funktionen.
Durch Phosphorylierung an spezifischen Stellen im Tau-Protein kann die Bindung an
verschiedene Interaktionspartner differentiell reguliert werden. So verhindert z.B. die
Phosphorylierung an Serin 262, das in der „Repeat“-Domäne lokalisiert ist, die Bindung an
Mikrotubuli (Drewes et al., 1995) und phosphoryliertes Tau fördert die Tubulin-
Polymerisation weniger als unphosphoryliertes Tau (Lindwall & Cole, 1984; Biernat et al.,
1993; Bramblett et al., 1993). In der Nervenzelle ist Tau im somatodendritischen
Kompartiment höher phosphoryliert als im Axon (Mandell & Banker, 1996). Dies könnte mit
einer kompartimentspezifischen Bindung an Mikrotubuli im Zusammenhang stehen. Es
konnte auch gezeigt werden, dass es sich bei zytosolischem und Plasmamembran-
assoziiertem Tau um unterschiedliche Phosphoisoformen handelt, wobei zytosolisches Tau
stärker phosphoryliert ist (Maas et al., 2000).
Die Phosphorylierung von Tau wird entwicklungsspezifisch reguliert. Generell ist Tau im
fötalen Gehirn höher phosphoryliert als im adulten Tier (Mawal-Dewan et al., 1994; Soulié et
al., 1996). Dies könnte dadurch erklärt werden, dass bei der neuronalen Entwicklung eine
6

Einleitung
dynamischere Tubulin-Population benötigt wird. Es wird angenommen, dass Phosphory-
lierungs- und Dephosphorylierungsereignisse an dem ständigen Umbau des neuronalen
Zytoskeletts beteiligt sind. Tau kann in vitro und in Zellen durch eine Vielzahl von Kinasen
phosphoryliert werden (für eine Übersicht siehe Billingsley & Kincaid, 1997). Die Phos-
phorylierung kann in verschiedenen Regionen des Tau-Proteins erfolgen, jedoch ist ein
Großteil der Phosphorylierungsstellen in der „Repeat“-Domäne und den flankierenden
Bereichen lokalisiert. Die Phosphorylierung erfolgt hauptsächlich an Serin oder Threonin-
Resten. Zudem wurde auch die Phosphorylierung von Tyrosin durch die src-Nicht-Rezeptor-
Tyrosine-Kinase Fyn gezeigt (Lee et. al, 1998; Lee et al., 2004). In vitro ist Tau Substrat
verschiedener Kinasen, wie z.B. der Ca2+/Calmodulin abhängigen Kinase II (CaMKII), der
MAP Kinase, der Glykogen Synthase Kinase-3β (GSK-3β) und der cAMP-abhängigen
Protein Kinase (PKA) (Pierre & Nunez, 1983; Steiner et al., 1990; Drewes et al., 1992;
Hanger et al., 1992). Als Phosphatasen sind z.B. die Protein Phosphatasen 2A und -2B
(Calcineurin) zu nennen (Wang et al., 1995; Gong et al., 1994b).
Allerdings ist noch weitgehend unklar, welche Kinasen und Phosphatasen Tau in vivo
phosphorylieren bzw. dephosphorylieren. Im engen Zusammenhang mit der Tau-Phosphory-
lierung steht eine weitere posttranslationale Modifikation, die O-Glykosilierung. Diese
zytosolische Anhängung von N-Acetylglukosamin tritt in verschiedenen zytoskeletalen und
nukleären Proteinen auf und konnte auch in Tau nachgewiesen werden (Arnold et al., 1996).
Häufig wird die O-Glykosilierung eines Proteins invers zu seiner Phosphorylierung reguliert,
da oft dieselben Aminosäure-Reste entweder durch O-Glykosylierung oder durch Phosphory-
lierung modifiziert werden (zur Übersicht siehe Comer et al., 2000). Somit könnte die O-
Glykosylierung von Tau, ähnlich wie die Phosphorylierung, die Bindung verschiedener
Interaktionspartner beeinflussen und eventuell dadurch seine Funktionen wie z.B. seine
Aktivität, die Mikrotubulipolymerisation zu fördern, verändern.
A.2.4 Tau in der Alzheimer-Krankheit
Im Gehirn von Patienten mit AD kommt es neben extrazellulären Amyloid-Plaques zu
intrazellulären Ablagerungen von NFTs, die aus abnormalen Tau-Filamenten bestehen. Unter-
suchungen von Braak et. al. (1993) konnten zeigen, dass es im Gehirn von AD-Patienten zu
einer charakteristischen Ausbreitung der Bildung der NFTs vom Enthorhinalen Kortex über
den Hippocampus zum Isokortex kommt. Diese Ausbreitung ist einteilbar in sechs
7

Einleitung
ansteigende Schädigungsgrade und korreliert mit einem Anstieg des Phosphorylierungsgrades
des in NFTs aggregierten Tau-Proteins.
NFTs kommen in der Abwesenheit von Amyloid-Plaques auch in anderen neurodegenerativen
Erkrankungen wie z.B. Morbus Pick, Progressiver supranukleäre Blickparese, Kortikobasaler
Degeneration, Erkrankung mit agryophilen Körperchen und Frontotemporaler Demenz mit
Parkinsonismus (FTDP-17) vor. Diese und über 20 weitere Erkrankungen werden unter dem
Begriff Tauopathien zusammengefasst. In Nervenzellen, die durch diese Tauopathien betrof-
fen sind, ist Tau abnormal phosphoryliert, in Filamente aggregiert und vom axonalen zum
somatodendritischen Kompartiment relokalisiert. Im Gegensatz zu AD, wo NFTs nur in
Neuronen auftreten, treten in verschiedenen Tauopathien, wie z.B. in der Kortikobasalen
Degeneration und der Progressiven supranukleären Blickparese, NFTs auch in Gliazellen auf.
Inwiefern die gliale Pathologie allerdings die neuronale Degeneration beeinflusst oder für das
Fortschreiten der Krankheit verantwortlich ist, ist vom heutigen Standpunkt der Forschung
ungeklärt (zur Übersicht siehe Komori, 1999). Kürzlich konnte in einer Studie von Forman et
al. (2005) in einem transgenen Mausmodell mit glialer Tau-Pathologie eine Degeneration von
Neuronen gezeigt werden. Dies deutet darauf hin, dass eine Aggregation von Tau in
Gliazellen hinreichend für eine neuronale Degeneration ist.
Die Hauptkomponenten der NFTs sind „Straight filaments“ (SF) und „Paired helical
filaments“ (PHF), die sich ultrastrukturell unterscheiden (Kidd, 1963). PHFs bestehen aus
zwei umeinander gewundenen Strängen, während SFs nicht helikal sind. Es wird vermutet,
dass SFs die Vorläufer der PHFs sind (Crowther & Wischik, 1985; Crowther, 1991; Ksiezak-
Reding et al., 1996). SFs sowie PHFs bestehen beide aus hyperphosphoryliertem Tau-Protein.
Tau, das aus PHFs isoliert wurde, ist ungefähr 3-4 Mal stärker phosphoryliert (6-8 mol
Phosphat/mol Tau) als Tau aus dem gesunden Gehirn (1,9 mol Phosphat/mol Tau) (Kenessey
& Yen, 1993). Die Hyperphosphorylierung erfolgt an 18 Stellen. Einige dieser Stellen sind
auch in Tau aus gesunden Gehirnen phosphoryliert, aber in geringerem Maße (Matsuo et al,
1994; Goedert et al., 1996). In PHF-Tau lassen sich 10 PHF-Hauptphosphorylierungsstellen
ausmachen, die alle in den die „Repeat“-Region flankierenden Bereichen liegen (Morishima-
Kawashima et al., 1995). Fünf davon befinden sich in der prolinreichen Region, und fünf C-
terminal von der Mikrotubuli-Binde-Domäne. Es wird angenommen, dass ein geändertes
Kinasen/Phosphatasen-Gleichgewicht zu der sehr hohen Phosphorylierung der einzelnen
Reste führt. Dies könnte aus einer krankhaft erhöhten Kinase-Aktivität oder einer krankhaften
Erniedrigung der Aktivität bestimmter Phosphatasen resultieren. In verschiedenen Studien
8

Einleitung
wurde eine erhöhte Aktivität von Kinasen im Hirngewebe von AD-Patienten gezeigt, wie z.B.
von der Cyclin-abhängigen Kinase Cdk5 und der cAMP-abhängigen Kinase PKA (Patrick et
al., 1999; Jicha et al., 1999b). Zudem gibt es Ergebnisse, die eine verringerte Aktivität
verschiedener Protein-Phosphatasen bestätigen (Gong et al., 1993). Wie es zu diesen
Veränderungen in der Kinase-/Phosphatase-Aktivität kommt, ist weitgehend unbekannt. Es
gibt Hinweise, dass Aβ, die Hauptkomponente der Amyloidplaques, eine erhöhte Tau-Phos-
phorylierung an PHF-spezifischen Tau-Phosphorylierungsstellen induzieren kann (Busciglio
et al., 1995; Rapoport & Ferreira, 2000). Allerdings ist das Auftreten von NFTs nur in einigen
Tauopathien von Senilen Plaques begleitet. Dies spricht dafür, dass nicht ausschließlich Aβ
für eine erhöhte Tau-Phosphorylierung verantwortlich sein kann.
Im Gegensatz zu Tau aus gesunden Gehirnen ist PHF-Tau nicht in der Lage an Mikrotubuli
zu binden oder die Mikrotubulipolymerisation zu fördern (Yoshida & Ihara, 1993; Alonso et
al.,1994). Durch Phosphatase-Behandlung können diese Funktionen allerdings wiederher-
gestellt werden. Das deutet darauf hin, dass die Hyperphosphorylierung von Tau in
neurodegenerativen Erkrankungen eine zentrale Rolle spielt (Lu & Wood, 1993; Iqbal et al.,
1994).
Die Rolle der Hyperphosphorylierung bei der Aggregation von Tau und der Bildung der
NFTs ist allerdings weitgehend ungeklärt. Kürzlich konnte gezeigt werden, dass hyper-
phosphoryliertes Tau aus dem Gehirn von AD-Patienten in PHFs und SFs assembliert, und
dass die Phosphorylierung entscheidend für die Tau-Aggregation ist. Außerdem konnte die
Aggregation in Filamente durch Dephosphorylierung inhibiert werden (Alonso et al., 2001).
Zudem konnte durch Überexpression von humanen Tau in Kombination mit dessen
Phosphorylierung durch das Drosophila GSK-3β-Homolog Shaggy eine Tau-induzierte
Neurodegeneration und eine Aggregation in NFT-ähnliche Tau-Filamente beobachtet werden
(Jackson et al., 2002). Demgegenüber stehen in vitro-Experimente, in denen mit phosphory-
liertem rekombinanten Tau entweder eine reduzierte Filamentbildung oder keine Änderung
in der Filamentbildung festgestellt werden konnte (Goedert et al., 1996; Schneider et al.,
1999). Dies würde gegen eine Rolle der Tau-Hyperphosphorylierung bei der Verstärkung der
Aggregation sprechen. Unterstützt wird dies durch eine Studie, in der die Hyperphosphory-
lierung von Tau durch ortsgerichtete Mutation simuliert wurde. Dieses pseudophosphorylierte
Tau (PHP-Tau) reduzierte die Filamentbildung im Vergleich zu wildtyp (wt)-Tau
(Eidenmüller et al., 2000). In einer Folgestudie durch Verwendung dieser Tau-Mutanten
konnte jedoch gezeigt werden, dass PHP-Tau einen zytotoxischen Effekt in humanen
9

Einleitung
Modellneuronen ausübt (Fath et al., 2002). Dieses Ergebnis weist darauf hin, dass eine
Aggregation in Filamente nicht notwendig für die neuronale Degeneration ist, und statt dessen
die Hyperphosphorylierung von Tau ausreicht. Eine Aggregation in NFTs könnte also auch
einen „Rescue“-Mechanismus darstellen, der die Menge an freiem löslichen hyperphosphory-
lierten Tau in der Zelle verringert.
Außerdem scheinen die Mikrotubuli-bezogenen Funktionen von Tau keine primäre Rolle in
der Neurodegeneration zu spielen. Die Expression von PHP-Tau in PC12-Zellen führte nicht
zu einer Destabilisierung der zellulären Mikrotubuli. Auch die Behandlung mit Taxol, einem
Mikrotubuli-stabilisierenden Agenz, hob den toxischen Effekt PHP-Taus nicht auf (Fath et al.,
2002). Dies argumentiert für einen „Toxic gain of function“ (pseudo)hyperphosphorylierten
Taus gegenüber einem ebenso hypothetisierten „Loss of function“-Mechanismus, der die
Funktion von Tau als neuronales MAP in Vordergrund stellt. Hierbei wird angenommen, dass
die Hyperphosphorylierung von Tau oder Mutationen im Tau-Gen eine Destabilisierung und
eventuelle Depolymerisierung von Mikrotubuli zur Folge haben könnten. Ein Zusammen-
bruch des axonalen Zytoskeletts würde demnach zum Zelltod der Nervenzellen führen
(Ebneth et al., 1998; Stamer et al., 2002). Diese Hypothese steht im Gegensatz zu der Beob-
achtung, dass Tau nicht essentiell notwendig für stabile Mikrotubuli ist (Tint et al., 1998).
Zudem ist Tau hauptsächlich im distalen Axon angereichert, also an einer Position, in der
axonale Mikrotubuli am wenigsten stabil sind (Black et al., 1996; Kempf et al., 1996; Mandell
et al., 1996). Diese Verteilung wäre von einem Mikrotubuli-stabilisierenden Agenz nicht zu
erwarten.
A.2.5 PHF-ähnliche Phosphorylierungsmutationen
Wie bereits oben kurz erläutert, eignet sich die Methode der Imitation von Phosphory-
lierungen, um die Rolle der Hyperphosphorylierung in Tauopathien näher zu untersuchen
(Lèger et al., 1997). Durch ortsgerichtete Mutagenese können die Aminosäuren, deren Phos-
phorylierung simuliert werden soll, durch Aminosäuren mit negativ geladenen Seitenketten
wie Glutamat oder Aspartat ersetzt werden. Der Vorteil zu einer in vitro-Phosphorylierung
von Tau mit Kinasen liegt zum einen darin, eine homogene Tau-Population mit definierten
Modifikationen zu erhalten, zum anderen wird der Phosphorylierungszustand anderer Proteine
nicht verändert. 1998 stellten Eidenmüller et al. (2000) Tau-Konstrukte her, bei denen die 10
identifizierten Serin- oder Threonin-Hauptphosphorylierungstellen in Tau von AD-Patienten,
durch Glutamat ersetzt wurden. Die Einführung der negativen Ladung dieser Phosphory-
10

Einleitung
lierungsstellen führt dazu, dass dieses Tau sich strukturell und funktionell ähnlich verhält, wie
aus Gehirnen von AD-Patienten isoliertes hyperphosphoryliertes PHF-Tau (Eidenmüller et
al., 2000, Maas et al., 2000). Aus diesem Grund werden die Tau-Phosphomutanten auch als
pseudohyperphosphoryliertes Tau (PHP-Tau) bezeichnet. Eines der von Dr. J. Eidenmüller
hergestellen PHP-Tau-Konstrukte, ist schematisch in Abb. A.3 dargestellt.
Abb. A.3: Schematische Darstellung eines über ortsgerichtete Mutation hergestellten PHP-Tau-
Konstrukts.
Die Hauptphosphorylierungstellen in PHF-Tau (Morishima-Kawashima, 1995) wurden durch Glu-tamat ersetzt und sind durch Sternchen gekennzeichnet. Gezeigt ist die längste adulte Tau-Isoform, nach der sich die Nummerierung der ausgetauschten Aminosäuren richtet. Die „Repeat“-Region ist als schwarzes Kästchen, die adultspezifischen Exone in grau unterschiedlicher Schattierung ein-gezeichnet.
In der Studie von Eidenmüller et al. (2000) konnte gezeigt werden, dass PHP-Tau ebenso wie
PHF-Tau Konformationsänderungen des Proteins bewirkt, und dass PHP-Tau nicht in der
Lage ist, Mikrotubuli de novo zu nukleieren. Die Fähigkeit von PHP-Tau in Filamente zu
aggregieren, ist im Vergleich zum Wildtyp reduziert, was auch für in vitro phosphoryliertes
Tau gilt (Eidenmüller et al., 2000). Desweiteren wird wie bei phosphoryliertem Tau die
Interaktion mit der Protein-Phosphatase 2A (PP2A) durch die Phosphorylierungsimitation
komplett aufgehoben. Außerdem wird PHP-Tau durch wt-Tau von den Mikrotubuli verdrängt,
d.h. die Affinität von PHP-Tau, an Mikrotubuli zu binden, ist geringer als die von wt-Tau. Es
existiert daher die Modellvorstellung, dass das abgelöste hyperphosphorylierte Tau, den Pool
von freien Tau-Molekülen erhöht, und so eine Filamentbildung begünstigt.
Wie bereits zuvor erwähnt, zeigten nachfolgende Studien, dass in NT2-N-Zellen und differen-
zierten Pheochromocytoma (PC12)-Zellen exprimiertes PHP-Tau begleitet von einer
Caspase-3-Aktivierung zu einem zytotoxischen Effekt führt (Fath et al., 2002). Die
Aktivierung von Caspase-3 deutet daraufhin, dass bei der PHP-Tau-induzierten Neuro-
degeneration apoptotische Vorgänge (programmierter Zelltod) involviert sind. Bei Caspase-3
handelt es sich um ein Schlüsselenzym der apoptotischen Enzymkaskade, die schließlich über
11

Einleitung
die Aktivierung von Endonukleasen zur DNA-Fragmentierung und Auflösung der gesamten
Zelle führt.
Es konnte weitergehend gezeigt werden, dass hyperphosphoryliertes Tau zur Auslösung der
Neurodegeneration keine weiteren Modifikationen benötigt, wie z.B. eine Aggregation in
Filamente. Allerdings ist noch ungeklärt, welche Auslöser direkt zu dem neurotoxischen
Effekt von PHP-Tau beitragen und welche anderen Faktoren den neurotoxischen Effekt von
PHP-Tau beeinflussen.
Auf genetischer Ebene wurden bereits verschiedene Faktoren ermittelt, die in der Pathogenese
der Alzheimer-Krankheit eine Rolle spielen. Hierzu zählen neben Mutationen im APP-Gen,
Mutationen in den Genen, die für die Preseniline 1 und 2 codieren sowie Mutationen im
Apolipoprotein E- (ApoE-) Gen (zur Übersicht siehe: Levy-Lahad et al., 1995; Steiner et
al.,1999; Finckh et al., 2000). Weiterhin ist bekannt, dass oxidativer Zellstress durch die
Produktion reaktiver Oxygen-Spezies (ROS) ebenso einen Risikofaktor für die Erkrankung
darstellt (zur Übersicht siehe: Behl, 1999).
Wichtig wäre zu untersuchen, inwieweit wt-Tau bzw. PHP-Tau mit diesen Faktoren wechsel-
wirkt, bzw. ob die Hyperphosphorylierung von Tau entscheidend ist für die molekulare
Wirkweise dieser Faktoren.
A.3 Das Amyloid-Vorläufer-Protein APP und sein Peptidfragment Aβ
Aβ, der Hauptbestandteil der Amyloid-Plaques, ist ein 39 bis 43 Aminosäuren langes Protein
mit einem Molekulargewicht von ca. 4 kDa. Die hauptsächlichen Aβ-Spezies in vivo sind Aβ
40, welches mit einem Valin-Rest endet und Aβ 42, welches zwei zusätzliche hydrophobe
Reste, Isoleucin und Alanin, besitzt. Durch diese erhöhte Hydrophobizität kann Aβ42 leichter
aggregieren. Aus diesem Grund wird es im Vergleich zu Aβ40 auch als die amyloidogenere
Variante bezeichnet. Dies wird bestätigt durch die Tatsache, dass Aβ42 die vorherrschende
Komponente in den Amyloid-Plaques im Gehirn von Alzheimer-Patienten ist (Miller et al.,
1993; Roher et al., 1993, Iwatsubo et al., 1994; Näslund et al., 1994; Tamaoka et al., 1994a;
Gravina et al., 1995; Shinkai et al., 1995). Aβ40 dagegen ist die hauptsächliche Spezies in der
Zerebrospinal-Flüssigkeit (CSF) und im sekretierten Medium von kultivierten Zellen (Dovey
et al., 1993; Vigo-Pelvrey et al., 1993; Asami-Odaka et al., 1995). Da Aβ42 die Haupt-
komponente der Amyloid-Plaques repräsentiert, wird angenommenen, dass die hydrophobere
Variante in der Entwicklung der Alzheimer-Pathologie eine wichtige Rolle spielt. Auch in
12

Einleitung
Experimenten mit transfizierten Zellkulturen, zeigten APP-Mutanten, die in familiären Alz-
heimer-Fällen auftreten, eine erhöhte Aβ42-Produktion, was ein weiterer Hinweis auf die
Verbindung von Aβ42 in der AD-Pathogenese ist (Suzuki et al., 1994; Tamaoka et al.,
1994b).
A.3.1 Die Prozessierung von APP
Aβ ist ein Peptidfragment des Amyloid-Vorläufer-Proteins APP. APP ist ein glykosyliertes
Typ-1 Transmembranprotein mit einer kurzen carboxyterminalen zytoplasmatischen Domäne.
Die biologische Funktion von APP ist nicht vollständig geklärt. Es existieren Hinweise, dass
APP eine Rolle bei der Aufrechterhaltung und Funktion von Synapsen hat. Zudem gibt es
einige Veröffentlichungen, die auf eine trophische Funktion von Aβ in physiologischen
Konzentrationen hindeuten (für eine Übersicht siehe Saitho & Mook-Jung, 1996; Atwood et
al., 2003). Aβ besteht aus der Ektodomäne von APP (28 Aminosäuren außerhalb der Mem-
bran) und der luminalen Hälfte der Transmembrandomäne (12 oder 14 Aminosäuren). APP,
welches in drei hauptsächlichen Isoformen (695, 751, 770) zellulär vorliegt, wird im
Endoplasmatischen Retikulum und im Golgi-Apparat N- bzw. O-glykosyliert und über
sekretorische Vesikel zur Plasmamembran transportiert. Ein Teil der APP-Moleküle wird
über Clathrin-abhängige Endozytose internalisiert. Während dieses Transportes wird APP in
verschiedenen Schritten proteolytisch prozessiert (siehe Abb. A.4). Das Schneiden von APP
durch die Typ-1 membrangebundene Aspartyl-Protease BACE-1 (β-Sekretase) resultiert in
einer löslichen Form von APP, solubleAPPβ (sAPPβ), und einem membrangebundenen C-
terminalen Fragment (CTF oder C99) (Hussain et al., 1999; Sinha et al., 1999; Vassar et al.,
1999; Yan et al. 1999). Die nachfolgende Prozessierung durch den γ-Sekretase-Komplex in
der Transmembrandomäne setzt Aβ40 bzw. Aβ 42 frei (für eine Übersicht siehe: Kimberly &
Wolfe, 2003). In einem alternativen Weg kann APP auch durch die α-Sekretase (TACE,
ADAM10) in der Mitte der luminalen Region geschnitten werden, was zu einem löslichen
sAPPα und einem anderem carboxyterminalen Fragment (C83) führt. Dieser Prozessierungs-
weg schließt die Produktion des amyloidogenen Aβ aus und führt letztlich zur Generierung
des p3-Fragmentes (Aβ 17-40 bzw. Aβ 17-42) (Buxbaum et al., 1998; Lammich et al., 1999;
Postina et al., 2004).
13

Einleitung
Abb. A.4: Prozessierung von APP.
APP wird entweder durch die α- oder die β-Sekretase prozessiert, welche an verschiedenen Stellen in der extrazellulären Domäne spalten. Es ist die Orientierung von APP in der Membran eines sekretorischen Vesikels gezeigt. Das Schneiden der α-Sekretase (TACE, ADAM10) resultiert in C83, welches die β-Amyloid (Aβ)-Generierung ausschließt. Nach der γ-Sekretase-Prozessierung von C83 wird ein kleines Peptidfragment p3 generiert. Das sequentielle Schneiden von β-Sekretase (BACE1) und γ-Sekretase führen zu der Produktion von vollständigem Aβ. AICD (APP intracellular domain) entsteht im amyloidogenen und nicht-amyloidogenen Weg.
Das C-terminale Produkt der γ-Sekretase AICD (APP intracellular domain) bindet das
Adaptor Protein Fe65 und wird in den Zellkern transloziert, um einen transkriptionsaktiven
Komplex mit der Histon-Acetyltransferase Tip60 einzugehen (Cao & Sudhof, 2001, 2004;
Kimberly et al., 2001). Vor kurzem wurde berichtet, dass die Fe65-mediierte Transkriptions-
aktivität von APP die Expression verschiedener Gene, darunter APP und BACE, hoch-
reguliert (Von Rotz et al., 2004). Deshalb wird angenommen, dass die γ-Proteolyse von APP,
die AICD freisetzt, in einer Hochregulierung der amyloidogenen Prozessierung resultiert.
Es ist noch ungeklärt, in welchem zellulären Kompartiment die APP-Prozessierung tat-
sächlich erfolgt. Nach dem bisherigen Stand der Forschung scheint Aβ sehr früh im
sekretorischen und/oder endosomalen Weg produziert zu werden. Das meiste Aβ,
insbesondere Aβ40 wird allerdings von Zellen sekretiert und kann im CSF und im Plasma von
normalen Individuen detektiert werden.
14

Einleitung
A.3.2 Aβ in familiären (FAD) und sporadischen Formen der Alzheimer-Krankheit
Aβ wird in senilen Amyloid-Plaques im Gehirn von Alzheimer-Patienten abgelagert.
Interessanterweise akkumuliert Aβ auch im Gehirn von alternden, normalen (nicht-dementen)
Individuen, was gegen eine zentrale Rolle der Amyloid-Ablagerungen in der Neurodege-
neration spricht (Funato et al., 1998; Morishima-Kawashima et al., 2000). Allerdings ist in
diesem Zusammenhang entscheidend, dass das Vorkommen von einzelnen Punktmutationen
im APP-Gen oder in den Presenilin (PS1 und 2)-Genen zu familären Formen von Alzheimer
(FAD) führt, die durch ein sehr frühes Auftreten der Erkrankung (<60 Jahren) und eine
aggressive Pathologie gekennzeichnet sind (Goate et al., 1991; Rogaev, 1995; Sherrington et
al., 1995). Diese FAD-Fälle machen jedoch nur ca. 3% aller Alzheimer-Erkrankungen aus
und werden von den sporadischen Formen unterschieden, die ohne einen kausalen
genetischen Zusammenhang auftreten.
FAD-Mutationen werden autosomal dominant vererbt, und führen alle zu einer stark erhöhten
Produktion von Aβ42 (Cai et al., 1993; Suzuki et al., 1994; Borchelt et al., 1996; Citron et al.,
1996; Murayama et al., 1999). Die meisten der FAD-Mutationen in APP treten in den
flankierenden Regionen des Aβ Moleküls auf, und es wird spekuliert, dass durch diese
Mutationen entweder die β- oder die γ-Sekretase so beeinfusst werden, dass es dadurch zu
einer erhöhten Gesamtproduktion von Aβ bzw. Produktion von Aβ42 kommt. Es exisitieren
verschiedene „Pools“ von Aβ im Gehirn von AD-Patienten. Durch die Spaltung von APP liegt
Aβ zunächst intrazellulär (hauptsächlich im ER) vor, dann extrazellulär im Gehirngewebe.
Für eine Aggregation in Aβ-Fibrillen tritt vermutlich vorerst eine Dimerisierung von Aβ auf,
dann eine Oligomerisierung und schließlich eine Multimerisierung. Extrazellulär wird Aβ in
Amyloid-Plaques abgelagert, welche in neuritische, primitive und diffuse Plaques unterteilt
werden. Neuritische und primitive Plaques bestehen aus Aβ-Fibrillen und begleiten dystro-
phische Neuriten und gliale Reaktionen, wie reaktive Astrozyten und aktivierte Mikroglia.
Die Hauptkomponente der diffusen Plaques dagegen, ist eher amorphes, granuläres Aβ als
Fibrillen.
Für sporadische Fälle der Alzheimer-Krankheit ist als hauptsächlicher Risikofaktor die
Gendosis der Apolipoprotein E4 (ApoE4)-Isoform entscheidend (Corder et al., 1993).
Apolipoprotein E ist mit High Density Lipoproteinen (HDL) assoziiert, welche wichtig für die
zelluläre Cholesterin-Homöostase sind (Rebeck, 1998). Zudem ist bekannt, dass ein hoher
Cholesteringehalt mit einem erhöhtem Risiko der Alzheimer-Krankheit korreliert (Kuo et al.,
1998). Auf der zellulären Ebene findet die Aβ-Generierung in speziellen Cholesterin-reichen
15

Einleitung
Subdomänen der Membranen, den DRMs (Detergenz-resistente Membranen), statt (Ehehalt et
al., 2003). Desweiteren zeigte die Behandlung mit Cholesterin-Synthese-Hemmern (Statinen)
ein starkes Abfallen der Aβ-Produktion in kultivierten Neuronen. In transgenen Tieren
verringerten Statine die Menge an Amyloid-Plaques (Simons et al., 1998; Fassbender et al.,
2001). Es wird postuliert, dass dies zu einem großen Teil durch die Verschiebung in Richtung
des nicht-amyloidogenen α-Sekretase-Wegs zu Stande kommt (Kojro et al., 2001). Vorläufige
Ergebnisse aus epidemiologischen Studien unterstützen die Hypothese, dass eine
Verringerung des Cholesteringehalts das Risiko der Alzheimer-Krankheit senken kann
(Wolozin et al., 2000; Simons et al., 2002).
A.3.3 Die Neurotoxizität von Aβ
Verschiedene Studien mit Zellkulturen belegen, dass die Expression von FAD mutanten APP
und PS Genen zu neuronalem Zelltod führt (Yamatsuji et al., 1996; Guo et al., 1996, 1999;
Wolozin et al., 1996; Zhao et al., 1997; Czech et al., 1998; Luo et al., 1999; Weihl et al.,
1999). Zudem wirken Aβ-Peptide in vitro toxisch auf Neurone (Loo et al., 1993; Gschwind &
Huber, 1995). Diese Zytotoxizität erfolgt durch unterschiedliche, definierte intrazelluläre
Mechanismen wie die Destabilisierung der Calcium-Homöostase (Mattson et al., 1992),
Calpain aktivierte Cdk5-Signalwege (Lee et al., 2000) und die Beteiligung des c-Jun N-
terminal Kinase-Weges (JNK) (Boyzysko-Coyne et al., 2001; Troy et al., 2001; Morishima et
al., 2001; Wei et al., 2002).
Es ist bekannt, dass im Gehirn von Alzheimer-Patienten pathologische Oxidations-Reaktionen
auftreten können. Eine erhöhte Protein-Oxidation konnte in Gehirnschnitten verstorbener
Patienten insbesondere im Hippokampus und parahippokampalen Gyrus detektiert werden
(Aksenov et al., 2001). In diesem Zusammenhang konnte nachgewiesen werden, dass Aβ
durch Akkumulation von Wasserstoffperoxid oxidativen Stress in kultivierten Neuronen
induzieren kann (Behl et al., 1994; Schubert et al., 1995; Behl, 1999). Zudem wurde
beschrieben, dass fibrilläres Aβ den Phosphorylierungszustand von Tau erhöht, gefolgt von
einer progressiven Degeneration neuronaler Fortsätze (Rapoport & Ferreira, 2000). Außer-
dem wurde berichtet, dass Tau notwendig für die Aβ-induzierte Neurotoxizität ist (Rapoport
et al., 2002). Schließlich führt Aβ über die Aktivierung der Caspase-Kaskade zur Auslösung
von Apoptose (Harada & Sugimoto, 1999; Nakagawa et al., 2000; Troy et al., 2000). Jedoch
ist die Relevanz der Amyloid-Plaques im Pathomechanismus der Alzheimer-Erkrankung
immer noch ungeklärt. In mehreren Studien mit transgenen Mäusen, die zwar durch die
16

Einleitung
Überproduktion von extrazelluärem Aβ Plaques entwickelten, konnte im Gehirn dieser Mäuse
kein Absterben von Neuronen beobachtet werden (La Ferla, 1995; Irizarry et al., 1997a, b;
Holocomb et al., 1999). Dies weist in vivo auf eine limitierte Rolle der extrazellulären Aβ-
Sekretion in der Neurodegeneration hin. Im Gegensatz dazu konnte ein Absterben von
Neuronen durch die Verwendung einer α-Sekretase-resistenten APP-Mutante in der Abwesen-
heit von Amyloid-Plaques beobachtet werden (Moechars et al., 1996). Desweiteren gibt es
verschiedene klinische Indizien, die gegen eine Beteiligung der Aβ-Ablagerungen am
neuronalen Zelltod im AD-Gehirn sprechen. Zum Beispiel verursacht die APP-FAD-Mutante
E618Q eine vererbbare Gefäßkrankheit mit starken Hirnblutungen. Trotz einem gehäuften
Auftreten von Amyloid-Plaques, führt diese Mutation allerdings in den meisten Fällen nicht
zu der typischerweise im AD-Gehirn beobachteten Neurodegeneration (Haan et al., 1992).
Diese Befunde decken sich mit in vitro-Ergebnissen, die zeigen konnten, dass nicht das
sekretierte Aβ verantwortlich für die APP-induzierte-Zytotoxizität ist (Irizarry et al., 1997a, b;
Sudo et al., 2001). Es wird diskutiert, dass möglicherweise intrazelluläres Aβ zum Absterben
von Neuronen führt. La Ferla et al. (1995) konnten zeigen, dass in transgenen Mäusen, die Aβ
intrazellulär exprimieren, neuronale Degeneration auftritt. Zudem wurde berichtet, dass die
Überexpression von FAD mutantem PS1 in Mäusen zum Neuronentod führt, begleitet von
einer intraneuronalen Aβ-Akkumulation ohne das Vorhandensein von extrazellulären Plaques
(Chui et al., 1999). Tabira et al. (2002) fanden eine signifikante Erhöhung von intrazellulärem
Aβ42-positiven Neuronen in sporadischen und familiären Formen von Alzheimer. Unterstützt
werden diese in vivo-Ergebnisse, durch eine Studie, die nachweist, dass intrazelluläres Aβ42
zur Apoptose von kortikalen Primärkulturen führt (Kienlen-Campard et al., 2002). Dement-
sprechend erfordert diese Hypothese weitere Nachforschungen.
Interessanterweise konnte auch für durch die γ-Sekretase produzierte intrazelluläre Domäne
AICD eine positive Regulation der Apoptose beschrieben werden (Passer et al., 2000).
A.3.4 Aβ und Tau im funktionellen Zusammenhang
Intronische und exonische Mutationen im Tau-Gen führen zu der Bildung von NFTs, nicht
aber von Amyloid-Plaques (Spillantini et al., 1998). Da Mutationen in APP dagegen aber in
beiden neuropathologischen Merkmalen resultieren, läßt dies den Schluß zu, dass die
Amyloid-Pathologie „upstream“ von der Tau-Pathologie erfolgt (Price et al., 1998; Selkoe,
1998). Allerdings zeigen Mutationen im Tau-Gen, dass eine Tau-Pathologie ausreicht, eine
Demenzerkrankung auszulösen. Obwohl diese beiden Pathologien in denselben
17

Einleitung
Gehirnregionen auftreten, fehlt immer noch der Nachweis eines klaren mechanistischen
Zusammenhangs. Wie oben bereits erwähnt, konnte in verschiedenen in vitro-Studien gezeigt
werden, dass Aβ einen direkten Einfluß auf den erhöhten Phosphorylierungszustand von Tau
hat (Busciglio et al., 1995; Greenberg et al., 1995; Takashima et al., 1998a; Rapoport &
Ferreira, 2000) und eine Aggregation in Tau-Filamente verursacht (Rank et al., 2002).
Desweiteren konnte in Studien mit Tau-„knock-out“-hippokampalen Neuronen nachgewiesen
werden, dass die Aβ-induzierte Tau-Phosphorylierung letztlich zum Neuronentod führt, und
Tau für die Neurotoxizität von Aβ essentiell ist (Rapoport et al., 2002; Zheng et al., 2002). Es
wurde vermutet, dass in diesen Neuronen, die kein Tau besitzen, die dadurch dynamischere
Mikrotubuli-Population eventuell die Neurotoxizität von Aβ verhindert. Für die
Überexpression von Tau in Fibroblasten (Ebneth et al., 1998) und neuralen Zellen (Stamer et
al., 2002) konnte bereits eine Störung des axonalen Transports durch Hemmung des Kinesin-
abhängigen Vesikeltransports und dem Transport von Neurofilamenten und Peroxisomen
gezeigt werden. Dies könnte die Zelle sensitiver für oxidativen Stress machen und letzlich zur
Neurodegeneration führen. Unter anderem wurde der Transport von APP gehemmt, wodurch
APP im Zellkörper akkumulierte, was eine hohe intrazelluläre Produktion von toxischem Aβ
begünstigen könnte (Xu, 1997; Stamer et al., 2002).
Auch in vivo konnte durch die Verwendung verschiedener FAD-transgener Tiermodelle ein
kausaler Zusammenhang zwischen Aβ und Tau nachgewiesen werden. So zeigten unter-
schiedliche FAD-transgene Mäuse, die Aβ überproduzierten, einen Anstieg in der Tau-Phos-
phorylierung und kognitive Defekte (Tomidokoro et al., 2001; Otth et al., 2002; Echeverria, et
al., 2004). In doppelt transgenen FAD-APP/-PS1-transgenen Mäusen wurden, im Gegensatz
zu einer früheren Studie von Xu et al. (2002), neben hyperphosphoryliertem Tau, PHF-
ähnliche Filamente nachgewiesen (Kurt et al., 2003). In diesem Zusammenhang sind auch die
Ergebnisse von Lewis et al. (2001) zu nennen. In einer FDTP-17-Tau/FAD-APP Doppel-
transgenen Maus, die Plaques und NFTs aufweist, konnte ein stärkeres Ausmaß an NFTs im
Vergleich zu einer Einzel-Tau mutanten Maus festgestellt werden. Zusätzlich wurden NFTs in
Regionen beobachtet, die im Einzel-transgenen Tier nicht betroffen sind. In einem parallelen
Ansatz von Götz et al. (2001) wurde in einer FDTP17-Tau-transgenen Maus durch die
Injektion von fibrillärem Aβ eine erhöhte Tau-Aggregation in NFTs nachgewiesen.
Trotz dieser vielfältigen Studien, die auf eine Korrelation von APP und Tau hinweisen, ist die
Rolle der Plaques in der Tau-Phosphorylierung und der Aggregation in Filamente unklar. Im
Gegensatz zu der oben beschrieben Studie von Kurt et al. (2003) existieren im Tiermodell von
18

Einleitung
Lewis et al. (2001) z.B. keine typischen Aβ-Plaques, welche NFT-positive dystrophische
Neuriten umgeben. Dagegen konnten von Tomidokoro et al. (2001) in einem FAD-APP-
Mausmodell eine erhöhte Tau-Phosphorylierung in dystrophischen Neuriten, umgeben von
senilen Plaques, nachgewiesen werden.
Eventuell ist auch hier intrazelluläres Aβ von Bedeutung. In transgenen Ratten, die Aβ
intrazellulär überexprimierten, konnte trotz fehlender extrazellulärer Plaques eine Hyper-
phosphorylierung von Tau aber keine Aggregation in Filamente nachgewiesen werden
(Echeverria et al., 2004). Allerdings scheint genau wie intrazelluläres Aβ, extrazelluläres Aβ
nicht ausreichend für eine NFT-Pathologie zu sein, da es auch durch die Aβ-Injektion in wt-
Tau überexprimierenden Mäusen nicht zur Generierung von NFTs kam (Götz et al., 2001).
Es ist klar ersichtlich, dass weitere zelluläre Modelle und Tiermodelle entwickelt werden
müssen, um die Rolle von Tau in der Neurodegeneration und der Interaktion mit anderen
krankheitsrelevanten Faktoren, insbesondere Aβ, zu analysieren.
A.4 Die Preseniline
A.4.1 Die biologische und pathologische Rolle der Preseniline
Wie bereits oben beschrieben, führen neben FAD-APP-Mutationen auch Mutationen in den
beiden sehr homologen Presenilin-Genen, die für Presenilin1 und 2 (PS1, PS2) codieren, zur
früh einsetzenden autosomal dominant vererbbaren Form der Alzheimer-Erkrankung. Bei fast
allen der über 120 Presenilin assoziierten FAD-Mutationen, die bis jetzt identifiziert wurden,
handelt es sich um den Austausch einzelner konservierter Aminosäuren.
Beide humanen Presenilin-Gene werden ubiquitär exprimiert, aber in einer geringen Konzen-
tration. Im Gehirn werden diese Gene stärker in Nervenzellen als in Gliazellen exprimiert
(Price et al., 1998). Subzellulär sind sie hauptsächlich im Endoplasmatischen Retikulum (ER)
und im Golgi-Apparat verteilt. Presenilin 1 und 2 sind Multitransmembran-Proteine, die die
Membran acht mal durchspannen, mit N- und C-terminalen zytoplasmatischen Enden und
einem großen zytosolischen „Loop“ zwischen Transmembransegment 6 und 7 (Haass & De
Strooper, 1999). Preseniline werden endoproteolytisch gespalten, was zur Generierung eines
stabilen N- und C-terminalen Fragments führt (Abb. A.5). Diese assoziieren wieder mit-
einander, um ein funktionelles Heterodimer zu formen (Thinakaran et al., 1996; Podlisny
et al., 1997).
19

Einleitung
Abb. A.5: Schematische Darstellung von Presenilin in der Membran.
Presenilin ist ein Membranprotein mit 8 Transmembrandomänen, einem zytosolischen N- und C-Terminus und einem großen zytosolischen Loop. Presenilin wird endoproteolytisch zwischen T291 und M298 (weiße Kästchen) gespalten und bildet stabile N- und C-terminale Fragmente (NTF, CTF). Die Transmembransegmente 6 und 7 besitzen zwei katalytische Aspartat-Reste (durch Sternchen markiert).
Beide Proteine weisen große Sequenzhomologien in der gesamten Proteinlänge auf. Am
Aminoterminus, sowie in der zweiten Hälfte des zytosolischen „Loops“ kommt es jedoch zu
hochdivergenten Bereichen. Daher wird angenommen, dass diese Bereiche zu den
unterschiedlichen Funktionen beider Preseniline führen könnten. Eine Untersuchung der
bisher analysierten Presenilin-assoziierten FAD-Mutationen zeigt, dass diese „Missense“-
Mutationen über das gesamte Protein verteilt in für beide Preseniline identischen Resten
vorliegen. Hierdurch ist anzunehmen, dass diese konservierten Reste wichtig für die Funktion
beider Proteine sind. Studien mit Presenilin-Homologen in Caenorhabditis elegans (Levitan
& Greenwald, 1995; Haass & De Strooper, 1999) und Drosophila melanogaster (Struhl &
Greenwald, 1999; Ye et al., 1999) haben gezeigt, dass Preseniline für die Entwicklung
essentiell sind. Auch in Mäusen führt ein „knock-out“ des Presenilin-1-Gens zum Tod des
Jungtieres kurz nach der Geburt. Eine Untersuchung dieser Maus-Embryonen ergab, dass
schwere Defekte im zentralen Nervensystem mit einer abnormalen Entwicklung des axialen
Skeletts und der Spinalganglien vorlagen (Shen et al., 1997). Im Gegensatz dazu zeigte eine
PS-2-„knock-out“-Maus keine Defekte, jedoch starben PS1/PS-2- Doppel-„knock-out“-
Mäuse bereits im Embryonalalter. Hier zeigte sich eine erhebliche Fehlexpression von
Proteinen, deren Expression durch Notch reguliert wird (Donoviel et al., 1999). Notch ist
Signalmolekül, welches während der Embryogenese das Zellschicksal determiniert.
20

Einleitung
Diese Ergebnisse deuten darauf hin, dass eine funktionelle Redundanz zwischen PS1 und PS2
besteht, nämlich dass PS1 einen Verlust von PS2 kompensieren kann, aber nicht umgekehrt.
Preseniline sind essentiell für die Prozessierung des Notch-Rezeptors und die Freisetzung der
Notch intrazellulären Domäne (NICD), die letzlich im Kern mit Transkriptionsfaktoren
interagiert (Schroeter et al., 1998). Die Freisetzung von NICD erfolgt durch Spaltung des
membranassoziierten C-Terminus in der Transmembran-Region. Dies erfolgt durch die
Aktivität der γ-Sekretase, welche Presenilin benötigt, um katalytisch aktiv zu sein (De
Strooper et al., 1999). Es existieren Parallelen zwischen der Prozessierung von Notch und
APP. Bei APP handelt es sich ebenso um ein Transmembran-Protein, welches durch den γ-
Sekretase-Komplex in der Transmembranregion zu den kleineren Fragmenten Aβ bzw. p3
und AICD hydrolysiert wird. Genauso wird für die γ-Sekretase-Spaltung die Beteiligung von
Presenilin benötigt, wie Presenilin 1- und 2- Knock-Out-Experimente zeigen. Diese
resultierten in einem völligen Verlust der APP-Prozessierung (Herreman et al., 2000; Zhang
et al., 2000). Kimberly et al. (2003a) konnten nachweisen, dass die Substrate APP und Notch
ihre Spaltung gegenseitig verhinderten, was darauf hinweist, dass an jedem Signalweg
dergleiche γ-Sekretase-Komplex beteiligt ist. Kürzlich konnten die Komponenten des γ-
Sekretase-Komplex vollständig identifiziert und charakterisiert werden. Neben PS1 und PS2,
sind drei weitere Proteine an der vollständigen γ-Sekretase-Funktion beteiligt: Nicastrin, Aph-
1 und Pen-2. Die Analyse in Säugerzellen zeigte, dass die 4 Komponenten die Reifung
voneinander kooperativ regulieren, und dass Nicastrin, Aph-1 und Pen-2 die postulierten
limitierenden Co-Faktoren von Presenilin repräsentieren (Edbauer, 2003, Kimberly et al.,
2003b; Takasugi et al., 2003). In der Studie von Edbauer et al. (2003) konnte erst durch
Expression der vier unterschiedlichen Proteine in Hefe eine funktionsfähige γ-Sekretase
hergestellt werden. Hefen besitzten kein apparentes Ortholog jedes dieser Proteine und somit
auch keine endogene γ-Sekretase-Aktivität. Es wird angenommen, dass die Preseniline das
katalytische Zentrum der γ-Sekretase ausmachen und eventuell der Familie der Aspartyl-
Proteasen angehören, die zwei katalytische Aspartat-Reste im Reaktionszentrum besitzen
(Wolfe et al., 1999). Kürzlich wurde von Steiner et al. (2000) festgestellt, dass es sich bei
Glyzin 384 (in PS) um eine konservierte Aminosäure im Sequenzvergleich mit bakteriellen
Aspartyl-Proteasen (type 4 prepilin peptidases, TFPP) handelt, und dass dieses Glyzin
eventuell entscheidenden funktionellen Einfluss auf den möglicherweise proteolytisch aktiven
Bereich von Presenilin hat (Steiner et al., 2000).
21

Einleitung
A.4.2 Die Rolle der Preseniline im Zelltod
Mutationen in Presenilin-Genen, die mit der familiären Form von AD in Verbindung stehen,
erhöhen die Produktion des 42-Aminosäure langen APP-Fragments Aβ42. Außerdem
aktivieren Mutationen in den Presenilin-Genen apoptotische Kaskaden und sensitivieren die
Zellen z.B. gegenüber Aβ- Zytotoxizität (Deng et al., 1996; Vito et al., 1996; Wolozin et al.,
1996; Guo et al., 1997; Mattson et al., 2000a; Alves da Costa, 2002). Dies erfolgt eventuell
über die Aktivität der Tau-Kinase GSK-3β, von der gezeigt wurde, dass sie in den Aβ-
induzierten Zelltod involviert ist (Hoshi et al., 1996). Es gibt Hinweise, dass die Expression
des Presenilin 1-Gens antiapoptotisch in Zellen wirken kann, eventuell unter Beteiligung des
antiapototischen Proteins Bcl-2 (Bursztajn et al., 1998; Araki et al., 2001; Alves da Costa,
2002; Terro et al., 2002). Außerdem wurde gezeigt, dass der PI3K/Akt-Signalweg hier
involviert ist. Phosphatidylinositol 3-Kinase (PI3K) sichert das Überleben der Zelle durch die
Aktivierung der Effektor-Kinase Akt, welche u.a. die Kinase GSK-3 (α/β) hemmt. Baki et al.
(2004) konnten zeigen, dass PS1 durch die Aktivierung des PI3K/Akt-Signalweges und einer
folgenden Hemmung der GSK-3 Apoptose verhindert. FAD-mutantes Presenilin dagegen
hemmte PI3K/Akt und führte zu einer GSK-3-induzierten Tau-Phosphorylierung.
A.2.3 Presenilin und Tau im funktionellen Zusammenhang
In kultivierten Neuronen assoziiert PS1 mit Mikrotubuli und Aktinfilamenten und einige
FAD-Mutationen verhindern diese Interaktionen (Pigino et al., 2001).
Durch Co-Immunpräzipitations-Experimente transfizierter Zellen konnte gezeigt werden, das
PS1 auch direkt mit GSK-3β und Tau interagiert (Takashima et al., 1998b). Die PS1/Tau-
Interaktion erfolgt in Presenilin zwischen den Resten 250-298 und in Tau über die
Mikrotubuli-Bindedomäne. Demnach erfolgt die Kolokalisation von Mikrotubuli und PS1
nicht über Tau. Während die Aminosäuren 250-263 in Presenilin in der Membran liegen, sind
die Reste 264-298 im Zytosol lokalisiert und machen somit eine Interaktion mit Tau möglich
(für eine Übersicht der Presenilin-Organisation siehe Abb. A.5 und Wolfe & Haass, 2001).
Nicht alle der getesteten FAD-Mutationen hatten einen Effekt auf die Interaktion mit Tau
(Takashima et al., 1998b). Auf der anderen Seite führten die FAD-Mutationen, die im Bereich
der Tau-Bindedomäne, 250-298, lokalisiert sind (C263R, P264L), zu einer stark erhöhten
Tau-Phosphorylierung. Eventuell ist dies auf die beobachtete drei mal stärkere PS1-GSK-3β-
Assoziation zurückzuführen. Da die Interaktionsdomäne von GSK-3β und Tau im selben
Bereich von Presenilin liegt, wird vermutet, dass Presenilin sich wie ein Ankerprotein verhält,
22

Einleitung
welches Tau und GSK-3β nahe an einander bindet, um so deren Enzym-Substrat-Reaktion zu
katalysieren. Versuche mit anderen FAD-Mutationen, die nicht in der Tau-Binde-Region
liegen (G384A und I143T), zeigten keine Änderungen der Tau-Phosphorylierung (Julliams et
al., 1999). Für die Mutation L392V konnte zudem eine geringere Assoziation von PS1 mit
GSK-3β nachgewiesen werden (Gantier et al., 2000). Allerdings konnte kürzlich in der bereits
oben beschriebenen Studie von Baki et al. (2004) gezeigt werden, dass die Mutationen
M146L (nicht in der Tau Binderegion), A246E und E280A die Aktivität von GSK-3 und die
Tau-Phosphorylierung an AD-relevanten Seiten erhöhen. Dies erfolgte durch eine FAD-PS1-
induzierte Hemmung des PS1-abhängigen PI3K/Akt-Signalweges, dem eine bedeutende
Funktion im Zellüberleben zukommt. Zudem konnte festgestellt werden, dass dies γ-
Sekretase-unabhängig erfolgt und somit eher von einem „Loss of function“ der PS1 FAD-
Mutationen gesprochen werden kann. Im Vergleich dazu wird der γ-Sekretase-abhängige
Anstieg der Aβ-Produktion als „Gain of function“ hypothetisiert.
Interessanterweise befindet sich die Seite, an der Preseniline proteolytisch in N- und C-
Terminale Fragmente prozessiert werden (T291-M298), auch in der Tau-Binderegion
(Podlisny et al., 1997). Es kann vermutet werden, dass Tau die proteolytische Spaltung von
Presenilin reguliert und dadurch die Formation eines funktionellen γ-Sekretase-Komplexes
beeinflusst.
Es ist unklar, ob die Tau-Pathologie und die Preseniline durch eine direkte funktionelle
Interaktion im Zusammenhang stehen. Möglich ist auch, dass die PS1 FAD-Mutationen einen
indirekten Effekt, z.B. durch eine erhöhte Tau-Phosphorylierung durch die gesteigerte
Produktion von neurotoxischen Aβ-Fragmenten, ausüben.
23

Einleitung
A.5 Ziel der Arbeit
Die Alzheimer-Krankheit ist gekennzeichnet durch ein massives Absterben von Nervenzellen
in selektiven Regionen im Gehirn. Nach Autopsie verstorbener Patienten werden fibrilläre
Proteinablagerungen in verschiedenen Gehirnregionen nachgewiesen. Es handelt sich zum
einen um extrazelluläre Ablagerungen des APP-Proteinfragments Aβ, zum anderen um in
Nervenzellen vorliegende Aggregate des Proteins Tau. In familiären Formen der
Alzheimerkrankheit (FAD) liegen Mutationen entweder im APP-Gen oder in den Presenilin
(PS)-Genen 1 und 2 vor. Alle bisher identifizierten FAD-Mutationen sind durch eine
Erhöhung der Produktion des neurotoxischen Aβ42-Fragments charakterisiert.
Das mikrotubuliassoziierte Protein Tau ist vermutlich ein für die Entwicklung und
Aufrechterhaltung der Zellstruktur entscheidendes Protein. Im Gehirn, welches an AD
erkrankt ist, liegt Tau in einem abnormalen stark phosphorylierten Zustand (Hyperphosphory-
lierung) vor. Um die pathologische Rolle des krankhaft veränderten Phosphorylierungs-
zustandes von Tau zu untersuchen, wurden mutante DNA-Konstrukte von Tau hergestellt,
deren Expression eine Hyperphosphorylierung von Tau simulieren kann. Es wurde fest-
gestellt, dass dieses pseudohyperphosphorylierte Tau (PHP-Tau) in humanen Modellneuronen
zur Degeneration führt. Allerdings sind die intrazellulären Mechanismen, durch die PHP-Tau
einen zytotoxischen Effekt ausübt, noch weitgehend ungeklärt. Bisher konnte gezeigt werden,
dass letztlich die Auslösung des programmierten Zelltods (Apoptose) involviert ist. Die
Zwischenschritte, die zur Auslösung der apoptotischen Kaskade führen, sind allerdings
unbekannt. Das Ziel dieser Arbeit bestand darin, die Neurotoxizität von PHP-Tau im
Zusammenhang mit weiteren AD-relevanten Proteinen zu untersuchen. Zur Analyse einer
funktionellen Interaktion sollten hierfür kortikale Primärkulturen aus FAD-mutanten
transgenen APP- und PS1-Mäusen bzw. PS1wt-transgenen Mäusen verwendet werden. Die
Überexpression von humanem wt- und PHP-Tau sollte in kultivierten Neuronen dieser Mäuse
über HSV-1-vermittelten Gentransfer erfolgen.
24

Material und Methoden
B Material und Methoden
B.1 Material und Geräte
B.1.1 Chemikalien
Alle verwendeten Chemikalien wurden, falls nicht anders vermerkt, von den Firmen
Chemicon (Hofheim), Merck (Darmstadt), Carl Roth GmbH & Co. KG (Karlsruhe), Serva
Feinbiochemica GmbH (Heidelberg), Sigma-Aldrich Chemie GmbH (Deishofen) und Riedel-
de Haen AG (Seelze) bezogen. Zellkultur-Produkte stammten von der Firma GIBCO BRL
Life Technologies GmbH (Karlsruhe). Reinstwasser (ddH2O) wurde mit dem Milli-Q Plus
Ultra-Pure Water System der Firma MILLIPORE GmbH (Eschborn) hergestellt (elektrischer
Widerstand 18,2 MegaOhm). Flüssiger Stickstoff wurde von dem physikalischen Institut der
Universität Osnabrück zur Verfügung gestellt.
B.1.2 Material-Molekularbiologie
B.1.2.1 Plasmide
pHSV-eGFP-352wt-Tau (Leschik, 2000)
pHSV-eGFP-352PHP-Tau (Leschik, 2000)
pHSV-eGFP (hergestellt von Dr. Jörg Piontek)
B.1.2.2 Primer
Alle verwendeten Primer wurden von der Firma MWG-Biotech AG (Ebersburg) hergestellt.
Konzentration: 100 pmol/µl
PS1 Primer: PS1 „aller“: 5’ - TAA TTG GTC CAT AAA AGG C - 3’
PS1 „retour“: 5’ - GCA CAG AAA GGG AGT CAC AAG - 3’
APP Primer : APP „aller“: 5’ - GTA GCA GAG GAG GAA GAA GTG - 3’
APP „retour“: 5’ - CAT GAC CTG GGA CAT TCT C - 3’
25

Material und Methoden
B.1.2.3 Bakterienstämme
DH5α supE44 ∆lacU169 (j180 lacZ∆M15) hsdR17 recA1 endA1
GyrA96 thi-1 relA1, Gabe von Dr. Gerdes (Heidelberg)
B.1.2.4 Viren
5d11.2 Helfer Viren stammen ab von HSV-1 Stamm KOS durch Deletion eines
Teils des IE2 Gens, Gabe von Prof. Huttner (Heidelberg)
B.1.2.5 Bakterienkultur
LB-Agar LB-Medium mit 15 g/l Bacto-Agar autoklavieren, nach
Abkühlen auf 45°C Zugabe des Antibiotikums
LB-Medium 10 g/l Bacto-Trypton (Difco), 5 g/l Hefe Extrakt (Gibco),
10 g/l NaCl, mit 1 M NaOH auf pH 7,5 eingestellt;
(autoklaviert)
SOB-Medium 2% (w/v) Bacto-Trypton, 0,5% (w/v) Hefe-Extrakt, 0,05%
(w/v) NaCl, 2,5 mM KCL, mit NaOH auf pH 7,0 eingestellt;
(autoklaviert)
B.1.2.6 Antibiotika
Ampicillin-Stammlösung 100 mg/ml in ddH2O, steril (Lagerung bei -20°C), Sigma
B.1.2.7 Enzyme und Größenmarker
Alle Enzyme und Größenmarker wurden soweit nicht anders vermerkt von der Firma MBI
Fermentas (St. Leon Roth) bezogen.
Restriktionsenzyme:
HindIII 10 U/µl
KpnI 10 U/µl
26

Material und Methoden
Weitere Enzyme:
Taq DNA-Polymerase 2-3 U/µl, Präparation von Dr. J. Piontek (nach Pluthero,
1993)
DNA-Größenmarker:
geneRuler™1kb DNA Ladder 0,5 µg DNA/µl
B.1.3 Material-Zellbiologie
B.1.3.1 Eukaryotische Zelllinien
NT2 Gabe von Dr. Koo (Boston)
PC12 Gabe von Dr. Wagner (Boston)
Vero 2-2 Gabe von Prof. Huttner (Heidelberg)
B.1.3.2 Tiere
Mäuse Mus musculus Inzuchtstamm C57BL/6 NCrl (Charles River)
Mus musculus Inzuchtstamm C57BL/6 Jico (Harlan Winkel-
mann)
Die Tierhaltung erfolgte unter SPF-Bedingungen (Deutsch/
Französische Richtlinien).
Transgene Mauslinien C57BL/6 Jico, heterozygot transgen für: humanes PS1 wt
(HMG-Promoter), humanes PS1(M146L) (HMG-Promoter),
humanes APP695 SDL mutant (PDGF-Promoter), Gabe von
Dr. Eckert (Frankfurt a.M./Basel), hergestellt von Dr. Pradier
(Aventis Pharma, Vitry-sur-Seine, F)
B.1.3.3 Zellkulturmedien
Alle Zellkulturmedien wurden vor Gebrauch steril filtriert.
Die Lagerung erfolgte bei 4°C. Soweit nicht anders beschrieben wurden die Nährmedien zur
Verwendung bei 37°C vorgewärmt.
27

Material und Methoden
DMEM Dulbecco’s Modifiziertes Eagle Medium (Highglucose),
Gibco BRLLife Technologies
DMEM-Ser(um) DMEM mit 10% (v/v) FCS (Fötales Kälberserum,
hitzeinaktiviert), 5% (v/v) HS (Pferdeserum, hitzeinaktiviert),
2 mM Glutamin, 1% Penicillin/Streptomycin
Die Hitzeinaktivierung der Seren erfolgte für 1 h bei 56°C.
Vero2-2-Medium DMEM mit 10% FCS, 4 mM Glutamin, 1% Penicillin/
Streptomycin
Primärkultur-Medium Neurobasal Medium mit 2% (v/v) B27-supplement, 2 mM
(NB/B27) Glutamin, 1% FCS, 1% HS, 25 µM β-Merca-
ptoethanol, 100 µg/ml Primocin™ (InvivoGen)
Präparationsmedien
(Primärkultur)
Dissektion: Hanks’ balanced salts solution (HBSS) ohne CaCl2/MgCl2
(PAA Laboratories)
Zell-Dissoziation: Earle’s Minimum essential medium (1 x MEM) mit Na-Bi-
Karbonat, ohne L-Glutamin (PAA Laboratories)
B.1.4 Material-Biochemie
ELISA Kit hAmyloid β40 ELISA-, hAmyloid β42 ELISA-Microtiter-
platten Enzym-Immunoassay der Firma The Genetics Com-
pany, bestehend aus: Testplatte, synthetischem Aβ40- bzw.
Aβ42-Standard, Standard- und Probenverdünnungslösung,
Antikörper-Konjugat (100x), Enzym-Konjugat (Streptavidin-
Peroxidase-Konjugat) (100x), Enzym-Konjugat-Verdünnungs-
lösung, Waschlösung (20x), Substratlösung (TMB-Was-
serstoffperoxidgemisch), Stopplösung (1 M Schwefelsäure)
Protein-Marker Benchmark™ prestained protein ladder (Invitrogen)
28
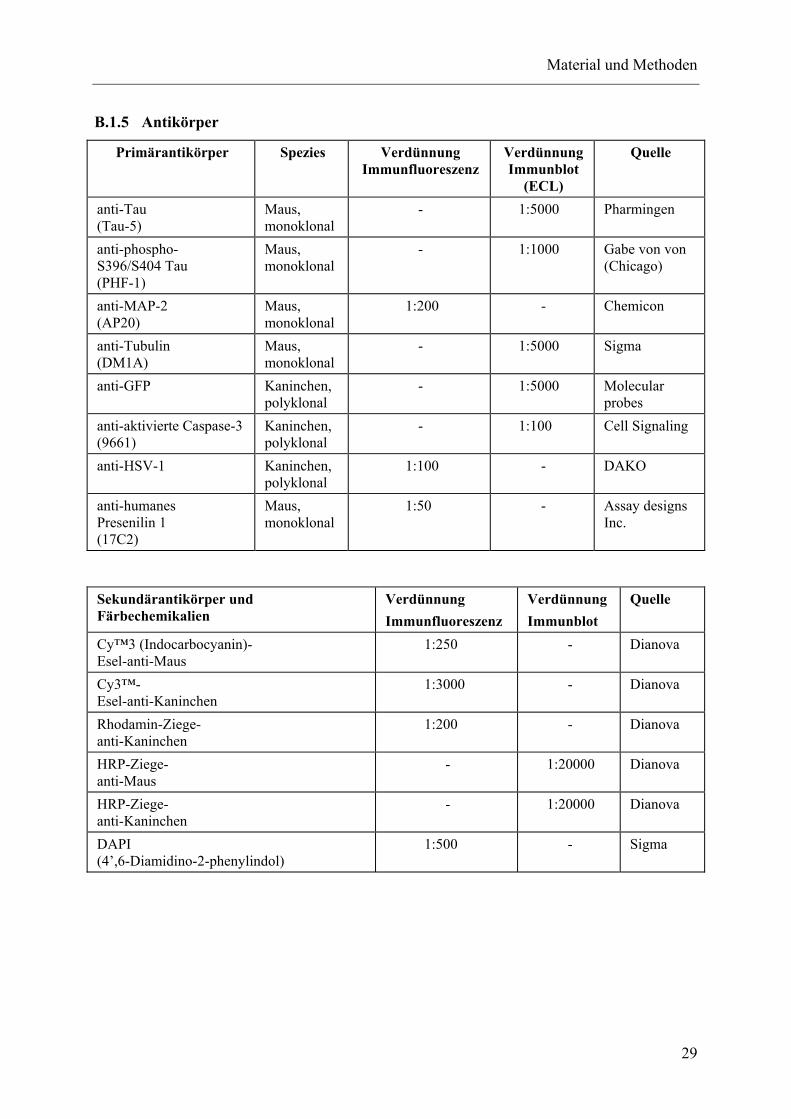
Material und Methoden
B.1.5 Antikörper
Primärantikörper Spezies Verdünnung Immunfluoreszenz
Verdünnung Immunblot
(ECL)
Quelle
anti-Tau (Tau-5)
Maus, monoklonal
- 1:5000 Pharmingen
anti-phospho- S396/S404 Tau (PHF-1)
Maus, monoklonal
- 1:1000 Gabe von von (Chicago)
anti-MAP-2 (AP20)
Maus, monoklonal
1:200 - Chemicon
anti-Tubulin (DM1A)
Maus, monoklonal
- 1:5000 Sigma
anti-GFP Kaninchen, polyklonal
- 1:5000 Molecular probes
anti-aktivierte Caspase-3(9661)
Kaninchen, polyklonal
- 1:100 Cell Signaling
anti-HSV-1 Kaninchen, polyklonal
1:100 - DAKO
anti-humanes Presenilin 1 (17C2)
Maus, monoklonal
1:50 - Assay designs Inc.
Sekundärantikörper und Färbechemikalien
Verdünnung Immunfluoreszenz
Verdünnung Immunblot
Quelle
Cy™3 (Indocarbocyanin)- Esel-anti-Maus
1:250 - Dianova
Cy3™- Esel-anti-Kaninchen
1:3000 - Dianova
Rhodamin-Ziege- anti-Kaninchen
1:200 - Dianova
HRP-Ziege- anti-Maus
- 1:20000 Dianova
HRP-Ziege- anti-Kaninchen
- 1:20000 Dianova
DAPI (4’,6-Diamidino-2-phenylindol)
1:500 - Sigma
29

Material und Methoden
B.1.6 Puffer und Stammlösungen
B.1.6.1 Molekularbiologie
Agaroselösung 1% in 1x TBE
DTT-Stammlösung 1M Dithiothreitol in ddH2O
Ethidiumbromid-Stammlösung 25 mg/ml in ddH2O
HTB 10 mM Hepes, 15 mM CaCl2, 250 mM KCl, pH mit KOH
auf 6,7 einstellen, dann 55 mM MnCl2 x 4 H2O zufügen;
steril filtriert
PCR-Nucleotid-Mix 10 mM je dNTP (Roche Diagnostics)
10 x PCR-Puffer 250 mM CHES/HCL pH 9,3, 20 mM MgCl2, 0,5 M KCl,
10 mM DTT
6x Probenpuffer 6 x Loading Dye Solution (MBI Fermentas)
RNase A-Lösung 100 mg/ml (Qiagen)
5x TBE 0,45M Tris, 0,45 M Borsäure, 1mM EDTA pH 8,0
TE-Puffer (pH 8.5) 10 mM Tris/HCl pH 8,5, 1mM EDTA
Qiagen® DNA-Extraktionskit (Präparative Plasmidpräparation)
P1 (Resuspendierungspuffer) 50 mM Tris/HCl, pH 8,0, 10 mM EDTA, 100 µg/ml RNAse
A
P2 (Lysierungspuffer) 200 mM NaOH , 1% (w/v) SDS
P3 (Neutralisierungspuffer) 3,0 M KAc, pH 5.5
QBT (Equilibrierungspuffer) 750 mM NaCl, 50 mM MOPS, pH 7,0, 15% (v/v) EtOH,
0,15% (v/v) Triton X-100
QC (Waschpuffer) 1,0 M NaCl, 50 mM MOPS, pH 7,0, 15% (v/v) EtOH
30

Material und Methoden
QF (Elutionspuffer) 1,25 M NaCl, 50 mM Tris/HCl, pH 8,5, 15% (v/v) EtOH
Qiagen® DNeasy Tissue Kit (Präparation genomischer DNA aus tierischem Gewebe)
hier: keine genauen Angaben des Herstellers über die Pufferzusammensetzungen
AE (Elutionspuffer) Kat. Nr. 19077, Qiagen
AL (Lysepuffer) Kat. Nr. 19075, Qiagen
ATL (Gewebe-Lysepuffer) Kat. Nr. 19076, Qiagen
AW1 (Waschpuffer 1) Kat. Nr. 19081, Qiagen
AW2 (Waschpuffer 2) Kat. Nr. 19072, Qiagen
Proteinase K-solution 20 mg/ml, Kat. Nr. 19131, Qiagen
B.1.6.2 Zellbiologie
Anti-Ausbleich-Medium 0,1% (w/v) p-Phenylendiamin in 90% (v/v) Glyzerin, 10%
(v/v) PBS, gepuffert auf pH 9,0 mit 0,5 M
Karbonat/Bikarbonat
Borat-Puffer 1,24 g Borsäure, 1,9 g Borax (Na-Tetraborat) ad 400 ml
ddH2O, pH 8,5
DAPI –Stammlösung 1 mg/ml 4’,6-Diamidino-2-phenylindol in DMSO
Extraktions-Lösung 0,5% (v/v) Triton X-100 in PBS
Fugene 6 Roche Diagnostics (Transfektions Reagenz)
Glyzin-Lösung 0,1 M Glyzin in PBS
Karbonat-Puffer 0,05 M Na2CO3 in ddH2O, pH 9,6
Kollagenlösung 50 µg/ml Kollagen einer Präparation aus Rattenschwänzen in
0.02 N Essigsäure, steril filtriert
31

Material und Methoden
Lamininlösung 4 µg/ml Laminin (Maus), (Chemicon) in Karbonat-Puffer,
steril filtriert
Paraformaldehyd-Lösung PBS 70°C erhitzt, Zugabe 4% Paraformaldehyd, Klärung mit
1 M NaOH, nach dem Abkühlen Zugabe von 4% Saccharose
PEM-Fixierungspuffer 0,1 M PIPES, 5 mM EGTA, 2 mM MgCl2 x 6 H2O in ddH2O,
pH 6,8
1 x PBS 8 g NaCl , 0,2 g KCL, 0,2 g KH2PO4, 1,15 g Na2HPO4, ad 1l
mit ddH2O, pH 7,4
PBS/BSA-Puffer 1% (w/v) BSA in PBS
PBS/BSA/Saccharose-Puffer 1% (w/v) BSA, 10% (w/v) Saccharose (ultrapure) in PBS
PBS/BSA/Tween-Puffer 1% (w/v) BSA in PBS, 0,1% (v/v) Tween 20, 0,02% (w/v)
Natriumazid
Poly-L-Lysin-Lösung 100 mg/ml Poly-L-Lysin in Borat-Puffer
Saccharoselösungen 10%-, 30%-, 60%- (w/v) Saccharose (ultrapure) in PBS
Trypanblau-Lösung 0,4% (v/v) Trypanblau (Sigma) in ddH2O
Trypsin/EDTA-Lösung 0,5 g Trypsin, 0,2 g EDTA ad 1l mit PBS, (PAA
Laboratories)
Vitamin A-Säure-Lösung 10 mM Vitamin A-Säure in DMSO, Lagerung lichtgeschützt
Zytostatika-Lösung 10 µM 5-Fluoro-2‘-deoxyuridin, 10 µM Uridin, 1 µM
Cytosin-β-D-arabinosid in ddH2O
B.1.6.3 Protein-Biochemie
Acrylamid/ Bisacrylamid- Rotiphorese® Gel 30 (Roth)
Stammlösung
32

Material und Methoden
Antikörper-„Strip“-Lösung 0,2% SDS (Natriumdodecylsulfat), 20 mm TCEP, in ddH2O,
frisch angesetzt
Blockierungspuffer 5% Magermilch-Pulver (Neuform, Zarrentin) in 1x TBS-
Tween
Elektrophorese-Laufpuffer 1,2 g Tris-Base, 5,76 g Glyzin, 0,4 g SDS, ad 400 ml mit
ddH2O
Entwickler ECL-Kit (Pierce), Luminol-Enhancer-Solution und Peroxi-
dase-Puffer im Verhältnis 1:1
Gelpuffer 4x Lower-Tris: 1,5 M Tris/HCl, pH 6,8, 0,4% (w/v) SDS 4x
Upper-Tris: 0,5 M Tris/HCl, pH 6,8, 0,4% (w/v) SDS
Katalysator-Stammlösungen APS (Ammoniumpersulfat): 10% (w/v) in ddH2O TEMED
(N,N,N`,N`-Tetramethyl-ethylendiamin)
Phosphataseinhibitoren 100 mM Natrium-Pyrophosphat in ddH2O
(Stocklösungen) 100 mM Natrium-Ortho-Vanadat in ddH2O
2 mM Natriumfluorid in ddH2O
5x Probenpuffer 300 mM Tris/HCl, pH 6,8, 10% SDS, 50% (v/v) Glyzerin,
6,25% (v/v) β-Mercaptoethanol, 0,005% (w/v)
Bromphenolblau
Proteaseinhibitoren 0,2 M PMSF (Phenylmethylsulfonylfluorid) in EtOH
(Stocklösungen) 2 mg/ml Leupeptin in ddH2O
1 mg/ml Pepstatin in DMSO
2x RIPA-Puffer 100 mM Tris/ HCl pH 7,5, 300 mM NaCl, 300 mM NaCl, 2
mM EDTA, 2% (v/v) NP-40, 1% (v/v) DOC, 0,2% SDS
Zugabe frisch (v/v): 1% PMSF-, 1% Leupeptin-, 2%
Pepstatin A- und je 2% Phosphataseinhibitoren-Stocklösung
10x TBS 9% (v/v) NaCl, 100 mM Tris, pH7,4
33

Material und Methoden
TBS-Tween 0,05% (v/v) Polyethylensorbitan Monolaurat (Tween 20) in
1x TBS
Transfer-Puffer 0,2 M Glyzin, 250 mM Tris, 20% (v/v) Methanol
B.1.7 Geräte und sonstige Materialien
Absaugsystem AF 204 (HLC Heaep Labor Consult, Bovenden)
Agarosegelkammer hergestellt in den Mechanikwerkstätten der Universitäten
Heidelberg und Osnabrück
Agarosegel-Dokumentations- Gel Jet Imager, UV-Kammer und optisches System (Intas
Einheit Science Imaging Instruments, GmbH, Göttingen)
Analyse- und Feinwaagen Type 1702 (Sartorius AG, Göttingen)
Blotkammer Blotkammer (Idea Scientific Company, Minneapolis, MN
USA)
CCD-Kamera Cool 1300 (VDI Vosskühler GmbH, Osnabrück)
ECL-Dunkelkammer EpiChemi II Darkroom (Intas Science Imaging Instruments
GmbH, Göttingen)
Einfriergefäße Cryo-Einfriergerät mit einer Abkühlrate von 1C°/min (Nunc
GmbH, Wiesbaden)
Eismaschine Scotsman MF22-AS (Enodis Deutschland GmbH, Herborn)
Etherkammer Glasgefäß mit Metall-Abdeckung
Filterpapier Whatman 3 MM (Whatman, Kent, UK)
Fotokopierfolie OHP-Folien A4-Copy/Laser Film (Durable Hunke & Joch-
heim GmbH & Co. KG, Iserlohn)
Fotopapier High Glossy Type, Thermal paper, Mitsubishi Electric
Europe, Ratingen)
34

Material und Methoden
Gas-Brenner Laborgaz 206 (C.G.I.D., Hattersheim) Fireboy (Tecnomara,
Zürich, CH)
Hämozytometer Neubauer-Zählkammer, 0,0025mm2, Tiefe: 0,1mm,
„Assistent“ (Glaswarenfabrik Karl Hecht, KG, Sondheim)
Heizblock Thermostat 5320 (Eppendorf-Netheler-Hinz GmbH, Ham-
burg)
Horizontalschüttler Schüttler MTS 2 (IKA Werke GmbH & Co. KG, Staufen)
Immobilion-PVDF- 0,45 µm Porengröße, Polyvinylidenfluorid-Membran:
Transfermembran Immobilion-P™ (Millipore GmbH, Eschborn)
Inkubatoren HeraCell (Kendro Laboratory Products GmbH, Langen-
selbold)
Kimwipe-Filterpapier Kimwipe Lite (bezogen über Neolab Migge, Heidelberg)
Kulturflaschen 75cm2 Flaschen (Nunclon™, Nunc GmbH, Wiesbaden)
Kulturplatten 4- und 24-Well-Platten; Schalen (∅ 4, 6, 10 und 15 cm)
(Nunclon™, Nunc GmbH Wiesbaden)
Küvetten Einmal-Küvetten, 2,5 ml, aus Polystyrol (Carl Roth GmbH &
Co. KG, Karlsruhe)
Luftverdrängungspipetten Eppendorf 10, 100, 1000 (Eppendorf AG, Hamburg) mit
adjustierbarem Volumen
Finnpipette 10, 50, 200, 1000 (Thermo Electron Oy, Vantaa,
FIN)
Gilson 0,1-2 (Gilson International BV Deutschland, Bad
Camberg)
Magnetrührer KMO 2 electronic (Janke & Kunkel GmbH & Co. KG - IKA-
Labortechnik, Staufen)
35

Material und Methoden
Mikroskope Fluoreszenzmikroskop Eclipse TE 2000-U (Nikon GmbH,
Düsseldorf)
Lichtmikroskop Leitz Labovert (Leica Microsystems AG,
Wetzlar)
Binokularmikroskop SMZ 645 (Nikon GmbH, Düsseldorf)
mit externer Lichtquelle, Highlight 3100 (Olympus Europa
GmbH, Hamburg)
Mikroskopische Deckgläser rund, ∅ 12 mm, „Assistent“ (Glaswarenfabrik Karl Hecht
KG, Sondheim)
Mikrotiterplattenreader Precision microplate reader E max (Molecular Devices,
GmbH, Ismaning)
Nagellack (klar) zur Versiegelung mikroskopischer Deckgläschen
Netzgeräte Electrophoresis Power Supply ST 305 (GIBCO, Invitrogen
GmbH, Karlsruhe)
Power Pac 1000, Power Pac 3000 (BioRad Laboratories
GmbH, München)
Objekträger 76 x 26 mm (LAB Euroline, Benzinger und Partner,
Walldorf)
Parafilm Parafilm (Pechiney Plastic Packaging, Menasha, WI, USA)
PCR-Gerät Eppendorf Mastercycler Gradient (Eppendorf AG, Hamburg)
pH-Meter Mikroprozessor-pH-Meter 761 (Knick Elektronische
Messgeräte GmbH, Berlin)
Photometer Eppendorf Bio-Photometer (Eppendorf AG, Hamburg)
Pipettierhilfen Pipetman 20, 200 (Gilson International, Bad Camberg)
Pipetboy Acu (IBS Integra Biosciences, Chur, CH)
Präparationsbesteck (Fine Science Tools GmbH, Heidelberg)
36

Material und Methoden
Rotierendes Lochscheibenrad Rollodrum (New Brunswick Scientific & Co., USA)
Sicherheits-Sterilwerkbänke HeraSafe (Kendro Laboratories Products GmbH,
Langenselbold)
semisteril: (Prettl Laflow Prozess-Technik GmbH)
Sterilfilter Steriflip, Steritop-GP (0,22µm Express™ Membrane)
(Millipore GmbH, Eschborn)
Thermomixer Eppendorf Thermomixer comfort (Eppendorf AG, Hamburg)
Thermo-Wasserbad Thermomix 1420 (B. Braun Melsungen AG, Melsungen)
Ultraschallbad Sonorex RK 100 H (Bandelin electronic GmbH & Co. KG,
Berlin)
Vakuumpumpe Vakuum Pump XF54230540 (Millipore GmbH, Eschborn)
Vortex-Geräte Vortex-Genie (Scientific Industries Inc., Bohemia, NY, USA)
Wippe hergestellt in der Mechanikwerkstatt der Universität
Osnabrück
Zellschaber Sarstedt AG & Co., Nürnbrecht
Zentrifugenröhrchen 40 ml, Polyallomer-Röhrchen (Beckmann, München)
Zentrifugen und Rotoren Biofuge Fresco, Megafuge 1.0 R, Multifuge 3-SR Heareus,
Sorvall evolution, Rotoren: SS34, SLA 1500, Sorvall
discovery 90 SE, Ausschwingrotor: Surespin 630, (Kendro
Laboratories Products GmbH, Langenselbold)
Falls nicht extra aufgelistet, wurden sonstige Glas- und Plastikwaren von folgenden Firmen
bezogen: Schott Glas (Mainz), Brand GmbH & Co. KG (Weinheim), Glaswarenfabrik Karl
Hecht KG (Sondheim), Fischer Scientific GmbH (Schwertheim), Poulton & Graf GmbH
(Wertheim), Hirschmann Laborgeräte GmbH & Co. KG (Eberstadt), Nunc GmbH & Co. KG,
Greiner Bio-One GmbH (Frickenhausen), Sarstedt AG & Co. (Nürnbrecht) und Eppendorf
AG (Hamburg). Alle für die PCR verwendeten sterilen RNase-freien Plastikwaren
37

Material und Methoden
(Reaktionsgefäße, gestopfte Pipettenspitzen) stammten von Biozyme Diagnostik GmbH
(Hess. Oldendorf).
B.1.8 Software
Agarosegel-Dokumentation Intas GDS imaging software
ECL-Dokumentation Image Pro® Plus, Version 4.5.1.27 (Media Cybernetics)
ECL-Auswertung Gel Pro™ Analyzer (Media Cybernetics Inc., Silver Spring,
USA)
Immunfluoreszenz- Lucia GF on MV 1500, Version 4.80 Build 090 (Laboratory
mikroskopische Aufnahmen Imaging s.r.o. Prag, CZ)
Immunfluoreszenz- Scion Image, Version Beta 4.0.2
mikroskopische Messungen
Analyse sämtlicher Daten Microcal Origin 7.0
Statistische Testauswertung Microcal Origin 7.0, Statview™SE+graphics,1998 (Abacus
concepts, Inc., Berkeley, CA, USA)
B.2 Methoden
B.2.1 Molekularbiologische Methoden
B.2.1.1 Präparation genomischer DNA aus embryonalem Lebergewebe bzw. adultem
Schwanzgewebe der Maus
Zur Bestimmung des Genotyps der verwendeten Mäuse mittels PCR-Analyse musste zunächst
genomische DNA aus verschiedenen Gewebetypen präpariert werden. Zur Herstellung der
unterschiedlich transgenen Primärkulturen wurde embryonales Lebergewebe verwendet. Für
die fortlaufende Zucht der Mauslinien wurde DNA aus Schwanzgewebe adulter Tiere
präpariert. Die Protokolle weichen geringfügig voneinander ab. Änderungen im Falle des
Schwanzgewebes werden in Klammern aufgeführt.
38

Material und Methoden
Die Präparation erfolgte durch die Anwendung des “DNeasy Tissue Kits” der Firma Qiagen®
und wurde nach Herstellerangaben wie folgt durchgeführt.
Protokoll:
Jedem Embryo wurden ca. 25 mg Lebergewebe entnommen (adulte Maus: 0,4-0,6 cm
Schwanzgewebe) und in flüssigem Stickstoff kurz Schock-gefroren. Nach dem Auftauen
wurden 180 µl ATL-Puffer und 20 µl Proteinase-K-Lösung zugefügt.
Es folgte eine Inkubation bei 55°C für ca. 1-2 h unter Schütteln auf einem Thermomixer
(Schwanzgewebe: über Nacht (ÜN)). Im Falle des Lebergewebes wurden die Proben
anschließend mit 4 µl RNase-Lösung behandelt und dann für 2 min bei RT inkubiert. Nach 15
s Vortexen wurde den Proben 200 µl AL-Puffer zugegeben, diese wieder kurz gemischt und
für 10 min bei 70°C inkubiert. Anschließend wurden 200 µl Ethanol (99%, p.A.) zugegeben.
(Bei Schwanzgewebe entfiel der zusätzliche Inkubations-Schritt: es wurden direkt 200 µl AL-
Puffer + 200 µl Ethanol zugeben, ohne weitere Inkubation). Die erhaltene Lösung wurde auf
die Säulen pipettiert, bei 6000 x g für 1 min abzentrifugiert und der Durchfluss verworfen.
Anschließend erfolgten Waschschritte der Säulen mit zunächst 500 µl AW1-Puffer und einer
Zentrifugation mit 6000 x g für 1 min, dann mit 500 µl AW2 und einer drei-minütigen
Zentrifugation mit 6000 x g. Die DNA wurde mit 75 µl (Schwanzgewebe: 100 µl) AE-Puffer
eluiert. Der Puffer wurde auf die Säule gegeben und für 1 min bei Raumtemperatur (RT)
inkubiert. Es folgte eine Zentrifugation mit 6000 x g. Die Elution wurde mit der bereits
eluierten Lösung wiederholt, um die DNA-Ausbeute zu erhöhen.
B.2.1.2 Polymerase-Kettenreaktion (PCR)
Mittels PCR lassen sich geringste Mengen spezifischer DNA-Sequenzen in vitro nachweisen.
Das Prinzip der PCR-Methode ist die enzymatische Vervielfältigung zwischen zwei
Oligonukleotid-Primern (sense- und antisense Primer), die gegenläufig an komplementäre
DNA-Stränge gebunden sind. Die PCR wird über 20-50 Zyklen durchgeführt, wobei jeder
Zyklus aus einem Denaturierungs-, einem „Annealing“- und einem Elongationsschritt besteht.
Im Denaturierungsschritt wird die DNA bei 94°C in Einzelstränge getrennt. Danach binden
im „Annealing“schritt die im Überschuss vorhandenen Primer an die Einzelstrang-DNA. Die
Temperatur liegt hierfür je nach Primer bei 50-70°C. Der Elongationsschritt erfolgt bei 72°C
und gewährleistet die Verlängerung der Primer durch Anknüpfen der zur DNA-Sequenz
komplementären Nukleotide. Dies erfolgt am 3’-OH Ende des jeweiligen Primers und wird
39

Material und Methoden
durch eine hitzestabile DNA-Polymerase (Taq- oder Pfu-Polymerase) katalysiert. In dieser
Arbeit wurde die PCR zur Genotypisierung der verwendeten Mäuse bzw. Mausembryonen
benutzt. Es wurden Primer gegen eine spezifische Sequenz humanen APPs bzw. humanen
Presenilin 1 verwendet. Die Annealingtemperatur der Primer betrug für beide Fälle 60°C.
Protokoll:
Nach folgendem Pipettier- und Temperatur-Zyklus-Schema wurde vorgegangen:
PCR-Mix-Pipettierschema (Volumen je Probe):
Template DNA 1 µl (~ 10 ng)
(genomische DNA)
sense-Primer 2,5 µl
(1:10 verdünnt)
antisense-Primer 2,5 µl
(1 :10 verdünnt)
dNTP-Mix (2mM) 2,2 µl
Taq-Polymerase 1,5 µl
ddH2O 12,8 µl
Temperatur-Zyklus-Schema:
1. Denaturierung bei 94°C, 5min 1 Zyklus
2. Denaturierung bei 94°C, 1min
3. „Annealing“ bei 60°C, 1min 35 Zyklen
4. Extension (Elongation) bei 72°C, 1,5 min
5. Extension (Elongation) bei 72°C, 5min 1 Zyklus
6. Aufbewahrung bei 4°C
40

Material und Methoden
B.2.1.3 Analyse des PCR-Produkts bzw. Restriktionsverdaus
Das Ergebnis einer PCR-Analyse bzw. eines Restriktionsenzymverdaus (siehe B.2.1.7) wird
durch Gelelektrophorese analysiert. Die DNA-Fragmente werden entsprechend ihrer Größe
aufgetrennt (umgekehrt proportional zum Logarithmus des Molekulargewichts). Durch den
Vergleich mit einer Referenz-DNA (DNA-Größenmarker) lässt sich die Fragmentgröße
bestimmen. Die Konzentration des Agarosegels bestimmt das Auflösungsvermögen und wird
in Abhängigkeit der zu erwartenden Fragmente gewählt. Die Visualisierung der DNA erfolgt
mit Ethidiumbromid, das in die DNA interkaliert und bei UV-Bestrahlung fluoresziert.
Protokoll:
Es wurden Agarosegele von 0.8% oder 1% gegossen. Hierzu wurde die entsprechende Menge
an Agarose in 1 x TBE eingewogen, aufgekocht und in die zuvor abgedichtete
Elektrophoresekammer mit Kamm gegossen.
Im noch flüssigen Zustand des Gels wurden 20 µl Ethidiumbromid zugefügt und gut
vermischt.
Vor Auftrag der Proben auf das Gel wurden die Proben mit 1/5 Volumen 6x Probenpuffer
versetzt. Zur Analyse von PCR-Produkten wurde jeweils der gesamte PCR-Ansatz von 25 µl
auf das Gel aufgetragen. Nach Pipettieren der Proben in die Taschen erfolgte die elektro-
phoretische Auftrennung bei 60-80 mV für ca. 45 min. Die Detektion und Dokumentation des
DNA/Ethidiumbromidkomplexes erfolgte mittels eines UV-Transilluminators und einer
Videokamera-Dokumentationseinheit. Im Falle der PCR gegen humanes APP wurde eine
Bande bei 326 bp detektiert, gegen humanes Presenilin 1 bei 427 bp. Die PCR-Proben von
Tieren mit negativem genetischen Hintergrund zeigten nur schwache Hintergrundbanden, die
durch unspezifisches Binden der PCR-Primer zustande kamen.
B.2.1.4 Herstellung von kompetenten Bakterien
Bakterien sind in der Lage, Plasmide aufzunehmen. Die Aufnahmerate lässt sich durch
vorherige Inkubation mit bestimmten Lösungen erhöhen. Man spricht dann von kompetenten
Bakterien.
Protokoll (nach Inoue et al., 1990):
100 ml SOB Medium wurden mit DH5α angeimpft (Klone wurden von einer frischen Platte
gepickt) und bei RT unter Schütteln bis zu einer OD600 von 0.45-0.5 herangezogen (13-15h).
41

Material und Methoden
Der Bakterienansatz wurde 10 min im Eisbad abgekühlt und anschließend 15 min bei 4°C mit
2700 x g abzentrifugiert. Das Pellet wurde in 16 ml HTB je 50 ml Falconröhrchen vorsichtig
resuspendiert. Nach 10 min auf Eis folgte eine Zentrifugation von 15 min bei 4°C mit 2700
rpm. Das entstandene Pellet wurde in 4 ml HTB je 50 ml Falconröhrchen vorsichtig
resuspendiert.
Danach wurden 300 µl DMSO zugefügt, die Suspension in 200 µl Aliquots in flüssigen
Stickstoff eingefroren und bei -80°C gelagert.
B.2.1.5 Transformation
Als Transformation wird das Einführen eines rekombinierten Plasmids in einen geeigneten
Wirt bezeichnet. Der wirtseigene Syntheseapparat amplifiziert die rekombinierte DNA.
Die Selektion erfolgreich transformierter Bakterien erfolgt über Antibiotika-Resistenzen der
jeweiligen Plasmide. Die Transformation und nachfolgende Maxi-Präparation der DNA
erfolgte in dieser Arbeit zur Vervielfältigung der für die Virusproduktion erforderlichen HSV-
1-Konstrukte.
Protokoll (nach Inoue et al.1990):
200µl kompetente Bakterien wurden bei RT aufgetaut, in ein 15 ml Polypropylengefäß
überführt und auf Eis gestellt. Dann wurde 1 µl Plasmid-DNA-Lösung (ca. 0,5-2 µg/µl)
zugegeben und vorsichtig gemischt. Die nachfolgende Inkubation auf Eis betrug 30 min,
wobei alle 10 min vorsichtig gemischt wurde. Danach wurde der Ansatz für 30 s bei 42°C
inkubiert und anschließend bei 37°C auf einem rotierenden Lochscheibenrad inkubiert. Es
folgte eine Zentrifugation für 10 min bei 2500 x g zur Pelletierung der Bakterienzellen. Der
Überstand wurde nun auf 100 µl abgenommen, resuspendiert und auf eine Agarplatte, die mit
dem entsprechendem Antibiotikum versetzt war, ausplattiert.
Nach der ÜN-Inkubation auf 37°C waren Bakterienkolonien gewachsen.
B.2.1.6 Präparative Plasmid-DNA-Präparation (Maxipräparation)
Durch diese Präparation wird eine große Ausbeute von reiner Plasmid-DNA ermöglicht.
Zudem ist der Reinheitsgrad ausreichend, um Zellen damit zu transfizieren. Das verwendete
eGFP-Konstrukt (hergestellt von Dr. J. Piontek) war in ausreichender Menge vorhanden, so
dass nur Maxi-Präparationen von HSV-1-eGFP-352wt-Tau und -PHP-Tau durchgeführt
wurden.
42

Material und Methoden
Die Präparation erfolgte durch die Anwendung des “Plasmid-Maxi-Kits” der Firma Qiagen®
und wurde nach Herstellerangaben wie folgt durchgeführt.
Protokoll:
200 ml LB/Antibiotika wurden angeimpft und bei 37°C ÜN auf dem Schüttler inkubiert. Zur
Zellernte betrug der erste Zentrifugationsschritt 15 min , bei 4600 x g und 4°C. Das Pellet
wurde in 10 ml P1-Puffer resuspendiert. Dann wurden 10 ml P2-Puffer zugegeben. Danach
wurde die Lösung 4-6 x invertiert und bei RT für 5 min inkubiert. Dann wurden 10 ml P3-
Puffer zugefügt, die Lösung sofort durch vorsichtiges Invertieren gemischt und 15-20 min auf
Eis inkubiert.
Es folgte ein zweiter Zentrifugationsschritt bei 20000 x g für 30 min bei 4°C. Der Plasmid-
DNA-enthaltende Überstand wurde erneut nach denselben Angaben zentrifugiert.
Der resultierende Überstand wurde auf die zuvor mit 10 ml QBT-Puffer äquilibrierte Säule
gegeben, und dann nach dem Durchlaufen 2 x mit je 30 ml QC-Puffer gewaschen.
Die anschließende Elution erfolgte mit 5 ml QF-Puffer.
Die DNA wurde mit 0,7 Volumen Isopropanol und einem weiteren Zentrifugationsschritt bei
15000 x g für 30 min bei 4°C präzipitiert.
Nach vorsichtigem Abgießen des Überstandes wurde das Pellet mit 10 ml 70% Ethanol bei
RT gewaschen (durch Zentrifugieren bei 13000 rpm für 10 min).
Der Überstand wurde unter der Sterilwerkbank abgenommen, das Pellet 5 min trocknen
gelassen und in 100-200 µl TE-Puffer aufgenommen.
B.2.1.7 Analytischer Restriktionsverdau
Zur Überprüfung der DNA des gepickten Klons wird mit dem in der Maxipräparation
aufgereinigten Plasmid ein analytischer Restriktionsenzymverdau durchgeführt. Die Restrik-
tionsenzyme für einen analytischen Restriktionsverdau werden so gewählt, dass das entste-
hende DNA-Fragmentmuster charakteristisch für das Plasmid ist. Ein analytischer Restrik-
tionsverdau wird mit ca. 200-500 ng DNA in einem Endvolumen von 10 µl durchgeführt.
Protokoll:
Ein analytischer Restriktionsverdau wurde mit 400-500 ng DNA und 3-5 U Enzym /µg DNA
angesetzt. Desweiteren wurde der vom Hersteller mitgelieferte Restriktionspuffer zugefügt
und mit ddH2O ein Endvolumen von 10 µl eingestellt. Die Inkubation betrug 1 h bei 37°C.
43

Material und Methoden
Die Analyse des Bandenmusters erfolgte durch elektrophoretische Auftrennung auf
Agarosegelen bei 60-80 mV.
Im Falle pHSV-1-eGFP-352wt-Tau bzw. -352PHP-Tau konnte durch das Schneiden mit
HindIII und KpnI ein charakteristisches Bandenmuster von drei Spaltprodukten mit ca. 5000
(4812) bp, ca. 1500 (1531) bp und ca. 300 (314) bp nachgewiesen werden.
B.2.1.8 Konzentrationsbestimmung von DNA
Nach der Aufnahme der DNA in TE-Puffer wurde die Konzentration über eine
photometrische Messung bei 260/280 nm bestimmt. Die DNA wurde dabei mit ddH2O auf ein
Volumen von 1 ml verdünnt und aus der gemessenen Extinktion die Konzentration nach
folgender Formel berechnet.
OD 260 x Verdünnung = X mg
20 ml
Das Verhältnis der OD260/280, d.h., das Verhältnis DNA/Protein sollte bei reiner DNA größer
als 1,5 sein, 1,7-1,8 ist optimal.
B.2.2 Zellbiologische Methoden
Alle Zellkultur-Arbeiten wurden unter biologischen Sicherheits-Sterilwerkbänken (Herasafe)
der Firma Kendro durchgeführt. Sämtliche Glaswaren wurden vor der Benutzung
autoklaviert. Die 37°C-Inkubatoren wurden für PC12- und NT2-N-Zellen auf 10% CO2, für
Vero 2-2-Zellen und kortikale Primärkulturen auf 5% CO2 eingestellt. Kulturmedien wurden,
soweit nicht anders vermerkt, auf 37°C vorgewärmt.
B.2.2.1 Kultur von Vero 2-2-Zellen
Vero 2-2-Zellen stammen von der Vero Nierenepithel Zelllinie aus afrikanischen Meerkatzen
ab. Diese Zelllinie wurde mit einem Plasmid transfiziert, welches das IE2 Gen exprimiert.
Das entstehende Genprodukt ist notwendig, um die vorhandene Replikationsunfähigkeit von
HSV-1 Helferviren zu kompensieren, die in dem IE2 Gen eine Deletion tragen. Diese Zellinie
wird zur Produktion und Amplifikation der HSV-1-Amplikons genutzt.
44

Material und Methoden
Protokoll:
Die Zellen wurden in Kulturschalen (∅10 cm) bis zur Konfluenz kultiviert. Zweimal pro
Woche wurde das Medium gewechselt. Zum Passagieren wurde nach Absaugen des Mediums
und Waschen mit PBS 0,5 ml Trypsin-EDTA zugefügt. Die Schale wurde zurück in den
Inkubator gestellt bis die Zellen sich abgelöst hatten. Dann wurden 4,5 ml Medium zugefügt
und 500 µl der Zellsuspension auf eine neue Kulturschalen (∅10 cm) gegeben. Anschließend
wurden 7,5 ml Medium zugefügt.
B.2.2.2 Kultur von Ratten Pheochromocytoma-Zellen (PC12)
Die adrenale Pheocytochroma-Zelllinie ist eine aus Ratten isolierte Zelllinie neuronalen
Ursprungs (Greene & Tischler, 1976). Durch Einwirkung des Nervenwachstumsfaktors NGF
zeigen PC12-Zellen verschiedene Eigenschaften sympathischer Nervenzellen, wie z. B. das
Einstellen der Proliferation und das Auswachsen langer Neuriten. In dieser Arbeit wurden
PC12-Zellen zur HSV-1-Titerbestimmung verwendet.
Protokoll:
PC12 Zellen wurden in Kollagen-beschichteten Schalen (∅ 10 cm) bis zur Konfluenz
kultiviert. Die Kollagenbeschichtung einer Platte erfolgte durch Inkubation mit Kollagen für 1
h bei 37°C.
Das Medium wurde alle 3 Tage gewechselt. Zum Passagieren wurde altes Kulturmedium
abgesaugt und 8 ml frisches DMEM-Serum zugegeben. Die auf der Kulturschale haftenden
Zellen wurden durch kräftiges Abspülen mit dem Medium abgelöst. 1 ml der Zellsuspension
wurde in eine neue Kollagen-beschichtete Zellkulturschale (∅ 10 cm) überführt und 4 ml
DMEM-Serum zugegeben.
B.2.2.3 Kultur von NT2-/NT2-N-Zellen
NT2-N-Zellen sind postmitotische, terminal differenzierte, polare Nervenzellen, die durch
Vitamin A-Säure-Behandlung aus NT2-Zellen differenzieren. Die NT2-Zelllinie wurde aus
einem humanen embryonalen Teratokarzinom isoliert. In ihrem Phänotyp entsprechen diese
Zellen zentralnervösen Vorläuferzellen (Pleasure & Lee, 1993).
45

Material und Methoden
Protokoll:
NT2-Zellen wurden bis zur Konfluenz auf Schalen (∅ 10 cm) mit 8 ml Serum-DMEM
kultiviert. Alle 2-3 Tage wurden sie 1 zu 5 passagiert. Zunächst wurde das alte Kulturmedium
abgesaugt, 1 x mit PBS gewaschen und schließlich 0,5 ml Trypsin-EDTA zugegeben.
Nachdem die Zellen sich abgelöst hatten (ca. 1 min auf 37°C) wurden 4,5 ml DMEM-Serum
zugefügt und 1 ml Zellsuspension erneut ausplattiert.
Zur Differenzierung wurden die Zellen in einer Dichte von 2 x 106 Zellen in einer 75 cm2
Kulturflasche ausplattiert und 10 ml DMEM-Serum zugefügt. Nach einem Tag wurde
Vitamin A-Säure (RA) mit einer Endkonzentration von 10 µM zugegeben. Das RA
enthaltende Medium wurde 2 x die Woche gewechselt.
5 Wochen nach der ersten RA-Zugabe wurden die Zellen das erste Mal replattiert:
Das Medium wurde abgesaugt, danach 1 x mit PBS gewaschen und 1 ml Trypsin-EDTA
zugegeben. Es folgte eine Inkubation bei 37 °C bis sich alle Zellen abgelöst hatten. Die Zellen
wurden in 19 ml DMEM-Serum aufgenommen und auf eine Kollagen beschichtete
Kulturschale (∅ 15 cm) überführt. Die Beschichtung einer Platte erfolgte durch Inkubation
mit Kollagen für 1 h bei 37°C.
Nach 2 weiteren Tagen folgte eine zweite Replattierung:
Das Medium wurde abgesaugt und die differenzierten Zellen mit 5 ml DMEM-Serum
vorsichtig von der Oberfläche der Kulturschale gespült, die Zellen im Hämozytometer gezählt
und in einer Dichte von 10000 Zellen /cm2 auf Kollagen beschichtete Deckgläschen in 4-
Well-Platten in 500 µl DMEM-Ser ausplattiert. Einen Tag später wurden 5 µl Zytostatika-
Stammlösung je Well zugesetzt. Das Medium wurde 2 x die Woche durch frisches Medium
inkl. Zytostatika ersetzt.
Nach 2 Wochen wurden die Zellen durch frisches, keine Zytostatika enthaltendes, Medium
ersetzt und die Zellen bis zur Infektion 4 Tage in DMEM-Ser inkubiert.
B.2.2.4 Einfrieren und Auftauen von Zellen
Mitotisch aktive Zellen (hier: PC12-, Vero 2-2- und NT2-Zellen) wurden bei -80°C gelagert,
wieder aufgetaut und weiter kultiviert.
46

Material und Methoden
Protokoll:
Zum Einfrieren wurden die Zellen auf Kulturschalen (∅ 15 cm) kultiviert. Nach erreichter
Konfluenz wurden die Zellen entweder durch Abspülen (PC12) oder 1 ml Trypsin (NT2, Vero
2-2) von der Platte gelöst. In 10 ml frischem Kulturmedium wurden die Zellen für 10 min bei
200 x g und 4°C abzentrifugiert. Zuvor wurde die Zellzahl bestimmt, da 1 x 106-107 je
Kryoröhrchen eingefroren werden sollten. Nach der Zentrifugation wurde das Zellsediment in
einem entsprechenden Volumen 4°C-kaltem Einfriermedium (normales Kulturmedium + 10%
(v/v) DMSO) aufgenommen. 1 ml der Zellsuspension wurde jeweils in ein Kryoröhrchen
überführt und in einem Einfriergefäß langsam bei -80°C eingefroren und nach mehreren
Tagen in flüssigen Stickstoff überführt.
Zum Auftauen der Zellen wurden die in flüssigem Stickstoff gelagerten Zellen unmittelbar
nach schnellem Auftauen im 37°C-Wasserbad in eine Kulturschale (∅ 10 cm) mit 9 ml
Kulturmedium überführt und kultiviert. Einen Tag nach dem Auftauen wurde das Medium
gewechselt und die Zellen bis zur Verwendung im Experiment mindestens 1 Woche
kultiviert.
B.2.2.5 Zellzahlbestimmung
Die Verwendung eines Hämozytometers ermöglicht die Bestimmung der genauen Anzahl
von Zellen in einer Zellsuspension. In dieser Arbeit wurde die Methode des Trypanblau-
Ausschlusses von Zellen verwendet. Sie basiert darauf, dass lebendige Zellen kein Trypan-
blau aufnehmen, während tote Zellen durch eine zerstörte Zellmembran Trypanblau inkor-
porieren. Diese erscheinen im Lichtmikroskop blau, während die lebendigen Zellen weiß
bleiben.
Protokoll:
10 µl Zellsuspension wurden mit 10 µl Trypanblaulösung vermischt und hiervon 10 µl in
einer Neubauer-Zählkammer angesaugt. Unter dem Lichtmikroskop wurden die weißen,
lebenden Zellen in einem Viertel des sichtbaren Rasters (16 Kästchen) ausgezählt. Die
erhaltene Zahl wurde mit 2 (Verdünnungsfaktor) und mit 104 multipliziert und ergab so die
Anzahl von Zellen/ml.
47

Material und Methoden
B.2.2.6 Präparation und Kultivierung von kortikalen Primärkulturen
Kortikale Primärkulturen wurden aus E 15-17 (Embryonaltag 15-17) zerebralem
Kortexgewebe der Maus hergestellt.
Protokoll (nach Terro et al., 2002):
1. Isolierung der Kortices aus Embryonen
Das schwangere Muttertier wurde durch irreversible Narkose in einer Ether-Kammer betäubt
und getötet. Nach Einstellen der spontanen Reflexe, wurden die Extremitäten der Maus fixiert
und nach Ethanol-Sterilisation des Fells drei Abdominalschnitte mit sterilem Präparations-
besteck durchgeführt. Der Uterus mit den Embryonen (bis zu 12 Individuen) wurde entfernt
und auf ein mit Ethanol getränktes Kimwipe-Tuch gelegt. Anschließend wurden die
Embryonen aus dem Gewebe freipräpariert und in eine Schale (∅ 10 cm) mit 4°C-kaltem
HBSS (ohne Ca2+- und Mg2+-Ionen) gelegt. Die Größe der Embryonen wurde dokumentiert
und variierte je Experiment zwischen 11 und 18 mm. Dies war konsistent mit der Embryo-
Größe während der letzten Schritte der Organogenese (für eine Übersicht siehe Nagy et al.,
2003). Die Dissektion der Embryonen wurde unter einer semi-sterilen Sicherheits-Werkbank
unter Benutzung eines Binokularmikroskops durchgeführt. Zunächst wurden die Embryonen
dekapitiert und der Kopf mit einer Pinzette fixiert. Mit einer zweiten scharfen Pinzette wurde
nun ein Schnitt durch die Haut und die Schädeldecke entlang der Mittellinie caudal zu rostral
durchgeführt. Die nun sichtbaren Kortex-Hälften wurden entfernt und von Hirnhautgewebe
freipräpariert. Das restliche Hirngewebe wurde verworfen, die Kortexhälften in 500 µl 4°C-
kaltes MEM überführt und bis zur Gewebedissoziation auf 4°C gelagert. Parallel dazu wurde
jedem Embryo ein Stück Lebergewebe entnommen, zur nachfolgenden Bestimmung des
Genotyps durch PCR-Analyse.
2. Dissoziation und Kultivierung
Während der gesamten nachfolgenden Vorgehensweise wurden die Proben auf 4°C gehalten
und alle Arbeitsschritte unter einer sterilen Sicherheits-Werkbank durchgeführt.
Die Kortexhälften jedes Embryos wurden mechanisch durch Triturierung mit einer Feuer-
polierten Pasteurpipette (20-30 Passagen) dissoziiert. Die Zellsuspension wurde für eine
Minute inkubiert bis sich die nicht-dissoziierten Fragmente abgesetzt hatten. Der Überstand
wurde nun für 15 min bei 700 rpm (Megafuge) und 4°C abzentrifugiert. Der resultierende
Überstand wurde anschließend verworfen und die sedimentierten Zellen in je 500 µl kaltem
48

Material und Methoden
NB/B27-Kulturmedium aufgenommen und während der Analyse des genetischen
Hintergrundes (siehe B.2.1.1,2) auf 4°C gelagert. Die Zellen wurden in einem Neubauer-
Hämozytometer gezählt und in 4- oder 24-Well-Platten mit einer Dichte von 1 x 105
Zellen/Well für die HSV-1-Infektion bzw. 3 x 105 Zellen/Well für die Aβ-ELISA-Assays
ausplattiert. Die Kultivierung erfolgte auf Laminin beschichteten Deckgläschen in 500 µl
Kulturmedium. Für die qualitative Immunblot-Analyse der Infektion (C.2.1) wurden die
Zellen mit einer Dichte von 6 x 105 Zellen auf fünf Laminin beschichtete Deckgläschen in
Kulturschalen (∅ 6 cm) ausplattiert (4 ml Medium).
Die Zellen wurden Genotyp-spezifisch ausplattiert. In einigen Experimenten wurden die
Zellen einzelner Embryonen mit gleichem Genotyp vor dem Ausplattieren vereint. Bis zur
Infektion nach 5 Tagen in vitro (DIV) (siehe B.2.2.8) wurde das Medium nicht gewechselt.
B.2.2.7 Herstellung der HSV-1-Amplikons
Herpes Simplex Virus Typ I ermöglicht einen Gentransfer in postmitotische Zellen. Zur
Herstellung der rekombinanten Virus-Vektoren wurde das “Amplikon”-System verwendet.
Dieses System umfasst ein Plasmid “Amplikon”, das den Replikationsstartpunkt (ori) und die
HSV-Verpackungssignale trägt, Helferviren, die eine Deletion in dem Immediate Early Gene
2 (IE2) tragen, und eine kompensierende Zelllinie (Vero 2-2). Diese Vero-Zellen ermöglichen
durch das IE2 Genprodukt die Replikation. Die Klonierung fremder Genstücke in das
Amplikon und die darauffolgende Verpackung in Viruspartikel ermöglicht durch anschlie-
ßende Infektion postmitotischer Zellen deren gentechnische Manipulation (Fath et al., 2000).
Protokoll:
1. Transfektion von Vero 2-2 Zellen
Die Transfektion ist eine Methode, mit der fremde DNA in Zellen eingeschleust werden kann.
Die Transfektion von Vero 2-2-Zellen mit HSV-1-Konstrukten ist der Anfangsschritt in der
HSV-1-Amplikon-Herstellung. In dieser Arbeit wurde das Transfektionsreagenz Fugene 6
verwendet, das einen Gentransfer durch Bindung der DNA an Liposomen-ähnliche Strukturen
und so ein Passieren der Zellmembran mitotisch aktiver Zellen ermöglicht.
Einen Tag vor der Transfektion wurden Vero 2-2-Zellen (ca. 5 x 104 Zellen) in Kulturschalen
(∅ 4 cm) so ausplattiert, dass sie am nächsten Tag 70-80% konfluent waren. Zusätzlich wurde
zum Testen der Transfektionseffizienz ein PLL beschichtetes Deckgläschen in jede
Kulturschale gelegt.
49

Material und Methoden
3,8 µl Fugene wurden mit 50 µl DMEM (serumfrei) vermischt und 5 min bei RT inkubiert.
Währenddessen wurden in einem anderen Eppendorfgefäß 0,8 µg DNA mit DMEM auf 10 µl
Gesamtvolumen verdünnt. Die 53,8 µl Fugene-Lösung wurden nun tropfenweise zugefügt,
vorsichtig gemischt und zur Liposomenbildung 15-20 min bei RT inkubiert.
Vor Zugabe der Transfektionslösung wurde ein Mediumwechsel durchgeführt: Das alte
Medium wurde abgesaugt und 1,5 ml frisches Kulturmedium zugegeben. Anschließend wurde
die Transfektionslösung vorsichtig in Tropfen zugefügt. Die Zellen wurden ungefähr 24 h bei
37°C inkubiert.
2. Infektion der transfizierten Vero 2-2-Zellen mit HSV-1-Helferviren
Vor der Infektion mit Helfer-Viren wurde zunächst die Transfektionseffizienz bestimmt, da
eine Transfektionsrate von mindestens 30% erforderlich ist, um eine effiziente Virus-
Herstellung zu gewährleisten. Hierzu wurden die Deckgläschen aus den Kulturschalen
genommen, die Zellen standardfixiert und die Zellkerne mit DAPI gefärbt. Anschließend
wurde fluoreszenzmikroskopisch die Anzahl GFP-positiver Zellen bestimmt. Lag eine
ausreichend hohe Transfektionseffizienz vor, wurde das Medium der Kulturschalen (∅ 4 cm)
durch 1,5 ml frisches Medium ersetzt und 2,3 x 105 Infektiöse Partikel oder Plaque forming
units (Ifps/pfu) 5d11.2 Helfervirus zugegeben. Die Zellen wurden anschließend so lange auf
37°C inkubiert bis sie sich abgerundet hatten (nach ca. 18-24 h).
3. Virus-Ernte
Durch Abschaben mit einem Zellschaber wurden die Zellen von der Kulturschale gelöst, 3 x
im Wechsel in flüssigem Stickstoff eingefroren und im 37°C Wasserbad aufgetaut, danach 2
min mit Ultraschall behandelt. Durch eine anschließende Zentrifugation bei 2300 x g
(Megafuge) und 4 °C für 5 min wurden die Zelltrümmer abzentrifugiert. Der resultierende
Überstand enthielt die Viren (p0).
4. Virus-Amplifikation
Erste Amplifikation (p1):
Einen Tag vor dem ersten Amplifikationsschritt wurden Vero 2-2-Zellen (ca. 2 x 105 Zellen)
je Schale (∅ 6 cm) in 5 ml Medium so ausplattiert, dass sie am nächsten Tag 70-80%
konfluent waren. Das alte Medium wurde abgenommen, 4 ml frisches Medium zugegeben
50

Material und Methoden
und 3 ml p0 zugesetzt. Die Zellen rundeten sich in einer Zeitspanne von 18-24 h ab und
wurden anschließend geerntet (siehe Abschnitt Viren Ernte).
Zweite Amplifikation (p2):
Einen Tag vor dem zweiten Amplifikationsschritt wurden Vero 2-2-Zellen (ca. 5 x 105 Zellen)
je Kulturschale (∅ 10 cm) in 10 ml Medium so ausplattiert, dass sie am folgenden Tag eine
Konfluenz von 70-80% erreichten (je Konstrukt wurden 2 Kulturschalen vorbereitet).
Das Medium wurde durch 6 ml frisches Medium ersetzt. In jede der beiden Schalen wurde die
Hälfte des Virenüberstands der Ernte p1 zugefügt. Nach ca. 20 h hatten sich die Zellen
abgerundet und die Viren wurden erneut geerntet.
Dritte Amplifikation (p3):
Einen Tag vor dem dritten Amplifikationsschritt wurden Vero 2-2-Zellen (ca. 5 x 105 Zellen)
je Kulturschale (∅ 10 cm) in 10 ml Medium so ausplattiert, dass sie am folgenden Tag eine
Konfluenz von 70-80% erreichten (je Konstrukt wurden 4 Kulturschalen vorbereitet).
Das alte Medium wurde durch 6 ml neues Medium ersetzt und in jede der beiden Schalen die
Hälfte des Überstandes aus der letzten Viren Ernte p2 zugegeben. Nach ca. 20 h wurden die
Viren geerntet.
Die Amplifikationsschritte können beliebig oft wiederholt werden, abhängig vom
erwünschten Titer.
Letzte Amplifikation (pX):
Bei der letzten Amplifikation war zu beachten, dass für die Virusaufreinigung (siehe Schritt
5) nur 23 ml Überstand pX auf einen Saccharose-Gradienenten geladen werden konnten.
Zudem sollte dieser Überstand von nicht mehr als vier Kulturschalen (∅ 15 cm) stammen.
Um das Volumen des Überstandes für die Aufreinigung zu verringern, wurde das Medium der
Platten entfernt und mit 800 rpm (Megafuge) für 5 min abzentrifugiert. Auf die Platten
wurden 5 ml frisches Medium gegeben und die Zellen mit einem Zellschaber abgeschabt. Die
pelletierten Zellen aus dem Zentrifugationsschritt wurden hiermit vermischt und nach
Schockgefrieren in flüssigem Stickstoff und späterem Einfrieren bei -80°C für maximal eine
Woche gelagert.
51

Material und Methoden
5. Virusaufreinigung:
In 40 ml Beckmann Polyallomer-Zentrifugenröhrchen wurde ein Saccharose-Gradient
vorbereitet. Dazu wurden folgende Lösungen übereinander geschichtet:
7 ml 60% (w/v) Saccharose, 6 ml 30% (w/v) Saccharose, 3 ml (w/v) 10% Saccharose. Die
fertigen Gradienten wurden auf Eis gelagert.
Der Überstand aus der letzten Virenernte (pX) wurde vorsichtig auf den Saccharosegradienten
gegeben. Danach folgte eine Zentrifugation für 1 h mit 125000 x g bei 4°C, wobei sich die
Viren zwischen der 30%igen und 60%igen Saccharoseschicht ansammelten. Die oberen
Saccharoseschichten wurden verworfen, die Viren enthaltende Trennschicht direkt oberhalb
der 60% Saccharoseschicht (ca. 4 ml) abgenommen und mit 35 ml PBS/BSA-Puffer verdünnt.
Es wurde erneut für 1 h mit 125000 x g und bei 4°C zentrifugiert.
Anschließend wurde der Überstand verworfen und das Pellet in PBS/BSA/Saccharose-Puffer
aufgenommen und in 10 µl Aliquots in flüssigem Stickstoff schockgefroren. Die Lagerung
erfolgte bei -80°C.
6. Titerbestimmung
Einen Tag vor der Titerbestimmung wurden 1,5 x 103 PC12-Zellen pro Well in 4-Well-
Platten mit PLL beschichteten Deckgläsern ausplattiert. Vor der Infektion wurde das Medium
abgenommen und 300 µl frisches DMEM-Serum je Well zugefügt. Zur Infektion wurden
jeweils 10 µl unterschiedlicher Verdünnungen in PBS/BSA-Puffer (1:10, 1:100, 1:1000) der
Virus-Stammlösung zugesetzt. 4 h nach der Infektion erfolgte erneut ein Mediumwechsel mit
500 µl frischem Kulturmedium. 24 h nach dem Zeitpunkt der Infektion wurden die Zellen
standardfixiert, eine Immunfärbung gegen ein Oberflächenprotein von HSV-1 und eine
Kernfärbung mit DAPI durchgeführt. Die Immunfärbung gibt Aufschluss über die Menge der
amplifizierten Helferviren in der erhaltenen Viruslösung. Alle infizierten GFP-positiven
Zellen sowie alle Helfervirus-infizierten Zellen wurden am Fluoreszenzmikroskop ausgezählt
und auf die Größe des Wells hochgerechnet. Das Verhältnis von HSV-1-GFP-Viren zu
Helferviren bewegte sich im Rahmen von 1:2 bis 1:20. Um den Einfluss einer Helfervirus-
Infektion auf die Ergebnisse zu verhindern, wurden je Experiment für alle verwendeten
Konstrukte Viruslösungen mit ähnlichem Verhältnis verwendet .
52

Material und Methoden
B.2.2.8 Infektion von NT2-N-Zellen und kortikalen Primärkulturen
Die HSV-1-Infektion wurde für NT2-N-Zellen und Primärkulturen gleichermaßen
durchgeführt. Die einzigen Unterschiede bestanden in dem eingesetzten Titer und in der
Kulturdauer vor der Infektion (NT2-N-Zellen: 4 Tage nach Zytostatika-Behandlung,
Primärkulturen: 5 DIV).
Protokoll:
Vor der HSV-1-Infektion wurde zunächst ein Mediumwechsel mit jeweils 300 µl frischem
Kulturmedium (NT2-N: DMEM-Ser, Primärkulturen: NB/B27) pro Well durchgeführt. Die
Virusstammlösungen wurden je nach eingesetztem Titer mit PBS/BSA-Puffer auf ein
Gesamtvolumen von 10 µl verdünnt und zugegeben. Die infizierten Zellen wurden zurück in
den Inkubator auf 37°C gestellt. 4 h nach der Infektion wurde das Medium abgesaugt und 500
µl frisches Kulturmedium pro Well zugegeben. Die Zellen wurden bis zur Analyse auf 37°C
und 10% bzw. 5% CO2 inkubiert.
NT2-N-Zellen wurden für die immunfluoreszenzmikroskopischen Analysen mit 1 x 104
Ifps/Well infiziert, kortikale Primärkulturen mit 5 x 103 Ifps/Well. Die Immunblotanalysen
von HSV-1-infizierten Primärkulturen wurden nach einer Infektion mit 2 x 104 Ifps/Well
(zum Untersuchen der Tau-Phosphorylierung) und 1 x 105 Ifps/Well (zur Untersuchung der
Caspase-3-Aktivität) durchgeführt.
Für die qualitative Immunblotanalyse der Infektion kortikaler Primärkulturen (siehe C.2.1)
wurde die Infektion in Kulturschalen (∅ 6 cm) durchgeführt. Vor der Infektion wurde das alte
Medium durch 3 ml frisches Medium ersetzt. Es folgte eine Infektion mit jeweils 137 µl
Virusüberstand (p3). Nach 4 h wurde das Medium abgesaugt und jeweils 4 ml frisches
Kulturmedium zugegeben.
B. 2.2.9 Immunfluoreszenzmikroskopie
Die Immunfluoreszenzmikroskopie dient als sensitive Methode zum indirekten Nachweis
subzellulärer Bestandteile und der Analyse ihrer Lokalisation. Hierfür müssen Zellen auf
mikroskopischen Deckgläschen kultiviert werden. Zur besseren Haftung der Zellen müssen
die Deckgläschen zuvor mit bestimmten Proteinen oder Peptiden z.B. Poly-L-Lysin oder Kol-
lagen beschichtet werden. Im ersten Schritt werden die Zellen fixiert und permeabilisiert. Es
folgt die Inkubation mit einem nicht-markierten Primärantikörper, dessen Antigen ein Epitop
des gewünschten Proteins ist. Der Primärantikörper kann monoklonal oder polyklonal sein.
53

Material und Methoden
Antigene des Sekundärantikörpers sind speziesspezifische Regionen des Primärantikörpers.
Der Sekundärantikörper ist mit einem Fluorochrom, z.B. Rhodamin kovalent verbunden.
Das Präparat kann mit der entsprechenden Filterkombination am Fluoreszenz-Mikroskop
ausgewertet werden.
B.2.2.9.1 Beschichtung von mikroskopischen Deckgläschen
1. Poly-L-Lysin (PLL) Beschichtung
Protokoll:
Die mikroskopischen Deckgläschen wurden drei mal durch die Flamme eines Gasbrenners
geführt und anschließend in 99% Ethanol für ca. 2 min inkubiert. Nach 1 x Waschen mit
ddH2O wurde Poly-L-Lysin zugegeben, so dass die Deckgläschen gut bedeckt waren (z.B. in
einer 24-Well-Platte: 350 µl). Es folgte eine Inkubation für 6 h bei 37 °C. Nach der
Inkubation wurden die Deckgläschen 2 x für 1 h mit je 500 µl (24-Well-Platte) ddH2O
gewaschen.
2. Kollagen Beschichtung
Protokoll:
Auf Poly-L-Lysin beschichtete Deckgläschen wurden 350 µl Kollagenlösung/Well gegeben
und für 1 h bei 37°C inkubiert. Danach wurde 1 x mit PBS gewaschen.
3. Laminin Beschichtung
Protokoll:
Auf Poly-L-Lysin beschichtete Deckgläschen wurde 350 µl Lamininlösung/Well gegeben und
für mindestens 5 h oder ÜN bei 37°C inkubiert. Danach wurde 1 x mit PBS gewaschen.
B.2.2.9.2 Fixierung und Färbung von Zellen
Es gibt verschiedene Möglichkeiten Zellen zu fixieren. Bei der Standardfixierung werden
Zellen mit Paraformaldehyd fixiert. Danach wird die Plasmamembran mit Detergenzien
permeabilisiert, sodass der Antikörper die Membran gut durchdringen kann und die
zellulären Proteine erhalten bleiben.
54

Material und Methoden
Bei der Ethanol-Fixierung werden die Zellen gleichzeitig extrahiert und fixiert.
1. Standardfixierung
Protokoll:
Die Deckgläschen wurden aus der Kulturschale entnommen und 1 x mit PBS gewaschen.
Danach wurden sie 20 min mit 50 µl Standardfixierungslösung inkubiert und anschließend 1
x mit PBS gewaschen. Zum Absättigen der unspezifischen Bindestellen wurde nun 20 min
mit 50 µl Glyzin-Lösung inkubiert. Nach 1 x Waschen mit PBS wurden die Zellen mit 0.5%
Triton X-100/PBS (5 min) permeabilisiert und dann nochmals mit PBS gewaschen.
2. Ethanol/PEM-Fixierung
Protokoll:
Die Deckgläschen wurden aus der Kulturschale entnommen und 1 x mit PBS gewaschen.
Dann wurden 50 µl PEM-Puffer auf die Zellen gegeben und 10 min bei RT inkubiert. An-
schließend wurden die Deckgläschen 2 x mit PBS gewaschen und dann mit 50 µl -20°C
kaltem Ethanol (99%, p.A.) für 5-10 min bei -20°C inkubiert. Es folgten 3 Waschschritte mit
PBS.
B.2.2.9.3 Immunfärbung und Einbettung
Protokoll:
Nach der Fixierung wurden die Zellen für 5 x 2 min mit PBS/BSA/Tween inkubiert. Danach
wurde die Primärantikörper-Lösung zugegeben und 1 h in einer feuchten Kammer bei RT
inkubiert. Dann wurde erneut 5 x 2 min mit PBS/BSA/Tween gewaschen und 50 µl der
Sekundärantikörperlösung (mit DAPI versetzt) zugefügt (30 min Inkubation in feuchter
Kammer). Anschließend wurde 5 x 2 min mit PBS/BSA/Tween gewaschen. 5 µl Anti-
Ausbleichmedium wurden auf einen Objektträger gegeben und das Deckgläschen mit der
Zellseite nach unten aufgelegt. Mit farblosem Nagellack wurde das Deckgläschen versiegelt.
B.2.2.9.4 Zellkernfärbung
Alternativ zur Immunfärbung kann auch nur eine Zellkernfärbung mit dem chemischen
Reagenz DAPI erfolgen.
55

Material und Methoden
Protokoll:
Die fixierten Zellen wurden direkt mit 50 µl DAPI-Lösung (verdünnt in PBS) für 5 min bei
RT inkubiert. Es folgten 5 Waschschritte mit PBS, das Einbetten wurde wie in Abschnitt
B.2.2.9.3 durchgeführt.
B.2.3 Protein-Biochemische Methoden
B.2.3.1 Präparation von Zelllysat aus kortikalen Primärkulturen
Protokoll:
Die Zellen wurden auf Laminin beschichteten Deckgläschen in 4-Well-Platten oder Kultur-
schalen (∅ 6 cm) kultiviert (siehe B.2.2.6). Die Kulturgefäße wurden zur Lysat-Herstellung
auf Eis transferiert und das Kulturmedium vorsichtig mit einer 1000 µl-Pipette abgenommen.
In einem weiteren Schritt wurden die Kulturgefäße im Eis für ca. 2 min schräggestellt, um das
restliche Medium aufzufangen und abzuziehen. Die Schalen wurden nun wieder waagerecht
auf Eis inkubiert und 25 µl RIPA-Puffer in die Mitte eines Wells bzw. einer Kulturschale (∅
6 cm) gegeben. Mit einer Pipettenspitze bzw. Zellschaber wurden nun die Zellen von dem
Deckgläschen abgekratzt und die Zelllösung in ein Eppendorf-Reaktionsgefäß überführt. Im
Anschluss wurden die Wells bzw. Kulturschalen mit je 10 µl RIPA-Puffer abgespült und dies
der jeweiligen Probe zugefügt. Es folgte eine 30minütige Inkubation auf 4°C unter Schütteln.
Anschließend wurden die Proben bei 10000 x g für 10 min bei 4°C zentrifugiert, um
Zelltrümmer zu sedimentieren. Der Überstand wurde entweder sofort für die gelelektro-
phoretische Analyse weiter verwendet oder in flüssigem Stickstoff schockgefroren und dann
bei -80°C gelagert. Je nach Experiment wurden, um die Lysatmenge zu erhöhen, zum Teil
Lysate aus 2 Wells gleicher Bedingung vereint.
B.2.3.2 Natriumdodecylsulfat-Polyacrylamidgel-Elektrophorese (SDS-PAGE)
Die Natriumdodecylsulfat-Polyacrylamidgel-Elektrophorese (SDS-PAGE, nach Laemmli) ist
ein hochauflösendes Trennverfahren zur Molekulargewichtsbestimmung von Proteinen. Die
Trennmatrix ist ein Polyacrylamidgel, das durch Kopolymerisation von Acrylamid und N,N‘-
Methylenbisacrylamid in wässriger Lösung gebildet wird. Die Polymerisationsreaktion wird
durch Ammoniumpersulfat, einem SO4 Radikal, als Initiator, gestartet. Als Mediator der
Polymerisation fungiert Tetramethylenethyldiamin (TEMED). Die Auftrennungsfähigkeit des
56
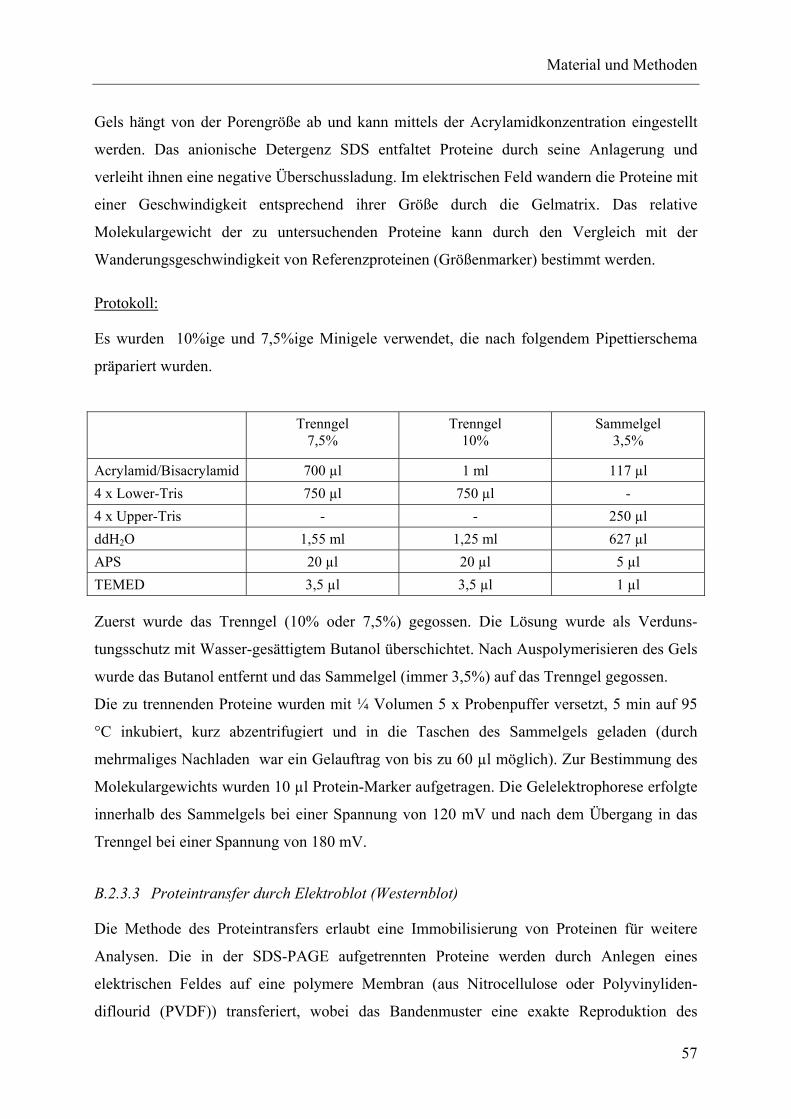
Material und Methoden
Gels hängt von der Porengröße ab und kann mittels der Acrylamidkonzentration eingestellt
werden. Das anionische Detergenz SDS entfaltet Proteine durch seine Anlagerung und
verleiht ihnen eine negative Überschussladung. Im elektrischen Feld wandern die Proteine mit
einer Geschwindigkeit entsprechend ihrer Größe durch die Gelmatrix. Das relative
Molekulargewicht der zu untersuchenden Proteine kann durch den Vergleich mit der
Wanderungsgeschwindigkeit von Referenzproteinen (Größenmarker) bestimmt werden.
Protokoll:
Es wurden 10%ige und 7,5%ige Minigele verwendet, die nach folgendem Pipettierschema
präpariert wurden.
Trenngel 7,5%
Trenngel 10%
Sammelgel 3,5%
Acrylamid/Bisacrylamid 700 µl 1 ml 117 µl 4 x Lower-Tris 750 µl 750 µl - 4 x Upper-Tris - - 250 µl ddH2O 1,55 ml 1,25 ml 627 µl APS 20 µl 20 µl 5 µl TEMED 3,5 µl 3,5 µl 1 µl
Zuerst wurde das Trenngel (10% oder 7,5%) gegossen. Die Lösung wurde als Verduns-
tungsschutz mit Wasser-gesättigtem Butanol überschichtet. Nach Auspolymerisieren des Gels
wurde das Butanol entfernt und das Sammelgel (immer 3,5%) auf das Trenngel gegossen.
Die zu trennenden Proteine wurden mit ¼ Volumen 5 x Probenpuffer versetzt, 5 min auf 95
°C inkubiert, kurz abzentrifugiert und in die Taschen des Sammelgels geladen (durch
mehrmaliges Nachladen war ein Gelauftrag von bis zu 60 µl möglich). Zur Bestimmung des
Molekulargewichts wurden 10 µl Protein-Marker aufgetragen. Die Gelelektrophorese erfolgte
innerhalb des Sammelgels bei einer Spannung von 120 mV und nach dem Übergang in das
Trenngel bei einer Spannung von 180 mV.
B.2.3.3 Proteintransfer durch Elektroblot (Westernblot)
Die Methode des Proteintransfers erlaubt eine Immobilisierung von Proteinen für weitere
Analysen. Die in der SDS-PAGE aufgetrennten Proteine werden durch Anlegen eines
elektrischen Feldes auf eine polymere Membran (aus Nitrocellulose oder Polyvinyliden-
diflourid (PVDF)) transferiert, wobei das Bandenmuster eine exakte Reproduktion des
57

Material und Methoden
Originalgels darstellt. Die in dieser Arbeit verwendeten PVDF-Membranen sind hydrophobe
Mikroporen-Membranen mit einer hohen Protein-Bindungskapazität und einer hohen
mechanischen Stabilität.
Protokoll:
Der Westernblot wurde unmittelbar im Anschluss an die SDS-PAGE durchgeführt. Dazu
wurden 2 Schwammtücher und 2 zurechtgeschnittene 3 MM Whatman-Papiere mit Transfer-
Puffer befeuchtet. Die PVDF-Membran wurde kurz in Methanol aktiviert und anschließend in
Transfer-Puffer getaucht. Der Blot-Rahmen des Tanks wurde in folgender Reihenfolge
luftblasenfrei belegt: Schwammtuch, 3 MM Papier, Gel, 3 MM Papier, Schwammtuch. Das
Gel wurde in Richtung Kathode und die Membran in Richtung Anode orientiert und in den
mit Transfer-Puffer gefüllten Tank eingebaut. Der Transfer erfolgte bei 100 mA ÜN.
B.2.3.4 Immundetektion
Die ECL-Immundetektion (verstärkte Chemolumineszenz) ist eine nicht-radioaktive, sensitive
Methode zum Nachweis geringer Mengen Protein, die auf eine Membran transferiert wurden.
Bei der Chemolumineszenz werden Substanzen durch eine chemische Reaktion in einen ange-
regten Energiezustand versetzt. Beim Rückfallen in den Energiegrundzustand wird diese
Energie in Form von Lichtquanten wieder frei und kann mit Hilfe einer ECL-Kamera, die sich
in einer ECL-Dunkelkammer befindet, detektiert und über einen Computer visualisiert
werden.
Bei der Immundetektion mittels ECL wird zunächst ein primärer Antikörper verwendet, der
das auf der Membran immobilisierte Protein spezifisch bindet. Danach wird ein Meerrettich-
Peroxidase (HRP)- gekoppelter Sekundärantikörper eingesetzt. Antigene des Sekundäranti-
körpers sind speziesspezifische Regionen des Primärantikörpers. Die Meerretich-Peroxidase
katalysiert die Oxidation von Luminol, einem zyklischen Diacylglyzerid, was zu einer
Lichtemission bei 428 nm führt. Die Reaktion wird bei der Chemolumineszenz durch Phenol
ca. 1000fach verstärkt.
Protokoll:
Nach dem ÜN-Transfer wurde die Membran mit TBS-Tween kurz abgespült und für 2 h mit
Blockierungspuffer auf dem Schüttler inkubiert. Danach wurde die Membran 3 x 10 min mit
TBS-Tween gewaschen und anschließend mit der Primärantikörper-Lösung für 1,5 h bei RT
58

Material und Methoden
oder ÜN bei 4°C auf der Wippe inkubiert. Monoklonale Antikörper und ihre Sekundäranti-
körper wurden in TBS-Tween, polyklonale Antikörper und die korrespondierenden Sekundär-
antikörper in Blockierungspuffer verdünnt. Nach Inkubation mit dem Primärantikörper wurde
die Membran erneut 3 x 10 min mit TBS-Tween gewaschen und der HRP-gekoppelte-
Sekundärantikörper zugegeben und 45 min auf der Wippe inkubiert. Anschließend wurde
wieder 3 x 10 min mit TBS-Tween gewaschen. Nun folgte eine 5minütige Inkubation mit
dem ECL-Entwickler (0,125 ml/cm2). Im Anschluss wurde die Membran zwischen zwei
Klarsicht-Fotokopierfolien in die ECL-Dunkelkammer gelegt. In dieser wurden mit der
Chemocam und unter Verwendung der Software „Image Pro Plus“ Aufnahmen der
chemolumineszierenden Proteinbanden und des Markers gemacht.
B.2.3.5 „Strippen“ einer PVDF- Membran
Um verschiedene Antikörper auf derselben Membran zu testen, kann nach der ECL-Reaktion
der zuvor benutzte Antikörper von den auf der Membran immobilisierten Proteinen wieder
abgelöst werden.
Protokoll:
Getrocknete Membranen (auf 4°C gelagert) wurden zuerst mit Methanol befeuchtet und kurz
mit TBS-Tween abgespült. Noch feuchte Membranen konnten direkt 3 x 10 min mit ddH2O
gewaschen und mit der Antikörper-„Strip“-Lösung für 1 h bei 37°C auf der Wippe inkubiert
werden. Anschließend wurde die Membran nochmals 3 x 10 min mit ddH2O gewaschen und
eine neue Immundetektion durchgeführt.
B.2.3.6 Densitometrische Quantifizierung der Immunblotanalyse
Die Auswertung der Immundetektion erfolgte mit dem Programm „Gel-Pro-Analyzer Version
4.0“ von Cybernetics. Hierbei wurde die Signal-Intensität der chemolumineszierenden
Proteinbanden unter Substraktion des Hintergrundwertes gemessen. Anschließend wurden die
Proteinbanden quantitativ miteinander verglichen.
B.2.3.7 ELISA (Enzyme-Linked-Immunosorbant-Assay)
Die ELISA-Methode dient der quantitativen Bestimmung von Proteinen und Peptidfrag-
menten in einer Lösung. Hierbei wird das Prinzip eines Festphasen-Enzym-Immunoassays
angewandt. Das zu testende Antigen (hier Aβ40 oder Aβ42) wird durch spezifische mono-
59

Material und Methoden
klonale Antikörper erfasst, die selektiv an zwei unterschiedlichen Epitopen binden und so
einen „Sandwich-Komplex“ bilden. An die Polysteroloberfläche einer Mikrotiterplatte ist
bereits ein sogenannter „Fangantikörper“ gekoppelt, der selektiv das Antigen (hier das N-
terminale Ende Aβs) erkennt. Während der Testdurchführung wird ein zweiter gegen das
Antigen gerichteter Antikörper, gekoppelt mit einem Biotinkonjugat, („Detektionsantikörper“
gegen das C-terminale Ende Aβs ) mit den Proben und den Standards inkubiert und bildet
einen Antikörper-Antigen-Antikörper-Komplex. Dieser Komplex wird in einem nächsten
Schritt indirekt über eine Biotin-Streptavidin-Bindung mit einem Enzym gekoppelt. Das
Enzym katalysiert in einer Folgereaktion die Umwandlung eines Substrats (Chromogen) zu
einem farbigen Produkt, dessen Farbintensität photometrisch bestimmt wird. Die Extinktion
korreliert direkt mit der Menge des zu messenden Proteins oder Peptids. Durch den Vergleich
mit einer Standardgerade mittels linearer Regression (hier aus synthetischem Aβ) wird eine
quantitative Auswertung der Proben möglich.
Die ELISA-Methode erfolgte durch die Anwendung des “hAmyloid β40 ELISA”- und des
“hAmyloid β42 ELISA”-Kits der Firma The Genetics Company, und wurde nach
Herstellerangaben wie folgt durchgeführt.
Protokoll:
APPSDL-transgene und nicht-transgene Primärkulturen (3 x 105 Zellen ausplattiert, um im
messbaren Bereich des ELISA-Kits zu liegen) wurden nach 5 DIV mit 10µl PBS/BSA-Puffer
behandelt. Nach 4 h wurde ein Mediumwechsel mit 500 µl frischem Kulturmedium
durchgeführt und 2 bzw. 3 Tage später das Medium abgenommen und bei -80°C eingefroren.
Zur Bestimmung des Aβ-Gehalts im Zellüberstand wurden die Proben am Tag der
Durchführung der ELISA auf Eis aufgetaut. Es wurden Doppelbestimmungen der Standards
und der Proben durchgeführt. Es wurden insgesamt 100 µl unverdünnte Probe eingesetzt.
Zunächst wurden in jede Kavität der bereitgestellten Testplatte 50 µl der Antikörper-
Konjugatverdünnung pro Kavität pipettiert. Anschließend wurden 50 µl Probe
beziehungsweise Standard je Kavität pipettiert. Diese Schritte erfolgten auf Eis. Nachdem die
Testplatte mit einer Abdeckfolie versehen war, wurde für ca. 5 min auf dem Schüttler
gemischt und ÜN bei 4°C inkubiert. Am nächsten Tag wurde die Testplatte mit 5 x 200 µl
Waschlösung pro Kavität gewaschen und vor jedem Waschschritt wurden die
Flüssigkeitsreste durch Ausklopfen der Platte auf einer saugfähigen Unterlage entfernt. Im
nächsten Schritt wurden 100 µl Enzym-Konjugatverdünnung in jede Kavität pipettiert, die
60

Material und Methoden
Testplatte mit Abdeckfolie versehen und für 30 min bei RT auf einem Schüttler inkubiert.
Danach wurde erneut 5 x mit 200 µl Waschlösung gewaschen. Anschließend wurde pro
Kavität 100 µl Substratlösung zugegeben und die Platte 15-30 min bei RT inkubiert. Zum
Stoppen der Enzymreaktion wurden 50 µl Stop Solution pro Kavität in der gleichen
Reihenfolge wie im Testschritt zuvor zugefügt. Bei einer Wellenlänge von 450 nm wurde die
Absorption im Mikrotiterplatten-Photometer gemessen.
61

Ergebnisse
C Ergebnisse
In früheren Arbeiten wurde eine erhöhte Neurotoxizität von exogen exprimiertem PHP-Tau
im Vergleich zu wt-Tau nachgewiesen (Fath et al., 2002). Es handelte sich hierbei um mit
einem Flag-Epitop markierte Konstrukte, die HSV-1-vermittelt in humanen Modellneuronen
(NT2-N-Zellen) exprimiert wurden. Die Analyse des neurodegenerativen Effekts erfolgte u.a.
durch Zählen fragmentierter und kondensierter Zellkerne infizierter Neurone, welches als
Merkmal apoptotischer Vorgänge in Zellen beschrieben ist.
In der vorliegenden Arbeit sollte zunächst versucht werden, die durch PHP-Tau ausgelöste
Neurodegeneration in NT2-N-Zellen durch Expression von HSV-1-eGFP-Tau-Fusions-
konstrukten zu bestätigen. GFP-markierte Konstrukte bieten den Vorteil, dass es möglich
wird, Vorgänge in Zellen im Lebendzustand zu analysieren, desweiteren kann auf Immun-
fluoreszenzanfärbungen fixierter Zellen verzichtet werden. Die Echtzeitanalyse der Tau-
abhängigen Degeneration in einzelnen Neuronen wurde in einer Parallelstudie von Welzel
(2005) durchgeführt.
Das Ziel dieser Arbeit bestand in der Analyse des neurodegenerativen Effekts exprimierten
eGFP-wt und -PHP fötalen Taus in kortikalen Primärkulturen. Als Kontrollkonstrukt wurde
eGFP verwendet. Die verwendeten Primärkulturen aus dem embryonalen Kortex der Maus
stammten von Tieren mit unterschiedlichem transgenen Hintergrund. Es handelte sich hierbei
um Mäuse, die entweder humanes mutiertes APP oder Presenilin 1 (wt bzw. M146L)
heterozygot stabil überexprimierten. Hierdurch sollte eine eventuelle funktionelle Interaktion
dieser beiden AD-relevanten Proteine auf die durch PHP-Tau ausgelöste Neurodegeneration
untersucht werden.
Die dabei verwendeten HSV-1-eGFP-Tau-Konstrukte wurden bereits in einer vorangehenden
Arbeit hergestellt. Zudem wurde die Expression auf Immunblot- bzw. fluoreszenzmikro-
skopischer Ebene im NT2-/NT2-N-System qualitativ und quantitativ getestet (Leschik, 2000).
C.1 EGFP-PHP-Tau ist neurotoxisch in NT2-N-Zellen
NT2-N-Zellen wurden mit den unterschiedlichen Virusstammlösungen HSV1-eGFP-352wt-
Tau, eGFP-352PHP-Tau sowie -eGFP als Kontrolle infiziert und standardfixiert. Zur Analyse
der Zellkernmorphologie wurde mit dem chemischen Reagenz DAPI eine Kernfärbung
durchgeführt. Die Zellen wurden 1, 2 und 4 Tage nach der Infektion fixiert, um einen
möglichen Einfluss der Expressionsdauer auf die Neurodegeneration zu ermitteln. Die
62

Ergebnisse
Auswertung erfolgte fluoreszenzmikroskopisch, wobei infizierte Neurone auf Fragmentierung
ihres Zellkernes untersucht wurden. Abb. C.1 zeigt exemplarisch standardfixierte HSV-1-
infizierte NT2-N-Zellen der drei verwendeten Konstrukte. Die subzelluläre Verteilung des
exogenen Proteins ist im Fall von eGFP-wt-Tau und eGFP-PHP-Tau auf das Zytosol
beschränkt (Abb. C.1A-D). EGFP hingegen ist in einigen Zellen auch im Kernbereich
sichtbar (Abb. C.1E,F). Da es ein geringes Molekulargewicht von 27 kD besitzt, kann es
durch die Poren der Kernmembran diffundieren, die ein ungehindertes Passieren von
Proteinen bis zu einer Größe von 44 kD erlauben (zur Übersicht siehe: Alberts et al., 2002).
Die Kernfärbung mit DAPI ermöglicht ein genaues Untersuchen der Integrität des Zellkernes.
Dabei gilt eine vorliegende Fragmentierung als Zeichen einer bereits vorangeschrittenen
zellulären Degeneration. In Abb. C.1C,D ist eine solche Zellkern-Fragmentierung eines
eGFP-PHP exprimierenden Neurons erkennbar. Im Gegensatz dazu sind die Zellkerne der
mit wt-Tau (Abb. C.1A,B) und eGFP (Abb. C.1E,F) infizierten NT2-N-Zellen intakt.
Die quantitative Analyse der fragmentierten Zellkerne erfolgte durch Auszählen und wurde
relativ zur Gesamtzahl aller infizierten Neurone berechnet. Abb. C.2 zeigt den prozentualen
Anteil fragmentierter Zellkerne nach Infektion mit den verschiedenen e-GFP-Konstrukten zu
den gewählten Zeitpunkten.
63
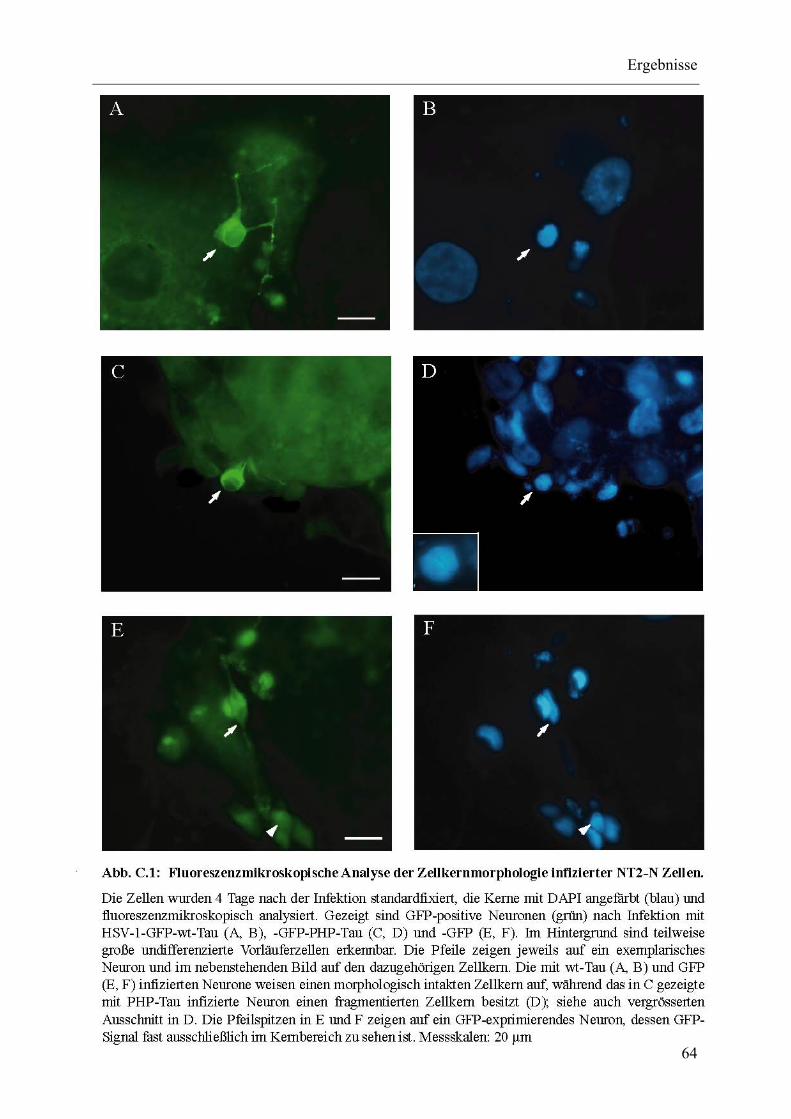
Ergebnisse
Abb. C. 1:
64
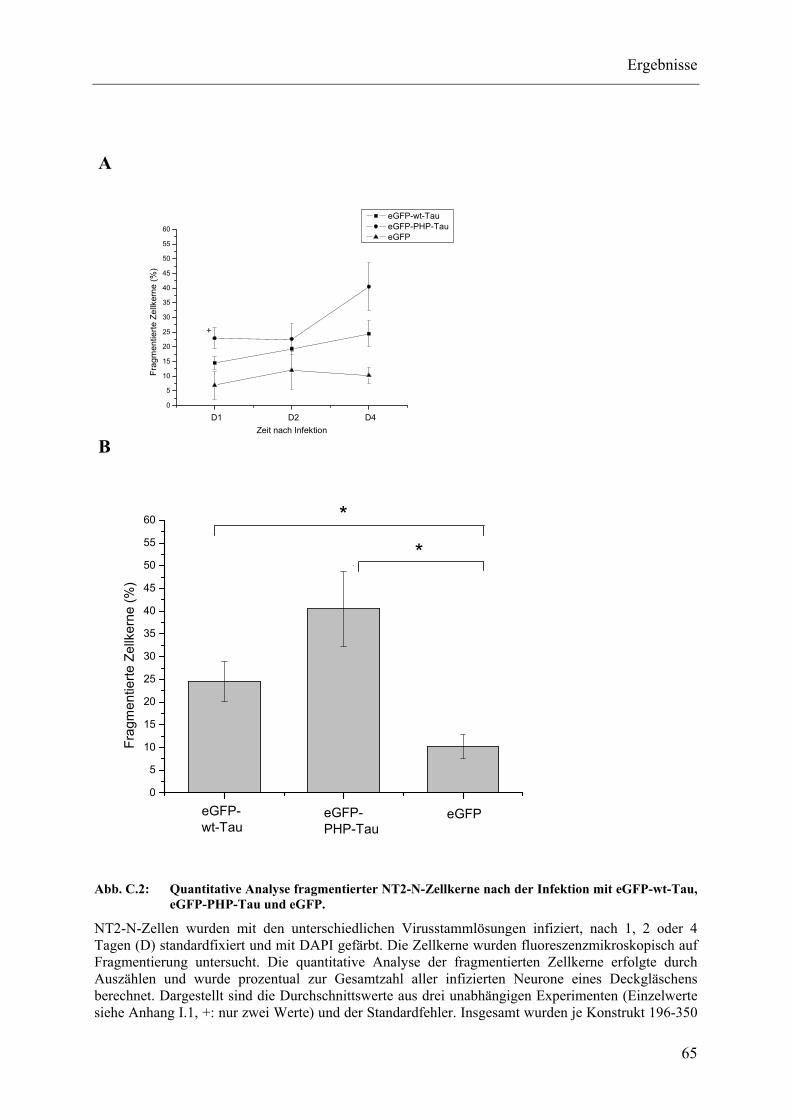
Ergebnisse
A
B
0
5
10
15
20
25
30
35
40
45
50
55
60
Fra
gmen
tierte
Zel
lker
ne (%
)
Zeit nach Infektion
eGFP-wt-Tau eGFP-PHP-Tau eGFP
+
D1 D2 D4
0
5
10
15
20
25
30
35
40
45
50
55
60
*
eGFPeGFP-PHP-Tau
Frag
men
tierte
Zel
lker
ne (%
)
eGFP-wt-Tau
*
Abb. C.2: Quantitative Analyse fragmentierter NT2-N-Zellkerne nach der Infektion mit eGFP-wt-Tau, eGFP-PHP-Tau und eGFP.
NT2-N-Zellen wurden mit den unterschiedlichen Virusstammlösungen infiziert, nach 1, 2 oder 4 Tagen (D) standardfixiert und mit DAPI gefärbt. Die Zellkerne wurden fluoreszenzmikroskopisch auf Fragmentierung untersucht. Die quantitative Analyse der fragmentierten Zellkerne erfolgte durch Auszählen und wurde prozentual zur Gesamtzahl aller infizierten Neurone eines Deckgläschens berechnet. Dargestellt sind die Durchschnittswerte aus drei unabhängigen Experimenten (Einzelwerte siehe Anhang I.1, +: nur zwei Werte) und der Standardfehler. Insgesamt wurden je Konstrukt 196-350
65

Ergebnisse
infizierte Neurone analysiert. Die Anzahl an fragmentierten Zellkernen nimmt von Tag 1 bis Tag 4 bei eGFP-wt-Tau und -PHP-Tau zu (A). Eine erhöhte Toxizität von PHP-Tau relativ zu wt-Tau und der Kontrolle eGFP ist zu jedem Zeitpunkt vorhanden und ist an Tag 4 am deutlichsten ausgeprägt (B). Für alle Bedingungen wurde der two-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 angewandt.
Generell ist durch die Expression von eGFP-wt-Tau und -PHP-Tau ein Anstieg in der Anzahl
an fragmentierten Zellkernen von Tag 1 zu Tag 4 sichtbar (Abb. C.2A). In der eGFP-
Kontrolle sinkt die Anzahl fragmentierter Zellkerne von Tag 2 zu Tag 4. Neurone, die eGFP-
PHP-Tau exprimieren, zeigen an allen drei Tagen eine höhere Degeneration im Vergleich zu
eGFP-wt-Tau und eGFP exprimierenden Zellen. Dieser Effekt ist an Tag 4 am deutlichsten
ausgeprägt (Abb. C.2B). Hier besitzen 40,5% aller PHP-Tau infizierten Neurone einen
fragmentierten Zellkern, mit wt-Tau infizierte 24,5%, mit eGFP infizierte hingegen nur
10,3%. Der neurodegenerative Effekt von eGFP-PHP-Tau ist 1,6-fach erhöht relativ zu
eGFP-wt-Tau und ist signifikant höher im Vergleich zur eGFP-Kontrolle. Auch mit eGFP-wt-
Tau infizierte Neurone zeigen einen signifikant erhöhten Anteil fragmentierter Zellkerne
relativ zur eGFP-Kontrolle. Daraus läßt sich schließen, dass auch die Expression von eGFP-
wt-Tau in NT2-N-Zellen einen geringen degenerativen Effekt hat. Wie in der Studie von Fath
et al. (2002) für Flag-Konstrukte beschrieben, zeigt auch das eGFP-Konstrukt mit PHP-Tau
einen degenerativen Effekt. Damit sind die eGFP-Konstrukte für weitere Analysen geeignet.
C.2 Expression der eGFP-Konstrukte in kortikalen Primärkulturen
Im Gehirn von Patienten mit der Alzheimer-Krankheit sind u.a. die Nervenzellen des
Neokortex besonders stark durch die neurofibrillären Ablagerungen betroffen. Nachdem die
Zytotoxizität von eGFP-PHP-Tau in humanen Modellneuronen nachgewiesen werden konnte,
sollte dies nun in kortikalen Primärneuronen der Maus getestet werden. Die Verwendung von
Nervenzellen aus der Maus bietet den Vorteil, dass in weiteren Experimenten durch die
Verwendung transgener Mauslinien ein zusätzlicher Effekt anderer AD-relevanter Proteine
untersucht werden kann. Zunächst sollte jedoch getestet werden, ob die Fusionskonstrukte in
diesem System korrekt exprimiert werden. Die Analyse erfolgte qualitativ auf Immunblot-
und quantitativ auf immunfluoreszenzmikroskopischer Ebene.
66

Ergebnisse
C.2.1 Protein-biochemische Analyse der Expression der eGFP-Konstrukte in
kortikalen Primärkulturen
Neben einer immunzytochemischen Charakterisierung wurde die Expression der Konstrukte
zunächst biochemisch untersucht.
Abb. C.3 zeigt die Expression der 3 verwendeten Kontrukte HSV-1-eGFP-352wt-Tau, HSV-
1-eGFP-352PHP-Tau und HSV-1-eGFP auf Westernblot-Ebene. Hierzu wurden Lysate HSV-
1-infizierter Primärkulturen angefertigt und im Immunblot mit Antikörpern gegen GFP, Tau
(Tau-5) und Tubulin (DM1A) als Referenzprotein detektiert. Im Fall der GFP-Tau-Konstrukte
werden die intakten Fusionskonstrukte exprimiert (Abb. C.3A,B). Beide Fusionsproteine
werden von dem αTau- und dem αGFP-Antikörper detektiert und zeigen ein Molekular-
gewicht im Bereich von 85 kD, was mit vorherigen Expressionsanalysen übereinstimmt
(Leschik, 2000). Dieses Molekulargewicht resultiert aus der Addition der apparenten
Molekulargewichte von fötalem Tau (48 kD) und GFP (27 kD) sowie von Konformations-
änderungen durch die Phosphorylierung in der Zelle. Zusätzlich erscheint im Tau-5-Blot bei
allen Lysaten eine Bande bei ca. 50 kD. Hierbei handelt es sich um endogenes Maus-Tau mit
einem Molekulargewicht von 48 kD (Abb. C.3B). EGFP wurde mit dem αGFP-Antikörper
nachgewiesen und zeigt, wie beschrieben, ein Molekulargewicht von 27 kD (Abb. C.3A). In
Abb. C.4 ist die Expression der einzelnen Konstrukte zu unterschiedlichen Zeitpunkten (Tag
1, 2 und 3) nach der Infektion dargestellt. Bei den GFP-Tau-Fusionskonstrukten
(Abb. C.4A,B) wurde mit Tau-5 und DM1A, im Fall des GFP-Konstrukts (Abb. C.4C) mit
dem αGFP-Antikörper und DM1A detektiert. Die Expression der 3 unterschiedlichen
Konstrukte kann an allen drei Tagen ohne erkennbaren Abbau nachgewiesen werden. Alle
Konstrukte zeigen ein vergleichbares Expressionsmuster mit einem leichten Anstieg an Tag 2
und einem Rückgang an Tag 3.
67
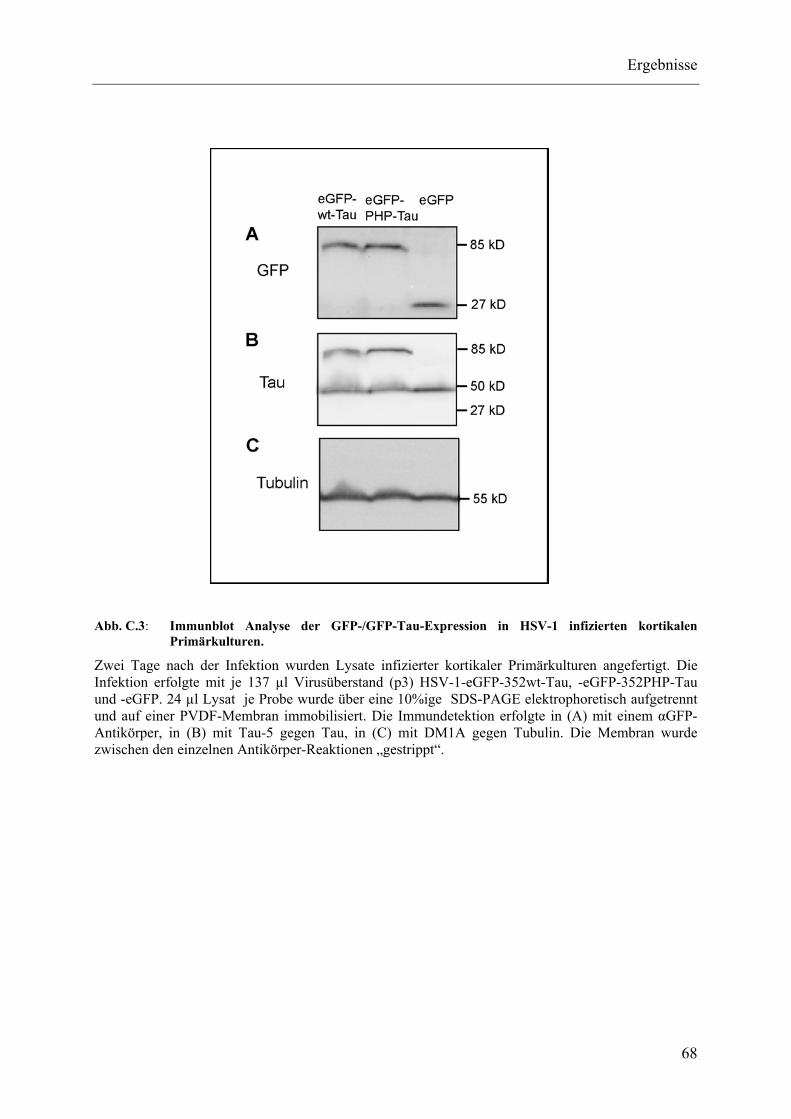
Ergebnisse
Abb. C.3: Immunblot Analyse der GFP-/GFP-Tau-Expression in HSV-1 infizierten kortikalen
Primärkulturen.
Zwei Tage nach der Infektion wurden Lysate infizierter kortikaler Primärkulturen angefertigt. Die Infektion erfolgte mit je 137 µl Virusüberstand (p3) HSV-1-eGFP-352wt-Tau, -eGFP-352PHP-Tau und -eGFP. 24 µl Lysat je Probe wurde über eine 10%ige SDS-PAGE elektrophoretisch aufgetrennt und auf einer PVDF-Membran immobilisiert. Die Immundetektion erfolgte in (A) mit einem αGFP-Antikörper, in (B) mit Tau-5 gegen Tau, in (C) mit DM1A gegen Tubulin. Die Membran wurde zwischen den einzelnen Antikörper-Reaktionen „gestrippt“.
68
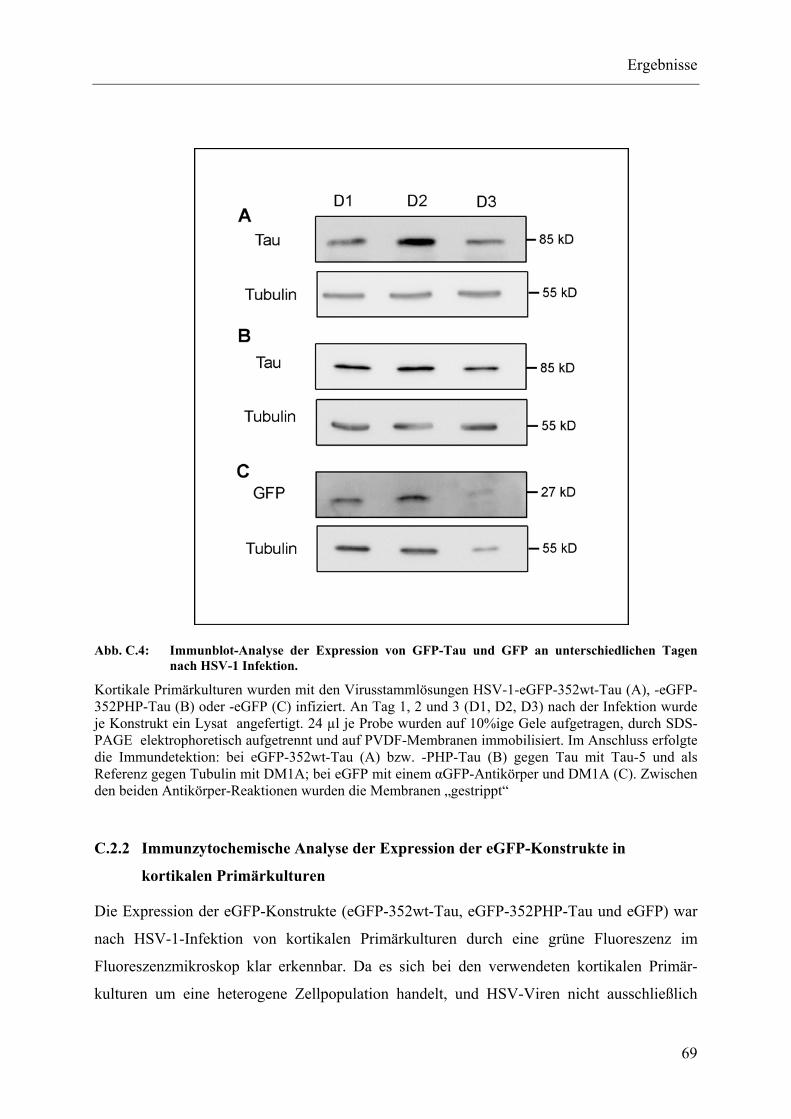
Ergebnisse
Abb. C.4: Immunblot-Analyse der Expression von GFP-Tau und GFP an unterschiedlichen Tagen nach HSV-1 Infektion.
Kortikale Primärkulturen wurden mit den Virusstammlösungen HSV-1-eGFP-352wt-Tau (A), -eGFP-352PHP-Tau (B) oder -eGFP (C) infiziert. An Tag 1, 2 und 3 (D1, D2, D3) nach der Infektion wurde je Konstrukt ein Lysat angefertigt. 24 µl je Probe wurden auf 10%ige Gele aufgetragen, durch SDS-PAGE elektrophoretisch aufgetrennt und auf PVDF-Membranen immobilisiert. Im Anschluss erfolgte die Immundetektion: bei eGFP-352wt-Tau (A) bzw. -PHP-Tau (B) gegen Tau mit Tau-5 und als Referenz gegen Tubulin mit DM1A; bei eGFP mit einem αGFP-Antikörper und DM1A (C). Zwischen den beiden Antikörper-Reaktionen wurden die Membranen „gestrippt“
C.2.2 Immunzytochemische Analyse der Expression der eGFP-Konstrukte in
kortikalen Primärkulturen
Die Expression der eGFP-Konstrukte (eGFP-352wt-Tau, eGFP-352PHP-Tau und eGFP) war
nach HSV-1-Infektion von kortikalen Primärkulturen durch eine grüne Fluoreszenz im
Fluoreszenzmikroskop klar erkennbar. Da es sich bei den verwendeten kortikalen Primär-
kulturen um eine heterogene Zellpopulation handelt, und HSV-Viren nicht ausschließlich
69

Ergebnisse
neurotrop sind, werden durch HSV-1-Infektion auch Glia-Zellen infiziert. Die spätere
Analyse des Zelltods sollte aber ausschließlich in Neuronen untersucht werden. Aus diesem
Grund erschien es notwendig, die quantitative Analyse der Expression auch auf
immunfluoreszenzmikroskopischer Ebene für die einzelnen Konstrukte zu quantifizieren.
Dies erfolgte durch Intensitätsmessungen der Fluoreszenz einzelner morphologisch klar
erkennbarer Nervenzellen. Auf eine MAP-2-Färbung als neuronalem Marker, wie sie in
späteren Analysen durchgeführt wurde, wurde hierbei verzichtet, um ein Verfälschen der
Fluoreszenzintensität durch „Cross-Talk“ (Erscheinen roter Fluoreszenz bei
Filterkombination für grüne Fluoreszenz) zwischen den einzelnen Kanälen zu vermeiden
(Abb. C.5A,C,E). Die Messungen der GFP-Fluoreszenz der einzelnen analysierten Neurone
wurden im Anschluss gemittelt (Einzelwerte siehe Anhang I.2). Die durchschnittliche
Fluoreszenz von eGFP-wt-Tau war niedriger als die von eGFP-PHP-Tau (eGFP-wt-Tau:
6,87+/-0,87 (+/-SE), eGFP-PHP-Tau: 12,79+/-1,97 , relative Einheiten (rel. E); gemessen mit
Scion Image). Das Kontrollkonstrukt eGFP zeigte eine deutlich geringere mittlere Fluores-
zenzintensität im Vergleich zu den beiden anderen Konstrukten (eGFP: 2,39+/-0,3 rel. E). Um
zu bestimmen, ob diese Fluoreszenzunterschiede durch eine unterschiedliche zelluläre
Proteinmenge oder durch eine unterschiedliche Faltung von GFP im jeweiligen Fusions-
protein verursacht werden, wurde in einem folgenden Experiment eine immunzytochemische
Antikörperfärbung gegen GFP durchgeführt (Abb. C.5B,D,F). Der Zweitantikörper war mit
einem im roten Kanal fluoreszierenden Fluorochrom (Cy3) gekoppelt, sodass die Intensität im
roten Kanal ohne Beeinträchtigung des grünen GFP-Signals gemessen werden konnte. Nach
Mittelung der einzelnen Werte (siehe Anhang I.2) zeigte sich, dass kein signifikanter
Intensitätsunterschied zwischen eGFP-wt-Tau und eGFP-PHP-Tau exprimierenden Neuronen
vorlag (8,17+/-0,7 rel. E (für eGFP-wt-Tau), 8,84+/-1,28 rel. E (für eGFP-PHP-Tau) und 5,95+/-
1,37 rel. E (für eGFP).
70
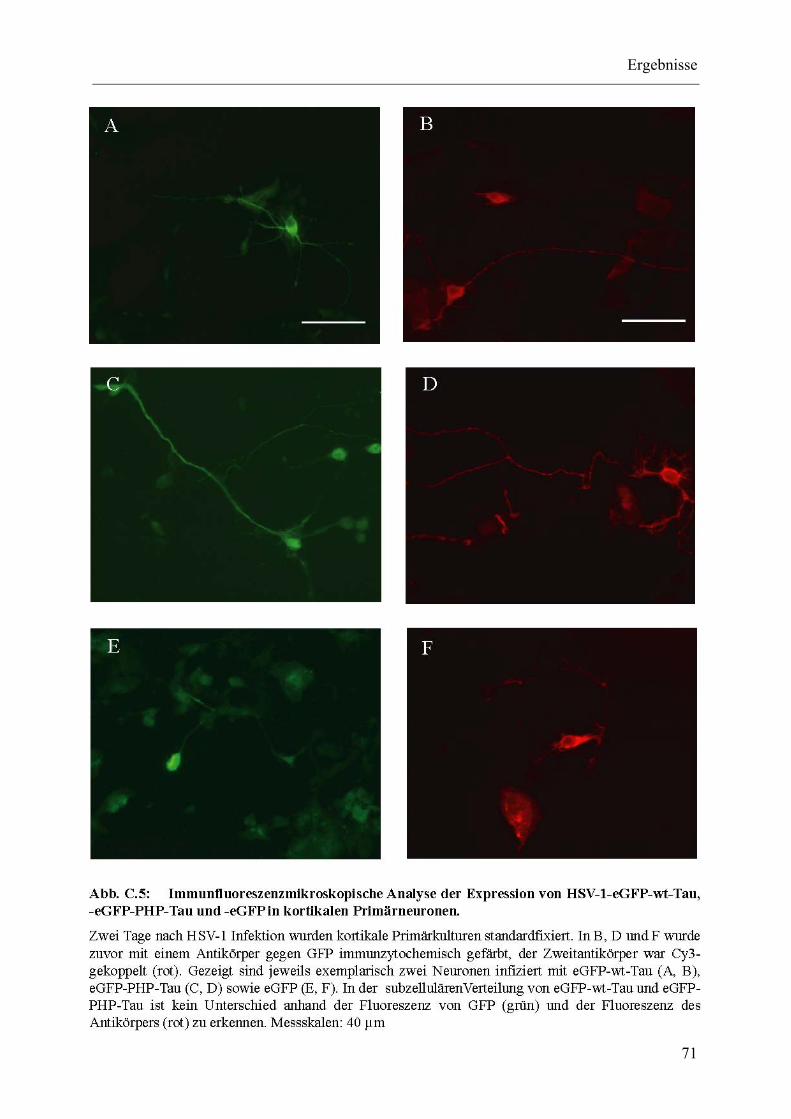
Ergebnisse
Abb. C. 5:
71
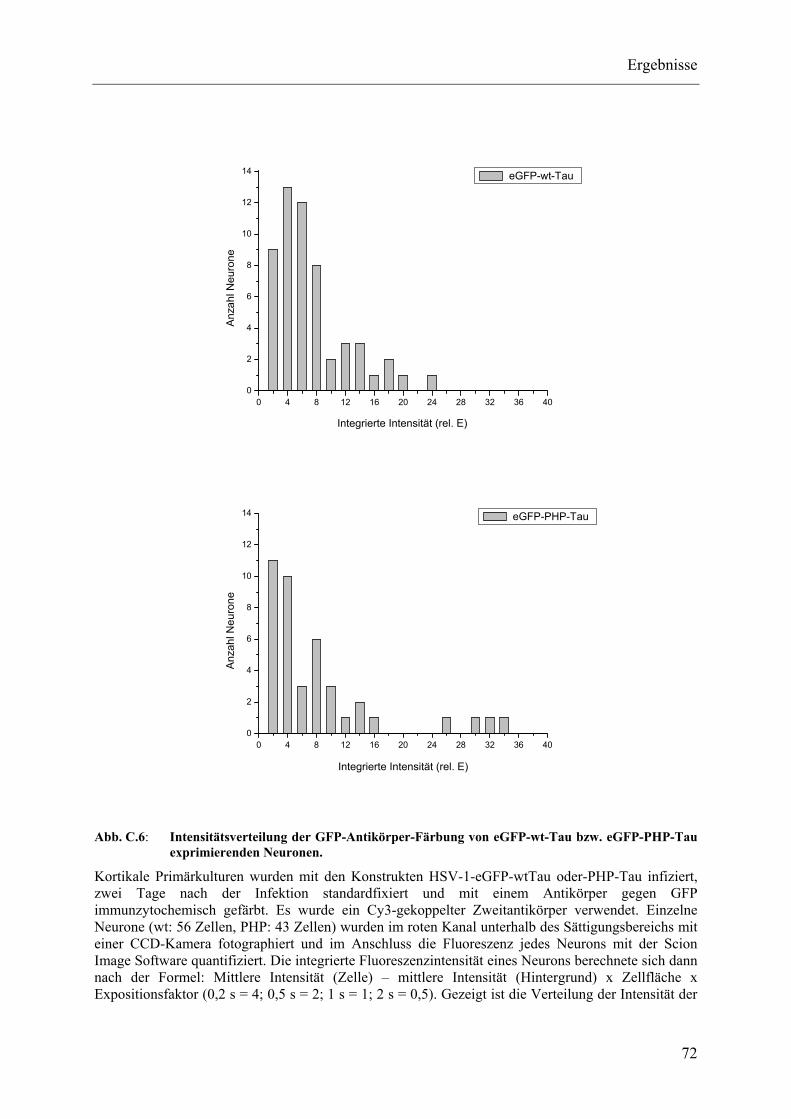
Ergebnisse
0 4 8 12 16 20 24 28 32 36 400
2
4
6
8
10
12
14
Anz
ahl N
euro
ne
Integrierte Intensität (rel. E)
eGFP-wt-Tau
0 4 8 12 16 20 24 28 32 36 400
2
4
6
8
10
12
14
Anz
ahl N
euro
ne
Integrierte Intensität (rel. E)
eGFP-PHP-Tau
Abb. C.6: Intensitätsverteilung der GFP-Antikörper-Färbung von eGFP-wt-Tau bzw. eGFP-PHP-Tau
exprimierenden Neuronen.
Kortikale Primärkulturen wurden mit den Konstrukten HSV-1-eGFP-wtTau oder-PHP-Tau infiziert, zwei Tage nach der Infektion standardfixiert und mit einem Antikörper gegen GFP immunzytochemisch gefärbt. Es wurde ein Cy3-gekoppelter Zweitantikörper verwendet. Einzelne Neurone (wt: 56 Zellen, PHP: 43 Zellen) wurden im roten Kanal unterhalb des Sättigungsbereichs mit einer CCD-Kamera fotographiert und im Anschluss die Fluoreszenz jedes Neurons mit der Scion Image Software quantifiziert. Die integrierte Fluoreszenzintensität eines Neurons berechnete sich dann nach der Formel: Mittlere Intensität (Zelle) – mittlere Intensität (Hintergrund) x Zellfläche x Expositionsfaktor (0,2 s = 4; 0,5 s = 2; 1 s = 1; 2 s = 0,5). Gezeigt ist die Verteilung der Intensität der
72

Ergebnisse
Fluoreszenz in relativen Einheiten (rel. E). Bei eGFP-wt-Tau und -PHP-Tau liegt eine ähnliche Verteilung der Fluoreszenzintensitäten vor.
Das Histogramm in Abb. C.6 zeigt, dass auch die Verteilung der Fluoreszenzintensitäten sehr
ähnlich ist. Dies bedeutet, dass nicht eine unterschiedliche Proteinmenge, sondern eine
differentielle Faltung von eGFP im jeweiligen Fusionsprotein zu dem oben genannten
Unterschied der GFP-Eigenfluoreszenz führt. Der hierbei festgestellte Intensitätsunterschied
des Kontrollkonstrukts eGFP im Vergleich zu den Fusionsproteinen kann möglicherweise
darauf zurückzuführen sein, dass eGFP in Primärneuronen durch Diffusion hauptsächlich im
Kernbereich verteilt ist (Abb. C.5E). Dies könnte die Fluoreszenzintensität erheblich ver-
fälschen, da eine Interaktion mit Kernkomponenten wie z.B. Chromatin und eine dadurch
bewirkte Faltungsveränderung möglich ist. Die Intensitätsmessung nach der Antikörper-
färbung ergab, dass der Unterschied zu den Fusionsproteinen mit einem Durchschnittswert
von 5,95+/-1,37 rel. E nach wie vor deutlich war. Auch nach Methanol-Fixierung (nicht
gezeigt), die zu einer vollständigen Zerstörung der Kernlamina führt, war kein deutliches
Signal im Kernbereich zu erkennen. Daraus lässt sich folgern, dass möglicherweise eine Inter-
aktion mit Kernkomponenten vorliegt, die die Antikörperbindung im Kernbereich verhindert.
Auf Grund seiner subzellulären Verteilung erscheint eGFP also nicht als optimales
Kontrollkonstrukt. Allerdings hatten frühere Experimente mit einem lac-Z-Kontrollkonstrukt
ergeben, dass dieses einen toxischen Effekt auf Primärneurone ausübt (Fath, 2002), also auch
schlecht geeignet ist. Als mögliche Alternative könnte sich die Herstellung eines GFP-
Konstrukts mit doppelter eGFP-Sequenz anbieten, welches aufgrund seines größeren
Molekulargewichts nicht in den Kern diffundieren kann. Allerdings wäre dieses dann aber
aufgrund seiner zwei Fluorophore pro Molekül wiederum nicht direkt mit den eGFP-Tau-
Konstrukten vergleichbar.
Da in dieser Arbeit grundsätzlich ein differentieller Effekt von wt-Tau und PHP-Tau auf die
Neurodegeneration untersucht werden sollte, und die neuronale Expression von eGFP-wt-Tau
und -PHP-Tau ähnlich ist, konnten die Konstrukte für weitere Experimente verwendet
werden.
73

Ergebnisse
C.3 PHP-Tau ist neurotoxisch in kortikalen Primärkulturen
Nachdem für eGFP-wt-Tau und eGFP-PHP-Tau eine vergleichbare Expression in
Primärkulturen festgestellt wurde, wurde nun ein möglicher neurodegenerativer Effekt
untersucht. Hierfür wurden Primärkulturen mit den 3 HSV-1-Konstrukten eGFP-wt-Tau,
eGFP-PHP-Tau sowie eGFP als Kontrollkonstrukt infiziert. Ähnlich wie in NT2-N-Zellen
wurde zunächst der optimale Zeitpunkt nach der Infektion für die Analysen ermittelt. Aus
diesem Grund wurden infizierte Primärkulturen zu unterschiedlichen Zeitpunkten nach der
Infektion standardfixiert und eine DAPI-Färbung zum Untersuchen der nukleären
Morphologie durchgeführt. Zusätzlich erfolgte mit einem Antikörper gegen MAP-2, als
neuronalem Marker, eine immunzytochemische Färbung (Abb. C.7). Die Analyse des
Zelltods erfolgte durch Auszählen fragmentierter Zellkerne infizierter und MAP-2 positiver
Zellen. In Abb. C.8 ist exemplarisch für jedes Konstrukt ein infiziertes Neuron dargestellt.
Das mit PHP-Tau (Abb. C.8C,D) infizierte Neuron besitzt in diesem Fall einen
fragmentierten Zellkern, nicht aber die gezeigten wt-Tau (Abb. C.8A,B) und eGFP
(Abb. C.8E,F) exprimierenden Nervenzellen.
74
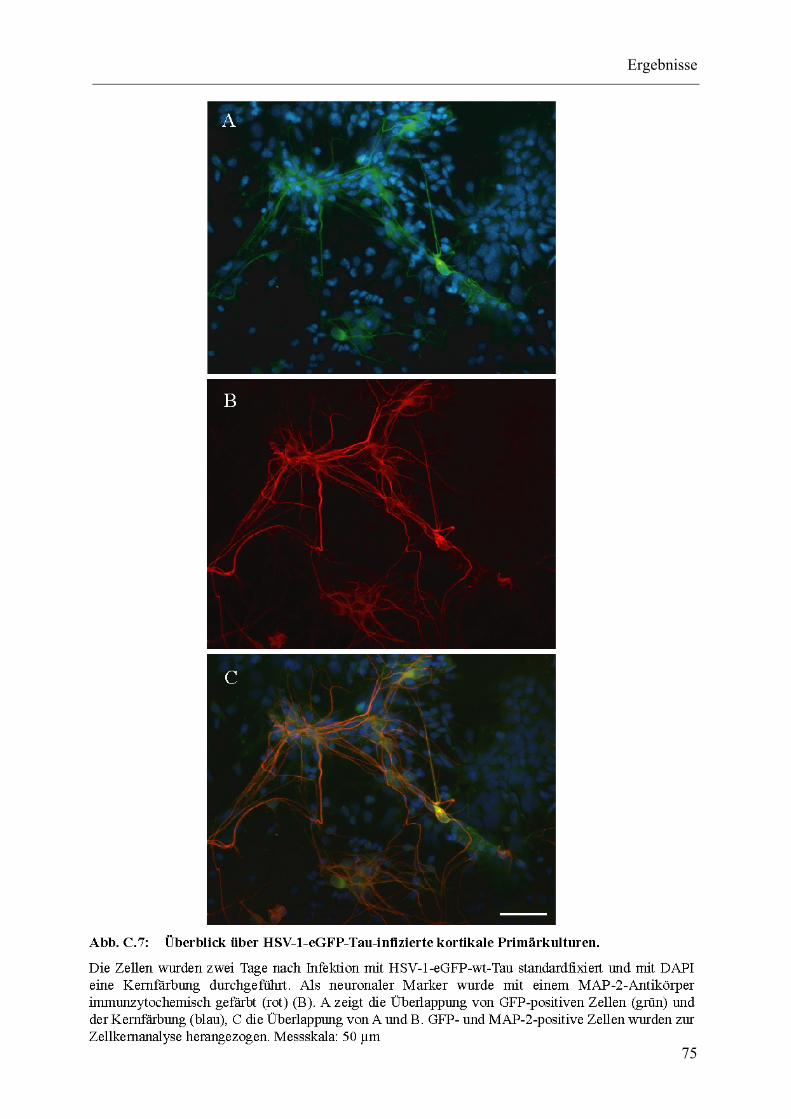
Ergebnisse
Abb. C. 7:
75
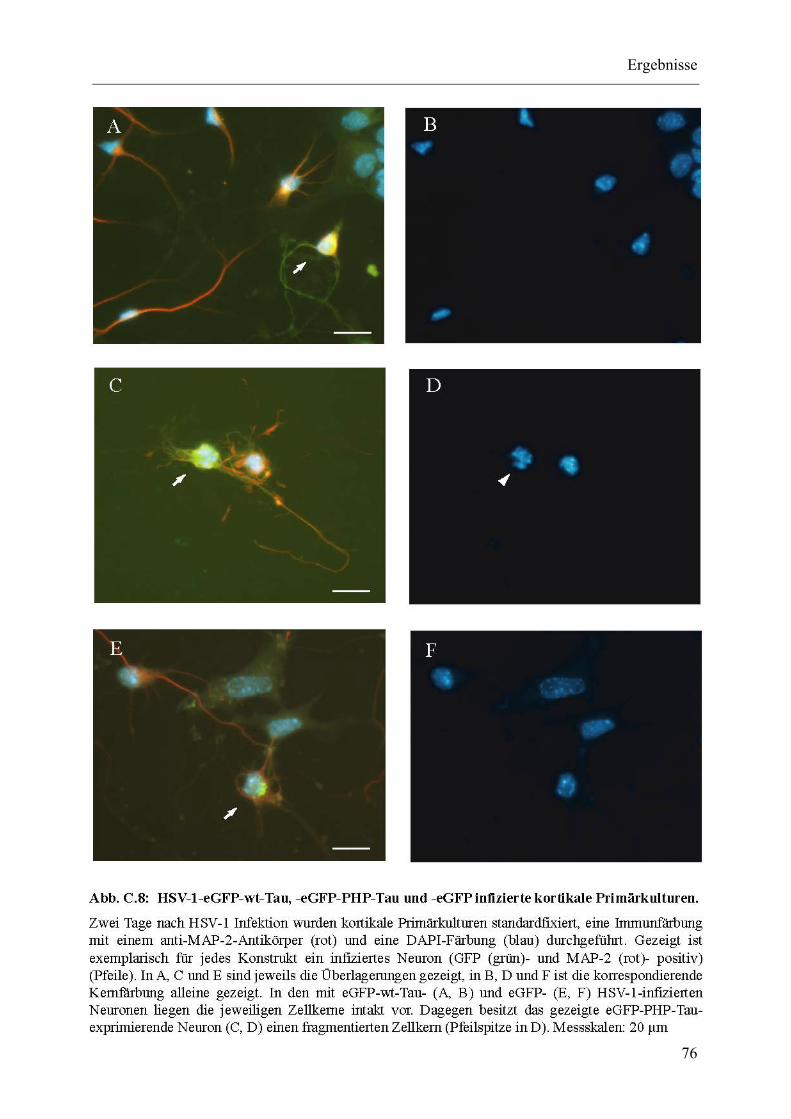
Ergebnisse
Abb. C. 8:
76

Ergebnisse
C.3.1 Quantitative Analyse des neuronalen Zelltods in Primärkulturen zu
unterschiedlichen Zeitpunkten nach HSV-1-Infektion
Da es sich bei kortikalen Primärkulturen aus der Maus um ein anderes neuronales Zellsystem
als bei humanen NT2-N-Zellen handelt, ist der Zeitpunkt (Post-Infektion) der Zelltod-Analyse
nicht direkt übertragbar. Aus diesem Grund wurden infizierte Primärkulturen verschiedene
Tage nach der Infektion mit den Konstrukten eGFP-wt-Tau, eGFP-PHP-Tau und eGFP auf
ihre Zellkernmorphologie untersucht (Abb. C.9). Die Anzahl der fragmentierten Zellkerne je
Konstrukt wurde prozentual zu allen analysierten infizierten Neuronen des jeweiligen
Experiments berechnet. Es zeigte sich, dass zu allen drei Zeitpunkten nach der Infektion die
Expression von PHP-Tau toxischer ist als wt-Tau oder eGFP (Abb. C.9A). An Tag 4 zeigten
über 66% aller analysierten PHP-Tau exprimierenden Neurone einen fragmentierten Kern.
Auch bei beiden anderen Konstrukten stieg der Anteil fragmentierter Kerne an Tag 4
erheblich an und erreichte bei der GFP-Kontrolle etwa 30%. Auf weitere Experimente an Tag
4 wurde deshalb verzichtet. Auch wt-Tau war zu jedem Zeitpunkt toxischer als die GFP-
Kontrolle. Ebenso wie bei den Experimenten mit NT2-N Zellen deutet dies auf einen leicht
neurodegenerativen Effekt wt-Taus hin. An Tag 3 war allerdings kein deutlicher Unterschied
zwischen wt-Tau und GFP zu erkennen, jedoch zwischen PHP-Tau und wt-Tau sowie GFP.
Die GFP-Kontrolle zeigte an Tag 2 den geringsten Anteil an fragmentierten Zellkernen (ca.
12%), sodass Tag 2 für die weiteren Experimente ausgewählt wurde.
77
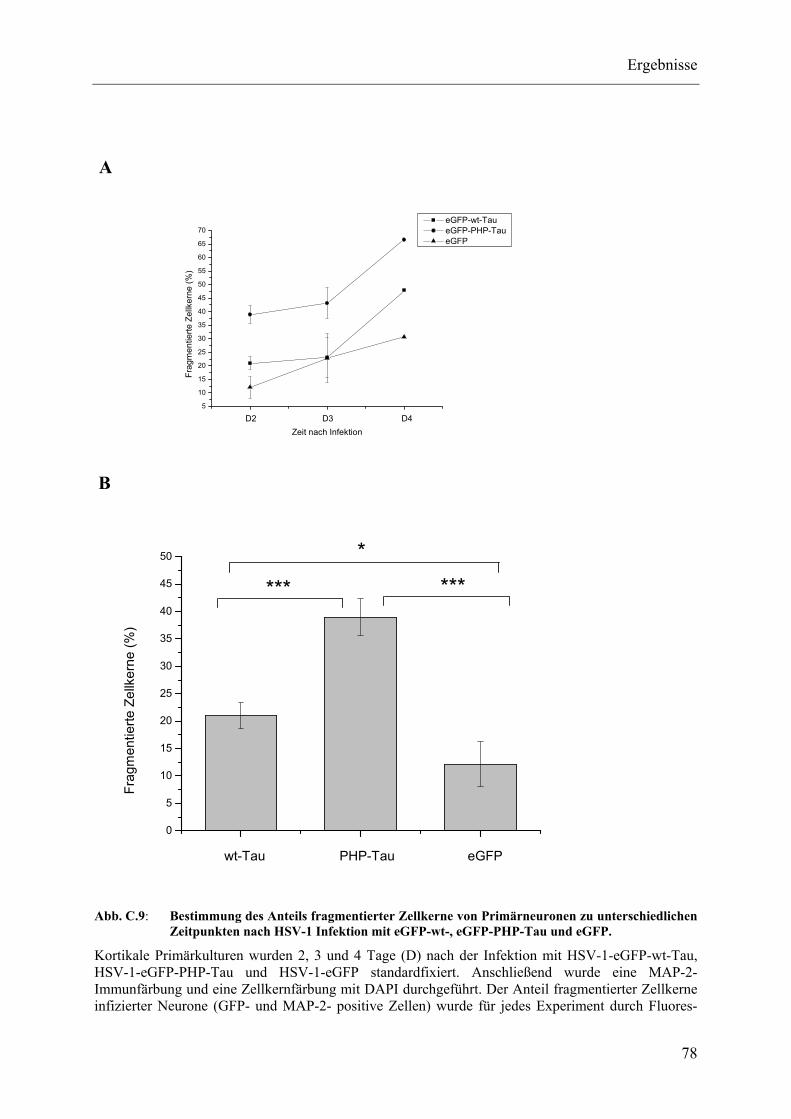
Ergebnisse
A
5
10
15
20
25
30
35
40
45
50
55
60
65
70
Frag
men
tierte
Zel
lker
ne (%
)
Zeit nach Infektion
eGFP-wt-Tau eGFP-PHP-Tau eGFP
D2 D3 D4
B
0
5
10
15
20
25
30
35
40
45
50
Frag
men
tierte
Zel
lker
ne (%
)
*
***
wt-Tau PHP-Tau eGFP
***
Abb. C.9: Bestimmung des Anteils fragmentierter Zellkerne von Primärneuronen zu unterschiedlichen Zeitpunkten nach HSV-1 Infektion mit eGFP-wt-, eGFP-PHP-Tau und eGFP.
Kortikale Primärkulturen wurden 2, 3 und 4 Tage (D) nach der Infektion mit HSV-1-eGFP-wt-Tau, HSV-1-eGFP-PHP-Tau und HSV-1-eGFP standardfixiert. Anschließend wurde eine MAP-2-Immunfärbung und eine Zellkernfärbung mit DAPI durchgeführt. Der Anteil fragmentierter Zellkerne infizierter Neurone (GFP- und MAP-2- positive Zellen) wurde für jedes Experiment durch Fluores-
78

Ergebnisse
zenzmikroskopie bestimmt. Insgesamt wurden je Konstrukt zwischen 365 und 902 Neurone untersucht. Dargestellt sind die Durchschnittswerte und Standardfehler aus sechs Experimenten an Tag 2, drei Experimenten an Tag 3 und einem Experiment an Tag 4 (Einzelwerte siehe Anhang I.3). PHP-Tau ist zu allen Zeitpunkten toxischer als wt-Tau und eGFP (A). An Tag 2 ist PHP-Tau hochsignifikant toxischer als wt-Tau und eGFP (B). Für alle Bedingungen wurde der two-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 und ***P < 0,001 angewandt.
Abb. C.9B zeigt, dass PHP-Tau im Vergleich zu wt-Tau und der Kontrolle an Tag 2 einen
hochsignifikant neurodegenerativen Effekt auf Primärneuronen ausübt. Im Durchschnitt ist
der Anteil an degenerierten Zellen nach Expression mit PHP-Tau fast doppelt so hoch wie bei
wt-Tau und mehr als 3 mal so hoch wie bei der GFP-Kontrolle.
C.3.2 Protein-biochemische Analyse des durch PHP-Tau ausgelösten toxischen Effekts
in kortikalen Primärkulturen
Im folgenden Abschnitt wurde der bereits immunzytochemisch analysierte neurotoxische
Effekt PHP-Taus auf Immunblot-Ebene getestet. Dies erfolgte durch Verwendung eines
Antikörpers gerichtet gegen ein 17kD-Fragment von Caspase-3, das die Aktivierung von
dieser Caspase während apoptotischen Vorgängen anzeigt. Ähnlich wie für die immun-
zytochemische Analyse wurden kortikale Primärkulturen mit gleichem HSV-1-Titer für jedes
Konstrukt infiziert. Basierend auf den Ergebnissen aus Abschnitt C.3.1 wurden Lysate an Tag
2 nach der Infektion angefertigt und auf Westernblot-Ebene die Aktivierung von Caspase-3
quantifiziert. Das erhaltene Signal wurde in Relation zu dem jeweiligen Tubulin-Signal der
Probe (Referenz) gesetzt. Abb. C.10 zeigt exemplarisch den Immunblot eines Experiments,
detektiert mit den Antikörpern gegen aktivierte Caspase-3 (A), gegen Tubulin (DM1A) (B)
und GFP (C). Da es sich bei den verwendeten kortikalen Primärkulturen um eine heterogene
Zellpopulation handelt, die neben postmitotischen Neuronen auch mitotisch aktive Gliazellen
enthält, kann nicht ausgeschlossen werden, dass dies einen Einfluß auf die Ergebnisse hat.
Zum einen ist es möglich, dass der analysierte Effekt glialen Ursprungs ist, zum anderen kann
außer durch die Verwendung eines gleichen Titers kein Einfluß auf eine gleiche Expression
der Konstrukte genommen werden. Es ist denkbar, dass PHP-Tau, welches Mikrotubuli
weniger stabilisiert als wt-Tau, die Zellteilung von Gliazellen begünstigt. Außerdem ist nicht
klar, ob in Gliazellen eGFP-PHP-Tau stärker als eGFP-wt-Tau oder eGFP exprimiert wird.
79

Ergebnisse
Abb. C.10: Immunblot-Analyse des durch PHP-Tau ausgelösten Zelltods durch Verwendung eines Antikörpers gegen aktivierte Caspase-3.
Kortikale Primärkulturen wurden mit 2 x 105 Ifps je Konstrukt (HSV-1-eGFP-wt-Tau, eGFP-PHP-Tau, -eGFP) infiziert und zwei Tage nach der Infektion lysiert. Die Lysate (48 µl je Konstrukt) wurden auf ein 7,5%iges Gel aufgetragen, über SDS-PAGE aufgetrennt und auf einer PVDF-Membran immobilisiert. Die Immunblotanalyse erfolgte mit einem Antikörper gegen aktivierte Caspase-3 (aktiv. Casp.-3 (A), DM1A gegen Tubulin (B) und einem αGFP-Antikörper gegen die exprimierten Proteine (C). Zwischen den einzelnen Antikörper-Reaktionen wurde die Membran „gestrippt“.
Der densitometrischen Analyse in Abb. C.11 ist zu entnehmen, dass PHP-Tau eine signifikant
stärkere Caspase-3-Aktivierung in Primärkulturen auslöst als wtTau und eGFP, der Effekt ist
in beiden Fällen ca. 2,7 mal so hoch. Die Aktivität von Caspase-3 ist in wt-Tau- und eGFP-
infizierten Primärkulturen nicht signifikant unterschiedlich. Die Ergebnisse deuten darauf hin,
dass apoptotische Mechanismen bei dem durch PHP-Tau induzierten neurodegenerativen
Effekt beteiligt sind.
80
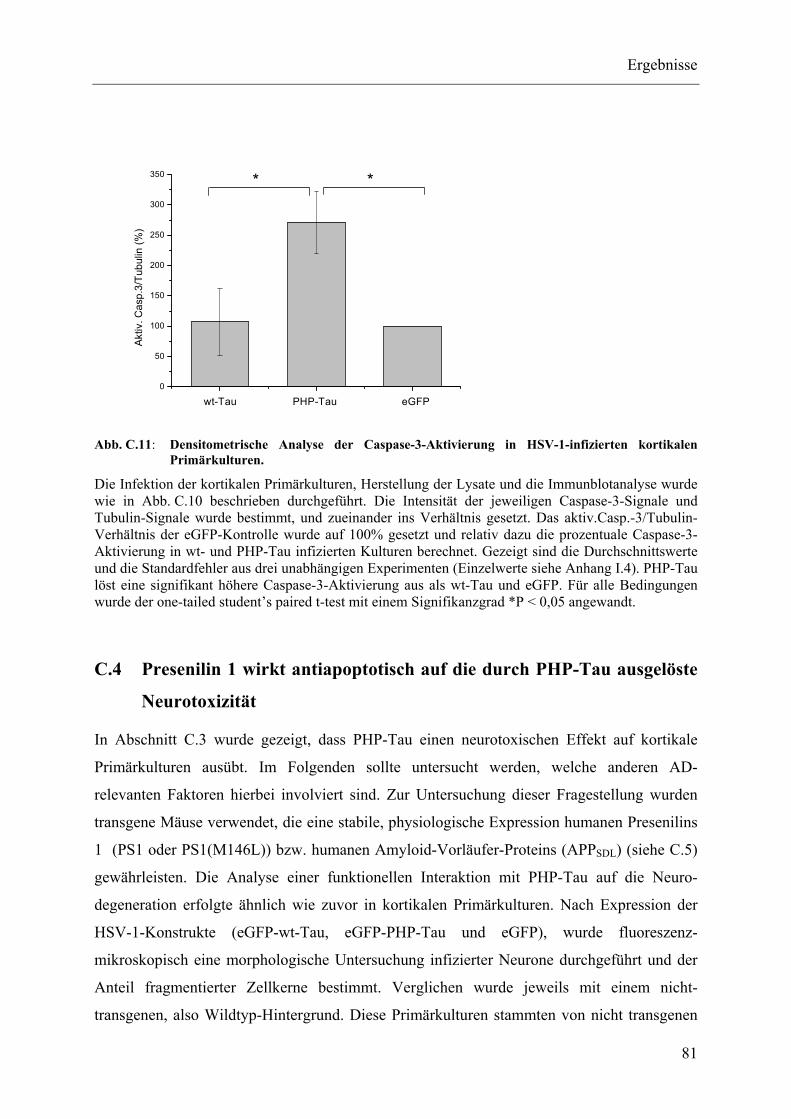
Ergebnisse
0
50
100
150
200
250
300
350
Akt
iv. C
asp.
3/Tu
bulin
(%)
* *
wt-Tau PHP-Tau eGFP
Abb. C.11: Densitometrische Analyse der Caspase-3-Aktivierung in HSV-1-infizierten kortikalen
Primärkulturen.
Die Infektion der kortikalen Primärkulturen, Herstellung der Lysate und die Immunblotanalyse wurde wie in Abb. C.10 beschrieben durchgeführt. Die Intensität der jeweiligen Caspase-3-Signale und Tubulin-Signale wurde bestimmt, und zueinander ins Verhältnis gesetzt. Das aktiv.Casp.-3/Tubulin-Verhältnis der eGFP-Kontrolle wurde auf 100% gesetzt und relativ dazu die prozentuale Caspase-3-Aktivierung in wt- und PHP-Tau infizierten Kulturen berechnet. Gezeigt sind die Durchschnittswerte und die Standardfehler aus drei unabhängigen Experimenten (Einzelwerte siehe Anhang I.4). PHP-Tau löst eine signifikant höhere Caspase-3-Aktivierung aus als wt-Tau und eGFP. Für alle Bedingungen wurde der one-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 angewandt.
C.4 Presenilin 1 wirkt antiapoptotisch auf die durch PHP-Tau ausgelöste
Neurotoxizität
In Abschnitt C.3 wurde gezeigt, dass PHP-Tau einen neurotoxischen Effekt auf kortikale
Primärkulturen ausübt. Im Folgenden sollte untersucht werden, welche anderen AD-
relevanten Faktoren hierbei involviert sind. Zur Untersuchung dieser Fragestellung wurden
transgene Mäuse verwendet, die eine stabile, physiologische Expression humanen Presenilins
1 (PS1 oder PS1(M146L)) bzw. humanen Amyloid-Vorläufer-Proteins (APPSDL) (siehe C.5)
gewährleisten. Die Analyse einer funktionellen Interaktion mit PHP-Tau auf die Neuro-
degeneration erfolgte ähnlich wie zuvor in kortikalen Primärkulturen. Nach Expression der
HSV-1-Konstrukte (eGFP-wt-Tau, eGFP-PHP-Tau und eGFP), wurde fluoreszenz-
mikroskopisch eine morphologische Untersuchung infizierter Neurone durchgeführt und der
Anteil fragmentierter Zellkerne bestimmt. Verglichen wurde jeweils mit einem nicht-
transgenen, also Wildtyp-Hintergrund. Diese Primärkulturen stammten von nicht transgenen
81

Ergebnisse
Geschwistertieren. Die Unterscheidung zwischen Wildtyp (wt) und transgenen Tieren erfolgte
durch PCR mit Primern gegen humanes Presenilin1 (identisch für PS1 und PS1(M146L)).
Zusätzlich konnte immunfluoreszenzmikroskopisch durch Verwendung eines gegen humanes
PS1 gerichteten Antikörpers dessen Expression nachgewiesen werden (Abb. C.12). Hierbei
war die ER-typische Verteilung von PS1 und PS1(M146L) erkennbar (Abb. C.12A). Bei der
Sensitivität des hier verwendeten monoklonalen Antikörpers zeigten ca. 50% der Zellen eine
Expression des jeweiligen Transgens. Die Expression der PS-1-Transgene liegt unter der
Kontrolle des humanen HMG-CR-Promoters, welcher ein „house-keeping“ Promoter ist, der
eine starke und ubiquitäre Expression mit einer hohen Expression in Neuronen zeigt (Czech et
al., 1997; Czech et al., 1998). Die Herstellung und Charakterisierung der hier verwendeten
PS1-transgenen Mäuse wurde bereits zuvor beschrieben (Czech et al., 1997; Czech et al.,
1998; Leutner et al., 2000; Terro et al., 2002). Es konnte bis zu einem Alter von 18 Monaten
keine Amyloid-Plaque-Entwicklung festgestellt werden (Moussaoui et al., 2000).
82
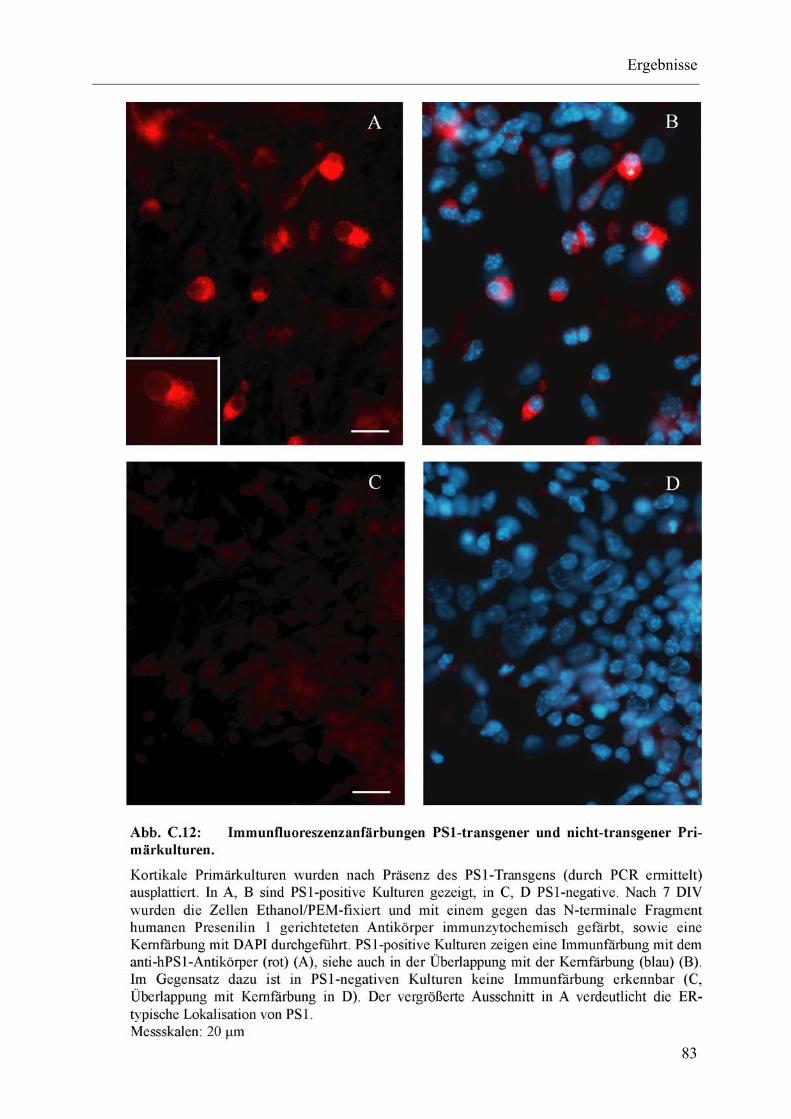
Ergebnisse
Abb. C. 12:
83

Ergebnisse
C.4.1 Quantitative Analyse der PHP-Tau induzierten Neurodegeneration in Presenilin
1 transgenen Primärkulturen
Zunächst wurde der Effekt von PS1 auf die Tau-vermittelte Degeneration untersucht. Wie
bereits im vorherigen Abschnitt erwähnt, wurden PS1-transgene und nicht-transgene (wt)
Primärkulturen mit den Konstrukten HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP
infiziert. Zwei Tage nach der Infektion wurden die Zellen fixiert, eine MAP-2-Immunfärbung
und eine Kernfärbung mit DAPI durchgeführt. Anschließend wurden die Zellkerne infizierter
Neurone fluoreszenzmikroskopisch auf Fragmentierung untersucht. In Abb. C.13 ist jeweils
der Anteil fragmentierter Zellkerne prozentual zur Gesamtzahl aller analysierten Neurone
dargestellt. Die in nicht-transgenen (wt) Primärkulturen durch PHP-Tau ausgelöste
Neurotoxizität ist in PS1-Primärkulturen signifikant erniedrigt. PHP-Tau zeigt auf PS1-
transgenem Hintergrund keine gegenüber wt-Tau erhöhte Toxizität (wt-Tau: 23,13%, PHP-
Tau: 26,15% fragmentierte Kerne). Dagegen zeigt sich weder bei wt-Tau- noch bei eGFP-
exprimierenden Neuronen eine wesentliche Reduktion im Anteil fragmentierter Zellkerne.
Durch mehrere Studien ist bekannt, dass die Expression von humanem PS1 in neuronalen
Zellsystemen eine antiapoptotische Wirkung besitzt (Bursztajn et al., 1998; Terro et al.,
2002). Dies könnte den Rückgang des neurodegenerativen Effekts von PHP-Tau erklären, da,
wie zuvor gezeigt, die PHP-Tau induzierte Degeneration von einer Caspase-3-Aktivierung
begleitet ist (siehe Abschnitt C.3.2).
84
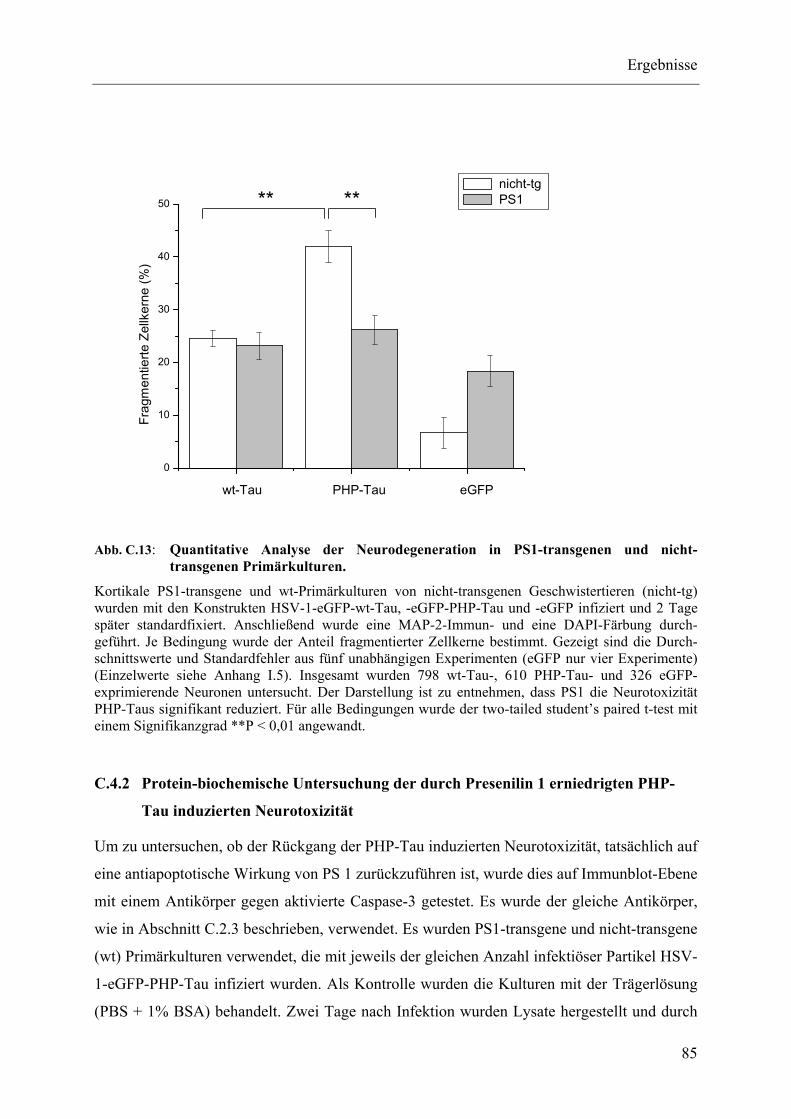
Ergebnisse
0
10
20
30
40
50
Frag
men
tierte
Zel
lker
ne (%
)
nicht-tg PS1
wt-Tau PHP-Tau eGFP
** **
Abb. C.13: Quantitative Analyse der Neurodegeneration in PS1-transgenen und nicht-transgenen Primärkulturen.
Kortikale PS1-transgene und wt-Primärkulturen von nicht-transgenen Geschwistertieren (nicht-tg) wurden mit den Konstrukten HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP infiziert und 2 Tage später standardfixiert. Anschließend wurde eine MAP-2-Immun- und eine DAPI-Färbung durch-geführt. Je Bedingung wurde der Anteil fragmentierter Zellkerne bestimmt. Gezeigt sind die Durch-schnittswerte und Standardfehler aus fünf unabhängigen Experimenten (eGFP nur vier Experimente) (Einzelwerte siehe Anhang I.5). Insgesamt wurden 798 wt-Tau-, 610 PHP-Tau- und 326 eGFP-exprimierende Neuronen untersucht. Der Darstellung ist zu entnehmen, dass PS1 die Neurotoxizität PHP-Taus signifikant reduziert. Für alle Bedingungen wurde der two-tailed student’s paired t-test mit einem Signifikanzgrad **P < 0,01 angewandt.
C.4.2 Protein-biochemische Untersuchung der durch Presenilin 1 erniedrigten PHP-
Tau induzierten Neurotoxizität
Um zu untersuchen, ob der Rückgang der PHP-Tau induzierten Neurotoxizität, tatsächlich auf
eine antiapoptotische Wirkung von PS 1 zurückzuführen ist, wurde dies auf Immunblot-Ebene
mit einem Antikörper gegen aktivierte Caspase-3 getestet. Es wurde der gleiche Antikörper,
wie in Abschnitt C.2.3 beschrieben, verwendet. Es wurden PS1-transgene und nicht-transgene
(wt) Primärkulturen verwendet, die mit jeweils der gleichen Anzahl infektiöser Partikel HSV-
1-eGFP-PHP-Tau infiziert wurden. Als Kontrolle wurden die Kulturen mit der Trägerlösung
(PBS + 1% BSA) behandelt. Zwei Tage nach Infektion wurden Lysate hergestellt und durch
85

Ergebnisse
Immunblot mit Antikörpern gegen das 17 kD-Fragment von Caspase-3, gegen Tubulin als
Referenzprotein und gegen Tau, zum Nachweis der Tau-Expression, getestet. In nicht
transgenen Primärkulturen wurde eine stärkere 17 kD-Bande (aktivierte Caspase-3) in PHP-
Tau infizierten im Vergleich zu nicht infizierten Zellen detektiert (Abb. C.14). Dies ist nicht
der Fall in PS1-positiven Primärkulturen. Dies ist konsistent mit der Annahme, dass PS1
antiapoptotisch auf die PHP-Tau-Toxizität wirkt. In der nachfolgenden densitometrischen
Analyse wurde jeweils das Caspase-3 Signal und das zugehörige Tubulin-Signal gemessen
und zueinander in Relation gesetzt. Verglichen wurde die Änderung des aktiv. Casp.-
3/Tubulin-Verhältnisses innerhalb einer Kultur, d.h. uninfizierte PS1-transgene bzw.
uninfizierte nicht-transgene Kulturen wurden auf 100% gesetzt, um mögliche Effekte der
Expression von PS1 auf die basale Caspase-3-Aktivität in uninfizierten Kulturen unberück-
sichtigt zu lassen. Abb. C.15 zeigt, dass PHP-Tau keine Aktivierung der Caspase-3 in PS1-
Kulturen auslöst. In nicht-transgenen-Primärkulturen, ohne PS1-Expression, führt die
Expression von PHP-Tau im Vergleich zur uninfizierten Kontroll-Kultur zu einer 1,7-fach
erhöhten Caspase-3-Aktivierung. Die Ergebnisse bestätigen die Annahme, dass PS1 einen
antiapoptotischen Effekt auf den durch PHP-Tau ausgelösten Zelltod hat.
Abb. C.14: Immunblot-Analyse der Caspase-3-Aktivierung in PHP-Tau-infizierten PS1-transgenen Primärkulturen.
86
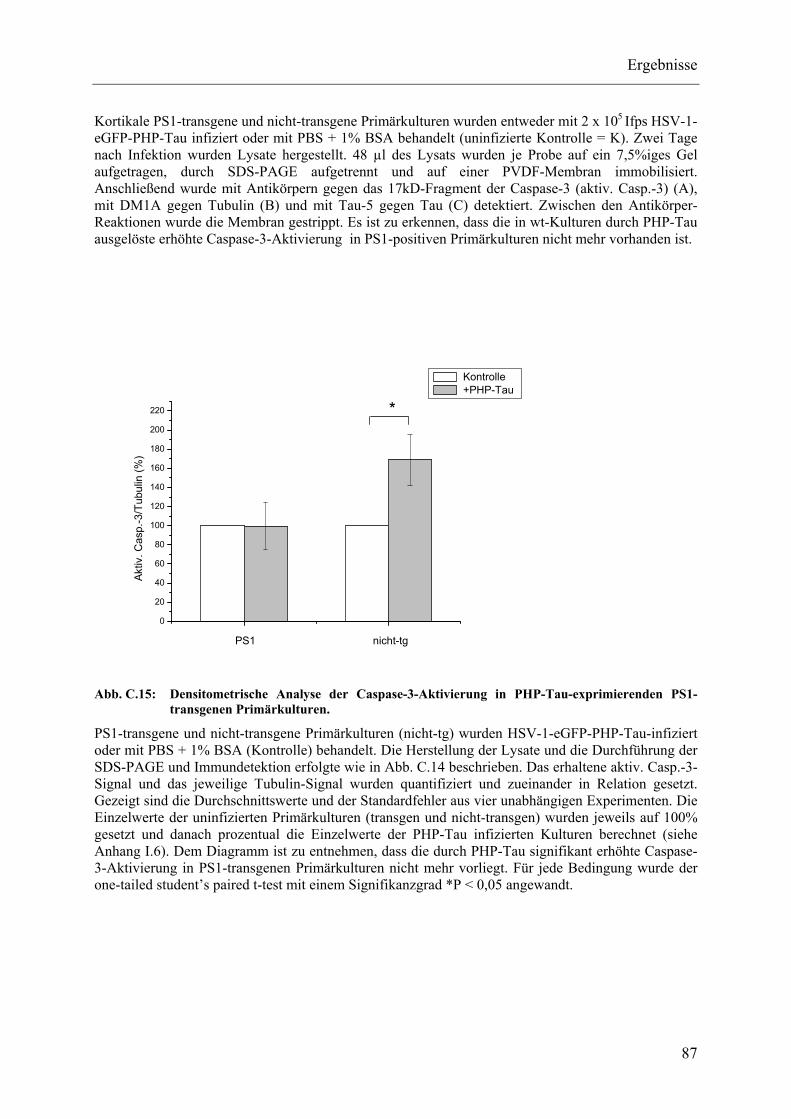
Ergebnisse
Kortikale PS1-transgene und nicht-transgene Primärkulturen wurden entweder mit 2 x 105 Ifps HSV-1-eGFP-PHP-Tau infiziert oder mit PBS + 1% BSA behandelt (uninfizierte Kontrolle = K). Zwei Tage nach Infektion wurden Lysate hergestellt. 48 µl des Lysats wurden je Probe auf ein 7,5%iges Gel aufgetragen, durch SDS-PAGE aufgetrennt und auf einer PVDF-Membran immobilisiert. Anschließend wurde mit Antikörpern gegen das 17kD-Fragment der Caspase-3 (aktiv. Casp.-3) (A), mit DM1A gegen Tubulin (B) und mit Tau-5 gegen Tau (C) detektiert. Zwischen den Antikörper-Reaktionen wurde die Membran gestrippt. Es ist zu erkennen, dass die in wt-Kulturen durch PHP-Tau ausgelöste erhöhte Caspase-3-Aktivierung in PS1-positiven Primärkulturen nicht mehr vorhanden ist.
0
20
40
60
80
100
120
140
160
180
200
220
Aktiv
. Cas
p.-3
/Tub
ulin
(%)
Kontrolle +PHP-Tau
PS1 nicht-tg
*
Abb. C.15: Densitometrische Analyse der Caspase-3-Aktivierung in PHP-Tau-exprimierenden PS1-transgenen Primärkulturen.
PS1-transgene und nicht-transgene Primärkulturen (nicht-tg) wurden HSV-1-eGFP-PHP-Tau-infiziert oder mit PBS + 1% BSA (Kontrolle) behandelt. Die Herstellung der Lysate und die Durchführung der SDS-PAGE und Immundetektion erfolgte wie in Abb. C.14 beschrieben. Das erhaltene aktiv. Casp.-3-Signal und das jeweilige Tubulin-Signal wurden quantifiziert und zueinander in Relation gesetzt. Gezeigt sind die Durchschnittswerte und der Standardfehler aus vier unabhängigen Experimenten. Die Einzelwerte der uninfizierten Primärkulturen (transgen und nicht-transgen) wurden jeweils auf 100% gesetzt und danach prozentual die Einzelwerte der PHP-Tau infizierten Kulturen berechnet (siehe Anhang I.6). Dem Diagramm ist zu entnehmen, dass die durch PHP-Tau signifikant erhöhte Caspase-3-Aktivierung in PS1-transgenen Primärkulturen nicht mehr vorliegt. Für jede Bedingung wurde der one-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 angewandt.
87

Ergebnisse
C.4.3 Die Presenilin 1 FAD-Mutation M146L wirkt nicht antiapoptotisch auf die PHP-
Tau induzierte Neurotoxizität
Im Folgenden wurde untersucht, inwiefern die Expression von humanem FAD-mutanten PS1
einen Einfluss auf den neurotoxischen Effekt PHP-Taus hat. Zudem wurde dies verglichen
mit den Ergebnissen von Wildtyp-PS1 (PS1wt) (Abschnitt C.4.1,2) verglichen. Es gibt
Hinweise, dass FAD-Mutationen in Presenilin 1 apoptotische Kaskaden aktivieren und
neurale Zellen gegenüber apoptotischen sowie nekrotischen Stimuli sensitivieren können
(Guo et al., 1997; Guo et al., 1999; Weihl et al., 1999; Terro et al., 2002). Demgegenüber
stehen Studien, die dies nicht bestätigen (Bursztjan et al., 1998; Siman et al., 2000). Um zu
untersuchen, wie sich in diesem Zusammenhang der Effekt von Presenilin 1 (M146L) auf
Tau-infizierte Primärneuronen auswirkt, wurden kortikale Primärkulturen aus PS1(M146L)-
transgenen Mäuse hergestellt und mit wt-Tau, PHP-Tau oder eGFP HSV-1-infiziert. Wie in
vorherigen Analysen wurde eine MAP-2-Immunfärbung sowie eine Kernfärbung mit DAPI
durchgeführt und das Verhältnis fragmentierter Zellkerne GFP-positiver Neurone fluoreszenz-
mikroskopisch bestimmt. Verglichen wurden die Ergebnisse mit infizierten Primärneuronen
nicht-transgener Geschwistertiere. In Abb. C16 ist zu erkennen, dass die antiapoptotische
Wirkung von Wildtyp-PS1 (Abschnitt C.4.1,2) auf die Wirkung von PHP-Tau in
PS1(M146L)-Primärkulturen nicht mehr vorhanden ist. Dagegen ist bei Expression jeden
Konstrukts eine leichte Zunahme im Anteil fragmentierter Zellkerne im mutanten PS1
Hintergrund gegenüber den Wildtypzellen zu erkennen. Da wt-Tau ebenso wie PHP-Tau
einen leicht erhöhten Anteil fragmentierter Zellkerne zeigt, könnte eine Phosphorylierung
weiterer Stellen als die in PHP-Tau mutierten Hauptphosphorylierungsstellen bei der
Neurodegeneration beteiligt sein. Alternativ könnte dieser Effekt komplett unabhängig von
einer erhöhten Tau-Phosphorylierung sein und auf einer generellen Sensitivierung der Tau-
Expression beruhen.
Tatsächlich wurde in verschiedenen Zellsystemen bereits gezeigt, dass FAD-mutantes PS1 die
Phosphorylierung von Tau erhöht (Takashima et al., 1998; Baki et al., 2004), jedoch gibt es
hierzu auch Gegenbeispiele (Irving & Miller, 1997; Julliams et al., 1999). Es wurde damit
begonnen, die Phosphorylierung wt-Taus in PS1(M146L)-Primärkulturen im Immunblot zu
testen, allerdings konnten bisher keine eindeutigen Ergebnisse erzielt werden.
88
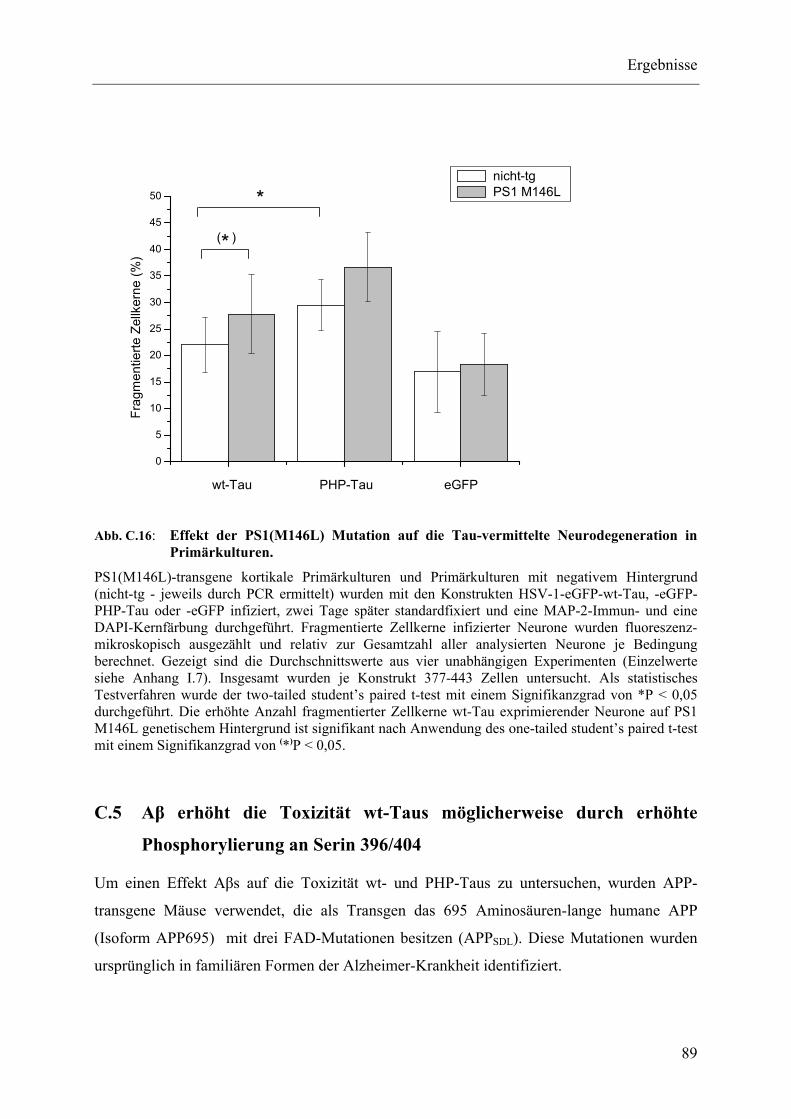
Ergebnisse
0
5
10
15
20
25
30
35
40
45
50
Frag
men
tierte
Zel
lker
ne (%
)
nicht-tg PS1 M146L
*
*
wt-Tau PHP-Tau eGFP
( )
Abb. C.16: Effekt der PS1(M146L) Mutation auf die Tau-vermittelte Neurodegeneration in
Primärkulturen.
PS1(M146L)-transgene kortikale Primärkulturen und Primärkulturen mit negativem Hintergrund (nicht-tg - jeweils durch PCR ermittelt) wurden mit den Konstrukten HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau oder -eGFP infiziert, zwei Tage später standardfixiert und eine MAP-2-Immun- und eine DAPI-Kernfärbung durchgeführt. Fragmentierte Zellkerne infizierter Neurone wurden fluoreszenz-mikroskopisch ausgezählt und relativ zur Gesamtzahl aller analysierten Neurone je Bedingung berechnet. Gezeigt sind die Durchschnittswerte aus vier unabhängigen Experimenten (Einzelwerte siehe Anhang I.7). Insgesamt wurden je Konstrukt 377-443 Zellen untersucht. Als statistisches Testverfahren wurde der two-tailed student’s paired t-test mit einem Signifikanzgrad von *P < 0,05 durchgeführt. Die erhöhte Anzahl fragmentierter Zellkerne wt-Tau exprimierender Neurone auf PS1 M146L genetischem Hintergrund ist signifikant nach Anwendung des one-tailed student’s paired t-test mit einem Signifikanzgrad von (*)P < 0,05.
C.5 Aβ erhöht die Toxizität wt-Taus möglicherweise durch erhöhte
Phosphorylierung an Serin 396/404
Um einen Effekt Aβs auf die Toxizität wt- und PHP-Taus zu untersuchen, wurden APP-
transgene Mäuse verwendet, die als Transgen das 695 Aminosäuren-lange humane APP
(Isoform APP695) mit drei FAD-Mutationen besitzen (APPSDL). Diese Mutationen wurden
ursprünglich in familiären Formen der Alzheimer-Krankheit identifiziert.
89

Ergebnisse
Es handelt sich hierbei um die Mutationen in einer schwedischen (S = KM670/671NL),
niederländischen (D = E693Q) und Londoner (L = V717I) Familie; die Nummern der
Mutationen beziehen sich auf die Aminosäuresequenz der APP-Isoform 770. Die Expression
des humanen APPSDL-Transgens liegt unter der Kontrolle des PDGF-Promoters, der ein hohes
Expressionsniveau des Transgens im Gehirn gewährleistet. Die hier verwendeten transgenen
Mäuse entwickeln ab einem Alter von 18 Monaten Amyloid-Plaques (Moussaoui et al., 2000;
Blanchard et al., 2003). FAD-Mutationen in APP führen zu einer erhöhten Produktion des
amyloidogeneren Aβ42-Fragments von APP (Suzuki et al., 1994; Citron et al., 1997;
Scheuner et al., 1996). Um einen Anstieg der Aβ-Produktion in dem hier verwendeten System
der kortikalen Primärkultur nachzuweisen, wurden zunächst ELISA-Tests mit Antikörpern,
gerichtet gegen humanes Aβ40 sowie gegen humanes Aβ42, durchgeführt. Die Analysen
wurden zu Zeitpunkten durchgeführt, an denen die Analysen der Toxizität nach der Infektion
durchgeführt werden würden (Tag 2 und 3, Zeitpunkt Tag 0 = fiktive Infektion). In nicht-
transgenen Primärkulturen konnte weder Aβ40 noch Aβ42 gemessen werden, da die gegen
humanes Aβ gerichteten Antikörper das endogen produzierte Maus-Aβ nicht detektieren
können. Wie erwartet konnte in den transgenen Primärkulturen, die positiv für humanes
APPSDL sind, die Konzentrationen beider Aβ-Spezies (ohne Infektion mit eGFP-Tau)
gemessen werden. Das Diagramm in Abb. C.17 zeigt, dass die Konzentration von Aβ40 an
Tag 2 etwas höher ist als die von Aβ42 ist. An Tag 3 dagegen ist kein Unterschied mehr
sichtbar, die Aβ40-Konzentration bleibt gleich, während die Aβ42-Konzentration leicht
ansteigt. Dies steht im Einklang mit Ergebnissen aus APP-transgenen Tieren, die zeigen
konnten, dass durch die schwedische Mutante APP verstärkt amyloidogen prozessiert wird
(Reaume et al., 1996), während normalerweise das Verhältnis Aβ42/Aβ40 weit auf der Seite
von Aβ40 liegt (Citron et al., 1997; Eckman et al., 1997; Araki et al., 2001). Eine genauere
Aussage über den Faktor der Erhöhung von Aβ42 in APPSDL mutanten Primärkulturen kann in
dieser Arbeit nicht gemacht werden, da diese Tiere nicht mit hAPPwt-Tieren verglichen wur-
den, sondern nur mit nicht-transgenen Tieren, deren Aβ-Produktion nicht detektierbar war.
90
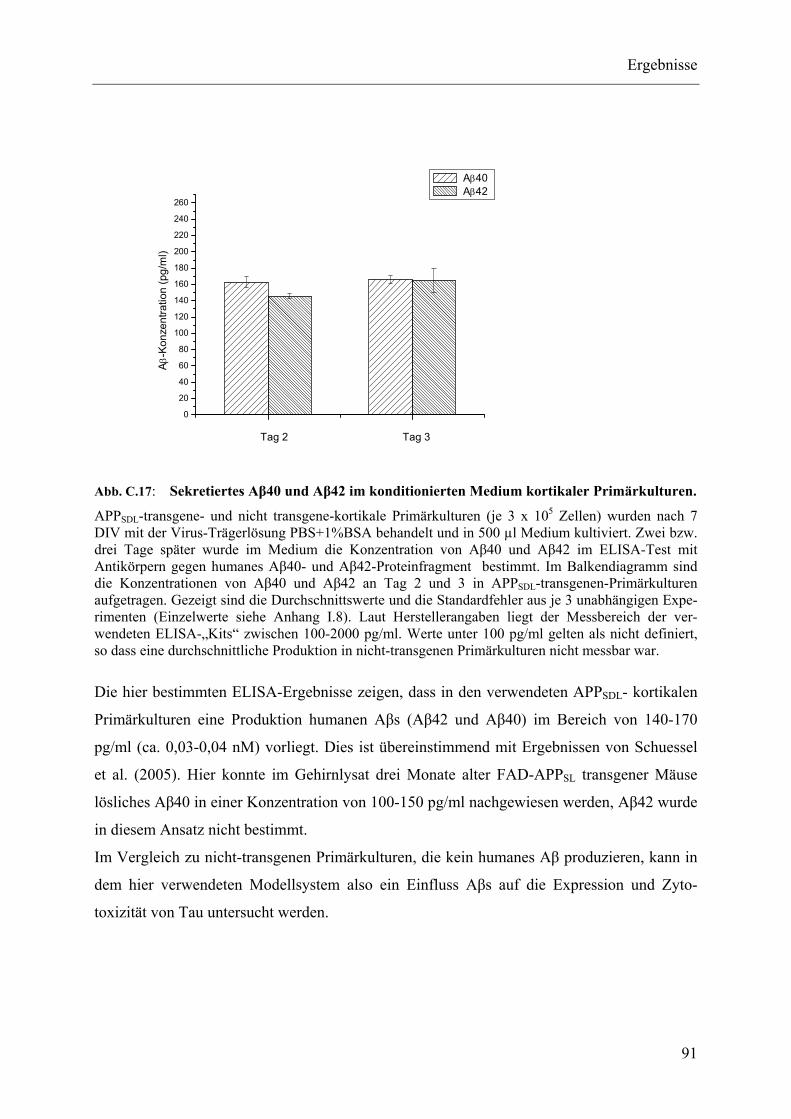
Ergebnisse
0
20
40
60
80
100
120
140
160
180
200
220
240
260
Aβ-
Konz
entra
tion
(pg/
ml)
Aβ40 Aβ42
Tag 2 Tag 3
Abb. C.17: Sekretiertes Aβ40 und Aβ42 im konditionierten Medium kortikaler Primärkulturen.
APPSDL-transgene- und nicht transgene-kortikale Primärkulturen (je 3 x 105 Zellen) wurden nach 7 DIV mit der Virus-Trägerlösung PBS+1%BSA behandelt und in 500 µl Medium kultiviert. Zwei bzw. drei Tage später wurde im Medium die Konzentration von Aβ40 und Aβ42 im ELISA-Test mit Antikörpern gegen humanes Aβ40- und Aβ42-Proteinfragment bestimmt. Im Balkendiagramm sind die Konzentrationen von Aβ40 und Aβ42 an Tag 2 und 3 in APPSDL-transgenen-Primärkulturen aufgetragen. Gezeigt sind die Durchschnittswerte und die Standardfehler aus je 3 unabhängigen Expe-rimenten (Einzelwerte siehe Anhang I.8). Laut Herstellerangaben liegt der Messbereich der ver-wendeten ELISA-„Kits“ zwischen 100-2000 pg/ml. Werte unter 100 pg/ml gelten als nicht definiert, so dass eine durchschnittliche Produktion in nicht-transgenen Primärkulturen nicht messbar war.
Die hier bestimmten ELISA-Ergebnisse zeigen, dass in den verwendeten APPSDL- kortikalen
Primärkulturen eine Produktion humanen Aβs (Aβ42 und Aβ40) im Bereich von 140-170
pg/ml (ca. 0,03-0,04 nM) vorliegt. Dies ist übereinstimmend mit Ergebnissen von Schuessel
et al. (2005). Hier konnte im Gehirnlysat drei Monate alter FAD-APPSL transgener Mäuse
lösliches Aβ40 in einer Konzentration von 100-150 pg/ml nachgewiesen werden, Aβ42 wurde
in diesem Ansatz nicht bestimmt.
Im Vergleich zu nicht-transgenen Primärkulturen, die kein humanes Aβ produzieren, kann in
dem hier verwendeten Modellsystem also ein Einfluss Aβs auf die Expression und Zyto-
toxizität von Tau untersucht werden.
91

Ergebnisse
C.5.1 Quantitative Analyse der Neurodegeneration in Tau-infizierten APPSDL-
transgenen Primärkulturen
Damit der Einfluss einer erhöhten Aβ-Produktion auf die PHP-Tau-induzierte Neurotoxizität
untersucht werden konnte, wurden aus den oben beschriebenen transgenen APPSDL-Mäusen
kortikale Primärkulturen hergestellt. Als Kontrolle dienten kortikale Primärkulturen aus nicht
transgenen Geschwistertieren (wt). Die Präsenz des Transgens wurde durch PCR mit Primern
gegen humanes APP ermittelt. Nach 5 DIV wurden die Kulturen jeweils mit den HSV-1-
Konstrukten eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP infiziert, zwei Tage später fixiert und
eine MAP-2- bzw. DAPI-Färbung durchgeführt. Wie auch in früheren Experimenten (siehe
oben) wurde der Anteil von infizierten Neuronen mit fragmentiertem Zellkern fluoreszenz-
mikroskopisch bestimmt. Die Toxizität von wt-Tau auf APPSDL-transgenem Hintergrund ist
im Vergleich zur nicht-transgenen-Kontrollkultur signifikant um etwa 50% erhöht
(Abb. C.18). Demgegenüber ist keine Zunahme in PHP-Tau oder eGFP infizierten Neuronen
festzustellen. Die Ergebnisse könnten darauf hindeuten, dass Aβ eine erhöhte Phospho-
rylierung von wt-Tau induziert und zwar an Phosphorylierungsstellen, die in PHP-Tau mutiert
sind. Diese Hypothese sollte durch Immunblot-Analyse getestet werden.
92
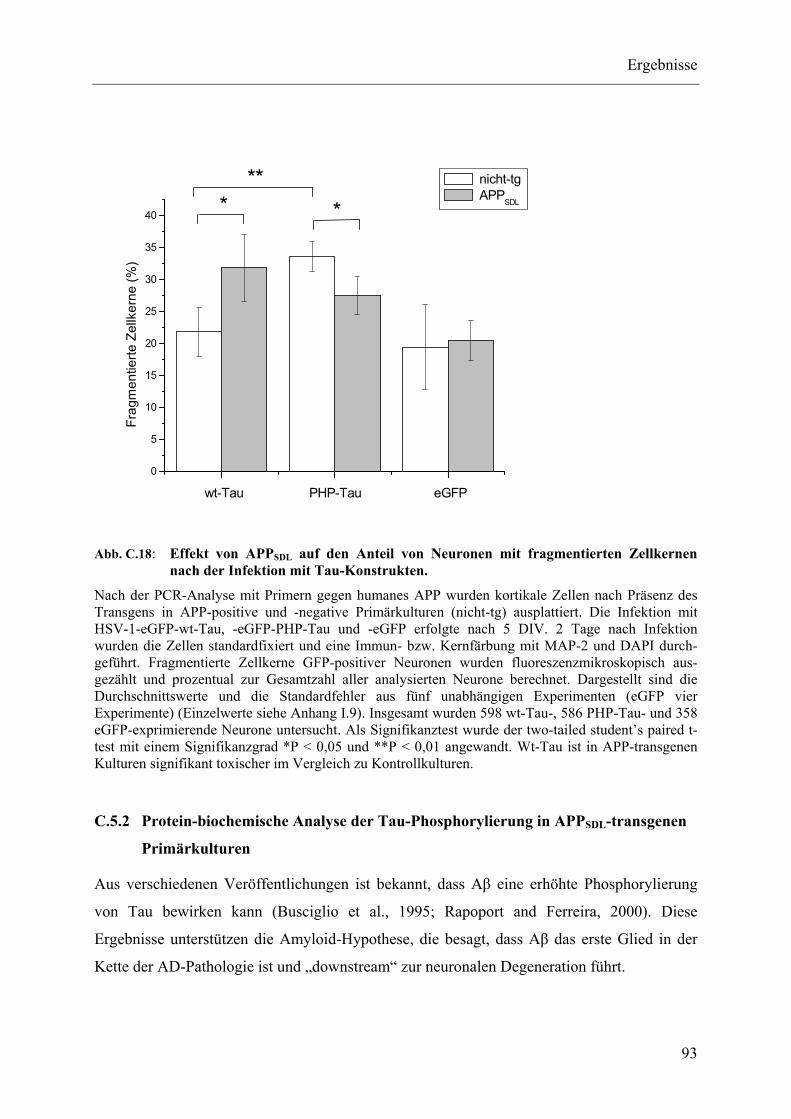
Ergebnisse
0
5
10
15
20
25
30
35
40
Frag
men
tierte
Zel
lker
ne (%
)
nicht-tg APPSDL
wt-Tau PHP-Tau eGFP
***
*
Abb. C.18: Effekt von APPSDL auf den Anteil von Neuronen mit fragmentierten Zellkernen nach der Infektion mit Tau-Konstrukten.
Nach der PCR-Analyse mit Primern gegen humanes APP wurden kortikale Zellen nach Präsenz des Transgens in APP-positive und -negative Primärkulturen (nicht-tg) ausplattiert. Die Infektion mit HSV-1-eGFP-wt-Tau, -eGFP-PHP-Tau und -eGFP erfolgte nach 5 DIV. 2 Tage nach Infektion wurden die Zellen standardfixiert und eine Immun- bzw. Kernfärbung mit MAP-2 und DAPI durch-geführt. Fragmentierte Zellkerne GFP-positiver Neuronen wurden fluoreszenzmikroskopisch aus-gezählt und prozentual zur Gesamtzahl aller analysierten Neurone berechnet. Dargestellt sind die Durchschnittswerte und die Standardfehler aus fünf unabhängigen Experimenten (eGFP vier Experimente) (Einzelwerte siehe Anhang I.9). Insgesamt wurden 598 wt-Tau-, 586 PHP-Tau- und 358 eGFP-exprimierende Neurone untersucht. Als Signifikanztest wurde der two-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 und **P < 0,01 angewandt. Wt-Tau ist in APP-transgenen Kulturen signifikant toxischer im Vergleich zu Kontrollkulturen.
C.5.2 Protein-biochemische Analyse der Tau-Phosphorylierung in APPSDL-transgenen
Primärkulturen
Aus verschiedenen Veröffentlichungen ist bekannt, dass Aβ eine erhöhte Phosphorylierung
von Tau bewirken kann (Busciglio et al., 1995; Rapoport and Ferreira, 2000). Diese
Ergebnisse unterstützen die Amyloid-Hypothese, die besagt, dass Aβ das erste Glied in der
Kette der AD-Pathologie ist und „downstream“ zur neuronalen Degeneration führt.
93

Ergebnisse
Um festzustellen, ob tatsächlich eine erhöhte Phosphorylierung von wt-Tau in APPSDL-
transgenen Primärkulturen mit dem Anstieg in der Toxizität Taus in Verbindung steht (siehe
Abschnitt C.5.1), wurden Lysate wt-Tau-infizierter Primärkulturen im Immunblot auf die
Phosphorylierung einzelner Serin-Reste untersucht. Da es nicht sicher ist, ob die
Phosphorylierung zeitlich mit der beobachteten Neurodegeneration auf Ebene fragmentierter
Zellkerne (Tag 2 nach der Infektion) korreliert, wurde außerdem ein früherer und ein späterer
Zeitpunkt der Analyse (Tag 1 und Tag 3) gewählt. Die Detektion erfolgte mit dem
phosphorylierungssensitiven Tau-Antikörper PHF-1. Dieser Antikörper bindet spezifisch
phosphorylierte Reste (Serin396/404) in PHF-Tau und wird u.a. histopathologisch zur
Erkennung der Alzheimer-Erkrankung verwendet. Das Epitop des Antikörpers befindet sich
an zwei Hauptphosphorylierungstellen, die in PHP-Tau mutiert sind. In Abb. C.19 ist exem-
plarisch ein Immunblot, detektiert mit PHF-1 gegen die an Serin 396 und Serin 404
phosphorylierte Tau-Population und mit Tau-5 gegen Gesamt-Tau, gezeigt. Verglichen
wurden jeweils wt-Tau-infizierte APPSDL-positive oder negative (nicht-transgene) Primär-
kulturen. In beiden Fällen wird wt-Tau von PHF-1 detektiert, und zwar im Vergleich zur
Gesamt-Tau-Expression (Tau-5) stärker in APP-transgenen Kulturen. Die densitometrische
Bestimmung der Signale von PHF-1 und Tau-5 (Abb. C.20) zeigt, dass zu allen Zeitpunkten
die Phosphorylierung von Tau im transgenen Hintergrund erhöht ist. Besonders deutlich ist
der Unterschied am dritten Tag, an dem die Tau-Phosphorylierung an S396/404 in APP-
transgenen Kulturen etwa doppelt so hoch wie in nicht-transgenen Kulturen ist.
94
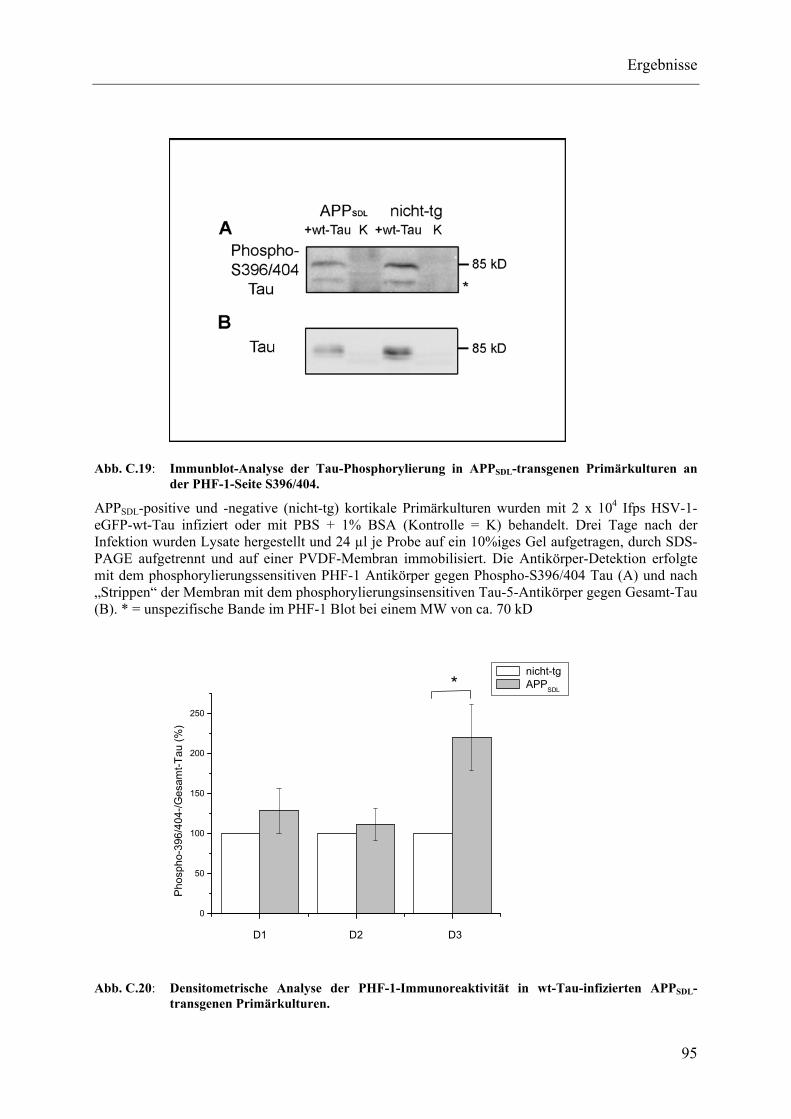
Ergebnisse
Abb. C.19: Immunblot-Analyse der Tau-Phosphorylierung in APPSDL-transgenen Primärkulturen an der PHF-1-Seite S396/404.
APPSDL-positive und -negative (nicht-tg) kortikale Primärkulturen wurden mit 2 x 104 Ifps HSV-1-eGFP-wt-Tau infiziert oder mit PBS + 1% BSA (Kontrolle = K) behandelt. Drei Tage nach der Infektion wurden Lysate hergestellt und 24 µl je Probe auf ein 10%iges Gel aufgetragen, durch SDS-PAGE aufgetrennt und auf einer PVDF-Membran immobilisiert. Die Antikörper-Detektion erfolgte mit dem phosphorylierungssensitiven PHF-1 Antikörper gegen Phospho-S396/404 Tau (A) und nach „Strippen“ der Membran mit dem phosphorylierungsinsensitiven Tau-5-Antikörper gegen Gesamt-Tau (B). * = unspezifische Bande im PHF-1 Blot bei einem MW von ca. 70 kD
0
50
100
150
200
250
Pho
spho
-396
/404
-/Ges
amt-T
au (%
)
nicht-tg APPSDL
D1 D2 D3
*
Abb. C.20: Densitometrische Analyse der PHF-1-Immunoreaktivität in wt-Tau-infizierten APPSDL-transgenen Primärkulturen.
95

Ergebnisse
APPSDL-positive und -negative kortikale Primärkulturen (nicht-tg) wurden mit je 2 x 104 Ifps HSV-1-eGFP-wt-Tau infiziert oder mit PBS + 1% BSA (Kontrolle = K) behandelt. 1, 2 oder 3 Tage (D) nach der Infektion wurden Lysate hergestellt und 24 µl je Probe auf ein 10%iges Gel aufgetragen, durch SDS-PAGE aufgetrennt und auf einer PVDF-Membran immobilisiert. Die Antikörper-Detektion erfolgte zunächst mit Tau-5 (gegen Gesamt-Tau), nach Strippen der Membran mit PHF-1 gegen Phospho-396/404-Tau. Die spezifischen Signale von PHF-1- und Tau-5 wurden densitometrisch mit der Intas Software bestimmt und zueinander in Relation gesetzt (PHF-1/Tau-5). Die Werte der wt-Kultur wurden jeweils auf 100% gesetzt. Danach berechnete sich die relative PHF-1-Intensität in der APP-transgenen Kultur. Im Diagramm sind die Durchschnittswerte und die Standardfehler aus sechs unabhängigen Experimenten an Tag 1, sieben an Tag 2, sowie vier unabhängigen Experimenten an Tag 3 dargestellt (Einzelwerte siehe Anhang I.10). An Tag 3 zeigt sich eine signifikant erhöhte Tau-Phosphorylierung in APPSDL-transgenen- im Vergleich zu wt-Kulturen. Als statistisches Testverfahren wurde der one-tailed student’s paired t-test mit einem Signifikanzgrad *P < 0,05 durchgeführt.
Die Ergebnisse unterstützen somit die Hypothese, dass wt-Tau auf Grund erhöhter
Phosphorylierung an AD-relevanten Hauptphosphorylierungsstellen ähnlich toxisch wird wie
PHP-Tau. Ob die Tau-Phosphorylierung auch an anderen Hauptphosphorylierungsstellen
erhöht ist, könnte durch weitere phosphorylierungssensitive Antikörper getestet werden.
Hierfür würde sich z.B. die Verwendung des AT8-Antikörpers (gegen phosphoryliertes Serin
202/Threonin205) oder des AT180-Antikörpers (gegen phosphoryliertes Threonin 231)
anbieten. Aus Zeitgründen konnten diese Experimente nicht mehr im Rahmen dieser Arbeit
durchgeführt werden.
96

Diskussion
D Diskussion
D.1 Hyperphosphoryliertes Tau ist neurotoxisch
In dieser Arbeit wurde gezeigt, dass die Expression von GFP-markierten Tau-Konstrukten,
die eine AD-ähnliche Hyperphosphorylierung durch Einführen von Glutamat-Mutationen
imitieren, einen zytotoxischen Effekt in NT2-N-Zellen und in kortikalen Primärneuronen
ausüben.
Das verwendete pseudohyperphosphorylierte Tau-Protein (PHP-Tau) simuliert in vitro struk-
turelle und funktionelle Eigenschaften von PHF-Tau in der Alzheimer-Krankheit (Eiden-
müller et al., 2000, 2001; Maas et al., 2000). Durch Transfektion neuraler Zellen wurde
gezeigt, dass im Vergleich zu wt-Tau, PHP-Tau eine reduzierte Mikrotubuli-Binde-Kapazität
besitzt und unfähig ist, Mikrotubuli zu stabilisieren. Zudem wurde gezeigt, dass PHP-Tau
zytotoxisch in transfizierten PC12-Zellen und Virus-infizierten ZNS Modellneuronen ist,
begleitet von der Induktion apoptotischer Mechanismen (Fath et al., 2002). In der Studie von
Fath et al. (2002) wurden Tau-Konstrukte mit einem Flag-Epitop verwendet, deren apopto-
tische Eigenschaften durch Chromatin Kondensation, DNA-Fragmentierung und Aktivierung
von Caspase-3, einem Schlüsselenzym der apoptotischen Kaskade, ermittelt wurden.
Die Fusion von wt-Tau und PHP-Tau mit eGFP ermöglicht die Echtzeitanalyse der Tau-
abhängigen Neurodegeneration. Dazu musste aber zunächst ermittelt werden, ob die eGFP-
Fusionskonstrukte sich ähnlich wie die Flag-Konstrukte verhalten. Die Echtzeitanalyse wurde
in einer Parallelstudie von Welzel (2005) durchgeführt.
Die Ergebnisse der vorliegenden Arbeit zeigen, dass die verwendeten eGFP-PHP-Tau-
Fusionsproteine sich ähnlich toxisch wie Flag-Tau in NT2-N-Zellen verhalten und auch auf
kortikale Primärneuronen der Maus einen zytotoxischen Effekt ausüben. Die Zytotoxizität
PHP-Taus wurde hier in einem ersten Schritt auf Ebene der Zellkernmorphologie
(Kernfragmentierung) HSV-1-infizierter Neuronen ermittelt. In kortikalen Primärkulturen
wurde in einem weiteren Schritt auf Immunblot-Ebene eine erhöhte Caspase-3-Aktivierung
durch die Expression PHP-Taus im Vergleich zu wt-Tau festgestellt.
Im Zusammenhang mit den zuvor veröffentlichten Arbeiten zeigen die Ergebnisse, dass die
Expression PHP-Taus unabhängig von der verwendeten Markierung (Flag-Epitop-Anhang
oder GFP-Fusion) in verschiedenen Zellsystemen neurotoxisch ist, und das unter der
Beteiligung apoptotischer Mechanismen.
97

Diskussion
Apoptose ist ein bedeutender Mechanismus im neuronalen Zelltod während der Entwicklung
und in neurodegenerativen Erkrankungen (für eine Übersicht siehe Mattson, 2000; Nihawan
et al., 2000; Yuan & Yanker, 2000; Eckert et al., 2003). Im mit Alzheimer erkrankten Gehirn
ist ein Verlust von Neuronen besonders im limbischen System und im zerebralen Kortex
ausgeprägt. Aus verschiedenen Studien gibt es Hinweise, dass Apoptose einer der
Mechanismen ist, der zu neuronalem Zelltod in der Alzheimer-Krankheit führt (Forloni et al.,
1993; Loo, 1993). Zum Beispiel konnte in postmortem Gehirngewebe von AD-Patienten
apoptotische DNA-Fragmentierung (Su et al., 1994; Lassmann et al., 1995; Smale et al.,
1995) und ein erhöhtes Level aktivierter Caspase-3 (Gervais, 1999; Su et al., 2001)
festgestellt werden. Caspase-3, -6 und -7 gehören zu den Effektorcaspasen, die durch die
Initiatorcaspasen -9 (mitochondrialer Stress), -4 und -12 (ER-Stress) sowie -2, -8, -10 und -11
(extrinsische apoptotische Stimuli) geschnitten und hierdurch aktiviert werden (für eine
Übersicht über die biochemischen Grundlagen der Apoptose siehe Hengartner, 2000). Es ist
bekannt, dass Caspase-3, aktiviert durch verschiedene apoptotische Stimuli, nötig und
ausreichend ist, Apoptose einzuleiten und wird daher auch als Zelltod-Exekutionsprotease
bezeichnet (Darmon et al., 1996; Datta et al., 1997).
Der relative Beitrag von Apoptose zum neuronalen Zelltod in der Alzheimer-Krankheit ist
unklar. Im mit Alzheimer erkrankten Gehirn exisistieren Neurone, die morphologische
Anzeichen von Apoptose aufweisen, allerdings zeigen auch viele degenerierende Nerven-
zellen keine apoptotischen Merkmale. Nekrose, der nicht programmierte Zelltod, der häufig
zu einer Entzündungsreaktion führt, ist im Gegensatz zur Apoptose u.a. gekennzeichnet durch
ein Anschwellen des Zellkörpers (Majno & Joris, 1995; Mills et al., 1999). In dieser Arbeit
wurde die Gegenwart nekrotischer Mechanismen nicht bestimmt. Allerdings konnten in der
parallel durchgeführten Echtzeitanalyse der Tau-abhängigen neuronalen Degeneration mit den
hier verwendeten GFP-Tau-Konstrukten, PHP-Tau-exprimierende Neuronen beobachtet
werden, die deutliche Anzeichen von Nekrose aufwiesen (Welzel, 2005). Demnach ist
anzunehmen, dass Apoptose nicht den einzigen neurodegenerativen Mechanismus in der
Alzheimer-Krankheit darstellt (Su et al., 1994; Troncoso, 1996).
In der Alzheimer-Krankheit sind die konkreten Auslöser der apoptotischen Kaskade umstrit-
ten, allerdings konnte für das Proteinfragment Aβ, der Hauptkomponente der Senilen Plaques,
in verschiedenen Studien gezeigt werden, dass Aβ Apoptose in kultivierten Zelllinien und
Neuronen induzieren bzw. verstärken kann (Estus et al., 1997; Troy et al., 2000; Mc Phie
et al., 2001). Zudem ist APP ein Substrat der Caspase-3, wodurch die intrazelluläre
98

Diskussion
Konzentration von Aβ erhöht wird (Gervais et al., 1999). Auch für FAD-mutante Preseniline,
die zu einer Erhöhung von Aβ führen, ist bekannt, dass diese Zellen für Apoptose
sensitivieren und apoptotische Kaskaden aktivieren können (Deng et al., 1996; Wolozin et al.,
1996; Vito et al., 1996; Guo et al., 1997; Mattson et al., 2000; Alves da Costa, 2002).
Die Rolle der Tau-Phosphorylierung in der Apoptose ist unklar. Es gibt Ergebnisse, die eine
Dephosphorylierung von Tau in apoptotischen Neuronen während der apoptotischen
Exekutionsphase zeigen (Lesort et al., 1997; Mills et al., 1998). Außerdem ist bekannt, dass
diese Dephosphorylierung entscheidend ist für die nachfolgende Proteolyse von Tau durch
Caspasen und Calpain, wodurch Tau-Fragmente entstehen, die unfähig sind, Mikrotubuli zu
binden (Canu et al., 1998; Rametti et al., 2004). Demgegenüber stehen Studien, die zeigen,
dass Tau während apoptotischen Vorgängen Seiten-spezifisch phosphoryliert wird und dies in
einer erniedrigten Bindung von Tau an Mikrotubuli resultiert (Zhang & Johnson, 2000;
Mookherjee & Johnson, 2001; Shelton & Johnson, 2001).
Die Daten der vorliegenden Arbeit zeigen, konsistent mit der Arbeit von Fath et al. (2002),
dass die Simulation einer Hyperphosphorylierung von Tau apoptotischen Zelltod in Neuronen
induzieren kann.
In vorherigen Analysen (Fath et al., 2002) wurde gezeigt, dass die durch PHP-Tau induzierte
Zytotoxizität nicht im Zusammenhang mit einer Filamentbildung steht, da ultrastrukturell in
degenerierenden PHP-Tau-exprimierenden Neuronen keine auffällige Aggregation in
Filamente nachgewiesen werden konnte. Auch in den hier beschriebenen Experimenten gab
es keine Hinweise auf eine mögliche Tau-Aggregation, wie sie sich in einem partikulären
Verteilungsmuster zeigen würde. Tatsächlich wurde in verschiedenen Veröffentlichungen
gezeigt, dass phosphoryliertes Tau eher die Aggregation in Filamente hemmt oder sich neutral
verhält (Goedert et al., 1996; Schneider et al., 1999; Eidenmüller et al., 2000), was im
Gegensatz zu der Annahme steht, dass die Aggregation von Tau einen primären Effekt auf die
Neurodegeneration hat. Vor kurzem konnte gezeigt werden, dass eine Inhibition des
proteasomalen Weges in kultivierten Oligodendrozyten neben Apoptose zu Thioflavin S-
positiven Tau-Aggregaten und schwerwiegenden Störungen des Mikrotubuli-Netzwerkes
führt (Goldbaum & Richter-Landsberg, 2004). Ein Verlust der proteosomalen Funktion ist
bereits in alternden Tieren beschrieben und könnte in neurodegenerativen Erkrankungen
ebenso eine zentrale Rolle spielen (zur Übersicht siehe Keller et al., 2002). Ob aber die
intrazelluläre Ablagerung von aggregiertem und oft ubiquitinierten Proteinen, wie Tau, Zellen
vor weiteren Schädigungen schützt oder toxisch ist und zu schweren zellulären Dysfunktionen
99

Diskussion
führt, muss weiterhin untersucht werden (für eine Übersicht siehe Layfield et al., 2001; Taylor
et al., 2002). Die Ergebnisse dieser Arbeit im Zusammenhang mit der Studie von Fath et al.
(2002) deuten eher auf eine Art „Rescue“-Mechanismus der neuronalen NFT-Bildung hin.
Auch ist es sehr wahrscheinlich, dass die Funktion von Tau auf die Mikrotubuli-
Polymerisation keine zentrale Rolle in der Neurodegeneration spielt, da gezeigt wurde, dass
PHP-Tau nicht aktiv Mikrotubuli destabilisiert und auch die Behandlung mit dem
Mikrotubuli-stabilisierenden Agenz Taxol in PHP-Tau exprimierenden PC12-Zellen keine
verringerte Toxizität bewirkt (Fath et al., 2002). Die Ergebnisse deuten insgesamt auf einen
„Toxic gain of function“ (pseudo)hyperphosphorylierten Taus hin, und es kann vermutet
werden, dass Konformationsänderungen Tau zu einem neurotoxischen Agenz werden lassen.
Tatsächlich zeigt PHP-Tau genau wie PHF-Tau in der SDS-PAGE eine reduzierte
elekrophoretische Mobilität, was auf das Vorhandensein SDS-resistenter Domänen hindeutet
(Grundke et al., 1986; Eidenmüller et al., 2000). Das Ausmaß der strukturellen Verände-
rungen PHP-Taus korreliert wahrscheinlich eng mit dem neurodegenerativen Effekt. In
Studien mit partiell pseudohyperphosphoryliertem Tau konnte auch eine partielle Toxizität im
Vergleich zu PHP-Tau festgestellt werden. Desweiteren zeigte sich, dass Glutamat-
Mutationen im C-Terminus eine strukturelle Veränderung im Tau-Protein bewirken und auch
neurotoxisch sind, sich aber nicht auf Mikrotubuli-bezogene Eigenschaften auswirken
(Eidenmüller et al., 2001). Die Ergebnisse stehen im Einklang damit, dass familäre FTDP-17-
Mutationen im Tau-Protein, die ebenso zu Tau-Aggregaten und neuronalem Zelltod führen,
strukturelle Veränderungen im Tau-Protein bewirken können (Jichia et al., 1999a).
D.2 Aβ führt zur Neurodegeneration über die Phosphorylierung von Tau
(Amyloid-Hypothese)
Verschiedene Studien haben gezeigt, dass Aβ Apoptose in kultivierten Zelllinien und
Neuronen induzieren oder verstärken kann (Estus et al., 1997; Troy et al., 2000; Mc Phie
et al., 2001). Aβ kann die intrazelluläre Ca2+-Konzentration erhöhen, oxidativen Stress indu-
zieren und hierdurch neurotoxisch wirken (Mattson et al.,1993; Behl et al., 1999). Es wird
vermutet, dass Aβ die apoptotische Kaskade anschaltet, z.B. durch die Interaktion mit
neuronalen Rezeptoren, wie dem RAGE-Rezeptor (Receptor for Advanced Glycation End
Products), der eine Produktion von freien Radikalen vermittelt (Yan et al., 1997), sowie dem
100

Diskussion
p75-Neurotrophin Rezeptor, für den bekannt ist, dass er nach Aktivierung den Neuronentod
induzieren kann (Yaar et al., 1997).
Die Amyloid-Hypothese (Hardy & Higgins, 1992; Yankner, 1996) besagt, dass abnormales
Prozessieren von APP und die dadurch erhöhte Produktion, Aggregation und Ablagerung von
Aβ primär in der AD-Pathologie sind. Nach dieser Hypothese sollen die Aggregate von Aβ
dann als neurotoxisches Agenz zur Hyperphosphorylierung von Tau, der Bildung von NFTs,
sowie zur synaptischen Degeneration und schließlich zum Neuronentod führen. Ergebnisse
aus Tiermodellen unterstützen die Amyloid-Hypothese. In zwei FAD-mutanten APP-
Mausmodellen konnte ein Verlust von Neuronen nachgewiesen werden. Hier führte die
Überexpression des FAD-mutanten APP-Gens zu massiven Amyloid-Ablagerungen im
Neokortex und Hippokampus, begleitet von hyperphosphoryliertem Tau und CA1-neuro-
nalem Zelltod (Sturchler-Pierrat et al., 1997; Calhoun et al., 1998).Weitere AD-ähnliche
Merkmale in den Gehirnen dieser Mäuse waren dystrophische Neuriten und Gliose (Sturchler-
Pierrat et al., 1997). Allerdings gab es auch Studien, die keinen Neuronentod in FAD-
mutanten transgenen Mäusen nachweisen konnten (La Ferla, 1995; Irizarry et al.,1997a, b;
Holocomb et al., 1999).
In der hier vorliegenden Arbeit wurde gezeigt, dass die Toxizität wt-Taus in APPSDL-
transgenen Primärkulturen im Vergleich zu nicht-transgenen Primärkulturen erhöht ist. Das
Ausmaß der Degeneration war ähnlich der Toxizität PHP-Taus. Diese war in APPSDL-
transgenen Neuronen nicht erhöht. Durch ELISA-Tests wurde im Vergleich zu nicht-
transgenen Primärkulturen eine erhöhte Produktion von Aβ im Medium nachgewiesen, was
mit Daten aus der Literatur übereinstimmt (Suzuki et al., 1994; Rogeav et al., 1995;
Sherrington et al., 1995; Duff et al., 1996; Scheuner et al., 1996; Citron et al., 1997).
Da aus anderen Studien bereits bekannt ist, dass Aβ den Phosphorylierungszustand von Tau
erhöhen kann (Busciglio et al., 1995; Greenberg et al., 1995; Takashima et al., 1998a;
Rapoport & Ferreira, 2000), könnte eine Hyperphosphorylierung die Verbindung zwischen
der Zytotoxizität von Aβ und der Neurodegeneration darstellen. Diese Hypothese wird
unterstützt durch die Beobachtung, dass Aβ eine Tau-Phosphorylierung mit nachfolgendem
Verlust von cholinergen Neuronen induziert (Zheng et al., 2002). Zudem konnte von
Rapoport et al. (2002) durch Verwendung von Tau-„knock-out“-Neuronen gezeigt werden,
dass Tau essentiell für die Aβ-induzierte Neurotoxizität ist. In Übereinstimmung wurde
gezeigt, dass die Aβ-induzierte Phosphorylierung von Tau und die Apoptose durch Inhibition
der Kinase Cdk5, die eine Tau-Phosphorylierung induzieren kann, verhindert wird. Ähnliche
101

Diskussion
Ergebnisse wurden zuvor mit Lithium, einem Inhibitor der Tau-Kinasen GSK-3α und β,
erzielt (Alvarez et al., 1999). In nanomolaren Konzentrationen wirkt Aβ auf undifferenzierte
hippokampale Neuronen, die eine niedrige Tau-Expression aufweisen, neurotroph bzw.
antiapoptotisch (Whitson et al., 1989; Yankner et al., 1990; Kaltschmidt et al., 1999; Liu et
al., 2004), in reifen, differenzierten Neuronen dagegen führt es in höheren Konzentrationen,
zur Apoptose. Dieses ambivalente Verhalten Aβs wird dadurch begründet, dass es während
einer Differenzierung von 4-8 Tagen in vitro zu einem Anstieg der Expression von Tau und
Cdk5, sowie zu einem erhöhten Niveau der Tau-Phosphorylierung kommt. Die Tau-
Phosphorylierung wurde hierbei zum selben Zeitpunkt wie die Zytotoxizität durch Aβ
beobachtet und die Verwendung von Tau-Antisense-Nukleotiden blockierte die Aβ-induzierte
Neurotoxizität (Liu et al., 2004). In dieser Arbeit wurde eine erhöhte wt-Tau Toxizität in
APPSDL-transgenen Primärkulturen beobachtet. Durch ELISA-Tests wurde eine sehr geringe
nanomolare Konzentration von Aβ im Medium nachgewiesen. Es ist möglich, dass
intrazelluläres Aβ an der erhöhten Toxizität wt-Taus beteiligt ist. Aus verschiedenen Studien
gibt es Hinweise, dass intrazelluläres Aβ in den neuronalen Zelltod involviert ist (La Ferla,
1995; Chui et al., 1999).
Die Daten in der Literatur könnten darauf hindeuten, dass der in dieser Arbeit beobachtete
Aβ-induzierte Anstieg der Toxizität wt-Taus über eine Aβ-induzierte Phosphorylierung von
Tau ausgelöst wird. In nachfolgenden Experimenten konnte dies unterstützt werden.
Tatsächlich zeigte sich eine signifikant erhöhte Phosphorylierung am AD-relevanten PHF-1-
Epitop, das in der pseudophosphorylierten Mutante modifiziert wurde. Demnach sind die
Ergebnisse der vorliegenden Arbeit konsistent mit der Amyloid-Hypothese.
Verschiedene Kinasen sind an der Phosphorylierung dieses Epitops beteiligt. In vitro konnte
gezeigt werden, dass diese Stelle durch Cdk2 (Baumann et al., 1993), Cdk5, Cdc2, MAPK,
GSK-3β (Illenberger et al., 1998; Godemann et al., 1999), GSK-3α, Casein kinase I und II
(Billingsley et al., 1997) sowie durch die JNK und p38 (Reynolds et al., 2000) phosphoryliert
wird. In COS- und SH-SY5Y-Zellen ist eine Phosphorylierung durch GSK-3β und SAPK
beschrieben (Michel et al., 1998; Xie et al., 1998; Buée-Scherrer et al., 2002). Eine
Phosphorylierung durch Cdk5/p25 aber nicht durch Cdk5/p35 wurde zudem in COS-Zellen
und transgenen Mäusen beschrieben (Michel et al., 1998; Van den Haute et al., 2001). In
Gehirnschnitten der Ratte wurde festgestellt, dass die Phosphatasen PP2B (Gong et al., 1994a)
und A (Gong et al., 2000) Tau an dieser Stelle dephosphorylieren.
102

Diskussion
Eine Aβ-induzierte Tau-Phosphorylierung am PHF-1-Epitop ist in verschiedenen zellulären
Ansätzen beschrieben. In der Arbeit von Busciglio & Yanker (1995) wurde durch Zusatz
fibrillären Aβ, im Gegensatz zu löslichem oder amorph aggregiertem Aβ, eine PHF-1-
Phosphorylierung in fötalen Ratten-hippokampalen bzw. humanen kortikalen Neuronen
nachgewiesen. Es wurde gezeigt, dass hyperphosphoryliertes Tau im somatodendritischen
Kompartiment akkumuliert und keine Assoziation mit Mikrotubuli aufweist. In vitro zeigte
sich, dass dieses Tau unfähig ist, Mikrotubuli zu binden und dieser Effekt durch eine
Dephosphorylierung von Tau aufgehoben werden konnte. In mutanten APPswe Mäusen
akkumulierte PHF-1-positives Tau in dystrophischen Neuriten, welche von Amyloid-Plaques
umgeben waren (Tomidokoro et al., 2001). Auch in transgenen Ratten, welche keine
Amyloid-Plaques aufweisen, aber intrazelluläres Aβ akkumulieren und Beeinträchtigungen
der kognitiven Fähigkeiten zeigen, konnte eine erhöhte Tau-Phosphorylierung am PHF-1-
Epitop nachgewiesen werden. Die erhöhte Tau-Phosphorylierung war begleitet von einer
erhöhten Aktivität der ERK-2 (Extracellular Regulated Kinase-2), was ein Hinweis darauf ist,
dass der MAP-Kinase-Signalweg in die Aβ-induzierte Tau-Phosphorylierung involviert sein
könnte. Im Gegensatz dazu zeigten sich keine Änderungen in der Aktivität der Kinasen p38,
GSK-3β und Cdk-5 (Echiverra et al., 2004). Allerdings sind die Signalwege und Kinasen,
durch die Aβ eine erhöhte Tau-Phosphorylierung auslöst, umstritten. So konnte in einer
Studie von Alvarez et al. (1999) gezeigt werden, dass Lithium, ein GSK-3-Inhibitor,
Neuronen gegen eine Aβ-induzierte Neurodegeneration schützt. Hierbei wurde außerdem eine
reduzierte Phosphorylierung von Tau am PHF-1-Epitop festgestellt. Auch Takashima et al.
(1998a) konnten zuvor in hippokampalen Neuronen nachweisen, dass eine erhöhte Tau-
Phosphorylierung am PHF-1-Epitpop und anderen AD-relevanten Stellen durch die
Behandlung mit dem Aβ-Fragment Aβ25-35 in einem direkten Zusammenhang mit einer
gesteigerten Aktivität von GSK-3β steht. In diesem Ansatz konnte aber keine erhöhte
Aktivierung von GSK-3α oder der MAP-Kinase (Mitogen aktivierte Protein Kinase) ermittelt
werden. Im Gegensatz dazu steht eine kürzlich veröffentlichte Studie von Ghribi et al.
(2003a), in der im Ratten-Hippokampus Lithium zwar Aβ-induzierten ER-Stress inhibierte
aber weder die oxidativen Zellschäden noch die Tau-Phosphorylierung verhinderte. Die
unterschiedlichen Ergebnisse deuten daraufhin, dass möglicherweise verschiedene
Signalwege beteiligt sind und komplex zusammenspielen.
Ebenso sind weitere Aβ-induzierte AD-relevante Tau-Phosphorylierungsstellen bekannt.
Neben der PHF-1-Phosphorylierungsstelle ist in zellulären oder transgenen Tiermodellen z.B.
103

Diskussion
eine Aβ-induzierte Phosphorylierung am AT8-Epitop (den Aminosäure-Resten Serin202 und
Threonin205) beschrieben (Rapoport & Ferreira, 2000; Stein et al., 2004; Otth et al., 2005;
Zheng et al., 2005). Ob eine erhöhte AT8-Reaktivität auch in den hier verwendeten APP-
transgenen Primärkulturen vorliegt, sollte in weiteren Experimenten überprüft werden.
Auch aus den Ergebnissen der oben erwähnten Studien (Rapoport & Ferreira, 2000; Stein et
al., 2004; Otth et al., 2005; Zheng et al., 2005) sind die beteiligten Signalwege unklar.
Rapoport & Ferreira (2000) konnten durch PD98059, einen Inhibitor der MAPK-Kinase
MEK1, eine Reduktion der Tau-Phosphorylierung durch fibrilläres Aβ nachweisen. Deswei-
teren wurde durch PD98059 die durch Aβ ausgelöste Degeneration reifer hippokampaler
Neurone verhindert.
Auch die erhöhte Tau-Phosphorylierung durch die Kinase Cdk5 scheint eine wichtige Rolle in
der Aβ-induzierten Tau-Phosphorylierung zu spielen. Unter Verwendung eines Cdk5-
Inhibitorpeptids wurde eine Reduktion der Tau-Phosphorylierung an Serin199/202, Serin404
sowie am AT8-Epitop und ein antiapoptotischer Effekt in Aβ-behandelten kortikalen
Neuronen gezeigt (Zheng et al., 2005). Zudem wurde in APPswe (Tg2576) Mäusen eine Cdk-5
abhängige Phosphorylierung am AT8- und PHF-1-Epitop nachgewiesen (Otth et al., 2002).
Die Rolle von Aβ bzw. der Amyloid-Plaques bei der Bildung von NFTs ist nach wie vor
ungeklärt. In den Mausmodellen von Sturchler-Pierrat et al. (1997) und Calhoun et al. (1998)
kam es neben Amyloid-Plaques zwar zu hyperphosphoryliertem Tau und Neuronentod, aber
nicht zur NFT-Bildung. Ebenso konnte durch intrazelluläre Aβ-Expression in transgenen
Ratten, die keine Amyloid-Plaques entwickelten, zwar eine erhöhte Tau-Phosphorylierung
aber keine NFT-Entwicklung nachgewiesen werden (Echeverria et al., 2004). In FTDP-17-
Tau transgenen Mausmodellen, die eine Bildung von NFTs aufweisen, wurde dagegen
festgestellt, dass entweder die Expression von mutantem APP oder die Injektion von Aβ die
NFT-Bildung verstärkt (Götz et al., 2001; Lewis et al., 2001). Desweiteren konnten, im
Gegensatz zu einer früheren Studie von Xu et al. (2002), in doppelt-transgenen FAD-APP/-
PS1 Mäusen neben hyperphosphoryliertem Tau PHF-ähnliche Filamente nachgewiesen
werden (Kurt et al., 2003). Jedoch scheint Aβ in vivo nicht ausreichend in Entwicklung einer
NFT-Pathologie zu sein, da eine Injektion von Aβ in das Gehirn wt-Tau-überexprimierender
Mäuse nicht zur Bildung von NFTs führte (Götz et al., 2001). In vitro dagegen konnte eine
Aβ-induzierte Aggregation in Tau-Filamente beobachtet werden (Rank et al., 2002).
104

Diskussion
Ein kritischer Aspekt der Amyloid-Hypothese ist, dass die senilen Amyloid-Plaques im
Gehirn von AD-Patienten im geringsten Ausmaß in der Amygdala, dem Hippokampus und
dem Gyrus parahippocampalis konzentriert sind. Das Vorkommen der NFTs tritt dagegen im
limbischen System verstärkt auf und korreliert eng mit den Orten der neuronalen
Degeneration (Braak et al., 1993). Zudem konnte, wie bereits oben erwähnt, nicht in jedem
FAD-APP-Tiermodell neuronaler Zelltod nachgewiesen werden. Es wird diskutiert, dass
möglicherweise intrazelluläres Aβ zum Absterben von Neuronen führt. La Ferla et al. (1995)
konnten zeigen, dass in transgenen Mäusen, die intrazelluläres Aβ exprimieren, neuronale
Degeneration auftritt. Dies wird unterstützt durch Studien mit anderen FAD transgenen
Mausmodellen, die Aβ intraneuronal akkumulieren ohne dass es zur Bildung extrazellulärer
Plaques kommt. Interessanterweise konnte in den Gehirnen dieser Tiere eine Hyperphos-
phorylierung von Tau (Echeverria et al., 2004) und ein Absterben von Neuronen nach-
gewiesen werden (Chui et al., 1999). Daraus könnte man schließen, dass die Bildung von
extrazellulären Amyloid-Plaques nicht notwendig ist, eine Hyperphosphorylierung von Tau
und eine damit verknüpfte Degeneration auszulösen, sondern dass dies durch intrazelluläres
Aβ erfolgt.
Dass pathologisch verändertes Tau ohne eine Überproduktion von Aβ oder das Auftreten von
Amyloid-Plaques neurotoxisch sein kann, wird auch durch andere neurodegenerative
Erkrankungen, insbesondere FTDP-17, unterstützt. Hier führen Mutationen im Tau-Gen zur
Hyperphosphorylierung von Tau, NFT-Bildung und neuronaler Degeneration. Dies lässt den
Schluß zu, dass in anderen Tauopathien, bei denen es nicht zu einer erhöhten Aβ-Produktion
kommt, auch andere Mechanismen zur Tau-Hyperphosphorylierung und Degeneration führen
können.
Es kann gegenwärtig auch nicht ausgeschlossen werden, dass bei der Alzheimer-Krankheit
auch weitere durch Aβ ausgelöste zelluläre Reaktionen eine zentrale Rolle im neuronalen
Zelltod spielen. So konnte z.B. in einer Studie von Ekinci & Shea (2000) nach Aβ25-35-
Behandlung kortikaler Maus-Neuronen zwar eine erhöhte Tau-Phosphorylierung am AT270-
Epitop und massive neuronale Degeneration festgestellt werden, erstaunlicherweise enthielten
aber die meisten degenerierenden Neuronen weniger phosphoryliertes Tau als die nicht
dgenerierenden Neuronen. Dagegen fand man aber in diesen Zellen eine erhöhte Produktion
reaktiver Sauerstoff-Spezies.
105

Diskussion
D.3 Presenilin 1 wt wirkt antiapoptotisch auf die Toxizität
hyperphosphorylierten Taus
In dieser Arbeit wurden neben der Analyse des funktionellen Zusammenhangs zwischen Tau
und Aβ, PS1 wt- und FAD mutante PS1-transgene Primärkulturen verwendet und ein Einfluss
auf die Zytotoxizität pseudophosphorylierten Taus untersucht.
Es konnte gezeigt werden, dass die Expression von wt-Presenilin 1, im Gegensatz zu FAD-
PS1(M146L), den neurotoxischen Effekt PHP-Taus signifikant reduziert. Dies wurde durch
Analyse der Tau-vermittelten Zellkernfragmentierung in HSV-1 infizierten Neuronen
bestimmt. Weitergehend wurde auf Immunblot-Ebene gezeigt, dass in HSV-1-PHP-Tau
infizierten PS1wt transgenen Primärkulturen die Caspase-3-Aktivierung reduziert. Zum einen
bestätigt dieses Ergebnis, dass der zytotoxische Effekt PHP-Taus von Apoptose begleitet ist,
zum anderen steht dieses Ergebnis im engen Zusammenhang mit Daten aus der Literatur, die
Presenilin 1 als antiapoptotisches Protein beschreiben (Burztajn et al., 1998; Terro et al.,
2002; Baki et al., 2004). Für FAD-mutantes PS1 gilt dies nicht, hier wurde in verschiedenen
Veröffentlichungen eine pro-apoptotische Rolle beschrieben (Kovacs et al., 1999; Weihl et
al., 1999; Vestling et al., 2001; Terro et al., 2002; Leroy et al., 2003).
Insgesamt gesehen sprechen die Veröffentlichungen für eine kritische Rolle von PS1 im
Zellüberleben. Dies wird dadurch unterstützt, dass die Gegenwart von PS1 in der Entwicklung
essentiell ist. PS1 Null-Embryonen sterben kurz nach der Geburt und zeigen erhöhten
neuronalen Zelltod und schwere Missbildungen (Shen et al., 1997). Lange Zeit war unklar,
wie PS1 die Entwicklung und das Zellüberleben begünstigt (Hong et al., 1999) und ob diese
Funktionen von PS1 in Relation stehen zu den Mechanismen, durch die PS1 FAD-Mutationen
zu einer erhöhten Tau-Phosphorylierung und Zelltod führen (Irving & Miller, 1997;
Takashima et al., 1998a; Pigino et al., 2001). In einer kürzlich veröffentlichten Studie von
Baki et al. (2004) konnte jedoch eine entscheidende Beteiligung des PI3K/Akt-Signalweges
nachgewiesen werden.
Die Phosphatidylinositol 3-Kinase (PI3K) hat in verschiedenen Zelltypen inklusive Neuronen
entscheidende Funktionen im Zellüberleben (für eine Übersicht siehe Chan et al., 1999;
Brunet et al., 2001). PI3K mediiert die Übertragung von extrinsischen Signalen wie
Wachstumsfaktoren, Insulin und Cadherin homophilen Zell-Zellinteraktionen (Brunet et al.,
2001). Durch PIK3-Phosphorylierung der Effektorkinase Akt, wird diese aktiviert und phos-
phoryliert wiederum eine große Anzahl an Substraten, wobei anti-apoptotische Signale
106

Diskussion
akiviert und pro-apoptotische Signale inaktiviert werden. Zum Beispiel inaktiviert Akt GSK-
3β und -α durch die Phosphorylierung an Serin21 und Serin9 (Cross et al., 1995; Kaytor &
Orr, 2002). Aktivierter GSK-3 (Tyr216 phosphoryliert) wurde eine Funktion im neuronalen
Zelltod (Pap & Cooper, 1998; Hetman et al., 2000; Cross et al., 2001; Lucas et al., 2001), in
der Tau-Hyperphosphosphorylierung (Hanger et al., 1992; Hong et al., 1997; Pei et al., 1999;
Lucas et al., 2001) und in der γ-Sekretase-unabhängigen-Produktion von Aβ (Phiel et al.,
2003) zugeschrieben.
In der Studie von Baki et al. (2004) wurde gezeigt, dass PS1, unabhängig von seiner γ-
Sekretase-Aktivität, durch Stimulierung von PI3K/Akt die Aktivität der Tau-Kinase GSK-3
reduziert, dadurch die Tau-Phosphorylierung an AD-relevanten Seiten hemmt und zudem
Apoptose verhindert. Im Gegensatz dazu inhibierten PS1-FAD Mutationen diesen Signalweg.
Die das Zellüberleben sichernden Funktionen von PS1, wie die Hemmung der GSK-3-
Aktivität sowie die hierdurch erniedrigte Tau-Phosphorylierung wurden unterdrückt. Aus
diesen Gründen ist es möglich, dass es sich bei FAD-mutantem Presenilin 1 um einen „Loss
of function“-Mechanismus unabhängig von dem „Gain of function“ in der γ-Sekretase-
Spaltung von APP handelt.
Die Ergebnisse dieser Arbeit weisen darauf hin, dass Presenilin 1 seinen antiapoptotischen
Effekt ausübt, wenn Tau bereits (pseudo)phosphoryliert ist. Dies ist kein Widerspruch zu der
oben genannten Studie von Baki et al. (2004). Es würde lediglich bedeuten, dass neben der
Hemmung der Tau-Phosphorylierung andere antiapoptotische Mechanismen aktiviert werden.
Alternativ könnten auch Phosphorylierungsereignisse an anderen als den mutierten Stellen
eine Rolle spielen, da in unserem Labor gezeigt wurde, dass die PHP-Mutationen andere
Phosphorylierungen „primen“ können (Shahani & Brandt, unveröffentlichte Ergebnisse).
Kürzlich wurde beschrieben, dass PS1 Cadherin bindet und Cadherin-Cadherin-Zell-Zell-
Adhäsions-Interaktionen begünstigt (Goergakopoulos et al., 1999; Baki et al., 2001).
Weiterhin ist bekannt, dass es durch Ca2+-Einstrom zur PS1/γ-Sekretase-Spaltung von
Cadherin kommt und dies in der Produktion von Gen-Expression-Signalen resultiert
(Marambaud et al., 2002, 2003). Hierbei könnte es sich um die Expression antiapoptotischer
Proteine handeln. Aber auch die Hemmung der GSK-3β-Aktivität könnte neben einem
Einfluß auf die Tau-Phosphorylierung in diesem Zusammenhang wichtig sein. Generell
scheint GSK-3β eine entscheidende Rolle bei der Neurodegeneration in AD zu spielen (für
eine Übersicht siehe Alvarez et al., 2002). Neben einer Erhöhung der Tau-Phosphorylierung
ist bekannt, dass GSK-3β β-Catenin phosphoryliert und dadurch für den proteasomalen Abbau
107

Diskussion
markiert, wodurch das zytosolische Level und die nukleare Translokation von β-Catenin
verringert werden (Ding et al., 2000; Patapoutian & Reichardet, 2000). β-Catenin ist ein
multifunktionales Protein, welches durch eine spezifische Gen-Expression verschiedener
Proteine insbesondere antiapoptotische Funktionen induziert (Zhang et al., 1998; Weihl et al.,
1999; Patapoutian & Reichardet, 2000). Es wäre also möglich, dass in den hier verwendeten
transgenen Primärkulturen PS1 durch β-Catenin und folglich durch die Expression anti-
apoptotischer Gene der Toxizität PHP-Taus entgegenwirkt.
Aktive GSK-3β ist an der Regulierung vieler verschiedener Prozesse in der Zelle beteiligt,
z.B. wurde eine kritische Rolle von GSK-3β in der Entwicklung der neuronalen Polarität
beschrieben (Jiang et al., 2005; Yoshimura et al., 2005). Interessanterweise reduzierte sich in
4-„Repeat“-hTau/GSK-3β doppelt transgenen Mäusen die Beeinträchtigung des axonalen
Systems und erleichterte die motorischen Dysfunktionen, welche in transgenen 4-„Repeat“-
hTau-Mäusen vorkommen (Spittaels et al., 1999; Spittaels et al., 2000). Dies steht im
Gegensatz zu der Annahme, dass GSK-3β eine zentrale Rolle in der Neurodegeneration bei
AD spielt.
D.4 Die Mutation PS1(M146L) vermittelt keinen anti-degenerativen
Effekt auf die Toxizität hyperphosphorylierten Taus
In dieser Arbeit wurde durch Bestimmung des Anteils fragmentierter Zellkerne gezeigt, dass
die Expression von PS1 FAD-transgenen M146L Primärkulturen keinen anti-degenerativen
Effekt auf die Neurotoxizität PHP-Taus ausübt. In wt-Tau und PHP-Tau infizierten
PS1(M146L)-Kulturen wurde ein Anstieg in der Tau-induzierten Zytotoxizität beobachtet.
Es ist bekannt, dass PS1 FAD Mutationen in apoptotische Vorgänge involviert sind. Zum
einen wurde beschrieben, dass mutantes PS1 Zellen gegenüber verschiedenen apoptotischen
Stimuli wie Staurosporin oder Aβ sensitiviert. Zum anderen haben in vitro-Experimente
gezeigt, dass überexprimierte und physiologisch exprimierte PS1 FAD Mutanten Apoptose
einleiten können (Kovacs et al., 1999; Weihl et al., 1999; Terro et al., 2002; Leroy et al.,
2003) und die Aktivität von Akt, einer im Zellüberleben involvierten Kinase, verringern
können (Weihl et al., 1999; Vestling et al., 2001; Baki et al., 2004).
Die hier beobachtete Erhöhung der Toxizität von wt- und PHP-Tau in PS1(M146L)-
transgenen Primärkulturen könnte durch zwei unterschiedliche Vorgänge bewirkt werden.
108

Diskussion
Entweder könnte sie abhängig oder unabhängig von einer erhöhten Tau-Phosphorylierung
sein, zudem könnte sie direkt oder indirekt über eine erhöhte Aβ42 Produktion erfolgen.
Es ist möglich, dass der Effekt der PS1 Mutation Neuronen generell gegenüber einer Tau-
Expression sensitiviert. Hierbei könnte durch die Überproduktion des endogenen
neurotoxischen Aβ42 z.B. oxidativer Zellstress (Behl et al., 1994; Schubert et al.,1995; Behl
et al.,1999) oder die Destabilisierung der Ca2+-Homöostase (Mattson et al., 1992) zum
erhöhten Neuronentod beitragen. Auch für PS1 FAD-Mutanten konnte gezeigt werden, dass
deren Überexpression zu Störungen in Ca2+-mediierten Signalwegen führt. Zum Beispiel ist
bekannt, dass FAD mutantes PS1 den IP3-regulierten Ausstrom von Ca2+ drastisch erhöht (Ito
et al., 1994; Gibson et al., 1996; Guo et al., 1996) und zur Inhibition des kapazitativen Ca2+-
Einstroms führt (Leissering et al., 2000). Insgesamt gesehen würde dies in einer reduzierten
Ca2+-Speicherung im ER resultieren. Störungen in der ER-Ca2+-Homöostase können einen
Effekt auf die Genexpression verschiedener Proteine haben, und es wird angenommen, dass
solche Deregulationen in einer Vielzahl neurodegenerativer Erkrankungen wie auch
Alzheimer vorkommen (Mattson et al., 2000b). Nicht auszuschließen ist auch eine Fehl-
regulation in der Transkription von z.B. anti-apoptotischen Proteinen in Relation zu dem in
Abschnitt D.3 genannten „Loss of function“ im GSK-3β/Catenin-Signalweg. Allerdings wird
in den hier verwendeten PS1 mutanten Primärkulturen noch endogenes funktionelles PS1 wt
exprimiert, so dass ein „Loss of function“ von PS1(M146L) in diesem System eher
unwahrscheinlich ist.
Die erhöhte Toxizität wt- und PHP-Taus in PS1 mutanten Primärkulturen könnte auch durch
eine Tau-vermittelte Sensitivierung der Zellen erklärt werden. Betrachtet man die Möglich-
keit, dass eine gesteigerte Tau-Phosphorylierung zu dem erhöhten neurotoxischen Effekt
führt, ist unklar, ob es sich hierbei tatsächlich um eine Phosphorylierung an den Haupt-
phosphorylierungsstellen handelt, da die Toxizität von PHP-Tau auch erhöht war. Dies müsste
durch weitere Experimente geklärt werden, bis jetzt kam es zu keinem eindeutigen Ergebnis.
Außerdem ist nicht eindeutig geklärt, ob eine eventuell erhöhte Tau-Phosphorylierung direkt
durch die Presenilin-1 Mutation oder indirekt über Aβ vermittelt wird. Wie bereits in
Abschnitt D.2 ausführlich beschrieben, kann Aβ GSK-3β und andere Kinasen aktivieren, was
schließlich zur Neurodegeneration durch hyperphosphoryliertes Tau führen könnte (Taka-
shima et al., 1996; Takashima et al., 1998; Alvarez et al., 1999). In einer kürzlich erschienen
Veröffentlichung wurde gezeigt, dass PS1 Mutationen die Aβ-induzierte Tau-Phosphory-
lierung steigern können (Piginio et al., 2001). Aus früheren Studien ist bekannt, dass PS1
109

Diskussion
direkt mit GSK-3β und Tau interagiert, und es konnte gezeigt werden, dass Mutationen
(C263R, P264L), die in der Tau-Binderegion (250-298) liegen, eine verstärkte PS1-Bindung
an GSK-3β und eine erhöhte Tau-Phosphorylierung induzieren (Takashima et al., 1998b). Da
die PS1-Interaktionsdomäne von GSK-3β und Tau im selben Bereich liegt, wird vermutet,
dass PS1 sich wie ein Ankerprotein verhält, welches Tau und GSK-3β für eine
Phosphorylierungsreaktion nahe zueinander bringt. Allerdings gibt es widerstreitende
Ergebnisse in Bezug auf die FAD-PS1-induzierte Tau-Phosphorylierung. So konnte z.B. in
Studien, die einen Einfluß von PS1 Mutationen untersuchten, die nicht in der Tau-Binde-
region (I143T, M146L, G384A) liegen, keine erhöhte Tau-Phosphorylierung nachgewiesen
werden (Irving & Miller, 1997; Julliams et al., 1999). Desweiteren erniedrigte die Mutation
L392V die Bindung von PS1 an GSK-3β (Gantier et al., 2000). Es wird diskutiert, dass
eventuell die einzelnen Mutationen im Bezug auf die Tau-Phosphorylierung unterschiedliche
Funktionen haben könnten. Dies erscheint möglich, da Mutationen in unterschiedlichen
funktionellen Domänen Presenilins auftreten. So könnte z.B. argumentiert werden, dass nur
Mutationen, die in der Tau- bzw. GSK-3β-Binderegion Presenilins liegen, einen direkten
Einfluß auf den Phosphorylierungszustand von Tau haben. Allerdings gibt es auch Studien,
die dies widerlegen (Pigino et al., 2001; Boutajangout et al., 2002; Baki et al., 2004). So
konnte z.B. in einer Tau/PS1(M146L) doppelt transgenen Maus (Mutation nicht in der
Tau/GSK-3β-Binderegion) eine erhöhte Tau-Phosphorylierung nachgewiesen werden
(Boutajangout et al., 2002).
Die Studien von Irving & Miller (1997), Boutajangout et al. (2002) und Baki et al. (2004)
zeigen, dass es unterschiedliche Ergebnisse der in dieser Arbeit verwendeten PS1 Mutation
M146L in Bezug auf eine erhöhte Tau-Phosphorylierung gibt.
Möglicherweise sind generell die verschiedenen Zellsysteme und Versuchsansätze entschei-
dend. Die oben erwähnten Studien (Irving & Miller, 1997; Takashima et al., 1998b) wurden
mittels Co-Transfektionen in nicht-neuronalen Zellen (COS-7) durchgeführt. Es ist möglich,
dass diese Ergebnissse in kultivierten Zellen nicht direkt mit Presenilin-Mutationen im
Tiermodell vergleichbar sind, da ebenso ein indirekter Effekt auf die Phosphorylierung durch
das Auftreten von Amyloid-Plaques vorliegen kann (Boutajangout et al., 2002). Eventuell
spielt auch die Höhe der Expression eine Rolle. In der Studie von Baki et al. (2004), in der
eine erhöhte Tau-Phosphorylierung durch die PS1 Mutationen M146L, A246E und E280A
festgestellt werden konnte, wurde PS1 Virus-vermittelt überexprimiert. Es handelt sich also
um eine stärkere PS1-Expression als bei einer transienten Transfektion. Das sehr hohe
110

Diskussion
Expressionlevel der PS1 Mutanten könnte hier also eine Tau-Phosphorylierung begünstigen.
Eventuell spielen alle Faktoren zusammen, und es sind weitere Versuchsmodelle notwendig,
diese unterschiedlichen Ergebnisse zu erklären.
D.5 Eine integrierte Sicht
Unterschiedliche Mechanismen und Signalwege können in der Pathologie der Alzheimer-
Krankheit eine Rolle spielen. Um diesen komplexen Zusammenhang zu verdeutlichen, sind
einige Schlüsselbefunde in einer schematischen Abbildung (Abb. D.1) zusammengefasst.
Gemäß der Amyloid-Hypothese ist die erhöhte Produktion von Aβ primär. Diese Über-
produktion wird in familiären Formen durch mutantes APP oder PS1 begünstigt. Aβ führt
durch die Aktivierung verschiedener Kinasen, insbesondere GSK3β, Cdk5 und MAPK, zu
hyperphosphoryliertem Tau, welches schließlich zur Neurodegeneration führt. Auch Aβ-
induzierter oxidativer Zellstress durch die Produktion Reaktiver Oxygen Spezies (ROS) kann
direkt zum Neuronentod beitragen. Der Kinase GSK-3β kommt möglicherweise eine zentrale
Rolle zu. Neben der Aβ-induzierten Phosphorylierung von Tau ist sie durch die Induktion des
Abbaus von β-Catenin entscheidend an der Genregulation beteiligt. Funktionelles PS1 wt
hemmt GSK-3β und dadurch den Abbau von β-Catenin. Es kommt zu der Transkription anti-
apoptotischer Proteine und zu einer Inhibition der Neurodegeneration. Im Gegensatz dazu
unterdrückt FAD mutantes PS1 diese Effekte. Zusätzlich sind eventuell Störungen der
Calcium-Homöostase durch mutantes PS1 oder Aβ an einer fehlerhaften Genexpression
beteiligt.
111
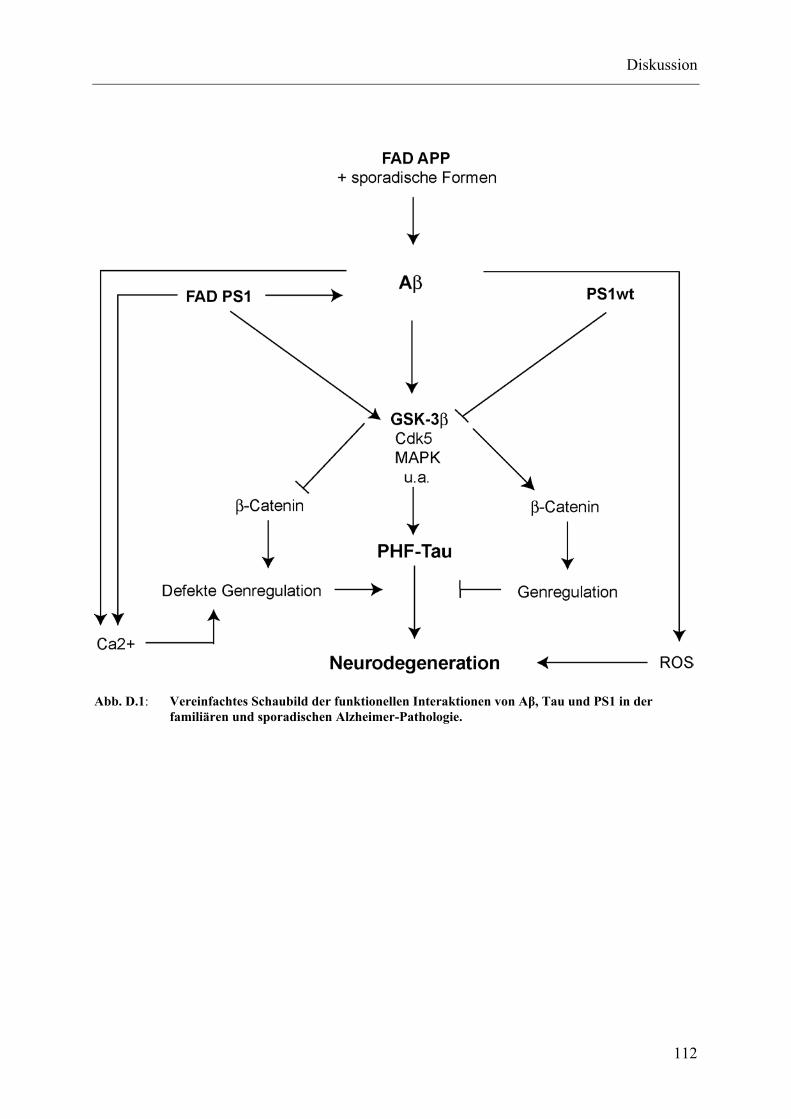
Diskussion
Abb. D.1: Vereinfachtes Schaubild der funktionellen Interaktionen von Aβ, Tau und PS1 in der familiären und sporadischen Alzheimer-Pathologie.
112

Zusammenfassung
E Zusammenfassung
In neurodegenerativen Erkrankungen vom Alzheimer-Typ kommt es zu einem massiven
selektiven Absterben von Neuronen in bestimmten Gehirnregionen. Die zwei charakte-
ristischen histopathologischen Hauptmerkmale der Alzheimer-Krankheit sind das Auftreten
von extrazellulären Ablagerungen des APP-Peptidfragments Aβ (Amyloid-Plaques) und
intrazelluläre Ablagerungen von hyperphosphoryliertem Tau-Protein (NFTs). Familiäre
Formen von AD (FAD) können durch Mutationen im APP-Gen oder in den beiden sehr
homologen Presenilin-Genen entstehen und führen zu einer erhöhten Produktion des
neurotoxischen Aβ42.
Durch die Herstellung von Tau mit Glutamat-Mutationen ist es möglich, den erhöhten
Phosphorylierungszustand von Tau zu simulieren, so dass dieses strukturelle und funktionelle
Eigenschaften von hyperphosphoryliertem Tau in der Alzheimer-Krankheit (PHF-Tau)
erwirbt. Aus früheren Arbeiten ist bekannt, dass pseudohyperphosphoryliertes Tau (PHP-Tau)
ohne Aggregation in Filamente einen neurotoxischen Effekt auf neurale Zellen ausübt, der
von einer Induktion apoptotischer Mechanismen begleitet ist. In dieser Arbeit konnte dies
durch die HSV-1-vermittelte Expression von GFP-Tau-Fusionsproteinen in kortikalen
Primärneuronen der Maus bestätigt werden. Durch die Verwendung transgener Mauslinien,
welche physiologisch entweder FAD-mutantes APP, FAD-mutantes PS1 oder wt PS1
exprimieren, wurde analysiert, wie andere AD-relevante Proteine die Zytotoxizität PHP-Taus
beeinflussen. Es konnte gezeigt werden, dass sich PS1 wt antiapoptotisch auf die PHP-Tau-
induzierte Neurodegeneration auswirkt. Im Gegensatz dazu vermittelte die PS1 FAD-
Mutation M146L nicht diesen anti-degenerativen Effekt. In APPSDL-transgenen Primär-
kulturen, in welchen ein Anstieg in der Aβ-Produktion nachgewiesen werden konnte, wurde
eine Erhöhung der wt-Tau-Toxizität beobachtet. Zudem wurde festgestellt, dass Aβ den
Phosphorylierungszustand wt-Taus an zwei AD-relevanten Phosphorylierungsstellen (dem
PHF-1-Epitop) erhöht. Die Ergebnisse dieser Arbeit unterstützen die sogenannte Amyloid-
Hypothese, nach welcher Aβ eine primäre Rolle in der AD-Pathologie hat. Sie geben Evidenz
dafür, dass eine erhöhte Tau-Phosphorylierung letzlich zur Neurodegeneration führt. Die
Hyperphosphorylierung von Tau spielt demnach eine zentrale Rolle bei den pathologischen
Mechanismen in der Alzheimer-Krankheit. Mutationen im PS1-Gen können entweder indirekt
über eine erhöhte Produktion von Aβ oder direkt über eine Störung der PS1-Funktion den
PHF-Tau induzierten Neuronentod bewirken.
113

Abkürzungen
F Abkürzungen
A Ampere Abb. Abbildung Aβ β-Amyloid AD engl.: Alzheimer’s disease (Alzheimer-Krankheit) ADAM engl.: a disintegrin and metalloprotease (α-Sekretase) AICD engl.: APP intracellular domain (intrazelluläre Domäne von APP) ALS Amyotrophe Lateralsklerose ApoE Apolipoprotein E APP engl.: Amyloid Precursor Protein (Amyloid-Vorläufer-Protein) APS Ammoniumpersulfat AS Aminosäure BACE engl.: β-amyloid converting enzyme (β-Sekretase) BSA engl.: bovine serum albumin (Rinderserumalbumin) bp Basenpaar bzw. beziehungsweise °C Grad Celsius Ca Calcium ca. circa CaMKII Ca2+/Calmodulin abhängige Kinase II Cdk2 engl.: cyclin-dependent kinase 2 (Cyclin abhängige Kinase 2) Cdk5 engl.: cyclin-dependent kinase 5 (Cyclin abhängige Kinase 5) Cl Chlor CSF engl.: cerebrospinal fluid (Zerebrospinal-Flüssigkeit) C-terminal carboxyterminal Cy3 Indocarbocyanin C83 membrangebundenes C-terminales Fragment von APP (83 AS
lang) C99 membrangebundenes C-terminales Fragment von APP (99 AS
lang) D Tag ddH2O doppelt destilliertes Wasser d.h. das heißt DIV Tage in vitro DNA Desoxy-Ribonukleinsäure DAPI 4’,6-Diamidino-2-phenylindol
114

Abkürzungen
DMEM Dulbecco’s Modified Eagle Medium DMEM-Ser DMEM-Serum DMSO Dimethylsulfoxid dNTPs Desoxy-Nukleosidtriphosphate E Einheit E Embryonaltag ECL engl.: enhanced chemoluminescence(verstärkte Chemolumin-
eszenz) EDTA Ethylendiaminotetraessigsäure eGFP engl.: enhanced Green Fluorescent Protein (Grünes Fluores-
zierendes Protein mit verstärkter Fluoreszenz) EGTA Ethylenglykol-bis-(β-aminoethylether)-N,N,N‘,N‘-Tetraessigsäure ER Endoplasmatisches Retikulum ERK extrazellulär regulierte Kinase EtOH Ethanol FCS engl.: fetal calf serum (fötales Kälberserum) FTDP-17 Frontotemporale Demenz mit Parkinsonismus g Gramm x g Gravitationskonstante (Erdbeschleunigung) GSK-3 Glykogen Synthase Kinase-3 H Wasserstoff HBSS Hanks Balanced Salt Solution HRP engl.: horseraddish peroxidase (Meerrettich-Peroxidase) HS engl.: horse serum (Pferdeserum) HSV-1 Herpes simplex Virus Typ-1 IE engl.: immediate early gene (frühes Gen) Ifps engl.: infectious particles (infektiöse Partikel) JNK Jun-Kinase k Kilo K Kalium kD Kilodalton KOH Kalilauge l Liter m Meter m milli M Molar MAPK Mitogen aktivierte Protein Kinase
115

Abkürzungen
MAP mikrotubuliassoziiertes Protein MEM Minimum Essential Medium MeOH Methanol Mg Magnesium µ mikro min Minute MW Molekulargewicht n nano Na Natrium NaOH Natronlauge n.d. nicht definiert NFTs engl.: neurofibrillary tangles (neurofibrilläre Ablagerungen) NICD engl.: Notch intracellular domain (Notch intrazelluläre Domäne) NCLK engl.: neuronal Cdc2-like protein kinase (neuronale Cdc2-ähnliche
Protein Kinase) N-terminal aminoterminal OD Optische Dichte p pico p3 α-/γ-Sekretase-prozessiertes APP-Peptidfragment PAGE Polyacrylamid Gelelektrophorese PBS Phosphatgepufferte Kochsalzlösung PCR engl.: polymerase chain reaction (Polymerase Kettenreaktion PFA Paraformaldehyd pfu engl.: plaque forming units (plaquebildende Einheiten) pH lat.: Potentia hydrogenii (negativer dekadischer Logarithmus der
Wasserstoffionenkonzentration einer Lösung) PHF-Tau engl.: paired helical filament tau (Tau in gepaarten helikalen
Filamenten) PHP-Tau pseudohyperphosphoryliertes Tau PI3K Phosphatidylinositol 3-Kinase PKA Protein Kinase A PLL Poly-L-Lysin PMSF Phenylmethylsulfonylfluorid PP2 Protein Phosphatase 2 PS1 Presenilin 1 PS2 Presenilin 2 % (v/v) Volumenprozent
116

Abkürzungen
% (w/v) Gewichtsprozent PVDF Polyvinyliden-Difluorid RA Retinolsäure (Vitamin A Säure) RAGE engl.: Receptor for Advanced Gycation End products (Rezeptor für
fortgeschrittene Glykierungsendprodukte) ROS engl.: Reactive Oxygen Species (Reaktive Sauerstoffspezies) rpm engl.: revolutions per minute (Umdrehungen pro minute) RT Raumtemperatur s Sekunde SAPK Stress aktivierte Protein Kinase sAPPα lösliches α-/γ-Sekretase-prozessiertes APP-Fragment sAPPβ lösliches β-/γ-Sekretase-prozessiertes APP-Fragment SDS engl.: sodium dodecylsulfate (Natriumdodecylsulfat) SE engl.: standard error (Standardfehler) S(er) Serin SF engl.: straight filament T Threonin TACE engl.: TNF-alpha converting enzyme (α-Sekretase) TBS Tris-gepufferte Kochsalzlösung TCA Trichloressigsäure TEMED N,N,N‘,N‘-Tetramethylendiamin TNX-100 Triton X-100 Tris 2-Amino-2-(hydroxymethyl)1-1,3-propandiol (Tris/Base) Tween-20 Polyoxyethylensorbitan Monolaurat Tyr Tyrosin U engl.: units (Einheiten) u.a. unter anderem ÜN über Nacht V Volt wt Wildtyp z.B. zum Beispiel ZNS zentrales Nervensystem
117

Die Veröffentlichung der Ergebnisse dieser Arbeit ist unter folgenden Titeln vorgesehen:
Leschik, J., Welzel, A. und Brandt, R. Amyloid-β peptide induces the toxicity of tau via
osphorylation of residues hyperphosphorylated in Alzheimer’s disease: Support for
e amyloid hypothesis. Manuskript in Vorbereitung
ph
th
pr
Leschik, J. und Brandt, R. Effects of wildtype and familial Alzheimer’s disease related mutant
esenilin 1 on tau-mediated neurodegeneration. Manuskript in Vorbereitung

Literatur
G Literatur
Agar-Mawal, A., Qureshi, H.Y., Cafferty, P.W., Yuan, Z., Han, D., Lind, R. et al. (2003) 14-3 connects gycogen-synthase-kinase-3 beta to tau within a brain microtubule-sociated tau phosphorylation complex. J. Biol. Chem., 278, 12722-12728
3-as
(2
BT
phN
Aksenov, M.Y., Aksenova, M. V., Butterfield, D.A. , Geddes, J. W. and Markesbery, W. R. 001) Protein oxidation in the brain in Alzheimer’s disease. Neurosci., 103, 373-383
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. (2002) Molecular iology of the Cell, Fourth Edition, New York, Garland Science, a member of the aylor & Francis Group
Alonso, A.C., Zaidi, T., Grundke-Iqbal, and Iqbal, K. (1994) Role of abnormally osphorylated tau in the breakdown of microtubules in Alzheimers disease. Proc.
atl.
Hfi
(1de
(2neD
Capm
N
(2do
mac
paA
beB
Acad. Sci. USA, 91, 5562-5566
Alonso, A., Zaidi, T., Novak, M., Grundke-Iqbal, I. and Grundke-Iqbal, K. (2001) yperphosphorylation induces self-assembly of tau into tangles of paired helical laments/straight filaments. Proc. Natl. Acad. Sci. U. S. A., 98, 6923-6928
Alvarez, G., Muñoz-Montaño, J.R., Satrústegui, J., Avila, J., Bogónez, E. and Diaz-Nido, J. 999) Lithium protects cultured neurons against beta-amyloid-induced neuro-generation. FEBS Lett., 453, 260-264
Alvarez, G., Muñoz-Montaño, J.R., Satrústegui, J., Avila, J., Bogónez, E. and Diaz-Nido, J. 002) Regulation of tau-phosphorylation and protection against β-amyloid-induced urodegeneration by lithium. Possible implications for Alzheimer’s disease. Bipolar isord., 4, 153-165
Alves da Costa, C., Paitel, E., Mattson, M.P., Amson, R., Telerman, A., Ancolio, K. and hecler, F. (2002) Wild-type and mutated presenilins 2 trigger p53-dependent optosis and down-regulate presenilin 1 expression in HEK293 human cells and in urine neurons. Proc. Natl. Acad. Sci. U. S. A., 6, 4043-4048
Alzheimer, A. (1911) Über eigenartige Krankheitsfälle des späteren Alters. Z. Gesamte eurol. Psychiat., 4, 356-385
Araki, W., Yuasa, K., Takeda, S., Takeda, K., Shirotani, K., Takahashi, K. and Tabira, T. 001) Pro-apoptotic effects of presenilin 2 (PS2) overexpression is associated with wn-regulation of Bcl-2 in cultured neurons. J. Neurochem., 79, 1161-1168
Arnold, C.S., Johnson, G.V.W., Cole, R.N., Dong, D.L., Le, M. and Hart, G.W. (1996) The icrotubule-associated protein tau is extensively modified with O-linked N-etylglucosamine. J. Biol. Chem., 271, 28741-28744
Arriagada, P.V., Marzloff, K. and Hyman, B.T (1992) Distribution of Alzheimer-type thologic changes in nondemented elderly individuals matches the pattern in lzheimer's disease. Neurology, 42, 631-639
Asami-Odaka, A., Ishibashi, Y., Kikuchi, T., Kitada, C. and Suzuki, N. (1995) Long amyloid ta-protein secreted from wild-type human neuroblastoma IMR-32 cells.
iochemistry, 34, 10272-10278
119

Literatur
Atwood, C.S., Obrenovich, M.E., Liu, T., Chan, H., Perry, G., Smith, M.A. and Martins, R.N. 003) Amyloid-β: a chameleon walking in two worlds: a review of the trophic and xic properties of amyloid-β. Brain Res. Brain Rev., 43, 1-16
(2to
m
J.cyst98
Rov
Acd
am
ap
an
G
Pbe
Billingsley, K.L. and Kincaid, R.L. (1997) Regulated phosphorylation and dephosphorylation
ne
Binder, distribution of tau in the mammalian
Black, M cher, I. (1996) Tau is enriched
Blancha .,
anw
Borchelt ky, T., Prada, C.M., Kim, G., Seekins, S., Yager, D., Slunt, H.H., Wang, R., Seeger, M., Levey, A.I., Gandy, S.E., Copeland, N.G., Jenkins, N.A., Price, D.L., Younkin, S.G. and Sisodia SS. (1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron, 17, 1005-1013
Avila, J., Brandt, R. and Kosik, K. (1997) Brain microtubule associated proteins-odifications in disease. Harwood academic publishers
Baki, L., Marambaud, P., Efthimiopoulos, S., Georgakopoulos, A., Wen, P., Cui, W., Shioi, , Koo, E., Ozawa, M., Friedrich Jr, V.L. and Robakis, N.K. (2001) Presenilin 1 binds toplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates
ability and function of the cadherin/catenin complex. Proc. Natl. Acad. Sci. U. S. A., , 2381-2386
Baki, L., Shioi, J., Wen, P., Shao, Z., Schwarzman, A., Gama-Sosa, M., Neve, R. and obakis, N.K. (2004) PS1 activates PI3K thus inhibiting GSK-3 activity and tau-erphosphorylation: effects of FAD mutations. EMBO J., 23, 2586-2596
Baumann, K., Mandelkow, E.M., Biernat, J., Piwnica-Worms, H. and Mandelkow, E (1993) bnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases k2 and cdk5. FEBS Lett., 336, 417-424
Behl, C., Davis, J.B., Lesley, R. and Schubert, D. (1994) Hydroxide peroxide mediates yloid β protein toxicity. Cell, 77, 817-827
Behl, C. (1999) Alzheimer’s disease and oxidative stress: Implications for novel therapeutic proaches. Prog. Neurobiol., 57, 301-323
Bickel, H. (2000) Dementia syndrome and Alzheimer disease: an assessment of morbidity and nual incidence in Germany. Gesundheitswesen, 62, 211-218
Bickel, H. (2001) Dementia in advanced age: estimating incidence and health care costs. Z. erontol. Geriatr., 34, 108-115
Biernat, J., Gustke, N., Drewes, G., Mandelkow, E.M. and Mandelkow, E. (1993) hosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction tween PHF-like immunoreactivity and microtubule binding. Neuron, 11, 153-163
of tau protein: effects on microtubule interaction, intracellular trafficking and urodegeneration. Biochem. J., 323, 577-591
L.I., Frankfurter, A., Rebhun, L.I. (1985) The central nervous system. J. Cell Biol., 101, 1371-1378
.M., Slaughter, T., Moshiach, S.,Obrocka, M. and Fison microtubules in the distal region of growing axons. J. Neurosci., 16, 3601-3619
rd, V., Moussaoui, S., Czech, C., Touchet, N., Bonici, B., Planche, M., Canton, TJedidi, I., Gohin, M., Wirths, O., Bayer, T.A., Langui, D., Duyckaerts, C., Tremp, G.
d Pradier, L. (2003) Time sequence of maturation of dystrophic neurites associated ith Abeta deposits in APP/PS1 transgenic mice. Exp. Neurol., 184, 247-263
, D.R., Thinakaran, G., Eckman, C.B., Lee, M.K., Davenport, F., Ratovits
120

Literatur
Boutajangout, A., Leroy, K., Touchet, N., Authelet, M., Blanchard, V., Tremp, G., Pradier, L. d Brion, J.P. (2002) Increased tau-phosphorylation but absence of neurofibrillary ngles in mice double transgenic for human tau and Alzheimer mutant (M146L) esenilin-1. Neurosci. Lett., 318, 29-33
o-Coyne, D., O'Kane, T.M., Wu, Z.L., Dobrzanski, P., Murthy, S., Va
antapr
Bozyczk ught, J.L. and
prne
Braak, H Alzheimer-related cortical destruction. Eur.
Bramble(1de ing. Neuron, 10, 1089-1099
Identifications of regions which affect microtubule growth, nucleation, and bundle
Scott, RW. (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, omotes survival and blocks multiple events associated with Aβ-induced cortical uron apoptosis. J. Neurochem., 77, 849-863
., Braak, E., Bohl, J. (1993) Staging of Neurol., 33, 403-408
tt, G.T., Goedert, M., Jakes, R., Merrick, S.E., Trojanowski, J.Q. & Lee, V.M.-Y. 993) Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates velopment and contributes to reduced microtubule bind
Brandt, R. and Lee,G.(1993) Functional organization of microtubule-associated protein tau.
formation in vitro. J.Biol.Chem., 268, 3414-3419
R., Léger, J. and Lee, G. (1995) Interaction of the neural plasma membran mediated tau’s aminoterminal projection domain. J. Cell Biol., 131, 1327-1340
Brandt, by
B
O
ta
RA t enhance apoptosis. J. Neurosci., 18, 9790-9799
ph
ar
Buxbaum K.N., Luo, Y., Slack, J.L., Stocking, K.L., Peschon, J.J., Johnson, R.S.,
A
ol 1-463
Brandt, R. (1996). The tau proteins in neuronal growth and development. Frontiers in ioscience, 1, d118-130
Brandt, R. and Leschik, J. (2004) Functional interaction of tau and their relevance for Alzheimer’s disease. Curr. Alzheimer Res., 1, 255-269
Brandt, R., Hundelt, M. and Shahani, N. (2005) Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim. Biophys. Acta, 1739, 331-354
Brunet, A., Datta, S.R. and Greenberg, M.E. (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr.
pin. Neurobiol., 11, 297-305
Buée-Scherrer, V. and Goedert, M. (2002) Phosphorylation of microtubule-associated protein u by stress-activated protein kinases in intact cells. FEBS Lett., 515, 151-154
Bursztajn, S., De Souza, R., Mc Phie, D.L., Berman, S.A., Shioi, J., Robakis, N.K. and Neve .L. (1998) Overexpression in neurons of human presenilin 1 or a presenilin 1 familial lzheimer disease mutant does no
Busciglio, J., Lorenzo, A., Yeh, J. and Yanker, B.A. (1995) Beta-amyloid fibrils induce tau osphorylation and loss of microtubule binding. Neuron, 14, 879-888
Butner, K.A. and Kirschner M.W. (1991) Tau protein binds to microtubules trough a flexible ray of distributed weak sites. J. Cell Biol., 131, 1327-1340
, J.D., Liu,Castner, B.J., Cerretti, D.P. and Black, R.A. (1998) Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of
lzheimer amyloid protein precursor. J. Biol. Chem., 273, 27765-27767
Caceres, A. and Kosik, K. (1990) Inhibition of neurite polarity by tau antisense igonucleotides in primary cerebellar neurons. Nature, 343, 46
121

Literatur
Caceres, A., Potrebic, S. and Kosik, K. (1991) The effect of tau antisense oligonucleotides in imary cerebellar neurons. J. Neurosci., 11, 1515-1523
., Golde, T.E. and Younkin, S.G. (1993) Release of excess amyloid beta protein from mutant amyloid beta protein precursor. Science, 259, 514-516
pr
Cai, X.Da
P n loss in APP
Canu, N Rinaldi, A.M., Novak, M.,
la sci., 18, 7061-7074
hi
Cao, X. and Sudhof, T.C. (2004) Dissection of APP-dependent transcriptional transactivation.
Chan, Tphphosphorylation. Annu. Rev. Biochem., 68, 965-1014
In Tabira, T. (1999) Transgenic
w at. Med., 5, 560-564
Ppr otein production. Nature, 360,
Citron, M(1am mice. Nat. Med., 3, 67-72
D29
Corder, E.H., Saunders, A.M, Strittmatter, W.J., Schmechel, D.E., Gaskell, P.C., Small,
apSc
Correas, I., Padilla, R. and Avila, J. (1990) The tubulin-binding-sequence of brain micro-
J.
Couchie, D., Mavilia, C., Georgieff, I.S., Liem, R.K., Shelanski, M.L. and Nunez, J. (1992)
sy
Calhoun, M.E., Wiederhold, K.H., Abramowski, D., Phinney, A.L., Probst, A., Sturchler-ierrat, C., Staufenbiel, M., Sommer, B. and Jucker, M. (1998) Neuro
transgenic mice. Nature, 395, 755-756
., Dus, L., Barbato, C., Ciotti, M.T., Brancolini, C., Cattaneo, A., Bradbury, A. and Calissano, P. (1998) Tau cleavage and dephosphory-
tion in cerebellar granule neurons undergoing apoptosis. J. Neuro
Cao, X. and Sudhof, T.C. (2001) A transcriptively active complex of APP with Fe65 and stone acetyltransferase Tip60. Science, 293, 115-120
J. Biol. Chem., 279, 24601-24611
.O., Rittenhouse, S.E. and Tsichlis, P.N. (1999) AKT/PKB and other D3 phos-oinositide regulated kinases: kinase activation by phosphoinositide-dependent
Chui, D.H., Tanahashi, H., Ozawa, K., Ikeda, S., Checler, F., Ueda, O., Suzuki, H., Araki, W., oue, H., Shirotani, K., Takahashi, K., Gallyas, F. and
mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration ithout amyloid plaque formation. N
Citron, M., Oltersdorf, T., Haass, C., Mc Conlogue, L., Hung, A.Y., Seubert, P., Vigo-elfrey, C., Lieberburg, I. and Selkoe, D.J. (1992) Mutation of the β-amyloid precursor otein in familial Alzheimer's disease increases β -pr
672-674
., Westaway, D., Xia, W., Carlson, G., Diehl, T., Levesque, G., Selkoe, D. et al. 997) Mutant presenilins of Alzheimer’s disease increase the production of 42-residue yloid beta-protein in both transfected cells and transgenic
Comer, F.I. and Hart, G.W. (2000) O-glycosylation of nuclear and cytosolic proteins: ynamic interplay between O-GlcNac and O-phosphate. J. Biol. Chem., 275, 29179-182
G.W., Roses, A.D., Haines, J.L. and Pericak-Vance, M.A. (1993) Gene dose of olipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. ience. 261, 921-923
tubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem. , 269, 61-64
Primary structure of high molecular weight tau present in the peripheral nervous stem. Proc. Natl. Acad. Sci. U. S. A., 89, 4378-4381
122

Literatur
Cross, D.A, Alessi, D.R., Cohen, P., Andjelkovich, M. and Hemmings, B.A. (1995) Inhibition gykogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 5-789
of78
Spr
he
Crowther, R.A. (1991) Straight and paired helical filaments in Alzheimer disease have a
Czech, CM ve, J.N., Beyreuther, K. and
pr rain Res., 47, 108-116
B , L. (1998)
Darmongr ion target of cell
Datta, RRse hem., 272, 1965-
Deng, Gcr 50-54
Eprintracellular domain. Nature, 398, 518-522
sy 32475-32481
wN
Drewes, andelkow, E.M., Biernat, J., Goris, J.,
tr
Drewes, G., Trinczek, B., Illenberger, S., Biernat, J., Schmitt-Ulms, G., Meyer, H.E.,
mre actions and dynamic instability by phosphorylation at the Alzheimer-specific site serine-262. J. Biol. Chem., 270, 7679-7688
Cross, , D.A., Culbert, A.A., Chalmers, K.A., Facci, L., Skaper, S.D. and Reith, A.D. (2001) elective small molecule inhibitors of glycogen synthase kinase-3 activity protect imary neurons from death. J. Neurochem., 77, 94-102
Crowther, R.A. and Wischik, C.M, (1985) Image reconstitution of the Alzheimer paired lical filament, EMBO J., 4, 3661-3665
commom structural unit. Proc. Natl. Acad. Sci. U. S. A., 88, 2288-2292
., Delaere, P., Macq, A.F., Reibaud, M., Dreisler, S., Touchet, N., Schombert, B., azadier, M., Mercken, L., Theisen, M., Pradier, L., Octa
Tremp, G. (1997) Proteolytical processing of mutated human amyloid precursor otein in transgenic mice. Brain Res. Mol. B
Czech, C., Lesort, M., Tremp, G., Blanchard, V., Schombert, B., Carpentier, N., Dreisler, S., onici, B., Takashima, A., Moussaoui, S., Hugon, J. and Pradier
Characterization of human presenilin 1 transgenic rats: increased sensitivity to apoptosis in primary neuronal cultures. Neuroscience, 87, 325-336
, A.J., Ley, T.J., Nicholson, R.C. and Bleakly, R.C. (1996) Cleavage of cpp32 by anenzyme b represents a critical role for granenzyme B in the induct
DNA fragmentation. J. Biol. Chem., 271, 21709-21712
., Kojima, H., Banach, D., Bump, N.J., Talanian, R.V., Alnemri, E.S., Weichselbaum, .R., Wong, W.W. and Kufe, D.W. (1997) Activation of a CrmA-insensitive, p35-nsitive pathway in ionising radiation-induced apoptosis. J. Biol. C
1969
., Pike, C. and Cotman, C. (1996) Alzheimer-associated presenilin-2 confers in-eased sensitivity to apoptosis in PC12 cells. FEBS Lett., 397,
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., Schroeter, . H., Schrijvers, V., Wolfe, M., S., Ray, W., J., Goate , A.. and Kopan, R. (1999) A esenilin-1-dependent gamma-secretase-like protease mediates release of Notch
Ding, V.W., Chen, R.-H. and McCormick, F. (2000) Differential regulation of gycogen nthase kinase-3beta by insulin and wnt signaling. J. Biol. Chem., 275,
Dovey, H. F., Suomesaari-Chrysler, S., Lieberburg, I., Sinha, S. and Kiem, P. S. (1993) Cells ith a familial Alzheimer's disease mutation produce authentic beta-peptide. euroReport, 4, 1039-1042
G., Lichtenberg-Kraag, B., Doring, F., MDoree, M. and Mandelkow, E. (1992) Mitogen activated protein (MAP) kinase
ansforms tau protein in an Alzheimer like state. EMBO J., 11, 2131-2138
Mandelkow, E. M. and Mandelkow, E. (1995) Microtubule-associated protein/ icrotubule affinity-regulating kinase (p110mark). A novel protein kinase that gulates tau-microtubule inter
123

Literatur
Donoviel, D.B., Hadjantonakis, A.K., Ikeda, M., Zheng, H., Hyslop, P.S., Bernstein, A. 999) Mice lacking both presenilin genes exhibit early embryonic patterning defects. enes Dev., 13, 2801-2810
(1G
Hbr
Ebneth, (1 g of vesicles,
B
Echeverria, V., Ducatenzeiler, A., Dowd, E., Janne, J., Grant, S.M., Szyf, M., Wandosell, F.,
(2an he beta-amyloid
Eckert, Ace60
NMin20
eaNatl. Acad. Sci. USA., 99, 8666-8671
prC
EidenmüStructural and functional implications of tau hyperphosphorylation: Information from
Eidenmüph the proline-rich region are sufficient to
75
Ekinci, F.J. and Shea, T.B. (2000) Beta-amyloid-induced tau-phosphorylation does not
Estus, S.R nd
Farias, Gpr31
Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C.M., Perez-Tur, J., Hutton, M., Buee, L., arigaya, Y., Yager, D., Younkin, S. et al. (1996) Increased amyloid-beta42(43) in ains of mice expressing mutant presenilin 1. Nature, 383, 710-713
A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B. and Mandelkow, E. 998) Overexpression of tau protein inhibits kinesin-dependent traffickin
mitochondria and endoplasmic reticulum: implications for Alzheimer’s disease. J. Cell iol., 143, 777-794
Avila, J., Grimm, H., Dunnett, S.B., Hartmann, T., Alhonen, L. and Cuello, A.C. 004) Altered mitogen-activared protein kinase signaling, tau hyperphosphorylation d mild spatial learning dysfunction in transgenic rats expressing t
peptide intracellularly in hippocampal and cortical neurons. Neurosci., 129, 583-592
., Marques, A., Keil, U., Schüssel, K. and Müller, W.E. (2003) Increased apoptotic ll death in sporadic and genetic Alzheimer’s disease. Ann. N. Y. Acad. Sci., 1010, 4-609
Eckmann, C.B., Mehta, N.D., Crook, R., Perez-Tur, J., Prihar, G., Pfeiffer, E., Graff-Radford, ., Hinder, P., Yager, D., Zenk, B., Refolo, L.M., Prada, C.M., Younkin, S., Hutton, . and Hardy, J. (1997) A new pathogenic mutation in the APP gene (I716V) creases the relative proportion of Aβ42(43). Human Molecular Genetics, 5, 2087-89
Edbauer, D., Winkler, E., Haass, C. and Steiner, H. (2002) Presenilin and nicrastrin regulate ch other and determine amyloid β-peptide production via complex formation. Proc.
Ehehalt, R., Keller, P., Haass, C., Thiele, C. and Simons, K. (2003) Amyloidogenic ocessing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. ell Biol., 160, 113–123
ller, J., Fath, T., Pool, M., Hellwig, A., Reed, J., Sontag, E. and Brandt, R. (2000)
phosphorylation-mimicking mutated tau proteins. Biochemistry, 39, 13166-13175
ller, J., Fath, T., Maas, T., Pool, M., Sontag, E. and Brandt, R. (2001) Phos-orylation-mimicking glutamate clusters in
simulate functional deficiencies of hyperphosphorylated tau protein. Biochem. J., 357, 9-767
correlate with degeneration in cultured neurons. J. Alzheimer Dis., 2, 7-15
, Tucker, H.M., Van Rooyen, C., Wright, S., Brigham, E.F., Wogulis, M. and Rydel, .E. (1997) Aggregated amyloid-beta protein induces cortical neuronal apoptosis a
concomitant “apoptotic” pattern of gene induction. J. Neurosci., 17, 7736-7745
.A., Munoz, J.P., Garrido, J. and Maccionoi, R.B. (2002) Tubuliun, actin and tau otein interactions and the study of their macromolar assemblies. J. Biochem., 85, 5-324
124

Literatur
Fassbender, K., Simons, M., Bergmann, C., Stroick, M., Lutjohann, D., Keller, P., Runz, H., uhl, S., Bertsch, T., Von Bergmann, K. et al. (2001) Simvastatin strongK ly reduces
Fath, T.
Fath, T. (2002) Die Rolle des Mikrotubuli-assoziierten Proteins Tau in neurodegenerativen
Fath, Thy
Finckh, iner, J., Meins, W., Binetti,
frY
(1 beta-
Forman, ki,
Funato, K., Okeda, R. and Ihara, Y. (1998) Quantitation of amyloid β-protein (Aβ) in the
Gantier, pa s the affinity to glycogen synthase
Gervais,D n der Ploeg, L.H.T.,
A39
enan
(1reR
Gibson, G.E., Zhang, H., Toral-Barza, L., Szolosi, S. and Tofel-Grehl, B. (1996) Ca2+-stores
levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA., 98, 5856-5861
, Eidenmüller J., Maas, T. and Brandt, R. (2000) Herpes simplex virus-mediated expression of the axonal protein tau in human model neurons (NT2-N cells). Microscopy Research and Technique, 48, 85-96
Erkrankungen. Inaugural-Dissertation, Ruprecht-Karls-Universität Heidelberg
., Eidenmüller, J. and Brandt, R. (2002) Tau-mediated cytotoxicity in a perphosphorylation model of Alzheimer’s disease. J. Neurosci., 15, 9733-9741
U., Muller-Thomsen, T., Mann, U., Eggers, C., MarksteG., Alberci, A., Sonderegger, P., Hock, C., Nitsch, R. M. and Gal, A. (2000) High
equency of mutations in four different disease genes in early onset dementia. Ann. N. . Acad. Sci. , 920, 100-106
Forloni, G., Chiesa, R., Smiroldo, S., Verga, L., Salmona, M., Tagliavini, F. and Angeretti, N. 993) Apoptosis mediated neurotoxicity induced by chronic application of
amyloid fragment 25-35, Neuroreport, 4, 523-526
M.S., Lal, D., Zhang, B., Dabir, D.V., Swanson, E., Lee, V.M.-Y. and TrojanowsJ.Q. (2005) Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. Neurobiol. Dis., 25, 3539-3550
H., Yoshimura, M., Kusui, K., Tamaoka, A., Ishikawa, K., Ohkoshi, N., Namekata,
cortex during aging and in Alzheimer's disease. Am. J. Pathol., 152, 1633-1640
R., Gilbert, D., Dumanchin, C., Campion, D., Davoust, D. and Toma, F. (2000) The thogenic L392V mutation of presenilin 1 decrease
kinase-3beta. Neurosci. Lett., 283, 217-220
F., Xu, D., Robertson, G.S., Vaillancourt, J., Zhu, Y., Huang, J., Le Blanc, A., Smith, ., Rigby, M., Shearman, M.S., Clarke, EE, Zheng, H., Va
Ruffolo, S.C. Thornberry, N.A., Xanthoudakis, S., Zamboni, R.J., Roy, S. and Nicholson, D.W. (1999) Involvement of caspases in proteolytic cleavage of
lzheimer’s amyloid precursor protein and amyloidogenic peptide formation. Cell, 97, 5-406
Ghribi, O., Herman, M.M. and Savory, J. (2003a) Lithium inhibits Abeta-induced stress in doplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage d tau phosphorylation. J. Neurosci. Res., 71, 853-863
Ghribi, O., Prammonjago, P., Herman, M.M., Spaulding, N.K. and Savory, J. (2003b) Abeta -42)-induced JNK and ERK activation in rabbit hippocampus is differentially gulated by lithium but is not involved in tau phosphorylation. Brain Res. Mol. Brain es., 119, 201-206
in cultured fibroblasts and their changes with Alzheimer’s disease. Biochim. Biophys. Acta, 1316, 71-77
125

Literatur
Goate, A., Chartier-Harlin, M.-C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, ., Haynes, A., Hardy, J. et al. (1991) Segregation of a missense mutation in the
yloid precursor protein gene with familial Alzheimer’s disease. Nature, 349, 704-6
Lam70
taP osphorylation at Ser262 in the repeat domain. FEBS Lett., 454,
Goedert,antandem repeats: differential expression of tau protein mRNAs in human brain. EMBO
Goedert,asSc 1987
(1in
Goedert, pillantini, M. G. (1998) Tau mutations cause frontotemporal
GeorgakM., Gordon, R., Holstein, G.R., Martinelli, G., Mehta, P., Friedrich Jr, V.L. and
ad89
induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive
Gong, Cph
Gong, Cab hosphorylated tau is dephosphorylated by protein phosphatase-2B
Gong, Cm -2C and its
Gong, C 2000)
phA 5-5544
in
Godemann, R., Biernat, J., Mandelkow, E. and Mandelkow, E.M. (1999) Phosphorylation of u protein by recombinant GSK-3beta: pronounced phosphorylation at select Ser/Thr-ro motifs but no ph
157-164
M., Spillantini, M.G., Poitier, M.C., Ulrich, J. and Crowther, R.A. (1989) Cloning d sequencing of an isoform of microtubule associsted protein tau containing four
J., 8, 393-399
M., Spillantini, M.G. and Crowther, R.A. (1992) Cloning of a big tau microtubule sociated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. i. U. S. A., 89, 1983-
Goedert, M., Jakes, R., Spillantini, M.G., Hasegawa, M., Smith, M.J., and Crowther, R.A. 996) Assembly of microtubule associated protein tau into Alzheimer-like filaments duced by sulphated glycosaminoglycans. Nature, 383, 550-553
M., Crowther, R. A., Sdementias. Neuron, 21, 955-958
opoulos, A., Marambaud, P., Efthimiopoulos, S., Shioi, J., Cui, W., Li, H.C., Schutte,
Robakis, N.K. (1999) Presenilin 1 forms complexes with the cadherin/catenin cell-cell hesion system and is recruited to intercellular and synaptic contacts. Mol. Cell, 4, 3-902
Goldbaum, O. and Richter-Landsberg, C. (2004) Proteolytic stress causes heat shock protein
aggregates in oligodendrocytes in culture. J. Neurosci., 24, 5748-5757
.X., Singh, T.J., Grundke-Iqbal, I. and Grundke-Iqbal, K. (1993) Phosphoprotein osphatase activities in Alzheimer disease brain. J. Neurochem., 61, 921-927
.X., Singh, T.J., Grundke-Iqbal, I. and Iqbal, K. (1994a) Alzheimer's disease normally p
(calcineurin). J. Neurochem., 62, 803-806
.X., Grundke-Iqbal, I., Damuni, Z. and Iqbal, K. (1994b) Dephosphorylation of icrotubule associated protein tau by Protein-Phosphatase-1 and
implication in Alzheimer’s disease. FEBS Lett., 341, 94-98
.X., Lidsky, T., Wegiel, J., Zuck, L., Grundke-Iqbal, I. and Iqbal, K. (Phosphorylation of microtubule-associated protein tau is regulated by protein
osphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in lzheimer’s disease. J. Biol. Chem., 275, 553
Götz, J., Chen, F., Van Dorpe, J. and Nitsch, R.M. (2001) Formation of neurofibrillary tangles P301L tau transgenic mice induced by Abeta fibrils. Science, 293, 1491-1495
126

Literatur
Gravina, S., Ho, L., Eckman, C.B., Long, K.E., Otvos, L., Younkin, L.H., Suzuki, N. and ounkin, S. G. (1995) Amyloidβ protein (Aβ) in Alzheimer’s disease brain. iochemical and Immunocytochemical analysis with antibodies specific for forms ding at Aβ40 or Aβ42. J. Biol. Chem., 270, 7013-701
YBen 6
A ted-
Greene, phU
Acy U.S.A., 83, 4913-4917
is
Guo, Q., Furukawa, K., Sopher, B.L., Pham, D.G., Xie, J., Robinson, N., Martin, G.M. and
se
Guo, Q., . and Mattson,
in d beta-peptide: involvement of
Guo, Q.(1presenilin-1 mutant knock-in mice. Nat. Med., 5, 101-106
wC
Haass, C heimer’s disease - proteolysis
Hanger, sy lation of tau: generation of
14
Harada, Oshima, T., Sato-Yoshitake, R.,
ca . Nature, 369, 488-91
of
Hardy, JScience, 256, 184-185
Greenberg, S.M., Qiu, W.Q., Selkoe, D.J., Ben-Itzhak, A. and Kosik, K.S. (1995) minoterminal region of beta-amyloid precursor protein activates mitogen-activa
protein kinase. Neurosci. Lett., 198, 52-56
L.A. and Tischler, A.S. (1976) Establishment of noradrenergic a clonal line of rat eochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci.
. S. A., 73, 2424-2428
Grundke, I.I., Iqbal, K., Tung, Y.C., Quinlan, M., Wisniewski, H.M. and Binder, L.I. (1986) bnormal phosporylation of the microtubule associated protein tau in Alzheimer toskeletal pathology. Proc. Natl. Acad. Sci.
Gschwind, M. and Huber, G. (1995) Apoptotic cell death induced by β-amyloid 1-42 peptide cell type dependent. J. Neurochem., 65, 292-300
Mattson, M.P. (1996) Alzheimer's PS-1 mutation perturbs calcium homeostasis and nsitizes PC12 cells to death induced by amyloid β-peptide. Neuroreport, 8, 379-383
Sopher, B.L., Furukawa, K., Pham, D.G., Robinson, N., Martin, G.MM.,P. (1997) Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis
duced by trophic factor withdrawal and amyloicalcium and oxyradicals. J. Neurosci., 17, 4212-4222
, Fu, W., Sopher, B.L., Miller, M.W., Ware, C.B., Martin, G.M. and Mattson, M.P. 999) Increased vulnerability of hippocampal neurons to excitotoxic necrosis in
Haan, J., Bakker, E., Jennekens-Schinkel, A. and Roos, R.A. (1992) Progressive dementia, ithout cerebral hemorrhage, in a patient with hereditary cerebral amyloid angiopathy. lin. Neurol. Neurosurg., 94, 317-318
. and De Strooper, B. (1999) The presenilins in Alzholds the key. Science, 286, 916-919
D.P., Hughes, K., Woodgett, J.R., Brion, J.P. and Anderton, B.H. (1992) Glykogen nthase kinase-3 induces Alzheimer disease-like phosphory
paired helical filament epitopes and neuronal lokalisation of the kinase. Neurosci. Lett., 7, 58-62
A., Oguchi, K., Okabe, S., Kuno, J., Terada, S., Takei, Noda, T. and Hirokawa, N. (1994) Altered microtubule organization in small-
libre axons of mice lacking tau protein
Harada, J. and Sugimoto, M. (1999) Activation of caspase-3 in β-amyloid-induced apoptosis cultured rat cortical neurons. Brain. Res., 842, 311-323
.A. and Higgins, G.A. (1992) Alzheimer’s disease: the amyloid cascade hypothesis.
Hengartner, M.O. (2000) The biochemistry of apoptosis. Nature, 407, 770-776
127

Literatur
Herreman, A., Serneels, L., Annaert, W., Collen, D., Schoonjans, L. and De Strooper, B. 000) Total inactivation of gamma-secretase activity in pres(2 enilin-deficient embryonic
Hetman, M., Cavanaugh, J.E., Kimelman, D. and Xia, Z. (2000) Role of glycogen synthase . J. Neurosci.,
Bpr yloid deposits. Behav. Genet., 29,
Hong, Mby inhibition of glycogen synthase kinase-3. J. Biol. Chem., 272, 25326-25332
IsdeProc. Natl. Acad. Sci. U.S.A., 93, 2719-2723
M F.S.,
as
IllenbergB elkow, E. (1998) The
im
Inoue, Hco
fi10
ne Exp. Neurol., 56, 965-973
Bneuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic
Irving, NA
Gpa
cl n, 13, 45-53
stem cells. Nat. Cell Biol., 2, 461-462
kinase-3beta in neuronal apoptosis induced by trophic factor withdrawel20, 2567-2574
Holcomb, L.A., Gordon, M.N., Jantzen, P., Hsiao, K., Duff, K. and Morgan, D. (1999) ehavioral changes in transgenic mice expressing both amyloid precursor protein and esenilin-1 mutations: lack of association with am
177-185
., Chen, D.C., Klein, P.S. and Lee, V.M. (1997) Lithium reduces tau phosphorylation
Hoshi, M., Takashima, A., Noguchi, K., Murayama, M., Sato, K., Kondo, S., Saitoh, Y., higuro, K., Hoshino, T. and Imahori, K. (1996) Regulation of mitochondrial pyruvate hydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain.
Hussain, I., Powell, D., Howlett, D.R., Tew, D.G., Meek, T.D., Chapman, C., Gloger, I.S., urphy, K.E., Southan, C.D., Ryan, D.M., Smith, T.S., Simmons, D.L., Walsh,
Dingwall, C. and Christie, G. (1999) Identification of a novel aspartic protease (Asp 2) β-secretase. Mol. Cell Neurosci., 14, 419-427
er, S., Zheng-Fischhofer, Q., Preuss, U., Stamer, K., Baumann, K., Trinczek, B., iernat, J., Godemann, R., Mandelkow, E.M. and Mand
endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: plications for Alzheimer's disease. Mol. Biol. Cell, 9, 1495-1512
., Nojima H. and Okayama, H. (1990) High efficient transformation of Escherichia li with plasmid. Gene, 96, 23-28
Iqbal, K., Zaidi, T., Bancher, C. and Grundke-Iqbal, I. (1994) Alzheimer paired helical laments. Restoration of the biological activity by dephosphorylation. FEBS Lett., 349, 4-108
Irizarry, M.C., McNamara, M., Fedorchak, K., Hsiao, K., Hyman, B.T. (1997a) APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no
uronal loss in CA1. J. Neuropathol.
Irizarry, M.C., Soriano, F., McNamara, M., Page, K.J., Schenk, D., Games, D. and Hyman, .T. (1997b) Aβ deposition is associated with neuropil changes, but not with overt
mouse. J. Neurosci., 17, 7053-7059
. and Miller, C. (1997) Tau phosphorylation in cells transfected with wild-type or an lzheimer’s disease mutant presenilin-1. Neurosci. Lett. 222, 71-74
Ito, E., Oka, K., Etcheberrigaray, R., Nelson, T.J., McPhie, D.L., Tofel-Grehl, B., Gibson, .E. and Aldon, D.L. (1994) Internal Ca2+ mobilization is altered in fibroblasts from tients with Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A., 90, 10836-10840
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N. and Ihara, Y. (1994) Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ mono-
onals: evidence that an initially deposited species is Aβ42(43). Neuro
128

Literatur
Jackson, G.R., Wiedau-Pazos, M., Sang, T.G., Wagle, N., Brown, C.A. et al. (2002) Human ild-type tau interacts with wingless pathway components and produces urofibrillary tangle pathology in Drosophila. Neuron, 34, 509-519
wne
neup
coN
(1di
va
ph
Kaltschmidt, B., Uherek, M., Wellmann, H., Volk, B. and Kaltschmidt, C. (1999) Inhibition
Sc
InY
di
Keller, J he proteasome in brain aging. Ageing Res. Rev., 1,
Kempf, axon early in development of polarity in a microtubule- and microfilament-dependent
Kenesse t of phosphorylation of fetal tau is
R 29, 40-46
de
Kidd, M. (1963) Paired helical filaments in electron microscopy of Alzheimer’s disease.
Kienlen-Campard, P., Miolet, S., Tasiaux, B. and Octave, J.N. (2002) Intracellular amyloid-β1-42, but not extracellular soluble amyloid- β peptides, induces neuronal apoptosis. J.
Jiang, H., Guo, W., Liang, X. and Rao, Y. (2005) Both the establishment and maintenance of uronal polarity require active mechanisms: critical roles of GSK-3beta and its stream regulators. Cell, 120, 123-135
Jichia, G.A., Rockwood, J.M., Berenfeld, B., Hutton, M. and Davies, P. (1999a) Altered nformation of recombinant frontotemporal dementia-17 mutated tau-proteins.
eurosci. Lett., 260, 153-156
Jicha, G.A., Weaver, C., Lane, E., Vianna, C., Kress, Y., Rockwood, J. and Davies, P. 999b) CAMP-dependent Protein Kinase phosphorylations on tau in Alzheimer’s sease. J. Neurosci., 19, 7486-7494
Jorm, A.F. (1991) Cross national comparison of the occurance of Alzheimer’s disease and scular dementia. Eur. Arch. Psychiatr. Clin. Neurosci., 240, 218-222
Julliams, A., Vanderhoeven, I., Kuhn, S., Van Broeckhoven, C. and De Jonghe, C. (1999) No influence of presenilin1 I143T and G384A mutations on endogenous tau
osphorylation in human mouse neuroblastoma cells. Neurosci. Lett., 2, 83-86
of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc. Natl. Acad. i. U.S.A., 96, 9409-9414
Katzman, R. and Kawas, C. (1994) The epidemiology of dementia and Alzheimer’s disease. : Terry, R.D., Katzman, R. and Bick, K.L.: Alzheimer’s Disease. Raven Press, New ork: 105-120
Kaytor, M.D. and Orr, H.T. (2002) The GSK3 beta signaling cascade and neurodegenerative sease. Curr. Opin. Neurobiol., 12, 275-278
.N., Gee, J. and Ding, Q. (2002) T279-293
M., Clement, A., Faissner, A., Lee, G. and Brandt, R. (1996) Tau binds to the distal
manner. J. Neurosci., 16, 5583-5592
y, A.A. and Yen, S.H. (1993) The extencomparable to that of PHF-tau from Alzheimer paired helical filaments. Brain
esearch, 6Kessler, J., Calabrese, P. et al. (2000) A new screening method to support diagnosis of
mentia. Psycho, 26, 343-347
Nature, 197, 192-193
Biol. Chem., 277, 15666-15670
129

Literatur
Kimberly, W.T., Zheng, J.B., Guenette, S.Y. and Selkoe, D. (2001) The intracellular domain the beta-amyloid preof cursor protein is stabilized by Fe65 and translocates to the
KimberlR
Wga
(2 s a membrane protein complex comprised of presenilin, nicastrin,
Kojro, Est ffect on the alpha-secretase ADAM
Komori,de
R1 n Neuroglioma Cells. J. Neurochem.,
Ksiezak- M. and Wall, J.S. (1996)
Kuo, Y.and Roher, A.E. (1998) Elevated low-density lipoprotein in Alzheimer's disease
Kurt, Mand paired helical filament-like structures in the brains of mice carrying mutant
La FerlaAtransgenic mice. Nat. Genet., 9, 21-29
oftaNeurosci., 21, 879-888
FA by a disintegrin metalloprotease. Proc. Natl.
LassmanWfr Neuropathol., 89, 35-41
nucleus in a Notch-like manner. J. Biol. Chem., 276, 40288-40292
y, W.T. and Wolfe, M.S. (2003) Identity and function of γ-secretase. J. Neurosci. es., 74, 353-360
Kimberly, W.T., Esler, W.P., Ye, W., Ostaszewski, B.L., Gao, J., Diehl, T., Selkoe, D.J. and olfe, M.S. (2003a) Notch and the amyloid precursor protein are cleaved by similar mma-secretase(s). Biochemistry, 1, 137-144.
Kimberly, W.T., La Voie, M.J., Ostaszewski, B.L., Ye, W., Wolfe, M.S. and Selkoe, D.J. 003b) γ-secretase i
aph-1 and pen-2. Proc. Natl. Acad. Sci. U.S.A., 100, 6382-6387
., Gimpl, G., Lammich, S., Marz, W. and Fahrenholz, F. (2001) Low cholesterol imulates the nonamyloidogenic pathway by its e
10. Proc. Natl. Acad. Sci. USA., 98, 5815-5820
T. (1999) Tau-positive glial inclusions in progressive nuclear palsy, corticobasal generation and Pick’s disease. Brain Pathol., 9, 663-679
Kovacs, D.M., Mancini, R., Henderson, J., Na, S.J., Schmidt, S.D., Kim, T.-W. and Tanzi, .E. (1999) Staurosporine-induced activation of caspase-3 is potentiated by presenilin familial Alzheimer’s disease mutations in huma
73, 2278-2285
Reding, H., Tracz, E., Yang, L.S., Dickson, D.W., Simon, Ultrastructural instability of paired helical filaments from corticobasal degeneration as examined by scanning transmission electron microscopy. Am. J. Pathol., 149, 639-652
M., Emmerling, M.R., Bisgaier, C.L., Essenburg, A.D., Lampert, H.C., Drumm, D.
correlates with brain abeta 1-42 levels. Biochem. Biophys. Res. Commun., 252, 711–715
.A., Davies, D.C., Kidd, Duff, K. and Howlett, DR. (2003) Hyperphosphorylated tau
amyloid-precursor protein and mutant presenilin transgenes. Neurobiol. Dis., 14, 89-97
, F.M., Tinkle, B.T., Bieberich, C.J., Haudenschild, C.C. and Jay, G. (1995) The lzheimer's Aβ peptide induces neurodegeneration and apoptotic cell death in
Lain, E., Penke, B., Delacourte, A., Gundisch, D., Schroder, H. and Witter, B. (2005) Effects Abeta1-42 fibrils and of the tetrapeptide Pr-IIGL on the phosphorylation state of u-protein and on the alpha7 nicotinic acetylcholine receptor in vitro. Eur. J.
Lammich, S., Kojro, E., Postina, R., Gibert, S., Pfeiffer, R., Jasionowski, M., Haass, C. and ahrenholz, F. (1999) Constitutive and regulated alpha-secretase cleavage of lzheimer’s amyloid precursor protein
Acad. Sci. U. S. A., 96, 3922-3927
n, H., Bancher, C., Breitschof, H., Wegiel, J., Bobinski, M., Jellinger, K. and iesniewski, H.M. (1995) Cell death in Alzheimer’s disease evaluated by DNA-
agmentation in situ. Acta
130

Literatur
Layfield, R., Alban, A., Mayer, R.J. and Lowe, J. (2001) The ubiquitin protein catabolic sorders. Neuropathol. Appl. Neurobiol., 27, 171-179
Neve R.L., and Kosik, K.S. (1989) The microtubule binding domain of tau protein. euron, 2, 1615-1624
di
Lee, G.,N
w
by , 24, 2304-12
Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature, 405, 360-364
im otubule-
Leisserin(2 ontent in
Leroy, K . and
de
Leschik,
U
Lesort, M 997) Cultured neurons
se
Leutner, d Müller, A.W. (2000)
pr
Levitan, D. and Greenwald, I. (1995) Facilitation of lin-12 mediated signalling by sel-12, a
Levy-LaA lzheimer’s disease. Ann.
Lewis, JE t Tau and
Lindwal mote
Liu, T., (2 toxicity of primary neurons is dependent upon differen-tiation-associated increases in tau and cyclin-dependent kinase 5 expression. J. Neurochem., 88, 554-563
Lee, G., Newman, S.T., Gard, D.L., Band, H. and Panchamoorthy, G. (1998) Tau interacts ith src-familiy non-receptor-tyrosine-kinases. J. Cell Sci., 111, 3167-3177
Lee, G., Thangavel, R., Sharma, V.M., Litersky, J.M., Bhaskar, K., Fang, S.M., Do, L.H., Andreadis, A., Van Hoesen, G. and Ksiezak-Reding, H. (2004) Phosphorylation of tau
fyn: implications for Alzheimer's disease. J. Neurosci.
Lee, M.S., Kwon, Y.T., Li, M., Peng, J., Friedlander, R.M. and Tsai, L.H. (2000)
Léger, J., Kempf, M., Lee, G. and Brandt, R.(1997) Conversion of Serine to Aspartate itates phosphorylation induced changes in the structure and function of micr
associated protein tau. J. Biol. Chem., 272, 8441-8446
g, M.A., Akbari, Y., Fanger, C.M., Cahalan, M.D., Mattson, M.P. and LaFerla, F.M. 000) Capacitative calcium entry deficits and elevated luminal calcium c
mutant presenilin 1 knockin mice. J. Cell Biol., 149, 793-797
., Boutajangout, A., Richardson, J., Octave, J.N., Lovestone, S., Anderton, B.HBrion, J.P. (2003) Mutant presenilin 1 proteins induce cell death and reduce tau-
pendent process outgrowth. Neurosci. Lett., 353, 226-230
J. (2000) Expression von GFP-markiertem Tau-Protein und PHF-ähnlichen Tau-Phosphomutanten in Hippocampus-Schnittkulturen. Diplomarbeit, Ruprecht-Karls-
niversität Heidelberg
., Blanchard, C., Yardin, C., Esclaire, F. and Hugon, J. (1expressing phosphorylated tau are more resistant to apoptosis induced by NMDA or
rum deprivation. Brain Res. Mol. Brain Res., 45, 127-132
S., Czech, C., Schindowski, K., Touchet, N., Eckert, A. anReduced antioxidant enzyme activity in brains of mice transgenic for human
esenilin-1 with single or multiple mutations. Neurosci. Lett., 292, 87-90
Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature, 377, 351-354
had, E., Lahad A., Wijsman E. M., Bird, T. D. and Schellenberg, G. D. (1995) polipoprotein E genotypes and age of onset in early-onset A
Neurol. 38, 678-80
., Dickson, D.W., Lin, W.L., Chisholm, L., Corral, A., Jones, G. et al. (2001) nhanced neurofibrillary degeneration in transgenic mice expressing mutan
APP. Science, 293, 1487-1491
l, G. and Cole, R.D. (1984) Phosphorylation affects ability of tau protein to promicrotubule assembly. J. Biol. Chem., 259, 5301-5305
Perry, G., Chan, H.W., Verdile, G., Martins, R.N., Smith, M.A. and Atwood, C. 004) Amyloid-β-induced
131

Literatur
Loo, D.T., Copani, A., Pike, C.J., Whittemore, E.R., Walencewicz, A.J. and Cotman, C.W. 993) Apoptosis is induced by β-amyloid in cultured central nervous system neurons. roc. Natl. Acad. Sci. U.S.A., 90,
(1P 7951-7955
N
DG
Luo, J.J. , Ingram, D.K., Roth, G.S. and Kusiak, J.W. (1999) Death
am
Maas, T f tau with the neural plasma
he hem., 275, 15733-15740
A
Mandell sphorylation in nascent
MarambE in-
di
Maramb ., Siman, R. and Robakis, N.K.
of PS1 FAD mutations. Cell, 114, 635-645
and Lee, V.M. (1994) Biopsy derived adult human brain tau is phosphorylated at many
MattsonAvu
deB
12
Mattsonneuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation
13
Mattson .N. and Geiger, J.D. (2000b) Calcium signaling in the ER: ist role in neuronal plasticity and neuro-degenerative disorders. Trends Neurosci., 23, 222-229
Lu Q. and Wood, J.G. (1993) Functional studies of Alzheimers disease in tau proteins. J. eurosci., 13, 508-515
Lucas, J.J., Hernandez, F., Gomez-Ramos, P., Moran, M.A., Hen, R. and Avila, J. (2001) ecreased nuclear beta-catenin, tau hyperphosphorylation and neurodegenration in SK-3beta conditional transgenic mice. EMBO J., 20, 27-39
, Wallace, W., Riccioni, T.of PC12 cells and hippocampal neurons induced by adenoviral-mediated FAD human
yloid precursor protein gene expression. J. Neurosci. Res., 55, 629-642
, Eidenmüller, J. and Brandt, R.(2000) Interaction omembrane cortex is regulated by phosphorylation sites that are modified in paired
lical filaments. J. Biol. C
Majno, G. and Joris, I. (1995) Apoptosis, oncosis and necrosis. An overview of cell death. m. J. Pathol., 146, 3-15
, J.W. and Banker, G.A. (1996) A spatial gradient of tau phoaxons. J. Neurosci., 16, 5727-5740
aud, P., Shioi, J., Serban, G., Georgapoulos, Sarner, S., Nagy, V., Baki, L., Wen, P., fthimiopoulos, S., Shao, Z., Wisniewski, T. and Robakis, N.K. (2002) A presenil
1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates sassembly of adherens junctions. EMBO J., 21, 1948-1956
aud, P., Wen, P.H., Dutt, A., Shioi, J., Takashima, A(2003) A CBD binding transcriptional repressor produced by the PS1/epsilon-cleavage
N-cadherin is inhibited by
Matsuo E.S., Shin, R., Billingsley, M.L., De Voorde, A.V., O’Connor, M., Trolanowski, J.Q.,
of the same sites as Alzheimer disease paired helical filaments. Neuron, 13, 989-1002
, M.P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I. and Rydel, R.E. (1992) β-myloid peptides destabilize calcium homeostasis and render human cortical neurons lnerable to excitotoxicity. J. Neurosci., 12, 376-389.
Mattson, M.P., Tomaselli, K.J. and Rydel, R.E. (1993) Calcium destabilization and neuro-generative effect of aggregate beta amyloid peptide are attenuated by basic FGF.
rain Res., 621, 35-49
Mattson, M.P. (2000) Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol., 1, 0-129
, M.P., Zhu, H., Yu, J. and Kindy, M.S. (2000a) Presenilin-1 mutation increases
in cell culture: involvement of perturbed calcium homeostasis. J. Neurosci., 20, 1358-64
, M.P., La Ferla, F.M., Chan, S.L., Leissering, M.A., Shepel, P
132

Literatur
Mawal-Dewan, M., Henley, J., Van de Voorde, A., Trojanowski, J.Q. and Lee, V.M.-Y. 994) The phosphorylation state of tau in the developing rat brain is regulated by osphoprotein kinases. J
(1ph . Biol. Chem., 269, 30981-30987
secretase cleavage of the amyloid precursor protein mediates neuronal apoptosis
Michel, T acterization of tau phosphorylation in glycogen synthase
Migheli, ight and electron microscope
Miller, D Y.Y., Biemann, K. and
Mills, J.dephosphorylation of tau protein characterize the execution phase of apoptosis. J. Cell
Mills, Jcy
Miyata, Y. and Sakai, H. (1986) Binding of the
fr
Moechars, D., Lorent, K., De Strooper, B., Dewachter, I. and Van Leuven, F. (1996)
diE
S
Morishim J., Barrett, T., Takano, H., Flavell, R., Davis, R.J., Shirasaki,
m nd the induction of Fas
Morishiman PHF-
MorishimSph mulation in the human brain. Am. J. Pathol., 157,
Mc Phie, D.L., Golde, T., Eckmann, C.B., Yager, D., Brant, J.B. and Neve, R.L (2001) Beta-
caused by familial Alzheimer’s disease mutations. Brain Res. Mol. Brain Res., 97, 103-113
G., Mercken, M., Murayama, M., Noguchi, K., Ishiguro, K., Imahori, K. and akashima, A. (1998) Char
kinase-3beta and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim. Biophys. Acta, 1380, 177-182
A., Butler M., Brown,K. & Shelanski, M.L. (1988) Llocalization of the microtubule-associated tau protein in rat brain. J. Neurosci., 8, 1846-51 (1988)
.L., Papayannopoulos, I.A., Styles, J., Bobin, S.A., Lin,Iqbal, K. (1993) Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch. Biochem. Biophys., 301, 41-52
, Lee, V.M. and Pittman, R.N. (1998) Activation of PP2A-like phosphatase and
Sci., 111, 625-636
.C., Stone, N.L. and Pittman, R.N. (1999) Extranuclear apoptosis. The role of toplasm in the execution phase. J. Cell Biol., 146, 703-708
Y., Hoshi, M., Nishida, E., Minami, microtubule-associated protein-2 and tau to the intermediate filament reassembled
om neurofilament 70 kDa-subunit protein. J. Biol. Chem., 261, 13026-13030
Expression in brain of amyloid precursor protein mutated in the α-secretase site causes sturbed behavior, neuronal degeneration and premature death in transgenic mice. MBO J., 15, 1265-1274
Mookherjee, P. and Johnson, G.V. (2001) Tau phosphorylation during apoptosis of human H-SY5Y neuroblastoma cells. Brain Res., 921, 31-43
a, Y., Gotoh, Y., Zieg, Y. and Greenberg, M.E. (2001) β-Amyloid induces neuronal apoptosis via a
echanism that involves the c-Jun N-terminal kinase pathway aligand. J. Neurosci., 21, 7551-7560.
a-Kawashima, M., Hasegawa, M., Takio, K., Suzuki, M., Yoshida, H., Titani, K. d Ihara, Y. (1995) Prolin-directed and non-Prolin directed phosphorylation of
tau. J. Biol.Chem., 270, 823-829
a-Kawashima, M., Oshima, N., Ogata, H., Yamaguchi, H., Yoshimura, M., ugihara, S. and Ihara, Y. (2000) Effect of apolipoprotein E allele ε4 on the initial ase of amyloid β-protein accu
2093-2099.
133

Literatur
Moussaoui, S., Tremp, G., Blanchard, V., Bonici, B., Touchet, N., Clavel, N., Pradier, L. and zech, C. (2000) Progression of neuropathology in transgenic mice expressing genes nked to familial Alzheimer’s disease : APP, PS1 or both. Neurobiol. Aging, 21 uppl.1), 89
Cli(S
anpr er's disease. Neurosci. Lett., 265, 61-63
emL
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yanker, B.A. and Yuan, J. (2000)
am
NäslundSilberring, J., Gandy, S. E., Winblad, B., Greengard, P., Nordstedt, C. and Terenius, L.
D
Neve, R id
Nijhawadi Neurosci., 23, 73-87
Rm , 417-430
in
Papazom
Passer, B ardo, M.J., Schettini, G., Bachmann,
A
m
N
(1 s
58
Pierre, MB -219
Murayama, O., Tomita, T., Nihonmatsu, N., Murayama, M., Sun, X., Honda, T., Iwatsubo, T. d Takashima, A. (1999) Enhancement of amyloid β42 secretion by 28 different esenilin 1 mutations of familial Alzheim
Nagy, A., Gertsenstein, M., Vintersten, K. and Behringer, R. (2003) Manipulating the mouse bryo: a laboratory manual. Third Edition, New York, Cold Spring Harbor
aboratory Press
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by yloid-β. Nature, 403, 98-103.
, J., Schierhorn, A., Hellman, U., Lannfelt, L., Roses, A. D., Tjernberg, L. O.,
(1994) Relative abundance of Alzheimer Aß amyloid peptide variants in Alzheimer isease and normal Aging. Proc. Natl. Acad. Sci. U. S. A., 91, 8378-8382
.L. and Robakis, N.K. (1998) Alzheimer’s disease: a re-examination of the amylohypothesis. Trends in Neuroscience, 21, 15-19
n, D., Honarpour, N. and Wang, X. (2000) Apoptosis in neuronal development and sease. Annu. Rev.
Otth, C., Concha, I.I, Arendt, T., Stieler, J., Schliebs, R., González-Billault, C. and Maccioni, .B. (2002) A/betaPP induces cdk5-dependent tau hyperphosphorylation in transgenic ice Tg2576. J. Alzheimer’s Dis., 4
Pap, M. and Cooper, G.M. (1998) The role of glycogen synthase kinase-3 in the phosphatidyl-ositol 3-kinase/Akt cell survival pathway. J. Biol. Chem., 273, 19929-19932
enos, S.C. and Binder, L.I. (1987) Phosphorylation of tau determines two distinct species of Tau in the central nervous system. Cell Motility Cytoskeleton, 8, 210-26
., Pellegrini, L., Russo, C., Siegel, R.M., LenM., Tabaton, M. and D'Adamio, L. (2000) Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's beta-amyloid precursor protein. J.
lzheimer's Dis., 2, 289-301
Patapoutian, A. and Reichardet, L.F. (2000) Roles of wnt proteins in neural development and aintenance. Curr. Opin. Neurobiol., 10, 392-399
Patrick, G.N., Zukerberg, L., Nicolic, M., De la Monte, S., Dikkes, P. and Tsai, L.H. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promtes neurodegeneration.
ature, 402, 614-622
Pei, J.J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B. and Cowburn, R.F. 999) Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brain
staged for Alzheimer’s disease neurofibrillary changes. J. Neuropathol. Exp. Neurol., , 1010-1019
. and Nunez, J. (1983) Multisite phosphorylation of tau-protein from rat brain. iophys. Res. Commun., 115, 212
134

Literatur
Pigino, G., Pelsman, A., Mori, H. and Busciglio, J. (2001) Presenilin-1 mutations reduce toskeletal association, deregulate neurite growth acy nd potentiate neuronal dystrophy
Pleasurecharacteristics expected of human commited neuronal progenitor cell line. J. Neurosci.
PlutheroR
Podlisny, M.B., Citron, M., Amarante, P., Sherrington, R., Xia, W. and Zhang, J. (1997)
and Ala299 and occur as stable N- and C-terminal fragments in normal and in
Postina, K A., Godaux, E., Van Leuven, F.
In
Rank, Kan uman recombinant Abeta 1-42
Rapoporfi
Rapoport, M., Dawson, H.N., Binder, L.I., Vitek, M.P. and Ferreira, A. (2002) Tau is
63
Slo or protein in gene-targeted mice bearing the Swedish familial Alzheimer’s
Rehbecked
Reynolds, C.H., Betts, J.C., Blackstock, W.P., Nebreda, A.R. and Anderton, B.H. (2000)
di -
and tau phosphorylation. J. Neurosci., 21, 834-842
S.J. and Lee V.M.-Y. (1993). NTera 2 cells: a human cell line which displays
Res., 35, 585-602
, G. (1993) Rapid purification of high activity Taq-DNA-Polymerase. Nucleic Acid esearch, 21, 4850-4851
Presenilin proteins undergo heterogenous proteolytic endoproteolysis between Thr291
Alzheimer brain tissue. Neurobiol. Dis., 3, 325-337
R., Schroeder, A., Dewachter, I., Bohl, J., Schmitt, U., Kojro, E., Prinzen C., Endres, ., Hiemke, C., Blessing, M., Flamez, P., Dequenne,
and Fahrenholz, F. (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin.
vest., 113, 1456-1464
Price, D.L., Tanzi, R.E., Borchelt, D.R. and Sisodia, S.S. (1998) Alzheimer’s disease: genetic studies and transgenic models. Annu. Rev. Genet., 32, 461-493
Rametti, A., Esclaire, F., Yardin, C. and Terro, F. (2004) Linking alterations in tau phos-phorylation and cleavage during neuronal apoptosis. J. Biol. Chem., 259, 54518-54528
.B., Pauley, A.M., Bhattacharya, K., Wang, Z., Evans, D.B., Fleck, T., Johnston, J.A. d Sharma, S.K. (2002) Direct interaction of soluble h
results in tau aggregation and hyperphosphoryltion by tau protein kinase II. FEBS Lett., 514, 263-268
t, M. and Ferreira, A. (2000) PD98059 prevents neurite degeneration induced by brillar beta-amyloid in mature hippocampal neurons. J. Neurochem., 74, 125-133
essentiell for beta-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U. S. A., 99, 64-6369
Reaume, A.G., Howland, D.S., Trusko, S.P., Savage, M.J., Lang, D.M., Greenberg, B.D., iman, R. and Scott, R.W. (1996) Enhanced amyloidogenic processing of the β-amy-id precurs
disease mutation and a humanized Aβ-sequence. J. Biol. Chem., 271, 23380-23388
, G.W. (1998) ApoE and its role in late onset Alzheimer’s disease. In: Haass, C., itor. Molecular biology of Alzheimer’s disease. Amsterdam: Harwood p. 291-305
Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: fferences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N
terminal kinase and P38, and glycogen synthase kinase-3beta. J. Neurochem., 74, 1587-1595
135

Literatur
Rogaev, E.I, Sherrington, R., Rogaeva, E.A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., in, C., Li, G., Holman, K and Tsuda, T. (1995) Familial Alzheimer’s disease in ndreds with missense mutations in a gene on chromosome 1 related to Alzheimer’s sease type 3 gene. Nature, 376, 775-778
Lkidi
Roher, A. E., Lowenson, J. D., Clarke, S., Woods, A. S., Cotter, R. J., Gowing, E. and Ball,
de
Saitoh, A
ScheuneS. et al. (1996) Secreted amyloid beta-protein similar to that in the senile plaques of
li
SchmidtH s in normal and Alzheimer’s disease
SchneidePpr n into paired helical filaments. Biochemistry, 38, 3549-
Schroeter, E.H., Kisslinger, J.A.. and Kopan, R. (1998) Notch-1 signalling requires ligand-
SchuberAU
Wox Neurobiol. Dis., 18, 89-99
re
Selkoe, D. (1998) The cell biology of beta-amyloid precursor protein and presenilin in
Shelton, lation and
Shen, J.,an -1 deficient mice. Cell, 89, 629-639
Lea
(1 - and intracranial blood vessels. Ann. Neurol., 38, 421-428
M. J. (1993) ß-Amyloid-(1-42) is a major component of cerebrovascular amyloid posits: Implications for the pathology of Alzheimer Disease. Proc. Natl. Acad. Sci.
U. S. A., 90, 10836-10840
T. and Inhee, M.J. (1996) Biological function of APP and Alzheimer’s disease. lzeimer’s Dis. Rev., 1, 30-36
r, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., Bird, T.D., Younkin,
Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations nked to familial Alzheimer’s disease. Nat. Med., 2, 864-870
, M.L., Lee, V.M. and Trojanowski, J.Q. (1989) Analysis of epitopes shared by irano bodies and neurofilament protein
hippocampus. Lab. Invest., 60, 513-522
r, A., Biernat, J., von Bergen, M., Mandelkow, E. and Mandelkow, E.M. (1999) hosphorylation that detaches tau protein from microtubules (Ser 262, Ser214) also otects it against aggregatio
3558
induced proteolytic release of intracellular domain. Nature, 393, 382-386
t, D., Behl, C., Lesley, R., Brack, A., Dargusch, R., Sagara, Y. and Kimura, H. (1995) myloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. .S.A., 92, 1989-1993
Schuessel, K., Schäfer, S., Bayer, T.A., Czech, C., Pradier, L., Müller-Spahn, F., Müller, .E. and Eckert, A. (2005) Impaired Cu/Zn-sod activity contributes to increased idative damage in APP transgenic mice.
Selden, C.S. and Pollard, T.D. (1983) Phosphorylation of microtubule-associated proteins gulates their interaction with actin filaments. J. Biol. Chem., 258, 7064-7071
Alzheimer’s disease. Trends Cell Biol., 8, 447-453
S.B. and Johnson, G.V.W. (2001) Tau and HMW Tau phosphorycompartimentalization in apoptotic neuronal PC12 cells. J.Neurosci. Res., 66, 203-213
Bronson, R.T., Chen, D.F., Xia, W., Selkoe, D.J. and Tonegawa, S. (1997) Skeletal d CNS defects in presenilin
Sherrington, R., Rogaev, E.I., Liang, Y., Rogaeva, E.A., Levesque, G., Ikeda, M., Chi, H., in, C., Li, G. and Holman, K. (1995) Cloning of a gene bearing missense mutations in rly onset familial Alzheimer’s disease. Nature, 375, 754-760
Shinkai, Y., Yoshimura, M., Ito, Y., Odaka, A., Suzuki, N., Yanagisawa, K. and Ihara, Y. 995) Amyloid beta-proteins 1-40 and 1-42(43) in the soluble fraction of extra
136

Literatur
Siman, R., Reaume, A.G., Savage, M.J., Trusko, S., Lin, Y.G., Scott, R.W. and Flood, D.G. 000) Presenilin 1 P264L knock-in mutation: differ(2 ential effect on abeta production,
Simons, Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons.
Simons, r, K., Dichgans, J.,
pl
LS c Coulogue, L. and John, V.
br
Smale, G ence for
Sobue, In icrotubule associated protein
Sontag, ofph
stN
Spillantipa some 17: a new group of tauopathies. Brain Pathol., 8,
Spittaels ., Laenen, I.,
ov
Mph xonopathy in the central nervous system of
Stamer, traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances
amyloid deposition and neuronal vulnerability. J. Neurosci., 20, 8717-8726
M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C.G. and Simons, K. (1998)
Proc. Natl. Acad. Sci. U.S.A., 95, 6460–6464
M., Schwarzler, F., Lutjohann, D., Von Bergmann, K., BeyreutheWormstall, H., Hartmann, T. and Schulz, J.B. (2002) Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized,
acebo-controlled, double-blind trial. Ann. Neurol., 52,346-350
Sinha, S., Anderson, J.P., Barbour, R., Basi, G.S., Caccavello, R., Davis, D., Doan, M., Dovey, H.F., Frigon, N., Hong, J., Jacobson-Croak, K., Jewett, N., Keim, P., Knops, J.,
ieberburg, I., Power, M., Tan, H., Tatsuno, G., Tung, J., Schenk, D., Seubert, P., uomensaari, S.M., Wang, S., Walker, D., Zhao, J., M
(1999) Purification and cloning of amyloid precursor protein β-secretase from human ain. Nature, 402, 537-540
., Nichols, N.R., Brady, D.R., Finch, C.E. and Horton, W.E.J. (1995) Evidapoptotic cell death in Alzheimer’s disease. Exp. Neurol., 133, 225-230
K., Agarwal-Mawal, A., Li, W., Sun, W., Miura, Y. and Paudel, H.K. (2000) teraction of neuronal Cdc2-like protein kinase with m
tau. J. Biol. Chem., 275, 16673-16680
E., Nunbhakdi-Craig, V., Lee, G., Bloom, G.S. and Mumby, M.C. (1996) Regulation the tau phosphorylation state and microtubule binding activity of tau by protein osphatase 2A. Neuron, 17, 1201-1207
Soulié, C., Lepagnol, J., Delacourte, A. and Caillet-Boudin, M.L. (1996) Dephosphorylation udies of SK N SH-SY 5Y cell tau proteins by endogenous phosphatase activity. eurosci. Lett., 206, 189-192
ni, M.G., Bird, T.D. and Ghetti, B. (1998) Frontotemporal dementia and rkinsonism linked to chromo
387-402
, K., Van den Haute, C., Van Dorpe, J., Bruynseels, K., Vandezande, KGeerts, H., Mercken, M., Sciot, R., Van Lommel, A., Loos, R. and Van Leuven, F. (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice
erexpressing four-repeat human tau protein. Am. J. Pathol., 155, 2153-2165
Spittaels, K., Van den Haute, C., Van Dorpe, J., Geerts, H., Mercken, M., Bruynseels, K., Lasrado, R., Vandezande, K., Laenen, I., Boon, T., Van Lint, J., Vandenheede, J.,
oechars, D., Loos, R. and Van Leuven, F. (2000) Glycogen synthase kinase-3beta osphorylates protein tau and rescues the a
human four-repeat tau transgenic mice. J. Biol. Chem., 275, 41340-41349
K., Vogel, R., Thies, E., Mandelkow, E. and Mandelkow, E.M. (2002) Tau blocks
oxidative stress. J. Cell Biol., 156, 1051-1063
137

Literatur
Stein, T.D., Nicholas, J.A., DeCarli, C., Chan, S.L., Mattson, M.P. and Johnson, J.A. (2004) eutralization of transthyretin reverses the neuroprotective seffects of secreted yloid precursor protien (APP) in APPswmice resulting in Tau phosphorylation and
ss of hippocampal neurons: support for the amyloiud hypothesis. J. Neurosc
Namlo i., 24,
Steiner, SPC ation in
Steiner, be sease. Eur. Arch. Psychiatry Clin.
Steiner, GfoC 48-851
N
SturchleprA
ev
A
Sudo, H ., Shao, Z., Yazukawa, T., Ito, Y., Yamada, M., Hata, M.,
B
(2m 33-11940
Y n secreted
Tabira, Tin
anN
7707-7717
B., Mandelkow, E.M., Biernat, J., Gustke, N., Meyer, H.E., Schmidt, B., Mieskes, G., oling, B., Mieskes, G., Soling, H.D., Drechsel, D., Kirschner, M.W. et al. (1990) hosphorylation of microtubule-associated protein tau: identification of the site for a2(+)-calmodulin dependent kinase and relationship with tau phosphoryl
Alzheimer tangles. EMBO J. , 9, 3539-3544
H., Capell, A., Leimer, U. and Haass, C. (1999) Genes and mechanisms involved in ta-amyloid generation and Alzheimer’s di
Neurosci., 249, 266-70
H., Kostka, M., Romig, H., Basset, G., Pesold, B., Hardy, J., Capell, A., Meyn, L., rim, M., Baumeister, R., Fechteler, K. and Haass, C. (2000) Glycine 384 is required r presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. ell Biol., 2, 8
Struhl, G. and Greenwald, I. (1999) Presenilin is required for activity and nuclear access of otch in Drosophila. Nature, 398, 522-525
r-Pierrat, P.C., Abramowski, D., Duke, M. et al. (1997) Two amyloid precursor otein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. cad. Sci. U.S.A., 94, 13287-13292
Su, J.H., Anderson, A.J., Cummings, B.J. and Cotman, C.W. (1994) Immunohistochemical idence for apoptosis in Alzheimer’s disease. Neuroreport, 5, 2529-2533
Su, J.H., Zhao, M., Anderson, A.J., Srinivasan, A. and Cotman, C.W. (2001) Activated Caspase-3 expression in Alzheimer’s and aged control brain: correlation with
lzheimer pathology. Brain Res., 898, 350-357
., Yashimoto, Y., Niikura, THiraki, T., Kawasumi, M., Kouyama, K. and Nishimoto, I. (2001) Secreted Aβ does not mediate neurotoxicity by antibody stimulated amyloid precursor protein. Biochem.
iophys. Res. Commun., 382, 548-556
Sun, W., Qureshi, H.Y., Cafferty, P.W., Sobue K., Agar-Mawal, A., Neufield, K.D. et al. 002) Glykogen synthase kinase-3 beta is complexed with tau protein in brain icrotubules. J. Biol. Chem., 277, 119
Suzuki, N., Cheung, T.T., Cai, X.D., Odaka, A., Otvos, L. Jr, Eckmann, C., Golde, T.E. and ounkin, S.G. (1994) An increased percentage of long amyloid beta protei
by familial amyloid beta precursor (betaAPP717) mutants. Science, 264, 1336-1340
., Chui, D.H. and Kuroda, S. (2002) Significance of intracellular Aβ42 accumulation Alzheimer's disease. Front. Biosci., 7, a44-a49.
Tagasugi, N., Tomita, T., Hayashi, I., Tsuruoka, Niimura, M., Takahashi, Y., Thinakaran, G. d Iwatsubo, T. (2003) The role of presenilin co-factors in the γ-secretase complex.
ature, 422, 438-441
138

Literatur
Takashima, A., Noguchi, K., Sato, K., Hoshino, T. and Imahori, K. (1996) Tau protein kinase is essential for amyloid-beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. .S.A., 90, 7789-7793
I U
Kki
Takashim , T., Honda, T., Yasutake, T.,
N
98
ofB
m40
Terro, F., Czech, C., Esclaire, F., Elyaman, W., Yardin, C., Baclet, M.-C., Touchet, N.,
S
Thinaka ., Kim, G., Ratovitsky, T.,
pr
Tint, I., has no
in
Troncos oliatsos, V.E. (1996) In situ labeling of
Troy, C.(21386-1392
Takashima, A., Honda, T., Yasutake, K., Michel, G., Murayama, M., Murayama, O., Ishiguro, . and Yamaguchi, H. (1998a) Activation of tau protein kinase I/glycogen synthase nase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in
hippocampal neurons. Neurosci. Res., 31, 317-323
a, A., Murayama, M., Murayama, O., KohnoNihonmatsu, N., Mercken, M., Yamaguchi, H., Sugihara., S. and Wolozin, B. (1998b) Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc.
atl. Acad. Sci. U.S.A., 95, 9637-9641
Takei, Y., Teng, J., Harada, A. and Hirokawa, N. (2000) Defects in axonal elongation and neuronal migration in mice with disrupted tau and map 1 b genes. J. Cell Biol., 150,
9-100
Tamaoka, A., Kondo, T., Odaka, A., Sahara, N., Sawamura, N., Ozawa, K., Suzuki, N., Shoji, S. and Mori, H. (1994a) Biochemical evidence for the long-tail form (A beta 1-42/43)
amyloid beta protein as a seed molecule in cerebral deposits of Alzheimer's disease. iochem. Biophys. Res. Commun., 205, 834-842
Tamaoka, A., Odaka, A., Ishibashi, Y., Usami, M., Sahara, N., Suzuki, N., Nukina, N., Mizusawa, H., Shoji, S., Kanazawa, I. and Mori, H. (1994b) APP717 missense
utation affects the ratio of amyloid beta protein species (A beta 1-42/43 and a beta 1-) in familial Alzheimer's disease brain. J. Biol. Chem., 269, 32721-32724
Taylor, J.P., Hardy, J. and Fischbeck, K.H. (2002) Toxic proteins in neurodegenerative disease. Science, 296, 1991-1995
Tremp, G., Pradier, L. and Hugon, J. (2002) Neurons overexpressing mutant presenilin-1 are more sensitive to apoptosis induced by Endoplasmic Reticulum-Golgi
tress. J. Neurosci. Res., 69, 530-53
ran, G., Borchelt, D.R., Lee, M.K., Slunt, H.H., Spitzer, LDavenport, F., Nordstedt, C., Seeger, M., Hardy, J., Levey, A.I., Gandy, S.E., Jenkins, N.A., Copeland, N.G., Price, D.L. and Sisodia, S.S. (1996) Endoproteolysis of
esenilin 1 and accumulation of processed derivatives in vivo. Neuron, 17, 181-190
Slaughter, T., Fischer, I. and Black, M.M. (1998) Acute inactivation of tau effect on dynamics of microtubules in growing axons of cultured sympathetic neurons. J. Neurosci., 18, 8660-8673
Tomidokoro, Y., Ishiguro, K., Harigara, Y., Matsubara, E., Ikeda, M., Park, J.M., Yasutake, K., Kawarabayashi, T, Okamoto and Shoji, M. (2001) Abeta amyloidosis induces the
itial stage of tau accumulation in APP(Sw) mice. Neurosci. Lett., 299, 169-172
o, J.C., Sukhov, R.R., Kawas, C.H. and Kdying cortical neurons in normal aging and in Alzheimer’s disease: correlation with senile plaques and disease progression. J. Neuropathol. Exp. Med., 55, 1134-1142
M., Rabacchi, S.A., Friedman, W.J., Frappier, T.F., Brown, K. and Shelanski, M.L. 000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J. Neurosci., 20,
139

Literatur
Troy, C.M., Rabacchi, S.A., Xu, Z., Maroney, A.C., Connors, T.J., Shelanski, M.L. and reene, L.A. (2001) β-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal nase activation. J. Neurochem., 77, 15
Gki 7-164
m
Van den , I.,
p3D
RJarosinski, M.A., Biere, A.L., Curran, E., Burgess, T., Louis, J.C., Collins, F., Treanor,
73
de
of19
A
Von RotUre scription of its own precursor. J. Cell Sci., 117, 4435-4448
of
Wei, W. de-induced
Weihl, CMutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J. Neurosci., 19,
Welzel, M
pr32
D odeling
B
Vale, R.D., Banker, G. and Hall Z.W. (1992) The neuronal cytoskeleton. In An introduction to olecular neurobiology Z.W. Hall, editors. Sinauer, Sunderland. 247-280
Haute, C., Spittaels, K., Van Dorpe, J., Lasrado, R., Vandezande, K., LaenenGeerts, H. and Van Leuven, F. (2001) Coexpression of human cdk5 and its activator
5 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol. is., 8, 32-44
Vassar, R., Bennett, B.D., Babu-Khan, S., Kahn, S., Mendiaz, E.A., Denis, P., Teplow, D.B., oss, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J.,
J., Rogers, G. and Citron, M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science, 286,
5-741
Vestling, M., Wiehager, B., Tanii, H. and Cowburn, R.F. (2001) Akt activity in presenilin 1 wild-type and mutation transfected human SY-SY5Y neuroblastoma cells after serum
privation and high glucose stress. J. Neurosci. Res., 66, 448-456
Vigo-Pelfrey, C., Lee, D., Keim, P., Lieberburg, I. and Schenk, D. B. (1993) Characterization beta-amyloid peptide from human cerebrospinal fluid. J. Neurochem., 61, 1965-68
Vito, P., Wolozin, B., Ganjei, J.K., Iwasaki, K., Lacana, E. and D'Adamio, L.J (1996) Requirement of the Alzheimer’s disease gene PS2 for apoptosis. Opposing effect of
LG-3. J. Biol. Chem., 49, 31025-31028
z, R.C., Kohli, B.M., Bosset, J., Meier, M., Suzuki, T., Nitsch, R.M. and Konietzko, . (2004) The APP intracellular domain forms nuclear multiprotein complexes and gulates the tran
Wang, J.Z., Gong, C.X., Zaidi, T., Grundke-Iqbal, I. and Iqbal, K (1995) Dephosphorylation Protein Phosphatase-2A and -2B. J Biol.Chem., 270, 4854-4860
, Wang, X. and Kusiak, J.W. (2002) Signaling events in amyloid β-peptineuronal death and IGF-I protection. J. Biol. Chem., 277, 17649-17656
.C., Ghadge, G.D., Kennedy, S.G., Hay, N., Miller, R.J. and Roos, R.P. (1999)
5360-5369
A. (2005) Echtzeitanalyse der Tau-abhängigen Degeneration in einzelnen Neuronen. asterarbeit, Universität Osnabrück
Whitson, J.S., Glabe, C.G., Shintani, E., Abcar, A. and Cotman, C.W. (1990) Beta-amyloid otein promotes neuritic branching in hippocampal cultures. Neurosci. Lett., 110, 319-4
Wolfe, M.S., Xia, W., Moore, C.L., Leatherwood, D.D., Ostaszewski, B., Rahmati, T., onkor, I.O. and Selkoe, D. J. (1999) Peptidomimetic probes and molecular m
suggest that Alzheimer’s γ-secretase is an intramembrane cleaving aspartyl protease. iochemistry, 38, 4720-4727
140

Literatur
Wolfe, M.S. and Haass, C. (2001) The role of presenilins in gamma-secretase activity. J. Biol. hem., 276, 5413-5416 C
Wolozin, B., Iwasaki, K., Vito, P., Ganjei, J.K., Lacana, E., Sunderland, T., Zhao, B., Kusiak,
en Alzheimer Mutation. Science, 274, 1710-1713
prco reductase inhibitors. Arch. Neurol., 57, 1439-1443
inB
Xu, G., 002) Abeta deposition does not cause the
19
Gab
Yaar, M., Zhai, S., Pilch, D.F., Doyle, S.M., Eisenhauer, P.B., Fine, R.E. and Gilchrest, B.A.
po
Yamatsu Suzuki, N., Asami-
neof 352
acpresence of phosphatidyl-inositol. Biochem. Biophys. Res. Commun., 190, 710-715
SL protease
Yan, S.DR 685-
Yankner16
Yankner 990) Neurotrophic and neurotoxic effects
Ye, Y., pr
fe11
J. W., Wasco, W. and D’Adamio, L. (1996) Participation of presenilin-2 in apoptosis: hanced basal activity conferred by an
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G.G. and Siegel, G. (2000) Decreased evalence of Alzheimer disease associated with 3-hydroxy-3-methylglutaryl enzyme A
Xie, H., Litersky, J.M., Hartigan, J.A., Jope, R.S. and Johnson, G.V. (1998) The terrelationship between selective tau phosphorylation and microtubule association. rain Res., 798, 173-183
Gonzales, V. and Borchelt, D.R. (2aggregation of endogenous tau in transgenic mice. Alzheimer Dis. Assoc. Disord., 16,
6-201
Xu, H., Sweeney, D., Wang, R., Thinakaran, G., Lo, A.C., Sisodia, S.S. et al. (1997) eneration of Alzheimer beta-amyloid protein in the trans-Gogi network in the sence of vesicle formation. Proc. Natl. Acad. Sci. U. S. A., 94, 3748-3752
(1997) Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A ssible mechanism for Alzheimer’s disease. J. Clin. Invest.,100, 2333-2340
ji, T., Okamoto, T., Takeda, S., Fukumoto, H., Iwatsubo, T., Okada, A., Ireland, S., Kinae, T.B. and Nishimoto, I. (1996) G-protein-mediated
uronal DNA fragmentation by familial Alzheimer's disease-associated V642 mutants APP. Science, 272, 1349-1
Yamauchi, P.S. and Purich, D.L. (1993) Microtubule-associated protein interactions with tin filaments: evidence for differential behavior of neuronal MAP-2 and tau in the
Yan, R., Bienkowski, M.J., Shuck, M.E., Miao, H., Tory, M.C., Pauley, A.M., Brashier, J.R., tratman, N.C., Mathews, W.R., Buhl, A.E., Carter, D.B., Tomasselli, A.G., Parodi, .A., Heinrikson, R.L. and Gurney, M.E. (1999) Membrane-anchored aspartyl
with Alzheimer's disease β-secretase activity. Nature, 402, 533-537
., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., Slattery, T., Zhao, L. et al. (1996) AGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature, 382,
691
, B.A. (1996) Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron, , 921-932
, B.D., Duffy, L.K. and Kirschner, D.A. (1of amyloid beta protein: reversal by tachykinin neuropeptides. Science, 250, 279-282
Lukinova, N. and Fortini, M.E. (1999) Neurogenic phenotypes and altered notch ocessing in Drosophila presenilin mutants. Nature, 398, 525-529
Yoshida, H. and Ihara, Y. (1993) Tau in paired helical filaments is functionally distinct from tal tau: assembly and incompetence of paired helical filament-tau. J. Neurochem., 61, 83-1186
141

Literatur
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A. and Kaibuchi, K. (2005) SK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell, 120, 7-149
G13
Zhang, J. and Johnson, G.V. (2000) Tau protein is hyperphosphorylated in a site-specific
Zhang, Z
tr
m47
peseptal cultures. Neurosci., 115, 201-211
ers,
Yuan, J. and Yanker, B.A. (2000) Apoptosis in the nervous system. Nature, 407, 802-809
manner in apoptotic neuronal PC12 cells. J. Neurochem., 75, 2346-2357
., Nadeau, P., Song, W., Donoviel, D., Yuan, M., Bernstein, A.. and Yanker, B.A. (2000) Presenilins are required for gamma-secretase cleavage of beta-APP and
ansmembrane cleavage of Notch-1. Nat. Cell Biol., 2, 463-465
Zhao, B., Chrest, F.J., Horton, W.E. Jr, Sisodia, S.S. and Kusiak, JW. (1997) Expression of utant amyloid precursor proteins induces apoptosis in PC12 cells. J. Neurosci. Res., , 253-263
Zheng, W.H., Bastianetto, S., Mennicken, F., Ma, W. and Kar, S. (2002) Amyloid beta ptide induces tau phosphorylation and loss of cholinergic neurons in rat primary
Zheng, Y.-L., Kesavapany, S., Gravell, M., Hamilton, R.S., Schubert, M., Amin, N., AlbW., Grant, P. and Pant, H.C. (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J., 24, 209-220
142

Danksagung
H Danksagung
Ganz besonderer Dank gilt meinem Doktorvater Herrn Prof. Dr. Roland Brandt, dass er mir
die Möglichkeit gegeben hat, so viel zu lernen. Durch seine ausgezeichnete Betreuung hat er
mich während der gesamten Zeit vollstens unterstützt und bestärkt.
Außerdem möchte ich allen Mitgliedern der Arbeitsgruppe für ihre Hilfs- und Diskussions-
bereitschaft danken. Insbesondere möchte ich mich für alle Arbeiten rund um den Tierstall
bedanken, ohne die meine Promotion in dieser Form nicht durchführbar gewesen wäre.
Auch bei Frau Dr. Anne Eckert möchte ich mich bedanken für ihre Kooperationsbereitschaft,
unserer Arbeitsgruppe die transgenen Mauslinien zur Verfügung zu stellen.
Ganz spezieller Dank gilt allen meinen lieben Freunden, die mir immer wieder Mut gemacht
haben und die immer für mich da waren.
Meinen Eltern möchte ich ganz besonders dafür danken, dass sie mir diesen Weg ermöglicht
haben. Sie waren während der gesamten Zeit für mich da und haben mir Rückhalt gegeben.
143
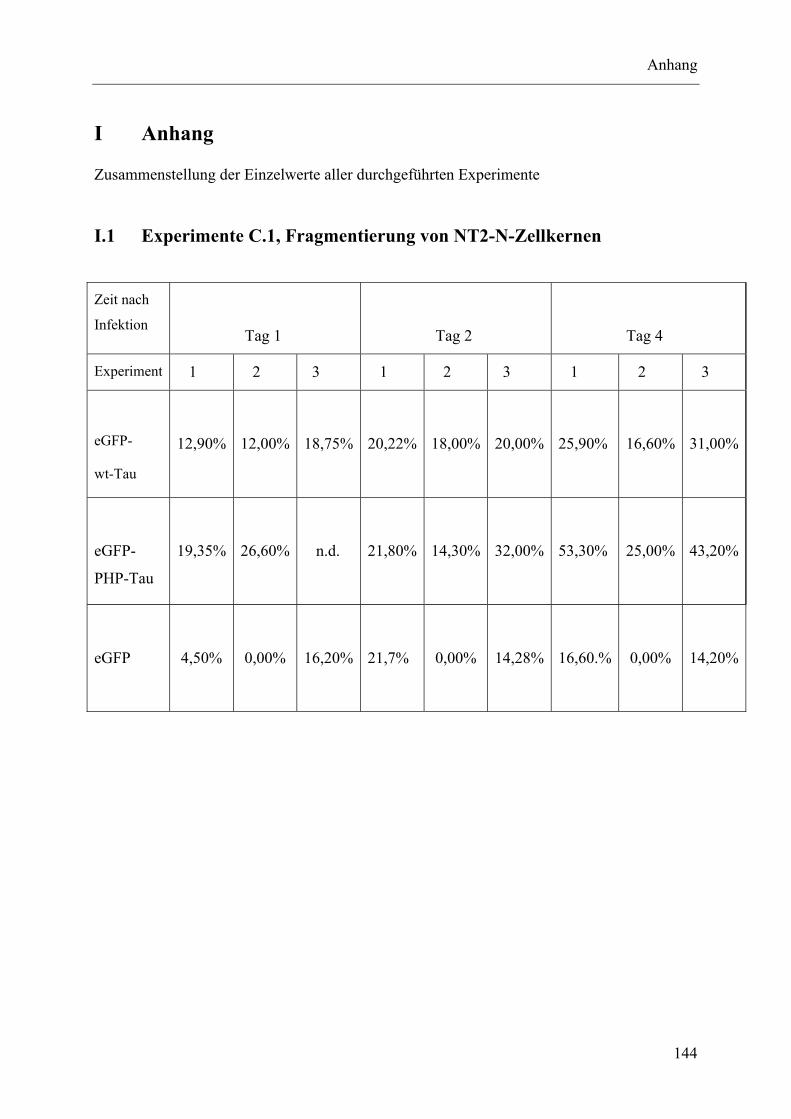
Anhang
I Anhang
Zusammenstellung der Einzelwerte aller durchgeführten Experimente
I.1 Experimente C.1, Fragmentierung von NT2-N-Zellkernen
Zeit nach
Infektion
Tag 1
Tag 2
Tag 4
Experiment 1 2 3 1 2 3 1 2 3
eGFP-
wt-Tau
12,90%
12,00%
18,75%
20,22%
18,00%
20,00%
25,90%
16,60%
31,00%
eGFP-
PHP-Tau
19,35%
26,60%
n.d.
21,80%
14,30%
32,00%
53,30%
25,00%
43,20%
eGFP
4,50%
0,00%
16,20%
21,7%
0,00%
14,28%
16,60.%
0,00%
14,20%
144
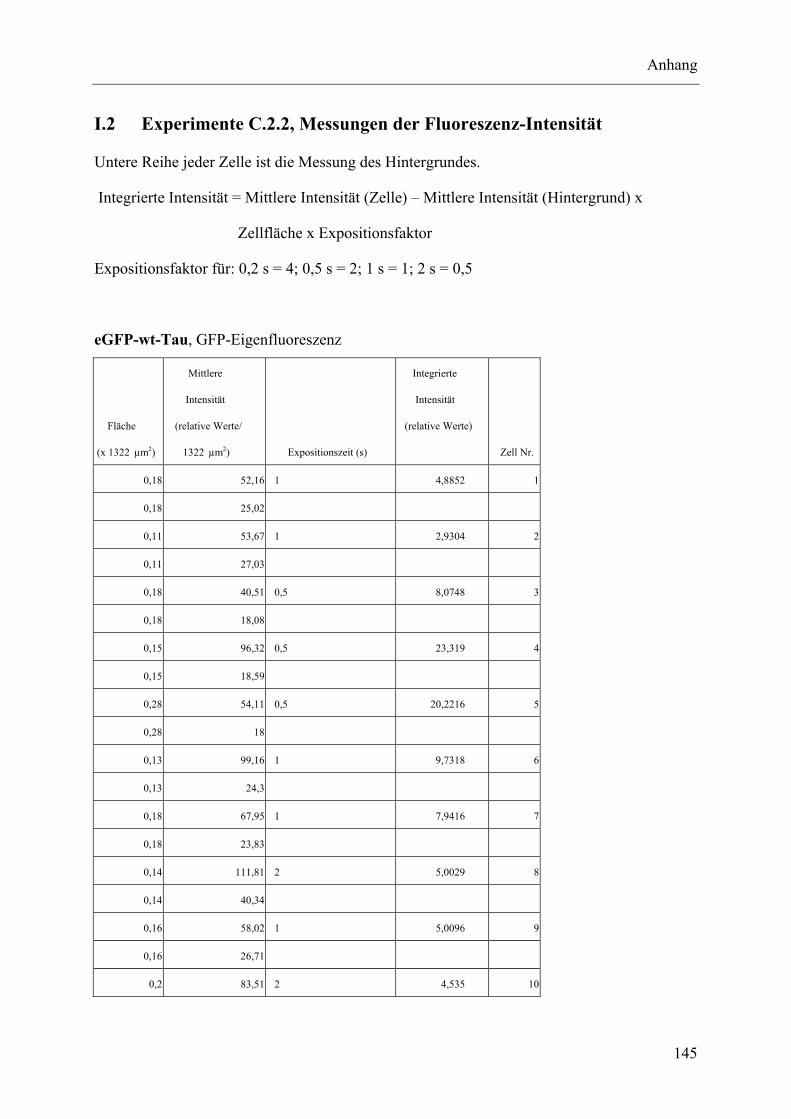
Anhang
I.2 Experimente C.2.2, Messungen der Fluoreszenz-Intensität
Untere Reihe jeder Zelle ist die Messung des Hintergrundes.
Integrierte Intensität = Mittlere Intensität (Zelle) – Mittlere Intensität (Hintergrund) x
Zellfläche x Expositionsfaktor
Expositionsfaktor für: 0,2 s = 4; 0,5 s = 2; 1 s = 1; 2 s = 0,5
eGFP-wt-Tau, GFP-Eigenfluoreszenz
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,18 52,16 1 4,8852 1
0,18 25,02
0,11 53,67 1 2,9304 2
0,11 27,03
0,18 40,51 0,5 8,0748 3
0,18 18,08
0,15 96,32 0,5 23,319 4
0,15 18,59
0,28 54,11 0,5 20,2216 5
0,28 18
0,13 99,16 1 9,7318 6
0,13 24,3
0,18 67,95 1 7,9416 7
0,18 23,83
0,14 111,81 2 5,0029 8
0,14 40,34
0,16 58,02 1 5,0096 9
0,16 26,71
0,2 83,51 2 4,535 10
145
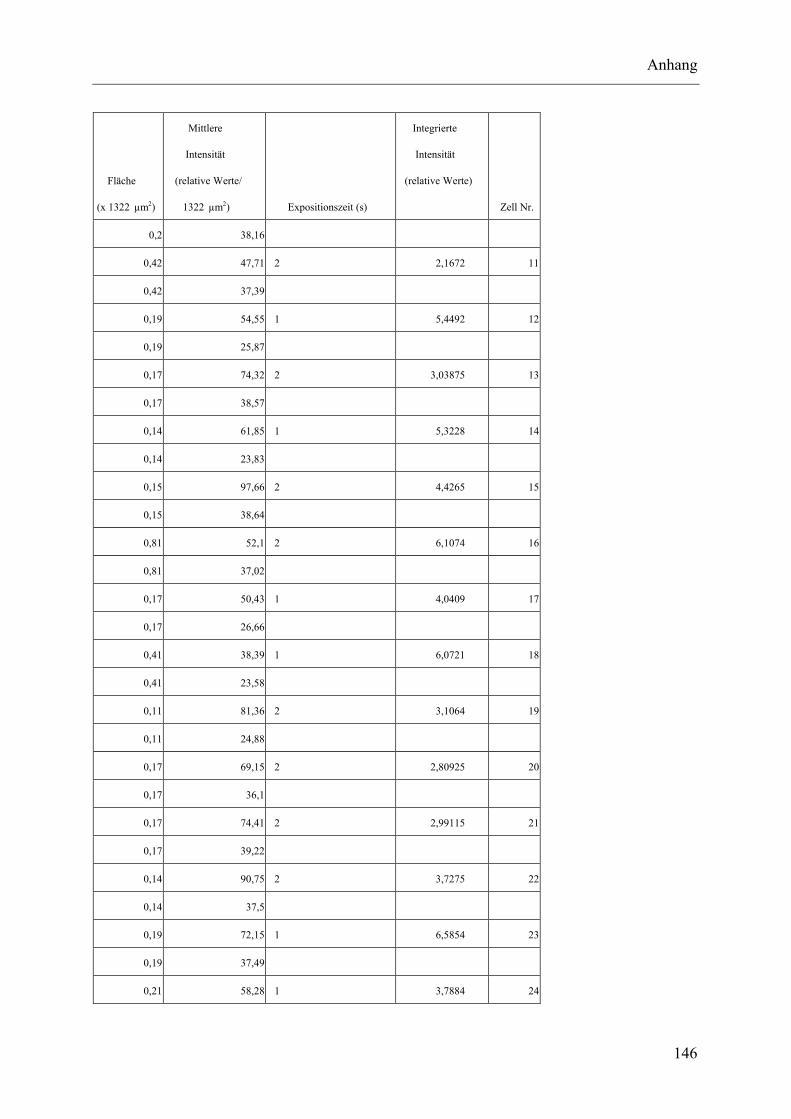
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,2 38,16
0,42 47,71 2 2,1672 11
0,42 37,39
0,19 54,55 1 5,4492 12
0,19 25,87
0,17 74,32 2 3,03875 13
0,17 38,57
0,14 61,85 1 5,3228 14
0,14 23,83
0,15 97,66 2 4,4265 15
0,15 38,64
0,81 52,1 2 6,1074 16
0,81 37,02
0,17 50,43 1 4,0409 17
0,17 26,66
0,41 38,39 1 6,0721 18
0,41 23,58
0,11 81,36 2 3,1064 19
0,11 24,88
0,17 69,15 2 2,80925 20
0,17 36,1
0,17 74,41 2 2,99115 21
0,17 39,22
0,14 90,75 2 3,7275 22
0,14 37,5
0,19 72,15 1 6,5854 23
0,19 37,49
0,21 58,28 1 3,7884 24
146
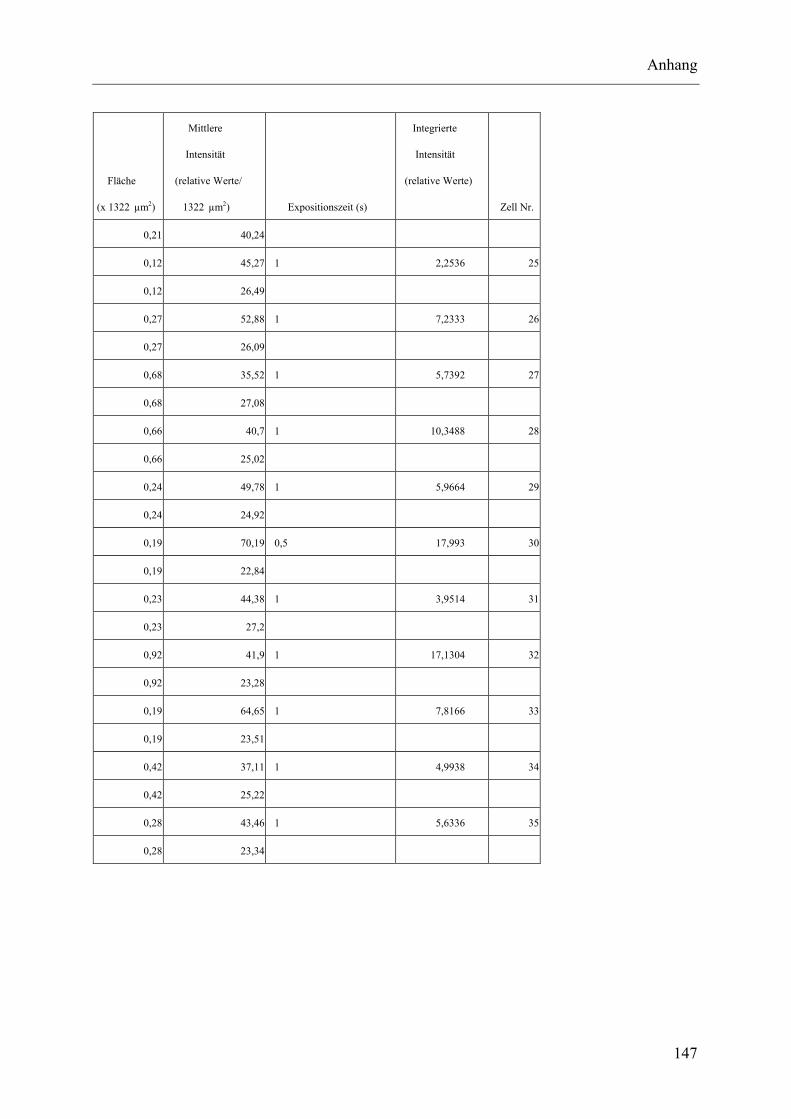
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,21 40,24
0,12 45,27 1 2,2536 25
0,12 26,49
0,27 52,88 1 7,2333 26
0,27 26,09
0,68 35,52 1 5,7392 27
0,68 27,08
0,66 40,7 1 10,3488 28
0,66 25,02
0,24 49,78 1 5,9664 29
0,24 24,92
0,19 70,19 0,5 17,993 30
0,19 22,84
0,23 44,38 1 3,9514 31
0,23 27,2
0,92 41,9 1 17,1304 32
0,92 23,28
0,19 64,65 1 7,8166 33
0,19 23,51
0,42 37,11 1 4,9938 34
0,42 25,22
0,28 43,46 1 5,6336 35
0,28 23,34
147
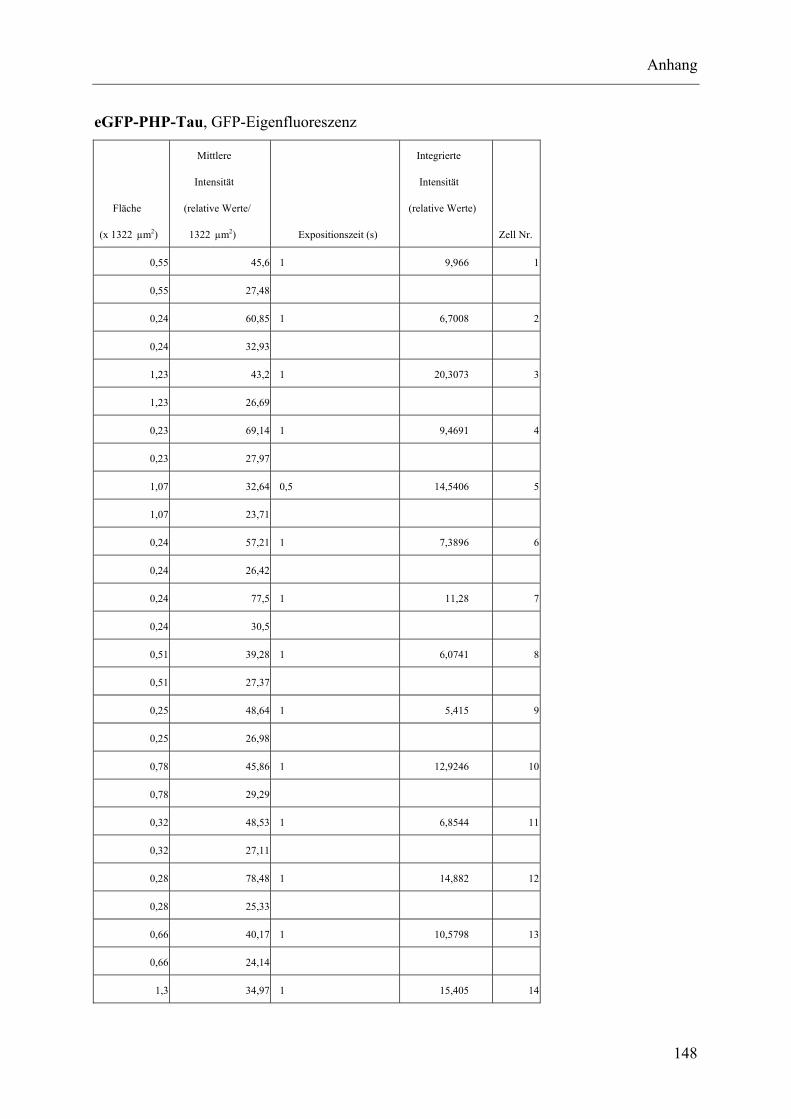
Anhang
eGFP-PHP-Tau, GFP-Eigenfluoreszenz
Fläche
(x 1322 µm ) 2
Mittlere
Intensität
1322 µm ) 2 Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,55 45,6 1 9,966 1
(relative Werte/
0,55
0,24 60,85 1 6,7008 2
0,24 32,93
1,23 43,2 1 20,3073 3
1,23 26,69
0,23 69,14 1 9,4691 4
0,23 27,97
1,07 32,64 0,5 14,5406 5
1,07 23,71
0,24 57,21 1 7,3896 6
0,24 26,42
0,24 77,5 1 11,28 7
0,24 30,5
0,51 39,28 1 6,0741 8
0,51 27,37
0,25 48,64 1 5,415 9
0,25 26,98
0,78 45,86 1 12,9246 10
0,78 29,29
0,32 48,53 1 6,8544 11
0,32 27,11
0,28 78,48 1 14,882 12
0,28 25,33
0,66 40,17 1 10,5798 13
0,66 24,14
1,3 34,97 1 15,405 14
27,48
148
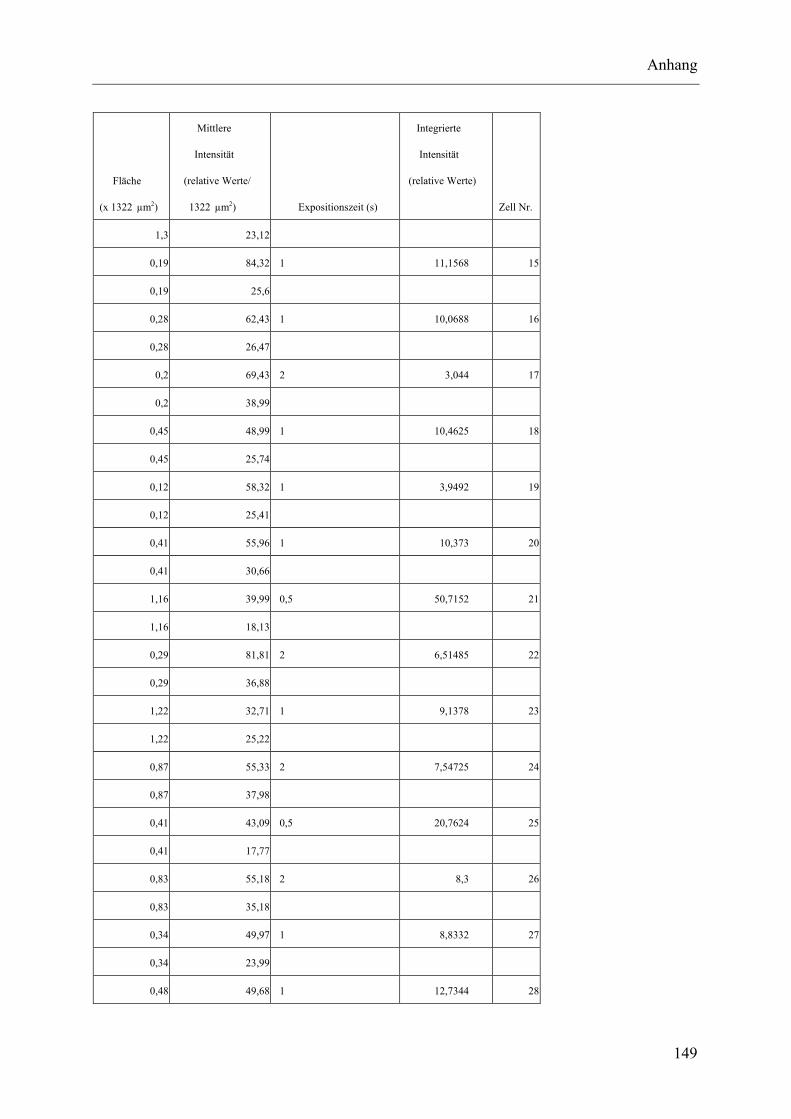
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
1,3 23,12
0,19 84,32 1 11,1568 15
0,19 25,6
0,28 62,43 1 10,0688 16
0,28 26,47
0,2 69,43 2 3,044 17
0,2 38,99
0,45 48,99 1 10,4625 18
0,45 25,74
0,12 58,32 1 3,9492 19
0,12 25,41
0,41 55,96 1 10,373 20
0,41 30,66
1,16 39,99 0,5 50,7152 21
1,16 18,13
0,29 81,81 2 6,51485 22
0,29 36,88
1,22 32,71 1 9,1378 23
1,22 25,22
0,87 55,33 2 7,54725 24
0,87 37,98
0,41 43,09 0,5 20,7624 25
0,41 17,77
0,83 55,18 2 8,3 26
0,83 35,18
0,34 49,97 1 8,8332 27
0,34 23,99
0,48 49,68 1 12,7344 28
149
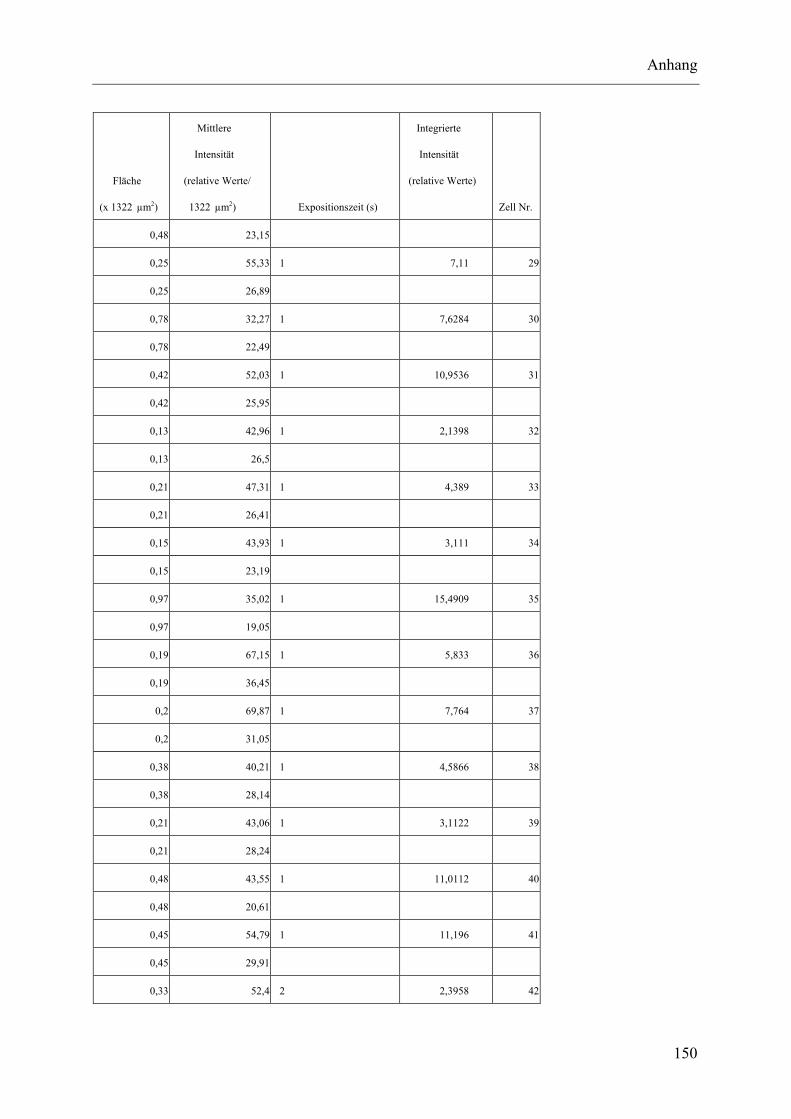
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,48 23,15
0,25 55,33 1 7,11 29
0,25 26,89
0,78 32,27 1 7,6284 30
0,78 22,49
0,42 52,03 1 10,9536 31
0,42 25,95
0,13 42,96 1 2,1398 32
0,13 26,5
0,21 47,31 1 4,389 33
0,21 26,41
0,15 43,93 1 3,111 34
0,15 23,19
0,97 35,02 1 15,4909 35
0,97 19,05
0,19 67,15 1 5,833 36
0,19 36,45
0,2 69,87 1 7,764 37
0,2 31,05
0,38 40,21 1 4,5866 38
0,38 28,14
0,21 43,06 1 3,1122 39
0,21 28,24
0,48 43,55 1 11,0112 40
0,48 20,61
0,45 54,79 1 11,196 41
0,45 29,91
0,33 52,4 2 2,3958 42
150
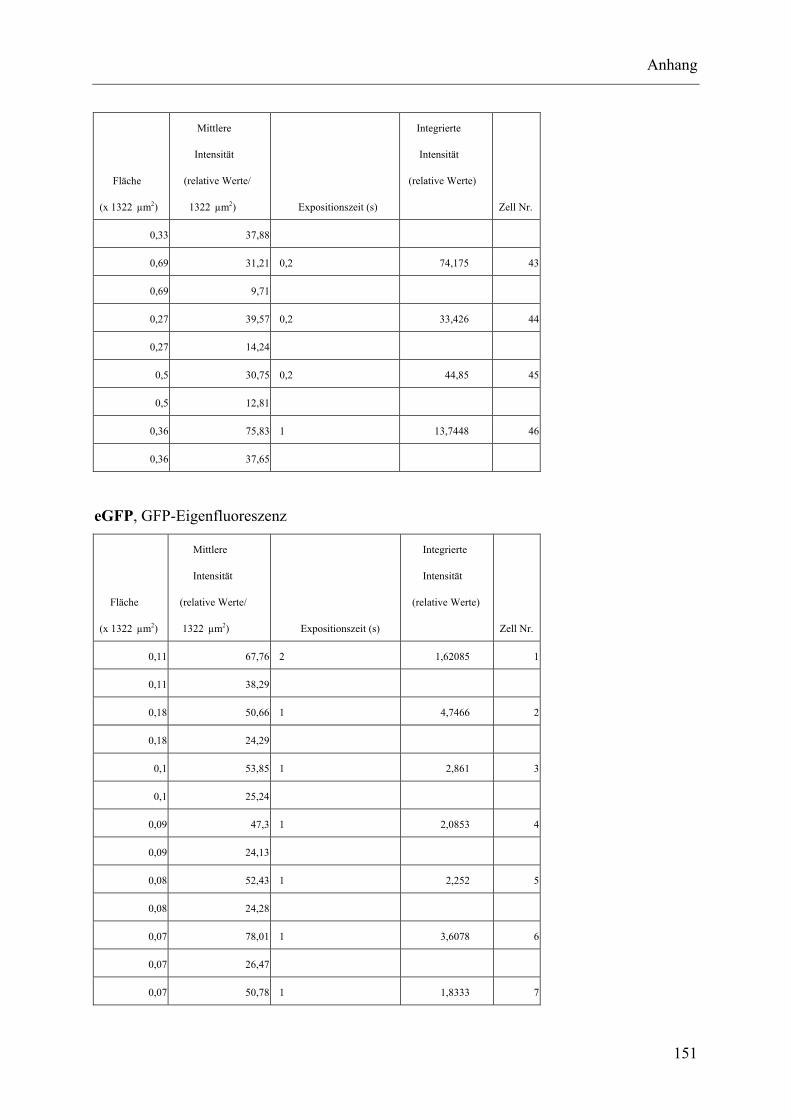
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,33 37,88
0,69 31,21 0,2 74,175 43
0,69 9,71
0,27 39,57 0,2 33,426 44
0,27 14,24
0,5 30,75 0,2 44,85 45
0,5 12,81
0,36 75,83 1 13,7448 46
0,36 37,65
eGFP, GFP-Eigenfluoreszenz
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,11 67,76 2 1,62085 1
0,11 38,29
0,18 50,66 1 4,7466 2
0,18 24,29
0,1 53,85 1 2,861 3
0,1 25,24
0,09 47,3 1 2,0853 4
0,09 24,13
0,08 52,43 1 2,252 5
0,08 24,28
0,07 78,01 1 3,6078 6
0,07 26,47
0,07 50,78 1 1,8333 7
151
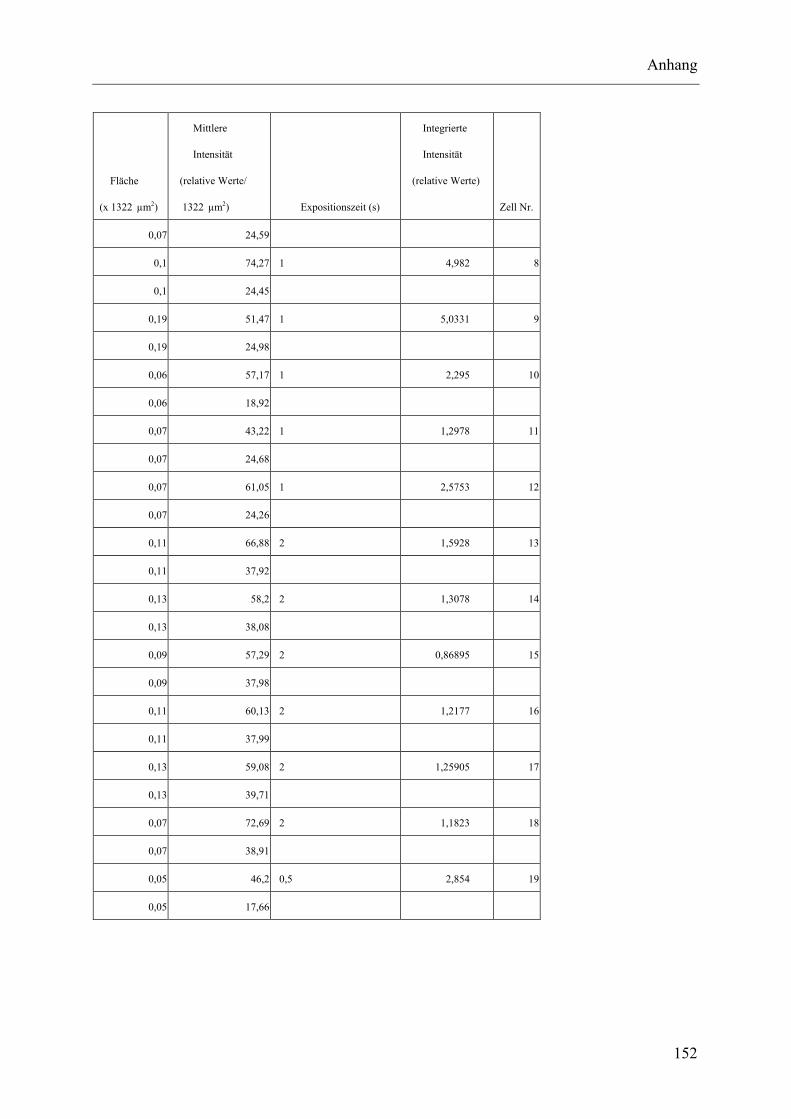
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,07 24,59
0,1 74,27 1 4,982 8
0,1 24,45
0,19 51,47 1 5,0331 9
0,19 24,98
0,06 57,17 1 2,295 10
0,06 18,92
0,07 43,22 1 1,2978 11
0,07 24,68
0,07 61,05 1 2,5753 12
0,07 24,26
0,11 66,88 2 1,5928 13
0,11 37,92
0,13 58,2 2 1,3078 14
0,13 38,08
0,09 57,29 2 0,86895 15
0,09 37,98
0,11 60,13 2 1,2177 16
0,11 37,99
0,13 59,08 2 1,25905 17
0,13 39,71
0,07 72,69 2 1,1823 18
0,07 38,91
0,05 46,2 0,5 2,854 19
0,05 17,66
152
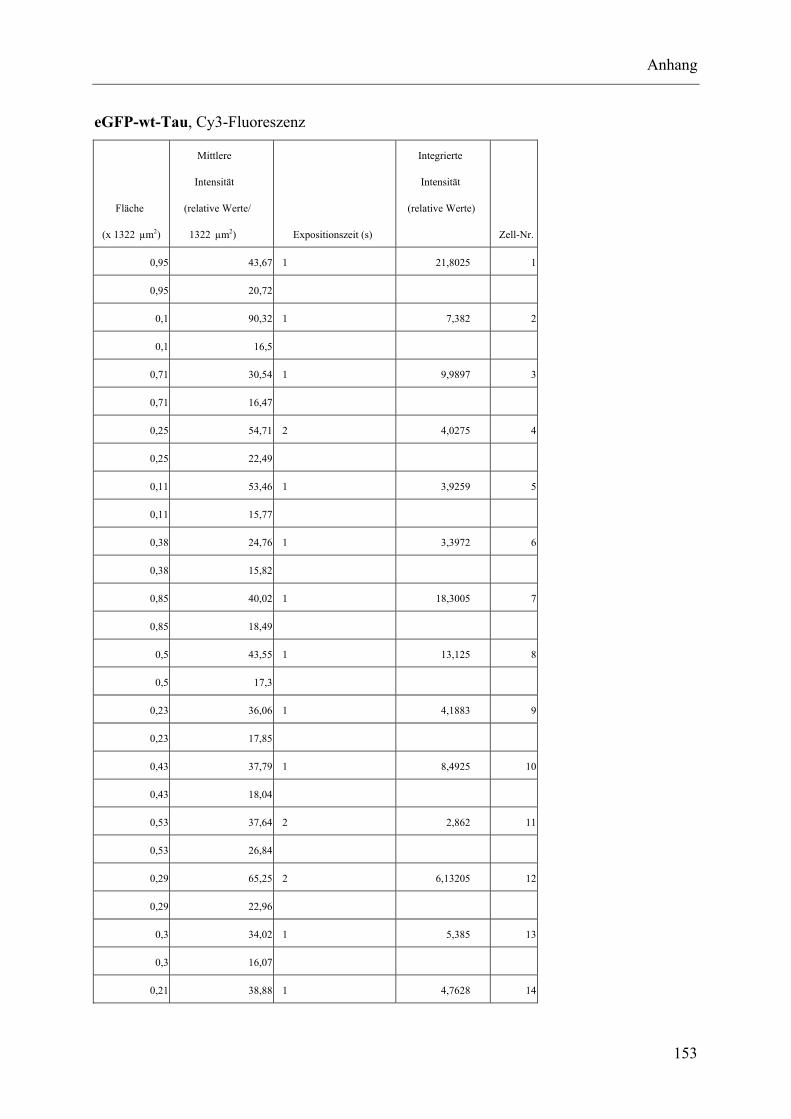
Anhang
eGFP-wt-Tau, Cy3-Fluoreszenz
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell-Nr.
0,95 43,67 1 21,8025 1
0,95 20,72
0,1 90,32 1 7,382 2
0,1 16,5
0,71 30,54 1 9,9897 3
0,71 16,47
0,25 54,71 2 4,0275 4
0,25 22,49
0,11 53,46 1 3,9259 5
0,11 15,77
0,38 24,76 1 3,3972 6
0,38 15,82
0,85 40,02 1 18,3005 7
0,85 18,49
0,5 43,55 1 13,125 8
0,5 17,3
0,23 36,06 1 4,1883 9
0,23 17,85
0,43 37,79 1 8,4925 10
0,43 18,04
0,53 37,64 2 2,862 11
0,53 26,84
0,29 65,25 2 6,13205 12
0,29 22,96
0,3 34,02 1 5,385 13
0,3 16,07
0,21 38,88 1 4,7628 14
153
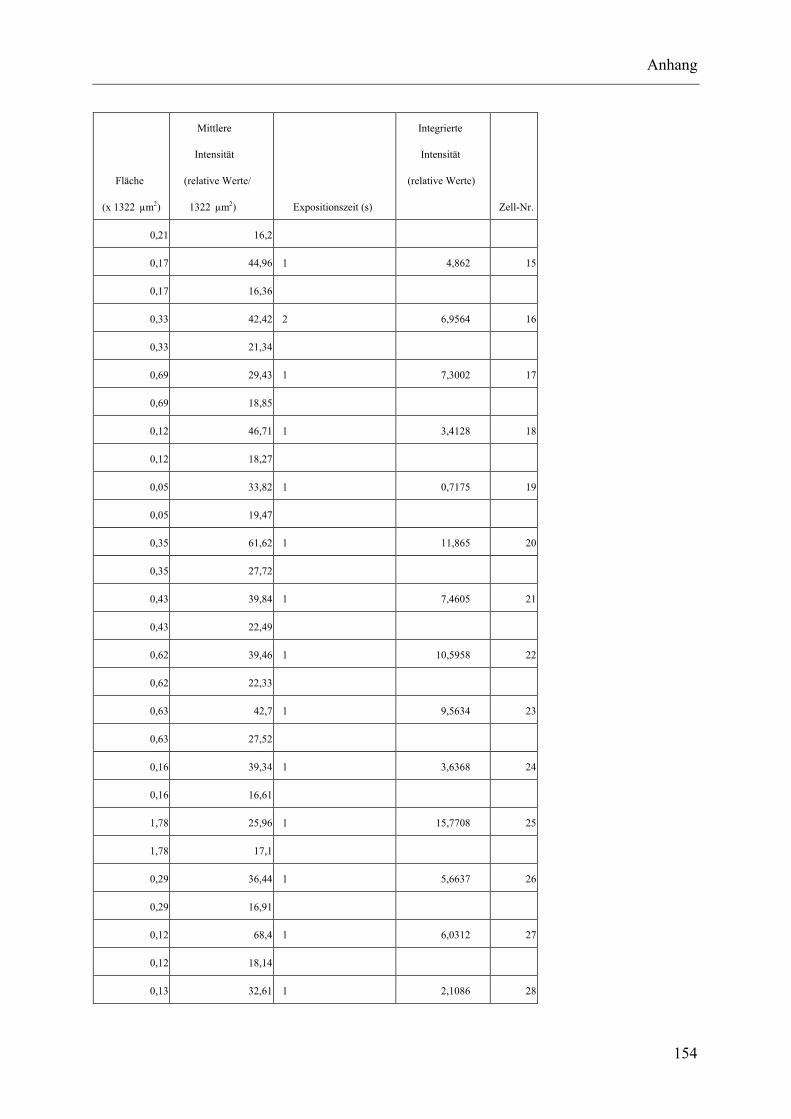
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell-Nr.
0,21 16,2
0,17 44,96 1 4,862 15
0,17 16,36
0,33 42,42 2 6,9564 16
0,33 21,34
0,69 29,43 1 7,3002 17
0,69 18,85
0,12 46,71 1 3,4128 18
0,12 18,27
0,05 33,82 1 0,7175 19
0,05 19,47
0,35 61,62 1 11,865 20
0,35 27,72
0,43 39,84 1 7,4605 21
0,43 22,49
0,62 39,46 1 10,5958 22
0,62 22,33
0,63 42,7 1 9,5634 23
0,63 27,52
0,16 39,34 1 3,6368 24
0,16 16,61
1,78 25,96 1 15,7708 25
1,78 17,1
0,29 36,44 1 5,6637 26
0,29 16,91
0,12 68,4 1 6,0312 27
0,12 18,14
0,13 32,61 1 2,1086 28
154
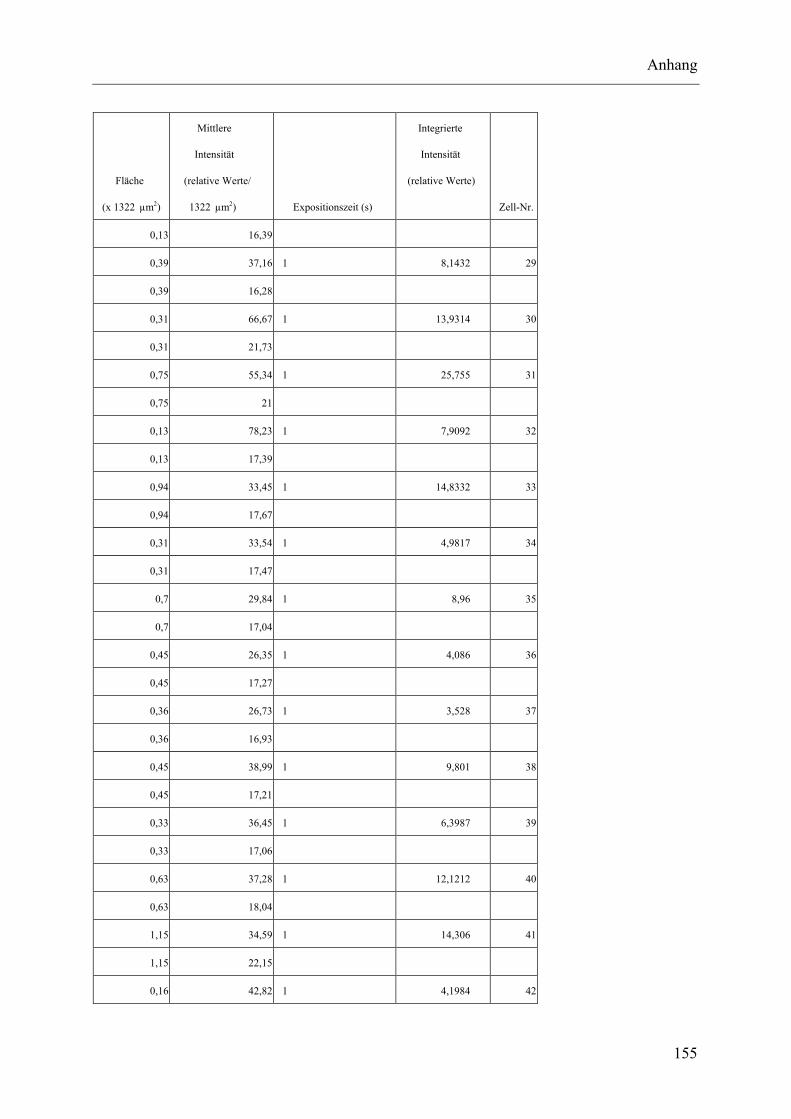
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell-Nr.
0,13 16,39
0,39 37,16 1 8,1432 29
0,39 16,28
0,31 66,67 1 13,9314 30
0,31 21,73
0,75 55,34 1 25,755 31
0,75 21
0,13 78,23 1 7,9092 32
0,13 17,39
0,94 33,45 1 14,8332 33
0,94 17,67
0,31 33,54 1 4,9817 34
0,31 17,47
0,7 29,84 1 8,96 35
0,7 17,04
0,45 26,35 1 4,086 36
0,45 17,27
0,36 26,73 1 3,528 37
0,36 16,93
0,45 38,99 1 9,801 38
0,45 17,21
0,33 36,45 1 6,3987 39
0,33 17,06
0,63 37,28 1 12,1212 40
0,63 18,04
1,15 34,59 1 14,306 41
1,15 22,15
0,16 42,82 1 4,1984 42
155
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell-Nr.
0,16 16,58
0,14 31,66 1 2,156 43
0,14 16,26
0,63 24,17 1 4,788 44
0,63 16,57
0,48 32,1 1 7,584 45
0,48 16,3
0,28 36,01 1 5,4712 46
0,28 16,47
0,16 56,11 1 5,928 47
0,16 19,06
0,42 35,01 1 7,1778 48
0,42 17,92
0,08 62,21 1 3,5144 49
0,08 18,28
0,2 48,4 1 6,352 50
0,2 16,64
0,23 50,07 1 7,7349 51
0,23 16,44
0,3 46,89 1 8,655 52
0,3 18,04
0,36 28,87 1 4,7808 53
0,36 15,59
0,39 39,8 1 8,8686 54
0,39 17,06
0,23 47,45 0,5 17,2592 55
0,23 9,93
0,26 47,81 0,5 18,8552 56
156
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell-Nr.
0,26
Fläche
(x 1322 µm ) 2
Mittlere
Intensität
(relative Werte/
2 Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
3,03 27,21 1 33,2694
3,03 16,23
0,41 42,03 1 9,2824
0,41 19,39
0,46 42,04 1 11,7806
0,46 16,43
0,85 24,87 1 4,9895
0,85 19
4,35 25,83 1 35,9745
4,35 17,56
2,77 25,94 1 27,3676
2,77 16,06
1,89 35,13 1 10,2438
1,89 29,71
0,46 38,45 1 9,6508
0,46 17,47
0,24 33,51 1 2,592
0,24 22,71
0,3 32,01 1 2,847
0,3 22,52
0,15 56,38 1 4,8165
11,55
eGFP-PHP-Tau, Cy3-Fluoreszenz
1322 µm ) Zell Nr.
1
2
3
4
5
6
7
8
9
10
11
157
Anhang
Fläche
(x 1322 µm ) 2
Mittlere
Intensität
(relative Werte/
2 Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
0,15 24,27
0,65 30,11 1 4,316
0,65 23,47
0,63 30,24 1 4,9518
0,63 22,38
0,37 31,49 1 3,3485
0,37 22,44
0,26 47,13 1 7,2176
0,26 19,37
0,67 45,21 1 15,1353
0,67 22,62
0,52 36,03 1 8,6268
0,52 19,44
0,49 26,11 1 3,6015
0,49 18,76
0,21 61,86 2 3,7023
0,21 26,6
1,07 32,18 2 3,51495
1,07 25,61
1,37 24,81 2 6,634191
1,37 18,64
0,3 58,22 1 12,225
0,3 17,47
2,02 34,43 1 31,6534
2,02 18,76
0,19 31,07 1 1,919
0,19 20,97
0,24 25,55 1 1,4496
1322 µm ) Zell Nr.
12
13
14
15
16
17
18
19
20
21
22
23
24
25
158
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,24 19,51
0,12 43,68 1 2,7528 26
0,12 20,74
0,38 27,91 1 3,5036 27
0,38 18,69
0,19 38,51 1 4,085 28
0,19 17,01
0,28 37,13 1 4,9952 29
0,28 19,29
0,72 23,58 1 3,6792 30
0,72 18,47
0,39 45,45 1 10,1946 31
0,39 19,31
0,51 27,07 1 4,59 32
0,51 18,07
0,75 28,97 1 8,445 33
0,75 17,71
0,37 34,3 1 5,9052 34
0,37 18,34
0,15 52,78 1 5,181 35
0,15 18,24
0,29 46,42 1 8,0997 36
0,29 18,49
0,41 29,66 1 4,8257 37
0,41 17,89
0,37 40,34 1 7,4888 38
0,37 20,1
0,75 30,2 1 3,75 39
159
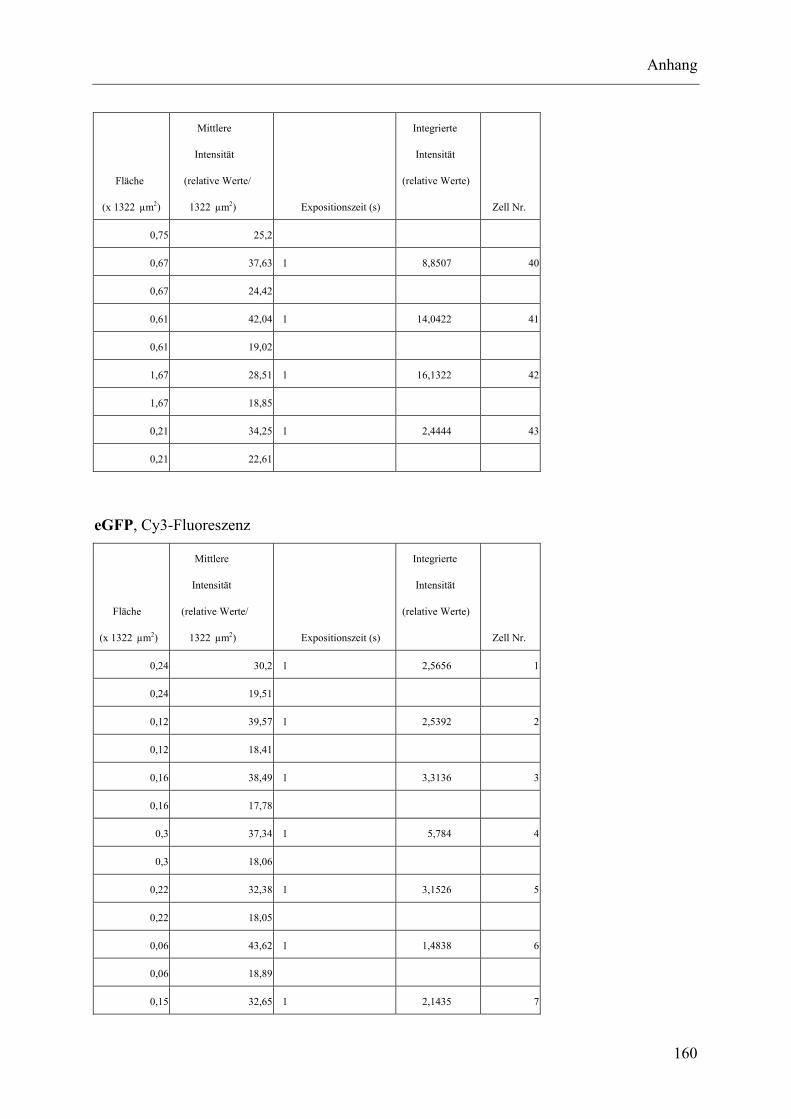
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,75 25,2
0,67 37,63 1 8,8507 40
0,67 24,42
0,61 42,04 1 14,0422 41
0,61 19,02
1,67 28,51 1 16,1322 42
1,67 18,85
0,21 34,25 1 2,4444 43
0,21 22,61
eGFP, Cy3-Fluoreszenz
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,24 30,2 1 2,5656 1
0,24 19,51
0,12 39,57 1 2,5392 2
0,12 18,41
0,16 38,49 1 3,3136 3
0,16 17,78
0,3 37,34 1 5,784 4
0,3 18,06
0,22 32,38 1 3,1526 5
0,22 18,05
0,06 43,62 1 1,4838 6
0,06 18,89
0,15 32,65 1 2,1435 7
160
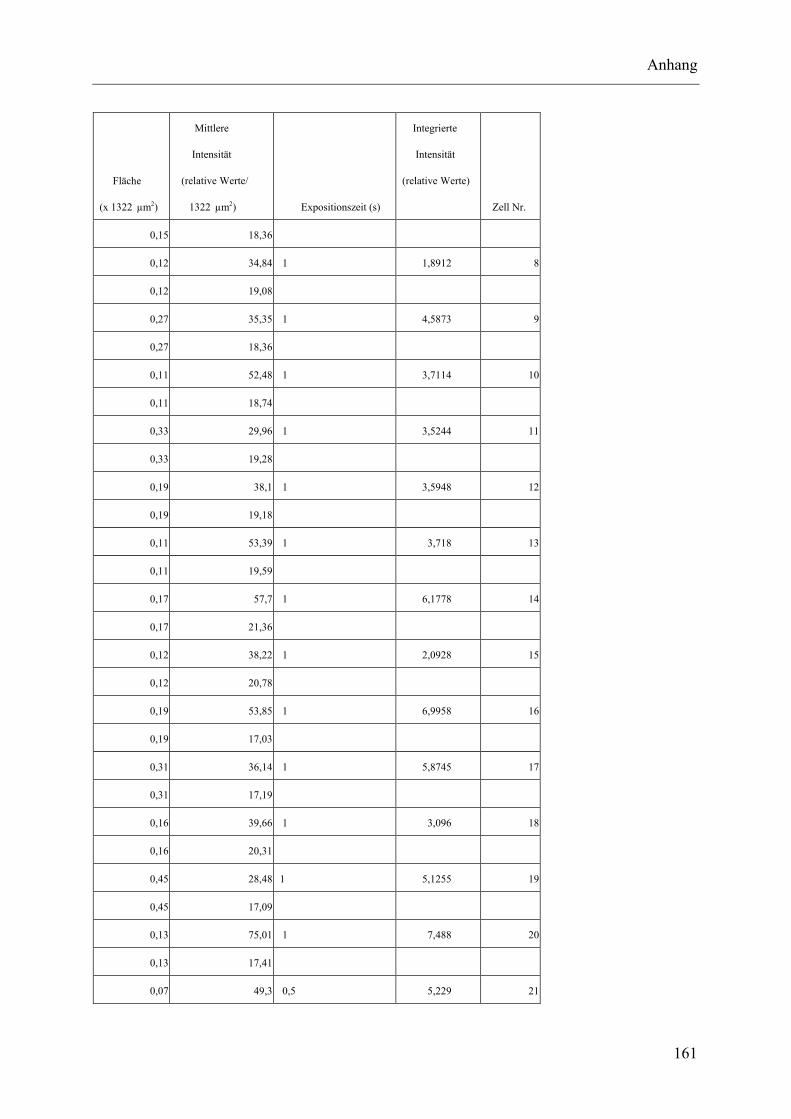
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,15 18,36
0,12 34,84 1 1,8912 8
0,12 19,08
0,27 35,35 1 4,5873 9
0,27 18,36
0,11 52,48 1 3,7114 10
0,11 18,74
0,33 29,96 1 3,5244 11
0,33 19,28
0,19 38,1 1 3,5948 12
0,19 19,18
0,11 53,39 1 3,718 13
0,11 19,59
0,17 57,7 1 6,1778 14
0,17 21,36
0,12 38,22 1 2,0928 15
0,12 20,78
0,19 53,85 1 6,9958 16
0,19 17,03
0,31 36,14 1 5,8745 17
0,31 17,19
0,16 39,66 1 3,096 18
0,16 20,31
0,45 28,48 1 5,1255 19
0,45 17,09
0,13 75,01 1 7,488 20
0,13 17,41
0,07 49,3 0,5 5,229 21
161
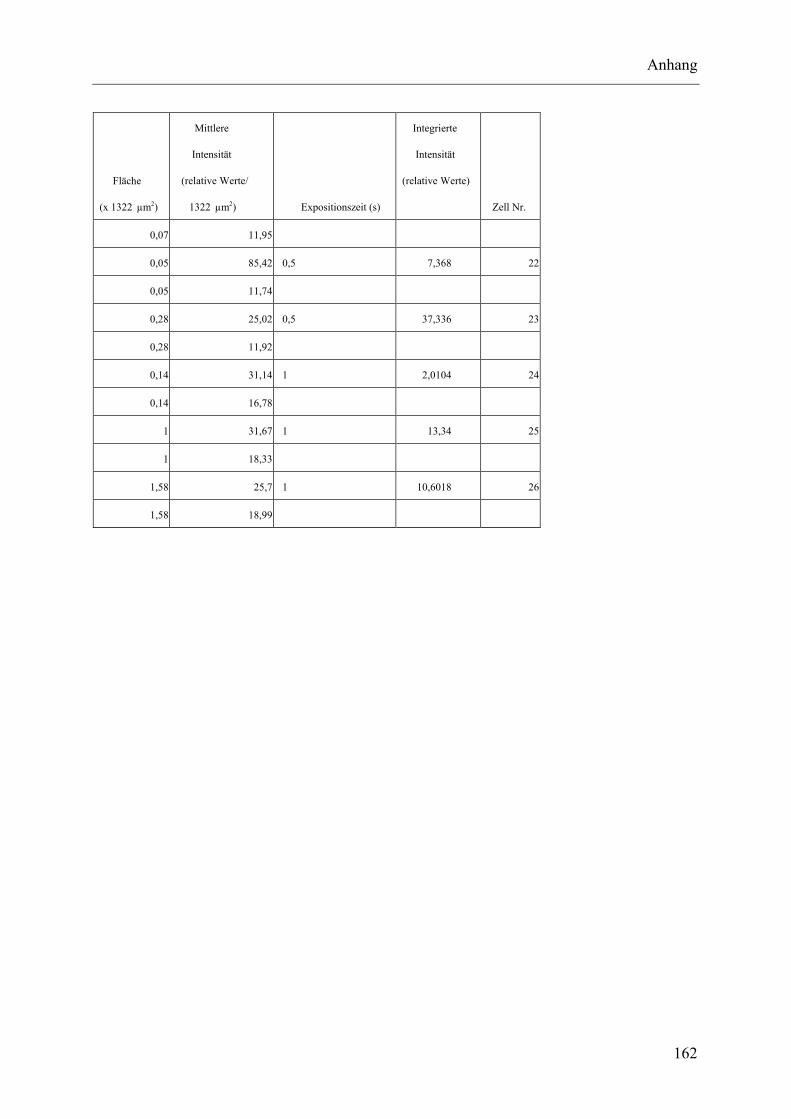
Anhang
Fläche
(x 1322 µm2)
Mittlere
Intensität
(relative Werte/
1322 µm2) Expositionszeit (s)
Integrierte
Intensität
(relative Werte)
Zell Nr.
0,07 11,95
0,05 85,42 0,5 7,368 22
0,05 11,74
0,28 25,02 0,5 37,336 23
0,28 11,92
0,14 31,14 1 2,0104 24
0,14 16,78
1 31,67 1 13,34 25
1 18,33
1,58 25,7 1 10,6018 26
1,58 18,99
162
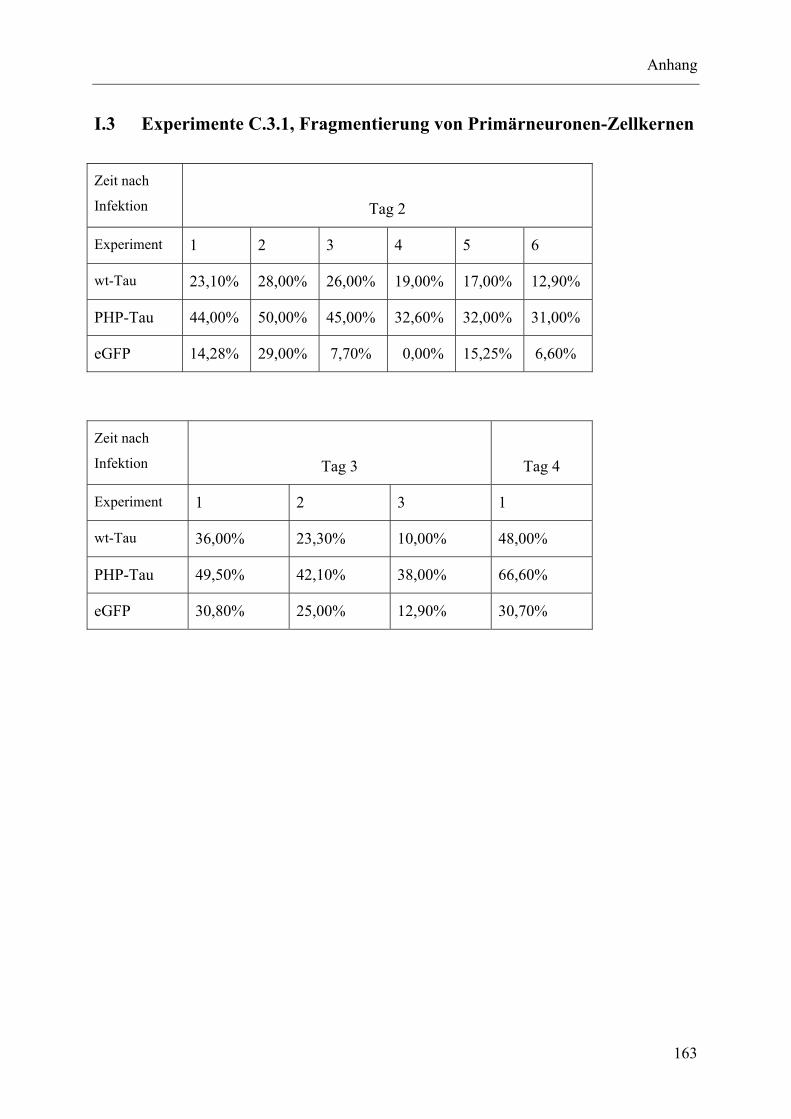
Anhang
I.3 Experimente C.3.1, Fragmentierung von Primärneuronen-Zellkernen
Zeit nach
Infektion
Tag 2
Experiment 1 2 3 4 5 6
wt-Tau 23,10% 28,00% 26,00% 19,00% 17,00% 12,90%
PHP-Tau 44,00% 50,00% 45,00% 32,60% 32,00% 31,00%
eGFP 14,28% 29,00% 7,70% 0,00% 15,25% 6,60%
Zeit nach
Infektion
Tag 3
Tag 4
Experiment 1 2 3 1
wt-Tau 36,00% 23,30% 10,00% 48,00%
PHP-Tau 49,50% 42,10% 38,00% 66,60%
eGFP 30,80% 25,00% 12,90% 30,70%
163
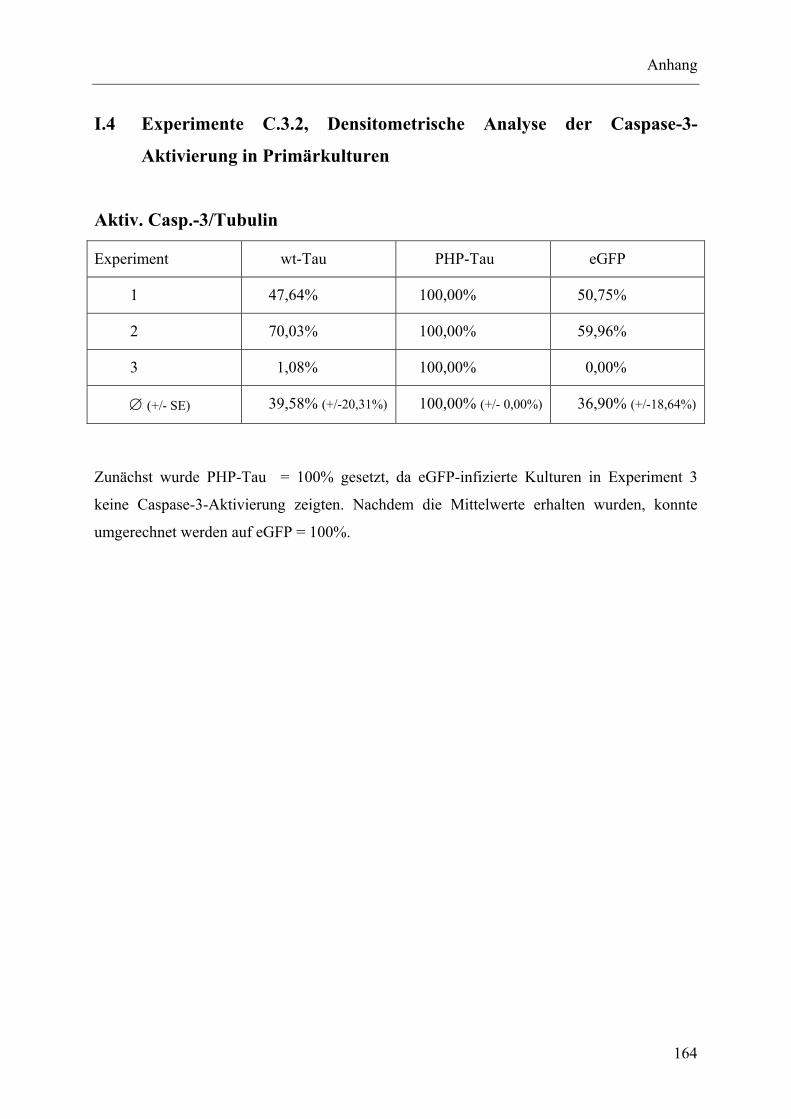
Anhang
I.4 Experimente C.3.2, Densitometrische Analyse der Caspase-3-
Aktivierung in Primärkulturen
Aktiv. Casp.-3/Tubulin
Experiment wt-Tau PHP-Tau eGFP
1 47,64% 100,00% 50,75%
2 70,03% 100,00% 59,96%
3 1,08% 100,00% 0,00%
∅ (+/- SE) 39,58% (+/-20,31%) 100,00% (+/- 0,00%) 36,90% (+/-18,64%)
Zunächst wurde PHP-Tau = 100% gesetzt, da eGFP-infizierte Kulturen in Experiment 3
keine Caspase-3-Aktivierung zeigten. Nachdem die Mittelwerte erhalten wurden, konnte
umgerechnet werden auf eGFP = 100%.
164
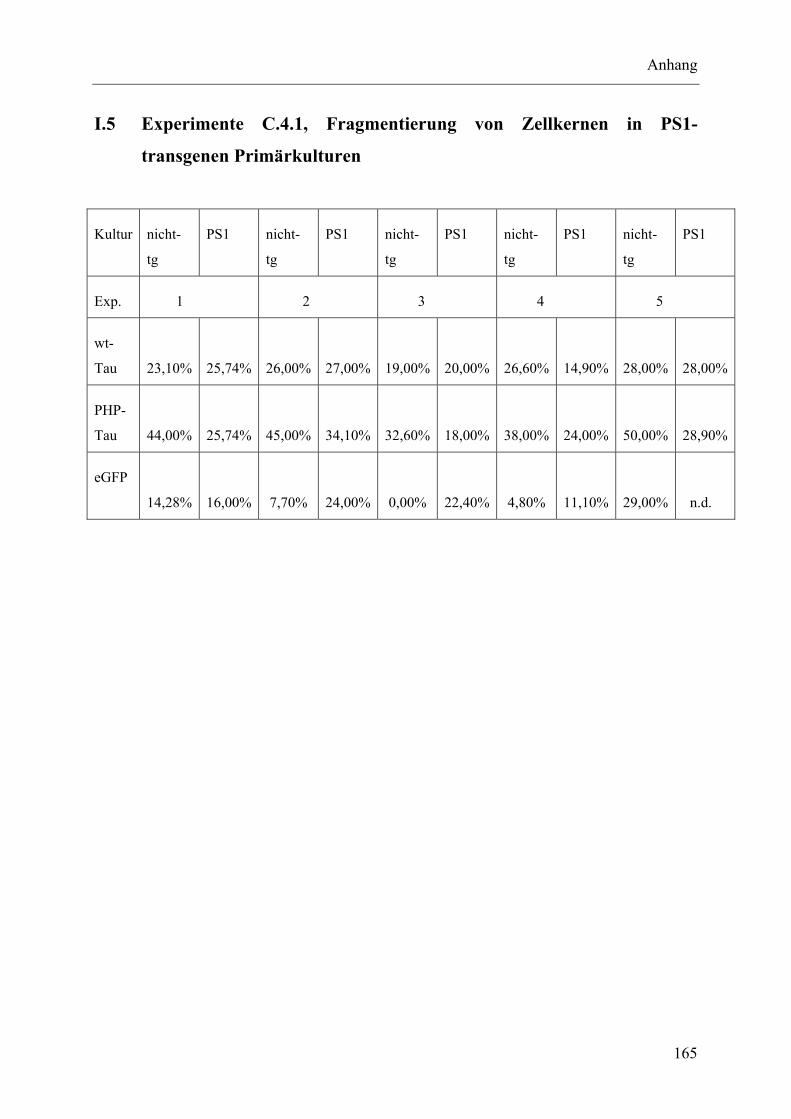
Anhang
I.5 Experimente C.4.1, Fragmentierung von Zellkernen in PS1-
transgenen Primärkulturen
Kultur nicht-
tg
PS1 nicht-
tg
PS1 nicht-
tg
PS1 nicht-
tg
PS1 nicht-
tg
PS1
Exp. 1 2 3 4 5
wt-
Tau
23,10%
25,74%
26,00%
27,00%
19,00%
20,00%
26,60%
14,90%
28,00%
28,00%
PHP-
Tau
44,00%
25,74%
45,00%
34,10%
32,60%
18,00%
38,00%
24,00%
50,00%
28,90%
eGFP
14,28%
16,00%
7,70%
24,00%
0,00%
22,40%
4,80%
11,10%
29,00%
n.d.
165
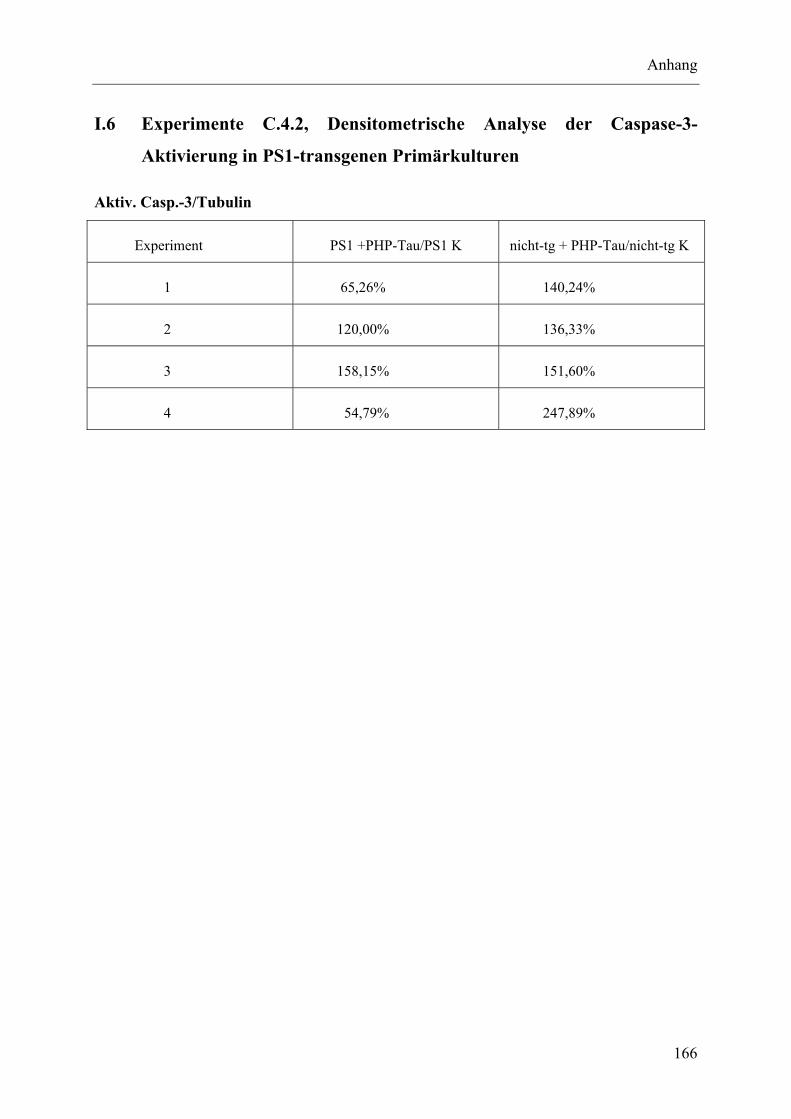
Anhang
I.6 Experimente C.4.2, Densitometrische Analyse der Caspase-3-
Aktivierung in PS1-transgenen Primärkulturen
Aktiv. Casp.-3/Tubulin
Experiment PS1 +PHP-Tau/PS1 K nicht-tg + PHP-Tau/nicht-tg K
1 65,26% 140,24%
2 120,00% 136,33%
3 158,15% 151,60%
4 54,79% 247,89%
166
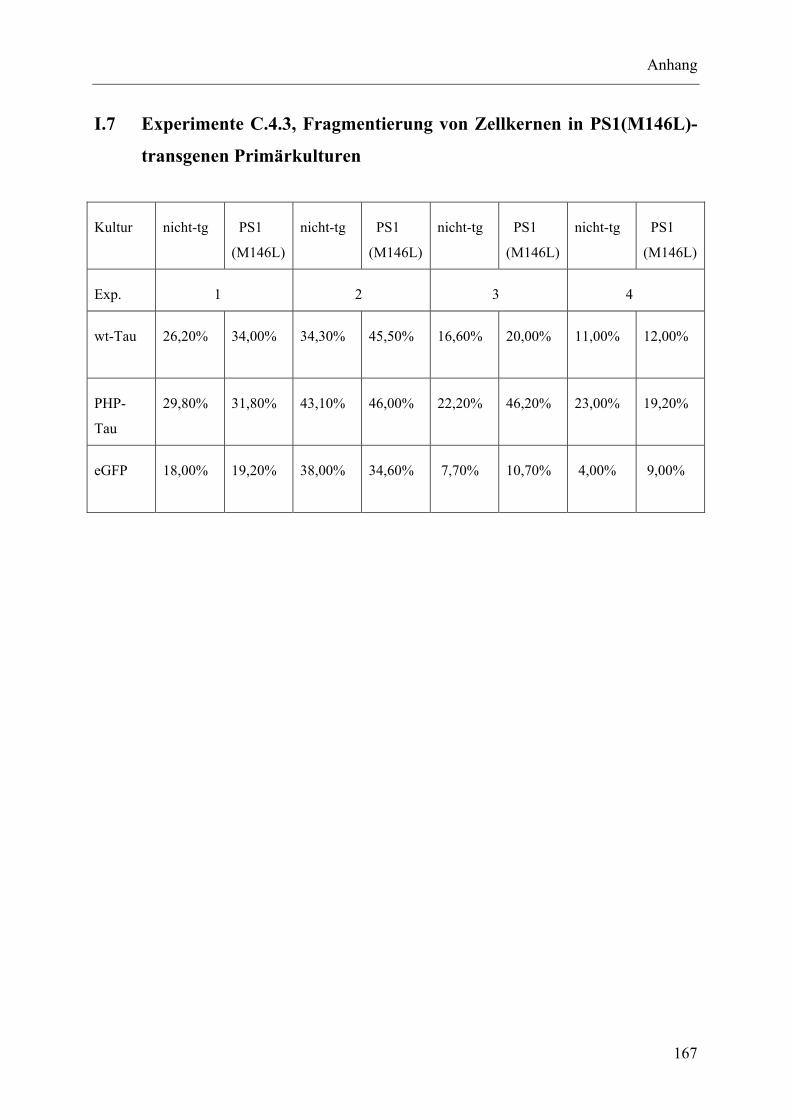
Anhang
I.7 Experimente C.4.3, Fragmentierung von Zellkernen in PS1(M146L)-
transgenen Primärkulturen
Kultur nicht-tg PS1
(M146L)
nicht-tg PS1
(M146L)
nicht-tg PS1
(M146L)
nicht-tg PS1
(M146L)
Exp. 1 2 3 4
wt-Tau
26,20% 34,00% 34,30% 45,50% 16,60% 20,00% 11,00% 12,00%
PHP-
Tau
29,80% 31,80% 43,10% 46,00% 22,20% 46,20% 23,00% 19,20%
eGFP
7,70% 18,00% 19,20% 38,00% 34,60% 10,70% 4,00% 9,00%
167
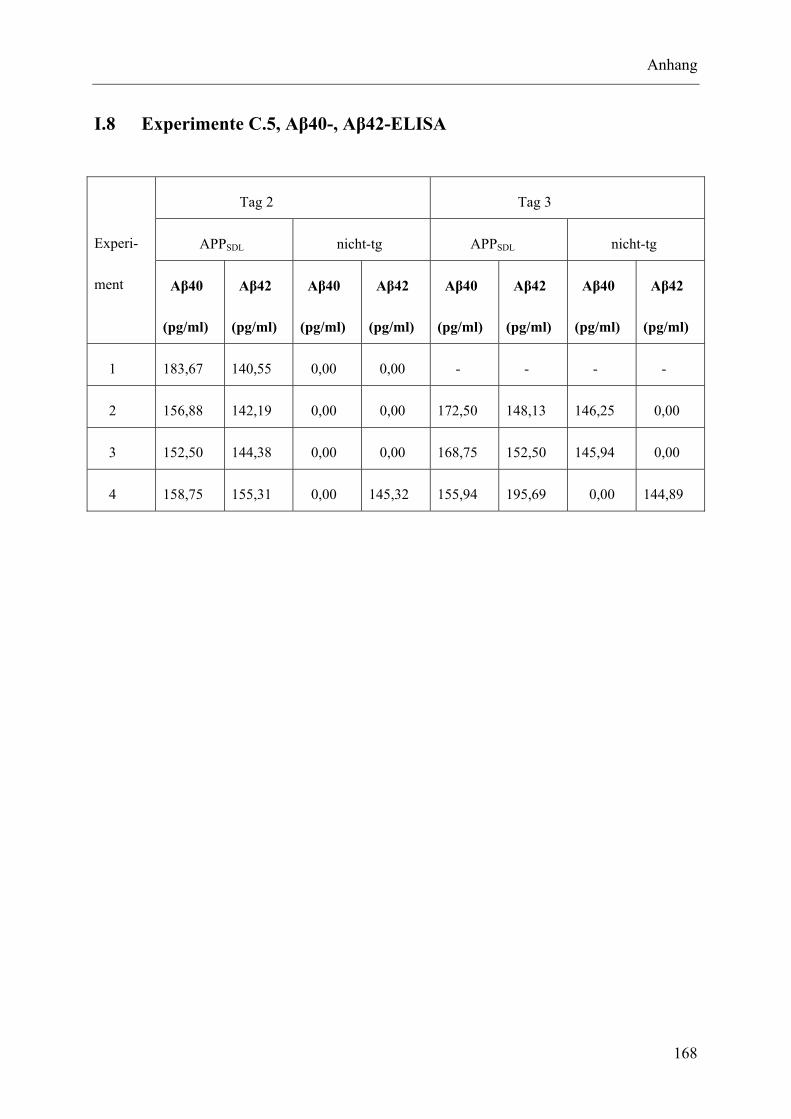
Anhang
I.8 Experimente C.5, Aβ40-, Aβ42-ELISA
Tag 2 Tag 3
SDL nicht-tg APPSDL nicht-tg
Experi-
ment
Aβ40
(pg/ml)
Aβ42
(pg/ml)
Aβ40
(pg/ml)
Aβ42
(pg/ml)
Aβ40
(pg/ml)
Aβ42
(pg/ml)
Aβ40
(pg/ml)
Aβ42
(pg/ml)
1 183,67 140,55 0,00 0,00 - - - -
2 156,88 142,19 0,00 0,00 172,50 148,13 146,25 0,00
3 152,50 144,38 0,00 0,00 168,75 152,50 145,94 0,00
195,69 0,00 144,89
APP
4 158,75 155,31 0,00 145,32 155,94
168
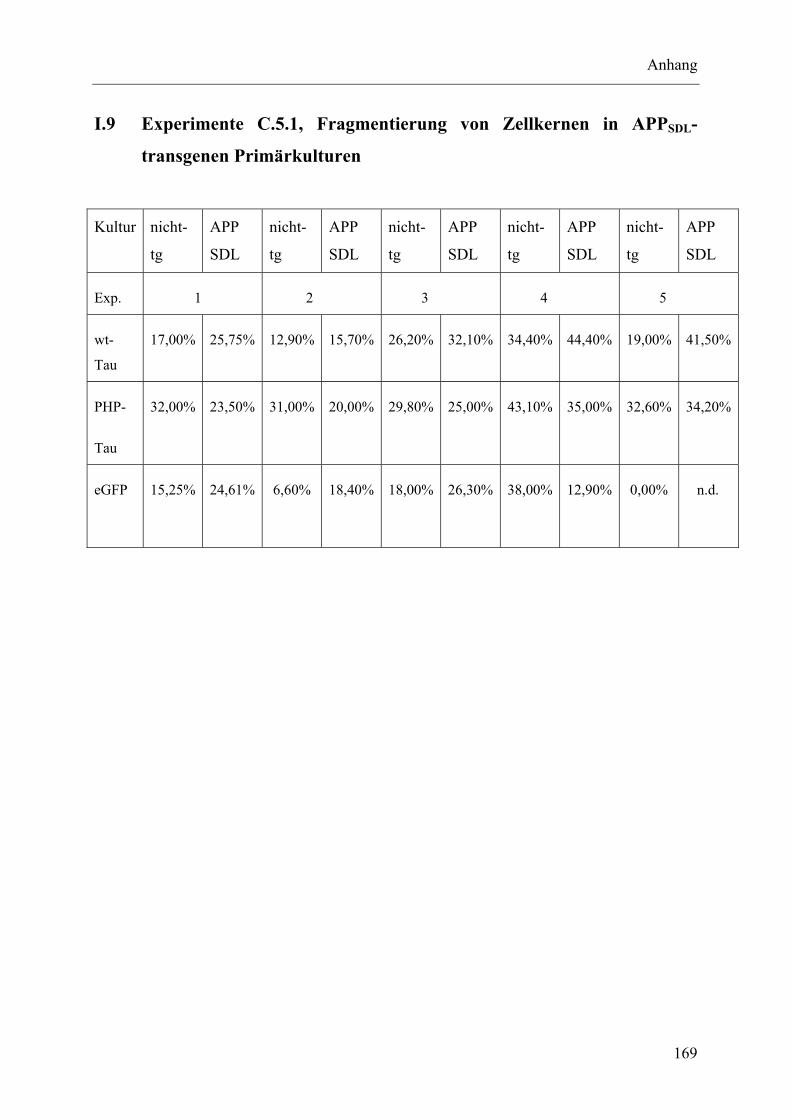
Anhang
I.9 Experimente C.5.1, Fragmentierung von Zellkernen in APP
transgenen Primärkulturen
APP
SDL
nicht-
tg
APP
SDL
nicht-
tg
APP
SDL
SDL-
Kultur nicht-
tg
nicht-
tg
APP
SDL
nicht-
tg
APP
SDL
Exp. 1 2 3 4 5
wt-
Tau
17,00% 12,90% 25,75% 15,70% 26,20% 32,10% 34,40% 44,40% 19,00% 41,50%
PHP-
Tau
32,00% 23,50% 31,00% 20,00% 29,80% 25,00% 43,10% 35,00% 32,60% 34,20%
eGFP
24,61% 18,00% 26,30% 38,00% 12,90% 0,00% n.d. 15,25% 6,60% 18,40%
169
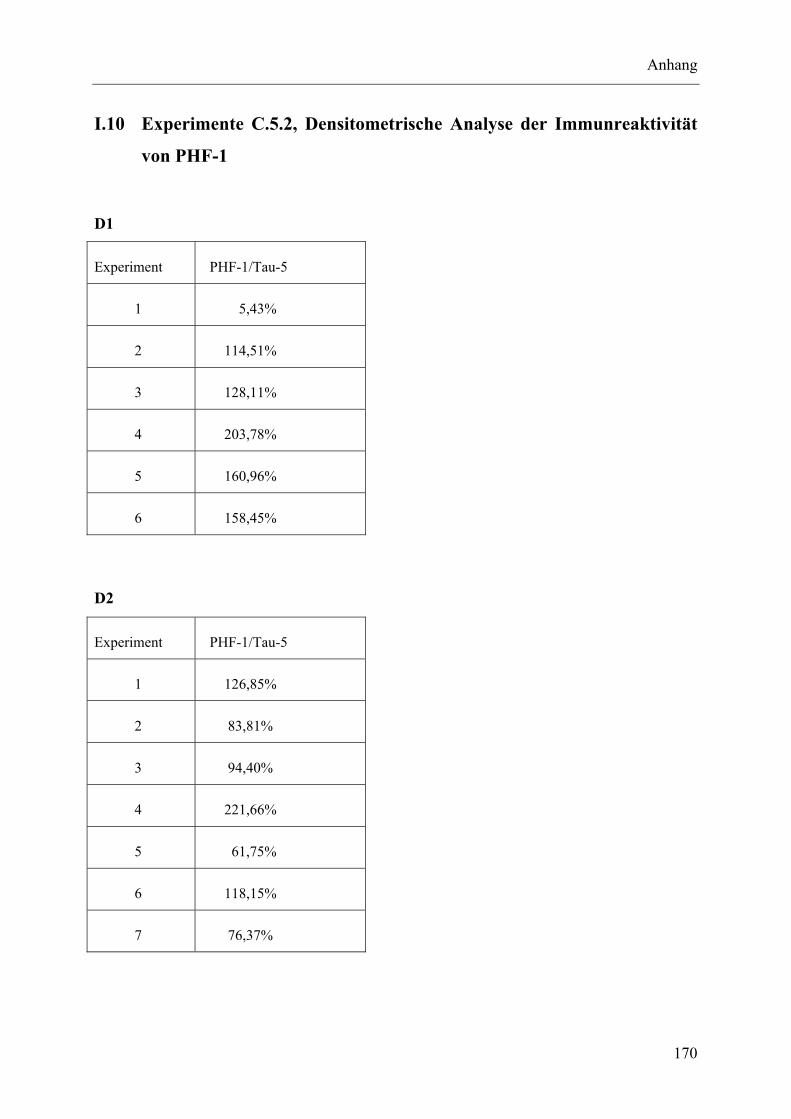
Anhang
I.10 Experimente C.5.2, Densitometrische Analyse der Immunreaktivität
von PHF-1
D1
Experiment PHF-1/Tau-5
1 5,43%
2 114,51%
4 203,78%
5
158,45%
3 128,11%
160,96%
6
D2
Experiment PHF-1/Tau-5
1 126,85%
2 83,81%
3 94,40%
4 221,66%
5 61,75%
6 118,15%
7 76,37%
170
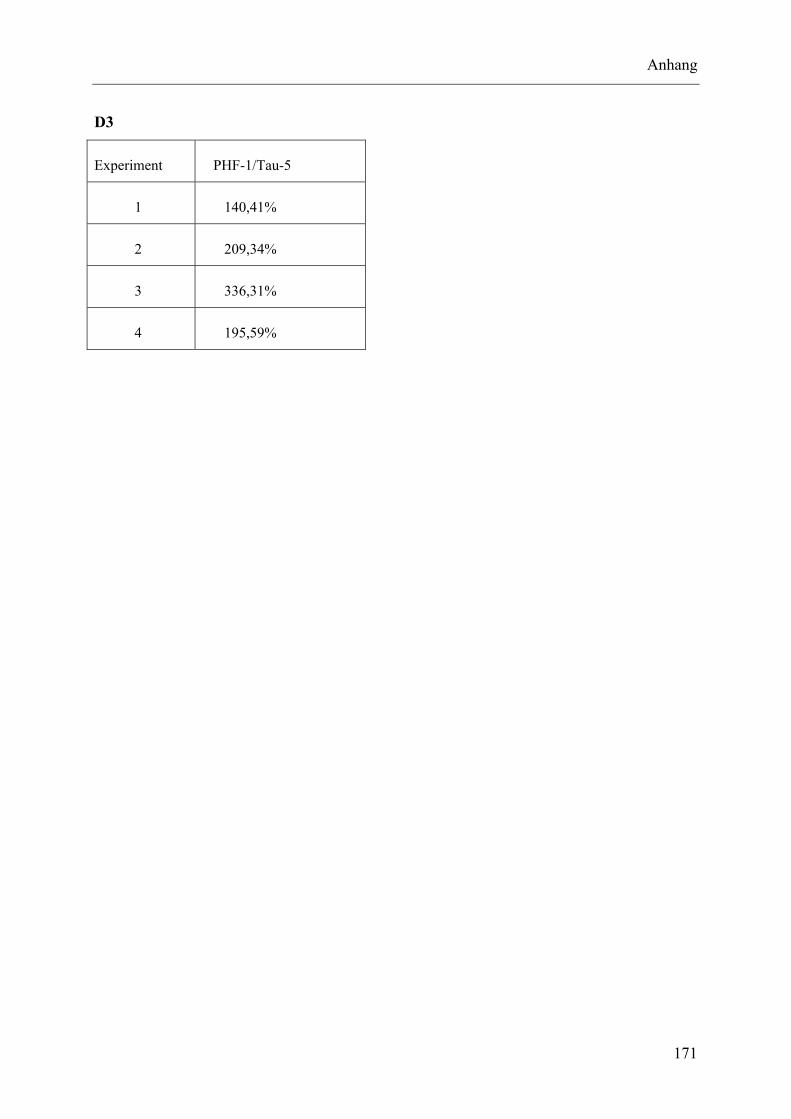
Anhang
171
Experiment PHF-1/Tau-5
D3
1 140,41%
2 209,34%
3 336,31%
4 195,59%
Recommended

















