
— —< <
Automated Flexible Ligand DockingMethod and Its Application for DatabaseSearch
SHINGO MAKINO,1,2 IRWIN D. KUNTZ1
1Department of Pharmaceutical Chemistry, School of Pharmacy, University of California, San Francisco,California 94143-04462Central Research Lab, Ajinomoto Co., Inc., Kawasaki, Japan
Received 13 February 1997; accepted 11 June 1997
ABSTRACT: We have developed a new docking program that explores ligandflexibility. This program can be applied to database searches. The program issimilar in concept to earlier efforts, but it has been automated and improved.The algorithm begins by selecting an anchor fragment of a ligand. This fragmentis protonated, as needed, and then placed in the receptor by the DOCKalgorithm, followed by minimization using a simplex method. Finally, theconformations of the remaining parts of the putative ligands are searched by alimited backtrack method and minimized to get the most stable conformation.To test the efficiency of this method, the program was used to regenerate tenligand]protein complex structures. In all cases, the docked ligands basicallyreproduced the crystallographic binding modes. The efficiency of this methodwas further tested by a database search. Ten percent of molecules from the
Ž .Available Chemicals Directory ACD were docked to a dihydrofolate reductaseŽ .structure. Most of the top-ranking molecules 7 of the top 13 hits are
dihydrofolate or methotrexate derivatives, which are known to be DHFRinhibitors, demonstrating the suitability of this program for screening moleculardatabases. Q 1997 John Wiley & Sons, Inc. J Comput Chem 18: 1812]1825,1997
Keywords: automated docking; flexible ligand; database search; protonation;minimization
Correspondence to: I. D. KuntzContractrgrant sponsor: NIGMS; grant number GM-31497,
GM-39552
( )Journal of Computational Chemistry, Vol. 18, No. 14, 1812]1825 1997Q 1997 John Wiley & Sons, Inc. CCC 0192-8651 / 97 / 141812-14

LIGAND DOCKING METHOD
Introduction
he development of new computer hardwareT and software and the dramatic growth in theavailable x-ray and NMR protein structures offermany opportunities for discoveries of ligandswhich bind to the macromolecular structures.1,2
The prediction of the geometric binding mode is acritical issue both for lead discovery from molecu-lar databases3 and for lead optimization.4 ] 6 Al-though there have been many reports dealing withrigid-body docking methods,7 ] 9 it is generally ex-pected that the correct docking modes cannot beobtained unless the probe conformation is quitesimilar to the active conformation. Thus, consider-ation of the flexibility of ligands has received muchrecent attention. Algorithms that take the confor-mational flexibility of ligands into account havebeen reported: docking fragments and joining themtogether10 ; simulated annealing11; Monte Carlosearch12 ; genetic algorithms13,14; and incrementalsearches.1, 15 ] 17 For carrying out database searches,a program has to satisfy the following conditions:Ž . Ž .1 all processes should be fully automated; 2 itshould identify the most stable conformations; andŽ .3 calculations should be rapid enough to searchthrough a database efficiently. Recently, two pro-grams, HAMMERHEAD16 and FLEXX,17 have beenreported, which place flexible ligands into macro-molecules speedily. Both methods use an heuristicsearch algorithm. The FLEXX program is not fullyautomated and manual selection of the basic frag-ments is necessary. The HAMMERHEAD programis fully automated. In one test, biotin was identi-fied as the top-scoring ligand in the ACD againststreptavidin. Although several other successes arereported, this search is based on the assumptionthat all partial structures should have near opti-mum energies. In addition, the scoring functionsare not reflections of physical forces. Here, wereport an automated flexible ligand docking pro-gram using a limited backtrack algorithm whichsearches broadly among partial structures. Com-putation time is reduced by skipping conforma-tions that are similar to conformations already
Ž .sampled. Other new features include: 1 Classifi-cation of flexible bonds and anchor fragment iden-
Ž .tifications are automated. 2 Multiple protonationstates of a ligand are treated simultaneously, sousers do not have to decide in advance whichprotonation model of a ligand is the best for dock-
Ž .ing. 3 Information from hydrogen bond donorand acceptor sites and ligand shape are used forthe initial docking, so a ligand with few or no
Ž .hydrogen bonds can also be docked. 4 Bindingmodes of ligands are predicted rapidly by trim-ming the conformational tree without loss of di-versity of the conformational sampling. Althoughsome previous studies have tried to reduce com-putation time by using partial structures of theligands,15 or by fixing the number of rotatablebonds,13 it is difficult to apply such a special
Ž .treatment to all molecules in a large database. 5The program also only estimates the necessarypartial energy in the backtrack search to reduce thecomputational time. In this article, we describe thetheory behind this method and test the programby regenerating x-ray complex structures. Finally,the program is applied to an ACD database search.
Methods
We describe the features of the algorithm in thefollowing order: force field scoring, flexible bondidentification, anchor identification, multistateprotonation, site-point generation and docking,limited backtrack search, partial energy estimationin the backtrack search, and simplex minimization.We also discuss the programming language andresource usage. All the molecular modeling andcharge calculations were performed using theSYBYL program from Tripos.18 SYBYL atom types19
used are shown in Table I.
FORCE-FIELD SCORE
We adopted an AMBER-type potential func-tion20,21 for measuring the affinity of ligands. Inearlier work22 with rigid ligands, we only usedthe intermolecular term. For this project, both interand intramolecular interactions are examined toidentify the best conformation of each ligand.However, for comparison of different ligands, onlythe intermolecular interaction is calculated, basedon the assumption that the selected best conforma-tion of a ligand has an intramolecular energy closeto the gas-phase conformational minimum. Theintermolecular interaction energy is calculated us-ing the grid-based force field23 and the receptorsare assumed to be rigid. For intramolecular inter-action, we used full AMBER potential on the fly.
JOURNAL OF COMPUTATIONAL CHEMISTRY 1813

MAKINO AND KUNTZ
TABLE I.List of SYBYL Atom Types Used in This Experiment.
C.3 carbon sp3C.2 carbon sp2C.1 carbon spC.ar carbon aromaticC.cat carbocationN.3 nitrogen sp3N.2 nitrogen sp2N.1 nitrogen spN.ar nitrogen aromaticN.am nitrogen amidN.pl3 nitrogen trigonal planerN.4 nitrogen sp3 positively chargedO.3 oxygen sp3O.2 oxygen sp2O.co2 oxygen in carboxylate and phosphateS.3 sulfur sp3S.2 sulfur sp2S.O sulfoxide sulfurS.O2 sulfone sulfurP.3 phosphorous sp3H hydrogen
( )H.spc special hydrogen in this work see text
In the conformational search before minimization,Ž .the Van der Waals vdw radii of the ligand atoms
are scaled to 0.8. This reduction in vdw radiimakes it easier to find low energy conformationsthrough a rapid conformational search. After find-ing the possible conformations, the vdw scale isreset to normal values and minimizations are per-formed with full-sized atoms.
FLEXIBLE BOND IDENTIFICATION
Flexible bonds are automatically identified bythe following method. First, all bonds not part of
ring systems are selected as candidates for flexiblebonds. If a selected bond has the SYBYL bond type
Ž .‘‘1’’ a single bond and is not prohibited fromŽ .being a flexible bond see Table II , it is identified
as a flexible bond.
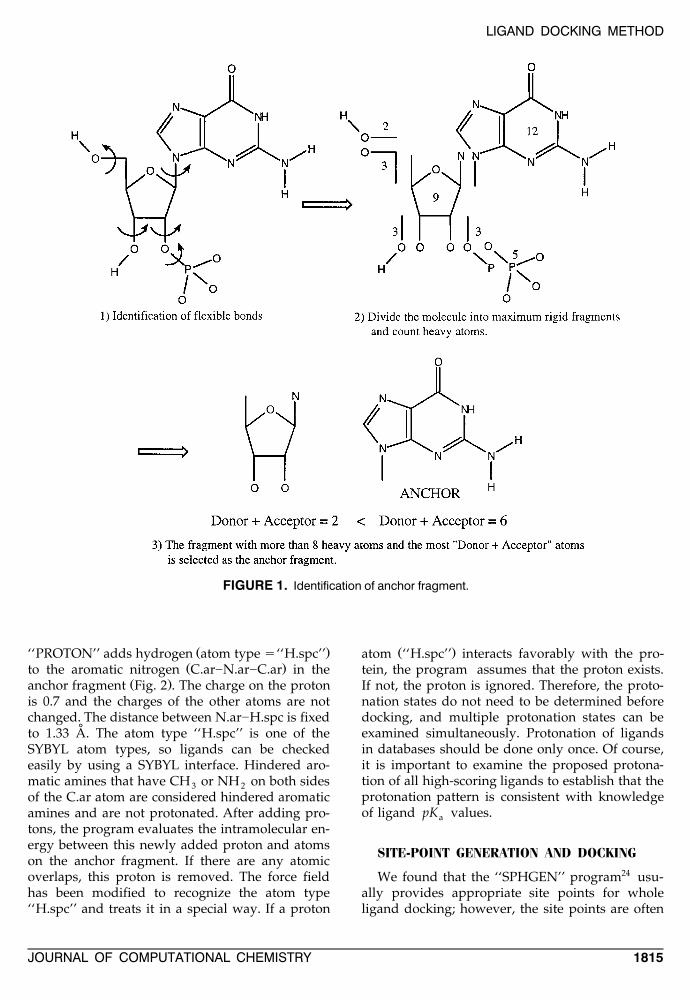
ANCHOR IDENTIFICATION
Anchor fragments are automatically identifiedby the ‘‘ANCHOR’’ program shown in Figure 1.First, ligands are divided into the largest rigidfragments. Next, fragments that contain more thaneight heavy atoms are selected. If a ligand does nothave any fragments with more than eight heavyatoms, then that ligand is skipped. Among theselected anchor fragment candidates, the fragmentwith the maximum ‘‘donor q acceptor’’ atoms isselected as the anchor fragment. The ANCHORprogram automatically adds the keyword² :‘‘ ANCHOR ’’ and an appropriate anchor identifi-
cation flag to SYBYL multiple mol2 format files.This anchor identification process only needs to beperformed once when the molecule is added to amolecular database.
MULTISTATE PROTONATION
In some cases, protonation states are critical fordocking. Although previous studies selected spe-cific protonation states in their models ofligands,15,17 it is still very difficult to determinewhich state of the protonation is the best for adocking study without knowing the real dockingmode. Instead of predetermining the protonationstates, multistate protonation options are treatedsimultaneously by our program. The program
TABLE II.Prohibited Flexible Bonds.
aNumber Atom Pattern 1 Atom Pattern 2 Pattern Name
( )( )( ) 5 5 51 C.3 H H H C.* N.* O.* Si.* Methyl2 N.2 N.2 Imine
5 5 53 C.2 C.2 N.am N.2 N.pl3 Double and amide4 C.1 C.1 Triple
a 5 ( ) ( )5 C. N.pl3 N.2 H H Planar amine5 ( < ) ( < ) ( )6 C.ar C.2 C.cat N.pl3 N.2 N.pl3 N.2 Amidine aromatic( ) ( ) ( ) ( )7 C.ar S.3 O.co2 O.co2 O.co2 Sulfonic acid aromatic( ) ( )8 C.ar N.* O.2 Nitro, nitroso aromatic5 ( ) ( ) ( )9 C.ar C.2 C.cat O.co2 O.co2 Carboxylic acid aromatic
a 5‘‘ ’’ means ‘‘or’’; ‘‘*’’ means ‘‘any symbols’’ following the UNIX convention.
VOL. 18, NO. 141814
LIGAND DOCKING METHOD
FIGURE 1. Identification of anchor fragment.
Ž .‘‘PROTON’’ adds hydrogen atom type s‘‘H.spc’’Ž .to the aromatic nitrogen C.ar]N.ar]C.ar in the
Ž .anchor fragment Fig. 2 . The charge on the protonis 0.7 and the charges of the other atoms are notchanged. The distance between N.ar]H.spc is fixed
˚to 1.33 A. The atom type ‘‘H.spc’’ is one of theSYBYL atom types, so ligands can be checkedeasily by using a SYBYL interface. Hindered aro-matic amines that have CH or NH on both sides3 2of the C.ar atom are considered hindered aromaticamines and are not protonated. After adding pro-tons, the program evaluates the intramolecular en-ergy between this newly added proton and atomson the anchor fragment. If there are any atomicoverlaps, this proton is removed. The force fieldhas been modified to recognize the atom type‘‘H.spc’’ and treats it in a special way. If a proton
Ž .atom ‘‘H.spc’’ interacts favorably with the pro-tein, the program assumes that the proton exists.If not, the proton is ignored. Therefore, the proto-nation states do not need to be determined beforedocking, and multiple protonation states can beexamined simultaneously. Protonation of ligandsin databases should be done only once. Of course,it is important to examine the proposed protona-tion of all high-scoring ligands to establish that theprotonation pattern is consistent with knowledgeof ligand pK values.a
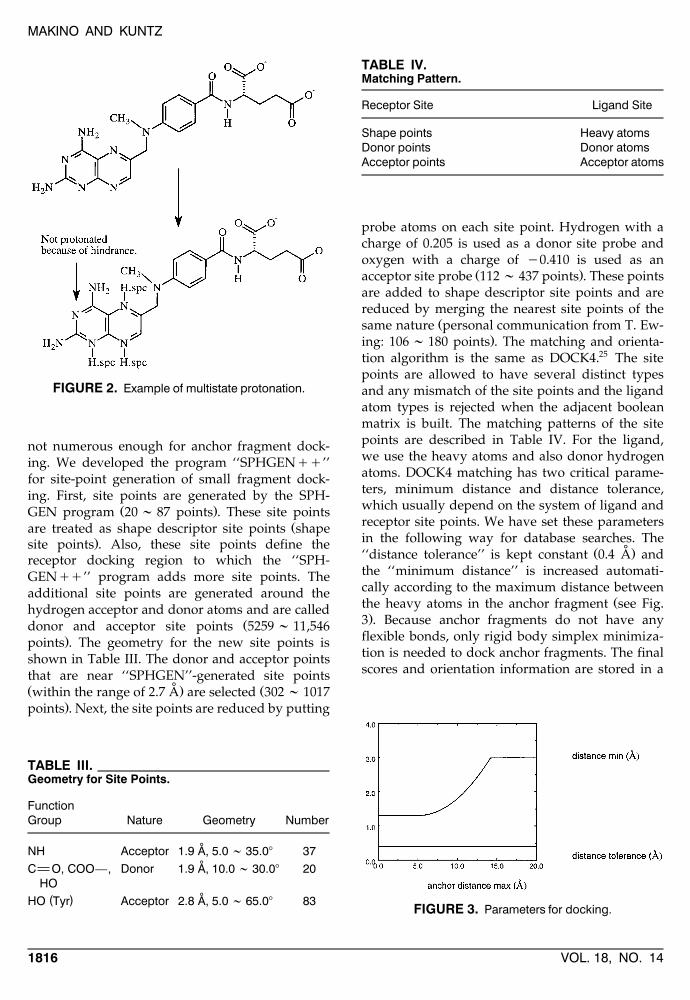
SITE-POINT GENERATION AND DOCKING
We found that the ‘‘SPHGEN’’ program24 usu-ally provides appropriate site points for wholeligand docking; however, the site points are often
JOURNAL OF COMPUTATIONAL CHEMISTRY 1815
MAKINO AND KUNTZ
FIGURE 2. Example of multistate protonation.
not numerous enough for anchor fragment dock-ing. We developed the program ‘‘SPHGENqq’’for site-point generation of small fragment dock-ing. First, site points are generated by the SPH-
Ž .GEN program 20 ; 87 points . These site pointsŽare treated as shape descriptor site points shape
.site points . Also, these site points define thereceptor docking region to which the ‘‘SPH-GENqq’’ program adds more site points. Theadditional site points are generated around thehydrogen acceptor and donor atoms and are called
Ždonor and acceptor site points 5259 ; 11,546.points . The geometry for the new site points is
shown in Table III. The donor and acceptor pointsthat are near ‘‘SPHGEN’’-generated site points
˚Ž . Žwithin the range of 2.7 A are selected 302 ; 1017.points . Next, the site points are reduced by putting
TABLE III.Geometry for Site Points.
FunctionGroup Nature Geometry Number
˚NH Acceptor 1.9 A, 5.0 ; 35.08 37˚C O, COO—, Donor 1.9 A, 10.0 ; 30.08 20
HO˚( )HO Tyr Acceptor 2.8 A, 5.0 ; 65.08 83
TABLE IV.Matching Pattern.
Receptor Site Ligand Site
Shape points Heavy atomsDonor points Donor atomsAcceptor points Acceptor atoms
probe atoms on each site point. Hydrogen with acharge of 0.205 is used as a donor site probe andoxygen with a charge of y0.410 is used as an
Ž .acceptor site probe 112 ; 437 points . These pointsare added to shape descriptor site points and arereduced by merging the nearest site points of the
Žsame nature personal communication from T. Ew-.ing: 106 ; 180 points . The matching and orienta-
tion algorithm is the same as DOCK4.25 The sitepoints are allowed to have several distinct typesand any mismatch of the site points and the ligandatom types is rejected when the adjacent booleanmatrix is built. The matching patterns of the sitepoints are described in Table IV. For the ligand,we use the heavy atoms and also donor hydrogenatoms. DOCK4 matching has two critical parame-ters, minimum distance and distance tolerance,which usually depend on the system of ligand andreceptor site points. We have set these parametersin the following way for database searches. The
˚Ž .‘‘distance tolerance’’ is kept constant 0.4 A andthe ‘‘minimum distance’’ is increased automati-cally according to the maximum distance between
Žthe heavy atoms in the anchor fragment see Fig..3 . Because anchor fragments do not have any
flexible bonds, only rigid body simplex minimiza-tion is needed to dock anchor fragments. The finalscores and orientation information are stored in a
FIGURE 3. Parameters for docking.
VOL. 18, NO. 141816

LIGAND DOCKING METHOD
binary tree structure. After exploring all of thepossible orientations, the program clusters orienta-tions and chooses one from each cluster. The pro-gram picks out the minimum score orientation andremoves other similar orientations. Then, it choosesthe second minimum score and the same cycle isrepeated. Finally, the program writes the orienta-tion information sorted by score into a file forfurther conformational search. Once an anchorfragment is docked in a specific location, that frag-ment can be displaced by the minimization proce-dure, but the anchor is not redocked as side chainsare added.
LIMITED BACKTRACK SEARCH
Although a systematic search is an efficient wayto explore the wide range of conformational space,the search space increases dramatically as the flexi-bility of a ligand increases. For example, if one
Žtorsion has N states N s 360.0rsearch step] ].angle , then a ligand with M flexible bonds has
N M conformations. However, a great amount ofthe conformational space can be pruned by trim-ming all the branches under bad nodes as shownin Figure 4a. This is called the backtrack method.26
This method is efficient, but it will still take toolong for a ligand with many flexible bonds, so weadded a limitation to this backtrack method. Thislimitation fixes the number of conformations ac-cepted below a certain level in the search tree asshown in Figure 4b. This version of the greedyalgorithm27 allows a reduction of sampling of simi-lar conformations without reducing the samplingof the distinct conformations. We call this a‘‘limited backtrack search.’’ The amount of sam-pling can be controlled by changing the level of
Žthe limitation. compare numbers of sampling be-.tween Fig. 4b and 4c . This limitation should be
better than taking the first N conformations, be-cause such a sampling tends to take similar sam-ples and skip distinct ones. For example, samplesin Figure 4b are more diverse than in Figure 4d,even though the number of conformations sam-pled is the same. Additionally, for increased speed,at each node of the search tree, it is necessary tocalculate matrices and vectors for rotation once.
PARTIAL ENERGY ESTIMATION IN THEBACKTRACK SEARCH
We tried to eliminate unnecessary calculationsto the extent possible to speed up the dockingcalculations. On each node of the search tree, only
FIGURE 4. Search trees to show efficiency of the limitedalgorithm.
the partial energy is calculated for both inter- andintramolecular energy. Figure 5 shows how theatoms in a ligand are categorized depending oneach search level. At every stage of the conforma-tional search, the atoms designated by verticallines and by a checkerboard pattern are not movedby changing the torsion angle, so the interactionenergy between these atoms and the atoms of thereceptor is constant and does not need to be recal-culated. The blank atoms will be moved by thedeeper flexible bond search, so their energies arenot considered at this search level. Because theatoms with horizontal lines are moved by thecurrent flexible bond and will not be moved by thedeeper flexible bonds, these atoms are the onlyreal variables at this level of the search. Thus, theenergy between the atoms with horizontal linesand the atoms of the receptor are calculated as
JOURNAL OF COMPUTATIONAL CHEMISTRY 1817

MAKINO AND KUNTZ
FIGURE 5. Categorization of atoms for partial energyestimation.
intermolecular energy terms and the energy be-tween atoms with horizontal lines and the atomswith vertical lines are calculated as intramolecularenergy terms on each node of the search tree.Notice that the intramolecular energy betweenatoms with the same pattern does not need to becalculated, because the distances between atomswith the same patern are always the same, thustheir contribution to intramolecular energy re-mains constant. These reductions in calculationtime are especially effective for intramolecular en-ergy, which cannot be calculated with a grid-basedforce field. In our current version, we use a ran-dom seed to generate the initial conformation.Thus, two different runs can yield slightly differ-
Ž .ent results data not shown .
SIMPLEX MINIMIZATION
Minimization28 is one of the most time-consum-ing steps in a ligand docking to a macromolecule.Thus, it is very important to accelerate a minimiza-
tion. For this purpose, each branch is treated inde-pendently in the minimization process, becausethe dependency of the branches is considered inthe search process. We will explain the efficiency
Žof this method with the example of glucose Fig..6 . In a backtrack search, flexible bonds are simply
Žsorted by the ‘‘bond weight’’ the number of atoms.moved by the rotation of the bond . In a minimiza-
tion, flexible bonds are categorized into branches.The simple branches from the anchor fragment areused and sub-branches or recursive branches arenot used because recursive branching tends tointroduce interdependence. We will compare theconformational space of independent branch mini-mization with the conformational space of the de-pendent branch minimization by this example. If
Žeach flexible bond has N status N s.minimization rangerstep degree , the conforma-] ]
tional space for minimization will normally be N 6.If each branch is treated independently, then the
2 Ž 2minimization space will be N q 4N N for. 6branch 1 . For example, if N s 12 then N s
2,985,984 and N 2 q 4N s 192. If there is only onebranch, this categorization is superfluous. A sim-plex minimizer generates many conformationsfrom the same conformation in each orientation, sothe ‘‘look-up matrix’’ does not have to be reset forchanging conformations. Each flexible bond has anindependent transformation object especially forminimization, so unnecessary recalculations formatrix and vector transformations are avoided,thus speeding up the calculations.
PROGRAMMING LANGUAGE AND RESOURCEUSAGE
All the programs except ‘‘SPHGEN’’ were writ-ten in Cqq and compiled by GNU gcc-2.7.2. The
FIGURE 6. Analysis of flexible bonds.
VOL. 18, NO. 141818
LIGAND DOCKING METHOD
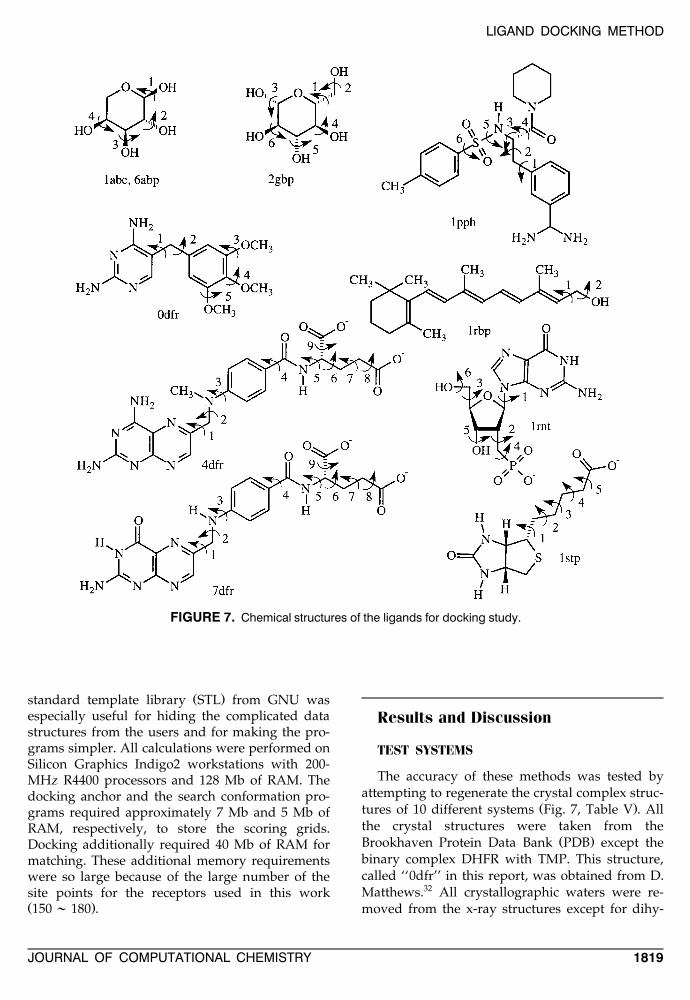
FIGURE 7. Chemical structures of the ligands for docking study.
Ž .standard template library STL from GNU wasespecially useful for hiding the complicated datastructures from the users and for making the pro-grams simpler. All calculations were performed onSilicon Graphics Indigo2 workstations with 200-MHz R4400 processors and 128 Mb of RAM. Thedocking anchor and the search conformation pro-grams required approximately 7 Mb and 5 Mb ofRAM, respectively, to store the scoring grids.Docking additionally required 40 Mb of RAM formatching. These additional memory requirementswere so large because of the large number of thesite points for the receptors used in this workŽ .150 ; 180 .
Results and Discussion
TEST SYSTEMS
The accuracy of these methods was tested byattempting to regenerate the crystal complex struc-
Ž .tures of 10 different systems Fig. 7, Table V . Allthe crystal structures were taken from the
Ž .Brookhaven Protein Data Bank PDB except thebinary complex DHFR with TMP. This structure,called ‘‘0dfr’’ in this report, was obtained from D.Matthews.32 All crystallographic waters were re-moved from the x-ray structures except for dihy-
JOURNAL OF COMPUTATIONAL CHEMISTRY 1819

MAKINO AND KUNTZ
TABLE V.X-Ray Condition and Affinities.
apdb Resolution Protein Ligand Source pK i
29,301abe 1.7 L-Arabinose-binding protein L-Arabinose E. coli 6.50316abp 1.67 L-Arabinose-binding protein M108L L-Arabinose E. coli 6.36
320dfr 2.3 Dihydrofolate reductase Trimethoprim E. coli 8.89334dfr 1.7 Dihydrofolate reductase Methotrexate E. coli 8.6034 q7dfr 2.5 Dihydrofolate reductase / NADP Folate E. coli —
352gbp 1.9 D-Galactose / D-glucose-binding protein D-Glucose E. coli 7.40361pph 1.9 Trypsin 3-TAPAP Bovine 5.90
371rbp 2.0 Retinol-binding protein Retinol Human serum 6.7038,391rnt 1.9 Ribonuclease T1 29-GMP Aspergillus 5.18
oryzae401stp 2.6 Streptavidin Biotin Streptomyces 13.4
avidinii
a ( )pK s ylog K molar .i 10 i
drofolate reductase. In the case of dihydrofolatereductase, two waters in the ligand-binding re-gion, which are known to bind to the protein
Žstrongly, were preserved WAT403, WAT405 in.4dfr and corresponding waters in 0dfr and 7dfr .
After the ligands were removed from the proteins,hydrogens were added to the proteins and all thecharges of the atoms were calculated byGasteiger]Marsili method.41,42 Then, all the hydro-gens were optimized with fixed heavy atoms us-ing MAXIMIN2. All these calculations were doneusing the SYBYL program. The ligand conforma-tions were taken from the x-ray complex struc-tures. Hydrogens were added and optimized inthe same manner as the proteins, but no furtherminimization was done. Then, the conformationsof the ligands were randomized by changing allthe rotatable bonds and the orientations. The an-chor fragments of the ligands were defined by the‘‘ANCHOR’’ program and the anchor fragments
were protonated by the ‘‘PROTON’’ program. Theanchor fragments were docked and the top eightscoring orientations were selected for further con-formational search. The search conditions were de-termined based on the maximum number of theflexible bonds on a branch of a ligand as shown inTable VI. As the bond depth is increased, thesearch step angle is also increased:
search step angle] ]
s initial angle q increment angle] ]
Ž . Ž .= bond depth y 1 1]
As a result of this conformational search, thetop 120 scoring conformations were stored in abinary tree structure for further minimization. Af-ter each rigid body minimization, each branchŽ .Fig. 6 was minimized separately in its torsionaldegrees of freedom. These minimization steps wererepeated three times. The top score conformations
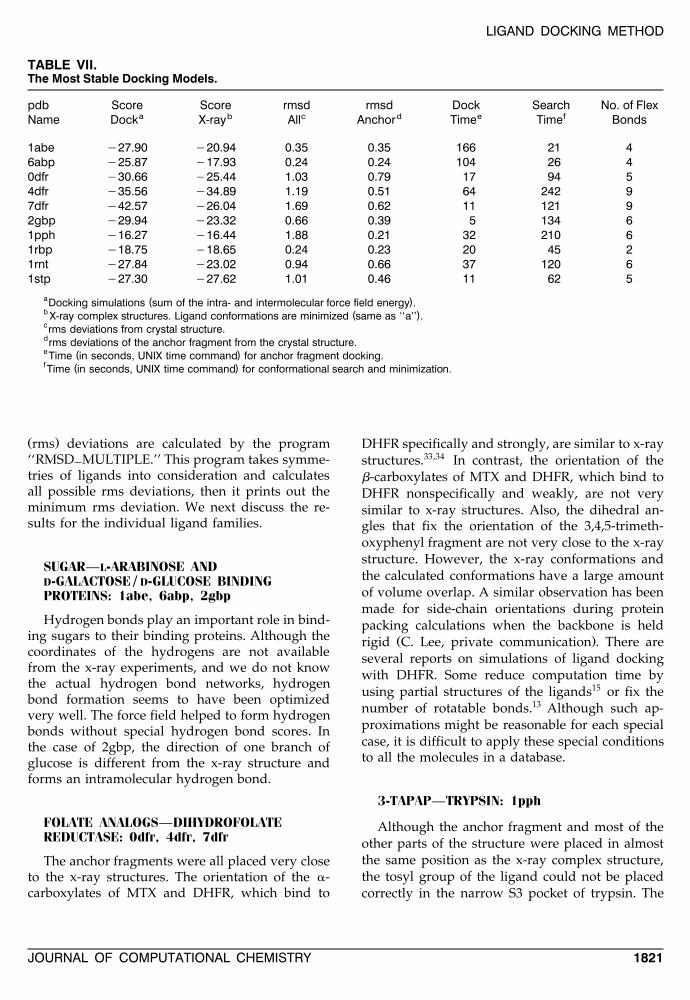
Ž .were then selected Table VII . Root-mean-square
TABLE VI.Search Conditions.
Conditions Initial Angle Increment Angle Limitation Level
Max. a of flexible bonds on a branch - 2 40.08 0.08 2Max. a of flexible bonds on a branch G 2 30.08 5.08 4
VOL. 18, NO. 141820
LIGAND DOCKING METHOD
TABLE VII.The Most Stable Docking Models.
pdb Score Score rmsd rmsd Dock Search No. of Flexa b c d e fName Dock X-ray All Anchor Time Time Bonds
1abe y27.90 y20.94 0.35 0.35 166 21 46abp y25.87 y17.93 0.24 0.24 104 26 40dfr y30.66 y25.44 1.03 0.79 17 94 54dfr y35.56 y34.89 1.19 0.51 64 242 97dfr y42.57 y26.04 1.69 0.62 11 121 92gbp y29.94 y23.32 0.66 0.39 5 134 61pph y16.27 y16.44 1.88 0.21 32 210 61rbp y18.75 y18.65 0.24 0.23 20 45 21rnt y27.84 y23.02 0.94 0.66 37 120 61stp y27.30 y27.62 1.01 0.46 11 62 5
a ( )Docking simulations sum of the intra- and intermolecular force field energy .b ( )X-ray complex structures. Ligand conformations are minimized same as ‘‘a’’ .crms deviations from crystal structure.drms deviations of the anchor fragment from the crystal structure.e ( )Time in seconds, UNIX time command for anchor fragment docking.f ( )Time in seconds, UNIX time command for conformational search and minimization.
Ž .rms deviations are calculated by the program‘‘RMSD MULTIPLE.’’ This program takes symme-]tries of ligands into consideration and calculatesall possible rms deviations, then it prints out theminimum rms deviation. We next discuss the re-sults for the individual ligand families.
SUGAR—L-ARABINOSE ANDD-GALACTOSE / D-GLUCOSE BINDINGPROTEINS: 1abe, 6abp, 2gbp
Hydrogen bonds play an important role in bind-ing sugars to their binding proteins. Although thecoordinates of the hydrogens are not availablefrom the x-ray experiments, and we do not knowthe actual hydrogen bond networks, hydrogenbond formation seems to have been optimizedvery well. The force field helped to form hydrogenbonds without special hydrogen bond scores. Inthe case of 2gbp, the direction of one branch ofglucose is different from the x-ray structure andforms an intramolecular hydrogen bond.
FOLATE ANALOGS—DIHYDROFOLATEREDUCTASE: 0dfr, 4dfr, 7dfr
The anchor fragments were all placed very closeto the x-ray structures. The orientation of the a-carboxylates of MTX and DHFR, which bind to
DHFR specifically and strongly, are similar to x-raystructures.33,34 In contrast, the orientation of theb-carboxylates of MTX and DHFR, which bind toDHFR nonspecifically and weakly, are not verysimilar to x-ray structures. Also, the dihedral an-gles that fix the orientation of the 3,4,5-trimeth-oxyphenyl fragment are not very close to the x-raystructure. However, the x-ray conformations andthe calculated conformations have a large amountof volume overlap. A similar observation has beenmade for side-chain orientations during proteinpacking calculations when the backbone is held
Ž .rigid C. Lee, private communication . There areseveral reports on simulations of ligand dockingwith DHFR. Some reduce computation time byusing partial structures of the ligands15 or fix thenumber of rotatable bonds.13 Although such ap-proximations might be reasonable for each specialcase, it is difficult to apply these special conditionsto all the molecules in a database.
3-TAPAP—TRYPSIN: 1pph
Although the anchor fragment and most of theother parts of the structure were placed in almostthe same position as the x-ray complex structure,the tosyl group of the ligand could not be placedcorrectly in the narrow S3 pocket of trypsin. The
JOURNAL OF COMPUTATIONAL CHEMISTRY 1821

MAKINO AND KUNTZ
rms deviation without the tosyl group is 0.57.Nevertheless, the score is very similar to the mini-mized x-ray complex score, so the program mighthave found the alternative binding mode.
RETINOL—RETINOL BINDING PROTEIN:1rbp
The program recognized the conjugated doublebonds and treated them as nonrotatable bonds. Asa result, the anchor fragment is relatively large inthis case and the possibility of alternative orienta-tions is reduced. This is the reason why the pro-gram regenerated the x-ray structure very pre-cisely.
29-GMP—RIBONUCLEASE T : 1rnt1
Both the anchor fragment and the rest of theconformation were predicted correctly.
BIOTIN—STREPTAVIDIN: 1stp
The anchor fragment was oriented very close tothe x-ray complex structure. There was some wob-ble in the methylene chain between the anchorfragment and the carboxylic acid, but because thisregion of the molecule lies in a hydrophobic pocket,this wobble seems acceptable.
ROBUSTNESS IN SAMPLING BY LIMITEDBACKTRACK SEARCH
We employed the limited backtrack search algo-rithm to reduce computation time. However, thissearch might fail to find the conformation with theminimum energy. To test if this search method
samples enough conformations, backtrack searchwith and without limitation were compared usingligands of DHFR. Because the approximate answershould be obtained within a reasonable computa-tion time, it is necessary to restrict the conforma-tional space for the search. Here, the results of theusual backtrack search with no limitations and thelimited backtrack search are compared. As TableVIII shows, the full search always resulted in bet-ter energy scores, as expected. However, we notethat the best scoring conformations from the lim-ited search are actually somewhat close to thex-ray structure in two cases. Thus, we judged thatthis approximation reduced the quality of the re-sults only marginally, while reducing the compu-tation time significantly. On the whole, the pro-gram successfully predicted the binding modes of
Žthe ligands with many flexible bonds five, nine,.and nine flexible bonds in a relatively short time.
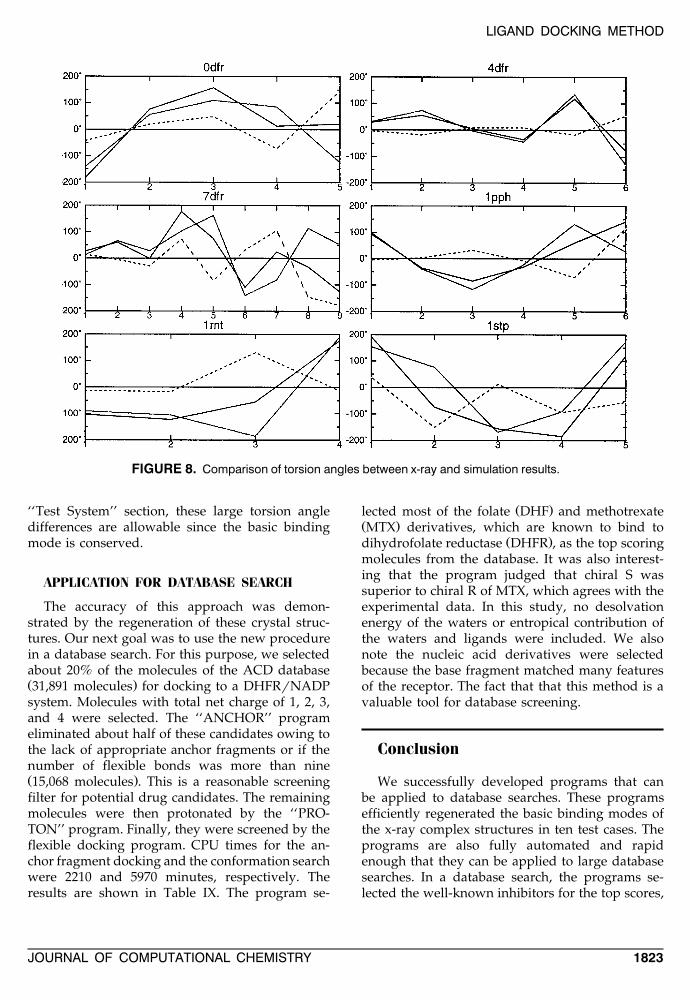
COMPARISON OF DIHEDRAL ANGLES
Torsion angles of tested results are comparedŽ .with x-ray crystal structures see Fig. 8 . The x-axes
are the torsion angle number and the y-axes arethe angle by degree. The solid lines represent ac-tual torsion angles of x-ray and tested results ofligands, the dashed lines represent the torsion an-gle differences between x-ray and tested results.In general, the dihedral angle differences aresmaller near the anchor fragment. The torsion an-gle differences display a ‘‘crankshaft’’ motion asreported in the previous study,13 thus x-ray andtested ligands occupy very similar volume. In thecase of the 1stp system, the torsion angle differ-ences are rather large and do not show a‘‘crankshaft’’ motion. However, as discussed in the
TABLE VIII.Comparisons of Backtrack Search.
No Limitation Limitationa b c a b cpdb Score rmsd Time Score rmsd Time
0dfr y31.71 0.89 200 y30.66 1.03 944dfr y38.65 1.43 100880 y35.56 1.19 2627dfr y42.74 2.45 32063 y42.58 1.69 121
aSum of the intra- and intermolecular force field energy.brms deviations between the tested results and the crystal structures.c ( )Time in seconds for conformational search and minimization.
VOL. 18, NO. 141822
LIGAND DOCKING METHOD
FIGURE 8. Comparison of torsion angles between x-ray and simulation results.
‘‘Test System’’ section, these large torsion angledifferences are allowable since the basic bindingmode is conserved.
APPLICATION FOR DATABASE SEARCH
The accuracy of this approach was demon-strated by the regeneration of these crystal struc-tures. Our next goal was to use the new procedurein a database search. For this purpose, we selectedabout 20% of the molecules of the ACD databaseŽ .31,891 molecules for docking to a DHFRrNADPsystem. Molecules with total net charge of 1, 2, 3,and 4 were selected. The ‘‘ANCHOR’’ programeliminated about half of these candidates owing tothe lack of appropriate anchor fragments or if thenumber of flexible bonds was more than nineŽ .15,068 molecules . This is a reasonable screeningfilter for potential drug candidates. The remainingmolecules were then protonated by the ‘‘PRO-TON’’ program. Finally, they were screened by theflexible docking program. CPU times for the an-chor fragment docking and the conformation searchwere 2210 and 5970 minutes, respectively. Theresults are shown in Table IX. The program se-
Ž .lected most of the folate DHF and methotrexateŽ .MTX derivatives, which are known to bind to
Ž .dihydrofolate reductase DHFR , as the top scoringmolecules from the database. It was also interest-ing that the program judged that chiral S wassuperior to chiral R of MTX, which agrees with theexperimental data. In this study, no desolvationenergy of the waters or entropical contribution ofthe waters and ligands were included. We alsonote the nucleic acid derivatives were selectedbecause the base fragment matched many featuresof the receptor. The fact that that this method is avaluable tool for database screening.
Conclusion
We successfully developed programs that canbe applied to database searches. These programsefficiently regenerated the basic binding modes ofthe x-ray complex structures in ten test cases. Theprograms are also fully automated and rapidenough that they can be applied to large databasesearches. In a database search, the programs se-lected the well-known inhibitors for the top scores,
JOURNAL OF COMPUTATIONAL CHEMISTRY 1823

MAKINO AND KUNTZ
TABLE IX.Results of ACD Database Search.
aName Compound Score
b1 MFCD00150465 L-Aminopterin y73.469c2 MFCD00150769 L-Folic acid y66.955
c( )3 MFCD00006707 L-N10- trifluoroacetyl pteroic acid y64.965f4 MFCD00167129 29-Deoxyinosine 59-monophosphate y61.371
5 MFCD00079526 3-TAPAP y58.941b6 MFCD00064370 L-Methotrexate y57.979
c7 MFCD00057319 39,59-dichlorofolic acid y57.970d8 MFCD00057016 Cordycepin 59-monophosphate y57.969
d9 MFCD00079147 29-Deoxyadenosine 59-mono phosphomorpholidate y55.112d10 MFCD00005753 29-Deoxyadenosin 59-monophosphate y54.838
g( )11 MFCD00065638 Fmoc-L-Cys Npys y53.782c12 MFCD00057318 L-39,59-dibromofolic acid y53.061
c13 MFCD00075823 Pteroic acid y51.45414 MFCD00056768 1,N6-etheno-29-deoxyadenosine 59-monophosphate y51.236
e15 MFCD00210875 29-Deoxyguanosine 59-monophosphate y50.531g( )16 MFCD00079516 Npys-L-Tyr tBu y50.345
e17 MFCD00057108 29-O-monosuccinylguanosin 39:59-cyclic monophosphate y50.25518 MFCD00070111 29-Deoxyadenosine 39-monophosphated y50.01319 MFCD00098907 Maybridge NRB 00318 y49.854
g( )20 MFCD00079512 Npys-L-His tosyl y49.83121 MFCD00184466 Salor S7,530-3 y49.704
( )22 MFCD00057601 N,O-di 2,4-DNP -L-tyrosine y49.362b[ ( ) ]23 MFCD00075773 4- N- 2,4-diamino-6-pteridinylmethyl amino benzoic acid y49.257
( )24 MFCD00038472 N,S-di 2,4-DNP -L-cysteine y48.895( )25 MFCD00057369 N,N-di 2,4-DNP -L-lysine y48.693
26 MFCD00099689 Maybridge KM 06664 y48.502d27 MFCD00058133 29-Deoxyadenosine 39-monophosphate y48.400
g( )28 MFCD00079492 Npys-L-Glu tBu y48.255( )29 MFCD00020220 3,39-hexamethylene diureido bis 2,4,6-triidobenzoic acid y48.223
30 MFCD00037966 Trp]Gly]Gly y48.18031 MFCD00064185 2-Naphthalene sulfonic acid y48.02232 MFCD00097525 Maybridge KM 06606 y47.87033 MFCD00118936 Maybridge CD 02454 y47.783
d34 MFCD00057021 29-Deoxyadenosine 39-monophosphate y47.601g35 MFCD00079489 Npys-L-Asp-NH y47.3552
36 MFCD00006710 D-Amethopterinb y47.249
aScores are only intermolecular energy.bMTX derivatives.cDHF derivatives.dAdenosine derivatives.eGuanosine derivatives.fInosine derivatives.g ( )Protected by 3-nitro-2-pyridinesulfenyl Npys .
showing the efficiency of the programs for use invirtual database screening.
Availability
Because this program is still under active devel-opment, interested readers should contact the au-thors concerning its availability.
Acknowledgments
We thank Greg Couch of the UCSF ComputerGraphics Laboratory for many suggestions aboutprogramming in Cqq. We also thank Dr. YaxiongSun, Todd Ewing, Dr. Connie Oshiro, and EricPettersen for helpful comments and discussions.Mr. Ewing was especially helpful in making avail-
VOL. 18, NO. 141824

LIGAND DOCKING METHOD
able the DOCK4 program source code in advanceof its public release. Tripos Associates providedthe SYBYL program and Molecular Design Ltd.provided the Available Chemicals Directory forwhich we are grateful.
References
1. A. R. Leach and I. D. Kuntz, J. Comput. Chem., 13, 730Ž .1992 .
2. Distributed by Molecular Design, Ltd., San Leandro, CA.3. E. Rutenber, E. B. Fauman, R. J. Keenan, S. Fong, P. S.
Furth, P. R. Ortiz de Montellano, E. Meng, I. D. Kuntz, D. L.DeCamp, R. Salto, J. R. Rose, C. S. Craik, and R. M. Stroud,
Ž .J. Biol. Chem., 268, 15343 1993 .4. P. Y. S. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge, Y.
Ru, L. T. Bacheler, J. L. Meek, M. J. Otto, M. M. Rayner, Y.N. Wong, C.-H. Chang, P. C. Weber, D. A. Jackson, T. R.
Ž .Sharpe, and S. Erickson-Viitanen, Science, 263, 380 1994 .5. K. R. Romines, K. D. Watenpaugh, W. J. Howe, P. K.
Tomich, K. D. Lavasz, J. K. Morris, M. N. Janakiraman, J. C.Lynn, M.-M. Horng, K.-T. Chong, R. R. Hinshaw, and L. A.
Ž .Dolak, J. Med. Chem., 38, 4463 1995 .Ž .6. T. L. Blundell, Nature, 384, 23 1996 .
7. I. D. Kuntz, J. M. Blaney, S. J. Oatley, R. Langridge, and T.Ž .Ferrin, J. Mol. Biol., 161, 269 1982 .
Ž .8. M. Lawrence and P. C. Davis, Proteins, 12, 31 1992 .9. N. Kasinos, G. A. Lilly, N. Subbarao, and I. Haneel, Pro-
Ž .teins, 5, 69 1992 .10. R. L. DesJarlais, R. P. Sheridan, J. S. Dixon, I. D. Kuntz, and
Ž .R. Venkataraghavan, J. Med. Chem., 29, 2149 1986 .Ž .11. D. S. Goodsell and A. J. Olson, Proteins, 8, 195 1990 .
12. G. Chang, W. C. Guida, and W. C. Still, J. Am. Chem. Soc.,Ž .111, 4379 1989 .
13. C. M. Oshiro, I. D. Kuntz, and J. S. Dixon, J. Comput.-AidedŽ .Mol. Design, 9, 113 1995 .
Ž .14. K. P. Clark, J. Comput. Chem., 16, 1210 1995 .15. M. Y. Mizutani, N. Tomioka, and A. Itai, J. Mol. Biol., 243,
Ž .310 1994 .16. W. Welch, J. Ruppert, and A. Jain, Chem. Biology, 3, 449
Ž .1996 .17. M. Rarey, B. Kramer, T. Lengauer, and G. Klebe, J. Mol.
Ž .Biol., 261, 470 1996 .
18. SYBYL, Version 6.0.2, Tripos Associates, St. Louis, MO,1993.
19. SYBYL Theory Manual 1, Tripos Associates, St. Louis, MO,1993.
20. S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C.Ghio, G. Alagona, S. Profeta Jr., and P. Weiner, J. Am.
Ž .Chem. Soc., 106, 765 1984 .21. S. J. Weiner, P. A. Kollman, D. T. Nguyen, and D. A. Case,
Ž .J. Comput. Chem., 7, 230 1986 .22. I. D. Kuntz, E. C. Meng, and B. K. Shoichet, Acc. Chem. Res.,
Ž .27, 117 1994 .23. E. C. Meng, B. K. Shoichet, and I. D. Kuntz, J. Comput.
Ž .Chem., 13, 505 1992 .24. I. D. Kuntz, J. M. Blaney, S. J. Oatley, R. Langridge, and T.
Ž .E. Ferrin, J. Mol. Biol., 161, 269 1982 .Ž .25. T. Ewing and I. D. Kuntz, J. Comput. Chem., 18, 1175 1997 .
Ž .26. R. M. Karp and Y. Zhang, J. ACM, 40, 756 1993 .27. M. Gondran, M. Minoux, and S. Vajda, Graphs and Algo-
rithms, John Wiley & Sons, New York, 1984.Ž .28. J. A. Nelder and R. Mead, J. Comput., 7, 308 1965 .
Ž .29. F. A. Quiocho and N. K. Vyas, Nature, 310, 381 1984 .30. M. E. Newcomer, D. M. Miller III, and F. A. Quiocho, J.
Ž .Biol. Chem., 254, 7529 1979 .31. F. A. Quiocho, D. K. Wilson, and N. K. Vyas, Nature, 340,
Ž .404 1989 .32. D. A. Matthews, J. T. Bolin, J. M. Burridge, D. J. Filman, K.
W. Volz, B. T. Kaufman, C. R. Beddell, J. N. Champness, D.Ž .K. Stammers, and J. Kraut, J. Biol. Chem., 260, 381 1985 .
33. J. T. Bolin, D. J. Filman, D. A. Matthews, R. C. Hamlin, andŽ .J. Kraut, J. Biol. Chem., 257, 13650 1982 .
34. C. Bystroff, S. J. Oatley, and J. Kraut, Biochemistry, 29, 3263Ž .1990 .
35. N. K. Vyas, M. N. Vyas, and F. A. Quiocho, Science, 242,Ž .1290 1988 .
36. D. Turk, J. Sturzebecher, and W. Bode, FEBS Lett., 287, 133Ž .1991 .
37. S. W. Cowan, M. E. Newcomer, and T. A. Jones, Proteins, 8,Ž .44 1990 .
38. R. Arni, U. Heinemann, M. Maslowska, R. Tokuoka, and W.Ž .Saenger, Acta Cryst., B43, 548 1987 .
Ž .39. K. Takahashi, J. Biochem., 72, 1469 1972 .40. P. C. Weber, J. J. Wendoloski, M. W. Pantoliano, and F. R.
Ž .Salemme, J. Am. Chem. Soc., 114, 3197 1992 .Ž .41. M. Marsili and J. Gasteiger, Chim. Acta, 52, 601 1980 .
Ž .42. J. Gasteiger and M. Marsili, Tetrahedron, 36, 3210 1980 .
JOURNAL OF COMPUTATIONAL CHEMISTRY 1825
Recommended

















