
AssemblyAssembly
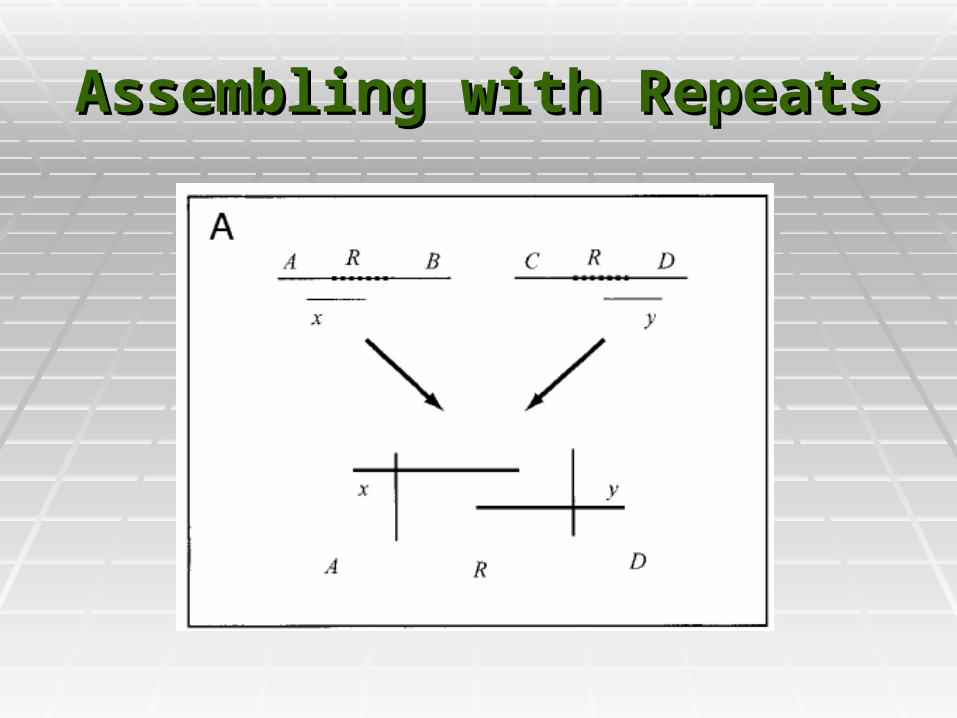
Assembling with RepeatsAssembling with Repeats
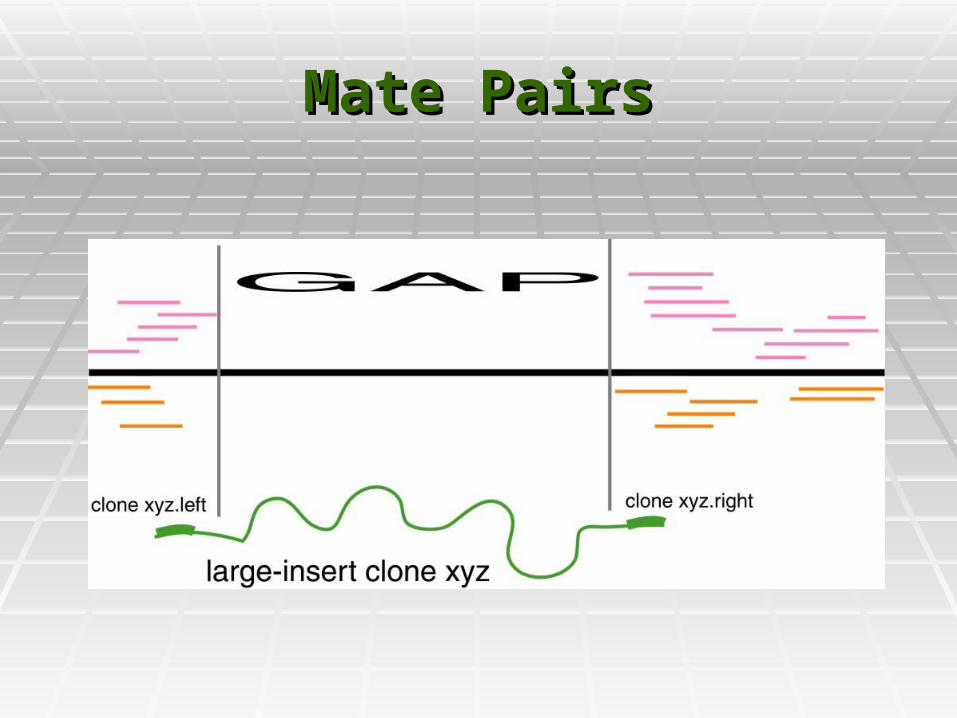
Mate PairsMate Pairs
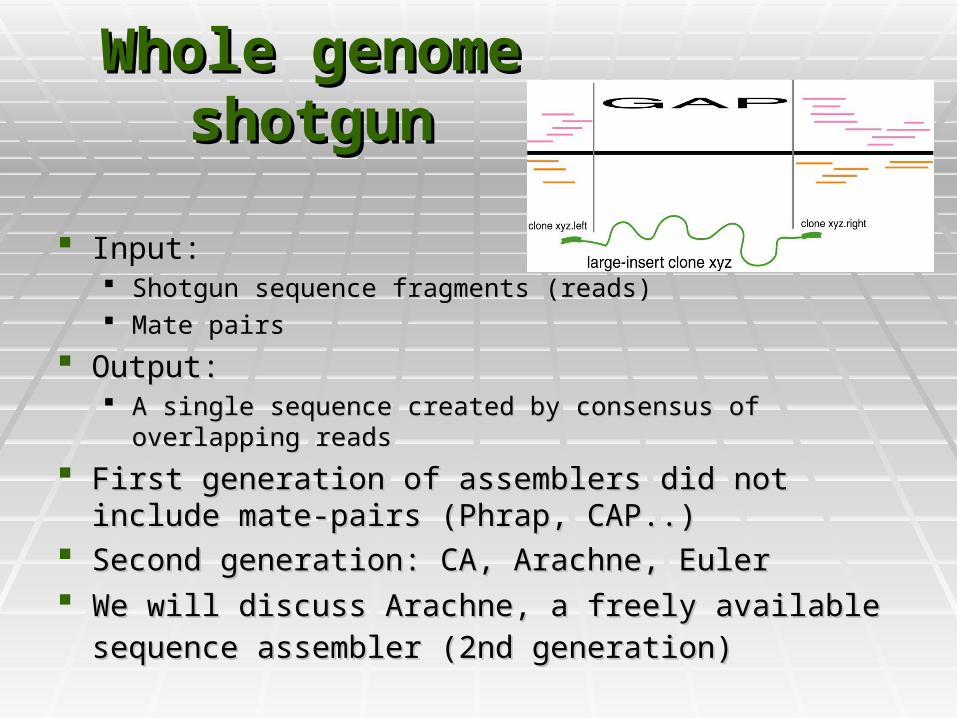
Whole genome Whole genome shotgunshotgun
Input: Input: Shotgun sequence fragments (reads)Shotgun sequence fragments (reads) Mate pairsMate pairs
Output:Output: A single sequence created by consensus of overlapping readsA single sequence created by consensus of overlapping reads
First generation of assemblers did not include mate-pairs First generation of assemblers did not include mate-pairs (Phrap, CAP..)(Phrap, CAP..)
Second generation: CA, Arachne, EulerSecond generation: CA, Arachne, Euler We will discuss Arachne, a freely available sequence We will discuss Arachne, a freely available sequence
assembler (2nd generation)assembler (2nd generation)

Arachne: DetailsArachne: Details
Initial processingInitial processing Alignment moduleAlignment module

Alignment ModuleAlignment Module
Input: Collection of DNA sequences of Input: Collection of DNA sequences of arbitrary lengtharbitrary length
Output: Pairwise alignments between Output: Pairwise alignments between them.them.

Overlap detectionOverlap detection
Option 1: Compute an alignment between Option 1: Compute an alignment between every pair.every pair. G = 150Mb, L=500G = 150Mb, L=500 Coverage LN/G = 10Coverage LN/G = 10 N = 10*150*10N = 10*150*1066/500 = 3*10/500 = 3*1066
Not good! (Only a small fraction are true Not good! (Only a small fraction are true overlaps)overlaps)

K-mer based overlapK-mer based overlap
A 25-bp sequence appears at most once A 25-bp sequence appears at most once in the genome!in the genome!
Two overlapping sequences should share Two overlapping sequences should share a 25-mera 25-mer
Two non-overlapping sequences should Two non-overlapping sequences should not!not!

Sorting k-mersSorting k-mers
Build a list of k-mers that appear in the Build a list of k-mers that appear in the sequences and their reverse complementssequences and their reverse complements
Create a record with 4 entries:Create a record with 4 entries: K-merK-mer Sequence numberSequence number Position in the sequencePosition in the sequence Reverse complementation flagReverse complementation flag
Sort a vector of these according to k-merSort a vector of these according to k-mer If number of records exceeds threshold, discard If number of records exceeds threshold, discard
(why?)(why?)
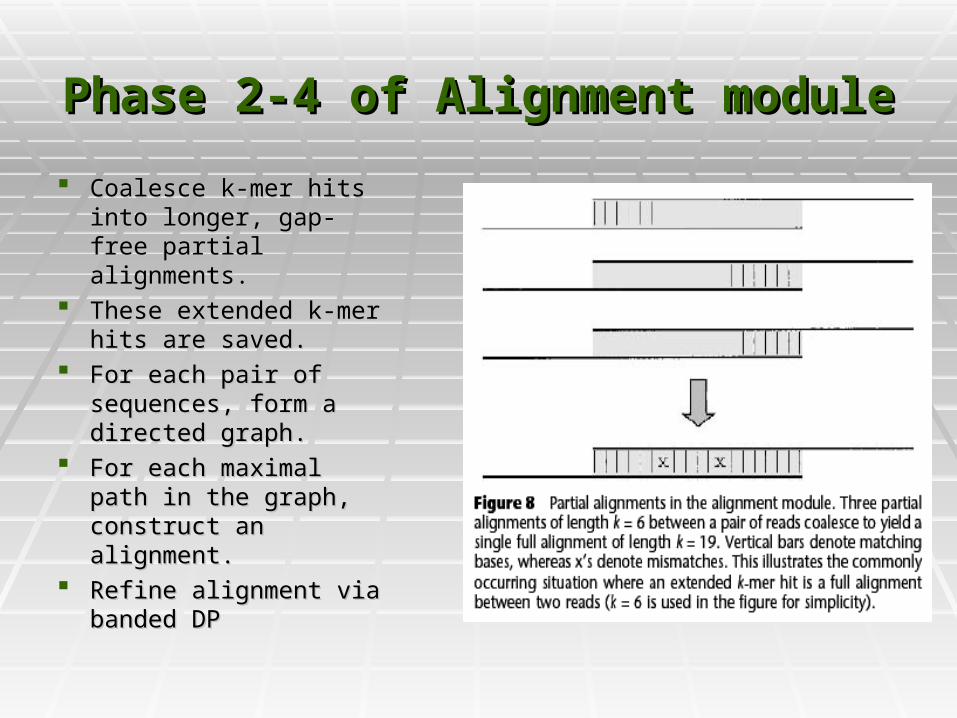
Phase 2-4 of Alignment modulePhase 2-4 of Alignment module
Coalesce k-mer hits into Coalesce k-mer hits into longer, gap-free partial longer, gap-free partial alignments.alignments.
These extended k-mer These extended k-mer hits are saved.hits are saved.
For each pair of For each pair of sequences, form a sequences, form a directed graph. directed graph.
For each maximal path For each maximal path in the graph, construct in the graph, construct an alignment.an alignment.
Refine alignment via Refine alignment via banded DPbanded DP

Detecting Chimeric readsDetecting Chimeric reads
Chimeric reads: Reads that Chimeric reads: Reads that contain sequence from two contain sequence from two genomic locations.genomic locations.
Good overlaps: G(a,b) if a,b Good overlaps: G(a,b) if a,b overlap with a high scoreoverlap with a high score
Transitive overlap: T(a,c) if Transitive overlap: T(a,c) if G(a,b), and G(b,c) G(a,b), and G(b,c)
Find a point x across which Find a point x across which only transitive overlaps occur. only transitive overlaps occur. X is a point of chimerismX is a point of chimerism
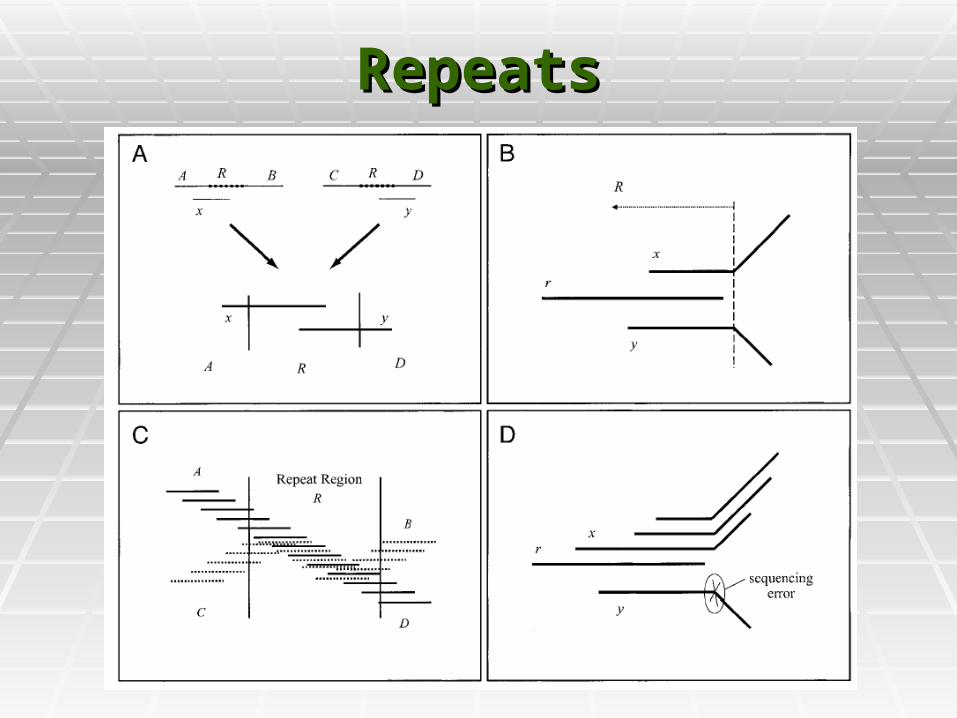
RepeatsRepeats

Contig assemblyContig assembly
Reads are merged into contigs Reads are merged into contigs upto repeat boundaries.upto repeat boundaries.
(a,b) & (a,c) overlap, (b,c) (a,b) & (a,c) overlap, (b,c) should overlap as well. Also, should overlap as well. Also, shift(a,c)=shift(a,b)+shift(b,c)shift(a,c)=shift(a,b)+shift(b,c)
Most of the contigs are unique Most of the contigs are unique pieces of the genome, and end pieces of the genome, and end at some Repeat boundary.at some Repeat boundary.
Some contigs might be entirely Some contigs might be entirely within repeats. These must be within repeats. These must be detecteddetected

Detecting Repeat Contigs 1: Read DensityDetecting Repeat Contigs 1: Read Density
Compute the log-odds Compute the log-odds ratio of two ratio of two hypotheses:hypotheses:
H1: The contig is from H1: The contig is from a unique region of the a unique region of the genome.genome.
The contig is from a The contig is from a region that is region that is repeated at least repeated at least twicetwice

Creating Super ContigsCreating Super Contigs

Supercontig assemblySupercontig assembly
Supercontigs are built incrementallySupercontigs are built incrementally Initially, each contig is a supercontig.Initially, each contig is a supercontig. In each round, a pair of super-contigs is In each round, a pair of super-contigs is
merged until no more can be performed.merged until no more can be performed. Create a Priority Queue with a score for Create a Priority Queue with a score for
every pair of ‘mergeable supercontigs’.every pair of ‘mergeable supercontigs’. Score has two terms:Score has two terms:
A reward for multiple mate-pair linksA reward for multiple mate-pair links A penalty for distance between the links.A penalty for distance between the links.

Supercontig mergingSupercontig merging
Remove the top scoring pair (S1,S2) from Remove the top scoring pair (S1,S2) from the priority queue.the priority queue.
Merge (SMerge (S11,S,S22) to form contig T.) to form contig T. Remove all pairs in Q containing SRemove all pairs in Q containing S11 or S or S22
Find all supercontigs W that share mate-Find all supercontigs W that share mate-pair links with T and insert (T,W) into the pair links with T and insert (T,W) into the priority queue.priority queue.
Detect Repeated Supercontigs and Detect Repeated Supercontigs and removeremove

Repeat SupercontigsRepeat Supercontigs
If the distance If the distance between two super-between two super-contigs is not contigs is not correct, they are correct, they are marked as marked as RepeatedRepeated
If transitivity is not If transitivity is not maintained, then maintained, then there is a Repeatthere is a Repeat
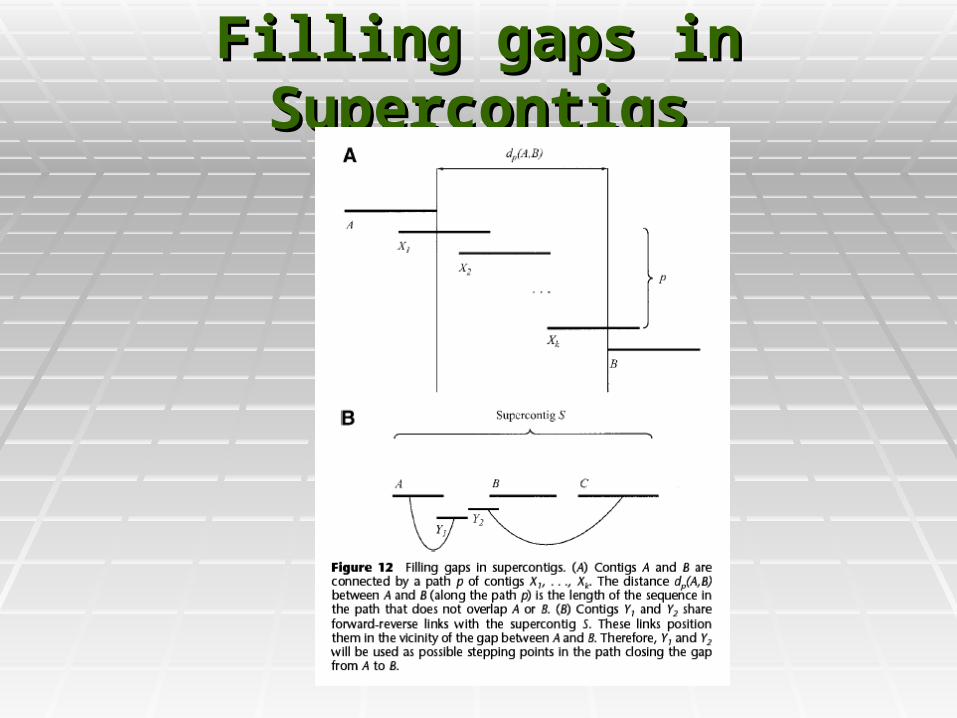
Filling gaps in SupercontigsFilling gaps in Supercontigs

Consenus DerivationConsenus Derivation
Consensus sequence is created by Consensus sequence is created by converting pairwise read alignments into converting pairwise read alignments into multiple-read alignmentsmultiple-read alignments

SummarySummary
Whole genome shotgun is now routine:Whole genome shotgun is now routine: Human, Mouse, Rat, Dog, Chimpanzee..Human, Mouse, Rat, Dog, Chimpanzee.. Many Prokaryotes (One can be sequenced in a day)Many Prokaryotes (One can be sequenced in a day) Plant genomes: Arabidopsis, Rice Plant genomes: Arabidopsis, Rice Model organisms: Worm, Fly, YeastModel organisms: Worm, Fly, Yeast
A lot is not known about genome structure, A lot is not known about genome structure, organization and function.organization and function. Comparative genomics offers low hanging fruitComparative genomics offers low hanging fruit
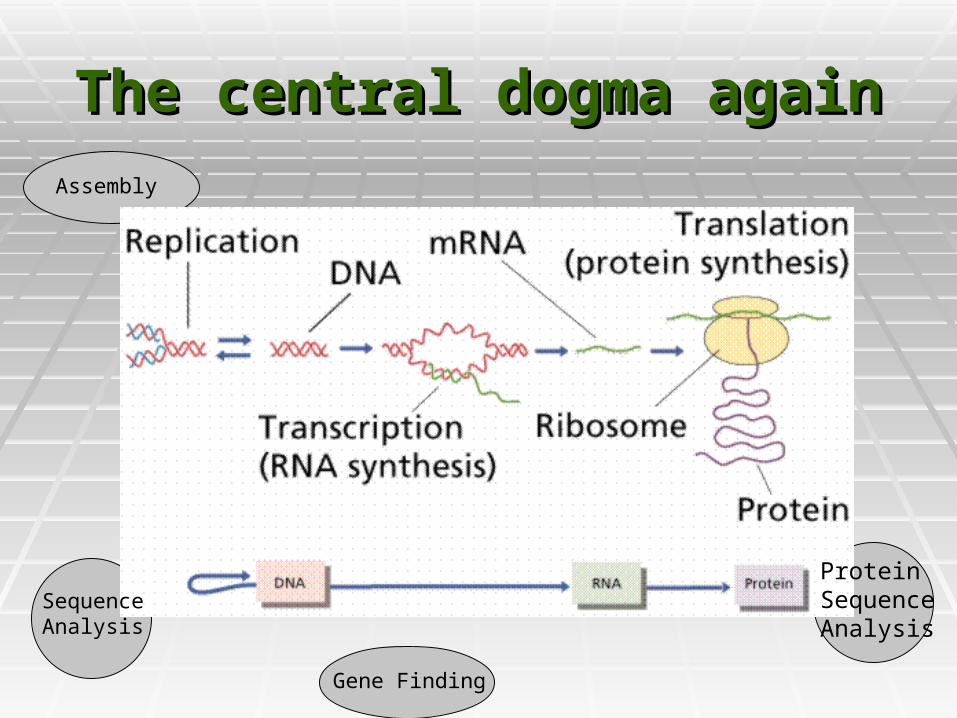
The central dogma againThe central dogma again
Protein SequenceAnalysis
Sequence Analysis
Gene Finding
Assembly
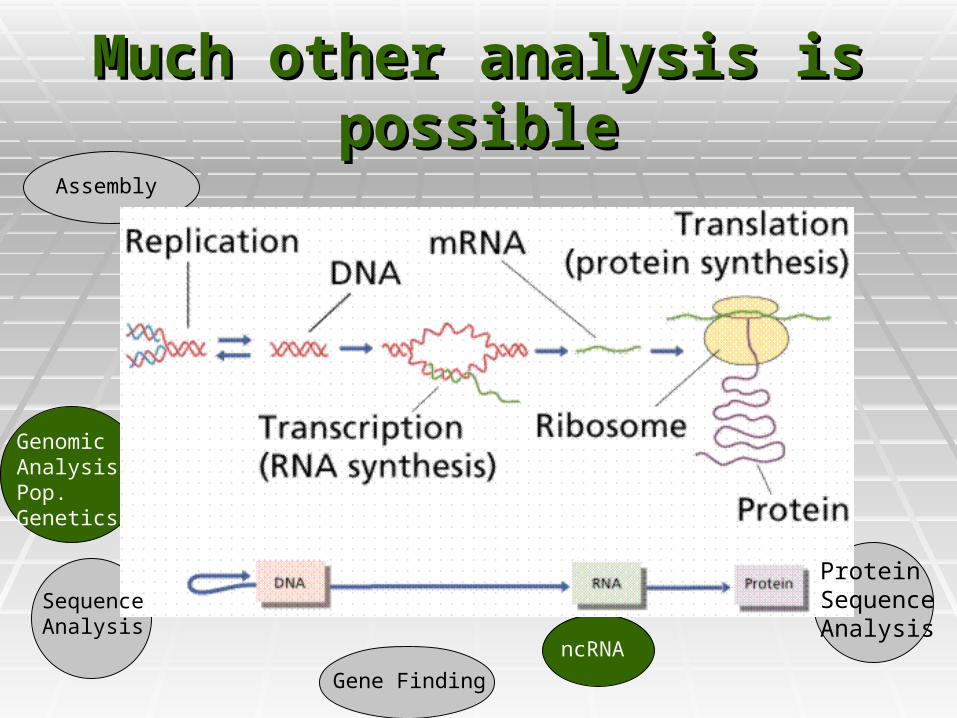
Much other analysis is Much other analysis is possiblepossible
Protein SequenceAnalysis
Sequence Analysis
Gene Finding
Assembly
ncRNA
GenomicAnalysis/Pop. Genetics

A Static picture of the cell is insufficientA Static picture of the cell is insufficient
Each Cell is continuously active, Each Cell is continuously active, Genes are being transcribed into RNAGenes are being transcribed into RNA RNA is translated into proteinsRNA is translated into proteins Proteins are PT modified and transportedProteins are PT modified and transported Proteins perform various cellular functionsProteins perform various cellular functions
Can we probe the Cell dynamicallyCan we probe the Cell dynamically
Recommended

















