Embed Size (px)
DESCRIPTION
novel
Citation preview


Vitrification in AssistedReproduction
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page i

REPRODUCTIVE MEDICINE & ASSISTED REPRODUCTIVETECHNIQUES SERIES
Series Editors
David K Gardner DPhil
Colorado Center for Reproductive Medicine, Englewood, CO, USA
Jan Gerris MD PhD
Professor of Gynecology, University Hospital Gheni, Ghent, Belgium
Zeev Shoham MD
Director, Infertility Unit, Kaplan Hospital, Rehovot, Israel
Published Titles
Gerris, Delvigne and Olivennes, Ovarian Hyperstimulation SyndromeISBN 978 1842143285
Sutcliffe, Health and Welfare of ART ChildrenISBN 9780415379304
Tan, Chian and Buckett, In-vitro Maturation of Human OocytesISBN 978 1842143322
Keck, Tempfer and Hugues, Conservation Infertility ManagementISBN 9780415384513
Pellicer and Simon, Stem Cells in Human ReproductionISBN 978 0 415 397 773
Elder and Cohen, Human Preimplantation Embryo SolutionISBN 978 0415399739
Forthcoming Titles
Aplin, Fazleabas, Glasser, Giudice, The Endometrium, second editionISBN 978 0415385831
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page ii

Vitrification in AssistedReproductionA User’s Manual andTrouble-shooting Guide
Edited by
Michael J Tucker PhD FIBiol HCLDScientific DirectorGeorgia Reproductive SpecialistsAtlanta, GAUSA
Juergen Liebermann PhD HCLDScientific DirectorFertility Centers of IllinoisChicago, ILUSA
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page iii

© 2007 Informa UK Ltd
First published in the United Kingdom in 2007 by Informa Healthcare, Telephone House, 69–77 Paul Street, LondonEC2A 4LQ. Informa Healthcare is a trading division of Informa UK Ltd. Registered Office: 37/41 Mortimer Street,London W1T 3JH. Registered in England and Wales number 1072954.
Tel: +44 (0)20 7017 5000Fax: +44 (0)20 7017 6699Website: www.informahealthcare.com
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in anyform or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior permission ofthe publisher or in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the termsof any licence permitting limited copying issued by the Copyright Licensing Agency, 90 Tottenham Court Road,London W1P 0LP.
Although every effort has been made to ensure that all owners of copyright material have been acknowledged inthis publication, we would be glad to acknowledge in subsequent reprints or editions any omissions brought to ourattention.
A CIP record for this book is available from the British Library.Library of Congress Cataloging-in-Publication Data
Data available on application
ISBN-10: 0 415 40882 2 ISBN-13: 978 0 415 40882 0
Distributed in North and South America byTaylor & Francis6000 Broken Sound Parkway, NW, (Suite 300)Boca Raton, FL 33487, USA
Within Continental USATel: 1 (800) 272 7737; Fax: 1 (800) 374 3401Outside Continental USATel: (561) 994 0555; Fax: (561) 361 6018Email: [email protected]
Distributed in the rest of the world byThomson Publishing ServicesCheriton HouseNorth WayAndover, Hampshire SP10 5BE, UKTel: +44 (0)1264 332424Email: [email protected]
Composition by C&M Digitals (P) Ltd, Chennai, IndiaPrinted and bound in India by Replika Press Pvt Ltd
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page iv

Michael J Tucker would like to thank his wife Megan and three children who make this allworthwhile.
Juergen Liebermann gives most sincere thanks to his wife Maike and his sons Richard,Lennart Martin, and Tobias Georg for providing unfailing support and encouragement tobring this work to reality.
Dedication
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page v

Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page vi

List of Contributors ix
Preface xiii
Acknowledgments xv
1A. Vitrification: an overview 1Gregory M Fahy and William F Rall
1B. Vitrification in small quenched volumes with a minimal amountof, or without vitrificants: basic biophysics and thermodynamics 21
Igor I Katkov, Vladimir Isachenko and Evgenia Isachenko
2. Disadvantages and benefits of vitrification 33Gábor Vajta, Masashige Kuwayama and Pierre Vanderzwalmen
3. Development of vitrification solutions 45Jaffar Ali and James Shelton
4A. Vitrification in animal reproduction: vitrification of embryosusing open pulled straws 65Gábor Vajta
4B. Vitrification in animal reproduction: vitrification of embryosusing conventional straws with ethylene glycol-based solutions 75Magosaburo Kasai and Keisuke Edashige
5. Cryoprotectant-free vitrification of spermatozoa 87Evgenia Isachenko, Vladimir Isachenko, Igor I Katkov, Raul Sanchez,Hans van der Ven, Markus Montag and Frank Nawroth
6. Potential developmental consequences of cryopreservation ofmammalian oocytes and embryos 107Gary D Smith and Luis G Villa-Diaz
7A. Vitrification of oocytes: general considerations and theuse of the Cryotop method 119Masahige Kuwayama, Ana Cobo and Gábor Vajta
Contents
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page vii

viii
7B. Vitrification of oocytes: various procedures 129Shee-Uan Chen and Yu-Shih Yang
7C. Vitrification of oocytes using gold grid and slush nitrogen 145Tae Ki Yoon, Dong Ryul Lee and Kwang Ryul Cha
7D. Vitrifying and warming of oocytes using cryotop 153Koichi Kyono, Yukiko Nakajo, Shima Kumagai, and Chikako Nishinaka
8. Vitrification of pronuclear embryos: research basis for aseptictechnology and its application to oocytes and blastocysts 163Hans van der Ven, Vladimir Isachenko, Evgenia IsachenkoMarkus Montag and Frank Nawroth
9. Vitrification of day 2–3 human embryos: using varioustechniques (Cryoloop, Cryotop and conventional cryostraw) 183Tetsunori Mukaida and Katsushiko Takahashi
10A. One decade of experience with vitrification of human embryosin straws, hemi-straws, and high security vitrification straws 195Pierre Vanderzwalmen, Thomas Ebner and Nicolas Zech
10B. Vitrification of blastocysts using the Cryoloop technique 219Tetsunori Mukaida and Katsuhiko Takahashi
10C. Vitrification of blastocysts using the electron microscope grid 239Weon-Young Son and Jin-Ho Lim
10D. Vitrifying and warming of human blastocysts using the Cryotop 253Juergen Liebermann and Michael J Tucker
11A Vitrification of ovarian tissue 261Frank Nawroth, Vladimir Isachenko, Evgenia Isachenko andGohar Rahimi
11B. Vitrification of ovarian tissues 273Ying C Song, Zhenzhen Chen, Carol Journey, Adelina M Emmi,Xiayang Xie and Rosemary L Song
12. Vitrification of human embryonic stem cells 293Yoel Shufaro, Gábor Vajta, Alan O Trounson and Benjamin E Reubinoff
Index 299
CONTENTS
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page viii

Jaffar Ali PhD
IVF UnitMaternity HospitalKing Fahad Medical CityRiyadhSaudi Arabia
Kwang Ryul Cha MD
Fertility Center of CHA General Hospital,CHA Research InstitutePochon Cha UniversityUniversity College of MedicineSeoulKorea
Shee-Uan Chen MD
Department of Obstetrics and GynecologyNational Taiwan University HospitalTaipeiTaiwan
Zhenzhen ChenOrgan Recovery Systems IncCharleston, SCUSA
Ana CoboInstitute of InfertilityValencia UniversitySchool of MedicineValenciaSpain
Thomas Ebner PhD
Landes-Frauen- und KinderklinikIVF Unit Linz,Austria
Keisuke Edashige PhD
Laboratory of Animal ScienceCollege of AgricultureKochi UniversityKochiJapan
Adelina M EmmiMedical College of GeorgiaAugusta, GAUSA
Gregory M Fahy PhD
21st Century Medicine IncRancho Cucamonga, CAUSA
Evgenia Isachenko PhD
Department of Endocrinology andReproductive MedicineUniversity of BonnBonnGermany
Vladimir Isachenko PhD
Department of Endocrinology andReproductive MedicineUniversity of BonnBonnGermany
Magosaburo Kasai PhD
Kochi Laboratory of Animal ScienceCollege of AgricultureKochi UniversityNankokuJapan
Contributors
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page ix

Igor I Katkov PhD
Department of Pediatrics,University of CaliforniaSan Diego, CAUSA
Carol JourneyMedical College of GeorgiaAugusta, GAUSA
Masashige Kuwayama PhD
Kato Ladies ClinicTokyoJapan
Koichi KyonoKyono ART ClinicJapan
Dong Ryul Lee PhD
Fertility Center of CHAGeneral HospitalCHA Research InstitutePochon Cha UniversityUniversity College of MedicineSeoulKorea
Juergen Liebermann PhD HCLD
Fertility Centers of IllinoisChicago, ILUSA
Jin-Ho Lim MD
PresidentMaria Infertility HospitalSeoulSouth Korea
Peter Mazur PhD
Fundamental and AppliedCryobiology GroupThe University of TennesseeKnoxville, TNUSA
Markus Montag MD PhD
Department of GynaecologicalEndocriniology and Reproductive MedicineUniversity of BonnBonn-VenusbergGermany
Tetsunori Mukaida MD
Hiroshima Hart ClinicHiroshimaJapan
Yukiko NakajoKyono ART ClinicJapan
Frank Nawroth MD PhD
Zentrum für Hormon- undStoffwechselerkrankungen,Reproduktionsmedizinund Gynäkologische EndokrinologieHamburgGermany
Chikako NishinakaKyono ART ClinicJapan
Gohar RahimiDepartment of Obstetrics and GynecologyUniversity of CologneCologneGermany
William F Rall PhD
National Center for Research ResourcesNational Institutes of HealthBethesda, MDUSA
Benjamin E Reubinoff MD PhD
Department of Obstetrics and GynecologyHadassah Embryonic StemCell Research CenterHadassah University HospitalJerusalemIsrael
LIST OF CONTRIBUTORS
x
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page x

xi
LIST OF CONTRIBUTORS
Raul Sanchez PhD MD
University of TemucoChile
Shima KumagaiKyono ART ClinicJapan
James Shelton DVSc PhD FACVSc
ScarboroughQueenslandAustralia
Yoel Shufaro MD
IVF UnitDepartment of Obsetrics and GnecologyThe Hadassah Human Embryonic Stem CellsResearch CenterHassadah Ein Kerem University HospitalJerusalemIsrael
Gary D Smith PhD HCLD
Department of Obstetrics and GynecologyUniversity of MichiganAnn Arbor, MIUSA
Weong-Yong Son PhD
McGill Reproductive CenterRoyal Victoria HospitalMontreal, QuebecCanada
Weon-Young Son PhD
Lead EmbryologistMcGill Reproductive CenterDepartment of Obstetrics and GynecologyRoyal Victoria HospitalMcGill UniversityMontrealCanada
Rosemary L SongMedical College of GeorgiaAugusta, GAUSA
Ying C Song MD PhD
Director of ResearchXytex ResearchAugusta, GAUSA
Katsuhiko Takahashi MD
Hiroshima HART ClinicHiroshimaJapan
Alan O Trounson BSc MSc PhD
Monash Immunology andStem Cell LaboratoriesMonash Medical CenterClayton, VictoriaAustralia
Michael J Tucker PhD FIBiol HCLD
Georgia Reproductive SpecialistsAtlanta, GAUSA
Gábor Vajta MD PhD DVSc
Department of Genetics and BiotechnologyDanish Institute of Agricultural SciencesResearch Center FoulumTjeleDenmark
Hans van der Ven MD
Zentrum für Geburtshilfe u. FrauenheilkundeRheinische Friedrich-Wilhelm-Universität BonnBonnGermany
Pierre Vanderzwalmen PhD
Institute for ReproductiveMedicine and EndocrinologyBregenzAustriaandCentre Hospitalier Inter Regional Cavell(CHIREC)BrusselsBelgium
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xi

Luis G Villa-Diaz PhD
University of MichiganAnn Arbor, MIUSA
Xiayang XieMedical College of GeorgiaAugusta, GAUSA
Yu-Shih Yang MD PhD
National Taiwan University Hospitaland College of MedicineTaipeiTaiwan
Tae Ki Yoon MD
Fertility Center of CHA General HospitalCHA Research InstitutePochon CHA UniversitySeoulKorea
Nicolas Zech MD
Institute for Reproductive Medicine andEndocrinologyBregenzAustria
LIST OF CONTRIBUTORS
xii
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xii

The impact of cryopreservation on the growth and improved efficiency of assisted reproductionin humans is becoming increasingly appreciated, with close to one-fifth of all births followingin vitro fertilization and embryo transfer worldwide arising from cryopreservation of super-numerary embryos. As culture techniques and related embryo quality improve, it is inevitablethat this ratio of fresh to cryopreserved embryo babies will become more equal, indicative of anincreased reliance on the use of embryos following cryostorage. This will be a consequence ofthe improved efficiencies inherent in this approach, including possibly improved uterine recep-tivity in unstimulated uterine transfer cycles, in conjunction with the recognition of the needfor more routine single embryo transfers, to reduce multiple implantations often seen withcycles where multiple embryos have been transferred, as is a common current practice.
Although initially reported in 1985 as a successful cryopreservation approach for mouseembryos, vitrification has taken a backseat to the much more widely adopted conventionalfreezing technology applied to both gametes and embryos in animal and human assisted repro-duction. Recent years have seen a resurgence of interest in this ultrarapid cryopreservationtechnology, as the limitations of slow-rate freezing have become more evident in the clinicalarena. Frustrations with mediocre cryosurvival, development, and ultimately compromisedimplantation rates have fuelled clinical embryologists in particular to seek alternative strategiesto improve outcomes. Vitrification, an ice-free form of cryopreservation, offers a level of con-sistency of performance, once mastered, that may achieve clinical results that rival outcomesusing fresh material. Additionally, vitrification offers certain benefits in the ease of its applica-tion that make current conventional freezing technology appear unpredictable, costly, andinflexible.
This book makes no pretence to be the definitive text on vitrification. It nonetheless attemptsto present in a straightforward manner the current and breaking vitrification technology avail-able to those in the animal reproduction industry, and to clinical practitioners in humanassisted reproduction. This includes discussion and guidance for cryoprotectant-free spermcryostorage, highly consistent oocyte cryopreservation, as well as consideration and explana-tion of successful protocols for vitrification of embryos at all stages of preimplantation devel-opment, with particular emphasis on in vitro derived human embryos. These various protocolsare discussed in the clear context of the type of vitrification carrier device used, with commenton how and why they were developed, and the relevance of cooling and warming rates that arecentral to achieving the vitrified state.
A wealth of easily understood background material is presented, as well as the extremelyuseful comparative discussion of vitrification protocols applied to mammalian oocytes and
Preface
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xiii

xiv
PREFACE
embryos, that enable greater appreciation of the nuances of vitrification technology, and thecrucial need in vitrification for close adherence to protocol per cell and tissue type. We areproud to have been able to have worked with so many of the pivotal researchers in this area ofcryobiology, and trust our collaboration here might prove as useful to others interested invitrification and assisted reproduction as it has done for us.
Michael J Tucker Juergen Liebermann
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xiv

Acknowledgments
The editors Michael J Tucker and Juergen Liebermann would like to thank the following indi-viduals for their efforts to bring this publication to fruition: Nick Dunton (formerly of InformaHealthcare) who had the vision to propose and initiate this project; also Lindsay CampbellRobert Peden, and Alexa Chamay Berrier who as current editors at Informa Healthcare, havebeen extremely helpful in keeping this publication on track throughout the vagaries of theprocess to provide a well-focused and timely book. All contributing authors are thanked for theircommitment to providing current and comprehensive chapters in this increasingly recognizedarea of cryobiology. Their enthusiasm and professionalism in their work is evident in their writ-ings, and we thank them enormously for allowing us to act as coordinating editors to put thisbook together on a subject that we think is both interesting and important. We hope that thisbook is enjoyed by, and of benefit to, all who read it. We are also grateful to all the IVF labora-tory staff at the Fertility Centers of Illinois, River North: Jill Matthews, Elissa Pelts, MandyErman, Becky Brohammer, Sara Sanchez, Yuri Wagner, and Andrew Barker whose clinical skillsfacilitated clinical application of routine vitrification within that laboratory – to all we say a veryspecial thank you. We also extend thanks to all the beautiful people out there who have ever beennice to us.
Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xv

Prelims Tucker 8029.qxd 8/23/2007 8:44 PM Page xvi

1
Vitrification: an overviewGregory M Fahy and William F Rall
1A
INTRODUCTION
Vitrification is an increasingly popularmethod of cryopreservation (Table 1A.1;Figure 1A.1). In the realm of reproduction,vitrification is currently used for the routinecryopreservation of human4,5 and animal5
embryos, and has been used successfully forthe cryopreservation of ovarian tissue,6 ova,7–9
and possibly even whole ovaries.10 In additionto solving an increasing variety of basic andapplied biological problems, vitrification hasattained much of its appeal by making cryo-preservation both simpler and more conve-nient than conventional freezing methods formany living systems.
Our present overview is not intended torecapitulate the scope of specific methods of
vitrification or the range of present applica-tions of vitrification. Instead, our goal is toprovide a broader background context for thechapters that follow by briefly considering theplace of vitrification within the natural world,the historical origins of the modern conceptof vitrification as a means of cryopreservation,and the fundamental theoretical basis of allmethods of natural and applied cryopreserva-tion by vitrification.
VITRIFICATION IN NATURE
The lowest terrestrial temperature everrecorded was apparently −89.3°C, at theRussian Vostok station in Antarctica.11 This iswell above the glass transition temperature
The information, opinions, data, and statements contained herein are not necessarily those of theUS Government or the National Institutes of Health (NIH) and should not be interpreted, acted onor represented as such.
Table 1A.1 Some successes in cell, tissue, organ, and organism cryopreservation by vitrification
Cells Tissues Organs and organisms
Ova (cow, human, mouse) Embryos (buffalo, cow, fly, human, Ovaries? (mouse)Oocyte cytoplasts llama, mouse, etc.) Embryonic hearts (chicken)Embryonic stem cells (human) Ovarian tissue (various) Embryonic brains (chicken)Spermatozoa? (no cryoprotectant) Embryonic kidney tissue? Veins (jugular, rabbit)Hematopoietic progenitors Corneas (rabbit, human) Arteries (rabbit)Hepatocytes (rat, microencapsulated) Heart valves (human) Kidney (rabbit)Islet substitute cells Organ slices (hippocampus, liver, kidney) Skin? (human)Monocytes (human) Islets, pancreatic (man, monkey, mouse) SchistosomesOsteoblasts Peripheral nerves TetrahymenaPlant cells (many varieties) Cartilage (rabbit, pig) Red blood cells (human) Plant tissues (many types)
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 1
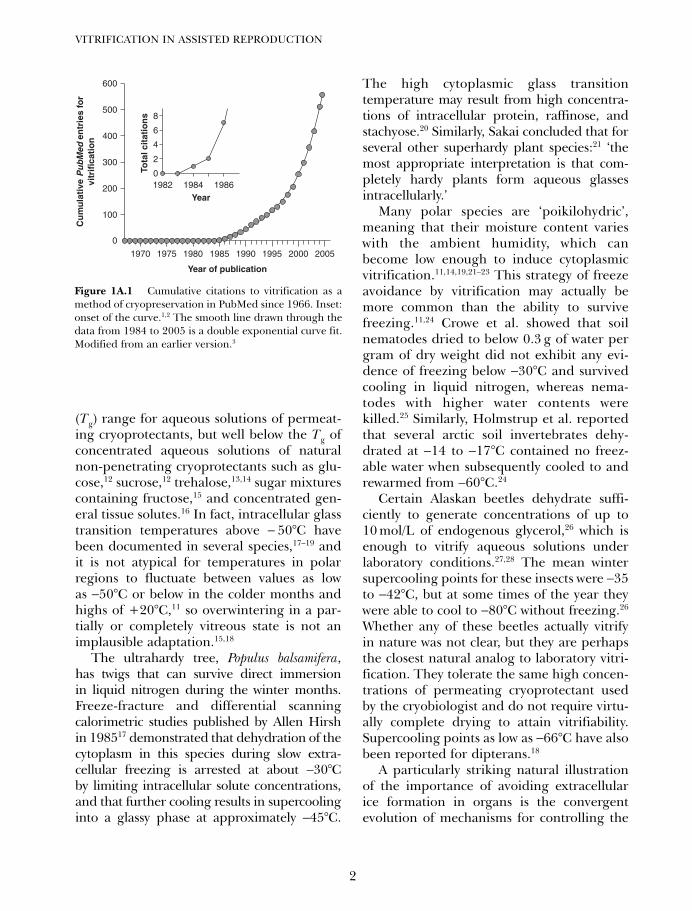
(Tg) range for aqueous solutions of permeat-ing cryoprotectants, but well below the Tg ofconcentrated aqueous solutions of naturalnon-penetrating cryoprotectants such as glu-cose,12 sucrose,12 trehalose,13,14 sugar mixturescontaining fructose,15 and concentrated gen-eral tissue solutes.16 In fact, intracellular glasstransition temperatures above − 50°C havebeen documented in several species,17–19 andit is not atypical for temperatures in polarregions to fluctuate between values as lowas −50°C or below in the colder months andhighs of +20°C,11 so overwintering in a par-tially or completely vitreous state is not animplausible adaptation.15,18
The ultrahardy tree, Populus balsamifera,has twigs that can survive direct immersionin liquid nitrogen during the winter months.Freeze-fracture and differential scanningcalorimetric studies published by Allen Hirshin 198517 demonstrated that dehydration of thecytoplasm in this species during slow extra-cellular freezing is arrested at about −30°Cby limiting intracellular solute concentrations,and that further cooling results in supercoolinginto a glassy phase at approximately −45°C.
The high cytoplasmic glass transitiontemperature may result from high concentra-tions of intracellular protein, raffinose, andstachyose.20 Similarly, Sakai concluded that forseveral other superhardy plant species:21 ‘themost appropriate interpretation is that com-pletely hardy plants form aqueous glassesintracellularly.’
Many polar species are ‘poikilohydric’,meaning that their moisture content varieswith the ambient humidity, which canbecome low enough to induce cytoplasmicvitrification.11,14,19,21–23 This strategy of freezeavoidance by vitrification may actually bemore common than the ability to survivefreezing.11,24 Crowe et al. showed that soilnematodes dried to below 0.3 g of water pergram of dry weight did not exhibit any evi-dence of freezing below −30°C and survivedcooling in liquid nitrogen, whereas nema-todes with higher water contents werekilled.25 Similarly, Holmstrup et al. reportedthat several arctic soil invertebrates dehy-drated at −14 to −17°C contained no freez-able water when subsequently cooled to andrewarmed from −60°C.24
Certain Alaskan beetles dehydrate suffi-ciently to generate concentrations of up to10 mol/L of endogenous glycerol,26 which isenough to vitrify aqueous solutions underlaboratory conditions.27,28 The mean wintersupercooling points for these insects were −35to −42°C, but at some times of the year theywere able to cool to −80°C without freezing.26
Whether any of these beetles actually vitrifyin nature was not clear, but they are perhapsthe closest natural analog to laboratory vitri-fication. They tolerate the same high concen-trations of permeating cryoprotectant usedby the cryobiologist and do not require virtu-ally complete drying to attain vitrifiability.Supercooling points as low as −66°C have alsobeen reported for dipterans.18
A particularly striking natural illustrationof the importance of avoiding extracellularice formation in organs is the convergentevolution of mechanisms for controlling the
VITRIFICATION IN ASSISTED REPRODUCTION
2
600
5008
6
4
2
01982 1984
Year
Tota
l cit
atio
ns
1986
400
300
200
100
0
1970 1975 1980
Year of publication
Cu
mu
lati
ve P
ub
Med
en
trie
s fo
rvi
trif
icat
ion
1985 1990 1995 2000 2005
Figure 1A.1 Cumulative citations to vitrification as amethod of cryopreservation in PubMed since 1966. Inset:onset of the curve.1,2 The smooth line drawn through thedata from 1984 to 2005 is a double exponential curve fit.Modified from an earlier version.3
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 2

location of ice in frogs and the silver fir, Abiessachalinensis. In the terrestrial frog, ice formsfirst in strategic locations within the relativelyhardy peripheral tissues, such as between theskin and the muscle and in the abdominalcavity.29 This ensures long freezing plateausand sufficient time for water to distill overmacroscopic distances from the sensitiveabdominal, thoracic, and intracranial organsonto ice crystals present external to theseorgans. The liver, heart, brain, intestines, andkidneys visibly shrink as adjacent ice massesexpand, thus allowing them to behave likesingle cells shrinking in response to extra-cellular ice formation rather than sufferinginjury from extensive invasion by ice.29 In thefir tree, the apical meristem survives by essen-tially the same mechanism. Ice is nucleated ina safe place adjacent to the apical meristem,and this results in distillation of meristemwater through a special barrier throughwhich water can diffuse but ice cannot grow inthe opposite direction to invade the meristem(Sakai, personal communication). Otherplants appear to have evolved essentially thesame mechanism.30
Several other very different and fascinat-ing freeze avoidance strategies have alsoevolved in both plants and animals. For ourpresent purposes, we simply note the ‘takehome’ message of these many diverse andelaborate examples, which is that ice avoid-ance is a winning natural strategy for thecryopreservation of complex living systems.Therefore, the experience of nature providesreassuring support for the pursuit of vitrifica-tion in the laboratory.
THE HISTORICAL DEVELOPMENTOF VITRIFICATION
Stiles
Although the possibility of vitrified water wasproposed by Brayley as long ago as 1860,31
the earliest suggestion we are aware of thatvitrification might be an appropriate strategy
for cryopreservation came from Walter Stilesin 1930.32 Although his suggestion could havebeen more clearly stated, he nevertheless def-initely proposed that biological vitrificationmight be both possible and desirable: ‘Ingeneral . . . protoplasm . . . is similar to . . .non-living colloidal systems . . . if a hydrosolor hydrogel is frozen very rapidly . . . thewhole will set into a finely crystalline or evenamorphous mass . . . Such a solid in thawing,might be expected to give again the originalsystem without change.’
Luyet and colleagues
Father Basile J Luyet independently envi-sioned and is widely acknowledged as beingthe first to take to heart the idea of vitrifica-tion of living cells.33 Luyet and his associatesexpended a vast amount of effort in the directpursuit of vitrification over the period from1937 to 195834 and much subsequent indirectwork that would later prove to be moreimportant for vitrification than his originaldirect efforts.34 Luyet’s fascination with thenature of life35 led him to seek clues about theliving state by attempting to arrest life andthen restart it, and vitrification seemed to bea promising means to this end.
Both Stiles and Luyet were inspired36 byTammann’s theory37 that it should be possibleto vitrify any liquid through the use of veryhigh cooling rates. As Luyet stated in his firstpaper on vitrification in 1937:33 ‘There are 2intrinsic factors which control the productionof the vitreous state, they are the velocityof crystallization and the size of the zone ofcrystallization temperatures. A third factoris extrinsic and depends on the methodemployed, it is the cooling velocity. Theessential problem of the vitrification tech-nique consists of . . . obtaining a coolingvelocity sufficient to prevent the formation ofcrystals.’
The freezing of water leads to the evolutionof a great deal of heat that slows the coolingof the remaining unfrozen water, and the
3
VITRIFICATION: AN OVERVIEW
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 3

attainable cooling rate depends also on thevolume of the material to be vitrified.Accordingly, Luyet and his colleagues oftendried or dehydrated their biological samplesprior to rapid cooling, sometimes using briefexposure to solutes such as ethylene glycol orglycerol to withdraw water just before cool-ing.34,36 This approach is very different fromthe modern method of deliberately allowingintracellular uptake of high concentrationsof cryoprotectant to slow the cooling ratesneeded for vitrification, but was a reasonableapproach to vitrification at the time.
Despite some triumphs,38,39 Luyet’s claimsof successful vitrification were directly andpowerfully challenged by Smith in 1954.36
This challenge probably prompted two spe-cific experiments that, as reported in 1958,directly contradicted Luyet’s conclusion thathis methods had generally achieved vitrifica-tion. The first experiment was to microscopi-cally examine putatively vitreous films ofgelatin gels using crossed polarizing filters.This examination showed the macroscopi-cally transparent films to be full of ice crystalsin the form of ‘evanescent spherulites’.40
The second experiment, by Meryman, was toexamine the same type of film using the par-allel technique of X-ray crystallography. Thisstudy showed an X-ray diffraction patterncharacteristic of the presence of ice, althoughonly one peak was observed instead of theexpected three.41 This suggested to Merymanthe presence of incompletely formed icecrystals (reflecting growth primarily in thedirection of the 001, or basal, plane of ice).However, Dowell et al.42 later disagreed, con-cluding, in reference to Meryman’s results: ‘itseems quite evident in the light of our studiesthat this was really . . . cubic ice and that ascan in the angular region 40–48° (2 θ) wouldhave shown the other two cubic lines.’
After 1962, all deliberate attempts byLuyet and his colleagues to vitrify livingsystems ceased,34 and all of their subsequentcryopreservation methods were referred toas freezing methods. Ironically, Rapatz and
Luyet successfully vitrified human red bloodcells almost by accident in 1968 in the courseof examining the relationship between iceformation and hemolysis,43 but seemed notto recognize the significance of this accom-plishment. In 1969, Luyet characterized hisattempts to achieve vitrification in retrospectas ‘mostly negative’.28 A more detailed reviewof Luyet’s views and experiments hasappeared elsewhere.34
The electron microscopists
Tokio Nei44 essentially reproduced the 1968Rapatz and Luyet result for vitrified redblood cells in 1976, for very similar reasons.He showed no ice inside or outside humanred cells vitrified in 4.1 mol/L (30%) glyceroland cooled at 105°C/min and obtained over95% survival of the cells after rewarming.However, Nei never referred to the concept ofvitrification or its desirability for cryopreser-vation, saying only that ‘as a cryotechniquefor electron microscopy, the addition of 30%glycerol and ultrarapid freezing at 105°C/minare minimum requirements for the inhibitionof ice formation and the prevention of thecorresponding artifacts in erythrocytes.’
Nei’s goal was to achieve cryofixation with-out ice artifacts, which was a serious objectivefor electron microscopists at the time.However, the real goal of the field was toaccomplish this without the use of cryoprotec-tants. Franks and Skaer took a major stepaway from the use of permeating cryoprotec-tants for morphological vitrification in thesame year, claiming vitrification or quasivitri-fication of a cell in the center of a 1 mm thickcockroach brain by infiltrating the specimenwith 50% w/w polyvinylpyrrolidone (PVP) andquenching in melting freon 22.45
In 1980, Bruggeller and Mayer publishedthe first reproducible demonstration of thevitrification of pure liquid water and a0.1 mol/L CuCl2 solution.46 They achieved anestimated cooling rate of 105–106°C/s. Oneyear later, Dubochet and McDowall claimed
VITRIFICATION IN ASSISTED REPRODUCTION
4
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 4

vitrification of layers up to 1 micron thick insmall water droplets based on electronmicroscopy.47 These methods, and others thatadded brief exposure to very high hydrostaticpressures, produced impressive results,achieving fields of view that were devoid ofdiscernible ice crystals even in the electronmicroscope. However, there was no way toconnect the kind of vitrification sought byelectron microscopists to the problem of pre-serving and recovering the viability of livingsystems.
Boutron
A major development in the history of vitrifi-cation began in the late 1970s, when PierreBoutron decided to give up his work on mag-netism and devote his considerable skills as aphysical scientist to the problems of cryobiol-ogy. His first paper in 197848 explained hisaims as follows: ‘It should be interesting tosee if the more stable the amorphous state ofa cryoprotective solution is, the better will beits protection of cells against freezing . . . Inthe extreme case of a solution which remainsentirely amorphous even at very slow coolingor warming rates, all cells should be protected. . . It should then be interesting to find cryo-protective solutions of very low toxicity . . . or,in other words, to find a very stable amor-phous state of the whole solution even fordiluted solutions.’ As this quote illustrates,Boutron clearly understood the ability ofcryoprotectants to enable vitrification and todo so even at low cooling rates, but there werethree significant problems with vitrification asBoutron pursued it.
First, it was assumed that low concentra-tions were necessary to avoid toxicity, whichharkens back to the approaches of Luyet andthe electron microscopists. Second, it wasabundantly clear from Boutron’s path-break-ing studies of the kinetics of glass formationon cooling and devitrification on warming48
that even at concentrations well above thosenormally used in cryobiology at the time, very
high cooling rates were generally required forcomplete vitrification and, even more prob-lematically, astronomical warming rates wererequired in theory to prevent devitrification.The combination of these two factors seemedto confirm Luyet’s conclusion that for allpractical purposes, complete vitrification wasuntenable, and relegated the practical signif-icance of Boutron’s initial work to theimprovement of the outcomes of freezing andthawing as he himself said was his objec-tive.48,49 Third, Boutron’s experiments onsolutions that were actually concentratedenough to demonstrably vitrify and remainamorphous on warming merely confirmedthat enormous concentrations were neededthat were, in his words, ‘toxic for most cells’.49
A key discovery of Boutron was the relativelymiraculous behavior of propylene glycol (PG,or 1,2-propanediol), which vitrifies at concen-trations as low as ~30–40% w/w, depending onthe cooling rate.50 The critical warming ratesto prevent devitrification of 35 and 40% w/wsolutions of PG were calculated to be nearly109 and 105°C/min, respectively. Nevertheless,in 1984, Boutron and Arnaud were able toshow that at cooling rates sufficient to vitrify30% and 35% PG, high levels of survival ofhuman erythrocytes were obtainable despitethe high critical warming rates for theseconcentrations and the existence of homoge-neous nucleation on cooling.51 Survival wasexplained on the basis of initial formation ofcubic ice and the inability of cubic ice to dam-age cells until it is converted to hexagonal ice,which was not expected to happen at the5000°C/min warming rates employed.51
Although these experiments recapitulatedthe prior studies of Rapatz and Luyet in 1968and of Nei in 1976, they took place in a muchmore useful conceptual context in which cryo-preservation in the amorphous state was clearlyrecognized as being valuable in its own right,and not just valuable as a means to some otherend. However, there was still no demonstrationthat nucleated cells could be vitrified andrewarmed successfully.
5
VITRIFICATION: AN OVERVIEW
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 5

Rall
Peter Mazur’s classic 1963 paper showing thatthe survival of slowly frozen cells is attribut-able to the avoidance of intracellular iceformation52 assumed that the remainingunfrozen intracellular water at very low tem-peratures is essentially unfreezable ‘boundwater’. The concept that significant freezablewater could remain at the end of slow coolingbut fail to freeze upon subsequent transferinto liquid nitrogen due to intracellular vitri-fication simply had not ‘crystallized’ at thetime. Vitrification had always been associatedwith rapid cooling, not slow cooling, and, 6years after Mazur’s seminal paper, Luyet28
noted that ‘the reaction of most cryobiolo-gists, when the notion of amorphous or glassystate is mentioned to them, is that it may be asubject of academic interest but not one ofpractical significance, since to obtain theamorphous state one needs to use extremelyhigh cooling rates difficult to attain.’ EvenBoutron did not explicitly state that slowlyfrozen cells survive by virtue of intracellularvitrification.
In 1980, Rall and colleagues53 reportedtheir cryomicroscopic observations of mouseembryos during slow freezing, subsequentrapid cooling, and rewarming. They described,for the first time, intracellular devitrificationupon rewarming, which clearly implied previ-ous intracellular vitrification.54,55 Rall, Reid,and Polge56 later used differential scanningcalorimetry to demonstrate that the extracellu-lar solution in contact with the embryos at thetime they were rapidly cooled does in fact vit-rify upon rapid cooling and devitrify upon slowwarming, as predicted from their cryomicro-scopic observations. They thereby convincinglysupported their view that slow freezing fol-lowed by rapid cooling causes the cytoplasm tovitrify, and generalized their observation byconcluding: ‘other cryopreservation methodsthat employ a protective additive and rapidcooling from an intermediate subzero temper-ature may rely on the ability of the residual
liquid to form a metastable glass during rapidcooling’.56
Although this insight did not suggest analternative to freezing as a means of cryo-preservation, the realization that embryossurvive cryopreservation in the final analysisas a result of at least intracellular vitrification,even when the cells are said to be ‘frozen’ dueto the presence of extracellular ice, was to bea key impetus toward the first truly influentialdemonstration of the complete vitrification ofliving cells.
Fahy
In 1965, John Farrant57 showed that wholeguinea pig uteri could fully recover after pre-vious cooling without freezing to −79°C in thepresence of 55% dimethyl-sulfoxide (DMSO).Elford and Walter,58 in 1972, successfullyextended Farrant’s observations to intestinalsmooth muscle. Inspired by these successes,Fahy began attempts to apply the same prin-ciples to other systems in 1972 as an under-graduate student59 and continued this effortin graduate school. However, his graduatestudies showed that even 50% DMSO wasoverwhelmingly toxic to kidney tissue.60
Discouraged by this result, Fahy returnedto freezing as a way of preserving kidneys inthe late 1970s, but soon found that frozenkidneys appeared to be severely damagedstructurally.61,62 Searching for an alternativeto both Farrant’s equilibrium freezing pointdepression method and freezing, he turnedto deep supercooling as a way of avoidingboth ice formation and Farrent’s high con-centrations of cryoprotectant, hoping to beable to preserve kidneys for a week at −80°C.He was disappointed to find that supercooledcryoprotectant systems stored at −80°C weretoo prone to freeze after one or more days,61
but in contemplating this situation, a keyinspiration dawned: it might be possible toextend supercooling all the way down to theglass transition temperature, enabling the
VITRIFICATION IN ASSISTED REPRODUCTION
6
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 6

same concentrations attempted for preservationby supercooling to allow vitrification andthereby stability against ice formation forindefinite periods of time. Farrant did notconnect his method to vitrification, statingthat cooling to lower temperatures wouldresult in freezing,57 but, as a graduate student,Fahy observed that such high concentrationsdo vitrify upon such cooling.
Fahy realized that in order to pursue hisnew vision of the possibility of preservingwhole organs indefinitely by vitrification hewould have to face the problem of the toxic-ity of vitrifiable concentrations of cryoprotec-tant head on.27,62–66
To control cryoprotectant toxicity, Fahycombined several different leads and newideas, including the use of ‘toxicity neutraliz-ers’;63,67b Boutron’s 1,2-propanediol;50,61,63 theuse of elevated hydrostatic pressure;27,61,63
non-penetrating cryoprotectants to reduceintracellular permeating cryoprotectantrequirements;1 mild osmotic dehydration tofacilitate intracellular vitrification, reducetoxicity, and facilitate cryoprotectantwashout;1 exponential addition and washoutof cryoprotectants;1,66 introduction of thehighest cryoprotectant concentrations atreduced temperatures;1,65 and determinationof the minimum amount of cryoprotectantneeded to vitrify at the cooling rates applica-ble to organs from both an empirical and atheoretical point of view, and restriction ofthe concentrations of cryoprotectant used tothose minimum levels.61,63–64 By 1984 Fahyand his colleagues were able to publish thefirst full and generally available descriptionof his new approach to vitrification, and todescribe a solution that was both capable ofpermitting vitrification at 10°C/min andcompatible with a 90% functional recovery ofrenal tissue.1 This paper also demonstratedthe avoidance of fracturing in a whole vitri-fied rabbit kidney. The stage was thereby setfor the first proof of principle of the newmethod and a demonstration that the resultsobtained using the rabbit kidney slice model
were not so idiosyncratic as to be inapplicableto other important biological systems.
Rall and Fahy
In 1985, we were fortunate enough to be ableto close the gap between theory and practice,and confirm the universality of the fundamen-tal principles of vitrification by successfullyapplying lessons learned from adult rabbit kid-neys and the freezing of mouse embryos to thevitrification of 8-cell mouse embryos.2 Mouseembryos vitrified in plastic straws using a widerange of cooling rates and rewarmed over awide range of warming rates survived in highproportions provided that warming was fastenough to prevent devitrification. Our papercoined the term ‘vitrification solution’ anddemonstrated that the first vitrification solu-tion attempted, VS1, while toxic to embryos ata concentration permitting vitrification at acooling rate of only 10°C/min and after longerexposure periods, permitted successful vitrifi-cation without appreciable toxicity whenslightly diluted.
Success was attained using a protocol foraddition and washout of VS1 that facilitateddiffusion and cellular uptake of lower concen-trations of the permeating cryoprotectants ofVS1 at room temperature and inhibited toxi-city and facilitated embryo dehydration bythe highest concentrations near 0°C. Survivalwas judged initially based on in vitro develop-ment to expanded blastocysts,2 but Rallet al. soon demonstrated that these embryoswere capable of developing to live young fol-lowing transfer to recipient females.68
The survival rates reported were equiva-lent to those obtained using optimized slowfreezing methods, but the time required tocarry out the vitrification procedure wasmuch less than the time required for freezingand thawing, and no expensive controlled ratefreezing device was required. Vitrification alsoeliminated the usual need to search for theoptimum cooling and warming rates whenfreezing and thawing. All of these practical
7
VITRIFICATION: AN OVERVIEW
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 7

advantages of vitrification soon caught theattention of many embryology laboratories,which proceeded to investigate the techniquefor their own purposes and in their own ways(Figure 1A.1). As even less toxic vitrificationsolutions were developed for selected sys-tems,21,69 interest in vitrification continued toaccelerate.
The ghost of Luyet
Although the modern advent of vitrificationarose from the realization that ultrarapidcooling is not required for vitrification andthat solutions concentrated enough for vitri-fication at slow cooling rates need notinevitably be extremely toxic, once the feasi-bility of vitrification became widely appreci-ated there was no longer any need to limitvitrification to low cooling rates for thosesystems that could be cooled and warmedrapidly, and lingering problems with thetoxicity of many alternative vitrification solu-tions, and particularly with chilling injury insome systems,70–72 accordingly inspired moreand more efforts to reduce damage by usinglower concentrations and faster coolingrates.
There is insufficient space in this shortoverview to recount in any adequate detailthe many ingenious approaches that haveemerged, and the reader must be referred tolater chapters for further information withinthe sphere of reproductive cryobiology. Herewe can only note the irony that, having beenlaunched by breaking free of Luyet’s tyrannyof ultrarapid cooling, vitrification methodshave now essentially turned back closer toLuyet’s original idea of cooling as quicklyas possible with minimal intracellular expo-sure to cryoprotectants, albeit this time usingat least marginally adequate concentrationsof intracellular solutes. The ghost of Luyetlives on in the form of this ongoing method-ological evolution, and we think he wouldhave been pleased to see how his ideas about
vitrification ultimately related to the nowwidespread use of vitrification as a practicaland successful method of cryopreservationlong after he, himself, had abandoned thisapproach.
THE PHYSICAL BASIS OFVITRIFICATION
The physics of ice formation and of the glasstransition have been summarized in depthin several excellent and cryobiologically ori-ented reviews to which the interested readeris referred for a more complete treatmentthan can be presented here.73,74 Here we focuson the essential issues of why the glass transi-tion takes place, how it stabilizes livingsystems, and what the limiting conditionsare for vitrification and survival.
The thermodynamic necessityof vitrification
Vitrification is ultimately the result of the factthat a liquid cannot have more order than itscorresponding crystal. As the temperature ofa liquid substance is lowered, its entropy isreduced more rapidly than the entropy of thecorresponding crystalline form of the sub-stance. If the liquid does not freeze, a thermo-dynamic conundrum is approached as theentropy of the liquid approaches the entropyof the crystal. Kauzmann75 recognized that tomaintain thermodynamic consistency, someevent must prevent the continued reductionof entropy of the liquid from creating a liquidwhose entropy is less than the entropy of thecrystal (a situation referred to as Kauzmann’sparadox). That event is the glass transition,and it must occur at a temperature above theKauzmann temperature, TK, at which the lawsof thermodynamics would be violated by theattainment of a liquid state with the sameentropy as the crystalline state of the samematerial. The glass transition prevents theparadox by eliminating the translational and
VITRIFICATION IN ASSISTED REPRODUCTION
8
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 8
rotational degrees of freedom that characterizethe liquid state and are responsible for thegreater dependence of the entropy of a liquidon temperature in comparison to that of thecrystal, which retains primarily vibrationaldegrees of freedom. Another way to view theglass transition is that the molecular mobilityof a liquid has an activation energy, and as theglass transition temperature is approached,that activation energy becomes unavailable.
The kinetic basis of vitrification
As the activation energy for translationalmolecular motions vanishes with continuedcooling, the time scale for structural reorgani-zations within the liquid stretches out towardinfinity. The temperature at which structuralrelaxation becomes impossible in principle, T0,can be empirically inferred, for example, from
curve fits of the Vogel–Tammann–Fulcher(VTF) equation:
η(T2) = η(T1)exp[B/(T2− T0)]
where η(T1) is a known reference viscosity attemperature T1, η(T2) is the viscosity at temper-ature T2, and T0 is the limiting temperature forstructural change, which approximates TK.
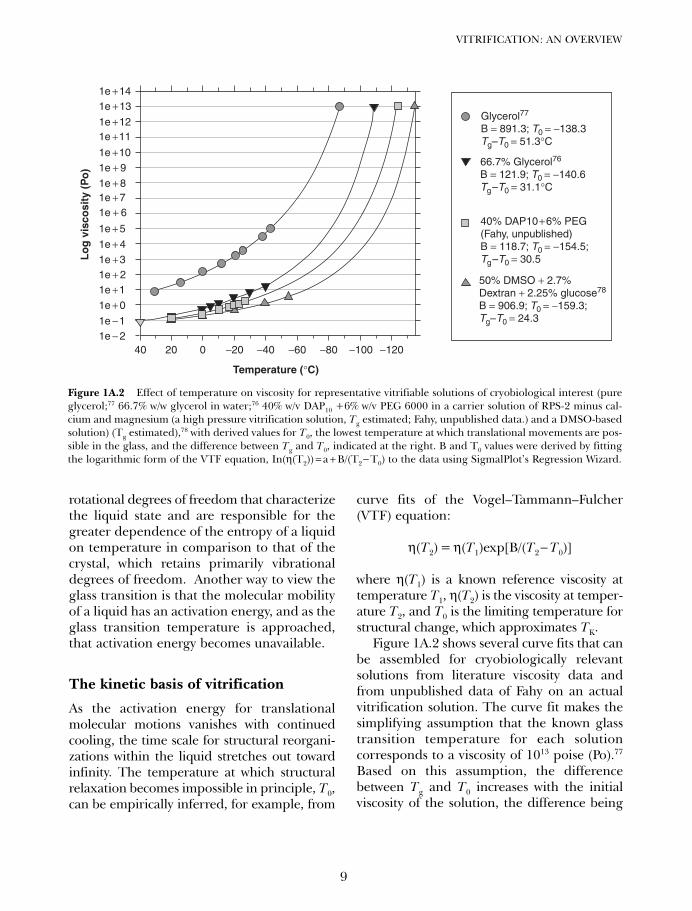
Figure 1A.2 shows several curve fits that canbe assembled for cryobiologically relevantsolutions from literature viscosity data andfrom unpublished data of Fahy on an actualvitrification solution. The curve fit makes thesimplifying assumption that the known glasstransition temperature for each solutioncorresponds to a viscosity of 1013 poise (Po).77
Based on this assumption, the differencebetween Tg and T0 increases with the initialviscosity of the solution, the difference being
9
VITRIFICATION: AN OVERVIEW
Glycerol77
B = 891.3; T0 = −138.3Tg–T0 = 51.3°C
66.7% Glycerol76
B = 121.9; T0 = −140.6Tg–T0 = 31.1°C
40% DAP10 +
6% PEG
(Fahy, unpublished)B = 118.7; T0 = −154.5;Tg–T0 = 30.5
50% DMSO + 2.7%Dextran + 2.25% glucose78
B = 906.9; T0 = −159.3;Tg–T0 = 24.3
Temperature (°C)
Lo
g v
isco
sity
(P
o)
1e +141e +13
1e +121e +11
1e +101e + 9
1e + 81e +71e + 6
1e + 5
1e + 41e +31e + 21e +1
1e + 0
1e −11e − 2
−120−100−80−60−40−2002040
Figure 1A.2 Effect of temperature on viscosity for representative vitrifiable solutions of cryobiological interest (pureglycerol;77 66.7% w/w glycerol in water;76 40% w/v DAP10 +6% w/v PEG 6000 in a carrier solution of RPS-2 minus cal-cium and magnesium (a high pressure vitrification solution, Tg estimated; Fahy, unpublished data.) and a DMSO-basedsolution) (Tg estimated),78 with derived values for T0, the lowest temperature at which translational movements are pos-sible in the glass, and the difference between Tg and T0, indicated at the right. B and T0 values were derived by fittingthe logarithmic form of the VTF equation, In(η(T2)) = a + B/(T2− T0) to the data using SigmalPlot’s Regression Wizard.
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 9

51°C for pure glycerol but only 24°C for acomparatively fluid dimethyl sulfoxide-basedsolution. In the parlance of glass physicists,cryoprotectant-water solutions form ‘fragile’glasses (glasses with steep slopes in suchplots), and Tg appears to come closer andcloser to T0 as the fragility of the glassincreases. Stated another way, the higher theglass transition temperature, the fartherbelow Tg one needs to go to reach T0. In stillother words, Tg as measured in the laboratoryis an artifact of the time scale of typical labo-ratory measurements. If it were possible tocool test solutions more and more slowly with-out limit, Tg would be seen at lower and lowertemperatures until, in principle, Tg wouldapproach TK as closely as possible as the cool-ing rate approached zero.
Optimal storage below Tg
From the point of view of cryopreservation,storage below TK (or T0) should be unneces-sary. Further, storage in liquid nitrogen maybe undesirable if sample contamination orsample fracturing is an issue,56,79 as mightoccur for vitrified ovaries intended for vascu-lar transplantation. Given that in many situa-tions T0 and TK will be unknown and that inmany cases the risk of fracturing will increaseas these critical temperatures are approached,we would like to have a clearer general idea ofhow the safe period of storage depends ontemperature below Tg, particularly given thepotential need for very long-term storage ofprecious genetic resources.
On the assumption that biological changeis generally diffusion limited, we can get auseful feeling for the time dependence of dif-fusion-mediated changes from the followingsimplified treatment. Although the kinetics ofrelaxational processes are nominally slowerbelow Tg than the simpler VTF equationwould indicate, we will see that the differencemay well be academic.
The time t required for a substance todiffuse a distance d is equivalent to d2/6D,
where D is the diffusion coefficient of thesubstance. At temperatures above Tg, theStokes–Einstein equation states thatD = kT/(6πη(T)), where η(T) is the viscosity attemperature T. Combining the equations fordiffusion, the temperature dependence ofthe diffusion coefficient, and the VTF equation,one obtains
t = t1(T1/T)exp[B[(1/(T−T0)) – 1/(T1− T0)]]
where t is the time required to diffuse dis-tance d at temperature T and t1 is the timerequired to diffuse the same distance atreference temperature T1.
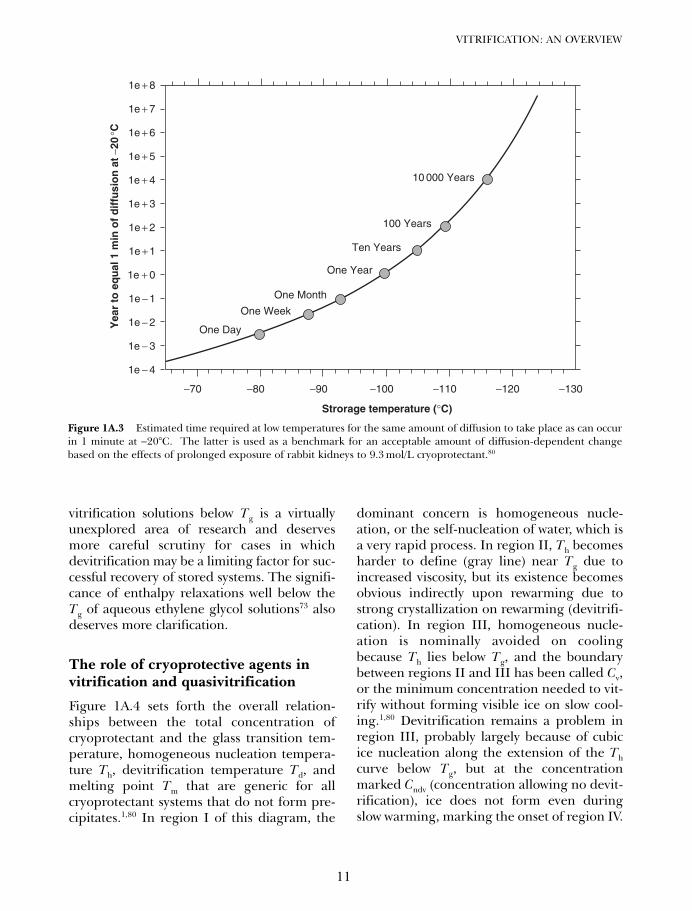
If diffusion times are related to biologicalchanges in or near the vitreous state, and ifwe take the injury onset time for the on set ofinjury as being 1 minute at −20°C (basedarbitrarily on the observation80 that perfus-ing a rabbit kidney with the M22 vitrificationsolution at −22°C requires an equilibrationtime that approaches the time associatedwith significant M22 toxicity), then we canderive the relationship shown in Figure 1A.3between storage temperature and the timerequired for storage injury to accumulate,using the curve fit parameters for thevitrification solution described in Figure1A.2. According to Figure 1A.3, storagetimes already begin to reach extreme valueseven at temperatures above Tg, which indi-cates that normal diffusional sources ofinjury are not likely to be important forsystems stored comfortably below Tg regard-less of the approximations underlying theprojections in Figure 1A.3.
On the other hand, ice nucleation is knownto take place on short time scales between− 90°C and about − 135°C in the VS41A/VS55vitrification solution (Tg approximately− 125°C).81 Nucleation can take place over avery short distance scale and evidentlyrequires very little diffusion since the temper-ature optimum for nucleation is very muchbelow the temperature optimum for ice crystalgrowth.34,81 At the present time, the time andtemperature dependence of ice nucleation in
VITRIFICATION IN ASSISTED REPRODUCTION
10
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 10
vitrification solutions below Tg is a virtuallyunexplored area of research and deservesmore careful scrutiny for cases in whichdevitrification may be a limiting factor for suc-cessful recovery of stored systems. The signifi-cance of enthalpy relaxations well below theTg of aqueous ethylene glycol solutions73 alsodeserves more clarification.
The role of cryoprotective agents invitrification and quasivitrification
Figure 1A.4 sets forth the overall relation-ships between the total concentration ofcryoprotectant and the glass transition tem-perature, homogeneous nucleation tempera-ture Th, devitrification temperature Td, andmelting point Tm that are generic for allcryoprotectant systems that do not form pre-cipitates.1,80 In region I of this diagram, the
dominant concern is homogeneous nucle-ation, or the self-nucleation of water, which isa very rapid process. In region II, Th becomesharder to define (gray line) near Tg due toincreased viscosity, but its existence becomesobvious indirectly upon rewarming due tostrong crystallization on rewarming (devitrifi-cation). In region III, homogeneous nucle-ation is nominally avoided on coolingbecause Th lies below Tg, and the boundarybetween regions II and III has been called Cv,or the minimum concentration needed to vit-rify without forming visible ice on slow cool-ing.1,80 Devitrification remains a problem inregion III, probably largely because of cubicice nucleation along the extension of the Thcurve below Tg, but at the concentrationmarked Cndv (concentration allowing no devit-rification), ice does not form even duringslow warming, marking the onset of region IV.
11
VITRIFICATION: AN OVERVIEW
10 000 Years
100 Years
Ten Years
One Year
One Month
One Week
One Day
Strorage temperature (°C)
Yea
r to
eq
ual
1 m
in o
f d
iffu
sio
n a
t −2
0 °C
1e + 8
1e + 7
1e + 6
1e + 5
1e + 4
1e + 3
1e + 2
1e + 1
1e + 0
1e − 1
1e − 2
1e − 3
1e − 4
−70 −80 −90 −100 −110 −120 −130
Figure 1A.3 Estimated time required at low temperatures for the same amount of diffusion to take place as can occurin 1 minute at −20°C. The latter is used as a benchmark for an acceptable amount of diffusion-dependent changebased on the effects of prolonged exposure of rabbit kidneys to 9.3 mol/L cryoprotectant.80
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 11
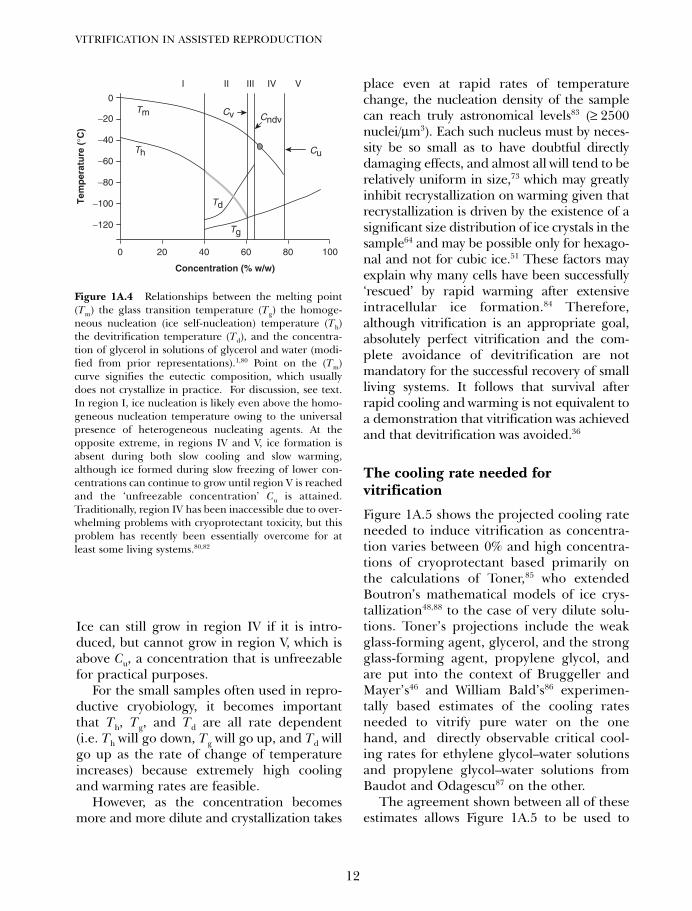
Ice can still grow in region IV if it is intro-duced, but cannot grow in region V, which isabove Cu, a concentration that is unfreezablefor practical purposes.
For the small samples often used in repro-ductive cryobiology, it becomes importantthat Th, Tg, and Td are all rate dependent(i.e. Th will go down, Tg will go up, and Td willgo up as the rate of change of temperatureincreases) because extremely high coolingand warming rates are feasible.
However, as the concentration becomesmore and more dilute and crystallization takes
place even at rapid rates of temperaturechange, the nucleation density of the samplecan reach truly astronomical levels83 (≥ 2500nuclei/µm3). Each such nucleus must by neces-sity be so small as to have doubtful directlydamaging effects, and almost all will tend to berelatively uniform in size,73 which may greatlyinhibit recrystallization on warming given thatrecrystallization is driven by the existence of asignificant size distribution of ice crystals in thesample64 and may be possible only for hexago-nal and not for cubic ice.51 These factors mayexplain why many cells have been successfully‘rescued’ by rapid warming after extensiveintracellular ice formation.84 Therefore,although vitrification is an appropriate goal,absolutely perfect vitrification and the com-plete avoidance of devitrification are notmandatory for the successful recovery of smallliving systems. It follows that survival afterrapid cooling and warming is not equivalent toa demonstration that vitrification was achievedand that devitrification was avoided.36
The cooling rate needed forvitrification
Figure 1A.5 shows the projected cooling rateneeded to induce vitrification as concentra-tion varies between 0% and high concentra-tions of cryoprotectant based primarily onthe calculations of Toner,85 who extendedBoutron’s mathematical models of ice crys-tallization48,88 to the case of very dilute solu-tions. Toner’s projections include the weakglass-forming agent, glycerol, and the strongglass-forming agent, propylene glycol, andare put into the context of Bruggeller andMayer’s46 and William Bald’s86 experimen-tally based estimates of the cooling ratesneeded to vitrify pure water on the onehand, and directly observable critical cool-ing rates for ethylene glycol–water solutionsand propylene glycol–water solutions fromBaudot and Odagescu87 on the other.
The agreement shown between all of theseestimates allows Figure 1A.5 to be used to
VITRIFICATION IN ASSISTED REPRODUCTION
12
0
Tem
per
atu
re (
°C)
−20
−40
−60
−80
−100
−120
0 20 40 60
Tm
I II III IV V
Th
Td
Cv Cndv
Cu
Tg
Concentration (% w/w)
80 100
Figure 1A.4 Relationships between the melting point(Tm) the glass transition temperature (Tg) the homoge-neous nucleation (ice self-nucleation) temperature (Th)the devitrification temperature (Td), and the concentra-tion of glycerol in solutions of glycerol and water (modi-fied from prior representations).1,80 Point on the (Tm)curve signifies the eutectic composition, which usuallydoes not crystallize in practice. For discussion, see text.In region I, ice nucleation is likely even above the homo-geneous nucleation temperature owing to the universalpresence of heterogeneous nucleating agents. At theopposite extreme, in regions IV and V, ice formation isabsent during both slow cooling and slow warming,although ice formed during slow freezing of lower con-centrations can continue to grow until region V is reachedand the ‘unfreezable concentration’ Cu is attained.Traditionally, region IV has been inaccessible due to over-whelming problems with cryoprotectant toxicity, but thisproblem has recently been essentially overcome for atleast some living systems.80,82
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 12
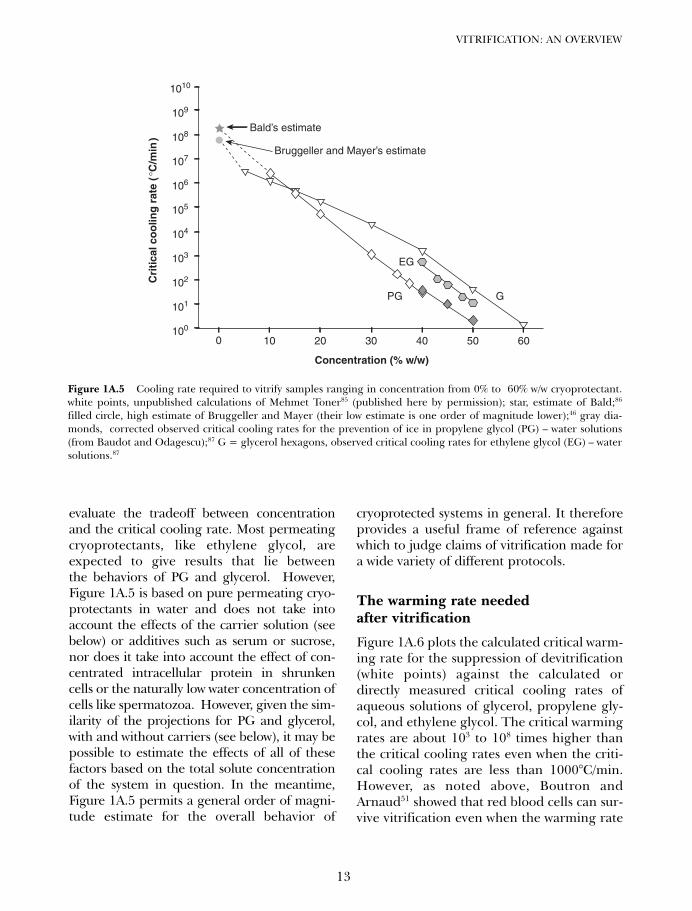
evaluate the tradeoff between concentrationand the critical cooling rate. Most permeatingcryoprotectants, like ethylene glycol, areexpected to give results that lie betweenthe behaviors of PG and glycerol. However,Figure 1A.5 is based on pure permeating cryo-protectants in water and does not take intoaccount the effects of the carrier solution (seebelow) or additives such as serum or sucrose,nor does it take into account the effect of con-centrated intracellular protein in shrunkencells or the naturally low water concentration ofcells like spermatozoa. However, given the sim-ilarity of the projections for PG and glycerol,with and without carriers (see below), it may bepossible to estimate the effects of all of thesefactors based on the total solute concentrationof the system in question. In the meantime,Figure 1A.5 permits a general order of magni-tude estimate for the overall behavior of
cryoprotected systems in general. It thereforeprovides a useful frame of reference againstwhich to judge claims of vitrification made fora wide variety of different protocols.
The warming rate neededafter vitrification
Figure 1A.6 plots the calculated critical warm-ing rate for the suppression of devitrification(white points) against the calculated ordirectly measured critical cooling rates ofaqueous solutions of glycerol, propylene gly-col, and ethylene glycol. The critical warmingrates are about 103 to 108 times higher thanthe critical cooling rates even when the criti-cal cooling rates are less than 1000°C/min.However, as noted above, Boutron andArnaud51 showed that red blood cells can sur-vive vitrification even when the warming rate
13
VITRIFICATION: AN OVERVIEW
Bald’s estimate
Cri
tica
l co
olin
g r
ate
( °C
/min
)Bruggeller and Mayer’s estimate
EG
Concentration (% w/w)
GPG
0100
101
102
103
104
105
106
107
108
109
1010
10 20 30 40 50 60
Figure 1A.5 Cooling rate required to vitrify samples ranging in concentration from 0% to 60% w/w cryoprotectant.white points, unpublished calculations of Mehmet Toner85 (published here by permission); star, estimate of Bald;86
filled circle, high estimate of Bruggeller and Mayer (their low estimate is one order of magnitude lower);46 gray dia-monds, corrected observed critical cooling rates for the prevention of ice in propylene glycol (PG) – water solutions(from Baudot and Odagescu);87 G = glycerol hexagons, observed critical cooling rates for ethylene glycol (EG) – watersolutions.87
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 13
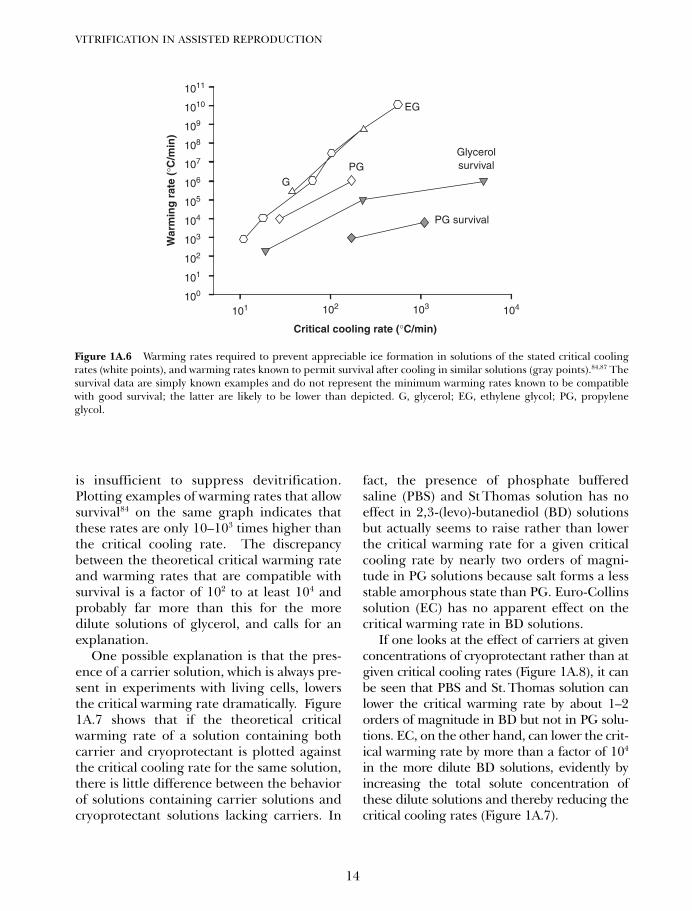
is insufficient to suppress devitrification.Plotting examples of warming rates that allowsurvival84 on the same graph indicates thatthese rates are only 10–103 times higher thanthe critical cooling rate. The discrepancybetween the theoretical critical warming rateand warming rates that are compatible withsurvival is a factor of 102 to at least 104 andprobably far more than this for the moredilute solutions of glycerol, and calls for anexplanation.
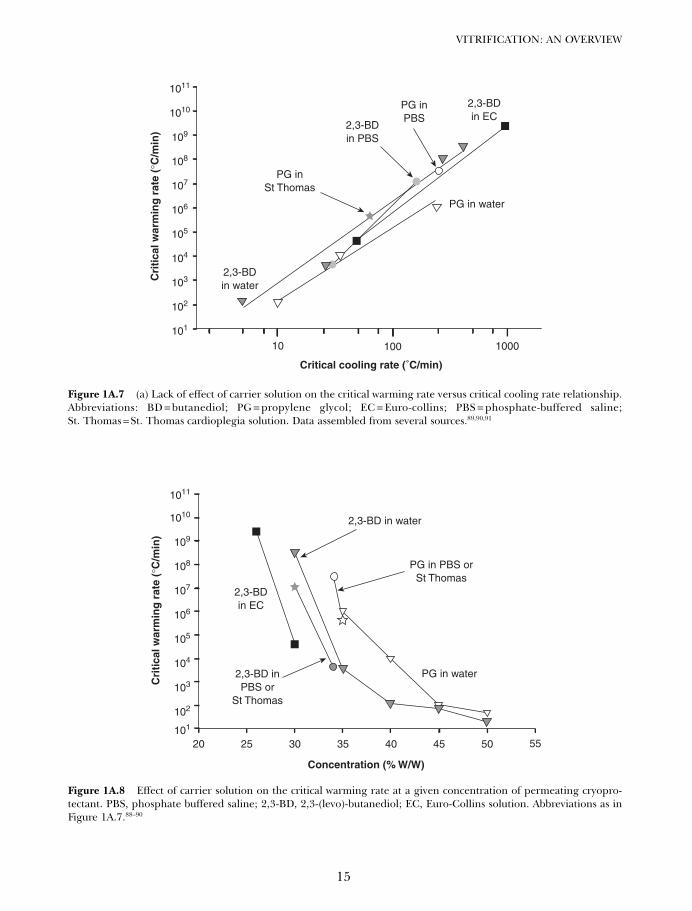
One possible explanation is that the pres-ence of a carrier solution, which is always pre-sent in experiments with living cells, lowersthe critical warming rate dramatically. Figure1A.7 shows that if the theoretical criticalwarming rate of a solution containing bothcarrier and cryoprotectant is plotted againstthe critical cooling rate for the same solution,there is little difference between the behaviorof solutions containing carrier solutions andcryoprotectant solutions lacking carriers. In
fact, the presence of phosphate bufferedsaline (PBS) and St Thomas solution has noeffect in 2,3-(levo)-butanediol (BD) solutionsbut actually seems to raise rather than lowerthe critical warming rate for a given criticalcooling rate by nearly two orders of magni-tude in PG solutions because salt forms a lessstable amorphous state than PG. Euro-Collinssolution (EC) has no apparent effect on thecritical warming rate in BD solutions.
If one looks at the effect of carriers at givenconcentrations of cryoprotectant rather than atgiven critical cooling rates (Figure 1A.8), it canbe seen that PBS and St. Thomas solution canlower the critical warming rate by about 1–2orders of magnitude in BD but not in PG solu-tions. EC, on the other hand, can lower the crit-ical warming rate by more than a factor of 104
in the more dilute BD solutions, evidently byincreasing the total solute concentration ofthese dilute solutions and thereby reducing thecritical cooling rates (Figure 1A.7).
VITRIFICATION IN ASSISTED REPRODUCTION
14
Critical cooling rate (°C/min)
War
min
g r
ate
(°C
/min
)
100
101
101
102
102 103
EG
PG
PG survival
Glycerolsurvival
G
104
103
104
105
106
107
108
109
1010
1011
Figure 1A.6 Warming rates required to prevent appreciable ice formation in solutions of the stated critical coolingrates (white points), and warming rates known to permit survival after cooling in similar solutions (gray points).84,87 Thesurvival data are simply known examples and do not represent the minimum warming rates known to be compatiblewith good survival; the latter are likely to be lower than depicted. G, glycerol; EG, ethylene glycol; PG, propyleneglycol.
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 14
15
VITRIFICATION: AN OVERVIEW
Cri
tica
l war
min
g r
ate
(°C
/min
)
Critical cooling rate (˚C/min)
PG inSt Thomas
2,3-BDin PBS
2,3-BDin water
PG in water
2,3-BDin EC
PG inPBS
101
10 100 1000
102
103
104
105
106
107
108
109
1010
1011
Figure 1A.7 (a) Lack of effect of carrier solution on the critical warming rate versus critical cooling rate relationship.Abbreviations: BD = butanediol; PG = propylene glycol; EC = Euro-collins; PBS = phosphate-buffered saline;St. Thomas = St. Thomas cardioplegia solution. Data assembled from several sources.89,90,91
Cri
tica
l war
min
g r
ate
(°C
/min
)
Concentration (% W/W)
2,3-BD in water
101
102
103
104
105
106
107
108
109
1010
1011
20 25 30 35 40 45 50 55
2,3-BDin EC
PG in water
PG in PBS orSt Thomas
2,3-BD inPBS or
St Thomas
Figure 1A.8 Effect of carrier solution on the critical warming rate at a given concentration of permeating cryopro-tectant. PBS, phosphate buffered saline; 2,3-BD, 2,3-(levo)-butanediol; EC, Euro-Collins solution. Abbreviations as inFigure 1A.7.88–90
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 15

The calculated critical warming rate is awarming rate sufficient to reduce ice formationto a very small volume fraction of the solu-tion. It is apparent that cells can at leastsometimes tolerate a much larger volumefraction of ice than the presumptive valueupon which the critical warming rate is calcu-lated.56,84 In the case of erythrocytes, the cyto-plasm is known to be more resistant to iceformation than the extracellular milieu,43,44
perhaps due to the high intracellular hemo-globin concentrations. On the other hand, inslowly frozen embryos, killing during slowwarming was associated with an invisibleevent that took place after devitrification butbefore recrystallization and ice melting,53 sug-gesting that at least in this case the traditionalexplanations for devitrification injury may beinvalid. Whether other explanations such asenrichment of cryoprotectant concentrationsto damaging levels92 could be involvedremains to be seen.
In summary, experimentally determiningand theoretically understanding the effect ofdevitrification on living cells and tissuesdeserves much more attention than it hasreceived. Excellent previous studies on thephysics of devitrification73,74,89 should facili-tate understanding of the biology of devitrifi-cation in future investigations.
BIOLOGICAL PHENOMENARELATED TO SUCCESSFULVITRIFICATION
The biological requirements for vitrificationare the subject of this book, and only afew general points can be touched on here. Westart by listing a few simple biological princi-ples that are easy to state and easy to accommo-date methodologically, but the implications ofwhich are not always kept firmly in mind. 1 Cells have limited tolerance to shrinkageand swelling.93 Therefore, due care mustbe taken not to exceed their osmotic limitsduring the introduction and removal ofcryoprotective agents, preferably using theguidance provided by appropriate mathe-
matical models of cellular dehydration andrehydration kinetics (although in practicesuch models are often dispensable for famil-iar systems). 2 Cell shrinkage before vitrification rendersthe cytoplasm more stable.1,94 Therefore, thefinal step of cryoprotectant exposure neednot be longer than the time required for thecells of interest to shrink osmotically, result-ing in reduced intracellular permeatingcryoprotectant to cause toxicity before vitrifi-cation and to cause osmotic damage duringcryoprotectant washout. 3 Cells are more permeable to cryoprotec-tants at higher temperatures, but more resistantto toxic effects at lower temperatures.2,94
Therefore, it is frequently advantageous tointroduce and remove lower, less toxic concen-trations at higher temperatures and to use lowertemperatures for the safe dehydration of thecell by higher concentrations of cryoprotectantsthe full permeation of which is less criticalfor and may even be counterproductive forthe vitrification of osmotically concentratedcytoplasm. 4 Cellular toxicity is time dependent.1,2,94
Therefore, variations in exposure time fromone cell to another, as can occur with manualmethods for vitrification of ova in which ovaare sequentially pipetted into a common vit-rification solution before vitrification is done,may contribute to variations in outcomeand should be minimized. In addition, manyreported procedures are unclear about thetime spent pipetting the ova or the timebetween the end of pipetting and vitrifica-tion, which may make reported results diffi-cult to reproduce.5 Cellular toxicity is concentration depen-dent.2 Therefore, the dilution caused bypipetting cells into a nominally vitrifiablemedium must be taken into account andreported if injury is to be linked to eitherthe toxicity of the vitrification solution or thestability of the amorphous state of the solutionafter dilution with the cell suspension.
Other biological principles governing thesuccess of vitrification are less well established
VITRIFICATION IN ASSISTED REPRODUCTION
16
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 16

and straightforward, such as chilling injuryand cellular toxicity exacerbated by strongglass-forming agents.1 A minority of living systems experienceserious ‘chilling injury’,70,71,80 or injurycaused by cooling per se, independent of iceformation. Separating chilling injury fromother sources of injury may be necessary ifsatisfactory results are not otherwise obtained.Because chilling injury has recently beenlinked to the tonicity of the vitrification solu-tion in the case of kidney slices and wholekidneys,80 unbridled shrinkage of the cellbefore vitrification may be counterproduc-tive for cells whose resistance to chillinginjury is dependent on their degree ofshrinkage.80
2 Cellular toxicity may be exacerbated bystrong glass-forming agents.82 A recent analy-sis of the toxicities of 21 different vitrificationsolutions showed that toxicity was linearlydependent on the ratio of the molarity ofwater in each solution to the molarity ofhydrogen bonding groups required to vitrifythe solution under standardized conditions.82
This ratio, called qv* for short, goes up as themean glass-forming tendency of the vitrifica-tion solution components increases, anddirectly correlates with injury after removingthe cryoprotectants. Fortunately, very low tox-icity solutions have been developed on thebasis of this observation and may have someadvantages in reproductive cryobiology. Forexample, mouse ova vitrified with a solution
known as 90% VM3 were able to be fertilizedand develop to blastocysts at 80% of the rateof untreated control ova without the need forICSI.82
SUMMARY AND CONCLUSIONS
Vitrification is a viable approach to cryo-preservation of a wide range of living systems.Its physical and biological principles are con-tinuing to become better understood, andthis is leading to more numerous and moresuccessful applications. Although the historyof the concept goes back more than three-quarters of a century, the field is probably stillin the infancy of its full potential. As always,nature may have preceded biologists in dis-covering viable approaches to vitrification,but for the most part nature’s examplesremain both recondite and difficult to emu-late directly. Nevertheless, reproductive cryo-biologists have ample means and ampleincentive to follow nature’s lead and developtheir own approaches to answering one ofbiology’s most interesting challenges, the goalof arresting life in a state of suspended anima-tion and restarting it at the right time toenable new lives to begin.
ACKNOWLEDGMENTS
We thank Stanley Leibo for his valued adviceand for his friendship over more than twodecades.
17
VITRIFICATION: AN OVERVIEW
References
1. Fahy GM, MacFarlane DR, Angell CA, et al.Vitrification as an approach to cryopreserva-tion. Cryobiology 1984; 21: 407–26.
2. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196oC by vitrification.Nature 1985; 313: 573–5.
3. Fahy GM, Wowk B, Wu J. Cryopreservationof complex systems: the missing link in theregenerative medicine supply chain. Rejuvena-tion Res 2006; 9: 279–91.
4. Michelmann HW, Nayudu P. Cryopreservationof human embryos. Cell Tissue Bank 2006; 7:135–41.
5. Kasai M, Mukaida T. Cryopreservation ofanimal and human embryos by vitrification.Reprod Biomed Online 2004; 9: 164–70.
6. Kagabu S, Umezu M. Transplantation ofcryopreserved mouse, chinese hamster, rab-bit, Japanese monkey and rat ovaries into ratrecipients. Exp Anim 2000; 49: 17–21.
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 17

7. Koutlaki N, Schoepper B, Maroulis G et al.Human oocyte cryopreservation: past, pre-sent and future. Reprod Biomed Online2006; 13: 427–36.
8. Kuwayama M, Vajta G, Kato O et al. Highlyefficient vitrification method for cryopreser-vation of human oocytes. Reprod BiomedOnline 2005;11: 300–8.
9. Leibo SP. Cryopreservation of mammalianoocytes. In: Tulandi T, Gosden RG, eds.Preservation of Fertility. London: Taylor &Francis, 2004: 141–55.
10. Migishima F, Suzuki-Migishima R, Song S-Y,et al. Successful cryopreservation of mouseovaries by vitrification. Biol Reprod 2003;68: 881–7.
11. Elster J, Benson EE. Life in the polar terres-trial environment with a focus on algae andcyanobacteria. In Fuller BJ, Lane N, BensonEE, eds. Life in the Frozen State. BocaRaton: CRC Press, 2004: 111–50.
12. Luyet B, Rasmussen D. Study by differentialthermal analysis of the temperatures of insta-bility of rapidly cooled solutions of glycerol,ethylene glycol, sucrose, and glucose.Biodynamica 1968; 10: 167–91.
13. Chen T, Fowler A, Toner M. Literaturereview: supplemented phase diagram of thetrehalose-water binary mixture. Cryobiology2000; 40: 277–82.
14. Acker JP, Chen T, Fowler A, et al. Engineeringdesiccation tolerance in mammalian cells:tools and techniques. In Fuller BJ, Lane N,Benson EE, eds. Life in the Frozen State.Boca Raton: CRC Press, 2004: 563–80.
15. Lee RE Jr, Denlinger DL. Insects at LowTemperature. New York: Chapman and Hall,1991.
16. Rasmussen D. A note about ‘phase diagrams’ offrozen tissues. Biodynamica 1969; 10: 333–9.
17. Hirsh AG, Williams RJ, Meryman HT. Anovel method of natural cryoprotection:intracellular glass formation in deeply frozenpopulus. Plant Physiol 1985; 79: 41–56.
18. Leather SR, Walters KFA, Bale JS. TheEcology of Insect Overwintering.Cambridge, UK: Cambridge UniversityPress, 1993.
19. Sun WQ, Leopold AC. Cytoplasmic vitrifica-tion and survival of anhydrobiotic organisms.Comp Biochem Physiol 1997; 117A: 327–33.
20. Hirsh AG. Vitrification in plants as a naturalform of cryoprotection. Cryobiology 1987;24: 214–28.
21. Sakai A. Plant cryopreservation. In Fuller BJ,Lane N, Benson EE, eds. Life in the FrozenState. Boca Raton: CRC Press, 2004: 329–45.
22. Burke MJ. The glassy state and survival ofanhydrous biological systems. In LeopoldAC, ed. Membranes, Metabolism, and DryOrganisms. Ithaca: Cornell University Press,1986: 358–64.
23. Crowe JH, Crowe LM, Tablin F, et al.Stabilization of cells during freeze-drying:the trehalose myth. In Fuller BJ, Lane N,Benson EE, eds. Life in the Frozen State.Boca Raton: CRC Press, 2004: 581–601.
24. Holmstrup M, Bayley M, Ramlov H.Supercool or dehydrate? An experimentalanalysis of overwintering strategies in smallpermeable arctic invertebrates. Proc NatlAcad Sci USA 2002; 99: 5716–20.
25. Crowe JH, Jackson S, Crowe LM. Non-freezable water in anhydrobiotic nematodes.Mol Physiol 1983; 3: 99–105.
26. Bennett VA, Sformo T, Walters K, et al.Comparative overwintering physiology ofAlaska and Indiana populations of the beetleCucujus clavipes (Fabricius): roles of antifreezeproteins, polyols, dehydration and diapause.J Exp Biol 2005; 208: 4467–77.
27. Fahy GM, Hirsh A. Prospects for organpreservation by vitrification. In Pegg DE,Jacobsen IA, Halasz NA, eds. OrganPreservation, Basic and Applied Aspects.Lancaster: MTP Press, 1982: 399–404.
28. Luyet B. On the amount of water remainingamorphous in frozen aqueous solutions.Biodynamica 1969; 10: 277–91.
29. Storey KB, Storey JM. Physiology, biochem-istry, and molecular biology of vertebratefreeze tolerance: the wood frog. In Fuller BJ,Lane N, Benson EE, eds. Life in the FrozenState. Boca Raton: CRC Press, 2004: 243–74.
30. Wisniewski M, Fuller M. Ice nucleation anddeep supercooling in plants: new insights usinginfared thermography. In Margesin R, SchinnerF, eds. Cold-Adapted Organisms – Ecology,Physiology, Enzymology and Molecular Biology.Berlin: Springer, 1999: 105–18.
31. Brayley E. Note on the apparent universalityof a principle analogous to regelation, on thephysical nature of glass, and on the probableexistence of water in a state corresponding tothat of glass. Proc R Soc 1860; 10: 450–60.
32. Stiles W. On the cause of cold death of plants.Protoplasma 1930; 9: 459–68.
33. Luyet B. The vitrification of organic colloids andof protoplasm. Biodynamica 1937; 1(29): 1–14.
34. Fahy GM. Vitrification. In McGrath JJ, DillerKR, eds. Low Temperature Biotechnology:Emerging Applications and EngineeringContributions. New York: American Societyof Mechanical Engineers, 1988: 113–46.
VITRIFICATION IN ASSISTED REPRODUCTION
18
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 18

19
VITRIFICATION: AN OVERVIEW
35. Luyet BJ. Working hypotheses on the natureof life. Biodynamica 1934(1); 1: 1–7.
36. Smith AU. Effects of low temperatures on livingcells and tissues. In Harris RJC, editor.Biological Applications of Freezing and Drying.New York: Academic Press, 1954: 1–53.
37. Tammann G. Kristallisieren und Schmelzen.Leipzig, 1903.
38. Gonzales F, Luyet B. Resumption of heartbeat in chick embryo frozen in liquid nitro-gen. Biodynamica 1950; 7: 1–5.
39. Luyet BJ, Gonzales F. Growth of nerve tissueafter freezing in liquid nitrogen.Biodynamica 1953; 7: 171–4.
40. Luyet B, Rapatz G. Patterns of ice formationin some aqueous solutions. Biodynamica1958; 8: 1–68.
41. Meryman HT. X-ray analysis of rapidly frozengelatin gels. Biodynamica 1958; 8: 69–72.
42. Dowell LG, Moline SW, Rinfret AP. A low-temperature X-ray diffraction study of icestructures formed in aqueous gelatin gels.Biochim Biophys Acta 1962; 59: 158–67.
43. Rapatz G, Luyet B. Electron microscopestudy of erythrocytes in rapidly cooled sus-pensions containing various concentrationsof glycerol. Biodynamica 1968; 10: 193–210.
44. Nei T. Freezing injury to erythrocytes. I.Freezing patterns and post-thaw hemolysis.Cryobiology 1976; 13: 278–86.
45. Franks F, Skaer HLB. Aqueous glasses asmatrices in freeze-fracture elecronmicroscopy. Nature 1976; 262: 323–5.
46. Bruggeller P, Mayer E. Complete vitrificationin pure liquid water and dilute aqueous solu-tions. Nature 1980; 288: 569–71.
47. Dubochet J, McDowall AW. Vitrification ofpure water for electron microscopy. J Microsc1981; 124: RP3–4.
48. Boutron P, Kaufmann A. Stability of the amor-phous state in the system water-glycerol-dimethylsulfoxide. Cryobiology 1978: 15:93–108.
49. Boutron P, Kaufmann A. Stability of theamorphous state in the system water-glycerol-ethylene glycol. Cryobiology 1979; 16: 83–9.
50. Boutron P. Stability of the amorphous state inthe system water-1,2-propanediol.Cryobiology 1979; 16: 557–68.
51. Boutron, P, Arnaud F. Comparison ofthe cryoprotection of red blood cells by1,2-propanediol and glycerol. Cryobiology1984; 21: 348–58.
52. Mazur P. Kinetics of water loss from cells atsubzero temperatures and the likelihood ofintracellular freezing. J Gen Physiol 1963;47: 347–69.
53. Rall WF, Reid DS, Farrant J. Innocuous bio-logical freezing during warming. Nature1980; 286: 511–4.
54. Rall WF. The role of intracellular ice in theslow warming injury of mouse embryos. InZeilmaker GH, ed. Frozen Storage ofLaboratory Animals. New York: GustavFischer Verlag, 1981: 33–44.
55. Lehn-Jensen H, Rall WF. Cryomicroscopicobservations of cattle embryos during freezingand thawing. Theriogenology 1983; 19: 263–77.
56. Rall WF, Reid DS, Polge C. Analysis of slow-warming injury of mouse embryos by cryomi-croscopical and physicochemical methods.Cryobiology 1984; 21: 106–21.
57. Farrant J. Mechanism of cell damage duringfreezing and thawing and its prevention.Nature 1965; 205: 1284–7.
58. Elford BC, Walter CA. Effects of electrolytecomposition and pH on the structure andfunction of smooth muscle cooled to −79oC inunfrozen media. Cryobiology 1972; 9: 82–100.
59. Fahy G. The effects of low temperatures andDMSO on the frog sciatic nerve. J UndergradRes Biol Sci 1972; 2: 411.
60. Fahy GM. Analysis of ‘solution effects’ injury:rabbit renal cortex frozen in the presence ofdimethyl sulfoxide. Cryobiology 1980; 17:371–88.
61. Fahy GM. Vitrification as an approach toorgan cryopreservation: past, present, andfuture. In Smit Sibinga CT, Das PC,Meryman HT, eds. Cryopreservation andLow Temperature Biology in BloodTransfusion. Boston: Kluwer, 1990: 255–68.
62. Fahy GM. Analysis of ‘solution effects’ injury:cooling rate dependence of the functionaland morphological sequelae of freezing inrabbit renal cortex protected with dimethylsulfoxide. Cryobiology 1981; 18: 550–70.
63. Fahy GM. Prospects for vitrification of wholeorgans. Cryobiology 1981; 18: 617.
64. MacFarlane DR, Angell CA, Fahy GM.Homogeneous nucleation and glass forma-tion in cryoprotective systems at high pres-sures. Cryo Letters 1981; 2: 353–8.
65. Fahy GM. Prevention of toxicity from highconcentrations of cryoprotective agents. InPegg DE, Jacobsen IA, Halasz NA, eds. OrganPreservation, Basic and Applied Aspects.Lancaster: MTP Press, 1982: 367–69.
66. Fahy GM. Cryoprotectant toxicity: biochemi-cal or osmotic? Cryo Letters 1984; 5: 79–90.
67. Baxter S, Lathe G. Biochemical effects onkidney of exposure to high concentrations ofdimethyl sulphoxide. Biochem Pharmacol1971; 30: 1079–91.
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 19

VITRIFICATION IN ASSISTED REPRODUCTION
20
67b. Fahy GM. Cytoprotectant toxicity neutraliz-ers reduce freezing damage. Cyro-letters1983; 4: 309–314.
68. Rall WF, Wood MJ, Kirby C, et al.Development of mouse embryos cryopre-served by vitrification. J Reprod Fertil 1987;80: 499–504.
69. Rall WF, Wood MJ. High in vitro and in vivosurvival of day 3 mouse embryos vitrified orfrozen in a non-toxic solution of glycerol andalbumin. J Reprod Fertil 1994; 101: 681–8.
70. Steponkus PL, Myers SP, Lynch DV, et al.Cryopreservation of Drosophila melanogasterembryos. Nature 1990; 345: 170–2.
71. Mazur P, Schneider U, Mahowald AP.Characteristics and kinetics of subzero chill-ing injury in Drosophila embryos.Cryobiology 1992; 29: 39–68.
72. Mazur P, Cole KW, Hall JW, et al.Cryobiological preservation of Drosophilaembryos. Science 1992; 258: 1896–7.
73. Mehl PM. Crystallization and vitrification inaqueous glass-forming solutions. Adv LowTemp Biol 1996; 3: 185–255.
74. MacFarlane DR, Forsyth M, Barton CA.Vitrification and devitrification in cryop-reservation. Adv Low Temp Biol 1991; 1:221–77.
75. Kauzmann W. The nature of the glassy stateand the behavior of liquids at low tempera-tures. Chem Rev 1948; 43: 219–56.
76. Segur JB. Physical properties of glycerol andits solutions. In Miner CS, Dalton NN, eds.Glycerol. New York: Reinhold PublishingCorporation, 1953: 238–334.
77. Angell CA, Sichina W. Thermodynamics ofthe glass transition: empirical aspects. AnnNY Acad Sci 1976; 279: 53–67.
78. Sherwood GJ, Flower JR. Engineeringaspects of equipment design for subzeroorgan preservation. In Pegg DE, editor.Organ Preservation. London: ChurchillLivingstone, 1973: 152–74.
79. Rall WF, Polge C. Effect of warming rate onmouse embryos frozen and thawed in glyc-erol. J Reprod Fertil 1984; 70: 285–92.
80. Fahy GM, Wowk B, Wu J, et al.Cryopreservation of organs by vitrification:perspectives and recent advances.Cryobiology 2004; 48: 157–78.
81. Mehl P. Nucleation and crystal growth ina vitrification solution tested for organcryopreservation by vitrification. Cryobiology1993; 30: 509–18.
82. Fahy GM, Wowk B, Wu J, et al. Improvedvitrification solutions based on predictabilityof vitrification solution toxicity. Cryobiology2004; 48: 22–35.
83. Dupuy J, Jal JF, Ferradou C, et al. Controllednucleation and quasi-ordered growth of icecrystals from low temperature electrolytesolutions. Nature 1982; 296: 135–40.
84. Fahy GM. Biological effects of vitrificationand devitrification. In Pegg DE, Karow AM,Jr, eds. The Biophysics of OrganCryopreservation. New York: Plenum Press,1987: 265–93.
85. Toner M, Cravalho EG, Chiang YM.Vitrification of biological cell suspensions:the importance of ultrarapid cooling andwarming. Cryobiology 1988; 25: 551.
86. Bald WB. Quantitative Cryofixation. Bristol:Hilger, 1987.
87. Baudot A, Odagescu V. Thermal propertiesof ethylene glycol and aqueous solutions.Cryobiology 2004; 48: 283–94.
88. Boutron P. Comparison with the theory of thekinetics and extent of ice crystallization and ofthe glass-forming tendency in aqueous cry-oprotective solutions. Cryobiology 1986; 23:88–102.
89. Boutron P, Mehl P. Theoretical prediction ofdevitrification tendency: determination ofcritical warming rates without using finiteexpansions. Cryobiology 1990; 27: 359–77.
90. Baudot A, Peyridieu JF, Boutron P, et al.Effects of saccharides on the glass-formingtendency and stability of solutions of 2,3-butanediol, 1,2-propanediol, or 1,3-butane-diol in water, phosphate-buffered saline,Euro-Collins solution, or Saint Thomas car-dioplegic solution. Cryobiology 1996; 33:363–75.
91. Boutron P. Glass-forming tendency and sta-bility of the amorphous state in solutions of a2,3-butanediol containing mainly the levoand dextro isomers in water, buffer, andEuro-Collins. Cryobiology 1993; 30: 86–97.
92. Fahy GM. The relevance of cryoprotectant‘toxicity’ to cryobiology. Cryobiology 1986; 23:1–13.
93. Meryman HT. Cryopreservation of livingcells: principles and practice. Transfusion2007; in press.
94. Rall WF. Factors affecting the survival ofmouse embryos cryopreserved by vitrifica-tion. Cryobiology 1987; 24: 387–402.
01a Tucker 8029.qxd 8/23/2007 8:03 PM Page 20

21
Vitrification in small quenchedvolumes with a minimal amount of,or without vitrificants: basicbiophysics and thermodynamicsIgor I Katkov, Vladimir Isachenko and Evgenia Isachenko
1B
INTRODUCTION: FIVE BASICMETHODS OF VITRIFICATIONOF CELLSIt is well established that storage of cells in aliquid milieu leads to degradation processes,and to eventual loss of cell viability. At thesame time, intracellular ice is lethal for themajority of cells.1 Thus, the only stage inwhich the cells can be stabilized would be asolid-like phase in which intracellular ice crys-tals are not formed, or have not grown to the‘critical lethal size’. Such a vitreous (‘glassy’)state has elevated viscosity (in the range of1012 Pa.s at the glass transition temperatureTg and up to 1013.5 Pa.s at the so called ‘strainpoint),2 so processes of chemical and physicaldegradation are essentially stopped for theduration of experiments or storage. We candistinguish five basic methods to achieveintracellular vitrification; all of which lead toa drastic decrease of the water activity.3
Equilibrium (slow) freezing causes the bulkof intracellular water to form ice outside thecell, while dehydrating the cell slowly enoughto prevent intracellular ice formation, andthus leading to vitrification of the intracellu-lar space. This is usually followed by storageat extremely low temperatures, usually −196°C,and more recently −130°C to −80°C. This iscurrently the conventional cryopreservation
process (CP), which in the majority of casesrequires the use of permeable and imperme-able cryoprotective agents (CPAs). While itremains the mainstream method of currentcryobanking, particularly of cryopreservedcell suspensions and some tissues, it holds itsown limitations described in other parts ofthis book (see, for example, Introduction byLeibo and Mazur). There are interesting sci-entific questions related mostly to this way ofvitrification and thawing, such as aging of vit-rified solutions, devitrification of overcooledsamples during slow warming, annealing,overshoot, etc.; however, these are not cov-ered in this chapter.
Lyophilization involves slow freezing tomoderately low (around −40°C) temperatures,sublimation of the bulk of ice at very high vac-uum, and secondary drying of the ‘cake’ at ele-vated (up to +30°C) temperatures until a stateclose to vitrification is achieved (note, that inpractical terms, one can not dry a samplehigher than temperature Tg − 10°C.4,5 Thusenabling stable long-term storage available ata temperature above 0°C, but one has toensure that it is still lower than the Tg of thesample.5 This method is widely used in foodproduction, microbiology, and the pharma-ceutical industry, but so far it has had very lim-ited applications for preservation of themajority of animal eukaryotic cells (see ourrecent review).3
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 21

‘High temperature’ (HT) vitrification of ahighly dehydrated sample and its stabiliza-tion by air/vacuum drying at temperatures>0°C.6–16 is similar to lyophilization but doesnot require the very damaging slow freezingstep to the cake formation temperatures, so itholds great potential for the future of long-term stabilization of mammalian cells atambient temperatures.3 However, so far bothlyophilization and ice-free HT drying ofmammalian cells have proved to be formida-ble tasks and need further investigation.
Ice-free vitrification of cell suspensions,tissues, and organs at very low temperaturesand relatively slow to moderate rates of freez-ing 17,18 requires the use of a high concentra-tion of vitrificants (historically but erroneouslyreferred to as ‘cryoprotectants’ in analogywith the slow freezing, see below), which ele-vates the viscosity of the milieu and preventsice formation during cooling and devitrifica-tion during warming. We will refer to thismethod as relatively slow to moderate rate offreezing–thawing with high concentrations ofthe vitrificants (SR–HC VF). It has hadlimited but notable successes in preservinganimal oocytes, embryos, and organs (seeChapters 4, 6, 7, 11, 12), as well as plant spec-imens. Moreover, it will probably be themethod of choice for cryopreservation oflarger samples, tissues, or even entire organs(see Chapter 1A and below). However, bothchemical toxicity of the vitrification solutionsand osmotic damage associated with thismethod have necessitated another approachfor vitrification in situations when possible,for example for vitrification of sensitivereproductive cells. It could also be useful inthe future for cryopreservation of certaintypes of stem cells and their therapeuticallyimportant derivatives such as embryoidbodies, differentiated progenitor lines, etc.(see Chapter 12 and references 3 and 19).
Ice-free vitrification of small sizes of thesamples at extremely high rates of coolingand especially warming (105 to 106°C/min)prevents ice formation during cooling and
devitrification during warming, and does notrequire the use of potentially toxic high con-centrations of vitrificants. For simplicity, wecall this method fast rate-low concentrationsvitrification (FR-LC VF). Although histori-cally this was the first method of vitrificationof cells (see below), it has not found wideapplication until recent work, mostly by theIsachenko group.20–25 The practical aspects ofthis method are discussed in Chapters 5, 8,and 11a. In this chapter, we concentrate onthe basic aspects of biophysics and thermo-dynamics of this method, with substantialoverview of the historical background of early‘fast rate-low concentration vitrification’ workin 1930s and 1940s when this method wasfirst introduced.
VITRIFICATION OF CELLS:HISTORICAL BACKGROUND ANDBASIC DEFINITIONS
The idea of using freezing and drying to pre-serve food and other perishable materialsgoes back thousands of years to prehistorictimes. However, long-term preservation ofliving objects, and particularly of cells ofanimals such as vertebrates, mollusks, andinsects, is a novel approach. Pioneering workon cryopreservation was performed by Italianpriest and scientist Lazzaro Spallanzani in1776, when he ‘froze’ stallion sperm in snow,noting the recovery of sperm motility uponwarming.26 Later, in the 19th century and thefirst half of the 20th century, scientists eluci-dated some aspects of the mechanisms ofcold adaptation of living matter, particularlythose used by plants and fungi. The mostimportant contribution was made by FatherLuyet, who has been rightfully called thefounder of the science of cryobiology. Fromthe outset, he recognized that ice damagemust be avoided, and that vitrification couldbe a method for preservation of cell viabil-ity.27 In 1938, Luyet and Hodapp achievedsurvival of frog spermatozoa vitrified withliquid nitrogen.28
VITRIFICATION IN ASSISTED REPRODUCTION
22
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 22

It should be mentioned that at almost thesame time several western European andAmerican groups reported their experienceswith attempts at cryopreservation of fowl,29
human,30–32 and rabbit30 spermatozoa. In con-trast to the widely cited work of Father Luyet,these efforts did not receive the recognitionthey deserved, hindered by the variability ofthe reported results and also by the SecondWorld War. Hoagland and Pincus,30 andJahnel31 used a microbiological loop for vitrifi-cation of cells.
At approximately the same time theresearch of another pioneer in the fieldvitrification of cells began in the USSR.Emmanuil Yakovlevich Graevsky started hiswork in 1946 by confirming in his PhD dis-sertation the observations of Luyet;33 he thenwent on to explore the mechanisms of vitrifi-cation and devitrification.34–36 In particular,he reported33,34 that following the Luyet’stechnique frog semen could be frozen witheven better results than in the original Luyet’sreport in 1938. Moreover, Graevsky andMedvedeva were two of the first cryobiologiststo use a microbiological loop to vitrify sus-pensions of bacteria.35,36 Only in the past fewyears has this ‘cryo-loop’ method been rein-vented and applied to the vitrification ofmammalian cells, particularly to the cryopro-tectant-free vitrification of human spermato-zoa, which is discussed in more detail below.
The approach to vitrification developed byLuyet and the other cryobiologists mentionedabove was based on the application of rela-tively fast cooling rates. Yet the sample viabil-ity was low and variable, mostly because, asthe authors recognized, sufficiently rapidcooling and warming which must be veryhigh for low concentrations of vitrificantswere not achievable. One of the reasons forthis was that very low temperature efficientcooling agents such as liquid nitrogen andliquid oxygen were not used in their research.As a result, the mainstream of cryobiologyturned to preservation methods based onslow freezing.
This method was facilitated by the discoveryof the protective properties of glycerol onhuman and animal sperm when spermatozoawere frozen to extremely low temperatures,made independently by Igor Smirnov in theUSSR37 and by Alan Parkes et al. in theUK.38,39 (Parkes also deserves mention forcoining the term ‘cryobiology’, the study of‘frosty life’.) The cryoprotective qualities ofglycerol and other low molecular weight,mostly non-electrolytic substances had beendescribed before. For example, Bernstein andPetropavlovski in 1937 used 0.3–0.5 mol/Lglycerol and other electrolytes for freezing ofbull, ram, stallion, boar, and rabbit spermato-zoa to a temperature of −21°C.40 However, itwas only after development of stable long-term cryopreservation at very low tempera-tures (−196°C and below), initiated bySmirnov’s and by Parkes’s groups, that the eraof practical applications of cryobiology, par-ticularly for animal breeding, began in theWest and in the Soviet Union. During the fol-lowing decades, the slow-freeze method dom-inated the field of cryopreservation, and itremains the basis of the majority of cryobio-logical techniques in use in production facili-ties and research laboratories. Progressivelymore sophisticated understandings of themechanisms underlying cell damage duringslow to moderate freezing, as well as methodsto prevent it, were contributed particularly byLovelock,41 Mazur,1,42 and Meryman,43 and bymany others in the later generations of cryo-biologists. In particular, Mazur developed aset of equations that allow calculation of theoptimal (equilibrium) regimens of freezingcells, which avoid formation of intracellularice. These equations also permit estimation ofthe probability of intracellular ice formationif the freezing has been done at higher thanequilibrium rates.
Despite the fact that equilibrium freezinghas been widely used and drawn a lot ofattention, there are many interesting scien-tific questions that remain. Particularly, thecentral dogma that ‘intracellular ice always
23
BASIC BIOPHYSICS AND THERMODYNAMICS OF VITRIFICATION
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 23

kills’ has recently undergone scrutiny byMcGann et al.44,45 Moreover, even the authorof this postulate has recently questioned itsabsolute applicability to all scenarios: when itis applied to spermatozoa, even the authorof the equations has admitted that the iceformation might not be damaging (or, as wethink, may even not exist at all!) at freezingrates much higher than his theory predicts.46
While ‘conventional’ slow freezing hasgiven satisfactory results for many suspendedcells, such as blood, sperm, and embryos,issues of ice formation and non-homogeneityof thermal profiles of samples have madeslow freezing and thawing a formidable taskfor tissues and organs. The breakthroughcame when Fahy et al. vitrified an entireorgan – a kidney.17 The presence of high con-centrations of vitrificants, particularly glyc-erol, allowed an ice-free bulk glassy state to beachieved both inside the organ (by perfusion)and in the external milieu. By tradition, glyc-erol and other vitrificants were still called‘cryoprotective agents’ (CPAs) as for slowfreezing, probably because the same chemi-cals were used for both applications.However, one has to remember that for ice-free vitrification, they play a completely dif-ferent role as the glass-formers than they dofor the slow freezing. In the latter case, espe-cially at suboptimal low rates of freezing,lower than the equilibrium, the CPAs (partic-ularly, the permeable ones) act mostly asosmotic buffers and ‘water holders’ prevent-ing osmotic damage, rehydration, increase ofconcentration of the ions, and other eventscollectively defined as the ‘solution effects’.1
However, some changes in the ice structure ofthe intracellular milieu might also play a pos-itive role at superoptimal rates of ice contentfreezing as the viscosity of CPAs such glycerolor dimethylsulfoxide (DMSO) increase at sub-zero temperatures, so the probability of thelethal amount of intracellular ice decreases.47,48
Thus, while used interchangeably, we thinkone has to be careful when equating the terms‘CPA’ with ‘vitrificants’, particularly, in the
case of small molecular weight permeablesubstances such as glycerol, DMSO, ethyleneglycol (EG), or propylene glycol (PG).Furthermore, it is important to note that theyall have glass transition temperatures (Tgs)much lower than 0°C, thus, by definition, inthe case of HT vitrification they would act asplasticizers by effectively decreasing the Tg ofthe blend. Thus making them unsuitablefor stable storage of vitrified samples above− 20°C. At the same time, they are widelyused for vitrification and following stablelong-term storage at ultra-low temperaturesof liquid nitrogen (LN2) and industrial freez-ers (−120°C and below).
Vitrification eliminated many of the prob-lems related to the slow freezing of liquid waterto an ice phase, particularly extensive rehydra-tion and osmotic damage, the increased ionicstrength of concentrated eutectic solutions, andshifts in pH, etc. However, vitrification in highconcentrations of vitrificants has introduced itsown set of problems. Notable among them areosmotic damage during addition and removalof vitrificants, CPA toxicity, mechanical crack-ing of glasses, and devitrification due to inad-vertent thermal cycling during storage, etc.(see Chapter 1A).
Nevertheless, practical cryobiologicalapplications of vitrification with the use ofhigh concentrations of vitrificants has contin-ued to grow, especially after Rall and Fahyreported the successful vitrification of mouseembryos in 1985.18 However, more wide-spread use of vitrification was slowed by theinherent pitfalls of high-CPA methods, andalso by the lack of understanding of themechanisms of vitrification and devitrifica-tion by most practitioners of the craft. By the1980s, the fundamental work of Luyet andthe other pioneers of vitrification had beenlargely forgotten. Unfortunately, the pioneer-ing work of the cryobiologists of the 1930s,1940s, and early 1950s is still under appreci-ated today.
In the 1990s, vitrification was applied tonew areas such as oocytes and ovarian tissues
VITRIFICATION IN ASSISTED REPRODUCTION
24
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 24
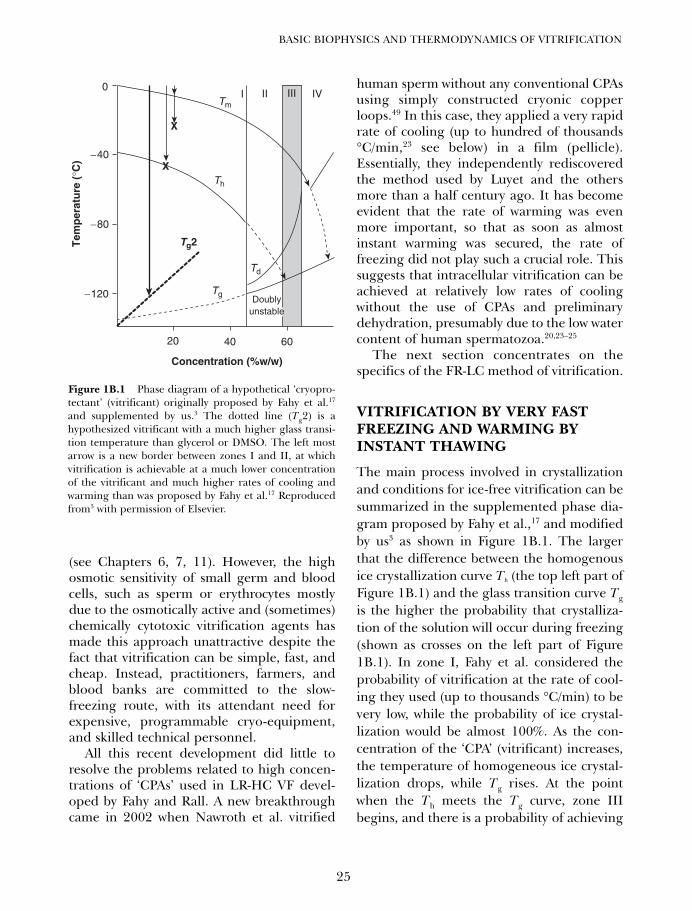
(see Chapters 6, 7, 11). However, the highosmotic sensitivity of small germ and bloodcells, such as sperm or erythrocytes mostlydue to the osmotically active and (sometimes)chemically cytotoxic vitrification agents hasmade this approach unattractive despite thefact that vitrification can be simple, fast, andcheap. Instead, practitioners, farmers, andblood banks are committed to the slow-freezing route, with its attendant need forexpensive, programmable cryo-equipment,and skilled technical personnel.
All this recent development did little toresolve the problems related to high concen-trations of ‘CPAs’ used in LR-HC VF devel-oped by Fahy and Rall. A new breakthroughcame in 2002 when Nawroth et al. vitrified
human sperm without any conventional CPAsusing simply constructed cryonic copperloops.49 In this case, they applied a very rapidrate of cooling (up to hundred of thousands°C/min,23 see below) in a film (pellicle).Essentially, they independently rediscoveredthe method used by Luyet and the othersmore than a half century ago. It has becomeevident that the rate of warming was evenmore important, so that as soon as almostinstant warming was secured, the rate offreezing did not play such a crucial role. Thissuggests that intracellular vitrification can beachieved at relatively low rates of coolingwithout the use of CPAs and preliminarydehydration, presumably due to the low watercontent of human spermatozoa.20,23–25
The next section concentrates on thespecifics of the FR-LC method of vitrification.
VITRIFICATION BY VERY FASTFREEZING AND WARMING BYINSTANT THAWING
The main process involved in crystallizationand conditions for ice-free vitrification can besummarized in the supplemented phase dia-gram proposed by Fahy et al.,17 and modifiedby us3 as shown in Figure 1B.1. The largerthat the difference between the homogenousice crystallization curve Th (the top left part ofFigure 1B.1) and the glass transition curve Tg
is the higher the probability that crystalliza-tion of the solution will occur during freezing(shown as crosses on the left part of Figure1B.1). In zone I, Fahy et al. considered theprobability of vitrification at the rate of cool-ing they used (up to thousands °C/min) to bevery low, while the probability of ice crystal-lization would be almost 100%. As the con-centration of the ‘CPA’ (vitrificant) increases,the temperature of homogeneous ice crystal-lization drops, while Tg rises. At the pointwhen the Th meets the Tg curve, zone IIIbegins, and there is a probability of achieving
25
BASIC BIOPHYSICS AND THERMODYNAMICS OF VITRIFICATION
0
60
Tg2
X
X
4020
−40
−80
−120
Concentration (%w/w)
Tem
per
atu
re (
°C)
I II III IVTm
Th
Tg
Td
Doublyunstable
Figure 1B.1 Phase diagram of a hypothetical ‘cryopro-tectant’ (vitrificant) originally proposed by Fahy et al.17
and supplemented by us.3 The dotted line (Tg2) is ahypothesized vitrificant with a much higher glass transi-tion temperature than glycerol or DMSO. The left mostarrow is a new border between zones I and II, at whichvitrification is achievable at a much lower concentrationof the vitrificant and much higher rates of cooling andwarming than was proposed by Fahy et al.17 Reproducedfrom3 with permission of Elsevier.
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 25

vitrification during appropriately fast cooling.However, the devitrification curve Td indi-cates that there would be a high possibility ofdevitrification during warming. The devitrifi-cation curve Td drastically decreases withhigher percentages of ‘CPA’ (vitrificant) dueto a dramatic rise of viscosity, and when Td
meets the non-homogeneous equilibriumcrystallization (melting) curve Tm zone IVbegins. At this point and higher, crystalliza-tion and devitrification will never occur atany reasonable rate of cooling and warming.Note, however, that for ‘conventional’ vitrifi-cants such as glycerol, this concentration is inthe range of 65–70%, which is extremelyhigh, and few known mammalian cells wouldwithstand such enormous (tens of Osm)osmotic pressure. Thus, vitrification shouldbe performed in a kinetic way, playing withconcentration of the vitrificant and rates ofcooling and warming. Fahy et al. defined thezone when practical vitrification is achievableas from 45% of ‘CPA’ (vitrificant) marking thisas zone II.
The critical speed of cooling and warm-ing, however, is negatively related to the con-centration of the CPA: the higher is thespeed of cooling/warming, the lower is theconcentration of the vitrificant needed, sothat the border between ‘non-achievable’ and‘achievable’ vitrification (zones I and II) isarbitrary. This means that if the speed ofcooling and warming are high enough, thenthe concentration of the glass-formers can bevery low (the dashed line is a new borderbetween zone I and zone II). Even pure watercan vitrify if special conditions, such assuperquenching are met (although there isstill a debate on the exact value of its Tg).
50,51
While there is still discussion among scien-tists about the minimal cooling rate that canbe achieved to vitrify water it must be at leastmillions of °C/min.52–54 In any case, loweringthe amount of vitrificants by increasingspeed of cooling and warming (as defined
above HR-LC VF), as well as by partialdehydration using impermeable osmoticallyactive compounds such as sucrose has beenthe main direction for the past decade in vit-rification cryobiology, particularly for repro-ductive cells. Note that the positive effect ofthe addition of sucrose is sometimes erro-neously attributed to ‘lowering toxicity’ ofglycerol or DMSO, while it is mostly due tothe osmotic and kinetic effects.
Several types of devices, such as openpulled and hemi-straws, microscopic grids,and cryoloops and cryotops have been inves-tigated to increase the speed of cooling andwarming and, thus, to decrease the CPA con-centrations to values of 25–30% (see details inChapters 4A, 10, 11, 12). While it has beenworking well for oocytes and embryos, smallcells like sperm and erythrocytes have provedto be practically intolerant even to such ‘mod-erate’ concentrations of CPA (e.g. 30% ofmost widely used CPAs such as glycerol, eth-ylene glycol, and propylene glycol translateto 15–20 times the isotonic value). However, amajor breakthrough came in 2001, when agroup from Germany managed successfullyto vitrify sperm without any conventional cry-oprotectants or vitrificants. The spermatozoawere cooled in very thin films (pellicles) incopper cryoloops.49 Basically, they rediscov-ered the original Luyet–Graevsky approach,but with the additional knowledge that hadbeen accumulated since the pioneering worksfrom 1939 to 1948. We have collaborated withthis group, and recently published severaljoint papers,20,23–25,49 in which we suggestedthat the extremely high rate of cooling (sev-eral hundred thousand °C/min) and almostinstant warming by dissolving in a warmmedia made vitrification during freezingwithout devitrification during rewarmingpractically feasible. Taking in to account thatthe culture medium contained proteins(serum albumin), and the internal cellularmilieu is abundant in high glass transitiontemperature (much above 0°C) componentssuch as proteins, polysaccharides, nucleic
VITRIFICATION IN ASSISTED REPRODUCTION
26
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 26

acids, and other biopolymers, then kineticvitrification can be achieved at very low con-centrations of non-osmotically active glassformers, so no conventional osmotically dam-aging and toxic ‘CPAs’ are needed (the far leftpart of Figure 1B.1). The typical glass transi-tion temperature of such intracellular compo-nents lies much higher than for glycerol orother ‘conventionally used’ vitrificants (shownas Tg2 in Figure 1B.1), so the probability ofvitrification is increased dramatically whenhigh rates of cooling and very high rates ofwarming are combined.
In experiments with cryonic loops, we esti-mated that the rate of cooling could be ashigh as a hundred thousand °C/min, so wespeculated that the border between zones Iand II could be shifted to very low values(Figure 1B.1, left arrow). In these conditionsof very fast cooling and practically instantwarming the cell’s natural high molecularweight intracellular vitrificants and presenceof albumin in the media would be enough toensure both intra- and (probably) extracellu-lar vitrification, as we have suggested.23 Even
cooling rates 100-fold slower than in cryonicloops immersed in LN2, e.g. in vapor ofliquid nitrogen, were sufficient to ensure thecell survival at the same level as for directplunging to LN2.
25 Moreover, the use of pel-licles in cryonic loops is not mandatory: thecells can be cryopreserved in an aseptic man-ner in small droplets in plastic straws withoutdirect contact with LN2.
20 One such modifi-cation is shown in Figure 1B.2. The crucialpoints are the small volume of the liquid tobe cooled and fast warmed with intensive agi-tation of the surrounding warm medium. Forsmall cells such as human spermatozoa novitrificants were needed, while larger andprobably ‘more watery’ cells need a certainamount of vitrificants (see Chapters 5, 8,and 11).
Application of fast cooling in combinationwith relatively small amounts of vitrificantscan be applied to human embryonic cellsobtained from cultured embryonic cell lines,which are proven to lose pluripotent abilityafter conventional slow freezing with DMSO,so vitrification seems a more favorable
27
BASIC BIOPHYSICS AND THERMODYNAMICS OF VITRIFICATION
2
431
2
2
2
Figure 1B.2 Images and scheme ofwarming a microdroplet in asepticconditions using the cut standardstraw (CSS) container for vitrification:(1) closed 0.5 ml straw, (2) CSS, (3)microdroplet of vitrification mediumwith the embryo, (4) tube with solu-tion for warming and removal of thevitrificant.
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 27

approach55–61 (see also Chapter 12). We havealso initiated thorough studies of the differencein human embryonic stem cell (hESC)pluripotency with respect to the mode ofcryopreservation: slow freezing versus vitrifi-cation, which has confirmed that slow freez-ing suppressed expression of pluripotencymarker Oct-4.19
CALCULATION OF THERATE OF COOLING/WARMINGOF SMALL SAMPLES
For small objects such as a thin pellet within acryoloop, or a very small droplet of cell sus-pension shown in Figure 1B.2, it is practicallyimpossible to measure directly the cooling orwarming rate with a thermometer or a ther-mocouple. Accurate measurement by visualobservation is also impossible because bothvitrification and warming occur very quickly.The only practical estimation of the range ofthe rate of cooling and warming can be doneby calculation. Here we give an example ofthe calculations in general, and the rate ofwarming of the droplet depicted in Figure1B.2.
The heat transfer in a thermal conductor isdescribed by the Biot-Fourier equation (alsocalled the heat conduction equation) describ-ing the variation of temperature T (heat flux)with position and time t in a thermal conduc-tor, and in the general form for isotropicpropagation with no internal heat sources itcan be written as follows:62,63
where
is the temperature diffusivity, κ is the thermalconductivity, c is the specific heat capacity, ρ isthe density of the conductor, and
are the operators for Cartesian, cylindrical,or spherical coordinates, respectively.
For a simple system such as a rod where itsside surface is thermo-isolated, so that theheat flow is in one direction, the equation canbe simplified to a one-dimensional form thatin Cartesian coordinates can be written asfollows:
If the initial temperature of the sample is T0and both its sides are kept at (immersed to)the same temperature Tf (i.e. in a thin film wehave estimated before),23 the solution of thisequation can be expressed as follows:
where τ = d2/DT is the characteristic time afterwhich all the processes are practically equili-brated, x is the distance from the middle tothe surface, and d is the thickness of the film.Thus, the rate of temperature change B canbe determined as (Tf −T0) /T.
For a homogeneous sphere with initial tem-perature T0 immersed into a highly thermocon-ductive medium with temperature Tf, the heat
T(x, t) =(
4
π
)(Tf − T0)
∞∑m=1
sin(2m + 1)π(
xd
)2m + 1
e−[(2m+1)π]2( tτ )
∂T
∂t= DT
∂2T
∂x2
∇2T = ∂2T
∂x2+ ∂2T
∂y2+ ∂2T
∂z2
∇2T = 1
r
∂
∂r
(r∂T
∂r
)+ 1
r2
∂2T
∂ϕ2+ ∂2T
∂z2
∇2T = 1
r2
∂
∂r
(r2 ∂T
∂r
)
+ 1
r2 sin θ
∂
∂θ
(sin θ
∂T
∂θ
)
+ 1
r2 sin2 θ
∂2T
∂ϕ2
DT = κ
cρ
∂T∂t
= DT∇2T
VITRIFICATION IN ASSISTED REPRODUCTION
28
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 28

flow distribution will be dependent on only thedistance from the center to the position in con-sideration (r) but will be independent of thespherical angles θ and ϕ, so a one-dimensionalform in the spherical coordinates is:
By substituting v = rT this can be simplified to:
This equation is completely analogous to theequation for a one-dimensional case discussedabove, but the thickness d must be substitutedby the radius of the sphere R, and r instead ofx would be the current distance from the core.Thus, both for a thin film, and a sphere, themathematical solution is equivalent and allconsiderations are the same. Practical differ-ence is due to the fact that v = rT, all processesin the center of the sphere are equilibrated ata time of about 0.5 τ, while for a pellicle theyare in range of its characteristic time.
Assuming for simplicity that the dropletshown in Figure 1B.2 is a sphere. The kinetics ofwarming as the function of the relative distancefrom the center of the sphere λ= x/r and the rel-ative time q = t/τ is shown in Figure 1B.3. At thefirst moment (q = 0) in all points of the dropletexcept its surface (λ=1), the temperature isequal to the temperature of the liquid nitrogen(T0= 77K), while we assume for the sake of sim-plicity that the temperature of the surface isequal to 0°C (Tf= 273K). So no warming abovezero (warming and dissolving) occurs until thewhole droplet is warmed to 0°C. At time τ/20,the core of the droplet is still very cold, so thetemperature at the center (λ= 0) is far below(122K) the freezing point. At the same time,close to the surface the temperature of the layerT is substantially higher (243K). The volume ofmore distant layers is the quadratic function ofthe radius that means a larger portion of thedroplet is warmed (in comparison with the thinfilm, where all layers have the same volumesregardless of their distance from the center).
After less than a half of the characteristic time,practically the whole droplet is in equilibriumwith the warming surroundings, so even thecenter of the droplet is warmed practically to243K at time q = 0.4 τ. After the temperature ofthe droplet reaches 0°C (Tf= 273K), it devitri-fies, detaches from the surface, and dissolves inthe surrounding medium.
Now we can estimate the characteristictime for a frozen spherical droplet immersedin a hot solution. Taking into account that forice κ = 0.005 cal/s/cm/K, c = 0.49 cal/g/K, andρ= 0.92 g/cm3 64 this gives the temperaturediffusivity DT to be equal to 0.011 cm2/s.
A spherical droplet of volume 0.75 µL willhave a radius rdr = 0.056 cm, so the character-istic time τ can be estimated as 0.29 s.
In reality, the shape of the sample is not aspherical droplet but rather a spherical seg-ment (cap) due to the low surface tension ofthe DMSO. The spherical surface of the cap isopened to the warming solution, while thebase surface can be considered to be thermo-isolated for a short time due to contact with theplastic. We estimated the height of the cap h isabout half of its base radius a. The volume ofthis cap is still 0.75 µL, so it can be shown thath = 0.048 cm, and c = 0.096 cm. The sphericalsurface of the cap would be 0.036 cm2, whilethe total surface of the spherical droplet dis-cussed above would be 0.040 cm2. In this case,the process of total equilibrium ends in 0.135 s.In such a scenario, the characteristic time τ ofthe process is about 0.27 s, and the process oftotal equilibrium occurs in 0.135 s. That meansthe rate of warming is in the range of87 000°C/ min. The presence of ‘cryoprotec-tants’ – vitrificants such as DMSO and sucrosewould affect viscosity, and thus the probabilityof ice formation, but c, κ, and ρ would not besignificantly different, so the time τ and therate of warming would be in the same range. Asimilar approach applied to a thin pellicle onthe cryoloop gave us an estimation of coolingat an order of magnitude higher,23 while warm-ing occurs in this case almost instantly.
In comparison, the use of the commonlyused open-pulled straws (calculated as a
∂ν
∂t= DT
∂2ν
∂r2
∂T
∂t= DT
r2
∂
∂r
(r2 ∂T
∂r
)
29
BASIC BIOPHYSICS AND THERMODYNAMICS OF VITRIFICATION
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 29
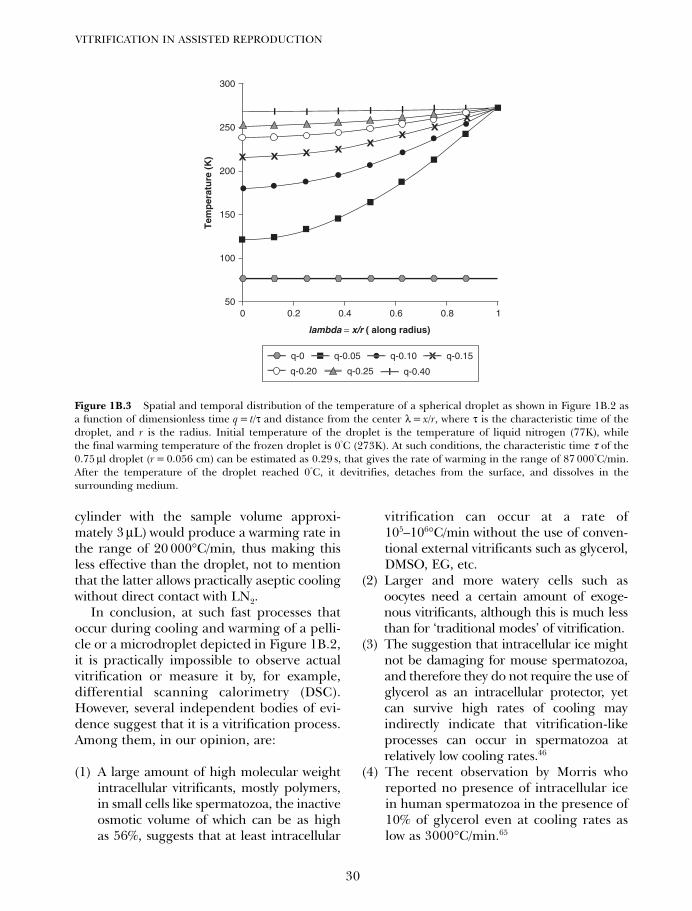
cylinder with the sample volume approxi-mately 3 µL) would produce a warming rate inthe range of 20 000°C/min, thus making thisless effective than the droplet, not to mentionthat the latter allows practically aseptic coolingwithout direct contact with LN2.
In conclusion, at such fast processes thatoccur during cooling and warming of a pelli-cle or a microdroplet depicted in Figure 1B.2,it is practically impossible to observe actualvitrification or measure it by, for example,differential scanning calorimetry (DSC).However, several independent bodies of evi-dence suggest that it is a vitrification process.Among them, in our opinion, are:
(1) A large amount of high molecular weightintracellular vitrificants, mostly polymers,in small cells like spermatozoa, the inactiveosmotic volume of which can be as highas 56%, suggests that at least intracellular
vitrification can occur at a rate of105–106°C/min without the use of conven-tional external vitrificants such as glycerol,DMSO, EG, etc.
(2) Larger and more watery cells such asoocytes need a certain amount of exoge-nous vitrificants, although this is much lessthan for ‘traditional modes’ of vitrification.
(3) The suggestion that intracellular ice mightnot be damaging for mouse spermatozoa,and therefore they do not require the use ofglycerol as an intracellular protector, yetcan survive high rates of cooling mayindirectly indicate that vitrification-likeprocesses can occur in spermatozoa atrelatively low cooling rates.46
(4) The recent observation by Morris whoreported no presence of intracellular icein human spermatozoa in the presence of10% of glycerol even at cooling rates aslow as 3000°C/min.65
VITRIFICATION IN ASSISTED REPRODUCTION
30
lambda = x/r ( along radius)
Tem
per
atu
re (
K)
050
100
150
200
250
300
0.2 0.4 0.6 0.8 1
X X X XX
XX
XI I I I I I I
q-0.05 q-0.10 q-0.15X
q-0.40Iq-0.20 q-0.25
q-0
Figure 1B.3 Spatial and temporal distribution of the temperature of a spherical droplet as shown in Figure 1B.2 asa function of dimensionless time q = t/τ and distance from the center λ = x/r, where τ is the characteristic time of thedroplet, and r is the radius. Initial temperature of the droplet is the temperature of liquid nitrogen (77K), whilethe final warming temperature of the frozen droplet is 0°C (273K). At such conditions, the characteristic time τ of the0.75 µl droplet (r = 0.056 cm) can be estimated as 0.29 s, that gives the rate of warming in the range of 87 000°C/min.After the temperature of the droplet reached 0°C, it devitrifies, detaches from the surface, and dissolves in thesurrounding medium.
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 30

References
31
BASIC BIOPHYSICS AND THERMODYNAMICS OF VITRIFICATION
1. Mazur P. Freezing of living cells: mechanismsand implications. Am J Physiol 1984; 247:C125–42.
2. Shelby JE. Viscosity of glass forming melts. In:Shelby JE (ed). Introduction to Glass Scienceand Technology, 2nd edn. Cambridge, UK:The Royal Society of Chemistry, 2005:111–37.
3. Katkov II, Isachenko V, Isachenko E et al.Low- and high-temperature vitrification as anew approach to biostabilization of reproduc-tive and progenitor cells. Int J Refrigeration2006; 29: 346–57.
4. Bronshtein V. ‘Good’ and ‘bad’ glasses forlong-term preservation. Cryobiology 2002;45: 213.
5. Katkov II, Levine F. Prediction of the glasstransition temperature of water solutions:comparison of different models. Cryobiology2004; 49: 62–82.
6. Lepeshinskaya O. The origin of the cell. Podznamenem marksizma (Rus) 1939(5):130–145. [in Russian]
7. Annear DI. Recoveries of bacteria after dryingin glutamate and other substances. Aust J ExpBiol Med Sci 1964; 42: 717–22.
8. Annear DI. Observations on drying bacteriafrom the frozen and from the liquid state. AustJ Exp Biol Med Sci 1958; 36: 211–21.
9. Annear DI. Recoveries of bacteria after dryingin vacuo at a bath temperature of 100 degreesC. Nature 1966; 211: 761.
10. Annear DI. Recoveries of bacteria after dryingand heating in glutamate foams. J Hyg (Lond)1970; 68: 457–9.
11. Bronshtein V, inventor Universal PreservationTechnologies, Inc., assignee. Preservation byfoam formation US Patent 5766520 USA. 1998.
12. Bronshtein V. Preservation of viruses andbacteria at ambient temperatures with methyl-glucoside. Cryobiology 2002; 45: 235.
13. Roser B, inventor Quadrant, assignee. Inter-national Application WO 96/40077. 1996.
14. Gordon SL, Oppenheimer SR, Mackay AM, etal. Recovery of human mesenchymal stemcells following dehydration and rehydration.Cryobiology 2001; 43: 182–7.
15. Guo N, Puhlev I, Brown DR, et al. Trehaloseexpression confers desiccation toleranceon human cells. Nat Biotechnol 2000; 18:168–71.
16. Puhlev I, Guo N, Brown DR, et al. Desiccationtolerance in human cells. Cryobiology 2001;42: 207–17.
17. Fahy GM, MacFarlane DR, Angell CA, et al.Vitrification as an approach to cryopreserva-tion. Cryobiology 1984; 21: 407–26.
18. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196 degrees C by vitri-fication. Nature 1985; 313: 573–5.
19. Katkov II, Kim MS, Bajpai R, et al.Cryopreservation by slow cooling with DMSOdiminished production of Oct-4 pluripotencymarker in human embryonic stem cells.Cryobiology 2006; 53: 194–205.
20. Isachenko V, Isachenko E, Montag M, et al.Clean technique for cryoprotectant-free vitri-fication of human spermatozoa. ReprodBiomed Online 2005; 10: 350–4.
21. Isachenko V, Montag M, Isachenko E, et al.Aseptic technology of vitrification of humanpronuclear oocytes using open-pulled straws.Hum Reprod 2005; 20: 492–6.
22. Isachenko V, Montag M, Isachenko E, et al.Aseptic vitrification of human germinal vesi-cle oocytes using dimethyl sulfoxide as acryoprotectant. Fertil Steril 2006; 85: 741–7.
23. Isachenko E, Isachenko V, Katkov II, et al.Vitrification of mammalian spermatozoa inthe absence of cryoprotectants: from pastpractical difficulties to present success.Reprod Biomed Online 2003; 6: 191–200.
24. Isachenko E, Isachenko V, Katkov II, et al.DNA integrity and motility of human sperma-tozoa after standard slow freezing versuscryoprotectant-free vitrification. Hum Reprod2004; 19: 932–9.
25. Isachenko V, Isachenko E, Katkov, II, et al.Cryoprotectant-free cryopreservation ofhuman spermatozoa by vitrification and freez-ing in vapor: effect on motility, DNA integrity,and fertilization ability. Biol Reprod 2004; 71:1167–73.
26. Spallanzani L. Dissertazioni di fisica animale evegetale. Modano 1780.
27. Luyet BE. The vitrification of organic colloidsand of protoplasm. Biodynamica 1937; 1: 1–14.
28. Luyet B, Hodapp A. Revival of frog’s sperma-tozoa vitrified in liquid air. Proc Meet Soc ExpBiol 1938; 39: 433–434.
29. Schaffner CS. Longevity of fowl spermatozoain frozen condition. Science 1942; 96: 337.
30. Hoagland H, Pincus GG. Revival of mam-malian sperm after immersion in liquid nitro-gen. J Genet Physiol 1942; 25: 337–344.
31. Jahnel F. Resistance of human spermatozoa todeep cold. Klinische Wochenschrift 1938; 17:1273–1274. [in German]
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 31

32. Parkes AS. Preservation of human spermatozoaat low temperatures. Br Med J 1945; 2:212–213.
33. Graevsky EY. Studies on deep cooling of theprotoplasm. PhD Thesis. Leningrad: LeningradState University, 1946: 112. [in Russian]
34. Graevsky EY. Living matter and low tempera-tures. Priroda (Rus) 1948; 5: 13–25. [in Russian]
35. Graevsky EY. Glassy state of protoplasm inconditions of deep cooling. Uspehi Sovr Biol(Rus) 1948; 14: 186–202. [in Russian]
36. Graevsky EY, Medvedeva YA. Causes of dam-age of protoplasm during deep freezing. ZhObschey Biologii (Rus) 1948; 9: 436–469. [inRussian]
37. Smirnov IV. Preservation of domestic animalsemen by deep cooling. Sovetskaja Zootechnia(Rus) 1949; 4: 63–65. [in Russian]
38. Polge C, Smith AU, Parkes AS. Revival of sper-matozoa after vitrification and dehydration atlow temperatures. Nature 1949; 164: 666–676.
39. Smith AU, Polge C, Parkes AS. Survival ofspermatozoa at low temperatures. Nature1950; 166: 668–9.
40. Bernstein AD, Petropavlovski VV. Influence ofnon-electrolytes on viability of spermatozoa.Buleten’ eksperimentalnoi biologii i medicini1937; III: 21–25. [in Russian]
41. Lovelock JE. The mechanism of the protectiveaction of glycerol against haemolysis by freez-ing and thawing. Biochim Biophys Acta 1953;11: 28–36.
42. Mazur P. Kinetics of water loss from cells atsubzero temperatures and the likelihood ofintracellular freezing. J Gen Physiol 1963; 47:347–69.
43. Meryman HT. Freezing injury and its preven-tion in living cells. Annu Rev Biophys Bioeng1974; 3: 341–63.
44. Acker JP, McGann LE. Innocuous intracellularice improves survival of frozen cells. CellTransplant 2002; 11: 563–71.
45. Acker JP, McGann LE. Protective effect ofintracellular ice during freezing? Cryobiology2003; 46: 197–202.
46. Mazur P, Koshimoto C. Is intracellular ice for-mation the cause of death of mouse spermfrozen at high cooling rates? Biol Reprod2002; 66: 1485–90.
47. Karlsson JO, Cravalho EG, Borel Rinkes IH,et al. Nucleation and growth of ice crystalsinside cultured hepatocytes during freezing inthe presence of dimethyl sulfoxide. Biophys J1993; 65: 2524–36.
48. Rall WF, Mazur P, McGrath JJ. Depression ofthe ice-nucleation temperature of rapidlycooled mouse embryos by glycerol anddimethyl sulfoxide. Biophys J 1983; 41: 1–12.
49. Nawroth F, Isachenko V, Dessole S, et al.Vitrification of human spermatozoa withoutcryoprotectants. Cryo Letters 2002; 23: 93–102.
50. Yue Y, Angell CA. Clarifying the glass-transi-tion behaviour of water by comparison withhyperquenched inorganic glasses. Nature2004; 427: 717–20.
51. Kohl I, Bachmann L, Mayer E, et al. Waterbehaviour: glass transition in hyperquenchedwater? Nature 2005; 435(7041): E1; discussionE1–2.
52. Angell CA. Amorphous water. Annu Rev PhysChem 2004; 55: 559–83.
53. Johari GP, Hallbrucker A, Mayer E. Twocalorimetrically distinct states of liquid waterbelow 150 Kelvin. Science 1996; 273: 90–2.
54. Velikov V, Borick S, Angell CA. The glass transi-tion of water, based on hyperquenching experi-ments. Science 2001; 294: 2335–8.
55. Fujioka T, Yasuchika K, Nakamura Y, et al. Asimple and efficient cryopreservation methodfor primate embryonic stem cells. Int J DevBiol 2004; 48: 1149–54.
56. Heng BC, Kuleshova LL, Bested SM, et al.The cryopreservation of human embryonicstem cells. Biotechnol Appl Biochem 2004.
57. Ji L, de Pablo JJ, Palecek SP. Cryopreservationof adherent human embryonic stem cells.Biotechnol Bioeng 2004; 88: 299–312.
58. Reubinoff BE, Pera MF, Vajta G, et al. Effectivecryopreservation of human embryonic stemcells by the open pulled straw vitrificationmethod. Hum Reprod 2001; 16: 2187–94.
59. Richards M, Fong CY, Tan S, et al. An efficientand safe xeno-free cryopreservation methodfor the storage of human embryonic stemcells. Stem Cells 2004; 22: 779–89.
60. Solberg S, Laerum OD. Cryobiology – freezepreservation and storage of living cells andtissues. Tidsskr Nor Laegeforen 2004; 124:2607–9. [in Norwegian]
61. Zhou CQ, Mai QY, Li T, et al. Cryopreservationof human embryonic stem cells by vitrification.Chin Med J (Engl) 2004; 117: 1050–5.
62. Zhou X. The Fourier Heat Flux Model andRelevant Associated Equations. 1999:http://www.msi.umn.edu/~xiangmin/paper/hhc97/node6.html.
63. Aramanovich IG, Levin VI. Equations ofThermal Conductivity. In: Equations ofMathematical Physics, 2nd edn. Moscow:Nauka, 1969: 145–216. [in Russian]
64. Nave CR. HyperPhysics. 2005: http://hyperphysics.phy-str.gsu.edu/hbase/hph.html.
65. Morris GJ. Rapidly cooled human sperm: noevidence of intracellular ice formation. HumReprod 2006; 21: 2075–83.
VITRIFICATION IN ASSISTED REPRODUCTION
32
01b Tucker 8029.qxd 8/23/2007 2:10 PM Page 32

33
Disadvantages and benefitsof vitrificationGábor Vajta, Masashige Kuwayama and Pierre Vanderzwalmen
2
CURRENT PROBLEMS,SHORTCOMINGS, AND POTENTIALPROBLEMS WITH VITRIFICATIONMETHODS
All cryopreservation methods expose mam-malian tissues and cells to an environmentthat they would not normally experience andhave no intrinsic genetically coded capacity tosurvive. The art of the work is to establish asituation where the injuries are minimal anddefensive-regenerative capacities are sup-ported. The most important known mecha-nisms of damage that occur during coolingto low subzero temperatures (without anyexternal intervention to increase chances ofsurvival) include chilling injury, ice crystalformation, and fracture damage.
Chilling occurs between around +15 and−5°C, induces at least partially irreversiblechanges in certain structures including lipiddroplets, lipid-rich membranes, and micro-tubuli of the mitotic or meiotic spindle.1–4
Oocytes and embryos from different speciesand developmental phases contain differentamounts of lipids that may partially explaintheir different sensitivities to chilling injury.According to our present knowledge, thereare very limited possibilities to avoid thisdamage including the mechanical or chemi-cal removal of lipid droplets from the cyto-plasm,5–11 or to pass oocytes and embryosextremely quickly through the critical tem-perature zone both at cooling and warming.
Ice crystal formation both in the mediumdirectly surrounding the cells, and inside the
cells, in the cytoplasm and nucleus is regardedas the major source of injury. The phenome-non may occur between −5 and −80°C (pre-dominantly between −5 and −40°C) and hasdeleterious, mostly mechanical effects on allstructures. Both major groups of successfulcryopreservation strategies try to minimizethese effects. The main elements are thesame, using cryoprotectant solutions andcontrolling the rate of temperature changes.Most important (but not the only) effect ofcryoprotectants is to minimize ice crystal for-mation by either removing water from thecytoplasm with osmotic effect (non-perme-able cryoprotectants), or interfering with icenucleation and growth inside the cytoplasmor in the extracellular space closely surround-ing the cells (extracellular cryoprotectants).Unfortunately the use of cryoprotectantsintroduces a new source of injuries into theprocess. None of the known cryoprotectants isentirely harmless. The permeable ones mayhave a considerable toxic effect, while thenon-permeable cryoprotectants may induceosmotic damage. The borders are, however,not sharply defined and most cryoprotectantsresult in both toxic and osmotic injury.Toxicity is usually proportional to the con-centration of the substance and to the time ofexposure and can be decreased by loweringthe temperature, while the osmotic effect inthe solution phase depends mostly on theconcentration, and also partially on the timeof exposure.
Between −50 and −150ºC, the mechanicaleffect of the solidified and occasionally broken
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 33

solution may cause fracture damage, especiallyin relatively large biological objects such asoocytes and embryos.12 By applying cryopre-servation methods developed in the past 30years, this type of damage may occur moreoften during the warming procedures, and maybe minimized by decreasing slightly the rate oftemperature changes in this temperature zone,although this approach is rather empirical andis without clear scientific explanation. It is quiteunlikely that zona fracture could occur as asimple consequence of osmotic stress assuggested by Smith and Silva.13
Storage at −196°C is probably the leastharmful part of the cryopreservation process,although accidental warming may destroy thestored samples. The effect of the background(cosmic) radiation is probably less harmfulthan previously supposed.14 On the otherhand, an existing – although frequentlyoverestimated – danger is the possibility ofliquid nitrogen-mediated cross contaminationof samples.
At warming, the same types of injuries mayoccur as at cooling, obviously in a reverse order.
Apart from these processes, there are somepartially understood injuries including damageof intracellular organelles, cytoskeleton, andcell-to-cell contacts.15–17
For successful cryopreservation, anattempt should be made to establish anoptimal balance, i.e. to keep the toxic andosmotic injury low while profiting maximallyfrom the ice preventing effects of thecryoprotectants. In spite of the considerableefforts in the past century including analysesof physical, chemical, and biological factors,and the establishment of mathematicalmodels and introduction of sophisticatedequipment, only limited success has beenachieved. In general, the size of the biologicalobject is critical: the smaller is the sample,the better are the chances of survival. Virusesand bacteria can be cryopreserved withrelatively simple methods, and good survivalrates have been achieved with masses ofsingle cells, for example, suspensions of
trypsinized monolayer cultures of somemammalian cells. From this point of view,embryology is in a privileged situation, as theearly phase of development offers apossibility to stop the biological time of life inmammals. The size, however, matters evenin the case of relatively small objects, forexample, oocytes, zygotes, and early stageembryos are generally more sensitive toinjuries than blastocysts that consist of cellswith a size comparable with a tissue culturemonolayer. However, shortly after hatching,with the increasing overall size and complexity,the chances of successful cryopreservationdecrease radically.
During the past decades, from the variousapproaches, two major strategies haveemerged that may fulfill the requirements forsuccessful cryopreservation of mammalianoocytes and embryos.
Chronologically the first was the strictlycontrolled, slow rate freezing,18–24 where theinitial toxic and osmotic injury is minimal, andthe induced ice crystal formation (seeding)results in a slow, stepwise concentration of thecryoprotectants around and in the cytoplasm.This increasing concentration occurs inparallel with the decreasing temperatures andcan cause relatively low levels of harm to thecells. When the temperature drops to the levelthat induces solidification in the wholesolution (around −30 or −40°C), solidificationoccurs with minimal or no ice formationin and around the cells, i.e. in themicroenvironment and in the cytoplasm thedominant phenomenon is the extremelyincreased viscosity, i.e. vitrification.
The other strategy is much more radical: itis an approach to avoid ice crystal formationin the entire solution containing the embryos.It is usually achieved with a drastic elevationof cryoprotectant concentration, and anincreased cooling rate. Basically, this latterapproach seems to be more straightforward,as it eliminates totally one source of injury;however, it exposes cells to a considerablyelevated toxic and osmotic effect. Both
VITRIFICATION IN ASSISTED REPRODUCTION
34
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 34

strategies have disadvantages and limitations.The subject of this chapter is to analyze thoserelated to vitrification.
ELIMINATION OF ICE FORMATIONAND THE REQUIRED COST
When we talk about the benefits of vitrifica-tion, the first and unquestionable one is thetotal elimination of the major source ofinjuries at cryopreservation, that is ice crystalformation. However, to ensure this benefit, wehave to establish technically difficult andpotentially harmful conditions. To achievevitrification of solutions a radical increase ofboth the cooling rates and the concentrationof cryoprotectants is required. The higher thecooling rate, the lower is the required cryopro-tectant concentration, and vice versa. However,none of the extremes can be used under prac-tical conditions. Although with the use ofdrastically high cryoprotectant concentrationsvitrification may be achieved, with relativelymoderate rates of temperature change, thisapproach would expose the samples to unrea-sonable toxic and osmotic injury. On the otherhand, although vitrification can also beachieved in pure water with a cooling rate ofapproximately 107°C/s,25 this theoretical limitcannot even be approached with the usualtechnology of an embryology laboratory. Theart of establishment of an efficient vitrificationmethod is to find a balance, i.e. to keep thetoxic-osmotic injury as low as possible whileproviding safely the required speed of coolingand warming.
There are several approaches to keep toxicand osmotic injury low, including (evidently)the application of cryoprotectants with lowtoxicity and high permeability (from thispoint of view ethylene glycol or propyleneglycol is almost indispensable for successfulvitrification); or to use two or more cryopro-tectants to decrease the specific toxicity ofeach (provided there is no synergistic toxiceffect between the different cryoprotectants);to use both permeable and non-permeable
cryoprotectants in a mixture; to add cryopro-tectants stepwise, in an increasing concentra-tion to the solutions; and to decrease thetemperature when the oocytes and embryosare exposed to the concentrated, final vitrifi-cation solution.25–28 Except for the latterstrategy (that may be beneficial in severalsituations, but may also increase the risk ofchilling injury), almost all listed approachesare now indispensable parts of a successfulvitrification method.
However, even the best cryoprotectantsand the most sophisticated strategies in com-bining and providing them may be insuffi-cient if the final required concentration ishigh. As mentioned above, according to ourpresent knowledge the only practicalapproach to keep this concentration at a levelthat is tolerated by oocytes and embryos is toincrease the cooling and warming rates.Traditional tools of cryopreservation areinsufficient for this purpose, as for examplethe maximum achievable cooling rate withdirect plunging of a standard 0.25 mLinsemination straw into liquid nitrogen is2500°C/min29 due to the relatively largeamount of solution (> 5 µL) required to forma stable column in the straw without dangerof dispersion during the pressure changescaused by cooling and warming, and also dueto the relatively thick plastic wall of the sealedstraw that presents a considerable thermo-insulating layer. On the other hand, even thisrelatively low rate was still hazardous to per-form as direct immersion into liquid nitrogenat cooling, and transfer to a water bath atwarming induced extreme pressure changesin the closed system, and frequently led to thecollapse or explosion of the straws and loss ofthe sample. Obviously, the achievable coolingrate is even lower in cryovials. Interestingly,however, for a relatively long period of timescientists have respected the limits deter-mined by these old tools. Even in 2005 and2006, there are many publications dealingwith vitrification experiments performed in0.25 mL straws, i.e. disregarding completely
35
DISADVANTAGES AND BENEFITS OF VITRIFICATION
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 35

the wide range of possibilities to overcomethe above-mentioned limitations.
The main principles of these innovativeapproaches are extremely simple and under-standable at an elementary level. The twoclear possibilities to increase cooling andwarming rates are to decrease the volume ofthe solution, and to minimize or completelyeliminate the thermoinsulating layer thatseparates the solution containing the samplefrom the cooling substance, in most casesliquid nitrogen. Apart from the high coolingrate, the small solution volume offers anotherbenefit as it decreases the chances of hetero-geneous ice formation,25 and provides theopportunity to decrease further the cryopro-tectant concentration required for safe andcomplete vitrification of the sample.
The most logical way to achieve these goalsseems to be to drop the solution directly intothe liquid nitrogen.30–34 However, the actualresult may not be as good as the idea itself. Toform a drop, a relatively large amount of solu-tion (approximately 5 µL) is required; accord-ingly the rate of cooling is limited. Moreover,the warm drop induces an extensive evapora-tion on the surface of the liquid nitrogen, andthe vapor coat not only isolates the samplefrom the rest of the liquid nitrogen but keepsit floating on the surface for a relatively longperiod of time (4–8 s) before sinking andreaching the final −196°C temperature;accordingly the cooling rate is far from whatis optimal. Finally, based on successfulattempts in Drosophila melanogaster embryo cry-opreservation35 copper grids manufactured tohold the ultrathin sections at electron micro-scopic investigations were used as carriertools to vitrify bovine oocytes.36–38 Themethod has some benefits that are almostunsurpassable: the solution surrounding thesample can be minimized by touching a filterpaper before submerging into liquid nitro-gen, only a thin, film-like layer remains thatprotects the sample from drying. Accordingly,the cooling and warming rate is close to thetheoretical maximum achievable with direct
immersion into liquid nitrogen. Moreover,the oocytes and embryos remain fixed safelyto the surface of the grid during the wholecryopreservation procedure, but are sepa-rated immediately at warming, when thegrid is simply immersed into the warmingsolution.
Unfortunately, there are some drawbacksof this approach, as well. Electron micro-scopic grids are tiny little tools, and requiredelicate handling during cooling, warming,and especially storage. Although a possiblesolution for the latter problem has been pub-lished by placing vitrified grids into cryovialsfilled with liquid nitrogen,39 this applicationis strongly opposed by producers of allcryovials because of the potential danger ofexplosion during accidental or intentionalwarming. Additionally, the direct contactbetween liquid nitrogen and the solution con-taining the sample may raise some concernsregarding the possibility of disease transmis-sion. As this problem is common in almostall recent vitrification techniques, it will bediscussed in a separate section below.
As a result of these problems, the use ofthe copper grids as carrier tools has beenmostly restricted to experimental purposes,and new tools had to be developed for prac-tical application. These tools are discussed indetail in other chapters of this book, so herewe provide only a short and incomplete listbeginning with the open pulled straw (OPS)technique40 and continuing with all its latersubclones: glass micropipettes, GMP;41
super-finely pulled OPS, SOPS;42 gel-loadingtips;43 sterile stripper tip;44 flexipet denudingpipettes, FDP;45 fine diameter plasticmicropipettes;46 100 µL pipetting tip;47 etc.The principles in all these methods were thesame: a narrow, thin walled plastic capillarythat is usually filled with a tiny (< 1 µL)amount of solution containing the sample byusing the capillary effect, and direct immer-sion of the tool into liquid nitrogen. Theachievable cooling and warming rates withthese tools may be as high as 20 000°C/min.
VITRIFICATION IN ASSISTED REPRODUCTION
36
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 36

This is almost an order of magnitudehigher than the limits of the standard 0.25 mLinsemination straw, but still considerably lowerthan that obtained with other advanced vitrifi-cation methods. Other versions of the originalOPS idea including the closed pulled straws(CPS),48 sealed open pulled straws,49 and themore professional and sophisticated form, theCryotip system50 hermetically isolated thesample from the liquid nitrogen, consequentlyeliminating the danger of cross contamina-tion. However, this modification does result ina decrease in the cooling and warming rates,and limits the efficiency of the technique incases of certain sensitive biological materialssuch as human oocytes.
The main benefit of application of thesenarrow tubes is the easy manipulation.Loading is performed by either using the cap-illary effect, or attaching the tool to a small vol-ume automatic pipette. The immersion intothe liquid nitrogen should be performed with
a rapid movement that also continues underthe level to remove the vapor coat that formsaround the tube, which may slow down thecooling rate. At warming, in open systemsthe tube should be simply immersed into theappropriate warming solution containing anosmotic buffer to counterbalance the effect ofthe cryoprotectants accumulated in the cyto-plasm. The vitrified solution becomes liquidalmost immediately after immersion, and atthe same moment, as the result of the capillaryeffect, the warming solution enters the strawand dilutes the concentrated cryoprotectants.Eventually, as a result of gravity, oocytes andembryos slowly float out of the straw andcan be subjected to stepwise dilution of boththe cryoprotectant and the osmotic buffer ofthe warming medium. Closed tubes are usuallywarmed in a water bath, then the end of thetube is opened with scissors or a blade, andfrom this point the dilution process is almostidentical to that of the open systems; however,
37
DISADVANTAGES AND BENEFITS OF VITRIFICATION
Table 2.1 Examples in mammalian embryology where the first success with cryopreservation was achieved by vitrifi-cation. Embryos and oocytes were not treated mechanically or chemically to prepare them for the vitrification. Fullterm developments were reported except where otherwise indicated.
Species, stage, system Reference
Bovine immature oocytes for IVF Vieria et al., 200280
Bovine in vitro matured oocytes for IVF Martino et al., 1996;87 Vajta et al., 1998a82
Bovine in vitro matured oocytes for somatic cell nuclear transfer Hou et al., 200583
Bovine cytoplasts for embryonic cell nuclear transfer Booth et al., 199984
Bovine early stage IVF embryos Vajta et al., 1998a, in vitro study40
Bovine zona-included blastocysts generated French et al., 200285
by somatic cell nuclear transfer Bovine zona-free blastocysts generated Tecirlioglu et al., 200486
by somatic cell nuclear transfer Bovine transgenic blastocysts generated French et al., 200387
by somatic cell nuclear transfer Ovine zona included embryos generated by nuclear transfer Peura et al., 200388
Porcine immature oocytes for ICSI Fujihira et al., 2004, in vitro study89
Porcine in vitro matured oocytes for ICSI Fujihira et al., 2005, in vitro study90
Porcine in vivo derived blastocysts Kobayashi et al., 199891
Porcine in vivo derived morulae Berthelot et al., 200191
Porcine in vitro produced blastocysts Men et al., 2005, in vitro study92
Equine in vivo matured oocytes Maclellan et al., 200293
European polecat in vivo derived morulae and blastocysts Piltty et al., 200494
Siberian Tiger in vivo derived embryos Crichton et al., 2003, in vitro study95
Minke whale immature oocytes for maturation Iwayama et al., 2004, in vitro study96
ICSI, intracytoplasmic sperm injection.
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 37

the required manipulation and the lackof the direct contact considerably slows downthe warming rate and the dilution of thecryoprotectants.
A third practical approach to increase thecooling and warming rates is the Cryolooptechnique,51–54 a small loop formed from athin nylon fiber. At vitrification, a thin solu-tion film bridging the hole of the loop isformed, and the oocytes and embryos areloaded onto this film. Although the systemseems to be very fragile and sensitive, it hasbeen widely acknowledged and used inhuman embryology, and its application seemsto be easy and practical. The film remainsintact during immersion into liquid nitrogen,the solution volume is negligible, accordinglythe cooling and warming rate may reach theestimated level of 700 000°C/min,55 and thestorage may be performed in cryovials.According to the latest data, cooling may alsobe performed in the vapor of liquid nitrogenwithout compromising results.56 The disad-vantages of this technique are that the tool isunusual, and the method seems to be techni-cally difficult to perform, although those whohave routinely tried it praise its simplicity andapplicability.
Apart from these approaches a fourth groupof methods has also been developed stepwiseduring the past 15 years using a commonprinciple, but entirely different tools. Firstdescribed by Arav57 as minimum drop size(MDS) technique, a very small (< 0.5 or even0.1 µL) droplet containing the sample is placedonto a solid surface and immersed into theliquid nitrogen. Depending on the tool ontowhich the drop is placed, different vitrificationmethods have been developed including theminimum volume cooling (MVC),58 the hemi-straw system,59 or the latest and probablymost practical approach, the Cryotop tech-nique.60,61 Loading, cooling, warming, anddilution procedures are quite similar to thoseapplied with the electron microscopic grids,but instead of touching a filter paper, theexcess solution is removed with a capillary.
Furthermore, these newer tools were developedfor the given purpose, accordingly their han-dling and storage is easier and requires lesstechnical skill than the grid approach; there-fore standardization and widespread practicalapplication is possible. The achievable coolingand warming rates are very difficult to measure,however, according to our estimation theyshould be between those achievable with theOPS analogs and the Cryoloop, i.e. between20 000 and 700 000°C/min.
Apart from the small volume and/or directcontact approach, there are some otherattempts to increase the cooling rates. A logi-cal approach is the elimination of the vaporcoat that arises around the sample in the liquidnitrogen at cooling, for example by usingliquid nitrogen slush instead of liquid nitrogenfor cooling (VitMaster),62–64 or to place smalldrops on pre-cooled metal surfaces instead ofliquid nitrogen for cooling. Originally, a metalblock immersed into liquid nitrogen wasused,65 but eventually a commercially availabletechnique has also been produced (CMV,Cryologic, Australia). The few comparativedata do not provide entirely convincing evi-dence regarding the superiority of thesevapor-minimizing or vapor-free approachescompared with the other vitrification proce-dures, although the use of metal surfacesinstead of liquid nitrogen for cooling maydecrease potential contamination problems(see below).
When talking about advantages and draw-backs of vitrification, we also have to mentionthat the broad choice of solutions, tools, andtechniques is actually a considerable disad-vantage that hampers the widespread appli-cation of vitrification. A routine practitionerboth in domestic animals and especially inthe human field needs a highly standardized,commercially available technology, a kit withready to use solutions and tools, and also abroad independent reference regarding thechances of successes and failures. In contrastto traditional freezing, very few (if any) ofthe current vitrification methods offer this
VITRIFICATION IN ASSISTED REPRODUCTION
38
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 38

option, the choices are abundant, but theavailability in most cases is difficult. Kits (ifthey exist) are incomplete, and most of theavailable data originate from the (potentiallycommercially interested) creators of themethod. Accordingly, this confusing situationdiscourages practitioners from entering thisnew field. Very recently, there have been signsof more professional handling of the matterby some commercial ventures: if this trendcontinues, it may be a decisive factor in thecommon acceptance of vitrification, to find itswell deserved place among cryopreservationtechniques in reproductive biology.
DECREASED CHILLING INJURY
The other unquestionable benefit of vitrifica-tion is the possibility to decrease dramaticallychilling injury. Compared with slow-rate freez-ing, the rate samples pass through the danger-ous temperature zones at vitrification areextremely high, and the very short exposure todangerous temperatures (15 to −5°C) radicallydecreases the injury of the sensitive structures.Accordingly, the only successful strategy that iscurrently available for cryopreservation ofintact porcine embryos (containing extremelyhigh amounts of chill-sensitive lipid droplets)is vitrification.66,67
THE POTENTIAL DANGER OFDISEASE TRANSMISSION
According to our knowledge, no infectionrelated to liquid nitrogen-mediated transmis-sion of infective agents has been describedafter application of embryo technologiesincluding all forms of cryopreservation, tradi-tional slow freezing, or vitrification, althoughthese techniques are applied in huge numbersworldwide in both humans and domestic andexperimental animals. However, the mostemphasized argument against the use of thenew and efficient vitrification techniques is thepotential risk of liquid nitrogen-mediated dis-ease transmission. The subject was discussed
in detail in a recent review,68 and here we sum-marize the arguments and facts to give anauthentic picture about the real risks.
The potential of infection in current repro-ductive techniques is inherent, as semen andembryo collection protocols are not sterile pro-cedures.69 Consequently, the contents of virtu-ally all stored straws and cryovials may besources of infection when transferred to recip-ients. In case an infection occurs in the future,it may be difficult to localize the source aseither infection before the cryopreservation orcross-contamination during storage.
On the other hand, in embryology practicethere are many other sources for contaminat-ing liquid nitrogen. In the everyday work, thesurface of straws, cryovials, racks, and othertools are not handled fully aseptically.According to our knowledge, the systematicand regular cleaning of containers and sam-ples in liquid nitrogen containers is not partof routine in any embryology laboratory, andit seems to be technically very demanding,maybe even impossible. Moreover, seeminglysterile containers may not be as safe as sup-posed, infection may occur through incom-plete sealing, pores of the plastic walls ofmost commonly used straws (except for somespecial ones produced to eliminate this prob-lem). As the result, all storage tanks maycontain a number of potentially pathogenicenvironmental microorganisms.69
Liquid nitrogen-mediated disease transmis-sion in other areas of human medicine or foodindustry is a documented fact, although a veryrare event.70–72 Also, the potential for transferof infectious agents between open tools usedfor vitrification has been proven under experi-mental conditions.73 Accordingly, the theoreti-cal danger of liquid nitrogen-mediated diseasetransfer exists, although it is not restricted toopen vitrification methods, as most traditionaltools and methods of cryopreservation (proba-bly at a lower level) may also be vulnerable.
However, it should be noted that the fewpublished disease transmissions happenedbetween blood specimens and carcasses,
39
DISADVANTAGES AND BENEFITS OF VITRIFICATION
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 39

between volumes approximately 103 to 104
larger than samples in embryology, as a resultof huge leaks of wrapping or when no wrap-ping was present at all. The fact that infectionfollowing embryo transfer (with or without cry-opreservation) is a rare event may also provethat the oviduct and uterus have an appropri-ate defense system to eliminate infectiousagents in the quantities in which they may betransferred during routine embryo transfer.
Both human and animal medicine usesmethods that may result in infections indifferent ways. Our duty is to eliminate allpossible sources of infections, but there aresome rational limitations to this approach.For example, nobody states that the facemasks of surgeons eliminate 100% of theexpired bacteria and viruses; however,according to the routine experience the risk isminimal, and reasonable compromises aremade, e.g. astronaut’s ‘scaphanders’ (space-suits) are fortunately rarely used by surgeonsperforming routine surgeries.
On the other hand, our duty is to usesystems that are resistant even to theoreticalhazards. In the past few years, a considerableeffort has been performed in this direction,and as the result of the advancements many vit-rification techniques now offer equally high oreven a higher level of aseptic handling of sam-ples than routine slow freezing procedures.
One approach is to separate the coolingand warming phase from storage: to use a rel-atively low amount of clean liquid nitrogenfor cooling, then to wrap the sample into aprecooled sterile container and seal it her-metically before placing it into the commonstorage tanks. Liquid nitrogen that is low orfree of infective agents can be obtained fromsome producers, or can also be obtained byUV irradiation,62 or by filtration throughsome 0.2 µm filters.74 Although the lattermanipulation does not remove viral particlesfrom the solution, the same is also applicableto most filter sterilized media. The probabil-ity that factory-derived, separately storedliquid nitrogen will contain pathogenic
viruses is extremely low. The effect of UVsterilization may depend on the actual situa-tion (volume, thickness, intensity, wave-length, and duration of irradiation) and canonly be regarded as reliable under standard-ized and controlled conditions.
The choice of storage container depends onthe vitrification tool. For the OPS analogs, aswell as for the Cryotop and some other tools,the best solution is probably to use large (0.5 or1 mL) plastic straws produced from materialimpermeable to any pathogenic agents.45,74–76
For safe loading, commercial kits are availableincluding the VitSet produced by Minitube.The safety of the OPS-vitset technique has beenrecently confirmed by an independent investi-gation.77 Cryoloop is equipped with a specialcryovial, and other tools including electronmicroscope grids can also be stored in commoncryovials, but for safe application, producers’suggestions should be strictly followed. Thesame is applicable for approaches where thecooling is performed on metal surfaces (SSFtechnique), or by dropping the sample into theliquid nitrogen.
The application of a sterile container forstorage of vitrified samples may offer anotherbenefit. Compared with the samples frozenby using the slow rate method, or vitrified in0.25 mL insemination straws, the small size ofsamples used in recent vitrification methodsmakes them extremely fragile to temporarywarming including transfer of the samplefrom one container to other. The wrappingapplied for sterility measures may also serveas a buffer to avoid accidental damage causedby such transitional warmings.
Based on an earlier description,77 recentlya new combination of OPS vitrification anddouble wrapping was applied by Isachenko etal.78 (discussed in detail in another chapter ofthis book).
Very recently, an aseptic vitrificationapproach based on the hemi-straw principlehas also been developed. This High SecurityVitrification kit (HSV kit)79 makes it possibleto place a microdroplet (< 0.5 µL) of
VITRIFICATION IN ASSISTED REPRODUCTION
40
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 40

cryoprotectant containing the embryos in thegutter of a capillary before inserting it in amini-straw. It is heat-sealed using a specialwelder which ensures a leak-proof seal and isresistant to pressures of up to 150 kg/cm2
before immersion in the liquid nitrogen.Future experiments are required to prove ifthe achievable cooling rates in the two lattersystems are appropriate for cryopreservationof chill-sensitive structures.
41
DISADVANTAGES AND BENEFITS OF VITRIFICATION
References
1. Aman RR, Parks JE. Effects of cooling andrewarming of the meiotic spindle and chro-mosomes of in vitro matured bovine oocytes.Biol Reprod 1994; 50: 103–10.
2. Leibo SP, Martino A, Kobayashi S, PollardJW. Stage-dependent sensitivity of oocytesand embryos to low temperatures. AnimReprod Sci 1996; 42: 45–53.
3. Martino A, Pollard JA, Leibo SP. Effect of chill-ing bovine oocytes on their developmental com-petence. Mol Reprod Dev 1996; 45: 503–12.
4. Zenzes MT, Bielecki R, Casper RF et al. Effectsof chilling to 0°C on the morphology of mei-otic spindles in human metaphase II oocytes.Fertility and Sterility 2001; 75: 769–77.
5. Nagashima H, Kashiwazaki N, Ashman RJet al. Removal of cytoplasmic lipid enhancesthe tolerance of porcine embryos to chilling.Biol Reprod 1994; 51: 618–22.
6. Nagashima H, Cameron R, Kuwayama M.Survival of porcine delipated oocytes andembryos after cryopreservation by freezingor vitrification. J Reprod Dev 1999; 45:167–76.
7. Dobrinsky JR, Nagashima H, Pursel VGet al. Cryopreservation of swine embryoswith reduced lipid content. Theriogenology1999; 51: 164.
8. Beebe LFS, Cameron RDA, Blackshaw AWet al. Piglets born from centrifuged and vitri-fied early and peri-hatching blastocysts.Theriogenology 2002; 57: 2155–65.
9. Esaki R, Ueda H, Kurome M et al.Cryopreservation of porcine embryos derivedfrom in vitro-matured oocytes. Biol Reprod2004; 71: 432–7.
10. Du Y, Kragh PM, Zhang X et al. Sucessful vitri-fication of parthenogenetic porcine blastocystsproduced from delipated in vitro maturedoocytes. Reprod Fertil Dev 2006; 18: 153.
11. Men H, Agca Y Riley LK, Critser JKImproved survival of vitrified porcine
embryos after partial delipation throughchemically stimulated lipolysis and inhibi-tion to apoptosis Theriogenology 2006;66(8) 2008–16.
12. Rall WF, Meyer TK. Zona fracture damageand its avoidance during the cryopreservationof mammalian embryos. Theriogenology1989; 31: 683–92.
13. Smith GD, Silva E, Silva CA. Developmentalconsequences of cryopreservation of mam-malian oocytes and embryos. ReprodBioMed Online 2004; 9: 171–78.
14. Rall WF. Cryopreservation of mammalianembryos, gametes and ovarian tissues.Current issues and progress. In: Wolf DP,Zelinski-Wooten M, eds. Assisted Fertilizationand Nuclear Transfer in Mammals. Totowa,NJ: Humana Press, 2001: 173–87.
15. Vincent C, Johnson JH. Cooling cryoprotec-tants and the cytoskeleton of the mammalianoocyte. Oxf Rev Reprod Biol 1992; 14:73–100.
16. Massip A, Mermillod P, Dinnyés A. Morphologyand biochemistry of in-vitro produced bovineembryos: implications for their cryopreserva-tion. Hum Reprod 1995; 10: 3004–11.
17. Dobrinsky JR. Cellular approach to cryo-preservation of embryos. Theriogenology1996; 45: 17–26.
18. Whittingham DG, Leibo SP, Mazur P.Survival of mouse embryos frozen to −196ºCand −269ºC. Science 1972; 178: 411–14.
19. Wilmut I. The effect of cooling rate, warmingrate, cryoprotective agent and stage of devel-opment on survival of mouse embryos duringfreezing and thawing. Life Sci 1972; 11:1071–79.
20. Wilmut I, Rowson LEA. Experiments on thelow temperature preservation of cowembryos. Vet Rec 1973; 92: 686–690.
21. Bank H, Maurer RR. Survival of frozen rabbitembryos. Exp Cell Res 1974; 89: 188–196.
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 41

22. Willadsen SM, Polge C, Rowson LE et al.Deep freezing of sheep embryos. J ReprodFertil 1976; 46: 151–54.
23. Trounson A, Mohr L. Human pregnancy fol-lowing cryopreservation, thawing, and trans-fer of an eight-cell embryo. Nature 1983;305: 707–9.
24. Zeilmaker G, Alberda A, van Gent I et al.Two pregnancies following transfer of intactfrozen-thawed embryos. Fertility andSterility 1984; 42: 293–96.
25. Rall WF. Factors affecting the survival ofmouse embryos cryopreserved by vitrifica-tion. Cryobiology 1987; 24: 387–402.
26. Vanderzwalmen P, Touati K, Ectors FJet al. Vitrification of bovine blastocysts.Theriogenology 1989; 31: 270.
27. Saha S, Takagi M, Boediono A et al. Direct rehy-dration of in vitro fertilised bovine embryosafter vitrification. Vet Rec 1994; 134: 276–77.
28. Széll A, Shelton NJ. Survival of vitrifiedsheep embryos in vitro and in vivo.Theriogenology 1994; 42: 881–9.
29. Palasz AT, Mapletoft RJ. Cryopreservation ofmammalian embryos and oocytes: recentadvances. Biotechnol Adv 1996; 14: 127–49.
30. Landa V, Tepla O. Cryopreservation ofmouse 8-cell embryos in microdrops. FoliaBiologica (Praha) 1990; 36: 153–8.
31. Riha J, Landa V, Kneissl J et al. Vitrification ofcattle embryos by direct dropping into liquidnitrogen and embryo survival after nonsurgi-cal transfer. Zivoc Viroba 1994; 36: 113–20.
32. Yang BS, Leibo SP. Viability of in vitro-derived bovine zygotes cryopreserved inmicrodrops. Theriogenology 1999; 51: 178.
33. Papis K, Shimizu M, Izaike Y. The effect ofgentle pre-equilibration on survival anddevelopment rates of bovine in vitro maturedoocytes vitrified in droplets. Theriogenology1999; 51: 173.
34. Papis K, Shimizu M, Izaike Y. Factors affect-ing the survivability of bovine oocytes vitri-fied in droplets. Theriogenology 2000; 54:651–58.
35. Steponkus PL, Myers SP, Lynch DV et al.Cryopreservation of Drosophila melanogasterembryos. Nature 1990; 10: 345, 170–2.
36. Martino A, Songsasen N, Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
37. Choi DH, Chung HM, Lim JM et al. Pregnancyand delivery of healthy infants developed fromvitrified blastocysts in an IVF-ET program.Fertil and Steril 2000; 74: 838–39.
38. Cho HJ, Son WY, Yoon SH et al. Animproved protocol for dilution of cryopro-tectants from vitrified human blastocysts.Hum Reprod 2002; 17: 2419–22.
39. Son WY, Lee SY, Chang MJ et al. Pregnancyresulting from transfer of repeat vitrifiedblastocysts produced by in vitro maturedoocytes in patient with polycystic ovary syn-drome. Reprod BioMed Online 10, 398–401.
40. Vajta G, Holm P, Kuwayama M, Booth PJ et al.Open Pulled Straw (OPS) vitrification: a newway to reduce cryoinjuries of bouine ova andembryos. Mol Reprod Dev 1998; 51(1) 53–8.
41. Kong IK, Lee SI, Cho SG et al. Comparisonof open pulled straw (OPS) vs glassmicropipette (GMP) vitrification in mouseblastocysts. Theriogenology 2000; 53:1817–26.
42. Isachenko V, Alabart JL, Vajta G et al.Double cryopreservation of rat embryos atdifferent developmental stages with identi-cal vitrification protocol: the not properlyunderstood phenomenon. In: Abstracts ofthe Winter Meeting of Society for the Studyof Fertility. Utrecht, Holland. J ReprodFertility 2000; 26 (Abstract Series): 10.
43. Tominaga K, Hamada Y. Gel-loading tips ascontainer for vitrification of in vitro-pro-duced bovine embryos. J Reprod Dev 2001;47: 259–65.
44. Kuleshova LL, Lopata A. Vitrification can bemore favorable than slow cooling. FertilSteril 2002; 78: 449–54.
45. Liebermann J, Tucker MJ, Graham JR et al.Blastocyst development after vitrification ofmultipronuclear zygotes using the Flexipetdenuding pipette. Reprod BioMed Online2002; 4: 146–50.
46. Cremades N, Sousa M, Silva J. Experimentalvitrification of human compacted morulae andearly blastocysts using fine diameter plasticmicropipettes. Hum Reprod 2004; 19: 300–5.
47. Hredzak R, Ostro A, Zdilova V. Clinical expe-rience with a modified method of humanembryo vitrification Ceska Gynekologica2005; 70: 99–103. [in Slovakian]
48. Chen SU, Lien YR, Cheng YY et al.Vitrification of mouse oocytes using closedpulled straws (CPS) achieves a high survivaland preserves good patterns of meioticspindles, compared with conventional straws,open pulled straws (OPS) and grids. HumReprod 2001; 11: 2350–56.
49. Lopez-Bejar M, Lopez-Gatius F.Nonequilibrium cryopreservation of rabbitembryos using a modified (sealed) open
VITRIFICATION IN ASSISTED REPRODUCTION
42
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 42

pulled straw procedure. Theriogenology2002; 58: 1541–52.
50. Kuwayama M, Vajta G, Ieda S, Kato O.Vitrification of human embryos using theCryoTip™ method. Reprod BioMed Online2005; 11: 608–14.
51. Lane M, Forest KT, Lyons EA et al. Livebirths following vitrification of hamsterembryos using a novel containerless tech-nique. Theriogenology 1999; 51: 167.
52. Lane M, Schoolcraft WB, Gardner DK.Vitrification of mouse and human blastocystsusing a novel cryoloop container-less tech-nique. Fertil Steril 1999; 72: 1073–78.
53. Mukaida T, Nakamura S, Tomiyama T et al.Successful birth after transfer of vitrified humanblastocysts with use of a cryoloop containerlesstechnique. Fertil Steril 2001; 76: 618–20.
54. Mukaida T, Takahashi K, Kasai M. Blastocystcryopreservation: ultrarapid vitrificationusing cryoloop technique. Reprod BioMedOnline 2003; 6: 221–25.
55. Isachenko E, Isachenko V, Katkov II et al.Vitrification of mammalian spermatozoa inthe absence of cryoprotectants: from pastpartial difficulties to present success. ReprodBioMed Online 2003; 6: 191–200.
56. Larman MG, Sheenan CB, Gardner D.Vitrification of mouse pronuclear oocyteswith no direct liquid nitrogen contact.Reprod Biomed Online 2006; 12: 66–69.
57. Arav A. Vitrification of oocytes and embryos.In: Lauria A, Gandolfi F, eds. New Trends inEmbryo Transfer. Cambridge, UK: PortlandPress, 1992: 255–64.
58. Hamawaki A, Kuwayama M, Hamano S.Minimum volume cooling method for bovineblastocyst vitrification. Theriogenology 1999;51: 165.
59. Vanderzwalmen P, Bertin G, Debauche Vet al. In vitro survival of metaphase IIoocytes (MII) and blastocysts after vitrifica-tion in an hemi-straw (HS) system. FertilSteril 2000; 74: S215–16.
60. Kuwayama M, Kato O. All-round vitrificationmethod for human oocytes and embryos. JAssist Reprod Genet 2000; 17: 477.
61. Kuwayama M, Vajta G, Kato O et al. Highlyefficient vitrification method for cryopreser-vation of human oocytes. Reprod BioMedOnline 2005; 11: 300–8.
62. Arav A, Zeron Y, Ocheretny A. A new deviceand method for vitrification increasesthe cooling rate and allows successfulcryopreservation of bovine oocytes.Theriogenology 2000; 53: 248.
63. Arav A, Yavin S, Zeron Y et al. New trendsin gamete’s cryopreservation. Mol CellEndocrinol 2002; 187: 77–81.
64. Huang et al., Successful pregnancy followingblastocyst cryopreservation using super-coolingultra-rapid vitrification. Hum Reprod 2005;20(1): 122–8.
65. Dinnyes A, Dai Y, Jiang S et al. High devel-opmental rates of vitrified bovine oocytesfollowing parthenogenetic activation, invitro fertilization, and somatic cell nucleartransfer. Biol of Reprod 2000; 63: 513–8.
66. Berthelot F, Martinat-Botté F, Perreau Cet al. Birth of piglets after OPS vitrificationand transfer of compacted morula stageembryos with intact zona pellucida. ReprodNutr Dev 2001; 41: 267–72.
67. Cuello C, Gil MA, Parrila I et al. In vitro devel-opment following one-step dilution of OPSvitrified porcine blastocysts. Theriogenology2004; 62: 1144–52.
68. Vajta G and Nagy ZP. Are programmablefreezers still needed in the embryo labora-tory? Review on vitrification. Reprod BiomedOnline 2006; 12(6): 779–96.
69. Bielanski A, Bergeron H, Lau PCK et al.Microbial contamination of embryos andsemen during long-term banking in liquidnitrogen. Cryobiology 2003; 46: 146–52.
70. Tedder RS, Zuckerman MA, Goldstone AH, et al.Hepatitis-B transmission from contaminated cry-opreservation tank. Lancet 1995; 346: 137–40.
71. Fountain DM, Ralston M, Higgins N et al.Liquid nitrogen freezers: a potential sourceof microbial contamination of hematopoieticstem cell components. Transfusion 1997; 37:585–91.
72. Berry ED, Dorsa WJ, Siragusa GR et al.Bacterial cross-contamination of meat duringliquid nitrogen immersion freezing. J Food Prot1998; 61: 1103–8.
73. Bielanski A, Nadin-Davis S, Sapp T et al.Viral contamination of embryos cryopre-served in liquid nitrogen. Cryobiology 2000;40: 110–16.
74. Vajta G, Lewis IM, Kuwayama M, Greve T,Callesen H. Sterile application of the OpenPulled Straw (OPS) vitrification method.Cryo Letters 1998b; 19: 389–392.
75. Jelinkova L, Selman HA, Arav A et al. Twinpregnancy after vitrification of 2-pronucleihuman embryos. Fertil and Steril 2002; 77:412–14.
76. Vanderzwalmen P, Bertin G, Debauche Cet al. Vitrification of human blastocysts withthe hemi-straw carrier: application of
43
DISADVANTAGES AND BENEFITS OF VITRIFICATION
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 43

assisted hatching after thawing. HumReprod 2003; 18: 1501–11.
77. Bielanski A. Hanniman A.Non-cross-contamination of bovine embryos withmicrobes using the OPS vitrification system.Reprod Fertil Dev 2007 19 (1): 232–233.
78. Isachenko V, Montag M, Isachenko E et al.Aseptic technology of vitrification of humanpronuclear oocytes using open-pulledstraws. Hum Reprod 2005; 20(2): 492–6.
79. Camus A, Clairaz P, Ersham A et al. (Thecomparison of the process of five differentvitrification devices) Gynecol Obstel Fertil2006; 34(9): 737–45. French.
80. Vieria AD, Mezzalira A, Barieri DP et al.Calves born after open pulled straw vitrifica-tion of immature bovine oocytes. Cryobiology45, 91–94.
81. Martino A, Songsasen N, Leibo SP.Development into blastocysts of bovine oocytescryopreserved by ultra-rapid cooling. Biologyof Reproduction 1996b; 54: 1059–1069.
82. Vajta G, Kuwayama M, Holm P et al. Openpulled straw vitrification; a new way toreduce cryoinjuries of bovine ova andembryos. Molecular Reproduction andDevelopment 1998a; 51: 53–58.
83. Hou YP, Dai YP, Zhu SE et al. Bovine oocytesvitrified by the open pulled straw methodand used for somatic cell cloning supporteddevelopment to term. Theriogenology 2005;64: 1381–91.
84. Booth PJ, Vajta G, HØj A, Holm P,Jacobsen H, Greve T, Callesen H. Full-termdevelopment of nuclear transfer-calves pro-duced from Open Pulled Straw (OPS) vitri-fied cytoplasts. Theriogenology 1999; 51:413.
85. French AJ, Hall VJ, Korfiatis NT et al. Viabilityof cloned bovine embryos following OPS vitri-fication. Theriogenology 2002; 57, 413.
86. Tecirlioglu RT, French AJ, Lewis JM et al.Birth of a cloned calf derived from a vitrifiedcloned embryo. Reproduction, Fertility andDevelopment 2004; 15: 361–366.
87. French AJ, Lewis IM, Ruddock NT et al.Generation of aS1 casein gene transgeniccalves by unclear transfer. Biology ofReproduction 2003; 68: 240.
88. Peura TT, Hartwich KM, Hamilton HM et al.No differences in sheep somatic cell nucleartransfer outcomes using serum starved oractively growing donor granulosa cells.Reproduction, Fertility and Development2003; 15, 157–165.
89. Fujihira T, Kishida R, Fukui Y. Developmentalcapacity of vitrified immature porcine oocytesfollowing ICSI: effects of cytochalasin B andcryoprotectants. Cryobiology 2004; 49,286–290.
90. Fujihira T, Nagai H, Fukui Y. Relationshipbetween equilibration times and the pres-ence of cumulus cells, and effect of Taxoltreatment for vitrification of in vitromatured porcine oocytes. Cryobiology 2005;51(3): 339–43.
91. Kobayashi S, Takei M, Kano M et al. Pigletsproduced by transfer of vitrified porcineembryos after stepwise dilution of cryopro-tectants. Cryobiology 1998: 36: 20–31.
92. Men H, Agca Y, Critser E et al. Beneficialeffects of serum supplementation during invitro production of porcine embryos on theirability to survive cryopreservation by the openpulled straw vitrification. Theriogenology2005; 64, 1340–1349.
93. Maclellan LJ, Carnevale EM, Coutinho daSilva MA et al. Pregnancies from vitrifiedequine oocytes collected from super-stimulated and non-stimulated mares.Theriogenology 2002; 58: 911–919.
94. Piltty K, Lindeberg H, Aalto J et al. Live cubsborn after transfer of OPS vitrified-warmedembryos in the farmed Europen polecat(Mustela putorius). Theriogenology 2004;61: 811–820.
95. Crichton EG, Bedows E, Miller-Lindholm AKet al. Efficacy of porcine gonadotropins forrepeated stimulation of ovarian activity foroocyte retrieval and in vitro embryo produc-tion and cryopreservation in Siberian tigers(Panthera tigris altaica). Biology ofReproduction 2003; 68: 105–113.
96. Iwayama H, Hochi S, Kato M et al. 2004Effects of cryodevice type and donor’s sexualmaturity on vitrification of minke whale(Balaenopter bonaerensis) oocytes at germi-nal vesicle stage. Zygote 12, 333–338.
VITRIFICATION IN ASSISTED REPRODUCTION
44
02 Tucker 8029.qxd 8/23/2007 8:06 PM Page 44

45
Development of vitrification solutionsJaffar Ali and James Shelton
3
INTRODUCTION
The development of the science and technol-ogy of vitrification of embryos and oocytes isdescribed elsewhere in this manual. Theessential requirements include a high concen-tration of cryoprotectant, non-toxicity at thisconcentration, and a rapid rate of cooling.The obvious practical way to obtain a rapidrate of cooling is by plunging into liquidnitrogen and this has been adopted almostuniversally as the cooling method in vitrifica-tion. As researchers began to realize that vit-rification may offer a simplified technique ofembryo and oocyte cryopreservation they uti-lized the cryoprotectants that had proved suc-cessful for conventional preservation by slowcooling. It is clear now that the requirementsof the cryoprotectant for successful vitrifica-tion of embryos are more demanding thanthey are for slow cooling. Practitioners ofconventional cryopreservation, in the main,choose the cryoprotectant arbitrarily andmainly use glycerol and dimethylsulfoxide.The work of various groups1–5 indicated thatthe common cryoprotectants vary in the rateof permeation of embryos, and in toxicity6–8
to embryos. Rate of permeation is also depen-dent on temperature.3,9 There are also speciesdifferences in the rate of permeation of cryo-protectants.3–5 With the necessity to use highconcentrations of cryoprotectants for vitrifica-tion it became obvious that toxicity was nowan important consideration in the selection ofcryoprotectants for this purpose.
Thus, a cryoprotectant suitable for use invitrification of embryos must vitrify at a con-centration that is not toxic to embryos duringthe cooling process and at the temperature of
the procedure. Temperature has a large effecton toxicity. Furthermore, it has been observedthat crystallization of water to form ice canoccur during the cooling as well as during thewarming of embryos.
It is clear that a cryoprotectant for use invitrification of embryos must have the follow-ing characteristics:
• It must enable vitrification when cooled byplunging into liquid nitrogen
• It must not devitrify to form ice or fractureon cooling or warming
• It must be non-toxic to embryos. Toxicity istemperature dependent; thus the selectionof a cryoprotectant must be undertaken atthe temperature at which embryos andmedia are held during the preparatorystages of cryopreservation.
A scientific approach to the design of vitrifi-cation solutions (VSs) requires an under-standing of the biophysical principles ofcryopreservation. It should take into consid-eration the cytotoxicity of the cryoprotectant;the role or effect of temperature; the inter-actions between various intracellular bio-molecules, organelles, cytoskeleton, and themembrane with the cryoprotectant; and othersupplements to the cryoprotectant solutionthat promote vitrification and/or protect thecell against cryoinjury such as serum proteins,osmolytes (to maintain membrane integrity),and buffers (to maintain pH of the cryopro-tectant solution). An appreciation of thesefactors and the physicochemical forces insidethe cell during dehydration, cooling, warm-ing, and rehydration will be particularlyuseful in designing efficacious VSs. These
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 45

interactions and the biophysical principles ofcryopreservation are discussed elsewhere inthis book.
A review of the literature on the variousVSs devised to cryopreserve embryos andoocytes revealed that most authors, withnotable exceptions, did not indicate how theyhad arrived at a particular formulation. Itis not clear whether such formulations areproducts of scientific enquiry. Capriciousselection of cryoprotectants does not conformto the standard norms of scientific practice. A‘quick-fix’ approach could have long-termnegative implications because the chosencryoprotectant solution may not be the idealsolution and it may be deficient in manyrespects. It could prove disastrous, especiallyin the healthcare industry, to utilize productsthat have been developed without thoroughinvestigation. The objective of this chapter isto describe ways of assessing the effects ofcryoprotectant properties and vitrificationmethodology on the formulation of VSs.
CRYOPROTECTANTS
Types of cryoprotectants
Cryoprotectants can be divided into twomain types: permeating and non-permeating.Both types have been used in VSs. The cryo-protectants used to date in vitrification arethose that have been previously used in con-ventional freezing of embryos. When used atan adequate concentration many will enablevitrification without formation of ice crystals.
There are various types of VSs. In the fieldof assisted reproductive technology (ART) thecommon VSs are usually binary or consist oftwo main cryoprotectants. The purpose of hav-ing more than one cryoprotectant in a VS is tolower the overall molarity at which vitrificationoccurs. Lowering the molarity is a strategy toreduce the toxicity of the vitrification solu-tion.10,11 Other less common and less investi-gated VSs consist of three (ternary), four(quaternary), or possibly more cryoprotectants.
Much work remains to be done investigatingternary, quaternary, and higher combinationsof cryoprotectant solutions that may provemore efficacious and less toxic than the cur-rently used binary cryoprotectant solutions.
High concentrations of cryoprotectants areneeded to achieve vitrification by plunginginto liquid nitrogen (Table 3.1). Because suchconcentrations can be toxic (Table 3.2), mix-tures of cryoprotectants have been used witha view to reducing toxicity of the cryoprotec-tant solution.
The first requirement in testing cryoprotec-tants is to determine the concentration atwhich they will vitrify. Plunging into liquidnitrogen has been adopted as the method ofcooling because of the simplicity of this methodand the ready availability of liquid nitrogen.There have been some experiments12–18 inwhich lower temperatures have been used toreduce cryoprotectant chemotoxicity.
Single cryoprotectants or mixtures of cryo-protectants may be tested for their ability tovitrify on plunging and to remain free of iceon both cooling and warming as shown inTables 3.1 and 3.3. Table 3.1 shows the mini-mum concentration of cryoprotectants thatwill vitrify under the conditions employed byAli and Shelton7 (loaded into 0.25 mL strawsand plunged into liquid nitrogen).
VITRIFICATION IN ASSISTED REPRODUCTION
46
Table 3.1 Minimum concentrations of cryoprotectantsin 0.25 mL straws that will vitrify when cooled by plung-ing into liquid nitrogen
Vitrification concentration
Cryoprotectant Molarity (mol/L) Percentage
Butylene glycol 3.0 27.04Propylene glycol 4.0 30.44Dimethyl sulfoxide 5.0 39.07Glycerol 5.0 46.05Ethylene glycol 6.5 40.35Methanol Crystallizes Crystallizes
at high at 99.8% concentrations
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 46
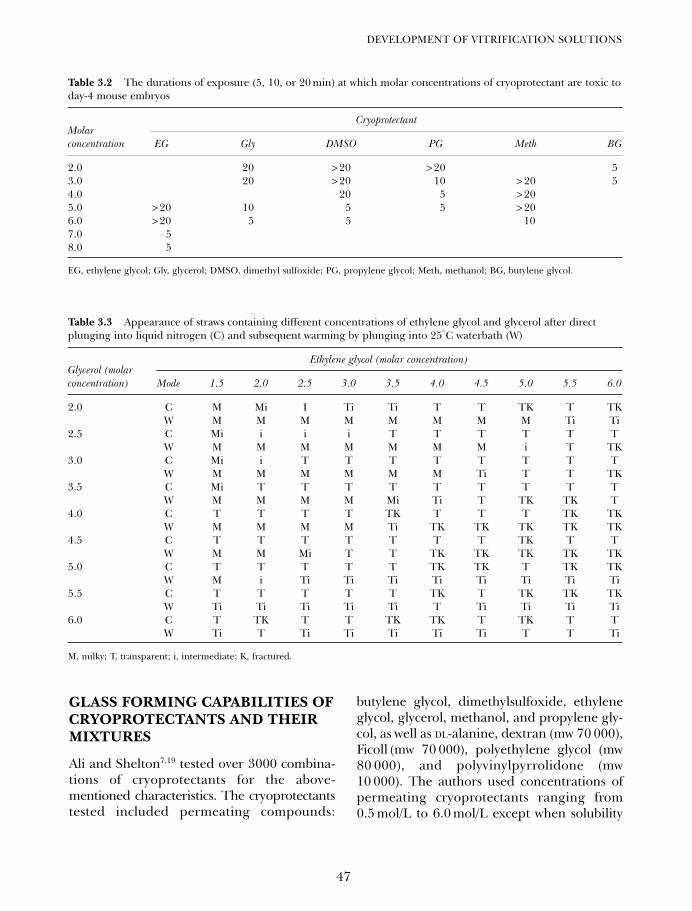
GLASS FORMING CAPABILITIES OFCRYOPROTECTANTS AND THEIRMIXTURES
Ali and Shelton7,19 tested over 3000 combina-tions of cryoprotectants for the above-mentioned characteristics. The cryoprotectantstested included permeating compounds:
butylene glycol, dimethylsulfoxide, ethyleneglycol, glycerol, methanol, and propylene gly-col, as well as DL-alanine, dextran (mw 70 000),Ficoll (mw 70 000), polyethylene glycol (mw80 000), and polyvinylpyrrolidone (mw10 000). The authors used concentrations ofpermeating cryoprotectants ranging from0.5 mol/L to 6.0 mol/L except when solubility
47
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Table 3.2 The durations of exposure (5, 10, or 20 min) at which molar concentrations of cryoprotectant are toxic today-4 mouse embryos
Cryoprotectant
EG Gly DMSO PG Meth BG
2.0 20 > 20 > 20 53.0 20 > 20 10 > 20 54.0 20 5 > 205.0 > 20 10 5 5 > 206.0 > 20 5 5 107.0 58.0 5
EG, ethylene glycol; Gly, glycerol; DMSO, dimethyl sulfoxide; PG, propylene glycol; Meth, methanol; BG, butylene glycol.
Molarconcentration
Table 3.3 Appearance of straws containing different concentrations of ethylene glycol and glycerol after directplunging into liquid nitrogen (C) and subsequent warming by plunging into 25°C waterbath (W)
Ethylene glycol (molar concentration)
Mode 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0
2.0 C M Mi I Ti Ti T T TK T TKW M M M M M M M M Ti Ti
2.5 C Mi i i i T T T T T TW M M M M M M M i T TK
3.0 C Mi i T T T T T T T TW M M M M M M Ti T T TK
3.5 C Mi T T T T T T T T TW M M M M Mi Ti T TK TK T
4.0 C T T T T TK T T T TK TKW M M M M Ti TK TK TK TK TK
4.5 C T T T T T T T TK T TW M M Mi T T TK TK TK TK TK
5.0 C T T T T T TK TK T TK TKW M i Ti Ti Ti Ti Ti Ti Ti Ti
5.5 C T T T T T TK T TK TK TKW Ti Ti Ti Ti Ti T Ti Ti Ti Ti
6.0 C T TK T T TK TK T TK T TW Ti T Ti Ti Ti Ti Ti T T Ti
M, milky; T, transparent; i, intermediate; K, fractured.
Glycerol (molarconcentration)
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 47

limited the concentration that could be used.The high molecular weight polymers, Ficoll,dextrose, and polyvinylpyrrolidone were usedat concentrations of 5–20%. Polyethylene gly-col was used at a maximum concentration of10% as higher concentrations were found tobe extremely toxic to embryos.
The authors loaded the cryoprotectantsolutions to be tested into 0.25 mL plasticstraws by suction with a syringe at room tem-perature (25oC). The straws were heat-sealedat both ends then plunged into liquid nitro-gen. The straw is suitable for this procedureas vitrification can be easily distinguishedfrom crystallization (ice formation) within thestraw. With vitrification on plunging the con-tents of the straw are clear and transparent,whereas they are white and opaque (milky)when ice formation occurs. With some solu-tions the glass (vitrified solution) will fractureduring vitrification. This produces a crack-ling noise within the straw and is easilydetected. Both ice formation and fracturingof the vitrified medium can be lethal toembryos contained therein.
Ice formation and fracture of the glass canoccur on cooling and warming of the VS. Ithas been found that it is more difficult toavoid ice formation on warming than on cool-ing. Very small crystals that are not easily vis-ible and are not harmful may form on coolingand these can act as nuclei for the formationof larger crystals during the warming process.Fractures formed during cooling may bereadily visible but those formed during warm-ing often are not visible to the naked eye.However, small bubbles often form at thefracture site and with crackling sounds areindicative of fracture formation. Solutions ofhigh concentrations of cryoprotectant appearto be more brittle and tend to fracture.Unfortunately good glass formers have agreater tendency to fracture than poor glassformers. Kroener and Luyet20 demonstratedthe factors responsible for the formation offractures during vitrification procedures.Fractures may be an indication of excess
solute concentration. A slight reduction insolute concentration may reduce fractureformation without compromising the vitrifi-cation properties of the solution.
The capacity to warm without devitrifyingis tested by plunging the straws of vitrifiedsolution into a water bath. Ali and Shelton7,19
chose to use a water bath at 25oC for warmingof straws as this is similar to the room andfield temperatures in most countries and thusobviates the need for an incubator and facili-tates work under field conditions. Duringwarming, solutions that do not devitrify aretransformed from the clear glass state to theliquid state without evidence of a milkyappearance.
Results of these tests for vitrificationand devitrification revealed the behavior ofindividual cryoprotectants and a number ofcombinations of cryoprotectants. Severalcombinations of cryoprotectants providedsatisfactory vitrification without crystalliza-tion or fracturing during cooling or warming.
Table 3.3 is adapted from Ali andShelton7 and shows results from one of themany combinations of cryoprotectantstested. They tested ethylene glycol and glyc-erol combinations up to a combined concen-tration of 12 mol/L. For each concentrationof cryoprotectants the table shows theappearance of straws during cooling (C) byplunging into liquid nitrogen and warming(W) by plunging the cooled straw into waterat 25oC. From these and similar observationswith other cryoprotectants and combina-tions thereof Ali and Shelton7 selected solu-tions for further testing. For example, it isobvious in Table 3.3 that 3.0 mol/L or3.5 mol/L ethylene glycol with 4.5 mol/Lglycerol is a solution that vitrifies on coolingand does not crystallize on warming. It isevident also that whilst vitrification isachieved with higher concentrations ofcryoprotectant, cracking of the glass alsobecomes a common occurrence. Anotherobservation was that brittleness and fractureformation appear to be more pronounced
VITRIFICATION IN ASSISTED REPRODUCTION
48
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 48

in solutions containing butylene glycol,propylene glycol, and dimethylsulfoxide,and to a lesser degree in those containingglycerol and methanol. While the firstthree were shown to be good glass formers,vitrified solutions of these compounds areprone to brittleness and fracture readilyduring cooling or warming. Ethylene glycolproved to be the cryoprotectant least proneto fracture.
The presence of high molecular weightpolymers, Ficoll, dextrose, polyethylene gly-col, and polyvinylpyrrolidone did not signifi-cantly reduce the vitrification concentrationof solutions.
Rate of cooling and warming
The rate of cooling is dependent on the vol-ume of liquid to be cooled and also its sur-face area. Therefore it is important that VSsbe tested in the container and at the samevolume that will be used for embryo vitrifica-tion. Whilst the results of testing in 0.25 mLstraws are a very good indicator of the capac-ity of cryoprotectant solutions to vitrify andsubsequently warm without ice formation orcracking, and thus are potentially useful forvitrification of embryos and oocytes, it isimportant to be aware that many variablesmay affect the behavior of any putative VS.In addition to concentration of cryoprotec-tant, factors that influence vitrificationinclude cooling rate, warming rate, volumeof solution, and shape of container. Ali19
compared direct plunging into liquid nitro-gen with plunging after pre-cooling in a boaton the surface of the nitrogen for 2 minutes.The latter method required the concentra-tion of the solution to be increased by at least0.5 mol/L to allow vitrification. Furthermore,the straw vitrified by direct plunging did notdevitrify on warming, whereas the straw vitri-fied by indirect plunging devitrified onwarming. It is also clear that volume of solu-tion and the shape of the container havea large influence on rate of cooling and
warming. For instance the cooling rate for a0.25 mL straw when plunged directly intoliquid nitrogen from room temperature isabout 2500oC/min.13 The warming rate fora 0.25 mL straw from − 196oC to 25oC isabout 1000oC/min.21 If the volume isreduced to about 1 µL it will be possible toincrease the cooling rate to about15 000–30 000oC/min.22–24 Higher coolingrates will allow VSs of lower solute concentra-tion to vitrify during cooling and warming.Also, the cytotoxicity of VSs with lower soluteconcentrations will be lower. The same ruleapplies to warming rates. Higher warmingrates will prevent the formation and growthof ice nuclei during warming. A warming ratein the order of 4460oC/min (when the speci-men is warmed from −196oC to 37oC) usedby some workers has resulted in acceptablesurvival of embryos.
Choice of carriers
The experiments of Ali and Shelton7,25,26 used0.25 mL straws as the container in which vit-rification was tested. Recently a number ofnovel containers have been trialed for vitrifi-cation of embryos.22,23,27–33 Because of theinfluence of volume, shape, and surface areaon cooling and warming rates it is importantthat any new cryoprotectant solution beassessed in the container intended for use inembryo vitrification.
The smaller the container or carrier, thelower the solute concentration required to vit-rify. For instance, a concentration of solutionthat barely allows vitrification when loaded ina 0.25 mL straw, devitrifies when loaded intoa 0.5 mL straw. This is probably due to thecreation of unequal thermal gradients at thesurface of the vehicle that is in direct contactwith the cooling or warming agent and thecore of the carrier. The cooling or warmingrates at the surface of the carriers are fasterthan at the core. This imbalance induces icenucleation in the VS and subsequently icecrystallization. Crystallization could prove
49
DEVELOPMENT OF VITRIFICATION SOLUTIONS
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 49

hazardous for the viability of cells. The directimplication of this observation is that it maybe possible to vitrify embryos and gameteswith VSs of significantly lower solute concen-tration if carriers that are capable of carryingvery low volumes, in the order of microliters,of cryoprotectants can be devised. The influ-ence of volume and shape on the incidence offracture formation is unclear but it is likelythat smaller volumes are less prone to frac-ture because of smaller temperature gradi-ents within the solution.
The use of VSs of significantly reducedsolute concentrations is advantageous becausesuch solutions may be non-toxic or of insignif-icant toxicity to embryos and gametes. Theuse of various carriers that allow the use of lowor ultralow volumes of VSs for vitrification,such as pulled straw, electron microscope cop-per grids, etc. are discussed elsewhere in thisbook. It must, however, be borne in mind thatsome microscopic carriers may not affordprotection against cross-infection by danger-ous pathogens during storage because thesecarriers are not sealed and may expose thecryopreserved specimen to the liquid nitrogenin storage tanks. Various strategies to preventcross-contamination during storage are alsodiscussed elsewhere in this book.
Toxicity of cryoprotectants andtheir mixtures
Most cryoprotectants are cytotoxic. A thor-ough investigation with various preimplanta-tion stages of the mouse embryo will providesome basic information on the cytotoxicity ofindividual cryoprotectants and mixtures ofcryoprotectant.25 Cytotoxicity is dependenton the operating temperature. Based on thisinformation, the most promising VSs can beselected for further investigation and if nec-essary further refinements can be made. Thestudy can then be extended to embryos ofhigher mammals and, finally, to humanembryos and possibly gametes. Viability ofembryos is dependent on whether intracellular
contents vitrified during cooling and remainedice-free during warming.
The cell will dehydrate immediately afterexposure to the high osmolar cryoprotectantsolution with the rapid passage of water out ofand shrinkage of the cell. The reduction involume of the embryo immediately after expo-sure to the high osmolar solution could be ashigh as 85–88%. Water leaves the cell at about5000 times faster than the macromoleculesand other solutes present in the cytoplasm9
and as a consequence, the intracellular soluteconcentration will increase many times, oftensufficient to promote intracellular vitrificationshould cooling occur. This high solute concen-tration can, however, be toxic. In the mousethe increase in intracellular solute concentra-tion after exposure to the VS appears to allowmost preimplantation stages to vitrify andsurvive the ultrarapid cooling and warming.Some higher preimplantation stages of themouse embryo can be cryopreserved by vitrifi-cation without prior partial equilibration withthe cryoprotectant. This is not the case forembryos of most higher mammals which seemto need prior partial equilibration.
It is evident from the preceding para-graphs that high concentrations of cryopro-tectant are required for vitrification at thecooling rate that can be achieved by plunginginto liquid nitrogen. Obviously the VS usedfor embryo vitrification must be non-toxic toembryos at the temperature and exposuretime employed throughout the whole proce-dure. In the testing of cryoprotectants forcapacity to vitrify, many combinations weretested as it is commonly believed that thehigh concentration attained by combinationof cryoprotectants may be less toxic than asingle cryoprotectant at the same concentra-tion. Ali and Shelton7 conducted a number oftrials to determine the toxicity of individualcryoprotectants and combinations thereof. Intheir initial tests they used day-4 Swiss out-bred mouse embryos to screen for toxicity.
Extracellular ice formation or devitrifica-tion during cooling or warming should
VITRIFICATION IN ASSISTED REPRODUCTION
50
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 50

theoretically not harm the specimen.However, should the growing ice nuclei con-tinue to grow rapidly in size, the potential todamage the cryopreserved cell is high. Forthis reason VSs that do not devitrify duringcooling and warming are selected for furtherinvestigation. The temperature and durationof exposure are variables which influence thetoxic effects of cryoprotectants on embryos.
For each molar concentration of cryoprotec-tant 60–78 morulae in 3–6 replicates wereexposed for 5, 10, or 20 min at 25oC. Followingexposure for the specified duration, themorulae were exposed to 1.0 mol/L sucrose for10 min at 25oC to dilute the cryoprotectant inthe morulae. The morulae were then placed inHepes buffered Whitten’s medium34 for 5 minat the same temperature after which they werecultured overnight in Whitten’s medium in anatmosphere of 5% CO2 in air at 37oC. The fol-lowing day the number of embryos that haddeveloped to the blastocyst stage was recorded.
Table 3.2 presents a summary of the testsfor toxicity of six cryoprotectants for day-4mouse morulae. It is clear that ethylene gly-col is the least toxic of the cryoprotectantstested. The order of toxicity from least tomost toxic was shown to be ethylene glycol,methanol, dimethyl sulfoxide, glycerol,propylene glycol, and butylene glycol. In theprevious section it was shown that butyleneglycol is a good vitrifier (see Table 3.1), but itshigh toxicity to embryos renders it unsuitablefor embryo cryopreservation. It must benoted that the butylene glycol studied by Aliand Shelton7 was a racemic mixture of iso-mers of butylene glycol, and the results maynot apply to each of the isomers. On theother hand, methanol is of relatively low tox-icity but does not vitrify even at very highconcentrations. Glycerol and dimethylsulfox-ide, both commonly used cryoprotectants,were quite toxic to mouse morulae after expo-sure for 5 min. Propylene glycol, also com-monly used as a cryoprotectant showedtoxicity to morulae after 10 min at a concen-tration of 3.0 mol/L. On comparing Tables
3.1 and 3.2 it is apparent that to achievevitrification with the commonly used cryopro-tectants it is necessary to use concentrationswhich at best (e.g. ethylene glycol) approacha level that is toxic to mouse morulae.
From examination of the data on vitrifica-tion of mixtures of cryoprotectants it is possi-ble to select mixtures that will vitrify oncooling and not devitrify on warming. Thus,Ali and Shelton7 selected for toxicity testing13 mixtures that had proven to be good vitri-fiers. These selections took into considerationthe individual cryoprotectant’s toxicity andthe mixture’s capacity to vitrify. The data pre-viously generated suggested that the molarconcentration required to vitrify and to be oflow toxicity might be achieved by using ethyl-ene glycol (low toxicity) to achieve the necessarymolar concentration in combination with glyc-erol, dimethylsulfoxide, or propylene glycolthat are good vitrifiers but of greater toxicity.Butylene glycol, a good vitrifier, could be usedonly at 1.0 mol/L concentration in mixturesbecause of toxicity at higher concentrations(Table 3.4).
51
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Table 3.4 Composition of vitrification solutionsdesignated VS1 to VS13
Mixture Composition
VS1 2.5 mol/L glycerol + 5.5 mol/L ethylene glycolVS2 3.0 mol/L methanol + 6.0 mol/L ethylene
glycolVS3 1.5 mol/L propylene glycol + 6.0 mol/L
ethylene glycolVS4 2.0 mol/L propylene glycol + 5.5 mol/L
ethylene glycolVS5 3.5 mol/L glycerol + 4.5 mol/L ethylene glycolVS6 3.0 mol/L glycerol + 3.0 mol/L propylene
glycol VS7 3.0 mol/L ethylene glycol + 4.0 mol/L
dimethylsulfoxideVS8 1.0 mol/L butylene glycol + 6.0 mol/L
ethylene glycolVS9 1.8 mol/L glycerol + 6.1 mol/L ethylene glycolVS10 1.5 mol/L glycerol + 6.5 mol/L ethylene glycolVS11 1.8 mol/L glycerol + 6.0 mol/L ethylene glycolVS12 1.5 mol/L glycerol + 6.3 mol/L ethylene glycolVS13 8.0 mol/L ethylene glycol
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 51
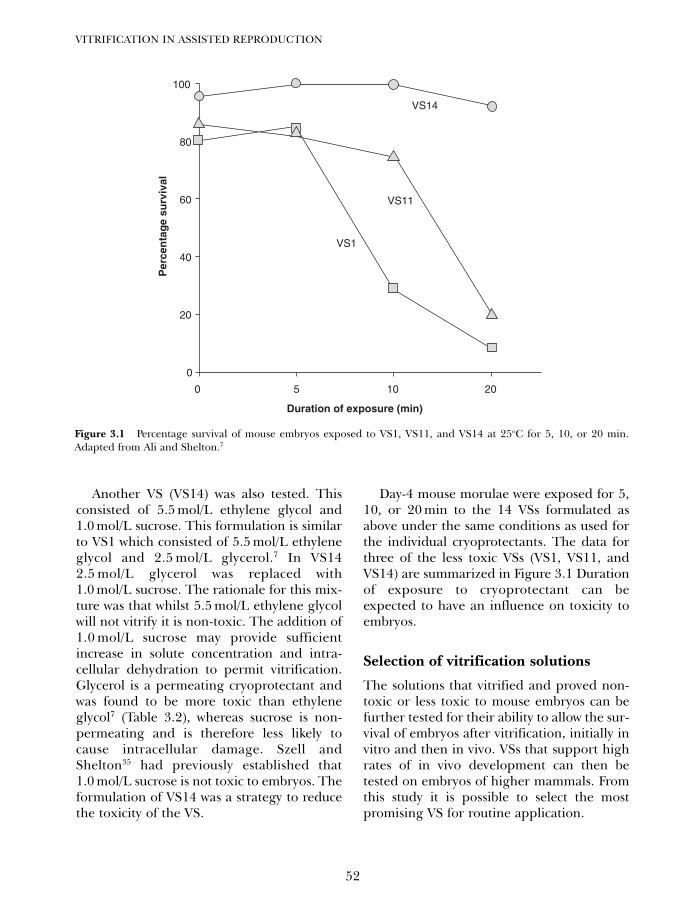
Another VS (VS14) was also tested. Thisconsisted of 5.5 mol/L ethylene glycol and1.0 mol/L sucrose. This formulation is similarto VS1 which consisted of 5.5 mol/L ethyleneglycol and 2.5 mol/L glycerol.7 In VS142.5 mol/L glycerol was replaced with1.0 mol/L sucrose. The rationale for this mix-ture was that whilst 5.5 mol/L ethylene glycolwill not vitrify it is non-toxic. The addition of1.0 mol/L sucrose may provide sufficientincrease in solute concentration and intra-cellular dehydration to permit vitrification.Glycerol is a permeating cryoprotectant andwas found to be more toxic than ethyleneglycol7 (Table 3.2), whereas sucrose is non-permeating and is therefore less likely tocause intracellular damage. Szell andShelton35 had previously established that1.0 mol/L sucrose is not toxic to embryos. Theformulation of VS14 was a strategy to reducethe toxicity of the VS.
Day-4 mouse morulae were exposed for 5,10, or 20 min to the 14 VSs formulated asabove under the same conditions as used forthe individual cryoprotectants. The data forthree of the less toxic VSs (VS1, VS11, andVS14) are summarized in Figure 3.1 Durationof exposure to cryoprotectant can beexpected to have an influence on toxicity toembryos.
Selection of vitrification solutions
The solutions that vitrified and proved non-toxic or less toxic to mouse embryos can befurther tested for their ability to allow the sur-vival of embryos after vitrification, initially invitro and then in vivo. VSs that support highrates of in vivo development can then betested on embryos of higher mammals. Fromthis study it is possible to select the mostpromising VS for routine application.
VITRIFICATION IN ASSISTED REPRODUCTION
52
100
80
60
Per
cen
tag
e su
rviv
al
VS1
VS11
VS14
40
20
0
0 5 2010
Duration of exposure (min)
Figure 3.1 Percentage survival of mouse embryos exposed to VS1, VS11, and VS14 at 25oC for 5, 10, or 20 min.Adapted from Ali and Shelton.7
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 52
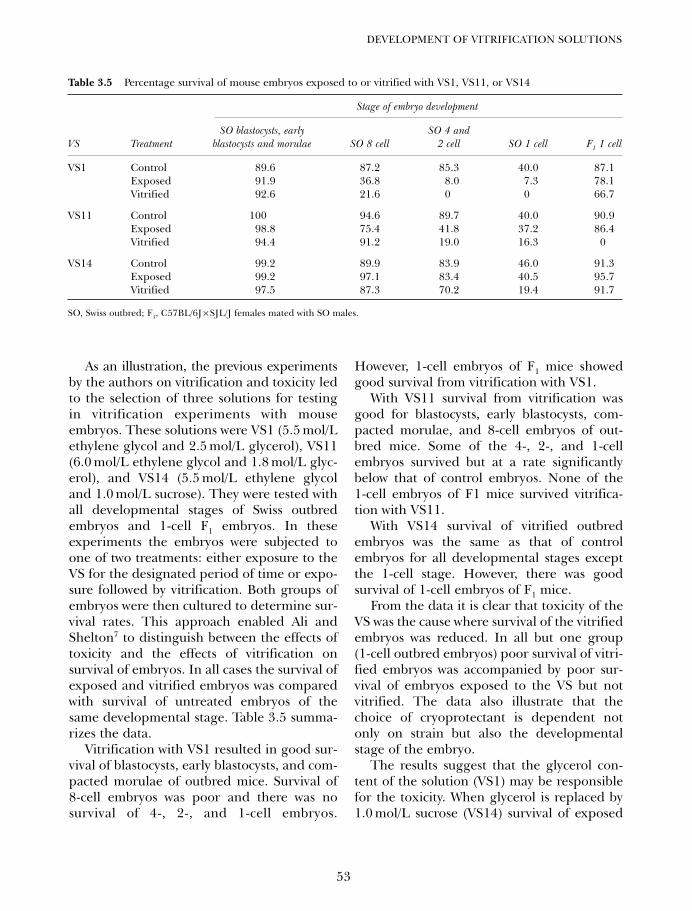
As an illustration, the previous experimentsby the authors on vitrification and toxicity ledto the selection of three solutions for testingin vitrification experiments with mouseembryos. These solutions were VS1 (5.5 mol/Lethylene glycol and 2.5 mol/L glycerol), VS11(6.0 mol/L ethylene glycol and 1.8 mol/L glyc-erol), and VS14 (5.5 mol/L ethylene glycoland 1.0 mol/L sucrose). They were tested withall developmental stages of Swiss outbredembryos and 1-cell F1 embryos. In theseexperiments the embryos were subjected toone of two treatments: either exposure to theVS for the designated period of time or expo-sure followed by vitrification. Both groups ofembryos were then cultured to determine sur-vival rates. This approach enabled Ali andShelton7 to distinguish between the effects oftoxicity and the effects of vitrification onsurvival of embryos. In all cases the survival ofexposed and vitrified embryos was comparedwith survival of untreated embryos of thesame developmental stage. Table 3.5 summa-rizes the data.
Vitrification with VS1 resulted in good sur-vival of blastocysts, early blastocysts, and com-pacted morulae of outbred mice. Survival of8-cell embryos was poor and there was nosurvival of 4-, 2-, and 1-cell embryos.
However, 1-cell embryos of F1 mice showedgood survival from vitrification with VS1.
With VS11 survival from vitrification wasgood for blastocysts, early blastocysts, com-pacted morulae, and 8-cell embryos of out-bred mice. Some of the 4-, 2-, and 1-cellembryos survived but at a rate significantlybelow that of control embryos. None of the1-cell embryos of F1 mice survived vitrifica-tion with VS11.
With VS14 survival of vitrified outbredembryos was the same as that of controlembryos for all developmental stages exceptthe 1-cell stage. However, there was goodsurvival of 1-cell embryos of F1 mice.
From the data it is clear that toxicity of theVS was the cause where survival of the vitrifiedembryos was reduced. In all but one group(1-cell outbred embryos) poor survival of vitri-fied embryos was accompanied by poor sur-vival of embryos exposed to the VS but notvitrified. The data also illustrate that thechoice of cryoprotectant is dependent notonly on strain but also the developmentalstage of the embryo.
The results suggest that the glycerol con-tent of the solution (VS1) may be responsiblefor the toxicity. When glycerol is replaced by1.0 mol/L sucrose (VS14) survival of exposed
53
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Table 3.5 Percentage survival of mouse embryos exposed to or vitrified with VS1, VS11, or VS14
Stage of embryo development
SO blastocysts, early SO 4 andVS Treatment blastocysts and morulae SO 8 cell 2 cell SO 1 cell F1 1 cell
VS1 Control 89.6 87.2 85.3 40.0 87.1Exposed 91.9 36.8 8.0 7.3 78.1Vitrified 92.6 21.6 0 0 66.7
VS11 Control 100 94.6 89.7 40.0 90.9Exposed 98.8 75.4 41.8 37.2 86.4Vitrified 94.4 91.2 19.0 16.3 0
VS14 Control 99.2 89.9 83.9 46.0 91.3Exposed 99.2 97.1 83.4 40.5 95.7Vitrified 97.5 87.3 70.2 19.4 91.7
SO, Swiss outbred; F1, C57BL/6J × SJL/J females mated with SO males.
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 53

and vitrified embryos is greatly enhanced.The beneficial effect of non-permeatingsucrose also suggests that dehydration is animportant contributor to conditions suitablefor vitrification. Furthermore, the short dura-tion of exposure to VS would not permit equi-libration of the embryos before vitrification.Ali and Shelton7,19,25,36 showed that embryosare in a very shrunken state when vitrifiedafter brief exposure to VSs. This observationleads to the suggestion that concentration ofthe solute within the embryonic cells is animportant contributor to vitrification.
Sucrose has been used extensively in dilu-tion of cryoprotectant from embryos thawedafter conventional cryopreservation. It mayalso be advantageous to use sucrose in dilu-tion of cryoprotectant after thawing of vitri-fied embryos. Ali and Shelton7,19,25 compareddilution of VS11 from all stages of Swiss out-bred embryos directly in medium with dilu-tion in 1.0 mol/L sucrose for 10 min. Survivalin vitro was not affected by the method of VSdilution.
The effects of species, stage ofdevelopment, and dilution procedureon toxicity
Testing on day-4 mouse morulae can beconsidered as a screening test to eliminatecryoprotectants and mixtures thereof that aretoxic. The susceptibility of embryos to thetoxic effects of cryoprotectants varies betweenspecies and between developmental stages ofembryos. From these data VS can be selectedfor further testing with embryos of differentstages and of different species. Ali andShelton7 selected VS11 for further testing andproceeded to test its toxicity on a range ofdevelopmental stages of mouse embryos.
The other important consideration is themethod of dilution of the cryoprotectant fromthe embryos after warming. Non-permeatingsucrose has been used successfully to removecryoprotectant from embryos prior to theirreturn to physiological medium. This can be
affected by the concentration of cryoprotectantin the embryo and the rate at which theparticular cryoprotectant permeates the cellmembrane. Whilst Szell et al.5 demonstratedthat ethylene glycol rapidly permeates cattleembryos, the concentration of ethylene glycolin VS11 is far greater than the 1.5 mol/L usedby those workers. Therefore, it is importantto investigate whether sucrose is helpful inremoving VS11 or other vitrifying solutionfrom embryos of different stages of develop-ment that have been exposed for specificdurations. Ali and Shelton7 exposed mouseembryos to VS11 for 1, 5, 10, and 20 min andsubsequently diluted the cryoprotectant fromthe embryos with or without 1.0 mol/L sucrosefor 10 min. After this treatment embryos werecultured to the blastocyst stage to determinerates of survival. The data in Table 3.6indicate that VS11 is well tolerated for shortdurations of exposure by all stages of mouseembryos except the 1- and 2-cell stages.Similar tests of VS11 toxicity on sheepembryos showed that compacted morulae aremuch more tolerant of exposure to VS11 thanuncompacted morulae (Table 3.7).
These data (Tables 3.6 and 3.7) demon-strate the effect of species and stage of devel-opment on response of embryos to exposureto VSs. Both these factors must be consideredwhen selecting or testing VSs. To extrapolatedirectly between species or between stagesof development of embryos is fraught withprobabilities of disappointment.
Preparation of vitrification solutions
Cryoprotectant and VSs are normally preparedin phosphate buffered saline. When mixingcryoprotectant solutions, calculation of theamount (volume) of cryoprotectant to beadded to the buffer solution should be basedon the specific gravity of the cryoprotectant.The pH of the solution must be adjusted toabout 7.25 prior to making up volume. This iscrucial as the pH will drop during coolingand this could affect subsequent viability of the
VITRIFICATION IN ASSISTED REPRODUCTION
54
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 54
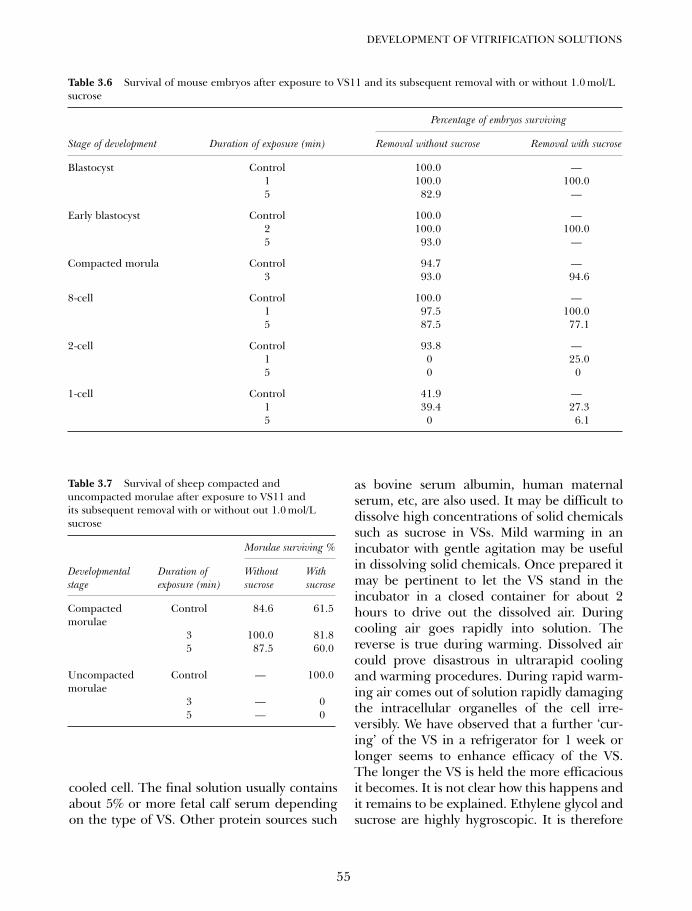
cooled cell. The final solution usually containsabout 5% or more fetal calf serum dependingon the type of VS. Other protein sources such
as bovine serum albumin, human maternalserum, etc, are also used. It may be difficult todissolve high concentrations of solid chemicalssuch as sucrose in VSs. Mild warming in anincubator with gentle agitation may be usefulin dissolving solid chemicals. Once prepared itmay be pertinent to let the VS stand in theincubator in a closed container for about 2hours to drive out the dissolved air. Duringcooling air goes rapidly into solution. Thereverse is true during warming. Dissolved aircould prove disastrous in ultrarapid coolingand warming procedures. During rapid warm-ing air comes out of solution rapidly damagingthe intracellular organelles of the cell irre-versibly. We have observed that a further ‘cur-ing’ of the VS in a refrigerator for 1 week orlonger seems to enhance efficacy of the VS.The longer the VS is held the more efficaciousit becomes. It is not clear how this happens andit remains to be explained. Ethylene glycol andsucrose are highly hygroscopic. It is therefore
55
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Table 3.6 Survival of mouse embryos after exposure to VS11 and its subsequent removal with or without 1.0 mol/Lsucrose
Percentage of embryos surviving
Stage of development Duration of exposure (min) Removal without sucrose Removal with sucrose
Blastocyst Control 100.0 —1 100.0 100.05 82.9 —
Early blastocyst Control 100.0 —2 100.0 100.05 93.0 —
Compacted morula Control 94.7 —3 93.0 94.6
8-cell Control 100.0 —1 97.5 100.05 87.5 77.1
2-cell Control 93.8 —1 0 25.05 0 0
1-cell Control 41.9 —1 39.4 27.35 0 6.1
Table 3.7 Survival of sheep compacted anduncompacted morulae after exposure to VS11 andits subsequent removal with or without out 1.0 mol/Lsucrose
Morulae surviving %
Developmental Duration of Without With stage exposure (min) sucrose sucrose
Compacted Control 84.6 61.5morulae
3 100.0 81.85 87.5 60.0
Uncompacted Control — 100.0morulae
3 — 05 — 0
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 55

important to ensure storage containers of VSare airtight to prevent progressive reduction ofthe solute concentration as a consequence ofhygroscopic dilution.
OSMOTIC EFFECTS OFVITRIFICATION SOLUTIONSON EMBRYOS
In the experiments described above and inpublished reports on vitrification of embryosof a number of species7,10–18,19,25,34 several cryo-protectant combinations have been used. Acommon component of these procedures is ashort duration of exposure to the VS beforeplunging in liquid nitrogen. As Ali andShelton7,19,25 explored a range of cryoprotec-tants they reduced the exposure time so asto minimize toxicity, and good survival oftransferred vitrified/warmed embryos wasobtained when the embryos were plungedinto liquid nitrogen after exposure to theVS for only a few minutes. It is obvious thatequilibration of VS concentrations intra- andextracellularly could not occur in this time.This is in contrast to the equilibration whichis considered desirable for conventionalcryopreservation of embryos.
To explore the degree of cryoprotectantequilibration that occurs when embryos aresubjected to vitrifying concentrations of cryo-protectants Ali and Shelton conducted a seriesof experiments to measure the volume ofembryos exposed to VS11 (6.0 mol/L ethyleneglycol and 1.8 mol/L glycerol) for varying dura-tions.7,19,25,36 Immediately after the embryowas immersed in the VS, microscopic observa-tions were commenced and were continuallyrecorded by a video camera connected to arecorder and a monitor screen. Measurementsfor calculation of volume were made with theaid of an image analyzer.
Study of the images showed that when sub-jected to the osmotic stress resulting fromimmersion in a high concentration of cryo-protectant, embryos shrank immediately anddid not retain a spherical shape (Figures 3.2
and 3.3) as seen with an image analyzer. Ifallowed to rest in its most stable position theshrunken embryo appeared approximatelyspherical but on closer examination was seento resemble a hollow ball that had been par-tially evacuated. One side had become con-cave and the other remained convex or wasslightly flattened. To calculate the approxi-mate volume of the shrunken embryos Aliand Shelton7,19,25,36 used two solid geometrymodels. One was the difference between thevolumes of two half spheres, and the otherwas the difference between the volumes of thesegments of two spheres (Figure 3.2). Theyconducted these experiments with threesheep and three mouse embryos. Here wedescribe the experiment performed on sheepembryos. Embryos of both species behaved ina similar manner. The sheep embryos wereobserved at 0, 3, 5, 10, 15, and 20 min. At3 min the embryos were 24.0%, 30.0%, and15.7% of original volume. Figure 3.2 showsthe responses of the individual embryos.Because of the irregularities in the solidgeometry of individual embryos the calcu-lated volumes can be considered as approxi-mations only. In view of the estimate37 that80–85% of the volume of ova consists ofwater, it is apparent that a high degree ofdehydration was induced immediately afterexposure to VS11 and there had been verylittle inflow of ethylene glycol. It is clear that,when plunged into liquid nitrogen after briefexposure to VS, embryos are in a shrunkenstate when they vitrify. The concentration ofcryoprotectant within the embryos is unknownbut in combination with the high concentra-tion of cell solutes it is sufficient to permitvitrification without ice formation.
SURVIVAL OF VITRIFIED EMBRYOS
Survival of vitrified mouse embryos
There appeared to be little differencebetween VS1, VS11, and VS14 when the end-point was in vitro survival after vitrificationand warming. The crucial test, however, is
VITRIFICATION IN ASSISTED REPRODUCTION
56
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 56
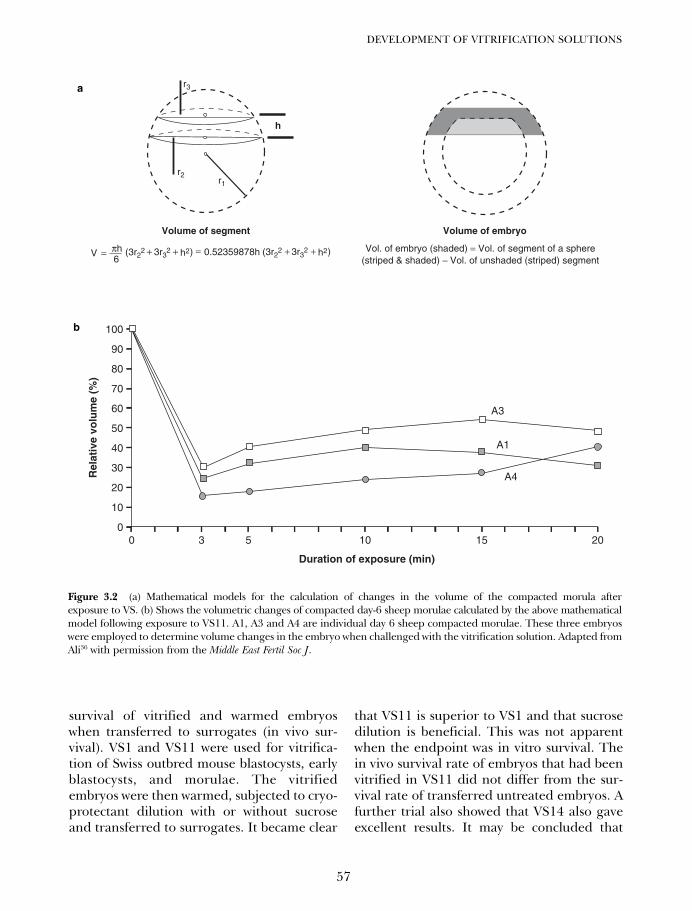
survival of vitrified and warmed embryoswhen transferred to surrogates (in vivo sur-vival). VS1 and VS11 were used for vitrifica-tion of Swiss outbred mouse blastocysts, earlyblastocysts, and morulae. The vitrifiedembryos were then warmed, subjected to cryo-protectant dilution with or without sucroseand transferred to surrogates. It became clear
that VS11 is superior to VS1 and that sucrosedilution is beneficial. This was not apparentwhen the endpoint was in vitro survival. Thein vivo survival rate of embryos that had beenvitrified in VS11 did not differ from the sur-vival rate of transferred untreated embryos. Afurther trial also showed that VS14 also gaveexcellent results. It may be concluded that
57
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Vol. of embryo (shaded) = Vol. of segment of a sphere(striped & shaded) − Vol. of unshaded (striped) segment
r3
r2r1
h
Volume of segment
0.52359878hV6πh= =+ +(3r2
2 3r3
2 h2) + +(3r22 3r3
2 h2)
Volume of embryo
100
90
80
70
60
Rel
ativ
e vo
lum
e (%
)
Duration of exposure (min)
50
40
30
20
10
00 3 5 10 15
A4
A1
A3
20
Figure 3.2 (a) Mathematical models for the calculation of changes in the volume of the compacted morula afterexposure to VS. (b) Shows the volumetric changes of compacted day-6 sheep morulae calculated by the above mathematicalmodel following exposure to VS11. A1, A3 and A4 are individual day 6 sheep compacted morulae. These three embryoswere employed to determine volume changes in the embryo when challenged with the vitrification solution. Adapted fromAli36 with permission from the Middle East Fertil Soc J.
a
b
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 57
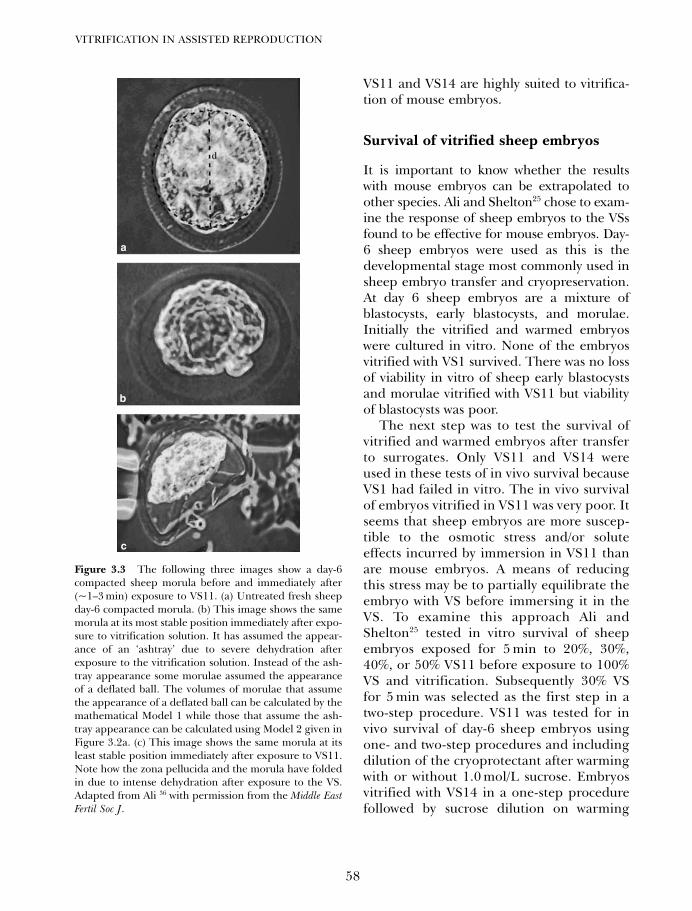
VS11 and VS14 are highly suited to vitrifica-tion of mouse embryos.
Survival of vitrified sheep embryos
It is important to know whether the resultswith mouse embryos can be extrapolated toother species. Ali and Shelton25 chose to exam-ine the response of sheep embryos to the VSsfound to be effective for mouse embryos. Day-6 sheep embryos were used as this is thedevelopmental stage most commonly used insheep embryo transfer and cryopreservation.At day 6 sheep embryos are a mixture ofblastocysts, early blastocysts, and morulae.Initially the vitrified and warmed embryoswere cultured in vitro. None of the embryosvitrified with VS1 survived. There was no lossof viability in vitro of sheep early blastocystsand morulae vitrified with VS11 but viabilityof blastocysts was poor.
The next step was to test the survival ofvitrified and warmed embryos after transferto surrogates. Only VS11 and VS14 wereused in these tests of in vivo survival becauseVS1 had failed in vitro. The in vivo survivalof embryos vitrified in VS11 was very poor. Itseems that sheep embryos are more suscep-tible to the osmotic stress and/or soluteeffects incurred by immersion in VS11 thanare mouse embryos. A means of reducingthis stress may be to partially equilibrate theembryo with VS before immersing it in theVS. To examine this approach Ali andShelton25 tested in vitro survival of sheepembryos exposed for 5 min to 20%, 30%,40%, or 50% VS11 before exposure to 100%VS and vitrification. Subsequently 30% VSfor 5 min was selected as the first step in atwo-step procedure. VS11 was tested for invivo survival of day-6 sheep embryos usingone- and two-step procedures and includingdilution of the cryoprotectant after warmingwith or without 1.0 mol/L sucrose. Embryosvitrified with VS14 in a one-step procedurefollowed by sucrose dilution on warming
VITRIFICATION IN ASSISTED REPRODUCTION
58
Figure 3.3 The following three images show a day-6compacted sheep morula before and immediately after(~1–3 min) exposure to VS11. (a) Untreated fresh sheepday-6 compacted morula. (b) This image shows the samemorula at its most stable position immediately after expo-sure to vitrification solution. It has assumed the appear-ance of an ‘ashtray’ due to severe dehydration afterexposure to the vitrification solution. Instead of the ash-tray appearance some morulae assumed the appearanceof a deflated ball. The volumes of morulae that assumethe appearance of a deflated ball can be calculated by themathematical Model 1 while those that assume the ash-tray appearance can be calculated using Model 2 given inFigure 3.2a. (c) This image shows the same morula at itsleast stable position immediately after exposure to VS11.Note how the zona pellucida and the morula have foldedin due to intense dehydration after exposure to the VS.Adapted from Ali 36 with permission from the Middle EastFertil Soc J.
a
b
c
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 58

were also included in this experiment. It wasconsidered that a two-step procedure wasnot necessary with VS14 as it had shown noevidence of toxicity when applied to theembryos in one step.
The results indicate that VS14 can be usedto vitrify day-6 sheep embryos using the one-step procedure with little loss of viability(Table 3.8). VS11 (6.0 mol/L ethylene glycoland 1.8 mol/L glycerol) was selected for vitri-fication experiments after intensive investiga-tion of cryoprotectants and mixtures ofcryoprotectants for capacity to vitrify and fortoxicity to mouse and sheep embryos.Observations had suggested that in the ethyl-ene glycol/glycerol combination toxicity waslargely attributable to the glycerol. When theglycerol is replaced with 1.0 mol/L sucroseembryo viability on vitrification is improvedparticularly when the embryos are transferredto surrogates for development. Furthermore,this was achieved with a lower concentration(5.5 mol/L) of ethylene glycol. Tests described
earlier had shown that 5.5 mol/L ethyleneglycol is well tolerated by embryos, and Szelland Shelton35 have shown that 1.0 mol/Lsucrose is not toxic to mouse embryos. It isinteresting that neither 5.5 mol/L ethyleneglycol nor 1.0 mol/L sucrose will vitrify on itsown but clearly the combination will vitrify.The results with vitrification of sheepembryos suggest that embryos of somespecies might be more sensitive to cryopro-tectants and may require partial equilibrationwith the cryoprotectant before cooling. It isapparent also that the necessity for this candiffer between cryoprotectant solutions.
Vitrification of oocytes, embryos,tissues, and cell lines of severalspecies with ethylene glycol andsucrose
Subsequent to the publication of the aboveresults with VS147,25,26 numerous workers22,38–56
have used this combination of ethylene glycol
59
DEVELOPMENT OF VITRIFICATION SOLUTIONS
Table 3.8 Viability of day-6 sheep embryos vitrified with VS11 or VS14 and transferred to surrogates
Vitrification Dilution Stage of Percentage developed Percentage of procedure procedure development to live fetuses surrogates pregnant
One-step VS11 With sucrose Morulae 7.9 15.8Early blastocysts 2.4 4.4Blastocysts 0 0
Without sucrose Morulae 13.3 26.7Early blastocysts 0 0Blastocysts 0 0
Total one-step VS11 (8/158) 5.1 (8/79) 10.1Two-step VS11 With sucrose Morulae 55.2 78.6
Early blastocysts 10.0 20.0Blastocysts 62.1 78.6Expanded blastocysts 50.0 50.0
Total two-step VS11 (37/72) 51.4 (25/35) 71.4One-step VS14 With sucrose Morulae 50.0 100.0
Blastocysts 100.0 100.0Expanded blastocysts 0 0
Total one-step VS14 (5/10) 50.0 (3/5) 60.0
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 59

and sucrose for successful vitrification ofembryos and oocytes of several species,including human, with live births. Moreover,immature oocytes, ovarian tissue, and celllines have also been cryopreserved withVS14.40,48,56 A minor modification27,57,58
(5.75 mol/L ethylene glycol + 0.6 mol/Lsucrose) of VS14 (5.5 mol/L ethylene glycol+1.0 mol/L sucrose)7,25,26 has also been usedto vitrify human zygotes and 8-cell embryoswith resultant live births. VS14 has also beencalled 5.5EG.22
Development of vitrifiedembryos
It is clear that merely demonstrating post-warming survival of vitrified embryos in cul-ture does not warrant an assumption thatequally good survival of embryos will followtransfer to surrogates. An examination ofembryos cultured after vitrification and post-warming dilution procedures provides a pos-sible clue to differences between embryos thatare not evident on conventional examinationbut may influence survival when transferredto surrogates. Ali and Shelton19,26 cultureduntreated and vitrified sheep embryos for24–48 h and then counted the nuclei afterfluorescent staining.
The method of Pursel et al.59 can be usedfor fluorescent staining of the nuclei. Thetechnique employs Hoescht 33342 stain withtrypan blue as a counter stain. The fluoresc-ing nuclei are counted on excitation withwavelengths between 351 and 364 nm. Therewas a strong trend for the treatment that pro-vided the better in vivo survival to result inembryos that had a greater number of nuclei.Day-6 sheep early blastocysts cultured aftervitrification by the one-step procedure hadan average of 60 nuclei, whereas controls had138 nuclei. Similar embryos cultured aftervitrification by the two-step procedure had184 nuclei compared with 147 for the controlembryos (unpublished work). It may beconcluded that a significant number of
blastomeres were irreversibly damagedduring the inferior procedure with the resultthat subsequent survival was affected in vivobut not in vitro. These data did not differen-tiate between trophoblastic and inner cellmass cells. It may be desirable also to countthe number of the inner cell mass cells by dif-ferential staining technique60,61 which willindicate the viability of the embryo.
NOVEL AND POTENTIALCRYOPROTECTANTS
Compounds other than those discussed hereshow promise as good vitrifiers. Butanediolhas been shown to be a good vitrifier6 but isalso quite toxic, at least in some of its iso-meric forms. Chemical substitutions in con-ventional cryoprotectants may have an effecton critical cooling rate and critical warmingrate necessary for avoidance of ice forma-tion. Methoxylation of 35% propylene glycoland 45% glycerol reduced the critical cool-ing rate for vitrification from approximately500oC to 50oC/min.62 The usefulness of sucha methoxylated compound in embryo vitri-fication would depend on its toxicity toembryos. While the mechanisms of cryopro-tectant toxicity may not be fully understood,it is clear that in the case of embryos theyvary between species and between develop-mental stages of the embryos. Identificationof compounds with superior vitrificationperformance might permit the use of slowercooling rates and/or lower concentrationof vitrifying solution. The latter would bebeneficial in reducing toxic effects of thecryoprotectant solution. The former mightallow the use of solid carbon dioxide (dryice) as the cooling agent in vitrification. Itmay be questionable whether this would beadvantageous.
The efficacy of some novel compounds inmaintaining the integrity of the cytoskeletonof the egg and embryo53 during vitrificationremains to be demonstrated conclusively. Theinvestigation of novel cryoprotectants for their
VITRIFICATION IN ASSISTED REPRODUCTION
60
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 60

potential in cryopreservation of embryos by vit-rification must follow sound experimental pro-cedures taking into account the effect of speciesand stage of embryonic development as out-lined above. They must also be assessed in com-bination with other cryoprotectants to arrive atVSs that result in optimal embryo survival.Researchers and clinicians must not neglect tostudy the possible genetic effects, on embryosand gametes of vitrification procedures. Whilstthere have been some reports of genetic dam-age from freezing,63,64 as discussed elsewhere inthis book, it is generally accepted that geneticanomalies subsequent to cryopreservation donot occur at detectable rates. Vitrification withVS14 does not appear to cause chromosomaldamage to human oocytes, cleavage stageembryos, and immature oocytes.45,65,66 There is,however, some evidence of long-term effectsof embryo freezing in mice.67 Possible subtlebehavioral effects of cryopreservation must beinvestigated in greater detail. Studies of thegenetic effects of embryo vitrification should
consider not only the effect of vitrification perse but also the effect of the cryoprotectants thatare used in the VS.
ACKNOWLEDGMENTS
The authors are grateful to The AustralianNational University, Canberra, Australia, forallowing the authors the use of their libraryfacilities and to Dr Basim Abu-Rafea andMrs Sandra A. Al-Abdulmunem of the KingFahad Medical City, Riyadh, Kingdom ofSaudi Arabia, for their assistance in literaturesearch. Jaffar Ali is grateful to the manage-ment of the King Fahad Medical City forallowing him to undertake this task. Theauthors gratefully acknowledge the assistanceof Ms Genesis I Salao and Mr Khalid AlHoute of the Audio Visual Department of theKing Fahad Medical City for their assistancein the preparation of the mathematicalmodels and graphics given in Figure 3.2.
61
DEVELOPMENT OF VITRIFICATION SOLUTIONS
References
1. Whittingham DG, Wales RG. Storage of two-cell mouse embryos in vitro. Aust J Biol Sci1969; 22: 1065–8.
2. Miyamoto H, Ishibashi T. The protectiveaction of glycols against freezing damage ofmouse and rat embryos. J Reprod Fertil 1978;54: 427–32.
3. Schneider U, Mazur P, Leibo S. The perme-ability of bovine embryos to DMSO or glyc-erol. Cryobiology 1983; 20: 741 abstr.
4. Leibo SP. A one-step method for direct non-surgical transfer of frozen-thawed bovineembryos. Theriogenology 1984; 21: 767–90.
5. Szell A, Shelton JN, Szell K. Osmotic charac-teristics of sheep and cattle embryos.Cryobiology 1989; 26: 297–301.
6. Bourton P. Levo- and dextro-2,3-butanediol andtheir racemic mixture: very efficient solutes forvitrification. Cryobiology 1990; 27: 55–9
7. Ali J, Shelton JN. Design of vitrification solu-tions for the cryopreservation of embryos. JReprod Fertil 1993; 99: 471–7.
8. Emiliani S, Van den Bergh M, Vannin AS et al.Comparison of ethylene glycol, 1,2-propane-diol, and glycerol for cryopreservation of slow-cooled mouse zygotes, 4-cell embryos, andblastocysts. Hum Reprod 2000; 15: 905–10.
9. Jackowski S, Leibo SP, Mazur P. Glycerolpermeabilities of fertilized and unfertilizedmouse ova. J Exp Zool 1980; 212: 329–41.
10. Scheffen B, Van der Zwalmen P, Massip A. Asimple and efficient procedure for preserva-tion of mouse embryos by vitrification. CryoLetters 1986; 7: 260–9
11. Massip A, Van der Zwalmen P, Scheffen B,et al. Pregnancies following transfer of cattleembryos preserved by vitrification. CryoLetters 1986; 7: 270–3
12. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196 degrees C by vitri-fication. Nature 1985; 313: 573–5.
13. Rall WF. Factors affecting the survival ofmouse embryos cryopreserved by vitrification.Cryobiology 1987; 24: 387–402.
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 61

14. Nakagata N. Survival of mouse morulae andblastocysts derived from in vitro fertilizationafter ultra rapid freezing. Jikken Dobutsu1993; 42: 229–31
15. Shaw PW, Bernard AG, Fuller BJ, et al.Vitrification of mouse oocytes using shortcryoprotectant exposure: effects of varyingexposure times on survival. Mol Reprod Dev1992; 33: 210–4.
16. Nakao K, Nakagata N, Katsuki M. Simple andefficient vitrification procedure for cryo-preservation of mouse embryos. Exp Anim1997; 46: 231–4
17. Nakao K , Nakagata N, Katsuki M. Productionof chimeric mice from cryopreserved blasto-cysts. Exp Anim 1998; 47: 167–71
18. Anzai M, Nishiwaki M, Yanagi M, et al.Application of laser-assisted zona drilling to invitro fertilization of cryopreserved mouse oocyteswith spermatozoa from a subfertile transgenicmouse. J Reprod Dev 2006; 52: 601–6.
19. Ali J. Factors affecting the ultrarapid vitrifica-tion and cryopreservation of embryos. PhDThesis Canberra: Australian NationalUniversity, 1992.
20. Kroener C, Luyet B. Formation of cracks dur-ing the vitrification of glycerol solutions anddisappearance of the cracks during rewarm-ing. Biodynamica 1966; 10: 47–52.
21. Rall WF, Meyer TK, Leibo SP. Effect of warmingconditions on the survival of mouse embryoscryopreserved and diluted by a one-step strawprocedure. Theriogenology 1986; 25: 186 (abstr)
22. Martino A, Songsasen N, Leibo SP. Developmentinto blastocysts of bovine oocytes cryopre-served by ultra-rapid cooling. Biol Reprod1996; 54: 1059–69.
23. Vajta G, Booth PJ, Holm P, et al. Successfulvitrification of early stage bovine in vitro pro-duced embryos with the open pulled straw(OPS) method. Cryo Lett 1997; 18: 191–5
24. Arav A, Zeron Y. Vitrification of bovineoocytes using modified minimum droplet sizetechnique (MDS) is affected by the composi-tion and concentration of the vitrificationsolution and by the cooling conditions.Theriogenology 1997; 47: 341 (abstr)
25. Ali J, Shelton JN. Vitrification of preimplan-tation stages of mouse embryos. J ReprodFertil 1993; 98: 459–65.
26. Ali J, Shelton JN. Successful vitrification ofday-6 sheep embryos. J Reprod Fertil 1993;99: 65–70.
27. Rama Raju GA, Haranath GB, Krishna KM,et al. Vitrification of human 8-cell embryos, amodified protocol for better pregnancy rates.Reprod Biomed Online 2005; 11: 434–7
28. Lane M, Gardner DK. Vitrification of mouseoocytes using a nylon loop. Mol Reprod Dev2001; 58: 342–7.
29. Papis K, Shimizu M, Izaike Y. Factors affectingthe survivability of bovine oocytes vitrified indroplets. Theriogenology 2000; 15: 651–8
30. Vanderzwalmen P, Bertin G, Debauche Ch.,et al. “In Vitro” survival of metaphase iioocytes (mii) and blastocysts after vitrificationin a hemi-straw (hs) system. Fertil Steril 2000:74: S215–S216
31. Matsumoto H, Jiang JY, Tanaka T, et al.Vitrification of large quantities of immaturebovine oocytes using nylon mesh.Cryobiology 2001; 42: 139–44
32. Liebermann J, Tucker M, Graham J, et al.Blastocyst development after vitrification ofmultipronucleate zygotes using the flexipetdenuding pipette (FDP). Reprod BiomedOnline 2002; 4: 148–52
33. Momozawa K, Fukuda Y. Vitrification ofbovine blastocysts on a membrane filterabsorbing extracellular vitrification solution.J. Mammalian Ova Res 2006; 23: 63–6
34. Ali J, Whitten WK, Shelton JN. Effect of cul-ture systems on mouse early embryo develop-ment. Hum Reprod 1993; 8: 1110–4.
35. Szell A and Shelton JN. Sucrose dilution ofglycerol from mouse embryos frozen rapidlyin liquid nitrogen vapour. J Reprod Fertil1986; 76: 401–8.
36. Ali J. Intense dehydration during cryopreser-vation by vitrification of the mammalianembryo is essential for subsequent embryosurvival and viability: a preliminary report.Middle East Fertil Soc J 2001; 6: 50–8
37. Leibo SP. Cryobiology: preservation of mam-malian embryos. Basic Life Sci 1986; 37: 251–72.
38. Papis K, Avery H, Holm P, et al. The effect ofvitrification solution, equilibration time, anddirect dilution method on survivability of equi-librated or vitrified bovine in vitro maturedoocytes. Theriogenology 1995; 43; 293 (abstr)
39. Ali J. Developmental competence of unipronu-clear and triploid day-2 human embryos aftervitrification with VS14. Med Sci Res 1996; 24:377–8.
40. Ali J. Highly efficient ultrarapid cryopreser-vation of established cell lines by vitrificationwith VS14. Med Sci Res 1996; 24: 837–8.
41. Hong SW, Hyung MS, Chung HM, et al.(1999) Improved human oocyte developmentafter vitrification: a comparison of thawingmethods. Fertil Steril 1999; 72: 142–6.
42. Chen SU, Lien YR, Chen HF, et al. Openpulled straws for vitrification of mature mouseoocytes preserve patterns of meiotic spindles
VITRIFICATION IN ASSISTED REPRODUCTION
62
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 62

and chromosomes better than conventionalstraws. Hum Reprod 2000; 15: 2598–603.
43. Chen SU, Lien YR, Chao KH, et al.Cryopreservation of mature human oocytes byvitrification with ethylene glycol in straws.Fertil Steril 2000; 74: 804–8.
44. Choi DH, Chung HM, Lim JM, et al.Pregnancy and delivery of healthy infantsdeveloped from vitrified blastocysts in an IVF-ET program. Fertil Steril 2000; 74: 838–9.
45. Chung HM, Seung WH, Hong MS, et al. Invitro blastocyst formation of human oocytesobtained from unstimulated and stimulatedcycles after vitrification at various matura-tional stages. Fertil Steril 2000; 73: 545–51.
46. Yoon TK, Chung HM, Lim JM, et al.Pregnancy and delivery of healthy infantsdeveloped from vitrified oocytes in a stimu-lated in vitro fertilization-embryo transferprogram. Fertil Steril 2000; 74: 180–1.
47. Yoon TK, Kim TJ, Park SE, et al. Live birthsafter vitrification of oocytes in a stimulated invitro fertilization-embryo transfer program.Fertil Steril 2003; 79: 1323–6.
48. Yin H, Kim SS, Fisher J, et al. Investigation ofoptimal conditions for equilibrating ovariantissue with ethylene glycol prior to vitrification.Fertil Steril 2001; 76(Suppl 1): S101 (abstr).
49. Kim, TJ, Hong SW, Park, SE, et al. Pregnancyafter vitrification of human oocytes and blas-tocysts using same cryoprotectant solution,ethylene glycol, and sucrose. Fertil Steril2003; 80 (Suppl 3): 143 abstr.
50. Kim T, Hong S, Cha K. Pregnancies from cryo-preserved oocytes using vitrification protocol.Fertil Steril 2005; 84: Suppl. 1, pp. S179 (abstr)
51. Kim SH, Ku SY, Sung KC, et al. Simplified EMgrid vitrification is a convenient and efficientmethod for mouse mature oocyte cryopreserva-tion. Yonsei Med J 2006; 30; 47: 399–404.
52. Kim, TJ, Hong SW, Chung HM, et al.Pregnancy and delivery after vitrification ofhuman oocytes. Fertil Steril 2005; 83: Suppl.5, pp. S13 (abstr)
53. Park SE, Chung HM, Cha KY, et al.Cryopreservation of ICR mouse oocytes:improved post-thawed preimplantationdevelopment after vitrification using Taxol, acytoskeleton stabilizer. Fertil Steril 2001; 75:1177–84.
54. Park SE, Kim TJ, Hong SW, et al. Vitrificationof human mature oocytes in a straw to preventthe risk of liquid nitrogen contamination dur-ing storage. Fertil Steril 80: Suppl. 3, pp. 64–5(abstr)
55. Hong S, Kim T, Lee S, et al. Cryopreservedblastocysts using vitrification protocol giveexcellent pregnancy and implantation ratesafter thawing. Fertil Steril 2005: 84: Suppl.1,pp. S178–S179 (abstr)
56. Martins RD, Costa EP, Chagas JSC, et al.Effects of vitrification of immature bovineoocytes on in vitro maturation. Anim Reprod2005; 2: 128–34
57. El-Danasouri I, Selman HA. Successful preg-nancies and deliveries after a simple vitrifica-tion protocol for day 3 human embryos. FertilSteril 2001; 76: 400–2.
58. Selman HA, El-Danasouri I. Pregnanciesderived from vitrified human zygotes. FertilSteril 2002; 77: 422–3.
59. Pursel VG, Wall RJ, Rexroad CE Jr, et al. Arapid whole-mount staining procedure fornuclei of mammalian embryos. Theriogenology1985; 24: 687–91.
60. Papaionnou VE, Ebert KM. The preimplanta-tion pig embryo: cell number and allocationto trophectoderm and inner cell mass of theblastocyst in vivo and in vitro. Development1988: 102: 793–803.
61. Iwasaki S, Yoshiba N, Ushijima H, et al.Morphology and proportion of inner cellmass of bovine blastocysts fertilized in vitroand in vivo. J Reprod Fertil 1990; 90: 279–84.
62. Wowk B, Darwin M, Harris SB, et al. Effects ofsolute methoxylation on glass-forming abilityand stability of vitrification solutions.Cryobiology 1999; 39: 215–27
63. Shaw JM, Kola I, McFarlane DR, et al. An asso-ciation between chromosomal abnormalitiesin rapidly frozen 2-cell mouse embryos andthe ice-forming properties of the cryoprotec-tive solution. J Reprod Fertil 1991; 91: 9–18
64. Bouquet M, Selva J, Auroux M. Cryo-preservation of mouse oocytes: mutagenic effectsin the embryo? Biol Reprod 1993; 49: 764–9
65. Ali J, Bongso A, Ratnam SS. Chromosomalanalysis of day-2 human embryos vitrifiedwith VS14. Med Sci Res 1995; 23: 539–40.[Erratum: Introduction section, paragraph 5,lines 2-3: the sentence should read: ‘VS14 hasbeen shown to be non-teratogenic.’]
66. Park SE, Hong SW, Lee SH, et al.Chromosome and spindle configurations ofhuman oocytes matured in vitro after vitrifica-tion at the germinal vesicle stage in stimulatedcycle. Fertil.Steril 2004; 82: Suppl. 2, pp. S114
67. Dulioust E, Toyama K, Busnel MC, et al.Long-term effects of embryo freezing in mice.Proc Natl Acad Sci USA 1995; 92: 589–93
63
DEVELOPMENT OF VITRIFICATION SOLUTIONS
03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 63

03 Tucker 8029.qxd 8/23/2007 2:11 PM Page 64

65
Vitrification in animal reproduction:vitrification of embryos using openpulled straws Gábor Vajta
4A
THE USE OF OPEN PULLEDSTRAWS IN ANIMALREPRODUCTION
Among various high-rate cooling approachesused for vitrification of mammalian oocytesand embryos (discussed in detail in Chapter2) two techniques have obtained relativelywide acceptance among embryologists: theCryotop with extensive and rapidly growingapplication in the human field, and the openpulled straw (OPS) in animal reproductivebiology. Although there is considerable over-lap, with the first baby born after oocyte vitri-fication using OPS being reported, followedby high pregnancy rates being achievedrecently after OPS use,1,2 and excellent resultsalso being achieved with the Cryotop in ani-mal reproduction,3,4 the field is more or lessevenly distributed. The reasons are partiallyunderstandable, as domestic animal embryol-ogists prefer to work with straws, and themore robust and simpler OPS also permits akind of semi-direct transfer after warming,5
an approach that is not only very simple andpractical, but also has not been found to com-promise pregnancy and calving rates at all incattle after transfer of in vitro produced orsomatic cell cloned, zona intact or zona freeembryos. This fact may provide considerablebenefits for on-farm applications (see below).On the other hand, the beauty of the delicateapproach of Cryotop, and even more theextremely convincing, and burgeoning statis-tics based on blastocyst cryopreservation (and
an increasing number of successful oocyte vit-rification) results achieved in the world’slargest human IVF clinic are particularlyattractive to human embryologists, and moti-vate them to attempt to replicate the pub-lished achievements. This distribution ofapplications seems to be established, and mayremain for some years to come, althoughvery few convincing statistics show the superi-ority of either of the two methods for anygiven purpose, provided the technique isapplied strictly according to the establishedguidelines.
THE OPEN PULLED STRAWMETHOD
Principles of the OPS method are described inChapter 2, the device is commercially avail-able, and the technique has been publishedseveral times and is now used in many labora-tories worldwide. Accordingly, here only ashort description of the technical procedure isprovided, along with some illustrations.
The term OPS refers to the preparationtechnology and its application. Standard0.25 ml insemination straws are warmed andpulled just like glass capillaries, to reduce wallthickness and diameter to approximately halfof the original (Figure 4A.1). Straws are thencut at the narrowest point with a sharp razorblade. Sterilization can be performed with gasor irradiation. The process seems to be quiteeasy, however, according to our past experi-ence, the toolmaking is still the most difficult
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 65

part of the whole OPS vitrification. From thispoint of view it is helpful that the straws arenow produced by several companies (includ-ing Minitüb, Germany), and may be pur-chased worldwide.The ‘openness’ mentionedin the name permits extremely simple load-ing and expelling, which is, to date, uniqueamong cryopreservation technologies. Incontrast to the standard 0.25 ml inseminationstraws, the narrow end of the OPS straw pre-disposes it for loading by the capillary effect.After equilibration (according to the specificrequirements of the oocyte or embryo) thesample is loaded into a small (approximately1 µl) droplet, which is immediately touchedwith the narrow end of the OPS straw (Figure4A.2a). As a consequence of the capillaryeffect, most of the solution including thesample will be loaded into the straw, which
VITRIFICATION IN ASSISTED REPRODUCTION
66
a
b
Figure 4A.1 An open pulled straw (a) with approximately50% of the diameter of a standard 0.25 ml inseminationstraw (b) Reproduced with permission from Vajta andNagy.6
a b c
Figure 4A.2 The process of loading, cooling, warming, and expelling at OPS vitrification. (a) Oocytes or embryos areplaced into an approximately 1 µl droplet of cryoprotectant medium; the drop is touched with the OPS straw; as a resultof the capillary effect, the medium with the oocytes or embryos enters the straw. (b) Subsequently, straws are immersedwith a continuous rapid movement into the liquid nitrogen. (c) At warming, straws are immersed into the warmmedium: the solid column melts immediately, and as the result of gravity, embryos or oocytes leave the straw. This lat-ter process can be facilitated by closing the wide end of the straw approximately 1s after immersion: the warming andexpanding air trapped in the straw will expel the solution column. Reproduced with permission from Vajta and Nagy.6
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 66

can subsequently be immersed immediatelyinto the liquid nitrogen (Figure 4A.2b). Atwarming, the straws are simply immersed intothe warming medium preferably containingan osmotic buffer to avoid excessive swelling,the vitrified column melts, and is also dilutedimmediately (again, as the result of the capil-lary effect), and, finally, embryos or oocytesslowly descend into the medium as the resultof gravity. This latter process can be facili-tated by closing the wide top end of the OPSstraw approximately 1 s after immersion:the warming and expanding air trapped in thestraw will expel the solution column into thewarming medium.
Exactly the same approach should be usedat semi-direct transfer, but the warmingmedium has to be placed into a 0.25 ml stan-dard insemination straw, and the narrow endof the OPS straw is inserted into this straw.Although expelling occurs almost alwaysautomatically, it may also be facilitated byclosing the other end of the OPS straw.Control with the naked eye is satisfactory: ifthe solution column disappears from the OPSstraw, the embryo will always be in the 0.25 mlstraw and can be transferred immediatelyinto the recipient animal. With this applica-tion, the two traumatic events (warming andtransfer) occur at the same time, which mayexplain the high pregnancy rates seen whenusing this technique.
APPLICATION IN CATTLE, SHEEP,AND THE GOAT
As successful vitrification of high quality invitro produced bovine blastocysts can besuccessfully performed by using relatively lowcooling rates, high cryoprotectant concentra-tions, and 0.25 ml standard inseminationstraws, the new vitrification methods in thisfield may not offer any real breakthrough.Accordingly, the first convincing evidenceregarding the usefulness of the OPS methodwas provided with cryopreservation of earlystage in vitro produced bovine embryos. Not
only was their in vitro survival impressivelyhigh (at and after day 3 vitrification, therewas no difference in developmental compe-tence compared with the control embryos),but also the day 3 vitrified embryos 24 hoursafter warming were successfully used asdonors for embryonic cell nuclear transfer.7,8
Even more convincing was the 25% blasto-cyst rate that was achieved by OPS vitrifica-tion of matured oocytes, followed by in vitrofertilization and embryo culture to day 7.9
A second OPS vitrification of these blasto-cysts, then transfer of the warmed embryosinto recipients, resulted in three calves beingborn (Figure 4A.3). OPS vitrified and warmedoocytes10 and cytoplasts11 were also used asrecipients for nuclear transfer, and develop-ment to term was achieved. However, by farthe greatest success has been achieved byVieira et al.,12 who used OPS vitrification ofthe extremely cryosensitive immature bovineoocyte, and obtained offspring after in vitromaturation, fertilization, and embryo culture,even after another OPS vitrification at theblastocyst stage (Figure 4A.4).
For logistical reasons, the possibility ofbeing able to cryopreserve cloned embryos
67
VITRIFICATION OF EMBRYOS USING OPEN PULLED STRAWS
Figure 4A.3 Calves born after OPS vitrification of invitro matured oocytes, in vitro fertilization, embryo cul-ture, and another OPS vitrification cycle at the blastocyststage. Reproduced with permission from Vajta et al.9
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 67

may have even more importance. OPS vitrifi-cation resulted in the first non-transgenicand transgenic calves born after somatic cellnuclear transfer, after cryopreservation of theblastocysts before transfer.13,14 Offspring werealso obtained after vitrification of zona-freeblastocysts made with a different nucleartechnology, handmade cloning (i.e. withoutmicromanipulators)15 (Figure 4A.5). In sheep,live births have been achieved after OPSvitrification of zona-included, somatic cellcloned blastocysts (Peura, personal communi-cation) (Figure 4A.6).
All these applications in the cow aremainly in the experimental and biotechno-logical area, as cryopreservation of transfer-able-stage bovine in vivo derived embryos isan established business, and pregnancy ratesachievable with fresh versus conventionallyfrozen embryos are less than 10%, a gap thatis difficult to narrow. Moreover, as empha-sized in Chapter 2, the often mentionedbenefits of vitrification in commercial cattleembryo transfer are not too significant.Vitrification of a single embryo can be per-formed in minutes, compared with the hours
VITRIFICATION IN ASSISTED REPRODUCTION
68
Figure 4A.4 Calves born after OPS vitrification ofimmature oocytes, in vitro maturation, fertilization,embryo culture, and (for the two calves on left) anotherOPS vitrification cycle at the blastocyst stage.Reproduced with permission from Vieira et al.12
Figure 4A.5 Handmade somaticcell cloned calf born after OPSvitrification at the blastocyst stagefollowed by semi-direct transfer.Reproduced with permission fromTecirlioglu et al.15
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 68

required for traditional freezing. However, inmost commercial situations freezing is notrestricted to one or two, but performed withtens of embryos, grouped in a maximum oftwo. Vitrification requires individual repeatsfor all (pairs of) embryos, while for the mostpart of the procedure in traditional freezing,it is performed in one run for an almostunlimited number of (groups of) embryos.The other benefit, the price of equipmentrequired for traditional freezing may not bea decisive factor for an established cattleembryo transfer practice. Roughly the same isapplicable to sheep,16 although the commer-cial aspects of embryo transfer in that speciesare less significant. However, it should bementioned that in the goat, OPS vitrificationhas significantly increased pregnancy ratescompared with traditional freezing.17
VITRIFICATION OF PORCINEEMBRYOS AND OOCYTES WITHTHE OPS METHOD
In pigs, however, the lack of an efficientembryo cryopreservation system profoundlylimits possibilities, regarding both local com-mercial embryo transfer and especially trans-portation including international exchange of
genetic material. The main reason for this isprobably the high lipid content. Porcineembryos are extremely sensitive to chillinginjury that is unavoidable during traditionalfreezing, and survival rates are extremely poor,so commercial application is almost entirelyout of the question. Vitrification was seen as areasonable opportunity to overcome the prob-lem. However, in spite of many attempts andsome promising achievements includingcytoskeleton relaxants and delipidation withcentrifugation followed by vitrification in nor-mal straws, the in vivo developmental ratesremained moderate, and the cumulative costsof the technology were not commensurate withthe achievements. Even initial approaches withOPS vitrification were controversial becausewhile the in vitro survival and developmentalrates were excellent, no pregnancies wereachieved. Finally, with a small modification ofparameters, e.g. extension of the equilibra-tion in the concentrated cryoprotectant solu-tion has resulted in the long-expectedbreakthrough. With these improved parame-ters,18 herds of pigs have been born after OPSvitrification of in vivo derived embryos,remarkable survival and offspring per trans-ferred embryo rates were achieved, and thetechnology is efficient for different develop-mental stages, from morulae to expandedblastocysts.19 Offspring have even beenreported after OPS vitrification and one-stepdilution combined with non-surgical trans-fer20,21 (Figure 4A.7). This latter achievementindicates the potential of this compact tech-nology similar to that widely applied in cattle,although the overall efficiency still can beincreased further.
Regarding the experimental application ofOPS vitrification in pigs, the first survivingembryos produced in vitro were first reportedby Men et al.22 Subsequently, results were fur-ther improved after chemical delipidation.23 Asignificant breakthrough from both thesomatic cell nuclear transfer and vitrificationpoint of view, was the production of healthypiglets after delipidation of oocytes with
69
VITRIFICATION OF EMBRYOS USING OPEN PULLED STRAWS
Figure 4A.6 Somatic cell cloned sheep born after OPSvitrification at the blastocyst stage. Published withpermission of T. T. Peura.
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 69

centrifugation, micromanipulation-basednuclear transfer, and OPS vitrification (Figure4A.8).24 Similar results have been achieved byDu et al. (unpublished) by using handmadecloned embryos, reconstructed from oocytesdelipidated by using a simplified procedure.As the latter work was performed by using theCryotop vitrification method, the achieve-ments provide further evidence that the two
methods may be equally efficient for anygiven purpose.
OTHER DOMESTIC ANDEXPERIMENTAL ANIMALS
In the rabbit, OPS vitrification of in vivoderived morulae was found to be superiorcompared with traditional freezing, and vitri-fication performed in 0.25 ml inseminationstraws in terms of in vitro survival and birthsof kittens.25 Similar results were achievedearlier with a modified version of theOPS, the ‘sealed open pulled straw proce-dure’, when the method was compared withvitrification in 0.25 ml straws.26
In the horse, both immature and matureoocyte vitrification was successfully per-formed with the OPS method, although thesurvival rates were lower than those in cat-tle.27 Later observations have revealed seriousspindle damage of matured oocytes after OPSvitrification, although for immature oocytes,the resumption of meiosis was higher afterOPS than after traditional freezing.28 Forequine embryos, traditional slow rate freezingand OPS vitrification were found to beequally efficient with respect to in vitrosurvival rates.29
In contrast to the equine results, in themouse a high proportion of oocytes preservedpatterns of meiotic spindles and chromo-somes after OPS vitrification. The results weresignificantly better than those achieved withvitrification in normal insemination straws.30
Survival rates with OPS were lower, but couldbe improved by modification of the appliedtechnique (closed pulled straws).31 For mouseblastocysts, high survival and in vitro develop-mental rates were reported both with the OPSand its modification, the glass micropipette(GMP) method.32 With further optimization ofparameters for OPS vitrification, a method wasestablished that was suitable to cryopreservemouse embryos from the 4-cell to the earlyblastocyst stage.33 An even more impressive
VITRIFICATION IN ASSISTED REPRODUCTION
70
Figure 4A.8 Piglets born after OPS vitrification ofsomatic cell cloned blastocysts produced from delipatedoocytes. Reproduced with permission from Li et al.
Figure 4A.7 Piglets born after OPS vitrification of invivo derived embryos and non-surgical transfer.Reproduced with permission from Cuello et al.
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 70

achievement was reported for rat embryos,where double vitrification was applied byusing a super-fine variation of the OPS straws,cryopreserving the embryos first at the earlymorula, then at various subsequent develop-mental stages.34
NON-DOMESTIC SPECIES
Although the low costs and the very simpleequipment predispose vitrification for cryo-preservation of oocytes and embryos of wildand endangered species permitting workunder compromised conditions, surprisinglyfew papers deal with this area, probablyreflecting the low level of attention given to,and the even lower funding of, this veryimportant field. Among the few publishedworks successful experiments involving threemammals should be mentioned. In Siberiantigers, OPS vitrification of early stageembryos resulted in further development, incontrast to traditional freezing.35 Minke whaleoocytes were successfully vitrified with boththe OPS and the Cryotop method, althoughresults were slightly better with Cryotop.36 Thegreatest promise for endangered carnivore
species is, however, the first reported birthafter OPS vitrification of in vivo derived car-nivore embryos, achieved in the Europeanpolecat (Figure 4A.9).37
REASONS TO USE THE OPSVITRIFICATION METHOD
As mentioned in the introduction, the OPStechnique – compared with the other high-rate cooling vitrification procedures – is rela-tively robust and is based on a simplemodification of the 0.25 ml inseminationstraw that is the most common tool for cryo-preservation in reproductive biology.Moreover, loading and expelling of samples issimple, does not require any special or delicatetools, and can be learned in minutes. The tech-nique, if properly applied, ensures appropriatecooling and warming rates (as shown above) fora wide variety of mammalian oocytes andembryos of different developmental stages andorigin. Storage and sample identification donot require any special approach as they arebased on the traditional tools and methods ofcryopreservation.
A unique benefit of OPS vitrification forapplication in the animal field is the possibilityof one-step dilution and semi-direct transfer.This option was described first for cattle embryovitrification,5 and was used afterwards for vari-ous purposes including transfer of in vitro fertil-ized, embryonic and somatic cell clonedembryos, both with and without their zonapellucida. Although no direct comparison isavailable, according to our experience the semi-direct transfer results in at least as high preg-nancy and calving rates as the stepwise dilutionand delayed transfer. This is an obvious benefit,and may open a wide scale practical applicationof OPS in cattle. Moreover, the method for suc-cessful one-step dilution and transfer of OPSvitrified embryos has also been published forsmall ruminants,16 and the mouse,38 wideningthe possibilities for practical application inmany mammalian species.
71
VITRIFICATION OF EMBRYOS USING OPEN PULLED STRAWS
Figure 4A.9 European polecat pups born after vitrifica-tion of in vivo derived embryos with the OPS method.Reproduced with permission from Piltti et al.37
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 71

1. Kuleshova L, Gianaroli L, Magli C, TrounsonA. Birth following vitrification of a small num-ber of human oocytes. Hum Reprod 1999; 14,3077–79.
2. Selman et al., 20063. Chian RC, Kuwayama M, Tan L et al. High
survival rate of bovine oocytes matured invitro following vitrification. J Reprod Dev2004; 50: 685–96.
4. Fujihira T, Nagai H, Fukui Y. Relationshipbetween equilibration times and the presenceof cumulus cells, and effect of Taxol treatmentfor vitrification of in vitro matured porcineoocytes. Cryobiology 2005; 51: 339–43.
5. Vajta G, Murphy C, Macháty Z et al. In strawdilution of in vitro produced bovine blastocystsafter vitrification with the Open Pulled Straw(OPS) method. Vet Rec 1999; 144: 180–81.
6. Vajta and Nagy, 2006.7. Vajta G, Booth PJ, Holm P, Jacobsen H, Greve T,
Callesen H. The use of vitrified Day 3embryos as donors in bovine nuclear transfer.Cryo Letters 1997; 18: 355–58.
8. Peura TT, Lane MW, Vajta G, Trounson AO.Post-thaw in vitro survival of vitrified clonedbovine embryos. Vet Rec 1997; 140: 404.
9. Vajta G, Kuwayama M, Holm P et al. Openpulled straw vitrification: a new way to reducecryoinjuries of bovine ova and embryos. MolReprod Dev 1998; 51, 53–58.
10. Hou YP, Dai YP, Zhu SE et al. Bovine oocytesvitrified by the open pulled straw method andused for somatic cell cloning supported devel-opment to term. Theriogenology 2005; 64,1384–91.
11. Booth PJ, Vajta G, Høj A et al. Full-term devel-opment of nuclear transfer calves producedfrom Open Pulled Straw (OPS) vitrified cyto-plasts. Theriogenology 1999; 51: 999–1006
12. Vieira AD, Mezzalira A, Barieri DP et al. Calvesborn after open pulled straw vitrification of
immature bovine oocytes. Cryobiology 2002;45, 91–94.
13. French AJ, Hall VJ, Korfiatis NT et al. Viabilityof cloned bovine embryos following OPS vitri-fication. Theriogenology 2002; 57, 413.
14. French AJ, Lewis IM, Ruddock NT et al.Generation of aS1 casein gene transgeniccalves by nuclear transfer. Biol Reprod 2003;68, 240
15. Tecirlioglu RT, French AJ, Lewis IM et al.2003 Birth of a cloned calf derived from avitrified cloned embryo. Reprod Fertil Dev 15,361–66.
16. Isachenko V, Alabart JL, Dattena M et al. Newtechnology for vitrification and field (micro-scope free) warming and transfer of smallruminant embryos. Theriogenology 2003; 59,1209–18
17. El-Gayar and Holtz, 200118. Berthelot et al. 2000.19. Berthelot F, Martinat-Botté F, Perreau C et al.
Birth of piglets after OPS vitrification andtransfer of compacted morula stage embryoswith intact zona pellucida. Reprod Nutr Dev2001; 41: 267–72.
20. Cuello C, Gil MA, Parrila I et al. In vitro devel-opment following one-step dilution of OPSvitrified porcine blastocysts. Theriogenology2004; 62, 1144–52.
21. Cuello et al., 200522. Men H, Agca Y, Critser E et al. Beneficial
effects of serum supplementation during invitro production of porcine embryos on theirability to survive cryopreservation by the openpulled straw vitrification. Theriogenology2005; 64, 1340–49.
23. Men et al., 2006.24. Li R, Lai L, Wax D, Hao Y, et al. Cloned
transgenic swine via in vitro production andcryopreservation. Biol Reprod. 2006; 75:226–30.
VITRIFICATION IN ASSISTED REPRODUCTION
72
Additionally, OPS vitrification offers asolution for disease transmission problemsthat are a major concern regarding the newvitrification procedures. With the application offiltered, separately stored liquid nitrogen forcooling and using a hermetically isolated dou-ble-straw technique for storage,39 the possibility
of cross-contamination is eliminated (as con-firmed by independent investigation),40 whileall the previously mentioned benefits (highcooling rate, low chilling injury, low cryoprotec-tant concentration, and consequently low toxicand osmotic damage, easy and safe handling,possibility for direct transfer) are still preserved.
References
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 72

25. Naik BR, Rao BS, Vagdevi R, Gnanprakash M,Amarnath D, Rao VH. Conventional slowfreezing, vitrification and open pulled straw(OPS) vitrification of rabbit embryos. AnimReprod Sci 2005; 86, 329–38.
26. Lopez-Bejar M, Lopez-Gatius F. Nonequilibriumcryopreservation of rabbit embryos using amodified (sealed) open pulled straw procedure.Theriogenology 2002; 58, 1541–52.
27. Hurtt AE, Landim-Alvarenga F, Seidel GE Jr,Squires EL. Vitrification of immature andmature equine and bovine oocytes in an ethyl-ene glycol, ficoll and sucrose solution usingopen-pulled straws. Theriogenology 2000; 54:119–128, 2000.
28. Tharasanit T, Colenbrander B, Stout TAE.Effect of maturation stage at cryopreservationof post-thaw cytoskeleton quality and fertiliz-ability of equine oocytes. Mol Reprod Dev2006; 73, 627–37.
29. Moussa M, Bersinger I, Doligez P, et al. Invitro comparisons of two cryopreservationtechniques for equine embryos: slow-coolingand open pulled straw (OPS) vitrification.Theriogenology 2005; 64, 1619–32.
30. Chen SU, Lien YR, Chen HF et al. Openpulled straws for vitrification of mature mouseoocytes preserve patterns of meiotic spindlesand chromosomes better than conventionalstraws. Hum Reprod 2000; 15: 2598–603.
31. Chen SU, Lien YR, Cheng YY et al.Vitrification of mouse oocytes using closedpulled straws (CPS) achieves a high survival andpreserves good patterns of meiotic spindles,compared with conventional straws, openpulled straws (OPS) and grids. Hum Reprod2001; 11: 2350–6.
32. Kong IK, Lee SI, Cho SG et al. Comparison ofopen pulled straw (OPS) vs glass micropipette(GMP) vitrification in mouse blastocysts.Theriogenology 2000; 53, 1817–26.
33. Zhou GB, Hou YP, Jin F, et al. Vitrification ofmouse embryos at various stages by open-pulled straw (OPS) method. Anim Biotechnol2005; 16, 153–63.
34. Isachenko V, Alabart JL, Vajta G et al. Doublevitrification of rat embryos at different devel-opmental stages using an identical protocol.Theriogenology 2002; 60, 445–52.
35. Crichton EG, Bedows E, Miller-Lindholm AKet al. Efficacy of porcine gonadotropinsfor repeated stimulation of ovarian activityfor oocyte retrieval and in vitro embryoproduction and cryopreservation in Siberiantigers (Panthera tigris altaica). Biol Reprod2003; 68, 105–13.
36. Iwayama H, Hochi S, Kato M et al. Effects ofcryodevice type and donor’s sexual maturityon vitrification of minke whale (Balaenopterbonaerensis) oocytes at germinal vesicle stage.Zygote 2004; 12, 333–38.
37. Piltti K, Lindeberg H, Aalto J et al. Live cubsborn after transfer of OPS vitrified-warmedembryos in the farmed European polecat(Mustela putorius). Theriogenology 2004; 61,811–20.
38. Yang QE, Hou YP, Zhou GB, Yang ZQ, ZhuSE. Stepwise in-straw dilution and directtransfer using open pulled straws (OPS) in themouse: a potential model for field manipula-tion of vitrified embryos. J Reprod Dev; inpress.
39. Vajta G, Lewis IM, Kuwayama M, Greve T,Callesen H. Sterile application of the OpenPulled Straw (OPS) vitrification method. CryoLetters 1998; 19, 389–92.
40. Bielanski et al. in press41. El-Gayar
73
VITRIFICATION OF EMBRYOS USING OPEN PULLED STRAWS
04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 73

04a Tucker 8029.qxd 8/23/2007 2:11 PM Page 74

75
Vitrification in animal reproduction: vitrification of embryos using conventional straws with an ethylene glycol-based solutionsMagosaburo Kasai and Keisuke Edashige
4B
VITRIFICATION
With conventional slow freezing, a process firstdeveloped by Whittingham et al.,1 embryossuspended in a solution containing a low con-centration (1–2 mol/L) of permeating cryopro-tectant are cooled slowly (0.3–0.5°C/min) beforethe sample is cooled with liquid nitrogen. Thismethod, derived deductively by Peter Mazur,was proven effective and is now used routinelywith the aid of a programmable freezer.However, it requires a long cooling stage and amachine for controlling the cooling rate. Inaddition, the formation of ice in the preserva-tion solution can lead to intracellular ice form-ing, probably the biggest obstacle to thesuccessful cryopreservation of embryos.
With slow freezing, embryos cooled slowlyto subzero temperatures between − 30°C and− 70°C before cooling with liquid nitrogen areplaced in a very concentrated solution, whichconstitutes small channels among ice. Undersuch conditions, ice forms in neither the solu-tion channel nor the embryo even after thecooling in liquid nitrogen. Therefore, even withslow freezing, embryos and the small concen-trated channels where they are located arevitrified. Vitrification is the solidification of asolution without crystallization. Therefore, if asolution similar to the one in the unfrozenchannel in slowly frozen samples is composedusing a very high concentration of cryoprotec-tant(s) and embryos are equilibrated in it, the
sample could be directly cooled in liquid nitro-gen from 0°C or above. Based on this idea, Ralland Fahy2 developed a simple method ofcryopreservation called vitrification, in whichsamples are cooled in liquid nitrogen withouta slow cooling stage.
Their idea was proven effective3 and vitri-fication has markedly simplified the coolingprocess. In addition, the method has thepotential to increase rates of embryonicsurvival because ice does not form duringcooling. Although extracellular ice can formduring warming in less concentrated solu-tions, its effect could be minimized by warm-ing the sample rapidly, which would helpprevent the formation of intracellular ice(devitrification). On the other hand, vitrifica-tion has a serious disadvantage in thatthe chemical toxicity of the solution is quitehigh, because the concentration of cryopro-tectant(s) is quite high (5–8 mol/L), and thetemperature at which embryos are exposed tothe solution is high.
VITRIFICATION USING AN ETHYLENE GLYCOL-BASED SOLUTION
The first vitrification solution, named VS1,2
contained three permeating cryoprotectants,dimethylsulfoxide (DMSO), acetamide, and pro-pylene glycol. This solution, or a modification
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 75
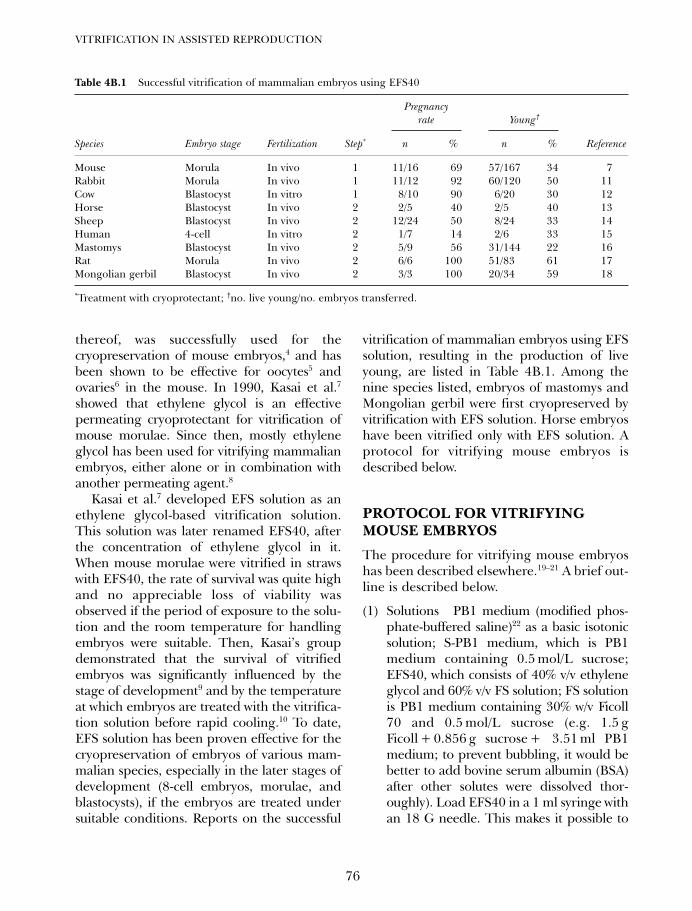
thereof, was successfully used for thecryopreservation of mouse embryos,4 and hasbeen shown to be effective for oocytes5 andovaries6 in the mouse. In 1990, Kasai et al.7
showed that ethylene glycol is an effectivepermeating cryoprotectant for vitrification ofmouse morulae. Since then, mostly ethyleneglycol has been used for vitrifying mammalianembryos, either alone or in combination withanother permeating agent.8
Kasai et al.7 developed EFS solution as anethylene glycol-based vitrification solution.This solution was later renamed EFS40, afterthe concentration of ethylene glycol in it.When mouse morulae were vitrified in strawswith EFS40, the rate of survival was quite highand no appreciable loss of viability wasobserved if the period of exposure to the solu-tion and the room temperature for handlingembryos were suitable. Then, Kasai’s groupdemonstrated that the survival of vitrifiedembryos was significantly influenced by thestage of development9 and by the temperatureat which embryos are treated with the vitrifica-tion solution before rapid cooling.10 To date,EFS solution has been proven effective for thecryopreservation of embryos of various mam-malian species, especially in the later stages ofdevelopment (8-cell embryos, morulae, andblastocysts), if the embryos are treated undersuitable conditions. Reports on the successful
vitrification of mammalian embryos using EFSsolution, resulting in the production of liveyoung, are listed in Table 4B.1. Among thenine species listed, embryos of mastomys andMongolian gerbil were first cryopreserved byvitrification with EFS solution. Horse embryoshave been vitrified only with EFS solution. Aprotocol for vitrifying mouse embryos isdescribed below.
PROTOCOL FOR VITRIFYINGMOUSE EMBRYOS
The procedure for vitrifying mouse embryoshas been described elsewhere.19–21 A brief out-line is described below.
(1) Solutions PB1 medium (modified phos-phate-buffered saline)22 as a basic isotonicsolution; S-PB1 medium, which is PB1medium containing 0.5 mol/L sucrose;EFS40, which consists of 40% v/v ethyleneglycol and 60% v/v FS solution; FS solutionis PB1 medium containing 30% w/v Ficoll70 and 0.5 mol/L sucrose (e.g. 1.5 gFicoll + 0.856 g sucrose + 3.51 ml PB1medium; to prevent bubbling, it would bebetter to add bovine serum albumin (BSA)after other solutes were dissolved thor-oughly). Load EFS40 in a 1 ml syringe withan 18 G needle. This makes it possible to
VITRIFICATION IN ASSISTED REPRODUCTION
76
Table 4B.1 Successful vitrification of mammalian embryos using EFS40
Pregnancyrate Young†
Species Embryo stage Fertilization Step* n % n % Reference
Mouse Morula In vivo 1 11/16 69 57/167 34 7Rabbit Morula In vivo 1 11/12 92 60/120 50 11Cow Blastocyst In vitro 1 8/10 90 6/20 30 12Horse Blastocyst In vivo 2 2/5 40 2/5 40 13Sheep Blastocyst In vivo 2 12/24 50 8/24 33 14Human 4-cell In vitro 2 1/7 14 2/6 33 15Mastomys Blastocyst In vivo 2 5/9 56 31/144 22 16Rat Morula In vivo 2 6/6 100 51/83 61 17Mongolian gerbil Blastocyst In vivo 2 3/3 100 20/34 59 18
*Treatment with cryoprotectant; †no. live young/no. embryos transferred.
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 76

use a minimal amount of fresh solution foreach sample (evaporation will make thesolution concentrated).
(2) Liquid nitrogen Pour liquid nitrogeninto a dewar flask with a large mouth orinto a Styrofoam box, and float a thinStyrofoam boat on it.
(3) Vitrification of mouse morulae Intoa 0.25 ml insemination straw (13 cm,including the cotton plug), load 65 mmS-PB1, 15 mm air, 3 mm EFS40, 4 mmair, and 13 mm EFS40, successively.Place the straw horizontally at 25°C.Place mouse morulae in the 13 mmEFS40 in the straw directly with a mini-mal amount of PB1 medium using a finepipette. If this is technically difficult,embryos can be washed in EFS40 quicklybefore being loaded in straw. Aspiratethe straw to seal the powder end andthen seal the open end with a heatsealer. After 30–60 s of exposure ofembryos to EFS40, place the straw onthe Styrofoam boat on liquid nitrogenhorizontally to cool the sample in thegas phase (this prevents fracture dam-age.)23 Leave it for 3 min or more beforethe straw is stored in liquid nitrogen.
(4) Vitrification of mouse 2-cell embryos andblastocysts For vitrifying 2-cell embryosand blastocysts, embryos need to havebeen pretreated with PB1 medium con-taining 10% v/v ethylene glycol for 5 min(or with EFS20, which is a mixture of 20%v/v ethylene glycol and 80% v/v FS solu-tion, for 2 min) before being transferred toEFS40 in straw. In this case, the exposuretime in EFS40 at room temperatureshould be limited to 30 s.
(5) Warming For warming, remove the strawfrom the liquid nitrogen and keep it in airat room temperature for 10 s (this also pre-vents fracture damage.)23 Then, immerseit into a 20–25°C water bath. After ~7 s,when the S-PB1 medium begins to melt,remove the straw, and wipe it quickly.Keeping the straw horizontal, cut off both
ends. Tilt the straw slightly with the EFSside down, and perfuse the straw slowlywith 1 ml of S-PB1 medium using a 1 mlsyringe into a watch glass. Shake the watchglass gently to dilute the EFS40. Recoverthe embryos with a pipette and place themin S-PB1 medium prepared in a culturedish. About 5 min after the perfusion,transfer the embryos to PB1 medium.
COMPONENTS OF THE VITRIFICATION SOLUTION
During the cryopreservation process, embryosare at risk of injury from chilling (this appliesto certain types of embryos, depending onthe species and the developmental stage), thetoxicity of the cryoprotectant, extracellularice, intracellular ice, fracture damage,osmotic swelling, and osmotic shrinkage.24 Toobtain high rates of survival, all these prob-lems must be circumvented. Among theinjuries, the damage caused by the formationof intracellular ice during cooling or warmingis surely the greatest obstacle to overcome. Inaddition, injury from osmotic swelling is alsoa major factor, which can occur duringremoval of the permeated cryoprotectantafter warming.25
For preventing intracellular ice from form-ing, rapidly permeating cryoprotectants aresuitable because permeation by the cryopro-tectant is essential to vitrify the embryo. Toprevent osmotic swelling during removal ofthe cryoprotectant(s) after warming, rapidlypermeating agents are also suitable, becausethe faster the diffusion of the intracellularcryoprotectant out of the cell, the lower therisk of osmotic over-swelling.
We examined the permeability of mouseembryos to various cryoprotectants from theapparent volume change in cryoprotectantsolutions.26 We found the permeability to varydepending on the developmental stage of theembryo and on the cryoprotectant (Figure4B.1). In mouse morulae, ethylene glycolpermeates quite rapidly. We have obtained
77
VITRIFICATION IN CONVENTIONAL STRAWS WITH AN ETHYLENE GLYCOL SOLUTION
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 77
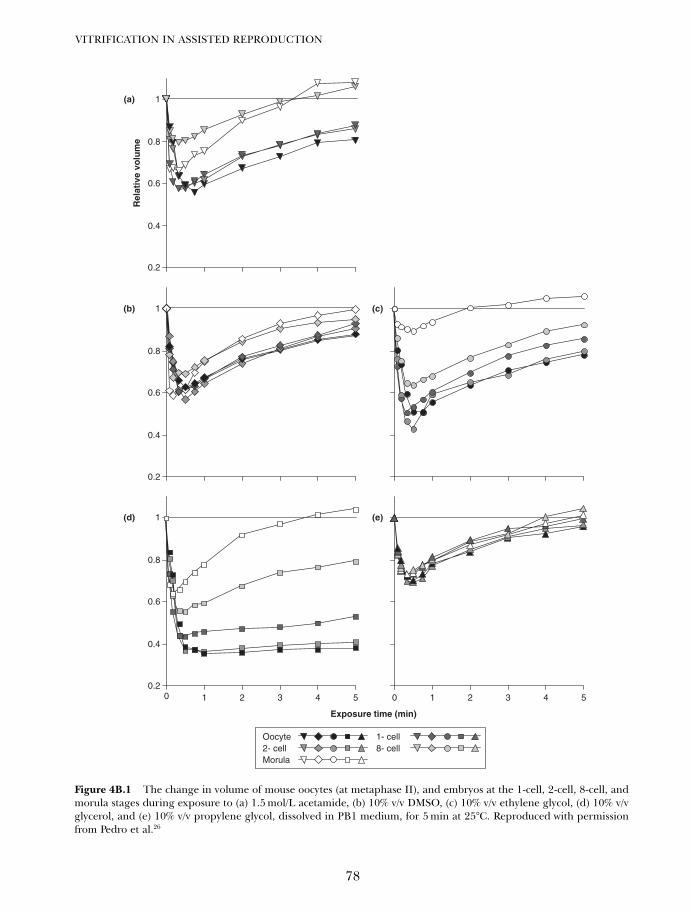
VITRIFICATION IN ASSISTED REPRODUCTION
78
Rel
ativ
e vo
lum
e
Exposure time (min)
0 1 2 3 40.2
0.4
0.6
0.8
1
55 0 1 2 3 4
0.2
0.4
0.6
0.8
1
0.2
0.4
0.6
0.8
1
Morula8- cell2- cell1- cellOocyte
(a)
(b) (c)
(d) (e)
Figure 4B.1 The change in volume of mouse oocytes (at metaphase II), and embryos at the 1-cell, 2-cell, 8-cell, andmorula stages during exposure to (a) 1.5 mol/L acetamide, (b) 10% v/v DMSO, (c) 10% v/v ethylene glycol, (d) 10% v/vglycerol, and (e) 10% v/v propylene glycol, dissolved in PB1 medium, for 5 min at 25°C. Reproduced with permissionfrom Pedro et al.26
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 78

similar results for bovine embryos,27 and rabbitembryos (unpublished observation). Therefore,it is suggested that the permeability of embryosto cryoprotectants is less species specific andmore stage specific.
In addition to the injuries caused by intra-cellular ice and osmotic swelling, the chemi-cal toxicity of the permeating cryoprotectantis a major obstacle to vitrification. The sensi-tivity of embryos to chemical toxicity woulddepend on the cryoprotectant and the speciesand developmental stage of the embryos.Kasai28 examined the toxicity of five majorpermeating cryoprotectants to mouse moru-lae and showed that ethylene glycol is theleast toxic (Table 4B.2). Therefore, in termsof ability to permeate and chemical toxicity,ethylene glycol is the most suitable cryopro-tectant, at least for mouse morulae.
Rall and Fahy2 suggested that the inclusionof a macromolecule promotes vitrification of asolution, and used polyethylene glycol. In EFSsolution, Ficoll is included as a non-permeatingpolymer, because it has high solubility, lowviscosity, and low toxicity. It has been shownthat even a large amount of Ficoll is virtuallynon-toxic.28 A large amount of polymer occu-pies a significant proportion of the solution vol-ume. Therefore, its inclusion must increase theproportion of permeating cryoprotectant perwater volume. This may be one mechanismthat promotes vitrification of the solution.
EFS solution contains not only ethyleneglycol and Ficoll, but also sucrose as a non-permeating small sugar which has consider-able osmotic effect. The inclusion of sucroseis quite effective at reducing the apparenttoxicity of ethylene glycol, probably becausesucrose promotes the shrinkage of embryos,which would restrict excess permeation ofethylene glycol and thus reduce its toxiceffect.7 In addition, sucrose helps preventover-swelling during the removal of ethyleneglycol after warming.29 Like Ficoll, sucrose isvirtually non-toxic.28,30
THE PATHWAY FOR THEMOVEMENT OF WATER ANDCRYOPROTECTANTS IN EMBRYOS
For the successful cryopreservation of embryos,the smooth movement of water and cryopro-tectants through the plasma membrane isessential. In most types of cells, water movesthrough the plasma membrane with limitedpermeability by simple diffusion through thelipid bilayer. However, the plasma membraneof some cells, e.g. human red blood cells andcells in renal proximal tubules, is extremelypermeable by water. In the 1990s, smallintrinsic membrane proteins that act as waterchannels, called aquaporins (AQPs), were dis-covered and characterized.31 The channelsoccur in two groups: one is highly selectivefor water and the other transports not onlywater but also neutral solutes with a smallmolecular weight, such as cell-permeatingcryoprotectants.
We have shown that mRNAs of AQP3 andAQP7 are present in metaphase II oocytes, 4-cellembryos, morulae, and blastocysts (Figure4B.2), and that mRNAs of AQP8 and AQP9 areexpressed in blastocysts in the mouse.32
Offenberg et al.33 also detected mRNAs of AQPs,and Barcroft et al.34 detected AQP proteins inmouse embryos. However, it is important toknow whether such a channel pathway actuallyplays a significant role in the movement ofwater and cryoprotectants in embryos.
79
VITRIFICATION IN CONVENTIONAL STRAWS WITH AN ETHYLENE GLYCOL SOLUTION
Table 4B.2 Survival of mouse morulae exposed to 30%v/v cryoprotectants for 20 min at 20° C. Data fromKasai28
Survival rate
n %
Ethylene glycol 48/49 98*
Glycerol 44/50 88*
DMSO 41/60 68†
Propylene glycol 8/48 17‡
Acetamide 0/60 0**
Values with different superscripts differ significantly(P < 0.05, χ2 analysis or Fisher’s probability test).
Permeatingcryoprotectant
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 79
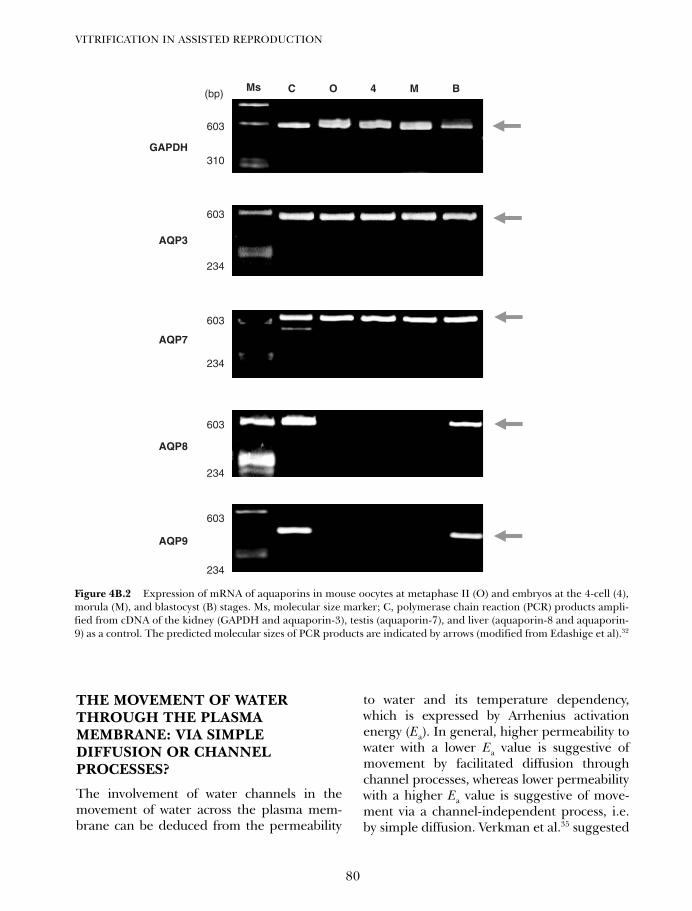
THE MOVEMENT OF WATERTHROUGH THE PLASMAMEMBRANE: VIA SIMPLEDIFFUSION OR CHANNELPROCESSES?
The involvement of water channels in themovement of water across the plasma mem-brane can be deduced from the permeability
to water and its temperature dependency,which is expressed by Arrhenius activationenergy (Ea). In general, higher permeability towater with a lower Ea value is suggestive ofmovement by facilitated diffusion throughchannel processes, whereas lower permeabilitywith a higher Ea value is suggestive of move-ment via a channel-independent process, i.e.by simple diffusion. Verkman et al.35 suggested
VITRIFICATION IN ASSISTED REPRODUCTION
80
603
234
AQP8
603
234
AQP9
C O 4 M B(bp)
603
310GAPDH
603
234
AQP7
603
234
AQP3
Ms
Figure 4B.2 Expression of mRNA of aquaporins in mouse oocytes at metaphase II (O) and embryos at the 4-cell (4),morula (M), and blastocyst (B) stages. Ms, molecular size marker; C, polymerase chain reaction (PCR) products ampli-fied from cDNA of the kidney (GAPDH and aquaporin-3), testis (aquaporin-7), and liver (aquaporin-8 and aquaporin-9) as a control. The predicted molecular sizes of PCR products are indicated by arrows (modified from Edashige et al).32
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 80
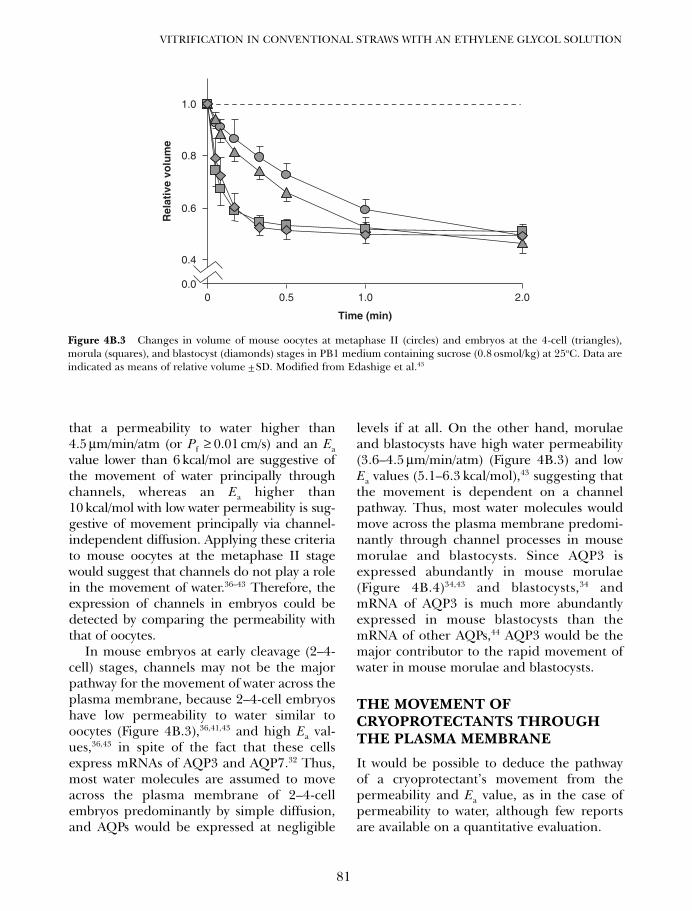
that a permeability to water higher than4.5 µm/min/atm (or Pf ≥ 0.01 cm/s) and an Eavalue lower than 6 kcal/mol are suggestive ofthe movement of water principally throughchannels, whereas an Ea higher than10 kcal/mol with low water permeability is sug-gestive of movement principally via channel-independent diffusion. Applying these criteriato mouse oocytes at the metaphase II stagewould suggest that channels do not play a rolein the movement of water.36–43 Therefore, theexpression of channels in embryos could bedetected by comparing the permeability withthat of oocytes.
In mouse embryos at early cleavage (2–4-cell) stages, channels may not be the majorpathway for the movement of water across theplasma membrane, because 2–4-cell embryoshave low permeability to water similar tooocytes (Figure 4B.3),36,41,43 and high Ea val-ues,36,43 in spite of the fact that these cellsexpress mRNAs of AQP3 and AQP7.32 Thus,most water molecules are assumed to moveacross the plasma membrane of 2–4-cellembryos predominantly by simple diffusion,and AQPs would be expressed at negligible
levels if at all. On the other hand, morulaeand blastocysts have high water permeability(3.6–4.5 µm/min/atm) (Figure 4B.3) and lowEa values (5.1–6.3 kcal/mol),43 suggesting thatthe movement is dependent on a channelpathway. Thus, most water molecules wouldmove across the plasma membrane predomi-nantly through channel processes in mousemorulae and blastocysts. Since AQP3 isexpressed abundantly in mouse morulae(Figure 4B.4)34,43 and blastocysts,34 andmRNA of AQP3 is much more abundantlyexpressed in mouse blastocysts than themRNA of other AQPs,44 AQP3 would be themajor contributor to the rapid movement ofwater in mouse morulae and blastocysts.
THE MOVEMENT OFCRYOPROTECTANTS THROUGHTHE PLASMA MEMBRANE
It would be possible to deduce the pathwayof a cryoprotectant’s movement from thepermeability and Ea value, as in the case ofpermeability to water, although few reportsare available on a quantitative evaluation.
81
VITRIFICATION IN CONVENTIONAL STRAWS WITH AN ETHYLENE GLYCOL SOLUTION
Rel
ativ
e vo
lum
e
0 0.5 1.0 2.0
Time (min)
0.4
0.6
0.8
1.0
0.0
Figure 4B.3 Changes in volume of mouse oocytes at metaphase II (circles) and embryos at the 4-cell (triangles),morula (squares), and blastocyst (diamonds) stages in PB1 medium containing sucrose (0.8 osmol/kg) at 25oC. Data areindicated as means of relative volume ± SD. Modified from Edashige et al.43
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 81
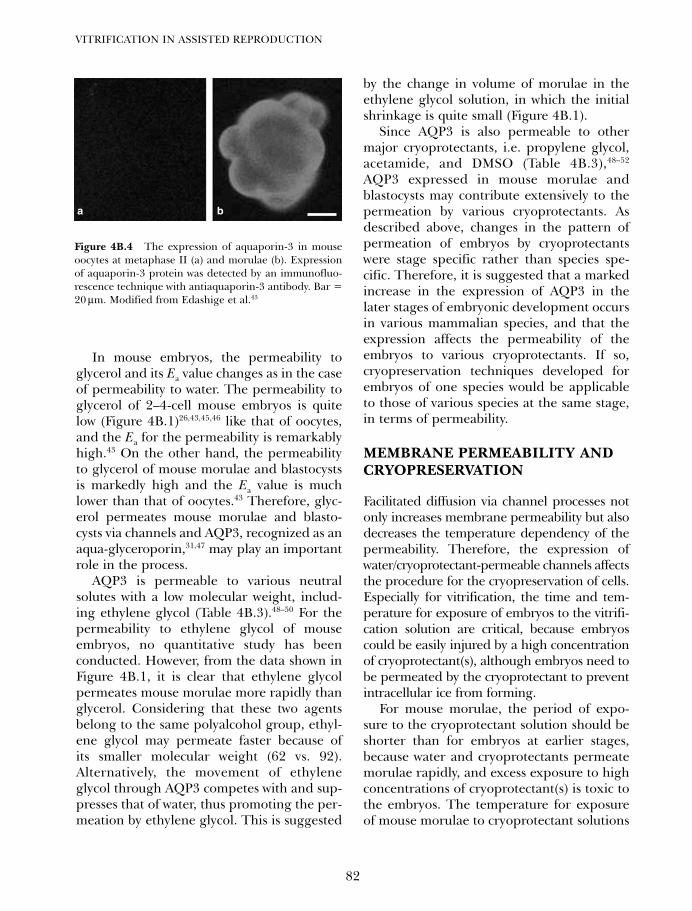
In mouse embryos, the permeability toglycerol and its Ea value changes as in the caseof permeability to water. The permeability toglycerol of 2–4-cell mouse embryos is quitelow (Figure 4B.1)26,43,45,46 like that of oocytes,and the Ea for the permeability is remarkablyhigh.43 On the other hand, the permeabilityto glycerol of mouse morulae and blastocystsis markedly high and the Ea value is muchlower than that of oocytes.43 Therefore, glyc-erol permeates mouse morulae and blasto-cysts via channels and AQP3, recognized as anaqua-glyceroporin,31,47 may play an importantrole in the process.
AQP3 is permeable to various neutralsolutes with a low molecular weight, includ-ing ethylene glycol (Table 4B.3).48–50 For thepermeability to ethylene glycol of mouseembryos, no quantitative study has beenconducted. However, from the data shown inFigure 4B.1, it is clear that ethylene glycolpermeates mouse morulae more rapidly thanglycerol. Considering that these two agentsbelong to the same polyalcohol group, ethyl-ene glycol may permeate faster because ofits smaller molecular weight (62 vs. 92).Alternatively, the movement of ethyleneglycol through AQP3 competes with and sup-presses that of water, thus promoting the per-meation by ethylene glycol. This is suggested
by the change in volume of morulae in theethylene glycol solution, in which the initialshrinkage is quite small (Figure 4B.1).
Since AQP3 is also permeable to othermajor cryoprotectants, i.e. propylene glycol,acetamide, and DMSO (Table 4B.3),48–52
AQP3 expressed in mouse morulae andblastocysts may contribute extensively to thepermeation by various cryoprotectants. Asdescribed above, changes in the pattern ofpermeation of embryos by cryoprotectantswere stage specific rather than species spe-cific. Therefore, it is suggested that a markedincrease in the expression of AQP3 in thelater stages of embryonic development occursin various mammalian species, and that theexpression affects the permeability of theembryos to various cryoprotectants. If so,cryopreservation techniques developed forembryos of one species would be applicableto those of various species at the same stage,in terms of permeability.
MEMBRANE PERMEABILITY ANDCRYOPRESERVATION
Facilitated diffusion via channel processes notonly increases membrane permeability but alsodecreases the temperature dependency of thepermeability. Therefore, the expression ofwater/cryoprotectant-permeable channels affectsthe procedure for the cryopreservation of cells.Especially for vitrification, the time and tem-perature for exposure of embryos to the vitrifi-cation solution are critical, because embryoscould be easily injured by a high concentrationof cryoprotectant(s), although embryos need tobe permeated by the cryoprotectant to preventintracellular ice from forming.
For mouse morulae, the period of expo-sure to the cryoprotectant solution should beshorter than for embryos at earlier stages,because water and cryoprotectants permeatemorulae rapidly, and excess exposure to highconcentrations of cryoprotectant(s) is toxic tothe embryos. The temperature for exposureof mouse morulae to cryoprotectant solutions
VITRIFICATION IN ASSISTED REPRODUCTION
82
Figure 4B.4 The expression of aquaporin-3 in mouseoocytes at metaphase II (a) and morulae (b). Expressionof aquaporin-3 protein was detected by an immunofluo-rescence technique with antiaquaporin-3 antibody. Bar =20 µm. Modified from Edashige et al.43
a b
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 82

and for the dilution of cryoprotectants wouldnot be a very important factor from the view-point of membrane permeability, because thechannel processes are less affected by tem-perature. However, in terms of the toxicity ofcryoprotectants, the exposure of embryos tocryoprotectant solutions at high temperatureshould be avoided, because cryoprotectantsare more toxic at higher temperatures.
Blastocysts are also highly permeable towater and cryoprotectants like morulae, butthey have a blastocoel. In mouse blastocysts, ithas been shown that the distribution of AQPsin the plasma membrane is not homogeneousamong the apical and basolateral sides of thetrophectoderm and inner cell mass.34 Thissuggests that blastocysts have compartmentsdiffering in membrane permeability, althoughoverall they are highly permeable to water andcryoprotectants. Moreover, expanded blasto-cysts contain a large amount of water, most ofwhich has to diffuse out before cryopreserva-tion. At the same time, the cryoprotectant(s) hasto permeate the blastocoel through trophoblas-tic cells. To minimize the toxic effect of the cry-oprotectant while promoting dehydration andpermeation, a modified procedure is required
for the cryopreservation of blastocysts havinga large blastocoel, in which ice forms easily.Usually, blastocysts are exposed to the cryopro-tectant in a stepwise manner: first in a solutioncontaining a low concentration of cryoprotec-tant for permeation, and then in vitrificationsolution to cause the embryos to shrink viarapid dehydration. In some cases, puncturingthe blastocoel or ultrarapid vitrification wouldbe effective. Such approaches will be describedfurther in other chapters in this book.
In mouse embryos at early cleavage stages,most water and cryoprotectant molecules per-meate slowly by simple diffusion across theplasma membrane. Thus, the stepwise treat-ment of embryos, as in the case of blastocysts, iseffective. The temperature for the treatment isvery important, because the movement of waterand cryoprotectants by simple diffusion isgreatly affected by temperature. After warming,the temperature at which embryos are dilutedis also important to prevent osmotic over-swelling during the recovery of cryopreservedembryos. Thus, permeability and the pathwayby which water and cryoprotectants move inembryos are closely related to the protocol usedfor cryopreservation.
83
VITRIFICATION IN CONVENTIONAL STRAWS WITH AN ETHYLENE GLYCOL SOLUTION
Table 4B.3 Permeability to cryoprotectants (Ps; 10−3cm/min) of AQP3 cRNA-injected Xenopus oocytes incryoprotectant solutions
Cryoprotectant dissolved in Barth’s solution
Oocytes 10% Glycerol 8% EG 10% PG 0.2 Osmol/kg AA 0.2 Osmol/kg DMSO
Injected with water 0.04 ± 0.01 0.11 ± 0.03 0.10 ± 0.02 6.50 ± 1.98 0.79 ± 0.40Injected with 36.13 ± 7.63* 33.50 ± 2.75* 31.45 ± 5.17* 24.60 ± 9.90* 6.33 ± 2.76*
AQP3 cRNA
*Significantly different from water-injected oocytes (Student’s t test, P < 0.01).Modified from Yamaji et al.50
EG, ethylene glycol; PG, propylene glycol; AA, acetamide; DMSO, dimethylsulfoxide
References
1. Whittingham DG, Leibo SP, Mazur P. Survivalof mouse embryos frozen to −196°C and−269°C. Science 1972; 178: 411–4.
2. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196°C by vitrification.Nature 1985; 313: 573–5.
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 83

3. Rall WF, Wood MJ, Kirby V et al.Development of mouse embryos cryopre-served by vitrification. J Reprod Fertil 1987;80: 499–504.
4. Nakao K, Nakagata N, Katsuki M. Productionof chimeric mice from cryopreserved blasto-cysts. Exp Anim 1998; 47: 167–71.
5. Nakagata N. High survival rate of unfertilizedmouse oocytes after vitrification. J ReprodFertil 1989; 87: 479–83.
6. Migishima F, Suzuki-Migishima R, Song SY,et al. Successful cryopreservation of mouseovaries by vitrification. Biol Reprod 2003; 68:881–7.
7. Kasai M, Komi JH, Takakamo A, et al. Asimple method for mouse embryo cryopreser-vation in a low toxicity vitrification solution,without appreciable loss of viability. J ReprodFertil 1990; 89: 91–7.
8. Ishimori H, Saeki K, Inai M, et al. Vitrificationof bovine embryos in a mixture of ethyleneglycol and dimethyl sulfoxide. Theriogenology1993; 40: 427–33.
9. Miyake T, Kasai M, Zhu SE, et al. Vitrification ofmouse oocytes and embryos at various stages inan ethylene glycol-based solution by a simplemethod. Theriogenology 1993; 40: 121–34.
10. Kasai M, Nishimori M, Zhu SE, et al. Survivalof mouse morulae vitrified in an ethyleneglycol-based solution after exposure to thesolution at various temperatures. Biol Reprod1992; 47: 1134–9.
11. Kasai M, Hamaguchi Y, Zhu SE, et al. Highsurvival of rabbit morulae after vitrification inan ethylene glycol-based solution by a simplemethod. Biol Reprod 1992; 46: 1042–6.
12. Tachikawa S, Otoi T, Kondo S, et al. Successfulvitrification of bovine blastocysts, derived byin vitro maturation and fertilization. MolReprod Dev 1993; 34: 266–71.
13. Hochi S, Fujimoto T, Braun J, et al.Pregnancies following transfer of equineembryos cryopreserved by vitrification.Theriogenology 1994; 42: 483–8.
14. Martinez AG, Matkovic M. Cryopreservationof ovine embryos: slow freezing and vitrifica-tion. Theriogenology 1998; 49: 1039–49.
15. Mukaida T, Wada S, Takahashi K, et al.Vitrification of human embryos based on theassessment of suitable conditions for 8-cellembryos. Hum Reprod 1998; 13: 2874–9.
16. Mochida K, Matsuda J, Noguchi Y, et al. Birthof pups by transfer of mastomys embryoscryopreserved by vitrification. In: Proc 31stAnnual Meeting Soc Study Reprod 1998:
180–1, abstr. Society for the Study ofReproduction, Inc, Madison, WI, USA.
17. Han M-S, Niwa K, Kasai M. Vitrification of ratembryos at various developmental stages.Theriogenology 2003; 59: 1851–63.
18. Mochida K, Wakayama T, Takano K, et al.Birth of offspring after transfer of Mongoliangerbil (Meriones unguiculatus) embryoscryopreserved by vitrification. Mol ReprodDev 2005; 70: 464–70.
19. Kasai M. Cryopreservation of mammalianembryos: vitrification. Methods Mol Biol1995; 38: 211–9.
20. Kasai M. Cryopreservation of mammalianembryos. Mol Biotechnol 1997; 7: 173–9.
21. Shaw JM, Kasai M. Embryo cryopreservationfor transgenic mouse lines. Methods Mol Biol2001; 158: 397–419.
22. Whittingham DG. Survival of mouse embryosafter freezing and thawing. Nature 1971; 233:125–6.
23. Kasai M, Zhu SE, Pedro PB, et al. Fracturedamage of embryos and its prevention duringvitrification and warming. Cryobiology 1996;33: 459–64.
24. Kasai M, Ito K, Edashige K. Morphologicalappearance of the cryopreserved mouse blas-tocyst as a tool to identify the type of cryo-injury. Hum Reprod 2002; 17: 1863–74.
25. Pedro PB, Zhu SE, Makino N, et al. Effects ofhypotonic stress on the survival of mouseoocytes and embryos at various stages.Cryobiology 1997; 35: 150–8.
26. Pedro PB, Yokoyama E, Zhu SE, et al.Permeability of mouse oocytes and embryos atvarious developmental stages to five cryopro-tectants. J Reprod Dev 2005; 51: 235–46.
27. Pedro PB, Kasai M, Mammaru Y, et al.Change in the permeability to different cryo-protectants of bovine oocytes and embryosduring maturation and development. In: Proc13th Int Congr Anim Reprod 1996; 3: abstrP15–9. The University of Sydney PrintingServece Sydney Australia.
28. Kasai M. Cryopreservation of mammalianembryos by vitrification. In: Mori T, Aono T,Tominaga T, Hiroi M, eds. Perspectives onAssisted Reproduction (Frontiers inEndocrinology) Rome, Ares-Serono SymposiaPublications, 1994; 4: 481–7.
29. Kasai M, Niwa K, Iritani A. Survival of mouseembryos frozen and thawed rapidly. J ReprodFertil 1980; 59: 51–6.
30. Kasai M, Niwa K, Iritani A. Protective effect ofsucrose on the survival of mouse and rat
VITRIFICATION IN ASSISTED REPRODUCTION
84
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 84

embryos stored at 0°C. J Reprod Fertil 1983;68: 377–80.
31. King LS, Kozono D, Agre P. From structure todisease: the evolving tale of aquaporin biol-ogy. Nat Rev Mol Cell Biol 2004; 5: 687–98.
32. Edashige K, Sakamoto M, Kasai M.Expression of mRNAs of the aquaporin familyin mouse oocytes and embryos. Cryobiology2000; 40: 171–5.
33. Offenberg H, Barcroft LC, Caveney A, et al.mRNAs encoding aquaporins 1–9 are presentduring murine preimplantation development.Mol Reprod Dev 2000; 57: 1–8.
34. Barcroft LC, Offenberg H, Thomsen P, et al.Aquaporin proteins in murine trophectodermmediate transepithelial water movements dur-ing cavitation. Dev Biol 2003; 256: 342–54.
35. Verkman AS, van Hoek AN, Ma T, et al. Watertransport across mammalian cell membranes.Am J Physiol 1996; 270: C12–30.
36. Leibo SP. Water permeability and its activationenergy of fertilized and unfertilized mouseova. J Membr Biol 1980; 53: 179–88.
37. Hunter J, Bernard A, Fuller B, et al.Measurements of the membrane water perme-ability (LP) and its temperature dependence(activation energy) in human fresh and failed-to-fertilize oocytes and mouse oocytes.Cryobiology 1992; 29: 240–9.
38. Benson CT, Critser JK. Variation of waterpermeability (LP) and its activation energy (Ea)among unfertilized golden hamster and ICRmurine oocytes. Cryobiology 1994; 31: 215–23.
39. Gao DY, Benson CT, Liu C, et al.Development of novel microperfusion cham-ber for determination of cell membrane trans-port properties. Biophys J 1996; 71: 443–50.
40. Litkouhi B, Marlow D, McGrath JJ, et al. Theinfluence of cryopreservation on murineoocyte water permeability and osmoticallyinactive volume. Cryobiology 1997; 34:23–35.
41. Pfaff RT, Liu J, Gao D, et al. Water and DMSOmembrane permeability characteristics of in-vivo and in-vitro derived and cultured murineoocytes and embryos. Mol Hum Reprod 1998;4: 51–9.
42. Toner M, Cravalho EG, Armant DR. Watertransport and estimated transmembranepotential during freezing of mouse oocytes.J Membrane Biol 1990; 115: 261–72.
43. Edashige K, Tanaka M, Ichimaru N, et al.Channel-dependent permeation of water andglycerol in mouse morulae. Biol Reprod 2006;74: 625–32.
44. Offenberg H, Thomsen PD. Functional chal-lenge affects aquaporin mRNA abundance inmouse blastocysts. Mol Reprod Dev 2005; 71:422–30.
45. Mazur P, Rigopoulos N, Jackowski SC, et al.Preliminary estimates of the permeability ofmouse ova and early embryos to glycerol.Biophys J 1976; 16: 232a.
46. Jackowski S, Leibo SP, Mazur P. Glycerolpermeability of fertilized and unfertilizedmouse ova. J Exp Zool 1980; 212: 329–41.
47. Ishibashi K, Sasaki S, Fushimi K, et al.Molecular cloning and expression of a mem-ber of the aquaporin family with permeabilityto glycerol and urea in addition to waterexpressed at the basolateral membrane of kidneycollecting duct cells. Proc Natl Acad Sci USA1994; 91: 6269–73.
48. Zeuthen T, Klaerke DA. Transport of waterand glycerol in aquaporin 3 is gated by H+.J Biol Chem 1999; 274: 21631–6.
49. Valdez DM Jr, Hara T, Miyamoto A, et al.Expression of aquaporin-3 improves thepermeability to water and cryoprotectants ofimmature oocytes in the medaka (Oryziaslatipes). Cryobiology 2006; 53: 160–8.
50. Yamaji Y, Valdez DM Jr, Seki S, et al.Cryoprotectant permeability of aquaporin-3expressed in Xenopus oocytes. Cryobiology2006; 53: 258–67.
51. Meinild AK, Klaerke DA, Zeuthen T.Bidirectional water fluxes and specificity forsmall hydrophilic molecular in aquaporins0–5. J Biol Chem 1998; 273: 32446–51.
52. Tsukaguchi H, Shayakul C, Berger UV, et al.Molecular characterization of a broad selectiv-ity neutral solute channel. J Biol Chem 1998;273: 24737–43.
85
VITRIFICATION IN CONVENTIONAL STRAWS WITH AN ETHYLENE GLYCOL SOLUTION
04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 85

04b Tucker 8029.qxd 8/23/2007 8:38 PM Page 86

87
Cryoprotectant-free vitrificationof spermatozoaEvgenia Isachenko, Vladimir Isachenko, Igor I Katkov, Raul Sanchez,Hans van der Ven and Frank Nawroth
5
INTRODUCTION
Cryopreservation and low-temperature storageof different cell types and tissues, includingmale and female gametes and embryos, areused worldwide and have become an integralpart of most human IVF programs. Sincethe late 1930–1940s1,2 spermatozoa of severalmammalian species, especially bovine andhuman, have been cryopreserved effectively.The empirical methods of cryopreservationdeveloped in the 1950s are still used today,and are very important as they allow thepreservation of male fertility before radiother-apy and/or chemotherapy.3 Such treatmentsand some kinds of surgical procedures maylead to testicular failure or ejaculatory dysfunc-tion.4 During conventional freezing, waterprecipitates as ice and thus separates from dis-solved substances. Both intracellular ice crystalformation and the high concentration of dis-solved substances pose problems. Slow coolingrates aim to maintain a very delicate balancebetween these factors, yet often lead to celldamage mostly because of ice crystallization,but also due to osmotic and chilling injury, cyto-plasm fracture, or even effects on the cytoskele-ton or genome-related structures. Due todamage associated with freezing, the motilityof cryopreserved spermatozoa after thawing issignificantly reduced in comparison with themotility before and shows a wide interindivid-ual variability.5 To date, the problem of cryo-protectant toxicity due to osmotic stress during
the addition and removal of cryoprotectants,and the possibility of its negative influence onthe genetic apparatus has, as yet, not beensolved,5,6 and cellular cryodamage can ariseduring slow thawing.7 At present the freezingprocedures for many species including humanshow acceptable results, but cryoprotectantsolutions and freezing equipment are neces-sary. Most IVF laboratories prefer programma-ble freezing devices. The whole freezingprocedure (equilibration, freezing, and dilu-tion of cryoprotectant) takes around 30–60minutes. Cryopreservation by direct plunginginto liquid nitrogen (vitrification) could bebeneficial when compared with the ‘slow’method, because it does not need any expen-sive equipment and takes only a fewseconds for freezing and warming. The vitrifi-cation technique arose as an alternative to slowconventional freezing, in order to avoid thecrystallization.8 The successful vitrification offrog9 and fowl spermatozoa10 has supportedLuyet’s proposal. However, the subsequentattempts to vitrify mammalian spermatozoausing this technique resulted in low or no sur-vival.11,12 Based on this, the vitrification tech-nique was initially unacceptable for routinework. While this method of vitrification usinghigh concentrations of permeable cryoprotec-tants was successfully applied in 1985 formouse embryos,13 nevertheless it was impossibleto perform this technique for spermatozoa cryo-preservation because of the resulting osmoticand cytotoxic effects.5,14
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 87

CRYOPRESERVATION OFSPERMATOZOA (CONVENTIONALMETHOD) AND CELL DAMAGEDURING FREEZING AND THAWING
The beneficial effects of glycerol and non-permeable cryoprotectants, including sucrose,on plant cryostability were described as earlyas 1908.15 In 1937 this was followed by thedemonstration of the positive effect of 1 mol/Lglycerol on rabbit, guinea pig, bull, ram, stal-lion, and boar sperm1 frozen to −21°C. In thelate 1940s, the results of experiments based onthe use of glycerol in the UK by Polge, Smithand Parkes2 as well as in the USSR by Smirnov16
and Milovanov17 were published. These empir-ical methods, which were subsequently devel-oped in the 1950s for use in many species, arestill applied today. The motility of cryopre-served/thawed spermatozoa normally drops toabout 50% of their pre-freezing value, withconsiderable intersample fluctuation.5
The question of diminished spermatozoalmotility after cryopreservation is crucial sincethis variable is known to be the first affected,18
although the mechanism of sperm impairmentand its mechanical and/or physical–chemicaletiology remain unclear. The mechanical cellinjury by conventional (ice-equilibrium)freezing is a consequence of intracellularor extracellular ice crystal formation, andosmotic damage due to extensive cell shrink-age. Subsequent re-warming and thawingof the cells can further deteriorate theirviability through possible excessive osmoticswelling.19,20 As a result, average velocity interms of the percentage of motile spermato-zoa drops significantly after cryopreservationwhen compared with that of fresh sperm.18
Conventional slow freezing may also causeextensive chemical and physical damage tosperm cell membranes due to changes in lipidphase transition and/or increased lipid perox-idation. It is well established that the produc-tion of reactive oxygen species leads to anincrease in lipid peroxidation after cryo-preservation21 and that this event is associated
with a loss of sperm motility.22,23 As previouslysuggested,23 the injury of human spermato-zoa induced by conventional cryopreserva-tion occurs mainly during thawing and hasbeen related to diminished antioxidantdefense activity during cooling, and/or struc-tural damage to the cytoskeleton, and/orantioxidant enzymes during cryopreserva-tion.23 All these findings suggest that slowcooling, and especially thawing of spermato-zoa, quite apart from ice crystal formation, isintrinsically deleterious.
The use of egg yolk as a non-permeablecryoprotectant provides indirect support forthis viewpoint. Egg yolk, a natural complexof lipids, proteins, lipoproteins, cholesterol,phospholipids, and antioxidants, has beenused in sperm preservation for many years toreduce the negative influence of freezing/thaw-ing, as well as cold and osmotic shock at tem-peratures above 0°C. How this protective effectis exerted is not entirely clear, but all the com-ponents of egg yolk may play a pivotal role inreducing the deleterious effects of hyper-osmotic salt solutions, membrane lipid phasetransition, and peroxidation by changing thecomposition of membranes, making themstronger (mechanism of membrane fortifica-tion) and less susceptible to peroxidationdamage.18,24
To prevent excessive cell shrinkage duringslow cooling, permeable cryoprotectiveagents (CPAs) are used. However, the effec-tiveness (prevention of intracellular ice for-mation) of permeable and non-permeablecryoprotectants during conventional freezingcan only be achieved with a low cooling rate,20
which as we indicated above can be damagingitself. In addition to this, the introduction(during freezing) and removal (after thawing)of CPAs can produce damage per se, evenat room temperature in the absence offreezing/thawing. The main mechanisms ofCPAs’ toxicity have been discussed by us else-where,25 and include osmotic damage as wellas chemical cell and membrane toxicity.5,19,26
All these negative effects of conventional slow
VITRIFICATION IN ASSISTED REPRODUCTION
88
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 88

(ice-equilibrium) freezing and thawing oncells can also lead to chromatin damage, andas has recently been noted,27 the chromatinabnormalities have repercussions on spermquality and male fertility status.
VITRIFICATION AS AN ALTERNATIVE TOCONVENTIONAL FREEZING
The vitrification technique has its own pecu-liarities. A decisive factor of cryopreservation isthe incompatibility of crystallization with livingsystems.8 It is possible to eliminate ice forma-tion and create instead a glass-like (vitreous)state by cooling of small liquid suspensions orjust water at ultra-high speeds of cooling.9
According to Fahy28 the physical definition ofvitrification is the solidification of a solution atlow temperature, not by ice crystallization, butby extreme elevation in viscosity during coolingthat provides stability and invariability of theentire solution, because the water does not pre-cipitate, so no ice crystals are formed. However,the glass-like solidification of water requires avery high cooling rate (> 300 000° C/min).Thus, to preserve living organisms stably for along period, they have to be placed in an envi-ronment where the viscosity of both intra- andextracellular water are raised to levels thatresult in the essential arrest of diffusion, sopractically all chemical processes includingdegradation and aging are effectively stopped.There are several ways to achieve vitrification.One approach is to slowly (equilibrium) freezeout the pure water as ice so that the remainingsolution becomes more and more concentrateduntil it reaches its point of vitrification, at atemperature usually referred to as Tg, or theglass transition temperature at equilibriumfreezing. Another method is to remove excesswater either by sublimation of the frozen por-tion of ice (lyophilization or freeze-drying),which is performed at subzero temperatures, orby evaporation from the liquid phase by directvacuum/air drying, or by softening and liquefy-ing the glassy material left after lyophilization
(secondary drying).29 Neither method neces-sarily implies a need for high initial concentra-tion of solutes. However, eventually the soluteconcentration dramatically increases due todehydration, that leads to a very low content ofresidual water. The next approach is vitrifica-tion of the bulk solution, which is free of a siz-able portion of ice during cooling and thawing,performed substantially faster than with equi-librium freezing (during which ice is formed).This can be done either by cooling at relativelyslow to moderate speed, but using high con-centrations of CPAs/vitrificants such asdimethylsulfoxide (DMSO), ethylene glycol,propylene glycol, or glycerol, or by solidifica-tion of the bulk solution by abrupt cooling atvery high speed to temperatures below theglass transition temperature of the solution (Tg,the temperature at which the transition to thevitreous condition begins) by plunging thespecimen directly into liquid nitrogen.According to our experience, this can beachieved in the absence of the ‘conventional’CPAs, provided that the cooling/warmingspeed is high enough to ensure both intra- andextracellular vitrification.
In general, the rate of cooling/warmingand the concentration of cryoprotectantsrequired to achieve vitrification are inverselyrelated. In other words, the higher the CPAconcentration, the lower the critical speed ofcooling and warming needed to avoid iceformation. Conversely, the faster cooling andwarming is undertaken, the lower the criticalsolute concentration necessary to obtain ice-free vitrification. Although the pioneer ofthe idea of vitrification, Luyet, initiallyemphasized the need for fast cooling andwarming of relatively dilute solutions,8 it isclear from the literature, that the use ofhighly concentrated vitrification solutionssoon became mainstream practice. Giventhe biological and physiochemical effects ofcryoprotectants and the high concentrationsused in vitrification, it is therefore not sur-prising that cryoprotectant toxicity hasbeen described as a key limiting factor in
89
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 89

the cryobiology of vitrification. In an attemptto avoid this toxicity, we achieved ultra-rapidcooling and warming rates in the range of0.5–1 ×106K/min using a relatively dilute vit-rification medium (around 12% total solutecontent), of similar solute concentration tosemen or blood; thus, ‘resurrecting’ the orig-inal vitrification approach of Luyet.
VITRIFICATION USING HIGHCRYOPROTECTIVE AGENTCONCENTRATIONS:ACHIEVEMENTS ANDPITFALLS
In contrast to the conventional slow freezingice-forming techniques, the protocols of vit-rification currently used for the cryopreser-vation of oocytes, embryos, and tissues as arule involve the use of very high concentra-tions (3.5–8 mol/L) of permeating cryopro-tectants and relatively high cooling rates (upto 105°C/min) compared with rates associatedwith conventional slow freezing. Accordingto the literature, the critical cooling speedfor the vitrification of pure water varies dras-tically, depending on the method used, from106 to 1010°C/min (see Figure 9 in Karlsson30
for references). Kanno et al.31 were able todemonstrate that the temperature of homo-geneous crystallization (Th) can be reducedthrough an increase in the hydrostatic pres-sure. Later, MacFarlane et al.32 observed thatTg rises with increasing pressure, allowingglass transition at lower cryoprotectant con-centrations. For example, for a 35% liquidDMSO solution at a hydrostatic pressure of1300 atm, Th is −80°C and the solution vitri-fies.33 A downside of this is that the increasedpressure can cause damage to the biologicalsystem. For example, dog kidneys can sur-vive 20 min of exposure to 1000 atm,34 whilerabbit kidneys show signs of severe damageafter only 20 min at 500 atm. This high pres-sure, however, is only needed during thevitrification process itself and atmospheric
pressure suffices for subsequent storage.Moreover, it has been possible to lower thecritical CPA concentration (Cv) by also includ-ing non-permeable polymers, which areunable to enter the cells.28 Cryoprotectanttoxicity can also be reduced by combiningtwo cryoprotectants, and/or exposing the tis-sue to a graded series of pre-cooled concen-trated solutions. These modifications of theconventional method reduce the toxic andosmotic effects of cryoprotectants. In addi-tion, if we increase the rate of cooling andwarming, the cryoprotectant concentrationcan be reduced even further, diminishing itstoxicity. It was recently shown that the highercooling rate provided by the nylon loop, per-mits an effective reduction in cryoprotectantconcentration such that 5.5 mol/L ethyleneglycol (EG) can be substituted by a 3.2 mol/LEG/3.2 mol/L DMSO mixture.14
The size of the sample to be cryopreservedshould be minimized such that most of thespecimen will be immersed in liquid ratherthan vapor leading to the maximal coolingrate. To achieve even higher cooling rates,the volume of vitrification solution shouldalso be kept to a minimum through the useof specially designed carriers such as openpulled straws (OPS),35 the flexipet-denudingpipette,14,36 micro drops,37 electron micro-scope copper grids,38 the hemi-straw sys-tem,39 small nylon coils,40 or nylon meshes,41
and the Cryoloop.36 These all have been usedas carriers or vessels to achieve higher cool-ing rates and have provided good results forthe vitrification of embryo species that areparticularly susceptible to freeze damage42
along with the highly sensitive humanoocytes.43 There have even been reports ofthe successful vitrification of human embry-onic stem cells using the OPS method.44
A further factor to consider is the changesin the properties of ethylene glycol-based vit-rification solutions after the addition of sugars(sucrose, glucose, fructose, sorbitol, trehalose,or raffinose).45 High molecular weight addi-tives such as disaccharides e.g. sucrose or
VITRIFICATION IN ASSISTED REPRODUCTION
90
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 90

trehalose, cannot pass through the cellmembrane, but can significantly reduce theamount of cryoprotectant required as well asthe toxicity of ethylene glycol by decreasingthe concentration needed to efficiently cryo-preserve human oocytes and embryos. Addingnon-permeating compounds to the vitrifyingsolution as well as pre-incubation in thissolution before vitrification to withdraw morewater from the cells, shortens the time ofexposure of the cells to the toxic effects of thecryoprotectants. Non-permeating sucrose alsoacts as an osmotic buffer, reducing the osmoticshock induced by diluting the cryoprotectantafter cryopreservation. However, even mole-cules as large as sucrose (MW = 342), tre-halose (MW = 378), or raffinose (MW = 504)can produce substantial osmotic damagewhen used at high concentrations. A furtheroption is to add high molecular weight poly-mers, both synthesized (polyvinyl pyrroli-done, polyethylene glycol) and of a biologicalorigin (proteins). This approach is discussedbelow.
BACKGROUND OF SPERM VITRIFICATION
After the successful vitrification of frog sper-matozoa by Luyet and Hodapp in 1938,9 4years later Shaffner10 vitrified fowl spermato-zoa. He modified Luyet’s sperm vitrificationtechnique which was considered an attrac-tive alternative to conventional slow freezing.However, early attempts at vitrifying mam-malian spermatozoa using this approach hadvery low or no survival11,12 mostly because, asshown below, critical speeds of freezing andwarming were unachievable at that time; thelow CPA concentration tolerated by spermrequiring high speeds. In spite of this, in theearly 1980s, Rall and Fahy13 managed tosuccessfully vitrify embryos using high CPAconcentrations and a relatively low speed ofcooling and warming and since then, themain approach to vitrification of spermato-zoa has been considered the same as the
methods used for other types of mammaliancells.46 Classical vitrification requires a highproportion of permeable cryoprotectants inthe medium (from 3.5 to 8 mol/L along orwith carbohydrates in combination with acooling rate from 2000 to 30 000°C/minachieved by direct plunging into liquid nitrogen(LN2) compared with 5–7% for slow-freezingwith 0.3°C/min cooling rate), and seems to beunsuitable for the vitrification of spermatozoadue to the lethal osmotic effects and possiblechemical alterations. In the opinion of someauthors, this is the main reason for the lack ofany significant practical results so far (seeHolt47 for a comprehensive analysis of thebackground). Indeed, using the ‘conven-tional’ methods of vitrifying human sperma-tozoa, our survival rates have been extremelylow or even lacking. This prompted our ideaof exploring vitrification methods that wouldnot require high concentrations of potentiallytoxic CPAs.
Do we always need cryoprotectants forsuccessful vitrification?
Still Luyet, in his vast number of papers oncryopreservation by vitrification, mentionedthat a small specimen cooled very rapidlycould be vitrified without substantial loss ofviability.48 Observations of Jahnel,49 who per-formed cryoprotectant-free cryopreservationof human spermatozoa cooled in liquid nitro-gen and liquid helium (−269.5°C), and Parkeswho also published in 194550 on freezing ofhuman spermatozoa without cryoprotectants.Large volumes (milliliters) of sperm werecooled in glass or metal tubes. Probably due tothe absence of quick thawing, motility of sper-matozoa after thawing was reduced. In 194211
the freezing of human and rabbit spermato-zoa using a bacteriological loop to rapidlycool small specimens was described. Theseauthors obtained up to 40% of viable humanspermatozoa after cooling of a sperm film inLN2 followed by quick warming of thesemicrovolumes. Later, however, they reported
91
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 91

low or no viability of mammalian sperm aftervitrification.11,12 Some years later, when inves-tigators turned their attention to the vitrifica-tion of large cells, tissues, and even organs,the dogma was established that vitrificationcould only be achieved using high concentra-tions of combinations of permeable (PCP) andimpermeable (PVP) cryoprotectants.48 Thetotal concentration of these protecting agentshad to be at least 50% w/w (if performed atatmospheric pressure) to achieve stable vitrifi-cation and enable the use of a relatively lowspeed of cooling and warming. Such highCPA concentrations can be very damaging tocells, causing both biochemical alterationsand lethal osmotic injury.51 It has been possi-ble to assess the relative contributions ofosmotic damage and true biochemical alter-ations to apparent ‘CPA toxicity’.26 Very often,some of the deleterious effects of exposure tocryoprotectants can be avoided by optimizingregimens for the addition and removal of theCPAs5,19,26 for human and animal sperm; wededicated a set of papers to this particularproblem.52 However, these methods also havetheir limitations. Regardless of the mecha-nism of damage, we could summarize that inthe majority of species, sperm cannot toleratethe high concentrations of cryoprotectantsconventionally used for vitrification.Therefore the obvious alternative is to usevery rapid cooling and warming rates on avery small sample size. Recently, Bischof ’sgroup52 predicted that one of the optimalcooling rates for spermatozoa theoreticallylies between 5000 and 7000°C/min. Accordingto other data53 the form and size of spermhead could be the factors which define thecryosensitivity of cells. Comparative investiga-tion54 of cryoproperties from different mam-malian species (boar, bull, ram, rabbit, cat,dog, horse, human) has shown a negativecorrelation between size of sperm head andcryostability. Among the above-mentionedmammalian species, the human spermatozoahave minimal size parameters and maximalcryostability.20
Taking into account all these data, we haveassumed that vitrification of human sperma-tozoa without permeable cryoprotectants canbe successful.
VITRIFICATION OFSPERMATOZOA: OUR EXPERIENCE
Our results agreed with the above-mentioneddata. In our preliminary investigation,25,55 thestudy performed in Italy and Germany whichwas approved by the University Review Board(Italy) and University Ethics Committees(Germany), we have shown that the successfulvitrification of spermatozoa is possible with-out permeable cryoprotectants. It was neces-sary to investigate not only the influence ofvitrification technique (vitrification usingCryoloops) on physiological spermatozoaparameters, but also to compare results ofthis technique with routinely used conven-tional programmable freezing in Frenchstraws.56 Earlier it was shown that the simple‘swim up’ procedure allows selection of sper-matozoa with progressive motility and nor-mal morphology before freezing.6,57,58 Takingthis into account we have investigated therole of such treatment on the physiologicalparameters of spermatozoa after both slowfreezing and vitrification. We found a 1.6-foldincrease in sperm motility and a 1.9-foldincrease in the percentage of morphologi-cally normal spermatozoa in swim up pre-pared spermatozoa compared with originalsamples. However, the negative influence ofthe routinely used cryoprotectant medium(TEST-egg-yolk-glycerol, TEYG) after only10 min exposure before freezing led to a sig-nificantly reduced motility by 12.6% in theoriginal semen sample and by 13.8% in swimup prepared spermatozoa, as well as to areduced percentage of living cells by 11.4%for the original sample and by 13.1% forswim up prepared spermatozoa comparedwith non-treated samples (P < 0.05). Theprogrammable freezing reduced the percent-age of living cells by 24.5% in the ejaculated
VITRIFICATION IN ASSISTED REPRODUCTION
92
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 92

semen and by 22.2% in swim up preparedsamples (P<0.05). The percentage of acrosome-reacted cells after TEYG-treatment beforecooling was increased by 10.9% in semen andby 15.7% for swim up prepared spermatozoa.After programmable freezing with cryopro-tectant compared with non-frozen samplesthe motility was reduced by 16.4% for semenand by 35.6% for swim up prepared sperma-tozoa; also the rate of spermatozoa withnormal morphology was decreased by 11.6%for semen samples and by 12.3% for swimup prepared ones (P < 0.05). Nevertheless,beneficial effects of cryoprotectant could beshown when comparing with sperm parame-ters after conventional freezing of TEYG-treated and non-treated spermatozoa. Slowfreezing without cryoprotectant reducedsperm motility dramatically for originalsemen samples (by 43.2%) and for swim upprepared spermatozoa (by 86.1%) comparedwith the values before freezing. It was shownthat slow freezing without cryoprotectantreduced the rate of normal sperm morphol-ogy by 7.3% for semen and by 25.0% for swimup prepared spermatozoa compared with thesame method after treatment with cryopro-tectant. In contrast to spermatozoa conven-tionally frozen with cryoprotectant thepercentage of motile spermatozoa recoveryfrozen without cryoprotectant was 1.3% forsemen and 0.2% for swim up prepared sper-matozoa (P < 0.05).
Completely opposite results were obtainedafter vitrification of spermatozoa on Cryoloops.To perform the vitrification procedure, wemanufactured our own cryoloops55 with a5 mm diameter and a loaded volume of20 ± 2 µl (Figure 5.1). The warming was per-formed by plunging the Cryoloops into acentrifuge tube containing 10 ml of spermpreparation medium (SPM; Scandinavian IVFScience, Gothenburg, Sweden) at 37°C underintense agitation. The same concentration ofcryoprotectant as for conventional freezinghad a highly toxic (possibly osmotic) effect.This is questionable because we do not know
what kind of effect glycerol has on thesperm membrane and intracellular structures,because the typical sperm osmotic reaction of‘coiled tail’ was not noted. Vitrification withoutpermeable cryoprotectant (only 1% humanserum albumin in preparation medium –medium + 1% HSA) has shown the best resultswith swim up prepared spermatozoa. In com-parison with the best post-thaw data afterconventional freezing with obligatory use ofpermeable cryoprotectants, vitrificationresulted in a significant increase of spermmotility after thawing (by 11.6%; P < 0.05).However, the difference in morphology, motilesperm recovery, viability after freezing, andacrosome reacted cells between the two cryo-preservation methods and sperm treatments(with or without cryoprotectant) was not statis-tically significant (P > 0.05). At present, it isnot clear why all these parameters for ejacu-lated spermatozoa after vitrification are lowerthan those of spermatozoa vitrified after swimup. We have found that, in contrast to vitrifica-tion, conventional freezing of ejaculated andswim up prepared spermatozoa without cryo-protectant resulted in 34.4% and 25.2% livingcells (P < 0.05), respectively, with nearly all ofthem being non-motile.
FREEZING IN LIQUIDNITROGENS VAPOR ANDINTRACELLULAR VITRIFICATION
To investigate the role of warming velocity forsuccessful viability of vitrified samples wedesigned an interesting experiment,60 anddeveloped an original technique (Figure 5.2).The experiment was conducted as follows. Apre-loaded Cryoloop with spermatozoa sus-pension was placed without agitation in a hor-izontal position into an N2 vapor at −160°C. Athin (27 gauge) injection needle was then usedto periodically (at ~1 s intervals) transfix thefilm at different locations on the loop (center,near the copper ring at the periphery). Whenthe film is liquid, it is possible to punchthrough it many times without disruption of
93
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 93

the film, and after the needle is removed thefilm remains intact. Upon freezing, the filmsolidifies (starting from the copper ring areatowards the center) and transfixion withoutdisruption of the film becomes impossibleand so the ring begins to move. The timeelapsed (visually indicated) from placing theloop in the box at room temperature (+23°C)to the beginning of solidification of the sus-pension (−4°C) allowed us to calculate thespeed of cooling of the spermatozoa. It wasfrom 6 s near the metal ring to 10 s at the cen-ter of it. Thus, the rate of cooling of sperma-tozoa in N2 vapor was in the range of162–270° C/min.
When the specimen is kept above the sur-face of the LN2 in its vapor at a temperatureof −160OC, given that the thermal conductiv-ity of the vapor is substantially lower than thatof the LN2; the cooling rate can beseveral orders of magnitude lower. The maincooling front will extend from the surface ofthe copper ring in a radial direction towardsthe center of the pellicle. The mathematical
calculations of this scenario are very complex.However, we were able to estimate the averagerate of cooling, by first measuring the time ofsolidification of the pellicle near the ring, andthen estimating the time taken for the surfaceof the film to completely solidify at the center.This gave an initial cooling rate in the rangeof 270°C/min near the copper ring to162°C/min at the center of the film. It is clearthat at this rate of cooling and in the absenceof any viscous vitrificants in the SPM (spermpreparation medium) medium, the extracel-lular milieu of the cells will not vitrify, but willstart freezing with the initiation of ice crystalformation. However, human spermatozoacontain large amounts of proteins, sugars, andother components that make the intracellularmatrix highly viscous and compartmental-ized. The quantity of high molecular weightmacromolecules and polymers diluted in thecytosol can be estimated from the fraction ofthe osmotically inactive volume, which isabout 20–25% for embryos and oocytes, andmuch higher (45–77%) for spermatozoa.21 As
VITRIFICATION IN ASSISTED REPRODUCTION
94
3
5
4
3
6
21
Figure 5.1 Scheme of ‘Cryoloop’ vitrification and warming of spermatozoa. (1) suspension of spermatozoa, (2) Petridish, (3) Cryo-loop, (4) film of sperm suspension, (5) tube for warming, (6) warming medium. With permission fromIsachenko et al.59
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 94

a result, we can speculate that we were able toachieve intracellular vitrification of thehuman spermatozoa even at such a low rate ofcooling. A further factor to consider is thesmall size and high degree of compartmental-ization of the sperm head, such that even ifsmall (non-lethal) crystals start to form duringthis relatively ‘slow’ cooling, there would beinsufficient time for substantial growth duringcooling. It is known that a major problem forsuch metastable systems can be the re-growthof crystals and devitrification during warming.The cooling rate of 160–250°C/min is 5–10times higher than the rate of conventionalslow freezing with the use of permeable cryo-protectants. At such a slow freezing speed,water has time to escape from the cells uponfreezing. As a result, the cell shrinks and canbe osmotically damaged unless an osmoticbuffer (permeable CPA) is used to preventhydration and excessive volume loss. In ourcase, however, the freezing rate is much faster,so the cell maintains its volume without theneed for a conventional cryoprotectant (per-meable osmotic buffer or non-permeableCPA).
Our data indicate essentially similarresults, in terms of sperm motility observedafter ‘instant vitrification’ by direct plunginginto LN2 and relatively ‘slow’ cooling in N2
vapor. It was shown that both regimens ofvitrification gave about a 40% reduction ofmotility of spermatozoa (P < 0.05) when com-pared with swim-up treated controls. No sta-tistically significant difference was found inthis parameter between the two regimens ofvitrification. The results from IVF, showapproximately equal fertilization potentialfor fresh human spermatozoa samples com-pared with swim up prepared CPA-free sam-ples vitrified by direct plunging into LN2 orinto N2 vapor (Table 5.1).
ROLE OF DEVITRIFICATION ANDIMPORTANCE OF FAST RATE OFWARMING
The founder of modern cryobiology empha-sized that devitrification and the growth of icecrystals formed during cooling could be a keyfactor promoting cell damage during re-warming and thawing procedures.9 Therefore,we directly placed the specimens in a warmsolution, ensuring a very high rate of warm-ing. In this process, the probability of sub-stantial devitrification (recrystallization) ofthe vitrified intracellular solution and re-growth of large lethal intracellular crystals islow, due to the high speed and very shorttime of warming. Our estimations showedthat, in general, during cooling and espe-cially during warming, the small specimensize, high viscosity of the freezing mediumand intracellular matrix, very high speed of
95
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
12
4
5
3
Figure 5.2 Scheme of estimation of spermatozoa cool-ing rate in vapor of liquid nitrogen. (1) Copper loop withfilm of spermatozoa suspension, (2) foam box, (3) liquidnitrogen, (4) foot for loop, (5) needle. With permissionfrom Isachenko et al.60
Table 5.1 Fertilization of oocytes with vitrifiedspermatozoa and subsequent development of embryos
Rate of Vitrification Vitrification fertilization with ‘rapid’ with ‘slow’ and cooling cooling development (n = 23) (%) (n = 22) (%)
Pronuclear formation 79 864–6 blastomeres 63 64Blastocysts formations 47 41
From Isachenko et al.60
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 95

warming, small size of the specimen and cells,their low water content, and high degree ofcompartmentalization would ensure thatdevitrification (especially intracellular) wouldnot occur.30,61
INTEGRITY OF CHROMOSOMEAPPARATUS OF SPERMATOZOA
All the negative influences of freezing oncells, that have been described earlier, canalso lead to chromatin damage and arestrongly correlated with mutagenic effects.62
It was shown that freezing/thawing in fertileand infertile men resulted not only in signifi-cantly changed sperm morphology andmembrane integrity, but also in significantchromatin damage. Futhermore, it was shownthat the fragmented DNA was negatively corre-lated with fertilization rates in IVF63 and ICSI.64
Although some deleterious cryoprotectant-exposure effects on mammalian sperm can beavoided using optimal regimens of additionand removal of CPA.26,65 However, such meth-ods do not work with very high concentra-tions of cryoprotectants. With the coolingparameters involved in vitrification such asvery high speed (up to 10 000°C/min at theinitial phase of cooling), short time (5–8 s),and low specimen size (20 µl), it seems thatnot many crystallization nuclei were formed,and their size was not large enough to dam-age human spermatozoa. The probability ofsubstantial devitrification (recrystallization)of the vitrified solution is also low due to thehigh speed, very short time of warming, andthe small size of the specimen (extracellularrecrystallization) and the cells (intracellularrecrystallization).
To investigate the occurrence of apoptosisin spermatozoa DNA after vitrification weperformed the comet assay (Figure 5.3). Ourestimations66 for albumin showed that, ingeneral, both during cooling and especiallyduring rewarming/resuscitation, the smallamount of the specimen and the cells, high
viscosity of the solution, and high speed ofcooling and warming25 would ensure thatdevitrification (especially intracellularly)would not occur.61 Seemingly, the presence ofrelatively high concentrations of albumin andsome amount of sugars substantially raisedthe viscosity of the solution, especially atlower temperatures, as well as the smallamount of cells and the small specimen size,made vitrification stable during both coolingand warming, and showed positive resultsafter warming. Using such an extreme cryo-protocol for sperm preservation it is reason-able to assume that sperm DNA may bedamaged. However, Evenson et al. found nodifference in sperm chromatin structure assayresults for both non- and cryopreservedsperm, and for slowly or flash-frozen speci-mens.67 Duty et al.68 have confirmed theseresults, and found that flash freezing in LN2without cryoprotectants most closely repro-duced the results obtained with freshly ejacu-lated human spermatozoa. They suggest thatthe unique packaging of sperm DNA protectsit from intracellular fluid shifts, and forma-tion of nuclei of crystallization during thecooling–warming cycle. Thus preventingDNA damage from intracellular fluid shiftsand ice crystal formation during cryopreser-vation.68 The substantial compartmentaliza-tion of their intracellular components mayalso contribute to the successful survival ofhuman spermatozoa. It is also known that theamount of osmotically inactive water ishigher in spermatozoa than in oocytes orembryos, since it is bound to several macro-molecular structures such as DNA, histones,hyaluronidase, etc. According to our calcula-tions25 the amount of high molecular weightcomponents can be 6–8 times higher thanin embryos, and this will probably affect thesubsequent increased viscosity and glass-transition temperature of the intracellularcytosol in sperm, yet the risk of lethal ice for-mation during cooling is likely to be higherfor embryos. This hypothesis is supported by
VITRIFICATION IN ASSISTED REPRODUCTION
96
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 96
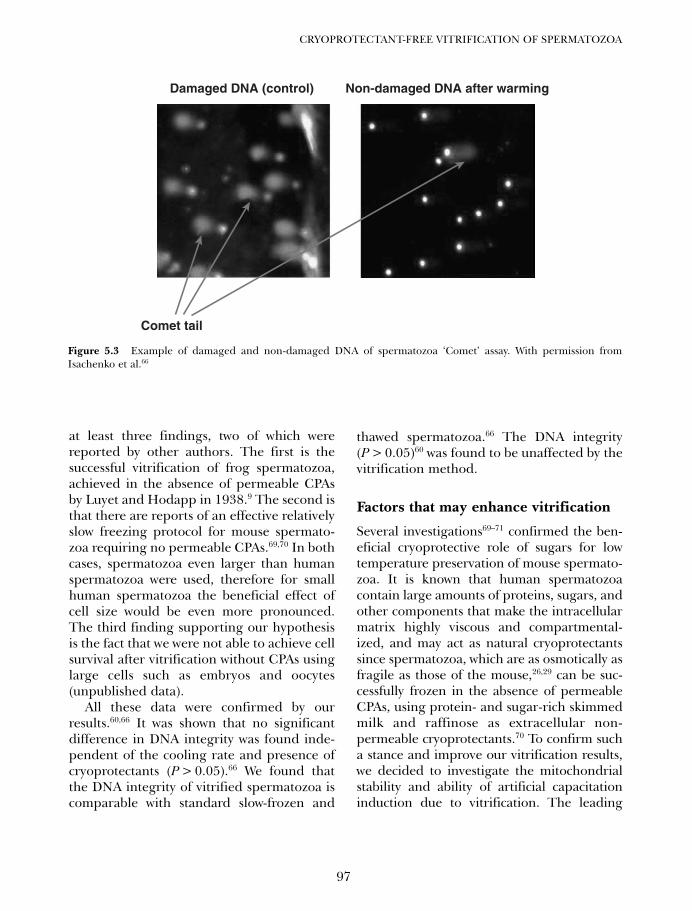
at least three findings, two of which werereported by other authors. The first is thesuccessful vitrification of frog spermatozoa,achieved in the absence of permeable CPAsby Luyet and Hodapp in 1938.9 The second isthat there are reports of an effective relativelyslow freezing protocol for mouse spermato-zoa requiring no permeable CPAs.69,70 In bothcases, spermatozoa even larger than humanspermatozoa were used, therefore for smallhuman spermatozoa the beneficial effect ofcell size would be even more pronounced.The third finding supporting our hypothesisis the fact that we were not able to achieve cellsurvival after vitrification without CPAs usinglarge cells such as embryos and oocytes(unpublished data).
All these data were confirmed by ourresults.60,66 It was shown that no significantdifference in DNA integrity was found inde-pendent of the cooling rate and presence ofcryoprotectants (P > 0.05).66 We found thatthe DNA integrity of vitrified spermatozoa iscomparable with standard slow-frozen and
thawed spermatozoa.66 The DNA integrity(P > 0.05)60 was found to be unaffected by thevitrification method.
Factors that may enhance vitrification
Several investigations69–71 confirmed the ben-eficial cryoprotective role of sugars for lowtemperature preservation of mouse spermato-zoa. It is known that human spermatozoacontain large amounts of proteins, sugars, andother components that make the intracellularmatrix highly viscous and compartmental-ized, and may act as natural cryoprotectantssince spermatozoa, which are as osmotically asfragile as those of the mouse,26,29 can be suc-cessfully frozen in the absence of permeableCPAs, using protein- and sugar-rich skimmedmilk and raffinose as extracellular non-permeable cryoprotectants.70 To confirm sucha stance and improve our vitrification results,we decided to investigate the mitochondrialstability and ability of artificial capacitationinduction due to vitrification. The leading
97
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
Damaged DNA (control)
Comet tail
Non-damaged DNA after warming
Figure 5.3 Example of damaged and non-damaged DNA of spermatozoa ‘Comet’ assay. With permission fromIsachenko et al.66
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 97
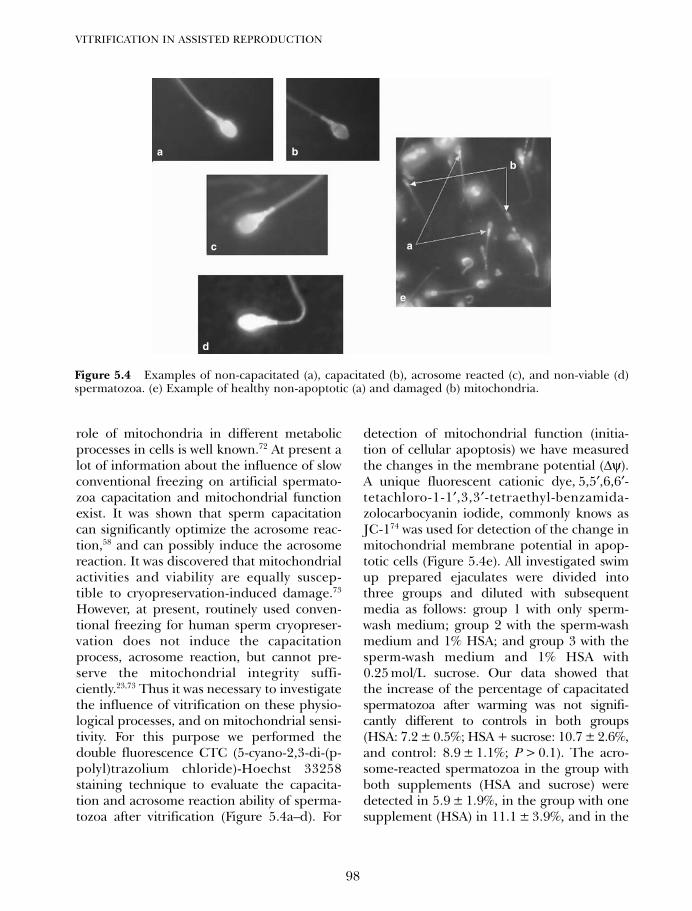
role of mitochondria in different metabolicprocesses in cells is well known.72 At present alot of information about the influence of slowconventional freezing on artificial spermato-zoa capacitation and mitochondrial functionexist. It was shown that sperm capacitationcan significantly optimize the acrosome reac-tion,58 and can possibly induce the acrosomereaction. It was discovered that mitochondrialactivities and viability are equally suscep-tible to cryopreservation-induced damage.73
However, at present, routinely used conven-tional freezing for human sperm cryopreser-vation does not induce the capacitationprocess, acrosome reaction, but cannot pre-serve the mitochondrial integrity suffi-ciently.23,73 Thus it was necessary to investigatethe influence of vitrification on these physio-logical processes, and on mitochondrial sensi-tivity. For this purpose we performed thedouble fluorescence CTC (5-cyano-2,3-di-(p-polyl)trazolium chloride)-Hoechst 33258staining technique to evaluate the capacita-tion and acrosome reaction ability of sperma-tozoa after vitrification (Figure 5.4a–d). For
detection of mitochondrial function (initia-tion of cellular apoptosis) we have measuredthe changes in the membrane potential (∆ψ).A unique fluorescent cationic dye, 5,5′,6,6′-tetachloro-1-1′,3,3′-tetraethyl-benzamida-zolocarbocyanin iodide, commonly knows asJC-174 was used for detection of the change inmitochondrial membrane potential in apop-totic cells (Figure 5.4e). All investigated swimup prepared ejaculates were divided intothree groups and diluted with subsequentmedia as follows: group 1 with only sperm-wash medium; group 2 with the sperm-washmedium and 1% HSA; and group 3 with thesperm-wash medium and 1% HSA with0.25 mol/L sucrose. Our data showed thatthe increase of the percentage of capacitatedspermatozoa after warming was not signifi-cantly different to controls in both groups(HSA: 7.2 ± 0.5%; HSA + sucrose: 10.7 ± 2.6%,and control: 8.9 ± 1.1%; P > 0.1). The acro-some-reacted spermatozoa in the group withboth supplements (HSA and sucrose) weredetected in 5.9 ± 1.9%, in the group with onesupplement (HSA) in 11.1 ± 3.9%, and in the
VITRIFICATION IN ASSISTED REPRODUCTION
98
b
a
Figure 5.4 Examples of non-capacitated (a), capacitated (b), acrosome reacted (c), and non-viable (d)spermatozoa. (e) Example of healthy non-apoptotic (a) and damaged (b) mitochondria.
a b
c
d
e
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 98

control in 9.7 ± 1.4% (P > 0.05). It wasobserved that supplements such as HSA alone(28.6 ± 4.7%) or in combination with sucrose(65.2 ± 2.6%) significantly preserved themitochondrial membrane integrity comparedwith controls (7.7 ± 0.7%, P < 0.05). However,the combination of two supplements had astronger cryoprotective effect (P < 0.05) com-pared with HSA alone.
VITRIFICATION METHODS
On the basis of these investigations, fourdifferent vitrification techniques using rapidwarming due to a small amount of cooling/warming medium, or due to a combination ofa relatively large volume of cooled suspensionand high warming rates in agitated mediumwere investigated. In this way, an aseptic tech-nology for spermatozoa vitrification was devel-oped, and the following vitrification techniqueswere tested.
(1) Cooling by direct plunging into LN2 or bycooling in N2 vapor using Cryoloops. Thesamples of both spermatozoa-preparedgroups were divided into the same foursubgroups as for the slow protocol. TEYG
with the same concentration as for slowfreezing was used for vitrification withcryoprotectant. Samples of spermatozoawere located onto copper loops of 5 mmdiameter (volume of drops 20 ± 2 µL), andthen the loops were plunged into LN2(Figure 5.1). The Cryoloops with samples,which were cooled for 3 min in N2 vapor(at −160°C), were placed into pre-cooledcryovials after 5 min and stored in LN2until the time of use.
(2) Cooling in N2 vapor at −160°C or bydirect plunging into LN2 using droplets.Spermatozoa were cooled as shown inFigure 5.5. Thirty microliters of samplewere dropped onto aluminum foil previ-ously cooled in vapor of N2 to −160°C ordirect into LN2. The temperature of foilwas determined using an electrical ther-mometer. After 5 min of cooling, thesolidified droplets of SPS were placedinto pre-cooled in LN2 cryovials andstored in LN2 until the time of use.
(3) Aseptic cooling in LN2 using open pulledstraws. Five microliters of SPS were drawninside the end of open pulled straws bycapillary effect.35 Straws were placedinside a sterile 0.5 mL insemination straw
99
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
2
45
36
1
75
7
Figure 5.5 Scheme of ‘droplet’ vitrification and warming of spermatozoa. (1) Foam box, (2) liquid nitro-gen, (3) foot for aluminum foil, (4) aluminum foil, (5) suspension of spermatozoa, (6) tube for warming,(7) warming medium. With permission from Isachenko et al.59
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 99

hermetically sealed using an ultrasoundhand-held sealer (Figure 5.6) and plungedinto LN2. It should be noted that there wasno contact between the wall of the 90 mmstraw and the suspension of spermatozoainside the open pulled straws due to thepresence of a meniscus of the spermatozoasample.
(4) Aseptic cooling in LN2 using inseminationstraws. One microliter of sample, using amicropipette, was drawn inside the endof an insemination straw. The straw was
placed inside a sterile 0.5 mL insemina-tion straw and sealed the same way asfor the open pulled straw method(Figure 5.7).75
The warming of Cryoloops and droplets wasperformed by plunging the Cryoloops anddroplets into a centrifuge tube with 10 mL ofsperm-preparation medium (SPM) at 37°Cunder intense agitation. The open pulled orinsemination straws were rapidly warmed byplunging into 1.5 mL microcentrifuge tubes
VITRIFICATION IN ASSISTED REPRODUCTION
100
7
3
55
55
2
1
4
8
2 2
Figure 5.6 Scheme of the method and photograph of container for ‘open-pulled straw’ vitrification andwarming of spermatozoa. (1) Open-pulled straw, (2) suspension of spermatozoa, (3) meniscus of suspen-sion, (4) 90 mm straw, (5) heat sealed end of 90 mm straw, (6) marked end of open-pulled straw, (7) tubefor warming, (8) warming medium. With permission from Isachenko et al.59
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 100

containing 1mL of SPM at 37°C after beingexpelled from their packaging.
Comparing these four vitrification tech-niques we have found that all the regimens ofcryopreservation gave about 40% reduction ofspermatozoa motility (P < 0.05) in comparisonwith non-treated swim up control. The qualityof spermatozoa increased dramatically 2–5 hlater, and decreased after 24 h of culture(Figure 5.8). No statistically significant differ-ence was found in these parameters between allthe regimens of cryopreservation tested.
CONCLUSIONS
(1) The cryoprotectant-free cryopreservationof human spermatozoa by fast or relatively
slow cooling, by either direct plunginginto LN2 or freezing in N2 vapor before-hand, followed in both cases by rapidthawing is feasible.
(2) The speed of warming plays a decisiverole in vitrification.
(3) The DNA integrity of vitrified spermato-zoa is comparable with that of standardslow-frozen spermatozoa and that of freshsperm.
(4) The use of a non-permeable cryoprotec-tant mixture (HSA and sucrose) can sig-nificantly enhance the mitochondrialintegrity and prevent initiation of capaci-tation and the acrosome reaction process.
101
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
7
3
3
2
55
5 5
8
5
4
2
223
2
4
1
Figure 5.7 Scheme of the method and photograph of container for ‘open straw’ vitrification and warm-ing of spermatozoa. (1) Tip of pipettor, (2) open straw, (3) drop of spermatozoa, (4) 90 mm straw, (5) heatsealed end of 90 mm straw, (6) marked end of open straw, (7) tube for warming, (8) warming medium.With permission from Isachenko et al.59
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 101
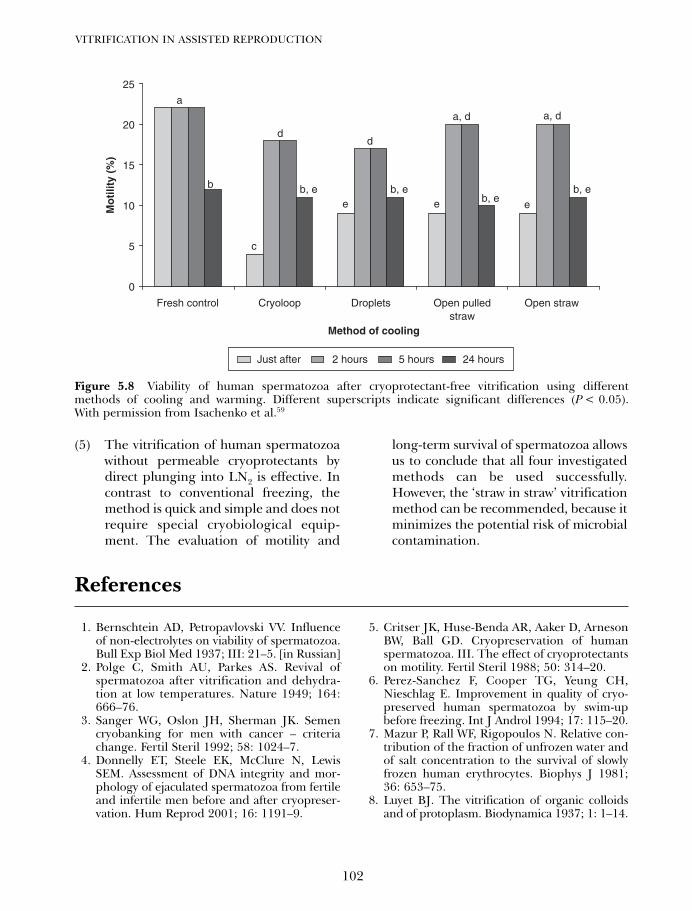
(5) The vitrification of human spermatozoawithout permeable cryoprotectants bydirect plunging into LN2 is effective. Incontrast to conventional freezing, themethod is quick and simple and does notrequire special cryobiological equip-ment. The evaluation of motility and
long-term survival of spermatozoa allowsus to conclude that all four investigatedmethods can be used successfully.However, the ‘straw in straw’ vitrificationmethod can be recommended, because itminimizes the potential risk of microbialcontamination.
VITRIFICATION IN ASSISTED REPRODUCTION
102
0
5
10
15
20
25
Fresh control Cryoloop Droplets Open pulledstraw
Open straw
Method of cooling
Mo
tilit
y (%
)a
b
c
dd
a, d a, d
b, e b, eb, e
b, ee e e
Just after 2 hours 5 hours 24 hours
Figure 5.8 Viability of human spermatozoa after cryoprotectant-free vitrification using differentmethods of cooling and warming. Different superscripts indicate significant differences (P < 0.05).With permission from Isachenko et al.59
References
1. Bernschtein AD, Petropavlovski VV. Influenceof non-electrolytes on viability of spermatozoa.Bull Exp Biol Med 1937; III: 21–5. [in Russian]
2. Polge C, Smith AU, Parkes AS. Revival ofspermatozoa after vitrification and dehydra-tion at low temperatures. Nature 1949; 164:666–76.
3. Sanger WG, Oslon JH, Sherman JK. Semencryobanking for men with cancer – criteriachange. Fertil Steril 1992; 58: 1024–7.
4. Donnelly ET, Steele EK, McClure N, LewisSEM. Assessment of DNA integrity and mor-phology of ejaculated spermatozoa from fertileand infertile men before and after cryopreser-vation. Hum Reprod 2001; 16: 1191–9.
5. Critser JK, Huse-Benda AR, Aaker D, ArnesonBW, Ball GD. Cryopreservation of humanspermatozoa. III. The effect of cryoprotectantson motility. Fertil Steril 1988; 50: 314–20.
6. Perez-Sanchez F, Cooper TG, Yeung CH,Nieschlag E. Improvement in quality of cryo-preserved human spermatozoa by swim-upbefore freezing. Int J Androl 1994; 17: 115–20.
7. Mazur P, Rall WF, Rigopoulos N. Relative con-tribution of the fraction of unfrozen water andof salt concentration to the survival of slowlyfrozen human erythrocytes. Biophys J 1981;36: 653–75.
8. Luyet BJ. The vitrification of organic colloidsand of protoplasm. Biodynamica 1937; 1: 1–14.
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 102

9. Luyet BJ, Hoddap A. Revival of frog’s sperma-tozoa vitrified in liquid air. Proc Meet Soc ExpBiol 1938; 39: 433–4.
10. Schaffner CS. Longevity of fowl spermatozoain frozen condition. Science 1942; 96: 337.
11. Hoagland H, Pincus G. Revival of mammaliansperm after immersion in liquid nitrogen.J Genet Physiol 1942; 25: 337–44.
12. Smith AU. Biological Effects of Freezing andSupercooling. London: Edward Arnold Ltd,1961: 196.
13. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at –196 °C by vitrification.Nature 1985; 313: 573–5.
14. Liebermann J, Tucker M, Graham J et al.Blastocyst development after vitrification ofmultipronucleate zygotes using the flexipetdenuding pipette (FDP). Reprod BiomedOnline 2002; 4: 148–52.
15. Maksimov NA. Chemical protection of plantsfrom being killed by frost. Influence of solu-tions of anorganic electrolytes of the anorganicsalts and salts of organic acids. Importance ofeutectic point of solutions protection. In:About being killed by frost and cold-resistantof plants. Experimental and critical investiga-tions. Sankt-Petersburg, printing-house ofFrolova, Galernaja 6, 1913: 242–278. [inRussian]
16. Smirnov IV. Preservation of domestic animals’semen by deep cooling. Sovetskaja Zootechnia1949; 4: 63–5. [in Russian]
17. Milovanov VK. Reproductive Biology andArtificial Insemination of Animals. Moskau,Publishers of domestic animals’ literature,journals and posters, MOSKau, udUSSR,1962: 696. [in Russian]
18. Watson PF. Recent developments and conceptsin the cryopreservation of spermatozoa andthe assessment of their post-thawing function.Reprod Fertil Dev 1995; 7: 871–91.
19. Gao DY, Liu C, McGann LE, Watson PF,Kleinhans FW, Mazur P et al. Prevention ofosmotic injury to human spermatozoa duringaddition and removal of glycerol. HumReprod 1995; 10: 1109–22.
20. Gao D, Mazur P, Critser J. Fundamentalcryobiology of mammalian spermatozoa. In:Karow AM, Critser JK, eds. ReproductiveTissue Banking. London: Academic Press,1997: 263–328.
21. Aitken RJ, Clarkson JS, Hargreave TB, IrvineDS, Wu FC. Analysis of the relationshipbetween defects sperm function and the gen-eration of reactive oxygen species in cases ofoligospermia. J Androl 1989; 10: 214–20.
22. Alvarez JG, Storey BT. Evidence for increasedlipid peroxidative damage and loss of super-oxide dismutase activity as a mode of sub-lethal cryodamage to human sperm duringcryopreservation. J Androl 1992; 13: 232–41.
23. O’Connell M, McClure N, Lewis SEM. The effectof cryopreservation on sperm morphology,motility and mitochondrial function. HumReprod 2002; 17: 704–9.
24. Holt WV, Morris GJ, Coulson G, North RD.Direct observation of cold shock effects inram spermatozoa with use of a programmablecryomicroscope. J Exp Zool 1988; 246: 305–14.
25. Isachenko E, Isachenko V, Katkov II, NawrothF. Vitrification of human spermatozoa withoutcryoprotectants: review of problems and prac-tical success. Reprod Biomed Online 2003; 6:191–200.
26. Katkov II, Katkova N, Critser JK, Mazur P.Mouse spermatozoa in high concentrations ofglycerol: chemical toxicity vs osmotic shock atnormal and reduced oxygen concentration.Cryobiology 1998; 37: 235–8.
27. Sakkas D, Tomlinson M. Assessment of spermcompetence. Semin Reprod Med 2000; 18:133–9.
28. Fahy GM. Vitrification: a new approach to organcryopreservation. In: Meryman HT editors.Transplantation: Approaches to Graft Rejection.New York: Alan R Liss, 1986: 305–35.
29. Kusakabe H, Szczygiel MA, Whittingham DG,Yanagimachi R. Maintenance of geneticintegrity in frozen and freeze-dried mousespermatozoa. Proc Natl Acad Sci USA 2001;98: 13501–6.
30. Karlsson JOM. A theoretical model of intra-cellular vitrification. Cryobiology 2001; 42:154–69.
31. Kanno H, Speedy RJ, Angell CA. Supercoolingof water to –90°C under pressure. Science1975; 189: 880–1.
32. MacFarlane DR, Scheirer J, Smedley SI.Pressure coefficient of conductance and ofglass transition temperatures in concentratedaqueous LiCl, LiI and AlCl3 solutions. J PhysChem 1986; 90: 2168–73.
33. MacFarlane DR. Physical aspects of vitrifica-tion in aqueous solutions. Cryobiology 1987;24: 181–95.
34. Karow AM, Liu WP, Humphries AL. Survival ofdog kidneys subjected to high pressures:necrosis of kidneys after freezing. Cryobiology1970; 7: 122–8.
35. Vajta G, Booth PJ, Holm P, Callesen H.Successful vitrification of early stage bovine invitro produced embryos with the Open Pulled
103
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 103

Straw (OPS) method. Cryo Letters 1997; 18:191–5.
36. Oberstein N, O’Donovan MK, Bruemmer JE.Cryopreservation of equine embryos by openpulled straws, cryoloop, or conventional coolingmethods. Theriogenology 2001; 15: 607–13.
37. Papis K, Shimizu M, Izaike Y. Factors affectingthe survivability of bovine oocytes vitrified indroplets. Theriogenology 2000; 15: 651–8.
38. Martino A, Pollard JA, Leibo SP. Effect of chill-ing bovine oocytes on their developmentalcompetence. Mol Reprod Dev 1996; 45:503–12.
39. Kuwayama M, Kato O. Successful vitrificationof human oocytes. Fertil Steril 2000; 74: 49abstr. 127.
40. Kurokawa T, Kinoshita T, Ito T, Sato H, Hotta T.Detection of minimal residual disease B celllymphoma by PCR mediated RNase protectionassay. Leukemia 1996; 10: 1222–31.
41. Matsumoto H, Jiang JY, Tanaka T, Sasada H,Sato E. Vitrification of large quantities ofimmature bovine oocytes using nylon mesh.Cryobiology 2001; 42: 139–44.
42. Lane M, Bavister BD, Lyons EA, Forest KT.Containerless vitrification of mammalianoocytes and embryos. Nat Biotechnol 1999;17: 1234–6.
43. Chen SU, Lien YR, Chao KH, Lu HF, Ho HN,Yang YS. Cryopreservation of mature humanoocytes by vitrification with ethylene glycol instraws. Fertil Steril 2000; 74: 804–8.
44. Reubinoff BE, Pera MF, Vajta G, Trounson AO.Effective cryopreservation of human embry-onic stem cells by the open pulled straw vitri-fication method. Hum Reprod 2001; 16:2187–94.
45. Kuleshova LL, MacFarlane DR, Trounson AO,Shaw JM. Sugars exert a major influence onthe vitrification properties of ethylene glycol-based solutions and have a low toxicity toembryos and oocytes. Cryobiology 1999; 38:119–30.
46. Rall WF. Prospects of the Cryopreservationof mammalian spermatozoa by vitrification.In: Johnson LA, Katz D, eds. Boar SemenPreservation. Berlin-Hamburg: PaureyScientific Publishers, 1991: 125–56.
47. Holt WV. Alternative strategies for the long-term preservation of spermatozoa. ReprodFertil Dev 1997; 9: 309–19.
48. Fahy GM. Vitrification. In: McGrath JJ, DillerKR, eds. Low Temperature Biotechnology:Emerging Applications and EngineeringContributions. Chicago: American Society ofMechanical Engineering 1988: 113–46.
49. Jahnel F. Über die Widerstandsfähigkeit vonmenschlichen Spermatozoen gegenüberstarker Kälte. Klin Wochenschrift 1938;37: 88–9. [in German]
50. Parkes AS. Preservation of human spermatozoaat low temperatures. Br Med J 1945; 2: 212–3.
51. Fahy GM, McFarlane DR, Angell CA, MerymanHT. Vitrification as an approach to cryopreser-vation. Cryobiology 1984; 21: 407–26.
52. Devireddy RV, Swanlund DJ, Roberts KP, PryorJL, Bishof JC. The effect of extracellular iceand cryoprotective agents on the water perme-ability parameters of human sperm plasmamembrane during freezing. Hum Reprod2000; 15: 1125–35.
53. Watson PR, Plummer JM. In deep freezing ofboar semen: the responses of boar spermmembranes to cold shock and cooling,Johnson LA, Larsson K editors. Swedish Univ.of Agricultural Sciences, Uppsala, Sweden;1985: 113–28.
54. Nauk VA. Structure and function of spermato-zoa from farm animal by cryopreservation.Furdui FI editor. Shtiinca, Kishineu, Modova;1991: 198.
55. Nawroth F, Isachenko V, Dessole S, Rahimi G,Farina M, Vargiu et al. Vitrification of humanspermatozoa without cryoprotectants. CryoLetters 2002; 23: 93–102.
56. Giraud MN, Motta C, Boucher D, Grizard G.Membrane fluidity predicts the outcome ofcryopreservation of human spermatozoa.Hum Reprod 2000; 15: 2160–4.
57. Holt WV. Fundamental aspects of sperm cry-obiology: the importance of species and indi-vidual differences. Theriogenology 2000; 53:47–58.
58. Esteves SC, Sharma RK, Thomas AJ Jr,Agarwal A. Effect of in vitro incubation onspontaneous acrosome reaction in fresh andcryopreserved spermatozoa. Int J FertilWomens Med 1998, 43: 235–42.
59. Isachenko V, Isachenko E, Montag M et al.Clean technique for cryoprotectant-free vitrifi-cation of human spermatozoa. Reprod BiomedOnline 2005; 10: 350–4.
60. Isachenko V, Isachenko E, Katkov II, MontagM, Dessole S, Nawroth F et al. Cryoprotectant-free cryopreservation of human spermatozoaby vitrification and freezing in vapor: effect onmotility, DNA integrity, and fertilization abil-ity. Biol Reprod 2004; 71: 1167–73.
61. Karlsson JOM, Cravalho EG. A model ofdiffusion-limited ice growth inside biologicalcells during freezing. J Appl Phys 1994; 75:4442–55.
VITRIFICATION IN ASSISTED REPRODUCTION
104
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 104

62. Hammadeh ME, Askari AS, Georg T,Rosenbaum P, Schmidt W. Effect of freezing-thawing procedure on chromatin stability,morphological alteration and membraneintegrity of human spermatozoa in fertile andsubfertile men. Int J Androl 1999; 22: 155–62.
63. Sun JG, Juriscova A, Casper RF. Detectionof deoxyribonucleic acid fragmentation inhuman sperm: correlation with fertilization invitro. Biol Reprod 1997; 56: 602–7.
64. Lopes S, Sun JG, Juriscova A, Meriano J,Casper RF. Sperm deoxyribonucleic acid frag-mentation in increased in poor-quality semensamples and correlated with failed fertilizationin intracytoplasmic sperm injection. FertilSteril 1998; 69: 528–32.
65. Katkov II. The point of maximum cell watervolume excursion in case of presence of animpermeable solute. Cryobiology 2002; 44:193–203.
66. Isachenko E, Isachenko V, Katkov II, Rahimi G,Schondorf T, Mallmann P et al. DNA integrityand motility of human spermatozoa after stan-dard slow freezing versus cryoprotectant-freevitrification. Hum Reprod 2004; 19: 932–9.
67. Evenson DP, Jost LK, Baer RK, Turner TW,Schrader SM. Individuality of DNA denatura-tion patterns in human sperm as measured bythe sperm chromatin structure assay. ReprodToxicol 1991; 5: 115–25.
68. Duty SM, Singh NP, Ruan L, Chen Z, Lewis C,Huang T et al. Reliability of the comet assay incryopreserved human sperm. Hum Reprod2002; 17: 1274–80.
69. Nakagata N, Takeshima T. High fertilizingability of mouse spermatozoa diluted slowlyafter cryopreservation. Theriogenology 1992;37: 1263–91.
70. Koshimoto C, Gamliel E, Mazur P. Effect ofosmolality and oxygen tension on the survivalof mouse sperm frozen to various tempera-tures in various concentrations of glycerol andraffinose. Cryobiology 2000; 41: 204–31
71. Nakagata N. Cryopreservation of mouse sper-matozoa. Mamm Genome 2000; 11: 572–6.
72. Van Blerkom J, Sinclair J, Davis P. Mitochondrialtransfer between oocytes: potential applicationsof mitochondrial donation and the issue of het-eroplasmy. Hum Reprod 1998; 12: 2857–68.
73. Meseguer M, Garrido N, Martinez-ConejeroJA, Simon C, Pellicer A, Remohi J. Role of cho-lesterol, calcium, and mitochondrial activity inthe susceptibility for cryodamage after a cycleof freezing and thawing. Fertil Steril 2004; 82:514–5.
74. Smiley ST, Reers M, Motolla-Hartshorn C, LinM, Chen A, Smith TW et al. Intracellularheterogeneity in mitochondrial membranepotentials revealed by a J-aggregate-forminglipophilic cation JC-1.Proc Natl Acad Sci USA1991; 88: 3671–5.
75. Isachenko V, Isachenko E, Montag M, Zaeva V,Krivokharchenko A, Nawroth F, et al. Cleantechnique for cryoprotectant-free vitrificationof human spermatozoa. Reprod BiomedOnline 2005; 10: 350–4.
105
CRYOPROTECTANT-FREE VITRIFICATION OF SPERMATOZOA
05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 105

05 Tucker 8029.qxd 8/23/2007 8:40 PM Page 106

107
Potential developmental consequencesof cryopreservation of mammalianoocytes and embryos Gary D Smith and Luis G Villa-Diaz
6
INTRODUCTION
The objective of prolonged storage of tissuesthat could be revitalized following suspendedanimation in cryostorage was envisioned byDr John Hunter over two centuries ago. Sincethen significant advances have been made incryopreservation of living cells, especially inthe 1940s when it was discovered that glycerolgreatly enhanced survival of cryopreservedliving cells.1 Around that time investigationsby Chang on low temperature storage of rab-bit oocytes, zygotes, and embryos2,3 paved theway for studies on cryopreservation of femalegametes and embryos. Subsequent experi-ments in the 1950s by Lin and Sherman4–6
demonstrated that mouse oocytes could alsobe cooled in glycerol, stored, and, subse-quently, fertilized in recipients, resulting inembryos that support pregnancies. In the1960s early investigations by Mazur7,8 formedthe foundation for understanding cell-specific optimal cooling and warming rateswhich today remain as pivotal keys to success-ful mammalian gamete and embryo cryo-preservation. Then in the 1970s thecombined strengths of Mazur, Leibo, andWhittingham resulted in successful cryop-reservation of mouse embryos.9 In 1977 thefirst successful IVF with live offspring fromcryopreserved mouse oocytes was reported,10
followed by the first human pregnancies afterembryo cryopreservation.11,12 Finally in 1986,Chen reported the first pregnancy afterhuman oocyte cryopreservation.13
OOCYTE CRYOPRESERVATIONMETHODS AND CRYODAMAGE
Currently mammalian oocyte cryopreserva-tion is performed in two different ways either‘slow-rate’ freezing or vitrification.14–16 ‘Slow-rate’ freezing attempts to minimize adversecellular events through control of the bio-physical properties of freezing, such as thecooling and warming rates in conjunctionwith cryoprotectants. With ‘slow-rate’ freez-ing, cells are cooled to very low temperatureswhile minimizing intracellular ice crystal for-mation, and at the same time it reduces thedetrimental influences caused by increasedsolute concentrations and osmotic stress.17
Therefore with ‘slow-rate’ freezing, cells aredehydrated through an equilibrium processwhere extracellular ice is formed. Alternatively,vitrification, a non-equilibrium approach tocryopreservation, utilizes high concentrationsof cryoprotectants that solidify without form-ing ice crystals, which are a major cause ofintracellular cryodamage. The vitrified solidstherefore contain the normal molecular andionic distributions of the original liquid stateand can be considered as an extremely vis-cous, supercooled liquid.18 During vitrifica-tion, cells are dehydrated by brief exposure toa concentrated solution of cryoprotectantsbefore plunging the samples directly intoliquid nitrogen. Vitrification comes from theLatin word vitrum, which means glass orresembling glass, and the technique wasoriginally developed for cryopreservation of
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 107

mammalian embryos.19 Currently vitrificationof oocytes and embryos is an area of focus formany clinical, rodent, and domestic animalproduction laboratories.
Cryopreservation of both germinal vesicle(GV)-stage20–22 and metaphase II (MII)23–25
oocytes has been reported in numerous species.With the use of ‘slow-rate’ freezing Erogluet al.22 found that 72% of GV-stage mouseoocytes survived the freezing process, 74% ofthose that survived progressed to MII, and 81%of MII oocytes underwent pronuclear forma-tion following insemination. Correspondingly,vitrification of GV-stage mouse oocytes resultedin similar rates of surviving oocytes and matu-ration to MII.26,27 Live offspring produced fromvitrified GV-stage mouse oocytes have alsobeen reported.27
Successful cryopreservation of human GV-stage oocytes has also been reported using‘slow-rate’ freezing, resulting in oocytes capa-ble of in vitro maturation, fertilization,28 andsupporting viable births.13,29 However, mostreported pregnancies after oocyte cryopreser-vation have been achieved with MII femalegametes. In spite of the fact that GV-intactoocyte cryopreservation is feasible, the ineffi-ciency of human oocyte in vitro maturationand subsequent ability to generate embryoswith embryonic developmental competenceand capacity to establish livebirths is amajor stumbling block in GV-intact oocytecryopreservation.30
Regardless of the methodology used forcryopreservation, effects on oocyte cellularfunctions can compromise the oocyte’s abilityto develop normally following the cryopreser-vation process. These compromised cellularevents can be collectively and generallytermed ‘cryodamage’. Experience in cryo-preservation of various cell types led to theappreciation that as cell size increases, diffi-culty in cryopreservation also increases.8
During cryopreservation, cells are exposed tonumerous stresses including mechanical,thermal, and chemical,8,31 which can lead tocompromised cell function and cell death.
This review addresses documented andtheoretical specific cellular structures andfunctions that are/may be compromised bycryopreservation and subsequent effects onoocyte developmental competence. The bio-physics of cryopreservation are not discussedhere; however, such information is availableelsewhere.32 Moreover, we do not attempt todelineate methodology-induced cryodamage,but view cryodamage from a cellular struc-tural/functional level, with attention directedtoward intracellular organelles and extracel-lular structures susceptible to cryodamage.Data from numerous mammalian species areconsidered concomitantly without differentia-tion between species.
INTRACELLULAR DAMAGE
Intracellular organelles within the nucleusand cytoplasm can be damaged during oocytecryopreservation; therefore we consider theirnormal developmental and homeostatic func-tions, and the potential developmental con-sequences of compromising their functionswith cryodamage.
Nucleus
The nucleus in the oocyte, also known as thegerminal vesicle, is composed of a nuclearenvelope (NE), and nuclear material such aschromatin, and the nucleolus. The nucleus ispresent during oocyte development and onceoocytes resume the first meiotic division, theNE is disintegrated allowing mixing of thenuclear material into the cytoplasm.
Nuclear envelope
When present the NE surrounds the nucleusand ensures a temporal and spatial separa-tion of events that take place within thenucleus and cytoplasm. Processes such as DNAreplication, transcription, RNA processing,and ribosomal subunit assembly occur within
VITRIFICATION IN ASSISTED REPRODUCTION
108
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 108

the nucleus, whereas most protein synthesistakes place in the cytoplasm.33–35 The NE iscomposed of the outer and inner nuclearmembranes, nuclear pore complexes, andlamina. Facing the cytoplasm is the outernuclear membrane, which is a specializedfunctional domain of the endoplasmic reticu-lum. The inner nuclear membrane is impor-tant both structurally and functionally since itcontains proteins that provide attachmentsites for both heterochromatin and thenuclear lamina. Both membranes are lipidbilayers that are connected to, and fenes-trated by, nuclear pore complexes which spanthe nuclear membrane.36 The nuclear porecomplexes are multiprotein assemblies dis-playing octagonal symmetry and form chan-nels that transport substrates with a diameterof approximately 26 nm.37 The nuclear porecomplexes also contain peripheral structuresprojecting into both the nucleoplasm andcytoplasm,38 and it is believed that these fila-mentous projections contain docking sitesfor transport substrates in association withspecific soluble receptors. Appropriate NEassembly with associated pore complexesprovides the correct nuclear and cytoplasmicsubstrates and signal trafficking necessary fornormal intracellular communication and func-tion. Finally, the lamina is a cage-like structurelocated within the nucleoplasm and in closeassociation with the inner nuclear membraneand nuclear pore complexes. Lamins, whichare type-V intermediate filaments, are themajor protein components of the lamina. Theimportance of lamina in determining nuclearshape, size, and ability to resist deformationand/or resume its initial shape after deforma-tion have been demonstrated in studies usinglamin knockout mice,39 dominant-negativemutants,40 and RNA interference (RNAi)‘knock-down’.41 In addition, it has been sug-gested that the lamina may also be importantregulators of gene expression,34 since it hasbeen found associated with heterochromatinat sites of DNA replication, RNA processing,replication proteins, and RNA polymerases.42
Therefore it could be hypothesized thatcompromising the structural integrity of theNE, and its associated lamina might influencesubsequent DNA replication, transcription,and normal cell function, and that both ‘slow-rate’ freezing and vitrification could disruptand/or distort the NE. Although the nucleushas the ability to reassemble morphologicallyfollowing oocyte cryopreservation,43 the futuredevelopment of the cell could be suboptimal.Recently it was demonstrated that recon-structed oocytes utilizing vitrified/warmedGVs were able to mature up to MII;44 however,their potential for further development wasnot tested.
Nucleolus
The nucleolus is the most prominent nuclearorganelle. In oocytes the nucleolus is presentin periods when protein synthesis is particu-larly important, such as during the oocytegrowth phase, and after the major activationof the embryonic genome following fertiliza-tion. These periods of profound stage-specificneed for protein synthesis also correlate wellwith elevated transcription; therefore ade-quate cytoplasmic pools of ribosomes mustexist during this time. Ribosomal subunits areformed in the nucleolus and subsequentlytransferred from the nucleus to the cytoplasmthrough the nuclear membrane pores of theNE, and associate to form cytoplasmic ribo-somes in conjunction with translation ofmRNAs into proteins.45
Van Blerkom et al.,20 using time-lapsephotography in vitrified/warmed oocytes,observed that the number and nuclear posi-tion of nucleoli as well as the majority ofnuclear structures returned to a normal statefollowing warming and rehydration. However,in some cases it was observed that nucleolarbodies were present in the cytoplasm afteroocyte warming and re-formation of the NE.Whether this aberrant intracellular nucleolilocalization influences subsequent develop-ment is unknown. Although these experiments
109
CONSEQUENCES OF CRYOPRESERVATION OF MAMMALIAN OOCYTES AND EMBRYOS
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 109
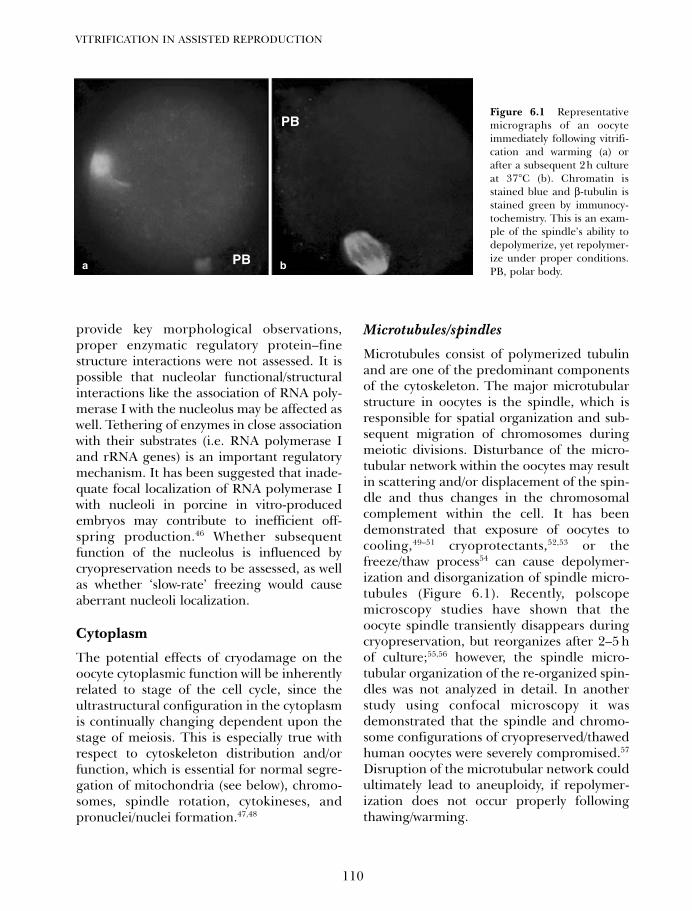
provide key morphological observations,proper enzymatic regulatory protein–finestructure interactions were not assessed. It ispossible that nucleolar functional/structuralinteractions like the association of RNA poly-merase I with the nucleolus may be affected aswell. Tethering of enzymes in close associationwith their substrates (i.e. RNA polymerase Iand rRNA genes) is an important regulatorymechanism. It has been suggested that inade-quate focal localization of RNA polymerase Iwith nucleoli in porcine in vitro-producedembryos may contribute to inefficient off-spring production.46 Whether subsequentfunction of the nucleolus is influenced bycryopreservation needs to be assessed, as wellas whether ‘slow-rate’ freezing would causeaberrant nucleoli localization.
Cytoplasm
The potential effects of cryodamage on theoocyte cytoplasmic function will be inherentlyrelated to stage of the cell cycle, since theultrastructural configuration in the cytoplasmis continually changing dependent upon thestage of meiosis. This is especially true withrespect to cytoskeleton distribution and/orfunction, which is essential for normal segre-gation of mitochondria (see below), chromo-somes, spindle rotation, cytokineses, andpronuclei/nuclei formation.47,48
Microtubules/spindles
Microtubules consist of polymerized tubulinand are one of the predominant componentsof the cytoskeleton. The major microtubularstructure in oocytes is the spindle, which isresponsible for spatial organization and sub-sequent migration of chromosomes duringmeiotic divisions. Disturbance of the micro-tubular network within the oocytes may resultin scattering and/or displacement of the spin-dle and thus changes in the chromosomalcomplement within the cell. It has beendemonstrated that exposure of oocytes tocooling,49–51 cryoprotectants,52,53 or thefreeze/thaw process54 can cause depolymer-ization and disorganization of spindle micro-tubules (Figure 6.1). Recently, polscopemicroscopy studies have shown that theoocyte spindle transiently disappears duringcryopreservation, but reorganizes after 2–5 hof culture;55,56 however, the spindle micro-tubular organization of the re-organized spin-dles was not analyzed in detail. In anotherstudy using confocal microscopy it wasdemonstrated that the spindle and chromo-some configurations of cryopreserved/thawedhuman oocytes were severely compromised.57
Disruption of the microtubular network couldultimately lead to aneuploidy, if repolymer-ization does not occur properly followingthawing/warming.
VITRIFICATION IN ASSISTED REPRODUCTION
110
PB
PB
Figure 6.1 Representativemicrographs of an oocyteimmediately following vitrifi-cation and warming (a) orafter a subsequent 2h cultureat 37°C (b). Chromatin isstained blue and β-tubulin isstained green by immunocy-tochemistry. This is an exam-ple of the spindle’s ability todepolymerize, yet repolymer-ize under proper conditions.PB, polar body.a b
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 110
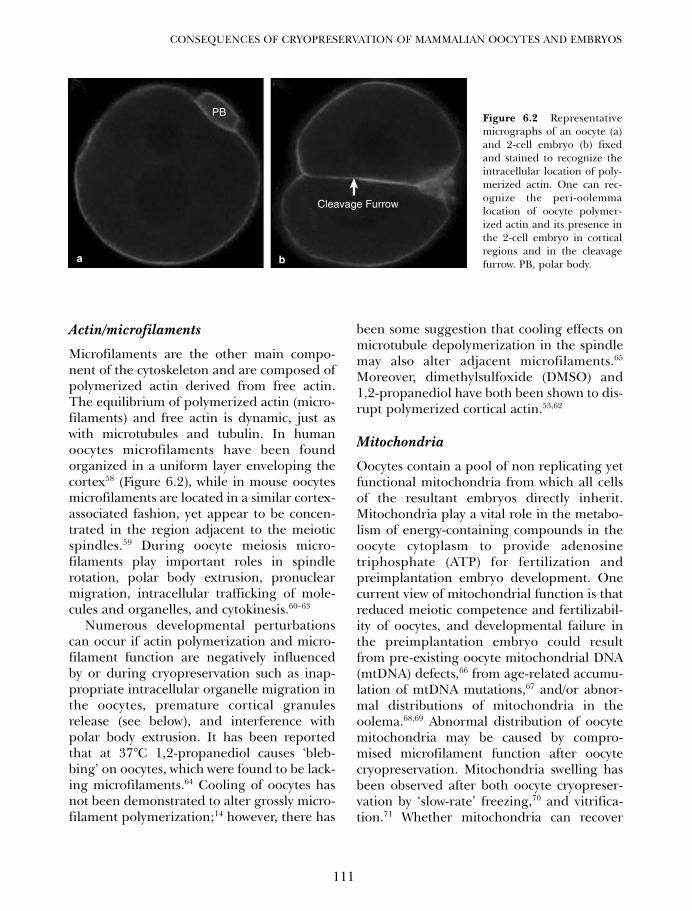
Actin/microfilaments
Microfilaments are the other main compo-nent of the cytoskeleton and are composed ofpolymerized actin derived from free actin.The equilibrium of polymerized actin (micro-filaments) and free actin is dynamic, just aswith microtubules and tubulin. In humanoocytes microfilaments have been foundorganized in a uniform layer enveloping thecortex58 (Figure 6.2), while in mouse oocytesmicrofilaments are located in a similar cortex-associated fashion, yet appear to be concen-trated in the region adjacent to the meioticspindles.59 During oocyte meiosis micro-filaments play important roles in spindlerotation, polar body extrusion, pronuclearmigration, intracellular trafficking of mole-cules and organelles, and cytokinesis.60–63
Numerous developmental perturbationscan occur if actin polymerization and micro-filament function are negatively influencedby or during cryopreservation such as inap-propriate intracellular organelle migration inthe oocytes, premature cortical granulesrelease (see below), and interference withpolar body extrusion. It has been reportedthat at 37°C 1,2-propanediol causes ‘bleb-bing’ on oocytes, which were found to be lack-ing microfilaments.64 Cooling of oocytes hasnot been demonstrated to alter grossly micro-filament polymerization;14 however, there has
been some suggestion that cooling effects onmicrotubule depolymerization in the spindlemay also alter adjacent microfilaments.65
Moreover, dimethylsulfoxide (DMSO) and1,2-propanediol have both been shown to dis-rupt polymerized cortical actin.53,62
Mitochondria
Oocytes contain a pool of non replicating yetfunctional mitochondria from which all cellsof the resultant embryos directly inherit.Mitochondria play a vital role in the metabo-lism of energy-containing compounds in theoocyte cytoplasm to provide adenosinetriphosphate (ATP) for fertilization andpreimplantation embryo development. Onecurrent view of mitochondrial function is thatreduced meiotic competence and fertilizabil-ity of oocytes, and developmental failure inthe preimplantation embryo could resultfrom pre-existing oocyte mitochondrial DNA(mtDNA) defects,66 from age-related accumu-lation of mtDNA mutations,67 and/or abnor-mal distributions of mitochondria in theoolema.68,69 Abnormal distribution of oocytemitochondria may be caused by compro-mised microfilament function after oocytecryopreservation. Mitochondria swelling hasbeen observed after both oocyte cryopreser-vation by ‘slow-rate’ freezing,70 and vitrifica-tion.71 Whether mitochondria can recover
111
CONSEQUENCES OF CRYOPRESERVATION OF MAMMALIAN OOCYTES AND EMBRYOS
PB
Cleavage Furrow
Figure 6.2 Representativemicrographs of an oocyte (a)and 2-cell embryo (b) fixedand stained to recognize theintracellular location of poly-merized actin. One can rec-ognize the peri-oolemmalocation of oocyte polymer-ized actin and its presence inthe 2-cell embryo in corticalregions and in the cleavagefurrow. PB, polar body. a b
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 111

from swelling, or what are its developmentalconsequences, has yet to be tested. Recently,Thouas et al.72 demonstrated that sublethalmitochondrial injury in mouse oocytes isheritable by resultant embryos, and results inpostimplantation pathologies similar to thosereported for clinically subfertile women,including recurrent implantation failure ormiscarriage, and decreased live birth weight.
EXTRACELLULAR DAMAGE
Zona pellucida
The zona pellucida is a glycoprotein mem-brane surrounding the plasma membrane ofoocytes and preimplantation embryos. Thezona pellucida is composed of three glyco-proteins termed ZP1, ZP2, and ZP3,73 and itis known to play a critical role in the entirefertilization process and in blockage ofpolyspermy following initial penetration byone spermatozoon. During the fertilizationprocess the spermatozoon binds to the zonapellucida and undergoes the acrosome reac-tion, it then penetrates the zona pellucidaand binds to a specific site on the oolemma ina ligand-receptor fashion.74 Binding of spermto the oolemma is believed to trigger the ‘cor-tical reaction’, which involves exocytosis ofcortical granules from the oocyte cortex intothe perivitelline space, and thus release ofcortical granule enzymes. These releasedenzymes result in a block to polyspermy bymodifying the zona pellucida (zona reaction),the oolemma, or both. The degree of zonareaction and plasma membrane block topolyspermy has been found to differ betweenspecies.75
Cortical granules are diffusely localizedthroughout the GV-intact cytoplasm, andmigrate to the oocyte cortex during matura-tion.76 Translocation and release of corticalgranules involves proper cytoskeleton functionand plasma membrane organization.It has been reported that DMSO77,78 and1,2-propanediol79 exposure during the cooling
process in oocyte cryopreservation causepremature cortical granule release and zonahardening, compromising sperm penetrationand fertilization. Recently, Gardner et al.80,81
demonstrated that the cryoprotectants men-tioned above, as well as ethylene glycol, causetransient calcium increases in mouse MIIoocytes, which were sufficient to cause zonahardening, presumably through triggeringcortical granule exocytosis, which is a cal-cium-dependent event.82 The authors alsoobserved that removal of calcium from thevitrification medium facilitated fertilizationby conventional insemination and develop-ment to the 2-cell stage at a rate approachingthat of control (non-vitrified) oocytes.
Zona hardening after oocyte cryopreserva-tion can also be avoided with the use ofbovine fetal serum;62,83 however, humanserum albumin has been shown not to pro-vide the same protection.84 Another negativeeffect of cryopreservation on oocytes is therapid changes in cell configuration. Such isthe case when oocytes are vitrified.43 This cell-shape alteration is observed as the cell fold-ing in upon itself and forming a crescentmoon or concave appearance. This can resultin fracture of the zona pellucida,85,86 and mostlikely contributes to polyspermic fertilizationfollowing oocyte cryopreservation.
OOCYTE CRYOSURVIVAL ANDPARTHENOGENETIC ACTIVATION
Obviously the future developmental compe-tency of a cryopreserved oocyte will be trun-cated if it does not survive, or if it becomesparthenogenetically activated after thaw-ing/warming. Although oocyte cryosurvivalhas yielded rates of ~90%,87,88 its variabilityrepresents a major obstacle for oocyte cryo-preservation, especially with ‘slow-rate’ freez-ing. Gardner and Lane have demonstratedthat ‘slow-rate’ freezing has detrimental effectson oocyte metabolism, and on subsequentembryo development and viability.88 Due to a
VITRIFICATION IN ASSISTED REPRODUCTION
112
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 112

significant rise in intracellular calcium, theauthors, in a separate set of experiments,observed low survival rates in oocytes cryo-preserved using ‘slow-rate’ freezing and1,2-propanediol.80 The correlation between ele-vated and sustained increases of intracellularcalcium, degeneration, and apoptosis inmature oocytes has been previously docu-mented.89,90 Elevated intracellular levels ofcalcium induced by slow-rate freezing with1,2-propanediol may also result in partheno-genetic activation of oocytes.91–94
Advances in technologies, such as mass spec-trometry, have allowed the ability to generateprotein profiles and identifying biomarkersfrom oocytes and embryos. Proteonomic analy-sis of MII oocytes following cryopreservationrevealed that ‘slow-rate’ freezing has asignificant effect on protein expression whencompared with non-cryopreserved MII oocytes.In contrast, vitrification had a minimal impactin protein expression.80 Now that this technol-ogy is available, it will be pertinent to study pro-teins and factors present in the nucleus andcytoplasm that affect oocyte maturation or itsfuture development. For example, it is knownthat factors present in the oocyte nucleus arerequired for male pronucleus formation.95
Whether cryopreservation would damage thosenuclear factors remains unknown; however, onecould speculate about its consequences if suchdamage occurs.
CONCLUSION
Oocyte cryopreservation has evolved sig-nificantly since the first achieved humanpregnancy reported by Chen13 in 1986.Accumulation of knowledge in basic biologyand physiology of mammalian oocytes andcryobiology has resulted in high survival ratesof cryopreserved oocytes, and moderate ratesof embryo development after fertilization.Healthy children have been born after bothslow-rate freezing and vitrification.
Continued accumulation of knowledgeregarding the intricacies of cryopreservationand related cell biology, such as the impor-tance of protein structural/functional rela-tionships to normal gene expression, proteintranslation, intracellular trafficking, epige-netic modifications, and cell development isnecessary. This knowledge will translate into abetter understanding of the potential devel-opmental consequences of cryopreservedoocytes, and will likely lead to the optimiza-tion of oocyte cryopreservation.
ACKNOWLEDGMENTS
The authors would like to express theirappreciation to Jeni Chapman for criticalreview of this manuscript. We apologize tothose whose work we have not cited owing tospace limitation.
113
CONSEQUENCES OF CRYOPRESERVATION OF MAMMALIAN OOCYTES AND EMBRYOS
References
1. Smith A. Biological Effects of Freezing andSupercooling. Edward Arnold, London, 1961
2. Chang MC. The effect of low temperature onfertilized rabbit ova in vitro, and the normaldevelopment of ova kept at low temperaturefor several days. J Gen Physiol 1948; 31:385–410.
3. Chang MC. Fertilizability of rabbit ova andthe effect of temperature in vitro on their sub-sequent fertilization and activation in vivo.J Exp Zool 1952; 121: 351–81.
4. Lin TP, Sherman JK, Willet EL. Survival of unfer-tilized mouse eggs in media containing glyceroland glycine. J Exp Zool 1957; 134: 275–97.
5. Sherman JK, Lin TP. Effect of glycerol and lowtemperature on survival of unfertilized mouseeggs. Nature 1958; 181: 785–6.
6. Sherman JK, Lin TP. Survival of unfertilizedmouse eggs during freezing and thawing. ProcSoc Exp Biol Med 1958; 98: 902–5.
7. Mazur P. Kinetics of water loss from cells atsubzero temperatures and the likelihood of
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 113

intracellular freezing. J Gen Physiol 1963; 47:347–69.
8. Mazur P, Leibo S, Chu E. A two-factor hypoth-esis of freezing injury – evidence fromChinese hamster tissue cells. Exp Cell Res1972; 71: 345–55.
9. Whittingham D, Leibo S, Mazur P. Survival ofmouse embryos frozen to −196o and −269oC.Science 1972; 178: 411–4.
10. Whittingham D. Fertilization in vitro a devel-opment to term of unfertilized mouse oocytespreviously stored at −196 degrees C.J ReprodFertil 1997; 49: 89–94.
11. Trounson A, Mohr L. Human pregnancy fol-lowing cryopreservation, thawing and transferof an eight-cell embryo. Nature 1983; 305:707–9.
12. Zeilmaker G, Alberda A, van Gent I, RijkmansC, Drogendijk A. Two pregnancies followingtransfer of intact frozen-thawed embryos.Fertil Steril 1984; 42: 293–6.
13. Chen C. Pregnancy after human oocyte cryo-preservation. Lancet 1986; 1: 884–6.
14. Bernard A, Fuller B. Cryopreservation ofhuman oocytes: a review of current problemsand perspectives. Hum Reprod Update 1996;2: 193–207.
15. Kuleshova L, MacFarlane D, Trounson A,Shaw J. Sugars exert a major influence on thevitrification properties of ethylene glycol-based solutions and have low toxicity toembryos and oocytes. Cryobiology 1999; 38:119–30.
16. Liebermann J, Nawroth F, Isachenko V et al.Potential importance of vitrification in repro-ductive medicine. Biol Reprod 2002; 67:1671–80.
17. Friedler S, Giudice L, Lamb E. Cryopreservationof embryos and ova. Fertil Steril 1988; 49:743–64.
18. Rall W. Factors affecting the survival of mouseembryos cryopreserved by vitrification.Cryobiol 1987; 24: 387–402.
19. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196 degrees C by vitri-fication. Nature 1985; 313: 573–5.
20. Van Blerkom J. Maturation at high frequencyof germinal-vesicle-stage mouse oocytes aftercryopreservation: alternations in cytoplasmic,nuclear, nucleolar and chromosomal structureand organization associated with vitrification.Hum Reprod 1989; 4: 883–98.
21. Candy C, Wood M, Whittingham D,Merriman J, Choudhury N. Cryopreservationof immature mouse oocytes. Hum Reprod1994; 9: 1738–42.
22. Eroglu A, Toner M, Leykin L, Toth TL.Cytoskeleton and polyploidy after maturationand fertilization of cryopreserved germinalvesicle-stage mouse oocytes. J Assist ReprodGenet 1998; 15: 447–54.
23. Martino A, Songsasen N, Leibo S.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
24. Eroglu A, Toth T, Toner M. Alterations of thecytoskeleton and polyploidy induced by cryo-preservation of metaphase II mouse oocytes.Fertil Steril. 1998; 69: 944–57.
25. Hong S, Chung H, Lim J, Ko J, Yoon T, Yee B,et al. Improved human oocyte developmentafter vitrification: a comparison of thawingmethods. Fertil Steril 1999; 72: 142–6.
26. Isachenko E, Nayudu P. Vitrification of mousegerminal vesicle oocytes: effect of treatmenttemperature and egg yolk on chromatin andspindle normality and cumulus integrity.Hum Reprod 1999; 14: 400–8.
27. Aono N, Abe Y, Hara K, Sasada H, Sato E,Yoshida H. Production of live offspring frommouse germinal vesicle-stage oocytes vitrifiedby a modified stepwise method, SWEID. FertilSteril 2005; 84 (Suppl 2): 1078–82.
28. Toth T, Baka S, Veeck L, Jones HJ, Muasher S,Lanzendorf S. Fertilization and in vitro devel-opment of cryopreserved human prophase Ioocytes. Fertil Steril 1995; 61: 891–4.
29. Tucker M, Morton P, Wright G, Sweitzer C,Massey J. Clinical application of human eggcryopreservation. Hum Reprod 1998; 13(11):3156–9.
30. Smith G. In vitro maturation of oocyte. CurrWomens Health Rep 2001: 143–51.
31. Meryman H. Cryoprotective agents.Cryobiology 1971; 8: 173–83.
32. Mazur P. Equilibrium, quasi-equilibrium, andnonequilibrium freezing of mammalianembryos. Cell Biophysics 1990; 17: 53–92.
33. Buendia B, Courvalin J, Collas P. Dynamics ofthe nuclear envelope at mitosis and duringapoptosis. Cell Mol Life Sci 2001; 58: 1781–89.
34. Hutchison C. Lamins: building blocks or reg-ulators of gene expression. Nature 2002; 3:848–58.
35. Rzepecki R. The nuclear lamins and thenuclear envelope. Cell Mol Biol Lett 2002; 7:1019–35.
36. Hinshaw J, Carragher B, Milligan R.Architecture and design of the nuclear porecomplex. Cell 1992; 69: 1133–41.
37. Feldherr C, Kallenbach E, Schultz N.Movement of a karyophilic protein through
VITRIFICATION IN ASSISTED REPRODUCTION
114
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 114

the nuclear pores of oocytes. J Cell Biol 1984;99: 2216–22.
38. Pante N, Aebi U. Towards understanding thethree-dimensional structure of the nuclearpore complex at the molecular level. CurrOpin Struct Biol 1994; 4: 187–96.
39. Sullivan T. Loss of A-type lamin expressioncompromises nuclear envelope integrity lead-ing to muscular dystrophy. J Cell Biol 1999;147: 913–20.
40. Schirmer E, Guam T, Gerace L. Involvementof the lamin rod domain in heterotypic lamininteractions important for nuclear organiza-tion. J Cell Biol 2001; 153: 479–89.
41. Liu YP, Dovzhenko OV, Garthwaite MA,Dambaeva SV, Durning M, Pollastrini LM,et al. Maintenance of pluripotency in humanembryonic stem cells stably over-expressingenhanced green fluorescent protein. StemCells Dev 2004; 13: 636–45.
42. Wilson K, Zastro M, Lee K. Lamins and dis-ease: insights into nuclear infrastructure. Cell2001; 104: 647–50.
43. Van Blerkom J, Davis P, Merriam J. The devel-opmental ability of human oocytes penetratedat the germinal vesicle stage after inseminationin vitro. Hum Reprod 1994; 9(4): 697–708.
44. Kren R, Fulka J, Fulka H. Cryopreservation ofisolated mouse germinal vesicles. J ReprodDev 2005; 51: 289–92.
45. Hyttel P, Viuff D, Fair T, Laurincik J, ThomsenP, Callesen H, et al. Ribosomal RNA geneexpression and chromosome aberrations inbovine oocytes and preimplantation embryos.Reproduction 2001; 122: 21–30.
46. Hyttel P, Viuff D, Laurincik J, Schmidt M,Thomsen P, Avery B, et al. Risks of in-vitroproduction of cattle and swine embryos: aber-rations in chromosome numbers, ribosomalRNA gene activation and perinatal physiol-ogy. Hum Reprod 2000; 15: 87–97.
47. Schatten G, Simerly C, Schatten H.Microtubule configurations during fertiliza-tion, mitosis, and early development in themouse and the requirement for egg micro-tubule-mediated motility during mammalianfertilization. Proc Natl Acad Sci USA 1985; 82:4152–6.
48. Maro B, Johnson M, Webb M, Flach G.Mechanism of polar body formation in themouse oocytes: an interaction between thechromosomes, the cytoskeleton and the plasmamembrane. J Embryol Exp Morphol 1986; 92:11–32.
49. Pickering S, Johnson M. The influence ofcooling on the organization of the meiotic
spindle of the mouse oocyte. Hum Reprod1987; 2: 207–16.
50. Pickering S, Braude P, Johnson M, Cant A,Currie J. Transient cooling to room tempera-ture can cause irreversible disruption of themeiotic spindle in the human oocyte. FertilSteril 1990; 54: 102–8.
51. Aman R, Parks J. Effects of cooling andrewarming on the meiotic spindle and chro-mosomes of in vitro matured bovine oocytes.Biol Reprod 1994; 50: 103–10.
52. Johnson M, Pickering S. The effect of dimethyl-sulphoxide on the microtubular system of themouse oocyte. Development 1987; 100: 313–24.
53. Vincent C, Garnier V, Heyman Y, Renard J.Solvent effects on cytoskeletal organizationand in-vivo survival after freezing of rabbitoocytes. J Reprod Fertil 1989; 87: 809–20.
54. Aigner S, Van der Elst J, Siebzehnrubl E, WildtL, Lang N, Van Steirteghem A. The influenceof slow and ultra-rapid freezing on the orga-nization of the meiotic spindle of the mouseoocyte. Hum Reprod 1992; 7: 857–64.
55. Rienzi L, Martinez F, Ubaldi F, Minasi MG,Iacobelli M, Tesarik J, et al. Polscope analysisof meiotic spindle changes in livingmetaphase II human oocytes during the freez-ing and thawing procedures. Hum Reprod2004; 19: 655–9.
56. Bianchi V, Coticchio G, Fava L, Flamigni C,Borini A. Meiotic spindle imaging in humanoocytes frozen with a slow freezing procedureinvolving high sucrose concentration. HumReprod 2005; 20: 1078–83.
57. Boiso I, Marti M, Santalo J, Ponsa M, Barri P,Veiga A. A confocal microscopy analysis of thespindle and chromosome configurations ofhuman oocytes cryopreserved at the germinalvesicle and metaphase II stage. Hum Reprod2002; 17: 1885–91.
58. Pickering S, Johnson M, Braude P, HoulstonE. Cytoskeletal organization in fresh aged andspontaneously activated human oocytes. HumReprod 1988; 3: 978–89.
59. Longo F. Actin-plasma membrane associa-tions in mouse eggs and oocytes. J Exp Zool1987; 243: 299–309.
60. Schatten G, Spector I, Cline C. Latrunculininhibits the microfilament mediated processesduring fertilisation, cytokinesis and earlydevelopment. Exp Cell Res 1986; 166:191–208.
61. Le Guen P, Crozet N, Huneau D, Gall L.Distribution of microfilaments during earlyevents of sheep fertilisation. Gamete Res1989; 22: 411–25.
115
CONSEQUENCES OF CRYOPRESERVATION OF MAMMALIAN OOCYTES AND EMBRYOS
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 115

62. Vincent C, Johnson M. Cooling cryoprotec-tants and the cytoskeleton of the mammalianoocyte. Ox Rev Reprod Biol 1992; 14:73–100.
63. Wang W, Abeydeera L, Prather R, Day B.Polymerization of nonfilamentous actin intomicrofilaments is an important process forporcine oocyte maturation and early develop-ment. Biol Reprod 2000; 62: 1177–83.
64. Joly C, Bchini O, Boulekbache H, Testart J,Maro B. Effects of 1,2-propanediol on thecytoskeletal organization of the mouse oocyte.Hum Reprod 1992; 7: 374–8.
65. Webb M, Howlet S, Maro B. Parthogenesisand cytoskeletal organization in ageing mouseeggs. J Embryol Exp Morphol 1986; 95:131–45.
66. Perez GI, Trbovich AM, Gosden RG, Tilly JL.Mitochondria and the death of oocytes.Nature 2000; 403: 500–1.
67. Keefe DL, Niven-Fairchild T, Powell S,Buradagunta S. Mitochondrial deoxyribonucleicacid deletions in oocytes and reproductive agingin women. Fertil Steril 1995; 64: 577–83.
68. Nagai S, Mabuchi T, Hirata S, Shoda T, KasaiT, Yokota S, et al. Correlation of abnormalmitochondrial distribution in mouse oocyteswith reduced developmental competence.Tohoku J Exp Med 2006; 210: 137–44.
69. Van Blerkom J, Davis P, Alexander S.Differential mitochondrial distribution inhuman pronuclear embryos leads to dispro-portionate inheritance between blastomeres:relationship to microtubular organization,ATP content and competence. Hum Reprod2000; 15: 2621–33.
70. Valojerdi MR, Salehnia M. Developmentalpotential and ultrastructural injuries ofmetaphase II (MII) mouse oocytes after slowfreezing or vitrification. J Assist Reprod Genet2005; 22: 119–27.
71. Hochi S, Kozawa M, Fujimoto T, Hondo E,Yamada J, Orugi N. In vitro maturation andtransmission electron microscopic observationof horse oocytes after vitrification.Cryobiology 1996; 33: 300–10.
72. Thouas GA, Trounson AO, Jones GM.Developmental effects of sublethal mitochon-drial injury in mouse oocytes. Biol Reprod2006; 74: 969–77.
73. Moos J, Faundes D, Kopf G, Schultz R.Composition of the human zona pellucidaand modifications following fertilization.Hum Reprod 1995; 10: 2467–71.
74. Wassarman P. Profile of a mammalian spermreceptor. Development 1990; 108: 1–17.
75. Wolf D. The mammalian egg’s block topolyspermy. In: Mastroianni L, Biggers JD, eds.Fertilization and Embryonic Development inVitro. New York: Plenum Press, 1981: 183–97.
76. Ducibella T, Anderson E, Albertini D, AalbergJ, Rangarajan S. Quantitative studies ofchanges in cortical granule number and dis-tribution in the mouse oocyte during meioticmaturation. Dev Biol 1988; 130: 184–97.
77. Johnson M. The effect on fertilisation ofexposure of mouse oocytes to dimethyl sulfox-ide: an optimal protocol. J In Vitro FertEmbryo Transf 1989; 6: 168–75.
78. Pickering S, Braude P, Johnson M. Cryo-protection of human oocytes: inappropriateexposure to DMSO reduces fertilization rates.Hum Reprod 1991; 6: 142–43.
79. Schalkoff M, Oskowitz S, Powers R.Ultrastructural observations of human andmouse oocytes treated with cryopreservation.Biol Reprod 1989; 40: 379–93.
80. Larman MG, Katz–Jaffe MG, Sheehan CB,Gardner DK. 1,2-propanediol and the type ofcryopreservation procedure adversely affectmouse oocyte physiology. Hum Reprod 2007;22: 250–9.
81. Larman MG, Sheehan CB, Gardner DK.Calcium-free vitrification reduces cryoprotec-tant-induced zona pellucida hardening andincreases fertilization rates in mouse oocytes.Reproduction 2006; 131: 53–61.
82. Kline D, Kline JT. Repetitive calcium tran-sients and the role of calcium in exocytosisand cell cycle activation in the mouse egg.Dev Biol 1992; 149: 80–9.
83. Carroll J, Wood M, Whittingham D. Normalfertilisation and development of frozen-thawedmouse oocytes: protective action of certainmacromolecules. Biol Reprod 1993; 48:606–12.
84. George MA, Johnson MH. Use of fetal bovineserum substitutes for the protection of themouse zona pellucida against hardening dur-ing cryoprotectant addition. Hum Reprod1993; 8: 1898–900.
85. Dhali A, Manik R, Das S, Singla S, PaltaP. Post-vitrification survival and in vitro matura-tion rate of buffalo (Bubalus bubalis) oocytes:effect of ethylene glycol concentration andexposure time. Anim Reprod Sci 2000; 63:159–65.
86. Wu C, Rui R, Dai J, Zhang C, Ju S, Xie B,et al. Effects of cryopreservation on the devel-opmental competence, ultrastructure andcytoskeletal structure of porcine oocytes. MolReprod Dev 2006; 73: 1454–62.
VITRIFICATION IN ASSISTED REPRODUCTION
116
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 116

87. Fosas N, Marina F, Torres PJ, Jove I, Martin P,Perez N, et al. The births of five Spanishbabies from cryopreserved donated oocytes.Hum Reprod 2003; 18: 1417–21.
88. Lane M, Gardner DK. Vitrification of mouseoocytes using a nylon loop. Mol Reprod Dev2001; 58: 342–7.
89. Gordo AC, Rodrigues P, Kurokawa M, JelleretteT, Exley GE, Warner C, et al. Intracellular cal-cium oscillations signal apoptosis rather thanactivation in in vitro aged mouse eggs. BiolReprod 2002; 66: 1828–37.
90. Takahashi T, Igarashi H, Doshida M,Takahashi K, Nakahara K, Tezuka N, et al.Lowering intracellular and extracellular cal-cium contents prevents cytotoxic effects ofethylene glycol-based vitrification solution inunfertilized mouse oocytes. Mol Reprod Dev2004; 68: 250–8.
91. Shaw JM, Trounson AO. Parthenogeneticactivation of unfertilized mouse oocytesby exposure to 1,2-propanediol is influenced
by temperature, oocyte age, and cumulusremoval. Gamete Res 1989; 24: 269–79.
92. Van Der Elst J, Van Den Abbeel E, Nerincks S,Van Steirteghem A. Parthenogenetic activationparttern and microtubular organization of themouse oocyte after exposure to 1,2-propane-diol. Cryobiology 1992; 29: 549–62.
93. Gook DA, Osborn SM, Johnston WI.Parthenogenetic activation of human oocytesfollowing cryopreservation using 1,2-propanediol. Hum Reprod 1995; 10: 654–8.
94. Ducibella T, Huneau D, Angelichio E, Xu Z,Schultz RM, Kopf GS, et al. Egg-to-embryotransition is driven by differential responses toCa(2+) oscillation number. Dev Biol 2002;250: 280–91.
95. Ogushi S, Fulka J, Jr., Miyano T. Germinalvesicle materials are requisite for male pro-nucleus formation but not for change in theactivities of CDK1 and MAP kinase duringmaturation and fertilization of pig oocytes.Dev Biol 2005; 286: 287–98.
117
CONSEQUENCES OF CRYOPRESERVATION OF MAMMALIAN OOCYTES AND EMBRYOS
06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 117

06 Tucker 8029.qxd 8/23/2007 2:13 PM Page 118

119
Vitrification of oocytes: generalconsiderations and the use of theCryotop method Masashige Kuwayama, Ana Cobo and Gábor Vajta
7A
WHY DO WE NEED TOCRYOPRESERVE HUMANOOCYTES?
In most papers dealing with this topic, a longlist of simple and obvious reasons to answerthis question can be found. These answers arealso listed below. However, this review startswith a more general and rarely mentionedargument: the handicapped situation of womenfrom the point of reproduction.
Although gender-based discrimination isless and less acceptable in most human soci-eties, nature still preserves the right to makea seemingly unfair but strong distinctionbetween males and females in certain areasincluding – obviously – reproduction. In mostmammalian species, females hold most of theweight of reproduction, including discomfortrelated to reproductive cycles and pregnancy,pain of labor, and nursing of babies. Inhumans, even with the best intentions, malepartners cannot share most of these sacrifices,and most women accept them as an inevitablepart of their full life, that is at least partiallycompensated by the most intimate relation-ship with their babies. However, there areadditional, even more frustrating differencesbetween males and females regarding thepossibility to distribute and preserve geneticmaterial. A healthy male can produce manymillions of sperm cells every day, while thenumber of oocytes is highly restricted, inmany mammals including human to one or
two per month. Moreover, males normallymay preserve their reproductive ability fortheir whole life, while the time for womenis rather limited. Theoretically they maybecome pregnant and have babies before themenopause – which also means almost halv-ing the available time compared with males –but in practice, even this period is drasticallyshortened by the fact that the quality ofoocytes decreases sharply after the age of 35,restricting the real freedom to reproducewithout concern and fear to approximately 15years, coinciding exactly with the time whichis the most critical to establish a professionalcareer for a lifetime.
We may be referring to the inevitableorder or laws of nature, but aging, loss ofteeth or hair may also be referred to as such,not to mention blindness, deafness, or otherserious handicaps. Considerable and fullyjustified efforts are made to eliminate theseunfair differences created by life and nature,and legislative and financial help are pro-vided to ensure equal opportunity in manyfields. More attention should probably befocused on this half of humankind to allevi-ate their handicapped situation in the field ofreproduction.
Unfortunately, although not at all accordingto the original intentions, modern reproduc-tive technologies in the human have made thesituation even worse. With the application ofintracytoplasmic sperm injection (ICSI), onegamete from both genders may be enough to
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 119

produce an offspring. Compared with thealmost infinite possibility, the 10-, sometimes20-fold artificial increase of the number ofavailable oocytes (as the result of an expensive,painful, and risky medical intervention) doesnot seem to be a real compensation. It is evenmore embarrassing than with ICSI, incom-pletely capacitated or matured, handicapped,immotile, or dying spermatozoa can still besuccessfully used for fertilization, while theslightest deviation in morphology, function ofoocytes (including those caused by the sup-portive medical intervention itself), or thesmallest inaccuracy of the stage of maturationmay seriously compromise their further devel-opmental competence.
Finally, when we consider storage possibil-ities of male and female gametes of mam-mals, the difference is even more frustrating.In many domestic species large commercialnetworks are dealing with collection, deep-freezing, and distribution of sperm of valu-able animals, by using highly standardizedprocedures and with an efficiency that almostcompletely eliminates the need for naturalmating or artificial insemination with freshsemen. The establishment of the ‘cryoprotec-tant-free vitrification’ method1 for spermato-zoa also proves the extreme tolerance of malegerm cells towards cryoinjuries, although theterm itself may raise some concerns. In themouse, a technology has also been developedenabling sperm transportation in a sealedenvelope by ordinary mail. On the otherhand, the recovery, laboratory handling, andespecially cryopreservation of mammalianoocytes from live animals is still regarded as achallenge and restricted (with a few excep-tions) to the experimental field. The situationis no better in humans: sperm can be easilycollected, frozen, stored, and utilized in smallaliquots, creating the commercial distributionof this supposedly valuable male genetic‘stuff ’ is a prosperous business in some coun-tries. In contrast, storage and use of thefemale gamete is seriously restricted by theabove-mentioned biological, technical, and
psychological problems related to collection;by legal measures restricting experimentaluse and/or donations in many countries; andby the poor and inefficient technologiesavailable for their cryopreservation. Untilrecently, due to the cumulative effect of thesefactors, the efficiency of the whole procedurewas so low that practically every baby bornafter oocyte cryopreservation deserved ascientific publication.
Establishment and widespread applicationof an efficient and safe cryopreservationmethod for oocytes would not eliminate dif-ferences in reproductive flexibility betweenfemales and males, but may provide a solu-tion for many specific problems and eventu-ally reverse the actual trend of thisunacceptable artificial widening of the gapbetween the two genders. Accordingly, thisarea deserves special attention, and should beregarded as more than just the subject of sci-entific ambition of a few, accidentally selectedscientists; making this an area that is benignlyand respectfully disregarded by the vastmajority of reproductive specialists.
The need for change in the general attitudetowards oocyte cryopreservation is even morejustified in view of the recent rapid advance-ment in technology now offering the prospectof a real breakthrough, and a definite andcurrently available solution in many impor-tant fields including the following.
• Malignant diseases where systemic anti-cancer treatment is required2
• Surgical procedures resulting in loss ofovarian function3
• Treatment of patients with polycystic ovar-ian syndrome4,5
• Patients with ovary hyperstimulationsyndrome3
• Poor responders to ovarian stimulation3
• Patients at risk of ovarian function lossthrough premature menopause3
• In cases of male factor infertility or prob-lems associated with difficulty of spermcollection, inadequate seminal samples, or
VITRIFICATION IN ASSISTED REPRODUCTION
120
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 120

non-viable spermatozoa at the time ofoocyte retrieval3
• To overcome ethical concerns and legalrestrictions in several countries associatedwith embryo cryopreservation6
• Cryobanking oocytes for young womenwho wish to delay motherhood for variousreasons (career, lack of appropriatepartner, etc.)7
• Cryobanking oocytes for egg donation pro-grams or for research purposes.8
WHY IS CRYOPRESERVATION OFOOCYTES DIFFICULT?
Some of the reasons why cryopreservation ofoocytes is difficult, including the size, shape,and cell number, are quite obvious. It iswell known that oocytes are the largest cellsof the human body. In cryobiology, the sizeor rather the mass is a decisive factor.Suspensions of somatic cell cultures can becryopreserved with high efficiency and with-out any sophisticated approach by using sim-ple media, a refrigerator, and a deep freezeror liquid nitrogen. For the even smaller bac-teria and viruses, we meet the frustrating evi-dence of this every day: they are present inalmost every liquid nitrogen tank, and pre-serve their viability without any protectionand in spite of our best intention (althoughapart from the size, some other factors, forexample, the simple structure may also play arole in this resistance). In reproductive biologywe just refer to the above-mentioneddifferences between cryotolerance of sperma-tozoa and oocytes that can also at least par-tially be attributed to the differences involume. Quite controversially, the cumulativemass of cells decreases exponentially duringthe first week of embryo development, and atthe expanded blastocyst stage the mass maybecome as low as 1/10 to 1/100 of that of theoocyte, with an obvious similar decrease inthe water content. Although the solutionaccumulated in the blastocoel may be apotential source of damage either by ice
crystal formation, or through the accumulationand slow dilution of toxic cryoprotectants,9–11
these mechanisms obviously cause more harmwhen they occur intracellularly in the oocyte.
Apart from the size, the shape of theoocyte is also most unfortunate. The almostperfect sphere slows down formation of anequal distribution of any substance, includingpermeable cryoprotectants coming from out-side or released from the oocyte. Accordingly,for a relatively long period of time a continu-ous concentration gradient from the periph-ery to the center or vice versa exists, resultingin toxic damage in one part while providingless than optimal protection in the other.From this point of view, the change in shapecaused by the osmotic effect at equilibrationmay offer some kind of benefit, but may alsocontribute to the damage of the cytoskeleton(see below).
The third major factor is the lowest possi-ble cell number. From this point of view theoocyte resembles a gambler who puts all hismoney on the very first bet: all or nothing.Multicellular embryos can survive and com-pensate for as much as 50% loss of their cells(and supposedly also some level of injury inthe remaining ones) as demonstrated bybiopsies, bisection of embryos, or just the lessthan optimal culture conditions apart fromthe cryopreservation experiences. The oocytehas only one chance, and there is no backupto regenerate from a serious injury. We haveto use an extremely careful approach to getout of the game as winners.
Unfortunately, apart from the factors listedabove, there are still many other factors thatcontribute to the sensitivity of oocytes tocryoinjuries. Chilling injury – that occurs atrelatively high temperatures and induces irre-versible damage of the cytoplasmic lipiddroplets, lipid-rich cell membranes, andmicrotubules – affects mostly the latter twostructures in the human oocyte, as (in con-trast for example to pigs) in humans cytoplas-mic lipid droplets are less abundant. On theother hand, the membranes are extremely
121
VITRIFICATION OF OOCYTES: GENERAL CONSIDERATIONS AND THE USE OF THE CRYOTOP METHOD
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 121

sensitive and rapidly undergo a transitionfrom the liquid state to the gel state, an irre-versible process that is detrimental for futuredevelopment. For unknown reasons, just onestep further on, after fertilization the mem-branes of zygotes are much less sensitive tothis type of injury.12
The depolymerization of microtubules,misalignment of the chromosomes, and thepossible increased risks of aneuploidy arefrequently emphasized and have a wideexperimental background,13–16 although inthe human comparative examinations maynot entirely confirm the seriousness of theseproblems,17 and the supposed beneficialeffect of some agents (cytoskeleton relaxantsor stabilizers) is not fully proven. Similar tosomatic cell nuclear transfer, spindle reorga-nization may occur surprisingly efficiently,and the number of chromosomal abnormalitiesin children born after oocyte vitrificationdoes not seem to show a significant increase.
A strange and not completely understoodphenomenon is the change in cryosensitivityof oocytes during the maturation process.Although there is only a minimal differencebetween the size and shape, immature oocytesare usually more sensitive to cryopreservationthan mature (metaphase II (MII) oocytes.12,18,19
The contrary might be supposed, based on theknown sensitivity of the meiotic spindle tochilling. More research is needed to under-stand the reasons for this difference; the alter-ation of sensitivity of membranes may be oneof the possible explanations.
The osmotic shock at equilibration mayresult in shrinking and misshaping of theoocytes, supposedly damaging the cytoskeleton.However, the effect of other agents (for exam-ple pronase digestion of the zona pellucida)induces much more serious deformation, fol-lowed by surprisingly rapid recovery andmaintenance of developmental competence.On the other hand, the osmotic shock thatcan occur during dilution may result in exten-sive swelling, rupture of the membrane, lysis,and immediate death of the oocytes.
Hardening of the zona pellucida, attributedby some authors to premature cortical granulerelease may cause decreased rates offertilization.6
Fracture is a common consequence of allcryopreservation procedures20 and does notseem to occur more frequently in oocytesthan in embryos. However, while the conse-quence for zona fracture may be similar forboth, embryos may survive some level of cellmembrane damage, while for the oocyte, anyinjury at this level is evidently fatal.
WHAT IS THE BEST APPROACH?
Based on the points listed above, the princi-ples of a successful cryopreservation strategycan be outlined. Although infrequent in bio-logical study, theory is mostly justified bypractice; however, it should be confessed thatthe sequence of events was (as usual)inverted: the empirically established meth-ods were retrospectively supported by thesubsequent detailed theoretical analyses ofevents.
First, we need a method that minimizeschilling injury. So far, in mammalian embryosand oocytes two approaches have been suc-cessfully applied for this purpose: theremoval of the lipid droplets (by high-speedcentrifugation and micromanipulation,although the latter step is not required withthe use of some recent techniques21) and byradically increasing the cooling and warmingrate to minimize the duration of exposureto the dangerous temperatures. As humanoocytes contain a relatively low amount oflipids, centrifugation does not significantlyimprove survival chances. On the other hand,all forms of traditional slow-rate freezing areobviously less appropriate to avoid chillingthan high-rate cooling vitrification strategies.
The large cell mass and spherical shape ofthe oocyte necessitate the use of highly per-meable cryoprotectants with low toxicity. As inmany areas of vitrification in mammalianembryology, ethylene glycol is the candidate
VITRIFICATION IN ASSISTED REPRODUCTION
122
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 122

of choice for this purpose. According toearlier investigations in rabbits,22 the perme-ability of ethylene glycol is facilitated bydimethylsulphoxide (DMSO). Further studieshave also demonstrated that DMSO may havea beneficial effect on spindle polymerization,and consequently a protective effect at oocytevitrification.3 Although various proportions ofDMSO and ethylene glycol were extensivelytested for vitrification of bovine oocytes andembryos, the best results were always achievedwith a 1:1 mixture (Vajta, unpublished). Tofacilitate dehydration, thus decreasing thechances of intracellular ice formation, theaddition of non-permeable cryoprotectants isalso required. Various substances includingpolymers with low toxicity were suggested forthe purpose, however, the traditionally usedsugars, i.e. sucrose or trehalose, seem to bemore appropriate. Curiously, although tre-halose has been reported many times to besuperior, in the past few years it has graduallydisappeared from the list of frequently usedcryoprotectants.
In the past few years, two basically differ-ent strategies of equilibration before coolingwere applied. Martino et al.23 suggested thatdehydration may even be more importantthan cryoprotectant concentration for pre-vention of ice crystal formation, and sug-gested extremely short equilibrations for boththe diluted and concentrated cryoprotectantsolutions. Subsequently, this strategy was suc-cessfully applied by many others for domesticanimal oocytes and embryos. Howeverrecently another approach has received moreattention, and seems to be more efficient formammalian oocytes: an extended equilibra-tion in a rather diluted first cryoprotectantsolution, followed by a short, but slightly pro-longed incubation in the second, relativelyconcentrated vitrification solution also con-taining a non-permeable cryoprotectant.24–26
Although the time of the exposure is signifi-cantly increased, the cumulative toxic effect(as a result of the lower concentration) may bethe same or even lower, and the prolonged
equilibration may ensure proper penetrationof cryoprotectant providing appropriate pro-tection to the entire oocyte.
The other way to minimize toxic andosmotic effects of cryoprotectant is todecrease the required concentration whilemaintaining the ice-free solidification pat-tern. Currently the only practical way toachieve this goal is with the extreme increasein cooling rates. Among the various toolsapplied for the purpose, electron microscopicgrids, Cryoloops, and Cryotops seem to bethe most appropriate ones; although veryrecently similar results were achieved with theopen pulled straw technique.27 Either directlyby the higher rate of cooling, or indirectly bythe decreased toxic and osmotic effect,Cryotop and Cryoloop vitrification with amixture of relatively low concentrations ofDMSO and ethylene glycol do not seem tocause serious anomalies in the spindle struc-ture, and may ensure relatively high develop-mental rates.26,28
As mentioned above, fracture damage isnot specific to oocyte cryopreservation,although the consequences may be moredetrimental in oocytes. Fortunately, the openvitrification systems have drastically reducedthe occurrence of this type of damage.Retrospectively, it may be supposed that in aclosed system, the extreme pressure changescaused by rapidly cooling or warming air bub-bles induce dislocations in the partially solid-ified solution, and with a scissor-like effect cutthe zona pellucida or the cell membranes. Inthe open systems, such mechanical forces arealmost completely avoided. The extremelysmall volume of solutions used also minimizesthe chance of fractures. Accordingly, this typeof damage is almost entirely eliminated bythe application of the ultrarapid open vitrifi-cation systems.
Finally, the problem of zona hardeningand subsequent low level of fertilization hasbeen eliminated in humans with the discoveryand subsequent widespread application ofICSI. Although not included in the original
123
VITRIFICATION OF OOCYTES: GENERAL CONSIDERATIONS AND THE USE OF THE CRYOTOP METHOD
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 123

goals, the application of ICSI after cryo-preservation has contributed much to theincrease of efficiency, and has opened thegate for widespread application of oocytecryopreservation. A road, that seems to beopen now, although, to date, very few humanembryologists have the courage to enter!
THE CRYOTOP TECHNIQUE
The technique and the tool for the Cryotoptechnique were briefly described in Chapter 2.Here we provide some technical detailsrequired for successful cryopreservation ofMII phase human oocytes. The commerciallyavailable kit (Kitazato, Tokyo, Japan) containsthe Cryotop device, a filmstrip attached to aplastic handle also equipped with a cap tocover the filmstrip for safe handling and stor-age (Figure 7A.1), and all media required forwashing, equilibration, vitrification, warming,and dilution. These solutions are based onTCM 199 medium supplemented with syn-thetic serum substitute (SSS), and containingethylene glycol, DMSO, and sucrose as per-meable and non-permeable cryoprotectants,respectively (see exact concentrations and
sources in Kuwayama et al.7). All media andmanipulations should be performed at25–27°C, except for thawing where mediumshould be warmed to 37°C. A pulled, firepolished glass pipette with 140–150 µminner diameter is suggested for all themanipulations.
Oocytes can be vitrified 2–6 hours afterthe ovum pick-up, immediately afterdenudation. A stepwise, very mild initialequilibration procedure can be carried outby making 20 µL droplets of washing andequilibration solutions (1 and 2 droplets,respectively) close to each other, and unify-ing droplets when oocytes seem to havecompletely recovered from the osmotic effect(a total of approximately 6 min). Finally,oocytes should be placed into an equilibra-tion drop and incubated until they arecompletely recovered (approximately in anadditional 9 min). Subsequently, one oocyteshould be placed into a large volume(4.5 mL) of vitrification solution, mixed well,and after 60 s loaded on the film strip ofthe Cryotop. All excess media should beremoved leaving only the oocyte coveredwith a thin layer of vitrification solution.Then the film part should be submerged intoliquid nitrogen with a quick and continuousvertical movement to ensure the maximumcooling rate (23 000°C/min). Finally, underthe liquid nitrogen, the cap should be fixedon the Cryotop with forceps to protect thefilm part from mechanical damage duringtransfer to the container and storage.
At warming, the film part of the Cryotopshould be submerged quickly into the 37°Cthawing solution to achieve the required42 000°C/min warming rate. After 10 s, theoocyte can be gently removed from the surfaceof the Cryotop with a pipette and kept sub-merged in the thawing solution. After 1 min,the dilution should be continued in dilutionsolution and washing solution 1 and 2 for 3, 5,and 5 min, respectively. Oocytes should becultured for an additional 2 hours before the
VITRIFICATION IN ASSISTED REPRODUCTION
124
a
d
b
c
Figure 7A.1 The Cryotop vitrification device. Thepolypropylene strip (a) is attached to a hard plastichandle (b). After vitrification, a hard plastic cover (c) isattached to protect the strip during storage in liquidnitrogen (d). Reproduced with permission fromKuwayama et al.7
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 124
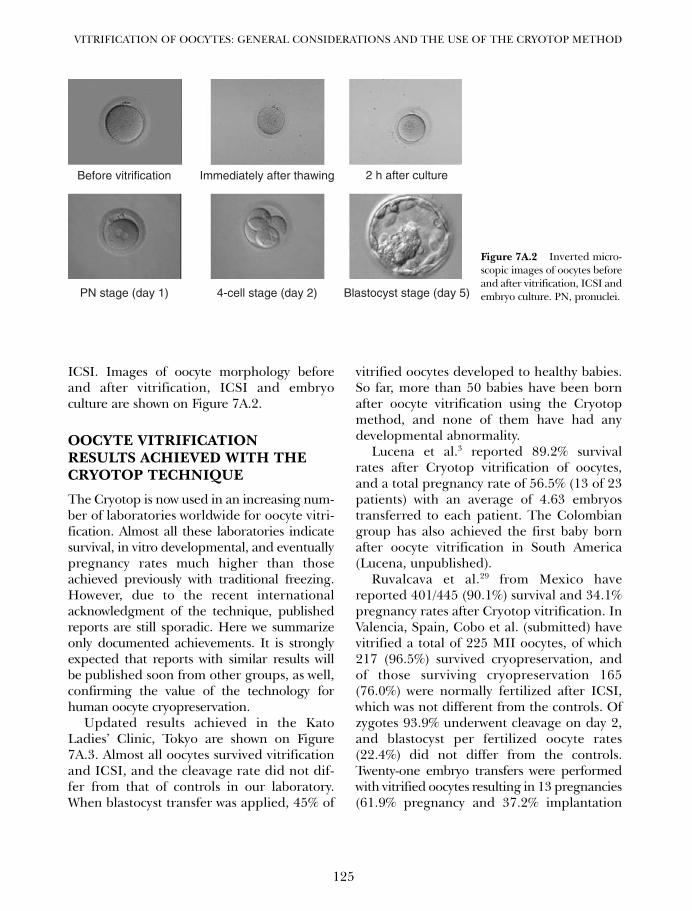
ICSI. Images of oocyte morphology beforeand after vitrification, ICSI and embryoculture are shown on Figure 7A.2.
OOCYTE VITRIFICATIONRESULTS ACHIEVED WITH THECRYOTOP TECHNIQUE
The Cryotop is now used in an increasing num-ber of laboratories worldwide for oocyte vitri-fication. Almost all these laboratories indicatesurvival, in vitro developmental, and eventuallypregnancy rates much higher than thoseachieved previously with traditional freezing.However, due to the recent internationalacknowledgment of the technique, publishedreports are still sporadic. Here we summarizeonly documented achievements. It is stronglyexpected that reports with similar results willbe published soon from other groups, as well,confirming the value of the technology forhuman oocyte cryopreservation.
Updated results achieved in the KatoLadies’ Clinic, Tokyo are shown on Figure7A.3. Almost all oocytes survived vitrificationand ICSI, and the cleavage rate did not dif-fer from that of controls in our laboratory.When blastocyst transfer was applied, 45% of
vitrified oocytes developed to healthy babies.So far, more than 50 babies have been bornafter oocyte vitrification using the Cryotopmethod, and none of them have had anydevelopmental abnormality.
Lucena et al.3 reported 89.2% survivalrates after Cryotop vitrification of oocytes,and a total pregnancy rate of 56.5% (13 of 23patients) with an average of 4.63 embryostransferred to each patient. The Colombiangroup has also achieved the first baby bornafter oocyte vitrification in South America(Lucena, unpublished).
Ruvalcava et al.29 from Mexico havereported 401/445 (90.1%) survival and 34.1%pregnancy rates after Cryotop vitrification. InValencia, Spain, Cobo et al. (submitted) havevitrified a total of 225 MII oocytes, of which217 (96.5%) survived cryopreservation, andof those surviving cryopreservation 165(76.0%) were normally fertilized after ICSI,which was not different from the controls. Ofzygotes 93.9% underwent cleavage on day 2,and blastocyst per fertilized oocyte rates(22.4%) did not differ from the controls.Twenty-one embryo transfers were performedwith vitrified oocytes resulting in 13 pregnancies(61.9% pregnancy and 37.2% implantation
125
VITRIFICATION OF OOCYTES: GENERAL CONSIDERATIONS AND THE USE OF THE CRYOTOP METHOD
Before vitrification Immediately after thawing 2 h after culture
PN stage (day 1) 4-cell stage (day 2) Blastocyst stage (day 5)
Figure 7A.2 Inverted micro-scopic images of oocytes beforeand after vitrification, ICSI andembryo culture. PN, pronuclei.
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 125
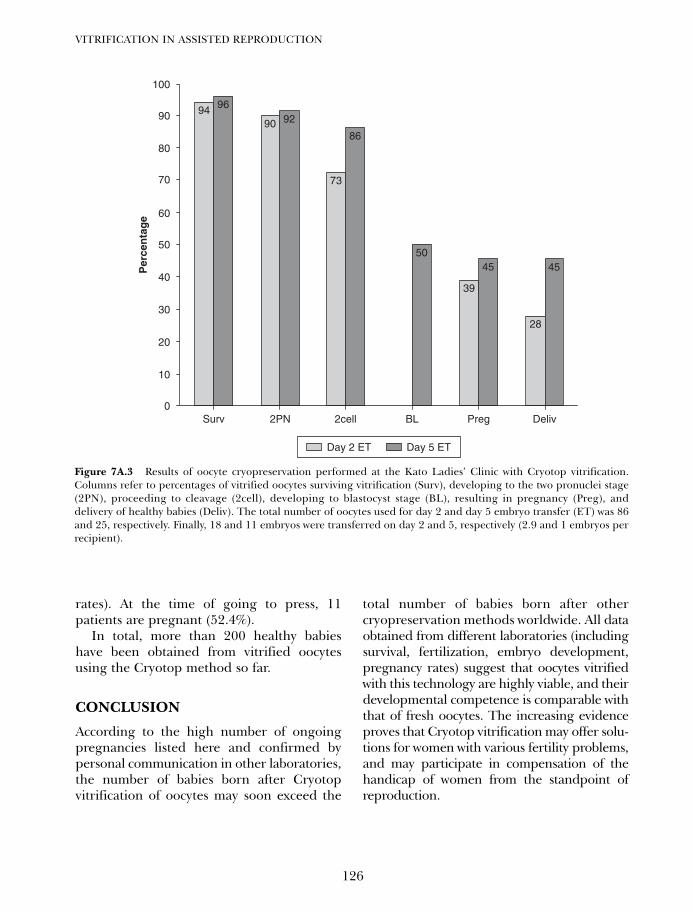
rates). At the time of going to press, 11patients are pregnant (52.4%).
In total, more than 200 healthy babieshave been obtained from vitrified oocytesusing the Cryotop method so far.
CONCLUSION
According to the high number of ongoingpregnancies listed here and confirmed bypersonal communication in other laboratories,the number of babies born after Cryotopvitrification of oocytes may soon exceed the
total number of babies born after othercryopreservation methods worldwide. All dataobtained from different laboratories (includingsurvival, fertilization, embryo development,pregnancy rates) suggest that oocytes vitrifiedwith this technology are highly viable, and theirdevelopmental competence is comparable withthat of fresh oocytes. The increasing evidenceproves that Cryotop vitrification may offer solu-tions for women with various fertility problems,and may participate in compensation of thehandicap of women from the standpoint ofreproduction.
VITRIFICATION IN ASSISTED REPRODUCTION
126
0
10
20
30
40
50
60
70
80
90
100
Surv 2PN 2cell
Per
cen
tag
e
BL Preg Deliv
94 96
90 92
73
86
50
39
45
28
45
Day 2 ET Day 5 ET
Figure 7A.3 Results of oocyte cryopreservation performed at the Kato Ladies’ Clinic with Cryotop vitrification.Columns refer to percentages of vitrified oocytes surviving vitrification (Surv), developing to the two pronuclei stage(2PN), proceeding to cleavage (2cell), developing to blastocyst stage (BL), resulting in pregnancy (Preg), anddelivery of healthy babies (Deliv). The total number of oocytes used for day 2 and day 5 embryo transfer (ET) was 86and 25, respectively. Finally, 18 and 11 embryos were transferred on day 2 and 5, respectively (2.9 and 1 embryos perrecipient).
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 126

1. Isachenko V, Isachenko E, Montag M, et al.2005c Clean technique for cryoprotectant-freevitrification of human spermatozoa. ReprodBiomed Online 10: 350–3544.
2. Meirow D 2000 Reproduction post-chemotherapyin young cancer patients. Mol Cell Endocrinol169, 123–131.
3. Lucena E, Bernal DP, Lucena C, Rojas A,Moran A, Lucena A 2006 Successful ongoingpregnancies after vitrification of oocytes.Fertility and Sterility 85: 108–11.
4. Cha KY, Han SY, Chung HM et al. Pregnanciesand deliveries after in vitro maturation culturefollowed by in vitro fertilization and embryotransfer without stimulation in women withpolycystic ovary syndrome. Fertil Steril 2000;73: 978–83.
5. Chung HM, Hong SW, Lim JM, et al. In vitroblastocyst formation of human oocytesobtained from unstimulated and stimulatedcycles after vitrification at various matura-tional stages. Fertil Steril 2000; 73: 545–51.
6. Stachecki JJ, Cohen J 2004 An overview ofoocyte cryopreservation. ReproductiveBiomedicine Online 9, 152–63.
7. Kuwayama M, Vajta G, Kato O et al. Highly effi-cient vitrification method for cryopreservationof human oocytes. Reproductive BioMedicineOnline 2005; 11: 300–308.
8. Paynter SJ. A rational approach to oocyte cryo-preservation. Reprod BioMed Online 2005;10: 578–86.
9. Vanderzwalmen P, Bertin G, Debauche Chet al. Births after vitrificaton at morula andblastocyst stages: effect of artificial reductionof the blastocoelic cavity before vitrification.Human Reproduction 2002; 17: 744–51.
10. Son WY, Yoon SH, Yoon, HJ. Pregnancy out-come following transfer of human blastocystsvitrified on electron microscopy grids afterinduced collapse of the blastocoele. HumanReproduction 2003; 18: 137–9.
11. Hiraoka K, Hiraoka K, Kinutani M et al.Blastocoele collapse by micropipetting prior tovitrification gives excellent survival and preg-nancy outcomes for human day 5 and 6expanded blastocysts. Human Reproduction2004; 19: 2884–8.
12. Ghetler Y, Yavin S, Shalgi R et al. The effect ofchilling on membrane lipid phase transition
in human oocytes and zygotes. HumanReproduction 2005; 20: 3385–9.
13. Magistrini M, Szollosi D. Effects of cold and ofisopropyl N-phenylcarbamate on the secondmeiotic spindle of mouse oocytes. Eur J. CellBiol 1980; 22: 699–707.
14. Sathananthan, 1998.15. Pickering S, Braude P, Hohnson M et al.
Transient cooling to room temperature cancause irreversible disruption of the meioticspindle in the human oocyte. Fertil Steril1990; 54:102–8.
16. Fabbri R, Porcu E, Marsella T et al. Humanoocyte cryopreservation: new perspectivesregarding oocyte survival. Hum Reprod 2001;16: 411–6.
17. Stachecki JJ, Munne S, Cohen J. Spindle orga-nization after cryopreservation of mouse,human, and bovine oocytes. ReproductiveBioMedicine Online 2004; 8: 664–72.
18. Leibo SP, Martino A, Kobayashi S, Pollard JW.Stage-dependent sensitivity of oocytes andembryos to low temperatures. Anim ReprodSci 1996 ; 42: 45–53.
19. Men H, Monson RL, Rutledge JJ. Effect ofmeiotic stage and maturation protocols onbovine oocyte’s resistance to cryopreservation.Theriogenology 2002; 57: 1095–103.
20. Kasai M, Zhu SE, Pedro PB et al. Fracturedamage of embryos and its prevention duringvitrification and warming. Cryobiology 1996;33: 459–64.
21. Esaki R, Ueda H, Kurome M et al. Cryopreser-vation of porcine embryos derived from invitro-matured oocytes. Biol Reprod 2004; 71:432–7.
22. Vicente JS, Garcia-Ximenez F. Osmotic andcryoprotective effects of a mixture of DMSOand ethylene glycol on rabbit morulae.Theriogenology 1994; 42: 1205–15.
23. Martino A, Songsasen N, Leibo SP. Develop-ment into blastocysts of bovine oocytes cryo-preserved by ultra-rapid cooling. Biol Reprod1996; 54: 1059–69.
24. Papis K, Shimizu M, Izaike Y. Factors affectingthe survivability of bovine oocytes vitrified indroplets. Theriogenology 2000; 54: 651–8.
25. Dinnyes A, Dai Y, Jiang S et al. High develop-mental rates of vitrified bovine oocytes follow-ing parthenogenetic activation, in vitro
127
VITRIFICATION OF OOCYTES: GENERAL CONSIDERATIONS AND THE USE OF THE CRYOTOP METHOD
References
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 127

fertilization, and somatic cell nuclear transfer.Biol Reprod 2000; 63: 513–8.
26. Kuwayama M. Vitrification of human oocytesand embryos (Japanese). In: IVF Update.Tokyo: Medical View, 2001: 230–4.
27. Selman H, Angelini A, Barnocchi N, BruscoGF, Pacchiarotti A, Aragona C. Ongoing preg-nancies after vitrification of human oocytes
using a combined solution of ethylene glycoland dimethyl sulfoxide. Fertility and Sterility2006; 86: 997–1000.
28. Liebermann et al. 2003.29. Ruvalcaba L, GarcíaM, MartínezR et al. Oocytes
vitrification success: first ongoing Pregnancy inMexico. Reprod Hum 2005; 3: 7–10.
VITRIFICATION IN ASSISTED REPRODUCTION
128
07a Tucker 8029.qxd 8/23/2007 2:14 PM Page 128

129
Vitrification of oocytes: various proceduresShee-Uan Chen and Yu-Shih Yang
7B
INTRODUCTION
Cryopreservation of mammalian and humanoocytes has been significantly improved bythe refined slow-freezing methods and newvitrification techniques.1–12 The slow-freezingmethod using a programmed cryo-machine istraditionally employed for the cryopreserva-tion of oocytes.13 This procedure usually takeseveral hours. Vitrification is an importantalternative method. With high concentrationsof cryoprotectants and a fast cooling rate, ittransforms cells into an amorphous glassystate, instead of ice crystal formation.14,15
Vitrification is time-saving and does notrequire special equipment. The vitrificationmethods may replace the slow-freezingmethod for cryopreservation of oocytes.16–19
The recent improvements of vitrificationinclude the concepts of reduction of concen-tration of cryoprotectants, increase of coolingand warming rates, recovery of meioticspindle, and timely fertilization.
REDUCTION OF CONCENTRATIONAND TOXICITY OF VITRIFICATIONSOLUTION
Rall and Fahy14 first successfully vitrifiedmouse 8-cell embryos using conventionalstraws with the medium consisting of 20.5%w/v dimethylsulfoxide (DMSO), 15.5% w/vacetamide, 10% w/v propylene glycol, and 6%w/v polyethylene glycol, which required a lowtemperature of 4°C during the treatment.Subsequent investigators made a significant
improvement by adjustment of cryoprotectantsto reduce toxicity, thus permitting the equili-bration steps to be performed at roomtemperature or 35–37°C.20,21 Ali and Shelton21
undertook a systematic and extensive investi-gation involving various combinations ofcryoprotectant solutions. They developed anethylene glycol (EG)-based solution consistingof 5.5 mol/L EG and 1.0 mol/L sucrose whichwas less toxic. This vitrification solution wasused to cryopreserve all preimplantationstages of in vivo generated mouse22 and day-6sheep embryos23 without significant loss ofviability in vitro or in vivo. Chen et al.24 usedthis formulation of vitrification solution forhuman oocytes and attained high survivalrates in conventional straws. Recently, theconcentration of vitrification solution hasbeen further decreased because the minimumvolume method with increased cooling andwarming rates achieves vitrification with lessconcentrated cryoprotectants (Table 7B.1).The vitrification solution consisting of 15%(v/v) EG, 15% (v/v) DMSO or 1,2-propanediol(PROH), and 0.5 mol/L sucrose can be vitri-fied with the minimum volume method.11,26,27
EG, with the characteristics of low toxicityand rapid permeation of the cell, is an impor-tant component of vitrification solutions.Some authors have mixed other permeatingagents, such as DMSO or PROH, to reducethe concentration of this single cryoprotectantin order to decrease the individual specifictoxicity.26–29 Non-permeable cryoprotectantscan facilitate dehydration and vitrification,and can be applied in combination to reduce
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 129

the required concentration of permeablecryoprotectants. This strategy further reducesthe toxicity of the vitrification solution.Sucrose has become almost a standard com-ponent of vitrification solutions. Other inves-tigators have added other macromolecules,such as Ficoll, in vitrification solutions thatare thought to stabilize the glass formationand form a protective coating aroundembryos or oocytes.20,25,30 Sera of various ori-gins, serum albumin preparations, recombi-nant albumin, or synthetic serum substitute(SSS) are common additives to vitrificationsolutions.
Another important strategy to reduce toxiceffects from vitrification solution is the stepwise
equilibration of cryoprotectants. A two-stepstrategy is most commonly used. The pretreat-ment (equilibration) solution contains 20–50%concentrations of permeating cryoprotectantsof the vitrification solution. The lower concen-tration of permeating cryoprotectants in theequilibration solution is much less toxic thanthe vitrification solution. Oocytes in the pre-treatment solution shrink initially and gradu-ally re-expand to their original volume. Thisobservation indicates the entry of the perme-ating cryoprotectants into the oocytes that mayfacilitate intracellular vitrification in the subse-quent procedures. It reduces the time neededfor exposure to the vitrification solution that ismore toxic for oocytes. This approach may
VITRIFICATION IN ASSISTED REPRODUCTION
130
Table 7B.1 Various regimens of cryoprotectants, exposure time, and temperature for vitrification and their devices
Equilibration Warming and solution Vitrification solutions dilution Devices Authors
25% VS1, 15 min, VS1: 20.5% w/v DMSO, 50% VS1, 4οC Straw Rall and Fahy, 198514
20οC; 50% VS1, 15.5% w/v acetamide, (mouse embryos)10 min, 4οC 10% w/v propylene glycol,
6% w/v polyethyleneglycol, 4οC
7.5% EG, 7.5% 16.5% EG, 16.5% DMSO, 0.25, 0.15 mol/L OPS Vajta et al., 19987
DMSO, 3 min, 0.5 mol/L sucrose, 25 s, sucrose, 37οC (bovine oocytes 34–36 oC 34–36οC and embryos)
1.5 mol/L EG, 5.5 mol/L EG, 1.0 mol/L 0.5, 0.25, Straw Chen et al., 200024
5 min, RT sucrose, 60 s, RT 0.125 mol/L (human oocytes)sucrose, RT
1.5 mol/L EG, 5.5 mol/L EG, 1.0 mol/L 1.0, 0.5, 0.25, Grid Yoon et al., 20039
2.5 min, 37οC sucrose, 20 s, 37οC 0.125 mol/L (human oocytes)sucrose, 37οC
7.5% EG, 7.5% 15% EG, 15% DMSO, 0.33, 0.2 mol/L Cryoloop Mukaida et al., 200325
DMSO, 2 min, 37oC 10 mg/mL Ficoll, sucrose, 37οC (human blastocysts)0.65 mol/L sucrose, 25–30 s, 37οC
1.6 mol/L EG, 5.0 mol/L EG, 1.0 mol/L 1.0, 0.5 mol/L Cryotop Kuwayama 5–15 min, 22οC sucrose, 30 s, 22οC sucrose, 37οC et al., 200510
(human oocytes)
7.5% EG, 7.5% 15% EG, 15% PROH, 1.0, 0.5, Cryoleaf Chian et al., 200511
PROH, 5 min, RT 0. 5 mol/L sucrose, 0.25 mol/L (human oocytes)45–60 s, RT sucrose, 37οC
VS, vitrification solution; RT, room temperature.
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 130

provide better protection for oocytes that havea low surface/volume ratio. In the vitrificationof human oocytes, it has been demonstratedthat oocytes pretreated with equilibration solu-tion had a significantly higher survival ratethan those without pretreatment.24 One-stepvitrification without pretreatment has the pos-sibility of insufficient permeation of the cryo-protectants that may result in intracellular iceformation during cooling or warming.
EQUILIBRATION AND VITRIFICATION
For oocyte vitrification, some investigators per-form exposure to cryoprotectants at room tem-perature,10,11,24 but other investigators operatethe procedures at 35–37°C7,9 (Table 7B.1). Thehigher temperature enhances the passage ofthe permeating cryoprotectants across the cellmembrane, but the toxicity is also increased.Therefore, at 37°C, 2–3 min are usually used forthe pretreatment solution, and 20–30 s forexposure to the vitrification solution.7,9 In con-trast, at room temperature, 5–15 min are com-monly used for the pretreatment solution, and30–60 s for exposure to the vitrification solu-tion.10,11,24 Prolonged exposure to the concen-trated cryoprotectants may induce toxic effects.The human oocytes treated in vitrification solu-tion for 120 s had a poorer fertilization outcomethan those vitrified in 60 s.24
Yoon et al.9 used electron microscope gridsto perform vitrification of human oocytes. At37°C, oocytes were pre-equilibrated for 2.5 minin Dulbecco’s phosphate buffered saline sup-plemented with 1.5 mol/L EG and 10% (v/v)fetal bovine serum. Oocytes were thenplaced for the final equilibration in5.5 mol/L EG and 1.0 mol/L sucrose vitrifica-tion solution for 20 s. They achieved a sur-vival rate of 69% (325/474) and a pregnancyrate of 21% (6/28).
Kuwayama et al.10 used Cryotops to carry outhuman oocyte vitrification. At 22°C, oocyteswere pretreated with 1.6 mol/L EG in tissueculture medium (TCM) for 5–15 min until the
oocytes had completely recovered their originalvolume. Oocytes were then treated with a vitri-fication solution of 6.8 mol/L EG and 1.0 mol/Lsucrose in TCM medium for 30 s. They achieveda survival rate of 90% (58/64) and a pregnancyrate of 41% (12/29).
Chian et al.27 used Cryotops to vitrify bovineoocytes. At room temperature, oocytes werepretreated in equilibration solution of 7.5%(v/v) EG and 7.5% (v/v) PROH for 5 min.Then the oocytes were transferred to a vitrifi-cation solution of 15% (v/v) EG, 15% (v/v)PROH, and 0.5 mol/L sucrose for 45–60 s.They found that 15% EG, 15% PROH, and0.5 mol/L sucrose had a higher rate of blasto-cyst development than 15% EG, 15% DMSOand 0.5 mol/L sucrose. This may be due tolower toxicity with PROH than DMSO. Theformula has been used for human oocytes bythe same group, and they accomplished goodresults with a survival rate of 94% (169/180)and a pregnancy rate of 47% (7/15).11
For vitrification, therefore, using minimumvolume methods instead of conventionalstraws achieves higher cooling and warmingrates, and leads to the reduction in concen-tration of cryoprotectants vitrified. The newlyformulated cryoprotectants allow the perfor-mance of equilibration at room temperatureor 37°C. The duration of exposure for equili-bration or vitrification solutions shoulddepend on operating temperature and con-centration of cryoprotectants.
WARMING AND DILUTION
The vitrified oocyte is sensitive to osmoticchanges after warming. Stepwise dilution inan osmotic buffer is commonly used toprevent excessive swelling and lysis of theoocyte as the permeating cryoprotectants areremoved. Two- to four-step dilutions withsucrose solutions have usually been used(Table 7B.1). Chen et al.24 found that therewere no differences in survival and fertiliza-tion for vitrified human oocytes diluted bythree or four steps. Therefore, the four-step
131
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 131

dilutions may not be necessary for humanoocytes. Although, one-step dilution withoutsignificant compromise of survival wasreported in vitrification of bovine blasto-cysts31 and porcine blastocysts.32
The speed of warming is important toprevent devitrification. For the conventionalstraw, warming is performed by direct immer-sion of the straw into a water bath. Holdingthe straw in air for 5 seconds before immer-sion can avoid bursting. After cutting thestraw, the oocytes are expelled into the dilu-tion solution. Minimum volume methodssuch as electron microscope grid, openpulled straws (OPS), Cryoloop, or Cryotopcan be directly submerged into the dilutionmedium. The oocytes come into contact withthe dilution medium immediately. Mostinvestigators performed warming and dilu-tion at 37°C7,9–11 (Table 7B.1).
For thawing of oocytes vitrified by theCryoleaf method, Chian et al.11 used three-stepdilutions and performed thawing with 1.0 mol/Lsucrose solution for 1 min at 37°C. The thawedoocytes were transferred to 0.5 mol/L and0.25 mol/L sucrose solutions for 3 min respec-tively, and then washed twice with culturemedium. Kuwayama et al.10 used two-step dilu-tions and performed thawing using 1.0 mol/Lsucrose for 1 min at 37°C. Oocytes were trans-ferred into 0.5 mol/L sucrose for 3 min, andthen washed twice with culture medium. It hasbeen shown that the meiotic spindle of humanoocytes depolymerized when cooled to roomtemperature for 10–30 min, and that the dam-age was time dependent.33 Therefore, dilutionfor vitrified oocytes with sucrose solutions at37°C may reduce spindle damage during theprocedures.
INCREASED COOLING AND WARMING RATES BY MINIMUMVOLUME METHODS
A 0.25 mL conventional straw was initially,commonly used for vitrification of mammalianoocytes or embryos.14,20,24 The cooling ratewas around 2500°C/min and the warming
rate was 1300°C/min.14 The limited speed ofthermal change of the conventional strawneeds more concentrated cryoprotectants toachieve vitrification during cooling and toprevent devitrification during warming. Thehigher concentrated cryoprotectants aremore toxic to oocytes or embryos. A highercooling rate can facilitate vitrification and ahigher warming rate will prevent devitrifica-tion with less concentrated cryoprotectants.This can reduce toxicity of vitrification solu-tions. Minimum volume methods can preventless concentrated cryoprotectants from icecrystal formation that had been observedmicroscopically.34 In addition, the high speedof cooling and warming of minimum volumemethods can rapidly pass through the dam-aging temperature zone liable to cause chill-ing injury, between 15 and − 15°C.6 Chillinginjury harms mainly the cytoplasmic lipiddroplets and meiotic spindle of oocytes.Minimum volume vitrification may also avoidfracture injury. Therefore, scientists havemade great efforts to find new methods usingminimum volume techniques to increasethermal change.
EVOLUTION OF MINIMUM VOLUME METHODS
Landa and Tepla35 performed vitrification bydropping mouse embryos directly into theliquid nitrogen. However, to form a droprequires a relatively large amount of solution(approximately 5 µL). When the drop reachesthe liquid nitrogen, it will not sink immedi-ately but will float on the surface for severalseconds. The drop induces a strong evapora-tion at its surface that decreases the coolingrate. Furthermore, it is difficult to find theembryos and to perform thawing. Speculativeadvantages in maximizing cooling and warm-ing rates cannot be attained by this method.Further refinements using various carriers tominimize the volume of vitrification solutionand to submerge the sample quickly into theliquid nitrogen have been comprehensivelystudied. The innovative devices for supporting
VITRIFICATION IN ASSISTED REPRODUCTION
132
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 132

oocytes or embryos for minimum volumevitrification include the electron microscopegrid, minimum drop size on a solid surface,open pulled straw (OPS), Cryoloop, hemi-straw system, or Cryotop, etc. (Table 7B.2).
Steponkus et al.15 first utilized electronmicroscope copper grids (Figure 7B.1) as acarrier for the minimum volume-direct con-tact approach. They successfully cryopre-served chill-sensitive Drosophila embryos byvitrification. The method overcame the prob-lem of chilling injury at subzero temperaturesassociated with conventional slow-freezingprocedures for Drosophila embryos. Martinoet al.6 applied this technique for bovineoocytes, and achieved higher growth poten-tial than using conventional straws. Honget al.8 and other authors9 achieved successfulpregnancies from vitrified human oocytesusing the grid method.
Arav36 developed the minimum drop size(MDS) method with a small droplet of vitrifi-cation solution containing the oocyte orembryo placed on a solid surface that wasthen immersed into liquid nitrogen. Theapproach was used later with some modifica-tions called the minimum volume cooling(MVC).37 Hamawaki et al.37 placed the mini-mum vitrification volume containing bovineblastocysts on the outside wall of a conven-tional straw and directly submerged it intoliquid nitrogen. They found that the survivalrate was higher for MVC than conventionalvitrification in straws. Vanderzwalmen et al.41
developed the hemi-straw system (HSS) usinga cut open straw as the carrier. Dinnyes et al.40
designed a pre-cooled metal surface (solid-surface vitrification; SSV) to vitrify bovineoocytes. A steel cube is partially submergedinto liquid nitrogen. Microdrops of vitrifica-tion solution containing oocytes are droppedonto the cold surface of aluminum foil on themetal cube for vitrification. Kuwayama andKato42 developed the Cryotop method froma modification of MVC procedure. Oocyteswere placed on the top of a fine polypropy-lene strip attached to a hard plastic handle.Chian et al.11 modified the Cryotop to create
the Cryoleaf with a different method ofapplying the protective sheath. Both Cryotopand Cryoleaf achieved high success rates invitrification of human oocytes.10,11
Vajta et al.7 developed the open pulledstraw (OPS) (Figure 7B.2) to hold bovineoocytes with a very small amount of vitrifica-tion solution. The idea was to reduce thevolume of the sample by diminishing thediameter of the conventional straw. Strawswere heated and pulled by hand, then cut atthe tapering end with a razor blade. As aresult, the diameter and the wall thickness ofthe straw decreased to approximately half ofthe original. The volume for keeping oocytesis reduced from 25 µL to 1 µL. They foundthat the OPS achieved better results than con-ventional straw vitrification. Kuleshova et al.48
applied the OPS for vitrification of humanoocytes and achieved a successful pregnancy.With a similar design, Kong et al.39 used glassmicropipettes (GMP) to perform vitrification
133
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
Table 7B.2 Various vitrification methods for oocytes orembryos
Vitrification method Authors
Straw Rall and Fahy, 198514
Direct dropping into Landa and Tepla, 199035
liquid nitrogenElectron microscopic grids Steponkus et al., 199015
Minimum drop size (MDS) Arav, 199236
Open pulled straw (OPS) Vajta et al., 19987
Minimum volume Hamawaki et al., 199937
cooling (MVC)Cryoloop Lane et al., 199938
Glass micropipettes (GMP) Kong et al., 200039
Solid surface Dinnyes et al., 200040
vitrification (SSV)Hemi-straw system (HSS) Vanderzwalmen
et al., 200041
Cryotop Kuwayama and Kate, 200042
Nylon mesh Matsumoto et al., 200143
Closed pulled straw (CPS) Chen et al., 200144
Flexipet denuding Liebermann pipette (FDP) et al., 200245
Cryotip Kuwayama et al., 200546
Cryoleaf Chian et al., 200511
Direct cover vitrification Chen et al., 200647
(DCV) (for ovarian tissues)
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 133

of mouse blastocysts. Liebermann et al.45
applied the Flexipet denuding pipette(FDP) for human pronuclear zygotes. Chenet al.44 modified the loading of the OPSturning it into a closed system, called closedpulled straw (CPS). CPS has the beneficialcharacteristics of the OPS such as a rapidthermal change method, and of conven-tional straws in that it is a non-contact mode.The vitrification medium containing theoocytes was isolated by two small segmentsof air and medium. Through this closedloading system of CPS, the oocytes do notdirectly contact the liquid nitrogen, whichmay occur with the OPS. Kuwayama et al.46
developed the Cryotip method. Principally,it is a heat-sealed pulled straw technique.The speed of cooling and warming is slowerwith the Cryotip than with the Cryotop.However, the thermal speed accompaniedby the Cryotip is still high enough to obtainadequate vitrification. Kuwayama et al.46
reported no difference for supporting blas-tocyst survival and pregnancies betweenthe Cryotip and the Cryotop. The use of theclosed Cryotip system may eliminate the
potential of contamination and maintain theefficacy.
Lane et al.38 used Cryoloops for vitrifyingmouse and human blastocysts. The Cryoloopconsists of a small nylon loop attached to aholder and is equipped with a vial as a con-tainer (Figure 7B.3). Oocytes or embryos aresuspended on a film of vitrification solutionbridging the hole of the loop that is thenplunged into liquid nitrogen. The Cryoloopwas originally designed for mounting of crystalsfor macromolecular cryo-crystallography. 49,50
Cryoloops have been confirmed to be highlysuccessful for human blastocyst vitrification byMukaida et al.25
With these minimum volume methods, theachievable cooling rate and warming ratewere significantly increased (Table 7B.3). Thecooling and warming rates were 23 000°C/min and 42 100°C/min, respectively, forthe Cryotop (Figure 7B.4).10 The coolingand warming rates are higher than thoseachievable with the OPS (16 700°C/min and13 900°C/min, respectively).7 Even for the non-contact method of the Cryotip, the coolingand warming rates are around 12 000 °C/min
VITRIFICATION IN ASSISTED REPRODUCTION
134
Figure 7B.1 The image shows an electron microscopecopper grid. Oocytes are transferred onto the grid. Toreduce the volume of vitrification solution, the undersideof the grid is blotted on a filter membrane. The grid sup-porting oocytes is plunged into liquid nitrogen using afine forceps. Scale bar = 0.3 cm.
Figure 7B.2 The image shows an open pulled straw(OPS). To produce a pulled straw, the 0.25 mL plasticstraw is heat-softened over a hot plate and pulled manu-ally. The pulled straw is cut at the tapered end. Oocytesare loaded into the tip (arrow) of the OPS by simplytouching a microdrop (1–2 µL) of vitrification solutioncontaining oocytes. Scale bar = 1 cm.
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 134

and 24 000°C/min, respectively.46 Rapidthermal changes are important to achievevitrification during cooling and to preventdevitrification during warming. Therefore,we can use less concentrated cryoprotectantto avoid toxicity from chemicals.
With the principle of minimum volume ofvitrification solution and direct contact withliquid nitrogen, Chen et al.47 developed aninnovative method to vitrify ovarian tissues bydirect cover vitrification (DCV). They used theless concentrated cryoprotectants (15% EG,15% DMSO, and 0.5 mol/L sucrose). After two-step equilibration with cryoprotectants, ovar-ian tissues were placed on a piece of gauze toremove the surrounding vitrification solution.The ovarian tissues were put in a cryovial.Liquid nitrogen was directly poured onto theovarian tissue for vitrification. The cooling ratewas maximized because there was no thermo-insulating layer between the tissues and the liq-uid nitrogen. The cap of the cryovial was thenclosed. The vial was placed into a liquid nitro-gen tank for storage. For thawing, the ovariantissues were moved into dilution solutions.
In the mice models of ovarian tissue cryo-preservation, the percentages of follicle viabil-ity and pregnancy potential from DCV werefound to be significantly greater than forconventional vitrification or slow freezing.
COMMON VITRIFICATION METHODS
Following pretreatment with the equilibrationsolution and treatment with the vitrificationsolution, the steps of loading oocytes for coolingare described for common vitrification meth-ods. The steps of warming with release ofoocytes are also included in the following.
Conventional straw
For the conventional straw method24 oocytesare loaded in a 0.25 mL plastic straw. Thestraw is filled with 1 cm of vitrificationmedium, 0.5 cm of air, 2 cm of vitrificationmedium containing the oocytes, 0.5 cm of air,and 3.5 cm of vitrification medium. The strawis then plunged into liquid nitrogen. Forthawing, the straw is taken out and held inthe air for 5 s, then it is plunged into waterfor 10 s. The vitrification solution in the strawshould remain transparent in the liquid
135
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
Figure 7B.3 The image shows a Cryoloop. TheCryoloop consists of a nylon loop (small arrow) mountedon a stainless steel rod that is inserted into the lid of acryovial. The Cryoloop is dipped into vitrification solu-tion to make a filmy layer of solution on the nylon loop.The oocytes are transferred onto the filmy layer. It isthen stored in a cryovial (large arrow) filled with liquidnitrogen. Scale bar = 1 cm.
Table 7B.3 Various procedures for vitrification ofoocytes and volumes of vitrification solution withestimated cooling and warming rates in the literature
Cooling Warming Vitrification Volume rate ratemethod (µL) (° C/min) (° C/min)
Straw (Rall and 25 2 500 1 300Fahy, 1987)14
OPS (Vajta 1 16 700 13 900et al., 1998)7
Cryotop (Kuwayama 0.1 23 000 42 100et al., 2005)10
Cryotip (Kuwayama 1 12 000 24 000et al., 2005)46
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 135

nitrogen, air, and water. Otherwise, a whitishdiscoloration of vitrification medium in thestraw indicates ice crystal formation or devit-rification. The straw is cut by scissors, and thecontents containing the oocytes are expelledinto the dilution solution.
Electron microscope copper grids
For the electron microscope grid procedure6
oocytes are transferred onto the electronmicroscope copper grid (Figure 7B.1). Toreduce the volume of vitrification solution,the underside of the grid is blotted on a filtermembrane. A small volume is critical to pre-vent fracture injury. The grid supporting theoocytes is plunged into liquid nitrogen usingfine forceps, and stored in a cryovial filledwith liquid nitrogen. The solidified vitrifica-tion solution fixes the oocytes to the gridduring cooling and storage. The vials areattached onto standard canes and stored inliquid nitrogen. At warming, the grid ispicked up and put directly into the dilutionsolution.
Open pulled straw
For making an OPS7 (Figure 7B.2), a 0.25 mLplastic straw is heat-softened over a hot plateand pulled manually. The pulled straw is cutat the tapered end. The inner diameter ofthe tip is approximately 0.8 mm with a wallthickness of 0.07 mm. Commercial sterilepulled straws have recently become available.Oocytes are loaded into the tip of the pulledstraw through the capillary effect by simplytouching a microdrop (1–2 µL) of vitrificationsolution containing the oocytes. Plunging theOPS into liquid nitrogen performs the cool-ing. For thawing, the tip of OPS is put intothe dilution solution, and the oocytes areexpelled spontaneously by the pressure of thewarming gas in the straw.
Cryoloop
The Cryoloop38 consists of a nylon loop(0.5–0.7 mm diameter) mounted on a stain-less steel rod that is inserted into the lidof a Cryovial (Hampton Research, LagunaNiguel, CA) (Figure 7B.3). While the oocytesare suspended in pretreatment solution, theCryoloop is dipped into vitrification solutionto make a filmy layer of solution on the nylonloop by surface tension. The oocytes are thentreated in vitrification solution and trans-ferred onto the filmy layer on the nylon loop.The volume of vitrification solution in theCryoloop method is especially confined. TheCryoloop is plunged into liquid nitrogen. Itis then sealed in a cryovial, which has beenpreviously submerged in liquid nitrogen. Whenwarming the oocytes, the vial is opened, andthe Cryoloop is placed directly into the dilutionsolution. Oocytes are immediately releasedfrom the loop into the solution.
Solid surface vitrification
For SSV40 groups of five to ten oocytes are drop-ped on the surface of aluminum foil on a steelcube that is cooled to around −150 to −180oC
VITRIFICATION IN ASSISTED REPRODUCTION
136
Figure 7B.4 The image shows a Cryotop. The Cryotopconsists of a fine polypropylene strip (small arrow),attached to a plastic holder. Oocytes are loaded on thestrip with minimal vitrification solution. The Cryotop isthen immersed into liquid nitrogen. The strip is coveredwith the plastic tube (large arrows) in liquid nitrogen toprotect it during storage. Scale bar = 1 cm.
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 136

by partial immersion in liquid nitrogen. Thedrop size is 1–2 µL. The oocytes are instanta-neously vitrified on the pre-cooled metalsurface, and do not contact liquid nitrogendirectly. This method also avoids nitrogenvapor. The vitrified droplets are moved with anitrogen-cooled forceps into 1 ml cryovials.For thawing, they are dropped into a 39oCdilution solution. A commercially availabledevice has also been manufactured (CMV;Cryologic, Australia).
Hemi-straw system
For the HSS method41,51 an estimated volumeof 0.3 µl of vitrification solution containing theoocytes is placed on the tip of the trough of thehemi-straw (CBS; Cryo Bio System, France).The hemi-straw is put into liquid nitrogen. It isthen inserted in a larger pre-cooled straw withthe aid of forceps. For thawing, the hemi-strawis pulled out of the larger straw under liquidnitrogen using forceps. The tip of the hemi-straw holding the samples is immediatelyplaced into the dilution solution.
Closed pulled straw
In the CPS method44 the oocytes are loaded intothe tip of a pulled straw with 2 mm of vitrifica-tion medium, 2 mm of air, 2 mm of vitrificationmedium containing the oocytes, 2 mm of air,and 2 mm of vitrification medium using asyringe. They are then plunged into liquidnitrogen for cooling and storage. For thawing,the CPS is removed from the liquid nitrogen.The tip is submerged into the dilution solution.The opposite end of the pulled straw is sealedusing the index finger. The contents are thenexpelled by using the increase in air pressure inthe tube caused by the thermal change.
Cryotop
The Cryotop10 has a fine polypropylene strip(0.4 mm wide × 20 mm long × 0.1 mm thick)(Kitazato Bio Pharma Co, Ltd, Shizuoka-ken,
Japan), attached to a plastic holder andequipped with a protective plastic tube (Figure7B.4). Oocytes are loaded on the strip withminimal solution, and the remaining solutionis almost completely removed by aspiration.The Cryotop is immersed into liquid nitrogen.Then, the strip is covered with the plastic tubein liquid nitrogen to protect it during storage.For warming, the protective cover is removedfrom the Cryotop while it is still submerged inliquid nitrogen. The strip is immersed directlyinto the dilution solution.
Cryotip
The Cryotip46 consists of a plastic straw with anarrow part (250 µm inner diameter, 20 µmwall thickness, and 3 cm length) connectedto a wide part. It is equipped with a movableprotective metal sleeve. Oocytes are loaded inapproximately 1 µL vitrification solution intothe narrow part of the Cryotip by aspirationwith a connected syringe. The straw is heat-sealed at both ends. The Cryotip is plungedinto liquid nitrogen. For warming, the Cryotipis taken out from liquid nitrogen and placedinto a 37°C water bath for 3 s. It is then decon-taminated with ethanol, and the sealed endsare cut with a pair of sterile scissors. The con-tents are expelled into the dilution solution.
Devices for vitrification of large numbers of oocytes
The number of oocytes per supporting devicesuch as electron microscope grid, OPS,Cryoloop, and Cryotop is limited. The electronmicroscope grid may hold 10–15 oocytes, whilean OPS can hold four to six, and a Cryoloopone to three. Using a Cryotop, Chian et al.reported that a maximum of 10–15 denudedoocytes can be loaded, however, a smaller num-ber of cumulus–oocyte complexes (COC) canbe loaded. For vitrification of large numbersof immature COCs in domestic animals,Matsumoto et al.43 developed a nylon meshtechnique. The nylon mesh design was a
137
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 137

modification of the electron microscope grid.The larger area of the supporting deviceallowed a large amount of oocytes to be held forvitrification at the same time; it could hold 40COCs. The thawed COCs were able to continuematuration in vitro and development.52 For thesame purpose, Chian et al.27 used a thin plas-tic sticker (20 ×5 ×0.2 mm), a modification ofthe Cryotop, to vitrify bovine oocytes. It allowed30–50 COCs to be held, and achieved a survivalrate of 90%.
INCREASED COOLING RATEUSING LIQUID NITROGEN SLUSH
Three main factors influence the possibilityof ice crystal formation during vitrificationincluding the volume of the sample, the vis-cosity of the solution, and the cooling rate. Forenhancing the cooling rate, one approach is toemploy liquid nitrogen slush instead of liquidnitrogen for cooling. Nitrogen slush can beproduced from liquid nitrogen by using a vac-uum so that part of the liquid nitrogen evapo-rates and the remainder cools down. Themixture of nitrogen slush and cooled liquidnitrogen reaches about −205°C. The sampleimmersed into liquid nitrogen slush has lessevaporation, and the cooling rate is relativelyfaster. The approach had been used very earlyon by Steponkus et al.15 It is commerciallyavailable in the form of the VitMaster (IMT,Israel), a device producing a vacuum in thetoughened liquid nitrogen container.53
However, the theoretical advantages forenhancing vitrification using the VitMasterhave not gained it popularity. Most investiga-tors achieve satisfactory results with vitrifica-tion of human oocytes or embryos usingliquid nitrogen only.10–12,35 Huang et al.54 usedCryoloops and the VitMaster to performvitrification of human blastocysts. Theyachieved a survival rate of 77% (74/96), apregnancy rate of 54% (7/13), with animplantation rate of 23% (14/60). Theirresults did not prove the advantage of theVitMaster, compared with the results from
Mukaida et al.25 or Hiraoka et al.26 who didnot use the VitMaster.
Cuello et al.55 used the OPS method forvitrifying porcine embryos with the VitMaster,or liquid nitrogen only. They found that, withthe same vitrification solution, using theVitMaster did not enhance the efficiency ofin vitro development of vitrified porcineembryos. In contrast, Cai et al.56 vitrifiedrabbit oocytes with Cryoloops and studiedmeiotic spindles with immunofluorescentstain and confocal microscopy. They foundthat the faster cooling rate with the VitMasterhad fewer adverse effects on the spindle con-figuration and embryo development thanusing liquid nitrogen alone. The superiority ofthe VitMaster compared with liquid nitrogenalone still needs further investigation.
PREVENTING POTENTIALCONTAMINATION FROM LIQUIDNITROGEN
Recent vitrification methods with a minimumvolume-direct contact technique significantlyimproved success rates. However, a majorconcern should be mentioned regarding thepotential risk to human oocytes or embryosfrom contaminated liquid nitrogen at the timeof vitrification or storage. Yet, no reports havedocumented cases of liquid nitrogen-mediateddisease transmission through embryo transferof frozen-thawed cycles. Nonetheless, basedon the experiments of Bielanski et al.57 cross-contamination may take place during storageamong open systems.
In order to avoid contamination fromvitrification of oocytes or embryos, effortsshould be made to reduce the risk with theprocedures of cooling and storage. Coolingcan be performed in aseptically treated liquidnitrogen. The liquid nitrogen is filteredthrough a 0.2 µm pore-size filter. UV illumi-nation can also be applied. Another strategyis to place the open carrier into a containerthat isolates it from the liquid nitrogen dur-ing storage. For aseptic storage of OPS, after
VITRIFICATION IN ASSISTED REPRODUCTION
138
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 138

cooling the pulled straws can be wrapped inpre-cooled 0.5 mL straws.7 For storage ofgrids, they can be placed into vials.9
Some minimum-volume vitrification tech-niques were developed to avoid direct contactwith the liquid nitrogen.40,44,46 These modi-fied techniques may have reduced thermalchange compared with direct contact meth-ods. The method of SSV using metal surfacesfor cooling does not expose samples to liquidnitrogen.40 Chen et al.44 modified the loadingof OPS into a closed system, called closedpulled straws (CPS). Kuwayama et al.46 devel-oped the Cryotip method using a heat-sealedpulled straw technique. These procedures havebeen described above.
DYNAMICS OF MEIOTIC SPINDLESOF FROZEN–THAWED OOCYTES
The meiotic spindles of oocytes consist ofmicrotubules that are constructed by poly-merization of tubulin dimers of α- and β-tubulin. Microtubules start from microtubularorganizing centers at both poles and anchorchromosomes at the kinetochores, forming abarrel shape. The chromosomes align at theequatorial plane of the meiotic spindles. Thetubulin dimers polymerize and depolymerizeat various stages of a cell cycle. The meioticspindles are crucial for the events followingfertilization at completion of meiosis, secondpolar body formation, migration of thepronuclei, and formation of the first mitoticspindle.58
The spindle is very sensitive to cryoprotec-tants and low temperature. Oocytes analyzedimmediately after thawing displayed severe dis-organization or disappearance of spindles usingeither slow-freezing or vitrification methods.59–61
Incubation for 1–5 h at 37oC resulted in recov-ery of spindles to varying degrees dependent onthe time interval after thawing, methods offreezing and thawing, and species.59–62 Eroglu etal.59 found complete recovery of spindles forslow-freezing mouse oocytes after incubation for1 h post-thawing.
With different distribution of pericentriolarmaterials and cytoplasmic asters, mouseoocytes are very distinct from humanoocytes.41,62 The former are stronger than thelatter with recuperation of temperature-induced microtubule disorganization. Gooket al.62 noticed that 60% of human oocyteswith slow cryopreservation were comprised ofnormal spindles after 1 h of incubation, com-pared with 81% of control specimens. Rienziet al.61 used a computer-assisted polarizationmicroscopy (Polscope) to observe meiotic spin-dle changes. Immediately after thawing, thespindle was visible in 36% of oocytes, but it dis-appeared in all of the thawed oocytes duringthe subsequent washing steps. The spindlereappeared in all surviving thawed oocytes, by3 hours of incubation at 37°C.
Bianchi et al.63 slowly froze human oocyteswith a 1.5 mol/L PROH and 0.3 mol/L sucrosesolution and evaluated meiotic spindlewith immunofluorescent stain and confocalmicroscopy. Immediately after thawing, only22.9% of oocytes showed a weak birefringencesignal, while only 1.2% of oocytes exhibited ahigh signal. Three hours after thawing, theproportion of oocytes displaying a weak orhigh intensity signal was 49.4% and 18.1%,respectively. After culture for 5 hours, a weakbirefringence signal was detected in 51.8% ofoocytes, while 24.1% showed a high signal.There was a significant increase in signalrecuperation after 3–5 h of culture.
Coticchio et al.64 performed human oocytecryopreservation using a slow-freezing methodwith 1.5 mol/L PROH and 0.1 or 0.3 mol/Lsucrose. After thawing, oocytes were culturedfor 3 h before examination. Spindle and chro-matin organization were significantly affectedafter cryopreservation using 0.1 mol/Lsucrose concentration, while these parame-ters were unchanged using 0.3 mol/L.Protocols with the higher sucrose concentra-tion (0.3 mol/L) in the freezing solution pre-served an intact chromosome segregationapparatus comparable with that of freshlycollected oocytes.
139
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 139

In vitrified mouse oocytes, Chen et al.44,60
observed that post-thawing incubation for1 h allowed recovery of normal spindle andchromosomes to varying degrees. The OPS,CPS, and electron microscope grids pre-served the spindle morphology and chromo-somal pattern better than in conventionalstraws. The rapid thermal change of mini-mum volume methods could traverse thetemperature range damaging to the spindle(assumed to be 15 to –15°C) more quickly.6
Moreover, oocytes in minimum volumemethods (< 1 µL) are directly warmed in thedilution solution and immediately diluted(~1 s), reducing exposure of oocytes to inap-propriate temperatures and concentratedcryoprotectants. In contrast, the conven-tional straw is warmed in water and thencut with scissors. The oocytes in vitrificationmedium (25 µL) are expelled into the dilu-tion solution and then placed into anotherdilution solution. Therefore, it takes moretime to pass through the unfavorable condi-tions (~45 s). Chen et al.44 further demon-strated that incubation for 2 or 3 h resultedin higher incidences of normal spindles thanincubation for 1 h.
Minimum volume methods for vitrificationof oocytes such as electron microscope grids,OPS, Cryotop, and Cryoleaf have been devel-oped to improve fertilization, development,and pregnancy potential.6–12 With more rapidcooling and warming rates, oocytes vitrifiedin a very small amount of vitrification solu-tion may better preserve the meiotic spindlesand expedite the recovery compared withconventional straws.44–60 These modificationscould be important for cold-sensitive oocytessuch as bovine and human oocytes, and maypartly explain why the developmental com-petence of vitrified bovine oocytes can beenhanced using OPS or grids, compared withconventional straws.6,7 Recent reports of vitri-fied human oocytes with minimum volumemethods such as the Cryotop or Cryoleafhave shown significantly improved successrates.10–12
SPINDLE STATUS OFFROZEN–THAWED OOCYTES ANDFERTILIZATION OUTCOME
The oocyte freezing and thawing by slow proto-col or vitrification inevitably leads to meioticspindle injury. The changes and recovery of thespindles have been linked to the functionaleffects of oocytes on fertilization and develop-ment.30,44,59 Eroglu et al.59 observed that slowlycryopreserved mouse oocytes inseminatedimmediately after thawing exhibited impair-ment of the spindle rotation, second polar bodyformation, pronuclear migration, and formationof the mitotic spindle. They found an increasedrate of digyny that was attributed to disorganiza-tion of the spindles, rather than to a primarymalfunction of the microfilaments, for thefailure of second polar body extrusion.Insemination of slowly cryopreserved mouseoocytes after 1h of incubation led to normal fer-tilization dynamics. Park et al.30 reported thatvitrified mouse oocytes inseminated at 2h afterwarming had a lower fertilization rate than thecontrol specimens. Chen et al.44 found that thepercentages of fertilization and blastocyst forma-tion of vitrified mouse oocytes inseminated at1h of incubation were significantly lower thanthe control specimens, but they were improvedwhen inseminated after 2 or 3h of incubation.
TIME SCHEDULE FOR VITRIFICATION OF OOCYTES ANDFERTILIZATION: CONSIDERINGASPECTS OF BOTH OOCYTEAGING AND SPINDLE RECOVERY
During ovulation of a dominant follicle, theoocyte at the diplotene stage of prophaseresumes meiosis and extrudes the first polarbody. The oocyte enters into metaphase ofmeiosis II and stays at this stage with an orga-nized spindle system. After fertilization byentry of a spermatozoon, the intracellular cal-cium increases, and the cytostatic factordecreases.65 The oocyte completes meiosis IIand extrudes the second polar body. After
VITRIFICATION IN ASSISTED REPRODUCTION
140
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 140

ovulation the oocyte must be fertilized at theappropriate time, and then continue develop-ment; otherwise, it will undergo apoptosis.Delayed insemination of mature oocytes resultsin compromised embryos and developmentfailure.66 Dozortsev et al.67 found that the opti-mal time for ICSI of human oocytes was from37 to 41 h after administration of human chori-onic gonadotropin (hCG). These fertilizedoocytes achieved the highest implantation rate.
However, inseminating oocytes shortly afterthawing when there is serious spindle disorgani-zation adversely affects fertilization outcomeand increases the rate of digyny.44,59 Choosingthe optimum time interval between oocytethawing and insemination is critical for normalfertilization and subsequent development.68
Therefore, considering the competing aspectsof oocyte aging and spindle recovery is essentialfor a successful oocyte cryopreservation pro-gram. We usually perform oocyte retrieval 34 hpost-hCG. Cryopreservation of oocytes is per-formed at 2 h after oocyte retrieval. The dura-tion of vitrification and dilution is about 10 minwhich is negligible. ICSI is performed after 3 hpost-thaw (post-hCG 39 h). Therefore, the tim-ing of insemination for frozen–thawed oocytesis within an optimal time frame. In the recentreports regarding vitrified human oocytes, ICSIis usually performed at 2 or 3 h after thawingand incubation.9–12
REMOVING CUMULUS CELLS ISBETTER FOR VITRIFICATION OFMATURE OOCYTES
For vitrification of mature human oocytes,some authors have removed cumulus cells
before exposure to cryoprotectants,11,12,48 butothers have not denuded the oocytes9 (Table7B.4). Some authors partially removed cumu-lus cells.10 Chian et al.27 reported that bovineoocytes vitrified without cumulus cells had ahigher survival rate after thawing, and asuperior embryonic developmental capacitycompared with oocytes vitrified with cumuluscells. For vitrification of immature oocytes,the cumulus cells should remained intact.The cumulus cells may help the in vitro mat-uration of oocytes after thawing of COCs.
SUPERIORITY OF VARIOUSVITRIFICATION METHODS
Several studies compared superiority of vitrifi-cation techniques regarding oocyte or embryosurvival and development. In vitrification ofmouse oocytes, Chen et al.44 found that signif-icantly more oocytes cryopreserved by the CPSor OPS methods survived than those by thegrid method. It is more difficult to attain theskill required to reach a consistent result withthe grid method. Using bovine oocytes,Kuwayama et al.10 found that more oocytesvitrified by the Cryotop method cleaved anddeveloped into blastocysts than those by con-ventional straw or OPS. With reference to thereports in the literature, for vitrification ofhuman oocytes, the Cryotop or Cryoleaf meth-ods appear to have a higher survival rate thanthe grid method9–12 (Table 7B.4). Kuwayamaet al.46 applied the Cryotip method for humanembryo vitrification. They found that theCryotip had the same efficiency as the Cryotop,although the cooling rates of the Cryotip areslightly lower than those of the Cryotop. The
141
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
Table 7B.4 Survival of vitrified human oocytes and pregnancy using various procedures
Method Cumulus Survival of oocytes Pregnancy Authors
OPS Removed 11/17 (65%) 1/3 (33%) Kuleshova et al., 199948
Grid Not removed 325/474 (69%) 6/28 (21%) Yoon et al., 20039
Cryotop Partially removed 58/64 (90%) 12/29 (41%) Kuwayama et al., 200510
Cryoleaf Removed 169/180 (94%) 7/15 (47%) Chian et al., 200511
Cryotop Removed 120/143 (84%) 13/23 (57%) Lucena et al., 200612
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 141

Cryotip method has the advantage of betteraseptic conditions.
CONCLUSIONS
The new vitrification techniques have sig-nificantly improved cryopreservation ofmammalian and human oocytes. These tech-niques transform cells into an amorphousglassy state, instead of ice crystal formation,thus preventing freezing–thawing injury dur-ing cryopreservation. The minimum volumemethods facilitate increased rates of coolingand warming, reduction of concentration ofcryoprotectants, and recovery of the meioticspindle. A time schedule for vitrification of
oocytes and fertilization should refer toaspects of both oocyte aging and spindlerecovery. Vitrification is time-saving and doesnot require special equipment. With increasesin survival, fertilization, and pregnancy rates,the vitrification of human oocytes wouldmake a significant contribution to infertilitytreatments, including its use for oocyte dona-tion and for patients about to lose ovarianfunction due to advancing age or prior tocancer therapy.5,69
ACKNOWLEDGMENTS
The authors would like to thank Ms Li-JungChang, Ms Yi-Yi Tsai, and Ms Li-Ting Lin fortheir technical assistance.
VITRIFICATION IN ASSISTED REPRODUCTION
142
References
1. Porcu E, Venturoli S. Progress with oocytecryopreservation. Curr Opin Obstet Gynecol2006; 18: 273–9.
2. Porcu E, Fabbri R, Damiano G et al. Clinicalexperience and applications of oocyte cryo-preservation. Mol Cell Endocrinol 2000; 169:33–7.
3. Fabbri R, Porcu E, Marsella T et al. Humanoocyte cryopreservation: new perspectivesregarding oocyte survival. Hum Reprod 2001;16: 411–6.
4. Boldt J, Cline D, McLaughlin D. Human oocytecryopreservation as an adjunct to IVF-embryotransfer cycles. Hum Reprod 2003; 18: 1250–5.
5. Chen SU, Lien YL, Chen HF et al.Observational clinical follow-up of oocytecryopreservation using a slow-freezing methodwith 1,2-propanediol plus sucrose followed byICSI. Hum Reprod 2005; 20: 1975–80.
6. Martino A, Songsasen N, Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
7. Vajta G, Holm P, Kuwayama M et al. Openpulled straw (OPS) vitrification: a new wayto reduce cryoinjuries of bovine ova andembryos. Mol Reprod Dev 1998; 51: 53–8.
8. Hong SW, Chung HM, Lim JM et al.Improved human oocyte development aftervitrification: a comparison of thawing meth-ods. Fertil Steril 1999; 72: 142–6.
9. Yoon TK, Kim TJ, Park SE et al. Live birthsafter vitrification of oocytes in a stimulated invitro fertilization-embryo transfer program.Fertil Steril 2003; 79: 1323–6.
10. Kuwayama M, Vajta G, Kato O et al. Highlyefficient vitrification method for cryopreserva-tion of human oocytes. Reprod BiomedOnline 2005; 11: 300–8.
11. Chian RC, Son WY, Huang JY et al. High sur-vival rates and pregnancies of human oocytesfollowing vitrification: preliminary report.Fertil Steril 2005; 84 (Suppl 1): S36.
12. Lucena E, Bernal DP, Lucena C et al.Successful ongoing pregnancies after vitrifica-tion of oocytes. Fertil Steril 2006; 85: 108–11.
13. Chen C. Pregnancy after human oocyte cryo-preservation. Lancet 1986; 1: 884–6.
14. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196°C by vitrification.Nature 1985; 313: 573–5.
15. Steponkus PL, Myers SP, Lynch DV et al.Cryopreservation of Drosophila melanogasterembryos. Nature 1990; 345: 170–2.
16. Kuleshova LL, Lopata A. Vitrification can bemore favorable than slow cooling. Fertil Steril2002; 78: 449–54.
17. Liebermann J, Dietl J, Vanderzwalmen P,Tucker MJ. Recent developments in humanoocyte, embryo and blastocyst vitrification:where are we now? Reprod Biomed Online2003; 7: 623–33.
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 142

18. Oktay K, Cil AP, Bang H. Efficiency of oocytecryopreservation: a meta-analysis. Fertil Steril2006; 86: 70–80.
19. Vajta G, Nagy ZP. Are programmable freezersstill needed in the embryo laboratory? Reviewon vitrification. Reprod Biomed Online 2006;12: 779–96.
20. Kasai M, Komi JH, Takakamo A et al. A sim-ple method for mouse embryo cryopreserva-tion in a low toxicity vitrification solution,without appreciable loss of viability. J ReprodFertil 1990; 89: 91–7.
21. Ali J, Shelton JN. Design of vitrification solu-tions for the cryopreservation of embryos.J Reprod Fertil 1993; 99: 471–7.
22. Ali J, Shelton JN. Vitrification of preimplanta-tion stages of mouse embryos. J Reprod Fertil1993; 98: 459–65.
23. Ali J, Shelton JN. Successful vitrification of day-6sheep embryos. J Reprod Fertil 1993; 99: 65–70.
24. Chen SU, Lien YL, Chao KH et al. Cryo-preservation of mature human oocytes by vit-rification with ethylene glycol in straws. FertilSteril 2000; 74: 804–8.
25. Mukaida T, Nakamura S, Tomiyama T et al.Vitrification of human blastocysts using cry-oloops: clinical outcome of 223 cycles. HumReprod 2003; 18: 384–91.
26. Hiraoka K, Hiraoka K, Kinutani M et al.Blastocoele collapse by micropipetting priorto vitrification gives excellent survival andpregnancy outcomes for human day 5 and 6expanded blastocysts. Hum Reprod 2004; 19:2884–8.
27. Chian RC, Kuwayama M, Tan L et al. Highsurvival rate of bovine oocytes matured invitro following vitrification. J Reprod Dev2004; 50: 685–96.
28. Ishimori H, Saeki K, Inai M et al. Vitrification ofbovine embryos in a mixture of ethylene glycoland dimethyl sulfoxide. Theriogenology 1993;40: 427–33.
29. Yokota Y, Sato S, Yokota M et al. Successfulpregnancy following blastocyst vitrification:case report. Hum Reprod 2000; 15: 1802–3.
30. Park SE, Chung HM, Cha KY et al. Cryo-preservation of ICR mouse oocytes: improvedpost-thawed preimplantation development aftervitrification using Taxol, a cytoskeleton stabilizer.Fertil Steril 2001; 75: 1171–84.
31. Vajta G, Holm P, Greve T et al. Survival anddevelopment of in vitro produced bovine blas-tocysts following assisted hatching, vitrifica-tion and in-straw direct rehydration. J ReprodFertil 1997; 111: 65–70.
32. Cuello C, Gil MA, Parrila I et al. In vitrodevelopment following one-step dilution of
OPS vitrified porcine blastocysts. Theriogenology2004; 62: 1144–52.
33. Pickering SJ, Cant A, Braude PR et al. Transientcooling to room temperature can cause irre-versible disruption of the meiotic spindle in thehuman oocytes. Fertil Steril 1990; 54: 102–8.
34. Arav A, Yavin S, Zeron Y et al. New trends ingamete cryopreservation. Mol Cell Endocrinol2002; 187: 77–81.
35. Landa V, Tepla O. Cryopreservation of mouse8-cell embryos in microdrops. Folia Biologica(Praha) 1990; 36: 153–8.
36. Arav A. Vitrification of oocytes and embryos.In: Lauria A, Gandolfi F, eds. New Trends inEmbryo Transfer. Cambridge: Portland Press,1992: 255–4.
37. Hamawaki A, Kuwayama M, Hamano S.Minimum volume cooling method for bovineblastocyst vitrification. Theriogenology 1999;51: 165.
38. Lane M, Schoolcraft WB, Gardner DK.Vitrification of mouse and human blastocystsusing a novel cryoloop container-less tech-nique. Fertil Steril 1999; 72: 1073–8.
39. Kong IK, Lee SI, Cho SG et al. Comparison ofopen pulled straw (OPS) vs. glass micropipette(GMP) vitrification in mouse blastocysts.Theriogenology 2000; 53: 1817–26.
40. Dinnyes A, Dai Y, Jiang S et al. High develop-mental rates of vitrified bovine oocytes follow-ing parthenogenetic activation, in vitrofertilization, and somatic cell nuclear transfer.Biol Reprod 2000; 63: 513–8.
41. Vanderzwalmen P, Benin G, Debauche Ch etal. In vitro survival of metaphase II oocytesand blastocysts after vitrification in a hemi-straw (HS) system. Fertil Steril 2000; 74:S215–6.
42. Kuwayama M, Kato O. All-round vitrificationmethod for human oocytes and embryos. JAssist Reprod Genet 2000; 17: 477.
43. Matsumoto H, Jiang JY, Tanaka T et al.Vitrification of large quantities of immaturebovine oocytes using nylon mesh. Cryobiology2001; 42: 139–44.
44. Chen SU, Lien YL, Cheng YY et al.Vitrification of mouse oocytes using closedpulled straws (CPS) achieves a high survivaland preserves good patterns of meiotic spin-dles, compared with conventional straws,open pulled straws (OPS), and grids. HumReprod 2001; 16: 2350–6.
45. Liebermann J, Tucker MJ, Graham JR et al.Blastocyst development after vitrification ofmulti-pronuclear zygotes using the Flexipetdenuding pipette. Reprod Biomed Online2002; 4: 146–50.
143
VITRIFICATION OF OOCYTES: VARIOUS PROCEDURES
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 143

46. Kuwayama M, Vajta G, Ieda S et al.Comparison of open and closed methods forvitrification of human embryos and the elim-ination of potential contamination. ReprodBiomed Online 2005; 11: 608–14.
47. Chen SU, Chien CL, Wu MY et al. Noveldirect cover vitrification for cryopreservationof ovarian tissues increases follicle viabilityand pregnancy capability in mice. HumReprod 2006; 21: 2794–800.
48. Kuleshova L, Gianaroli L, Ferraretti A et al.Birth following vitrification of a small numberof human oocytes: case report. Hum Reprod1999; 14: 3077–9.
49. Teng TY. Mounting of crystals for macromol-ecular cryocrystallography in a free-standingthin film. J Appl Crystallogr 1990; 23:387–91.
50. Parkin S, Hope H. Macromolecular Cryocry-stallography: cooling, mounting, storage andtransportation of crystals. J Appl Crystallogr1998; 31: 945–53.
51. Vanderzwalmen P, Bertin G, Debauche Chet al. Vitrification of human blastocysts withthe Hemi-Straw carrier: application ofassisted hatching after thawing. Hum Reprod2003; 18: 1504–11.
52. Abe Y, Hara K, Matsumoto H et al. Feasibilityof a nylon-mesh holder for vitrification ofbovine germinal vesicle oocytes in subsequentproduction of viable blastocysts. Biol Reprod2005; 72: 1416–20.
53. Arav A, Zeron Y, Ocheretny A. A new device andmethod for vitrification increases the coolingrate and allows successful cryopreservation ofbovine oocytes. Theriogenology 2000; 53: 248.
54. Huang CC, Lee TH, Chen SU et al. Successfulpregnancy following blastocyst cryopreserva-tion using super-cooling ultra-rapid vitrifica-tion. Hum Reprod 2005; 20: 122–8.
55. Cuello C, Gil MA, Parrilla I et al. Vitrificationof porcine embryos at various developmentalstages using different ultra-rapid cooling pro-cedures. Theriogenology 2004; 62: 353–61.
56. Cai XY, Chen GA, Lian Y et al. Cryoloopvitrification of rabbit oocytes. Hum Reprod2005; 20: 1969–74.
57. Bielanski A, Nadin-Davis S, Sapp T et al. Viralcontamination of embryos cryopreserved inliquid nitrogen. Cryobiology 2000; 40: 110–6.
58. Schatten G, Simerly C, Schatten H. Microtubuleconfigurations during fertilization, mitosis, andearly development in the mouse and therequirement for egg microtubule-mediatedmotility during mammalian fertilization. ProcNatl Acad Sci USA 1985; 82: 4152–6.
59. Eroglu A, Toth TL, Toner M. Alterations ofthe cytoskeleton and polyploidy induced bycryopreservation of metaphase II mouseoocytes. Fertil Steril 1998; 69: 944–57.
60. Chen SU, Lien YL, Chen HF et al. Openpulled straws for vitrification of mature mouseoocytes preserve patterns of meiotic spindlesand chromosomes better than conventionalstraws. Hum Reprod 2000; 15: 2598–603.
61. Rienzi L, Martinez F, Ubaldi F et al. Polscopeanalysis of meiotic spindle changes in livingmetaphase II human oocytes during the freez-ing and thawing procedures. Hum Reprod2004; 19: 655–9.
62. Gook DA, Osborn SM, Johnston WIH.Cryopreservation of mouse and humanoocytes using 1,2-propanediol and the config-uration of the meiotic spindle. Hum Reprod1993; 8: 1101–9.
63. Bianchi V, Coticchio G, Fava L et al. Meioticspindle imaging in human oocytes frozen witha slow freezing procedure involving highsucrose concentration. Hum Reprod 2005; 20:1078–83.
64. Coticchio G, De Santis L, Rossi G et al.Sucrose concentration influences the rate ofhuman oocytes with normal spindle and chro-mosome configurations after slow-coolingcryopreservation. Hum Reprod 2005; 21:1771–6.
65. Edwards RG, Brody SA. Oocyte growth, matura-tion, and fertilization. In: Principles and Practiceof Assisted Human Reproduction. Philadelphia:WB Saunders Company, 1995: 322–3.
66. Gook DA, Osborn SM, Bourne H et al.Fertilization of human oocytes followingcryopreservation: normal karyotypes andabsence of stray chromosomes. Hum Reprod1994; 9: 684–91.
67. Dozortsev D, Nagy P, Abdelmassih S et al. Theoptimal time for intracytoplasmic sperminjection in the human is from 37 to 41 hoursafter administration of human chorionicgonado- tropin. Fertil Steril 2004; 82: 1492–6.
68. Chen SU, Lien YL, Chao KH et al. Effects ofcryopreservation on meiotic spindles ofoocytes and its dynamics after thawing: clini-cal implications in oocyte freezing- a reviewarticle. Mol Cell Endocrinol 2003; 202:101–7.
69. Li XH, Chen SU, Zhang X et al. Cryopreser-ved oocytes of infertile couples undergoingassisted reproductive technology could be animportant source of oocyte donation: a clini-cal report of successful pregnancies. HumReprod 2005; 20: 3390–4.
VITRIFICATION IN ASSISTED REPRODUCTION
144
07b Tucker 8029.qxd 8/23/2007 2:14 PM Page 144

145
Vitrification of oocytes using goldgrid and slush nitrogenTae Ki Yoon, Dong Ryul Lee and Kwang Ryul Cha
7C
OVERVIEW
Cryopreservation is a process of long-term low-temperature storage of biological samples with-out morphological and functional destructionsuch that the samples can be recovered in anintact state. Recent advances in reproductivebiology have resulted in the cryopreservation ofhuman oocytes, zygotes, early cleavage-stageembryos, and blastocysts becoming an integralpart of the human in vitro fertilization embryotransfer (IVF-ET) program.
The establishment of an egg bank usingoocyte cryopreservation techniques wouldprovide a number of benefits. First, it couldprevent ethical and legal problems associatedwith embryo freezing, particularly in certaincountries where embryo freezing is bannedor limited by law. Second, the age at whichpeople marry is rising, resulting in infertilityissues, and an egg bank would provide anoption for older women to have children laterin life. Third, it would allow for a quarantineperiod for genetic and infectious diseasescreening in donor oocyte programs. Oocytefreezing also increases the convenience forsynchronization of procedures. Finally, oocytefreezing gives the option of fertility preserva-tion for patients who receive anticancer treat-ments or oophorectomy.
Although cryopreservation of humanoocytes has been carried out successfully,clinical outcomes remain unsatisfactory dueto low pregnancy and implantation ratesresulting from decreased survival rates andhigh levels of chromosome abnormalities in
thawed oocytes. Many researchers haveintroduced several changes to improve theviability and quality of oocytes after thawing.However, oocyte cryopreservation remainsone of the most elusive tasks in the field ofassisted reproductive technology (ART).
APPLICATION OF VITRIFICATIONFOR OOCYTE CRYOPRESERVATION
Vitrification is the solidification of a solutioninto a glassy, vitrified state from the liquidphase due to an extreme elevation in viscositywhile cooling at low temperature.1 Duringvitrification the solution remains unchangedand the water does not crystallize, and henceno ice crystals are formed.2 In the early1970s, this technique was used for long-termpreservation of various cell types,3,4 and wasalso applied to mammalian embryos as itavoided ice crystallization during cooling andwarming.5
Although the first successful cryopreser-vation of unfertilized mammalian oocyteswas achieved in 1977,6 the efficiency wasextremely low until the mid-1990s becauseoocytes contain a large amount of water in thecytoplasm resulting in ice crystal formationwhen using conventional freezing protocols.Critser et al.7 were the first to apply the vitri-fication method to oocyte cryopreservation.Subsequently, new cryoprotective agents and/or cryovehicles have resulted in improvedsurvival and developmental rates of oocytesafter vitrification.8–12
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 145

CLINICAL APPLICATION OFVITRIFICATION FOR HUMANOOCYTES
Following the first reported pregnancy fromfrozen human mature oocytes by the applica-tion of slow cooling,13 numerous studies haveinvestigated optimal methods for oocytecryopreservation. The studies utilized surplusmature oocytes from patients undergoing invitro fertilization embryo transfer (IVF-ET),or all the oocytes from patients who mightlose their gonadal function during ther-apy.14–21 The oocytes were stored for futureuse by slow cooling or vitrification methods.When the patients wanted to have babiesafter their fresh IVF-ET cycles or therapies,stored oocytes were recovered and providedfor an additional IVF-ET.
A meta-analysis of reports in the literatureup to June 2005 gave the number of clinicalpregnancies as 117 and ten for cryopreserva-tion using slow cooling and vitrification,respectively, and the number of live births as85 and ten, respectively. These resulted in thebirth of 107 and 11 children, respectively, forslow cooling and vitrification.22 In that report,the fertilization rate, live-birth rate perinjected oocyte, and live-birth rate per ETfor vitrified oocytes were 70.6%, 4.5%, and29.4%, respectively. Up to 2002, our groupachieved six pregnancies from 28 ET casesusing vitrified/warmed oocytes, and the preg-nancies resulted in the delivery of sevenhealthy babies.19 However, the clinical out-comes for the thawed oocyte cycles using sur-plus eggs remain inferior to those for IVF usingunfrozen oocytes. Hence, many researchershave introduced new techniques and tools inorder to improve the viability and quality ofoocytes after warming.
ELEVATING COOLING SPEED CANIMPROVE THE CLINICAL RESULTS
Generally, vitrification requires a high con-centration of cryoprotectants and an elevated
cooling speed to avoid ice crystal formation,which is a major cause of cryoinjury. A varietyof cryo-containers and cryo-mediators wererecently introduced to improve the results ofoocyte and embryo freezing.
The use of a high cooling rate during vit-rification using an electron microscope (EM)grid has resulted in the freezing of chilling-sensitive Drosophila embryos and bovineoocytes.10,23 This system has been successfullyapplied to the human IVF-ET program andseveral pregnancies have resulted.18,19 Theuse of EM grids may provide rapid heat con-ductivity from the outside into the oocytes. Inparticular, the gold grid may provide low tox-icity with extremely high heat transmission,and may reduce the damage caused by longexposure to high cryoprotective agent (CPA)concentrations.
Immersion of a sample into liquid nitrogen(LN2) results in boiling and the formation ofgas bubbles around the specimen, which inturn results in poor heat transfer. By applyingnegative pressure with a vacuum, LN2 willfreeze and convert into a slush state (SN2). SN2has a lower internal temperature of −210°Cwithout vaporization.23 Thus, SN2 may offerhigh-speed cooling rates, which may increasethe oocyte survival rate. In our preliminarystudies, the survival of mouse metaphase II(MII) oocytes and human fertilization-failedoocytes was highly improved after vitrificationusing SN2 and warming. The analysis ofcumulus complex (CC) obtained from imma-ture oocytes showed that the number of apop-totic cells was lower in the super-rapid coolinggroup (using SN2) compared with the rapidcooling group (using LN2) (unpublished data).However, whether the short-term exposure ofapoptotic CC after freezing/thawing influ-ences fertilization or further embryonic devel-opment of the warmed oocytes remains to beinvestigated.
In clinical trials involving 30 cycles usinga gold grid and SN2, the fertilization rateof vitrified/warmed oocytes was 77.4 ± 3.5%(168/218), the cleavage rate on day 2 was
VITRIFICATION IN ASSISTED REPRODUCTION
146
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 146

94.3% ± 2.1% (158/168), and ET was success-ful in all patients. The pregnancy rate per ETand the implantation rate for SN2-vitrifiedIVF-ET were 43.3% (13/30) and 14.2%(17/120), respectively. Moreover, higher preg-nancy (72.7% (8/11)) and implantation(22.2% (12/54)) rates were observed afterSN2-vitrification in patients who did notundergo ET for various reasons, even thoughthere was no difference in the indication forIVF-ET.24 These data suggest that the use ofthe super-rapid cooling vitrification proce-dure was not limited to specific patients, andmay be an option for fertility preservationand avoiding the ethical problems of embryofreezing or the risk of ovarian hyperstimula-tion syndrome (OHSS).
During two periods of time (phase I:October 1997 to December 2002; phase II:December 2003 to August 2005), the preg-nancy rates in fresh cycles of conventionalIVF-ET for patients who had similar indica-tions (with more than 15 retrieved oocytes) atour center were 50.3% (731 pregnancies/1454cycles), and 47.2% (376 pregnancies/797cycles); there was no significant differencebetween the two periods. In order to analyzethe efficiency of super-rapid cooling, we com-pared these clinical results (phase II)24 withdata from our previous oocyte cryopreserva-tion program performed in phase I using LN2vitrification.19 The survival and fertilizationrates after warming of oocytes using SN2 wereimproved when compared with those usingLN2. Furthermore, the embryonic develop-ment, pregnancy, and implantation ratesafter warming and ICSI were significantlyimproved (Figure 7C.1).
CRYOPROTECTIVE AGENT
While CPAs are essential for cell cryopreserva-tion, they are usually damaging at high con-centrations. As such, researchers have used alow toxicity CPA such as ethylene glycol(EG)16,19 or a combination of several CPAs.25
EG has a rapid diffusion rate into the cell
through the zona pellucida and the cellularmembrane, and equilibrates rapidly.26,27 Also,combinations of permeating and non-perme-ating CPAs (sucrose, trehalose, or Ficoll) havebeen used for human oocyte cryopreservation.
STORAGE DEVICES USINGNITROGEN (N2) VAPOR
Vitrification has several advantages comparedwith slow cooling, including control of solutepenetration, dehydration rate, and exposuretime to outside environmental conditions.While the LN2 system equipment and run-ning costs are inexpensive,28 this system isbased on direct contact with LN2, and thereis a potential risk of disease transmissionthrough contaminated LN2 during storage.29
Thus, various sealing methods have beenintroduced to eliminate the potential risk ofcontamination.30–32
In 2005, Bielanski et al.33 reported that N2vapor could be a useful tool since no trans-mission of bacterial or viral microbes toembryos and semen stored in the vapor phaseof LN2 in dry shippers had been observed.Indeed, our experience is that over 6 monthsthe survival and development rates ofembryos stored in N2 vapor were similar tothose in LN2 (unpublished data). The survivaland developmental capacity of oocytes storedin the vapor phase of LN2 remains underinvestigation.
PROCEDURE OF OOCYTEVITRIFICATION USING ANELECTRON MICROSCOPE GRID
Yoon et al.24 described the vitrification of sur-plus oocytes from patients who yielded morethan 15 oocytes, or all oocytes from patientswho did not undergo fertilization for variousreasons. Recovered cumulus–oocyte complexes(COCs) were briefly incubated for 10 with80 IU/mL hyaluronidase to remove excesscumulus cells, and were then pre-equilibrated
147
VITRIFICATION OF OOCYTES USING GOLD GRID AND SLUSH NITROGEN
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 147
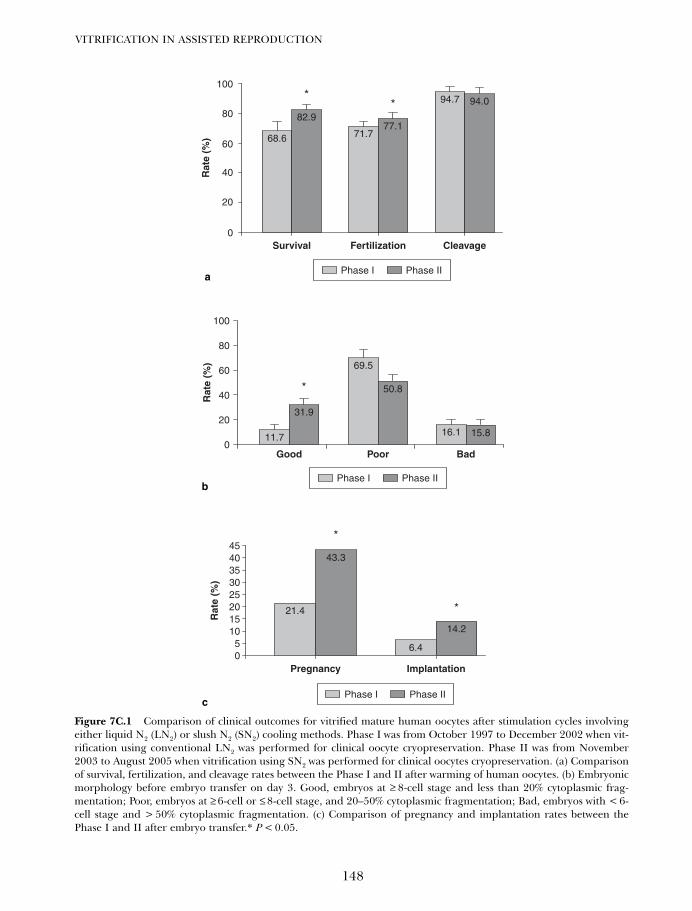
VITRIFICATION IN ASSISTED REPRODUCTION
148
100
80
60
40
20
011.7
31.9
69.5
50.8
16.1 15.8
*
Rat
e (%
)
Good Poor Bad
Phase I Phase II
68.6
100
80
60
40
20
0
82.9
71.777.1
94.7 94.0*
*
Survival
Rat
e (%
)
Fertilization Cleavage
Phase I Phase II
4540353025201510
50
*
*
14.2
6.4
43.3
21.4
Rat
e (%
)
Pregnancy Implantation
Phase I Phase II
Figure 7C.1 Comparison of clinical outcomes for vitrified mature human oocytes after stimulation cycles involvingeither liquid N2 (LN2) or slush N2 (SN2) cooling methods. Phase I was from October 1997 to December 2002 when vit-rification using conventional LN2 was performed for clinical oocyte cryopreservation. Phase II was from November2003 to August 2005 when vitrification using SN2 was performed for clinical oocytes cryopreservation. (a) Comparisonof survival, fertilization, and cleavage rates between the Phase I and II after warming of human oocytes. (b) Embryonicmorphology before embryo transfer on day 3. Good, embryos at ≥ 8-cell stage and less than 20% cytoplasmic frag-mentation; Poor, embryos at ≥ 6-cell or ≤ 8-cell stage, and 20–50% cytoplasmic fragmentation; Bad, embryos with < 6-cell stage and > 50% cytoplasmic fragmentation. (c) Comparison of pregnancy and implantation rates between thePhase I and II after embryo transfer.* P < 0.05.
a
b
c
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 148
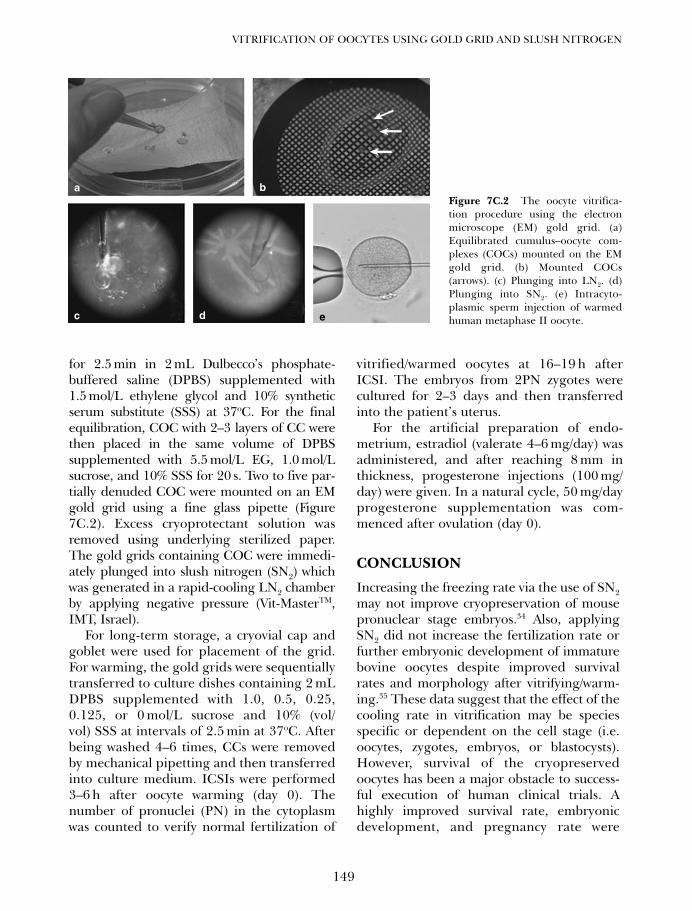
for 2.5 min in 2 mL Dulbecco’s phosphate-buffered saline (DPBS) supplemented with1.5 mol/L ethylene glycol and 10% syntheticserum substitute (SSS) at 37oC. For the finalequilibration, COC with 2–3 layers of CC werethen placed in the same volume of DPBSsupplemented with 5.5 mol/L EG, 1.0 mol/Lsucrose, and 10% SSS for 20 s. Two to five par-tially denuded COC were mounted on an EMgold grid using a fine glass pipette (Figure7C.2). Excess cryoprotectant solution wasremoved using underlying sterilized paper.The gold grids containing COC were immedi-ately plunged into slush nitrogen (SN2) whichwas generated in a rapid-cooling LN2 chamberby applying negative pressure (Vit-MasterTM,IMT, Israel).
For long-term storage, a cryovial cap andgoblet were used for placement of the grid.For warming, the gold grids were sequentiallytransferred to culture dishes containing 2 mLDPBS supplemented with 1.0, 0.5, 0.25,0.125, or 0 mol/L sucrose and 10% (vol/vol) SSS at intervals of 2.5 min at 37oC. Afterbeing washed 4–6 times, CCs were removedby mechanical pipetting and then transferredinto culture medium. ICSIs were performed3–6 h after oocyte warming (day 0). Thenumber of pronuclei (PN) in the cytoplasmwas counted to verify normal fertilization of
vitrified/warmed oocytes at 16–19 h afterICSI. The embryos from 2PN zygotes werecultured for 2–3 days and then transferredinto the patient’s uterus.
For the artificial preparation of endo-metrium, estradiol (valerate 4–6 mg/day) wasadministered, and after reaching 8 mm inthickness, progesterone injections (100 mg/day) were given. In a natural cycle, 50 mg/dayprogesterone supplementation was com-menced after ovulation (day 0).
CONCLUSION
Increasing the freezing rate via the use of SN2may not improve cryopreservation of mousepronuclear stage embryos.34 Also, applyingSN2 did not increase the fertilization rate orfurther embryonic development of immaturebovine oocytes despite improved survivalrates and morphology after vitrifying/warm-ing.35 These data suggest that the effect of thecooling rate in vitrification may be speciesspecific or dependent on the cell stage (i.e.oocytes, zygotes, embryos, or blastocysts).However, survival of the cryopreservedoocytes has been a major obstacle to success-ful execution of human clinical trials. Ahighly improved survival rate, embryonicdevelopment, and pregnancy rate were
149
VITRIFICATION OF OOCYTES USING GOLD GRID AND SLUSH NITROGEN
Figure 7C.2 The oocyte vitrifica-tion procedure using the electronmicroscope (EM) gold grid. (a)Equilibrated cumulus–oocyte com-plexes (COCs) mounted on the EMgold grid. (b) Mounted COCs(arrows). (c) Plunging into LN2. (d)Plunging into SN2. (e) Intracyto-plasmic sperm injection of warmedhuman metaphase II oocyte.
a b
c d e
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 149

1. Fahy GM, MacFarlane DR, Angell CA et al.Vitrification as an approach to cryopreserva-tion. Cryobiology 1987; 24: 387–402.
2. Fahy GM. Vitrification: a new approach toorgan cryopreservation. In: Meryman HT, ed.Transplantation: Approaches to GraftRejection. New York: Alan R Liss, 1986:305–35.
3. Wilmut I. The effect of cooling rate, warmingrate, cryoprotective agent and stage of devel-opment on survival of mouse embryos duringfreezing and thawing. Life Sci II 1972; 11:1071–9.
4. Leibo SP, Mazur P. Methods for the preserva-tion of mammalian embryos by freezing. In:Daniel JC Jr, ed. Methods in MammalianReproduction. New York: Academic Press,1978: 179–201.
5. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at –196°C by vitrification.Nature 1985; 313: 573–5.
6. Whittingham D. Fertilization in vitro anddevelopment to term of unfertilized mouseoocytes previously stored at –196oC. J ReprodFertil 1977; 49: 89–94.
7. Critser JK, Arneson BW, Aaker DW et al.Cryopreservation of hamster oocytes: effectsof vitrification or freezing on human spermpenetration of zona-free hamster oocytes.Fertil Steril 1986; 46: 277–84.
8. Nakagata N. High survival rate of unfertilizedmouse oocytes after vitrification. J ReprodFertil 1989; 87: 479–83.
9. Hotamisligil S, Toner M, Powers RD. Changesin membrane integrity, cytoskeletal structure,and developmental potential of murineoocytes after vitrification in ethylene glycol.Biol Reprod 1996; 55: 161–8.
10. Martino A, Songsasen N, Leibo SP. Develop-ment into blastocysts of bovine oocytes cryo-preserved by ultrarapid cooling. Biol Reprod1996; 54: 1059–69.
11. Vajta G, Holm P, Kuwayama M et al. Openpulled straw (OPS) vitrification: a new wayto reduce cryoinjuries of bovine ova andembryos. Mol Reprod Dev 1998; 51: 53–8.
12. Isachenko V, Alabart JL, Nawroth F et al. Theopen pulled straw vitrification of ovine GV-oocytes: positive effect of rapid cooling orrapid thawing or both? Cryo Letters 2001; 22:157–62.
13. Chen C. Pregnancy after human oocyte cryo-preservation. Lancet 1986; 1: 884–6.
14. Tucker M, Wright G, Morton P et al.Preliminary experience with human oocytecryopreservation using 1,2-propanediol andsucrose. Hum Reprod 1996; 11: 1513–5.
15. Porcu E, Fabbri R, Seracchioli R et al. Birth ofhealthy female after intracytoplasmic sperminjection of cryopreserved human oocytes.Fertil Steril 1997; 68: 724–6.
16. Kuleshova L, Gianoroli L, Magli C et al. Birthfollowing vitrification of small number ofhuman oocytes. Hum Reprod 1999; 14:3077–9.
17. Porcu E, Fabbri R, Damiano G et al. Clinicalexperience and applications of oocytecryopreservation. Mol Cell Endocrinol 2000;169: 33–7.
18. Yoon TK, Chung HM, Lim JM et al.Pregnancy and delivery of healthy infantsdeveloped from vitrified oocytes in a stimu-lated in vitro fertilization-embryo transferprogram. Fertil Steril 2000; 74: 180–1.
19. Yoon TK, Kim TJ, Park SE et al. Live birthafter vitrification of oocytes in a stimulated invitro fertilization-embryo transfer program.Fertil Steril 2003; 79: 1323–6.
20. Borini A, Bonu MA, Coticchio G et al.Pregnancies and births after oocyte cryo-preservation. Fertil Steril 2004; 82: 601–5.
21. Chen ZJ, Li M, Li Y et al. Effects of sucroseconcentration on the developmental potentialof human frozen-thawed oocytes at different
VITRIFICATION IN ASSISTED REPRODUCTION
150
obtained by increasing the cooling rate usingSN2 and a gold grid, which may have a signif-icant positive effect on the undertaking ofclinical trials. Such advances may lead tooocyte cryopreservation being a valuable toolin human ART.
ACKNOWLEDGMENTS
The authors thank Ms Soo Kyung Cha fortechnical support, and staff at the FertilityCenter of CHA General Hospital, Seoul,Korea.
References
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 150

stages of maturity. Hum Reprod 2004; 19:2345–9.
22. Oktay K, Cil AP, Bang H. Efficiency of oocytecryopreservation: a meta-analysis. Fertil Steril2006; 86: 70–80.
23. Steponkus PL, Caldwell S. An optimized pro-cedure for the cryopreservation of Drosophilamelanogaster embryos. Cryo Letters 1993; 14:375–80.
24. Yoon TK, Lee DR, Cha SK et al. Survival rate ofhuman oocytes and pregnancy outcome aftervitrification using slush nitrogen in assistedreproductive technology. Fertil Steril 2007; inpress. (electronic version published Mar 8, 20)
25. Katayama KP, Stehlik J, Kuwayama M et al.High survival rate of vitrified human oocytesresults in clinical pregnancy. Fertil Steril 2003;80: 223–4.
26. Zhu SE, Kasai M, Otoge H et al.Cryopreservation of expanded mouse blasto-cysts by vitrification in ethylene glycol basedsolutions. J Reprod Fertil 1993; 98: 139–45.
27. Yokota Y, Sato S, Yokota M et al. Successfulpregnancy following blastocyst vitrification.Hum Reprod 2000; 15: 1802–3.
28. Kuleshova LL, Lopata A. Vitrification can bemore favorable than slow cooling. Fertil Steril2002; 78: 449–54.
29. Bielanski A, Nadin-Davis S, Sappi T et al.Viral contamination of embryos cryopre-served in liquid nitrogen. Cryobiology 2000;40: 110–6.
30. Kuleshova LL, Shaw JM. A strategy for rapidcooling of mouse embryos within a doublestraw to eliminate the risk of contaminationduring storage in liquid nitrogen. HumReprod 2000; 15: 2604–9.
31. Isachenko V, Montag M, Isachenko E et al.Aseptic technology of vitrification of humanpronuclear oocytes using open-pulled straws.Hum Reprod 2005; 20: 492–6.
32. Kuwayama M, Vajta G, Kato O et al. Highlyefficient vitrification method for cryopreserva-tion of human oocytes. Reprod BiomedOnline 2005; 11: 300–8.
33. Bielanski A. Non-transmission of bacterial andviral microbes to embryos and semen stored inthe vapour phase of liquid nitrogen in dry ship-pers. Cryobiology 2005; 50: 206–10.
34. Nowshari MA, Brem G. Effect of freezing rateand exposure time to cryoprotectant on thedevelopment of mouse pronuclear stageembryos. Hum Reprod 2001; 16: 2368–73.
35. Khanna S, Lee DR, Parks JE. Effect of coolingrate on vitrification of immature bovine oocytes.Biol Reprod 2002; 67(Suppl 1): 338.
151
VITRIFICATION OF OOCYTES USING GOLD GRID AND SLUSH NITROGEN
07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 151

07c Tucker 8029.qxd 8/23/2007 2:14 PM Page 152

153
Vitrifying and warming ofoocytes using CryotopKoichi Kyono, Yukiko Nakajo, Shima Kumagai and Chikako Nishinaka
7D
INTRODUCTION
As of March 2006, a total of 51 pregnancies(28 live births, 23 ongoing, and 40 childrenborn) after vitrification of a human matureoocyte had been reported worldwide.1 Wereported the first delivery in Japan followingcryopreservation of a human mature oocytein 2001.2 This was achieved by using the slowfreezing method (SFM) with 1,2-propanediol(PROH) and mature oocytes. We alsoreported the first successful pregnancy anddelivery after transfer of a single blastocystderived from a vitrified human oocyte in2005.3
PROCEDURES
Intracellular osmotic pressure at −30°C in theSFM is about 13 000 mOsmol/L, comparedwith 8000 mOsmol/L for the vitrificationmethod (VM). Intracellular condition duringcooling and warming is thus better with theVM than with the SFM.
VMs have been greatly improved over theyears. Initially, 30 or 40% ethylene glycol (EG)and 1 or 0.5 mol/L sucrose were used as cryo-protectants for vitrification of human matureoocytes. In 2000, Kuwayama and Katoreported the first pregnancy of a maturehuman oocyte using this protocol, althoughunfortunately the pregnancy did not persist.4
Currently, 15% EG and 15% dimethylsulfoxide(DMSO) are used as cryoprotectants in vitrifi-cation solutions (VSs),3,5,6 as the toxicity ofthese lower concentration VSs is reduced andpermeability is more prompt. Cryotop,Cryotip, Cryoloop, Cryoleaf, electronmicroscopy (EM) grid, open or closed pulledstraw, and the like have all been usedas cryotools3,5–11 (Table 7D.1). Cooling andwarming procedures have also differedbetween reporters (Tables 7D.2, and 7D.3).Further accumulation of outcome data at aninternational level is needed. However, theCryotop devised by Kuwayama offers numer-ous advantages, including that it is a simpleand easy procedure (cooling and warming);
Table 7D.1 Delivery after vitrification of mature human oocytes
Year Cryotool Country
Kuleshova7 1999 OPS ItalyYoon8 2003 EM grid South KoreaKatayama9 2003 Cryotop USAKyono3 2005 Cryotop JapanKuwayama6 2005 Cryotop JapanChian11 2005 Cryoleaf CanadaMukaida10 2005 Cryoloop JapanSun 2006 Cryoloop USA
OPS, open pulled straw; EM, electron microscope.
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 153
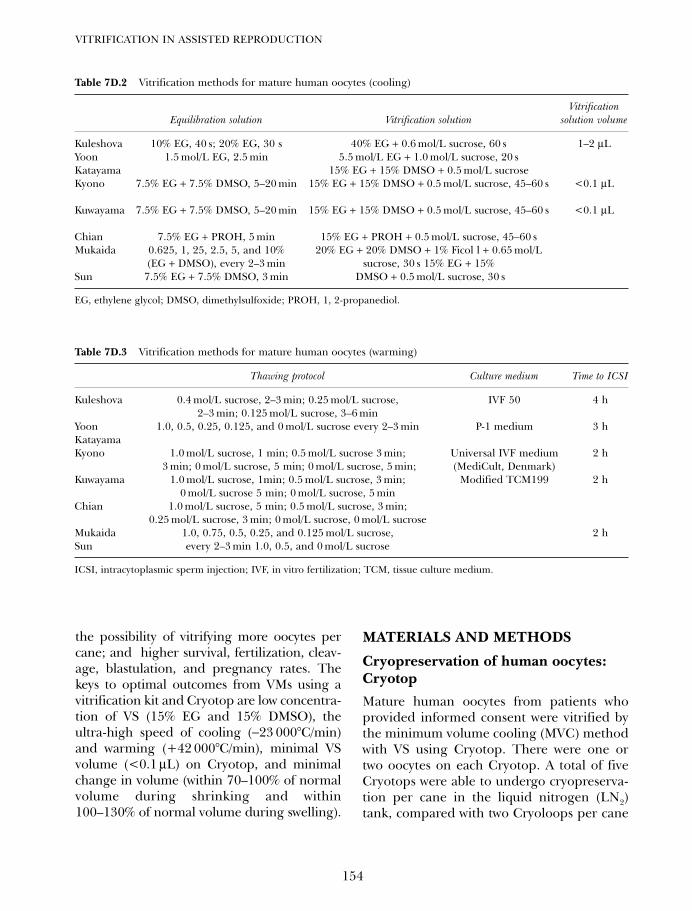
the possibility of vitrifying more oocytes percane; and higher survival, fertilization, cleav-age, blastulation, and pregnancy rates. Thekeys to optimal outcomes from VMs using avitrification kit and Cryotop are low concentra-tion of VS (15% EG and 15% DMSO), theultra-high speed of cooling (−23 000°C/min)and warming (+42 000°C/min), minimal VSvolume (<0.1 µL) on Cryotop, and minimalchange in volume (within 70–100% of normalvolume during shrinking and within100–130% of normal volume during swelling).
MATERIALS AND METHODS
Cryopreservation of human oocytes:CryotopMature human oocytes from patients whoprovided informed consent were vitrified bythe minimum volume cooling (MVC) methodwith VS using Cryotop. There were one ortwo oocytes on each Cryotop. A total of fiveCryotops were able to undergo cryopreserva-tion per cane in the liquid nitrogen (LN2)tank, compared with two Cryoloops per cane
VITRIFICATION IN ASSISTED REPRODUCTION
154
Table 7D.2 Vitrification methods for mature human oocytes (cooling)
VitrificationEquilibration solution Vitrification solution solution volume
Kuleshova 10% EG, 40 s; 20% EG, 30 s 40% EG + 0.6 mol/L sucrose, 60 s 1–2 µLYoon 1.5 mol/L EG, 2.5 min 5.5 mol/L EG + 1.0 mol/L sucrose, 20 sKatayama 15% EG + 15% DMSO + 0.5 mol/L sucroseKyono 7.5% EG + 7.5% DMSO, 5–20 min 15% EG + 15% DMSO + 0.5 mol/L sucrose, 45–60 s <0.1 µL
Kuwayama 7.5% EG + 7.5% DMSO, 5–20 min 15% EG + 15% DMSO + 0.5 mol/L sucrose, 45–60 s <0.1 µL
Chian 7.5% EG + PROH, 5 min 15% EG + PROH + 0.5 mol/L sucrose, 45–60 sMukaida 0.625, 1, 25, 2.5, 5, and 10% 20% EG + 20% DMSO + 1% Ficol l + 0.65 mol/L
(EG + DMSO), every 2–3 min sucrose, 30 s 15% EG + 15%Sun 7.5% EG + 7.5% DMSO, 3 min DMSO + 0.5 mol/L sucrose, 30 s
EG, ethylene glycol; DMSO, dimethylsulfoxide; PROH, 1, 2-propanediol.
Table 7D.3 Vitrification methods for mature human oocytes (warming)
Thawing protocol Culture medium Time to ICSI
Kuleshova 0.4 mol/L sucrose, 2–3 min; 0.25 mol/L sucrose, IVF 50 4 h2–3 min; 0.125 mol/L sucrose, 3–6 min
Yoon 1.0, 0.5, 0.25, 0.125, and 0 mol/L sucrose every 2–3 min P-1 medium 3 hKatayamaKyono 1.0 mol/L sucrose, 1 min; 0.5 mol/L sucrose 3 min; Universal IVF medium 2 h
3 min; 0 mol/L sucrose, 5 min; 0 mol/L sucrose, 5 min; (MediCult, Denmark)Kuwayama 1.0 mol/L sucrose, 1min; 0.5 mol/L sucrose, 3 min; Modified TCM199 2 h
0 mol/L sucrose 5 min; 0 mol/L sucrose, 5 minChian 1.0 mol/L sucrose, 5 min; 0.5 mol/L sucrose, 3 min;
0.25 mol/L sucrose, 3 min; 0 mol/L sucrose, 0 mol/L sucroseMukaida 1.0, 0.75, 0.5, 0.25, and 0.125 mol/L sucrose, 2 hSun every 2–3 min 1.0, 0.5, and 0 mol/L sucrose
ICSI, intracytoplasmic sperm injection; IVF, in vitro fertilization; TCM, tissue culture medium.
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 154
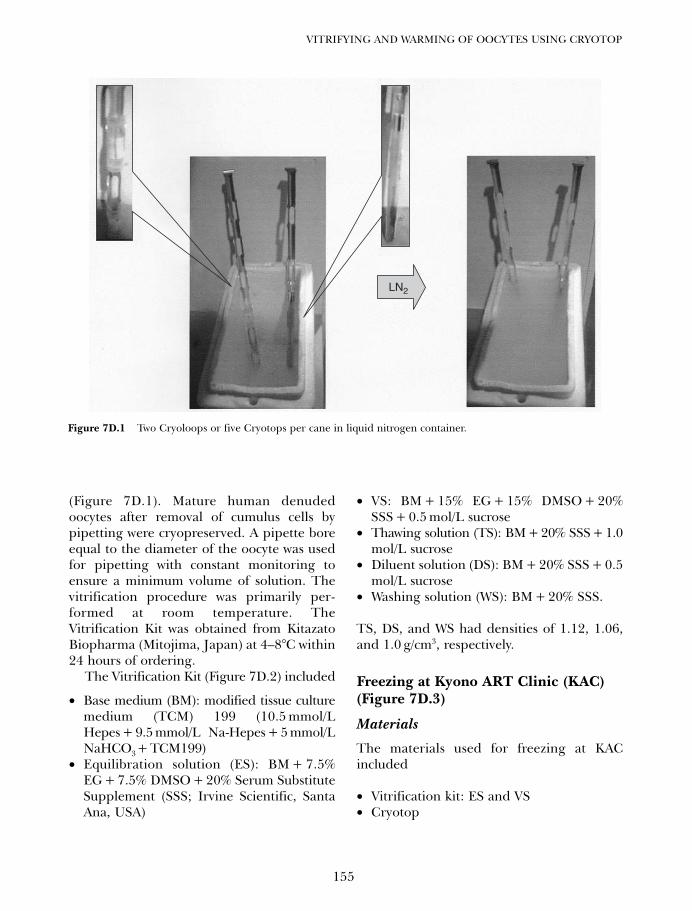
(Figure 7D.1). Mature human denudedoocytes after removal of cumulus cells bypipetting were cryopreserved. A pipette boreequal to the diameter of the oocyte was usedfor pipetting with constant monitoring toensure a minimum volume of solution. Thevitrification procedure was primarily per-formed at room temperature. TheVitrification Kit was obtained from KitazatoBiopharma (Mitojima, Japan) at 4–8°C within24 hours of ordering.
The Vitrification Kit (Figure 7D.2) included
• Base medium (BM): modified tissue culturemedium (TCM) 199 (10.5 mmol/LHepes + 9.5 mmol/L Na-Hepes + 5 mmol/LNaHCO3+ TCM199)
• Equilibration solution (ES): BM + 7.5%EG + 7.5% DMSO + 20% Serum SubstituteSupplement (SSS; Irvine Scientific, SantaAna, USA)
• VS: BM + 15% EG + 15% DMSO + 20%SSS + 0.5 mol/L sucrose
• Thawing solution (TS): BM + 20% SSS + 1.0mol/L sucrose
• Diluent solution (DS): BM + 20% SSS + 0.5mol/L sucrose
• Washing solution (WS): BM + 20% SSS.
TS, DS, and WS had densities of 1.12, 1.06,and 1.0 g/cm3, respectively.
Freezing at Kyono ART Clinic (KAC)(Figure 7D.3)
Materials
The materials used for freezing at KACincluded
• Vitrification kit: ES and VS• Cryotop
155
VITRIFYING AND WARMING OF OOCYTES USING CRYOTOP
LN2
Figure 7D.1 Two Cryoloops or five Cryotops per cane in liquid nitrogen container.
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 155
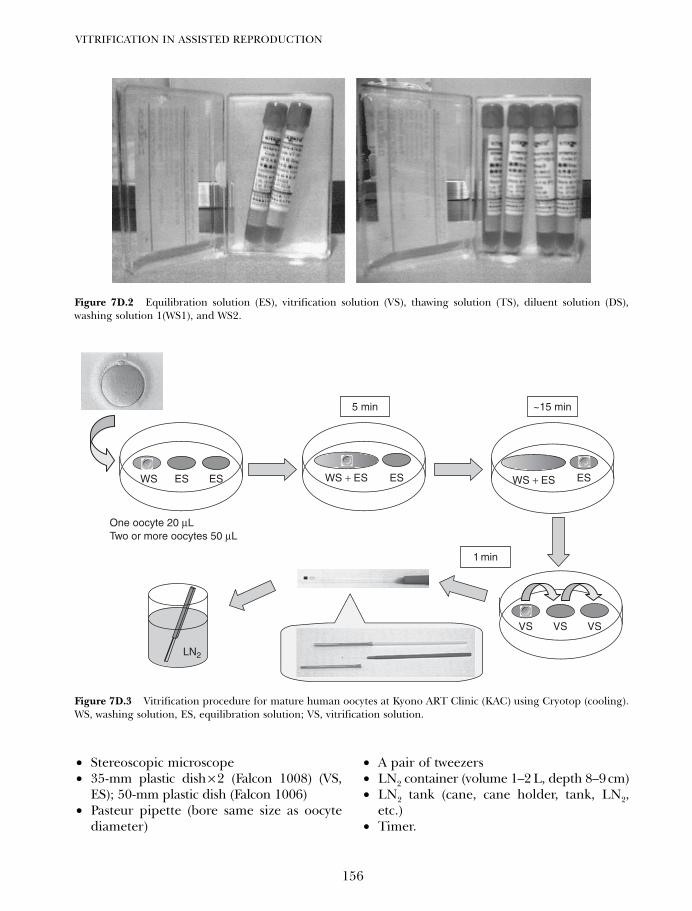
• Stereoscopic microscope• 35-mm plastic dish × 2 (Falcon 1008) (VS,
ES); 50-mm plastic dish (Falcon 1006)• Pasteur pipette (bore same size as oocyte
diameter)
• A pair of tweezers• LN2 container (volume 1–2 L, depth 8–9 cm)• LN2 tank (cane, cane holder, tank, LN2,
etc.)• Timer.
VITRIFICATION IN ASSISTED REPRODUCTION
156
Figure 7D.2 Equilibration solution (ES), vitrification solution (VS), thawing solution (TS), diluent solution (DS),washing solution 1(WS1), and WS2.
5 min
1 min
~15 min
LN2
One oocyte 20 µLTwo or more oocytes 50 µL
WS WS + ES WS + ESES ES
VSVSVS
ES ES
Figure 7D.3 Vitrification procedure for mature human oocytes at Kyono ART Clinic (KAC) using Cryotop (cooling).WS, washing solution, ES, equilibration solution; VS, vitrification solution.
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 156

Procedure
(1) ES, VS, and WS were kept for more than30 min at room temperature, and thenpoured onto a 35-mm dish (Falcon 1008)from the tube. On the bottom of 50-mmdish were written one ‘WS’ and two ‘ES’s.One 20 µL drop of WS and two 20 µLdrops of ES were placed onto the 50-mmdish in that order. In case there weremore than two oocytes, one 50 µL dropwas needed. The LN2 was poured intothe container (vessel or bottle, volume1–2 L) at the 90% level. The sterilizedpouch of the Cryotop was opened andthe date of cryopreservation, name, andgrade of oocytes were noted.
(2) Culture dishes with denuded oocytesfrom the incubator (atmosphere: 6%CO2, 5% O2, 89% N2 at 37°C) were placedin air at room temperature for 5 min.Oocytes were judged by rolling using thepipette microscopically. Oocytes with apoor grade, irregular contours, or darkcoloration were excluded from cryo-preservation.
(3) Denuded oocytes with minimum vol-ume of previous medium were movedinto the center of the WS drop.
(4) WS including denuded oocytes wasmixed with the adjacent first drop of ESand then placed in mixed medium atroom temperature for 5 min.
(5) Denuded oocytes were then placed onthe surface of the second drop of ES. Wethen waited for ES to permeate into theinside of the oocyte and for perfectrecovery of size and shape within 15 min.Recovery time is dependent on the via-bility of each oocyte. During this time,three ‘VS’s were written on the bottom of50-mm dish. Three 20 µL drops of VSwere placed in the marked postions ontothe 50-mm dish.
(6) Perfectly recovered oocytes with mini-mum volume of ES after the end ofequilibration were placed on the surfaceof the first VS drop. ES in the pipette
was discarded in the dish and the insideof the pipette was washed in the VSdrop.
(7) After 5 s, floating oocytes were sunk in asecond VS drop to exchange water withcryoprotectants (EG and DMSO) insidethe oocyte. The procedure was repeatedtwice in a new third VS drop.
(8) Oocytes that had shrunk due to dehy-dration after perfect exchange ofoocytes were confirmed. Perfectlyexchanged oocytes were picked up witha minimum volume (<0.1 µL) of VS,then placed on the end of the Cryotop.
(9) The tip of Cryotop loading oocytes wasimmediately plunged into LN2. The timerequired from steps (6) to (9) was 45–60 s.Cooling rate was 23 000°C/min.
(10) The tip of the Cryotop was covered bythe cap and cryopreserved in LN2.
Thawing at KAC (Figure 7D.4)
Materials
The materials used for thawing at KAC(Figure 7D.5) included
• Vitrification kit: TS, DS, WS1, and WS2• Culture medium for recovery (Universal
IVF medium; MediCult, Denmark)• Stereoscopic microscopy• 35-mm dish × 4 (TS, DS, WS1, WS2) (Falcon
1008)• LN2• LN2 container (volume 1–2 L; depth 8–9 cm)• Timer• Pasteur pipette • A pair of tweezers.
Procedure
(1) TS tube with closed cap and 35-mm dishon which ‘TS’ was written on the bottomwere warmed in an incubator at 37°Covernight.
(2) Cryotop was taken from the cane in theLN2 tank and promptly moved into thecontainer 90% filled with new LN2.
157
VITRIFYING AND WARMING OF OOCYTES USING CRYOTOP
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 157

(3) On the bottom of the three 35-mm disheswas labeled ‘DS’, ‘WS1’, and ‘WS2’.
(4) The TS tube and the 35-mm dish weretaken from the incubator. The TS tube
was mixed by rotating twice, and then thecontents were poured over the 35-mmdish, which was then placed on the micro-scope stage. Focus was adjusted to the
VITRIFICATION IN ASSISTED REPRODUCTION
158
LN2
For warming
1 min37°C
TS(1.0 mol/L sucrose)
ICSI 37°C, CO2 6%, O2 5%, N2 89
DS(0.5 mol/L sucrose)
WS1, WS2
3 minRT
5 min × 2RT
2 h
Figure 7D.4 Vitrification procedure for mature human oocytes at KAC using Cryotop (warming). TS, thawing solu-tion; RT, room temperature; DS, diluent solution, WS, washing solution; ICSI, intracytoplasmic sperm injection; RT,room temperature.
LN2 container
Figure 7D.5 Preparation for thawing procedure.
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 158

bottom of the 35-mm dish at minimummagnification.
(5) The cap of the Cryotop in the LN2 con-tainer was removed using the pair oftweezers, and the tip of the Cryotop wasdipped into 37°C TS within 1 s. Warmingspeed was +42 000°C/min. The Cryotopwith oocyte was placed in TS at the centerof the dish for 1 min. Oocytes sponta-neously separated from the end of theCryotop. Oocytes were found by focusingon the tip of the Cryotop and aspiratedby the pipette tip after separation fromthe tip of the Cryotop, and placed in thecenter of the 35-mm dish. During thistime, we confirmed that the shrunkenoocyte increased in volume due to thereturn of water into the inside of theoocyte after thawing.
(6) Oocytes were moved to the bottom centerof the DS dish and kept at room temper-ature for about 3 min. The most impor-tant point was to aspirate the oocyte witha TS volume in accordance with the 3 mmlength of tip of pipette and to make a TSlayer in accordance with the 2 mm lengthof tip of pipette on the bottom of the 35-mm dish, followed by placement of theoocyte into a TS layer in the DS. All TSremaining in the pipette was poured overthe oocyte. Allowing the oocyte to adaptgradually from TS to DS is very impor-tant. Oocytes start to shrink after brieflyswelling. A DS layer in accordance withthe 2 mm length of tip of pipette as abovewas made in WS1 on the bottom of a 35-mm dish and the oocyte was placed intothe DS layer in the WS1 drop. DS remain-ing in the pipette was poured over theoocyte and kept at room temperature for5 min.
(7) The oocyte was aspirated by pipette andplaced on the surface of the center of theWS2 dish. Minimizing the volume of WS1is important when moving the oocytefrom WS1 to WS2 at the end of thepipette. The oocyte was warmed from
room temperature to 37°C by turning thehot plate ‘on’ for 5 min. Re-expandedoocytes were considered to have survived.
(8) Intracytoplasmic sperm injection (ICSI)was performed after culturing in media(Universal IVF medium; MediCult,Denmark) in an atmosphere of 6% CO2,5% O2, 89% N2 at 37°C for 2 hours.3,6
INDICATIONS FORHUMAN OOCYTECRYOPRESERVATION
Indications for human oocyte preservationinclude
• Oocyte donation• Preservation of own fertile ability for aging• Malignant disease (leukemia, etc.)• Premature ovarian failure (Turner syndrome,
etc.)
OUTCOME OF VITRIFICATION
Our data showed that survival (82.2% = 60/73vs. 100% = 12/12), fertilization (63.3% = 38/60vs. 41.7% = 5/12), cleavage (97.4% = 37/38 vs.100% = 5/5) and blastulation rates (26.7% =4/15 vs. 33.3% = 1/3) differed significantlybetween SFM and vitrification using cryotop(VF), respectively.13 The survival rate was50–80% for SFM, compared with more than80% for VF in some studies. Fertilizationrate, clinical pregnancies per thawed oocyte,clinical pregnancies per injected oocyte, clin-ical pregnancies per transfer, and implanta-tion rate for SFM were 64.9% (2478/3818),2.3% (153/6720), 4.0% (153/3818), 2.1%(153/7429), and 10.1% (185/1828), respec-tively. Fertilization rate, clinical pregnanciesper thawed oocytes, clinical pregnancies pertransfer, live births per transfer, and implan-tation rate for VF were 74.2% (637/859),4.5% (61/1354), 45.5% (61/134), 36.6%(49/134), and 17.1% (81/473), respectively.1
Pregnancy rates with VF thus appear to haveimproved.
159
VITRIFYING AND WARMING OF OOCYTES USING CRYOTOP
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 159

SEX RATIO
We examined sex ratio in all references intro-duced by Oktay et al.1 More females thanmales have been born following cryopreserva-tion of human mature oocytes (female : male,52 : 28).
SPINDLE DAMAGE
Some studies have suggested spindle distrup-tion.14–16 Several studies have shown that thespindle reforms after thawing and rewarm-ing.17–1 In particular, some studies have sug-gested that vitrification forms morphologicallynormal metaphase spindles with properlyaligned chromatin.19–21.
Outcome for children born from slowfrozen oocytes
A total of 52 pregnancies have been reportedin two studies,22,23 with 32 live births. Follow-up has been reported for 30 of the 32 livebirths, with abnormalities limited to ventricu-lar septal defect (VSD) in one case andtriploidy in one case.
In our clinic, the one child born followingSFM in 2001 and the child born followingvitrification in 2004 were normal as ofNovember 2006.
An international registry is needed tocompile data regarding pregnancy/birth out-comes especially from vitrified oocytes.
IMMATURE OOCYTECRYOPRESERVATION
Immature oocytes have been suggested tobe more suitable than mature oocytes for cryo-preservation, as the chromatin and spindlesof immature oocytes are not damaged by cryo-preservation. However, only one case ofpregnancy and delivery achieved using cryo-preserved immature human oocytes has beenreported.24 Recent reports have suggestedthat cryopreservation of immature oocytes
damages the cytoplasm. The use of in vitromaturation of oocytes from germinalvesicle (GV) to metaphase II has not been estab-lished, as associated problems of cytoplasmicmaturation have been identified.
TaxolR, a cytoskeletal stabilizer, appearseffective in the cryopreservation of GVoocytes. Park et al.25 reported that addition ofTaxol significantly improved post-thaw devel-opment of cumulus-enclosed ICR mouseoocytes vitrified at the mature stage. We havereported that addition of Taxol to vitrificationsolution greatly promoted post-thaw preim-plantation development of human immatureoocytes.26 Damage to the cytoskeleton andalteration of gene/protein expression or mito-chondrial function due to oocyte freezing willbe investigated in the future.
CONCLUSIONS
Oocyte cryopreservation remains experimen-tal, but the techniques utilized are rapidlybeing refined and improved, particularly invitrification. Further follow-up data areneeded on children born after oocyte cryo-preservation.
ACKNOWLEDGMENTS
The authors would like to thank Ms ChiharuOnuma and Ms Wakako Funakoshi.
REFERENCES1. Oktay K, Cil AP, Bang H. Efficiency of oocyte
cryopreservation: a meta-analysis. Fertil Steril2006; 86: 70–80.
2. Kyono K, Fukunaga N, Haigo K et al.Pregnancy and delivery of a healthy femaleinfant after intracytoplasmic sperm injectioninto cryopreserved human oocytes. Jpn J FertilSteril 2001; 46: 171–7.
3. Kyono K, Fuchinoue K, Yagi A et al. Successfulpregnancy and delivery after transfer of a sin-gle blastocyst derived from a vitrified maturehuman oocyte. Fertil Steril 2005; 84: 1017.
4. Kuwayama M, Kato O. Successful vitrificationof human oocytes. Fertil Steril 2000; 74: S49.
VITRIFICATION IN ASSISTED REPRODUCTION
160
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 160

5. Okimura T, Kato K, Zhan Q et al. Update onclinical efficiency of the vitrification methodfor human oocytes in an in vitro fertilizationprogram. Fertil Steril 2005; 84: S174.
6. Kuwayama M, Ieda S. Vitrification of mouseoocytes. J Mamm Ova Res 2005; 22: 71–5.
7. Kuleshova K, Gianaroli L, Magli C et al. Birthfollowing vitrification of a small number ofhuman oocytes: case report. Hum Reprod1999; 14: 3077–9.
8. Yoon TK, Kim TJ, Park SE et al. Live birthsafter vitrification of oocytes in a stimulated invitro fertilization-embryo transfer program.Fertil Steril 2003; 79: 1323–6.
9. Kayatama KP, Stehlik J, Kuwayama M et al.High survival rate of vitrified human oocytesresults in clinical pregnancy. Fertil Steril 2003;80: 223–4.
10. Mukaida T, Matsubara T, Takahashi K et al.Birth after vitrified oocytes using cryolooptechnique. Fertil Steril 2005; 84: S454.
11. Chian RC, Son WY, Huang JY et al. High sur-vival rates and pregnancies of human oocytesfollowing vitrification: preliminary report.Fertil Steril 2005; 84: S36.
12. Personal Communication Sun 2006.13. Kyono K, Fuchinoue K, Nakajo Y et al.
Comparing vitrification and slow freezing pro-cedures in cryopreservation of mature humanoocytes. Fertil Steril 2004; 82: S135.
14. Boiso I, Marti M, Santalo J et al. A confocalmicroscopy analysis of the spindle and chromo-some configurations of human oocytes cryopre-served at the germinal vesicle and metaphase IIstage. Hum Reprod 2002; 17: 1885–91.
15. Pickering SJ, Braude PR, Johnson MH et al.Transient cooling to room temperature cancause irreversible disruption of the meioticspindle in the human oocyte. Fertil Steril1990; 54: 102–8.
16. Wang WH, Meng L, Hackett RJ et al. Limitedrecovery of meiotic spindles in living humanoocytes after cooling-rewarming observedusing polarized light microscopy. Hum Reprod2001; 16: 2374–8.
17. Gook DA, Osborn SM, Johnston WI.Cryopreservation of mouse and humanoocytes using 1,2-propanediol and the config-uration of the meiotic spindle. Hum Reprod1993; 8: 1101–9.
18. Stachecki JJ, Munne S, Cohen J. Spindle orga-nization after cryopreservation of mouse,human, and bovine oocytes. Reprod BiomedOnline 2004; 8: 664–77.
19. Silva CA, Swain J, Acevedo N et al. Influenceof vitrification on metaphase II spindledynamics, spindle morphology, and chromatinalignment. Fertil Steril 2004; 82: S111.
20. Chen CK, Wang CW, Tsai WJ et al. Evaluationof meiotic spindles in thawed oocytes after vit-rification using polarized light microscopy.Fertil Steril 2004; 82: 666–72.
21. Huang JY, Chen HY, Wang Y et al. Meioticspindle and chromosome alignment of in vitromatured oocytes following vitrification. FertilSteril 2006; 86: S66.
22. Porcu E, Fabbri R, Seracchioli R et al.Obstetric, perinatal outcome and follow upof children conceived from cryopreservedoocytes. Fertil Steril 2000; 74: S48.
23. Winslow KL , Yang D, Blohm PL et al. Oocytecryopreservation/a three year follow up of six-teen births. Fertil Steril 2001; 76: S120–1.
24. Tucker MJ, Wright G, Morton PC et al. Birthafter cryopreservation of immature oocyteswith subsequent in vitro maturation. FertilSteril 1998; 70: 578–9.
25. Park SE, Chung HM, Cha KY et al.Cryopreservation of ICR mouse oocytes:improved post-thawed preimplantation devel-opment after vitrification using Taxol, acytosleleton stabilizer. Fertil Steril 2001; 75:1177–84.
26. Kyono K, Haigo K, Fukunaga N et al.Comparison of two freezing methods forhuman immature oocytes with cumulus orcumulus free: Slow freezing versus vitrification(including the effect of Taxol). Fertil Steril2002; 76: S13.
161
VITRIFYING AND WARMING OF OOCYTES USING CRYOTOP
07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 161

07d Tucker 8029.qxd 8/23/2007 2:15 PM Page 162

163
Vitrification of pronuclear embryos:research basis for aseptic technologyand its application to oocytes and blastocystsHans van der Ven, Vladimir Isachenko, Evgenia Isachenko, Markus Montag andFrank Nawroth
8
Development of a refined method for cryo-preservation of human pronuclear embryos isan important topic, as it is illegal to cryopre-serve an oocyte after fusion of the pronucleidue to ethical difficulties in some countries.To date, in some centers, numerous babieshave been born after transfer of embryosdeveloped from frozen pronuclear embryos.1
However, with regard to the low efficacy ofthe oocyte freezing technique, the ‘frozenoocyte’ pregnancy rate is lower than thatresulting from fresh pronuclear embryos.2
Conventional (slow) freezing of humanpronuclear and developing embryos hasbeen the most widely used method of preser-vation to date.2,3 However, there have beenseveral reports of the successful cryopreser-vation of human pronuclear embryos bydirect plunging into liquid nitrogen (socalled vitrification).5–13 This method is nowthe object of intensive investigation innumerous laboratories taking into accountthat the protocol of vitrification includes twoparameters: the vitrification process occupiesonly a few minutes in contrast to the longtime-consuming conventional method, andthis method needs no special equipmentcompared with conventional freezing.
Our protocol of vitrification of humanpronuclear embryos includes the followingparameters: step-wise saturation by andremoval of permeable cryoprotectants, using a
small amount of vitrification medium thatallows quick warming of cells; full isolation ofvitrification medium from contact with liq-uid nitrogen; and short contact of cells withpermeable cryoprotectant dimethylsulfoxide(DMSO) at decreased temperature for reduc-tion of parthenogenetic and toxic effects. Theresearch basis of the technology is describedbelow.
STEP-WISE SATURATION BYAND REMOVAL OF PERMEABLECRYOPROTECTANTS
According to the most common protocols ofvitrification, human oocytes and embryos,once thawed, are placed in hypertonic disac-charide solution (normally sucrose) to removepermeable cryoprotectants before transfer-ring the cells to an isotonic culture medium.This rehydration process is generally con-ducted by gradual dilution of the permeablecryoprotectants through exposure in threesteps to 0.5, 0.25, and 0.125 mol/L sucrose5,6
or to 1.0, 0.5, and 0.25 mol/L sucrose.9 Four-step dilution protocols involving exposureto 1.0, 0.5, 0.25, and 0.125 mol/L sucrosehave also been described.8,10 In contrast, ithas recently been possible to directly rehy-drate vitrified ovine and bovine oocytes afterthawing without the need for gradual dilutionof the cryoprotectants.14–16
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 163
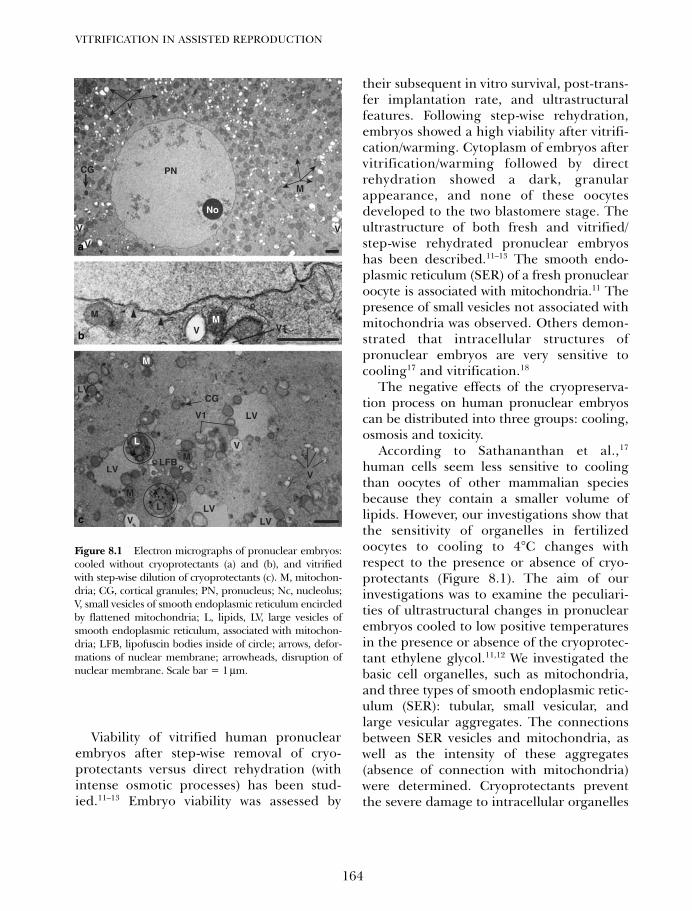
Viability of vitrified human pronuclearembryos after step-wise removal of cryo-protectants versus direct rehydration (withintense osmotic processes) has been stud-ied.11–13 Embryo viability was assessed by
their subsequent in vitro survival, post-trans-fer implantation rate, and ultrastructuralfeatures. Following step-wise rehydration,embryos showed a high viability after vitrifi-cation/warming. Cytoplasm of embryos aftervitrification/warming followed by directrehydration showed a dark, granularappearance, and none of these oocytesdeveloped to the two blastomere stage. Theultrastructure of both fresh and vitrified/step-wise rehydrated pronuclear embryoshas been described.11–13 The smooth endo-plasmic reticulum (SER) of a fresh pronuclearoocyte is associated with mitochondria.11 Thepresence of small vesicles not associated withmitochondria was observed. Others demon-strated that intracellular structures ofpronuclear embryos are very sensitive tocooling17 and vitrification.18
The negative effects of the cryopreserva-tion process on human pronuclear embryoscan be distributed into three groups: cooling,osmosis and toxicity.
According to Sathananthan et al.,17
human cells seem less sensitive to coolingthan oocytes of other mammalian speciesbecause they contain a smaller volume oflipids. However, our investigations show thatthe sensitivity of organelles in fertilizedoocytes to cooling to 4°C changes withrespect to the presence or absence of cryo-protectants (Figure 8.1). The aim of ourinvestigations was to examine the peculiari-ties of ultrastructural changes in pronuclearembryos cooled to low positive temperaturesin the presence or absence of the cryoprotec-tant ethylene glycol.11,12 We investigated thebasic cell organelles, such as mitochondria,and three types of smooth endoplasmic retic-ulum (SER): tubular, small vesicular, andlarge vesicular aggregates. The connectionsbetween SER vesicles and mitochondria, aswell as the intensity of these aggregates(absence of connection with mitochondria)were determined. Cryoprotectants preventthe severe damage to intracellular organelles
VITRIFICATION IN ASSISTED REPRODUCTION
164
PN
M
M M
No
CG
V
V
V
V V1
LV
CG
L
LFB M
M
M
LV
LV
L LVLV
V
V
V
V1
v
Figure 8.1 Electron micrographs of pronuclear embryos:cooled without cryoprotectants (a) and (b), and vitrifiedwith step-wise dilution of cryoprotectants (c). M, mitochon-dria; CG, cortical granules; PN, pronucleus; Nc, nucleolus;V, small vesicles of smooth endoplasmic reticulum encircledby flattened mitochondria; L, lipids, LV, large vesicles ofsmooth endoplasmic reticulum, associated with mitochon-dria; LFB, lipofuscin bodies inside of circle; arrows, defor-mations of nuclear membrane; arrowheads, disruption ofnuclear membrane. Scale bar = 1 µm.
a
b
c
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 164

such as multiple vacuolizations, clustering ofmitochondria, and deformation and disrup-tion of nucleolar and plasma membranes.After vitrification, thawing, and 1 h of expo-sure in CO2 incubator, the presumably viablepronuclear embryos showed large SER vesi-cles associated with mitochondria, and smallvesicles close to flattened, crescent-shapedmitochondria (Figure 8.1). Human pronu-clear embryos are highly sensitive to coolingand low positive temperatures. Adding thecryoprotectant provides significant protec-tion against damage of the intracellularorganelles (Figure 8.1).
We believe that osmosis plays a central rolethroughout all negative effects of the cryo-preservation process of human pronuclearembryos. Figure 8.2 shows images of humanpronuclear embryos that were vitrified,warmed, and directly rehydrated with intenseosmotic processes. The cytoplasm was seen tobe filled with a finely granulated substance withdark inclusions. Note the remains of mem-brane structures and a dense dark pulp withtransparent (low density) membrane-bearingand membrane-free vesicles. The dark pulp isenveloped by membrane and is characteristicof the deformation and destruction provokedby the intense osmotic effects of direct rehydra-tion. The cytoplasm shows transparent (lowdensity) spots which are probably the result ofthe disruption of lysosomes, that release theirproteolytic contents, inducing lysis of the cyto-plasmic matrix. The image shows the disrup-tion of the pronucleus followed by rupture ofthe membranes first and the nucleoli later.
The method of direct post-thaw rehydra-tion induces lethal osmotic effects in humanpronuclear embryos. Accordingly, the vitrifi-cation protocol for these embryos mustinclude the step-wise dilution of the cryopro-tectant. Taking into account that the satura-tion by cryoprotectants is also accompaniedby osmotic processes, our aseptic technologyincludes a step-wise method of saturation bycryoprotectants.
FULL ISOLATION OF A SMALLAMOUNT OF VITRIFICATIONMEDIUM FROM CONTACT WITHLIQUID NITROGEN (ASEPTICMODE)
Rall and Fahy,19 and others have publishedintensely on vitrification including theinfluence of cooling and warming rates onsurvival of vitrified objects. Investigations ofthese authors provided evidence that the rateof cooling during vitrification is secondary aslong as it is fast enough to prevent crystal-lization. For example, for vitrification of8-cell mouse embryos using solutions of dif-ferent combinations of DMSO, acetamide,propylene glycol, and glycerol in total con-centrations from 5.5 to 6.54 mol/L plus 6%polyethylene glycol, equally high survivalrates were obtained at cooling rates rangingfrom about 15 to 2500°C/min.20 Differentialscanning calorimetry compared the appear-ance of the solutions using phase-contrastcryomicroscopy. No crystallization wasobserved when these solutions were cooled atrates greater than 10° C/min.21 High survivalrates of mouse embryos after vitrificationwith a cooling rate of at least 10° C/min usingvitrification solution 6.5 glycerol plus 6%polyethylene glycol were reported.20
In practice there are two different tech-nologies of cooling of cells before storage inliquid nitrogen: cooling by direct plunginginto liquid nitrogen (decreasing of tempera-ture from some thousands to some tens ofthousands °C/min;15,22–27 and cooling in thevapor above liquid nitrogen before long-termstorage in liquid nitrogen (at about200°C/min).28–34 This methodology of ‘slow’cooling of cells before storage in liquid nitro-gen is evidence for the secondary role of theparameter ‘speed of cooling’ for vitrificationof oocytes and embryos.
Another important parameter of vitrificationis the speed of warming after storage in liquidnitrogen. Formation of crystals in vitrified
165
VITRIFICATION OF PRONUCLEAR EMBRYOS
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 165
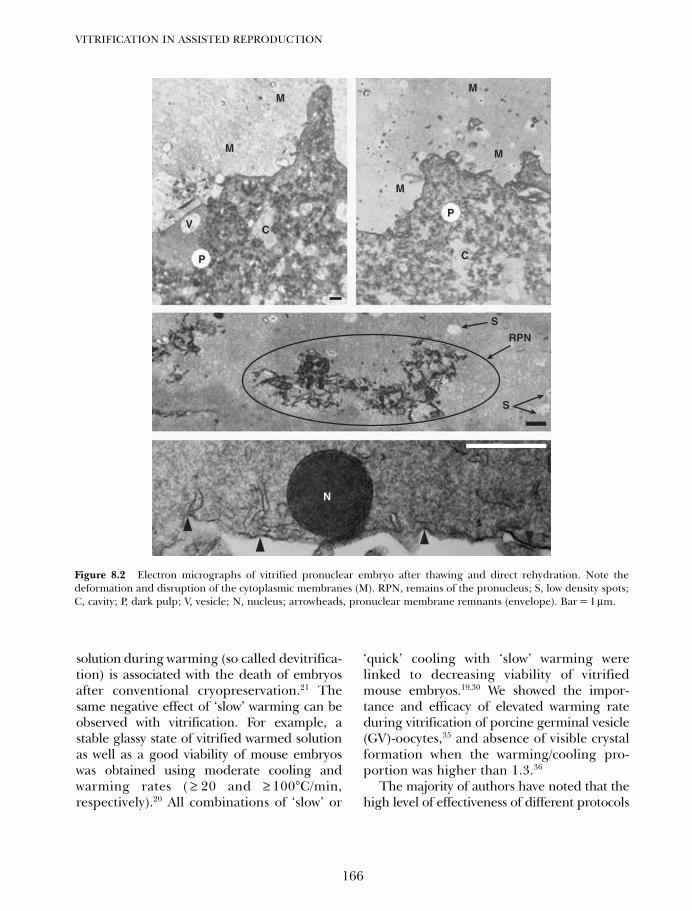
solution during warming (so called devitrifica-tion) is associated with the death of embryosafter conventional cryopreservation.21 Thesame negative effect of ‘slow’ warming can beobserved with vitrification. For example, astable glassy state of vitrified warmed solutionas well as a good viability of mouse embryoswas obtained using moderate cooling andwarming rates ( ≥ 20 and ≥ 100°C/min,respectively).20 All combinations of ‘slow’ or
‘quick’ cooling with ‘slow’ warming werelinked to decreasing viability of vitrifiedmouse embryos.19,30 We showed the impor-tance and efficacy of elevated warming rateduring vitrification of porcine germinal vesicle(GV)-oocytes,35 and absence of visible crystalformation when the warming/cooling pro-portion was higher than 1.3.36
The majority of authors have noted that thehigh level of effectiveness of different protocols
VITRIFICATION IN ASSISTED REPRODUCTION
166
MM
M
M
P
C
S
S
RPN
M
V
P
N
C
Figure 8.2 Electron micrographs of vitrified pronuclear embryo after thawing and direct rehydration. Note thedeformation and disruption of the cytoplasmic membranes (M). RPN, remains of the pronucleus; S, low density spots;C, cavity; P, dark pulp; V, vesicle; N, nucleus; arrowheads, pronuclear membrane remnants (envelope). Bar = 1 µm.
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 166

for the vitrification of oocytes and embryos withdecreased volumes of cooled medium, can beexplained by the combination of high speeds ofcooling and warming.22,23,33,37–40 The results ofour investigations with human oocytes do notsupport this point of view. For human GV-oocytes and pronuclear embryos a relativelyslow cooling rate in combination with rapidwarming is an efficient ‘conventional’ vitrifica-tion process with respect to survival rates andembryo development.41–43
We have shown that effective vitrification ofovine GV-oocytes is dependent on a combina-tion of high speed of cooling and warming.24
However, using the regimen ‘slow cooling –rapid warming’ we observed no developmentof oocytes but excellent expansion of cumulusof these oocytes, in contrast to ‘slow cooling –slow warming’ when oocytes as well ascumulus cells were dead after warming.24 Thisphenomenon can be explained by differencesof cryoproperties (cryostability) of oocytesand cumulus cells within of the same mam-malian species.
Vitrification of human embryos using finediameter plastic micropipettes, which werecooled in the vapor above liquid nitrogenbefore their placement into a precooledcryotube, was reported. Similarly to previousfindings, this methodology also includedthe parameter ‘slow cooling’ and ‘rapidwarming’.44
‘Slow’ cooling of biological objects can alsobe used for cooling of relatively large volumesof cooled solution. To prevent the 0.25 mLstraw from coming into direct contact withliquid nitrogen and eliminating the potentialcontamination risk associated with storagein liquid nitrogen, Kuleshova and Shaw45
reported about ‘straw-in-straw’ vitrification ofmouse embryos. A standard 0.25 mL strawwith column of vitrification medium waslocated into a 0.5 mL straw, which was her-metically closed before plunging into liquidnitrogen. However, this method, in contrastwith the one described in this chapter, does
not allow rapid warming of small volumesof vitrification medium with simultaneousremoval of cryoprotectant.
Our conclusion about the minor importanceof the speed of cooling for vitrifying oocytesand embryos, can probably be applied to allreproductive human cells including spermato-zoa. We recently reported that vitrification ofhuman spermatozoa by fast (20 000°C/min)or relatively slow (200°C/min) cooling withfast warming, results in similar post-thawcharacteristics.46
In many publications about vitrification ofcells in a small amount of vitrificationmedium, the described technique includesthe direct contact between liquid nitrogenand the cells. However, any other technologyin reproductive biology, and especially inmedicine, must ensure and guarantee full iso-lation of biological objects from microorgan-isms.47 Liquid nitrogen, which is used forstorage of frozen materials, can be a source ofcontamination by these microorganisms.47,48
Filtration or ultraviolet treatment of liquidnitrogen cannot guarantee the absence ofcontamination of biological materials byviruses including the HIV and mycoplasma.For example, the contamination of bloodprobes by hepatitis virus during the time ofstorage of probes in liquid nitrogen has beenreported.48 Different types of viruses, whichare simple and very cryostable structures, mayincrease their virulence after direct plungingand storage in liquid nitrogen: hepatitisvirus,49 papova (?) virus,50 vesicular stomatitisvirus,51 and herpes virus.52
The aim of our investigations was to testwhether the method of cryopreservation ofhuman pronuclear embryos in straws, whichare placed inside a hermetically closed con-tainer (larger straw), guarantees complete iso-lation of embryos from liquid nitrogen andavoids potential contamination by pathogenicmicroorganisms. We found that vitrificationof human pronuclear embryos (Figure 8.3)in open straws, which are placed inside a
167
VITRIFICATION OF PRONUCLEAR EMBRYOS
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 167
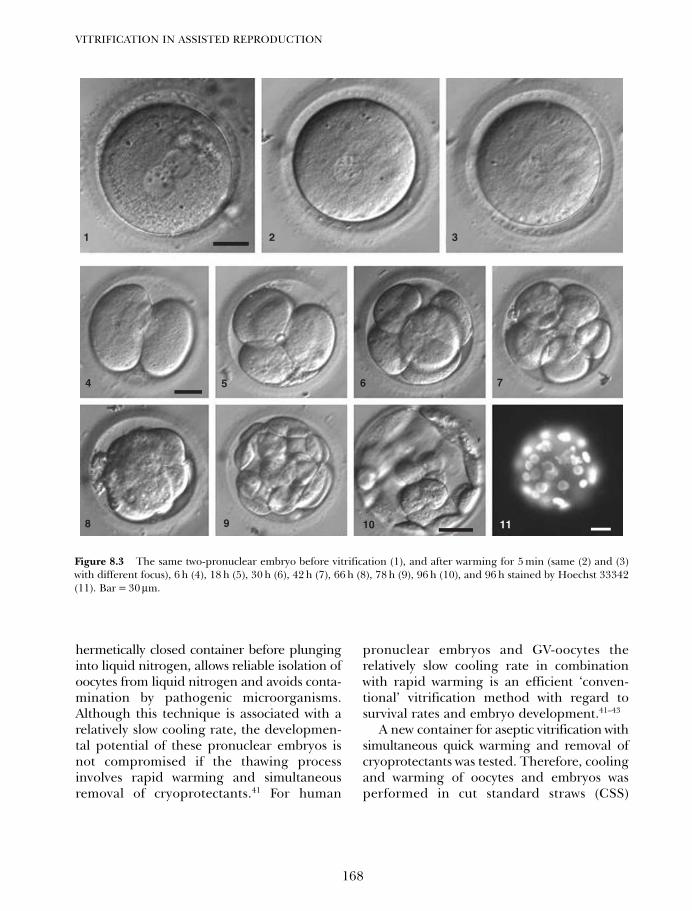
hermetically closed container before plunginginto liquid nitrogen, allows reliable isolation ofoocytes from liquid nitrogen and avoids conta-mination by pathogenic microorganisms.Although this technique is associated with arelatively slow cooling rate, the developmen-tal potential of these pronuclear embryos isnot compromised if the thawing processinvolves rapid warming and simultaneousremoval of cryoprotectants.41 For human
pronuclear embryos and GV-oocytes therelatively slow cooling rate in combinationwith rapid warming is an efficient ‘conven-tional’ vitrification method with regard tosurvival rates and embryo development.41–43
A new container for aseptic vitrification withsimultaneous quick warming and removal ofcryoprotectants was tested. Therefore, coolingand warming of oocytes and embryos wasperformed in cut standard straws (CSS)
VITRIFICATION IN ASSISTED REPRODUCTION
168
1 2 3
7
111098
654
Figure 8.3 The same two-pronuclear embryo before vitrification (1), and after warming for 5 min (same (2) and (3)with different focus), 6 h (4), 18 h (5), 30 h (6), 42 h (7), 66 h (8), 78 h (9), 96 h (10), and 96 h stained by Hoechst 33342(11). Bar = 30 µm.
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 168

(Figure 8.4). CSS were produced from standardinsemination 0.25 mL (so called French)straws, which are cut at an angle of approxi-mately 45°. Vitrification medium holding theembryo is placed onto the cut part of thestraw. After exposure to vitrification medium,one embryo with 0.25–0.75 µL of thismedium is aspirated into the tip of a pipettorand transferred to CSS (Figure 8.4). The CSSare loaded into 0.5 mL straws, which areclosed at both sides and plunged into liquidnitrogen with a cooling speed of 600°C/min.For rapid warming, the CSS is first removedfrom the larger straw, which is still half sub-merging in liquid nitrogen, prior to plunging
into sucrose solution (Figure 8.4). Thismethod allows for the simultaneous removalof cryoprotectant and rapid warming.
SHORT CONTACT OF CELLS WITHPERMEABLE CRYOPROTECTANTDIMETHYLSULFOXIDE ATDECREASED TEMPERATURE
The cryoprotectant DMSO is widely used forvitrification of human oocytes and embryos. Itwas reported that vitrification of blastocystsusing ethylene glycol and DMSO resulted in a33.3% pregnancy rate and the birth of ahealthy baby.53 Later, in the same clinic two
169
VITRIFICATION OF PRONUCLEAR EMBRYOS
2
431
2
2
2
Figure 8.4 Photographs and scheme of warming using the cut standard straw (CSS) container for vitrification: (1)closed 0.5 mL straw, (2) CSS, (3) vitrification medium with embryo, and (4) tube with solution for warming and removalof cryoprotectant.
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 169
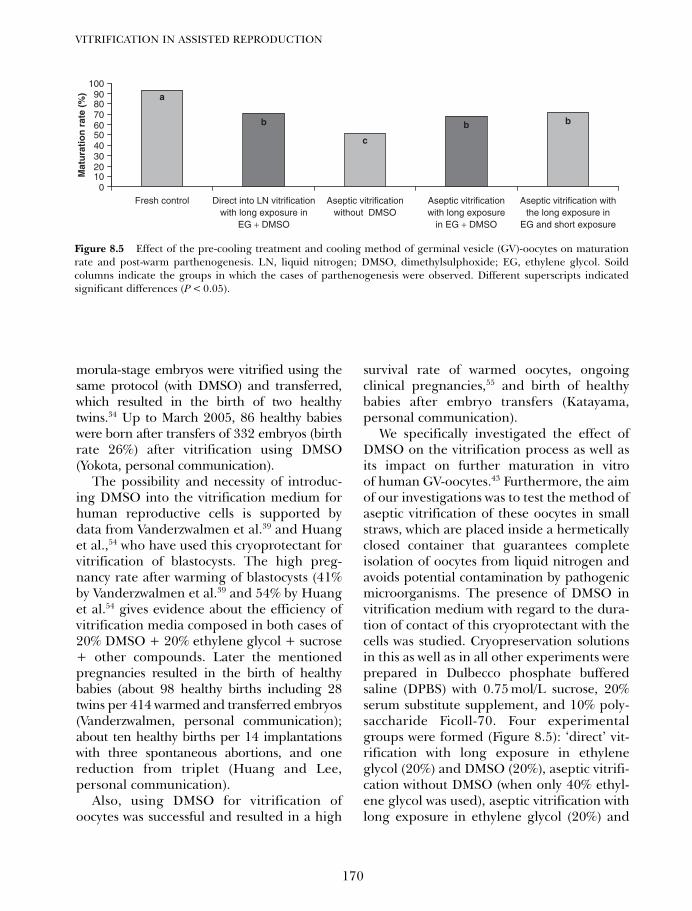
morula-stage embryos were vitrified using thesame protocol (with DMSO) and transferred,which resulted in the birth of two healthytwins.34 Up to March 2005, 86 healthy babieswere born after transfers of 332 embryos (birthrate 26%) after vitrification using DMSO(Yokota, personal communication).
The possibility and necessity of introduc-ing DMSO into the vitrification medium forhuman reproductive cells is supported bydata from Vanderzwalmen et al.39 and Huanget al.,54 who have used this cryoprotectant forvitrification of blastocysts. The high preg-nancy rate after warming of blastocysts (41%by Vanderzwalmen et al.39 and 54% by Huanget al.54 gives evidence about the efficiency ofvitrification media composed in both cases of20% DMSO + 20% ethylene glycol + sucrose+ other compounds. Later the mentionedpregnancies resulted in the birth of healthybabies (about 98 healthy births including 28twins per 414 warmed and transferred embryos(Vanderzwalmen, personal communication);about ten healthy births per 14 implantationswith three spontaneous abortions, and onereduction from triplet (Huang and Lee,personal communication).
Also, using DMSO for vitrification ofoocytes was successful and resulted in a high
survival rate of warmed oocytes, ongoingclinical pregnancies,55 and birth of healthybabies after embryo transfers (Katayama,personal communication).
We specifically investigated the effect ofDMSO on the vitrification process as well asits impact on further maturation in vitroof human GV-oocytes.43 Furthermore, the aimof our investigations was to test the method ofaseptic vitrification of these oocytes in smallstraws, which are placed inside a hermeticallyclosed container that guarantees completeisolation of oocytes from liquid nitrogen andavoids potential contamination by pathogenicmicroorganisms. The presence of DMSO invitrification medium with regard to the dura-tion of contact of this cryoprotectant with thecells was studied. Cryopreservation solutionsin this as well as in all other experiments wereprepared in Dulbecco phosphate bufferedsaline (DPBS) with 0.75 mol/L sucrose, 20%serum substitute supplement, and 10% poly-saccharide Ficoll-70. Four experimentalgroups were formed (Figure 8.5): ‘direct’ vit-rification with long exposure in ethyleneglycol (20%) and DMSO (20%), aseptic vitrifi-cation without DMSO (when only 40% ethyl-ene glycol was used), aseptic vitrification withlong exposure in ethylene glycol (20%) and
VITRIFICATION IN ASSISTED REPRODUCTION
170
0102030405060708090
100
Fresh control Direct into LN vitrificationwith long exposure in
EG + DMSO
Aseptic vitrificationwithout DMSO
Mat
ura
tio
n r
ate
(%)
Aseptic vitrificationwith long exposure
in EG + DMSO
Aseptic vitrification withthe long exposure in
EG and short exposure
a
b
c
b b
Figure 8.5 Effect of the pre-cooling treatment and cooling method of germinal vesicle (GV)-oocytes on maturationrate and post-warm parthenogenesis. LN, liquid nitrogen; DMSO, dimethylsulphoxide; EG, ethylene glycol. Soildcolumns indicate the groups in which the cases of parthenogenesis were observed. Different superscripts indicatedsignificant differences (P < 0.05).
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 170
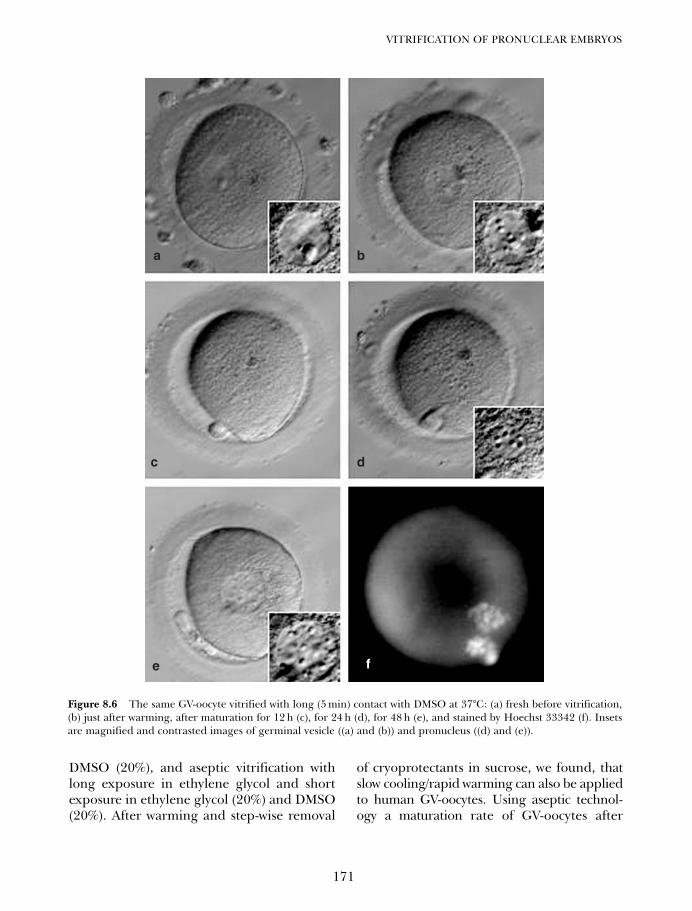
DMSO (20%), and aseptic vitrification withlong exposure in ethylene glycol and shortexposure in ethylene glycol (20%) and DMSO(20%). After warming and step-wise removal
of cryoprotectants in sucrose, we found, thatslow cooling/rapid warming can also be appliedto human GV-oocytes. Using aseptic technol-ogy a maturation rate of GV-oocytes after
171
VITRIFICATION OF PRONUCLEAR EMBRYOS
a b
d
fe
c
Figure 8.6 The same GV-oocyte vitrified with long (5 min) contact with DMSO at 37°C: (a) fresh before vitrification,(b) just after warming, after maturation for 12 h (c), for 24 h (d), for 48 h (e), and stained by Hoechst 33342 (f). Insetsare magnified and contrasted images of germinal vesicle ((a) and (b)) and pronucleus ((d) and (e)).
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 171

vitrification of approximately 85% wasobserved. The short-term presence of DMSOat room temperature in the vitrificationmedium can improve the maturation rate ofGV-oocytes and does not cause spontaneousparthenogenesis.43
In one experiment we studied the possibil-ity of using a vitrification solution which wascompletely free of DMSO. One reason to doso was that DMSO was reported to affect theorganization of microfilaments in mouseoocytes,56 as well as to induce chromosomalabnormalities (increase in the rates of degen-eration and digynic polyploid embryos) aftercryopreservation of mouse oocytes in thepresence of DMSO.57
However, in our experiments the absenceof DMSO in the vitrification medium causeda significant decrease in maturation rates. Onthe other hand, a 5 min contact of oocyteswith DMSO at 37°C was enough to causespontaneous oocyte activation and partheno-genetic development. The protocol which wefound to be recommendable for use in med-ical practice is a compromise between thepresence of DMSO in vitrification mediumand parthenogenetic activation. Based on ourresults we propose to use short (1 min) con-tact with DMSO which is only included in thefinal portion of vitrification medium, andcontact with DMSO at room temperature thattheoretically can decrease the negative effecton oocytes. The high maturation rates andthe absence of parthenogenesis are a directproof that this way of using DMSO (decreas-ing time and temperature of contact) is suit-able for vitrification of human oocytes andembryos.
USE OF ASEPTIC TECHNOLOGYFOR LATE HUMAN EMBRYOS ANDPRONUCLEAR EMBRYOS OFOTHER SPECIES
In most human in vitro fertilization programsembryo transfer is routinely performed on
day 2 or 3. However, implantation rates arerelatively low.58 One attempt to increase thisrate might be the prolonged culture ofhuman embryos up to the blastocyst stageand to select the best embryos at that stagefor transfer. To achieve this goal, sequentialculture media were developed which fulfillthe differential requirements of the embryoduring early preimplantation development.59
Consequently, these media enabled blastocystformation rates of up to 50% and due toextended culture those embryos with the bestdevelopmental potential were recognizedmore readily on day 5.60 Based on thisapproach, several studies reported higherimplantation rates following transfer ofselected blastocysts.
Comparative investigations on freezingand vitrification of human late blastocystswere performed. Liebermann and Tucker61
have evaluated implantation of day 5 and day6 vitrified and conventionally (slowly) frozenblastocysts. Day 5 and day 6 blastocysts werevitrified or frozen and transferred afterwarming or thawing. In 508 transfer cycles,embryonic implantation rates for day 5 andday 6 vitrified blastocysts were 33% and 26%,respectively, and after conventional freezingwere 30% and 28%, respectively.
Blastocysts with laser opening of zonapellucida were the object of our investiga-tions. They are more difficult to cryopreservethan early blastocysts or blastocysts withintact zona pellucida for two reasons. It wasreported that the survival rate of expandedblastocysts after vitrification is relatively low.62
These authors explain this fact by the pres-ence of a large volume of water in the blasto-coel which, in addition, can be crystallized oncooling and these crystals can destroy theembryo. Also, the laser opening of the zonapellucida decreases the cryostability of blasto-cysts and reports about vitrification of thistype of blastocyst are limited.63
The aim of this study was to compare theviability of vitrified human blastocysts, usingdecreased concentrations of cryoprotectants,
VITRIFICATION IN ASSISTED REPRODUCTION
172
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 172

by direct submerging of cut standard straws(CSS) into liquid nitrogen, with that aftervitrification by cooling of CSS loaded insidea 0.5 mL straw (aseptic system). Six-day blas-tocysts were obtained after culture of pro-nuclear embryos and laser assisted hatching.These blastocysts were cryopreserved in CSSby vitrification in ethylene glycol + DMSO +0.3 mol/L sucrose. In the first experiment,embryos were vitrified using vitrificationmedium with 20% ethylene glycol + 20%DMSO and 15% ethylene glygol + 15%DMSO. In the second experiment, when vit-rification medium included 15% ethyleneglycol + 15% DMSO + sucrose, embryoswere cooled rapidly and slowly (with isolationfrom liquid nitrogen). Embryos in bothexperiments (four groups) were rapidlythawed at a speed of 30 000– 85 000°C/minusing an identical protocol. Embryos of allexperimental groups had the same rate of re-expansion (80–85%) after in vitro culture(Figure 8.7). It was noted that the methodinvolving vitrification of expanded blasto-cysts with decreased concentrations of per-meable cryoprotectants and completeisolation of embryos from liquid nitrogen inCSS is efficient.
The described aseptic technology can beapplied to pronuclear embryos of otherspecies including mouse. Pronuclear mouseembryos attracted special interest for cryo-investigation for two reasons. First, thisstage is used for production of transgenicmouse, which is a promising model forpresent and future biological investigation.Second, in some European countries, cryo-preservation of zygotes after fusion ofpronuclei or later stage embryos is prohib-ited. For these two reasons, cryopreserva-tion of mouse pronuclear embryos by directplunging into liquid nitrogen is a goodmodel for investigation.
The aim of our investigations was to testwhether the method of cryopreservation ofbiopsied mouse pronuclear embryos insmall straws, which are placed inside an
hermetically closed container, guaranteescomplete isolation of oocytes from liquidnitrogen and avoids potential contaminationby pathogenic microorganisms. It was shownthat the developmental potential of mousepronuclear oocytes is not compromised if thethawing process involves rapid warming andsimultaneous removal of cryoprotectants.42
Aseptic vitrification can be applied tohuman pronuclear embryos and oocytes aswell as for embryos of any developmentalstage; the possibility of using this technologyfor other mammalian species is promising.
Recentely, several reports about vitrificationof human embryos using cooling with isola-tion from liquid nitrogen were published.Kuwayama et al.64 performed 16 000 humanembryo cryopreservations and proved thatvitrification is a good alternative to conven-tional freezing and resulted in high survivaland in vitro developmental rates for pro-nuclear, multicellular, and blastocyst stagehuman embryos. The authors described a vit-rification method that eliminates the risk ofcontamination of oocytes and embryos inliquid nitrogen.64 This method includes isola-tion of the vitrification medium from liquidnitrogen at cooling. Vanderzwalmen et al.65
proposed the method of small volume vitrifi-cation in hemi-straws, tested two methods ofcooling 3- and 5-day embryos using directplunging into liquid nitrogen (non-asepticconditions) and cooling with isolation fromliquid nitrogen (aseptic conditions). Theresults show that post-thaw survival rates ofmorulae and blastocysts vitrified at a lowercooling rate inside a hermetic sealed straware comparable with those of the control non-aseptic group.
Recently, it was shown that the previouslyreported method of vitrification usingCryoloopTM can be used to vitrify and storemouse embryos without direct liquid nitrogencontact (during cooling and storage). Whensuch vitrified embryos are warmed, they arecapable of subsequent development compa-rable with that of non-vitrified embryos.66
173
VITRIFICATION OF PRONUCLEAR EMBRYOS
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 173
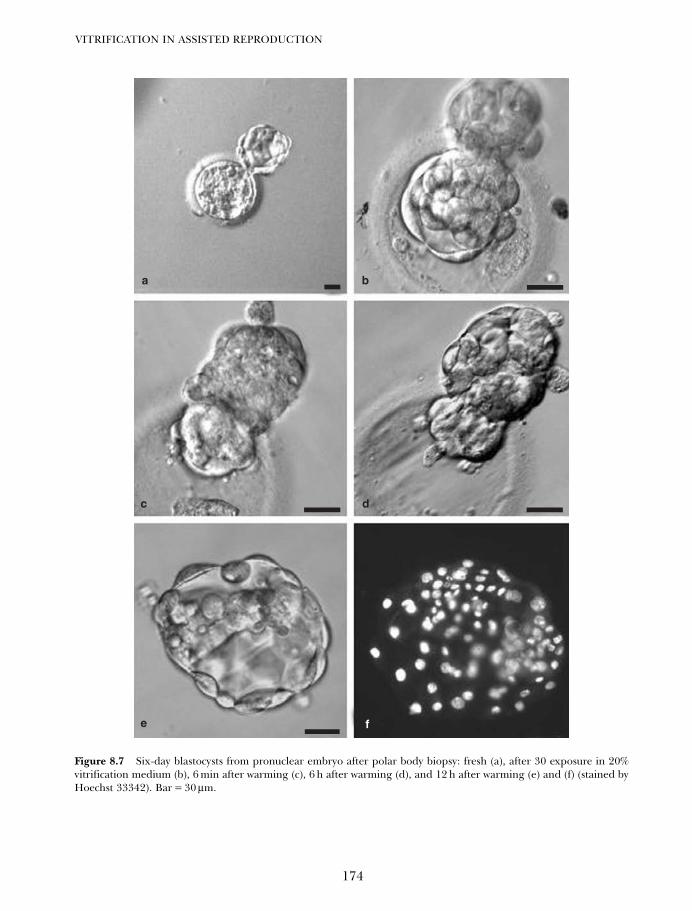
VITRIFICATION IN ASSISTED REPRODUCTION
174
a b
dc
e f
Figure 8.7 Six-day blastocysts from pronuclear embryo after polar body biopsy: fresh (a), after 30 exposure in 20%vitrification medium (b), 6 min after warming (c), 6 h after warming (d), and 12 h after warming (e) and (f) (stained byHoechst 33342). Bar = 30 µm.
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 174

We believe that these investigations ofaseptic vitrification arose following our ownresults.11–13,26,27,41,43
DIFFERENCES WITH HUMAN ANDANIMAL PRONUCLEAR EMBRYOSDURING CRYOPRESERVATION:INTRACELLULAR LIPIDS
As a rule, cryoinvestigations in humanembryos are based on successfully used proto-cols emerging from similar experiments per-formed on embryos from laboratory animals.Before it was possible to vitrify human pro-nuclear embryos, animal embryos at thisstage had been successfully cryopreservedusing the method of direct plunging into liq-uid nitrogen, and an effective protocol for vit-rification of mouse pronuclear embryos wasdeveloped. These data proved useful indeveloping protocols for the vitrification ofhuman pronuclear embryos.
Aseptic technology of vitrification ofhuman oocytes and embryos can be success-fully used for cryopreservation of mammalianoocytes of other species. However, it is neces-sary to take into account that human cellshave their own peculiarities. One suchattribute is the presence of intracellularlipids.
Intracellular lipids are a ‘stumbling block’for cryopreservation including vitrification.Data demonstrating the role of these intracel-lular structures during cryopreservation havebeen published.67 The method proposedinvolves polarization and removal of cytoplas-mic lipids from oocytes or embryos before vit-rification. Nagashima et al.67 were the first tosuccessfully grow embryos from GV-porcineoocytes that were vitrified following delipid-ization. Using this method the authorsavoided negative effects caused by cooledintracellular lipids. According to the dataprovided by the authors, removal of intra-cellular lipids does not lead to a worsening of
further development of oocytes and embryos.Successful oocyte vitrification after removal ofcytoplasmic lipids leads to the question ofpossible changes in the physiochemical prop-erties of cytoplasmic membrane lipids arisingat low temperatures68 which were discussed asa significant cause of cryobiological problemsduring experiments.
We believe that it is impossible to dismissclassic data about the role of intracellularlipids as energetic materials of oocytes andbuilding materials for membranes of futureembryos. The fact that the volume of mito-chondria as well as lipid vesicles increasesduring oocyte development to metaphase II(MII) stage69 indirectly confirms this point.Moreover, Sathananthan et al.70 have shownthat in the cell complex called ‘smooth endo-plasmic reticulum–lipid globules–mitochondria’reticulum–globules–mitochondria connectionsdo exist. They have also shown that theseconnections may be damaged after oocytecooling or freezing.
In the overwhelming majority of workstudying the effect of cooling on mammalianoocytes and embryos, a negative cryo-influ-ence is explained in terms of the effect oncytoskeletal elements. For example, cooling ofhuman oocytes causes depolymerization ofcytoskeletal protein structures and most mouseoocytes cooled to 25°C for 10 min had anabnormal cytoskeleton.71 After exposure to 4°Cfor 20 min, completely disassembled spindleswere observed. This process of depolymeriza-tion is, however, reversible. Spindles of mouseoocytes returned to normal appearance afterwarming at 37°C for 60 min. Spindles of abouthalf of human oocytes exposed to roomtemperature for 10 min returned to normalafter 4 h of cultivation at 37°C.71 The negativeeffect of cooling is also explained as depoly-merization of microtubules and microfila-ments in other works performed on humanoocytes. Bovine oocytes are also sensitive todecreasing temperatures.72 It has been shown
175
VITRIFICATION OF PRONUCLEAR EMBRYOS
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 175

that 56% of oocytes exposed to 25°C and 90% ofoocytes exposed to 4°C for 1 min had abnormalspindles.
Data on the sensitivity of porcine oocytes,which probably are a more ‘difficult’ entity tocryopreserve at low temperatures, are limited.Didion et al.73 who examined the viability ofpig GV-oocytes following cooling or freezingby conventional methods found that thecumulus-intact GV-porcine oocytes do notsurvive cooling to temperatures at or below15°C. As the authors noted, this was notsurprising considering that porcine embryosfrom the 8-cell to blastocyst stage were killedwhen cooled below 15°C.
Publications on problems of mammalianoocyte and embryos cryopreservation containinformation on the negative effects of low tem-perature including the cytoskeletal depolymer-ization by permeable cryoprotectants.74
We suppose that the negative effect ofcooling on oocytes can be explained by theeffect of cooling lipids on cytoskeletal struc-tures. Whilst performing our investigationson porcine oocytes, we found that followingcentrifugation, redistribution of lipids occurswithin 48 h of in vitro culture in oocytes notexposed to vitrification/warming (data notpublished). However, when polarized oocytesare vitrified/warmed, the lipid polarizationis irreversible. This, in our opinion, suggeststhat the vitrification/warming process inducesan alteration to the physiochemical propertiesof intracellular lipids.
It is known that MII oocytes are more resis-tant to freeze damage than GV-stage oocytes.We consider that this may be due to differencesin the properties of cytoskeletal elements. Oneimportant difference is that the configurationof microtubules and microfilaments is differ-ent at these two stages of oocyte maturation.Cytoskeletal elements in GV-oocytes appearstraight and rigid, while the appearance ofmicrofilaments and microtubules in MII stageoocytes is undulating and flexible. Based onthe hypothesis of a complex interactionbetween the lipid phase of cells and the
elements of the cytoskeleton, hardening ofthese lipids might cause deformation and dis-ruption of the cytoskeleton. In the case of therigid GV-oocyte cytoskeleton this apparentlyresults in permanent damage while in themore flexible MII-oocyte cytoskeleton, per-manent damage is absent. Cytochalasins havea specific, reversible effect on cytoskeletalelements making them more flexible and lesssusceptible to the effect from cooled lipids.Testing this substance for vitrification of GV-porcine oocytes in combination with elevatedtemperature was effective.35 For the successfulvitrification of GV-porcine oocytes,35 the fol-lowing points could be important. An optimalprotocol of vitrification must prevent thealterations of the physiochemical propertiesof cooled lipids; avoid irreversible damage tothe lipid globules membranes; and protectthe cooling reticulum-lipids connection fromdestruction. Further investigations areneeded to verify these assumptions.
It is known that bovine oocytes are to a con-siderable extent more cryostable than porcineoocytes. There is also information suggestingthat the diameters of bovine and porcineintracellular lipid vesicles are different. Thecharacteristics of the intracellular lipid gran-ule membranes are also a topic of discussion.
We compared the ultrastructure of lipiddroplets, and the effect of cooling on intra-cellular lipid vesicles of bovine and porcineGV-oocytes.75 It was shown that lipid dropletsof bovine GV-oocytes have a homogenousstructure. The utilization of lipids takes placedirectly from these vesicles without forma-tion of interim lipid compounds. In contrast,there are two kinds of lipid droplets inporcine GV-oocytes: ‘dark’ and homogenousvesicles next to ‘gray’ vesicles with electron-lucent streaks. Vesicles of each specific groupare connected to each other. After 12 hourculture, the formation of the cisternalsmooth endoplasmic reticulum layer isalways associated with ‘gray’ lipid vesicles.This is evidence that during oogenesis lipo-lysis takes place only in ‘gray’ vesicles. It is
VITRIFICATION IN ASSISTED REPRODUCTION
176
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 176
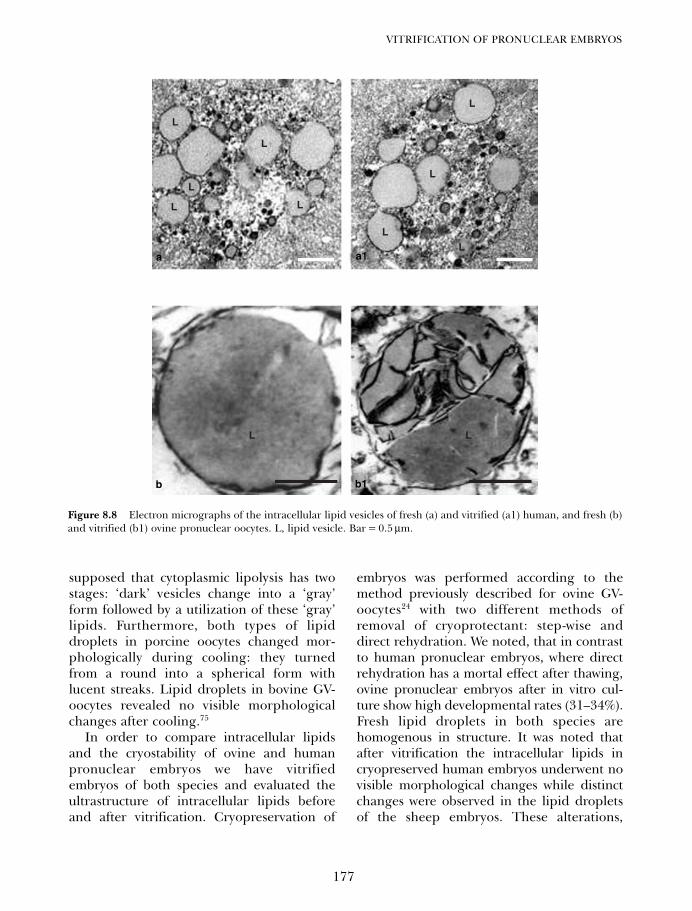
supposed that cytoplasmic lipolysis has twostages: ‘dark’ vesicles change into a ‘gray’form followed by a utilization of these ‘gray’lipids. Furthermore, both types of lipiddroplets in porcine oocytes changed mor-phologically during cooling: they turnedfrom a round into a spherical form withlucent streaks. Lipid droplets in bovine GV-oocytes revealed no visible morphologicalchanges after cooling.75
In order to compare intracellular lipidsand the cryostability of ovine and humanpronuclear embryos we have vitrifiedembryos of both species and evaluated theultrastructure of intracellular lipids beforeand after vitrification. Cryopreservation of
embryos was performed according to themethod previously described for ovine GV-oocytes24 with two different methods ofremoval of cryoprotectant: step-wise anddirect rehydration. We noted, that in contrastto human pronuclear embryos, where directrehydration has a mortal effect after thawing,ovine pronuclear embryos after in vitro cul-ture show high developmental rates (31–34%).Fresh lipid droplets in both species arehomogenous in structure. It was noted thatafter vitrification the intracellular lipids incryopreserved human embryos underwent novisible morphological changes while distinctchanges were observed in the lipid dropletsof the sheep embryos. These alterations,
177
VITRIFICATION OF PRONUCLEAR EMBRYOS
L
L
L
L
L
L
L
L
L
LL
Figure 8.8 Electron micrographs of the intracellular lipid vesicles of fresh (a) and vitrified (a1) human, and fresh (b)and vitrified (b1) ovine pronuclear oocytes. L, lipid vesicle. Bar = 0.5 µm.
a a1
b b1
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 177

attributable to the vitrification process, reflectchanges in the physical and chemical proper-ties of the lipids such as hardening. Figure 8.8shows intracellular lipid droplets in bothhuman and sheep pronuclear embryos andillustrates the differences between the intra-cellular lipids.
The protocols used for vitrifying humanpronuclear embryos are based on methodsdeveloped for animal embryos at this stage.In animal models, vitrification may some-times be followed by effective post-thawrehydration, without the need for step-wisedilution of permeable cryoprotectants inhypertonic solution. Thus, the vitrified andthawed cells are directly plunged into iso-tonic (culture) medium. In our investigationswe used a vitrification/direct post-warm rehy-dration protocol that had been previouslytested in our laboratory on GV-ovineoocytes24 and, after slight modification, inrat early morulae, early blastocysts, andexpanded blastocysts.25 With the subsequentstep-wise dilution of cryoprotectants, thesame vitrification protocol was found to behighly efficient for human pronuclearembryos yet was completely ineffective whenthese human embryos were directly rehy-drated after warming.
Thus, it appears that the ideal protocol forvitrification of pronuclear embryos withdirect rehydration may have to be a compro-mise between the two conditions of maximalbinding of intracellular water by permeablecryoprotectants and the presence of minimalpost-thaw quantities of these cryoprotectantsin the cytoplasm. Prolonged exposure to thecryoprotectant seems to require extra-rehydration due to the increased quantity ofintracellular cryoprotectant. In contrast, aninsufficient amount of intracellular cryopro-tectant during pre-cooling exposure mayresult in insufficient binding of intracellularwater. The step-wise dilution procedure that weused after warming indicates that both expo-sure times to the permeable cryoprotectant aresufficient for intracellular water binding. We,
therefore, also tested the two time periods of10 s or 1 min exposure in vitrification solutionwhen the vitrified embryos were directly rehy-drated after thawing. All attempts to vitrifyhuman pronuclear oocytes with direct rehy-dration were unsuccessful.
We suggest that the specificity of humanpronuclear embryos, reflected by their highsensitivity to osmotic processes, is related to thespecificity of both intracellular lipids and cyto-plasmic and organelle membranes. Lipids arethe most cryo-labile intracellular compoundsof oocytes and embryos. Indeed, the specificnature of intracellular lipids in pig oocytesmakes them practically unsuitable for cryo-preservation, particularly vitrification. Figure8.8 shows the appearance of intracellular lipidsof fresh human pronuclear embryos before andafter vitrification in which no changes can beobserved. We compared these lipids with thoseof pronuclear ovine embryos subjected to thesame protocol which was used for humanoocytes with direct rehydration in our previousinvestigations. These lipids of viable embryosshowed ultrastructural changes after vitrifica-tion not noted in the human oocytes (Figure8.8). Bearing in mind the resistance of ovinepronuclear embryos to direct post-thaw rehy-dration, we were able to observe hardening(increase in density) and morphological alter-ations in the intracellular lipids of all cooledovine oocytes. These alterations were absent inall human oocytes, which were clearly unable totolerate direct rehydration. It may be assumedthat within the same cell, the structure of intra-cellular and membrane lipids are the same.Given the detrimental role of lipids duringcryopreservation, the lipid cryostability yetosmotic instability of pronuclear humanembryos is still far from being fully understood.
Direct post-thaw rehydration induceslethal osmotic effects in human pronuclearoocytes but is successful for ovine oocytes. Acorrelation between the cryostability ofmammalian oocytes and the ultrastructureof intracellular lipids is proposed for fur-ther investigations. Taking into account that
VITRIFICATION IN ASSISTED REPRODUCTION
178
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 178

the ultrastructure of intracellular lipids oflaboratory and agricultural animals aredifferent from human intracellular lipids, webelieve that using animal oocytes as a modelfor human oocytes in cryo-investigations isquestionable.
In conclusion, aseptic vitrification can besuccessfully applied to human pronuclearembryos, oocytes, and embryos of any devel-opmental stage; the possibility of using thistechnology for other mammalian species ispromising.
179
VITRIFICATION OF PRONUCLEAR EMBRYOS
References
1. Porcu E, Fabbri R, Seracchioli et al. Birth of ahealthy female after intracytoplasmic sperminjection of cryopreserved human oocytes.Fertil Steril 1997; 68: 724–6.
2. Mohr LR, Trounson AO. Cryopreservation ofhuman embryos. Ann N Y Acad Sci 1985; 442:536–43.
3. Siebzehnrübl ER. Cryopreservation ofgametes and cleavage stage embryos. HumReprod 1989; 4: 105–10.
4. Al-Hasani S, Demirel LC, Schöpper B et al.Pregnancies achieved after frozen-thawedpronuclear oocytes obtained by intracyto-plasmic sperm injection with spermatozoaextracted from frozen-thawed testicular tis-sues from non-obstructive azoospermic men.Hum Reprod 1999; 14: 2031–5.
5. Park SP, Kim EY, Oh JH et al. Ultra-rapidfreezing of human multipronuclear zygotesusing electron microscope grids. Hum Reprod2000; 15: 1787–90.
6. Liebermann J, Tucker MJ, Graham JR, Han T,Davis A, Levy MJ. Blastocyst developmentafter vitrification of multipronuclear zygotesusing the Flexipet denuding pipette. ReprodBiomed Online 2002; 4: 148–52.
7. Liebermann J, Nawroth F, Isachenko V,Isachenko E, Rahimi G, Tucker M. Potentialimportance of vitrification in reproductivemedicine. Biol Reprod 2002; 67: 1671–80.
8. Liebermann J, Tucker MJ. Effect of carriersystem on the yield of human oocytes andembryos as assessed by survival and develop-mental potential after vitrification. Reproduction2002; 124: 483–9.
9. Selman HA, El-Danasouri I. Pregnanciesderived from vitrified human zygotes. FertilSteril 2002; 77: 422–3.
10. Jelinkova L, Selman HA, Arav A, Strehler E,Reeka N, Sterzik K. Twin pregnancy after vit-rification of 2-pronuclei human embryos.Fertil Steril 2002; 77: 412–4.
11. Isachenko V, Selman H, Isachenko E, MontagM, Al-Danasuri I, Nawroth F. Modified vitrifi-cation of human pronuclear oocytes: efficacyand effect on ultrastructure. Reprod BiomedOnline 2003; 7: 211–6.
12. Isachenko V, Selman H, Isachenko E, Montag M,El-Danasouri I, Nawroth F. Effect of cryo-protectants on the ultrastructure of cooled humanpronuclear oocytes. Fertil Steril 2004; 81: 720–2.
13. Isachenko V, Montag M, Isachenko E, NawrothF, Dessole S, Van der Ven H. Developmentalrate and ultrastructure of vitrified humanpronuclear oocytes after step-wise versus directrehydration. Hum Reprod 2004; 19: 660–5.
14. Papis K, Shimizu M, Izaike Y. Factors affectingthe survivability of bovine oocytes vitrified indroplets. Theriogenology 2000; 54: 651–8.
15. Isachenko V, Isachenko E, Ostashko F,Grishchenko V. Ultra-rapid freezing of ratembryos with rapid dilution of permeable cryo-protectants. Cryobiology 1997; 34: 157–64.
16. Isachenko V, Alabart JL, Isachenko E,Michelmann HW, Bezugly N. Ultra-rapidfreezing and storage of rat embryos in an elec-tric refrigerator at –130°C without liquid cryo-agents with ultra-short exposure in thefreezing medium and direct rehydration afterthawing. Cryo Letters 2000; 21: 13–8.
17. Sathananthan AH, Trounson A, Freemann L,Brady T. The effects of cooling humanoocytes. Hum Reprod 1988; 3: 968–77.
18. Fuku E, Xia L, Downey BR. Ultrastructuralchanges in bovine oocytes cryopreserved byvitrification. Cryobiology. 1995; 32: 139–56.
19. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196 °C by vitrification.Nature 1985; 313: 573–5.
20. Rall WF. Factors affecting the survival of mouseembryos cryopreserved by vitrification. Cryo-biology 1987; 24: 387–402.
21. Rall WF, Reid DS, Polge C. Analysis of slow-warming injury of mouse embryos by cryo-
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 179

microscopical and physiochemical methods.Cryobiology 1984; 21: 106–21.
22. Martino A, Songansen N, Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
23. Vajta G, Kuwayama M, Holm P, Booth PJ,Jacobsen H, Greve T et al. Open pulled straw(OPS) vitrification: a new way to reduce cryo-injuries of bovine ova and embryos. MolReprod Dev 1998; 51: 53–8.
24. Isachenko V, Alabart JL, Nawroth F, IsachenkoE, Vajta G and Folch J. The open pulled strawvitrification of ovine GV-oocytes: positiveeffect of rapid cooling or rapid thawing orboth? Cryo Letters 2001; 22: 157–62 .
25. Isachenko V, Folch J, Nawroth F,Krivokharchenko A, Vajta G, Dattena M, et al.Double vitrification of rat embryos at differentdevelopmental stages using an identificationprotocol. Theriogenology 2003; 60: 445–52.
26. Isachenko V, Selman H, Isachenko E, MontagM, El-Danasouri I, Nawroth F. Modified vitri-fication of human pronuclear oocytes: efficacyand effect on ultrastructure. Reprod BiomedOnline 2003; 7: 211–6.
27. Isachenko V, Montag M, Isachenko E,Nawroth, Dessole S, Van der Ven H. Devel-opmental rate and ultrastructure of vitrifiedhuman pronuclear oocytes after step-wiseversus direct rehydration. Hum Reprod 2004;19: 660–5.
28. Szell A, Zhang J, Hudson R. Rapid cryo-preservation of sheep embryos by direct trans-fer into liquid nitrogen vapour at –180°C.Reprod Fertil Dev 1990; 2: 613–8.
29. Rall WF. Advances in the cryopreservation ofembryos and prospects for application to theconservation of salmonid fishes. In: Cloud JG,Thorgaard GH, eds. Genetic Conservation ofSalmonid Fishes. New York: Plenum Press,1993: 137–58.
30. Rall WF, Wood MJ. High in vitro and in vivosurvival of day 3 mouse embryos vitrified in anon-toxic solution of glycerol and albumin.J Reprod Fertil 1994; 101: 681–8.
31. Dinnyes A, Dai Y, Jiang S, Yang X. High devel-opmental rates of vitrified bovine oocytesfollowing parthenogenetic activation, in vitrofertilization, and somatic cell nuclear transfer.Biol Reprod 2000; 63: 513–8.
32. Vajta G, Holm P, Greve T, Callesen. Factorsaffecting survival rates of in vitro producedbovine embryos after vitrification and directin-straw rehydration. Anim Reprod Sci 1996;45: 191–200.
33. Mukaida T, Nakamura S, Tomiyama T et al.Wada S, Oka C, Kasai M et al. Vitrification ofhuman blastocysts using cryoloops: clinicaloutcome of 223 cycles. Hum Reprod 2003; 18:384–91.
34. Yokota Y, Yokota H, Yokota M, Sato S, Araki Y.Birth of healthy twins from in vitro develop-ment of human refrozen embryos Fertil Steril2001; 76: 1063–5.
35. Isachenko V, Perez-Sanchez F, Isachenko E,Grishchenko V, Soler C. Vitrification of GV-porcine oocytes with intact intracellular lipids:effect of the cryoprotectant saturation/dilutionstepping, elevated temperature and cytoskele-tal inhibitor. Cryobiology 1998; 36: 250–3.
36. Isachenko V, Gorbunov L, Isachenko E,Ostashko F, Bezugly N. Some physical andtechnological aspects of GV-porcine oocytevitrification. Cryobiology 1999; 39: 35.
37. Lane M, Bavister BD, Lyons EA, Forest KT.Containerless vitrification of mammalianoocytes. Nat Biotechnol 1999; 17: 1234–6.
38. Lane M, Gardner DK. Vitrification of mouseoocytes using a nylon loop. Mol Reprod Dev2001; 58: 342–7.
39. Vanderzwalmen P, Bertin G, Debauche Ch,Standaert V, Bollen N, Rosendaal van E et al.Vitrification of human blastocysts with hemi-straw carrier: application of assisted hatchingafter thawing. Hum Reprod 2003; 18: 1504–11.
40. Son WY, Yoon SH, Yoon HJ, Lee SM, Lim JH.Pregnancy outcome following transfer ofhuman blastocysts vitrified on electronmicroscopy grids after induced collapse of theblastocoele. Hum Reprod 2003; 18: 137–9.
41. Isachenko V, Montag M, Isachenko E, Zaeva V,Krivokharchenko I, Shafei R, et al. Aseptictechnology of vitrification of human pronu-clear oocytes using open-pulled straws. HumReprod 2005; 20: 492–6.
42. Isachenko V, Montag M, Isachenko E, Van derVen H. Vitrification of mouse pronuclearembryos after polar body biopsy withoutdirect contact with liquid nitrogen. FertilSteril 2005; 84: 1011–6.
43. Isachenko V, Montag M, Isachenko E, DessoleS, Nawroth F, Van der Ven H. Aseptic vitrifica-tion of human germinal vesicle oocytes usingdimethyl sulfoxide as a cryoprotectant FertilSteril 2006; 85: 741–7.
44. Cremades N, Sousa M, Silva J, Viana P,Sousa S, Olivera C, et al. Experimental vitri-fication of human compacted morulae andearly blastocysts using fine diameter plasticmicropipettes. Hum Reprod 2004; 19:300–5.
VITRIFICATION IN ASSISTED REPRODUCTION
180
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 180

45. Kuleshova LL, Shaw JM. A strategy for rapidcooling of mouse embryos within a doublestraw to eliminate the risk of contaminationduring storage in liquid nitrogen. HumReprod 2000; 15; 2604–9.
46. Isachenko V, Isachenko E, Katkov II, MontagM, Dessole S, Nawroth F, et al. Cryoprotectant-free cryopreservation of human spermatozoaby vitrification and freezing in vapor: effect onmotility, DNA integrity, and fertilization abil-ity. Biol Reprod 2004; 71: 1167–73.
47. Bielanski A, Nadin-Davis S, Sapp T, Lutze-Wallace C. Viral contamination of embryoscryopreserved in liquid nitrogen. Cryobiology2000; 40: 110–16.
48. Tedder RS. Hepatitis B nucleotide sequenceanalysis: linking an outbreak of acute hepati-tis B to contamination of a cryopreservationtank. J Virol Methods 1996; 60: 818.
49. Hawkins AE, Zuckerman MA, Briggs M,Gilson RJ, Goldstone AH, Brink NS et al.Hepatitis B nucleotide sequence analysis:linking an outbreak of acute hepatitis B tocontamination of a cryopreservation tank. JVirol Methods 1996; 60: 81–8.
50. Charles GN, Sire DJ. Transmission of papovavirus by cryotherapy applicator. JAMA 1971;218: 1435.
51. Schaffer TW, Everett J, Silver GH, Came PE.Biohazard potential: recovery of infectiousvirus from the liquid nitrogen of a virus repos-itory. Health Lab Sci. 1976; 13: 23–4.
52. Jones SK, Darville JM. Transmission of virus-particles by cryo-therapy and multi-use causticpencils: a problem to dermatologist? Br JDermatol 1989; 121: 481–6.
53. Yokota Y, Sato S, Yokota M, Araki Y. Birth of ahealthy baby following vitrification of humanblastocysts. Fertil Steril 2001; 75: 1027–9.
54. Huang CC, Lee TH, Chen SU, Chen HH,Cheng TC, Liu CH et al. Successful pregnancyfollowing blastocyst cryopreservation usingsuper-cooling ultra-rapid vitrification. HumReprod 2005; 20: 122–8.
55. Katayama KP, Stehlik J, Kuwayama M, KatoO, Stehlik E. High survival rate of vitrifiedhuman oocytes results in clinical pregnancy.Fertil Steril 2003; 80: 223–4.
56. Vincent C, Pickering SJ, Johnson MH, QuickSJ. Dimethylsulphoxide affects the organisa-tion of microfilaments in the mouse oocytes.Mol Reprod Dev 1990; 26: 227–35.
57. Bouquet M, Selva J, Auroux M. Effect of cool-ing and equilibration in DMSO, and cryo-preservation of mouse oocytes, on the ratesof in vitro fertilization, development and
chromosomal abnormalities. Mol Reprod Dev1995; 40: 110–5.
58. Edwards RG, Brody SA. Principles andPractice of Assisted Human Reproduction.Philadelphia: WB Saunders 1995: 425–518.
59. Jones GM, Trounson AO, Gardner DK.Evolution and culture protocol for successfulblastocyst development and pregnancy. HumReprod 1998; 13: 169–77.
60. Gardner DK. Development of serum-freemedia for the culture and transfer of humanblastocysts. Hum Reprod 1998; 13: 218–85.
61. Liebermann J, Tucker MJ. Comparison of vit-rification and conventional cryopreservationof day 5 and day 6 blastocysts during clinicalapplication. Fertil Steril 2006; 86: 20–6.
62. Vanderzwalmen P, Bertin G, Debauche Ch,Standaert V, van Roosendaal E, Vandervorst Met al. Births after vitrification at morula andblastocyst stages: effect of artificial reductionof the blastocoelic cavity before vitrification.Hum Reprod 2002; 17: 744–51.
63. Zech NH, Lejeune B, Zech H, VanderzwalmenP. Vitrification of hatching and hatchedhuman blastocysts: effect of an opening in thezona pellucida before vitrification. ReprodBiomed Online 2005; 11: 355–61.
64. Kuwayama M, Vajta G, Ieda S, Kato O.Comparison of open and closed methods forvitrification of human embryos and the elimi-nation of potential contamination. ReprodBiomed Online 2005; 11: 608–14.
65. Vanderzwalmen P, Lejeune A, Stecher N, ZechN, Delval A, Zech H. Survival of day 3 and day5 embryos following vitrification in asepticand non-aseptic conditions: a prospectiverandomised analysis. Hum Reprod 2005; 84(Suppl 1): 185.
66. Larman MG, Sheehan CB, GardnerDK.Vitrification of mouse pronuclear oocyteswith no direct liquid nitrogen contact. ReprodBiomed Online 2006; 12: 66–9
67. Nagashima H, Kashiwazaki N, Ashman R J,Grupen CG, Nottle MB. Cryopreservation ofporcine embryos. Nature 1995; 374: 416.
68. Quinn P. Principles of membrane stability andphase behaviour under extreme conditions. JBioenerg Biomembr 1989; 21: 3–19.
69. Dvorak M. Ultrastructure and quantitativeanalysis of mouse and human oocytes. In: LissAR, ed. Developments in Ultrastructure ofReproduction. New York: Academic Press,1989: 273–280.
70. Sathananthan AH, Kirby C, Peura A,Trounson A. Mouse oocyte cooling. J AssistReprod Genet 1992; 9: 139–48.
181
VITRIFICATION OF PRONUCLEAR EMBRYOS
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 181

71. Pickering SJ, Braude PR, Johnson MH, CantA, Currie J. Transient cooling to roomtemperature can cause irreversible disruptionof the meiotic spindle in the human oocyte.Fertil Steril 1990; 54: 102–8.
72. Aman RR, Parks JE. Effects of cooling andrewarming on the meiotic spindle and chro-mosomes of in vitro-matured bovine oocytes.Biol Reprod 1994; 50: 103–10.
73. Didion BA, Pom D, Martin MJ, Homanics GE,Markert CL. Observation on the cooling andcryopreservation of pig oocytes at the germinalvesicle stage. J Anim Sci 1990; 68: 2803–10.
74. Johnson MH, Pickering SJ. The effect ofdimethylsulfoxide on the microtubular systemof the mouse oocyte. Development 1987; 100:313–24.
75. Isachenko V, Michelmann HW, Alabart JL,Vazquez I, Isachenko E, Bezugly N, et al.Lipolyse and ultra-structural changes of anintracellular lipid vesicles after cooling ofbovine and porcine GV-oocytes. Anat HistolEmbryol 2001; 30: 333–8.
VITRIFICATION IN ASSISTED REPRODUCTION
182
08 Tucker 8029.qxd 8/23/2007 8:39 PM Page 182

183
Vitrification of day 2–3 human embryos:using various techniques (Cryoloop,Cryotop, and conventional cryostraw)Tetsunori Mukaida and Katsuhiko Takahashi
9
INTRODUCTION
The definition of vitrification is the solidifica-tion of a solution at a low temperature with-out the formation of ice crystals, by increasingthe viscosity using high cooling rates.1 Therapid cooling process can minimize chillinginjury and osmotic shock to the embryos.Vitrification, with recent improvements, hasbecome a reliable strategy, not only because itis very simple but also because it can lead tohigh survival rates. To induce vitrification inliquid nitrogen, the solution must contain ahigh concentration of cryoprotectant.2
In vitrification, the selection of cryoprotec-tants requires extreme care because their con-centration can be as high as 6 mol/L, whichcan make the toxicity of these compounds akey limiting factor in cryobiology. The mostappropriate characteristics of a penetratingcryoprotectant are low toxicity and highpermeability. For cryopreservation of humanembryos, 1,2-propanediol (PROH) and di-methylsulfoxide (DMSO) have been used asthe dominant cryoprotectants, although glyc-erol is used when embryos are frozen at theblastocyst stage.3 As a less toxic cryoprotec-tant, ethylene glycol is commonly and widelyused.2 However, few comparative studies haveexamined the effect of the cryoprotectant onthe survival of vitrified embryos. In 1998, weperformed an investigation to find a suitablecryoprotectant and suitable conditions for
exposing embryos to the vitrification solutionusing 8-cell mouse embryos.4 The survivalrates of 8-cell embryos vitrified in varioussolutions after exposure to the solutions for0.5 and 2 min at 20 and 25°C are summarizedin Figure 9.1. The highest levels of survivalwere obtained, regardless of the time andtemperature, with ethylene glycol-based solu-tions. Although none of the vitrified embryoswere morphologically normal when embryoswere vitrified after 0.5 min exposure to anymixture of 30% cryoprotectant, the survivalrate was over 90% when embryos were treatedfor a longer time (2 min) at a higher temper-ature (25°C), or when embryos were treatedwith a higher concentration of ethylene glycol(EFS40) at a higher temperature (25°C).
In addition, a small saccharide (e.g. sucrose)and a macromolecule (e.g. Ficoll 70, bovineserum albumin (BSA), or polyvinylpyrrolidone(PVP)) are frequently included in vitrificationsolutions. These non-permeating agents aremuch less toxic, and are known to promotevitrification of the solution.5 Therefore, theirinclusion can reduce the toxicity of the solu-tion by decreasing the concentration of thepermeating agent required for vitrification.In addition, inclusion of a saccharide promotesshrinkage of embryos, and thus reducesthe amount of intracellular cryoprotectant,which will also reduce the toxic effect of thepermeating cryoprotectant.5 At the same time,the osmotic action of a saccharide plays an
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 183
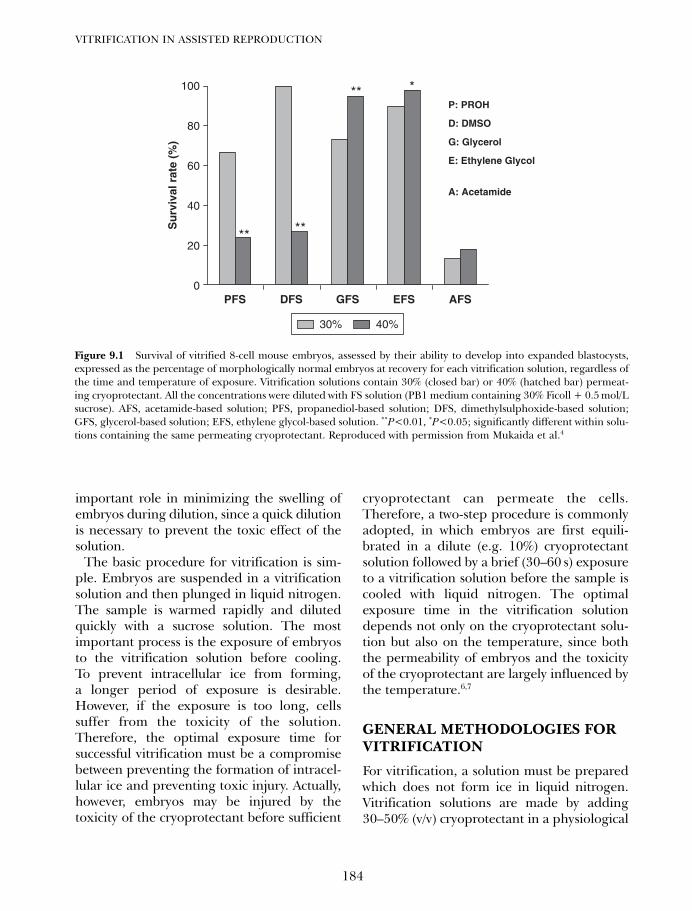
important role in minimizing the swelling ofembryos during dilution, since a quick dilutionis necessary to prevent the toxic effect of thesolution.
The basic procedure for vitrification is sim-ple. Embryos are suspended in a vitrificationsolution and then plunged in liquid nitrogen.The sample is warmed rapidly and dilutedquickly with a sucrose solution. The mostimportant process is the exposure of embryosto the vitrification solution before cooling.To prevent intracellular ice from forming,a longer period of exposure is desirable.However, if the exposure is too long, cellssuffer from the toxicity of the solution.Therefore, the optimal exposure time forsuccessful vitrification must be a compromisebetween preventing the formation of intracel-lular ice and preventing toxic injury. Actually,however, embryos may be injured by thetoxicity of the cryoprotectant before sufficient
cryoprotectant can permeate the cells.Therefore, a two-step procedure is commonlyadopted, in which embryos are first equili-brated in a dilute (e.g. 10%) cryoprotectantsolution followed by a brief (30–60 s) exposureto a vitrification solution before the sample iscooled with liquid nitrogen. The optimalexposure time in the vitrification solutiondepends not only on the cryoprotectant solu-tion but also on the temperature, since boththe permeability of embryos and the toxicityof the cryoprotectant are largely influenced bythe temperature.6,7
GENERAL METHODOLOGIES FORVITRIFICATION
For vitrification, a solution must be preparedwhich does not form ice in liquid nitrogen.Vitrification solutions are made by adding30–50% (v/v) cryoprotectant in a physiological
VITRIFICATION IN ASSISTED REPRODUCTION
184
0
20
40
60
80
100
PFS DFS GFS EFS AFS
Su
rviv
al r
ate
(%)
30% 40%
P: PROH
D: DMSO
G: Glycerol
E: Ethylene Glycol
A: Acetamide
** **
** *
Figure 9.1 Survival of vitrified 8-cell mouse embryos, assessed by their ability to develop into expanded blastocysts,expressed as the percentage of morphologically normal embryos at recovery for each vitrification solution, regardless ofthe time and temperature of exposure. Vitrification solutions contain 30% (closed bar) or 40% (hatched bar) permeat-ing cryoprotectant. All the concentrations were diluted with FS solution (PB1 medium containing 30% Ficoll + 0.5 mol/Lsucrose). AFS, acetamide-based solution; PFS, propanediol-based solution; DFS, dimethylsulphoxide-based solution;GFS, glycerol-based solution; EFS, ethylene glycol-based solution. **P<0.01, *P<0.05; significantly different within solu-tions containing the same permeating cryoprotectant. Reproduced with permission from Mukaida et al.4
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 184

solution such as Hepes HTF or phosphatebuffered saline (PBS).
In a few cases, embryos are suspended ina vitrification solution directly from a physio-logical solution (one-step method).5 In mostcases, however, embryos are first suspended ina solution containing a lower concentration(10–20%) of permeating cryoprotectant topromote permeation of the cryoprotectantunder less toxic conditions; then they are sus-pended in the vitrification solution (two-stepmethod). Embryos are loaded in a container,cooled with liquid nitrogen, and preserved init. The time embryos are suspended in thevitrification solution before cooling is critical.During the suspension, embryos need to bedehydrated and concentrated, but this usuallyoccurs quickly. Thus the exposure time mustbe minimized (~30 s) to prevent injury fromthe toxicity of the cryoprotectant. As the con-tainer, 0.25 mL cryostraws have been conven-tionally used.4 However, to intensify the ratesof cooling and warming, various minute toolshave been used, such as small grids,8 thin cap-illaries,9 tiny loops,10 and small plastic sticks.11
For embryo recovery, vitrified embryos arewarmed rapidly and the embryos are sus-pended in a solution containing sucrose. Insolution, sucrose prevents over-swelling ofembryos as the permeated cryoprotectant dif-fuses out. After this diffusion has been con-firmed by embryo shrinkage, the embryos arerecovered in a physiological solution. In thischapter, three different approaches aredescribed including the conventional Cryo-straw, Cryotop, and Cryoloop techniques.
PROTOCOLS AND RESULTS
Vitrification using conventionalcryostraws
A two-step protocol for straw vitrificationusing ethylene glycol-based solutions, EFS20and EFS40, is described.12 This method hasbeen proven available for human embryos onday 2–34 (Mukaida et al., unpublished data).
The two solutions (EFS20 and EFS40) areused for pretreatment and vitrification, respec-tively. The base medium used for vitrificationof embryos is modified phosphate-bufferedsaline (PB1), in which BSA is replaced withhuman serum albumin (HSA). Ethylene glycolis diluted to 20% (v/v) or 40% (v/v) with Ficoll-sucrose (FS) solution; the components of theFS solution are 30% (w/v) Ficoll 70 (averagemolecular weight 70 000; Amersham PharmaciaBiotech, Buckinghamshire, UK), and 0.5 mol/Lsucrose in PB1 medium. The respective vitrifi-cation solutions are designated EFS20 andEFS40. The final concentrations of Ficoll 70and sucrose are 24% (w/v) and 0.4 mol/L,respectively, in EFS20, and 18% (w/v) and0.3 mol/L, respectively, in EFS40. For dilution,PB1 medium containing 0.5 mol/L sucrose(S-PB1) is prepared.
All the solutions are placed in a roomat 25–27°C, at which temperature embryosare manipulated. A 0.25 mL plastic straw(~132 mm including the cotton plug) is pre-pared for embryo loading by drawing S-PB1medium up to a depth of ~60 mm, followedby air (~25–30 mm), EFS40 (~5 mm),another volume of air (~5 mm), and finallymore EFS40 (~12 mm). First, embryos arepretreated by being suspended in a drop ofEFS20 in the lid of a culture dish (or a dish)for 2 min. Then, embryos are transferredinto the larger column of FES40 near themouth of the straw. The contents of the straware aspirated until the first column of S-PB1medium is in contact with the cotton plug,and the straw is sealed with the heat-sealer.After exposure of embryos to EFS40 for~30 s, the straw is positioned in the liquidnitrogen vapor phase by placing it horizon-tally on a ~1 cm thick Styrofoam boat float-ing on the surface of the liquid nitrogen in aDewar vessel (inner diameter, 140 mm). After3 min or more, the straw is placed in a canis-ter and stored in liquid nitrogen. EFS40 andEFS20 are prepared in 1 mL syringesequipped with 18 G needles, and new smalldrops are placed on the lid of a dish just
185
VITRIFICATION OF DAY 2–3 HUMAN EMBRYOS
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 185

before use for each sample, to preventconcentration of the solution by evaporation.
For embryo recovery, the straw is kept inair for 10 s and then immersed in water at25–28°C. When the crystallized S-PB1medium in the straw begins to melt (afterabout 7 s), the straw is removed from thewater, quickly wiped dry, and cut at bothends. The contents of the straw are expelledinto a watch glass (or a culture dish) by flush-ing the straw with 0.8mL of S-PB1 mediumusing a 1 mL syringe attached with an 18 Gneedle. After gently agitating the watch glassto promote mixing of the contents, theembryos are pipetted into fresh S-PB1medium. About 5 min after being flushed out,the embryos are transferred to fresh PB1medium. Embryos are further washed withfresh PB1 medium, and are transferred to aculture medium for culture until transfer.
In 1998, Mukaida et al.4 reported theeffectiveness of this vitrification method forday 2–3 human embryos, more trials on thisstraw vitrification were performed in theHART Clinic group (Hiroshima HARTClinic, Osaka HART Clinic, and TokyoHART Clinic), and its effectiveness was con-firmed. In our unpublished data for day 2–3embryos, a total of 661 embryos were vitri-fied, and 486 (74%) of them had 50% or moremorphologically intact blastomeres afterwarming which was confirmed by furtherdevelopment of these vitrified embryos onthe day after warming. A total of 335 vitrifiedembryos were transferred in 127 cycles at 1 or2 days after warming, resulting in 34 (26.8%)women becoming pregnant, and 22 (17%)women delivering babies.
Vitrification using the Cryoloop
Here we describe an ultrarapid vitrificationapproach using the Cryoloop.13–15 Thismethod is available not only for embryos onday 2–3, but also for blastocysts, for whichstraw vitrification was found to be less effec-tive. The protocol for vitrification using the
Cryoloop can be found in the chapter ofvitrification of blastocysts (Chapter 10B),which is basically the same.
The Cryoloop consists of a tiny nylon loop(20 µm wide, 0.5–0.7 mm in diameter)mounted on a small stainless steel tubeinserted into the lid of a cryovial (HamptonResearch, Laguna Niguel, CA, USA).16 A metalinsert on the lid enables the use of a stainlesssteel handling rod with a small magnet(CrystalWand With Tab; Hampton Research) formanipulation of the loop at low temperature(liquid nitrogen).
Cryoprotectant solution I and cryoprotec-tant solution II are used for pretreatment andvitrification, respectively. The cryoprotectantsolutions are made with a base medium con-sisting of HEPES-buffered human tubal fluid(HTF) medium containing 5 mg/mL of HSA.Cryoprotectant solution I consists of basemedium containing 1.07 mol/L DMSO and1.36 mol/L ethylene glycol, and cryoprotectantsolution II consists of base medium containing2.08 mol/L DMSO, 2.64 mol/L ethylene glycol,10 mg/mL Ficoll 70, and 0.65 mol/L sucrose.Both cryoprotectant solutions (1–1.2 mL) areplaced in a 4-well culture dish and warmed inan incubator at 37°C for ~30 min.
Embryos are manipulated on a platewarmed at 37°C in a room at 25–27°C, andthus presumably at ~37°C. Initially, embryosare suspended in cryoprotectant solution I. At2 min after suspension, the embryos arewashed quickly in small drops of solution IIon the lid of a culture dish. Quickly, aCryoloop is dipped into cryoprotectant solu-tion II to create a thin filmy layer of the solu-tion on the nylon loop, and the embryos aretransferred onto the thin filmy layer on thenylon loop using a micropipette. Within 30 sof suspension in solution II, the Cryoloop isplunged into liquid nitrogen. The time theembryos are kept on the loop with the thinlayer of solution in the air should be as shortas possible, because evaporation will makethe solution more concentrated. Using thestainless steel rod, the loop containing the
VITRIFICATION IN ASSISTED REPRODUCTION
186
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 186

embryos is sealed in a cryovial, which hasbeen previously submerged in liquid nitro-gen. The vial is attached to a standard caneand stored in liquid nitrogen. The whole pro-cedure is completed within 5 min.
For recovery, the cryovial is opened withthe aid of the stainless steel rod. Then theloop containing the embryos is removed fromthe liquid nitrogen and placed quickly anddirectly into a well of the base medium con-taining 0.33 mol/L sucrose at ~35°C. Theembryos never fail to float off the loop intothe solution; thus, the embryos are almostalways successfully recovered. After 2 min,embryos are transferred to the base mediumcontaining 0.2 mol/L sucrose. After an addi-tional 3 min, embryos are suspended in basemedium for 5 min. Finally, they are transferredinto culture medium for further culturing untiltransfer.
At the HART Clinic group, Cryoloop vitri-fication is adopted for cryopreservation ofsupernumerary human embryos on day 2–3for a short period of time, and mainly for
blastocysts on day 5–6 that were obtainedfrom culture in sequential media. Availabledata show that for embryos on day 2–3, a totalof 269 embryos have been vitrified, and 188(70%) of them had 50% or more morpholog-ically intact blastomeres which was confirmedby further development on the day afterwarming. A total of 112 vitrified embryoswere transferred into 44 patients, and 14(32%) of these women conceived (Mukaidaet al., unpublished data).
In 2007, Desai et al.17 reported the postvit-rification development, pregnancy outcomes,and live birth rates for Cryoloop vitrificationof human day 3 cleavage-stage embryos.Table 9.2 includes their results that representconsecutive vitrification-warming cycles per-formed over a 2.5-year interval. A total of 236embryos were warmed, and the average num-ber of embryos transferred per patient was2.66 ± 0.86. They reported that the clinicalpregnancy rate was 44% (34/77), and theimplantation rate was 20% (40/201). Thepostwarming survival rate was 85% (201/236).
187
VITRIFICATION OF DAY 2–3 HUMAN EMBRYOS
Table 9.1 Summary of the protocols regarding concentration, time, and properties of the vitrification solution forday 2–3 human embryo cryopreservation
Cryostraw Cryoloop17 Cryoloop18 Cryotop
Temperature Room (25–27°C) Warm stage (37°C) Warm stage (37°C) Room (25–27°C)
Equilibration EGFS 20: 20% 7.5% EG + 7.5% 10% EG (5 min) 7.5% EG + 7.5% step EG + F + S (2 min) DMSO (2 min) DMSO (5–10 min*)
Vitrification EGFS 40: 40% 15% EG + 15% 40% EG + S (30 s) 15% EG + 15% step EG + F + S (1 min) DMSO + F + S(35 s) DMSO + S (1 min)
Cooling Vapor phase above Plunged into LN2 Plunged into LN2 Plunged into LN2
system LN2 (3 min), then directly (ultra-rapid directly (ultra-rapid directly (ultra-rapid plunged into LN2 cooling) cooling) cooling)
Warming One step: Two steps: Four steps: Two steps:step 0.5 mol/L S 0.25 mol/L S (2 min) 1 mol/L S (2.5 min), 1 mol/L S (1 min)
(5 min) 0.12 mol/L S (3 min) 0.5 mol/L S (2.5 min), 0.5 mol/L S (3 min)0.25 mol/L S (2.5 min),0.125 mol/L S (2.5 min)
*Duration of equilibration is adjusted according to the time needed for re-expansion of the vitrified embryos.EG, ethylene glycol; F, Ficoll; S, sucrose.Data from Desai et al.17 and Rama Raju et al.18
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 187
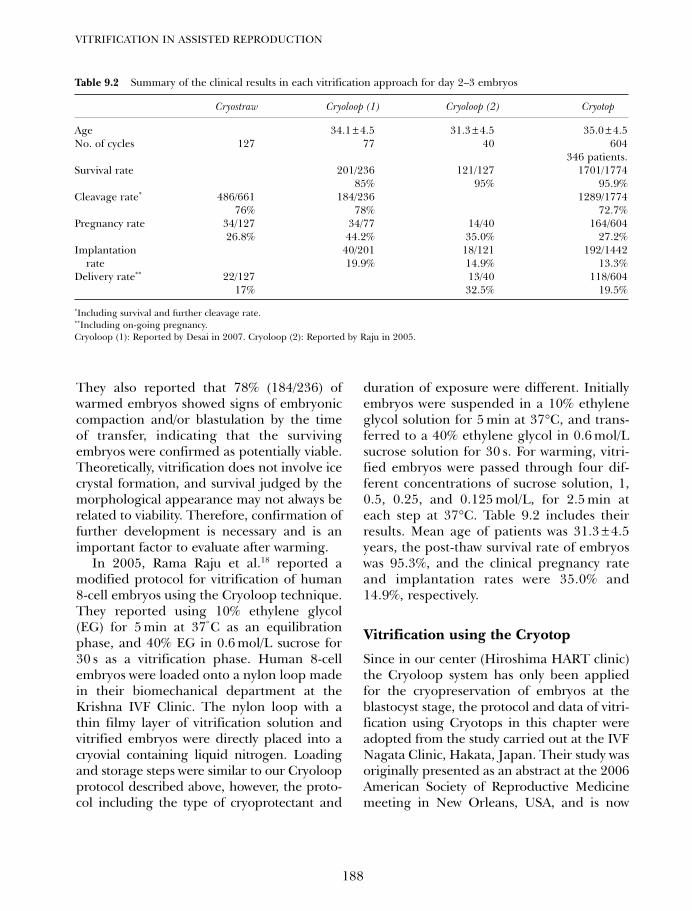
They also reported that 78% (184/236) ofwarmed embryos showed signs of embryoniccompaction and/or blastulation by the timeof transfer, indicating that the survivingembryos were confirmed as potentially viable.Theoretically, vitrification does not involve icecrystal formation, and survival judged by themorphological appearance may not always berelated to viability. Therefore, confirmation offurther development is necessary and is animportant factor to evaluate after warming.
In 2005, Rama Raju et al.18 reported amodified protocol for vitrification of human8-cell embryos using the Cryoloop technique.They reported using 10% ethylene glycol(EG) for 5 min at 37°C as an equilibrationphase, and 40% EG in 0.6 mol/L sucrose for30 s as a vitrification phase. Human 8-cellembryos were loaded onto a nylon loop madein their biomechanical department at theKrishna IVF Clinic. The nylon loop with athin filmy layer of vitrification solution andvitrified embryos were directly placed into acryovial containing liquid nitrogen. Loadingand storage steps were similar to our Cryoloopprotocol described above, however, the proto-col including the type of cryoprotectant and
duration of exposure were different. Initiallyembryos were suspended in a 10% ethyleneglycol solution for 5 min at 37°C, and trans-ferred to a 40% ethylene glycol in 0.6 mol/Lsucrose solution for 30 s. For warming, vitri-fied embryos were passed through four dif-ferent concentrations of sucrose solution, 1,0.5, 0.25, and 0.125 mol/L, for 2.5 min ateach step at 37°C. Table 9.2 includes theirresults. Mean age of patients was 31.3 ± 4.5years, the post-thaw survival rate of embryoswas 95.3%, and the clinical pregnancy rateand implantation rates were 35.0% and14.9%, respectively.
Vitrification using the Cryotop
Since in our center (Hiroshima HART clinic)the Cryoloop system has only been appliedfor the cryopreservation of embryos at theblastocyst stage, the protocol and data of vitri-fication using Cryotops in this chapter wereadopted from the study carried out at the IVFNagata Clinic, Hakata, Japan. Their study wasoriginally presented as an abstract at the 2006American Society of Reproductive Medicinemeeting in New Orleans, USA, and is now
VITRIFICATION IN ASSISTED REPRODUCTION
188
Table 9.2 Summary of the clinical results in each vitrification approach for day 2–3 embryos
Cryostraw Cryoloop (1) Cryoloop (2) Cryotop
Age 34.1 ± 4.5 31.3 ± 4.5 35.0 ± 4.5No. of cycles 127 77 40 604
346 patients.Survival rate 201/236 121/127 1701/1774
85% 95% 95.9%Cleavage rate* 486/661 184/236 1289/1774
76% 78% 72.7%Pregnancy rate 34/127 34/77 14/40 164/604
26.8% 44.2% 35.0% 27.2%Implantation 40/201 18/121 192/1442
rate 19.9% 14.9% 13.3%Delivery rate** 22/127 13/40 118/604
17% 32.5% 19.5%
*Including survival and further cleavage rate.**Including on-going pregnancy.Cryoloop (1): Reported by Desai in 2007. Cryoloop (2): Reported by Raju in 2005.
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 188

being prepared for publication includingtheir intensive analysis.19
Their protocol and data of vitrification werebased on 346 women who needed to have theirembryos cryopreserved on day 2 of develop-ment to avoid either ovarian hyperstimulationsyndrome (OHSS), or as supernumeraryembryos for subsequent transfer attempts, whoagreed to cryopreservation of the embryos byvitrification. A total of 1774 day-2 embryoswere vitrified with equilibration solution, anequal mixture of 7.5% DMSO and 7.5% ethyl-ene glycol (EG) in HTF supplemented with20% HSA, and a vitrification solution, using amixture of 15% DMSO, 15% EG, and 0.5 mol/Lsucrose in HTF/HSA. Initially, embryos wereexposed to the equilibration solution for5–10 min, and to the vitrification solution for1 min at room temperature (25–27°C). Theduration of the equilibration time was adjustedby morphological changes that indicatedshrinkage for dehydration and re-expansionfor cryoprotectant (CPA) permeation, and wasindividually recorded for further analysis.Embryos were then loaded onto a minutenylon sheet (Cryotop), and plunged into liquidnitrogen immediately. For warming, vitrifiedembryos on the tip of a Cryotop were dippedand kept in 1 mol/L sucrose solution for 1 minand then diluted in 0.5 mol/L sucrose solutionfor 3 min. Embryos with 70% or more intactblastomeres were considered as indicative ofsurvival and were kept in culture until transferon the following day.
Table 9.2 includes the results from the useof the Cryotops at Nagata Clinic. Briefly, 346patients with day-2 embryos that were to becryopreserved to avoid either OHSS, or assupernumerary embryos for subsequenttransfer attempts, were entered in this inves-tigation. A total of 1774 day 2 embryos werewarmed in 604 transfer cycles. Of the vitrifiedembryos 1701 survived, and 1442 of themwere transferred. Pregnancy was confirmed in164 cycles and 192 embryos implanted asconfirmed by the presence of a gestational
sac. Forty six cycles ended in miscarriage and67 deliveries were achieved; other cases areongoing pregnancy.
Total survival and cleavage rates were95.9% (1701/1774) and 75.8% (1289/1701),respectively. The mean number of embryostransferred was 2.4 ± 0.7, the pregnancyrate was 27.2% (164/604), the implantation ratewas 13.3% (192/1442), and the abortion ratewas 28.0% (46/164). Ninety nine healthybabies were born following 84 deliveries (49boys and 50 girls).
TROUBLE SHOOTING
Conventional vitrification
(1) Room temperature other than 25°C. Thetemperature for handling embryos in acryoprotectant solution has significanteffects. When the room temperature ishigher, the duration of exposure ofembryos to EFS40 must be shortened andvice versa. Elevating the temperature ispreferable for promoting the permeationof the cryoprotectant, but the optimalrange of the exposure time decreasesbecause the toxicity of the cryoprotectantincreases considerably. For the protocoldescribed (at 25°C), keep the lights off soas not to elevate the temperature of themicroscope stage when embryos are notbeing manipulated under the micro-scope. Avoid warming the EFS40 columnof the straw with one’s fingers.
(2) Cooling in a small dewar flask. If only asmall dewar flask is available, immersehalf of the straw containing the embryosin liquid nitrogen first, and then cool therest of the straw slowly in nitrogen vapor.If the whole straw is immersed in liquidnitrogen in one step, it may rupturebecause of the rapid increase in the vol-ume of the freezing sucrose solution. Bythis method, a small percentage of vitri-fied embryos may suffer fracture damage.
189
VITRIFICATION OF DAY 2–3 HUMAN EMBRYOS
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 189

Ultrarapid vitrification: Cryoloop
(1) During the step of equilibration, morpho-logical change can be an important factorto understand the degree of permeationof cryoprotectant and the degree of dehy-dration. Two minutes duration is approxi-mate, and the duration of equilibrationmay need to be adjusted depending onthe morphological changes similar towhen using the Cryotop protocol.
(2) Preventing evaporation of the vitrificationsolution. Because the amount of vitrifica-tion solution on the Cryoloop is very small,the solution is liable to be concentrated byevaporation, which increases its toxicity. Toprevent this, the Cryoloop should beloaded with the vitrification solution justbefore the loading of embryo(s) on theloop. The amount of vitrification solutionon the loop need not be minimal, as long asthe drop is kept on the loop by surface ten-sion. A dewar flask containing liquid nitro-gen should be placed nearby to enablequick cooling.
(3) Quick warming. At warming, the loopcontaining the embryo(s) should besoaked in the sucrose solution as quicklyas possible, making sure the sample isnever held in the air. Again, a dewar flaskcontaining liquid nitrogen and thesucrose solution should be placed nearby.When the loop is dipped in the sucrosesolution, the steel pipe portion should notbe immersed to prevent bubbling.
(4) Morphology of the recovered blastocyst.At recovery in an isotonic solution, theblastocoel of the blastocyst will be col-lapsed and the embryo may look like a‘morula’. However, the blastocoelic cavityshould reform within 1–3 h of culture.
Ultrarapid vitrification: Cryotop
(1) When the Cryotop system is used to loadthe embryos for vitrification, the loaded
side of the tip should be clearly marked inorder to identify its side at the time ofwarming.
(2) Tip of the Cryotop should be cappedtightly into the capping part in a dewarflask containing liquid nitrogen, and con-firmation of tight capping should alwaysbe made before transferring back to thestorage tank.
(3) Size and volume of vitrification on the tipof Cryotop should be minimized, how-ever, if the volume is too small, the vitri-fied embryo may occasionally be stuckedon the surface of the tip.
(4) At the step of warming, the tip of theCryotop should always be inside thewarming solution.
(5) Prevention of evaporation of the vitrifica-tion solution on the tip. The drop with theembryos should be loaded on the tip ofCryotop just before plunging into liquidnitrogen. This is the same precaution as forCryoloop vitrification. Because the amountof vitrification solution on the tip ofCryotop is very small, evaporation of thesolution increases its toxicity. A dewar flaskcontaining liquid nitrogen should beplaced nearby to enable quick cooling andwarming.
DISCUSSION
Numerous protocols for the cryopreservationof mammalian embryos have been reported.The protocols can be classified into fourmethods, original slow freezing, conven-tional slow freezing, conventional vitrifica-tion using the conventional cryostraw, andultrarapid vitrification using a minute tool.Although strategies to circumvent variousinjuries (especially from the formation ofintracellular ice) are different, the principleof cryopreservation is the same. The mostsuitable protocol should be adopted for eachcase. For certain types of embryos, such as
VITRIFICATION IN ASSISTED REPRODUCTION
190
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 190

human blastocysts and bovine embryos atearlier stages, ultrarapid vitrification will bethe preferred choice, because the survivalrates of embryos cryopreserved by othermethods have been low. For other embryos,e.g. mouse embryos, bovine blastocysts, andhuman embryos at 2–8-cell stages, both slowfreezing and conventional vitrification havebeen proven effective. However, vitrificationhas a potential advantage in that higher sur-vival rates can be obtained if conditions, suchas temperature and duration of exposure ofembryos to the cryoprotectant, as well as theskill of pipetting, are optimized. Therefore,vitrification would be a preferred method ofcryopreservation compared with the slow-cooling method because of the lack of icecrystal formation and its convenience.
In this chapter, three different approachesof vitrification were compared for the cryo-preservation of human day 2–3 embryos. Table9.1 summarizes the differences in characteris-tics with each vitrification approach. In theCryoloop approach, temperature is always setat 37° C using a stage warmer. On the otherhand, the cryostraw and Cryotop techniquesare performed at room temperature. Highertemperatures can stimulate the plasma mem-brane permeability to permeating cryoprotec-tants and water. For the equilibration andvitrification phase, either EG only or anEG + DMSO mixture is used as the permeat-ing cryoprotectant. The cryostraw approachhad the highest concentration of cryoprotec-tant (20%, 40%) compared with otherapproaches, because the cooling rate in thevapor phase of liquid nitrogen might not behigh enough to avoid ice crystal formation withrelatively low concentrations of cryoprotectants.Theoretically, the flexible time needed accord-ing to the morphological changes during theequilibration and vitrification steps may notbe realistic, as it might become highly subjec-tive due to the individual judgment of eachembryologist, and may not achieve adequatefull equilibration. Furthermore, in clinicalART laboratories consistent and reproducible
protocols are important. This might be one ofthe reasons why fixed durations are commonlyapplied. Figure 9.1 shows that EG-based solu-tions obtained the highest level of survival,regardless of the time and temperature.However, this result was not obtained withhuman embryos, but with mouse 8-cellembryos. The equilibration phase (10% EG for5 min) of Rama Raju’s group18 approach to vit-rification may be the most appropriate strategybased on that study. The cryostraw has a disad-vantage in terms of cooling rate because of thethermal insulating layer of the cryostraw, andthe larger volume of vitrification solutionrequired. However, direct contact of liquidnitrogen with minimal volume of vitrificationsolution as with the Cryoloop and Cryotopsystem can create extremely high cooling ratesthat can avoid ice crystal formation, even witha relatively lower level of concentration ofcryoprotectant that would cause devitrificationin classical cooling steps. Warming is the stepfor removing the permeating cryoprotectantinside the vitrified embryos and rehydratingthe embryos. In order to facilitate this step, anosmotic gradient of sucrose concentration isnecessary. Rama Raju’s vitrification18 using aCryoloop has four steps of sucrose concentra-tion. The concentration of sucrose at 1.0 mol/Lis relatively high compared with other proto-cols that have been reported. However, ahigher concentration of sucrose would be bet-ter in order to prevent osmotic swelling duringthe warming steps, unless warmed embryosstart collapsing. Ideally, the duration of thewarming steps can also be recommended toadjust the morphological appearance of thevitrified embryos, and the sucrose concentra-tion should be maintained until the embryosstart to shrink. We believe more frequent gra-dient steps of sucrose with adjustment of theduration according to individual evaluationcan improve the survival and viability afterwarming; however, this may not be practicalduring daily routine clinical ART work.
Table 9.2 shows the comparison of clinicalresults from each vitrification protocol. The
191
VITRIFICATION OF DAY 2–3 HUMAN EMBRYOS
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 191

protocols of Rama Raju et al.18 using theCryoloop and Cryotop vitrification had similarhigh survival rates (95%). However, vitrifica-tion theoretically does not involve ice crystalformation, and morphological intactness(survival) is not equivalent to viability. Withvitrification, vitrified embryos with sufficientequilibration of high concentration of cryo-protectant generally show a clear and trans-parent appearance because no ice crystalformation has occurred, and confirmation offurther development is an important factor toevaluate after warming. Therefore, cleavagerate is related to real viability after warming.All of the protocols except for that of RamaRaju et al.18 using the Cryoloop showed72–78% survival rates which are clinicallyacceptable. In the protocol of Rama Rajuet al.18 using the Cryoloop, vitrified embryosafter warming were cultured only for3–4 hours before transfer. In this case, it isdifficult to evaluate further growth ofembryos through cleavage rate. The preg-nancy rate varied from 27% to 44% depend-ing on the embryo scoring, and criteria forvitrification and patient background. It seemsthat the pregnancy rates using the Cryoloopwere higher than those using the cryostrawand Cryotop. Implantation and delivery rateswere similar among Cryoloop and Cryotopprotocols except for the cryostraw protocol.This suggests that ultrarapid cooling withdirect contact of liquid nitrogen seems to bemore effective than conventional cooling withuse of the vapor phase of liquid nitrogen withthe cryostraw as a carrier. An extremely highcooling rate is one of the most important fac-tors for improving the effectiveness of vitrifi-cation, because it allows a reduction in theconcentration of cryoprotectants to evenlower levels that will create ice crystal forma-tion (devitrification) during conventionalcooling. In this way, lowering the concentra-tion of the cryoprotectants will reduce theirtoxicity. The volume of the vitrification solu-tion should be minimized by using specialcarriers such as the Cryoloop, Cryotop, and
other comparable carriers in order to achievethis extremely high cooling rate.
We have already reported4 a simple vitrifica-tion method using an ethylene glycol-basedsolution for 4–8-cell human embryos frozen inconventional cryostraws. Moreover, the successof vitrification procedures has recently beenincreased by techniques that substantiallyreduce the volume of the vitrification solution.Among such techniques, the Cryoloop andCryotop are the most refined strategies. Amajor difference between the Cryoloop andCryotop, and the conventional cryostraw forvitrification is the cooling/ warming rate. TheCryoloop and Cryotop enable ultrarapid cool-ing and warming, and this may prevent intra-cellular ice formation more consistently, sincewe have observed that human embryos aredehydrated and concentrated more slowlythan other types of embryos, suggesting thatintracellular ice is more likely to form. The dif-ference between the Cryoloop and Cryotop isonly in the way that vitrified embryos are held.In the Cryoloop system, the vitrified embryo isalmost floating in the thin filmy layer of thedroplet on the nylon loop, and heat conduc-tion to the embryos becomes homogenous. Inthe Cryotop, the vitrified embryo is placed onthe surface of a nylon sheet, and heat conduc-tion might not be so homogenous especiallyfrom the sides of the tip. For the cryopreserva-tion of multiple cells or small sizes of tissue,this may cause uneven heat conduction, how-ever, for a smaller number of eggs or embryos,it does not seem to make any difference.
In conclusion, for day 2–3 humanembryos, vitrification through ultrarapidcooling achieved by direct contact with liquidnitrogen seems preferable, and either EGonly or EG + DMSO as cryoprotectants areacceptable. For equilibration prior to vitrifi-cation, a two-step approach is enough toobtain acceptable clinical results. Moreover,individual adjustment of cryoprotectantexposure times depending on the morpho-logical change is always better for equilibra-tion compared with the fixed duration
VITRIFICATION IN ASSISTED REPRODUCTION
192
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 192

protocol. Theoretically, the Rama Raju et al.18
protocol for equilibration and warming,adding individual adjustment in each step,will be most appropriate approach based onour review. Finally, maximizing the coolingrate and minimizing the concentration of
cryoprotectants is critical to establish theprotocol for vitrification with any stage ofembryo, and vitrification of day 2–3 humanembryos is more effective than using the slowcooling approach.
193
VITRIFICATION OF DAY 2–3 HUMAN EMBRYOS
References
1. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at −196°C by vitrification.Nature 1985; 313: 573–5.
2. Kasai M. Simple and efficient methods forvitrification of mammalian embryos. AnimReprod Sci 1996; 42: 67–75.
3. Menezo Y, Sakkas D, Veiga A. Cryopreservationof blastocysts. In: Gardner DK, Lane M, eds.ART and the Human Blastocyst. New York:Springer-Verlag, 2000: 188–95.
4. Mukaida T, Takahashi K, Kasai M et al.Vitrification of human embryos based on theassessment of suitable conditions for 8-cellmouse embryos. Hum Reprod 1998; 13: 2874–9.
5. Kasai M, Komi JH, Machida T et al. A simplemethod for mouse embryo cryopreservationin a low toxicity vitrification solution, withoutappreciable loss of viability. J Reprod Fertil1990; 89: 91–7.
6. Miyake T, Kasai M, Machida T et al.Vitrification of mouse oocytes and embryos atvarious stages of development in an ethyleneglycol-based solution by a simple method.Theriogenology 1993; 40: 121–34.
7. Zhu SE, Kasai M, Machida T et al.Cryopreservation of expanded mouse blastocystsby vitrification in ethylene glycol-based solutions.J Reprod Fertil 1993; 98: 139–45.
8. Martino A, Songsasen N, Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
9. Vajta G, Kuwayama M, Callesen H et al. OpenPulled Straw (OPS) vitrification: a new wayto reduce cryoinjuries of bovine ova andembryos. Mol Reprod Dev 1998; 51: 53–8.
10. Lane M, Bavister BD, Forest KT et al.Containerless vitrification of mammalianoocytes and embryos. Nat Biotechnol 1999;17: 1234–36.
11. Kuwayama M, Vajta G, Kato O et al.Comparison of open and closed methods forvitrification of human embryos and the elimi-nation of potential contamination. ReprodBiomed Online 2005; 11: 300–8.
12. Kasai M. Vitrification: refined strategy for thecryopreservation of mammalian embryos.J Mamm Ova Res 1997; 14: 17–28.
13. Mukaida T, Kasai M, Takahashi K et al.Successful birth after transfer of vitrifiedhuman blastocysts with use of a cryoloop con-tainerless technique. Fertil Steril 2001; 76:618–20.
14. Mukaida T, Oka C, Takahashi K et al.Vitrification of human blastocysts using cry-oloops: clinical outcome of 223 cycles. HumReprod 2003; 18: 384–91.
15. Mukaida T, Takahashi K, Kasai M et al.Blastocyst cryopreservation: ultrarapid vitrifi-cation using cryoloop technique. ReprodBiomed Online 2003; 6: 221–5
16. Lane M, Schoolcraft WB, and Gardner DK.Vitrification of mouse and human blastocystsusing a novel cryoloop container-lesstechnique. Fertil Steril 1999; 72: 1073–78.
17. Desai N, Blackmon H, Goldfarb J et al.Cryoloop vitrification of human day 3 cleav-age-stage embryos: post-vitrification develop-ment, pregnancy outcomes and live births.Reprod Biomed Online 2007; 14: 208–13.
18. Rama Raju GA, Haranath GB, Madan K et al.Vitrification of human 8-cell embryos, amodified protocol for better pregnancy rates.Reprod Biomed Online 2005; 11: 434–37.
19. Tomari Y, Nagata K, Honjo K, Takahara K.Kunitake. IVF Nagata Clinic, Fukuoka, Japan.Data presented to American Society forReproductive Medicine 2006 annual meetingat New Orleans, LA.
09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 193

09 Tucker 8029.qxd 8/23/2007 2:15 PM Page 194

195
One decade of experience with vitrification of human embryos instraws, hemi-straws, and high security vitrification strawsPierre Vanderzwalmen, Thomas Ebner and Nicolas Zech
10A
WHY IS THERE A RENEWEDINTEREST IN CRYOPRESERVATION?
In the past few decades assisted reproductivetechnologies (ART) have been used to helpcouples with infertility problems. With this inview, many techniques have been developedor improved, including ovary stimulationmethods, criteria and selection processes toassess the quality and viability of gametes andembryos, as well as culture conditions, andculture media. This has led to being able toobtain significantly more viable and transfer-able embryos nowadays. The result has beenthat the proportion of viable and transferableembryos has increased significantly in thepast few years. Moreover, awareness of riskslinked to multiple pregnancies has requiredthe implementation of standards limiting thenumber of embryos transferred at any onetime. One of these standards, namely cryo-preservation of embryos between the zygoteand blastocyst stages has become commonpractice when attempting ART.
In 1984, Zeilmaker et al.1 reported the firstbirth of a healthy child following slow freezingof a 4-cell embryo. Since then, slow-rate freezingtechniques have become widely used for thecryopreservation of zygotes and early cleavagestage embryos. For blastocysts, however, thesituation looks quite different. In 1985, Cohen
et al.2 reported the first pregnancy after con-trolled slow freezing of blastocysts using glyc-erol as a permeable cryoprotectant in a seriesof ten increasing solution concentrations.This initial protocol modified by Ménézoet al.3 by adding sucrose as a non-permeablecryoprotectant was considered popular forslow cooling of blastocysts obtained afterco-culture.4
In the mid-1990s, interest in blastocyst cryo-preservation emerged due to the introductionof commercial sequential and non-sequentialmedia allowing more and more fresh embryotransfers on a large scale at the blastocyst stage.Even though several manuscripts report accept-able implantation rates ranging from 16% to34%,5–8 it became apparent that, in spite of thefact that high implantation and pregnancy rateswere advocated after transfer of fresh blasto-cysts, slow-freezing strategies for blastocysts hadlimited applications and were rather inconve-nient due to the relatively poor survival ratesreported by several ART units.9–11 The decreaseof blastocysts available after the slow-freezingprocedure resulted in a reduction in the cumu-lative pregnancy rate per retrieval following thetransfer of both fresh and frozen blastocysts.This is one of the main reasons why embryo cul-ture to the blastocyst stage has never becomepopular. Such variable results with conventionalslow-freezing methods justified investigationsinto alternative approaches.
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 195

VITRIFICATION: AN ALTERNATIVETO SLOW FREEZING
During the various stages of a cryopreservationprocess, including exposure to cryoprotectants,cooling, storage in liquid nitrogen, re-warming,and return to a physiological solution, it issolely the skill of being able to prevent ice crys-tal formation that determines the viability ofan embryo after re-warming. It is obvious thatcrystallization is incompatible with any livingorganism and must be avoided as far as possi-ble. As a consequence, the primary factor whichinfluences whether a cell or an embryo survivescryopreservation is the prevention of intra-cellular ice formation. There are two generalapproaches to achieve this aim: the slow-ratefreezing12 and vitrification.13
Before initiating slow cooling in a pro-grammable controlled system (0.3°C/min),embryos have to be equilibrated in a solutioncontaining a low concentration (1.5 mol/L) ofpermeable cryoprotectant. During the follow-ing cooling step, a progressive dehydration,shrinkage, and increase in intra-cellularsolute concentration occurs due to the risingosmolarity in the extracellular solution aswater freezes out. In the most favorable situa-tion, the intracellular concentration of cryo-protectant becomes high enough so that theremaining intracellular unfrozen fraction willvitrify, preventing intracellular ice formation.However, during the cooling process, the for-mation of intracellular ice crystals is frequentand is thought to be the major cause of ‘cryo-injury’ during re-warming due to aggregationof small ice crystals that can induce irre-versible cell damage.
In this context vitrification is another cryo-preservation strategy which, unlike pro-grammed slow freezing, does not theoreticallyinvolve the formation of ice crystals either inthe intracellular or in the extracellular space,because the whole sample turns directly to‘glass’. In contrast to slow freezing, theembryos are exposed to high concentrationsof cryoprotectant solutions before being
plunged into liquid nitrogen (LN2) at veryhigh cooling rates (2000–20 000°C/min).
As a consequence, the physical injuriescaused by the formation of extracellular aswell as intracellular ice crystals during thecontrolled slow-freezing procedure are elimi-nated. Moreover, and in addition to theabsence of ice formation, this technique iscarried out quickly and thus makes it possibleto avoid problems caused by transitionsbetween non-physiological temperaturezones, known as ‘chilling’. However, thetoxicity caused by the high concentration ofcryoprotectants is considered by many scien-tists to be a drawback of this technique.Another beneficial aspect of this techniqueis that it does not require the use ofprogrammed freezing equipment.
WHAT ARE THE REQUIREDCONDITIONS TO ACHIEVE AVITRIFIED STATE?
The combination of high concentration ofcryoprotectants and an extremely fast coolingand warming rate are two conditions thatsupport the formation of a glassy state. Otherparameters that influence the formation ofan amorphous state include temperature con-duction, the concentration of the cryoprotec-tant, the volume of the solution, and thebiophysical parameters of the embryos.
First condition: high concentration ofcryoprotectants in the final solution
The type of cryoprotectants
Since the development of the first vitrifica-tion solution,13 which contained dimethyl-sulfoxide (DMSO), acetamide, and propyleneglycol, numerous cryoprotectant solutionshave been tested and reported to be effective.At the present time, the cryoprotectant solu-tions most widely used for vitrification arecomposed of agents that penetrate the cell(ethylene glycol (EG), DMSO, 1,2-propanediol
VITRIFICATION IN ASSISTED REPRODUCTION
196
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 196

(PROH), or glycerol), and non-penetratingagents of low molecular weight (sucrose ortrehalose). In some cases, polymers with highmolecular weights such as polyethylene glycol(PEG, MW 8000), Ficoll (MW 70 000 or400 000; Sigma), or polyvinyl pyrrolidone (MW360 000) are also added to the vitrificationmedium.14
The concentration of cryoprotectants
Usually, with vitrification, embryos are exposedto increasing concentrations of permeable andnon-permeable cryoprotectants in two steps.When the embryos are immersed in the liquidnitrogen the medium, which has a high viscos-ity, solidifies so quickly that the molecules donot have time to rearrange themselves into acrystalline structure. It is this that inducesthe formation of an intra- and extracellularvitreous state. Furthermore, the extreme cyto-toxicity resulting from the high concentrationof required cryoprotectants with highermembrane permeability can be reduced byaddition of an appropriate concentration ofnon-permeable cryoprotectant.15,16
Exposure to or equilibration of thecryoprotectants
The success of cryopreservation protocolsdepends on an optimal dehydration processwhen cells are exposed to hypertonic con-ditions and an optimal penetration rate ofcryoprotectant. This is determined by severalbiophysical factors such as the properties ofthe membrane (cellular permeability), thetype and concentration of cryoprotectiveadditives (permeability property, concentra-tion, and temperature of the cryoprotectants),and the surface–volume ratio of the cells.
Nearly all vitrification methods (coolingwithout equilibrium) consist of exposing theembryos for short periods and in two steps toincreasing concentrations of cryoprotectants.
In the first step (Figure 10A.1), the embryosare exposed to a non-vitrifying solution (VS1)
of permeable cryoprotectants (2.3–3.2 mol/L)for a period of from 2 to 4 min dependingon the stage of embryonic growth, each cellof which has a well-determined surface tovolume ratio and permeability coefficient.Following a rapid dehydration phase (< 30 s)at the beginning of exposure to the solution,a certain quantity of cryoprotectants (e.g.only about 30% of the initial cryoprotectantconcentration will enter the cytoplasm of azygote) penetrates the cells during the2–4 min exposure.
Another pre-incubation strategy17,18 con-sists of exposing the embryos to the firstsolution until the moment when the embryoregains its original volume. This prolongedexposure in the first bath may last between5 and 15 min, but this increases the risk ofinducing toxicity problems. However, as sug-gested by Vatja and Nagy,19 this approach,even though it may increase slightly the toxiceffect, could provide much better protectionfor the whole cell, and may be especiallybeneficial in the case of large objects with alow surface/volume ratio including oocytesor early stage embryos such as zygotes.
In the second step (Figure 10A.1) theembryos are put in contact with a vitrifying (VS2)solution of permeable and non-permeablecryoprotectants (4.8–6.4 mol/L) for a shortperiod lasting 30–45 s before being placed onthe carrier maximum of two embryos andplunged into liquid nitrogen. During thiswhole step a second phase of dehydrationoccurs.
Applying these two steps, an intracellularvitrifying state is obtained as a result of dehy-dration of the embryos in VS2, which con-centrates the intracellular solutions of salts,proteins, and cryoprotectant that have pene-trated the cell in the course of exposure toVS1. The extracellular vitrifying state isobtained by the high concentration of VS2cryoprotectants that encapsulate the embryoin a vitrifying sheath. Consequently, celldehydration is carried out before cooling, asis the achieving of an intra- and extracellular
197
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 197
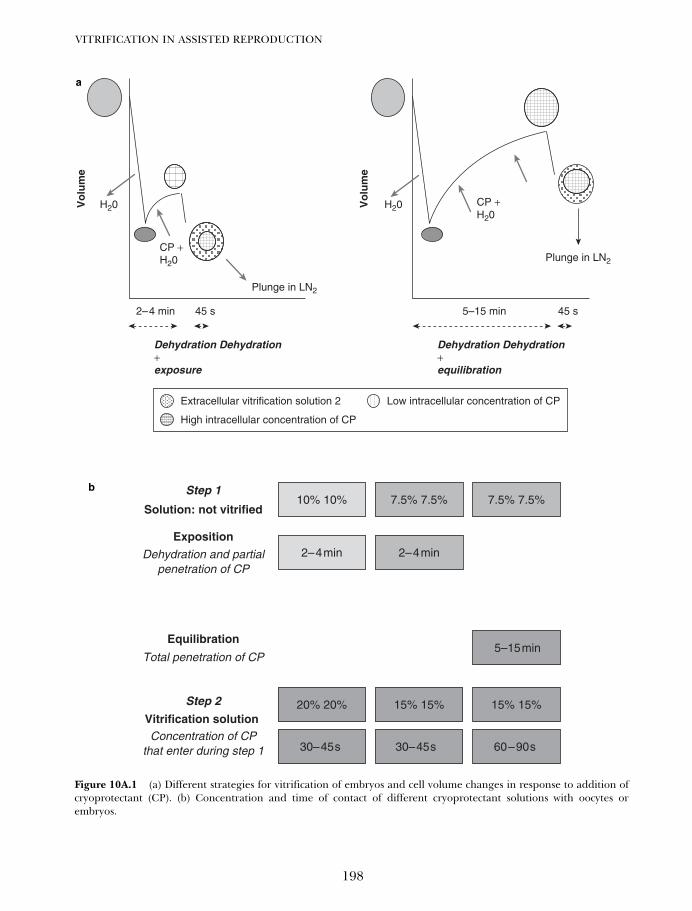
VITRIFICATION IN ASSISTED REPRODUCTION
198
Dehydration Dehydration+exposure
H20 H20
CP +H20
CP +H20
2–4 min 45 s 5–15 min 45 s
Dehydration Dehydration+equilibration
Vo
lum
e
Vo
lum
e
Plunge in LN2
Plunge in LN2
Extracellular vitrification solution 2 Low intracellular concentration of CP
High intracellular concentration of CP
10% 10% 7.5% 7.5%
Exposition
Dehydration and partialpenetration of CP
7.5% 7.5%
Equilibration
Total penetration of CP5–15 min
Solution: not vitrified
Step 1
2– 4 min2– 4 min
30– 45 s
15% 15% 15% 15%Step 2
Vitrification solution Concentration of CP
that enter during step 1
20% 20%
30– 45 s 60 – 90 s
Figure 10A.1 (a) Different strategies for vitrification of embryos and cell volume changes in response to addition ofcryoprotectant (CP). (b) Concentration and time of contact of different cryoprotectant solutions with oocytes orembryos.
b
a
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 198

vitrifiable state. When the embryos areimmersed in LN2 the whole fluid (intra- andextracellular) has gained such a high viscositythat it solidifies so quickly that the moleculesdo not have time to rearrange themselvesinto a crystalline structure. Thus, an intra-and extracellular vitreous state can beachieved.
Addition in one step or several steps
The method of addition of cryoprotectant isalso an important step to consider especiallyfor oocytes. The cytoskeleton and themetaphase plate are particularly sensitive toosmotic fluctuation and excessive variation involume. For this reason, addition of the cryo-protectants in several steps or by dilution isproposed.17
Second condition: fast coolingand warming
The first successful results were obtained usinga 0.25 mL French mini-straw (IMV-France) asthe storage method for mouse embryos.13
The thickness of the straw walls, the volumeof liquid therein (250 µL), and the formationof a thermal insulating screen (Leidenfrosteffect) around the straw during immersionin the liquid nitrogen made it impossibleto obtain cooling speeds of more than2000°C/min.
Thereafter, particular attention has beenpaid to the cooling rate. Martino et al.,20 Vajtaet al.,21,22 and Lane et al.23 have shown thebeneficial effect of a significant increaseof cooling speed on the survival rate ofcattle oocytes and hamster blastocysts. Suchincreases in the cooling rates up to 20 000°C/min, make it possible to reduce the probabil-ity of ice crystal formation during cooling toeven under insufficient permeation of cryo-protectant. In particular, a high cooling ratehelps to reduce problems linked to ‘chilling’(mainly observed with cryopreservation of
oocytes). The speed of cooling depends onthe type of carrier as well as on the volumeof the sample. The smaller volume a samplehas, the less surface area will hinder contactwith the LN2. In ultrarapid vitrification tech-niques the smallest possible volumes (< 1 µl)of vitrification solutions are deposited on spe-cial supports, giving cooling speeds of around20 000°C/min.
With the aim of reducing the thermal gra-dient, one of the approaches consists of usingopen pulled straws, the walls of which aremade thinner,21,24,25 or to encourage directcontact of the whole microdroplet of cryopro-tectant solution with LN2.
20,23,26–30 One impor-tant step that has not to be underestimated, isthe rate of warming. Devices allowing a directcontact of the droplet with LN2 permit also toattain very high warming rates (> 20 000°C)because the small droplet is directly placed incontact with the dilution medium solution.
DESCRIPTION OF DIFFERENTVITRIFICATION STRATEGIESDEVELOPED DURING THE PASTDECADE IN OUR ART UNITS
Rall and Fahy13 reported on the first ‘in vitro’development of mice embryos after vitrifica-tion in a solution of DMSO, PROH, acetamide,and PEG. The following year, the first birthsafter vitrification of mice and calf morulaeand blastocysts treated with a mixture of glyc-erol and PROH were reported.31,32 Based onour previous experiences in animal mod-els,31,32 and referring to several reports frommammalian species,33–35 in 1996 we decidedto apply vitrification as an alternative tech-nique for handling day 4 and day 5 humanembryos.36 Later a whole spate of papers onthe subject were published, and this tech-nique is now considered to be the best methodfor preserving embryos of certain species ofmammal (e.g. those threatened with extinc-tion) or embryos produced from transgenicstrains.16 In spite of a certain coldness with
199
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 199

which the idea is received in some ARTcenters, nevertheless, mounting interest isbeing shown for this technique among someunits and companies.
Randomized studies comparing slow freez-ing with vitrification have concluded that thevitrification technique is better in terms ofembryo survival rates and pregnancy in mice37
as well as in humans.11,38,39
Over the past decade, several embryo sup-ports have been used and designed with theaim of first improving the survival rate of vit-rified embryos and second to achieve coolingand storage in aseptic conditions.
First approach: vitrification in 0.25 mLFrench mini-straw as embryo carrier
Our first vitrification approach36,40 for cryo-preservation of morulae and blastocysts wasbased on a known and already publishedmethod for vitrification of mouse33 and cattleembryos.35
Description of the carrier system
As mentioned above, the 0.25 mL French mini-straw was the first device that we utilized tovitrify morulae and human blastocysts in anaseptic manner. However, the drawback of suchan approach concerns the low cooling andwarming rates due to the amount of liquid inthe straw, the volume of air inside the straw, andthe large surface area of the straw creating for-mation of a thermal insulating vapor coat. Onepossibility to eliminate the vapor coat that arisesaround the sample is to use a system (VitMaster)that decreases the temperature of LN2 to as lowas −210°C, thereby eliminating the formation ofvapor around the straw.
Moreover, during the direct immersioninto LN2 and/or during the warming step in awater bath collapsing or explosion of thestraws and loss of the sample can occurbecause of extreme pressure changes inpoorly sealed straws.
Methodology of vitrificationand warming
After an exposure of 3 min at room tempera-ture to a solution of EG (20%) in PBS + 20%HSA, the embryos were placed in the vitrifi-cation solution consisting of 40% (v/v) EG +18% (w/v) Ficoll (MW 70 000), and 0.3 mol/Lsucrose. A maximum of two embryos wereloaded directly into a 0.25 mL French strawcontaining vitrification medium. The timeinterval between exposure of the embryos tothe vitrification solution and plunging thestraw into a container filled with LN2 rangedbetween 45 and 60 s. After storage, the strawswere taken out of the LN2 tank and warmedin a water bath at 40°C. The contents of thestraw were expelled into a Petri dish contain-ing 3 mL of 0.5 mol/L sucrose solution atroom temperature. Five minutes after beingflushed out, the embryos were transferred to0.25 mol/L sucrose solution before washingand subsequent culture for 24 h.
Second approach: ultrarapidvitrification using the hemi-straw asembryo carrier
Description of the carrier system
According to different documents reportingthe beneficial effect of a significant increaseof the cooling speed, a method using thesmall volume–direct contact principle wasdeveloped. Therefore, in order to circumventthe problem of intracellular ice formation inless-permeable embryos such as with blasto-cysts, we developed an embryo carrier device(hemi-straw) that allows extremely fast cool-ing and warming rates30 (Figure 10A.2).
The tip of the hemi-straw is designedto hold the embryos (maximum of two) in avery small volume of vitrification solution(< 0.5 µL) thus reducing the thermoinsulat-ing effect of a conventional straw. The advan-tage of the hemi-straw is that the microdropof cryoprotectant is loaded in a gutter and
VITRIFICATION IN ASSISTED REPRODUCTION
200
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 200
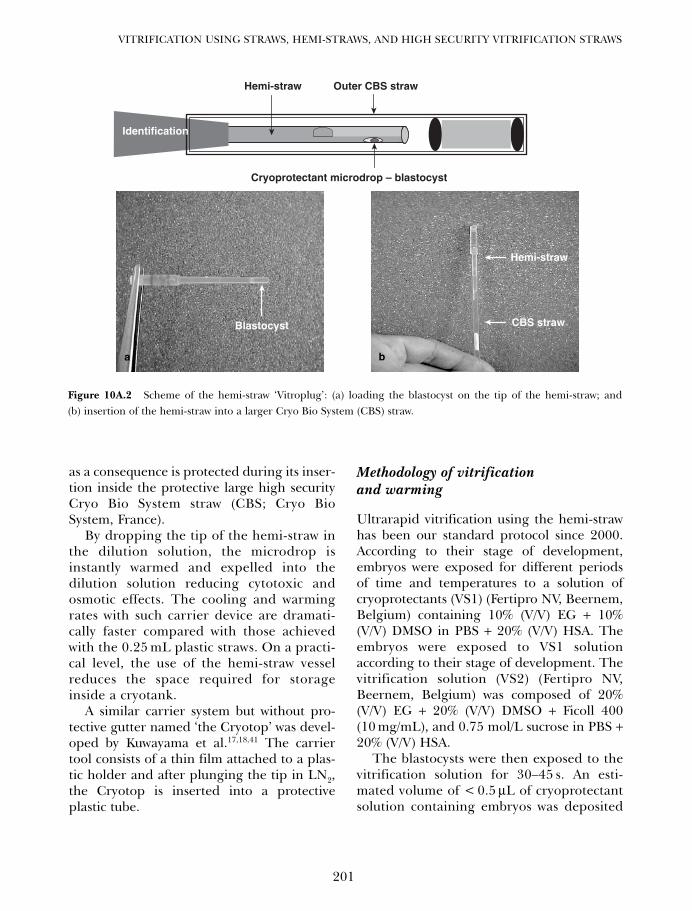
as a consequence is protected during its inser-tion inside the protective large high securityCryo Bio System straw (CBS; Cryo BioSystem, France).
By dropping the tip of the hemi-straw inthe dilution solution, the microdrop isinstantly warmed and expelled into thedilution solution reducing cytotoxic andosmotic effects. The cooling and warmingrates with such carrier device are dramati-cally faster compared with those achievedwith the 0.25 mL plastic straws. On a practi-cal level, the use of the hemi-straw vesselreduces the space required for storageinside a cryotank.
A similar carrier system but without pro-tective gutter named ‘the Cryotop’ was devel-oped by Kuwayama et al.17,18,41 The carriertool consists of a thin film attached to a plas-tic holder and after plunging the tip in LN2,the Cryotop is inserted into a protectiveplastic tube.
Methodology of vitrificationand warming
Ultrarapid vitrification using the hemi-strawhas been our standard protocol since 2000.According to their stage of development,embryos were exposed for different periodsof time and temperatures to a solution ofcryoprotectants (VS1) (Fertipro NV, Beernem,Belgium) containing 10% (V/V) EG + 10%(V/V) DMSO in PBS + 20% (V/V) HSA. Theembryos were exposed to VS1 solutionaccording to their stage of development. Thevitrification solution (VS2) (Fertipro NV,Beernem, Belgium) was composed of 20%(V/V) EG + 20% (V/V) DMSO + Ficoll 400(10 mg/mL), and 0.75 mol/L sucrose in PBS +20% (V/V) HSA.
The blastocysts were then exposed to thevitrification solution for 30–45 s. An esti-mated volume of < 0.5 µL of cryoprotectantsolution containing embryos was deposited
201
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
Blastocyst
Outer CBS straw
Cryoprotectant microdrop – blastocyst
Hemi-straw
Hemi-straw
CBS straw
Identification
Figure 10A.2 Scheme of the hemi-straw ‘Vitroplug’: (a) loading the blastocyst on the tip of the hemi-straw; and(b) insertion of the hemi-straw into a larger Cryo Bio System (CBS) straw.
a b
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 201

VITRIFICATION IN ASSISTED REPRODUCTION
202
using a fine pipette into the tip of the troughof the hemi-straw. The hemi-straw was theninstantly plunged into a dewar of LN2 andwith the aid of forceps inserted in a largerpre-cooled straw (CBS) before closing it(Figure 10A.3). Before warming, a dewar ofLN2 containing the hemi-straw was placedclose to the microscope. Under LN2 andwith forceps, the hemi-straw was pulled outof the larger straw and the tip of the strawholding the embryos was immediatelyimmersed and stirred into a Petri dish con-taining 3 mL of 0.5 mol/L sucrose at 37°C.After 2–3 min, the blastocysts were trans-ferred to different bath of 0.25 mol/L sucroseat intervals of 3–4 min (Table 10A.1). Theblastocysts were then washed several times inPBS + HSA solution, then placed in culturemedium (Figure 10A.3B).
Third approach: asepticvitrification using High SecurityVitrification carrier
Problems linked with an open system
Vitrification has suffered from differentdrawbacks that have hampered its widerapplication. Although successful resultswere reported with the previous carrier sys-tems, one of the main remaining drawbacksconcerns the possible risk of contaminationby bacteria, fungi, and viruses from the LN2,or by contamination with another sampleeven if the probability of the latter is low.The different methods do not provide aleak proof environment for cryopreservedembryos.
Liquid nitrogen in storage tanks probablycontains several pathogenic environmentalmicro-organisms.42 These could be acquiredat the time of distribution of LN2, or throughstorage by deposition of ice and microbesfrom the air, through inadequate asepsis of sur-faces of straws or cryovials, or as a result of cross-contamination with unprotected samples.42–45
For clinical application, it is therefore manda-tory to cool and store the embryos in asepticconditions, to minimize the likelihood ofmicrobial transmission during storage, whileat the same time retaining acceptable survivalrates. Various aseptic approaches such as theuse of sterile LN2, storage in nitrogen vapor,and hermetically sealed and isolated contain-ers have been proposed and designed.46
Mouse embryos have been vitrified using Cry-oloops which were put into the vapor of liquidnitrogen. However, there is an obvious risk ofaccidental warming and the greater fluctua-tions in storage temperature should not beunderestimated. Various aseptic closed sys-tems such as the ‘open pulled straw (OPS) instraw’,47 or the sealed OPS ‘Cryotip’18 havebeen recently developed.
Description of the High SecurityVitrification kit
In response to the demand stated above, withrespect to European regulation, our ARTunit, in collaboration with Cryo Bio SystemCompany, developed an aseptic vitrificationkit based on the hemi-straw principle (Figure10A.4). The system is composed of two dis-tinct parts, a high security thermal autogenicsealed clear mini-straw, and a split capillarytube with its extremity in the form of a gutter.This High Security Vitrification kit (HSV kit)makes it possible to place a microdroplet(< 0.5 µL) of cryoprotectant containing theembryos in the gutter of a small capillary tubebefore inserting it in a mini-straw. It is heat-sealed using a special welder which ensures aleak-proof seal, before immersion in the LN2.Cooling rates of 1627°C/min have beenrecorded.48
Methodology of vitrificationand warming
The methodology is similar to that applied withthe hemi-straw carrier system. The embryos
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 202
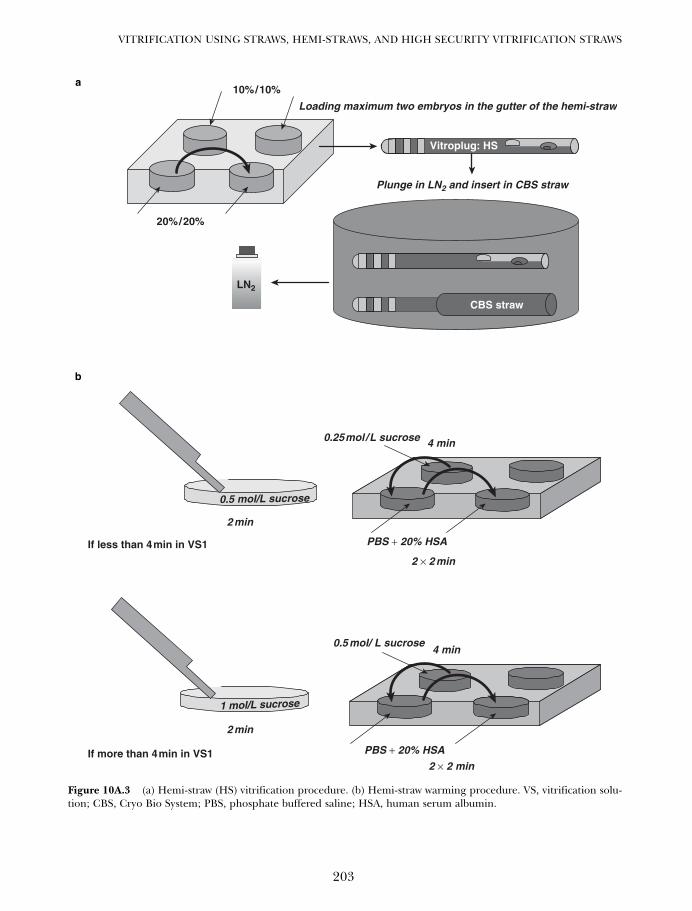
203
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
CBS straw
Vitroplug: HS
Loading maximum two embryos in the gutter of the hemi-straw
20% / 20%
10% / 10%
Plunge in LN2 and insert in CBS straw
LN2
0.5 mol/ L sucrose
PBS + 20% HSA
1 mol/L sucrose
0.25 mol / L sucrose
PBS + 20% HSA
0.5 mol/L sucrose
2 min
2 min
4 min
4 min
2 × 2 min
2 × 2 minIf more than 4 min in VS1
If less than 4 min in VS1
Figure 10A.3 (a) Hemi-straw (HS) vitrification procedure. (b) Hemi-straw warming procedure. VS, vitrification solu-tion; CBS, Cryo Bio System; PBS, phosphate buffered saline; HSA, human serum albumin.
a
b
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 203

VITRIFICATION IN ASSISTED REPRODUCTION
204
2
1
3
4
Figure 10A.4 High Security Vitrification Kit: 1 Mini hemi-straw, 2 250ml high security straw; 3 mini hemi-strawinside the high security straw; and 4 complete sealed system (VHS).
Table 10A.1 Protocols for vitrification of embryos at different stages of development with the hemi-straw.
Before cooling Warming dilution
VS1 VS2 Sucrose (mol/L)
10/10% (min) Temperature 20/20% (s) Temperature 1 (min) 0.5 (min) 0.25 (min)
Zygotes 6–8 RT 45 RT 2–3 3–4 3–46–8 cells 3 RT 45 RT 2–3 3–4Morulae 2 RT 45 RT 2–3 3–4Early blastocysts 2 RT 45 RT 2–3 3–4Blastocysts 3 37° C 45 37° C 2–3 3–4Full blastocyst + AS 2–3 37° C 45 37° C 2–3 3–4Expanded blastocyst + AS 2–3 37° C 45 37° C 2–3 3–4Intact full blastocysts 7–10 RT 45 RT 2–3 3–4 3–4Intact expanded blastocysts 7–10 RT 45 RT 2–3 3–4 3–4
AS, artifical shrinkage before vitrification; RT, room temperature ~ 25°C; Cryoprotectant, ethylene glycol + DMSO.
were vitrified after exposure to VS1 solutionsas presented in Table 10A.1. After 15 s in thevitrification solution (VS2), an extremely smallamount (< 0.5 µL) of solution containing the
embryos was deposited at the tip of thetrough of a HSV using a fine pipette. TheHSV was inserted inside a small straw, sealed,and then plunged into LN2. For warming, the
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 204
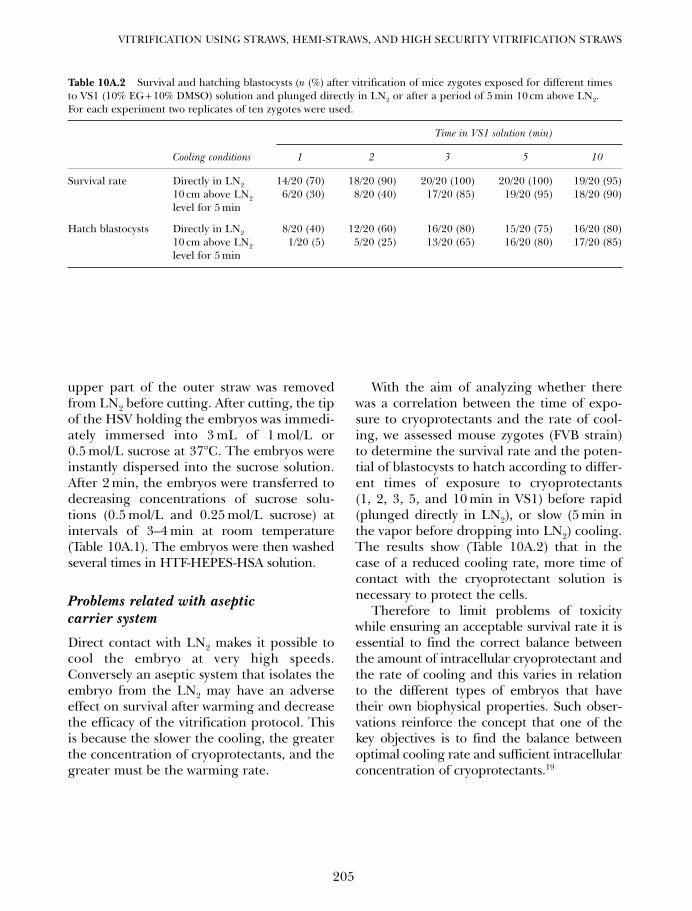
upper part of the outer straw was removedfrom LN2 before cutting. After cutting, the tipof the HSV holding the embryos was immedi-ately immersed into 3 mL of 1 mol/L or0.5 mol/L sucrose at 37°C. The embryos wereinstantly dispersed into the sucrose solution.After 2 min, the embryos were transferred todecreasing concentrations of sucrose solu-tions (0.5 mol/L and 0.25 mol/L sucrose) atintervals of 3–4 min at room temperature(Table 10A.1). The embryos were then washedseveral times in HTF-HEPES-HSA solution.
Problems related with asepticcarrier system
Direct contact with LN2 makes it possible tocool the embryo at very high speeds.Conversely an aseptic system that isolates theembryo from the LN2 may have an adverseeffect on survival after warming and decreasethe efficacy of the vitrification protocol. Thisis because the slower the cooling, the greaterthe concentration of cryoprotectants, and thegreater must be the warming rate.
With the aim of analyzing whether therewas a correlation between the time of expo-sure to cryoprotectants and the rate of cool-ing, we assessed mouse zygotes (FVB strain)to determine the survival rate and the poten-tial of blastocysts to hatch according to differ-ent times of exposure to cryoprotectants(1, 2, 3, 5, and 10 min in VS1) before rapid(plunged directly in LN2), or slow (5 min inthe vapor before dropping into LN2) cooling.The results show (Table 10A.2) that in thecase of a reduced cooling rate, more time ofcontact with the cryoprotectant solution isnecessary to protect the cells.
Therefore to limit problems of toxicitywhile ensuring an acceptable survival rate it isessential to find the correct balance betweenthe amount of intracellular cryoprotectant andthe rate of cooling and this varies in relationto the different types of embryos that havetheir own biophysical properties. Such obser-vations reinforce the concept that one of thekey objectives is to find the balance betweenoptimal cooling rate and sufficient intracellularconcentration of cryoprotectants.19
205
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
Table 10A.2 Survival and hatching blastocysts (n (%) after vitrification of mice zygotes exposed for different timesto VS1 (10% EG + 10% DMSO) solution and plunged directly in LN2 or after a period of 5 min 10 cm above LN2.For each experiment two replicates of ten zygotes were used.
Time in VS1 solution (min)
Cooling conditions 1 2 3 5 10
Survival rate Directly in LN2 14/20 (70) 18/20 (90) 20/20 (100) 20/20 (100) 19/20 (95)10 cm above LN2 6/20 (30) 8/20 (40) 17/20 (85) 19/20 (95) 18/20 (90)level for 5 min
Hatch blastocysts Directly in LN2 8/20 (40) 12/20 (60) 16/20 (80) 15/20 (75) 16/20 (80)10 cm above LN2 1/20 (5) 5/20 (25) 13/20 (65) 16/20 (80) 17/20 (85)level for 5 min
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 205

CLINICAL APPLICATION OFVITRIFICATION OF HUMANEMBRYOS – WHERE DO WESTAND?
In spite of a certain reluctance in some ARTcenters as mentioned above, nevertheless,mounting interest is being shown for this tech-nique among some units and companies.19,49,50
Vitrification of zygotes
High rates of survival (81–100%), and growthup to the blastocyst stage have been recordedfor zygotes exposed for short periods to cryo-protectant solutions and using non-hermeti-cally sealed supports such as the flexibledenuding pipette,51 copper grid,52 OPS,53,54
and Cryotop.18,41 Using aseptic supports(straw in straw) Isachenko et al.47 reported asurvival rate of 75%, and 25% of the zygoteswent on to grow to the blastocyst stage. Theyconcluded that reducing the cooling speeddoes not have any major negative effect onthe later growth of an embryo.
Zygotes like oocytes are the largest cell in thebody, have a high water content, and one singlemembrane. Such large cells have a low surfaceto volume ratio, hence they are less efficient attaking up cryoprotectant and at losing water.Isachenko et al. reported that out of 5881 vitri-fied zygotes using the Cryotop as embryo sup-port, that were equilibrated in 7.5% EG + 7.5%DMSO, 93% survived and 52% developed tothe blastocyst stage.
Vitrification of day 3 embryos
The first pregnancy following vitrificationof embryos at the 8-cell stage was reportedby Mukaida et al.55 after exposure of theembryos to a mixture of ethylene glycol +Ficoll and sucrose, and loading them into0.25 mL french mini-straws as support. ElDanasouri and Selman56 using the OPS ascarrier system, vitrified 215 embryos on day
3 of culture. Survival rates of 79% and40% were observed following vitrificationof embryos with 8 and 6–7 blastomeres,respectively. After transfer, a pregnancy rateof 30% and implantation rate of 10% wereobserved. More recently, Gottumukkalaet al.57 cryopreserved day 3 embryos both byvitrification using a Cryoloop, and by slowfreezing in a randomized manner. Thestudy showed significantly higher rates ofsurvival (95% vs. 60%), pregnancies (37% vs.17%), and implantation (15% vs. 4%) follow-ing vitrification as compared with slowfreezing. After vitrifying more than 8904-cell embryos using a Cryotop, Kuwayamaet al.18 recorded significantly higher rates ofsurvival and pregnancies compared withslow cryopreservation (98% vs. 91%).Liebermann and Tucker24 reported that thehemi-straw support was easy to use and suc-cessful for vitrification of different stages ofdevelopment such as day 3 embryos.
Our experience with vitrification ofday 3 embryos
Table 10A.3 shows the results of vitrificationof day 3 embryos using different cryoprotec-tant solutions and the hemi-straw as embryosupport. We observed that the results were inrelation to the concentration of the vitrifica-tion solution (VS2) suggesting that the vit-rifcation state is probably not fully obtainedwhen loading the embryos in the 15% EG +15% DMSO solution. It may be that an incu-bation time of 2–3 min in the first solution istoo short to allow sufficient intra-cellularpermeation of cryoprotectant that has to beconcentrated during the second dehydrationphase in the vitrification VS2 solution. Usinga more concentrated cryoprotectant VS2 solu-tion such as 20% EG + 20% DMSO allowsmore rapid dehydration of the cells, andas a consequence a threshold level of soluteis reached that has the property of anamorphous state.
VITRIFICATION IN ASSISTED REPRODUCTION
206
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 206
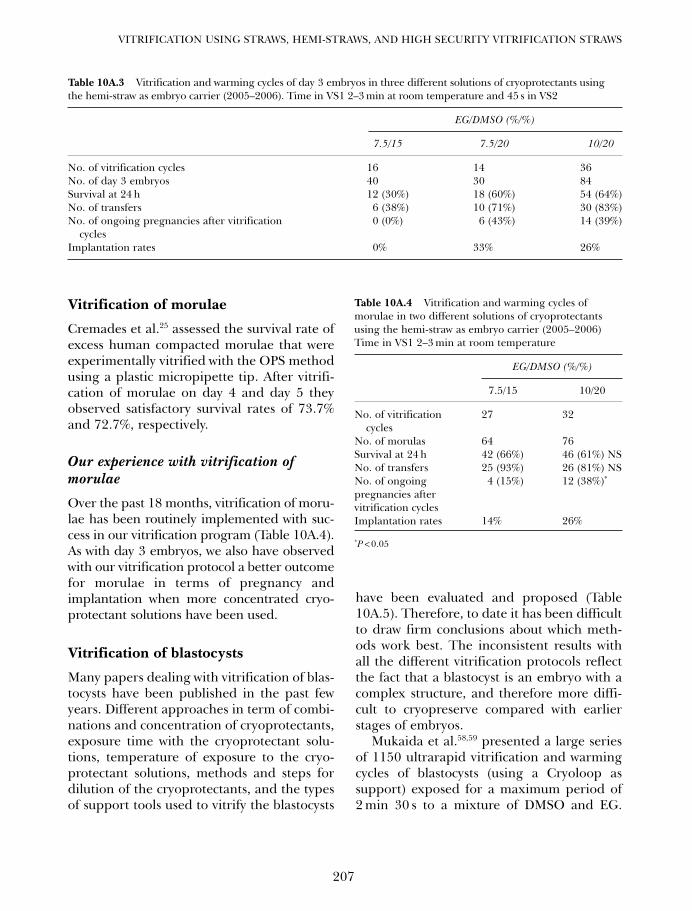
Vitrification of morulae
Cremades et al.25 assessed the survival rate ofexcess human compacted morulae that wereexperimentally vitrified with the OPS methodusing a plastic micropipette tip. After vitrifi-cation of morulae on day 4 and day 5 theyobserved satisfactory survival rates of 73.7%and 72.7%, respectively.
Our experience with vitrification ofmorulae
Over the past 18 months, vitrification of moru-lae has been routinely implemented with suc-cess in our vitrification program (Table 10A.4).As with day 3 embryos, we also have observedwith our vitrification protocol a better outcomefor morulae in terms of pregnancy andimplantation when more concentrated cryo-protectant solutions have been used.
Vitrification of blastocysts
Many papers dealing with vitrification of blas-tocysts have been published in the past fewyears. Different approaches in term of combi-nations and concentration of cryoprotectants,exposure time with the cryoprotectant solu-tions, temperature of exposure to the cryo-protectant solutions, methods and steps fordilution of the cryoprotectants, and the typesof support tools used to vitrify the blastocysts
have been evaluated and proposed (Table10A.5). Therefore, to date it has been difficultto draw firm conclusions about which meth-ods work best. The inconsistent results withall the different vitrification protocols reflectthe fact that a blastocyst is an embryo with acomplex structure, and therefore more diffi-cult to cryopreserve compared with earlierstages of embryos.
Mukaida et al.58,59 presented a large seriesof 1150 ultrarapid vitrification and warmingcycles of blastocysts (using a Cryoloop assupport) exposed for a maximum period of2 min 30 s to a mixture of DMSO and EG.
207
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
Table 10A.3 Vitrification and warming cycles of day 3 embryos in three different solutions of cryoprotectants usingthe hemi-straw as embryo carrier (2005–2006). Time in VS1 2–3 min at room temperature and 45 s in VS2
EG/DMSO (%/%)
7.5/15 7.5/20 10/20
No. of vitrification cycles 16 14 36No. of day 3 embryos 40 30 84Survival at 24 h 12 (30%) 18 (60%) 54 (64%)No. of transfers 6 (38%) 10 (71%) 30 (83%)No. of ongoing pregnancies after vitrification 0 (0%) 6 (43%) 14 (39%)
cyclesImplantation rates 0% 33% 26%
Table 10A.4 Vitrification and warming cycles ofmorulae in two different solutions of cryoprotectantsusing the hemi-straw as embryo carrier (2005–2006)Time in VS1 2–3 min at room temperature
EG/DMSO (%/%)
7.5/15 10/20
No. of vitrification 27 32cycles
No. of morulas 64 76Survival at 24 h 42 (66%) 46 (61%) NSNo. of transfers 25 (93%) 26 (81%) NSNo. of ongoing 4 (15%) 12 (38%)*
pregnancies aftervitrification cyclesImplantation rates 14% 26%
*P < 0.05
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 207
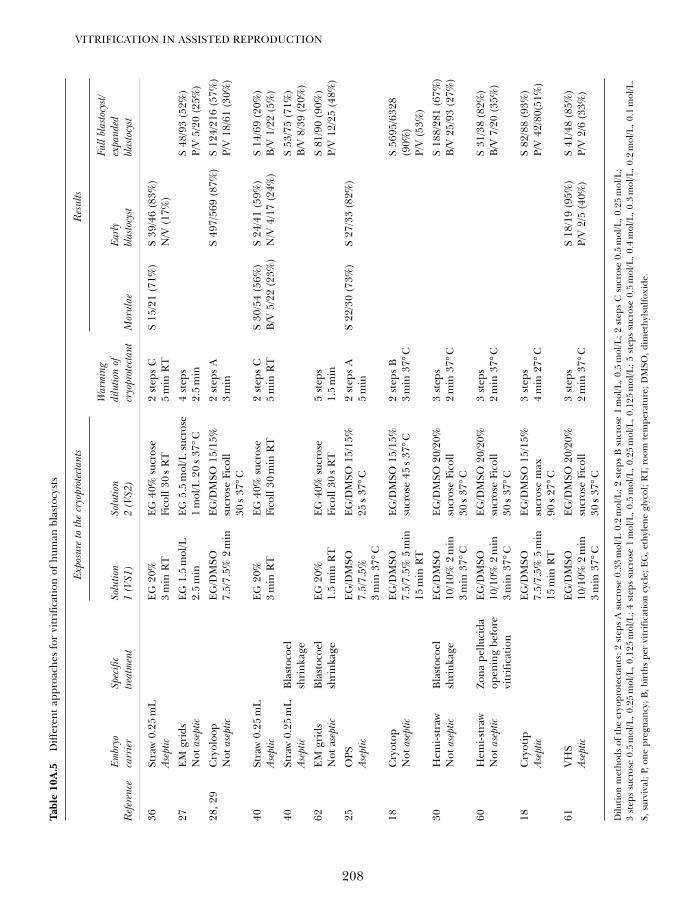
VITRIFICATION IN ASSISTED REPRODUCTION
208
Tabl
e 10
A.5
Diff
eren
t ap
proa
ches
for
vitr
ifica
tion
of h
uman
bla
stoc
ysts
Exp
osur
e to
the
cryo
prot
ecta
nts
Res
ults
War
min
gFu
ll bl
asto
cyst/
Em
bryo
Sp
ecifi
c So
lutio
n So
lutio
n di
lutio
n of
Ear
ly
expa
nded
Ref
eren
ceca
rrie
rtre
atm
ent
1 (V
S1)
2 (V
S2)
cryo
prot
ecta
ntM
orul
aebl
asto
cyst
blas
tocy
st
36St
raw
0.2
5m
LE
G 2
0%E
G 4
0% s
ucro
se
2 st
eps
CS
15/2
1 (7
1%)
S 39
/46
(83%
)As
eptic
3m
in R
TFi
coll
30s
RT
5m
in R
TN
/V (
17%
)
27E
M g
rids
EG
1.5
mol
/LE
G 5
.5m
ol/L
suc
rose
4
step
sS
48/9
3 (5
2%)
Not
ase
ptic
2.5
min
1m
ol/L
20
s 37
°C2.
5m
inP/
V 5
/20
(25%
)
28, 2
9C
ryol
oop
EG
/DM
SO
EG
/DM
SO 1
5/15
%
2 st
eps
AS
497/
569
(87%
)S
124/
216
(57%
)N
ot a
sept
ic7.
5/7.
5% 2
min
sucr
ose
Fico
ll 3
min
P/V
18/
61 (
30%
)30
s 37
°C40
Stra
w 0
.25
mL
EG
20%
E
G 4
0% s
ucro
se
2 st
eps
CS
30/5
4 (5
6%)
S 24
/41
(59%
)S
14/6
9 (2
0%)
Asep
tic3
min
RT
Fico
ll 30
min
RT
5m
in R
TB
/V 5
/22
(23%
)N
/V 4
/17
(24%
)B
/V 1
/22
(5%
)
40St
raw
0.2
5m
LB
last
ocoe
lS
53/7
5 (7
1%)
Asep
ticsh
rink
age
B/V
8/3
9 (2
0%)
62E
M g
rids
Bla
stoc
oel
EG
20%
E
G 4
0% s
ucro
se
5 st
eps
S 81
/90
(90%
)N
ot a
sept
icsh
rink
age
1.5
min
RT
Fico
ll 30
s R
T1.
5m
inP/
V 1
2/25
(48
%)
25O
PSE
G/D
MSO
E
G/D
MSO
15/
15%
2 st
eps
AS
22/3
0 (7
3%)
S 27
/33
(82%
)As
eptic
7.5/
7.5%
25
s 37
°C5
min
3m
in 3
7°C
18C
ryot
opE
G/D
MSO
E
G/D
MSO
15/
15%
2 st
eps
BS
5695
/632
8 N
ot a
sept
ic7.
5/7.
5% 5
min
sucr
ose
45s
37°C
3m
in 3
7°C
(90%
)15
min
RT
P/V
(53
%)
30H
emi-s
traw
Bla
stoc
oel
EG
/DM
SO
EG
/DM
SO 2
0/20
%3
step
sS
188/
281
(67%
)N
ot a
sept
icsh
rink
age
10/1
0% 2
min
su
cros
e Fi
coll
2m
in 3
7°C
B/V
25/
93 (
27%
)3
min
37°
C30
s 37
°C60
Hem
i-str
awZo
na p
ellu
cida
E
G/D
MSO
E
G/D
MSO
20/
20%
3 st
eps
S 31
/38
(82%
)N
ot a
sept
icop
enin
g be
fore
10
/10%
2m
in
sucr
ose
Fico
ll 2
min
37°
CB
/V 7
/20
(35%
)vi
trifi
catio
n3
min
37°
C30
s 37
°C18
Cry
otip
EG
/DM
SO
EG
/DM
SO 1
5/15
%3
step
s S
82/8
8 (9
3%)
Asep
tic7.
5/7.
5% 5
min
su
cros
e m
ax
4m
in 2
7°C
P/V
42/
80(5
1%)
15m
in R
T90
s 27
°C61
VH
SE
G/D
MSO
E
G/D
MSO
20/
20%
3 st
eps
S 18
/19
(95%
)S
41/4
8 (8
5%)
Asep
tic10
/10%
2m
in
sucr
ose
Fico
ll 2
min
37°
CP/
V 2
/5 (
40%
)P/
V 2
/6 (
33%
)3
min
37°
C30
s 37
°C
Dilu
tion
met
hods
of
the
cryo
prot
ecta
nts:
2 s
teps
A s
ucro
se 0
.33
mol
/L 0
.2m
ol/L
; 2 s
teps
B s
ucro
se 1
mol
/L, 0
.5m
ol/L
; 2 s
teps
C s
ucro
se 0
.5m
ol/L
, 0.2
5m
ol/L
;3
step
s su
cros
e 0.
5m
ol/L
, 0.2
5m
ol/L
, 0.1
25m
ol/L
; 4 s
teps
suc
rose
1m
ol/L
, 0.5
mol
/L, 0
.25
mol
/L, 0
.125
mol
/L; 5
ste
ps s
ucro
se 0
.5m
ol/L
, 0.4
mol
/L, 0
.3m
ol/L
, 0.2
mol
/L, 0
.1m
ol/L
.
S, s
urvi
val;
P, o
ne p
regn
ancy
; B, b
irth
s pe
r vi
trifi
catio
n cy
cle;
EG
, eth
ylen
e gl
ycol
; RT,
roo
m t
empe
ratu
re; D
MSO
, dim
ethy
lsul
foxi
de.
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 208

209
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
After warming 2749 blastocysts they recordeda survival rate of 89.7%. Out of a total of543 early pregnancies (49.1%), 174 devel-oped gestations and 247 deliveries (295babies including 153 boys and 142 girls)were recorded. An implantation rate of 34.5%was observed. It should be noted, as wedescribe below, that the blastocoelic cavity ofblastocysts and expanding blastocysts weresystematically artificially collapsed.
Also, presenting a large series of vitrifica-tion using a different approach, Kuwayamaet al.18 obtained pregnancy rates of 59% and51%, using a Cryotop (open, non-asepticsystem) and Cryotip (sealed, aseptic system)supports, respectively. In their protocol theblastocysts were no longer exposed to cryo-protectant solutions, but instead equilibratedfor an extended period of time (rangingbetween 5 and 15 min) in a DMSO + EG solu-tion until the moment when they return toalmost their original volume.
Our experience with vitrificationof blastocysts
In 1995 prolonged embryo culture to day 5was implemented in our ART activities. As aconsequence, for one decade, the greater part
of our experience with vitrification has beenwith embryos that have developed in culturefor 5 days.
During these past 10 years, a total of 664vitrification and warming cycles of blastocystshave been undertaken using the 0.25 mLFrench straw (n = 78), the hemi-straw(n = 554), and recently the aseptic HSV kit(n = 22) as embryo support systems.
Vitrification of blastocysts in 0.25 mL Frenchstraw Our results showed that applicationof the vitrification method using EG + Ficoll +sucrose solution and loading the embryos in a0.25 mL French straw was effective for thecryopreservation of day 4 and day 5 earlyblastocysts. With this method we obtained thefirst pregnancy after vitrification of a humanblastocyst.36 However, it was questionablewhether this simple aseptic method wasapplicable to blastocysts and expandedblastocysts. In fact, our preliminary resultsshowed that the efficiency of the vitrificationmethod was dependent on the stage ofembryo development, and was negatively cor-related to the expansion of the blastocoel(Table 10A.6). Only 20% of the blastocystsand expanded blastocysts survived the vitrifi-cation and warming procedure with the
Table 10A.6 Overall results after vitrification and warming of morulae (n = 22) and blastocysts (n = 78) in 0.25 mLFrench mini-straw: effect of the developmental stage and artificial shrinkage of the blastocoel.
Without artificial shrinkage With artificial shrinkage
Blastocysts BlastocystsEarly expanded expanded
Morulae blastocysts blastocysts blastocysts
No. of vitrification–warming cycles 22 17 22 39No. of vitrified–warmed embryos 55 41 71 75Survival after 24 hours (%) 54.5 58.5 20.3* 74.6Mean embryos transferred 1.7 1.5 2.0 1.4Births per vitrification–warming cycle (n (%)) 5 (22.7%) 4 (23.5%) 1 (4.5%) 8 (20.5%)Implantation per (%)
transferred embryo 20.0 16.7 7.1 18.4vitrified embryos 10.9 9.8 1.4 12.0
P < 0.001 (χ2 test)
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 209

French 0.25 mL straw as compared with a50% survival rate for day 4 morulae and day5 early blastocysts.
Vitrification of blastocysts with the hemi-strawsupport Between the years 2000 and 2006,a total of 554 vitrification and warming cycleswith the hemi-straw carrier system wereundertaken in a group of women agedbetween 21 and 44 years. After 505 transfers3 h or 24 h after the warming step of an aver-age of 2.1 embryos, a total of 179 patientsbecame pregnant, out of which 141 ongoingpregnancies resulted (with 101 deliveries atthe time of going to press). The ongoingpregnancy rates were 25.4% and 27.9% pervitrification and warming cycle, and embryotransfer attempts, respectively. The implanta-tion rates per transferred embryo and pervitrified and warmed blastocysts were 15.8%and 10.2%, respectively.
Vitrification of blastocysts with the HighSecurity Vitrification kit A prospective ran-domized analysis of the survival rates onsibling day 5 embryos vitrified at a lowercooling rate inside a hermetically sealedstraw demonstrated comparable results withthe control non-aseptic group in hemi-straws.61 With both techniques, the warmingrate is extremely fast because the tip of bothsystems is plunged directly into a bath ofsucrose at 37°C/min. This reinforces the con-cept that the warming rate is as important asthe cooling rate especially in case of lowcooling rate or low exposure time with thecryoprotectant.
Twenty-two vitrification and warmingcycles of blastocysts in the HSV kit have beenperformed. After warming of 106 blastocysts,91 survived. Fifty-eight were transferred in afirst group to 18 patients after 3 h of addi-tional culture thereby establishing four(22.2%) ongoing pregnancies. In a secondgroup, 13 blastocysts re-expanded after 24 hin culture, and after four transfers of ten
embryos, two ongoing pregnancies wereobtained.
Factors affecting the outcome ofblastocyst vitrification
Several factors whether or not they are associ-ated with vitrification may influence the out-come, and may explain differences in theresults between published studies.
Time of embryo transfer: 3 h or 24 hafter warming
Significantly higher pregnancy rates wereobserved when embryo transfers were per-formed 24 h as compared with embryo trans-fers 3 h after warming.
During a period of 14 months, 64 and 95blastocyst transfers were performed at 3 or24 h after warming, respectively. The percent-ages of blastocysts showing signs of survivalin the 3 h group (blastocoel cavity starting todevelop) and in the 24 h culture group (fullre-expansion or hatching) were 86.1% and79.4%, respectively (no significant differ-ence). Nevertheless, the pregnancy rate pervitrified and warming cycle in the 3 h culturegroup was 14.7% (14/95) compared with29.7% (19/64) when transfers occurred 1 daylater. The implantation rate per transferredembryo in the 3 h culture group was 8.9%(16/179) compared with 20.9% (19/91) afteran additional 24 h of in vitro culture.
Such results are very intriguing, but it isdifficult to find a good explanation. After24 h culture, it is possible to detect more accu-rately which embryos have the higher capac-ity to further develop. Immediately afterwarming and dilution of the cryoprotectant,it is difficult to assess exactly the viability ofthe embryos at the stereomicroscopic leveleven though some signs of re-expansion mayoccur. Under high magnification (× 400), ifonly minor morphological changes aredetectable, a prolonged culture period may
VITRIFICATION IN ASSISTED REPRODUCTION
210
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 210
be necessary to evaluate the re-expansion andviability of the embryos.
Selection of blastocysts for vitrification
An important parameter to consider beforestarting the vitrification process concernsembryo selection. We can conclude from ourdata (Table 10A.7) that the quality of the blas-tocysts selected before vitrification has animportant impact on the capacity of the blas-tocyst to survive the vitrification and todevelop further. If poor quality blastocysts arevitrified, then the probability for them to sur-vive the vitrification and warming procedureis rather low.
When compared with day 3 embryo selec-tion, the selection of blastocysts is more diffi-cult to perform. In fact, classification ofblastocysts between ‘good’ and ‘moderate’can vary between biologists. It is not always aneasy task to judge the quality of a blastocystaccording to the type and the quality of eitherthe inner cell mass or the trophectoderm
cells. This part of the work is vital for theoutcome of the embryo after warming.
Moreover, impaired embryonic develop-ment may also contribute to a reduction inthe capacity to produce an optimal blastocystthat will survive the vitrification procedure.After vitrification of blastocysts that originatefrom good quality early stage embryos, thesurvival rates observed immediately afterthawing, the percentage of blastocysts thathatch and re-expand after 24 h culture, andthe proportion of blastocysts available forembryo transfer, are all considerably higherwhen compared with those already exhibitingpoor embryo quality during cleavage stage.We observed a reduced likelihood of successafter vitrification of morphologically normal-looking blastocysts that originated fromcleavage stage embryos with suboptimal mor-phology. The post-thaw survival rates dependon the cleavage characteristics during the5 days of culture. Only those developing fromgood quality cleavage stage embryos couldbe cryopreserved and thawed with goodresults.30
211
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
Table 10A.7 Overall results after vitrification and warming of blastocysts exhibiting different quality beforevitrification. Embryo transfers 24 h after warming step.
Quality of blastocysts before vitrification using the hemi-straw
Bad Good Mixedquality quality quality Total
No. of vitrification–warming cycles 113 211 135 459No. of vitrified–warmed embryos 361 537 481 1379Survival after 24 h (n) 194 414 320 928Survival after 24 h (%) 53.7* 77.1 66.5* 67.3Transfers (n (%)) 88 (77.9) 205 (97.1) 121 (89.6) 414 (90.2)No. of embryos transferred 194 393 261 848Mean no. of embryos transferred 2.2 1.9 2.2 2.0Birth/Ongoing pregnancy per
vitrification–warming cycle (n (%)) 12 (10.6) 76 (36.0) 36 (26.6) 124 (27.0)transfers (%) 13.6 37.1 30.0 30.0
Implantation per (%)transferred embryos 6.2 24.0 15.3 17.2vitrified embryos 3.3 17.5 8.3 10.6
Twins 0 18 4 22
*P < 0.001.
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 211

This underlines the need for good scoringand selection of embryos through all wholestages of in vitro development. Only with suchan approach, can the best embryos be identifiedand selected, not only for a fresh embryo trans-fer but also for future cryopreserved cycles.
Degree of blastocoelic expansion
The surface to volume ratio of blastomeres inthe blastocysts is higher and even increaseswith an increase in the degree of expansion ofthe blastocyst compared with that of blas-tomeres from embryos in their early stage ofdevelopment. As a consequence, we mayexpect a faster rate of entry of the permeatingcryoprotectant, and more rapid intracellularprotection in the expanded blastocyst. Inaddition, the cytoplasm of the blastomerescontains various intrinsic macromoleculesthat increase during the dehydration phasecontributing to the formation of an amor-phous state. Nevertheless, our experience isthat the ability of blastocysts to survive the vit-rification procedure depends on the degreeof expansion of the blastocoel, and was nega-tively correlated with the expansion of theblastocoel (Table 10A.6). Full blastocysts andexpanded blastocysts are more likely to sur-vive the vitrification procedure.
We postulated40 that a large blastocoel mightdisturb cryopreservative potential due to icecrystal formation during the cooling step. Dueto the short exposure time to the cryoprotec-tant solutions (maximum exposure time in VS14 min), the large blastocoelic cavity is not suffi-ciently protected against formation of ice crys-tals. Therefore, in order to verify whether theblastocoel is the source of the problem, we ana-lyzed in the first instance the effect of reducingthe cavity, and subsequently the effect of alonger exposure to the cryoprotectant.
Artificial shrinkage of the blastocoel Weinvestigated the impact that a reduction inblastocoel fluid before vitrification has on
post-thaw embryo survival, and demonstratedan increase in the survival rates after artificialshrinkage of the blastocoel cavity using amicroneedle.40
The beneficial effect of this procedureprior to vitrification was later confirmed byother ART units by applying a 29-gaugeneedle,62 a hand-drawn Pasteur pipette,63 andmore recently also by using a laser pulse.64
They reported survival and pregnancy ratesof almost 90% and 45%, respectively.Artificial shrinkage prior vitrification hasshown no adverse effects on the developmentof blastocysts and on the health status onbabies born to date.
Full equilibration with the cryoprotectantsolution If the reduced viability of blastocystsis truly related to an insufficient permeationof cryoprotectant inside the cavity, otherapproaches such as increasing the time ofexposure to cryoprotectant solutions can beproposed to prevent ice crystal formation. Withconventional slow-freezing procedures, theblastocysts are equilibrated first in a solution ofcryoprotectant for a period ranging from 10 to20 min. If blastocysts still survived after slow-cooling steps to –30°C to –40°C, and thenplunging in LN2, we could postulate that thereason for those embryos to survive wasbecause an amorphous state in the blastomeresand inside the cavity had formed. Based on thisdeduction, recently we started to equilibrateblastocysts for a longer time period in a lowconcentration of cryoprotectants in order toallow full equilibration inside the cavity. Thelow concentration of cryoprotectants is morebeneficial when considering the cytotoxic effectof high concentration of cryoprotectants at pro-longed exposure time. After this initial equili-bration step, blastocysts were exposed for ashorter period to VS1 and VS2, before loadingthem on to the hemi-straw or HSV, and plung-ing into LN2. The preliminary results demon-strate, after vitrification and warming of 40intact full and expanded blastocysts, are-expansion rate of 97.5% 24 h after warming.
VITRIFICATION IN ASSISTED REPRODUCTION
212
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 212

In vitro culture conditions: reduction ofcryotolerance
One factor that can have an effect on the via-bility of blastocysts lies in the in vitro cultureconditions. The introduction of sequentialculture media allows for production of blasto-cysts without the need for co-culture usingfeeder cells. Notwithstanding the fact thatfresh blastocysts give good implantationrates, it has been shown65 that cryopreservedIVF human embryos obtained in sequentialmedia are less viable than cryopreserved blas-tocysts after co-culture. According to Massipet al.66 inappropriate in vitro culture condi-tions can also explain the low pregnancy ratesobtained after blastocyst freezing. It isincreasingly confirmed that culture condi-tions influence the cryotolerance of theembryo, and that a high survival rate of cryo-preserved, in vitro cultured embryos requiresimprovement in the techniques of maturationand culture, rather than simple changes incryopreservation methods.67,68
WHY HAS THERE BEEN A LACK OFINTEREST IN VITRIFICATION?
Several articles have been published claimingthat vitrification is an optimal method for thecryopreservation of embryos of several mam-malian species.11,16,50,69 Yet despite the publi-cation of encouraging results,18,28,39 there hasbeen a notable lack of enthusiasm for intro-ducing this technique to ART programs.
The lack of an aseptic embryo supportsystem has been one reason for this lack ofinterest. Another cause of concerns is the highconcentrations of cryoprotectants utilized; butis this the reality? The need to expose embryosto high concentrations of cryoprotectants(30–50% v/v) has aroused a certain mistrust inmany ART centers about using this method ofcryopreservation for humans. Yet there are var-ious methods of limiting the toxicity of cryo-protectants. Use of a solution containing twodifferent cryoprotectants lowers the relative
toxicity of each one. Another alternative consistsof adding macromolecules to the cryoprotec-tant solution. These polymers, with high mol-ecular weights, are generally less toxic and canprotect embryos against the effects of cold byincreasing the viscosity of the extracellularsolution.14 It is also possible to reduce thetoxicity of vitrification solutions by loweringthe temperature of cryoprotectants or theexposure time before cooling. Penetrationspeed of the cryoprotectant is affected by tem-perature, and it has been shown that priorequilibration of mouse blastocysts with a mix-ture of PROH and glycerol at 4°C has a benefi-cial effect.70 A disadvantage of this approach liesin the difficulty of maintaining the solutionsat 4°C under the microscope.
The crucial question remains about thequantity of cryoprotectant that may jeopardizeembryonic survival and growth. It is essentialto determine a balance between prevention ofintracellular ice crystal formation and preven-tion of toxic effects. As in most vitrificationmethods, exposures to cryoprotectant solu-tions are of short duration (Table 10A.1), andthe quantity of intracellular cryoprotectants isprobably similar or even lower than those pre-sent in slow freezing. Indeed we have observedthat one can achieve an acceptable survivalrate by placing the vitrified embryos directlyinto a sucrose solution at 0.33 mol/L, and evendirectly into the PBS. Following immersion ofblastocysts in a 0.5 mol/L sucrose solution (63vitrification cycles) or directly into the PBS (55vitrification cycles) survival rates of 60% and71% were obtained, respectively. Followingtransfer of on average 1.7 embryos, pregnancyrates of 27% and 24%, and implantation ratesof 18% and 16% were obtained in each group,respectively.
Observations of similar survival rates in theabsence or presence of sucrose, suggest thatlow levels of cryoprotectants are present inthe cell. This is because rapid penetration ofthe cell by water, due to a high intracellularconcentration of cryoprotectants, would makethe cell swell beyond its critical volume before
213
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 213

the cryoprotectants are removed. Such astrategy cannot be used after slow freezing,because direct dilution in an isotonic solutionwould lead to immediate cell lysis followingtoo rapid hydration of the cell well beforeremoval of the cryoprotectants.
Over 15 years ago Kasai et al.71 showedthat EG can be considered to be a cryoprotec-tant of very low toxicity in terms of embryosurvival, and is therefore now considered as areference cryoprotectant for vitrifying oocytesand embryos. On the other hand, a study byKlug et al.72 recorded an embryo-toxic effectof EG and more particularly of its metabolitesin contact with rat embryos (on days 9.5–11.5of gestation). According to Vatja and Nagy19
no study has shown any growth abnormalityin animal or human embryos following vitri-fication using EG. Despite these findings onemust remain cautious.
Other paths remain to be explored, such asthe use of other permeable cryoprotectants(PROH), or addition of polymers at higherconcentration in order to reduce the amountof permeating cryoprotectants.14 In a recentstudy Takahashi et al.73 did not record anydifference in terms of malformations after thebirth of children produced by transfers offresh and vitrified blastocysts.
CONCLUSIONS AND PROSPECTS
Are programmable freezers still needed inthe embryo laboratory? It seems evident thatvitrification methods will continue to improvein the coming years, thanks to a resurgence of
interest of scientists in this field.19 It may bethat in future vitrification will prove to beeffective for all stages of embryo development.However, in spite of the promising resultsobtained so far, the procedure will have to beadapted first to each type of embryo carriersystem. The higher is the cooling rate, thelower is the required cryoprotectant concen-tration and vice versa.19 Also, it will be neces-sary to adjust our methods according to thedifferent stages of embryo development inorder to find the most favorable conditions ofexposure, in terms of time and temperature,to cryoprotectant solutions in relation to therate of cooling that depends on the type ofembryo carrier device used. For zygotes, orblastocysts with a large blastocoel, a longerexposure time with the cryoprotectant solu-tion seems a better approach to obtainacceptable survival and pregnancy rates. Forday 3 embryos, morulae, and blastocysts withan artificial reduced cavity shorter exposureto the cryoprotectant will be sufficient to protectthe embryos.
In order to optimize the technique, vitrifi-cation has undergone several changes. Theuse of ultrarapid cooling has made it possibleto improve results, but because of the everpresent risk of contamination, alternative sys-tems of embryo supports, such as the Cryotipor the high security vitrification straw, thateliminate contact with LN2, have recentlybeen developed. The results, expressed interms of embryo survival rates and pregnan-cies, do not seem to be adversely affected inany way by this security adjustment.
VITRIFICATION IN ASSISTED REPRODUCTION
214
References
1. Zeilmaker G, Alberda A, van Gent I et al. Twopregnancies following transfer of intact frozen-thawed embryos. Fertil Steril 1984; 2: 293–6.
2. Cohen J, Simons R, Fehilly CB et al. Birthafter replacement of hatching blastocyst cryo-preserved at expanded blastocyst stage. Lancet1985; 1: 647.
3. Ménézo Y, Nicollet B, Herbaut N et al.Freezing cocultured human blastocysts. FertilSteril 1992; 58: 977–80.
4. Kaufman R, Ménézo Y, Hazout A et al.Cocultured blastocysts cryopreservation; expe-rience of more than 500 transfer cycles. FertilSteril 1995; 64: 1125–9.
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 214

5. Langley M, Marek D, Gardner D et al.Extended embryo culture in human assistedreproduction treatment. Hum Reprod 2001;16: 902–8.
6. Behr B, Gebhardt J, Lyon J et al. Factors relat-ing to a successful cryopreserved blastocysttransfer program. Fertil Steril 2002; 77:697–9.
7. Veeck L, Bodine R, Clarke R et al. High preg-nancy rates can be achieved after freezing andthawing human blastocysts. Fertil Steril 2004;82: 1418–27.
8. Virant- Klun I, Tomazevic T, Bacer-KermavnerL et al. Successful freezing and thawing ofblastocysts cultured in sequential media usinga modified method. Fertil Steril 2003; 79:1428–33.
9. Ludwig M, Al-Hasani S, Felderbaum R et al.New aspect of cryopreservation of oocytes andembryos in assisted reproduction and futureperspectives. Hum Reprod 1999; 14 (Suppl 1):162–85.
10. Plachot M, Belaisch-Allart J, Mayenga J et al.Blastocyst stage transfer: the real benefitscompared with early embryo transfer. HumReprod 2000; 15 (Suppl 6): 24–30.
11. Stehlik E, Stehlik J, Katayama K et al.Vitrification demonstrates significant improve-ment versus slow freezing of human blastocysts.Reprod Biomed Online 2005; 11: 53–7.
12. Whittingham D, Leibo S, Mazur P. Survival ofmouse embryos frozen to −196° and −269°C.Science 1972; 178: 411–14.
13. Rall W, Fahy G. Ice-free cryopreservation ofmouse embryos at −196°C by vitrification.Nature 1985; 313: 573.
14. Kuleshova L, Shaw J, Trounson A. Studies onreplacing most of the penetrating cryoprotec-tant by polymers for embryo cryopreservation.Cryobiology 2001; 43: 21–31.
15. Rall W. Factors affecting the survival of mouseembryos cryopreserved by vitrification.Cryobiology 1987; 24: 387–402.
16. Kuleshova L, Lopata A Vitrification can bemore favorable than slow cooling. Fertil Steril2002; 78: 449–54.
17. Kuwayama M, Vajta G, Kato O et al.Highly efficient vitrification method forcryopreservation of human oocytes. ReprodBiomed Online. 2005; 11: 300–8.
18. Kuwayama M, Vajta G, Leda S et al.Comparison of open and closed methods forvitrification of human embryos and theelimination of potential contamination.Reprod Biomed Online. 2005b; 11: 608–14.
19. Vajta G, Nagy Z. Are programmable freezersstill needed in the embryo laboratory? Review
on vitrification. Reprod Biomed Online 2006;7: 623–33.
20. Martino A, Songsasen N, Leibo S.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
21. Vajta G, Holm P, Greve T et al. Vitrification ofporcine embryos using the Open Pulled Straw(OPS) method. Acta Vet Scand 1997; 38:349–52.
22. Vajta G, Kuwayama M, Holm P et al. Openpulled straw vitrification: a new way to reducecryoinjuries of bovine ova and embryos. MolReprod Dev 1998; 51: 53–8.
23. Lane M, Schoolcraft WB, Gardner D.Vitrification of mouse and human blastocystsusing a novel cryoloop container-less tech-nique. Fertil Steril 1999; 72: 1073–78.
24. Liebermann J, Tucker M. Effect of carriersystem on the yield of human oocytes andembryos as assessed by survival and devel-opmental potential after vitrification. Reprod2002; 124: 483–9.
25. Cremades N, Sousa M, Silva J et al.Experimental vitrification of human com-pacted morulae and early blastocysts usingfine diameter plastic micropipettes. HumReprod 2004; 19: 300–5.
26. Hamawaki A, Kuwayama M, Hamano S.Minimum volume cooling method for bovineblastocyst vitrification. Theriogenology 1999;51: 165.
27. Choi D, Chung H, Lim J et al. Pregnancy anddelivery of healthy infants developed from vit-rified blastocysts in an IVF-ET program. FertilSteril 2000; 74: 838–39.
28. Mukaida T, Nakamura S, Tomiyama T et al.Vitrification of human blastocysts using cryo-loops: clinical outcome of 223 cycles. HumReprod 2003a; 18: 384–91.
29. Mukaida T, Nakamura S, Tomiyama T et al.Successful birth after transfer of vitrified humanblastocysts with use of a cryoloop containerlesstechnique. Fertil Steril 2001; 76: 618–20.
30. Vanderzwalmen P, Bertin G, Debauche C et al.Vitrification of human blastocysts with theHemi-Straw carrier: application of assistedhatching after thawing. Hum Reprod 2003;18: 1504–11.
31. Scheffen B, Vanderzwalmen P, Massip A et al. Asimple and efficient procedure for preservationof mouse embryos by vitrification. CryoLetters; 1986; 7: 260–9.
32. Massip A, Vanderzwalmen P, Scheffen B et al.Pregnancies following transfer of cattleembryos preserved by vitrification. Cryo-Letters1986; 7: 270–3.
215
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 215

33. Kasai M, Komi JH, Takakamo A. A simplemethod for mouse embryo cryopreservation ina low toxicity vitrification solution, withoutappreciable loss of viability. J Reprod Fertil1990; 89: 91–7.
34. Kasai M, Nishimori M, Zhu SE et al. Survivalof mouse morulae vitrified in an ethyleneglycol-based solution afer exposure to thesolution at various temperatures. Biol Reprod1992; 47: 1134–39.
35. Mahmoudzadeh A, Van Soom A, Ysebaert Met al. Comparison of two-step vitrificationversus controlled freezing on survival ofin vitro produced cattle embryos.Theriogenology, 1994; 42: 1387–97.
36. Vanderzwalmen P, Delval A, ChatziparasidouA et al. Pregnancies after vitrification ofhuman day 5 embryos. Hum Reprod 1997; 12(Suppl): 98.
37. Walker D. Vitrification versus programmablerate freezing of late stage murine embryos: arandomised comparison prior to applicationin clinical IVF. Reprod Biomed Online 2004;8: 558–68.
38. Zheng W, Zhuang G, Zhou C et al. Comparisonof the survival of human biopsied embryosafter cryopreservation with four differentmethods using non-transferable embryos.Hum Reprod 2005; 20: 1615–18.
39. Liebermann J, Tucker MJ. Comparison of vit-rification and conventional cryopreservationof day 5 and day 6 blastocysts during clinicalapplication. Fertil Steril 2006; 86: 20–6.
40. Vanderzwalmen P, Bertin G, Debauche C et al.Births after vitrification at morula and blasto-cyst stages: effect of artificial reduction of theblastocoelic cavity before vitrification. HumReprod 2002; 17: 744–51.
41. Kuwayama M. Highly efficient vitrification forcryopreservation of human oocytes andembryos: the Cryotop method. Theriogenology2006; 67: 73–80.
42. Bielanski A, Bergeron H, Lau PCK et al.Microbial contamination of embryos andsemen during long-term banking in liquidnitrogen. Cryobiology 2003; 46: 146–52.
43. Tedder R, Zuckerman M, Goldstone A et al.Hepatitis-B transmission from contaminatedcryopreservation tank. Lancet 1995; 346:137–40.
44. Bielanski A, Nadin-Davis S, Sapp T et al. Viralcontamination of embryos cryopreserved inliquid nitrogen. Cryobiology 2000; 40: 110–1.
45. Fountain D, Ralston M, Higgins N et al.Liquid nitrogen freezers: a potential source ofmicrobial contamination of hematopoietic
stem cell components. Transfusion 1997; 37:585–91.
46. Larman M, Sheenan C, Gardner D.Vitrification of mouse pronuclear oocytes withno direct liquid nitrogen contact. ReprodBiomed Online 2006; 12: 66–9.
47. Isachenko V, Montag M, Isachenko E et al.Aseptic technology of vitrification of humanpronuclear oocytes using open-pulled straws.Hum Reprod 2005; 20: 492–6.
48. Camu A, Clairaz P, Ersham A et al. Thecomparison of the process of five differentvitrification devices. Gynécol Obstét Fertil2006; 34: 737–45.
49. Kasai M, Mukaida T Cryopreservation of ani-mal and human embryos by vitrification.Reprod Biomed Online 2004; 9: 164–70.
50. Lieberman J, Dietl J, Vanderzwalmen P et al.Recent developments in human oocytes,embryo and blastocyst vitrification: where arewe now? Reprod Biomed Online 2003; 7:623–33.
51. Liebermann J, Tucker M, Graham J et al.Blastocyst development after vitrification ofmultipronucleate zygotes using the Flexipetdenuding pipette. Reprod Biomed Online2002; 4: 146–50.
52. Park S, Kim E, Oh J et al. Ultra-rapid freezingof human multipronuclear zygotes usingelectron microscope grids. Reprod 2000; 15:1787–90.
53. Jelinkova L, Selman H, Arav A et al. Twinpregnancy after vitrification of 2 pronucleihuman embryos. Fertil Steril 2002; 77: 412–4.
54. Selman H, El-Danasouri I. Pregnanciesderived from vitrified human zygotes. FertilSteril 2002; 77: 422–3.
55. Mukaida T, Wada S, Takahashi K et al.Vitrification of human embryos based on theassessment of suitable conditions for 8-cellmouse embryos. Hum Reprod 1998; 13:2874–9.
56. El-Danasouri I, Selman HA. Successful preg-nancies and deliveries after a simple vitrifica-tion protocol for day 3 human embryos. FertilSteril 2001; 76: 400–2.
57. Gottumukkala A, Geddam B, Kota K et al.,Vitrification of human 8-cell embryos, a mod-ified protocol for better pregnancy rates.Reprod Biomed Online 2005; 11: 434–7.
58. Mukaida T, Takahashi K, Kasai M. Blastocystcryopreservation: ultrarapid vitrification usingcryoloop technique. Reprod Biomed Online2003; 6: 221–5.
59. Mukaida T, Takahashi K, Goto T et al. Ultra-rapid vitrification using cryoloop technique
VITRIFICATION IN ASSISTED REPRODUCTION
216
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 216

for human blastocyst cryopreservation. FertilSteril 2005; 84 (Suppl) S454.
60. Zech N, Lejeune B, Zech H et al. Vitrificationof hatching and hatched human blastocysts:effect of an opening in the zona pellucidabefore vitrification. Reprod Biomed Online2005; 11: 355–61.
61. Vanderzwalmen P, Lejeune B, Stecher A et al.Survival of day 3 and day 5 embryos followingvitrification in aseptic and non-aseptic condi-tions: a prospective randomized analysis.Fertil Steril 2005; 84 (Suppl), S175.
62. Son W, Yoon S, Yoon H et al. Pregnancy out-come following transfer of human blastocystsvitrified on electron microscopy grids afterinduced collapse of the blastocoele. HumReprod 2003; 18: 137–9.
63. Hiraoka K, Hiraoka K, Kinutani M, KinutaniK. Blastocoele collapse by micropipettingprior to vitrification gives excellent survivaland pregnancy outcomes for human day 5 and6 expanded blastocysts. Hum Reprod 2004;19: 2884–8.
64. Mukaida T, Oka C, Goto T et al. Artificialshrinkage of blastocoeles using either a micro-needle or a lazer pulse prior to the coolingsteps of vitrification improves survival rate andpregnancy outcome of vitrified human blasto-cysts. Hum Reprod 2006; 21: 3246–52.
65. Dumont-Hassan, M, Aubriot, F, Cohen, J et al.Viability of cryopreserved blastocysts: sequen-tial medium versus coculture. 11th WorldCongress on In Vitro Fertilization and HumReprod Genetics 1999; 129: 230, abstr.
66. Massip, A, Mermillod P. Dinnyes A. Morphologyand biochemistry of in-vitro produced bovine
embryos: implications for their cryopreservation.Hum Reprod 1995; 10: 3004–11.
67. Rizos D, Gutierrez-Adan A, Perez-Garnelo Set al. Bovine embryo culture in the presence orabsence of serum: implication for blastocystsdevelopment, cryotolerance, and messengerRNA expression. Biol Reprod 2003; 68:236–43.
68. Mucci N, Aller J, Kaiser G et al. Effect ofestrous cow serum during bovine embryoculture on blastocyst development and cryo-tolerance after slow freezing or vitrificationTheriogenology 2006; 65: 1551–62.
69. Shaw J, Jones G. Terminology associated withvitrification and other cryopreservation proce-dures for oocytes and embryos. Hum ReprodUpdate 2003; 9: 583–605.
70. Vanderzwalmen P, Gaurois B, Ectors F et al.Some factors affecting successful vitrificationof mouse blastocysts. Theriogenology 1988;30: 1177–83.
71. Kasai M, Nishimori M, Zhu S et al. Survival ofmouse morulae vitrified in an ethylene glycol-based solution after exposure to the solution atvarious temperatures. Biol Reprod 1992; 47:1134–39.
72. Klug S, Merker H, Jackh R. Effects of ethyleneglycol and metabolites on in vitro develop-ment of rat embryos during organogenesis.Toxicology In Vitro 2001; 15: 635–42.
73. Takahashi K, Mukaida T, Goto T et al.Perinatal outcome of blastocyst transfer withvitrification using cryoloop: a 4-year follow-upstudy. Fertil Steril 2005; 84: 88–92.
217
VITRIFICATION USING STRAWS, HEMI-STRAWS, AND HIGH SECURITY VITRIFICATION STRAWS
10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 217

10a Tucker 8029.qxd 8/23/2007 2:16 PM Page 218

219
Vitrification of blastocysts using theCryoloop techniqueTetsunori Mukaida and Katsuhiko Takahashi
10B
INTRODUCTION
In assisted reproductive technology (ART),cryopreservation of embryos has provedimportant to enable the best use of super-numerary embryos. In the cryopreservationof embryos, there is a risk of various types ofinjury.1,2 Among them, the formation of intra-cellular ice appears to be the most damaging.The first strategy to prevent intracellular icefrom forming was to adopt a lower concentra-tion of cryoprotectant and a long slow-coolingstage. This slow freezing method has provedeffective for embryos of a wide range of mam-malian species. Unlike embryos of laboratoryanimals and domestic animals, in whichdimethylsulfoxide (DMSO), glycerol, or eth-ylene glycol (EG) is commonly used as thecryoprotectant, human embryos at earlycleavage stages have most often been frozenin a solution of propanediol supplementedwith sucrose,3 although those at the blastocyststage have more frequently been frozen withglycerol and sucrose.4–6 With slow freezing,however, it is difficult to eliminate completelyinjuries occurring from ice formation.Furthermore, the slow freezing methodrequires a long period of time before embryosare stored in liquid nitrogen.
In 1985, Rall and Fahy reported7 an inno-vative approach called vitrification, in whichinjuries related to ice formation are mini-mized by using very high concentrations ofcryoprotectant. This approach simplifies thecooling process, because embryos can berapidly cooled directly in liquid nitrogen.Although embryos subjected to vitrification
are liable to be injured by the toxicity of thehigh concentration of cryoprotectant, themethod has been refined and proved effectivefor the cryopreservation of embryos at vari-ous stages of development in laboratory anddomestic species. In 1998, we showed thatvitrification using an EG-based vitrificationsolution (EFS20–408) with conventionalcryostraws is effective for human embryos atthe 4–8-cell stage.9 The effectiveness of vitri-fication was confirmed for human embryos atthe 8–16-cell stage10 and the morula stage,11
also using EG-based solutions.Recent advances in culture systems with
sequential media have made it possible todevelop human IVF embryos to the blastocyststage quite easily. Because the blastocyst isbetter suited to the uterine environment, andblastocyst formation is a form of selection formore viable embryos, blastocyst transfer hasbecome a promising option to raise the over-all pregnancy rate.12,13 Accordingly, the needto cryopreserve human blastocysts is increas-ing. Menezo et al.14 cryopreserved humanblastocysts which were developed in a co-culturesystem using the slow freezing method withglycerol and obtained reasonable clinicalresults (27% pregnancy rate, 17% implanta-tion rate). However, results reported by otherclinics have not been consistent.15–17 Menezoet al.14 speculated that the cryopreservationoutcome is influenced by the culture condi-tions, such as the co-culture system.
Recently, human blastocysts were success-fully vitrified in straws.18 However, our ownattempts to vitrify human blastocysts usingstraws resulted in only 45% survival (39/86,
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 219

unpublished data). Vanderzwalmen et al.19
also reported a low pregnancy rate withhuman blastocysts vitrified in straws. This isprobably because human blastocysts are muchless permeable to cryoprotectant and water,since we have observed that they shrink moreslowly than mouse and bovine blastocysts inthe cryoprotectant solution. This suggeststhat human blastocysts are more likely to beinjured by intracellular ice.
Increased rates of cooling and warming canhelp circumvent the problem of intracellularice formation in less permeable embryos.Faster rates of cooling and warming can beachieved by minimizing the volume of the solu-tion with which embryos are vitrified, i.e. byusing minute tools such as electron microscopegrids,20 open pulled straws,21 or Cryoloops22 (seeKasai2 for a review). We demonstrated the firstsuccessful birth of a baby following transfer ofhuman blastocysts vitrified using Cryoloops.23
Since our original report, we have continued touse this vitrification approach in our group oftwo clinics (Tokyo, Osaka, Hiroshima HARTclinic) for the cryopreservation of blastocysts onday 5 and day 6. This chapter includes oursummary of the clinical outcomes from ourblastocyst vitrification program for the past 6years, which confirms the effectiveness of theCryoloop technique for the cryopreservation ofhuman blastocysts.
As previously mentioned, the first success-ful outcome of blastocyst vitrification using aCryoloop was originally reported in 2001,and in 2003 the summary of the clinicalresults with 223 warming cycles confirmedthe effectiveness of the Cryoloop techniquefor the cryopreservation of human blasto-cysts. However, the report in 2003 revealedthe survival rates were dependent on thedevelopmental stage of blastocysts, and werenegatively correlated with the expansion ofthe blastocoel.24 The survival rate of earlyblastocysts with a smaller blastocoelic cavity,which was scored 1 and 2 according toGardner’s criteria25 were 87% (48/55) and97% (62/64), respectively. Also, full blastocysts
lacking an expanded blastocoelic cavity,which were scored 3, had a survival rate of89% (99/111). The total survival rate ofblastocysts scored 1–3 was 91% (209/230).However, the survival rate of both expandedand hatching blastocysts, scored 4 and 5,respectively, was 85.0% (288/339), which wassignificantly lower than that of the score 1–3group (P < 0.05). We therefore postulatedthat a large blastocoel might lessen cryo-preservative potential due to ice crystal for-mation during the rapid cooling phase ofvitrification. In order to overcome this prob-lem, shrinkage of the blastocoel was thoughtto be the appropriate approach. Several stud-ies reported an increase in the survival rate ofblastocysts when the volume of the blastocoelwas artificially reduced with glass micro-needle,26 29-gauge needle,27 or micropipettingwith a hand-drawn Pasteur pipette.28
Since 2003, therefore, we have added arti-ficial shrinkage (AS) after puncturing theblastocoel with a microneedle, or laser pulseusing a Cryoloop technique prior to vitrifica-tion to improve the survival rate and clinicaloutcome of vitrified blastocyst transfer pro-grams. In 2006, we reported the effectivenessof AS prior to vitrification, including the con-firmation of the safety of this procedure.29
VITRIFICATION OF BLASTOCYSTS
The protocol for the Cryoloop vitrification ofblastocysts was adopted from a previousreport,22,30 albeit with slight modifications, andhas been described previously (Figures 10B.1and 10B.2).22,23,28 The Cryoloop consisted of anylon loop (20 µm wide; 0.5–0.7 mm in diam-eter) mounted on a stainless steel pipeinserted into the lid of a cryovial (HamptonResearch, Laguna Niguel, CA, USA). A metalinsert on the lid enables the use of a stainlesssteel handling rod with a small magnet(Crystalwand, Hampton Research) for manip-ulation of the loop at low temperature.31
One to three blastocysts were vitrified in aCryoloop after a two-step procedure to load the
VITRIFICATION IN ASSISTED REPRODUCTION
220
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 220

blastocysts with cryoprotectants at ~37°C. Asthe base medium, HEPES-buffered modifiedHTF medium containing 5 mg/mL humanserum albumin (HSA) was used. Initially, blas-tocysts were placed in the base medium con-taining 7.5% (v/v) DMSO and 7.5% (v/v) EG(cryoprotectant solution I). After 2 min, theblastocysts were transferred into cryoprotectantsolution II, which is the base medium contain-ing 15% (v/v) DMSO, 15% (v/v) EG, 10 mg/mLof Ficoll 70 (average molecular weight 70 000;Pharmacia Biotech, Uppsala, Sweden), and0.65 mol/L sucrose (cryoprotectant solution II).Both cryoprotectant solutions had beenwarmed briefly in an incubator at 37°C andblastocysts were handled on the stage warmerof a dissecting microscope at 37°C.
While the blastocysts were suspended incryoprotectant solution II, a Cryoloop wasdipped into cryoprotectant solution II inorder to create a thin, filmy layer of solution,by surface tension, on the nylon loop. Theblastocysts were then washed quickly in solu-tion II and transferred onto the filmy layeron the nylon loop using a micropipette.
Immediately after the loading of blastocysts,the Cryoloop was plunged into liquid nitro-gen. The time blastocysts were exposed tosolution II before cooling was limited to25–30s. Using a stainless steel rod, the loopcontaining the blastocysts was sealed in acryovial, which was previously submerged inliquid nitrogen. The vials were attached tostandard canes and stored in liquid nitrogen.The entire procedure was completed within5 min. Vitrified blastocysts were kept in the liq-uid nitrogen tank for from 1 month to 5 yearsdepending on the patient’s background.
WARMING OF BLASTOCYSTS,ASSISTED HATCHING, ANDASSESSMENT OF SURVIVAL
In a 4-well multidish, ~1 mL of base mediumcontaining 0.33 mol/L sucrose in no. 1 well,base medium containing 0.2 mol/L sucrose inno. 2 well, and base medium in no. 3 well werewarmed briefly in an incubator at 37°C andthen placed on the stage warmer of a
221
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
Freeze loops Cap
Figure 10B.1 (a) Capping portion of the cryovial, which consists of a minute nylon loop (20 µm wide, 0.5–0.7 mm indiameter) mounted on a stainless steel tube inserted into the lid of a cryovial. (b) Container part of the cryovial. Theshape and the size are similar to those of Nunc Cryovials used for semen cryopreservation. (c) The capping portionattached to a stainless steel handling rod with a small magnet for manipulation of the loop at low temperature.
a b
c
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 221
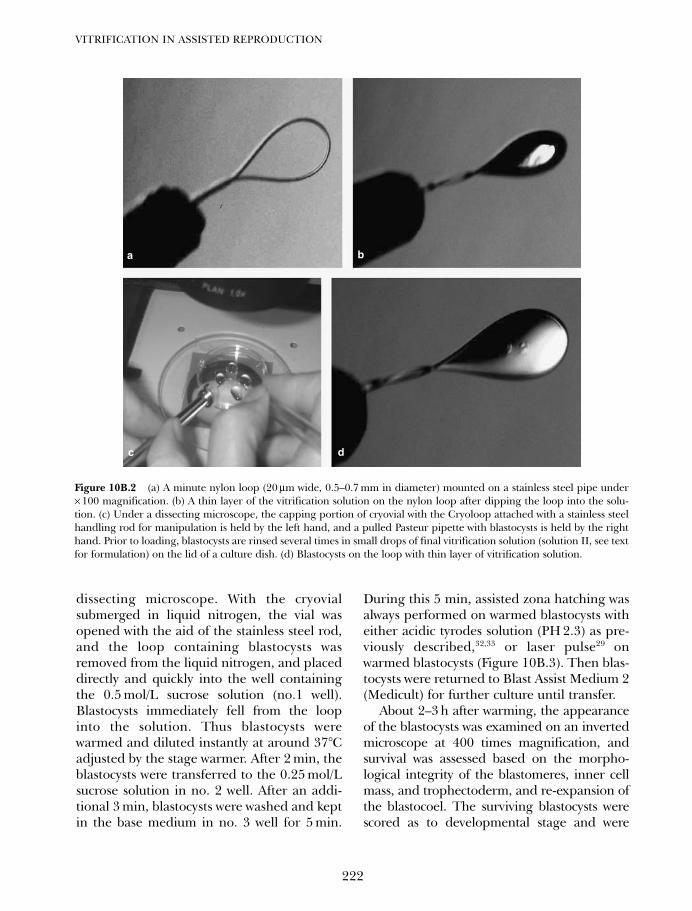
dissecting microscope. With the cryovialsubmerged in liquid nitrogen, the vial wasopened with the aid of the stainless steel rod,and the loop containing blastocysts wasremoved from the liquid nitrogen, and placeddirectly and quickly into the well containingthe 0.5 mol/L sucrose solution (no.1 well).Blastocysts immediately fell from the loopinto the solution. Thus blastocysts werewarmed and diluted instantly at around 37°Cadjusted by the stage warmer. After 2 min, theblastocysts were transferred to the 0.25 mol/Lsucrose solution in no. 2 well. After an addi-tional 3 min, blastocysts were washed and keptin the base medium in no. 3 well for 5 min.
During this 5 min, assisted zona hatching wasalways performed on warmed blastocysts witheither acidic tyrodes solution (PH 2.3) as pre-viously described,32,33 or laser pulse29 onwarmed blastocysts (Figure 10B.3). Then blas-tocysts were returned to Blast Assist Medium 2(Medicult) for further culture until transfer.
About 2–3 h after warming, the appearanceof the blastocysts was examined on an invertedmicroscope at 400 times magnification, andsurvival was assessed based on the morpho-logical integrity of the blastomeres, inner cellmass, and trophectoderm, and re-expansion ofthe blastocoel. The surviving blastocysts werescored as to developmental stage and were
VITRIFICATION IN ASSISTED REPRODUCTION
222
Figure 10B.2 (a) A minute nylon loop (20 µm wide, 0.5–0.7 mm in diameter) mounted on a stainless steel pipe under× 100 magnification. (b) A thin layer of the vitrification solution on the nylon loop after dipping the loop into the solu-tion. (c) Under a dissecting microscope, the capping portion of cryovial with the Cryoloop attached with a stainless steelhandling rod for manipulation is held by the left hand, and a pulled Pasteur pipette with blastocysts is held by the righthand. Prior to loading, blastocysts are rinsed several times in small drops of final vitrification solution (solution II, see textfor formulation) on the lid of a culture dish. (d) Blastocysts on the loop with thin layer of vitrification solution.
a b
dc
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 222

graded according to quality as described in thesection on grading of blastocysts.
ARTIFICIAL SHRINKAGE OFEXPANDED BLASTOCYSTS
When we cryopreserve human blastocystsusing the above vitrification technique at full ormore advanced stages including expanded andhatching blastocysts, the artificial shrinkage(AS) procedure is always performed prior tovitrification (Figures 10B.4 and 10B.5).
The technique of shrinkage using micro-needle puncture has been described previ-ously.25 Briefly, about 10 min before thevitrification, the expanded blastocysts wereplaced in 50 µL drops of pre-equilibrated BlastAssist Medium 2. The expanded blastocyst washeld with a holding pipette connected to themicromanipulator, and the inner cell mass(ICM) was placed at the 6 or 12 o’clock posi-tion; a glass microneedle was pushed throughthe cellular junction of the trophectoderm intothe blastocoel cavity until it shrank (Figure10B.4). After removing the microneedle, con-traction of the blastocoel was observed within afew minutes. After complete shrinkage of the
blastocoel, the blastocyst was vitrified andstored in the liquid nitrogen tank.
Since September of 2004, a laser pulsegenerated by laser system ZILOS-tkTM
(Hamilton Thorn Bioscience Inc., Beverly,MA, USA) has been introduced to performthe artificial shrinkage, instead of micro-needle puncture. The ICM should be locatedaway from the targeted point of the laserpulse. One single laser pulse (200 ms) tar-geted at the cellular junction of the trophec-toderm creates a hole to induce collapsing ofthe blastocoelic cavity. The blastocoel of theexpanded blastocyst shrank immediately(Figure 10B.5). With the use of this laser sys-tem, it is not necessary to hold and locate theexpanded blastocyst with a holding pipetteconnected to micromanipulator. The lasertechnique makes the procedures simple andconvenient.
LABORATORY PROTOCOL
In Appendix 10B.1 of this chapter, our labo-ratory manual of vitrification for human blas-tocysts is detailed in order to help establishthis technique in the clinical ART laboratory.
223
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
Zona hatching bylaser pulse
After 2–3 hr
Figure 10B.3 Assisted hatching using laser pulses on warmed blastocyst prior to transfer.
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 223
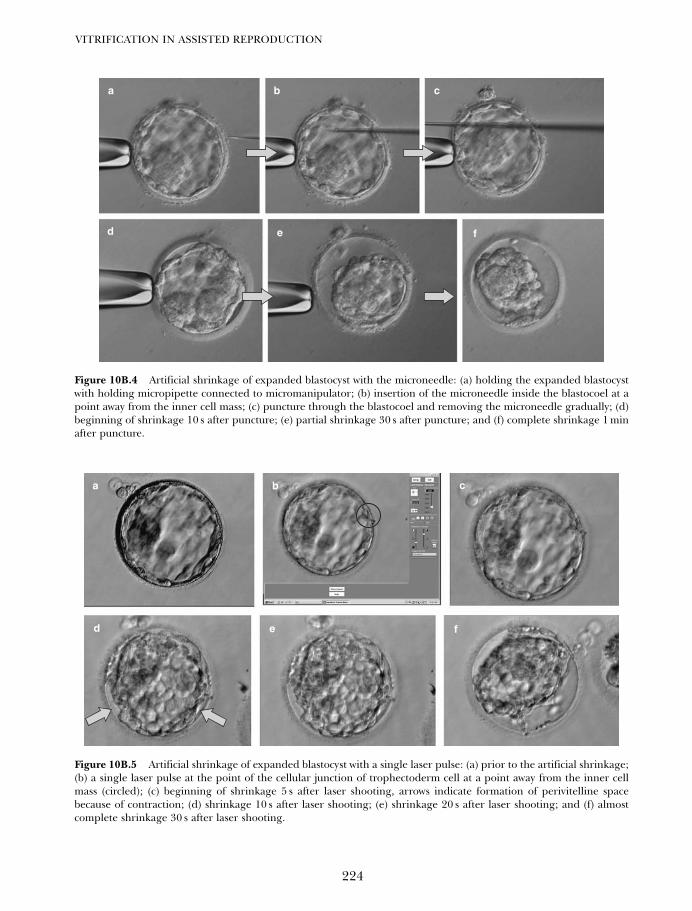
VITRIFICATION IN ASSISTED REPRODUCTION
224
Figure 10B.4 Artificial shrinkage of expanded blastocyst with the microneedle: (a) holding the expanded blastocystwith holding micropipette connected to micromanipulator; (b) insertion of the microneedle inside the blastocoel at apoint away from the inner cell mass; (c) puncture through the blastocoel and removing the microneedle gradually; (d)beginning of shrinkage 10 s after puncture; (e) partial shrinkage 30 s after puncture; and (f) complete shrinkage 1 minafter puncture.
Figure 10B.5 Artificial shrinkage of expanded blastocyst with a single laser pulse: (a) prior to the artificial shrinkage;(b) a single laser pulse at the point of the cellular junction of trophectoderm cell at a point away from the inner cellmass (circled); (c) beginning of shrinkage 5 s after laser shooting, arrows indicate formation of perivitelline spacebecause of contraction; (d) shrinkage 10 s after laser shooting; (e) shrinkage 20 s after laser shooting; and (f) almostcomplete shrinkage 30 s after laser shooting.
a b c
d e f
a b c
d e f
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 224

TROUBLESHOOTING
(1) Warming solutions: Instead of warmingsolutions after preparation in a 4-wellmultidish, it is possible to warm the solu-tions and then prepare them in the dish.
(2) Preventing evaporation of the vitrificationsolution: Because the amount of vitrifica-tion solution on the Cryoloop is very small,the solution is liable to be concentrated byevaporation, which increases its toxicity. Toprevent this, the Cryoloop should be loadedwith the vitrification solution just before theloading of embryo(s) on the loop. Theamount of vitrification solution on the loopneed not be minimal, as long as the drop iskept on the loop by surface tension. A dewarflask containing liquid nitrogen should beplaced nearby to enable quick cooling.
(3) Quick warming: At warming, the loop con-taining embryo(s) should be soaked in thesucrose solution as quickly as possible, and toensure that the loop with vitrified embryo(s)is never held in the air. Again, liquid nitrogenand the sucrose solution should be placednearby. When the loop is dipped in thesucrose solution, the steel pipe portionshould not be immersed to prevent bubbling.
(4) Morphology of the recovered blastocyst:At recovery in an isotonic solution, theblastocoel of the blastocyst will be col-lapsed and the embryo may look like a‘morula’. However, the blastocoel shouldreform within 1–3 h of culture.
(5) Cryoprotectant equilibration: With thisvitrification protocol complete equilibra-tion of cryoprotectant is not expected.Whenever the blastocoelic cavity is large,it is recommended to collapse the cavityin order to improve blastocoel survival.
(6) Temperature equilibration: With thisvitrification protocol equilibration time isjust 2min and exposure of vitrification solu-tion is 30 sec. This means the status insidethe vitrified blastocyst may not be fully
equilibrated and will be metastable situation.Therefore, vitrified embroys should alwaysbe kept under the phase transition (liquid/solid) tempertature (approximately −1300C)in order to avoid fracture damage. When thesamples are transferred between tanks, anunexpected rising in temperature canoccur; it is critical to avoid such transienttemperature fluctuations.
PATIENTS, EMBRYO CULTURE,AND GRADING OF BLASTOCYSTS
We perform blastocyst transfer programs onpatients who have had previous multiple fail-ures of conventional day 2 or day 3 embryotransfer and who have agreed to use of theCryoloop vitrification method to cryopreservetheir supernumerary blastocysts obtained 5 or6 days after oocyte retrieval. Women weretreated with gonadotropin releasing hormone(GnRH) agonist and human menopausalgonadotropin (hMG) using either a long or ashort treatment protocol. An injection ofhuman chorionic gonadotropin (hCG) wasgiven when dominant follicles reached adiameter of ~18 mm. Oocytes were collected36 h after hCG administration using thevaginal ultrasound-guided procedure. Theoocytes were inseminated by either conven-tional IVF or intracytoplasmic sperm injection(ICSI), and were incubated in HTF mediumcontaining 5 mg/mL of HSA or Blast AssistMedium 1, respectively, in a 4-well multidishunder mineral oil in a CO2 incubator at 37°C.
Fertilization was assessed 15–18 h afterinsemination by the presence of two pronu-clei. Zygotes were washed well and cultured ingroups of two or three in Blast Assist Medium1 for 48 h, and then in Blast Assist Medium 2for another 48–72 h. All culturing of embryoswas performed in a CO2, O2, N2 environment(6:5:89%, respectively). In conventional cases,one or two 4–8-cell embryos were transferredto the patient after 24–48 h of culture, andonly supernumerary embryos were extendedto culture in Blast Assist Medium 2.
225
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 225

On day 5 after the oocyte pick up, blastocystdevelopment was examined. Only on day 5,each embryo developed to the blastocyststage was scored depending on the develop-mental stage and graded according to qualitycriteria25, 34 with slight modifications.24
Briefly, blastocysts were first given a numer-ical score from 1 to 6 on the basis of theirdegree of development. Second, the blasto-cysts were graded in three ranks based on mor-phological appearance. For example, the ICMwas graded as A (many tightly packed cells), B(several loosely grouped cells), or C (few cells);and the trophectoderm was graded as A (manycells forming a cohesive epithelium), B (fewercells forming a loose epithelium), or C (veryfew large cells).
When patients had their fresh embryos trans-ferred on day 2–3, all the remaining embryoswere cultured to allow those that developed intoblastocysts to be vitrified. Patients who receivedtransfers of fresh blastocysts had all theirremaining supernumerary blastocysts vitrified.On day 5, if at least one supernumerary blasto-cyst was graded as A or B, all the blastocysts ofthe patient were vitrified regardless of the devel-opmental stage and the grading. In a few cases,compacted morulae were also vitrified with theblastocysts. If all the blastocysts of the patientwere graded C, they were not cryopreserved.On day 6, if at least one blastocyst had a largeblastocoel (i.e. scored as 3–6) and was graded asA or B, all the developed blastocysts scored as3–6 were vitrified.
TRANSFER OF VITRIFIEDBLASTOCYSTS AND ASSESSMENTOF PREGNANCY
All women received transdermal estradiol(Estrana 0.4mg/day; Hisamitsu, Tokyo, Japan)with GnRH agonists for preparation of theendometrium. Administration of both pro-gesterone (50 mg in oil, daily) and hCG10000IU was initiated when the endometrialthickness achieved more than 10mm. On day 5after the initiation of progesterone treatment, the
blastocysts were warmed and surviving blastocystswere transferred into the patient’s uterus. Mostpatients received one or two blastocysts; occa-sionally three blastocysts were transferreddepending on the patient’s background (i.e. mul-tiple failures of ART). In potential pregnancy,serum hCG levels were assessed 14 days afteroocyte pick up (OPU), then implantation andclinical pregnancy was confirmed by the presenceof fetal heart activity or a gestational sac.
CLINICAL RESULTS OF VITRIFIEDBLASTOCYST TRANSFER
Table 10B.1 summarizes the clinical results ofa vitrified blastocyst transfer program usingthe Cryoloop technique between November1999 and April 2006. A total of 3496 blasto-cysts originating from 1549 cycles of oocytecollection from 1258 patients were vitrifiedand warmed. Mean age was 35.2 years.Vitrified blastocysts were generated in threecategories of patient groups. Group 1 hadtheir fresh embryos transferred on day 2–3,and all the remaining embryos were culturedto allow those that developed into blastocyststo be vitrified. Group 2 received transfers offresh blastocysts and had all their remainingsupernumerary blastocysts vitrified. Group 3had no fresh embryo transfer due to eitherovarian hyperstimulation syndrome (OHSS)symptoms or attempting vitrified blastocysttransfer intentionally along with controlledendometrial cycle supplemented by exogenous
VITRIFICATION IN ASSISTED REPRODUCTION
226
Table 10B.1 Clinical outcome of vitrified blastocysttransfer program (November 1999–April 2006)
Attempted cycles (n) 1549 Warmed vitrified blastocysts (n) 3496Survived blastocysts (n) 3176Survival rate (%) 90.8Transferred cycles (n) 1496 Average no. of blastocysts transferred 1.82Clinical pregnancies/blastocyst 750 (50.1)transfer (n (%)Implantation (n %) 945 (38.5)Ongoing pregnancies (n) 213Miscarriages (n %) 174 (23.2)
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 226

female hormones, in order to overcomemultiple implantation failures, because uter-ine receptivity under controlled endometrialcycles was indicated as better than that understimulated cycles. In HART clinics, fresh blas-tocyst transfer is intentionally avoided aftertwo or three failures of fresh transfer. One ofthe reasons why fresh transfer is intentionallyavoided is that ovarian hyperstimulation doesnot always create a suitable uterine receptivityand environment for implantation comparedwith controlled endometrial preparationusing exogenous hormone. Group 1 included580 cycles (37.4% of total), group 2 included540 cycles (34.9% of total), and group 3included 429 cycles (27.7% of total).
After warming of vitrified blastocysts fortransfer, 3176 (90.8%) of the embryos sur-vived. In 45 cycles, no blastocysts survived andembryo transfer was not conducted. In fivecycles, viable blastocysts were obtained butembryo transfer was cancelled because thenumber of cells that survived and the quality ofthe embryos were low. A total of 2722 blasto-cysts were transferred into 1213 patients in1496 cycles. The mean number of blastocyststransferred per cycle was 1.8. Of 1496 trans-fers, 750 resulted in clinical pregnancy (con-firmed by gestational sac in the uterus); thepregnancy rate was 48.4% per warming cycle,and 50.1% per transfer. A total of 174 preg-nant cycles ended in miscarriage (23.2%), with455 healthy babies being born in 361 deliver-ies, and with 213 pregnancies ongoing. Nobias in the sex ratio was observed since 232babies were boys and 223 were girls. In com-parison of 935 pregnancies established fromfresh blastocyst transfer in our group of clinicsduring the same period, 178 (19%) resulted inmiscarriages.
CLINICAL RESULTS OFARTIFICIAL SHRINKAGEPROCEDURES
In order to show the effectiveness of AS, 270cycles in 245 patients who had only expanded
and/or hatching blastocysts vitrified were retro-spectively evaluated. The average age of thepatients was 35.6 years (range 27–41).In total 266 cycles had vitrified blastocyst trans-fer with artificial shrinkage. In four cycles, noblastocysts survived and embryo transfer wascancelled. There were 502 vitrified blastocystswarmed for transfer, and 488 survived. Survivalrate was 97.2%. Of the vitrified blastocysts 448were transferred, and the mean number of blas-tocysts transferred per cycle was 1.7. Of 266transfers, 160 resulted in clinical pregnancy, witha pregnancy rate of 59.3% per warming cycle(160/270) and 60.2% per transfer (160/266).
Results of vitrified expanded and hatchingblastocysts in our previous study reported in2003 served as a control group. Survival rate ofboth expanded and hatching blastocysts, scored4 and 5, respectively, was 85.0% (288/339). Astatistical difference was noted between thestudy and control groups (P< 0.05). When thepregnancy rate of the study group was com-pared with the control group, a statistically sig-nificant improvement was noticed in the ASgroup (60.2% vs. 34.1%; P < 0.01).
Furthermore, we performed a preliminarycomparisons between the results achieved byusing microneedle or laser pulse for blastocoelshrinkage to show the differences betweenmethodologies for AS. AS using a microneedlewas performed in 240 cycles with 462 blasto-cysts, and AS using a laser pulse was per-formed in 26 cycles with 40 blastocysts. Thesurvival rates achieved with the two methodswere similar (microneedle 97.2% vs. laser pulse97.5%). The mean number of survived blasto-cysts transferred was also similar. Clinical preg-nancy, implantation, and miscarriage rateswere also similar. No statistical difference wasobserved in the results achieved with the twomethods (Table 10B.3).
PERINATAL OUTCOME OFVITRIFIED BLASTOCYSTS
We reported the perinatal outcome of our vit-rified blastocyst transfer program in 2004.35
227
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 227
This report summarized the comparisonbetween perinatal outcome after fresh blasto-cyst transfer and that of vitrified blastocyststransfer. Between April 2000 and June 2003,we performed 435 cycles of vitrified-warmedblastocyst transfer in our clinics. Of which,138 were IVF and 297 were ICSI. During thesame period, we performed 662 fresh blasto-cyst transfers. Of which, 184 were IVF and478 were ICSI. Many patients were included
in both groups because they received vitrifiedblastocyst transfer due to failure of concep-tion, or were having their second baby after asuccessful birth following fresh blastocysttransfer. The mean age of women was34.5 ± 4.2 years (range 25–40) in the vitrifiedgroup, and 35.7 ± 4.4 years (range 24–43) inthe fresh group. The vitrified group was sig-nificantly younger (P< 0.05), indicating thatyounger women had a high number of goodquality supernumerary blastocysts vitrified.There were 34 patients in the vitrificationgroup (23.9%) and 43 in the fresh group(21.6%) lost to follow-up (either no responsesor changed address).
The survival rate of vitrified blastocystswas 85.7% (967/1129). We transferred 716warmed-vitrified blastocysts in 413 cycles outof 435 attempted cycles (mean number ofembryo transfers 1.7). Of the cycles trans-ferred, 182 resulted in pregnancy (44.1% pertransfer), and the implantation rate was29.0%. Of which, 40 resulted in miscarriage(22.0%) and 108 deliveries were reported.
In fresh blastocyst transfer, of 662 cycles,transfer was carried out in 602 cycles with1252 blastocysts (mean number of embryostransferred 2.1) and 267 became pregnant(44.4% per transfer), and the implantationrate was 23.4%. Of the pregnancies, 68resulted in miscarriage (25.5%) and 153deliveries were reported. There were nostatistical differences between the groups.
We performed retrospective analysis ofthe data obtained from questionnaires sentto the parents, who were asked to send usinformation after delivery, and/or fromreferring physicians in cases of abnormaloutcome. We obtained 108 responses out of142 parents (76.1%). Of 108 deliveries, weevaluated 147 live-born infants, and com-pared these with 205 live-born infants bornfrom 153 (78.1%) responding parents out of196 who conceived after fresh blastocysttransfer during the same period. From 108deliveries with vitrified blastocysts, 147babies (74 boys and 73 girls) were born. The
VITRIFICATION IN ASSISTED REPRODUCTION
228
Table 10B.2 Perinatal outcome of infants conceivedafter vitrified blastocyst transfer (November1999–April 2006)
n %
Live born 455 (361 deliveries)Male 232 50.9Female 223 49.1Vaginal delivery 164 45.4Cesarean section 197 54.6 Mean gestational 37.5 ± 3.4
age (weeks)Preterm birth (< 37 weeks) 74 20.5Mean birth weight (g) 2663 ± 779
< 1500 151500–2500 180>2 500 260
Twins 76 21.0Triplets 10 2.7
Table 10B.3 Clinical outcome of artificial shrinkageusing either a microneedle or a laser pulse
Microneedle Laser pulse
Cycles with vitrified 240 26blastocyst transfer (n)
Blastocysts vitrified (n) 462 40Survived blastocyst (n) 449 39Survival rate (%) 97.2 97.5Mean no. of blastocysts 1.6 1.4
transferredClinical pregnancies (%) 144 16
60.0 61.5Implantation (n) 191 18Implantation rate (%) 46.5 48.6Cycles miscarried (%) 32 3
22.2% 18.8
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 228

mean gestational age was 38.1 weeks and themean birth weight was 2601 ± 709 g. Therewere 20 preterm deliveries (18.5%); 40 setsof twins (27.8%); and four sets of triplets(5.1%). From 153 deliveries following freshblastocyst transfer, 205 babies (120 boys and85 girls) were born. The male to female ratiowas significantly higher (P < 0.05). Themean gestational age was 37.9 weeks and themean birth weight was 2593 ± 629 g. Therewere 19 preterm deliveries (12.4%); 40 setsof twins (26.1%); and six sets of triplets(3.9%). No statistical differences were seenbetween the groups except for the sex ratiodifference.
With respect to congenital birth defects,two were observed following vitrification(1.4%), and four in the fresh transfer group(2.0%). In the vitrification group, one dizy-gotic twin had Treacher–Collins syndrome ormandibulofacial dystosis, and the other hadpatent ductus arteriosus (PDA). In the freshtransfer group, a baby from a 39-year-oldmother died with 21 trisomy at 2 days post-partum, and three minor defects, includinganal atresia, PDA, and defect of 4th lumbarspine were noted.
The latest results arising betweenNovember 1999 and April 2006 for perinataloutcome are shown in Table 10B.2. A total of455 children were born in 361 deliveries. Ofwhich, 232 babies were male, and 223 babieswere female, therefore no bias in the sex ratiowas observed. Cesarean section was per-formed in 197 deliveries and the mean gesta-tional age was 37.5 weeks. Mean birth weightwas 2663 g. There were 86 (23.7%) multiplepregnancies and 74 births were preterm(20.5%). Twelve babies in 12 deliveries hadeither congenital birth defects or neonatalcomplication (2.7%), including four chromo-somal abnormalities (one trisomy 18, threetrisomy 21), two multiple anomalies, onedeath of unknown cause during delivery,one malformation of the skull, two congeni-tal heart malformations, and two minoranomalies.
DISCUSSION
Numerous protocols for the cryopreservationof mammalian embryos have been reported.The protocols can be classified into four meth-ods: original slow freezing, conventional slowfreezing, conventional vitrification, and ultrarapid vitrification. Although the principles ofcryopreservation are the same, strategies tocircumvent various injuries (especially fromthe formation of intracellular ice) are different.The most suitable protocol should be adoptedfor each case. For certain types of embryossuch as human blastocysts and bovine embryosat earlier stages, ultrarapid vitrification wouldbe a preferable choice, because the survivalrates of embryos cryopreserved by other meth-ods have been low. For other embryos, such asmouse embryos, bovine blastocysts, andhuman embryos at 2–8-cell stages, both slowfreezing and conventional vitrification hasproved effective. Slow freezing produces moreconsistent results, because embryos are lesslikely to be injured by the toxicity of the cryo-protectant and thus can be handled under lessstrict conditions. However, vitrification has apotential advantage in that higher survivalrates can be obtained if conditions, such astemperature and period of exposure ofembryos to the cryoprotectant, as well as theskill of pipetting, are optimized.
Therefore, vitrification would be a preferredmethod of cryopreservation to the slow-coolingmethod because of both the lack of ice crystalformation and the convenience. We havealready reported a simple vitrification methodusing an ethylene glycol-based solution for 4–8-cell human embryos frozen in conventionalcryostraws.8 Moreover, the success of vitrifica-tion procedures has recently been increased bytechniques that substantially reduce the volumeof the vitrification solution. Among such tech-niques, the Cryoloop can be considered tobe the most refined strategy.21,22 A majordifference between the Cryoloop and theconventional straw for vitrification are thecooling/warming rates.
229
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 229

With the development of sequential cul-ture media, based upon the physiology of thehuman reproductive tract and the changingphysiology of the developing embryos, it ispossible to grow viable blastocysts easily invitro; and because of the resulting highimplantation rates, blastocyst transfer willnecessarily be a good approach in ART.Accordingly, the need to cryopreserve humanblastocysts is increasing.
The Cryoloop enables ultrarapid coolingand warming, and this may consistently pre-vent intracellular ice formation, since we haveobserved that human blastocysts are dehy-drated and concentrated more slowly than inearlier stage embryos, suggesting that intra-cellular ice is more likely to form in blasto-cysts. Furthermore, the technique using theCryoloop is easier and simpler than thatusing the cryostraw. In this chapter, we haveshown that vitrification using the Cryoloop isa clinically useful method for the cryopreser-vation of human blastocysts.
ADDING ARTIFICIAL SHRINKAGEAS PRIOR TO VITRIFICATION
Achievement of pregnancy is related to manydifferent factors. Obviously one of the mostimportant factors to achieve pregnancy isembryo viability. Developing more blastocystsby day 5 should indicate that these havebetter potential developmental competencyand higher viability. Theoretically, the moredeveloped blastocysts should give a betterpregnancy rate than that of early stage blas-tocysts. However, we have observed that thesurvival rate of expanded (86%) and hatching(82%) blastocysts were significantly lowerthan that of early stage blastocysts (97%),even after we had established the vitrificationsteps. This is in accordance with the observa-tion of Vanderzwalmen et al.,19,26 that the effi-ciency of vitrification of human blastocystswas negatively correlated with the expansionof the blastocoel. Increase in survival ratemight contribute to the improvement of the
viability of survived blastocysts in the ASgroup, and may facilitate implantation.
Other reports showing the effectiveness ofblastocoel shrinkage contained 25 cycles,26 29cycles,27 and 49 cycles of transfers. However,in our study (above), 270 cycles of transferwith artificial shrinkage were used to confirmthe clinical value of this technique.
A possible explanation for the lower survivalrate is that the large blastocoel of more devel-oped blastocysts may disturb the efficacy ofvitrification due to inappropriate dehydrationand permeation of cryoprotectant, whichmight cause ice crystal formation in the rapidcooling and warming steps during vitrification.
A study on mouse blastocysts also reportedthat survival rates of vitrified blastocystsafter a one-step exposure to EFS40 decreasedas the volume of the blastocoelic cavityincreased.36 In mouse blastocysts, a two-steploading of cryoprotectant was effective inpreventing the decrease in post-warming sur-vival of fully expanded blastocysts.37 Bovineblastocysts were also successfully vitrified aftera two-step loading of cryoprotectant.38
However, human blastocysts appear to be dif-ferent25 and are thought to have specific char-acteristics related to their lower cryoresistance.Such characteristics could be attributable tothe permeability of the membrane, whichwould decrease as the blastocyst develops.Therefore, it seems preferable to cryopreservehuman blastocysts on day 5 before the blasto-coel fully develops, or even earlier on day 4.
Recently, it has been suggested thatmechanical damage caused by ice crystal for-mation can be avoided by reducing the fluidcontent of the blastocoel of more developedstage blastocysts.25 One of the strategiesreported was reducing the size of the blasto-coel mechanically. Vanderzwalmen et al.26
reported making a small hole in the trophec-toderm with a needle, so causing the blastocoelto shrink. They vitrified shrunken blastocystsin straws in EFS40, and reported that thisartificial shrinkage dramatically raised thepost-warming survival rate, which resulted
VITRIFICATION IN ASSISTED REPRODUCTION
230
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 230

in eight pregnancies from 35 transfers. Sonet al.27 reported using a 29-gauge needle forartificial shrinkage of the blastocoele.Hiraoka et al.28 reported collapsing the blas-tocoel by micropipetting without puncturingthe zona and trophectoderm.
An alternative method is to increase theconcentration of, and exposure to, the cryo-protectant. The use of a high concentrationand the increase in the duration of exposureto the cryoprotectant might be effective forpreventing injuries from intracellular icewhile allowing sufficient dehydration.However, the risk of an injury from the chem-ical toxicity of the cryoprotectant increasesas the concentration and the duration ofexposure is increased. For these reasons,reducing the fluid content of the blastocoel ofmore developed stage blastocysts is preferredto instead of increasing the concentration ofthe cryoprotectant.
Thus, these authors have reported that therate of survival was improved after artificialshrinkage of the blastocoel. Similarly, weapplied the artificial shrinkage technique inour vitrification protocol using the Cryoloopfor human blastocyst cryopreservation.
Initially, we collapsed the blastocoel bypuncturing it with a fine needle using micro-manipulation procedures. Occasionally, wecame across cases where collapsing of theblastocoel occurred partially or incompletely,because the elastic plasma membrane of thetrophectoderm cells adhered to the openingsite of puncture with a microneedle. In thosecases, puncturing was performed repeatedlyat a different site of the trophectoderm untilcomplete collapse of the blastocoel wasachieved. However, since we have appliedlaser pulse for artificial shrinkage sinceSeptember 2004, incomplete collapse of theblastocoel has seldom been observed. Wespeculated that the single laser pulse createsan instant heat effect, that causes cellulardegeneration of the trophectoderm, andallows a large enough hole to be collapsed.Also, collapsing the blastocoel by laser pulse
did not require time for preparation prior tothe procedure, such as setting the holdingpipette and microneedle for puncturing nee-dle pipette, as well as holding the blastocyston the micromanipulation stage. It is onlynecessary to locate the peripheral junction oftrophectoderm cells of the expanded blasto-cyst, and administer a single laser pulse. Thesafety of the laser application to humanembryos and blastocysts such as for blas-tomere biopsy has already been reported.39,40
When we compared the various techniquesof artificial shrinkage in other reports,blastocoel collapse by micropipetting,27 and29-gauge needle26 do not require the microma-nipulation technique. Both techniques can beperformed under the dissecting microscope.Micropipetting is much the same as removal ofcumulus cells from oocytes for ICSI. However,the size of human oocytes is quite similar toblastocysts depending on the size of the blasto-coel relative to the developmental stage of theblastocyst. Thus, for micropipetting it is neces-sary to prepare several sizes of hand-drawnPasture pipettes according to the expansion ofthe blastocoel. Also, shrinkage by 29-gaugeneedle requires a very precise hand movement,because the edge of the 29-gauge needle isquite large compared with a glass microneedle.The final goal of this technique is completecollapse of the blastocoel in order to facilitatethe vitrification steps. Among these techniques,laser pulse is the simplest and most convenientfor artificial shrinkage. The only drawback isthat it is expensive to purchase the equipmentinitially.
According to our observation, the averagetime for re-expansion of surviving blastocystsin the AS group tends to be shorter than forthe control group. This also might be relatedto the viability of the surviving blastocyst.
In conclusion, we have shown that artificialshrinkage of human expanded and hatchingblastocysts prior to vitrification statisticallyimproves the survival and pregnancy rate,and that delivery of healthy babies confirmsthe safety of this technique.
231
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 231

SUMMARY
For embryo cryopreservation, the vitrificationmethod has many advantages over the slowfreezing method: injuries related to ice are lesslikely to occur; survival of embryos can bemaintained at a higher level if conditions forembryo treatment are optimized; and embryoscan be cryopreserved by a simple method in ashort period without a programmable freezer.Therefore, vitrification is suitable for humanembryos, in which a small number of embryos
are cryopreserved frequently. Human embryosat early cleavage stages can be cryopreservedby conventional vitrification using cryostraws,or by ultrarapid vitrification using Cryoloops.Human blastocysts are more efficiently cryo-preserved by the ultrarapid approach. Ourclinical outcomes show that vitrification ofblastocysts using the Cryoloop techniqueresults in high survival and high pregnancyrates, and confirms the safety of this procedureas seen in our perinatal evaluation.
VITRIFICATION IN ASSISTED REPRODUCTION
232
References
1. Kasai M. Simple and efficient methods forvitrification of mammalian embryos. AnimReprod Sci 1996; 42: 67–75.
2. Kasai M. Advances in the cryopreservation ofmammalian oocytes and embryos: develop-ment of ultra rapid vitrification. Reprod MedBiol 2002; 1: 1–9.
3. Lassalle B, Testart J, Renard JP. Humanembryo features that influence the success ofcryopreservation with the use of 1,2 propane-diol. Fertil Steril 1985; 44: 645–51.
4. Fehilly CB, Cohen J, Edwards RG et al.Cryopreservation of cleaving embryos andexpanded blastocysts in the human: acomparative study. Fertil Steril 1985; 44:638–44.
5. Hartshorne GM, Elder K, Edwards RG et al.The influence of in-vitro development uponpost-thaw survival and implantation of cryo-preserved human blastocysts. Hum Reprod1991; 6: 136–41.
6. Menezo Y, Nicollet B, Andre D, et al. Freezingcocultured human blastocysts. Fertil Steril,1992; 58: 977–80.
7. Rall WF and Fahy GM. Ice-free cryopreserva-tion of mouse embryos at −196°C by vitrifica-tion. Nature 1985; 313: 573–5.
8. Kasai M, Komi JH, Machida T, et al. A simplemethod for mouse embryo cryopreservationin a low toxicity vitrification solution, withoutappreciable loss of viability. J Reprod Fertil1990; 89: 91–7.
9. Mukaida T, Takahashi K, Kasai M, et al.Vitrification of human embryos based on the
assessment of suitable conditions for 8-cellmouse embryos. Hum Reprod 1998; 13:2874–9.
10. Saito H, Kaneko T, Hiroi M, et al. Applicationof vitrification to human embryo freezing.Gynecol Obstet Invest 2000; 49: 145–9.
11. Yokota Y, Yokota H, Araki Y, et al. Birth ofhealthy twins from in vitro development ofhuman refrozen embryos. Fertil Steril 2001;76: 1063–5.
12. Gardner DK, Schoolcraft WB, Stevens J, et al.A prospective randomized trial of blastocystculture and transfer in in vitro fertilization.Hum Reprod 1998; 13: 3434–40.
13. Cruz JR, Dubey AK, Gindoff PR, et al. Is blas-tocyst transfer useful as an alternative treat-ment for patients with multiple in vitrofertilization failures? Fertil Steril 1999; 72:218–20.
14. Menezo Y, Sakkas D, and Veiga A. Cryo-preservation of blastocysts. In: Gardner DK,Lane M, eds. ART and the Human Blastocyst.New York: Springer-Verlag 2000: 188–95.
15. Troup SA, Matson PL, Lieberman BA, et al.Cryopreservation of human embryos at thepronucleate, early cleavage, or expanded blas-tocyst stages. Eur J Obstet Gynecol ReprodBiol 1990; 38: 133–9.
16. Nakayama T, Goto Y, Noda Y, et al.Developmental potential of frozen-thawedhuman blastocysts. J Assist Reprod Genet1995; 12: 239–43.
17. Ludwig M, Al-Hasani S, Diedrich K, et al. Newaspects of cryopreservation of oocytes and
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 232

embryos in assisted reproduction and futureperspectives. Hum Reprod 1999; 14: (Suppl1): 162–85.
18. Yokota Y, Sato S, Araki Y, et al. Birth of ahealthy baby following vitrification of humanblastocysts. Fertil Steril 2001; 75: 1027–9.
19. Vanderzwalmen P, Zech H, Van Roosendaal E,et al. Pregnancy and implantation rates aftertransfers of fresh and vitrified embryos on day4 or 5. J Assist Reprod Genet 1999; 16: 147.
20. Martino A, Songsasen N. and Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
21. Vajta G, Kuwayama M, Callesen H, et al. Openpulled straw (OPS) vitrification: a new wayto reduce cryoinjuries of bovine ova andembryos. Mol Reprod Dev 1998; 51: 53–8.
22. Lane M, Bavister BD, Forest KT, et al.Containerless vitrification of mammalianoocytes and embryos. Nat Biotechnol 1999;17: 1234–6.
23. Mukaida T, Kasai M, Takahashi K, et al.Successful birth after transfer of vitrified humanblastocysts with use of a cryoloop containerlesstechnique. Fertil Steril 2001; 76: 618–20.
24. Mukaida T, Oka C, Takahashi K, et al.Vitrification of human blastocysts usingcryoloops: clinical outcome of 223 cycles.Hum Reprod 2003; 18: 384–91.
25. Gardner DK, Schoolcraft WB. In-vitro cultureof human blastocyst. In Jansen R. Mortimer D,eds, Towards Reproductive Certainty:Infertility and Genetics Beyond 1999Carnforth: Parthenon Publishing, 1999:378–88.
26. Vanderzwalmen P, Mukaida T, Schoysman R,et al. Births after vitrification at morula andblastocyst stages: effect of artificial reductionof the blastocoelic cavity before vitrification.Hum Reprod 2002; 17: 744–51.
27. Son WY, Yoon SH, Lim JH, et al. Pregnancyoutcome following transfer of human blasto-cysts vitrified on electron microscopy gridsafter induced collapse of the blastocoele.Hum Reprod 2003; 18: 137–9.
28. Hiraoka K, Hiraoka K, Kinutani K, et al.Blastocoele collapse by micropipetting priorto vitrification gives excellent survival andpregnancy outcomes for human day 5 and 6expanded blastocysts. Hum Reprod 2004; 19:2884–8.
29. Mukaida T, Takahashi K, Oka C, et al.Artificial shrinkage of blastocoele using
either micro–needle or laser pulse prior tothe cooling steps of vitrification improves sur-vival rate and pregnancy outcome of vitrifiedhuman blastocysts. Hum Reprod 2006; 21:3246–52.
30. Lane M, Schoolcraft WB, Gardner DK.Vitrification of mouse and human blastocystsusing a novel cryoloop container-less tech-nique. Fertil Steril 1999; 72: 1073–8.
31. Mukaida T, Takahashi K, Kasai M, et al.Blastocyst cryopreservation: ultra rapid vitrifi-cation using cryoloop technique. ReprodBioMed Online 2003; 6: 221–5
32. Cohen J, Alikani M, Rosenwaks Z, et al.Implantation enhancement by selectiveassisted hatching using zona drilling ofhuman embryos with poor prognosis. HumReprod 1992; 7: 685–91.
33. Obruca A, Strohmer H, Sakkas D. Use oflasers in assisted fertilization and hatching.Hum Reprod 1994; 9: 1723–6.
34. Gardner DK, Lane M, Schoolocraft WB, et al.Blastocyst score affects implantation andpregnancy outcome: towards a single blasto-cyst transfer. Fertil Steril 2000; 73: 1155–8.
35. Takahashi K, Mukaida T, Oka C, et al.Perinatal outcome of blastocyst transfer withvitrification using cryoloop: a 4 year follow-upstudy. Fertil Steril 2005; 84: 88–92.
36. Miyake T, Kasai M, Machida T, et al.Vitrification of mouse oocytes and embryos atvarious stages of development in an ethyleneglycol-based solution by a simple method.Theriogenology 1993; 40: 121–34.
37. Zhu SE, Kasai M, Machida T, et al.Cryopreservation of expanded mouseblastocysts by vitrification in ethylene glycol-based solutions. J Reprod Fertil 1993; 98:139–45.
38. Mahmoudzadeh AR, Van Soom A, de Kruif A,et al. Optimization of a simple vitrificationprocedure for bovine embryos produced invitro: effect of developmental stage, two-stepaddition of cryoprotectant and sucrose dilu-tion on embryonic survival. J Reprod Fertil1995; 103: 33–39.
39. Veiga A, Sandalinas M, Menezo Y, et al. Laserblastocyst biopsy for preimplantation diagno-sis in the human. Zygote 1997; 5: 351–4
40. Ebner T, Moser M, Tews G. Possible applica-tions of non-contact 1.48 micron wavelengthdiode laser in assisted reproduction tech-nologies. Hum Repord Update 2005; 11:425–35.
233
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 233

VITRIFICATION IN ASSISTED REPRODUCTION
234
Appendix 10B.1 Laboratory manual of vitrification for human blastocyst vitrification
Protocol: Ultra-rapid vitrification for HUMAN BLASTOCYSTS using cryoloop
PRINCIPLE:
Cryopreserved embryos will be vitrified and warmed at the request of the patient andphysician for the purpose of embryo transfer. Cryoprotectants will be added andremoved stepwise and post warmed embryos will be cultured for a period of two to fourhours depending on the embryo stage at the time of freezing. All embryos will be eval-uated prior to transfer to the patient. The words ‘Vitrified and Warmed’ are used in thisprotocol, instead of ‘freezing and thawing,’ because no ice formation is involved in thisapproach.
MATERIALS AND CHEMICALS:
– Blastocyst vitrification mediaEthylene Glycol: SIGMA E-9129 (250 ml), DMSO: SIGMA D-2650 (5 × 5 ml glass vial)
– Blastocyst warming mediaFicoll: Ficoll 70 Amersham Pharmacia Biotech AB 17-0410-01, 100 g
– pulled 9″ Pasteur pipet– 1 ml pipet– Culture media for blastocyst stage (Ex. G2.2, Blast Assist, KSBM, Life Global, etc.)– 10 × 35 Primaria dish– Mineral oil for embryo culture grade (Minoil, Ovoil, etc)– 4 well Nunc plate– Mounted crystal loops: Catalog No. HR4 –963 (20 microns, 0.5-0.7mm, 25 pieces)– Crystal cap: Catalog No. HR4 – 913 (60 pieces), HR4 – 911
(Sampler pack)– Crystal wand with Tab: Catalog No. HR4 – 619
www.hamptonresearch.com
To Prepare the Cryoloop by yourself: Use forceps to place a mounted cryoloop into thecap of Crystal Cap manually. Not necessary to use glue for this. If that is loose and easyto come off, heat the forceps with the alcohol or Gas burner and squeeze the stainlesspipe with the loop at the base of the cap until it is tight.
EQUIPMENT:
– Olympus Inverted microscope (IX-71)– Nikon stereo microscope or Wild dissecting microscope– Forma or Heracell Incubator– Forma hood– Thermometer– Stage warmer
(Continued)
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 234

235
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
Appendix 10B.1 (Continued)
VITRIFICATION PROCEDURE:
There are two ways to prepare the solutions for vitrifying and warming:
(A) Preparing the solution A,B,C just for use once. Do not keep it more than 3 days.
(B) Making stock solutions 1,2,3 every two weeks and adding DMSO and Ethylene glycol whenyou vitrify.
Either (A) or (B) can be chosen depending on the volume of the cases.
(A) Prepare the vitrification solution: (Vitrification solution B: VB, Vitrification solution C: VC)
Solution A Pipet 29 ml Hepes (with penicillin) into a 50 ml flask. Add 1 ml H.S.A. or SSS,mix.
Solution VB Pipet 8.5 ml solution A into a 50 ml flask, add 0.75 ml DMSO, 0.75 ml ethyleneglycol, mix. Filter sterilize with 0.2 µ syringe filter into a 50 ml flask. Wrap withfoil.
Solution VC Weigh 2.4 g sucrose into a tube. Weigh 0.07 g ficoll into a tube.Add 7 ml solution A to the ficoll tube, vortex to completely dissolve. Combinewith the sucrose, allow to dissolve for a while. Add 1.5 ml DMSO and 1.5 mlethylene glycol, mix. Filter sterilize with 0.2 µ syringe filter into a 50 ml flask.Wrap with foil.
(A) Prepare the warming solution: (Warming solution B: WB, Warming solution C: WC)
Solution WB Weigh 1.14 g sucrose into a 17 × 100 tube. Add 10 ml solution A. Filter sterilize with 0.2 µ syringe filter into a 50 ml flask.
Solution WC Weigh 0.68 g sucrose into a 17 × 100 tube. Add 10 ml solution A. Filter sterilize with 0.2 µ syringe filter into a 50 ml flask
The day before the warming preparing & incubate rinse and culture disheswith blastocyst culture medium.
(Continued)
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 235

VITRIFICATION IN ASSISTED REPRODUCTION
236
Appendix 10B.1 (Continued)
(B) Another way to prepare the stock solution and media for cryoloop vitrification
Base Medium
Hepes HTF (Hepes G1.2 & G2.2 or any other hepes medium)
Solution 1,
To 10 ml of base medium add 5 mg/ml of H.S.A
Solution 2,To 10 ml of base medium add
3.4 g of sucrose (dissolve) 5 mg/ml of H.S.A
Solution 3,To 10 ml of base medium0.1 g of Ficoll (dissove)3.4g of sucrose (dissolve) 5 mg/ml of H.S.A
Solution for Vitrification (Vitrification solution B: VB, Vitrification solution C: VC)
Solution A is equal to Solution 1
Solution VB850 µl of Solution 175 µl of DMSO75 µl of Ethylene glycol
Solution VC700 µl of Solution 3150 µl of DMSO150 µl of Ethylene glycol
(Continued)
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 236

237
VITRIFICATION OF BLASTOCYSTS USING THE CRYOLOOP TECHNIQUE
Appendix 10B.1 (Continued)
Solution for warming (Warming solution B: WB, Warming solution C: WC)
Solution WB800 of Solution 1400 of Solution 2
Solution WC800 of Solution 1200 of Solution 2
Solution 1, 2, 3 will be kept as a stock solution for two weeks. At the day of vitrification/warming, solution VB, VC, WB, WB will be prepared with Solution 1, 2, 3.
VITRIFICATION STEPS:
Fill liquid nitrogen dewar (Styro-foam Box is enough)
1. At 370C temperature, add 0.7 ml of A, VB, and VC in each of the well of a 4 well plate.
2. Place the dish in an incubator at 370C for ~30 min
3. Check under the microscope that the loop is not broken.
4. Attach an open cryovial (without loop portion) to the labeled cane and immerse it in theliquid nitrogen in a dewer flask.
5. Take the warmed 4-well dish out of the incubator and place it on the stage warmer at 370C
6. Use a large pulled pipet to move the blast to A for 1-2 minutes. And ready to cryopreserve it.
< Transfer it to VB for 2 minutes. >
7. Empty pipet into the empty well, load with solution VB, pick the blast up to transfer themat the bottom of the solution VB well. They will collapse in solution VB.
8. At the almost end of this 2 minutes, prepare two ~20 µl droplets of solution VC on the ridof petri dish.
9. Attach the crystal wand to cap and carefully remove the cap with loop. Just before the2 minutes timer dip the loop portion into VC to create a thin, filmy layer of solution onthe nylon loop by surface tension.
< Transfer it to VC for 30 seconds. Be careful of time >
(Continued)
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 237

VITRIFICATION IN ASSISTED REPRODUCTION
238
Appendix 10B.1 (Continued)
10. Empty pipet into the empty well, load with solution VC, pick up the blasts from solutionVB well to transfer to one of the droplets of solution VC. They will more collapse insolution C.
11. Pick up and release the blasts several times in the droplet of solution VC and move toanother droplet.
12. At the almost end of this 30 seconds, pick the blast up in the droplet of solution VC.
< Loading it onto the loop >
13. Load the blast into the tip of the pipet and pipet it onto the loop. In order to confirm if allthe blasts are in the loop, expel out the media in the pipet to check whether nothing is leftbehind. You do not need to force the blasts on the loop, they will move almostautomatically by surface tension. (Do practice with any discarded embryos plenty timesto feel confident this step prior to clinical application. This is easy step but really critical step!)
14. Immediately after loading the blastocyst, plunged the loop portion into the LN2 by hold-ing the cap with crystal wand (at 30 s)
15. Screw and seal tightly the cap with the loop into cryovial with a labeled cane by crystalwand under LN2. Place on the cane and move into the storage tanks.
16. If there are more blastocyst to freeze, empty the pipet and fill with A and repeat above.
WARMING STEPS:
1. At 370C temperature, add 0.7 ml of A, WB, and WC in each of the well of a 4 well plate.(make 2 wells of A)
2. Place the dish in an incubator at 37°C for ~30 min
3. Take the warmed 4-well dish out of the incubator and place it on the stage warmer at 37°C
4. Remove the cap with a loop from cryovial under LN2, immediately dip the loop directly intosolution WB well. The vitrified blastocyst will sink to the bottom of the well, leave for 2 minutes.
5. Transfer it to WC then leave in WC for 3 minutes.
6. Rinse it through first well of A then leave in 2nd A well for 5 minutes.
7. After 5 min, transfer the embryo to culture medium, which has been equilibrated in a CO2incubator at 370C overnight, for further culture until transfer.
8. About 2 h after warming, observe the appearance of the blastocyst under a dissectingmicroscope to assess the survival.
10b Tucker 8029.qxd 8/23/2007 8:24 PM Page 238

239
Vitrification of blastocysts using theelectron microscope gridWeon-Young Son and Jin-Ho Lim
10C
INTRODUCTION
Cryopreservation of human embryos hasbecome a routine procedure to increasecumulative pregnancy rates, to help avoid therisk of multiple pregnancies after the transferof many embryos, and to avoid unnecessaryadditional stimulation procedures. Recently,blastocyst transfer based on an improvedculture system has been proven effective forincreasing the pregnancy rate in assistedreproductive technology (ART).1,2 Therefore,a reliable procedure for the cryopreservationof supernumerary blastocysts is neededbecause usually only a small number of blasto-cysts after transfer are likely to be available forcryopreservation. Freezing of human blasto-cysts has been carried out with the slow-cool-ing method, but clinically satisfactory resultshave not been obtained.3,4 In addition, theslow-freezing method requires expensiveequipment, and is time-consuming. Therefore,it is essential to establish a simple, fast, andreliable procedure to optimize clinical out-comes of cryopreservation at the blastocyststage. Vitrification has become more widelyused, and is now regarded as a potentialalternative to conservative slow freezing.Vitrification takes only a few seconds to coolembryos, and there is no extracellular crystal-lization, which is one of the major causes ofcell injury.5 It also offers lower osmotic andtoxic effects, and less severe chilling injuryresulting from the rapid passage through the‘dangerous’ temperature zone.6 Furthermore,it does not require specialized programmable
freezers. Initially, most vitrification methodsused standard French mini-straws for holdingthe embryos during cooling, storage, andthawing. Recently, to overcome the disadvan-tages of straws that have low cooling andwarming rates, techniques using either elec-tron microscope (EM) grids,7,8 Cryotop,9
hemi-straw,10 Cryoloop,11,12 or cryotip13 thatsubstantially increase the cooling rate havebeen applied, and significant improvementsin the success rate of human blastocyst vitri-fication have been reported.7–13 Among them,it has been shown that an ultrarapid freezingmethod using EM grids was efficient for thecryopreservation of bovine oocytes.14 Sincethen, several researchers have applied EMgrids successfully for the vitrification ofhuman oocytes15 and blastocysts.7,8,16
Embryo cryopreservation using EM grids wasoriginally designedfor the vitrification of exceed-ingly chill-sensitive Drosophila embryos.17,18 TheEM grid has about 3-fold higher cooling ratesthan those obtained with straws. It is known thatthe increased rate of cooling and thawing mayconsiderably decrease the chilling injury ofembryos. In addition, the EM grid is muchcheaper than another apparatus that is beingused regularly for vitrification.
In this article, we discuss our experience of1000 cycles of human blastocyst vitrificationusing EM grids from 1999–2006. The humanblastocyst vitrification system using EM gridswas set up in different periods.
In period I: ‘vitrification of human blasto-cysts on EM grids’, we examined the possibilityof clinical application of human blastocyst
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 239

vitrification using EM grids based on theexperience of bovine blastocysts,19 and weexamined the effect of a six-step serial dilutionafter vitrification and thawing using EM grids.
In period II: ‘artificial shrinkage ofexpanded blastocysts before vitrification’, weapplied ‘artificial shrinkage’ (i.e. induced col-lapse of the blastocoel) before vitrification ofhuman blastocysts and examined the effectson blastocyst survival and subsequent preg-nancy rate.
In period III: ‘modified two-step dilutionmethod’, we modified dilution method to atwo-step method after thawing vitrified blas-tocysts using EM grids where blastocysts weresubjected to artificial shrinkage.
Finally, we applied the established vitrifica-tion method to blastocysts derived fromimmature oocytes in our in vitro maturation(IVM) program.
METHODOLOGY
Patients and IVF treatment
Women were treated with gonadotropin releas-ing hormone (GnRH) agonist and humanmenopausal gonadotropin (hMG) in either along- or a short-treatment protocol. Whentwo and more follicles reached 18 mm indiameter, a dose of 10 000 IU human chorionicgonadotropin (hCG) (IVF-C; LG Chemical,Seoul, Korea) was administered. Oocytes wereretrieved transvaginally 36–38 h after hCGinjection, and the oocytes were inseminated byeither conventional in vitro fertilization (IVF)or intracytoplasmic sperm injection (ICSI).Fertilization was examined17–19 h after insem-ination for the appearance of two distinctpronuclei (PN) and two polar bodies.
Embryo culture procedure
The procedure used to culture fertilizedoocytes was the sameas described in a previousstudy.2 All zygotes were co-cultured with cumu-lus cells in a 10 µl YS medium supplemented
with 10% human follicular fluid (hFF) for 5 or6 days at 37°C in an atmosphere of 5% CO2,5% O2, and 90% N2.
2 The hFF was preparedusing a previously reported method.20 Thecumulus cells for co-culture were preparedusing the method reported by Yoon et al.2
The embryos were transferred on either day 3or day 5. The date of the embryo transfer wasdetermined by the number of zygotes and thequality of the embryo on day 2, according toestablished criteria.2 After transferring theembryos, surplus embryos were further cul-tured until day 6, regardless of the embryotransfer date. The developed blastocysts wereclassified according to their degree of expan-sion (Figure 10C.1). In early blastocysts (ErB)the blastocoel is less than half the volume ofthe embryo and diameter < 140 µm; in earlyexpanding blastocysts (EEB) the blastocoel isgreater than or equal to half of the volumeof the embryo and diameter 140–160 µm; inmiddle expanding blastocysts (MEB) the blas-tocoel completely fills the embryo and diame-ter 160–180 µm; and in expanded blastocysts(EdB) the blastocoel volume is larger thanthat of the early embryo with thinning zonaand diameter > 180 µm. The blastocysts wereassigned one of four grades: grade A blasto-cysts have a clear inner cell mass (ICM) andtrophectoderm cells; grade B a clear ICMbut poor trophectoderm development;grade C a poor ICM but good trophecto-derm cells; and grade D a poor ICM andpoor trophectoderm cells. Only the embryosthat developed to the expanded blastocyststage (diameter is > 160 mm and grade A/B)were cryopreserved by vitrification on EMgrids.
Vitrification of blastocysts on electronmicroscope grid and warming procedure
Materials
Chemicals were obtained from SigmaChemical Company (St Louis, MO, USA) and
VITRIFICATION IN ASSISTED REPRODUCTION
240
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 240
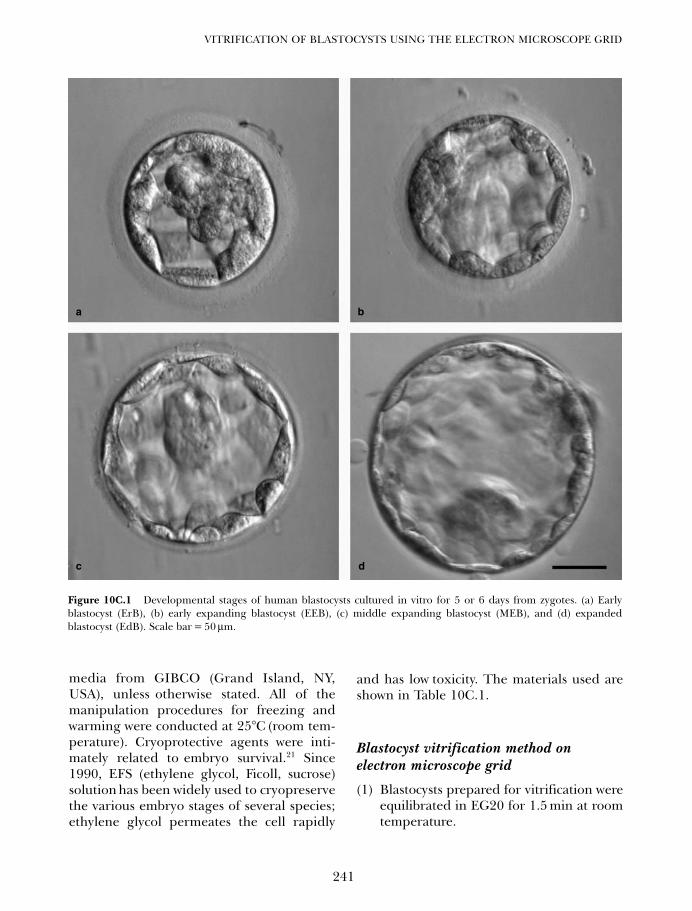
media from GIBCO (Grand Island, NY,USA), unless otherwise stated. All of themanipulation procedures for freezing andwarming were conducted at 25°C (room tem-perature). Cryoprotective agents were inti-mately related to embryo survival.21 Since1990, EFS (ethylene glycol, Ficoll, sucrose)solution has been widely used to cryopreservethe various embryo stages of several species;ethylene glycol permeates the cell rapidly
and has low toxicity. The materials used areshown in Table 10C.1.
Blastocyst vitrification method onelectron microscope grid
(1) Blastocysts prepared for vitrification wereequilibrated in EG20 for 1.5 min at roomtemperature.
241
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
Figure 10C.1 Developmental stages of human blastocysts cultured in vitro for 5 or 6 days from zygotes. (a) Earlyblastocyst (ErB), (b) early expanding blastocyst (EEB), (c) middle expanding blastocyst (MEB), and (d) expandedblastocyst (EdB). Scale bar = 50 µm.
a b
dc
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 241

(2) Blastocysts were incubated in EFS40 atroom temperature, loaded onto the EMgrid using a Pasteur pipette, and excess cry-oprotectant was removed using sterilizedfilter paper (Figure 10C.2a). Mean numberof blastocysts loaded on one grid was 2–3.
(3) EM grid containing blastocysts wasdirectly plunged in LN2 within 30 s.
(4) EM grid was scaled in a cryovial that hadpreviously been submerged under LN2(Figure 10C.2b).
(5) Cryovial was attached on canes and storedin LN2 storage tank (Figure 10C.2c).
Warming methods after thawingof vitrified blastocyst on electronmicroscope grid
Two-step dilution method:
(1) EM grids stored in LN2 were transferreddirectly into 1 mL of 0.3 mol/L sucrosesolution in a 4-well dish as soon as possi-ble, and the blastocysts were recovered.
(2) Blastocysts were quickly transferred intofresh 0.3 mol/L sucrose and incubated for1.5 min at room temperature.
(3) Blastocysts were transferred into DPBScontaining 10% serum for 1.5 min atroom temperature.
Modified two-step dilution method:
(1) EM grids stored in LN2 were transferreddirectly into 1 mL of 0.5 mol/L sucrosesolution in a 4-well dish as soon as possi-ble, and the blastocysts were recovered(Figure 10C.2d and e).
(2) Blastocysts were quickly transferred intofresh 0.5 mol/L sucrose and incubatedfor 5 min at room temperature (Figure10C.2f).
(3) Blastocysts were transferred into DPBScontaining 10% serum for 5 min at roomtemperature.
Six-step serial dilution method:
(1) EM grids stored in LN2 were transferreddirectly into 1 mL of 0.5 mol/L sucrosesolution as soon as possible, and the blas-tocysts were recovered.
(2) Blastocysts were quickly transferred intofresh 0.5 mol/L sucrose and incubated for3 min at room temperature.
VITRIFICATION IN ASSISTED REPRODUCTION
242
Table 10C.1 Materials used for blastocyst vitrification using EM grids
• Four hundred mesh copper EM grids (IGC 400; Pelco International, CA, USA)• Filter paper (sterilized)• Modified-Dulbecco’s phosphate-buffered saline (m-DPBS; GIBCO, Grand Island, NY, USA)• Serum (human follicular fluid (hFF) or human serum albumin (HSA)) • Ethylene glycol (EG; Sigma Chemical Co, St Louis, USA)• Ficoll (Ficoll 70, average MW 70 000 Da; Pharmacia Biotech, Uppsala, Sweden)• Sucrose (Sigma Chemical Co, St Louis, USA)• 4-well nunc dish• Cryovial • Cane • Liquid nitrogen (LN2)• Storage LN2 Tank • Pretreatment solution (EG20): 20% (v/v) EG in m-DPBS containing 10% hFF (or HSA) • Vitrification solution (EFS40): 40% (v/v) EG, 18% (w/v) Ficoll, and 0.3 mol/L sucrose in m-DPBS containing
10% hFF (or HSA)• Dilution solutions: 0.5 mol/L, 0.4 mol/L, 0.3 mol/L, 0.2 mol/L, 0.1 mol/L, and 0.0 mol/L sucrose in m-DPBS con-
taining 10% hFF (or HSA)
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 242

243
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
Figure 10C.2 Vitrification and warming process of blastocysts on electron microscope (EM) grids. (a) Blastocysts wereloaded onto the EM grid on sterilized filter paper using a Pasteur pipette, (b) directly plunged the EM gridcontaining blastocysts in LN2, (c) the cryovial was attached in canes and stored in LN2 storage tank, (d) EM grid storedin LN2 was transferred into 0.5 mol/L sucrose solution, (e) the blastocysts on EM grid were recovered, (f) blastocystswere tranferred into fresh 0.5 mol/L sucrose.
a
c d
e f
b
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 243

(3) Blastocysts were transferred sequentiallyinto 1 mL of 0.4, 0.3, 0.2, and 0.1 mol/Lsucrose at intervals of 1.5 min at roomtemperature.
(4) Blastocysts were transferred into m-DPBScontaining 10% serum for 1.5 min atroom temperature.
Assessment of survivalafter thawing
After thawing the blastocysts were washedthree times in culture medium and co-culturedwith cumulus cells in 10 µL YS mediumcontaining 10% hFF. The post-thawing sur-vival of blastocysts was observed ~18–20 hafter warming under a microscope, and blasto-cysts with a morphologically intact ΙCM andtrophectoderm, and re-expanding blastocoelwere judged to have survived.
Embryo transfer and endometrium preparation
Vitrified day 5 and 6 blastocysts were thawedin the afternoon of the day before embryotransfer. Embryo transfer was scheduled onday 5 after ovulation in the spontaneouscycles, or on days 19–20 in artificial cyclesprepared with exogenous estrogen and prog-esterone. One to three surviving blastocystswere transferred into the patient’s uterus.Pregnancy was first assessed by serum βhCG 9days after blastocyst transfer, and then clinicalpregnancy was determined by the presence offetal heart activity 30 days after blastocysttransfer.
Statistical analysis
The significance of difference between treat-ment groups in each experiment was com-pared with the χ2 test using the StatisticalAnalysis System (SAS) Institute software pack-age (SAS Institute Inc, 1985).
RESULTS
Vitrification of human blastocysts onelectron microscope grid
Previously, Park et al. reported that highersurvival (78.1%) and hatching (65.4%) ratesof vitrified–thawed bovine blastocysts couldbe obtained using EM grids and a two-stepcryoprotectant dilution.19 Initially, we appliedthe same protocol to human blastocysts, butthis resulted in disappointing survival rates(30%, 6/20). Cryopreserved blastocyst survivalusually depends on the freezing–thawingprocedure. Thus, the low percentage ofblastocysts that survived after thawing in atwo-step dilution method might have beendue to the dramatic osmotic shock.
Following some trial experiments, weobtained a good survival rate by changing thefirst concentration of sucrose to 0.5 mol/L incryoprotectant dilution after thawing. Thus,we compared a thawing protocol using a six-step serial dilution with the two-step dilutionused for bovine blastocysts19 on the survival ofvitrified human blastocysts (n = 293) derivedfrom 3PN before trying to apply the methodclinically. Figure 10C.3 shows the survival rateof blastocysts after thawing. As shown in Figure10C.3, the survival rate of blastocysts followingthe six-step dilution method was significantlyhigher than following the two-step dilutionmethod (P < 0.01). It could be speculated thatthe higher survival rateof human blastocysts ina six-step cryoprotectant dilution was due to adecrease in the osmotic shock.
Based on the above results, we applied thesix-step thawing methodclinically. Table 10C.2shows the clinical results. A total of 92 patients(mean age ± SD = 34.2 ± 3.5) and 382 blasto-cysts were included in this study. Out of 287(75.1%) survived blastocysts, 42 (14.6%)hatched at the time of the embryo transfer.After transfer, 29 clinical pregnancies (31.5%)were achieved. Although we obtained a reason-able clinical pregnancy rate (31.5%) followingtransfer, this pregnancy outcome was lower
VITRIFICATION IN ASSISTED REPRODUCTION
244
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 244
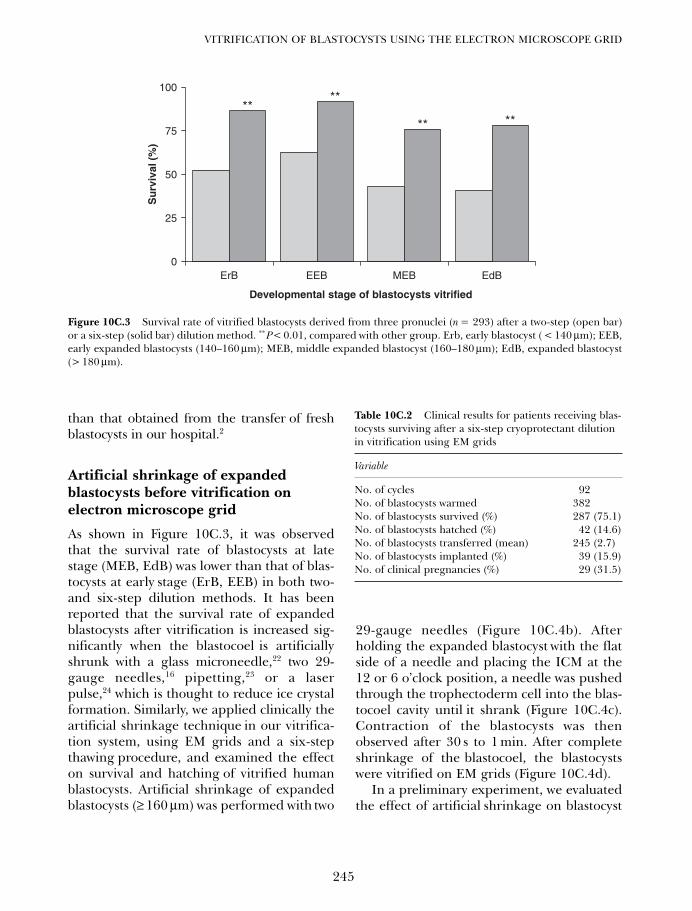
than that obtained from the transfer of freshblastocysts in our hospital.2
Artificial shrinkage of expandedblastocysts before vitrification onelectron microscope grid
As shown in Figure 10C.3, it was observedthat the survival rate of blastocysts at latestage (MEB, EdB) was lower than that of blas-tocysts at early stage (ErB, EEB) in both two-and six-step dilution methods. It has beenreported that the survival rate of expandedblastocysts after vitrification is increased sig-nificantly when the blastocoel is artificiallyshrunk with a glass microneedle,22 two 29-gauge needles,16 pipetting,23 or a laserpulse,24 which is thought to reduce ice crystalformation. Similarly, we applied clinically theartificial shrinkage technique in our vitrifica-tion system, using EM grids and a six-stepthawing procedure, and examined the effecton survival and hatching of vitrified humanblastocysts. Artificial shrinkage of expandedblastocysts (≥ 160 µm) was performed with two
29-gauge needles (Figure 10C.4b). Afterholding the expanded blastocyst with the flatside of a needle and placing the ICM at the12 or 6 o’clock position, a needle was pushedthrough the trophectoderm cell into the blas-tocoel cavity until it shrank (Figure 10C.4c).Contraction of the blastocysts was thenobserved after 30 s to 1 min. After completeshrinkage of the blastocoel, the blastocystswere vitrified on EM grids (Figure 10C.4d).
In a preliminary experiment, we evaluatedthe effect of artificial shrinkage on blastocyst
245
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
0
25
50
75
100
ErB EEB MEB EdB
Developmental stage of blastocysts vitrified
Su
rviv
al (
%)
****
** **
Figure 10C.3 Survival rate of vitrified blastocysts derived from three pronuclei (n = 293) after a two-step (open bar)or a six-step (solid bar) dilution method. **P< 0.01, compared with other group. Erb, early blastocyst ( < 140 µm); EEB,early expanded blastocysts (140–160 µm); MEB, middle expanded blastocyst (160–180 µm); EdB, expanded blastocyst(> 180 µm).
Table 10C.2 Clinical results for patients receiving blas-tocysts surviving after a six-step cryoprotectant dilutionin vitrification using EM grids
Variable
No. of cycles 92No. of blastocysts warmed 382No. of blastocysts survived (%) 287 (75.1)No. of blastocysts hatched (%) 42 (14.6)No. of blastocysts transferred (mean) 245 (2.7)No. of blastocysts implanted (%) 39 (15.9)No. of clinical pregnancies (%) 29 (31.5)
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 245
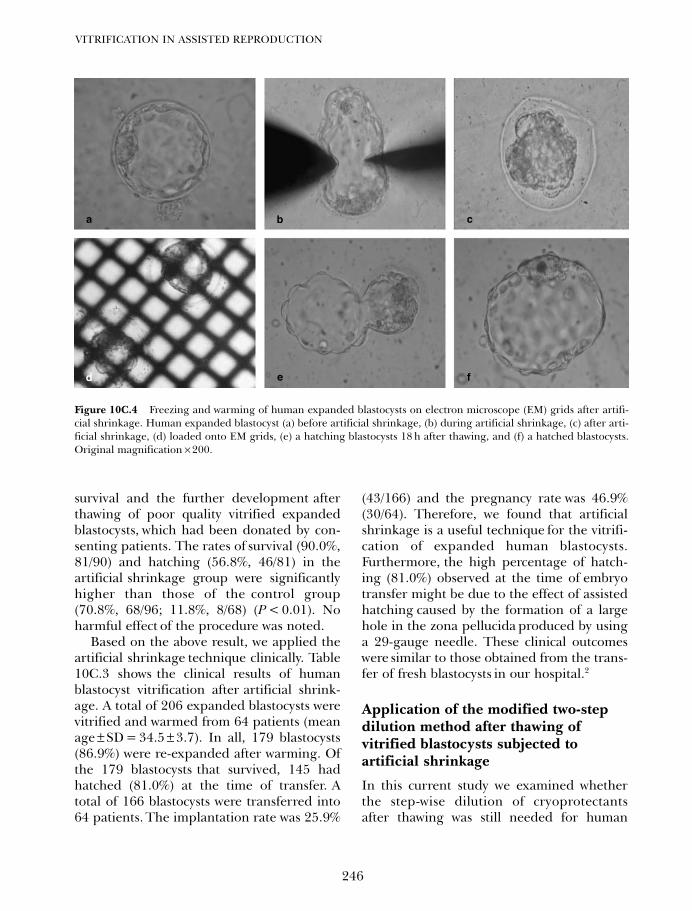
survival and the further development afterthawing of poor quality vitrified expandedblastocysts, which had been donated by con-senting patients. The rates of survival (90.0%,81/90) and hatching (56.8%, 46/81) in theartificial shrinkage group were significantlyhigher than those of the control group(70.8%, 68/96; 11.8%, 8/68) (P < 0.01). Noharmful effect of the procedure was noted.
Based on the above result, we applied theartificial shrinkage technique clinically. Table10C.3 shows the clinical results of humanblastocyst vitrification after artificial shrink-age. A total of 206 expanded blastocysts werevitrified and warmed from 64 patients (meanage ± SD = 34.5 ± 3.7). In all, 179 blastocysts(86.9%) were re-expanded after warming. Ofthe 179 blastocysts that survived, 145 hadhatched (81.0%) at the time of transfer. Atotal of 166 blastocysts were transferred into64 patients. The implantation rate was 25.9%
(43/166) and the pregnancy rate was 46.9%(30/64). Therefore, we found that artificialshrinkage is a useful technique for the vitrifi-cation of expanded human blastocysts.Furthermore, the high percentage of hatch-ing (81.0%) observed at the time of embryotransfer might be due to the effect of assistedhatching caused by the formation of a largehole in the zona pellucida produced by usinga 29-gauge needle. These clinical outcomeswere similar to those obtained from the trans-fer of fresh blastocysts in our hospital.2
Application of the modified two-stepdilution method after thawing of vitrified blastocysts subjected to artificial shrinkage
In this current study we examined whetherthe step-wise dilution of cryoprotectantsafter thawing was still needed for human
VITRIFICATION IN ASSISTED REPRODUCTION
246
Figure 10C.4 Freezing and warming of human expanded blastocysts on electron microscope (EM) grids after artifi-cial shrinkage. Human expanded blastocyst (a) before artificial shrinkage, (b) during artificial shrinkage, (c) after arti-ficial shrinkage, (d) loaded onto EM grids, (e) a hatching blastocysts 18 h after thawing, and (f) a hatched blastocysts.Original magnification × 200.
a b c
fed
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 246
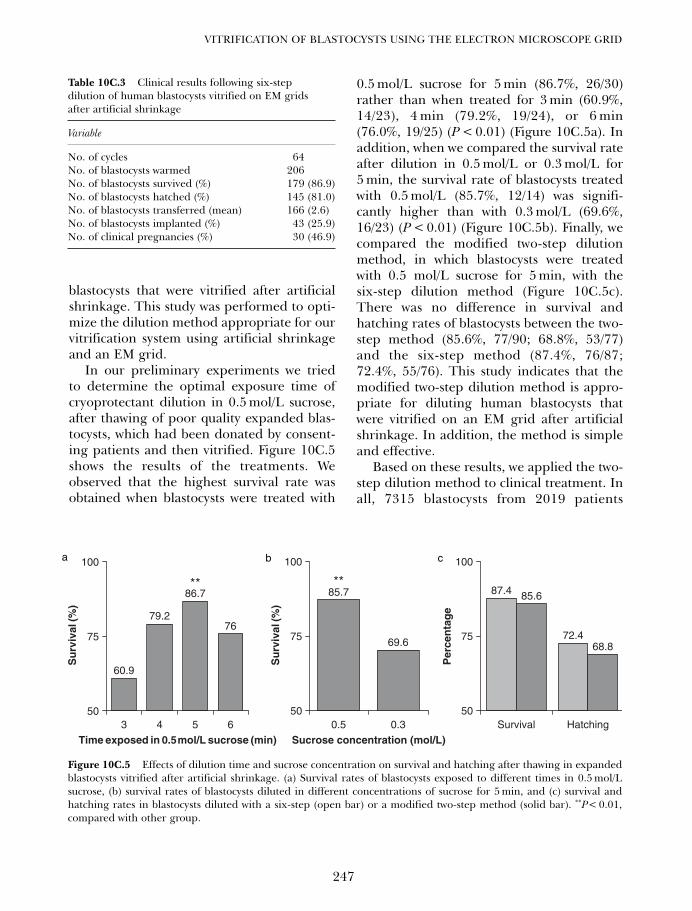
blastocysts that were vitrified after artificialshrinkage. This study was performed to opti-mize the dilution method appropriate for ourvitrification system using artificial shrinkageand an EM grid.
In our preliminary experiments we triedto determine the optimal exposure time ofcryoprotectant dilution in 0.5 mol/L sucrose,after thawing of poor quality expanded blas-tocysts, which had been donated by consent-ing patients and then vitrified. Figure 10C.5shows the results of the treatments. Weobserved that the highest survival rate wasobtained when blastocysts were treated with
0.5 mol/L sucrose for 5 min (86.7%, 26/30)rather than when treated for 3 min (60.9%,14/23), 4 min (79.2%, 19/24), or 6 min(76.0%, 19/25) (P < 0.01) (Figure 10C.5a). Inaddition, when we compared the survival rateafter dilution in 0.5 mol/L or 0.3 mol/L for5 min, the survival rate of blastocysts treatedwith 0.5 mol/L (85.7%, 12/14) was signifi-cantly higher than with 0.3 mol/L (69.6%,16/23) (P < 0.01) (Figure 10C.5b). Finally, wecompared the modified two-step dilutionmethod, in which blastocysts were treatedwith 0.5 mol/L sucrose for 5 min, with thesix-step dilution method (Figure 10C.5c).There was no difference in survival andhatching rates of blastocysts between the two-step method (85.6%, 77/90; 68.8%, 53/77)and the six-step method (87.4%, 76/87;72.4%, 55/76). This study indicates that themodified two-step dilution method is appro-priate for diluting human blastocysts thatwere vitrified on an EM grid after artificialshrinkage. In addition, the method is simpleand effective.
Based on these results, we applied the two-step dilution method to clinical treatment. Inall, 7315 blastocysts from 2019 patients
247
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
Table 10C.3 Clinical results following six-stepdilution of human blastocysts vitrified on EM gridsafter artificial shrinkage
Variable
No. of cycles 64No. of blastocysts warmed 206No. of blastocysts survived (%) 179 (86.9)No. of blastocysts hatched (%) 145 (81.0)No. of blastocysts transferred (mean) 166 (2.6)No. of blastocysts implanted (%) 43 (25.9)No. of clinical pregnancies (%) 30 (46.9)
60.9
79.2
86.7
76
50
75
100
3 4 5 6
Time exposed in 0.5 mol/L sucrose (min)
Su
rviv
al (%
)
85.7
69.6
50
75
100
0.5 0.3
Sucrose concentration (mol/L)
Su
rviv
al (%
)
Per
cen
tag
e
87.4
72.4
85.6
68.8
50
75
100
Survival Hatching
** **
Figure 10C.5 Effects of dilution time and sucrose concentration on survival and hatching after thawing in expandedblastocysts vitrified after artificial shrinkage. (a) Survival rates of blastocysts exposed to different times in 0.5 mol/Lsucrose, (b) survival rates of blastocysts diluted in different concentrations of sucrose for 5 min, and (c) survival andhatching rates in blastocysts diluted with a six-step (open bar) or a modified two-step method (solid bar). **P< 0.01,compared with other group.
a b c
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 247

(mean age ± SD = 34.8 ± 4.2) were vitrifiedon EM grids between January 1999 andDecember 2005. For 4 years, from March2002 to December 2005, we vitrified andwarmed 2626 blastocysts from 884 cyclesusing the modified two-step dilution method,2366 of the vitrified blastocysts (90.1%) sur-vived and 2066 of the surviving blastocysts(87.3%) hatched at the time of transfer. Atotal of 2287 survived blastocysts were thentransferred into patients. The implantationrate was 26.8% (612/2287) and the pregnancyrate was 49.7% (439/884) (Table 10C.4). Thelive birth rate was 40.3% (356/884). Therewere no triplet pregnancies. Birth weights ofthe infants were within the range of1540–4220 g, and at the time of going topress all delivered infants had normal physi-cal profiles.
Vitrification of blastocysts derivedfrom in vitro maturation of immaturehuman oocytes using the established vitrification method
In vitro maturation (IVM) is an attractiveoption to eliminate several problems associ-ated with controlled ovarian hyperstimula-tion (COH) used for conventional in vitrofertilization (IVF). Recent studies have shownan improved pregnancy rate per embryotransfer in small numbers of cycles.25,26 Inaddition, several IVF centers have reportedthat acceptable rates of oocyte maturationand pregnancies were achieved in patientswith polycystic ovary syndrome (PCOS).26
However, they reported that the implantationrate was less than 15%. Therefore, moreembryos in an IVM program have been trans-ferred to obtain acceptable pregnancy ratesthan in COH cycles. In addition, althoughcryopreservation is now a routine procedurein human IVF, there have been only a fewcase reports in which embryos generatedfrom an IVM program were frozen at 2PN27
and cleavage stage28 using the slow-cooling
method. One of the reasons is that considerabledifferences of efficiency exist depending onthe origin of mature oocytes (in vivo or invitro produced). Generally, in vitro producedembryos were much more sensitive to freezingthan their in vivo derived counterparts.29
Suikkari et al. also found that the cryosurvivalof in vitro matured zygotes and cleavedembryos was very poor compared withembryos generated from in vivo maturedoocytes using the slow-cooling method.30 Intheir study, eight out of 24 cleaved embryosand 14 out of 25 zygotes survived after thaw-ing, suggesting that cryopreservation of invitro matured embryos might not be an opti-mal procedure. Therefore, clinically satisfac-tory results from embryos generated fromIVM oocytes have not yet been obtained.30
Recently, we found that the oocytesretrieved after hCG priming in women withhigh risk of OHSS in the IVM program candevelop to blastocyst stage, and pregnanciescan be established by transfer of these blasto-cysts.31–34 Therefore, a reliable procedure forthe cryopreservation of supernumerary blas-tocysts generated from IVM oocytes is alsoneeded. Although we have already reportedthat successful pregnancies can be achievedafter vitrification of blastocyst stages gener-ated from IVM cycles,35,36 it is still unclearwhether the established vitrification methodcan be applied to the blastocysts produced
VITRIFICATION IN ASSISTED REPRODUCTION
248
Table 10C.4 Clinical results following a modifiedtwo-step dilution of human blastocysts vitrified onEM grids after artificial shrinkage
Variables
No. of cycles 884No. of blastocysts warmed 2626No. of blastocysts survived (%) 2366 (90.1)No. of blastocysts hatched (%) 2066 (87.3)No. of blastocysts transferred (mean) 2287 (2.6)No. of blastocysts implanted (%) 612 (26.8)No. of clinical pregnancies (%) 439 (49.7)
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 248

by in vitro matured oocytes. Therefore, weobserved the clinical outcome after vitrifica-tion and warming of the blastocysts producedfrom IVM cycles using the method estab-lished in our IVF program. A total of 233blastocyst-stage embryos produced from 66IVM cycles were vitrified through 2005.Among them 32 cycles that had transfersafter warming were examined. Out of the 125vitrified blastocysts a total of 100 expandedblastocysts were warmed from 32 cycles.Ninety-two blastocysts (92.0%) were re-expanded after warming. Of the 92 blasto-cysts that survived, 68 had hatching orhatched blastocysts (73.9%) at the time oftransfer. A total of 89 blastocysts were trans-ferred in 32 cycles, 14 clinical pregnancies(43.8%, 14/32) and a 23.1% implantation rate(21/89) were established.
DISCUSSION
After being warmed, the vitrified humanembryos must be separated from the perme-able cryoprotectants, which were used in thecooling process. This is usually achieved byimmersing embryos in a graded series ofsucrose solutions until isotonic conditions aremet. This gradual replacement of cryoprotec-tants with sucrose solution serves to re-hydrate the thawed embryos, and helps toreduce osmotic pressure during rehydra-tion.9,37 Generally, three- or four-step dilutionprocedures have been used in thisprocess.10,22,23,38,39 As shown in previousresults, we also improved the success rate ofhuman blastocyst vitrification by increasingthe number of cryoprotectant dilution stepsafter thawing by using a six-step dilutionmethod. However, we found that there was arelatively poor survival of the expanded blas-tocysts after vitrification. It was thought thatlate blastocysts consist of a well-developedblastocoel, which may disturb effective cryo-preservation due to ice crystal formation, andthat this may be caused by inadequate perme-ation of the cryoprotectants during the cool-
ing step. Therefore, we introduced an artifi-cial shrinkage technique into our vitrificationsystem, and we have noted a dramaticincrease in the survival and pregnancy ratesof human expanded blastocysts.16,40 After thepotential for intrablastocoelic ice formationwas eliminated in expanded blastocysts bysuch an artificial shrinkage technique, wequestioned whether the step-wise dilution ofcryoprotectants after thawing is still neededfor human blastocysts that were vitrified afterartificial shrinkage. Finally, we found that thetwo-step dilution method could be a simplerand more effective protocol for humanexpanded blastocysts that are vitrified usingEM grids following artificial shrinkage, thanthe six-step method. When the blastocyst isvitrified after artificial shrinkage, the fastexchange of cryoprotectant seems to be moreimportant than reducing the osmotic stressduring the cryoprotectant dilution step. Todate, we have been achieving reasonable clin-ical outcomes in our IVF and IVM programsusing our established vitrification system.
Choi and her colleagues observed that only51.6% (48/93) of human blastocysts survivedafter warming in an EM grid vitrificationprogram.8 The vitrification and warmingmethods were different from our methods.Therefore, the difference in survival rates ofblastocysts after warming may depend on theconcentrations and times they are exposed tocryoprotectant solutions, rather than on theuse of the same EM grids. Another possibleexplanation could be the different cultureconditions used for producing the blastocysts,and the quality of the expanded blastocystscryopreserved.
New culture systems and media are cur-rently being developed for IVF programs,which are helping in the production of goodquality blastocysts. Variations caused by dif-ferent media batches can be minimized byusing qualified products that are commer-cially controlled. Therefore, blastocyst cultureand transfer are becoming routine proce-dures in ART programs. This implies that the
249
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 249

current culture system is relatively good toculture blastocysts from embryos generatedin IVF cycles. However, it is still questionablewhether the commercially available mediacan produce blastocysts from embryosderived from in vitro matured oocytes.Although, Barnes et al. were the first toreport successful development and preg-nancy to the blastocyst stage from in vitromatured oocytes in sequential culturemedium designed specifically to optimizeblastocyst development,41 since then, therehave not been any reports about the success-ful clinical application following transfer ofblastocysts in IVM programs using a sequen-tial culture system similar to that used in con-ventional IVF. Therefore, there may still besuboptimal conditions for support of laterdevelopment of the embryos generated inIVM cycles.
The other option for culturing embryosgenerated from IVM cycles is a co-culturesystem. It has been shown that co-culture sys-tems significantly enhance the percentage ofembryos developing to the blastocyst stagein IVF42,43 and IVM44,45 cycles. These reportsimply that to date the co-culture method isthe best option to obtain blastocysts in anIVM program. An improved survival rateafter cryopreservation of in vitro producedblastocysts may be attributed to theimproved culture conditions, as well aschanges in the cryopreservation technology.We have previously reported high clinicalpregnancy (51.9%, 55/106) and implantation(26.8%, 84/313) rates without assisted hatch-ing of blastocysts produced from IVMcycles.34 After vitrification of blastocysts pro-duced from IVM cycles we observed a clinicalpregnancy of 43.8%, and an implantationrate of 23.6% similar to that obtained in freshcycles. These results imply that this vitrifica-tion method is also an efficient cryo-preservation method for blastocysts producedfrom IVM programs. In addition, theseresults highlight the use of three advancedassisted reproduction techniques, namely
IVM, blastocyst culture, and vitrification, toovercome infertility problems.
CONCLUSION
This study showed that vitrification of humanblastocysts at the expanded blastocyst stageusing EM grids and artificial shrinkage tech-nique is a clinically useful cryopreservationmethod. Additionally, the modified two-stepdilution method could be a simpler andmore effective protocol for human expandedblastocysts that are vitrified using EM gridsfollowing artificial shrinkage, when com-pared with the six-step method. Furthermore,blastocyst-stage embryos produced from invitro matured human oocytes can be safelycryopreserved by the established vitrificationmethod, and successful pregnancy can beachieved following embryo transfer. However,a disadvantage of vitrification on EM grids isthe risk of contamination by pathogens suchas viruses, prions, and bacteria caused whenthe vitrification solution comes into directcontact with LN2 during cooling or storage.46
Although to date there are no cases of conta-mination occurring via LN2 in our vitrifica-tion system, the development of safetystrategies for reducing the risk of contamina-tion by larger pathogens may be necessary.Nevertheless, our results, based on morethan 500 live births, prove that the vitrifica-tion method using an EM grid is asimple, inexpensive, and efficient techniquefor cryopreservation of expanded humanblastocysts.
ACKNOWLEDGMENTS
We owe great thanks to the physicians andembryologists working at the Maria InfertilityHospital, Korea. We are very grateful to BelenHerrero and Jin-Tae Chung, Department ofObstetrics and Gynecology, McGill University,Montreal, Quebec, Canada, for critical reviewof this manuscript.
VITRIFICATION IN ASSISTED REPRODUCTION
250
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 250

1. Gardner DK, Vella P, Lane M et al. Cultureand transfer of human blastocysts increasesimplantation rates and reduces the need formultiple embryo transfers. Fertil Steril 1998;69: 84–8.
2. Yoon HG, Yoon SH, Son WY et al. Alternativeembryo transfer of day 3 or day 5 for reducingthe risk of multiple gestations. J Assist ReprodGenet 2001; 18: 262–7.
3. Ménézo Y, Nicollet B, Herbaut N et al.Freezing cocultured human blastocysts. FertilSteril 1992; 58: 977–80.
4. Kaufman RA, Ménézo Y, Hazout A et al.Cocultured blastocyst cryopreservation: expe-rience of more than 500 transfer cycles. FertilSteril 1995; 64: 1125–9.
5. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at –196°C by vitrification.Nature 1985; 313: 573–5.
6. Vajta G, Holm P, Kuwayama M et al. Openpulled straw (OPS) vitrification: a new way toreduce cryoinjuries of bovine ova and embryos.Mol Reprod Dev 1998; 51: 53–8.
7. Cho HJ, Son WY, Yoon SH et al. An improvedprotocol for dilution of cryoprotectants fromvitrified human blastocysts. Hum Reprod2002; 17: 2419–22.
8. Choi DH, Chung HM, Lim JM et al. Pregnancyand delivery of healthy infants developed fromvitrified blastocysts in an IVF–ET program.Fertil Steril 2000; 74: 838–44.
9. Kuwamaya M, Hamano S, Nagai T.Vitrification of bovine blastocysts obtained byin vitro culture of oocytes matured and fertil-ized in vitro. J Reprod Fertil 1992; 96: 187–93.
10. Vanderzwalmen P, Bertin G, Debauche CH et al.Vitrification of human blastocysts with theHemi-Straw carrier: application of assistedhatching after thawing. Hum Reprod 2003; 18:1504–11.
11. Yokota Y, Sato S, Yokota M et al. Successfulpregnancy following blastocyst vitrification:case report. Hum Reprod 2000; 15: 1802–3.
12. Mukaida T, Nakamura S, Tomiyama T et al.Successful birth after transfer of vitrifiedhuman blastocysts with use of a cryoloop con-tainerless technique. Fertil Steril 2001; 76:618–20.
13. Kuwayama M, Vajta G, Ieda S et al.Comparison of open and closed methods forvitrification of human embryos and the elimi-nation of potential contamination. RBMOnline 2005; 11: 608–14.
14. Martino A, Songsasen N, Leibo SP.Development into blastocysts of bovineoocytes cryopreserved by ultra-rapid cooling.Biol Reprod 1996; 54: 1059–69.
15. Yoon TK, Chung HM, Lim JM et al.Pregnancy and delivery of healthy infantsdeveloped from vitrified oocytes in a stimu-lated in vitro fertilization–embryo transferprogram. Fertil Steril 2000; 74: 180–1.
16. Son WY, Yoon SH, Yoon HJ et al. Pregnancyoutcome following transfer of human blasto-cysts vitrified on electron microscopy gridsafter induced collapse of the blastocoele.Hum Reprod 2003; 18: 137–9
17. Mazur P, Cole KW, Hall JW et al.Cryobiological preservation of Drosophilaembryos. Science 1992; 258: 1932–5.
18. Steponkus PL and Caldwell S. An optimizedprocedure for the cryopreservation ofDrosophila melanogaster embryos. Cryo-Letters 1993; 14: 375–80.
19. Park SP, Kim EY, Kim DI et al. Simple, effi-cient and successful vitrification of bovineblastocysts using electron microscope grids.Hum Reprod 1999; 14: 2838–43.
20. Chi HJ, Kim DH, Koo JJ et al. The suitabilityand efficiency of human follicular fluid as aprotein supplement in human in vitro fertiliza-tion programs. Fertil Steril 1998; 70: 871–7.
21. Tachikawa S, Otoi T, Kondo S et al. Successfulvitrification of bovine blastocysts derived by invitro maturation and fertilization. Mol ReprodDev 1993; 34: 266–71.
22. Vanderzwalmen P, Bertin G, Debauche CHet al. Birth after vitrification at morula andblastocyst stage: effect of artificial reductionof the blastocoelic cavity before vitrification.Hum Reprod 2002; 17: 744–51.
23. Hiraoka K, Kiraoka K, Kinutani M et al.Blastocoele collapse by micropipetting priorto vitrification gives excellent survival andpregnancy outcomes for human day 5 and 6expanded blastocysts. Hum Reprod 2004; 19:2884–8.
24. Mukaida T, Oka C, Goto T et al. Artificialshrinkage of blastocoeles using either a micro-needle or a laser pulse prior to the coolingsteps of vitrification improves survival rateand pregnancy outcome of vitrified humanblastocysts. Hum Reprod 2006; 21: 3246–52.
25. Mikkelsen AL, Smith SD, Lindenberg S. In-vitromaturation of human oocytes from regularlymenstruating women may be successful without
251
VITRIFICATION OF BLASTOCYSTS USING THE ELECTRON MICROSCOPE GRID
References
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 251

follicle stimulating hormone priming. HumReprod 1999; 14: 1847–51.
26. Chian RC. In-vitro maturation of immatureoocytes for infertile women with PCOS. RBMOnline 2004; 8: 547–52.
27. Chian RC, Gülekli B, Buckett WM et al.Pregnancy and delivery after cryopreservationof zygotes produced by in-vitro maturedoocytes retrieved from a woman with polycys-tic ovarian syndrome. Hum Reprod 2001; 16:1700–2.
28. Godin PA, Gaspard O, Thonon F et al. Twinpregnancy obtained with frozen-thawedembryos after in vitro maturation in a patientwith polycystic ovarian syndrome. J AssistReprod Genet 2003; 20: 347–50.
29. Leibo SP. Piglets produced by transfer of vitri-fied porcine embryos after stepwise dilution ofcryoprotectants. Cryobiology 1998; 36: 20–31.
30. Suikkari AM, Tulppala M, Tuuri T et al.Luteal phase start of low-dose FSH priming offollicles results in an efficient recovery, matu-ration and fertilization of immature humanoocytes. Hum Reprod 2000; 15: 747–51.
31. Son WY, Park SJ, Hyun CS et al. Successfulbirth after transfer of blastocysts derived fromoocytes of unstimulated woman with regularmenstrual cycle after IVM Approach. J AssistReprod Genet 2002; 19: 541–3.
32. Son WY, Yoon SH, Lee SW et al. Blastocystdevelopment and pregnancies after IVF ofmature oocytes retrieved from unstimulatedpatients with PCOS after in-vivo HCG prim-ing. Hum Reprod 2002; 17: 134–6.
33. Son WY, Lee SY, Lim JH. Fertilization, cleavageand blastocyst development according to thematuration timing of oocytes in in vitro matu-ration cycles. Hum Reprod 2005; 20: 3204–7.
34. Son WY, Lee SY, Yoon SH et al. Pregnanciesand deliveries after transfer of human blasto-cysts derived from in vitro matured oocytes inIVM cycles. Fertil Steril 2007; in press.
35. Son WY, Yoon SH, Park SJ et al. Ongoing twinpregnancy after vitrification of blastocysts pro-duced by in vitro matured oocytes retrievedfrom a woman with polycystic ovary syndrome.Hum Reprod 2002; 17: 2963–6.
36. Son WY, Lee SY, Chang MJ et al. Pregnancyresulting from transfer of repeat vitrified
blastocysts produced by in-vitro maturedoocytes in patient with polycystic ovarysyndrome. RBM online 2005; 10: 398–401.
37. Isachenko V, Montag M, Isachenko E et al.Developmental rate and ultrastructure ofvitrified human pronuclear oocytes after step-wise versus direct rehydration. Hum Reprod2004; 19: 660–5.
38. Mukaida T, Nakamura S, Tomiyama T et al.Vitrification of human blastocysts usingcryoloops: clinical outcome of 223 cycle. HumReprod 2003; 18: 384–91.
39. Takahashi K, Kukaida T, Goto T et al. Perinataloutcome of blastocyst transfer with vitrificationusing cryoloop: a 4-year follow-up study. FertilSteril 2004; 84: 88–92.
40. Lee SY, Kim HJ, Park SJ et al. Optimization ofa dilution method for human expanded blas-tocysts vitrified using EM grids after artificialshrinkage. J Assist Reprod Genet 2006; 23:87–91.
41. Barnes FL, Crombie A, Gardner DK et al.Blastocyst development and birth after in-vitromaturation of human primary oocytes, intra-cytoplasmic sperm injection and assistedhatching. Hum Reprod 1995; 10: 3243–7.
42. Menezo YJR, Guerin JF, Czyba JC.Improvement of human early embryo devel-opment in vitro by co-culture on monolayersof Vero cells. Biol Reprod 1990; 42: 301–7.
43. Bongso A, Ng SC, Fong CY et al. Cocultures:a new lead in embryo quality improvement forassisted reproduction. Fertil Steril 1991; 56:179–91.
44. Hwu YM, Lee RK, Chen CP et al.Development of hatching blastocysts fromimmature human oocytes following in vitromaturation and fertilization using a co-culturesystem. Hum Reprod 1998; 13: 1916–21.
45. Cobo AC, Requena A, Neuspiller F et al.Maturation in vitro of human oocytes fromunstimulated cycles: selection of the optimalday for ovum retrieval based on follicular size.Hum Reprod 1999; 14: 1864–8.
46. Bielanski A, Nadin-Davis S, Sapp T et al. Viralcontamination of embryos cryopreserved inliquid nitrogen. Cryobiology 2000; 40: 110–6.
VITRIFICATION IN ASSISTED REPRODUCTION
252
10c Tucker 8029.qxd 8/23/2007 2:17 PM Page 252

253
Vitrifying and warming of humanblastocysts using the CryotopJuergen Liebermann and Michael J Tucker
10D
PURPOSE
Cryopreservation of human oocytes, embryos,or blastocysts allows us to maximize thepotential for conception from any one in vitrofertilization cycle, and prevents wastage ofembryos. Furthermore, this enables best uti-lization of a patient’s supernumerary oocytesafter retrieval, and maximization of the useof embryos from a single stimulation cycle.In other words, at completion of acycle a patient’s supernumerary embryos maybe used beyond the fresh transfer, and canlead to later opportunities for pregnancy.Cryopreservation has been shown to increasepregnancy rates while allowing for furtherselection of embryos. In addition, it is possi-ble to achieve implantation and pregnancyrates with frozen–thawed embryos closeto those achieved with fresh embryos.Blastocysts have been shown to increase preg-nancy rates while allowing for improvedselection of potentially viable embryos. At thislate stage of development lower numbers ofembryos are transferred, resulting in fewerhigh-order multiple pregnancies andincreased implantation rates. Decreasednumbers of embryos are transferred whichresults in lower multiple implantations andthe potential for more blastocysts to be placedin frozen storage with good potential post-thaw, also this reduces the overall number ofoocyte retrieval procedures to which patientsare exposed. Blastocyst cryopreservation is
clearly an optimization of the assisted repro-ductive technologies (ART) process.
MATERIALS AND METHODS
The materials and reagents used are shown inTables 10D.1 and 10D.2, respectively.
Table 10D.1 Materials used for blastocyst vitrificationusing the Cryotop
• Cryotop (Kitazato BioPharma Co, Ltd, Japan)• Polycarbonate micropipettes, 175 and 150 µm
internal diameter (MidAtlanticDiagnostics)• 60 × 15 mm Tissue culture dish (Nunclon 150362)• 60 × 15 mm Center-well organ culture dish
(Falcon 353037)• Weigh paper (Fisher 09-898 12A)• 0.22 µm Syringe filter (Millipore Millex SLGP033RS)• 1 mL Serological pipets (Falcon 7521)• 5 mL Serological pipets (Falcon 7543)• 10 mL Serological pipets (Falcon 7551)• 50 mL Tissue culture flasks (Falcon 3014) • Indelible marker (Sharpie pen, Sanford 33000)• 9” Glass Pasteur pipettes (Sigma S6143)• 10 mL Syringe (Airtite A-10)• 5 mL cryovial (Nalgene 5000-0050)
Table 10D.2 The reagents used for blastocyst vitrifica-tion using the Cryotop
• Synthetic serum substitute (SSS) (Irvine Scientific99193P)
• Modified human tubal fluid (mHTF) (Sage)• Ethylene glycol (EG) (Sigma E-9129)• Dimethylsulfoxide (DMSO) (Sigma D-2650)• Sucrose (Sigma #S1888)
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 253

Solutions were prepared as follows:
(1) All solutions were filtered to ensure steril-ity using a 60 mL syringe and a 0.2 µmsyringe filter into clean labeled flasks.The first few drops of solution were dis-carded to help remove residual contami-nation in the filters.
(2) Aliquots of both solutions were dispensedinto 1.0 mL vials and labeled appropri-ately with data.
(3) All solutions were refridgerated when notin use.
(4) All quality control procedures for the use ofin-house prepared media were followed.
The step-wise vitrification procedure wasperformed as follows:
(1) The benchwarmer was turned off for 2 minto cool closer to room temperature (∼25oC).
(2) Aseptic techniques were required at allstages. Equilibration and vitrificationwere performed at room temperature of22–25°C.
(3) Reagents were removed from the refrig-erator and allowed to warm to roomtemperature.
(4) A tissue culture dish lid (upside down)was labeled with the patient’s name asfollows: stock solution A, ES, and VS.Aliquots of 1 × 50 µL of solution A,4 × 50 µL of ES, and 4 × 50 µL of VS wereprepared (see Figure 10D.1).
(5) The Cryotop was labeled using a cryo-marker with the patient’s last name, firstname, the date of vitrifying, accessionnumber, and the number and develop-mental stage of blastocysts (place no morethan two blastocysts per Cryotop). Note:Cryotops were pre-labeled on the sameside on which the blastocysts were placed.Before vitrification, a micropipette with adiameter of 175–200 µm (‘Stripper’ tip) wasused to load the blastocysts on the top.
(6) A styroform container was filled withliquid nitrogen (LN2).
(7) Procedures for each patient that had blas-tocysts vitrified were performed in sepa-rate hoods and all details were verified bya second embryologist before proceed-ing. Good expanded/hatching blastocystswere vitrified on day 5/6.
(8) Blastocysts were removed from their cul-ture dish using a 200 µm Stripper tip andplaced into the drop of solution A, anytraces of culture media were removed bygentle aspiration.
(9) Blastocysts were pipetted from mHTF tothe 7.5% ES, and the 15% VS as follow:
(a) The blastocysts were placed in solu-tion A (#1 droplet) and this was con-nected with droplet #2 (ES) at roomtemperature. The blastocysts wereallowed to dehydrate appropriately.
(b) After shrinkage and re-expansiondroplet #2 was connected with droplet#3 of ES, and then with droplet #4 ofES to close the ‘circle’.In detail: Exposure to ES occurreduntil blastocysts re-expanded to ~80%of their original volume. Blastocystswere placed, one to three at a time,into a single drop of ES (see Figure10D.1), with three drops of ES (A) ona plain 60 mm dish lid surface. Thedrops were connected 1–2, 2–3, 3–4,and finally 4–1 approximately every1–2 min, so exposing the blastocysts toa gently increasing concentration
VITRIFICATION IN ASSISTED REPRODUCTION
254
Table 10D.3 Formulations of equilibration andvitrification solutions
• Stock solution A: 40 mL mHTF + 10 mL SSS(mHTF + 20% SSS) – final total volume of 50 mL
• Stock solution B: 6.0 ml EG + 6.0 mL DMSO + 28 mLA (15% EG/DMSO) − final total volume of 40 mL
• Equilibration solution: 15 mL A + 15 mL B (7.5%EG/DMSO) − final total volume of 30 mL
• Vitrification solution: 5 mL B + 1.71 g sucrose(15% EG/DMSO + 0.5 mol/L sucrose) – bring to afinal total volume of 10 mL with solution B*
* Always consider 10 mL as the final total volume allowing fortotal dissolution of sucrose.
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 254

gradient until they were in nearly‘pure’ equilibration solution (ES). Notethat the blastocysts remained in theoriginal drop #1 area, and were influ-enced by influx of higher concentra-tions of ES over a period of 5–7 min.These stages should not be rushed,and care was taken not to introduceincreasing ES concentrations until theblastocysts were partially re-expanded.
(c) Blastocysts were placed into the topof drop #5 and allowed to sink to thebottom for 1–2 min depending onre-expansion of the blastocysts whichshould have returned to ~75–85% oforiginal volume by which time theywere ready to pass into the vitrifica-tion solution (VS).
(10) The blastocysts were loaded in a newVS back loaded Stripper tip, and rinsedthrough the four droplets of VS,between each droplet the tip of thepipette was rinsed.
In detail: With four drops (#6–9) of theVS aligned next to the ES drop #5 (seeFigure10D.1), the re-expanded blastocystswere moved into the first of the four drops(#6–9), and up through the VS drops of
15% EG + 15% DMSO plus 0.5 mol/Lsucrose by a process of joining the firstthree drops (#6–8), so that the blastocysts‘rose’ up incrementally through into thepurest VS droplet; then were moved intodrop #9. Blastocysts shrinkage was quiteextreme as was to be expected at this highconcentration of vitrificants. Re-expansionshould not be allowed.
(11) Placement into the VS and loading ofthe Cryotop took less than 1 min,preferably the total exposure time in VSwas approximately 30 s. After 30 s, theywere gently transferred to the tip of theCryotop by using a Stripper tip to loadthe blastocysts in as small volume(0.5 µL) as possible onto the edge of theCryotop (the ‘loading’ side, which isthe side labeled with ‘CRYOTOP’ plusthe patient information, was used). Thetip of the Cryotop was marked withblack marker to help visualize the tipunder the liquid nitrogen.
(12) Placement of the blastocyst(s) was visu-ally confirmed.
(13) The loaded Cryotop was plungeddirectly and vertically in liquid nitrogen(LN2). Under LN2 the Cryotops werecapped with the flared blue straw cover
255
VITRIFYING AND WARMING OF HUMAN BLASTOCYSTS USING THE CRYOTOP
Stock solution A ES
6.2.
(Merge drops)
(Transferto drop)
3.
1.
4.
5.
7.
8.
9.
VS
ES
Figure 10D.1 Setup for the vitrification procedure on a plain 60 mm dish lid surface ES, equilibration solution;VS, vitrification solution.
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 255

before they were placed in an enclosedlabeled cane holder. The Cryotop wasplaced on a precooled aluminum canefor further storage.
(14) The cane was stored in a LN2 dewar.(15) The cane location was recorded on the
freezing worksheet and in the cryoin-ventory log.
The warming solutions used are shown inTable 10D.5 (see also Table 10D.4).
Solutions were prepared as follows:
(1) All solutions were filtered to ensuresterility using a 60 mL syringe and a0.2 µm syringe filter into clean labeledflasks. The first few drops of solutionwere discarded to remove residual cont-amination from the filters.
(2) Aliquots of both solutions were dis-pensed into 1.0 mL vials and labeledappropriately with data.
(3) All solutions were refridgerated whennot in use.
(4) All quality control procedures for theuse of in-house prepared media werefollowed.
The step-wise warming procedure was per-formed as follows:
(1) Reagents were removed from the refrig-erator and allowed to warm to roomtemperature. All cryoprotectants wereremoved at 25°C.
(2) 1 mL of TS was placed into an organculture dish and warmed for 5 min at37°C.
(3) A 60 mm Petri dish was labeled with thepatient’s name under the lid as follows:TS, DS, and HS. Aliquots of 1 × 50 µL ofTS, 4 × 50 µL of DS, and 6 × 50 µL of HSwere prepared (see Figure 10D.2).
(4) Before warming, a micropipette with a200 µm bore was used for removing theblastocysts from the Cryotop.
(5) A styroform container was filled withLN2.
(6) The location and identification of blasto-cysts was verified with a second embryol-ogist before warming any Cryotop. OneCryotop was warmed at a time.
(7) A separate hood was used for each patientfor whom Cryotops are to be warmed, andverification by a second embryologistoccurred before proceeding.
(8) With the Cryotop under LN2, the tip wasopened by removing the cap.
(9) The Cryotop was submerged direct in thepre-warmed organ culture dish (~33oC)containing ~0.5mL TS. As soon as theCryotop contents liquefied (within 15 s),the blastocysts were located before beingremoved with a Stripper pipette.Blastocysts were located as they floated freefrom the Cryotop surface, and moved intodrop A (see Figure 10D.2). Immediatelydrops B and A were connected together,allowing the 0.5 mol/L sucrose solution
VITRIFICATION IN ASSISTED REPRODUCTION
256
Table 10D.4 Summary of vitrification and warmingsolutions
Ethyleneglycol DMSO Sucrose
Equilibration 7.5% 7.5% 0.0 mol/Lsolution (ES)Vitrification 15% 15% 0.5 mol/Lsolution (VS)Thawing solution (TS) 0.0% 1.0 mol/L 0.0 mol/LDiluent solution (DS) 0.0% 0.5 mol/L 0.0 mol/LHolding solution (HS) 0.0% 0.0 mol/L 0.0 mol/L
Modified human tubal fluid (mHTF) (commercially avail-able) + 20% SSS (Holding solution – HS)
Table 10D.5 Warming solutions used for blastocyst
• Holding solution (HS): 16 mL mHTF + 4 mL SSS(mHTF + 20% SSS)
• Thawing solution (TS): Initial starting with 5 mlHS + 3.423 g sucrose (1.0 mol/L sucrose + HS) – afterdissolution of sucrose add an additional HS to finaltotal volume of 10 mL.
• Diluent solution (DS): 5 mL HS + 5 ml TS (0.5 mol/Lsucrose + HS)
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 256

(DS) at room temperature to wash intodrop A (TS) for ~2min.
(10) Drops were connected C–B, D–C, andthen finally drop D directly with A wherethe blastocysts still sat. All connectionswere performed such that blastocystover-expansion was limited, to ensurethat they were not allowed to re-expandto their full 100% state, reconnectingoccurred when approximately 75–85%of full size. Blastocysts were moved todrop E, and the gradual dilutionrepeated by connecting drops F–E, G–F,H–G, and finally H directly to E (‘the cir-cle of life’). The benchwarmer wasturned on, and finally dilution occurredthrough a series of three wash drops ofHS (I–K).
(11) Blastocysts were moved into culturecompatible media and put in the incu-bator for subsequent culture.
(12) Survival and appearance of all blasto-cysts were recorded. The log wasupdated with data following warming ofblastocyst(s), and the physician notifiedof results.
Definitions
A positive pregnancy was defined as a positiveβhCG of ≥ 20 IU 10 days after blastocysttransfer. Implantation rate was defined by thenumber of gestational sacs per embryo num-ber transferred. Clinical pregnancy refers tothe identification of a pregnancy sac in theuterus, whereas ongoing/delivered pregnancydecribes pregnancies that continued beyond20 weeks. Statistical analysis was carried outby means of a χ2 test using Microsoft Excel2001 for Mac (Redmond, WA, USA).Statistical significance was defined asP < 10.05.
RESULTS
Table 10D.6 shows the mean age and clinicaloutcome of patients who completed the vitri-fied blastocyst transfer program. The meanage of the women was 34.1 ± 5.1 years in thevitrified group. The blastocyst post-warmingsurvival rate after using the vitrification tech-nique is shown in Table 10D.2. A total of 1140vitrified blastocysts (day 5 and day 6) were
257
VITRIFYING AND WARMING OF HUMAN BLASTOCYSTS USING THE CRYOTOP
(Merge drops)
(Merge drops)
TS
DS
HSDS
60 mm Organ culture dishwith 1.0 mL TS
(Transferto drop)
A.
D.
B. I.
J.
K.
C.
G.
F.E.
H.
Figure 10D.2 Setup for the warming procedure on a plain 60 mm dish lid surface. TS, thawing solution; DS, diluentsolution; HS, holding solution.
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 257

warmed, of which 1085 survived warming(95.2%).
In the vitrified group 1073 vitrifiedwarmed blastocysts were transferred in 541cycles out of 548 attempted cycles (mean 2.0blastocysts per frozen embryo transfer).Overall the implantation and clinical
pregnancy rates per transfer were 28.9% and42.1%, respectively. To date, 93 deliverieshave occurred with no reported abnormali-ties (117 babies: 65 girls and 52 boys).
When the vitrified warmed blastocysts weredivided into blastocysts vitrified on day 5 andday 6 groups, 95.5% (611/640) of day 5 blas-tocysts and 94.8% (474/500) of day 6 blasto-cysts survived after warming (Table 10D.7),but this difference was not significant. Asshown in Table 10D.7, implantation and clin-ical pregnancy rates per transfer occurring inthe day-5 blastocyst group were 32.3% and45.8%, respectively, which were significantlyhigher (P < 0.01) than the day-6 blastocystgroup (24.6% and 37.8%, respectively).
CONCLUSIONS
In conclusion, although some problemsremain to be fully addressed regarding vitrifi-cation as a routine cryopreservation tech-nique, we believe that it shows much promiseas a successful alternative to conventionalfreezing technology. Even without significantclinical improvement, the evident advantagesof vitrification are that cryosurvival seems
VITRIFICATION IN ASSISTED REPRODUCTION
258
Table 10D.6 Retrospective data from the blastocystcryopreservation program at the Fertility Centers ofIllinois, Chicago, where vitrification technology wasapplied from January 2004 to December 2006
Technique
Patient’s age (years) 34.1 ± 5.1No. of thawed cycles 548No. of transfers 541No. of blastocysts thawed 1140No. of blastocysts survived (%) 1085(95.2)No. of blastocysts transferred 1073Mean no. of blastocysts transferred 2.0No. of implantations (%) 310 (28.9)No. of positive pregnancy/thaw (%) 270 (49.3)No. of positive pregnancy/FET (%) 270 (50.0)No. of clinical pregnancy/thaw (%) 228 (41.6)No. of clinical pregnancy/FET (%) 228 (42.1)Ongoing pregnancies (%) 198 (73.3)No. of livebirths 117 (65 girls
and 52 boys)
FET, frozen embryo transfer.
Table 10D.7 A comparison of retrospective data from the blastocyst cryopreservation program at the FertilityCenters of Illinois, Chicago of vitrified on day 5 and day 6 from January 2004 to December 2006
Day of development
Day 5 Day 6
Patient’s age (years) 34.0 ± 5.0 34.2 ± 5.2No. of thawed cycles 297 251No. of transfers 295 246No. of blastocysts thawed 640 500No. of blastocysts survived (%) 611 (95.5) 474 (94.8)No. of blastocysts transferred 601 472Mean no. of blastocysts transferred 2.0 1.9No. of implantations (%) 194 (32.3)* 116 (24.6)*
No. of positive pregnancy/thaw (%) 164 (55.2) 106 (42.2)No. of positive pregnancy/FET (%) 164 (55.6)* 106 (43.0)*
No. of clinical pregnancy/thaw (%) 135 (45.5) 93 (37.0)No. of clinical pregnancy/FET (%) 135 (45.8)* 93 (37.8)*
Ongoing/delivered pregnancies (%) 120 (73.2) 78 (73.6)
*P < 0.01FET, frozen embryo transfer.
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 258

more consistent, allowing greater ease ofpatient management with transfers beingalmost certain to occur. Concerns with vit-rification are well defined and limited innumber, and to our way of thinking easilysurmountable. In general, with much shorterprotocols, vitrification is able to be under-taken on a more flexible basis by laboratorystaff, allowing for the potential reductionin personnel time needed during the entirevitrification process, simplifies laboratorytechniques for cryopreservation in humanART, and may enable more optimal timing ofembryo cryopreservation, e.g. individual blas-tocysts may be cryopreserved at their optimalstage of development and expansion. Interestlevels will inevitably rise given the potentialbenefits of vitrification. This in turn willdrive its development to higher levels ofclinical efficiency and utilization.
SPECIAL NOTES FOR THEPRACTITIONER
Special care must be given to the selecting ofthe carriers. It is necessary to use types of car-rier or vessel material with rapid heat transferthat also support the process of uniform heatexchange to achieve higher cooling rates.
To minimize the toxicity of the cryoprotec-tant a step-wise exposure of cells to pre-cooledconcentrated solutions is recommended.
Higher concentrations of cryoprotectantshould be utilized that allow shorter exposure
times to the cryoprotectant – but be careful –the toxicity of the cryoprotectants will beraised with their concentration. Becausealmost all cryoprotectants are toxic, it isimportant to watch the duration of exposureto the final cryoprotectant very carefullybefore plunging into liquid nitrogen.
To facilitate vitrification by even higher cool-ing rates, it is also necessary to minimize thevolume of the vitrification solution as muchas practical. From this point of view it is veryimportant to use a micropipette with a smalldiameter (< 150 µm). Furthermore, by collect-ing the embryos on one place, and loading notmore than two embryos at the same time in thepipette it is possible to keep the volume small.
To make sure that the cells have been loadedon the carrier, perform the loading processunder a stereo-microscope. Check the numberof loaded embryos, and check the pipette isempty after loading.
Submerge the carrier loaded with the cellsdirectly in liquid nitrogen by passing quicklythrough the vapor phase (nitrogen gas).
Before moving the carrier quickly from theLN2 in to the warming solution, prepare amicropipette. Fill the pipette with a smallamount of the first warming solution.
Even when switching the embryos betweendifferent concentrations of warming solu-tions, fill up the pipette tip with the nextlower concentration of warming solutionbefore picking up the embryos for moving into the following concentration.
259
VITRIFYING AND WARMING OF HUMAN BLASTOCYSTS USING THE CRYOTOP
10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 259

10d Tucker 8029.qxd 8/23/2007 2:17 PM Page 260

261
Vitrification of ovarian tissueFrank Nawroth, Vladimir Isachenko, Evgenia Isachenko and Gohar Rahimi
11A
INTRODUCTION
Secondary ovarian failure could for a varietyof reasons be an indication to cryopreserveovarian tissue. Such incidences haveincreased since malignant diseases can nowbe successfully treated, but often result inirreversible fertility loss. At the moment, forlater fertility preservation the extraction andstorage of larger amounts of ovarian cortexusing unilateral ovarectomy is favored.1
The protocols used for the cryopreserva-tion of ovarian tissue are based on those formature oocytes.2 However, additional prob-lems exist with the cellular heterogeneity ofovarian tissue, giving rise to different diffu-sion rates of the cryoprotectants throughoutthe tissue. The optimal protocol with regard tosuitable cryoprotectant and freezing techniquefor ovarian tissue has yet to be found,2 but atpresent ‘slow cooling and rapid thawing’ isfavored.3
Ovarian tissue can be auto- or xenotrans-planted after thawing. At the time of ovariantissue harvest careful examination sometimesreveals the presence of free immature oocytesthat can be gathered during tissue prepara-tion before freezing, and can additionally befrozen at the immature stage or after in vitromaturation (IVM).4–6
The conventional cryopreservation ofhuman ovarian tissue is developing from anexperimental procedure into daily practice.Although further research must focus oninvestigating current options and new alterna-tives to identify the best way of using ovariantissue after thawing, a consultation on the pos-sibility of this procedure for different patients
(for example before chemotherapy/radiation)is now more commonly recommended.7,8
Several pregnancies after conventional slowfreezing of human ovarian tissue have beenreported since 2004 (Table 11A.1).
At the same time, the method should beroutinely performed with minimal equipment,staff, and time requirements. This is the reasonfor the increased interest in vitrification notonly of cells but also of tissue.13 The problemsrelated to successful cryopreservation increasewith the complexity of the sample intended forvitrification (cell–tissue–entire organ).
A renaissance of interest in vitrificationstarted in 1980. At this time the AmericanRed Cross initiated studies investigating thepotential of vitrification as an alternative toclassic cryopreservation. Viability assays havedemonstrated successful application of vitri-fication in a variety of single cells or smallcell aggregates including human islets ofLangerhans, monocytes, red blood cells, livercells in culture, certain plants and plant tis-sues, a variety of animal embryos, and rodentegg cells.14 It has been performed partiallysuccessfully with human corneas and rabbitkidney slices.
When vitrification is used with tissue, dueto the high concentration of cryoprotectants,the heterogeneity of the cells with long diffu-sion times and the resulting potential toxicitycan be problematic. The main problems withvitrification of large samples are fracturing aswell as crystallization during cooling and/orwarming. Fracturing can mostly be preventedthrough careful handling of the sample, sothat crystallization remains the more seriousproblem.15
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 261

VITRIFICATION OF OVARIANTISSUE IN ANIMALS
Various research groups have reported thesuccessful vitrification of ovarian tissuefrom mice, rats, Chinese hamsters, rabbits,Japanese apes, monkey, cows, sheep, dog,and human fetuses.15–24 The problem of thetransfer of data from animal studies to thehuman was the focus of a study dealing withthe determination of the best experimentalmodel for developing new protocols of equi-librium cooling and vitrification.25 Theauthors compared adult cow and pig withhuman ovaries and found that human andbovine follicles responded in the same way tothe two equilibrium cooling protocols used,whereas pig tissue was more cryoresistant.Both vitrification protocols caused extensivedamage to the tissue of all species. Humantissue showed a response to vitrification that
was different from that of both cow and pig.Their conclusion was the cow is a good ani-mal model for the development of equilib-rium cooling procedures but at present,neither cow nor pig can be considered rele-vant animal models for the vitrification ofhuman ovarian tissue.
Specific experiences with vitrificationof ovarian tissue in some animalspecies
Vitrification of ovarian tissue in ratsand mice
Miyamoto and Sugimoto16 vitrified rat ovariesand removed the cryoprotectant step-wise.Histological examination of the folliclesyielded positive results regarding surface area,but revealed degenerative changes, such aspyknosis, vacuolization, and cell swelling, in
VITRIFICATION IN ASSISTED REPRODUCTION
262
Table 11A.1 Published pregnancies after autotransplantation of cryopreserved/thawed human ovarian tissue (up toNovember 2006)
Age beforefreezing Location of Reproductive
Reference (years) Indication transplantation outcome
Donnez et al., 20049 25 Hodgkin’s Orthotopic: peritoneum Spontaneous lymphoma of the ovarian fossa pregnancy, live birth
Meirow et al., 200510 28 Non-Hodgkin’s Orthotopic: ovary Modified menstrual lymphoma cycle, IVF-ICSI,
ET, live birth
Demeestere 24 Hodgkin’s Orthotopic: ovary + Spontaneous et al., 200611 lymphoma peritoneum of the ovarian pregnancy, abortion
fossa combined with at 7 weeksHeterotopic: abdominal wall subcutaneously
Rosendahl 28 Hodgkin’s Orthotopic: ovary + FSH stimulation, et al., 200612 lymphoma peritoneum of the ovarian IVF-ICSI after
fossa combined with oocyte retrieval from Heterotopic: subperitoneal the heterotopic graft, tissue beneath the abdominal ET, biochemical fascia between the umbilicus pregnancyand the pubic bone
IVF-ICSI, in vitro fertilization – intracytoplasmic sperm injection;ET, embryo transfer; FSH, follicle stimulating hormone.
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 262

the remaining tissue. Therefore, ‘slow cooling’was considered to be superior, even thoughthe tissue showed partial vitality.
Vital follicles were still detected 4 daysafter the warming of vitrified fetal ratovaries.18 Sugimoto et al.19 vitrified infantilerat ovaries (left side) and assessed their via-bility after warming and autotransplantationunder the kidney capsule. The right ovary ofeach rat was removed. For the control ani-mals, the left ovary was dissected and imme-diately transplanted without priorvitrification. The time course of developmentof endocrine function of cryopreserved graftswas similar to that of fresh grafts. In ovariantransplants recovered on postnatal day 84,antral follicles and corpora lutea wereobserved in addition to small follicles,although the number of antral follicles in cry-opreserved grafts was smaller than in thefresh grafts. The authors concluded that vitri-fication of ovarian tissue can be used for thepreservation of fertility and endocrine func-tion of ovaries.
Salehnia et al.26 vitrified ovaries from 8–10-week-old mice using a vitrification solutioncontaining 30% (w/v) Ficoll 70, 0.5 mol/Lsucrose, 10.7% (v/v) acetamide, and 40% (v/v)ethylene glycol (EG). After warming with1 mol/L sucrose solution the vitrified and freshovarian tissues were fixed and histological andelectron microscopic investigations were per-formed. Under the electron microscope, theintegrity of cell organelles, nuclei, andmicrovilli of oocyte and follicular cells were wellpreserved but some mitochondria were swollenand their cristae had partially disappeared.Vitrification did not cause any harmful damageto follicular cells and oocytes. The samegroup27 reported interesting results if mouseovaries, vitrified/warmed using the same solu-tions, were autografted intraperitoneally andrecovered after one and two estrous cycles.Light microscope studies after 5 days showedthat the grafted ovarian tissues were invaded bymany fat and fibrous cells. Many large pre-antral and antral follicles were degenerated;
however, after 10 days the stroma of trans-planted ovaries was devoid of necrotic cells andcontained normal primordial and primary fol-licles. Non-frozen ovaries (control) had normalfollicles at all developmental stages.
Kim et al.28 investigated whether oocyteswithin preantral follicles isolated from vitri-fied mouse ovaries are viable and can be res-cued to undergo growth, maturation,fertilization, and embryo development invitro. EG, dimethylsulfoxide (DMSO), andsucrose were used as cryoprotectants.Equilibration time was either 5 or 10 min.Survival and maturation rates were signifi-cantly higher in the 5 min compared with the10 min exposure groups. This led to the con-clusion that mouse oocytes within preantralfollicles isolated from the vitrified ovary canachieve full maturation and normal fertiliza-tion and embryo development.
Takahashi et al.29 reported the first suc-cessful vitrification of adult mouse ovariesusing the method of Rall and Fahy30 with VS1,a combination of 20.5% (w/v) DMSO, 15.5%(w/v) acetamide, 10% (w/v) propylene glycol(PG) and 6% (w/v) polyethylene glycol (PEG).
Migishima et al.31 developed a new methodof cryopreservation of whole mouse ovaries byvitrification using DAP213 (2 mol/L DMSO,1 mol/L acetamide, and 3 mol/L PG) as acryoprotectant. Cryopreserved or fresh ovarieswere orthotopically transplanted (experimen-tal or control group). Histologically, normaldevelopment of follicles and formation of cor-pora lutea were observed in frozen/thawedgrafts. However, estimated number of folliclesdecreased in frozen/thawed ovaries comparedwith fresh ovaries showing that further studiesare required to overcome the possibleinhibitory effects of this method on the growthof the ovarian graft.31
Segino et al.32 isolated and cultured cumulus-enclosed oocyte complexes (COC) and pre-antral folicles from vitrified mouse ovariantissue. The survival rate of the follicles obtainedfrom the cooled/warmed ovaries was 66.4%.Comparison of the follicles isolated from fresh
263
VITRIFICATION OF OVARIAN TISSUE
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 263

and frozen/thawed ovaries after 12 days ofculture showed significant differences regard-ing the diameter of the isolated follicles(620.2 ± 11.3 µm vs. 518.7 ± 15.1 µm) andestradiol concentrations (3474.2 ± 159 pg/mLvs. 1508.2 ± 134 pg/mL). After in vitro ovula-tion, metaphase II (MII) stage oocytes wereobserved in 84.5% of the fresh group and60.5% of the cooled/warmed group, while thefertilization rates were 74.2% and 53.5%,respectively. The study demonstrated that vit-rification of mouse ovarian tissues did notaffect the oocyte’s ability to undergo meiosis.Follicles isolated from vitrified/warmed mouseovarian tissues reached the mature folliclestage on the 12th day of culture. However, thedevelopmental ability was lower than that offresh ovarian tissue. The same group33 cul-tured preantral follicles isolated from vitri-fied/warmed ovarian tissues for 12–16 days.Then the follicles were stimulated with humanchorionic gonadotropin. They developedslowly compared with the freshly prepared pre-antral follicles. However, during prolongedculture from 12 to 16 days, these folliclesobtained the potential to fertilize and developto the blastocyst stage. This confirmed theresults from another study published 1 yearearlier.34 COC were retrieved from vitrifiedmouse ovaries by enzymatic treatment and invitro fertilized after in vitro growth (IVG) andIVM. EG and DMSO were used as cryoprotec-tants. Following the procedure including IVM,75.9% of oocytes in COC matured to the MIIstage compared with 75.2% from fresh ovaries.After in vitro fertilization (IVF) the fertilizationrate of these oocytes was 57.5% as comparedwith 69.5% for fresh ovaries. Both differenceswere not statistically significant. The conclu-sion was that oocytes enclosed in preantralfollicles from vitrified/warmed mouse ovariespreserved capacity for fertilization and devel-opment to preimplantation embryos.
Recently an interesting paper was publisheddealing with a new vitrification method usingless concentrated cryoprotectants and directapplication of liquid nitrogen to the ovarian
tissue in mice (direct cover vitrification,DCV).35 In the first step the ovary (size of anovary approximately 1.2 × 1.5 × 1.5 mm) waspretreated with an equilibration solution(0.8 mL) consisting of 7.5% (v/v) EG and 7.5%DMSO in Dulbecco’s phosphate-buffered solu-tion (DPBS) with 20% fetal bovine serum (FBS)for 10 min at room temperature. It was thentransferred to a vitrification solution (0.8 mL)consisting of 15% EG, 15% DMSO, and0.5 mol/L sucrose for 2 min. After removal ofthe surrounding vitrification medium theovary was placed on a piece of gauze, put in a1.8 mL plastic standard cryovial (Nunc,Roskilde, Denmark), and liquid nitrogen wasdirectly applied onto the ovary. Following thisprocedure the vial was placed into a liquidnitrogen tank. For thawing, the ovary wasmoved into 1 mL of 1 mol/L sucrose, keptthere for 5 min and then put into 0.5 mol/Lfollowed by 0.25 mol/L sucrose and DPBSmedium for 5 min each. One of the results ofthe study was a significantly higher pregnancyrate of DCV compared with conventional vitri-fication after orthotopic transplantation(P < 0.01).
Hani et al.36 showed that vitrification ofmouse ovaries is possible for the preservationof female germ cells from mice of variousages. They used the above mentionedDAP213 as vitrification solution31 and com-pared the viability of cryopreserved adultmouse ovaries with that of immature ovaries.Both were viable and able to produce youngafter orthotopic transplantation to 4- or 10-week-old mice.
Vitrification of bovineovarian tissue
The comparison of conventional freezing andvitrification of bovine ovarian tissue demon-strated that a vitrification protocol using EGand equilibration with 5.5 mol/L EG at 22°Cfor 20 min may be just as effective as ‘slowfreezing’.15
VITRIFICATION IN ASSISTED REPRODUCTION
264
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 264

Vitrification of ovarian tissuein sheep
Al-Aghbari and Menino37 removed and cutovaries from 15 pubertal ewes. They usedequilibration medium consisting of 4% (v/v)EG and 20% (v/v) FBS in TCM-199 on ice for30 min and transferred the pieces to vitrifica-tion solution (35% EG, 5% polyvinylpyrroli-done, 0.4 mol/L trehalose, and 20% FBS inTCM-199) for 5 min. They also collected andvitrified COC. After 2–3 weeks storage in liq-uid nitrogen, ovarian tissues and COC werethawed at 37°C. Vitrified COC and freshlycollected (mechanically and enzymaticallyisolated from vitrified tissue after thawing)COC were cultured for 23–25 h. Significantlyfewer (P < 0.05) oocytes obtained from vitri-fied ovarian tissue (70%) reached MII com-pared with vitrified oocytes (88%) andnon-vitrified control oocytes (90%). In con-trast, when oocytes with at least 3–5 layers ofcumulus cells were considered from each ofthe three groups, no significant differenceswere observed due to treatment in the per-centages of oocytes developing to MII.Hence, these authors demonstrated thatsheep oocytes can be successfully cryopre-served by vitrification of ovarian tissue andexhibit in vitro maturation rates similar tothose of vitrified and non-vitrified oocytes.
A further step was the attempt to vitrifywhole sheep ovaries with vascular pedicle.38
Their study based on results from a rabbitmodel,39 where whole rabbit kidneys were per-fused with a vitrification solution containing2.75 mol/L DMSO, 2.76 mol/L formamide,and 1.97 mol/L PG as cryoprotectants (VS4).The kidneys were evaluated using an auto-graft model with immediate contralateralnephrectomy. Using the protocol with thehighest concentrations and perfusion at about−3°C, the survival rate was 100%, serum crea-tinine returned to a normal baseline aftertransient elevation, other clinical chemistryresults normalized, and no histological dam-age was apparent 3 weeks after autografting.
Courbiere et al.38 used the same VS4 in com-parison with VS1 (2.62 mol/L DMSO,2.60 mol/L acetamide, 1.31 mol/L PG, and0.0075 mol/L PEG as cryoprotectants dilutedin BM1 medium (Eurobio, Les Ulis, France))in their sheep model. They described nostatistically significant differences in follicleviability or normal primordial follicle ratesbetween ovaries exposed or not exposed tocryoprotectant solutions, before and after vit-rification with the two cryoprotectant solu-tions. VS4 was beneficial regarding nuclearanomalies and general follicular anomalies.However, vascular pedicle fractures occurredin most ovaries during thawing (11/15).38 Ina later published study the same authors24
focused on the question of whether wholesheep ovaries can really be totally vitrifiedusing VS4. The difference of follicle viabilityfor ovaries exposed to VS4, without vitrifica-tion and for ovaries vitrified with VS4 wasnot statistically significant (70.6% ± 4.7% vs.61.4% ± 2.2%). However, they showed that thecritical cooling rate for the impregnated ovar-ian cortex exceeded −300°C/min, suggestingthat, under the studied experimental condi-tions, the ovarian tissue is unlikely to be totallyvitrified at the end of cooling.
After vitrification using VS1, warming andorthotopic autotransplantation of hemi-ovaries into sheep three pregnanciesoccurred. One of the four lambs that wereborn had a malformation of the left leg andthe esophagus, with a questionable link to thevitrification.40
Vitrification of ovarian tissue in monkey
Co-culture of vitrified/warmed or slow-frozen monkey ovarian tissue on mouse fetalfibroblast monolayers supplemented withfollicle stimulating hormone (FSH), insulin,transferrin, and selenium resulted in a sig-nificantly increased rate of viable follicles invitrified/warmed as well as slow-frozen tis-sue. Co-culture could therefore be a benefi-cial approach to improve graft survival after
265
VITRIFICATION OF OVARIAN TISSUE
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 265

vitrification, just as it can be as effective asafter slow freezing.23
Vitrification of ovarian tissue in rabbit
Ishijima et al.41 vitrified canine ovaries.Warmed tissue which showed normal mor-phology in histology was transplanted intothe ovarian bursa of the severe combinedimmune deficiency (SCID) mice and recov-ered 4 weeks after the operation. Antral folli-cle formation did not occur after grafting butproliferating cell nuclear antigen immunore-activity was detectable in many of the granu-losa cells in the primary follicles of the grafts.The authors concluded that vitrification hasthe potential to restore endocrine functionand ovulation potential in canine.
VITRIFICATION OF OVARIANTISSUE IN HUMAN
It is known from other areas of humanresearch that the vitrification of cornea42 andvessels14,43 is possible. Practical knowledgeregarding virtification of human ovarian tis-sue is limited. Nevertheless, some initialstudies have dealt with vitrification of humanovarian tissue.44–46
Vitrification of human ovarian tissueand investigation/in vitro culture afterwarming
Human fetal17,22,47 and adult ovarian tissuesamples20 have been vitrified successfullyusing EG and sucrose. In a computerized pic-ture analysis of the cell nuclei the results werecomparable after vitrification or slow freezingof human ovarian tissue.44 They removedovarian cortical slices (10 × 2 × 1 mm) from sixconsenting women (aged 29–36 years) duringsecond Cesarean section and cut each sliceinto five pieces. The aim was to compare afresh sample (scraped with a scalpel, smearedon a microscopic slide, and air-dried at room
temperature), a necrotic sample (put on agauze moisturized with 0.9% NaCl for 24 h),a slow-frozen sample (automated freezer incryovials in α-MEM + 10% patient serum+1.5 mol/L DMSO), and two vitrified samples(vitrified in cryovials in 0.5 mL of PBS + 20%fetal cord blood serum + 5.5 mol/L EG +1 mol/L sucrose for 1 and 5 min, respectively,and then plunged into liquid nitrogen). Afterthawing, 15 nuclear parameters includingmorphometric descriptors (nuclear area,perimeter, form factor) and nuclear texturefeatures (contrast, entropy, variance, andMarkovian features 4, 5, 6, 7, 10, 11, 16, 17,and 18) were assessed for each nuclear image.Preliminary results of computerized imageanalysis of nuclear features including textureindicated a promising model for post-thawviability evaluation. In their study, 1 min vitri-fication in 5.5 mol/L EG appeared to be com-parable with slow freezing for human ovariantissue.
During our own first histological studiesof vitrified human adult ovarian tissue sam-ples (maximum size, 1 mm3) (unpublisheddata) we found that freezing and warmingwith EG + saccharose + egg yolk in combina-tion with direct plunging of straws or grids inliquid nitrogen did not significantly influencethe ovarian tissue morphology or the folliclemorphology. In combination with suitablelong-term cultures of human ovarian tissue,the subsequent IVM could complement treat-ment for example, in planned transplants. Ina long-term culture of native human ovariantissue, we were already able to show that nosignificant increase in apoptosis occurredafter 6 weeks compared with control tissueson day 1.48
We further evaluated the effect of differentvitrification protocols on reactive oxygenspecies (ROS) and apoptosis in human ovariantissue.46 Ovarian tissue pieces (1 ±0.5 mm3) wererandomly distributed into three treatmentgroups and exposed at 0–1°C to differentvitrification solutions as follows: group 1,
VITRIFICATION IN ASSISTED REPRODUCTION
266
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 266

control (without treatment); group 2, 40% EG(v/v) + 0.35 mol/L sucrose + 10% egg yolkextract (v/v)49 for 6 min; and group 3, 40% EG +18% Ficoll 70 (w/v) + 0.35 mol/L sucrose50 for6 min. Egg yolk was used as cryoprotectant.51 Allexposures in vitrification solutions started with1 min shaking at 5 Hz. After equilibration withthe cryoprotectants, ovarian tissue pieces wereplaced with a 1cm column of vitrification solu-tion in 0.25 mL standard insemination straws(IMV, L’Aigle, France) and plunged into liquidnitrogen, inserted into the metallic powdercooled previously in liquid nitrogen,52 insertedinto liquid nitrogen vapor (−120°C) for 15 minand subsequently plunged into liquid nitrogen,or plunged into liquid nitrogen after beingtransferred onto copper grids.53 Very rapidlycooled tissue (plunged directly into liquid nitro-gen in straws or on grids, or plunged directlyinto metal filings precooled to −196°C) showedno statistically significant increase in either tis-sue ROS levels or the number of apoptotic cellsafter warming. In contrast, cooling using a lessrapid method (nitrogen vapor at −120°C)resulted in significantly elevated ROS levels andapoptosis after warming. There were no signifi-cant differences between the two vitrificationsolutions. This indicates that human ovarian tis-sue pieces should be vitrified using very rapidcooling rates.46
Our results indicated that vitrification ofhuman ovarian tissue should involve onlypermeable cryoprotectants and substancesthat prevent ice formation. The addition ofthe disaccharide Ficoll 70 resulted in anincreased osmotic injury.54
In another study, we tried to furtherdevelop the vitrification protocol. Humanovarian biopsies from 20 patients (cut into~ 0.5 mm3 pieces) were exposed to 40% EG +0.35 mol/L sucrose + 5% egg yolk, 40% EG +18% Ficoll 70 + 0.35 mol/L sucrose, or 20%EG + 20% DMSO. Cryopreservation of pieceswas accomplished by plunging 0.25 mL strawsor copper grids into liquid nitrogen, or0.25 mL straws into precooled (−196°C)
metallic powder. Thawed pieces were trans-ferred to sucrose solution for incrementaldilution of cryoprotectants. Histologicalobservation of the tissue was performed aftercryopreservation, and in vitro culture wasundertaken to study the ability to producehormones after cryopreservation. The onlyvitrification solution which protected bothfollicles and stroma was 40% EG + 0.35 mol/Lsucrose + 5% egg yolk using standard0.25 mL straws or copper grids, with directplunging into liquid nitrogen.45
Recently a modified method of in vitro cul-ture of vitrified human ovarian tissue waspublished.55 Different groups of in vitro cul-ture after warming during 2 and 6 weeks werecompared with respect to follicle growth: in2 mL of culture medium which was regularlyrenewed (group 1), in 30 mL of culturemedium without agitation (group 2), and in30 mL of culture medium with agitation (75oscillations/min using a rotation shaker)(group 3). After 2 weeks of culture, the meannumber of non-degenerated follicles permm2 of tissue was significantly higher ingroup 3 when compared with groups 1 and 2(P < 0.05). The conclusion was that agitationduring culture of ovarian tissue is beneficial(Figures. 11A.1 and 11A.2).
Kagawa et al.56 vitrified human ovarian tissueusing different sample sizes (0.5 ×0.5 ×0.5 mm(small), 10 × 10 × 1 mm or 20 × 10 × 1 mm(large)) with the Cryotop as carrier. After incu-bating the tissues in the equilibration solution(7.5% EG + 7.5% DMSO) at room temperature,they transferred the samples to five different vit-rification solutions (EG and DMSO: 15 + 15%;17.5 +17.5%; 20 + 20%; 22.5 + 22.5% and25 + 25%, respectively), all supplemented with0.5 mol/L sucrose for 5, 10, 15, 20, 25, 30, 35,and 40 min, respectively. Warming was per-formed by immersing in a 37°C thawing solu-tion containing 1 mol/L sucrose for 3 min,followed by 0.5 mol/L sucrose solution for5 min, and 30 min incubation in isotonic solu-tions. The authors reported that both large and
267
VITRIFICATION OF OVARIAN TISSUE
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 267
small samples could be successfully vitrified: thelarge tissues with >15 min incubation and inconcentrations of cryoprotectants of17.5 +17.5% or higher in the vitrification solu-tion, and the small tissues with a 91% survivalrate after 10 min incubation in the 15 +15%group. Survival rates of oocytes were in theranged between 0 and 91%, and negatively cor-related with the size of the ovarian tissue.
Vitrification of human ovarian tissueand xenografting after warming
We investigated and compared the necroticareas after subcutaneous transplantation
of vitrified/warmed and slow cryopreserved/thawed human ovarian tissue into SCID micefor 6 weeks.57 Solutions for vitrification wereprepared in DPBS medium supplementedwith 15% fetal calf serum. The cryoprotec-tants used were glycerol, EG, and thesynthetic ice blocking agent Supercool® X-100 (Cooltechnica, New York, USA). Thestudy showed that the size of the necroticareas in human ovarian tissue were com-parable between the different methods(Figure 11A.3).
In a further study of ours (unpublisheddata) in SCID mice, we observed neovascular-ization of human ovarian tissue after slow
VITRIFICATION IN ASSISTED REPRODUCTION
268
70 µm
0.5 mm 1 mm
0.5 mm
Figure 11 A.1 Follicles developed in vitrified/warmed human ovarian tissue after 3 weeks of culture: (a) primordial,(b) and (c) early antral, and (d) antral.
a
c d
b
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 268
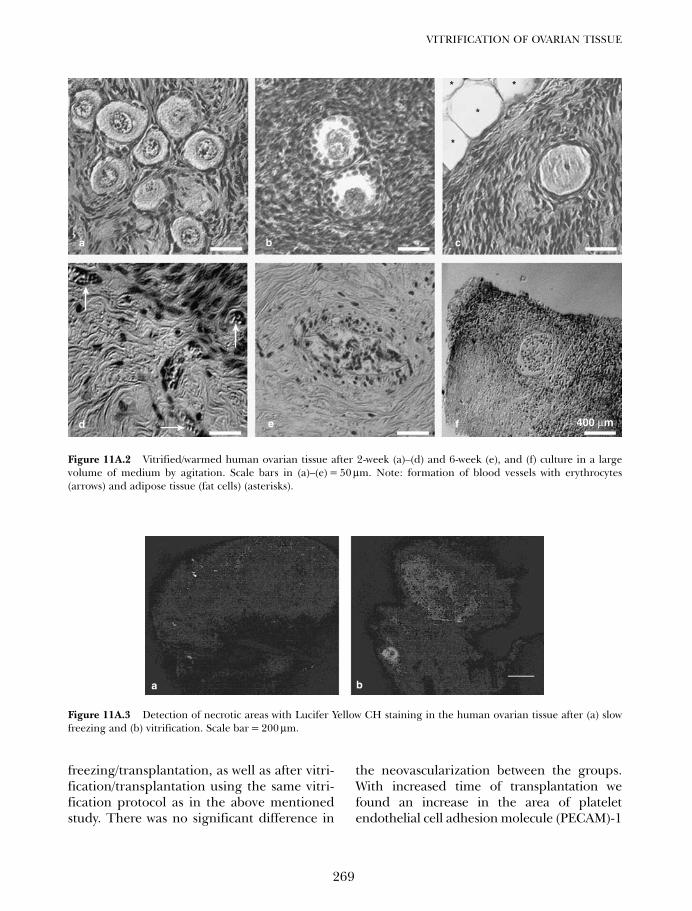
freezing/transplantation, as well as after vitri-fication/transplantation using the same vitri-fication protocol as in the above mentionedstudy. There was no significant difference in
the neovascularization between the groups.With increased time of transplantation wefound an increase in the area of plateletendothelial cell adhesion molecule (PECAM)-1
269
VITRIFICATION OF OVARIAN TISSUE
400 µm
*
*
*
*
Figure 11A.2 Vitrified/warmed human ovarian tissue after 2-week (a)–(d) and 6-week (e), and (f) culture in a largevolume of medium by agitation. Scale bars in (a)–(e) = 50 µm. Note: formation of blood vessels with erythrocytes(arrows) and adipose tissue (fat cells) (asterisks).
Figure 11A.3 Detection of necrotic areas with Lucifer Yellow CH staining in the human ovarian tissue after (a) slowfreezing and (b) vitrification. Scale bar = 200 µm.
a b
a b c
fed
11a Tucker 8029.qxd 8/23/2007 2:17 PM Page 269

positive blood vessels in the transplantedhuman ovarian tissue. The area increasedfrom 2.85 ± 0.27 mm² (slow freezing), 2.19 ±0.48 mm² (vitrification), and 1.96 ± 0.89 mm²(control = fresh transplanted tissue) after 1week, to 92.5 ± 24 mm², 104.1 ± 22 mm² and72.3 ± 17 mm², respectively, after 4 weeks(Figure 11A.4).
Vitrification of human ovarian tissueand autografting after warming
To our knowledge there have been no pub-lished papers dealing with vitrification of
human ovarian tissue and autografting untilnow.
CONCLUSION
At the moment the conventional cryopreser-vation of human ovarian tissue seems to befavored due to considerably greater experi-ence. Vitrification, nevertheless, could becomea realistic alternative, however, further studiesregarding optimization of cryoprotectantsolutions and protocols are necessary toachieve the same results as with equilibriummethods.
VITRIFICATION IN ASSISTED REPRODUCTION
270
Figure 11A.4 The platelet endothe-lial cell adhesion molecule (PECAM)-1immunofluorescence in the humanovarian tissue after vitrification andtransplantation for 1 week (a) and 4weeks (b). Scale bar = 20 µm.a b
References
1. Kim SS, Battaglia DE, Soules MR. The futureof human ovarian cryopreservation and trans-plantation: fertility and beyond. Fertil Steril2001; 75: 1049–56.
2. Newton H, Fisher J, Arnold JRP et al.Permeation of human ovarian tissue with cryo-protective agents in preparation for cryo-preservation. Hum Reprod 1998; 13: 376–80.
3. Gosden RG. Low temperature storage andgrafting of human ovarian tissue. Mol CellEndocrinol 2000; 163: 125–9.
4. Revel A, Koler M, Simon A et al. Oocyte col-lection during cryopreservation of the ovariancortex. Fertil Steril 2003; 79: 1237–9.
5. Revel A, Safran A, Benshushan A et al. In vitromaturation and fertilization of oocytes froman intact ovary of a surgically treated patientwith endometrial carcinoma: case report.Hum Reprod 2004; 19: 1608–11.
6. Isachenko E, Rahimi G, Isachenko V et al. In-vitro maturation of germinal-vesicle oocytesand cryopreservation in metaphase I/II: apossible additional option to preserve fertilityduring ovarian tissue cryopreservation.Reprod Biomed Online 2004; 8: 553–7.
7. Lightman A, Werner-Kimel N, Solt I et al. Aprogram of ovarian tissue cryopreservationfor women with malignant disease: lessonsfrom 5 years’ experience. Fertil Steril 2001;76(Suppl 1): 81.
8. Donnez J, Martinez-Madrid B, Jadoul P et al.Ovarian tissue cryopreservation and trans-plantation: a review. Hum Reprod Update2006; 12: 519–35.
9. Donnez J, Dolmans MM, Demylle D et al.Livebirth after orthotopic transplantation ofcryopreserved ovarian tissue. Lancet 2004;364: 1405–10.
11a Tucker 8029.qxd 8/23/2007 2:18 PM Page 270

10. Meirow D, Levron J, Eldar-Geva T et al.Pregnancy after transplantation of cryopre-served ovarian tissue in a patient with ovarianfailure after chemotherapy. N Engl J Med2005; 353: 318–21.
11. Demeestere I, Simon P, Buxant F et al.Ovarian function and spontaneous pregnancyafter combined heterotopic and orthotopiccryopreserved ovarian tissue transplantationin a patient previously treated with bone mar-row transplantation: case report. Hum Reprod2006; 21: 2010–14.
12. Rosendahl M, Loft A, Byskov AG et al.Biochemical pregnancy after fertilization ofan oocyte aspirated from a heterotopic auto-transplant of cryopreserved ovarian tissue:case report. Hum Reprod 2006; 21: 2006–9.
13. Liebermann J, Nawroth F, Isachenko V et al.Potential importance of vitrification in reproduc-tive medicine. Biol Reprod 2002; 67: 1671–80.
14. Brockbank KGM, Song YC, Khirabadi BSet al. Storage of tissues by vitrification.Transplant Proc 2000; 32: 3–4.
15. Yin H, Kim SS, Fisher J et al. Investigation ofoptimal conditions for equilibrating ovariantissue with ethylene glycol prior to vitrifica-tion. Fertil Steril 2001; 76(Suppl 1): S101.
16. Miyamoto H, Sugimoto M. Assessment of follicledevelopment and histocytological examinationof neonatal rat ovaries cryopreserved by vitrifica-tion technique. Cryobiology 1994; 31: 614–5.
17. Zhang J, Liu J, Xu KP et al. Extracorporaldevelopment and ultrarapid freezing ofhuman fetal ova. J Assist Reprod Genet 1995;12: 361–8.
18. Sugimoto M, Miyamoto H, Kabasawa T et al.Follicle survival in neonatal rat ovariescryopreserved by vitrification. Cryo Letters1996; 17: 93–8.
19. Sugimoto M, Maeda S, Manabe N et al.Development of infantile rat ovaries auto-transplanted after cryopreservation by vitrifi-cation. Theriogenology 2000; 53: 1093–103.
20. Lee SH, Shin CS, Ko JJ et al. In vitro culture ofthe human adult ovarian tissues after vitrification:comparison among detection methods of the cul-ture effect. Fertil Steril 2000; 74(Suppl 1): 161.
21. Kagabu S, Umezu M. Transplantation ofcryopreserved mouse, Chinese hamster, rab-bit, Japanese monkey, and rat ovaries into ratrecipients. Exp Anim 2000; 49: 17–21.
22. Van den Broecke R, Liu J, Handyside A et al.Follicular growth in fresh and cryopreservedhuman ovarian cortical grafts transplanted toimmunodeficient mice. Eur J Obstet GynecolReprod Biol 2001; 97: 193–201.
23. Yeoman RR, Wolf DP, Lee DM. Coculture ofmonkey ovarian tissue increases survival aftervitrification and slow-rate freezing. FertilSteril 2005; 83(Suppl 1): 1248–54.
24. Courbiere B, Odagescu V, Baudot A et al.Cryopreservation of the ovary by vitrificationas an alternative to slow-cooling protocols.Fertil Steril 2006; 86(Suppl 4):1243–51.
25. Gandolfi F, Paffoni A, Papasso Brambilla Eet al. Efficiency of equilibrium cooling and vit-rification procedures for the cryopreservationof ovarian tissue: comparative analysis betweenhuman and animal models. Fertil Steril 2006;85(Suppl 1): 1150–6.
26. Salehnia M, Abbasian Moghadam E,Rezazadeh Velojerdi M. Ultrastructure of folli-cles after vitrification of mouse ovarian tissue.Fertil Steril 2002; 78: 644–5.
27. Salehnia M, Moazzeni SM. Autograft of vitri-fied mouse ovarian tissue using ethylene gly-col as cryoprotectant. Hum Reprod 2001;16(Suppl 1):159.
28. Kim DH, Lee HC, Ko DS et al. In vitro growthand maturation of preantral follicles isolatedfrom vitrified mouse ovaries. Fertil Steril2002; 78(Suppl 1): S268.
29. Takahashi E, Miyoshi I, Nagasu T. Rescue of atransgenic mouse line by transplantation of afrozen-thawed ovary obtained postmortem.Contemp Top Lab Anim Sci 2001; 40: 28–31.
30. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryo at −196 degrees C by vitrifi-cation. Nature 1985; 313: 573–5.
31. Migishima F, Suzuki-Migishima R, Song SYet al. Successful cryopreservation of mouseovaries by vitrification. Biol Reprod 2003; 68:881–7.
32. Segino M, Ikeda M, Aoki S et al. In vitroculture of mouse GV oocytes and preantralfollicles isolated from ovarian tissues cryopre-served by vitrification. Hum Cell 2003; 16:109–16.
33. Segino M, Ikeda M, Hirahara F et al. In vitrofollicular development of cryopreservedmouse ovarian tissue. Reproduction 2005;130: 187–92.
34. Hasegawa A, Hamada Y, Mehandjiev T et al.In vitro growth and maturation as well as fer-tilization of mouse preantral oocytes from vit-rified ovaries. Fertil Steril 2004; 81(Suppl 1):824–30.
35. Chen SU, Chien CL, Wu MY et al. Noveldirect cover vitrification for cryopreservationof ovarian tissues increases follicle viabilityand pregnancy capability in mice. HumReprod 2006; 21: 2794–800.
271
VITRIFICATION OF OVARIAN TISSUE
11a Tucker 8029.qxd 8/23/2007 2:18 PM Page 271

36. Hani T, Tachibe T, Shingai S et al. Fertility ofmice receiving vitrified adult mouse ovaries.Reproduction 2006; 131: 681–7.
37. Al-Aghbari AM, Menino AR. Survival ofoocytes recovered from vitrified sheep ovariantissues. Anim Reprod Sci 2002; 71: 101–10.
38. Courbiere B, Massardier J, Salle B et al.Follicular viability and histological assessmentafter cryopreservation of whole sheep ovarieswith vascular pedicle by vitrification. FertilSteril 2005; 84(Suppl 2): 1065–71.
39. Kheirabadi BS, Fahy GM. Permanent life sup-port by kidneys perfused with a vitrifiable (7.5molar) cryoprotectant solution. Transplantation2000; 70: 51–7.
40. Bordes A, Lornage J, Demirci B et al. Normalgestations and live births after orthotopicautograft of vitrified-warmed hemi-ovariesinto ewes. Hum Reprod 2005; 20: 2745–8.
41. Ishijima T, Kobayashi Y, Lee DS et al.Cryopreservation of canine ovaries by vitrifi-cation. J Reprod Dev 2006; 52: 293–9.
42. Armitage WJ, Rich SJ. Vitrification of orga-nized tissues. Cryobiology 1990; 27: 483–91.
43. Song YC, Khirabadi BS, Lightfoot F et al.Vitreous cryopreservation maintains the func-tion of vascular grafts. Nat Biotechnol 2000;18: 296–9.
44. Duru NK, Öngürü Ö, Celasun B et al. Post-thaw texture analysis of slowly frozen and vit-rified human ovarian cortex. Hum Reprod2001; 16(Suppl 1):180.
45. Isachenko EV, Isachenko E, Rahimi G et al.Cryopreservation of human ovarian tissue bydirect plunging into liquid nitrogen. Eur JObstet Gynecol Reprod Biol 2003; 108:187–94.
46. Rahimi G, Isachenko E, Sauer H et al. Effectof different vitrification protocols for humanovarian tissue on reactive oxygen species andapoptosis. Reprod Fertil Dev 2003; 15: 343–9.
47. Lee KA, Lee SH, Yoon SJ et al. Resumption ofthe human primordial follicle growth inxenografts after vitrification of the ovariantissues. Fertil Steril 2000; 74(Suppl 1): 214.
48. Rahimi G, Isachenko E, Sauer H et al.Measurement of apoptosis in long-term cultures
of human ovarian tissue. Reproduction 2001;122: 657–63.
49. Isachenko E, Nayudu PL. Vitrification ofmouse germinal vesicle oocytes: effect oftreatment temperature and egg yolk on chro-matin and spindle normality and cumulusintegrity. Hum Reprod 1999; 14: 400–8.
50. Miyake T, Kasai M et al. Vitrification of mouseoocytes and embryos at various stages ofdevelopment in an ethylene glycol-based solu-tion by a simple method. Theriogenology1993; 40: 121–34.
51. Hallak J, Sharma RK, Wellstead C et al.Cryopreservation of human spermatozoa:comparison of TEST-yolk buffer and glycerol.Int J Fertil Womens Med 2000; 45: 38–42.
52. Isachenko V, Alabart J, Isachenko E et al. Ultra-rapid freezing and storage of rat embryos in anelectric refrigerator at −130ºC without liquidcryo-agents, with ultra-short exposure in thefreezing medium and direct rehydration afterthawing. Cryo Letters 2000; 21: 13–8.
53. Steponkus PL, Myers SP, Lynch DV et al.Cryopreservation of Drosophila melanogasterembryos. Nature 1990; 345: 170–2.
54. Isachenko V, Isachenko E, Rahimi G et al.Cryopreservation of human ovarian tissue bydirect plunging into liquid nitrogen: negativeeffects of disaccharides in vitrification solu-tion. Cryo Letters 2002; 23: 333–44.
55. Isachenko V, Montag M, Isachenko E et al.Effective method for in-vitro culture of cryo-preserved human ovarian tissue. ReprodBiomed Online 2006; 13: 228–34.
56. Kagawa N, Kuwayama M, Silber SJ et al.Vitrification may be a promising approach forcryopreservation of human ovarian tissue forauto- and xenotransplantation. Fertil Steril2006; 86(Suppl 2): S403.
57. Rahimi G, Isachenko E, Isachenko V. et al.Comparison of necrosis in human ovarian tis-sue after conventional slow freezing or vitrifi-cation and transplantation in ovarectomizedSCID mice. Reprod Biomed Online 2004; 9:187–93.
VITRIFICATION IN ASSISTED REPRODUCTION
272
11a Tucker 8029.qxd 8/23/2007 2:18 PM Page 272

273
Vitrification of ovarian tissuesYing C Song, Zhenzhen Chen, Carol Journey, Adelina M Emmi, Xiayang Xie andRosemary L Song
11B
INTRODUCTION
It was estimated that by the year 2006 1.4 mil-lion people would be diagnosed with cancerin the United States, of which 49% would bewomen.1 It was estimated that 8% of thesewomen would be under the age of 40.1–3 Withmodern improvements in treatment regi-mens, which include aggressive chemo- andradiotherapy, as well as bone marrow trans-plantation, cure rates can exceed 90%.4
However, alkylating agents commonly used inchemotherapy regimens, as well as ionizingradiation, induce premature ovarian failurein a majority of these patients. This is a signif-icant consequence of cancer treatment aspatients are not only rendered infertile butalso undergo premature menopause.5
Ovarian tissue banking and transplantation,and other means of fertility preservation arepromising prophylactic options for thesepatients. Patients with a low to intermediateprobability of ovarian involvement are goodcandidates for an autografting procedure.6
Ovarian tissue banking allows time to screentissue samples to avoid reseeding cancer cellsto the patients. For patients with a high risk ofovarian metastasis, ovarian tissue banking, invitro follicle maturation, and in vitro fertiliza-tion could be an option for preserving theirfertility.7,8 The opportunities in the area ofhuman infertility are immense for cryopreser-vation technologies.8,9
At present, techniques used for ovariancryopreservation have a serious limitation.Extracellular ice formation directly damagescells and tissues through mechanical disruption
and high salt concentration. This damage pre-cludes normal cellular organization in both thematuring oocyte and the surrounding cell layers(theca and granulosa), conditions which arenecessary for fertilization. Secondary injury tomicrovasculature by mechanical disruption isdetrimental to revascularization of cryopre-served implants. Ischemic damage, induced bydelayed revascularization, is caused by freeradicals and apoptosis. There is clear evidenceto show that only about 50% of the follicularpopulation survives after re-implantation.Therefore, cryopreserved ovarian tissue islimited by the current technology.
Several groups have demonstrated thecompetence of cryopreserved ovarian grafts inewes,10 non-human primates,11 andhumans.12–14 Each of the human studiesresulted in a live birth, demonstrating theconcept of ovarian autograft implantationusing cryopreserved tissues, but the live birthrate is extremely low (only three live births).One of the problems is the conventional cellcryopreservation protocols used. These con-ventional approaches to cryopreservationcannot be successfully extrapolated to morecomplex multicellular tissues primarily due tothe destructive effect of extracellular ice for-mation. Tissues are much more than simpleaggregates of different cell types; they have ahighly organized and often complex struc-ture, which undoubtedly influences theirresponse to freezing and thawing. The forma-tion of extracellular ice, in particular, which isgenerally innocuous for cells in suspension, isa prime hazard to structured tissues andorgans. There are many questions that have
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 273

not yet been answered in ovarian tissuecryopreservation. Whether sufficient primor-dial and growing follicles survive in theovaries after cryopreservation, and whethersurviving oocytes resume meiosis, fertilize,and develop to the blastocyst stage after invitro maturation and fertilization have yet tobe investigated. Communication between anoocyte and its surrounding granulosa cells isessential for the physiological function anddevelopment of the follicle.15 Extracellularmatrix is very important for the survival ofprimordial and primary follicles in long-termculture.16 Gosden et al. suggested thatmicrovasculature damage by mechanical dis-ruption during ice formation is a criticalproblem in ovarian tissue cryopreservation.17
Poor development rates (37%) of frozen andthawed oocytes have been observed after invitro maturation.18 The reason for the poorresults of conventional cryopreservation hasbeen linked to spindle damage,19 chromoso-mal abnormalities,20 pathogenesis of oocytes,zona hardening, and low oocyte survival ratesafter thawing. Post-thaw recovery of folliclesin cryopreserved mouse ovarian tissue hasranged from 5%21 to 30%22 with vitrification.Extremely low post-thaw survival rates ofhuman oocytes necessitate banking of ovariantissues for patients who will soon receiveradiotherapy or chemotherapy.23 In thisregard, better survival may be expected froman ice-free cryopreservation method.17
Restricting the amount and size of ice crystalformation during cryopreservation can beachieved by using sufficiently high concen-trations of cryoprotectants to promoteamorphous solidification rather than crys-tallization, that is vitrification rather thanfreezing.24–27 Vitrification is an alternative toconventional freezing of living biologicalmaterials with ubiquitous applications in cell,tissue, and organ storage.
Employing a rabbit jugular vein model, acomparison of the effects of vitrification andconventional cryopreservation upon venouscontractility showed that the maximum
contractions achieved by the vitrified bloodvessel rings, in response to a panel of fouragonists, were greater than 80% of freshmatched controls. In contrast, the maximumcontractions achieved by conventionally cryo-preserved frozen veins were less than 30% offresh matched controls.28 During a 4-weekfollow-up of vitrified and fresh grafts in anautologous transplant model, there were nosignificant differences between the fresh andvitrified groups with patency rates of ~90%.29
Furthermore, there was no evidence of tunicamedia disruption, loss of endothelialintegrity, aneurysm development, or graftstenosis in either group.29 Feasibility studiesusing vitrification have been reported for thepreservation of arteries,30 heart valves,31,32
articular cartilage,33–36 tissue engineered pan-creas substitutes,37,38 and tissue engineeredblood vessels.39,40 The results combine todemonstrate feasibility of vitrification as astorage method for living tissue implants.41
Cryopreservation technologies represent apotential long-term storage method to pre-serve tissues. Ovary banking provides the fol-lowing options and advantages: it allows thebanking of the patient’s own ovarian tissuesfor future autologous grafting and long-termfecundity; it allows time for adequate qualityassurance and safety measures to be appliedto avoid reseeding cancer cells via trans-planted tissue; and it provides the means forbanking ovarian tissues for future in vitro fol-licle maturation for patients with high risk ofovarian metastasis. If the current cryopreser-vation method can be improved and incorpo-rated within fertility restoration programs, itwill be possible to create various options bywhich fertility can be successfully preserved.
In this chapter, the current trend in fertil-ity preservation and the current methods forcryopreservation of ovarian tissues are givenin introductory sections. The introductoryparts are followed by a section that explainshow to avoid ice formation using vitrificationincluding properties of vitrification solutions,critical cooling and warming rates, ice crystal
VITRIFICATION IN ASSISTED REPRODUCTION
274
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 274

growth, cryosubstitution, and vitrificationsolution design. The final part deals with thetransplantation of vitrified ovarian tissues inan animal model, follicle growth in explanttissues, and cell proliferation and apoptosis.
CRYOPRESERVATION OPTIONSFOR FERTILITY PRESERVATION
Chemotherapy and radiotherapy can severelydeplete the follicular store, often compromis-ing ovarian function. Ovarian damage willresult in loss of fertility, prematuremenopause, loss of sexual interest, and in thelong term, an enhanced risk of osteoporosisand arterial vascular disease. While inyounger patients amenorrhea is often tempo-rary, hormone replacement therapy decreasesthe severity of the symptoms. However,restoration of fertility is more difficult, and atpresent lies with embryo cryopreservationprior to treatment. Although in some casesembryo cryopreservation may be consideredbefore chemotherapy and radiotherapy, thisis a far from optimal solution, because manyof the patients are premenarchal girls,women without male partners, and patientsfor whom chemotherapy and radiotherapycannot be delayed. Embryo preservation isindeed not an option for these women,because delaying treatment for at least 2months of in vitro fertilization cycles is inap-propriate or life-threatening. Another optionis oocyte cryopreservation. Cryopreservationof oocytes can be performed in single womenwho can undergo a stimulation cycle, althoughthe effectiveness of this technique is very low,with pregnancy and delivery rates rangingfrom 1 to 5% per frozen oocyte.42–44 There aretwo main reasons for these poor results: zonapellucida hardening during freezing,45–48 andspindle apparatus damage by intracellular iceformation.49
Ovarian cryopreservation may overcomethese problems. In addition, ovarian tissuecan be collected simply by laparoscopy at anytime during the menstrual cycle. It is possible
to bank ovarian tissue from cancer patientsprior to therapy and re-implant the cryopre-served tissue once the patients are cured torestore fertility. In the most idealized situa-tion, ovarian tissue transplantation wouldrestore both the synthesis of sex steroids tomaintain phenotype and the cyclical produc-tion of oocytes. Steroidogenic function wouldbe restored in these patients, thereby elimi-nating the need for exogenous hormonereplacement therapy. The most reliable andpressing need appears to be for those womenwith premature sterility as a result of certaincancer treatments, including high-dosechemotherapy and abdominal irradiation. Ifovarian tissue could be cryopreserved beforepatients received cancer treatment and thenreturned successfully after remission, the nat-ural state could potentially be restored andconception could even be achieved. Althoughwhole ovaries from mice and rats survivefreezing,17 successful cryopreservation ofwhole ovaries from other mammalian species(human and non-human primates, and live-stock species) is difficult due to their largesize and low survival rate of vasculature.Effective cryopreservation would enabletransplantation of intact ovaries with pre-served vasculature and immediate revascular-ization by vascular anastomosis. In vitromaturation of ovarian tissue is an alternativefor patients with high risk of ovarian metasta-sis. Success has been demonstrated in freshtissue.7 However, successful in vitro matura-tion using cryopreserved ovarian tissue hasnot yet been reported.50,51 Communicationbetween the oocyte and surrounding granu-losa cells is essential for correct function anddevelopment of the follicle.15 Extracellularmatrix is very important for the survival ofprimordial and primary follicles in long-termculture.16 The destructive effects of extra-cellular ice formation caused by conventionalcryopreservation (freezing) and ischemiaafter transplantation are the major obstaclesfor the development of in vivo maturationusing cryopreserved tissue. We anticipate that
275
VITRIFICATION OF OVARIAN TISSUES
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 275

these hurdles can be overcome using ice-freecryopreservation techniques.
CONVENTIONALCRYOPRESERVATION
Cryopreservation is a complex process ofcoupled heat and mass transfer, generallyexecuted under non-equilibrium condi-tions.52 Simply freezing cells or tissues resultsin dead, non-functional materials. Duringslow freezing, water is removed from cells andtissues as ice begins to form in the extracellu-lar environment. Widespread formation ofintracellular ice in the cell has been uni-formly associated with lethal cell injury.53,54
For most cell types, optimal cooling ratesexist that result in maximum cell viability.These cooling rates provide sufficient cellulardehydration to prevent the formation ofintracellular ice without causing excessivewater loss that may lead to cell injury via‘solution effects’. Thawing cells from cryopre-served states is typically performed with max-imal warming rates to prevent therecrystallization of ice from smaller ice crys-tals as the sample temperature is raised fromcryogenic levels to the melting point of thesolution. Recrystallization during rewarmingis deleterious and results in lower cell viabil-ity; exact mechanisms of injury are not fullyunderstood. Viability is greater when cells arethawed rapidly through the conditions inwhich recrystallization occurs.55,56
Advances in the field of cryopreservationhad been modest until Polge et al,57 discov-ered the cryoprotective properties of glycerol.Subsequent research by Lovelock and Bishop58
showed that dimethylsulfoxide (DMSO) wasalso a cryoprotectant agent (CPA). These chem-icals are usually divided into two classes:intracellular cryoprotectants with low molecu-lar weights that permeate into cells; and,extracellular cryoprotectants with relativelyhigh molecular weights (greater than or equalto sucrose e.g. 342 Da) which do not pene-trate cells.59 The primary mode of protection
for permeating cryoprotectants is the dis-placement of intracellular water by thecryoprotectant. Regulated removal of intra-cellular water is essential to inhibiting thelethal formation of intracellular ice. Intra-cellular cryoprotectants, such as glyceroland DMSO at concentrations from 0.5 to3.0 mol/L, are effective in minimizing celldamage in many small biological systemsfrozen with slow cooling rates. Extracellularcryoprotective agents such as polyvinylpyrroli-done or hydroxyethyl starch are more effec-tive at protecting biological systems cooledat rapid rates. Such agents are often largemacromolecules that affect the properties ofthe solution to a greater extent than wouldbe expected from their colligative properties.The primary mechanism of action appears tobe the induction of extracellular glassformation. However, non-penetrating cryopro-tectants also condition the intracellular com-partment by causing the osmotic efflux ofintracellular water, thereby preventing theformation of intracellular ice. Some of thesenon-permeating cryoprotective agents are alsothought to have direct protective effects on thecell membrane. When cryoprotectants areused in extremely high concentrations, ice for-mation can be eliminated during cooling toand warming from cryogenic temperatures.
As discussed above, ovarian tissue has beencryopreserved and transplanted into rodents,rabbits, sheep, and marmoset monkeys.60–63
In a recent review of their experimentalstudies, Baird et al.64 observed a significantdecrease of primordial follicles in conven-tionally cryopreserved ovarian tissues. Experi-mental studies have indicated that the fall inthe number of primordial follicles in graftedtissue is due to hypoxia and the delay thatoccurs before reimplanted cortical tissuebecomes revascularized. The loss of primor-dial follicles in cryopreserved ovarian tissueafter transplantation was estimated to be50–65% in some studies65,66 and > 90% in onestudy.67,68 Kim et al.69 showed that a correla-tion exists between ischemic tissue damage
VITRIFICATION IN ASSISTED REPRODUCTION
276
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 276

and the duration of ischemia, and found thatthe ovarian cortex can tolerate ischemia for3 h at 4°C. Significant ischemic damage wasevidenced by decreased oxygen consumptionand increased apoptosis. Moreover, it appearedthat stromal cells were more vulnerable toischemia than primordial follicles. The apop-tosis rate of stromal cells was higher in thefrozen–thawed group than in the fresh group,regardless of the duration of incubation,which may reflect a degree of ice damage tothe ovarian cortex.
ICE-FREE CRYOPRESERVATION –VITRIFICATION
Introduction
When a sufficiently high concentration of CPA isused, the formation of ice is avoided com-pletely.52 The rate of cooling and warming isthen unimportant because there is no drivingforce for trans-membrane water movement andno ice to recrystallize during warming. The con-centration of CPA necessary to avoid freezing isvery high (typically ~60%) and ‘compatibility’(the absence of deleterious effects of the soluteitself) is the essential problem: the concentra-tion of solute required is unattainable at tem-peratures above 0°C.
When materials are vitrified, ice formationis prevented, even at cryogenic temperatures,by the presence of high concentrations ofchemicals that interact strongly with waterand, therefore, prevent water molecules frominteracting to form ice. Depressing the homo-geneous nucleation temperature until itequals the glass transition temperature per-mits vitrification of macroscopic biologicalsystems. Prevention of freezing means thatthe water in a tissue remains liquid duringcooling. As cooling proceeds, however, themolecular motions in the liquid permeatingthe tissue decrease. Eventually, an ‘arrestedliquid’ state known as a glass is achieved. Aglass is a liquid that is too cold or viscous toflow. A vitrified liquid is essentially a liquid in
molecular stasis. Vitrification does not haveany of the biologically damaging effects asso-ciated with freezing because no appreciabledegradation occurs over time in living mattertrapped within a vitreous matrix. It provideseffective preservation for a number of cells,including monocytes, ova, and early embryos,and pancreatic islets.27,70–75 Vitrification ispotentially applicable to all biological systems.
Cryopreservation by complete vitrificationof the tissue suspension offers several impor-tant advantages compared with proceduresthat allow or require crystallization of the sus-pension. First, complete vitrification elimi-nates concerns for the known damagingeffects of intra- and extracellular crystalliza-tion. Second, tissues cryopreserved by vitrifi-cation are exposed to less concentratedsolutions of CPAs for shorter periods of time.For example, during a typical cryopreserva-tion protocol involving slow freezing to−40°C, or −70°C, cells are exposed to solu-tions the concentration of which increasesgradually to 21.5 and 37.6 osmol/L, respec-tively. In contrast, cells dehydrated in vitrifi-cation solutions are exposed for much shorterperiods of time to < 18 osmol/L, solution,although the temperature of exposure ishigher.71 Third, unlike conventional proce-dures that employ freezing, vitrification doesnot require controlled cooling and warmingat optimum rates – cooling and warmingneed only be rapid enough to prevent crystal-lization, and this can generally be achievedwithout the need for specialist equipment.Vitrification offers a number of practicaladvantages that will be attractive in tissueprocessing, as indeed they have been forembryo banking.71
Critical cooling rates and criticalwarming rates
The development of ice-free cryopreservationtechniques applicable to ovarian tissues andoocytes requires careful attention to avoiddamaging ice growth during both cooling
277
VITRIFICATION OF OVARIAN TISSUES
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 277
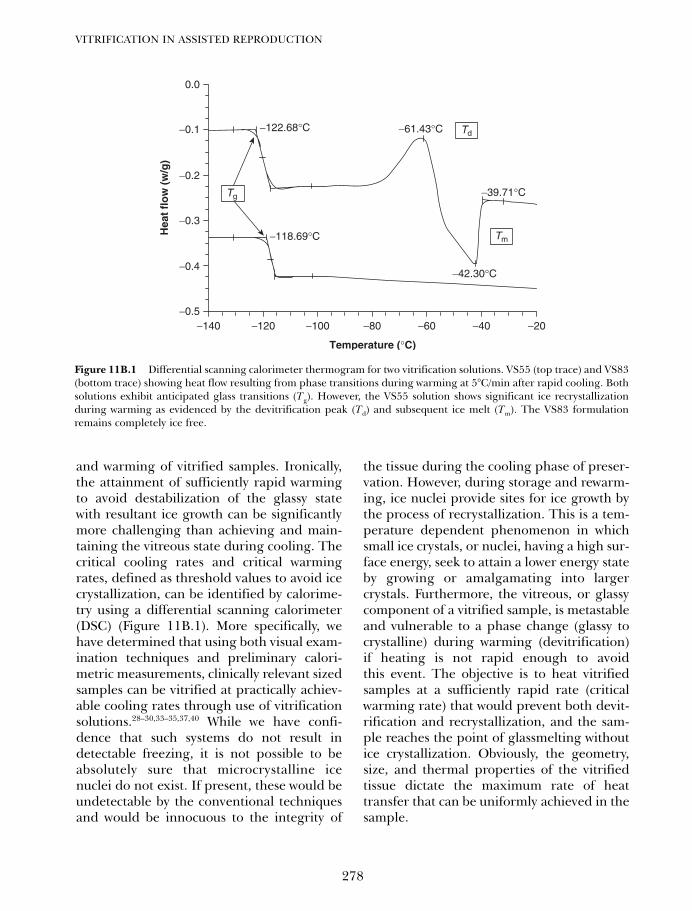
and warming of vitrified samples. Ironically,the attainment of sufficiently rapid warmingto avoid destabilization of the glassy statewith resultant ice growth can be significantlymore challenging than achieving and main-taining the vitreous state during cooling. Thecritical cooling rates and critical warmingrates, defined as threshold values to avoid icecrystallization, can be identified by calorime-try using a differential scanning calorimeter(DSC) (Figure 11B.1). More specifically, wehave determined that using both visual exam-ination techniques and preliminary calori-metric measurements, clinically relevant sizedsamples can be vitrified at practically achiev-able cooling rates through use of vitrificationsolutions.28–30,33–35,37,40 While we have confi-dence that such systems do not result indetectable freezing, it is not possible to beabsolutely sure that microcrystalline icenuclei do not exist. If present, these would beundetectable by the conventional techniquesand would be innocuous to the integrity of
the tissue during the cooling phase of preser-vation. However, during storage and rewarm-ing, ice nuclei provide sites for ice growth bythe process of recrystallization. This is a tem-perature dependent phenomenon in whichsmall ice crystals, or nuclei, having a high sur-face energy, seek to attain a lower energy stateby growing or amalgamating into largercrystals. Furthermore, the vitreous, or glassycomponent of a vitrified sample, is metastableand vulnerable to a phase change (glassy tocrystalline) during warming (devitrification)if heating is not rapid enough to avoidthis event. The objective is to heat vitrifiedsamples at a sufficiently rapid rate (criticalwarming rate) that would prevent both devit-rification and recrystallization, and the sam-ple reaches the point of glassmelting withoutice crystallization. Obviously, the geometry,size, and thermal properties of the vitrifiedtissue dictate the maximum rate of heattransfer that can be uniformly achieved in thesample.
VITRIFICATION IN ASSISTED REPRODUCTION
278
−140−0.5
−0.4
−0.3
−0.2
−0.1 −122.68°C
−118.69°C
−61.43°C
−39.71°C
−42.30°C
0.0
−120 −100 −80
Temperature (°C)
Hea
t fl
ow
(w
/g)
−60 −40 −20
Tm
Td
Tg
Figure 11B.1 Differential scanning calorimeter thermogram for two vitrification solutions. VS55 (top trace) and VS83(bottom trace) showing heat flow resulting from phase transitions during warming at 5°C/min after rapid cooling. Bothsolutions exhibit anticipated glass transitions (Tg). However, the VS55 solution shows significant ice recrystallizationduring warming as evidenced by the devitrification peak (Td) and subsequent ice melt (Tm). The VS83 formulationremains completely ice free.
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 278

Ice crystal growth
A variety of microscopic and macroscopictechniques are applied to measure the physi-cal parameters associated with ice nucleationand growth (Figure 11B.2), the efficacy of arange of prospective ice control moleculeshave been evaluated. These include naturalantifreeze proteins, existing (and newly syn-thesized) synthetic ice blocker molecules,76
and other macromolecules such as polyethyl-ene glycol (PEG) known to promote a vitreousstate.77 The selection of CPAs was based uponour extensive experience and knowledge ofthe choice and efficacy of CPAs in other bio-logical systems. For example, DMSO has beenwidely used as a cryophylactic agent and is stillregarded as the single most effective CPAavailable. Attempts to understand the require-ments of vitrification solutions at the molecu-lar level have led to the identification of newsolutes with physical properties that wouldpromote the vitreous state during cooling.
One promising class of compounds is thepolyalcohols such as 1,2-propanediol (PROH)and butane-2,3-diol, the optical isomers ofwhich have been shown to vitrify at signifi-cantly lower concentrations than any otherknown CPA.78,79 This is important to minimizethe toxic effects of high concentrations ofCPAs. Moreover, it is now established thatboth the concentration needed to vitrify, andthe toxicity can be reduced by incorporating awide variety of non-permeating disaccharides,or polymeric compounds.79,80
Individual ice control molecules were com-bined in the baseline vitrification medium,VS55, or the alternative DP6 (3 mol/L DMSOand 3 mol/L PROH) medium and examinedfor ice nucleation and growth during coolingto, and warming from, the glass transitiontemperature.76,77 Critical cooling and warmingrates were determined by DSC (Figure 11B.1).Ice growth kinetics can be measured byvideo-cryomicroscopy (Fig 11B.2). The pres-ence of ice in bulk samples (20 mL glass
279
VITRIFICATION OF OVARIAN TISSUES
0 sec 30 sec 1 min
Figure 11B.2 Ice crystals formed from vitrification solution with and without ice control at −60°C. Ice crystal growthcan be recorded by video-cryomicroscopy. Kinetics of linear ice crystallization growth in vitrification solutions can becalculated as a function of temperature. Bar = 100 µm.
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 279

vials; transparent polyester freezer bags thatcan be fabricated to any desired form; andthe 75 mL plexiglass cassette) was deter-mined initially by visual inspection andquantified in a custom-built plexiglass cas-sette using digital photography and imageanalysis.77
Design of a vitrification solution usingbulk samples
The effect of these compounds on the modu-lation of ice recrystallization kinetics duringthe thawing of deliberately frozen solutionscan be determined by cryomicroscopy.Compounds that promote ice nucleation arecommercially available as products usedprincipally for snowmaking (Snowmax). Thisproduct can be used as a tool to test the effi-cacy of the best combinations of ice-controlmolecules emerging from these physical stud-ies. This will be achieved by determining thethreshold concentration of Snowmax neces-sary to promote crystallization in the vitrifica-tion cryoprotectant cocktails.
The effects of sample volume and geome-try on critical cooling and warming rates canbe determined using the best ice-controlcocktails as a prelude to attempting vitrifica-tion of biological tissues suspended in sam-ples of similar dimensions and materials.
The vitrification solution design will thendefine the combinations of CPA type andCPA concentration of vitrification solutionswith cooling and heating conditions that willguarantee ice-free systems. The objective is tooptimize the interaction of the solution for-mulations with the cooling and heating ratesto determine the conditions necessary tomaintain stable vitreous systems during cool-ing, storage and warming of bulk samples.After physical measurement, the bulk vitrifi-cation solution can be tested in biologicalsamples to evaluate ice formation using acryosubstitution technique. The most favor-able systems will finally be evaluated forbiocompatibility and efficacy in sustaining
biological integrity (structure and function)during vitrification of bulk samples.
Vitrification process
To achieve optimal cryoprotection, it is essen-tial that the protocols allow uniform penetra-tion of CPAs throughout the ovarian tissue.Thus, the rate of CPA permeation is an impor-tant determining factor in developing bettercryopreservation protocols for ovarian tissues.Some studies used the more sophisticatedtechnique of 1H nuclear magnetic resonance(NMR) spectroscopy to measure DMSO per-meation in human and porcine ovarian tissue(3–5 mm3). Newton et al.81 have investigatedDMSO, PROH, 1,2-ethanediol (ED), and glyc-erol diffusion into human tissue at both 4ºCand 37ºC, and have observed that at 4ºC,PROH and glycerol penetrate the tissue signif-icantly more slowly than either ED or DMSO.At the higher temperature (37ºC), however, allfour CPAs penetrate at a faster rate. They haveshown that bathing the tissue (4 mm in diame-ter and 2 mm in thickness) for 30 min at 4ºCin a 1.5 mol/L solution of DMSO produced amean tissue CPA concentration approaching80% of that in the bathing medium. Thomaset al.82 reported that by the end of the 20 minexposure at 0–2ºC, the mean tissue concentra-tion reaches 0.68 mol/L and 0.76 mol/L in theporcine and human ovarian tissue, respectively,indicating that CPA permeation into bothtissues was incomplete using their protocols.
A method was adapted from vitrification ofvein segments, in which a baseline vitrifica-tion medium, designated VS55 to reflect thatit comprises 55% (w/v) total cryoprotectivesolute, was used to replace at least 50% of thetissue water with a combination of CPAs. TheVS55 solution consisted of 3.1 mol/L DMSO,3.1 mol/L formamide, and 2.2 mol/L PROHin EuroCollins solution, and VS70 and VS83are concentrated VS55 solutions.35 The fullstrength of the vitrification solutions wereadded and removed in incremental steps.28–30
Based on the NMR study on ovarian tissue81,82
VITRIFICATION IN ASSISTED REPRODUCTION
280
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 280

and the experience in blood vessels,28–30 aprotocol used for blood vessel (15 min at eachstep at 4ºC) was adapted for addition andremoval of cryoprotectant for ovarian tissue.This protocol has shown minimal toxicity tobiological materials and optimal viability inblood vessel, heart valve, cartilage and tissueengineered constructs.28–30,30–35,37,40,83
The vitrification procedure employed theuse of a closed sample container alongside adummy sample that had been fitted with athermocoupler for temperature monitoring.The procedure has been used on bloodvessels, other tissues, and engineered con-structs.28–30,33–35,37,40,83 The packaging shouldeliminate excessive contamination of thesample during the vitrification process.
As discussed above, steps for loading andunloading the VS55 and VS83 solutions werederived from the kinetics of CPA permeationstudies.81,82 After addition of the final vitrifi-cation solution, ovarian tissues were cooledrapidly (43°C/min) to −100°C, followed byslow cooling (3°C/min) to −135°C, and finallystored in a freezer at −135°C for a minimumof 24 h. These samples were then eitherwarmed for further study, or cryosubstitutedto determine the location and distribution ofice, if any, within the ovaries. A thermocouplewas inserted into a separate dummy sampleof the same vitrification solution, and its out-put was monitored via a digital thermometer.Vitrified samples were rewarmed in twostages: first, slow warming to −100°C(30°C/min) and then rapid warming to melt-ing (225°C/min). A slow warming rate wasachieved by moving the sample to the top ofthe −135°C freezer. The fast warming rate wasgenerated by placing the glass vial in a mix-ture of 30% DMSO/water at room tempera-ture. This technique prevents ice fromforming on the outside surface of the glassvial, thereby allowing visualization of themelting process. After rewarming, the vitrifi-cation solution was removed in a step-wisemanner using a mannitol solution for osmoticbuffering.28–30
A clinical procedure (1.5 mol/L DMSO)84
has been used as a classic protocol for humanovarian tissue cryopreservation in the field.However, a less toxic CPA, 1.5mol/L PROHand 0.2 mol/L sucrose85 has been selected byothers as the conventional cryopreservationmethod. The protocol used by Gosden et al.was selected as a ‘freezing’ control to comparewith the ice-free cryopreservation methods. Ithas to be noted that there will be ice forma-tion in conventional cryopreservation proto-cols regardless of CPA type and concentrationused. The study focused on comparing theprotocols with and without ice formation onpost-thaw cell survival.
For conventional cryopreservation, ovarieswere equilibrated for 30 min at 0ºC in a cryo-genic container containing Leibovitz mediumwith 10% bovine calf serum and 1.5 mol/LDMSO. The container was transferred to aprogrammable freezer (Planar products) andcooled at 2ºC/min to −9ºC for seeding. Thesecond cooling ramp was cooled at 0.3ºC/minto −40ºC, and subsequently at 10ºC/min to−140ºC. Finally, the containers were plungedinto liquid nitrogen and stored in vapor phaseliquid nitrogen (less than −160ºC). The tissueswere thawed rapidly by swirling in a water bathat room temperature. They were immediatelytransferred to fresh medium and washed threemore times to remove the cryoprotectant.
Cryosubstitution
Cryosubstitution can be used to identify ice for-mation in cryopreserved tissues. Cryosubstitu-tion is a technique in which tissue is preserved atsubzero temperatures using organic solvents todissolve ice.28,29,40,86 In this way the profile of anyice domains can be revealed in the frozen orvitrified tissue samples.
Cryosubstitution of the cryopreserved ovar-ian tissues took place at −90°C using a substitu-tion media of methanol and osmium tetroxide.Vials containing samples and the substitutionmixture were placed in a heat sink to maintaina constant temperature. The substitution
281
VITRIFICATION OF OVARIAN TISSUES
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 281
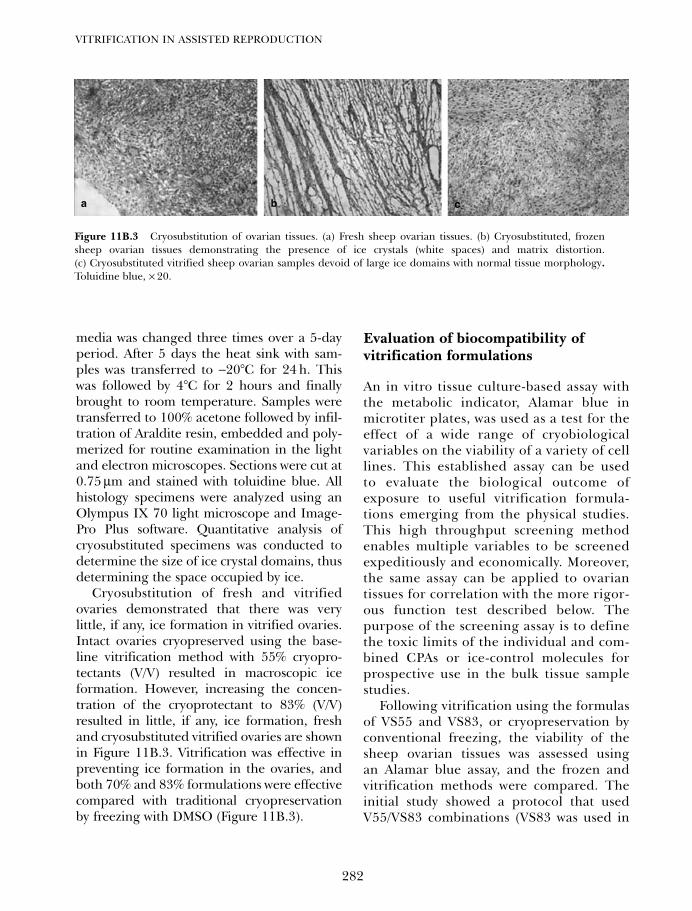
media was changed three times over a 5-dayperiod. After 5 days the heat sink with sam-ples was transferred to −20°C for 24 h. Thiswas followed by 4°C for 2 hours and finallybrought to room temperature. Samples weretransferred to 100% acetone followed by infil-tration of Araldite resin, embedded and poly-merized for routine examination in the lightand electron microscopes. Sections were cut at0.75 µm and stained with toluidine blue. Allhistology specimens were analyzed using anOlympus IX 70 light microscope and Image-Pro Plus software. Quantitative analysis ofcryosubstituted specimens was conducted todetermine the size of ice crystal domains, thusdetermining the space occupied by ice.
Cryosubstitution of fresh and vitrifiedovaries demonstrated that there was verylittle, if any, ice formation in vitrified ovaries.Intact ovaries cryopreserved using the base-line vitrification method with 55% cryopro-tectants (V/V) resulted in macroscopic iceformation. However, increasing the concen-tration of the cryoprotectant to 83% (V/V)resulted in little, if any, ice formation, freshand cryosubstituted vitrified ovaries are shownin Figure 11B.3. Vitrification was effective inpreventing ice formation in the ovaries, andboth 70% and 83% formulations were effectivecompared with traditional cryopreservationby freezing with DMSO (Figure 11B.3).
Evaluation of biocompatibility ofvitrification formulations
An in vitro tissue culture-based assay withthe metabolic indicator, Alamar blue inmicrotiter plates, was used as a test for theeffect of a wide range of cryobiologicalvariables on the viability of a variety of celllines. This established assay can be usedto evaluate the biological outcome ofexposure to useful vitrification formula-tions emerging from the physical studies.This high throughput screening methodenables multiple variables to be screenedexpeditiously and economically. Moreover,the same assay can be applied to ovariantissues for correlation with the more rigor-ous function test described below. Thepurpose of the screening assay is to definethe toxic limits of the individual and com-bined CPAs or ice-control molecules forprospective use in the bulk tissue samplestudies.
Following vitrification using the formulasof VS55 and VS83, or cryopreservation byconventional freezing, the viability of thesheep ovarian tissues was assessed usingan Alamar blue assay, and the frozen andvitrification methods were compared. Theinitial study showed a protocol that usedV55/VS83 combinations (VS83 was used in
VITRIFICATION IN ASSISTED REPRODUCTION
282
Figure 11B.3 Cryosubstitution of ovarian tissues. (a) Fresh sheep ovarian tissues. (b) Cryosubstituted, frozensheep ovarian tissues demonstrating the presence of ice crystals (white spaces) and matrix distortion.(c) Cryosubstituted vitrified sheep ovarian samples devoid of large ice domains with normal tissue morphology.Toluidine blue, × 20.
a b c
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 282

the last minute before vitrification) producedan adequate result. The specimens vitrifiedusing this protocol had a mean of 66.55%cell viability compared with 52.8% for thefrozen tissue. Statistical analysis using theKruskal–Wallis, non-parametric Dunn’s posttest demonstrated that the vitrified group(n = 42) was significantly different com-pared with the frozen group (n = 28)(P < 0.01). However, cell viability in boththe vitrified and frozen groups was signifi-cantly less than in the fresh group (P < 0.05and P < 0.001, respectively) where n = 6.The ovary samples were obtained fromeight sheep (16 ovaries). Therefore, furtheroptimization of vitrification in ovarian tis-sues is required to achieve a result equiva-lent to that of fresh tissues.
Aspects of reproductive biology andphysiology that relate to ovarian tissuepreservation and transplantation
Before discussing the results on transplan-tation of ovarian tissues, it is necessary todiscuss some aspects of reproductive biologyand physiology that are directly related tothe protocol of ovarian tissue transplanta-tion. As we know, gonadotropin releasinghormone (GnRH) is synthesized in thehypothalamus and released from neuronsin pulses. These pulses, which increase infrequency as a female begins puberty,stimulate the biosynthesis and release ofluteinizing hormone (LH) and follicle stim-ulating hormone (FSH).87 As the concentra-tion of these two hormones rises, primordialfollicles (in which the oocytes were arrested
in meiosis at the female’s birth) begin togrow.88 Growth from the primordial to thesecondary stage has been shown to be FSHindependent, while follicle maturation fromthe preantral stage is FSH/LH dependent.Figure 11B.4 below shows the timeline of fol-licular growth as stated by Gougeon et al.89 in1986. Not shown is the 21-week intervalbetween primordial and secondary follicle.However, as the timeline of follicle growth iscurrently under debate, we should take intoaccount other investigators’ proceduresregarding the timing of gonadotropinstimulation.
Several investigators use injections ofFSH in an effort to stimulate the growth ofantral follicles in ovarian tissue xenografts.There are many different doses and timeintervals that have been used by variousgroups. Van den Broecke et al.90 found thata 5 IU daily injection of FSH for 2 weeksbeginning 14 weeks after grafting resultedin a ‘significant shift from primordial toprimary follicles’. Van den Broecke et al.90
also used another procedure in whichSCID mice were injected every other daywith 5 IU of FSH/LH for 3 months. In thisinstance, antral stage development wasseen in the xenografted tissues. Kim et al.91
stimulated with 4 IU pregnant mare’sserum gonadotropin (PMSG) every otherday for 4 weeks beginning 16 weeks afterxenografting. Then in 2005, Kim et al.92
stimulated with 5 IU PMSG every otherday for 2 weeks beginning 20 weeks aftergrafting. In both instances, Kim et al. alsostimulated with 10 IU human chorionicgonadotropin (hCG) and this resulted
283
VITRIFICATION OF OVARIAN TISSUES
Figure 11B.4 Timeline of follicular growth. The timeline shows the stages of follicular growth, beginning with thesecondary stage, along with the approximate number of weeks to reach each stage. FSH dependency is also shown.Reproduced with permission from Gougeor.89
17 weeks 9 weeks 3 weeksPreovulatorySecondary AntralPreantral
FSH Independent FSH dependent
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 283

in antral follicle development. Finally,Weismann et al.93 used 4 IU FSH/LH dailyfor 2 weeks beginning 12 weeks aftergrafting. Results from this study showedpreantral or larger follicle development.
Oocytes are surrounded by one or morelayers of granulosa cells (GC) depending onthe stage of the follicle in which it resides.These granulosa cells promote oocyte growththrough junctions, which allow cell–cell com-munication,87 thus enabling the oocyte toreceive signals from growth factors. Theoocytes, in turn, synthesize proteins such asgrowth differentiation factor (GDF)-9 andGDF-9b (members of the tranforming growthfactor (TGF-β) superfamily), which allow thegranulosa cells to proliferate.87,94–96 Oocytesproduce proliferating cell nuclear antigen(PCNA) during follicle growth, thereforeimmunostaining of PCNA has been used toassess the growth of follicles in human ovar-ian explants.97 PCNA is an auxiliary proteinto DNA polymerase delta, which is involvedin DNA synthesis and repair. Ki-67 is anotherindicator of cell viability.98 Unlike PCNA, it isutilized only if nuclear DNA is functioning atthe time the stain is incorporated, but Ki-67has a very short life span.
Animal model
Although it is important to evaluate whetherovarian tissues and follicles are viable andcompetent after the cryopreservation proce-dure, the current in vitro models used tostudy preantral follicle growth are not wellestablished.99 One way to evaluate the func-tion of cryopreserved tissues is to transplantthe grafts and then monitor the develop-mental competence in host animals.However, to avoid immune rejection of thegraft in hetero- or xenotransplantation, allo-geneic transplantation into a recipient witha suitable histocompatibility complex or intoan immunodeficient recipient is necessary.Immunodeficient mice have been used effec-tively as in vivo research models for studying
xenogeneic follicle development.68 Currentlytwo of these animal models, athymic nude(nu/nu) mice, which are lacking mature T cells,and severe combined immunodeficient (SCID)mice, which are lacking both mature B and Tcells, are being used to assess in vivo xeno-geneic follicular development. Immuno-deficient rodents are promising researchmodels for ovarian cortex xenografting.Ovarian grafts appeared to take better incastrated or hypogonadal mice.65,93,97
Several groups92,93,100,101 working withxenografts of cryopreserved human ovariantissue use male SCID mice instead of femaleSCID mice as graft recipients, as this results innot only a higher frequency of developing fol-licles (13 of 17 vs. 6 of 20),93 but also in folliclesof increased diameter (15 mm).101 Aubard et al.demonstrated a high follicular survival ratein grafts placed under the kidney capsule.68
Transplants grafted subcutaneously above theflanks (several on each side) of the recipienthave shown results equivalent to kidney cap-sule grafts.91,93,101 A very recent study in miceconcluded that the graft site affects the num-ber and quality of oocytes produced from ovar-ian grafts.102 The study used a mouse ovariangrafting model to investigate whether the graftsite (bursal cavity or kidney capsule) influencesthe number, fertilization rate, and develop-mental potential of oocytes recovered fromgrafts. The number of 2-cell embryos pro-duced was significantly higher with oocytesfrom grafts to the bursa, compared with graftsto the other sites.
Fresh, vitrified, or frozen sheep ovariantissues have been implanted beneath therenal capsule or in an orthotopic location.The renal capsule was used as a graft locationdue to its highly vascularized nature and itsability to hold a graft in place. However, notonly does the size of the kidneys limit theamount of tissue that can be transplanted,but the surgery is also difficult and highlyinvasive for the animal. An optimal alterna-tive graft site that would allow an increasedtissue sample number as well as a less difficult
VITRIFICATION IN ASSISTED REPRODUCTION
284
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 284

and less invasive surgery is a subcutaneousgraft site. Many investigators have used asubcutaneous graft site above the flanks inSCID mouse models, as discussed above.101
This site has been used to graft up to six1–2 mm3 tissue blocks on dorsal areas of themouse. This site has also been shown to allowfor larger follicle growth due to its expand-ing nature.
Logistics of a research design for ovariantissue vitrification
Human ovarian tissues could be obtainedfrom a hospital or a tissue compositoryagency. The tissues can be shipped to thevitrification facility overnight at 4ºC. Tissueacquisition begins in the operating roomwhere a section of ovarian tissue is removed.The tissues are then transported to the labo-ratory on ice in a covered container contain-ing Leibovitz-15 (L-15) medium. The warmischemia time is limited to 30 min and coldischemia time to 24 h. Once in the laboratory,manipulation of the tissue including trim-ming and vitrification can be conductedunder sterile conditions inside a tissue culturehood.
After the tissues are transported to the lab-oratory, the gross anatomy of the acquiredtissue is assessed for retrieving tissue blocks.Cauliflower-like protrusions (which areknown to contain follicles) are trimmed out ofthe stroma bridges and further cut into 3 mm3
blocks. This is a way to ensure there are folli-cles in tissue samples before vitrification andimplantation. There have been concernsabout low follicle density in a 1 mm3 tissueblock that is used for in vivo or in vitro matu-ration. Hovatta et al. published a study in2004 in which they counted approximately100 primordial and primary follicles in a1–2 mm3 tissue block.103 Figure 11B.5 showsfollicles that were seen in human ovarian tis-sue following the tissue dissection procedure.Oktay et al.104 developed an isolation tech-nique for human primordial follicles using
enzymatic digestion and microdissection, andobtained high follicular viability rates withboth fresh and frozen ovarian tissues.However, for clinical utility of vitrified ovar-ian tissue blocks, it may be not practical forsurgical handling during transplantation.Serum anti-Müllerian hormone levels havebeen found to reflect the size of the primor-dial follicle pool.105,106 Incubation of ovariantissue blocks before vitrification and/or trans-plantation and detection of anti-Müllerianhormone levels may be useful for selection offollicle-rich blocks for vitrification and subse-quent transplantation.
After trimming, the vitrification processbegins. The tissue blocks are evenly distrib-uted into small, glass specimen vials (approx-imately ten blocks per vial) and the first of thevitrification solutions is added. After the vitri-fication process has begun, the sample vialsare kept on ice at all times and shaken on anorbital shaker under the tissue culture hood.The addition and removal of vitrificationsolutions and vitrification process have beendescribed in previous sections. After the lastrewarming solution (pure medium) is added,the samples are kept in L-15 medium on ice
285
VITRIFICATION OF OVARIAN TISSUES
Figure 11B.5 Follicle pool of human ovarian tissue.Human ovarian tissue from a 32-year-old patient showingprimordial follicles in 1 mm3 cortex block. H&E, × 25.
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 285
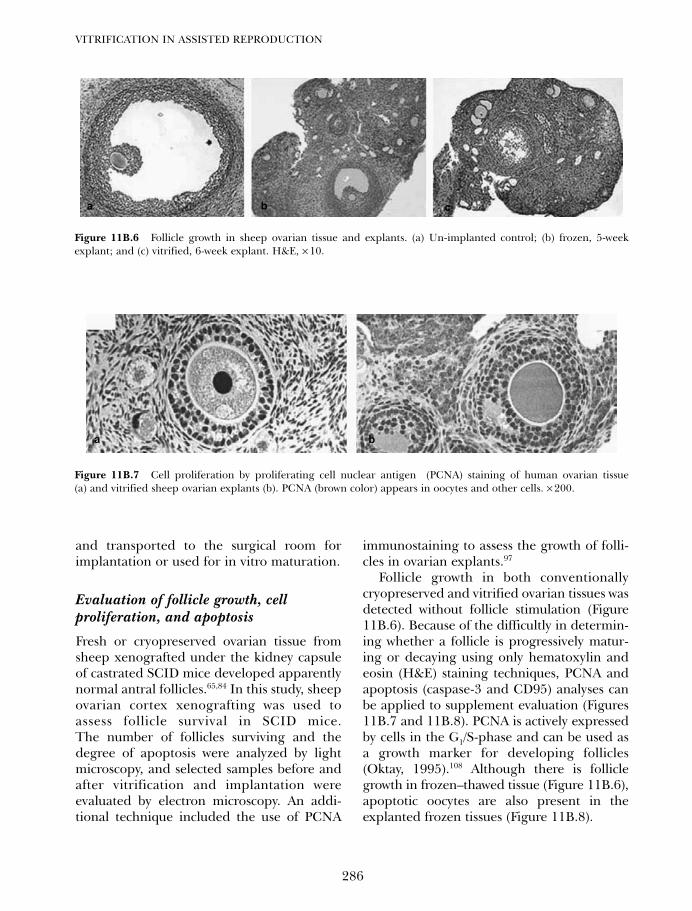
and transported to the surgical room forimplantation or used for in vitro maturation.
Evaluation of follicle growth, cellproliferation, and apoptosis
Fresh or cryopreserved ovarian tissue fromsheep xenografted under the kidney capsuleof castrated SCID mice developed apparentlynormal antral follicles.65,84 In this study, sheepovarian cortex xenografting was used toassess follicle survival in SCID mice.The number of follicles surviving and thedegree of apoptosis were analyzed by lightmicroscopy, and selected samples before andafter vitrification and implantation wereevaluated by electron microscopy. An addi-tional technique included the use of PCNA
immunostaining to assess the growth of folli-cles in ovarian explants.97
Follicle growth in both conventionallycryopreserved and vitrified ovarian tissues wasdetected without follicle stimulation (Figure11B.6). Because of the difficultly in determin-ing whether a follicle is progressively matur-ing or decaying using only hematoxylin andeosin (H&E) staining techniques, PCNA andapoptosis (caspase-3 and CD95) analyses canbe applied to supplement evaluation (Figures11B.7 and 11B.8). PCNA is actively expressedby cells in the G1/S-phase and can be used asa growth marker for developing follicles(Oktay, 1995).108 Although there is folliclegrowth in frozen–thawed tissue (Figure 11B.6),apoptotic oocytes are also present in theexplanted frozen tissues (Figure 11B.8).
VITRIFICATION IN ASSISTED REPRODUCTION
286
Figure 11B.6 Follicle growth in sheep ovarian tissue and explants. (a) Un-implanted control; (b) frozen, 5-weekexplant; and (c) vitrified, 6-week explant. H&E, × 10.
a b c
Figure 11B.7 Cell proliferation by proliferating cell nuclear antigen (PCNA) staining of human ovarian tissue(a) and vitrified sheep ovarian explants (b). PCNA (brown color) appears in oocytes and other cells. × 200.
ba
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 286

CONCLUSION
The reliability of tissue transport logistics inboth animal and human ovarian tissue as wellas tissue viability after exposure to vitrificationsolutions have been discussed. The feasibilityof vitrification of ovarian tissue has beendemonstrated in vivo in the SCID mousemodel. More large-size follicles can be pro-duced through use of FSH stimulation andlonger term maturation. Immunocytochemicaltechniques used to supplement morphologi-cal analysis for better evaluations of vitrifiedexplants help distinguish between prolifer-ated cells and apoptotic cells. In conclusion,vitrification has no apparently negative effecton follicle formation or follicle growth. Themain problem of using a high concentrationof vitrification solutions, as opposed to lowconcentration vitrification solutions in eggvitrification, is the chemical toxicity of CPAs.Low concentrations may cause problems withrecrystalization and devitrification duringrewarming.
This chapter deals with a challengingproblem, namely the vitrification of three-dimensional tissue in comparison with con-ventional, ice-forming cryopreservationprocesses. Vitrification involves several keyparameters, including chemical identity andconcentration of CPA(s), and rates of cooling
and warming. Ovarian tissues are also com-plex systems in respect to reproductive biol-ogy and physiology. This chapter, therefore,can not possibly represent a comprehensiveevaluation and discussion of the entire para-metric space of such a problem. For instance,oocyte competence, aspects of reproductivephysiology, and surgical model cannot bedealt with in a comprehensive fashion. Oocyteretrieval from the matured ovarian explantshas not been covered in the chapter. Graftinglocations, male or female recipients, castratedor non-castrated animals, and exogenous fol-licle stimulation protocols (timing and dose)are mentioned, but not in detail. Nevertheless,the study offers a multitude of opportunitiesfor further clinical investigations, such as can-cer cell detection in ovarian tissue biopsies, invitro maturation of vitrified tissues, and manyother topics, which will all bring contributionsfrom various disciplines to the field.
ACKNOWLEDGMENTS
Partly supported by US Public Health Grant,R43HD047060. The authors would like tothank John Wash and Fred G Lightfoot fortheir work on the physical measurements ofice growth kinetics and cryosubstitution ofovarian tissues.
287
VITRIFICATION OF OVARIAN TISSUES
Figure 11B.8 Apoptosis assay in frozen sheep ovarian explants. Immunocytochemistry of (a) Caspase-3 and (b) CD95showing apoptotic oocytes in brown. There was no apoptosis in other cells. × 200.
ba
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 287

1. Jemal A, Siegel R, Ward E, et al. Cancerstatistics, 2006. CA Cancer J Clin 2006; 56:106–30.
2. Jemal A, Murray T, Ward E, et al. Cancer statis-tics, 2005. CA Cancer J Clin 2005; 55: 10–30.
3. NIH/NCI publication: http://www.seer.cancer.gov/statfacts
4. Ries LG, Percy CL, Bunin GR. In: Ries. LAG,Smith MA, Gurney JG, et al, eds. CancerIncidence and Survival among Children andAdolescents: United States SEER Program1975–1995: Bethesda, MD: National CancerInstitute, SEER program, 1999 NIH pub.No. 99–4649: 1–15.
5. Lee SJ, Schover LR, Partridge AH, et al.American Society of Clinical Oncology rec-ommendations on fertility preservation incancer patients J Clin Oncol 2006; 24:2917–31.
6. Oktay K, Yih, M. Preliminary experiencewith orthotopic and heterotopic transplanta-tion of ovarian cortical strips. Semin ReprodMed 2002; 20: 63–74.
7. Son WY, Yoon SH, Park SJ, Yoon HJ, LeeWD, Lim JH. Ongoing twin pregnancy aftervitrification of blastocysts produced by in-vitro mature oocytes retrieved form a womanwith polycystic ovary syndrome: case report.Hum Reprod 2002; 17: 2963–6.
8. Marcus AD. In vitro fertilization offeredwithout drugs to lower costs. The Wall StreetJournal 2003; 6: 30–32.
9. Nugent D, Meirow D, Brook PE, Aubard Y,and Gosden RG. Transplantation in repro-ductive medicine; previous experience,present knowledge and future prospects.Human Reproduction 1999; 3, pp 267–80.
10. Bordes A, Lornage J, Demirci B, Franck M,Courbiere B, Guerin JF, Salle B. Normal ges-tations and live births after orthotopic auto-graft of vitrified-warmed hemi-ovaries intoewes. Hum Reprod 2005: 20: 2745–8.
11. Lee DM, Yeoman RR, Battaglia DE, et al.Live birth after ovarian tissue transplant.Nature 2004; 428: 137–8.
12. Donnez J, Domans MM, Demylle D, et al.Livebirth after orthotopic transplantion ofcyropreserved ovarian tissue. Lancet 2004;364: 1405–10.
13. Devi KP, Kumar L. Live-birth after trans-plantation of cryopreserved ovarian tissue.Nat Med J India 2005; 18: 257–8.
14. Meirow D, Levron J, Eldar-Geva T, et al.Pregnancy after transplantation of cryopre-served ovarian tissue in a patient with ovar-ian failure after chemotherapy. N Engl JMed 2005; 353: 318–21.
15. Wright CS, Becker DL, Lin JS, Warner AE,Hardy K, Stage-specific and differentialexpression of gap junctions in the mouseovary: connexin-specific roles in follicularregulation. Reproduction 2001; 121: 77–88.
16. Hovatta O, Silye R, Abir R, Krausz T,Winston RM. Extracellular matrix improvessurvival of both stored and fresh humanprimordial and primary ovarian follicles inlong-term culture. Hum Reprod 1997; 12:1032–6.
17. Wang X, Chen H, Yin H, Kim SS, Tan SL,Gosden RG. Fertility after intact ovary trans-plantation. Nature 415: 2002.
18. Cooper A, Paynter SJ, Fuller BJ, Shaw RW.Differential effects of cryopreservation onnuclear or cytoplasmic maturation in vitroin immature mouse oocytes from stimulatedovaries. Hum Reprod 1998; 13: 971–8.
19. Harp R, Leibach J, Black J, Kelsadl C, KarowA. Cryopreservation of mature ovarian tis-sue. Cryobiology 1994; 31: 336–43.
20. Men H, Monson RL, Parrishe JJ, RutledgeJJ. Detection of DNA damage in bovinemetaphase II oocytes resulting from cryo-preservation. Mol Reprod Dev 2003; 64:245–50.
21. Green SH, Smith AU, Zuckerman, S. Thenumber of oocytes in ovarian autograftsafter freezing and thawing. J Endocrinol1956; 13: 330–34.
22. Sugimoto M, Miyamoto H, Kabasawa T,Manabe N. Follicle survival in neonatalrat ovaries cryopreserved by vitrification.Cryo Lett 1996; 17: 93–98.
23. Oktay K, Newton H, Aubard Y, Salha O,Gosden RG. Cryopreservation of immaturehuman oocytes and ovarian tissue: an emerg-ing technology? Fertil Steril 1998; 69: 1–7.
24. Fahy GM, MacFarlane DR, Angell CA,Meryman HT. Vitrification as an approachto cryopreservation. Cryobiology 1984; 21:407–26.
25. Fahy GM. Biological effects of vitrification anddevitrification. In: Biophysics of OrganCryopreservation. Pegg, D.E. and Karow, A.M.Jr. (eds): New York, Plenum, 1987; pp 265–97
VITRIFICATION IN ASSISTED REPRODUCTION
288
References
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 288

26. Fahy GM, Levy D, Ali SE. Some principlesunderlying the physical properties, biologi-cal actions and utility of vitrification solu-tions. Cryobiology 1987b; 24: 196–213.
27. Fahy GM. “Vitrification.” In: LowTemperature Biotechnology: EmergingApplications and Engineering Contributions.McGrath JJ, Diller KR, (eds): New York, TheAmerican Society of Mechanical Engineers,1988; pp 113–146.
28. Song YC, Khirabadi BS, Lightfoot FG,Brockbank KGM, Taylor MJ. Vitreous cryop-reservation maintains the function of vascu-lar grafts. Nat Biotechnol 2000; 18: 296–9.
29. Song YC, Hagen P-O, Lightfoot FG, TaylorMJ, Smith AC, Brockbank KGM. In vivoevaluation of the effects of a new ice-freecryopreservation process on autologousvascular grafts. J Invest Surg 2000; 13:279–88.
30. Song YC, Taylor MJ, Brockbank KGM. Ice-free cryopreservation of arterial grafts.Cryobiology 41: 2000c: 370.
31. Brockbank KGM, Song YC. Mechanismsof bioprosthetic heart valve calcification.Transplantation 2003; 75: 1133–5.
32. Brockbank KGM, Song YC. Morphologicalanalyses of ice-free and frozen cryopreservedheart valve explants. Heart Valve Dis 2004;13: 297–301.
33. Song Ying C, Lightfoot FG, Chen Z, TaylorMJ, Brockbank KGM. Vitreous preservationof rabbit articular cartilage. Cell PreservTechnol 2004; 2: 67–74.
34. Song Ying C, An YH, Kang QK, et al.Vitreous preservation of articular cartilagegrafts. J Invest Surg 2004; 17: 65–70.
35. Song YC, ZZ, Chen MJ, Taylor KGB,Brockbank KCM. Successful preservation ofporcine articular cartilage using a higherconcentration vitrification formulation.Cyobiology 2004; 49: 326.
36. Li X, An YH, Wu YD, Song YC, Chao YJ,Chien CH. Microindentation test for assess-ing the mechanical properties of cartilagi-nous tissues. J Biomed Mater Res B ApplBiomater 2007; 80: 25–31.
37. Song YC, ZZ, Chen N, Mukherjee FG et al.Vitrification of tissue engineered pancreaticsubstitute (TEPS). Transplant Proc 2005; 37:253–5.
38. Mukherjee N, Chen Z, Sambanis A and SongY. Effects of cryopreservation on cell viabilityand insulin secretion in a model tissue engi-neered pancreatic substitute (TEPS). CellTransplant 2005; 14: 449–56.
39. Elder EN, Chen Z, Ensley AE, Nerem RM,Brockbank KGM, Song YC. Enhanced tissuestrength in cryopreserved collagen-basedblood vessel constructs. Transplantation Proc2005; 37: 4625–9.
40. Dahl S, Chen Z, Solan A, Brockbank KGM,Niklason LE, Song YC. Feasibility of vitrifi-cation as a storage method for tissue engi-neered blood vessels. Tissue Eng 2006; 12:291–300.
41. Taylor MJ, Song YC, and Brockbank KGM.Vitrification in tissue preservation: new devel-opments. In: Benson E, Fuller B, and Lane N,(eds.), Life in the Frozen State. Taylor andFrancis: London, 2004; pp. 603–41.
42. Stachecki JJ, Cohen J. An overview of oocytecryopreservation. Reprod Biomed Online2004; 9: 152–63.
43. Borini A, Sciajno R, Bianchi V, Sereni E,Flamigni C, Coticchio G. Clinical outcome ofoocyte cryopreservation after slow coolingwith a protocol utilizing a high sucrose con-centration. Hum Reprod 2006; 21: 512–7.
44. Levi Setti PE, Albani E, Novara PV, Cesana Aand Morreale G. Cryopreservation of super-numerary oocytes in IVF/ICSI cycles. HumReprod 2006; 21: 370–5.
45. Gook DA, Osborn SM, Johnston WI. Cry-opreservation of mouse and human oocytesusing 1,2-propanediol and the configurationof the meiotic spindle. Hum Reprod 1993; 8:1101–9.
46. Gook DA, Schiewe MC, Osborn SM, Asch RH,Jansen RP, Johnston WI. Intracytoplasmicsperm injection and embryo developmentof human oocytes cryopreserved using1,2-propanediol. Hum Reprod 1995; 10:2637–41.
47. Porcu E, Fabbri R, Seracchioli R, Ciotti PM,Magrini O, Flamigni C. Birth of a healthyfemale after intracytoplasmic sperm injec-tion of cryopreserved human oocytes. FertilSteril 1997; 68: 724–6.
48. Fabbri R, Porcu E, Marsella T, Rocchetta G,Venturoli S, Flamigni C. Human oocyte cry-opreservation: new perspectives regardingoocyte survival. Hum Reprod 2001; 16:411–16.
49. Pickering SJ, Braude PR, Johnson MH,Cant A, Currie J. Transient cooling to roomtemperature can cause irreversible disrup-tion of the meiotic spindle in the humanoocyte. Fertil Steril 1990; 54: 102–8.
50. Paynter SJ, Cooper A, Fuller BJ, Shaw RW.Cryopreservation of bovine ovarian tissue:structural normality of follicles after thawing
289
VITRIFICATION OF OVARIAN TISSUES
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 289

and culture in vitro. Cryobiology 1999;3894: 301–9.
51. Moomjy M, Rosenwaks Z. Ovarian tissue cryo-preservation: the time is now. Transplantationor in vitro maturation: the time awaits. Fertilityand Sterility 1998; 69: 999–1000.
52. Karow A. Cryobiology 2001 for mammalianembryologists. www. xytexinternational.com.
53. Meryman HT, Williams RJ. Basic principlesof freezing injury to plant cells; naturaltolerance and approaches to cryopreserva-tion. In: Cryopreservation of Plant Cells.(Kartha, K.K. eds), CRC Press, New York,1985; 13–47.
54. Karlsson JOM, Cravalho EG, Toner M.:Intracellular ice formation: causes and con-sequences. CryoLetters 1993; 14: 323–34.
55. Song YC, Hunt CJ, and Pegg DE Cryo-preservation of the common carotid artery ofthe rabbit. Cryobiology 1994; 31: 317–29.
56. Song YC, Pegg DE, Hunt CJ. Cryo-preservation of the common carotid artery ofthe rabbit: optimization of dimethyl sulfox-ide concentration and cooling rate.Cryobiology 1995; 32: 405–21.
57. Polge C, Smith AY, Parkes AS. Revivalof spermatozoa after vitrification and de-hydration at low tem-peratures. Nature1949; 164: 666.
58. Lovelock JE, Bishop MWH. Prevention offreezing damage to living cells by dimethylsulfoxide. Nature 1959; 183: 1349–59.
59. McGrath JJ. Membrane transport properties.In: Low Temperature Biotechnology:Emerging Applications and EngineeringContributions. (McGrath, J. J. and Diller, K. R.eds), The American Society of MechanicalEngineers, New York, 1988; 273–330.
60. Candy CJ, Wood MJ, Whittingham DG.Follicular development in cryopreservedmarmoset ovarian tissue after transplanta-tion. Hum Reprod 1995; 10: 2334–8.
61. Candy CJ, Wood MJ, Whittingham DG.Restoration of a normal reproductive life-span after grafting of cryopreserved mouseovaries. Hum Reprod 2000; 15: 1300–4.
62. Salle B, Demirci B, Franck M, Rudigoz RF,Guerin JF, Lornage J. Normal pregnanciesand live births after autograft of frozen-thawed hemiovaries into ewes. Fertil Steril2002; 77: 403–8.
63. Almodin CG, Minguetti-Câmara VC,Meister H et al. Recovery of fertility aftergrafting of cryopreserved germinative tissuein female rabbits following radiotherapy.Hum Reprod 2004; 19: 1287–3.
64. Baird DT, Campbell BK, Souza C, Telfer EE.Long-term ovarian function in sheep afterovariectomy and autotransplantation ofcryopreserved cortical strips. Eur J ObstetGynecol Reprod Biol 2004; 113: 55–9.
65. Baird DT, Webb R, Cambell K, HarknessLM, Gosden RG. Long-term ovarian func-tion in sheep after ovarectomy and trans-plantation of autografts stored at −196oC.Endocrinology 1999; 140: 462–71.
66. Nisolle M, Godin PA, Casanas-Roux F, Qu J,Motta P, Donnez J. Histological and ultra-structural evaluation of fresh and frozen-thawed human ovarian xenografts in nudemice. Fertil Steril 2000; 74: 122–9.
67. Aubard Y, Piver P, Cogni Y, et al. Orthotopicand heterotopic autografts of frozen-thawedovarian cortex in sheep. Hum Reprod 1999;14: 2149–54.
68. Aubard Y. Ovarian tissue xenografting. Eur JObstet Gynecol Reprod Biol 2003; 108: 14–8.
69. Kim SS, Yang HW, Kang HG, et al.Quantitative assessment of ischemic tissuedamage in ovarian cortical tissue with orwithout antioxidant (ascorbic acid) treat-ment. Fertil Stertil 2004; 82: 679–85.
70. Bodziony et al., 199471. Rall WF. Factors affecting the survival of
mouse embryos cryopreserved by vitrification.Cryobiology 1987; 24: 387–402.
72. Jutte NHPM, Heyse P, Jansen HG, BruiningGJ, Zeilmaker GH. Vitrification of mouseislets of Langerhans: comparison with amore conventional freezing method.Cryobiology 1987; 24: 292–302.
73. Jutte NHPM, Heyse P, Jansen HG,Bruining GJ, Zeilmaker GH. Vitrification ofhuman islets of Langerhans. Cryobiology1987; 24: 403–11.
74. Takahashi T, Hirsh AG, Erbe EF, Bross JB,Steere RL, Williams RJ. Vitrification of humanmonocytes. Cryobiology 1986; 23: 103–15.
75. Rall WF, Fahy GM. Ice-free cryopreservationof mouse embryos at –196°C by vitrification.Nature 1985; 313: 573–5.
76. Brockbank KGM, Song Ying C, Walsh JR,Taylor MJ. Vitrification, the new frontierin preservation of tissues. In: Vossoughi J,ed. Biomedical Engineering: Recent Deve-lopments. Washington, DC: Medical andEngineering Publishers, Inc., 2002; 197–8.
77. Brockbank KGM, Walsh JR, Song YC,Taylor MJ. Vitrification: preservation ofcellular implants. In: Ashammakhi, N.and Ferretti, P. (eds.), Topics in TissueEngineering, Published on the Web: www.
VITRIFICATION IN ASSISTED REPRODUCTION
290
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 290

tissue-engineering-c.com/ebook_topics_in_t_e/ contents.html, 2003; Chapter 19,1–26.
78. Boutron P. Levo- and dextro-2,3butanediol andtheir racemic mixture: very efficient solutes forvitrification. Cryobiology 1990; 27: 55–69.
79. Sutton RL. Critical cooling rates for aqueouscryoprotectants in the presence of sugarsand polysaccharides. cryobiology 1992; 29:585–98.
80. Boutron P, Peyrdieu J-F. Reduction in toxicityfor red blood cells in buffered solutions con-taining high concentrations of 2,3-butane-diol by trehalose, sucrose, sorbitol, ormannitol. Cryobiology 1994; 31: 367–73.
81. Newton H, Fisher J, Arnold JRP, Pegg DE,Faddy MJ, Gosden RG. Permeation ofhuman ovarian tissue with cryoprotectiveagents in preparation for cryopreservation,Hum Reprod 1998; 13 pp. 376–80.
82. Thomas N, Busza A, Cooper A, et al.Measurement of permeating levels of cryo-protectant during ovarian tissue cryopreser-vation using 1H NMR spectroscopy in humanporcine ovaries. Cryo Letters 1997; 18:179–84.
83. Song Ying C, Chen ZZ, Taylor MJ,Brockbank KGM. Successful vitrification ofarticular cartilage in a large animal model.Presented at the Sixth Annual TESiInternational Conference and Exposition,Orlando FL, 2003, 11–13.
84. Gosden GR, Baird DT, Wade JC, Webb R.Restoration of fertility in oophorectomisedsheep by ovarian autografts stored at−196ºC. Hum Reprod 1994; 9: 597–603.
85. Fabbri R, Pasquinelli G, Bracone G, OrricoC, Tommaso BD, Venturoli S. Cryopreserva-tion of human ovarian tissue. Cell TissueBank 2006; 7: 123–33.
86. Brockbank KGM, Lightfoot FG, Song YC,Taylor MJ. Interstitial ice formation in cryo-preserved homografts: a possible cause oftissue deterioration and calcification in vivo.J Heart Valve Dis 2000; 9: 200–206
87. Fauser BCJM. Reproductive Medicine: Mole-cular, Cellular and Genetic Fundamentals.Lancaster, UK: Parthenon Publishing, 2003.
88. Erickson GF. Female ReproductiveEndocrinology: Morphology and Physiologyof the Ovary. www.endotext.com.
89. Gougeon A. Dynamics of follicular growth inthe human: a model from preliminaryresults. Hum Reprod 1986; 1: 81–87.
90. Van den Broecke R, Liu J, Handyside A, et al.Follicular growth in fresh and cryopreserved
human ovarian cortical grafts transplanted toimmunodeficient mice. Eur J Obs GynecolReprod Biol 2001; 97: 193–201.
91. Kim SS, Soules MR, Battaglia DE. Folliculardevelopment, ovulation, and corpus luteumformation in cryopreserved human ovariantissue after xenotransplantation. Fertil Steril2002; 78: 77–82.
92. Kim SS, Kang HG, Kim NH, Lee HC, LeeHH. Assessment of the integrity of humanoocytes retrieved from cryopreserved ovar-ian tissue after xenotransplantion. HumReprod 2005; 20: 2502–8.
93. Weissman A, Gotlieb L, Colgan T, JuriscovaA, Greenblatt EM, Casper RF. Preliminaryexperience with subcutaneous humanoavrian cortex transplantationin the NON-SCID mouse. Biol Reprod 1999; 60: 1462–7.
94. Juengel JL, Hudson NL, Heath DA, et al.Growth differentiation factor 9 and bonemorphogenetic protein 15 are essential forovarian follicular development in sheep. BiolReprod 2002; 67: 1777–89.
95. McNatty KP, Juengel JL, Reader KL, et al.Bone morphogenic protein 15 and growth dif-ferentiation factor 9 co-operate to regulategranulose cell function in ruminants.Reproduction 2005; 129: 481–7.
96. Bodensteiner KJ, Clay CM, Moeller CL,Sawyer HR. Molecular cloning of the ovinegrowth/differentiation factor-9 gene andexpression of growth/differentiation factor-9in ovine and bovine ovaries. Biol Reprod1999; 60: 381–6.
97. Oktay K, Newton H, Mullan J, Gosden RG.Development of human primordial folliclesto antral stages in SCID/hpg mice stimulatedwith follicle stimulatiing hormone. HumReprod 1998; 13: 1133–8.
98. Nubani R, Hughes FF, Virdi A, Leven RWood-Molo M and Rawlins RG. Viabilitytesting of cryopreserved ovarian tissue. FertilSteril 86 (Suppl 2): S209, 2006
99. Donnez J, Martinez-Madrid B, Jadoul P, VanLangendonckt A, Demylle D, Dolmans MM.Ovarian tissue cryopreservation and trans-plantation: a review. Hum Reprod Update2006; 5: 519–35.
100. Van den Broecke R. Timing of FSH-stimula-tion and follicular development in cryopre-served human ovarian grafts. ReproductiveBioMedicine Online 2001; 4: 21–6.
101. Hernandez-Fonseca H, Bosch P, Sirisathien S,Wininger JD, Massey JB, Brackett BG.Effect of site of transplantation on folliculardevelopment of human ovarian tissue
291
VITRIFICATION OF OVARIAN TISSUES
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 291

transplanted into intact or castratedimmunodeficient mice. Fertil Steril 2004;81: 888–92.
102. Yang HY, Cox SL, Jenkin G, Findlay J,Trounson A, Shaw J. Graft site andgonadotrophin stimulation influences thenumber and quality of oocytes from murineovarian tissue grafts. Reproduction 2006;131(Suppl): 851–9.
103. Zhang P, Henna L, Timo T. In vitro effect ofcyclic adenosine 3′, 5′-monophosphate (cAMP)on early human ovarian follicles. J AssistReprod Genet 2004; 21: 301–6.
104. Oktay K, Nugent D, Newton H, Salha O,Chatterjee P, Gosden RG. Isolation andcharacterization of primordial follicles fromfresh and cryopreserved human ovarian tis-sue. Fertil Steril 1997; 67: 481–6.
105. Kevenaar ME, Mohamed F, Meerasahib PietKramer, et al. Serum anti-Müllerian hormonelevels reflect the size of the primordial folliclepool in mice Endocrinology 2006; 147:3228–34.
106. Visser JA, Frank H de Jong, Joop S E et al.Anti-Müllerian hormone: a new marker forovarian function. Reproduction 2006; 131:1–9.
107. Fabbri R, Venturoli S, D’Errico A, et al. Ovariantissue banking and fertility preservation in can-cer patients: histological and immunohisto-chemical evaluation. Gynecologic Oncology2003; 89: 259–66.
108. Oktay K, Schenken R, Nelson J. Prolife-rating cell nuclear antigen marks the inita-tion of follicular growth in the rat. BiolReprod 1995; 53: 295–301.
VITRIFICATION IN ASSISTED REPRODUCTION
292
11b Tucker 8029.qxd 8/23/2007 2:18 PM Page 292

293
Vitrification of human embryonicstem cellsYoel Shufaro, Gábor Vajta, Alan O Trounson and Benjamin E Reubinoff
12
BACKGROUND
Human embryonic stem cells (hESCs), arederived from early embryos and typically theblastocyst’s inner cell mass, can be propa-gated indefinitely in vitro, have a normalkaryotype, and are pluripotent. Thus, thesestem cells can differentiate into progeny fromall three germ layers both in vitro and in vivo.When engrafted into immune deficient mice,hESCs develop into teratoma tumors withmesoderm, endoderm, and ectoderm tissuecomponents.1,2 Given their unique proper-ties, hESCs can be utilized for the study ofearly human development, drug discovery,and as a renewable source of human cells fortransplantation therapy.3
Safe and robust cryopreservation is requiredfor the development and clinical applicationof hESC lines. Cryopreservation enables thepreservation of stocks of early passage cells,and is insurance for situations in which the cul-ture is lost or damaged. Early passage stocksmay be needed after prolonged cultivation inorder to return to the properties of the origi-nal cells, which might be genetically or perma-nently altered for development due toprolonged culture and repeated passaging.Efficient freezing and thawing methods arealso required for the conservation of specificESCs such as genetically modified clones, orpatient and disease specific autologous hESClines originating from embryos created bysomatic nuclear transplantation. Moreover,these technologies will be essential if hESCbanks are to be established.3,4 Finally, effective
cryopreservation technologies will allow effi-cient transfer of cells between research centers,promoting scientific collaboration, and facili-tating widespread use of the hESCs forresearch and clinical applications.
FREEZING METHODS
Slow cooling rate freezing and rapid thawingmethods are most commonly used for thecryopreservation of cell lines.5 While thesestandard methods are efficient for the cryo-preservation of mouse ESCs,6 the survival ofundifferentiated hESCs following slow cool-ing rate freezing are variable. They are par-ticularly poor when the hESCs are cultured inthe presence of serum, where after thawingmost of the cells differentiate or die.7
Since embryonic stem cells originate fromthe pluripotent cells of the blastocyst and inculture retain the properties of these cellssuch as morphology, gene expression, andpluripotence, it is plausible that methodswhich have been specifically developed forthe cryopreservation of human blastocystsmay be also efficient for hESCs.
Vitrification, which is solidification of solu-tions at low temperature without ice forma-tion, has been extensively studied as amethod of cryopreservation of embryos8 andoocytes.9–11 It was found to be highly efficientin cattle12,13 as well as in other species suchas pigs and hamsters,14,15 in which slow cool-ing freezing methods usually cause lethalcryoinjuries to most embryos. In addition,
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 293

vitrification methods may also be beneficialfor the cryopreservation of human oocytesand blastocysts.11,15–17
With the slow cooling conventional freez-ing methods, the cells are transferred into astandard cryovial containing freezingmedium supplemented with a cryoprotectant.The vials are slowly cooled (~1°C/min) in afreezing container or in a programmedfreezer to −80°C and then plunged into andstored in liquid nitrogen. Thawing is conven-tionally performed by immersion of the vialsin a water bath at 37°C, followed by a gradualdilution and replacement of the freezingmedium by the culture medium and transferof the cells into the appropriate cultureconditions.5
With the vitrification approach, a glass-likesolidification of solutions is achieved by usinghigh solute concentrations of cryoprotectantingredients and rapid cooling. While thisapproach can eliminate cell injury due to icecrystal formation, the high concentration ofcryoprotectants may induce significant toxicand osmotic damage. The concentrations ofcryoprotectants that are required to achievevitrification are inversely related to the rateof cooling. Therefore, an increased speed ofcooling can lessen the cryoprotectant inducedtoxicity, as it minimizes the time of exposureto these toxic compounds at liquid concen-trations. Increasing the cooling rate has beenachieved by plunging samples in small vol-ume held on electron microscope grids,9 thin-walled open pulled straws, (OPS)12 and smallnylon loops15 directly into liquid nitrogen.
Vitrification is simple to perform; cells aretransferred into thin straws which are plungeddirectly into liquid nitrogen. OPS are mostcommonly used.7,12–14,18 Common 0.25 mLinsemination straws are heat-softened andthen pulled manually to a thin diameter.The hESC clusters, incubated briefly in thefreezing medium are then loaded into thenarrow end of the straw by capillary action.For vitrification of hESCs we used two typesof vitrification solutions (VS1 and VS2), both
based on a holding medium (HM) whichincluded DMEM containing HEPES buffersupplemented with 20% fetal bovine serum(FBS). The cryoprotectants used are 10%dimethylsulfoxide (DMSO) and 10% ethyleneglycol (EG) for VS1, and 20% DMSO, 20%EG, and 0.5 mol/L sucrose for VS2. All proce-dures are performed on a heated stage at37°C. Four to six clusters of hESCs are firstincubated in VS1 for 1 min followed byincubation in VS2, containing a higher con-centration of cryoprotectants, for 25 s. Theyare then washed in a 20 µL droplet of VS2and placed within a droplet of 1–2 µL of VS2.The small hESC clusters are loaded into thenarrow end of the straw from the droplet bycapillary action (Figure 12.1a). The narrowend is immediately submerged into liquidnitrogen. Warming is performed on a heatedstage at 37°C. Three seconds after removalfrom liquid nitrogen, the narrow end of thestraw is submerged into the holding mediumwith 0.2 mol/L sucrose. When the drop ofmedium at the tip of the straw has liquified,and medium begins to fill up the straw, thewide opening of the straw is sealed by theoperator’s finger. The expansion of cold airin the straw caused by the gradual rise of itstemperature leads to the expulsion of thehESC clusters into the dish (Figure 12.1b).After 1 min of incubation the clusters aretransferred to and further incubated for5 min in holding medium with 0.1 mol/Lsucrose, followed by two 5 min washoutincubations in holding medium withoutsucrose before being plated on a fresh feederlayer.7
RESULTS OF VITRIFICATION OFHUMAN EMBRYONIC STEM CELLS
The efficacy of hESC vitrification can beassessed by several in vitro and in vivo quan-titative and qualitative parameters. A primaryparameter is the rate of recovery, i.e. the rateof hESC clusters successfully recovered fromthe vitrification straws after warming. Two
VITRIFICATION IN ASSISTED REPRODUCTION
294
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 294

parameters are growth rate and the level ofbackground spontaneous differentiation. Inaddition to quantitative criteria, preservationof the embryonic stem cell chacteristic statehas to be evaluated by morphological criteria,karyotyping, immunophenotyping, and fluo-rescent activated cell sorting (FACS) analysisof the expression of markers of pluripotentcells, such as the cell surface markers stagespecific embryonic antigen 3 (SSEA-3), SSEA-4, TRA 1–60, TRA 1–81, etc., and analysis ofthe expression of transcriptional factorscharacteristic of hESCs such as Oct4, Nanog,and Sox2. The pluripotent potential of thethawed hESCs should be confirmed bydemonstrating their capability to differenti-ate into progeny representing the threeembryonic germ layers both in vitro and invivo within teratoma tumors after xenograft-ing the devitrified cells into severe combinedimmunodeficiency (SCID) mice.7,19
Human ESCs from various cell lines can besuccessfully recovered and propagated aftercryopreservation with conventional slow rate
freezing and rapid thawing methods.5
However, in two comparative analyses, vitrifi-cation was superior to slow freezing.7,19 Theefficiency of the standard slow freezing meth-ods with hESCs was relatively low. It was pos-sible to recover only 23–70% of the hESCclusters after freezing by slow cooling andthawing, and only 16% developed furtherafter plating into hESC colonies; these wereundersized compared with controls. In con-trast, an improved outcome post-warmingwas observed when the OPS vitrificationmethod was used. A total of 82–100% of thevitrified hESC cell clusters were recoveredafter warming and all generated coloniesafter plating.7,19
Vitrification has been reported to be asso-ciated with some cell death at the first dayafter plating and with a significantly reducedmean area of the colonies at day 2 and 7 afterplating, compared with control non-frozenthawed colonies.7 However, an additional dayin culture was sufficient to overcome the vitri-fication induced cell deficit, and at day 8 after
295
VITRIFICATION OF HUMAN EMBRYONIC STEM CELLS
37 °C
a b
Figure 12.1 (a) Vitrification by the open pulled straw method. The cells to be vitrified, which are held at this stagein a cryoprotectant supplemented medium, are pulled into the thin end of an open pulled straw by capillary action.The straw is then plunged into liquid nitrogen. (b) Warming of vitrified cells. The straw is plunged in holding mediumsupplemented with sucrose heated to 37°C. The vitrified-warmed cell clusters are expelled out of the straw once thetemperature rises and the large open end is sealed by the operator's finger. The cells are then washed out of the sucrosebefore further culture.
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 295

plating the area of the vitrified warmedcolonies was similar to that of the controlcolonies at day 7.7 Vitrification was also asso-ciated with a significant increase in the levelof background differentiation as evaluated atday 7 compared with controls.7,19 Nevertheless,the morphological appearance of the coloniesimproved with additional time in culture,probably due to the proliferation of undif-ferentiated cells.7
After vitrification hESCs retain their char-acteristic morphology and other key proper-ties of human pluripotent cells.7,19 Vitrifiedwarmed hESCs can be propagated for pro-longed periods, express alkaline phosphataseactivity, and retain their normal karyo-type.2,7,19 Immunophenotyping of the vitri-fied ESCs can be carried out using a seriesof antibodies that detect cell surface carbo-hydrates and associated proteins found onhuman pluripotent cells.1,2 The vitrifiedwarmed hESCs remain immunoreactive inindirect immunofluorescence assays withantibodies against the SSEA-4 and TRA 1-60carbohydrate epitopes, and with the mono-clonal antibody GCTM-2, which detects anepitope on the protein core of a keratan sul-fate/chondroitin sulfate pericellular matrixproteoglycan found in human embryonalcarcinoma cells.7,19–21
Devitrified hESC colonies express Oct4, anessential factor for the establishment of thepluripotent stem cell population of the innercell mass, which governs the fate of ESCs.1,2,22
A critical level is required to sustain stem cellself-renewal. Increased expression of Oct4induces differentiation into primitive endo-derm and mesoderm, while down regulationof Oct4 levels results in dedifferentiation totrophectoderm.22 Oct4 is expressed in hESCsand its expression is down regulated whenthese cells differentiate.2 Reverse transcrip-tase polymerase chain reaction (RT-PCR)analysis of mRNA isolated from coloniesconsisting mainly of devitrified stem cells,showed that these hESCs retained the expres-sion of Oct4 after vitrification.7,19 The
retention of pluripotency after vitrificationand warming of hESCs was also evaluated invivo by xenografting the cells into SCID mice.Benign teratomas containing tissues repre-sentative of all three germ layers wereformed, and embryonal carcinoma was notobserved in any lesion.7,19
CONCLUDING REMARKS
The data published in the literature indicatethat vitrification by the OPS method is a verysimple and effective approach for the cryo-preservation of hESCs. A high proportion ofthe vitrified hESC clusters can be recoveredafter warming, and develop into hESCcolonies after plating. The vitrified warmedhESCs retain the key properties of pluripotentcells as demonstrated by their normal kary-otype, marker expression, and the potential todifferentiate to derivatives of the three germlayers in xenografts.7 Nevertheless, it shouldbe noted that vitrification of hESCs is associ-ated with some cell injury. A significantincrease in the levels of cell death and sponta-neous differentiation after warming do occur,however, an additional proliferation of the EScells in culture for a further 2 days is sufficientto overcome these effects.7 Therefore, it seemsthat for the purpose of cryopreservation of celllines, the practical significance of this cryo-injury is probably negligible. It is possible thatalteration of the composition of the vitrifica-tion solution in favor of low toxicity cryopro-tectants such as ethylene glycol,23 use of nylonloops instead of the OPS,15 and increasing therate of cooling may further reduce the vitrifi-cation induced cryoinjury. Increased coolingrate requires direct contact between the cellcontaining medium and liquid nitrogen.9,12,15
This direct contact may carry a potential haz-ard for transmission of infective agents.24 Thepotential hazard of contamination may beeliminated by utilizing the OPS vitrificationmethod under sterile conditions.13
While vitrification is a highly effectivemethod for hESC cryopreservation, and is an
VITRIFICATION IN ASSISTED REPRODUCTION
296
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 296

ideal method for the efficient storing of newhESC lines and clones and for their safe andexpeditious transfer between research labora-tories, it has limitations with regard to thenumber of cells that are cryopreserved.Current methods of vitrification do not allowfreezing of large cell pools or volumes. This isa significant limitation since cryopreservationof large scale bulk cultures may be required inthe future when clinical grade hESC lines areto be developed for clinical purposes.Additional methodologies will be required toallow the exploitation of vitrification methodsfor the preservation of hESC bulk cultures forclinical purposes.
The currently published successful results ofhESC vitrification may further enhance thecryopreservation of human blastocysts byvitrification, since it has been demonstratedthat human pluripotent inner cell mass-likecells retain a normal karyotype and pluripotentstate following vitrification and warming.
Cryopreservation of hESCs may serve as an invitro model to test and compare the efficacy ofvarious cryopreservation protocols beforeexploring their use for cryopreservation ofhuman blastocysts in clinical infertility.Nevertheless, while cryopreservation of hESCsmay serve as a reliable model for the pluripotentcells of the inner cell mass, this model will not betotally predictive of the outcome of cryopreser-vation of the total blastocyst which has a criticallayer of trophectoderm cells as well.
In conclusion, the results published in theliterature indicate that hESCs may be effec-tively cryopreserved by using the vitrificationmethod. The key properties of pluripotentcells were maintained after warming and theassociated cryoinjury was mild and transient.Cryopreservation by vitrification improvesthe handling of hESC lines and may be usedfor storage of stocks of cells and for the estab-lishment of ESC banks, facilitating efficientinterlaboratory transfer of cells.
297
VITRIFICATION OF HUMAN EMBRYONIC STEM CELLS
References
1. Thomson JA, Itskovitz-Eldor J, Shapiro SS et al.Embryonic stem cell lines derived from humanblastocysts. Science 1998; 282: 1145–7.
2. Reubinoff BE, Pera MF, Fong CY et al.Embryonic stem cell lines from human blasto-cysts: somatic differentiation in vitro. NatBiotechnol 2000; 18: 399–404.
3. Shufaro Y, Reubinoff BE. Therapeutic applica-tions of embryonic stem cells. Best Pract ResClin Obstet Gynaecol 2004; 18: 909–27.
4. Gearhart J. New potential for human embry-onic stem cells. Science 1998; 282: 1061–2.
5. Freshnny R. Culture of Animal Cells; a Manualof Basic Technique. New York, Wiley-Liss Inc,1994: 255–65.
6. Robertson EJ. Embryo derived stem cell lines.In: Robertson EJ, ed. Teratocarcinomas andEmbryonic Stem Cells: A Practical Approach.Oxford: IRL Press, 1987: 71–112.
7. Reubinoff BE, Pera MF, Vajta G, Trounson AO.Effective cryopreservation of human embryonicstem cells by the open pulled straw vitrificationmethod. Hum Reprod 2001; 16: 2187–94.
8. Rall WF, Fahy GM. Ice-free cryopreservation ofmouse embryos at −196 degrees C by vitrifica-tion, in Nature. 1985; 313: 573–5.
9. Martino A, Songsasen N, Leibo S. Develop-ment into blastocysts of bovine oocytes cryo-preserved by ultra-rapid cooling. Biol Reprod1996; 54: 1059–69.
10. Chen SU, Lien YR, Chen HF et al. Openpulled straws for vitrification of maturemouse oocytes preserve patterns of meioticspindles and chromosomes better than con-ventional straws. Hum Reprod 2000; 15:2598–603.
11. Kuleshova L, Gianaroli L, Magli C et al. Birthfollowing vitrification of a small number ofhuman oocytes: case report. Hum Reprod1999; 14: 3077–9.
12. Vajta G, Holm P, Greve T, Callesen H.Vitrification of porcine embryos using theopen pulled straw (OPS) method Acta VetScand 1997; 38: 349–52.
13. Vajta G, Holm P, Kuwayama M et al. OpenPulled Straw (OPS) vitrification: a new way to
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 297

reduce cryoinjuries of bovine ova and embryos.Mol Reprod Dev 1998; 51: 53–8.
14. Vajta G, Hyttel P, Callesen H. Morphologicalchanges of in-vitro-produced bovine blastocystsafter vitrification, in-straw direct rehydration,and culture. Mol Reprod Dev. 1997; 48: 9–17.
15. Lane M, Schoolcraft WB, Gardner DK.Vitrification of mouse and human blastocystsusing a novel cryoloop container-less tech-nique. Fertil Steril. 1999; 72: 1073–8.
16. Lane M, Bavister BD, Lyons EA, Forest KT.Containerless vitrification of mammalianoocytes and embryos. Nat Biotechnol 1999;17: 1234–6.
17. Yokota Y, Sato S, Yokota M et al. Successfulpregnancy following blastocyst vitrification:Case report. Hum Reprod 2000; 15: 1802–3.
18. Karlsson JO. Cryopreservation: freezing andvitrification. Science 2002; 296: 655–6.
19. Zhou CQ, Mai QY, Li T et al. Cryopreservationof human embryonic stem cells by vitrification.Chin Med J (Engl) 2004; 117: 1050–5.
20. Pera MF, Blasco-Lafita MJ, Cooper S et al.Analysis of cell-differentiation lineage inhuman teratomas using new monoclonal anti-bodies to cytostructural antigens of embryonalcarcinoma cells. Differentiation 1988; 39:139–49.
21. Badcock G, Pigott C, Goepel J, Andrews PW.The human embryonal carcinoma marker anti-gen TRA-1-60 is a sialylated keratan sulfate pro-teoglycan. Cancer Res 1999; 59: 4715–9.
22. Niwa H, Miyazaki Smith AG. Quantitativeexpression of Oct-3/4 defines differentiation,dedifferentiation or self-renewal of ES cells.Nat Genet 2000; 24: 372–6.
23. Palasz AT, Mapletoft RJ. Cryopreservation ofmammalian embryos and oocytes: recentadvances. Biotechnol Adv 1996; 14: 127–49.
24. Tedder RS, Zuckerman MA, Goldstone AH etal. Hepatitis B transmission from contami-nated cryopreservation tank. Lancet 1995;346: 137–40.
VITRIFICATION IN ASSISTED REPRODUCTION
298
12 Tucker 8029.qxd 8/23/2007 2:18 PM Page 298

299
Index
actin 111American Red Cross 261American Society of
Reproductive Medicine 188animal reproduction, vitrification
using EG-based solutions 75–85apoptosis 266, 286vit
assay 287in spermatozoa 96
aqua-glyceroporin 82aquaporin-3 in mouse oocytes 82aquaporins (AQPs) 79, 80–81
bacteria cryopreservation 34banking
ovarian tissue 273ovary 274
biocompatibility,BS evaluation 282–6
Biot-Fournier equation 28blastocoels 83
artificial shrinkage 212expansion degree 212fluid content reduction 230shrinkage effectiveness 230volume reduction 220
blastocystsCryoloop vitrification 220–1cryopreservation 65, 195early expanding (EEB) 240, 245EM grid vitrification, six-step
dilution, clinical results 245expanded
artificial shrinkage 223–4before EM vitrification
245–6effects of dilution and sucrose
concentration 247survival and pregnancy rate
increase 249full equilibrium with
cryoprotectant solution 212hatching using laser pulses 223human
developmental stages 241EM grid vitrification 239–252
materials 242
embryo transfer 244freezing
with glyceroland sucrose 219
and vitrification 172survival 244–5
assessment afterthawing 244
transfer 239vitrification, manual of 234–8warming methods after thawing
242, 243–4human expanded, EM grid
freezing and warming 246middle expanding
(MEB) 240, 245mouse
survival and hatching 205survival rates 230
permeability 83recovered, morphology 225selection for vitrification 211transfer 153vitrification 207, 208–210
factors affectingoutcome 210–213
vitrifiedinfant outcome 228perinatal outcome 227–9survival rate after
dilution methods 245transfer 226
outcome 226–7in vitro culture, cryotolerance
reduction 213warming, hatching and survival
assessment 221–3Boutron, Pierre 5bovine serum albumin (BSA) 183butanediol 60butane-2,3–diol 279butylene glycol 49, 51
calorimeterdifferential scanning 278thermogram 278
calorimetry 165
calvesborn after OPS vitrification 67
of immature oocytes 68somatic cell cloned, born
after OPS process 68capillary effect 36carrier
selection 259solution, lack of effect on
warming vs cooling rates 15system 200–201
aseptic, problems 205see also container
cattle, OPS method 67–9, 71cattle embryos, cryoprotectant toxic
effects 54cells
concentration dependent toxicity16–17
cooling techniques 165cryodamage 87cryopreservation by vitrification 1cryosensitivity 92damage in freezing and
thawing 88–9dehydration 197, 198–9glass-forming agents toxicity 17ice formation 33osmotic limits 15–16permeability 197
to cryoprotectants 16proliferation by PCNA staining of
human ovarian tissue 286shrinkage 16
during cooling 88surface-volume ratio 197thawing 276time dependent toxicity 16vitrification 22–5
children born from slow frozenoocytes, outcome 160
chilling injury 17, 77, 87, 121, 122avoidance 196decrease 39EM copper grid use
for avoidance 133chromatin damage 89
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 299

chromosomes 139–140spermatozoal 96–9
closed pulled straw (CPS) 134, 137congenital birth defects 229container
aseptic 168–9for OPS vitrification and
spermatozoa warming 100–1see also carrier
controlled ovarian hyperstimulation(COH) 248
cooling rate 49calculation of small samples
28–30critical 277–8fast 196, 199high, improved
clinical results 146–7increased 138liquid nitrogen 45’slow’ 165, 167for vitrification 13
Cryo Bio System 201–3cryo-injury 196cryodamage 107–8Cryoleaf method
superiority 141thawing 132vitrification 133, 140
Cryoloop 90, 92–3, 123, 155, 202advantages 229–230blastocysts 207
human, cryopreservation 231vitrification 219–238
day 3 embryos 206manipulation 222modified protocol 188with spermatozoa 93–5use without LN contact 173vitrification 133–135,
136, 186–8, 191–2ultrarapid 190
vitrification scheme 94cryomicroscopy, phase-contrast 165cryopreservation 33–4
animal model 284–5conventional 276–7early work 22–4embryo and oocyte successes 37ice-free 277–286mammalian developmental
consequences 107–117mammalian embryo protocols
190–3process (CP) 21see also vitrification
cryoprotectant,hypothetical 25
cryoprotectantequilibration 56
cryoprotectants 29causing osmotic damage 33combinations 48concentration 197effect on embryos 213equilibration 225exposure to or
equilibration of 197glass forming capabilities 47–56for hESCs 29increased concentration of and
exposure to 231, 276–7intracellular concentration 196mouse embryo toxicity 47movement through
plasma membrane 81–2regimens 130required characteristics 45saturation by and
removal of 163–5selection 259stepwise equilibration 130temperature rate
change control 33toxicity 33, 50–2, 89, 90types 46, 196–7for vitrification 91vitrification concentrations 46
cryoprotective agents (CPAs) 21,24–5, 26–7, 88, 92
combination 147ice crystal growth 279–282role 11–12
cryostability 167cryostorage 107cryostraws 185–6
disadvantage 191cryosubstitution 281–2Cryotip 37, 202
blastocysts 209method 134, 137
cryotolerance reduction 213Cryotop 123, 155, 201
advantages 153–4blastocysts 209
human, vitrifyingand warming 253–9
definitions 257equilibrium solution 254–6materials used 253reagents used 253storage container choice 40
vitrification 124–125, 133–4,188–9, 191–2
device 124, 136human application 65mouse and human
blastocysts 134oocytes 131–2procedure 137, 254, 255–6superiority 141ultrarapid 190
vitrification solution 254–6warming
procedure 256–7solutions 256
zygotes 206cryovial capping portion 221cumulus cells removal 141cumulus complex 146cumulus-oocyte complexes (COC)
137–8, 147, 149, 263–4cut standard straws
(CSS) 168–9, 173cytochalasins 176cytoplasm 110–112
fracture 87
dehydration 197depolymerization 175devitrification 11–12
and fast warming rate 95–6dewar flask, cooling in 189differential scanning calorimetry 30dilution method
six-step, human blastocystsEM vitrified clinicalresults 247–8
two-step 240, 242, 244after thawing of vitrified
shrunk blastocysts 246–8EM vitrified human
blastocysts 248–9dimethylsulfoxide
(DMSO) 24, 29, 45, 49, 123cell contact at decreased
temperature 169–172as CPA 276
direct cover vitrification(DCV) 135
disease transmissionliquid nitrogen-mediated 40potential danger 39–41
DNAdamaged and non-damaged 97fragmented 96–7
droplet, spherical,temperature distribution 30
INDEX
300
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 300

egg bank 145egg yolk 88electron microscope
(EM) grids 36, 146blastocyst vitrification 239–252copper 133–4, 136high cooling rate 239human blastocyte vitrification,
results 244–9oocyte vitrification
procedure 147, 149electron microscopists 4–5embryos
animal, vitrification usingEG-based solutions 75–85
carrier 200–1cryoprotectant toxic effects 54day 3
vitrification 206–7and warming cycles 207
dehydration 198freezing, legal problems 145human
cryopreservation negativeeffects 164–5
culture for EM gridprocedure 240
frozen with propanedioland sucrose 219
transfer 172viability 164
mammalian, cryopreservationdevelopmentalconsequences 107–117
mouse 8–cell, vitrification 183–4pronuclear
blastocysts 174human and animal
differences 175–9human intracellular lipids 178micrographs 164, 168vitrification 163–182vitrified 166
shrinkage in cryoprotectant 56transfer, time after
warming 210–211vitrification
with EG and sucrose 59–60methods 133using OPS 65–73
vitrifieddevelopment 60survival 56–60
rate 213water and cryoprotectants
movement pathway 79–80
endometrium preparation 149, 244equilibration
solution 156strategies 123and vitrification 131
ethylene glycol 35, 122–3, 129with glycerol 48, 53low toxicity 214, 219and sucrose
embryo vitrification 59–60oocyte vitrification 59–60
toxicity 91vitrification 47–48
ethylene glycol-based solutions,animal embryovitrification 75–85
evanescent spherulites 4extracellular damage 112extracellular vitrifying
state 197, 199
FahyJohn 6–8, 24, 165
embryo vitrification 91Fertility Centers of Illinois,
Chicago, retrospective data 258fertility preservation 273–4
cryopreservation options 275–6fertilization assessment 225–6Flexipet denuding
pipette (FDP) 134fluorescent activated cell sorting
(FACS) 295follicle stimulating
hormone (FSH) 283–4follicles
development 284growth
evaluation 286in sheep ovarian tissue 286timeline 283
human ovarian tissue 285survival 274, 276
fracturein cryopreservation 122–3formation 48
freezers, industrial 24freezing
at Kyono ART Clinic 155–9conventional 87genetic damage 61for hESCs 293–4in liquid nitrogen vapor 93–5prevention 277slow 21, 23–24, 89, 219, 229
method (SFM) 153
French mini-straw 199, 200blastocyst vitrification 209
French straws 92
glass, fracture 48glass micropipette
method (GMP) 70, 133glass transition temperatures 24, 89glycerol 23–24, 26, 45, 60
cryoprotective properties 276vitrification 47–8
goat, OPS method 67–69gold grid 146, 149–150Graevsky, Emmanuil Y 23granulosa cells 284growth differentiation
factor (GDF) 284
HART Clinic group 186–7, 227heat conduction equation 28hemi-straw system 133, 137, 200–2
blastocyst vitrification 210vitrification procedure 203vitrification protocols 204warming procedure 203
High Security Vitrification(HSV) kit 41, 202, 204
for blastocysts 210horse, OPS vitrification 70human embryonic stem cells
(hESCs) 28, 90vitrification 293–7
results 294–6human serum albumin (HSA) 185hydrostatic pressure 90
iceavoidance 34crystallization 49dry 60formation 48, 50–1
elimination 35–39extracellular 273–5, 276–7
intracellular 30seeding 34
ice crystals 279growth 279–280
in vitro fertilization embryo transfer(IVF-ET) 146–7
in vitro fertilization(IVF) 248–9, 250
in vitro maturation (IVM) 248–9,250, 261, 266
ovarian tissue 275program 240
infections elimination 40
301
INDEX
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 301

intracellular damage in oocytecryopreservation 108–112
intracytoplasmic sperm injection(ICSI) 119–120, 123–4, 149
IVF Nagata Clinic, Japan 188–9
Kato Ladies’ Clinic, Tokyo,Cryotop results 125–6
Kauzman’s paradox 8
laser pulse 231for shrinkage 223–4
lipids 164cryostability 178droplet removal 122gray vesicles 176intracellular 175–9
micrographs 177–8liquid nitrogen 24,
36–38, 39, 89, 205contamination 167
prevention 138–9cooling 45vapor, freezing in 93–5
liquid nitrogen slush (SN2) 138, 146–7
luteinizing hormone (LH) 283Luyet
BJ 3–6, 8, 22–23, 24–5, 48frog spermatozoa
vitrification 97sperm vitrification
technique 89–90, 91lyophilization 21, 89
magnetic resonancespectroscopy (NMR) 280
meiotic spindles 139injury 140
melting point relationships 12membrane permeability and
cryopreservation 82–3microdroplet, aseptic warming 27microfilaments 111microneedle for shrinkage 223, 224micropipettes 167micropipetting 231microtubules 110, 139microvacular damage 273–4minimum drop size (MDS) 133
technique 38minimum volume cooling
(MVC) 38, 133, 154minimum volume vitrification 132
evolution 132–5techniques 139
Minke whale oocytes,OPS vitrification 71
mitochondria 98, 111–112monkey, ovarian tissue
vitrification 265–6morulae
vitrification andwarming cycles 207
results 209mouse
OPS vitrification 70–1ovarian tissue vitrification 263–4
mouse embryossurvival
after exposure to VS 52, 53after VS11 55
vitrification protocol 76–7volume change with VS 78
mouse morulae,survival after VS 79
mouse oocytesvolume change with PB1 81volume change with VS 78
mRNA in AQPs 79, 80–1
necrosis 268–29Nei, Tokio 4nitrogen (N2) vapor,
storage devices 147non-vitrifying
solution (VS1) 197nuclear envelope 108–9nucleation, ice 10nucleolus 109–110nucleus, in oocyte 108
oocytesaging 140–1bovine, lipid granules 176cryopreservation 145, 275
methods 107–8problems 121–122reasons for 120–1
cryoprotectant addition 199cryosensitivity change 122cryosurvival and parthenogenetic
activation 112–113frozen-thawed
fertilization outcome 140meiotic spindle dynamics
139–140GV
after long contactwith DMSO 171
cooling treatment effects 170cytoskeletal elements 176
humanclinical outcomes 148cryopreservation
indications 159Cryotop 154–5delivery after vitrification 153vitrification 146
at Kyono ARTClinic 156, 158
methods 154immature, cryopreservation 160inverted microscopic images 125large numbers, vitrification
devices 137–8mammalian, cryopreservation
developmental consequences107–117
meiosis 140micrograph 110–111pig, sensitivity 176vitrification
and Cryotop method 119–128Cryotop results 125–6with EG and sucrose 59–60methods 133time schedule 140–1
vitrified, survivaland pregnancy 141
open pulled straw (OPS) 66, 90animal reproduction use 65–73application in cattle, sheep and
goat 67–9aseptic 202
storage 138–9for hESCs 294method 65–7
glass micropipette (GMP) 70reasons to use 71–2vitrification 295
technique 36–8, 40–1vitrification 133–4, 136
process 66organs, cryopreservation
by vitrification 1osmosis 165osmotic shock 122osmotic stress 34, 87ovarian autograft implantation 273ovarian hyperstimulation
syndrome 147, 189, 226ovarian tissues
autotransplantation,pregnancies 262
bovine, vitrification 264cryopreservation,
limitations 273–4
INDEX
302
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 302

Cryotop vitrified human 267human
follicles in 268vitrification
and autograftingafter warming 270
and xenografting afterwarming 268, 269–270
reproductive biologyand physiology 283–4
sheepcryosubstitution 282implantation 284–5
transplanting 261vitrification 261–272, 273–292
in animals 262–6in human 266–8in monkeys 265–6in rabbit 266in rats and mice 262–4research design logistics 285–6in sheep 265
ovarybanking 274cryopreservation 275–6
Parkes, Alan 23, 88, 91patients
and IVF treatment 240preparation 225–6
pellicles 26–7, 29–30permeability property 197pig embryos and oocytes,
vitrification with OPSmethod 69–70
pigletsborn after OPS vitrification
of cloned blastocysts 70born after OPS vitrification
of in vivo embryos 70plasma membrane
cryoprotectant movementthrough 81–2
water movement through 80–81platelet endothelial cell adhesion
molecule (PECAM) 269polecat pups born after OPS
vitrification of in vivoembryos 71
Polscope 139polycystic ovary syndrome 248polyethylene glycol (PEG) 279polyvinylpyrrolidone 183pregnancy
assessment 226multiple 195
pregnancy rateafter DMSO vitrification 169–170Cryoloop vitrification 187–8Cryotop vitrification 189
proliferating cell nuclear antigen(PCNA) 284, 286
1,2–propanediol 153, 279propylene glycol 35, 49, 60
quarantine 145quasivitrification, cryoprotective
agents and 11–12
rabbitOPS vitrification 70ovarian tissue vitrification 266
Raju, Rama 191–3Rall
Peter 6, 7–8, 24, 129, 165embryo vitrification 91
rat, ovarian tissue vitrification 262–3reactive oxygen species (ROS) 266–7rehydration
post-thaw 165osmosis 176
RNA interference 109room temperature 189
severe combined immunodeficiency(SCID) mice 268, 284, 295–6
sex ratio after cryopreservation 160sheep
OPS method 67–9ovarian tissue vitrification 265
sheep embryoscryoprotectant toxic
effects 54–5, 57–8vitrified
survival 58–9viability 59
shrinkageartificial 220, 223, 240
before EM grid vitrification245–6
before vitrification 230–1clinical results 227–8
Siberian tigers, OPS vitrification 71Smirnov, Igor 23, 88smooth endoplasmic reticulum
(SER)-lipidglobules-mitochondria 175
smooth endoplasticreticulum (SER) 164–5
solid-surface vitrification(SSV) 133, 136–7
Spallanzani, Lazzaro 22
spermatozoachromosome apparatus
integrity 96–9cooling rate estimation 95cryopreservation 88–9diminished motility 88examples 98human
viability aftercryoprotectant-freevitrification 102
vitrification 25, 26‘swim up’ selection 92vitrification
background 91cryprotectant-free 87–105
vitrified, oocyte fertilization andembryo development 95
warming 94and ’droplet’ vitrification 99and OPS vitrification
container 100–1spindles 110, 139–140
damageafter cryopreservation 160by ice formation 275
recovery 141SSF technique 41statistical analysis 244, 283stereo-microscope 259Stiles, Walter 3Stokes-Einstein equation 9storage
containers 40optimal, below Tg 9–11
strain point 21straw vitrification method,
conventional 135–6straw-in-straw vitrification 167
method 102sucrose 53–4, 130, 139, 163, 183
gradient steps 191as osmotic buffer 91
sugars 123cryoprotective role 97
Supercool X-100268
Tammann’s theory 3Taxol 160temperature
distribution inspherical droplet 30
effect on viscosity 10equilibration 225low, diffusion time 11and toxicity 45
303
INDEX
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 303

TEST-egg-yolk-glycerol(TEYG) 92–3
thawingat Kyono ART Clinic 157preparation for 158preparation procedure 158six-step method 243–4
tissue, cryopreservation byvitrification 1
toxicity 197cryoprotectants 50–2dilution procedure effects 54species effects 54
transplantation, ischemia after 275trehalose 123trophectoderm cells 240
viability assays 261virus cryopreservation 34VitMaster 138, 200vitreous state 21vitrificants 22, 24vitrification
achievement 89as alternative to freezing 89–90animal embryos
with EG-based solutions 75–85method 75using EFS40 76
in animal reproduction 65–73aseptic 173, 202, 204cryoprotectant intracellular
concentration 196cumulative citations 2day 2–3 embryos,
clinical results 188definition 89enhancement factors 97–9and equilibration 131fast rate-low concentration
(FR-HC VF) 22, 25–8
high temperature 22historical development 3–8human blastocyst, laboratory
manual 234–8human embryonic stem cells
(hESC) 293–8ice-free 22kinetic basis 9kits 39medium isolation from liquid
nitrogen contact 165–9methods 99–101, 184–5, 280–1minimum volume methods 132–5in nature 1–3outcome 159ovarian tissue 261–272overview 1–20problems 24–5procedures with cooling
and warming rates 135strategies 199–205successful, biological
phenomena 15–17superior methods 141–2testing container choice 49–50thermodynamic necessity 8–9ultrarapid 200–202using EG-based solutions 75–6using high CPA
concentrations 90–1warming rate after 13–15see also cryopreservation
Vitrification Kit 155vitrification solutions (VS) 45–6
biocompatibility evaluation 282–6components 77, 79composition 51concentration reduction
and toxicity 129–131design using bulk samples 280for hESCs 294
osmotic effects on embryos 56preparation 54–6protocols 187regimens 130selection 52–4toxicity reduction 213
vitrification solutions (VS2) 197day 3 embryos 206
vitrified solutions (VS),evaporation prevention 225
Vitroplug 201VitSet 40Vogel-Tammann-Fulcher
equation 9, 10
warming rate 49calculation of
small samples 28–30critical 277–8and dilution 131–2fast 196, 199, 225
and devitrification 95–6increase 35‘rapid’ 167‘slow’ 166to prevent ice formation 14
warming solutions 225water
critical cooling speed 90diffusion 80–1evaporation 89
water channels 80–1
Xenopus oocytes, permeabilityto cryoprotectants 83
zona pellucida 112, 122–3hardening 275laser opening 172
zygotes, vitrification 206
INDEX
304
Index Tucker 8029.qxd 8/23/2007 2:20 PM Page 304






























