Embed Size (px)
Citation preview

University of Groningen
Acetylcholine beyond bronchoconstriction: a regulator of inflammation and remodelingKistemaker, Loes
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2015
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Kistemaker, L. (2015). Acetylcholine beyond bronchoconstriction: a regulator of inflammation andremodeling. [S.l.]: [S.n.].
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 10-03-2020

Acetylcholine beyond bronchoconstriction:
a regulator of inflammation and remodeling
Loes E.M. Kistemaker

The studies described in this thesis were performed within the framework of the
Groningen University Institute for Drug Exploration (GUIDE) and the Groningen Research
Institute for Asthma and COPD (GRIAC), and financially supported by the Netherlands Lung
Foundation.
Printing of this thesis was financially supported by:
Univeristy of Groningen
Groningen Graduate School of Science
Netherlands Lung Foundation
Boehringer Ingelheim
Holaira
Paranimfen: Anita Spanjer ‐ van Dijk
Els Vernooij ‐ Kistemaker
ISBN: 978‐90‐367‐7618‐9
ISBN: 978‐90‐367‐7617‐2 (electronic version)
Cover: The bronchial tree of fetal pig lung, stained for nerves and smooth muscle;
previously published in The Lung: Development, Aging and the Environment, MP Sparrow,
Development of the Airway Innervation, 33‐53, Copyright Elsevier (2003).
Cover design: Bart Leferink
Printed by: Ipskamp Drukkers BV, Enschede
Copyright © L.E.M. Kistemaker, 2015
All rights reserved. No part of this book may be reproduced in any manner or by any
means without permission.

Acetylcholine beyond bronchoconstriction A regulator of inflammation and remodeling
Proefschrift
ter verkrijging van de graad van doctor aan de Rijksuniversiteit Groningen
op gezag van de rector magnificus prof. dr. E. Sterken,
en volgens het besluit van het College voor Promoties.
De openbare verdediging zal plaats vinden op
vrijdag 6 maart 2015 om 16.15 uur
door
Loes Elisabeth Maria Kistemaker
geboren op 4 september 1986 te Oldenzaal

Promotores
Prof. dr. R. Gosens
Prof. dr. H. Meurs
Prof. dr. H.A.M. Kerstjens
Beoordelingscommissie
Prof. dr. A.J. Halayko
Prof. dr. W. Kummer
Prof. dr. D.S. Postma

Table of contents
Chapter 1
General introduction
7
Chapter 2 Regulation of airway inflammation and remodeling by muscarinic
receptors: perspectives on anticholinergic therapy in asthma and
COPD
Life Sci, 2012, 91(21‐22):1126‐33
35
Chapter 3 Muscarinic receptor subtype‐specific effects on cigarette smoke‐
induced inflammation in mice
Eur Respir J, 2013, 42(6):1677‐88
57
Chapter 4 Muscarinic M3 receptors on structural cells regulate cigarette
smoke‐induced neutrophilic airway inflammation in mice
Am J Physiol Lung Cell Mol Physiol, 2015, 308(1):L96‐L103
81
Chapter 5 Anti‐inflammatory effects of acetylcholine inhibition after
targeted lung denervation in patients with COPD
Submitted to Thorax, 2015
101
Chapter 6 Muscarinic M₃ receptors contribute to allergen‐induced airway
remodeling in mice
Am J Respir Cell Mol Biol, 2014, 50(4):690‐8
111
Chapter 7 Murine precision‐cut lung slices as a model to study TGF‐β and
bronchoconstriction‐induced remodeling
135
Chapter 8 Tiotropium attenuates IL‐13‐induced goblet cell metaplasia of
human airway epithelial cells
Thorax, 2015, In revision
155
Chapter 9 General discussion and summary
Trends Pharmacol Sci, 2014, Epub
175
Nederlandse samenvatting
199
Dankwoord
211
Curriculum vitae
217
List of publications 219


CHAPTER 1
GENERAL INTRODUCTION

Chapter 1
8
Preface
The objective of this thesis is to establish the role of acetylcholine and individual
muscarinic receptor subtypes in inflammation and remodeling of the airways.
Inflammation and remodeling are two important pathophysiological processes in asthma
and chronic obstructive pulmonary disease (COPD), affecting the decline in lung function
and severity of the disease. In the airways, acetylcholine acts as a parasympathetic
neurotransmitter, but also as an autocrine and/or paracrine hormone. Muscarinic
receptors are target receptors for acetylcholine and muscarinic receptor antagonists are
used as a therapy for COPD, and to a lesser extent also for asthma. Consequently, a
potential role for acetylcholine in inflammation and remodeling in COPD and asthma could
have important therapeutic implications.
Acetylcholine, a neurotransmitter and a hormone
Acetylcholine – a neurotransmitter
Acetylcholine is the primary parasympathetic neurotransmitter of the airways.
Acetylcholine was detected by Otto Loewi and Sir Henry Dale in the early 1920s, for which
they received the Nobel prize in 1936. As a neurotransmitter, acetylcholine is synthesized
in nerve endings from the substrates choline and acetyl‐CoA by the enzyme choline acetyl
transferase (ChAT) (1). The uptake of choline from the extracellular space is the rate‐
limiting step in this process (2). Synthesized acetylcholine is translocated into synaptic
vesicles via the vesicular acetylcholine transporter (VAChT), and released by exocytosis.
Exocytosis is triggered by a depolarizing stimulus and modulated by several regulatory
mechanisms in the neuroeffector junction (1). This results in the release of acetylcholine
from airway parasympathetic nerve endings in the extracellular space, where it interacts
with postsynaptic target receptors, but also with presynaptic receptors on cholinergic
nerve terminals themselves. Target receptors of acetylcholine include muscarinic
receptors and nicotinic receptors, which will be introduced in more detail below.
Acetylcholine in the synaptic cleft is rapidly degraded into acetate and choline by the
highly expressed enzyme acetylcholine esterase (AChE). Alternatively, acetylcholine can be
degraded by butyrylcholinesterase (BChE) (1). Choline can be taken up again by the nerve
for acetylcholine synthesis. The acetylcholine synthesis pathway in neurons is summarized
in figure 1A, together with the non‐neuronal synthesis pathway, which will be elaborated
on later.

General introduction
9
Figure 1. Schematic representation of acetylcholine synthesis, release, action and breakdown in neuronal cells at
a cholinergic nerve terminal (A) and in non‐neuronal cells, such as epithelial cells (B). In neurons, there is an
efficient metabolism of acetylcholine (ACh), involving the high‐affinity choline transporter‐1 (CHT1), choline
acetyltransferase (ChAT), the vesicular ACh transporter (VAChT) and ACh release via exocytosis. Although some
non‐neuronal cells also express these components, they are not common to all cell types and probably
alternative, less efficient, mechanisms predominate. Perhaps as a consequence, acetylcholine content is lower. In
non‐neuronal cells, choline is taken up not only via CHT1, but also via choline transporter‐like proteins (CTL),
including CTL1, CTL2 and CTL4. Carnitine acetyltransferase (CarAT) has been identified as an alternative
synthesizing enzyme of ACh. Organic cation transporters (OCT), including OCT1 and OCT2, have been shown to
transport ACh in and out of cells (3). MR: muscarinic receptor, NR: nicotinic receptor, AChE: acetylcholinesterase,
BChE: butyrylcholinesterase.
Airway nerves
Airway nerves regulate many aspects of airway function. One of the most prominent
effects is the regulation of airway smooth muscle tone, the neural component of which is
almost solely controlled by the parasympathetic nervous system (4, 5). Although airways
also express sympathetic and non‐adrenergic, non‐cholinergic (NANC) neural pathways,
focus of this thesis is on the parasympathetic neural pathway, which is the dominant
neural pathway in the airways and uses acetylcholine as a neurotransmitter. Airway vagal
nerve fibers can be divided into three major components: the primary afferent nerve
fibers, the integrating centers in the brain and the parasympathetic efferent nerve fibers
(see figure 2) (6). Afferent nerve fibers in the airways include stretch‐sensitive myelinated
nerve fibers or A‐fibers, and unmyelinated C‐fibers (7). A‐fibers conduct action potentials

Chapter 1
10
at a relatively high velocity and consist of rapidly adapting receptors (RARs) in the mucosal
layer, which respond to the dynamic phase of inspiration, and slowly adapting receptors
(SARs) in the smooth muscle layer, which respond to maintained inflation (7‐9). However,
most of the airway afferent nerves are unmyelinated C‐fibers, which are present
throughout the airways and conduct action potentials at a much slower velocity compared
to A‐fibers (6, 10). C‐fibers respond to noxious stimuli such as heat, cold or mechanical
forces, but also to mediators released upon tissue damage and inflammation (7).
Activation of these C‐fibers results in bronchoconstriction, mucus secretion and cough
(11). This can be a local reflex in the airways at the level of the parasympathetic ganglia,
which involves the release of peptide neurotransmitters, including substance P and other
tachykinins, directly from the nerve terminals (12). Next to this local reflex, a central reflex
arch via the brain stem exists (9). Parasympathetic efferent nerve fibers consist of
preganglionic neurons, which innervate the parasympathetic ganglia in or near the airway
wall of larger airways, and postganglionic neurons (6). Postganglionic neurons innervate
airway smooth muscle, mucus glands and the microvasculature throughout the airway
tree (figure 2). In addition, postganglionic parasympathetic neurons, which do not use
acetylcholine as a transmitter, exist. Instead, these NANC pathways use nitric oxide and/or
neuropeptides such as vasoactive intestinal peptide as a transmitter, and are in fact
bronchodilatory (6). However, the parasympathetic cholinergic nervous system is the
major neural regulator of airway smooth muscle tone (4, 9, 13).
Figure 2. The vagal nervous system in the airways. Mechanical forces, inhaled irritants and endogenous
inflammatory mediators activate afferent nerve fibres, which send signals to the central nervous system (CNS).
This results in the release of acetylcholine (ACh) in the airways. Parasympathetic, post‐ganglionic neurons are
located within the airway wall and innervate airway smooth muscle and submucosal glands to induce contraction
and mucus secretion, respectively. Blue: afferent nerve fibers; red: efferent nerve fibers. RAR: rapidly adapting
receptors; SAR: slowly adapting receptors.

General introduction
11
Acetylcholine ‐ a hormone
During the last decades, it has become clear that acetylcholine production in the airways is
not only restricted to the nerves. Bacteria, algae, protozoa, tubellariae and primitive
plants, all lacking a nervous system, express acetylcholine. This suggests an early and
widely distributed role of acetylcholine (14). Indeed, cells of higher organisms, including
cells of the human airway, have been shown to produce acetylcholine (14, 15). Thus,
acetylcholine can act as a local signaling molecule in an autocrine or paracrine fashion,
which is referred to as non‐neuronal acetylcholine. RNA expression of ChAT has been
detected in almost all cell types of the airways, including epithelial cells, airway smooth
muscle cells and inflammatory cells (16). Additionally, carnitine acetyltransferase (CarAT)
has been detected as an alternative, albeit less efficient, route for acetylcholine synthesis
in airway cells (2). In particular epithelial cells have been shown to express relatively high
levels of acetylcholine (17, 18), and the epithelial non‐cholinergic system has been
characterized in detail (2). Evidence for the actual release of non‐neuronal acetylcholine
from other airway cell types is still limited (15). Part of the biosynthesis pathway of non‐
neuronal acetylcholine overlaps with that used by the neuronal system; however, clear
differences exist, the former being less efficient (2). For example, neurons use the highly
efficient CHT1 transporter for choline uptake, whereas non‐neuronal cells mainly use
choline transporter‐like proteins (CTLs) and organic cation transporters (OCTs) for choline
uptake (2). In addition, in neurons, acetylcholine is stored and subsequently released from
vesicles, whereas in non‐neuronal cells acetylcholine is mainly released directly via OCTs
(2, 3) (figure 1B). As a consequence, acetylcholine levels seem to be lower in non‐neuronal
cells compared to neuronal cells. The functional relevance of non‐neuronal acetylcholine
in the airways still has to be established and it is unclear whether non‐neuronal
acetylcholine contributes to the classical cholinergic driven effects as described above (3).
However, the concept that acetylcholine is not only a neurotransmitter but also a
hormone has dramatically broadened the view on the role of acetylcholine in the airways,
and holds therapeutic promises for the future, which will be further elaborated on in
chapter 2 (19‐21).
Targets of acetylcholine
Acetylcholine can act via two different classes of receptors, namely muscarinic and
nicotinic receptors. Muscarinic receptors are G‐protein coupled receptors, characterized
by a seven transmembrane domain (1). Five different subtypes have been identified (M1‐
M5), of which M1‐M3 receptors are expressed abundantly in the airways (1, 16, 21). Almost
all cell types in the airways express muscarinic receptors, as discussed in detail below.
Activation of muscarinic receptors in the airways leads to bronchoconstriction and mucus
secretion (21). Nicotinic receptors are ligand gated ion channels, which are comprised of

Chapter 1
12
one to five different types of subunits (α, β, γ, δ and ε) (1). Multiple different nicotinic
receptor isotypes exist and at least ten different α‐subunits and four different β‐subunits
have been identified. In the airways, almost all cell types express nicotinic receptors (1,
15). Nicotinic receptors are involved in neurotransmission in the airways, and activation of
nicotinic receptors causes an influx of positively charged ions, leading to membrane
depolarization (1, 22). The nicotinic α7 receptor is highly expressed on multiple cell types
in the airways and plays an immunomodulatory role (15). Moreover, the nicotinic α7
receptor is thought to play a regulatory role in lung cancer (23). Current therapy for
obstructive airway diseases, however, is selectively directed to muscarinic receptors.
Therefore, the focus of this thesis is on muscarinic receptors, and nicotinic receptors will
not be discussed in more detail here.
Muscarinic receptors
Expression of muscarinic receptors
Expression of muscarinic M1, M2 and M3 receptors has been detected in the airways.
Although muscarinic receptor antibodies have been proven not to be very useful in the
detection of subtype specific expression of muscarinic receptors because of lack of
specificity (24, 25), other techniques, including binding studies and gene expression
analyses, have elucidated the expression pattern of muscarinic receptors throughout the
airways. From these studies, it is clear that M1 receptors appear to be expressed
particularly in peripheral lung tissue, M2 receptors are expressed throughout the airways,
whereas expression of M3 receptors is highest in the larger airways (1, 26). Almost all cells
of the airways express muscarinic receptors, with high expression on airway smooth
muscle cells, epithelial cells and submucosal glands. Moreover, in parasympathetic
ganglia, M1 receptors are expressed, as well as autoinhibitory prejunctional M2 receptors
(21, 22). Expression of muscarinic receptors per cell type, and their major functional
effects, are summarized in table 1.
Signal transduction of muscarinic receptors
Muscarinic receptors couple to heterotrimeric G proteins. G proteins are composed of an
α‐, β‐ and γ‐subunit, and are classified according to the α‐subunit. Four different families
have been identified: Gαs, Gαi/o, Gαq/11 and Gα12/13 (30, 31). M2 receptors couple primarily
to Gαi and M1 and M3 receptors couple primarily to Gαq (32). Receptor activation
promotes the exchange of guanosine diphosphate (GDP), bound to the α‐subunit, with
guanosine triphosphate (GTP). This will lead to dissociation of the α‐subunit from the βγ‐
heterodimer and subsequent activation of second messengers (30). Activation of Gαi via
M2 receptors results in inhibition of adenylyl cyclase (AC), which in turn inhibits the second
General introduction
13
messenger cyclic AMP (cAMP), and Gβγ mediated inhibition of potassium channels (33,
34). Activation of Gαq via M1 and M3 receptors results in activation of phospholipase C
(PLC). PLC converts phosphatidylinositol‐4,5‐bisphosphate (PIP2) into inositol 1,4,5‐
trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces the release of calcium from
internal endoplasmic reticulum stores, whereas DAG activates protein kinase C (PKC). The
initial calcium release is followed by a more sustained calcium release mediated via
ryanodine receptors on the endoplasmic reticulum, and via an increase in the open
probability of calcium channels in the cell membrane, mediated via PKC (35).
Functional roles in the airways
Neurotransmission
M1 receptors expressed in parasympathetic ganglia contribute to neurotransmission, by
facilitating depolarization via inhibition of potassium currents. The magnitude of
muscarinic depolarization may not be sufficient to initiate an action potential by itself, but
rather enhances or facilitates nicotinic‐induced depolarization (1). It should be noted
therefore that there is still debate about the functional relevance of M1‐mediated
neurotransmission (1). Evidence for the auto‐inhibitory role of prejunctional M2 receptors
is more convincing, since M2 selective antagonists enhance electrical field stimulation‐
induced acetylcholine release from guinea pig and human trachea (36, 37). The
autoinhibitory M2 receptor is dysfunctional in asthma, which contributes to the enhanced
cholinergic tone in this disease (16). No such mechanism has been observed in COPD (38).
Table 1. Expression of muscarinic receptors on airway cells and their major effects (16, 27‐29).
Cell Muscarinic receptor
expression
Functional effect
Neuron M1, M2 Neurotransmission
Airway smooth muscle cell M2, M3 Bronchoconstriction via M3
Epithelial cell M1, M2, M3 Mucus secretion via M3
Submucosal gland M1, M3 Mucus secretion via M3
Fibroblast M1, M2, M3 Proliferation, ECM production
Mast cell M1, M3 Inhibition of histamine release
Macrophage M1, M2, M3 Cytokine production
Lymphocyte M1, M2, M3 Cytokine production
Neutrophil M1, M2, M3 Cytokine production
Eosinophil M1, M2, M3 ?

Chapter 1
14
Airway smooth muscle contraction
Muscarinic receptors on airway smooth muscle mediate airway smooth muscle
contraction, and therefore play an important role in bronchoconstriction associated with
COPD and asthma. Although M2 receptors represent the majority of muscarinic receptors
expressed on airway smooth muscle, airway smooth muscle contraction is primarily
mediated by M3 receptors (39). This is clear from affinity profiles of selective muscarinic
antagonists in different species, including humans (16). Moreover, the introduction of
specific muscarinic receptor subtype deficient animals confirmed that this is an M3‐
mediated effect, as M3 receptor deficient mice (M3R‐/‐), and not M2R
‐/‐ mice, lack
methacholine and vagally‐induced bronchoconstriction in vivo (40).
Mucus secretion
Muscarinic receptors regulate mucus secretion in the airways. Mucus secretion is
enhanced in COPD and asthma, which contributes to airflow obstruction of the airways
(41). Mucus is secreted by airway submucosal glands, which are in connection to the
airway lumen, and by goblet cells, which are embedded in the airway epithelium.
Submucosal glands are innervated by parasympathetic nerves (see also figure 2), and
mucus secretion from glands is under cholinergic control, mediated via M3 receptors.
Thus, electrical field stimulation increases mucus secretion from airway preparations,
which can be inhibited by the M3 antagonist 4‐DAMP (1,1‐Dimethyl‐4‐diphenylacetoxy‐pi‐
peridinium iodide) (42, 43). Acetylcholine can also induce mucus secretion from goblet
cells, again mediated primarily via M3 receptors (21, 41). There is still debate whether
acetylcholine‐induced mucus secretion from goblet cells is mediated by neuronal or non‐
neuronal acetylcholine, or by a combination of both. M1 receptors are also expressed on
epithelial cells and submucosal glands, and mediate electrolyte and water secretion in
cooperation with M3 receptors (21, 42).
Vagal dysregulation
Activity of the neuronal system is altered in disease, including asthma and COPD, via
several mechanisms, leading to exaggerated acetylcholine release and airway
hyperresponsiveness (9, 13, 44). This can be via [1] changes in activity of afferent nerves,
[2] changes in synaptic transmission and [3] changes in neurotransmitter content in the
synaptic cleft. First, changes in activity of afferent nerves have been observed in response
to inflammatory mediators, present in the airways of patients with COPD or asthma. This
results in enhanced release of acetylcholine from vagal nerve endings and an increase in
cholinergic tone (6). This effect can be mediated via several inflammatory mediators,
including histamine, tachykinins, prostaglandins and thromboxane A2, which stimulate

General introduction
15
sensory nerve fibers directly (6, 45). Moreover, epithelial damage will expose afferent
sensory nerve endings in the subepithelial layer to the airway lumen (46). Second,
inflammation can also change synaptic transmission, by increasing synaptic efficacy and by
increasing acetylcholine release, via the release of mediators including tachykinins, which
interact with receptors on nerve terminals (6, 12). Moreover, autoinhibitory prejunctional
M2 receptors, which limit acetylcholine release under healthy conditions, are
dysfunctional in asthmatic airways. This has been shown in animal models of allergen
exposure, viral infection and ozone exposure (47), but also in patients with asthma (16,
48). M2 receptor dysfunction is probably mediated via several mechanisms, but convincing
evidence exists for the involvement of major basis protein. Major basic protein is secreted
by eosinophils that are recruited to airway nerves, and acts as an allosteric antagonist for
the M2 receptor (49). In support, treatment with an antibody against major basic protein
prevents M2 receptor dysfunction in guinea pigs (50). Third, acetylcholine content in the
synaptic cleft is altered. In part this is due to dysfunctional M2 receptors as described
above, but it has also been shown that activity of AChE is reduced in tracheal smooth
muscle homogenates obtained from ragweed pollen‐sensitized dogs compared to sham
controls (51). Together, this results in enhanced acetylcholine concentrations in the
synaptic cleft and prolonged effects on postjunctional target receptors.
There is little evidence for altered muscarinic receptor expression in asthma or COPD.
Using radioligand binding studies, no significant changes were observed in affinity or
density of muscarinic receptors in the central or peripheral airways of patients compared
to healthy controls (52, 53). Moreover, until now, there is no evidence for genetic
polymorphisms of muscarinic receptors, as at least M2 and M3 receptors seem to be highly
conserved, and no significant differences between healthy and asthmatic subjects were
detected (54).
Besides these acute effects on vagal nerves, there is also evidence for neural remodeling.
Growth and development of airway nerves continues well into adolescence, and exposure
to allergens might alter airway nerve structure (55). Pan et al. demonstrated that allergen
exposure in guinea pigs results in a shift from a NANC phenotype, which does not use
acetylcholine as a neurotransmitter, towards a cholinergic phenotype (56). In addition,
alterations in airway nerve structure might start very early in life, which may affect the
incidence of airway diseases later in life (57). A recent paper demonstrated that early‐life
exposure of mouse neonates to ovalbumin leads to a two‐fold increase in airway smooth
muscle innervation in adulthood (58). Furthermore, exposure of infant rhesus monkeys to
ozone and/or house dust mite results in changes in airway innervation in the epithelial
region (59). Thus, exposure to allergen or environmental stress, either early in life or in

Chapter 1
16
adulthood, may lead to dysregulated vagal innervation, and might thereby affect airways
diseases.
Although most of these studies focus on asthmatic airways, evidence suggests that
innervation is also altered in the airways of COPD patients (7). The inflammatory response
in the airways of patients with COPD might trigger acetylcholine release via above
described mechanisms. Moreover, altered breathing patterns and changes in gas tensions
in patients with COPD activate RAR‐fibers and SAR‐fibers (7), and the ability of SARs to
respond to lung inflation seems to be be altered in patients with COPD (13). Therefore,
dysregulation of vagal nerves, leading to an increased cholinergic tone, largely contributes
to both COPD and asthma pathophysiology. Strikingly, increased cholinergic tone is the
major reversible component of airflow limitation in COPD (60, 61).
Muscarinic receptors in the treatment of COPD and asthma
Therapeutic targets in COPD and asthma
As described above, muscarinic receptors play a key role in regulating bronchoconstriction
and mucus secretion, and cholinergic tone is increased in patients with COPD and asthma.
For these reasons, muscarinic receptors represent key therapeutic targets for COPD and
asthma. Muscarinic receptor antagonists, referred to as anticholinergics, have already
been used for centuries for the treatment of obstructive airway diseases. This started with
the use of naturally occurring medicinal plants containing anticholinergic alkaloids such as
atropine. Fumes of these medicinal plants were inhaled, and later on even smoked via
cigarettes, until the middle of the 20th century (62, 63). Since then, more safe and effective
synthetic anticholinergics have been introduced to the market.
Anticholinergics
Ipratropium was the first synthetic anticholinergic introduced to the market. Because of
the quaternary ammonium structure, ipratropium is much safer compared to the natural
occurring anticholinergic atropine. Atropine can easily pass the blood‐brain barrier leading
to numerous side effects, which is prevented by the quaternary structure of ipratropium.
The safety profile of ipratropium is further enhanced by applying the drug via inhalation,
thereby limiting systemic exposure. Besides dry mouth, ipratropium, but also other
anticholinergics, have limited side effects (64). Thereafter, oxitropium was introduced.
Both drugs have relatively short durations of action (4‐8 hours), and there was a need for
long‐acting anticholinergics. Tiotropium is the first long‐acting anticholinergic introduced
to the market in the early 2000s. Tiotropium has been shown to be a potent muscarinic
General introduction
17
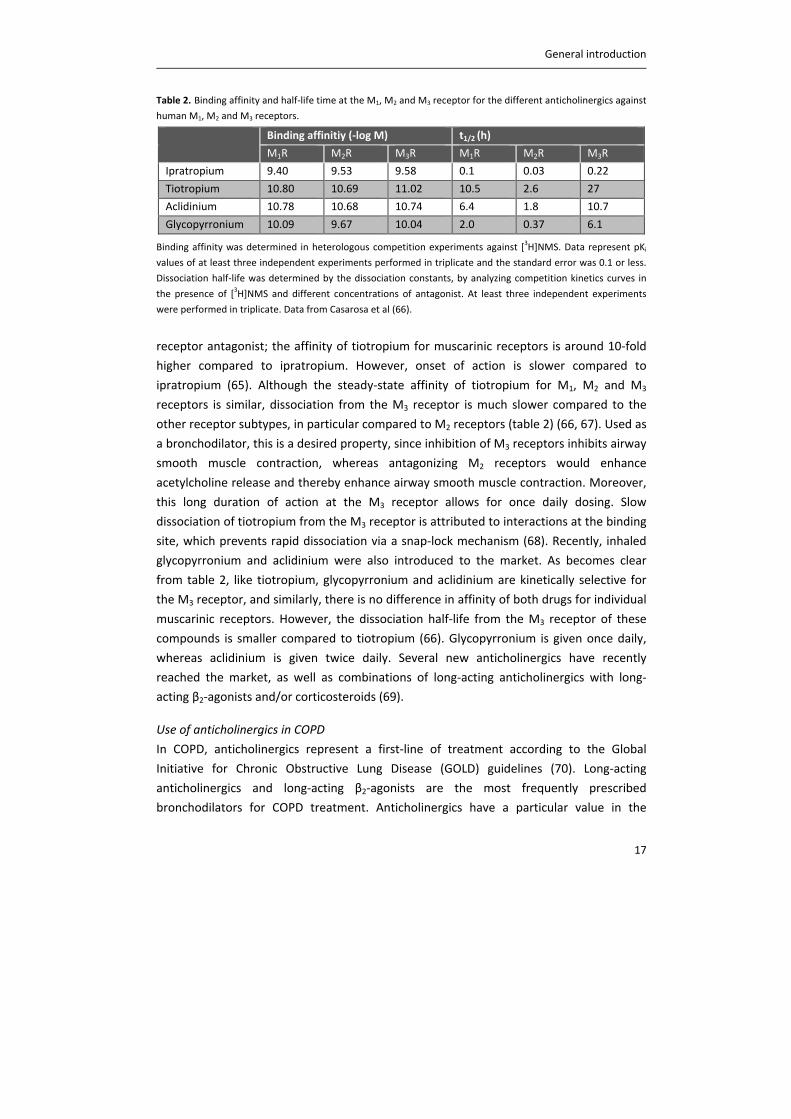
Table 2. Binding affinity and half‐life time at the M1, M2 and M3 receptor for the different anticholinergics against
human M1, M2 and M3 receptors.
Binding affinitiy (‐log M) t1/2 (h)
M1R M2R M3R M1R M2R M3R
Ipratropium 9.40 9.53 9.58 0.1 0.03 0.22
Tiotropium 10.80 10.69 11.02 10.5 2.6 27
Aclidinium 10.78 10.68 10.74 6.4 1.8 10.7
Glycopyrronium 10.09 9.67 10.04 2.0 0.37 6.1
Binding affinity was determined in heterologous competition experiments against [3H]NMS. Data represent pKi
values of at least three independent experiments performed in triplicate and the standard error was 0.1 or less.
Dissociation half‐life was determined by the dissociation constants, by analyzing competition kinetics curves in
the presence of [3H]NMS and different concentrations of antagonist. At least three independent experiments
were performed in triplicate. Data from Casarosa et al (66).
receptor antagonist; the affinity of tiotropium for muscarinic receptors is around 10‐fold
higher compared to ipratropium. However, onset of action is slower compared to
ipratropium (65). Although the steady‐state affinity of tiotropium for M1, M2 and M3
receptors is similar, dissociation from the M3 receptor is much slower compared to the
other receptor subtypes, in particular compared to M2 receptors (table 2) (66, 67). Used as
a bronchodilator, this is a desired property, since inhibition of M3 receptors inhibits airway
smooth muscle contraction, whereas antagonizing M2 receptors would enhance
acetylcholine release and thereby enhance airway smooth muscle contraction. Moreover,
this long duration of action at the M3 receptor allows for once daily dosing. Slow
dissociation of tiotropium from the M3 receptor is attributed to interactions at the binding
site, which prevents rapid dissociation via a snap‐lock mechanism (68). Recently, inhaled
glycopyrronium and aclidinium were also introduced to the market. As becomes clear
from table 2, like tiotropium, glycopyrronium and aclidinium are kinetically selective for
the M3 receptor, and similarly, there is no difference in affinity of both drugs for individual
muscarinic receptors. However, the dissociation half‐life from the M3 receptor of these
compounds is smaller compared to tiotropium (66). Glycopyrronium is given once daily,
whereas aclidinium is given twice daily. Several new anticholinergics have recently
reached the market, as well as combinations of long‐acting anticholinergics with long‐
acting β2‐agonists and/or corticosteroids (69).
Use of anticholinergics in COPD
In COPD, anticholinergics represent a first‐line of treatment according to the Global
Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines (70). Long‐acting
anticholinergics and long‐acting β2‐agonists are the most frequently prescribed
bronchodilators for COPD treatment. Anticholinergics have a particular value in the

Chapter 1
18
treatment of COPD because they block the increased cholinergic tone, which is the major
reversible component of the disease (60, 61). The first randomized controlled clinical trial
into the efficacy and safety of tiotropium in patients with COPD was published in 2000
(71). In this study, tiotropium was compared to ipratropium. Both tiotropium and
ipratropium induced an increase in lung function over a 13 week period, and the long‐
acting tiotropium was more effective compared to the short‐acting ipratropium. The
safety profile of both drugs was similar (71). Since then, a number of studies have been
undertaken, of which the UPLIFT (Understanding Potential Long‐term Impacts on Function
with Tiotropium) trial was the most extensive study. In this study, a total of 5993 patients
were randomly assigned to placebo or tiotropium, and followed for 4 years. Tiotropium
significantly improved lung function and quality of life, and reduced the number of
exacerbations compared to placebo. Tiotropium did not alter the rate of decline in forced
expiratory volume in 1 second (FEV1) (72). More recently, new anticholinergics were
introduced, including glycopyrronium and aclidinium. Glycopyrronium (NVA237) was
shown to improve lung function, quality of life and dyspnoea, and to reduce the risk of
exacerbations compared to placebo in patients with moderate to severe COPD (73), with
efficacy comparable to tiotropium (74). Similarly, aclidinium was shown to improve lung
function, quality of life and dyspnoea compared to placebo in moderate to severe COPD
patients (75, 76), which was comparable to the effects of tiotropium over a 15 day period
(77). Fixed‐dose combinations of several long‐acting anticholinergics and long‐acting β2‐
agonists are currently under development or have recently reached the market, and large
trials are evaluating the efficacy and safety of fixed‐dose combinations (35, 78). First
evidence suggests that the fixed‐dose combinations of tiotropium and olodaterol (79),
glycopyrronium and indacaterol (80, 81), aclidinium and formoterol (82, 83), and
umeclidinium and vilanterol (84, 85) are more effective than monotherapy.
Use of anticholinergics in asthma
In asthma, the use of anticholinergics is limited to the treatment of exacerbations, and
anticholinergics are not approved for chronic treatment (86). Short‐acting anticholinergics
have a significant but small effect on peak expiratory flow (PEF) compared to placebo in
patients with stable asthma (87). The effects of short‐acting anticholinergics are smaller
than the effects of short‐acting β2‐agonists in patients with asthma (64, 87). According to
the Global Initiative for Asthma (GINA) guidelines, ipratropium can be used in stable state
as an alternative bronchodilator to short‐acting β2‐agonists, but is usually less effective. In
case of an exacerbation, the addition of a short‐acting anticholinergic to the short‐acting
β2‐agonists is recommended (86). More recently, clinical trials into the effects of the long‐
acting anticholinergic tiotropium on lung function in patients with asthma have started. In
one of the first studies, Peters et al. demonstrated that the addition of tiotropium to

General introduction
19
glucocorticoid therapy in patients with mild to moderate asthma was more effective than
doubling of the glucocorticoid dose by means of morning peak expiratory flow, the
proportion of asthma‐control days, FEV1 and daily symptom scores, and very similar to the
effects of adding salmeterol (88). Moreover, it was shown in a phase 2 study in patients
with severe uncontrolled asthma that addition of tiotropium to standard therapy with
inhaled corticosteroids and long‐acting β2‐agonists improved lung function over an 8 week
period (89). These findings were confirmed by two replicate phase 3 randomized
controlled trials involving 912 severe asthma patients over a period of 48 weeks.
Tiotropium induced bronchodilation on top of the use of inhaled corticosteroids and long‐
acting β2‐agonists. Furthermore, tiotropium reduced exacerbations and episodes of
worsening of asthma (90). Two phase 2 studies in patients with moderate persistent
asthma demonstrated that tiotropium can also induce bronchodilation in this patient
group, on top of the use of corticosteroids (91, 92). These results have been confirmed in
two large replicate phase 3 trials in 2100 patients with uncontrolled asthma on medium
dose corticosteroids, of which the results have only been presented as abstracts (93).
Tiotropium induced sustained bronchodilation after 24 weeks, accompanied by
improvements in asthma control, all very similar to salmeterol (93). Together these data
indicate that asthma patients might benefit from addition of tiotropium to standard
therapy.
Regulation of inflammation and remodeling by muscarinic receptors
COPD and asthma are associated with airway inflammation and remodeling. Increasing
evidence suggests a role for acetylcholine in inflammation and remodeling in both
obstructive airway diseases (16, 21). This suggests that the role of acetylcholine in patients
with COPD or asthma might be much broader than previously appreciated.
Inflammation and remodeling in COPD
COPD is associated with persistent inflammation and remodeling of the airways.
Inflammation is present throughout the airways (94), and the degree of inflammation
correlates to the severity of the disease (95). Upon inhalation of cigarette smoke, which is
the main risk factor for COPD, epithelial cells are activated and macrophages are
attracted, resulting in the release of chemotactic factors. Macrophages are thought to
orchestrate the inflammatory response in COPD (96). Amongst others, macrophages
release CC‐chemokine ligand 2 (CCL2), also known as monocyte chemotactic protein
(MCP1), to attract more monocytes to the lung, and CXC‐chemokine ligand (CXCL)‐1 and
CXCL‐8, to attract neutrophils (94, 96). Moreover, both macrophages and epithelial cells
release CXCL9, CXCL10 and CXCL11, which attract T‐cells to the lung (94, 97). In COPD, T‐

Chapter 1
20
cells are mainly T‐helper type 1 (TH1) and type 1 cytotoxic T (TC1) CD8+ cells (94). The
inflammatory response is thought to contribute to the remodeling of the airways, by the
release of TGF‐β and proteases such as matrix metalloproteinase 9 (MMP9) (94, 98).
Fibrosis, by means of peribronchiolar deposition of extracellular matrix proteins, mainly
occurs around the small airways, and is an important factor contributing to the irreversible
airway narrowing observed in COPD (99). Moreover, alveolar walls in the lung parenchyma
are destructed due to the chronic inflammation, which is called emphysema. Emphysema
results in enlargement of parenchymal airspaces and loss of elastic recoil, and is a major
contributor to morbidity and mortality in COPD (100, 101). Remodeling of the epithelium
also occurs, leading to squamous and mucous metaplasia (100). Together with mucus
gland hypertrophy, this leads to mucus hypersecretion in patients with COPD (102, 103).
Remodeling of the airways starts later in life in patients with COPD, but is progressive and
irreversible, and is the major contributor to the decline in lung function (100).
Inflammation and remodeling in asthma
Like COPD, asthma is characterized by airway inflammation and remodeling. However,
there are marked differences between the pattern of inflammation and remodeling in
asthma compared to COPD. In asthma, inflammation and remodeling is mainly present in
the larger conducting airways. Depending on the severity of disease, small airways can
also be affected, however, the lung parenchyma is generally not affected in most patients
with asthma (94). The nature of the inflammation and the cell types involved in the
inflammatory response in asthma are different compared to COPD. Upon allergen
challenge, which is the main trigger for an inflammatory response in allergic asthma, mast
cells are activated. Mast cells release bronchoconstricting agents, including histamine and
lipid mediators. Moreover, CD4+ T‐helper type 2 (TH2) cytokines IL‐4, IL‐5 and IL‐13 are
released by mast cells (94, 97). In addition to mast cells, TH2 cells also release these
cytokines, and TH2 cells play a central role in orchestrating the inflammatory response in
asthma (94). IL‐4 release stimulates B cells to synthesize IgE, the driver of allergic
inflammation that binds to high‐affinity Fc receptors for IgE (FcεRI) on mast cells. The
release of IL‐5 attracts eosinophils, whereas the release of IL‐13 contributes to an
enhancement of airway hyperresponsiveness and an increase in goblet cell number (104,
105). There is also an increase in the size of submucosal glands (106), and together this
leads to excessive mucus production. Enhanced mucus secretion significantly contributes
to airflow limitation in asthma by obstructing the airways (41, 107). Furthermore, there is
thickening of the basement‐membrane, as a result of collagen deposition under the
epithelium, and thickening of the airway smooth muscle layer, as a result of airway
smooth muscle hyperplasia and hypertrophy (100). The former is observed in all patients
with asthma, whereas the latter is mainly observed in patients with severe asthma (108).

General introduction
21
Airway smooth muscle thickness is related to severity of the disease, but not to its
duration, since airway smooth muscle thickening is already present in children with
asthma (109, 110). Pulmonary vascular remodeling and increased angiogenesis are also
observed, which is mediated by various angiogenic proteins, including several growth
factors, like vascular endothelial growth factor (VEGF) (100, 111). Structural changes in the
airways of patients with asthma are considered an important component of airflow
limitation and may accelerate the decline in lung function (112, 113).
Muscarinic receptor regulation of inflammation and remodeling
Increasing evidence suggests a role for acetylcholine in regulating airway inflammation
and remodeling. From in vitro studies it is known that muscarinic receptor stimulation can
enhance the release of cytokines from inflammatory cells and structural cells of the
airways, either alone or in concerted action with stimuli including cigarette smoke and
growth factors. Moreover, acetylcholine has been shown to enhance remodeling
parameters in vitro. Thus, muscarinic receptor stimulation of airway smooth muscle cells
and fibroblast enhances proliferation and the production of extracellular matrix proteins
(16, 21). This is further supported by evidence from in vivo studies, demonstrating that
airway inflammation and remodeling can be inhibited by anticholinergic intervention. It
has been shown that cigarette smoke‐induced inflammation in mice, and LPS‐induced
inflammation and remodeling in guinea pigs can be inhibited by tiotropium, indicating the
potential relevance of anticholinergic treatment for patients with COPD (114, 115). In
addition, allergen‐induced inflammation and remodeling can be inhibited by tiotropium in
both mice and guinea pigs, indicating the potential relevance of anticholinergic treatment
for patients with asthma (116‐118). The role of acetylcholine in inflammation and
remodeling is extensively reviewed in chapter 2, and will therefore not be elaborated on
in more detail here.
Clinical implications
As briefly described above and reviewed in chapter 2, there is convincing evidence from in
vitro and in vivo animal studies that acetylcholine contributes to airway inflammation and
remodeling. This might be relevant for patients with COPD and asthma, especially since
acetylcholine release is enhanced in these disease states. However, this hypothesis still
needs to be proven by clinical studies investigating the effect of acetylcholine or
anticholinergic therapy on airway inflammation and remodeling. In the UPLIFT study,
COPD patients treated with tiotropium for 4 years showed improvements in lung function,
quality of life and exacerbation frequency (72). The latter might suggest an anti‐
inflammatory effect of tiotropium, as the inflammatory response is enhanced during
exacerbations, with increased expression of pro‐inflammatory cytokines, including IL‐8

Chapter 1
22
(119, 120). However, Powrie et al. were not able to demonstrate a reduction in sputum IL‐
6 or IL‐8 levels after tiotropium use for one year, even though the exacerbation frequency
was reduced (121). In this study, tiotropium reduced the amount of sputum, which might
result in a concentration of cytokines in the sputum of patients treated with tiotropium, as
proposed by the authors to explain this discrepancy. Therefore, further studies are needed
to elucidate the mechanism by which tiotropium reduces the number of exacerbations in
patients with COPD and whether a direct anti‐inflammatory effect of tiotropium is
involved. Structural changes in the airways of COPD patients may accelerate the decline of
lung function. Initially, it was thought that tiotropium might affect this process, since a
retrospective study suggested that the use of tiotropium was associated with a reduction
in decline of lung function after 1 year (122). However, this was not replicated in the large,
prospective UPLIFT trial, in which no significant effect on the rate of decline in lung
function was observed in the overall study population (72). Pre‐specified post‐hoc
subgroup analysis revealed that tiotropium did inhibit the accelerated decline in lung
function in young patients and in patients with moderate disease (123, 124). Future
studies are needed to understand the effects of tiotropium on lung function decline in
patients with COPD in these subgroups.
Recently, clinical studies into the effects of tiotropium in patients with asthma have
started. Addition of tiotropium to standard therapy for severe asthma patients with long‐
acting β‐agonists and corticosteroids increased the time to the first exacerbation and
reduced the risk of a severe exacerbation (90). Moreover, it has been shown that repeated
inhalation of the muscarinic receptor agonist methacholine induces airway remodeling in
mild asthma patients (125). This suggests that acetylcholine might also affect airway
inflammation and remodeling in patients with asthma, but clearly more studies are
needed to further elucidate the effects of acetylcholine or anticholinergic therapy on
airway inflammation and remodeling in patients with asthma.
Scope of the thesis
The above mentioned findings suggest that acetylcholine is not only a neurotransmitter
involved in bronchoconstriction and mucus secretion in the airways, but might also be a
mediator of airway inflammation and remodeling, which possibly involves non‐neuronal
acetylcholine. This will have implications for therapy of obstructive airways diseases like
COPD and asthma, in which muscarinic antagonists are frequently used. This thesis will
further elucidate the role of acetylcholine in inflammation and remodeling in COPD and
asthma. Specifically, the aim of this thesis is to investigate the role of individual muscarinic

General introduction
23
receptor subtypes, as well as the contribution of neuronal versus non‐neuronal
acetylcholine, to airway inflammation and remodeling in COPD and asthma.
Chapter 2 provides a comprehensive review on the regulation of airway inflammation and
remodeling in COPD and asthma by muscarinic receptors. In addition, therapeutic
implications of muscarinic receptor regulation of inflammation and remodeling are
discussed.
The following chapters deal with the role of acetylcholine in inflammation in COPD. As
stated above, there is evidence for a pro‐inflammatory role of acetylcholine in
inflammation. However, it is not known which individual muscarinic receptor subtypes are
involved in this response. There is a focus for anticholinergics on M3 receptor selectivity,
because of involvement of this receptor subtype in bronchoconstriction, however, there is
no evidence that the pro‐inflammatory effects of acetylcholine are also mediated via this
receptor subtype. Therefore, in chapter 3, we investigated the contribution of individual
muscarinic receptor subtypes to cigarette smoke‐induced inflammation. Because of
limited selectivity of available muscarinic receptor antagonists, muscarinic receptor
subtype specific knock‐out animals were used. In this way, the contribution of individual
muscarinic receptors can be investigated in detail. Knock‐out animals were exposed to
cigarette smoke for four days and the inflammatory response in the airways was analyzed.
The contribution of the M3 receptor to cigarette smoke‐induced inflammation was
investigated in more detail in chapter 4. As discussed above, practically all cells in the
airways express muscarinic receptors. It is not known which cell types contribute to the
pro‐inflammatory effect of the M3 receptor as described in chapter 3. To distinguish
between effects of structural cells and inflammatory cells, bone marrow chimeric animals
were generated and exposed to cigarette smoke, using a similar protocol as described in
chapter 3. In this way, the contribution of structural cells versus inflammatory cells to the
pro‐inflammatory effect of the M3 receptor can be defined.
In chapter 5, we further elucidated the role of acetylcholine in inflammation in patients
with COPD. In this chapter, we investigated the effects of targeted lung denervation (TLD)
on inflammation in patients with COPD. TLD is a novel potential therapy for patients with
COPD, in which parasympathetic airway nerves are ablated by locally applying radio‐
frequency energy in the main bronchi using bronchoscopy, to inhibit acetylcholine‐
induced bronchoconstriction. We hypothesized that TLD would also inhibit airway
inflammation. Specifically, we studied whether TLD affects inflammatory cell number and
pro‐inflammatory cytokine expression in bronchial wash fluid and gene expression of pro‐
inflammatory cytokines in bronchial brush specimen.

Chapter 1
24
The second half of the thesis is focused on the role of acetylcholine in inflammation and
remodeling in asthma. As is described above for COPD, it is not known which muscarinic
receptor subtypes mediate inflammation and remodeling in asthma. Therefore, in chapter
6, we investigated the effects of individual muscarinic receptor subtypes on allergen‐
induced inflammation and remodeling. To this aim, muscarinic receptor subtype specific
knock‐out animals were exposed to ovalbumin, and lungs were collected for analysis of
inflammation and remodeling.
Increasing evidence suggests that airway remodeling might not only be a consequence of
inflammation, but might also or alternatively be a consequence of bronchoconstriction,
which triggers TGF‐β release via biomechanical activation. In chapter 7, we aimed to
investigate the effect of TGF‐β and bronchoconstriction on remodeling, and the
involvement of the M3 receptor in this response, by using murine precision cut lung slices
(PCLS). TGF‐β was used to induce remodeling in PCLS from wild‐type animals. In addition,
PCLS were stimulated with methacholine, to investigate the effect of bronchoconstriction
on remodeling. Moreover, PCLS from M3R‐/‐ mice, in which bronchoconstriction is
abolished, were exposed to TGF‐β and methacholine, to investigate the involvement of
the M3 receptor in this response. After two days, expression of remodeling parameters, as
well as the functional impact on bronchoconstriction, was analyzed.
In chapter 8, we investigated the effects of endogenous non‐neuronal acetylcholine on
epithelial cell differentiation. There is evidence from in vivo studies which suggests an
indirect role for acetylcholine in epithelial cell differentiation and goblet cell metaplasia.
Here, we aimed to investigate direct effects of endogenous non‐neuronal acetylcholine on
epithelial cell differentiation and possible mechanisms involved. Human airway epithelial
cells were isolated from healthy donors and cultured at an air‐liquid interface. During
differentiation at the air‐liquid interface, cells were exposed to tiotropium, to investigate
the effects of acetylcholine on epithelial cell differentiation following air exposure, and to
IL‐13 and tiotropium, to investigate the effects of acetylcholine on IL‐13‐induced goblet
cell metaplasia.
Finally, in chapter 9, the results of this thesis are summarized and discussed. Perspectives
for future studies are provided.

General introduction
25
References
(1) Racke K, Matthiesen S. The airway cholinergic system: physiology and pharmacology. Pulm
Pharmacol Ther 2004;17:181‐198.
(2) Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol
2008;130:219‐234.
(3) Kummer W, Krasteva‐Christ G. Non‐neuronal cholinergic airway epithelium biology. Curr Opin
Pharmacol 2014;16C:43‐49.
(4) Colebatch HJ, Halmagyi DF. Effect of Vagotomy and Vagal Stimulation on Lung Mechanics and
Circulation. J Appl Physiol 1963;18:881‐887.
(5) Widdicombe JG, Nadel JA. Airway Volume, Airway Resistance, and Work and Force of Breathing:
Theory. J Appl Physiol 1963;18:863‐868.
(6) Undem B, Myers A. Cholinergic and noncholinergic parasympathetic control of airway smooth
muscle. In: Zaagsma J, Meurs H and Roffel AF, editors. Muscarinic receptors in airways diseases.
Basel: Birkhauser; 2001. p. 1‐‐24.
(7) Undem BJ, Kollarik M. The role of vagal afferent nerves in chronic obstructive pulmonary disease.
Proc Am Thorac Soc 2005;2:355‐60; discussion 371‐2.
(8) Sant'Ambrogio G, Widdicombe J. Reflexes from airway rapidly adapting receptors. Respir Physiol
2001;125:33‐45.
(9) Undem BJ, Carr MJ. The role of nerves in asthma. Curr Allergy Asthma Rep 2002;2:159‐165.
(10) Agostoni E, Chinnock JE, De Daly MB, Murray JG. Functional and histological studies of the vagus
nerve and its branches to the heart, lungs and abdominal viscera in the cat. J Physiol 1957;135:182‐
205.
(11) Coleridge JC, Coleridge HM. Afferent vagal C fibre innervation of the lungs and airways and its
functional significance. Rev Physiol Biochem Pharmacol 1984;99:1‐110.
(12) Verhein KC, Fryer AD, Jacoby DB. Neural control of airway inflammation. Curr Allergy Asthma
Rep 2009;9:484‐490.
(13) Canning BJ, Fischer A. Neural regulation of airway smooth muscle tone. Respir Physiol
2001;125:113‐127.
(14) Wessler I, Kirkpatrick CJ, Racke K. The cholinergic 'pitfall': acetylcholine, a universal cell
molecule in biological systems, including humans. Clin Exp Pharmacol Physiol 1999;26:198‐205.
(15) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008;154:1558‐1571.
(16) Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology
of asthma and COPD. Respir.Res. 2006;7:73.
(17) Klapproth H, Reinheimer T, Metzen J, Munch M, Bittinger F, Kirkpatrick CJ, Hohle KD, Schemann
M, Racke K, Wessler I. Non‐neuronal acetylcholine, a signalling molecule synthezised by surface cells
of rat and man. Naunyn Schmiedebergs Arch Pharmacol 1997;355:515‐523.
(18) Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is
an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells.
Endocrinology 2004;145:2498‐2506.

Chapter 1
26
(19) Gwilt CR, Donnelly LE, Rogers DF. The non‐neuronal cholinergic system in the airways: an
unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208‐222.
(20) Pieper MP. The non‐neuronal cholinergic system as novel drug target in the airways. Life Sci
2012;91:1113‐1118.
(21) Kistemaker LE, Oenema TA, Meurs H, Gosens R. Regulation of airway inflammation and
remodeling by muscarinic receptors: perspectives on anticholinergic therapy in asthma and COPD.
Life Sci 2012;91:1126‐1133.
(22) Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol
2006;533:57‐68.
(23) Schuller HM. Regulatory role of the alpha7nAChR in cancer. Curr Drug Targets 2012;13:680‐687.
(24) Jositsch G, Papadakis T, Haberberger RV, Wolff M, Wess J, Kummer W. Suitability of muscarinic
acetylcholine receptor antibodies for immunohistochemistry evaluated on tissue sections of
receptor gene‐deficient mice. Naunyn Schmiedebergs Arch Pharmacol 2009;379:389‐395.
(25) Pradidarcheep W, Stallen J, Labruyere WT, Dabhoiwala NF, Michel MC, Lamers WH. Lack of
specificity of commercially available antisera against muscarinergic and adrenergic receptors.
Naunyn Schmiedebergs Arch Pharmacol 2009;379:397‐402.
(26) Ikeda T, Anisuzzaman AS, Yoshiki H, Sasaki M, Koshiji T, Uwada J, Nishimune A, Itoh H,
Muramatsu I. Regional quantification of muscarinic acetylcholine receptors and beta‐adrenoceptors
in human airways. Br J Pharmacol 2012;166:1804‐1814.
(27) Profita M, Bonanno A, Siena L, Bruno A, Ferraro M, Montalbano AM, Albano GD, Riccobono L,
Casarosa P, Pieper MP, Gjomarkaj M. Smoke, choline acetyltransferase, muscarinic receptors, and
fibroblast proliferation in chronic obstructive pulmonary disease. J Pharmacol Exp Ther
2009;329:753‐763.
(28) Radosa J, Dyck W, Goerdt S, Kurzen H. The cholinergic system in guttate psoriasis with special
reference to mast cells. Exp Dermatol 2011;20:677‐679.
(29) Wallon C, Persborn M, Jonsson M, Wang A, Phan V, Lampinen M, Vicario M, Santos J, Sherman
PM, Carlson M, Ericson AC, McKay DM, Soderholm JD. Eosinophils express muscarinic receptors and
corticotropin‐releasing factor to disrupt the mucosal barrier in ulcerative colitis. Gastroenterology
2011;140:1597‐1607.
(30) Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science
1991;252:802‐808.
(31) Neves SR, Ram PT, Iyengar R. G protein pathways. Science 2002;296:1636‐1639.
(32) Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic
acetylcholine receptors. Pharmacol Rev 1998;50:279‐290.
(33) Meurs H, Roffel AF, Elzinga CRS, Zaagsma J. Muscarinic receptor‐beta‐adrenaceptor cross‐talk in
airway smooth muscle. In: Zaagsma J, Meurs H and Roffel AF, editors. Muscarinic receptors in
airways diseases. Basel: Birkhauser; 2001. p. 121‐‐158.
(34) Zhou XB, Wulfsen I, Lutz S, Utku E, Sausbier U, Ruth P, Wieland T, Korth M. M2 muscarinic
receptors induce airway smooth muscle activation via a dual, Gbetagamma‐mediated inhibition of
large conductance Ca2+‐activated K+ channel activity. J Biol Chem 2008;283:21036‐21044.
(35) Pera T, Penn RB. Crosstalk between beta‐2‐adrenoceptor and muscarinic acetylcholine
receptors in the airway. Curr Opin Pharmacol 2014;16C:72‐81.

General introduction
27
(36) Kilbinger H, Schneider R, Siefken H, Wolf D, D'Agostino G. Characterization of prejunctional
muscarinic autoreceptors in the guinea‐pig trachea. Br J Pharmacol 1991;103:1757‐1763.
(37) ten Berge RE, Zaagsma J, Roffel AF. Muscarinic inhibitory autoreceptors in different generations
of human airways. Am J Respir Crit Care Med 1996;154:43‐49.
(38) On LS, Boonyongsunchai P, Webb S, Davies L, Calverley PM, Costello RW. Function of pulmonary
neuronal M(2) muscarinic receptors in stable chronic obstructive pulmonary disease. Am J Respir
Crit Care Med 2001;163:1320‐1325.
(39) Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in
bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol
1988;153:73‐82.
(40) Fisher JT, Vincent SG, Gomeza J, Yamada M, Wess J. Loss of vagally mediated bradycardia and
bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J
2004;18:711‐713.
(41) Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol 2001;125:129‐144.
(42) Ramnarine SI, Haddad EB, Khawaja AM, Mak JC, Rogers DF. On muscarinic control of neurogenic
mucus secretion in ferret trachea. J Physiol 1996;494 ( Pt 2):577‐586.
(43) Baker B, Peatfield AC, Richardson PS. Nervous control of mucin secretion into human bronchi. J
Physiol 1985;365:297‐305.
(44) Spina D, Shah S, Harrison S. Modulation of sensory nerve function in the airways. Trends
Pharmacol Sci 1998;19:460‐466.
(45) Undem BJ, Carr MJ. Pharmacology of airway afferent nerve activity. Respir Res 2001;2:234‐244.
(46) Gleich GJ, Flavahan NA, Fujisawa T, Vanhoutte PM. The eosinophil as a mediator of damage to
respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol 1988;81:776‐
781.
(47) ten Berge RE, Santing RE, Hamstra JJ, Roffel AF, Zaagsma J. Dysfunction of muscarinic M2
receptors after the early allergic reaction: possible contribution to bronchial hyperresponsiveness in
allergic guinea‐pigs. Br J Pharmacol 1995;114:881‐887.
(48) Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits
reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol (1985)
1989;67:2461‐2465.
(49) Coulson FR, Fryer AD. Muscarinic acetylcholine receptors and airway diseases. Pharmacol Ther
2003;98:59‐69.
(50) Evans CM, Fryer AD, Jacoby DB, Gleich GJ, Costello RW. Pretreatment with antibody to
eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal M2 muscarinic
receptors in antigen‐challenged guinea pigs. J Clin Invest 1997;100:2254‐2262.
(51) Mitchell RW, Kelly E, Leff AR. Reduced activity of acetylcholinesterase in canine tracheal smooth
muscle homogenates after active immune‐sensitization. Am J Respir Cell Mol Biol 1991;5:56‐62.
(52) Haddad EB, Mak JC, Belvisi MG, Nishikawa M, Rousell J, Barnes PJ. Muscarinic and beta‐
adrenergic receptor expression in peripheral lung from normal and asthmatic patients. Am J Physiol
1996;270:L947‐53.

Chapter 1
28
(53) van Koppen CJ, Lammers JW, Rodrigues de Miranda JF, Beld AJ, van Herwaarden CL, van
Ginneken CA. Muscarinic receptor binding in central airway musculature in chronic airflow
obstruction. Pulm Pharmacol 1989;2:131‐136.
(54) Fenech AG, Ebejer MJ, Felice AE, Ellul‐Micallef R, Hall IP. Mutation screening of the muscarinic
M(2) and M(3) receptor genes in normal and asthmatic subjects. Br J Pharmacol 2001;133:43‐48.
(55) Nockher WA, Renz H. Neurotrophins and asthma: novel insight into neuroimmune interaction. J
Allergy Clin Immunol 2006;117:67‐71.
(56) Pan J, Rhode HK, Undem BJ, Myers AC. Neurotransmitters in airway parasympathetic neurons
altered by neurotrophin‐3 and repeated allergen challenge. Am J Respir Cell Mol Biol 2010;43:452‐
457.
(57) Hunter DD, Wu Z, Dey RD. Sensory neural responses to ozone exposure during early postnatal
development in rat airways. Am J Respir Cell Mol Biol 2010;43:750‐757.
(58) Aven L, Paez‐Cortez J, Achey R, Krishnan R, Ram‐Mohan S, Cruikshank WW, Fine A, Ai X. An
NT4/TrkB‐dependent increase in innervation links early‐life allergen exposure to persistent airway
hyperreactivity. FASEB J 2014;28:897‐907.
(59) Larson SD, Schelegle ES, Walby WF, Gershwin LJ, Fanuccihi MV, Evans MJ, Joad JP, Tarkington
BK, Hyde DM, Plopper CG. Postnatal remodeling of the neural components of the epithelial‐
mesenchymal trophic unit in the proximal airways of infant rhesus monkeys exposed to ozone and
allergen. Toxicol Appl Pharmacol 2004;194:211‐220.
(60) Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to
emphysema. N Engl J Med 1984;311:421‐425.
(61) Barnes PJ. The role of anticholinergics in chronic obstructive pulmonary disease. Am J Med
2004;117 Suppl 12A:24S‐32S.
(62) Jackson M. "Divine stramonium": the rise and fall of smoking for asthma. Med Hist 2010;54:171‐
194.
(63) Meurs H, Oenema TA, Kistemaker LE, Gosens R. A new perspective on muscarinic receptor
antagonism in obstructive airways diseases. Curr Opin Pharmacol 2013;13:316‐323.
(64) Gross NJ. Anticholinergic agents in asthma and COPD. Eur J Pharmacol 2006;533:36‐39.
(65) Barnes PJ. The pharmacological properties of tiotropium. Chest 2000;117:63S‐6S.
(66) Casarosa P, Bouyssou T, Germeyer S, Schnapp A, Gantner F, Pieper M. Preclinical evaluation of
long‐acting muscarinic antagonists: comparison of tiotropium and investigational drugs. J Pharmacol
Exp Ther 2009;330:660‐668.
(67) Disse B, Speck GA, Rominger KL, Witek TJ,Jr., Hammer R. Tiotropium (Spiriva): mechanistical
considerations and clinical profile in obstructive lung disease. Life Sci 1999;64:457‐464.
(68) Tautermann CS, Kiechle T, Seeliger D, Diehl S, Wex E, Banholzer R, Gantner F, Pieper MP,
Casarosa P. Molecular basis for the long duration of action and kinetic selectivity of tiotropium for
the muscarinic M3 receptor. J Med Chem 2013;56:8746‐8756.
(69) Matera MG, Rogliani P, Cazzola M. Muscarinic receptor antagonists for the treatment of chronic
obstructive pulmonary disease. Expert Opin Pharmacother 2014;15:961‐977.
(70) Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis,
Management and Prevention of COPD (GOLD) 2013. Available from:
http://www.goldcopd.org/uploads/users/files/GOLD_Report_2014_Jan23.pdf. ;June 4, 2014.

General introduction
29
(71) van Noord JA, Bantje TA, Eland ME, Korducki L, Cornelissen PJ. A randomised controlled
comparison of tiotropium nd ipratropium in the treatment of chronic obstructive pulmonary
disease. The Dutch Tiotropium Study Group. Thorax 2000;55:289‐294.
(72) Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M, UPLIFT Study
Investigators. A 4‐year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med
2008;359:1543‐1554.
(73) D'Urzo A, Ferguson GT, van Noord JA, Hirata K, Martin C, Horton R, Lu Y, Banerji D, Overend T.
Efficacy and safety of once‐daily NVA237 in patients with moderate‐to‐severe COPD: the GLOW1
trial. Respir Res 2011;12:156‐9921‐12‐156.
(74) Kerwin E, Hebert J, Gallagher N, Martin C, Overend T, Alagappan VK, Lu Y, Banerji D. Efficacy and
safety of NVA237 versus placebo and tiotropium in patients with COPD: the GLOW2 study. Eur
Respir J 2012;40:1106‐1114.
(75) Rennard SI, Scanlon PD, Ferguson GT, Rekeda L, Maurer BT, Garcia Gil E, Caracta CF. ACCORD
COPD II: a randomized clinical trial to evaluate the 12‐week efficacy and safety of twice‐daily
aclidinium bromide in chronic obstructive pulmonary disease patients. Clin Drug Investig
2013;33:893‐904.
(76) Jones PW, Singh D, Bateman ED, Agusti A, Lamarca R, de Miquel G, Segarra R, Caracta C, Garcia
Gil E. Efficacy and safety of twice‐daily aclidinium bromide in COPD patients: the ATTAIN study. Eur
Respir J 2012;40:830‐836.
(77) Fuhr R, Magnussen H, Sarem K, Llovera AR, Kirsten AM, Falques M, Caracta CF, Garcia Gil E.
Efficacy of aclidinium bromide 400 mug twice daily compared with placebo and tiotropium in
patients with moderate to severe COPD. Chest 2012;141:745‐752.
(78) Bateman ED, Mahler DA, Vogelmeier CF, Wedzicha JA, Patalano F, Banerji D. Recent advances in
COPD disease management with fixed‐dose long‐acting combination therapies. Expert Rev Respir
Med 2014;8:357‐379.
(79) Derom E, Westerman J, Groenke L, Hamilton A, Li C, Beeh K. The 24‐Hour Lung Function Profile
Of Once‐Daily Tiotropium And Olodaterol Fixed‐Dose Combination Compared With Placebo And
Monotherapies In Chronic Obstructive Pulmonary Disease [abstract]. Am J Respir Crit Care Med
2014;189:A6727.
(80) Bateman ED, Ferguson GT, Barnes N, Gallagher N, Green Y, Henley M, Banerji D. Dual
bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. Eur Respir J
2013;42:1484‐1494.
(81) Frampton JE. QVA149 (Indacaterol/Glycopyrronium Fixed‐Dose Combination): A Review of Its
Use in Patients with Chronic Obstructive Pulmonary Disease. Drugs 2014;74:465‐488.
(82) Cazzola M, Rogliani P, Matera MG. Aclidinium bromide/formoterol fumarate fixed‐dose
combination for the treatment of chronic obstructive pulmonary disease. Expert Opin Pharmacother
2013;14:775‐781.
(83) Singh D, Jones PW, Bateman ED, Korn S, Serra C, Molins E, Caracta C, Leselbaum A.
Aclidinium/formoterol Fixed‐Dose Combination Improves Bronchodilation And Is Well Tolerated In
Patients With COPD: Results From The Acliform/COPD Study [abstract]. Am J Respir Crit Care Med
2014;189:A5987.

Chapter 1
30
(84) Decramer M, Anzueto A, Kerwin E, Kaelin T, Richard N, Crater G, Tabberer M, Harris S, Church A.
Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium
monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from
two multicentre, blinded, randomised controlled trials. Lancet Respir Med 2014;2:472‐486.
(85) Gras J. Umeclidinium/vilanterol fixed‐dose combination for COPD. Drugs Today (Barc)
2014;50:231‐238.
(86) Global Initiative for Asthma (GINA). GINA Report, Global Strategy for Asthma Management and
Prevention. Available from: www.ginasthma.org. 2014;June 4, 2014.
(87) Westby M, Benson M, Gibson P. Anticholinergic agents for chronic asthma in adults. Cochrane
Database Syst Rev 2004;(3):CD003269.
(88) Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT, Boushey HA, Calhoun
WJ, Castro M, Cherniack RM, Craig T, Denlinger L, Engle LL, DiMango EA, Fahy JV, Israel E, Jarjour N,
Kazani SD, Kraft M, Lazarus SC, Lemanske RF,Jr, Lugogo N, Martin RJ, Meyers DA, Ramsdell J,
Sorkness CA, Sutherland ER, Szefler SJ, Wasserman SI, Walter MJ, Wechsler ME, Chinchilli VM,
Bleecker ER, National Heart, Lung, and Blood Institute Asthma Clinical Research Network.
Tiotropium bromide step‐up therapy for adults with uncontrolled asthma. N Engl J Med
2010;363:1715‐1726.
(89) Kerstjens HA, Disse B, Schroder‐Babo W, Bantje TA, Gahlemann M, Sigmund R, Engel M, van
Noord JA. Tiotropium improves lung function in patients with severe uncontrolled asthma: a
randomized controlled trial. J Allergy Clin Immunol 2011;128:308‐314.
(90) Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, Sigmund R, Seibold W,
Moroni‐Zentgraf P, Bateman ED. Tiotropium in asthma poorly controlled with standard combination
therapy. N Engl J Med 2012;367:1198‐1207.
(91) Bateman ED, Kornmann O, Schmidt P, Pivovarova A, Engel M, Fabbri LM. Tiotropium is
noninferior to salmeterol in maintaining improved lung function in B16‐Arg/Arg patients with
asthma. J Allergy Clin Immunol 2011;128:315‐322.
(92) Beeh KM, Moroni‐Zentgraf P, Ablinger O, Hollaenderova Z, Unseld A, Engel M, Korn S.
Tiotropium Respimat(R) in asthma: a double‐blind, randomised, dose‐ranging study in adult patients
with moderate asthma. Respir Res 2014;15:61.
(93) Kerstjens HAM, Bleecker E, Meltzer E, Casale T, Pizzichini E, Schmidt O, Engel M, Bour LJ, Verkleij
CB, Moroni‐Zentgraf PM, Bateman ED. Tiotropium as add‐on therapy to inhaled corticosteroids for
patients with symptomatic asthma: Lung function and safety. ERJ 2013;42: Suppl 57:4629.
(94) Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol
2008;8:183‐192.
(95) Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp CE, Fabbri LM,
Donner CF, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation
in smokers. Am J Respir Crit Care Med 1998;158:1277‐1285.
(96) Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD 2004;1:59‐70.
(97) Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin
Invest 2008;118:3546‐3556.
(98) Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 2000;343:269‐280.

General introduction
31
(99) Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM,
Sciurba FC, Coxson HO, Pare PD. The nature of small‐airway obstruction in chronic obstructive
pulmonary disease. N Engl J Med 2004;350:2645‐2653.
(100) Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care
Med 2001;164:S28‐S38.
(101) Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez‐
Roisin R, van Weel C, Zielinski J, Global Initiative for Chronic Obstructive Lung Disease. Global
strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease:
GOLD executive summary. Am J Respir Crit Care Med 2007;176:532‐555.
(102) Caramori G, Di Gregorio C, Carlstedt I, Casolari P, Guzzinati I, Adcock IM, Barnes PJ, Ciaccia A,
Cavallesco G, Chung KF, Papi A. Mucin expression in peripheral airways of patients with chronic
obstructive pulmonary disease. Histopathology 2004;45:477‐484.
(103) Saetta M, Turato G, Baraldo S, Zanin A, Braccioni F, Mapp CE, Maestrelli P, Cavallesco G, Papi
A, Fabbri LM. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers
with both symptoms of chronic bronchitis and chronic airflow limitation. Am J Respir Crit Care Med
2000;161:1016‐1021.
(104) Corren J. Role of interleukin‐13 in asthma. Curr Allergy Asthma Rep 2013;13:415‐420.
(105) Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med 2012;18:684‐692.
(106) Ordonez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y,
Novikov A, Dolganov G, Fahy JV. Mild and moderate asthma is associated with airway goblet cell
hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med 2001;163:517‐
523.
(107) Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med 2006;12:1‐6.
(108) Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations
selectively associated with severe asthma. Am J Respir Crit Care Med 2003;167:1360‐1368.
(109) James AL, Bai TR, Mauad T, Abramson MJ, Dolhnikoff M, McKay KO, Maxwell PS, Elliot JG,
Green FH. Airway smooth muscle thickness in asthma is related to severity but not duration of
asthma. Eur Respir J 2009;34:1040‐1045.
(110) Payne DN, Rogers AV, Adelroth E, Bandi V, Guntupalli KK, Bush A, Jeffery PK. Early thickening of
the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med
2003;167:78‐82.
(111) Hoshino M, Nakamura Y, Hamid QA. Gene expression of vascular endothelial growth factor
and its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol 2001;107:1034‐1038.
(112) An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman
DH, Fabry B, Fairbank NJ, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gosens R, Gunst SJ,
Halayko AJ, Ingram RH, Irvin CG, James AL, Janssen LJ, King GG, Knight DA, Lauzon AM, Lakser OJ,
Ludwig MS, Lutchen KR, Maksym GN, Martin JG, Mauad T, McParland BE, Mijailovich SM, Mitchell
HW, Mitchell RW, Mitzner W, Murphy TM, Pare PD, Pellegrino R, Sanderson MJ, Schellenberg RR,
Seow CY, Silveira PS, Smith PG, Solway J, Stephens NL, Sterk PJ, Stewart AG, Tang DD, Tepper RS,
Tran T, Wang L. Airway smooth muscle dynamics: a common pathway of airway obstruction in
asthma. Eur Respir J 2007;29:834‐860.

Chapter 1
32
(113) Pare PD, Roberts CR, Bai TR, Wiggs BJ. The functional consequences of airway remodeling in
asthma. Monaldi Arch Chest Dis 1997;52:589‐596.
(114) Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J,
Meurs H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of
COPD. Eur Respir J 2011;38:789‐796.
(115) Wollin L, Pieper MP. Tiotropium bromide exerts anti‐inflammatory activity in a cigarette smoke
mouse model of COPD. Pulm Pharmacol Ther 2010;23:345‐354.
(116) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T,
Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling
in a mouse model of asthma. Clin Exp Allergy 2010;40:1266‐1275.
(117) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(118) Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the
progression of airway smooth muscle remodeling. Am J Respir Crit Care Med 2005;171:1096‐1102.
(119) Wedzicha JA, Donaldson GC. Exacerbations of chronic obstructive pulmonary disease. Respir
Care 2003;48:1204‐13; discussion 1213‐5.
(120) Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, Jeffery PK. Biopsy neutrophilia,
neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive
pulmonary disease. Am J Respir Crit Care Med 2003;168:968‐975.
(121) Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K, Kesten S, Wedzicha JA. Effect
of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J
2007;30:472‐478.
(122) Anzueto A, Tashkin D, Menjoge S, Kesten S. One‐year analysis of longitudinal changes in
spirometry in patients with COPD receiving tiotropium. Pulm Pharmacol Ther 2005;18:75‐81.
(123) Decramer M, Celli B, Kesten S, Lystig T, Mehra S, Tashkin DP. Effect of tiotropium on outcomes
in patients with moderate chronic obstructive pulmonary disease (UPLIFT): a prespecified subgroup
analysis of a randomised controlled trial. Lancet 2009;374:1171‐1178.
(124) Morice AH, Celli B, Kesten S, Lystig T, Tashkin D, Decramer M. COPD in young patients: a pre‐
specified analysis of the four‐year trial of tiotropium (UPLIFT). Respir Med 2010;104:1659‐1667.
(125) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH.
Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med 2011;364:2006‐2015.



CHAPTER 2
REGULATION OF AIRWAY INFLAMMATION AND REMODELING BY
MUSCARINIC RECEPTORS: PERSPECTIVES ON ANTICHOLINERGIC
THERAPY IN ASTHMA AND COPD
Loes E.M. Kistemaker
Tjitske A. Oenema
Herman Meurs
Reinoud Gosens
Life Sci, 2012, 91(21‐22):1126‐33

Chapter 2
36
Abstract
Acetylcholine is the primary parasympathetic neurotransmitter in the airways and an
autocrine/paracrine secreted hormone from non‐neuronal origins including inflammatory
cells and airway structural cells. In addition to the well‐known functions of acetylcholine in
regulating bronchoconstriction and mucus secretion, it is increasingly evident that
acetylcholine regulates inflammatory cell chemotaxis and activation, and also participates
in signaling events leading to chronic airway wall remodeling that is associated with
chronic obstructive airways diseases including asthma and COPD. As muscarinic receptors
appear responsible for most of the pro‐inflammatory and remodeling effects of
acetylcholine, these findings have significant implications for anticholinergic therapy in
asthma and COPD, which is selective for muscarinic receptors. Here, the regulatory role of
acetylcholine in inflammation and remodeling in asthma and COPD will be discussed
including the perspectives that these findings offer for anticholinergic therapy in these
diseases.
Introduction
Acetylcholine is the primary parasympathetic neurotransmitter in the airways and a
paracrine/autocrine hormone released from non‐neuronal origins. The role of
acetylcholine in the regulation of bronchomotor tone and mucus secretion from airway
submucosal glands is well established (1). More recent findings suggest that acetylcholine,
acting on muscarinic receptors, regulates additional functions in the airways, including
inflammation and remodeling in obstructive airways diseases such as asthma and COPD
(2‐4). Based on these findings, we have previously questioned the traditional view on the
role of acetylcholine, and suggested new possibilities for therapeutic targeting of
muscarinic receptors in asthma and COPD (2). In this review, we will discuss the role of
muscarinic receptors in obstructive airways disease further and update the discussion in
view of these recent research papers and trials. In view of the selectivity of currently used
anticholinergics for muscarinic receptors, we will not elaborate on the role of nicotinic
receptors in this review. Nicotinic receptors are, however, expressed in the airways and
mediate anti‐inflammatory effects of acetylcholine. For excellent reviews on the anti‐
inflammatory role of nicotinic receptors, we would like to refer to recently published
reviews (5‐7).

Muscarinic receptors in inflammation and remodeling
37
Acetylcholine and muscarinic receptors in the airways
Biosynthesis, metabolism and mode of action of acetylcholine
Acetylcholine is synthesized from choline and acetyl‐CoA mainly by the enzyme choline
acetyltransferase (ChAT) (7). Airway neurons and non‐neuronal cells such as airway
epithelial cells express ChAT and release acetylcholine (8). Further, macrophages, mast
cells, lymphocytes, granolucytes, fibroblasts and smooth muscle cells all have been
suggested to express ChAT (7), although the release of acetylcholine from these cells has
not yet been demonstrated directly. Acetylcholine can bind to and activate a family of G
protein coupled muscarinic receptors, but also a family of nicotinic receptors, which are
ligand gated cation channels (9). Most inflammatory and airway structural cells express
muscarinic and/or nicotinic receptors (2,7). The individual receptor subtypes and subunits
expressed by these cells have been reviewed extensively by Wessler and Kirkpatrick (7).
The mechanisms that regulate the metabolism of non‐neuronal acetylcholine by airway
epithelial cells are still not fully established, although recent studies have yielded
important new insights. The uptake of choline is the rate‐limiting step in the synthesis of
acetylcholine. Choline uptake in airway epithelial cells is regulated by the high affinity
choline transporter (CHT1) and by choline‐specific transporter‐like proteins (CTL) (10,11).
Organic cation transporter (OCT) subtypes 1 and 2 play a dominant role in the release of
acetylcholine by airway epithelial cells (10,11). Furthermore, the expression of the
vesicular acetylcholine transporter (VAChT) by some epithelial cell types, including
secretory cells, neuroendocrine cells and brush cells has been reported, suggesting that
storage and release of acetylcholine via vesicles may mediate acetylcholine release by
non‐neuronal cell types (10,11). The expression of muscarinic receptors, nicotinic
receptors, synthesizing enzymes such as ChAT and the release of acetylcholine from non‐
neuronal cells is solid evidence for the existence of a non‐neuronal cholinergic system in
the airways next to the well‐established neuronal cholinergic system.
Muscarinic receptor expression and function in the airways
Muscarinic receptors are the target for anticholinergic therapy in obstructive airways
diseases as asthma and COPD and are the focus of this review. Muscarinic receptors are
expressed by structural cells in the airways, predominantly airway smooth muscle, airway
epithelium and airway fibroblasts. The parasympathetic neural network penetrates deep
into the airway wall, and regulates bronchoconstriction, the release of mucus from
submucosal glands, and to a lesser degree from goblet cells in the airway epithelium (2).
The functional role of non‐neuronal acetylcholine released from the airway epithelium is
less well described, although recent studies suggest a role in airway smooth muscle
contraction (12). It should be noted however that this finding is still controversial (13,14).

Chapter 2
38
Additionally, acetylcholine, either neuronal or non‐neuronal, may modulate airway
inflammation and remodeling, as will be discussed further on.
The distribution of muscarinic receptor subtypes throughout the bronchial tree is mainly
restricted to muscarinic M1, M2 and M3 receptors (2). M1 receptors are expressed by
epithelial cells, where they play a modulatory role in electrolyte and water secretion, and
in the ganglia, where they facilitate parasympathetic neurotransmission. M2 receptors are
expressed by neurons, where they function as autoreceptors, inhibiting the release of
acetylcholine from both preganglionic nerves and from parasympathetic nerve terminals.
M2 autoreceptors are dysfunctional in allergic asthma due to eosinophil‐derived release of
major basic protein which acts as an allosteric antagonist of the M2 receptor (15),
augmenting acetylcholine release. Furthermore, M2 receptors are widely expressed by
airway mesenchymal cells such as fibroblasts and smooth muscle cells (2). Recent studies
suggest that they may modulate cellular responses associated with airway remodeling
(16). Also, a role in inhibition of Gs mediated airway smooth muscle relaxation has been
proposed (1). M3 receptors are probably the best characterized subtype and are the
dominant receptor subtype in the regulation of mucus secretion from submucosal glands
and airway smooth muscle contraction (2). As a result, M3 receptors are the primary target
for anticholinergics, and M3 subtype‐selectivity has been advocated for by several
research groups (17‐22).
Muscarinic receptors as therapeutic targets for asthma and COPD
Anticholinergic therapy in COPD, and to a lesser extent asthma, is mainly aimed at
inhibition of bronchoconstriction by inhibition of muscarinic receptors. Although the term
anticholinergic is most commonly used, all available anticholinergics used for the
treatment of asthma and COPD are in fact specific antimuscarinics as they lack binding
affinity at the nicotinic receptor. Clinically available anticholinergics are the short‐acting
ipratropium and the long‐acting tiotropium. In addition to its longer duration of action,
tiotropium has a considerably slower rate of dissociation from the M1 and the M3 receptor
than from the M2 receptor, making the drug ‘kinetically selective’ for M1 and M3 receptors
(21). It is conceivable that this functional selectivity of tiotropium is beneficial, as smooth
muscle contraction is primarily mediated by M3 receptors, whereas M2 receptor blockade
facilitates acetylcholine release from parasympathetic nerves (2). However, direct
evidence for a beneficial clinical effect of this functional M3 selectivity of tiotropium is still
lacking and the major difference between these drugs appears to be the duration of
action.

Muscarinic receptors in inflammation and remodeling
39
The Understanding the Potential Long‐term Impacts of on Function with Tiotropium
(UPLIFT) trial has demonstrated that treatment with tiotropium provides a significant and
sustained improvement in lung function and quality of life in COPD patients, and reduces
exacerbations and hospitalizations (23). Currently available anticholinergics are the short‐
acting ipratropium and the long‐acting tiotropium. These can be used either as
monotherapy or in combination with β2‐agonists and provide significant improvement in
FEV1 in both asthma and COPD patients (24‐27). The combination therapy with β2‐agonists
is more effective than anticholinergic treatment alone; nonetheless, monotherapy is
already markedly effective (28). The explanation for this relatively large effect of
monotherapy may lie within the role that mediators of inflammation (e.g. thromboxane
A2, histamine) have in activating the airway cholinergic system. Airway inflammation has
several ways to increase the output of neuronally released acetylcholine, as it results in
exposure and activation of afferent C‐fibres that facilitate ganglionic and central
parasympathetic neurotransmission. Further, the release of acetylcholine can be
facilitated directly via excitatory receptors for inflammatory mediators (e.g.
prostaglandins, tachykinins) present on parasympathetic nerve terminals, and indirectly
via inhibition of the M2 autoreceptor through the release of eosinophil derived major basic
protein that acts as an allosteric M2 receptor antagonist (1,2). As a result, the
bronchoconstrictor response (and perhaps additional responses) induced by pro‐
inflammatory mediators such as thromboxane A2 is for a large part mediated by
neuronally released acetylcholine (29). Further, bronchoconstriction induced by histamine
after the early asthmatic response can be inhibited by ipratropium in a guinea pig model
of asthma (30). This advocates for the use of anticholinergic therapy not only in COPD –
where parasympathetic tone is the primary reversible component of airway obstruction
(31) – but also in asthma. Indeed, recent clinical trials indicate significant improvements in
lung function in asthma patients on top of usual care, and show that tiotropium therapy is
non‐inferior to β2‐agonist therapy when combined with corticosteroids in severe asthma
patients (25,26,32). The additional observations that next to FEV1 also exacerbation rate
and lung function decline in subgroups of COPD patients are improved by treatment with
tiotropium (33,34) has prompted speculations on the possible beneficial effects of
anticholinergics on airway inflammation and remodeling (35).
Airway inflammation
Asthma and COPD are both characterized by chronic airway inflammation, albeit that the
patterns of inflammation are markedly different. Different subtypes of T cells are involved
in asthma and COPD: in asthma there is an increase in TH2 (CD4+) cells, whereas in COPD

Chapter 2
40
CD8+ T cells predominate. Furthermore, the inflammation that occurs in asthma can be
described as eosinophilic, whereas that occurring in COPD is mainly neutrophilic.
However, when disease severity increases these differences become less pronounced (36).
Inflammation and the non‐neuronal cholinergic system
Increasing evidence suggests that acetylcholine contributes to airway inflammation. In
2004, Wessler et al. found that in patients with atopic dermatitis, a condition
characterized by TH2 type inflammation and often associated with bronchial asthma,
expression of ChAT is increased in skin biopsies, with a consequent increase in
acetylcholine (37). Further, Profita et al. (2011) demonstrated that cigarette smoke extract
upregulated the non‐neuronal cholinergic system in bronchial epithelial cells, by showing
that expression of M2 and M3 receptors and ChAT mRNA and protein were increased,
whereas M1 receptor levels were not affected. Consequently, acetylcholine levels in cell
extracts were significantly higher after stimulation with cigarette smoke extract. This
increase could be reduced by tiotropium (38). In contrast, lungs of ovalbumin challenged
rats and mice show a significant decrease in ChAT and other components of the
cholinergic system, including the functionally relevant choline transporter CHT1 (39).
Future studies are clearly warranted within this area to better understand the complex
mechanism of regulation of the cholinergic system by inflammation and the significance of
this process in asthma and COPD.
Inflammatory cells
Acetylcholine has been shown to affect inflammatory cells involved in asthma and COPD
directly, by inducing proliferation or cytokine release from these cells. Carbachol can
induce the proliferation of macrophages from mice in vitro (40). Also T‐cell proliferation
can be observed ex vivo after treatment of rats with the muscarinic agonist oxotremorine,
whereas atropine suppresses the proliferation of T‐cells (41). These anti‐inflammatory
properties of atropine were also demonstrated in rats in vivo, were it suppressed the
turpentine‐induced infiltration of leukocytes (41). Moreover, bovine alveolar macrophages
exhibit neutrophil, eosinophil and monocyte chemotactic activity in response to
acetylcholine, which is likely explained by cholinergic induction of leukotriene B4 (LTB4)
release (42). Recently, this was confirmed for primary human macrophages (43).
Moreover, it was shown that acetylcholine‐induced release of chemotactic activity from
monocytes, macrophages and epithelial cells could be inhibited by tiotropium (43). It has
also been shown that acetylcholine can induce the release of LTB4 from sputum cells of
COPD patients (44). These results are consistent with a study demonstrating that
tiotropium and also acetylcholinesterase, the degrading enzyme of acetylcholine, inhibited
alveolar macrophage mediated migration of neutrophils from COPD patients (45). Using
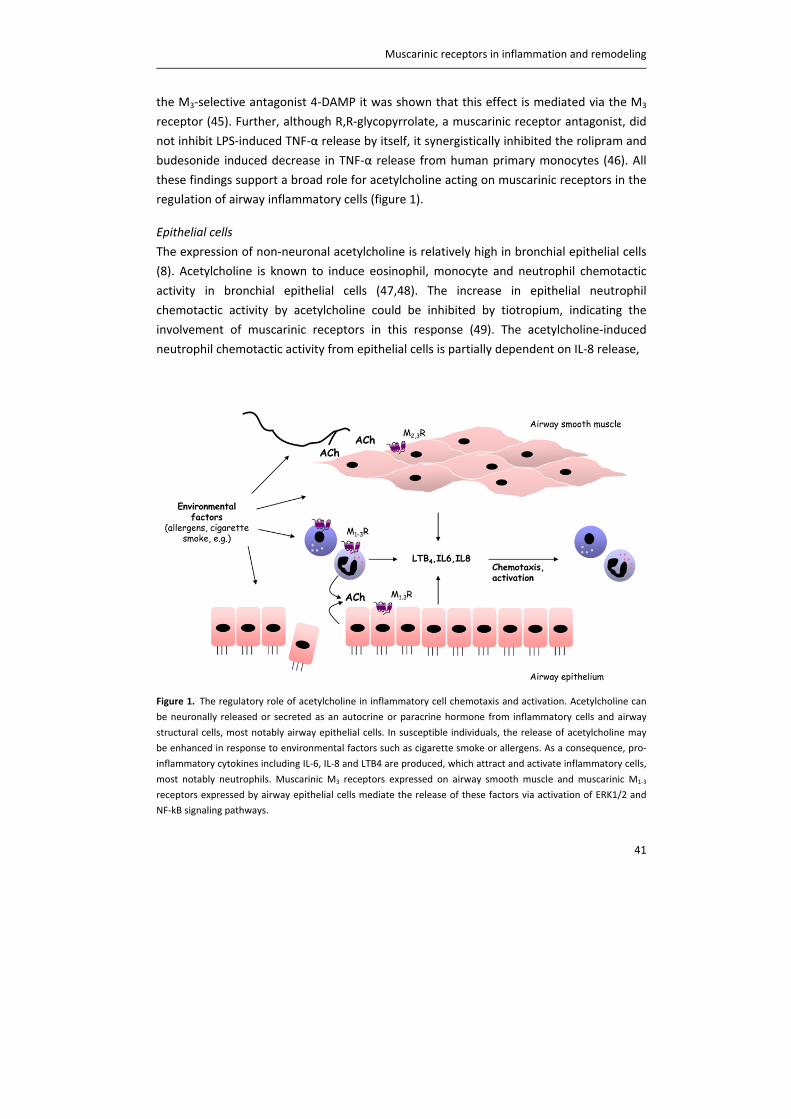
Muscarinic receptors in inflammation and remodeling
41
the M3‐selective antagonist 4‐DAMP it was shown that this effect is mediated via the M3
receptor (45). Further, although R,R‐glycopyrrolate, a muscarinic receptor antagonist, did
not inhibit LPS‐induced TNF‐α release by itself, it synergistically inhibited the rolipram and
budesonide induced decrease in TNF‐α release from human primary monocytes (46). All
these findings support a broad role for acetylcholine acting on muscarinic receptors in the
regulation of airway inflammatory cells (figure 1).
Epithelial cells
The expression of non‐neuronal acetylcholine is relatively high in bronchial epithelial cells
(8). Acetylcholine is known to induce eosinophil, monocyte and neutrophil chemotactic
activity in bronchial epithelial cells (47,48). The increase in epithelial neutrophil
chemotactic activity by acetylcholine could be inhibited by tiotropium, indicating the
involvement of muscarinic receptors in this response (49). The acetylcholine‐induced
neutrophil chemotactic activity from epithelial cells is partially dependent on IL‐8 release,
Figure 1. The regulatory role of acetylcholine in inflammatory cell chemotaxis and activation. Acetylcholine can
be neuronally released or secreted as an autocrine or paracrine hormone from inflammatory cells and airway
structural cells, most notably airway epithelial cells. In susceptible individuals, the release of acetylcholine may
be enhanced in response to environmental factors such as cigarette smoke or allergens. As a consequence, pro‐
inflammatory cytokines including IL‐6, IL‐8 and LTB4 are produced, which attract and activate inflammatory cells,
most notably neutrophils. Muscarinic M3 receptors expressed on airway smooth muscle and muscarinic M1‐3
receptors expressed by airway epithelial cells mediate the release of these factors via activation of ERK1/2 and
NF‐kB signaling pathways.

Chapter 2
42
since it is inhibited by an anti‐IL‐8 monoclonal antibody (49). In line with this contention,
the increase in IL‐8 release in response to acetylcholine could be partially inhibited by
tiotropium. In addition, acetylcholine induced LTB4 release from bronchial epithelial cells
in a tiotropium sensitive manner (38). Both IL‐8 and LTB4 release from bronchial epithelial
cells is mediated via ERK1/2 and NF‐κB signaling pathways and dependent on multiple
muscarinic receptor subtypes (M1/M2/M3) (38,49). Taken together, these studies implicate
an important role for epithelial acetylcholine in airway inflammation, via the activation of
muscarinic receptors (figure 1).
Another potential mechanism by which tiotropium could inhibit inflammation induced by
epithelial cells is by attenuating respiratory syncytial virus (RSV) replication in these cells
(50). RSV is one of the major causes of acute lower respiratory tract infection and has
been detected in patients with exacerbations of asthma and COPD (51). In an in vitro
study, Iesato et al. demonstrated that the attenuation of virus replication by tiotropium
was partially due to inhibition of RhoA activity. Moreover, tiotropium inhibited epithelial
IL‐6 and IL‐8 production induced by RSV infection (50). In vivo studies are needed to
investigate the importance of inhibition of infection‐induced airway inflammation by
tiotropium.
Airway smooth muscle cells
The airway smooth muscle is increasingly recognized for its role in modulating
inflammation by secreting cytokines and chemokines (52), and it has been shown that
muscarinic receptors on airway smooth muscle cells are involved in these responses.
Stimulation of bovine airway smooth muscle strips with the muscarinic agonist carbachol
induces pro‐inflammatory gene expression, including IL‐6, IL‐8 and cyclo‐oxygenase‐2 (53).
Furthermore, carbachol augmented the cyclic stretch‐induced expression of these genes
(53). Stimulation of airway smooth muscle cells with carbachol also induces the protein
release of IL‐6 and IL‐8 via M3 receptors (54). Furthermore, methacholine strongly
augmented cigarette smoke extract (CSE) induced IL‐8 release (54). In line with findings in
epithelial cells, IL‐8 release induced by stimulation with methacholine and CSE in airway
smooth muscle is ERK1/2 and NF‐κB dependent (55).
In vivo studies
The regulatory role of muscarinic receptor signaling in inflammatory processes involved in
asthma and COPD has been confirmed by in vivo studies, using animal models of these
diseases.

Muscarinic receptors in inflammation and remodeling
43
Wollin and Pieper were the first to report anti‐inflammatory properties of tiotropium in an
animal model of cigarette smoke induced COPD (56). Total cell number and neutrophils in
the bronchoalveolar lavage fluid (BALF) were concentration‐dependently decreased after
treatment with tiotropium. Furthermore, tiotropium inhibited the increase of several
cytokines in the BALF, including IL‐6, KC, TNF‐α and LTB4 (56). Similar inhibitory effects of
tiotropium on airway neutrophilia were observed in a guinea pig model of LPS‐induced
COPD (57). Moreover, neutrophilia was inhibited by ipratropium in a cadmium‐induced rat
model of pulmonary inflammation (58), by tiotropium in a HCl‐induced rat model of
gastro‐oesophageal reflux (59) and by bilateral vagotomy or treatment with atropine in a
diesel particle‐induced rat model of pulmonary inflammation (60). Of interest, the latter
study found that atropine was more effective in inhibiting pulmonary inflammation than
bilateral vagotomy, suggesting a role for non‐neuronal acetylcholine in this response (60).
These findings may also be relevant for asthma. Our group has shown that tiotropium
partially inhibits eosinophilia in a guinea pig model of asthma (61), which has been
confirmed by Buels et al. (62). In line with these findings, infiltration of macrophages and
eosinophils in the BALF was significantly inhibited by tiotropium treatment in a murine
model of asthma. Furthermore, expression levels in BALF of IL‐4, IL‐5 and IL‐13 were
decreased by tiotropium treatment (63). In addition, aclidinium, a novel muscarinic
receptor antagonist, inhibited infiltration of eosinophils in BALF in a mouse model of
Aspergillus fumigatus‐induced asthma (64). A recent study also suggested that M3
receptors regulate these inflammatory responses, although the selectivity profile of the
antagonist bencycloquidium that was used in this study precludes firm conclusions on the
involvement of other receptor subtypes (65,65). Since both tiotropium and aclidinium are
kinetically selective for the M3 receptor, this suggests predominant involvement of this
receptor subtype in the observed anti‐inflammatory effects in asthma and COPD models
described above. This is supported by our own data on M3R‐/‐ mice, in which neutrophilia
and cytokine release in BALF were inhibited compared to wild‐type mice after exposure to
cigarette smoke (chapter 3).
Clearly, all these in vivo studies indicate a profound role for acetylcholine in inflammation
in asthma and COPD, which is in accordance with results of in vitro studies that report pro‐
inflammatory effects of muscarinic receptors (figure 1). The implication of these findings is
that treatment with anticholinergics may have beneficial effects that exceed their
bronchodilatory properties, a contention confirmed in several models of pulmonary
inflammation. However, the exact mechanism responsible for the regulatory role of
acetylcholine in inflammation is far from understood.

Chapter 2
44
Airway remodeling
Airway inflammation in chronic airway diseases such as asthma and COPD is often
associated with cellular and structural alterations in the airways, referred to as airway
remodeling (66). Airway remodeling is considered a major component of irreversible
airflow limitation in these diseases (67), is progressive, and correlates with disease
severity (68,69). Airway remodeling in asthma and COPD is characterized by mucus gland
hypertrophy, goblet cell hyperplasia and pulmonary vascular remodeling (66). In addtion,
in asthma the basement membrane is thickened, there is subepithelial fibrosis, and there
is considerable thickening of the airway smooth muscle bundle (67). In contrast, in COPD
the fibrosis is mostly peribronchial, and although increased airway smooth muscle mass
may occur, this appears restricted to severe stages of COPD (68). Airway structural
alterations may accelerate decline of lung function (70).
Epithelial cells and mucus production
The airway epithelial layer is in continuous interaction with the external environment. To
protect itself from exogenous stimuli, mucus is secreted under the control of the
cholinergic system by muscarinic receptors (71). Mucus secretion can be increased by
electrical field stimulation of the vagal nerve in bronchial preparations, predominantly via
M3 receptors on the submucosal glands (71). In addition, electrolyte and water secretion
are regulated by M1 and M3 receptors (72,73). Neuronal M2 autoreceptors appears to
regulate the extent of the secretory response, by limiting neuronally released
acetylcholine (73). In response to acetylcholine, glandular goblet cells also produce mucus
(71).
Mucus hypersecretion is an important pathological feature of chronic airway diseases
contributing to airway obstruction (71). MUC5AC expression in airway epithelial cells and
airway submucosal glands is directly correlated to airway obstruction in smokers (74) and
in smokers, COPD patients and asthma patients, the expression of the MUC5AC gene is
augmented (75). Also, the expression of MUC5B and the insoluble MUC2 are increased,
particularly in COPD. The ratio of mucus cells to serous cells in the submucosal glands is
also increased in COPD patients (76). In vitro studies demonstrated that aclidinium
suppressed carbachol‐induced MUC5AC overexpression in human bronchial tissue.
Additionally, the increased expression of MUC5AC by the co‐stimulation of cigarette
smoke extract and carbachol could be attenuated by the use of aclidinium or atropine
(77). Moreover, epidermal growth factor (EGF) stimulation enhanced the ACh‐induced
response on mucus cell activation in airway submucosal glands (78). In vivo studies
confirm the role of acetylcholine in mucus hypersecretion and demonstrate that
tiotropium reduces allergen‐induced mucus gland hypertrophy and MUC5AC‐positive

Muscarinic receptors in inflammation and remodeling
45
goblet cell number in guinea pigs (61). Further, it has been reported that tiotropium
inhibits neutrophil elastase‐induced goblet cell metaplasia in mice (79) and that treatment
with tiotropium inhibited the increased MUC5AC expression and mucus gland hypertrophy
in a guinea pig model of COPD (57). This demonstrates the important role of acetylcholine
in the regulation of mucus secretion, both in vitro and in animal models of asthma and
COPD in vivo (figure 2).
Acetylcholine may also regulate the proliferative and pro‐fibrotic responses of airway
epithelial cells. Bronchoconstriction induced by repeated challenges with methacholine
induced epithelial cell proliferation and an increase in the expression of the profibrotic
cytokine TGF‐β by these cells in mild asthmatic subjects (80). In line with these findings,
airway constriction induced by methacholine significantly increased the phosphorylation
of the EGF receptor in airway epithelial cells (81). Moreover, in rat tracheal epithelial cells,
acetylcholine induces proliferation mediated by M1 receptors (82) and autocrine release of
acetylcholine is sufficient to induce monkey airway epithelial cell proliferation (8). Thus,
the cholinergic system is able to regulate epithelial cell proliferation, either through the
induction of mechanical strain or in an autocrine/paracrine manner, which is required for
the repair of the airway epithelial layer.
Mesenchymal cells
Airway mesenchymal cells (e.g. fibroblasts, airway smooth muscle cells) contribute to
airway remodeling by means of proliferation, contractile protein expression and the
release of components such as mediators, extracellular matrix proteins and matrix
metalloproteinases (MMPs) (83,84). In vitro studies showed that the stimulation of
muscarinic receptors on lung fibroblasts induces cell proliferation and the synthesis of
collagen (16,85) through the activation of the mitogen‐activated protein kinase pathway
(85,86). This effect was mediated by the activation of M2 receptors (16). Interestingly,
acetylcholine‐induced cell proliferation is enhanced in human lung fibroblasts from COPD
patients compared with healthy non‐smokers and healthy smokers without COPD (87).
The higher activation of cell proliferation in fibroblasts from COPD patients was due to
enhanced ERK1/2 and NF‐κB phosphorylation. Notably, the synthesizing enzyme ChAT was
also increased in lung fibroblasts from healthy smokers and COPD patients (87).
MMPs play a key role in airway remodeling, inflammation and emphysema (88). In COPD
patients, increased expression levels of MMP‐1, MMP‐2 and MMP‐9 have been reported
(89,90). The activity of the MMPs can be inhibited by tissue inhibitor of matrix
metalloproteinases (TIMPs) (88). Recently, it was demonstrated that tiotropium inhibited
TGF‐β‐induced protein expression of both MMP‐1 and MMP‐2 in human lung fibroblasts,

Chapter 2
46
but had no effect on the TGF‐β‐induced TIMP‐1 and TIMP‐2 expression (91,92). Therefore,
these data suggest that treatment with tiotropium improves the balance between MMPs
and TIMPs, inhibiting pro‐fibrotic responses. As MMPs also play important roles in the
infiltration of inflammatory cells, this effect could also contribute to the anti‐inflammatory
properties of anticholinergics.
Airway smooth muscle thickening is a characteristic pathological feature of asthma, and to
a lesser extent of COPD. The induction of airway smooth muscle cell proliferation by
growth factors, including PDGF and EGF, can be enhanced by the stimulation of muscarinic
receptors (93‐96). Specifically, Gβγ subunits derived from Gq protein coupled receptors
cooperate with receptor tyrosine kinases (e.g. the PDGF/EGF receptor) to induce
synergistic activation of PI3K/Akt/p70S6K signaling leading to cell proliferation (93,95,96).
Figure 2. The regulatory role of acetylcholine in airway wall remodeling. Acetylcholine is neuronally released and
secreted as an autocrine or paracrine hormone from airway structural cells and inflammatory cells. In the
inflamed airway, inflammatory cells and airway epithelial cells also secrete growth factors that in concerted
action with acetylcholine activate cell proliferation and matrix production by airway mesenchymal cells, including
airway fibroblasts and airway smooth muscle cells. Furthermore, acetylcholine activates smooth muscle
contraction leading to airway wall compression, which activates inflammatory cells and promotes remodeling
responses by airway epithelial cells. Acetylcholine also directly promotes mucus production by and cell
proliferation of airway epithelial cells.

Muscarinic receptors in inflammation and remodeling
47
Moreover, the activation of conventional PKC isoenzymes, likely via M3 receptor mediated
Gαq stimulation, leads to GSK‐3 inactivation, which potentiates both translational and
transcriptional processes (94). These pathways are also involved in the acquisition of
contractile protein expression by TGF‐β via transcriptional and translational processes (97‐
99) and can be activated by muscarinic receptor stimulation (100). Indeed, the expression
of myosin light‐chain kinase was augmented by carbachol in human airway smooth muscle
cells exposed to cyclical mechanical strain (101). Additionally, we recently described that
muscarinic receptor stimulation enhanced the TGF‐β1‐induced contractile protein
expression in human airway smooth muscle cells (102). Collectively, these findings suggest
an important role of muscarinic receptor stimulation in the proliferation and maturation
of mesenchymal cells (figure 2).
In vivo studies
Inhibitory effects of anticholinergics on airway mesenchymal cell remodeling have indeed
been reported in animal models of asthma and COPD. Treatment with tiotropium
significantly inhibited airway smooth muscle remodeling in a guinea‐pig model of chronic
asthma using repeated challenges with ovalbumin (103). This was associated with the
inhibition of increased contractile protein expression and of airway smooth muscle
thickening. In a murine model of asthma, it was shown that tiotropium could also
significantly inhibit smooth muscle thickening and the expression of TGF‐β1 in BALF (63).
Similar effects have been described for the M3 receptor selective antagonist
bencycloquidium bromide (65). Furthermore, bencycloquidium bromide reduced mucus
production, globet cell metaplasia and collagen deposition and inhibited the upregulation
of MMP‐9, but not of TIMP‐1 mRNA (65). Treatment with tiotropium also inhibited the
increased peribronchial collagen deposition in a guinea pig model of COPD (57). Similarly,
in a chronic gastro‐oesophageal reflux model, tiotropium treatment prevented the
increase in airway fibrosis (59). Taken together, these in vivo studies confirm in vitro
studies showing that anticholinergics have anti‐remodeling properties in asthma and
COPD (figure 2).
Clinical implications
The above mentioned in vitro and in vivo studies indicate significant pro‐inflammatory and
remodeling effects for acetylcholine via muscarinic receptors, suggesting that
anticholinergics may have anti‐inflammatory and anti‐remodeling properties in asthma
and COPD patients. This hypothesis still needs to be proven in clinical studies, however. In
the UPLIFT study, COPD patients treated with tiotropium during a 4 year period showed
an improved quality of life and lung function, and a reduction in the frequency of

Chapter 2
48
exacerbations. Although tiotropium did not reduce FEV1 decline in the overall study
population (23), in pre‐specified post‐hoc studies, GOLD stage II and young COPD patients
with rapid lung function decline had a significant improvement in the accelerated post‐
bronchodilator FEV1 decline (33,34). No notable reduction in exacerbation frequency was
reported for ipratropium (104,105). This suggests a beneficial role for tiotropium as a long‐
acting anticholinergic or a possible role for M3 receptor subtype selectivity, as tiotropium
is kinetically selective for M3 receptors compared with ipratropium. Moreover, it also
indicates anti‐inflammatory effects of tiotropium, since patients who have more
exacerbations demonstrate increased levels of inflammatory markers at stable state (106).
However, Powrie et al. (2007) were not able to demonstrate a reduction in sputum IL‐6 or
IL‐8 levels in patients treated with tiotropium during one year, even though the number of
exacerbations was significantly decreased (107). A possible explanation for this
discrepancy proposed by the authors is that the reduction in amount of sputum after
tiotropium treatment might result in an increase in cytokine concentrations.
Measurement of cytokine concentrations in sputum might therefore not be the optimal
method. Also, Perng et al. did not find a decrease in sputum IL‐8 levels after tiotropium
treatment (108). However, the treatment group in their study was small and patients only
received tiotropium for 12 weeks. Further studies are therefore needed to elucidate the
mechanisms by which tiotropium reduces exacerbations and FEV1 decline in subgroups of
COPD patients and whether this is based on the anti‐inflammatory effects of tiotropium
discussed in this paper or by other effects, including a reduction in dyspnea or mucus
hypersecretion. Likewise, further studies on the beneficial effects of anticholinergics in
asthma patients are warranted. In patients with severe, uncontrolled asthma it has
recently been shown that treatment with tiotropium improves lung function (25).
Furthermore, a recent clinical trial showed that repeated inhalations with the muscarinic
receptor agonist methacholine induces airway remodeling in asthma patients, including
the expression of TGF‐β and collagen I in bronchial biopsies (80). Therefore, although a
rationale for beneficial effects of anticholinergics beyond the well‐described
bronchodilator properties in asthma and COPD certainly exists, it is evident that this still
needs to be confirmed in clinical studies.
Acknowledgements
The authors would like to thank the Netherlands Lung Foundation (grant 3.2.08.014) and
the Groningen Center for Drug Research for financial support.

Muscarinic receptors in inflammation and remodeling
49
References
(1) Belmonte KE. Cholinergic pathways in the lungs and anticholinergic therapy for chronic
obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2(4):297‐304.
(2) Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology
of asthma and COPD. Respir Res 2006; 7:73.
(3) Racke K, Matthiesen S. The airway cholinergic system: physiology and pharmacology. Pulm
Pharmacol Ther 2004; 17(4):181‐198.
(4) Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol 2006;
533(1‐3):57‐68.
(5) Cui WY, Li MD. Nicotinic modulation of innate immune pathways via alpha7 nicotinic
acetylcholine receptor. J Neuroimmune Pharmacol 2010; 5(4):479‐488.
(6) Rosas‐Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med 2009; 265(6):663‐
679.
(7) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008; 154(8):1558‐1571.
(8) Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J et al. Acetylcholine is an
autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells.
Endocrinology 2004; 145(5):2498‐2506.
(9) Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new
insights for drug development. Nat Rev Drug Discov 2007; 6(9):721‐733.
(10) Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol
2008; 130(2):219‐234.
(11) Lips KS, Volk C, Schmitt BM, Pfeil U, Arndt P, Miska D et al. Polyspecific cation transporters
mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol 2005;
33(1):79‐88.
(12) Moffatt JD, Cocks TM, Page CP. Role of the epithelium and acetylcholine in mediating the
contraction to 5‐hydroxytryptamine in the mouse isolated trachea. Br J Pharmacol 2004;
141(7):1159‐1166.
(13) Kummer W, Wiegand S, Akinci S, Wessler I, Schinkel AH, Wess J et al. Role of acetylcholine and
polyspecific cation transporters in serotonin‐induced bronchoconstriction in the mouse. Respir Res
2006; 7:65.
(14) Kummer W, Wiegand S, Akinci S, Schinkel AH, Wess J, Koepsell H et al. Role of acetylcholine and
muscarinic receptors in serotonin‐induced bronchoconstriction in the mouse. J Mol Neurosci 2006;
30(1‐2):67‐68.
(15) Jacoby DB, Gleich GJ, Fryer AD. Human eosinophil major basic protein is an endogenous
allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest 1993; 91(4):1314‐1318.
(16) Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C et al. Muscarinic
receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol 2006;
35(6):621‐627.

Chapter 2
50
(17) Decramer M, Celli B, Tashkin DP, Pauwels RA, Burkhart D, Cassino C et al. Clinical trial design
considerations in assessing long‐term functional impacts of tiotropium in COPD: the UPLIFT trial.
COPD 2004; 1(2):303‐312.
(18) Lu S, Parekh DD, Kuznetsova O, Green SA, Tozzi CA, Reiss TF. An oral selective M3 cholinergic
receptor antagonist in COPD. Eur Respir J 2006; 28(4):772‐780.
(19) Barnes PJ. The role of anticholinergics in chronic obstructive pulmonary disease. Am J Med
2004; 117 Suppl 12A:24S‐32S.
(20) Campbell SC. Clinical aspects of inhaled anticholinergic therapy. Respir Care 2000; 45(7):864‐
867.
(21) Disse B, Speck GA, Rominger KL, Witek TJ, Jr., Hammer R. Tiotropium (Spiriva): mechanistical
considerations and clinical profile in obstructive lung disease. Life Sci 1999; 64(6‐7):457‐464.
(22) Barnes PJ, Belvisi MG, Mak JC, Haddad EB, O'Connor B. Tiotropium bromide (Ba 679 BR), a novel
long‐acting muscarinic antagonist for the treatment of obstructive airways disease. Life Sci 1995;
56(11‐12):853‐859.
(23) Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S et al. A 4‐year trial of tiotropium in
chronic obstructive pulmonary disease. N Engl J Med 2008; 359(15):1543‐1554.
(24) Kerstjens HA, Bantje TA, Luursema PB, Sinninghe Damste HE, de Jong JW, Lee A et al. Effects of
short‐acting bronchodilators added to maintenance tiotropium therapy. Chest 2007; 132(5):1493‐
1499.
(25) Kerstjens HA, Disse B, Schroder‐Babo W, Bantje TA, Gahlemann M, Sigmund R et al. Tiotropium
improves lung function in patients with severe uncontrolled asthma: A randomized controlled trial. J
Allergy Clin Immunol 2011; 128(2):308‐314.
(26) Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT et al. Tiotropium
bromide step‐up therapy for adults with uncontrolled asthma. N Engl J Med 2010; 363(18):1715‐
1726.
(27) Tashkin D, Kesten S. Long‐term treatment benefits with tiotropium in COPD patients with and
without short‐term bronchodilator responses. Chest 2003; 123(5):1441‐1449.
(28) Vogelmeier C, Kardos P, Harari S, Gans SJ, Stenglein S, Thirlwell J. Formoterol mono‐ and
combination therapy with tiotropium in patients with COPD: a 6‐month study. Respir Med 2008;
102(11):1511‐1520.
(29) Allen IC, Hartney JM, Coffman TM, Penn RB, Wess J, Koller BH. Thromboxane A2 induces airway
constriction through an M3 muscarinic acetylcholine receptor‐dependent mechanism. Am J Physiol
Lung Cell Mol Physiol 2006; 290(3):L526‐L533.
(30) ten Berge RE, Santing RE, Hamstra JJ, Roffel AF, Zaagsma J. Dysfunction of muscarinic M2
receptors after the early allergic reaction: possible contribution to bronchial hyperresponsiveness in
allergic guinea‐pigs. Br J Pharmacol 1995; 114(4):881‐887.
(31) Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to
emphysema. N Engl J Med 1984; 311(7):421‐425.
(32) Bateman ED, Kornmann O, Schmidt P, Pivovarova A, Engel M, Fabbri LM. Tiotropium is
noninferior to salmeterol in maintaining improved lung function in B16‐Arg/Arg patients with
asthma. J Allergy Clin Immunol 2011; 128(2):315‐322.

Muscarinic receptors in inflammation and remodeling
51
(33) Decramer M, Celli B, Kesten S, Lystig T, Mehra S, Tashkin DP. Effect of tiotropium on outcomes
in patients with moderate chronic obstructive pulmonary disease (UPLIFT): a prespecified subgroup
analysis of a randomised controlled trial. Lancet 2009; 374(9696):1171‐1178.
(34) Morice AH, Celli B, Kesten S, Lystig T, Tashkin D, Decramer M. COPD in young patients: a pre‐
specified analysis of the four‐year trial of tiotropium (UPLIFT). Respir Med 2010; 104(11):1659‐1667.
(35) Bateman ED, Rennard S, Barnes PJ, Dicpinigaitis PV, Gosens R, Gross NJ et al. Alternative
mechanisms for tiotropium. Pulm Pharmacol Ther 2009; 22(6):533‐542.
(36) Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol
2008; 8(3):183‐192.
(37) Wessler I, Reinheimer T, Kilbinger H, Bittinger F, Kirkpatrick CJ, Saloga J et al. Increased
acetylcholine levels in skin biopsies of patients with atopic dermatitis. Life Sci 2003; 72(18‐19):2169‐
2172.
(38) Profita M, Bonanno A, Montalbano AM, Ferraro M, Siena L, Bruno A et al. Cigarette smoke
extract activates human bronchial epithelial cells affecting non‐neuronal cholinergic system
signalling in vitro. Life Sci 2011; 89(1‐2):36‐43.
(39) Lips KS, Luhrmann A, Tschernig T, Stoeger T, Alessandrini F, Grau V et al. Down‐regulation of the
non‐neuronal acetylcholine synthesis and release machinery in acute allergic airway inflammation of
rat and mouse. Life Sci 2007; 80(24‐25):2263‐2269.
(40) de la Torre E, Genaro AM, Ribeiro ML, Pagotto R, Pignataro OP, Sales ME. Proliferative actions
of muscarinic receptors expressed in macrophages derived from normal and tumor bearing mice.
Biochim Biophys Acta 2008; 1782(2):82‐89.
(41) Razani‐Boroujerdi S, Behl M, Hahn FF, Pena‐Philippides JC, Hutt J, Sopori ML. Role of muscarinic
receptors in the regulation of immune and inflammatory responses. J Neuroimmunol 2008; 194(1‐
2):83‐88.
(42) Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages
to release inflammatory cell chemotactic activity. Am J Physiol 1998; 274(6 Pt 1):L970‐L979.
(43) Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine‐
induced release of chemotactic mediators in vitro. Respir Med 2007; 101(11):2386‐2394.
(44) Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F et al. Muscarinic receptors,
leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy 2005;
60(11):1361‐1369.
(45) Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide.
Respir Res 2011; 12:24.
(46) Pahl A, Bauhofer A, Petzold U, Cnota PJ, Maus J, Brune K et al. Synergistic effects of the anti‐
cholinergic R,R‐glycopyrrolate with anti‐inflammatory drugs. Biochem Pharmacol 2006; 72(12):1690‐
1696.
(47) Koyama S, Rennard SI, Robbins RA. Acetylcholine stimulates bronchial epithelial cells to release
neutrophil and monocyte chemotactic activity. Am J Physiol 1992; 262(4 Pt 1):L466‐L471.
(48) Koyama S, Sato E, Nomura H, Kubo K, Nagai S, Izumi T. Acetylcholine and substance P stimulate
bronchial epithelial cells to release eosinophil chemotactic activity. J Appl Physiol 1998; 84(5):1528‐
1534.

Chapter 2
52
(49) Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F et al. Acetylcholine
mediates the release of IL‐8 in human bronchial epithelial cells by a NFkB/ERK‐dependent
mechanism. Eur J Pharmacol 2008; 582(1‐3):145‐153.
(50) Iesato K, Tatsumi K, Saito K, Ogasawara T, Sakao S, Tada Y et al. Tiotropium bromide attenuates
respiratory syncytial virus replication in epithelial cells. Respiration 2008; 76(4):434‐441.
(51) Johnston NW. The similarities and differences of epidemic cycles of chronic obstructive
pulmonary disease and asthma exacerbations. Proc Am Thorac Soc 2007; 4(8):591‐596.
(52) Tliba O, Panettieri Jr RA. Noncontractile Functions of Airway Smooth Muscle Cells in Asthma.
Annu Rev Physiol 2008.
(53) Kanefsky J, Lenburg M, Hai CM. Cholinergic receptor and cyclic stretch‐mediated inflammatory
gene expression in intact ASM. Am J Respir Cell Mol Biol 2006; 34(4):417‐425.
(54) Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J et al. Muscarinic M3 receptor
stimulation increases cigarette smoke‐induced IL‐8 secretion by human airway smooth muscle cells.
Eur Respir J 2009; 34(6):1436‐1443.
(55) Oenema TA, Kolahian S, Nanninga JE, Rieks D, Hiemstra PS, Zuyderduyn S et al. Pro‐
inflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir Res
2010; 11:130.
(56) Wollin L, Pieper MP. Tiotropium bromide exerts anti‐inflammatory activity in a cigarette smoke
mouse model of COPD. Pulm Pharmacol Ther 2010; 23(4):345‐354.
(57) Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R et al. Tiotropium inhibits
pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur Respir J 2011.
(58) Zhang W, Fievez L, Cheu E, Bureau F, Rong W, Zhang F et al. Anti‐inflammatory effects of
formoterol and ipratropium bromide against acute cadmium‐induced pulmonary inflammation in
rats. Eur J Pharmacol 2010; 628(1‐3):171‐178.
(59) Cui Y, Devillier P, Kuang X, Wang H, Zhu L, Xu Z et al. Tiotropium reduction of lung inflammation
in a model of chronic gastro‐oesophageal reflux. Eur Respir J 2010; 35(6):1370‐1376.
(60) McQueen DS, Donaldson K, Bond SM, McNeilly JD, Newman S, Barton NJ et al. Bilateral
vagotomy or atropine pre‐treatment reduces experimental diesel‐soot induced lung inflammation.
Toxicol Appl Pharmacol 2007; 219(1):62‐71.
(61) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H et al. Inhibition of allergen‐
induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J 2007;
30(4):653‐661.
(62) Buels KS, Jacoby DB, Fryer AD. Non‐bronchodilating mechanisms of tiotropium prevent airway
hyperreactivity in a guinea pig model of allergic asthma. Br J Pharmacol 2011.
(63) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y et al. Effect of tiotropium bromide
on airway inflammation and remodelling in a mouse model of asthma. Clin Exp Allergy 2010;
40(8):1266‐1275.
(64) Damera G, Jiang M, Zhao H, Fogle HW, Jester WF, Freire J et al. Aclidinium bromide abrogates
allergen‐induced hyperresponsiveness and reduces eosinophilia in murine model of airway
inflammation. Eur J Pharmacol 2010; 649(1‐3):349‐353.

Muscarinic receptors in inflammation and remodeling
53
(65) Cao R, Dong XW, Jiang JX, Yan XF, He JS, Deng YM et al. M(3) muscarinic receptor antagonist
bencycloquidium bromide attenuates allergic airway inflammation, hyperresponsiveness and
remodeling in mice. Eur J Pharmacol 2011; 655(1‐3):83‐90.
(66) Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care
Med 2001; 164(10 Pt 2):S28‐S38.
(67) An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V et al. Airway smooth muscle dynamics: a
common pathway of airway obstruction in asthma. Eur Respir J 2007; 29(5):834‐860.
(68) Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L et al. The nature of small‐airway
obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350(26):2645‐2653.
(69) James AL, Bai TR, Mauad T, Abramson MJ, Dolhnikoff M, McKay KO et al. Airway smooth muscle
thickness in asthma is related to severity but not duration of asthma. Eur Respir J 2009; 34(5):1040‐
1045.
(70) Pare PD, Roberts CR, Bai TR, Wiggs BJ. The functional consequences of airway remodeling in
asthma. Monaldi Arch Chest Dis 1997; 52(6):589‐596.
(71) Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol 2001; 125(1‐2):129‐
144.
(72) Ishihara H, Shimura S, Satoh M, Masuda T, Nonaka H, Kase H et al. Muscarinic receptor subtypes
in feline tracheal submucosal gland secretion. Am J Physiol 1992; 262(2 Pt 1):L223‐L228.
(73) Ramnarine SI, Haddad EB, Khawaja AM, Mak JC, Rogers DF. On muscarinic control of neurogenic
mucus secretion in ferret trachea. J Physiol 1996; 494 ( Pt 2):577‐586.
(74) Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC et al. Epithelial
mucin stores are increased in the large airways of smokers with airflow obstruction. Chest 2006;
130(4):1102‐1108.
(75) Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med 2006; 12(1):1‐6.
(76) Rogers DF. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin
Pharmacol 2004; 4(3):241‐250.
(77) Cortijo J, Mata M, Milara J, Donet E, Gavalda A, Miralpeix M et al. Aclidinium inhibits cholinergic
and tobacco smoke‐induced MUC5AC in human airways. Eur Respir J 2011; 37(2):244‐254.
(78) Iwase N, Sasaki T, Oshiro T, Tamada T, Nara M, Sasamori K et al. Differential effect of epidermal
growth factor on serous and mucous cells in porcine airway submucosal gland. Respir Physiol
Neurobiol 2002; 132(3):307‐319.
(79) Arai N, Kondo M, Izumo T, Tamaoki J, Nagai A. Inhibition of neutrophil elastase‐induced goblet
cell metaplasia by tiotropium in mice. Eur Respir J 2010; 35(5):1164‐1171.
(80) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S et al. Effect of bronchoconstriction on
airway remodeling in asthma. N Engl J Med 2011; 364(21):2006‐2015.
(81) Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK et al. Mechanotransduction
through growth‐factor shedding into the extracellular space. Nature 2004; 429(6987):83‐86.
(82) Metzen J, Bittinger F, Kirkpatrick CJ, Kilbinger H, Wessler I. Proliferative effect of acetylcholine
on rat trachea epithelial cells is mediated by nicotinic receptors and muscarinic receptors of the M1‐
subtype. Life Sci 2003; 72(18‐19):2075‐2080.
(83) Kelly EA, Jarjour NN. Role of matrix metalloproteinases in asthma. Curr Opin Pulm Med 2003;
9(1):28‐33.

Chapter 2
54
(84) Parks WC, Shapiro SD. Matrix metalloproteinases in lung biology. Respir Res 2001; 2(1):10‐19.
(85) Haag S, Matthiesen S, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of
collagen synthesis in human lung fibroblasts. Eur Respir J 2008; 32(3):555‐562.
(86) Matthiesen S, Bahulayan A, Holz O, Racke K. MAPK pathway mediates muscarinic receptor‐
induced human lung fibroblast proliferation. Life Sci 2007.
(87) Profita M, Bonanno A, Siena L, Bruno A, Ferraro M, Montalbano AM et al. Smoke, Choline‐
Acetyl‐Transferase, Muscarinic Receptors and fibroblast proliferation in COPD. J Pharmacol Exp Ther
2009.
(88) Lagente V, Boichot E. Role of matrix metalloproteinases in the inflammatory process of
respiratory diseases. J Mol Cell Cardiol 2010; 48(3):440‐444.
(89) Cataldo D, Munaut C, Noel A, Frankenne F, Bartsch P, Foidart JM et al. MMP‐2‐ and MMP‐9‐
linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive
pulmonary disease. Int Arch Allergy Immunol 2000; 123(3):259‐267.
(90) Imai K, Dalal SS, Chen ES, Downey R, Schulman LL, Ginsburg M et al. Human collagenase (matrix
metalloproteinase‐1) expression in the lungs of patients with emphysema. Am J Respir Crit Care Med
2001; 163(3 Pt 1):786‐791.
(91) Asano K, Shikama Y, Shibuya Y, Nakajima H, Kanai K, Yamada N et al. Suppressive activity of
tiotropium bromide on matrix metalloproteinase production from lung fibroblasts in vitro. Int J
Chron Obstruct Pulmon Dis 2008; 3(4):781‐789.
(92) Asano K, Shikama Y, Shoji N, Hirano K, Suzaki H, Nakajima H. Tiotropium bromide inhibits TGF‐
beta‐induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways
in vitro. Int J Chron Obstruct Pulmon Dis 2010; 5:277‐286.
(93) Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA, Jr., Chan TO et al.
Cooperative regulation of p70S6 kinase by receptor tyrosine kinases and G protein‐coupled
receptors augments airway smooth muscle growth. Biochemistry 2005; 44(44):14595‐14605.
(94) Gosens R, Dueck G, Rector E, Nunes RO, Gerthoffer WT, Unruh H et al. Cooperative regulation
of GSK‐3 by muscarinic and PDGF receptors is associated with airway myocyte proliferation. Am J
Physiol Lung Cell Mol Physiol 2007; 293(5):L1348‐L1358.
(95) Kong KC, Billington CK, Gandhi U, Panettieri RA, Jr., Penn RB. Cooperative mitogenic signaling by
G protein‐coupled receptors and growth factors is dependent on G(q/11). FASEB J 2006; 20(9):1558‐
1560.
(96) Krymskaya VP, Orsini MJ, Eszterhas AJ, Brodbeck KC, Benovic JL, Panettieri RA, Jr. et al.
Mechanisms of proliferation synergy by receptor tyrosine kinase and G protein‐coupled receptor
activation in human airway smooth muscle. Am J Respir Cell Mol Biol 2000; 23(4):546‐554.
(97) Deng H, Dokshin GA, Lei J, Goldsmith AM, Bitar KN, Fingar DC et al. Inhibition of glycogen
synthase kinase‐3beta is sufficient for airway smooth muscle hypertrophy. J Biol Chem 2008;
283(15):10198‐10207.
(98) Goldsmith AM, Bentley JK, Zhou L, Jia Y, Bitar KN, Fingar DC et al. Transforming growth factor‐
beta induces airway smooth muscle hypertrophy. Am J Respir Cell Mol Biol 2006; 34(2):247‐254.
(99) Zhou L, Goldsmith AM, Bentley JK, Jia Y, Rodriguez ML, Abe MK et al. 4E‐binding protein
phosphorylation and eukaryotic initiation factor‐4E release are required for airway smooth muscle
hypertrophy. Am J Respir Cell Mol Biol 2005; 33(2):195‐202.

Muscarinic receptors in inflammation and remodeling
55
(100) Halayko AJ, Tran T, Gosens R. Phenotype and functional plasticity of airway smooth muscle:
role of caveolae and caveolins. Proc Am Thorac Soc 2008; 5(1):80‐88.
(101) Fairbank NJ, Connolly SC, Mackinnon JD, Wehry K, Deng L, Maksym GN. Airway smooth muscle
cell tone amplifies contractile function in the presence of chronic cyclic strain. Am J Physiol Lung Cell
Mol Physiol 2008; 295(3):L479‐L488.
(102) Oenema TA, Smit M, Racke K, Halayko AJ, Meurs H, Gosens R. Muscarinic receptor stimulation
augments TGF‐B1 induced contractile protein and fibronectin stimulation in airway smooth muscle
cells. Am J Respir Crit Care Med 2010; 181:A5314.
(103) Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the
progression of airway smooth muscle remodeling. Am J Respir Crit Care Med 2005; 171(10):1096‐
1102.
(104) Anthonisen NR, Connett JE, Kiley JP, Altose MD, Bailey WC, Buist AS et al. Effects of smoking
intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1.
The Lung Health Study. JAMA 1994; 272(19):1497‐1505.
(105) Van Den BA, Gailly J, Neyt M. Does tiotropium lower exacerbation and hospitalization
frequency in COPD patients: results of a meta‐analysis. BMC Pulm Med 2010; 10:50.
(106) Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory
markers to symptoms and lung function changes in COPD exacerbations. Thorax 2000; 55(2):114‐
120.
(107) Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K et al. Effect of tiotropium on
sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J 2007; 30(3):472‐
478.
(108) Perng DW, Tao CW, Su KC, Tsai CC, Liu LY, Lee YC. Anti‐inflammatory effects of
salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur Respir J 2009; 33(4):778‐
784.


CHAPTER 3
MUSCARINIC RECEPTOR SUBTYPE‐SPECIFIC EFFECTS ON
CIGARETTE SMOKE‐INDUCED INFLAMMATION IN MICE
Loes E.M. Kistemaker
I. Sophie T. Bos
Machteld N. Hylkema
Martijn C. Nawijn
Pieter S. Hiemstra
Jürgen Wess
Herman Meurs
Huib A.M. Kerstjens
Reinoud Gosens
Eur Respir J, 2013, 42(6):1677‐88

Chapter 3
58
Abstract
Rationale
Cholinergic tone contributes to airflow obstruction in COPD. Accordingly, anticholinergics
are effective bronchodilators by blocking the muscarinic M3 receptor on airway smooth
muscle. Recent evidence indicates that acetylcholine also contributes to airway
inflammation. However, which muscarinic receptor subtype(s) regulate(s) this process is
unknown.
Methods
Muscarinic receptor subtype deficient mice were exposed to cigarette smoke for four days
to investigate the contribution of the M1, M2 and M3 receptor subtypes to cigarette
smoke‐induced airway inflammation.
Results
In wild‐type mice, cigarette smoke induced an increase in macrophages, neutrophils and
lymphocytes in bronchoalveolar lavage fluid. Neutrophilic inflammation was higher in
M1R‐/‐ and M2R
‐/‐ mice compared to wild‐type mice, but lower in M3R‐/‐ mice. Accordingly,
the release of KC, MCP‐1 and IL‐6 was higher in M1R‐/‐ and M2R
‐/‐ mice and reduced in
M3R‐/‐ mice. Markers of remodeling were not increased after cigarette smoke exposure.
However, M3R‐/‐ mice had reduced expression of TGF‐β1 and matrix proteins. Cigarette
smoke‐induced inflammatory cell recruitment and KC release were also prevented by the
M3 receptor selective antagonist 4‐DAMP in wild‐type mice.
Conclusions
Our data indicate a pro‐inflammatory role for the M3 receptor in cigarette smoke‐induced
neutrophilia and cytokine release, yet an anti‐inflammatory role for M1 and M2 receptors.
Introduction
Chronic obstructive pulmonary disease (COPD) is an inflammatory disease characterized
by a progressive airflow limitation that is not fully reversible (1). The most common cause
of COPD in the Western world is tobacco smoking. Inflammation plays a central role in the
disease, and contributes to airway fibrosis, mucus hypersecretion and emphysema that is
observed in patients with COPD (1). Macrophages and neutrophils are particularly
increased in patients with COPD and this increase is related to an increased production of
specific cytokines and chemokines, including interleukin (IL)‐8, IL‐6, IL‐1β, IL‐17, monocyte
chemotactic protein‐1 (MCP‐1) and tumor necrosis factor‐α (TNF‐α) (2). Moreover, COPD
is associated with an increased production of growth factors, including transforming
growth factor (TGF)‐β and vascular endothelial growth factor (VEGF), which are thought to
contribute to the remodeling of the airways (3).

M3 receptors promote smoke‐induced inflammation
59
Acetylcholine is the primary parasympathetic neurotransmitter in the airways that induces
bronchoconstriction. Parasympathetic activity is increased in patients with COPD, and this
appears to be the major reversible component of airway obstruction (4). Therefore,
treatment with anticholinergics, inhibiting muscarinic receptor activation, is an effective
bronchodilator therapy in COPD.
Five muscarinic receptor subtypes are present in the human genome (M1‐M5). The
muscarinic M1, M2 and M3 receptors are abundantly expressed in the lungs and have been
extensively studied in the context of vagal neurotransmission. Their primary roles are
bronchoconstriction and mucus secretion, which are mainly regulated via M3 receptors on
airway smooth muscle and glands, respectively. Further, the M1 receptor facilitates
neurotransmission in the parasympathetic ganglia and regulates electrolyte and water
secretion by mucus producing cells. The M2 receptor is an auto‐inhibitory prejunctional
receptor on vagal nerves inhibiting acetylcholine release, and an abundant postjunctional
receptor on airway smooth muscle (chapter 2, 5, 6).
It is now known that acetylcholine can exert many additional, non‐neuronal effects in the
airways. Muscarinic receptors are expressed by almost all cell types in the lungs, including
epithelial and inflammatory cells (7, 8). Strikingly, these cells express all necessary
components to synthesize and release acetylcholine by themselves, including ChAT, the
synthesizing enzyme of acetylcholine. This is referred to as non‐neuronal acetylcholine
and may contribute to airway inflammation (8, 8‐10). Indeed, in vitro studies revealed a
variety of effects of acetylcholine on these cell types (10), including an induced release of
the potent neutrophil chemoattractants IL‐8 and leukotriene B4 (LTB4) from airway
epithelial, smooth muscle and inflammatory cells (11‐14). Recent evidence from in vivo
studies also demonstrated a pro‐inflammatory role for acetylcholine under
pathophysiological conditions. In a cigarette smoke‐induced mouse model of COPD,
tiotropium partly prevented the increase in total cells and neutrophils in the
bronchoalveolar lavage fluid (BALF). Furthermore, the release of various cytokines,
including IL‐6, keratinocoyte‐derived chemokine (KC, the mouse orthologue of IL‐8), LTB4
and MCP‐1, was inhibited by tiotropium (15). Our group recently demonstrated that LPS‐
induced neutrophilic inflammation could be completely prevented by tiotropium in a
guinea pig model of COPD (16). Similar findings indicating a pro‐inflammatory role for
acetylcholine have been observed in animal models of asthma, acute lung injury and
fibrosis (see chapter 2 for review).
Together, these studies clearly indicate a role for acetylcholine in inflammation, which
may have implications for anticholinergic therapy in patients with COPD. Therapy with

Chapter 3
60
anticholinergics is presently focused on the M3 receptor, since this receptor subtype
mediates bronchoconstriction. Although in vitro studies suggest a role for the M3 receptor
in cytokine release (11, 17), no information is available with respect to the muscarinic
receptor subtypes involved in the pro‐inflammatory effects of acetylcholine in vivo.
Therefore, the aim of this study was to investigate the role of the M1, M2 and M3 receptor
subtypes in cigarette smoke‐induced airway inflammation using muscarinic receptor
subtype‐deficient mice. We hypothesized that the M3 receptor plays a predominant pro‐
inflammatory role. In order to study this, we assessed inflammatory cell counts and
mediator release in the lavage fluid. Furthermore, we analyzed the expression of genes
associated with remodeling.
Methods
Animals
Homozygous, inbred, specific‐pathogen‐free breeding colonies of M1R‐/‐, M2R
‐/‐ and M3R‐/‐
mice and C57Bl/6NTac wild‐type (WT) mice with the same genetic background were
obtained from Taconic (Cambridge City, Indiana, USA). The M1R‐/‐, M2R
‐/‐ and M3R‐/‐ mice
used were generated on a 129 Sv/J background and backcrossed for at least 10
generations onto the C57Bl/6NTac background (18‐20). Knock‐out animals did not differ
from WT controls in overall health, fertility and longevity (18‐20), although the weight of
M3R‐/‐ mice was less compared to WT mice (table S1). Exposure to cigarette smoke (CS) did
not affect the weight of the mice (table S1). Animals were housed conventionally under a
12‐h light‐dark cycle and received food and water ad libitum. All experiments were
performed in accordance with the national guidelines and approved by the University of
Groningen Committee for Animal Experimentation.
Animal model
Male mice (n=8‐9 per group, 10 – 12 weeks old) were exposed to CS from Kentucky 3R4F
research cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, USA)
on 4 consecutive days by whole body exposure. Each cigarette was smoked without a filter
in 5 minutes at a rate of 5L/hr in a ratio with 60L/hr air using a peristaltic pump (45 rpm,
Watson Marlow 323 E/D, Rotterdam, The Netherlands). CS was directly distributed into a
6‐liter perspex box. On day 1, mice were exposed to the mainstream smoke of one
cigarette in the morning and three cigarettes in the afternoon. On day 2 to 4, mice were
exposed to five cigarettes in the morning and five in the afternoon (figure 1). Control
animals were handled in the same way but exposed to fresh air only. Because of the
capacity of the experimental set‐up, not all animals used for this study could be included
in a single experiment. Therefore, experiments were performed on 3 occasions (n=19‐24

M3 receptors promote smoke‐induced inflammation
61
animals per experiment). Air and cigarette smoke exposed WT mice were included in
every experiment to minimize variability. Sixteen hours after the last CS exposure, animals
were euthanized by intraperitoneal pentobarbital injection (400 mg/kg, hospital
pharmacy, University Medical Center Groningen), after which the lungs were immediately
lavaged, resected and snap frozen in liquid nitrogen.
Muscarinic antagonist administration
In a sub‐study, the M3 receptor selective antagonist 4‐DAMP (1mg/kg) was administered
to WT mice (n=7) by intraperitoneal injection, 30 minutes prior to each CS exposure. The
same experimental protocol was used as described above.
Analysis of the bronchoalveolar lavage fluid
After euthanizing the mice, the lungs were gently lavaged through a tracheal cannula with
1 ml PBS containing 5% BSA and protease inhibitors (inhibiting chymotrypsin, thermolysin,
papain, pronase, pancreatic extract and trypsin; F. Hoffman‐La Roche, Basel, Switzerland)
and another 4 times with 1 ml PBS. Cells were pelleted and the supernatants of the first
fraction were stored at ‐20◦C for measurement of cytokines and growth factors by ELISA.
For each animal individually, BAL cells of the different fractions were combined,
resuspended in 500 µl PBS, and total cell numbers were determined. For cytological
examination, cytospin‐preparations were stained with May–Grünwald and Giemsa (both
Sigma, St. Louis) and a differential cell count was performed by counting at least 400 cells
in duplicate in a blinded fashion. KC, MCP‐1, IL‐6, IL‐1β, IL‐17, TNF‐α and VEGF release was
determined in BALF supernatants by a MILLIPLEX assay (Millipore, Billerica, USA). TGF‐β in
BALF was determined by an ELISA kit (R&D Systems, Minneapolis, USA) according to the
manufacturer’s instructions.
Figure 1. Experimental procedure. Male
C57Bl/6NTac mice were exposed to cigarette
smoke twice daily on 4 consecutive days by
whole body exposure. Sixteen hours after the
last smoke exposure a bronchoalveolar lavage
is performed and lungs are harvested for lung
tissue homogenates and cryo‐sections.

Chapter 3
62
ChAT staining
To identify ChAT expression, 5‐µm‐thick cryo‐sections (n=4) were stained with a specific
rabbit anti‐ChAT‐antibody provided by Prof. Kummer (Institute for Anatomy and Cell
Biology, Justus‐Liebig‐University, Giessen, Germany). The antibody was visualized using a
horseradish peroxidase‐linked secondary antibody and diaminobenzidine (1 mg/ml).
Airways within each section were digitally photographed.
Analysis of gene expression in lung tissue
Total RNA was extracted from lung tissue (right superior lobe) or BAL cells using the
RNeasy mini kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's
instructions. Lung homogenates were prepared by pulverizing the tissue under liquid
nitrogen. Equal amounts of total mRNA were then reverse transcribed and cDNA was
subjected to real‐time qPCR (Westburg, Leusden, The Netherlands). Real time PCR was
performed with denaturation at 94°C for 30 seconds, annealing at 59°C for 30 seconds and
extension at 72°C for 30 seconds for 40 cycles followed by 10 minutes at 72°C. Real‐time
PCR data were analyzed using the comparative cycle threshold (Ct: amplification cycle
number) method. The amount of target gene was normalized to the endogenous
reference gene 18S ribosomal RNA. Several other housekeeping genes, including β2‐
microglobulin and GAPDH, were tested for the influence of the experimental procedure on
the expression. The expression of all housekeeping genes, including 18S, was stable in the
tested conditions. The specific forward and reverse primers used are listed in table S2.
Statistical analysis
Data are presented as mean ± s.e. of the mean. Statistical differences between means
were calculated using one‐ or two‐way ANOVA, followed by Newman Keuls multiple
comparison tests. Differences were considered significant at p<0.05.
Results
Characterization of the mice
The genotypes of the knock‐out mice were confirmed by PCR analysis of mouse ear DNA
(figure 2A, table S3). We also examined whether the deletion of one muscarinic receptor
gene affected the expression levels of the two other muscarinic receptors expressed in the
lung. As shown in figure 2B, no such compensatory changes were observed. Muscarinic
receptors were highly expressed in lung tissue homogenates, with M2R>M3R>M1R (figure
2B).
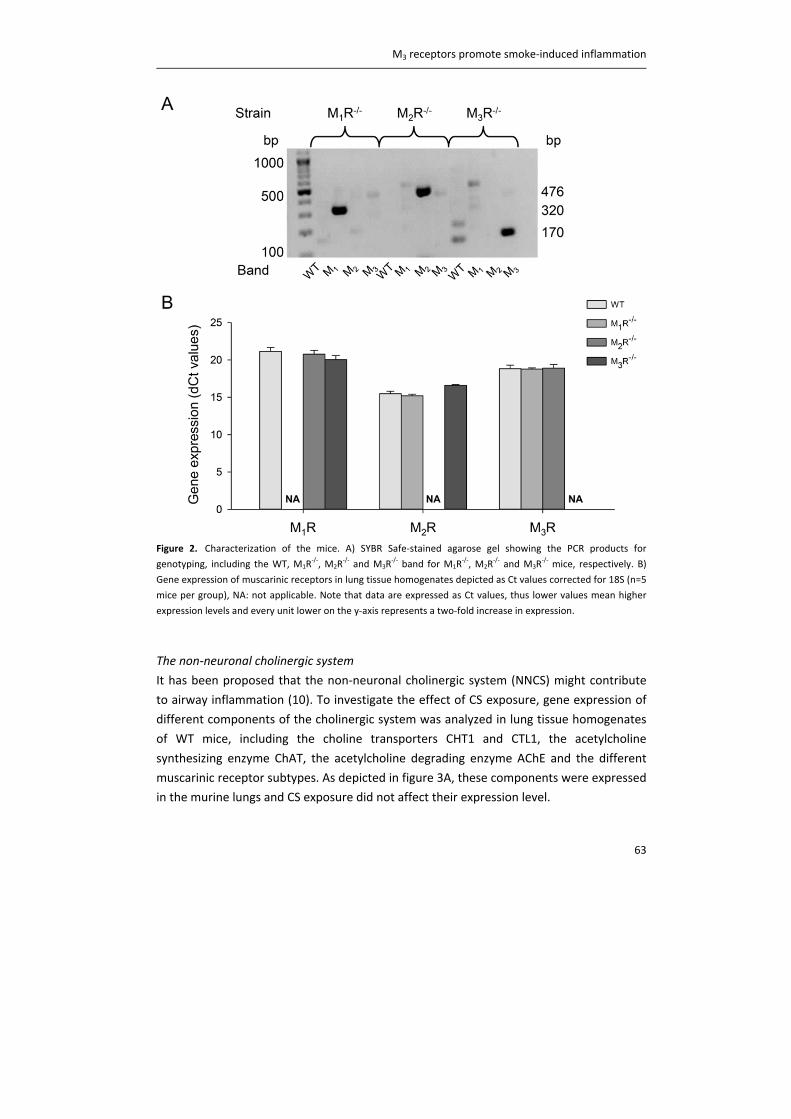
M3 receptors promote smoke‐induced inflammation
63
Figure 2. Characterization of the mice. A) SYBR Safe‐stained agarose gel showing the PCR products for
genotyping, including the WT, M1R‐/‐, M2R
‐/‐ and M3R
‐/‐ band for M1R
‐/‐, M2R
‐/‐ and M3R
‐/‐ mice, respectively. B)
Gene expression of muscarinic receptors in lung tissue homogenates depicted as Ct values corrected for 18S (n=5
mice per group), NA: not applicable. Note that data are expressed as Ct values, thus lower values mean higher
expression levels and every unit lower on the y‐axis represents a two‐fold increase in expression.
The non‐neuronal cholinergic system
It has been proposed that the non‐neuronal cholinergic system (NNCS) might contribute
to airway inflammation (10). To investigate the effect of CS exposure, gene expression of
different components of the cholinergic system was analyzed in lung tissue homogenates
of WT mice, including the choline transporters CHT1 and CTL1, the acetylcholine
synthesizing enzyme ChAT, the acetylcholine degrading enzyme AChE and the different
muscarinic receptor subtypes. As depicted in figure 3A, these components were expressed
in the murine lungs and CS exposure did not affect their expression level.
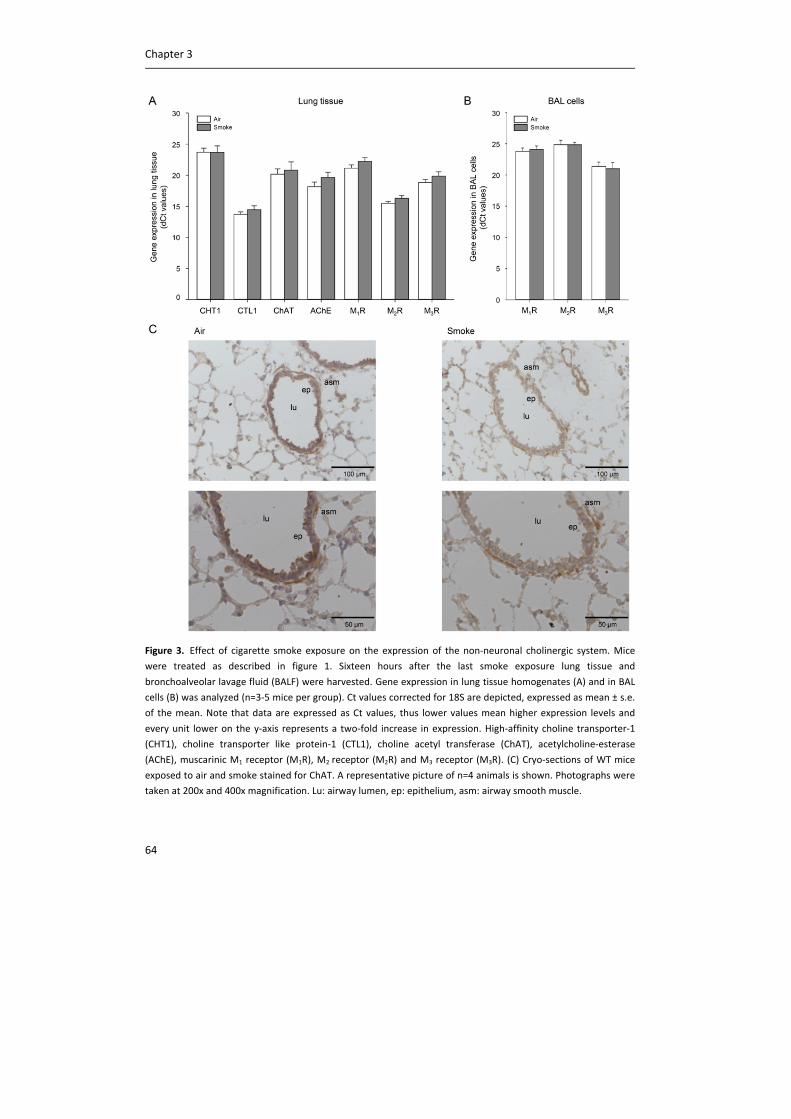
Chapter 3
64
Figure 3. Effect of cigarette smoke exposure on the expression of the non‐neuronal cholinergic system. Mice
were treated as described in figure 1. Sixteen hours after the last smoke exposure lung tissue and
bronchoalveolar lavage fluid (BALF) were harvested. Gene expression in lung tissue homogenates (A) and in BAL
cells (B) was analyzed (n=3‐5 mice per group). Ct values corrected for 18S are depicted, expressed as mean ± s.e.
of the mean. Note that data are expressed as Ct values, thus lower values mean higher expression levels and
every unit lower on the y‐axis represents a two‐fold increase in expression. High‐affinity choline transporter‐1
(CHT1), choline transporter like protein‐1 (CTL1), choline acetyl transferase (ChAT), acetylcholine‐esterase
(AChE), muscarinic M1 receptor (M1R), M2 receptor (M2R) and M3 receptor (M3R). (C) Cryo‐sections of WT mice
exposed to air and smoke stained for ChAT. A representative picture of n=4 animals is shown. Photographs were
taken at 200x and 400x magnification. Lu: airway lumen, ep: epithelium, asm: airway smooth muscle.

M3 receptors promote smoke‐induced inflammation
65
Similarly, the muscarinic receptor expression in the BAL cells from WT mice was not
altered after CS exposure figure 3B). In lung tissue, M2 receptors were expressed at the
highest levels (M2R>M3R>M1R), whereas in the cells from the lavage fluid M3 receptors
were expressed at the highest levels (M3R>M1R>M2R; figure S1). ChAT expression,
detected by immunohistochemical staining, was localized to the epithelium and smooth
muscle layer of the airway wall in WT mice (figure 3C). Exposure to CS did not alter the
expression or localization of ChAT (figure 3C).
Cigarette smoke‐induced inflammatory cell recruitment
To study the contribution of muscarinic receptor subtypes to CS‐induced airway
inflammation, cell counts were determined in the BALF of WT, M1R‐/‐, M2R
‐/‐ and M3R‐/‐
mice. All mice exposed to CS had a 2‐fold increase in the number of inflammatory cells
compared to air‐exposed control animals (figure 4A). The predominant cell type after CS
exposure was the macrophage, which almost doubled in number in all strains (figure 4B).
Only small increases in lymphocytic infiltration were observed after CS exposure, which
were comparable in all strains (figure 4C). Neutrophilic infiltration however, showed
significant differences among the strains (figure 4D). A 4‐fold increase in the amount of
neutrophils was observed in WT mice compared to air exposed animals. In M1R‐/‐ and
M2R‐/‐ mice however, the increase in neutrophil number upon CS exposure was much
higher, up to a 24‐fold in M1R‐/‐ mice. In striking contrast, no significant increase in
neutrophil numbers was observed in M3R‐/‐ mice after CS exposure compared to air‐
exposed control animals (figure 4D).
Cigarette smoke‐induced cytokine release
Subsequently, inflammatory cytokine release in BALF was determined. Levels of KC (the
mouse orthologue of IL‐8) in BALF of WT mice exposed to CS were 15‐fold higher
compared to air‐exposed animals (figure 5A). No significant differences in MCP‐1 and IL‐6
release were observed in CS‐exposed WT mice (figure 5B and C). In M1R‐/‐ mice, CS‐
exposed mice had higher levels of KC, MCP‐1 and IL‐6 compared to air‐exposed mice.
Interestingly, the concentration of all these cytokines was significantly higher when
compared to WT CS‐exposed mice. In CS‐exposed M2R‐/‐ mice, KC release was also
significantly higher, both compared to air‐exposed mice and to WT CS‐exposed mice,
whereas MCP‐1 and IL‐6 release were not significantly different. In M3R‐/‐ mice, no
increase in the release of any of these cytokines was observed after CS exposure, and KC
release was significantly lower compared to WT CS‐exposed mice (figure 5). Levels of IL‐
17, IL‐1β and TNF‐α were below detection limit in all strains (not shown).
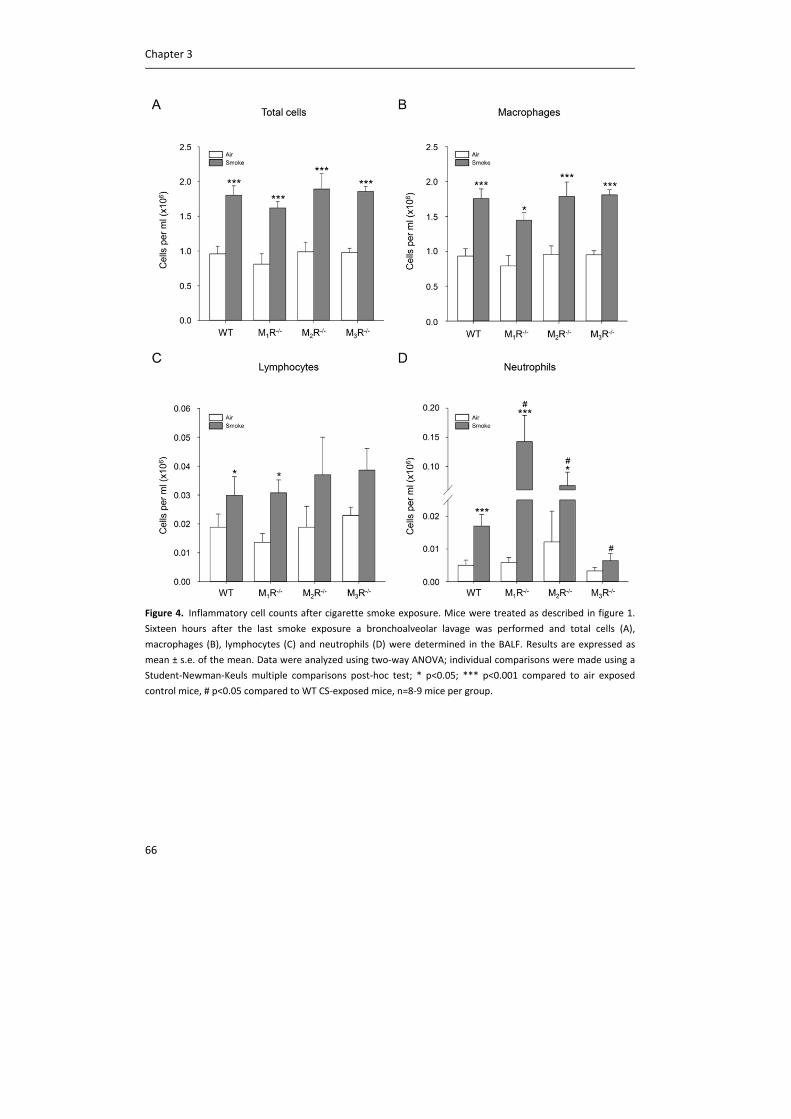
Chapter 3
66
Figure 4. Inflammatory cell counts after cigarette smoke exposure. Mice were treated as described in figure 1.
Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed and total cells (A),
macrophages (B), lymphocytes (C) and neutrophils (D) were determined in the BALF. Results are expressed as
mean ± s.e. of the mean. Data were analyzed using two‐way ANOVA; individual comparisons were made using a
Student‐Newman‐Keuls multiple comparisons post‐hoc test; * p<0.05; *** p<0.001 compared to air exposed
control mice, # p<0.05 compared to WT CS‐exposed mice, n=8‐9 mice per group.
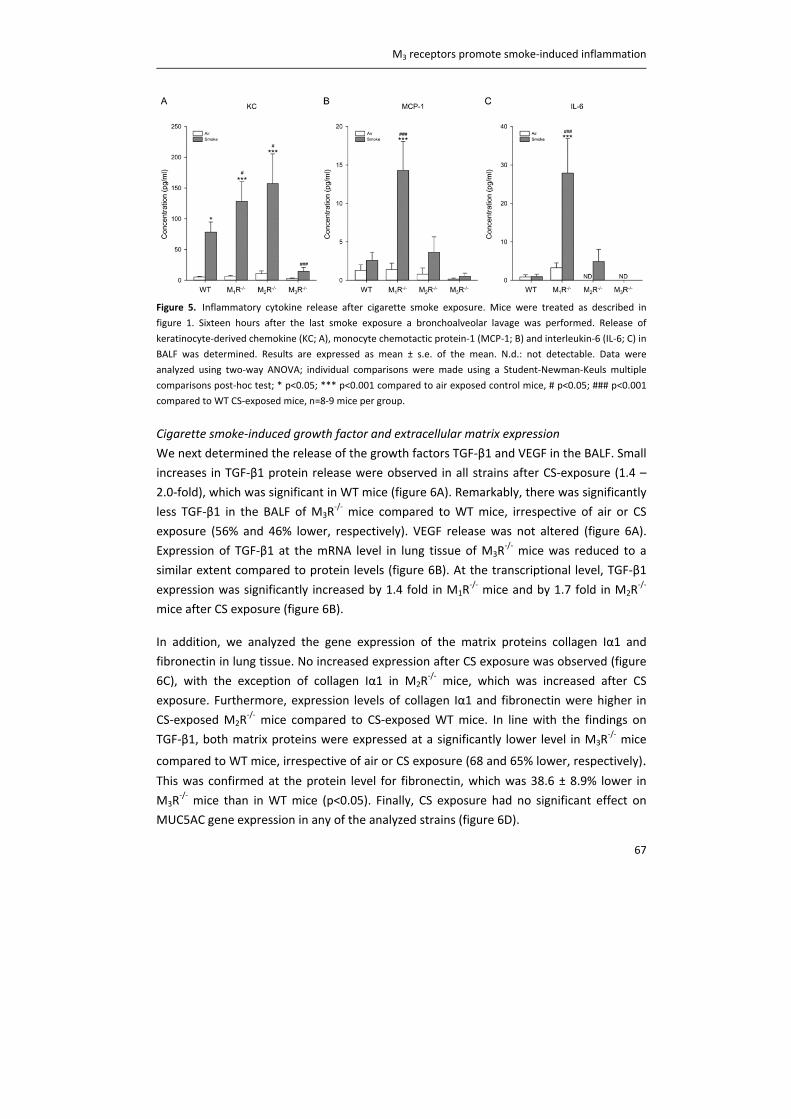
M3 receptors promote smoke‐induced inflammation
67
Figure 5. Inflammatory cytokine release after cigarette smoke exposure. Mice were treated as described in
figure 1. Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed. Release of
keratinocyte‐derived chemokine (KC; A), monocyte chemotactic protein‐1 (MCP‐1; B) and interleukin‐6 (IL‐6; C) in
BALF was determined. Results are expressed as mean ± s.e. of the mean. N.d.: not detectable. Data were
analyzed using two‐way ANOVA; individual comparisons were made using a Student‐Newman‐Keuls multiple
comparisons post‐hoc test; * p<0.05; *** p<0.001 compared to air exposed control mice, # p<0.05; ### p<0.001
compared to WT CS‐exposed mice, n=8‐9 mice per group.
Cigarette smoke‐induced growth factor and extracellular matrix expression
We next determined the release of the growth factors TGF‐β1 and VEGF in the BALF. Small
increases in TGF‐β1 protein release were observed in all strains after CS‐exposure (1.4 –
2.0‐fold), which was significant in WT mice (figure 6A). Remarkably, there was significantly
less TGF‐β1 in the BALF of M3R‐/‐ mice compared to WT mice, irrespective of air or CS
exposure (56% and 46% lower, respectively). VEGF release was not altered (figure 6A).
Expression of TGF‐β1 at the mRNA level in lung tissue of M3R‐/‐ mice was reduced to a
similar extent compared to protein levels (figure 6B). At the transcriptional level, TGF‐β1
expression was significantly increased by 1.4 fold in M1R‐/‐ mice and by 1.7 fold in M2R
‐/‐
mice after CS exposure (figure 6B).
In addition, we analyzed the gene expression of the matrix proteins collagen Iα1 and
fibronectin in lung tissue. No increased expression after CS exposure was observed (figure
6C), with the exception of collagen Iα1 in M2R‐/‐ mice, which was increased after CS
exposure. Furthermore, expression levels of collagen Iα1 and fibronectin were higher in
CS‐exposed M2R‐/‐ mice compared to CS‐exposed WT mice. In line with the findings on
TGF‐β1, both matrix proteins were expressed at a significantly lower level in M3R‐/‐ mice
compared to WT mice, irrespective of air or CS exposure (68 and 65% lower, respectively). This was confirmed at the protein level for fibronectin, which was 38.6 ± 8.9% lower in
M3R‐/‐ mice than in WT mice (p<0.05). Finally, CS exposure had no significant effect on
MUC5AC gene expression in any of the analyzed strains (figure 6D).
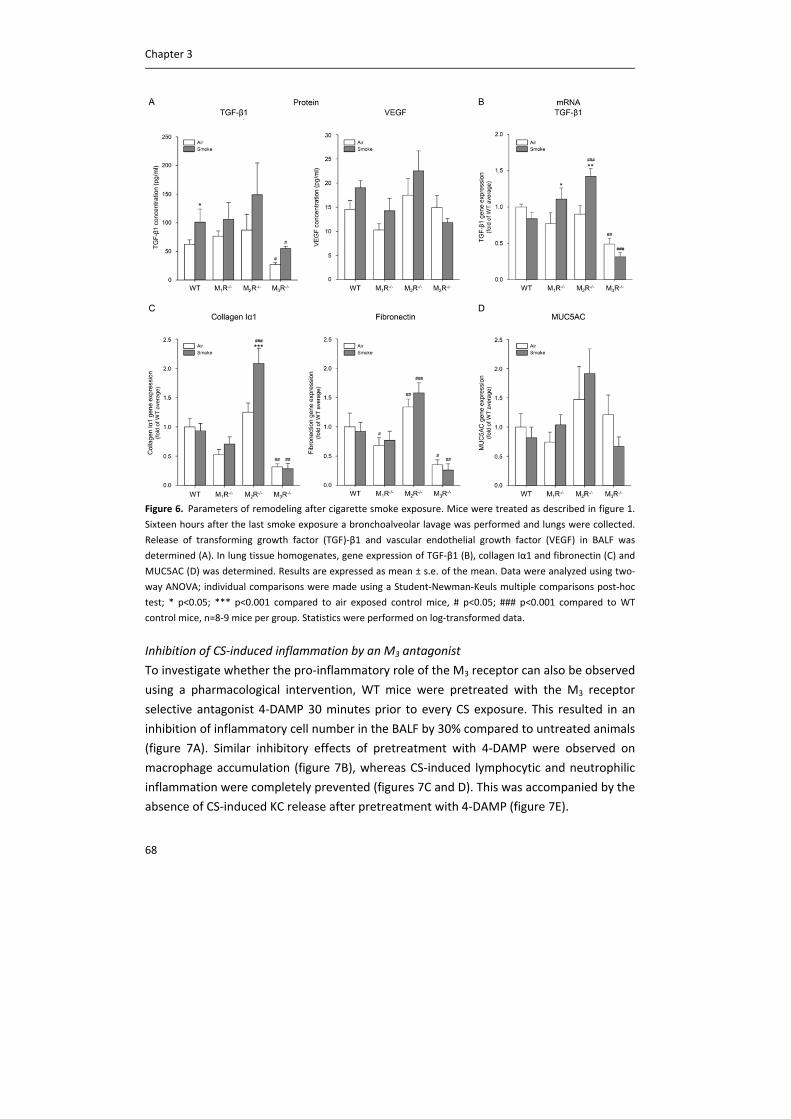
Chapter 3
68
Figure 6. Parameters of remodeling after cigarette smoke exposure. Mice were treated as described in figure 1.
Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed and lungs were collected.
Release of transforming growth factor (TGF)‐β1 and vascular endothelial growth factor (VEGF) in BALF was
determined (A). In lung tissue homogenates, gene expression of TGF‐β1 (B), collagen Iα1 and fibronectin (C) and
MUC5AC (D) was determined. Results are expressed as mean ± s.e. of the mean. Data were analyzed using two‐
way ANOVA; individual comparisons were made using a Student‐Newman‐Keuls multiple comparisons post‐hoc
test; * p<0.05; *** p<0.001 compared to air exposed control mice, # p<0.05; ### p<0.001 compared to WT
control mice, n=8‐9 mice per group. Statistics were performed on log‐transformed data.
Inhibition of CS‐induced inflammation by an M3 antagonist
To investigate whether the pro‐inflammatory role of the M3 receptor can also be observed
using a pharmacological intervention, WT mice were pretreated with the M3 receptor
selective antagonist 4‐DAMP 30 minutes prior to every CS exposure. This resulted in an
inhibition of inflammatory cell number in the BALF by 30% compared to untreated animals
(figure 7A). Similar inhibitory effects of pretreatment with 4‐DAMP were observed on
macrophage accumulation (figure 7B), whereas CS‐induced lymphocytic and neutrophilic
inflammation were completely prevented (figures 7C and D). This was accompanied by the
absence of CS‐induced KC release after pretreatment with 4‐DAMP (figure 7E).
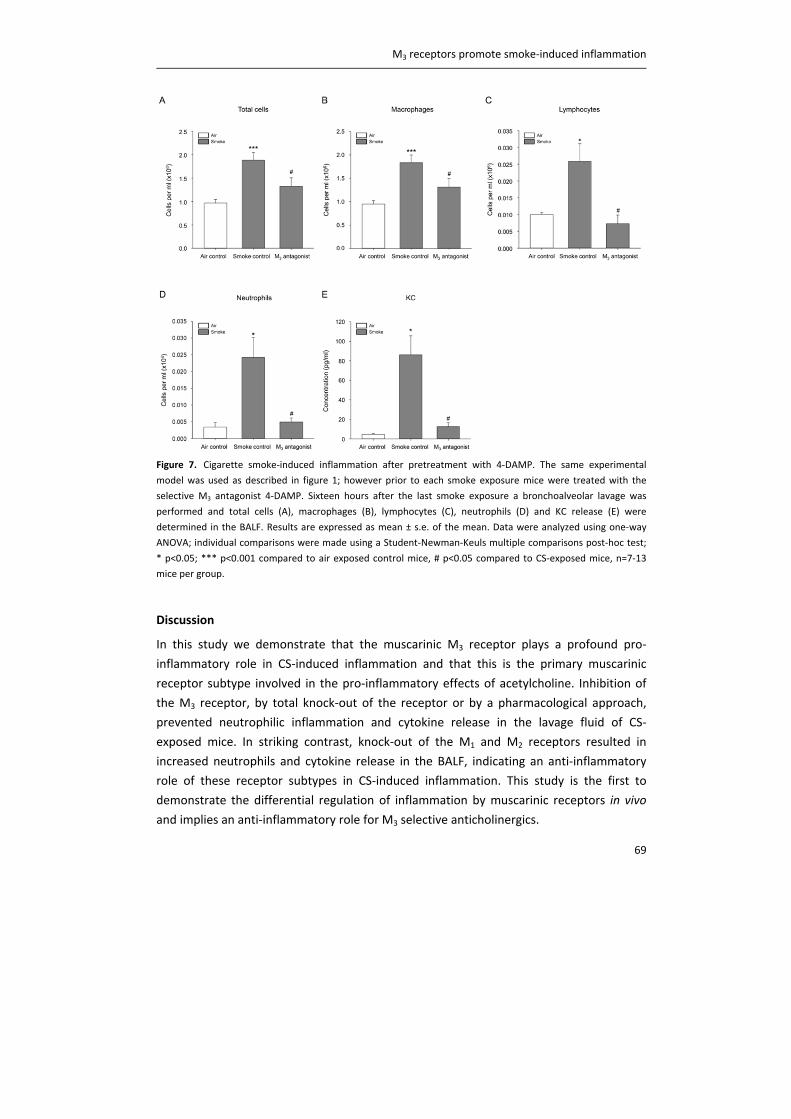
M3 receptors promote smoke‐induced inflammation
69
Figure 7. Cigarette smoke‐induced inflammation after pretreatment with 4‐DAMP. The same experimental
model was used as described in figure 1; however prior to each smoke exposure mice were treated with the
selective M3 antagonist 4‐DAMP. Sixteen hours after the last smoke exposure a bronchoalveolar lavage was
performed and total cells (A), macrophages (B), lymphocytes (C), neutrophils (D) and KC release (E) were
determined in the BALF. Results are expressed as mean ± s.e. of the mean. Data were analyzed using one‐way
ANOVA; individual comparisons were made using a Student‐Newman‐Keuls multiple comparisons post‐hoc test;
* p<0.05; *** p<0.001 compared to air exposed control mice, # p<0.05 compared to CS‐exposed mice, n=7‐13
mice per group.
Discussion
In this study we demonstrate that the muscarinic M3 receptor plays a profound pro‐
inflammatory role in CS‐induced inflammation and that this is the primary muscarinic
receptor subtype involved in the pro‐inflammatory effects of acetylcholine. Inhibition of
the M3 receptor, by total knock‐out of the receptor or by a pharmacological approach,
prevented neutrophilic inflammation and cytokine release in the lavage fluid of CS‐
exposed mice. In striking contrast, knock‐out of the M1 and M2 receptors resulted in
increased neutrophils and cytokine release in the BALF, indicating an anti‐inflammatory
role of these receptor subtypes in CS‐induced inflammation. This study is the first to
demonstrate the differential regulation of inflammation by muscarinic receptors in vivo
and implies an anti‐inflammatory role for M3 selective anticholinergics.

Chapter 3
70
Neutrophils are considered as one of the major cell types involved in COPD (2).Various
studies suggest an important role for acetylcholine in regulating neutrophilic
inflammation. Activation of muscarinic receptors can contribute to neutrophil influx by
inducing neutrophil chemotactic activity from macrophages (21, 22) and LTB4 release
from sputum cells of COPD patients (14). Furthermore, IL‐8 is released from epithelial and
airway smooth muscle cells in response to muscarinic receptor stimulation (11, 13). These
findings are supported by in vivo studies in which CS‐ and LPS‐induced neutrophilia was
inhibited by the muscarinic receptor antagonist tiotropium (15, 16).
The regulatory effects of muscarinic receptors appeared to be specific for neutrophils in
the muscarinic receptor deficient mice, whereas macrophages and lymphocytes were not
altered. Our results on neutrophilic inflammation are in line with various in vitro studies
demonstrating that the pro‐inflammatory effects of muscarinic receptor activation are
mainly dependent on the M3 receptor subtype. Thus, it has been shown that 4‐DAMP and
DAU5884, M3 receptor selective antagonists, inhibited methacholine and CS‐induced IL‐8
release from airway smooth muscle cells (11). In addition, alveolar macrophage mediated
migration of neutrophils from COPD patients was inhibited by 4‐DAMP (17). Moreover,
the M3 receptor was the primary receptor subtype expressed by inflammatory cells within
the BALF as shown in our study. It is also known that inflammatory cells express M3
receptors and that this receptor mediates pro‐inflammatory effects (7). We therefore
believe that the pro‐inflammatory effect of the M3 receptor as found in our study is
dependent on regulation of cytokine release by structural cells, in combination with direct
activation of M3 receptors on inflammatory cells.
In contrast to the findings in M3R‐/‐ mice, neutrophilic inflammation was increased in M2R
‐/‐
mice compared to WT mice. The M2 receptor is located prejunctionally on pre‐ and
postganglionic nerves and acts as an inhibitory autoreceptor limiting acetylcholine release
(5). Furthermore, the M2 receptor is expressed postjunctionally by smooth muscle cells
and fibroblasts (chapter 2, 23). With acetylcholine acting as a pro‐inflammatory mediator
inducing chemokine release from structural and inflammatory cells via the M3 receptor,
increased levels of acetylcholine in the M2R‐/‐ mice, due to loss of its autoinhibitory role,
may therefore explain the observed aggravated neutrophilia. In support of such a role, M2
receptor expression appeared low on inflammatory cells in the BALF, but high in lung
tissue, suggesting that the effects of M2 receptor deficiency are not due to direct effects
on inflammatory cells. The literature supports this notion, indicating that the pro‐
inflammatory effects of acetylcholine in macrophages, epithelial cells and airway smooth
muscle cells are not mediated by M2 receptors (11, 22, 24).

M3 receptors promote smoke‐induced inflammation
71
A role for M2 receptors as prejunctional autoreceptors driving exaggerated acetylcholine
release and inflammation has significant implications. Acetylcholine has long been known
as a classical neurotransmitter. More recent findings suggest that acetylcholine can also
be released from non‐neuronal origins, including epithelial, airway smooth muscle and
inflammatory cells (8, 9). It is not yet known to which extent this non‐neuronal
acetylcholine affects airway inflammation (10). The release of non‐neuronal acetylcholine
is not known to be affected by M2 receptors in an auto‐inhibitory way. Our data therefore
imply an important role for neuronal acetylcholine in CS‐induced inflammation. Further,
we did not find any upregulation of expression of components of the NNCS in the lungs of
CS‐exposed mice, in contrast to the previously reported increase in cultured human airway
epithelial cells after exposure to CS extract (12). Future studies investigating the
contribution of neuronal and non‐neuronal acetylcholine to inflammation are clearly
warranted.
Surprisingly, neutrophilic inflammation was also enhanced in M1R‐/‐ mice. It is well known
that M1 receptors facilitate neurotransmission in the parasympathetic ganglia (5). Based
on this however, one would expect that M1 receptor deficiency causes reduced
acetylcholine release leading to inhibition of inflammation. Reinheimer et al. reported that
the M1 receptor selective antagonist pirenzepine can antagonize the inhibitory effect of
acetylcholine on histamine release from human mast cells (25). Lack of this inhibitory M1
receptor might explain the increased neutrophil chemotaxis, since mast cell numbers are
increased upon smoking and higher in patients with COPD (26, 27). Alternatively, as the
M1 receptor also controls electrolyte and water secretion by airway epithelial cells (28),
lack of M1 receptor expression may result in a reduced ability of M1R‐/‐ mice to clear their
lungs of smoke particles after CS exposure, leading to aggravated inflammatory and injury
responses. The sharp induction of the damage response‐associated cytokines IL‐6 and
MCP‐1 in M1R‐/‐ mice, which is absent in WT mice, supports this hypothesis.
The results of this study on transgenic mice suggest a primary role for the M3 receptor in
regulating CS‐induced inflammation, implying that M3 receptor subtype selectivity of
anticholinergics would be beneficial. Indeed, we show that pharmacological inhibition of
the M3 receptor using 4‐DAMP partly prevented accumulation of inflammatory cells in
BALF, accompanied by a strong inhibition of CS‐induced KC release. 4‐DAMP is selective
for M3 receptors over M2 (~16‐fold) (6) and to a lesser extent M1 receptors (~3‐fold) (29).
Interestingly, the anti‐inflammatory effects of pharmacological inhibition of the M3
receptor are more pronounced than effects of knock‐out of this receptor. Similar
discrepant data are reported on muscarinic receptor mediated contraction ex vivo, which
can be fully inhibited with a M3 receptor selective antagonist in wild‐type mice, whereas

Chapter 3
72
only partial inhibition of contraction is observed in M3R knock‐out mice (30, 31). Although
the mechanism behind this discrepancy is unclear it thus appears that compensating
mechanisms are operative in the knock‐out mice that limit the impact of the M3 receptor
deficiency.
Although tiotropium is known to be kinetically selective for the M3 receptor (dissociation
half‐life = 27h), it still has a dissociation half‐life from the M1 receptor of 10.5 hours (32).
Steady‐state binding affinity of tiotropium for the M1, M2 and M3 receptors is not different
(33). The half‐life ratio of ipratropium and of aclidinium and glycopyrrolate, two
anticholinergics under development, for the M3 versus M1 receptor is comparable to
tiotropium (32). Our study suggests that an even more selective compound solely
inhibiting M3 receptors is desirable and may lead to improved effects on CS‐induced
inflammation.
Interestingly, in our study basal expression of TGF‐β1, collagen Iα1 and fibronectin was
significantly lower in M3R‐/‐ mice compared to WT mice. In M2R
‐/‐ mice, expression of these
components was increased after CS exposure. This suggests that in addition to
inflammation, acetylcholine regulates important aspects of lung structure via the M3
receptor. Indeed, M3 receptors are expressed by structural cells in the airways, including
epithelial cells and airway smooth muscle cells (23). Moreover, in vitro studies have
demonstrated a role for the M3 receptor in the regulation of airway smooth muscle
proliferation (34) and in vivo studies have shown a protective effect of anticholinergics on
matrix protein deposition (16, 35). The exact roles of the individual muscarinic receptor
subtypes in this process are not yet clear and remain to be elucidated (chapter 2).
Evidence for the pro‐inflammatory role of acetylcholine from in vitro and in vivo studies is
increasing (chapter 2). However, the translational utility of these observations is not yet
clear, since in patients with COPD, effects on inflammation or on the rate of decline in lung
function after anticholinergic therapy have not been demonstrated. It is known from the
UPLIFT (Understanding Potential Long‐Term Impacts on Function with Tiotropium) study
that the use of tiotropium is associated with a reduction in the number of exacerbations,
generally seen as inflammatory events (36). Patients who have more exacerbations
demonstrate increased levels of inflammatory markers at stable state (37, 38). In two
recent studies however, no direct evidence for an anti‐inflammatory effect was found,
since IL‐6 and IL‐8 levels in the sputum of patients with COPD were not decreased after
anticholinergic therapy (39, 40). However, both studies have substantial limitations as
discussed by the authors. In the study of Perng et al., the treatment group was small and
patients were only treated with tiotropium for 12 weeks (40). In the study of Powrie et al.,

M3 receptors promote smoke‐induced inflammation
73
the amount of sputum was reduced after tiotropium treatment, which might have
resulted in increased cytokine concentrations (39). Future studies using different methods
to assess inflammation could therefore resolve the question whether anticholinergics
indeed have anti‐inflammatory properties in patients with COPD. At present, there is no
evidence for such a role.
In conclusion, the results of our study demonstrate that inhibition of the M3 receptor
prevents inflammation in response to CS‐exposure in mice. This confirms the previously
established pro‐inflammatory role of acetylcholine in the pathophysiology of airway
diseases, and demonstrates that this is solely mediated via M3 receptors, since knock‐out
of the M1 and M2 receptor aggravated inflammation compared to WT mice. This study
therefore opens new perspectives on M3 receptor selective anticholinergics to specifically
target airway inflammation.
Acknowledgements
The authors would like to thank the Netherlands Lung Foundation for financial support
(grant: 3.2.08.014).
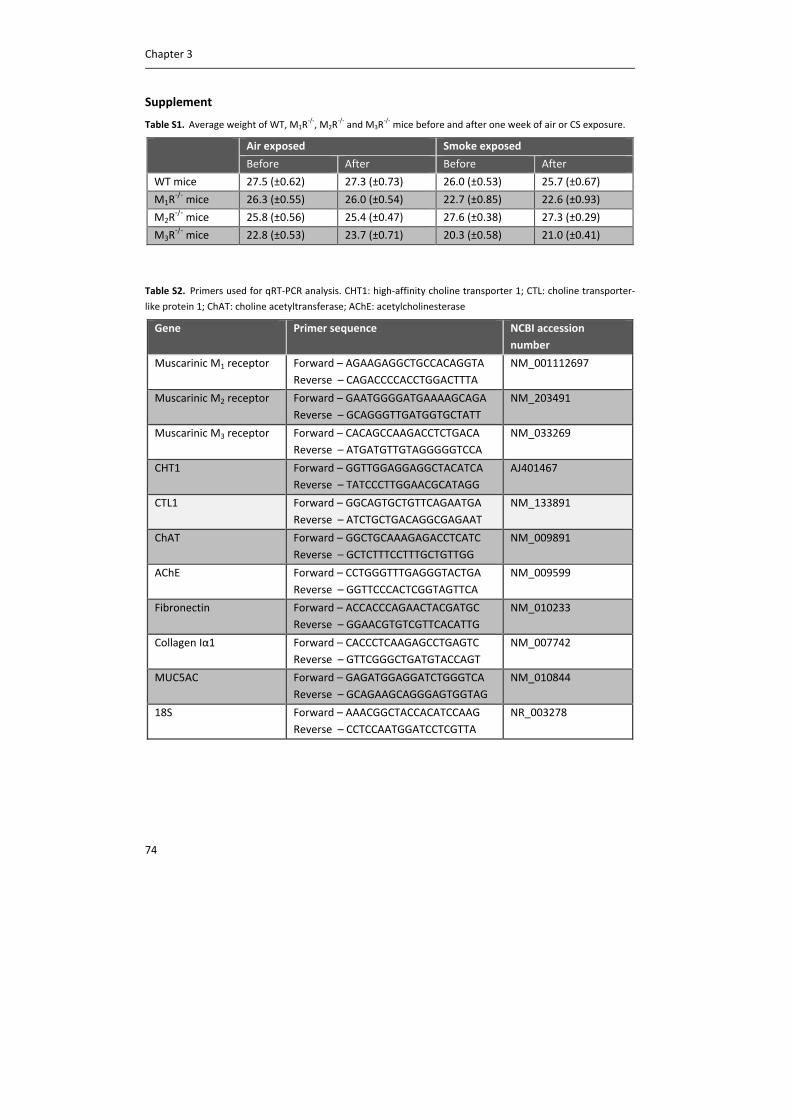
Chapter 3
74
Supplement
Table S1. Average weight of WT, M1R‐/‐, M2R
‐/‐ and M3R
‐/‐ mice before and after one week of air or CS exposure.
Air exposed Smoke exposed
Before After Before After
WT mice 27.5 (±0.62) 27.3 (±0.73) 26.0 (±0.53) 25.7 (±0.67)
M1R‐/‐ mice 26.3 (±0.55) 26.0 (±0.54) 22.7 (±0.85) 22.6 (±0.93)
M2R‐/‐ mice 25.8 (±0.56) 25.4 (±0.47) 27.6 (±0.38) 27.3 (±0.29)
M3R‐/‐ mice 22.8 (±0.53) 23.7 (±0.71) 20.3 (±0.58) 21.0 (±0.41)
Table S2. Primers used for qRT‐PCR analysis. CHT1: high‐affinity choline transporter 1; CTL: choline transporter‐
like protein 1; ChAT: choline acetyltransferase; AChE: acetylcholinesterase
Gene Primer sequence NCBI accession
number
Muscarinic M1 receptor Forward – AGAAGAGGCTGCCACAGGTA
Reverse – CAGACCCCACCTGGACTTTA
NM_001112697
Muscarinic M2 receptor Forward – GAATGGGGATGAAAAGCAGA
Reverse – GCAGGGTTGATGGTGCTATT
NM_203491
Muscarinic M3 receptor Forward – CACAGCCAAGACCTCTGACA
Reverse – ATGATGTTGTAGGGGGTCCA
NM_033269
CHT1 Forward – GGTTGGAGGAGGCTACATCA
Reverse – TATCCCTTGGAACGCATAGG
AJ401467
CTL1 Forward – GGCAGTGCTGTTCAGAATGA
Reverse – ATCTGCTGACAGGCGAGAAT
NM_133891
ChAT Forward – GGCTGCAAAGAGACCTCATC
Reverse – GCTCTTTCCTTTGCTGTTGG
NM_009891
AChE Forward – CCTGGGTTTGAGGGTACTGA
Reverse – GGTTCCCACTCGGTAGTTCA
NM_009599
Fibronectin Forward – ACCACCCAGAACTACGATGC
Reverse – GGAACGTGTCGTTCACATTG
NM_010233
Collagen Iα1 Forward – CACCCTCAAGAGCCTGAGTC
Reverse – GTTCGGGCTGATGTACCAGT
NM_007742
MUC5AC Forward – GAGATGGAGGATCTGGGTCA
Reverse – GCAGAAGCAGGGAGTGGTAG
NM_010844
18S Forward – AAACGGCTACCACATCCAAG
Reverse – CCTCCAATGGATCCTCGTTA
NR_003278

M3 receptors promote smoke‐induced inflammation
75
Table S3. Primers used for genotyping.
Gene Primer sequence
M1‐S1 5'‐CCAACATCACCGTCTTGGCAC‐3'
M1‐A1 5'‐AGTGCCAATGATGAGATCAGC‐3'
M2‐A6 5'‐GCTATTACCAGTCCTTACAAGACA‐3'
M2‐B5 5'‐CCAGAGGATGAAGGAAAGAACC‐3'
M3‐A3 5'‐AAGACCACAGTAGCAGTG‐3'
M3‐B 5'‐CTCTCTACATCCATAGTCCC‐3'
NEO‐1 5'‐CAGCTCATTCCTCCCACTCATGAT‐3'
M3‐NEO9 5'‐TGGATGTGGAATGTGTGCGAGG‐3'
Figure S1. Muscarinic receptor expression in lung tissue and BAL cells. Mice were treated as described in figure
1. Sixteen hours after the last smoke exposure lung tissue and bronchoalveolar lavage fluid (BALF) were
harvested. Gene expression in lung tissue homogenates (light grey bars) and in BAL cells (dark grey bars) was
analyzed (n=3‐5 mice per group). Ct values corrected for 18S are depicted, expressed as mean ± s.e. of the mean.
Note that low Ct values mean high expression levels. Muscarinic M1 receptor (M1R), M2 receptor (M2R) and M3
receptor (M3R). Data were analyzed using two‐way ANOVA; individual comparisons were made using a Student‐
Newman‐Keuls multiple comparisons post‐hoc test; *** p<0.001 (adjusted p‐value). Statistics were performed on
log‐transformed data.

Chapter 3
76
References
(1) Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 2000;343:269‐280.
(2) Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol
2008;8:183‐192.
(3) Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease.
Proc.Am.Thorac.Soc. 2006;3:434‐439.
(4) Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to
emphysema. N Engl J Med 1984;311:421‐425.
(5) Lee AM, Jacoby DB, Fryer AD. Selective muscarinic receptor antagonists for airway diseases. Curr
Opin Pharmacol 2001;1:223‐229.
(6) Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in
bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol
1988;153:73‐82.
(7) Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology
of asthma and COPD. Respir.Res. 2006;7:73.
(8) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008;154:1558‐1571.
(9) Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol
2008;130:219‐234.
(10) Gwilt CR, Donnelly LE, Rogers DF. The non‐neuronal cholinergic system in the airways: an
unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208‐222.
(11) Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J, Kolahian S, Halayko AJ, Hiemstra
PS, Zuyderduyn S. Muscarinic M3 receptor stimulation increases cigarette smoke‐induced IL‐8
secretion by human airway smooth muscle cells. Eur Respir J 2009;34:1436‐1443.
(12) Profita M, Bonanno A, Montalbano AM, Ferraro M, Siena L, Bruno A, Girbino S, Albano GD,
Casarosa P, Pieper MP, Gjomarkaj M. Cigarette smoke extract activates human bronchial epithelial
cells affecting non‐neuronal cholinergic system signalling in vitro. Life Sci 2011;89:36‐43.
(13) Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP,
Gjomarkaj M. Acetylcholine mediates the release of IL‐8 in human bronchial epithelial cells by a
NFkB/ERK‐dependent mechanism. Eur J Pharmacol 2008;582:145‐153.
(14) Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F, Gjomarkaj M, Bonsignore G,
Bousquet J, Vignola AM. Muscarinic receptors, leukotriene B4 production and neutrophilic
inflammation in COPD patients. Allergy 2005;60:1361‐1369.
(15) Wollin L, Pieper MP. Tiotropium bromide exerts anti‐inflammatory activity in a cigarette smoke
mouse model of COPD. Pulm Pharmacol Ther 2010;23:345‐354.
(16) Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs
H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur
Respir J 2011;38:789‐796.
(17) Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide.
Respir Res 2011;12:24.

M3 receptors promote smoke‐induced inflammation
77
(18) Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J. Muscarinic induction of
hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents.
Neuron 2002;33:615‐624.
(19) Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H, Wess J.
Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc
Natl Acad Sci U S A 1999;96:1692‐1697.
(20) Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ,
Xia B, Crawley JN, Felder CC, Deng CX, Wess J. Mice lacking the M3 muscarinic acetylcholine receptor
are hypophagic and lean. Nature 2001;410:207‐212.
(21) Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine‐
induced release of chemotactic mediators in vitro. Respir Med 2007;101:2386‐2394.
(22) Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages
to release inflammatory cell chemotactic activity. Am J Physiol 1998;274:L970‐L979.
(23) Meurs H, Dekkers BG, Maarsingh H, Halayko AJ, Zaagsma J, Gosens R. Muscarinic receptors on
airway mesenchymal cells: novel findings for an ancient target. Pulm Pharmacol Ther 2013;26:145‐
155.
(24) Koyama S, Rennard SI, Robbins RA. Acetylcholine stimulates bronchial epithelial cells to release
neutrophil and monocyte chemotactic activity. Am J Physiol 1992;262:L466‐L471.
(25) Reinheimer T, Mohlig T, Zimmermann S, Hohle KD, Wessler I. Muscarinic control of histamine
release from airways. Inhibitory M1‐receptors in human bronchi but absence in rat trachea. Am J
Respir Crit Care Med 2000;162:534‐538.
(26) Lamb D, Lumsden A. Intra‐epithelial mast cells in human airway epithelium: evidence for
smoking‐induced changes in their frequency. Thorax 1982;37:334‐342.
(27) Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax
1998;53:129‐136.
(28) Ishihara H, Shimura S, Satoh M, Masuda T, Nonaka H, Kase H, Sasaki T, Sasaki H, Takishima T,
Tamura K. Muscarinic receptor subtypes in feline tracheal submucosal gland secretion. Am J Physiol
1992;262:L223‐8.
(29) Boddeke HW, Buttini M. Pharmacological properties of cloned muscarinic receptors expressed
in A9 L cells; comparison with in vitro models. Eur J Pharmacol 1991;202:151‐157.
(30) Schlenz H, Kummer W, Jositsch G, Wess J, Krasteva G. Muscarinic receptor‐mediated
bronchoconstriction is coupled to caveolae in murine airways. Am J Physiol Lung Cell Mol Physiol
2010;298:L626‐36.
(31) Garssen J, Van Loveren H, Gierveld CM, Van der Vliet H, Nijkamp FP. Functional characterization
of muscarinic receptors in murine airways. Br J Pharmacol 1993;109:53‐60.
(32) Casarosa P, Bouyssou T, Germeyer S, Schnapp A, Gantner F, Pieper M. Preclinical evaluation of
long‐acting muscarinic antagonists: comparison of tiotropium and investigational drugs. J Pharmacol
Exp Ther 2009;330:660‐668.
(33) Barnes PJ. The pharmacological properties of tiotropium. Chest 2000;117:63S‐6S.
(34) Gosens R, Nelemans SA, Grootte Bromhaar MM, McKay S, Zaagsma J, Meurs H. Muscarinic M3‐
receptors mediate cholinergic synergism of mitogenesis in airway smooth muscle. Am J Respir Cell
Mol Biol 2003;28:257‐262.

Chapter 3
78
(35) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(36) Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M, UPLIFT Study
Investigators. A 4‐year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med
2008;359:1543‐1554.
(37) Liesker JJ, Bathoorn E, Postma DS, Vonk JM, Timens W, Kerstjens HA. Sputum inflammation
predicts exacerbations after cessation of inhaled corticosteroids in COPD. Respir Med
2011;105:1853‐1860.
(38) Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers
to symptoms and lung function changes in COPD exacerbations. Thorax 2000;55:114‐120.
(39) Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K, Kesten S, Wedzicha JA. Effect
of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J
2007;30:472‐478.
(40) Perng DW, Tao CW, Su KC, Tsai CC, Liu LY, Lee YC. Anti‐inflammatory effects of
salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur Respir J 2009;33:778‐784.



CHAPTER 4
MUSCARINIC M3 RECEPTORS ON STRUCTURAL CELLS
REGULATE CIGARETTE SMOKE‐INDUCED NEUTROPHILIC
AIRWAY INFLAMMATION IN MICE
Loes E.M. Kistemaker
Ronald P. van Os
Albertina Dethmers‐Ausema
I. Sophie T. Bos
Machteld N. Hylkema
Maarten van den Berge
Pieter S. Hiemstra
Jürgen Wess
Herman Meurs
Huib A.M. Kerstjens
Reinoud Gosens
Am J Physiol Lung Cell Mol Physiol, 2015, 308(1):L96‐L103

Chapter 4
82
Abstract
Rationale
Anticholinergics, blocking the muscarinic M3 receptor, are effective bronchodilators for
patients with COPD. Recent evidence from M3 receptor deficient mice (M3R‐/‐) indicates
that M3 receptors also regulate neutrophilic inflammation in response to cigarette smoke
(CS). M3 receptors are present on almost all cell types and in this study we investigated
the relative contribution of M3 receptors on structural cells versus inflammatory cells to
CS‐induced inflammation using bone‐marrow chimeric mice. Methods
Bone‐marrow chimeras (C56Bl/6 mice) were generated and engraftment was confirmed
after 10 weeks. Thereafter, irradiated and non‐irradiated control animals were exposed to
CS or fresh air for four consecutive days.
Results
CS induced a significant increase in neutrophil numbers in non‐irradiated and irradiated
control animals (4‐35 fold). Interestingly, wild‐type animals receiving M3R‐/‐ bone marrow
showed a similar increase in neutrophil number (15‐fold). In contrast, no increase in the
number of neutrophils was observed in M3R‐/‐ animals receiving wild‐type bone marrow.
The increase in KC levels was similar in all smoke‐exposed groups (2.5‐5.0 fold). Micro‐
array analysis revealed that fibrinogen α and CD177, both involved in neutrophil
migration, were downregulated in CS‐exposed M3R‐/‐ animals receiving wild‐type bone
marrow compared to CS‐exposed wild‐type animals, which was confirmed by RT‐qPCR
(1.6‐2.5 fold).
Conclusions
These findings indicate that the M3 receptor on structural cells plays a pro‐inflammatory
role in CS‐induced neutrophilic inflammation, whereas the M3 receptor on inflammatory
cells does not. This effect is probably not mediated via KC release, but may involve altered
adhesion and transmigration of neutrophils via fibrinogen α and CD177.
Introduction
Acetylcholine is the primary parasympathetic neurotransmitter in the airways. It induces
bronchoconstriction and mucus secretion via M3 receptors (chapter 2). Cholinergic tone is
increased in patients with chronic obstructive pulmonary diseases (COPD), and this is the
major reversible component of airflow obstruction in COPD (1, 2). Therefore,
anticholinergics, which block M3 receptors, are effective bronchodilators for patients with
COPD (2).

M3 receptors on structural cells regulate inflammation
83
In addition to inducing bronchoconstriction and mucus secretion, acetylcholine has been
shown to affect airway inflammation (chapter 2), which is a hallmark feature of COPD (3).
In animal models of COPD, pretreatment with anticholinergics inhibits cigarette smoke‐
induced and LPS‐induced neutrophilic inflammation (4, 5). More recently, we
demonstrated that these pro‐inflammatory effects of acetylcholine are mediated via M3
receptors. Total knock‐out of the M3 receptor, or inhibition of the M3 receptor by a
pharmacological approach using the muscarinic antagonist 4‐DAMP, which is selective for
M3 receptors over M2 receptors, prevented cigarette smoke‐induced neutrophilic
inflammation in an early smoke induced‐inflammation model in mice mice (chapter 3).
Together, these data suggest a pro‐inflammatory role for acetylcholine via M3 receptors.
M3 receptors are expressed abundantly throughout the airways and almost all cell types
express M3 receptors. This includes structural cells, such as airway smooth muscle,
epithelial and endothelial cells, but also inflammatory cells, such as macrophages and
neutrophils (6). From in vitro studies it is known that muscarinic receptor stimulation of
both structural and inflammatory cells might contribute to the pro‐inflammatory effects of
acetylcholine. For instance, stimulation of airway smooth muscle cells and epithelial cells
with methacholine or acetylcholine induces the release of IL‐8 (7, 8). Furthermore,
acetylcholine activates human monocytes and alveolar macrophages to promote
neutrophil migration, which is also observed when using macrophages from COPD
patients (9, 10). It is not known whether the pro‐inflammatory effects of acetylcholine
observed in vivo are primarily mediated via M3 receptors on structural cells or via M3
receptors on inflammatory cells.
Therefore, in this study, we investigated the relative contribution of the M3 receptor on
structural cells versus inflammatory cells to cigarette smoke‐induced inflammation. In
order to distinguish between structural cells and inflammatory cells, bone marrow
chimeric mice were generated. Here, we demonstrate that the pro‐inflammatory effects
of acetylcholine on neutrophilic inflammation are primarily mediated via M3 receptors on
structural cells, which may involve altered adhesion and transmigration of neutrophils.
Methods
Animals
Homozygous, inbred, specific‐pathogen‐free breeding colonies of muscarinic M3 receptor
deficient (M3R‐/‐) mice and C57Bl/6NTac wild‐type mice with the same genetic background
(CD45.2) were obtained from Taconic (Cambridge City, Indiana, USA). The M3R‐/‐ mice
were on a 129 Sv/J background and had been backcrossed for at least 10 generations onto
Chapter 4
84
the C57Bl/6NTac background (11). CD45.1 congenic C57Bl/6 mice were obtained from
Jackson Laboratories (Bar Harbor, ME). All animals were maintained in our own facility.
Animals were housed conventionally under a 12‐h light‐dark cycle and received food and
water ad libitum. All experiments were performed in accordance with the national
guidelines and approved by the University of Groningen Committee for Animal
Experimentation (number: 5463G).
Generation of bone marrow chimeras
Bone marrow cells were collected from femurs and tibia of M3R‐/‐ mice and wild‐type
CD45.2 or CD45.1 mice, and retro‐orbitally injected into irradiated male mice (9 Gy),
within 24 hours after irradiation. Per mouse, 8 × 106 bone marrow cells were transplanted.
Non‐irradiated wild‐type animals were used as additional controls; see table 1 for an
overview of the groups. After 10 weeks, engraftment was analyzed by flow cytometry of
peripheral blood. Seventy microliter of heparinized peripheral blood was collected 10
weeks after bone marrow transplantation and at the end of the study when the mice were
sacrificed. Red blood cells were lysed and the remaining cells were stained with antibodies
directed against the following markers: CD45.2‐PE (CD45.2), CD45.1‐Pacific Blue (CD45.1),
CD3‐APC (T cells), B220‐FITC (B cells), GR‐1‐PE/Cy7 and Mac1‐PE/Cy7 (monocytes and
granulocytes, respectively) (Biolegend, San Diego, CA, USA). The stained cells were
analyzed with a LSR‐II flow cytometer (BD Biosciences, San Diego, CA, USA). Engrafment
was calculated as a percentage of CD45.1 versus CD45.2 cells. Engraftment was high in all
animals (>90% donor derived cells in all lineages), and mice were included in the study 3
months after bone marrow reconstitution (figure 1).
Table 1. Overview of the experimental groups included in this study. Back‐
ground
Recipient Irradiation Donor Groups
CD45.1 CD 45.1 ‐ ‐ Non‐irradiated control
CD 45.1 + CD45.2 Irradiated control
CD 45.1 + M3R‐/‐ M3R inflammatory cells
CD45.2 CD45.2 ‐ ‐ Non‐irradiated control
CD45.2 + CD45.1 Irradiated control
M3R‐/‐ + CD45.1 M3R structural cells

M3 receptors on structural cells regulate inflammation
85
Cigarette smoke exposure
Mice (n=5‐12 per group) were exposed to cigarette smoke from Kentucky 3R4F research
cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, USA) on 4
consecutive days by whole body exposure in an early smoke induced‐inflammation model,
as described previously (chapter 3). Mice were exposed to mainstream smoke of one
cigarette in the morning and three cigarettes in the afternoon on the first day. On day 2 to
4, mice were exposed to five cigarettes in the morning and five in the afternoon (figure 1).
Each cigarette was smoked without a filter in 5 minutes at a rate of 5L/hr in a ratio with
60L/hr air using a peristaltic pump (45 rpm, Watson Marlow 323 E/D, Rotterdam, The
Netherlands). Control animals were handled in the same way but instead exposed to fresh
air only. Sixteen hours after the last CS exposure, animals were euthanized by
intraperitoneal pentobarbital injection (400 mg/kg, hospital pharmacy, University Medical
Center Groningen), after which the lungs were immediately lavaged, resected and snap
frozen in liquid nitrogen. Bronchoalveolar lavage (BAL) fluid was stored at ‐20°C and lung
tissue was stored at ‐80°C.
Analysis of bronchoalveolar lavage cells and cytokines
A BAL was performed as described previously (chapter 3). Briefly, lungs were lavaged 5
times with 1 ml PBS. The first fraction was used for measurement of cytokines by ELISA.
Levels of KC, IL‐6 and MCP‐1 were determined by a multiplex assay (R&D Systems,
Minneapolis, USA). Total cell numbers were determined and cytospins were prepared and
stained with May–Grünwald and Giemsa (both Sigma, St. Louis) after which a differential
cell count was performed by counting at least 400 cells in duplicate in a blinded fashion.
Figure 1. Experimental protocol. Chimeric animals were generated by sublethal irradiation (9 Gy) of recipient
male C57Bl/6 mice (n=5‐12) and subsequent transplantation of 8 x 106 donor bone marrow cells via retro‐orbital
injection within 24h after irradiation. To allow for complete engraftment, animals were included in the protocol
three months after bone marrow reconstitution. Animals were exposed to cigarette smoke twice daily on 4
consecutive days by whole body exposure. Sixteen hours after the last smoke exposure a bronchoalveolar lavage
was performed and lungs were harvested for lung tissue homogenates.
Chapter 4
86
Analysis of gene expression in lung tissue
Total RNA was extracted from lung tissue (right superior lobe) using the RNeasy mini kit
(Qiagen, Venlo, The Netherlands), as described previously (chapter 3). Equal amounts of
total mRNA were then reverse transcribed and cDNA was subjected to real‐time qPCR
(Westburg, Leusden, The Netherlands) or to micro‐array analysis using an Illumina chip
and GeneSpring software. The specific forward and reverse primers used for real‐time
qPCR are listed in table 2.
Statistical analysis
Data are presented as mean ± s.e. of the mean. Statistical differences between means
were calculated using one‐ or two‐way ANOVA, followed by Newman Keuls multiple
comparison tests, or by a Student’s t‐test with two‐tailed distribution where appropriate.
Differences were considered significant at p<0.05.
Table 2. Primers used for qRT‐PCR analysis. KC: keratinocyte‐derived chemokine, the mouse orthologue of IL‐8,
also known as CXCL1 in mice. MIP‐2 is also known as CXCL2, LIX is also known as CXCL5.
Gene Primer sequence NCBI accession
number
KC Forward – GCTGGGATTCACCTCAAGAA
Reverse – AGGTGCCATCAGAGCAGTCT
NM_008176.3
IL‐6 Forward – CCGGAGAGGAGACTTCACAG
Reverse – TCCACGATTTCCCAGAGAAC
NM_031168.1
MIP‐2 Forward – AAGTTTGCCTTGACCCTGAA
Reverse – AGGCACATCAGGTACGATCC
NM_009140.2
LIX Forward – GAAAGCTAAGCGGAATGCAC
Reverse – GGGACAATGGTTTCCCTTTT
NM_009141.3
Fibrinogen α Forward – AACTTCAGGTGCCCAAAATG
Reverse – AACTGAGCCCCTCAGAGTGA
NM_001111048.1
CD177 Forward – GGGAGGTGGACTGACTACCA
Reverse – GGGTTCAGCAGAGAGGACAG
NM_026862.3
18S Forward – AAACGGCTACCACATCCAAG
Reverse – CCTCCAATGGATCCTCGTTA
NR_003278
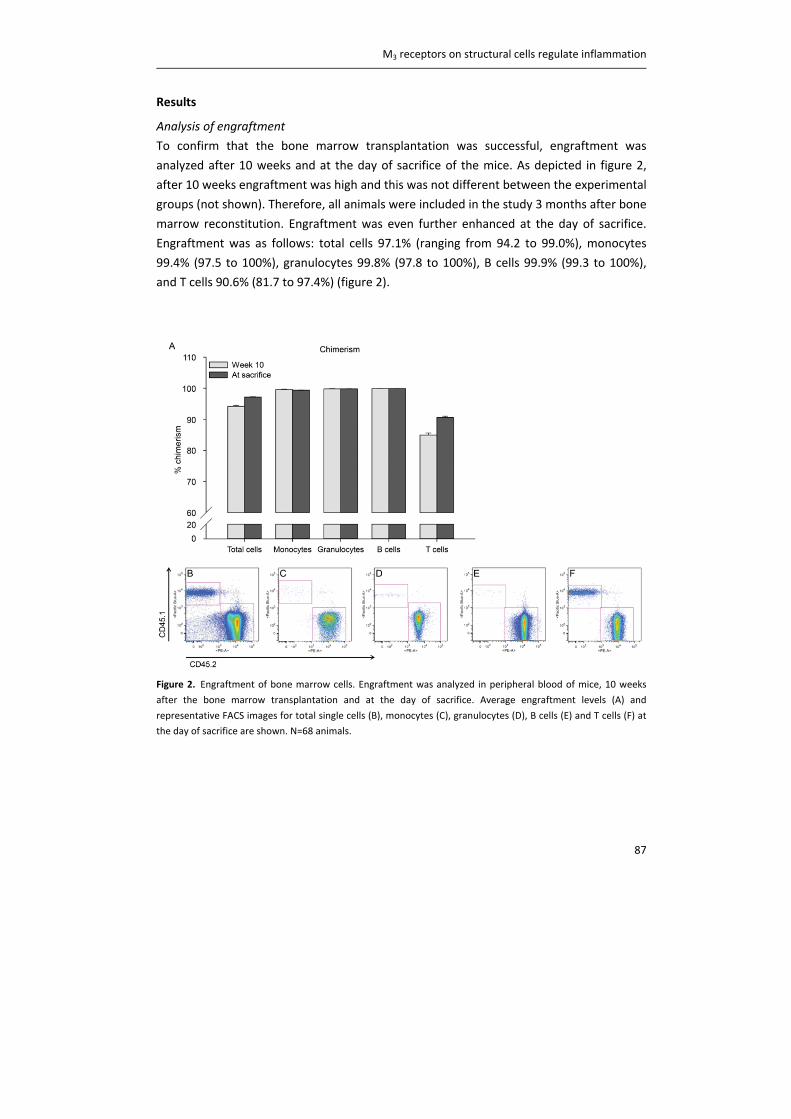
M3 receptors on structural cells regulate inflammation
87
Results
Analysis of engraftment
To confirm that the bone marrow transplantation was successful, engraftment was
analyzed after 10 weeks and at the day of sacrifice of the mice. As depicted in figure 2,
after 10 weeks engraftment was high and this was not different between the experimental
groups (not shown). Therefore, all animals were included in the study 3 months after bone
marrow reconstitution. Engraftment was even further enhanced at the day of sacrifice.
Engraftment was as follows: total cells 97.1% (ranging from 94.2 to 99.0%), monocytes
99.4% (97.5 to 100%), granulocytes 99.8% (97.8 to 100%), B cells 99.9% (99.3 to 100%),
and T cells 90.6% (81.7 to 97.4%) (figure 2).
Figure 2. Engraftment of bone marrow cells. Engraftment was analyzed in peripheral blood of mice, 10 weeks
after the bone marrow transplantation and at the day of sacrifice. Average engraftment levels (A) and
representative FACS images for total single cells (B), monocytes (C), granulocytes (D), B cells (E) and T cells (F) at
the day of sacrifice are shown. N=68 animals.

Chapter 4
88
Contribution of the M3 receptor on inflammatory cells
To study the contribution of the M3 receptor on inflammatory cells to early cigarette
smoke‐induced airway inflammation, inflammatory cells in the lavage fluid of CD45.1
animals were determined after exposure to cigarette smoke. The following groups were
compared: CD45.1 non‐irradiated control animals, irradiated CD45.1 control animals
receiving wild‐type (CD45.2) bone marrow cells and irradiated CD45.1 animals receiving
M3R‐/‐ (CD45.2) bone marrow cells (table 1). Cigarette smoke exposure had no significant
effect on macrophage and lymphocyte numbers in all groups (figure 3). Cigarette smoke
induced a significant increase in neutrophil numbers in non‐irradiated (figure 3A; 28‐fold)
and irradiated control animals receiving wild‐type bone marrow cells (figure 3B; 18‐fold).
Interestingly, wild‐type animals receiving M3R‐/‐ bone marrow cells showed a similar
increase in neutrophil number (figure 3C; 15‐fold), suggesting that the M3 receptor on
inflammatory cells is not involved in cigarette smoke‐induced neutrophilic inflammation.
Contribution of the M3 receptor on structural cells
To study the contribution of the M3 receptor on structural cells to early cigarette smoke‐
induced airway inflammation, inflammatory cells in the lavage fluid of CD45.2 animals
were determined after exposure to cigarette smoke (figure 4). The following groups were
compared: CD45.2 non‐irradiated control animals, irradiated CD45.2 control animals
receiving CD45.1 bone marrow cells and irradiated M3R‐/‐ (CD45.2) animals receiving
CD45.1 bone marrow cells (table 1). Cigarette smoke induced a small increase in
macrophage numbers in the lavage fluid in all groups (1.3‐1.4 fold). No increase in
lymphocyte numbers was observed. Cigarette smoke induced a significant increase in
neutrophil numbers in non‐irradiated (figure 4A; 8‐fold) and irradiated control animals
receiving wild‐type bone marrow cells (figure 4B; 4‐fold). In contrast, no increase in the
number of neutrophils was observed in M3R‐/‐ animals receiving wild‐type bone marrow
(figure 4C), suggesting that the M3 receptor on structural cells is involved in cigarette
smoke‐induced neutrophilic inflammation.
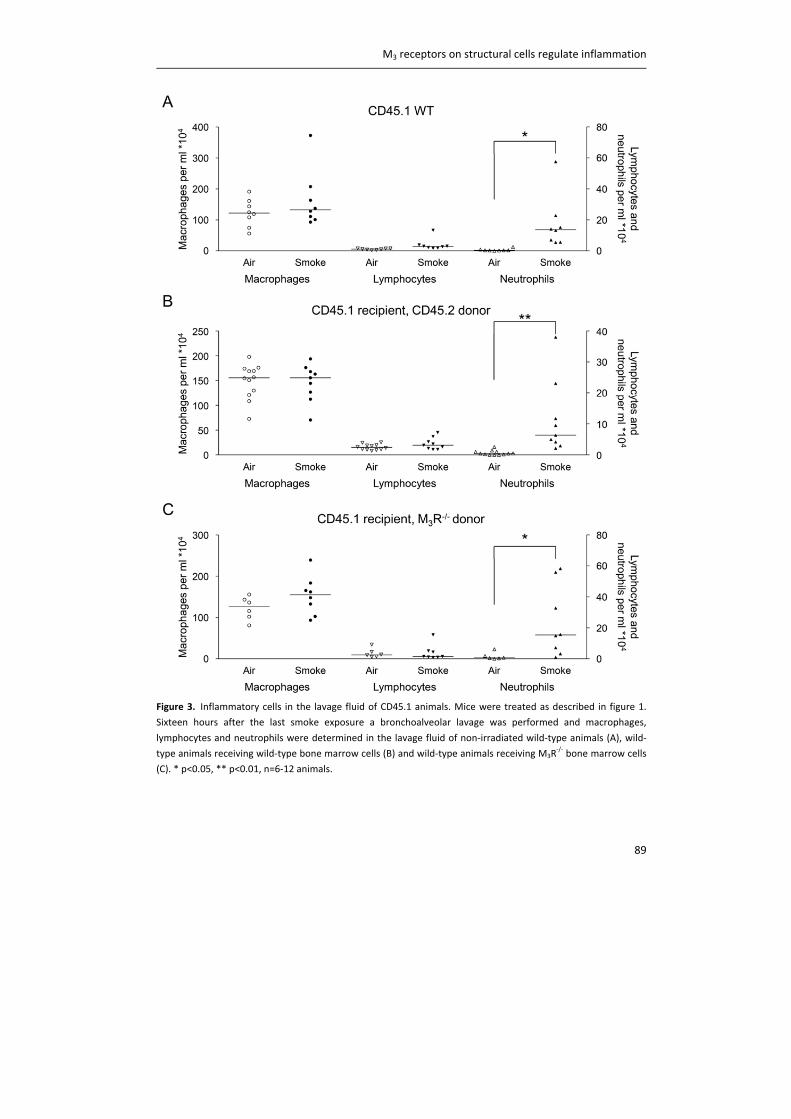
M3 receptors on structural cells regulate inflammation
89
Figure 3. Inflammatory cells in the lavage fluid of CD45.1 animals. Mice were treated as described in figure 1.
Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed and macrophages,
lymphocytes and neutrophils were determined in the lavage fluid of non‐irradiated wild‐type animals (A), wild‐
type animals receiving wild‐type bone marrow cells (B) and wild‐type animals receiving M3R‐/‐ bone marrow cells
(C). * p<0.05, ** p<0.01, n=6‐12 animals.
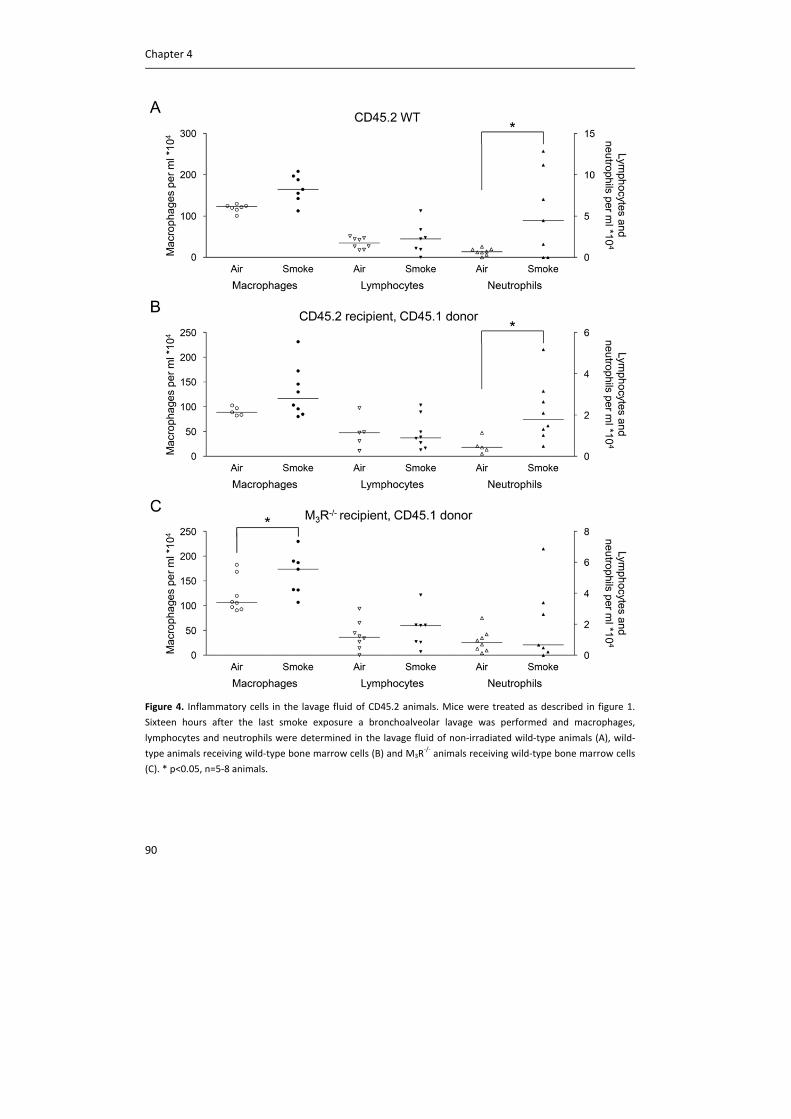
Chapter 4
90
Figure 4. Inflammatory cells in the lavage fluid of CD45.2 animals. Mice were treated as described in figure 1.
Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed and macrophages,
lymphocytes and neutrophils were determined in the lavage fluid of non‐irradiated wild‐type animals (A), wild‐
type animals receiving wild‐type bone marrow cells (B) and M3R‐/‐ animals receiving wild‐type bone marrow cells
(C). * p<0.05, n=5‐8 animals.
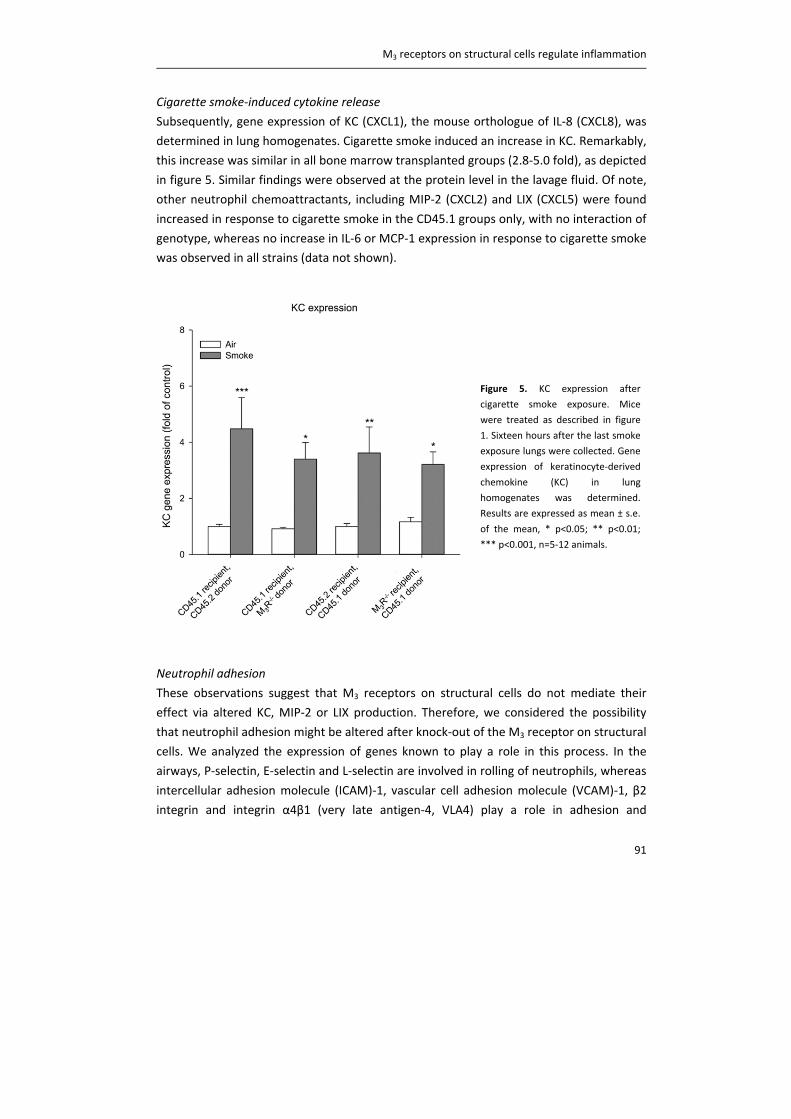
M3 receptors on structural cells regulate inflammation
91
Cigarette smoke‐induced cytokine release
Subsequently, gene expression of KC (CXCL1), the mouse orthologue of IL‐8 (CXCL8), was
determined in lung homogenates. Cigarette smoke induced an increase in KC. Remarkably,
this increase was similar in all bone marrow transplanted groups (2.8‐5.0 fold), as depicted
in figure 5. Similar findings were observed at the protein level in the lavage fluid. Of note,
other neutrophil chemoattractants, including MIP‐2 (CXCL2) and LIX (CXCL5) were found
increased in response to cigarette smoke in the CD45.1 groups only, with no interaction of
genotype, whereas no increase in IL‐6 or MCP‐1 expression in response to cigarette smoke
was observed in all strains (data not shown).
Neutrophil adhesion
These observations suggest that M3 receptors on structural cells do not mediate their
effect via altered KC, MIP‐2 or LIX production. Therefore, we considered the possibility
that neutrophil adhesion might be altered after knock‐out of the M3 receptor on structural
cells. We analyzed the expression of genes known to play a role in this process. In the
airways, P‐selectin, E‐selectin and L‐selectin are involved in rolling of neutrophils, whereas
intercellular adhesion molecule (ICAM)‐1, vascular cell adhesion molecule (VCAM)‐1, β2
integrin and integrin α4β1 (very late antigen‐4, VLA4) play a role in adhesion and
Figure 5. KC expression after
cigarette smoke exposure. Mice
were treated as described in figure
1. Sixteen hours after the last smoke
exposure lungs were collected. Gene
expression of keratinocyte‐derived
chemokine (KC) in lung
homogenates was determined.
Results are expressed as mean ± s.e.
of the mean, * p<0.05; ** p<0.01;
*** p<0.001, n=5‐12 animals.
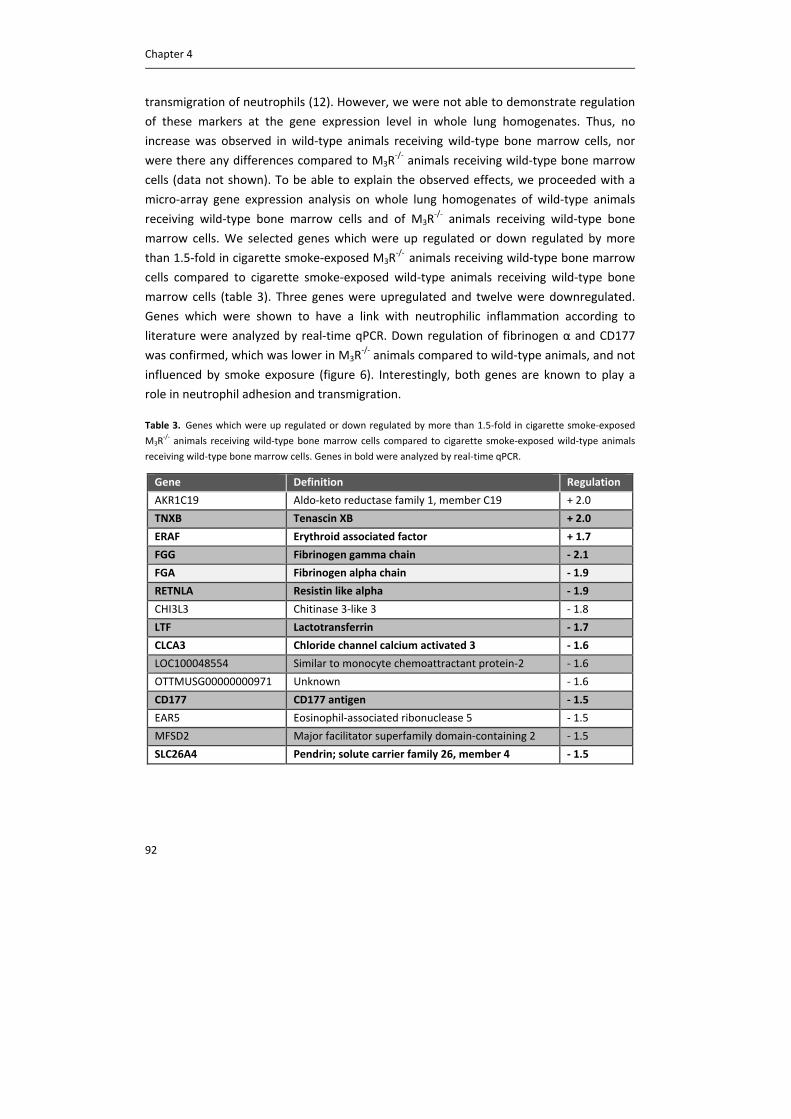
Chapter 4
92
transmigration of neutrophils (12). However, we were not able to demonstrate regulation
of these markers at the gene expression level in whole lung homogenates. Thus, no
increase was observed in wild‐type animals receiving wild‐type bone marrow cells, nor
were there any differences compared to M3R‐/‐ animals receiving wild‐type bone marrow
cells (data not shown). To be able to explain the observed effects, we proceeded with a
micro‐array gene expression analysis on whole lung homogenates of wild‐type animals
receiving wild‐type bone marrow cells and of M3R‐/‐ animals receiving wild‐type bone
marrow cells. We selected genes which were up regulated or down regulated by more
than 1.5‐fold in cigarette smoke‐exposed M3R‐/‐ animals receiving wild‐type bone marrow
cells compared to cigarette smoke‐exposed wild‐type animals receiving wild‐type bone
marrow cells (table 3). Three genes were upregulated and twelve were downregulated.
Genes which were shown to have a link with neutrophilic inflammation according to
literature were analyzed by real‐time qPCR. Down regulation of fibrinogen α and CD177
was confirmed, which was lower in M3R‐/‐ animals compared to wild‐type animals, and not
influenced by smoke exposure (figure 6). Interestingly, both genes are known to play a
role in neutrophil adhesion and transmigration.
Table 3. Genes which were up regulated or down regulated by more than 1.5‐fold in cigarette smoke‐exposed
M3R‐/‐ animals receiving wild‐type bone marrow cells compared to cigarette smoke‐exposed wild‐type animals
receiving wild‐type bone marrow cells. Genes in bold were analyzed by real‐time qPCR.
Gene Definition Regulation
AKR1C19 Aldo‐keto reductase family 1, member C19 + 2.0
TNXB Tenascin XB + 2.0
ERAF Erythroid associated factor + 1.7
FGG Fibrinogen gamma chain ‐ 2.1
FGA Fibrinogen alpha chain ‐ 1.9
RETNLA Resistin like alpha ‐ 1.9
CHI3L3 Chitinase 3‐like 3 ‐ 1.8
LTF Lactotransferrin ‐ 1.7
CLCA3 Chloride channel calcium activated 3 ‐ 1.6
LOC100048554 Similar to monocyte chemoattractant protein‐2 ‐ 1.6
OTTMUSG00000000971 Unknown ‐ 1.6
CD177 CD177 antigen ‐ 1.5
EAR5 Eosinophil‐associated ribonuclease 5 ‐ 1.5
MFSD2 Major facilitator superfamily domain‐containing 2 ‐ 1.5
SLC26A4 Pendrin; solute carrier family 26, member 4 ‐ 1.5
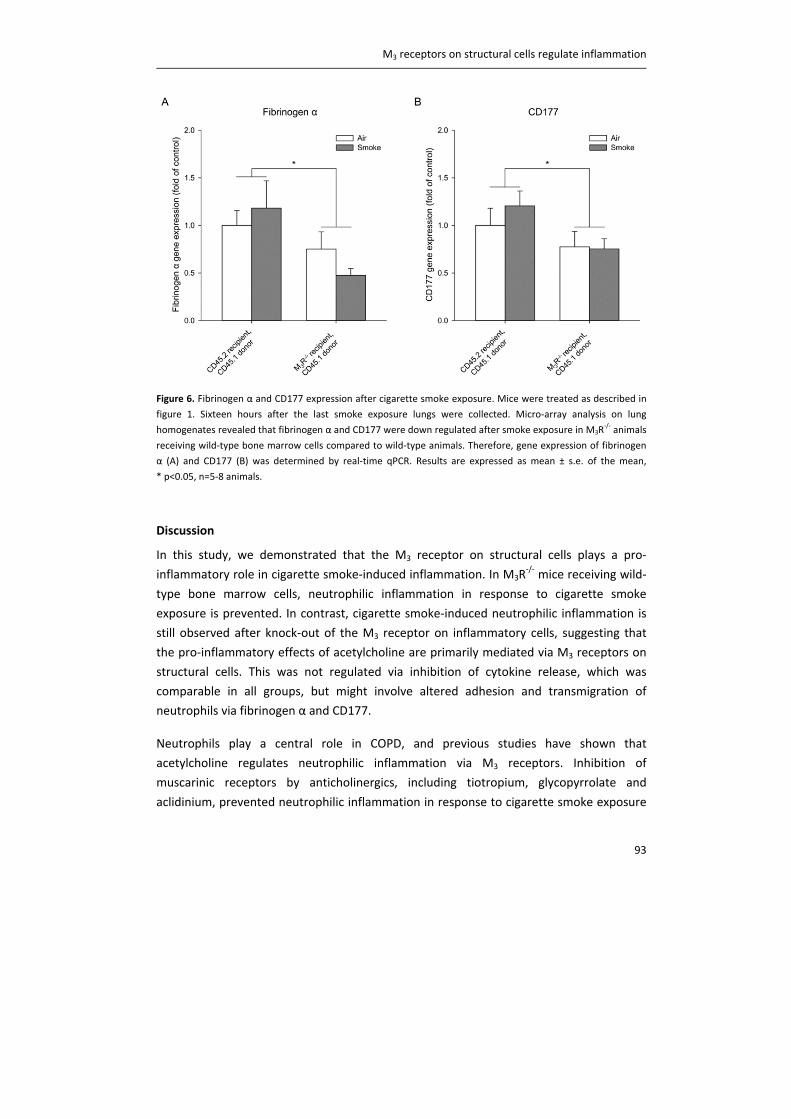
M3 receptors on structural cells regulate inflammation
93
Figure 6. Fibrinogen α and CD177 expression after cigarette smoke exposure. Mice were treated as described in
figure 1. Sixteen hours after the last smoke exposure lungs were collected. Micro‐array analysis on lung
homogenates revealed that fibrinogen α and CD177 were down regulated after smoke exposure in M3R‐/‐ animals
receiving wild‐type bone marrow cells compared to wild‐type animals. Therefore, gene expression of fibrinogen
α (A) and CD177 (B) was determined by real‐time qPCR. Results are expressed as mean ± s.e. of the mean,
* p<0.05, n=5‐8 animals.
Discussion
In this study, we demonstrated that the M3 receptor on structural cells plays a pro‐
inflammatory role in cigarette smoke‐induced inflammation. In M3R‐/‐ mice receiving wild‐
type bone marrow cells, neutrophilic inflammation in response to cigarette smoke
exposure is prevented. In contrast, cigarette smoke‐induced neutrophilic inflammation is
still observed after knock‐out of the M3 receptor on inflammatory cells, suggesting that
the pro‐inflammatory effects of acetylcholine are primarily mediated via M3 receptors on
structural cells. This was not regulated via inhibition of cytokine release, which was
comparable in all groups, but might involve altered adhesion and transmigration of
neutrophils via fibrinogen α and CD177.
Neutrophils play a central role in COPD, and previous studies have shown that
acetylcholine regulates neutrophilic inflammation via M3 receptors. Inhibition of
muscarinic receptors by anticholinergics, including tiotropium, glycopyrrolate and
aclidinium, prevented neutrophilic inflammation in response to cigarette smoke exposure

Chapter 4
94
or LPS exposure in guinea pigs and mice (4, 5, 13, 14). Since long‐acting anticholinergics
are kinetically selective for M3 receptors, these findings suggest the involvement of this
specific muscarinic receptor subtype. A recent study from our lab using M3R‐/‐ mice
showed that the pro‐inflammatory effects of acetylcholine are indeed mediated via the M3
receptor. We demonstrated that cigarette smoke‐induced neutrophilic inflammation was
prevented in M3R‐/‐ mice, whereas opposite effects were observed in M1R
‐/‐ and M2R‐/‐
mice (chapter 3). In the present study we have extended these findings by demonstrating
that the pro‐inflammatory effects of acetylcholine are primarily mediated via M3 receptors
on structural cells in an early smoke induced‐inflammation model in mice. The
involvement of the M3 receptor on structural cells in the inflammatory response is also
supported by evidence from in vitro studies. Methacholine and cigarette smoke‐induced
IL‐8 release from airway smooth muscle cells can be inhibited by the M3 antagonists 4‐
DAMP and DAU5884 (7). Moreover, acetylcholine‐induced IL‐8 release from bronchial
epithelial cells can be inhibited by 4‐DAMP (8). Strikingly, acetylcholine‐induced effects on
inflammatory cells were also shown to be mediated via M3 receptors. 4‐DAMP inhibited
acetylcholine‐induced neutrophil chemotactic activity from macrophages (15), as well as
neutrophil chemotaxis from LPS‐activated macrophages from COPD patients (10). These in
vitro studies suggest a direct effect of acetylcholine on both structural cells and
inflammatory cells. Apparently, the observed effects of acetylcholine on inflammatory
cells in vitro are not relevant to the in vivo situation, as we demonstrate that cigarette
smoke‐induced inflammation is still observed after knock‐out of the M3 receptor on
inflammatory cells.
Although there was no increase in neutrophil numbers in M3R‐/‐ animals receiving wild‐
type bone marrow cells after exposure to cigarette smoke compared to wild‐type animals,
the increase in the neutrophil chemoattractant KC was similar to the increase observed in
wild‐type animals. This suggests that neutrophil recruitment is altered after knock‐out of
the M3 receptor. Neutrophil recruitment is a complex cellular process which occurs
through a cascade of multiple steps, including tethering, rolling, adhesion, crawling and
subsequent transmigration of neutrophils (12). An initial screen on genes known to be
involved in the process of neutrophil recruitment, including selectins, ICAM‐1, VCAM‐1, β2
integrin and VLA4, could not explain the observed increase in KC in M3R‐/‐ animals
receiving wild‐type bone marrow cells, without an increase in neutrophil numbers.
However, using a micro‐array analysis, two genes which were recently linked to the
process of neutrophil recruitment, fibrinogen α and CD177 (16, 17), were shown to be
down regulated in M3R‐/‐ animals compared to wild‐type animals.

M3 receptors on structural cells regulate inflammation
95
Fibrinogen α, besides its role in blood hemostasis, regulates the adhesive behavior of
neutrophils. Fibrinogen can bind to β2 integrins and thereby alter the recruitment of
neutrophils (16, 18). Neutrophil recruitment in response to platelet activating factor (PAF)
is altered in fibrinogen α knock‐out mice. Specifically, the number of rolling neutrophils
and the number of adhering neutrophils was decreased in fibrinogen α knock‐out mice
compared to wild‐type mice (16). Therefore, a reduction in fibrinogen α levels in M3R‐/‐
mice might affect neutrophil recruitment in response to cigarette smoke exposure, by
reducing the rolling and adhesion of neutrophils.
CD177 is a neutrophil specific antigen, expressed by a subpopulation of neutrophils.
CD177 can bind to platelet endothelial cell adhesion molecule (PECAM)‐1, and thereby
alter the recruitment of neutrophils (17, 19). PECAM‐1 is known to be involved in
neutrophil transmigration (12). In vitro, transmigration of neutrophils through endothelial
cells in response to IL‐8 or fMLP was inhibited after inhibition of the CD177‐PECAM‐1
interaction (17). Interestingly, the same study showed that CD177‐positive neutrophils
transmigrated more efficiently than CD177‐negative neutrophils, indicating the potential
relevance of CD177 for transmigration. These findings were confirmed by Kuckleburg et
al., who demonstrated that inhibition of CD177 or inhibition of the interaction between
CD177 and PECAM‐1 inhibits neutrophil transmigration under both static and flow
conditions (20). Therefore, a reduction in CD177 levels in M3R‐/‐ mice might affect
neutrophil recruitment in response to cigarette smoke exposure, by reducing the
transmigration of neutrophils.
The gene expression of neither fibrinogen α nor CD177 was regulated by cigarette smoke,
indicating that this is a constitutive difference between the wild‐type and M3R‐/‐ mice.
Expression levels of both genes were lower in the M3R‐/‐ strain compared to wild‐type
mice, which further supports the observation that neutrophillic inflammation is altered in
M3R‐/‐ mice receiving wild‐type bone marrow, and not in wild‐type animals receiving M3R
‐/‐
bone marrow. Interestingly, both genes were recently identified as potential inflammatory
biomarkers for COPD. Fibrinogen was identified as a biomarker in the Evaluation of COPD
Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study (21), and an
elevated level of fibrinogen was independently and significantly associated with mortality
in patients with COPD (22). CD177 levels were increased in the airways of Cynomolgus
monkeys after ozon challenge (23), indicating the potential relevance of both genes for
COPD pathophysiology. It is beyond the scope of the current manuscript to investigate the
functional roles of CD177 and fibrinogen α in detail; however, our results and the above
mentioned clinical data certainly provide strong rationale for future studies in this area.

Chapter 4
96
The fact that KC levels were increased in M3R‐/‐ mice receiving wild‐type bone marrow cells
in our study also has other implications. Previously, we have shown that increased KC
release in response to cigarette smoke exposure is prevented after total knock‐out of the
M3 receptor (chapter 3). KC can be released from airway structural cells, including airway
smooth muscle cells and epithelial cells, and from inflammatory cells, including
macrophages. The finding that KC release is prevented after total knock‐out of the M3
receptor, but not after knock‐out of the M3 receptor on structural cells only, suggests a
role for inflammatory cells in this response. The suggestion that acetylcholine induces the
release of KC from inflammatory cellsis supported by in vitro studies showing enhanced
neutrophil recruitment after stimulation of macrophages with acetylcholine (10, 15).
However, knock‐out of the M3 receptor only on inflammatory cells does not prevent
neutrophilic inflammation, whereas knock‐out of the M3 receptor on structural cells does.
Therefore, the contribution of the M3 receptor on inflammatory cells to the pro‐
inflammatory effect of acetylcholine is only small, and this is mainly dependent on M3
receptors on structural cells.
Acetylcholine is not only released as a neurotransmitter, but can also be released from
non‐neuronal cells. Almost all cell types in the airways have been shown to express
synthesizing enzymes for acetylcholine, suggesting acetylcholine production (24). It has
been proposed that this non‐neuronal acetylcholine might contribute to inflammation,
although direct evidence is still limited (chapter 2, 25). Results from our study indicate
that non‐neuronal acetylcholine acting on inflammatory cells does not significantly
contribute to the pro‐inflammatory effects of acetylcholine. There might be a role for non‐
neuronal acetylcholine released from airway structural cells including epithelial cells,
however, future studies are needed to investigate this in more detail.
In conclusion, we demonstrate that acetylcholine regulates inflammation via M3 receptors
on structural cells. This emphasizes the important role of airway structural cells in
neutrophilic inflammation and the pathophysiology of COPD, and the contribution of
acetylcholine to this response. Whether anticholinergic therapy also affects inflammation
in patients with COPD still needs to be elucidated, but our results indicate that this might
be an additional beneficial effect of M3 selective anticholinergics.
Acknowledgements
The authors would like to thank the Netherlands Lung Foundation for financial support
(grant: 3.2.08.014).

M3 receptors on structural cells regulate inflammation
97
References
(1) Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to
emphysema. N Engl J Med 1984;311:421‐425.
(2) Barnes PJ. The role of anticholinergics in chronic obstructive pulmonary disease. Am J Med
2004;117 Suppl 12A:24S‐32S.
(3) Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 2000;343:269‐280.
(4) Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs
H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur
Respir J 2011;38:789‐796.
(5) Wollin L, Pieper MP. Tiotropium bromide exerts anti‐inflammatory activity in a cigarette smoke
mouse model of COPD. Pulm Pharmacol Ther 2010;23:345‐354.
(6) Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology
of asthma and COPD. Respir.Res. 2006;7:73.
(7) Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J, Kolahian S, Halayko AJ, Hiemstra
PS, Zuyderduyn S. Muscarinic M3 receptor stimulation increases cigarette smoke‐induced IL‐8
secretion by human airway smooth muscle cells. Eur Respir J 2009;34:1436‐1443.
(8) Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP,
Gjomarkaj M. Acetylcholine mediates the release of IL‐8 in human bronchial epithelial cells by a
NFkB/ERK‐dependent mechanism. Eur J Pharmacol 2008;582:145‐153.
(9) Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine‐
induced release of chemotactic mediators in vitro. Respir Med 2007;101:2386‐2394.
(10) Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide.
Respir Res 2011;12:24.
(11) Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J. Muscarinic induction of
hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents.
Neuron 2002;33:615‐624.
(12) Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat
Rev Immunol 2013;13:159‐175.
(13) Shen LL, Liu YN, Shen HJ, Wen C, Jia YL, Dong XW, Jin F, Chen XP, Sun Y, Xie QM. Inhalation of
glycopyrronium inhibits cigarette smoke‐induced acute lung inflammation in a murine model of
COPD. Int Immunopharmacol 2014;18:358‐364.
(14) Dominguez‐Fandos D, Ferrer E, Puig‐Pey R, Carreno C, Prats N, Aparici M, Musri MM, Gavalda A,
Peinado VI, Miralpeix M, Barbera JA. Effects of aclidinium bromide in a cigarette smoke‐exposed
Guinea pig model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2014;50:337‐
346.
(15) Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages
to release inflammatory cell chemotactic activity. Am J Physiol 1998;274:L970‐L979.
(16) de Almeida VV, Silva‐Herdade A, Calado A, Rosario HS, Saldanha C. Fibrinogen modulates
leukocyte recruitment in vivo during the acute inflammatory response. Clin Hemorheol Microcirc
2012.

Chapter 4
98
(17) Sachs UJ, Andrei‐Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ,
Preissner KT, Chavakis T, Santoso S. The neutrophil‐specific antigen CD177 is a counter‐receptor for
platelet endothelial cell adhesion molecule‐1 (CD31). J Biol Chem 2007;282:23603‐23612.
(18) Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte
engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac‐1 is critical for host
inflammatory response in vivo. J Clin Invest 2004;113:1596‐1606.
(19) Stroncek DF. Neutrophil‐specific antigen HNA‐2a, NB1 glycoprotein, and CD177. Curr Opin
Hematol 2007;14:688‐693.
(20) Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 contributes to transendothelial
migration of NB1‐positive neutrophils. J Immunol 2012;188:2419‐2426.
(21) Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal‐Singer R, Evaluation of COPD
Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. COPD association and
repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res 2011;12:146‐9921‐12‐146.
(22) Celli BR, Locantore N, Yates J, Tal‐Singer R, Miller BE, Bakke P, Calverley P, Coxson H, Crim C,
Edwards LD, Lomas DA, Duvoix A, MacNee W, Rennard S, Silverman E, Vestbo J, Wouters E, Agusti A,
ECLIPSE Investigators. Inflammatory biomarkers improve clinical prediction of mortality in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med 2012;185:1065‐1072.
(23) Hicks A, Kourteva G, Hilton H, Li H, Lin TA, Liao W, Li Y, Wei X, March T, Benson J, Renzetti LM.
Cellular and molecular characterization of ozone‐induced pulmonary inflammation in the
Cynomolgus monkey. Inflammation 2010;33:144‐156.
(24) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008;154:1558‐1571.
(25) Gwilt CR, Donnelly LE, Rogers DF. The non‐neuronal cholinergic system in the airways: an
unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208‐222.



CHAPTER 5
ANTI‐INFLAMMATORY EFFECTS OF ACETYLCHOLINE INHIBITION
AFTER TARGETED LUNG DENERVATION IN PATIENTS WITH COPD
Loes E.M. Kistemaker
Dirk‐Jan Slebos
Herman Meurs
Huib A.M. Kerstjens
Reinoud Gosens
Submitted to Thorax, 2015

Chapter 5
102
Abstract
Rationale
Acetylcholine does not only induce bronchoconstriction, but is also identified as a
regulator of airway inflammation in animal models of COPD. This has not been
demonstrated in patients with COPD. Therefore, the effect of targeted lung denervation
(TLD) on inflammation in patients with COPD was investigated in this study. TLD is a novel
bronchoscopic therapy for COPD, in which airway nerves are ablated by locally applying
radiofrequency energy.
Methods
Markers of inflammation in bronchial washings and brushes were investigated 30 days
after TLD.
Results
TLD attenuated airway inflammation, with a significant decrease in CCL4 (MIP‐1β) protein
levels in the bronchial wash and CXCL8 and TGF‐β gene expression levels in the bronchial
brush.
Conclusions
These findings suggest that parasympathetic denervation reduces inflammation in
patients with COPD.
To the editor
Acetylcholine is the primary parasympathetic neurotransmitter in the airways, and
induces bronchoconstriction. Animal models of COPD revealed that acetylcholine also
promotes airway inflammation, which can be inhibited by anticholinergic intervention
(chapter 2). This could be clinically relevant, but has never been demonstrated in COPD
patients.
We investigated the effect of targeted lung denervation (TLD) on airway inflammation in
COPD. TLD is a novel potential therapy for COPD, in which parasympathetic airway nerves
are bronchoscopically ablated by applying radiofrequency energy. Clinical evidence in
COPD suggests that TLD improves lung function and quality of life (1). We hypothesized
that TLD would inhibit airway inflammation.
Seven patients with moderate to severe COPD were recruited as part of a safety and
technical feasibility study for TLD (NCT01483534) (1). TLD of the right lung was performed
on day 0 (see supplement). A bronchial wash and brush were collected from the lung
distal to the site of denervation before (day 0) and after denervation (day 30). We
Lung denervation attenuates inflammation
103
analyzed inflammatory cells and cytokines in the bronchial wash, and gene expression of
inflammatory markers in the brush.
Patient characteristics are presented in Table 1, the main results in Figure 1, and all
inflammatory cells and cytokines in Table S1. The percentage of neutrophils in the
bronchial wash was decreased after TLD in 5/7 patients, CXCL8 decreased in 4/7 patients,
and CCL4 decreased in 6/7 patients (p=0.047). Gene expression of CXCL8 in the brush
decreased in 6/7 patients (p=0.031), as did IL‐6 in 5/7 patients, TGF‐β in 6/7 patients
(p=0.047), and MUC5AC in 5/7 patients.
Our findings suggest that TLD attenuates airway inflammation. Evidence on the role of
acetylcholine in inflammation from patients is limited. Although the use of tiotropium
bromide is associated with a reduction in exacerbation frequency (2), methodological
problems have hampered the evaluation of inflammation in the available drug study (3).
This bronchoscopic targeting of the parasympathetic system enabled us to examine the
effects on airway inflammation in a completely novel, direct manner. Although
denervation also targets sensory nerve fibers that release pro‐inflammatory
neuropeptides, such as substance P and calcitonin gene‐related peptide, these nerves
have been shown to regenerate, in contrast to cholinergic nerve fibers. We believe that
these findings suggest a pro‐inflammatory role for acetylcholine in the airways.
Acetylcholine is not only a neurotransmitter, but is also produced by non‐neuronal cells. It
has been proposed that this non‐neuronal acetylcholine is responsible for the pro‐
inflammatory effects of acetylcholine (chapter 2). The current study suggests that
neuronal acetylcholine may in fact contribute to inflammation in COPD.
Table 1. Baseline characteristics of subjects (n=7 subjects).
Age (years) 56.4 (10.4)
Male (n, %) 2 (29%)
History of Smoking (n, %) 7 (100%)
Pack‐Years 35.8 (13.7)
FEV1 (L) – Pre‐bronchodilator 0.70 (0.13)
FVC (L) – Pre‐bronchodilator 2.19 (0.27)
Data are mean (SD) unless stated otherwise. FEV1=forced expiratory volume in 1 s.
FVC=forced vital capacity.

Chapter 5
104
Figure 1. Percentage change at day 30 compared to day 0 in levels of neutrophils and protein expression of
CXCL8 and CCL4 in bronchial wash, and in gene expression of CXCL8, IL‐6, TGF‐β and MUC5AC in bronchial brush.
Baseline values are set to 100%, represented by the dashed line. * p<0.05 (Wilcoxon signed rank test). CXCL8: C‐
X‐C chemokine 8, also known as IL‐8; CCL4: chemokine C‐C ligand 4, also known as MIP‐1β; IL‐6: interleukin‐6;
TGF‐β: transforming growth factor‐β. In this small study population, a repeated measures analysis of covariance
did not show significant interactions of these parameters with changes in FEV1 in response to TLD.
Furthermore, TLD might also have effects on airway remodeling, since a significant
reduction in TGF‐β was observed. This aligns well with the reduction in TGF‐β we have
previously found in muscarinic M3 receptor knock‐out mice (chapter 3), and with the
human data from the UPLIFT trial, where tiotropium reduced lung function decline in a
subgroup of patients (4).
There are limitations to this small cohort pilot study, including the limited number of
subjects and the lack of control of successful denervation. However, we believe that the
findings are promising, and a large‐scale multicenter sham‐controlled study of the
effectiveness of TLD is planned and will include similar analyses. This should provide more
evidence for the role of neuronal acetylcholine as a pro‐inflammatory mediator.
Moreover, it would be interesting to perform similar analyses in patients treated with
anticholinergic drugs.
In conclusion, our findings constitute explorative evidence for a role of acetylcholine in
airway inflammation in patients with COPD. This is a novel and sparsely explored
mechanism that could be relevant for treatment strategies, but first asks for confirmation.

Lung denervation attenuates inflammation
105
Acknowledgements
The authors would like to thank the Netherlands Lung Foundation (grant: 3.2.08.014) and
Holaira Inc. for financial support.

Chapter 5
106
Supplement
Methods
Details of TLD Procedure
Targeted lung denervation (TLD) was performed using a bronchoscopically guided catheter
based lung denervation system (Holaira, Inc., Plymouth, MN, USA) delivered during two
rigid bronchoscopic procedures 30 days apart, performed under general anesthesia in an
outpatient setting. The lung denervation system includes a catheter with a stainless steel
internally cooled electrode, thermocouple, expandable balloon, and coolant entry/exit
tubing that are attached to a separate radiofrequency generator, cooling console, and
coolant pump. The cooled electrode is designed to generate therapeutic lesions at a
sufficient depth from the inner surface of the main right and left bronchus to ablate the
airway nerves that travel parallel to and outside of the main bronchi and into the lungs.
The expandable balloon provided protective cooling to minimize airway wall effects of the
main bronchi during radiofrequency ablation of the nerves. The electrode was placed and
activated as prescribed in up to 8 positions per bronchus to ensure complete
circumferential treatment. Bronchoscopic and fluoroscopic visualization was used to guide
electrode positioning.
Analysis on bronchial wash and brush
On the bronchial wash, a differential cell count was performed, and cytokine
concentrations were measured. For a differential cell count, cytospin‐preparations were
stained with May–Grünwald and Giemsa (both Sigma, St. Louis) and a cell count was
performed by counting at least 400 cells in duplicate in a blinded fashion. Levels of 26
cytokines in the wash were determined using a multiplex assay (Millipore, Billerica, MA,
USA). The following cytokines were assessed: eotaxin, G‐CSF, GM‐CSF, IFN‐α2, IFN‐γ, IL‐1α,
IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, IL‐10, IL‐12 (p40), IL‐12 (p70), IL‐13, IL‐15, IL‐17, IP‐
10, MCP‐1, MIP‐1α, MIP‐1β, TNF‐α, and TNF‐β. From the bronchial brush, total RNA was
extracted (Qiagen, Venlo, The Netherlands), reverse transcribed, and subjected to qRT‐
PCR. qRT‐PCR was performed with denaturation at 94°C for 30 seconds, annealing at 59°C
for 30 seconds and extension at 72°C for 30 seconds for 40 cycles followed by 10 minutes
at 72°C. Real‐time PCR data were analyzed using the comparative cycle threshold (Ct:
amplification cycle number) method. The amount of target gene was normalized to the
endogenous reference gene 18S ribosomal RNA.

Lung denervation attenuates inflammation
107
Table S1. Levels of inflam
matory cells and cytokines in
bronchial w
ash fluid. C
CL2: also known as MCP‐1, CCL4: also known as MIP‐1β; C
CL11: also known as eotaxin;
CXCL8: also known as IL‐8; C
XCL10: also known as IP‐10. O
ther cytokines were below the detection limit of 3.2 pg/ml.
Subject 1
Subject 2
Subject 3
Subject 4
Subject 5
Subject 6
Subject 7
0
30
0
30
0
30
0
30
0
30
0
30
0
30
Cells
in wash
(% of
total
cells)
Neu
trophils
32.45
17.19
18.40
20.13
22.48
19.19
60.21
39.88
10.87
32.50
70.63
39.00
49.75
29.75
Macrophages
66.30
81.63
80.66
78.94
73.83
78.88
38.35
58.38
87.82
66.31
28.44
59.19
46.50
67.29
Lymphocytes
1.06
1.19
0.69
0.94
2.06
1.75
0.25
1.00
1.25
0.31
0.31
0.19
0.19
0.23
Eosinophils
0.19
0.00
0.25
0.00
1.62
0.19
1.19
0.75
0.06
0.88
0.63
0.50
3.56
2.15
Proteins
in wash
(pg/ml)
CCL2
80.98
27.70
47.02
13.71
73.73
262.28
49.02
29.71
14.78
35.45
131.51
214.44
85.48
106.10
CCL4
32.23
5.80
10.71
0.00
12.12
0.00
12.65
5.61
12.61
14.99
20.31
19.20
16.36
13.49
CCL11
12.46
5.19
12.71
4.73
5.20
4.41
7.83
6.39
12.20
19.80
32.32
27.05
25.10
23.69
CXCL8
382.55
98.26
303.31
98.65
146.73
89.49
334.34
253.24
177.80
274.15
517.86
568.10
458.91
899.18
CXCL10
231.51
80.04
195.21
42.55
79.32
65.51
145.77
135.46
34.71
52.81
193.74
319.31
869.57
710.37
G‐CSF
108.24
39.21
94.48
38.91
33.00
42.11
119.97
66.87
79.01
165.35
294.60
237.03
51.75
80.11
IFNα2
21.36
13.78
14.82
9.81
6.36
13.09
13.17
11.79
10.05
4.03
7.72
16.25
20.44
14.84
IL‐6
19.42
5.59
21.18
4.81
4.67
3.91
21.49
17.39
1.18
8.75
15.89
15.06
16.26
15.98
IL‐7
11.70
4.00
5.18
1.19
2.74
2.74
4.00
5.53
3.55
2.80
10.40
7.34
10.80
9.43

Chapter 5
108
References
(1) Slebos DJ, Klooster K, Koegelenberg CFN, Theron J, Steyn D, Mayse M. Efficacy of targeted lung
denervation on patients with moderate to severe COPD [abstract]. ERJ 2014: 44; Suppl. 58: P1774.
(2) Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M, UPLIFT Study
Investigators. A 4‐year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med
2008;359:1543‐1554.
(3) Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K, Kesten S, Wedzicha JA. Effect of
tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J
2007;30:472‐478.
(4) Morice AH, Celli B, Kesten S, Lystig T, Tashkin D, Decramer M. COPD in young patients: a pre‐
specified analysis of the four‐year trial of tiotropium (UPLIFT). Respir Med 2010;104:1659‐1667.



CHAPTER 6
MUSCARINIC M3 RECEPTORS CONTRIBUTE TO ALLERGEN‐
INDUCED AIRWAY REMODELING IN MICE
Loes E.M. Kistemaker
I. Sophie T. Bos
Willemieke M. Mudde
Machteld N. Hylkema
Pieter S. Hiemstra
Jürgen Wess
Herman Meurs
Huib A.M. Kerstjens
Reinoud Gosens
Am J Respir Cell Mol Biol, 2014, 50(4):690‐8

Chapter 6
112
Abstract
Rationale
Asthma is a chronic obstructive airway disease, characterized by inflammation and
remodeling. Acetylcholine contributes to symptoms by inducing bronchoconstriction via
the muscarinic M3 receptor. Recent evidence suggests that bronchoconstriction can
regulate airway remodeling and therefore implies a role for the M3 receptor. In this study,
the contribution of the M3 receptor to allergen‐induced remodeling was investigated using
M3 receptor subtype deficient (M3R‐/‐) mice. Wild‐type, M1R
‐/‐ and M2R‐/‐ mice were used
as controls.
Methods
C57Bl/6 mice were sensitized and challenged with ovalbumin (twice weekly, for 4 weeks).
Control animals were challenged with saline.
Results
Allergen exposure induced goblet cell metaplasia, airway smooth muscle thickening (1.7‐
fold), pulmonary vascular smooth muscle remodeling (1.5‐fold) and deposition of collagen
type I (1.7‐fold) and fibronectin (1.6‐fold) in the airway wall of wild‐type mice. These
effects were absent or markedly lower in M3R‐/‐ mice (30‐100%), whereas M1R
‐/‐ and M2R‐/‐
mice responded similar to wild‐type mice. In addition, airway smooth muscle and
pulmonary vascular smooth muscle mass were 35‐40% lower in saline‐challenged M3R‐/‐
mice compared to wild‐type mice. Interestingly, allergen‐induced airway inflammation,
assessed as infiltrated eosinophils and TH2‐cytokine expression, was similar or even
enhanced in M3R‐/‐ mice.
Conclusions
Our data indicate that acetylcholine contributes to allergen‐induced remodeling and
smooth muscle mass via the M3 receptor, and not via M1 or M2 receptors. No stimulatory
role for M3 receptors in allergic inflammation was observed, suggesting that the role of
acetylcholine in remodeling is independent of the allergic inflammatory response and may
involve bronchoconstriction.
Introduction
Asthma is an obstructive lung disease, characterized by airway inflammation and
remodeling. In the inflammatory process eosinophils, mast cells and T cells play an
important role. Inflammation is mainly regulated by the T‐helper type 2 (TH2) cytokines
interleukin (IL)‐4, IL‐5 and IL‐13 (1). The inflammatory response may contribute to
remodeling of the airways, inducing structural changes such as goblet cell metaplasia,
airway smooth muscle thickening and subepithelial fibrosis (2). Furthermore, pulmonary

M3 receptors promote allergen‐induced remodeling
113
vascular remodeling and increased angiogenesis are observed, associated with higher
vascular endothelial growth factor (VEGF) release (2, 3).
The parasympathetic neurotransmitter acetylcholine induces bronchoconstriction via M3
receptor stimulation and can thereby contribute to symptoms and loss of lung function in
asthma (chapter 2). Parasympathetic nerve activity is increased in patients with asthma
and the airways are hyperresponsive to cholinergic stimuli (4, 5). Indeed, a recent
randomized controlled trial demonstrated that the long‐acting anticholinergic tiotropium
induces bronchodilation in symptomatic asthma patients, when used on top of standard
combination therapy of inhaled glucocorticosteroids and long‐acting β2‐agonists (6),
indicating a role for muscarinic receptors in airflow obstruction in asthma (7‐9).
Recent evidence indicates that acetylcholine also regulates airway inflammation and
remodeling, as indicated by the protective effects of anticholinergics on these parameters
in animal models of asthma (10‐12). Inhibition of remodeling by anticholinergics may be a
consequence of reduced inflammation, but might also be caused by direct inhibitory
effects on bronchoconstriction. This latter hypothesis is supported by a recent study from
Grainge et al., who demonstrated that repeated methacholine challenges induced airway
remodeling in asthma patients, with no effect on inflammation (13).
The lung expresses multiple muscarinic receptor subtypes that have differential roles in
regulating bronchoconstriction, inflammation and remodeling (chapter 2). This limits the
interpretation of the effects of non‐selective anticholinergics and muscarinic receptor
agonists on these parameters. Since the M3 receptor is the muscarinic receptor subtype
that causes bronchoconstriction in the airways (14), we studied the specific role of the M3
receptor in allergen‐induced inflammation and remodeling using M3 receptor subtype
deficient (M3R‐/‐) mice. We investigated the role of the M3 receptor on ovalbumin‐induced
goblet cell metaplasia, airway smooth muscle thickening, pulmonary vascular remodeling,
extracellular matrix deposition, airway eosinophilia and cytokine expression. M1R‐/‐ and
M2R‐/‐ mice were used to verify that the effects were specific for the M3 receptor. In this
study we demonstrate the specific involvement of the M3 receptor in allergen‐induced
airway remodeling in vivo.

Chapter 6
114
Methods
Animals
Homozygous, inbred, specific‐pathogen‐free breeding colonies of M1R‐/‐, M2R
‐/‐ and M3R‐/‐
mice and C57Bl/6NTac wild‐type (WT) mice with the same genetic background were
obtained from Taconic (Cambridge City, Indiana, USA). The M1R‐/‐, M2R
‐/‐ and M3R‐/‐ mice
used were on a 129 Sv/J background and backcrossed for at least 10 generations onto the
C57Bl/6NTac background (15). Knock‐out animals did not differ from their WT littermates
in overall health, fertility and longevity (15‐17). Animals were housed conventionally
under a 12‐h light‐dark cycle and received food and water ad libitum. All experiments
were performed in accordance with the national guidelines and approved by the
University of Groningen Committee for Animal Experimentation (number: 5463A).
Animal model
Female mice (n=8‐10 per group, 10–12 weeks old) were sensitized to ovalbumin (OVA;
Sigma‐Aldrich, Zwijndrecht, The Netherlands) on days 1, 14 and 21 by an intraperitoneal
injection of 10 µg OVA emulsified in 1.5 mg aluminium hydroxide (Aluminject; Pierce
Chemical, Etten‐Leur, The Netherlands) and diluted to 200 µl with phosphate buffered
saline (PBS). Subsequently, mice were challenged with saline or OVA aerosols (1% in PBS)
for 20 minutes twice weekly on consecutive days for 4 weeks (figure 1). The aerosol was
delivered to a Perspex exposure chamber (9 l) by a De Vilbiss nebulizer (type 646; De
Vilbiss, Somerset, PA) driven by an airflow of 40 l/min providing an aerosol with an output
of 0.33 ml/min as described previously (18).
Figure 1. Experimental procedure. Female C57Bl/6NTac mice (n=8‐10 per group) were sensitized to ovalbumin
on day 1, 14 en 21. Subsequently, mice were challenged with saline or ovalbumin aerosols for 20 minutes twice
weekly for 4 weeks. Twenty‐four hours after the last challenge mice were sacrificed and lungs were harvested.
Tissue collection
Twenty‐four hours after the last challenge, animals were euthanized by intraperitoneal
pentobarbital injection (400 mg/kg, hospital pharmacy, University Medical Center
Groningen) and lungs were harvested. The smallest lower left lobe was snap frozen for
mRNA analysis, the upper left lobe was snap frozen for multiplex ELISA and protein

M3 receptors promote allergen‐induced remodeling
115
analysis, the two lower right lung lobes were snap frozen for immunohistochemistry and
the upper right lobe was formalin fixed and embedded in paraffin for
immunohistochemistry. A complete description of the methods for mRNA analysis,
Western Blot analysis, cytokine analysis and immunohistochemistry can be found in the
supplement.
Statistical analysis
Data are presented as mean ± s.e. of the mean. Statistical differences between means
were calculated using two‐way ANOVA, followed by Newman Keuls multiple comparison
tests. Differences were considered significant at p<0.05.
Results
Goblet cell metaplasia
In order to investigate the effect of ovalbumin challenge on mucus‐producing cells,
sections were stained with PAS to identify goblet cells and MUC5AC gene expression was
analyzed in lung homogenates. Ovalbumin challenge increased the number of goblet cells
in the airways of WT mice, as well as in M3R‐/‐ mice (figure 2A and B). However, the
increase in goblet cells was 30% lower in M3R‐/‐ mice compared to that in WT mice
(p<0.05). This inhibitory effect was not seen in mice deficient in the M1 or M2 receptor
(figure S1). In line with findings on goblet cell number, ovalbumin challenge increased
MUC5AC expression in the lungs of WT mice and M3R‐/‐ mice, but the increase was 35%
lower in M3R‐/‐ mice compared to WT mice (p<0.05; figure 2C).
Airway smooth muscle remodeling
To assess airway smooth muscle remodeling, lung sections were stained for α‐smooth
muscle (sm)‐actin. Ovalbumin challenge induced a 1.7‐fold increase in α‐sm‐actin positive
area around the airways of WT mice (figure 3). This increase was fully absent in M3R‐/‐
mice (figure 3). Interestingly, sm‐α‐actin positive area was lower in M3R‐/‐ mice compared
to WT mice, irrespective of saline or ovalbumin treatment (p<0.001 by two‐way ANOVA).
Mice deficient in the M1 or M2 receptor responded similar to WT mice, although the
increase in α‐sm‐actin deposition in response to ovalbumin was not significant in M2R‐/‐
mice (figure S2). This indicates that these effects are specific for the M3 receptor.
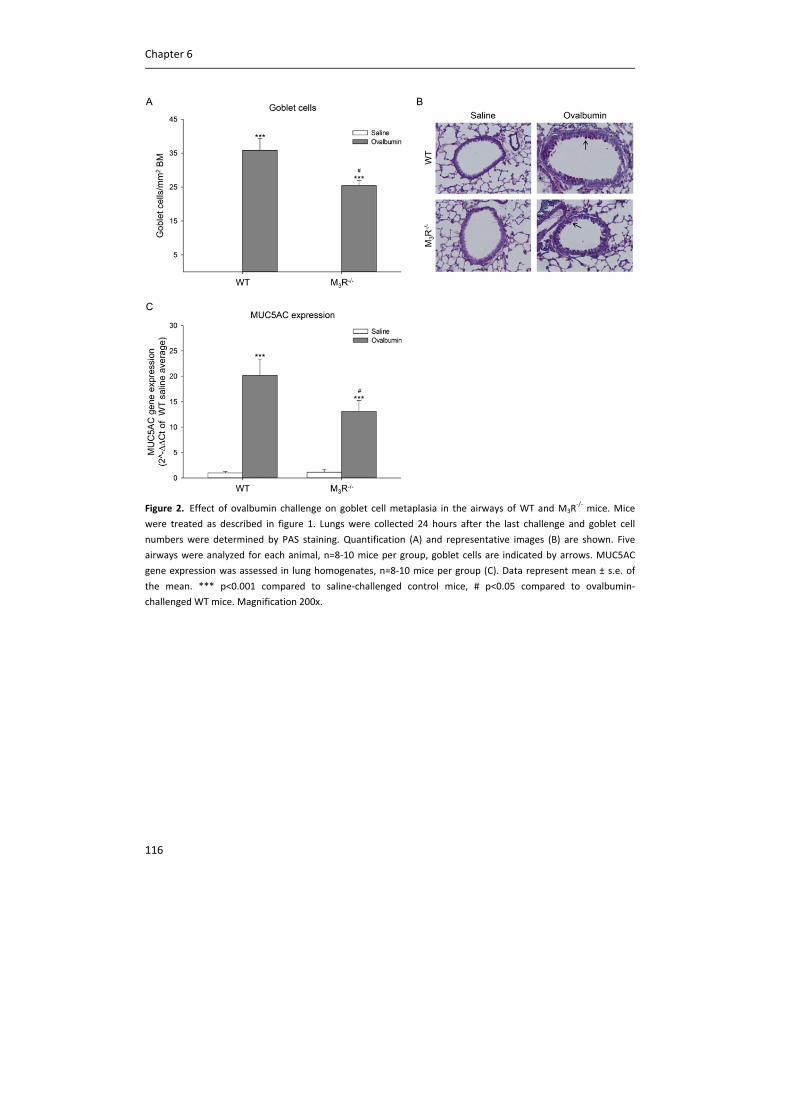
Chapter 6
116
Figure 2. Effect of ovalbumin challenge on goblet cell metaplasia in the airways of WT and M3R
‐/‐ mice. Mice
were treated as described in figure 1. Lungs were collected 24 hours after the last challenge and goblet cell
numbers were determined by PAS staining. Quantification (A) and representative images (B) are shown. Five
airways were analyzed for each animal, n=8‐10 mice per group, goblet cells are indicated by arrows. MUC5AC
gene expression was assessed in lung homogenates, n=8‐10 mice per group (C). Data represent mean ± s.e. of
the mean. *** p<0.001 compared to saline‐challenged control mice, # p<0.05 compared to ovalbumin‐
challenged WT mice. Magnification 200x.
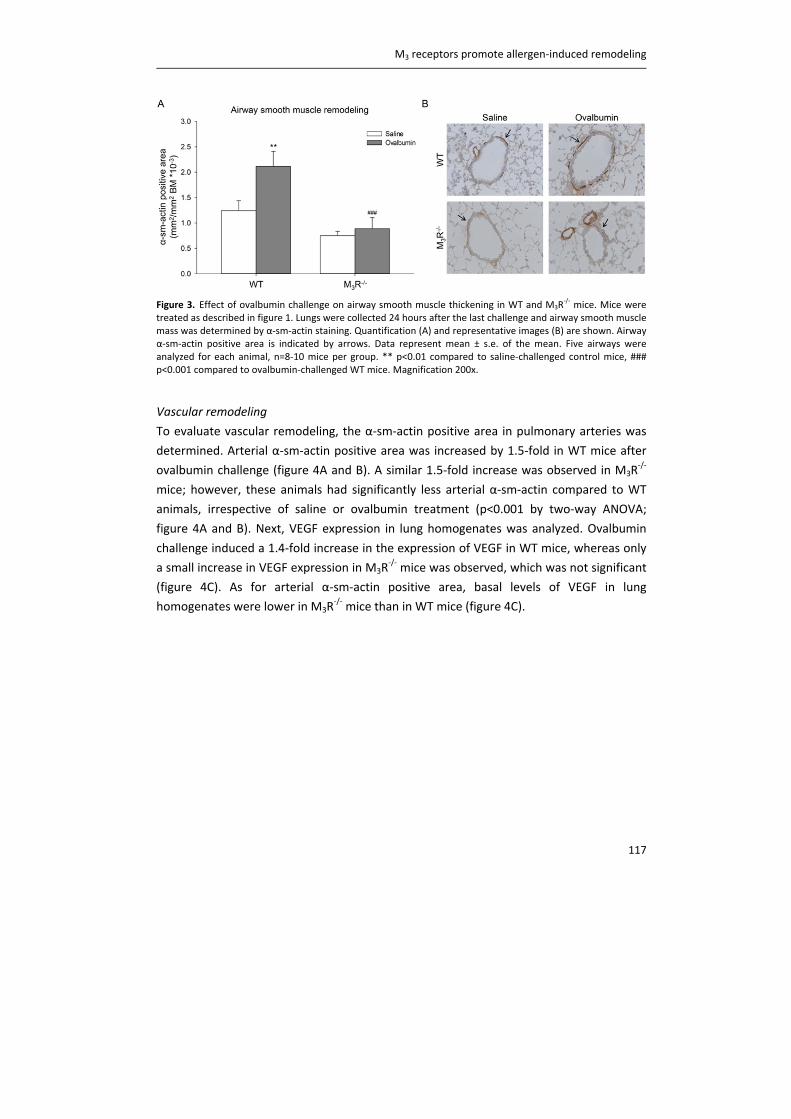
M3 receptors promote allergen‐induced remodeling
117
Figure 3. Effect of ovalbumin challenge on airway smooth muscle thickening in WT and M3R‐/‐ mice. Mice were
treated as described in figure 1. Lungs were collected 24 hours after the last challenge and airway smooth muscle mass was determined by α‐sm‐actin staining. Quantification (A) and representative images (B) are shown. Airway α‐sm‐actin positive area is indicated by arrows. Data represent mean ± s.e. of the mean. Five airways were analyzed for each animal, n=8‐10 mice per group. ** p<0.01 compared to saline‐challenged control mice, ### p<0.001 compared to ovalbumin‐challenged WT mice. Magnification 200x.
Vascular remodeling
To evaluate vascular remodeling, the α‐sm‐actin positive area in pulmonary arteries was
determined. Arterial α‐sm‐actin positive area was increased by 1.5‐fold in WT mice after
ovalbumin challenge (figure 4A and B). A similar 1.5‐fold increase was observed in M3R‐/‐
mice; however, these animals had significantly less arterial α‐sm‐actin compared to WT
animals, irrespective of saline or ovalbumin treatment (p<0.001 by two‐way ANOVA;
figure 4A and B). Next, VEGF expression in lung homogenates was analyzed. Ovalbumin
challenge induced a 1.4‐fold increase in the expression of VEGF in WT mice, whereas only
a small increase in VEGF expression in M3R‐/‐ mice was observed, which was not significant
(figure 4C). As for arterial α‐sm‐actin positive area, basal levels of VEGF in lung
homogenates were lower in M3R‐/‐ mice than in WT mice (figure 4C).
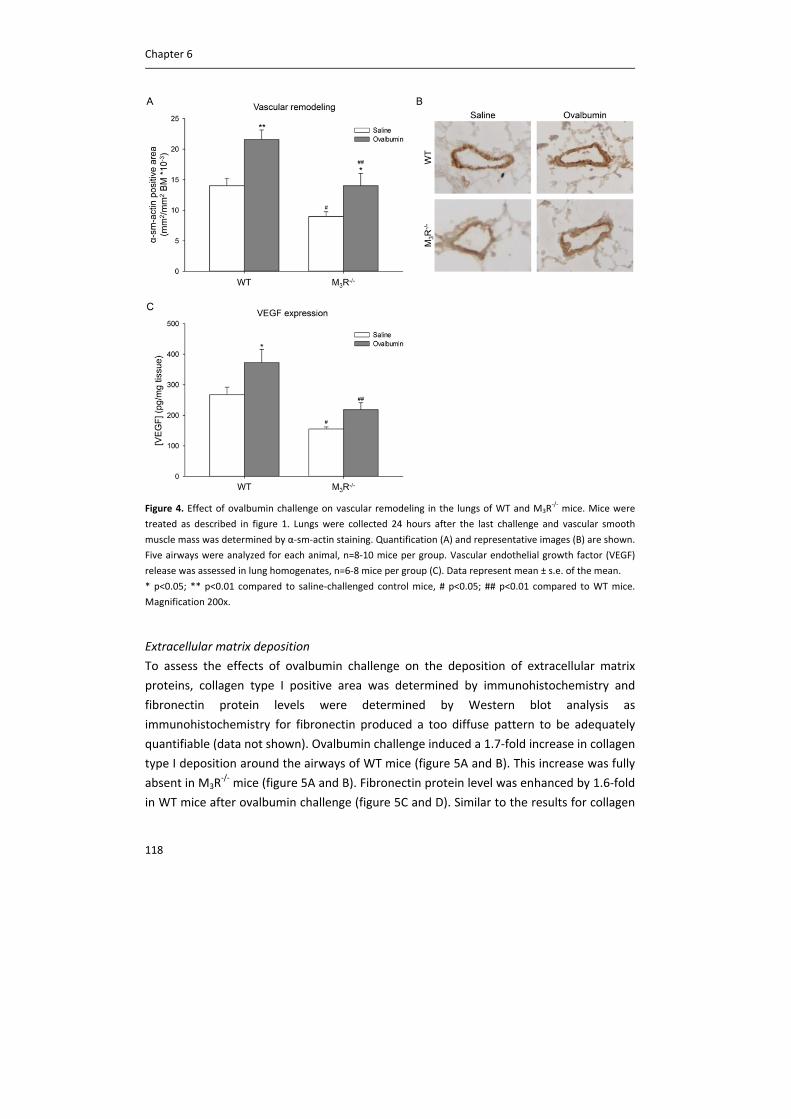
Chapter 6
118
Figure 4. Effect of ovalbumin challenge on vascular remodeling in the lungs of WT and M3R‐/‐ mice. Mice were
treated as described in figure 1. Lungs were collected 24 hours after the last challenge and vascular smooth
muscle mass was determined by α‐sm‐actin staining. Quantification (A) and representative images (B) are shown.
Five airways were analyzed for each animal, n=8‐10 mice per group. Vascular endothelial growth factor (VEGF)
release was assessed in lung homogenates, n=6‐8 mice per group (C). Data represent mean ± s.e. of the mean.
* p<0.05; ** p<0.01 compared to saline‐challenged control mice, # p<0.05; ## p<0.01 compared to WT mice.
Magnification 200x.
Extracellular matrix deposition
To assess the effects of ovalbumin challenge on the deposition of extracellular matrix
proteins, collagen type I positive area was determined by immunohistochemistry and
fibronectin protein levels were determined by Western blot analysis as
immunohistochemistry for fibronectin produced a too diffuse pattern to be adequately
quantifiable (data not shown). Ovalbumin challenge induced a 1.7‐fold increase in collagen
type I deposition around the airways of WT mice (figure 5A and B). This increase was fully
absent in M3R‐/‐ mice (figure 5A and B). Fibronectin protein level was enhanced by 1.6‐fold
in WT mice after ovalbumin challenge (figure 5C and D). Similar to the results for collagen
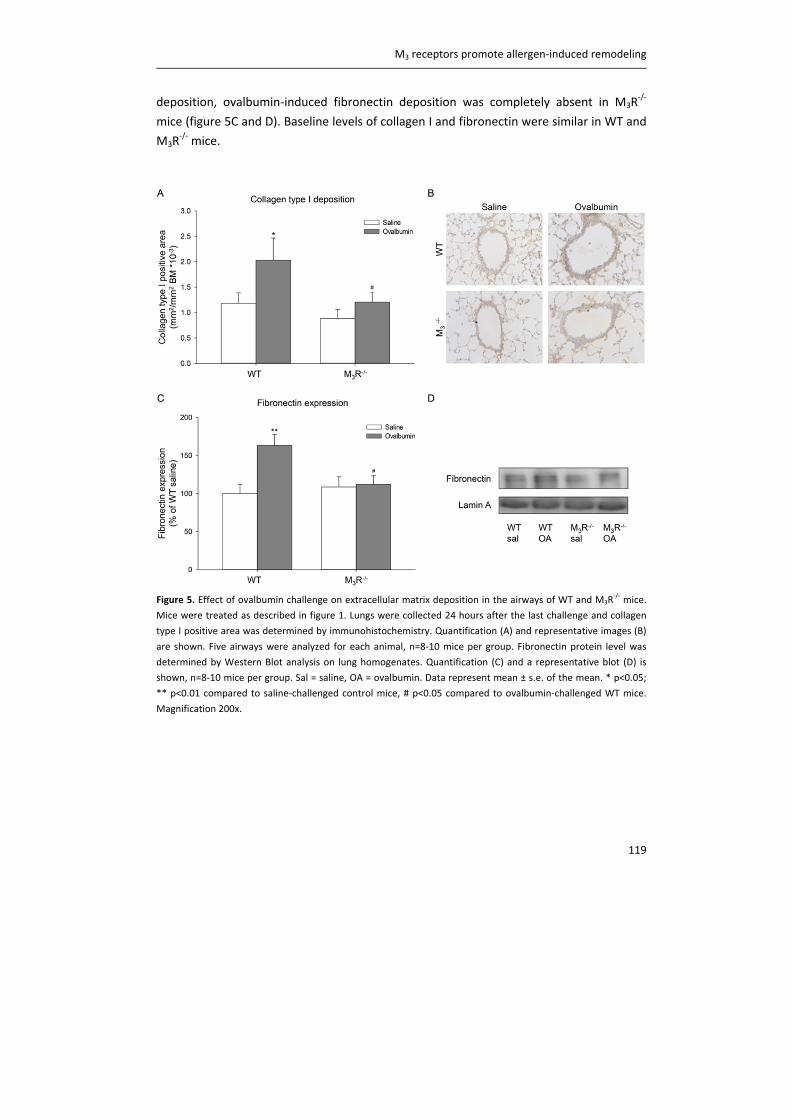
M3 receptors promote allergen‐induced remodeling
119
deposition, ovalbumin‐induced fibronectin deposition was completely absent in M3R‐/‐
mice (figure 5C and D). Baseline levels of collagen I and fibronectin were similar in WT and
M3R‐/‐ mice.
Figure 5. Effect of ovalbumin challenge on extracellular matrix deposition in the airways of WT and M3R
‐/‐ mice.
Mice were treated as described in figure 1. Lungs were collected 24 hours after the last challenge and collagen
type I positive area was determined by immunohistochemistry. Quantification (A) and representative images (B)
are shown. Five airways were analyzed for each animal, n=8‐10 mice per group. Fibronectin protein level was
determined by Western Blot analysis on lung homogenates. Quantification (C) and a representative blot (D) is
shown, n=8‐10 mice per group. Sal = saline, OA = ovalbumin. Data represent mean ± s.e. of the mean. * p<0.05;
** p<0.01 compared to saline‐challenged control mice, # p<0.05 compared to ovalbumin‐challenged WT mice.
Magnification 200x.
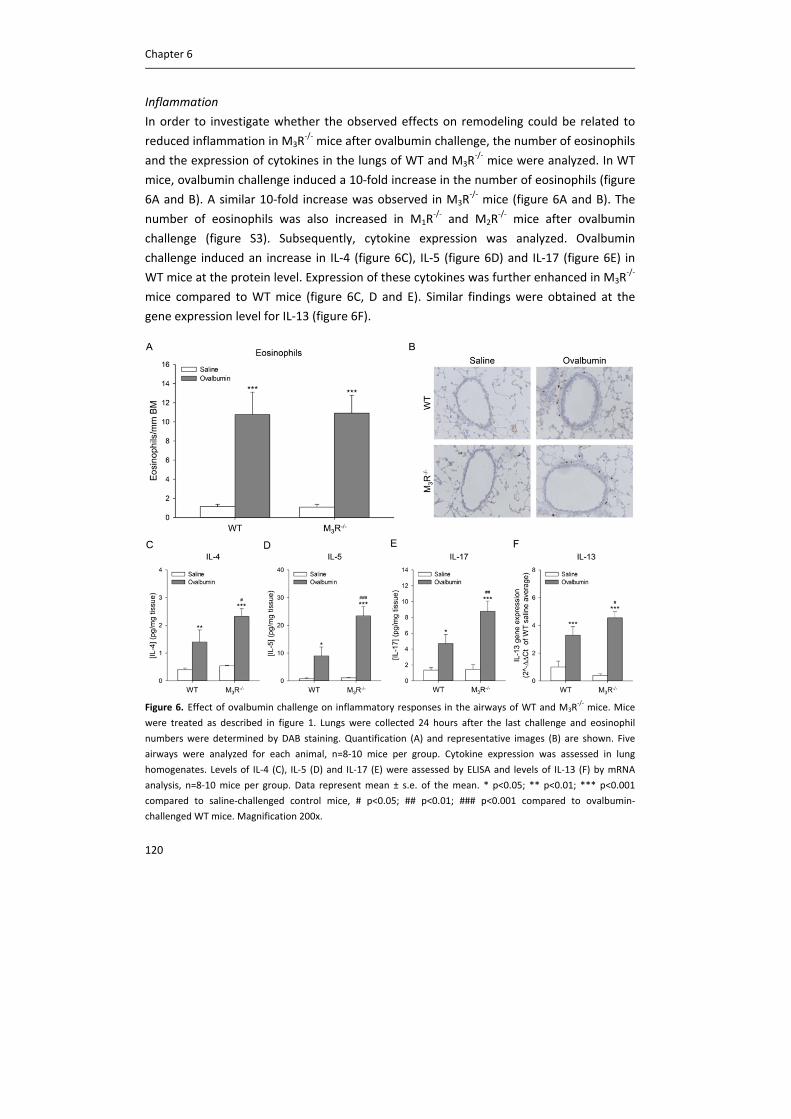
Chapter 6
120
Inflammation
In order to investigate whether the observed effects on remodeling could be related to
reduced inflammation in M3R‐/‐ mice after ovalbumin challenge, the number of eosinophils
and the expression of cytokines in the lungs of WT and M3R‐/‐ mice were analyzed. In WT
mice, ovalbumin challenge induced a 10‐fold increase in the number of eosinophils (figure
6A and B). A similar 10‐fold increase was observed in M3R‐/‐ mice (figure 6A and B). The
number of eosinophils was also increased in M1R‐/‐ and M2R
‐/‐ mice after ovalbumin
challenge (figure S3). Subsequently, cytokine expression was analyzed. Ovalbumin
challenge induced an increase in IL‐4 (figure 6C), IL‐5 (figure 6D) and IL‐17 (figure 6E) in
WT mice at the protein level. Expression of these cytokines was further enhanced in M3R‐/‐
mice compared to WT mice (figure 6C, D and E). Similar findings were obtained at the
gene expression level for IL‐13 (figure 6F).
Figure 6. Effect of ovalbumin challenge on inflammatory responses in the airways of WT and M3R
‐/‐ mice. Mice
were treated as described in figure 1. Lungs were collected 24 hours after the last challenge and eosinophil
numbers were determined by DAB staining. Quantification (A) and representative images (B) are shown. Five
airways were analyzed for each animal, n=8‐10 mice per group. Cytokine expression was assessed in lung
homogenates. Levels of IL‐4 (C), IL‐5 (D) and IL‐17 (E) were assessed by ELISA and levels of IL‐13 (F) by mRNA
analysis, n=8‐10 mice per group. Data represent mean ± s.e. of the mean. * p<0.05; ** p<0.01; *** p<0.001
compared to saline‐challenged control mice, # p<0.05; ## p<0.01; ### p<0.001 compared to ovalbumin‐
challenged WT mice. Magnification 200x.

M3 receptors promote allergen‐induced remodeling
121
The non‐neuronal cholinergic system
It has been proposed that the non‐neuronal cholinergic system (NNCS) might contribute
to airway inflammation and remodeling (chapter 2, 19). In order to investigate effects of
ovalbumin challenge on the NNCS, gene expression of different components of the NNCS
was analyzed in lung tissue homogenates of WT mice, including the choline transporters
CHT1 and CTL1, the acetylcholine synthesizing enzyme ChAT, the acetylcholine degrading
enzyme AChE and the different muscarinic receptor subtypes. As shown in figure S4,
ovalbumin challenge did not influence expression levels of these components of the NNCS.
Discussion
In this study we demonstrate that the muscarinic M3 receptor plays a profound role in
allergen‐induced airway remodeling. In M3R‐/‐ mice, goblet cell metaplasia is partly
prevented compared to WT mice after ovalbumin challenge, whereas airway smooth
muscle thickening and excessive extracellular matrix deposition are completely absent.
These effects are specific for the M3 receptor, since M1R‐/‐ and M2R
‐/‐ mice responded
similar to ovalbumin as WT mice. Surprisingly, no stimulatory role for the M3 receptor in
allergic inflammation was observed, indicating that the role of acetylcholine in remodeling
does not involve an increased TH2 type inflammatory response. This study is the first to
demonstrate the specific role of the M3 receptor in regulating allergen‐induced
remodeling in vivo and implies that alternative mechanisms to induce remodeling,
independent of eosinophilic inflammation, play an important role.
Remodeling of the airways, including goblet cell metaplasia, airway smooth muscle
thickening and excessive extracellular matrix deposition, is an important feature of
asthma. It is known from in vitro studies that acetylcholine can induce proliferation of and
mucus secretion from epithelial cells (20), which can be inhibited by aclidinium, a novel
long‐acting anticholinergic (21). Furthermore, muscarinic receptor stimulation can
enhance growth factor‐induced proliferation of airway smooth muscle cells and fibroblasts
(22, 23) and enhances extracellular matrix deposition (24, 25) and contractile protein
expression in these cells (26). Moreover, fibroblast to myofibroblast transition appears to
be regulated by muscarinic receptors (27). This is further supported by in vivo studies in
mice and guinea pigs, in which ovalbumin‐induced structural changes, including mucus
gland hypertrophy, airway smooth muscle remodeling and excessive extracellular matrix
deposition, can be inhibited by the anticholinergic tiotropium (10‐12). Acetylcholine
contributes to the asthmatic response in these models as indicated by profound inhibitory
effects of tiotropium (10, 28). Here, we demonstrate that the remodeling‐promoting

Chapter 6
122
effects of acetylcholine in vivo in response to allergen exposure are solely mediated via M3
receptors, and not via M1 or M2 receptors.
Furthermore, our study also demonstrates that the M3 receptor is involved in smooth
muscle mass development and/or maintenance, since basal levels of α‐sm‐actin are lower
in M3R‐/‐ mice compared to WT mice, both in the airways and arteries. For the airways, this
may imply that parasympathetic regulation of for example bronchoconstriction is needed
to fully develop and/or maintain the smooth muscle, as this is lacking in M3R‐/‐ mice. For
the arteries this is less easily explained. Possibly, the lower levels of pro‐angiogenic factors
such as VEGF, or cardiovascular effects secondary to M3 deficiency might account for the
decreased actin levels. M3R‐/‐ mice show intact bradycardia in response to vagal
stimulation but have reduced peripheral vasodilation in response to methacholine
stimulation (29).
There are some limitations to our study. Using an animal model we recapitulated different
features of human asthma, including eosinophilia, goblet cell metaplasia, airway smooth
muscle thickening and enhanced extracellular matrix deposition. However, some features
of asthma, including cough and mucus gland hypertrophy, are not observed in mice,
whereas these may be sensitive to cholinergic inhibition as demonstrated recently (10,
28). Furthermore, to study the contribution of the individual muscarinic receptor
subtypes, we used muscarinic receptor knock‐out mice. Although these mice do not differ
from WT mice in overall health, fertility and longevity, they do have some known, and
maybe also unknown, developmental differences. There is no clear alternative however,
as subtype‐selective agonists are not available and the subtype selective M3 receptor
antagonists that are available have significant affinity for M1 receptors. The same accounts
for tiotropium, which is known to be kinetically selective for M3 receptors, but also has a
substantial dissociation half‐life for the M1 receptor (30). Despite the limitations, the use
of muscarinic receptor knockout mice does provide a unique strength and novel view on
the role of muscarinic receptor subtypes in allergic airways disease, as we now present
strong evidence that M3 receptors, and not M1 or M2 receptors, are involved in allergen‐
induced airway remodeling in vivo.
We did not observe a stimulatory role for individual muscarinic receptor subtypes on
inflammation in our study. If anything, the inflammatory response was enhanced in M3R‐/‐
mice. Previously, it has been shown that acetylcholine exerts pro‐inflammatory effects
mediated by muscarinic receptors, both in vitro and in vivo (chapter 2). Ovalbumin‐
induced eosinophilia can be inhibited by tiotropium in guinea pigs and mice (10, 12).
Similar inhibitory effects of tiotropium are found on ovalbumin‐induced cytokine release

M3 receptors promote allergen‐induced remodeling
123
in mice, including IL‐4, IL‐5 and IL‐13 (12). The absence of an inhibitory effect on
eosinophilia and increased cytokine production as observed in M3R‐/‐ mice in our study
were therefore unexpected. A possible explanation for this observation is that multiple
subtypes may be involved in the inflammatory response induced by acetylcholine.
Tiotropium is kinetically selective for M3 receptors; however, also has a substantial
dissociation half‐life for the M1 receptor (30). As shown in figure S3, there is a trend
towards lower eosinophil numbers in M1R‐/‐ mice after ovalbumin challenge compared to
WT mice. It is therefore possible that the pro‐inflammatory effect of acetylcholine
depends on the simultaneous activation of both M1 and M3 receptors, and that a single
muscarinic receptor is not sufficient to induce these effects. Anti‐inflammatory effects of
acetylcholine in certain experimental settings (chapter 2, 31) and relaxation of bronchi by
acetylcholine‐induced NO release (32), may also help to explain the differential outcomes
of these studies. Furthermore, the antimuscarinic agents were administered after allergen
sensitization in the above mentioned studies, whereas the mice used in our protocols lack
specific muscarinic receptors throughout development. Moreover, since cytokines
responsible for recruiting eosinophils and inducing goblet cell metaplasia are increased in
M3R‐/‐ mice compared to WT mice, whereas eosinophil numbers are similar and goblet cell
numbers are even reduced in M3R‐/‐ mice compared to WT mice, M3R
‐/‐ mice may actually
be less sensitive to inflammatory mediators.
The finding that ovalbumin‐induced remodeling is greatly inhibited in M3R‐/‐ mice, with no
inhibitory effect on inflammation, has significant implications. Generally, airway structural
changes after allergen challenge are attributed to eosinophilic inflammation (33).
However, Grainge et al. recently demonstrated that repeated methacholine challenges in
asthma patients, inducing bronchoconstriction, are sufficient to induce remodeling
without causing an inflammatory response (13). Thus, methacholine challenge induced
epithelial TGF‐β staining, collagen deposition, goblet cell metaplasia and epithelial cell
proliferation, with no effect on eosinophil numbers (13). In vitro, muscarinic receptor
stimulation in combination with mechanical strain can induce α‐sm‐actin and sm‐myosin
mRNA in intact bovine tracheal smooth muscle strips and myosin light‐chain kinase
expression in human airway smooth muscle cells (34, 35). Moreover, methacholine‐
induced bronchoconstriction induces epithelial epidermal growth factor receptor
activation in mice (36) and promotes TGF‐β release and airway smooth muscle remodeling
in guinea pig lung slices (37). The M3 receptor is the muscarinic receptor subtype that
mediates bronchoconstriction and in M3R‐/‐ mice bronchoconstriction is abolished (29).
Therefore our current findings are in line with the above mentioned findings and suggest
that inhibition of bronchoconstriction may underlie the inhibition of allergen‐induced
remodeling after knock‐out of the M3 receptor. Our hypothesis is that acetylcholine‐

Chapter 6
124
induced bronchoconstriction via M3 receptors results in mechanical stress and the
subsequent release of growth factors (e.g. TGF‐β) and activation of mechanosensitive
transcription factors (e.g. myocardin). These events drive remodeling of the airways, both
in the airway smooth muscle and the airway submucosal compartment, suggesting
activation of both airway smooth muscle cells and fibroblasts by acetylcholine. This is
supported by the findings from Grainge et al., who report that bronchoconstriction
activates remodeling processes in the epithelial and submucosal compartment,
presumably related to mechanical compression by the airway smooth muscle (13). This
hypothesis implies that prevention of bronchoconstriction in patients with asthma may be
an important means of preventing remodeling later on.
Currently, anticholinergic controller therapy is not included in the guidelines for the
treatment of asthma. However, recently it has been shown that tiotropium can induce
broncodilation in patients with severe asthma on top of the use of inhaled glucocorticoids
and long‐acting β2‐agonists (6). Furthermore, tiotropium reduced severe exacerbations
and episodes of worsening of asthma (6). Moreover, adding tiotropium to glucocorticoid
therapy is more effective than doubling the glucocorticoid dose by means of morning peak
expiratory flow, the proportion of asthma‐control days, the forced expiratory volume in 1
second and daily symptom scores (9). Together, these observations indicate that asthma
patients may benefit from anticholinergic therapy. Our data suggests that M3 receptor
antagonism may also affect airway remodeling in asthma patients, however, clinical
studies are clearly required to confirm this interesting hypothesis.
In the last decade, it has become clear that non‐neuronal cells can also produce and
release acetylcholine and it has been suggested that the observed effects of acetylcholine
on inflammation and remodeling might be, at least in part, attributed to this non‐neuronal
cholinergic system (NNCS) (chapter 2, 38, 39). Direct evidence for such a role is still lacking
however. In contrast to the previously reported decreased expression in components of
the NNCS after ovalbumin challenge (40), we did not find any regulation of these
components after ovalbumin challenge. Similarly, such type of regulation was also not
observed after cigarette smoke‐induced inflammation (chapter 3). Based on our studies,
which are limited to measurement of mRNA expression levels, we cannot confirm or
exclude a role for the NNCS. Until now, evidence for a role of the NNCS is limited to the
fact that non‐neuronal cells can produce acetylcholine (20, 39). The functional
contribution of the NNCS to airway inflammation and remodeling therefore warrants
further research.

M3 receptors promote allergen‐induced remodeling
125
In conclusion, the results of the present study demonstrate that the M3 receptor
contributes to allergen‐induced airway remodeling in mice. These findings provide novel
insight into the previously established role of acetylcholine in airway diseases and
demonstrate that the remodeling‐promoting effect of acetylcholine is solely mediated via
M3 receptors. This study may therefore open new perspectives on the role of M3 selective
anticholinergics in the treatment of asthma.
Acknowledgements
The authors would like to thank N. de Haas, F. Sinkeler and A. Al Awwadi for technical
assistance. The authors would like to thank the Netherlands Lung Foundation for financial
support (grant: 3.2.08.014).

Chapter 6
126
Supplement
Methods
Immunohistochemistry
Transverse cross‐sections of 5 µm thick were used for morphometric analyses.
Goblet cells were stained with Periodic Aced Schiff’s (PAS, Sigma‐Aldrich, Zwijndrecht, The
Netherlands) in paraffin‐embedded sections. PAS‐positive cells were counted and
expressed per mm2 basement membrane.
Collagen type I and α‐sm‐actin were stained on cryo‐sections with a goat anti‐type‐I
collagen antibody (SBA, Birmingham, AL, USA) and a rabbit anti‐alpha smooth muscle actin
antibody (Abcam, Cambridge, UK), respectively. Primary antibodies were visualised using
horseradish‐peroxidase‐linked secondary antibodies and diaminobenzidine (Sigma‐Aldrich,
Zwijndrecht, The Netherlands). Airways within sections were digitally photographed after
which collagen I and α‐sm‐actin presence around the airway was quantified using Image J
(National Institute of Health). The surface of positively stained tissue was expressed as
mm2 per mm2 basement membrane.
Eosinophils were determined by staining of cryo‐sections for cyanide resistant
endogenous peroxidase activity with diaminobenzidine (Sigma‐Aldrich, Zwijndrecht, The
Netherlands). The number of eosinophils around the airways was counted and expressed
as number of cells per mm basement membrane.
mRNA analysis
Lung homogenates were prepared by pulverizing the tissue under liquid nitrogen and total
RNA was extracted using the RNeasy mini kit (Qiagen, Venlo, The Netherlands) according
to the manufacturer's instructions. Equal amounts of total mRNA were then reverse
transcribed and cDNA was subjected to real‐time qPCR (Westburg, Leusden, The
Netherlands). Real time PCR was performed with denaturation at 94°C for 30 seconds,
annealing at 59°C for 30 seconds and extension at 72°C for 30 seconds for 40 cycles
followed by 10 minutes at 72°C. Real‐time PCR data were analyzed using the comparative
cycle threshold (Ct: amplification cycle number) method. The amount of target gene was
normalized to the endogenous reference gene 18S ribosomal RNA. The specific forward
and reverse primers used are listed in table S1.
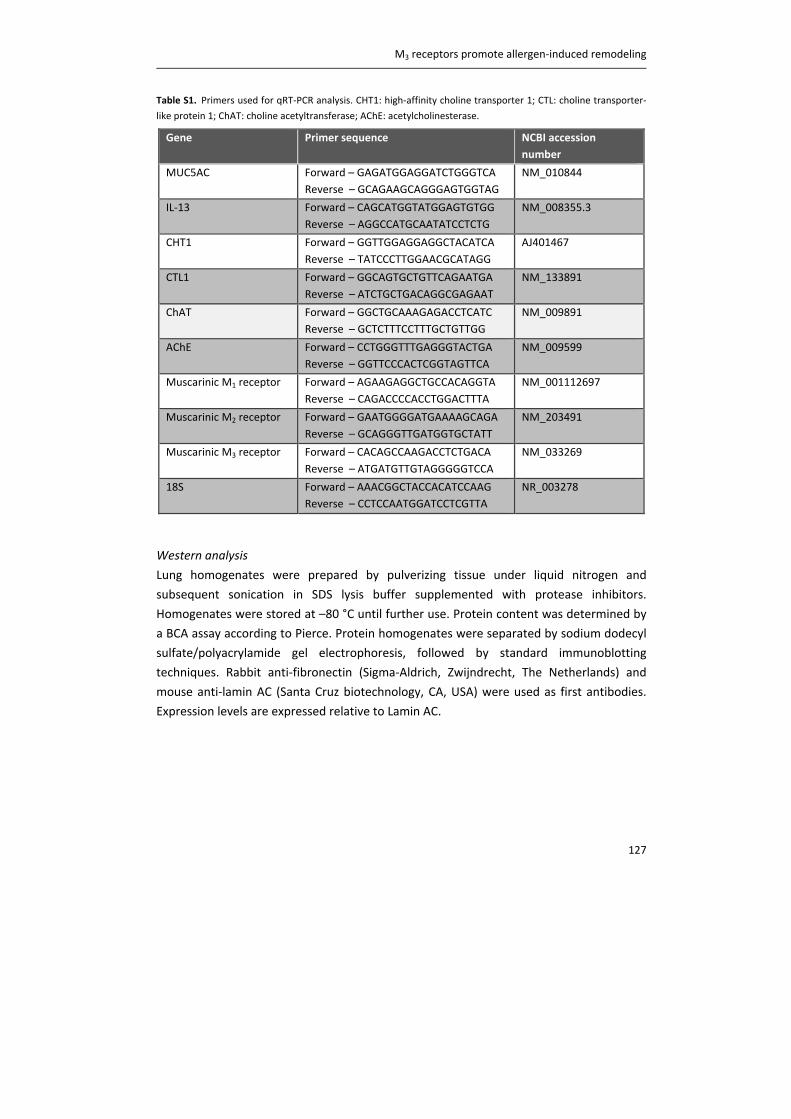
M3 receptors promote allergen‐induced remodeling
127
Table S1. Primers used for qRT‐PCR analysis. CHT1: high‐affinity choline transporter 1; CTL: choline transporter‐
like protein 1; ChAT: choline acetyltransferase; AChE: acetylcholinesterase.
Gene Primer sequence NCBI accession
number
MUC5AC Forward – GAGATGGAGGATCTGGGTCA
Reverse – GCAGAAGCAGGGAGTGGTAG
NM_010844
IL‐13 Forward – CAGCATGGTATGGAGTGTGG
Reverse – AGGCCATGCAATATCCTCTG
NM_008355.3
CHT1 Forward – GGTTGGAGGAGGCTACATCA
Reverse – TATCCCTTGGAACGCATAGG
AJ401467
CTL1 Forward – GGCAGTGCTGTTCAGAATGA
Reverse – ATCTGCTGACAGGCGAGAAT
NM_133891
ChAT Forward – GGCTGCAAAGAGACCTCATC
Reverse – GCTCTTTCCTTTGCTGTTGG
NM_009891
AChE Forward – CCTGGGTTTGAGGGTACTGA
Reverse – GGTTCCCACTCGGTAGTTCA
NM_009599
Muscarinic M1 receptor Forward – AGAAGAGGCTGCCACAGGTA
Reverse – CAGACCCCACCTGGACTTTA
NM_001112697
Muscarinic M2 receptor Forward – GAATGGGGATGAAAAGCAGA
Reverse – GCAGGGTTGATGGTGCTATT
NM_203491
Muscarinic M3 receptor Forward – CACAGCCAAGACCTCTGACA
Reverse – ATGATGTTGTAGGGGGTCCA
NM_033269
18S Forward – AAACGGCTACCACATCCAAG
Reverse – CCTCCAATGGATCCTCGTTA
NR_003278
Western analysis
Lung homogenates were prepared by pulverizing tissue under liquid nitrogen and
subsequent sonication in SDS lysis buffer supplemented with protease inhibitors.
Homogenates were stored at –80 °C until further use. Protein content was determined by
a BCA assay according to Pierce. Protein homogenates were separated by sodium dodecyl
sulfate/polyacrylamide gel electrophoresis, followed by standard immunoblotting
techniques. Rabbit anti‐fibronectin (Sigma‐Aldrich, Zwijndrecht, The Netherlands) and
mouse anti‐lamin AC (Santa Cruz biotechnology, CA, USA) were used as first antibodies.
Expression levels are expressed relative to Lamin AC.
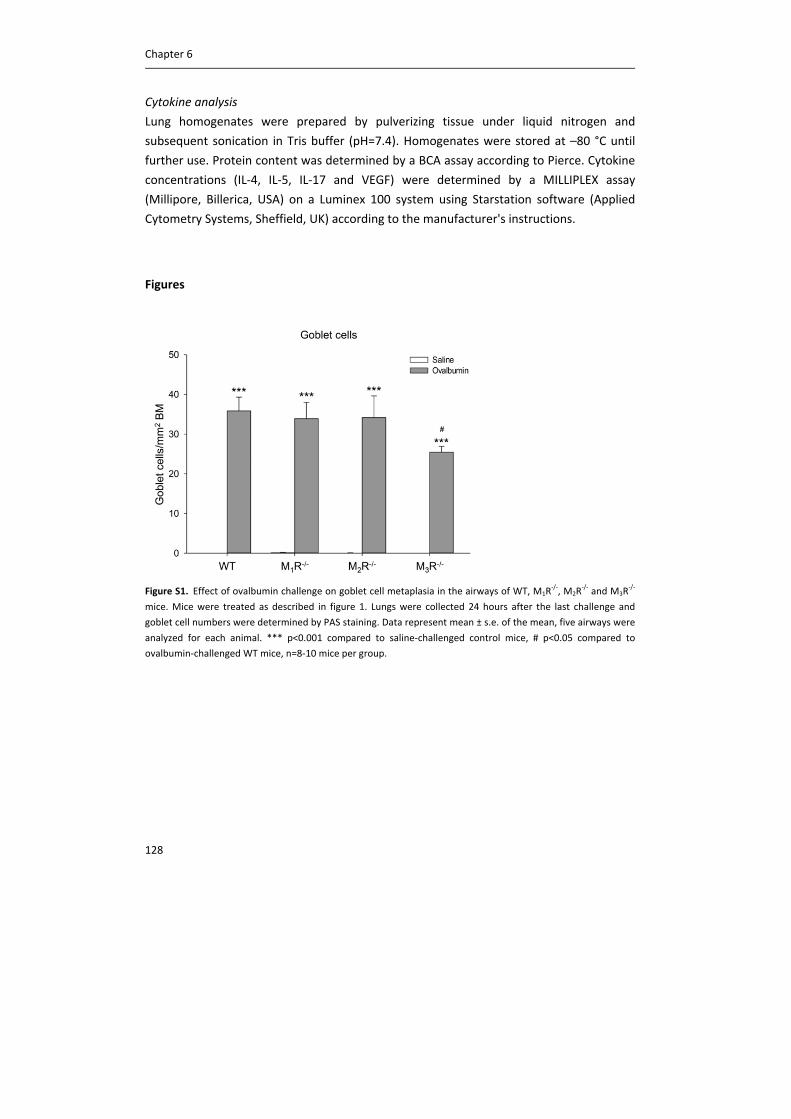
Chapter 6
128
Cytokine analysis
Lung homogenates were prepared by pulverizing tissue under liquid nitrogen and
subsequent sonication in Tris buffer (pH=7.4). Homogenates were stored at –80 °C until
further use. Protein content was determined by a BCA assay according to Pierce. Cytokine
concentrations (IL‐4, IL‐5, IL‐17 and VEGF) were determined by a MILLIPLEX assay
(Millipore, Billerica, USA) on a Luminex 100 system using Starstation software (Applied
Cytometry Systems, Sheffield, UK) according to the manufacturer's instructions.
Figures
Figure S1. Effect of ovalbumin challenge on goblet cell metaplasia in the airways of WT, M1R‐/‐, M2R
‐/‐ and M3R
‐/‐
mice. Mice were treated as described in figure 1. Lungs were collected 24 hours after the last challenge and
goblet cell numbers were determined by PAS staining. Data represent mean ± s.e. of the mean, five airways were
analyzed for each animal. *** p<0.001 compared to saline‐challenged control mice, # p<0.05 compared to
ovalbumin‐challenged WT mice, n=8‐10 mice per group.
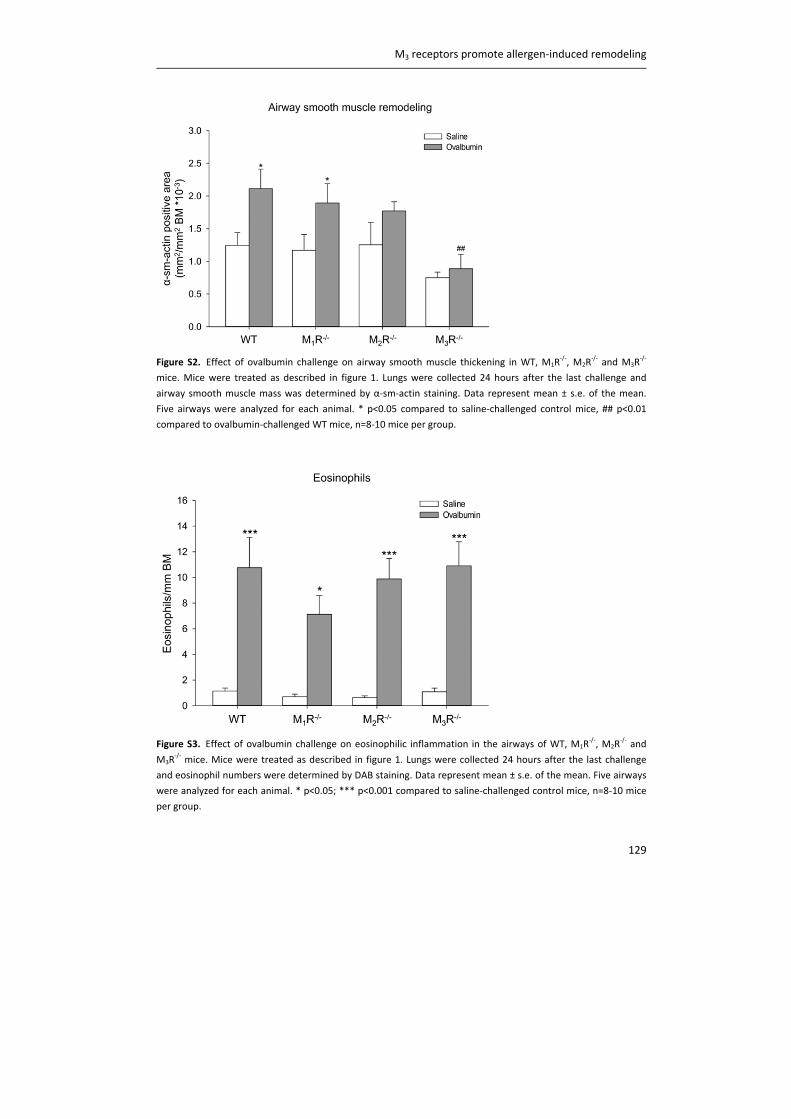
M3 receptors promote allergen‐induced remodeling
129
Figure S2. Effect of ovalbumin challenge on airway smooth muscle thickening in WT, M1R‐/‐, M2R
‐/‐ and M3R
‐/‐
mice. Mice were treated as described in figure 1. Lungs were collected 24 hours after the last challenge and
airway smooth muscle mass was determined by α‐sm‐actin staining. Data represent mean ± s.e. of the mean.
Five airways were analyzed for each animal. * p<0.05 compared to saline‐challenged control mice, ## p<0.01
compared to ovalbumin‐challenged WT mice, n=8‐10 mice per group.
Figure S3. Effect of ovalbumin challenge on eosinophilic inflammation in the airways of WT, M1R‐/‐, M2R
‐/‐ and
M3R‐/‐ mice. Mice were treated as described in figure 1. Lungs were collected 24 hours after the last challenge
and eosinophil numbers were determined by DAB staining. Data represent mean ± s.e. of the mean. Five airways
were analyzed for each animal. * p<0.05; *** p<0.001 compared to saline‐challenged control mice, n=8‐10 mice
per group.
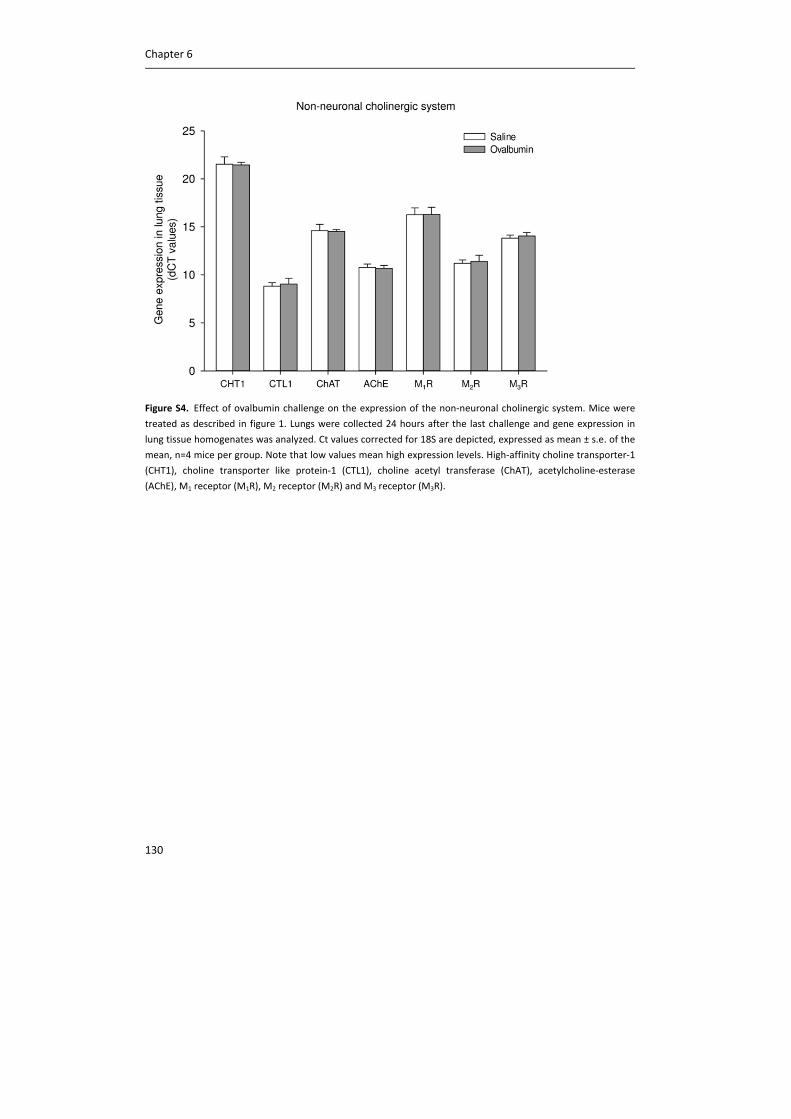
Chapter 6
130
Figure S4. Effect of ovalbumin challenge on the expression of the non‐neuronal cholinergic system. Mice were
treated as described in figure 1. Lungs were collected 24 hours after the last challenge and gene expression in
lung tissue homogenates was analyzed. Ct values corrected for 18S are depicted, expressed as mean ± s.e. of the
mean, n=4 mice per group. Note that low values mean high expression levels. High‐affinity choline transporter‐1
(CHT1), choline transporter like protein‐1 (CTL1), choline acetyl transferase (ChAT), acetylcholine‐esterase
(AChE), M1 receptor (M1R), M2 receptor (M2R) and M3 receptor (M3R).

M3 receptors promote allergen‐induced remodeling
131
References
(1) Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin
Invest 2008;118:3546‐3556.
(2) Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med
2001;164:S28‐38.
(3) Hoshino M, Nakamura Y, Hamid QA. Gene expression of vascular endothelial growth factor and
its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol 2001;107:1034‐1038.
(4) Gross NJ. Anticholinergic agents in asthma and COPD. Eur J Pharmacol 2006;533:36‐39.
(5) Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits
reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol 1989;67:2461‐
2465.
(6) Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, Sigmund R, Seibold W,
Moroni‐Zentgraf P, Bateman ED. Tiotropium in asthma poorly controlled with standard combination
therapy. N Engl J Med 2012;367:1198‐1207.
(7) Bateman ED, Kornmann O, Schmidt P, Pivovarova A, Engel M, Fabbri LM. Tiotropium is
noninferior to salmeterol in maintaining improved lung function in B16‐Arg/Arg patients with
asthma. J Allergy Clin Immunol 2011;128:315‐322.
(8) Kerstjens HA, Disse B, Schroder‐Babo W, Bantje TA, Gahlemann M, Sigmund R, Engel M, van
Noord JA. Tiotropium improves lung function in patients with severe uncontrolled asthma: a
randomized controlled trial. J Allergy Clin Immunol 2011;128:308‐314.
(9) Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT, Boushey HA, Calhoun
WJ, Castro M, Cherniack RM, Craig T, Denlinger L, Engle LL, DiMango EA, Fahy JV, Israel E, Jarjour N,
Kazani SD, Kraft M, Lazarus SC, Lemanske RF,Jr, Lugogo N, Martin RJ, Meyers DA, Ramsdell J,
Sorkness CA, Sutherland ER, Szefler SJ, Wasserman SI, Walter MJ, Wechsler ME, Chinchilli VM,
Bleecker ER, National Heart, Lung, and Blood Institute Asthma Clinical Research Network.
Tiotropium bromide step‐up therapy for adults with uncontrolled asthma. N Engl J Med
2010;363:1715‐1726.
(10) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(11) Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the
progression of airway smooth muscle remodeling. Am J Respir Crit Care Med 2005;171:1096‐1102.
(12) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T,
Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling
in a mouse model of asthma. Clin Exp Allergy 2010;40:1266‐1275.
(13) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH.
Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med 2011;364:2006‐2015.
(14) Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in
bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol
1988;153:73‐82.

Chapter 6
132
(15) Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J. Muscarinic induction of
hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents.
Neuron 2002;33:615‐624.
(16) Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H, Wess J.
Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc
Natl Acad Sci U S A 1999;96:1692‐1697.
(17) Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ,
Xia B, Crawley JN, Felder CC, Deng CX, Wess J. Mice lacking the M3 muscarinic acetylcholine receptor
are hypophagic and lean. Nature 2001;410:207‐212.
(18) Melgert BN, Postma DS, Geerlings M, Luinge MA, Klok PA, van der Strate BW, Kerstjens HA,
Timens W, Hylkema MN. Short‐term smoke exposure attenuates ovalbumin‐induced airway
inflammation in allergic mice. Am J Respir Cell Mol Biol 2004;30:880‐885.
(19) Gwilt CR, Donnelly LE, Rogers DF. The non‐neuronal cholinergic system in the airways: an
unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208‐222.
(20) Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is
an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells.
Endocrinology 2004;145:2498‐2506.
(21) Cortijo J, Mata M, Milara J, Donet E, Gavalda A, Miralpeix M, Morcillo EJ. Aclidinium inhibits
cholinergic and tobacco smoke‐induced MUC5AC in human airways. Eur Respir J 2011;37:244‐254.
(22) Gosens R, Dueck G, Rector E, Nunes RO, Gerthoffer WT, Unruh H, Zaagsma J, Meurs H, Halayko
AJ. Cooperative regulation of GSK‐3 by muscarinic and PDGF receptors is associated with airway
myocyte proliferation. Am J Physiol Lung Cell Mol Physiol 2007;293:L1348‐L1358.
(23) Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C, Juergens UR, Racke
K. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell
Mol Biol 2006;35:621‐627.
(24) Oenema TA, Mensink G, Smedinga L, Halayko AJ, Zaagsma J, Meurs H, Gosens R, Dekkers BG.
Cross‐talk between transforming growth factor‐beta(1) and muscarinic M(2) receptors augments
airway smooth muscle proliferation. Am J Respir Cell Mol Biol 2013;49:18‐27.
(25) Haag S, Matthiesen S, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of
collagen synthesis in human lung fibroblasts. Eur Respir J 2008;32:555‐562.
(26) Oenema TA, Smit M, Smedinga L, Racke K, Halayko AJ, Meurs H, Gosens R. Muscarinic receptor
stimulation augments TGF‐beta1‐induced contractile protein expression by airway smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol 2012;303:L589‐97.
(27) Milara J, Serrano A, Peiro T, Gavalda A, Miralpeix M, Morcillo EJ, Cortijo J. Aclidinium inhibits
human lung fibroblast to myofibroblast transition. Thorax 2012;67:229‐237.
(28) Raemdonck K, de Alba J, Birrell MA, Grace M, Maher SA, Irvin CG, Fozard JR, O'Byrne PM, Belvisi
MG. A role for sensory nerves in the late asthmatic response. Thorax 2012;67:19‐25.
(29) Fisher JT, Vincent SG, Gomeza J, Yamada M, Wess J. Loss of vagally mediated bradycardia and
bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J
2004;18:711‐713.
(30) Barnes PJ. The pharmacological properties of tiotropium. Chest 2000;117:63S‐6S.
(31) Rosas‐Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med 2009;265:663‐679.

M3 receptors promote allergen‐induced remodeling
133
(32) Matera MG, Calzetta L, Passeri D, Facciolo F, Rendina EA, Page C, Cazzola M, Orlandi A.
Epithelium integrity is crucial for the relaxant activity of brain natriuretic peptide in human isolated
bronchi. Br J Pharmacol 2011;163:1740‐1754.
(33) Kelly MM, O'Connor TM, Leigh R, Otis J, Gwozd C, Gauvreau GM, Gauldie J, O'Byrne PM. Effects
of budesonide and formoterol on allergen‐induced airway responses, inflammation, and airway
remodeling in asthma. J Allergy Clin Immunol 2010;125:349‐356.e13.
(34) Fairbank NJ, Connolly SC, Mackinnon JD, Wehry K, Deng L, Maksym GN. Airway smooth muscle
cell tone amplifies contractile function in the presence of chronic cyclic strain. Am J Physiol Lung Cell
Mol Physiol 2008;295:L479‐88.
(35) Wahl M, Eddinger TJ, Hai CM. Sinusoidal length oscillation‐ and receptor‐mediated mRNA
expression of myosin isoforms and alpha‐SM actin in airway smooth muscle. Am J Physiol Cell
Physiol 2004;287:C1697‐708.
(36) Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, Haley KJ, Lilly CM, So PT,
Lauffenburger DA, Kamm RD, Drazen JM. Mechanotransduction through growth‐factor shedding
into the extracellular space. Nature 2004;429:83‐86.
(37) Oenema TA, Maarsingh H, Smit M, Groothuis GM, Meurs H, Gosens R. Bronchoconstriction
Induces TGF‐beta Release and Airway Remodelling in Guinea Pig Lung Slices. PLoS One
2013;8:e65580.
(38) Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol
2008;130:219‐234.
(39) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008;154:1558‐1571.
(40) Lips KS, Luhrmann A, Tschernig T, Stoeger T, Alessandrini F, Grau V, Haberberger RV, Koepsell H,
Pabst R, Kummer W. Down‐regulation of the non‐neuronal acetylcholine synthesis and release
machinery in acute allergic airway inflammation of rat and mouse. Life Sci 2007;80:2263‐2269.


CHAPTER 7
MURINE PRECISION‐CUT LUNG SLICES AS A MODEL TO STUDY
TGF‐β AND BRONCHOCONSTRICTION‐INDUCED REMODELING
Loes E.M. Kistemaker
I. Sophie T. Bos
Cécile M. Bidan
Jürgen Wess
Huib A.M. Kerstjens
Herman Meurs
Reinoud Gosens

Chapter 7
136
Abstract
Rationale
Asthma is a chronic obstructive airway disease, characterized by airway remodeling.
Airway remodeling is conventionally thought to be the result of airway inflammation, with
inflammatory cells giving rise to the production of pro‐remodeling factors, such as TGF‐β.
However, new insights suggest that airway remodeling might also or alternatively be a
consequence of bronchoconstriction, which triggers TGF‐β release via biomechanical
activation. Previously, we developed a model to study this phenomenon, using guinea pig
lung slices treated with bronchoconstrictors in culture. As the mouse offers species
advantages with respect to the availability of molecular and genetic tools, we aimed to
develop a similar model using murine precision cut lung slices. In addition, we aimed to
investigate the involvement of the M3 receptor in bronchoconstriction‐induced
remodeling.
Methods
Airway constriction in response to methacholine was assessed in lung slices from wild‐
type and M3R‐/‐ mice. In subsequent experiments, lung slices from wild‐type and M3R
‐/‐
mice were stimulated with TGF‐β1 (2 ng/ml) or methacholine (10 μM) for two days to
induce airway remodeling. Thereafter, slices were used for gene expression analysis,
Western Blot analysis, 2‐photon imaging, or for constriction experiments.
Results
Slices were viable for at least three days. Methacholine induced a dose dependent airway
constriction in wild‐type mice, with a maximal effect of 80%. In M3R‐/‐ mice, a 35% airway
constriction was observed. TGF‐β induced a 2‐fold increase in the mRNA expression of
fibronectin, collagen Iα1 and α‐sm‐actin in both wild‐type and M3R‐/‐ mice. Similar findings
were observed on the protein level. 2‐Photon imaging revealed that collagen and α‐sm‐
actin expression was localized around the airways. No effect of methacholine on
parameters of remodeling was observed. Stimulation with TGF‐β or methacholine did not
alter airway constriction induced by the thromboxane A2 agonist U46619 compared to
control conditions in both wild‐type and M3R‐/‐ mice.
Conclusions
Murine lung slices are suitable to study TGF‐β‐induced airway remodeling, which was
comparable in wild‐type and M3R‐/‐ mice. Methacholine did not induce remodeling in
murine lung slices, suggesting that the mouse, in contrast to the guinea pig, is not a good
model to study bronchoconstriction‐induced remodeling. Despite effects of TGF‐β on the
expression of fibronectin, collagen Iα1 and α‐sm‐actin, this did not translate into changes
in airway contractility. Further studies, using a pharmacological approach, are needed to

Lung slices as a model to study remodeling
137
investigate the potential involvement of the M3 receptor in bronchoconstriction‐induced
remodeling.
Introduction
Asthma is a chronic obstructive airway disease, which is characterized by structural
changes in the airways, referred to as airway remodeling. Airway remodeling
encompasses airway smooth muscle thickening and excessive deposition of extracellular
matrix proteins (1). These airway structural changes are considered to be an important
component of airflow limitation in asthma and may contribute to an accelerated decline in
lung function (2, 3). In addition, asthma is characterized by airway hyperresponsiveness,
which consists of an excessive airway narrowing in response to bronchoconstricting
stimuli, and by airway inflammation (4).
It is believed that airway remodeling is the result of ongoing airway inflammation. For
example, eosinophils are a rich source of the pro‐remodeling factor TGF‐β (5). However,
the severity of airway remodeling in patients with asthma is not related to the severity of
inflammation, and treatment with corticosteroids, which modify airway inflammation,
does not affect the decline in lung function or the natural history of asthma (6‐8).
New insights suggest that airway remodeling might also or alternatively be a consequence
of bronchoconstriction. In patients with asthma, repeated challenges with methacholine,
inducing bronchoconstriction, induced features of airway remodeling, without affecting
eosinophilic inflammation. Interestingly, these effects could be prevented when
methacholine was combined with the β2‐agonist albuterol, suggesting that
bronchoconstriction is importantly involved (9). Furthermore, in a mouse model of
asthma, we demonstrated that ovalbumin‐induced remodeling is prevented in muscarinic
M3 receptor deficient (M3R‐/‐) mice, in which bronchoconstriction is almost completely
abolished, without affecting the inflammatory response (chapter 6). Together, these
studies indicated that airway remodeling is not merely induced by inflammation, but may
also be a consequence of mechanical forces applied to the airways during
bronchoconstriction.
The notion that mechanical forces could promote airway remodeling is also supported by
in vitro evidence. For example, compression of epithelial cells increases the activation of
TGF‐β (10), the epithelial thickness (11), and the expression of fibronectin and collagen in
a co‐culture system with fibroblasts (12). Moreover, in airway smooth muscle cells,
muscarinic receptor stimulation and mechanical strain induces TGF‐β activation (13) and

Chapter 7
138
myosin expression (14). Clearly, mechanical forces have an impact on multiple cell types in
the airway, and might thereby enhance remodeling of the airways.
Recently we developed a model to study bronchoconstriction‐induced remodeling ex vivo,
using guinea pig precision‐cut lung slices (PCLS). Oenema et al. demonstrated that
stimulation of guinea pig PCLS with methacholine or other bronchoconstrictors induces
airway remodeling (15). The effects of methacholine were comparable to the effects of
TGF‐β, and were shown to be mediated via TGF‐β (15). In this study, we aimed to develop
a model to study bronchoconstriction‐induced remodeling using murine PCLS as the
mouse offers species advantages with respect to the availability of molecular and genetic
tools. In addition, we aimed to investigate the potential involvement of the M3 receptor in
these responses, for which PCLS from M3R‐/‐ mice were used.
Methods
Animals
Homozygous, inbred, specific‐pathogen‐free breeding colonies of M3R‐/‐ mice and
C57Bl/6NTac wild‐type mice with the same genetic background were obtained from
Taconic (Cambridge City, Indiana, USA). The M3R‐/‐ mice used were on a 129 Sv/J
background and backcrossed for at least 10 generations onto the C57Bl/6NTac
background. M3R‐/‐ mice did not differ from their wild‐type littermates in overall health,
fertility and longevity (16). Animals were housed conventionally under a 12‐h light‐dark
cycle and received food and water ad libitum. All experiments were performed in
accordance with the national guidelines and approved by the University of Groningen
Committee for Animal Experimentation (DEC5463I and DEC6792A).
Precision‐cut lung slices
Mouse PCLS were prepared, according to a protocol described previously for guinea pig
PCLS by our lab (15). Male mice (6‐8 weeks old) were euthanized by intraperitoneal
pentobarbital injection (400 mg/kg, hospital pharmacy, University Medical Center
Groningen), after which the lungs were filled with 1.5 ml low melting‐point agarose
solution (1.5% final concentration (Gerbu Biotechnik GmbH, Wieblingen, Germany) in
CaCl2 (0.9 mM), MgSO4 (0.4 mM), KCl (2.7 mM), NaCl (58.2 mM), NaH2PO4 (0.6 mM),
glucose (8.4 mM), NaHCO3 (13 mM), Hepes (12.6 mM), sodium pyruvate (0.5 mM),
glutamine (1 mM), MEM‐amino acids mixture (1:50), and MEM‐vitamins mixture (1:100),
pH=7.2). The agarose was solidified for 15 minutes by placing the lungs on ice. Lungs were
harvested and individual lobes were sliced at a thickness of 250 µm in medium composed
of CaCl2 (1.8mM), MgSO4 (0.8 mM), KCl (5.4 mM), NaCl (116.4 mM), NaH2PO4 (1.2 mM),

Lung slices as a model to study remodeling
139
glucose (16.7 mM), NaHCO3 (26.1 mM), Hepes (25.2 mM), pH = 7.2, using a tissue slicer
(Compresstome™ VF‐300 microtome, Precisionary Instruments, San Jose CA, USA).
Thereafter, slices were kept at 37°C in a humidified atmosphere of 5% CO2 and washed
every 30 minutes for four times to remove the agarose and cell debris in medium
composed of CaCl2 (1.8mM), MgSO4 (0.8 mM), KCl (5.4 mM), NaCl (116.4 mM), NaH2PO4
(1.2 mM), glucose (16.7 mM), NaHCO3 (26.1 mM), Hepes (25.2 mM), sodium pyruvate
(1mM), glutamine (2 mM), MEM‐amino acids mixture (1:50), MEM‐vitamins mixture
(1:100,) penicillin (100 U/mL) and streptomycin (100 µg/mL), pH = 7.2. Average diameter
of the airways was 179 µM ± 82 µM.
Mitochondrial activity assay
Viability of lung slices was assessed by a mitochondrial activity assay performed after
culturing the slices for different time periods in Dulbecco’s Modification of Eagle’s
Medium (DMEM) supplemented with sodium pyruvate (1 mM), non‐essential amino acid
mixture (1:100), gentamycin (45 µg/mL), penicillin (100 U/mL), streptomycin (100 µg/mL)
and amphotericin B (1.5 µg/mL) at 37°C‐5% CO2. Slices were incubated with Hanks’
balanced salt solution (pH 7.4), containing 10% Alamar blue solution (BioSource,
Camarillo, CA), for 30 min at 37°C‐5% CO2. Mitochondrial activity was assessed by
conversion of Alamar blue into its reduced form, as described previously (17).
Treatment of lung slices
Lung slices were incubated in Dulbecco’s Modification of Eagle’s Medium (DMEM)
supplemented with sodium pyruvate (1 mM), non‐essential amino acid mixture (1:100),
gentamycin (45 µg/mL), penicillin (100 U/mL), streptomycin (100 µg/mL) and
amphotericin B (1.5 µg/mL) at 37°C‐5% CO2 in a 12‐well plate, using 3 slices per well. The
slices were treated with vehicle (CTR), 2 ng/ml TGF‐β1 (R&D systems, Abingdon, UK) or 10
μM methacholine (MCh; ICN Biomedicals, Zoetermeer, the Netherlands) for two days.
After two days, slices were collected for contraction studies, gene expression analysis,
Western Blot analysis, or for 2‐photon imaging.
Airway narrowing studies
Airway narrowing studies were performed on naïve lung slices and on slices treated with
vehicle, TGF‐β or methacholine (MCh) for two days. On naïve lung slices, dose response
curves for MCh (10‐9M ‐ 10‐3M) were recorded, as well as constriction after a single dose of
MCh (10‐4M), after which the airways were dilated with the bitter taste receptor agonist
chloroquine (10‐3M, Sigma‐Aldrich, Zwijndrecht, The Netherlands). On slices treated with
vehicle, TGF‐β or MCh for two days, dose response curves towards the thromboxane A2
agonist U‐46619 (10‐10M ‐ 10‐5M, Sigma‐Aldrich) were recorded. As described previously, a
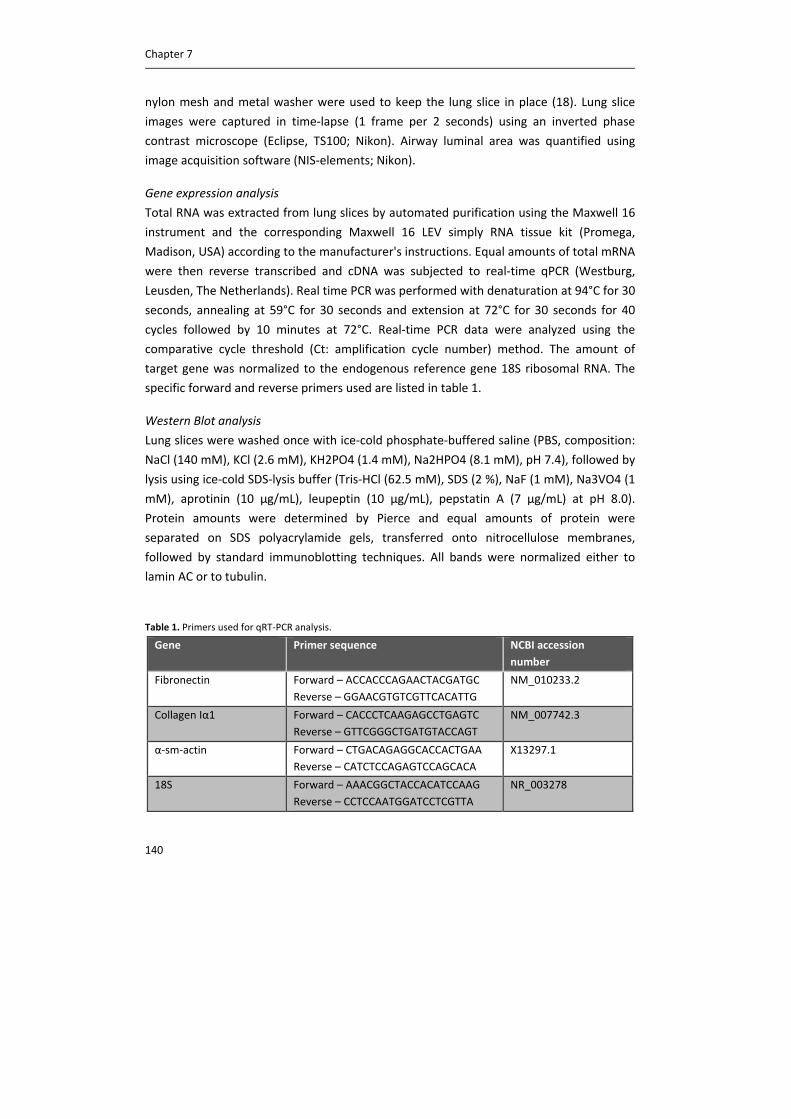
Chapter 7
140
nylon mesh and metal washer were used to keep the lung slice in place (18). Lung slice
images were captured in time‐lapse (1 frame per 2 seconds) using an inverted phase
contrast microscope (Eclipse, TS100; Nikon). Airway luminal area was quantified using
image acquisition software (NIS‐elements; Nikon).
Gene expression analysis
Total RNA was extracted from lung slices by automated purification using the Maxwell 16
instrument and the corresponding Maxwell 16 LEV simply RNA tissue kit (Promega,
Madison, USA) according to the manufacturer's instructions. Equal amounts of total mRNA
were then reverse transcribed and cDNA was subjected to real‐time qPCR (Westburg,
Leusden, The Netherlands). Real time PCR was performed with denaturation at 94°C for 30
seconds, annealing at 59°C for 30 seconds and extension at 72°C for 30 seconds for 40
cycles followed by 10 minutes at 72°C. Real‐time PCR data were analyzed using the
comparative cycle threshold (Ct: amplification cycle number) method. The amount of
target gene was normalized to the endogenous reference gene 18S ribosomal RNA. The
specific forward and reverse primers used are listed in table 1.
Western Blot analysis
Lung slices were washed once with ice‐cold phosphate‐buffered saline (PBS, composition:
NaCl (140 mM), KCl (2.6 mM), KH2PO4 (1.4 mM), Na2HPO4 (8.1 mM), pH 7.4), followed by
lysis using ice‐cold SDS‐lysis buffer (Tris‐HCl (62.5 mM), SDS (2 %), NaF (1 mM), Na3VO4 (1
mM), aprotinin (10 μg/mL), leupeptin (10 μg/mL), pepstatin A (7 µg/mL) at pH 8.0).
Protein amounts were determined by Pierce and equal amounts of protein were
separated on SDS polyacrylamide gels, transferred onto nitrocellulose membranes,
followed by standard immunoblotting techniques. All bands were normalized either to
lamin AC or to tubulin.
Table 1. Primers used for qRT‐PCR analysis.
Gene Primer sequence NCBI accession
number
Fibronectin Forward – ACCACCCAGAACTACGATGC
Reverse – GGAACGTGTCGTTCACATTG
NM_010233.2
Collagen Iα1 Forward – CACCCTCAAGAGCCTGAGTC
Reverse – GTTCGGGCTGATGTACCAGT
NM_007742.3
α‐sm‐actin Forward – CTGACAGAGGCACCACTGAA
Reverse – CATCTCCAGAGTCCAGCACA
X13297.1
18S Forward – AAACGGCTACCACATCCAAG
Reverse – CCTCCAATGGATCCTCGTTA
NR_003278

Lung slices as a model to study remodeling
141
2‐photon imaging
An immunofluorescent antibody staining was used to visualize α‐smooth muscle‐actin,
and 2‐photon imaging was used to visualize collagen. For α‐smooth muscle‐actin staining,
lung slices were washed two times with cytoskeleton buffer (CB: MES (10 mM), NaCl (150
mM), EGTA (5 mM), MgCl2 (5 mM), and glucose (5 mM) at pH=6.1) and fixed with CB
containing 3% paraformaldehyde (PFA) for 15 min. Thereafter, slices were incubated with
CB buffer containing 3% PFA and 0.3% Triton‐X‐100 for 5 min, followed by two washes
with CB buffer. Lung slices were blocked for 1 h in cyto‐TBS (Tris‐base (20 mM), NaCl (154
mM), EGTA (2.0 mM) and MgCl2 (2.0 mM), pH=7.2), supplemented with BSA (1%) and
normal donkey serum (2%). Thereafter, lung slices were stained with rabbit anti‐alpha
smooth muscle actin antibody (Abcam, Cambridge, UK) overnight at 4°C. After washing 3
times with cyto‐TBS containing 0.1% Tween‐20 (cyto‐TBS‐T) for 10 min, incubation with
the secondary antibody (FITC) was performed during 2 hour at room temperature. Lung
slices were then washed 3 times for 15 min in cyto‐TBS‐T, followed by 3 washes with ultra‐
pure water. The slides were mounted with ProLong Gold anti‐fade reagent (Invitrogen,
Breda, The Netherlands). The slides were then analyzed using a fluorescence confocal
microscope (Leica) equipped with a multiphoton laser. The α‐sm‐actin staining was excited
at 100 nm and collected at 500 nm, whereas the collagen bundles excited at 820 nm
naturally emitted a second harmonic generation signal collected around 410 nm.
Statistical analysis
Data are presented as mean ± s.e. of the mean. Statistical differences between means
were calculated using a paired Student’s t‐test with two‐tailed distribution. Differences
were considered significant at p<0.05.
Results
Viability of lung slices
Viability of the lung slices was assessed by a mitochondrial activity assay after 1, 2 and 3
days of culturing. As depicted in figure 1, no significant differences were observed in
mitochondrial activity for up to three days after culturing. This indicates that the lung
slices are viable for at least three days and can be used for experiments within this period.
Methacholine‐induced constriction
MCh induced a dose‐dependent airway constriction in PCLS from wild‐type mice, with a
maximal effect of 80% airway closure compared to the initial airway lumen (figure 2A). In
PCLS from M3R‐/‐ mice, the airways were more sensitive to MCh compared to wild‐type
PCLS, but the maximal airway narrowing was only 35% (figure 2A).

Chapter 7
142
Figure 1. Viability of lung slices is preserved for three days. Mitochondrial activity, assessed by conversion of
Alamar blue into its reduced form, was not significantly different in the first three days of culture. Data represent
mean ± s.e. of the mean of 4 animals, 2 slices per animal.
As depicted in figure 2B, MCh induced 40% airway closure in PCLS from wild‐type mice
after a single dose of MCh (10‐4M), and airway dilation was obtained after subsequent
addition of chloroquine (10‐3M). In contrast to PCLS from wild‐type animals, in PCLS from
M3R‐/‐ mice there was a lag time after addition of MCh before the airway constriction
started. However, the constriction rate was higher, and a plateau phase was reached
earlier in slices from M3R‐/‐ mice compared to wild‐type mice. The maximal effect of MCh
in PCLS from M3R‐/‐ mice was 30% closure, and the airways similarly dilated after addition
of chloroquine (figure 2B). Oscillations in airway constriction were observed in the PCLS
from M3R‐/‐ mice, as is demonstrated in figure 2C for a single experiment in which strong
oscillations were observed. The oscillations can also be observed in the plateau phase of
constriction in figure 2B; however, the effect of the strong oscillations observed in single
experiments is leveled out by averaging multiple experiments.
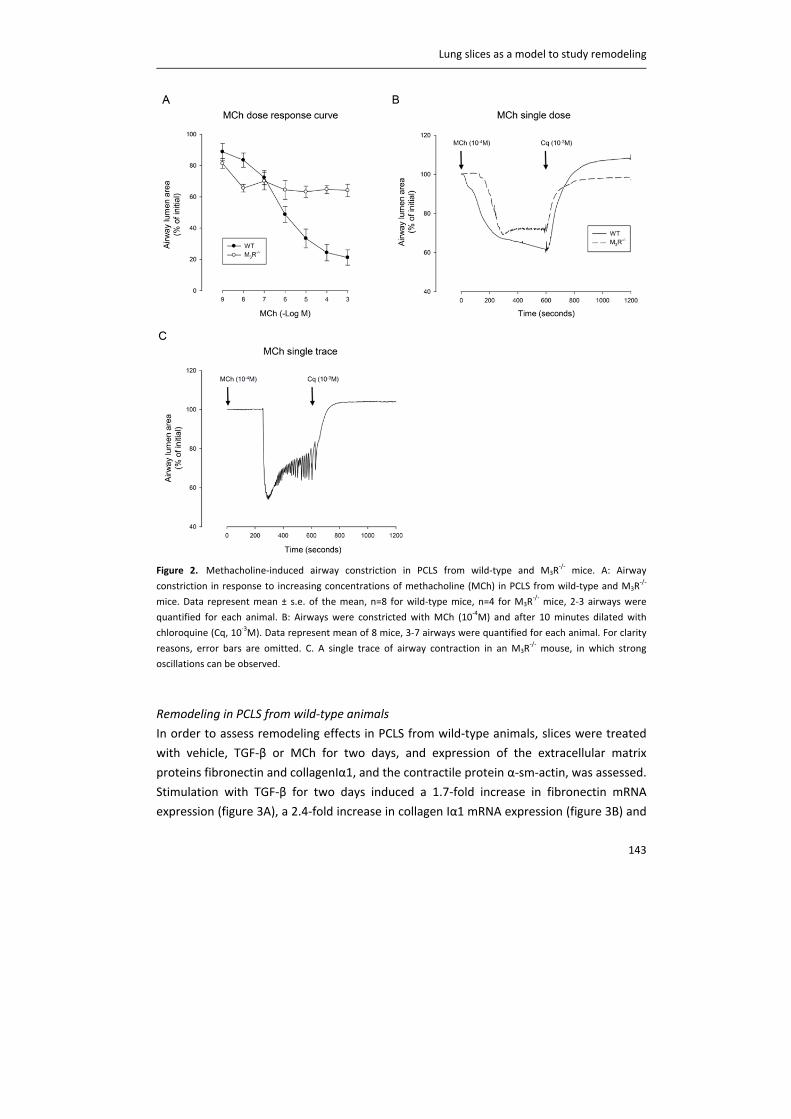
Lung slices as a model to study remodeling
143
Figure 2. Methacholine‐induced airway constriction in PCLS from wild‐type and M3R
‐/‐ mice. A: Airway
constriction in response to increasing concentrations of methacholine (MCh) in PCLS from wild‐type and M3R‐/‐
mice. Data represent mean ± s.e. of the mean, n=8 for wild‐type mice, n=4 for M3R‐/‐ mice, 2‐3 airways were
quantified for each animal. B: Airways were constricted with MCh (10‐4M) and after 10 minutes dilated with
chloroquine (Cq, 10‐3M). Data represent mean of 8 mice, 3‐7 airways were quantified for each animal. For clarity
reasons, error bars are omitted. C. A single trace of airway contraction in an M3R‐/‐ mouse, in which strong
oscillations can be observed.
Remodeling in PCLS from wild‐type animals
In order to assess remodeling effects in PCLS from wild‐type animals, slices were treated
with vehicle, TGF‐β or MCh for two days, and expression of the extracellular matrix
proteins fibronectin and collagenIα1, and the contractile protein α‐sm‐actin, was assessed.
Stimulation with TGF‐β for two days induced a 1.7‐fold increase in fibronectin mRNA
expression (figure 3A), a 2.4‐fold increase in collagen Iα1 mRNA expression (figure 3B) and
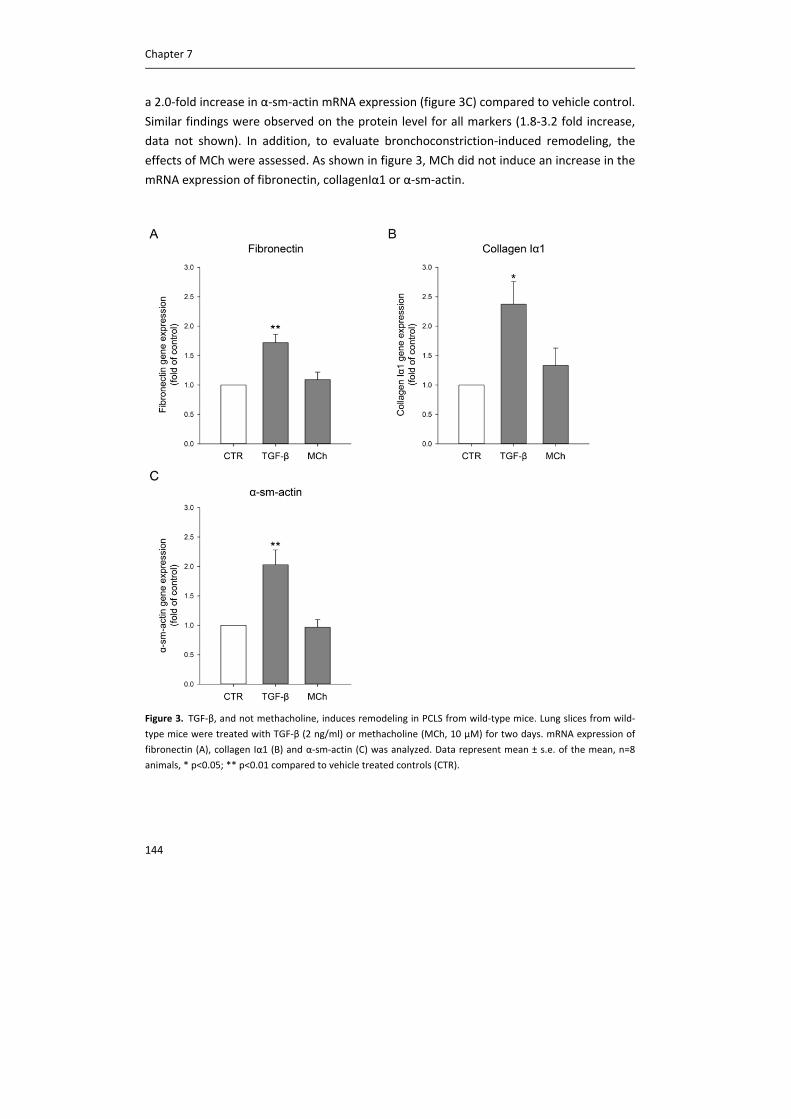
Chapter 7
144
a 2.0‐fold increase in α‐sm‐actin mRNA expression (figure 3C) compared to vehicle control.
Similar findings were observed on the protein level for all markers (1.8‐3.2 fold increase,
data not shown). In addition, to evaluate bronchoconstriction‐induced remodeling, the
effects of MCh were assessed. As shown in figure 3, MCh did not induce an increase in the
mRNA expression of fibronectin, collagenIα1 or α‐sm‐actin.
Figure 3. TGF‐β, and not methacholine, induces remodeling in PCLS from wild‐type mice. Lung slices from wild‐
type mice were treated with TGF‐β (2 ng/ml) or methacholine (MCh, 10 µM) for two days. mRNA expression of
fibronectin (A), collagen Iα1 (B) and α‐sm‐actin (C) was analyzed. Data represent mean ± s.e. of the mean, n=8
animals, * p<0.05; ** p<0.01 compared to vehicle treated controls (CTR).
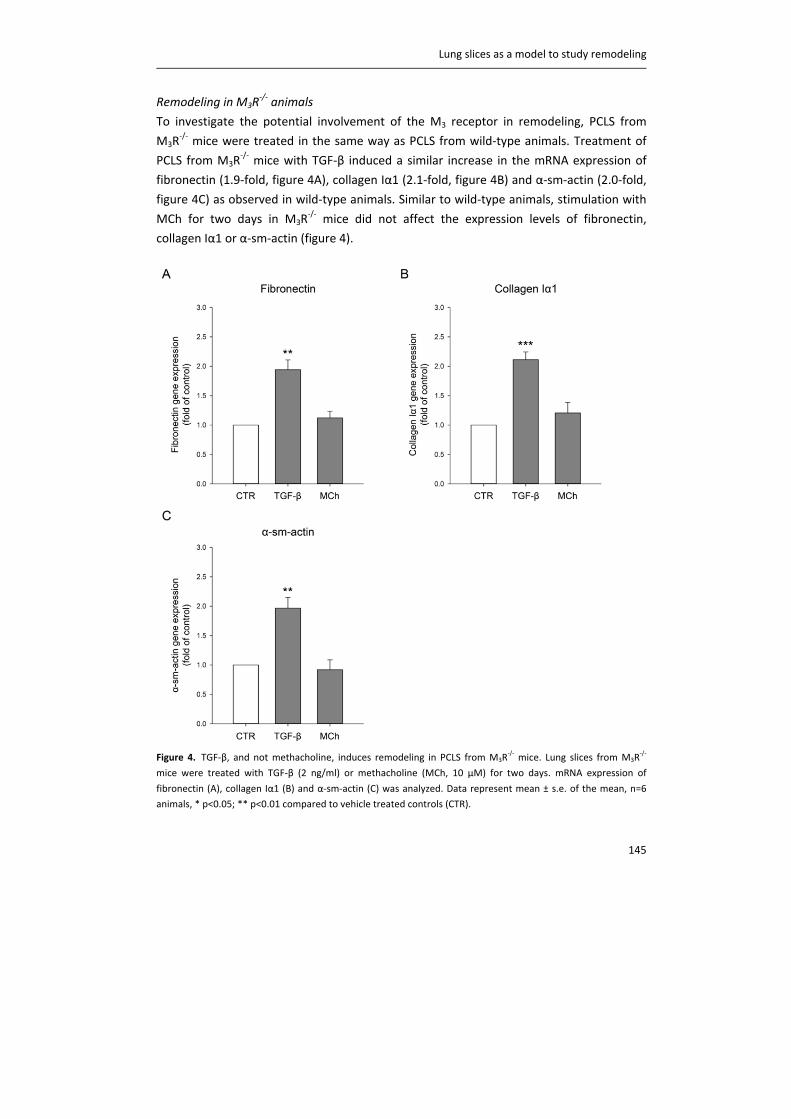
Lung slices as a model to study remodeling
145
Remodeling in M3R‐/‐ animals
To investigate the potential involvement of the M3 receptor in remodeling, PCLS from
M3R‐/‐ mice were treated in the same way as PCLS from wild‐type animals. Treatment of
PCLS from M3R‐/‐ mice with TGF‐β induced a similar increase in the mRNA expression of
fibronectin (1.9‐fold, figure 4A), collagen Iα1 (2.1‐fold, figure 4B) and α‐sm‐actin (2.0‐fold,
figure 4C) as observed in wild‐type animals. Similar to wild‐type animals, stimulation with
MCh for two days in M3R‐/‐ mice did not affect the expression levels of fibronectin,
collagen Iα1 or α‐sm‐actin (figure 4).
Figure 4. TGF‐β, and not methacholine, induces remodeling in PCLS from M3R‐/‐ mice. Lung slices from M3R
‐/‐
mice were treated with TGF‐β (2 ng/ml) or methacholine (MCh, 10 µM) for two days. mRNA expression of
fibronectin (A), collagen Iα1 (B) and α‐sm‐actin (C) was analyzed. Data represent mean ± s.e. of the mean, n=6
animals, * p<0.05; ** p<0.01 compared to vehicle treated controls (CTR).
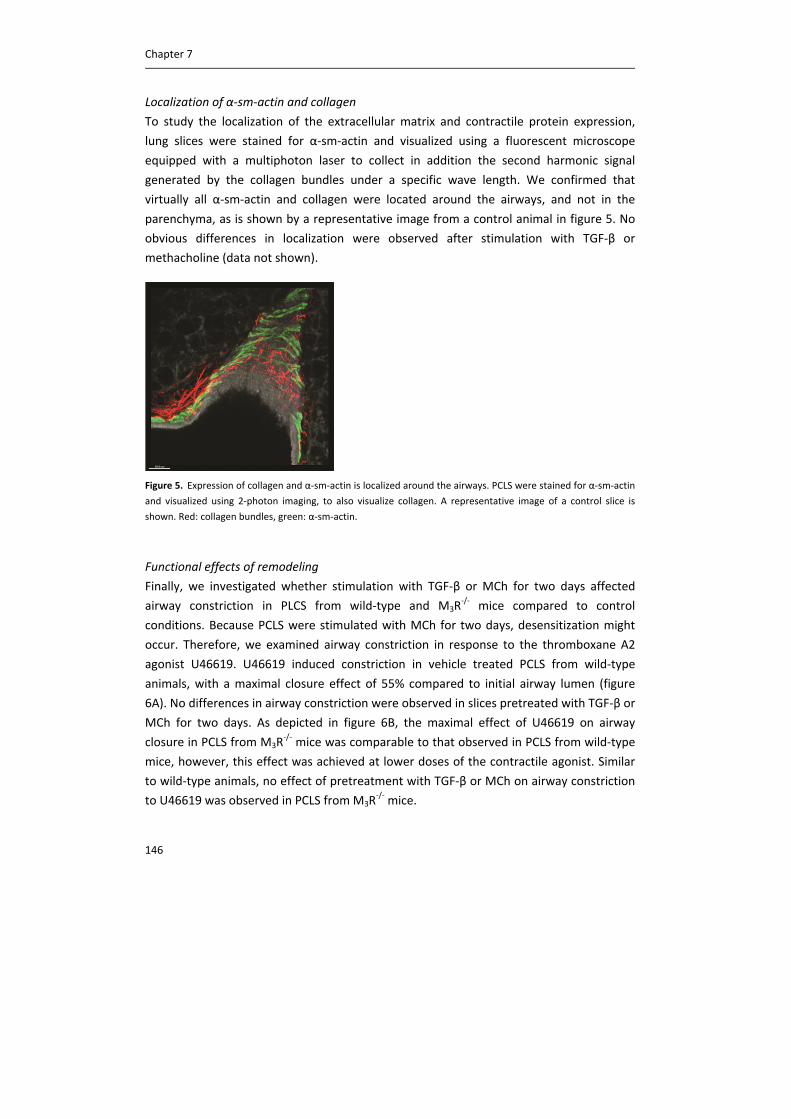
Chapter 7
146
Localization of α‐sm‐actin and collagen
To study the localization of the extracellular matrix and contractile protein expression,
lung slices were stained for α‐sm‐actin and visualized using a fluorescent microscope
equipped with a multiphoton laser to collect in addition the second harmonic signal
generated by the collagen bundles under a specific wave length. We confirmed that
virtually all α‐sm‐actin and collagen were located around the airways, and not in the
parenchyma, as is shown by a representative image from a control animal in figure 5. No
obvious differences in localization were observed after stimulation with TGF‐β or
methacholine (data not shown).
Figure 5. Expression of collagen and α‐sm‐actin is localized around the airways. PCLS were stained for α‐sm‐actin
and visualized using 2‐photon imaging, to also visualize collagen. A representative image of a control slice is
shown. Red: collagen bundles, green: α‐sm‐actin.
Functional effects of remodeling
Finally, we investigated whether stimulation with TGF‐β or MCh for two days affected
airway constriction in PLCS from wild‐type and M3R‐/‐ mice compared to control
conditions. Because PCLS were stimulated with MCh for two days, desensitization might
occur. Therefore, we examined airway constriction in response to the thromboxane A2
agonist U46619. U46619 induced constriction in vehicle treated PCLS from wild‐type
animals, with a maximal closure effect of 55% compared to initial airway lumen (figure
6A). No differences in airway constriction were observed in slices pretreated with TGF‐β or
MCh for two days. As depicted in figure 6B, the maximal effect of U46619 on airway
closure in PCLS from M3R‐/‐ mice was comparable to that observed in PCLS from wild‐type
mice, however, this effect was achieved at lower doses of the contractile agonist. Similar
to wild‐type animals, no effect of pretreatment with TGF‐β or MCh on airway constriction
to U46619 was observed in PCLS from M3R‐/‐ mice.
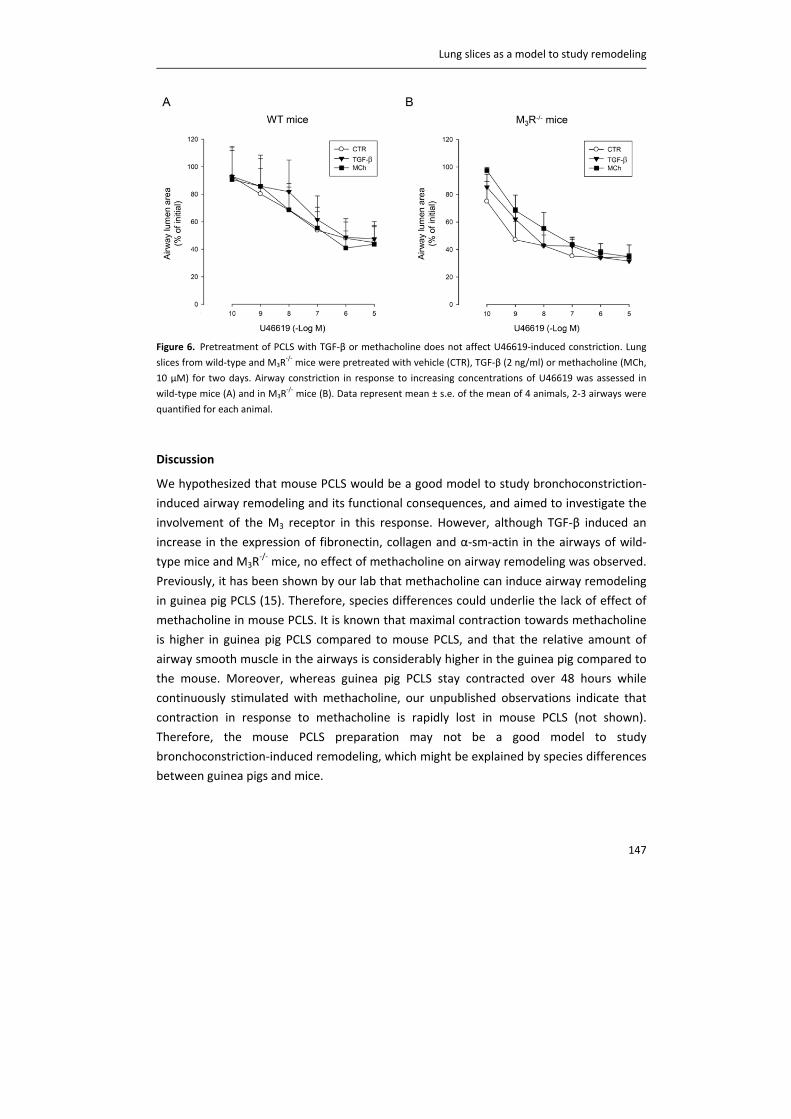
Lung slices as a model to study remodeling
147
Figure 6. Pretreatment of PCLS with TGF‐β or methacholine does not affect U46619‐induced constriction. Lung
slices from wild‐type and M3R‐/‐ mice were pretreated with vehicle (CTR), TGF‐β (2 ng/ml) or methacholine (MCh,
10 µM) for two days. Airway constriction in response to increasing concentrations of U46619 was assessed in
wild‐type mice (A) and in M3R‐/‐ mice (B). Data represent mean ± s.e. of the mean of 4 animals, 2‐3 airways were
quantified for each animal.
Discussion
We hypothesized that mouse PCLS would be a good model to study bronchoconstriction‐
induced airway remodeling and its functional consequences, and aimed to investigate the
involvement of the M3 receptor in this response. However, although TGF‐β induced an
increase in the expression of fibronectin, collagen and α‐sm‐actin in the airways of wild‐
type mice and M3R‐/‐ mice, no effect of methacholine on airway remodeling was observed.
Previously, it has been shown by our lab that methacholine can induce airway remodeling
in guinea pig PCLS (15). Therefore, species differences could underlie the lack of effect of
methacholine in mouse PCLS. It is known that maximal contraction towards methacholine
is higher in guinea pig PCLS compared to mouse PCLS, and that the relative amount of
airway smooth muscle in the airways is considerably higher in the guinea pig compared to
the mouse. Moreover, whereas guinea pig PCLS stay contracted over 48 hours while
continuously stimulated with methacholine, our unpublished observations indicate that
contraction in response to methacholine is rapidly lost in mouse PCLS (not shown).
Therefore, the mouse PCLS preparation may not be a good model to study
bronchoconstriction‐induced remodeling, which might be explained by species differences
between guinea pigs and mice.

Chapter 7
148
TGF‐β did induce features of airway remodeling in mouse PCLS, which is in line with
previous studies (15, 19). However, despite the clear effect of TGF‐β on the expression of
fibronectin, collagen Iα1 and α‐sm‐actin, this did not translate into changes in airway
contractility. Generally, it is assumed that airway remodeling affects airway contractility.
Here, we were not able to demonstrate such a direct link. Chew et al. demonstrated that
although ovalbumin challenge, leading to airway remodeling, induced airway
hyperresponiveness in vivo, airway constriction of mouse PCLS in response to
acetylcholine ex vivo was not altered (20). Using a similar approach, Donovan et al.
demonstrated airway hyperresponiveness to methacholine in vivo and in isolated trachea
in vitro after ovalbumin challenge, however, in PCLS, airway narrowing was slower and the
potency of methacholine was reduced in ovalbumin‐challenged mice compared to
controls (21). Although PCLS come closer to the in vivo situation than single cells or
smooth muscle strips, it may still be possible that mechanical differences exist. It might
therefore be difficult to demonstrate a direct link between airway remodeling and airway
contractility.
Muscarinic receptor stimulation can augment TGF‐β‐induced expression of contractile
proteins and extracellular matrix proteins in airway smooth muscle cells (22‐24).
Moreover, Oenema et al. demonstrated in guinea pig PCLS that methacholine can induce
remodeling via enhanced release of TGF‐β (15). Furthermore, the muscarinic antagonist
tiotropium has been shown to inhibit ovalbumin‐induced TGF‐β release, and also airway
remodeling, in a mouse model of asthma (25). Morevoer, TGF‐β release is decreased in
M3R‐/‐ animals (chapter 3). Methacholine has also been shown to increase TGF‐β and
collagen levels in biopsies of asthma patients (9). Therefore, methacholine‐induced
bronchoconstriction, and subsequent TGF‐β release, may enhance airway remodeling.
Since we have shown here that murine PCLS may not be an adequate model to study
bronchoconstriction‐induced remodeling, future studies to investigate mechanisms
involved in this response and the potential involvement of the M3 receptor are better
performed using guinea pig or human lung slices.
In our study, we found limited contraction towards methacholine in M3R‐/‐ mice, which is
in line with previous studies using lung slices from M3R‐/‐ mice (26). Although the M3
receptor is the dominant receptor in mediating airway smooth muscle contraction (27), it
has been shown that after knock out of the M3 receptor, some contraction can be elicited
via M2 receptors, and that airway contraction is only completely abolished after knock out
of both M2 and M3 receptors (26, 28, 29). Therefore, the observed contraction in the
M3R‐/‐ mice might be mediated via M2 receptors in our study. Clearly, methacholine‐
induced contraction is regulated via at least partially different mechanisms in M3R‐/‐

Lung slices as a model to study remodeling
149
animals than in wild‐type animals. Not only is there a lag time in effect, the contraction in
M3R‐/‐ mice also seemed to be sensitive to lower doses of methacholine compared to
contraction in WT animals. Using airway smooth muscle strips, Kume et al. demonstrated
that contraction in wild‐type animals at low concentrations of methacholine could be
completely inhibited by the M2 antagonist AF‐DX 116 and by pertussis toxin, whereas
contraction at higher concentrations was only partially inhibited by pertussis toxin, and
almost completely mediated via M3 receptors (30). Similar findings were observed using
ileal smooth muscle strips, in which 80% of the contraction was inhibited at low
concentrations of agonist by pertussis toxin, whereas contraction at higher concentrations
was not altered by pertussis toxin and completely mediated via M3 receptors (28). This
might explain the effect of methacholine at low concentrations via M2 receptors after
knock‐out of the M3 receptors compared to wild‐type animals. Future studies are needed
to elucidate the exact mechanisms underlying airway contraction in M3R‐/‐ mice. For
example, experiments under calcium free conditions, or pretreatment with an M2 receptor
selective antagonist or pertussis toxin, to uncouple Gi proteins, might help to explain the
mechanisms involved.
Finally, strong oscillations in airway constriction were observed in M3R‐/‐ mice after a
single dose of methacholine, which was not observed in wild‐type animals. Previously, we
have shown that levels of fibronectin and collagen are lower in the airways of M3R‐/‐ mice
(chapter 3). Decreased levels of these extracellular matrix proteins might result in altered
mechanical stability of the airways and thereby affect the capacity of the airway to
maintain a sustained contraction in M3R‐/‐ mice. Altered mechanical stability might also
explain the enhanced contraction rate in M3R‐/‐ mice compared to wild‐type mice after a
single dose of methacholine. Moreover, this might explain the enhanced airway
constriction in M3R‐/‐ mice compared to wild‐type mice observed at low concentrations of
methacholine, but also at low concentrations of U46619. Increased sensitivity for both
methacholine and U46619 might suggest that this is not a receptor driven process, but is
rather caused by intrinsic differences in the airways of M3R‐/‐ mice compared to wild‐type
mice, involving reduced mechanical stability.
In conclusion, murine PCLS are suitable to study TGF‐β‐induced airway remodeling, but are
most likely not an appropriate model to study bronchoconstriction‐induced remodeling.
This is in contrast to guinea pig PCLS, which are well suited to study this response.

Chapter 7
150
Acknowledgements
We would like to thank the Netherlands Lung Foundation (grant: 3.2.08.014) and
Boehringer Ingelheim (contract number: 43054530) for financial support. Part of the work
has been performed at the University Medical Center Groningen Imaging and Microscopy
Center (UMIC), which is sponsored by NWO‐grants 40‐00506‐98‐9021 and 175‐010‐2009‐
023.

Lung slices as a model to study remodeling
151
References
(1) Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med
2001;164:S28‐S38.
(2) An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman
DH, Fabry B, Fairbank NJ, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gosens R, Gunst SJ,
Halayko AJ, Ingram RH, Irvin CG, James AL, Janssen LJ, King GG, Knight DA, Lauzon AM, Lakser OJ,
Ludwig MS, Lutchen KR, Maksym GN, Martin JG, Mauad T, McParland BE, Mijailovich SM, Mitchell
HW, Mitchell RW, Mitzner W, Murphy TM, Pare PD, Pellegrino R, Sanderson MJ, Schellenberg RR,
Seow CY, Silveira PS, Smith PG, Solway J, Stephens NL, Sterk PJ, Stewart AG, Tang DD, Tepper RS,
Tran T, Wang L. Airway smooth muscle dynamics: a common pathway of airway obstruction in
asthma. Eur Respir J 2007;29:834‐860.
(3) Pare PD, Roberts CR, Bai TR, Wiggs BJ. The functional consequences of airway remodeling in
asthma. Monaldi Arch Chest Dis 1997;52:589‐596.
(4) Lemanske RF,Jr, Busse WW. Asthma: clinical expression and molecular mechanisms. J Allergy Clin
Immunol 2010;125:S95‐102.
(5) Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, Ernst P, Hamid Q. Eosinophil‐associated
TGF‐beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol
1997;17:326‐333.
(6) Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations
selectively associated with severe asthma. Am J Respir Crit Care Med 2003;167:1360‐1368.
(7) Guilbert TW, Morgan WJ, Zeiger RS, Mauger DT, Boehmer SJ, Szefler SJ, Bacharier LB, Lemanske
RF,Jr, Strunk RC, Allen DB, Bloomberg GR, Heldt G, Krawiec M, Larsen G, Liu AH, Chinchilli VM,
Sorkness CA, Taussig LM, Martinez FD. Long‐term inhaled corticosteroids in preschool children at
high risk for asthma. N Engl J Med 2006;354:1985‐1997.
(8) Murray CS, Woodcock A, Langley SJ, Morris J, Custovic A, IFWIN study team. Secondary
prevention of asthma by the use of Inhaled Fluticasone propionate in Wheezy INfants (IFWIN):
double‐blind, randomised, controlled study. Lancet 2006;368:754‐762.
(9) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH. Effect
of bronchoconstriction on airway remodeling in asthma. N Engl J Med 2011;364:2006‐2015.
(10) Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal
epithelial cells to transmembrane pressure. Am J Physiol Lung Cell Mol Physiol 2000;278:L1264‐72.
(11) Choe MM, Sporn PH, Swartz MA. An in vitro airway wall model of remodeling. Am J Physiol Lung
Cell Mol Physiol 2003;285:L427‐33.
(12) Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated
between different cell types to elicit matrix remodeling. Proc Natl Acad Sci U S A 2001;98:6180‐
6185.
(13) Tatler AL, John AE, Jolly L, Habgood A, Porte J, Brightling C, Knox AJ, Pang L, Sheppard D, Huang
X, Jenkins G. Integrin alphavbeta5‐mediated TGF‐beta activation by airway smooth muscle cells in
asthma. J Immunol 2011;187:6094‐6107.

Chapter 7
152
(14) Wahl M, Eddinger TJ, Hai CM. Sinusoidal length oscillation‐ and receptor‐mediated mRNA
expression of myosin isoforms and alpha‐SM actin in airway smooth muscle. Am J Physiol Cell
Physiol 2004;287:C1697‐708.
(15) Oenema TA, Maarsingh H, Smit M, Groothuis GM, Meurs H, Gosens R. Bronchoconstriction
Induces TGF‐beta Release and Airway Remodelling in Guinea Pig Lung Slices. PLoS One
2013;8:e65580.
(16) Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ,
Xia B, Crawley JN, Felder CC, Deng CX, Wess J. Mice lacking the M3 muscarinic acetylcholine receptor
are hypophagic and lean. Nature 2001;410:207‐212.
(17) Gosens R, Baarsma HA, Heijink IH, Oenema TA, Halayko AJ, Meurs H, Schmidt M. De novo
synthesis of {beta}‐catenin via H‐Ras and MEK regulates airway smooth muscle growth. FASEB J
2010;24:757‐768.
(18) Rosner SR, Ram‐Mohan S, Paez‐Cortez JR, Lavoie TL, Dowell ML, Yuan L, Ai X, Fine A, Aird WC,
Solway J, Fredberg JJ, Krishnan R. Airway contractility in the precision‐cut lung slice after
cryopreservation. Am J Respir Cell Mol Biol 2014;50:876‐881.
(19) Halwani R, Al‐Muhsen S, Al‐Jahdali H, Hamid Q. Role of transforming growth factor‐beta in
airway remodeling in asthma. Am J Respir Cell Mol Biol 2011;44:127‐133.
(20) Chew AD, Hirota JA, Ellis R, Wattie J, Inman MD, Janssen LJ. Effects of allergen on airway
narrowing dynamics as assessed by lung‐slice technique. Eur Respir J 2008;31:532‐538.
(21) Donovan C, Royce SG, Esposito J, Tran J, Ibrahim ZA, Tang ML, Bailey S, Bourke JE. Differential
effects of allergen challenge on large and small airway reactivity in mice. PLoS One 2013;8:e74101.
(22) Goldsmith AM, Bentley JK, Zhou L, Jia Y, Bitar KN, Fingar DC, Hershenson MB. Transforming
growth factor‐beta induces airway smooth muscle hypertrophy. Am J Respir Cell Mol Biol
2006;34:247‐254.
(23) Oenema TA, Mensink G, Smedinga L, Halayko AJ, Zaagsma J, Meurs H, Gosens R, Dekkers BG.
Cross‐talk between transforming growth factor‐beta(1) and muscarinic M(2) receptors augments
airway smooth muscle proliferation. Am J Respir Cell Mol Biol 2013;49:18‐27.
(24) Oenema TA, Smit M, Smedinga L, Racke K, Halayko AJ, Meurs H, Gosens R. Muscarinic receptor
stimulation augments TGF‐beta1‐induced contractile protein expression by airway smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol 2012;303:L589‐97.
(25) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T,
Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling
in a mouse model of asthma. Clin Exp Allergy 2010;40:1266‐1275.
(26) Struckmann N, Schwering S, Wiegand S, Gschnell A, Yamada M, Kummer W, Wess J,
Haberberger RV. Role of muscarinic receptor subtypes in the constriction of peripheral airways:
studies on receptor‐deficient mice. Mol Pharmacol 2003;64:1444‐1451.
(27) Roffel AF, Elzinga CR, Van Amsterdam RG, De Zeeuw RA, Zaagsma J. Muscarinic M2 receptors in
bovine tracheal smooth muscle: discrepancies between binding and function. Eur J Pharmacol
1988;153:73‐82.
(28) Unno T, Matsuyama H, Sakamoto T, Uchiyama M, Izumi Y, Okamoto H, Yamada M, Wess J,
Komori S. M(2) and M(3) muscarinic receptor‐mediated contractions in longitudinal smooth muscle
of the ileum studied with receptor knockout mice. Br J Pharmacol 2005;146:98‐108.

Lung slices as a model to study remodeling
153
(29) Ehlert FJ. Pharmacological analysis of the contractile role of M2 and M3 muscarinic receptors in
smooth muscle. Receptors Channels 2003;9:261‐277.
(30) Kume H, Mikawa K, Takagi K, Kotlikoff MI. Role of G proteins and KCa channels in the muscarinic
and beta‐adrenergic regulation of airway smooth muscle. Am J Physiol 1995;268:L221‐9.


CHAPTER 8
TIOTROPIUM ATTENUATES IL‐13‐INDUCED GOBLET CELL
METAPLASIA OF HUMAN AIRWAY EPITHELIAL CELLS
Loes E.M. Kistemaker
Pieter S. Hiemstra
I. Sophie T. Bos
Susanne Bouwman
Maarten van den Berge
Machteld N. Hylkema
Herman Meurs
Huib A.M. Kerstjens
Reinoud Gosens
Thorax, 2015, In revision

Chapter 8
156
Abstract
Rationale
It has been demonstrated that acetylcholine is not only a neurotransmitter, but also acts
as a local mediator produced by airway cells, including epithelial cells. In vivo studies
demonstrate an indirect role for acetylcholine in epithelial cell differentiation and goblet
cell metaplasia. Here, we aimed to investigate direct effects of endogenous non‐neuronal
acetylcholine on epithelial cell differentiation and possible mechanisms involved.
Methods
Human airway epithelial cells were isolated from healthy donors and cultured at an air‐
liquid‐interface (ALI). During differentiation at the ALI, cells were exposed to the
muscarinic antagonist tiotropium (10 nM), IL‐13 (1 ng/ml), or the combination of IL‐13 and
tiotropium.
Results
Human airway epithelial cells expressed all components of the non‐neuronal cholinergic
system, suggesting acetylcholine production. Tiotropium had no effects on epithelial cell
differentiation following air exposure, as assessed by qPCR, histological analysis and
micro‐array. Differentiation towards goblet cells was barely induced following air
exposure. Therefore, IL‐13 was used to induce goblet cell metaplasia. IL‐13 induced
MUC5AC positive cells (5‐fold) and goblet cells (14‐fold), as assessed by histochemistry,
and MUC5AC gene expression (105‐fold). These effects were partly prevented by
tiotropium (47‐92%). IL‐13 decreased FoxA2 expression (1.6‐fold) and increased FoxA3
(3.6‐fold) and SPDEF (5.2‐fold) expression. Tiotropium prevented the effects on FoxA2 and
FoxA3, but not on SPDEF.
Conclusions
We demonstrate that tiotropium has no effects on epithelial cell differentiation following
air exposure, but inhibits IL‐13‐induced goblet cell metaplasia, possibly via FoxA2 and
FoxA3. This indicates that non‐neuronal acetylcholine contributes to goblet cell
differentiation by a direct effect on epithelial cells.
Introduction
Acetylcholine is long known as a classical neurotransmitter in the airways, released from
parasympathetic nerve fibers. It induces bronchoconstriction and mucus secretion via
muscarinic receptors. For this reason, anticholinergic therapy has been used for many
centuries for the treatment of obstructive airway diseases.
In addition, recent evidence indicates that acetylcholine can also be synthesized and
released from non‐neuronal origins, acting as an autocrine and/or paracrine mediator on

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
157
airway cells (1). In particular, airway epithelial cells have been shown to express relatively
high levels of acetylcholine (2, 3). Epithelial cells express enzymes to synthesize
acetylcholine, including choline acetyltransferase (ChAT), and have also been shown to
release non‐neuronal acetylcholine via organic cation transporters (OCTs) (2, 4).
Epithelial cells play an important role in obstructive airway diseases including asthma.
Epithelial differentiation is altered in the disease as there is an increase in goblet cell
numbers, leading to excessive mucus production (5). IL‐13 is the main driver of this
response and has been shown to play a central role in asthma (6). From animals models it
is known that IL‐13 is sufficient to induce goblet cell hyperplasia (7), which can be
reproduced in air‐liquid interface (ALI) cell cultures in vitro (8, 9). IL‐13‐induced MUC5AC
gene expression is mediated via a complex transcriptional network, in which expression of
the repressor forkhead box protein A2 (FoxA2) is reduced in response to STAT6 activation,
whereas the expression of SAM pointed domain containing ETS transcription factor
(SPDEF) and forkhead box protein A3 (FoxA3) are increased and promote MUC5AC gene
expression (10, 11).
Interestingly, several studies have indirectly demonstrated a role for acetylcholine in
epithelial cell differentiation and goblet cell metaplasia. Tiotropium, an anticholinergic
drug, has been shown to inhibit ovalbumin‐induced goblet cell metaplasia in guinea pigs
and mice (12, 13), an effect also observed after knock‐out of the M3 receptor in mice
(chapter 6). Furthermore, the increased MUC5AC expression observed after allergen
exposure was inhibited by tiotropium in guinea pigs and M3 knock‐out mice (chapter 6,
12). Moreover, in mild asthmatic patients, repeated challenges with methacholine
induced epithelial cell proliferation and goblet cell metaplasia (14). Although these studies
point to a critical role for acetylcholine in goblet cell metaplasia, it remains unresolved if
this is via direct regulation of goblet cell differentiation of airway epithelial cells, or via the
indirect regulation of pro‐inflammatory cells and cytokines that drive this response, since
tiotropium also inhibited the inflammatory response in the animal studies described
above.
Therefore, in this study, we investigated the role of endogenous non‐neuronal
acetylcholine in the differentiation of primary human airway epithelial cells, differentiated
in an ALI cell culture. We demonstrate that inhibition of endogenous acetylcholine by
tiotropium has no effects on epithelial cell differentiation following air exposure, but
inhibits IL‐13‐induced goblet cell metaplasia and MUC5AC expression, which might be
mediated via FoxA2 and FoxA3.

Chapter 8
158
Methods
Culture of human airway epithelial cells
The immortal human bronchial epithelial cell line 16HBE14o− was kindly donated by Dr DC
Gruenert, University of Vermont, Burlington, VT, USA (15). Cells were cultured as
described previously (16) and briefly described in the supplement.
Primary human airway epithelial (HAE) cells were obtained from healthy lung transplant
donors. Selection criteria for transplant donors are listed in the Eurotransplant guidelines
and include the absence of primary lung disease, such as asthma and COPD, and no more
than 20 pack years of smoking history. Cells were isolated by enzymatic digestion and
cultured as described previously (9), which is also briefly explained in the supplement and
summarized in figure 1. Cells were exposed to tiotropium (10 nM; provided by Boehringer
Ingelheim), IL‐13 (1ng/ml; Peprotech, Rocky Hill, NJ), or the combination of IL‐13 and
tiotropium during differentiation of two weeks (figure 1).
RNA analysis
Total RNA was extracted from freshly isolated airway epithelial cells, or from cultured
16HBE140‐ or HAE cells, either after submerged or ALI culture (HAE cells), using the
RNeasy mini kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's
instructions. Equal amounts of total RNA were then reverse transcribed and cDNA was
subjected to real‐time qPCR (Westburg, Leusden, The Netherlands) or to micro‐array
analysis. Details concerning real‐time qPCR and primers used can be found in the
supplement (text and table S1). Data are normalized to 18S ribosomal RNA. Micro‐array
analysis was performed using the llumina HT12 version 4 chip. Data was analyzed using R
software and subjected to gene set enrichment analysis using the Kyoto Encyclopedia of
Genes and Genomes (KEGG), Biocarta and Reactome definitions.
Figure 1. Schematic representation of the air‐liquid interface culture model. Human airway epithelial (HAE) cells
were seeded onto transwell insert and grown to confluence. Thereafter, apical medium was removed to create
an air‐liquid interface (ALI). Medium and stimuli were refreshed three times per week. After 14 days, cells were
harvested for PCR analysis or morphology and medium was collected for ELISA.

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
159
Histochemistry
The morphology of the cultures was assessed using light microscopy. After two weeks,
transwell inserts were embedded in paraffin according to the manufacturer’s instructions.
Transverse cross‐sections of 5 µm thick were used for morphometric analyses. Cells were
stained with H&E staining (Sigma‐Aldrich, Zwijndrecht, The Netherlands), MUC5AC
positive cells were stained with a MUC5AC antibody staining (Neomarkers, Fremont, CA,
USA) and mucin‐producing goblet cells were stained with Periodic Acid Schiff’s (PAS,
Sigma‐Aldrich, Zwijndrecht, The Netherlands). Cells were counted in duplicate in a blinded
fashion.
Cytokine analysis
Cytokine concentrations in basal medium were determined by a MILLIPLEX assay
(Millipore, Billerica, USA) on a Luminex 100 system using Starstation software (Applied
Cytometry Systems, Sheffield, UK) according to the manufacturer's instructions. A screen
for 26 cytokines was performed (see supplement for complete list).
Statistical analysis
Data are presented as mean ± standard error of the mean. Statistical differences between
means were calculated using one‐way ANOVA, followed by Newman Keuls multiple
comparison tests. Differences were considered significant at p<0.05.
Results
Differentiation of epithelial cells
First, the differentiation state of epithelial cells was characterized. Therefore, gene
expression of the differentiation markers MUC5AC, a marker for goblet cells, and tektin, a
marker for ciliated cells, was compared in different cells. We compared submerged
cultures of the cell line 16HBE14o‐, submerged cultures of HAE cells, freshly isolated HAE
cells, and ALI‐cultured HAE cells. Gene expression of MUC5AC and tektin was higher in
freshly isolated and ALI‐cultured HAE cultures compared to submerged cultures (figure 2).
Whereas tektin levels were comparable in freshly isolated cells and ALI‐cultures, MUC5AC
levels were considerably higher in freshly isolated cells compared to ALI‐cultures. Together
this indicates that differentiation of an ALI culture following air exposure induces epithelial
cell differentiation compared to submerged cultures, which mimics ciliated cell
differentiation compared to freshly isolated epithelial cells, and to a lesser extent goblet
cell differentiation (figure 2).
Chapter 8
160
Figure 2. Differentiation state of epithelial cells. Expression of the differentiation markers MUC5AC, a marker for
goblet cells (A), and tektin, a marker for ciliated cells (B), was analyzed by real‐time qPCR in submerged
16HBE14o‐ and submerged HAE cell cultures, in freshly isolated HAE cells and in ALI‐cultured HAE cells. Data
represent means ± s.e. of the mean, n=3‐5, ** p<0.01; *** p<0.001 compared to submerged culture, ## p<0.01;
### p<0.001 compared to HBE culture, $$$ p<0.001 compared to freshly isolated cells.
Expression of the non‐neuronal cholinergic system
Next, the expression of genes involved in the non‐neuronal cholinergic system in ALI‐
cultured HAE cells was characterized and compared to 16HBE14o‐ cells. In view of the
rationale of the study, we focused on genes involved in acetylcholine metabolism and
muscarinic receptors. HAE cells expressed all components of the non‐neuronal cholinergic
system needed for acetylcholine production and release (table 1). This comprises choline
transporters for uptake of choline, including the high‐affinity choline transporter 1 (CHT1),
the choline transporter‐like protein 1 (CTL1), and CTL4. Unexpectedly, the synthesizing
enzyme choline acetyltransferase (ChAT) was not detected in any of the epithelial cell
cultures, whereas the synthesizing enzyme carnitine acetyltransferase (CarAT) was
abundantly expressed. In addition, acetylcholinesterase (AChE) and butyrylcholinesterase
(BChE), two enzymes for degradation of acetylcholine, were expressed. Finally, muscarinic
M1, M2 and M3 receptors were expressed on the HAE cells as a target for acetylcholine
(table 1). Compared to 16HBE14o‐ cells, most components were expressed at similar or
higher levels in ALI‐cultured HAE cells. The most prominent higher expression in ALI‐
cultured HAE cells was observed for the M3 receptor and the choline transporters CHT1
and CTL4 (table 1).
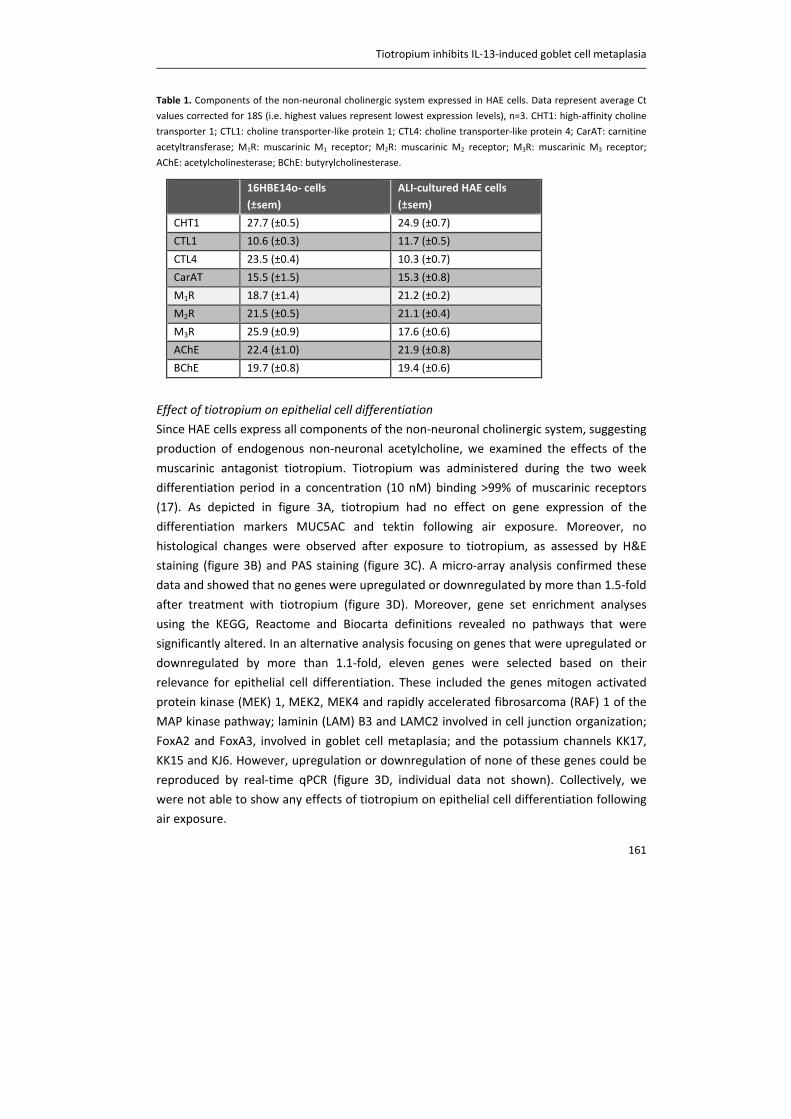
Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
161
Table 1. Components of the non‐neuronal cholinergic system expressed in HAE cells. Data represent average Ct
values corrected for 18S (i.e. highest values represent lowest expression levels), n=3. CHT1: high‐affinity choline
transporter 1; CTL1: choline transporter‐like protein 1; CTL4: choline transporter‐like protein 4; CarAT: carnitine
acetyltransferase; M1R: muscarinic M1 receptor; M2R: muscarinic M2 receptor; M3R: muscarinic M3 receptor;
AChE: acetylcholinesterase; BChE: butyrylcholinesterase.
Effect of tiotropium on epithelial cell differentiation
Since HAE cells express all components of the non‐neuronal cholinergic system, suggesting
production of endogenous non‐neuronal acetylcholine, we examined the effects of the
muscarinic antagonist tiotropium. Tiotropium was administered during the two week
differentiation period in a concentration (10 nM) binding >99% of muscarinic receptors
(17). As depicted in figure 3A, tiotropium had no effect on gene expression of the
differentiation markers MUC5AC and tektin following air exposure. Moreover, no
histological changes were observed after exposure to tiotropium, as assessed by H&E
staining (figure 3B) and PAS staining (figure 3C). A micro‐array analysis confirmed these
data and showed that no genes were upregulated or downregulated by more than 1.5‐fold
after treatment with tiotropium (figure 3D). Moreover, gene set enrichment analyses
using the KEGG, Reactome and Biocarta definitions revealed no pathways that were
significantly altered. In an alternative analysis focusing on genes that were upregulated or
downregulated by more than 1.1‐fold, eleven genes were selected based on their
relevance for epithelial cell differentiation. These included the genes mitogen activated
protein kinase (MEK) 1, MEK2, MEK4 and rapidly accelerated fibrosarcoma (RAF) 1 of the
MAP kinase pathway; laminin (LAM) B3 and LAMC2 involved in cell junction organization;
FoxA2 and FoxA3, involved in goblet cell metaplasia; and the potassium channels KK17,
KK15 and KJ6. However, upregulation or downregulation of none of these genes could be
reproduced by real‐time qPCR (figure 3D, individual data not shown). Collectively, we
were not able to show any effects of tiotropium on epithelial cell differentiation following
air exposure.
16HBE14o‐ cells
(±sem)
ALI‐cultured HAE cells
(±sem)
CHT1 27.7 (±0.5) 24.9 (±0.7)
CTL1 10.6 (±0.3) 11.7 (±0.5)
CTL4 23.5 (±0.4) 10.3 (±0.7)
CarAT 15.5 (±1.5) 15.3 (±0.8)
M1R 18.7 (±1.4) 21.2 (±0.2)
M2R 21.5 (±0.5) 21.1 (±0.4)
M3R 25.9 (±0.9) 17.6 (±0.6)
AChE 22.4 (±1.0) 21.9 (±0.8)
BChE 19.7 (±0.8) 19.4 (±0.6)
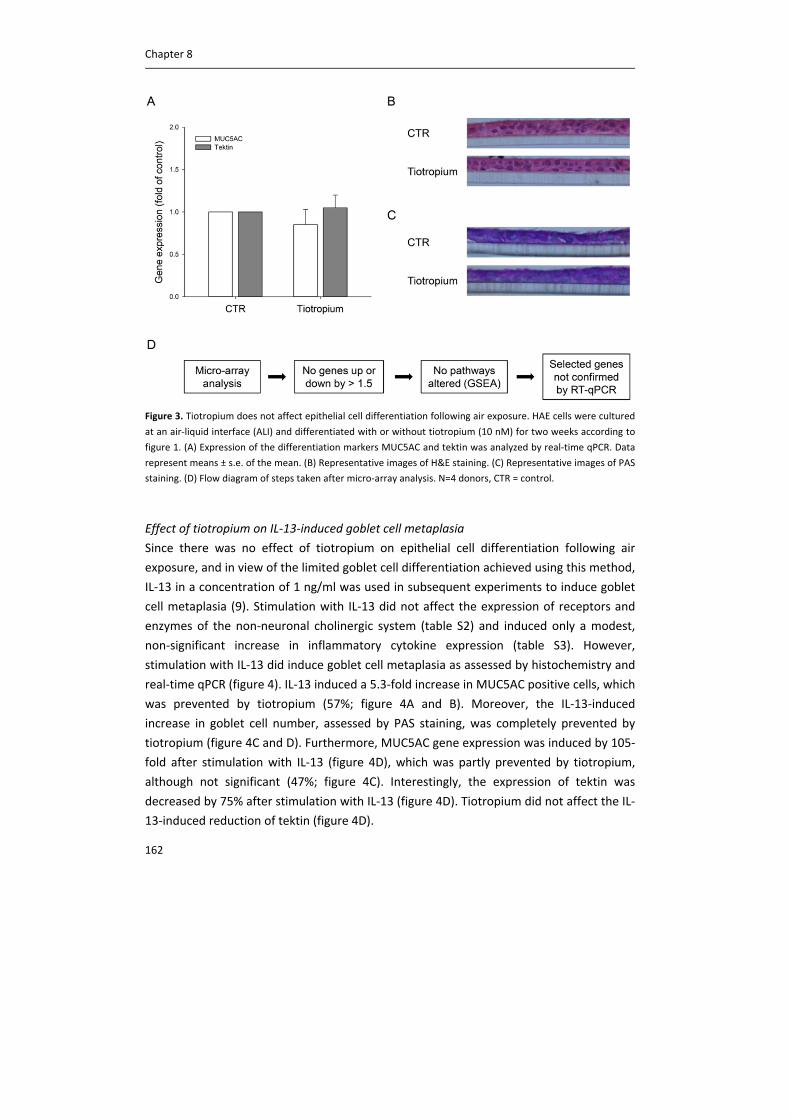
Chapter 8
162
Figure 3. Tiotropium does not affect epithelial cell differentiation following air exposure. HAE cells were cultured
at an air‐liquid interface (ALI) and differentiated with or without tiotropium (10 nM) for two weeks according to
figure 1. (A) Expression of the differentiation markers MUC5AC and tektin was analyzed by real‐time qPCR. Data
represent means ± s.e. of the mean. (B) Representative images of H&E staining. (C) Representative images of PAS
staining. (D) Flow diagram of steps taken after micro‐array analysis. N=4 donors, CTR = control.
Effect of tiotropium on IL‐13‐induced goblet cell metaplasia
Since there was no effect of tiotropium on epithelial cell differentiation following air
exposure, and in view of the limited goblet cell differentiation achieved using this method,
IL‐13 in a concentration of 1 ng/ml was used in subsequent experiments to induce goblet
cell metaplasia (9). Stimulation with IL‐13 did not affect the expression of receptors and
enzymes of the non‐neuronal cholinergic system (table S2) and induced only a modest,
non‐significant increase in inflammatory cytokine expression (table S3). However,
stimulation with IL‐13 did induce goblet cell metaplasia as assessed by histochemistry and
real‐time qPCR (figure 4). IL‐13 induced a 5.3‐fold increase in MUC5AC positive cells, which
was prevented by tiotropium (57%; figure 4A and B). Moreover, the IL‐13‐induced
increase in goblet cell number, assessed by PAS staining, was completely prevented by
tiotropium (figure 4C and D). Furthermore, MUC5AC gene expression was induced by 105‐
fold after stimulation with IL‐13 (figure 4D), which was partly prevented by tiotropium,
although not significant (47%; figure 4C). Interestingly, the expression of tektin was
decreased by 75% after stimulation with IL‐13 (figure 4D). Tiotropium did not affect the IL‐
13‐induced reduction of tektin (figure 4D).
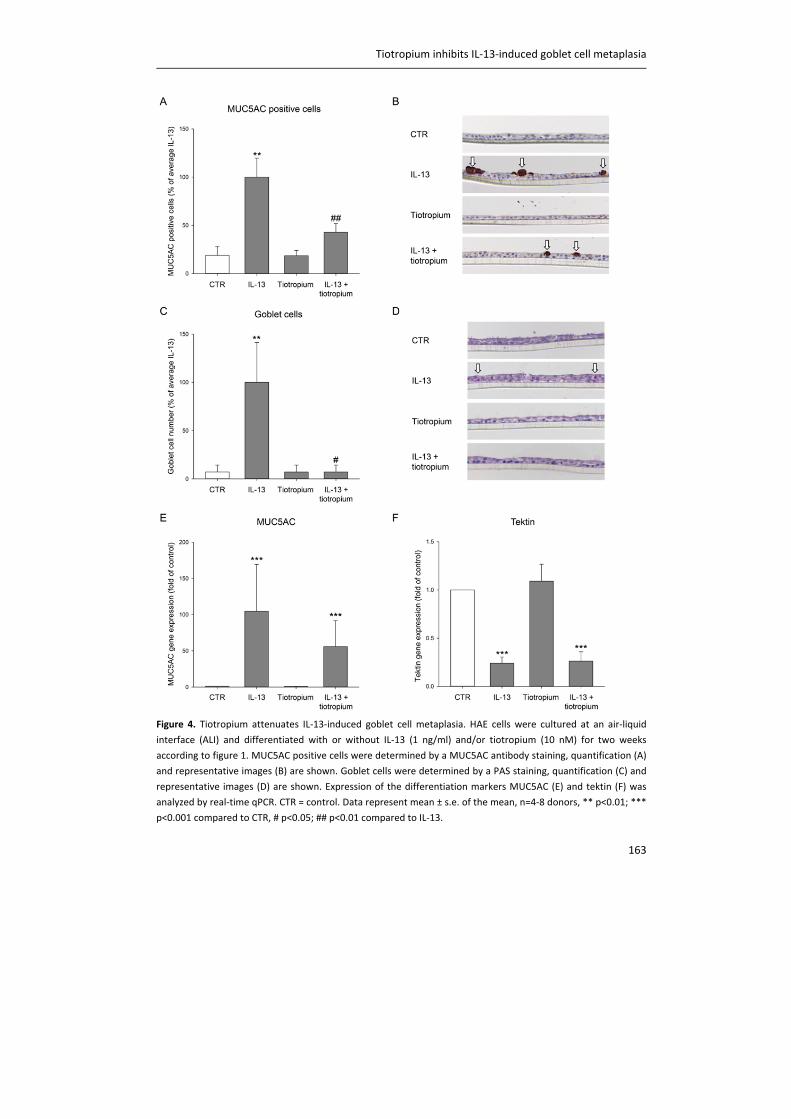
Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
163
Figure 4. Tiotropium attenuates IL‐13‐induced goblet cell metaplasia. HAE cells were cultured at an air‐liquid
interface (ALI) and differentiated with or without IL‐13 (1 ng/ml) and/or tiotropium (10 nM) for two weeks
according to figure 1. MUC5AC positive cells were determined by a MUC5AC antibody staining, quantification (A)
and representative images (B) are shown. Goblet cells were determined by a PAS staining, quantification (C) and
representative images (D) are shown. Expression of the differentiation markers MUC5AC (E) and tektin (F) was
analyzed by real‐time qPCR. CTR = control. Data represent mean ± s.e. of the mean, n=4‐8 donors, ** p<0.01; ***
p<0.001 compared to CTR, # p<0.05; ## p<0.01 compared to IL‐13.
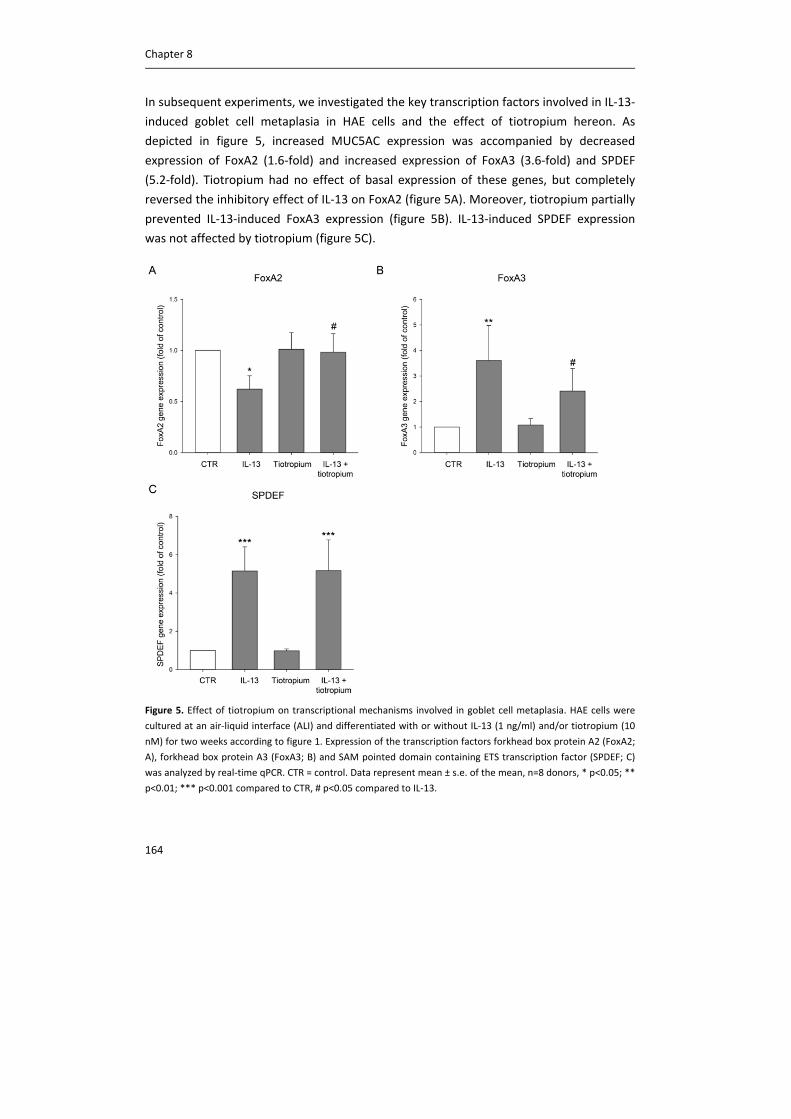
Chapter 8
164
In subsequent experiments, we investigated the key transcription factors involved in IL‐13‐
induced goblet cell metaplasia in HAE cells and the effect of tiotropium hereon. As
depicted in figure 5, increased MUC5AC expression was accompanied by decreased
expression of FoxA2 (1.6‐fold) and increased expression of FoxA3 (3.6‐fold) and SPDEF
(5.2‐fold). Tiotropium had no effect of basal expression of these genes, but completely
reversed the inhibitory effect of IL‐13 on FoxA2 (figure 5A). Moreover, tiotropium partially
prevented IL‐13‐induced FoxA3 expression (figure 5B). IL‐13‐induced SPDEF expression
was not affected by tiotropium (figure 5C).
Figure 5. Effect of tiotropium on transcriptional mechanisms involved in goblet cell metaplasia. HAE cells were
cultured at an air‐liquid interface (ALI) and differentiated with or without IL‐13 (1 ng/ml) and/or tiotropium (10
nM) for two weeks according to figure 1. Expression of the transcription factors forkhead box protein A2 (FoxA2;
A), forkhead box protein A3 (FoxA3; B) and SAM pointed domain containing ETS transcription factor (SPDEF; C)
was analyzed by real‐time qPCR. CTR = control. Data represent mean ± s.e. of the mean, n=8 donors, * p<0.05; **
p<0.01; *** p<0.001 compared to CTR, # p<0.05 compared to IL‐13.

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
165
Discussion
In this study we demonstrate that non‐neuronal acetylcholine contributes to goblet cell
differentiation. Human airway epithelial cells expressed all components needed for
acetylcholine synthesis, and although the muscarinic antagonist tiotropium did not affect
epithelial cell differentiation following air exposure, tiotropium did inhibit IL‐13‐induced
goblet cell metaplasia. Furthermore, we provide evidence for the involvement of the
transcriptional regulators FoxA2 and FoxA3 in this effect. This study is the first to
investigate the role of tiotropium in ALI‐cultured primary epithelial cells and the results
imply that tiotropium might affect mucus hypersecretion.
Data from this study clearly indicates that all components needed for acetylcholine
synthesis are present in cultured human airway epithelial cells derived from central
airways. The fact that epithelial cells express components of the non‐neuronal cholinergic
system is in line with previous studies (3, 4). However, in contrast to previous studies and
different from the neuronal cholinergic system where ChAT is the most prominent
synthesizing enzyme of acetylcholine (2, 4), we were unable to detect significant
expression of ChAT in any of the epithelial cell cultures. Rather, we demonstrate abundant
expression of the synthesizing enzyme CarAT. The enzyme CarAT was identified already 40
years ago as an alternative way to synthesize acetylcholine in rabbit heart by White and
Wu (18) and has been detected in multiple non‐neuronal cholinerigc systems since then,
including rat skeletal muscle (19), cat esophageal mucosa (20), mouse bladder and
abraded urothelium, and human mucosal bladder biopsies (21).
The fact that acetylcholine is not only a neurotransmitter, but also a local signaling
molecule produced by non‐neuronal cells, has dramatically broadened the view on the
role of acetylcholine. It has been suggested that the non‐neuronal cholinergic system
might play a role in lung physiology and/or pathophysiology, however, direct evidence for
such a role is still limited. In this study, human airway epithelial cells, cultured at an ALI,
were used as a model system. In this way, the role of non‐neuronal acetylcholine can be
investigated, since no neuronal system is present. Moreover, by using primary cells
cultured at an ALI, the human physiology is reflected more closely compared to cell lines
like 16HBE14o‐. Instead of only one basal cell type, an ALI‐culture is a pseudostratified
mucociliary cell culture with different epithelial cells present, as is also observed in vivo.
Our data indicate that ciliated cell marker expression in ALI‐cultured HAE cells is
comparable to freshly isolated airway epithelial cells, whereas MUC5AC expression, a
marker for goblet cells, is moderately expressed after ALI‐culture, but can be markedly
increased by IL‐13 exposure. Of specific relevance to our study is that components of the

Chapter 8
166
non‐neuronal cholinergic system are expressed at higher levels in primary ALI‐cultured
cells compared to 16HBE14o‐ cells.
IL‐13 is a central mediator in asthma pathology leading to goblet cell metaplasia and
mucus hypersecretion (6). Mucus secretion and MUC5AC expression are enhanced in
asthma and contribute significantly to airflow limitation in asthma by obstructing the
smaller airways (22, 23). Stimulation with IL‐13 increased MUC5AC expression in our
study, which is in line with previous studies using ALI‐cultures (8, 9). Moreover, expression
of tektin, a marker of ciliated cells, was reduced at the same time by IL‐13. Indeed goblet
cell metaplasia was induced, as MUC5AC antibody staining and PAS staining revealed an
increase in mucin‐producing cells compared to control sections. Furthermore, expression
of the transcription factors SPDEF and FoxA3 was enhanced, whereas expression of FoxA2
was reduced. This is in line with previous studies aimed at unraveling the transcriptional
network involved in goblet cell metaplasia (10, 11). Interestingly, tiotropium completely
prevented the reduced expression of FoxA2 and partly prevented the increase in FoxA3,
whereas increased expression of SPDEF was not affected. This may explain the partial
effect observed on goblet cell metaplasia after tiotropium exposure.
Mucus secreting cells in the airways include epithelial goblet cells and submucosal glands.
It has been convincingly shown that the latter are under cholinergic neural control (22).
Vagal stimulation of ferret trachea in vitro or in vivo enhanced mucus secretion, which
was inhibited by the M3 antagonist 4‐DAMP (24), and electrical field stimulation also
enhanced mucus release from human bronchi (25). This neuronal acetylcholine‐induced
mucus release is probably derived from airway submucosal glands. However, increasing
evidence suggests that acetylcholine can also induce mucus secretion from goblet cells
(22, 26, 27). Whether goblet cells can release mucus in response to neuronally released
acetylcholine is still a matter of debate, but our data do suggest a role for non‐neuronal
acetylcholine in the differentiation towards this cell type. While submucosal glands
predominate in healthy human airways, goblet cells undergo metaplasia under disease
conditions and predominate in asthma (22), indicating the potential relevance for
inhibition of this differentiation process.
Therefore, the finding that tiotropium partly prevents IL‐13‐induced goblet cell metaplasia
in our model implies that this might be beneficial for asthma patients. This is in line with
evidence from in vitro and in vivo studies, indicating a role for muscarinic antagonists in
mucus hypersecretion. The muscarinic antagonist oxitropium dose‐dependently inhibits
histamine‐induced mucus secretion from airway goblet cells (28). Furthermore, Cortijo et
al. demonstrated that muscarinic receptor stimulation by carbachol induced an increase in

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
167
MUC5AC expression in human bronchus and in cultured epithelial cells, which was
inhibited by the muscarinic antagonist aclidinium (29). Similar findings were observed in
mild asthma patients, in which repeated challenges with methacholine induced epithelial
cell proliferation and goblet cell metaplasia (14). Interestingly, these effects can be
prevented when bronchoconstriction is prevented, suggesting that preventing
bronchoconstriction with tiotropium might have an indirect inhibitory effect on mucus
hypersecretion (14). Animal studies have also provided indirect evidence for a role of
muscarinic receptors in allergen‐induced goblet cell metaplasia. In an ovalbumin‐induced
guinea pig model, tiotropium inhibited MUC5AC expression and mucus gland hypertrophy
(30). Moreover, goblet cell metaplasia was inhibited by tiotropium in an ovalbumin‐
induced mouse model (13). Interestingly, knock‐out of the muscarinic M3 receptor also
inhibits ovalbumin‐induced goblet cell metaplasia and MUC5AC expression (chapter 6).
Notably, IL‐13 expression was enhanced in these same animals, indicating that this effect
was not via a reduction of the inflammatory response (chapter 6).
Our findings extend these previous observations as we now demonstrate that the effects
of tiotropium are not only the result of reduced bronchoconstriction or reduced
inflammatory cytokine expression, but that tiotropium also has a direct effect on the
epithelium. We performed a multiplex cytokine analysis to verify the absence of the
involvement of an inflammatory response. Only a modest, non‐significant increase in
inflammatory cytokines was observed after stimulation with IL‐13, and also tiotropium
had no significant effect on inflammatory cytokines. Therefore we can conclude from our
study that airway epithelial cells express non‐neuronal acetylcholine and inhibition of this
component by anticholinergics is sufficient to prevent IL‐13‐induced goblet cell
differentiation. This direct effect of tiotropium on airway epithelial cells as observed in our
study might act synergistically with indirect effects of tiotropium on epithelial cells in vivo,
including reduced mechanical strain and reduced inflammation. However, future clinical
studies are clearly needed to confirm this hypothesis.
Tiotropium is currently not included as a chronic therapy for patients with asthma. Recent
evidence suggests that addition of tiotropium to standard therapy with long‐acting β‐
agonists and inhaled corticosteroids might be beneficial for asthma patients (31). In this
randomized controlled trial it was shown that tiotropium induces an increase in FEV1 in
severe asthma patients, on top of the use of β2‐agonists and corticosteroids. Furthermore,
severe exacerbations and episodes of worsening of asthma were reduced after add‐on of
tiotropium (31). Results from our study imply that mucus hypersecretion might also be
affected after tiotropium treatment. Previously, tiotropium has been shown to be
effective in reducing sputum levels in patients with chronic airway mucus hypersecretion

Chapter 8
168
(32). Although it has been proposed that anticholinergic treatment might impair sputum
clearance, there is now increasing evidence that reducing the volume of sputum without
altering viscoelasticity by anticholinergics is beneficial for patients with chronic mucus
hypersecretion (33). Future studies are needed to directly address this hypothesis in more
detail.
In conclusion, we demonstrated that tiotropium can prevent IL‐13‐induced goblet cell
metaplasia, indicating that non‐neuronal acetylcholine contributes to goblet cell
differentiation. Modulation of FoxA2 and FoxA3 expression is likely one of the
mechanisms involved. These data imply that this direct effect of tiotropium on the airway
epithelium, in combination with its protective effects on bronchoconstriction and
inflammation, may protect from mucus hypersecretion.
Acknowledgements
The authors would like to thank Renate M. Verhoosel and Willemieke M. Mudde for
technical assistance, and Tinne C.J. Mertens for expert advice on analysis of IL‐13 induced
mucin production. The authors would like to thank the Netherlands Lung Foundation for
financial support (grant 3.2.08.014). Previous research on the ALI‐model in the laboratory
of P.S. Hiemstra was supported by a grant from Boerhringer Ingelheim.

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
169
Supplement
Methods
Culture of human airway epithelial cells
The immortal human bronchial epithelial cell line 16HBE14o− was cultured in minimum
essential medium (ME)), supplemented with 10% heat‐inactivated foetal bovine serum,
20 U/ml penicillin and 20 μg/ml streptomycin and 2 mM L‐glutamine (Gibco, Grand Island,
NY). Cells were collected for RNA analysis.
Primary human airway epithelial (HAE) cells were obtained from healthy lung transplant
donors and isolated by enzymatic digestion. Cells from passage 2 were grown submerged
on collagen‐coated Transwell inserts until confluence, after which they were cultured at
an air‐liquid interface (ALI) and differentiated for two weeks in B/D medium (1:1 mixture
of DMEM [Gibco, Grand Island, NY] and bronchial epithelial growth medium [BEGM;
Lonza, Walkersville, MD, USA], supplemented with 0.4% [w/v] bovine pituitary extract
[BPE], 0.5 ng/ml epidermal growth factor [EGF], 5 μg/ml insulin, 10 μg/ml transferrin,
1 μM hydrocortisone, 6.5 ng/ml T3, 0.5 μg/ml epinephrine [all from Lonza], 15 ng/ml
retinoic acid [Sigma Chemical, St. Louis, MO], 1.5 μg/ml bovine serum albumin [Sigma
Chemical, St. Louis, MO], 0.5 mM sodium pyruvate [Gibco, Grand Island, NY ], 20 U/ml
penicillin and 20 μg/ml streptomycin [Gibco, Grand Island, NY]). Cells were exposed to IL‐
13 (1 ng/ml; Peprotech, Rocky Hill, NJ), tiotropium (10 nM; provided by Boehringer
Ingelheim) or the combination of IL‐13 and tiotropium that was added to the basal
medium during the differentiation process. ALI cultures were maintained for 14 days at
37°C in a humidified atmosphere of 5% CO2. Medium and stimuli were refreshed three
times per week. The apical side of the epithelial cells was washed with PBS at the same
time. After two weeks, cells were collected for RNA analysis or fixed for histochemistry,
and basal medium was collected for cytokine analysis.
Real time PCR
Real time PCR was performed with denaturation at 94°C for 30 seconds, annealing at 59°C
for 30 seconds and extension at 72°C for 30 seconds for 40 cycles followed by 10 minutes
at 72°C. Real‐time PCR data were analyzed using the comparative cycle threshold (Ct:
amplification cycle number) method. The amount of target gene was normalized to the
endogenous reference gene 18S ribosomal RNA. The specific forward and reverse primers
used are listed in Table S1.
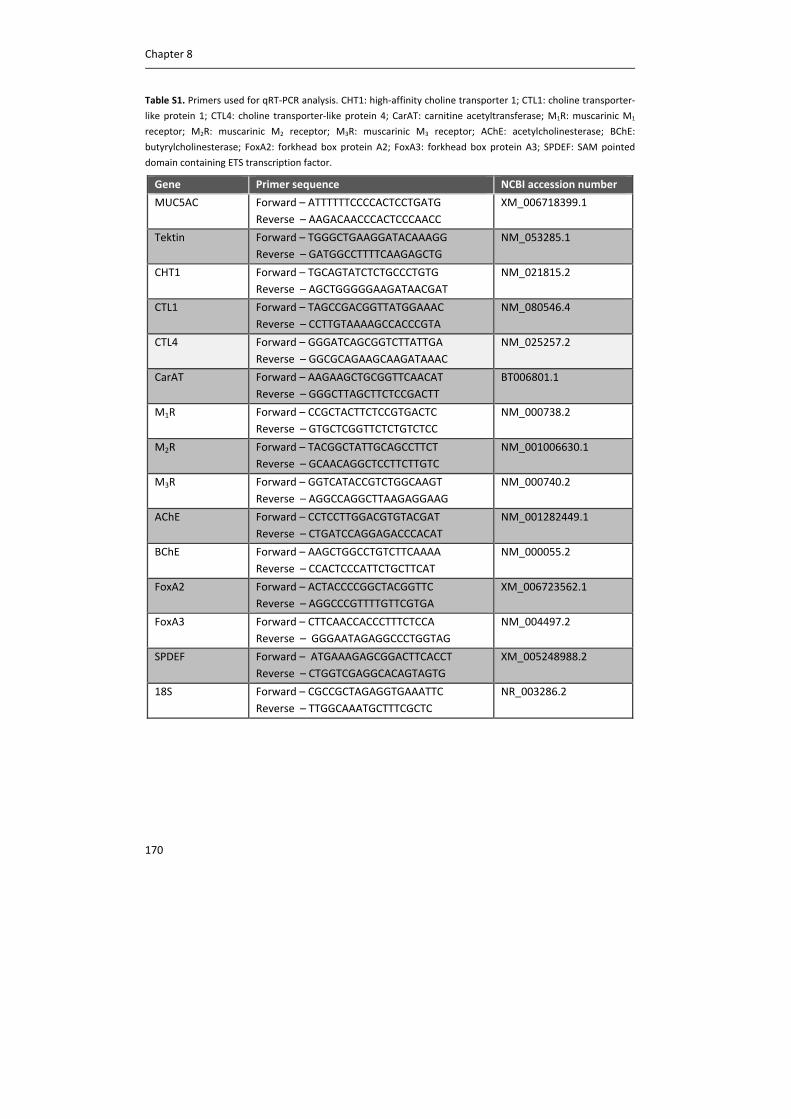
Chapter 8
170
Table S1. Primers used for qRT‐PCR analysis. CHT1: high‐affinity choline transporter 1; CTL1: choline transporter‐
like protein 1; CTL4: choline transporter‐like protein 4; CarAT: carnitine acetyltransferase; M1R: muscarinic M1
receptor; M2R: muscarinic M2 receptor; M3R: muscarinic M3 receptor; AChE: acetylcholinesterase; BChE:
butyrylcholinesterase; FoxA2: forkhead box protein A2; FoxA3: forkhead box protein A3; SPDEF: SAM pointed
domain containing ETS transcription factor.
Gene Primer sequence NCBI accession number
MUC5AC Forward – ATTTTTTCCCCACTCCTGATG
Reverse – AAGACAACCCACTCCCAACC
XM_006718399.1
Tektin Forward – TGGGCTGAAGGATACAAAGG
Reverse – GATGGCCTTTTCAAGAGCTG
NM_053285.1
CHT1 Forward – TGCAGTATCTCTGCCCTGTG
Reverse – AGCTGGGGGAAGATAACGAT
NM_021815.2
CTL1 Forward – TAGCCGACGGTTATGGAAAC
Reverse – CCTTGTAAAAGCCACCCGTA
NM_080546.4
CTL4 Forward – GGGATCAGCGGTCTTATTGA
Reverse – GGCGCAGAAGCAAGATAAAC
NM_025257.2
CarAT Forward – AAGAAGCTGCGGTTCAACAT
Reverse – GGGCTTAGCTTCTCCGACTT
BT006801.1
M1R Forward – CCGCTACTTCTCCGTGACTC
Reverse – GTGCTCGGTTCTCTGTCTCC
NM_000738.2
M2R Forward – TACGGCTATTGCAGCCTTCT
Reverse – GCAACAGGCTCCTTCTTGTC
NM_001006630.1
M3R Forward – GGTCATACCGTCTGGCAAGT
Reverse – AGGCCAGGCTTAAGAGGAAG
NM_000740.2
AChE Forward – CCTCCTTGGACGTGTACGAT
Reverse – CTGATCCAGGAGACCCACAT
NM_001282449.1
BChE Forward – AAGCTGGCCTGTCTTCAAAA
Reverse – CCACTCCCATTCTGCTTCAT
NM_000055.2
FoxA2 Forward – ACTACCCCGGCTACGGTTC
Reverse – AGGCCCGTTTTGTTCGTGA
XM_006723562.1
FoxA3 Forward – CTTCAACCACCCTTTCTCCA
Reverse – GGGAATAGAGGCCCTGGTAG
NM_004497.2
SPDEF Forward – ATGAAAGAGCGGACTTCACCT
Reverse – CTGGTCGAGGCACAGTAGTG
XM_005248988.2
18S Forward – CGCCGCTAGAGGTGAAATTC
Reverse – TTGGCAAATGCTTTCGCTC
NR_003286.2
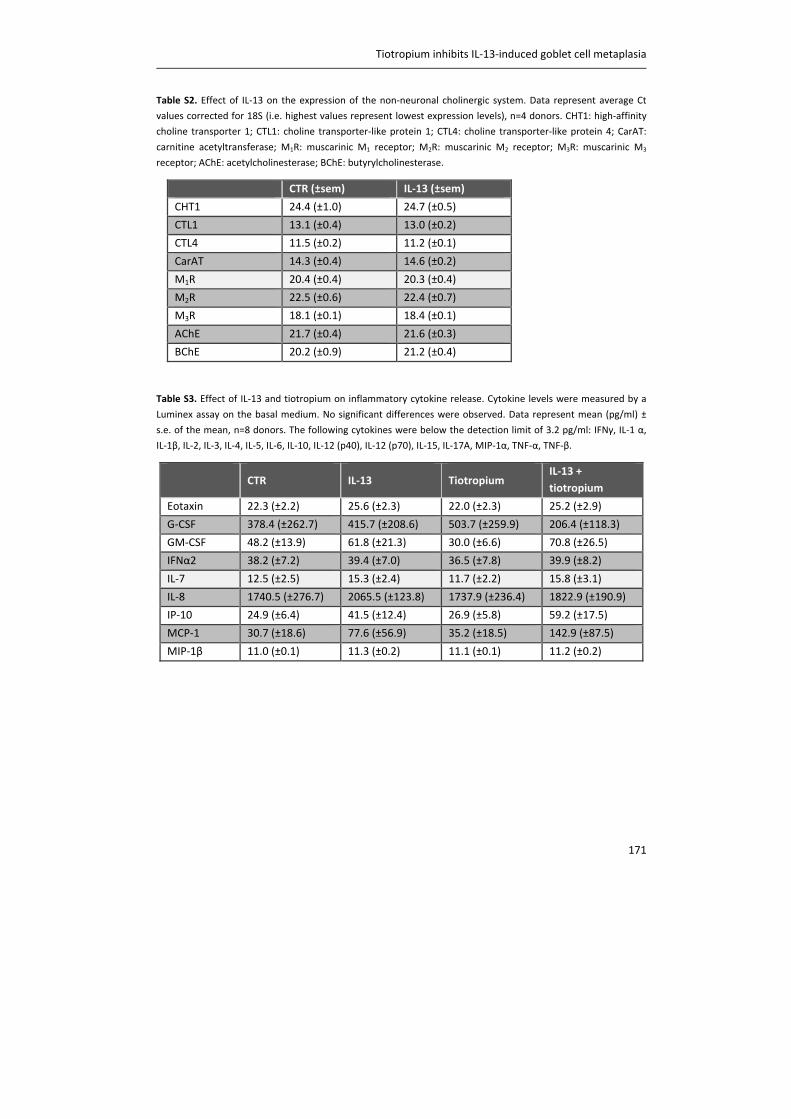
Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
171
Table S2. Effect of IL‐13 on the expression of the non‐neuronal cholinergic system. Data represent average Ct
values corrected for 18S (i.e. highest values represent lowest expression levels), n=4 donors. CHT1: high‐affinity
choline transporter 1; CTL1: choline transporter‐like protein 1; CTL4: choline transporter‐like protein 4; CarAT:
carnitine acetyltransferase; M1R: muscarinic M1 receptor; M2R: muscarinic M2 receptor; M3R: muscarinic M3
receptor; AChE: acetylcholinesterase; BChE: butyrylcholinesterase.
Table S3. Effect of IL‐13 and tiotropium on inflammatory cytokine release. Cytokine levels were measured by a
Luminex assay on the basal medium. No significant differences were observed. Data represent mean (pg/ml) ±
s.e. of the mean, n=8 donors. The following cytokines were below the detection limit of 3.2 pg/ml: IFNγ, IL‐1 α,
IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐10, IL‐12 (p40), IL‐12 (p70), IL‐15, IL‐17A, MIP‐1α, TNF‐α, TNF‐β.
CTR
IL‐13
Tiotropium IL‐13 +
tiotropium
Eotaxin 22.3 (±2.2) 25.6 (±2.3) 22.0 (±2.3) 25.2 (±2.9)
G‐CSF 378.4 (±262.7) 415.7 (±208.6) 503.7 (±259.9) 206.4 (±118.3)
GM‐CSF 48.2 (±13.9) 61.8 (±21.3) 30.0 (±6.6) 70.8 (±26.5)
IFNα2 38.2 (±7.2) 39.4 (±7.0) 36.5 (±7.8) 39.9 (±8.2)
IL‐7 12.5 (±2.5) 15.3 (±2.4) 11.7 (±2.2) 15.8 (±3.1)
IL‐8 1740.5 (±276.7) 2065.5 (±123.8) 1737.9 (±236.4) 1822.9 (±190.9)
IP‐10 24.9 (±6.4) 41.5 (±12.4) 26.9 (±5.8) 59.2 (±17.5)
MCP‐1 30.7 (±18.6) 77.6 (±56.9) 35.2 (±18.5) 142.9 (±87.5)
MIP‐1β 11.0 (±0.1) 11.3 (±0.2) 11.1 (±0.1) 11.2 (±0.2)
CTR (±sem) IL‐13 (±sem)
CHT1 24.4 (±1.0) 24.7 (±0.5)
CTL1 13.1 (±0.4) 13.0 (±0.2)
CTL4 11.5 (±0.2) 11.2 (±0.1)
CarAT 14.3 (±0.4) 14.6 (±0.2)
M1R 20.4 (±0.4) 20.3 (±0.4)
M2R 22.5 (±0.6) 22.4 (±0.7)
M3R 18.1 (±0.1) 18.4 (±0.1)
AChE 21.7 (±0.4) 21.6 (±0.3)
BChE 20.2 (±0.9) 21.2 (±0.4)

Chapter 8
172
References
(1) Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non‐neuronal cholinergic system in
humans. Br J Pharmacol 2008;154:1558‐1571.
(2) Klapproth H, Reinheimer T, Metzen J, Munch M, Bittinger F, Kirkpatrick CJ, Hohle KD, Schemann
M, Racke K, Wessler I. Non‐neuronal acetylcholine, a signalling molecule synthezised by surface cells
of rat and man. Naunyn Schmiedebergs Arch Pharmacol 1997;355:515‐523.
(3) Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is
an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells.
Endocrinology 2004;145:2498‐2506.
(4) Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol
2008;130:219‐234.
(5) Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med 2012;18:684‐692.
(6) Corren J. Role of interleukin‐13 in asthma. Curr Allergy Asthma Rep 2013;13:415‐420.
(7) Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of
interleukin‐13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic
abnormalities, and eotaxin production. J Clin Invest 1999;103:779‐788.
(8) Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, Coste A, Jorissen M, Marano F,
Caput D, Tournier F. IL‐13 alters mucociliary differentiation and ciliary beating of human respiratory
epithelial cells. J Clin Invest 2001;108:1817‐1824.
(9) Zuyderduyn S, Ninaber DK, Schrumpf JA, van Sterkenburg MA, Verhoosel RM, Prins FA, van
Wetering S, Rabe KF, Hiemstra PS. IL‐4 and IL‐13 exposure during mucociliary differentiation of
bronchial epithelial cells increases antimicrobial activity and expression of antimicrobial peptides.
Respir Res 2011;12:59‐9921‐12‐59.
(10) Whitsett JA, Haitchi HM, Maeda Y. Intersections between pulmonary development and disease.
Am J Respir Crit Care Med 2011;184:401‐406.
(11) Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, Gregorieff A, Clevers H, Whitsett JA.
SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes
associated with mucus production. J Clin Invest 2009;119:2914‐2924.
(12) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(13) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T,
Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling
in a mouse model of asthma. Clin Exp Allergy 2010;40:1266‐1275.
(14) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH.
Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med 2011;364:2006‐2015.
(15) Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH,
Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial
epithelial cells. Am J Respir Cell Mol Biol 1994;10:38‐47.

Tiotropium inhibits IL‐13‐induced goblet cell metaplasia
173
(16) Gkoumassi E, Dekkers BG, Droge MJ, Elzinga CR, Schmidt M, Meurs H, Zaagsma J, Nelemans SA.
Virodhamine and CP55,940 modulate cAMP production and IL‐8 release in human bronchial
epithelial cells. Br J Pharmacol 2007;151:1041‐1048.
(17) Disse B, Speck GA, Rominger KL, Witek TJ,Jr., Hammer R. Tiotropium (Spiriva): mechanistical
considerations and clinical profile in obstructive lung disease. Life Sci 1999;64:457‐464.
(18) White HL, Wu JC. Choline and carnitine acetyltransferases of heart. Biochemistry 1973;12:841‐
846.
(19) Tucek S. The synthesis of acetylcholine in skeletal muscles of the rat. J Physiol 1982;322:53‐69.
(20) Wolf‐Johnston AS, Hanna‐Mitchell AT, Buffington CA, Shinde S, Roppolo JR, Mayer E, Birder LA.
Alterations in the non‐neuronal acetylcholine synthesis and release machinery in esophageal
epithelium. Life Sci 2012;91:1065‐1069.
(21) Lips KS, Wunsch J, Zarghooni S, Bschleipfer T, Schukowski K, Weidner W, Wessler I, Schwantes
U, Koepsell H, Kummer W. Acetylcholine and molecular components of its synthesis and release
machinery in the urothelium. Eur Urol 2007;51:1042‐1053.
(22) Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol 2001;125:129‐144.
(23) Morcillo EJ, Cortijo J. Mucus and MUC in asthma. Curr Opin Pulm Med 2006;12:1‐6.
(24) Ramnarine SI, Haddad EB, Khawaja AM, Mak JC, Rogers DF. On muscarinic control of neurogenic
mucus secretion in ferret trachea. J Physiol 1996;494 ( Pt 2):577‐586.
(25) Baker B, Peatfield AC, Richardson PS. Nervous control of mucin secretion into human bronchi. J
Physiol 1985;365:297‐305.
(26) Bolduc P, Reid L. The effect of isoprenaline and pilocarpine on mitotic index and goblet cell
number in rat respiratory epithelium. Br J Exp Pathol 1978;59:311‐318.
(27) Steel DM, Hanrahan JW. Muscarinic‐induced mucin secretion and intracellular signaling by
hamster tracheal goblet cells. Am J Physiol 1997;272:L230‐7.
(28) Takeyama K, Tamaoki J, Nakata J, Konno K. Effect of oxitropium bromide on histamine‐induced
airway goblet cell secretion. Am J Respir Crit Care Med 1996;154:231‐236.
(29) Cortijo J, Mata M, Milara J, Donet E, Gavalda A, Miralpeix M, Morcillo EJ. Aclidinium inhibits
cholinergic and tobacco smoke‐induced MUC5AC in human airways. Eur Respir J 2011;37:244‐254.
(30) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(31) Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, Sigmund R, Seibold W,
Moroni‐Zentgraf P, Bateman ED. Tiotropium in asthma poorly controlled with standard combination
therapy. N Engl J Med 2012;367:1198‐1207.
(32) Saito Y, Azuma A, Morimoto T, Fujita K, Abe S, Motegi T, Usuki J, Kudoh S. Tiotropium
ameliorates symptoms in patients with chronic airway mucus hypersecretion which is resistant to
macrolide therapy. Intern Med 2008;47:585‐591.
(33) Bateman ED, Rennard S, Barnes PJ, Dicpinigaitis PV, Gosens R, Gross NJ, Nadel JA, Pfeifer M,
Racke K, Rabe KF, Rubin BK, Welte T, Wessler I. Alternative mechanisms for tiotropium. Pulm
Pharmacol Ther 2009;22:533‐542.


CHAPTER 9
GENERAL DISCUSSION AND SUMMARY
Trends Pharmacol Sci, 2014, Epub

Chapter 9
176
Preface
The objective of this thesis was to establish the role of acetylcholine in airway
inflammation and remodeling in COPD and asthma. We aimed to investigate the role of
individual muscarinic receptor subtypes, as well as the contribution of neuronal versus
non‐neuronal acetylcholine to inflammation and remodeling. The studies described in this
thesis reveal that there is a critical regulatory role for the M3 receptor in these processes,
which is mediated via neuronal acetylcholine, but also involves non‐neuronal
acetylcholine.
Anticholinergics as bronchodilators in COPD and asthma
COPD and asthma are chronic obstructive airway diseases, and the incidence of both
diseases is increasing (1). COPD is a leading cause of morbidity and mortality worldwide
and is expected to become the third leading cause of death in 2020. This results in a
substantial economic and social burden (2). With 180,000 deaths in the world each year,
mortality from asthma is considerably lower, but around 300 million people in the world
currently have asthma and its greatest burden lies in the morbidity and invalidity it causes,
also in children (3). Inflammation and remodeling are hallmark features of both diseases,
which contribute to the decline in lung function and the severity of the disease (4‐6).
Acetylcholine is the primary parasympathetic neurotransmitter in the airways, and
induces bronchoconstriction and mucus secretion via M3 receptors (chapter 2). The
activity of the neuronal system is altered in asthma and COPD via several mechanisms,
which can originate early in life or develop as an acute or chronic response after allergen
challenge or stimuli such as cigarette smoke, leading to exaggerated acetylcholine release
and airway narrowing (chapter 1). Strikingly, the increased cholinergic tone is the major
reversible component of airflow limitation in COPD (7, 8). Therefore, anticholinergics are
effective bronchodilators in this disease, and represent a first line of treatment (2). In
asthma, use of anticholinergics is currently limited to the treatment of exacerbations (9),
however, recent clinical trials indicate that anticholinergic therapy might also be beneficial
for chronic treatment of moderate and severe asthma patients (10, 11).

General discussion and summary
177
Muscarinic receptor subtypes in the lung – is there a need for M3 selective
anticholinergics?
Increasing evidence suggests that the role of acetylcholine in the airways is not limited to
bronchoconstriction and mucus secretion. In animal models of COPD and asthma,
anticholinergics inhibit both airway inflammation and airway remodeling (chapter 2).
However, it is not known which muscarinic receptor subtypes are involved in these
responses. Multiple muscarinic receptor subtypes are expressed in the airways, that have
differential roles in regulating bronchoconstriction, mucus secretion, inflammation and
remodeling (chapter 2). Muscarinic receptor agonists and antagonists have limited
selectivity towards individual muscarinic receptors, which limits the interpretation of the
effects of these agents on functional parameters. Therefore, in this thesis, we used
muscarinic receptor specific knock‐out animals to investigate the role of individual
muscarinic receptor subtypes in inflammation and remodeling. Here, we present evidence
that the potentiating effects of acetylcholine on airway inflammation and remodeling are
solely mediated via M3 receptors in animal models of both COPD (chapter 3) and asthma
(chapter 6).
COPD
In chapter 3, we investigated the contribution of individual muscarinic receptor subtypes
to cigarette smoke‐induced inflammation. Exposure to cigarette smoke results in
neutrophilic inflammation, as is observed in patients with COPD. From different animals
models of COPD, it was already known that neutrophilic inflammation, induced by
cigarette smoke or LPS, can be prevented by pretreatment with anticholinergics, including
tiotropium (12, 13), glycopyrrolate (14) and aclidinium (15). Here, we demonstrated that
the pro‐inflammatory effects of acetylcholine are mediated via the M3 receptor. Inhibition
of the M3 receptor, by total knock‐out of the receptor or by a pharmacological approach
using the M3 antagonist 4‐DAMP, prevented the inflammatory response induced by
cigarette smoke. Thus, whole body exposure of wild‐type animals for four days to an
increasing number of cigarettes induced neutrophilic inflammation in the airways of wild‐
type animals, which was accompanied by an increase in the release of pro‐inflammatory
cytokines, including IL‐8. This inflammatory response was almost completely prevented by
inhibition of the M3 receptor, suggesting that acetylcholine potentiates airway
inflammation via this receptor subtype. This is further supported by the fact that the
inflammatory response was enhanced after knock‐out of the M2 receptor. Loss of this
autoinhibitory receptor results in enhanced acetylcholine release. With acetylcholine
acting as a pro‐inflammatory mediator, increased levels of acetylcholine in M2 receptor
knock‐out (M2R‐/‐) animals can explain the observed aggravated inflammatory response.
Surprisingly, inflammation was also enhanced after knock‐out of the M1 receptor. This

Chapter 9
178
might be explained via the role of the M1 receptor on the airway epithelium, where it
contributes to mucus production, by controlling electrolyte and water secretion. Impaired
clearance of detrimental smoke particles from the airways after knock‐out of the M1
receptor could therefore underlie the enhanced inflammatory response observed in M1R‐/‐
animals. This is further supported by a marked induction in the release of the cytokines IL‐
6 and MCP‐1 in M1R‐/‐ animals compared to wild‐type animals. Taken together, the results
described in chapter 3 indicate a pro‐inflammatory role for acetylcholine via M3 receptors.
The fact that acetylcholine has a pro‐inflammatory effect on neutrophilic inflammation via
M3 receptors is supported by various in vitro studies. Muscarinic receptor stimulation
induced the release of IL‐6 and IL‐8 from airway smooth muscle cells, which has been
shown to be mediated via M3 receptors, and not via M2 receptors (16). Moreover,
tiotropium and the M3 selective antagonists 4‐DAMP and DAU5884 inhibited
methacholine and cigarette smoke‐induced IL‐8 release from airway smooth muscle cells,
whereas there was no effect of the M2 antagonist gallamine (16). Furthermore,
acetylcholine can induce IL‐8 release from bronchial epithelial cells, which can be inhibited
by tiotropium and 4‐DAMP, and not by the M1 antagonist telenzepine or the M2
antagonist gallamine (17). In addition, acetylcholine‐induced neutrophil chemotactic
activity from macrophages can be inhibited by 4‐DAMP, and not by the M1 antagonist
pirenzepine or the M2 antagonist gallamine (18). More recently, this was confirmed in
macrophages from COPD patients. Thus, neutrophil chemotaxis induced by LPS‐activated
alveolar macrophages could be inhibited by tiotropium and 4‐DAMP, and not by
telenzepine, gallamine or tubocurarine (19). These in vitro findings, suggesting an
important role for the M3 receptor in inflammation, are now supported by conclusive in
vivo findings, confirming the pro‐inflammatory role of the M3 receptor.
M3 receptors are expressed on almost all cell types in the airways, including structural
cells, like airway smooth muscle cells and epithelial cells, as well as inflammatory cells, like
macrophages and neutrophils (table 1, chapter 1). As is evident from various in vitro
studies, both structural cells and inflammatory cells can contribute to the pro‐
inflammatory effects of acetylcholine. Studies using airway structural cells demonstrated
that muscarinic receptor stimulation can induce IL‐8 release from airway smooth muscle
cells and epithelial cells (16, 17). Moreover, acetylcholine can induce neutrophil and
monocyte chemotactic activity from bronchial epithelial cells (20), as well as neutrophil
migration by activating alveolar epithelial cells (21). In the latter study, acetylcholine also
induced neutrophil migration by activating monocytes and alveolar macrophages,
suggesting a direct effect of muscarinic receptor stimulation on inflammatory cells (21).
Similar effects of muscarinic receptor stimulation on macrophages were observed in the

General discussion and summary
179
studies described above (18, 19). Moreover, proliferation of macrophages and T‐cells has
also been shown to be regulated by muscarinic receptors (22, 23). Thus, both structural
and inflammatory cells can contribute to the pro‐inflammatory effects of acetylcholine
and it is not known whether this effect is primarily mediated via M3 receptors on
structural cells or via M3 receptors on inflammatory cells.
Therefore, in chapter 4, we investigated the contribution of the M3 receptor on structural
cells versus inflammatory cells to cigarette smoke‐induced inflammation. To distinguish
between both cell types, bone marrow chimeric mice were generated. Bone marrow cells
from M3R‐/‐ animals were transplanted into irradiated wild‐type animals, to investigate the
contribution of the M3 receptor on inflammatory cells, and bone marrow cells from wild‐
type animals were transplanted into irradiated M3R‐/‐ animals, to investigate the
contribution of the M3 receptor on structural cells. Irradiated and transplanted wild‐type
animals and non‐irradiated wild‐type animals were used as controls. Mice were exposed
to cigarette smoke, using a similar protocol as described in chapter 3. Exposure to
cigarette smoke induced neutrophilic inflammation in non‐irradiated and irradiated
control animals. Interestingly, wild‐type animals receiving M3R‐/‐ bone marrow cells
showed a similar increase in neutrophil number, suggesting that the M3 receptor on
inflammatory cells is not involved in the pro‐inflammatory effect of acetylcholine. In
contrast, no increase in the number of neutrophils was observed in M3R‐/‐ animals
receiving wild‐type bone marrow cells, suggesting a critical role for airway structural cells
in the pro‐inflammatory effect of acetylcholine. Surprisingly, the increase in the release of
IL‐8 was similar among all smoke‐exposed groups, both on mRNA and protein level. Thus,
although the neutrophil chemoattractant is present in the airways of M3R‐/‐ animals
receiving wild‐type bone marrow cells, no increase in neutrophil number is observed when
compared to control animals. This suggests that neutrophil adhesion might be altered
after knock‐out of the M3 receptor. Using a micro‐array analysis, fibrinogen α and CD177
were identified as genes which might be involved in this process. Both genes are recently
linked to neutrophil adhesion and transmigration (24‐27). Interestingly, fibrinogen α and
CD177 are both identified as inflammatory biomarkers for COPD, indicating the potential
relevance of both genes for COPD pathophysiology (28‐30). IL‐8 can be released from
structural cells, including epithelial cells, and from inflammatory cells, including
macrophages. Our results suggest that the release of IL‐8 from both structural cells and
inflammatory cells is mediated via M3 receptors, since IL‐8 release is prevented after total
knock‐out of the M3 receptor as demonstrated in chapter 3, but not after knock‐out of the
M3 receptor on structural cells or inflammatory cells only. Moreover, this indicates a role
for M3 receptors on inflammatory cells, and suggests that acetylcholine can induce the
release of IL‐8 from inflammatory cells via M3 receptors, as is described for macrophages

Chapter 9
180
in vitro (18, 19). However, knock‐out of the M3 receptor on inflammatory cells in not
sufficient to prevent neutrophilia, whereas knock‐out of the M3 receptor on structural
cells only is sufficient to prevent neutrophilia. This suggest that the contribution of the M3
receptor on inflammatory cells to the pro‐inflammatory effect of acetylcholine is only
small, and that this is mainly mediated via M3 receptors on structural cells. Taken
together, the results from chapter 4 highlight the important role of airway structural cells
in neutrophilic inflammation and the pathophysiology of COPD, and the involvement of
acetylcholine in this response.
Asthma
In chapter 6, we demonstrated that the effects of acetylcholine in response to allergen
exposure are also mediated via the M3 receptor. From animal models of asthma it was
already known that acetylcholine plays a role in allergen‐induced airway inflammation and
remodeling. Pretreatment of guinea pigs with tiotropium partly prevented airway smooth
muscle thickening, mucous gland hypertrophy and goblet cell metaplasia, as well as
eosinophilic inflammation in response to allergen exposure (31, 32). Similar findings were
observed in allergen‐exposed mice, in which tiotropium partly prevented features of
airway remodeling and airway inflammation. In addition to the findings in guinea pigs,
tiotropium was also shown to inhibit excessive extracellular matrix deposition and to
inhibit TH2 cytokine release in mice (33). To investigate the effects of individual muscarinic
receptor subtypes on allergen‐induced inflammation and remodeling, muscarinic receptor
subtype specific knock‐out animals were exposed to ovalbumin, and lungs were collected
for analysis of inflammation and remodeling. Allergen exposure induced features of
remodeling, including goblet cell metaplasia, airway smooth muscle thickening, pulmonary
vascular smooth muscle remodeling and enhanced deposition of extracellular matrix
proteins in the airway wall of wild‐type mice. These effects were absent or markedly lower
in M3R‐/‐ mice, whereas M1R
‐/‐ and M2R‐/‐ mice responded similar to wild‐type mice with
respect to remodeling. This suggests that the remodeling‐promoting effects of
acetylcholine in vivo in response to allergen exposure are solely mediated via M3
receptors, and not via M1 or M2 receptors.
Evidence from in vitro studies supports a role for acetylcholine in remodeling of the
airways. It is known that muscarinic receptor stimulation can induce MUC5AC expression
in epithelial cells, which can be inhibited by aclidinium (34). Moreover, tiotropium can
inhibit IL‐13‐induced goblet cell metaplasia, as demonstrated in chapter 8. Muscarinic
receptor stimulation is also shown to be involved in airway smooth muscle thickening and
airway fibrosis, as it can enhance growth factor‐induced proliferation (35, 36) contractile
protein expression (37), and extracellular matrix deposition of airway smooth muscle cells

General discussion and summary
181
and fibroblasts (38, 39). Furthermore, aclidinium has been shown to inhibit fibroblast to
myofibroblast transition (40). Although some of these in vitro studies point to an
important role for the M2 receptor in remodeling responses, we did not observe any
effects of knock‐out of the M2 receptor on allergen‐induced remodeling. It is known that
presynaptic M2 receptors on parasympathetic nerves are dysfunctional in asthmatic
airways. This has been shown in animal models of allergen exposure, viral infection and
ozone exposure, and also in patients with asthma (41‐43). Although this effect is less well
described for postsynaptic M2 receptors, dysfunction of M2 receptors after allergen
challenge might explain why we did not observe any effects of knock‐out of the M2
receptor. M2‐mediated responses observed in vitro might therefore be less relevant to the
in vivo situation. Moreover, M2 receptor dysfunction in response to allergen might also
explain why there is no effect of M2 receptor knock‐out on allergen‐induced inflammation,
whereas there is an effect of M2 receptor knock‐out on cigarette smoke‐induced
inflammation. Furthermore, we did not observe any effects of knock‐out of the M1
receptor on allergen‐induced airway inflammation or remodeling. Whereas in the
cigarette smoke model used in chapter 3, detrimental smoke particles had to be cleared
from the airways in which the M1 receptor might be involved, this effect is likely less
pronounced after inhalation of an allergen. Together, this might explain why there is no
role for M1 or M2 receptors in allergen‐induced airway remodeling, which is shown to be
solely mediated by M3 receptors in vivo.
Next to its role in airway remodeling, the M3 receptor also seems to be involved in basal
maintenance of airway structure. In chapter 3, we observed a decrease in basal gene
expression of TGF‐β1 and the extracellular matrix proteins collagen Iα1 and fibronectin in
lung homogenates of M3R‐/‐ mice compared to WT mice. Moreover, in chapter 6, we
observed a decrease in basal levels of α‐sm‐actin in the airways and arteries of M3R‐/‐ mice
compared to WT mice. This may suggest that cholinergic regulation of airway tone, which
is almost completely absent in M3R‐/‐ mice, is important for the development and/or
maintenance of airway structure, including the airway smooth muscle and the
extracellular matrix.

Chapter 9
182
Bronchoconstriction as a driver of airway remodeling
Lack of bronchoconstriction in M3R‐/‐ mice might also have other, far‐reaching
consequences in our model. We observed a marked inhibition of airway remodeling in
M3R‐/‐ mice compared to wild‐type mice, without inhibition of the allergic inflammatory
response (chapter 6). Generally, airway structural changes after allergen challenge are
attributed to eosinophilic inflammation (44), and tiotropium has previously been shown to
inhibit eosinophilic inflammation in animal models of asthma (32, 33). The observation
that there is no inhibition of the eosinophilic inflammation after knock‐out of the M3
receptor might be explained by the fact that this is orchestrated by both M1 and M3
receptors. Tiotropium also has a substantial dissociation half‐life for the M1 receptor (45),
and we observed a trend towards lower eosinophil numbers in M1R‐/‐ mice. However, the
finding that remodeling can be inhibited after knock out of the M3 receptor, without
affecting inflammation, has significant implications. Recently, Grainge et al. demonstrated
that repeated methacholine challenges in mild asthmatic patients is sufficient to induce
airway remodeling, without affecting inflammation. Thus, methacholine challenge
promoted TGF‐β release, collagen deposition, goblet cell metaplasia and epithelial cell
proliferation in airway biopsies, with no effect on eosinophil numbers (46). Interestingly,
these effects on airway remodeling were similar to those induced by house dust mite,
administered in an equi‐effective dose with respect to bronchoconstriction but also
causing eosinophilic inflammation. Moreover, effects on remodeling were prevented
when methacholine was administered together with the β2‐agonist albuterol, to prevent
methacholine‐induced bronchoconstriction (46). These findings, together with our
observations from chapter 6, raise the hypothesis that bronchoconstriction by itself might
be sufficient to induce airway remodeling, and that the origin of airway remodeling is not
inflammatory, but mechanical in nature.
Therefore, in chapter 7, we aimed to investigate the effects of bronchoconstriction on
airway remodeling, and the involvement of the M3 receptor in this response, by using
mouse precision cut lung slices (PCLS). PCLS are a valuable tool to study
bronchoconstriction‐induced remodeling, since all lung cell types are present and cell‐cell
contacts and cell‐matrix interactions are preserved, in contrast to cell cultures of single
airway cell types. This was confirmed previously by our lab using guinea pig PCLS (47). In
this study, Oenema et al. demonstrated that the effects of bronchoconstriction induced by
methacholine on airway remodeling were similar to those of TGF‐β, a pleiotropic cytokine
known to be an important mediator of airway remodeling. Thus, stimulation of guinea pig
PCLS for two days with TGF‐β induced features of airway remodeling, including enhanced
contractile protein expression, as assessed by Western Blot analysis and

General discussion and summary
183
immunohistochemistry. Interestingly, stimulation of guinea pig PCLS for two days with
methacholine had similar effects on airway remodeling, which was shown to be mediated
via enhanced release of TGF‐β (47). In chapter 7, we aimed to investigate the involvement
of the M3 receptor in this response, by exposing PCLS from wild‐type and M3R‐/‐ mice to
TGF‐β and methacholine. First, we confirmed that bronchoconstriction in response to
methacholine is almost completely abolished in M3R‐/‐ mice, which is in line with the
literature (48, 49). Next, PCLS from wild‐type and M3R‐/‐ mice were exposed to TGF‐β and
methacholine for two days. Exposure of PCLS from wild‐type mice to TGF‐β induced an
increase in the expression of the contractile protein α‐sm‐actin and the extracellular
matrix proteins fibronectin and collagen type I. A similar increase in TGF‐β‐induced airway
remodeling was observed in M3R‐/‐ mice, suggesting that the muscarinic receptor
regulation of airway remodeling occurs upstream of TGF‐β signaling. In contrast to
findings in guinea pigs PCLS, exposure to methacholine did not induce airway remodeling
in murine PCLS. Species differences might underlie the differences observed in guinea pig
PCLS versus murine PCLS. Whereas airway constriction in guinea pig PCLS is maintained
over days in the presence of methacholine, unpublished observations from our lab
indicate that the methacholine‐induced constriction is rapidly lost in murine PCLS.
Moreover, the contraction rate in response to methacholine is also greater in guinea pig
PCLS compared to murine PCLS. Further studies, using different experimental approaches,
are therefore needed to fully elucidate the effect of M3 receptor activation on airway
remodeling. Next, we analyzed whether TGF‐β‐induced remodeling altered the airway
contractility. We did not observe any functional effects of exposure of PCLS to TGF‐β or
methacholine, since contraction against the thromboxane A2 agonist U46619 was not
altered in wild‐type or M3R‐/‐ mice after stimulation for two days. Thus, despite clear
effects of TGF‐β on the expression of fibronectin, collagen Iα1 and α‐sm‐actin, this did not
translate into changes in airway contractility, suggesting that it is difficult to demonstrate
a direct link between airway remodeling and airway contractility using lung slices.
The hypothesis that bronchoconstriction by itself might be sufficient to induce
remodeling, independent of the inflammatory response, is still ongoing, and an increasing
number of studies support this hypothesis. In vitro, it has been shown that muscarinic
receptor stimulation, in combination with mechanical strain, can induce α‐sm‐actin and
sm‐myosin mRNA expression in bovine tracheal smooth muscle strips, and myosin light‐
chain kinase expression in human airway smooth muscle cells (50, 51). Moreover,
contraction of airway smooth muscle cells induces TGF‐β activation (52). In intact airways,
the epithelium is compressed by bronchoconstriction, and this induces the activation of
epidermal growth factor (EGF) (53). Compression of epithelial cells in vitro results in
enhanced TGF‐β expression (54) and an increase in epithelial thickness (55). Moreover,

Chapter 9
184
mechanical stress applied to the epithelium has been shown to increase the expression of
fibronectin, collagen and matrix metalloproteinase type 9 (MMP‐9) in airway fibroblasts in
a co‐culture system (56). Taken together, the studies outlined above suggest that
bronchoconstriction may lead to airway remodeling in asthma via the release of growth
factors from epithelial and smooth muscle cells in response to mechanical stress. This is
important from a therapeutic perspective, as this would imply that prevention of
bronchoconstriction, probably already in an early stage of disease, might prevent airway
remodeling in asthma.
Neuronal and non‐neuronal acetylcholine
In the last decades, it has become clear that acetylcholine is not only released as a
neurotransmitter from nerve terminals, but also as a hormone from non‐neuronal cells,
including cells in the airways, acting in an autocrine and/or paracrine manner. However, it
is not known to which extent the pro‐inflammatory and remodeling‐promoting effects of
acetylcholine in the airways as discussed above are mediated by neuronal or by non‐
neuronal acetylcholine. It has been suggested that non‐neuronal acetylcholine contributes
to these effects, however, evidence for such a role is still limited (chapter 2, 57, 58). In this
thesis, we present evidence that both neuronal (chapter 5) and non‐neuronal (chapter 8)
acetylcholine contribute to airway inflammation and remodeling in COPD and asthma.
COPD
In chapter 5, we investigated the effect of targeted lung denervation (TLD) on
inflammation in patients with COPD. TLD is a novel potential therapy for patients with
COPD, in which parasympathetic airway nerves are ablated by locally applying
radiofrequency energy in the main bronchi using bronchoscopy. Inhibition of acetylcholine
by ablating airway nerves is expected to inhibit bronchoconstriction and the first
experimental data support this notion. In a small group of patients, TLD was shown to
increase FEV1, 6 minute walk‐test distance and the St. George’s Respiratory Questionnaire
score (59). We hypothesized that TLD would also inhibit airway inflammation. We
demonstrated that 30 days after TLD of the right lung, airway inflammation is attenuated.
TLD resulted in a reduction of inflammatory cells and cytokine release in the bronchial
wash, and a reduction in gene expression of inflammatory mediators, including the
expression of IL‐6, IL‐8, TGF‐β and MUC5AC, in bronchial brush specimen. This is the first
study reporting a direct inhibitory effect of acetylcholine on inflammation in patients with
COPD and suggests that this is mediated by neuronally released acetylcholine. The fact
that TGF‐β levels were significantly reduced after TLD suggests that TLD might also affect
remodeling in COPD. This implies that airway remodeling in COPD is also mediated by

General discussion and summary
185
neuronal acetylcholine. However, evidence for such a role is still limited, and clearly
further studies are needed to elucidate the role of neuronal versus non‐neuronal
acetylcholine in airway remodeling in COPD.
A role for neuronal acetylcholine in inflammation in COPD is further supported by the
results from chapter 3, in which the cigarette smoke‐induced inflammatory response was
enhanced in M2R‐/‐ mice compared to wild‐type mice. Knock‐out of prejunctional
autoinhibitory M2 receptors results in enhanced acetylcholine release. With acetylcholine
acting as a pro‐inflammatory mediator, knock‐out of the autoinhibitory receptors
enhances the inflammatory response. A similar autoinhibitory mechanism for the release
of non‐neuronal acetylcholine has not been demonstrated until now, thereby suggesting
that neuronally released acetylcholine is the main driver of this inflammatory response.
Furthermore, we did not observe any regulation of expression of components of the non‐
neuronal cholinergic system after exposure to cigarette smoke. Moreover, the importance
of structural cells over inflammatory cells in cigarette smoke‐induced inflammation, as
observed in chapter 4, might further support a role for neuronal acetylcholine in airway
inflammation in COPD.
Asthma
The hypothesis that bronchoconstriction might be the driver of allergen‐induced airway
remodeling suggests that also in allergic asthma, neuronal acetylcholine plays an
important role. Moreover, we did not observe any regulation of expression of components
of the non‐neuronal cholinergic system after allergen challenge. Thereby, we cannot
confirm a role for non‐neuronal acetylcholine in allergen‐induced remodeling (chapter 6).
However, we did observe a clear role for non‐neuronal acetylcholine in goblet cell
metaplasia in chapter 8. In this chapter, we investigated the direct effects of endogenous
non‐neuronal acetylcholine on epithelial cell differentiation. It was already known that
tiotropium can inhibit allergen‐induced goblet cell metaplasia and MUC5AC expression in
vivo (32, 33), an effect also observed after knock‐out of the M3 receptor in chapter 6.
Epithelial cells have been shown to produce and release acetylcholine (60). Therefore, the
observed effects on epithelial cell differentiation might be mediated by neuronal
acetylcholine, or by a direct effect of non‐neuronal acetylcholine on the airway
epithelium. To investigate the latter hypothesis, human airway epithelial (HAE) cells were
cultured at an air‐liquid‐interface (ALI) and exposed to tiotropium and/or IL‐13 during
differentiation. IL‐13 is a main driver of goblet cell metaplasia, and plays a central role in
the pathogenesis of asthma (61). Epithelial cells used for this study expressed all
components of the non‐neuronal cholinergic system, suggesting production of
acetylcholine by these cells. Previously, it has been shown that epithelial cells can indeed

Chapter 9
186
release acetylcholine (60). Interestingly, carnitine acetyltransferase (CarAT) was identified
as the synthesizing enzyme of acetylcholine. Tiotropium had no effects on epithelial cell
differentiation following air exposure, however, significantly inhibited IL‐13‐induced
goblet cell metaplasia. Tiotropium inhibited the increase in MUC5AC positive cells and
goblet cells, and the increase in MUC5AC gene expression. Moreover, we demonstrated
that the effects of tiotropium might be mediated via effects on the transcription factors
FoxA2 and FoxA3, whereas increased expression of SPDEF by IL‐13 was not affected by
tiotropium. This indicates that non‐neuronal acetylcholine contributes to goblet cell
metaplasia by a direct effect on epithelial cells. In the airways, mucus is secreted by goblet
cells and submucosal glands. Submucosal glands are innervated by airway nerves (chapter
1) and the release of mucus from glands is under cholinergic neural control (62). It is still a
matter debate whether goblet cells can also release mucus in response to neuronal
acetylcholine, but data from our study suggest at least a role for non‐neuronal
acetylcholine in this response.
Based on the in vivo studies described in this thesis, I propose that the potentiating effects
of acetylcholine on airway inflammation and remodeling are mainly mediated by
neuronally released acetylcholine. However, a number of in vitro studies, including the
study described in chapter 8, indicate that non‐neuronal acetylcholine might contribute to
the effects of acetylcholine. Tiotropium and 4‐DAMP have been shown to inhibit alveolar
macrophage mediated migration of neutrophils from COPD patients (19). Moreover,
tiotropium inhibited TGF‐β‐induced MMP‐1 and MMP‐2 expression in human lung
fibroblasts (63), and aclidinium has been shown to inhibit TGF‐β and cigarette smoke‐
induced fibroblast to myofibroblast differentiation (40, 64). These studies indicate that
non‐neuronal acetylcholine might contribute to airway inflammation and remodeling of
airways cells in an autocrine or paracrine manner. However, the relative contribution of
these effects to the effects of neuronal acetylcholine in vivo is still uncertain. Vagotomy of
animals under controlled experimental conditions might definitely establish the
contribution of neuronal versus non‐neuronal acetylcholine to inflammation and
remodeling in disease models. Bilateral vagotomy is, however, not possible in the mouse
(unpublished observations). In the literature, bilateral vagotomy of bigger laboratory
animals has been reported, and this might be a way to answer this question. Moreover,
targeted lung denervation in patients with asthma can answer whether neuronal
acetylcholine is also the main contributor to airway inflammation in asthma.

General discussion and summary
187
Clinical implications
COPD
Evidence for a role of acetylcholine as a driver of airway inflammation and remodeling in
patients with COPD or asthma is still limited. In this thesis, we demonstrate for the first
time that acetylcholine might act as a pro‐inflammatory mediator in patients with COPD,
since airway inflammation is attenuated after TLD (chapter 5). There are some limitations
to this study, of which the major limitations are the limited number of subjects included in
the study and the lack of a control arm. However, we believe that the findings from this
small study are very promising. A large‐scale, multicenter, sham‐controlled study into the
effectiveness of TLD is planned and this will provide more conclusive evidence on the role
of neuronal acetylcholine as a pro‐inflammatory mediator. There is no additional evidence
until now that demonstrates direct effects of acetylcholine on inflammation in patients
with COPD. From different trials, including the UPLIFT (Understanding Potential Long‐term
Impacts on Function with Tiotropium) trial, it is known that tiotropium reduces the
number of exacerbations (65). Treatment with glycopyrrolate or aclidinium, albeit for a
shorter period, was also shown to affect exacerbations, as the time to the first
exacerbation was increased (66, 67). This might suggest an anti‐inflammatory effect of
anticholinergic therapy, since the inflammatory response is enhanced during
exacerbations, with increased expression of pro‐inflammatory cytokines like IL‐8 (68, 69).
Moreover, patients who have more exacerbations demonstrate increased levels of
inflammatory markers at stable state (70, 71). Until now, methodological problems
complicated the evaluation of airway inflammation in drug studies. In a study by Powrie et
al., treatment with anticholinergics reduced the amount of sputum, which might result in
increased cytokine concentrations in the sputum. This has been suggested to explain why
no reduction in IL‐6 or IL‐8 sputum levels was observed in patients with COPD after
tiotropium treatment (72, 73). In chapter 5, we used a bronchoscopic intervention, which
enabled us to collect a bronchial wash and brush and examine the effects of acetylcholine
on airway inflammation in a direct manner. Here, we demonstrate that acetylcholine
might indeed affect airway inflammation in patients with COPD, as is expected from the
increasing body of evidence from in vitro and in vivo studies reviewed in chapter 2, and
from the studies described in this thesis. Moreover, TLD reduced the levels of TGF‐β,
suggesting effects of acetylcholine on airway remodeling in patients with COPD. In the
overall study population of the UPLIFT trial, tiotropium did not affect the rate of decline in
lung function (65). However, tiotropium did inhibit the accelerated decline in lung function
in specific subgroups of the trial, including young patients and patients with moderate
disease (74, 75). Interestingly, young patients are also highly represented in the TLD study.
Clearly, future studies are needed to understand the effects of acetylcholine on airway

Chapter 9
188
remodeling in patients with COPD. Accelerated decline in lung function might not be the
optimal outcome parameter for future studies to analyze remodeling, since it is known
from the ECLIPSE study that this is highly variable between patients with COPD, and lung
function might even increase in a subset of patients, irrespective of treatment (76).
Instead, more direct measurements of remodeling parameters, using airway biopsies from
patients, taken before and after long‐term anticholinergic therapy, either after TLD or
anticholinergic treatment, might help to elucidate the role of acetylcholine in airway
remodeling in patients with COPD.
Asthma
Anticholinergics are currently not included as controller therapy for the treatment of
asthma. However, recent trials suggest that patients with asthma might benefit from
anticholinergic controller therapy, since tiotropium has been shown to induce
bronchodilation in moderate and severe asthma patients (10, 11). In severe asthma
patients, addition of tiotropium to standard therapy with inhaled glucocorticosteroids and
long‐acting β2‐agonists did not only induce bronchodilation, but was also shown to
increase the time to the first exacerbation, and reduce the risk of a severe exacerbation
(10). This suggests that acetylcholine also exerts pro‐inflammatory effects in patients with
asthma. However, direct evidence for such a role is still lacking. Moreover, long‐term
studies into the effects of anticholinergic therapy on airway remodeling in asthma are
needed to confirm the remodeling‐promoting effects of acetylcholine as described in this
thesis. Repeated challenges with methacholine in mild asthma patients did demonstrate
that muscarinic receptor stimulation can induce airway remodeling (46). Moreover,
methacholine challenge induced epithelial cell proliferation and goblet cell metaplasia,
suggesting a role for acetylcholine in mucus hypersecretion. This is supported by results
from chapter 8, in which tiotropium inhibited goblet cell metaplasia of human airway
epithelial cells. Tiotropium has been shown to reduce sputum levels in patients with
chronic mucus hypersecretion (77). Although the concern has been expressed that
anticholinergics desiccate mucus, thereby increasing the viscosity and making the mucus
more difficult to clear, there is now some data to support that anticholinergics are
beneficial for patients with mucus hypersecretion (78). This is relevant for both COPD and
asthma, in which mucus hypersecretion can occur, which contributes to airflow
obstruction of the smaller airways and increases the risk of exacerbations.

General discussion and summary
189
M3 selective anticholinergics
The studies described in this thesis point to a critical regulatory role for the M3 receptor in
airway inflammation and remodeling. Although current anticholinergics are kinetically
selective for the M3 receptor, they also have a substantial dissociation half‐life from the
M1 receptor (table 2, chapter 1). For example, dissociation half‐life of tiotropium from the
M1 receptor is 10.5 hours (45). Our studies suggest that an even more selective muscarinic
antagonist, solely inhibiting M3 receptors, is desirable and may lead to improved effects
on airway inflammation and remodeling. Inhibition of acetylcholine release by TLD might
also further enhance outcomes on inflammation and remodeling, however, this does not
inhibit non‐neuronal acetylcholine release.
Conclusions
In conclusion, the described studies have revealed that acetylcholine contributes to airway
inflammation and remodeling in COPD and asthma, which is mediated via M3 receptors.
This involves neuronally released acetylcholine, but also non‐neuronal acetylcholine,
which was shown to contribute to goblet cell metaplasia (figure 1). Together, this suggests
that patients with COPD or asthma might benefit from anticholinergic therapy to a much
larger extent than previously appreciated. Moreover, as bronchodilators, anticholinergics
might also affect airway remodeling by preventing mechanical stress. Therefore, I believe
that the combined effects of anticholinergic therapy on bronchoconstriction, mucus
secretion, inflammation and remodeling may together lead to a positive outcome for
patients with COPD or asthma.
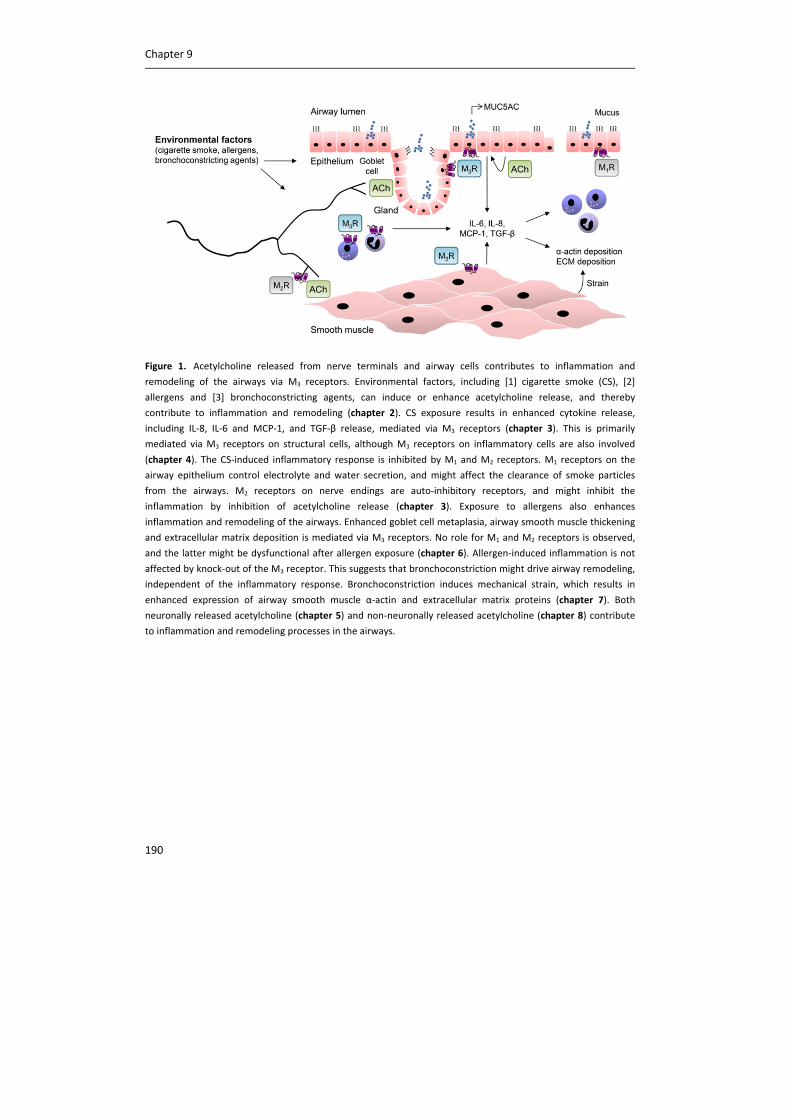
Chapter 9
190
Figure 1. Acetylcholine released from nerve terminals and airway cells contributes to inflammation and
remodeling of the airways via M3 receptors. Environmental factors, including [1] cigarette smoke (CS), [2]
allergens and [3] bronchoconstricting agents, can induce or enhance acetylcholine release, and thereby
contribute to inflammation and remodeling (chapter 2). CS exposure results in enhanced cytokine release,
including IL‐8, IL‐6 and MCP‐1, and TGF‐β release, mediated via M3 receptors (chapter 3). This is primarily
mediated via M3 receptors on structural cells, although M3 receptors on inflammatory cells are also involved
(chapter 4). The CS‐induced inflammatory response is inhibited by M1 and M2 receptors. M1 receptors on the
airway epithelium control electrolyte and water secretion, and might affect the clearance of smoke particles
from the airways. M2 receptors on nerve endings are auto‐inhibitory receptors, and might inhibit the
inflammation by inhibition of acetylcholine release (chapter 3). Exposure to allergens also enhances
inflammation and remodeling of the airways. Enhanced goblet cell metaplasia, airway smooth muscle thickening
and extracellular matrix deposition is mediated via M3 receptors. No role for M1 and M2 receptors is observed,
and the latter might be dysfunctional after allergen exposure (chapter 6). Allergen‐induced inflammation is not
affected by knock‐out of the M3 receptor. This suggests that bronchoconstriction might drive airway remodeling,
independent of the inflammatory response. Bronchoconstriction induces mechanical strain, which results in
enhanced expression of airway smooth muscle α‐actin and extracellular matrix proteins (chapter 7). Both
neuronally released acetylcholine (chapter 5) and non‐neuronally released acetylcholine (chapter 8) contribute
to inflammation and remodeling processes in the airways.

General discussion and summary
191
Taken together, the studies described in this thesis have revealed that:
The neurotransmitter acetylcholine is not only involved in bronchoconstriction
and mucus secretion in the airways, but also contributes to airway inflammation
and remodeling (chapter 2). Like the effects on bronchoconstriction and mucus
secretion, effects on inflammation and remodeling are primarily mediated via M3
receptors (chapter 3 and chapter 6).
The M3 receptor plays a pro‐inflammatory role in cigarette smoke‐induced
inflammation, whereas M1 and M2 receptors exert an anti‐inflammatory effect
(chapter 3). This suggests that M3 selective antagonists can reduce neutrophilia.
The pro‐inflammatory effect of acetylcholine on cigarette smoke‐induced
inflammation is primarily mediated via M3 receptors located on structural cells
(chapter 4).
The M3 receptor contributes to allergen‐induced airway remodeling, whereas no
role for M3 receptors in allergen‐induced inflammation is observed (chapter 6).
This suggests that bronchoconstriction might drive airway remodeling,
independent of the inflammatory response (chapter 6 and chapter 7).
M1 and M2 receptors are not involved in allergen‐induced inflammation and
remodeling (chapter 6).
Targeted lung denervation attenuates inflammation in patients with COPD,
providing the first direct evidence that acetylcholine might affect inflammation in
humans. This implies an important role for neuronal acetylcholine in the
inflammatory response (chapter 5).
Tiotropium prevents IL‐13‐induced goblet cell metaplasia of human airway
epithelial cells, which indicates that non‐neuronal acetylcholine contributes to
epithelial cell differentiation (chapter 8).
Patients with COPD and asthma may benefit from anticholinergic therapy to a
much broader extent than previously appreciated, since anticholinergic therapy
might also inhibit inflammation and remodeling of the airways. This can be
achieved via TLD or via anticholinergic treatment. Treatment with an even more
selective muscarinic antagonist, solely inhibiting M3 receptors, might further
enhance this beneficial effect (this thesis).

Chapter 9
192
References
(1) Ferkol T, Schraufnagel D. The global burden of respiratory disease. Ann Am Thorac Soc
2014;11:404‐406.
(2) Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis,
Management and Prevention of COPD (GOLD) 2013. Available from:
http://www.goldcopd.org/uploads/users/files/GOLD_Report_2014_Jan23.pdf. ;June 4, 2014.
(3) Global Initiative for Asthma (GINA). GINA Report, Global Burden of Asthma. Available from:
www.ginasthma.org. May, 2004;June 4, 2014.
(4) Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med
2001;164:S28‐S38.
(5) Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez‐
Roisin R, van Weel C, Zielinski J, Global Initiative for Chronic Obstructive Lung Disease. Global
strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease:
GOLD executive summary. Am J Respir Crit Care Med 2007;176:532‐555.
(6) Pare PD, Roberts CR, Bai TR, Wiggs BJ. The functional consequences of airway remodeling in
asthma. Monaldi Arch Chest Dis 1997;52:589‐596.
(7) Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to
emphysema. N Engl J Med 1984;311:421‐425.
(8) Barnes PJ. The role of anticholinergics in chronic obstructive pulmonary disease. Am J Med
2004;117 Suppl 12A:24S‐32S.
(9) Global Initiative for Asthma (GINA). GINA Report, Global Strategy for Asthma Management and
Prevention. Available from: www.ginasthma.org. 2014;June 4, 2014.
(10) Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, Sigmund R, Seibold W,
Moroni‐Zentgraf P, Bateman ED. Tiotropium in asthma poorly controlled with standard combination
therapy. N Engl J Med 2012;367:1198‐1207.
(11) Beeh KM, Moroni‐Zentgraf P, Ablinger O, Hollaenderova Z, Unseld A, Engel M, Korn S.
Tiotropium Respimat(R) in asthma: a double‐blind, randomised, dose‐ranging study in adult patients
with moderate asthma. Respir Res 2014;15:61.
(12) Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs
H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur
Respir J 2011;38:789‐796.
(13) Wollin L, Pieper MP. Tiotropium bromide exerts anti‐inflammatory activity in a cigarette smoke
mouse model of COPD. Pulm Pharmacol Ther 2010;23:345‐354.
(14) Shen LL, Liu YN, Shen HJ, Wen C, Jia YL, Dong XW, Jin F, Chen XP, Sun Y, Xie QM. Inhalation of
glycopyrronium inhibits cigarette smoke‐induced acute lung inflammation in a murine model of
COPD. Int Immunopharmacol 2014;18:358‐364.
(15) Dominguez‐Fandos D, Ferrer E, Puig‐Pey R, Carreno C, Prats N, Aparici M, Musri MM, Gavalda A,
Peinado VI, Miralpeix M, Barbera JA. Effects of aclidinium bromide in a cigarette smoke‐exposed
Guinea pig model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2014;50:337‐
346.

General discussion and summary
193
(16) Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J, Kolahian S, Halayko AJ, Hiemstra
PS, Zuyderduyn S. Muscarinic M3 receptor stimulation increases cigarette smoke‐induced IL‐8
secretion by human airway smooth muscle cells. Eur Respir J 2009;34:1436‐1443.
(17) Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP,
Gjomarkaj M. Acetylcholine mediates the release of IL‐8 in human bronchial epithelial cells by a
NFkB/ERK‐dependent mechanism. Eur J Pharmacol 2008;582:145‐153.
(18) Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages
to release inflammatory cell chemotactic activity. Am J Physiol 1998;274:L970‐L979.
(19) Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide.
Respir Res 2011;12:24.
(20) Koyama S, Rennard SI, Robbins RA. Acetylcholine stimulates bronchial epithelial cells to release
neutrophil and monocyte chemotactic activity. Am J Physiol 1992;262:L466‐L471.
(21) Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine‐
induced release of chemotactic mediators in vitro. Respir Med 2007;101:2386‐2394.
(22) de la Torre E, Genaro AM, Ribeiro ML, Pagotto R, Pignataro OP, Sales ME. Proliferative actions
of muscarinic receptors expressed in macrophages derived from normal and tumor bearing mice.
Biochim Biophys Acta 2008;1782:82‐89.
(23) Razani‐Boroujerdi S, Behl M, Hahn FF, Pena‐Philippides JC, Hutt J, Sopori ML. Role of muscarinic
receptors in the regulation of immune and inflammatory responses. J Neuroimmunol 2008;194:83‐
88.
(24) de Almeida VV, Silva‐Herdade A, Calado A, Rosario HS, Saldanha C. Fibrinogen modulates
leukocyte recruitment in vivo during the acute inflammatory response. Clin Hemorheol Microcirc
2012.
(25) Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte
engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac‐1 is critical for host
inflammatory response in vivo. J Clin Invest 2004;113:1596‐1606.
(26) Sachs UJ, Andrei‐Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ,
Preissner KT, Chavakis T, Santoso S. The neutrophil‐specific antigen CD177 is a counter‐receptor for
platelet endothelial cell adhesion molecule‐1 (CD31). J Biol Chem 2007;282:23603‐23612.
(27) Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 contributes to transendothelial
migration of NB1‐positive neutrophils. J Immunol 2012;188:2419‐2426.
(28) Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal‐Singer R, Evaluation of COPD
Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. COPD association and
repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res 2011;12:146‐9921‐12‐146.
(29) Celli BR, Locantore N, Yates J, Tal‐Singer R, Miller BE, Bakke P, Calverley P, Coxson H, Crim C,
Edwards LD, Lomas DA, Duvoix A, MacNee W, Rennard S, Silverman E, Vestbo J, Wouters E, Agusti A,
ECLIPSE Investigators. Inflammatory biomarkers improve clinical prediction of mortality in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med 2012;185:1065‐1072.
(30) Hicks A, Kourteva G, Hilton H, Li H, Lin TA, Liao W, Li Y, Wei X, March T, Benson J, Renzetti LM.
Cellular and molecular characterization of ozone‐induced pulmonary inflammation in the
Cynomolgus monkey. Inflammation 2010;33:144‐156.

Chapter 9
194
(31) Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the
progression of airway smooth muscle remodeling. Am J Respir Crit Care Med 2005;171:1096‐1102.
(32) Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of
allergen‐induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J
2007;30:653‐661.
(33) Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T,
Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling
in a mouse model of asthma. Clin Exp Allergy 2010;40:1266‐1275.
(34) Cortijo J, Mata M, Milara J, Donet E, Gavalda A, Miralpeix M, Morcillo EJ. Aclidinium inhibits
cholinergic and tobacco smoke‐induced MUC5AC in human airways. Eur Respir J 2011;37:244‐254.
(35) Gosens R, Dueck G, Rector E, Nunes RO, Gerthoffer WT, Unruh H, Zaagsma J, Meurs H, Halayko
AJ. Cooperative regulation of GSK‐3 by muscarinic and PDGF receptors is associated with airway
myocyte proliferation. Am J Physiol Lung Cell Mol Physiol 2007;293:L1348‐L1358.
(36) Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C, Juergens UR, Racke
K. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell
Mol Biol 2006;35:621‐627.
(37) Oenema TA, Smit M, Smedinga L, Racke K, Halayko AJ, Meurs H, Gosens R. Muscarinic receptor
stimulation augments TGF‐beta1‐induced contractile protein expression by airway smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol 2012;303:L589‐97.
(38) Oenema TA, Mensink G, Smedinga L, Halayko AJ, Zaagsma J, Meurs H, Gosens R, Dekkers BG.
Cross‐talk between transforming growth factor‐beta(1) and muscarinic M(2) receptors augments
airway smooth muscle proliferation. Am J Respir Cell Mol Biol 2013;49:18‐27.
(39) Haag S, Matthiesen S, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of
collagen synthesis in human lung fibroblasts. Eur Respir J 2008;32:555‐562.
(40) Milara J, Serrano A, Peiro T, Gavalda A, Miralpeix M, Morcillo EJ, Cortijo J. Aclidinium inhibits
human lung fibroblast to myofibroblast transition. Thorax 2012;67:229‐237.
(41) ten Berge RE, Santing RE, Hamstra JJ, Roffel AF, Zaagsma J. Dysfunction of muscarinic M2
receptors after the early allergic reaction: possible contribution to bronchial hyperresponsiveness in
allergic guinea‐pigs. Br J Pharmacol 1995;114:881‐887.
(42) Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits
reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol (1985)
1989;67:2461‐2465.
(43) Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology
of asthma and COPD. Respir.Res. 2006;7:73.
(44) Kelly MM, O'Connor TM, Leigh R, Otis J, Gwozd C, Gauvreau GM, Gauldie J, O'Byrne PM. Effects
of budesonide and formoterol on allergen‐induced airway responses, inflammation, and airway
remodeling in asthma. J Allergy Clin Immunol 2010;125:349‐356.e13.
(45) Casarosa P, Bouyssou T, Germeyer S, Schnapp A, Gantner F, Pieper M. Preclinical evaluation of
long‐acting muscarinic antagonists: comparison of tiotropium and investigational drugs. J Pharmacol
Exp Ther 2009;330:660‐668.
(46) Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH.
Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med 2011;364:2006‐2015.

General discussion and summary
195
(47) Oenema TA, Maarsingh H, Smit M, Groothuis GM, Meurs H, Gosens R. Bronchoconstriction
Induces TGF‐beta Release and Airway Remodelling in Guinea Pig Lung Slices. PLoS One
2013;8:e65580.
(48) Fisher JT, Vincent SG, Gomeza J, Yamada M, Wess J. Loss of vagally mediated bradycardia and
bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J
2004;18:711‐713.
(49) Struckmann N, Schwering S, Wiegand S, Gschnell A, Yamada M, Kummer W, Wess J,
Haberberger RV. Role of muscarinic receptor subtypes in the constriction of peripheral airways:
studies on receptor‐deficient mice. Mol Pharmacol 2003;64:1444‐1451.
(50) Fairbank NJ, Connolly SC, Mackinnon JD, Wehry K, Deng L, Maksym GN. Airway smooth muscle
cell tone amplifies contractile function in the presence of chronic cyclic strain. Am J Physiol Lung Cell
Mol Physiol 2008;295:L479‐88.
(51) Wahl M, Eddinger TJ, Hai CM. Sinusoidal length oscillation‐ and receptor‐mediated mRNA
expression of myosin isoforms and alpha‐SM actin in airway smooth muscle. Am J Physiol Cell
Physiol 2004;287:C1697‐708.
(52) Tatler AL, John AE, Jolly L, Habgood A, Porte J, Brightling C, Knox AJ, Pang L, Sheppard D, Huang
X, Jenkins G. Integrin alphavbeta5‐mediated TGF‐beta activation by airway smooth muscle cells in
asthma. J Immunol 2011;187:6094‐6107.
(53) Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, Haley KJ, Lilly CM, So PT,
Lauffenburger DA, Kamm RD, Drazen JM. Mechanotransduction through growth‐factor shedding
into the extracellular space. Nature 2004;429:83‐86.
(54) Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal
epithelial cells to transmembrane pressure. Am J Physiol Lung Cell Mol Physiol 2000;278:L1264‐72.
(55) Choe MM, Sporn PH, Swartz MA. An in vitro airway wall model of remodeling. Am J Physiol Lung
Cell Mol Physiol 2003;285:L427‐33.
(56) Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated
between different cell types to elicit matrix remodeling. Proc Natl Acad Sci U S A 2001;98:6180‐
6185.
(57) Gwilt CR, Donnelly LE, Rogers DF. The non‐neuronal cholinergic system in the airways: an
unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208‐222.
(58) Pieper MP. The non‐neuronal cholinergic system as novel drug target in the airways. Life Sci
2012;91:1113‐1118.
(59) Slebos DJ, Klooster K, Koegelenberg CFN, Theron J, Steyn D, Mayse M. Efficacy of targeted lung
denervation on patients with moderate to severe COPD [abstract]. ERJ 2014:1774.
(60) Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is
an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells.
Endocrinology 2004;145:2498‐2506.
(61) Corren J. Role of interleukin‐13 in asthma. Curr Allergy Asthma Rep 2013;13:415‐420.
(62) Rogers DF. Motor control of airway goblet cells and glands. Respir Physiol 2001;125:129‐144.
(63) Asano K, Shikama Y, Shoji N, Hirano K, Suzaki H, Nakajima H. Tiotropium bromide inhibits TGF‐
beta‐induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways
in vitro. Int J Chron Obstruct Pulmon Dis 2010;5:277‐286.

Chapter 9
196
(64) Milara J, Serrano A, Peiro T, Artigues E, Gavalda A, Miralpeix M, Morcillo EJ, Cortijo J. Aclidinium
inhibits cigarette smoke‐induced lung fibroblast‐to‐myofibroblast transition. Eur Respir J
2013;41:1264‐1274.
(65) Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M, UPLIFT Study
Investigators. A 4‐year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med
2008;359:1543‐1554.
(66) D'Urzo A, Ferguson GT, van Noord JA, Hirata K, Martin C, Horton R, Lu Y, Banerji D, Overend T.
Efficacy and safety of once‐daily NVA237 in patients with moderate‐to‐severe COPD: the GLOW1
trial. Respir Res 2011;12:156‐9921‐12‐156.
(67) Jones PW, Rennard SI, Agusti A, Chanez P, Magnussen H, Fabbri L, Donohue JF, Bateman ED,
Gross NJ, Lamarca R, Caracta C, Gil EG. Efficacy and safety of once‐daily aclidinium in chronic
obstructive pulmonary disease. Respir Res 2011;12:55‐9921‐12‐55.
(68) Wedzicha JA, Donaldson GC. Exacerbations of chronic obstructive pulmonary disease. Respir
Care 2003;48:1204‐13; discussion 1213‐5.
(69) Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, Jeffery PK. Biopsy neutrophilia,
neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive
pulmonary disease. Am J Respir Crit Care Med 2003;168:968‐975.
(70) Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers
to symptoms and lung function changes in COPD exacerbations. Thorax 2000;55:114‐120.
(71) Liesker JJ, Bathoorn E, Postma DS, Vonk JM, Timens W, Kerstjens HA. Sputum inflammation
predicts exacerbations after cessation of inhaled corticosteroids in COPD. Respir Med
2011;105:1853‐1860.
(72) Perng DW, Tao CW, Su KC, Tsai CC, Liu LY, Lee YC. Anti‐inflammatory effects of
salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur Respir J 2009;33:778‐784.
(73) Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K, Kesten S, Wedzicha JA. Effect
of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J
2007;30:472‐478.
(74) Decramer M, Celli B, Kesten S, Lystig T, Mehra S, Tashkin DP. Effect of tiotropium on outcomes
in patients with moderate chronic obstructive pulmonary disease (UPLIFT): a prespecified subgroup
analysis of a randomised controlled trial. Lancet 2009;374:1171‐1178.
(75) Morice AH, Celli B, Kesten S, Lystig T, Tashkin D, Decramer M. COPD in young patients: a pre‐
specified analysis of the four‐year trial of tiotropium (UPLIFT). Respir Med 2010;104:1659‐1667.
(76) Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, Calverley PM, Celli B, Coxson HO,
Crim C, Lomas DA, MacNee W, Miller BE, Silverman EK, Tal‐Singer R, Wouters E, Rennard SI, ECLIPSE
Investigators. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med
2011;365:1184‐1192.
(77) Saito Y, Azuma A, Morimoto T, Fujita K, Abe S, Motegi T, Usuki J, Kudoh S. Tiotropium
ameliorates symptoms in patients with chronic airway mucus hypersecretion which is resistant to
macrolide therapy. Intern Med 2008;47:585‐591.
(78) Bateman ED, Rennard S, Barnes PJ, Dicpinigaitis PV, Gosens R, Gross NJ, Nadel JA, Pfeifer M,
Racke K, Rabe KF, Rubin BK, Welte T, Wessler I. Alternative mechanisms for tiotropium. Pulm
Pharmacol Ther 2009;22:533‐542.



199
Nederlandse samenvatting
Introductie
Chronisch obstructief longlijden (COPD) en astma zijn veel voorkomende longziekten,
waarvan de incidentie de afgelopen decennia sterk is toegenomen. Het is voorspeld dat
COPD wereldwijd de derde doodsoorzaak zal zijn in 2020. Astma heeft veel minder vaak
een dodelijke afloop, maar kan een grote impact hebben op iemands leven en is één van
de meest voorkomende chronische ziekten in kinderen. COPD en astma worden beide
gekenmerkt door toegenomen vernauwing van de luchtwegen, wat leidt tot ademhalings‐
problemen.
Er zijn een aantal factoren die kunnen leiden tot een vernauwing van de luchtwegen:
voorbeelden daarvan zijn belemmering van de luchtstroom door aanwezig sputum,
verdikking van de luchtwegwand, verlies van elastische kracht om de luchtweg van buiten
opengetrokken te houden en tenslotte samentrekking van spieren in de luchtwegwand.
Deze samentrekking van de spieren rondom de luchtwegen kan het gevolg zijn van het
rechtstreekse aangrijpen van een stof uit de zenuwuiteinden op het spierweefsel. Zo’n
stof heet een neurotransmitter. Acetylcholine is de primaire neurotransmitter in de
luchtwegen die zorgt voor vernauwing van de luchtwegen door te binden aan haar
aangrijpingspunten; de zogenaamde muscarinereceptoren. Veranderingen in het
neuronale systeem, met verhoogde acetylcholine afgifte als gevolg, dragen bij aan de
toegenomen vernauwing van de luchtwegen. Patiënten met COPD, en in mindere mate
ook die met astma, worden daarom behandeld met anticholinergica, welke de werking
van acetylcholine tegengaan door middel van het blokkeren van de muscarinereceptoren.
Tot voor kort werd aangenomen dat acetylcholine uitsluitend luchtwegvernauwing en
slijmsecretie veroorzaakt. Er zijn echter aanwijzingen dat acetylcholine ook bijdraagt aan
chronische luchtwegontsteking en structurele veranderingen in de luchtwegen. Dit laatste
wordt ook wel ‘luchtwegremodeling’ genoemd. Luchtwegontsteking en remodeling zijn
twee belangrijke pathologische processen die zowel bij COPD als astma een rol spelen en
bijdragen aan de afname van de longfunctie en aan de ernst van de ziekte. In
diermodellen voor zowel COPD als astma is aangetoond dat anticholinergica ontsteking en
remodeling van de luchtwegen kunnen remmen. De rol van acetylcholine in longziekten
zou daarom veel breder kunnen zijn dan aanvankelijk werd gedacht. Het doel van dit
proefschrift was om dat te onderzoeken.

200
Muscarine receptor subtypes
In de luchtwegen worden verschillende muscarinereceptorsubtypes tot expressie
gebracht, te weten muscarine M1, M2 en M3 receptoren. Luchtweg‐gladde spiercontractie
en slijmsecretie worden voornamelijk gemedieerd via M3 receptoren. Dit is de reden
waarom men anticholinergica het liefst zo selectief mogelijk voor M3 receptoren
ontwerpt. Het is echter niet bekend of de effecten van acetylcholine op ontsteking en
remodeling ook gemedieerd worden via M3 receptoren. Daarom hebben we in dit
proefschrift onderzocht wat de bijdrage is van individuele muscarinereceptorsubtypes aan
luchtwegontsteking en luchtwegremodeling. Dit hebben we gedaan met behulp van
muizen welke één van de muscarinereceptoren niet tot expressie brengen, zogenaamde
muscarinereceptor knock‐out muizen. In hoofdstuk 3 hebben we de rol van individuele
muscarinereceptorsubtypes onderzocht in een muismodel voor COPD, en in hoofdstuk 6
hebben we dit gedaan in een muismodel voor astma.
COPD
In hoofdstuk 3 hebben we controle muizen, zogenaamde wild‐type muizen, en
muscarinereceptor knock‐out muizen blootgesteld aan sigarettenrook, omdat dit de
belangrijkste oorzaak is voor het ontwikkelen van COPD. Net als in COPD patiënten
ontstaat er ontsteking in de longen van wild‐type muizen na blootstelling aan
sigarettenrook. Deze ontsteking wordt gekenmerkt door een toename in neutrofiele
granulocyten (een type witte bloedcellen) en ontstekingsbevorderende stoffen, zoals het
cytokine interleukine (IL)‐8 dat neutrofielen aantrekt in de long. Eerder was al aangetoond
dat voorbehandeling met anticholinergica neutrofiele ontsteking kan remmen in
diermodellen van COPD. In hoofdstuk 3 laten we zien dat deze effecten gemedieerd
worden via M3 receptoren. Knock‐out van de M3 receptor kon voorkomen dat
sigarettenrook neutrofiele ontsteking en cytokine afgifte veroorzaakte. Daarnaast hebben
we aangetoond dat vergelijkbare effecten verkregen worden wanneer de M3 receptor
geblokkeerd wordt met een selectieve M3‐receptorblokker. Dit suggereert dat
acetylcholine luchtwegontsteking bevordert via de M3 receptor. Dit wordt verder
ondersteund door het feit dat de ontsteking was verhoogd na knock‐out van de M2
receptor. De M2 receptor functioneert als een receptor met een remmende werking op de
afgifte van acetylcholine. Verlies van deze receptor resulteert dus in verhoogde afgifte van
acetylcholine, wat leidt tot verhoogde luchtwegontsteking. Opvallend was dat de
ontsteking ook verhoogd was na knock‐out van de M1 receptor. Dit zou verklaard kunnen
worden door de rol van de M1 receptor bij de productie van slijm. De M1 receptor draagt
bij aan de afgifte van water en elektrolyten door slijmproducerende cellen, en kan zo de
samenstelling van het slijm veranderen. Slijm is belangrijk om deeltjes in de luchtwegen
op te ruimen. Het opruimen van de schadelijke rookdeeltjes zou minder goed kunnen

Nederlandse samenvatting
201
verlopen na knock‐out van de M1 receptor, bijvoorbeeld doordat het slijm dikker is.
Samengevat laat hoofdstuk 3 zien dat acetylcholine de luchtwegontsteking veroorzaakt
door roken bevordert via M3 receptoren, en niet via M1 en M2 receptoren.
Studies die gebruik maken van luchtwegcellen in kweek ondersteunen onze bevindingen
dat acetylcholine luchtwegontsteking bevordert via M3 receptoren. M3 receptoren worden
op bijna alle cellen in de luchtwegen tot expressie gebracht (zie tabel 1 in hoofdstuk 1).
Dit omvat ontstekingscellen, zoals neutrofielen en macrofagen, alsmede structurele
cellen, zoals luchtweg‐gladde spiercellen en epitheelcellen en endotheelcellen welke
respectievelijk de luchtwegen en de bloedvaten bekleden. In kweekcondities kan activatie
van muscarinereceptoren op zowel ontstekingscellen als structurele cellen de
luchtwegontsteking bevorderen door het vrijzetten van ontstekingsbevorderende
cytokines uit deze cellen. Het is echter niet bekend wat de relatieve bijdrage is van
ontstekingscellen ten opzichte van structurele cellen aan het ontstekingsbevorderende
effect van acetylcholine in het levende organisme. Dit hebben we daarom onderzocht in
hoofdstuk 4. Om onderscheid te kunnen maken tussen ontstekingscellen en structurele
cellen hebben we beenmergtransplantaties uitgevoerd in muizen. Door transplantatie van
beenmergcellen van M3 receptor knock‐out dieren in bestraalde wild‐type dieren kan de
bijdrage van de M3 receptor op ontstekingscellen, die ontstaan in het beenmerg,
onderzocht worden. Deze dieren hebben namelijk normale structurele cellen en
ontstekingscellen zonder M3 receptor. Omgekeerd kan door de transplantatie van
beenmergcellen van wild‐type dieren in bestraalde M3 receptor knock‐out dieren de
bijdrage van de M3 receptor op structurele cellen onderzocht worden. Deze dieren hebben
juist structurele cellen zonder M3 receptor en normale ontstekingscellen. Bestraalde en
niet bestraalde wild‐type dieren werden gebruikt als controle dieren. De muizen werden
blootgesteld aan sigarettenrook, op dezelfde manier als in hoofdstuk 3. Dit leidde tot een
toename van het aantal neutrofielen in controle dieren. Dieren met ontstekingscellen
zonder M3 receptor hadden een vergelijkbare toename in neutrofielen. Dit suggereert dat
ontstekingscellen geen bijdrage leveren aan het ontstekingsbevorderende effect van
acetylcholine. In dieren met structurele cellen zonder M3 receptor werd geen toename
van neutrofielen waargenomen. Dit suggereert dat de M3 receptor op structurele cellen
bijdraagt aan het ontstekingsbevorderende effect van acetylcholine. De afgifte van het
ontstekingsbevorderende cytokine IL‐8, dat zorgt voor het aantrekken van neutrofielen
naar de plek van ontsteking, was vergelijkbaar in alle groepen. Dit suggereert dat hoewel
neutrofielen wel aangetrokken konden worden door dit cytokine, de migratie vanuit de
bloedbaan naar de luchtwegen verhinderd was na knock‐out van de M3 receptor. Hiervoor
was inderdaad bewijs. Twee genen die betrokken zijn bij de migratie van neutrofielen,
fibrinogeen α en CD177, kwamen lager tot expressie in M3 receptor knock‐out dieren in

202
vergelijking met wild‐type dieren. Samengevat laat hoofdstuk 4 zien dat de
ontstekingsbevorderende effecten van acetylcholine worden gemedieerd via M3
receptoren op structurele cellen, doordat er verminderde migratie van neutrofielen
optreedt. Dit wijst op een belangrijke rol voor structurele cellen in de pathofysiologie van
COPD, en een bijdrage van acetylcholine aan dit proces.
Astma
Vervolgens hebben we in hoofdstuk 6 de rol van individuele muscarinereceptorsubtypes
in astma onderzocht. Hiervoor zijn wild‐type en muscarinereceptor knock‐out muizen
blootgesteld aan een allergeen, om zo een allergische reactie te induceren vergelijkbaar
met die bij allergische astmapatiënten. Blootstelling van wild‐type dieren aan het
allergeen resulteerde in luchtwegremodeling in de vorm van een toename in
slijmproducerende cellen, verdikking van de spierlaag rondom de luchtwegen en de
bloedvaten in de long en toegenomen afzetting van bindweefseleiwitten in de
luchtwegwand. Deze effecten werden niet of slechts in beperkte mate waargenomen in
M3 receptor knock‐out muizen. De remodeling in M1 en M2 receptor knock‐out muizen
was vergelijkbaar met die in wild‐type muizen. Dit suggereert dat de remodelingbevor‐
derende eigenschappen van acetylcholine na blootstelling aan allergeen gemedieerd
worden via M3 receptoren, en niet via M1 en M2 receptoren. Dit is dus vergelijkbaar met
de ontstekingsbevorderende eigenschappen van acetylcholine na rookblootstelling
beschreven in hoofdstuk 3, welke ook via de M3 receptor gemedieerd werden. In
tegenstelling tot de resultaten in hoofdstuk 3 hebben we in hoofdstuk 6 geen effect
gevonden van knock‐out van de M2 receptor. Het is bekend dat de M2 receptor niet meer
functioneel is in astma; dit is zowel voor diermodellen als voor patiënten met astma
gerapporteerd. Dit zou kunnen verklaren waarom de remodeling vergelijkbaar is in wild
type en M2 receptor knock‐out dieren. Daarnaast hebben we ook geen effect gevonden
van knock‐out van de M1 receptor. Terwijl in hoofdstuk 3 schadelijke rookdeeltjes uit de
longen verwijderd moesten worden, wordt zo’n effect niet verwacht na inhalatie van een
allergeen. Dit zou kunnen verklaren waarom de remodeling vergelijkbaar is in wild type en
M1 receptor knock‐out dieren. Samengevat laat hoofdstuk 6 zien dat acetylcholine
luchtwegremodeling bevordert via M3 receptoren, en niet via M1 en M2 receptoren.
Naast het effect van allergeen op luchtwegremodeling, hebben we in hoofdstuk 6 ook het
effect van allergeen op luchtwegontsteking onderzocht. Blootstelling van wild‐type dieren
aan het allergeen resulteerde in luchtwegontsteking, gekenmerkt door een toename in
eosinofielen (een type witte bloedcellen) en ontstekingsbevorderende cytokines zoals IL‐
13. Eenzelfde mate van ontsteking werd waargenomen in muscarinereceptor knock‐out
dieren, inclusief M3 receptor knock‐out dieren. Dit is opvallend, omdat deze dieren

Nederlandse samenvatting
203
nauwelijks luchtwegremodeling vertonen. Er werd altijd gedacht dat luchtwegremodeling
het gevolg is van luchtwegontsteking. Onze resultaten suggereren echter dat hier ook een
ander mechanisme aan ten grondslag zou kunnen liggen. Het is bekend dat mechanische
compressie, zoals veroorzaakt door gladde spiercontractie, kan leiden tot remodeling. Dit
is aangetoond voor luchtwegepitheelcellen in kweek, maar ook in astmapatiënten.
Wanneer in deze patiënten gladde spiercontractie veroorzaakt wordt door de stof
methacholine leidt dit tot remodeling zonder dat er effecten zijn op de ontsteking. Gladde
spiercontractie wordt gemedieerd via M3 receptoren en in M3 receptor knock‐out dieren
treedt er minder luchtwegvernauwing op, wat we ook laten zien in hoofdstuk 7.
Samengevat suggereert dit dus dat luchtwegvernauwing, door gladde spiercontractie via
M3 receptoren, een belangrijke oorzaak zou kunnen zijn van het ontstaan van
luchtwegremodeling bij allergisch astma. Dit impliceert dat het zinvol zou kunnen zijn om
toegenomen luchtwegvernauwing in patiënten met astma al in een vroeg stadium te
behandelen, om zo luchtwegremodeling gedeeltelijk te voorkomen.
In hoofdstuk 7 hebben we daarom verder onderzocht of luchtwegvernauwing via M3
receptoren kan leiden tot luchtwegremodeling. Hiertoe hebben we gebruik gemaakt van
longplakjes van muizen. Eerder was al aangetoond dat luchtwegvernauwing in longplakjes
van cavia’s kan leiden tot luchtwegremodeling. In hoofdstuk 7 wilden we de bijdrage van
de M3 receptor in deze respons onderzoeken met behulp van de M3 receptor knock‐out
muizen. Hoewel luchtwegremodeling veroorzaakt werd door de potente groeifactor TGF‐
β, vonden we geen effect van luchtwegvernauwing op remodeling. Dit suggereert dat de
cavia een beter model is om het effect van luchtwegvernauwing op luchtwegremodeling
te onderzoeken, en dat de muis hier niet geschikt voor is.
Neuronaal en non‐neuronaal acetylcholine
Acetylcholine functioneert als een neurotransmitter in de luchtwegen, maar kan daarnaast
ook aangemaakt worden door verschillende cellen in de luchtwegen, zoals epitheelcellen
en ontstekingscellen. Dit wordt non‐neuronaal acetylcholine genoemd. In dit proefschrift
hebben we onderzocht wat de bijdrage is van non‐neuronaal acetylcholine aan ontsteking
en remodeling. In hoofdstuk 5 hebben we de rol van neuronaal versus non‐neuronaal
acetylcholine onderzocht in ontsteking bij COPD, en in hoofdstuk 8 hebben we de bijdrage
van non‐neuronaal acetylcholine aan slijmbekerceldifferentiatie onderzocht.

204
COPD
In hoofdstuk 5 hebben we het effect onderzocht van denervatie op ontsteking in
patiënten met COPD. Denervatie is een potentiële nieuwe therapie voor patiënten met
COPD. Door het uitschakelen van de zenuwen komt er geen acetylcholine meer vrij uit de
zenuwuiteinden, waardoor er minder luchtwegvernauwing optreedt. Wij hebben
onderzocht of dit ook effect heeft op de ontsteking in de luchtwegen. We zagen inderdaad
een afname van de ontsteking na denervatie in patiënten, in de vorm van verminderde
neutrofielen en ontstekingsbevorderende cytokines zoals IL‐8. Dit is slechts in zeven
patiënten uitgevoerd, omdat deze therapie voor het eerst werd toegepast in mensen,
maar toch suggereert dit dat acetylcholine luchtwegontsteking bevordert, en dat dit
geremd kan worden door het remmen van de afgifte van acetylcholine door denervatie.
Daarnaast namen we ook een vermindering van TGF‐β waar, een remodelingbevorderen‐
de groeifactor. Dit suggereert dat denervatie op de lange termijn ook effect zou kunnen
hebben op remodeling. Uit dit hoofdstuk blijkt dat de effecten van acetylcholine in COPD
in ieder geval gedeeltelijk gemedieerd worden via neuronaal acetylcholine. Dit wordt
verder ondersteund door de resultaten uit hoofdstuk 3, waar we een toegenomen
ontsteking waarnamen na knock‐out van de M2 receptor, welke neuronaal acetylcholine
remt, terwijl een soortgelijk mechanisme voor non‐neuronaal acetylcholine niet bekend is.
Astma
In hoofdstuk 8 hebben we de rol van acetylcholine bij de vorming van slijmbekercellen in
het luchtwegepitheel onderzocht. Epitheelcellen maken zelf acetylcholine aan, zodat de
bijdrage van non‐neuronaal acetylcholine hierbij onderzocht kan worden. We hebben
hiertoe gebruik gemaakt van epitheelcellen uit luchtwegen van gezonde donorlongen, wat
als restmateriaal overblijft bij een longtransplantatie. Door het blootstellen van deze
cellen aan IL‐13 ontstaat er een toename in het aantal slijmbekercellen. In hoofdstuk 8
tonen we aan dat het blokkeren van muscarinereceptoren deze toename kan voorkomen.
Hieruit blijkt dat non‐neuronaal acetylcholine bijdraagt aan de toename in
slijmbekercellen. De resultaten uit hoofdstuk 6 suggereren dat neuronaal acetylcholine
een belangrijke rol speelt bij luchtwegremodeling in astma, omdat er hier waarschijnlijk
remodeling optreedt door luchtwegconstrictie, wat gemedieerd wordt via neuronaal
acetylcholine. Samengevat laat dit proefschrift dus zien dat er een belangrijke rol is voor
neuronaal acetylcholine in luchtwegontsteking en remodeling, maar dat ook non‐
neuronaal acetylcholine hier aan bij kan dragen. Denervatie in dieren onder
gecontroleerde omstandigheden zou uitsluitsel kunnen geven over de relatieve bijdrage
van non‐neuronaal acetylcholine in het levende organisme. In het kader van dit
proefschrift hebben we geprobeerd dit uit te voeren in muizen, maar dit bleek niet
mogelijk. Uit de literatuur blijkt echter dat denervatie in grotere proefdieren wel mogelijk

Nederlandse samenvatting
205
is, zodat door deze dieren bloot te stellen aan bijvoorbeeld sigarettenrook of allergeen de
bijdrage van neuronaal versus non‐neuronaal acetylcholine aan ontsteking en remodeling
onderzocht kan worden. Daarnaast zouden studies waarin denervatie in grotere groepen
patiënten met COPD of astma wordt uitgevoerd ook meer informatie kunnen geven.
Klinische implicaties
COPD
De resultaten in dit proefschrift tonen aan dat acetylcholine bijdraagt aan ontsteking en
remodeling in modellen voor COPD en astma. Er is echter nog weinig bekend over de rol
van acetylcholine bij ontsteking en remodeling in patiënten. In dit proefschrift tonen we
voor de eerste keer aan dat het remmen van de afgifte van acetylcholine, door middel van
denervatie, ontsteking remt bij COPD patiënten. Een geplande vervolgstudie, waarin meer
patiënten geïncludeerd zullen worden, zal meer licht werpen op de vraag of acetylcholine
in de luchtwegen ontstekingsbevorderend werkt. Het is wel bekend dat behandeling van
COPD patiënten met anticholinergica het optreden van exacerbaties remt. Een
exacerbatie is een toename van ziektesymptomen, welke gepaard gaat met een
toegenomen ontsteking. Dit suggereert dat anticholinergica de ontstekingsreactie zouden
kunnen remmen. De resultaten uit hoofdstuk 5 zijn de eerste directe aanwijzing dat
acetylcholine effect heeft op de ontsteking bij deze patiënten. Er zijn echter nog meer
studies nodig om dit onomstotelijk aan te tonen. Daarnaast zijn er nog studies nodig die
het effect van acetylcholine op remodeling bij COPD onderzoeken. In hoofdstuk 5 zagen
we een afname van TGF‐β na denervatie. Ook is bekend dat behandeling met
anticholinergica de versnelde afname in longfunctie in beperkte mate kan remmen bij
bepaalde patiëntengroepen met COPD. Studies waarin remodeling op een meer directe
manier gemeten wordt, bijvoorbeeld door remodeling in biopten van COPD patiënten na
langdurige behandeling met anticholinergica te onderzoeken, zouden helpen bij het
ophelderen van de rol van acetylcholine bij remodeling bij COPD.
Astma
Het anticholinergicum tiotropium is recent geregistreerd voor de chronische behandeling
van astmapatiënten. Er zijn steeds meer studies die aangeven dat anticholinergica wel
degelijk een positief effect hebben op longfunctie in deze patiëntengroep. Uit deze studies
blijkt ook dat, net als bij COPD, het aantal exacerbaties geremd wordt na behandeling met
anticholinergica. Dit is dus opnieuw indirect bewijs voor een rol van acetylcholine bij
astma, en mogelijk bij ontsteking. Daarnaast zijn er ook indicaties dat acetylcholine een rol
zou kunnen spelen bij remodeling in patiënten met astma. Een studie waarin
muscarinereceptoren geactiveerd werden in astmapatiënten toonde aan dat dit leidde tot

206
meer remodeling. Ook hier geldt dat er in de toekomst meer studies nodig zullen zijn om
definitief te bewijzen wat de rol is van acetylcholine in ontsteking en remodeling in
patiënten met astma.
M3 selectieve anticholinergica
Klinisch beschikbare anticholinergica binden aan zowel M1, M2 als M3 receptoren. Ze zijn
selectief doordat ze langer binden aan M3 receptoren dan aan M2 receptoren, en in
mindere mate M1 receptoren. Uit de resultaten van dit proefschrift blijkt dat het gewenst
is om een anticholinergicum te ontwikkelen dat nog selectiever is, en alleen bindt aan M3
receptoren. Dit zou, afhankelijk van de aandoening, kunnen leiden tot verbeterde effecten
op ontsteking en remodeling.
Conclusies
Samengevat laat dit proefschrift zien dat acetylcholine bijdraagt aan ontsteking en
remodeling bij COPD en astma. Dit omvat zowel neuronaal als niet‐neuronaal
acetylcholine. De effecten van acetylcholine op ontsteking en remodeling worden
gemedieerd via M3 receptoren. Samen suggereert dit dat patiënten met COPD of astma
meer baat zouden kunnen hebben bij anticholinergica dan aanvankelijk gedacht.
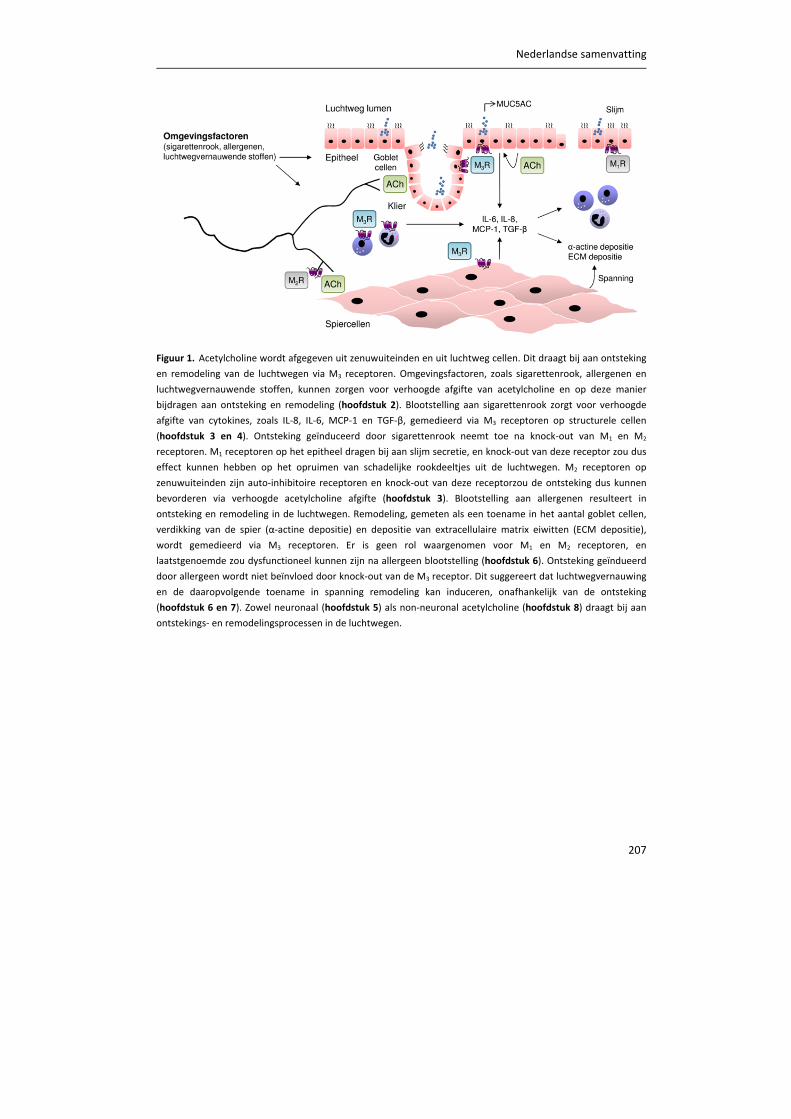
Nederlandse samenvatting
207
Figuur 1. Acetylcholine wordt afgegeven uit zenuwuiteinden en uit luchtweg cellen. Dit draagt bij aan ontsteking en remodeling van de luchtwegen via M3 receptoren. Omgevingsfactoren, zoals sigarettenrook, allergenen en
luchtwegvernauwende stoffen, kunnen zorgen voor verhoogde afgifte van acetylcholine en op deze manier
bijdragen aan ontsteking en remodeling (hoofdstuk 2). Blootstelling aan sigarettenrook zorgt voor verhoogde
afgifte van cytokines, zoals IL‐8, IL‐6, MCP‐1 en TGF‐β, gemedieerd via M3 receptoren op structurele cellen
(hoofdstuk 3 en 4). Ontsteking geïnduceerd door sigarettenrook neemt toe na knock‐out van M1 en M2
receptoren. M1 receptoren op het epitheel dragen bij aan slijm secretie, en knock‐out van deze receptor zou dus
effect kunnen hebben op het opruimen van schadelijke rookdeeltjes uit de luchtwegen. M2 receptoren op
zenuwuiteinden zijn auto‐inhibitoire receptoren en knock‐out van deze receptorzou de ontsteking dus kunnen
bevorderen via verhoogde acetylcholine afgifte (hoofdstuk 3). Blootstelling aan allergenen resulteert in
ontsteking en remodeling in de luchtwegen. Remodeling, gemeten als een toename in het aantal goblet cellen,
verdikking van de spier (α‐actine depositie) en depositie van extracellulaire matrix eiwitten (ECM depositie),
wordt gemedieerd via M3 receptoren. Er is geen rol waargenomen voor M1 en M2 receptoren, en
laatstgenoemde zou dysfunctioneel kunnen zijn na allergeen blootstelling (hoofdstuk 6). Ontsteking geïndueerd
door allergeen wordt niet beïnvloed door knock‐out van de M3 receptor. Dit suggereert dat luchtwegvernauwing
en de daaropvolgende toename in spanning remodeling kan induceren, onafhankelijk van de ontsteking
(hoofdstuk 6 en 7). Zowel neuronaal (hoofdstuk 5) als non‐neuronal acetylcholine (hoofdstuk 8) draagt bij aan
ontstekings‐ en remodelingsprocessen in de luchtwegen.

208
De belangrijkste conclusies van dit proefschrift zijn de volgende:
Acetylcholine is niet alleen betrokken bij luchtwegconstrictie en slijmsecretie,
maar draagt ook bij aan luchtwegontsteking en remodeling (hoofdstuk 2). Net als
de effecten op luchtwegconstrictie en slijmsecretie worden de effecten op
ontsteking en remodeling voornamelijk gemedieerd via M3 receptoren
(hoofdstuk 3 en 6).
De M3 receptor speelt een ontstekingsbevorderende rol bij ontsteking
veroorzaakt door sigarettenrook, terwijl de M1 en M2 receptoren
ontstekingsremmend werken (hoofdstuk 3). Dit suggereert dat M3 selectieve
anticholinergica de ontsteking bij COPD kunnen remmen.
Het ontstekingsbevorderende effect van acetylcholine bij ontsteking veroorzaakt
door sigarettenrook wordt voornamelijk gemedieerd door M3 receptoren
aanwezig op structurele cellen van de luchtwegen (hoofdstuk 4).
De M3 receptor draagt bij aan remodeling veroorzaakt door allergeen, terwijl de
M3 receptor geen rol speelt bij ontsteking veroorzaakt door allergeen (hoofdstuk
6). Dit suggereert dat luchtwegconstrictie remodeling kan veroorzaken,
onafhankelijk van de ontsteking (hoofdstuk 6 en 7).
M1 en M2 receptoren zijn niet betrokken bij ontsteking en remodeling
veroorzaakt door allergeen (hoofdstuk 6).
Denervatie vermindert de luchtwegontsteking in patiënten met COPD. Dit is de
eerste directe aanwijzing dat acetylcholine bijdraagt aan ontsteking in patiënten
en impliceert een belangrijke rol voor neuronaal acetylcholine (hoofdstuk 5).
Anticholinergica kunnen de differentiatie naar slijmbekercellen in het epitheel
voorkomen. Dit suggereert dat non‐neuronaal acetylcholine bijdraagt aan
remodeling bij astma (hoofdstuk 8).
Patiënten met COPD of astma zouden meer baat kunnen hebben bij
anticholinerge therapie dan aanvankelijk gedacht, omdat anticholinerge therapie
luchtwegontsteking en luchtwegremodeling zou kunnen remmen. Dit kan
bewerkstelligd worden via denervatie of via inhalatie van anticholinergica.
Behandeling met selectievere M3 receptor‐specifieke anticholinergica zou nog
meer voordeel op kunnen leveren (dit proefschrift).



211
Dankwoord
Ik kijk met een ontzettend goed gevoel terug op de afgelopen vier jaren en ben trots op
het werk beschreven in dit proefschrift. Dit had ik natuurlijk nooit kunnen bereiken zonder
de hulp van alle mensen om me heen. Aan elk hoofdstuk ligt een andere samenwerking
ten grondslag en ik heb veel geleerd van de verschillende mensen met wie ik heb kunnen
samen werken. Graag wil ik daarom iedereen bedanken die, op welke wijze dan ook, heeft
geholpen bij het tot stand komen van dit proefschrift. Een aantal mensen wil ik graag in
het bijzonder noemen.
Als eerste wil ik graag mijn promotores bedanken.
Reinoud, het moment waarop jij me vroeg voor dit project kan ik me nog herinneren als
de dag van gisteren. Ik was enigszins verrast en had mijn keuze voor het onderzoek dan
wel de apotheek eigenlijk nog niet gemaakt. Na enig beraad koos ik voor de uitdagingen
van het onderzoek en achteraf kan ik met zekerheid stellen dat dit de juiste keuze is
geweest. Niet in de laatste plaats omdat dit project en onze samenwerking zo goed
verliep. Hoewel we elkaar niet goed kenden op het moment dat ik aan dit project begon,
merkte ik al snel dat dit wel goed zat. Ik wil je bedanken voor de prettige begeleiding, je
positieve kijk op de dingen, de ruimte die je me hebt gegeven binnen het project, maar
ook voor de gezelligheid en leuke gesprekken tijdens onder andere de congressen die we
hebben bezocht en op de vrijdagmiddagborrels in de Pintelier. Je was een perfecte
dagelijkse begeleider, maar ik ben natuurlijk helemaal trots dat je uiteindelijk mijn eerste
promotor bent geworden. Ik kijk met vertrouwen uit naar de toekomst en hoop dat we
nog lang zullen samenwerken.
Herman, als promotor was jij ook betrokken bij mijn project. Nadat jij jouw kritische blik
op mijn stukken had geworpen wist ik zeker dat er geen fouten meer in zouden zitten.
Bovendien kon je mij door al je kennis en ervaring anders naar de data laten kijken,
bedankt daarvoor.
Huib, ook jij keek met een totaal andere blik naar mijn data, wat erg nuttig was. Na onze
maandelijkse overleggen was ik soms minder zeker over mijn data of de betekenis hiervan
dan voordat ik je kamer binnen stapte, maar ik denk dat zowel ik als mijn stukken hierdoor
alleen maar beter zijn geworden. Bedankt.
I would also like to thank the members of the reading committee, Prof. dr. Andrew
Halayko, Prof. dr. Wolfgang Kummer and Prof. dr. Dirkje Postma. Thank you for your time
and effort to critically review my thesis.

212
Martina, jij bent degene die mij kennis heeft laten maken met het onderzoek. Jouw passie
en enthousiasme voor het onderzoek hebben er zeker aan bijgedragen dat ik heb gekozen
voor deze weg, bedankt daarvoor.
Hans, hoewel je officieel niet betrokken was bij mijn project, kwam je toch regelmatig
even polsen hoe het er voor stond en of ik al nieuwe resultaten had, bijvoorbeeld over de
rol van de M1 receptor. Bedankt dat je altijd voor me klaar stond en staat met advies.
Machteld, ook jij was betrokken bij mijn project en hebt me veel kunnen leren. Bedankt
voor je bijdrage. Hopelijk ben je enigszins tevreden met de hoeveelheid puntengrafieken
die er uiteindelijk in mijn proefschrift terecht zijn gekomen.
Sophie, mijn project begon direct met proefdierwerk, en onveraren (en onhandig) als ik
was, kon ik jouw hulp hierbij goed gebruiken. Doordat we veel tijd doorbrachten op het
dierenlab leerden we elkaar snel beter kennen, en werd het dus ook snel gezellig. Vooral
onze eerste ’opofferdag‘ zal ik niet gauw vergeten; hoewel alles veel langer duurde dan
gepland heb ik nog nooit zo hard gelachen op het lab. Bedankt voor je hulp gedurende
mijn hele project, maar zeker ook voor de gezelligheid.
Ronald en Bertien, zonder jullie had hoofdstuk 4 nooit bewerkstelligd kunnen worden. We
hadden het idee opgepakt om de rol van de M3 receptor verder uit te zoeken met behulp
van beenmerg chimeren. Vervolgens moesten we op zoek naar iemand die dit
daadwerkelijk kon, en daar waren jullie. De samenwerking verliep vanaf het begin erg
prettig en ik wil jullie bedanken voor jullie bijdrage. Zowel in de opzet als in de uitvoering
was jullie hulp onmisbaar en ook op jullie lab voelde ik me altijd erg welkom, en dat terwijl
elke mogelijke deur van jullie lab op slot zat.
Dirk‐Jan en Karin, bedankt voor jullie bijdrage aan de totstandkoming van hoofdstuk 5.
Hoewel het voor jullie de normaalste zaak van de wereld is, had ik bij aanvang van mijn
project nooit verwacht dat ik een patiëntenstudie in mijn proefschrift kon opnemen. Voor
mij was dit een unieke kans en het bijwonen van de ingrepen was een hele ervaring.
Bedankt voor de prettige samenwerking en ik kijk erg uit naar onze toekomstige
samenwerking waarbij we een vervolg zullen geven aan de resultaten uit hoofdstuk 5.
Cécile, you were of great help for chapter 7. Although your stay in the Netherlands and
our lab was only short, we had a really good time, bot scientifically and socially. We just
started experiments using lung slices, and it was really fun to work together with an
engineer, and to share our totally different views on the lung slice. But also at all the

Dankwoord
213
different drinks you were really good company. It was great to visit you in Boston and I
wish you all the best in Grenoble.
Pieter, Ik ben jou en je lab ook dank verschuldigd. Hoewel op afstand was je altijd erg
geïnteresseerd in mijn project en gaf je waardevolle feedback. Dit bleef zeker niet beperkt
tot hoofdstuk 8, het hoofdstuk over epitheelcellen, waar de samenwerking op gebaseerd
was. Voor het opzetten van de zogenaamde ALI‐kweken kwam ik al direct bij aanvang van
mijn project regelmatig naar jullie lab om deze techniek te leren. Suzanne en Renate,
bedankt voor jullie hartelijke ontvangst en de fijne samenwerking. Ik denk graag dat dit
ook te maken had met mij als persoon, en niet alleen met het kostbare stukje gezond
longweefsel dat ik bij me had. Tinne, jij werd in een later stadium betrokken bij dit project
en ook jou wil ik graag bedanken voor je hulp. Ik wens je veel succes bij het afronden van
je promotie.
Anita, het leek me direct gezellig toen ik hoorde dat jij mijn collega zou worden, en niets
bleek minder waar. Je enthousiasme werkt aanstekelijk, het is erg prettig om zo’n collega
te hebben. Daarnaast was je een erg leuke kamergenoot tijdens congresbezoeken en dan
met name de bezoeken aan de ATS. Ook onze daaropvolgende USA tripjes, inclusief
pannenkoeken als ontbijt en bergwandelingen (het leven bestaat uit compromissen
sluiten), zal ik me altijd blijven herinneren. Bedankt dat je mijn paranimf wilt zijn.
Ook al mijn andere (oud‐)collega’s van de afdeling Moleculaire Farmacologie wil ik graag
bedanken. Annet, Anouk, Bart, Bing, Boy, Carolina, Christa, David, Eline, Hana, Haoxiao,
Harm, Hoeke, Jacques, Janneke, Kuldeep, Marieke, Mark, Pablo, Sara, Sepp, Tim, Tjitske,
Tonio, Vessa en Wilfred; bedankt voor de gezelligheid. Sara, thank you for introducing me
to science and the department of Molecular Pharmacology. Tonio, net als Sara duurde het
niet lang of je was vertrokken nadat ik met mijn promotie was begonnen, maar ik wil je
bedanken voor de gezelligheid op de toch nog vele vrijdagmiddagborrels. Gelukkig zien we
elkaar nog af en toe in Amerika. Anouk, Eline, Marieke; hoewel theepauzes vaak wat
langer duren dan de bedoeling is, zijn ze een heerlijke onderbreking van de middag,
bedankt daarvoor. Mark, bedankt voor je flauwe grappen, ik hou er van. Tjitske, samen
werkten wij aan muscarine receptoren en in navolging van jou werkte ik later ook aan
slices. Ik kan zeggen dat ik je miste toen je weg was, bedankt voor de prettige
samenwerking en je eerlijke mening.
Daarnaast wil ik ook graag alle collega’s binnen GRIAC bedanken. De GRIAC lezingen
hebben ervoor gezorgd dat ik een brede kijk op longziekten heb kunnen ontwikkelen en
hier wil ik iedereen voor bedanken. Ook wil ik jullie bedanken voor de goede sfeer tijdens
vrijdagmiddagborrels en op congressen.

214
Tijdens mijn project ben ik geholpen door verschillende studenten. Susanne en
Willemieke, jullie hebben beiden je masteronderzoek onder mijn begeleiding uitgevoerd.
Bedankt voor jullie inzet, toewijding en enthousiasme. Ook de bachelor studenten Nienke,
Esma, Fleur, Karen, Rosalie, Virginia en Sjoerd wil ik graag bedanken voor hun bijdrage.
Jullie bleven wat minder lang, maar jullie inzet wordt hierom niet minder gewaardeerd.
Ook wil ik graag de Centrale Dienst Proefdieren (CDP) noemen. Bedankt voor jullie
uitstekende zorg voor de proefdieren en de prettige samenwerking. Het Longfonds wil ik
graag bedanken voor hun financiële steun.
Als laatste wil ik graag iedereen uit mijn persoonlijke omgeving bedanken. Gelukkig lukte
het goed om naast mijn werk tijd vrij te maken voor sport, vrienden en familie. Er was
voor mij geen betere manier om mijn werk even los te laten dan door te hockeyen en
daarna een welverdiend biertje te drinken. Teamgenoten, bedankt voor de gezelligheid.
Daarnaast wil ik ook al mijn vrienden bedanken, van de basisschool tot aan mijn
studententijd, jullie vriendschap betekent veel voor me. Bedankt voor alle gezellige
borrels, weekendjes weg, dagjes sauna en avondjes stappen.
Ook mijn familie is erg belangrijk voor me. Pap en mam, bedankt voor jullie steun. Het is
erg prettig om te weten dat jullie altijd in me vertrouwen en dit heeft mij ook van het
begin af aan het vertrouwen gegeven dat ik dit tot een goed einde zou kunnen brengen.
Jullie zijn de beste ouders die ik me kan wensen. Jos en Hans, ik vind het erg stoer dat we
alle drie promotieonderzoek doen. Ik wens jullie veel succes met de laatste loodjes en met
jullie keuzes voor de toekomst. Ik heb er alle vertrouwen in dat dit goed komt. Els, ik vind
het leuk om te merken hoe we in de afgelopen jaren naar elkaar zijn toegegroeid. Waren
we vroeger erg verschillend, tegenwoordig lijken we steeds meer hetzelfde te worden.
Bedankt dat je er altijd voor me bent. Ik ben trots op je en vind het erg leuk dat je mijn
paranimf wilt zijn.
En tot slot Coen. Zonder jouw onvoorwaardelijke steun was het voor mij niet mogelijk
geweest om te promoveren. Er zullen ongetwijfeld moment zijn geweest waarop jij je hebt
afgevraagd waar je aan begonnen bent door voor mij naar Groningen te verhuizen en of
dit het allemaal wel waard is. Ik denk dat we inmiddels zeker weten dat het dat absoluut
was en ik ben je erg dankbaar dat je deze stap hebt genomen voor mij. Ik kijk met veel
vertrouwen en plezier uit naar de toekomst samen met jou.
Loes
Groningen, januari 2015



217
Curriculum Vitae
The author of this thesis was born in Oldenzaal, the Netherlands, on the 4th of September
1986. After finishing her pre‐university education (Twents Carmel Lyceum, Oldenzaal) in
2004, she studied Pharmacy at the University of Groningen. During her studies she was a
student assistant for the pharmacological practical course for year 3 pharmacy students at
the University of Groningen. She obtained her BSc degree in 2007 and her MSc degree
(PharmD) in 2010 (cum laude). Her master thesis on the effects of bradykinin and cyclic
AMP on interleukin‐8 production in human airway smooth muscle cells, and the role of
Epac and PKA hereon, was completed at the Department of Molecular Pharmacology,
University of Groningen. After her graduation, she initiated her PhD‐study at the same
department, where she worked on a research project funded by the Netherlands Lung
Foundation (grant: 3.2.08.014) on the role of acetylcholine in chronic inflammation and
remodeling of the airways, the results of which are presented in this thesis.


219
List of publications
Full papers
LEM Kistemaker, DJ Slebos, H Meurs, HAM Kerstjens, R Gosens. Anti‐inflammatory effects
of targeted lung denervation in patients with COPD. Submitted to Thorax, 2015.
LEM Kistemaker, PS Hiemstra, S Bouwman, M van den Berge, MN Hylkema, H Meurs,
HAM Kerstjens, R Gosens. Tiotropium attenuates IL‐13‐induced goblet cell metaplasia of
human airway epithelial cells. Thorax, 2015: In Revision.
LEM Kistemaker, R Gosens. Acetylcholine beyond bronchoconstriction: roles in
inflammation and remodeling. Trends Pharmacol Sci, 2014: Epub.
LEM Kistemaker, R van Os, A Ausema, IST Bos, MN Hylkema, PS Hiemstra, J Wess, H
Meurs, HAM Kerstjens, R Gosens. Muscarinic M3 receptors on structural cells regulate
neutrophilic inflammation in mice. Am J Physiol Lung Cell Mol Physiol, 2014: 308(1); L96‐
L103.
PB Noble, CD Pascoe, B Lan, S Ito, LEM Kistemaker, AL Tatler, T Pera, BS Brook, R Gosens,
AR West. Airway smooth muscle in asthma: linking contraction and mechanotransduction
to disease pathogenesis and remodelling. Pulmonary Pharmacology and Therapeutics,
2014: 29(2); 96‐107.
LEM Kistemaker, IST Bos, WM Mudde, MN Hylkema, PS Hiemstra, J Wess, H Meurs, HAM
Kerstjens, R Gosens. Muscarinic M₃ receptors contribute to allergen‐induced airway remodeling in mice. Am J Respir Cell Mol Biol, 2014: 50(4); 690‐8.
H Meurs, TA Oenema, LEM Kistemaker, R Gosens. A new perspective on muscarinic
receptor antagonism in obstructive airways diseases. Curr Opin Pharmacol, 2013: 13(3);
316‐23.
LEM Kistemaker, IST Bos, MN Hylkema, MC Nawijn, PS Hiemstra, J Wess, H Meurs, HAM
Kerstjens, R Gosens. Muscarinic receptor subtype‐specific effects on cigarette smoke‐
induced inflammation in mice. Eur Respir J, 2013: 42(6); 1677‐88.
LEM Kistemaker, TA Oenema, H Meurs, R Gosens. Regulation of airway inflammation and
remodeling by muscarinic receptors: perspectives on anticholinergic therapy in asthma
and COPD. Life Sci, 2012: 91(21‐22); 1126‐33.

220
SS Roscioni, LEM Kistemaker, MH Menzen, CRS Elzinga, R Gosens, AJ Halayko, H Meurs, M
Schmidt. PKA and Epac cooperate to augment bradykinin‐induced interleukin‐8 release
from human airway smooth muscle cells. Respir Res, 2009: 10; 88‐105.
Published abstracts
LEM Kistemaker, DJ Slebos, H Meurs, HAM Kerstjens, R Gosens. Anti‐inflammatory effects
of targeted lung denervation in patients with COPD. Eur Respir J, 2014: 44; Suppl. 58:
P3333.
LEM Kistemaker, R van Os, A Ausema, IST Bos, MN Hylkema, PS Hiemstra, J Wess, H
Meurs, HAM Kerstjens, R Gosens. Muscarinic M3 receptors on structural cells regulate
neutrophilic inflammation in mice. Eur Respir J, 2014: 44; Suppl. 58: P869.
LEM Kistemaker, PS Hiemstra, TCJ Mertens, MN Hylkema, H Meurs, HAM Kerstjens, R
Gosens. Effects Of Tiotropium On Il‐13‐Induced Differentiation Of Human Airway Epithelial
Cells. Am J Respir Crit Care Med, 2014: 189; A2050.
LEM Kistemaker, IST Bos, MN Hylkema, PS Hiemstra, J Wess, H Meurs, HAM Kerstjens, R
Gosens. The muscarinic M3 receptor regulates allergen‐induced airway remodeling in
mice. Eur Respir J, 2013: 42; Suppl. 57: P875.
LEM Kistemaker, IST Bos, MN Hylkema, PS Hiemstra, J Wess, H Meurs, HAM Kerstjens, R
Gosens. Allergen Induced Remodeling Requires The Muscarinic M3 Receptor In Mice. Am J
Respir Crit Care Med, 2013: 187; A5267.
LEM Kistemaker, IST Bos, H Meurs, MN Hylkema, MC Nawijn, PS Hiemstra, J Wess, HAM
Kerstjens, R. Gosens. Cigarette Smoke Induced Inflammation And Remodeling In
Muscarinic Receptor Subtype Deficient Mice. Am J Respir Crit Care Med, 2012: 185;
A1306.
SS Roscioni, LEM Kistemaker, CRS Elzinga, R Gosens, M Schmidt. NAUNYN‐
SCHMIEDEBERGS ARCHIVES OF PHARMACOLOGY, 2009: 380; 3: 270‐271.
SS Roscioni, LEM Kistemaker, MH Menzen, CRS Elzinga, R Gosens, AJ Halayko, H Meurs, M
Schmidt. A Role for Epac and PKA in GPCR−Mediated IL−8 Release from Airway Smooth
Muscle Cells. Am J Respir Crit Care Med, 2009: 179; A3899.































