Embed Size (px)
Citation preview

Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Development of nanoformulations of pyrazinoic
acid prodrugs for treatment of Mycobacterium
avium infections
Mariana Cordeiro Matoso
Mestrado em Microbiologia Aplicada
2012

Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Development of nanoformulations of pyrazinoic
acid prodrugs for treatment of Mycobacterium
avium infections
Mariana Cordeiro Matoso
Dissertação orientada por Doutora Manuela Gaspar (FFUL) e
Prof. Doutora Sandra Chaves (FCUL)
Mestrado em Microbiologia Aplicada
2012

Development of nanoformulations of pyrazinoic
acid prodrugs for treatment of Mycobacterium
avium infections
Mariana Cordeiro Matoso
Master Thesis
2012
This thesis was fully performed at the Departamento de Farmácia
Galénica e Tecnologia Farmacêutica of the Faculty of Pharmacy
(University of Lisbon), Campus do Lumiar, under the direct
supervision of Doutora Manuela Gaspar and Prof. Doutor Luís
Constantino.

I
Acknowledgments
A realização desta Dissertação de Mestrado só foi possível graças à colaboração e ao
contributo, de forma directa ou indirecta, de várias pessoas e instituições, às quais gostaria de
exprimir algumas palavras de agradecimento e profundo reconhecimento, em particular:
À Doutora Manuela Gaspar pela disponibilidade manifestada para orientar este
trabalho. Agradeço todo o apoio, todas as horas perdidas de sono e todo o emaranhado de
pensamentos que tornaram possível a obtenção de boas soluções para alguns pequenos
contratempos que foram surgindo durante a realização deste trabalho e que, por certo,
causaram alguns cabelos brancos. Por todos os conhecimentos que me foram transmitidos e
que me ajudaram a tornar numa pequena adulta e por toda a amizade que foi sendo construída
durante este último ano, um muito obrigada.
Ao Prof. Doutor Luís Constantino pela oportunidade que me deu em poder trabalhar
neste projecto bem como toda a disponibilidade concedida enquanto co-orientador deste
trabalho. Por todas as discussões científicas que protagonizámos em conjunto com a Doutora
Manuela Gaspar e a Prof. Doutora Emília Valente e por todas as criticas que daí surgiram e
que me guiaram no bom caminho. Gostaria ainda de expressar o meu agradecimento pelo
fornecimento dos pró-fármacos utilizados durante esta dissertação.
Á Prof. Doutora Emília Valente agradeço a disponibilidade para as discussões
científicas realizadas e todas as críticas relativas ao trabalho.
À Doutora Eugénia Cruz, investigadora do grupo Nanomedicine & Drug Delivery
Systems da Faculdade de Farmácia da Universidade de Lisboa, pioneira na tecnologia de
lipossomas em Portugal, pela oportunidade concedida em poder trabalhar no laboratório do
Campus do Lumiar.
À Prof. Doutora Sandra Chaves por ter aceite ser minha orientadora da Faculdade de
Ciências, por toda a disponibilidade e pela revisão critica desta tese.
À Carla Eleutério pela paciência que teve em me educar e me ensinar a funcionar com
o HPLC. Por todo o carinho e amizade com que me recebeu e me aturou alguns devaneios e
crises existenciais.
À Alexandra Borges por ter cuidado tão bem dos “meus” ratinhos e por toda a alegria e
boa disposição que tornou os dias menos bons em dias excepcionais.
À Susana Calado e à Joana Pereira de Almeida pelo apoio e amizade que criámos e
que tão bem me fez à saúde e por todos aqueles momentos que às vezes parecem pequenos
mas que se tornam enormes.

II
Aos colegas e amigos Filipa Fontes e Rui Lopes por toda a amizade e apoio
demonstrados.
Às investigadoras e amigas Sandra Simões e Manuela Colla obrigada pela
disponibilidade e apoio prestados quando necessário.
Aos amigos de Sintra, Bruno, FT, Jises, Sandro, Sheila, Guida e Rute, por toda a
preocupação e pelas cafezadas tão boas que me permitiram desanuviar das semanas de
trabalho.
Ao meu namorado, João, um muito obrigada pelas longas horas de atenção
relativamente às minhas dúvidas, inquietações, desânimos e subvalorizações. Por todo o
apoio, confiança, valorização do meu trabalho e da minha pessoa e por toda a coragem que
me deu para ultrapassar a culpa pelo tempo que a cada dia lhe subtraía.
Aos meus pais, Lina e João, e à minha mana Madalena, por todos os conselhos e
incentivos que me deram e que me permitiram fazer mais e melhor nunca desistindo perante os
vários obstáculos e muros que se colocaram à minha frente. Obrigada!
Igualmente expresso o meu agradecimento à Fundação para a Ciência e Tecnologia
(FCT) que através do projecto A new life for old antimycobacterial drugs: development of
prodrugs of pyrazinoic acid activated by mycobacterial esterases as a way to circumvent
resistance to pyrazinamide, PTDC/SAU-FCF/101950/2008, contribuiu para o suporte financeiro
da presente dissertação.

III
Communications in Scientific Meetings
Stable Prodrugs Liposomes for Treatment of Mycobacterial Infections, Matoso M., Eleutério C.,
Constantino L., Cruz M.E.M., Gaspar M.M. 3rd iMed.UL Post--Graduate Students Meeting,
Faculdade de Fármacia Universidade de Lisboa, Lisboa, Portugal, p60

IV
Resumo
As micobactérias são bactérias Gram+ cujo diverso número de espécies actualmente
descritas (cerca de 130) partilham as mesmas características no que diz respeito à espessura
da parede celular e à sua composição única que as torna uma forte barreira à permeabilidade
de antibióticos. Algumas destas micobactérias têm um papel preponderante como agentes
patogénicos em indivíduos imunocompetentes como acontece com Mycobacterium
tuberculosis, Mycobacterium leprae and Mycobacterium ulcerans, os agentes causadores da
tuberculose (TB), lepra e úlcera de Buruli, respectivamente. A grande maioria das outras
espécies de micobactérias está presente no ambiente como microrganismos saprófitas,
podendo alguns ser considerados patogénicos oportunistas e causadores de morte em
indivíduos imunocomprometidos. Deste grupo de micobactérias ambientais oportunistas faz
parte o complexo Mycobacterium avium (MAC), composto por M. avium, subespécie avium,
paratuberculosis, silvaticum, e M. intracellulare, responsável por uma grande percentagem de
doenças micobacterianas não tuberculosas (NTM). As infecções provocadas por este complexo
são adquiridas através do ambiente, especialmente pela água, solo e alimentos. Apesar da
infecção poder ocorrer por inalação a principal via de infecção, especialmente em indivíduos
imunocomprometidos, é o tracto gastrointestinal, já que um sistema imunitário
imunocompetente consegue controlar estas micobactérias.
Os problemas associados aos tratamentos contra M. avium e M. tuberculosis são
provocados, entre outras causas, pela incapacidade dos antibióticos atravessarem a parede
celular complexa das micobactérias, atingindo concentrações sub-terapêuticas no interior dos
macrófagos, local de proliferação das micobactérias. A necessidade de ultrapassar esta
desvantagem utilizando doses mais elevadas dá origem ao aparecimento de efeitos tóxicos.
Uma vez que os esquemas de tratamento destas infecções são muito longos a adesão á
terapia é muitas vezes quebrada contribuindo para o desenvolvimento de resistências à grande
maioria dos antibióticos.
A pirazinamida (PZA) além de ser um antibiótico de primeira linha no tratamento da TB
é um pró-fármaco que requer activação pela enzima pirazinamidase (PZase) para ser
transformado em ácido pirazinóico (POA), a sua forma activa. Esta enzima é sintetizada pela
própria micobactéria que hidrolisa o pró-fármaco apenas dentro desta. No entanto, esta
activação está dependente apenas de uma enzima tendo sido observada a emergência de
estirpes resistentes à PZA. A origem destas resistências poderão ser devidas a mutações no
gene que codifica a PZase podendo não ocorrer a formação do POA uma vez que a enzima
não reconhece o seu substacto. De modo a ultrapassar estes problemas, têm sido investigadas
formas alternativas ao uso da PZA. Assim, a síntese de novos pró-fármacos que possam ser
activados por esterases micobacterianas e que, em virtude da sua elevada abundância,
possam contornar o desenvolvimento de resistências à PZA constitui uma excelente estratégia
terapêutica.
Nesta linha de investigação, novos pró-fármacos de POA foram investigados por
diversos autores tendo demonstrado actividade in vitro superior à PZA contra estirpes de M.

V
tuberculosis, M. avium e M. kansasii. No entanto, estes compostos apresentaram estabilidades
reduzidas na presença de fluidos biológicos. Recentemente, no grupo de Química Medicinal da
Faculdade de Farmácia da Universidade de Lisboa, foram sintetizados pró-fármacos de amidas
e ésteres de POA, com cadeias alcóxido de diferentes tamanhos. Os ésteres de POA
apresentaram elevada estabilidade em plasma, particularmente os de cadeia alcóxido longa.
Para além disso, estes pró-fármacos são facilmente activados pelas esterases
micobacterianas. Estudos in vitro demonstraram que estes ésteres são activos contra estirpes
sensíveis de M. tuberculosis em concentrações 10 vezes mais baixas do que as necessárias
para a PZA.
Na presente dissertação foram desenvolvidas formulações lipossomais de dois ésteres
de POA e o seu efeito terapêutico avaliado num modelo murino infectado com uma estirpe de
M. avium. A associação destes ésteres aos lipossomas é baseada no facto destes sistemas
lipídicos conseguirem contornar os problemas de solubilidade associados a estas moléculas e
ainda no facto de, após administração parentérica, poderem atingir o fígado e o baço, os
principais orgãos infectados por M. avium.
Na primeira parte desta tese foram desenvolvidas e caracterizadas formulações
lipossomais de dois ésteres de POA contendo 4 e 12 carbonos na cadeia alquílica linear,
designados por C4 e C12. Todas as formulações lipossomais de C12 apresentaram eficácias
de incorporação superiores a 90%. No entanto, para as formulações de C4 os valores obtidos
foram inferiores a 6% o que pode ser explicado pelo maior carácter lipofílico do C12 em
comparação com o C4.
Com base nestes resultados, o pró-fármaco C12 foi seleccionado para todos os
estudos efectuados a posteriori. Sendo assim, foram preparadas dois tipos de formulações
lipossomais: extrusadas e não extrusadas. Os resultados obtidos indicam que a razão final
entre o C12 incorporado e o lípido foi superior a 71 nmol/µmol de lípido para todas as
suspensões, independentemente do diâmetro e da composição lipídica, o que apoia a hipótese
do pró-fármaco estar incorporado na bicamada lipídica. Para fortalecer esta afirmação, foram
realizados estudos de calorimetria com lipossomas vazios e com C12 incorporado. Não se
observaram alterações nas temperaturas de transição de fase para as duas formulações. No
entanto, o aumento de entalpia registado na formulação lipossomal com C12 é o resultado da
interacção deste pró-fármaco com as cadeias lipídicas. Estas observações estão relacionadas
com o carácter lipofílico do C12, contribuindo para a rigidez do sistema lipossomal e
confirmando a sua localização na bicamada lipídica.
Na segunda parte deste trabalho foi realizado um estudo sistemático da estabilidade
das formulações de C12 na presença de tampão HEPES pH 7.4 e de plasma humano,
avaliando a velocidade de hidrólise de C12 e a respectiva formação de POA. Assim, a
quantificação destas duas moléculas foi realizada por HPLC, tendo-se procedido previamente à
optimização desta metodologia. A estabilidade em tampão foi realizada em duas condições
distintas: temperatura ambiente e 37ºC. A evolução do diâmetro das vesículas foi avaliada
durante um mês, para garantir a homogeneidade dos lipossomas aquando da sua utilização,

VI
não se verificando alterações nestas propriedades para as formulações em estudo. A
realização da estabilidade em tampão a 37ºC pretendeu avaliar se a percentagem de C12
incorporado se mantém ao longo de 24 horas. Os resultados obtidos demonstraram uma
elevada estabilidade de todos as suspensões relativamente ao pró-fármaco incorporado. Para
uma das formulações testadas (DMPC:DSPE-PEG) foi, ainda, analisada a concentração de
C12 e a possível formação de POA, tendo-se verificando que o pró-fármaco incorporado não
se encontrava na forma hidrolisada. A estabilidade das formulações na presença de plasma
humano foi também realizada a 37ºC a fim de avaliar a velocidade de hidrólise do pró-fármaco,
na forma livre e lipossomal, em função da composição lipidica, e de modo a estimar o seu
comportamento in vivo. Os resultados obtidos indicaram que a hidrólise do C12 foi influenciada
principalmente pelo diâmetro médio e rigidez das formulações testadas. De facto, os tempos de
semi-vida mais elevados foram obtidos para os lipossomas preparados com fosfolípidos
neutros e não submetidos a extrusão. A existência de um elevado número de bicamadas
lipídicas que caracterizam estas vesículas está na origem destes resultados. A incubação de
C12 na forma livre apresentou uma estabilidade superior comparativamente a algumas
formulações, particularmente para as vesículas extrusadas. No entanto, esta estabilidade foi
dependente da concentração inicial do pró-fármaco uma vez que, utilizando a dose terapêutica
usada no modelo animal infectado com M. avium testado neste trabalho, a velocidade de
hidrólise do C12 foi mais lenta nos lipossomas extrusados do que na forma livre.
Embora na literatura esteja descrito que as estirpes de M. avium são intrinsecamente
resistentes à PZA pretendeu-se avaliar se era possível com este pró-fármaco ultrapassar estas
desvantagens tendo em consideração os resultados promissores obtidos in vitro para os
ésteres de POA, contra a estirpe de M. tuberculosis. Foi a primeira vez que lipossomas deste
pró-fármaco foram testados num modelo murino infectado com M. avium. O estudo preliminar
permitiu demonstrar que após administração i.v. do inóculo os principais orgãos infectados são
o fígado e o baço. Tendo em consideração estes resultados foi avaliado o efeito terapêutico
das diferentes formulações em estudo através da contagem de unidades formadoras de
colónias (CFU) nestes dois orgãos. Paralelamente às formulações de C12 foi também avaliada
a actividade terapêutica de POA e PZA na forma livre. Os grupos de animais infectados e não
tratados evidenciaram valores de carga bacteriana mais elevados. O maior efeito terapêutico
foi observado para os grupos de animais tratados com C12 nas formas livre ou incorporado em
lipossomas extrusados, particulamente no fígado. Tendo em consideração que a estirpe de M.
avium utilizada neste modelo apresenta uma resistência superior à maioria dos antibióticos
comparativamente a algumas estirpes de M. tuberculosis os resultados obtidos são bastante
promissores.
Palavras-chave
Mycobacterium avium, pirazinamida, pró-fármacos, lipossomas, modelo animal

VII
Abstract
Pyrazinamide is a first line agent for tuberculosis treatment. Being a prodrug requires
activation by Mycobacterium enzyme pyrazinamidase to be converted into its active form,
pyrazinoic acid (POA). To overcome the emergence of resistant strains the synthesis of
prodrugs, esters of POA, which may be transformed in its active form, through mycobacterial
esterases were performed. Their high abundance contributes to a reduction in the emergence of
resistances.
In the present thesis, the development of liposomal formulations of two POA esters, with
different alkoxy chain lengths, and their therapeutic potential in an in vivo model infected
with Mycobacterium avium were performed. The association of POA esters to liposomes was
based on the ability of these lipidic systems to circumvent some solubility problems associated
to these molecules and, after parenteral administration, they can passively target liver and
spleen, the main affected organs in M. avium infections.
The incorporation parameters for these two prodrugs were dependent on their chain
length. Selecting the prodrug with higher incorporation efficiencies, in vitro and in vivo studies
were performed. A systematic study was performed in the presence of HEPES buffer pH 7.4
and biological fluids. In the buffer medium all liposomal formulations evidenced high stability in
terms of percentage of incorporated prodrug and preservation of non-hydrolyzed form. In
human plasma, the prodrug hydrolysis rate was mainly influenced by mean size and rigidity of
liposomal formulations: higher half-lives were obtained for neutral and non-extruded
liposomes. In vivo studies represent the first biological evaluation and comparison of new
synthesized esters of POA either in free or in liposomal forms. Extruded liposomes, although in
in vitro stability studies have shown a higher prodrug hydrolysis rate, leading to lower CFU/g of
liver than large sized vesicles. The use of other treatment schedules and mycobacterial strains
should be further considered.
Keywords
Mycobacterium avium, pyrazinamide, prodrugs, liposomes, animal model

VIII
List of Contents
Acknowledgments………………………………………………………………………………….…....I
Communications in Scientific Meetings……………………………………………………….......III
Resumo……………………………………………………………………………………………….....IV
Abstract……………………………………………………………………………………………........VII
List of Contents……………………………………………………………………………………….VIII
List of Figures…………………………………………………………………………………….……..X
List of Tables………………………………………………………………………………………...…XII
List of Abbreviations……………………………………………………………………..………….XIV
1. Introduction ................................................................................................................ - 1 -
1.1. Mycobacterial Infections........................................................................................ - 1 -
1.1.1. Nontuberculous Mycobacterial Diseases – The particular case of MAC ......... - 1 -
1.2. Conventional treatment of mycobacterial infections ............................................... - 2 -
1.3. Strategies to overcome antibiotic resistance .......................................................... - 3 -
1.3.1. Pyrazinamide ................................................................................................ - 3 -
1.3.2. Pyrazinoic Acid Prodrugs .............................................................................. - 4 -
1.3.3. Liposomes .................................................................................................... - 5 -
1.4. Mycobacterial murine models of infection .............................................................. - 6 -
1.5. Objectives of the thesis ......................................................................................... - 7 -
2. Materials and Methods ............................................................................................... - 8 -
2.1. Materials ............................................................................................................... - 8 -
2.1.1. Chemical products ........................................................................................ - 8 -
2.1.2. Animals ......................................................................................................... - 8 -
2.1.3. Mycobacterium avium strain .......................................................................... - 8 -
2.2. Methods ............................................................................................................... - 8 -
2.2.1. Preparation of prodrug liposomal formulations ............................................... - 8 -
2.2.1.1. Characterization of prodrug liposomal formulations .................................... - 9 -
2.2.1.1.1. Liposomal size measurements ......................................................... - 9 -
2.2.1.1.2. Zeta Potential Determination .......................................................... - 10 -
2.2.1.1.3. Prodrugs quantification .................................................................. - 10 -
2.2.1.1.4. Phospholipid quantification ............................................................. - 10 -
2.2.2. Differential scanning calorimetry (DSC) studies ........................................... - 11 -
2.2.3. HPLC System ............................................................................................. - 11 -
2.2.4. Stability of C12 formulations in presence of HEPES buffer........................... - 11 -

IX
2.2.5. Stability of C12 formulations in presence of human plasma ......................... - 12 -
2.2.5.1. Half-Life quantification of C12...................................................................... - 12 -
2.2.5.2. Stability of liposome structure in presence of human plasma ....................... - 12 -
2.2.6. Murine model .............................................................................................. - 13 -
2.2.6.1. Mycobacterium avium inocula quantification ................................................ - 13 -
2.2.6.2. In vivo evolution of mycobacterial infection .................................................. - 13 -
2.2.6.3. Biological evaluation of antimycobacterial formulations ................................ - 13 -
2.2.6.4. Evaluation of M. avium growth in mice ......................................................... - 14 -
2.3. Statistical analysis .............................................................................................. - 14 -
3. Results and discussion ............................................................................................ - 15 -
3.1. Physicochemical properties of esters of POA ...................................................... - 15 -
3.2. Incorporation of C12 and C4 in liposomes ........................................................... - 16 -
3.3. Optimization of HPLC procedures for quantification of C12 and POA .................. - 21 -
3.4. Stability of C12 formulations................................................................................ - 23 -
3.4.1. Stability of C12 formulations in HEPES buffer at room temperature ............. - 23 -
3.4.2. Stability of C12 formulations in HEPES buffer at 37ºC ................................. - 24 -
3.4.3. Stability of C12 formulations in human plasma at 37ºC ................................ - 25 -
3.4.3.1. Stability of C12 in the free form ................................................................ - 26 -
3.4.3.2. Stability of C12 liposomes – influence of lipid composition ....................... - 28 -
A) C12 incorporated in PC liposomes .............................................................. - 28 -
B) C12 incorporated in DMPC liposomes ......................................................... - 29 -
C) C12 incorporated in DMPC:DMPG liposomes .............................................. - 31 -
D) C12 incorporated in DPPC .......................................................................... - 32 -
E) C12 incorporated in DPPC:DPPG ............................................................... - 34 -
F) C12 incorporated in DMPC:DSPE-PEG ....................................................... - 35 -
3.4.3.3. Stability of C12 formulations – Influence of C12 concentration ................. - 37 -
3.4.3.4. Stability of liposome structure in presence of human plasma ................... - 39 -
3.5. Mycobacterium avium murine model of infection ................................................. - 40 -
3.5.1. Biological evaluation of antimycobacterial formulations ................................ - 40 -
3.5.2. In vivo evolution of mycobacterial infection .................................................. - 40 -
3.5.3. Influence of antimycobacterial formulations on M. avium murine model ....... - 41 -
4. Conclusions and Future Perspectives .................................................................... - 45 -
5. References ................................................................................................................ - 47 -

X
List of Figures
Figure 1. Action sites of the principal anti-TB drugs…………………………………………….……2
Figure 2. Zhang hypothesis for the mode of action of PZA……………………………………….…4
Figure 3. Cross section view of a liposome structure………………………………………………..5
Figure 4. Schematic representation of the experimental M. avium murine model of
infection…………………………………………………………………………………………….…….14
Figure 5. DSC thermograms. Influence of C12 on thermotropic behavior of DPPC
liposomes………………………………………………………………………………………………...20
Figure 6. Typical chromatogram of a C12 (a) and POA (b) freshly prepared solutions………...21
Figure 7. Calibration curves for C12 (a) and for POA (b)…………………….……………….……22
Figure 8. C12 hydrolysis over time…………………………………………………………………...23
Figure 9. POA formation over time due to C12 hydrolysis…………………………………………23
Figure 10. Stability on storage of C12 formulations: variation of mean size of C12 liposomes
during one month at room temperature……………………………………………………………….24
Figure 11. Stability in buffer, at 37ºC, of C12 formulations: influence of lipid
composition………………………………………………………………………………………..…….24
Figure 12. Concentration of C12 and POA after incubation in buffer at 37ºC..………………….25
Figure 13. Hydrolysis of C12 in free form and formation of POA in the presence of human
plasma 50% (v/v)………………………………………………………………………………….…….26
Figure 14. Hydrolysis of C12 in the free form in the presence of plasma for an initial
concentration of 60 µM…………………………………………………………………………………26
Figure 15. Hydrolysis of C12 incorporated in PC liposomes and formation of POA in the
presence of human plasma 50% (v/v)………………………………………………………………...29

XI
Figure 16. Hydrolysis of C12 incorporated in DMPC liposomes and formation of POA in the
presence of human plasma 50% (v/v)………………………………………………………………...30
Figure 17. Hydrolysis of C12 incorporated in DMPC:DMPG (7:3) liposomes and formation of
POA in the presence of human plasma 50% (v/v)…………………………………………………..31
Figure 18. Hydrolysis of C12 incorporated in DPPC liposomes and formation of POA in the
presence of human plasma 50% (v/v) ………………………………………………………….…….33
Figure 19. Hydrolysis of C12 incorporated in DPPC:DPPG (7:3) non-extruded liposomes and
formation of POA in the presence of human plasma 50% (v/v)………………………..…………..34
Figure 20. Hydrolysis of C12 incorporated in DMPC:DSPE-PEG (2.85:0.15) extruded liposomes
and formation of POA in the presence of human plasma 50% (v/v)…………………………. …..35
Figure 21. Half-life of C12 formulations after incubation in human plasma 50% (v/v)................36
Figure 22. Hydrolysis of C12 and formation of POA after incubation with human plasma 50%
(v/v)……………………………………………………………………………………………………….38
Figure 23. Absorbance at 420 nm of eluted C12 DMPC:DMPG extruded liposomes following
their application on the top of a Sephadex G200 column ……………………………………….…39
Figure 24. Evolution of CFU per g of organ in liver, spleen and lung of BALB/c mice 15 days
after infection induction ……………………………………………………………………………...…41
Figure 25. Evolution of M. avium infection in mice. ………………………...………………………42
Figure 26. . Evolution of M. avium infection in mice. Influence of administered formulations on
CFU per g of organ.…………………………………………………………………………………..…43

XII
List of Tables
Table 1. Structure and physicochemical properties of C4 and C12……………………………….15
Table 2. Physicochemical characterization of C12 and C4 extruded liposomes. Influence of
alkoxy chain length of prodrugs on incorporation parameters………………………...…………...17
Table 3. Physicochemical characterization of extruded and non-extruded C12 liposomal
formulations……………………………………………………………………………………………...19
Table 4. Detection and quantification limits for C12 and POA according to the respective
standards for each calibration curve…………………………………………………………………..22
Table 5. . kobs and half-life of free C12 in the presence of human plasma. Influence of initial
concentration…………………………………………………………………………………………….27
Table 6. kobs and half-life of free C12 in the presence of human plasma. Initial concentration: 60
µM……………………………………………………………………………………………………...…27
Table 7. kobs and half-life of C12 incorporated in PC liposomes in the presence of human
plasma: influence of mean vesicle size ………………………………………………………………29
Table 8. kobs and half-life of C12 incorporated in DMPC liposomes in the presence of human
plasma: influence of mean vesicle size …………………..…………………………………………30
Table 9. . kobs and half-life of C12 incorporated in DMPC:DMPG (7:3) liposomes in the presence
of human plasma: influence of mean vesicle size……………………….…………………………..32
Table 10. kobs and half-life of C12 incorporated in DPPC liposomes in the presence of human
plasma: influence of mean vesicle size……………………………………………………………….33
Table 11. kobs and half-life of C12 incorporated in DPPC:DPPG (7:3) non-extruded liposomes in
the presence of human plasma……………………………………………………………………..…34
Table 12. kobs and half-life of C12 incorporated in DMPC:DSPE-PEG (2.85:0.15) liposomes in
the presence of human plasma………………………………………………………………………..36
Table 13. kobs and half-life of C12 in free form and incorporated in DMPC:DMPG (7:3) extruded
liposomes in the presence of human plasma………………………………………………………...39

XIII
Table 14. Stability of liposome structure in the presence of human plasma - Mean size of
liposomes at time zero and 1 and 3h post-incubation following their application on the top of a
Sephadex G200 column………………………………………………………………………………..40
Table 15. C12 liposomal formulations used for the treatment of M. avium murine model of
infection.…………………………………………………………………………………………..……...41

XIV
List of Abbreviations
Ø Mean size
[(Cx/Lip)f Final Cx to Lipid ratio
[(Cx/Lip)i Inicial Cx to Lipid ratio
ΔH Enthalpy variation
ACN Acetonitrile
AIDS Acquired immune deficiency syndrome
ATCC American Type Culture Collection
CFU Colony forming units
D.L. Detection Limit
DMPC Dimyristoyl Phosphatidylcholine
DMPG Dimyristoyl Phosphatidylglycerol
DPPC Dipalmitoyl Phosphatidylcholine
DPPG Dipalmitoyl Phosphatidylglycerol
DSC Differential Scanning Calorimetry
DSMZ Deutsche Sammlung von Mikroorganismen und Zellkulturen
DSPE Distearoyl Phosphatidyl Ethanolamine
ETB Ethambutol
h hour
HIV Human immunodeficiency virus
HPLC High Performance Liquid Chromatography
I.E. Incorporation efficiency
INH Isoniazid
i.v. Intravenous
kobs First order rate constant
Lip Lipid
ln neperian logarithm
Ltd Limited
log Poct log octanol/water partition coefficient

XV
M. avium Mycobacterium avium
M. tuberculosis Mycobacterium tuberculosis
MAC Mycobacterium avium complex
min minute
MPS Mononuclear phagocytic system
NAD Nicotinamide adenine dinucleotide
non-MDR-TB non-Multi-Drug-Resistant-Tuberculosis
NTM Nontuberculous mycobacterial
OADC Oleic acid-albumin-dextrose-catalase
PC Egg Phosphatidylcholine
PdI Polydispersity index
PEG Polyethyleneglycol
POA Pyrazinoic Acid
PZA Pyrazinamide
PZase Pyrazinamidase
RIF Rifampicin
RT Retention time
s second
S.D Standard deviation
t1/2 Half-life time
Tc Phase transition temperature
TB Tuberculosis
UK United Kingdom
USA United States of America

- 1 -
1. Introduction
1.1. Mycobacterial Infections
Mycobacteria are Gram positive, nonspore-forming, aerobic bacteria. They are
considered facultative intracellular microorganisms presenting intracellular and extracellular
multiplication phases. Macrophages, resident phagocytes in different organs, are the main cells
that engulf invading microorganisms. Some pathogens that are internalized by macrophages
are transferred to lysosomal organelles followed by degradation. However, pathogenic
mycobacteria evade innate immunity by manipulating host, ensuring long-term survival and
proliferation that can occur in the phagosome (Gaspar et al., 2008a).
There are now over 130 known species of mycobacteria. However, only a few play an
important role as pathogenic agents for immunocompetent human hosts, namely
Mycobacterium tuberculosis, Mycobacterium leprae and Mycobacterium ulcerans, the causative
agents of tuberculosis (TB), leprosy and Buruli ulcer, respectively. The majority of other
Mycobacterium species are present in the environment as saprophytes, some of which can be
opportunistic and often deadly pathogens for immunocompromised human hosts. From this
group of environmental opportunistic mycobacteria, those from Mycobacterium avium complex
(MAC) are responsible for a large percentage of nontuberculous mycobacterial (NTM) diseases
(Reed et al., 2006).
1.1.1. Nontuberculous Mycobacterial Diseases – The particular case of
MAC
NTM diseases are acquired from environmental (water, soil) reservoirs and are not
transmitted between humans or between animals and humans. NTM infection progression to
clinical disease requires one or more predisposing host conditions. NTM species that cause
pulmonary disease vary by geographic region being MAC the predominant pathogen (Cook,
2010). In pre-AIDS (acquired immune deficiency syndrome) era, MAC, predominantly caused
pulmonary mycobacteriosis, that were difficult to distinguish radiologically from tuberculosis and
were rarely associated with extrapulmonary sites or disseminated diseases (Wolinsky, 1979).
Disseminated MAC became a frequent diagnosis in late-stage HIV (human immunodeficiency
virus) infected patients, usually occurring in patients with CD4+ T-cell counts below 50/mm
3.
Food-stuffs and water are the source of these organisms and the portal of entry in hosts is likely
to be the gastrointestinal tract. The syndrome is characterized by fever, shills, sweats, malaise,
weight loss, abdominal pain, and diarrhea (Cynamon and DeStefano, 1999). MAC refers to a
group of two closely related environmental mycobacteria: M.avium and M. intracellulare (Cosma
et al., 2003). Unlike TB, MAC disease is not required to be reported to public health entities in
most countries, and therefore, precise incidence and prevalence data are not available (Gaspar
et al., 2008a).

- 2 -
1.2. Conventional treatment of mycobacterial infections
Mycobacterial species share a characteristic thick cell wall, with a unique composition
that confers an exceptionally strong barrier to antibiotics, leading to resistances to a wide variety
of antimycobacterial agents. The lipid content of this cell wall gives rise to the common
characteristics of the Mycobacterium genus, which is the ability to retain basic dyes in presence
of an acid-alcohol solution (Cosma et al., 2003). The cell wall of mycobacteria is composed of
four layers: the first consists of peptidoglycan and the other three of a variety of soluble
proteins, carbohydrates, lipids and insoluble macromolecular components like arabinogalactan,
peptidoglycan and mycolic acid. The outer lipid barrier is extremely hydrophobic due to the
covalent bond between molecules of mycolic acid and arabinogalactan (Gaspar et al., 2008a).
The thick cell wall of mycobacterial species is in the origin of the high difficulty on the treatment
of mycobacterial infections. The most effective drugs to control M. tuberculosis infection are
Isoniazid (INH), Ethambutol (ETB), Pyrazinamide (PZA) and Rifampicin (RIF). These drugs are
first line therapeutic drugs and they represent the basis of tuberculosis therapeutic regiments. In
Figure 1 are shown their action sites in mycobacterial membrane: RIF inhibits bacterial RNA
synthesis by binding to the β-subunit of bacterial DNA-dependent RNA polymerase and
blocking the initiation chain formation in RNA synthesis; PZA is converted to Pyrazinoic Acid
(POA) and by decreasing the pH inside the mycobacterial cell prevents the microorganism
growth. PZA may also function as an antimetabolite of nicotinamide and interferes with the
synthesis of nicotinamide adenine dinucleotide (NAD), inhibiting the synthesis of short-chain,
fatty-acid precursors; ETB inhibits mycobacterial arabinosyl transferases involved in the
polymerization of D-arabinofuranose to arabinoglycan, an essential cell wall component; INH is
a prodrug activated by the mycobacterial catalase-peroxidase enzyme KatG inhibiting the
synthesis of mycolic acids, an essential component of mycobacterial cell walls (du Toit et al.,
2006).
Figure 1. Action sites of the principal anti-TB drugs. Adapted from du Toit et al., (2006)
Pyrazinamide
Isoniazid
Ethambutol
Rifampicin

- 3 -
In contrast with TB therapy, which is nearly uniformly effective when correctly administered
to non-Multi-Drug-Resistant-TB (non-MDR-TB), the therapy of MAC infections has historically
been less efficient and there are no standardized treatments (French et al., 1997; Georgiev,
1994; Nuermberger and Grosset, 2004). Therapeutic regimens include drugs such as
clarithromycin, RIF, Amikacin, Ciprofloxacin, ETB, Azithromycin, Rifabutin. However, an
increased number of mycobacterial strains have shown to be resistant to the available drugs. A
combinatory therapy for the treatment of these infections is recommended since monotherapy
gives rise to an enhanced occurrence of drug resistance and clinical failure.
1.3. Strategies to overcome antibiotic resistance
In an attempt to overlap the emergence of resistance strains to the available first-line
drugs, a viable strategy may be achieved by synthesis of new molecules or chemical
modification of already available ones. The design of new molecules such as prodrugs, entities
that need to undergo an enzymatic and/or chemical transformation in vivo to release the active
parent drug constitutes a good alternative. The development of prodrugs has become an
established tool for improving physicochemical, biopharmaceutical or pharmacokinetic
properties of pharmacologically active molecules and represent an increased tendency in
pharmaceutical industry (Rautio et al., 2008). In particular, the prodrug approach via activation
inside mycobacterial cells is a relevant alternative especially considering the problematic
penetration of drugs into these cells (Valente et al., 2011).
1.3.1. Pyrazinamide
PZA, a first line agent for the treatment of TB is also a prodrug. PZA plays an important
role in shortening TB therapy. This ability is related to its activity against a population of semi-
dormant mycobacteria residing in an acidic pH environment and that are not killed by other TB
drugs (Wade and Zhang, 2004; Zhang and Mitchison, 2003). In fact in vitro tests revealed a
lower activity in normal culture conditions close to neutral pH, while higher activity is observed in
acidic media (Wade and Zhang, 2004).
In order to have antimycobacterial activity, PZA requires activation by the
Mycobacterium enzyme pyrazinamidase (PZase) to be transformed into its active form, the
pyrazinoic acid (POA). This enzyme conveniently activates the drug only inside the bacteria
(Pires, 2011). The hypothesis for the mode of action of PZA according to Zhang and Mitchison,
(2003) is shown in Figure 2. PZA diffuses into the bacterial cell being then deaminated to form
POA. This molecule can only leave the cell again when it is excreted by an inefficient
mycobacterial efflux pump requiring energy (Mitchison and Fourie, 2010; Zhang et al., 2003).

- 4 -
Figure 2. Zhang hypothesis for the mode of action of PZA. Adapted from Mitchison and Fourie, (2010)
However, since the activation of PZA is dependent on only one enzyme, it has been
observed the emergence of resistant strains to this prodrug. As reviewed by Zhang and
Mitchison, (2003) the acquired PZA resistance in M. tuberculosis occurs by pncA gene mutation
(Raynaud et al., 1999).In other mycobacteria infections, in particular by M. avium, the natural
resistance to PZA may be due to an efficient POA efflux mechanism (Zhang and Mitchison,
2003). On the other hand the administration of POA, the active form of PZA, is not
recommended for treatment of mycobacteriosis due to its poor absorption and significantly
serum binding (Konno et al., 1967).
1.3.2. Pyrazinoic Acid Prodrugs
To overcome the already mentioned drawbacks, the synthesis of new POA prodrugs
were conducted by several authors (Cynamon et al., 1992; Cynamon et al., 1995; Fernandes et
al., 2010; Yamamoto et al., 1995). Cynamon et al., (1992) prepared series of POA esters and
evaluated their activity in vitro. Compared with PZA some compounds displayed higher activity
than PZA against M. tuberculosis, M. avium and M. kansasii (Cynamon et al., 1995). However,
these new compounds had lower stability in presence of biological fluids (Bergmann et al.,
1996).
Recently Simões et al. (2009) have synthesized series of pyrazinoic lipophilic prodrugs
esters with different alkoxy groups, and amide (Simões, 2005; Simões et al., 2009; Valente et
al., 2011). They observed that lipophilic esters with long alkoxy chains were more resistant to
plasma hydrolysis than the short chain esters (Simões et al., 2009). The selection of long chain
esters over amide prodrugs was based on the fact that lipophilic esters are easily activated by
mycobacterial esterases. This may contribute to a reduction in resistances to these prodrugs.
These esters were active in concentrations 10-fold lower than those needed for PZA to kill
sensitive M. tuberculosis in in vitro tests. On the other hand, it seems that amide prodrugs
cannot easily be activated by mycobacterial enzymes and thus have low in vitro activity (Simões
et al., 2009).

- 5 -
Besides the development of prodrugs with high in vitro activity, when applied to in vivo,
their pharmacological performance will be dependent on several factors, such as clearance or
metabolization after administration; distribution to infected sites, possible binding to plasmatic
proteins and the toxicity resulting from localization in non-affected sites, restricting the amount
of drug that can be administered (Gaspar et al., 2008a). A good strategy to overcome some of
these possible drawbacks could be the association of therapeutically promising molecules to
drug delivery systems. Ideally, these delivery systems should allow a preferential targeting of a
specific compound within a therapeutic concentration range at the affected sites; should be non-
toxic and non-immunogenic; should be biodegradable or easily excreted after exerting its effect;
and should be cheap and stable upon storage (Gaspar et al., 2008a). One of the most
extensively studied drug delivery systems are liposomes.
1.3.3. Liposomes
Liposomes, by definition, are submicron lipidic particles consisting of one or more
concentric lipid bilayers, separated by aqueous compartments (Gaspar et al., 2008a). It has
been more than 4 decades since the first report of the successful preparation of liposomes by
Bangham et al., (1965). There are several types of liposomes according to their lipid
compositions, number of lipid bilayers, superficial charges and mean sizes, ranging from few
nanometers to several microns. These liposomes can also be prepared using different methods
(Crommelin et al., 1994). Liposomes have been widely used for targeted drug delivery due to
their structural versatility, biodegradability, innocuous nature and resemblance to biological
membranes (Gregoriadis and Ryman, 1972). They can entrap drugs of different sizes and
solubility properties since the water-soluble compounds will be encapsulated in the aqueous
spaces while lipid-soluble ones will be incorporated in the lipid bilayer (Cruz et al., 2009;
Torchilin, 2005). In Figure 3 is shown the schematic structure of a liposome.
Figure 3. Cross section view of a liposome structure: (a) Hydrophilic head; (b) Hydrophobic tail; (c)
Internal aqueous space.
(Available at 22-08-2012 in http://www.britannica.com/EBchecked/media/92244/Phospholipids-can-be-
used-to-form-artificial-structures-called-liposomes )
Liposomes can be prepared according to the physicochemical properties of the molecule to
be incorporated and in order to reach the target either for therapy or for diagnosis (Cruz et al.,
2009). The structural and functional diversity of liposomes has been explored for the design of

- 6 -
drug carrier systems in particular for the treatment of mycobacterial infections (Fielding and
Lasic, 1999). After intravenous (i.v.) administration, some liposomes, depending on their
properties, are rapidly removed from blood circulation by the phagocytic cells of the
mononuclear phagocyte system (MPS), particularly by macrophages in liver and spleen. This
natural tendency has been exploited for treatment of infectious diseases localized in these
organs (Gaspar et al., 2008a). Intracellular infections caused by M. avium and M. tuberculosis
may be envisioned as a preferential target for liposomes The therapeutic advantages by
incorporating antibiotics in liposomes over the free respective molecules has been
demonstrated in several animal models of infection (Fielding and Lasic, 1999; Gaspar et al.,
2008a; Pinto-Alphandary et al., 2000). This superior therapeutic effect may be explained by the
different biodistribution profile of liposomal formulations in comparison with the free antibiotic
(Allen and Hansen, 1991; Allen and Stuart, 1999; Bakker-Woudenberg et al., 1993; Gaspar et
al., 2008b). However, the use of these type of liposomes, that are rapidly cleared from
bloodstream, may not be the right choice when other organs than liver and spleen are the
target. For inflammation and tumor pathological situations, that are not localized in liver and
spleen, the use of other type of liposomes with longer blood circulation half-lives represent
better solutions. Thus, the selection of lipid components is essential for the design of a
liposomal formulation. One successful hypothesis has been achieved by including in the lipid
composition the polyethyleneglycol (PEG), a polymer covalently linked to a phospholipid. The
presence of PEG at liposome surface is able to reduce plasmatic proteins adsorption and
consequently to decrease MPS clearance (Allen and Cullis, 2004; Torchilin, 2005). The
increased circulation time of liposomes has improved the delivery of therapeutic molecules to
infections localized in other organs than liver and spleen or even to inflammation and tumor
sites (Bakker-Woudenberg et al., 1993; Corvo et al., 2002; Gaspar et al., 1996; Gaspar et al.,
2007; Liu et al., 2006).
Therefore, the incorporation of esters of POA within liposomes appears as a good strategy
to enhance their in vivo antimycobacterial properties.
1.4. Mycobacterial murine models of infection
During the past years various animal models of MAC infection have been explored,
namely pigs, sheep, fowl, rabbits, goats, mice and guinea-pigs. However, the mouse is the most
widely used model of infection to evaluate the chemotherapeutic effect of antimycobacterial
drugs (Cynamon and DeStefano, 1999). Mice require small laboratory space, their purchase
and maintenance are relatively inexpensive and handling of these animals is easy if performed
by trained personal.
The induction of the infection may be perforrmed by a variety of routes including
intraperitoneal, intranasal, oral, intrarectal, aerogenic and i.v. The i.v. route is the most
commonly used as provides a consistent disseminated disease model with viable organisms
recovered from spleen, liver, blood, lung and lymph nodes. Usually for i.v. induction, mice are
infected in the tail vein. Each experiment consists of an early control group that is sacrificed at

- 7 -
the beginning of treatment and a later control that is sacrificed at the end of treatment schedule,
together with all treated groups. Primary cultures of mycobacteria species to be used for
infection may be obtained from clinical isolates or from bioresource centers such as American
Type Culture Collection, (ATCC) or from German Collection of Microorganisms and Cell Culture.
1.5. Objectives of the thesis
The main objectives in the present work were the development and characterization of
liposomal formulations of prodrugs of pyrazinoic acid (POA), particularly esters of POA, that in
previous in vitro tests have demonstrated to be active against M. tuberculosis (Simões et al.,
2009). The possible therapeutic effect of some of these liposomal formulations was tested in a
preliminary in vivo model infected with M. avium.
In order to fulfill the objectives of the present master thesis the work was executed
according to the following activities:
Development of best liposome methodologies for incorporating POA prodrugs in
liposomes. The influence of lipid composition, mean size, superficial charge and of the
physicochemical properties of POA prodrugs on incorporation parameters were studied.
Optimization of HPLC methodologies for evaluating the stability of the prodrug in buffer
and biological media. The comparison of chemical stability of the prodrug in free and
liposomal forms was performed.
Establishment of a M. avium model of infection that may be suitable for testing the
chemotherapeutic effect of POA prodrugs was carried out. Microbiological techniques
were used.

- 8 -
2. Materials and Methods
2.1. Materials
2.1.1. Chemical products
The prodrugs used C4 and C12 (esters of pyrazinoic acid) were previously synthesized
by the Medicinal Chemistry group of the Faculty of Pharmacy of the University of Lisbon
according to Simões et al., (2009). The following pure phospholipids were purchased from
Avanti Polar Lipids (USA): Dimyristoyl Phosphatidylcholine (DMPC), Dimyristoyl
Phosphatidylglycerol (DMPG), Dipalmitoyl Phosphatidylcholine (DPPC), Dipalmitoyl
Phosphatidylglycerol (DPPG), egg Phosphatidylcholine (PC) and Distearoyl Phosphatidyl
Ethanolamine covalently linked to Polyethyleneglycol (DSPE-PEG). Deionized water (Milli-Q
system; Millipore, Japan) was used for the preparation of all experimental solutions.
Middlebrook 7H9 broth and 7H10 agar, and Bacto Middlebrook albumin-dextrose-catalase and
oleic acid-albumin-dextrose-catalase (OADC) enrichments were obtained from Difco
Laboratories (USA). Nuclepore Track-Etch Membranes were purchased from Whatman Ltd,
USA. Kolliphor ELP from BASF, Germany, was kindly supplied by DS Produtos Químicos, Lda,
Portugal. All other reagents were of analytical grade.
2.1.2. Animals
Male BALB/c mice (5 to 7 weeks old, weight 25-30 g) were obtained from the
Gulbenkian Institute of Science (Portugal). The animals were kept under hygienic conditions,
fed commercial chow, and given acidified drinking water ad libitum. All the experimental
procedures were carried out with the permission of the local laboratory animal committee.
2.1.3. Mycobacterium avium strain
The bacterial strain used for the mouse model was M. avium 44157 from DSMZ
depository (Deutsche Sammlung von Mikroorganismen und Zellkulturen) and was kindly
provided by Unidade dos Retrovírus e Infecções Associadas of de Faculty of Pharmacy of the
University of Lisbon.
2.2. Methods
2.2.1. Preparation of prodrug liposomal formulations
Multilamellar vesicles composed of selected phospholipids were prepared as
previously described (Gaspar et al., 2000). The phospholipids and each prodrug used, in a
molar ration of 1:10, were previously solubilized in chloroform and dried by rotary evaporation

- 9 -
(Buchi, Switzerland) under nitrogen stream, to eliminate the organic solvent, until the formation
of a thin lipid film. The film was dispersed in deionized water, frozen and lyophilized (Edwards,
USA) overnight. Rehydration of lyophilized powder was made in two steps of 30 minutes (min)
each, at a temperature above the phase transition temperature (Tc) of the correspondent lipid
mixture. First rehydration was done in a volume of 2/10 of the final volume with 150 mM NaCl in
10 mM HEPES buffer, pH 7.4 (HEPES buffer, pH 7.4). After 30 min, rehydration was completed
with the same buffer. In order to reduce and homogenize the diameters of prodrug liposomes,
some formulations were submitted to an extrusion technique using an Extruder device (Lipex
Biomembranes Inc., Canada). Liposomal suspensions were sequentially filtered through
polycarbonate membranes of different porosities (0.8, 0.6, 0.4 and 0.2 µm) using a nitrogen
pressure ranging from 100 to 500 lb/in2 , until an average vesicle size between 0.1 – 0.2 µm was
achieved. The separation of non-incorporated prodrug from liposomes was performed by
ultracentrifugation (300 000 g for 2 hour (h) at 17ºC in a Beckman LM-80 ultracentrifuge
(Beckman Instruments, USA). The obtained pellet was ressuspended in the desired volume of
HEPES buffer, pH 7.4.
2.2.1.1. Characterization of prodrug liposomal formulations
Prodrug liposomal formulations were characterized in terms of lipid composition, lipid
and prodrug concentration, zeta potential, mean vesicle size and polydispersity index. The
following abbreviations and equations were used to determine these incorporation parameters:
(equation 1)
2.2.1.1.1. Liposomal size measurements
Liposome mean diameter was determined by dynamic light scattering in a
hydrodynamic sizing system (Zetasizer Nano S, Malvern Instruments, UK). This technique was
based on Brownian motion and was related to the size of the particles. For viscosity and
refractive index, the values of pure water were used. As a measure of particle size distribution
of the dispersion, the system reports the polydispersity index (PdI). The index ranges from 0.0
for an entirely monodisperse sample up to 1.0 for a polydisperse suspension. To determine the

- 10 -
mean diameter and PdI of liposomal preparations, a dilution to a final lipid concentration of 0.3
mM in HEPES buffer, pH 7.4 was previously performed, and samples were placed in an
appropriate polycarbonate cell. To ensure that appropriate mean diameter and PdI were
achieved, besides the measurements done for final liposomal preparations, these parameters
were also determined during the extrusion procedure.
2.2.1.1.2. Zeta Potential Determination
Zeta potential of liposomal formulations was measured in a hydrodynamic sizing system
(Zetasizer Nano Z, Malvern Instruments, UK). Zeta potential is defined as an electric potential
between the membrane surface and the ionic dispersion medium. This method determines how
fast a particle moves in a liquid when an electrical field is applied. For viscosity and refractive
index, the values of pure water were used.
Before determination of the zeta potential of liposomal formulations, an initial check of
the apparatus was made with a standard of a known zeta potential value (standard DTS5050,
Malvern Instruments, Ltd., UK). Dilutions of liposomal formulations were made in HEPES buffer,
pH 7.4 for a final lipid concentration of 0.3 mM. Samples were slowly introduced into a clear
disposable zeta cell with a syringe to avoid air bubbles. The zeta potential of samples at a
temperature of 25ºC was recorded. Three independent dilutions were prepared for each
liposomal formulation under study.
2.2.1.1.3. Prodrugs quantification
The prodrugs were quantified spectrophotometrically at 267 nm after disruption of the
liposomes with ethanol. Briefly, samples containing an amount of prodrug between 6.25 and
31.25 µM were pipetted in triplicate into 1.5 mL tubes and completed to 1 mL with ethanol. In
parallel, a calibration curve using a 2.5 mM stock prodrug solution and standards ranging from
6.25 to 31.25 µM were used. All tubes were shaken for 10 min and absorbances at 267 nm
were recorded in a UV 160 Spectrophotometer (Shimadzu, Japan).
The amount of prodrug in samples was obtained using a linear regression. The
calibration curve was linear up to absorbance values of at least 0.364.
2.2.1.1.4. Phospholipid quantification
The method for phospholipid quantification was based on the colorimetric determination
of PO43-
according to Rouser et al., (1970). Briefly, samples in triplicate containing a phosphate
amount between 20 and 80 nmol (sample volume less than 100 µL) were pipetted into 15 mL
glass tubes. In parallel, a calibration curve with phosphate amounts ranging from 20 to 80 nmol
was prepared using a 0.5 mM phosphate stock solution. All samples were then heated (180ºC)
in a heating block until their dryness. After cooling, 0.3 mL of perchloric acid (70-72%) was
added to all tubes. Marbles were placed on the top of all tubes and heated at 180ºC for 1 h to
convert all the organic lipid phosphate to inorganic form. After cooling samples to room

- 11 -
temperature, 1 mL of distilled water, 0.4 mL of hexa ammonium heptamolybate solution [1.25%
(w/v)], followed by 0.4 mL of ascorbic acid solution [5% (w/v)] were added to all glass tubes. All
tubes were heated in a boiling water bath for 5 min. During heating, the inorganic phosphate is
converted to phosphomolybdic acid due to reduction of ascorbic acid and a blue color is
developed. After cooling, the absorbance at 797 nm of all samples was recorded in a UV 160
Spectrophotometer (Shimadzu, Japan) against the blank of the calibration curve. The amount of
phosphate in samples was obtained using a linear regression. The calibration curve was linear
up to absorbance values of at least 1.000.
2.2.2. Differential scanning calorimetry (DSC) studies
The phase transition behavior of POA esters in liposomal formulations was performed in
a calorimeter DSC Q200 (TA Instruments, USA). Approximately 10 µL of liposomal
formulations (lipid concentration ca. 60 mM) were accurately measured into aluminum pans
which were hermetically sealed and then measured against an empty reference pan. The pans
were heated and the thermograms were recorded at a temperature ranging from 15 to 65ºC at
a heating rate of 3ºC/min.
2.2.3. HPLC System
All stability studies were made using C12. The stability of C12 was evaluated by HPLC
following an optimization of best conditions for quantifying the prodrug C12 and the
corresponding hydrolysis product, the POA, according to Simoes et al., (2009). The Beckman
System Gold HPLC system consisted of a 126 Pump Direct Control, the model detector 166
from Beckman Instruments, Inc and the Midas Spark 1.1 autoinjector. The wavelenght of the
detector was set at 267 nm. The analytical column was a LiChroCart® 125-4 Purospher Star
RP-8 (5 µm) (Merck, Germany). The system was attached to a computer with the software,
Katarat 7.7 for integration and treatment of chromatograms. The mobile phase, in an isocratic
solvent system, consisted in 75% of acetonitrile (ACN) for C12 and in 2% for POA in
KH2PO4/H3PO4 25 mM, (phosphate buffer, pH 2.0) with a flow rate of 1 mL/min. Calibration
curves for each compound, C12 and POA, were constructed with different standards ranging
from 5 to 25 µM. The limits of detection and quantification were calculated based on the
different standards used. All the quantifications were performed using calibration curves
constructed freshly prepared C12 and POA solutions.
2.2.4. Stability of C12 formulations in presence of HEPES buffer
The stability of C12 liposomes in HEPES buffer was evaluated by incubating the
formulations at 37ºC. At defined times, aliquots of suspensions were taken and applied onto the
top of a PD10 column to separate non incorporated C12. The fraction correspondent to
liposomes was analyzed for C12 and lipid contents. The mean size and superficial charge were
also analyzed along the incubation period. Different lipid compositions were characterized
(DMPC, DMPC:DSPE-PEG (2.85:0.15), DMPC:DMPG (7:3)). The stability was defined as the

- 12 -
ratio in percentage between C12 to lipid ratio at each studied time and the C12 to lipid ration at
time zero as shown in the following equation:
(equation 2)
,where tx are the ratios obtained at 0.5, 1, 4 and 24 h after incubation and t0 is the ratio at time
zero.
2.2.5. Stability of C12 formulations in presence of human plasma
The stability in plasma of C12 in free and liposomal forms was evaluated by incubating
the formulations in the presence of plasma diluted with HEPES buffer, pH 7.4 (50% v/v). The
hydrolysis of C12 and the formation of POA were determined by HPLC. At defined times 100 µL
of the reaction medium were 10 times diluted in ACN or in phosphate buffer, pH 2.0,
respectively for C12 and POA. Samples were centrifuged (Bench Centrifuge 202 MK, Sigma,
Germany) for 15 min, at 13 000 g at 17ºC, and the supernatant was filtrated with a 0.2 µm filter
and injected onto the HPLC system.
2.2.5.1. Half-Life quantification of C12
Apparent pseudo first-order kinetics and rate constants were determined, according to
Gupta et al., (2009), by using initial rates of hydrolysis. The apparent pseudo first-order
hydrolysis rate constants of C12 at 37ºC were determined by plotting the logarithm of C12
concentration as a function of time according to the equation:
(equation 3)
The hydrolysis half-live (in min) was then calculated by the equation:
(equation 4)
2.2.5.2. Stability of liposome structure in presence of human plasma
The evaluation of liposome structure in presence of human plasma was performed by
incubating liposomal formulations at 37ºC. At defined times aliquots of the suspension were
taken and applied onto the top of a Sephadex G200 column to separate liposomes from human
plasma. The fraction correspondent to liposomes was analyzed in terms of turbidity and
recorded by spectrophotometry at 420 nm. The mean size was also analyzed along the

- 13 -
incubation period. A single lipid composition was tested – DMPC:DMPG (7:3) extruded
liposomes.
2.2.6. Murine model
2.2.6.1. Mycobacterium avium inocula quantification
Inocula were prepared as previously described (Silva et al., 1987). Briefly, transparent
colonies of M. avium DSMZ 44157 were subcultured in Middlebrook 7H9 broth with albumin-
dextrose-catalase supplement and 0.04% Tween 80 (w/v) and allowed to grow at 37ºC on an
orbital shaker for 2 weeks. The bacteria were harvested by centrifugation (2 000 g, 10 min) in a
GPR Beckman centrifuge (Beckman Instruments, USA), suspended in a small volume of saline
with 0.04% Tween 80 (w/v), sonicated at low energy for 90 second (s) in a bath-type sonicator
(Bandelin Sonorex RK156, Germany) to disrupt bacterial clumps, diluted in the same medium to
an optical density of 0.8 at 600 nm and stored frozen at -70ºC until use. When needed, aliquots
were diluted to the desired concentration, and inoculated.
2.2.6.2. In vivo evolution of mycobacterial infection
BALB/c mice were injected intravenously with 5x105 colony forming units (CFU) of M.
avium DSMZ 44157. Fifteen days after infection, mice were sacrificed and their livers, spleens
and lungs were aseptically removed and homogenized. Homogenates were suspended and
serially diluted in 0.04% Tween 80 (w/v) and plated onto Middlebrook 7H10 agar medium
enriched with OADC for CFU counting after incubation at 37ºC for 10 to 15 days (Gaspar et al.,
2000).
2.2.6.3. Biological evaluation of antimycobacterial formulations
A murine model of M.avium infection previously described (Pedrosa et al., 1994) was
used. Each animal was infected by i.v. injection in a lateral tail vein with 1x106 CFU per mouse
in 200 µL of a M. avium suspension.
The therapeutic treatment started 2 weeks after the infection. On each week, treated
animals received three i.v. injections in a lateral tail vein during 2 weeks. The formulations under
study were C12 in the free form (solubilized in HEPES buffer pH 7.4 and Kolliphor ELP 5%
(w/v)) or incorporated in DMPC:DMPG (7:3) extruded and non-extruded liposomes. POA and
PZA previously solubilized in HEPES buffer pH 7.4 were also tested. The administered dose
was 12mg/kg of body weight related to POA.

- 14 -
2.2.6.4. Evaluation of M. avium growth in mice
Two days after the last treatment mice were killed by cervical dislocation and livers and
spleens were removed according to the method described in 2.2.6.2.
In Figure 4 a schematic representation of M. avium murine model of infection used in
this work is shown.
Figure 4. Schematic representation of the experimental M. avium murine model of infection
2.3. Statistical analysis
Data presented are expressed as mean (±) and standard deviation (S.D.). Statistical
analysis was performed using Single Factor ANOVA. The acceptable probability for a significant
difference between mean values was p<0.05 (Zar, 2009).

- 15 -
3. Results and discussion
3.1. Physicochemical properties of esters of POA
In previous work, developed by Simões et al. (2009) series of esters of POA, with a linear
alkoxy chain, ranging from 4 to 16 carbon atoms have been synthesized and stability studies performed
either in plasma or in macrophage cell cultures have demonstrated to be good candidates as
antituberculous drugs. Two of those compounds were selected for the present work to be incorporated
in liposomes: the esters containing 4 and 12 carbons linear chain length (C4 and C12, respectively).
The rational for associating these two esters of POA to liposomes was based on the fact that these lipid
systems are able to circumvent some solubility problems associated to these molecules. As widely
described in literature after parenteral administration liposomes can passively target their incorporated
material to infected cells in liver and spleen as is the case of M. avium infections (Cruz et al., 2009;
Gaspar et al., 2008a).
C12 and C4 are two prodrugs with different properties, which may influence their incorporation
in liposomes as well as their retention. In Table 1 are shown some physicochemical properties of these
two low molecular weight prodrugs, in particular the log octanol/water partition coefficient (log Poct.). The
increase on the carbon linear chain resulted on a concomitant enhancement on the log Poct from 0.9 to
4.55 for C4 and C12, respectively. The log Poct is a measure of the equilibrium concentration of a solute
between two immiscible phases: octanol and water. It may predict the potential for partitioning into
hydrophobic compartments such as lipid bilayers and hydrophilic compartments such as the aqueous
core of liposomes (Ribeiro et al., 2010). Generally, we can say that molecules with a log Poct higher than
1 present a lipophilic character while solutes with a log Poct below 1 are considered to be hydrophilic.
Moreover, log Poct > 5 correspond to high lipophilic compounds (Allen and Stuart, 1999; Defrise-
Quertain et al., 1984).
Table 1. Structure and physicochemical properties of C4 and C12
Data from Simões et al., (2009)
Prodrug R log Poct Molecular Weight
(MW)
C4 n-Butyl - (CH2)3CH3 0.9 180.2
C12 n-Dodecyl - (CH2)11CH3 4.55 292.4

- 16 -
3.2. Incorporation of C12 and C4 in liposomes
Due to the hydrophobic properties of C12 and C4, the liposome method for incorporating these
molecules in liposomes was the lipid film method, where both phospholipids and prodrugs were
solubilized in chloroform, submitted to an evaporation step, rehydrated with water, lyophilized overnight
and then rehydrated with a suitable buffer. This methodology has been widely used for incorporating low
molecular weight molecules of hydrophobic properties (Carvalheiro et al., 2009; Constantino et al.,
1993; Gaspar et al., 2000)
As described in materials and methods, two types of liposomes were used in the present work:
extruded and non-extruded. This technique constitutes one of the best methodologies to reduce and
homogenize the mean size of liposomes as it is suitable for the preparation of lipid systems in a scale
ranging from one to hundreds of mL (Cruz et al., 2009).
The influence of different lipid compositions on incorporation parameters was studied. Taking
this into consideration, the influence of phase transition temperature (Tc)* of the phospholipids as well
as the presence of negatively charged phospholipids in the lipid composition on the incorporation
efficiencies (I.E.) was compared for both prodrugs. The phospholipids tested were Dimiristoyl
Phosphatidylcholine (DMPC), Dimiristoyl Phosphatidylglycerol (DMPG) with a Tc of +23ºC and
Dipalmitoyl Phosphatidylcholine (DPPC) and Dipalmitoyl Phosphatidylglycerol (DPPG) with a Tc of
+41ºC. Liposomal formulations of C4 and C12 were prepared with neutral phospholipids (DMPC or
DPPC) and lipid mixtures containing also negatively charged phospholipids (DMPG or DPPG). In
addition, zeta potential, mean vesicle sizes and PdI were determined for all liposomal formulations.
In Table 2 are shown the physicochemical properties of extruded C12 and C4 liposomal
formulations.
*Tc values for the phospholipids referred above were obtained from Gunstone et al., (1986)

17
Table 2. Physicochemical characterization of C12 and C4 extruded liposomes. Influence of alkoxy chain length of prodrugs on incorporation parameters
Formulation nº
Lipid
Composition
(Molar Ratio)
Prodrug (Cx/Lip)i
(nmol/µmol)
(Cx/Lip)f
(nmol/µmol) I.E. (%)
Ø (µm)
(PdI)
Zeta Potential
(mV)
1 DMPC C12 117 ± 24 107 ± 16 92 ± 6 0.19
(0.4 - 0.5) -4 ± 1
2 DMPC C4 107 ± 1 8 ± 2 5 ± 2 0.18
(<0.2) -3 ± 1
3 DPPC C12 135 ± 11 103 ± 18 96 ± 10 1.3
(0.4 - 0.5) -2 ± 1
4 DPPC C4 129 ± 10 5 ± 1 4 ± 1 1.2
(0.5 - 0.6) -2 ± 2
5 DMPC:DMPG (7:3) C12 93 ± 5 85 ± 5 91 ± 4 0.16
(<0.2) -41 ± 4
6 DMPC:DMPG (7:3) C4 153 ± 4 7 ± 1 5 ± 1 0.16
(<0.2) -42 ± 4
7 DPPC:DPPG (7:3) C12 87 ± 2 95 ± 2 110 ± 6 0.17
(<0.2) -35 ± 3
8 DPPC:DPPG (7:3) C4 141 ± 3 6 ± 1 5 ± 1 0.19
(0.2 - 0.3) -39 ± 3
Initial lipid concentration – 20 mM; Initial prodrug concentration – 2 mM; [(Cx/Lip)f – Final Cx to Lipid ratio; [(Cx/Lip)i – Inicial Cx to Lipid ratio; Incorporation Efficiency I.E. (%) - [(Cx/Lip)f / (Cx/Lip)i)] * 100; Ø – mean size of liposomes; PdI – Polydispersity index.. Values correspond to mean ± S.D. of at least three independent preparations

18
C12 and C4 liposomal formulations presented a mean size below 0.2 µm with the
exception of liposomes prepared with the phospholipid DPPC (Formulations 3 and 4). For these
two formulations the mean vesicle sizes were higher than 1.2 µm after final preparation of
liposomes even using the extrusion step. DPPC is a neutral and rigid phospholipid with a Tc of
+41ºC and the possibility of aggregation due to its low superfical charge may be one of the
reasons of these high mean diameters (Crommelin and Schreier, 1994). For the other neutral
formulations (Formulations 1 and 2), prepared with a less rigid phospholipid (DMPC), this
aggregation effect was not observed. The zeta potential observed for Formulations 1, 2, 3 and 4
ranged from -2 to -4 mV. C12 and C4 liposomes containing in the lipid composition the
negatively charged phospholipids DMPG or DPPG, gave rise to formulations with a superficial
charge in agreement with their constituints: zeta potential values ranged from -35 to -41 mV. In
terms of loading capacity, a direct influence on the length chain of POA esters was observed.
While for C12 liposomal formulations an I.E. higher than 90% was achieved for all tested lipid
compositions (neutral and negatively charged) independently from Tc of the phospholipids used,
C4 formulations presented I.E. below 6% irrespectively from the lipid mixture used. This can be
explained by the less lipophilic character of C4 in comparison to C12. As above presented in
Table 1, a high difference on log Poct for these two prodrugs was observed: 0.9 and 4.55 for C4
and C12, respectively. The most favorable conditions for the maintenance of a solute within
liposomes correspond to extremely low or extremely high log Poct , while molecules with an
intermediate log Poct, such as C4, are poorly retained in liposomes (Allen and Stuart, 1999;
Defrise-Quertain et al., 1984). Based on these results the following studies were only performed
with C12. In conditions where molecules are poorly retained in liposomes, other liposome
preparation methods may be used particularly in case of weak acids or weak bases. The
incorporation of these molecules may be carried out in pre-formed liposomes in response to a
pH or salt gradient due to the higher permeability of neutral over charged molecules through
lipid bilayers. This strategy has been successfully used for weak bases such as doxorubicin or
vincristine (Allen et al., 1995; Bolotin et al., 2012).
A physicochemical comparison for extruded and non-extruded liposomes was
performed for C12 and the obtained results are shown in Table 3. The main interest was to
evaluate the influence of extruded and non-extruded liposomes on hydrolysis rate of this
prodrug in presence of human plasma. These studies were a first screening and that is the
reason why some formulations were not prepared in triplicate.
Besides the lipid compositions already tested and shown in Table 2, C12 was also
incorporated in liposomes prepared with a more fluid phospholipid, PC (Tc of -6ºC) and with a
lipid mixture of DMPC with DSPE-PEG. In addition, for the lipid mixture DMPC:DMPG three
different formulations (Formulation 5, 13 and 14) in terms of mean size were prepared and the
respective incorporation parameters compared.
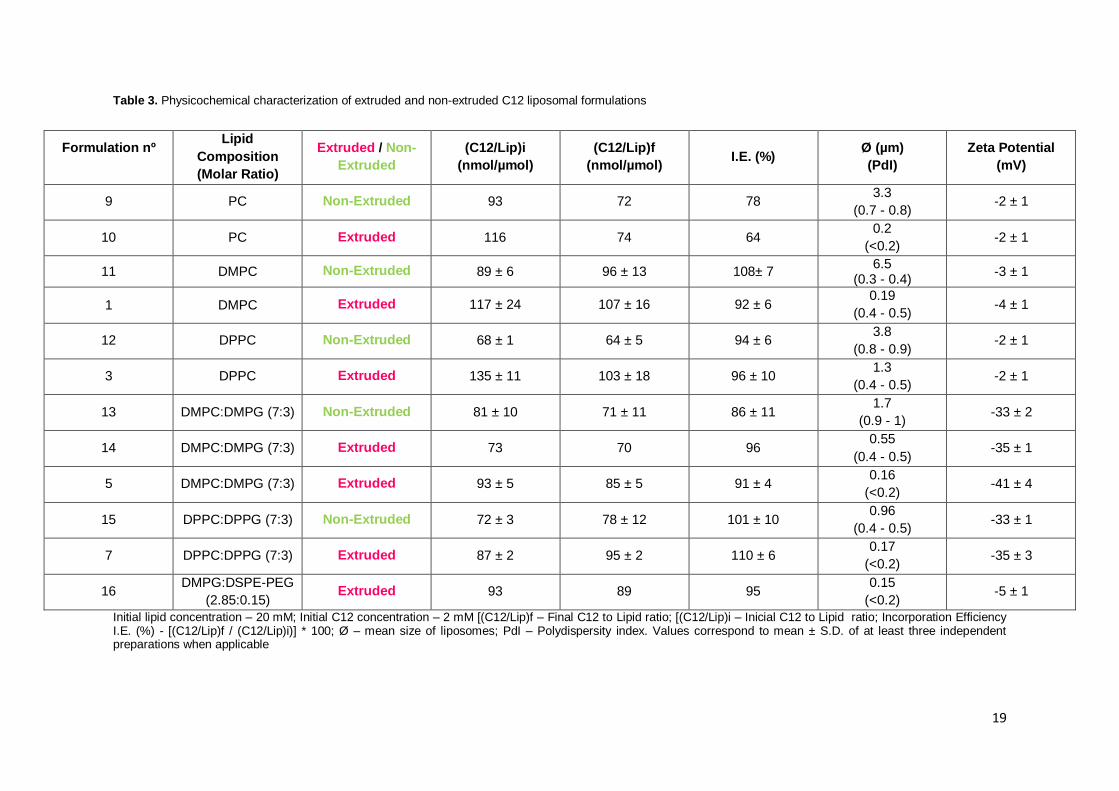
19
Table 3. Physicochemical characterization of extruded and non-extruded C12 liposomal formulations
Formulation nº
Lipid
Composition
(Molar Ratio)
Extruded / Non-
Extruded
(C12/Lip)i
(nmol/µmol)
(C12/Lip)f
(nmol/µmol) I.E. (%)
Ø (µm)
(PdI)
Zeta Potential
(mV)
9 PC Non-Extruded 93 72 78 3.3
(0.7 - 0.8) -2 ± 1
10 PC Extruded 116 74 64 0.2
(<0.2) -2 ± 1
11 DMPC Non-Extruded 89 ± 6 96 ± 13 108± 7 6.5
(0.3 - 0.4) -3 ± 1
1 DMPC Extruded 117 ± 24 107 ± 16 92 ± 6 0.19
(0.4 - 0.5) -4 ± 1
12 DPPC Non-Extruded 68 ± 1 64 ± 5 94 ± 6 3.8
(0.8 - 0.9) -2 ± 1
3 DPPC Extruded 135 ± 11 103 ± 18 96 ± 10 1.3
(0.4 - 0.5) -2 ± 1
13 DMPC:DMPG (7:3) Non-Extruded 81 ± 10 71 ± 11 86 ± 11 1.7
(0.9 - 1) -33 ± 2
14 DMPC:DMPG (7:3) Extruded 73 70 96 0.55
(0.4 - 0.5) -35 ± 1
5 DMPC:DMPG (7:3) Extruded 93 ± 5 85 ± 5 91 ± 4 0.16
(<0.2) -41 ± 4
15 DPPC:DPPG (7:3) Non-Extruded 72 ± 3 78 ± 12 101 ± 10 0.96
(0.4 - 0.5) -33 ± 1
7 DPPC:DPPG (7:3) Extruded 87 ± 2 95 ± 2 110 ± 6 0.17
(<0.2) -35 ± 3
16 DMPG:DSPE-PEG
(2.85:0.15) Extruded 93 89 95
0.15
(<0.2) -5 ± 1
Initial lipid concentration – 20 mM; Initial C12 concentration – 2 mM [(C12/Lip)f – Final C12 to Lipid ratio; [(C12/Lip)i – Inicial C12 to Lipid ratio; Incorporation Efficiency I.E. (%) - [(C12/Lip)f / (C12/Lip)i)] * 100; Ø – mean size of liposomes; PdI – Polydispersity index. Values correspond to mean ± S.D. of at least three independent preparations when applicable

20
Although the initial conditions were the same for the preparation of all liposomes, the
experimental values obtained presented high variations (from 68 ± 1 to 135 ± 11). These may
be attributed to problems during weighing process of liposome constituents, loss of material
during lyophilization step or due to inexistence of triplicates. Nevertheless, the preparation of
extruded and non-extruded liposomes presented similar incorporation parameters for the same
lipid composition. Taking into account the formulations that were prepared in triplicate and,
consequently, presenting more reliable values, the final C12 to lipid ratio ranged from 71 ± 11 to
107 ± 16 either for extruded or non-extruded liposomes independently from lipid composition.
These results support the hypothesis that C12 was incorporated in the lipid bilayer.
To confirm the above results, calorimetric studies were preformed with unloaded and loaded
liposomes (liposomes without C12 and with C12 incorporated, respectively) to evaluate the
influence of C12 on thermal behavior of lipid bilayers. Differential Scanning Calorimetry (DSC)
has been extensively used to evaluate possible changes on conformational properties of
phospholipid membranes (Taylor and Morris, 1995). DPPC, the highest rigid phospholipid used
in these studies, was selected as even after submitting this formulation to extrusion procedures
it was not possible to obtain vesicles with a mean size below 0.2 µm. In Figure 5 are shown the
thermograms obtained with the respective Tc and enthalpies (ΔH).
Figure 5. DSC thermograms. Influence of C12 on thermotropic behavior of DPPC liposomes. Tc and ΔH for unloaded and loaded liposomes
According to obtained results, no differences were observed in terms of Tc for unloaded
and loaded liposomes. Both formulations presented a Tc of +41ºC; that corresponds to the
transition from a gel state to a liquid crystal state of the constituent phospholipid. However, an
increase on the enthalpy was observed when C12 was incorporated in the lipid bilayer. This
may be due to an interaction of the prodrug with the lipid chains that is related to its lipophilic
properties, leading to a higher rigidity of the lipid bilayer and confirming the localization of C12 in
the lipid matrix. These observations are in accordance with literature (Castelli et al., 2003;
Demetzos, 2008) and may explain the increase on the mean size of C12 DPPC liposomes,
even using an extrusion step, since the rigidity influences the mean size of the vesicles (see
Formulation 3 in Tables 2 and 3). These calorimetric studies were only performed with

21
liposomes prepared with DPPC. Nevertheless, we may expect that the same effect should be
observed using other phospholipids.
3.3. Optimization of HPLC procedures for quantification of C12 and
POA
The quantification of C12 for the physicochemical characterization of liposomes was
performed by spectrophotometric techniques.
The chemical stability studies of C12 formulations in the presence of buffer and human
plasma were intended to evaluate the hydrolysis rate of C12 and the respective formation of
POA. In order to perform these studies, the quantification of C12 and POA was performed by
HPLC. Thus, an optimization of this methodology was carried out as described by Simões et al.,
(2009). In Figure 6 is shown a typical chromatogram for C12 and for POA from freshly prepared
solutions.
Figure 6. Typical chromatogram of a C12 (a) and POA (b) freshly prepared solutions. The eluents consisted in 75% of ACN for C12 and in 2% for POA in phosphate buffer, pH 2.0 with a flow rate of 1 mL/min. C12 retention time (RT) – 5.24 min; POA RT – 4.31 min
At least three independent calibration curves were constructed for each compound. The
graphical representations of the different sets of calibration curves are shown in Figure 7 as well
as the respective values of the linear regressions.

22
(a) (b)
Figure 7. Calibration curves for C12 (a) and for POA (b). The results represent mean ± S.D. of all standard curves. The linear regression was calculated using the method of least squares
The obtained results evidence a high linearity and reproducibility in the selected ranges
from batch to batch as a variation below 10% was observed from experiment to experiment.
For both molecules, and regarding the respective calibration curves, it was determined
the detection and quantification limits according to Miller, (1991). Results are shown in Table 4.
Table 4. Detection and quantification limits for C12 and POA according to the respective standards for each calibration curve
Molecule Detection Limit (µM) Quantification Limit (µM)
C12 0.173 0.523
POA 1.069 3.241
The HPLC optimization allowed the evaluation of C12 stability and the formation of POA
over time. In Figure 8 a) is represented the chromatogram of a freshly prepared C12 solution.
The reduction of C12 and the formation of POA are shown in Figures 8 b), c) and 9 b), c),
respectively.

23
Figure 8. C12 hydrolysis over time: a) Freshly C12 solution; b) and c) C12 solution along hydrolysis process
Figure 9. POA formation over time due to C12 hydrolysis: a) Freshly C12 solution; b) and c) increase of POA formation due to C12 hydrolysis
3.4. Stability of C12 formulations
The stability of some C12 liposomal formulations above mentioned was evaluated
following their incubation in HEPES buffer and biological fluids. The stability in buffer was
analyzed using two different conditions: room temperature and 37ºC. The first one was
performed in order to analyze the stability of liposome mean size over time. The incubation at
37ºC had the purpose of evaluating if the properties of liposomal formulations, particularly the
percentage of C12 incorporated, were maintained even after being submitted to severe
conditions. These stability studies intent to select the formulations that are able to preserve their
properties in terms of mean size and incorporated prodrug for further evaluation in in vivo
animal models. On the other hand, the stability studies in biological fluids were performed in
order to evaluate the hydrolysis rate of the prodrug and thus to estimate its in vivo behavior.
3.4.1. Stability of C12 formulations in HEPES buffer at room temperature
The stability on storage at room temperature of C12 liposomes was evaluated during
one month by the determination of mean sizes, as described in materials and methods. The
selected lipid compositions in this study were DMPC, DPPC, DMPC:DMPG and DPPC:DPPG
and all of them were submitted to extrusion procedures. Figure 10 shows the obtained results.

24
0 10 20 300,0
0,1
0,2
1,0
1,2
1,4
1,6
1,8
2,0
Me
an
Siz
e (
µm
)
Time (days)
DMPC
DPPC
DPPC:DPPG
DMPC:DMPG
Figure 10. Stability on storage of C12 formulations: variation of mean size of C12 liposomes during one month at room temperature. Lipid compositions under study: DMPC, DPPC, DMPC:DMPG (7:3) and DPPC:DPPG (7:3)
In the beginning of the study all formulations presented a diameter below 0.2 µm with
the exception for DPPC liposomes, with a mean size around 1.6 µm, regardless of being
extruded.
During the analyzed period, all C12 liposomal formulations presented high stability as
no particular differences in terms of mean size were observed. The highest variation was
observed for the lipid composition DPPC but in the beginning of the experiment this formulation
was already very heterogeneous.
3.4.2. Stability of C12 formulations in HEPES buffer at 37ºC
The stability of C12 liposomes, in the presence of HEPES buffer, was evaluated
following their incubation at 37ºC. For this study C12 was incorporated in DMPC, DMPC:DSPE-
PEG and DMPC:DMPG (with two different mean sizes). At defined times aliquots of each
suspension were taken and applied onto the top of a PD10 column to separate non-
incorporated C12. The fraction correspondent to liposomes was determined in terms of C12 and
lipid contents. The influence of lipid composition and mean size, particularly for DMPC:DMPG
formulations, on the preservation of C12 liposomes properties was analyzed. The obtained
values for C12 to lipid ratio related to initial conditions expressed in percentage, according to
equation 2 in materials and methods, are represented in Figure 11.
Figure 11. Stability in buffer, at 37ºC of C12 liposomal formulations: influence of lipid composition. Lipid compositions tested: DMPC - 0.19 µm, DMPC:DSPE-PEG (2.85:0.15) - 0.15 µm, DMPC:DMPG A (7:3) - 0.16 µm, DMPC:DMPG B (7:3) - 0.55 µm

25
Taking into account these results, for all lipid compositions under study, a high stability
of C12 when incorporated in liposomes was demonstrated, as 24 h post-incubation more than
95% of C12 was still associated to these vesicles.
Mean size and zeta potential were also determined in the beginning and at the end of
incubation period for all formulations and no differences were observed (data not shown).
The quantification of C12 (Figure 11) was performed by spectrophotometry and so it
was not possible to evaluate if part of the prodrug was already hydrolyzed. Thus, a liposomal
formulation was chosen randomly (DMPC:DSPE-PEG) to conduct a more detailed study by
HPLC. The C12 concentration and the possible formation of POA were determined using
aliquots recovered from the PD10 column. These quantifications meant to evaluate if the
incubation of C12 liposomes, at 37ºC, was able to induce the hydrolysis of the prodrug. The
obtained results are shown in Figure 12.
Figure 12. Concentration of C12 and POA after incubation in buffer at 37ºC. Lipid Composition – DMPC:DSPE-PEG (2.85:0.15)
The amount of POA obtained by HPLC was below detection limit (D.L.) while a constant
value for C12 was achieved for all analyzed samples proving that, under these experimental
conditions, C12 incorporated in liposomes did not undergo hydrolysis.
Data obtained shows a high stability of C12 incorporated in liposomes for the lipid
composition DMPC:DSPE-PEG both in terms of percentage of incorporated prodrug (Figure 11)
and in the ability to preserve the non-hydrolyzed form (Figure 12).
3.4.3. Stability of C12 formulations in human plasma at 37ºC
The stability in plasma was performed, as already mentioned, to evaluate the hydrolysis
rate of the prodrug and the formation of POA and thus to estimate its in vivo behavior. The
obtained results were compared to the respective profiles after prodrug incorporation in
liposomes. In addition, the influence of lipid composition and mean size of liposomes on C12
hydrolysis rate was analyzed.

26
3.4.3.1. Stability of C12 in the free form
A stock solution of C12 was prepared and the hydrolysis rate of this prodrug and the
formation of POA were monitored using two different initial concentrations: 24 and 60 µM.
Figure 13 shows the hydrolysis of C12 and the consequent formation of POA for these two
experimental conditions.
(a) (b)
Figure 13. Hydrolysis of C12 in free form and formation of POA in the presence of human plasma 50% (v/v). (a) Initial concentration: 24 µM. (b) Initial concentration: 60 µM. The results represent mean ± S.D. of two independent experiments
According to obtained results, the reduction of C12 was always followed by a
concomitant increase on POA in both experimental conditions. Particularly, when using the
highest C12 initial concentration, at time zero, this value was approximately 50 µM and 180 min
post-incubation the amount of POA determined was very similar (46 µM).
The apparent first-order hydrolysis rate constants of C12 at 37ºC were determined by
plotting the ln of C12 concentration as a function of time according to equations 3 and 4 in
materials and methods. As an example, in Figure 14 is represented data obtained for hydrolysis
of C12 in free form. The same procedure was executed for all the other formulations studied in
the present work.
Figure 14. Hydrolysis of C12 in free form in the presence of plasma for an initial concentration of 60 µM. Evolution of ln of C12 concentration over time
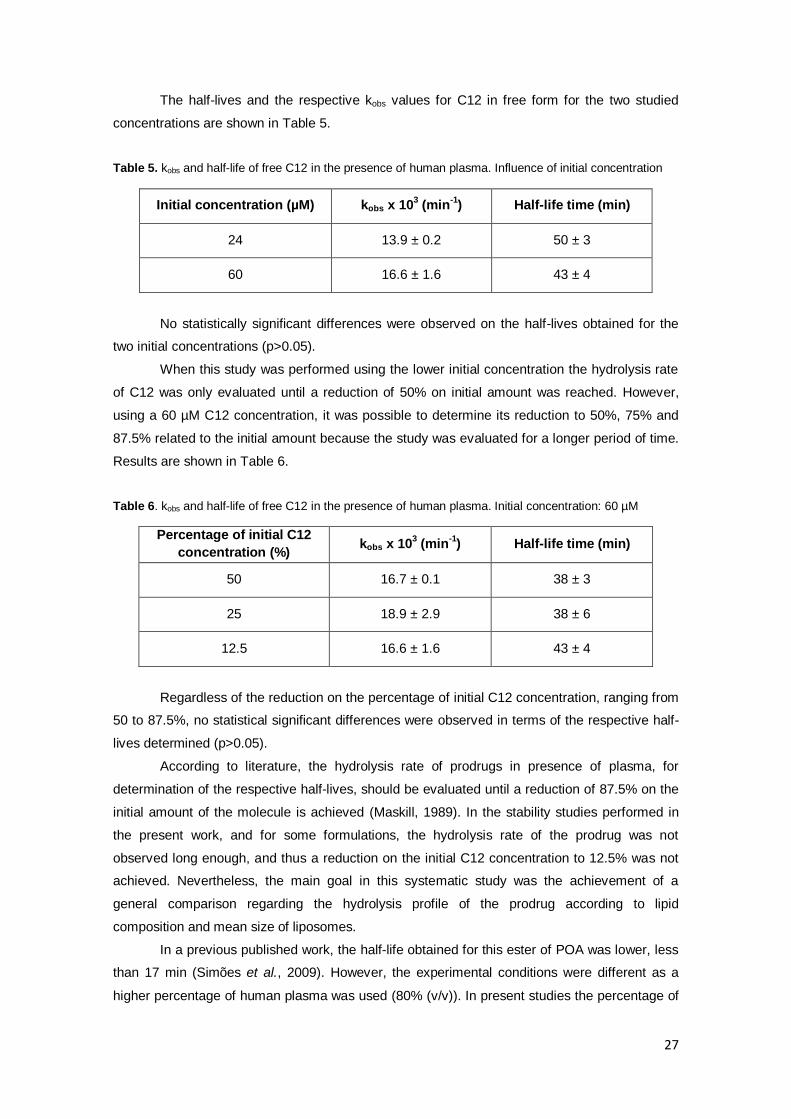
27
The half-lives and the respective kobs values for C12 in free form for the two studied
concentrations are shown in Table 5.
Table 5. kobs and half-life of free C12 in the presence of human plasma. Influence of initial concentration
Initial concentration (µM) kobs x 103 (min
-1) Half-life time (min)
24 13.9 ± 0.2 50 ± 3
60 16.6 ± 1.6 43 ± 4
No statistically significant differences were observed on the half-lives obtained for the
two initial concentrations (p>0.05).
When this study was performed using the lower initial concentration the hydrolysis rate
of C12 was only evaluated until a reduction of 50% on initial amount was reached. However,
using a 60 µM C12 concentration, it was possible to determine its reduction to 50%, 75% and
87.5% related to the initial amount because the study was evaluated for a longer period of time.
Results are shown in Table 6.
Table 6. kobs and half-life of free C12 in the presence of human plasma. Initial concentration: 60 µM
Percentage of initial C12
concentration (%) kobs x 10
3 (min
-1) Half-life time (min)
50 16.7 ± 0.1 38 ± 3
25 18.9 ± 2.9 38 ± 6
12.5 16.6 ± 1.6 43 ± 4
Regardless of the reduction on the percentage of initial C12 concentration, ranging from
50 to 87.5%, no statistical significant differences were observed in terms of the respective half-
lives determined (p>0.05).
According to literature, the hydrolysis rate of prodrugs in presence of plasma, for
determination of the respective half-lives, should be evaluated until a reduction of 87.5% on the
initial amount of the molecule is achieved (Maskill, 1989). In the stability studies performed in
the present work, and for some formulations, the hydrolysis rate of the prodrug was not
observed long enough, and thus a reduction on the initial C12 concentration to 12.5% was not
achieved. Nevertheless, the main goal in this systematic study was the achievement of a
general comparison regarding the hydrolysis profile of the prodrug according to lipid
composition and mean size of liposomes.
In a previous published work, the half-life obtained for this ester of POA was lower, less
than 17 min (Simões et al., 2009). However, the experimental conditions were different as a
higher percentage of human plasma was used (80% (v/v)). In present studies the percentage of

28
human plasma was 50% (v/v) and consequently the possibility of C12 hydrolysis is also
diminished. Nevertheless, the stability studies in human plasma, in the present work, were
always performed using the same percentage of this biological fluid and thus the validity of the
comparisons made between the free and the liposomal forms remains. The hydrolysis rate of
C12 in the liposomal form, and the respective influence of different lipid compositions and
concentrations will be compared with the obtained C12 half-life presented in Table 6 due to the
superior number of analyzed samples.
3.4.3.2. Stability of C12 liposomes – influence of lipid composition
In a previous work (Simões, 2005) C12 was already incorporated in liposomes.
However the mean size of those vesicles was not determined and according to microscope
visualization those lipid structures presented very high and heterogeneous sizes (> 3 µm). In the
present work we intended to study the influence of mean size and lipid composition of C12
liposomes on hydrolysis rate. The reason for studying these two parameters is based on the
fact that mean size and lipid composition, among others, are able to influence liposomes in vivo
fate. With this purpose a systematic study was performed using several C12 liposomal
formulations prepared with phospholipids of different phase transition temperatures (Tc). For this
purpose phospholipids with increased Tc were used: A) PC, (Tc -6ºC); B) DMPC and D) DPPC.
In addition, for phospholipids B) and D) the presence of negatively charged phospholipids with
similar Tc (C) DMPG (Tc- +23ºC) and (E) DPPG (Tc - +41ºC) was also analyzed.
A) C12 incorporated in PC liposomes
C12 was incorporated in PC, a neutral and fluid phospholipid with Tc below 0ºC (-6ºC).
Two different liposome suspensions were prepared: non-extruded and extruded, with a mean
size of 3.3 µm and 0.2 µm, respectively. The stability of these two C12 formulations was
performed after incubation in human plasma and the hydrolysis of the prodrug and the formation
of POA were determined.

29
(a) (b)
Figure 15. Hydrolysis of C12 incorporated in PC liposomes and formation of POA in the presence of human plasma 50% (v/v). Initial concentration: 60 µM. (a) Non-extruded liposomes. (b) Extruded liposomes. The results represent mean ± S.D. of two independent experiments
According to obtained results a faster depletion of C12 and the corresponding formation
of POA was observed for extruded liposomes (Figure 15 (b)) in comparison with non-extruded
vesicles (Figure 15 (a)). Moreover, the hydrolysis profile for extruded liposomes presented a
more linear variation over time than non-extruded vesicles.
The half-lives and the respective kobs values were determined according to equations 3
and 4 in materials and methods. Results are shown in Table 7.
Table 7. kobs and half-life of C12 incorporated in PC liposomes in the presence of human plasma: influence of mean vesicle size
Formulation kobs x 103 (min
-1)
Half-life time
(min)
Ø (µm)
(PdI)
Non-Extruded 18.7 ± 1.5 37 ± 3 3.3
(0.7 - 0.8)
Extruded 29.5 ± 0.1 24 ± 1 0.2
(<0.2)
A higher half-life was observed for non-extruded (37 ± 3 min) in comparison to extruded
liposomes (24 ± 1 min). This may be explained due to the high number of lipid bilayers for non-
extruded vesicles. In contact with plasma, liposomes undergo adsorption of opsonins leading to
destabilization of lipid bilayers and release of incorporated material (Crommelin and Schreier,
1994; Moghimi and Hunter, 2001). In the present work, the release of C12 was followed by its
hydrolysis and consequent formation of POA. The higher number of lipid bilayers in non-
extruded liposomes will protect C12 from a faster hydrolysis rate.
B) C12 incorporated in DMPC liposomes
C12 was incorporated in DMPC, a neutral phospholipid with a moderate Tc of +23ºC and
thus presenting a higher rigidity than PC liposomes.
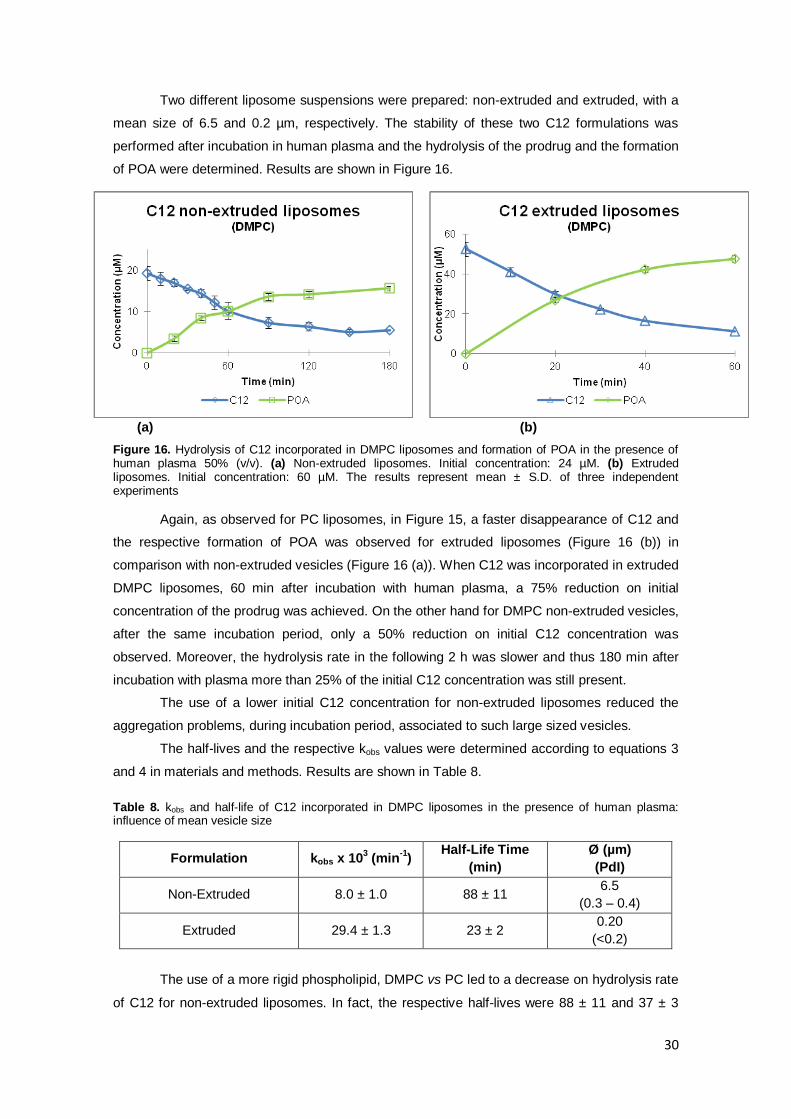
30
Two different liposome suspensions were prepared: non-extruded and extruded, with a
mean size of 6.5 and 0.2 µm, respectively. The stability of these two C12 formulations was
performed after incubation in human plasma and the hydrolysis of the prodrug and the formation
of POA were determined. Results are shown in Figure 16.
(a) (b)
Figure 16. Hydrolysis of C12 incorporated in DMPC liposomes and formation of POA in the presence of human plasma 50% (v/v). (a) Non-extruded liposomes. Initial concentration: 24 µM. (b) Extruded liposomes. Initial concentration: 60 µM. The results represent mean ± S.D. of three independent experiments
Again, as observed for PC liposomes, in Figure 15, a faster disappearance of C12 and
the respective formation of POA was observed for extruded liposomes (Figure 16 (b)) in
comparison with non-extruded vesicles (Figure 16 (a)). When C12 was incorporated in extruded
DMPC liposomes, 60 min after incubation with human plasma, a 75% reduction on initial
concentration of the prodrug was achieved. On the other hand for DMPC non-extruded vesicles,
after the same incubation period, only a 50% reduction on initial C12 concentration was
observed. Moreover, the hydrolysis rate in the following 2 h was slower and thus 180 min after
incubation with plasma more than 25% of the initial C12 concentration was still present.
The use of a lower initial C12 concentration for non-extruded liposomes reduced the
aggregation problems, during incubation period, associated to such large sized vesicles.
The half-lives and the respective kobs values were determined according to equations 3
and 4 in materials and methods. Results are shown in Table 8.
Table 8. kobs and half-life of C12 incorporated in DMPC liposomes in the presence of human plasma: influence of mean vesicle size
Formulation kobs x 103 (min
-1)
Half-Life Time
(min)
Ø (µm)
(PdI)
Non-Extruded 8.0 ± 1.0 88 ± 11 6.5
(0.3 – 0.4)
Extruded 29.4 ± 1.3 23 ± 2 0.20
(<0.2)
The use of a more rigid phospholipid, DMPC vs PC led to a decrease on hydrolysis rate
of C12 for non-extruded liposomes. In fact, the respective half-lives were 88 ± 11 and 37 ± 3

31
min. The mean sizes of these two non-extruded formulations were also very different: 6.5 and
3.3 µm for DMPC and PC liposomes, respectively. The obtained results confirm the hydrolysis
rate dependence on liposome mean sizes. For extruded vesicles no statistically significant
differences were observed between PC and DMPC formulations (p>0.05).
C) C12 incorporated in DMPC:DMPG liposomes
In order to study the influence of superficial charge of liposomes on C12 hydrolysis rate,
this prodrug was also incorporated in a lipid mixture containing, besides DMPC, a phospholipid
negatively charged, DMPG, with a Tc of +23ºC. Moreover, for this lipid composition,
DMPC:DMPG, the influence of mean size was studied using three different liposome
suspensions with diameters of 1.8 µm; 0.16 µm and 0.55 µm. The stability of these three C12
formulations was performed after incubation in human plasma and the hydrolysis of the prodrug
and the formation of POA were determined. Results are shown in Figure 17.
(a) (b)
(c)
Figure 17. Hydrolysis of C12 incorporated in DMPC:DMPG (7:3) liposomes and formation of POA in the presence of human plasma 50% (v/v). Initial concentration: 60 µM. (a) Non-Extruded liposomes with a mean size of 1.8 µm, (b) Extruded liposomes with a mean size of 0.16 µm, (c) Extruded liposomes with a mean size of 0.55 µm. The results represent mean ± S.D. of three independent experiments

32
A faster hydrolysis for C12 was observed for extruded liposomes as compared to non-
extruded vesicles. Moreover, the hydrolysis of C12 incorporated in both sized liposomes
showed similar profiles (Figures 17 (b) and (c)). Sixty min post-incubation these two
formulations have already hydrolyzed more than 75% of initial C12 while for non-extruded
vesicles the percentage of non-hydrolyzed C12 was higher than 36%.
The half-lives and the respective kobs values were determined according to equations 3
and 4 in materials and methods. Results are shown in Table 9.
Table 9. kobs and half-life of C12 incorporated in DMPC:DMPG (7:3) liposomes in the presence of human plasma: influence of mean vesicle size
Formulation kobs x 103 (min
-1)
Half-life time
(min)
Ø (µm)
(PdI)
Non-Extruded 19.4 ± 2.0 36 ± 4 1.7
(0.9 – 1)
Extruded 29.0 ± 1.0 24 ± 1 0.16
(<0.1)
Extruded 26.4 ± 0.2 26 ± 1 0.55
(0.4 – 0.5)
The overall results obtained for DMPC and DMPC:DMPG extruded formulations (Tables
8 and 9) did not reveal statistically significant differences in half-lives (p>0.05).
For non-extruded vesicles prepared with DMPC:DMPG a lower half-life was observed
when comparing to DMPC formulation. The presence of a negatively charged phospholipd in
the lipid composition enabled the achievement of smaller sized vesicles: 1.7 vs 6.5 µm. These
results are in accordance with literature, where the presence of negatively charged
phospholipids, such as DMPG, in the lipid composition reduces the aggregation of the vesicles
(Crommelin and Schreier, 1994). Data in Tables 8 and 9 clearly demonstrate a relationship
between mean size and hydrolysis rate.
D) C12 incorporated in DPPC
C12 was incorporated in DPPC, a neutral phospholipid with a Tc of +41ºC and
consequently presenting a higher rigidity than PC and DMPC previously tested. Two liposome
suspensions were prepared: non-extruded and extruded, with mean sizes of 3.8 µm and 1.8
µm, respectively. The stability of these two C12 formulations was performed after incubation in
human plasma and the hydrolysis of the prodrug and the formation of POA were determined.
Results are shown in Figure 18.

33
(a) (b)
Figure 18. Hydrolysis of C12 incorporated in DPPC liposomes and formation of POA in the presence of
human plasma 50% (v/v). (a) Non-extruded liposomes; Initial concentration: 24 µM (b) Extruded
liposomes. Initial concentration: 60 µM. The results represent mean ± S.D. of three independent
experiments
Again a faster decrease of C12 and the corresponding formation of POA was observed
for extruded liposomes (Figure 18 (b)) in comparison with non-extruded vesicles (Figure 18 (a)).
Sixty min post-incubation, extruded formulations already presented a 75% reduction of the initial
C12 concentration while for non-extruded vesicles the percentage of non-hydrolyzed C12 was
higher than 52%. Even 2 h post-incubation there were still more than 25% of the initial C12
concentration for non-extruded liposomes.
For extruded formulations, at the end of the analyzed stability period, the concentration
obtained for POA was higher than initial C12 values. The HPLC determinations of POA and C12
were performed separately and so these differences may be attributed to problems during
pippeting of samples.
The half-lives and the respective kobs values were determined according to equations 3
and 4 in materials and methods. Results are shown in Table 10.
Table 10. kobs and half-life of C12 incorporated in DPPC liposomes in the presence of human plasma: influence of mean vesicle size
Formulation kobs x 103 (min
-1)
Half-life time
(min)
Ø (µm)
(PdI)
Non-Extruded 9.7 ± 1.7 75 ± 9 3,8
(0.9 - 1)
Extruded 26.5 ± 0.5 26 ± 1 1.8
(0.4 – 0.5)
For extruded vesicles no statistically significant differences were observed in terms of
half-life for PC, DMPC and DPPC formulations (p>0.05). Besides the superior mean size of
DPPC extruded formulation, 1.8 µm, they presented a very high PdI (around 0.5).
Non-extruded DPPC liposomes presented a C12 half-life of 75 ± 9 min. This value is
similar to the one obtained for DMPC 88 ± 11 min (p>0.05) and higher for PC 37 ± 3 min.

34
Statistically significant differences were observed between PC vs DMPC and DPPC liposomes
(p<0.05).
Taking into account the overall results, two main factors are responsible for hydrolysis
rate of C12: the mean size and the rigidity of liposomes.
E) C12 incorporated in DPPC:DPPG
The influence of superficial charge of liposomes on C12 hydrolysis rate was also
evaluated by incorporating this prodrug in a lipid mixture containing, besides DPPC, a
negatively charged phospholipid, the DPPG, with a Tc of +41ºC. In this study the stability of C12
formulations was only evaluated for non-extruded vesicles. Liposomes presented a mean size
of 0.96 µm. The stability of this formulation was performed after incubation in human plasma
and the hydrolysis of the prodrug and the formation of POA were determined. Results are
shown in Figure 19.
Figure 19. Hydrolysis of C12 incorporated in DPPC:DPPG (7:3) non-extruded liposomes and formation of POA in the presence of human plasma 50% (v/v). Initial concentration: 24 µM. The results represent mean ± S.D. of three independent experiments
According to the obtained results, the reduction of C12 was followed by a concomitant
increase on POA. One hundred and eighty min post-incubation less than 3 % of C12 was in
non-hydrolyzed form while around 97% was converted in POA.
The half-life and the respective kobs value were determined according to equations 3
and 4 in materials and methods. Results are shown in Table 11.
Table 11. kobs and half-life of C12 incorporated in DPPC:DPPG (7:3) non-extruded liposomes in the presence of human plasma
Formulation kobs x 103 (min
-1)
Half-life time (min)
Ø (µm) (PdI)
Non-Extruded 22.2 ± 2.1 32 ± 3 0.96
(0.4 – 0.5)

35
The inclusion of DPPG in the lipid composition resulted in a faster C12 hydrolysis when
compared to DPPC: 32 ± 3 min and 75 ± 9 min (Tables 10 and 11). The same effect has been
already observed for DMPC vs DMPC:DMPG with a reduction from 88 ± 11 min to 36 ± 4 min.
The presence of a negatively charged phospholipid in the lipid bilayer has lead to a reduction on
the mean size of liposomes and concomitant decrease on the C12 half-life in presence of
human plasma.
Although extruded vesicles were not performed for the lipid composition DPPC:DPPG
we may expect that an hydrolysis rate for C12 should be similar to the one observed for
DMPC:DMPG extruded liposomes (24 ± 1 min).
F) C12 incorporated in DMPC:DSPE-PEG
The presence of PEG in the lipid composition on C12 hydrolysis rate was also
evaluated. As widely described in literature, PEG is able to reduce plasmatic protein adsorption
at liposome surface when included in lipid mixtures (Allen and Cullis, 2004; Dos Santos et al.,
2007; Torchilin, 2005). The stability of this formulation was performed after incubation in human
plasma and the hydrolysis of the prodrug and the formation of POA were determined. Results
are shown in Figure 20.
Figure 20. Hydrolysis of C12 incorporated in DMPC:DSPE-PEG (2.85:0.15) extruded liposomes and formation of POA in the presence of human plasma 50% (v/v). Initial concentration: 60 µM. Extruded liposomes. The results represent mean ± S.D. of three independent experiments
According to the obtained results the reduction of C12 was followed by a concomitant
increase on POA. Sixty min post-incubation a reduction of more than 75% of the initial C12
concentration was achieved. The percentage of non-hydrolyzed C12 was 21% while around
79% was converted in POA.
The half-life and kobs were determined according to equations 3 and 4 in materials and
methods. Results are shown in Table 12.

36
Table 12. kobs and half-life of C12 incorporated in DMPC:DSPE-PEG (2.85:0.15) liposomes in the presence of human plasma
Formulation kobs x 103 (min
-1)
Half-life time (min)
Ø (µm) PdI
Extruded 26.8 ± 0.4 26 ± 1 0.15
(<0.2)
The half-life of C12 obtained for liposomes containing DSPE-PEG, in the lipid
composition, presented similar results to all the formulations submitted to extrusion procedures
(see tables 7-10).
In order to summarize all obtained data related with C12 stability in the presence of
plasma in Figure 21 are shown half-lives for all C12 studied liposomal formulations.
A B C D E F G H I J
100
80
60
40
20
0
Non-extruded liposomes Extruded liposomes
Ha
lf-l
ife
(m
in)
Figure 21. Half-life of C12 formulations after incubation in human plasma 50% (v/v): A – non-extruded PC
liposomes; B – non-extruded DMPC liposomes; C – non-extruded DPPC liposomes; D – non-extruded
DMPC:DMPG (7:3) liposomes; E – non-extruded DPPC:DPPG liposomes; F –extruded PC liposomes; G –
extruded DMPC liposomes; H – extruded DPPC liposomes;I – DMPC:DMPG (7:3) extruded liposomes; J –
DMPC:DSPE-PEG (2.85:0.15) extruded liposomes
In order to evaluate the parameters able to influence the hydrolysis rate of C12 in the
presence of human plasma, a systematic study was performed. In this context several
conditions were studied: composition and rigidity of the lipid bilayer, mean size and superficial
charge of the vesicles.
Taking into account the overall results it is clear that the main factors able to influence
the hydrolysis rate of C12 liposomal formulations in the presence of human plasma are the
mean size, the rigidity and the presence of negatively charged phospholipids in the lipid mixture.
The higher half-life values were observed for non-extruded formulations prepared with
neutral phospholipids that also represented the larger liposomes: DMPC and DPPC with 88 ±
11 min and 75 ± 9 min, respectively. In a previous work, similar results were observed (Simões,
2005). However, non-extruded liposomes are very heterogeneous and thus from a

37
pharmaceutical point of view are not acceptable for parenteral administration. In addition, their
heterogeneity does not allow the achievement of a reproducible formulation from batch to batch.
Intermediate half-life values ranging from 32 to 37 min were obtained for non-extruded
liposomes prepared with fluid (PC) or negatively charged phospholipid (DMPC:DMPG and
DPPC:DPPG).
For extruded liposomes the half-life ranged from 23 to 26 min independent of rigidity
and of the presence of negatively charged phospholipids.
In the presence of biological fluids liposomes suffer the adsorption of plasmatic proteins
at their surface leading to destabilization of their lipid bilayers. The high number of lamella as it
occurs in non-extruded liposomes is the main reason why these type of liposomes presented
lower hydrolysis rates for the incorporated prodrug. These observations are in accordance with
previous reports (Hernandezcaselles et al., 1993; Ishida et al., 2002). When C12 in free form
was incubated in human plasma, the half-life value determined ranged from 38 to 43 min (Table
6) evidencing a lower hydrolysis rate as compared to C12 extruded liposomes. Nevertheless,
the parenteral administration of a prodrug with hydrophobic properties, such as C12 in free
form, requires the use of solubilizing agents that may lead to toxic side effects. Liposomes,
particularly extruded vesicles, are able to solve solubility problems and thus they are preferable
for being tested in in vivo models, as a high reproducibility between batches is achieved
(Crommelin and Schreier, 1994). Moreover, sometimes in vitro studies are not able to correlate
exactly what happens in in vivo (Yardley and Croft, 2000).
3.4.3.3. Stability of C12 formulations – Influence of C12 concentration
C12 Hydrolysis rate
All stability studies above presented were performed using C12 concentrations ranging
from 24 to 60 µM. In order to simulate the in vivo conditions, the C12 hydrolysis rate was also
evaluated using the same therapeutic dose that was administered in the M. avium murine model
of infection: 1300 µM in human plasma 50% (v/v). A C12 liposomal formulation prepared with
the lipid mixture DMPC:DMPG with a mean size below 0.2 µm was used. The hydrolysis rate of
C12 in the free form and incorporated in liposomes was compared. Results are shown in Figure
22.

38
(a) (b)
Figure 22. Hydrolysis of C12 and formation of POA after incubation with human plasma 50% (v/v). Tested formulations: C12 in the free form (a) and incorporated in DMPC:DMPG (7:3) extruded liposomes (b) Initial concentration: 1300 µM. The results represent mean ± S.D. of three independent experiments
A lower hydrolysis rate of C12 and the respective formation of POA was observed for
both formulations as compared to the above tested formulations (Figures 13 (b) and 17 (b))
using lower initial C12 concentrations. The decrease of C12 was observed for more than 400
min.
Although the initial C12 concentration was the same for both formulations, for C12
liposomes this value was around 1200 µM, while for free C12 was below 1000 µM. This
decrease may be explained due to a possible precipitation of free C12 in the presence of
plasma proteins. The use of a high concentration of a lipophilic molecule such as C12, even
using 20% of ACN in the reaction medium, may not be enough to avoid precipitation. In fact, the
quantification of POA for free C12 reached higher values than the correspondent initial C12
concentration: approximately1200 µM 400 min post-incubation in human plasma. In addition,
the same effect was observed for C12 in the liposomal form.
Transposing these results for an in vivo model and using this lipid composition, 400 min
after parenteral administration a high fraction of this formulation will no longer be in
bloodstream. The removal from blood circulation by the cells of MPS and accumulation,
particularly in liver and spleen, is widely described in literature for this type of liposomes (Allen
and Hansen, 1991; Allen and Cullis, 2004; Torchilin, 2005;). This natural accumulation has been
exploited for efficiently delivering of antibiotics to these tissues for treatment of intracellular
infections (Fielding and Lasic, 1999; Gaspar et al., 2008a; Gaspar et al., 2008b; Pinto-
Alphandary et al., 2000).
Half-lives and kobs were determined according to equations 3 and 4 in materials and
methods (Table 13).

39
Table 13. kobs and half-life of C12 in free form and incorporated in DMPC:DMPG (7:3) extruded liposomes in the presence of human plasma
C12 kobs x 103 (min
-1)
Half-life time
(min)
Free form 4.9 ± 0.5 95 ± 5
Liposomal formulation 7.3 ± 0.4 144 ± 13
As already mentioned, using an initial concentration of C12 ranging from 24 to 60 µM
only for non-extruded liposomes prepared with DMPC and DPPC a higher half-life for C12 was
observed as compared to free prodrug (see Figure 21 and Table 6): 75 ± 9 and 88 ± 11 min vs
43 ± 4 min, respectively. On the other hand, when C12 was incorporated in extruded liposomes
prepared with DMPC:DMPG the respective half-life obtained was 24 ± 1 min (Table 9). An
increase on the initial C12 concentration in the reaction medium led to the achievement of a
higher half-life for C12 in DMPC:DMPG extruded liposomes than the one obtained for C12 in
the free form: 144 ± 13 and 95 ± 5 min, respectively. In this latter experimental condition plasma
proteins were not enough to destabilize the total amount of liposomes present in the reaction
medium (Hernandezcaselles et al., 1993; Ishida et al., 2002).
3.4.3.4. Stability of liposome structure in presence of human plasma
The evaluation of physicochemical properties of liposomes in the presence of human
plasma using a 1300 µM C12 concentration, the same as later used in the M. avium murine
model of infection, was performed. These studies intended to extrapolate if liposomes maintain
their physicochemical properties after i.v. administration to mice. For this study, C12 was
incorporated in DMPC:DMPG extruded liposomes following their incubation in human plasma at
37ºC. At defined times aliquots of suspensions were taken and applied onto the top of a
Sephadex G200 column. Eluted fractions (1 mL) were recovered and the absorbance of all
samples was recorded by spectrophotometry at 420 nm. In Figure 23 is shown the graphical
representation of the fractions eluted from columns at time zero and 1 and 3 h after liposomes
incubation.
Figure 23.Absorbance at 420 nm of eluted C12 DMPC:DMPG extruded liposomes following their application on the top of a Sephadex G200 column. The results represent mean ± S.D. of three independent experiments

40
As shown in Figure 23 a slight reduction on the fraction of liposomes was observed for
samples recovered 1 and 3 h post-incubation. This slight decrease on the absorbance is in
accordance with the C12 hydrolysis using the higher concentration (1300 µM) (Figure 22 (b)).
Moreover, the determination of mean size for the eluted fractions corresponding to liposomes
demonstrated a slight decrease from 0.16 to 0.12 µm (Table 14). These results evidence that
the incubation of liposomes in human plasma led to a reduction on the number of lipid bilayers
and a concomitant release of incorporated C12 followed by its hydrolysis. Nevertheless, this
study confirms the high stability of this lipid composition when in presence of human plasma.
Table 14. Stability of liposome structure in the presence of human plasma - Mean size of liposomes at time zero and 1 and 3h post-incubation following their application on the top of a Sephadex G200 column
Time post incubation (h) Ø (µm) / (PdI)
0 0.16 / (<0.2)
1 0.12 / (<0.2)
3 0,12 / (<0.2)
Lipid composition – extruded DMPC:DMPG
3.5. Mycobacterium avium murine model of infection
3.5.1. Biological evaluation of antimycobacterial formulations
Previous in vitro studies using C12 evidenced antimycobacterial activity against M.
tuberculosis (Simões, 2005; Simões et al., 2009). With the aim to evaluate the possible
antimycobacterial effect of C12 against M. avium some preliminary studies were performed. To
our knowledge it was the first time that this in vivo experiment was assessed. According to
literature, M. avium is intrinsically resistant to PZA (Zhang and Mitchison, 2003) and thus it is of
great interest to elucidate if new synthesized esters of POA are able to overlap this handicap.
The M. avium strain (DSMZ 44157) was chosen as this microorganism is a level 2
pathogenic biological agent, whereas M. tuberculosis must be handled in facilities with biosafety
level P3.
Before the establishment of the animal model the exact quantification of the inoculum of
M. avium DSMZ 44157 and the in vivo evolution of mycobacterial infection were carried out.
3.5.2. In vivo evolution of mycobacterial infection
Mycobacterial murine models of infection can affect different organs such as lungs,
spleens, livers and lymph nodes. This is dependent not only on the properties of the inocula
used but also on the route selected for the establishment of the infection (Cynamon and
DeStefano, 1999).

41
In order to evaluate which are the main infected organs, a preliminary study was
performed. For this purpose, BALB/c mice were injected intravenously with an inoculum of M.
avium DSMZ 44157 corresponding to 5x105 colony forming units (CFU). Fifteen days after
infection mice were sacrificed and liver, spleen and lungs were removed and the number of
CFU were determined (Gaspar et al, 2000). In Figure 24 are represented the CFUs per g of
organ in the analyzed time point.
15 days after infection induction
108
106
104
102
CF
U /
g o
rga
nLiver Spleen Lung
Figure 24. Evolution of CFU per g of organ in liver, spleen and lung of BALB/c mice 15 days after infection induction. Mice received an i.v. injection of an inoculum of M. avium DSMZ 44157. Infection dose: 5x10
5
CFU per mouse. Results are expressed as mean ± S.D. of 4 mice
The obtained results indicate that the main infected organs are liver and spleen. The
lung was the less affected organ with a number of CFU per g of organ below 1x105.
Taking these results into account the biological evaluation exerted by the selected
formulations was determined in liver and spleen.
3.5.3. Influence of antimycobacterial formulations on M. avium murine
model
The murine model of infection intended to investigate if in vitro stability studies of C12
formulations, performed in the presence of plasma, could be correlated with their therapeutic
response. With this purpose, the lipid composition DMPC:DMPG was chosen to be tested in the
M. avium murine model of infection. The prodrug C12 was incorporated in extruded and non-
extruded liposomes. In Table 15 are shown some physicochemical properties of the liposomal
formulations used in treatment schedule.
Table 15. C12 liposomal formulations used for the treatment of M. avium murine model of infection
Formulation
nº
Lipid Composition
(Molar Ratio)
Extruded / Non-
Extruded
Ø (µm)
(PdI)
Zeta Potential
(mV)
17 DMPC:DMPG (7:3) Extruded 0.15
(<0.2) -29 ± 3
18 DMPC:DMPG (7:3) Non-Extruded* 1.2
(0.7 – 0.8) -32 ± 2
Ø – mean size of liposomes; PdI – Polydispersity index. *This formulation was extruded through filter of 2 µm of porosity

42
Two control groups were used: infected and non treated animals (Cont.I and Cont.II)
Cont.I corresponds to the bacterial loads in liver and spleen of mice in the beginning of the
treatment, while Cont.II represents the bacterial loads at the end of treatment schedule. Two
other groups received PZA or POA in the free form. In Figure 25 are represented the number of
CFU per g of organ for these tested groups.
**statistically significant from Cont.II
Figure 25. Evolution of M. avium infection in mice. Mice received an i.v. injection of an inoculum of M. avium strain DSMZ 44157. Infection dose: 1x10
6 CFU per mouse. Treatment started two weeks after
infection. Mice received i.v. injections of PZA and POA three times a week for two weeks. Administered dose: 12 mg/kg of body weight. Control I (Cont.I) corresponds to the bacterial load in the beginning of treatment; the other groups correspond to the bacterial loads at the end of treatment: Control II (Cont.II) – untreated animals; POA – Pyrazinoic Acid; PZA – Pyrazinamide. Results are expressed as mean ± S.D. of at least 5 mice per group.
The results indicate a high progression on the mycobacterial infection. Cont.I presented
a number of CFU/g of organ of around 108, while two weeks after infection a 10 fold increase on
the bacterial load was observed (Cont.II). For mice receiving PZA and POA no statistically
significant differences in bacterial loads in liver were detected in comparison with Cont.II
(p>0.4). One the other hand, in spleen a therapeutic effect for both molecules was achieved.
Statistically significant differences were observed (p<0.05) in both cases.
C12 was administered in free and liposomal forms. In Figure 26 are shown the number
of CFU per g of organ of all analyzed animal groups two weeks after treatment: untreated
animals (Cont.II) and treated animals (POA, C12, LIP1 and LIP2).

43
* Statistically significant from Cont.II; ** Statistically significant from Cont.II
Figure 26. Evolution of M. avium infection in mice. Influence of administered formulations on CFU per g of organ. Mice received an i.v. injection of an inoculum of M. avium strain DSMZ 44157. Infection dose: 1x10
6
CFU per mouse. Treatment started two weeks after infection. Mice received i.v. injections of formulations under study three times a week for two weeks. Administered dose: 12 mg/kg of body weight. Studied formulations: Control II (Cont.II) – untreated animals; POA – Pyrazinoic Acid; C12 – Prodrug in the free form; LIP1 – C12 incorporated in DMPC:DMPG (7:3) non-extruded liposomes; LIP2 – C12 incorporated in DMPC:DMPG (7:3) extruded liposomes. Results are expressed as mean ± S.D. of at least 4 mice per group
The results show, as already mentioned, that for mice treated with POA the degree of
infection in liver was very similar to Cont.II (p>0.05) while in spleen a significant reduction was
observed (p<0.05). The administration of C12 in the free and liposomal forms (extruded
liposomes – LIP2) reduced significantly the bacterial loads either in liver or in spleen (around 10
fold) when compared to Cont.II (p<0.05). On the opposite, the use of non-extruded liposomes
(LIP1) did not show a therapeutic response, as no statistical differences over Cont.II were
observed (p>0.4 and p>0.1 for spleen and liver, respectively). These results demonstrate that in
vitro studies are not always able to correlate exactly what happens in in vivo (Yardley and Croft,
2000). Indeed, the increase of around 30% in the half-life observed for LIP1 vs LIP2 (Figure 21)
in plasma stability assays was not accomplished by an increase on the therapeutic effect.
When comparing bacterial loads in liver for mice treated with POA vs LIP2, the
differences in the number of CFU/g organ were statistically significant (p<0.05), but not in
spleen (p>0.08). Moreover, for mice groups treated with POA, C12 and LIP2, no statistically
significant differences were observed in infection degree in spleen (p>0.4). Based on these
results, the optimization of a liposomal formulation able to exert a superior therapeutic effect in
this organ should be assayed.
In this in vivo model the degree of infection of all animals was very high. Thus, the
possibility of achieving higher reductions on bacterial loads is more difficult. In addition the M.
avium strain used in the present thesis is described in literature as being more resistant to
several antibiotics than some M. tuberculosis strains (Inderlied et al., 1993; Luo et al., 2011).
The performance of other M. avium murine models of infection using more susceptible strains
should be considered in order to evaluate if the therapeutic effect is even higher than the one
already obtained.

44
These in vivo studies exhibit very promising results. As already mentioned, M. avium is
intrinsically resistant to PZA and this may be due to an efficient POA efflux mechanism (Wade
and Zhang, 2004; Zhang and Mitchison, 2003). However, a therapeutic effect of POA and PZA
was achieved for the spleen. For C12 formulations no statistically significant differences were
observed, in terms of bacterial loads either in liver or in spleen. The use of lower administered
doses should be considered in further in vivo tests in order to clarify these therapeutic
responses.
The similar antimycobacterial effect of C12 formulations (C12 vs LIP2) could be a result
of a plateau in terms of dose response. According to literature, in other animal models this effect
has also been observed when high therapeutic doses are administered (Lala et al., 2006;
Sarkar et al., 2002). Taking this into account, the use of lower doses could lead to statistically
significant differences between free and liposomal C12, showing the therapeutic advantages of
using liposomes, based on the assumption of different biodistribution profiles.

45
4. Conclusions and Future Perspectives
Mycobacterial species resist to a variety of antimycobacterial agents because of their
thick cell wall that confers an exceptionally strong barrier to antibiotics. In an attempt to overlap
these drawbacks a viable strategy may be achieved by the synthesis of new molecules or
chemical modification of the already available ones. In particular, the prodrug approach via
activation inside mycobacterial cells is a relevant alternative especially considering the
problematic penetration of drugs into these cells (Valente et al., 2011).
PZA, a first line agent for treatment of TB, is also a prodrug. In order to exert
antimycobacterial activity, PZA requires activation by the Mycobacterium enzyme
pyrazinamidase (PZase) to be transformed into its active form, the POA. This enzyme
conveniently activates the drug only inside the bacteria (Pires, 2011). However, since the
activation of PZA is only dependent on one enzyme, it has been observed the emergence of
resistant strains to this prodrug. To overcome these drawbacks the synthesis of new POA
prodrugs were conducted by several authors where some compounds displayed higher activity
than PZA against M. tuberculosis, M. avium and M. kansasii (Cynamon et al., 1992; Cynamon
et al., 1995). However, these new molecules displayed low stability in presence of biological
fluids (Bergmann et al., 1996). Simões and co-workers have synthesized series of POA
prodrugs with different alkoxy chain groups (Simões, 2005; Simões et al., 2009; Valente et al.,
2011). They found that longer fatty acid chains were more resistant to plasma hydrolysis. As
these prodrugs are easily activated by mycobacterial esterases, this may contribute to a
reduction on the occurrence of resistances against mycobacterial strains. In in vitro tests some
POA esters were found to be 10 times more active against sensitive M. tuberculosis than PZA
(Simões et al., 2009).
Liposomal formulations of two POA esters, C4 and C12, were developed and their
therapeutic potential, in a preliminary in vivo model infected with Mycobacterium avium DSMZ
44157, was evaluated. The rational for associating these POA esters to liposomes was based
on the ability of these lipid systems to circumvent some solubility problems associated to these
molecules and, after parenteral administration, they can passively target liver and spleen, the
main affected organs in M. avium infections.
The physicochemical characterization of liposomal formulations enabled the
achievement of incorporation efficiencies higher than 90% for C12 while very low values were
obtained for C4 (below 6%). C12 was then selected for further in vitro and in vivo studies. The
log Poct of C12 is around 4.55, owning a hydrophobic character. Due to this property, it is
hypothesized that this prodrug may be accommodated in the lipid bilayer (Allen and Stuart,
1999; Defrise-Quertain et al., 1984;). This assumption was confirmed by calorimetric studies as
an increase on the enthalpy was observed for loaded liposomes in comparison with the
unloaded ones.
Stability studies of C12 liposomes in HEPES buffer evidenced the high stability of these
formulations considering the percentage of incorporated prodrug and the preservation of non-

46
hydrolyzed form. A systematic study of C12 formulations in human plasma was also performed.
C12 in free form presented a half-life of around 43 ± 4 min. Almost all liposomal formulations
presented higher hydrolysis rates than C12 in the free form, with the exception for neutral and
non-extruded liposomes. However, from the pharmaceutical point of view, it is crucial the
achievement of liposomes with a high homogeneous mean size and the preservation of all
physicochemical properties as they will influence the in vivo profile of the correspondent
formulation. Taking this into account, extruded liposomes appear more attractive as the
maintenance of their characteristics and the reproducibility from batch to batch was observed.
The influence of C12 initial concentration on the hydrolysis rate was also evaluated, evidencing
that, when similar experimental conditions as those tested in the in vivo model were used,
higher stability for C12 in extruded liposomes was achieved when compared to the respective
free form. These observations are in accordance with literature as plasma proteins are not able
to destabilize the total amount of liposomes in reaction medium (Hernandezcaselles et al.,
1993; Ishida et al., 2002).
With the aim to evaluate the possible antimycobacterial effect of C12 against a M.
avium strain DSMZ 44157, some preliminary studies were performed. From our knowledge, it
was the first time that this in vivo experiment was assayed. Taking into consideration the
promising in vitro results obtained for POA ester prodrugs against M. tuberculosis (Simões et
al., 2009) we intended to evaluate whether the selected C12 formulations could also overcome
the intrinsic resistance of M. avium to PZA (Zhang and Mitchison, 2003). Even using a M. avium
strain resistant to most antibiotics (Inderlied et al., 1993; Luo et al., 2011), it was possible to
observe a therapeutic effect for mice treated with C12, both in free form and incorporated in
extruded liposomes. Inexplicably, mice groups receiving POA and PZA also demonstrated a
reduction on the bacterial loads in spleen. The effect may be due to the high administered dose.
These preliminary in vivo tests constitute good future prospects and so the use of other
treatment schedules and mycobacterial strains may be considered for future work. Moreover,
the performance of biodistribution studies should also be assayed to evaluate the influence of
doses and lipid compositions, to select the ones that would lead to a higher prodrug
accumulation within the infected organs. Nevertheless, these preliminary in vivo results may be
envisioned as an alternative strategy for treatment of mycobacterial infections.

47
5. References
Allen T.M. and Cullis P.R. (2004) Drug Delivery Systems: Entering the Mainstream. Science 303: pp 1818-1822. Allen T.M. and Hansen C. (1991) Pharmacokinetics of Stealth Versus Conventional Liposomes - Effect of Dose. Biochimica et Biophysica Acta 1068: pp 133-141. Allen T.M., Newman M.S., Woodle M.C., Mayhew E. and Uster P.S. (1995) Pharmacokinetics and Antitumor-Activity of Vincristine Encapsulated in Sterically Stabilized Liposomes. International Journal of Cancer 62: pp 199-204. Allen T.M. and Stuart D.D. (1999) Liposome Pharmacokinetics: classical, sterically stabilized, cationic liposomes and immunoliposomes, in Liposomes: Rational Design (Janoff A.S. ed) pp 63-87, Marcel Dekker, Inc, New York, Basel. Bakker-Woudenberg I.A.J.M., Lokerse A.F., Tenkate M.T., Mouton J.W., Woodle M.C. and Storm G. (1993) Liposomes With Prolonged Blood-Circulation and Selective Localization in Klebsiella-Pneumoniae Infected Lung-Tissue. Journal of Infectious Diseases 168: pp 164-171. Bangham A.D., Standish M.M. and Watkins J.C. (1965) Diffusion of Univalent Ions Across Lamellae of Swollen Phospholipids. Journal of Molecular Biology 13: pp 238-&. Bergmann K.E., Cynamon M.H. and Welch J.T. (1996) Quantitative Structure-Activity Relationships for the in Vitro Antimycobacterial Activity of Pyrazinoic Acid Esters. Journal of Medicinal Chemistry 39: pp 3394-3400. Bolotin E.M., Cohen R., Bar L.K., Emanuel S.N., Ninio S., Lasic D.D. and Barenholz Y. (2012) Ammounium Sulfate Gradients for Efficient and Stable Remote Loading of Amphipathic Weak Bases into Liposomes and Ligandoliposomes. Journal of Liposome Research 4: pp 455-479. Carvalheiro M., Jorge J., Eleuterio C., Pinhal A.F., Sousa A.C., Morais J.G. and Cruz M.E.M. (2009) Trifluralin Liposomal Formulations Active Against Leishmania Donovani Infections. European Journal of Pharmaceutics and Biopharmaceutics 71: pp 292-296. Castelli F., Caruso S. and Uccella N. (2003) Effect of Simple Olive Biophenols and Analogues on the Thermotropic Behavior of Biological Model Membranes. European Journal of Lipid Science and Technology 105: pp 260-265. Constantino L., Cruz M.E.M., Mehta R. and Lopez-Berestein G. (1993) Formulation and Toxicity of Liposomes Containing Rifampicin. Journal of Liposome Research 2: pp 275-301. Cook J.L. (2010) Nontuberculous Mycobacteria: Opportunistic Environmental Pathogens for Predisposed Hosts. British Medical Bulletin 96: pp 45-59. Corvo M.L., Jorge J.C.S., van't Hof R., Cruz M.E.M., Crommelin D.J.A. and Storm G. (2002) Superoxide Dismutase Entrapped in Long-Circulating Liposomes: Formulation Design and Therapeutic Activity in Rat Adjuvant Arthritis. Biochimica et Biophysica Acta-Biomembranes 1564: pp 227-236. Cosma C.L., Sherman D.R. and Ramakrishnan L. (2003) The Secret Lives of the Pathogenic Mycobacteria. Annual Review of Microbiology 57: pp 641-676. Crommelin D.J.A., Grit M., Talsma H. and Zuidam N.J. (1994) Liposomes As Carriers for Drugs and Antigens - Approaches to Preserve Their Long-Term Stability. Drug Development and Industrial Pharmacy 20: pp 547-556. Crommelin D.J.A. and Schreier H. (1994) Liposomes, in Colloidal Drug Delivery Systems (Kreuter J. ed) pp 73-190, Marcel Dekker, Inc, New York. Cruz M.E.M., Simões S.I., Corvo M.L., Martins M.B.F. and Gaspar M.M. (2009) Formulation of NPDDS for Macromolecules, in Drug Delivery Nanoparticles: Formulation and Characterization (Yashwant Pathak DT ed) pp 35-49, Informa Healthcare. Cynamon M.H. and DeStefano M.S. (1999) Beige Mouse Model of Disseminated Mycobacterium avium Complex Infection, in Handbook of Animal Models of Infection Academic Press.

48
Cynamon M.H., Gimi R., Gyenes F., Sharpe C.A., Bergmann K.E., Han H.J., Gregor L.B., Rapolu R., Luciano G. and Welch J.T. (1995) Pyrazinoic Acid-Esters With Broad-Spectrum In-Vitro Antimycobacterial Activity. Journal of Medicinal Chemistry 38: pp 3902-3907. Cynamon M.H., Klemens S.P., Chou T.S., Gimi R.H. and Welch J.T. (1992) Antimycobacterial Activity of A Series of Pyrazinoic Acid-Esters. Journal of Medicinal Chemistry 35: pp 1212-1215. Defrise-Quertain F., Chatelain P., Delmelle M. and Ruysschaert M.J. (1984) Model Studies for Drug Entrapment and Liposome Stability, in Liposome Technology - Incorporation of Drugs, Proteins and Genetic Materials (Gregoriadis G ed) pp 1-17, CRC Press, Inc, Boca Raton, Florida, USA. Demetzos C. (2008) Differential Scanning Calorimetry (DSC): A Tool to Study the Thermal Behavior of Lipid Bilayers and Liposomal Stability. Journal of Liposome Research 18: pp 159-173. Dos Santos N., Allen C., Doppen A.M., Anantha M., Cox K.A.K., Gallagher R.C., Karlsson G., Edwards K., Kenner G., Samuels L., Webb M.S. and Bally M.B. (2007) Influence of Poly(Ethylene Glycol) Grafting Density and Polymer Length on Liposomes: Relating Plasma Circulation Lifetimes to Protein Binding. Biochimica et Biophysica Acta-Biomembranes 1768: pp 1367-1377. du Toit L.C., Pillay V. and Danckwerts M.P. (2006) Tuberculosis Chemotherapy: Current Drug Delivery Approaches. Respiratory Research 7. Fernandes J.P.S., Pasqualoto K.F.M., Felli V.M.A., Ferreira E.I. and Brandt C.A. (2010) QSAR Modeling of a Set of Pyrazinoate Esters As Antituberculosis Prodrugs. Archiv der Pharmazie 343: pp 91-97. Fielding R.M. and Lasic D.D. (1999) Liposomes in the Treatment of Infectious Diseases. Expert Opinion on Therapeutic Patents 9: pp 1679-1688. French A.L., Benator D.A. and Gordin F.M. (1997) Nontuberculous Mycobacterial Infections. Medical Clinics of North America 81: pp 361-&. Gaspar M.M., Boerman O.C., Laverman P., Corvo M.L., Storm G. and Cruz M.E.M. (2007) Enzymosomes With Surface-Exposed Superoxide Dismutase: In Vivo Behaviour and Therapeutic Activity in a Model of Adjuvant Arthritis. Journal of Controlled Release 117: pp 186-195. Gaspar M.M., Cruz A., Fraga A.G., Castro A.G., Cruz M.E.M. and Pedrosa J. (2008a) Developments on Drug Delivery Systems for the Treatment of Mycobacterial Infections. Current Topics in Medicinal Chemistry 8: pp 579-591. Gaspar M.M., Cruz A., Penha A.E., Reymao J., Sousa A.C., Eleuterio C.V., Domingues S.A., Fraga A.G., Filho A.L., Cruz M.E.M. and Pedrosa J. (2008b) Rifabutin Encapsulated in Liposomes Exhibits Increased Therapeutic Activity in a Model of Disseminated Tuberculosis. International Journal of Antimicrobial Agents 31: pp 37-45. Gaspar M.M., Neves S., Portaels F., Pedrosa J., Silva M.T. and Cruz M.E.M. (2000) Therapeutic Efficacy of Liposomal Rifabutin in a Mycobacterium Avium Model of Infection. Antimicrobial Agents and Chemotherapy 44: pp 2424-2430. Gaspar M.M., PerezSoler R. and Cruz M.E.M. (1996) Biological Characterization of L-Asparaginase Liposomal Formulations. Cancer Chemotherapy and Pharmacology 38: pp 373-377. Georgiev S.V. (1994) Developmental Therapeutics of Mycobacterium Avium Complex (MAC) Infections. International Antimicrobial Agents 4: pp 157-173. Gregoriadis G. and Ryman B.E. (1972) Fate of Protein-Containing Liposomes Injected Into Rats - Approach to Treatment of Storage Diseases. European Journal of Biochemistry 24: pp 485-&. Gunstone F.D., Padley F.B. and Harwood J.L. (1986) In the Lipid Handbook. Chapman and Hall, London. Gupta D., Gupta S.V., Lee K.D. and Amidon G.L. (2009) Chemical and Enzymatic Stability of Amino Acid Prodrugs Containing Methoxy, Ethoxy and Propylene Glycol Linkers. Molecular Pharmaceutics 6: pp 1604-1611. Hernandezcaselles T., Villalain J. and Gomezfernandez J.C. (1993) Influence of Liposome Charge and Composition on Their Interaction With Human Blood-Serum Proteins. Molecular and Cellular Biochemistry 120: pp 119-126.

49
Inderlied C.B., Kemper C.A. and Bermudez L.E.M. (1993) The Mycobacterium-Avium Complex. Clinical Microbiology Reviews 6: pp 266-310. Ishida T., Harashima H. and Kiwada H. (2002) Liposome Clearance. Bioscience Reports 22: pp 197-224. Konno K., Feldmann F.M. and McDermot.W. (1967) Pyrazinamide Susceptibility and Amidase Activity of Tubercle Bacilli. American Review of Respiratory Disease 95: pp 461-&. Lala S., Gupta S., Sahu N.P., Mandal D., Mondal N.B., Moulik S.P. and Basu M.K. (2006) Critical Evaluation of the Therapeutic Potential of Bassic Acid Incorporated in Oil-in-Water Microemulsions and Poly-D,L-Lactide Nanoparticles Against Experimental Leishmaniasis. Jounal of Drug Targeting 14 (4): pp 171-179. Liu J.B., Lee H., Huesca M., Young A.P. and Allen C. (2006) Liposome Formulation of a Novel Hydrophobic Aryl-Imidazole Compound for Anti-Cancer Therapy. Cancer Chemotherapy and Pharmacology 58: pp 306-318. Luo X., Pires D., Ainsa J.A., Gracia B., Mulhovo S., Duarte A., Anes E. and Ferreira M.J.U. (2011) Antimycobacterial Evaluation and Preliminary Phytochemical Investigation of Selected Medicinal Plants Traditionally Used in Mozambique. Journal of Ethnopharmacology 137: pp 114-120. Maskill H. (1989) The Physical Basis of Organic Chemistry. Press, U.K. Miller J.N. (1991) Basic Statistical-Methods for Analytical-Chemistry .2. Calibration and Regression Methods - A Review. Analyst 116: pp 3-14. Mitchison D.A. and Fourie P.B. (2010) The Near Future: Improving the Activity of Rifamycins and Pyrazinamide. Tuberculosis 90: pp 177-181. Moghimi S.M. and Hunter A.C. (2001) Recognition by Macrophages and Liver Cells of Opsonized Phospholipid Vesicles and Phospholipid Headgroups. Pharmaceutical Research 18: pp 1-8. Nuermberger E, and Grosset J. (2004) Pharmacokinetic and Pharmacodynamic Issues in the Treatment of Mycobacterial Infections. European Journal of Clinical Microbiology & Infectious Diseases 23: pp 243-255. Pedrosa J., Florido M., Kunze Z.M., Castro A.G., Portaels F., Mcfadden J., Silva M.T. and Appelberg R. (1994) Characterization of the Virulence of Mycobacterium-Avium Complex (Mac) Isolates in Mice. Clinical and Experimental Immunology 98: pp 210-216. Pinto-Alphandary H., Andremont A. and Couvreur P. (2000) Targeted Delivery of Antibiotics Using Liposomes and Nanoparticles: Research and Applications. International Journal of Antimicrobial Agents 13: pp 155-168. Pires, D. Role of mycobacteria porins and efflux pumps on the intracellular survival within macrophages. 2011. Faculdade de Ciências da Universidade de Lisboa . Rautio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Jarvinen T. and Savolainen J. (2008) Prodrugs: Design and Clinical Applications. Nature Reviews Drug Discovery 7: pp 255-270. Raynaud C., Laneelle M.A., Senaratne R.H., Draper P., Laneelle G. and Daffe M. (1999) Mechanisms of Pyrazinamide Resistance in Mycobacteria: Importance of Lack of Uptake in Addition to Lack of Pyrazinamidase Activity. Microbiology-Uk 145: pp 1359-1367. Reed C., von Reyn C.F., Chamblee S., Ellerbrock T.V., Johnson J.W., Marsh B.J., Johnson L.S., Trenschel R.J. and Horsburgh C.R. (2006) Environmental Risk Factors for Infection With Mycobacterium Avium Complex. American Journal of Epidemiology 164: pp 32-40. Ribeiro M.M.B., Melo M.N., Serrano I.D., Santos N.C. and Castanho M.A.R.B. (2010) Drug-Lipid Interaction Evaluation: Why a 19th Century Solution? Trends in Pharmacological Sciences 31: pp 449-454. Rouser G., Fleische S. and Yamamoto A. (1970) 2 Dimensional Thin Layer Chromatographic Separation of Polar Lipids and Determination of Phospholipids by Phosphorus Analysis of Spots. Lipids 5: pp 494-&. Sarkar S., Mandal S., Sinha J., Mukhopadhyay S., Das N. and Basu M.K. (2002) Quercetin: Critical Evaluation As an Antileishmanial Agent In Vivo in Hamsters Using Different Vesicular Delivery Modes. Journal of Drug Targeting 10 (8): pp 573-578.

50
Silva M.T., Appelberg R., Silva M.N.T. and Macedo P.M. (1987) Invivo Killing and Degradation of Mycobacterium-Aurum Within Mouse Peritoneal-Macrophages. Infection and Immunity 55: pp 2006-2016. Simões, M. F. Estudo da estabilidade e actividade de pró-fármacos derivados da pirazinamida sob a forma livre e encapsulada em lipossomas. 2005. Universidade Lusófona de Humanidades e Tecnologia. Simões M.F., Valente E., Gomez M.J.R., Anes E. and Constantino L. (2009) Lipophilic Pyrazinoic Acid Amide and Ester Prodrugs Stability, Activation and Activity Against M. Tuberculosis. European Journal of Pharmaceutical Sciences 37: pp 257-263. Taylor K.M.G. and Morris R.M. (1995) Thermal-Analysis of Phase-Transition Behavior in Liposomes. Thermochimica Acta 248: pp 289-301. Torchilin V.P. (2005) Recent Advances With Liposomes As Pharmaceutical Carriers. Nature Reviews Drug Discovery 4: pp 145-160. Valente E., Simoes M.F., Testa B. and Constantino L. (2011) Development of a Method to Investigate the Hydrolysis of Xenobiotic Esters by a Mycobacterium Smegmatis Homogenate. Journal of Microbiological Methods 85: pp 98-102. Wade M.M. and Zhang Y. (2004) Anaerobic Incubation Conditions Enhance Pyrazinamide Activity Against Mycobacterium Tuberculosis. Journal of Medical Microbiology 53: pp 769-773. Wolinsky E. (1979) Non-Tuberculous Mycobacteria and Associated Diseases. American Review of Respiratory Disease 119: pp 107-159. Yamamoto S., Toida I., Watanabe N. and Ura T. (1995) In-Vitro Antimycobacterial Activities of Pyrazinamide Analogs. Antimicrobial Agents and Chemotherapy 39: pp 2088-2091. Yardley V. and Croft S.L. (2000) A Comparison of the Activities of Three Amphotericin B Lipid Formulations Against Experimental Visceral and Cutaneous Leishmaniasis. International Journal of Antimicrobial Agents 13: pp 243-248. Zar, J.H. (2009) Biostatistical Analysis. 5
Th Ed., Prentice Hall, London
Zhang Y. and Mitchison D. (2003) The Curious Characteristics of Pyrazinamide: a Review. International Journal of Tuberculosis and Lung Disease 7: pp 6-21. Zhang Y., Wade M.M., Scorpio A., Zhang H. and Sun Z.H. (2003) Mode of Action of Pyrazinamide: Disruption of Mycobacterium Tuberculosis Membrane Transport and Energetics by Pyrazinoic Acid. Journal of Antimicrobial Chemotherapy 52: pp 790-795.





























