Embed Size (px)
Citation preview

Translational Bioinformatics Approachesto Drug Development
Ben Readhead1,2 and Joel Dudley1–3,*1Center for Biomedical Informatics, 2Institute for Genomics and Multiscale Biology, and 3Department of Genetics
and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York.
Significance: A majority of therapeutic interventions occur late in the patho-logical process, when treatment outcome can be less predictable and effective,highlighting the need for new precise and preventive therapeutic developmentstrategies that consider genomic and environmental context. Translationalbioinformatics is well positioned to contribute to the many challenges inherentin bridging this gap between our current reactive methods of healthcare de-livery and the intent of precision medicine, particularly in the areas of drugdevelopment, which forms the focus of this review.Recent Advances: A variety of powerful informatics methods for organizingand leveraging the vast wealth of available molecular measurements availablefor a broad range of disease contexts have recently emerged. These includemethods for data driven disease classification, drug repositioning, identifica-tion of disease biomarkers, and the creation of disease network models, eachwith significant impacts on drug development approaches.Critical Issues: An important bottleneck in the application of bioinformaticsmethods in translational research is the lack of investigators who are versed inboth biomedical domains and informatics. Efforts to nurture both sets ofcompetencies within individuals and to increase interfield visibility will help toaccelerate the adoption and increased application of bioinformatics in trans-lational research.Future Directions: It is possible to construct predictive, multiscale networkmodels of disease by integrating genotype, gene expression, clinical traits, andother multiscale measures using causal network inference methods. This canenable the identification of the ‘‘key drivers’’ of pathology, which may representnovel therapeutic targets or biomarker candidates that play a more direct rolein the etiology of disease.
SCOPE AND SIGNIFICANCE
A majority of medical interven-tions occur late in the pathologicalprocess, when treatment outcomecan be less predictable and effective,highlighting the need for new preciseand preventive therapeutic develop-ment strategies that consider geno-mic and environmental context.Translational bioinformatics is wellpositioned to contribute to the manychallenges inherent in bridging this
gap between our current reactivemethods of healthcare delivery andthe intent of precision medicine,particularly in the areas of drug de-velopment, which forms the focus ofthis review.
TRANSLATIONAL RELEVANCE
A range of powerful informaticsmethods have been developed to or-ganize and capitalize on the deluge ofmolecular level information avail-
Joel Dudley, PhD
Submitted for publication December 30,
2012. Accepted in revised form April 7, 2013.
*Correspondence: Department of Genetics
and Genomic Sciences, Icahn School of Medicine
at Mount Sinai, One Gustave L. Levy Pl., Box
1498, New York, NY 10029-6574 (e-mail: joel
Abbreviationsand Acronyms
ALK = anaplastic lymphomareceptor tyrosine kinase
CSS = cultured skin substitute
DNA = deoxyribonucleic acid
eQTL = expression quantitativetrait loci
GEO = Gene ExpressionOmnibus
GOBP = gene ontologybiological process
ICD = International Classificationof Disease
MEMN = macrophage enrichedmetabolic network
MET = mesenchymal epithelialtransition factor
mGlu = metabotropic glutamate(receptor)
(continued)
470 j ADVANCES IN WOUND CARE, VOLUME 2, NUMBER 9Copyright ª 2013 by Mary Ann Liebert, Inc. DOI: 10.1089/wound.2012.0422

able in a range of disease contexts, inareas as diverse as disease classifi-cation, drug repositioning, identifi-cation of disease biomarkers and thecreation of disease network models,each with significant impacts on drugdevelopment approaches.
CLINICAL RELEVANCE
The approaches discussed are rele-vant to multiple domains of clinicalpractice, and exist as a diverse arrayof potential and realized translationalendeavors. While some methods (e.g.,the use of disease biomarkers to drivetreatment choices) already reflect astandard of care within certain areasof medicine, other areas (e.g., themodeling of causal disease networks)which do not yet routinely impactclinical care, are expected to add in-sight to the identification of high-quality drug targets to inform drugdevelopment, as well as guiding theinterpretation of individual geneticvariation in creating tailored thera-peutic strategies.
BACKGROUND
Healthcare in the 21st century fa-ces a unique set of challenges in facingrising healthcare costs and decliningresearch and development productiv-ity in therapeutic discovery and de-velopment. Until 2010, costs havebeen rising faster than economicgrowth in many Organization forEconomic Cooperation and Develop-ment countries,1 which has positionedhealthcare expenditure as a key eco-nomic focus. As innovation of newtreatments, rising provider costs, andan increased prevalence of expensive,chronic conditions press treatmentcosts upwards, the expanding, ageingpopulation in many developed nationsis reflected in an increased demandfor medical care.
An important driver of this costgrowth is the partial success of manyexisting treatments; medical inter-
ventions which lead to so-called‘‘chronification’’ of complex, previ-ously fatal diseases which can now bemodified or at least managed symp-tomatically for many years (e.g., is-chemic heart disease, type 2 diabetes,cerebrovascular disease and chronicobstructive pulmonary disease), withthe incidence of such diseases in-creasing with age.2 Despite the bene-fits of being able to offer suchtreatments to patients, the interven-tion and maintenance of treatment atthis level of disease progression is in-efficient and expensive. Of course,historically there has been little al-ternative but for a patient becomesymptomatic before seeking clinicalreview, as the combination of clini-cally observable signs and reportedsymptoms have been our primaryview into a patient’s physiology, and itis usually only once a significant sys-tem perturbation has occurred that anindividual will notice anything un-usual. However, in many patholo-gies, predisease conditions can nowbe identified (e.g., prehypertension,prediabetes, mild cognitive impair-ment), states which imply increasedrisk of progression to full-blown dis-ease for a patient, and in some cases,with an accompanying opportunityto engage in early therapeutic strat-egies (e.g., exercise prescriptions,dietary changes, medications) whichcan reduce the risk of progression.Some of the key challenges in thisparadigm of disease screening are inidentifying predisease states whichrepresent significantly increasedrisk of future adverse health out-comes, identifying reliable markersof these predisease states, and de-veloping useful therapeutic strate-gies to offer individuals should theyreceive a predisease diagnosis. Withhealthcare costs amounting toUS$2.6 trillion in 2010, and threeout of four treatment dollars beingspent on the management of chronicconditions, the need for new approaches
NSCLC = non small cell lungcancer
PAH = pulmonary artery hyper-tension
R&D = research and develop-ment
RNA = ribonucleic acid
SNOMED-CT = SystematizedNomenclature of MedicalTerms-Clinical Terms
SNP = single nucleotide poly-morphism
TTC = tissue-tissue coexpres-sion
Abbreviationsand Acronyms (continued)
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 471
to address these issues has never been morepressing.3
The concept of precision medicine, an approachwhere a patient’s health traits are interpretedalongside state-of-the-art molecular profiles, todevelop accurate diagnostic, prognostic and ther-apeutic strategies which are tailored to reflect in-dividual physiological context, has been put forthas a model for transitioning towards a more safe,effective, and efficient healthcare paradigm.4 Pre-cision medicine offers an expectation of an im-proved power to diagnose disease at earlier stages,facilitating treatment initiation at more stablephases of pathogenesis. Recent advances in meth-ods for gathering and analyzing genomic andother molecular level data across many technologyplatforms, and different biological contexts (e.g.,different diseases, populations, tissues, or timeperiods5) serves as the groundwork for enablingprecision medicine.
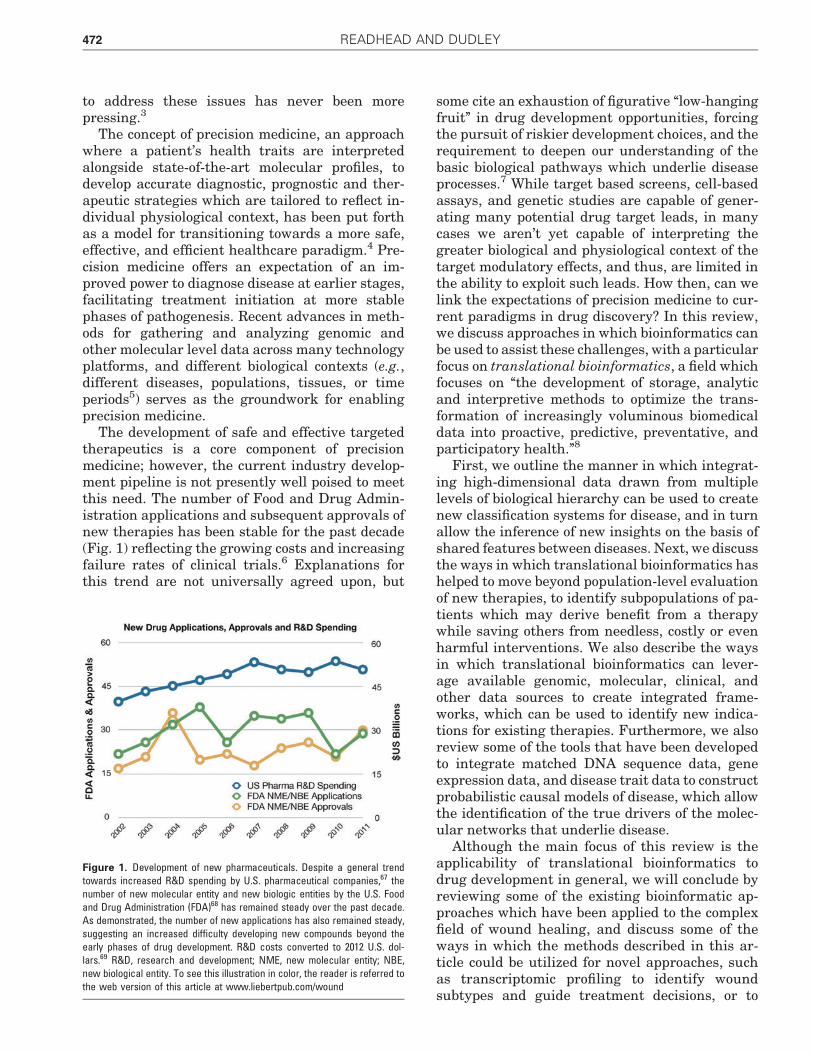
The development of safe and effective targetedtherapeutics is a core component of precisionmedicine; however, the current industry develop-ment pipeline is not presently well poised to meetthis need. The number of Food and Drug Admin-istration applications and subsequent approvals ofnew therapies has been stable for the past decade(Fig. 1) reflecting the growing costs and increasingfailure rates of clinical trials.6 Explanations forthis trend are not universally agreed upon, but
some cite an exhaustion of figurative ‘‘low-hangingfruit’’ in drug development opportunities, forcingthe pursuit of riskier development choices, and therequirement to deepen our understanding of thebasic biological pathways which underlie diseaseprocesses.7 While target based screens, cell-basedassays, and genetic studies are capable of gener-ating many potential drug target leads, in manycases we aren’t yet capable of interpreting thegreater biological and physiological context of thetarget modulatory effects, and thus, are limited inthe ability to exploit such leads. How then, can welink the expectations of precision medicine to cur-rent paradigms in drug discovery? In this review,we discuss approaches in which bioinformatics canbe used to assist these challenges, with a particularfocus on translational bioinformatics, a field whichfocuses on ‘‘the development of storage, analyticand interpretive methods to optimize the trans-formation of increasingly voluminous biomedicaldata into proactive, predictive, preventative, andparticipatory health.’’8
First, we outline the manner in which integrat-ing high-dimensional data drawn from multiplelevels of biological hierarchy can be used to createnew classification systems for disease, and in turnallow the inference of new insights on the basis ofshared features between diseases. Next, we discussthe ways in which translational bioinformatics hashelped to move beyond population-level evaluationof new therapies, to identify subpopulations of pa-tients which may derive benefit from a therapywhile saving others from needless, costly or evenharmful interventions. We also describe the waysin which translational bioinformatics can lever-age available genomic, molecular, clinical, andother data sources to create integrated frame-works, which can be used to identify new indica-tions for existing therapies. Furthermore, we alsoreview some of the tools that have been developedto integrate matched DNA sequence data, geneexpression data, and disease trait data to constructprobabilistic causal models of disease, which allowthe identification of the true drivers of the molec-ular networks that underlie disease.
Although the main focus of this review is theapplicability of translational bioinformatics todrug development in general, we will conclude byreviewing some of the existing bioinformatic ap-proaches which have been applied to the complexfield of wound healing, and discuss some of theways in which the methods described in this ar-ticle could be utilized for novel approaches, suchas transcriptomic profiling to identify woundsubtypes and guide treatment decisions, or to
Figure 1. Development of new pharmaceuticals. Despite a general trendtowards increased R&D spending by U.S. pharmaceutical companies,67 thenumber of new molecular entity and new biologic entities by the U.S. Foodand Drug Administration (FDA)68 has remained steady over the past decade.As demonstrated, the number of new applications has also remained steady,suggesting an increased difficulty developing new compounds beyond theearly phases of drug development. R&D costs converted to 2012 U.S. dol-lars.69 R&D, research and development; NME, new molecular entity; NBE,new biological entity. To see this illustration in color, the reader is referred tothe web version of this article at www.liebertpub.com/wound
472 READHEAD AND DUDLEY

identify new therapeutic leads to inform drugdevelopment. The methods described here arealso relevant to the extent that they can be usedto improve diagnostic and therapeutic strategiesfor the range of systemic diseases which have animpact on wound healing, such as peripheralvascular disease, type 2 diabetes and venoushypertension.
DISCUSSION OF FINDINGSAND RELEVANT LITERATUREDisease taxonomy and drug development
The value of creating a framework by which toclassify diseases was apparent in the time of Hip-pocrates, though wasn’t developed in a systematicway until the Enlightenment, initially by Sau-vages9 and then built upon by Linnaeus.10 Thecomplexity of modern disease taxonomies have in-creased significantly since then, though the un-derlying principles of classification on the basis ofobservable attributes remains similar. Our currentdisease classification systems reflect the diverserequirements of stakeholders, including classifica-tion for processing of reimbursement (e.g., Inter-national Classification of Disease [ICD]), researchpurposes (e.g., Medical Subject Headings), special-ist reporting (e.g., Systematized Nomenclature ofMedical Terms-Clinical Terms [SNOMED-CT]),and clinical classification (ICD, Diagnostic andStatistical Manual IV). These taxonomies tend toreflect the attributes of disease that are routinelymeasured, which is itself a function of convenienceand utility. Therefore, current systems classifydiseases according to constellations of clinical, an-atomical or investigative findings. Such methodshave undoubtedly been beneficial, but can also bemisleading, by clustering essentially dissimilarpathologies (which may share organs systems orclinical manifestations) or separating similar ones(which may appear distinct at a clinical level).Loscalzo et al.11 discuss the examples of diseaseswhich form clinically distinct syndromes yetemerge from the same molecular basis, and con-versely, diseases that have multiple genetic bases,yet converge into a common phenotype. Sicklecell anemia, a monogenic disease, with a well-characterized, single genetic basis, can clinicallymanifest along a spectrum of clinical severity rang-ing from mild anemia to veno-occlusive stroke, de-pendent on additional genetic and environmentalrisks.11 This is contrasted with diseases, such asfamilial pulmonary artery hypertension (PAH),which are associated with many different mutationsaffecting members of the transforming growth fac-
tor beta receptor superfamily, yet tending towardsa common phenotype.12
There have been multiple efforts to classifysystems of diseases according to their molecularlevel correlates, including investigation by theNational Academy of Science into creating aknowledge network of disease to underpin a newtaxonomy of disease.13 At a disease class level,Sirota et al.14 examined Genome Wide Associa-tion Study results to determine genetic variationprofiles associated with a range of autoimmunedisease, identifying sets of diseases which ap-peared to cluster together, conferring some pro-tective effects against diseases in other classes.They reported individual single nucleotide poly-morphism (SNP) and gene associations that ap-pear to drive the similarities and differencesbetween different clusters of diseases. Interest-ingly, they found that some disease pairs (e.g.,Crohn’s disease and rheumatoid arthritis), whichshare some treatments, and clinical features,possess different underlying genetic profiles sug-gesting a differing molecular basis. Suthramet al.15 used protein interaction networks toidentify functional modules, which were then as-sessed in the context of gene expression micro-array data for 54 different diseases. Each diseasewas comprised of a signature of ‘‘module responsescores’’, which were used to cluster diseases. In-terestingly, sets of modules relating to key bio-logical functions were found to be perturbed inmany diseases. This same set of pan-diseasemodules was also enriched for proteins identifiedas pluripotent drug targets (i.e., drugs targetingthese proteins tended to have many indications),suggesting the relevance of targets within thesemodules for a wide range of therapeutic uses.
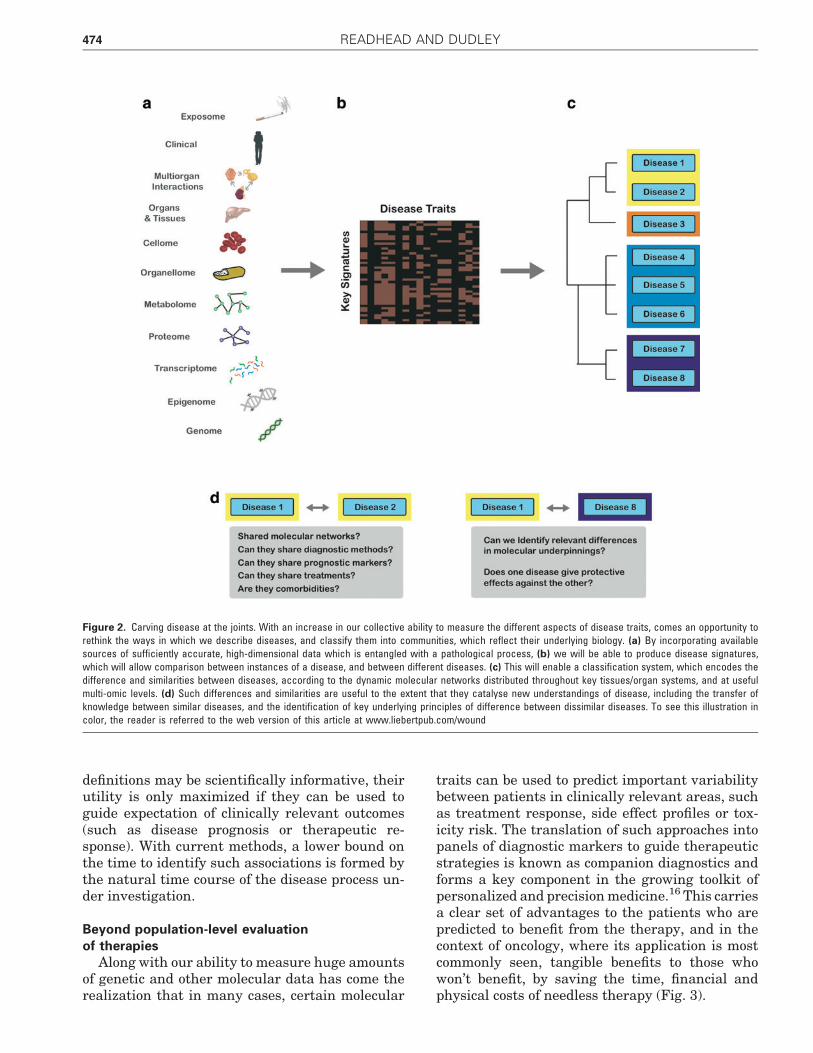
The highlighted works demonstrate an impor-tant principle—the potential for data-driven mo-lecular taxonomy approaches to challenge andimprove existing models of disease taxonomy.Such studies offer the possibility to share insightsbetween ‘‘similar’’ diseases with the potential ofidentifying common disease pathways (with as-sociated drug repositioning opportunities), as wellas by looking at dissimilar pairs, which can leadto the identification of exclusive pathways, withassociated opportunities for further hypothesis-led studies to identify novel treatment targets(Fig. 2).
Despite an increasing ability to compare andcontrast different disease entities at a range ofbiological levels of organization, an important chal-lenge lies in translating this into clinical decision-making tools. While new disease descriptions and
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 473
definitions may be scientifically informative, theirutility is only maximized if they can be used toguide expectation of clinically relevant outcomes(such as disease prognosis or therapeutic re-sponse). With current methods, a lower bound onthe time to identify such associations is formed bythe natural time course of the disease process un-der investigation.
Beyond population-level evaluationof therapies
Along with our ability to measure huge amountsof genetic and other molecular data has come therealization that in many cases, certain molecular
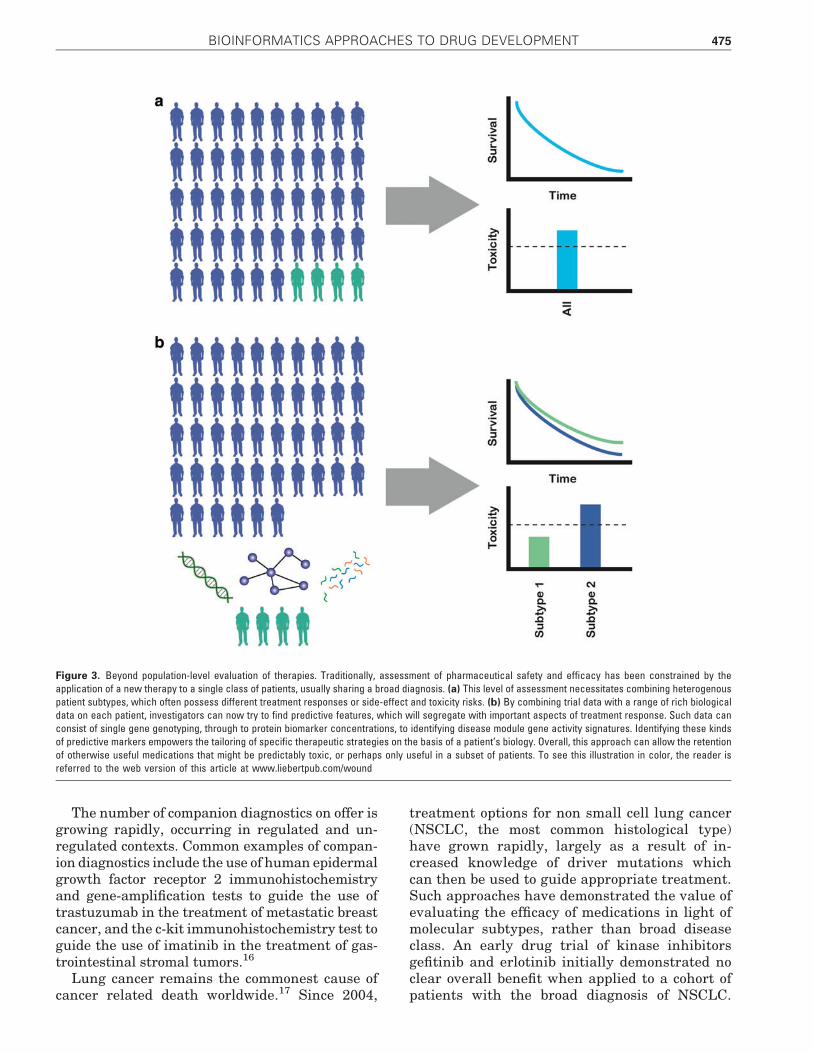
traits can be used to predict important variabilitybetween patients in clinically relevant areas, suchas treatment response, side effect profiles or tox-icity risk. The translation of such approaches intopanels of diagnostic markers to guide therapeuticstrategies is known as companion diagnostics andforms a key component in the growing toolkit ofpersonalized and precision medicine.16 This carriesa clear set of advantages to the patients who arepredicted to benefit from the therapy, and in thecontext of oncology, where its application is mostcommonly seen, tangible benefits to those whowon’t benefit, by saving the time, financial andphysical costs of needless therapy (Fig. 3).
Figure 2. Carving disease at the joints. With an increase in our collective ability to measure the different aspects of disease traits, comes an opportunity torethink the ways in which we describe diseases, and classify them into communities, which reflect their underlying biology. (a) By incorporating availablesources of sufficiently accurate, high-dimensional data which is entangled with a pathological process, (b) we will be able to produce disease signatures,which will allow comparison between instances of a disease, and between different diseases. (c) This will enable a classification system, which encodes thedifference and similarities between diseases, according to the dynamic molecular networks distributed throughout key tissues/organ systems, and at usefulmulti-omic levels. (d) Such differences and similarities are useful to the extent that they catalyse new understandings of disease, including the transfer ofknowledge between similar diseases, and the identification of key underlying principles of difference between dissimilar diseases. To see this illustration incolor, the reader is referred to the web version of this article at www.liebertpub.com/wound
474 READHEAD AND DUDLEY
The number of companion diagnostics on offer isgrowing rapidly, occurring in regulated and un-regulated contexts. Common examples of compan-ion diagnostics include the use of human epidermalgrowth factor receptor 2 immunohistochemistryand gene-amplification tests to guide the use oftrastuzumab in the treatment of metastatic breastcancer, and the c-kit immunohistochemistry test toguide the use of imatinib in the treatment of gas-trointestinal stromal tumors.16
Lung cancer remains the commonest cause ofcancer related death worldwide.17 Since 2004,
treatment options for non small cell lung cancer(NSCLC, the most common histological type)have grown rapidly, largely as a result of in-creased knowledge of driver mutations whichcan then be used to guide appropriate treatment.Such approaches have demonstrated the value ofevaluating the efficacy of medications in light ofmolecular subtypes, rather than broad diseaseclass. An early drug trial of kinase inhibitorsgefitinib and erlotinib initially demonstrated noclear overall benefit when applied to a cohort ofpatients with the broad diagnosis of NSCLC.
Figure 3. Beyond population-level evaluation of therapies. Traditionally, assessment of pharmaceutical safety and efficacy has been constrained by theapplication of a new therapy to a single class of patients, usually sharing a broad diagnosis. (a) This level of assessment necessitates combining heterogenouspatient subtypes, which often possess different treatment responses or side-effect and toxicity risks. (b) By combining trial data with a range of rich biologicaldata on each patient, investigators can now try to find predictive features, which will segregate with important aspects of treatment response. Such data canconsist of single gene genotyping, through to protein biomarker concentrations, to identifying disease module gene activity signatures. Identifying these kindsof predictive markers empowers the tailoring of specific therapeutic strategies on the basis of a patient’s biology. Overall, this approach can allow the retentionof otherwise useful medications that might be predictably toxic, or perhaps only useful in a subset of patients. To see this illustration in color, the reader isreferred to the web version of this article at www.liebertpub.com/wound
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 475

However, it was recognized at the time that asubpopulation of *10% of patients actually ex-perienced dramatic antitumor results, which wasa sufficient basis for Food and Drug Adminis-tration approval in 2003, despite the fact thatthe specific underlying mechanism conferringsensitivity wasn’t yet known.18 Later, it wasfound that positive treatment response corre-lated with activating epidermal growth factorreceptor mutations, which could then be used asa distinct biomarker to guide initiation of treat-ment.19–22 This biomarker enabled a more ef-fective trial design, with optimization oftherapeutic indications; thus, reducing unneces-sary side effects and costs in patients thatwouldn’t benefit from the therapy.
A more recent example, centers on the story ofcrizotinib, another kinase inhibitor (particularlyMET [mesenchymal epithelial transition factor]and ALK [anaplastic lymphoma kinase] activity)used in the treatment of NSCLC. Part way througha clinical trial assessing its efficacy on a broadpopulation of lung cancer patients,23 an indepen-dent team published findings of a particular chro-mosomal translocation affecting the ALK genewith an association to tumor growth in a subset ofNSCLC patients.24 This allowed modification of theclinical trial to assess treatment efficacy within theframe of this particular variant, with dramaticallydifferent results, showing an increase in mediansurvival time to about a year, as opposed to a fewmonths with the standard of care. Such targetedapproaches also offer the potential benefit of iden-tifying useful therapeutic responses on smallercohorts of patients.
It is clear that historically, a level of serendipityhas contributed to these discoveries, suggestingthat many additional opportunities exist, and haveperhaps been missed. The increased use of sys-tematic, bioinformatics approaches to correlatemolecular and genomic level patient data withtreatment response and adverse outcomes anddrug toxicity is likely to enable more routine andaccelerated discovery in relevant patient sub-populations. Such approaches could potentiallyleverage any accessible, meaningful biological ele-ments, from single gene genotyping,25 to non-coding RNA expression,26 to multi-module diseasegene expression signature.27
Computational drug repositioningThe practice of identifying additional thera-
peutic indications for existing drug compounds isreferred to as drug repositioning, and has somekey benefits over traditional methods of drug
development. Estimating the average costs ofbringing a drug to market is a complicated un-dertaking, though estimates placed capitalizedcosts between approximately US$800 million28
and US$1.8 billion29 with an accompanyingtimeline of 15 years.30 A large portion of thesecosts are associated with early stages of develop-ment and toxicity testing, with over 90% of testedcompounds failing to progress beyond this stage.31
The identification of additional indications forexisting medications offers clear time and costbenefits, thereby avoiding many of these earlyhurdles.
Traditional approaches to drug developmentusually focus on identification of a novel treatmenttarget, followed by a search for a compound capableof appropriately modulating the target. A lengthyand costly target validation process then followstarget identification. For a given compound, addi-tional targets are not usually investigated, andadditional clinical applications, are not routinelyexplored.32 This represents a huge opportunity forthe systematic identification of new indications forexisting therapeutics—some of the most commer-cially successful medications are administered fordifferent indications than envisioned in their ini-tial development. Prominent examples amongstmany include sildenafil (an antiangina medicationnow used for treatment of erectile dysfunction andPAH), minoxidil (anti-hypertensive, now used foralopecia) and thalidomide (antinausea, now usedfor multiple myeloma and erythema nodosumleprosum).
Multiple approaches have been used in drugrepositioning efforts, ranging from blind screeningof libraries of drug compounds against modelsystems, to data-driven computational approachesthat integrate and search across links betweendrugs and diseases using various forms of biomo-lecular and clinical data. Here we focus on com-putational approaches, which offer substantialadvantages in cost and speed of discovery com-pared to experimental screening based ap-proaches.
In general, computational approaches exploitthe known links between diseases and drugs, withthe accompanying possibility that shared attri-butes between drugs used to treat the same dis-ease, or diseases which share treatments, implies adegree of meaningful similarity between some as-pects of the linked objects, which can be used togeneralize existing treatments into new clinicalcontexts. Dudley et al.32 describe a means to clas-sify computational approaches to drug reposition-ing according to whether the mode of inference is
476 READHEAD AND DUDLEY

drug-based or disease-based (Fig. 4). We will brieflyoutline these approaches, giving examples of each.
Drug-based approaches to drug repositioning
Approaches using drug similarity. Drug simi-larity approaches leverage known properties ofexisting drug compounds to compare and classifyon the basis of similarity in their chemical prop-erties. Such approaches use a variety of chemicaldescriptors, such as QSAR models (quantitativestructure-activity models) and pharmacophores toallow comparison and clustering of drugs whichshare similar properties.33 Such clustering canthen allow the inference of similarity betweendrugs, and identification of potential novel thera-peutic applications.
Noeske et al.34 used this conceptual approachto identify new treatment targets for a group ofknown metabotropic glutamate (mGlu) receptoragonists by preparing pharmacophore descriptorsfor the antagonists as well as a range of addi-tional known compounds, and used self-organizingmaps to create a compound map. They reportedsubclusters containing mGlu antagonists, withproximity to additional compounds known tobind to dopamine and histamine receptors. Thiswas then used as the basis for further experi-ments, which confirmed the predicted interac-tions between mGlu receptor antagonists andHistamine Receptor 1 and dopamine receptor 2targets.
Keiser et al.35 developed a quantitiative ap-proach for clustering protein targets on the basis of
similarity amongst their associated ligands. Their‘‘Similarity Ensemble Approach’’ used 65,000 li-gands grouped into sets on the basis of known in-teractions with hundreds of protein targets. Apairwise ligand similarity score was calculatedbetween every member of each set and every othermember of every other set, to derive the inter-setsimilarity, which reflected the similarity of theprotein targets at the core of each set. In this way,protein targets can be linked even when they don’tshare any common ligands. Despite utilizing aprotein similarity score derived entirely from che-mical similarity of ligands, biologically relatedprotein clusters nevertheless emerged, alongsideunexpected links, such as methadone antagonizingmuscarinic M3 receptors and loperamide antago-nizing neurokinin N2 receptors (these predictionswere then confirmed experimentally). With its po-tential to link proteins which are functionally re-lated, yet share nothing in the way of structural orsequence similarity, this ligand-centric approachrepresents a valuable tool for deriving knowledgeabout biological function which can circumventsome of the limitations of approaches which relysolely on protein similarity metrics.
Drug similarity approaches are limited by anumber of factors, including the requirement foraccurate surveys of chemical properties for anydrug under consideration and closed access tothese types of information, often as part of the in-tellectual property strategy of a pharmaceuticalcompany which has invested heavily in the devel-opment of its compounds. In addition, the effect of a
Figure 4. Drug repositioning strategies. Many computational approaches to drug repositioning capitalize on known relationships between diseases anddrugs, with the accompanying possibility that shared features between drugs used to treat the same disease, or diseases that share treatments, implies adegree of meaningful similarity between some aspects of the linked entities, which can then be used to generalize existing treatments into new clinicalcontexts. A number of emerging methods integrate multiple sources of disease and drug based knowledge, which can enable even more sophisticatedinferences about new therapeutic opportunities. (Figure reprinted with permission from Dudley et al.32)
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 477

drug is much more complex than its initial chemi-cal properties. Many drugs undergo extensivemetabolic transformations into their active com-ponents, and such processes aren’t always wellcharacterized, or well accounted for in pharmaco-phore descriptions.36
Approaches using molecular activity similarity.Another exciting approach to drug repositioningarises by characterizing drug compounds accordingto their impact on molecular activity. Such an ap-proach frames a compound as a perturbation to thesystem it is introduced to, which can be used toidentify a characteristic signature for that com-pound. This can then be used to compare manymedications, with many opportunities to general-ize therapeutic indications between ‘‘similar’’drugs.
One of the most widely used resources in this areais the Connectivity Map,37,38 a repository for thewhole genome transcriptional response of 1,309small bioactive molecules applied to a range of cul-tured human cells. Iorio et al.39 used the connectivitymap to perform pairwise comparisons between allcompounds, followed by clustering to identify drugcommunities with cluster co-location, implying pos-sible repositioning opportunities. By leveragingthese associations, they predicted previously un-known autophagy activity for fasudil, a Rho-kinaseinhibitor, which was later validated experimentally.
Sirota et al.40 used an integrated approachwhich compared drug signatures, with analogousdisease signatures to identify repositioning oppor-tunities. Leveraging publicly available microarraygene expression data, the authors looked at 100diseases, and 164 drug compounds, hypothesizingthat if a given disease is characterized by a geneexpression signature, then drugs which induce aninverse signature may have therapeutic value,while drugs with similar signatures may exacer-bate the disease. Using this approach, the authorsrederived many known therapeutic applications,such as the efficacy of prednisolone in treatment ofCrohn’s disease and ulcerative colitis. In addition,they predicted therapeutic benefit for the over-the-counter H2-receptor antagonist cimetidine(commonly used in gastric reflux and peptic ulcerdisease to reduce gastric acid secretion) in thetreatment of lung adenocarcinoma. They thenvalidated this prediction in-vitro using cell lines,and in-vivo using xenograft mouse models.
It is clear that such approaches aren’t con-strained to the use of gene expression data. Anymethod which provides meaningful, high-dimensionaldescription of a drug compound can be considered.
Chen et al.41 used PubChem42 to create signatureson the basis of bioassay results for PubChem com-pounds. They then constructed a bipartite networkthat mapped PubChem profiles onto biologicalnetworks (protein interaction networks and meta-bolic pathways) on the basis of sequence similaritybetween compound protein targets and the pro-teins represented in the biological networks. Thisallowed interpretation of compound efficacy or ad-verse effects to occur in a rich biological context,which can form the basis of further hypothesis ledexperiments.
The primary limitations of molecular similarityapproaches are their reliance on the quality of datathat are used to form the disease and drug signa-tures, as well as the simplifications which must bemade in forming the disease signatures. For ex-ample, Connectivity Map reference signatures arederived from specific cell lines, which may be ir-relevant to the actions of certain compounds, aswell as neglecting the often complex metabolismwhich compounds undergo from the time of theiradministration to their arrival in distant cells.Additional simplifications occur in the transcrip-tional classification of many diseases, which areknown to affect multiple tissue types and organsystems (such as peripheral vascular disease), ordiseases where the relevant tissue types may notbe obvious (hypertension) or amenable to easysampling (Alzheimer’s disease).
Approaches using molecular docking. Moleculardocking is a simulation-based approach that uses3D structural protein information to character-ize the interaction interface between drug com-pounds and targets. This can then be used as thebasis for making predictions about whether drugcompounds will interact with novel treatmenttargets. If a compound is predicted to interactwith a novel treatment target, then an inferencecan be made that the compound may be useful inany diseases in which the treatment target isimplicated.
In an interesting molecular docking approach,Kinnings et al.42 started with approved drugs, andidentified binding sites from the 3D structural dataof their known targets, using these sites as thebasis for a similarity search with other proteins.Additional proteins that were found to possesssimilar sites were highlighted as predicted addi-tional targets for the drug compound, allowing therepositioning of entacapone (most frequently usedin Parkinson’s disease to inhibit the metabolism oflevodopa) as a treatment for multi-drug resistanttuberculosis.
478 READHEAD AND DUDLEY

Such approaches rely heavily on having access to3D structural information of drugs and their li-gands. Although many such structures are avail-able and growing rapidly, some very significantomissions exist, including many members of theG-protein coupled receptor superfamily.
Disease-based approaches to drugrepositioning
Approaches using associative indication trans-fer. Another approach to drug repositioningwhich has been recently explored is one which le-verages known associations between drugs anddiseases, on the assumption that drugs whichshare many disease indications may share mech-anisms of action; thus, allowing the generalizationof putatively similar drugs into additional clinicalcontexts.43 As has been noted previously,32 thisinteresting approach is currently constrained bylack of systematic knowledge of the therapeuticintent which links a particular drug to a particulardisease. For example, the treatment of an infec-tious disease may require concomitant adminis-tration of paracetamol for relief of fever and anantibiotic for the underlying infection—two treat-ments which clearly engage with the underlyingpathology at different levels.
Approaches using shared molecular pathology.Some repositioning strategies proceed on theassumption that diseases that share drugs maydo so because of common aspects in their mo-lecular pathophysiology, implying a shared re-sponsiveness to the therapeutic functions of thedrug. On the basis of this, drugs may be reposi-tioned into additional diseases that look quitedifferent at the phenotypic level. The approach isanalogous in many ways to methods which makeuse of shared molecular similarity, in fact anumber of interesting studies have emergedwhich combine the two approaches to clusterdiseases and drugs into communities to identifyrepositioning opportunities, including some al-ready discussed.15,40 Hu and Agarwal44 usedGene Expression Omnibus (GEO) data to for-mulate disease signatures, and then clusteredthese using a correlation based approach. Thesesignatures were then integrated with Con-nectivity Map data to cluster disease molecularpathology and drug mechanisms.
Understandably, such approaches are limited bythe technologies that are used to characterize apathophysiological state (e.g., gene expression mi-croarray). Additional current limitations reside incapturing and meaningfully comparing the multi-
tissue, multi-organ and dynamic nature of manydiseases over time.
Approaches using side effect similarity. Anothernovel repositioning approach consists of linkingdrugs together on the basis of their side effectsimilarity. On the assumption that such side effectprofiles provide a read-out of the physiological ef-fect of a compound on a host system, Campilloset al.45 mapped medication side-effects from pack-age leaflets, and mapped the listed terms to theUnified Medical Language System, creatingweighted associations between side effects anddrugs on the basis of side-effect frequency. Inter-drug similarity was then calculated by the sum ofcommon weights, allowing formation of drug com-munities, from which repositioning opportunitiescould be inferred.
Approaches that rely on the integration ofdata represented as a structured language aresubject to the constraints of codifying such acomplex, heterogeneous, and ambiguous domainas healthcare delivery into a formalized system.Some of the more common issues include dis-crepancies in categorization of concepts and thedevelopment of multiple representations of thesame concept (e.g., SNOMED-CT possesses aconcept for ‘‘Apgar score at 1 minute’’ as an ob-servable entity, as well as a separate concept for‘‘Apgar score at 1 minute’’ as a finding entity)46
which can lead to ambiguities in interpreting thesemantic context of a given term.
Such limitations are also compounded by theprecision with which side effect profiles are re-corded. Many clinical signs and symptoms arequite non-specific, and the level of granularity withwhich side-effects are elicited could have signifi-cant effects on characterizing a drug. For example,a reported side-effect of abdominal pain, if notcharacterized further may imply mechanisms asdiverse as gastric reflux, hepatotoxicity or drug-induced diarrhea.
Many promising computational drug re-positioning approaches exhibiting quantifiablesuccess have emerged in the past decade. It’s alsoclear from the described approaches that tech-niques can be devised which exploit any suffi-ciently high-dimensional description of a disease orits known efficacious treatments. Some of the mostpromising approaches have been ones which inte-grate multiple strategies to various degrees: uti-lizing high throughput data describing therapies,as well as diseases to create communities of drug-disease interactions, with the possibility of linkingthese to other forms of rich biological data, such as
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 479

protein interaction networks, DNA and proteinsequence data, transcriptional and metabolic net-works. Future approaches may also see the use ofmuch higher-level phenotypic data, extractingfeatures from imaging studies, electroencephalo-gram, or wireless monitoring devices as the basisfor comparing disease and drug similarity.
Causal network models of diseaseAn emerging perspective in systems medicine
is that to understand biology, and identify thetrue drivers of pathology in individual patientswill require the construction of high-dimensionalnetworks representing multiple levels of biolog-ical hierarchy.47 In this view, core biologicalprocesses are mediated by networks of interact-ing molecules (DNA, RNA, proteins, metabolites,etc.) in a manner which is dynamically coordi-nated within and between tissues, and in ahighly context dependent manner. This systemsmedicine view frames these molecular networksas key sensors to environmental and geneticperturbation,48 shifting their state in response toa myriad of context dependent cues, and in theframe of pathogenesis, representing intermedi-ate molecular phenotypes which mediate theflow of risk between disease associated DNAvariants and disease states.49
One of the great challenges implicit in devel-oping such an understanding is the extraction ofthe relevant networks from an immensely noisymolecular background, to identify the molecularunderpinnings of normal and pathogenic pro-cesses. Some exciting computational approacheshave been developed, which integrate matchedDNA variation, gene expression, and clinical datato construct probabilistic causal networks50—models which capture functional relationshipsbetween key biological objects (such as gene reg-ulatory networks) and phenotypic data (such asdisease traits), to approach a mechanistic under-standing of disease and allow the differentiation ofreactive elements from the true drivers of disease(Fig. 5). Bayesian network approaches are com-monly employed to learn the conditional proba-bility distributions of molecular and clinicalvariables, and then used to solve conditionalprobability equations to infer probable causalnetworks.51 Bayesian networks often infer manyequally possible network graphs as solutions.51
This quickly leads to nontrivial computationaland methodological challenges in exploring thecomplete space of all probable network graphs.Other popular approaches for network recon-struction are gene coexpression networks, or
methods that first group genes and loci into func-tionally coherent groups called ‘‘modules’’.51 Cau-sal inference is then performed to infer networksof functional gene modules.
The Macrophage Enriched Metabolic Network(MEMN) is an example of a disease network de-rived by integrating gene expression, genotype andclinical data from adipose and liver tissue, in miceand humans, which was found to be causally as-sociated with phenotypic traits of metabolic dis-ease, such as diabetes, heart disease and obesity.49
The authors combined co-expression networkanalysis, with a statistical procedure called ‘‘like-lihood-based causality model selection’’50 to iden-tify whether patterns of gene expression werecausal for, or reactive to the phenotypic diseasetraits. The MEMN was shown to be strongly causalfor the disease traits, facilitating the identificationand validation of three previously unknown type 2diabetes associated genes.
A key approach in constructing such networks,builds on weighted gene co-expression networkanalysis52 to identify sets of highly co-expressedgenes in a biological context of interest. Thesegene modules can then be converted into a net-work through consideration of pair-wise geneco-expression thresholds. Schadt et al.50 demon-strated that these correlation-derived associationnetworks could be converted into causal networks(where the direction of an interaction can beknown) by integrating them with a systematic formof biologically meaningful perturbation—DNA se-quence variation. By utilizing genetic loci that arecorrelated with mRNA transcript abundance (ex-pression quantitative trait loci [eQTL]), the au-thors developed a statistical approach thatconsiders an eQTL alongside a pair of genes withtranscript levels that correlate with sequence var-iation of eQTL. By analyzing conditional correla-tions between these three variables, it is possible toinfer the most likely relationship between the genepair (either gene A regulates gene B, gene B isregulated by gene A, or gene A and B do not regu-late each other).
Once such directed networks of gene regulatoryrelationships are available, it becomes possible toidentify the key drivers of that network, and thus,the disease trait it has been constructed to reflect.50
By functionally annotating the network members,and identifying the enriched functional pathwayswhich it is associated with, it becomes possible towork upstream into the network to identify geneswhich regulate many downstream members im-plicated in this particular function (local drivers)and ultimately to identify the most upstream genes
480 READHEAD AND DUDLEY

(global drivers).53 This set of drivers represents avery high leverage set of targets that can then beinvestigated for disease diagnosis, monitoring anddrug targeting purposes.
Such networks also offer the opportunity forin-silico screening of drug compounds by simulat-ing the extended effects a given drug may imposeon the network. In this way, unfavorable side effectprofiles, or worthwhile therapeutic potential can beanticipated. Utilizing the network comprising theMEMN, Chen et al.49 predicted that Ppm1l (at the
time, a poorly characterized protein phosphatase)had the potential to improve insulin resistance(highlighting its potential as a treatment target forType 2 diabetes); however, due to its network po-sition, it was also likely to increase weight gain,and increase blood pressure (therefore worseningoverall cardiovascular risk). These predictionswere then verified in Ppm1l knock-down mice,which demonstrated an improved diabetic profile,but with increased weight gain and blood pressure.This method of prioritizing leads for streamlined
Figure 5. Building causal models of disease. One of the exciting possibilities in systems medicine is the capacity to build models of disease which reflect thecausal interactions underlying disease at the molecular level. These models can then be used to identify new diagnostic, prognostic and therapeuticapproaches. One approach utilizes (a, b) gene expression microarray data from disease tissue to (c) identify modules of genes which are coexpressed, (d)
and for each of these modules, derive an undirected network which best accounts for the pair-wise gene expression correlations. (e) By layering in matchedDNA variation data for each patient, it becomes possible to transform the undirected network, into a causal network, which encodes the direction of functionalregulatory relationships between its genes. By considering expression quantitative trait loci (genomic locations where sequence variation correlates withmRNA transcript abundance of one or more genes) alongside a pair of its associated genes, it is possible of look at conditional correlations to find the mostlikely causal model connecting the eQTL and the gene pair. (f) By iterating over all relevant gene pairs (with their associated eQTLs) in all the modules, itbecomes possible to create a probabilistic causal network, and identify ‘‘local drivers’’ (blue)—genes which regulate many relevant downstream genes andeven the most upstream, so-called global drivers (dark blue). This allows the identification of potentially high leverage molecular targets, facilitating theprioritisation of high-value validation experiments. eQTL, expression quantitative trait locus. To see this illustration in color, the reader is referred to the webversion of this article at www.liebertpub.com/wound
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 481
experimental validation represents a very promis-ing advance in early drug development.
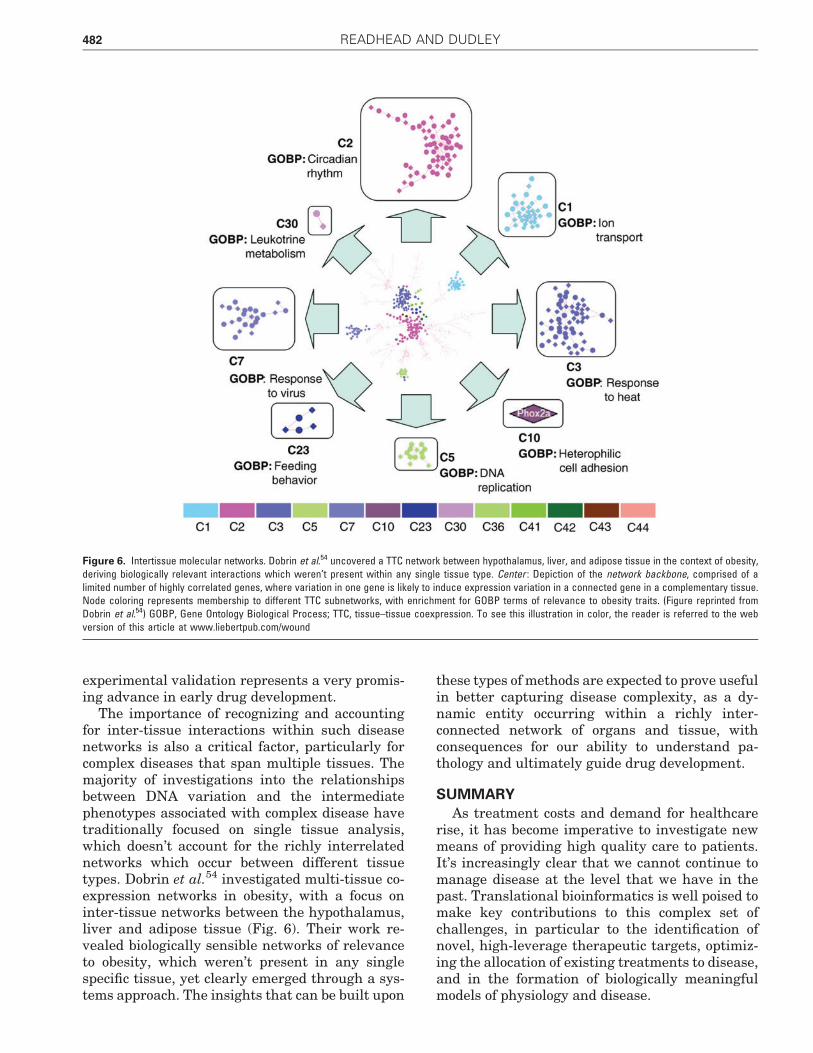
The importance of recognizing and accountingfor inter-tissue interactions within such diseasenetworks is also a critical factor, particularly forcomplex diseases that span multiple tissues. Themajority of investigations into the relationshipsbetween DNA variation and the intermediatephenotypes associated with complex disease havetraditionally focused on single tissue analysis,which doesn’t account for the richly interrelatednetworks which occur between different tissuetypes. Dobrin et al.54 investigated multi-tissue co-expression networks in obesity, with a focus oninter-tissue networks between the hypothalamus,liver and adipose tissue (Fig. 6). Their work re-vealed biologically sensible networks of relevanceto obesity, which weren’t present in any singlespecific tissue, yet clearly emerged through a sys-tems approach. The insights that can be built upon
these types of methods are expected to prove usefulin better capturing disease complexity, as a dy-namic entity occurring within a richly inter-connected network of organs and tissue, withconsequences for our ability to understand pa-thology and ultimately guide drug development.
SUMMARY
As treatment costs and demand for healthcarerise, it has become imperative to investigate newmeans of providing high quality care to patients.It’s increasingly clear that we cannot continue tomanage disease at the level that we have in thepast. Translational bioinformatics is well poised tomake key contributions to this complex set ofchallenges, in particular to the identification ofnovel, high-leverage therapeutic targets, optimiz-ing the allocation of existing treatments to disease,and in the formation of biologically meaningfulmodels of physiology and disease.
Figure 6. Intertissue molecular networks. Dobrin et al.54 uncovered a TTC network between hypothalamus, liver, and adipose tissue in the context of obesity,deriving biologically relevant interactions which weren’t present within any single tissue type. Center : Depiction of the network backbone, comprised of alimited number of highly correlated genes, where variation in one gene is likely to induce expression variation in a connected gene in a complementary tissue.Node coloring represents membership to different TTC subnetworks, with enrichment for GOBP terms of relevance to obesity traits. (Figure reprinted fromDobrin et al.54) GOBP, Gene Ontology Biological Process; TTC, tissue–tissue coexpression. To see this illustration in color, the reader is referred to the webversion of this article at www.liebertpub.com/wound
482 READHEAD AND DUDLEY

We have discussed a range of translationalbioinformatics approaches which can contributeto these challenges at multiple levels, includingthe appropriate characterization and classifica-tion of diseases on the basis of their underlyingmolecular features, enabling the transfer of in-sights between pathologies deemed to be similar,and aiding the search for underlying principleswhich may separate pathologies deemed to bedistant. One implication of this type of diseasemap is the possibility to identify specific bio-markers that correlate with disease subtypes,with relevance for choice of therapy, and allowingprediction and prevention of unfavorable side-effect profiles or toxicity effects.
We have reviewed some of the translationalbioinformatics approaches for computational drugrepositioning, which has the potential to power-fully leverage from pre-existing drug developmentefforts to expand and reshape the catalog of ther-apies which are currently available. We can see
that current data-driven methods can be readilyadapted to utilize increasingly voluminous, high-quality biological data to identify ever more pre-cise attributes of disease and therapy, which canbe used to generalize between therapeutic con-texts.
We have also provided a brief overview of theevolving area of probabilistic causal networkmodeling of disease, which integrates genotype,expression and clinical data to provide deep in-sights into disease physiology and facilitating thedevelopment of powerful new therapeutic options.We expect that causal network modeling of diseasewill play a key role in building truly predictivemodels of natural disease progression in the nearfuture. As the cost of molecular profiling technolo-gies continues to decrease, it will become tractableto score multiscale molecular traits from individ-uals over time, and thus, become possible to char-acterize the dynamic changes driving networkstates from health to disease.55
Table 1. Upregulated genes
Gene Symbol Adjusted p-Value Fold Change Biological Function
PTX3 6.73 · 10 - 7 31 Zymosan bindingIGF2BP3 1.79 · 10 - 6 26 RNA binding; mRNA 3¢-UTR binding; mRNA 5¢-UTR binding; nucleotide binding; protein binding; translation
regulator activityS100A12 4.31 · 10 - 6 22 RAGE receptor binding; calcium ion binding; protein binding; zinc ion bindingMMP1 7.90 · 10 - 6 20 Calcium ion binding; metalloendopeptidase activity; peptidase activity; zinc ion bindingPI3 7.90 · 10 - 6 20 Endopeptidase inhibitor activity; serine-type endopeptidase inhibitor activityMME 8.03 · 10 - 6 20 Metal ion binding; metalloendopeptidase activity; metallopeptidase activity; peptidase activity; peptide binding;
protein binding; zinc ion bindingMMP3 1.33 · 10 - 5 19 Calcium ion binding; metalloendopeptidase activity; peptidase activity; zinc ion bindingPI3 1.33 · 10 - 5 18 Endopeptidase inhibitor activity; serine-type endopeptidase inhibitor activityWISP1 1.61 · 10 - 5 18 Insulin-like growth factor binding; protein bindingSLC16A1 1.64 · 10 - 5 18 Mevalonate transmembrane transporter activity; monocarboxylic acid transmembrane transporter activity;
protein binding; secondary active monocarboxylate transmembrane transporter activity; symporter activityTCN1 1.74 · 10 - 5 18 Cobalamin bindingADAM12 1.84 · 10 - 5 17 SH3 domain binding; metal ion binding; metalloendopeptidase activity; metallopeptidase activity; peptidase
activity; protein binding; zinc ion bindingKCNJ15 1.89 · 10 - 5 17 Inward rectifier potassium channel activity; potassium channel activity; protein binding; voltage-gated ion
channel activityHMGA2 2.56 · 10 - 5 16 AT DNA binding; DNA binding; protein bindingKRT6B 2.79 · 10 - 5 16 Structural constituent of cytoskeletonKRT16 4.92 · 10 - 5 15 Protein binding; structural constituent of cytoskeletonIL6 5.08 · 10 - 5 14 Cytokine activity; cytokine activity; growth factor activity; interleukin-6 receptor binding; contributes_to
interleukin-6 receptor binding; interleukin-6 receptor bindingTMEM158 5.23 · 10 - 5 14 Peptide bindingSLC6A14 5.73 · 10 - 5 14 Amino acid transmembrane transporter activity; neurotransmitter:sodium symporter activity; symporter activityMME 6.42 · 10 - 5 14 Metal ion binding; metalloendopeptidase activity; metallopeptidase activity; peptidase activity; peptide binding;
protein binding; zinc ion bindingSERPINB3 7.22 · 10 - 5 14 Peptidase inhibitor activity; protein binding; serine-type endopeptidase inhibitor activityDSC2 7.22 · 10 - 5 14 calcium ion bindingSLC16A1 7.31 · 10 - 5 13 Mevalonate transmembrane transporter activity; monocarboxylic acid transmembrane transporter activity;
protein binding; secondary active monocarboxylate transmembrane transporter activity; symporter activityDSC2 7.31 · 10 - 5 13 Calcium ion bindingANPEP 7.40 · 10 - 5 13 Aminopeptidase activity; metal ion binding; metallopeptidase activity; peptidase activity; peptide binding;
receptor activity; zinc ion binding
Upregulated genes arranged according to fold change, with adjusted p-values (Benjamini-Hochberg corrected70) and associated Gene Ontology biologicalfunctions.71
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 483
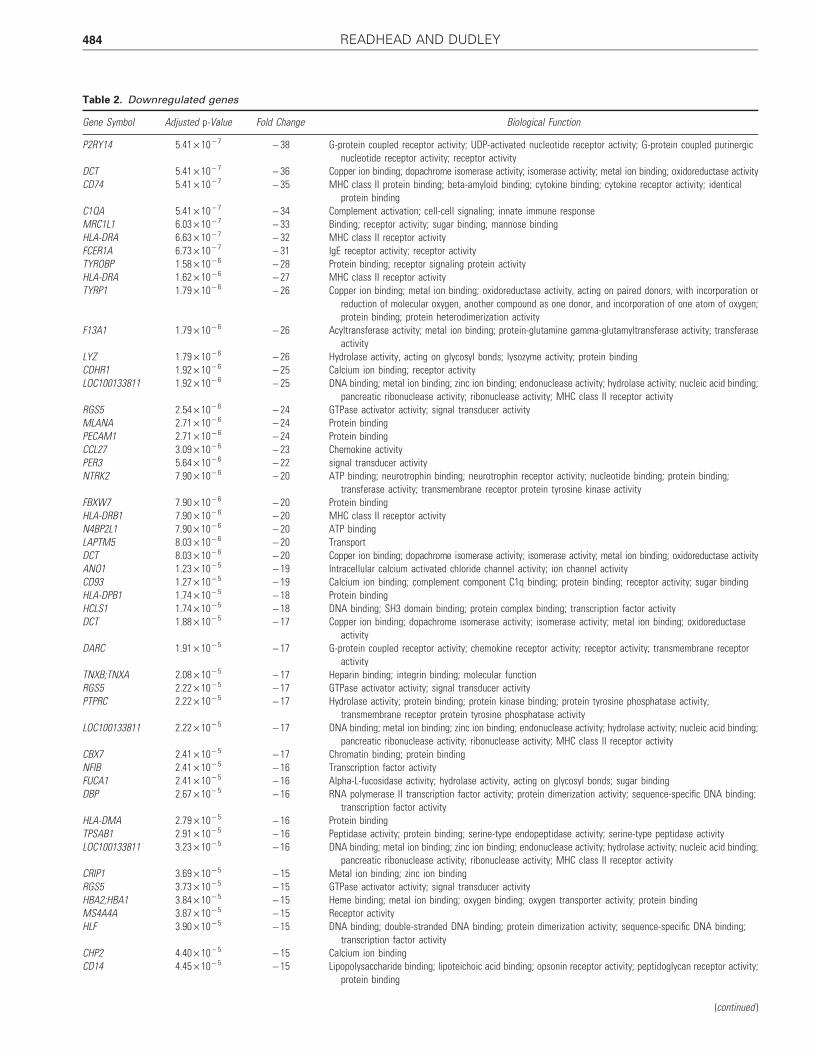
Table 2. Downregulated genes
Gene Symbol Adjusted p-Value Fold Change Biological Function
P2RY14 5.41 · 10 - 7 - 38 G-protein coupled receptor activity; UDP-activated nucleotide receptor activity; G-protein coupled purinergicnucleotide receptor activity; receptor activity
DCT 5.41 · 10 - 7 - 36 Copper ion binding; dopachrome isomerase activity; isomerase activity; metal ion binding; oxidoreductase activityCD74 5.41 · 10 - 7 - 35 MHC class II protein binding; beta-amyloid binding; cytokine binding; cytokine receptor activity; identical
protein bindingC1QA 5.41 · 10 - 7 - 34 Complement activation; cell-cell signaling; innate immune responseMRC1L1 6.03 · 10 - 7 - 33 Binding; receptor activity; sugar binding; mannose bindingHLA-DRA 6.63 · 10 - 7 - 32 MHC class II receptor activityFCER1A 6.73 · 10 - 7 - 31 IgE receptor activity; receptor activityTYROBP 1.58 · 10 - 6 - 28 Protein binding; receptor signaling protein activityHLA-DRA 1.62 · 10 - 6 - 27 MHC class II receptor activityTYRP1 1.79 · 10 - 6 - 26 Copper ion binding; metal ion binding; oxidoreductase activity, acting on paired donors, with incorporation or
reduction of molecular oxygen, another compound as one donor, and incorporation of one atom of oxygen;protein binding; protein heterodimerization activity
F13A1 1.79 · 10 - 6 - 26 Acyltransferase activity; metal ion binding; protein-glutamine gamma-glutamyltransferase activity; transferaseactivity
LYZ 1.79 · 10 - 6 - 26 Hydrolase activity, acting on glycosyl bonds; lysozyme activity; protein bindingCDHR1 1.92 · 10 - 6 - 25 Calcium ion binding; receptor activityLOC100133811 1.92 · 10 - 6 - 25 DNA binding; metal ion binding; zinc ion binding; endonuclease activity; hydrolase activity; nucleic acid binding;
pancreatic ribonuclease activity; ribonuclease activity; MHC class II receptor activityRGS5 2.54 · 10 - 6 - 24 GTPase activator activity; signal transducer activityMLANA 2.71 · 10 - 6 - 24 Protein bindingPECAM1 2.71 · 10 - 6 - 24 Protein bindingCCL27 3.09 · 10 - 6 - 23 Chemokine activityPER3 5.64 · 10 - 6 - 22 signal transducer activityNTRK2 7.90 · 10 - 6 - 20 ATP binding; neurotrophin binding; neurotrophin receptor activity; nucleotide binding; protein binding;
transferase activity; transmembrane receptor protein tyrosine kinase activityFBXW7 7.90 · 10 - 6 - 20 Protein bindingHLA-DRB1 7.90 · 10 - 6 - 20 MHC class II receptor activityN4BP2L1 7.90 · 10 - 6 - 20 ATP bindingLAPTM5 8.03 · 10 - 6 - 20 TransportDCT 8.03 · 10 - 6 - 20 Copper ion binding; dopachrome isomerase activity; isomerase activity; metal ion binding; oxidoreductase activityANO1 1.23 · 10 - 5 - 19 Intracellular calcium activated chloride channel activity; ion channel activityCD93 1.27 · 10 - 5 - 19 Calcium ion binding; complement component C1q binding; protein binding; receptor activity; sugar bindingHLA-DPB1 1.74 · 10 - 5 - 18 Protein bindingHCLS1 1.74 · 10 - 5 - 18 DNA binding; SH3 domain binding; protein complex binding; transcription factor activityDCT 1.88 · 10 - 5 - 17 Copper ion binding; dopachrome isomerase activity; isomerase activity; metal ion binding; oxidoreductase
activityDARC 1.91 · 10 - 5 - 17 G-protein coupled receptor activity; chemokine receptor activity; receptor activity; transmembrane receptor
activityTNXB;TNXA 2.08 · 10 - 5 - 17 Heparin binding; integrin binding; molecular functionRGS5 2.22 · 10 - 5 - 17 GTPase activator activity; signal transducer activityPTPRC 2.22 · 10 - 5 - 17 Hydrolase activity; protein binding; protein kinase binding; protein tyrosine phosphatase activity;
transmembrane receptor protein tyrosine phosphatase activityLOC100133811 2.22 · 10 - 5 - 17 DNA binding; metal ion binding; zinc ion binding; endonuclease activity; hydrolase activity; nucleic acid binding;
pancreatic ribonuclease activity; ribonuclease activity; MHC class II receptor activityCBX7 2.41 · 10 - 5 - 17 Chromatin binding; protein bindingNFIB 2.41 · 10 - 5 - 16 Transcription factor activityFUCA1 2.41 · 10 - 5 - 16 Alpha-L-fucosidase activity; hydrolase activity, acting on glycosyl bonds; sugar bindingDBP 2.67 · 10 - 5 - 16 RNA polymerase II transcription factor activity; protein dimerization activity; sequence-specific DNA binding;
transcription factor activityHLA-DMA 2.79 · 10 - 5 - 16 Protein bindingTPSAB1 2.91 · 10 - 5 - 16 Peptidase activity; protein binding; serine-type endopeptidase activity; serine-type peptidase activityLOC100133811 3.23 · 10 - 5 - 16 DNA binding; metal ion binding; zinc ion binding; endonuclease activity; hydrolase activity; nucleic acid binding;
pancreatic ribonuclease activity; ribonuclease activity; MHC class II receptor activityCRIP1 3.69 · 10 - 5 - 15 Metal ion binding; zinc ion bindingRGS5 3.73 · 10 - 5 - 15 GTPase activator activity; signal transducer activityHBA2;HBA1 3.84 · 10 - 5 - 15 Heme binding; metal ion binding; oxygen binding; oxygen transporter activity; protein bindingMS4A4A 3.87 · 10 - 5 - 15 Receptor activityHLF 3.90 · 10 - 5 - 15 DNA binding; double-stranded DNA binding; protein dimerization activity; sequence-specific DNA binding;
transcription factor activityCHP2 4.40 · 10 - 5 - 15 Calcium ion bindingCD14 4.45 · 10 - 5 - 15 Lipopolysaccharide binding; lipoteichoic acid binding; opsonin receptor activity; peptidoglycan receptor activity;
protein binding
(continued )
484 READHEAD AND DUDLEY
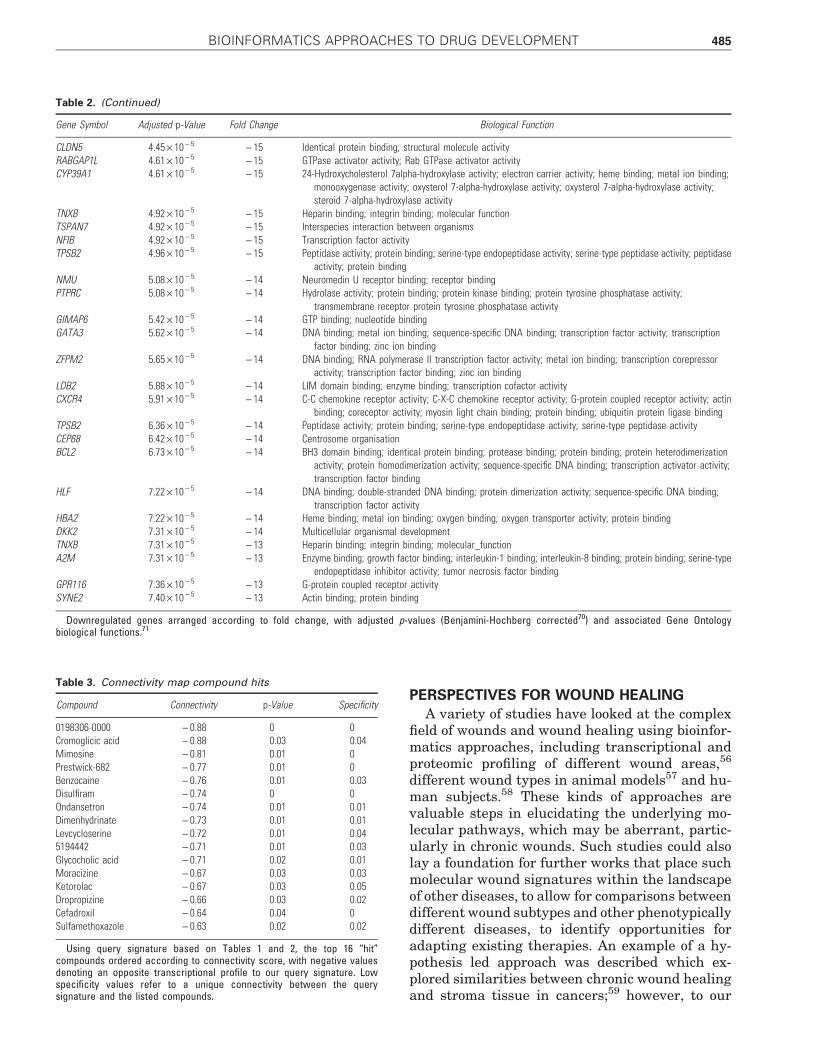
PERSPECTIVES FOR WOUND HEALING
A variety of studies have looked at the complexfield of wounds and wound healing using bioinfor-matics approaches, including transcriptional andproteomic profiling of different wound areas,56
different wound types in animal models57 and hu-man subjects.58 These kinds of approaches arevaluable steps in elucidating the underlying mo-lecular pathways, which may be aberrant, partic-ularly in chronic wounds. Such studies could alsolay a foundation for further works that place suchmolecular wound signatures within the landscapeof other diseases, to allow for comparisons betweendifferent wound subtypes and other phenotypicallydifferent diseases, to identify opportunities foradapting existing therapies. An example of a hy-pothesis led approach was described which ex-plored similarities between chronic wound healingand stroma tissue in cancers;59 however, to our
Table 2. (Continued)
Gene Symbol Adjusted p-Value Fold Change Biological Function
CLDN5 4.45 · 10 - 5 - 15 Identical protein binding; structural molecule activityRABGAP1L 4.61 · 10 - 5 - 15 GTPase activator activity; Rab GTPase activator activityCYP39A1 4.61 · 10 - 5 - 15 24-Hydroxycholesterol 7alpha-hydroxylase activity; electron carrier activity; heme binding; metal ion binding;
monooxygenase activity; oxysterol 7-alpha-hydroxylase activity; oxysterol 7-alpha-hydroxylase activity;steroid 7-alpha-hydroxylase activity
TNXB 4.92 · 10 - 5 - 15 Heparin binding; integrin binding; molecular functionTSPAN7 4.92 · 10 - 5 - 15 Interspecies interaction between organismsNFIB 4.92 · 10 - 5 - 15 Transcription factor activityTPSB2 4.96 · 10 - 5 - 15 Peptidase activity; protein binding; serine-type endopeptidase activity; serine-type peptidase activity; peptidase
activity; protein bindingNMU 5.08 · 10 - 5 - 14 Neuromedin U receptor binding; receptor bindingPTPRC 5.08 · 10 - 5 - 14 Hydrolase activity; protein binding; protein kinase binding; protein tyrosine phosphatase activity;
transmembrane receptor protein tyrosine phosphatase activityGIMAP6 5.42 · 10 - 5 - 14 GTP binding; nucleotide bindingGATA3 5.62 · 10 - 5 - 14 DNA binding; metal ion binding; sequence-specific DNA binding; transcription factor activity; transcription
factor binding; zinc ion bindingZFPM2 5.65 · 10 - 5 - 14 DNA binding; RNA polymerase II transcription factor activity; metal ion binding; transcription corepressor
activity; transcription factor binding; zinc ion bindingLDB2 5.88 · 10 - 5 - 14 LIM domain binding; enzyme binding; transcription cofactor activityCXCR4 5.91 · 10 - 5 - 14 C-C chemokine receptor activity; C-X-C chemokine receptor activity; G-protein coupled receptor activity; actin
binding; coreceptor activity; myosin light chain binding; protein binding; ubiquitin protein ligase bindingTPSB2 6.36 · 10 - 5 - 14 Peptidase activity; protein binding; serine-type endopeptidase activity; serine-type peptidase activityCEP68 6.42 · 10 - 5 - 14 Centrosome organisationBCL2 6.73 · 10 - 5 - 14 BH3 domain binding; identical protein binding; protease binding; protein binding; protein heterodimerization
activity; protein homodimerization activity; sequence-specific DNA binding; transcription activator activity;transcription factor binding
HLF 7.22 · 10 - 5 - 14 DNA binding; double-stranded DNA binding; protein dimerization activity; sequence-specific DNA binding;transcription factor activity
HBA2 7.22 · 10 - 5 - 14 Heme binding; metal ion binding; oxygen binding; oxygen transporter activity; protein bindingDKK2 7.31 · 10 - 5 - 14 Multicellular organismal developmentTNXB 7.31 · 10 - 5 - 13 Heparin binding; integrin binding; molecular_functionA2M 7.31 · 10 - 5 - 13 Enzyme binding; growth factor binding; interleukin-1 binding; interleukin-8 binding; protein binding; serine-type
endopeptidase inhibitor activity; tumor necrosis factor bindingGPR116 7.36 · 10 - 5 - 13 G-protein coupled receptor activitySYNE2 7.40 · 10 - 5 - 13 Actin binding; protein binding
Downregulated genes arranged according to fold change, with adjusted p-values (Benjamini-Hochberg corrected70) and associated Gene Ontologybiological functions.71
Table 3. Connectivity map compound hits
Compound Connectivity p-Value Specificity
0198306-0000 - 0.88 0 0Cromoglicic acid - 0.88 0.03 0.04Mimosine - 0.81 0.01 0Prestwick-682 - 0.77 0.01 0Benzocaine - 0.76 0.01 0.03Disulfiram - 0.74 0 0Ondansetron - 0.74 0.01 0.01Dimenhydrinate - 0.73 0.01 0.01Levcycloserine - 0.72 0.01 0.045194442 - 0.71 0.01 0.03Glycocholic acid - 0.71 0.02 0.01Moracizine - 0.67 0.03 0.03Ketorolac - 0.67 0.03 0.05Dropropizine - 0.66 0.03 0.02Cefadroxil - 0.64 0.04 0Sulfamethoxazole - 0.63 0.02 0.02
Using query signature based on Tables 1 and 2, the top 16 ‘‘hit’’compounds ordered according to connectivity score, with negative valuesdenoting an opposite transcriptional profile to our query signature. Lowspecificity values refer to a unique connectivity between the querysignature and the listed compounds.
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 485

knowledge an approach integratingprofiles from a wider range of dis-ease has not yet been performed. Otherinteresting bioinformatic wound stud-ies have included the use of tran-scriptomic profiling to identify woundedge biomarkers which could then beused to guide surgical debridement.60
Such approaches could be adapted toidentify topical therapeutics that couldhelp guide enzymatic debridement, or toidentify therapeutics with com-plemetary transcriptional profiles. Ad-ditional interesting directions mayinclude the use of transcriptomic orproteomic profiling to identify differentmolecular subtypes of wound, to guidetherapy choice, or a data-driven com-parison between wound signatures andcompound molecular signatures toidentify new topical or systemic treat-ments.
Tables 1–3 provide an example of abioinformatics approach to woundcare. We used publicly available mi-croarray data, collected by Smileyet al.,61 (stored in the GEO reposito-ry,62 GSE3204) in an investigation ofthe transcriptome of cultured skinsubstitute (CSS), an autologous cul-ture of patient keratinocytes and fi-broblasts which can facilitate healingin large burns. Although useful, suchCSS only contain two cell types, andare thus, limited in their capacity toemulate normal skin. We hypothe-sized that identifying the differencesignature between CSS and normalskin may highlight an opportunityfor an existing drug compound to actas an adjunctive therapy alongsideCSS, to augment its transcriptionalresponse in the direction of normalskin.
We used the web-based tool GEO2R5
to rederive the most differentially ex-pressed genes, reflecting the differencesignature between CSS and normalskin cells. We extracted the 100 most differen-tially expressed genes, which comprised 25 upre-gulated (Table 1) and 75 downregulated genes(Table 2), which we used as the basis of a querysignature, for use with Connectivity Map.37,38 Wepresent the top 16 compounds (Table 3) whichwere found to have an ‘‘opposite’’ transcriptional
signature to the CSS signature, comprising aselection of well characterized, approved medica-tions, as well as a number of experimental com-pounds. We suggest that these results could formthe basis of hypothesis led studies investigatingthe role of adjuvant therapies in the application ofCSS to large healing wounds.
TAKE-HOME MESSAGES� A key driver of healthcare expenditure is the treatment of chronic disease,
accounting for three out of four healthcare dollars spent in the UnitedStates.
� Most interventions for chronic conditions occur late in the pathologicalprocess, at a time when a patient is symptomatic, when intervention isless predictable, less effective, and thus, more costly.
� Translational bioinformatics is concerned with ‘‘the development of stor-age, analytic and interpretive methods to optimize the transformation ofincreasingly voluminous biomedical data into proactive, predictive, pre-ventative, and participatory health.’’
� Precision medicine represents an approach where a patient’s disease traitsare interpreted alongside state-of-the-art molecular profiles, to developaccurate diagnostic, prognostic and therapeutic strategies that are tailoredto reflect individual biological contexts.
� A key milestone in the progress of medicine toward the ideals of precisionmedicine will be the organization of a new classification system for dis-ease, which reflects the set of clinically meaningful and exploitable sim-ilarities and difference between disease traits. Such a map will likelyintegrate high-dimensional signatures reflecting network level activity for arange of key biological networks (gene regulatory networks, RNA net-works, protein networks, etc.), as well as higher-level data, such as clinicaland investigative findings.
� Translational bioinformatics can exploit known associations between dis-eases and treatments, to reposition existing medications into new thera-peutic contexts.
� Translational bioinformatics can use molecular level patient data (DNAsequence or structural variation, protein concentrations, etc.) to helpidentify subpopulations within groups of patients that share a broad di-agnosis. These subpopulations can then be stratified and/or assessedseparately when testing new therapies, to identify useful markers that arepredictive for important clinical outcomes, such as treatment responsive-ness, risks of toxicity, etc.
� By integrating genotype, gene expression and clinical data collected ongroups of animals or humans with a disease trait, scientists can buildcausal models of the molecular underpinnings of disease, effectivelyseparating its causal and reactive components.
� This can enable the identification of the true drivers of pathology, whichmay represent high-leverage treatment targets, and help prioritize time-consuming experimental validation.
� An important bottleneck in the application of bioinformatic methods toclinical domains is the presence of scientists conversant in both fields.Creative solutions are required to nurture both sets of competencies withinindividuals, as well as to increase interfield visibility, to facilitate cross-pollination between domain experts within each field.
486 READHEAD AND DUDLEY

Additional ways in which translational bioin-formatic approaches are being applied to the field ofwound care, is in the contribution of better treat-ments of the diseases which predispose to chronicwounds, such as type 2 diabetes,49 venous hyper-tension and peripheral arterial disease.63,64
One of the key challenges facing the systematicintegration of bioinformatic approaches with clin-ical medicine is the relative paucity of ‘‘bilingual’’practitioners—computer scientists with a sense offront-line clinical priorities, and clinicians familiarwith a broad range of computational tools and ap-proaches. To remain current in any one field is afull-time undertaking, so dual trainees are still alimited resource. An ideal scenario would be onein which individuals possess enough biologicalknowledge to ask clinically useful questions, andalso possess a command of existing computationaland bioinformatic approaches to be able to addressthis question in a scientific way. Indeed, one of themajor challenges presented by large-scale, inte-grative informatics approaches is the fact thatthese approaches enable discovery and evaluationof large number of hypotheses, and it can requiresignificant clinical domain expertise to reduce orfocus the set of hypotheses evaluated by informat-ics approaches.
Some universities have begun integratingbioinformatic content into their curriculum, in-cluding units on translational bioinformatics65 andinterpretation of whole genome sequencing,66 forbioinformaticists, PhDs, genetic counselors andmedical practitioners alike. An initial approachmay also lie in broad, cross-field educational mod-ules, for example, a series of reviews directed to-wards clinicians to provide outlines of the field ofbioinformatics, without necessarily focusing ondeveloping the computational skills to build spe-cific solutions. A value inflection point will occurwhen clinicians possesses some fluency in thecapabilities and limits of the available bioinfor-matics tools, and are able to recognize and articu-late some broad applications to their areas ofclinical expertise. Likewise, a similar approachcould be envisioned where a survey of key clinicalneeds within medical domains are made available
to bioinformaticians. This may then help catalyzeconversations and collaboration with domain ex-perts in both fields, informing the next iteration oftool-building and biological inquiry.
AUTHOR DISCLOSURE AND GHOSTWRITING
J.D. declares that he owns an equity interestin NuMedii, Inc. and has served as a consultantto GSK and Janssen Pharmaceuticals. B.R. de-clares that he has no competing financial inter-ests. The content of this article was expresslywritten by B.R. and J.D. No ghostwriters wereused to write this article.
ABOUT THE AUTHORS
Dr. Ben Readhead, MBBS, is an Australiantrained medical practitioner with a long-stand-ing interest in innovation within medicine andthe life sciences. This has included the designand exploration of novel technologies for thedelivery of advanced wound care in difficult toheal wounds. More recent work has looked atnetwork level features of disease genes to gainbiological insight into the mechanisms of patho-genesis in certain disease types. He has recentlyjoined the Institute for Genomics and MultiscaleBiology (Icahn School of Medicine at Mount Sinai)as a Biomedical Informatician with a focus ontranslational bioinformatic approaches to drugrepositioning, gene co-expression network analysisand causal disease modeling. Dr. Joel Dudley,PhD, is a veteran bioinformatics and genomicsresearcher with more than 10 years of profes-sional experience studying the genomic basis ofspecies evolution and human disease. He haspublished more than 40 peer-reviewed researcharticles pertaining to personal genomics, genomicmedicine, pharmacogenomics, drug discovery,bioinformatics, and evolutionary genomics. Joel isDirector of Informatics and Assistant Professor ofGenetics and Genomics Sciences at Icahn Schoolof Medicine at Mount Sinai in New York. Heearned a BS in Microbiology from Arizona StateUniversity and a PhD in Biomedical Informaticsfrom Stanford University.
REFERENCES
1. OECD Statistics. (GDP, unemployment, in-come, population, labour, education, trade,finance, prices, health, debt). http://stats.oecd.org
2. Manton KG, Corder L, and Stallard E: Chronicdisability trends in elderly United States popula-tions: 1982–1994. Proc Natl Acad Sci USA 1997;94: 2593.
3. National Health Expenditure Projections 2010–2020.https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthExpendData/downloads/proj2010.pdf
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 487

4. Mirnezami R, Nicholson J, and Darzi A: Preparingfor precision medicine. N Engl J Med 2012; 366:489.
5. Barrett T, Troup DB, Wilhite SE, Ledoux P, Evan-gelista C, Kim IF, Tomashevsky M, Marshall KA,Phillippy KH, Sherman PM, Muertter RN, Holko M,Ayanbule O, Yefanov A, and Soboleva A: NCBIGEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res 2010; 39: D1005.
6. Hu M, Schultz K, Sheu J, and Tschopp D: Theinnovation gap in pharmaceutical drug discovery& new models for R&D success. Kellogg Schoolof Management (2007). https://ksmb02.kellogg.northwestern.edu/research/biotech/faculty/articles/NewRDModel.pdf
7. Cockburn IM: The changing structure of the phar-maceutical industry. Health Affairs 2004; 23: 10.
8. Butte AJ: Translational bioinformatics: coming ofage. J Am Med Inform Assoc 2008; 15: 709.
9. Sauvages F: Nosologia methodica, sistens mor-borum classes, genera [et] species. (de Tournes,1763). http://books.google.com.au/books?hl = en&lr = &id = SD6AjVv4Nk4C&oi = fnd&pg = PA1&dq =Nosologia + methodica + sistens + morborum +classes&ots = 3geJwbc6o9&sig = WiNUZS2kHLan8-O-Qz715vwnas4&redir_esc = y
10. Linnaeus C: Genera morborum. (1763). http://books.google.com.au/books?id = ZIMZAAAAYAAJ&printsec = frontcover&source = gbs_ge_summary_r&cad = 0#v = onepage&q&f = false
11. Loscalzo J, Kohane I, and Barabasi A-L: Humandisease classification in the postgenomic era: acomplex systems approach to human pathobi-ology. Mol Syst Biol 2007; 3: 124.
12. Farber HW and Loscalzo J: Pulmonary arterialhypertension. N Engl J Med 2004; 351: 1655.
13. National Research Council Committee on a Fra-mework for Developing a New Taxonomy of Dis-ease: Toward Precision Medicine: Building aKnowledge Network for Biomedical Research anda New Taxonomy of Disease. Washington, DC:National Academies Press (U.S.), 2011. Availableonline at: www.ncbi.nlm.nih.gov/books/NBK91503
14. Sirota M, Schaub MA, Batzoglou S, Robinson WH,and Butte AJ: Autoimmune disease classificationby inverse association with SNP alleles. PLoSGenet 2009; 5: e1000792.
15. Suthram S, Dudley JT, Chiang AP, Chen R, HastieTJ, and Butte AJ: Network-based elucidation ofhuman disease similarities reveals commonfunctional modules enriched for pluripotent drugtargets. PLoS Comput Biol 2010; 6: e1000662.
16. Hamburg MA and Collins FS: The path to per-sonalized medicine. N Engl J Med 2010; 363: 301.
17. GLOBOCAN: Country Fast Stat. http://globocan.iarc.fr/factsheets/populations/factsheet.asp?uno =900
18. Memorial Sloan-Kettering Cancer Center: Whysome lung cancers stop responding to tarceva andiressa. www.sciencedaily.com/releases/2005/02/050223164218.htm
19. Kris MG, Natale RB, Herbst RS, Lynch TJ Jr,Prager D, Belani CP, Schiller JH, Kelly K, Spir-idonidis H, Sandler A, Albain KS, Cella D, WolfMK, Averbuch SD, Ochs JJ, and Kay AC: Efficacyof gefitinib, an inhibitor of the epidermal growthfactor receptor tyrosine kinase, in symptomaticpatients with non-small cell lung cancer. JAMA2003; 290: 2149.
20. Lynch TJ, Bell DW, Sordella R, GurubhagavatulaS, Okimoto RA, Brannigan BW, Harris PL, HaserlatSM, Supko JG, Haluska FG, Louis DN, ChristianiDC, Settleman J, and Haber DA: Activating mu-tations in the epidermal growth factor receptorunderlying responsiveness of non-small-cell lungcancer to gefitinib. N Engl J Med 2004; 350: 2129.
21. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H,Gabriel S, Herman P, Kaye FJ, Lindeman N, Bog-gon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, SellersWR, Johnson BE, and Meyerson M: EGFR muta-tions in lung cancer: correlation with clinical re-sponse to gefitinib therapy. Sci Signal 2004; 304:1497.
22. Pao W, Miller V, Zakowski M, Doherty J, Politi K,Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L,Mardis E, Kupfer D, Wilson R, Kris M, and VarmusH: EGF receptor gene mutations are common inlung cancers from ‘never smokers’ and are asso-ciated with sensitivity of tumors to gefitinib anderlotinib. Proc Natl Acad Sci USA 2004; 101:13306.
23. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solo-mon B, Maki RG, Ou SH, Dezube BJ, Jðnne PA,Costa DB, Varella-Garcia M, Kim WH, Lynch TJ,Fidias P, Stubbs H, Engelman JA, Sequist LV, TanW, Gandhi L, Mino-Kenudson M, Wei GC, ShreeveSM, Ratain MJ, Settleman J, Christensen JG,Haber DA, Wilner K, Salgia R, Shapiro GI, ClarkJW, and Iafrate AJ: Anaplastic lymphoma kinaseinhibition in non-small-cell lung cancer. N Engl JMed 2010; 363: 1693.
24. Soda M, Choi YL, Enomoto M, Takada S, Yama-shita Y, Ishikawa S, Fujiwara S, Watanabe H,Kurashina K, Hatanaka H, Bando M, Ohno S,Ishikawa Y, Aburatani H, Niki T, Sohara Y, Su-giyama Y, and Mano H: Identification of thetransforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448: 561.
25. Druker BJ, Talpaz M, Resta DJ, Peng B, Buch-dunger E, Ford JM, Lydon NB, Kantarjian H,Capdeville R, Ohno-Jones S, and Sawyers CL:Efficacy and safety of a specific inhibitor of theBCR-ABL tyrosine kinase in chronic myeloid leu-kemia. N Engl J Med 2001; 344: 1031.
26. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, LambJ, Peck D, Sweet-Cordero A, Ebert BL, Mak RH,Ferrando AA, Downing JR, Jacks T, Horvitz HR,and Golub TR: MicroRNA expression profilesclassify human cancers. Nature 2005; 435: 834.
27. Golub TR, Slonim DK, Tamayo P, Huard C, Gaa-senbeek M, Mesirov JP, Coller H, Loh ML,Downing JR, Caligiuri MA, Bloomfield CD, andLander ES: Molecular classification of cancer:class discovery and class prediction by gene ex-pression monitoring. Science 1999; 286: 531.
28. DiMasi JA, Hansen RW, and Grabowski HG:The price of innovation: new estimates ofdrug development costs. J Health Econ 2003; 22:151.
29. Paul SM, Mytelka DS, Dunwiddie CT, PersingerCC, Munos BH, Lindborg SR, and Schacht AL: Howto improve R&D productivity: the pharmaceuticalindustry’s grand challenge. Nat Rev Drug Discov2010; 9: 203.
30. DiMasi JA: New drug development in the UnitedStates from 1963 to 1999. Clin Pharmacol Ther-apeut 2001; 69: 286.
31. Chong CR and Sullivan DJ: New uses for olddrugs. Nature 2007; 448: 645.
32. Dudley JT, Deshpande T, and Butte AJ: Exploitingdrug-disease relationships for computational drugrepositioning. Brief Bioinform 2011; 12: 303.
33. Lill MA: Multi-dimensional QSAR in drug discov-ery. Drug Discov Today 2007; 12: 1013.
34. Noeske T, Sasse BC, Stark H, Parsons CG, Weil T,and Schneider G: Predicting compound selectivityby self-organizing maps: cross-activities of meta-botropic glutamate receptor antagonists. ChemMed Chem 2006; 1: 1066.
35. Keiser MJ, Roth BL, Armbruster BN, Ernsberger P,Irwin JJ, and Shoichet BK: Relating proteinpharmacology by ligand chemistry. Nat Biotechnol2007; 25: 197.
36. Guha R and Jurs PC: Development of QSARmodels to predict and interpret the biologicalactivity of artemisinin analogues. J Chem InformComput Sci 2004; 44: 1440.
37. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC,Wrobel MJ, Lerner J, Brunet JP, Subramanian A,Ross KN, Reich M, Hieronymus H, Wei G, Arm-strong SA, Haggarty SJ, Clemons PA, Wei R, CarrSA, Lander ES, and Golub TR: The connectivitymap: using gene-expression signatures to connectsmall molecules, genes, and disease. Sci Signal2006; 313: 1929.
38. Lamb J: The connectivity map: a new tool forbiomedical research. Nat Rev Cancer 2007; 7: 54.
39. Iorio F, Bosotti R, Scacheri E, Belcastro V, Mith-baokar P, Ferriero R, Murino L, Tagliaferri R,Brunetti-Pierri N, Isacchi A, and di Bernardo D:Discovery of drug mode of action and drug re-positioning from transcriptional responses. ProcNatl Acad Sci USA 2010; 107: 14621.
40. Sirota M, Dudley JT, Kim J, Chiang AP, MorganAA, Sweet-Cordero A, Sage J, and Butte AJ:Discovery and preclinical validation of drug indi-cations using compendia of public gene expres-sion data. Sci Transl Med 2011; 3: 96ra77.
41. Chen B, Wild D, and Guha R: PubChem as asource of polypharmacology. J Chem InformModel 2009; 49: 2044.
42. Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L,and Bourne PE: A machine learning-based methodto improve docking scoring functions and its ap-plication to drug repurposing. J Chem InformModel 2011; 51: 408.
488 READHEAD AND DUDLEY

43. Chiang AP and Butte AJ: Systematic evaluation ofdrug-disease relationships to identify leads fornovel drug uses. Clin Pharmacol Ther 2009; 86:507.
44. Hu G and Agarwal P: Human disease-drug net-work based on genomic expression profiles. PLoSONE 2009; 4: e6536.
45. Campillos M, Kuhn M, Gavin AC, Jensen LJ, andBork P: Drug target identification using side-effectsimilarity. Science 2008; 321: 263.
46. Qamar R and Rector A: Semantic issues in inte-grating data from different models to achievedata interoperability. Stud Health Technol Inform2007; 129: 674.
47. Schadt EE and Bjorkegren JLM: NEW: network-enabled wisdom in biology, medicine, and healthcare. Sci Transl Med 2012; 4: 115rv1.
48. Schadt EE: Molecular networks as sensors anddrivers of common human diseases. Nature 2009;461: 218.
49. Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeilDJ, Zhang C, Lamb J, Edwards S, Sieberts SK,Leonardson A, Castellini LW, Wang S, ChampyMF, Zhang B, Emilsson V, Doss S, Ghazalpour A,Horvath S, Drake TA, Lusis AJ, and Schadt EE:Variations in DNA elucidate molecular networksthat cause disease. Nature 2008; 452: 429.
50. Schadt EE, Lamb J, Yang X, Zhu J, Edwards S,Guhathakurta D, Sieberts SK, Monks S, ReitmanM, Zhang C, Lum PY, Leonardson A, Thieringer R,Metzger JM, Yang L, Castle J, Zhu H, Kash SF,Drake TA, Sachs A, and Lusis AJ: An integrativegenomics approach to infer causal associationsbetween gene expression and disease. Nat Genet2005; 37: 710.
51. Dudley JT, Schadt E, Sirota M, Butte AJ, andAshley E: Drug discovery in a multidimensionalworld: systems, patterns, and networks. J Cardi-ovasc Transl Res 2010; 3: 438.
52. Zhang B and Horvath S: A general framework forweighted gene co-expression network analysis.Stat Appl Genet Mol Biol 2005; 4: 1128.
53. Tran L, Zhang B, Zhang Z, Zhang C, Xie T, Lamb J,Dai H, Schadt E, and Zhu J: Inferring causal ge-nomic alterations in breast cancer using geneexpression data. BMC Syst Biol 2011; 5: 121.
54. Dobrin R, Zhu J, Molony C, Argman C, Parrish ML,Carlson S, Allan MF, Pomp D, and Schadt EE:Multi-tissue coexpression networks reveal unex-pected subnetworks associated with disease.Genome Biol 2009; 10: R55.
55. Zhu J, Chen Y, Leonardson AS, Wang K, Lamb JR,Emilsson V, and Schadt EE: Characterizing dy-namic changes in the human blood transcriptionalnetwork. PLoS Comput Biol 2010; 6: e1000671.
56. Roy S, Patel D, Khanna S, Gordillo GM, Biswas S,Friedman A, and Sen CK: Transcriptome-wideanalysis of blood vessels laser captured fromhuman skin and chronic wound-edge tissue. ProcNatl Acad Sci USA 2007; 104: 14472.
57. Soulet F, Kilarski W, Antczak P, Herbert J, BicknellR, Falciani F, and Bikfalvi A: Gene signatures inwound tissue as evidenced by molecular profilingin the chick embryo model. BMC Genomics 2010;11: 495.
58. Eming SA, Koch M, Krieger A, Brachvogel B, KreftS, Bruckner-Tuderman L, Krieg T, Shannon JD, andFox JW: Differential proteomic analysis distin-guishes tissue repair biomarker signatures inwound exudates obtained from normal healing andchronic wounds. J Proteome Res 2010; 9: 4758.
59. Chang HY, Sneddon JB, Alizadeh AA, Sood R,West RB, Montgomery K, Chi J-T, van de Rijn M,Botstein D, and Brown PO: Gene expression sig-nature of fibroblast serum response predicts hu-man cancer progression: similarities betweentumors and wounds. PLoS Biol 2004; 2: e7.
60. Brem H, Stojadinovic O, Diegelmann RF, Entero H,Lee B, Pastar I, Golinko M, Rosenberg H, andTomic-Canic M: Molecular markers in patientswith chronic wounds to guide surgical debride-ment. Mol Med 2007; 13: 30.
61. Smiley AK, Klingenberg JM, Aronow BJ, BoyceST, Kitzmiller WJ, and Supp DM: Microarrayanalysis of gene expression in cultured skin
substitutes compared with native human skin. JInvest Dermatol 2005; 125: 1286.
62. Edgar R, Domrachev M, and Lash AE: Gene ex-pression omnibus: NCBI gene expression and hy-bridization array data repository. Nucleic AcidsRes 2002; 30: 207.
63. Cooke JP and Wilson AM:. Biomarkers of pe-ripheral arterial disease. J Am Coll Cardiol 2010;55: 2017.
64. Masud R, Shameer K, Dhar A, Ding K, and KulloIJ: Gene expression profiling of peripheral bloodmononuclear cells in the setting of peripheralarterial disease. J Clin Bioinform 2012; 2: 6.
65. BIOMEDIN217 Translational Bioinformatics:Stanford University Online. at http://scpd.stanford.edu/search/publicCourseSearchDetails.do?method =load&courseId = 1165711
66. Mount Sinai School of Medicine Offers First-EverCourse with Whole Genome Sequencing: MountSinai School of Medicine. www.mssm.edu/departments-and-institutes/genetics-and-genomic-sciences/news/mount-sinai-school-of-medicine-offers-first-ever-course-with-whole-genome-sequencing
67. PhRMA 2012 Profile. www.phrma.org/research/publications/profiles-reports
68. Center for Drug Evaluation and Research and FDABasics—Is it true FDA is approving fewer newdrugs lately? www.fda.gov/AboutFDA/Transparency/Basics/ucm247348.htm
69. CPI Inflation Calculator. http://data.bls.gov/cgi-bin/cpicalc.pl
70. Benjamini Y and Hochberg Y: Controlling the falsediscovery rate: a practical and powerful approachto multiple testing. J R Stat Soc Ser B 1995; 289.
71. Ashburner M, Ball CA, Blake JA, Botstein D,Butler H, Cherry JM, Davis AP, Dolinski K, DwightSS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L,Kasarskis A, Lewis S, Matese JC, Richardson JE,Ringwald M, Rubin GM, Sherlock G; The GeneOntology Consortium: Gene ontology: tool for theunification of biology. Nat Genet 2000; 25: 25.
BIOINFORMATICS APPROACHES TO DRUG DEVELOPMENT 489





























