Embed Size (px)
Citation preview

Macromol. Chem. Phys. 2001, 202, 495–511 495
Theoretical and Spectroscopic UV Study of the
EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 and
EtInd2ZrCl2/MAO-Ethyl Benzoate Interaction
M. L. Ferreira,* P. G. Belelli, D. E. Damiani
PLAPIQUI-UNS-CONICET-Camino La Carrindanga-Km 7 CC 717, 8000 Bahıa Blanca-Prov. Bs As-R. ArgentinaE-mail: [email protected]
Introduction
Metallocene catalysts find special value in the homo and
copolymerization of a-olefins. Since the discovery of
Kaminsky-Sinn, the reaction mechanism of olefin poly-
merization by metallocenes has been the subject of con-
tinuous study. The best-investigated system is the original
one composed of Cp2TiCl2 and EtAlCl2. The hypothesis
of a cationic, methylated active species is accepted. Since
1984/1985, when the stereoselective polymerization of
propene with zirconocene catalysts was first reported
using compounds such as EtInd2ZrCl2, basic and applied
research on metallocene catalysts has been growing.[1 a, b]
One of the most important facts is that zirconocene
complexes require the action of a cocatalyst. The devel-
opment of metallocene catalysis for olefin polymerization
is closely tied to the discovery and use of the cocatalyst
methylaluminoxane (MAO). Technically MAO is made
from the direct reaction of water with AlMe3. From its
synthesis MAO contains residual AlMe3 (about 5 up to
30%) partly free and in part associated to MAO, which is
important for the solubility of MAO in aromatic hydro-
carbons. The agreement seems to be that the MAO oligo-
mers are fluxional molecules with a dynamic equilibrium
among them that changes their size and structure.[2, 3] The
average molecular weight depends on the preparative
conditions (about 900–1100 g/mol for the commercial 10
wt.-% toluene solution). The full cocatalytic functionality
of MAO towards zirconocenes is also unknown. Aside of
formation of a [R2ZrMe]+ cation and being a scavenger
for impurities, the cocatalytic function of MAO is best
described as establishing an environment for the metallo-
cene cation or cation-anion pair in the form of a crown
alumoxane complex.[4] Many zirconocene systems
described in the literature require Al/Zr ratios about
1000:1 up to 10000:1 to achieve a reasonable activity.[5]
Economically such high excess ratios of the expensive
MAO are very unfavorable because it makes the cocata-
lyst the cost factor and research is carried out to reduce
MAO concentration with similar productivities. Appar-
ently up to 90% of the MAO can be replaced by TMA
Full Paper: This work presents a theoretical and experi-mental study of the active sites formation in the systemEtInd2ZrCl2/MAO. Results about the effect of additives asa Lewis acid (AlCl3) or a Lewis base (EB) are also ana-lyzed. The theoretical study takes into account all theknown steps in the active site formation: alkylation of zir-conocene, formation of a polarized complex metallocene:MAO, formation of a contact ion pair and finally a solventseparated ion pair. Other reactions of the zirconocene withother aluminium alkyls (AlCl3 and AlMe3) are also pre-sented and studied. UV-vis experimental results of theadditive effects are reported and explained taking intoaccount the theoretical results. A short review of ourresults and others is presented to complete and discuss theideas. The most probable sites are loosened contact ionpairs, not the solvent separated ion pairs.
Macromol. Chem. Phys. 2001, 202, No. 4 i WILEY-VCH Verlag GmbH, D-69451 Weinheim 2001 1022-1352/2001/0402–0495$17.50+.50/0
V/Visble spectrum of zirconocene, zirconocene withMAO at Al/Zr = 3363 with AlCl3/Zr = 33, zirconocenewith MAO at Al/Zr = 8400, zirconocene with MAO atAl/Zr = 8400 with AlCl3/Zr = 13 and zirconocene withMAO at Al/Zr = 3363 and EB/Zr = 5. [Zr] = 3 E-05 M.

496 M. L. Ferreira, P. G. Belelli, D. E. Damiani
without a significant loss in ethylene polymerization
activity.[6] The often high TMA content in MAO and the
role of TMA in the activation process was addressed in a
separate way in the literature.[7] MAO seems to be a better
alkylating agent and has a greater capacity for producing
and stabilizing cationic complexes. At low Al/Zr ratio
two types of ion pairs have been detected.[7]
The active site in homogeneous polymerization is
cationic, and Cp2ZrCH3+ is of special interest. In fluid
solution it may be generated by oxidation or protonation
of Cp2ZrMe2 but it has been characterized only in the sol-
vated form Cp2ZrMe+ (S) where S = THF or CH3CN. It is
an open question as to whether Cp2ZrMe+ is stable in
solution without coordinating solvent.[8] Some authors
report that Cp2ZrMe2 readily exchanges methyl groups
with Al2Me6 and that it reacts with (Me2AlEt)2 in neat
Al2Me6 to form Cp2Zr(nCH3)CH2CH2AlMe2.[9]
The interaction Cp2ZrCl2/MAO was studied by 1H
NMR spectroscopy. It was found that Cp2ZrCl2 is mono-
alkylated as a first step in the reaction mechanism.[10]
Cp2ZrClCH3*MAO highly polarized complex is formed
with an excess of MAO. These authors point out that
TMA is the real alkylating agent.
Looking at the literature, several conformational studies
have been carried out to study the site control in the homo-
geneous isotactic specific polimerization.[11, 12] Until now,
almost all theoretical papers concerning a quantum-
mechanical study of homogeneous Ziegler Natta polymeri-
zation have focused on the propagation mechanism, assum-
ing a cationic model for the catalytic species, but little atten-
tion has been dedicated to the formation of the active spe-
cies (initiation step) and to the possible role played by the
counterion or by the solvent.[13–19] These papers deal with
the idea of a solvent separated ion pair, an idea that has sev-
eral drawbacks now, but the solvent is not included in the
calculation in several of them. One of the main problems of
the cationic model concerns the energetics involved in the
dissociation of the ion pairs in weakly polar solvents, such
as toluene.[20] Fusco et al.[20] evaluated for the first time the
energy produced to form separated ion pairs in vacuum
from Cp2TiR.AlRnCl4-n. Its value, more than 100 kcal/mol
(=419 kJ/mol), can hardly be compensated for by solvating
energy. With regard to the MAO– zirconocene+ ion pair, the
energy would be even higher than 100 kcal/mol because the
polarization is high due the presence of oxygens. Brintzin-
ger et al. in a recent review suggested the formation of olefin
separated ion pairs as active intermediates of the polymeri-
zation.[21] The inactivity of titanocene/chloroalkylalumium
systems in the polymerization of propene and higher olefins
is thus justified on the basis of the insufficient capability of
more weakly coordinating substituted olefins to displace
the anion and to coordinate to the transition metal in tight
ion pairs.
Methylaluminoxane appears to form an unusually
weakly interactive anion. It is known that MAO is less
coordinating than MeB(C6F5)3–. Therefore MAO appears
to be an excellent zirconocenium stabilizer. We believe
that this situation arises from the capability of MAO to
accommodate a negative charge, especially due to the
presence of oxygen atom.[23]
The reactions of Cp2MtMeCl with AlMe3 and with
MeAl[OAl2H4]2O as a model of the catalytic site of
methylalumoxane, were studied by means of DFT quan-
tum-mechanical calculations by Fusco et al.[24] They con-
clude that ion-pair dissociation is a difficult process
whose energetics is incompatible with experimental poly-
merization data. The authors point out that even when the
DFT study shows that negative charge dispersion in MAO
macroanions strongly affects the ion-pair dissociation
energy, this feature cannot justify by itself the formation
of free cationic species.
The objective of the present work is to study theoreti-
cally and experimentally the active site formation in the
EtInd2ZrCl2/MAO system, without olefin. We present the
effect of toluene as solvent through a model of ion-pair-
solvent interaction. The method used was an improved
extended Huckel.
In our laboratory we have studied experimentally the
effect of additives in ethylene and propylene polymeriza-
tion with EtInd2ZrCl2/MAO with toluene as solvent. The
additives used were AlCl3 and ethyl benzoate (EB). There-
fore we present here the comparison in the active sites for-
mation taking account of EtInd2ZrCl2/MAO EtInd2ZrCl2/
MAO-AlCl3 EtInd2ZrCl2/MAO-EB results. We include a
spectroscopic UV-vis study of the same systems, at the used
conditions in ethylene and propylene polymerization, and
we present a coherent explanation of our results.
Theoretical Part
EHMO Method
Molecular orbital calculations were carried out by means
of the extended Huckel method (EHMO). This semiempir-
ical procedure provides a useful preliminary approach to
changes in the electronic structure. This formalism is use-
ful to obtain qualitative trends in adsorption processes.
The program used (ICONC) was developed by Calza-
ferri et al.[25] It includes repulsive terms to the total
energy. The repulsive coulombic energy is taken into
account in a pairwise term. The change in the energies in
the presented reactions (DE) was calculated as the sum of
the product energies minus the sum of the reactant ener-
gies as an approximation to the reaction energies. The
atomic parameters are listed in Table 1.
The total energy (Et) of our adsorbate/substrate system
is expressed as:
Et ¼Xi imj
niEi þ 1þ 2XX
Erepði;jÞ

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 497
The first term corresponds to the valence electrons con-
tribution (ni) and the second term to the pairwise intera-
tomic repulsion. Each valence level i has an energy Ei with
occupancy ni. The repulsion energy of a nucleus i in the pre-
sence of a fixed atom j is calculated as an electrostatic term.
The summation is extended to all possible atom pairs (Erep).
The last is partitioned into a perfectly following (with
respect to the nuclei or internal cores) atom part and a non-
perfectly following bond charge part. More details about
the calculation can be found in literature[26, 27] In the modi-
fied version (ICONC) used in the present work, repulsive
terms are included; thus besides the determination of rela-
tive energies, optimization of bond-length and geometry
may be performed with an accuracy of l10%.[28 a] Hoffman
and Juan have used ICONC to carry out the optimization of
the bond length of H to an Fe surface.[28 b]
Higher levels of theory (HF or DFT) which are known
to produce reliable bond energies should be performed on
smaller models, especially of the methylaluminoxane
(MAO). This leads us use to smaller molecules, which
raises several problems. In a HF-SCF procedure time is
spent improving the distribution of electronic density of
the contours at the expense of the description of the active
site. We can resolve this problem when dangling bonds
are saturated. A distribution of point charges can also
account for remote atoms. The results from EHMO, we
repeat, are only valid in relative terms. Even in the case
of DFT or H–F calculations, no experimental results are
available. Remember that bond distances from zircono-
cene to MAO are difficult to obtain, especially with the
system zirconocene-MAO. Generally the studies have
been carried out in the solid state. No easy experiments
are yet reported in these systems. Very recent application
of EH support the idea of a useful method that could pro-
vide “understanding” rather than qualitative results. We
are using the ASED or ICONC methods which compute
the electronic structure “like” the traditional EHM, but
has a distance dependence for the Hi, j matrix elements
and include explicit repulsive (coulombic) terms to the
energy[28] so better bond distances could be obtained.
However the energies are not correct in an absolute sense
but reliable in “relative” terms which is really important
in the design of a catalyst. In previous papers we have
explored Ziegler Natta systems with the ICONC method.
The predictions were reliable including some experimen-
tal support of our findings.[29–31]
Mechanism of Ion Pair Formation
The mechanism of ion pair formation in EtInd2ZrCl2/
MAO includes basically:
Stage 1 – Alkylation of EtInd2ZrCl2 to produce EtInd2-
ZrClCH3
Stage 2 – Polarized complex formation EtInd2ZrCl-
CH3*MAO
Stage 3 – Ion-pair formation
Stage 4 – Dissociation of ion pair to produce solvent se-
parated ion pair.
We perform the calculations for the three MAO sys-
tems considered and stage 2 was avoided because it leads
easily to stage 3 following Kaminsky proposal.[1 b]
MAO Model
Based on the famous work of Barron et al.,[32] Sinn and
co-workers have performed excellent work on MAO pre-
paration and structure.[33, 34] In their paper they pointed
out that MAO may be in distinct forms in toluene solu-
tions and these forms include “rod”-like macromolecules
(linear) and shell-like (similar to Barron ball-like). In
consideration of that work we selected the form that it is
considered to be better: the linear one. Sinn and co-work-
ers claim that they found a small difference in energy
content of the different MAO molecules. Therefore equi-
librium will be present between the different forms,
which naturally lies on the left-hand side:
MAO (rod) gggsaggg MAO (shell)
In view of the high TMA content of MAO, the role of
TMA in the activation process was considered in the
EHMO calculation. The linear MAO*TMA model
includes 11 Al, 9O, 3CH3 groups, one AlMe2H group
coordinated to an O and 9H to saturate the three-coordi-
nation of Al. The distances used in the model are pre-
sented in Table 2. The structure is distorted (especially
O1Al1C angles), taking account of the possible coordi-
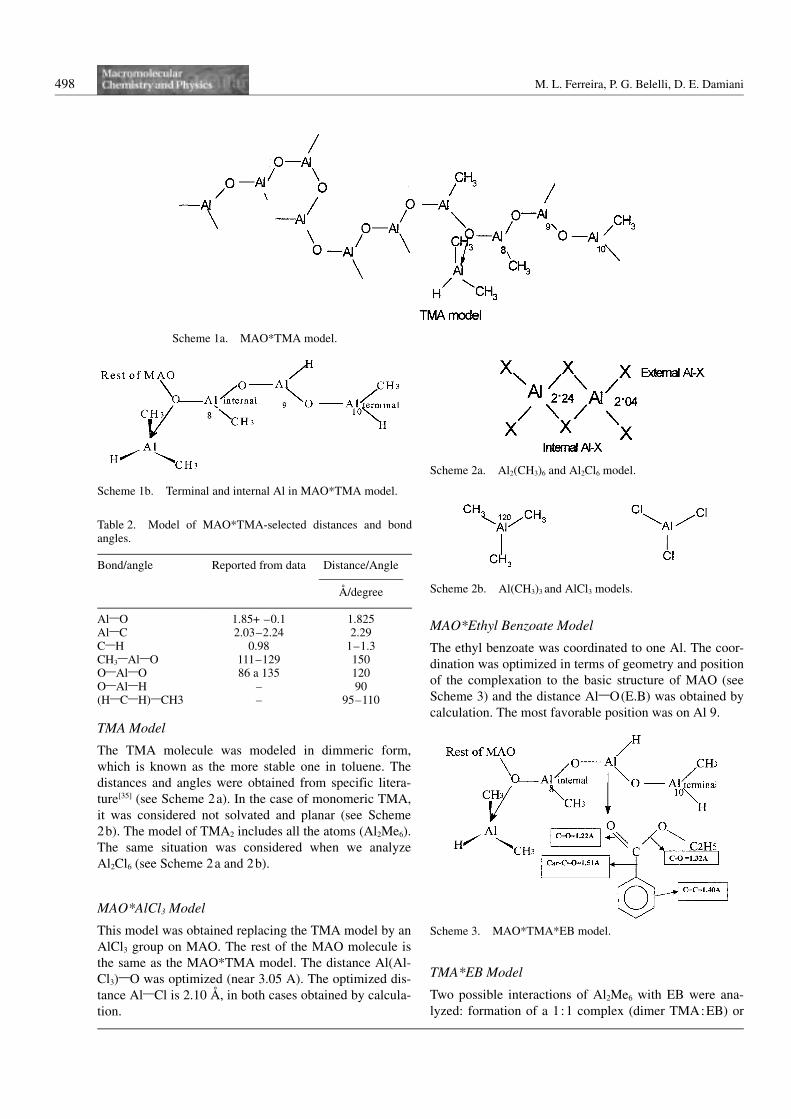
nation to others MAO molecules[34] (see Scheme 1a and
Scheme 1b). The figures in the following sections will
include a simplified version of this full molecule, show-
ing the last portion of the MAO molecule, from Al 8 to
Al 10 of the structure.
Table 1. EHMO parameters.
Atom Orbital Orbital exponents Ionization potentials————————–
eV
H 1s 1.000 –13.60C 2s 1.154 –19.65
2p 1.451 –11.13O 2s 2.163 –31.6
2p 2.750 –16.78Al 3s 1.670 –12.30
3p 1.383 –6.50Cl 3s 2.356 –26.03
3p 2.039 –14.20Zr 5s 1.817 –9.870
5p 1.776 –6.7604d 3.835 (0.6211)a) –11.18
1.505 (0.5796)a) –11.18
a) Coefficients used in the double n expansion of the d orbitals.
498 M. L. Ferreira, P. G. Belelli, D. E. Damiani
TMA Model
The TMA molecule was modeled in dimmeric form,
which is known as the more stable one in toluene. The
distances and angles were obtained from specific litera-
ture[35] (see Scheme 2a). In the case of monomeric TMA,
it was considered not solvated and planar (see Scheme
2b). The model of TMA2 includes all the atoms (Al2Me6).
The same situation was considered when we analyze
Al2Cl6 (see Scheme 2a and 2b).
MAO*AlCl3 Model
This model was obtained replacing the TMA model by an
AlCl3 group on MAO. The rest of the MAO molecule is
the same as the MAO*TMA model. The distance Al(Al-
Cl3)1O was optimized (near 3.05 A). The optimized dis-
tance Al1Cl is 2.10 A, in both cases obtained by calcula-
tion.
MAO*Ethyl Benzoate Model
The ethyl benzoate was coordinated to one Al. The coor-
dination was optimized in terms of geometry and position
of the complexation to the basic structure of MAO (see
Scheme 3) and the distance Al1O(E.B) was obtained by
calculation. The most favorable position was on Al 9.
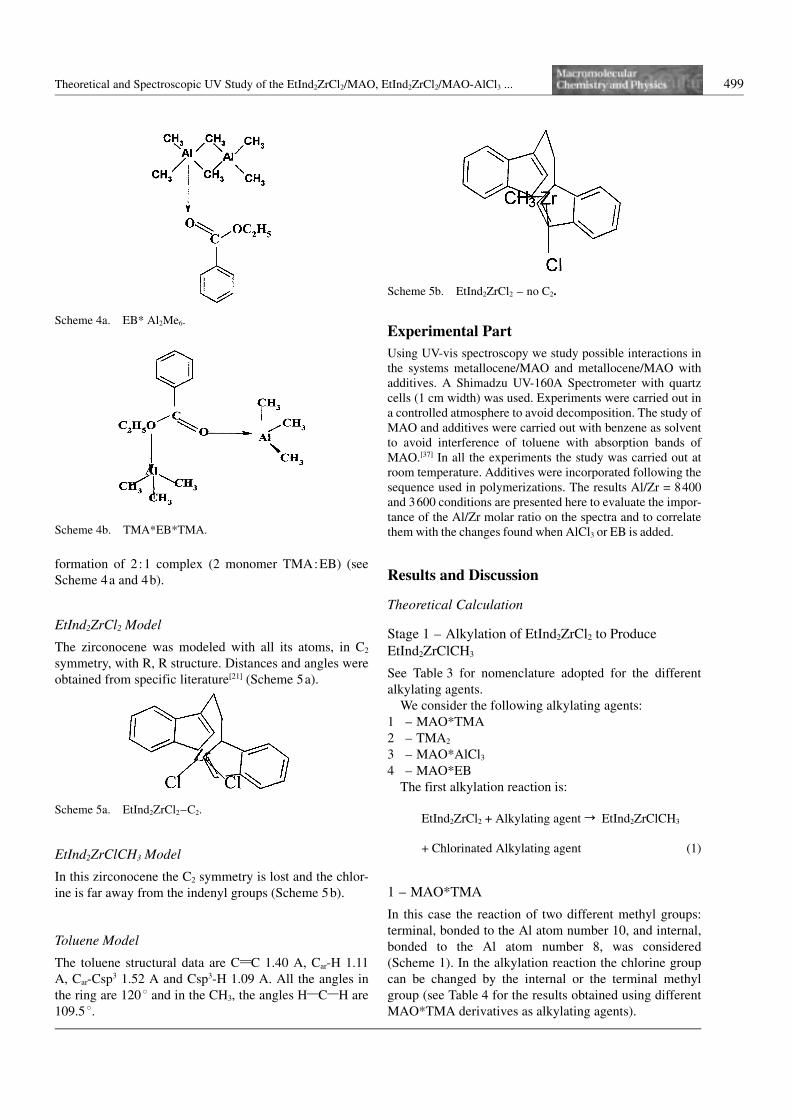
TMA*EB Model
Two possible interactions of Al2Me6 with EB were ana-
lyzed: formation of a 1:1 complex (dimer TMA:EB) or
Table 2. Model of MAO*TMA-selected distances and bondangles.
Bond/angle Reported from data Distance/Angle———————
A/degree
Al1O 1.85+ –0.1 1.825Al1C 2.03–2.24 2.29C1H 0.98 1–1.3CH31Al1O 111–129 150O1Al1O 86 a 135 120O1Al1H – 90(H1C1H)1CH3 – 95–110
Scheme 1a. MAO*TMA model.
Scheme 1b. Terminal and internal Al in MAO*TMA model.
Scheme 2a. Al2(CH3)6 and Al2Cl6 model.
Scheme 2b. Al(CH3)3 and AlCl3 models.
Scheme 3. MAO*TMA*EB model.
Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 499
formation of 2:1 complex (2 monomer TMA:EB) (see
Scheme 4a and 4b).
EtInd2ZrCl2 Model
The zirconocene was modeled with all its atoms, in C2
symmetry, with R, R structure. Distances and angles were
obtained from specific literature[21] (Scheme 5a).
EtInd2ZrClCH3 Model
In this zirconocene the C2 symmetry is lost and the chlor-
ine is far away from the indenyl groups (Scheme 5b).
Toluene Model
The toluene structural data are C2C 1.40 A, Car-H 1.11
A, Car-Csp3 1.52 A and Csp3-H 1.09 A. All the angles in
the ring are 1208 and in the CH3, the angles H1C1H are
109.58.
Experimental Part
Using UV-vis spectroscopy we study possible interactions inthe systems metallocene/MAO and metallocene/MAO withadditives. A Shimadzu UV-160A Spectrometer with quartzcells (1 cm width) was used. Experiments were carried out ina controlled atmosphere to avoid decomposition. The study ofMAO and additives were carried out with benzene as solventto avoid interference of toluene with absorption bands ofMAO.[37] In all the experiments the study was carried out atroom temperature. Additives were incorporated following thesequence used in polymerizations. The results Al/Zr = 8400and 3600 conditions are presented here to evaluate the impor-tance of the Al/Zr molar ratio on the spectra and to correlatethem with the changes found when AlCl3 or EB is added.
Results and Discussion
Theoretical Calculation
Stage 1 – Alkylation of EtInd2ZrCl2 to Produce
EtInd2ZrClCH3
See Table 3 for nomenclature adopted for the different
alkylating agents.
We consider the following alkylating agents:
1 – MAO*TMA
2 – TMA2
3 – MAO*AlCl3
4 – MAO*EB
The first alkylation reaction is:
EtInd2ZrCl2 + Alkylating agent e EtInd2ZrClCH3
+ Chlorinated Alkylating agent (1)
1 – MAO*TMA
In this case the reaction of two different methyl groups:
terminal, bonded to the Al atom number 10, and internal,
bonded to the Al atom number 8, was considered
(Scheme 1). In the alkylation reaction the chlorine group
can be changed by the internal or the terminal methyl
group (see Table 4 for the results obtained using different
MAO*TMA derivatives as alkylating agents).
Scheme 4a. EB* Al2Me6.
Scheme 4b. TMA*EB*TMA.
Scheme 5a. EtInd2ZrCl2–C2.
Scheme 5b. EtInd2ZrCl2 – no C2.
500 M. L. Ferreira, P. G. Belelli, D. E. Damiani
2 – Al2Me6
The alkylation reaction is modeled replacing one methyl
by chlorine until all the methyl groups are replaced. The
terminal methyl groups are the first to be replaced and
later the internal ones in the Al2Me6 molecule (see Table 5
for the results).
In these cases, the Al1X (X2CH3 or Cl) distance is
2.24 A for the internal Al1X and 2.04 A for the external
Al1X, obtained from literature. From here to the end
Al2Me6 is dimeric TMA.
3 – MAO*AlCl3
When AlCl3 is present the following reaction was consid-
ered:
a) Replacing TMA by AlCl3 (Scheme 1)
MAO*TMA + AlCl3 e MAO*AlCl3 + TMA
DE = –3.881 eV (2)
b) In this case AlCl3 and TMA were considered as
plane molecules (see Scheme 2b). The addition of AlCl3
to MAO*TMA is not so probable.
MAO*TMA + AlCl3 e AlCl3*MAO*TMA
DE = –1.229 eV (3)
The interchanging of Cl by a methyl group is possible
in the MAO*TMA interaction with AlCl3, as several
authors propose.[38] In the following reactions Cl is placed
at Al 8 for MAOCl*TMA and at Al 8 and Al 10 for
MAOCl2*TMA.
MAO*TMA + Al2Cl6 e MAOCl*TMA + Al2MeCl5
DE = –3.68 eV (4)
MAO*TMA + Al2Cl6 e MAOCl2*TMA+Al2Me2Cl4
DE = –5.75 eV (5)
This reaction has been reported for alkyls with t-butyl
as lateral chains.[38] With the methyl group in methylalu-
minoxane, the reaction is probably fast and displaced to
the right.
4 – MAO*EB
The possible reactions of EB with MAO*TMA are:
a) Addition
MAO*TMA + EB e MAO*TMA*EB
DE = –0.80 eV (Scheme 3) (6a)
b) Coordination of dimer TMA to EB (1:1, complex of
EB with dimer)
TMA2 + EB e EB*TMA2
DE = –0.733 eV (Scheme 4a) (6b)
c) Coordination of TMA monomers to the two O of EB
TMA2 + EB e TMA*EB*TMA
DE = –8.14 eV (Scheme 4b) (6c)
Table 3. Nomenclature.
MeHAl-(Al9O10H7Me2)*AlMe2H MAO*TMAMeHAl-(Al9O10H7Me2)*AlCl3 MAO*AlCl3
MeHAl-(Al9O10H7Me2)*AlMe2HCOOC2H5C6H5
MAO*EB
MeHAl-(Al9O10H7MeCl)*AlMe2HCOOC2H5C6H5
MAO*EBCl in Al 8
ClHAl-(Al9O10H7Me2)*AlMe2HCOOC2H5C6H5
MAO*EBCl in Al 10
ClHAl-(Al9O10H7Me2)*AlMe2H MAO*TMA (Cl in Al 10)CH3HAl-(Al9O10MeH7Cl)*AlMe2H MAO*TMA (Cl in Al 8)ClHAl-(Al9O10H7Me2)*AlMe2H MAO*AlCl3 (Cl in Al 10)MeHAl-(Al9O10H7MeCl)*AlCl3 MAO*AlCl3 (Cl in Al 8)Al2Me6 Dimer TMAMe2AlMe2AlMeCl TMA2ClClMeAlMe2AlMeCl TMA2Cl2
ClMeAlMe2AlClCl TMACl3
ClClAlMe2AlClCl TMA2Cl4
ClClAlMeClAlClCl TMA2Cl5
Al2Cl6 Al2Cl6
ClHAl-(Al9O10H7MeCl)*AlMe2H,Cl in Al 10 and 8
MAOCl2*TMA
ClHAl-(Al9O10H7MeCl)*AlCl3
in Al 10 and 8MAOCl2*AlCl3
ClHAl-(Al9O10H7MeCl)*AlMe2H*COOC2H5C6H5, Cl in Al 10 and 8
MAOCl2*EB
Table 4. Results of EHMO calculation with MAO*TMA andderivatives. Reaction 1.
Alkylating agent Product DE
eV
MAO*TMA MAO*TMACl in Al 8 –15.69MAO*TMA MAO*TMACl in Al 10 –12.50MAO*TMA Cl in Al 8 MAO*TMACl2 –10.84MAO*TMACl in Al 10 MAO*TMACl2 –14.03
Table 5. Results of EHMO calculation in reaction 1-TMA2 andderivatives.
Alkylating agent Product DE
eV
TMA2 TMA2Cl –9.76TMA2Cl TMA2Cl2 –9.20TMA2Cl2 TMACl3 –9.48TMACl3 TMA2Cl4 –8.68TMA2Cl4 TMA2Cl5 –8.756TMA2Cl5 Al2Cl6 –11.768

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 501
From the EHMO calculation it is possible to say that
the data confirm that MAO*TMA is a better alkylating
agent than TMA (see Table 4 and 5). An internal CH3
group near to an O coordinated to TMA, is more easily
exchanged by chlorine from zirconocene than CH3 on a
terminal Al (see Table 4 entries 1 and 2 and Scheme 1),
although CH3 on a terminal Al is able to alkylate the zir-
conocene, too. Probably in the actual MAO solution, the
1AlMe2 group concentration is higher than internal,
available, non-coordinated Al1CH3 groups concentra-
tion. Once MAO*TMA has a chlorine atom on a terminal
Al, the alkylation reaction using the internal Al1CH3
group is even easier (see entry 4 – Table 4). In the case of
dimer TMA, exchanging a Cl by a methyl group is not
favored when the number of chlorine increases up to 3
(see entries 1–5 of Table 5). An almost fully chlorinated
molecule of dimer TMA (see entry 6) is the best alkylat-
ing agent of this series, although not better than
MAO*TMA.
The presence of AlCl3 changes the MAO*TMA nature.
AlCl3 replaces TMA and is not added to MAO*TMA (see
Equation (2) and (3)) following our modeling. New alky-
lating species in high concentration, compared to
MAO*TMA, are present as mainly mono and bichlori-
nated species of MAO (see Equation (4) and (5)). These
reactions produce modified MAO*TMA and mono and
dialkylated species of Al2Cl6. Especially the latter is a
better alkylating agent than TMA2 (see Table 5).
The alkylation step is favored in almost –0.6 eV when
MAO*AlCl3 or MAO*EB are used as alkylating agent. If
the alkylating agent carries one chlorine on Al 8 or Al 10,
the Equation (1) is more probable in almost –2.2 eV
(compare Table 4, 6, and 7, entries 3 and 4).
Stage 2 – Polarized Complex Formation
EtInd2ZrClCH3*MAO
Over the past several years, many groups have presented
different evidence about the nature of the active site.
Many of them think that the most active site is a solvent
separated ion pair[39–41] while others think that it is a loo-
sened contact ion pair.[7, 22] Others support the idea that
the precursor of the active site is a loosened contact ion
pair (with a non-coordinating anion) that becomes olefin
separated when the olefin is added.[20, 24]
The existence of contact ion pairs and solvent sepa-
rated ion pairs can be summarized in the following way:
A+B– (contact ion pair) e A+/solvent/B–
(solvent separated ion pair) e A+/solvent
+ B-/solvent (completely dissociated ion pairs)
An additional reaction can be depicted in this picture:
the formation of cation solvated ion pairs from solvent
separated ion pairs, especially when the solvent does not
coordinate the anion.
Generally the theoretical studies about mechanistic
aspects of ethylene and propylene polymerization have
considered completely dissociated ion pairs. Neither the
solvent nor MAO are considered in the calculation in sev-
eral studies of these kind[42, 43, 12] and the initial structure
(as C2 symmetry) in terms of the way olefins coordinate is
maintained. This situation must change if the solvent is
included because the zirconocene changes from tetrahe-
dral (R2ZrMe+ olefin) to pentacoordinated (R2ZrMe+ ole-
fin/solvent). Recent studies of Fusco et al. attempt to
model the interaction of MAO-zirconocene, taking
account of the local structure of MAO, leading to the
active sites.[24] Karol et al.[44] showed that Me(MAO)9– has
greater importance in the angle Centroid Cp A Zr A Cen-
troid Cp than Me B(C6F5)3– although the former is twice
as far away from the cation as the latter. This is the only
report about counterion consideration in calculations.
First we present a model of contact ion pair formation,
second a model of the solvent separated ion pair, and
finally a model of the cation solvated separated ion pair.
Stage 3 – Contact Ion-Pair Formation
The contact ion pair formation is considered in two reac-
tions:
a) the ion pair formation from the initial zirconocene
dichloride with two molecules of the alkylating agent:
EtInd2ZrCl2 + 2 alkylating agent e EtInd2Zr+CH3*
Counterion + chlorinated alkylating agent (7)
Here the counterion is one alkylating agent with the
basic structure of MAO shown in Scheme 1 with a nega-
Table 6. Results of EHMO calculation in reaction 1-MAO*AlCl3 and derivatives.
Alkylating agent Product DE
eV
MAO*AlCl3 MAO*AlCl3Cl in Al 8 –16.14MAO*AlCl3 MAO*AlCl3Cl in Al 10 –12.71MAO*AlCl3Cl in Al 8 MAO*AlCl3Cl2 –12.7MAO*AlCl3Cl in Al 10 MAO*AlCl3Cl2 –16.21
Table 7. Results of EHMO calculation in reaction 1- MAO*EBand derivatives.
Alkylating agent Product DE
eV
MAO*EB MAO*EBCl in Al 8 –16.19MAO*EB MAO*EBCl in Al 10 –12.71MAO*EBCl in Al 8 MAO*EBCl2 –12.78MAO*EBCl in Al 10 MAO*EBCl2 –16.25

502 M. L. Ferreira, P. G. Belelli, D. E. Damiani
tive charge localized on an oxygen atom of the structure
of MAO. The chlorinated alkylating agent (product) has
the chlorine in Al 8 (see Scheme 6b), not in Al 10 (see
Scheme 6a), from the first alkylation reaction.
b) The ion pair formation from the initial zirconocene
dichloride with only one alkylating agent:
EtInd2ZrCl2 + Alkylating agent
e EtInd2Zr+CH3*counterion (8)
Here the counterion contains two chlorines in the struc-
ture, one in Al 8 from the zirconocene first alkylation,
and the other on Al 10 from the zirconocene.
Taking into account recent work[7, 45 a, 24] we considered
the formation of three different contact ion pairs, shown
in Scheme 7. One presents a close interaction Zr+1O–
(site 1) (Scheme 7a) and the others are loosened contact
ion pairs (Sites 21 and 22) (Scheme 7b). The latter inter-
acts with MAO through a chlorine bridge between Zr and
Al from MAO. Looking at Scheme 7b, it can be seen that
this site has two possible forms varying the position of
CH3 (by Zr1Cl bond rotation): on MAO “internal” struc-
tures (Site 21) and on MAO “terminal structures” (Site
22). The negative charge is always localized on O from
MAO. The stabilization energies were calculated as the
total energy of the contact ion pair minus the energy
when the ion pair is completely dissociated (cation energy
plus anion energy) following the reaction:
Free Cation + Free Anion e Contact Ion Pair
DE = Contact ion pair Energy –
(Free Cation energy + Free Anion Energy)
The reaction presented above is the inverse of:
Contact Ion Pair e Free Cation + Free Anion
The energy of this reaction is an approximation to the
energy necessary to produce free ions in the gas phase.
You can consider that this energy is (– stabilization
energy) the energy that we include in the tables.
The structure and chemistry of bis(cyclopentadie-
nyl)MLn complexes has been extensively studied by
Hoffmann et al.[45 b] They pointed out that the best candi-
date for a polymerization catalyst would be a coordina-
tively unsaturated d0 complex. They presented also exam-
ples of Cp2MX3 and an alternative mechanism of olefin
polymerization when a pentacoordinated complex is
requiered, with an aluminium alkyl coordinated to the
active site. They realize that some of the intermediates
have been coordinately saturated, 18-electron complexes;
but they mention that some other metallocenes (Cp2ReH,
Cp2MoH2) are 18-electron complexes that readily react to
give the vinyl insertion products. The transition complex
involves an olefin with a polarized bond and the build up
of a negative charge at the uncomplexed carbon terminus
and a positive charge at the metal.
MAO*TMA
We considered that the alkylating agents in Equation (7)
and (8) is MAO*TMA:
Case a) When two alkylating agents are considered.
We considered 2 MAO*TMA because the chlorinated
Scheme 6a. MAO*TMA with Cl in Al 10.
Scheme 6b. MAO*TMA with Cl in Al 8.
Scheme 7a. Site 1 – Tight contact ion pair (TCIP).
Scheme 7b. Sites 2 – Loosened contact ion pair (LCIP).

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 503
MAO*TMA are present in very low concentrations (see
Scheme 8 for the structure of the contact ion pairs) in the
real systems Zirconocene//MAO. This means that we sup-
pose that the relative concentration of MAO*TMA is
very much higher than MAO*TMACl in Al 8. Remember
that in this case the chlorine is provided only by the zirco-
nocene and the Zr concentration is very much lower than
MAO. Only the first alkylation is possible when
EtInd2ZrCl2 is analyzed.
2 MAO*TMA + EtInd2ZrCl2 e MAO*TMACl
(in Al 8) + EtInd2ZrCH3+ ClMAO*TMA– (9)
In Scheme 8 the optimized distances Al1Cl and Zr1O
can be seen found by calculation for site 1, 21 and 22.
Case b)
MAO* TMA + EtInd2ZrCl2 e
EtInd2ZrCH3+ ClMAO*TMACl– (in Al 8) (10)
Table 8 shows reaction DE and stabilization energy for
the 6 contact ion pairs modeled in the presence of
MAO*TMA.
MAO*AlCl3
Next we considered that the alkylating agents in Equation
(7) and (8) as MAO*AlCl3:
Case a) When two alkylating agents are considered.
We considered MAO*AlCl3 and MAOCl2*TMA. The cal-
culations show that the presence of this species is very
probable when AlCl3 is added to MAO*TMA (see Equa-
tion (5)).
2 MAO*AlCl3 + EtInd2ZrCl2 e MAO*AlCl3Cl
(in Al 8) + EtInd2ZrCH3+ClMAO*AlCl3
– (11)
MAO*TMA + MAO*TMACl2 + EtInd2ZrCl2 e
MAO*TMACl (in Al 8)
+ EtInd2ZrCH3+ ClMAOCl2*TMA– (12)
Case b) When one alkylating agent is considered we
propose that one molecule of MAO*AlCl3 alkylates the
zirconocene and the same molecule reacts to act as the
counterion, finally leaving two chlorine atoms.
MAO*AlCl3 + EtInd2ZrCl2 e
EtInd2ZrCH3+ ClMAO*AlCl3Cl– (in Al 8) (13)
Table 9 shows the reaction DE and stabilization energy
for the 9 contact ion pairs modeled in the presence of
MAO*AlCl3.
MAO*EB
We considered that the alkylating agent in Equation (7) and
(8) is MAO*EB From reactions 6a, 6b and 6c it is clear that
EB can be added to MAO*TMA, but the more probable
reaction is with free TMA (dimeric or monomeric).
Table 8. Reaction DE, stabilization energies and anion struc-ture with reactions 9 and 10.
Reaction Site Anion structure ReactionDE
eV
Stabilizationenergies
9 1 MAO*TMACl– –14.33 –8.799 21 MAO*TMACl– –18.18 –12.259 22 MAO*TMACl– –18.06 –12.21
10 1 ClMAO*TMA–Cl in Al8 –14.58 –8.2510 21 ClMAO*TMA–Cl in Al8 –18.84 –12.2410 22 ClMAO*TMA–Cl in Al8 –18.99 –12.18
Scheme 8. Sites with MAO*TMA.
Table 9. Reaction DE, stabilization energies and anion struc-ture with reactions 11, 12 and 13.
Reaction Site Anion structure ReactionDE
Stabilizationenergies
11 1 MAO*AlCl3Cl– –14.72 –8.7311 21 MAO* AlCl3Cl– –18.63 –12.2511 22 MAO* AlCl3Cl– –18.51 –12.2112 1 ClMAO* TMACl2 –16.02 –8.7712 21 ClMAO* TMACl2 –20.52 –12.1212 22 ClMAO*TMACl2 –20.38 –11.9813 1 ClMAO* AlCl3Cl2 –15.19 –8.3713 21 ClMAO*AlCl3Cl2 –19.43 –12.2413 22 ClMAO*AlCl3Cl2 –19.32 –12.21

504 M. L. Ferreira, P. G. Belelli, D. E. Damiani
Case a) When two alkylating agents MAO*EB are con-
sidered.
2 MAO* EB + EtInd2ZrCl2 e EB*MAOCl
(in Al 8) + EtInd2ZrCH3+ ClMAO*EB– (14)
Case b) When one alkylating agent is considered we
propose that one molecule of MAO*EB alkylates the zir-
conocene and the same molecule reacts to become the
counterion, finally leaving two chlorine atoms (see
Scheme 10a).
MAO*EB + EtInd2ZrCl2 e
EtInd2ZrCH3+ ClMAO*EBCl– (in Al 8) (15a)
Table 10 presents reaction DE and stabilization energy
for the 6 contact ion pair modeled in the presence of
MAO*EB.
Another reactions has been considered, once the sites
are formed:
Site 1:
EtInd2ZrCH3+ ClMAO*EBCl– (in Al 8) e
EtInd2ZrCl :CH3EB + MAO*TMACl–
DE = –0.84 eV (15b)
Site 21:
EtInd2ZrCH3+ ClMAO*EBCl– (in Al 8) e
EtInd2ZrCl :CH3EB + MAO*TMACl–
DE = +3.29 eV (15c)
Site 22:
EtInd2ZrCH3+ ClMAO*EBCl– (in Al 8) e
EtInd2ZrCl :CH3EB + MAO*TMACl–
DE = +3.81 eV (15d)
Another possible reaction was the coordination of EB
without destruction of the positive charge on Zr (see
Scheme 10b).
EtInd2ZrCH3+ Cl–MAO*EB e
ClMAO– EtInd2ZrCH3+ EB
DE = 1.56 eV (15e)
The Lewis acidity of the zirconocenium ion would be
higher than the Lewis acidity of MAO*TMA. Therefore,
once the cation is formed, the “free” EB reacts with site 1
to produce a new neutral compound EtInd2ZrCH3 :EB
(see Scheme 9). If EB is coordinated to MAO, the basi-
city changes and probably EB would coordinate to zirco-
nocenium without reaction, as a “soft” base.
From the results shown in Table 8, we conclude that
the most probable contact ion pair at a higher concentra-
tion would be those whose counterion carries two chlor-
ine atoms. Therefore, the same MAO*TMA molecule
would alkylate the zirconocene and produce CIP. The dif-
ference varies between Equation (9) and (10) from –0.3
up to near –1 eV for site 21 (compare entries 3 and 6).
Sites 21 and 22 seem to be the most probable, but the site
1 can not be ruled out.
In the case of using AlCl3, the energies for Equation
(11) and (13) are almost the same as for MAO*TMA. In
these cases, they are even more probable than with
MAO*TMA in –0.6 eV. The results from Equation (12)
are interesting. The more chlorine atoms carries
MAO*TMA, the stronger its alkylating power (see
Table 9 entries 4, 5 and 6). For Equation (12), the ener-
gies for sites 1, 21 and 22 formation are 2 eV more nega-
tive. Therefore, the new alkylating agents due to the addi-
tion of AlCl3 are even better than MAO*AlCl3.
On consideration of EB, the chemistry of the solution
becomes more complicated. Besides the interaction of
MAO*TMA with EB, the interaction with the CIP, once
formed, can not be ruled out. Equation (14) and (15a–d)
show these supposed interactions. The results permit us
to conclude that EB can react with the CIP site 1 and
destroy it to produce a neutral adduct, as Jordan proposes
for zirconocenes in CH3CN as solvent.[51] Site 21 and 22
are not so easily deactivated. Looking at Table 10 in the
Table 10. Reaction DE, stabilization energies and anion struc-ture with reactions 14 and 15a.
Reaction Site Anion structure ReactionDE
eV
Stabilizationenergies
14 1 MAO*EBCl– –14.57 –8.8314 21 MAO*EBCl– –18.69 –12.4114 22 MAO*EBCl– –19.22 –13.0115 a 1 ClMAO* EBCl– –14.60 –8.4115 a 21 ClMAO*EBCl– –19.13 –12.415 a 22 ClMAO*EBCl– –19.66 –13.00
Scheme 9. Neutral compound EtInd2ZrCH3Cl: EB.

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 505
case of EB addition, the problem is not the CIP formation
but the reaction of it with EB.
Stage 4 – Dissociation of ion pair to produce solvent
separated ion pairs
In order to consider the reaction of solvent separated ion
pair formation, we modeled this situation in the case of
MAO*TMA as alkylating agent. Table 11 resumes the
results of EHMO calculations.
The reactions considered were:
EtInd2ZrCH3+ ClMAO*TMA– + Toluene e
EtInd2ZrCH3+ /Toluene/ClMAO*TMA– (16)
EtInd2ZrCH3+ ClMAO*TMACl– (in Al 8)
+ Toluene e EtInd2ZrCH3+/Toluene/
ClMAO*TMACl– (in Al 8) (17)
Table 11 shows the results of reactions 16 and 17.
When the counterion is double chlorinated for site 1, the
reaction is probable. In the other situations, the reaction
of SSIP formation is unfavourable. However, site 1 would
prefer to change to Site 2 with the same counterion struc-
ture and not to a SSIP (–4.26 eV vs –3.32 eV Table 11
entry 3).Site 1 + Toluene e cation/Toluene/anion
DE = –3.32 eV
Site 1 e Site 2 DE = –4.26 eV
A point to consider here is that the solvent separated
ion pair is an ion pair where the solvent acts as a neutral
spacer. Therefore the distance cation-anion increases; per-
haps the electrophilicity of Zr too, but it is not the same
to “free” solvated.[1c]
Stage 5 – Formation of cation solvated ion pairs
We considered the following reactions:
EtInd2ZrCH3+/Toluene/ClMAO*TMA– e
EtInd2ZrCH3+/Toluene + ClMAO*TMA– (18)
EtInd2ZrCH3+/Toluene/ClMAO*TMACl–
(in Al 8) e EtInd2ZrCH3+/Toluene
+ ClMAO*TMACl– (in Al 8) (19)
Schemes 11 and 12 show the structures of anion and
cation, resulting from reactions 18 and 19. Table 12
resumes the reaction DEs shown in Equation (18) and
(19). The reaction is not favored whatever the situation
considered (see Table 12).
Table 11. Reaction DE for solvent separated ion pairs.
Reaction Site Anion structure Reaction DE
16 1 ClMAO*TMA– + 4.8316 21 ClMAO*TMA– +9.7517 1 ClMAO*TMACl in Al 8– –3.3217 21 ClMAO*TMACl in Al 8– +9.76
Table 12. Reaction DE for cation solvated ion pairs.
Reaction Site Anion structure Reaction DE
18 1 ClMAO*TMA– +1.2718 21 ClMAO*TMA– +0.2519 1 ClMAO*TMACl– +8.8819 21 ClMAO*TMACl– –0.25
Scheme 10a. Coordination of EB to counterion MAO*TMA ofSite 21.
Scheme 10b. Direct coordination of EB to the zircocation ofSite 21.
506 M. L. Ferreira, P. G. Belelli, D. E. Damiani
6 – Other reactions
Considering the different alkylating agents (TMA2,
Al2Cl6) and recent reports about the importance of binuc-
lear cations in the zirconocene/MAO systems we
included the following reactions:
Al2Me6 + EtInd2ZrCl2 e
EtInd2ZrCH3+ClAl2Me5Cl– (20)
Al2Cl6 +EtInd2ZrCl2 e EtInd2ZrCl+ClAl2Cl5– (21)
EtInd2ZrCH3+MAO*TMACl– (Site 1)
+ EtInd2ZrCl2 e EtInd2ZrCH3+(lCl)2EtInd2Zr
+ MAO*TMACl– (22a)
EtInd2ZrCH3+MAO*TMACl– (Site 21)
+ EtInd2ZrCl2 e EtInd2ZrCH3+(lCl)2EtInd2Zr
+ MAO*TMACl– (22b)
The formation of binuclear cations from CIP would be
non-probable. Schemes 13, 14 and 15 show the product
structures modeled from Equation (20), (21) and (22).
Table 13 resumes the results for these equations.
7 – Formation of binuclear cations from
EtInd2ZrCH3+ClAl2Me5
– and EtInd2ZrCl+ClAl2Cl5–
The formation of binuclear cations (see Scheme 15)
would be possible from the interaction between zircono-
cene and TMA2.
EtInd2ZrCH3+ClAl2Me5
– + Toluene e
EtInd2ZrCH3+/Toluene + ClAl2Me5
– (23)
EtInd2ZrCl+ClAl2Cl5– + Toluene e
EtInd2ZrCl+/Toluene + ClAl2Cl5– (24)
EtInd2ZrCH3+/Toluene + EtInd2ZrCl2 e
Toluene + EtInd2ZrCH3+(lCl)2EtInd2Zr (25)
CSIP would be transformed to binuclear cations at high
concentrations of zirconocene (see Table 14).
Scheme 11. A: Anion MAO*TMACl2– B: MAO*TMACl–.
Scheme 12. Solvated cation.
Scheme 13. Contact ion pair model with Al2Me6.
Scheme 14. Contact ion pair model with Al2Cl6.
Table 13. Reaction DE for reactions 20, 21 and 22.
Reaction Product ReactionDE
eV
Stabilizationenergies
eV
20 EtInd2ZrCH3+ClAl2Me5
- –2.32 –1.5921 EtInd2ZrCl+ClAl2Cl5
- –0.14 –0.3122 Binuclear cation from site 1 2.87 –5.42
Binuclear cation from site 2 6.82 –5.42
Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 507
UV Spectroscopy
Deffieux and co-workers[46–47] studied the system
EtInd2ZrCl2/MAO in two different solvents (toluene and
dichloromethane) by means of UV-vis spectroscopy.
They used two Al/Zr molar ratio in the reaction media for
the 1-hexene polymerization. Different zirconocenium
species are proposed to be present. They depend first on
Al/Zr molar ratios but also on other components present
in the reaction media e.g. TMA. Kim and co-workers[48]
used this technique to study changes in the catalyst titano-
cenes/MAO. They used the styrene polymerization as a
test reaction. Gianetti and co-workers[37] found a kmax for
MAO in benzene at 286 mn. This band was assigned to
the interaction of O non-bonding electrons with Al atoms
near to O. They propose dative bonds between O and Al.
Our results show a band at 282 nm for MAO. This band
is not concentration dependent ([MAO] = 0.015–5610–3
M).
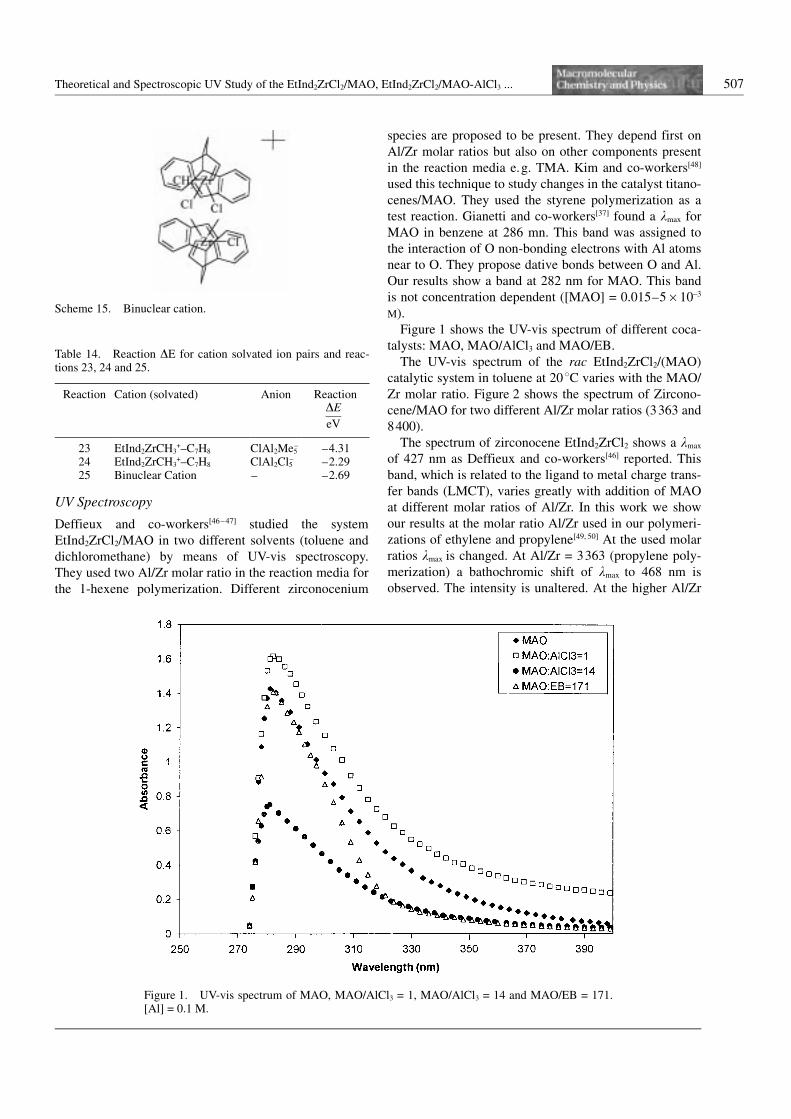
Figure 1 shows the UV-vis spectrum of different coca-
talysts: MAO, MAO/AlCl3 and MAO/EB.
The UV-vis spectrum of the rac EtInd2ZrCl2/(MAO)
catalytic system in toluene at 208C varies with the MAO/
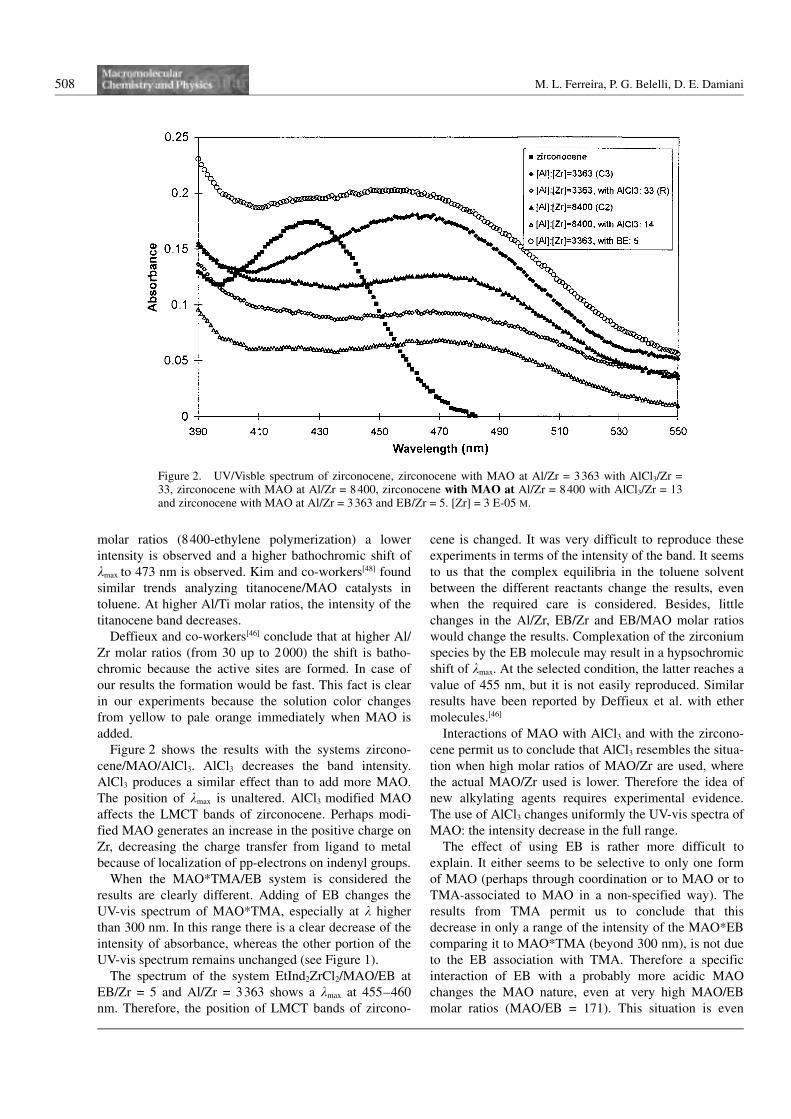
Zr molar ratio. Figure 2 shows the spectrum of Zircono-
cene/MAO for two different Al/Zr molar ratios (3363 and
8400).
The spectrum of zirconocene EtInd2ZrCl2 shows a kmax
of 427 nm as Deffieux and co-workers[46] reported. This
band, which is related to the ligand to metal charge trans-
fer bands (LMCT), varies greatly with addition of MAO
at different molar ratios of Al/Zr. In this work we show
our results at the molar ratio Al/Zr used in our polymeri-
zations of ethylene and propylene[49, 50] At the used molar
ratios kmax is changed. At Al/Zr = 3363 (propylene poly-
merization) a bathochromic shift of kmax to 468 nm is
observed. The intensity is unaltered. At the higher Al/Zr
Scheme 15. Binuclear cation.
Table 14. Reaction DE for cation solvated ion pairs and reac-tions 23, 24 and 25.
Reaction Cation (solvated) Anion ReactionDE
eV
23 EtInd2ZrCH3+–C7H8 ClAl2Me5
– –4.3124 EtInd2ZrCH3
+–C7H8 ClAl2Cl5– –2.29
25 Binuclear Cation – –2.69
Figure 1. UV-vis spectrum of MAO, MAO/AlCl3 = 1, MAO/AlCl3 = 14 and MAO/EB = 171.[Al] = 0.1 M.
508 M. L. Ferreira, P. G. Belelli, D. E. Damiani
molar ratios (8400-ethylene polymerization) a lower
intensity is observed and a higher bathochromic shift of
kmax to 473 nm is observed. Kim and co-workers[48] found
similar trends analyzing titanocene/MAO catalysts in
toluene. At higher Al/Ti molar ratios, the intensity of the
titanocene band decreases.
Deffieux and co-workers[46] conclude that at higher Al/
Zr molar ratios (from 30 up to 2000) the shift is batho-
chromic because the active sites are formed. In case of
our results the formation would be fast. This fact is clear
in our experiments because the solution color changes
from yellow to pale orange immediately when MAO is
added.
Figure 2 shows the results with the systems zircono-
cene/MAO/AlCl3. AlCl3 decreases the band intensity.
AlCl3 produces a similar effect than to add more MAO.
The position of kmax is unaltered. AlCl3 modified MAO
affects the LMCT bands of zirconocene. Perhaps modi-
fied MAO generates an increase in the positive charge on
Zr, decreasing the charge transfer from ligand to metal
because of localization of pp-electrons on indenyl groups.
When the MAO*TMA/EB system is considered the
results are clearly different. Adding of EB changes the
UV-vis spectrum of MAO*TMA, especially at k higher
than 300 nm. In this range there is a clear decrease of the
intensity of absorbance, whereas the other portion of the
UV-vis spectrum remains unchanged (see Figure 1).
The spectrum of the system EtInd2ZrCl2/MAO/EB at
EB/Zr = 5 and Al/Zr = 3363 shows a kmax at 455–460
nm. Therefore, the position of LMCT bands of zircono-
cene is changed. It was very difficult to reproduce these
experiments in terms of the intensity of the band. It seems
to us that the complex equilibria in the toluene solvent
between the different reactants change the results, even
when the required care is considered. Besides, little
changes in the Al/Zr, EB/Zr and EB/MAO molar ratios
would change the results. Complexation of the zirconium
species by the EB molecule may result in a hypsochromic
shift of kmax. At the selected condition, the latter reaches a
value of 455 nm, but it is not easily reproduced. Similar
results have been reported by Deffieux et al. with ether
molecules.[46]
Interactions of MAO with AlCl3 and with the zircono-
cene permit us to conclude that AlCl3 resembles the situa-
tion when high molar ratios of MAO/Zr are used, where
the actual MAO/Zr used is lower. Therefore the idea of
new alkylating agents requires experimental evidence.
The use of AlCl3 changes uniformly the UV-vis spectra of
MAO: the intensity decrease in the full range.
The effect of using EB is rather more difficult to
explain. It either seems to be selective to only one form
of MAO (perhaps through coordination or to MAO or to
TMA-associated to MAO in a non-specified way). The
results from TMA permit us to conclude that this
decrease in only a range of the intensity of the MAO*EB
comparing it to MAO*TMA (beyond 300 nm), is not due
to the EB association with TMA. Therefore a specific
interaction of EB with a probably more acidic MAO
changes the MAO nature, even at very high MAO/EB
molar ratios (MAO/EB = 171). This situation is even
Figure 2. UV/Visble spectrum of zirconocene, zirconocene with MAO at Al/Zr = 3363 with AlCl3/Zr =33, zirconocene with MAO at Al/Zr = 8400, zirconocene with MAO at Al/Zr = 8400 with AlCl3/Zr = 13and zirconocene with MAO at Al/Zr = 3363 and EB/Zr = 5. [Zr] = 3 E-05 M.

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 509
more complex when the zirconocenium ion is present.
The EB effect on it is very difficult to reproduce. There is
evidence about hypsochromic shifts of the zirconocenium
ion when EB is present, and destruction of the charge on
it for, at least, an important fraction of the zirconocenes.
The main differences between MAO*TMA and dimer
TMA and Al2Cl6 results would be the low CIP energy for-
mation and stabilization for the last. In the case of dimer
TMA, the solvation energy for toluene could stabilize the
SSIP, but in the case of CIP formed with MAO*TMA, the
SSIP are very difficult to produce.
MAO*AlCl3 is a better alkylating agent than MAO*
TMA. Besides, it would produce new and efficient alky-
lating agents and alternative counterions (dichlorinated
MAO*TMA) in higher concentration than MAO*TMA.
EB interacts with MAO*TMA and once it is formed,
with zirconocenium. Therefore EB would act as a poison,
as we have noticed in our own experimental results.
With dimer TMA, following EHMO results, the forma-
tion of CIP is possible. Experimentally Cp2ZrMeCl shows
no activity in ethylene polymerization with dimer TMA.
Therefore the lack of activity must be explained. In the
case of the TMA2Cl anion, the negative charge is on Cl
and it can not be dispersed as in the MAOCl– anion to the
oxygens of the structure. The counterion from dimer
TMA is very much coordinative compared to that of
MAO and the deactivated Al1CH21Zr bonds are very
difficult to reactivate with dimer TMA. In the case of
MAO, this group can be methylated to generate Zr1Me
and Al1CH21Al. MAO would have a higher reactivating
power than dimer TMA. This point is in agreement with
recent reports about the increased activity of zirconocene
using chelating organoalanes as coactivators.[44] These
compounds would poorly coordinate to the cation.
The idea of CIP as the most active and stereospecific
sites in systems EtInd2ZrCl2//MAO, is supported by the
structure of MAO. Sometimes MAO is compared to the
macrocyclic polyethers used as very powerful complex-
ing agents for alkali metal cations in aprotic solvent.
These ethers are able to break up CIP, hindering the
aproximation cation-anion in the “coronates” complexes
or in the “cryptates”.[1a] But the point is that these ethers
are used like neutral spacers. In case of MAO, the anion
is the same spacer. When the ion pairs of radical anions
of o-diaroylbenzenes and of 1, 2, 4, 5 tetraaroylbenzenes
are considered, due to their chelating properties and to
the high concentration of negative charge on the O atoms,
the CIP of these anions are extremely difficult to break
up.[1a] The other remarkable feature of the crown ethers is
that they are only able to increase the interionic distance
by a very small amount by placing itself near the cation
and opposed to the anion (“pulling off” effect). If several
O (neutral) surround the cation, the ionic association is
expected to be weaker. The increase in temperature
makes the interionic distance higher.
The conclusions presented here correlates well with
activity. Results from our laboratory show that the activ-
ities in ethylene and propylene polymerization increase
when AlCl3 is added and decrease when EB is present.[49, 50]
Even at molar ratio EB/Zr much higher than 1 the activ-
ities drastically decrease, but are not eliminated. There-
fore, CIP with Al2Cl7– is not formed because these species
are claimed to be inactive. Some of the zirconocenium
cation remains unpoisoned although EB is present. Simu-
lation performed on the kinetics of propylene polymeriza-
tion with all these soluble systems revealed that at least
two sites with different deactivation (one of second order
and the other of first) must be considered to fit the data.[51–
52] Following our EHMO results we can propose the equili-
brium presented later. In the real situation the picture
would be very much complicated[54–56] but this simplified
picture could be associated to the results in our lab with
Lewis acid and bases and they could give us enough tools
to explain our experimental data. Site 2 (LCIP) would be
preferred to site 1 (TCIP) and this could be transformed to
SSIP only. Sites 2 (both 21 and 22) LCIP could not be
transformed to SSIP, but can react with EB or coordinate
it. Free ions would not be possible at these conditions. In
this sense, the overall picture for both additives (AlCl3 and
EB) agree with the EHMO results.
Simulation of propylene semibatch polymerization
kinetics from our group[51, 52] show that the deactivation
constant of the site with first order deactivation is
strongly affected by additives, whereas the deactivation
constant of the site with second order deactivation is not.
If the sites of second order deactivation are considered to
be LCIPs and the additives change the Zr electrophilicity,
it is easy to think as these sites as the most active. TCIP
would have the highest electrophilicity, but it is highly
hindered for propylene coordination and insertion. The
SSIPs have been considered as the most active ones. With
MAO, this would be not the case looking at our calcula-
tion. The SSIPs would be poorly active and poorly stereo-
selective.[57–58]
We believe that the activity for different contact ion
pairs in systems EtInd2ZrCl2//MAO would decrease
LCIP S CIP A SSIP

510 M. L. Ferreira, P. G. Belelli, D. E. Damiani
taking into account the EHMO calculation and experi-
mental results from our own and others.[49, 50, 57]
Conclusions
Our results permit us to say:
– Two different groups of CIP could be present in the
EtInd2ZrCl2//MAO//toluene system with and without
modifiers. Loosened contact ion pairs (LCIP) are more
probable than tight contact ion pair (TCIP).
– Site 1 would produce SSIP easily but sites 21 and 22
would not. The formation of CSIP would be non-prob-
able.
– Site 1 is a tight CIP (TCIP) with close interaction
with the counterion. This type of site would suffer second
order deactivation by bimolecular reaction with methane
evolution and Zr1CH21Zr bond formation with other
site 1. We checked this point with results from modeling
of our experimental kinetics curves (see the litera-
ture[51, 52]).
– Site 2 has another, more open, structure (see Scheme
7b). This site would have a first order decay by reaction
with the MAO anion, as we checked from modeling of
our experimental kinetics curves. Interchanging between
Sites 21 and 22 must be considered and also with sites 1.
Acknowledgement: We acknowledge support from the Con-sejo Nacional de Investigaciones Cientıficas y Tecnicas (CONI-CET, Argentina) and the Universidad Nacional del Sur (UNS,Bahıa Blanca-Argentina).
Received: February 8, 2000Revised: August 3, 2000
[1] (a) A. Togni, R. L. Halterman, “Metallocenes”, WileyVCH 1998, 2, 548; (b) W. Kaminsky, A. Bark, R. Steiger,J. Mol. Catal. 1992, 74, 109. (c) B. J. Herold, “V SimposioIberoamericano de Catalysis”, Lisboa, Portugal 1976.
[2] C. Janiak, B. Rieger, R. Voelkel, H. G. Braun, J. Polym.Sci., Part A: Polym. Chem. 1993, 31, 2959.
[3] I. Tritto, M. C. Sacci, P. Locatelli, S. X. Li, Macromol.Chem. Phys. 1996, 197, 1537.
[4] J. C. W. Chien, R. Sugimoto, J. Polym. Sci., Part A: Polym.Chem. 1991, 29, 459.
[5] N. Herbert, G. Fink, Makromol. Chem. 1992, 193, 1359.[6] J. C. W. Chien, B. P. Wang, J. Polym. Sci., Part A: Polym.
Chem. 1988, 26, 3089.[7] I. Tritto, S. X. Li, M. C. Sacchi, P. Locatelli, G. Zannoni,
Macromolecules 1995, 28, 5358.[8] A. R. Siedle, R. A. Newmark, W. M. Lamanna, J. N.
Schroepfer, Polyhedron 1990, 9, 2/3, 301.[9] A. R. Siedle, R. A. Newmark, J. N. Schroepfer, P. A. Lyon,
Organometallics 1991, 10, 400.
[10] D. Cam, U. Giannini, Makromol. Chem. 1992, 193, 1049.[11] T. K. Woo, L. Fan, T. Ziegler, “Ziegler Catalysts”, Eds. G.
Fink, R. Mulhaupt, H. H. Brintzinger, 1995, 291.[12] N. Koga, T. Yoshida, K. Morokuma, “Ziegler Catalysts”,
Eds G. Fink, R. Mulhaupt, H. H. Brintzinger, 1995, 276.[13] C. A. Jolly, D. J. Marynick, J. Am. Chem. Soc. 1989, 11,
7968.[14] H. Kawamura-Kuribayashi, N. Koga, K. Morokuma, J.
Am. Chem. Soc. 1992, 114, 2359.[15] P. Margl, J. C. W. Lohrenz, T. Zieglers, P. E. Bilochli, J.
Am. Chem. Soc. 1996, 118, 4434.[16] J. C. W. Lohrenz, T. K. Woo, L. Fan, T. Ziegler, J. Organo-
met. Chem. 1995, 497, 92.[17] V. L. Cruz, A. Munoz Escalona, J. Martınez Salazar, Poly-
mer 1996, 37, 1663.[18] H. Kawamura-Kuribayashi, N. Koga, K. Morokuma, J.
Am. Chem. Soc. 1992, 14, 8687.[19] H. Fujimoto, T. Yamasaki, H. Mizutani, N. Koga, J. Am.
Chem. Soc. 1985, 107, 6157.[20] R. Fusco, L. Longo, F. Masi, F. Garbassi, Macromol.
Rapid. Commun. 1997, 18, 433–441.[21] H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger, R.
M. Waymouth, Angew. Chem. Int. Ed. Engl. 1995, 34,1143.
[22] A. R. Siedle, R. A. Newmark, W. M. Lamanna, R. A. New-mark, J. Stevens, D. E. Richardson, M. Ryan, Makromol.Chem. Macromol. Symp. 1993, 66, 215.
[23] A. R. Siedle, B. Hangkl, R. A. Newmark, K. R. Mann, T.Wilson, Macromol. Symp. 1995, 89,299–305.
[24] R. Fusco, L. Longo, F. Masi, F. Garbassi, Macromolecules1997, 30, 7673.
[25] J. Kamber, L. Forrs, G. Calzaferri, J. Phys. Chem. 1989,93, 5366.
[26] W. Lotz, J. Opt. Soc. Am. 1970, 60, 206.[27] A. Vela, L. Gazquez , J. Phys. Chem. 1985, 92, 5688.[28] (a) R. Hoffmann, J. Am. Chem. Soc. 1985, 107, 4440; (b)
A. Juan, R. Hoffmann, Surf. Sci. 1999, 421, 1.[29] M. L. Ferreira, N. J. Castellani, D. E. Damiani, A. Juan, J.
Mol. Catal. A: Chem. 1997, 122, 25.[30] (a) M. L. Ferreira, A. Juan, M. M. Branda, D. E. Damiani,
J. Mol. Catal. A: Chem. 1997, 122, 51; (b) M. L. Ferreira,A. Juan, D. E. Damiani, React. Kinet. Catal. Lett. 1998, 64,139.
[31] M. L. Ferreira, D. E. Damiani, A. Juan, Comp. Mat. Sci.1998, 9, 357.
[32] M. R. Mason, J. M. Smith, S. G. Bott, R. Barron, J. Am.Chem. Soc. 1993, 115, 4971–4984.
[33] H. Sinn, I. Schimmel, M. C. N. von Thienen, A. Harder, W.Hagendorf, B. Heitmann, E. Haupt, Z. Yu, “MetalorganicCatalysts for Synthesis and Polymerization”, 105, Ed. W.Kaminsky, Springer, 1999.
[34] H. A. R. Siedle, R. A. Newmark, W. B. Gleaason, U. N.Lamama, J. Bleimeister, W. Hagendorf, H. Harder, B. Heit-mann, I. Schimmel, E. Schmedt, W. Schnuckel, H. Sinn, L.Tikwe, N. von Thienen, K. Urlass, H. Winter, O. Zarncke,“Ziegler Catalysts”, 57, Ed. Fink/Mulhaupt/Brintzinger1995, 57.
[35] D. F. Shriver, P. W. Atkins, C. H. Langsford, InorganicChemistry, Oxford Uni. Press, 1994.
[36] Handbook of Chemistry and Physics, David R. Lide Ed.76th Edition, 1995–1996, CRC Press.
[37] E. Giannetti, G. M. Nicoletti, R. Mazzocchi, J. Polym. Sci.,Part A: Polym. Chem. 1985, 23, 2117.
[38] G. Belov, N. Kornnev, Macromol. Symp. 1995, 97, 63.[39] J. J. Eisch, K. R. Caldwell, S. Werner, C. Kruger, Organo-
metallics 1991, 10, 3417.

Theoretical and Spectroscopic UV Study of the EtInd2ZrCl2/MAO, EtInd2ZrCl2/MAO-AlCl3 ... 511
[40] J. J. Eisch, S. I. Pombrik, G. Xu Zheng, Organometallics1993, Vol. 12, 3856.
[41] S. L. Borbowsky, R. F. Jordan, G. D. Hinck, Organometal-lics 1991, 10, 1268.
[42] P. Corradini, G. Guerra, L. Cavallo, G. Masardi, M. Vaca-tello, Zarncke, “Ziegler Catalysts”, Ed. Fink/Mulhaupt/Brintzinger 1995, 238.
[43] K. Angermund, a. Hanuschik, M. Nolte, “Ziegler Cata-lysts”, Ed. Fink/Mulhaupt/Brintzinger 1995, 253.
[44] F. Karol, S. C. Kao, E. P. Wassermann, Z. Yu, “Metalor-ganic Catalysts for Synthesis and Polymerization”, 629,Ed. W. Kaminsky, Springer, 1999.
[45] (a) C. J. Harlan, S. G. Bott, A. R. Barron J. Am. Chem. Soc.1995, 117, 6465; (b) J. W. Lauher, R. Hoffmann, J. Am.Chem. Soc. 1976, 98, N7, 1729.
[46] D. Coevoet, H. Cramail, A. Deffieux, Macromol. Chem.Phys. 1998, 199, 1459.
[47] D. Coevoet, H. Cramail, A. Deffieux, Macromol. Chem.Phys. 1998, 199, 1451.
[48] J. Kim, K. H. Kim, J. Cho, S. Kwak, K. U. Kim, W. Jo, H.Yoon, D. Lim, J. Polym. Sci., Part A: Polym. Chem. 1998,36, 1733.
[49] P. G. Belelli, M. L. Ferreira, D. E. Damiani, Macromol.Chem. Phys., accepted.
[50] P. G. Belelli, M. L. Ferreira, D. E. Damiani, Macromol.Chem. Phys., accepted.
[51] P. G. Belelli, M. L. Ferreira, D. E. Damiani, M. H. Lacunza,A. Brandolın, IVArgentinian, Polym. Symp. 1999.
[52] P. G. Belelli, M. L. Ferreira, M. H. Lacunza, D. E.Damiani, A. Brandolın, Polym. Eng. Sci., submitted.
[53] R. Jordan, J. Chem Ed. 1985, 65, 285.[54] T. Haselwander, S. Beck, H. H. Brintzinger, “Ziegler Cata-
lysts”, Ed. Fink/Mulhaupt/Brintzinger 1995, 182.[55] J. J. Eisch, S. I. Pombrik, S. Gurtzen, B. Rieger, W. Uzick,
“Catalyst Design for Tailor Made Polyolefins”, Ed. Kodan-sha 1994, 221.
[56] C. Sishta, R. M. Hathorn, T. J. Marks, J. Am. Chem. Soc.1992, 114, 1112.
[57] J. C. Vizzini, J. C. W. Chien, G. N. Babu, R. A. Newmark,J. Polym. Sci., Part A: Chem. 1994, 32, 2049.
[58] J. C. W. Chien, B. Rieger, R. Sugimoto, D. T. Mallin, M.Rausch, “Catalytic Olefin Polymerization”, “Studies inSurface Science and Catalysis”, Ed. T. Keii, K. Soga, Ed.Kodansha, 1990, 535.





























