Embed Size (px)
Citation preview

The role of Syk in airway hyperresponsiveness and remodeling in house dust mite induced murine models of allergic airways inflammation
by Sepehr Salehi
A thesis submitted in conformity with the requirements for the degree of Master in Science
Institute of Medical Science University of Toronto
© Copyright by Sepehr Salehi 2013

ii
The role of Syk in airway hyperresponsiveness and remodeling in house dust mite induced murine models of allergic airways inflammation
Sepehr Salehi Master of Science
Institute of Medical Science University of Toronto
2013
Abstract
Spleen tyrosine kinase (Syk) plays a critical role in regulation of immune and
inflammatory responses. This thesis investigated the role of Syk in the development of the
asthma phenotype in acute and chronic mouse models of allergic airways inflammation.
Airway hyperresponsiveness (AHR) to methacholine and inflammation increased
significantly in HDM-induced compared with the saline control mice. We demonstrated that in
vivo inhibition of Syk by selective Syk inhibitors, and genetic deletion of Syk using conditional
Syk knockout mice attenuated AHR despite of inflammatory cell influx in the lung. Histological

iii
analysis showed airway remodeling in the chronic model, which was attenuated to some degree
by deletion of Syk.
This study identified a role of Syk in airway hyperresponsiveness and remodeling without
significantly affecting leukocyte recruitment in HDM model of airways disease. My results
support the improvement of therapeutic strategies in asthma by targeting the Syk pathway.

iv
Acknowledgments
First of all, I would like to express my gratitude to Dr. Chung-Wai Chow who has been my
supervisor and provided me the opportunity to accomplish my MSc thesis in the Institute of
Medical Science. I also want to thank Dr. Jeremy Scott for his support and encouragement during
my study.
In addition, I would like to thank my Program Advisory Committee members, Dr. Jane Batt, Dr.
Haibo Zhang and Dr. Hartmut Grasemann for sharing their expertise in the field of airway
inflammation and asthma.
I would like to thank the members of Dr. Chow’s Laboratory, who have assisted me in this work
in many ways. In particular, I would like to acknowledge Xiaomin Wang who helped me with
different experiments and also provided me Sykflox/flox mice during my Study. I want to thank Dr.
Nivedita Khanna who trained me in pulmonary function testing and also special thank to Patricia
Castellanos for all her help and support. I also would like to thank Dr. Krystal Godri and
Josephine Cooper who helped me with Luminex assay and editing of my thesis.
I would like to thank Dr. Jamila Chakir from Département de médecine, Université Laval for her
help in assessment of airway remodeling.
Many thanks to my friends Mahtab Mehrabi, Dr. Dariush Davani and Dr. Mohammad
Bahmanyar for all the support and help.
Last but not the least, I would express my deepest gratitude to my family, especially to my
father, mother and sister who have always been the greatest support and encouragement for me.
Sykfl/fl-Syk//rosa26-CreERT2 mouse was a valuable gift from Boehringer Ingelheim, Germany.
This study was funded by the Canadian Institutes of Health Research (CIHR), and The Queen
Elizabeth II- Graduate Scholarships in Science and Technology (QEII-GSST) programs.

v
Table of Contents ACKNOWLEDGMENTS .................................................................................................................................. IV
TABLE OF CONTENTS ..................................................................................................................................... V
LIST OF ABBREVIATIONS ......................................................................................................................... VIII
LIST OF TABLES ............................................................................................................................................... XI
LIST OF FIGURES ........................................................................................................................................... XII
1 CHAPTER 1: LITERATURE REVIEW .................................................................................................. 1 1.1. ASTHMA ............................................................................................................................................................................ 1
1.1.1. Definition and prevalence .................................................................................................................................... 1 1.1.2. Airway inflammation in asthma ........................................................................................................................ 6 1.1.3. Airway hyperresponsiveness in asthma ....................................................................................................... 11 1.1.4. Airway remodeling in asthma .......................................................................................................................... 14
1.2. ANIMAL MODELS OF ASTHMA .............................................................................................................................. 20 1.2.1. Mouse models of asthma .................................................................................................................................... 21 1.2.2. House dust mite (HDM) model of asthma .................................................................................................. 21
1.3. SPLEEN TYROSINE KINASE (SYK) ........................................................................................................................ 23 1.3.1. Structure and cellular expression of the Syk family of tyrosine kinases ...................................... 24 1.3.2. Role and function of Syk .................................................................................................................................... 26 1.3.3. Syk and disease pathogenesis ........................................................................................................................... 32 1.3.4. Syk and asthma ...................................................................................................................................................... 32
1.4. SYK DEFICIENT MICE ............................................................................................................................................... 33 1.5. INHIBITION OF SYK ................................................................................................................................................... 35
1.5.1. Mechanisms of action .......................................................................................................................................... 35 1.5.2. Syk selective inhibitors in clinical studies .................................................................................................. 36 1.5.3. Syk selective inhibitors in asthma and allergic rhinitis ........................................................................ 37
2 CHAPTER 2: HYPOTHESIS AND RESEARCH OBJECTIVES .................................................... 41 2.1. OVERALL HYPOTHESIS ............................................................................................................................................ 41 2.2. RESEARCH OBJECTIVES .......................................................................................................................................... 41
3 CHAPTER 3: MATERIAL AND METHODS ...................................................................................... 42 3.1. ANIMALS ........................................................................................................................................................................ 42 3.2. MURINE HDM-SENSITIZATION AND -CHALLENGE MODEL OF AIRWAYS INFLAMMATION ......... 42

vi
3.2.1. Acute model of HDM-induced airways inflammation .......................................................................... 42 3.2.2. Chronic model of HDM-induced airways inflammation ..................................................................... 43
3.3. IN VIVO INHIBITION OF SYK ................................................................................................................................... 44 3.4. KNOCKDOWN OF SYK IN INDUCIBLE SYK KNOCKOUT MICE ................................................................... 45 3.5. PULMONARY FUNCTION TESTS (PFTS) AND METHACHOLINE CHALLENGE ...................................... 46 3.6. BRONCHOALVEOLAR LAVAGE FLUID (BALF) ............................................................................................... 47 3.7. ANALYSIS OF INFLAMMATORY MEDIATORS IN BALF ............................................................................... 47 3.8. HISTOLOGY OF LUNG TISSUE SECTIONS ......................................................................................................... 48 3.9. RNA EXTRACTION AND QUANTITATIVE PCR ................................................................................................ 48 3.10. QUANTIFICATION OF HDM-SPECIFIC IGG1 AND IGE IN SERUM ........................................................ 49 3.11. STATISTICAL ANALYSIS ........................................................................................................................................ 50
4 CHAPTER 4. RESULTS ............................................................................................................................ 51 4.1. ACUTE MODEL OF HDM-INDUCED AIRWAYS INFLAMMATION .............................................................. 51
4.1.1. A 10 day course of HDM sensitization and challenge enhanced AHR to methacholine, a
response that is abrogated by treatment with a single dose of Syk inhibitor ........................................... 51 4.1.2. HDM-induced AHR was abrogated by deletion of Syk ........................................................................ 53 4.1.3. Deletion of Syk reduced HDM-specific IgG1 and IgE levels in serum ......................................... 56 4.1.4. BALF total cell was not affected by either Syk knock-down or pharmacological inhibition57 4.1.5. Effect of Syk deletion on the production of IL-6, KC, VEGF and MMP-9 in the acute model
of HDM-induced airways inflammation .................................................................................................................. 60 4.1.6. Effect of Syk deletion on expression levels of inflammatory mediators in lung tissue ........... 61 4.1.7. Histologic evidence of airway inflammation and goblet cell hyperplasia is evident following
a 10-day HDM sensitization and challenge period ............................................................................................. 63 4.2. CHRONIC MODEL OF HDM-INDUCED ALLERGIC AIRWAYS INFLAMMATION ................................... 66
4.2.1. The development of HDM-induced AHR was abrogated by deletion of Syk .............................. 66 4.2.2. Deletion of Syk reduced HDM-specific IgG1 and IgE levels in serum ......................................... 70 4.2.3. Deletion of Syk did not affect BALF total cell counts ........................................................................... 71 4.2.4. Deletion of Syk did not affect blood total cell counts nor did it affect the bone marrow
response to HDM ............................................................................................................................................................... 72 4.2.5. Syk mediates HDM-induced expression of IL-6, IL-17, KC and RANTES in the chronic
model of HDM-induced airways inflammation .................................................................................................... 74 4.2.6. Effect of Syk deletion on expression levels of inflammatory mediators in lung tissue ........... 76 4.2.7. Deletion of Syk attenuated the degree of epithelium modification, mucus cell hyperplasia
and increase in smooth muscle mass ........................................................................................................................ 78

vii
CHAPTER 5: DISCUSSION ............................................................................................................................ 81
5 CHAPTER 6: CONCLUSION .................................................................................................................. 97
CHAPTER 7: FUTURE DIRECTIONS ........................................................................................................ 98
REFERENCES ................................................................................................................................................. 100
COPYRIGHT ACKNOWLEDGEMENTS ................................................................................................ 117

viii
List of abbreviations AHR airway hyperresponsiveness
APCs antigen-presenting cells
ASM airway smooth muscle
ASOs antisense oligonucleotides
BAL bronchoalveolar lavage
BALF bronchoalveolar lavage fluid
BCRs B cell receptors
BMMCs bone marrow mast cells
CAMs cellular adhesion molecule
CCAC Canadian Council on Animal Care
CCL2 chemokine (C-C motif) ligand 2
cDNA complementary DNA
CLL B cell chronic lymphatic leukemia
COPD chronic obstructive pulmonary disease
DCs dendritic cells
Df Dermatophagoides pteronyssinus
DMSO dimethyl sulfoxide
Dp Dermatophagoides farinae
E elastance
ECM extracellular matrix
ECs endothelial cells
ELISA enzyme linked immunosorbent assay
eNO exhaled nitric oxide
ERK extracellular-signal-regulated kinase
ET-1 endothelin-1
FcRs Fc receptors
FEV1 forced expiratory volume in one second
FGF-2 fibroblast growth factor-2
FVC forced vital capacity
G tissue damping

ix
GM-CSF granulocyte macrophage colony-stimulating factor
GSC Global Symptom Complex
H&E hematoxylin and eosin
HDM house dust mite
HRV human rhinovirus
i.t. intratracheal
IC50 inhibitory concentration 50
ICAM-1 intracellular adhesion molecule 1
Ig immunoglobulin
INF-ϒ gamma interferon ITAMs immunoreceptors tyrosine based activation motifs
ITP immune thrombocytopenic purpura
KSHV Kaposi’s sarcoma-associated herpes virus
LAT linker for activation of T-Cells
LPS lipopolysaccharide
MAP mitogen activation protein
MBP major basic protein
MHC major histocompatibility complex
MMPs matrix metalloproteinases
NFAT nuclear factor of activated T-cells
NHBE normal human bronchial epithelial
NHL non-Hodgkin lymphoma
NK natural killer cells
NO nitric oxide
NOS nitric oxide synthase
ONOO- perroxynitrite
OVA ovalbumin
PAS periodic acid-schiff
PBS phosphate buffered saline
PCR polymerase chain reaction
PFT pulmonary function testing
PGD2 prostaglandin D2

x
PGE2 prostaglandin E2
PGI2 prostacyclin
PI3K phosphatidylinositol 3-kinases
PKC protein kinase C
PLCγ phospholipase Cγ
PM2.5 fine particles with an aerodynamic diameter less than 2.5 µm
PTK protein tyrosine kinase
RA rheumatoid arteritis
RANTES regulated on activation, normal T cell expressed and secreted
RICS RNA-induced silencing complex
RN Newtonian resistance
RNAi RNA interference
ROS reactive oxygen species
Rrs total respiratory resistance
RSV respiratory syncytial virus
SH2 SRC homology 2
SiRNA small interfering RNA
SLE systemic lupus erythematous
Syk spleen tyrosine kinase
TCRs T cell receptors
TGF-β1 transforming growth factor beta 1
Th T helper
TIMPs tissue inhibitor metalloproteinase
TLRs toll like receptors
TNF-α tumor necrosis factor-alpha
VCAM-1 vascular cell adhesion molecule 1
VEGF vascular endothelial growth factor
ZAP-70 zeta-chain associated protein kinase 70

xi
List of Tables
Table 3.1. List of Applied Biosystems qPCR primer sets for the gene of interest ....................... 49
Table 4.1. BALF total and differential leukocyte counts in the 10-day acute model of HDM-
induced airways inflammation ...................................................................................................... 59
Table 4.2. BALF total and differential leukocyte counts in the 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 72
Table 4.3. Blood total and differential leukocyte counts in the 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 73
Table 4.4. The quantitative findings of histopathological examination in the 8-week chronic
model of HDM induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice by
Hematoxylin and Eosin, Masson Trichrome, and PAS staining ................................................... 80

xii
List of Figures
Figure 1.1. Molecular structure of Syk, Syk B and ZAP70 .......................................................... 25
Figure 1.2. The general scheme of signal transduction through Syk ............................................ 26
Figure 1.3. Recruitment of Syk or ZAP70 to plasma membrane receptors .................................. 28
Figure 1.4. Schematic diagram of the knockout strategy .............................................................. 35
Figure 3.1 Experimental timeline for developing of acute (A) and chronic (B) models of HDM-
induced allergic airways inflammation ......................................................................................... 44
Figure 3.2. In vivo ventilator-based assessment of respiratory mechanics by the flexiVent®
system ........................................................................................................................................... 45
Figure 3.3. Experimental timeline for Tamoxifen treatment for deletion of Syk in inducible Syk
knockout, Sykfl/fl-Syk//rosa26-CreERT2 mouse ............................................................................ 46
Figure 4.1. Effect of HDM on airways responsiveness to methacholine and attenuation by NVP-
QAB-205 in 10-day acute model of HDM-induced airways inflammation in Balb/c mice ......... 52
Figure 4.2. Airways responsiveness to methacholine and abrogation by deletion of Syk in the 10-
day acute model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice 54
Figure 4.3. Dose-response curve to increasing doses of methacholine for RN and G and
abrogation by deletion of Syk in the 10-day acute model of HDM-induced airways inflammation
in Sykfl/fl-Syk//rosa26-CreERT2 mice ............................................................................................ 55
Figure 4.4. Serum levels of HDM-specific IgG1 and IgE in the 10-day acute model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 56
Figure 4.5. Level of inflammatory mediators in the BALF in the 10-day acute model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 61
Figure 4.6. The expression levels for inflammatory mediators in the lung samples in HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice ........................................ 62
Figure 4.7. Histological analysis of airways inflammation and abnormalities in the 10-day acute
model of HDM-induced airways inflammation in Balb/c and Sykfl/fl-Syk//rosa26-CreERT2 mice
....................................................................................................................................................... 65
Figure 4.8. Airways responsiveness to methacholine and abrogation by deletion of Syk in the 8-
week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2
mice ............................................................................................................................................... 68

xiii
Figure 4.9. Increase in Newtonian resistance (RN) and tissue damping (G) to methacholine and
abrogation by deletion of Syk in the 8-week chronic model of HDM-induced airways
inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice ..................................................................... 69
Figure 4.10. Serum levels of HDM-specific IgG1 and IgE in the 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 70
Figure 4.11. Level of inflammatory mediators in the BALF in 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice .......................................... 75
Figure 4.12. The level of IL-6 and KC expression in the lung samples in HDM-induced airways
inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice ..................................................................... 77
Figure 4.13. Histological analysis of airways inflammation and abnormalities in the 8-week
chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice ... 80

1
1
1 Chapter 1: Literature review
1.1. Asthma
1.1.1. Definition and prevalence
Asthma is a chronic inflammatory disorder of the lung, characterized by airway
hyperresponsiveness, airway inflammation and airway remodeling [1-3]. The clinical presentation
of asthma manifests as repeated, variable and episodic attacks of dyspnea, chest tightness,
coughing and wheezing [3]. Asthma is an important healthcare issue worldwide. The prevalence of
asthma has increased significantly over the past three decades, affecting 300 million people
worldwide [1, 4]. The highest prevalence of asthma has been reported in the United Kingdom,
New Zealand, Australia, Canada, and the United States [5]. In Canada, 8.5% of the nation’s adult
(approximately 2.5 million people) and 10% of the child population have been diagnosed with
asthma [6].
The recent increase in asthma prevalence that affect all age and ethnic groups, presents a
global health burden [7]. Worldwide, deaths from asthma have reached over 250,000 annually. In
Canada, approximately 20 children and 500 adults die each year from asthma [8]. In addition,
asthma poses a significant economic burden due to direct medical costs such as hospital
admissions, and indirect costs, such as absenteeism from school. The financial burden on patients
with asthma in Western countries varies from $300 to $1,300 per patient annually, proportionally
affecting those with the most severe disease [5].

2
2
1.1.1.1. Etiology and triggers
The factors that determine the risk of developing asthma, asthma severity and response to
therapy can include host and environmental factors [7, 9, 10]. The host factors include genetics,
sex and obesity. Important environmental factors include allergens, airborne pollutants and
respiratory infections [10].
Family and twin studies have indicated that genetics plays an important role in the
development of asthma and allergy [11]. These studies demonstrated that asthma has significant
genetic contribution. Variation at the 17q21 asthma locus, encoding the ORMDL3 and GSDML
genes, is specifically associated with risk for childhood onset asthma [7]. Individuals of African
descent have been reported to suffer from more severe asthma than individuals of European
descent; IgE levels in this population group are lower and associated with a higher degree of
corticosteroid dependency [12].
Asthma has a higher prevalence in boys than in girls. This gender difference has been
demonstrated for children younger than 14 years old (i.e. prior to puberty). However, once
adulthood has been reached, a greater prevalence of asthma has been found in women than men
[10, 13].
Additional studies suggest that obesity is associated with increased severity of asthma and
allergic inflammation [14-16]; obesity related asthma phenotype appears to be predominantly non-
Th2 mediated [15].
With respect to external factors, exposure to allergens has been shown to be one of the
strongest determinants of asthma [17]. Different studies suggest a relationship between exposure to
aeroallergens associated with domestic cat and dog [18, 19], cockroaches [20], house dust mite
[21], molds [22] and pollen [23, 24] in development of asthma, especially in children. Infections in
early childhood have also been shown to be associated with development of asthma later in life.

3
3
Colonization of the airways with Streptococcus pneumoniae and Haemophilus influenzae, in
neonates without symptoms at 1 month of age has been associated with increases in reversibility of
airway resistance and development of asthma by the age of 5 years [25]. Published studies have
found a potential association of early respiratory syncytial virus (RSV) and human rhinovirus
(HRV) infections and subsequently risk for asthma [26-29].
There is emerging evidence that air pollution is an environmental factor that influences
pathogenesis of asthma. Many epidemiology studies have revealed that exposure to air pollutants,
such as particulate matter (PM) and ozone (O3), increases the risk of developing asthma de novo
[30, 31] and exacerbations of established asthma [32, 33]. A recent study in our laboratory showed
that a combined exposure of PM2.5 and O3 significantly increased airways hyperresponsiveness
(AHR) to methacholine (MCh) in a chronic murine model of asthma [34]. Cigarette smoking is an
important air pollutant risk factor for the development of asthma cases in adults with allergic
rhinitis. Also, older children and adults who are active smokers, have more severe symptoms
compared with asthmatic non-smokers [17, 35]. Children or young adults who have smoker
parents or friends (i.e. passive smokers) are more likely to have asthma symptoms[35, 36].
The likelihood of developing asthma might be determined in utero by maternal diet and
lifestyle. Previous studies evaluating the relationship between maternal diet during pregnancy and
asthma in the children have found a protective effect of higher intake of vitamin E, vitamin D,
zinc, selenium and iron, and higher consumption of fish and apples [37]. Breast-fed children have
been reported to be less likely to develop asthma compared to children fed with alternative milk
products [38].

4
4
1.1.1.2. Diagnosis
A diagnosis of asthma is made on the basis of the respiratory signs and symptoms
including cough, wheeze, chest tightness and dyspnea, in addition to laboratory tests such as
spirometry or peak expiratory flow monitoring. Spirometry is recommended for all patients to
confirm the diagnosis of asthma before initiation of therapy; however, it is especially relevant for
patients whose symptoms are not typically characteristic of asthma [39]. This technique evaluates
airflow limitation and reversibility using a forced expiratory maneuver using a spirometer to
measure forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) [39, 40].
Flow reversibility is used to characterize rapid improvements in FEV1 (i.e. minutes) following
inhalation of a rapid-acting bronchodilator. FEV1 reversibility may be used to diagnose asthma; a
≥12% or 200 ml change in FEV1 from the pre-bronchodilator value is generally accepted to be
indicative of an asthmatic response [40]. However, most asthmatic patients will not exhibit
reversibility at each assessment, particularly those on a treatment. For these patients, repeated
testing at different visits is conducted [40, 41]. Reduction in FEV1 can be seen in many lung
diseases. Therefore it is important in asthma to assess the ratio of FEV1 to FVC. The FEV1/FVC
ratio is considered to be normal if it is greater than 0.75 to 0.80, and greater than 0.90 in children.
Any values less than these suggest airflow limitation [40].
For patients with normal lung function but asthma symptoms, diagnosis can be confirmed
by airway responsiveness to direct or indirect airway challenges. Inhaled methacholine and
histamine [39, 41] are traditionally used for direct challenges, while inhaled mannitol [42] or
exercise are used for indirect challenges [40, 43].
The strong association between asthma and allergic rhinitis has suggested that patients with
respiratory syndromes may be more susceptible to developing asthma [40]. Consequential,
allergen levels may be evaluated using commercial assay kits in an effort to diagnosis asthma.

5
5
Endotoxin and proteases may also be screened but these screens are not generally used [44].
1.1.1.3 . Treatment
The main goal for asthma treatment is to reach and maintain clinical control. Asthma
medications are classified in two groups: controllers and relievers. Controllers are used daily over
a long period of time and generally have anti-inflammatory properties, while relievers act quickly
to reverse bronchoconstriction and are used as needed [40]. These medications can be delivered by
inhalation, orally or by injection. Inhaled therapy is preferred due to lower risk of adverse
reactions [40].
Glucocorticosteroids, potent anti-inflammatory agents, are the most effective drugs for
treatment of asthma [40, 45, 46]. Inhaled corticosteroids are the mainstay of asthma therapy which
act to decrease asthma symptoms, control airway inflammation, decrease airway
hyperresponsiveness, reduce frequency and severity of exacerbations and improve lung function
[40].
Bronchodilator therapies include β2-agonists and anticholinergic agents are also effective
for asthma [40]. Anticholinergics such as tiotropium bromide act as antagonists of muscarinic
receptors, and block the effects of endogenous acetylcholine. Although not as an effective
bronchodilator as β2-agonists, anticholinergics may have some additive bronchodilator effect,
especially in patients with severe asthma [45].
Leukotriene modifiers such as cysteinyl-leukotriene 1 receptor antagonists and 5-
lipoxygenase inhibitor reduce asthma symptoms, airway inflammation, and exacerbations and
improve lung function. These medications are used for mild asthma or as add-on to inhaled
glucocorticosteroids [40, 45], particularly for patients whose symptoms are triggered by allergies.
These drugs are used orally and appear to be safe [45].

6
6
Theophylline is a bronchodilator that has moderate anti-inflammatory effect when used in
lower dose [40]. Theophylline has been used for many years in the treatment of asthma.
Theophylline is still widely used in developing countries because of the low price. However, the
frequency of adverse effects and the low potency of theophylline have recently led to decreased
usage in many countries [45]. Theophylline still remains a very useful add-on therapy in patients
with severe asthma [40, 45].
Recently Omalizumab has been licensed for use in severe allergic asthma. Omalizumab is
used as an adjunctive therapy for patients ≥12 years of age who have allergies and severe
persistent asthma. It is a recombinant DNA-derived humanized IgG1 monoclonal antibody that
selectively binds to free and membrane-bound immunoglobulin E (IgE) antibodies [47].
Unfortunately, the high cost associated with this drug has limited its applicability in many
countries.
1.1.2. Airway inflammation in asthma
Airway inflammation contributes to the pathogenesis of asthma by affecting airflow
through the production of mucus, release of inflammatory mediators, and by enhancing
susceptibility to bronchospasm [48]. A variety of cells such as dendritic cells (DCs), T
lymphocytes, eosinophils, mast cells, neutrophils, and macrophages are involved in the
development of airway inflammation in asthma [49, 50]. These cells are reviewed below.
1.1.2.1. Dendritic cells
The role of antigen-presenting cells (APCs) such as DCs in the pathogenesis of asthma has
been well studied [2]. Resident dendritic cells in the airway epithelium and mucosa recognize and

7
7
bind to inhaled antigens, and subsequently migrate to lymph nodes to initiate an antigen-induced
immune response [51]. DCs present antigens to naive T-cells and induce expression of selective
cytokines. The presence of DCs consequentially leads to activation and differentiation of T-cells to
T helper 2 (Th2) subtype and contributes indirectly to the development of airway inflammation [2,
49].
1.1.2.2. T lymphocytes
There is considerable evidence to suggest elevated levels of T lymphocytes are
characteristic of asthma [48-50]. CD4+ T-cells, particularly the Th2 subtype, play a pivotal role in
pathogenesis of asthma by producing interleukin (IL)-4, IL-5, IL-9 and IL-13 [2, 48, 49]. Of these
mediators, IL-4 and IL-13 are implicated to play special role in development of airway
hyperresponsiveness, eosinophilic inflammation [2, 52], goblet cell metaplasia, macrophage
activation, smooth muscle remodeling, and airways fibrosis [52].
The Th17 subtype is another CD4+ T helper that has been shown to have a role in the
development and pathogenesis of asthma. IL-17A is expressed in the airways of asthmatic patients,
and has been correlated with neutrophil recruitment in the airways and severity of asthma [53, 54].
Another cytokine produced by Th17 lymphocytes, IL-23, has been shown to be crucial for the
maintenance of Th17 cells and enhances Th2 cell–mediated eosinophilic airway inflammation
[54].
1.1.2.3. B lymphocytes
B lymphocytes are involved in the humoral immune response through production of IgE,
an important effector of the allergic response [55]. This cell type is also capable of recognizing

8
8
specific antigens which are then presented to T cells via the major histocompatibility complex
(MHC) class II molecule [55]. The production of IL-4 and IL-13 by T lymphocytes stimulates
switching class from IgG antibodies to IgE antibodies [56].
1.1.2.4. Mast cells
The numbers of mast cells present in airway wall and airway smooth muscle of asthmatic
patients are elevated compared to non-asthmatics [2]. Mast cells express the FcεRI receptor, which
is a high affinity surface receptor for IgE [2, 57]. Binding of IgE to FcεRI receptor initiates mast
cell activation [2, 58]. Inducing cascades that lead to degranulation and cytokine release from these
mast cells contribute to the development and maintenance of allergic inflammation [2].
1.1.2.5. Basophils
Basophils, the least abundant of circulating granulocytes, are elevated in sites of allergic
inflammation. This cell type is similar to mast cells in many ways: both cells express IgE receptor
FcεRI and upon receptor activation both cells rapidly produce cytokines, histamine, and lipid
mediators [59, 60]. Basophils, however, also produce IL-4 and IL-13 after allergen challenge and
thus promote the development of the Th2 based immune response [60].
1.1.2.6. Eosinophils
Increased numbers of eosinophils in peripheral blood and in airway secretions are
characteristics of asthma [49, 61]. Th2 cytokines such as IL-4, IL-5, and IL-13 induce airway
eosinophilia in animal models of asthma [61]. Eosinophils are an abundant source of granule basic

9
9
proteins like major basic protein (MBP), eosinophil peroxidase, eosinophil cationic protein and
eosinophil-derived neurotoxin [58, 60]. In addition, this cell type is also capable of producing
eicosanoids such as prostacyclin (PGI2) and leukotrienes, superoxide, and a range of cytokines and
chemokines [60].
1.1.2.7. Macrophages
Macrophages are derived from circulating monocytes [58, 60, 62] and play a role in
enhancing inflammatory responses [60]. Although these cells are an important source of
leukotrienes, reactive oxygen intermediates, and a variety of lysosomal enzymes, their role in
mediating tissue damage and the overall airway pathology in asthma is largely unknown [60].
1.1.2.8. Airway epithelium
The airway epithelium plays an important role as a physical barrier as the first line of direct
contact of inhaled particles, toxins and other triggers [60, 63]. In addition, it is now recognized that
airway epithelium has a major role in orchestrating the inflammation that develops in asthmatic
airways [63]. Environmental or mechanical stress factors can trigger the airway epithelial to
release mediators such as chemokines, cytokines and growth factors [57, 64, 65]. The airway
epithelium also facilitates the selective migration of leukocytes into the airway lumen [63]. It is
also the primary source of inflammatory mediators those are produced in response to HRV, a cause
for asthma exacerbations. The Chow group described the mechanisms that regulates entry of HRV
into airway epithelium and moreover has demonstrated the airway epithelial inflammatory
response to HRV [66, 67]. HRV has been shown to activate spleen tyrosine kinase (Syk) with
subsequent Syk-mediated cell signaling that then leads to the expression of inflammatory

10
10
mediators, such as IL-8 and VEGF [29, 66, 68].
1.1.2.9. Airway smooth muscle
Airway smooth muscle (ASM) cells are the main structural cells within the bronchi [64].
These cells regulate airway resistance through contraction [64, 69]. In some conditions, airway
smooth muscle cells have been shown to synthesize cytokines, growth factors, and adhesion
molecules, which may contribute to the inflammatory responses [69].
1.1.2.10. Neutrophils and asthma
Neutrophils play a key role in the innate immune response and act as the first line of
protection against microorganisms [70]. Neutrophils accumulate in the airway in more severe
forms of chronic asthma. Elevated levels of this cell type are associated with chronic airway
narrowing and are characteristic of acute severe asthma exacerbations [60, 61].
Neutrophils role in the inflammation was once thought to be limited to phagocytosis and
the release of enzymes and other cytotoxic agents. It is believed now that these cells can release
diverse mediators that have effects on the airways of asthmatic individuals. Metalloproteinases,
elastase, lactoferrin, myeloperoxidase, adhesion molecules, reactive oxygen species, and Lipid
Mediators are considered to be some of important ones [70, 71].
In addition, like other cell types such as lymphocytes, macrophages, and NK cells,
neutrophils are able to synthesize and release a great variety of cytokines that play a key role in the
development of the immune response including IL-1, IL-3, IL-6, IL-8, TNF-α, IL-12, and TGF-ß
[70]. IL-8 production from them contributes to an additional recruitment of these cells in the
airway [71]. It is also suggested that neutrophils may be responsible for the activation/release of

11
11
eosinophils in IgE-mediated processes [72].
In a recent study, it has been shown that both neutrophils and eosinophils are key cellular
players in a mouse model of chemical-induced asthma. They showed that depletion of both cells
prevented AHR, lung epithelial damage and also significant reduced in airway inflammation [73].
1.1.3. Airway hyperresponsiveness in asthma
AHR, the excessive response of the airways to a variety of stimuli [74], is seen in nearly all
individuals with asthma and is characterized as a functional abnormality [75]. Asthma severity is
commonly taken to be proportional to AHR [40, 76]. AHR results in airway narrowing in an
asthmatic patient in response to a stimulus that is harmless in a non-asthmatic person [40]. The
mechanisms of AHR are not completely understood [74, 77]; ASM abnormalities, airway
inflammation, airway remodeling, neural control and involvement of mediators have all been
reported to have some involvement in the response [74]. There are two components of AHR,
which may have different regulation/pathogenesis mechanisms. These two components are
baseline and variable AHR [77]. Baseline AHR is relatively persistent and is seen in the majority
cases of chronic asthma. It can be influenced by intermittent exposure to environmental factors
such as allergens, air pollutants or viral respiratory tract infections [77]. Baseline AHR is
suspected to reflect structural changes in the airways or airway remodeling. In contrast, variable
AHR relates to current airway inflammation linked to asthma activity and severity [75-77].
It is believed that both airway remodeling and chronic AHR are the results of chronic or
prolonged airway inflammation [77]. The link between variable AHR and inflammation of the
airways appears to be well established. In allergic asthma, many studies have shown a direct
association of allergen-induced acute changes in airway responsiveness with Th2-driven
eosinophilic airway inflammation [77].

12
12
Airway remodeling or structural alteration in the airways occurs during growth or in
response to injury and/or inflammation [78]. Structural changes lead to overall thickening of the
airway walls in asthma [74]. Increase in airway vascularity [79], basement membrane thickness
[80] and total wall thickness [80] have been shown to be associated with increase in AHR.
The smooth muscle mass that surrounds the airways and controls the luminal diameter is
thicker in asthmatics [77, 81]. The thickness of the ASM has been suggested to be related with the
level of asthma severity [82]. The exact relation between ASM thickening and AHR is not entirely
clear; however, increases in ASM mass resulting from allergen sensitization and challenge are
associated with AHR in mouse models in vivo [75]. In addition, studies show that ASM mass is
likely the most important factor influencing the increased resistance seen in response to broncho-
constricting stimuli in asthmatic patients [83]. These alterations in airway structure are exacerbated
as the disease progresses, which show the chronic increase in severity of airways narrowing [84].
Elevated ASM cell numbers (hyperplasia) and cell size (hypertrophy) has been extensively
studied in cell culture and animal models of asthma. In vitro models of bronchial smooth muscle
hypertrophy have shown increases in smooth muscle α-actin content after induction by
transforming growth factor-β1 (TGF-β1) or cardiotropin-1. This rise in smooth muscle mass
corresponds to increased cell shortening in response to acetylcholine [85].
Increased contractility of bronchial smooth muscle has been considered as one of the
causes of the AHR. Two pathways mediate contraction of airway smooth muscles: Ca2+-dependent
and –independent. Contraction of airway smooth muscle is initially regulated by Ca2+-dependent
mechanisms, initiated by a rapid increase in intracellular Ca2+ concentration, followed by the
formation of Ca2+ complexes that activate myosin light chain (MLC) kinase, eventually resulting
in phosphorylation of the 20-kDa regulatory MLC [77]. A monomeric GTP-binding protein,
RhoA, and a downstream target Rho-kinase regulates the Ca2+-independent pathway. In animal

13
13
models of allergic bronchial asthma, an augmented agonist-induced, RhoA-mediated contraction
of bronchial smooth muscle has been proposed. RhoA/Rho-kinase signaling has been suggested as
a novel target for the treatment of AHR in asthma [86, 87]. Moreover, a strong relationship
between exhaled nitric oxide (eNO) and AHR to methacholine has been demonstrated in various
studies [88].
The metabolism of L-arginine plays a critical homeostatic role in the airways by synthesis
of bronchodilating nitric oxide (NO) from L-arginine by enzymatic activity of the [89] nitric oxide
synthase (NOS) [90]. The release of NO is decreased during airway inflammation as a result of
attenuated L-arginine bioavailability to NOS in the airway epithelium. Furthermore, low
concentration of L-arginine results in elevated formation of pro-contractile and pro-inflammatory
perroxynitrite (ONOO-). Two major mechanisms have been involved in allergen induced NO
deficiency [77].
Reduced L-arginine bioavailability can be the result of increase in expression of arginase in
response to Th2 cytokines: IL-4 and IL-13 [77]. The arginase isozymes containing arginase 1 and
arginase 2, convert L-arginine into L-ornithine and urea, and compete with the NOS for the
substrate [90, 91]. These enzymes are both expressed in the airways, primarily by epithelial cells,
fibroblasts and alveolar macrophages [92]. It has been shown that expression of arginase is up-
regulated in human asthma [93] and that the arginase isozymes, particularly arginase 1, play a
functional role in the AHR in animal models of asthma, using ovalbumin (OVA) [93] and house
dust mite [1]. In addition, animal studies have demonstrated that polycationic proteins such as
MBP from eosinophils interfere with cellular uptake of L-arginine by amino acid transporters [94].
The parasympathetic nervous system controls a major bronchoconstrictor pathway. Both
ganglionic and post-ganglionic cholinergic nerve activity mediate basal airway smooth muscle
tone. Acetylcholine is the major neurotransmitter in both activities. The fidelity with pre-

14
14
ganglionic impulses are translated into post-ganglionic action potentials is relatively low. The
filtering function of these ganglia keep the translation of pre-ganglionic impulses into post
ganglionic relatively low [95]. Select inflammatory mediators, such as histamine, prostaglandin D2
(PGD2) and bradykinin, decrease this filtering function, and as a result, increase ganglionic
cholinergic transmission [77].
In vitro and in vivo studies have investigated the mechanisms of acute and chronic AHR.
Many mechanisms are associated with the release of mediators, growth factors and
neurotransmitters that change airway smooth muscle function.
1.1.4. Airway remodeling in asthma
1.1.4.1. Features of airway remodeling
Airway remodeling causes changes in structural cells and tissue in patients with asthma
compared with healthy individuals. These alterations were found to contribute in the pathogenesis
of asthma [64, 96]. These modifications include changes in the composition and organization of its
cellular and molecular constituents [64, 96].
Structural changes of the airway include epithelial alterations, subepithelial fibrosis,
increased ASM mass, mucous gland and goblet cell hyperplasia, vascular changes mostly around
large airways and edema [64, 96, 97]. It has been shown that patients who suffer from severe
asthma (5% to 10% of asthma cases) experience an earlier disease onset and develop chronic
persistent level of airflow limitation [98]. These findings provide further evidence that airway
remodeling plays a critical role in the impairment of lung function [64, 96].
Morphologic alterations in airway epithelium are considered to be a key feature of airway
remodeling in patients with asthma. These alterations include epithelium shedding, loss of ciliated
cells, goblet cell hyperplasia, and also up-regulation of cytokines, chemokines and growth factors

15
15
[64]. It has been suggested that barrier function of the airway among asthmatic patients is
impaired, and shows breakdown in epithelial tight junction [64].
Increased mucin secretion, such as MUC5AC and MUC5B, by goblet cells is a feature of
airway remodeling in asthmatic patients. The origin of the goblet cells is not well known; however,
Clara and ciliated cells have been implicated in goblet cell origin [99]. Production of IL-9, IL-13
and IL-1β, in addition to their associated intracellular signaling pathways are involved in the
process of developing goblet cells and also up-regulation of mucin synthesis [64].
Sub-epithelial fibrosis is another structural alteration associated with airway remodeling
found in asthmatics. Fibroblasts, which reside close to the basal epithelium, are large and flat cells.
They are activated and differentiated to myofibroblasts during inflammation. Myofibroblasts
secrete pro-inflammatory mediators and also extracellular matrix (ECM) proteins in such
inflammatory environment [64, 89]. The ECM compartment of the airways are regulated by matrix
metalloproteinases (MMPs) and tissue inhibitor metalloproteinase (TIMPs) [89]. A shift in balance
between these enzymes alters the structure and increases matrix deposition that leads to fibrosis.
There is evidence to support that persistent activation of fibroblasts results in subepithelial fibrosis
in asthmatic patients [100].
ASM cells are the main structural cells in the airways. Remodeling in ASM is counted to
be the main reason for obstruction of airways in asthma [64, 101]. ASM cell proliferation
(hyperplasia) and cell size (hypertrophy) are considered to be reasons for increase in ASM mass
[101]. Remodeling of the ASM is also associated with changes in the phenotype of the airway
muscle cells that facilitate proliferative, synthetic and contractile functions [101]. In addition, the
migration of these cells towards the epithelium is another important factor [64, 102]. Additionally,
ASM cells express cellular adhesion molecule (CAMs), toll like receptors (TLRs), cytokines,
chemokines and also cytokines receptors [64]. Numerous mediators, including TNF-α, IL-1 and

16
16
INF-ϒ, can increase expression of intracellular adhesion molecule 1 (ICAM-1) and vascular cell
adhesion molecule 1 (VCAM-1) on ASM. Using these adhesion molecules, ASM cells can
regulate the interaction between different inflammatory cells [64].
Different studies have indicated unusual increases in size and number of microvessels or
angiogenesis within airway tissue in remodeled airways, especially in the space between the
muscle layer and the encircled parenchyma [64, 103]. Studies have shown that vascular endothelial
growth factor (VEGF) and angiopoietin-1 are involved in angiogenesis. VEGF has further been
shown to increase the permeability of unusual blood vessels. This results in vessel dilation and
edema, which are involved in airway narrowing. These affected blood vessels can be the source for
inflammatory cells and also mediators [103].
1.1.4.2. Mechanisms of airway remodeling
Inflammation is considered to be the main reason behind most features of airway
remodeling in asthma. A variety of chemokines, cytokines and growth factors are released from
inflammatory and structural cells leads to a complex signaling environment in airway tissue and
consequently airway remodeling [104].
It has been shown that IgE and mast cells are involved in the acute response and
eosinophils with their highly basic granules in the late response, along with T cells, especially Th2
cells, which regulate responses through producing and releasing of cytokines, like IL-4, IL-5, IL-9,
and IL-13 [64]. Th2 cells are considered to be central to inducing airway inflammation in
asthmatic patients. These cells are important for IgE synthesis, chemokines and cytokine
production, eosinophilia in airways, ASM hyperplasia and production of mucus [64, 105]. In
addition to the central role of Th2 cells in pathogenesis of mild to moderate asthma, Th1 cells also
play a role in asthma progression; the presence of Th1 cells are associated with more chronic and

17
17
severe forms of asthma, likely due to the secretion of IFN-γ, which inhibits Th2 cell proliferation
[106]. Another subset of T helper cells, Th17 has been found in asthmatic patients with severe
disease. Studies have shown over-expression of IL-17 mRNA in a mouse model of asthma [107].
Eosinophils are another important cell type involved in tissue remodeling in asthma. This
cell type is considered to be the main source of the TGF-β, a pro-fibrotic cytokine, which
orchestrates remodeling [108]. In addition, eosinophils are also involved in activities such as
proliferation of fibroblasts, maturation of myofibroblasts and collagen synthesis [64]. In subjects
with asthmatic airways, eosinophils develop from CD34+ bone marrow precursor cells. IL-13,
granulocyte macrophage colony-stimulating factor (GM-CSF) and eotaxins modulate their
development while IL-5 increases their maturation and recruitment into the airways. Moreover,
eosinophils are an abundant source of granule basic proteins, eicosanoids, cysteinyl leukotrienes,
tissue-damaging reactive oxygen species, and different cytokines and chemokines [64].
Epithelial injury also happens as part of remodeling in asthmatic patients. The epithelium
layer in bronchi is considered to be a physical barrier, which keeps the internal milieu of the lung
safe against external factors [105, 109]. Several studies have suggested that severe injuries in the
airway epithelial layer allow environmental microorganisms, allergens, and toxins to have access
to airways [109, 110]. An injured epithelial layer has been shown to be associated with a reduced
repair process that drives inflammatory and consequently remodeling responses in the underlying
sub-mucosa in asthmatic patients [111]. Environmental factors such as pathogen microorganisms,
allergens, air pollutants, cigarette smoke, and/or physical stress can cause injury in the epithelial
layer and trigger release of inflammatory mediators that contribute to airway remodeling from
epithelial cells [111].
Different inflammatory mediators, involved in remodeling, such as TGF-β [112] and
chemokines, are released from repairing/damaged epithelium or in response to other inflammatory

18
18
mediators. It has been shown that these mediators play a crucial role in the formation of
subepithelial fibrosis and also increase in ASM mass [108].
Cell-cell interactions mediate some structural changes of airways and activate
inflammation, which initiates airway tissue remodeling. Mast cells have been shown to activate the
release of IL-6 from fibroblasts by direct contact of the cells [64]. Additionally, it is suggested that
CD4+ T cells increase ASM proliferation through cell-cell interactions in vivo [64]. However,
Lazaar et al. showed that an interaction between activated T lymphocytes and cultured ASM,
which mediated by ICAM-1, VCAM-1, and CD44 on ASM cells, resulted in up-regulation of cell
adhesion molecules and increasing of DNA synthesis in ASM cells [64].
Moreover interactions between inflammatory cells, such as mast cells, eosinophils, and
neutrophils with ASM cells through ICAM-1 and VCAM-1 have been shown [64]. These studies
have suggested that interactions between ASM and inflammatory cells by CAMs play part in
airway remodeling in patients with asthma.
Inflammatory mediators, including growth factors, cytokines, and chemokines produced
and released by structural and inflammatory cells are considered to have key roles in inducing and
synchronizing remodeling of airway. Different remodeling inflammatory mediators, such as
profibrotic cytokines (TGF-β and IL-11), Th2 cytokines (IL-4, IL-5, IL-9, and IL-13), Th17
cytokines (IL-17A, IL-17E {IL-25}, and IL-17F), epithelium-derived chemokines (RANTES,
macrophage inflammatory protein-1α {MIP-1α}, IL-8, and eotaxins), and MMPs have been
identified [113].
TGF-β, a pleiotropic cytokine, has different functions depending on the environment and/or
cellular conditions. These functions include differentiation, apoptosis, survival and proliferation.
Moreover, this cytokine has been involved in the development of several diseases such as cancer

19
19
and asthma [114]. Although TGF-β is produced and released by many cell types, eosinophils are
considered as one of the most important sources for that in patients with asthma [64, 112, 114].
TGF-β has effect on many structural cells both in vitro and in vivo and is involved in remodeling
process in asthma or other inflammatory lung diseases [115]. TGF-β assists fibroblast
differentiation to myofibroblasts and thus promotes expression of MMPs and TIMPs, which are
involved in ECM turn over. It also affects the proliferation of ASM cells through activation of
MAP3 kinase pathway [64] and the migration of ASM cells towards the epithelium [116].
IL-11, another profibrotic cytokine, is also involved in remodeling in asthmatic patients
such as airway wall thickening, sub epithelial fibrosis and proliferation of ASM cells [64].
Th2 cytokines have been shown to be involved in asthmatic patients airway remodeling.
Each of these cytokines has different functions. For example, IL-4 regulates synthesis of allergen
specific IgE, IL-5 is involved in eosinophil recruitment [112], IL-9 helps recruitment of mast cells
while IL-13 regulates AHR [117]. IL-6 and TGF-β are both involved in the differentiation of T
helper cells to Th17 cells. IL-17, especially IL-17A, IL-17F, and IL-17 E, is critical in the immune
response in patients with severe asthma [64]. Additionally, other cells such as natural killer T cells
(NK cells), neutrophils and macrophages have ability to produce IL-17, which is a powerful
neutrophil chemotactic agent [118]. IL-25, or IL-17E, is produced by epithelial cells [118], as well
as activated eosinophils, mast cells and basophils, after exposure to aeroallergens [119].
In addition, chemokines have been shown to play a main role in the development of airway
remodeling. They can be expressed and released by different cell types in the lung, such as
epithelial and ASM cells in asthmatic patients. The main role of these chemokines is to recruit
inflammatory cells to the site of inflammation. They can also mobilize airway structural cells and
consequently result in airway remodeling during asthma [64]. Epithelium-derived CC and CXC
chemokines can be bind to their receptor on ASM cells, which stimulate migration of these cells

20
20
toward the epithelium [120]. Studied which have used mouse model of allergic asthma have shown
that neutralization of certain chemokines, such as RANTES, decrease leukocyte recruitment as
well as AHR [120]. CCR-3 is a chemokine receptor, which is expressed by eosinophils. This
receptor mediates chemotaxis in response to chemokines such as RANTES and eotaxin. Studies
have shown that CCR-3 and eotaxin deficient mice exhibit lower levels of airway eosinophilia and
also production of mucus [112]. However, more research is needed to determine the physiologic
contributions of the epithelium in the pathogenesis of asthma and to identify novel therapeutic
targets able to protect the airways from asthma triggering environmental factors.
1.2. Animal models of asthma
For many years, animal models have been used extensively to study the cause and
treatment of asthma. These can be utilized for the identification of potential drug targets and
evaluate effectiveness and safety of new drugs [77].
In vitro systems for studies on AHR, airways remodeling, and inflammation in asthma,
typically involve mammalian airway smooth muscle or airway epithelial cells. [121]. In vivo
animal models of asthma utilize both non-rodent and rodent species. Non-rodent models are not
commonly used as they are expensive and pose increased technological and ethical challenges.
Prior to the rapid increase of mouse molecular technologies, the guinea pig and rat were the most
commonly used animal models of asthma [122]. Caveats associated with these older models
include: the extreme nature of responses (guinea pig), and the need for high doses of broncho-
constrictors to elicit reactions (rat) [123].

21
21
1.2.1. Mouse models of asthma
Mouse models of allergic asthma have been developed to study the key features of asthma:
inflammation, airways remodeling, and AHR [77, 91, 124]. The availability of transgenic animals,
as well as the variety of mouse-specific immunological tools that can be used for phenotypic and
functional analysis of cells and mediators, are partly responsible for their use in this field. In
addition, lower maintenance costs and shorter gestation periods have further contributed to mice
being the most used species in asthma model research [77].
Mice can be easily sensitized to variety of antigens, such as ovalbumin [57, 91] and also
human allergens, including house dust mite [1, 50, 125]. Intraperitoneal or intranasal sensitization,
and subsequent inhalational or intranasal challenge with these antigens, results in a Th2 response
in the airways, characterized by the production of antigen-specific IgE, eosinophilia and AHR.
These responses are considerably varied between different strains of mice [123, 125] and exposure
protocols. Different exposure protocols determine development of different features of the asthma
phenotype; for instance, short-term exposures (acute models) are useful for studying the immune-
mediated features, while long-term exposures (chronic models) result in airway remodeling [126].
Therefore, it is essential to choose the appropriate murine model for the specific human phenotype
or pathogenic mechanism of interest.
1.2.2. House dust mite (HDM) model of asthma
1.2.2.1. HDM and asthma
Exposure to different allergens in sensitized individuals can trigger asthma attacks. House
dust includes a variety of different allergens; however, the main allergen is derived from mites
[127], which belong to taxonomical subclass Acari, with more than 50,000 species [128]. The
European HDM, Dermatophagoides pteronyssinus (Dp), and American HDM, D. farinae (Df), are

22
22
considered the most common aeroallergens [128]. They typically reside in indoor environments
such as houses; especially beds where pillows, duvets, and mattresses often serve as allergen
sources. Carpets and furniture also contain high levels of mite [127]. A male HDM lives for
between 10 and 19 days, while a mated female HDM lives up to 70 days and has the ability to lay
60 to 100 eggs in its last 5 weeks of the life. The HDM thrives in all climates, even at high altitude,
but grow better in a relative humidity of 50-60% [129].
An allergy to HDM is considered one of the most prevalent causes of allergic sensitization
and asthma. More than 20 proteins from HDM have been identified as allergens, including
structural proteins and various enzymes. However, these allergens are inhaled with a variety of
bacterial products, such as lipopolysaccharide (LPS), which may function as adjuvants for the
development of sensitization to HDM allergens [130].
In spite of their geographical differences, up to 85% of asthmatic patients in highly
populated areas of North and South America, Europe, South East Asia, and Australasia are allergic
to HDM. Diagnosis and immunological treatment of HDM allergy is carried out using HDM
extracts from their bodies, feces and other mite products[131].
1.2.2.2. HDM models of allergic airways inflammation
Findings from house dust mite (HDM) mouse models of airway inflammation have shown
airway eosinophilic inflammation, with increases in BALF levels of Th2-associated cytokines, and
increases in serum levels of Th2-associated immunoglobulins [50, 125, 132], proving its aptness
for modeling the physiological features of airways inflammation.
There are a number of key differences between the HDM model of allergic airway
inflammation, and allergic airway inflammation in humans. Generally, ovalbumin (OVA) has been
used in most experimental models of allergic airway inflammation, although it is not considered a

23
23
common human airways allergen. Most experimental animal models of allergic asthma have been
developed by systemic sensitization, using either intraperitoneal or subcutaneous injection with
adjuvant, which is not how human allergen elicits a response [125].
Recently, researchers have developed a mouse model of allergic asthma using local
exposure of HDM, a clinically relevant allergen, without adjuvant to mimic the sensitization route
in humans. This model of asthma uses an intranasal HDM exposure route, and has been used to
characterize airway eosinophilia and AHR [50, 133]. A study by Shibamori et al, induced allergic
asthma-like responses in mice by short-term administration of Df and Dp intranasally. In the acute
model, they observed BALF eosinophilia, increase in AHR, and increase in Th2 chemokines and
cytokines following intranasal instillation of Df and Dp in 5 different mice strains; of those, the
NC/Nga mice strain showed the strongest response [125]. Continuous and intermittent exposure to
HDM was also investigated, and they observed chronic airway inflammation, AHR, and structural
remodeling [50, 75]. Takahashi et al. developed a HDM acute model of airway inflammation to
investigate the role of arginase in AHR using arginase inhibitor; nor-NOHA [1]. The role of Th2 in
asthmatic responses was also investigated by intranasal instillation of Df in mice. Significant
increases in recruitment of inflammatory cells, AHR, and Th2 cytokine and chemokine levels were
found in the HDM group, relative to the control group [132].
1.3. Spleen tyrosine kinase (Syk)
Spleen tyrosine kinase (Syk), a member of the cytoplasmic protein tyrosine kinase (PTK)
family, is an important signaling mediator in biological pathways. Syk was purified and described
initially by Geahlen et al. [134, 135] and Yamamura et al. [136]. During the 1990s, it was reported
that classical immunoreceptors such as Fc receptors (FcRs), B cell receptors (BCRs), and T cell

24
24
receptors (TCRs) signal by a common mechanism via Syk. They associate with transmembrane
proteins that contain immunoreceptors tyrosine based activation motifs (ITAMs) in their
cytoplasmic domain. This short peptide sequence contains two tyrosine residues that are 6-12
amino acids apart, and is the primary signaling domain used by classical immunoreceptors. [137].
Tyrosine residues of ITAMs are phosphorylated quickly after engagement of these receptors, and
lead to recruitment and activation of either Syk or zeta-chain associated protein kinase 70
(ZAP70), another Syk family protein, consequently orchestrating different cellular processes
[137]. It has been shown that the lack of expression of ZAP70 results in reduced T lymphocyte-
mediated immunity, highlighting the pivotal role of this tyrosine kinase in T cell development and
function [138]. In addition to its expression in hematopoietic cells, Syk is also highly expressed in
non-hematopoietic cells, such as osteoclasts, epithelial cells, fibroblasts, hepatocytes, and neuronal
and vascular endothelial cells [137, 139].
1.3.1. Structure and cellular expression of the Syk family of tyrosine
kinases
Syk contains two SRC homology 2 (SH2) domains, and a carboxy terminal tyrosine kinase
domain, which are connected by Two linker regions: interdomain A and interdomain B,
respectively. An alternative spliced form of Syk, known as SykB, has 23 amino acids less than
normal Syk in its interdomain B; however, the regulatory mechanism that leads to SykB
expression is not completely understood. ZAP70 is a homologue of Syk, which is mostly
expressed by T and NK cells (Figure 1.1.). The tandem SH2 domains are responsible for binding
to ITAM, which initiates a series of cellular responses (Figure 1.2) [137, 140].

25
25
Figure 1.1. Molecular structure of Syk, Syk B and ZAP70
Syk contains two SH2 domains and a kinase domain, connected by two linker regions: interdomains A and B. Alternatively spliced Syk, Syk B, has 23 fewer amino acids in its interdomain B than Syk. The molecular structure of ZAP70 has a similar number of total amino acids to Syk (618 amino acids versus 608, respectively). Adapted from “The SYK 31 tyrosine kinase: a crucial player in diverse biological functions” by Mocsai, A., J. Ruland, and V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature reviews. Immunology, 2010. 10(6): p. 387-402.
26
26
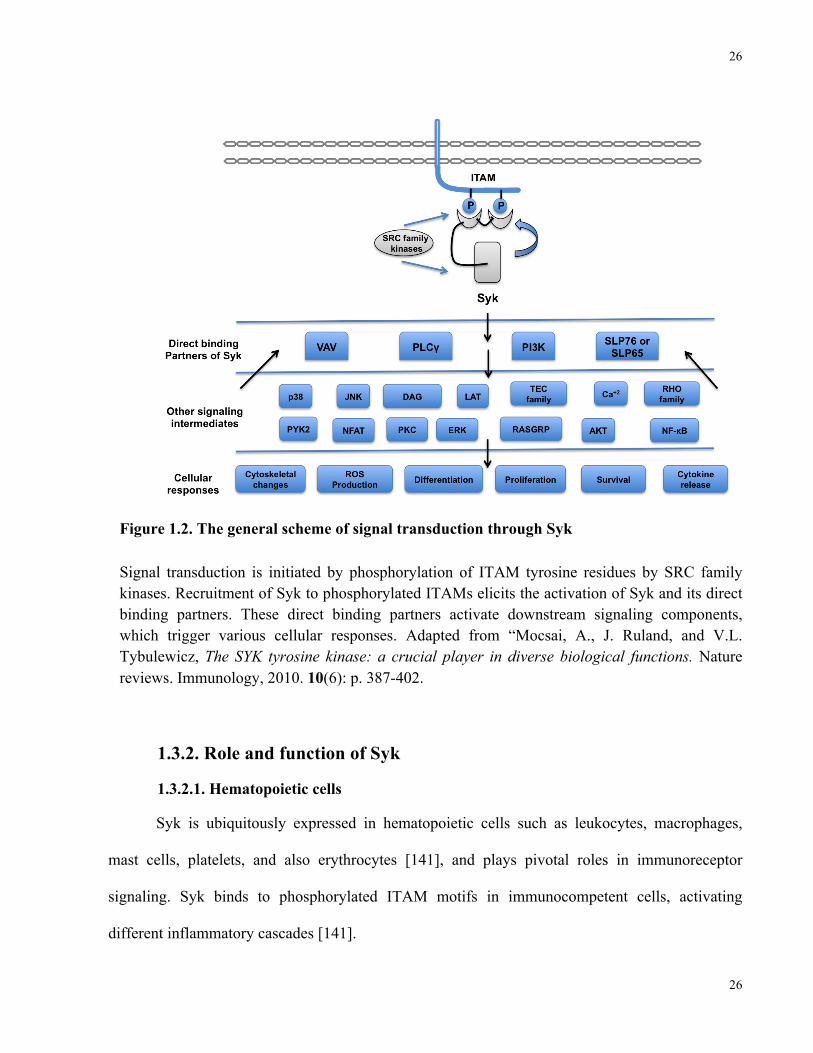
Figure 1.2. The general scheme of signal transduction through Syk
Signal transduction is initiated by phosphorylation of ITAM tyrosine residues by SRC family kinases. Recruitment of Syk to phosphorylated ITAMs elicits the activation of Syk and its direct binding partners. These direct binding partners activate downstream signaling components, which trigger various cellular responses. Adapted from “Mocsai, A., J. Ruland, and V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature reviews. Immunology, 2010. 10(6): p. 387-402.
1.3.2. Role and function of Syk
1.3.2.1. Hematopoietic cells
Syk is ubiquitously expressed in hematopoietic cells such as leukocytes, macrophages,
mast cells, platelets, and also erythrocytes [141], and plays pivotal roles in immunoreceptor
signaling. Syk binds to phosphorylated ITAM motifs in immunocompetent cells, activating
different inflammatory cascades [141].

27
27
Inflammatory responses in allergies have been associated with the ability of locally
produced immune complexes to stimulate inflammatory effector cells, and subsequently, the
production of inflammatory mediators [142]. Activation of Syk causes cell activation by the
increased synthesis and release of inflammatory mediators, which are responsible for acute allergic
responses [141]. Allergies are characterized by increased production of IgE antibodies against
antigens. Binding of IgE to the presented FcεR1 receptors on the cell membrane of basophils and
mast cells aggregates receptors in the presence of antigens, and consequently activates Lyn, a
cytoplasmic Src kinase. The FcεR1 receptor consists of a single α subunit (IgE-binding), a β
subunit, and two γ chains. The cytoplasmic tails of both β and γ subunits contain ITAM motifs,
which can phosphorylate the β subunit of the FcεR1 receptor when bound by Lyn, and recruited by
two SH2 domains of Syk [141].
As well as inflammatory mediator activation, Syk can activate B and T cells by interacting
with antigens on BCRs and TCRs [141]. Syk is crucial for B cell development and function. B
cells are important for producing antibodies against foreign antigens and also presenting antigen
coupled with major histocompatibility (MHC) to T cells as the reason of interaction between BCRs
and antigens [141]. Activation of BCRs leads to development and maturation of B cells, which
express unusual receptors for immunoglobulin, FcγRIIB, a single subunit containing an inhibitory
motif that lacks ITAM. Phosphorylation of this inhibitory motif inhibits activation of Syk (Figure
1.3) [141].
In addition, T cells express an unusual TCR, which is responsible for recognizing antigens
bound to MHC. Antigen and MHC complexes trigger a series of events, which activate resting T
cells, inducing their differentiation to effector T cells. The ITAM motif of the CD3ζ subunit of
TCR interacts with the tandem SH2 domain of ZAP70 (Figure 1.3) [141].

28
28
Figure 1.3. Recruitment of Syk or ZAP70 to plasma membrane receptors
Recruitment of SYK or ZAP70 to plasma membrane receptors takes place through binding of the tandem SH2 domains of SYK or ZAP70 to two phosphorylated tyrosine residues in the receptor complex. The two phosphorylated tyrosine residues are located in a single ITAM or two hemITAMs (separate receptor peptide chains). The ITAMs are present in the cytoplasmic tail of the receptor chain itself or the receptor-associated transmembrane adaptors. Adapted from “The SYK 31 tyrosine kinase: a crucial player in diverse biological functions” by Mocsai, A., J. Ruland, and V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature reviews. Immunology, 2010. 10(6): p. 387-402.
The DCs, as the major antigen-presenting cells, are necessary for differentiation and
activation of T cells. On the surface of immature DCs, FcαR, FcγR and FcεR bind to their
respective antibodies IgA, IgG, and IgE, resulting in their internalization to process the antigens.
Studies have shown that Syk is not involved in dendritic cell differentiation, but it is necessary for
internalization of immune complexes followed by the presentation of antigens to T cells [141].
Macrophages and neutrophils are considered the first inflammatory cells to migrate
towards microorganisms during infections. In macrophages, it has been shown that Syk is crucial
for phagocytosis induced by the IgG-induced activation of FcγR; whereas neutrophils generate

29
29
reactive oxygen substances in response to IgG [141]. Syk is also important for platelet function, in
particular, the induction of some of the receptors on the platelets such as integrin, C-type lectin
CLEC-2, and GPVI receptors [143].
1.3.2.2. Non-hematopoietic cells
1.3.2.2.1. Airway epithelia
In addition to the role Syk plays in leukocyte activation and signal transduction, recent
studies have found that Syk function affects other cell types including the airway epithelium.
These cells cover more than 95% of the respiratory tract surface, and are considered to be the first
line of defense against air pollutants, allergens and microorganisms [68]. Airway epithelia directly
participate in the regulation of the immune response in many ways such as formation of the
mucous ciliary coating, secretion of inflammatory chemokines and cytokines, and expression of
adhesion molecules [144]. Syk is believed to have roles in regulation of the inflammatory
responses of the airway epithelium [145, 146].
Work in our lab on the BEAS-2B airway epithelial cell line and normal human bronchial
epithelial (NHBE) cells suggest that Syk is recruited and phosphorylated after engagement of
ICAM-1 by HRV. Syk activation leads to the downstream signaling through the p38 mitogen
activation protein (MAP) kinase pathway and PI3K resulting in the expression of IL-8 [66] and
VEGF induced by HRV [29].
Other studies using primary human bronchial epithelial cell lines and the HS-24 cell line
showed that engagement of β1-integrin can activate Syk and increase production of NO [146], as
well as upregulation of IL-6 and TNF-α mediated ICAM-1 [145, 146].

30
30
1.3.2.2.2. Epithelia in other organs
Syk is expressed in epithelia of other organs, including small and large intestine, breast,
glomerulus, renal tubular, bile duct, and esophagus stratified squamous epithelium [147].
It has been demonstrated that oxidative stress originating from H2O2 activates Syk. This is
followed by the phosphorylation of Iκβα, which consequently activates the DNA binding activity
of NF-κβ in IEC6 intestinal epithelial cells. A similar cause of events happens in IEC6 epithelial
restitution following scrape wounding, which shows that Syk has a role in proliferation and
migration of intestinal epithelial cell [148].
Sung et al. demonstrated that the presence of Syk in normal breast epithelial cells is
pivotal for suppression of proliferation and invasion. They also showed that loss of Syk, even if
only partially, could induce hyperplasia resulting in the formation of mammary tumors in vivo
[149].
1.3.2.2.3. Endothelia
Some studies suggested that Syk is expressed in endothelial cells [139, 141, 144]. Yanagi
et al. hypothesized that the unusual micro-vascular structure observed in Syk-deficient mice was
mainly caused by the dysfunction of endothelial cells (ECs) and that Syk plays a pivotal role in
their physiologic regulation, maintaining the vessel integrity in vivo. These Syk deficient mice
show a lethal phenotype associated with severe petechiae and hemorrhaging, usually dying shortly
after birth [150]. Cheng et al. investigated the role of Syk in vivo using a mouse strain with a
targeted mutation in the Syk gene, finding that homozygous mutants of Syk suffered from fatal
perinatal hemorrhages [151]. These findings indicate the critical role of Syk during embryogenesis
[57].
Research findings have also suggested that tyrosine phosphorylation mediated by reactive

31
31
oxygen species (ROS) can result in high glucose induction of NF-κβ and Syk, playing an important
role in Iκβα phosphorylation of human glomerular endothelial cells [152].
1.3.2.3. Syk signaling pathways
Syk mediated signal transduction usually follows a distinctive pathway. Briefly, Src family
kinases are recruited and activated after engagement of immunoreceptors, leading to the
phosphorylation of two tyrosine residues, which are either in a single ITAM or in two hem-ITAM.
ITAMs can be presented in either the cytoplasmic tail of the receptor or in receptor associated
transmembrane adaptors [137, 153]. Syk is recruited to the dual phosphorylated ITAM, which
triggers activation of Syk, followed by various downstream pathways. Syk then directly binds to
members of the VAV family, phospholipase Cγ (PLCγ) family, the p85α subunit of PI3K, and
SH2 domain-containing leukocyte protein 76 (SLP76) and SLP65 [137, 144]. Other mediators,
such as JNK, p38, LAT, NFAT, PKC, AKT, and NF-κβ may be involved in Syk downstream
pathways as well. Cellular response by activation of these various downstream signaling
components is dependent on the binding partners and cell type. However, activation of Syk by
hemITAM-containing receptors, such as CLEC7A (dectin 1) or CLEC2, likely proceeds via
similar mechanisms (Figure 1.3) [137, 144].
Signaling mediated by Syk through ITAM-independent receptors, which do not carry
ITAM, include β-integrins [137, 144] and the α-subunit of IL-15 receptors [154]. The activation of
Syk by integrins is mediated by an interaction between the non-phosphorylated tyrosine binding
area in the amino terminal of the Syk SH2 domain and the intracellular domain of the integrin β-
chain [137]. Furthermore, it has been shown in some cell types, such as neutrophils and
macrophages, that an ITAM containing adaptor can bind to the β2 subunit of integrins, triggering
Syk activation. In the proposed model, ligation of integrins with adaptor links activates Src family

32
32
kinases, which phosphorylate the ITAM containing adaptor link, resulting in Syk recruitment by
its SH2 domain [155]. Consequently, activation of Syk triggers different cellular responses such as
cytokine release, survival, proliferation, differentiation, migration, adhesion, and cytoskeletal
changes.
1.3.3. Syk and disease pathogenesis
It has been suggested that Syk plays an essential role in many acute and chronic
inflammatory disorders. As discussed previously, activation of Syk occurs primarily through
extracellular antigen-induced crosslinking of FcεR1 and FcγRs, and propagates to downstream
signaling molecules, resulting in initiation of both allergic and autoimmune responses [141, 156].
Many studies have supported a role for Syk in the pathogenesis of several inflammatory diseases
such as rheumatoid arteritis (RA), systemic lupus erythematous (SLE), immune thrombocytopenic
purpura (ITP), lymphoma, leukemia, anaphylactic shock, and allergic rhinitis and asthma [141].
Syk is involved in some hematopoietic malignancies such as leukemia and lymphoma
[157]. This is thought to be a result of the role of Syk in lymphatic proliferation and
differentiation. Signaling in B-cell receptors can be responsible for the pathogenesis of many B-
cell malignancies, including chronic lymphocytic leukemia (CLL), diffuse large B-cell lymphoma,
follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, and B-lineage acute
lymphoblastic leukemia [158]. In preclinical and early clinical studies for treatment of such
disorders, agents that inhibit Syk kinase appear to show particularly promising outcomes [158].
1.3.4. Syk and asthma
Recent studies and research within in our own laboratory have identified Syk’s role in

33
33
airway inflammation and asthma.
Observations made in animal models of asthma propose that Syk plays a role in mediating
allergic airways inflammation [2, 159, 160]. Stenton et al. used Brown Norway rats sensitized with
OVA to demonstrate that aerosolized Syk anti-sense oligodeoxynucleotides (ASO), administered
before OVA challenge, significantly attenuated airways inflammation and acetylcholine-induced
contractility of isolated tracheas ex vivo. In another model of OVA-induced allergic airways
inflammation using BALB/c mice, administration of Syk inhibitor R406 concomitant with OVA
challenge inhibited the development of airways inflammation [160]. These early studies suggest
that Syk is involved in the development of asthma.
Studies in our laboratory have revealed that Syk is involved in regulating virus cell entry,
and the HRV-induced inflammatory response in airway epithelium, both causes of asthma
exacerbation [66, 67]. In addition, we have shown that HRV can induce Syk activation and Syk-
mediated cell signaling, which leads to the expression of inflammatory mediators such as IL-8 and
VEGF [29, 66, 68]. In another study, we showed increase in Syk expression in the airway epithelia
of OVA challenged mice compared with saline controls. We also showed that administration of
Syk inhibitor, NVP-QAB-205, attenuated the augmented AHR in OVA challenged mice to the
level observed in saline control mice [34].
1.4. Syk deficient mice
The analysis of gene function based on the generation of mutant mice by homologous
recombination in embryonic stem cells is restricted if gene disruption is lethal. In this case,
conditional gene targeting is the method of choice. One such system depends on the inducible
activation of a bacterial enzyme, the Cre recombinase. Cre targets a specific DNA sequence, the

34
34
loxP site, and deletes the segment of DNA flanked by two loxP sequences [161].
Syk deficient mice have been studied for many years. Investigation into the role of Syk in
vivo was restricted as result of the perinatal fatality of Syk deficient mice [151, 162]. Cheng et al.
investigated the function of Syk in vivo by generating a mouse strain with a targeted mutation in
the syk gene. Homozygous syk mutants suffered severe hemorrhaging as embryos and died
perinatally, indicating that Syk has a critical role in wound healing or in maintaining vascular
integrity during embryogenesis. Analysis of syk −/− lymphoid cells also revealed that syk mutation
impairs the differentiation of B-lineage cells [151].
To prevent the occurrence of perinatal fatality in Syk-/- mice, an inducible Syk knockout
mouse strain (Sykflox/flox) was generated by the crossbreeding of Rosa26-CreERT2 mice with mice
that carried exon 2 of the Syk gene edged by loxP-sites [57]. Oral administration of tamoxifen
causes deletion of exon 2 of the Syk gene, consequently knocking out Syk. This occurs by
induction of fusion protein (CreERT2), which is expressed constitutively under control of the
Rosa26-promoter, resulting in the nuclear translocation of CreERT2, followed by deletion of exon 2
in the Syk gene (Figure 1.4) [57]. These mice are valuable tool for investigating the role of Syk in
different disease-associated animal models.

35
35
Figure 1.4. Schematic diagram of the knockout strategy
Fusion protein, CreERT2, carries Cre recombinase and a mutated form of the ligand-binding domain of the estrogen receptor, which are constitutively expressed under control of the Rosa26-promoter. Nuclear translocation of the fusion protein, and subsequent deletion of exon 2 of the Syk gene, is induced by tamoxifen. Adapted from Wex, E., et al., Induced Syk deletion leads to suppressed allergic responses but has no effect on neutrophil or monocyte migration in vivo. European Journal of Immunology, 2011. 41(11): p. 3208-18.
1.5. Inhibition of Syk
1.5.1. Mechanisms of action
Another way to study Syk is the use of pharmacological inhibitors. Thus far, Syk kinase
inhibitors for therapeutic purposes include the use of small molecule inhibitors and the suppression
of Syk gene expression. In the past decade, many ATP-competitive Syk inhibitors have been
recognized and isolated from natural or synthetic sources [163]. These Syk inhibitors are small
molecules, with a high affinity for the ATP-binding site of Syk. Many groups have developed
these highly selective, potent Syk inhibitors that compete with ATP. Different inhibitors were co-
crystalized with the catalytic domain of Syk; using its crystalized structure, useful information on

36
36
the inhibitors’ structure is revealed [164]. The Syk inhibitors used presently in clinical trials are
those that have been developed at the Rigel Pharmaceuticals, Inc. Laboratories including R112
[165], R788 [166]; and its water soluble prodrugs R406 [167], and R343 [168]. Other Syk
inhibitors include Bay61-3606 [169] and NVP-QAB-205.
Currently, Syk expression is targeted to inhibit synthesis of Syk. The most frequently used
methods to silence gene expression are antisense oligonucleotides (ASOs) or RNA interference
(RNAi) [141].
To prevent protein synthesis of the cell, ASOs identify and attach tightly to complementary
mRNA [170]. Use of Syk-ASO as inhibitor for Syk kinase expression decreased FcγRIIA
phagocytosis mediated in macrophages [171]. Unfortunately, there has been great debate over the
last decade as to whether ASOs are specific enough [172], so other methods are in development to
silence gene expression.
RNAi, double stranded RNA, is considered another tool for inhibiting expression of genes
in cultured cells and animal models [173]. When RNAi is delivered into the cells, an endogenous
RNAse-III named Dicer cleaves the dsRNA and generates short fragments of RNA (approximately
20-23 nucleotides long) called small interfering RNA (SiRNA). A protein complex, RNA-induced
silencing complex (RISC), recognizes and uncoils the two strands of SiRNA, incorporating them
into one strand that identifies and cleaves mRNA molecules [174]. Use of RNAi to silence Syk
mRNA expression has been reported in different Syk related inflammatory conditions, preventing
degranulation of mast cells by interfering with the PLCγ and PI3K signaling pathways [175].
1.5.2. Syk selective inhibitors in clinical studies
Syk has been the center of attention as a therapeutic target for treatment of various different
inflammatory diseases including RA, SLE, and ITP. Syk inhibitor 406 was used in research and

37
37
clinical trials for RA; observations made in both mouse [167] and rat [176] models of RA showed
abrogated inflammation and degradation of bone. R406 was also effective in a murine model of
ITP syndrome [167]. Oral administration of Syk inhibitor R788 has shown impressive success in
the phase II clinical trial of RA, leading to a reduction in the clinical signs and symptoms of the
disease [177]. R788 showed therapeutic effects in an animal model of SLE, delaying signs of the
disease such as renal dysfunction, proteinuria, kidney infiltrates, and increased mouse survival
[178]. R788 was found to decrease the severity of experimental antibody mediated
glomerulonephritis in the rat [179]. It has also been shown that R788 inhibits tumor growth in B
cell chronic lymphatic leukemia (CLL) by blocking B cell receptor signalling [157] and was
successfully used for clinical trial of CLL and non-Hodgkin lymphoma (NHL) by disrupting the
signaling induced by BCR [180].
1.5.3. Syk selective inhibitors in asthma and allergic rhinitis
The role of Syk in asthma has been evaluated in different studies. Syk mediates the main
signaling pathways in asthma operated by mast cells [159, 181] and basophils [182] after
activation through FcεRI, which induces a release of inflammatory mediators. Mastubara et al.
determined the activity of R406 on the activation of mast cells, and the development of AHR and
inflammation induced by OVA in a mouse model of allergic inflammation and asthma. The results
revealed prevention of bone marrow mast cell (BMMC) degranulation in vitro [159], as well as
AHR, eosinophilia, and goblet cell hyperplasia in vivo [2, 159]. A potent Syk inhibitor, BAY 61-
3606, was used in another study to evaluate the role of Syk in asthma. Observations showed that
oral administration of Bay 61-3606, include significant suppression of bronchoconstriction,
bronchial edema, and airways inflammation induced by OVA in rat [169]. In addition, Stenton et
al. used Syk kinase ASOs to inhibit allergic inflammation in the airways in an OVA rat model of

38
38
allergic inflammation. They showed suppression of tracheal contraction induced by antigen and
pulmonary inflammation in a rat ovalbumin model of allergic asthma [160].
Meltzer et al. investigated the efficacy of Syk inhibitor R112 in 160 human volunteers with
allergic rhinitis. Intranasal administration of R112 reduced symptoms of patients with allergic
rhinitis in comparison with those who received a vehicle control [183]

41
2 Chapter 2: Hypothesis and research objectives
2.1. Overall hypothesis
Syk mediates HDM-induced airways hyperresponsiveness, inflammation and remodeling
in murine models of allergic airways inflammation
2.2. Research objectives
2.2.1. To establish murine models of acute and chronic HDM-induced allergic airway
inflammation in the transgenic Sykfl/fl/rosa26-CreERT mice.
2.2.2. To investigate the role of Syk in the pathogenesis of HDM-induced allergic airway
inflammation with respect to:
a) airway hyper-responsiveness
b) airway inflammation and
c) airway remodeling.

42
3 Chapter 3: Material and methods
3.1. Animals
Eight-week old female BALB/c mice, purchased from Charles River Laboratories
Canada (Saint-Constant, PQ, Canada; average body weight 19.5 g) and inducible Syk knockout
mice, Sykfl/fl-Syk//rosa26-CreERT2 (a gift from Boehringer Ingelheim Pharma GmbH & Co.,
Biberach, Germany; average body weight 20.9 g) were used. Mice were kept in ventilated plastic
cages with a 12-hour light-dark cycle and fed standard laboratory diet. Experiments were
approved by University of Toronto Faculty Advisory Committee on Animal Services based on
Canadian Council on Animal Cares (CCAC) ‘guide to the care and use of experimental animals’.
3.2. Murine HDM-sensitization and -challenge model of airways inflammation
3.2.1. Acute model of HDM-induced airways inflammation
Mice were sensitized to Dermatophagoides farinae (GREER® Laboratories, Inc. Lenoir,
NC) crude extract based on previously established protocol [125]. Briefly, mice were
anesthetized with isoflurane (Baxter Co., Mississauga, ON, Canada) using an anesthesia
vaporizer (Benson Medical Industries Inc., Markham, ON, Canada) and sensitized intranasally
with 100 µg of D.farinae crude extract in 50 µl of normal saline for 5 consecutive days from day
0-4. On day 11, the same dose of D.farinae was administered intranasally as a single challenge.
An aliquot 50 µl of normal saline was used as control in Df-sensitized mice. Twenty-four hours
after the single challenge with D.farinae or saline, pulmonary function testing (PFT) and

43
assessment of methacholine-induced airway hyperresponsiveness (AHR) were performed (Figure
3.1.A) using the flexiVent® system for respiratory function (Scireq Inc. PQ).
3.2.2. Chronic model of HDM-induced airways inflammation
Chronic model of HDM-induced airways inflammation was established by long-term
intranasal instillation of D. pteronyssinus in the inducible Syk knockout mouse strain. In this
model, mice were sensitized intranasally to 25 µg of D. pteronyssinus crude extract in 35 µl of
normal saline for 5 consecutive days, followed by exposures every other day for 8 weeks [75].
Control groups received 35µl saline. The role of Syk in airways remodeling, AHR and airways
inflammation was assessed in HDM-treated mice in the presence or absence of Syk. Physiologic
responses were evaluated 24 hours after the last challenge using the flexiVent® system to
measure airway resistance and methacholine responsiveness (Figure 3.1.B).

44
Figure 3.1 Experimental timeline for developing of acute (A) and chronic (B) models of HDM-induced allergic airways inflammation
3.3. In vivo inhibition of Syk
NVP-QAB-205 (a gift from GlaxoSmithKline Incorporated) is a competitive ATP-
inhibitor of Syk with an IC50 of 10 nM [141]. After determination of baseline respiratory
mechanics in HDM or saline sensitized and challenged BALB/c mice, NVP-QAB-205 (0.3
mg/kg body weight; dissolved in DMSO and diluted in PBS) or vehicle were administered
directly into the ventilator circuit using an aeroneb laboratory nebulizer, and allowed to
equilibrate for 15 minutes. Respiratory mechanics were reassessed after the equilibration prior to
assessment of methacholine responsiveness.

45
Figure 3.2. In vivo ventilator-based assessment of respiratory mechanics by the flexiVent® system
3.4. Knockdown of Syk in inducible Syk knockout mice
Inducible Syk knockout mice were generated by crossing Rosa26-CreERT2 mice with
mice in which exon 2 of the Syk gene had been flanked by loxP-site. Oral administration of
tamoxifen is resulted in deletion of exon 2 of Syk gene [57]. Syk knockout was induced by oral
administration of 22mg/kg tamoxifen in 10% v/v ethanol in sunflower oil (3.6 ml/kg) for 5
consecutive days. Ten days after the first set of treatment, the same dose of tamoxifen was used
for 5 more consecutive days. Control group of mice received 10% v/v ethanol in 3.6 ml/kg of
sunflower oil. Ten days after the second set of treatment with tamoxifen, mice are Syk knocked
out and ready to be used for sensitization and challenge with HDM. Animals in vehicle-treated
control groups received 10% v/v ethanol in sunflower oil (3.6 ml/kg) instead of tamoxifen
(Figure 3.3).

46
Figure 3.3. Experimental timeline for Tamoxifen treatment for deletion of Syk in inducible Syk knockout, Sykfl/fl-Syk//rosa26-CreERT2 mouse
3.5. Pulmonary function tests (PFTs) and methacholine challenge
PFTs and methacholine challenge were performed based on previously used protocol
[93]. In brief, 24 hours after the single intranasal challenge with HDM or saline, mice were
anesthetized with intraperitoneal injection of ketamine-xylazine (50 mg/kg xylazine, Bioniche,
Belleville, ON, Canada and 10 mg/kg ketamine, Bayer, Toronto, ON, Canada). The trachea was
surgically exposed, intubated with an 18-gauge stainless steel cannula (BD Biosciences Canada,
Mississauga, ON, Canada), and connected to the FlexiVent System (Scireq Inc., Montreal, PQ)
for measurement of methacholine responsiveness. At first, baseline airway resistance was
determined and then increasing doses of methacholine (0–100 mg/ml in sterile PBS; Sigma) was
nebulized directly into the ventilator circuit. Anesthesia was maintained during methacholine
challenge by intraperitoneal injection of 25% of the starting dose of ketamine-xylazine every 10
minutes until the end of challenge. All data points were exported to Microsoft Excel (Microsoft,
Redmond, WA) using the FlexiVent software and analyzed offline.

47
3.6. Bronchoalveolar lavage fluid (BALF)
Mouse lungs were lavaged to evaluate inflammatory cell numbers immediately after
methacholine induced AHR measurements were complete. The lungs were washed with 2 ml of
PBS via tracheal cannula; initially flushed with 1 ml of PBS followed by two further 0.5 ml PBS
washes. The mouse BALF from each of the washes was pooled in a sterile plastic tube and kept
on ice. Tubes were then centrifuged for 10 minutes at 350g at 4°C. The volume of recovered
BALF was recorded for each mouse; typical recoveries were greater than 85%. The cell-free
supernatant was divided in cryovials and kept in -80°C degree freezers. The cell pellet was
washed with H2O to lyse the red blood cells and then resuspended in 500 µl of PBS. Total
leukocyte counts in the BALF pellet for each mouse were measured with a hemocytometer using
trypan blue (Wisent, St. Bruno, PQ).
Differential cell counts were performed manually by spinning down the cells onto a slide
using a cytospin (StatSpin Cytophuge, Westwood, MA) and staining with Diff-Quick (Invitrogen
Life Technologies) [91]. Different cell types were then counted under light microscope.
3.7. Analysis of inflammatory mediators in BALF
The levels of different cytokines and chemokines in the cell-free supernatant of BALF
were determined by enzyme linked immunosorbent assay (ELISA) and the Luminex System.
The concentrations of IL-6, chemokine (C-X-C motif) ligand 1 (CXCL1-KC), vascular
endothelial growth factor (VEGF), and matrix metallopeptidase 9 (MMP-9) were assessed in
BALF obtained from acute model mice by ELISA (CXCL1-KC, IL-6, VEGF and MMP-9 from
Life Technologies, Invitrogen, Burlington, ON). Also, the level of IL-4, IL-6, IL-10, IL-17A,
KC, RANTES, VEGF and TNF-α were evaluated in BALF obtained from chronic model mice

48
by Luminex System. Luminex assays were performed in triplicate according to manufacturer's
instructions; using an 8-plex Milliplex MAP mouse cytokine/chemokine magnetic based kit
(Millipore, Billerica, MA). Data was obtained on a Luminex 200 with the Luminex XY plate
handling platform and the Luminex SD sheath fluid delivery system, and analyzed with 3.1
xPonent and MILLIPLEX Analyst software.
3.8. Histology of lung tissue sections
Immediately after the assessment of AHR by methacholine, the lungs were extracted and
inflated with 4% paraformaldehyde solution (Canemco, Quebec, Canada) to a pressure of 20cm
of H2O. Sections (5 µm thickness) were prepared and stained at pathology core facility of the
Centre for Modeling Human Disease, Toronto Centre for Phenogenomics, Toronto, ON, Canada.
Hematoxylin and eosin (H&E) was used to assess the degree of inflammation [125]. Periodic
acid-schiff (PAS) and masson trichrome staining were used to observe the degree of goblet cell
hyperplasia and to assess the degree of collagen deposition [1, 125], respectively.
3.9. RNA extraction and quantitative PCR
Total RNA was extracted and purified from lung tissues using RNeasy Mini Kit (Qiagen,
Maryland, USA) according to manufacture’s protocol. One microgram of total RNA was reverse
transcribed into cDNA using the QuantiTech® Reverse Transcription Kit (Qiagen, Maryland,
USA). Aliquots of cDNA (2.5 µl) contain 15 ng of cDNA, LightCycler® 480 Probe Master and
Primer-probe mix were then mixed in a total reaction volume of 10 µl. qPCR was performed in
triplicate, using the Roche LightCycler® 480 instrument. Expression of IL-6, CXCL-1, VEGF,
MMP-9, TGF-β1 and FGF-2 were determined at copy number level and normalized against Ppia.
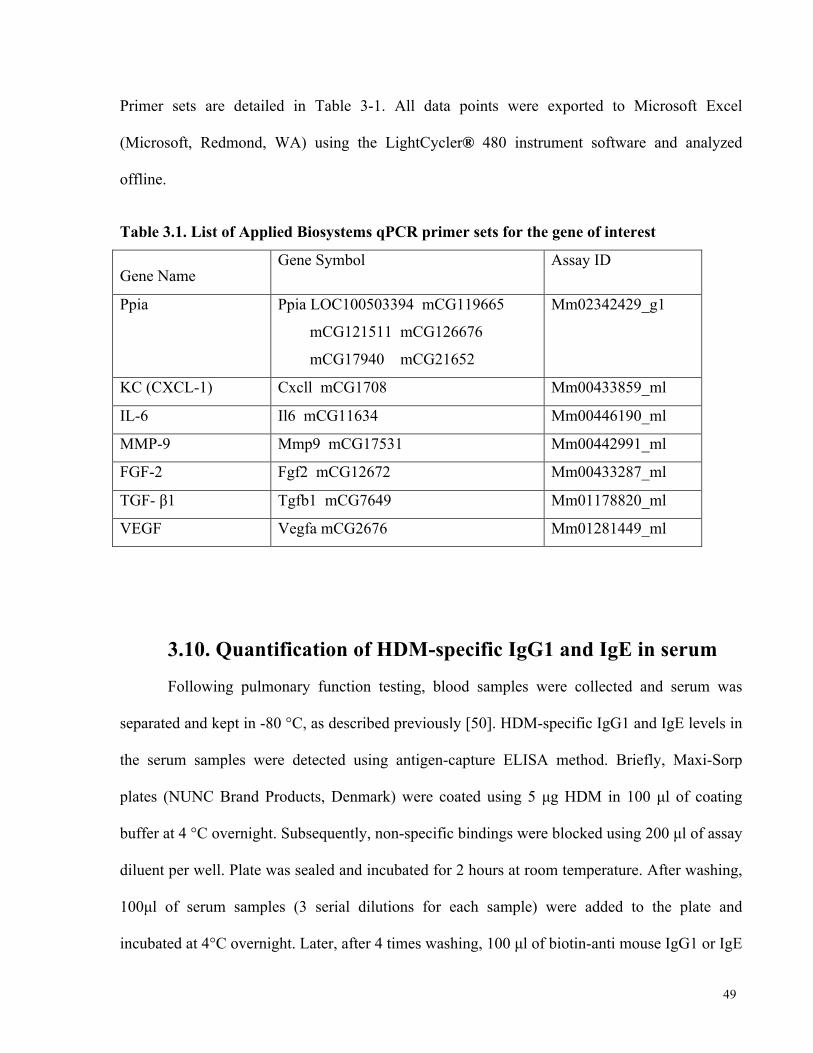
49
Primer sets are detailed in Table 3-1. All data points were exported to Microsoft Excel
(Microsoft, Redmond, WA) using the LightCycler® 480 instrument software and analyzed
offline.
Table 3.1. List of Applied Biosystems qPCR primer sets for the gene of interest
Gene Name Gene Symbol Assay ID
Ppia Ppia LOC100503394 mCG119665
mCG121511 mCG126676
mCG17940 mCG21652
Mm02342429_g1
KC (CXCL-1) Cxcll mCG1708 Mm00433859_ml
IL-6 Il6 mCG11634 Mm00446190_ml
MMP-9 Mmp9 mCG17531 Mm00442991_ml
FGF-2 Fgf2 mCG12672 Mm00433287_ml
TGF- β1 Tgfb1 mCG7649 Mm01178820_ml
VEGF Vegfa mCG2676 Mm01281449_ml
3.10. Quantification of HDM-specific IgG1 and IgE in serum
Following pulmonary function testing, blood samples were collected and serum was
separated and kept in -80 °C, as described previously [50]. HDM-specific IgG1 and IgE levels in
the serum samples were detected using antigen-capture ELISA method. Briefly, Maxi-Sorp
plates (NUNC Brand Products, Denmark) were coated using 5 µg HDM in 100 µl of coating
buffer at 4 °C overnight. Subsequently, non-specific bindings were blocked using 200 µl of assay
diluent per well. Plate was sealed and incubated for 2 hours at room temperature. After washing,
100µl of serum samples (3 serial dilutions for each sample) were added to the plate and
incubated at 4°C overnight. Later, after 4 times washing, 100 µl of biotin-anti mouse IgG1 or IgE

50
(Biolegend, CA, USA) was added in each well and incubated at room temperature for 1 hour.
The plate was washed and incubated with Avidin-HRP (Biolegend) at room temperature for 30
minutes. Finally to develop color reaction, 100 µl of TMB substrate solution (Biolegend) was
added to each well and incubated for 30 minutes at room temperature in dark. Optical densities
were read at 450 nm with the reference at 540 nm.
3.11. Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0c (GraphPad Software, La
Jolla, CA). All data were presented as mean±SEM. Student t-test was used for binary
comparison, and 1-way or 2-way ANOVA were used for multiple comparisons. A P<0.05 were
considered significantly different.

51
4 Chapter 4. Results
4.1. Acute model of HDM-induced airways inflammation
4.1.1. A 10 day course of HDM sensitization and challenge enhanced
AHR to methacholine, a response that is abrogated by treatment with a
single dose of Syk inhibitor
To ensure that a previously published protocol of an acute model of HDM-induced airways
inflammation [125] works in our laboratory, the effect of a 10-day HDM sensitization and
challenge protocol in Balb/c mice was first examined. We used the ventilator-based FlexiVent
system to measure respiratory mechanics and AHR to methacholine (MCh). We found that total
respiratory resistance (Rrs) (Figure 4.1.A) and maximum respiratory resistance to methacholine
(Figure 4.1.B) were significantly increased in HDM-exposed mice compared with Saline control
group.
We then assessed the effect of administration of Syk inhibitor, NVP-QAB-205, in MCh
responsiveness after establishment of the model. Following sensitization and challenge with HDM
or Saline, mice from both groups were selected randomly to receive NVP-QAB-205 as a single
dose (0.3 mg/kg body weight) administered intratracheally 15 minutes prior to assessment of
airways responsiveness. NVP-QAB-205 caused a significant reduction in the MCh-induced
increase in Rrs (Figure 4.1.A), particularly at the maximum concentration of MCh (Figure 4.1.B)
in the HDM mice, bringing Rrs to levels observed in the naïve Saline controls.
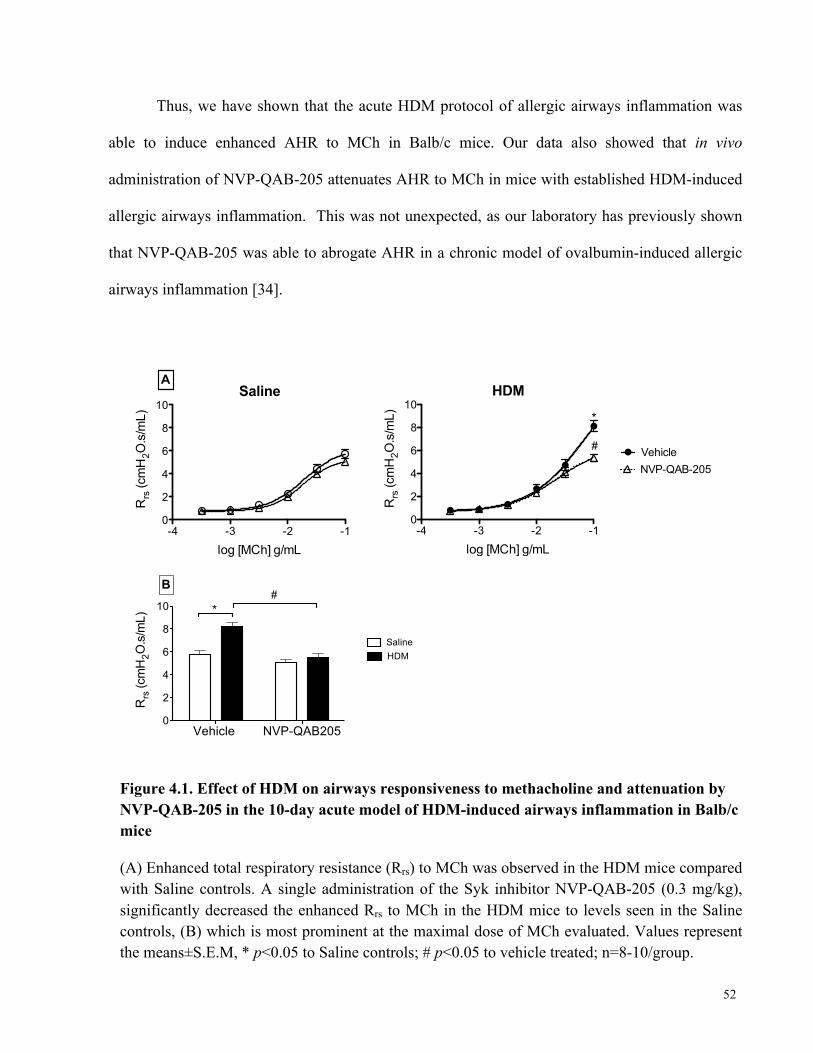
52
Thus, we have shown that the acute HDM protocol of allergic airways inflammation was
able to induce enhanced AHR to MCh in Balb/c mice. Our data also showed that in vivo
administration of NVP-QAB-205 attenuates AHR to MCh in mice with established HDM-induced
allergic airways inflammation. This was not unexpected, as our laboratory has previously shown
that NVP-QAB-205 was able to abrogate AHR in a chronic model of ovalbumin-induced allergic
airways inflammation [34].
Figure 4.1. Effect of HDM on airways responsiveness to methacholine and attenuation by NVP-QAB-205 in the 10-day acute model of HDM-induced airways inflammation in Balb/c mice
(A) Enhanced total respiratory resistance (Rrs) to MCh was observed in the HDM mice compared with Saline controls. A single administration of the Syk inhibitor NVP-QAB-205 (0.3 mg/kg), significantly decreased the enhanced Rrs to MCh in the HDM mice to levels seen in the Saline controls, (B) which is most prominent at the maximal dose of MCh evaluated. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to vehicle treated; n=8-10/group.
-4 -3 -2 -10
2
4
6
8
10Saline
A
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)
Vehicle NVP-QAB2050
2
4
6
8
10 *
B#
SalineHDM
Rrs
(cm
H2O
.s/m
L)
-4 -3 -2 -10
2
4
6
8
10
Vehicle NVP-QAB-205
*
#
HDM
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)

53
4.1.2. HDM-induced AHR was abrogated by deletion of Syk
To address the role of Syk in AHR to MCh, inducible Syk knockout mice were used.
Following treatment of Sykfl/fl-Syk//rosa26-CreERT2 mice with tamoxifen to induce deletion of the
Syk (Sykdel/del mice) or sunflower oil for the control group (Syk-expressing Sykflox/flox mice), the
mice were subjected to the acute HDM (or Saline) sensitization and challenge protocol described
above, and respiratory mechanics were assessed using the FlexiVent system.
In the control Sykflox/flox mice, we observed significantly enhanced MCh-induced total
respiratory resistance (Rrs) in the HDM mice compared with Saline control group (Figure 4.2.A).
On the other hand, in the Sykdel/del mice, HDM sensitization and challenge did not result in
enhanced Rrs to MCh (Figure 4.2.A). In the Sykflox/flox mice, the maximal MCh-responsiveness of
Rrs (Figure 4.2.B), and the slope of the Rrs dose-response curve (Figure 4.2.C) were significantly
increased in the HDM-exposed mice compared with Saline control mice. We also observed that
tissue elastance (E) was significantly increased in Sykflox/flox HDM-mice compared with Saline
controls. (Figure 4.2.D). In contrast, no differences between the HDM and Saline groups were
observed in the Rrs dose-responsiveness, maximal Rrs, and E in the Sykdel/del mice (Figure 4.2.A-D).
In addition, we also observed the central airway Newtonian resistance (RN) and peripheral
tissue damping/resistance to be significantly increased in HDM-treated Sykflox/flox compared with
Saline controls, whereas no differences in these two parameters following HDM sensitization and
challenge were observed in the Syk-deleted state (Figures 4.3.A and 4.3.B).
Thus, in this acute model, deletion of Syk resulted in reduction of both the central and
peripheral airways responsiveness to MCh.
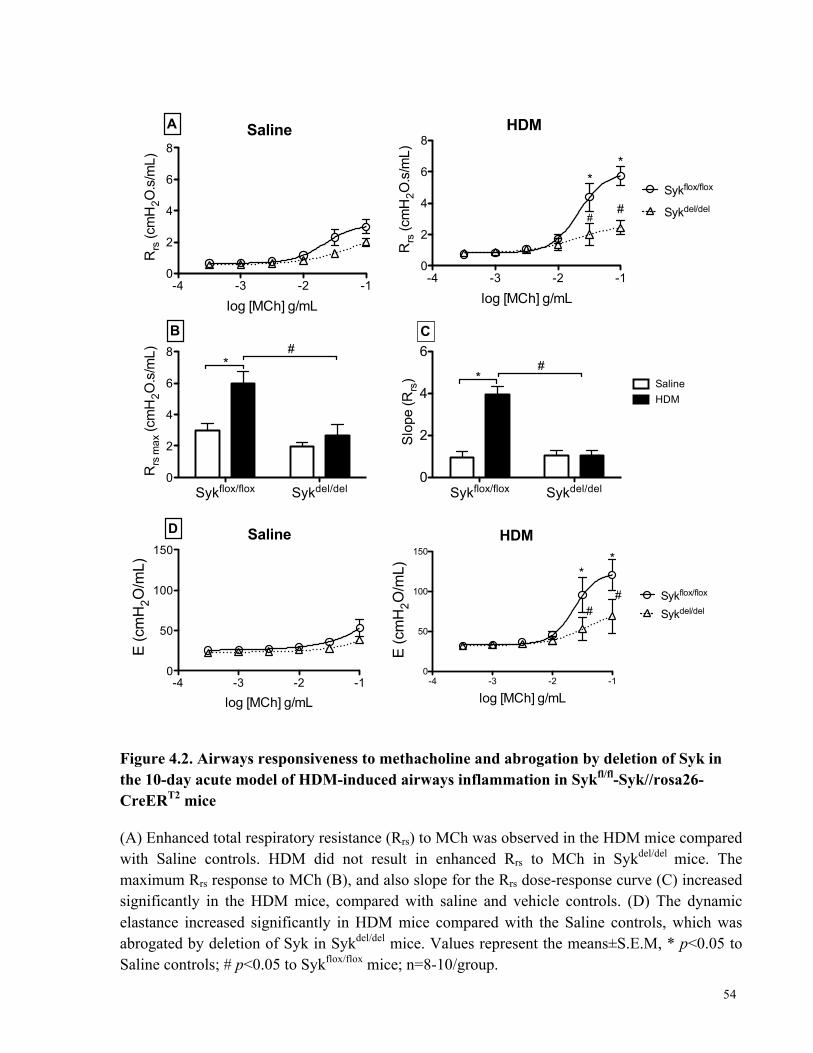
54
Figure 4.2. Airways responsiveness to methacholine and abrogation by deletion of Syk in the 10-day acute model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
(A) Enhanced total respiratory resistance (Rrs) to MCh was observed in the HDM mice compared with Saline controls. HDM did not result in enhanced Rrs to MCh in Sykdel/del mice. The maximum Rrs response to MCh (B), and also slope for the Rrs dose-response curve (C) increased significantly in the HDM mice, compared with saline and vehicle controls. (D) The dynamic elastance increased significantly in HDM mice compared with the Saline controls, which was abrogated by deletion of Syk in Sykdel/del mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=8-10/group.
-4 -3 -2 -10
2
4
6
8
A Saline
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)
Sykflox/flox Sykdel/del0
2
4
6
8*
B#
Rrs
max
(cm
H2O
.s/m
L)
-4 -3 -2 -10
2
4
6
8
Sykflox/flox
Sykdel/del
* *
##
HDM
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)
Sykflox/flox Sykdel/del0
2
4
6#
*HDMSaline
C
Slo
pe (R
rs)
-4 -3 -2 -10
50
100
150
D Saline
log [MCh] g/mL
E (c
mH
2O/m
L)
-4 -3 -2 -10
50
100
150
Sykflox/flox
Sykdel/del
**
HDM
##
log [MCh] g/mL
E (c
mH
2O/m
L)
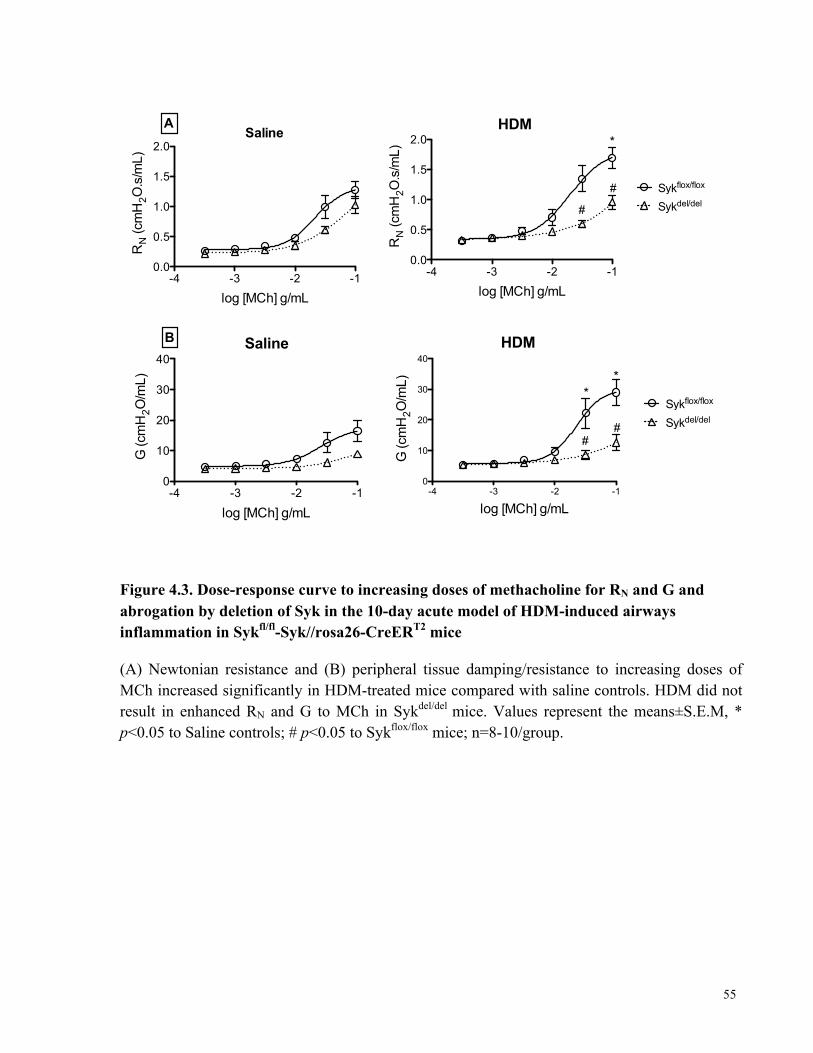
55
Figure 4.3. Dose-response curve to increasing doses of methacholine for RN and G and abrogation by deletion of Syk in the 10-day acute model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
(A) Newtonian resistance and (B) peripheral tissue damping/resistance to increasing doses of MCh increased significantly in HDM-treated mice compared with saline controls. HDM did not result in enhanced RN and G to MCh in Sykdel/del mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=8-10/group.
-4 -3 -2 -10.0
0.5
1.0
1.5
2.0
ASaline
log [MCh] g/mL
RN (c
mH
2O.s
/mL)
-4 -3 -2 -10.0
0.5
1.0
1.5
2.0
Sykflox/flox
Sykdel/del#
*
#
HDM
log [MCh] g/mL
RN (c
mH
2O.s
/mL)
-4 -3 -2 -10
10
20
30
40
B Saline
log [MCh] g/mL
G (c
mH
2O/m
L)
-4 -3 -2 -10
10
20
30
40
Sykflox/flox
Sykdel/del
**
##
HDM
log [MCh] g/mL
G (c
mH
2O/m
L)

56
4.1.3. Deletion of Syk reduced HDM-specific IgG1 and IgE levels in
serum
Syk plays an important role in innate immunity and allergic responses [57, 137]. Therefore,
it is possible that the failure of the Sykdel/del mice to develop enhanced AHR to MCh is a result of
an inability to mount an immune response to HDM in the Syk-deleted state. To evaluate this
possibility, we measured the levels of HDM-specific IgG1 and IgE.
The serum HDM-specific IgG1 and IgE levels were significantly elevated in the HDM-
exposed Sykflox/flox mice compared with Saline control group (Figure 4.4). In the Syk-deleted state,
HDM sensitization and challenge was able to induce significantly increased levels of HDM-
specific IgG1 and IgE when compared with Saline controls, although not to the levels observed in
the Syk-expressing mice (Figure 4.4)
Figure 4.4. Serum levels of HDM-specific IgG1 and IgE in the 10-day acute model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
The levels of HDM-specific IgG1 and IgE were increased significantly in HDM-treated mice compared with Saline controls in both Sykflox/flox and Sykdel/del mice. Deletion of Syk significantly reduced HDM-specific IgG1 and IgE levels in serum. Values represent the means±S.E.M, *p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=4-5/group.
HDM Specific IgG1
Sykflox/flox Sykdel/del0
1
2
3
4 * #
*
OD(450-570)
HDM Specific IgE
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0*
#
SalineHDM*
OD(450-570)

57
4.1.4. BALF total cell was not affected by either Syk knock-down or
pharmacological inhibition
Airway inflammation is a key feature of the acute model of allergen-induced airways
inflammation [1, 91, 125]. To evaluate the role of Syk in mediating airway inflammation, we
assessed the total and differential cell counts in bronchoalveolar lavage fluid (BALF) in both
Balb/c and Sykfl/fl //rosa26-CreERT2 mice following the acute HDM sensitization and challenge
protocol.
The total cell counts in the BALF samples of HDM-exposed mice were significantly
elevated compared with Saline controls in the Balb/c mice (69.15±13.31 × 103 vs. 275.50±24.30 ×
103, p<0.05), the Syk-expressing Sykflox/flox mice (27.85±6.73 × 103 vs. 188.05±16.10 × 103,
p<0.05), and the Sykdel/del mice (45.31±7.77 × 103 vs. 140.15±24.56 × 103, p<0.05) (Tables 4.2.A
and 4.2.B). Differential cell counts showed that these were primarily due to significant increases in
the number of neutrophils, lymphocytes and eosinophils in HDM-exposed mice compared to
Saline controls.
In the Balb/c mice, administration of 0.3 mg/kg NVP-QAB-205 before assessment of
respiratory function did not impact the BALF total and differential leukocyte counts in the HDM
sensitized and challenged group (220.33±74.35 × 103 vs. 275.50±24.30 × 103) compared with
vehicle control group (Table 4.1.A).
In the Sykfl/fl //rosa26-CreERT2 mice, deletion of Syk did not affect leukocyte recruitment
to the airways as the total BALF cells counts were similar to those observed in Sykflox/flox
(140.15±24.56 × 103 vs. 188.05±16.10 × 103). However, differential BALF cell counts in the
Sykdel/del mice revealed that deletion of Syk preferentially decreased the number of eosinophils
without affecting the number of neutrophils, macrophages and lymphocytes in HDM-exposed
mice when compared to HDM-exposed Sykflox/flox mice (Table 4.1.B). These results showed that

58
deletion of Syk did not affect recruitment of lymphocytes, neutrophils and macrophage to the
airways in vivo. Our findings revealed that Syk inhibitor nor deletion of Syk had no effect on
leukocyte recruitment to the airways following HDM sensitization and challenge.
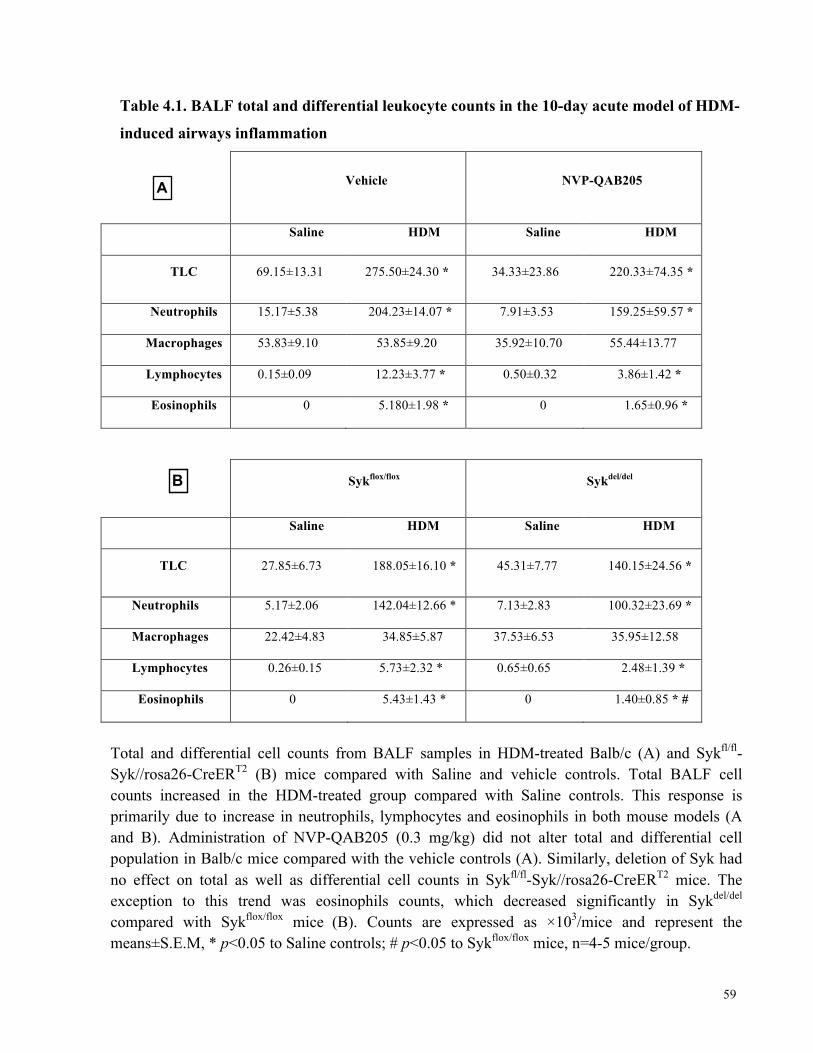
59
Table 4.1. BALF total and differential leukocyte counts in the 10-day acute model of HDM-
induced airways inflammation
Vehicle NVP-QAB205
Saline HDM Saline HDM
TLC 69.15±13.31 275.50±24.30 * 34.33±23.86 220.33±74.35 *
Neutrophils 15.17±5.38 204.23±14.07 * 7.91±3.53 159.25±59.57 *
Macrophages 53.83±9.10 53.85±9.20 35.92±10.70 55.44±13.77
Lymphocytes 0.15±0.09 12.23±3.77 * 0.50±0.32 3.86±1.42 *
Eosinophils 0 5.180±1.98 * 0 1.65±0.96 *
Sykflox/flox Sykdel/del
Saline HDM Saline HDM
TLC 27.85±6.73 188.05±16.10 * 45.31±7.77 140.15±24.56 *
Neutrophils 5.17±2.06 142.04±12.66 * 7.13±2.83 100.32±23.69 *
Macrophages 22.42±4.83 34.85±5.87 37.53±6.53 35.95±12.58
Lymphocytes 0.26±0.15 5.73±2.32 * 0.65±0.65 2.48±1.39 *
Eosinophils 0 5.43±1.43 * 0 1.40±0.85 * #
Total and differential cell counts from BALF samples in HDM-treated Balb/c (A) and Sykfl/fl-Syk//rosa26-CreERT2 (B) mice compared with Saline and vehicle controls. Total BALF cell counts increased in the HDM-treated group compared with Saline controls. This response is primarily due to increase in neutrophils, lymphocytes and eosinophils in both mouse models (A and B). Administration of NVP-QAB205 (0.3 mg/kg) did not alter total and differential cell population in Balb/c mice compared with the vehicle controls (A). Similarly, deletion of Syk had no effect on total as well as differential cell counts in Sykfl/fl-Syk//rosa26-CreERT2 mice. The exception to this trend was eosinophils counts, which decreased significantly in Sykdel/del compared with Sykflox/flox mice (B). Counts are expressed as ×103/mice and represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice, n=4-5 mice/group.
A
B

60
4.1.5. Effect of Syk deletion on the production of IL-6, KC, VEGF and
MMP-9 in the acute model of HDM-induced airways inflammation
We also evaluated airway inflammation by assessing production of inflammatory
mediators in the BALF samples. Previous studies in our laboratory revealed that Syk regulates
expression of IL-8, IL-6 and VEGF in response to HRV infection in BEAS-2B airway epithelial
and also primary airway epithelial cell [29, 66]. HRV infection is common cause of asthma
exacerbations [29, 66, 68]. For this reason, our initial studies focused on these Syk-regulated
inflammatory mediators. In the Sykflox/flox mice, BALF analysis showed significant increases in the
level of KC (a murine homolog of human IL-8), IL-6, VEGF and MMP-9 in HDM-exposed mice
compared with Saline control (Figure 4.5). Deletion of Syk significantly decreased production of
VEGF, but had no effect on production of other mediators.
These results showed that deletion of Syk had no effect on the production of inflammatory
mediators such as IL-6, KC and MMP-9, with the exception of VEGF, in this acute model of
airways inflammation.
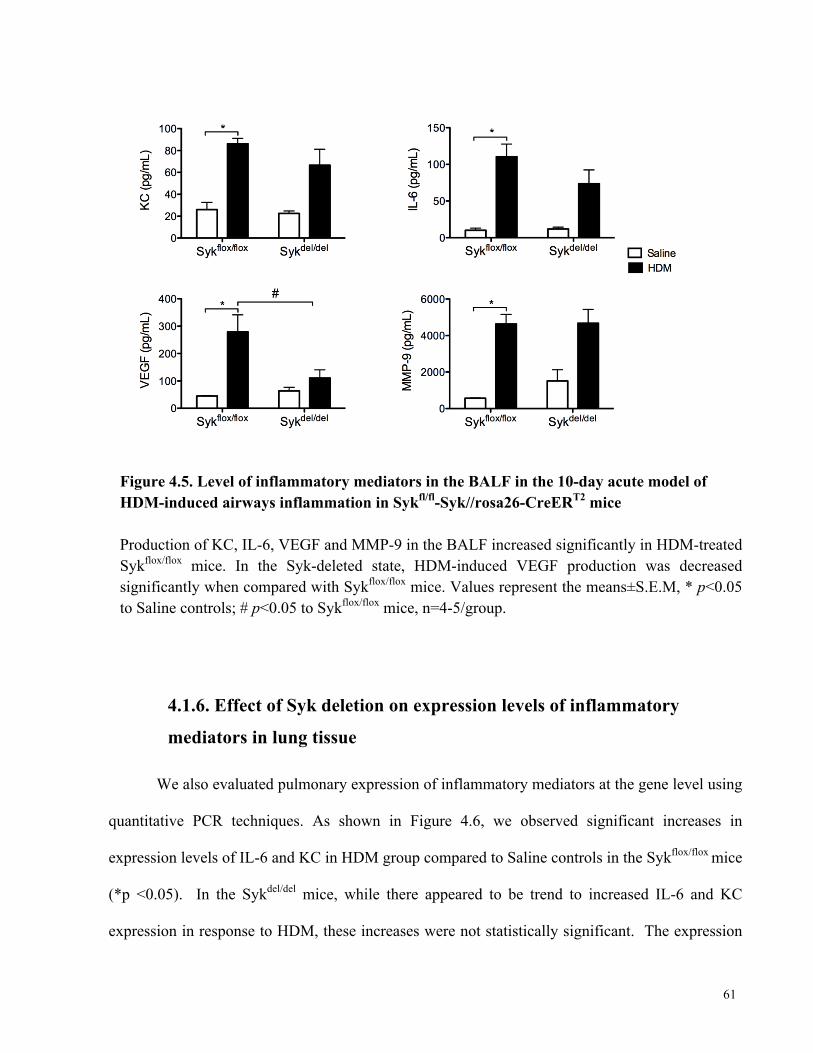
61
Figure 4.5. Level of inflammatory mediators in the BALF in the 10-day acute model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice Production of KC, IL-6, VEGF and MMP-9 in the BALF increased significantly in HDM-treated Sykflox/flox mice. In the Syk-deleted state, HDM-induced VEGF production was decreased significantly when compared with Sykflox/flox mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice, n=4-5/group.
4.1.6. Effect of Syk deletion on expression levels of inflammatory
mediators in lung tissue
We also evaluated pulmonary expression of inflammatory mediators at the gene level using
quantitative PCR techniques. As shown in Figure 4.6, we observed significant increases in
expression levels of IL-6 and KC in HDM group compared to Saline controls in the Sykflox/flox mice
(*p <0.05). In the Sykdel/del mice, while there appeared to be trend to increased IL-6 and KC
expression in response to HDM, these increases were not statistically significant. The expression
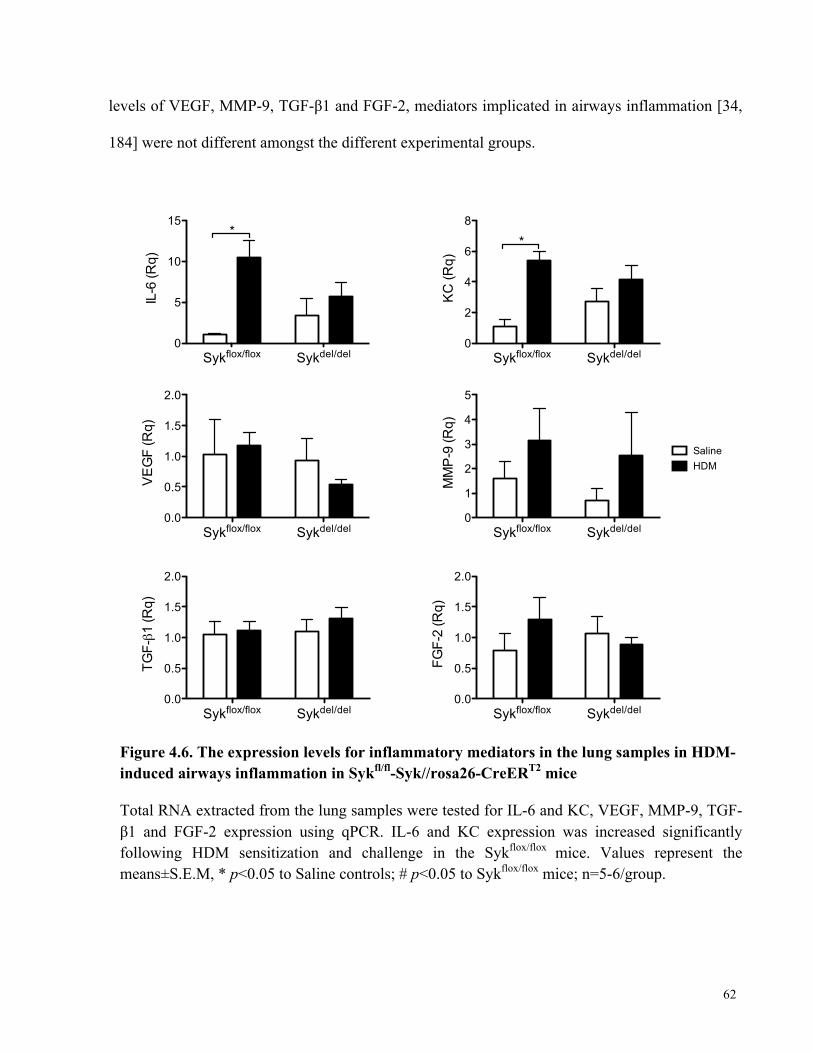
62
levels of VEGF, MMP-9, TGF-β1 and FGF-2, mediators implicated in airways inflammation [34,
184] were not different amongst the different experimental groups.
Figure 4.6. The expression levels for inflammatory mediators in the lung samples in HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
Total RNA extracted from the lung samples were tested for IL-6 and KC, VEGF, MMP-9, TGF-β1 and FGF-2 expression using qPCR. IL-6 and KC expression was increased significantly following HDM sensitization and challenge in the Sykflox/flox mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=5-6/group.
Sykflox/flox Sykdel/del0
5
10
15*
IL-6
(Rq)
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0
VE
GF
(Rq)
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0
TGF-!1
(Rq)
Sykflox/flox Sykdel/del0
2
4
6
8
*
KC
(Rq)
Sykflox/flox Sykdel/del0
1
2
3
4
5
SalineHDM
MM
P-9
(Rq)
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0
FGF-
2 (R
q)

63
4.1.7. Histologic evidence of airway inflammation and goblet cell
hyperplasia is evident following a 10-day HDM sensitization and
challenge period
Lastly, we also evaluated airway inflammation by histologic examination of lung sections
from Balb/c and Sykfl/fl-Syk//rosa26-CreERT2 mice following the 10-day protocol of HDM
sensitization and challenge. Histological analysis demonstrated peribronchial (solid arrows) and
perivascular infiltration (solid pentagons) with inflammatory cells in the HDM-exposed compared
with naive Saline control groups in the Balb/c, Sykflox/flox and Sykdel/del mice (Figure 4.7.A). PAS
staining of the lung sections revealed increased number of PAS-positive cells (open pentagons)
that are indicative of Goblet cells in the HDM-exposed mice compared with Saline controls for all
three groups of mice (Figure 4.7.B).
The fact that we did not see obvious differences in the number of inflammatory infiltrates
(Figure 4.7.A) and PAS-positive cells (Figure 4.7.B) between Sykflox/flox and Sykdel/del mice
following HDM treatment supports our observations in the BALF; Syk did not appear to have a
role in mediating of leukocyte recruitment to the lung and airways in response to HDM
sensitization and challenge.
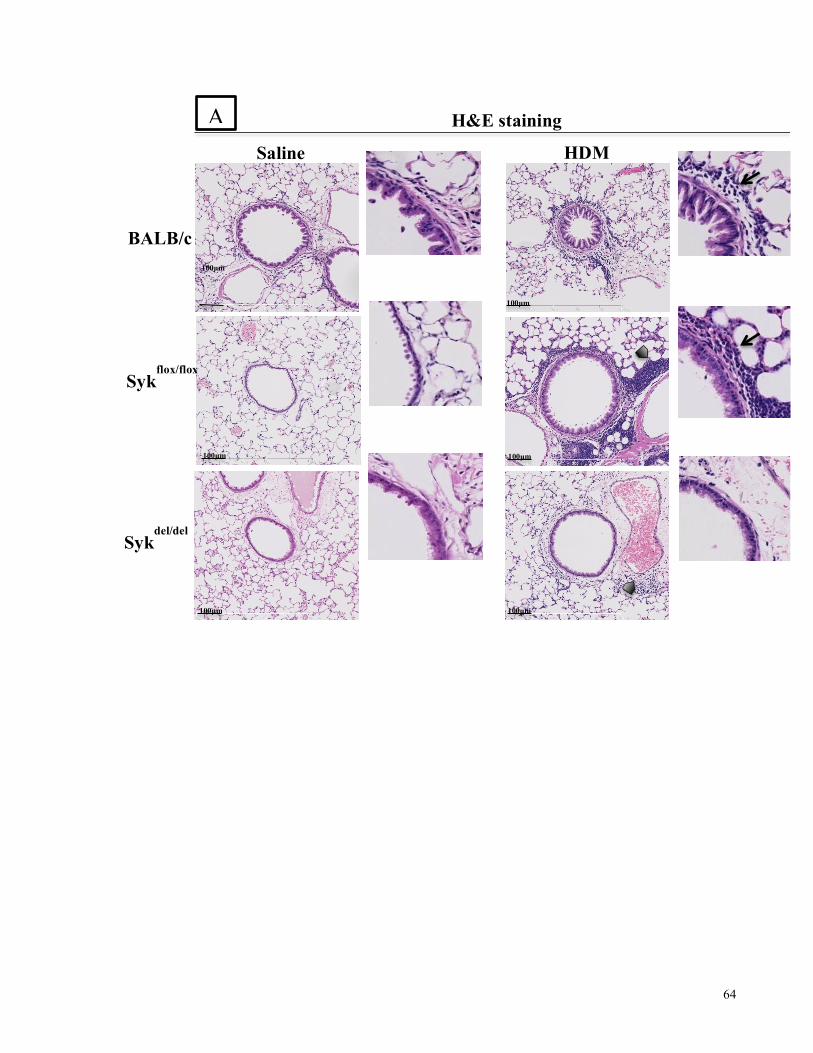
64
100µm
100µm
100µm 100µm
100µm 100µm
BALB/c
HDM Saline
Sykflox/flox
Sykdel/del
H&E staining A

65
Figure 4.7. Histological analysis of airways inflammation and abnormalities in 10-day acute model of HDM-induced airways inflammation in Balb/c and Sykfl/fl-Syk//rosa26-CreERT2 mice
H&E (A) and PAS (B) images of HDM-exposed vs. Saline control mice revealed increased in peribronchial (solid arrows), perivascular (solid pentagons) inflammatory infiltrates and PAS-positive cells (open pentagons), compared with Saline and vehicle controls (representative of n=4-5/group, magnification X200).
100µm
100µm
100µm
100µm
100µm
100µm
BALB/c
PAS staining
HDM Saline
Sykflox/flox
Sykdel/del
B

66
4.2. Chronic model of HDM-induced allergic airways
inflammation
4.2.1. The development of HDM-induced AHR was abrogated by deletion
of Syk
The acute model of airways inflammation is characterized by significant airways
inflammation, mucus plugging of the airways and robust AHR, while the chronic model displays
less marked airways inflammation, moderate levels of AHR and also prominent remodeling [34].
In the previous studies we showed the role of Syk in AHR and inflammation in two
different acute 10-day models. To further assess the effect of Syk in airways responsiveness,
inflammation and remodeling, Sykdel/del and Sykflox/flox mice were exposed to HDM (or Saline) in
an 8-week chronic model, a model that closely mimics the human asthma.
In the control Syk-expressing Sykflox/flox mice, we observed significantly increased MCh-
induced total respiratory resistance (Rrs) in the HDM mice compared with Saline control group
(Figure 4.8.A). On the contrary, in the Sykdel/del mice, HDM sensitization and challenge did not
result in enhanced Rrs to MCh (Figure 4.8.A). In the Syk-expressing Sykflox/flox mice, the maximal
MCh-responsiveness of Rrs (Figure 4.8.B), and the slope of the Rrs dose-response curve (Figure
4.8.C) were significantly increased in the HDM-exposed mice compared with Saline control mice.
We also showed that tissue elastance (E) was significantly elevated in Sykflox/flox HDM-mice
compared with Saline controls. (Figure 4.8.D). On the other hand, no differences between the
HDM and Saline groups were observed in the maximal Rrs, Rrs dose-responsiveness and E in the
Syk-deleted state (Figure 4.8.A-D).
Additionally, we also noticed the central airway Newtonian resistance (RN) and peripheral
tissue damping/resistance to be significantly increased in HDM-treated Sykflox/flox compared with

67
Saline controls, whereas no differences in these two parameters following HDM sensitization and
challenge were observed in the Sykdel/del mice (Figures 4.9.A and 4.9.B).
These results from the 8-week chronic model of airway inflammation similar to acute
model showed that deletion of the Syk abrogates AHR to methacholine in HDM group.
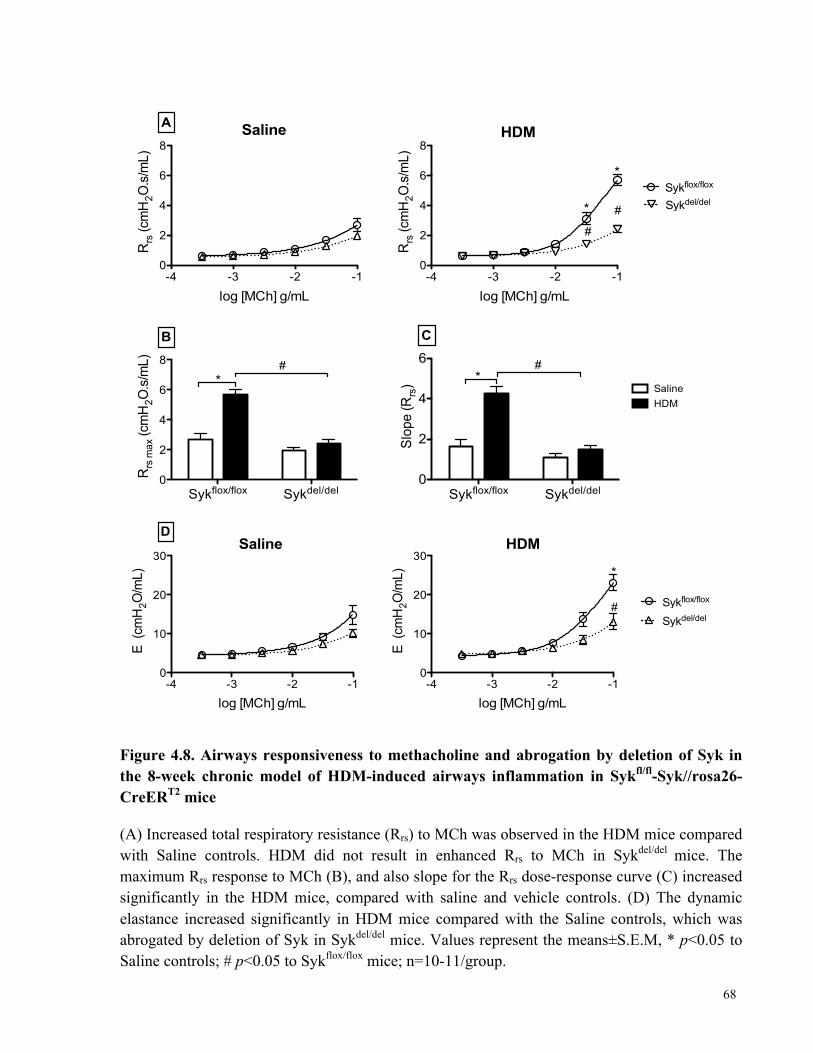
68
Figure 4.8. Airways responsiveness to methacholine and abrogation by deletion of Syk in the 8-week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
(A) Increased total respiratory resistance (Rrs) to MCh was observed in the HDM mice compared with Saline controls. HDM did not result in enhanced Rrs to MCh in Sykdel/del mice. The maximum Rrs response to MCh (B), and also slope for the Rrs dose-response curve (C) increased significantly in the HDM mice, compared with saline and vehicle controls. (D) The dynamic elastance increased significantly in HDM mice compared with the Saline controls, which was abrogated by deletion of Syk in Sykdel/del mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=10-11/group.
-4 -3 -2 -10
2
4
6
8
A Saline
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)
Sykflox/flox Sykdel/del0
2
4
6
8
*
B
#
Rrs
max
(cm
H2O
.s/m
L)
-4 -3 -2 -10
2
4
6
8
Sykflox/flox
Sykdel/del*
#
#
*
HDM
log [MCh] g/mL
Rrs
(cm
H2O
.s/m
L)
Sykflox/flox Sykdel/del0
2
4
6 #*
HDMSaline
C
Slop
e (R
rs)
-4 -3 -2 -10
10
20
30
DSaline
log [MCh] g/mL
E (c
mH
2O/m
L)
-4 -3 -2 -10
10
20
30
Sykflox/flox
Sykdel/del#
*
HDM
log [MCh] g/mL
E (c
mH
2O/m
L)
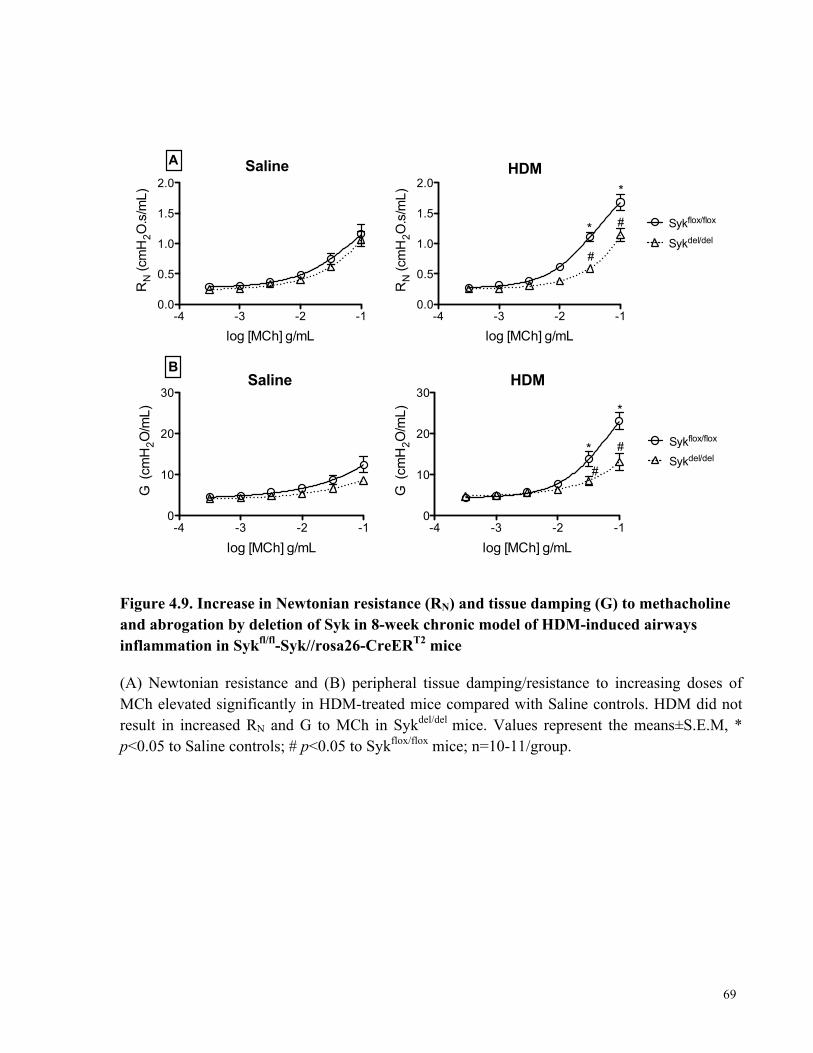
69
Figure 4.9. Increase in Newtonian resistance (RN) and tissue damping (G) to methacholine and abrogation by deletion of Syk in 8-week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
(A) Newtonian resistance and (B) peripheral tissue damping/resistance to increasing doses of MCh elevated significantly in HDM-treated mice compared with Saline controls. HDM did not result in increased RN and G to MCh in Sykdel/del mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=10-11/group.
-4 -3 -2 -10.0
0.5
1.0
1.5
2.0
A Saline
log [MCh] g/mL
RN (c
mH
2O.s
/mL)
-4 -3 -2 -10.0
0.5
1.0
1.5
2.0
Sykflox/flox
Sykdel/del*
*
#
#
HDM
log [MCh] g/mL
RN (c
mH
2O.s
/mL)
-4 -3 -2 -10
10
20
30
BSaline
log [MCh] g/mL
G (
cmH
2O/m
L)
-4 -3 -2 -10
10
20
30
Sykflox/flox
Sykdel/del#
#*
*
HDM
log [MCh] g/mL
G (
cmH
2O/m
L)

70
4.2.2. Deletion of Syk reduced HDM-specific IgG1 and IgE levels in serum
Based on the role of Syk in innate immunity and allergic responses [57, 137], it is possible
that failure to develop increased AHR to MCh in Sykdel/del mice is due to their inability to mount an
immune response to HDM. The levels of HDM-specific IgG1 and IgE were then measured to
evaluate this possibility.
The serum HDM-specific IgG1 and IgE levels were significantly increased in the HDM-
exposed Sykflox/flox mice compared to Saline control group (Figure 4.10). Similar results observed
in Syk-deleted state as HDM sensitization and challenge was able to induce significantly elevated
levels of HDM-specific IgG1 and IgE when compared with Saline controls, although not to the
levels observed in the Syk-expressing mice (Figure 4.10)
These data showed that deletion of Syk resulted in significant reduction in production of
HDM-specific IgG1 and IgE, even though the production was not completely blocked.
Figure 4.10. Serum levels of HDM-specific IgG1 and IgE in the 8-week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
The levels of HDM-specific IgG1 and IgE were elevated significantly in HDM-treated mice compared with Saline controls in both Sykflox/flox and Sykdel/del mice. Deletion of Syk significantly decreased HDM-specific IgG1 and IgE levels in serum. Values represent the means±S.E.M, *p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=4/group.
HDM Specific IgE
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0*
#
HDMSaline
*
OD(450-570)
HDM Specific IgG1
Sykflox/flox Sykdel/del0
2
4
6*
#
OD(450-570)
*

71
4.2.3. Deletion of Syk did not affect BALF total cell counts
Airway inflammation is a feature of the chronic model of allergen-induced airways
inflammation [34, 75, 91]. To evaluate the role of Syk in mediating airway inflammation, we
evaluated the total and differential cell counts in BALF of Sykflox/flox and Sykdel/del mice following
the 8 weeks of chronic HDM sensitization and challenge protocol.
As expected, HDM sensitization and challenge resulted in an increase in the BALF total
leukocyte cell counts with predominant increase in the neutrophils, lymphocytes and eosinophils
population in both Sykflox/flox and Sykdel/del mice.
The total numbers of inflammatory cells in BALF samples of HDM-exposed mice were
significantly increased compared with Saline control in both Sykflox/flox mice (45.64±11.2 × 103 vs.
206.18±46.99 × 103, p<0.05) and Sykdel/del mice (55.19±10.13 × 103 vs. 91.59+3.88 × 103) (Table
4.2). In addition, number of neutrophils, lymphocytes and eosinophils were significantly increased
in HDM-exposed mice compared with Saline control (Table 4.2).
We also showed that deletion of Syk did not have an effect on leukocyte recruitment to the
airways as the total BALF cells counts were similar to those observed in Sykflox/flox (206.18±46.99
× 103 vs. 91.59±3.88 × 103). Deletion of Syk decreased the number of eosinophils and
lymphocytes in BALF while there is no significant difference in number of neutrophils and
macrophages between HDM-exposed Sykflox/flox and Sykdel/del mice (Table 4.2). These findings
show that deletion of Syk did not affect recruitment of leukocytes to the airways following HDM
sensitization and challenge.
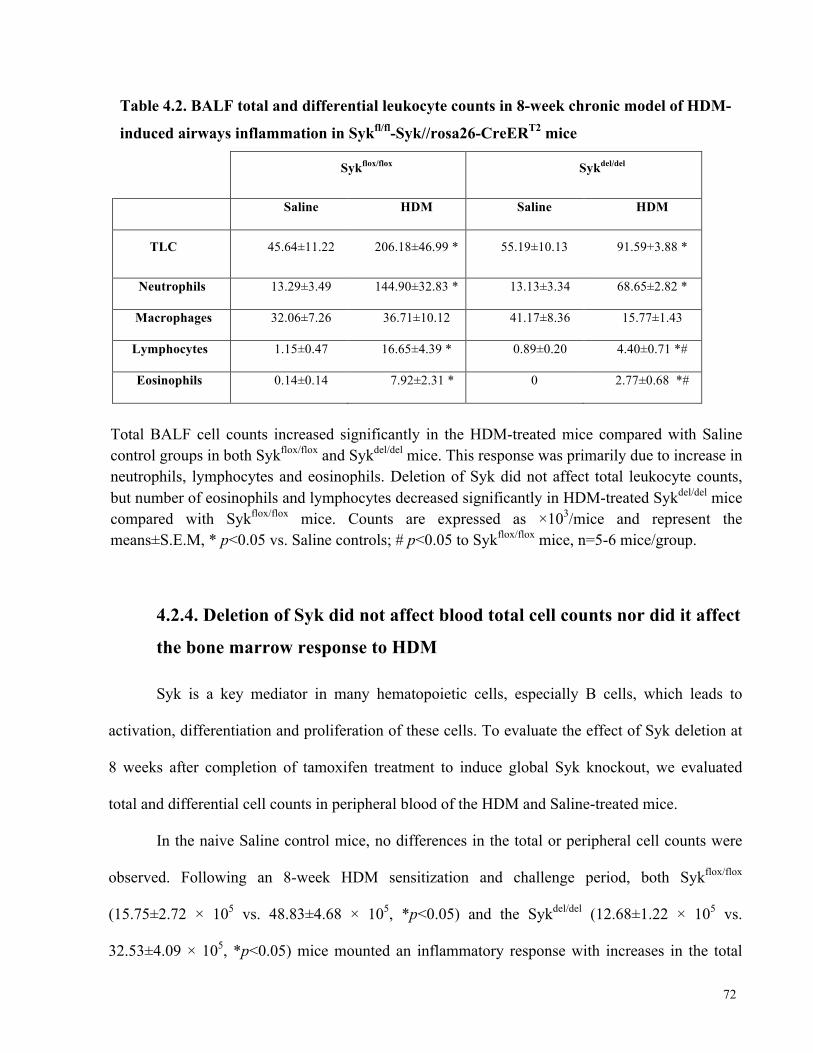
72
Table 4.2. BALF total and differential leukocyte counts in 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
Sykflox/flox Sykdel/del
Saline HDM Saline HDM
TLC 45.64±11.22 206.18±46.99 * 55.19±10.13 91.59+3.88 *
Neutrophils 13.29±3.49 144.90±32.83 * 13.13±3.34 68.65±2.82 *
Macrophages 32.06±7.26 36.71±10.12 41.17±8.36 15.77±1.43
Lymphocytes 1.15±0.47 16.65±4.39 * 0.89±0.20 4.40±0.71 *#
Eosinophils 0.14±0.14 7.92±2.31 * 0 2.77±0.68 *#
Total BALF cell counts increased significantly in the HDM-treated mice compared with Saline control groups in both Sykflox/flox and Sykdel/del mice. This response was primarily due to increase in neutrophils, lymphocytes and eosinophils. Deletion of Syk did not affect total leukocyte counts, but number of eosinophils and lymphocytes decreased significantly in HDM-treated Sykdel/del mice compared with Sykflox/flox mice. Counts are expressed as ×103/mice and represent the means±S.E.M, * p<0.05 vs. Saline controls; # p<0.05 to Sykflox/flox mice, n=5-6 mice/group.
4.2.4. Deletion of Syk did not affect blood total cell counts nor did it affect
the bone marrow response to HDM
Syk is a key mediator in many hematopoietic cells, especially B cells, which leads to
activation, differentiation and proliferation of these cells. To evaluate the effect of Syk deletion at
8 weeks after completion of tamoxifen treatment to induce global Syk knockout, we evaluated
total and differential cell counts in peripheral blood of the HDM and Saline-treated mice.
In the naive Saline control mice, no differences in the total or peripheral cell counts were
observed. Following an 8-week HDM sensitization and challenge period, both Sykflox/flox
(15.75±2.72 × 105 vs. 48.83±4.68 × 105, *p<0.05) and the Sykdel/del (12.68±1.22 × 105 vs.
32.53±4.09 × 105, *p<0.05) mice mounted an inflammatory response with increases in the total
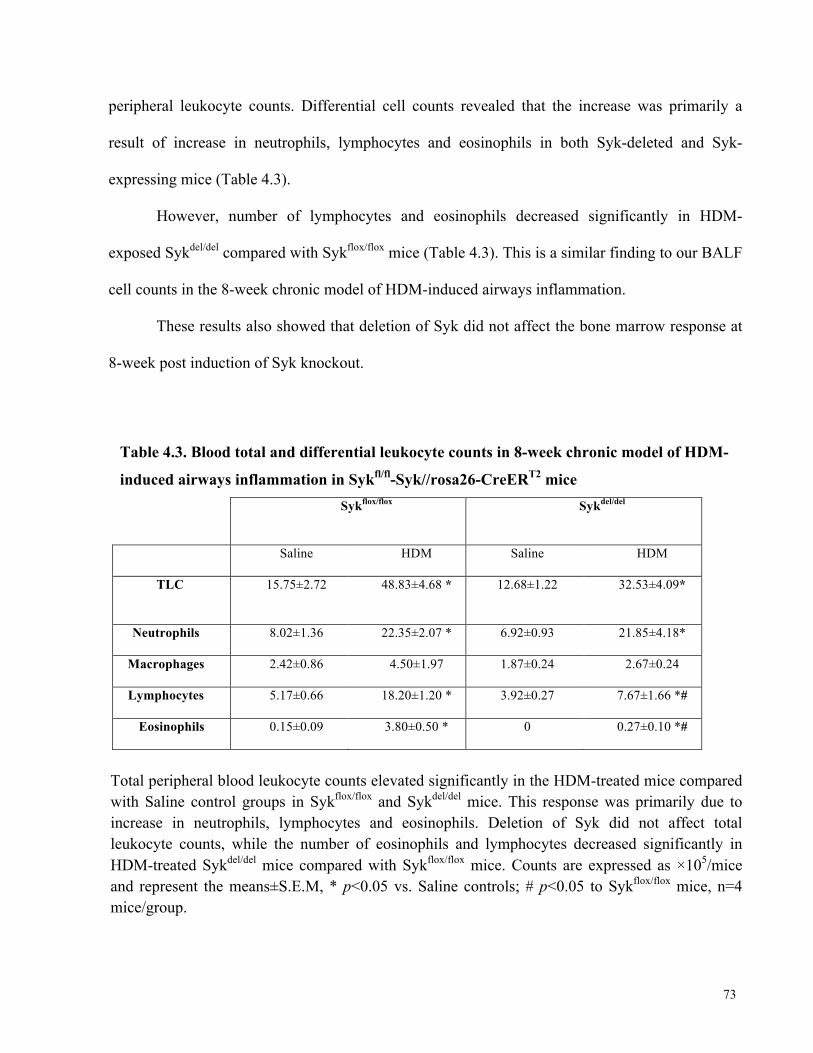
73
peripheral leukocyte counts. Differential cell counts revealed that the increase was primarily a
result of increase in neutrophils, lymphocytes and eosinophils in both Syk-deleted and Syk-
expressing mice (Table 4.3).
However, number of lymphocytes and eosinophils decreased significantly in HDM-
exposed Sykdel/del compared with Sykflox/flox mice (Table 4.3). This is a similar finding to our BALF
cell counts in the 8-week chronic model of HDM-induced airways inflammation.
These results also showed that deletion of Syk did not affect the bone marrow response at
8-week post induction of Syk knockout.
Table 4.3. Blood total and differential leukocyte counts in 8-week chronic model of HDM-
induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
Sykflox/flox Sykdel/del
Saline HDM Saline HDM
TLC 15.75±2.72 48.83±4.68 * 12.68±1.22 32.53±4.09*
Neutrophils 8.02±1.36 22.35±2.07 * 6.92±0.93 21.85±4.18*
Macrophages 2.42±0.86 4.50±1.97 1.87±0.24 2.67±0.24
Lymphocytes 5.17±0.66 18.20±1.20 * 3.92±0.27 7.67±1.66 *#
Eosinophils 0.15±0.09 3.80±0.50 * 0 0.27±0.10 *#
Total peripheral blood leukocyte counts elevated significantly in the HDM-treated mice compared with Saline control groups in Sykflox/flox and Sykdel/del mice. This response was primarily due to increase in neutrophils, lymphocytes and eosinophils. Deletion of Syk did not affect total leukocyte counts, while the number of eosinophils and lymphocytes decreased significantly in HDM-treated Sykdel/del mice compared with Sykflox/flox mice. Counts are expressed as ×105/mice and represent the means±S.E.M, * p<0.05 vs. Saline controls; # p<0.05 to Sykflox/flox mice, n=4 mice/group.

74
4.2.5. Syk mediates HDM-induced expression of IL-6, IL-17, KC and
RANTES in the chronic model of HDM-induced airways inflammation
We also assessed airway inflammation by evaluating the production of inflammatory
mediators in the BALF samples.
We found significant increase in the BALF concentration of IL-6, IL-17, KC, and RANTES
in HDM-exposed Sykflox/flox mice compared with Saline control group (Figure 4.11). In the
Sykdel/del mice, HDM sensitization and challenge failed to increase production of IL-6, IL-17, KC
and RANTES (# p<0.05). The levels of TNF-α and VEGF were not different amongst different
experimental groups. We also studied IL-4 and IL-10 production; however, concentration of these
two mediators was below detectable levels in BALF samples.
Similar to acute model, these results show that the levels of IL-6 and KC increased
significantly in HDM-treated Sykflox/flox compared with saline controls, while oppose to acute
model, the increase in levels of these two mediators was not affected by Syk deletion.

75
Figure 4.11. Level of inflammatory mediators in the BALF in 8-week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice Production of IL-6, IL-17, KC and RANTES in the BALF increased significantly in HDM-treated Sykflox/flox mice. In the Syk-deleted state, their production was decreased significantly when compared with Sykflox/flox mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice, n=4-5/group.

76
4.2.6. Effect of Syk deletion on expression levels of inflammatory
mediators in lung tissue
We also assessed the effects of Syk on expression levels of IL-6, KC (CXCL-1), VEGF,
MMP-9, TGF-β1 and FGF-2 at the gene level using qPCR techniques. As shown in figure 4.12, we
observed significant increases in expression levels of IL-6 and KC in HDM-treated group
compared to Saline control group in the Sykflox/flox mice in 8-week chronic model (*p <0.05). We
observed difference between the Sykflox/flox and Sykdel/del mice following HDM exposure in the
levels of KC, which was significantly reduced in the Syk deleted state (# p<0.05). In the Sykdel/del
mice, while there appeared to be trend to increased IL-6 expression in response to HDM, this
increase was not statistically significant. The expression levels of VEGF, MMP-9, TGF-β1 and
FGF-2 were not different amongst different experimental groups.
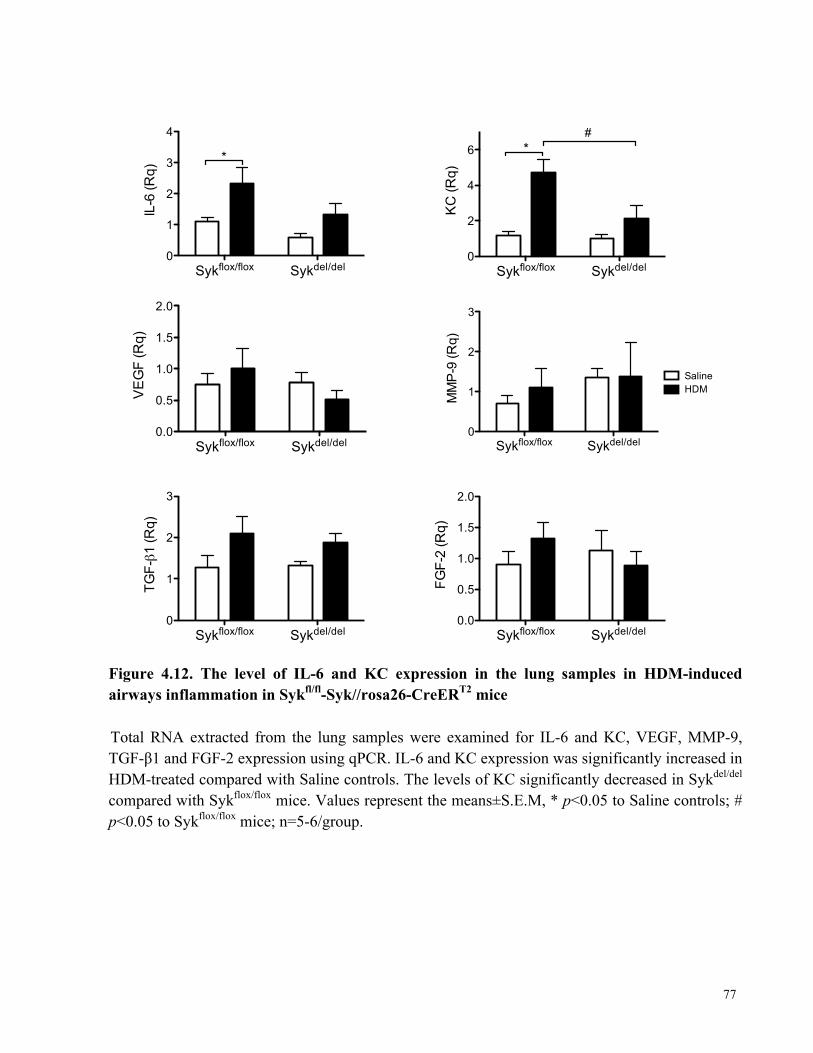
77
Figure 4.12. The level of IL-6 and KC expression in the lung samples in HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice Total RNA extracted from the lung samples were examined for IL-6 and KC, VEGF, MMP-9, TGF-β1 and FGF-2 expression using qPCR. IL-6 and KC expression was significantly increased in HDM-treated compared with Saline controls. The levels of KC significantly decreased in Sykdel/del compared with Sykflox/flox mice. Values represent the means±S.E.M, * p<0.05 to Saline controls; # p<0.05 to Sykflox/flox mice; n=5-6/group.
Sykflox/flox Sykdel/del0
1
2
3
4
*IL
-6 (R
q)
Sykflox/flox Sykdel/del0
1
2
3
TGF-!1
(Rq)
Sykflox/flox Sykdel/del0
2
4
6 *#
KC
(Rq)
Sykflox/flox Sykdel/del
0.0
0.5
1.0
1.5
2.0
VE
GF
(Rq)
Sykflox/flox Sykdel/del0.0
0.5
1.0
1.5
2.0
FGF-
2 (R
q)Sykflox/flox Sykdel/del
0
1
2
3
SalineHDM
MM
P-9
(Rq)

78
4.2.7. Deletion of Syk attenuated the degree of epithelium modification,
mucus cell hyperplasia and increase in smooth muscle mass
The chronic mouse models of allergen-induced airways inflammation typically show
evidence of chronic airway inflammation and airway remodeling. We evaluated these features in
our 8-week chronic HDM model in lung sections stained with H&E, masson trichrome and PAS.
Histologic evaluation of lung inflammation using H&E staining subjected to HDM
exposure corroborated our findings in the BALF total leukocyte counts. Histological analysis
demonstrated peribronchial and perivascular airway inflammation (solid pentagons) in HDM-
treated Sykflox/flox and Sykdel/del mice compared with Saline controls (Figure 4.13.A).
Histological analysis by masson trichrome staining demonstrated an increase in
subepithelial fibrosis (solid arrows), increase in smooth muscle mass (open arrows) and epithelial
modification (open pentagons) in both HDM-exposed Sykflox/flox and Sykdel/del mice compared with
Saline controls. 4.14.B). In addition, PAS staining of the lung sections showed increase in number
of PAS-positive cells (solid chevron) that are indicative of Goblet cells in the HDM group in both
Sykflox/flox and Sykdel/del compared to Saline controls. (Figure 4.14.C).
We performed semi-quantitative analysis of remodeling on lung sections from different
experimental groups. The analysis was performed on 5-6 mice per experimental group and the
lung sections were assigned a score of normal to ++++ (normal, no remodeling; +, mild
remodeling; ++, moderate remodeling; +++, severe remodeling; ++++, extreme remodeling).
Table 4.4 summarizes remodeling features in different experimental groups.
Histological analysis revealed airway remodeling, i.e., fibrosis, epithelial thickening, mucus cell
hyperplasia and increase in smooth muscle cell mass, in this chronic model of HDM-induced
allergic airway inflammation, which was attenuated in some degree by deletion of Syk.
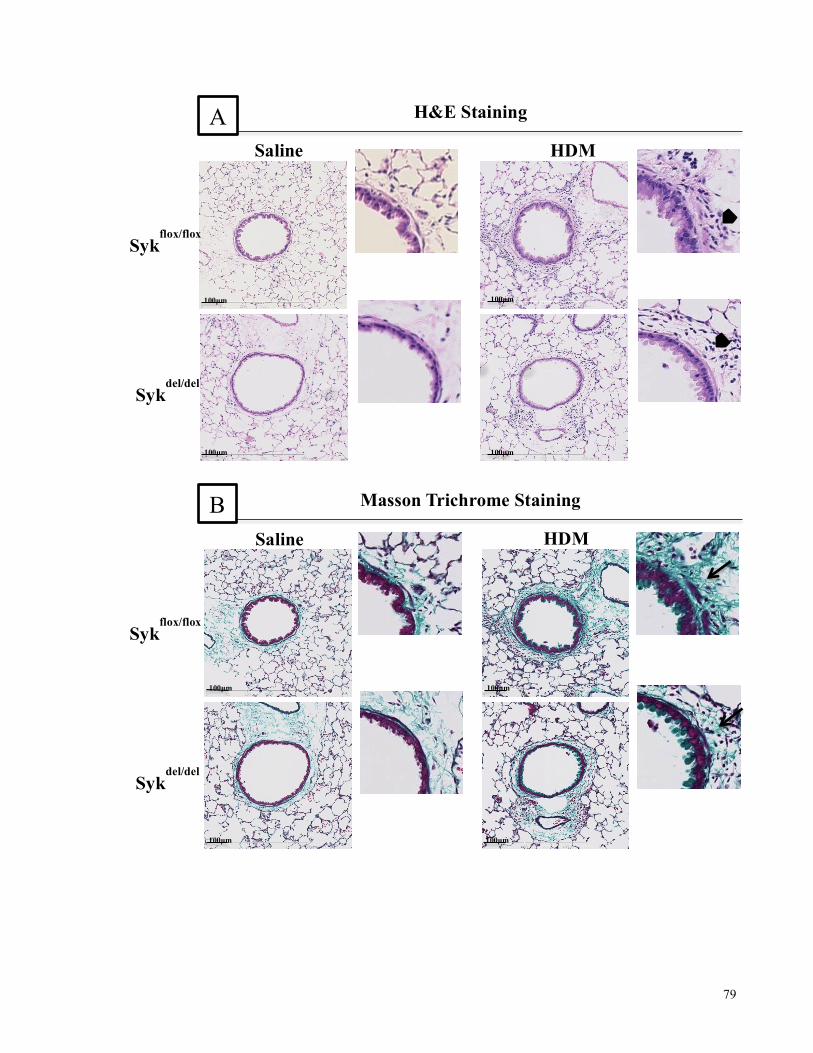
79
100µm
100µm 100µm
100µm
Sykflox/flox
HDM Saline
Sykdel/del
H&E Staining A
100µm
100µm 100µm
100µm
Sykflox/flox
Sykdel/del
Saline HDM
Masson Trichrome Staining B
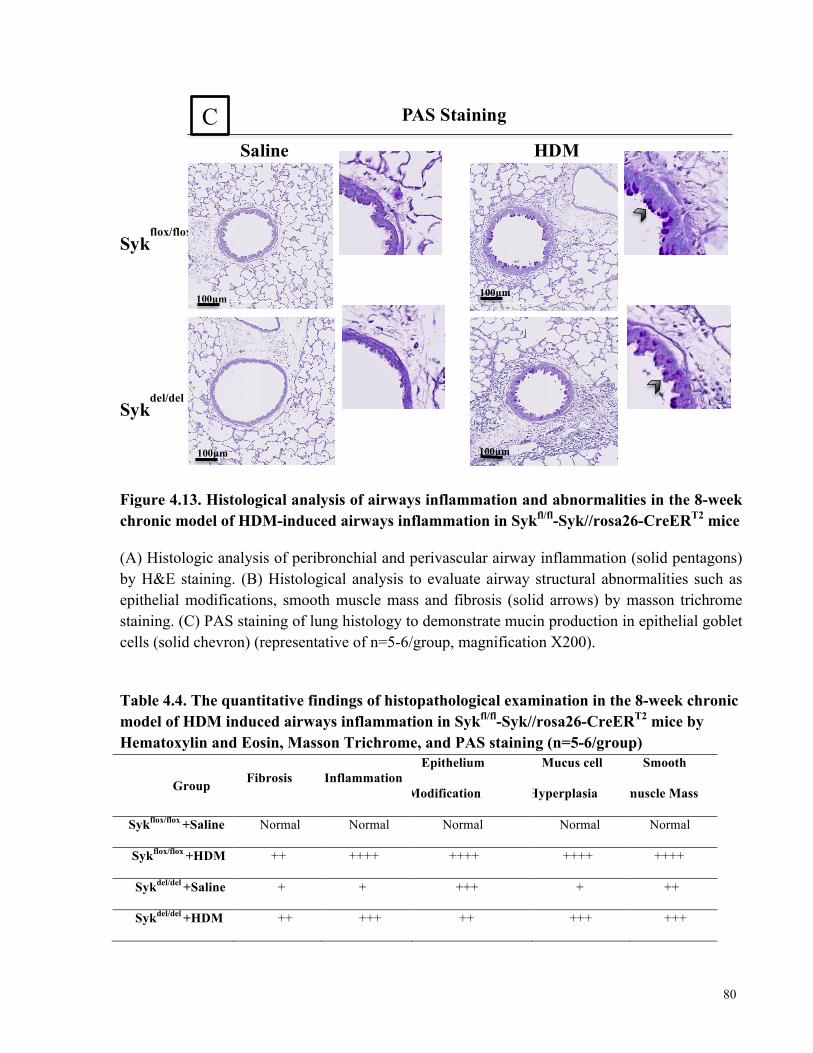
80
Figure 4.13. Histological analysis of airways inflammation and abnormalities in the 8-week chronic model of HDM-induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice
(A) Histologic analysis of peribronchial and perivascular airway inflammation (solid pentagons) by H&E staining. (B) Histological analysis to evaluate airway structural abnormalities such as epithelial modifications, smooth muscle mass and fibrosis (solid arrows) by masson trichrome staining. (C) PAS staining of lung histology to demonstrate mucin production in epithelial goblet cells (solid chevron) (representative of n=5-6/group, magnification X200).
Table 4.4. The quantitative findings of histopathological examination in the 8-week chronic model of HDM induced airways inflammation in Sykfl/fl-Syk//rosa26-CreERT2 mice by Hematoxylin and Eosin, Masson Trichrome, and PAS staining (n=5-6/group)
Group Fibrosis Inflammation Epithelium
Modification
Mucus cell
Hyperplasia
Smooth
muscle Mass
Sykflox/flox +Saline Normal Normal Normal Normal Normal
Sykflox/flox +HDM ++ ++++ ++++ ++++ ++++
Sykdel/del +Saline + + +++ + ++
Sykdel/del +HDM ++ +++ ++ +++ +++
Sykdel/del
Sykflox/flox
PAS Staining
Saline HDM
100µm
100µm 100µm
100µm
C

81
Chapter 5: Discussion
Asthma is an important health problem worldwide with a prevalence that continues to
increase despite the availability of effective anti-inflammatory and bronchodilator drugs for asthma
control. However, there is no permanent cure. In vivo animal models, especially those employing
mice, have been widely used to investigate the cause and identify potential therapies for asthma.
Although they do not recapitulate all aspects of the disease, animal studies are nonetheless, very
useful for the understanding the pathogenic mechanisms that lead to asthma for the evaluation of
novel drugs as therapies [121, 122].
Syk was initially identified as a mediator involved in innate immunity, especially in B cell
function, and later, in allergic immunity [144]. Consequently, several studies have focused on Syk
as a potential target to suppress allergic responses in airway inflammation and in allergic asthma
[2, 159, 160].
In this thesis, we assessed the effect of Syk inhibition on specific features of asthma that
include AHR, remodeling and inflammation in two mouse models of HDM-induced allergic
airways inflammation.
In order to analyze the role of Syk in different asthma phenotypes, we designed two
different models of HDM-induced airway inflammation, the acute and chronic models. The acute
model is characterized by AHR, prominent airway inflammation and minimal degree of airway
remodeling, while the chronic model is characterized by AHR, minimal airways inflammation and
remodeling [34]. The results from our acute model revealed AHR, significant increase in
inflammatory cell influx into the airways, and increased in numbers of mucus producing cells [1,
125], while the chronic model showed AHR, airway inflammation and as expected features of

82
remodeling such as fibrosis [125], epithelium modification, mucus cell hyperplasia and increase in
smooth muscle cell mass [50, 75].
We demonstrated that in vivo inhibition of Syk by selective Syk inhibitors, and genetic
deletion of Syk using conditional Syk knockout mice attenuated AHR despite of inflammatory cell
influx in the lung. We also showed for the first time that deletion of Syk attenuated development
of key features of remodeling in the chronic mouse model of airway inflammation.
Our results revealed the importance of Syk in airway hyperresponsiveness and remodeling
in HDM mouse model of allergic inflammation and asthma. More interestingly, it also showed the
importance of Syk in non-inflammatory cells in the AHR and remodeling in asthma. This has not
been previously reported.
The effect of Syk on airway hyperresponsiveness
Earlier reports have suggested that administration of the Syk inhibitors during allergen
challenge prevent development of AHR [2, 159, 160]. Stenton et al. reported the effect of Syk
antisense oligodeoxynucleotides (ASO) delivered in liposome complexes on AHR of Brown-
Norway rat model of OVA-induced allergic airways inflammation. They showed inhibition of Ag-
induced contraction of isolated trachea in mice pretreated with Syk-ASO 3 days before the antigen
challenge [160]. Matsubara et al. showed the effect of Syk inhibitor, R406, on development of
allergen-induced AHR in 10-day OVA-induced mouse model. AHR was detected as a change in
the contractile response of tracheal smooth muscle ring to electric field stimulation (EFS) [2, 159].
In another model system, which showed to be independent of B cell and mast cells, they revealed
that administration of R406 during antigen challenge prevents the development of AHR [2, 159].
A previous study in our laboratory revealed the in vivo effect of the Syk inhibitor NVP-
QAB-205 on AHR. In contrast to previous studies, Syk inhibitor was given after establishment of

83
airway inflammation in a 12-week OVA-sensitized and -challenged mouse model. This study
showed the airway epithelial cell expression of Syk was most prominent in the asthmatic airways
and demonstrate the importance of Syk activity in airway epithelium in the development of AHR
[34].
In a different model described in this thesis, we used a 10-day acute model of HDM-
induced allergic airways inflammation. We showed for the first time that inhibition of Syk in vivo
in mice after establishment of disease phenotype abrogated AHR. A treatment-based protocol,
which is applicable for the management of asthma, was used for administration of 0.3 mg/kg of
Syk inhibitor NVP-QAB-205. Treatment with single intratracheal (i.t.) dose of NVP-QAB-205 in
HDM-sensitized and -challenged mice decreased the total airway resistant (Rrs) to the levels
similar to Saline control mice (Figure 4.1). Our observations are significantly different from the
previous studies that used Syk inhibitor concurrent with the allergen sensitization and challenge [2,
159, 160].
In addition, we investigated the role of Syk in HDM-induced 10-day acute and 8-week
chronic models of airway inflammation using inducible Syk knockout mice, Sykfl/fl-Syk//rosa26-
CreERT2, which express a Syk knockout phenotype based on the tamoxifen-induced CreERT2
system. It has been shown in short term experiments that deletion of Syk after tamoxifen treatment
had no adverse effects on basic body functions and mice exhibited healthy normal behavior [57].
We observed that the development of AHR in HDM-exposed Sykdel/del mice was abrogated
to the level similar to Saline control mice in both acute (Figure 4.2) and chronic models (Figure
4.8). Airway elastance was also abrogated by Syk deletion, which showed that Syk knockout
decrease elastic rigidity of the lung (Figures 4.2 and 4.8).
In addition, we also observed abrogation of airways central Newtonian resistant (RN) and
peripheral tissue damping (G) in Sykdel/del mice. These observations support that deletion of Syk

84
results in reduction of both the central and peripheral airways responsiveness to MCh. (Figures 4.3
and 4.9).
The role of Syk on airway inflammation
One distinctive feature of allergic asthma is the presence of inflammatory cell infiltration in
bronchoalveolar space. Increase in number of neutrophils, lymphocytes and eosinophils in the
bronchoalveolar lavage fluid have been previously reported in HDM models of allergic airways
inflammation [1, 75, 125, 132]. In our studies, we showed that HDM sensitization and challenge
by the intranasal instillation induced leukocyte recruitment to the BALF. Both acute and chronic
mouse models were characterized by increased numbers of neutrophils, eosinophils and
lymphocytes infiltrates in the BALF. Thus, our protocols of HDM sensitization and challenge
worked effectively in producing models of allergic inflammation.
Not unexpectedly, the single dose of NVP-QAB-205 had no effect on the BALF total or
differential cell counts in the HDM-treated mice. We showed that the in vivo intratracheal
administration of 0.3 mg/Kg of NVP-QAB-205 for 15 minutes did not affect inflammatory cells
influx in 10-day acute model of HDM-induced allergic inflammation and asthma (Table 4.1.A).
Earlier report by our laboratory also showed similar findings after administration of 0.3 and 3
mg/kg NVP-QAB-205 in a 12-week chronic OVA-sensitized and –challenged mouse model of
airway inflammation [34].
To further investigation for the role of Syk in airway inflammation, blood and BALF total
and differential leukocyte count was measured in both 10-day acute and 8-week chronic models of
HDM- induced allergic inflammation using inducible Syk knockout mice.
The total and differential leukocyte counts under basal condition (Saline treated) were
similar between Sykflox/flox and Sykdel/del mice in the peripheral blood from the chronic model. The

85
total leukocyte counts increased significantly in peripheral blood in HDM-sensitized and -
challenged mice compared with the control Saline treated in both Sykflox/flox and Sykdel/del groups
(Table 4.3). These observations support that even 12 weeks after induction of Syk knockout, the
bone marrow was still responsive in the Sykdel/del mice. Although deletion of Syk did not affect
total leukocyte counts in HDM-treated groups, Sykdel/del mice exhibited significantly lower number
of eosinophils and lymphocytes in the peripheral blood compared with Sykflox/flox mice (Table 4.3).
These observations are in keeping with those of Wex et al. that showed long-term deletion of Syk
leads to a gradual reduction of B lymphocytes in peripheral blood [185].
The results also showed that deletion of Syk did not affect the total leukocyte number of
inflammatory infiltrates in BALF in both acute and chronic models. Our findings revealed that
only the number of eosinophils and lymphocytes dropped significantly in the BALF of Sykdel/del
mice compared to Sykflox/flox (Tables 4.1.B and 4.2). The lower number of eosinophils and
lymphocytes in the BALF of Sykdel/del mice may be the result of decrease in bone marrow
production of these cells rather than impaired leukocyte recruitment as these two cell population
were also lower in the peripheral blood. Our results are consistent with the study Wex et al. that
reported normal neutrophil and monocyte migration in Sykdel/del mice in different models of
airway inflammation. They did not observe an effect of Syk on total cell counts from BALF, but
reported significant decrease in the number of eosinophils in OVA model of Sykdel/del mice
compared with Sykflox/flox [57].
Histological examination of lung sections using H&E staining also showed peribronchial and
perivascular infiltration with inflammatory cells in the HDM-exposed compared with Saline
control groups in the Balb/c, Sykflox/flox and Sykdel/del mice. Neither treatment with NVP-QAB-
205 nor deletion of Syk showed any effect on the presence of inflammatory infiltrates.

86
Together, results of our study using conditional Syk knockout mice suggest that Syk
does not play a role in leukocyte recruitment.
The role of Syk on serum immunoglobulin production
Syk is crucial mediator of humoral and allergic responses [49, 50]. Our data shows that
HDM exposure resulted in increased serum levels of HDM-specific immunoglobulins such as
IgG1 and IgE Syk dependently. The serum HDM-specific IgG1 and IgE levels were significantly
increased in HDM-exposed Sykflox/flox mice compared to Saline control groups. In the Syk-deleted
state, HDM sensitization and challenge was also able to induce significantly elevated levels of
HDM-specific IgG1 and IgE when compared with Saline controls, even though not at levels
observed in the Syk-expressing mice. In addition, deletion of Syk decreased the levels of both
HDM-specific immunoglobulins in HDM-exposed Sykdel/del mice significantly compared to
Sykflox/flox mice (Figures 4.4 and 4.10). In other words, despite loss of Syk, the Sykdel/del mice were
stay able to mount the antibody response.
The effects of Syk on inflammatory mediator production
Although histologic features of airway remodeling have been well characterized in asthma,
the underlying responsible inflammatory and immunologic mechanisms are still not completely
understood. Studies have shown that several inflammatory and structural cell types and their
mediators are involved in airway remodeling. Additionally, different characteristics of airway
remodeling are likely mediated by different inflammatory pathways [186]. Increased levels of
inflammatory mediators such as IL-6, IL-8, IL-17, VEGF and also RANTES are considered to be
recognized hallmark of asthmatic airways [64].
To understand the cellular mechanisms that are regulated by Syk following HDM

87
sensitization and challenge, we investigated inflammatory mediator production in BALF and gene
expression in the whole lung. We found significant increases in the levels of KC (or CXCL-1, the
murine homologue of human IL-8), IL-6, IL-17 and RANTES in BALF of the chronic model of
HDM-induced airway inflammation. Production of these mediators was abrogated by the loss of
Syk in Sykdel/del mice. A trend was also observed toward reduction of VEGF and TNF-α after loss
of Syk (Figure 4.11).
Many studies have implicated a role of these inflammatory mediators in asthma
pathogenesis.
IL-8 plays a key role in the activation of neutrophils and is considered as a potent
chemoattractant that recruits neutrophils to the airway [29, 187]. IL-6 is a cytokine, which is
produced by inflammatory cells, and lung epithelial cells in response to a variety of different
stimuli including allergens and respiratory virus [188, 189] and is thus implicated in pathogenesis
of asthma.
Increased in the levels of IL-8 and IL-6, and associated signal molecules were also
observed in primarily cultured nasal epithelial cells from patients with allergic rhinitis after HDM
stimulation [190]. Lee et al. also observed increased in expression of IL-8 and IL-6 mRNA after
exposing the human monocyte cell line to HDM [191]. This group showed that HDM increased
IL-6 and IL-8 expression, and transduces its signal through Src family tyrosine kinase, protein
kinase C (PKC), extracellular-signal-regulated kinase (ERK) and p38 mitogen-activated protein
kinase [191].
Previous in vitro studies in our laboratory revealed that Syk regulates expression of IL-8,
IL-6 and VEGF in response to HRV infection following engagement of intercellular adhesion
molecule-1 (ICAM-1) in BEAS-2B airway epithelial [66] and primary airway epithelial cells [29].
In addition, in an in vivo study in our laboratory, the higher levels of IL-8 and VEGF were

88
observed in OVA/OVA mice compared with OVA/PBS group in an OVA murine model of
airways inflammation [34].
A growing body of evidence reveals that the levels of IL-6 are elevated in blood, BALF,
and lung tissues of asthmatic patients [189]. A study investigating IL-6 in BALF showed higher
levels of IL-6 in active asthmatic patients compared with healthy non-smoker subjects, stable
asthmatic and non-asthmatic patients receiving mechanical ventilation [192]. In addition,
comparing increased levels of IL-6 in BALF from 18 mild-moderate allergic asthmatic subjects
and 16 healthy controls have also been reported, suggesting that IL-6 may play a role beyond
patients with allergic asthma [193].
A recent study in mild allergy showed the selective presence of IL-6 in the sputum of
asthmatic patients without active inflammation compared with healthy subjects [193], suggesting
that the presence of IL-6 in the airways may be independent of inflammation. In addition, another
study has revealed high levels of IL-6 mRNA in mouse primary lung epithelial cells, but not in
immune cells resident in the lung. They also showed that the p38 mitogen-activated protein kinase
pathway mediates regulation of IL-6 in response to fungal β-glucan extracts [194].
There is also growing evidence for involvement of IL-17 in the pathogenesis of asthma.
IL-17 organizes airway neutrophilic influx [195] and increases eosinophilic airway inflammation
mediated by Th2 in asthma [196].
The levels of IL-17 in sputum, BALF, bronchial biopsies and serum has been shown to be
increased in asthmatic patients compared with healthy control subjects [195]. Chakir et al. showed
that patients with moderate to severe asthma exhibit an increased number of IL-17-positive cells in
bronchial biopsies compared to patients with mild asthma and healthy controls [197]. In a study on
OVA challenged mice, increase in IL-17 protein and mRNA were observed in macrophages [198].
Another study demonstrated that the average concentrations of IL-17 mRNA is higher in bronchial

89
biopsies from patients with severe asthma than in healthy controls [107]. Sun et al. found increase
in levels of IL-17 protein in sputum from patients with severe asthma in compare with healthy
controls [199].
In an animal model of asthma, Wang et al. showed that the influx of Th17 cells happen
within the first 3 hours after the last challenge They suggested that the rapid influx of Th17 cells
may be part of the inflammatory processes triggered by the injured epithelial cells stimulated by
environmental stimuli or invaded pathogens at the acute phase of allergic asthma [200].
RANTES is a chemokine family, which activates inflammatory cells such as eosinophils,
T lymphocytes, basophils, monocytes/ macrophages, and mast cells, and is a potent
chemoattractant for these cells [201]. Accumulating evidence suggests that many cell types present
in asthmatic airways produce RANTES, which supports the potential role of this chemokine in
asthma [202]. In a current study of 60 Egyptians, the serum levels of RANTES were significantly
higher in the asthmatic patients compared to the controls. This increase was higher in patients with
moderate and severe asthma compared to patients with mild asthma. Levels of serum RANTES
corresponded to eosinophil count as well as total serum IgE [203]. In addition, Zietkowski et al.
showed that anti-IgE therapy in a group of 19 patients with severe persistent allergic asthma,
decreased activation of inflammatory cells and release of RANTES [201].
Increased levels of VEGF and its receptor have been reported in asthmatic airways [204,
205] as well as animal models [206]. It is increased in sputum, bronchial biopsies and BALF of
asthmatic patients [204].
A number of other studies demonstrated increased levels of VEGF in biologic fluids from
patients with asthma, where they correlate with disease severity [206, 207]. In a study on human
asthma, it was shown that vascular remodeling and increased expression of VEGF are features of
moderate to severe persistent asthma and non-asthmatic eosinophilic bronchitis [205, 208]. Lee et

90
al. used VEGF lung transgenic mice and showed that VEGF is a potent stimulator of
inflammation, airways and vascular remodeling. This leads to physiologic dysregulation that
increases antigen sensitization, Th2 inflammation and also number and activation of DCs [206].
Taken together, results of our study using conditional Syk knockout mice confirmed the
role of Syk in production of inflammatory mediators such as KC, IL-6, IL-17, and also
RANTES that have been implicated in asthma pathogenesis. These observations offer some
insight for the potential mechanisms of how Syk can regulate airways reactivity and also
airway epithelial-smooth muscle interaction.
The effects of Syk on airway remodeling
Asthma is a chronic inflammatory disease, which results in airway remodeling that affects
both epithelial and subepithelial components and contributes to clinical symptoms [50, 64]. These
changes include a wide range of pathophysiologic features, such as epithelial changes, increase in
smooth muscle mass, subepithelial fibrosis and vascular changes [50, 64, 97].
In this study, we used the chronic HDM model specially to evaluate the role of Syk in
airway remodeling. We showed for the first time that Syk is involved in some features of airway
remodeling such as epithelial modifications, mucus cell hyperplasia, and increase in smooth
muscle mass. These features were attenuated to some degree in Sykdel/del mice exposed to HDM
compared with Sykflox/flox.
We observed epithelial modifications in Sykflox/flox mice that exposed to HDM
chronically, while deletion of Syk attenuated the degree of modifications (Figure 4.13).
A previous study by our laboratory revealed a role of Syk in airway epithelial cell
migration, proliferation, and survival. Using an in vitro wounding model with the BEAS-2B cell
line, we showed that Syk plays a role in wound repair by mediating cell proliferation and

91
migration [29]. It suggests a role for Syk in maintaining the homeostatic integrity of the epithelial
cells in response to injuries and also viral infections. Another group, Cheng et al., used
homozygous Syk mutants to investigate the role of Syk in vivo. They suggested that Syk has a
critical role in maintaining the wound healing and vascular integrity during embryogenesis [151].
In addition, studies have also showed a role of Syk in regulation of cell proliferation and
migration in breast epithelial cells [209, 210]. Syk also regulates cell proliferation and
differentiation in hematopoietic cells. Its activity is pivotal for regulating of B cell homeostasis,
while dysregulation of Syk activity causes abnormal proliferation, results in different types of B
cell lymphoma [211].
Goblet cell hyperplasia has been consistently shown in mild, moderate and severe asthma,
with the most significant increases in severe fatal asthma [212]. We observed significant increase
in the number of goblet cells in both the acute 10-day and chronic 8-week models of HDM-
induced airways inflammation. Goblet cell hyperplasia was observed using PAS staining in both
models. We noticed extensive distribution of goblet cells in the bronchial epithelium of HDM
exposed mice. In contrast, knocking out Syk reduced number of goblet cells (Figure 4.7.B and
Figure 4.13.C).
Different studies showed significant increase in the number of goblet cells in the lung
airway epithelium of HDM-exposed group compared with the Saline control group [1, 50, 75, 125,
132]. Southam et al. observed increase in mucin-containing goblet cells 1, 2, 3, 6, and 8 weeks
following intranasal exposure to HDM compared with the control group. They suggested that this
elevation was independent of the duration of allergen exposure as there was no significant increase
among different weeks of HDM exposure [75]. They observed that attenuation in maximum
airway resistance is associated with the attenuation in goblet cells [75]. Mucus hyper-secretion
from goblet cells results in airway mucus plugging, particularly in the peripheral airways. Mucus

92
plugging has been associated with severity of acute asthma [132]. Furthermore, they also
suggested that D.farinae transforms ciliated epithelial cells into goblet cells [132]. Mastubara et al.
observed increase in PAS positive or goblet cells after 10 days of repeated exposure to OVA
compared to Saline control group, while treatment with 3 mg/kg of Syk inhibitor R406 twice a day
decreased the number of goblet cells [159]. This Syk regulation of goblet cells differentiation and
hyperplasia may be a mechanism in pathogenesis of asthma.
We observed increase in subepithelial fibrosis in HDM-exposed mice compared with the
Saline controls using Masson’s Trichrome staining. Results also reveal that knocking out of the
Syk did not affect formation of subepithelial fibrosis in this model of allergic inflammation and
asthma (Figure 4.13.B).
Airway biopsies from asthmatic patients suggested that extent of subepithelial fibrosis is
directly related to the severity of the disease [213]. Wakahara et al. showed increase in the area of
subepithelial fibrosis after repeated instillation of D.farinae dose dependently. These alterations
were mainly observed in the central large airways, though most of the peripheral airways were
intact [132].
During inflammation such as asthmatic airways, fibroblasts are first activated and then
differentiated into myofibroblasts that release proinflammatory mediators, α-smooth muscle actin
as well as extracellular matrix (ECM) [64, 96].
Matrix metalloproteinases (MMPs) are enzymes capable of extracellular matrix molecules
degradation [64, 212]. MMP-9 and it’s inhibitor, tissue inhibitor of metalloproteinase-1 (TIMP-1),
are critical for determination of matrix remodeling in asthma [214]. It is also suggested that
profibrotic balance between two MMP-9 and TIMP-1 is important for determination of fibrosis
[104]. TGF-β expression is also increased in asthmatic airways compared to controls and
correlates with number of fibroblasts in airway, subepithelial fibrosis, and severity of the disease

93
[215, 216]. We did not observe any changes in expression of MMP-9, TGF-β, and also fibroblast
growth factor-2 (FGF-2) in the 8-week chronic model of HDM-induced airway inflammation and
asthma, suggesting that other mechanisms might be involved in subepithelial fibrosis in asthma.
Airway smooth muscle (ASM) cells are the main structural cells in the bronchi and are
considered to be primary cells affected the airway remodeling. ASM mass increases as a result of
ASM cell proliferation (hyperplasia) and increase in cell size (hypertrophy) in the airways of
asthmatic patients [64, 212, 217]. ASM mass is correlated with severity and duration of the disease
as it is bigger in fatal cases and older patients compared with non-fatal and younger patients [64,
212].
We observed increased ASM mass in 8-week chronic model of HDM-induced airway
inflammation compared with the Saline control group. We also showed that deletion of Syk
attenuates the area of smooth muscle in some degree (Figure 4.13).
In a study on a HDM 8-week chronic model, increased in ASM mass was observed one
week after allergen exposure, and persisted at least 4 weeks after 8 weeks of exposure. Increase in
ASM area correlated with increase in airway sensitivity and reactivity such as maximum airway
resistance (Rrs) [75]. Johnson et al. showed increase in the area of smooth muscle by
immunohistochemistry using α-smooth muscle staining. They showed increase in the smooth
muscle mass 5 weeks after HDM exposure, which decreased somewhat 9 weeks after cessation of
HDM exposure [50]. Another study on the acute 3-week and chronic 12-week OVA model of
airway inflammation revealed ASM proliferation with decreased apoptosis in the acute phase, and
development of ASM hypertrophy and increased muscle area in the chronic challenge, which was
related to airway resistance [83]. Syk mediated ASM hypertrophy and hyperplasia may be a
mechanism for pathogenesis of asthma.

94
Chemokines play an important role in development of airway remodeling. They are
involved in recruiting of inflammatory cells to the side of inflammation as well as mobilizing of
airway structural cells and hence play a part in airway remodeling in asthma. ASM cells express
chemokine receptors for CC (such as RANTES) and CXC (such as IL-8) chemokines, which
induce ASM cells migration toward the airway epithelium [218]. In addition, It has been shown
that mitogens such as IL-6 play a crucial role in the increase of ASM mass in asthmatic patients
[97]. Regulation of mitogenesis by the receptor system is primarily controlled throughout the
phosphoinositide 3’-kinase (PI3K) and extracellular signal-regulated kinase (ERK) signaling
pathways [97]. Increased levels of different mitogens have been observed in BALF taken from
asthmatic patients and also asthmatic airway cell cultures [219].
Previous studies in our laboratory on BEAS-2B cells revealed that Syk activate two
different signaling pathways following engagement of intercellular adhesion molecule-1 (ICAM-1)
by human rhinovirus (HRV) [66, 68]. The first pathway, ERK 1/2 and p38 mitogen activated
protein kinase (p38 MAPK) that results in expression of IL-8 [66] and the PI3K pathway [68].
Previous studies have been shown that HDM enhance the surface expression of ICAM-1 in
airway epithelial cell lines [220, 221]. In an in vitro study using primary airway epithelial cell line
we showed that HDM could increase surface expression of ICAM-1. Thus a potential signaling
pathway after exposing to HDM can be recruitment of Syk to ICAM-1 and consequently activation
of Syk and downstream signaling for regulating of cell proliferation, migration and also production
of inflammatory mediators, enzymes and growth factors.
All things considered, these results show involvement of Syk in regulation of airways
remodeling in a mouse model of asthma, in addition of its function in leukocytes. Therefore, Syk
may represent a target for modulating of airway hyper-responsiveness and remodeling in asthma.

95
Syk is critically involved in regulation of immunoreceptor signaling and inflammatory
responses, and has been implicated in autoimmune disease and allergic disorders such as asthma,
allergic rhinitis and rheumatoid arthritis [141]. Syk has been target in a number of clinical trials of
various immune mediated diseases [140].
The selective Syk inhibitor, R112, was used in a double-blinded placebo-controlled study
in volunteers with symptomatic seasonal allergic rhinitis in a park. The study examined efficacy
and safety of intranasal R112 in 160 patients compared with 159 patients who received vehicle
control during spring season [183]. Patients rated the severity of their symptoms on a possible
maximum scale of 32 using a Global Symptom Complex (GSC); these symptoms include
sneezing, runny, itchy and stuffy nose. R112 was found to significantly decrease the GSC
compared with placebo after 8 hours. The adverse effects were not distinguishable between the
groups. The result of the study looks promising as a new treatment for seasonal rhinitis [183].
There are several clinical trials completed or under investigation for the safety and efficacy
of Syk inhibitors, especially R788, for treatment of rheumatoid arthritis, lymphoma and
anaphylaxis. A phase II study for evaluation of efficiency and safety of an oral Syk inhibitor,
R788, was performed on 457 patients with active rheumatoid arthritis for the coarse of 6 months
[222]. The results of the study revealed that Syk inhibitor R788 decreased disease activity in
patients. Some adverse effect such as diarrhea, hypertension, and neutropenia were observed [222].
A significant drop in Syk expression was first noticed during breast cancer development,
but an abnormal expression of Syk has now also been showed in many other tumor types. Studies
demonstrated suppressive function of Syk in tumorigenesis and metastasis formation [210].
Constitutional and inducible Syk activation in chronic lymphatic leukemia (CLL) and B cell
lymphomas makes it well suited for treatment of such malignancies [223].

96
In a phase 1/2 clinical trial, the Syk inhibitor fostamatinib disodium (R788) was used in
patients with recurrent B-cell non-Hodgkin lymphoma (B-NHL). R788 was found to be efficient
and safe in the treatment of B-NHL and also small lymphocytic leukemia/ chronic lymphocytic
leukemia (SLL/CLL) [180]. Side effects including diarrhea, fatigue and cytopenias [180] were
similar to those observed in rheumatoid arthritis studies [222].
Rigel Pharmaceuticals announced that it has initiated phase 2 clinical for 270 patients with
allergic asthma using R343. They are randomized into the three groups for eight weeks of
treatment with either of two different doses of the R343 or placebo. The result has not been
reported yet. They expect to complete the study in 2013 [224].
The results of our study revealed the efficacy of single dose of Syk inhibitor, NVP-QAB-
205, for attenuating of AHR in an acute mouse model of HDM-induced allergic inflammation. In
addition Syk inhibitor may have a role in regulating airway remodeling when used during a
prolonged period.

97
5 Chapter 6: Conclusion
The work described in this thesis demonstrates the importance of Syk in several key
aspects of allergic airways inflammation. The studies presented show that in vivo inhibition of
Syk by selective Syk inhibitors completely reversed airway hyperresponsiveness to methacholine
in an established acute model of HDM-induced airway inflammation. I additionally revealed that
deletion of Syk abrogates airway hyperresponsiveness, which occurred independent of
inflammatory cell influx in both acute and chronic models of airways inflammation. Moreover, I
showed that genetic deletion of Syk attenuates certain key features of remodeling such as goblet
cell hyperplasia, increase in airway smooth muscle cell mass, and epithelial modification in a
chronic murine model HDM-induced allergic airway inflammation. Down-regulation of
inflammatory mediators such as KC, IL-6, IL-17, and RANTES by deletion of Syk may represent
mechanisms by which Syk mediates airways hyperresponsiveness and remodeling in allergic
airways inflammation. These findings increased the body of evidence supporting Syk targeting for
treatment of allergic airways inflammation and asthma.

98
Chapter 7: Future directions
Syk and airways remodeling
Despite the multiple studies investigating the role of Syk in airways remodeling, many
questions remain. Previous studies in the Chow laboratory revealed that Syk mediates airway
epithelial cell proliferation, migration and apoptosis in response to human rhinovirus infection. I
demonstrated in my thesis that deletion of Syk attenuates certain features of remodeling, regardless
of inflammatory cell influx. However, this has not been directly demonstrated using chronic
pharmacologic inhibition of Syk in a chronic model of allergic airways inflammation and asthma.
Chronic administration of a Syk inhibitor during allergen challenges is more applicable for drug
development in the clinical phase. Long-term Syk inhibitor treatment in an establish disease model
would demonstrate the therapeutic potential of this approach to be able to either attenuate or
reverse remodeling features. In addition, a more vigorous approach using the American Thoracic
Society (ATS) guidelines for assessment of structural changes in the lung will allow better
evaluation of degree of remodeling in future studies.
Airway epithelial- smooth muscle communications
It is previously shown that Syk is expressed at below detectable or very low levels by the
airway smooth muscle cells in mice [145]. Hence, the rapid bronchodilator effect of the Syk
inhibitor may rely on indirect action rather than a direct effect on airway smooth muscle cells.
Both inflammatory and airways epithelial cells may indirectly affect airway smooth muscle cells.
Given that the deletion of Syk or use of a Syk inhibitor did not affect airways inflammation in
these airways inflammation models, ex vivo studies on lung sections can be helpful for future

99
studies to eliminate the effect of inflammatory cells. Use of lung section model would further
provide an improved insight in the communication between airway epithelial and airway smooth
muscle cells.

100
References
1. Takahashi, N., et al., Direct inhibition of arginase attenuated airway allergic reactions and inflammation in a Dermatophagoides farinae-induced NC/Nga mouse model. American journal of physiology. Lung cellular and molecular physiology, 2010. 299(1): p. L17-24.
2. Matsubara, S., et al., Syk activation in dendritic cells is essential for airway hyperresponsiveness and inflammation. American journal of respiratory cell and molecular biology, 2006. 34(4): p. 426-33.
3. Murphy, D.M. and P.M. O'Byrne, Recent advances in the pathophysiology of asthma. Chest, 2010. 137(6): p. 1417-26.
4. Kim, H.Y., R.H. DeKruyff, and D.T. Umetsu, The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nature immunology, 2010. 11(7): p. 577-84.
5. Braman, S.S., The global burden of asthma. Chest, 2006. 130(1 Suppl): p. 4S-12S.
6. Thomas, E.M., Recent trends in upper respiratory infections, ear infections and asthma among young Canadian children. Health reports / Statistics Canada, Canadian Centre for Health Information = Rapports sur la sante / Statistique Canada, Centre canadien d'information sur la sante, 2010. 21(4): p. 47-52.
7. Ober, C. and T.C. Yao, The genetics of asthma and allergic disease: a 21st century perspective. Immunological reviews, 2011. 242(1): p. 10-30.
8. Canada, T.A.S.o. Asthma Facts & Statistics. 2010; Available from: http://www.asthma.ca/corp/newsroom/pdf/asthmastats.pdf.
9. Choudhry, S., et al., Dissecting complex diseases in complex populations: asthma in latino americans. Proceedings of the American Thoracic Society, 2007. 4(3): p. 226-33.
10. van Merode, T., et al., Gender-specific differences in the prevention of asthma-like symptoms in high-risk infants. Pediatric allergy and immunology : official publication of the European Society of Pediatric Allergy and Immunology, 2007. 18(3): p. 196-200.
11. Willemsen, G., et al., Heritability of self-reported asthma and allergy: a study in adult Dutch twins, siblings and parents. Twin research and human genetics : the official journal of the International Society for Twin Studies, 2008. 11(2): p. 132-42.
12. Barnes, K.C., et al., African Americans with asthma: genetic insights. Proceedings of the American Thoracic Society, 2007. 4(1): p. 58-68.
13. Ober, C., D.A. Loisel, and Y. Gilad, Sex-specific genetic architecture of human disease. Nature reviews. Genetics, 2008. 9(12): p. 911-22.

101
14. Fitzpatrick, S., R. Joks, and J.I. Silverberg, Obesity is associated with increased asthma severity and exacerbations, and increased serum immunoglobulin E in inner-city adults. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2012. 42(5): p. 747-59.
15. Holguin, F., Obesity as a risk factor for increased asthma severity and allergic inflammation; cause or effect? Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2012. 42(5): p. 612-3.
16. Farah, C.S. and C.M. Salome, Asthma and obesity: a known association but unknown mechanism. Respirology, 2012. 17(3): p. 412-21.
17. Polosa, R., et al., Cigarette smoking is associated with a greater risk of incident asthma in allergic rhinitis. The Journal of allergy and clinical immunology, 2008. 121(6): p. 1428-34.
18. Chen, C.M. and J. Heinrich, Re: Exposure to furry pets and the risk of asthma and allergic rhinitis: a meta-analysis. Allergy, 2009. 64(3): p. 494-5.
19. Takkouche, B., et al., Exposure to furry pets and the risk of asthma and allergic rhinitis: a meta-analysis. Allergy, 2008. 63(7): p. 857-64.
20. Chew, G.L., et al., Cockroach allergen levels and associations with cockroach-specific IgE. The Journal of allergy and clinical immunology, 2008. 121(1): p. 240-5.
21. Celedon, J.C., et al., Exposure to dust mite allergen and endotoxin in early life and asthma and atopy in childhood. The Journal of allergy and clinical immunology, 2007. 120(1): p. 144-9.
22. Jones, R., et al., Association between indoor mold and asthma among children in Buffalo, New York. Indoor air, 2011. 21(2): p. 156-64.
23. Hayden, T.J. and D.J. Muscatello, Increased presentations to emergency departments for asthma associated with rye grass pollen season in inland NSW. New South Wales public health bulletin, 2011. 22(7-8): p. 154-8.
24. Krmpotic, D., et al., Effects of traffic air pollution and hornbeam pollen on adult asthma hospitalizations in Zagreb. International archives of allergy and immunology, 2011. 156(1): p. 62-8.
25. Bisgaard, H., et al., Childhood asthma after bacterial colonization of the airway in neonates. The New England journal of medicine, 2007. 357(15): p. 1487-95.
26. Jackson, D.J., et al., Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. American journal of respiratory and critical care medicine, 2008. 178(7): p. 667-72.

102
27. Kusel, M.M., et al., Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. The Journal of allergy and clinical immunology, 2007. 119(5): p. 1105-10.
28. Piedimonte, G., Respiratory syncytial virus and asthma: speed-dating or long-term relationship? Current opinion in pediatrics, 2013. 25(3): p. 344-9.
29. Wang, X., et al., Spleen tyrosine kinase mediates BEAS-2B cell migration and proliferation and human rhinovirus-induced expression of vascular endothelial growth factor and interleukin-8. The Journal of pharmacology and experimental therapeutics, 2012. 340(2): p. 277-85.
30. Clark, N.A., et al., Effect of early life exposure to air pollution on development of childhood asthma. Environmental health perspectives, 2010. 118(2): p. 284-90.
31. Penard-Morand, C., et al., Long-term exposure to close-proximity air pollution and asthma and allergies in urban children. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology, 2010. 36(1): p. 33-40.
32. Dales, R., et al., The influence of neighborhood roadways on respiratory symptoms among elementary schoolchildren. Journal of occupational and environmental medicine / American College of Occupational and Environmental Medicine, 2009. 51(6): p. 654-60.
33. Liu, L., et al., Acute effects of air pollution on pulmonary function, airway inflammation, and oxidative stress in asthmatic children. Environmental health perspectives, 2009. 117(4): p. 668-74.
34. Penton, P.C., et al., Spleen tyrosine kinase inhibition attenuates airway hyperresponsiveness and pollution-induced enhanced airway response in a chronic mouse model of asthma. The Journal of allergy and clinical immunology, 2013. 131(2): p. 512-520 e10.
35. Mak, K.K., R.C. Ho, and J.R. Day, The Associations of Asthma Symptoms With Active and Passive Smoking in Hong Kong Adolescents. Respiratory care, 2012.
36. Gilliland, F.D., et al., Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. American journal of respiratory and critical care medicine, 2002. 166(4): p. 457-63.
37. Willers, S.M., et al., Maternal food consumption during pregnancy and the longitudinal development of childhood asthma. American journal of respiratory and critical care medicine, 2008. 178(2): p. 124-31.
38. Sonnenschein-van der Voort, A.M., et al., Duration and exclusiveness of breastfeeding and childhood asthma-related symptoms. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology, 2012. 39(1): p. 81-9.

103
39. D'Urzo, A.D., Diagnosis of asthma. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne, 2010. 182(1): p. 64.
40. GLOBAL STRATEGY FOR ASTHMA MANAGEMENT AND PREVENTION. 2011.
41. Busse, W.W., Asthma diagnosis and treatment: filling in the information gaps. The Journal of allergy and clinical immunology, 2011. 128(4): p. 740-50.
42. Mertz, T.L., Recent developments in asthma research, diagnosis, and treatment. The Journal of the American Osteopathic Association, 2011. 111(11 Suppl 7): p. S27-9, S32.
43. Lowhagen, O., Diagnosis of asthma - a new approach. Allergy, 2012. 67(6): p. 713-7.
44. Reed, C.E., Asthma in the elderly: diagnosis and management. The Journal of allergy and clinical immunology, 2010. 126(4): p. 681-7; quiz 688-9.
45. Barnes, P.J., Biochemical basis of asthma therapy. The Journal of biological chemistry, 2011. 286(38): p. 32899-905.
46. Barnes, P.J., Glucocorticosteroids: current and future directions. British journal of pharmacology, 2011. 163(1): p. 29-43.
47. Babu, K.S., R. Polosa, and J.B. Morjaria, Anti-IgE--emerging opportunities for Omalizumab. Expert opinion on biological therapy, 2013. 13(5): p. 765-77.
48. Guidelines for the Diagnosis and Management of Asthma. 2007.
49. Nakagome, K. and M. Nagata, Pathogenesis of airway inflammation in bronchial asthma. Auris, nasus, larynx, 2011. 38(5): p. 555-63.
50. Johnson, J.R., et al., Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. American journal of respiratory and critical care medicine, 2004. 169(3): p. 378-85.
51. Veres, T.Z., et al., Spatiotemporal and functional behavior of airway dendritic cells visualized by two-photon microscopy. The American journal of pathology, 2011. 179(2): p. 603-9.
52. Finkelman, F.D., et al., Importance of cytokines in murine allergic airway disease and human asthma. Journal of immunology, 2010. 184(4): p. 1663-74.
53. Nakajima, H. and K. Hirose, Role of IL-23 and Th17 Cells in Airway Inflammation in Asthma. Immune network, 2010. 10(1): p. 1-4.
54. Wakashin, H., et al., IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. American journal of respiratory and critical care medicine, 2008. 178(10): p. 1023-32.
55. Poulsen, L.K. and L. Hummelshoj, Triggers of IgE class switching and allergy development. Annals of medicine, 2007. 39(6): p. 440-56.

104
56. Bonilla, F.A. and H.C. Oettgen, Adaptive immunity. The Journal of allergy and clinical immunology, 2010. 125(2 Suppl 2): p. S33-40.
57. Wex, E., et al., Induced Syk deletion leads to suppressed allergic responses but has no effect on neutrophil or monocyte migration in vivo. European journal of immunology, 2011. 41(11): p. 3208-18.
58. Suzuki, T., C.W. Chow, and G.P. Downey, Role of innate immune cells and their products in lung immunopathology. The international journal of biochemistry & cell biology, 2008. 40(6-7): p. 1348-61.
59. Shen, T., et al., T cell-derived IL-3 plays key role in parasite infection-induced basophil production but is dispensable for in vivo basophil survival. International immunology, 2008. 20(9): p. 1201-9.
60. Buc, M., et al., Immunopathogenesis of bronchial asthma. Archivum immunologiae et therapiae experimentalis, 2009. 57(5): p. 331-44.
61. Fahy, J.V., Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proceedings of the American Thoracic Society, 2009. 6(3): p. 256-9.
62. Mosser, D.M. and J.P. Edwards, Exploring the full spectrum of macrophage activation. Nature reviews. Immunology, 2008. 8(12): p. 958-69.
63. Erjefalt, J.S., The airway epithelium as regulator of inflammation patterns in asthma. The clinical respiratory journal, 2010. 4 Suppl 1: p. 9-14.
64. Al-Muhsen, S., J.R. Johnson, and Q. Hamid, Remodeling in asthma. The Journal of allergy and clinical immunology, 2011. 128(3): p. 451-62; quiz 463-4.
65. Vareille, M., et al., The airway epithelium: soldier in the fight against respiratory viruses. Clinical microbiology reviews, 2011. 24(1): p. 210-29.
66. Wang, X., et al., Syk is downstream of intercellular adhesion molecule-1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. Journal of immunology, 2006. 177(10): p. 6859-70.
67. Proud, D. and C.W. Chow, Role of viral infections in asthma and chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology, 2006. 35(5): p. 513-8.
68. Lau, C., et al., Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. Journal of immunology, 2008. 180(2): p. 870-80.
69. Lin, W.N., et al., Involvement of MAPKs and NF-kappaB in LPS-induced VCAM-1 expression in human tracheal smooth muscle cells. Cellular signalling, 2007. 19(6): p. 1258-67.

105
70. Monteseirin, J., Neutrophils and asthma. Journal of investigational allergology & clinical immunology, 2009. 19(5): p. 340-54.
71. Maneechotesuwan, K., et al., Loss of control of asthma following inhaled corticosteroid withdrawal is associated with increased sputum interleukin-8 and neutrophils. Chest, 2007. 132(1): p. 98-105.
72. Zuurbier, A.E., et al., Neutrophils enhance eosinophil migration across monolayers of lung epithelial cells. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2001. 31(3): p. 444-52.
73. De Vooght, V., et al., Neutrophil and eosinophil granulocytes as key players in a mouse model of chemical-induced asthma. Toxicological sciences : an official journal of the Society of Toxicology, 2013. 131(2): p. 406-18.
74. Berend, N., C.M. Salome, and G.G. King, Mechanisms of airway hyperresponsiveness in asthma. Respirology, 2008. 13(5): p. 624-31.
75. Southam, D.S., et al., Components of airway hyperresponsiveness and their associations with inflammation and remodeling in mice. The Journal of allergy and clinical immunology, 2007. 119(4): p. 848-54.
76. Busse, W.W., The relationship of airway hyperresponsiveness and airway inflammation: Airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest, 2010. 138(2 Suppl): p. 4S-10S.
77. Meurs, H., R. Gosens, and J. Zaagsma, Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology, 2008. 32(2): p. 487-502.
78. Jeffery, P.K., Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society, 2004. 1(3): p. 176-83.
79. Orsida, B.E., et al., Vascularity in asthmatic airways: relation to inhaled steroid dose. Thorax, 1999. 54(4): p. 289-95.
80. Kasahara, K., et al., Correlation between the bronchial subepithelial layer and whole airway wall thickness in patients with asthma. Thorax, 2002. 57(3): p. 242-6.
81. Leguillette, R. and A.M. Lauzon, Molecular mechanics of smooth muscle contractile proteins in airway hyperresponsiveness and asthma. Proceedings of the American Thoracic Society, 2008. 5(1): p. 40-6.
82. James, A.L., et al., Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology, 2009. 34(5): p. 1040-5.

106
83. Plant, P.J., et al., Hypertrophic airway smooth muscle mass correlates with increased airway responsiveness in a murine model of asthma. American journal of respiratory cell and molecular biology, 2012. 46(4): p. 532-40.
84. Bai, T.R., et al., The effect of age and duration of disease on airway structure in fatal asthma. American journal of respiratory and critical care medicine, 2000. 162(2 Pt 1): p. 663-9.
85. Goldsmith, A.M., et al., Transforming growth factor-beta induces airway smooth muscle hypertrophy. American journal of respiratory cell and molecular biology, 2006. 34(2): p. 247-54.
86. Kume, H., RhoA/Rho-kinase as a therapeutic target in asthma. Current medicinal chemistry, 2008. 15(27): p. 2876-85.
87. Chiba, Y., K. Matsusue, and M. Misawa, RhoA, a possible target for treatment of airway hyperresponsiveness in bronchial asthma. Journal of pharmacological sciences, 2010. 114(3): p. 239-47.
88. Downie, S.R., et al., Ventilation heterogeneity is a major determinant of airway hyperresponsiveness in asthma, independent of airway inflammation. Thorax, 2007. 62(8): p. 684-9.
89. Royce, S.G., et al., The regulation of fibrosis in airway remodeling in asthma. Molecular and cellular endocrinology, 2012. 351(2): p. 167-75.
90. Maarsingh, H., J. Zaagsma, and H. Meurs, Arginine homeostasis in allergic asthma. European journal of pharmacology, 2008. 585(2-3): p. 375-84.
91. North, M.L., et al., Augmentation of arginase 1 expression by exposure to air pollution exacerbates the airways hyperresponsiveness in murine models of asthma. Respiratory research, 2011. 12: p. 19.
92. Lindemann, D. and K. Racke, Glucocorticoid inhibition of interleukin-4 (IL-4) and interleukin-13 (IL-13) induced up-regulation of arginase in rat airway fibroblasts. Naunyn-Schmiedeberg's archives of pharmacology, 2003. 368(6): p. 546-50.
93. North, M.L., et al., Functionally important role for arginase 1 in the airway hyperresponsiveness of asthma. American journal of physiology. Lung cellular and molecular physiology, 2009. 296(6): p. L911-20.
94. Maarsingh, H., et al., Heparin normalizes allergen-induced nitric oxide deficiency and airway hyperresponsiveness. British journal of pharmacology, 2004. 142(8): p. 1293-9.
95. Myers, A.C., Transmission in autonomic ganglia. Respiration physiology, 2001. 125(1-2): p. 99-111.
96. Halwani, R., S. Al-Muhsen, and Q. Hamid, Airway remodeling in asthma. Current opinion in pharmacology, 2010. 10(3): p. 236-45.

107
97. Tagaya, E. and J. Tamaoki, Mechanisms of airway remodeling in asthma. Allergology international : official journal of the Japanese Society of Allergology, 2007. 56(4): p. 331-40.
98. Kaminska, M., et al., Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. The Journal of allergy and clinical immunology, 2009. 124(1): p. 45-51 e1-4.
99. Turner, J. and C.E. Jones, Regulation of mucin expression in respiratory diseases. Biochemical Society transactions, 2009. 37(Pt 4): p. 877-81.
100. Araujo, B.B., et al., Extracellular matrix components and regulators in the airway smooth muscle in asthma. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology, 2008. 32(1): p. 61-9.
101. Dekkers, B.G., et al., Airway structural components drive airway smooth muscle remodeling in asthma. Proceedings of the American Thoracic Society, 2009. 6(8): p. 683-92.
102. Girodet, P.O., et al., Airway remodeling in asthma: new mechanisms and potential for pharmacological intervention. Pharmacology & therapeutics, 2011. 130(3): p. 325-37.
103. Kanazawa, H., VEGF, angiopoietin-1 and -2 in bronchial asthma: new molecular targets in airway angiogenesis and microvascular remodeling. Recent patents on inflammation & allergy drug discovery, 2007. 1(1): p. 1-8.
104. Zhang, W.X. and C.C. Li, Airway remodeling: a potential therapeutic target in asthma. World journal of pediatrics : WJP, 2011. 7(2): p. 124-8.
105. Siddiqui, S. and J.G. Martin, Structural aspects of airway remodeling in asthma. Current allergy and asthma reports, 2008. 8(6): p. 540-7.
106. Bhavsar, P., T. Ahmad, and I.M. Adcock, The role of histone deacetylases in asthma and allergic diseases. The Journal of allergy and clinical immunology, 2008. 121(3): p. 580-4.
107. Al-Ramli, W., et al., T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. The Journal of allergy and clinical immunology, 2009. 123(5): p. 1185-7.
108. Halwani, R., et al., Role of transforming growth factor-beta in airway remodeling in asthma. American journal of respiratory cell and molecular biology, 2011. 44(2): p. 127-33.
109. Holgate, S.T., The airway epithelium is central to the pathogenesis of asthma. Allergology international : official journal of the Japanese Society of Allergology, 2008. 57(1): p. 1-10.
110. Holgate, S.T., A look at the pathogenesis of asthma: the need for a change in direction. Discovery medicine, 2010. 9(48): p. 439-47.

108
111. Davies, D.E., The role of the epithelium in airway remodeling in asthma. Proceedings of the American Thoracic Society, 2009. 6(8): p. 678-82.
112. Cho, J.Y., Recent advances in mechanisms and treatments of airway remodeling in asthma: a message from the bench side to the clinic. The Korean journal of internal medicine, 2011. 26(4): p. 367-83.
113. Doherty, T. and D. Broide, Cytokines and growth factors in airway remodeling in asthma. Current opinion in immunology, 2007. 19(6): p. 676-80.
114. Makinde, T., R.F. Murphy, and D.K. Agrawal, The regulatory role of TGF-beta in airway remodeling in asthma. Immunology and cell biology, 2007. 85(5): p. 348-56.
115. Johnson, J.R., et al., Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PloS one, 2011. 6(1): p. e16175.
116. Ito, I., et al., Platelet-derived growth factor and transforming growth factor-beta modulate the expression of matrix metalloproteinases and migratory function of human airway smooth muscle cells. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2009. 39(9): p. 1370-80.
117. Holgate, S.T. and R. Polosa, Treatment strategies for allergy and asthma. Nature reviews. Immunology, 2008. 8(3): p. 218-30.
118. Roussel, L., et al., IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. Journal of immunology, 2010. 184(8): p. 4531-7.
119. Dolgachev, V., et al., Pulmonary IL-17E (IL-25) production and IL-17RB+ myeloid cell-derived Th2 cytokine production are dependent upon stem cell factor-induced responses during chronic allergic pulmonary disease. Journal of immunology, 2009. 183(9): p. 5705-15.
120. Takeda, N., et al., Epithelium-derived chemokines induce airway smooth muscle cell migration. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2009. 39(7): p. 1018-26.
121. Riesenfeld, E.P. and C.G. Irvin, Asthma treatment through the beta receptor: lessons from animal models. Frontiers in bioscience, 2011. 3: p. 1201-8.
122. Holmes, A.M., R. Solari, and S.T. Holgate, Animal models of asthma: value, limitations and opportunities for alternative approaches. Drug discovery today, 2011. 16(15-16): p. 659-70.
123. Zosky, G.R. and P.D. Sly, Animal models of asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2007. 37(7): p. 973-88.

109
124. Bao, Z.S., et al., Inhibition of airway inflammation, hyperresponsiveness and remodeling by soy isoflavone in a murine model of allergic asthma. International immunopharmacology, 2011. 11(8): p. 899-906.
125. Shibamori, M., et al., Intranasal mite allergen induces allergic asthma-like responses in NC/Nga mice. Life sciences, 2006. 78(9): p. 987-94.
126. Locke, N.R., et al., Comparison of airway remodeling in acute, subacute, and chronic models of allergic airways disease. American journal of respiratory cell and molecular biology, 2007. 36(5): p. 625-32.
127. Guo, W., et al., [Preparation and evaluation of mouse model of house dust mite-induced asthma]. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics, 2008. 10(5): p. 647-50.
128. Gregory, L.G. and C.M. Lloyd, Orchestrating house dust mite-associated allergy in the lung. Trends in immunology, 2011. 32(9): p. 402-11.
129. Thomas, W.R., Geography of house dust mite allergens. Asian Pacific journal of allergy and immunology / launched by the Allergy and Immunology Society of Thailand, 2010. 28(4): p. 211-24.
130. Gandhi, V.D., et al., House dust mite interactions with airway epithelium: role in allergic airway inflammation. Current allergy and asthma reports, 2013. 13(3): p. 262-70.
131. Thomas, W.R., B.J. Hales, and W.A. Smith, House dust mite allergens in asthma and allergy. Trends in molecular medicine, 2010. 16(7): p. 321-8.
132. Wakahara, K., et al., Repeated instillations of Dermatophagoides farinae into the airways can induce Th2-dependent airway hyperresponsiveness, eosinophilia and remodeling in mice: effect of intratracheal treatment of fluticasone propionate. European journal of pharmacology, 2008. 578(1): p. 87-96.
133. Cates, E.C., et al., Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. Journal of immunology, 2004. 173(10): p. 6384-92.
134. Zioncheck, T.F., et al., Generation of an active protein-tyrosine kinase from lymphocytes by proteolysis. The Journal of biological chemistry, 1988. 263(35): p. 19195-202.
135. Zioncheck, T.F., M.L. Harrison, and R.L. Geahlen, Purification and characterization of a protein-tyrosine kinase from bovine thymus. The Journal of biological chemistry, 1986. 261(33): p. 15637-43.
136. Kobayashi, T., et al., Purification and characterization of a cytosolic protein-tyrosine kinase from porcine spleen. European journal of biochemistry / FEBS, 1990. 188(3): p. 535-40.

110
137. Mocsai, A., J. Ruland, and V.L. Tybulewicz, The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature reviews. Immunology, 2010. 10(6): p. 387-402.
138. Fischer, A., et al., ZAP70: a master regulator of adaptive immunity. Seminars in immunopathology, 2010. 32(2): p. 107-16.
139. Singh, R., E.S. Masuda, and D.G. Payan, Discovery and Development of Spleen Tyrosine Kinase (SYK) Inhibitors. Journal of medicinal chemistry, 2012.
140. Ruzza, P., B. Biondi, and A. Calderan, Therapeutic prospect of Syk inhibitors. Expert opinion on therapeutic patents, 2009. 19(10): p. 1361-76.
141. Riccaboni, M., I. Bianchi, and P. Petrillo, Spleen tyrosine kinases: biology, therapeutic targets and drugs. Drug discovery today, 2010. 15(13-14): p. 517-30.
142. Huang, Z.Y., et al., Effect of locally administered Syk siRNA on allergen-induced arthritis and asthma. Molecular immunology, 2013. 53(1-2): p. 52-9.
143. Spalton, J.C., et al., The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. Journal of thrombosis and haemostasis : JTH, 2009. 7(7): p. 1192-9.
144. Sanderson, M.P., et al., Syk: a novel target for treatment of inflammation in lung disease. Inflammation & allergy drug targets, 2009. 8(2): p. 87-95.
145. Ulanova, M., et al., Syk tyrosine kinase participates in beta1-integrin signaling and inflammatory responses in airway epithelial cells. American journal of physiology. Lung cellular and molecular physiology, 2005. 288(3): p. L497-507.
146. Ulanova, M., et al., Involvement of Syk kinase in TNF-induced nitric oxide production by airway epithelial cells. Biochemical and biophysical research communications, 2006. 351(2): p. 431-7.
147. Duta, F., et al., Differential expression of spleen tyrosine kinase Syk isoforms in tissues: Effects of the microbial flora. Histochemistry and cell biology, 2006. 126(4): p. 495-505.
148. Zou, L., N. Sato, and B.C. Kone, Alpha-melanocyte stimulating hormone protects against H2O2-induced inhibition of wound restitution in IEC-6 cells via a Syk kinase- and NF-kappabeta-dependent mechanism. Shock, 2004. 22(5): p. 453-9.
149. Sung, Y.M., et al., Tumor suppressor function of Syk in human MCF10A in vitro and normal mouse mammary epithelium in vivo. PloS one, 2009. 4(10): p. e7445.
150. Yanagi, S., et al., Syk expression in endothelial cells and their morphologic defects in embryonic Syk-deficient mice. Blood, 2001. 98(9): p. 2869-71.
151. Cheng, A.M., et al., Syk tyrosine kinase required for mouse viability and B-cell development. Nature, 1995. 378(6554): p. 303-6.

111
152. Yang, W.S., et al., High glucose-induced NF-kappaB activation occurs via tyrosine phosphorylation of IkappaBalpha in human glomerular endothelial cells: involvement of Syk tyrosine kinase. American journal of physiology. Renal physiology, 2008. 294(5): p. F1065-75.
153. Underhill, D.M. and H.S. Goodridge, The many faces of ITAMs. Trends in immunology, 2007. 28(2): p. 66-73.
154. Ratthe, C. and D. Girard, Interleukin-15 enhances human neutrophil phagocytosis by a Syk-dependent mechanism: importance of the IL-15Ralpha chain. Journal of leukocyte biology, 2004. 76(1): p. 162-8.
155. Mocsai, A., et al., Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nature immunology, 2006. 7(12): p. 1326-33.
156. Galli, S.J., M. Tsai, and A.M. Piliponsky, The development of allergic inflammation. Nature, 2008. 454(7203): p. 445-54.
157. Suljagic, M., et al., The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling. Blood, 2010. 116(23): p. 4894-905.
158. Efremov, D.G. and L. Laurenti, The Syk kinase as a therapeutic target in leukemia and lymphoma. Expert opinion on investigational drugs, 2011. 20(5): p. 623-36.
159. Matsubara, S., et al., Inhibition of spleen tyrosine kinase prevents mast cell activation and airway hyperresponsiveness. American journal of respiratory and critical care medicine, 2006. 173(1): p. 56-63.
160. Stenton, G.R., et al., Inhibition of allergic inflammation in the airways using aerosolized antisense to Syk kinase. Journal of immunology, 2002. 169(2): p. 1028-36.
161. Rawlins, E.L. and A.K. Perl, The a"MAZE"ing world of lung-specific transgenic mice. American journal of respiratory cell and molecular biology, 2012. 46(3): p. 269-82.
162. Turner, M., et al., Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature, 1995. 378(6554): p. 298-302.
163. Levitzki, A. and E. Mishani, Tyrphostins and other tyrosine kinase inhibitors. Annual review of biochemistry, 2006. 75: p. 93-109.
164. Vulpetti, A. and R. Bosotti, Sequence and structural analysis of kinase ATP pocket residues. Farmaco, 2004. 59(10): p. 759-65.
165. Rossi, A.B., et al., Identification of the Syk kinase inhibitor R112 by a human mast cell screen. The Journal of allergy and clinical immunology, 2006. 118(3): p. 749-55.

112
166. Weinblatt, M.E., et al., Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis and rheumatism, 2008. 58(11): p. 3309-18.
167. Braselmann, S., et al., R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. The Journal of pharmacology and experimental therapeutics, 2006. 319(3): p. 998-1008.
168. Norman, P., A novel Syk kinase inhibitor suitable for inhalation: R-343(?)--WO-2009031011. Expert opinion on therapeutic patents, 2009. 19(10): p. 1469-72.
169. Yamamoto, N., et al., The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. The Journal of pharmacology and experimental therapeutics, 2003. 306(3): p. 1174-81.
170. Crooke, S.T., Progress in antisense technology. Annual review of medicine, 2004. 55: p. 61-95.
171. Masuda, E.S. and J. Schmitz, Syk inhibitors as treatment for allergic rhinitis. Pulmonary pharmacology & therapeutics, 2008. 21(3): p. 461-7.
172. Fisher, A.A., et al., Evaluating the specificity of antisense oligonucleotide conjugates. A DNA array analysis. The Journal of biological chemistry, 2002. 277(25): p. 22980-4.
173. McManus, M.T. and P.A. Sharp, Gene silencing in mammals by small interfering RNAs. Nature reviews. Genetics, 2002. 3(10): p. 737-47.
174. Zamore, P.D., et al., RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 2000. 101(1): p. 25-33.
175. RNAi-mediated inhibition of spleen tyrosine kinase-related inflammatory conditions., L. Alcon Manufactering organization, Editor 2007.
176. Pine, P.R., et al., Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clinical immunology, 2007. 124(3): p. 244-57.
177. Sheridan, C., Small molecule challenges dominance of TNF-alpha inhibitors. Nature biotechnology, 2008. 26(2): p. 143-4.
178. Bahjat, F.R., et al., An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis and rheumatism, 2008. 58(5): p. 1433-44.
179. Smith, J., et al., A spleen tyrosine kinase inhibitor reduces the severity of established glomerulonephritis. Journal of the American Society of Nephrology : JASN, 2010. 21(2): p. 231-6.

113
180. Friedberg, J.W., et al., Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood, 2010. 115(13): p. 2578-85.
181. Havard, S., et al., Characterization of syk expression in human lung mast cells: relationship with function. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2011. 41(3): p. 378-88.
182. MacGlashan, D.W., Jr., Relationship between spleen tyrosine kinase and phosphatidylinositol 5' phosphatase expression and secretion from human basophils in the general population. The Journal of allergy and clinical immunology, 2007. 119(3): p. 626-33.
183. Meltzer, E.O., R.B. Berkowitz, and E.B. Grossbard, An intranasal Syk-kinase inhibitor (R112) improves the symptoms of seasonal allergic rhinitis in a park environment. The Journal of allergy and clinical immunology, 2005. 115(4): p. 791-6.
184. Bottoms, S.E., et al., Tgf-Beta isoform specific regulation of airway inflammation and remodelling in a murine model of asthma. PloS one, 2010. 5(3): p. e9674.
185. Wex, E., characterization of inducible Syk knockout mice, in Biochemistry2011, University of Konstanz: Konsantz, Germany.
186. Desai, D. and C. Brightling, Cytokines and cytokine-specific therapy in asthma. Advances in clinical chemistry, 2012. 57: p. 57-97.
187. Yalcin, A.D., A. Bisgin, and R.M. Gorczynski, IL-8, IL-10, TGF-beta, and GCSF levels were increased in severe persistent allergic asthma patients with the anti-IgE treatment. Mediators of inflammation, 2012. 2012: p. 720976.
188. Rincon, M. and C.G. Irvin, Role of IL-6 in asthma and other inflammatory pulmonary diseases. International journal of biological sciences, 2012. 8(9): p. 1281-90.
189. Doganci, A., et al., Pathological role of IL-6 in the experimental allergic bronchial asthma in mice. Clinical reviews in allergy & immunology, 2005. 28(3): p. 257-70.
190. Shi, J., et al., Induction of IL-6 and IL-8 by house dust mite allergen Der p1 in cultured human nasal epithelial cells is associated with PAR/PI3K/NFkappaB signaling. ORL; journal for oto-rhino-laryngology and its related specialties, 2010. 72(5): p. 256-65.
191. Lee, J.S., et al., House dust mite, Dermatophagoides pteronissinus increases expression of MCP-1, IL-6, and IL-8 in human monocytic THP-1 cells. Cytokine, 2008. 42(3): p. 365-71.
192. Tillie-Leblond, I., et al., Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. American journal of respiratory and critical care medicine, 1999. 159(2): p. 487-94.

114
193. Neveu, W.A., et al., Elevation of IL-6 in the allergic asthmatic airway is independent of inflammation but associates with loss of central airway function. Respiratory research, 2010. 11: p. 28.
194. Neveu, W.A., et al., Fungal allergen beta-glucans trigger p38 mitogen-activated protein kinase-mediated IL-6 translation in lung epithelial cells. American journal of respiratory cell and molecular biology, 2011. 45(6): p. 1133-41.
195. Silverpil, E. and A. Linden, IL-17 in human asthma. Expert review of respiratory medicine, 2012. 6(2): p. 173-86.
196. Hsu, H.C., et al., Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nature immunology, 2008. 9(2): p. 166-75.
197. Chakir, J., et al., Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. The Journal of allergy and clinical immunology, 2003. 111(6): p. 1293-8.
198. Song, C., et al., IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. Journal of immunology, 2008. 181(9): p. 6117-24.
199. Sun, Y.C., Q.T. Zhou, and W.Z. Yao, Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chinese medical journal, 2005. 118(11): p. 953-6.
200. Wang, Y.H., et al., A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. The Journal of experimental medicine, 2010. 207(11): p. 2479-91.
201. Zietkowski, Z., et al., RANTES in exhaled breath condensate of patients with severe persistent allergic asthma during omalizumab therapy. International archives of allergy and immunology, 2011. 154(1): p. 25-32.
202. Fang, Q., F. Wang, and D. Zhao, Association between regulated upon activation, normal T cells expressed and secreted (RANTES) -28C/G polymorphism and asthma risk--a meta-analysis. International journal of medical sciences, 2010. 7(1): p. 55-61.
203. Saad-El-Din Bessa, S., G.H. Abo El-Magd, and M.M. Mabrouk, Serum chemokines RANTES and monocyte chemoattractant protein-1 in Egyptian patients with atopic asthma: relationship to disease severity. Archives of medical research, 2012. 43(1): p. 36-41.
204. Hoshino, M., Y. Nakamura, and Q.A. Hamid, Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. The Journal of allergy and clinical immunology, 2001. 107(6): p. 1034-8.

115
205. Siddiqui, S., et al., Vascular remodeling is a feature of asthma and nonasthmatic eosinophilic bronchitis. The Journal of allergy and clinical immunology, 2007. 120(4): p. 813-9.
206. Lee, C.G., et al., Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nature medicine, 2004. 10(10): p. 1095-103.
207. Lee, C.G., et al., Studies of vascular endothelial growth factor in asthma and chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society, 2011. 8(6): p. 512-5.
208. Kanazawa, H., S. Nomura, and J. Yoshikawa, Role of microvascular permeability on physiologic differences in asthma and eosinophilic bronchitis. American journal of respiratory and critical care medicine, 2004. 169(10): p. 1125-30.
209. Coopman, P.J., et al., The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature, 2000. 406(6797): p. 742-7.
210. Coopman, P.J. and S.C. Mueller, The Syk tyrosine kinase: a new negative regulator in tumor growth and progression. Cancer letters, 2006. 241(2): p. 159-73.
211. Gobessi, S., et al., Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia, 2009. 23(4): p. 686-97.
212. Shifren, A., et al., Mechanisms of remodeling in asthmatic airways. Journal of allergy, 2012. 2012: p. 316049.
213. Pepe, C., et al., Differences in airway remodeling between subjects with severe and moderate asthma. The Journal of allergy and clinical immunology, 2005. 116(3): p. 544-9.
214. Visse, R. and H. Nagase, Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circulation research, 2003. 92(8): p. 827-39.
215. Ierodiakonou, D., et al., TGF-beta1 polymorphisms and asthma severity, airway inflammation, and remodeling. The Journal of allergy and clinical immunology, 2013. 131(2): p. 582-5.
216. Mabalirajan, U., et al., Mepacrine inhibits subepithelial fibrosis by reducing the expression of arginase and TGF-beta1 in an extended subacute mouse model of allergic asthma. American journal of physiology. Lung cellular and molecular physiology, 2009. 297(3): p. L411-9.
217. Larsson, K., Monitoring airway remodeling in asthma. The clinical respiratory journal, 2010. 4 Suppl 1: p. 35-40.

116
218. Yahiaoui, L., et al., CC family chemokines directly regulate myoblast responses to skeletal muscle injury. The Journal of physiology, 2008. 586(16): p. 3991-4004.
219. Pascual, R.M. and S.P. Peters, Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. The Journal of allergy and clinical immunology, 2005. 116(3): p. 477-86; quiz 487.
220. Osterlund, C., et al., The non-proteolytic house dust mite allergen Der p 2 induce NF-kappaB and MAPK dependent activation of bronchial epithelial cells. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2009. 39(8): p. 1199-208.
221. Bossios, A., et al., Rhinovirus infection and house dust mite exposure synergize in inducing bronchial epithelial cell interleukin-8 release. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology, 2008. 38(10): p. 1615-26.
222. Weinblatt, M.E., et al., An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. The New England journal of medicine, 2010. 363(14): p. 1303-12.
223. Hoellenriegel, J., et al., Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia, 2012. 26(7): p. 1576-83.
224. Rigel Initiates Phase 2 Clinical Studies with R343 for Asthma and R333 for Discoid Lupus. 2012; Available from: http://www.prnewswire.com/news-releases/rigel-initiates-phase-2-clinical-studies-with-r343-for-asthma-and-r333-for-discoid-lupus-168537986.html.

117
Copyright Acknowledgements















![Occupational asthma: Pathogenesis - UFPR...eosinophilic bronchitis) [1]. Occupational asthma is a disease characterized by variable airflow limitation, airway hyperresponsiveness,](https://img.dokumen.tips/doc/110x75/5e5df342484c5e16a546eb09/occupational-asthma-pathogenesis-eosinophilic-bronchitis-1-occupational.jpg)


![Guideline-oriented perioperative management of patients ... · Asthma (GINA) [1] as a collaborative effort to defi ne Abstract Increased airway hyperresponsiveness is a major concern](https://img.dokumen.tips/doc/110x75/5fb3c94e19b64f0bb5481ab3/guideline-oriented-perioperative-management-of-patients-asthma-gina-1-as.jpg)










