Embed Size (px)
Citation preview

“The FDA Inspector Cometh”
Inspection Process for Clinical Trials

FDA “Good Clinical Practice” Inspections
What has happened at UMHS? Who conducts the inspections? Who and what are inspected? What is the inspection process? How can the process go better?

What has happened at UMHS?
Some Metrics

FDA Inspection History at UMHS
Hematology/Oncology Endocrinology/Metabolism Neurosurgery Rheumatology Human Genetics Gastroenterology Dermatology
Opthamology Pharmacology Urology Cardiology Radiology Anaesthesiology Pulmonary
>24 investigators and/or drug trials since 1981
http://www.fda.gov/cder/regulatory/investigators/default.htm

FDA Inspection History at UMHS
FDA has inspected IRBs since 1980 Per IRBMED:
– Inspections occur about every 5 years– Known inspections
1992 1997 Latest in Oct, 2001

Who conducts inspections?
Hint:
Motto: Compliance, Science, Protection
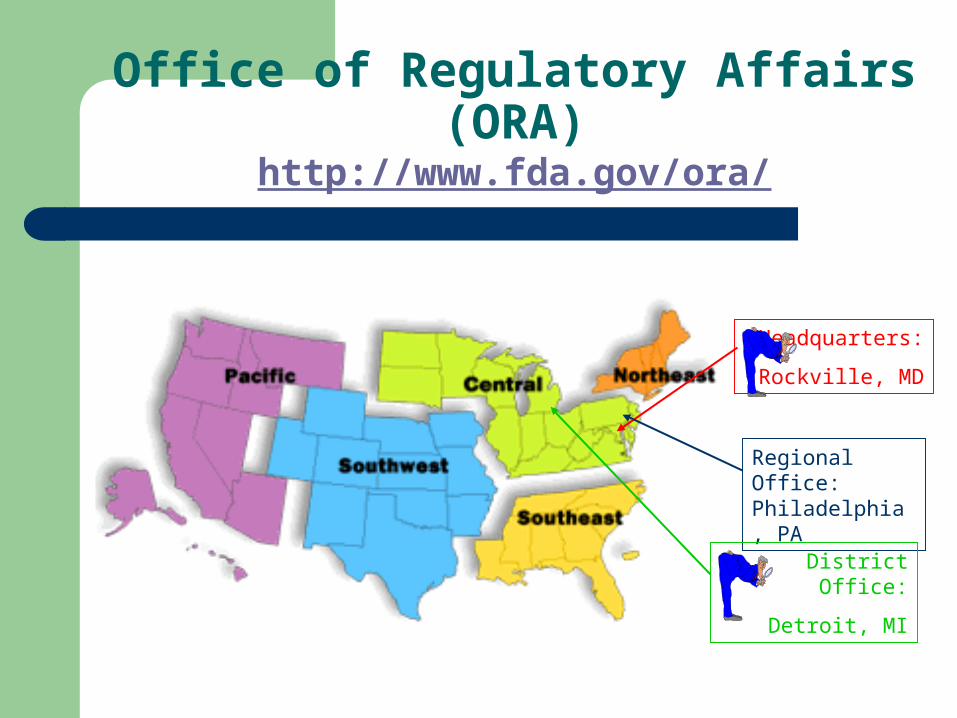
Office of Regulatory Affairs (ORA)http://www.fda.gov/ora/
Headquarters:
Rockville, MD
Regional Office: Philadelphia, PA
District Office:
Detroit, MI

FDA Field Investigators
Conduct inspections to enforce the Food, Drug and Cosmetic Act
Train themselves in “evidence collection”– “If it’s not documented, it didn’t happen.”

Who and What are Inspected?
Today’s Focus:
GCP-Related People and Places

Who?
Investigators (Doctors) and Study Coordinators IRB (IRBMED) Sponsor, if applicable (Industry) Contract Research Organization, if involved Laboratories (e.g., Mlabs) Pharmacy (e.g., Investigational Drug Services) Devices (e.g., Biomedical Engineering)

What studies?
Usual Emphasis: Phase 3– Adequate and well controlled
Blinded Safety and Efficacy
– Multi-site High patient enrolling sites
– Recent marketing application (e.g. New Drug Application) filed to an Investigational New Drug (IND)

What is the inspection process?
10 Steps
for
Investigators

1. FDA selects Site(s)
• FDA selects site for inspection:• Usually within 6 months of marketing application
[NDA] (Data Audit)• Selects 3 sites (average) per study, if multi-site• May concurrently inspect the associated IRB:
• If no previous inspection; or • Last inspection >5 years
OR• May conduct a “For Cause” Audit

Reasons: “For Cause” Inspections
Study of “singular importance” in product approval
Study has major impact on medical practice
Sponsor reports concerns about investigator
Patient complaint
Investigator conducts too many studies
Investigator works outside of specialty area
Safety or efficacy findings are inconsistent with other investigators
Lab results are outside range of biological expectations

2. FDA Investigator contacts Site
• FDA investigator from local District Office contacts responsible person at site:
• Gives short advance notice or no notice of visit • Becomes suspicious on attempts to delay visit
(e.g., >10 days without valid reason)• Previews internally following subject related data:
• Number of total subjects, dropouts and evaluable subjects
• List of AEs and deaths (with description and cause)

3. FDA and Site agree on Visit Date
• FDA investigator and site person agree on appointment for site visit:
• Averages 3-5 days for appointment• Targets typically one study, but may review other
studies performed by same investigator
Preparation Tips for Site
Notify all staff involved in AND/OR knowledgeable about the study:– Key staff, “information providers” are on standby– Office of General Counsel– Industry sponsor, if any
Review UMHS procedures– http://www.med.umich.edu/i/policies/umh/01-0
1-020.html for unannounced inspections
– http://www.med.umich.edu/irbmed/ae/oriotwoc.htm#audit for IRBMED notification


Preparation Tips for Site
Assign a site escort/facilitator Assemble all study documents in 1 place
– Include list of staff responsibilities and training– Request all patient charts
Prepare a list of investigator’s studies Reserve adequate work space for field
investigator for entire inspection Assure accessible photocopier

4. FDA presents Notice of Inspection
• Upon arrival FDA displays credentials (eg., photo ID) and FDA Form 482, Notice of Inspection
• Conducts inspection during routine business hours• May “meet and greet” 1-3 FDA investigators

FDA Form 482

5. FDA requests data and documents
• FDA investigator requests related trial data and documents during site visit
• May need copies of documents• Make 2 copies:
• Give 1 copy to FDA• Keep 1 copy at site to facilitate future communications

Tips on Document Requests
Do not provide or copy these information for FDA:– Financial data (salary information, budgets)
(except financial disclosure of clinical investigators)
– Personnel data (performance appraisals) (except qualifications [job descriptions] and training
records)

6. FDA interviews Site Staff
• FDA investigator interviews site staff directly involved in trial activities and processes
• May question any staff member during inspection• May use Compliance Program Guidance Manual
as interview guide

Tips for Anticipating FDA Questions
Compliance Program Guidance Manuals (CPGMs)http://www.fda.gov/ora/cpgm/default.htm
In Vivo Bioequivalence 7348.001
IRBs 7348.809
Sponsors, CROs and Monitors 7348.810
Clinical Investigators 7348.811

Tips for Handling FDA Questions
Answer– Politely, cooperatively, understanding them (ask for
clarification), factually, briefly, within one’s expertise (seek expert), directly (remain within scope), without speculation or guesswork
Avoid– Unsolicited questions, hypothetical questions, long
delays to requests, affidavits

7. FDA conducts “Exit Interview”
• [Review findings with FDA investigator at end of each inspection day]
• At site visit completion, FDA investigator conducts “exit interview” with responsible site personnel to:
• Review findings• Clarify misunderstandings• Describe any deviations from current regulations• Suggest corrective action, if appropriate

8. FDA presents Notice of Observations
• If deviations, FDA investigator leaves a FDA Form 483, Notice of Observations
• Submits findings to local District Office for any additional needed actions

FDA Form 483

Most Common Observations (for Investigators)
Protocol non-adherence Inadequate and inaccurate records Failure to report adverse events Failure to report concomitant therapy Inadequate drug accountability IRB/IEC problems Informed consent issues

9. FDA writes Inspection Report
• Upon return to local District Office, FDA investigator:
• Writes an Establishment Inspection Report (EIR); and
• Forwards to headquarters for evaluation

10. FDA classifies Inspection
• When evaluation is completed, FDA classifies inspection and sends a letter to site
Classification Type of Letter
NAI (No Action Indicated) Notice of no significant deviations
VAI (Voluntary Action Indicated)
Informational
OAI (Official Action Indicated)
Warning
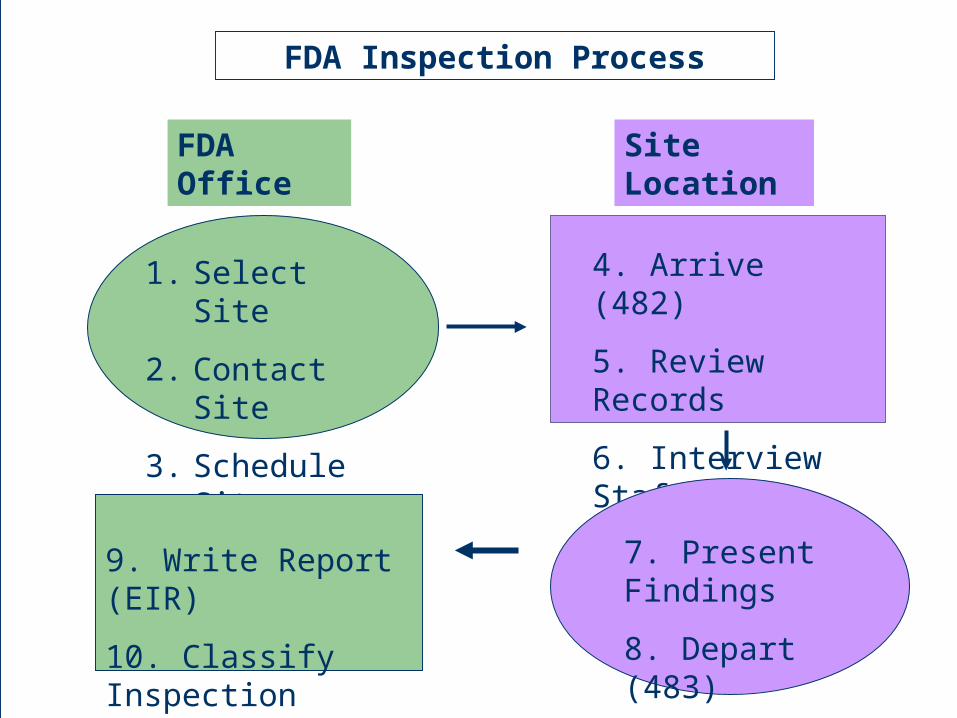
1. Select Site
2. Contact Site
3. Schedule Site
4. Arrive (482)
5. Review Records
6. Interview Staff
7. Present Findings
8. Depart (483)
9. Write Report (EIR)
10. Classify Inspection
FDA Office Site Location
FDA Inspection Process





























