Embed Size (px)
DESCRIPTION
tec frm
Citation preview

Tecnologia Farmaceutica
Come si fa in modo che la sostanza attiva possa essere somministrata? Di ciò si occupa la
tecnologia farmaceutica. Circa ¼ di molecole che vengono scoperte ad altissima attività
farmacologica, non sono poi commercializzate perché non si trova una formulazione funzionale per
queste sostanze.
Forma farmaceutica, ci sono due definizioni:
Categoria delle preparazioni formulate cui appartiene il prodotto;
Presentazione del farmaco conseguente una trasformazione che lo rende idoneo a un certo
tipo di somministrazione.
Quindi nella prima definizione, entra il concetto di formulazione che, nella seconda definizione, in
cui entra il concetto di trasformazione, e quindi un processo formulativo (di trasformazione) che
rende il principio attivo idoneo alla somministrazione. Quindi sono tutte le sostanze e tutti quei
processi che fanno sì che il principio attivo possa essere somministrato. La classificazione delle
forme farmaceutiche è varia, e può essere fatta in base:
Alla forma “fisica”: si possono avere forme farmaceutiche solide, liquide e semisolide.
Unidose/multidose: unidose per le forme farmaceutiche che sono suddivise, all'atto della
preparazione, nella singola unità posologica (compresse, supposte), quindi quando la
suddivisione nell'unità posologica è fatta da chi produce la forma farmaceutica. Multidose
quella in cui la suddivisione della singola unità posologica è fatta dal paziente (gocce,
sciroppi, colliri).
Via di somministrazione, che sono: 1) orale, 2) rettale, 3) parenterale, 4) topica, 5)
polmonare, 6) nasale, 7) oculare.
Un’altra suddivisione delle forme farmaceutiche è in base all'uso: l'uso sistemico e l'uso locale.
Spesso ci si confonde: spesso s’intende per somministrazione sistemica quella che prevede che la
forma farmaceutica arrivi all'interno dell'organismo mentre per somministrazione locale s’intende
una forma farmaceutica che si applica all'esterno; non è sempre così perché in realtà quello che fa la
differenza è se viene assorbito e quindi se viene trasportato dal torrente circolatorio si parla di uso
sistemico.
Nella forma farmaceutica, escludendo il principio attivo, ci sono degli eccipienti. Sono definiti
come qualsiasi materiale contenuto nella forma farmaceutica finale che non sia il principio attivo.
Gli eccipienti devono rientrare nella categoria di sostanze definite GRAS, generally recognized as
safe, ovvero, sicure dal punto di vista farmaceutico e tossicologico. Inoltre, nella definizione di
eccipiente, entra il fatto che queste sostanze devono essere inerti da tutti i punti di vista; devono
essere inerti chimicamente, fisicamente, farmacologicamente e dal punto di vista tossicologico. Per
ogni singolo eccipiente deve essere valutata e giustificata in sede di Autorizzazione all'immissione
in commercio la funzione, e quindi deve essere giustificato perché l'eccipiente è stato aggiunto e
deve essere dichiarata o validata la sua funzione nella forma farmaceutica.
Nella definizione della Ph Eur: un qualsiasi componente, al di fuori del principio attivo, presente in

una preparazione farmaceutica o utilizzata per la fabbricazione. Esiste un’istituzione internazionale
per il controllo e per la qualità degli eccipienti per uso farmaceutico che ha dato una sua
definizione. Nella definizione dell'IPEC(international Pharmaceutical Eccipients control) c'è una
classificazione degli eccipienti che devono essere inerti. La definizione secondo l'IPEC degli
eccipienti è: una qualsiasi sostanza,diversa dal principio attivo,adeguatamente valutata nella
sicurezza, che faccia parte di un sistema di azione del principio attivo, per una delle seguenti
ragioni:
1. Aiutare la lavorazione del sistema durante la preparazione;
2. Proteggere,supportare o aumentare la stabilità, la biodisponibilità e la patient compliance;
3. Aiutare l'identificazione del medicinale potenziare la sicurezza e l'efficacia del medicinale
durante lo stoccaggio e l'uso.
I primi eccipienti, utilizzati nelle preparazioni farmaceutiche, erano tutte sostanze alimentari e di
uso corrente, come il miele, il vino, l'uovo, le mele, etc. ovviamente si è passati dall'uso di queste
sostanze, poco standardizzate, all'uso di sostanze riconosciute come eccipienti farmaceutici. Questo
perché la composizione deve essere standardizzata come anche le caratteristiche chimico-fisiche.
Esiste, redatta e controllata dall'IPEC, una guida per la qualità degli eccipienti per uso farmaceutico
( good manufacting guide ok buik pharmaceutical excipient); in queste norme di buona
preparazione degli eccipienti sono state standardizzate le tecniche e i controlli, e vengono definite le
fonti degli eccipienti(che possono essere naturali o sintetici). Le industrie farmaceutiche speravano
di poter utilizzare o sostanze già utilizzate da altri (così si risparmiavano studi di inerzia e
tossicologia) o sostanze utilizzate nell'industria alimentare. Infatti si sono tantissime sostanze che
entrano nelle confezioni farmaceutiche che sono già in uso nei prodotti alimentari soprattutto del
mondo orientale.
Nella standardizzazione degli eccipienti deve essere validata la presenza di impurezze ( devono
essere identificate e/o analizzate) e possono essere:
Tossiche,che devono essere sempre controllate;
Funzionali,che devono essere accertate nella composizione della sostanza perché altrimenti
quella sostanza non riesce a esplicare la sua funzione di eccipiente(es. Cellulosa cristallina
ha una piccola frazione di emicellulosa che è fondamentale in quanto in assenza di questa
sostanza non si ha una modificazione della viscosità).
È difficile valutare la purezza e la stabilità degli eccipienti perché la maggior parte degli
eccipienti non sono specie chimiche pure, ma sono miscele di specie diverse. Per molti
eccipienti, la valutazione della stabilità non è fatta da un'analisi di tipo chimico ma è fatta da
un'analisi di tipo fisico. Ad esempio, la gelatina che è un eccipiente di derivazione animale,
e quindi non ha composizione chimica semplice, la sua stabilità nel tempo viene valutata
misurando una capacità fisica cioè il potere gelificante. Quindi si misura il tempo 0 di potere
gelificante e si misura, poi, dopo un anno e da questo si valuta se l'eccipiente è ancora
stabile oppure no. Questo è ovviamente valido per gli eccipienti di sintesi generalmente
hanno una composizione standard e quindi la stabilità viene fatta con altri metodi. Gli
eccipienti possono essere classificati in base al ruolo:
Costitutivo , la cui presenza è fondamentale per realizzare la forma farmaceutica; nella
quasi totalità delle forme farmaceutiche, gli eccipienti oscillano tra il 60 e l'80% della massa

della forma farmaceutica. Servono quindi per ottenere una “massa lavorabile”;
Produttivo, che non sono fondamentali, in assoluto ma sono legati alla tecnica di
produzione. Servono quindi per facilitare i processi tecnologici di produzione;
biofarmaceutico, ovvero che sono in grado di modificare la biodisponibilità o piu' in
generale, il “destino” del principio attivo nell'organismo;
Conservazione, che servono per la stabilità;
presentazione , per la patient compliance(es associazione odore-aroma come per esempio
nello sciroppo all'aroma di arancio con colore blu).
Gli eccipienti devono essere inerti ma possono influenzare il rilascio della forma farmaceutica, e la
presenza di un eccipiente è il responsabile delle allergie e delle reazioni avverse al 70%, ai farmaci.
Tra gli eccipienti che sono sicuramente presenti in maggiori quantità nella forma farmaceutica, ci
sono quelli che hanno un ruolo costitutivo, ovvero quelli che consentono la lavorabilità del
principio attivo. Nell'ambito degli eccipienti con il ruolo costitutivo si trovano i DILUENTI. Questi
possono essere: solidi, liquidi e semisolidi( per le preparazioni ad uso dermatologico). Uno dei
diluenti piu' utilizzati,nonostante il problema legato alle intolleranze, è il lattosio. Le informazioni
sul lattosio dalla farmacopea europea.
Le principali sono:monografie:anidro,monoidrato (2 forme di lattosio); caratteristiche
specifiche:potere rotatorio specifico,perdita all'essiccamento,metalli pesanti. Forme farmaceutiche
solide:diluente(65-85%); In soluzione ( + saccarosio 1/3): confettura; Lattosio spray dried (SP) è un
tipo particolare di lattosio,che può essere usato per ottenere compresse con la tecnica della
compressione diretta e quindi è un eccipiente facilmente comprimibile. Quindi il lattosio che può
essere utilizzato nelle preparazioni di uso farmaceutico deve rispondere ai requisiti di farmacopea,
in farmacia non si può utilizzare qualsiasi lattosio, ma bisogna utilizzare il lattosio FU. Se non esiste
per quella sostanza la monografia sulla farmacopea italiana si fa rifermento alla monografia che fa
riferimento alla sostanza sulla farmacopea europea.
Nell'utilizzo del lattosio cosa bisogna che sia valutato: l'intolleranza da parte del paziente e le
incompatibilità di tipo chimico o fisico. Di tipo chimico possono essere: da condensazione con
ammine primarie; può dare prodotti con colorazione scura. Quindi non è solo un problema legato al
fatto che il principio attivo non è piu' attivo una volta che il lattosio si è condensato, ma è anche che
la forma farmaceutica ha una colorazione non corretta. Durante il riscaldamento per la
nebulizzazione per ottenere il lattosio spray dried o se nella lavorazione della forma farmaceutica
c'è una fase di riscaldamento, il lattosio può dare luogo alla 5-idrossimetil-2-furfale che ha sempre
un colore scuro.
Tra gli eccipienti con ruolo costitutivo, oltre ai diluenti ci sono gli assorbenti e gli adsorbenti: gli
assorbenti, che sono generalmente dei silicati, che sono aggiunti nelle preparazioni farmaceutiche
per assorbire l'eventuale umidità residua che ci può essere nel processo produttivo. Gli adsorbenti
sono delle sostanze, come per esempio il caolino o il talco, che hanno la caratteristica di avere
elevata capacità assorbente e questo serve, ad esempio, a formulare in forma solida dei principi
attivi liquidi. Quindi il principio attivo liquido viene fatto adsorbire sul solito e poi viene formulato
in una forma farmaceutica solida. Ciò che però bisogna stare attenti è di non alterare la
biodisponibilità del principio attivo,quindi che non influenzi il rilascio in vivo del principio attivo.

Oltre a questi ci sono anche gli eccipienti con ruolo produttivo: sono una classe decisamente piu'
ampia e sono: i lubrificanti, i glicanti, i leganti, i elasticizzanti, i tensioattivi e i viscosizzanti. Quelli
più interessanti sono i lubrificanti e i glicanti: queste due classi fanno parte della classe
generalmente definita come lubrificanti. I glicanti agiscono sulle particelle delle polveri e quindi
aumentano la scorrevolezza di una polvere agendo sulla porosità; i lubrificanti aumentano la
scorrevolezza della polvere modificando o agendo sulle interazioni tra la polvere e le parti della
macchina che produce la forma farmaceutica.
Ci sono anche gli eccipienti con ruolo farmaceutico cioè quelli che influenzano il rilascio del
principio attivo della forma farmaceutica. In questo caso sono: i disgreganti, i polimeri e i bagnanti.
La classe preponderante è quella dei disgreganti o disaggreganti. Uno dei disgreganti più utilizzato
è l’amido, ma esistono anche dei polimeri particolari come la croscaramellosio il cui nome
commerciale è l'explotab, che porta proprio all'esplosione della compressa una volta che questa è
messa in contatto con l'acqua.
Da notare bene è che le forme farmaceutiche sono distinte in: convenzionali e in non
convenzionali. Le forma farmaceutica convenzionale non deve avere effetto sull'assorbimento del
principio attivo, quindi generalmente la forma farmaceutica convenzionale è realizzata per fare in
modo che il principio attivo sia rilasciato nella maggiore quantità e il più velocemente possibile.
Quindi nella forma farmaceutica convenzionale, la velocità di rilascio della forma farmaceutica è
molto maggiore rispetto alla velocità di assorbimento. Quando il principio attivo è formulato con la
forma farmaceutica convenzionale, lo stadio limitante della sua azione è l'assorbimento e non la
liberazione del principio attivo. La forma farmaceutica non convenzionale, invece, viene realizzata
per modulare il rilascio del principio attivo e la caratteristica è che la velocità di rilascio del
principio attivo è minore rispetto alla velocità di assorbimento, e quindi lo stadio limitante non è il
passaggio attraverso le membrane biologiche bensì la liberazione del principio attivo. Nelle forme
farmaceutiche convenzionali , il ruolo del disgregante è fondamentale. Uno dei disaggreganti più
utilizzato è l'amido; in farmacopea ci sono 4 monografie a seconda dell'azione dell'amido e non ha
un solo ruolo( come la maggior parte degli eccipienti); infatti funziona sia da diluente, che da
legante (pasta d'amido al 5-25%) e da disgregante(3-17%). Quindi indipendentemente dalla sua
composizione può funzionare da disgregante o da legante nella forma farmaceutica. Inoltre l'amido
può essere utilizzato anche come principio attivo:entra come principio attivo in numerose
preparazioni per uso cutaneo come assorbenti o come anti irritanti.
Un'altra classe degli eccipienti, presenti in quasi tutte le forme farmaceutiche industriali è la classe
degli stereati, in particolare lo stearato di magnesio (Ph. Eur) ha delle incompatibilità chimiche, che
vanno valutate; il problema degli stearati è che sono totalmente insolubili in acqua e quindi la loro
presenza è fondamentale per favorire lo scorrimento delle forme per uso farmaceutico nelle
macchine produttrici, ma bisogna valutare negli studi di pre-formulazione in maniera estremamente
accurata, la loro quantità perché possono influenzare in maniera drammatica la liberazione del
principio attivo, ovvero si potrebbero produrre delle compresso che non rilasciano il principio
attivo. Il loro ruolo è di lubrificanti.
Indipendentemente che le forme farmaceutiche siano solido o liquide, i conservanti avranno una
funzione diversa. Tra i conservanti ci sono:

antiossidanti:metabisolfito, acido ascorbico;
chelanti: EDTA;
antimicrobici: parabeni, acido benzoico.
Il processo ossidativo nasce sostanzialmente con i doppi legami di O2, catalizzati dalla luce o da
alcuni metalli; per la conservazione di moltissime preparazioni è richiesto il vetro scuro; il risultato
del processo ossidativo sono altri prodotti. Sono catalizzati da alcuni ioni, per questo vengono
aggiunti degli agenti chelanti che non sono dei protettori, ovvero non inibiscono il processo
ossidativo, ma eliminando i metalli rallenta il processo ossidativo. Oppure , gli antiossidanti veri e
proprio sono delle sostanze che si ossidano e quindi impediscono l'ossidazione della forma
farmaceutica.
Ci sono degli eccipienti con il ruolo di presentazione e sono:
aromatizzanti
edulcoranti: aspartame, saccarosio,saccarina
coloranti: idrosolubili, ossidi di ferro
opacizzanti: biossido di titanio.
Per gli aromatizzanti e per i coloranti ci sono degli studi impegnativi; c'è infatti, un istituto in
Svizzera che studia questi( sono un team di psicologi,chimici) che studia l'effetto psicologico del
colore nelle forme farmaceutiche. Ci sono degli effetti dei coloranti impensabili, per esempio
nell'anti aritmico se è di colore delle tonalità del rosso potenzia l'effetto del principio attivo. Un'altro
problema dei coloranti è che molti di questi(come gli ossidi di ferro) sono insolubili in acqua e
quindi con gli stearati, la loro presenza nella forma farmaceutica deve essere valutata molto
accuratamente perché possono avere effetti sul rilascio del principio attivo.

Polveri
Le polveri hanno diverse classificazioni: sono classificate come polveri delle forme allo stato solido
costituite da particelle che vanno dall'ordine del micron ai 100-4000 μm, ma in questo caso si parla
di aggregati, e quindi si passa da dimensioni non visibili ad occhio nudo a dimensioni visibili. Le
dimensioni delle particelle solide, che costituiscono una polvere, sono responsabili di quasi tutte le
caratteristiche delle polveri. Le dimensioni delle particelle influenzano l'area superficiale di una
polvere. Per area superficiale si intende la superficie della polvere/volume(viene anche chiamata
area superficiale specifica). Delle riduzioni, non caratteristiche, delle dimensioni (in questo caso
sono considerati dei gruppetti di polvere) o di lunghezza del lato portano a variazioni della
superficie specifica sostanziali. Se un cubetto, che ha 1 cm di lato, ha un'area specifica di 6
cm2/cm3, se riduciamo di 1μm si ha un incremento dell'area superficiale. L'area superficiale è
importante perché influenza numerose caratteristiche delle polveri in particolare la velocità di
dissoluzione, e quindi la capacità che la forma farmaceutica ha di liberare il principio attivo. Le
polveri sono preparate con:
un approccio di tipo meccanico(nella quasi totalità dei casi) con delle macchine dette
molini, in cui viene ridotta la dimensione delle particelle, in cui quindi si ha la riduzione di
materiale grossolano in particelle più piccole;
un approccio di tipo chimico-fisico(raramente perché la tecnica è costosa), generalmente si
utilizza per produrre delle polveri colloidali,cioè a dimensioni inferiori al micron.
Nell'approccio meccanico ci possono essere dei problemi sia per i principi attivi che per le polveri,
perché in un processo di riduzione meccanica si sviluppa sicuramente calore, e quindi bisogna
valutare la stabilità alle variazioni di calore del principio attivo e degli eccipienti e bisogna valutare
accuratamente l'energia che bisogna fornire al processo di macinazione; questo perché una parte di
energia servirà a ridurre le dimensioni delle particelle mentre una frazione sarà assorbita dal
sistema(dalla polvere e dalla macchina). Quindi vanno fatti degli studi di pre- formulazione in cui
deve essere valutata l'energia da fornire nel processo di macinazione, che deve essere l'energia
sufficiente alla riduzione delle dimensioni delle particelle ma non eccessiva per danneggiare, dal
punto di vista chimico-fisico, il principio attivo e gli eccipienti o addirittura l'apparecchiatura. Se ,
per esempio, bisogna macinare o trattare delle sostanze che sono fortemente ossidabili all'aria, nel
processo di macinazione ci potrebbe essere un'esplosione. Le macchine vengono scelta in base alla
tecnica, perché la tecnica che sfruttano influenza le dimensioni delle particelle. Quindi ci sono i
processi di:
frantumazione: è un processo grossolano, ed è la riduzione del materiale grezzo in pezzi
grossolani(mm);
macinazione: riduzione dimensionale dei pezzi grossolani in particelle più piccole
(centinaia/decine di μm)
micronizzazione: riduzione delle dimensioni di particelle o dimensioni inferiori a 10μm
fino a particelle colloidali( è la tecnica che si utilizza per produrre il lattosio spray dried,
molto costoso). Sostanzialmente il costo produttivo dipenda da numerosi fattori ma il
principale è l'energia assorbita, ovvero più energia ci vuole per realizzare il processo più il
processo è costoso.

Per la macinazione vera e propria si usano i molini, che hanno caratteristiche costruttive diverse
perché sfruttano dei meccanismi di polverizzazione diversi. I principali molini sono:a martello, a
cilindri, a coltelli, colloidale, a compressione e a impatto. Per esempio,sui molini a coltello il
meccanismo è il meccanismo di taglio. A seconda del tipo di molino e quindi a seconda del
meccanismo di macinazione,si otterranno polveri di dimensioni diverse.
Quindi il meccanismo principale è la macinazione,che si esegue attraverso i molini i quali sfruttano
meccanismi di macinazione diversi e questo meccanismo influenza la dimensione della polvere. La
scelta del molino viene fatta da:
caratteristiche di materiali di partenza ( termolabilità, dimensioni, degradabilità e struttura
fisica);
dimensioni del prodotto che si vuole ottenere;
fattori economici, definiti dal costo del processo e dal tempo del processo; perché se un
processo assorbe meno energia ma dura 6 giorni,si tiene l'impianto per 6 giorni per produrre
la base di uno dei costituenti della formulazione;
facilità di pulizia dell'apparecchiatura (anch'esso un punto cruciale della produzione) perché
lavare l'impianto vuol dire fermarlo e quindi non produrre. C'è una ricerca enorme, da parte
dell'ingegneria chimica, in quelle che si chiamano le tecniche CIP, ovvero in clean in place,
realizzati,cioè, degli impianti che possono essere lavati senza smontare nulla,riducendo,
così, in maniera drastica i tempo in cui l'impianto è fermo.
Possibilità, quando è necessario, di operare in ambiente di sterilità;
versatilità dell'operazione, macinazione o a secco o a umido, differenza della velocità di
rotazione degli elementi macinanti.
La polvere per uso farmaceutico è la base di tutte le preparazioni successive oppure può essere la
forma farmaceutica finale, e quindi la caratterizzazione secondo farmacopea sarà diversa a seconda
che la polvere per uso farmaceutico sia intermedia o sia la forma farmaceutica finita. La prima
caratterizzazione che viene richiesta dalla farmacopea è l'analisi granulometrica della polvere,
cioè la classificazione delle polveri in base alle loro dimensioni. A seconda di quali siano le
dimensioni delle particelle delle polveri, si avranno delle tecniche di dimensionamento diverse, che
daranno informazioni diverse. Le polveri possono essere caratterizzate dal diametro medio, ovvero
la dimensione media delle particelle. Un'altra informazione è data dal diametro mediano,ovvero
che tiene conto anche della distribuzione dei valori(si dice che la mediana è il valore che divide a
unità la popolazione che tiene conto anche di quante volte il valore è ripetuto, e quindi non solo il
valore medio ma della distribuzione dei valori).
Il metodo della farmacopea prende il nome di metodo dei setacci o degli stacci: i setacci sono di
alluminio e sono caratterizzati da un numero che indica l'apertura della maglia in μm. I setacci, della
farmacopea ufficiale italiana, sono a maglia quadrata. I setacci sono costituiti da materiali adatti ed
hanno maglie quadrate, in cui non deve avvenire alcuna reazione tra il materiale del setacci e della
sostanza da setacciare: servono per determinare il grado di finezza della polvere che viene descritto
nelle singole monografie utilizzando il numero del setaccio che indica l'apertura della maglia in μm.
Come si esegue il saggio di farmacopea: questo saggio dà informazioni sia sulle dimensioni che
sulla distribuzione. Vengono impilati i setacci, secondo un valore decrescente di apertura della
maglia. Vengono posti, per esempio, 100 g di polvere sul primo setaccio. La pila di setacci viene

sotto l'azione meccanica di un apparecchiatura che imprime degli scorrimenti. Dopo un certo
periodo di tempo, si pesa la quantità di polvere che si è fermata sui vari setacci. Per esempio, 80 g si
sono fermati sul setaccio 4000 e 20 g si sono fermati sul setaccio 3000. quindi 80 g della polvere
avranno un diametro medio di 4500, ovvero l'80% della polvere è certamente inferiore a 5000 ma
superiore a 4000(la farmacopea chiede il diametro medio), il 20% della polvere avrà un diametro
medio di 3500. Con questo saggio si hanno, come si è visto, sia informazioni sulle dimensioni che
sulla distribuzione; ciò perché, in farmacopea, la classificazione delle polveri non è fatta solo sulle
dimensioni ma è fatta anche sulla distribuzione delle dimensioni(che è
l'indice dell’omogeneità della polvere). Quindi le polveri sono
classificate in:
polvere grossolana: è una polvere che ha una dispersione delle
dimensioni tra 1400 μm e 355 μm, e quindi non è solo una polvere
grande, ma è molto disomogenea. La farmacopea, inoltre, guarda la %
della polvere che è passata attraverso i setacci->non meno del 95% in
massa della polvere attraverso il setaccio 1400 e non piu' del 40% in
massa della polvere attraverso il setaccio 355;
polvere moderatamente fine: non meno del 95% in massa della polvere passa attraverso il setaccio
355 e non piu' del 40% in massa della polvere attraverso il setaccio 180;
polvere fine: non meno del 95% in massa della polvere attraversa il setaccio 180 e non piu' del 40%
in massa della polvere attraversa il setaccio 125;
polvere molto fine: non meno del 95% in massa della polvere passa attraverso il setaccio 125 e non
piu' del 40% in massa della polvere passa attraverso il setaccio novanta.
Le polveri vengono classificate da dei numeri, vuol dire che la polvere ha un intervallo di
dimensioni, o da un solo numero, e vuol dire che la polvere è omogenea quando non meno del 97%
della polvere passa attraverso il setaccio di quel numero. Le dimensioni delle particelle influenzano
un'altra caratteristica importante, ovvero la porosità. La porosità delle polvere è un valore % che
esprime:
ε = volume dei pori/volume apparente x 100
e quindi esprime il rapporto tra il volume degli spazi vuoti e il volume occupato dalle particelle
delle polveri piu' gli spazi vuoti. La presenza o meno e l'entità degli spazi vuoti dipende
dall’impaccamento della polvere. Ci sono
due impaccamenti:
a) cubico, che ha una maggiore quantità
di spazi vuoti
b) romboedrico, in cui gli spazi vuoti
sono minori

Le caratteristiche della polvere che definiscono la porosità sono due: a) volume apparente,che è il
volume che tiene conto del volume delle particelle piu' gli spazi vuoti, b) volume reale,cioè il
volume della polvere una volta che si sono eliminati gli spazi tra le particelle, quindi il volume
occupato solo dalle particelle. La porosità quindi diventerà:
La farmacopea chiama il volume apparente e il volume reale: a) volume apparente prima
dell'impaccamento;b) volume apparente dopo l'impaccamento. Come si esegue il saggio: si
pongono 100 g di polvere all'interno di un cilindro, si misura il volume a impaccamento, il cilindro
è posto su un motore che imprime dei colpi al cilindro( un numero di colpi sempre fissato dalla
farmacopea): i valori che si raccolgono da questo saggio è il volume apparente prima
dell'impaccamento, che la farmacopea indica con Vo, e il volume apparente dopo l'impaccamento,
caratterizzato da Vn, dove n è il n° di colpi che sono stati dati al cilindro, e la capacità di
impaccamento, ovvero quanto la polvere è impaccabile.
Dal volume apparente,dice la farmacopea, si può rilevare la densità apparente o Vo e la densità
apparente dopo impacchettamento: è fondamentale conoscere la porosità di una polvere perché
dipende dalla dimensione delle particelle e dell'impaccamento della polvere, ma anche perché la
porosità della polvere dipendono numerose altre caratteristiche tra cui la scorrevolezza di una
polvere, che è fondamentale perché la suddivisione nelle singole unità posologiche della massa, sia
nella produzione industriale ma anche nella preparazione galenica, è fatta a volume. Quindi anche
se le macchine lavorano con dei solidi, i dosatori sono dosatori di volume e quindi la polvere si
deve muovere sulla macchina simile ad un fluido( che non viene dosato a peso ma a volume).
Il saggio di scorrimento: i solidi suddivisi in farmacopea( le polveri e i granulati) devono essere in
grado di scorrere verticalmente perché tutte le suddivisioni sulle singole unità posologiche vengono
fatte a volume e non a peso. Quindi anche se le polveri sono solidi devono scorrere e comportarsi
come un fluido. Sulla farmacopea italiana viene riportato un solo saggio come ufficiale, per
determinare la velocità di scorrimento che misura in realtà un tempo di scorrimento. 100 g di
polvere vengono fatti passare dentro un imbuto in cui è stato tappato il fondo; si apre e si misura il
tempo necessario a che la polvere defluisca attraverso questo imbuto. L'imbuto non è casuale ma
deve avere delle dimensioni in mm dettate dalla farmacopea, così come gli angoli. Ciò per avere un
sistema di riferimento uguale per tutti.
Nelle versioni precedenti della farmacopea italiana ed è stato
inserito nuovamente nella farmacopea europea. C'è un altro
saggio per definire la scorrevolezza di una polvere che si indica
con il concetto di angolo di riposo. In cosa consiste: un piatto
circolare di raggio noto che viene posto su un sostegno. Su
questo piano circolare,viene fatta cadere attraverso un imbuto la
polvere;questa, cadendo sul piano circolare, formerà un cono. Il
saggio è considerato terminato ossia quando sarà il momento in
cui è possibile eseguire la misura, quando per aggiunte
successive di polvere, l'altezza del cono non varia più. Questo

perché: la polvere che si aggiunge incomincia a cadere dai lati,che dal punto di vista fisico significa
che si è formato un equilibrio tra la forza peso e le forze di attrito e di tensione tra le particelle di
polvere,che sono quelle che mi interessa determinare(ovvero la capacità o meno delle particelle di
scorrere, quanto l'attrito influenza questa proprietà). Si misura l'angolo α che si definisce come
angolo di riposo e che dà una misura della scorrevolezza o meno della polvere.
In realtà non si misura l'α ma misuro la tag α.
si considera un valore di scorrevolezza ottimale di una polvere quando 35°< α <45°, perché polveri
poco scorrevoli sono un problema perché possono bloccare le macchine mentre polveri troppo
scorrevoli possono essere altrettanto un problema perché una polvere che scorre troppo rapidamente
viene difficilmente dosata dal sensore di volume, e quindi può dare problemi di dosaggio tanto
quanto una polvere poco scorrevole.
Le caratteristiche di una polvere che possono influenzare la scorrevolezza sono:
dimensione delle particelle; le polveri più sono piccole e più scorrono ma anche maggiore è
la loro uniformità e maggiore è la scorrevolezza;
forma delle particelle; più è regolare la forma, e quindi più assimilabile a una sfera,
maggiore sarà la scorrevolezza;
porosità: una polvere poco porosa ovvero con poco spazio vuoto tra le particelle scorre
meno rispetto a quella più porosa per un problema di ingombro fisico.
Un' altra caratteristica che viene misurata per le polveri, i cui saggi sono presenti in farmacopea, è la
misura della densità della polvere che viene fatta utilizzando un picnometro. Altra caratteristica
che viene misurata è la misura della superficie specifica della polvere. La superficie specifica è la
superficie su unità di volume ed è un parametro fondamentale per valutare la biodisponibilità di un
principio attivo perché è un parametro che influenza in maniera sostanziale la velocità di
dissoluzione delle forme farmaceutiche e quindi la capacità di liberare il principio attivo. Ci sono
moltissime tecniche per misurare la superficie specifica, ma c'è un concetto che le accomuna: sono
tutte tecniche che sfruttano l'adsorbimento (o di gas o di mercurio) di sostanze che hanno un volume
noto sulla superficie della polvere. Quindi si fa in modo che ci sia un adsorbimento
monomolecolare ( ad esempio di gas) sulla superficie della polvere e si misura quanto gas è stato
adsorbito; dato che è nota l'area che ogni molecola di gas occupa e quindi moltiplicando la
superficie occupata da una molecola per il numero di molecole che sono state adsorbite si misura la

superficie della polvere.
Le caratteristiche e i saggi visti sono quelli che la farmacopea chiede quando la polvere è
l'intermedio della lavorazione della forma farmaceutica. Però le polveri possono anche essere la
forma farmaceutica finita. Le polveri possono essere impiegate:
come polveri per uso orale;
come polveri per uso iniettabile;
come polveri per applicazione cutanea.
Sulla monografia delle polveri per uso orale, le polveri per uso orale sono preparazioni costituite
da particelle solide, non aggregate, asciutte e di vari gradi di finezza. Contengono una o più principi
attivi, con o senza eccipienti. Sono generalmente somministrate in acqua o altro liquido adatto.
Possono anche essere ingerite direttamente. Sono presentate come preparazioni a dose unica o
multidose. La farmacopea in questo caso, indica anche le caratteristiche del processo produttivo e
dà delle indicazioni sui saggi da eseguire sulla forma farmaceutica; cioè per dire che la forma
farmaceutica può essere autorizzata all'immissione in commercio deve rispondere ai saggi di
farmacopea.
Quando si hanno le polveri per applicazione cutanea si nota che la definizione è identica a quella
delle polveri per uso orale, ma aggiunge una caratteristica: se le polveri per applicazione cutanea
sono pensate per essere applicate sulle ferite e sulla cute lesa, il preparato deve essere STERILE. Le
polveri possono essere anche formulate per essere somministrate attraverso dei contenitori
pressurizzati.
Nell'ambito delle polveri per uso orale ci sono le polveri per gocce orali o per sciroppi. Quindi la
polvere può essere la forma farmaceutica finita oppure può essere venduta come polvere ma al
momento dell'assunzione l'utilizzatore deve ricostituire la vera forma farmaceutica: le gocce o lo
sciroppo. Per i problemi legati alle polveri, di fatto, raramente l'azienda farmaceutica le polveri
vengono utilizzate come forme farmaceutiche finite;oggi si aggiunge un passaggio che è quello
della granulazione. Da una miscela di polveri si ottengono i granulati. I granulati vengono fatti
sostanzialmente per ovviare a due problematiche fondamentali :innanzitutto alla regolarità delle
particelle della polvere, perché nella polvere sono polverizzate con i molini e quindi non c'è una
grande attenzione alla forma geometrica della particella di polvere. Con i granulati, invece, si
ottengono sempre delle particelle sferiche, e quindi il granulato è, a parità di composizione, più
scorrevole della polvere.
Esempio: si ha un principio attivo più il lattosio. Si miscelano insieme tutte le polveri; si attua la
misura della velocità di scorrimento e queste due componenti avranno due velocità di scorrimento
(che dipenderanno dalla granulometria di entrambi,dalla porosità di entrambi e dalla miscela).
Invece passando attraverso la formazione del granulato si miscela,preventivamente il lattosio e il
principio attivo e da questa miscela si prepara il granulato e si avranno dei granuli perfettamente
sferici che saranno composti, in maniera omogenea, da eccipiente e principio attivo. Questo anche
perché risolve il problema che si definisce di de miscelamento delle polveri: perché in un impianto
che fa mini compresse ora si mescolano tonnellate di eccipienti e tonnellate di principio attivo.
Dopo averli miscelati si metteranno nei silos da carico. Dipendentemente dal tempo in cui la

miscela è ferma;siccome le polveri che sono state miscelate non hanno la stessa densità, queste si
demiscelano, ovvero si depositano in fondo le polveri più pesanti. Quindi si correrebbe il rischio di
fare le prime compresse solo di principio attivo e tutte le restanti di eccipienti.
I metodi di granulazione sono sostanzialmente distinti:
1. granulazione a secco;
2. granulazione ad umido:
- granulazione per estrusione- sfendiziazione;
- granulazione a letto fluido
- granulazione per spray drying.
Il metodo della granulazione a secco, cioè che non prevede la
presenza di solventi, si realizza attraverso un processo di
supercompressione, cioè le polveri sono mescolate insieme e
vengono formate attraverso 2 nuclei e quindi vengono
supercompresse insieme e poi, questo nastro che viene fuori dal
granulatore a secco viene frantumato, setacciato e vengono poi
realizzate le compresse. È un processo che non richiede la presenza
di acqua sì la presenza di calore. I vantaggi sostanziali di questo processo sono: va bene per principi
attivi che possono subire idrolisi e per sostanze che sono termolabili ed inoltre costa poco. Ci sono
però molti svantaggi: innanzitutto il fatto che il granulato a secco ha comunque un elevato grado di
polverosità, cioè la possibilità “volare” porta a quello che vengono chiamate contaminazioni
crociate, cioè si possono mescolare all'interno dell'impianto produttivo e può essere rischiosa per il
personale, perché la polvere può essere malata. Inoltre ci sono dei tempi di pulizia molto lunghi.
Un altro problema è quello di distribuire in modo uniforme i coloranti.
Nella granulazione ad umido c'è un numero maggiore di fasi. L'agente legante viene sciolto o in
acqua o generalmente in una miscela di acqua ed etanolo (miscele sono alcoliche). Le polveri sono
impastate con la soluzione di agenti leganti e si ottiene una massa pastosa che viene forzata
attraverso un setaccio. I granuli ottenuti vengono seccati e vengono poi setacciati per ottenere delle
dimensioni omogenee di granulato; una tipica forma farmaceutica che si ferma a questa fase è la
citrosodina, che è un esempio di granulato non setacciato. La setacciatura finale è detta
calibrazione dei granuli.
Il problema fondamentale della granulazione ad umido è la scelta dell'agente legante e la sua
concentrazione. Che anche qui deve essere un compromesso tra una quantità sufficiente ad ottenere
un granulato che sia resistente ma che poi sia facilmente solubile o disgregabile. Deve essere un
buon agente legante ovvero che dia un prodotto resistente ma che deve essere in grado di liberare il
principio attivo in maniera efficace una volta che è stato assunto. Le soluzioni leganti sono o
soluzioni zuccherine (gelatina e amido) ma l'agente che viene utilizzato principalmente è il polivinil
pirrolidone (PVP).
Le tecniche di granulazione ad umido possono essere o in continuo o in fase discontinua, ovvero
che il processo produttivo può avvenire passaggio per passaggio e quindi la miscela di polveri viene

bagnata con la soluzione di agente legante e poi avviene l'essiccamento ed infine avviene la
setacciatura per uniformare i granulati. Sono delle tecniche che si chiamano granulatori a letto
fluido o flusso continuo in cui il processo viene fatto in continuo e quindi c'è una sola macchina
dove dalla miscela di polveri si arriva direttamente al granulato setacciato.
La definizione della Farmacopea dei Granulati: i granulati sono delle preparazioni solide costituite
da aggregati solidi, secchi, di particelle di polvere, sufficientemente resistenti a manipolazioni
energiche.
Lo stadio più complesso nell'operazione di granulazione ad umido è la scelta dell'agente legante e
della sua concentrazione, ciò torna nella definizione di farmacopea, perché deve essere resistente a
manipolazioni energiche (dal punto di vista meccanico) del granulato e quindi non si deve
ripolverizzare nel processo produttivo, ma a sua volta non deve essere eccessivamente resistente dal
non liberare il principio attivo. I granulati, secondo Farmacopea, sono destinati alla
somministrazione orale. Possono essere deglutiti tal quale,come per esempio i granulati
effervescenti, masticati oppure sciolti o dispersi in acqua o in altro liquido adatto prima di essere
somministrati(di questo tipo usiamo l'Aulin e l'Oki). I granulati possono essere a dose unica(Aulin)
o a multidose(citrosodina).
I saggi che prevede la farmacopea per i granulati sono saggi relativi al granulato come forma
farmaceutica finita unidose. Sarà obbligatorio il saggio di uniformità di massa e il saggio di
uniformità di contenuto. I granulati previsti in farmacopea sono distinti in:
- effervescenti
- rivestiti: ovvero che ogni singolo granulo che costituisce la forma farmaceutica finale
avrà un rivestimento di natura polimerica. In questo ambito si possono trovare i
granulati gastroresistenti, in cui ogni singolo granulo è ricoperto con un polimero che
gli conferisce delle caratteristiche gastroresistenti (non si sciolgono a pH acido ma a pH
intestinale); generalmente, questi polimeri, hanno dei residui bicarbonilici, con uno dei
residui carbossilici sono legati al polimero naturale e l'altro conferisce la sensibilità a pH
diversi. Uno dei più utilizzati è l'acetoftalato cellulosa. Quindi è la cellulosa
devitalizzata con l'acido ftalico(che è un derivato bicarbonilico) che ha un COOH che
esterifica con un OH della cellulosa e l'altro COOH libero.

Compresse
Il primo riferimento a delle forme farmaceutiche compresse, risalgono a dei trattati di medicina
araba del X sec. , in cui proprio le polveri erano poste in delle forme e venivano compresse a
martellate. Il processo produttivo della compressione, però, è abbastanza giovane; è alla fine del
1800 che compare, per la prima volta, in terapia, la parola compressa. Sempre verso la fino dell'800
compare la prima monografia (sulla farmacopea inglese) sulle compresse, che rimane l'unica fino al
1945. Da notare che i primi studi sull'influenza della formulazione sul principio sistematico sono
della fine degli anni '70. Le compresse sono delle preparazioni solide contenenti ciascuna una dose
unica di uno o più principi attivi e ottenuti usualmente per compressione di volumi uniformi di
particelle. La maggior parte delle compresse sono per somministrazione orale, in quanto ci sono
anche le compresse per uso rettale. Si possono distinguere varie categorie di compresse per uso
orale: compresse non rivestite,rivestite, effervescenti, solubili, dispersibili, orodispersibili,a rilascio
modificato, gastroresistenti e da utilizzare nella cavità buccale. Esistono due tipi di comprimitrici:
alternativa e rotativa.
In che cosa consiste il processo di compressione: (1) riempimento della camera di compressione
,che ha un dosatore a volume nella comprimitrice; ci sono due parti dette punzoni, uno superiore e
l'altro inferiore. Nella comprimitrice alternativa i due punzoni si muovono in maniera alternata; (2)
il punzone superiore è quello che comprime e quindi è quello che applica la forza di compressione
e (3) il punzone inferiore è quello che fa espellere la compressa. La forma della camera di
compressione, che si chiama anche matrice , determina la forma e la sezione trasversale della
compressa; i punzoni superiori ed inferiori , determinano la forma alla linea di separazione visibile
delle compresse e questo dipende dalle facce dei punzoni. La comprimitrice rotativa è fatta in modo
che i punzoni si muovano contemporaneamente, cioè scorre tra il punzone superiore e il punzone
inferiore un letto di polvere; ad altezza di questi rulli i punzoni vengono spinti inseme e quindi
l'operazione di compressione viene fatta in continuo(che non viene fatta in quella alternativa). Nelle
industrie, di fatto, ci sono solo le comprimitrici rotative, che arrivano a produrre fino a 1000000 di
compresse/ora; per la formulazione galenica invece, le poche farmacie che fanno le compresse
hanno comprimitrici alternative, in quanto più piccole e maneggevoli. Le compresse possono essere
ottenute per: compressione diretta o compressione dopo granulazione(la maggior parte).
La forza di compressione porta ad una deformazione plastica della particelle o ad una deformazione
elastica, ovvero dipendentemente dalla forza che si applica si può avere:

Quindi si ha (1) la polvere che inizialmente si trova nel suo stato di volume apparente (particelle +
spazi vuoti); iniziando ad applicare una forza di compressione si arriva alla (2) alla condizione di
volume reale, detto impaccamento denso, in cui si elimina l'aria tra le particelle. Continuando ad
applicare la forza si ottiene (3) la deformazione elastica: questo vuol dire che se in questo punto si
smette di applicare la forza di compressione , la polvere torna alla sua situazione di partenza.
Continuando ad applicare la forza si arriva (4) alla deformazione plastica, che è quella si che vuole
ovvero la formazione di una compressa e vuol dire che se in questo punto si smette di applicare la
forza di compressione la compressa rimane tale( la polvere rimane coesa). Continuando ad applicare
una forza eccessiva si ha la rottura della compressa(5). Quindi negli studi di questo tipo ovvero in
base alla polvere o al granulato che si deve comprimere dovranno essere fatti degli studi di forza di
compressione, per ogni singola miscela.
La compressione diretta è quella che si fa in farmacia ed è possibile quando la polvere ( o la
miscela di polveri di principio attivo e degli eccipienti) ha buona proprietà di scorrimento e di
comprimibilità; quindi la polvere deve essere scorrevole e deve essere possibile, applicando una
forza di compressione eccessiva, ottenere una compressa resistente. Come si prepara una compressa
per compressione diretta:
glicante
(2)
lubrificante
(3)
Principio Attivo
miscelazione
compressione
disgregante
(4)
diluenti
(1)
Si prende il principio attivo o i principi attivi e li si miscela, prima di tutto, con un diluente (1), che
sono gli eccipienti che rendono la massa lavorabile, e poi si aggiunge un glicante (2) e un
lubrificante (3), fanno parte della classe che generalmente si chiama dei lubrificanti ma la differenza
è che i glicanti influenzano la scorrevolezza vera e propria della polvere cioè agiscono sulle forze di
attrito e di frizione delle particelle della polvere e quindi modificano l'angolo di riposo. Un glicante
che viene usato anche in galenica è la silice colloidale ( il cui nome commerciale è aerosil) ed è
praticamente una polvere impalpabile che aumenta la scorrevolezza delle polveri. I lubrificanti,
propriamente detti, hanno la funzione di modificale le interazioni della polvere con le parti
meccaniche della comprimitrice e quindi evitare i fenomeni di pitching e stiching, che sono
rispettivamente la possibilità che la compressa resti attaccata al punzone superiore( se la polvere
non è sufficientemente scorrevole può rimanere adesa al punzone superiore e quindi quando risale
la compressa non si stacca), oppure può rimanere attaccata al punzone inferiore(cioè quando il
punzone superiore, dopo la compressione, risale per espellere la compressa ma questa non si
stacca). Quindi i glicanti agiscono sulla polvere, i lubrificanti agiscono sul processo di

compressione. A questa miscela dovrà essere aggiunto un disaggregante (4) che è un eccipiente con
ruolo biofarmaceutico perchè modifica o comunque influenza la disaggregazione della compressa e
quindi la liberazione del principio attivo.
I vantaggi della compressione diretta sono:
tempi minori: se la miscela è comprimibile e scorrevole.
No acqua e nessun contatto con i solventi.
No calore.
Non essendoci riscaldamento, è un processo energicamente meno costoso.
Gli svantaggi sono:
le polveri difficilmente sono facilmente comprimibili
demiscelamento polveri
costo elevato eccipienti, perchè gli eccipienti che possono essere utilizzati nella compressione
diretta sono estremamente costosi(si utilizza infatti il lattosio spray dried).
Generalmente le aziende, per la produzione delle compresse per la compressione diretta, in farmacia
vendono delle miscele di eccipienti di cui raramente interessa la composizione reale.
La compressione dopo granulazione ad umido è più complicata della compressione diretta( mi
basta miscelare le polveri evitando la loro demiscelazione e poi comprimere).
diluente Miscelazione Principio Attivo
Legante Granulazione H2O
Essiccamento
Setacciatura
Glicante 2° miscelazione Lubrificante
Disgregante
Compressione
Inizialmente verrà miscelato il principio attivo con il diluente; dopo di che, questa polvere, verrà
bagnata con la soluzione di agente legante ( H2O o miscele idroalcoliche) e avverrà il processo vero
e proprio di granulazione e ci sarà poi la fase di essiccamento. Successiva alla fase di essiccamento
c'è la setacciatura, ovvero rendere omogenee le dimensioni del granulato. Sul granulato, essiccato e
setacciato, avverrà la seconda miscelazione e quindi saranno aggiunti il glicante, il lubrificante e
l'agente disaggregante, dopo la seconda setacciatura avverrà la compressione.
La disaggregazione e la disgregazione sono due processi diversi: la disaggregazione è la fase
iniziale della rottura della compressa mentre la disgregazione avviene dopo, ovvero dalla rottura

grossolana(disaggregazione) si ha la rottura in frammenti più piccoli(disgregazione). L'eccipiente è
sempre lo stesso(amido, cellulosa, derivati o zuccheri), che influenza in realtà il processo dalla
compressa al principio attivo solubile; è sempre chiamato disaggregante ma influenza tutti e due
questi processi.
Il processo di spray drying è quello che consente di ottenere delle polveri finemente suddivise, e
quindi con delle dimensioni ridotte e estremamente omogenee, oppure consente di ottenere da
principi attivi liquidi una formulazione solida. La miscela, o il principio attivo, viene disciolta, e
quindi si trova sotto forma di soluzione; sull'impianto c'è un atomizzatore che spruzza, dall'alto, la
miscela( e che viene quindi nebulizzata, ed entra nella camera(sotto forma di goccioline finissime)).
Queste goccioline vengono investite, generalmente dal basso, da una corrente di aria calda:quindi,
le micro goccioline vengono immediatamente essiccate dalla corrente di aria calda. L'aria calda, non
solo essicca, ma mantiene in movimento, e quindi rende il processo ancora più rapido. Una volta
che le particelle sono diventate solide, chiaramente pesano di più e quindi scendono verso il basso e
vengono portate nel ciclone, dove sono setacciate in maniera raffinata, per forza centrifuga: in base
alle dimensioni verranno separate all'interno del ciclone. Alla fine della setacciatura verrà raccolto il
prodotto.
E' un processo molto costoso e visto le temperature elevate che si raggiungono nella camere(180°-
200°C) è ovvio che questo è un processo che non si può utilizzare per principi attivi termolabili.
Una volta appurato come si ottengono le compresse e quali sono i problemi produttivi delle
compresse, quali sono i saggi che la farmacopea chiede per le compresse; sono dei saggi che vanno
eseguiti obbligatoriamente dalle aziende prima di poter licenziare il tutto. Viene prodotto così un
lotto di compresse, che deve rispondere a tutti i saggi della farmacopea, e solo dopo che l'ufficio di
controllo di qualità ha dato il permesso, in azienda si “licenzia” il lotto, ovvero si mette in vendita
quel lotto.
Saggio dell’uniformità di massa, obbligatorio per tutte le forme farmaceutiche solide a dose unica.
Il saggio si esegue su 20 unità, nel caso delle compresse sono 20 compresse,che vengono pesate
singolarmente, su questi 20 pesi si calcola il peso medio delle compresse. Il saggio dice: non più di
due di tali masse individuali, ovvero dei singoli pesi, possono presentare uno scarto rispetto alla
media superiore allo scarto percentuale ammesso e nessuna unità può presentare uno scarto
maggiore del doppio di tale scarto percentuale ammesso.

Nella tabella è riportata la deviazione percentuale
della massa media. Cioè i pesi singoli delle 20 unità
devono essere confrontate al peso medio, più o meno
con lo scarto percentuale ammesso. Lo scarto
percentuale ammesso o deviazione percentuale,
come si vede dalla tabella, varia a seconda della
forma farmaceutica(dato che è obbligatorio per le
forme farmaceutiche solide) e quindi si hanno dei
valori di deviazione percentuale(che si indica con K)
diversi. All'interno della stessa forma farmaceutica K
varia al variare del peso medio. Come varia: minore
è il peso maggiore è la deviazione percentuale
ammessa.
Es: peso medio 80 mg; il valore di K=10; ovvero
intorno a questo peso medio la deviazione
percentuale ammessa è +/- 10%. quindi si avrà un
intervallo di 80+/- 8; la farmacopea dice che non più
di 2 unità possono essere fuori da questo intervallo,
cioè si possono discostare di 1 valore di deviazione percentuale ammessa dal peso medio, e nessuna
delle 20 unità si deve discostare dal peso medio di 2 deviazioni percentuali cioè dall'intervallo 80+/-
16. Il secondo intervallo è più ampio e quindi 18 compresse devono rientrare nell'intervallo 72-88, e
2 possono uscire ma non oltre i 16; devono essere rispettati entrambi questi criteri e quindi anche
se tutte e 20 rientrano in 80+/-16 ma 17 rientrano nel primo limite comunque il lotto non può essere
validato. È ovvio che se tutte e 20 le compresse rientrano nel primo intervallo, rientreranno anche
nel secondo.
K diminuisce all'aumento del peso medio. Per esempio se il peso medio è 250 mg, K = 5 → 250+/-
12,5, questo perchè, in questo modo, l'intervallo intorno al peso medio è più o meno lo stesso.
Il saggio di uniformità di contenuto è ' il saggio in cui viene dosato il principio attivo(il saggio di
uniformità di massa è quello che dà per l'appunto l'uniformità di massa). Il saggio di uniformità di
contenuto si esegue solo quando il principio attivo è inferiore al 2% in peso dell'intera forma
farmaceutica o a 2 mg. In tutti gli altri casi, cioè quando il principio attivo è in quantità superiore al
2 % o a 2 mg, l'uniformità di contenuto è ottemperata dall'uniformità di massa;quindi se si sono
fatte delle compresse omogenee in peso certamente queste saranno omogenee nel contenuto. Il
criterio di fabbricazione sarà sempre un range ammesso percentuale intorno al contenuto dichiarato
del principio attivo. Ci sono saggi diversi(saggio A,B,C) in cui ciò che cambia è la tecnica analitica
per i principi attivi. È importante vedere che il range intorno al contenuto medio ammesso deve
essere compreso tra i 85 e il 115%; quindi se il contenuto nominale, in mg, è X, l'uniformità di
contenuto si considera ottemperata se quello che si trova analiticamente si trova nell'intervallo 85%
< X < 115%. Questa è la base di buona parte del dibattito sugli equivalenti, perchè il range è
abbastanza ampio del dichiarato e su questo si basa quello che i produttori di generici cercano come
l'ammissibilità nelle prove di rilascio, ovvero fanno un po' di confusione. In questo saggio si dice
che la quantità nominale deve essere +/- 15%; allora i produttori, sulle prove di dissoluzione, dicono
che c'è l'oscillazione del +/- 15% perchè lo dice la farmacopea; ma la farmacopea non parla del

rilascio del principio attivo ma ciò che c'è realmente.
Il Saggio della friabilità è un saggio obbligatorio per le compresse non rivestite. Esiste un
apparecchio, il friabilometro, costituito da un tamburo di plexiglas che viene posto verticalmente
su una macchina. All'interno di questo tamburo c'è un braccio, e questo tamburo viene fatto ruotare
dalla macchina. Nella rotazione del tamburo, le compresse sono sollevate e poi ricadono. Con
questo saggio si misura la resistenza meccanica superficiale delle compresse. Il saggio,
generalmente si esegua una volta sola.
Il numero di compresse da utilizzare nel
saggio, anche in questo caso, dipende dal
peso delle compresse. Per compresse di
massa unitaria fino a 0,75 g è di 20
compresse, per compresse maggiori di
0,75 g sono 10. Si pongono le compresse
all'interno del friabilometro, prima di porle
all'interno si pesano, si esegue il saggio, si
spolverano le compresse alla fine del
saggio e si ripesano. Quindi si valuta se c'è
una perdita di peso e quindi una scarsa
resistenza superficiale delle compresse. È
ovvio che questo saggio non si esegue per
le compresse rivestite perchè hanno un
rivestimento e dovrebbero essere
resistenti.
Il saggio della rottura delle compresse è quello proprio in cui si misura la resistenza meccanica
alla rottura della compressa. Prima in farmacopea erano descritti degli apparecchi, ma nelle XII non
c'è un apparecchio specifico per determinare la rottura. La farmacopea dice solo che si misura come
forza necessaria a rompere le compresse. Uno degli apparecchi che può essere usato( i risultati
devono essere espressi in N) è l'apparecchio di monsanto
si pone la compressa lungo la sezione longitudinale e girando la vite si calcola la forza necessaria
alla frantumazione. L'apparecchio di Monsanto è calibrato in Kg.
Saggio di disaggregazione delle compresse e delle capsule: Un cestello, di plexiglas o di vetro,
che contiene 6 cilindri di vetro la cui base, di questo cilindro, è una rete di acciaio, è agganciato ad
un motore che fa muovere il cestello dal basso verso l'alto. Il cestello viene immerso in una
soluzione di H2O e serve a misurare il tempo necessario a che le forme farmaceutiche solide siano
disaggregate

la compressa si considera disaggregata quando sulla rete metallica sul fondo non c'è nessun
frammento oppure ci sono dei frammenti ma che sono completamente bagnati dall'acqua; la
farmacopea dice che non deve resistere nessun nucleo non bagnato. Il saggio si esegue su 6
compresse: la farmacopea dice che la compressa si considera disaggregata quando non rimane
nessun residuo oppure c'è un residuo costituito da una massa molle. Ci sono dei saggi diversi per le
compresse e le capsule, per quelle grandi, e per le supposte e per gli ovuli; questi saggi sono
obbligatori per tutte le forme farmaceutiche solide unidose.
Fattori che influenzano la velocità di disaggregazione di una forma farmaceutica. L'influenza
delle caratteristiche della compressa e dell'ambiente sulla velocità di disaggregazione si misurano
con profondità di ingresso del solvente all'interno della compressa. La velocità di disaggregazione
sarà:
dove L è la velocità di disaggregazione, r è il raggio dei pori della compressa, γ cos θ è la
bagnabilità ( la tensione superficiale per il coseno dell'angolo di contatto, ed è la capacità di un
solido di essere bagnato da un liquido ed è quindi una caratteristica del soluto). Η è la viscosità
del liquido(con cui la compressa entra in contatto) e t è il tempo. Questa relazione vale per le
compresse non rivestite. Un parametro che può avere un effetto sul raggio dei pori è la grandezza
delle particelle ma soprattutto la forza di compressione, ed è per questo che bisogna valutarla con
attenzione perchè bisogna arrivare alla deformazione plastica e non alla rottura, ma bisogna anche
fare in modo che la compressa sia disaggregabile. Infatti, il meccanismo di azione principale dei
disaggreganti è quello di creare dei canali all'interno della compressa;cioè generalmente sono
delle sostanze immediatamente solubili in H2O che a contatto con il fluido si dissolvono
rapidamente e aumentano r sia come dimensione che come numero. Un altro meccanismo di azione

dei disaggreganti, di quelli che vengono chiamati super disaggreganti, è la caratteristica che
hanno alcuni polimeri di assorbire l'H2O rapidamente e di rigonfiarsi. I fenomeno si chiama
swelling, e in questo caso le compresse meccanicamente esplodono( explotab, o sodio
caramellosio).
Il saggio della velocità di dissoluzione è un saggio non obbligatorio, ma per le forme
farmaceutiche non convenzionali sì perchè serve per dimostrare che abbiamo un effetto in quanto
forma farmaceutica nella liberazione del principio attivo. Il macchinario che si utilizza è costituito
da 1 contenitore, nel quale viene posto il liquido che viene utilizzato come solvente, e da un
agitatore meccanico, che può essere a cestello rotante, ovvero un cestello di acciaio inossidabile che
è legato direttamente al motore che imprime la rotazione, o a paletta(che invece agita il solvente). In
questo caso la forma farmaceutica è posta nel cestello e ruota in maniera solidale con il motore; nel
caso dell'agitatore a paletta la forma farmaceutica è posta nel contenitore dove c'è il liquido ed il
movimento è imposto al liquido. Le palette sono generalmente di teflon o di acciaio inossidabile. Il
saggio di dissoluzione si esegue a 37° C. Nel caso della determinazione della velocità di
dissoluzione con questi dissolutori, la forma farmaceutica viene messa nel fluido(generalmente 1 L)
si impone una certa velocità di rotazione, a intermedi di tempo predefiniti si fanno dei prelievi,si
misura la concentrazione di principio attivo e, indipendentemente dal volume che si preleva, si
aggiunge solvente nuovo. Quello che si calcola è un incremento della quantità di principio attivo
che si sta analizzando nel tempo,che si chiama quantità cumulativa, che dovrà tenere conto delle
singole diluizioni, perchè ad ogni prelievo si aggiunge solvente nuovo. Si aggiunge solvente nuovo
per fare in modo che comunque il volume sia costante, perchè sennò si avrebbe una concentrazione
crescente che può essere legata al fatto che il volume sia diminuito.
Il saggio si fa su 6 compresse ma valutate singolarmente, in cui poi il profilo di rilascio si fa
facendo la media. Nel caso del cestello rotante, se si dice che è stata applicata una velocità di
rotazione che si misura in giramento o RPM, si è sicuri che la forma farmaceutica ruota a quella
velocità. Nel caso del dissolutore a paletta il movimento che ha la la forma farmaceutica non è
controllato ,perchè la paletta imprime il movimento al solvente e il movimento del solvente trascina
la forma farmaceutica che potrebbe trovarsi nella parte bassa( e quindi è agitata più velocemente)
oppure potrebbe spostarsi nella parte più alta e quindi imprime una velocità di rotazione minore. Se
però la forma farmaceutica ha una composizione particolare, il cestello si può otturare(come per es.
polimeri che rigonfiano) e quindi il principio attivo è intrappolato nel cestello. Lo stesso problema
si può avere utilizzando delle compresse di natura polimerica, con il dissolutore a paletta, e bisogna
essere certi che la forma farmaceutica non aderisca al recipiente, per cui in questo casi si imprime la
velocità la velocità di rotazione ma la forma farmaceutica non si muove. Mettere appunto il saggio
di dissoluzione funzionale non è facile, perchè bisogna scegliere il dissolutore, il solvente(che
solitamente sono tamponi a pH 1-2, ed alcuni utilizzano il fluido gastrico intestinale simulato), la
velocità di rotazione, il modello di dissolutore, i tempi di prelievo e i volumi dei prelievi. Per
simulare le condizioni in vivo è stata introdotta, nella XII edizione della farmacopea, una nuova
apparecchiatura: il dissolutore a flusso continuo. Perchè in questi dissolutori, non cambiando se
non di pochi ml il volume nel tempo, non si simulano le condizioni in vivo; la compressa nello
stomaco viene in contatto in continuo con liquido gastrico diverso. Quindi è previsto un
apparecchio a flusso continuo in cui c'è una camera di dissoluzione in cui viene messa una
compressa(o capsula) che è termostatata, viene inviato in questa in continuo, solvente riscaldato a
37°C e si raccolgono poi dei campioni che vengono analizzati. Questi saggi non sono obbligatori

per tutte le forme farmaceutiche solida ma sono consigliati per le forme farmaceutiche a rilascio
continuo.

Compresse
Le compresse sono preparazioni solide contenenti ciascuna una dose unica di uno o più principi
attivi e ottenute usualmente per compressione di volumi uniformi di particelle. Si possono
distinguere varie categorie di compresse per uso orale:
compresse non rivestite(con dei saggi obbligatori);
compresse rivestite
compresse effervescenti;
compresse disperdibili ed orodisperdibili;
compresse a rilascio modificato;
compresse gastroresistenti;
compresse da utilizzare nella cavità buccale.
Queste compresse devono avere una sufficiente resistenza meccanica, che tiene conto di due fattori:
la resistenza meccanica superficiale(i) che si misura con il saggio della friabilità, e la resistenza alla
rottura. Se le compresse sono realizzate e dispensate come compresse divisibili( quelle con il
“taglio” al centro), la farmacopea dice che, le aziende devono dimostrare che in ciascuna delle unità
in cui è divisibile la compressa ci sia uniformità di contenuto. I saggi obbligatori sono l'uniformità
di massa, l'uniformità di contenuto se il principio attivo è inferiore a 2 mg o al 2% della massa
totale, altrimenti l'uniformità di massa garantisce anche l'uniformità di contenuto.
Le compresse non rivestite possono essere:
1. a strato singolo
2. a multistrato
quindi si avrà una compressa in cui una miscela di polveri, nella maggior parte dei casi di
granulato,oppure nelle compresse multistrato si ottengono per compressione successiva e possono
gli strati essere paralleli o concentrici. Le compresse multistrato, indipendentemente dal disegno,
sono ottenute per compressioni successive di miscele diverse.
Le compresse multistrato vengono fatte o per avere il rilascio modificato o si può avere, nelle
compresse a multistrato quelle concentriche,concentrazioni diverse dello stesso principio attivo. Per
esempio la parte iniziale libera più rapidamente,etc, ma questo è soprattutto per le compresse a
rilascio modificato ovvero per forme farmaceutiche non convenzionali in cui la forma farmaceutica
controlla l'assorbimento. In genere viene fatta quando c'è una incompatibilità chimico-fisica dei
principi attivi; se la formulazione, quindi, contiene più principi attivi che presentano tra loro
incompatibilità chimico-fisica, vengono granulate separatamente e poi vengono compresse le
miscele diverse.
Il saggio, oltre quelli obbligatori, obbligatorio per queste compresse è il saggio della

disaggregazione: si fa in H2O R( secondo farmacopea ovvero inclusa, con delle caratteristiche, nei
reattivi della farmacopea). Anche per le compresse rivestite è obbligatorio questo saggio, ma ciò che
cambia è il tempo, ovvero una compressa non rivestita è conforme alle caratteristiche di farmacopea
se disaggrega in H2O in 15 minuti, viceversa, le compresse rivestite, se hanno un rivestimento di
tipo zuccherino si chiamano confetti, come quelli per uso alimentare in quanto la tecnica è stata
importata dall'industria dolciaria, devono disaggregare in 60 minuti, mentre le compresse rivestite
con film devono disaggregare in 30 minuti.
Le compresse rivestite sono compresse ricoperte con uno o più strati di miscele di varie sostanze;
la rivestitura si fa senza compliance del paziente, perchè generalmente servono per coprire l'odore
sgradevole e il colore sgradevole. La confettura è un processo complesso, perchè per esempio
richiede che la soluzione sia di una composizione giusta e bisogna ottenere una confettatura che non
influenzi troppo la disaggregazione della compressa; fino a 5-6 anni fa la confettatura veniva fatta a
mano ovvero c'erano degli artigiani, importati dall'industria dolciaria, e operavano bagnando le
compresse, in cui stavano in un grande contenitore di rame e venivano fatte girare, con la soluzione
zuccherina fino alla consistenza voluta. La confettura in genere viene fatta per aiutare la
deglutabilità; infatti in genere i confetti sono per quelle compresse per uso pediatrico o geriatrico, o
per qualche patologia in cui il paziente ha difficoltà a deglutire(perchè sempre perfettamente liscio).
Il saggio di disaggregazione non si attua sulle compresse effervescenti.
Il tipo di rivestimento divide le compresse in:
compresse con rivestimento zuccherino(confetti) costituito un gran parte da saccarosio(che
può costituire fino la metà del peso totale)
compresse con rivestimento filmogeno, il film molto sottile è costituito da polimeri, questo
rivestimento può essere anche applicato a secco(non come il confetto,in cui la compressa e
bagnata della sostanza zuccherina), anche se ci sono delle compresse che vengono bagnate
con la soluzione polimerica.
Compresse a doppio strato o con rivestimento a secco.
Le compresse effervescenti sono compresse non rivestite contenenti generalmente sostanze acide e
carbonati o bicarbonati che reagiscono rapidamente in presenza di H2O sviluppando anidride
carbonica. Sono destinate ad essere disciolte o disperse in acqua prima della somministrazione.
Differiscono dalla compresse solubili in quanto devono essere sciolte in acqua prima della
somministrazione, mentre le compresse effervescenti hanno l'aggiunta che una volta sciolte in acqua
devono sviluppare rapidamente CO2. Il saggio di disaggragazione non si esegue sulle compresse
effervescenti ma si pone la compressa in un recipiente con acqua a una temperatura diversa da
quella corporea(37°C) e deve sviluppare numerose bolle di gas in 6 minuti, quindi il saggio di
disaggregazione è obbligatorio ma è diverso da quello delle altre compresse. Dalla reazione
dell'acido debole con il bicarbonato si sviluppa l'anidride carbonica.
3NaHCO3 + H3C6H2O7 → 3H2O + 3CO2 + Na3C6H2O7
252 g 192 g 54 g 132 g 258 g

da notare la quantità in grammi degli eccipienti utilizzati: le compresse effervescenti sono molto più
grandi di quelle normali, perchè per sviluppare 3 moli di CO2 si ha bisogno di quasi 3 etti di
eccipienti.
Le compresse disperdibili sono compresse non rivestite o rivestite con film destinate ad essere
disperse in acqua prima della somministrazione. La differenza con le compresse solubili è che la
compressa solubile dà luogo ad una soluzione, non necessariamente trasparente, mentre la
compressa disperdibile dà luogo ad una dispersione omogenea. I saggi che si fanno sulle compresse
disperdibili sono: 1) saggio della disaggregazione e 2) finezza della dispersione; quindi non solo
viene valutato il tempo in cui(a temperatura ambiente) disaggrega ma viene anche misurata la
dimensione della dispersione, che deve avere una dispersione tale da passare attraverso un setaccio
di 710 μm.
Le compresse oro disperdibili sono le compresse non rivestite destinate ad essere poste nella
bocca dove si disperdono rapidamente prima di essere inghiottite; sono quelle definite fast. Una
caratteristica formulativa particolare è che sono formulate con il mannitolo , perchè questo quando
si scioglie assorbe calore e quindi lascia un senso di freschezza perchè la dissoluzione è un processo
che libera calore.
Le compresse gastroresistenti sono le compresse a rilascio ritardato preparate per resistere al
fluido gastrico e rilasciare il o i loro principi attivi nel fluido intestinale. Sono preparate rivestendo
le compresse con una sostanza gastroresistente. Queste compresse devono resistere 2 ore in HCl 0,1
M e poi devono disaggregare in 1 ora in un tampone a pH= 6,8.
Sempre nell'ambito delle forme farmaceutiche solide per uso orale ci sono le gomme da masticare
medicate: sono preparazioni solide a dose unica con una base costituita essenzialmente da gomma,
destinate ad essere masticate ma non inghiottite. Sono per esempio il travelgum. Dopo dissoluzione
o dispersione dei principi attivi nella saliva, le gomme da masticare sono destinate:
al trattamento locale di affezioni della cavità buccale
all'azione sistemica dopo assorbimento attraverso la mucosa buccale o attraverso il tratto
gastro intestinale.
La compressa:
La compressa viene quindi prima (1) deaggregata, poi (2) disaggregata in particelle fini, poi avviene
il (3) processo di dissoluzione ed infine (5) l'assorbimento. Il principio attivo viene assorbito solo
dopo essere stato solubilizzato. È stato già visto quali sono i fattori che influenzano la
disaggregazione. I fattori che influenzano la dissoluzione di un solido: esistono molti modelli
matematici per descrivere il processo di dissoluzione di un solido. Si considera una sola teoria che è
quella che descrive la quasi totalità dei processi di dissoluzione.

La teoria del film lega la velocità della dissoluzione ai processi di diffusione, e quindi lega la
velocità di dissoluzione alla legge di Fick, che regola i processi diffusivi. Se si considera una
particella solida, questa entra in contatto con il solvente; la teoria del film dice che intorno alla
compressa di forma uno strato, detto strato idrodinamico diffusionale, in cui la concentrazione del
solido è pari alla sua solubilità.
Il solido incomincia a dissolversi nel solvente
e la porzione iniziale del solido disciolto
rimane a formare un film; quindi in questo
film che si crea intorno alla particella di
solido si ha una concentrazione di solido pari
a cs. Si avrà un volume di solvente in cui c'è
poco solido, in cui si avrà una concentrazione(ct), ovvero che dipende dal
tempo al quale si misura la concentrazione. Questa teoria richiama la teoria di
Fick perché in questo modello il processo di dissoluzione è considerato
costituito da due stadi: (1) passaggio solido nel film e (2) diffusione del solido
disciolto verso solvente puro.
In questo modello, il primo stadio è considerato lo stadio veloce mentre lo stadio lento è la
diffusione attraverso lo strato idrodinamico diffusionale e quindi sarà quello che influenza la
velocità di dissoluzione. La velocità di dissoluzione, che si misura come variazione della quantità
del solido che si trova in soluzione in funzione del tempo
dove A è la superficie specifica del solido(e quindi al grado di finezza dopo la disaggregazione), D è
il coefficiente di diffusione(attraverso le strato idrodinamico diffusionale), (cs - ct) è il gradiente di
concentrazione(tra lo strato idrodinamico diffusionale e la massa del solvente), h è lo spessore dello
strato idrodinamico diffusionale e V è il volume. Il gradiente di concentrazione tiene conto del
percorso che la molecola fa:
la differenza tra cs e ct al punto (1) è diverso dal punto (2) in quanto
dipende del percorso.
Per aumentare la velocità di dissoluzione di una forma farmaceutica in vitro si aumenta la superficie
specifica(evitando che la polvere eccessivamente fissa, non è facilmente lavorabile fino quindi al
limite della lavorabilità), h(aumentando la velocità di agitazione, in quanto gli agitatori sono
sottoposti ad agitazione si diminuisce h, perchè agitando lo strato idrodinamico diffusionale intorno
al solido si assottiglia); una delle tecniche, infatti, che si utilizza per discriminare il meccanismo di
dissoluzione è fare dei test al dissolutore variando la velocità di rotazione. Se la velocità di
dissoluzione aumenta all'aumentare della velocità di rotazione è plausibile che il meccanismo
coinvolto sia un meccanismo di questo tipo cioè legato a dei fenomeni diffusivi; se aumentando la

velocità di rotazione la velocità di dissoluzione rimane uguale si deve utilizzare un altro modello
matematico per valutare il processo di dissoluzione.
In vivo la situazione è molto più semplice, perchè ci sono delle situazioni sink cioè il solido viene in
contatto in continuo con il fluido gastrointestinale fresco quindi cs è sempre molto maggiore con ct.
quindi la variazione di concentrazione sarà legata al tempo attraverso una costante nel quale
rientrano dei parametri che sono costitutivi della forma farmaceutica. Questi grafici rappresentano
come gli eccipienti e i processi formulativi possono influenzare in maniere determinante la velocità
di dissoluzione e quindi la sua biodisponibilità.
In questo grafico è riportato il profilo di dissoluzione(riportato
come % disciolto in funzione del tempo) del fenobarbital,
formulato in polvere(1), in granuli(2) e in compresse(3).
Inspiegabilmente si vede che il fenobarbital in compresse si
scioglie più velocemente della polvere, quindi la polvere è già
disaggregata e quindi ha un processo in meno così come i
granuli, ma ciò nonostante, facendo il profilo di dissoluzione si
nota che le compresse si sciolgono più velocemente. Ciò
perchè nella compressione sono stati aggiunti degli eccipienti
che hanno favorito, ad esempio, la bagnabilità della polvere; prima di essere disciolto, il principio
attivo deve essere bagnato dal solvente e quindi se si aumenta la capacità della polvere di essere
bagnata si aumenta anche la velocità di dissoluzione. In genere, questa potrebbe essere
un'indicazione che comunque la granulazione è stata eseguita correttamente, perchè la presenza di
agente legante non ha ridotto, rispetto alla polvere (perchè la parte finale delle curve si
sovrappongono), la velocità di dissoluzione e che quindi la sua scelta e la sua concentrazione è
ottimale.
questo(1) invece è un grafico che fa vedere l'effetto del colorante; i coloranti hanno un effetto
sostanziale, soprattutto quelli insolubili in acqua come gli ossidi di ferro, nella biodisponibilità del
principio attivo. In questo caso viene studiata la velocità di dissoluzione della lipoflavina in
compresse rivestite con idrossipropil metil cellulosa. In questi due casi la velocità di dissoluzione è
stata eseguita sui succhi gastrici simulati: questo(*) è il profilo di dissoluzione in succo gastrico
simulato, mentre l'altro prifilo di rilascio è stato eseguito in fluido intestinale simulato. Nel fluido

gastrico simulato e nel fluido intestinale simulato non cambia solo il pH ma sono aggiunte delle
proteine e avvolte, nel fluido intestinale simulato, anche i sali biliari. In questo caso, la presenza di
colorante, nel succo gastrico simulato, rallenta in maniera sostanziale il profilo di dissoluzione
mentre i due profili si avvicinano e sono perfettamente sovrapponibili nel fluido intestinale
simulato. Questo è proprio legato al fatto che nel fluido intestinale simulato sono presenti i sali
biliari, che sono delle sostanze che formano micelle , e quindi in questo caso la presenza del
colorante che rallenta in maniera sostanziale il profilo di dissoluzione della vitamina(liposolubile) è
contrastata dal fatto che la presenza di sali biliari favorisce il processo di dissoluzione attraverso la
solubilizzazione micellare del colorante. Quindi è ovvio che in un saggio normale di
dissoluzione,eseguito in H2O o in un tampone non sarebbe possibile discriminare l'effetto del
colorante sul profilo della velocità di dissoluzione.
La solubilità, fino ad ora è sempre stata espressa come una concentrazione e quindi con dei valori
numerici(g/l, n/l o % p/v); in realtà, la farmacopea, esprime la solubilità delle sostanze utilizzando
una modalità completamente diversa. Infatti, divide le sostanze con delle indicazioni di solubilità
che vanno da solubilissimo a praticamente insolubile, dando la quantità in ml di H2O che solubilizza
1g di sostanza. Per esempio, solubilissimo meno di 1 vuol dire che 1g della sostanza si solubilizza
in 1ml di H2O(tra i 15-25°C ovvero a temperatura ambiente). Praticamente insolubile vuol dire che
1g di sostanza si scioglie in 10 l di H2O; quindi rispetto all'indicazione numerica in cui si esprime la
solubilità, la modalità della farmacopea di valutare la solubilità è completamente diversa. Wagner,
nel 1970, ideò uno schema per valutare gli stadi che portavano dalla forma farmaceutica solida al
farmaco in soluzione, che è la condizione necessaria perchè avvenga l'assorbimento del farmaco.
Forma farmaceutica solida → disintegrazione → granuli + aggregati → deaggregazione → polveri
fini
dalla forma farmaceutica solida inizia il processo di disintegrazione che porta a granuli e aggregati,
la deaggregazione, poi, porta alle particelle fini. Il processo di dissoluzione maggiore(ovvero in %
maggiore) avviene a carico ovviamente delle particelle fini anche se inizia un moderato processo di
dissoluzione anche nella forma farmaceutica solida. Qualche anno dopo, nel 1982, Cartensen
elabora uno schema più complesso e aggiunge, agli stadi che erano stati ipotizzati da Wagner, degli
stadi che precedono il processo di disaggregazione della forma farmaceutica solida. Inserisce un
tempo di lagging , necessario a che la forma farmaceutica sia bagnata dal solvente(in vivo dal
fluido biologico) per poi iniziare il processo di
disintegrazione.
Se si pone a grafico l'andamento del processo
considerato da Cartensen, si ottiene il tipico
diagramma ad S, che caratterizza il profilo di
dissoluzione delle forme farmacutiche solide.
La forma del grafico è ad S perchè: nella fase
iniziale (1) si avrà un tempo di lagging iniziale
in cui non c'è interazione tra la forma
farmaceutica e il fluido biologico; quindi la
forma farmaceutica dovrà essere bagnata dal

fluido biologico, il quale dovrà entrare all'interno della forma farmaceutica e solo a questo punto
inizierà il processo di disintegrazione(2) e il processo di disaggregazione(3). Nella fase centrale
della curva(4) si ha la fase di dissoluzione e andando avanti nel tempo non si arriva alla
dissoluzione totale perchè si genera uno stadio, secondo Cartensen, di occlusione. Si arriva così ad
uno stadio in cui il solido non solubilizza più e quindi la quantità disciolta rimane costante(5). Da
notare che la parte centrale del grafico è una retta; questo per i parametri che influenzano la velocità
di dissoluzione
In questi tre diagrammi A,B,C, si nota che tutti hanno la fase di
occlusione. L'andamento ad S è tipico del processo di
disintegrazione- dissoluzione: nel grafico A c'è solo il processo di
dissoluzione e quindi la compressa è a disaggregazione
immediata. Nelle curve B e C, il profilo di dissoluzione e quindi il
passaggio in soluzione del farmaco è influenzato dalla parte
iniziale. In queste forme farmaceutiche, quello che cambia è la
fase di disaggregazione; quindi la B rappresenta una forma
farmaceutiche in cui la disaggregazione è rapida mentre la C è
una forma farmaceutica poco bagnabile ( perchè il tempo di
lagging è lungo) e che ha una velocità di disaggregazione lenta.
Costruire questi grafici,utilizzando lo schema di Cartensen consente di evidenziare quale è lo stadio
limitante del processo di dissoluzione e quindi, indipendentemente da quello che si vuole ottenere
dalla forma farmaceutica, si modifica la formulazione. Da notare, che non è detto che una velocità
di disaggregazione elevata corrisponde a una velocità di dissoluzione elevata. Esistono una serie
infinità di modelli matematici che mettono in relazione, per le diverse formulazioni, la velocità di
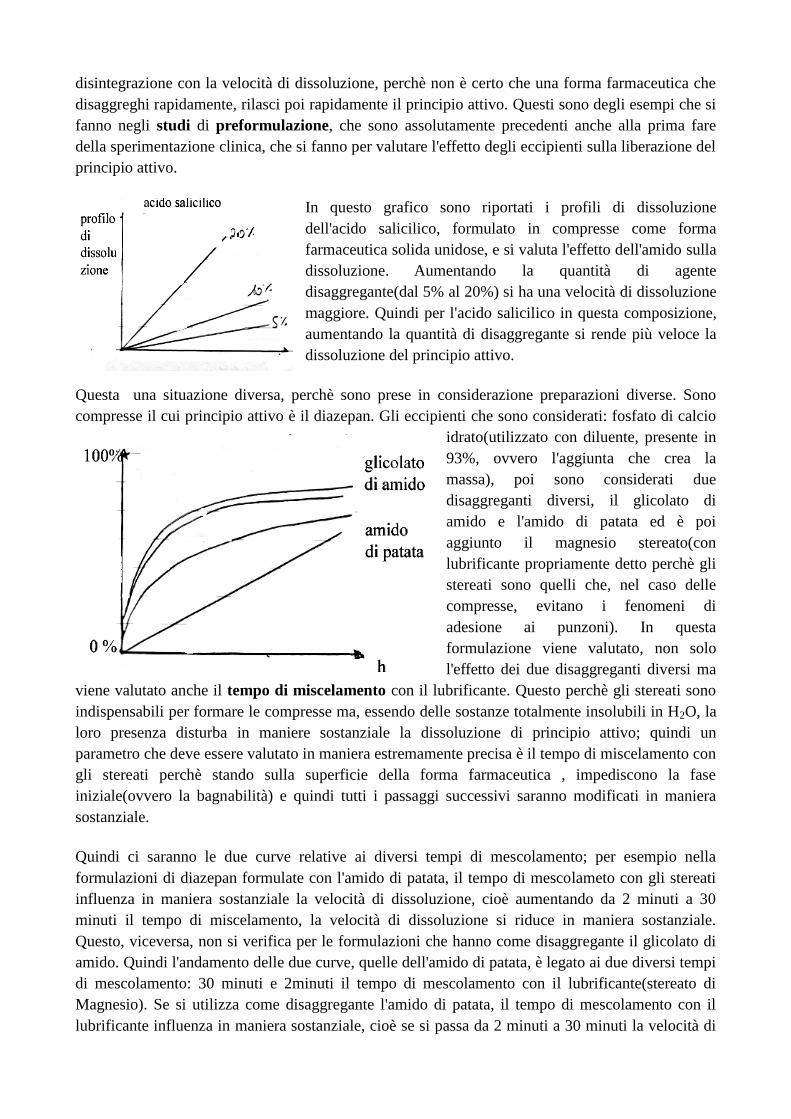
disintegrazione con la velocità di dissoluzione, perchè non è certo che una forma farmaceutica che
disaggreghi rapidamente, rilasci poi rapidamente il principio attivo. Questi sono degli esempi che si
fanno negli studi di preformulazione, che sono assolutamente precedenti anche alla prima fare
della sperimentazione clinica, che si fanno per valutare l'effetto degli eccipienti sulla liberazione del
principio attivo.
In questo grafico sono riportati i profili di dissoluzione
dell'acido salicilico, formulato in compresse come forma
farmaceutica solida unidose, e si valuta l'effetto dell'amido sulla
dissoluzione. Aumentando la quantità di agente
disaggregante(dal 5% al 20%) si ha una velocità di dissoluzione
maggiore. Quindi per l'acido salicilico in questa composizione,
aumentando la quantità di disaggregante si rende più veloce la
dissoluzione del principio attivo.
Questa una situazione diversa, perchè sono prese in considerazione preparazioni diverse. Sono
compresse il cui principio attivo è il diazepan. Gli eccipienti che sono considerati: fosfato di calcio
idrato(utilizzato con diluente, presente in
93%, ovvero l'aggiunta che crea la
massa), poi sono considerati due
disaggreganti diversi, il glicolato di
amido e l'amido di patata ed è poi
aggiunto il magnesio stereato(con
lubrificante propriamente detto perchè gli
stereati sono quelli che, nel caso delle
compresse, evitano i fenomeni di
adesione ai punzoni). In questa
formulazione viene valutato, non solo
l'effetto dei due disaggreganti diversi ma
viene valutato anche il tempo di miscelamento con il lubrificante. Questo perchè gli stereati sono
indispensabili per formare le compresse ma, essendo delle sostanze totalmente insolubili in H2O, la
loro presenza disturba in maniere sostanziale la dissoluzione di principio attivo; quindi un
parametro che deve essere valutato in maniera estremamente precisa è il tempo di miscelamento con
gli stereati perchè stando sulla superficie della forma farmaceutica , impediscono la fase
iniziale(ovvero la bagnabilità) e quindi tutti i passaggi successivi saranno modificati in maniera
sostanziale.
Quindi ci saranno le due curve relative ai diversi tempi di mescolamento; per esempio nella
formulazioni di diazepan formulate con l'amido di patata, il tempo di mescolameto con gli stereati
influenza in maniera sostanziale la velocità di dissoluzione, cioè aumentando da 2 minuti a 30
minuti il tempo di miscelamento, la velocità di dissoluzione si riduce in maniera sostanziale.
Questo, viceversa, non si verifica per le formulazioni che hanno come disaggregante il glicolato di
amido. Quindi l'andamento delle due curve, quelle dell'amido di patata, è legato ai due diversi tempi
di mescolamento: 30 minuti e 2minuti il tempo di mescolamento con il lubrificante(stereato di
Magnesio). Se si utilizza come disaggregante l'amido di patata, il tempo di mescolamento con il
lubrificante influenza in maniera sostanziale, cioè se si passa da 2 minuti a 30 minuti la velocità di

dissoluzione a ora si riduce dall'80% a meno del 40%, ovvero più della metà di principio attivo
rimane non disciolto.
Le due curve superiori sono quelle relative alla formulazione che usano un derivato dell'amido, il
glicolato di amido, ma in questo caso il tempo di mescolamento non influenza per niente la velocità
di dissoluzione; dopo 1 ora ,questa formulazione, arriva all'80% di dissoluzione di principio attivo
sia che il tempo di mescolamento sia di 2 minuti sia che sia di 30 minuti. Se il tempo di
mescolamento con lo stereato non influenza la velocità di dissoluzione, vuol dire che il glicolato di
amido è un ottimo disaggregante e che quindi consente di eliminare l'effetto negativo degli stereati.
Quindi se si ha bisogno, per motivi formulativi, di usare un tempo lungo di mescolamento con gli
stereati non si sceglierà un moderato disaggregante(come l'amido di patata) ma si utilizzerà un
disaggregante migliore(come il glicolato d'amido).

Capsule
Le capsule sono preparazioni solide con involucri duri o molli di varie forme e capacità, contenenti
usualmente una dose di principio attivo. Sono destinate alla somministrazione orale. È la forma
farmaceutica di cui esiste la traccia più antica: si parla di capsule in alcuni documenti egiziani(del
1800 a.C); le capsule come si conoscono sono nate da un brevetto pensato e realizzato da una
studente della facoltà di farmacia dell'università di Parigi agli inizi dell' '800.
Esistono sostanzialmente due tipi di capsule:
capsule rigide o opercolare o opercoli;
capsule molli
I costituente delle capsule, in generale, sono: la gelatina, è il costituente principale, è un polimero
di derivazione naturale ed esistono due tipi di gelatina, in farmacopea, la gelatina A e la gelatina B e
questa distinzione dipende dalla tecnica che si utilizza per fare la gelatina. La gelatina si ottiene da
resti di animali(cartilagine, gelatine e farina di osso), bovina ma nella maggior parte dei casi suina;
la gelatina A e la gelatina B si distinguono dal fatto che l'idrolisi di queste parti di animali viene
fatta o in ambiente acido o in ambiente basico. Le caratteristiche che vengono richieste da valutare
nella farmacopea(ovvero che permetta di dire che la gelatina che si sta utilizzando è gelatina FU)
sono le caratteristiche di viscosità e la blouse strenght (cioè la capacità di gelificare): queste due
caratteristiche fisiche danno un'indicazione nel tempo della stabilità di questo eccipiente; dato che
non ha una composizione chimica definita né omogenea e quindi la stabilità nel tempo della
gelatina si misura utilizzando questi parametri fisici.
Il plasticizzante, solo in alcune capsule(insieme alla gelatina e all'H2O); nella maggior parte dei
casi è glicerolo, ma può essere anche sorbitolo, una gomma (raramente) o il PEG ( polietilenglicol o
glicol propilenico). Per evitare che l'involucro della capsula, fatto solo di gelatina ed acqua, nel
tempo diventi secco e che si frantumi, si aggiungono queste sostanze, come il glicerolo e il
sorbitolo, chiamati plasticizzanti perchè mantengono una frazione di acqua all'inteno dell'involucro;
quindi fanno in modo che, nel tempo, l'involucro della capsula non si frantumi.
Come si sceglie il rapporto glicerina / gelatina da utilizzare per realizzare una capsula rigida: si
sceglie in base al tipo di utilizzo e dal tipo di impiego della capsula. Ad esempio, considerando che
il rapporto è espresso come parti di glicerolo anidro per una parte di gelatina secca, per le capsule
contenenti olii il rapporto è di 0,35(cioè per una parte di gelatina ci sono 0,35 parti di glicerolo). Per
le capsule che contengono liquidi idrofili il rapporto aumenta(cioè per ogni parte di gelatina secca
0,55-0,65 parti di glicerolo). Il rapporto 0,35 nelle capsule contenenti oli è basso perchè non ci sono
interazioni tra l'involucro e il contenuto e quindi la soluzione acquosa del contenuto non può
richiamare l'acqua dall'involucro. Altri costituenti sono
I coloranti, i conservanti che vanno aggiunti successivamente perchè l'involucro, essendo di
gelatina, è sempre una matrice che può essere attaccata dal punto di vista microbiologico.
Secondo la farmacopea: Gli involucri delle capsule sono fatti di gelatina o altre sostanze, la cui
consistenza può essere regolata per aggiunte di sostanze come glicerolo e sorbitolo.

La capsula è una forma farmaceutica estremamente versatile perchè consente di veicolare al suo
intendo diluizioni, granulati,piccole compresse, granulati e compresse, polveri. Di fatto è la forma
farmaceutica più facile da preparare perchè per riempire una capsula serve il principio attivo, il
diluente e qualcosa che aiuti la scorrevolezza della polvere intrinseca alla polvere. Non si
aggiungono disaggreganti e lubrificanti. In tutti gli studi di sperimentazione clinica, il principio
attivo è formulato in capsule perchè è la forma farmaceutica più facile da realizzare dal punto di
vista produttivo( in galenica si fanno le capsule piuttosto che le compresse). In farmacopea, le
capsule sono distinte:
capsule rigide
capsule molli
capsule a rilascio modificato
capsule gastroresistenti
cialdini (sono rientrati in farmacopea dalla X edizione perchè sono costituiti da amido e
quindi non hanno una composizione diversa dalla gelatina animale).
Essendo forme farmaceutiche solide unidose i saggi obbligatori saranno l'uniformità di contenuto e
l'uniformità di massa. Nella conservazione è specificato che deve essere conservato a temperature
non superiore a 30 °C perchè è la temperatura di transizione da solida a gel delle soluzioni di
gelatina e quindi a T > 30° C la capsula si scioglie( ed è proprio quello che viene sfruttato nel
meccanismo d'azione). Le differenze tra le capsule rigide e quelle molli sono:composizione , lo
spessore dell'involucro; la farmacopea infatti dice che l'involucro delle capsule molli(pur costituito
da gelatina, glicerolo e acqua) è generalmente più spesso.
Capsule rigide: sono composte da due parti: una parte nella quale viene inserita la polvere o il
granulato (ovvero quella che viene riempita) che si chiama corpo della capsula e poi la parte con
cui viene chiusa la capsula che si chiama testa, che ha un diametro leggermente superiore a quello
del corpo.
Quindi la caratteristica delle capsule opercolate è che le capsule vengono
preparate da aziende specializzate e poi vengono riempite o dal farmacista
o dall'azienda produttrice con la forma farmaceutica; quindi la preparazione
della capsula e il riempimento avvengono in 2 tempi distinti del processo
produttivo. Le capsule molli, invece, sono preparate e riempite
contemporaneamente con delle tecniche, che sono state brevettate dalle
aziende che preparano solo capsule molli. Quindi le aziende produttrici
inviano alle aziende che fanno le capsule le soluzioni del principio attivo e queste preparano le
capsule molli. La farmacopea dice che le capsule rigide hanno involucri costituiti da due sezioni
cilindriche preformate, un'estremità delle quali è arrotondata e chiusa, l'altra è aperta. Le capsule a
rilascio modificato e quelle gastroresistenti sono simili come caratteristiche.
I cialdini sono simili alle capsule, come concetto ma
non come forma, il cui involucro è costituito da pane
azzimo:infatti sono due parti che vengono riempite e
poi chiuse in cui le due parti che vengono messe insieme hanno questo aspetto

I due bordi, poi, vengono saldati insieme. Le dimensioni sono generalmente più grandi delle
capsule. I cialdini sono rientrati in farmacopea sostanzialmente per venire incontro a problemi legati
all'ambito religioso perchè per alcune perso e è un problema assumere derivati animali, in
particolare dei suini.
Esistono in commercio delle capsule preparate proprio per questi motivi, quindi non i cialdini fatti
con il pane azzimo,ma delle capsule vere e proprie fatte con delle sostanze diverse dalla gelatina:
come ad esempio dei derivati della cellulosa, in particolare sono preparati con
idrossipropilmetilcellulosa (HPMC) ; le capsule di gelatina e le capsule di
idrossipropilmetilcellulosa hanno delle caratteristiche di rilascio del principio attivo perfettamente
sovrapponibili e quindi la variazione del costituente della capsula non modifica il rilascio del
principio attivo e che quindi l'HPMC può sostituire l'eccipiente di derivazione animale nella
realizzazione delle capsule.
Le capsule rigide hanno varie dimensioni (non casuali), standardizzate, che sono contraddistinte da
un numero, che va da 000 a 5, e questi numeri sono inversamente proporzionali alla capacità della
capsula in ml. Anche per le capsule la capacità è espressa in ml e quindi la polvere, che viene
realizzata per riempire le capsule, deve essere scorrevole tanto quando un fluido.
bisogna preparare
0,3 g di acido acetilsalicilico
0,025 g di caffeina
eccipienti q.b.
Per 1 cps t. 30 (una capsula e tali 30)
Quali tipo di capsula si
utilizza? Si hanno, quindi, le
quantità degli eccipienti, si
miscelano con una quantità
di diluenti che sono stati
scelti, e si misura il valore
che si ottiene dai due
principi attivi e dagli
eccipienti. Si arriva così ad
un valore di 18 ml. Si
utilizzano i grafici di

riempimento. Bisogna quindi preparare 30 cps, da 18 ml: sul grafico non c'è una misura che faccia
al caso nostro. Quindi si osserva il valore che si avvicina a quella quantità: è il n° di capsula 0 ma
per riempirla correttamente non sono sufficienti 19 ml ma bisogna farne 20 ml. Dal punto di vista
formulativo sarebbe opportuno avere dei volumi ottimali perchè se bisogna suddividere in capsule
grandi delle quantità di polvere piccole è più facile fare errori perchè bisogna fare anche
l'uniformità di massa delle capsule: in teoria il valore che si suddivide deve essere proporzionale in
maniera precisa al volume delle capsule che si utilizzano perchè questo fa in modo che non si
facciano errori ed è il motivo per cui esistono tanti tipi di capsule.
Le capsule molli vengono fatte perchè il principio attivo è assorbito prima(rispetto alle compresse)
in quanto il principio attivo è già in soluzione e quindi tutti gli step( la disaggregazione, l'effetto
degli eccipienti sulla velocità di dissoluzione) non avvengono, e quindi il principio attivo viene, una
volta rilasciato dalla capsula(almeno teoricamente), immediatamente assorbito. Le forme che
possono assumere le capsule molli:
La maggior parte delle capsule molli ha la forma ovale, quando le capsule molli sono rotonde si
chiamano perle( come le perle di bagnoschiuma , e hanno anche la stessa tecnica di preparazione).
Quando si utilizzano le capsule molli:
per avere il principio attivo già in soluzione e quindi per aumentare la biodisponibilità;
nella maggior parte dei casi è quando si hanno dei principi attivi con un ristretto intervallo
terapeutico, cioè in cui la dose efficace è la dose tossica sono vicine, perchè è più facile
dosare uniformemente una soluzione che un solido.
La differenza sostanziale tra le capsule rigide e quelle molli è nella composizione quantitativa del
rivestimento, perchè comunque la composizione qualitativa di base è la stessa e nella tecnica di
preparazione: le capsule opercolate esistono come tali e vengono riempite poi, generalmente, con
una polvere o un granulato, mentre nel caso delle capsule molli le capsule vengono riempite e e
formate contemporaneamente. Ci sono due nastri di gelatina, che vengono scaldati e quindi si
ammorbidisce leggermente la gelatina, e passando attraverso gli stampi: la capsula viene riempita e
formata contemporaneamente. Tutti questi sistemi sono brevettati, cioè le tecniche di ottenimento
delle capsule molli sono brevettate e quindi generalmente ci sono delle aziende specializzate alle
quali le aziende produttrici mandano i principi attivi.

Un altro motivo per cui vengono realizzate le capsule molli è la possibilità di realizzare delle forme
farmaceutiche in cui sia elevata la stabilità del principio attivo: questo perchè, ad esempio, il guscio
di gelatina delle capsule molli protegge dal contatto con l'O2 e quindi per principi attivi che sono
facilmente ossidabili(e che quindi devono essere conservati in assenza di ossigeno si realizzano
delle capsule molli in cui il principio attivo, ad esempio, viene sciolto in un solvente oleoso.
Si può misurare il coefficiente di permeabilità del guscio di gelatina delle capsule molli come:
dove P è il coefficiente di permeabilità, q la quantità di O2 che passa attraverso la parete di gelatina,
h è lo spessore delle capsule molli, A è la superficie della capsula( e quindi attraverso la quale
avviene il processo di diffusione, t è il tempo, e la differenza(dato che si parla di un gas) di pressioni
parziali all'interno e all'esterno della capsula.
In questi due grafici si può vedere l'effetto proteggente, nella degradazione della riboflavina (1) e
della vitamina A (2). Sono confrontate le concentrazioni, misurate nel tempo, per le compresse e per
le capsule: si osserva che la variazione di concentrazione nel tempo rimane costante nelle capsule.
Quindi sia che la vitamina sia idrosolubile(riboflavina) che liposolubile(vitamina A) l'inclusione
nelle capsule molli diventa, in maniera sostanziale, la stabilità di principio attivo nel tempo
Quindi riassumendo, la funzione delle capsule molli:
aumento della biodisponibilità
aumento della stabillità del principio attivo
formulazione di principi attivi che abbiano un ristretto indice terapeutico

Nell'ambito delle preparazioni per uso orale si trovano le forme farmaceutiche, che la farmacopea
definisce, preparazioni oromucosali, cioè dove il sito di assunzione e di assorbimento è la bocca e
quindi la mucosa buccale è il sito attraverso il quale avviene l'assorbimento, quindi sono assunte per
via orale ma non vanno deglutite. Quindi, in queste preparazioni, l'assorbimento non avverrà nel
tratto gastroenterico, ma avverrà nel cavo orale. Le preparazioni oromucosali, secondo
farmacopea, sono preparazioni solide,semi-solide o liquide, contenenti una o più sostanze attive,
destinate alla somministrazione alla cavità orale e/o alla gola per ottenere un effetto locale o
sistemico. Il formulatore, da notare bene, per realizzare la forma farmaceutica deve studiare
l'anatomia del sito dove vuole che la sua forma farmaceutica sia assorbita; questo perchè, in base
alla costituzione dei tessuti si dovrà cambiare il tipo di formulazione e il tipo di interazione tra la
formulazione e il sito di assorbimento. Attraverso questo tipo di forme farmaceutiche e quindi con
l'assorbimento atttraverso la mucosa orale si possono avere delle formulazioni a rilascio locale; cioè
in cui il sito target è proprio la mucosa orale, oppure un'azione sistemica , che sarà ottenuta in
maniera differente a seconda che la frazione di assorbimento sia la frazione sublinguale della
bocca o la mucosa buccale vera e propria.
L'epitelio ha uno spessore di 40-50 strati di cellule, con un turnover di 5-6 giorni: alcune parti
dell'epitelio, come nelle gengive e nel palato, è cheratinizzato ed è ricco di ceramidi, in altre zone
della bocca c'è l'epitelio non cheratinizzato, ed è caratterizzato da un elevata frazione di colesterolo .
Quindi è possibile formulare delle preparazioni che di fermano in questi strati o si può pensare o
delle formulazioni che portino il principio attivo nella sottomucosa dove ci sono i vasi sanguigni e
quindi avere un effetto sistemico. Le caratteristiche della mucosa buccale è che ha una capacità di
permeabilità fino a 4000 volte superiore di quella della pelle; la capacità di assorbimento è massimo
nella zona sublinguale. La velocità di assorbimento è inversamente proporzionale allo spessore e al
grado di cheratizzazione.
sublinguale buccale palatale
assorbimento
spessore + cheratinizzazione
Uno degli ostacoli principali all'assorbimento attraverso la mucosa buccale è il materiale
intercellulare costituito da dei granuli che rivestono la membrana della cellule degli strati più alti
dell'epitelio della buccale; la presenza di questi granuli rallentano entrambi i processi di
assorbimento, che possono essere trans cellulare, cioè attraverso le cellule dell'epitelio, o
paracellulare , cioè degli spazi tra le cellule dell'epitelio. I meccanismi di assorbimento sono solo
all'interno del cavo orale, meccanismi di diffusione passiva. I componenti della mucosa buccale
sono:
il muco, componente principale, il quale è costituito al 90%(fino al 90%) di acqua, e il
restante è costituito da una glicoproteina ( la mucina) e poi sali numerali; la caratteristica
del muco è quello di avere una carica negativa, responsabile di queste sono i residui liberi di
acido sialico.
La saliva, costituita anch'essa sostanzialmente di acqua, ed è una delle fonti in cui il
principio attivo, qua, ed è una delle fonti in cui il principio attivo, rilasciato nella mucosa
buccale, si perde perchè ha un V flusso, che indipendentemente dalle condizioni fisiologiche

e patologiche, che varia da 0,5- 0,2 l/ al giorno. Infatti, indipendentemente da dove la forma
farmaceutica va commercializzata, bisogna valutare il tipo di dieta che segue la popolazione;
cioè la formulazione di una compressa, nel mondo occidentale, in cui c'è una dieta
principalmente a base di carne e di grassi, e quindi ha un tempo di residenza nell'intestino,
questa è diversa dalla formulazione che viene commercializzata nei paesi asiatici, dove la
dieta è principalmente vegetariana, e quindi sono diversi i tempi di residenza nell'intestino
delle forme farmaceutiche.
Il vantaggio attraverso la mucosa buccale è che si evita l'effetto di primo passaggio, perchè
l'assorbimento del principio attivo viene versato all'interno della giugulare interna e quindi è poi
un sangue che non passa attraverso il fegato, cioè non subisce l'effetto di primo passaggio. Le
caratteristiche chimico – fisiche del principio attivo, necessarie affinchè sia possibile attraverso la
mucosa buccale:
PM adeguato, anche se ci sono alcune molecole grandi come l'insulina che possono essere
assorbite attraverso la mucosa con dei meccanismi di interazione con il muco e con l'aiuto
dei “promotori di assorbimento”( delle sostanze che favoriscono il passaggio attraverso le
membrane) perchè disorganizzano in in maniera momentanea, la membrana da attraversare
facendo tornare la membrana alle condizioni iniziali dopo aver fatto passare il principio
attivo.
pKa della molecola, perchè con concetto generale l'assorbimento avviene a carico delle
molecole non ionizzate in quanto il pH della cavità orale è un pH fisiologico, e quindi
essendo la maggior parte dei farmaci acidi o basi deboli saranno più o meno assorbiti
all'interno della mucosa buccale perchè la maggior parte ha il pH della saliva(6,4);
coefficienti di ripartizione: il muco e la saliva sono costituiti principalmente da acqua e di
piccole frazioni lipoproteiche, e quindi la mucosa buccale è di costituzione essenzialmente
idrofila e quindi saranno preferenzialmente assorbiti i farmaci coi valori di coefficiente di
ripartizione basso( quindi per principi attivi idrofili). Il coefficiente di ripartizione è
all'equilibrio e a temperatura costante, il rapporto tra le concentrazioni di soluto che si
ripartisce tra due solventi immiscibili Ae B (P = ottanolo /H2O); il coefficiente di
ripartizione alto, quindi, indica lipofilia mentre un coefficiente di ripartizione basso indica
idrofilia.
Le dimensioni dell'area di somministrazione sono estremamente ridotte nella mucosa orale( 100
cm2, non di tutta la bocca ma delle zone non cheratinizzate della mucosa); quindi le dimensioni del
sistema di rilascio devono essere estremamente piccolo e, proprio per questo, la quantità di farmaco
che può essere somministrata è estremamente ridotta perchè le piccole dimensioni della forma
farmaceutica sono in limitate quantità. Una frazione del principio attivo disciolto nel cavo orale, che
ha come target l'assorbimento attraverso la mucosa buccale, viene inghiottito: quindi ci sarà una
frazione di principio attivo che esplica la sua azione all'interno del cavo orale ma hanno una
frazione che viene inghiottita e che quindi sarò assorbita a livello del tratto gastroenterico.

PA disciolto PA disciolto PA nel circolo
nei fluidi buccali nella membrana linfatico
buccale
PA inghiottito PA nel circolo
sanguigno
Infatti, sia a livello di biodisponibilità sia a livello di farmacocinetica bisogna tenere conto che una
frazione viene assorbita attraverso la mucosa orale ma comunque una frazione viene inghiottita, e
quindi anche questa esplicherà nel tempo la sua azione.
I siti per il rilascio di farmaci sono:
a) forme farmaceutiche sublinguali ( in commercio ed è quello che viene utilizzato
principalmente); si utilizzano perchè hanno un'elevata biodisponibilità, un assorbimento rapido,
facilmente accessibile a qualsiasi persona e presenta un'alta patient compliance. Si utilizza la forma
farmaceutica sublinguale, principalmente per i principi attivi di pronto soccorso o comunque per i
principi attivi da utilizzare per un attacco acuto. Un'altra applicazione sono quelli che si definiscono
a regimi di dosaggio infrequenti ovvero quando non c'è una terapia cronica. La maggior parte delle
formulazioni in commercio sono capsule molli o quelle che si chiamano compresse delitescenti:
sono delle compresse, per esempio ci sono degli antidolorifici, molto sottili che non si devono
toccare con le mani in quanto si scioglierebbero, infatti vengono chiamate in inglese “ a rapida
disintegrazione e dissoluzione”. Questi sono alcuni esempio di formulazioni sublinguali con
principi attivi particolari: nel caso ad esempio del MINIRIM/DDAIP è stato valutato che la
biodisponibilità è maggiore del 60 % rispetto alle compresse. Altri tipi di farmaci formulati in forme
farmaceutiche sublinguali sono:
farmaci peptidici: insulina, LHRH, α- interferone;
farmaci per la terapia del dolore: buprenorfinatil.
Comunque questi farmaci non sono commercializzati in Italia. Da notare bene gli eccipienti:
lattosio: è un diluente
mannitolo: per la freschezza(perchè la sua dissoluzione è un processo fortemente
endotermico e quindi assorbendo calore rilascia una sensazione di freschezza);
amido di mais: disaggregante( l'amido di patata, al contrario di quello di mais, non è un
potentissimo disaggregante).
PVP, o polivinilpolidone, K30(che dà un' indicazione di polimero non ad altissimo PM):
legante. Da notare bene che la presenza di un legante indica che la compressa è stata
ottenuta non per compressione diretta ma per compressione dopo granulazione:
Acido citrico e citrato di sodio: tampone
Stereato di magnesio: lubrificante propriamente detto e quindi la presenza degli stearati
indicano che la forma farmaceutica è una compressa, anziché una capsula, perchè vengono
usati solo se si utilizza la comprimitrice.
Quindi si è visto l'assorbimento sublinguale, e quindi formulazioni a rilascio immediato. Si può

avere anche un rilascio sistemico transmucosale, quindi attraverso la mucosa buccale quindi di
fatto sostanzialmente le guance e la zona posteriore del palato; per questo tipo di assorbimento, è
imprescindibile l'uso di promotori di assorbimento ( è penetration enhaceis) perchè la
biodisponibilità è bassa ed è legata al fatto che la velocità di assorbimento non è altissima e
soprattutto perchè la superficie assorbente è estremamente limitata. Un utilizzo, che ovviamente si
può fare, è quello per le patologie dentali e paradentali, in cui invece l'azione che si deve avere è
locale e , quindi il fatto che la superficie capace di assorbimento è ridotta non è importante.
Questi sono degli esempi di sostanze che vengono usate come promotori di assorbimento:
– ciclodestrine
– acido laurico( o acido laurico/ propilen glicol)
– mentolo
– acido oleico
– Na EDTA
– Na taurocolato
– Na taurodeossiedato
– fosfatildelcoline
– poliossietilene
– polisorbato 80
I promotori di assorbimento sono delle sostanze che hanno la capacità di interagire con le
membrane e di disorganizzare le membrane, facilitando il passaggio attraverso queste. Tutte le
sostanze che si utilizzano come promotori di assorbimento devono produrre una disorganizzazione
rapida ma totalmente reversibile; è ovvio che tutti i promotori di assorbimento per le formulazioni
oromucosali devono avere un odore e un sapore non sgradevoli. Un promotore di assorbimento
classico è il dimetilsolfossido che ha un odore sgradevole e quindi non può essere utilizzato per le
formulazioni oromucosali. Molte di queste sostanze hanno la caratteristica di essere dei tensioattivi,
alcuni naturali come gli acidi biliari ed altri sintetici.
Nell'ambito della mucosa buccale, la superficie capace dell'assorbimento è ridotta e i tempi di
permanenza sono ridotti, data la presenza di saliva che rimuove rapidamente il principio attivo, una
delle tecniche che si utilizza per questo tipo di formulazioni è l'utilizzo di polimeri mucoadesivi,
cioè delle forme farmaceutiche che si “mescolano” nella mucosa o all'interno della guancia o sopra
la mucosa sita sopra i denti. Le sostanze utilizzate come sostanze mucoadesive sono delle sostanze
di tipo polimerico, sintetico, oppure sono dei polimeri di origine naturale:
acidi poliacrilici → di sintesi
idrossipropil metilcellulosa → di sintesi
derivati polimetacrilati → di sintesi
acido ialuronico → naturale e quasi biocompatibili
chitosano → naturale e quasi biocompatibili
xanthum gum(xantano)
locust bean gum (carrubbo)
Gli ultimi due, sciolti in acqua, formano degli hydrogel, ovvero delle strutture polimeriche a forma

di gel.
Come funziona una sostanza mucoadesiva? Innanzitutto questo varia, in generale, per le sostanze
che sono definite bioadesive, cioè delle sostanze capaci di interagire con i tessuti; all'interno delle
sostanze bioadesive ci sono le sostanze mucoadesive, cioè capaci di interagire e di aderire alle
mucose. Ci sono molte teorie per spiegare la loro adesione, quelle che sono più sfruttate nella scelta
dei polimeri mucoadesivi:
teoria della bagnabilità, quindi scegliere un gel polimerico che sia capace di bagnare il
muco e quindi di interagire
mechanical theory, superficie ruvida
teoria “elettronica”:electron tranfert;
teoria dell'assorbimento. Hydrogel bonding- van der Waals force
diffusion theory: diffusione delle catene polimeriche dell'interfaccia
queste ultime 3 teorie sono legate perchè basano la mucoadesione sull'interazione delle catene
polimeriche con la struttura della mucina, quindi un'interazione di tipo fisico:le catene polimeriche
entrano nel reticolo polimerico della mucina e interagiscono fisicamente. Quasi tutti i polimeri
bioadesivi presentano dei gruppi -COOH o -OH liberi che sono capaci di formare legami ad H con
le proteine dei tessuti a cui aderiscono.
Sono delle forme farmaceutiche abbastanza giovani: il primo prodotto in commercio è stato l'aftach
(1985), di una compressa a doppio strato. Il primo strato è il principio attivo in presenza di carbopol
(un poliacrilato) più HPC( idrossipropilcellulosa) e quindi c'è il principio attivo disperso nei
polimeri mucoadesivi. Il secondo strato è costituito solo da lattosio, che è quello che rimane a
contatto all'interno della bocca e quindi quello che viene fatto aderire è lo strato polimerico,
all'interno del quale è presente il principio attivo, quello che resta a contatto con la saliva è il
lattosio. In questo caso il principio attivo diffonde solo attraverso la mucosa buccale; lo strato di
lattosio serve da “ protezione” e fa sì che il principio attivo venga rilasciato solo attraverso la
mucosa buccale e non attraverso il cavo orale. Nel 1996 è stato commercializzato il denti patch®,
uno strip adesivo che contiene lidocaina, da usare come pre-anestetico per i piccoli interventi
all'interno della bocca. L'effetto anestetico si instaura in 2 minuti, e rimane effettivo per 45 minuti e
non c'è assorbimento sistemico dell'anestetico, perchè le gengive sono cheratinizzate(e quindi c'è un
lentissimo assorbimento quasi nullo). Un'altra preparazione è l’orobase®,il cui principio attivo è la
benzocaina al 20 %, ed è formulato sottoforma di gomma da masticare(medicate): in questo caso è
un anestetico locale. Un'altra formulazione è il gel glair ®, in bustine (e quindi monodose) in cui si
trova il gel concentrato viscoso e mucoadesivo; quindi viene posto all'interno della bocca e il
polivinilpinolidone e l'acido ialuronico funzionano da agenti mucoadesivi e quindi fanno in modo,
anche se la formulazione non è una compressa o patch, di farla aderire in modo diffuso nella
mucosa buccale e quindi di esplicare un'azione locale nelle ulcere diffuse o sulle stomatiti da
radiochemioterapia, quindi non a livello locale ma su tutta la bocca.
In farmacopea, le preparazioni oromucosali sono definite come: sono preparazioni solide, semi-
solide o liquide destinate alla somministrazione alla cavità orale e/o alla gola per ottenere un effetto
locale o sistemico. Le preparazioni mucoadesive sono destinate ad essere trattenute nella cavità
buccale per adesione all'epitelio mucosale e possono modificare l'assorbimento sistemico del

farmaco al sito di applicazione. Si possono distinguere parecchie categorie di preparazioni per uso
oromucosale:
gargarismi
colluttori
gengivari
soluzioni e sospensioni oromucosali
preparazioni oromucosali semi-solide
gocce oromucosali, spray oromucosali sublinguali
pastiglie e paste
tavolette
compresse sublinguali e buccali
capsule oromucosali
preparazioni mucoadesive
La differenza tra i gargarismi e il colluttorio è che i gargarismi vanno fatti in gola mentre il
colluttorio nella bocca.
Se si parla di forme farmaceutiche solide unidose i saggi richiesti dalla farmacopea saranno
l'uniformità di massa e l'uniformità di contenuto; per gli spray oromucosali e sublinguali è previsto
un saggio particolare ed aggiuntivo, che è il saggio della dose rilasciata, perchè essendo gli spray
delle forme farmaceutiche multidose(cioè suddivisa alla singola unità posologica da parte del
paziente) deve essere garantito che ogni dose rilasciata abbia una concentrazione di principio attivo
uniforme.
I gargarismi sono soluzioni acquose destinate ad essere gargarizzate per ottenere un effetto locale.
Non devono essere inghiottiti. I colluttori sono soluzioni acquose destinate a venire in contatto con
la membrana mucosa della cavità orale, usualmente dopo diluizioni in acqua. Non devono essere
inghiottiti. I gengivari sono destinati ad essere somministrati alle gengive per mezzo di un
opportuno applicatore. Le gocce oromucosali, gli spray oromucosali e sublinguali sono soluzioni,
emulsioni o sospensioni destinati ad avere effetto locale o sistemico. Sono applicate per
instaurazione o nebulizzazione nella cavità orale o su una parte specifica della cavità orale come
nebulizzazione sotto la lingua(spray sublinguare) o nella gola(spray orofaringeo). Le pastiglie e le
paste sono preparazioni solide, a dose unica, destinate ad essere succhiate per ottenere, disciolto, un
effetto locale nella cavità buccale e della gola. Quindi la loro caratteristica è di sciogliere in bocca e
quindi anche la formulazione sarà legata e questo. Infatti contengono una o più sostanze attive,
usualmente in base aromatizzata e dolcificate e sono destinate o disciogliersi o disaggregarsi
lentantente nella bocca quando vengono succhiati.

Forme farmaceutiche liquide
Alcol etilico: In galenica farmaceutica, quando in una prescrizione si trova la parola alcool si
intende alcool etilico a 96°. Il grado alcolico, si definisce con le parti di alcool etillico in 100 parti
di soluzione idroalcolica: i volumi di acqua e di etanolo non sono- additivi, perchè quando la
soluzione si mescola(ovvero la soluzione di acqua e alcool) si ha una leggera contrazione di volume
pari area al 3%. Quindi la definizione di grado alcolico, che è una definizione volumetrica, è ml di
etanolo in 100 ml di soluzione idroalcolica. Nella farmacopea italiana non c'è più la monografia
dell'alcol ma si fa riferimento alla monografia sulla farmacopea europea;quindi l'alcool di
riferimento è l'etanolo a 96°. esiste l'etanolo anidro ma al farmacista non gli serve perchè non si
utilizza l'alcool assoluto nelle preparazioni farmaceutiche ma dovrà sempre operare delle
diluizioni(nelle forme farmaceutiche entrano sempre alcool diluiti). Si utilizza l'alcool al 96° perchè
la miscela di acqua e l'acool è una miscela azeotropica, che a quella percentuale non possono
essere separate per distillazione e si comportano come una fase singola. È possibile ottenere l'alcool
anidro ma con delle tecniche che sono più costose della distillazione, ovvero i setacci molecolari.
Determinazione del grado alcolico
il grado alcolico è , dal punto di vista fisico, una misura di densità e quindi non si misura
direttamente il grado alcolico ma si misura la densità della miscela di acqua e di alcool e da questo
valore di densità si estrapola il valore di grado alcolico. La densità è il rapporto tra la massa e il
volumetrica , d20
= m/V dove il 20 fa riferimento alla temperatura. Come si misura la densità dei
liquidi:
picnometro Questi due sfruttano principi diversi
densimetro
Picnometro: la densità è misurata sempre il relazione alla densità dell'H2O a 20°C.
Densitometro: ha una parte di vetro riempita da due piombini metallici, ed ha la scala
delle densità e il termometro(perchè la determinazione della densità dipende dalla
temperatura). Il densimetro sfrutta il principio di Archimede: un solido immerso in
un liquido riceve una spinta dal basso verso l'alto pari alla massa del volume di fluido
spostato. La misura viene fatta mettendo il densimetro nella soluzione, di cui si vuole
calcolare la densità,si misura sulla scala delle densità, al punto di affioramento, la
densità. Esistono dei densimetri che sono tarati direttamente a gradi alcolici che si
chiamano alcolometri. Sia l'alcolometro che il densimetro sono tra le apparecchiature
obbligatorie in farmacia( tabella VI della F.U).
In farmacopea viene scritto il metodo picnometrico oppure viene detto che può essere
anche fatto con il densimetro; essendo una misura di densità si potrebbe eseguire la

misura nella soluzione idroalcolica tal quale(se si avesse solo H2O e alcool nella miscela). Nella
maggior parte dei casi(nelle preparazioni farmaceutiche) non c'è solo H2O e alcool quindi prima di
effettuare la misura di grado alcolico,bisogna effettuare una distillazione, per separare H2O e alcool
e su questa miscela si farà la misura di grado alcolico. In farmacopea, poi, viene descritto il metodo
per distillare la miscela e sulla miscela di H2O e alcool che si raccoglie, all'interno del pallone, si
determina il grado alcolico. Dalla misura di densità si risale al grado alcolico attraverso le tabelle
alcolimetriche.
Quale è la relazione tra il grado alcolico e la densità? Per esempio un alcool a 20° è più denso di
quello a 96° perchè(l'alcool più è diluito più e denso) l'acqua ha densità 1 mentre l'etanolo circa 0,8
e quindi a parità di T essendo l'acqua più densa dell'alcool , più è diluito l'alcool maggiore sarà la
densità. Miscelando acqua e alcool si ha una contrazione di volume per preparare degli alcoli
diluiti non si può utilizzare la percentuale volumica(in ml) ma si deve utilizzare la percentuale
ponderale e le quantità espresse in PESO. Si utilizza la formula delle diluizioni
c1v1 = c2v2 → Aa = Bb
dove le lettere maiuscole corrispondono ai grammi di alcol a 96°(A) e la quantità in grammi di alcol
diluito(B), e le lettere minuscole sono le percentuali ponderali(ovvero la percentuale peso/peso). Le
percentuali ponderali si trovano in farmacopea, dove ci sono le tabelle alcolimetriche, nelle tabelle
alcolimetriche ci sono: grado alcolico (% volumica) | % p/p | ρ20
Per l'alcool a 96° la % peso/peso è 93,84.

Preparazioni liquide a base di alcool
Gli alcoliti si ottengono per dissoluzione diretta di sostanze in etanolo, e sono soluzioni. Possono
essere
salini
zuccherini, che si chiamano elisir
ammoniacali che si chiamano balsami.
Il nome tra alcoliti e alcolati, la diversità è nella tecnica di preparazione. Gli alcolati , invece, si
ottengono per macerazione e per distillazione di droghe fresche o secche( grado alcolico circa 80°).
Gli alcolaturi sono prodotti per macerazione di droghe fresche in un alcool 80-90°. Sono
aromatizzanti.
Nell'ambito delle preparazioni a base alcolica ci sono anche le forme farmaceutiche estrattive;
sono classificate secondo varie caratteristiche, ma fondamentalmente sono classificati in base al tipo
di processo per estrarre la droga. Diversi tipi di:
1) per infusione(o tisana)
2) per decozione
3) per digestione
4) per macerazione
5) per percolazione
Una delle tecniche estrattive è l'infusione, la cui forma farmaceutica si chiama infuso o tisana ed è
quando non si ha solo una pianta ma una miscela di piante da estrarre. La differenza tra infusi e
decotti è che nell'infuso si porta l'acqua ad ebollizione e poi si aggiunge la droga da estrarre(come
con il thè), mentre nel decotto la pianta da estrarre si mette in acqua e si fa bollire insieme all'acqua.
Un'altra tecnica che si può utilizzare è la digestione, cioè il trattamento della droga con acqua calda
ma a una temperatura inferiore alla temperatura di ebollizione dell'acqua(mentre gli infusi e i
decotti si ottengono con acqua portata alla temperatura di ebollizione). Le due tecniche complesse
sono la macerazione e la percolazione: la macerazione è un “processo statico” nel senso che la
droga, o le droghe, da estrarre vengono messe a macerare in un solvente. La percolazione, invece, è
un “processo dinamico” perchè in realtà la droga(o le droghe) da macerare viene posta in una specie
di colonna cromatografica ed attraverso la droga viene fatto passare in continuo solvente fresco;
quindi la macerazione è “statica” perchè ad un certo livello il solvente satura e non può estrarre
mentre nella percolazione si può arrivare all’esaurimento della droga, cioè all'estrazione completa
di tutti i principi attivi della droga di partenza.
Con la tecnica di macerazione si ottengono le tinture, che sono delle forme farmaceutiche estrattive,
con la percolazione di ottengono le tinture e gli estratti(fluidi, molli e secchi). Le tinture possono
essere classificate in base al solvente con cui viene effettuata l'estrazione: nella maggior parte dei
casi sono delle tinture alcoliche, perchè si utilizzano miscele idroalcoliche con una graduazione
media intorno ai 60-70°. possono essere anche classificate in base alla composizione; quindi si
possono avere delle tinture semplici o tinture composte. La classificazione più utile è quella relativa
alla droga di partenza: tinture eroiche e tinture non eroiche .Si chiama tintura eroica se si ottiene

da una droga eroica, ovvero quelle droghe in cui i principi attivi hanno un ristretto intervallo
terapeutico(digitale). Quello che cambia è il grado di diluizione rispetto alla droga di partenza.
Nelle tinture eroiche, il principio attivo è diluito 1 a 10 rispetto alla droga di partenza, mentre nelle
tinture non eroiche il principio attivo è diluito 1 a 5 rispetto alla droga di partenza.
Un'altra classificazione che può essere fatta è in base alla tecnica di preparazione e quindi per
macerazione e per percolazione. Come si sceglie la tecnica: per esempio una tintura composta,
ovvero che si realizza da una miscela di droghe diverse, non può essere fatta per percolazione
perchè non tutte le droghe hanno la capacità di estrazione dei principi attivi e quindi nel percolatore
si hanno delle droghe con una maggiore capacità di estrazione e altre no(e quindi si preparano per
macerazione). La scelta della tecnica dipende anche dalla natura della parte della droga da estrarre:
per esempio se si ha una corteccia è intuitivo che si utilizza la macerazione. Quando delle droghe da
estrarre sono delle resine o comunque delle parti di piante che in acqua rigonfiano(danno delle
mucillagini, per esempio) non si può utilizzare la percolazione perchè si bloccherebbe il processo
estrattivo.
La definizione di tintura, secondo farmacopea: le tinture sono delle preparazioni liquide ottenute
generalmente usando una parte di droga vegetale o materiali animali(essiccati) e dieci parti di
solvente di estrazione o una parte di droga vegetale o materiale di origine animale e cinque parti di
solvente di estrazione. I saggi sono: a) la densità relativa, b) il contenuto in etanolo(ovvero il grado
alcolico e per le tinture deve essere fatto prima della distillazione), c) metanolo e 2-propanolo,
ovvero la loro ricerca perchè sono due alcool tossici che devono essere sempre assenti dalle
preparazioni per uso farmaceutico. Le tinture sono più diluite rispetto alla droga di partenza in
principio attivo(o 1 a 10 o 1 a 5).
Gli estratti sono realizzati a partire da una tecnica diversa: cioè c'è una fase estrattiva, che
generalmente viene eseguita per percolazione, e una fase successiva di concentrazione, che deve
essere rapida, a pressione ridotta e a una temperatura che non deve superare mai i 60°C. Quindi le
tinture derivano da un processo di estrazione e sono sempre più diluite in principio attivo rispetto
alla droga di partenza, gli estratti sono preparati attraverso una tecnica di estrazione e una
successiva fase, però, di concentrazione. Gli estratti sono preparazioni di consistenza liquida
(estratti madri e tinture), semisolida( estratti madri e olio resine) o solida( estratti secchi), ottenute
da droghe vegetali o da materiali di origine animale generalmente allo stato essiccato.
I concentratori a vuoto , sono anch'essi presenti nella tabella 6 della farmacopea con la nota” solo
per farmacie che preparano estratti”. In base a quanto si rimuove il solvente usato per l'estrazione, si
hanno 3 tipi di estratti: estratti fluidi, molli e secchi. Cambia la consistenza e cambia soprattutto la
concentrazione del principio attivo; l'estratto fluido è liquido ed ha la stessa concentrazione di
principio attivo della droga di partenza. Gli estratti molli e gli estratti secchi sono più concentrati in
principio attivo rispetto alla droga di partenza. Generalmente gli estratti molli sono due volte
concentrati rispetto alla droga di partenza, negli estratti secchi la concentrazione di principio attivo
è 6 volte maggiore rispetto alla droga di partenza. L'estratto secco ha la consistenza di una polvere
mentre l'estratto molle ha la consistenza semisolida.
Anche per gli estratti fluidi e molli ma non secchi, i controlli sono il grado alcolico e il metanolo e il
2-propanolo. Poiché gli estratti,soprattutto quelli fluidi, vengono utilizzati generalmente per essere

diluiti e per realizzare altre preparazioni bisogna valutare quelle che sono le incompatibilità:
innanzitutto se si deve diluire l'estratto fluido in acqua, si deve considerare che si possono essere
delle sostanze poco solubili in H2O e che potrebbero precipitare diluendo. Così come con l'elevata
presenza di tannini nelle forme farmaceutiche estrattive potrebbe causare la precipitazione di alcuni
principi attivi(come per esempio gli alcaloidi); infatti, generalmente, si acquistano degli estratti
fluidi de-tannizzati, che servono a preparare le relative tinture perchè, a parte il processo estrattivo,
un'altro modo per ottenere una tintura è tramite la diluizione dell'estratto fluido( perchè ha la stessa
concentrazione in principio attivo della droga di partenza). Gli estratti liquidi sono preparazioni
nelle quali, in generale, una parte in massa o in volume è equivalente ad una parte in massa della
droga vegetale o del materiale di origine animale essiccati. Gli estratti molli sono preparazioni
semisolide ottenute per evaporazione o parziale evapora del solvente usato per l'estrazione. Gli
estratti secchi sono preparazioni ottenute per evaporazione del solvente usato per la loro
preparazione.
Da notare che nei saggi sia degli estratti molli che di quelli secchi non c'è più la determinazione del
grado alcolico e del metanolo e del 2-propanolo, ma soltanto la determinazione del residuo secco e
in questo caso la perdita all'essiccamento; nell'estratto molle c'è scritto, infatti, che hanno un residuo
secco non inferiore al 70% peso/peso.
Sono stati reintrodotti, in farmacopea, gli infusi e i decotti. Gli infusi sono preparazioni liquide
ottenute, estemporaneamente, versando sulle droghe, ridotte ad un grado di suddivisione e delle
quali si vogliono estrarre i principi attivi, acqua R alla temperatura di ebollizione e lasciando poi in
contatto con l'acqua stessa per un tempo più o meno lungo. I decotti sono preparazioni liquide
ottenute estemporaneamente facendo bollire in acqua le droghe opportunatamente polverizzate,
dalle quali si vogliono estrarre i principi attivi. In farmacia si può tenere soltanto alcool etilico a
96°, solo in bottiglie da 2l; in farmacia è possibile tenere fino ad un massimo di 50l di etanolo a 96°
in bottiglie da 2l. Se fosse necessario una quantità superiore a 50l è obbligatorio in farmacia un
registro di carico/scarico dell'alcool etilico. Si può tenere una sola bottiglia aperta per volta,dove per
aperta si intende la fascetta rosa dell'UTIF (ufficio delle imposte) rotta (quindi non è necessario che
la bottiglia sia aperta, ma se la fascetta è rotta la bottiglia si considera aperta).
Non è possibile tenere in farmacia alcol diluiti pronti, ma devono essere preparati
estemporaneamente: questo perchè sull'alcool c'è una tassa governativa, l'alcool a 96° non si può
utilizzare come sostanza da abuso mentre gli alcool diluiti possono essere utilizzati per preparare
liquori, profumi sui quali non ci sarebbe la tassa di imposta. Si può tenere, quindi, solo l'alcol a 96°
oppure l'alcol a graduazione più bassa, ovvero l'acool “rosa” che è alcool denaturato che ha una
graduazione in genere di circa 60°, che si chiama denaturato per aggiunta di colorante e di sostanze
che rendono impossibile l'uso alimentare o per uso cosmetico.
In farmacopea, sulla parte delle monografie specifiche sono riportati alcuni esempi di tinture e di
estratti: in realtà l'unica tintura ancora prescritta in farmacopea è la tintura di Ratania. La
monografia dice quale è la parte della pianta che si utilizza(la radice) e dice anche quale è la
percentuale di tannini presenti e indica la preparazione(dalla droga polverizzata 710 per trattamento
con alcool a 70° impiegando un metodo appropriato secondo le prescrizioni della monografia
tinture). 710 è l'indicazione granulometrica della polvere, in particolare 710 fa riferimento alla
maglia che ha un'apertura di 710 μm e deve essere omogenea(oltre il 96% della polvere è passata

dal molino 710) e lo deve essere perchè essendo un processo estrattivo le dimensioni devono essere
più piccole possibili; con una dispersione di dimensioni ampia si avrà una capacità estrattiva diversa
perchè cambia la superficie specifica.
Un esempio di estratto fluido è la Genzian: viene detta quale parte della pianta utilizzare(la radice),
la droga polverizzata è 1000 per trattamento con alcol 1 40°. Il grado alcolico si sceglie in base alla
solubilità del principio attivo, se basta alcol 30°, i principi attivi della genziana saranno più
idrosolubili, e quindi dipende dalla solubilità delle sostanze da portare in soluzione. Il rapporto,
nella genziana, della droga estratta è 1 a 1. Un altro esempio è l'estratto secco di Camomilla, la cui
droga sono i fiori, si utilizza alcol a 70°(con un metodo appropriato) operando una sgrassatura dei
percolati parzialmente concentrati: evidentemente perchè il processo di estrazione porta in
soluzione dei principi attivi che non si vogliono nell'estratto secco e quindi vengono eliminati prima
della concentrazione. Nei caratteri della polvere viene specificato che è una polvere amorfa
idroscopica(che assorbe facilmente umidità); questo tipo di indicazione serve per dare delle
considerazioni sulle modalità di conservazione della polvere. In etichetta, la farmacopea dice che
dovrà essere indicato il solvente usato per la sgrassatura, di cui non deve rimanere traccia nella
preparazione finale ma comunque in etichetta deve essere modificato.
Le droghe vegetali che si utilizzano in farmacia devono rispondere a dei requisiti precisi in
farmacopea, ovvero un farmacista deve comprare solo droghe vegetali FU, ovvero che rispondano a
dei requisiti di una precisa monografia,nel capitolo delle monografie generali, che si trova in
farmacopea. Sempre nel capitolo delle monografie generali c'è una monografia anche per le piante
per tisane; quindi la miscela per tisane preparata in farmacia è preparata con piante che rispondono
a dei requisiti precisi di farmacopea, e anche la semplice suddivisione dalla confezione grande alle
singole confezioni deve essere fatta utilizzando gli stessi criteri delle norme di buona preparazione
per preparare i galenici. Quindi la vendita in farmacia garantisce che le piante rispondono ai
requisiti della farmacopea ufficiale e che anche la ripartizione è fatta secondo le norme di buona
preparazione dei medicinali.
Gli oli grassi vegetali sono sostanzialmente degli eccipienti particolarmente importante perchè si
utilizzano per le formulazioni di tipo parenterale, cioè per le forme farmaceutiche iniettabili. Gli oli
grassi vegetali sono principalmente dei trigliceridi di acidi grassi in fase solida o liquida. Per
trigliceridi di acidi grassi si tratta di glicerolo i cui -OH sono esterificati con acidi grassi a lunga
catena; il glicerolo è un polialcool con tre funzioni alcoliche e si chiama 1,2,3 propantriolo. La
farmacopea, inoltre, dice da quale fonte si ottengono(semi, frutto o nocciola/drupa/nucleo). Si
possono avere secondo farmacopea:
olio vergine: olio ottenuto dalla materia prima di qualità speciale mediante procedure
meccaniche; questa è la caratteristica dell'olio di oliva. La differenza tra l'olio vergine e l'olio
extravergine è invece legata all'acidità dell'olio.
Olio raffinato: olio ottenuto mediante spremitura e/o estrazioni con solvente e successiva
raffinazione, in quanto il solvente estrae anche del materiale che non interessa e quindi
questo materiale deve essere allontanato.
Olio idrogenato: negli oli, essendo presenti dei doppi legami, il processo di idrogenazione
porta ad un dio che sia completamente saturo(quindi senza doppi legami). Nelle
preparazioni per uso parenterale sono usate solo oli raffinati con acido fosforico e alcali.

Le forme farmaceutiche parenterali sono quello forme farmaceutiche che non presentano il
passaggio attraverso il tratto gastrointestinale; all'interno delle forme farmaceutiche parenterali ci
sono le intravascolari( per cui sono previsti i veicoli oleosi), le endovenose, le intravenose, le
intradermiche. Per raffinazione si intende l'eliminazione delle impurezze e dei contaminanti
preservando il più possibile i trigliceridi(quindi i componenti essenziali dell'olio) e riducendo al
minimo la perdita di olio. Le sostanze allontanate per raffinazione sono:
acidi grassi liberi che possono essere facilmente ossidati, perchè gli acidi grassi esterificati
subiscono meno il processo di ossidazione;
l'acqua;
coloranti, come la clorofilla e i carotenoidi;
i metalli;
le proteine che possono provocane reazioni allergiche;
la frazione in saponificabile, cioè tutte le sostanze(lignine,steroli) che non possono essere
saponificate cioè sostanze che non subiscono idrolisi(in presenza di una base forte) che è il
processo che scinde i trigliceridi in glicerolo e acidi grassi.
In etichetta deve essere riportato:
– se l'olio è ottenuto mediante spremitura o estrazione;
– se l'olio è appropriato per le preparazioni di forme farmaceutiche per uso orale;
– il nome e la concentrazione di ogni antiossidante aggiunto (questo perchè gli oli subiscono il
processo di irrancidimento, che è una reazione di ossidazione con meccanismo radicalico in
cui l'O2 viene trasformato dalla luce in ossigeno tripletto, specie ad elevata energia, che
catalizza l'addizione radicalica di O2 alle catene degli acidi grassi con la formazione di
perossidi(stadio di propagazione)e nella fase di terminazione si hanno i prodotti finali come
chetoni e aldeidi; questi sono nella maggior parte dei casi i responsabili dell'odore
sgradevole.
Per alcuni oli a questa fase terminale del processo di irrancidimento si aggiunge una fase in cui le
aldeidi e i chetoni finali, polimerizzano e quindi l'olio non prende più la consistenza liquida ma
solida(cioè sono quelli utilizzati per dipingere) e sono detti oli siccativi. Gli oli saturi e i grassi
idrogenati perchè sono più stabili, in quanto in assenza di doppi legami è chiaro che l'ossigeno non
reagisce. Inoltre, il processo di irrancidimento è catalizzato dalla presenza di metalli; è per questo
che sostanze facilmente ossidabili sono conservati in contenitori di vetro scuri, in assenza di
ossigeno e, oltre agli antiossidanti classici che sono delle sostanze che si ossidano al posto dell'olio,
si aggiungono anche dei chelanti dei metalli (EDTA), che eliminano così i catalizzatori.
Alle forme farmaceutiche estrattive si ricollega l’omeopatia. I due principi sulla quale si basa
l'omeopatia sono:
1) il principio dell’analogia , ovvero l'agente che sull'uomo sano genera la malattia,sull'uomo
malato la cura;
2) principio delle diluizioni, dove il simile cura il simile se diluito in dosi infinitesimali;

L'Omeopatia è un metodo ideato da un metodo tedesco, Sanuel Hahnemann, alla fine del 18°
secolo. Le materie prime delle preparazioni omeopatiche sono quasi sempre di derivazione
vegetale(e a differenza della medicina tradizionale utilizza sempre la droga fresca raccolta nel
periodo balsamico della pianta ovvero nel periodo in cui la pianta è alla massima crescita), oppure
possono essere di derivazione animale(o animali interi vivi che sono estratti in soluzioni
idroalcoliche o animali essiccati), che in Italia sono vietati in quanto partono da polveri di organo o
di veleno. Le preparazioni base che sono tipiche nell'omeopatia sono: le tinture madri, i
macerati glicerici o gemmo derivati e le triturazioni.
Le tinture madri sono preparate in base ai requisiti della farmacopea tedesca(in quanto in
Germania c'è una doppia farmacopea) o in base alla farmacopea francese: sia in Francia che in
Germania l'omeopatia è riconosciuta nel sistema sanitario nazionale e in particolare in Germania ci
sono degli ospedali e ristretti medici in cui si utilizzano solo ed esclusivamente i prodotti
omeopatici. I gemmoderivati o macerati gliceridi sono ottenuti per macerazione in acqua, alcool e
glicerina e la caratteristica è che sono realizzati a partire dalla parte della pianta in crescita( le
gemme, le radici giovani o i semi). Le triturazioni sono preparazioni solide che si ottengono per
diluizione di polvere in lattosio. Nell'omeopatia ci sono due diluizioni diverse: quella decimale e
quella centesimale. Le triturazioni quindi possono essere preparate o diluendo una parte di materiale
di partenza in 9 parti di lattosio, oppure diluendo una parte di materiale di partenza in 99 parti di
lattosio. Le triturazioni non vengono utilizzate come forme farmaceutiche ma vengono utilizzate per
preparare le successive diluizioni. Le differenze tra la tintura FU e la tintura madre
Tintura FU Tinture madri
1:5, 1:10 10%(1:10)
pianta secca pianta fresca
macerazione,percolazione solo per macerazione
decozione,infusione,digestione
Le tinture madri sono utilizzate in quanto tale nella medicina omeopatica, ma sono poco utilizzate
se non per preparare le diluizioni. Le diluizioni possono essere preparate secondo 2 tecniche:
1) diluizioni Hahnemanniane, o per flaconi separati
2) diluizioni Korsakoviane o per flacone unico
Nelle diluzioni hahnemanniane,
le diluizioni sono
decimali(indicate con D o DH)
o centesimali(indicate con C o
CH). La prima diluizione,sia
quella decimale che quella
centesimale, viene fatta a peso.
Tutte le diluizioni successive
vengono fatte a volume. Già dalla terza diluizione, il contenuto in grammi della droga di partenza è
uno su un milione.
aumento
potenza
terapeutica
D1 10-1
D2 C1 10-2
D3 10-3
D4 C2 10-4
D5 10-5
D6 C3 10-6

Nelle diluizioni korsacoviane, indicate con la lettera K, invece si eseguono le diluizioni in un
flacone unico, ovvero si esegue la diluizione, si dinamizza poi si butta una frazione e poi si
aggiunge solvente nello stesso flacone. Ci sono poi le potenze cinquatesimali, dal n° latino
LM,che sono preparati a partire da un solido alla terza centesimale(solido diluito con lattosio) e poi
si prepara una soluzione 1 a 50000. Le diluizioni sono preparate generalmente in alcool a 30°.
Le forme farmaceutiche della terapia omeopatica sono:
compresse,fiale orali,suppositori,colliri,pomati,gocce → forme farmaceutiche comuni
sciroppo,granuli,globuli → tipiche della terapia omeopatica
In Italia, le preparazioni omeopatiche iniettabili sono vietate, i granuli e i globuli sono delle sfere di
saccarosio o di lattosio, i granuli sono più grandi mentre i globuli sono più piccoli(ed in genere
assunti come dose unica, ed infatti vengono chiamate anche dose globuli). Solitamente 20 granuli
sono 1 g, mentre 200 globuli sono 1g di preparazione. La tecnica di preparazione che si usa è la
tecnica dell’impregnazione, cioè i granuli vengono bagnati con la diluizione
richiesta(generalemente 5g di globuli con 32 gocce ovvero 0,5 ml) e vengono poi fatti essiccare.
Questo è un motivo per cui, globuli e granuli, non possono essere toccati con le mani. Nella terapia
omeopatica vengono assunti, per via orale, direttamente dal tubetto e posti sotto la lingua senza
toccarli, perchè sono impregnati superficialmente.
Secondo la farmacopea, le preparazioni omeopatiche sono ottenute da sostanze,prodotti o
preparazioni chiamati”materiale di partenza”, in accordo con un procedimento di produzione
omeopatico. Si nota che la definizione è data dalla materia di partenza e dalla tecnica di
preparazione (al contrario del medicinale che aveva la definizione per funzione e presentazione).
Sulle preparazioni omeopatiche, infatti, non è indicata l'indicazione terapeutica. I materiali di
partenza sono le tinture madri e il macerato glicerico.

Preparazioni nasali
Sempre nell'ambito delle forme farmaceutiche specifiche ci sono le preparazioni nasali , ovvero
preparazioni che servono e che vengono instillate nella cavità nasale e possono essere utilizzate sia
per avere un effetto locale sia per avere un effetto sistemico. Nella definizione di farmacopea, sono
preparazioni liquide,semisolide o solido da somministrare nelle cavità nasali per ottenere un effetto
sistemico o locale. Contengono uno o più principi attivi. Le preparazioni nasali, sono, per quanto
possibile non irritanti e non esercitano alcun effetto indesiderato sulle funzioni della mucosa nasale
e delle sue ciglia. Quindi non devono interagire con le mucose nasali e con la clearance mucociliare.
Le preparazioni per uso nasale, possono essere multidose o a dose unica e, se necessario, devono
essere dispensate insieme a un dispositivo di somministrazione,costituito in modo da evitare
l'introduzione di contaminanti. Si possono distinguere varie categorie di preparazioni nasali:
gocce nasali e spray nasali liquidi:sono quelli di uso comune
polveri nasali
preparazioni semisolide nasali
lavaggi nasali
bastoncini nasali
Nel caso che le preparazioni nasali siano a dose unica è il saggio obbligatorio è l'uniformità di
massa e l'uniformità di contenuto, che si eseguiranno in maniera diversa rispetto a quello per le
forme farmaceutiche solide unidose. In questo caso si pesa il contenuto dei singoli
contenitori,vuotati il più quantitativamente possibile e per determinare la massa media: quindi 10
contenitori si svuotano, si pesa il contenuto(non il contenitore) e si ricava la massa media dei
contenuti. Non più di due delle singole masse possono presentare uno scarto superiore al 10%,
nessuna uno scarto maggiore del 20%. Un saggio simile viene fatto sugli spray nasali, per quelli che
hanno la valvola dosatrice, cioè che per ogni singolo spruzzo erogano una dose fissa di farmaco.
Infatti, per questi spray nasali è previsto il saggio di uniformità di dose erogata.
Le polveri nasali devono rispettare le specifiche(dice la farmacopea) delle polveri per applicazione
cutanea ed inoltre la farmacopea dà indicazione sulle dimensioni delle particelle, cioè devono essere
tali da permettere la loro deposizione nella cavità nasale. La deposizione nella cavità nasali presenta
diverse serie di problemi: innanzitutto la via nasale ha la presenza della clearance mucocigliare,e
quindi l'utilizzo di forme farmaceutiche nasali per somministrazione locali è abbastanza diffuso ma
quello che è più complicato da ottenere è una forma farmaceutica nasale per uso sistemico. La
forma farmaceutica nasale è una forma farmaceutica che sicuramente ha una maggiore accettabilità
del paziente(rispetto all'ago) però c'è anche un fortissimo problema legato alle condizioni di salute,
ovvero mentre l'assorbimento attraverso l'ago garantisce una costanza di dose, la via di
somministrazione nasale no, perchè per esempio,raffreddore,allergia modificano in maniera
sostanziale l'assorbimento del principio attivo. Ci sono molti studi sulle polveri per via inalatoria
per via sistemica ma di fatto sul mercato le uniche forme farmaceutiche per uso nasale sono quelle
per uso topico(per il raffreddore o il sanguinamento del naso,etc.).
Ci sono, tra le preparazioni farmaceutiche specifiche alcune preparazioni galeniche(di gocce nasali)
in cui la più famosa è il protargolo,cioè l'argento proteinato gocce nasali che viene utilizzato come
disinfettante. La farmacopea non dice come è preparato ma dice solo cosa contiene(argento

proteinato in acqua depurata) e per come si prepara si fa riferimento al vecchio formulario;inoltre
dice quale deve essere il contenuto in argento(che è la sostanza che esplica l'azione antisettica e
astringente) e dice quali sono le concentrazioni che si possono preparare, in quanto è un galenico
officinale e quindi tenuto in farmacia, ovvero 0,5 per uso pediatrico, l'1 o 2% e queste sono le
concentrazioni che si possono tenere già preparate in farmacia. Parlando della terapia inalatoria, ci
sono problemi sui dispositivi inalatori. I dispositivi inalatori sono distinti in:
– pressurizzati (pressurized metered-dose inhalers o MDI)
– non pressurizzati o nebulizzatori.
Nell'ambito dei sistemi per la terapia inalatoria, oltre ai sistemi pressurizzati e ai nebulizzatori, ci
sono anche i sistemi erogatori di polveri (PI), perchè i sistemi pressurizzati erogano soluzioni.
Quali sono i parametri che devono essere tenuti presenti nel realizzare un sistema per la
terapia inalatoria:innanzitutto si deve considerare quali sono le modalità con cui una particella,
introdotta nell'albero respiratorio, si muove in questo. Nelle prime vie aeree, in cui c'è un regime di
moto vorticoso, le particelle si muovono per impatto inerziale ovvero vengono spostate dal vortice
di aria respirata, si muovono poi per deposito gravitazionale, e questo dipende dal fatto che si riduce
il diametro delle vie aeree e che si riduce il flusso di aria nelle basse vie aeree ed infine la
diffusione, perchè nella parte terminale dell'albero respiratorio(cioè nei bronchioli e negli alveoli)
c'è un'assenza di flusso di aria e quindi le particelle che riescono ad arrivare in questa parte
dell'albero respiratorio si muovono solo per diffusione.
Quindi riassumendo la cinetica dell’aerosol:
1) impatto iniziale: prime vie aeree che per la loro struttura creano un regime fortemente
vorticoso
2) deposito gravitazionale: per progressiva riduzione di:
- particelle
- flusso
- diametro delle vie aeree
3) diffusione dell'aerosol: per l'assenza di flusso sui bronchioli preterminali e terminali.
Per realizzare una particella che arrivi nelle alte vie respiratorie o sulla basse vie respiratorie
bisogna tenere presente una serie di parametri che sono il diametro aerodinamico della particella e
diametro aerodinamico mediano di massa; sono due parametri diversi ma che devono essere
misurati e valutati nella preparazione di una particella che viene aerosolizzata o che comunque
viene formulata per essere inalata. Il diametro aerodinamico tiene conto della geometria della
particella: attraverso la densità della particella, fa in modo che ogni particella inalata può essere
assimilata ad una sfera. È definito come il diametro di una sfera g/cm3 con la stessa velocità delle
particelle sotto l'azione della forza di gravità in aria calma, nelle stesse condizioni di
temperatura,pressione e umidità relativa. È quindi diametro geometrico della particella per densità
della particella. In questo modo si assimilano tutte le particelle a delle sfere. Il diametro
aerodinamico mediano di massa è la misura del diametro medio del 50% della frazione che si è

aerosolizzato cioè il valore che divide a metà la popolazione delle particelle che devono essere
somministrate. Quindi il diametro aerodinamico è una dimensione della particella(ovvero che
assimila una particella ad una sfera), mentre il diametro aerodinamico mediano di massa dice la
dimensione che divide a metà la popolazione cioè un diametro aerodinamico al di là del quale si
trova il 50% della popolazione delle particelle aerosolizzate.
Un altro parametro,molto importante,che dà un'indicazione sull'omogeneità della dispersione che si
realizza è la deviazione geometrica standard (GSD);differenzia sostanzialmente gli aerosol in due
classi: monodispersi e etero dispersi. I monodispersi classificano gli aerosol che hanno una
deviazione geometrica standard inferiore a 1,22,mentre gli eterodispersi hanno una GDS superiore a
1,22. E' fondamentale nella quasi totalità dei casi,avere un sistema monodisperso perchè garantisce
l'omogeneità delle particelle e quindi garantisce l'uniformità di dose.
Frazione
Di particelle
penetranti
Le particelle possono essere distinte in:
– frazione respirabile;
– frazione toracica;
– frazione inalabile.
La frazione inalabile,che corrisponde alle particelle più grandi, è quella che viene inalata attraverso
il naso e la bocca e quindi si deposita sostanzialmente nel primo tratto dell'albero respiratorio. La
frazione respirabile è la frazione che raggiunge gli alveoli polmonari,ovvero dove avviene lo
scambio gassoso. Quando si realizza una forma farmaceutica inalatoria, i principi attivi che si
possono veicolare sono:
se il target sono le alte vie respiratorie, si realizzano questo tipo di forme
farmaceutiche per quei principi attivi che hanno un'azione

locale(broncodilatatori,glucocorticoidi come antiinfiammatori e sostanze antinfettive).
Se il target sono le basse vie respiratorie, la forma farmaceutica è fatta per avere un
effetto sistemico e possono essere degli anestetici generali con tensioattivi polmonari nei
bambini prematuri, o per la terapia genica.
I sistemi utilizzati per realizzare queste forme farmaceutiche si dividono in pressurizzati e non
pressurizzati. I sistemi pressurizzati sono caratterizzati dal fatto che l'espulsione della forma
farmaceutica dal contenitore dipende all'azione di un gas compresso e liquefatto presente nel
contenitore stesso. Quindi gli spray per uso farmaceutico pressurizzati implicano la presenza di un
gas propellente o compresso o liquefatto. Sono utilizzati per applicazioni topiche e hanno il
vantaggio di avere un'elevata accettabilità da parte del paziente e soprattutto,se si utilizzano per la
protezione delle ferite o come antibatterici il farmaco non è suscettibile alla contaminazione. Gli
spray possono essere formulati anche per applicazioni orali e sublinguali, per applicazioni vaginali
o rettali e per applicazioni nasali. Ci sono anche dei farmaci per uso sistemico, ma che in Italia non
sono commercializzati, per la nicotina e la vasopressina. I componenti delle forme farmaceutiche
pressurizzate sono:
– propellente
– contenitore
– principio attivo ed eccipienti(soluzioni,emulsioni,sospensioni)
– valvola e tasto di erogazione.
Le valvole erogatrici possono essere di due tipi: a) una valvola continua(e vuol dire che il prodotto
viene erogato finchè viene tenuto premuto) e b) le valvole pre-dosate(che comunque alla pressione
segue l'emissione di una dose fissa). Uno dei problemi principali legati alle forme farmaceutiche
spray per uso inalatorio è la non capacità degli utilizzatori nel maneggiare i dispenser. Gli errori
che si fanno più frequentemente nell'uso di sistemi pressurizzati:
– il 54% dimentica di scuotere lo spray prima dell'uso;
– il 66% dimentica di espirare prima di erogare;
– il 58% ha problemi di coordinamento tra l'erogazione e l'aspirazione;
– il 68% inala troppo rapidamente.
Per ovviare a molti degli inconvenienti legati agli erogatori pressurizzati, sono stati realizzati degli
inalatori a polvere secca. Questi inalatori sono attivati direttamente dal respiro del paziente e
quindi non c'è bisogno della coordinazione pressione e respiro, e questo è il vantaggio principale,di
tipo ecologico, perchè non richiede la presenza di gas propellente e quindi meno inquinante ma ha
una serie di inconvenienti. Un inconveniente di tipo tecnologico cioè che non è adatto alle polveri
che sono igroscopiche perchè si aggregano e quindi cambia il diametro aerodinamico mediano di
massa(DAMM), che è il parametro che fa valutare l'altezza del tratto respiratorio in cui si
depositeranno le particelle. Da parte dell'accettabilità del paziente, invece, c'è il fatto che sostanze
che abbiano un odore o un sapore sgradevole restano comunque più in bocca come polvere secca
che lo spray.
Nei sistemi di erogazione delle polveri ci sono delle differenze: in alcuni, il farmaco è presente
come polveri e non dosate e viene dosata dall'apparecchio inalatore al momento dell'utilizzo,

azionando una leva, ma nella maggior parte, invece, di questi inalatori la quantità di polvere da
erogare è predosata(o sottoforma id capsule o in compresse). I nebulizzatori si dividono in:
nebulizzatori a membrana;
nebulizzatori a ultrasuoni;
nebulizzatori a pistone.
I nebulizzatori a membrana (o comunemente aerosol) sono gli strumenti più economici, che però
hanno lo svantaggio di dare un flusso non sufficiente e non costante nel tempo e di avere tempi di
nebulizzazione lunghi. Però riescono a dare una micronizzazione elevata, ovvero diminuendo la
dimensione delle particelle,aumenta la frazione di particelle che arrivano alle vie aeree basse, ma il
flusso non è costante nel tempo e quindi non c'è una garanzia della dose erogata. Viene chiamato a
membrana, perchè la pompa che si attiva per attivare il processo di nebulizzazione è la membrana e
il fluido viene forzato da una pompa nella camera di nebulizzazione.
I nebulizzatori ad ultrasuoni, non fanno rumore, sono rapidissimi in quanto la nebulizzazione dura
al massimo 5-6 minuti(contro la mezz'ora del nebulizzatore a membrana);la nebulizzazione viene
ottenuta sfruttando gli ultrasuoni tramite la vibrazione della testina piezoelettrica. Gli svantaggi
sono dovuti agli ultrasuoni che scaldano la soluzione del farmaco e quindi ci può essere
denaturazione del farmaco o addirittura una denaturazione della struttura molecolare del principio
attivo ed inoltre c'è una difficoltà di pulizia e di sterilizzazione della testina.
I nebulizzatori a pistone in cui la pressione esercitata per nebulizzare è ottenuta da una pompa a
pistone. Anche questi hanno un tempo di erogazione veloce, perchè è di circa 5 minuti e hanno le
dimensioni delle particelle che garantiscono che una larga parte che viene nebulizzata arriva nelle
basse vie aeree(il DAMM con soluzione fisiologica è inferiore a 2 micron e circa il 95% delle
particelle nebulizzate presentano 5 micron).

Tensione superficiale
La tensione superficiale è un fenomeno che considera una fase liquida e l'aria e in particolare i
fenomeni che avvengono all'interfaccia cioè alla superficie di separazione tra due fasi non
omogenee. Quindi la tensione superficiale è un fenomeno interfacciale e quindi avviene
all'interfaccia liquido-aria. Quindi si considera l'interfaccia,cioè la superficie di separazione tra il
liquido e l'aria. Si devono considerare quali sono le forze in gioco che determinano la presenza della
tensione superficiale. Le forze sono:
– forze coesive, si intendono le forze tra molecole uguali;
– forze adesive, le forze che si esercitano tra molecole di fasi diverse.
Se si considera un bicchiere d'acqua, le molecole che si trovano all'interno della massa dell'acqua
saranno soggette solo a forze tra molecole uguali(cioè a forze coesive) che saranno uguali in tutte le
direzioni. Le molecole, invece, che si trovano all'interfaccia sono soggette a forze di coesione,cioè
le molecole uguali e a forze adesive con l'aria che sono, nel caso specifico dell'acqua, molto minori
rispetto alle forze di coesione tra le molecole di acqua
Il risultato delle molecole che sono all'interfaccia è che tendono ad essere tirate verso l'interno;
questo però non è un fenomeno che avviene contemporaneamente allo stesso modo per tutte le
molecole all'interfaccia e quindi il risultato non è una pressione(e quindi non c'è una superficie e
una forza che agisce perpendicolarmente) ma una tensione. Se si considera che una molecola ogni
tot viene tirata verso l'interno il risultato è una contrazione della superficie di contatto per ridurre
il contenuto energetico del sistema.
La tensione superficiale si definisce come la tendenza che un liquido ha a contrarre la sua
superficie messa in contatto con l'aria. La direzione della tensione superficiale è tangente alla
superficie. Più sono alte le forze coesive maggiore sarà la tensione superficiale del liquido: il
liquido che ha la più alta tensione superficiale è il mercurio in quanto ha delle forze coesive di tipo
ionico e quindi sono dei legami estremamente forti. La forma della sfera che assume il liquido ha
un'elevata tensione superficiale è legata al fatto che a parità di volume,la sfera è il solido che ha la
minore superficie specifica, perchè non avendo gli spigoli è la forma che energeticamente ha la
minore superficie di contatto con l'aria(a parità di volume). Le dimensioni fisiche della tensione
superficiale sono definite considerando un'apparecchiatura, il telaio di Duprez; questo telaio è
costituito da 3 fissi e da un lato mobile in cui le dimensioni del lato sono note. Se si pone questo
telaio all'interno di un liquido e poi si estrae il telaio, si formerà un velo di liquido a contatto con
l'aria che si contrae e quindi sposta in basso il lato mobile del telaio.

Per riportare il telaio nella posizione di partenza si deve
applicare una forza. La forza che si deve applicare
dipenderà dalla tensione superficiale (γ) applicata sulla
lunghezza(l) del lato mobile. F = γ ∙ l → γ = F/l = dine/cm
, quindi γ è una forza su uno spostamento.
Se si vuole definire la tensione superficiale in termini
energetici e quindi si vuole considerare il lavoro fatto per
riportare il lato mobile nella condizione di partenza, bisogna
considerare l'area.
L = F ∙ dx → L = γ ∙l ∙ dx → γ = L / l ∙dx = erg/ cm2
anche se ha le dimensioni di una pressione, la tensione superficiale ha una direzione diversa perchè
è tangente alla superficie. Per esempio la tensione superficiale di varie sostanze è:
– Hg 476 dine/cm
– H2O 72,80 dine/cm
– etere etilico(sostanza volatile e quindi le forze di coesione sono estremamente basse) è 17,01
dine/cm.
La tensione interfacciale è la tensione che si genera all'interfaccia tra 2 liquidi diversi e immiscibili
mentre la tensione superficiale è la tensione liquido-aria e quindi la tendenza che il liquido ha a
contrarre la superficie di contatto con l'aria. Inoltre la tensione superficiale è dipendente dalla
temperatura, perchè all'aumentare della temperature aumenta l'energia cinetica delle molecole e
quindi le forze di coesione diminuiscono e quindi la tensione superficiale diminuisce.
Il concetto di tensione superficiale è legato a due caratteristiche: la bagnabilità e la spandibilità.
La bagnabilità si definisce come la tendenza di un solido ad essere bagnato da un liquido,e quindi
la bagnabilità è una caratteristica del solido. La bagnabilità si indica numericamente attraverso un
parametro, l' angolo di contatto (θ). Se si considerano le due situazioni estreme di bagnabilità
massima:
Il solido, quando è completamente bagnabile sarà coperto da un velo di liquido ed in questo caso θ
= 0. La condizione invece del solido che non è affatto bagnato dal liquido; si considera che un
solido non è bagnato dal liquido per un valore di θ > 90°. Come si misura l'angolo di contatto: se
si considera un solido ,si fa cadere su questo una goccia del liquido che si vuole bagnare. La goccia
si allargherà sul solido fino all'equilibrio

All'equilibrio, il punto triplo, perchè sono presenti tutte le fasi coinvolte (solido, aria e liquido) è
all'equilibrio. Nella rappresentazione grafica ce ne sono due ma nella realtà ce ne sono infiniti. Il
punto triplo è all'equilibrio quando le tensioni che agiscono su questo punto sono all'equilibrio:
quindi si considera la tensione superficiale del liquido ( γl ), che è tangente alla superficie, la
tensione interfacciale solido-liquido( γs/l ) e la tensione superficiale del solido(impropriamente
definita così perchè in realtà si chiama tensione superficiale critica e non è un valore reale, ovvero
non è un valore che descrive un fenomeno reale, perchè un solido a contatto con l'aria non contrae
la superficie). L'angolo di contatto è quello che si forma quando la goccia incontra il solido. Quindi,
il punto è all'equilibrio e le tensioni che agiscono su questo punto sono all'equilibrio. Poiché le
tensioni superficiale sono delle grandezze vettoriali, e quindi hanno un modulo, un verso e una
direzione, si può dire che γs e γs/l hanno una direzione uguale perchè agiscono sullo stesso piano
ma hanno il verso opposto, γl ha una direzione diversa,rispetto agli altri due vettori, perchè agisce
su un piano diverso. Per lavorare quindi con queste grandezze si deve scomporre il vettore γl nelle
due componenti che sono: γl sen θ e γl cos θ.
La componente γl sen θ della tensione superficiale è quella che cerca di alzare la goccia dal piano
del tavolo ed è,innanzitutto, vincolata dal tavolo e fondamentalmente bilanciata dalla forza peso e
quindi si può considerare nulla o meglio che non ha azione sull'equilibrio del punto. Il punto è
all'equilibrio quando:
γs = γs/l + γl cos θ → ovvero la componente di γl che agisce sullo stesso piano
La bagnabilità si esprime come cos θ.

Quindi la bagnabilità espressa come cos θ ( θ è l'angolo di contatto) dipende dalle tensioni che
agiscono nel sistema e per avere la bagnabilità massima, ovvero con θ = 0°. come bisogna operare
sull'equazione:bisogna tendere a un valore di coseno 1, che è il valore massimo che il coseno può
raggiungere. Quindi per aumentare la bagnabilità bisogna cercare di far arrivare questa equazione al
valore massimo.
γs è un numero ma non descrive un fenomeno, ma si può abbassare γl e abbassandola si abbassa
anche la tensione interfacciale solido-liquido e quindi γs/l aumenta: questo perchè abbassando
l'interfaccia solido-liquido e se al valore di γs, che è fisso, si sottrae un numero più piccolo aumenta.
Un buon valore di bagnabilità è indicato con θ intorno a 30°(il solido,quindi è bagnato bene).
La spandibilità , invece, riguarda un sistema costituito da due liquidi non miscibili tra loro; quindi
la bagnabilità è la proprietà di un solido di essere bagnata da un liquido, la spandibilità è la capacità
di un liquido di spandere su un altro liquido con il quale è miscibile. Se si considera un sistema
costituito da un parallelepipedo si olio con aria superficiale unitaria (1cm2)
Se si fa a metà, si distinguono due cubetti di olio e quindi generando, rispetto alla condizione
iniziale, due nuove superfici di contatto olio/aria. Se si deve considerare il lavoro fatto contro le
forze di coesione per separare i 2 cubetti olio bisognerà considerare la differenza dal punto di vista
energetico tra lo stato finale e quello iniziale. La tensione superficiale sarà:
γ = L / S dove il L = S ∙ γ
In questo caso la superficie è unitaria e quindi il lavoro che si fa sarà dato da
Lc = EII – EI = 2 γo – O → in questa condizione non si ha un contributo energetico poiché le
molecole sono in contatto solo tra loro. Se suppongo, invece, di avere un parallelepipedo costituito
da olio e acqua con superficie sempre unitaria e si separa anche questa in 2 cubetti

Il lavoro contro le forze di adesione sarà(La)
La = EII – EI = ( γa + γo) – γa/o
γa/o è la condizione di partenza ed in questo caso c'è la tensione perchè c'è una superficie di
contatto tra due liquidi non miscibili tra loro. Il COEFFICIENTE DI SPANDIBILITA'(S) sarà:
S = La – Lc dove La = lavoro contro le forze di adesione e
Lb = lavoro contro le forze di coesione
S = |( γa + γo) - γa/o | - 2 γo = γa – (γo + γa/o)
Bisogna però tendere a θ = 0 e quindi a cos θ = 1 e quindi la misura della bagnabilità è legata al
valore numerico dell'angolo. Come si fa a capire, avendo due liquidi immiscibili tra loro se è l'H2O
che spande sull'olio o è l'olio che spande sull'H2O? Si consideri la rappresentazione grafica del
fenomeno con una goccia di olio posta sull'acqua.
Quando la goccia di olio spande sull'acqua? Quando la somma γo + γa/o < γa, perchè γa trascina
la goccia di olio e fa in modo che spanda. Per valutare quale liquido spande sull'altro si deve partire
da un'ipotesi che si deve verificare presupponendo ciò: si dice che γa > γo + γa/o e quindi si
inseriscono i numeri. Se la relazione è confermata allora l'olio spande sull'acqua, se ciò non è vero è
l'acqua che spande sull'olio.
Metodo di Zisman: La tensione superficiale del solido non descrive un fenomeno fisico reale ma è
un valore che si determina per estrapolazione. Si costruisce un grafico( e si chiama metodo di
Zisman) in cui si bagna un solido con liquidi diversi, con γl diversi, bagneranno con angolo di
contatto diverso il solido che si considera e così si ottiene una retta. Γs si ottiene per estrapolazione
quando θ = 0, cioè al valore di bagnabilità massima in uni γl = γs.
Questi sono dei valori di bagnabilità di alcune sostanze di natura plastica.

γs
Nylon 46
Pralon 43
PVC 39
PVA 37
Polistirene 33
Politene 31
Teflon 18,5
γs diminuisce al diminuire della polarità dei solidi, dove i solidi più polari sono facilmente più
bagnabili. Questi invece sono dei valori di tensione superficiale interfacciale di alcune fasi oleose
ed è un'indicazione numerica del coefficiente di spandibilità di due sistemi.
γo/a γo γH2O = 72,8
Decano 54,3 23,9
Benzene 35 28,9
Acido oleico 15,1 32,2
Esano 51 18,4
Nel sistema decano/H2O: 72,8 -(23,9 + 52,3) = -3,4 è l'H2O che spande sul decano. Invece nel
sistema acido oleico/H2O: 72,8 – (32,9 + 15,1) = 24,7 ed è quindi l'olio che spande sull'H2O. Un
esempio di come si usano questi parametri in tecnologia, sono per esempio negli angoli di contatto
tra alcuni principi attivi nelle loro rispettive soluzioni sature, perchè la bagnabilità del principio
attivo nella sua soluzione satura influenzerà la velocità di dissoluzione di quel principio attivo.
Equazione di Gibbs
Uno dei parametri che agiscono sulla tensione superficiale e che la modifica è l'effetto del soluto
sulla tensione superficiale del liquido. L'effetto del soluto sulla tensione superficiale è definito
dall'equazione di Gibbs, che misura l'eccesso superficiale di concentrazione.
l'attività di soluzioni molto diluite si approssima alleconcentrazioni.
Γ è la differenza di concentrazioni del soluto tra la superficie e la massa interna del liquido. Ci
possono essere 3 situazioni:
1. se ΔC = 0 → dγ = 0; per aggiunta di soluto non si ha una variazione di tensione superficiale
(γ). Quindi il soluto si distribuisce nella soluzione e la concentrazione all'interfaccia è uguale
concentrazione della massa;
2. se ΔC > 0 → dγ è negativo, per aggiunta di un soluto diminuisce la tensione superficiale(γ);
quindi la concentrazione all'interfaccia è maggiore della concentrazione della massa cioè il
solido si distribuisce preferibilmente all'interfaccia;
3. se ΔC < 0 → dγ è positivo, quindi l'aggiunta di soluto aumenta la tensione superficiale(γ) e
la concentrazione all'interfaccia è minore della concentrazione della massa,quindi il soluto si
distribuisce preferibilmente nella massa del liquido.

Se si riporta in un grafico la variazione di tensione superficiale, in questo caso dell'H2O in funzione
della variazione di concentrazione di soluto, si avrà che per alcune sostante, come NaCl e glucosio,
si distribuiscono preferenzialmente nella massa avendo un ΔC < 0 e quindi per aggiunte successive
si avrà un incremento di γ. Per altre sostanze, come l'etanolo, si distribuisce preferenzialmente
all'interfaccia in quanto ha un ΔC > 0 e quindi un abbassamento della γ. Ci sono, inoltre, delle
sostanze (tensioattivi), che si dispongono solo all'interfaccia (ΔC >>0) e quindi si otterrà un
decremento notevole della tensione superficiale, ovviamente fino ad un valore di concentrazione
che è la concentrazione micellare critica.
I metodi per determinare la tensione superficiale sono:
a) metodo del piatto di Wilhelmy
b) metodo dell'anello (tensiometro di Nouy)
c) metodo del capillare
d) metodo dello stalagmometro
a) metodo del piatto di Wilhelmy
un piatto di platino viene immerso nel liquido di cui si vuole
misurare la tensione superficiale. Il piatto è legato ad una
bilancia che misura la forza necessaria a staccare il piatto subito
prima del distacco, quindi misura la forza che il liquido oppone
all'interfaccia al distacco del piatto; cioè misura le forze che

trattengono all'interfaccia il piatto. Questa forza sarà data dal valore letto sulla bilancia prima del
distacco meno il peso del piatto, e la forza è uguale alla tensione superficiale per l, cioè la lunghezza
su cui agisce la tensione superficiale. Quindi la tensione superficiale sarà legata alla lunghezza del
piatto e allo spessore del piatto, ovviamente in questo caso moltiplicato per due, perchè lo spessore
del piatto non è infinitesimo( il piatto ha 2 dimensioni e per quanto di lavori per avere una
dimensione unica); quindi sarà valutato il distacco della prima e della seconda faccia ma per quanto
lo spessore sia minimo, non si può avere un piatto in una dimesione.
Wl – W = 2 (L + T) γ
dove: Wl = valore letto sulla bilancia subito prima del distacco, W = peso del piatto, L = lunghezza
del piatto, T = spessore del piatto e quindi la forza di distacco è uguale a γ della superficie staccata.
Sullo stesso principio si basa b) il tensiometro di Novy (metodo dell'anello)
Si ha un anello di platino e si misura la forza di distacco di questo anello, nel caso della tensione
superficiale all'interfaccia liquido-aria, nel caso della misura interfacciale il distacco dall'interfaccia
fase idrofila - fase lipofila. La forza di distacco sarà data da γ ∙ l, in cui l in questo caso sarà data
dalla differenza tra raggio interno e raggio esterno, nell'altro caso si aveva la dimensione intera
perchè il piatto è pieno mentre questo invece è un anello, quindi il piatto è una superficie che si
distacca in questo caso forma un velo di liquido all'interno dell'anello(che è cavo); quindi la
superficie del liquido sarà data da 2π ( R1 + R2). Si deve calcolare la differenza dei due raggi
perchè non si può fare un anello di spessore
infinitesimale
il metodo misura la forza di distacco di un anello di
platino da una superficie o da un'interfaccia.
Forza di distacco = γ ∙ 2π ( R1 + R2) , dove R1 e
R2 sono il raggio interno e il raggio esterno e 2π ( R1
+ R2) è il perimetro del liquido distaccato.
c) il metodo del capillare
è legato al fatto che se si immerge un capillare in un liquido, il liquido sale
all'interno del capillare fino a quando la forza legata alla tensione
superficiale del liquido è bilanciata dalla forza peso, quindi il liquido salirà
all'interno del capillare fino ad un'altezza che sarà legata all'equilibrio alla
forza peso e la forza legata alla tensione superficiale data da γ ∙ L, dove L =
2πr (perimetro del capillare)
m ∙ g = γ 2πr

poiché si deve arrivare a grandezze utilizzabili, si moltiplica e si divide per il volume della
colonnina di liquido.
questo è vero quando la parete del capillare è perfettamente bagnata dal liquido; quindi quando si ha
un menisco concavo perfettamente emisferico vuol dire che il liquido sale all'interno del capillare
in base a una forza legata solo alla tensione superficiale non è limitata, o comunque condizionata
dalla bagnabilità del vetro da parte del liquido di cui si vuole misurare la tensione superficiale. Vale
solo quando la bagnabilità del capillare da parte del liquido è completa, quindi quando la
bagnabilità è massima,cioè quando l'angolo di contatto θ = 0, quando l'unica entità che agisce su
questa forza è la tensione superficiale del liquido perchè si implica che il liquido bagni
completamente il capillare. Nelle condizioni iniziali il menisco non è perfettamente emisferico,
quindi il menisco si forma per la tensione superficiale. Il menisco è il liquido che sale all'interno,
minori sono le dimensioni e più si percepisce(infatti in un bicchiere d'acqua non si percepisce).
Nelle condizioni reali, il menisco non è perfettamente emisferico e quindi si deve tenere conto della
bagnabilità del vetro da parte del liquido in esame e quindi anche in questo caso dell'angolo di
contatto e si devono considerare le 2 componenti:
ovviamente γsinθ è vincolata dalla parete quindi non può
tirare il liquido all'esterno del capillare → l'unica componente
da considerare è γcosθ. γ cosθ è quella che agisce sulla forza
ascenzionale, che consente al liquido di salire all'interno del
capillare ed in questo all'equilibrio si deve raggiungere cosθ.
quindi quando la bagnabilità è massima, cioè θ = 0 e cosθ = 1 quasto non compare nell'equazione e
quindi non c'è l'effetto della bagnabilità(cosθ è il parametro legato alla bagnabilità). Nel caso del
mercurio(che ha come valore di tensione superficiale 400 cm/dine) se si pone al suo interno un
capillare,il liquido non sale all'interno del capillare e dà un menisco convesso al di sotto del
normale.
c)metodo dello stalagmometro
si sfrutta l'utilizzo di un contagocce e viene misurata la tensione superficiale utilizzando un
contagocce basandosi sul fatto che la goccia si staccherà dal contagocce quando la forza che la tiene

attaccata al contagocce sarà bilanciata dalla forza peso, la forza che la tiene legata al contagocce è
una forza legata alla tensione superficiale F = γ ∙ L → L = 2πr (circonferenza)
γ2πrφ = mg è la forza che tiene attaccata la goccia al contagocce(legata alla tensione superficiale)
si aggiunge φ che è un fattore di correzione, che tiene conto del fatto che il bordo del contagocce
possa presentare delle irregolarità quindi che il vetro non sia perfettamente integro. M è la massa
della goccia, V è il volume della goccia, d è la densità del liquido, r è il raggio del capillare e g è
l'accelerazione di gravità. Con il metodo dello stalagmometro si fa una misura relativa, ovvero si fa
una misura di comparazione tra il liquido in esame e l'acqua cioè si raccolgono un numero definito
di gocce dell'acqua e del liquido in esame (incognito), si pesano queste gocce e si ottiene il rapporto
tra le tensioni superficiali, dove la tensione superficiale dell'acqua è nota ad una temperatura, così
come il rapporto tra i pesi delle gocce e si possono ricavare i valori di tensioni superficiali
incognite.
γH2O = m1g/2πr m1 e m2 hanno il peso medio di una gtt;
γx = m2g/ 2πr
γH2O/γx = m1/m2 → γx = γH2O m2/m1
γ 2πr = p
20 gtt di H2O 1g più H2O c'è minore è il numero di gocce
34 gtt di alcool 50° 1g perchè maggiore è il contenuto di H2O
55 gtt si alcool 70° 1g quindi maggiore è la tensione superficiale
64 gtt di alcool 90° 1g più grande è la goccia.
62 gtt di alcool assoluto 1g
Il contagocce da FU viene misurato mettendo all'interno 1g di acqua (si devono contare 20 gtt).
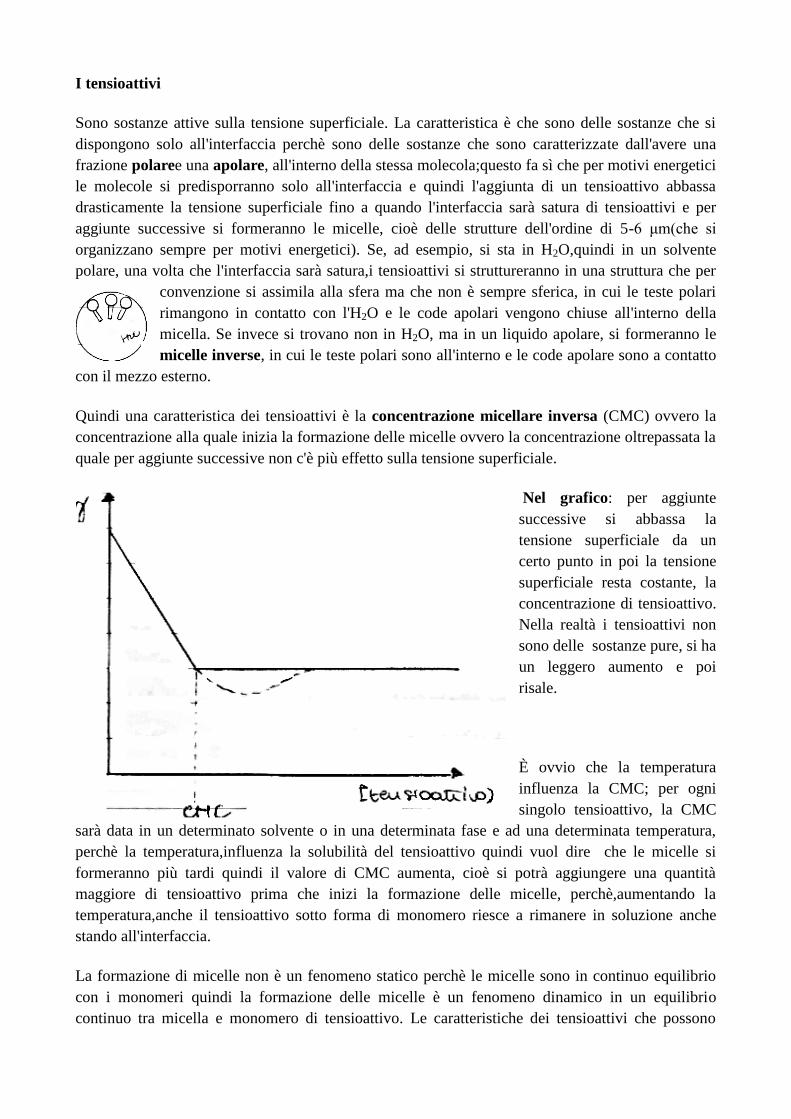
I tensioattivi
Sono sostanze attive sulla tensione superficiale. La caratteristica è che sono delle sostanze che si
dispongono solo all'interfaccia perchè sono delle sostanze che sono caratterizzate dall'avere una
frazione polaree una apolare, all'interno della stessa molecola;questo fa sì che per motivi energetici
le molecole si predisporranno solo all'interfaccia e quindi l'aggiunta di un tensioattivo abbassa
drasticamente la tensione superficiale fino a quando l'interfaccia sarà satura di tensioattivi e per
aggiunte successive si formeranno le micelle, cioè delle strutture dell'ordine di 5-6 μm(che si
organizzano sempre per motivi energetici). Se, ad esempio, si sta in H2O,quindi in un solvente
polare, una volta che l'interfaccia sarà satura,i tensioattivi si struttureranno in una struttura che per
convenzione si assimila alla sfera ma che non è sempre sferica, in cui le teste polari
rimangono in contatto con l'H2O e le code apolari vengono chiuse all'interno della
micella. Se invece si trovano non in H2O, ma in un liquido apolare, si formeranno le
micelle inverse, in cui le teste polari sono all'interno e le code apolare sono a contatto
con il mezzo esterno.
Quindi una caratteristica dei tensioattivi è la concentrazione micellare inversa (CMC) ovvero la
concentrazione alla quale inizia la formazione delle micelle ovvero la concentrazione oltrepassata la
quale per aggiunte successive non c'è più effetto sulla tensione superficiale.
Nel grafico: per aggiunte
successive si abbassa la
tensione superficiale da un
certo punto in poi la tensione
superficiale resta costante, la
concentrazione di tensioattivo.
Nella realtà i tensioattivi non
sono delle sostanze pure, si ha
un leggero aumento e poi
risale.
È ovvio che la temperatura
influenza la CMC; per ogni
singolo tensioattivo, la CMC
sarà data in un determinato solvente o in una determinata fase e ad una determinata temperatura,
perchè la temperatura,influenza la solubilità del tensioattivo quindi vuol dire che le micelle si
formeranno più tardi quindi il valore di CMC aumenta, cioè si potrà aggiungere una quantità
maggiore di tensioattivo prima che inizi la formazione delle micelle, perchè,aumentando la
temperatura,anche il tensioattivo sotto forma di monomero riesce a rimanere in soluzione anche
stando all'interfaccia.
La formazione di micelle non è un fenomeno statico perchè le micelle sono in continuo equilibrio
con i monomeri quindi la formazione delle micelle è un fenomeno dinamico in un equilibrio
continuo tra micella e monomero di tensioattivo. Le caratteristiche dei tensioattivi che possono

influenzare la concentrazione micellare critica sono:
– temperatura;
– dipende dalla fase liquida che si considera, quindi un tensioattivo polare(cioè con una
maggiore frazione lipofila) in acqua ci si mette più tensioattivo e quindi si avrà una CMC
alta perchè il tensioattivo è polare in una fase polare e dal punto di vista energetico, anche la
presenza del monomero nella massa, non sarà così energeticamente sfavorita;viceversa un
tensioattivo polare in fase lipofila avrà una bassissima CMC, perchè non appena si satura
l'interfaccia il tensioattivo non può esistere sotto forma di monomero.
La formazione delle micelle inizia quando si satura l'interfaccia liquido-aria o liquido-liquido se le
due fasi sono immiscibili, quindi quale altro parametro può influenzare la CMC? Lo spazio e che
occupa la molecola di tensioattivo quindi l'area superficiale della testa
polare e il volume della molecola di tensioattivo cioè si considera un
parametro che tiene conto dell'area superficiale della testa;si assimila la
molecola di tensioattivo ad un cono e si considera un parametro detto
parametro critico di impacchettamento (IPP) che considera il rapporto tra
l'area della testa polare ed il volume occupato dalla molecola di
tensioattivo. In base al valore che assume questo parametro critico
d'impaccamento si formeranno strutture micellari piuttosto che altre
strutture e questo parametro influenza anche la tensione superficiale perchè influenza il numero di
molecole di tensioattivo all'interfaccia;maggiore è lo spazio che occupano,minore sarà il numero di
molecole interessate. I tensioattivi possono essere classificati in base a varie caratteristiche. La
prima classificazione è in base alla natura chimica dei tensioattivi ovvero: ionici e non ionici.
La differenza tra questi 2 classi è che i tensioattivi ionici presentano sulla frazione polare una carica
netta, costituita da una testa polare e una coda apolare; quindi saranno distinti in:
– tensioattivi cationici quando la carica netta sulla testa è positiva;
– tensioattivi anionici quando la carica netta sulla testa è negativa.
I tensioattivi anionici sono i cosiddetti saponi, tutti i detergenti sono a base di tensioattivi anionici;
in genere i saponi veri e propri sono sali di acidi grassi a lunga catena oppure sono esteri di acidi
alchensolfonici. Un esempio di tensioattivo anionico è il sodio laurilsolfato o il sodio
dodecilsolfato, perchè l'acido grasso è l'acido laurico che è un acido grasso a 12 atomi di carbonio.
L'utilizzo di questi nei saponi e nei detersivi è molto dibattuto per la tossicità e la cancerogenicità;
infatti sono poco utilizzati in campo tecnologico farmaceutico ma usati in campo cosmetico ma
sono comunque sostanze fortemente inquinanti
I tensioattivi cationici sono i disinfettanti come per esempio la citrimmidee il cloruro di
acetilpiridinio che si utilizza per esempio nelle pastiglie come antisettico del cavo orale. La carica
positiva è conferita da un N quaternario quindi anche sulla tossicità di queste sostanze c'è un ampio
dibattito. Quelli più usati in tecnologia farmaceutica sono i tensioattivi non ionici e non hanno una
carica netta sulla testa polare. Su questi tensioattivi non devono essere valutate le incompatibilità di
tipo elettrostatico(un tensioattivo cationico, in presenza di una sostanza carica positivamente può
dare interazioni); quindi partendo dal presupposto che il tensioattivo deve essere inerte nella quasi

totalità delle preparazioni per uso farmaceutico, i tensioattivi usati sono i tensioattivi non ionici. I
tensioattivi non ionici si dividono in:
• esteri
• eteri
• esteri - eteri
• anfoliti
gli esteri sono quelli più rappresentativi
sono gli SPAN, la cui base è l'anello del
SORBITOLO, un polialcol a 5 atomi di
carbonio. Gli SPAN sono caratterizzati da
un numero che è l'indicazione dell'acido
grasso con cui questi tensioattivi sono
esterificati. Il numero più basso è il 20
(SPAN20) ; quindi SPAN20 dice la
composizione chimica, cioè l'anello di sorbitolo esterificato con l'acido laurico cioè l'acido grasso a
12 atomi di carbonio. Lo SPAN80 è l'acido oleico, che a differenza degli altri presenta una
insaturazione(un doppio legame). Il sucrodets ha come poliolo il saccarosio con una o due radici di
acido grasso.
Gli eteri sono i brij, pluronic e polixaleni.. Sono alchilossialcol, quindi alcol a lunga catena
esterificati con polietilglicol o con polipropilen glicol. Non ci sono acidi grassi e la frazione lipofila
sono gli alcoli a lunga catena esterificati con PAG O PEG.
Gli eteri – esteri sono i TWEEN chiamati anche polisorbati e la composione chimica è simile a
quella degli SPAN: si parte sempre dall'anello di sorbitolo e l'ossidrile primario(come negli span) è
esterificato con gli acidi grassi mentre gli ossidrili secondari sono eterificati con polietilenglicol. In
questa classe di tensioattivi cambia l'acido grasso e il numero di molecole di PEG presenti sulla
molecola. Per esempio il TWEEN20 l'acido grasso è l'acido laurico, cioè a 20 atomi di carbonio e la
somma delle unità di polietilene glicol che formano gli eteri è 20,
quindi ci sono 20 unità di polietilene glicol che eterificano gli
ossidrili secondari.
Nel TWEEN21 l'acido grasso è sempre l'acido laurico ma le
catene di PEG sono 4:quindi nel caso dei TWEEN, il numero non
indica solo l'acido grasso ma la variazione dei numeri darà
un'indicazione anche del numero di PEG presenti sulla molecola.
All'interno dei tensioattivi non ionici si trovano anche i tensioattivi come le lecitine, acidi colici e
aminoacidi, che si chiamano anfoliti, perchè a seconda del pH dell'ambiente avranno una carica
positiva o negativa sulla testa polare ma al loro pH isoelettrico non presentano una carica netta.
I tensioattivi possono essere classificati: o per fuzione o in base al valore di HLB. L'HLB è il
bilancio idrofilo lipofilo; questo concetto è stato introdotto nel '49 da Griffin ed è un numero che
indica il rapporto tra la frazione idrofila e la frazione lipofila della molecola e quindi è un numero
esterificato con
acido grasso

da 0 a 20 e dà un'indicazione dell'affinità del tensioattivo per la fase idrofila o per la fase lipofila.
Quindi i tensioattivi con
1 < HLB < 10 saranno lipofili e 10< HLB < 20 saranno idrofili
Griffin, nel '49, aveva creato il concetto di HLB solo per gli SPAN e misurava delle caratteristiche
dei grassi; poi ha introdotto delle modificazioni anche per i TWEEN e per gli altri tensioattivi, dove
però, avendo una carica netta sulla testa polare preferiscono ovviamente la fase idrofila e quindi i
tensioattivi ionici hanno un valore di HLB che si calcola con il metodo di Davies(che è un metodo
numerico perchè ha dato per convenzione un numero ad ogni gruppo chimico che costituisce le
molecole e dalla somma di questi numeri si calcola l'HLB).

I sistemi dispersi sono dei sistemi tendenzialmente stabili, dal punto di vista termodinamico, e
quindi presentano maggiore difficoltà dal punto di vista formulativo. Possono essere classificati in
base alle dimensioni della fase dispersa;cioè si hanno 2 o più fasi che non sono miscibili tra loro in
cui una delle due fasi è dispersa finemente nell'altra.
Dispersioni molecolari < 1 nm
dispersioni colloidali(pseudosoluzioni) da 1 a 0,5 μm
dispersioni grossolane > 0,5 μm
Si ha una fase finemente dispersa quando le particelle della fase dispersa vanno da 0,5 μm a 1 nm
quindi hanno delle dimensioni comprese tra il micron e il nanometro. Le dispersioni con un
diametro inferiore sono le dispersioni vere, dispersioni con delle particelle di diametro maggiore si
chiamano dispersioni grossolane,cioè costituite da particelle visibili al microscopio ottico, e i
sistemi dispersi per uso farmaceutico fanno parte di questa classe. Quindi la prima classificazione è
in base alle dimensioni della fase dispersa;un'altra classificazione può essere fatto in base alla fase
dispersa e alla fase disperdente. Ci sono possibili combinazioni:
Fase disperdente Fase dispersa Tipo
Gas Liquido
solido
aerosol(nebbia)
aerosol(fumo)
Liquido Gas
liquido
solido
Schiuma
emulsione
sospensione
Solido Gas
liquido
solido
Schiuma solida
emulsione solida
sospensione solida
Le emulsioni sono un sistema disperso costituito da due fasi liquide non miscibili tra loro, una delle
quali è dispersa finemente nell'altra. Le due fasi si chiamano in un sistema semplice:
– emulsione acqua in olio A/O
– emulsione olio in acqua O/A

Le fasi dell'emulsione si chiamano:
– fase esterna o disperdente o continua;
– fase (del liquido disperso) interna o dispersa o discontinua
I metodi per stabilire che tipo di emulsione abbiamo: il primo metodo è la misura della
conduvibilità elettrica, in cui la conducibilità della fase acquosa sarà più alta della conducibilità
della fase oleosa e quindi se la conducibilità dell'emulsione è elevata, la fase esterna sarà
l'acqua(viceversa sarà l'olio). Si possono usare dei coloranti o delle sostanze fluorescenti: se si
aggiunge all'emulsione un colorante idrofilo e l'emulsione è tutta colorata, la fase esterna sarà
l'acqua perchè il colorante sarà solubilizzato nella fase esterna e viceversa se la colorazione si
osserva a puntini, l'acqua sarà la fase interna( e lo stesso vale per le sostanze fluorescenti). Gli
esempi che si possono avere di emulsione in natura sono il latte e il burro,dove infatti la parola
emulsione viene dal latino che vuol dire “mungere”: il latte è un emulsione olio in acqua e il burro è
un emulsione acqua in olio. Non bisogna immaginare l'emulsione come una fase liquida, nonostante
le due fasi siano due fasi liquide, la consistenza dell'emulsione può essere anche semisolida o
solida(come il burro). Un altro metodo per capire che emulsione si sta trattando è la tecnica della
diluizione; se si prede un po' di latte e si aggiunge acqua il latte si diluisce,quindi la fase esterna è
l'acqua, invece nel burro, se si aggiunge acqua, rimane l'olio come fase esterna.
Quelle di uso più comune sono le macroemulsioni in cui il diametro della fase dispersa è superiore
a 1000 nm; in queste c'è una stabilità”cinetica”. Le miniemulsioni hanno un diametro tra 100 e
1000 nm e hanno più stabilità termodinamica; le microemulsioni sono micelle e c'è stabilità
termodinamica. Le emulsioni sono dei sistemi termodinamicamente instabili e quindi si può agire
sulla stabilità cinetica cioè si può rallentare il processo di degradazione del sistema disperso:ciò può
essere fatto diminuendo la dimensione della fase dispersa fino ad arrivare, nel caso delle
microemulsioni, alla stabilità termodinamica.
Perchè le emulsioni sono dei sistemi termodinamicamente instabili? Perchè sul
sistema disperso è estremamente elevata l'energia libera di superficie, cioè si deve
tenere in contatto, su una superficie estremamente elevata, 2 fasi immiscibili tra loro e
quindi l'energia che c'è alla superficie di contatto è elevata ed è legata alla tensione
interfacciale olio/acqua. Se si considera di avere due cubetti di olio e di acqua di
spigolo 1 cm e quindi 1 cm3 di H2O e 1 cm3 di olio, la superficie di contatto tra i due cubetti è 1
cm2. Invece, ottenendo un sistema disperso assimilando le goccioline disperse a delle sfere con un
diametro di 0,1 cm la superficie di contatto tra le due fasi diventa di 60 m2. Quindi un parametro su
cui bisogna agire per rendere stabile un sistema disperso(e in particolare le emulsioni) sarà la
tensione interfacciale olio/acqua quindi uno degli agenti che stabilizzerà le emulsioni saranno i
tensioattivi, cioè delle sostanze capaci di agire sull'energia libera di superficie,perchè l'intento è di
mantenere elevato il ΔS perchè altrimenti abbassare l'energia libera di superficie(dopo aver agitato e
disperso una fase nell'altra) quella tende a ridurre la fase di contatto, cioè appunto l'energia legata
alla superficie di contatto.
Poiché l'energia di superficie di contatto è molto elevata, il sistema tenderà a ridurre al minimo la

superficie di contatto; quindi le goccioline della fase dispersa tenderanno a riunirsi andando a
ricostruire la fase disperdente, quindi i tensioattivi agiscono sulla fase esterna diminuendo la
tensione superficiale, così da far rimanere le goccioline.
Energia libera di superficie:
W(erg/cm2) = γo/a ∙ ΔS
Agendo sulla tensione interfacciale diminuisce W.
I criteri che fanno formare un'emulsione rispetto e un'altra(cioè se si ha una fase acquosa e una
oleosa come si fa predire che si formerà un'emulsione A/O o O/A): la prima regola è il volume di
fase,cioè generalmente la fase presente in volume maggiore sarà la fase esterna. La seconda regola
è quella della viscosità di fase, generalmente la fase che presenta viscosità maggiore è la fase
esterna dell'emulsione. Un altro parametro è quello che si definisce regola di Bancroft o del cuneo
orientato,che dice che la fase per cui il tensioattivo è più affine sarà la fase esterna dell'emulsione.
Perchè? La fase dispersa deve rimanere come goccioline disperse, in teoria la tensione superficiale
della fase dispersa dovrebbe aumentare ovvero si dovrebbe conservare la forma di goccioline(come
il Hg messo in contatto con l'aria); allora la fase per cui il tensioattivo è più affine diventa quella
esterna perchè la fase di cui si deve abbassare la tensione superficiale per far sì che la fase interna
rimanga dispersa nella fase esterna.
La fase esterna si deve deformare per ospitare le goccioline di fase dispersa quindi chi deve
diminuire la tensione superficiale è la fase esterna perchè più si abbassa la tensione superficiale
della fase esterna più la fase esterna sarà in grado, dal punto di vista energetico, di contrastare il
fatto che le goccioline di fase interna tendono a ricombinarsi e a separarsi perchè nell'equazione(
quella di W) il valore energetico è legato al fatto che l'incremento di superficie è altissimo e quindi
per abbassare l'energia del sistema, il sistema tende a ridurre al minimo la superficie di contatto;
quindi le goccioline di fase disperse, lente ma inesorabili, tendono a rincontrarsi man mano,
aumentano le dimensioni delle goccioline di fase dispersa con un sistema detto di coalescenza e poi
c'è la separazione dell'emulsione cioè le due fasi distinte in cui la superficie di contatto è minima.
C'è bisogno di valutare, nella fase preformulativa, l'instabilità del sistema e poi bisognerà scegliere i
tensioattivi da aggiungere perchè generalmente non si usa un solo tensioattivo ma si usa una
miscela di tensioattivi e si dovrà aggiungere un agente viscosizzante, perchè si può agire sulla
viscosità della fase esterna dell'emulsione.
legge di Stockes
La velocità di sedimentazione è proporzionale al quadrato del diametro delle particelle, alla
differenza delle densità tra la fase interna e la fase esterna, all'accelerazione di gravità ed è
inversamente proporzionale alla viscosità della fase esterna. Se si riduce questa velocità, si
stabilizza il sistema disperso, perchè rallentando il movimento delle particelle di fase interna si
rallenta la possibilità che si incontrino e quindi la possibilità che possano formare una fase
completamente separata dalla fase esterna.

Nelle emulsioni si parla di creaming perchè può capitare che la fase esterna abbia una densità
maggiore della fase interna e quindi si ha il segno negativo a V,quindi non si parla di velocità di
sedimentazione ma di velocità di affioramento. Un esempio di creaming è la panna nel latte in cui
la parte grassa affiora.
Nelle emulsioni gli agenti viscosizzanti usati come stabilizzanti delle emulsioni sono i colloidi
idrofili e generalmente si usano le gomme o i derivati della cellulosa, ovviamente se l'emulsione è
un' emulsione O/A perchè bisogna aumentare la viscosità della fase esterna. Se la fase esterna è
l'olio generalmente l'agente viscosizzante che si utilizza è la vaselina filante, chimicamente
costituita da miscele di idrocarburi con catene di lunghezza diversa ed è un prodotto secondario che
si ottiene generalmente dalla raffinazione del petrolio ed è presente in 3 forme(si chiama anche
paraffina, proprio per la sua scarsa reattività chimica); esiste anche la paraffina liquida o olio di
vaselina, anche la paraffina semisolida o vaselina filante o vaselina bianca e poi c'è la paraffina
solida che si usa poco in tecnologia farmaceutica(ma ha comunque un impiego nell'ambito sanitario
in cui si usa per la risoluzione della sonda ecografica). Tutte le fasi intermedie, cioè la fase del
creaming e la fase della flocculazione sono dei fenomeno reversibili, cioè per agitazione si è in
grado di ricostituire l'emulsione,quando l'emulsione si rompe cioè quando le due fasi sono
completamente separate non è più possibile ricostituire l'emulsione.
Quindi un modo per stabilizzare le emulsioni è quello di agire sulla viscosità della fase esterna
aggiungendo dei modificatori di viscosità che si chiamano agenti viscosizzanti o agenti ispessenti.
Gli stabilizzanti delle emulsioni possono essere:
agenti emulsionanti: come tensioattivi che diminuiscono la tensione interfacciale
diminuendo l'energia libera del sistema.
Agenti viscosizzanti: come gomme o colloidi idrofili che aumentano la viscosità della fase
esterna,cioè creano una fase protettiva
Per scegliere il tensioattivo da aggiungere per stabilizzare l'emulsione si usa un parametro, l'HLBr
(richiesto) ;l'HLB è una caratteristica dei tensioattivi e indica il bilancio idrofilo-lipofilo e può
assumere un valore da 0 a 20. Da 0 a 10 il tensioattivo è lipofilo, da 10 a 20 il tensioattivo è idrofilo.
L'HLBr, invece, fa riferimento alla fase oleosa dell'emulsione, dove infatti l'HLBr è definito come
l'HLB richiesto dalla fase oleosa per ottenere l'emulsione più stabile; quindi esistendo la regola di
Bancroft cioè che il tensioattivo deve essere affine per la fase estena, una fase oleosa, che può
essere interna o esterna, avrà 2 valori di HLBr con un valore da 0-10 quando la fase oleosa sarà la
fase esterna e con un valore 10-20 se è la fase interna dell'emulsione. Quindi per scegliere il
tensioattivo che stabilizza l'emulsione si fa riferimento al valore di HLBr della fase oleosa e
dipendentemente se l'emulsione che si deve preparare è O/A o A/O si sceglierà un tensioattivo con
un valore di HLB simile al valore di HLBr.
Generalmente non si usa un solo tensioattivo ma una miscela di tensioattivi e per calcolare quanto
tensioattivo di A e quanto tensioattivo di B bisogna aggiungere ci sono 3 metodi, di cui uno in cui il
valore di HLBr è uguale(scegliendo i tensioattivi A e B):

oppure si può utilizzare il metodo grafico.
Si scelgono 2 tensioattivi che hanno 2 valori di HLB uno maggiore dell'HLBr e uno minore
dell'HLBr.
Non bisogna però solo valutare l'instabilità fisica ma anche l'instabilità chimica. L'instabilità può
essere legata all'utilizzo di tensioattivi ionici perchè i tensioattivi che presentano una carica netta
possono reagire ed interagire con altri costituenti del sistema disperso. La presenza di elettroliti può
dare instabilità chimica perchè gli elettroliti causano la rottura dell'emulsione. I saponi sono dei
tensioattivi ionici che funzionano creando delle emulsioni e riescono ad eliminare il
grasso(emulsionandolo). Quindi l'instabilità chimica può essere data da:
– presenza di tensioattivi ionici;
– presenza di elettroliti;
– particolari condizioni di pH che si trovano all'interno del sistema disperso;
– fenomeni ossidativi che possono interessare la fase oleosa dell'emulsione.
Inoltre, essendo comunque un sistema in cui è presente una fase acquosa e soprattutto quando la
fase acquosa è la fase esterna ci sono anche dei problemi di contaminazione microbica quindi
problemi di conservazione(dal punto di vista microbiologico) e quindi, oltre gli agenti ispessenti e i
tensioattivi, si dovranno aggiungere dei conservanti ovvero degli agenti antimicrobici, tenendo
conto che l'agente antimicrobico, seppur in ridotta quantità, si ripartirà tra le 2 fasi e,che proprio la
presenza di tensioattivi, ci potranno essere delle zone disomogenee nella concentrazione del
tensioattivo e quindi si potrà avere un equilibrio monomero-micella e il conservante potrebbe
restare incluso all'interno di una micella e quindi la quantità di conservante che si aggiunge non sarà
perfettamente alle concentrazioni di conservante attivo, perchè una frazione sarà riportata nella fase
oleosa. Una parte sarà inclusa in una micella quindi la concentrazione attiva del conservante sarà
data da una serie di parametri che agiscono sulla concentrazione iniziale che si è aggiunta; quindi

utilizzando questi parametri in cui si ha il rapporto tra fase lipofila e idrofila. Φ,P il coefficiente di
ripartizione tra la fase idrofila e lipofila del conservante e R il rapporto emulsionante-conservante
ovvero considera la quantità di conservante inclusa nella micella, si può definire: Concentrazione
attiva del conservante
P e R sono dei dati che si ricavano sperimentalmente. La solubilizzazione micellare quindi fa
variare P. Per preparare un'emulsione stabile, gli eccipiente da aggiungere sono:
1) tensioattivi
2) conservanti
3) viscosizzanti
Generalmente soprattutto se l'emulsione è O/A si aggiungeranno delle sostanze come il sorbitolo e
il glicerolo, che si chiamano
4) umettanti, che servono per ritardare l'evaporazione dell'acqua.
La stabilità di un sistema disperso si fa con due prove: a) prove scaffale e b) prove accelerate. Le
prove scaffale valutano la stabilità di un sistema disperso nelle normali condizioni d'uso,cioè
appoggiate sullo scaffale. Consta di:
esame visivo(macroscopico), ed è evidente perchè se l'emulsione si rompe,si vede;
dimensione dei globuli(fase dispersa) nel tempo per vedere se le dimensioni sono rimaste
omogenee(con microscopio, coulter counter,diffrazione e raggi x);
variazione di viscosità perchè generalmente una variazione di viscosità del sistema è inoltre
una modificazione fisica del sistema disperso.
Nelle prove accelerate si fanno le stesse prove di dimensione dei globuli e variazione di viscosità
ma nelle prove accelerate si sottopone l'emulsione a degli stress; questi stress sono degli stress
tecnici cioè il campione si conserverà a 0 °C e poi si porta a 30 °C e poi si riporta a 0° e poi a 30° o
alle temperature scelte nell'analisi di stabilità e si sottopone a centrifugazione (200-300 rpm).
Quindi chiaramente se l'emulsione risulterà stabile in queste prove stressate allora risulterà stabile in
condizioni di conservazione normale. La preparazione delle emulsioni(qui è visualizzata una
provetta ma è alla base degli omogenizzatori industriali) è
legata alle 2 fasi: la prima fase si chiama
omogenizzazione primaria in cui si formano i primi nuclei
più grandi e l'omogeneizzazione secondaria che porta al
sistema comunemente disperso ed è basata sull'agitazione
meccanica. Maggiori sono i volumi,maggiore dovrà essere
l'energia fornita
Omogenizzatori

1) high spread blander → miscelatore ad alta velocità. La maionese fatta con il frullatore ad
immersione è un esempio di omogenizzatore high spread blander, cioè un miscelatore ad alta
velocità, per la formazione dell'emulsione perchè la maionese è un'emulsione.
2) Negli omogeneizzatori a valvole, il sistema dopo l'omogenizzazione primaria viene forzato
attraverso un sistema in cui viene mandato ad impattare contro la valvola e la sua forza di impatto
costituisce l'omogenizzazione secondaria;
3) Negli omogeneizzatori ad ultrasuoni, l'omogenizzazione primaria è fatta per agitazione
meccanica e poi la riduzione delle dimensioni alle dispersione colloidale è fatta per azione degli
ultrasuoni.
Le emulsioni multiple sono sistemi in cui le fasi sono 3 o addirittura 4, quindi per esempio
un'emulsione A/O|O/A e viceversa. Le microemulsioni sono sistemi stabili; la formazione delle
microemulsioni, scegliendo accuratamente le 2 fasi, è spontanea quindi presentano stabilità
termodinamica perchè se il sistema si forma spontaneamente vuol dire che ha un contenuto
energetico basso e non bisogna sottoporlo all'azione dell'omogenizzatore ed ha un colore limpido e
trasparente(preferibile nella cosmetica per il suo aspetto gradevole). Le emulsioni sono
rappresentate in quasi tutte le forme farmaceutiche liquide per uso orale, quelle che si chiamano
impropriamente “sciroppi”(infatti lo sciroppo è solo acqua più saccarosio). Sono presenti come
liquidi per applicazione cutanea oppure anche come preparazioni semisolide per applicazione
cutanea quando la fase esterna è una fase grassa,viscosa come le creme. Sono presenti come liquidi
per la nebulizzazione,come preparazioni nasali o come preparazioni per uso oftalmico e sono
possibili anche nella realizzazione di forme farmaceutiche parentali. Per tutte le forme
farmaceutiche,tranne i parenterali, nelle monografie ci sarà scritto che le emulsioni possono
presentare segni di separazione di fase ma l'emulsione deve essere perfettamente ricostituibile al
momento dell'uso per semplice agitazione ma nelle parenterali le emulsioni devono essere
omogenee.
Un altro tipo di fase dispersa è quella della sospensione in cui un solido è sospeso in un liquido
essendo il solido insolubile nella fase liquida in esame mentre è possibile realizzare delle emulsioni
che rimangono tali, cioè una fase liquida finemente suddivisa in un altro liquido mentre è ovvio che
con le sospensioni, trattandosi di un solido, si può interagire solo con la cinetica della separazione
delle fasi, e quindi non si può evitare che il solido sedimenti sul fondo della sospensione(perchè c'è
g). Una delle tecniche che si può utilizzare è quella di aumentare la viscosità della fase esterna(nel
caso delle emulsioni) e la fase liquida( nel caso delle sospensioni) perchè la velocità di
sedimentazione è descritta dalla legge di stockes
Quindi la velocità di sedimentazione dipende dal diametro al quadrato delle particelle, per la
differenza di densità del solido e del liquido e per l'accelerazione di gravità ed è inversamente
proporzionale alla viscosità del sistema. Fermo restante che il diametro delle particelle può essere
ridotto per diminuire la velocità di sedimentazione ma non si può scendere sotto un certo limite

perchè è proprio nella definizione di sistema disperso; si potrebbe cambiare la densità ma se il
sistema che si deve preparare è quello si può fare poco, e quindi il fattore sul quale si agisce è
aumentare la viscosità della fase liquida. Nel caso delle sospensioni, quindi, una prima classe di
eccipienti che si useranno per stabilizzare le sospensioni è quella chiamata degli agenti ispessenti o
modificatori di viscosità; nel caso delle sospensioni, quasi sempre, si usano le gomme oppure i
derivati della cellulosa che sono colloidi idrofili, perchè nella quasi totalità delle formulazioni per
uso farmaceutico il veicolo disperdente è di natura acquosa. Se si parte da un sistema iniziale in cui
le particelle di solido sono omogeneamente disperse nel liquido, lente ma inesorabili, le particelle
sedimentano sul fondo a formare il “fondello” o Cake , cioè un sedimento non ridisperdibile per
semplice agitazione.
Se si fa il rapporto tra il volume del fondello e il volume iniziale, cioè quello in cui il solido è
disperso omogeneamente, si avrà un valore molto inferiore a 1 perchè 1 è il valore di riferimento in
cui non si è formato il fondello,cioè il sistema è rimasto omogeneo nella condizione iniziale; visto
che non si può impedire che il sedimento si formi, si deve cercare di arrivare ad una condizione che
si definisce di sedimento flocculato
cioè la formazione di un sedimento che sia però facilmente ridisperdibile per semplice agitazione
per tornare nella condizione iniziale di dispersione omogenea. Nel caso del sedimento flocculato si
può avere che il rapporto potrà essere leggermente inferiore a 1, uguale a 1,cioè se il sedimento
occupa il volume iniziale della sospensione o in alcuni casi maggiore di 1 perchè il sedimento che si
ottiene occupa un volume maggiore rispetto al volume iniziale della sospensione. Il formulatore
vuole che il rapporto sia leggermente inferiore a 1. La qualità della sospensione che si prepara viene
definita dal grado di flocculazione G, ovvero il rapporto tra il volume del sedimento non flocculato
e il volume del sedimento flocculato.

La formazione del cake(che non si vuole) o del sedimento flocculato dipende
sostanzialmente dalle caratteristiche elettriche superficiali delle particelle di solido.
Per capire quali sono i fenomeni che intervengono nel processo di flocculazione si
deve definire il potenziale Z. Se si considera la superficie della particella solida che
si è dispersa nel liquido, la particella avrà aulla sua superficie una carica esterna (-),
che non è una carica netta, ma è una carica che dipende o dalle cariche adsorbite sulla
superficie delle particelle oppure da delle cariche di tipo dipolo che si dispongono sulla superficie
della particella. Se si pone la particella solida in H2O, sulla superficie della particella si
predisporranno per attrazione elettrostatica delle cariche(+) o come cariche (+) nette oppure come i
dipoli dell'H2O che si orientano a seconda della carica della particella.
Se si considera la particella rivestita da vari gusci, si avrà la particella (-), un
guscio di cariche(+) e un nuovo guscio di cariche opposte o legate agli ioni
presenti nell'H2O o legate all'orientamento opposto del dipolo dell'H2O.
Man mano che ci si allontana dalla particella si avrà una zona a prevalenza di cariche (+), una zona
a prevalenza di cariche(-) e si arriverà alla zona di H2O neutra in cui l'orientamento delle cariche
nell'H2O non risente più della carica superficiale della particella.
*
Il piano di taglio separa la zona a carica simile a quella della particella dal resto della soluzione; ci
sono due gusci: 1) il primo guscio di cariche opposte alla particella e 2) il secondo guscio di
cariche uguali che si chiama zona dell’acqua legata, perchè se questa particella in sospensione
viene posta in un campo elettroforetico, la particella si muove non da sola ma con questo guscio di
acqua legata che ha la sua stessa carica. La differenza di potenziale tra il piano di taglio e la zona
dell'acqua neutra si definisce potenziale Z. La differenza di potenziale è la misura del lavoro
necessario a spostare una carica elettrica. Definire il potenziale Z serve; per ottenere un sedimento
flocculato bisogna ridurre il potenziale Z, ovvero bisogna ridurre la differenza di potenziale tra il
piano di taglio e la zona dell'acqua neutra.
Intorno agli anni '50 è stata elaborata, da 2 gruppi di danesi, la teoria DLV0, che studia le
interazioni elettrostatiche tra le particelle in funzione della distanza. In particolare le forze disperse
variano con la distanza, mentre le forze attrattive variano inversamente alla distanza. All'aumentare
della distanza, variano più velocemente le forze repulsive.

Quindi è possibile costruire un grafico che sia la somma delle
forze attrattive e repulsive di una particella carica in funzione
della distanza A distanza 0 delle particelle, le forze repulsive
tendono a ∞ così come a distanza infinita, sia le forze repulsive
che attrattive tendono a 0, perchè non ci sarà interazione tra le
particelle.
Si ha una condizione di minimo primario quando le forze
attrattive, seppur a breve distanza, sono elevate e una
condizione di minimo secondario quando le particelle si
attraggono ma con forze attrattive molto deboli; quindi, visto
che non si può impedire che agisca la forza di gravità, si deve
realizzare un sistema in cui le particelle si trovano nella
condizione di minimo secondario,cioè che ci sia attrazione tra
le particelle e che quindi ci sia la formazione del sedimento ma che l'attrazione tra le particelle sia
debole e che, quindi, il sedimento sia facilmente ridi sperdibile (cioè per semplice agitazione si
vincono le forze attrattive tra le particelle e che si riesce a disperdere il sedimento). Per portare il
sistema nella condizione di minimo secondario si deve abbassare il potenziale Z delle particelle,
perchè abbassandolo si fa in modo che il sistema si trovi in questa condizione. Il potenziale Z si
abbassa agendo sulle cariche, quindi sulla condizione di carica presunta della particella ed in
particolare aggiungendo dei polielettroliti; gli agenti flocculanti sono dei polielettroliti, in cui
generalmente per un sistema in cui la particella è carica ( + ) si usano gli ioni fosfato, mentre
quando la particella è carica(-) si usa l'ossido di alluminio e quindi l'agente flocculante è l'Al +3
.
perchè l'Al+3
e non il 3Na+? Perchè serve un polielettrolità? È una questione di nuvola carica e
devono arrivare nel guscio intorno alla particella e quindi una singola carica magari non riesce ad
arrivare al guscio vicino alle particelle e viceversa una densità di carica magari riesce ad agire
meglio fisicamente e ad avvicinarsi alla superficie della particella.
Se si mette in grafico l'andamento del
valore di sedimentazione e del potenziale Z
in funzione della concentrazione di agente
flocculante: il potenziale Z, man mano che
si aggiunge l'agente flocculante, diminuisce
fino ad arrivare a 0 e per aggiunte
successive di agente flocculante si ha
l'inversione della carica superficiale della
particella e quindi il potenziale Z, in valore
assoluto, aumenta; nella zona intorno al
valore minimo di potenziale Z si avrà il
massimo valore di volume di sedimentazione.
Per formulare una sospensione quali sono gli eccipienti da aggiungere? Nella definizione di sistema
disperso si cerca di lavorare con particelle che abbiano delle dimensioni ridotte e omogenee e che
quindi rallentino il volume di sedimentazione secondo la legge di Stokes. Si possono aggiungere
degli agenti ispessenti o che modificano la viscosità del sistema e si possono anche aggiungere degli
agenti bagnati quindi dei tensioattivi che migliorano la bagnabilità del solido da parte del liquido; si

aggiungeranno degli agenti flocculanti per avere la formazione del sedimento flocculato e
disperdibile;si aggiungeranno anche dei conservanti perchè essendo in fase acquosa, almeno per le
sospensioni per uso farmaceutico,sarà necessario anche l'aggiunta di antimicrobici. Dal punto di
vista industriale, i sistemi per produrre le sospensioni sono simili a quelli per produrre
l'emulsione(omogenizzatori), in cui la miscelazione delle 2 fasi avviene utilizzando alte energie. Per
quanto riguarda le sospensioni ci sono 2 tecniche; dal punto di vista galenico le emulsioni possono
essere preparate con due tecniche: per sospensione o per soluzione.
Il metodo per sospensione prevede la formazione di un nucleo primario in cui il rapporto olio –
acqua - gomma(agente stabilizzante) è 4-2-1,quindi si pongono nel mortaio, in questo caso, le fasi.
Si ottiene un nucleo primario dell'emulsione al quale poi si aggiunge la restante H2O. Leggendo
questa formulazione, che tipo di formulazione si forma e perchè?
Olio di ricino g 40
Gomma arabica g 10
gomma g 1
vanillina mg 50
alcool g 2
sciroppo g 10
H2O q.b a 100g
è da ricordare il volume di fase, la viscosità di fase, la regola di Bancroft o del cuneo orientato. Il
volume di fase = la fase in volume maggiore è l'olio, e quindi aiuta poco perchè i volumi sono più o
meno simili. Quindi la cosa che aiuta è la regola di Bancroft in quanto, non essendoci i tensioattivi,
ma essendo presenti come agenti ispessenti le gomme(che sono dei colloidi idrofili) che modificano
la viscosità(aumentandola) dell'H2O, ovvero della fase acquosa, quindi fa presupporre che questa è
un'emulsione olio in H2O; quindi è un'emulsione O/A perchè vince la regola della viscosità di fase,
perchè, per aggiunta della gomma, la fase acquosa diventa altamente viscosa e ed è la fase estena
dell'emulsione. Infatti, questa è un'emulsione lassativa in cui l'olio di ricino viene disperso in una
fase acquosa,dolcificata dallo sciroppo e aromatizzata dalla vanillina.
Il metodo per soluzione parte dalla formulazione di un nucleo primario costituito,questa volta,
dalla gomma e dall'acqua, che formano una mucillagine; a questa mucillagine si aggiunge il resto
della formulazione. La fase esterna sarà l'H2O(avendo le gomme).
La preparazione delle sospensioni è fatta secondo 2 tecniche:
1) Il metodo continentale o della gomma secca
2) Il metodo inglese
Il metodo continentale si chiama anche della gomma secca perchè prevede che alle gomme o
all'agente ispessente(in questo caso il Na argillato) si aggiunge l'H2O e lo sciroppo; si forma così il
veicolo al quale si aggiunge il principio attivo. Nel metodo inglese si mescolano tutti gli ingredienti
e quindi la sospensione del farmaco viene fatta contemporaneamente alla formazione della
mucillagine. Infatti, il metodo continuentale forma prima il veicolo H2O - mucillagine e , nel

veicolo formato, ingloba il principio attivo; nel metodo inglese,invece, si mescola tutto insieme e
quindi si ha contemporaneamente la dispersione del principio attivo e della mucillagine. Nel
metodo della bottiglia (che usavano i vecchi farmacisti) si mettevano tutti i componenti all'interno
della bottiglia, veniva tappata e poi veniva usata come un mattarello sul piano della farmacia.
I test che si sfruttano per valutare al stabilità delle sospensioni sono dei test fisici in cui si valuta:
1) variazione di velocità di sedimentazione;
2) variazione nel tempo della flocculazione,cioè il rapporto dei 2 volumi nel tempo, partendo
dal presupposto che al tempo 0, il rapporto tra il volume del sedimento e il volume della
sospensione è uguale a 1 (perchè nel sistema al tempo 0,cioè in cui la dispersione è
perfettamente omogenea, il volume delle particelle corrisponde al volume di
sedimentazione);
3) facilità della ridispersione in cui si valuta il tempo di ridispersione del sedimento: anche in
questo caso si fanno delle prove scaffale cioè nelle normali condizioni di conservazione, e le
prove stressanti, sottoponendo la sospensione a cicli tecnici a 0°C, a 70°C o 60°C(dipende
dai protocolli) e dopo la centrifugazione.
Un esempio di monografia di una preparazione farmaceutica che fisicamente è una
sospensione(nella fattispecie è un collirio): le sospensioni, infatti, si trovano nelle preparazioni
farmaceutiche per uso orale(sciroppi, ma non necessariamente solo questi), colliri e , in alcuni casi,
anche nelle forme farmaceutiche parenterali. Il formulario nazionale dà indicazioni sulle
caratteristiche del collirio, cioè che deve essere isotonico e sterile, sul principio attivo e del
rapporto in cui devono esistere in H2O depurata e sugli adeguati eccipienti:quindi la farmacopea
non dà indicazioni formulative ma lascia ai farmacisti preparatori la scelta degli eccipienti. Dice che
si deve presentare sotto forma di una sospensione uniforme e lattescente dopo agitazione, perchè è
ovvio che la formazione di una sospensione non sarà mai limpida(come può capitare nelle
emulsioni) perchè c'è comunque un solido disperso. I saggi per la formulazione di un collirio sono:
il pH e la dimensione delle particelle, perchè è sia una sospensione che un collirio. La dimensione
delle particelle deve essere omogenea perchè una dimensione delle particelle omogenea (99%)
garantisce una velocità di sedimentazione uguale per ambedue i farmaci, perchè altrimenti si
avrebbe una distribuzione non omogenea del principio attivo all'interno delle gocce che saranno poi
istillate nell'occhio.

Viscosità
La viscosità, in realtà, è la viscosità dinamica. Si presupponga di considerare un cubetto di fluido e
questo cubetto lo si può pensare come costituito da lamine di fluido di spessore
infinitesimo,sovrapposte l'una sull'altra,se sulla superficie del fluido viene applicata una forza. Le
lamine di fluido si muoveranno secondo un gradiente di velocità diverso, quindi con una velocità
che sarà sempre più bassa man mano che ci si allontana dalla superficie sulla quale è stata
applicata(tangenzialmente) la forza. È come un mazzo di carte sul tavolo:spingendo solo la prima
carta, le carte non si muoveranno tutte insieme ma scivolano l'una sull'altra con le carte,che stanno
con in basso nel mazzo a contatto con il tavolo, che non si muoveranno proprio. Questa è la
condizione del fluido
Lo sforzo di taglio è la forza applicata tangenzialmente alla superficie di fluido e la velocità di
taglio (D),che in realtà è un gradiente di velocità e quindi una variazione di velocità;questi sono
messi in relazione tra loro dal parametro η che è la viscosità dinamica che dà la misura della
resistenza che il fluido oppone allo scorrimento una volta che a questo fluido è stata applicata la
forza.
τ = η ∙ D → η = τ / D dove τ è lo sforzo di taglio e D è la velocità di taglio
e η è la viscosità dinamica.
Le dimensioni di τ sono forza/superficie ma è diversa dalla pressione perchè non è perpendicolare
alla superficie ma è tangente. D, ovvero la velocità di taglio, corrisponde a sec-1
ma in realtà è una
variazione di velocità in funzione dello spazio.η può avere diverse misure: 1) N ∙ m -2
x sec;2) Pa ∙
sec (nel SI) e 3) dine ∙ cm-1
∙ sec (cgs) che corrisponde a poise (P). Per lavorare con i numeri interi
si usa il centipoise (cP), il sui valore di riferimento è per l'acqua a 20°C, con η= 1cP.
Esiste e viene definita la viscosità cinematica che è il rapporto tra la viscosità dinamica e la densità
del fluido(con dimensioni m2 ∙ sec
-1
V = η/ρ V = m2 ∙sec
-1 (SI)
V = cm2 ∙sec
-1 (cgs) = stokes (SI)
ST= 10-4
m2 ∙ sec
-1 → cST = 10
-6 m
2 ∙ sec
-1
Per lavorare con numeri interi si utilizza il centiStokes (cST), per l'acqua a 20°C V = 1cST. Questo
è vero quando si considera un fluido puro però se si vuole misurare la viscosità di una soluzione, si
misurerà la viscosità relativa che è il rapporto tra la viscosità della soluzione e la viscosità del fluido
che funziona da solvente(ηo): ηr = η/ηo

Nel caso di una dispersione colloidale bisogna tenere conto della viscosità del fluido che funziona
da fase disperdente e da Φ, che è la frazione di volume della fase colloidale; nel caso della
sospensione basterà fare il rapporto tra la viscosità e quanto volume occupa la fase colloidale: η =
ηo (1+2,5Φ).
I fluidi si distinguono in fluidi newtoniani e non newtoniani . I fluidi newtoniani hanno la
caratteristica di avere un valore di viscosità costante ad una certa temperatura, e per ogni coppia di
valori: sforzo di taglio/velocità di taglio.
Se si costruisce un reogramma ,cioè l'andamento del fluido
newtoniano ad una determinata temperatura, si avrà una retta che
passa per l'origine. τ = η ∙ D, dove η corrisponde alla pendenza
della retta,che è costante per tutte le coppie di valori di sforzo di
taglio/velocità di taglio. L'esempio è l'acqua e le soluzioni
idroalcoliche. Dal grafico si vede come la velocità di flusso
aumenta linearmente con l'aumentare della forza applicata.
I fluidi non newtoniani presentano 3 diversi tipi di flussi:
1) fluido plastico(Birgham fluids)
2) pseudoplastico
3) fluido dilatante
Se si disegna il reogramma(chiamato così perchè lo studio delle variazioni di viscosità si chiama
reologia, da reo scorrere), si misura il ritardo; questo si misura misurando l'angolo di deviazione del
cilindro trascinato
dove k dipende dal viscosimetro, M è la deviazione del cilindro trascinato
e ω è la velocità angolare del cilindro che è libero di muoversi.
1) nel fluido plastico, all'inizio si applica uno sforzo di
taglio ma il fluido non scorre;inizia a scorrere lentamente fino
a un certo valore di sforzo di taglio in cui scorre e si comporta
come un fluido newtoniano. Il valore soglia si chiama valore
di scorrimento χ. Un esempio sono le sospensioni in cui sono
aggiunti dei polimeri come agenti ispessenti;questo perché
all'inizio lo sforzo di taglio servirà ad ordinare le catene
polimeriche e,da un certo valore, saranno ordinate e il fluido
si comporterà come uno newtoniano.

2) nel fluido pseudo plastico non c'è mai un momento in cui
la viscosità è costante, cioè la viscosità cambia istante per istante
per ogni coppia di valori sforzo di taglio/velocità di taglio. In
particolare, aumentando lo sforzo di taglio, la viscosità
diminuisce,cioè la resistenza che il fluido oppone allo scorrimento
diminuisce perchè la viscosità si definisce anche come l'inverso
della fluidità.
3) nel fluido dilatante, la viscosità di taglio e lo sforzo di
taglio, il valore di η cambiano istante per istante ma in maniera
opposta, cioè aumentando lo sforzo di taglio la viscosità aumenta.
Per esempio, se si mescola H2O e sabbia in un secchietto, la paletta
si ferma. Aumentando la velocità di taglio, la viscosità del sistema
si ferma. Nel caso dei fluidi dilatante, maggiore è lo sforzo di
taglio, maggiore è η e quindi aumenta la resistenza che il fluido
oppone allo scorrimento(un esempio è il tubetto di crema, in cui
aumentando lo sforzo di taglio la crema non esce più).
Ci sono, per esempio, delle preparazioni di uso dermatologico, le paste , che sono costituite da una
fase grassa in cui è dispersa una grande quantità di solido (fino al 50%), che è l'esempio tipico di
flusso dilatante;ciò è importante per capire quale contenitore scegliere in quanto la pasta non può
essere dispensata nel tubetto ma deve essere dispensata nel barattolo(dato che è un flusso dilatante)
perchè aumentando lo sforzo di taglio la viscosità aumenta e quindi la pasta non uscirà dal tubetto.
La viscosità si misura tramite 2 tipi di strumenti che possono essere utilizzati a seconda ci si trovi di
fronte a un fluido newtoniano o non newtoniano. In farmacopea viene definita la viscosità come la
forza tangenziale riferita all'unità di superficie, detta forza di taglio, necessaria a spostare
parallelamente al piano di scorrimento uno strato di liquido di 1m2 alla velocità di 1m/s, rispetto ad
un piano parallelo situato alla distanza di 1m. Il rapporto dV / dx costituisce il gradiente di V di
taglio espressa in sec-1
. La farmacopea dice che il viscosimetro a capillare può essere utilizzato solo
per determinare la viscosità di fluidi newtoniani, perchè ciò che si misura è il tempo di deflusso che
il liquido impiega per andare da un punto ad un altro nel capillare e da tempio di deflusso, dalla
densità del liquido e dalla costante k(che è un parametro che dipende dal viscosimetro) si può
determinare η. Quindi solo per i fluidi newtoniani perchè η è costante e quindi viene ricavata dal
tempo di scorrimento. L'altra apparecchiatura è il viscosimetro a corpo rotante che è costituito da
due cilindri coassiali; un cilindro è pieno ed uno è cavo. Uno dei due è mosso da un motore, mentre
l'altro viene trascinato nella rotazione;quando i 2 cilindri coassiali non hanno nulla in mezzo,questi
si muovono alla stessa velocità. Quando tra i 2 si pone un liquido,questo,opponendo una resistenza,
tarda la rotazione. Il ritardo si misura con l'angolo di deviazione del cilindro trascinato. Quindi la
viscosità dipende da μ(deviazione del cilindro trascurato) su ω (velocità angolare del cilindro che
trascina il fluido). η = k ( μ/ω).

Parlando di viscosità sono stati classificati i fluidi in newtoniani e non newtoniani; i fluidi non
newtoniani sono i fluidi in cui la viscosità varia dipendentemente dalla coppia forza di taglio-
velocità di taglio;nell'ambito dei fluidi non newtoniani si hanno i fluidi puramente viscosi(cioè nei
quali la viscosità, ovvero la fluidità, varia al variare della forza di taglio), poi c'è un'altra classe in
cui la viscosità varia al variare del tempo di applicazione della forza di taglio e sono i fluidi con
viscosità dipendente dal tempo. Questi fluidi, a loro volta, possono essere distinti in:
– fluidi tissotropici
– fluidi reo pectici
Questi due comportamenti reologici si distinguono mantenendo costante l'intensità della forza
applicata, valutando la variazione del comportamento reologico al variare del tempo di applicazione
della forza. In particolare, i fluidi tissotropici , per mantenere costante la resistenza allo
scorrimento(η costante) hanno bisogno che nel tempo si riduca l'intensità della forza
applicata;mentre i fluidi reopectici, per mantenere costante η,richiedono che nel tempo l'intensità
della forza sia gradualmente aumentata. Si parte dal presupposto che si vuole un sistema in cui la
viscosità rimanga costante, cioè mantenga lo stesso valore: nel caso dei fluidi newtoniani ciò è
semplice perchè lo fanno da soli, infatti per ogni coppia di valori sforzo di taglio e v di taglio(ad una
determinata temperatura), la η rimane costante,cioè il fluido oppone allo scorrimento la stessa
resistenza. Nel caso in cui, invece, si ha a che fare con dei fluidi non newtoniani si vuole che η
rimanga costante, cioè che il fluido imponga allo scorrimento la stessa resistenza. Se ci si trova di
fronte a dei fluidi tissotropici si deve diminuire, nel tempo, l'intensità della forza applicata e nel
caso dei fluidi reopectici si deve aumentare nel tempo la forza applicata. Lo scopo è quindi di
mantenere costante η in un sistema in cui η non sarebbe costante.
La tissotropia è una traformazione gel-sol reversibile e isoterma. Un esempio sono i dentifrici o il
ketchup che vengono conservati a testa in giù,che sono costruiti come dei gel tissotropici(ciò che
quindi permette a questi di non uscire dal recipiente). Nella tissotropia, quando si aumenta la forza
di taglio non si riforma il sistema. All'aumentare dello sforzo di taglio, la η diminuisce; quando si
smette di applicare lo sforzo di taglio si avrà una curva non sovrapponibile con quella di prima ed il
sistema torna indietro, sempre in modo che η rimanga costante. In pratica, quando si smette di
applicare la forza di taglio, la velocità di flusso è maggiore, cioè la viscosità è minore. Questo ciclo
si chiama ciclo di isteresi e la quantità del fluido tissotropico è data dall'area all'interno del ciclo, e
quindi si valuta con i reogramma, calcolando l'area all'interno del ciclo di isteresi

Il fluido dilatante è il comportamento reologico per le vernici, in quanto la vernice deve avere una
fluidità utile per poter dipingere, ma quando si smette di applicare la forza di taglio deve diminuire
il limite di scorrimento;quindi deve essere fluido quando si applica lo sforzo di taglio ma deve
aumentare la sua η immediatamente quando si smette di applicare lo sforzo di taglio per evitare che
la vernice, prima di seccarsi, coli. La reologia, dal punto di vista farmaceutico, è uno studio
fondamentale sui tipi ti fluidi, perchè può influenzare e può dare una misura sulla stabilità, perchè in
un'emulsione in cui le due fasi si separano,cambia il comportamento reologico. È una misura
fondamentale per valutare, nella forma farmaceutica per uso dermatologico, la spalmabilità. Un
altro parametro che può essere influenzato e che deve essere valutato negli studi reologici, è per le
fasi di confezionamento ; si consideri di avere una preparazione di tipo dermatologico o una
preparazione per uso orale; è fondamentale valutare il tipo di flusso,ad esempio nel riempimento dei
flaconi, in quanto si potrebbe avere un fluido, che all'aumentare dello sforzo di taglio può
aumentare la sua η e quindi potrebbe causare problemi nel riempimento uniforme dei flaconi.
I gel sono dei sistemi costituiti da due fasi liquido-solido in particolare un sistema polimerico
reticolato che a contatto con l'H2O dà,generalmente perchè esistono i gel lipofili, un reticolo
tridimensionale in cui si strutturano le catene polimeriche e hanno quindi la caratteristica di avere
un sistema altamente strutturato e con un elevato contenuto di H2O. I gel si possono distinguere in:
– gel chimici
– gel fisici
a seconda che il processo di reticolazione,cioè la formazione della struttura dimensionale sia
ottenuta per aggiunta di agenti reticolanti (che formano il reticolo tridimensionale reagendo
chimicamente) oppure il reticolo tridimensionale può essere ottenuto attraverso metodi fisici come
l'irraggiamento e la variazione di temperatura. I gel sono distinti in: a) di tipo I e b) di tipo II. I gel
di tipo I sono dei sistemi irreversibili, in cui la reticolazione è irreversibile perchè il reticolo è
formato da legami chimici covalenti; si utilizzano,ad esempio, per le lenti a contatto o in impianti ad
uso chirurgico(come quelli auricolari) perchè si fa avvenire la reazione di reticolazione di una
soluzione polimerica più un agente reticolante all'interno di un contenitore che ha una forma
predefinita. Il processo di reticolazione è irreversibile e rompendo la forma il gel mantiene la forma
del contenitore. Quelli che vengono utilizzati in tecnologia farmaceutica sono i gel di tipo II: sono
dei sistemi in cui la reticolazione è reversibile, che può essere chimica(con legami meno forti dei
legami covalenti) o fisica. Sono lentamente solubilizzabili. Inoltre sono caratterizzati da 2 parametri
che sono:
– la concentrazione critica , cioè la concentrazione al di sotto della quale il gel non si forma,
oppure una volta ottenuto il gel ritorna allo stato di sol;
– la temperatura di gelificazione,temperatura al di sotto della quale il sistema torna dallo
stato di sol allo stato di gel.
Quindi sono dei gel reversibili e i fattori che possono spostare l'equilibrio dal gel al solo sono la
concentrazione critica e la temperatura di gelificazione. La maggior parte sono dei derivati della
cellulosa oppure del PEG o del polivinilpirrolidone. Esempi di geli presenti in farmacopea sono: i

geli idrofobi, che sono costituiti da polietileni a basso PM in olio minerale (olio di vaselina),oppure
i geli idrofili, in cui una formulazione è presente in farmacopea ed è del glicerolato d'amido in cui il
gel è costituito dalla gelatina(gel di derivazione animale).
Gli agenti gelificanti naturali sono:
– le gomme
– gli alginati (polimeri che derivano dalle alghe)
– i carragenati (polimeri che derivano dalle alghe)
– pectine, presenti nella frutta
– xantani
tranne le gomme, le altre classi di polimeri sono tutti polisaccaridi.
Gli agenti gelificanti chimici sono:
– carbameri, polimeri dell'acido acrilico;
– derivati della cellulosa;
– polietilene e suoi capolimeri;
– silice;
– argina e caolina;
– PVP (polivinilpirrolidone) e il polivininalcol;
– paloxamer(capolimeri a blocchi).
Nella farmacopea è descritta la composizione del gel base per preparazione semisolida per
applicazione cutanea: la composizione è: caramellosa sodica59 (o explotab) ,glicerolo 85%
g10,acqua depurata q.b a g 100. Inoltre dice che la caramellosa sodica può essere sostituita dalla
idrossietilcellulosa. Il glicerolo è presente in questa preparazione perchè è un plasticizzante: cioè fa
in modo che non si perda(o si perda in misura ridotta) l'H2O all'interno della preparazione. Invece la
caramellosa sodica o l'idrossietilcellulosa è un polimero,e viene messa se, dopo la formazione del
gel, l'H2O evapora(perchè c'è una concentrazione critica di gelificazione),cambia il rapporto tra il
polimero/quantità di H2O e cambia anche la struttura e la natura del gel.
Questi sono degli esempi di gel
Gel per capelli:
carbopol(agente gelificante)
polivinilpirrolidone(agente gelificante)
alcol al 95 %
trietanolammina( è un conservante)
H2O depurata
Gel antivaricoso:
ippocastano ES (PA)
hannamelis ES (PA)

carbopol 9110(agente gelificante)
glicolpropilenico(cosolvente che serve a solubilizzare i conservanti e i PA)
Nipagina(conservante)
Nipasolo(conservante)
olio di ricino
profumazione
NaOH sol 10%(serve a realizzare il gel)
H2O depurata
Essaven gel
escina, eparina sodica, fosfatidilcolina(PA)
isopropanolo (solvente tossico,che dovrebbe essere assente ma in questa formulazione è
indispensabile per solubilizzare)
glicerolo (plasticizzante)
trietanolammina (agente gelificante)
H2O di colonia
H2O depurata
rosmarino essenza lavanda essenza

Preparazioni per uso dermatologico
Nelle preparazioni per uso dermatologico la struttura anatomica interessata è l'epidermide,
costituita(andando dall'esterno verso l'interno):
1) strato corneo, che è uno strato di cellule morte e che serve per difendere l'organismo,per
impedire che sostanze esterne possano aggredire la pelle;
2) altri strati
3) strato sottocutaneo nel quale si trovano i vasi sanguigni e i bulbi piliferi.
Quindi una preparazione per uso dermatologico potrà agire sulla superficie della pelle oppure avere
come target i singoli strati successivi. Si potranno avere delle sostanze che si devono fermare sulla
superficie senza nessun assorbimento, e questo è quello che per legge viene chiesto al
cosmetico(come ad esempio i coloranti non devono essere assorbiti). Ci sono delle sostanze che
devono raggiungere lo strato corneo, e che comunque devono essere assorbite attraverso lo strato
corneo ed hanno però come target la parte superficiale dell'epitelio (emollienti e gli esfolianti).
Potranno essere realizzate delle forme farmaceutiche(in quanto è il tipo di forma farmaceutica che
fa in modo che il principio attivo raggiunge uno strato piuttosto che un altro) che hanno come target
gli annessi cutanei(ghiandole sebacee o bulbi piliferi); si possono fare delle preparazioni per
veicolare principi attivi che esplichino un'azione a livello locale oppure realizzare delle
formulazioni che consentano al principio attivo di arrivare nello strato sottocutaneo e quindi al
torrente circolatorio per avere un'azione sistemica. Tutto questo si fa modificando il veicolo e quindi
scegliendo l'adeguata composizione del veicolo si può fare in modo che il PA si fermi alla superficie
o che possa essere assorbito.
Il processo di passaggio attraverso la pelle è un processo di tipo diffusivo, quindi dalla prima legge
di Fick:
J= flusso, D= coefficiente di diffusione(cm2/sec), dc/dx è una forza. Cioè un flusso viene generato
da una forza attraverso un coefficiente,che si chiama coefficiente fenomenologico, che in questo
caso è un coefficiente di diffusione; cioè il flusso si genera a spese della forza(e si nota dal segno (-)
del coefficiente). La diffusione attraverso la pelle è una diffusione unidirezionale(anche se nella
pelle c'è una diffusione laterale), nel caso più semplice in cui il gradiente di concentrazione viene
considerato solo sull'asse x ovvero una diffusione solo attraverso la pelle, l'assorbimento attraverso
la pelle avrà un andamento di questo tipo:
Si avrà quindi un tempo di latenza in cui si ha l'interazione della
forma farmaceutica con la pelle, una fase iniziale in cui
l'assorbimento è più rapido perchè avviene anche attraverso le
ghiandole sebacee e i bulbi piliferi(questo assorbimento si
chiama di derivazione pilosebacea; quando gli annessi cutanei,
sopo un breve lasso di tempo, saranno saturi, l'assorbimento
dipenderà soltanto da una costante perchè la velocità di

assorbimento sarà data dal coefficiente di diffusione attraverso la pelle, K il coefficiente di
ripartizione, Co è la concentrazione iniziale e h lo spessore della pelle. Quindi
I fattori che influenzano la diffusione: il coefficiente di ripartizione, cioè la capacità del principio
attivo di ripartirsi tra una fase idrofila e una lipofila, ed ovviamente le variazioni di pH, perchè
secondo la legge di ripartizione secondo pH, la forma che viene assorbita è la forma indissociata e
quindi, dipendentemente dal pka della sostanza, in base al pH in cui la sostanza si trova sarà nella
forma dissociata o indissociata;bisogna considerare che il pH di una pelle sana è intorno a
5,5,quindi leggermente acido. Ci sono poi dei fattori relativi alla pelle che influenzeranno la
diffusione del principio attivo. Uno dei fattori è lo spessore della pelle, e il distretto dell'organismo
sul quale si applica la preparazione per uso dermatologico(le piante delle mani e dei piedi sono
totalmente cheratinizzate e quindi sono delle zone del corpo in cui non avviene l'assorbimento,
mentre la pelle dietro l'orecchio è quella con meno spessore). Un altro fattore è lo stato di
idratazione della pelle, perchè una pelle più idratata ha più H2O e quindi indipendentemente da K
del principio attivo, l'assorbimento attraverso la pelle risulterà aumentato o rallentato;inoltre
l'idratazione della pelle è correlata all'età,con la pelle anziana che è meno idratata di quella di un
bambino. La composizione della pelle varia anche al variare delle diverse etnie e quindi ci sono
anche delle caratteristiche di popolazione che influenzano l'assorbimento cutaneo così come la cute
sana rappresenta una barriera per l'assorbimento, una cute infiammata o una cute lesa è una cute che
assorbe molto di più, una cute perturbata(come nel caso della psoriasi) in cui cambia la
composizione della cute ha delle caratteristiche di assorbimento diverse.
Per fare in modo che il principio attivo superi la barriera dello strato corneo si utilizzano I
promotori di assorbimento , che devono essere inerti dal punto di vista farmacologico e
tossicologico e che esplicano la loro azione disorganizzando i lipidi dello strato corneo e quindi
favorendo l'assorbimento. Lo strato corneo funziona da barriera, che essendo cellule morte, sono
completamente prive di H2O e quindi è una barriera fisica all'assorbimento. Questa azione di
disorganizzazione deve essere rapida e completamente reversibile: sono l'alcol(etanolo), DMSO,
DMF(dimetilformaldeide), 2-pirrolidone. Un'altra classe di assorbimento sono i tensioattivi ionici.
Quindi,dato che si modificherà la profondità di penetrazione del principio attivo modificando il
veicolo, si dovrà valutare come l'interazione veicolo – principi attivi influenza l'assorbimento del
principio attivo. Ci sono due possibilità da considerare:
1) se lo stadio lento è l'assorbimento percutaneo;quindi il processo che determina la velocità
della permeabilizzazione è l'assorbimento attraverso la pelle. Questo vuol dire che il veicolo non ha
nessun effetto, ovvero il veicolo rilascia immediatamente il PA, e sarà invece la diffusione
attraverso la pelle a modulare la velocità del processo. In questo caso la velocità di assorbimento
dipenderà dal coefficiente di ripartizione (P) del PA, dal coefficiente di diffusione della pelle (D),
dall'area dell'applicazione (A) e dalla concentrazione del PA nel veicolo e inversamente
proporzionale allo spessore della pelle da attraversare(h):

2) se invece lo stadio lento è il rilascio della forma farmaceutica, l'assorbimento percutaneo
sarà influenzato dalla diffusione dal PA nel veicolo, più che attraverso la pelle. In questo caso ci
sono 2 possibili situazioni:
a) quello in cui il veicolo sia una soluzione , e quindi il PA è già pronto per essere assorbito. La
velocità di assorbimento corrisponderà
Co = concentrazione iniziale che dipenderà dal coefficiente di ripartizione attraverso il
veicolo(DV), h è lo spessore.
b) se il veicolo sia una sospensione , si dovrà valutare anche la dissoluzione del PA che poi
diffonderà solo sotto forma di soluzione
Quindi la velocità dipenderà dalla quantità totale di PA sospeso nel veicolo(A), dalla solubilità nel
veicolo(Cs), dalla diffusione attraverso il veicolo (Dv);anch'essa varia al variare della radice
quadrata. Poiché generalmente la quantità totale del PA nel veicolo è molto maggiore di Cs
(A>>Cs),si può utilizzare la forma semplificata in cui non compare Cs, ovvero la solubilità è
trascurabile rispetto alla quantità iniziale di PA. Queste 3 leggi sono le leggi di Higuchi che
regolano la diffusione attraverso la pelle di un PA disperso in una preparazione per uso
dermatologico. Come possono essere classificate le preparazioni per uso dermatologico

Le preparazioni per uso dermatologico sono costituite da 3 fasi (la polvere, la fase grassa e la fase
acquosa) che possono essere mescolate tra loro per dare le diverse forme farmaceutiche per uso
dermatologico. Quella più semplice è la forma costituita solo da polveri; in questo caso si hanno lo
polveri aspensorie come il talco. Se si mescola una fase grassa con delle polveri(come la pasta
all'ossido di zinco) sono le paste, che sono caratterizzate dall'avere una quantità di polvere
inglobata in una singola fase grassa fino al 50%: è per questo che la pasta ha un comportamento
reologico dilatante. Poi si hanno gli unguenti, che sono costituiti da PA dispersi in una fase grassa;
l'unguento che avrà una η elevata rispetto alle altre formulazioni, si utilizza quando la preparazione
per uso dermatologico deve restare a contatto con la cute. Ci sono le basi di assorbimento che sono
costituite da una fase grassa e da una frazione emulsionante(come la lanolina): si chiama base di
assorbimento perchè riesce ad inglobare un'elevata quantità di H2O. Quando sono presenti la fase
grassa, la fase acquosa e gli emulsionanti si hanno le creme,che sono, dal punto di vista fisico delle
emulsioni, che possono essere A/O o O/A: le creme O/A sono dette anche lavabili perchè essendo
l'H2O la fase esterna, lavorando con l'acqua si diluisce l'emulsione e quindi si elimina. Quando la
fase acquosa è preponderante rispetto alla fase grassa e all'emulsione si hanno le lozione, mentre
quando si hanno i PA dispersi in H2O si hanno i Bagni; quando il sistema torna bifasico ma si ha
una polvere in H2O si hanno le lozioni per frizioni.
Gli eccipienti per uso dermatologico possono essere classificati in lipofili e in idrofili e possono
anche essere classificati a seconda del grado di penetrazione all'interno della pelle. Si avranno così
gli eccipienti epidermici, che fanno in modo che il PA resti nello strato superiore dell'epidermide,
gli endodermici, quando si raggiungono gli strati sotto quello corneo ma senza l'assorbimento, e gli
eccipienti diadermici. La classificazione dal punto di vista chimico (lipofili ed idrofili) e all'interno
di queste classi li suddivido in a)monofasibi e b) bifasici.
Eccipienti lipofili
La vaselina idrofila è costituita da una fase grassa(vaselina bianca, cera bianca e alcol steratico) e il
colesterolo come agente emulsionante(tensioattivo che permette di incorporare H2O)
a)
monofasici che non incorporano Grasso
H2O
vaselina siliconi
(dimeticone)
naturali che incorporano lanolina anidra
H2O
vaselina idrofila lanovaselina alcoli di lanolina

b) bifasici (emulsioni O/A) → lanolina e cold cream
eccipienti idrofili: a) monobasici → PEG e idrocolloidi e b) bifasici (creme lavabili) → a base di
stereati (evanescenti) e emulsionanti complessi.
Le preparazioni per applicazioni cutanee possono essere classificate in base ai sostituenti e
quindi alle fasi dei costituenti (la fase solida, grassa, acquosa e l’emulsionante ovvero lo
stabilizzante principale di questo tipo di formulazione); in farmacopea la suddivisione viene fatta in
base alla natura fisica delle preparazioni quindi si avranno le preparazioni liquide, le polveri per
applicazione cutanea e le preparazioni semi solide per applicazione cutanea (che veniva chiamata
anche pomata). Nella definizione di preparazione per applicazione cutanea secondo la farmacopea
entra il concento di viscosità: nella definizione si dice che sono preparazioni liquide diversa
viscosità da applicare sulla cute, sul cuoio capelluto e sulle unghie. Ci sono molti studi per le
preparazioni che riguardano le unghie per l’assorbimento trans unguale, sia per la medicina umana
(come antimicotici) sia per i farmaci veterinari, per le infezioni degli animali per alimenti perché la
normativa che riguarda questa classe di animali è estremamente restrittiva. Questo tipo di
applicazione può avere un effetto locale o un effetto sistemico attraverso la diffusione trans
dermica, il target può essere la superficie della cute o la cute oppure può prevedere l’effetto
sistemico attraverso l’assorbimento attraverso lo strato immediatamente sottostante l’epidermide.
Le preparazioni liquide per applicazione cutanea invece possono essere soluzioni, emulsioni o
sospensioni. Le preparazioni liquide per applicazione cutanea sono preparazioni di diversa viscosita'
destinate al rilascio locale o transdermico delle sostanze attive. Sono soluzioni, emulsioni o
sospensioni che possono contenere uno o più principi attivi in un adatto veicolo. Possono
contenere idonei antimicrobici, antiossidanti e altri eccipienti, quali stabilizzanti, emulsionanti
e addensanti. Le emulsioni possono presentare segni di separazione di fase, ma sono facilmente
ricostituite per agitazione. Le sospensioni possono presentare un sedimento che si disperde
facilmente dopo agitazione per dare una sospensione che e' sufficientemente stabile da permettere la
somministrazione di una preparazione omogenea. Quindi in queste preparazioni bisogna fare in
modo che nell’agitazione il sistema abbia una riduzione di viscosità che consenta di versare una
dose omogenea di principio attivo e che invece nel tempo, quando si deve conservare, si abbia un
incremento di viscosità che renda stabile la preparazione. Le preparazioni destinate
specificatamente all’applicazione su cute gravemente lesa sono sterili. Si possono distinguere varie
categorie di preparazioni liquide per applicazione cutanea, per esempio:
shampoo, è una preparazione medicata che deve agire sulla superficie della cute e quindi
non devono presentare assorbimento
schiume cutanee.
Le polveri per applicazione cutanea sono preparazioni costituite da particelle solide, non aggregate,
secche, di vari gradi di finezza. Contengono uno o più principi attivi, con o senza eccipienti e, se
necessario, coloranti autorizzati dall’autorità' competente. Le polveri per applicazione cutanea si
presentano come polveri a dose unica o come multidose; sono prive di granulosità. Le polveri
indicate specificamente per l’uso su larghe ferite aperte o su cute gravemente lesa sono sterili.

Hanno delle caratteristiche che devono essere valutate quali la finezza della polvere e , nel caso che
la polvere deve essere applicata sulla cute lesa o sulle ferite, deve essere sterile e quindi deve essere
formulata in modo da sopportare la sterilizzazione. Infatti una polvere granulata può risultare
abrasiva per la pelle. Le applicazioni semisolide per applicazione cutanea Le preparazioni
semisolide per applicazione cutanea sono destinate al rilascio locale o transdermico di principi
attivi, oppure hanno azione emolliente o protettiva. Hanno aspetto omogeneo. Le preparazioni
semisolide per applicazione cutanea sono costituite da una base semplice o composta in cui,
usualmente, sono disciolti o dispersi uno o piu' principi attivi. Secondo la sua composizione, la base
può influenzare l’azione della preparazione. Le basi possono essere costituite da sostanze naturali o
sintetiche e possono essere sistemi ad una fase o multifase. Secondo la natura della base, la
preparazione può avere carattere idrofilo o idrofobo (lipofilo), può contenere additivi adatti come
antimicrobici, antiossidanti, stabilizzanti, emulsionanti, addensanti e sostanze che aumentano
l’assorbimento.
Le preparazioni semisolide per applicazione cutanea destinate all’uso su pelle gravemente
danneggiata sono sterili. Si possono distinguere varie categorie di preparazioni semisolide per
applicazione cutanea: unguenti, creme, gel, paste, cataplasmi, impiastri medicati. Le ultime
due sono rientrate in farmacopea come forme farmaceutiche della vecchia galenica. Secondo la loro
struttura, unguenti, creme e gel generalmente hanno un comportamento viscoelastico ed un carattere
non-newtoniano, per esempio un flusso di tipo plastico, pseudoplastico o tissotropico ad alte
velocità di taglio. Le paste spesso mostrano un flusso dilatante. I cataplasmi consistono di una
base idrofila, che trattiene il calore, in cui sono dispersi principi attivi solidi o liquidi. Sono
usualmente spalmati in strato spesso su una tela adatta e scaldati prima dell’applicazione alla pelle.
Una base che si utilizza come base per i cataplasmi sono i semi di lino riscaldati; in questi viene
dispersa il principio attivo (generalmente si utilizzano per le problematiche respiratorie) e hanno
una consistenza sgradevole. Li hanno rimessi in uso perché la medicina tradizionale e quella
“naturale” sono tornati di moda. Al contrario dei cataclasmi, gli impiastri medicati sono una
preparazione flessibile in quanto il cataplasmo è una sacca che viene applicata l’impiastro medicato
può assume nere anche le caratteristiche di un cerotto e quindi si adatta alla parte che deve essere
trattata ed è a contatto diretto con la pelle (al contrario del cataplasma). Le schiume medicate sono
preparazioni costituite da grandi volumi di gas disperso in un liquido generalmente contenente uno
o piu' principi attivi, un tensioattivo che assicuri la loro formazione e vari altri eccipienti. Sono di
solito destinate ad essere applicate sulla cute o sulle mucose. Sono anch’esse un sistema disperso
dove la fase dispersa è un gas dispersa in un liquido, generalmente si formano al momento della
somministrazione: quindi nella bomboletta c’è la preparazione liquida in cui è disperso il gas e
sottoponendola all’azione della pressione della valvola dosatrice si attua la formazione della
schiuma. Nel caso in cui le schiume devono essere applicate sulle ferite, la formulazione deve
garantire la sterilità. Generalmente le schiume medicate sono per un’azione locale e non sistemica
(come antidolorifici, anticontusioni, rilassanti).
Come si valuta l’assorbimento percutaneo in vitro: sostanzialmente gli studi preliminari vengono
fatti in vitro utilizzando dei sistemi a stato stazionario in cui c’è la presenza di due compartimento:
uno donatore e uno accettore, e le celle possono essere verticali o orizzontali. La caratteristica
comune è quella di avere un compartimento donatore e uno accettore separati da una membrana che
simula la pelle. Si possono usare delle membrane sintetiche che sono della membrana lipofile, che
hanno una valenza relativa. Così come l’utilizzo di pelle animale o umana, che però ha la

caratteristica di essere il tessuto più simile (anziché della membrana) ma è un organo che è morto:
in vivo infatti sono presenti enzimi, fluidi biologici, il flusso sanguigno e comunque sono degli
esperimenti che sono fatti per uno screening iniziale che comunque hanno bisogno della prova in
vivo. La pelle più simile a quella umana come composizione è la pelle di maiale, dove infatti la
maggior parte degli esperimenti sull’assorbimento percutaneo viene fatta sulla pelle dei maiali.
I cerotti transdermici sono preparazioni farmaceutiche flessibili di varie dimensioni, contenenti
uno o piu' principi attivi, da applicare sulla pelle integra per rilasciare il o i principi attivi alla
circolazione sistemica, dopo aver attraversato la barriera cutanea. Sono costituiti normalmente da
una protezione esterna che serve da supporto a una preparazione contenente il o i principi attivi. La
loro superficie di rilascio è protetta da una copertura che è rimossa prima di applicare il cerotto alla
pelle. La preparazione contiene il o i principi attivi insieme con eccipienti come stabilizzanti,
solubilizzanti o sostanze destinate a modificare la velocità di rilascio o ad aumentare l’assorbimento
trans dermico. I cerotti trans dermici sono utilizzati per la somministrazione di sostanze che sono
particolarmente attive (come per le crisi di angina) e poi perché sono d’immediata applicazione e
soprattutto perché sono a immediata rimozione. Quindi il cerotto si toglie l'effetto indesiderato e
l'effetto collaterale finisce rapidamente.
I vantaggi della via transdermica rispetto a quella orale:
L'eliminazione della variabilità dell'assorbimento che può avvenire a livello gastrico
enterico; quindi sono dei principi attivi che devono essere immediatamente disponibili e in
maniera costante;
eliminazione dell'effetto di primo passaggio da parte del fegato;
Somministrazione costante e controllata del farmaco;
Eliminazione di picchi plasmatici di farmaco.
Il cerotto transdermico è realizzato con :
1. Una protezione esterna che funziona da sostegno e da protezione (generalmente) ed è
costituito da materiale impermeabile
2. Pellicola protettiva, che ha un foglio di materiale plastico metallico, rimosso
dall'applicazione del cerotto sulla pelle e serve per l'integrità del cerotto prima
dell'applicazione;
3. La parte centrale del cerotto che la formulazione delle proprie in cui è incluso il principio
attivo ed è definita preparazione o deposito del farmaco.
I cerotti transdermici possono essere realizzati secondo una classificazione: si hanno i sistemi
riserva o i sistemi a matrice, costituiti da una matrice polimerica nella quale sarà incluso il
principio attivo.
Sistema transdermico a riserva: si nota la protezione esterna (1), il deposito di farmaco, lo strato
adesivo, la pellicola protettiva e tra il deposito di farmaco e la parte adesiva c'è una membrana
polimerica che è quella che controlla il rilascio. Quindi nei sistemi transdermici a riserva la velocità
di rilascio del principio attivo è controllata dalla diffusione attraverso una membrana polimerica,
quindi la natura di questa membrana polimerica consentirà al formulatore di avere una velocità

fusione più o meno elevata o più o meno costante nel tempo e sarà questo passaggio a influenzare la
biodisponibilità del principio attivo. Un esempio in commercio è il transdermnitro in cui il principio
attivo è la glicerina e quindi utilizza per gli attacchi di angina: il deposito è costituito dal principio
attivo in un diluente in un mezzo diffuso e la membrana polimerica , che è la parte fondamentale di
questo sistema è una membrana costituita da un copolimero di polietilene e di vinilacetato.
Nei sistemi invece a matrice la diffusione del principio attivo è controllata da una matrice
polimerica: quindi principio attivo può essere disperso in una matrice polimerica e sarà funzione
attraverso questa matrice polimerica a regolare il rilascio del principio attivo oppure si avrà una
membrana polimerica che controlla il rilascio del principio attivo. Le caratteristiche del principio
attivo che consentono l'ipotesi di formularlo in un cerotto transdermico sono: innanzitutto deve
essere un principio attivo con un'alta potenza ovvero una dose inferiore a 2 mg pro die quindi deve
essere un farmaco molto vivo. Non deve essere irritante sulla pelle e deve avere un adatto
coefficiente di ripartizione che consente la diffusione attraverso la pelle ( anche adatte dimensioni).
Esempi sono: nitroglicerina, la scopolamina, gli ormoni (perché esistono delle formulazioni di
ormoni estrogeni per la terapia sostitutiva per le donne in menopausa), fentanyl (che necessita, per
essere somministrato come forma farmaceutica transdermica, di accortezze formulative particolari).
Non solo le caratteristiche del principio attivo devono essere valutate per ipotizzare l'uso di un
transdermico ma conosce anche in base alla regione del corpo su cui applicare il cerotto: questo è
l'esempio che riguarda l’idrocortisone. Quindi non solo bisogna valutare le caratteristiche del
principio attivo ma deve essere valutata anche la zona su cui deve essere applicato il cerotto; la zona
è scelta in base alla permeabilità del principio attivo in zone diverse dell'organismo. Esistono delle
tecniche per aumentare l'assorbimento percutaneo: una di queste è la iontoforesi cioè applicare una
corrente elettrica sulla cute che favoriscono il passaggio del principio attivo attraverso la pelle.
Sono stati realizzati dei sistemi miniaturizzati all'interno dei quali viene costruito un sistema di
iontoforesi e quindi un cerotto transdermico che ha all'interno del sistema di ionoforesi, che si attiva
attraverso una pila che applica una corrente quindi favorisce il passaggio del principio attivo
attraverso la pelle. Un altro esempio sono i cerotti con i micro aghi; sono un cerotto dalle
dimensioni normali la cui parte centrale nella quale viene inglobato il principio attivo ha dei micro
aghi generalmente di titanio e applicandoli, e quindi forano la pelle, si facilita assorbimento
attraverso la pelle.

Preparazioni liquide per l'uso orale
come vantaggi presenta un campo di applicazione vasto, una maggiore biodisponibilità del
principio attivo e una applicabilità superiore in campo pediatrico e geriatrico, perché non c'è
bisogno dell'atto della deglutizioni . Come inconvenienti si ha l'instabilità, perché rispetto alla
polvere o alle forme farmaceutiche solide, la reattività chimica e i processi di degradazione sono
estremamente più evidenti in forma liquida che forma solida. In farmacopea, Le preparazioni
liquide per uso orale sono generalmente soluzioni, emulsioni o sospensioni che contengono uno o
piu' principi attivi in un veicolo adatto; possono tuttavia essere costituiti da principi attivi liquidi
usati come tali (liquidi orali).
La differenza dal punto di vista normativo tra preparazione liquida per uso orale e il liquido orale
che il liquido orale è solo il principio attivo, dal punto di vista farmaceutico la preparazione liquida
per uso orale implica la presenza di eccipienti (veicolo); cioè il liquido orale è una formulazione
costituita da soli principi attivi e le preparazioni liquide per uso orale implica la presenza di uno o
più principi attivi ma anche di eccipienti. In questo caso la farmacopea indica come veicolo adatto.
La farmacopea dice anche quali sono le modalità di preparazione di queste forme farmaceutiche:
o per diluizione di preparazioni liquide concentrate;
per la preparazione di soluzioni o sospensioni a partire da polveri o granulati; quindi la
preparazione liquida per uso orale si può presentare come proprio una preparazione liquida
cioè già nello stato fisico di liquido oppure si può presentare fisicamente con un solido che
verrà portato in soluzione o in sospensione al momento dell'utilizzo dall'utilizzatore per
aggiunta, generalmente, di acqua; quindi sarà la persona che segue l'ultimo passaggio nella
preparazione liquida.
Il veicolo, dice la farmacopea, sarà scelto in base alla natura dei principi attivi per dare
caratteristiche organolettiche adatte all'uso previsto della preparazione. Quali sono le caratteristiche
da prendere in considerazione del principio attivo per scegliere gli adeguati eccipienti: Se si vuole
preparare una soluzione le caratteristiche del principio attivo da valutare è la solubilità; se la
solubilità in acqua è praticamente nulla si dovrà realizzare una sospensione. Un'altra caratteristica è
il sapore. Dipendentemente dall'odore dal sapore, come l’emulzione lassativa preparata con l'olio di
vaselina che ha un odore e un sapore sgradevole, questo viene coperto preparando un’emozione in
acqua dove l'acqua è dolcificato con lo zucchero. Una caratteristica che non è legata alla chimica
del principio attivo, nel caso per esempio di una sospensione andrà valutata la granulometria della
polvere.
Si possono distinguere parecchie categorie di preparazioni:
soluzioni, emulsioni e sospensioni orali,
polveri e granulati per soluzioni e sospensioni orali,
gocce per uso orale,
polveri per gocce orali,
sciroppi,
polveri e granulati per sciroppi.

Tranne le soluzioni, emulsioni e sospensioni orali e gli sciroppi tutte le altre forme farmaceutiche
solide che poi vengono ricostituite al momento all'uso dall'utilizzatore finale. Le soluzioni,
emulsioni e sospensioni orali sono fornite in contenitori unidose o multidose (unidose vuol dire
che la forma farmaceutica dispensata e divisa in una singola unità posologica mentre la
preparazione multidose indica che la suddivisione in unità posologica viene fatta dall'utilizzatore
finale). Ciascuna dose da un contenitore multidose e' somministrata per mezzo di un dispositivo
adatto a misurare il volume prescritto. Il dispositivo e' in genere un cucchiaio o una tazza per
volumi di 5 ml o multipli oppure una siringa orale per altri volumi.
Gli sciroppi sono preparazioni acquose caratterizzate da gusto dolce e viscosita' elevata. Possono
contenere saccarosio (in genere) ad una concentrazione di almeno il 45 per cento m/m o p/p. Il
gusto dolce può essere ottenuto anche usando altri polioli o dolcificanti. Generalmente gli sciroppi
contengono sostanze aromatiche o aromatizzanti. Ciascuna dose da un contenitore multidose viene
somministrata per mezzo di un dispositivo adatto a misurare il volume prescritto. Il dispositivo e'
usualmente un cucchiaio o una tazza per volumi di 5 ml o multipli. L’etichetta indica il nome e la
concentrazione del poliolo o del dolcificante. Se in etichetta quindi non c'è scritto niente oltre le
indicazioni di sciroppo vuol dire che il gusto dolce e la viscosità elevata sono impartite dalla
presenza di elevate percentuali di saccarosio. Quando saccarosio è sostituito da un altro dolcificante
deve essere specificato in etichetta.
La solubilità in farmacopea è la quantità massima di sostanza richiesta per questo saggio e' di 111
mg (per ogni solvente) e il volume massimo di solvente di 30 ml. La solubilità può essere espressa
con varie unità di misura come la concentrazione, %p/p, %p/v, %v/v, non sono usati nelle
preparazioni farmaceutiche in maniera casuale: viene detto nella farmacopea che generalmente le
soluzioni per uso orale e le soluzioni di gas nei liquidi vengono classificate in base alla % p/p, che
le soluzioni per uso parenterali vengono indicati come % p/v e le soluzioni liquide in liquidi
vengono indicate con la % v/v. Questo fa capire perché idroalcolico indicato con % v/v perché è una
soluzione di un liquido in un liquido. In farmacopea le indicazioni di solubilità non vengono date
con il Ks o con Cs ma sono delle indicazioni di tipo pratico (perché la farmacopea è un aiuto a chi
prepara) infatti
Il termine ``parzialmente solubile'' è usato nel caso di
miscele di cui solo alcuni dei componenti si
disciolgono. Il termine ``miscibile'' è usato per
descrivere un liquido che è miscibile in tutte le
proporzioni con il solvente indicato.
Solventi
Quando in farmacopea viene scritta la parola
soluzione implica che sia una soluzione acquosa;
vuol dire che il solvente di bisogni utilizzare è
l'acqua. Se non è espressamente detto l'acqua di bisogni utilizzare l'acqua depurata che risponde a
una specifica monografia di farmacopea e quindi da indicazione sulle caratteristiche di
composizione dell'acqua. L'acqua distillata invece indica acqua depurata ottenuta per distillazione.
Quanto viene detto alcol si indica alcol al 96% v /v. quindi in farmacopea ci sono due monografie di

acqua:
acqua depurata in grande volume
l'acqua depurata ripartita in contenitori
l'acqua depurata si ottiene da acqua potabile che viene deionizzata, generalmente quasi sempre con
una tecniche a scambio ionico e che nel caso dell'acqua sterile e le preparazioni iniettabili viene
distillata. In Italia l'unica che sta ammessa è la distillazione; in altri paesi è ammessa anche le
tecniche di micro filtrazione e di osmosi inversa. L'acqua depurata secondo farmacopea possiede i
requisiti specifici come:
assenza di sali minerali;
assenza di metalli pesanti;
assenza di sostanze ossidabili (perché indicano la contaminazione di microorganismi);
assenza di sostanze alcaline e acide residui;
pH 5,5-6, ovvero deve essere più vicino alla neutralità anche se l'acqua distillata ha un pH
leggermente acido.
Quali sono le soluzioni per uso farmaceutiche propriamente dette:
idroliti, che vuol dire soluzioni in acqua; in questo ambito si trovano le limonate, le
pozioni, i gargarismi, decotti, infusi e tisane.
Gli alcoliti sono le soluzioni a base di alcol.
I gliceriti sono utilizzati nella medicina omeopatica.
Le soluzioni in olio detti oleoliti o oli medicinali o medicali;
Enoliti e acetoliti, cui i principi attivi vengono disciolti in vino o aceto.
Degli esempi di preparazione sono: negli idroliti semplici nelle soluzioni acquose ci sono le
limonate e questo è un esempio di limonata presente nella farmacopea XI ma può essere lo stesso
preparata, in farmacia come galenico officinale, perché riportata in una farmacopea antecedente
alla XII.
Acido citrico
Magnesio carbonato sono aggiunti per sviluppare la CO2
H2O
Sciroppo di limone.
Come si prepara la limonata citro magnesiaca: si scioglie l'acido Citrico in acqua, portata 60°C,
si aggiunge sotto agitazione il carbonato di magnesio e nel recipiente che contiene già lo sciroppo di
limone si introduce la soluzione che si ottenuta filtrando sul cotone. La conservabilità di questa
formulazione, dice la farmacopea, è di due o tre giorni. Questa preparazione zuccherina è una
preparazione altamente viscosa e quindi filtrarla con tutte le sostanze è faticoso, inoltre lo zucchero
caramellizza e imbrunisce quindi se l'acqua viene riscaldata a 60°C e quindi se la preparazione è
eccessivamente calda e questa viene a contatto con una soluzione molto densa di zucchero si può
attivare il processo di caramellizzazione ; invece questo modo, filtrando sul cotone, gocciolando si
raffredda e viene in contatto poco a poco con lo zucchero.

Gli idroliti semplici prendono il nome di pozioni; nelle farmacopea più vecchie c'era un elevato
numero di pozioni: un esempio è la pozione gassosa che, così come la limonata citro magnesiaca, ha
funzioni digestive, per la presenza dell’aroma di limone che per lo sviluppo di anidride carbonica.
Pozione gassosa
Soluzione A: Sodio bicarbonato, sciroppo semplice e H2O depurata
Soluzione B: acido citrico anidro, sciroppo semplice e H2O depurata
queste due soluzioni sono preparate mantenute e separate perché al momento dell'utilizzo vengono
mescolate e l'acido Citrico e il sodio bicarbonato, miscelate insieme, producono una quantità di
anidride carbonica.
Alcoliti: sono soluzioni il veicolo alcolico; possono essere distinti in salini, ammoniacali ed elisir,
che sono soluzioni medicate idroalcoliche edulcorate ed aromatizzate, con l'alcol che ha una
percentuale tra il 20 e il 35% e c'è un'elevata percentuale di dolcificante che più alta se il
dolcificante è il saccarosio si abbassa se è un poliolo perché il potere dolcificante negli edulcorate
sintetici maggiore rispetto a quelli del saccarosio. Come si prepara l'elisir:
1. dato che è costituita da acqua e da alcol, la prima operazione da fare è individuare le
solubilità preferenziali cioè se la sostanza (con le sostanze) sono più solubili in acqua o in
alcol quindi bisogna preparare le diluizioni
2. miscelarle mettendo il volume minore nel volume maggiore, sotto agitazione. Anche questa
è una tecnica che serve a rendere la preparazione più omogenea possibile.
3. Quando è presente il saccarosio si scioglie in acqua;
se si scioglie tutto il saccarosio in tutta l'acqua che bisogna usare per tutta la preparazione potrebbe
darsi che poi quando si miscela la fase acquosa con la fase alcolica, la fase alcolica si separa perché
l'elevata percentuale di saccarosio riduce la miscibilità di acqua in alcol. Quindi si prepara prima lo
sciroppo semplice (acqua e saccarosio), si aggiunge la soluzione alcolica e poi si diluisce con la
restante acqua in modo da riportare la formulazione nella condizione di omogeneità qualora ci fosse
stata separazione delle fasi.
Per sciroppo semplice si intende , in farmacopea, una preparazione costituita da 1/3 di acqua e 2/3
di saccarosio. La caratteristica dello sciroppo semplice è che nonostante abbia un'elevata quantità di
zucchero si definisce una soluzione auto conservante perché, nonostante sia una soluzione di
zucchero in acqua (quindi con possibile crescita di microorganismi ), per l'elevata viscosità
impedisce la crescita dei microrganismi. Si prepara a caldo quindi si porterà all'ebollizione una
determinata quantità di acqua, più alta di quella che sarebbe per la preparazione dello sciroppo
semplice, con l'acqua bollita di fresco si scioglie il saccarosio stando attenti che l'acqua non sia
eccessivamente calda perché lo zucchero potrebbe caramellizzarsi. Si prepara quindi a caldo,
facendo però abbassare la temperatura dell'acqua a 50-60°C e poi si filtra a caldo su garza. La
filtrazione va effettuata quindi su garza e a caldo altrimenti, proprio per elevata viscosità della
preparazione, questa si incolla sull'imbuto.
Un esempio preso della farmacopea è l'elisir di paracetamolo

Alcol etilico ml 20
Glicerina ml 10
EF arancio ml 20
Sciroppo semplice ml 20
H2O depurata ml 45
Il paracetamolo solubile in acqua in rapporto 1:70, quindi avendo 2 g di paracetamolo dovrebbero
servire, per sensibilizzare il paracetamolo, 140 ml di H2O ma nella preparazione ce ne sono solo 45
ml. Quindi come si prepara: L'alcol etilico, in questo caso, serve da cosolvente per il
paracetamolo, in quanto la solubilità in acqua non è elevata ed inoltre l'acqua che è 1/3 di 20 ml è
presente nell'sciroppo semplice quindi la presenza dell'alcol etilico è indispensabile perché funziona
da cosolvente.
1. Paracetamolo in alcol; naturalmente avendo valutato la solubilità del paracetamolo in alcol
2. estratto fluido (che è di natura alcolica) viene inserito nella soluzione alcolica perché così
non precipita niente; l'estratto fluido di arancio è l’ aromatizzante. L'estratto fluido ha
riferito troppo perché è meglio che sia il principio attivo ad essere solubilizzato tutto che
l'estratto fluido, perché la solubilizzazione viene fatta per porzioni successive.
3. L’etanolo può causare la separazione della fase alcolica quindi si mette lo sciroppo semplice
e la glicerina
4. si diluisce con la restante acqua per avere una formulazione omogenea.
Da notare che generalmente si aggiunge il volume maggiore e il volume minore.
In farmacopea gli estratti sono distinti, a parte i fluidi, molli e secchi, in estratti titolati ed estratti
quantificati. L'estratto titolato vuol dire che la concentrazione del principio attivo o dei principi
attivi, che la farmacopea indica come costituenti con l'attività terapeutica nota, è definito.
Generalmente il titolo viene dato, dipende dalle sostanze attive, più o meno 5% o più o meno 10%,
con quindi un intervallo più o meno ristretto intorno al titolo che viene dato a quell'estratto. Gli
estratti quantificati sono aggiustati in un intervallo di costituenti; quindi non basta dire estratto ma
bisogna specificare che, nella quasi totalità degli estratti usati per le preparazioni farmaceutiche, gli
estratti utilizzati sono estratti titolati cioè al titolo noto. Degli esempi presenti nelle vecchie
farmacopea sono ad esempio la tintura di arnica, di mirra che si preparano per macerazione mentre
per percolazione gli estratti di bella donna, digitale e di rabarbaro: questo perché per esempio la
mirra perché è una resina e quindi in acqua dà luogo ad una mucillagine e quindi preparandola per
percolazione si arriva all'otturazione del percolatore. Invece nell'arnica si estrae il fiore e quindi è
facilmente estraibile.
I gliceriti possono essere preparati anche come preparazioni allopatiche (oltre che nella medicina
omeopatica) e sono medicamenti ottenuti per dissoluzione di sostanze medicamentose in glicerina:
possono essere utilizzati per uso esterno e quindi per preparazioni oromucosali (che sono
preparazioni che devono esser utilizzate all'interno della cavità boccale o per avere un'azione locale
o per avere un'azione sistemica), nasali e otologiche , o per uso interno per alcune preparazioni
generalmente da ripartirsi in gocce. Questo è un esempio di glicerita in gocce per uso orale
Gocce di digitossina

Digitossina (PA) mg 50
Metil p idrossibenzoato, g 0.1: o parabene è un conservante.
Glicerina 12.5 ml:
Alcol ml 50
H2O depurata q.b a ml 100;
Come si prepara:
1. per prima cosa bisogna vedere la solubilità di principio attivo, che è solubile in alcol anche
perché la quantità di alcol è molta in confronto all’H2O depurata.
2. Metil p idrossi benzonato con glicerina in H2O;
3. si mescola tutto insieme.
Un esempio di preparazione industriale di glicerita sono le gocce di Valium, il cui principio attivo è
la benzodiazepina ed anche in questo caso è presente acqua, alcol, glicerina e glicolpropilenico.
Sempre tra i veicoli nominati ci sono gli oli, che possono essere usati come principi attivi oppure
come solvente. Il problema dell’ utilizzo degli oli è il l’irrancidimento dell'olio, ovvero processo
ossidativo catalizzato da metalli e dalla luce che avviene a carico degli oli, e quindi quando si
utilizza un olio e necessario utilizzare un antiossidante. Gli oleoliti o gli oli medicati sono delle
soluzioni di uno o più farmaci in veicolo oleoso. Gli oli medicati sono per esempio l'olio di ricino o
di fegato di merluzzo.
I vini medicati ( o enoliti): si usavano gli aperitivi diuretici in cui si metteva in principi attivi nel
vino bianco e i digestivi o i tonici, come per esempio il vino chinato, in cui invece la base era il vino
rosso. Un esempio di enolita è il Vermouth, che normalmente sarebbe un vino medicato in quanto
presenta un principio attivo (che non è più considerato come medicinale ma come sostanza di
abuso), l'Assenzio, che veniva preparato in vino bianco. Può essere anche utilizzato l'aceto
(acetoliti).
Gli emulsionanti possono essere classificati impropriamente in tre classi, in quanto gli
emulsionanti veri e propri sono i tensioattivi:
1. tensioattivi sono le sostanze che favoriscono o comunque mantengono l'emulsione perché
riducono l'energia del sistema;
nella galenica però, quindi non con una terminologia che ha a che fare con la terminologia che fa
riferimento alla corretta azione delle sostanze, si possono avere (però non danno indicazioni sul
meccanismo di azione):
2. gli emulsionanti insolubili: sono storie insolubili in acqua e in olio con una elevata
superficie specifica, capaci di farsi bagnare da acqua ed olio, formano una pellicola
protettiva intorno le goccioline della fase dispersa. Quindi proteggono l'emulsione dal punto
di vista fisico e meritano quindi che le goccioline si avvicinano per arrivare alla rottura
dell'equazione. Sono la bentonite ( un silicato), il idrossido di magnesio e la grafite;
3. quasi emulsionanti che in realtà sono dei modificatori di viscosità (dal punto di vista
fisico), e sono le gomme alchil cellulosa, idrocolloidi o CMC.

Si possono utilizzare come stabilizzanti queste due gomme, che sono di origine naturale, e sono la
gomma adragante (E413, nome commerciale) e la gomma arabica (E4141). Le soluzioni di
queste gomme hanno un pH leggermente acido (la gomma adragante un pH di 5, la gomma arabica
un pH 5-7); anche questo importante per valutare le incompatibilità e le interazioni eventuali, in
particolare sono suscettibili di ossidazione (tramite le per ossidasi) e presentano incompatibilità con
gli ioni di valenti (calcio e magnesio). Ci sono anche altri agenti come: l’agar (anche questo di
derivazione naturale), la metilcellulosa (o methocel), carbossimetilcelluloca (o blanose),
carbossipolimetilene (o carbomer o carbopol, con un pH di 6 -11); le ultime si utilizzano in
percentuale minore rispetto agli altri agenti di derivazione naturale quindi hanno una capacità
viscosizzante più elevata rispetto alle gomme.
I tensioattivi utilizzati sono quelli che sono compatibili con il tratto gastrointestinale perché sono
all'interno di preparazioni per uso orale, e quindi verranno in contatto con la mucosa del tratto
gastrointestinale. Vengono utilizzati:
lecitine: sono di derivazione naturale. Quello più usato è quello denominato Epicuron 130
con edulcorante diverso dal saccarosio.
esteri del saccarosio con acidi grassi: saccarosio mono palmitato o sucrestere.
Le fasi oleose hanno due valori di HLB richiesto, a seconda che l'emulsione e si deve preparare sia
un emulsione A/O o O/A. qui la tabella degli HLBr
Sostanze A/O O/A
Acido stearico / 17
Alcol cetilico / 15
Alcol stearilico / 14
Lanolina anidra 8 10
Oli vegetali 4 10
Oli minerali 4 12
Vaselina 4 8
Cera d’api 4 12
Paraffina 4 11
Alcuni tensioattivi hanno un solo valore di HLBr, perché secondo la regola di Bancroft il
tensioattivo deve essere af ine per la parte esterna e quindi le sostanze con un solo valore di HLBr
non ha un'affinità della fase oleosa e che possono dare solo emulsioni O/A. I conservante da
aggiungere nelle emulsioni O/A, perché i battericidi e i batteriostatici vanno aggiunti nell'emulsione
quando la parte esterna è l'acqua, sono:
acido benzoioco, pH acido, per uso orale;

acido sorbico e Sali, pH acido, ossidazione, per uso orale;
cloro cresolo, pH acido, uso esterno;
fenil mercurio nitrato e acido, pH acido, colliri;
parabeni, pH 7 – 9, per uso orale;
Bisogna tener conto di pH di compatibilità di questi conservanti e quali possono essere usati per le
formulazioni per uso orale. Quando le emulsioni sono A/O si devono aggiungere gli antiossidanti,
utilizzati in minima concentrazione e quelli utilizzati sono il nutil idrossianisolo (BHA) e il butil
idrossi toluene (BHT) (sono definiti degli antiossidanti veri in quanto si ossidano preferenzialmente,
al contrario dei chelanti dei metalli che “chelano” di metalli che catalizzano i processi ossidativi).
Gli antiossidanti possono essere anche distinti in:
- idrofili: acido ascorbico, sodio bisolfito;
- lipofili: estere acido ascorbico, BHA, BHT, tocoferoli e gallati.
Sospensioni
presentano dal punto di vista formativo le stesse problematiche delle emulsioni. Per formulare le
sospensioni, gli eccipienti da aggiungere sono:
agenti viscosizzanti, che diminuiscono la velocità di sedimentazione;
agenti bagnanti, perché essendo un solido disperso in un liquido si favorisce il contatto tra
solido e liquido;
agenti flocculizzanti: che sono tipici della formulazione della sospensione perché fanno si
che si riottenga un sedimento facilmente ridisperdibile.
Analizzando i comportamenti reologici , quindi le variazioni di viscosità in funzione allo sforzo di
taglio, che si deve realizzare un fluido che abbia un comportamento reologico di compromesso tra
la possibilità di versare questa formulazione, la possibilità di deglutirla e la stabilità della
formulazione. Quindi si deve avere un sistema che abbia una viscosità elevata nella fase di
conservazione, viscosità che si deve abbassare per poter versare la formulazione e per poterla
diluire, e che poi deve rapidamente salire perché una volta che è stato rimesso il flacone sullo
scaffale la viscosità deve aumentare per aumentare rapidamente la stabilità della formulazione.
Metil cellulosa (o methocel)
Idrossietilcellulosa
CMC (anionico)
Il Methocel è quello che si utilizza di più, e può essere methocel A 15 o A4C, in cui i numeri e
lettere fanno riferimento alle caratteristiche fisiche legate al peso molecolare e alle capacità
viscosizzanti. Un potere viscosizzante buono degli agenti tranquillizzanti nei sistemi dispersi è un
potere viscosizzante compreso tra 1 – 1.5%. Poiché nelle sospensioni si deve disperdere un solido
in un liquido c'è il problema anche legato alla quantità di principio attivo che si deve inserire nella
sospensione; nell'emulsione sono due soluzioni e quindi nelle due fasi (acquosa o idrofila) il
principio attivo è disciolto e quindi a una concentrazione omogenea, nella sospensione invece
essendo un solido, se la quantità di principio attivo è molto bassa bisogna prima di diluire (ovvero a

concentrazioni di farmaco tra 100 mg – 1 g e quindi si ha una dispersione non omogenea)
omogeneamente in un recipiente inerte e poi lo si può disperdere. Quali sostanze si utilizzano
come diluenti (non si può utilizzare il lattosio perché è solubile in acqua e quindi non si avrà una
sospensione): fosfato di calcio, caolino (argilla) e bentonite (silicato) che, oltre a funzionare da
diluenti funzionano anche da strutturanti perché sono degli agenti che modificano la viscosità della
fase acquosa. Quindi non va valutata solo la solubilità del principio attivo ma si deve considerare
anche la quantità stessa del principio attivo.
Ci sono due metodi per la preparazione:
1) sospensione del principio attivo che avviene contemporaneamente alla formazione
della mucillagine; quindi viene mescolato il principio attivo con un agente ispessente e
questa miscela viene bagnata dall’agente umettante (o la glicerina o lo sciroppo semplice) e
poi si aggiunge la restante fase acquosa in piccole frazioni, agitando vigorosamente.
Preparazione
Caolino g 20
Carbonato di magnesio g 5
Sciroppo semplice g 20
Essenza di menta gtt 3
Idrossietilcellulosa 1% q.b.
H2O depurata q.b. a g 100
L’idrossietil cellulosa quanto basta vuol dire che per calcolare la quantità di agente viscosizzante
ispessente da aggiungere si deve preventivamente valutare la viscosità della preparazione di base,
ovvero c'è in un certo quantitativo di acqua 25 g di polvere, non solubile in acqua, ed in particolare
una di queste polveri è il caolino (che si può utilizzare come agente ispessente); quindi la prima
cosa da fare, non avendo il viscosimetro in una farmacia, non si può avere una misura esatta, ma si
può, disperdendo le polveri nell'opportuna quantità di acqua, valutare la viscosità della
preparazione. Una volta che è stata valutata la viscosità della preparazione si sceglierà se e quanto
agente stabilizzante aggiungere perché si potrebbe avere anche una preparazione con una viscosità
tale che garantisce una stabilità sufficiente alla formulazione. Anche in questo caso, essendo la parte
esterna acquosa e essendo in presenza di sciroppo semplice e comunque un mezzo zuccherino
diluito (e quindi non è più auto conservante) si aggiungerà gli antimicrobici. Il magnesio carbonato
ha un'azione blanda da agente flocculante (dato che il magnesio ha due cariche positive).
2) Un'altra tecnica nella preparazione degli emulsioni è che viene sospeso il principio
attivo e poi si forma la mucillagine.
Preparazione
Calcio carbonato g 5
Magnesio trisilicato g 10
Sciroppo semplice g 10
Essenza di menta gtt 3

Veicolo mucillaginoso di idrossietil cellulosa q.b. a 100 g
il calcio carbonato e il magnesio trisilicato solo degli agenti ispessenti e in particolare la presenza di
questi ioni bivalenti potrebbe favorire la modificazione del potenziale Z; lo sciroppo semplice può
la glicerina come agenti umettanti. Quando si utilizzano dei derivati della cellulosa bisogna valutare
la possibilità di interazione tra polimeri e gli ioni bivalenti, in particolare il calcio; infatti non è stato
scelta la carbossimetilcellulosa che interagisce con gli ioni calcio ma è stata utilizzata la
idrossietilcellulosa: questi ioni bivalenti interferiscono con la capacità di gelificare. Anche questo
quindi è un problema che deve essere valutato nella preparazione delle sospensioni: ovvero la
incompatibilità degli agenti ispessenti con gli ioni bivalenti, in particolare con lo ione calcio.
Sospensione pediatrica di spironolattone al 0.5% m/V in veicolo acquoso per uso orale
Spironolattone g 0,5
Fosfato di calcio g 10, diluente (quando il principio attivo è compreso tra 500 mg e 1 g
bisogna utilizzare un diluente)
Na CMC g 1
Sciroppo semplice g 30
Essenza di anice gtt 2
Metil p idrossibenzoato g 0,1
Etanolo 1 ml
H2O depurata q.b. a 100 ml
questo formulatore ha fatto l'errore della scelta dell'agente ispessente in quanto il calcio interagisce
con la CMC; in questa preparazione è utile sottolineare la necessità di diluente che è norma
interagiscano con la gente viscosizzante.
Rifamicina sciroppo (FU XII) ,Lo sciroppo di rifampicina contiene Rifampicina in un adeguato
veicolo sciropposo aromatizzato. Contenuto di rifampicina (C43H58N4O12): non meno del 95,0 per
cento e non piu' del 105,0 per cento della quantità indicata in etichetta. Lo sciroppo contiene il 2 per
cento m/V di rifampicina. Sospensione sciropposa, omogenea dopo agitazione, di colore rosso.
Anche in questo caso la farmacopea non da indicazione formulativa ma dice soltanto che la forma
farmaceutica, una sospensione sciropposa omogenea dopo agitazione, dice il contenuto di principio
attivo (ovvero il 2%) e al farmacista viene data la scelta del veicolo sciropposa da usare.
Sciroppo semplice
È costituito da 1/3 di acqua e 2/3 di saccarosio. La caratteristica dello sciroppo semplice è quello di
essere auto conservante, cioè che è un liquido altamente viscoso. Può essere preparato a caldo (che
ha come svantaggi la dissoluzione veloce e la filtrazione ma come svantaggi la caramellizzazione) o
a freddo ed ha una conservabilità di tre mesi. Essendo una preparazione presente nelle monografie
delle forme farmaceutiche specifiche ed essendo quindi una galenico officinale può essere tenuto
già rotto in farmacia e può essere usato in una preparazione galenico magistrale tenendo conto che
la sua conservabilità è di tre mesi.
Preparazione: scaldare all’ebollizione, per 20 min, una quantità sufficiente di Acqua depurata;

mantenendo la temperatura a 80-85 °C, sciogliervi il Saccarosio, agitando bene per disciogliere
completamente lo zucchero. Mescolare per omogeneizzare e filtrare subito a caldo su garza, posta in
un imbuto precedentemente riscaldato. Mescolare e portare a peso con Acqua depurata,
precedentemente bollita per 20 min. I saggi sono:
Aspetto. Deve essere limpido e non piu' intensamente colorato della soluzione di riferimento
Densita' relativa Da 1,32 a 1,33, è proprio della caratterizzazione dello sciroppo semplice (quindi
alla domanda di che cosa lo sciroppo semplice è una miscela di acqua e saccarosio in rapporto 3 a 2
corna densità relativa di 1.32 – 1.33)
Indice di rifrazione Da 1,448 a 1,458.
Gli sciroppi meditati devono contenere una concentrazione di saccarosio almeno il 45% e
contengono uno o più principi attivi; ovviamente essendo delle soluzioni zuccherine devono
contenere degli antimicrobici come i parabeni, sorbato di potassio (come conservante) che deriva
dall'industria alimentare, l’acido benzoico e il benzoato di sodio.
Come si preparano gli sciroppi medicati: la prima tecnica prevede la dissoluzione del principio
attivo o direttamente nell'sciroppo semplice, e questa tecnica può essere utilizzata per i principi
attivi molto solubili come principi attivi poco attivi perché dissolvendo una sostanza in un veicolo
molto viscoso si avrà una distribuzione non omogenea del principio attivo, oppure miscelando con
lo sciroppo semplice di estratti fluidi per sciroppi per dissoluzione in diretta, perché le tinture
possono essere preparate per diluizione di estratti fluidi in quanto la caratteristica dell'estratto fluido
è quello di avere la stessa concentrazione del principio attivo della droga di partenza: quindi si
avranno degli estratti fluidi nella maggior parte dei casi privati di tannini per la preparazione di
tinture e si avranno invece degli estratti fluidi per sciroppi che saranno stati precedentemente
chiarificati. Un'altra tecnica utilizzata per la dissoluzione del farmaco è disciogliere il farmaco in
una determinata quantità di acqua, che nella soluzione di farmaco, scioglie il saccarosio e questo
ovviamente sarà fatto che i principi attivi poco solubili o molto attivi. Un esempio è
Sciroppo di tiocolo
Tiocolo g 2
Sciroppo semplice g 98
per scegliere la tecnica di dissoluzione bisogna valutare la solubilità del principio attivo in acqua, e
questo caso è 1 : 8, quindi si solubilizzerà il tiocolo in H2O ( 2 g in 16 e poi si miscelerà), e poi si
miscelerà con lo sciroppo semplice.
Un altro esempio che viene dalla farmacopea è lo sciroppo di in cui sono contenuti la poligala e la
narceina, che sono degli antitossivi, e quindi è uno sciroppo per la tosse in cui sono miscelati
l'estratto fluido di poligala ( 2.5 g ) e la narceina ( 0.05 g) e la Farmacopea dice in veicolo
sciropposo aromatizzato q.b.
Questo è un esempio di veicolo sciropposo aromatizzato

Arancio EF, è un aromatizzante che può essere sostituito con l'aroma di lampone
Poligala EF
Nipagina
Nipasolo
Narceina
Saccarosio
H2O depurata
La nipagina e il napisolo sono 2 conservanti. Se nella descrizione c'è scritto 100 ml contengono tot
grammi di poligala e di narceina, per calcolare la quantità di veicolo sciropposo aromatizzato si
deve ricordare che la densità dello sciroppo semplice è di 1.32 – 1.33. Possono essere preparati
sciroppi con zuccheri diversi dal saccarosio tenendo presente che sia il sorbitolo che il levulosio
possono avere un effetto lassativo.
Secondo la Farmacopea, le basi per preparazioni liquide per uso orale ( sono due) hanno le
seguenti composizioni:
Sorbitolo 7,35 g 28 g
Glicerolo 85 per cento 10 g 10 g
Saccarosio 46,5 g -
Acqua depurata q.b. a 100 ml 100 ml
Il sorbitolo e il saccarosio sono gli agenti dolcificanti, il glicerolo come agente umettante.
Preparazione: disciogliere, agitando, i componenti solidi in un uguale peso di Acqua depurata
riscaldata a 50 °C. Lasciar raffreddare a temperatura ambiente e aggiungere, mescolando, i
componenti liquidi, portando a volume con Acqua depurata: se necessario filtrare su garza o colino.
Le basi possono contenere anche il 7,0 per cento m/V di Etanolo 96 per cento.
Edulcoranti sintetici: ci sono due classi di dolcificanti sintetici: gli edulcoranti intensivi tra i quali
si trovano la saccarina,il ciclammato e l'aspartame. Quindi sono presenti
- Acesulfame K
- Aspartame E951; è fonte di fenilalanina e quindi deve essere specificato e che quindi non
può essere somministrato a chi presenta patologie come la fenilchetonuria
- Ciclammato ( acido ciclamico e i suoi sali di Na e di Ca); portare le reazioni di
sensibilizzazione allergica (dermatite, prurito, eczema) e foto sensibilizzazione.
- Saccarina e i suoi sali di Na e Ca.
ci sono poi gli edulcoranti che sono diversi chimicamente dalla classe precedente e sono i polioli
(zuccheri - alcool)
- Sorbitolo;
- Mannitolo;
- Xilitolo;
- Isomalto;
- Lacticolo.

La caratteristica di tutte queste sostanze è che possono essere lassative. Per mettere in relazione con
gli edulcoranti di tutte le sostanze, viene definito come potere edulcorante di riferimento (uguale
a 1) quello del saccarosio; gli edulcoranti intensivi, a parità di potere calorico hanno un potere
edulcoranti di molto maggiore (quasi 100 volte di più a quello del saccarosio) mentre i polioli hanno
un potere dolcificante addirittura inferiore a quello del saccarosio con un potere calorico pressocché
paragonabile ma si utilizza quando gli zuccheri semplici non possono essere presi.
Nelle preparazioni liquide per uso orale vanno aggiunti gli aromatizzanti, che possono essere
naturali o di sintesi. Gli aromatizzanti naturali sono i succhi di frutta concentrati, le essenze e
idrolati e alcolati. In genere gli aromatizzanti andrebbero scelti in base al tipo di composto che deve
essere coperto. Se il principio che deve essere coperto ha un gusto:
Gusto Aromi consigliati
Amaro Ciliegia, cioccolato, menta, anice, noce
Salino Pesca, albicocca, vaniglia
Dolce Succhi acidi di frutta
acido Ananas, agrumi, liquirizia, lampone
Ovviamente l’aroma è scelto in base al colore che presenta la preparazione.

Pressione osmotica
due soluzioni separate da una membrana ideale o semipermeabile, ovvero permeabile al solo
solvente. Le concentrazione ai due lati della membrana saranno delle concentrazioni diverse (per
esempio C1 > C2); quindi due concentrazioni diverse di soluto da una parte e dall'altra parte
membrana permeabile al solo solvente. Lenta ed inesorabile l'acqua, o il solvente, si sposterà dalla
soluzione meno concentrata alla più concentrato fino ad ottenere una concentrazione di equilibrio
tra le due parti della membrana. La pressione che si deve esercitare per contrastare questo
movimento di acqua dalla soluzione diluita a quella più concentrato si definisce pressione osmotica.
Le proprietà colligative perché sono collegate tra loro: sono la pressione osmotica, ∆t
ebullioscopico, ∆t crioscopico e sono collegate tra loro perché sono determinate perché la presenza
di un soluto in un solvente fatta variare le caratteristiche del sistema in particolare facendo
riferimento al grado di dissociazione del soluto. Quindi non è soltanto che la sostanza è presente in
soluzione ma è soprattutto il suo grado di dissociazione, cioè le specie degli ioni che sono presenti
in soluzione e sono questi che avranno un effetto sulle proprietà colligative. In particolare, per
quanto riguarda la pressione osmotica, si possono definire in maniera quantitativa la concentrazione
delle specie attive sulla variazione o sul valore della pressione osmotica assume. Si può definire
osmole che è uguale al peso in grammi di un soluto che è in soluzione, come le molecole (e/o ione,
macromolecole, aggregati), che è osmoticamente equivalente al peso molecolare di un non
elettrolita (ovvero la sostanza che non è dissociata) che si comporta idealmente. La definizione di
osmole consente di definire la:
- Osmolalità: p/p, n° di osmoli di un soluto in 1 Kg di solvente;
- Osmolarità: p/v, n° di osmoli di un soluto in 1 l di solvente.
Quindi quando si vuole dare un'indicazione della capacità di una sostanza, in particolare di una
soluzione di questa sostanza, di avere un effetto sulla pressione osmotica si hanno delle indicazioni
di tipo quantitativo, ovvero ci sono dei numeri che danno la misura di quanto quella sostanza a
quella concentrazione agisce sulla pressione osmotica (una pressione misurata in presenza di un
membrana ideale).
La tonicità tiene conto ("misura") l'effetto sulla variazione di pressione
osmotica di un soluto e in condizioni reali, ovvero in presenza di
membrane reali che siano permeabili sia all'solvente ma anche a qualche
soluto. Quindi valutare la tonicità vuol dire valutare l'effetto di una
soluzione su un volume cellulare. Quindi, dato che l’osmolarità e la
osmolarità sono dei numeri e che sono delle indicazioni di concentrazione
di un soluto che ha effetto sulla pressione osmotica in condizioni ideali, la
tonicità è una misura di variazione di volume cellulare quando le cellule
messe in contatto con una determinata soluzione, e infatti si parla di tonicità in quanto si parla di
membrane reali. La cellula di riferimento che si utilizza per fare queste misure è il globulo rosso,
che viene utilizzato come cellula perché è una cellula anucleata e quindi è più facile seguire la
variazione di volume.
Isotonia e Isoosmia

Quando la soluzione è isosmotica non vuol dire necessariamente che la
soluzione sia isotonica con i liquidi biologici come dire che una soluzione è
isotonica non vuol dire che sia isoosmotica. Isotonia e isoosmia sono due
concetti diversi. Iso osmotico vuol dire che la concentrazione in milliosmoli di
soluto è uguale all'interno e all'esterno dell'globulo rosso e quindi le due
soluzioni sono Iso osmotiche e quindi non c'è movimento di acqua perché non
si deve diluire la soluzione più concentrata e quindi la soluzione è isosomotica
ed in particolare è anche isotonica, perché il globulo rosso immerso in questa
soluzione non modifica il suo volume cellulare. Isotonico si riferisce così al
volume cellulare mentre isoosmotico al valore numerico della concentrazione
in milliosmoli al di là ed al di qua della membrana. Questo è vero perché la
membrana cellulare è impermeabile alla sostanza che si utilizza per generare
questa concentrazioni in milliosmoli (NaCl) e quindi essendo la membrana
impermeabile all'unico ione che è stato messo in soluzione, quindi questa
membrana si comporta come una membrana ideale.
Iperosmia e ipertonia
ci può essere il caso in cui la cellula vengono posti in una concentrazione iperosmotica, in cui la
concentrazioni in milliosmoli del soluto sia maggiore rispetto a quella che c'è all'interno della
cellula. C'è quindi un movimento di acqua dall'interno della cellula verso l'esterno e in questo caso
quindi la soluzione è iperosmotica ed era anche ipertonica perché si arriva alla modificazione del
volume cellulare. Se la soluzione è iper osmotica, vuol dire che la concentrazione è maggiore
all'interno e quindi ci sarà un movimento di acqua per bilanciare le due concentrazioni; quando la
soluzione all'esterno è ipoosmotica ci sarà movimento al contrario e quindi la cellula avrà una
variazione di volume cellulare sia perché c'è movimento di acqua ma perché c'è quindi una
variazione di concentrazione.
È più pericolosa di una soluzione ipertonica o ipotonica? È più pericolosa quella ipotonica
perché c'è un rigonfiamento dell'globulo rosso e che può portare alla sua lisi (scoppia), mentre nella
ipertonica non c’è la lisi cellulare, non necessariamente quindi. Infatti ci sono delle soluzioni
volutamente ipertoniche di glucosio che vengono somministrate per endovena nei casi di shock
anafilattico, quando c'è un edema nell'organismo si somministra delle soluzioni ipertonica per
richiamare l'acqua dai tessuti e diminuire l’edema. Quando una soluzione può essere isoosmotica
ma non isotonica con il sangue? Ad esempio la soluzione di urea alla 1,8% è isoosmotica con il
sangue, cioè ha 290 milliosmole, ma non è isotonica. Se la membrana impermeabile alla sostanza
che si considera (come ad esempio l'urea) non si sposta solo l'acqua ma si sposta tutto il soluto e
quindi si trascina dietro tutta l'acqua e quindi, non ci sarebbe movimento netto di acqua se la
membrana fosse impermeabile, ma siccome la membrana è impermeabile al soluto si sposta il
soluto che si trascina dietro anche una frazione di acqua. Ci sono delle soluzioni, come ad esempio
l'acido borico, che è isotonico con il sangue ma non è isotonico con gli altri liquidi biologici come
ad esempio il liquido lacrimare. Isoosmotico vuol dire che la concentrazione di soluto è uguale al di
qua e al di là della membrana e questo indica che si può considerare la membrana come una
membrana ideale, e la si può approssimare ad una membrana ideale quanto quella membrana è
impermeabile al soluto che si sta considerando. Quindi la membrana dell'globulo rosso è
impermeabile al cloruro di sodio, e quindi se si ha la stessa concentrazione al di qua ed al di là della

membrana le due soluzioni hanno uguale concentrazione e quindi non c'è movimento di acqua, in
quanto l'unica cosa che si può passare attraverso la membrana. Ma se la membrana è permeabile al
soluto, come ad esempio per l'urea, attraverso la membrana non c'è movimento di acqua perché la
concentrazione della specie attive sulla pressione osmotica sono uguali però si sposta il soluto
cambia la concentrazione tra le due parti separate dalla membrana e il soluto che si sposta si porta
con sé anche l'acqua e quindi c'era una variazione di concentrazione.
La soluzione è definita soluzione fisiologica cioè perfettamente isotonica con il sangue è una
soluzione allo 0,9% in NaCl p/v. Quindi la soluzione di riferimento è la soluzione fisiologica.
Quindi per preparare le soluzioni isotoniche si deve fare riferimento al cloruro di sodio e alla sua
concentrazione pari a 0,9% p/v. Quali strumenti numerici si hanno per preparare una soluzione
isotonica:
1) il primo è quello che si definisce equivalente in NaCl; si definisce come le grandi di cloruro
di sodio che hanno lo stesso effetto osmotico di 1 g di farmaco in 100 ml. Viene che un
farmaco ha un equivalente di NaCl di 0,5 vuol dire se si mette 1 g di farmaco in 100 ml è
come se si avesse aggiunto 0,5 g di NaCl. Quindi si trasforma in farmaco, come effetto della
pressione osmotica, in cloruro di sodio.
2) ∆t crioscopico: la soluzione fisiologica cioè allo 0,9% di cloruro di sodio ha un valore di ∆t
crioscopico varia 0,52 °C; motivo per cui quando c'è la si mette il sale per le strade perché si
alza la temperatura di congelamento dell'acqua;
3) volume isotonico che si definisce come il volume nel quale, scegliendo 0,3 g di farmaco si
ottiene una soluzione isotonica. Questo vuol dire che se il volume isotonico del farmaco A è
9 vuol dire che 0.3 g in 9 ml di acqua danno una soluzione sicuramente isotonica. Il volume
isotonico può essere anche riferito ad 1 g di farmaco ma deve essere detto espressamente.
Le preparazioni per le quali la farmacopea richiede il requisito di isotonia sono: i colliri e le
preparazioni parenterali, e quindi devono essere isotoniche con i fluidi biologici, in specie i colliri
devono essere isotonici con il liquido lacrimare. Le preparazioni oftalmiche oltre ad essere
isotoniche devono anche essere sterili e quindi le caratteristiche di farmacopea dei colliri sono la
sterilità e l’isotonia.
Preparazioni oftalmiche
Le preparazioni oftalmiche sono preparazioni liquide, semisolide o solide da applicare sul bulbo
oculare e/o sulla congiuntiva o da introdurre nel sacco congiuntivale. Le preparazioni oftalmiche si
preparano utilizzando materiali e metodi in grado di assicurare la sterilita' e di evitare l’introduzione
di contaminanti e la crescita di microrganismi; Nella produzione di preparazioni oftalmiche
contenenti particelle disperse, sono prese misure atte ad assicurare una idonea e controllata
dimensione delle particelle in relazione all’uso previsto (quindi si deve vedere anche il volume
particellare e delle dimensioni particellare).
Colliri
I colliri sono soluzioni acquose od oleose oppure sospensioni sterili, di uno o piu' principi attivi, da
instillare nell’occhio. I colliri possono contenere eccipienti, per esempio per regolare la tonicità o la

viscosita' della preparazione, per aggiustare o stabilizzare il pH, per aumentare la solubilità del
principio attivo o per stabilizzare la preparazione. Il pH deve essere stabilizzato e deve essere un pH
di compromesso tra la stabilità del principio attivo e la compatibilità del pH dell'occhio. Il
parametro della viscosità deve essere accuratamente valutato per due motivi:
1) innanzitutto perché la soluzione deve essere sufficientemente viscosa per avere un tempo di
residenza utile per l'occhio, perché una delle caratteristiche che gli occhio ha è quello di
liberarsi degli elementi estranei; anche perché il volume distillato e quindi la concentrazione
del principio attivo è estremamente bassa (una goccia ha un volume di circa 50 µl).
2) Inoltre un altro parametro è di non avere interferenza con la visione, perché un materiale
altamente viscosa può avere un alto indice di rifrazione, e quindi una volta che è stato
instillato nell'occhio da problemi di visione.
I colliri possono essere formulati sia multidose che unidose, e in quelli multi dose deve essere
contenuto un antimicrobico a meno che la preparazione stessa abbia una sufficiente proprietà
antimicrobica; i colliri unidose sono formulati senza antimicrobici, e la farmacopea specifica che i
colliri destinati ad un uso di interventi chirurgici devono essere formulati come preparazione
unidose perché non devono contenere agenti conservanti. Le soluzioni possono essere limpide e
prive di particelle, le sospensioni possono presentare un sedimento facilmente ridisperso per
agitazione. La farmacopea dice che deve dare una formulazione sufficientemente stabile per
consentire l'applicazione della corretta dose. La farmacopea dice che le preparazioni multi dose
possono avere al massimo una capacità di 10 ml, mentre i colliri industriali dall'apertura possono
essere utilizzati per un massimo di quattro settimane mentre i colliri galenici, dal momento
dell'apertura, possono essere utilizzati al massimo per 15 giorni.
I bagni oculari sono soluzioni acquose sterili destinate a lavare o bagnare gli occhi, o per impacchi.
Hanno le stesse caratteristiche dei colliri c'è solo la differenza che hanno dei volumi maggiori
perché servono per la detersione dell'occhio.
Le preparazioni oftalmiche semi solide hanno le stesse composizioni delle preparazioni semi
solide per uso dermatologico e quindi sono unguenti, creme e gel e devono essere sterili.
Nel vecchio formulario è presente una preparazione di atropina solfato (principio attico), una
soluzione oftalmica 0.5 – 1 %, che è un collirio di atropina, in cui c’è
Atropina solfato
NaCl, agente isotonizzante principale a meno che non ci siano delle incompatibilità
Cloro butanolo, è uno dei conservanti che si aggiunge nella preparazione dei colliri in quanto
liquido;
acqua depurata sterile q.b.
i colliri si sterilizzano per filtrazione; quindi si utilizza un filtro sterilizzate con una dimensione di
pori pari a 0,22 o meno in contenitori sterilizzati quindi il contenitore sterile, l'acqua e si divide
questa preparazione non è quella depurata ma è quella sterile e la soluzione che deve essere
preparata deve essere sterilizzata per filtrazione su filtri sterilizzati. Il nitrato di fenil mercurio è un
altro stabilizzante che nei colliri.

Sterilizzazione e disinfezione
tra le caratteristiche dei colliri, oltre l’isotonia, c'è anche la sterilità. Inoltre i parenterali dovranno
essere isotonici e sterili ma anche apirogeni (quindi un ulteriore passaggio nella realizzazione del
controllo di queste forme farmaceutiche). La sterilizzazione consiste nell'uccisione di tutti i
microorganismi patogeni e non presenti in un oggetto o di un ambiente e comprende l'inattivazione
di tutti i virus. La disinfezione è l'utilizzo di particolari sostanze chimiche, utilizzate per un certo
tempo di applicazione, le quali uccidono i microorganismi patogeni attraverso molteplici
meccanismi. Quindi differisce dalla sterilizzazione poiché questa consiste nell'uccisione dei soli
microorganismi patogeni.
I metodi di sterilizzazione possono essere: fisici o chimici. I metodi fisici prevedono il calore, la
radiazione e la filtrazione (utilizzando i filtri da 0,22 micron). I metodi chimici invece, che
nell'industria farmaceutica vengono utilizzati per sterilizzare gli ambienti, sono l'uso di acidi e basi
concentrati , sostanzialmente sostanze che sviluppano cloro, le bombe ad acetilene per sterilizzare
gli ambienti.
Per sterilizzare le forme farmaceutiche le tecniche privilegiate sono la filtrazione ed il calore, in
quanto con la filtrazione si sterilizza la forma farmaceutica che deve però essere suddivisa in unità
posologica ancora, e quindi si sterilizza la forma farmaceutica priva di contenitore e quindi poi
bisogna sterilizzare il contenitore, ed infine bisogna riempire il contenitore con la forma
farmaceutica sterile in un ambiente in cui avviene la contaminazione. Invece con il calore, si può
fare la sterilizzazione delle forme farmaceutiche già suddivise nella singola unità posologica e già
chiuse (per esempio per i parenterali al calore si sterilizzano iniziale); è un processo che deve essere
studiato accuratamente perché deve tenere conto delle caratteristiche e dei componenti della forma
farmaceutica ma è, quando è possibile, privilegiato perché consente di eliminare passaggi che
potrebbero compromettere la qualità del prodotto finale. I parametri che influenzano una
sterilizzazione ottimale sono: la temperatura e il tempo. Questi due parametri devono essere
adeguati a garantire la sterilizzazione, non degradare componenti che si trova nell'interno della
forma farmaceutica, e deve infine costare il meno possibile (in quanto riscaldare bollire consuma
energia e quindi un costo maggiore): quindi deve essere un processo efficiente ed il meno costoso
possibile.
Il concetto di sterilità è strettamente collegato ad un concetto di probabilità statistica, che viene
indicato con la sigla LAS ovvero il livello di assicurazione di sterilità: è la probabilità di trovare un
certo numero di unità contaminate sulla totalità delle unità che sono state sterilizzate. Secondo
farmacopea, si realizza una sterilizzazione ottimale se si ha un LAS di 10-6
cioè una unità
contaminate su un 1 milione di unità sterilizzate.
Nella sterilizzazione al calore c'è la possibilità di sterilizzare alla fiamma ma questa è una tecnica
che ovviamente si utilizza il laboratorio; quindi quello che si utilizza in realtà sono due forme di
sterilizzazione al calore secco o al calore umido. Nel calore secco (forno pasteur) l'agente
sterilizzante è l'aria calda che però ha le caratteristiche di essere un cattivo conduttore di calore e
avere uno scarso potere penetrante; inoltre, per avere un LAS accettabile bisogna sterilizzare per
un'ora o un'ora e mezza a temperature intorno ai 180°C ( o 200°C). Quindi sono delle temperature
alte e tempi lunghi: quindi di fatto nell'ambito dell'industria farmaceutica il calore secco viene

utilizzato per sterilizzare i contenitori di vetro (quando non si utilizzano le radiazioni, ma dato che
costano di più nelle stufe, ma generalmente vengono utilizzate per sterilizzare le parti di plastica per
i presìdi chirurgici) e gli strumenti metallici. Nella sterilizzazione a calore umido l'agente
sterilizzante è il vapore acqueo che ha una buona conducibilità termica ed un elevato potere
penetrante che consente una sterilizzazione più efficiente a temperature più basse e a tempi minori.
Nell'ambito della sterilizzazione calore umido quella che viene usata nella quasi totalità delle
industrie farmaceutiche è l'utilizzo del vapore sotto pressione (sterilizzazione sotto autoclave o
pentola a pressione); gli altri due tipi di sterilizzazione al calore umido sono a vapore fluente e la
sterilizzazione frazionata.
Come si misura la qualità del processo di sterilizzazione e come
si scelgono il tempo e la temperatura di compromesso tra un
LAS convincente e consumo energetico minore: Innanzitutto si
deve definire il decadimento decimale che si indica con D e che
indica i minuti necessari a ridurre una popolazione di del 90% a
determinate condizioni di temperatura; quindi, si riporta sulle
ordinate le frazioni di unità sopravvissute alla sterilizzazione in
funzione del tempo di esposizione, si può ricavare il valore di
decadimento decimale cioè i minuti necessari ad abbattere la
popolazione microbica del 90%.
un altro parametro che viene valutato è il coefficiente di temperatura integrato con Z ed è il
numero di gradi che fa variare di 10 volte il valore D, cioè il tempo necessario ad una determinata
temperatura di ridurre al 90% la concentrazione microbica.
Un altro fattore che viene valutato per scegliere la condizione ottimale di sterilizzazione è il
fattore di letalità che si indica con F0; si paragonano i processi di sterilizzazione che si vuole
mettere appunto con il processo di sterilizzazione a 121°C, che è la temperatura della sterilizzazione
ad umido e sotto pressione: prendendo questo valore, cioè 121°, come valore di riferimento, si
calcola il fattore di letalità cioè quanto è efficace il nuovo processo di sterilizzazione che si vuole
realizzare considerando la differenza di tempo. Per esempio si vuole paragonare il ciclo standard a
121° con un ciclo a temperatura più bassa (per esempio 115°) per otto minuti. Se si sterilizza 115°
per otto minuti è come avesse sterilizzato per due minuti a 121°; questo consente di valutare
l'effetto, cioè la letalità del processo di sterilizzazione che si vuole applicare, paragonandolo alla
sterilizzazione standard. Abbassare la temperatura vuol dire consumare di meno ma riscaldare per
due minuti a 121° quanto può spendere rispetto otto minuti a 115°? Se fosse uguale allora sterilizza
121°, se invece fosse conveniente si sterilizza 115°: tutti questi studi vengono fatti perché
appartengono alla documentazione che si deve produrre al ministero per richiedere l'autorizzazione
al ciclo di riproduzione e all'immissione in commercio del prodotto. Si parla di 121° perché, del
diagramma di stato dell'acqua, se si aumenta la pressione la temperatura di ebollizione non è più a
100° ma è più alta (121°); quindi un processo che si instaura prima dal punto di vista temporale, con
una temperatura superiore e quindi il processo sotto pressione è più rapido perché si arriva
all'ebollizione prima ma ad una temperatura superiore e quindi proporre che si produce ha una
temperatura più elevata e quindi una maggiore capacità di sterilizzazione nel caso della autoclave.
Le autoclavi nell'azienda farmaceutica sono delle stanze; alcune farmacie che fanno sterili, per

sterilizzare hanno aperto la pressione. Invece nei laboratori sono presenti le autoclavi; il ciclo
classico è di 121°C per 16 minuti. Il ciclo overkill e il ciclo bioburden sono dei cicli di utilizzano
nella sterilizzazione al calore, soprattutto usati nelle aziende. Il ciclo bioburden si utilizza quando la
flora microbica risulta più resistente ai cibi standard sterilizzazione.
In farmacopea esiste tra gli argomenti generali un capitolo sulla sterilità e sui mezzi di preparare
prodotti sterili; dove è possibile si sceglie un processo in cui il prodotto è sterilizzato nel suo
contributo risale (sterilizzazione terminale) e viene anche definito il livello di assicurazione di
sterilità: il LAS è la probabilità di persistenza di un elemento non sterile in questo complesso.
Quando non è possibile sterilizzare, per esempio per i farmaci proteici e per la quasi totalità dei
farmaci biotecnologici in cui la sterilizzazione al calore è impossibile, si utilizza un tipo di
procedura che si chiama produzione in asepsi, cioè tutto il processo produttivo e il processo di
condizionamento viene fatto in ambiente sterili. Le strutture in cui si opera in condizioni di totale
sterilità si chiamano clean room. In test che vengono fatti su queste camere si indicano come:
1) As built: in cui l'azienda valuta se il costruttore ha rispettati i requisiti richiesti per;
2) As rest: test a riposo, e vengono effettuati per valutare l'efficacia e la qualità soprattutto dell'aria
di queste stanze a macchine ferme e quindi in assenza di persone;
3) As use: quando nelle clean room si lavora e quindi con le macchine funzionanti e le persone
all'interno e si vanta, sostanzialmente, la qualità dell'aria.
Nelle clean room non si protegge sul prodotto farmaceutico dalla contaminazione atmosferica ed
umana ma si protegge soprattutto l'operatore dal materiale che sta manovrando (per esempio nelle
aziende che fanno antitumorali, o che usano derivati di virus o di sostanze particolarmente tossiche).
Le clean room si classificano in base a degli standard americani in classi e le classi misurano il
numero di particelle per metro cubo. La classificazione è appunto per numero di particelle per metro
cubo perché quello che viene valutato, sostanzialmente, è la contaminazione particellare dell'aria
che c'è all'interno di queste stanze; in questi ambienti entra dell'aria filtrata, su filtri HEPA (high
efficient particle air) o ULPA (ultra low penetration air). Un altro parametro che viene valutato è il
tipo di flusso, e quindi sono di filtri che filtrano all'aria l'ingresso e che assicurano dei moti laminari
di aria, in modo che l'aria dall'alto verso il basso. Un altro parametro che è presente in queste stanze
è la pressione, infatti in queste stanze c'è una pressione maggiore rispetto all'ambiente esterno e
quindi ogniqualvolta che la porta è aperta l'aria filtrata esce e l'aria che viene da fuori che potrebbe
essere contaminata non entra. In genere intorno alla clean room ci sono degli ambienti intermedi,
anche perché le persone che lavorano all'interno di queste hanno degli abbigliamenti particolari: lo
spogliatoio e dove si lavano gli operatori sono diversi da quelli delle altre aree dell'azienda. Inoltre
gli operatori hanno delle tute che li ricoprono totalmente, non possono avere i capelli lunghi e la
barba mentre le operatrice non devono portare gioielli ed essere toccate: molte hanno dei costi
estremamente ridotti perché sono delle condizioni di lavoro altamente stressanti.
I pirogeni sono delle sostanze tossiche, dei lipopolisaccaridi prodotti dalla degradazione della
membrana dei gram - , e si chiamano pirogeni e che agiscono sui centri di termoregolazione, quindi
alzano la temperatura a livello centrale e sono quindi molto pericolosi. Per valutare la presenza di
questi contaminanti all'interno di una preparazione, viene utilizzato un test che si chiama il LAL
test (lisato di amebocita di limulus –polifemus- perché è un granchio di dimensioni notevoli) , da

questa granchio si ottiene una lisato proteico e si metta a contatto con la soluzione che si vuole
valutare la pirogenicità ; il LAL test può dare un'indicazione di tipo qualitativo (con il metodo del
coagulo) , perché se mettendo a contatto il lisato con la soluzione che si vuole testare si forma un
coagulo da indicazioni di tipo qualitativo, o di tipo quantitativo o con il saggio del substrato
cromogenico (con l'aggiunta di un colorante al lisato proteico e quindi poi misurando l'intensità del
colore, con il test colorimetrico, si può misurare quanti pirogeni ci sono) o con il saggio
torbidimetrico, in cui si misura nell'eventuale presenza del coagulo l'incremento di torbidità. Le
tecniche di de pirogenazione sono:
rimozione dell’ endotossina: sfruttano sostanzialmente delle tecniche di ultra filtrazione (per
grandi volumi) o di osmosi inversa , in queste pagine che il controllo di fatto sulle dimensioni
dei poveri della membrana, oppure con tecniche come la filtrazione con filtri modificati o la
cromatografia che sfruttano l'interazione elettrostatica tra la membrana e la colonna
elettrostatica perché i lipopolissacaridi sono carichi negativamente e quindi utilizzando la
cromatografia per affinità, o a scambio ionico e la filtrazione su membrane cariche
positivamente si sfrutta l'interazione elettrostatica. Si può utilizzare anche un processo al calore
come la distillazione e nel caso di contenitori o di tappi in elastomero (in gomma) vengono de
pirogenati per diluizione ovvero la mando in maniera estensiva i contenitori ed i tappi prima con
acqua sterile e poi con acqua de pirogenata. Si può utilizzare anche il carbone attivo.
inattivazione o distruzione della endotossina: prevedono che qui metodi fisici (come il calore
sia secco che umido) sia agenti chimici; queste tecniche possono essere usate solo per i
contenitori perché le condizioni sono veramente drastiche: in un contenitore di vetro viene
mantenuto per un minuto 427° C mentre gli agenti chimici possono essere:
acidi e basi, NaOH 0.1 – 1 N/ HCl 0.1 N – 1N;
sostanze ossidative; H2O2
agenti chelanti anidride carbonica o succinica.
Un altro processo che interessa le forme farmaceutiche, soprattutto quelli per uso parenterali è la
liofilizzazione, perché la presenza di acqua in generale favorisce la degradazione dei principi attivi,
perché una forma farmaceutica in soluzione si degrada più rapidamente rispetto alla forma
farmaceutica solida. Per migliorare quindi la conservabilità e la stabilità di queste forme
farmaceutiche si realizzano delle forme farmaceutiche si che e quindi si possono avere o delle
polveri o dei liofilizzati. La caratteristica dei liofilizzati, rispetto alla polvere, è quella di disperdersi
in maniera rapida ed omogenea una volta messa a contatto con il solvente. La polvere liofilizzata o
liofilo (“amico dell’acqua”) si trasforma rapidamente e in modo omogeneo con un solvente perché
il processo di liofilizzazione non avviene attraverso un processo di essiccamento, cioè il passaggio
dal liquido a vapore, ma perché sfrutta il passaggio da solido a vapore. Quindi il processo di
liofilizzazione prevede una prima fase di congelamento, in cui l'acqua viene trasformata in ghiaccio,
e si sfrutta la sublimazione e non l'evaporazione come per l'essiccamento.
Diagramma di stato dell'acqua : viene tracciato usando l'equazione di Clausius clapeyron:

Il diagramma di stato dell'acqua la caratteristica di avere la curva di equilibrio solido-liquido con
una diversa inclinazione (tendenza negativa) poiché il solido, rispetto al liquido presenta un
incremento di volume.
Il processo di liofilizzazione ha come fasi: fase
preparatoria (1) in cui le sostanze sono portato in
soluzione, sono filtrate e ripartite nei contenitori perché
anche in questo caso la liofilizzazione viene effettuata
sull'unità posologica già ripartita; a volte è opportuno
aggiungere delle sostanze che si chiamano crioprotettori
che servono a stabilizzare le strutture che vengono
liofilizzate, soprattutto ad esempio quando si ha a che fare
con farmaci proteici o peptidici:generalmente le sostanze
che si aggiungono come crioprotettori sono degli zuccheri
(o il mannitolo o il trealosio);
le sostanze ripartite vengono inserite nella liofilizzazione che nelle industrie sono delle camere;
quindi la fase di liofilizzazione vera e propria inizia con il processo di congelamento (2) , in
quanto bisogna fare in modo che il congelamento sia rapido e quindi più veloce possibile e che
avvenga su uno strato sottile possibile; quindi quando è necessario le fiale sono fatte ruotare su se
stesse durante il processo di liofilizzazione oppure vengono posizionati intorno alle rastrelliere nelle
posizioni tali per fare in modo che lo strato di liquido sulle pareti sia più sottile possibile; questo
perché bisogna fare in modo che il congelamento sia il più uniforme e il più rapido di perché così in
questo modo si formano dei cristalli di ghiaccio piccoli e di dimensioni omogenee in tutta la massa
che dovrà essere liofilizzata. Il sistema di congelamento comprende vari metodi su come
possono essere posizionate le fiale
1) congelamento statico: in quella fiala viene tenuta ferma ma in piedi;
2) congelamento depositato, in cui la fiala viene messa quasi orizzontale ma sempre ferma;
3) congelamento per lenta rotazione o Shell freezing;
4) congelamento della rotazione veloce o Spin freezing: in cui grazie alla forza centrifuga il
liquido si attacca alle pareti laterali, sempre su strato sottile;
5) congelamento con stratificazione a conchiglia.
Dopo che il liquido è stato congelato inizia la fase della sublimazione (3) del ghiaccio; il processo
di sublimazione dell'avvenire a temperatura costante e quindi questo significa che per rimanere su
questa linea di equilibrio solido-vapore, man mano che avviene il processo di sublimazione che
assorbe calore si dovrebbe lentamente ma costantemente riscaldare il sistema perché si deve
continuare a ragionare calore in modo tale che il sistema resti nella fase solido-vapore. Questa è la

fase che viene definita essiccamento primario, che viene fatto sotto vuoto con un incremento
costante di temperatura e quindi la pressione è costante ma man mano questo processo sarebbe
calore e quindi bisogna sempre continuare a riscaldarlo. C'è inoltre la fase dell'essiccamento
secondario che non sarebbe più al processo di sublimazione ma serve ad eliminare le eventuali
umidità residua che è presente all'interno del sistema.
Se si fa un grafico in cui si riporta la
variazione di temperatura del processo di
liofilizzazione in funzione del tempo e si
osserva che si ha un inizio un abbassamento
di temperatura rapido, veloce e su strato
sottile; poi si ha processo di liofilizzazione
delle proprie in cui si mantiene costante la
temperatura e poi si ha la fase finale, di
essiccamento primario in cui la realizzazione
è terminata e si elimina l'umidità residua,
infatti in questa fase si fornisce calore alla
temperatura rimane costante perché è
assorbita dal processo di liofilizzazione.
Invece nella fase di essiccamento secondario,
il calore che viene fornito invece serve per
eliminare la frazione parziale di umidità
residua e quindi fornendo calore si aumenta
anche la temperatura.
Il prodotto liofilizzato è un prodotto che ha una consistenza della massa spugnosa e da un'elevata
idroscopicità, cioè a un'elevata tendenza a riacquistare acqua perché idroscopico vuol dire che
assorbe acqua. Quindi una volta terminato il processo di liofilizzazione, in contenitori devono
venire esclusi rapidamente utilizzando generalmente delle gomme dette elastomere e delle ghiere di
alluminio che chiudono ermeticamente la fiala. Se il preparato è iniziale la chiusura della fiala è
stata saldando la fiala alla fiamma, che sono dei flaconi come la maggior parte dei casi viene messo
il tappo di elastomero che deve essere perforabile da un ago.

forme farmaceutiche parenterali
parenterali vuol dire che va oltre l'intestino e quindi nel caso del principio attivo non subisce
l'effetto di primo passaggio cioè non passa attraverso il fegato come accade per i principi attivi che
sono assorbiti nel tratto gastroenterico. Avviene attraverso l'uso dell'ago, e asseconda delle zone
dell'organismo in cui avviene l'iniezione si possono distinguere le forme: epidermiche,
intradermiche (ID), sottocutanea (SC), intramuscolare (IM) e endovenosa (EV), in cui quest'ultima
il farmaco viene inserito direttamente nel torrente circolatorio e quindi non c'è nessuna barriera e di
nessuna assorbimento e quindi non c'è nessuna difesa da parte dell'organismo. Queste sono le vie
parenterali più comuni, infatti ci sono delle definite vie parenterali specializzate, che sono più
invasive rispetto alle altre, come la via intrarteriosa, la intratecale, intrcardiaca e intrasinovale.
Gli aghi hanno un calibro: diametro del ago è indicato con un numero detto gauge; tanto maggiore
è questo numero tanto più sottile largo. Ci sono diversi sistemi per indicare il Gauge , come il
sistema francese, ma quelli di uso più comune prende in considerazione il calibro da 7 (il più
grosso) a 33 (più sottile).
La via sottocutanea consiste nella iniezione della sottocute di volumi di liquido piccolo; la ago
strato sottile e corto. I volumi somministrati quindi sono molto piccoli e possono andare da 0,5- 1
ml di soluzione. Siti per la somministrazione sottocutanea sono il braccio, la parte anteriore della
coscia e l'addome. Possono essere per esempio i vaccini, insulina e l’eparina. Questi sono dei
distretti dell'organismo in cui è stato studiato che c'è il massimo assorbimento del principio attivo.
L’ago va inserito con un'angolazione di 90° rispetto all'superficie cutanea. Nella via intradermica
cambia la votazione dell'angolo che diventa 10-15° rispetto al sito di iniezione; i siti di
somministrazione sono gli stessi dello sottocutanea ma anche la scapola ma è una via usata
soprattutto per scopi diagnostici o per la somministrazione di anestetici locali.
Nel caso della somministrazione per via intramuscolare si possono somministrare volumi
maggiori da 2-5 ml. Si possono utilizzare due distretti dell'organismo: i glutei e il deltoide (se viene
fatta di queste più rapida perché è stretto più irrorato rispetto i glutei). I veicoli che possono essere
utilizzati per queste forme farmaceutiche possono essere acquosi o oleosi; l'utilizzo di un veicolo
oleoso viene utilizzato in genere quando si vuole realizzare una forma deposito perché se si inietta
una forma acquosa il farmaco in soluzione passera nel torrente circolatorio ma al contrario se viene
iniettata una forma oleosa, o generalmente il principio attivo è solo liposolubile oppure perché si
vuole avere un'azione di rilascio lento e quindi c'è il nucleo che è stato iniettato oleoso e il principio
attivo si dovrà ripartire a seconda delle sue caratteristiche tra il veicolo oleoso e i fluidi biologici.
Ovviamente, essendo il punto di inoculazione il muscolo un parametro che influenzano fortemente
l'assorbimento e l'effetto di una somministrazione intramuscolare è lo spessore del pannicolo
adiposo, in quanto si basa la costituzione l'effetto di questa forma farmaceutica sarà diversa.
Per esempio per scegliere la lunghezza dell’ago nel deltoide per avere una somministrazione
efficiente:
- uomini 25 mm;
- donna < 60 Kg, 16 mm;
- donna tra 60 – 90 Kg, 25 mm;

- donna > 90 Kg, 38 mm.
Nell'ambito delle preparazioni parenterali ci sono le forme farmaceutiche iniettabili. Le
somministrazioni per via endovenosa sono quelle che devono essere controllate di più in assoluto
perché sono quelle che non prevedono la fase di assorbimento del farmaco viene iniettato
direttamente nel torrente circolatorio e quindi l'organismo non ha difese da opporre alla presenza del
farmaco; ci sono due tipi di somministrazione:
- lenta, che è quella che si definisce per infusione (flebo o con siringa), può arrivare fino a 50
ml per unica dose;
- rapida, in cui si inietta la dose di farmaco e si chiama anche in volo si può iniettare un
massimo di 20 ml.
I veicoli per le preparazioni iniettabili sono l'acqua, le preparazioni iniettabili (acqua pi) che deve
essere sterile e apirogena, ed è possibile anche l'utilizzo di solventi non acquosi come l'alcol etilico,
il glicol propilenico o polietilen glicol e anche l'utilizzo di veicoli oleosi, solo per le
somministrazioni intradermiche, sottocutanea e intramuscolare in quanto si utilizza come forma di
deposito e possono essere utilizzati oli sintetici ma in generale quelli vegetali (come l'olio di
sesamo) oppure oli semisintetici (come l’oleato di etile).
Le preparazioni parenterali secondo farmacopea: Le preparazioni parenterali sono preparazioni
sterili destinate alla somministrazione per iniezione, infusione o impianto nel corpo umano o
animale. Si possono distinguere varie categorie di preparazioni parenterali:
- preparazioni iniettabili,
- infusioni, preparazioni in grande volume per infusione generalmente endovenosa;
- concentrati per preparazioni iniettabili o infusioni, quindi delle soluzioni concentrate che
vengono diluite al momento della somministrazione;
- polveri per preparazioni iniettabili o infusioni,
- gel per preparazioni iniettabili,
- impianti.
La caratteristica comune di queste preparazioni è quella della sterilità,della isotonia nel caso delle
preparazioni liquide e della assenza di pirogeni. Vengono specificati i contenitori: Le preparazioni
parenterali sono fornite in contenitori di vetro o in altri contenitori come contenitori in plastica e
siringhe preriempite. La tenuta del contenitore e' assicurata con opportuni accorgimenti. Le chiusure
garantiscono l’ereticità' , impediscono l’accesso di microrganismi e altri contaminanti e di norma
consentono il prelievo di una parte o di tutto il contenuto senza rimuoverle. I materiali plastici o
elastomeri di cui e' fatta la chiusura, sono sufficientemente compatti ed elastici da permettere il
passaggio di un ago con il minor distacco possibile di particelle. Le chiusure per contenitori
multidose sono sufficientemente elastiche da garantire che il foro si richiuda quando si toglie l’ago.
per queste preparazioni le caratteristiche dei materiali cioè del vetro e della plastica sono più
stringenti rispetto alle altre preparazioni. In particolare, il vetro per uso farmaceutico si distingue in
tre classi (che il vetro è un derivato della sabbia che si ottiene per la fusione di essa): la
classificazione del vetro dal tipo I al IV è legata alle sue caratteristiche di resistenza idrolitica. Per
preparare il vetro, nel processo di fusione della sabbia, vengono aggiunti dei sali per rendere la

massa fusa nella parte più lavorabile. Quindi un problema del vetro, come materiale, è quella della
cessione idrolitica cioè il vetro può cedere, in presenza di soluzioni acquose, degli ioni che possono
cambiare il pH della soluzione contenuta all'interno del contenitore.
- Il Vetro di tipo I, è il vetro migliore (boro – silicato) a cui sono stati aggiunti dei sali che
conferiscono una resistenza idrolitica a tutta la massa del vetro e quindi inteso che viene
preparato e quindi contenitori che vengono preparati con il vetro di tipo I si dice che hanno
una resistenza idrolitica in massa. Come si esegue il saggio: la fiala o il contenitore viene
frantumato e viene posta all'interno di una soluzione acquosa e si rivaluta la cessione
alcalina di ioni da parte del vetro frantumato; il vetro di tipo I è il più costoso ed è quello che
si usa per gli emo - derivati per i contenitori quindi in cui verranno poste le preparazioni
iniettabili a base di di sangue umano o di derivati del sangue;
- il vetro di tipo II invece ha quella che si definisce resistenza idrolitica superficiale, cioè le
pareti interne del contenitore e solo queste sono trattate costruendo un velo di ioni che
impediscono la cessione idrolitica ma solo dalla superficie interna. Il caso viene fatto
riempiendo di acqua al contenitore e valutando le variazioni sia di pH che di ioni presenti in
soluzione; questo tipo di vetro si utilizza per le forme farmaceutiche parenterali in veicolo
acquoso;
- il vetro di tipo III è il vetro classico con delle caratteristiche particolari di resistenza e si
utilizza per le altre forme farmaceutiche come i liquidi acquosi ma per esempio anche per
sciroppi o parenterali (ovvero iniettabili) in veicolo oleoso;
- il vetro di tipo IV è il vetro di tipo alimentare.
I saggi sono previsti in farmacopea per le preparazioni parenterali sono: contaminazione
particellare, saggio di sterilità ma un saggio importante è quello della Contaminazione particellare:
In questo saggio devono essere assenti particelle sia visibili che non visibili (che prevederà delle
tecniche analitiche che permettano di evidenziare queste particelle). Il saggio delle particelle
invisibili viene anche chiamata sperlatura delle fiale che ormai viene fatta utilizzando dei
macchinari ma nella quasi totalità delle aziende qualche anno fa le faceva a mano solo dalle donne.
Le preparazioni iniettabili secondo farmacopea: Le preparazioni iniettabili sono soluzioni,
emulsioni o sospensioni sterili. Sono preparate disciogliendo, emulsionando o sospendendo il o i
principi attivi e qualunque altro eccipiente aggiunto in acqua, in un adatto liquido non acquoso che
puo' essere non sterile se giustificato o in una miscela di questi veicoli. Le soluzioni per
preparazioni iniettabili, esaminate in condizioni di luce adatta, sono limpide e praticamente prive di
particelle. Le emulsioni per preparazioni iniettabili non mostrano segni di separazione di fase. Le
sospensioni per preparazioni iniettabili possono presentare un sedimento che e' facilmente disperso
all’agitazione per dare una sospensione che rimane sufficientemente stabile per permettere di
prelevare la corretta dose. Preparazioni multidose. Le iniezioni acquose multidose contengono un
adatto antimicrobico ad una concentrazione appropriata eccetto quando la preparazione ha di per sé
adeguate proprieta' antimicrobiche. Quandouna preparazione per uso parenterale e' fornita in un
contenitore multidose, sono indicate le precauzioni da prendere per la sua somministrazione ed in
particolare per la sua conservazione tra prelievi successivi. Antimicrobici. Le preparazioni acquose
che vengono preparate usando condizioni asettiche e che non possono essere sterilizzate al termine
del procedimento possono contenere un adatto antimicrobico in concentrazione appropriata.

Non viene aggiunto alcun antimicrobico quando:
- il volume da iniettare in una dose unica supera i 15 ml, se non diversamente giustificato,
- la preparazione e' destinata alla somministrazione per vie dove, per ragioni mediche, non e'
accettabile un antimicrobico, quali quella intracisternale, epidurale, intratecale o per
qualsiasi altra via che dia accesso al liquido cerebrospinale, o intra- o retrooculare.
Tali preparazioni vengono presentate in contenitori a dose unica. Nelle preparazioni a dose unica
deve essere valutato il volume estraibile dal contenitore e quindi la fiala (o il flacone) devono
permettere il prelievo della dose nominale; quindi il volume all'interno della fiala o del flacone sarà
sempre leggermente maggiore rispetto al volume e si definisce estraibile e quindi del volume
nominale del farmaco. Quindi la preparazione deve essere realizzate in modo che, all'interno della
fiala del flacone resti sempre un'uguale quantità di liquido e che il volume che si riesce prelevare
contenga la dose corretta di principio attivo.
Infusioni: Le infusioni sono soluzioni acquose o emulsioni, con acqua come fase continua, sterili;
esse vengono generalmente rese isotoniche con il sangue (perché ci sono preparazioni che sono
realizzate ipertoniche soprattutto nello shock anafilattico). Sono principalmente destinate alla
somministrazione in grande volume. Le infusioni non contengono alcun antimicrobico aggiunto. Le
soluzioni per infusione, esaminate in condizioni adatte di visibilita' , sono limpide e praticamente
esenti da particelle.
Le polveri per preparazioni iniettabili o infusioni sono sostanze solide, sterili, ripartite nei loro
contenitori finali e che, quando vengono agitate con il volume prescritto di un dato liquido sterile,
danno luogo rapidamente o a soluzioni limpide e praticamente prive di particelle o a sospensioni
uniformi. Dopo dissoluzione o sospensione, esse soddisfano alle specifiche per le iniezioni o per le
infusioni.
I prodotti liofilizzati per uso parenterale sono considerati come polveri per preparazioni iniettabili
o infusioni. Infatti da ricordare che la differenza tra le polveri e liofilizzati è che le polveri sono
preparati per le miscelazione secca di varie polveri mentre il liofilizzato è un prodotto che si
ricostituisce immediatamente in acqua. Quando si congela si mantiene la struttura della
preparazione, invece quando si attua la sublimazione la struttura rimane intatta ed è per questo che
il liofilizzato è immediatamente ricostituito perché non c'è un processo di dissoluzione ma sarà un
processo di ricostituzione ed infatti per le polveri, nel caso in cui si deve realizzare una soluzione
parenterale si parla di soluzione e quindi è un processo di dissoluzione, mentre nel caso delle
utilizzato si parla di ricostituzione ovvero non viene sciolto nell'acqua ma è "l'acqua che entra”. Le
polveri per uso iniettabile in dose unica dovranno rispondere al saggio di uniformità di massa,
obbligatorio per tutte le forme farmaceutiche solite unidose, al passaggio di uniformità di contenuto,
qualora il principio attivo o i principi attivi sia inferiore a 2 mg o al 2%.
Gli impianti sono preparazioni solide, sterili, di dimensione e forma adatte per l’impianto e il
rilascio del/dei principi attivi per un periodo di tempo prolungato. Ciascuna dose viene fornita in un
contenitore sterile. Vengono impiantati sottocute, la loro caratteristica formulativa è quella di
garantire un rilascio dei principi attivi prolungato nel tempo. Perché è un piccolo intervento
chirurgico per essere immessa sottocute.

Nella farmacopea che il capitolo dei materiali e dei contenitori e nel capitolo dei contenitori viene
data la definizione di contenitori unidose e multi dose e viene data anche la definizione del tipo di
chiusura; quindi se la farmacopea dice che contenitore deve essere ben chiuso, significa che il
farmacista deve acquistare dal fornitore il contenitore come ben chiuso e sulle specifiche nel
certificato di analisi del contenitore della prescritto che risponde a questi requisiti di farmacopea.
Ermeticamente chiuso vuol dire che è impermeabile a soldi, liquidi e gas in condizioni normali di
manipolazione e di conservazione di trasporto. Il contenitore con chiusura inviolabile vuol dire che
la chiusura deve rilevare se il contenitore è stato aperto; questa tipologia di chiusura viene data della
farmacia ospedaliera in cui si deve essere certi, che sulla richiesta di reparto per i farmaci della sala
operatoria, che questi non siamo mai stati utilizzati. I contenitori a prova di bambino hanno una
chiusura particolare.

Farmaci proteici e biotecnologici
hanno dei problemi formativi in quanto hanno generalmente delle dimensioni maggiori rispetto ai
farmaci di reazione chimica; mediamente vanno dai 600 - 10.000 dalton di peso molecolare e questa
caratteristica influenza la capacità di permeabilità di questi farmaci. Sono anche i farmaci che
vengono degradati dai sistemi enzimatici, e quindi anche la dose che deve essere somministrata
deve tenere conto che una frazione di questi farmaci viene immediatamente digerito da parte degli
enzimi. Quindi questi farmaci hanno un problema di biodisponibilità, ma hanno anche altri
problemi formativi rispetto agli altri farmaci perché per esempio sono sicuramente tutti termolabili
perché il calore, presente in quasi tutti i processi produttivi, degrada le proteine. Poiché la quasi
totalità delle preparazioni di natura proteica in particolare i farmaci biotecnologici sono
somministrati per via iniettiva, il processo di sterilizzazione e di isotonicizzazione sono molto
complessi perché generalmente, la sterilizzazione viene fatta al calore, mentre l’isotonicizzazione
viene fatta con il cloruro di sodio: l'aggiunta di cloruro di sodio può dare problemi di stabilità delle
proteine con il fenomeno di salting in e di salting out. Quindi la sterilizzazione del prodotto finito è
praticamente impossibile se non in alcuni casi che viene fatta per filtrazione, ma nella quasi totalità
dei casi si lavora in asepsi, e quindi la produzione di questi farmaci viene fatta in sepsi
generalmente in classe di stanze 100 (in cui la suddivisione di queste clean room fatta in base al
numero di particelle presenti in 1 m³) e significa che ci sono al massimo 100 particelle con una un
diametro non maggiore di 0,5 micron in 1 m³ di aria; l'aria viene filtrata con tappe a flusso è
laminare (i filtri HEPA e ULPA). Devono naturalmente essere eliminati i pirogeni e deve essere
ridotta al minimo la presenza di eccipienti a rischio cioè eccipienti di derivazione umana (emo –
derivati o albumina serica); l'albumina serica deve essere eliminata per problemi di shock
anafilattico, ed era presente quasi sempre nella formulazione di questi tipi di farmaci perché i
farmaci proteici si adsorbono sull'contenitori di plastica, sulle pareti, e quindi contenitori in genere
venivano trattati con l'albumina che si adsorbe sulle pareti e quindi contrastava fisicamente la
perdita di principio attivo. Per ridurre al minimo l'uso di questo recipiente sono stati studiati classi
diverse che non di una interazione con i farmaci peptidici e proteici e una derivazione di farmaci
che inibisce questo tipo di interazione.
Quali sono gli eccipienti che si aggiungono:
- agenti stabilizzanti: per il pH, tensioattivi e aminoacidi (arginina e lisina) che funzionano
da agenti solubilizzanti
- agenti antiaggreganti e antiadsorbenti: tensioattivi e quando proprio non se ne può fare a
meno albumina.
I sistemi tampone che si utilizza per mantenere il pH sono in genere tamponi fosfati o tamponi
citrati, e gli antiossidanti che si aggiungono qualora sono richiesti sono generalmente i più naturali
possibili e quindi l'acido ascorbico (vitamina C) e la cisteina.
Un esempio di farmaco biotecnologico dell'insulina, ha un problema formulativo aggiuntivo
rispetto agli altri in quanto funziona come monomero ma tende a dare dei aggregati legati tra di loro
con legami covalenti che rendono l'insulina inattiva; quindi in più, dell'insulina, viene aggiunto lo
zinco che faccio formare gli aggregati che però non siano con legami covalenti e quindi poi in vivo
liberano il monomero di insulina. L'insulina è stato il primo farmaco biotecnologico immesso nel

mercato perché l'insulina che prima era utilizzato era un insulina di derivazione animale (ovina ma
soprattutto suina). Gli equivalenti dei farmaci biotecnologici sono definiti biosimilari.
La via orale è alquanto complicata per i farmaci proteici perché tendono ad essere digeriti del tratto
gastroenterico anche se c'era uno studio enorme per la somministrazione dei vaccini per via orale,
soprattutto per le vaccinazioni non è paesi industrializzati ma per le vaccinazioni di massa nei paesi
meno industrializzati perché si è stabilito dell'acqua e quindi si può arrivare un numero molto
maggiore di persone vaccinate. Le vie preferenziali sono la Via intramuscolare e la via sottocutanea,
considerando ovviamente la presenza delle peptidasi tissutali: generalmente si aggiungono nelle
preparazioni degli inibitori di questi enzimi come per esempio l'antibiotico della bacitracina.
Raramente sono somministrati per via endovenosa in quanto hanno un emivita estremamente
ridotta.
Degli esempi di farmaci proteici sottolineando le caratteristiche cumulative e non un principio
attivo: una classi di farmaci proteici biotecnologici è quella dei fattori di crescita emopoietici, che
comprende due classi
- fattore di stimolazione delle colonie di granulociti; rG - CSF
- eritropoietina: rEPO
l’indicazione r davanti al nome del farmaco indica che è stato prodotto con tecniche di DNA
ricombinante. Infatti nella definizione di farmaco biotecnologico rientra il fatto di essere prodotte
con tecniche biotecnologiche ovvero attraverso tecniche di di combinazione del materiale genetico.
Dal punto di vista formativo possono essere soluzioni pronte o liofilizzate che vengono in questo
caso somministrati per via endovenosa, dato che sono dei componenti del sangue e quindi non sono
metabolizzati e hanno una stabilità limitata dopo diluizione o ricostituzione.
Un altro esempio sono le interleuchine e gli interferoni che sono somministrate generalmente gli
interferoni in via sottocutanea o intramuscolare mentre l'interleuchina anche per via endovenosa;
sono anche questi commercializzati come liofilizzate e la soluzione per ricostituire questa
preparazione è una soluzione al destrosio al 5 % dove il destrosio funziona da isotonicizzante.
Generalmente queste formulazioni, poiché sono prive totalmente di conservanti, devono essere
inviati al massimo entro 48 ore dalla ricostituzione (per esempio l'interferone β liofilizzato può
essere utilizzato al massimo entro tre ore dalla ricostituzione). I prodotti in commercio sono:
- interleuchina IL 2 ricombinante;
- interferone α ricombinante
- interferone β liofilizzato
- interferone γ Ricombinante
un altro esempio è l'ormone della crescita:
- secreto dalle cellule somatotrope (ipofisi anteriore);
- rhGH, dove rh significa ricombinante umano
- hGH da ipofisi di cadavere.
Anche in questo caso le preparazioni sono liofilizzate, utilizzando come crioprotettori la glicina o il

mannitolo e sono ricostituite in acqua sterile o in soluzione salina batteriostatica multi dose. Un
esempio di ormone della crescita è il vutropin AQ® (soma tropina rDNA) ottenuto con tecniche di
DNA ricombinante con zie di somministrazione non convenzionali; tutti questi farmaci sono
iniettabili e le persone che devono fare uso di questi farmaci per tutta la vita l'uso dell’ago può
risultare l'uso e quindi sono di studio dei dispositivi che consentono di realizzare la stessa via di
somministrazione in assenza di dialogo; ci sono infatti delle penne che sono ad aria compressa in
cui il farmaco viene sparato sottocute con aria compressa. Questi dispositivi sono stati fatti per
aumentare la patient compliance ma queste penne non sono comunque molto accettati perché
lasciano degli ematomi.
Per quanto riguarda l'insulina, dal punto di vista formativo ci sono diversi tipi di insulina che si
distinguono in base all'insorgenza dell'azione cioè il tempo necessario per far restaurare l'azione
della formulazione e il tempo di durata dell'azione. Una classe di questa è l'insulina regolare ad
azione rapida: sono delle soluzioni limpide e l'azione di stabilizzazione dell'insulina che tende a
formare degli aggregati covalenti ottenuta o per aggiunta di zinco, che coordina 6 esameri distinti di
insulina che vengono poi liberati in vivo, e un altro tipo di insulina umana monomerica è quella che
viene chiamata LIS PRO perché all'interno della struttura vengono invertiti, con tecniche
ricombinanti, un residuo di prolina e un residuo di lisina e questo impedisce l'aggregazione in
strutture sovramonomeriche: quest'inversione consente di ottenere insulina monometrica e quindi di
avere un picco d'azione dopo un'ora dalla somministrazione (con una durata d'azione due-tre ore).
Poi ci sono le insulina ad azione intermedia in cui si ha un instaurarsi dell'adesione e della durata
dell'azione più lunga del tempo, e sono realizzate dal punto di vista formativo utilizzando tecniche
diverse. Si ha l'insulina NPH (neutral protamine Hagedom) in cui l'insulina viene coordinata non
dallo zinco ma da un complesso di protamina, ovvero l'insulina viene cocrisallizzata con la
protamina, e si ottiene una sospensione con un picco a sei-12 ore con una durata d'azione di 18 a 24
ore perché questo complesso cristallino si dissolve lentamente nell'organismo mantenendo una
concentrazione di insulina utile per un tempo più lungo. Nel insulina lenta, invece, l'azione più
lunga è realizzata mescolando due forme di insulina: 70% di forma cristallina e 30% di forma
amorfa, e questo fa in modo che l'insulina cristallina si dissolva più velocemente e poi più
lentamente si dissolva l'insulina amorfa. Esiste poi il insulina ultra lenta in cui c'è una sospensione
di insulina cristallina al 100%. Il tempo di picco di questa insulina lenta è di 8:20 ore e la durata
d'azione è tra le 24 e le 48 ore. Quindi le caratteristiche di biodisponibilità variano e quindi di
farmaco cinetica in vivo variando solo le caratteristiche formative del farmaco; si sceglie in base al
tipo di diabete e al tipo di funzione che serve al diabetico (se il diabetico fatto un passo
particolarmente abbondanti si preferirà un farmaco rispetto all'altro).













![[Prista] - Tecnologia Farmaceutica v.2](https://img.dokumen.tips/doc/110x75/56d6bf2e1a28ab301695374f/prista-tecnologia-farmaceutica-v2.jpg)















