Embed Size (px)
Citation preview

SUT-1 enables tau-induced neurotoxicity inC. elegans
Brian C. Kraemer1,2,* and Gerard D. Schellenberg1,2,3,4
1Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA
98108, USA, 2Division of Gerontology and Geriatric Medicine, Department of Medicine, 3Division of Neurogenetics,
Department of Neurology and 4Department of Pharmacology, University of Washington, Seattle, WA 98195, USA
Received March 29, 2007; Revised and Accepted June 3, 2007
We previously reported a transgenic Caenorhabditis elegans model for tauopathies in which expression ofhuman tau in neurons caused insoluble phosphorylated tau accumulation, neurodegeneration and uncoor-dinated movement (Unc). To identify genes participating in tau neurotoxicity, we conducted a forward geneticscreen for mutations that ameliorate tau-induced uncoordination. The recessive mutation sut-1(bk79) par-tially suppresses the Unc phenotype, tau aggregation and neurodegenerative changes caused by tau. Weidentified the sut-1 gene and found it encodes a novel protein. We conducted a yeast two hybrid screen toidentify SUT-1 binding partners and found UNC-34, the C. elegans homolog of the cytoskeletal regulatoryprotein Enabled (ENA). In vitro protein binding assays and genetic studies validated the interaction betweenSUT-1 and UNC-34. The SUT-1/UNC-34 protein–protein interaction plays a role in both the normal function ofUNC-34 and in the tau-induced phenotype. Thus, we have found a conserved molecular pathway participatingin tau neurotoxicity in C. elegans.
INTRODUCTION
The mammalian neuronal cytoskeleton contains tubulinprotein polymerized into microtubules (MTs), actin proteinpolymerized into microfilaments (MFs) and three distinct neu-rofilament protein subunits polymerized into neurofilamentfibrils (NFs). Tau is a microtubule-associated protein foundin neuronal axons and is involved in regulating MT dynamics(reviewed in 1). Normally, unphosphorylated tau protein bindsto MTs stimulating MT polymerization and promoting MTstabilization (2). In cooperation with other cytoskeletal com-ponents, neuronal MTs function to transport cargo from thecell body to synapses along axons, to develop and maintainaxonal structure, and to support growth cone and neuronalmigration (reviewed in 3). Also, tau may participate indynamic actin-mediated processes (e.g. neuronal processextension) either by directly binding to actin (4), or by inter-acting with specific kinases (5) that control these processes,or by binding to proteins that in turn bind actin directly. TheCaenorhabditis elegans protein closest to tau is the proteinwith tau-like repeats-1 (ptl-1). In adult worms, PTL-1 isonly expressed in 6 touch neurons, but is not required for
the mechanosensory function of these neurons (6,7) or forlocomotion.
In a number of neurodegenerative disorders, tau poly-merizes into abnormal filaments forming neurofibrillary andglial tangles (2,8). These disorders, collectively called tauopa-thies, include Alzheimer’s disease (AD), Down syndrome,corticobasal degeneration (CBD), progressive supranuclearpalsy (PSP), Pick’s disease, Guam amyotrophic lateral scler-osis/Parkinson’s dementia complex and frontotemporaldementia with parkinsonism chromosome 17 type with taupathology (FTDP-17T) (2,9). While the role tau-containinglesions play in most of these disorders remains unclear, inFTDP-17T autosomal dominant mutations in the gene encod-ing tau (MAPT ) cause the disease (10–12). These mutationsdemonstrate changes in tau can directly cause neurode-generation. However, the precise mechanism of tau-inducedneurodegeneration remains unclear. One hypothesis is thatabnormal tau disrupts axonal MT function resulting in synap-tic loss and neuronal dysfunction. As neurons accumulateaggregated tau deposits, MT function becomes increasinglyimpaired until MT axonal transport is disrupted causing neuro-degeneration (13,14). Alternatively, tau aggregates could be
Published by Oxford University Press 2007.
*To whom correspondence should be addressed at: Seattle Veterans Affairs Puget Sound Health Care System, S182, 1660 South Columbian Way,Seattle, WA 98108, USA. Tel: þ1 2062773275; Fax: +1 2067642569; Email: [email protected]
Human Molecular Genetics, 2007, Vol. 16, No. 16 1959–1971doi:10.1093/hmg/ddm143Advance Access published on June 18, 2007
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

intrinsically toxic, killing neurons directly. This paradigmholds that FTDP-17T mutations cause disease by promotingtau self-aggregation either by reducing the affinity of tau forMTs resulting in an increase in free tau concentrations (15),or by accelerating the rate of tau self-aggregation (16,17). Fur-thermore, tau in its pathological state is hyperphosphorylatedsuggesting tau phosphorylation state could play an essentialrole in neurodegeneration (18).
To explore the possible causes of tau-induced neurodegen-eration, we developed a transgenic C. elegans model of humantauopathy diseases using either normal human tau codingsequences, or tau with FTDP-17 mutations P301L or V337M
as transgenes (19). In this model, the pan neuronal aex-3promoter (20) drives expression of tau in all neurons. Theinitial phenotype seen in the transgenic lines is uncoordinatedlocomotion (Unc), a phenotype characteristic of a variety ofC. elegans nervous system defects. As the Unc phenotypebecomes more severe with age, phosphorylated, insolubletau begins to accumulate, which is a hallmark of human tauo-pathies. A progressive neurodegenerative phenotype occurswith gaps and bulges developing in GABAergic nerve cordaxons and subsequent neuronal loss. By age 9 days, abnormaltau aggregates are observed by transmission EM in axons thatshow severe degenerative changes that include axoplasmicclearing, dilated regions and onionskin membranous infoldstypical of other C. elegans neurodegenerative phenotypes.For these phenotypes, the FTDP-17 mutant transgenic linesare more severe than lines that express the normal humantau sequence. Thus, the model recapitulates several key fea-tures seen in human tauopathy disorders. To identify genesthat participate in tau neurotoxicity, we carried out aforward genetic screen to identify mutations that prevent thetau-induced Unc phenotype. We isolated a recessive mutationthat suppresses the strong Unc phenotype induced by tau. Wecall the mutated gene suppressor of tau 1 (sut-1). Animals car-rying a loss of function mutation in sut-1(allele bk79) areresistant to the toxic effects of tau indicating that theproduct of this gene, the SUT-1 protein, is essential for tauneurotoxicity in C. elegans.
RESULTS
The C. elegans tauopathy model used here is transgenic for thehuman tau protein with the V337M FTDP-17 mutation (strainT337), and the transgene is expressed in all neurons. Theresult is an uncoordinated worm with reduced motility and aneurodegenerative phenotype. To identify genes required forthis phenotype, we mutagenized strain T337 and screenedfor mutants with normal (non-Unc) locomotion. We isolated72 putative suppressor mutants with near-normal locomotion,11 of which bred true. Here we describe the characterizationof one mutant line called bk79. The tau transgene was notaltered by mutagenesis in bk79 because out-crossed mutantlines had Unc progeny similar to those of the T337 parentalstrain. When allele bk79 is present in the homozygous statein the parental T337 line, motility is restored to near wildtype levels as measured by liquid thrashing assays (Fig. 1A).In the absence of the tau transgene, bk79 has normal motilityessentially identical to N2, the wild type C. elegans strain used(Fig. 2E).
We used single nucleotide polymorphism mapping (21) tolocalize the bk79 mutation to chromosome II between poly-morphic markers uCe2-577 and uCe2-725, an interval of�298 kb (Fig. 1B). To localize the mutated gene more pre-cisely, we injected the bk79 mutant line with pools ofcosmid clones spanning the interval between uCe-2-577 anduCe2-725. We found a single cosmid, T13B5 (Fig. 1B), thatwhen present as an extrachromasomal transgene, preventedthe bk79 suppression of the tau induced Unc phenotype.T13B5 contains seven complete genes. A genomic PCR frag-ment containing the gene T13B5.8 relieved bk79 suppressionof tau when expressed as a transgene. We sequenced theexons of T13B5.8 in bk79 and found an early stop codon inexon 3 encoding a Y39X change (Fig. 1C). Thus, bk79 containsa mutation in gene T13B5.8, and we renamed this gene sut-1.
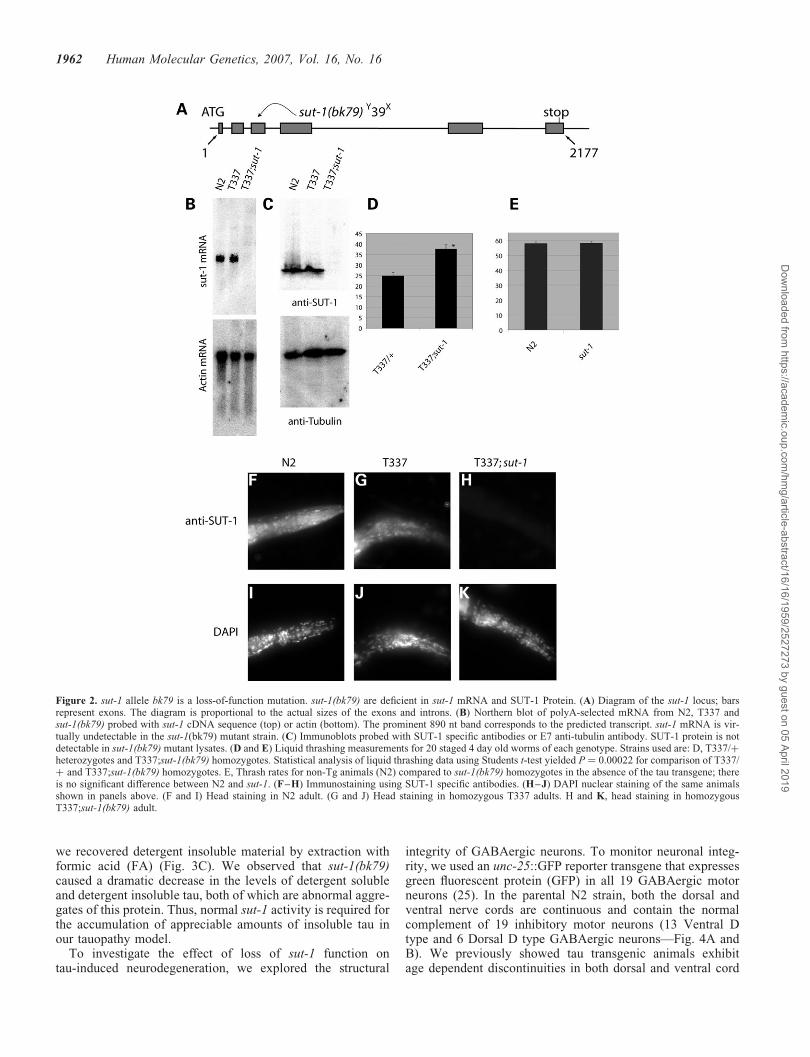
The predicted gene product encoded by the sut-1 gene is anovel 234 amino acid long protein (SUT-1). The gene has sixpredicted exons and covers �2 kb of genomic sequence(Fig. 2A). We sequenced three cDNAs (yk459d2, yk636d5and yk657g8) confirming the predicted open reading framefor sut-1. Northern blot analysis shows a single sut-1 transcriptconsistent in size with the 890 nucleotide transcript (Fig. 2B).sut-1 mRNA is essentially undetectable in sut-1(bk79) homo-zygotes, presumably because the nonsense mutation early inthe coding sequence causes sut-1(bk79) mRNA degradationvia nonsense mediated decay (22,23). To study SUT-1protein, antibodies were raised against full-length recombinantSUT-1. Immunoblotting shows that SUT-1 protein is abun-dantly expressed in N2 and T337, but is not detected in thesut-1(bk79) mutant (Fig. 2C). The SUT-1 protein migrates at�26 kD consistent with the predicted size of 26.3 kD.Human tau expression does not appear to dramatically influ-ence the protein expression level or mobility of SUT-1protein (Fig. 2C).
To determine the expression pattern of SUT-1 protein, westained worms with SUT-1 specific antibodies. N2 animalsshowed prominent specific SUT-1 staining within most cellsand tissues including neurons (Fig. 2F–K). SUT-1 is presentboth in the nuclear and cytoplasmic compartments, withnuclear staining being predominant. SUT-1 protein exhibitednucleo-cytoplasmic expression from mid embryogenesis toadulthood. The expression and distribution of SUT-1 proteinappears unchanged in non-transgenic when compared withtau transgenic animals. In contrast, the sut-1(bk79) mutantdid not show staining with SUT-1 antibodies, consistentwith immunoblotting and Northern blotting results (Fig. 2Band C), showing the bk79 allele is a null mutation.
One of the pathological features shared between tau trans-genic worms and authentic human tauopathy is aggregationof phosphorylated, insoluble tau. To examine how loss ofsut-1 function affects tau, we examined the expression level,phosphorylation and the formation of insoluble tau proteinin worms homozygous for the human tau transgene and thesut-1(bk79) mutation. Transgene expression levels wereevaluated in four independent worm lysates by immunoblot-ting using b-tubulin levels as an internal control (Fig. 3A).Tau protein is reduced by 28% relative to the parental tautransgenic strains, as determined by quantitative western blot-ting using tau specific primary and 125I labeled secondary anti-bodies. This decrease is not sufficient to alleviate the
1960 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

tauopathy phenotype, as heterozygous tau transgenic animalsthat express 50% of the tau found in the homozygous strainsstill exhibit a tauopathy phenotype (Fig. 2D). To assess tauphosphorylation, we probed total protein worm lysate immu-noblots with phosphorylation-specific tau antibodies PHF-1,AT8 and 12E8. These antibodies recognize tau phosphorylatedat S396/S404, S199/S202 and S262, respectively (Fig. 3B).We saw no change in tau phosphorylation state in the
T337;sut-1(bk79) line relative to the parental T337 line. Todetermine how the sut-1(bk79) mutant effects tau aggregation,we sequentially extracted worm lysates using buffers ofincreasing solublizing strength (24). We initially homogenizedT337 or T337;sut-1(bk79) worm pellets in RAB, a high saltbuffer, yielding the soluble tau fraction. We re-extractedmaterial insoluble in RAB with RIPA, a detergent containingbuffer yielding the detergent soluble fraction. Subsequently,
Figure 1. sut-1(bk79) suppresses tau induced locomotion defects. (A) Liquid thrashing assays for staged 1, 3 and 5 day old worms. Each data point is the mean(+SEM) thrashing rate for 20 worms. (B) sut-1 genomic region. The minimum sut-1 region as determined by genetic mapping was between SNP markersuCe2-571 and uCe2-725. Predicted genes are shown as boxed arrows. Cosmid coverage is shown below the transcripts. Cosmid T13B5 is sufficient torescue the sut-1 phenotype. (C) Sequence of sut-1 cDNA and SUT-1 protein. Shown is the early stop found in the sut-1(bk79) mutant.
Human Molecular Genetics, 2007, Vol. 16, No. 16 1961
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019
we recovered detergent insoluble material by extraction withformic acid (FA) (Fig. 3C). We observed that sut-1(bk79)caused a dramatic decrease in the levels of detergent solubleand detergent insoluble tau, both of which are abnormal aggre-gates of this protein. Thus, normal sut-1 activity is required forthe accumulation of appreciable amounts of insoluble tau inour tauopathy model.
To investigate the effect of loss of sut-1 function ontau-induced neurodegeneration, we explored the structural
integrity of GABAergic neurons. To monitor neuronal integ-rity, we used an unc-25::GFP reporter transgene that expressesgreen fluorescent protein (GFP) in all 19 GABAergic motorneurons (25). In the parental N2 strain, both the dorsal andventral nerve cords are continuous and contain the normalcomplement of 19 inhibitory motor neurons (13 Ventral Dtype and 6 Dorsal D type GABAergic neurons—Fig. 4A andB). We previously showed tau transgenic animals exhibitage dependent discontinuities in both dorsal and ventral cord
Figure 2. sut-1 allele bk79 is a loss-of-function mutation. sut-1(bk79) are deficient in sut-1 mRNA and SUT-1 Protein. (A) Diagram of the sut-1 locus; barsrepresent exons. The diagram is proportional to the actual sizes of the exons and introns. (B) Northern blot of polyA-selected mRNA from N2, T337 andsut-1(bk79) probed with sut-1 cDNA sequence (top) or actin (bottom). The prominent 890 nt band corresponds to the predicted transcript. sut-1 mRNA is vir-tually undetectable in the sut-1(bk79) mutant strain. (C) Immunoblots probed with SUT-1 specific antibodies or E7 anti-tubulin antibody. SUT-1 protein is notdetectable in sut-1(bk79) mutant lysates. (D and E) Liquid thrashing measurements for 20 staged 4 day old worms of each genotype. Strains used are: D, T337/þheterozygotes and T337;sut-1(bk79) homozygotes. Statistical analysis of liquid thrashing data using Students t-test yielded P ¼ 0.00022 for comparison of T337/þ and T337;sut-1(bk79) homozygotes. E, Thrash rates for non-Tg animals (N2) compared to sut-1(bk79) homozygotes in the absence of the tau transgene; thereis no significant difference between N2 and sut-1. (F–H) Immunostaining using SUT-1 specific antibodies. (H–J) DAPI nuclear staining of the same animalsshown in panels above. (F and I) Head staining in N2 adult. (G and J) Head staining in homozygous T337 adults. H and K, head staining in homozygousT337;sut-1(bk79) adult.
1962 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

axons (see also Fig. 4C and D) (19). Likewise, neurons are lostin an age dependent fashion. The sut-1(bk79) mutation par-tially ameliorates the tau induced neurodegenerative disrup-tion of axons and loss of neurons (Fig. 4E and F). Thispartial suppression of neurodegeneration is consistent withthe incomplete suppression of the locomotion phenotype insut-1(bk79) mutant (Fig. 1A).
We conducted a yeast two hybrid screen (26) using alexA-SUT-1 fusion protein as bait to identify proteins thatbind to SUT-1. Briefly, we transformed yeast reporter strainL40ura2 containing the lexA-SUT-1 bait construct with theC. elegans cDNA library pRB-1. From 14,600,000 transfor-mants, we recovered 43 colonies that activated both HIS3and lacZ reporter genes under control of lexA operator
Figure 3. Effects of sut-1 allele bk79 on tau protein. (A) Immunoblot of quadruplicate T337 and T337;sut-1 lysates probed for tau and tubulin. (B) Immunoblotof T337 and T337;sut-1(bk79) lysates probed with tau specific phosphorylation dependent antibodies. (C) Sequential extraction of insoluble tau protein fromT337 and T337;sut-1(bk79) mutant lysates. RAB is the soluble fraction; RIPA contains detergent soluble tau, while FA contains the detergent insoluble materialformic-acid (FA) solubilized protein.
Human Molecular Genetics, 2007, Vol. 16, No. 16 1963
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

Figure 4. Loss of sut-1 ameliorates tau-induced neurodegeneration. All strains shown contain a reporter transgene that marks GABAergic neurons with GFP(unc-25::GFP) from strain CZ1200. Left is anterior, down is ventral. (A) Reporter strain CZ1200 in a non-transgenic wild type background at 4 days of age.(B) Higher magnification view of CZ1200. No neurodegenerative changes are evident. (C) Four day old T337 animal exhibits loss of neurons (arrows), andgaps or interruptions of both dorsal and ventral nerve cords (arrowheads) in all animals. Bright head fluorescence (in panels C-F) is myo-2:GFP, the markertransgene used to identify tau transgenic worms. (D) Higher magnification view of T337. (E) Four day old T337;sut-1(bk79) animal showing that loss ofsut-1 ameliorates many of the defects seen in C and D. (F) Higher magnification of T337;sut-1(bk79). (G) Measurement of extent of neurodegenerativeevents for N2, T337 or T337;sut-1(bk79). Neurodegenerative events are broken down into three categories: neuronal loss (neurons), interruption of ventralcord continuity (VC axons), or interruption of dorsal cord continuity (DC axons). Measurements are the average from 50 animals for each strain and errorbars are SEM. Using the Students t-test, T337;sut-1(bk79) is significantly different from the parental T337 strain for neuronal loss (P ¼ 1.6 � 10215),ventral cord gaps (P ¼ 1.1 � 10211) and dorsal cord gaps (P ¼ 4.5 � 10223).
1964 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

sequences. From these, we isolated four cDNA clonesencoding the UNC-34 protein, which interact specificallywith the SUT-1 bait but not the control bait (MS2 phagecoat protein). To test whether SUT-1 and UNC-34 proteinscan bind in the absence of yeast proteins, we employed anin vitro protein-binding assay. Briefly, we expressed inEscherichia coli SUT-1 as a fusion protein with glutathione S-transferase (GST) and purified the recombinant protein usingglutathione sepharose affinity chromatography. We mixedrecombinant GST-SUT-1 fusion protein coated sepharosebeads with 35S labeled UNC-34 protein generated by in vitrotranslation of a full-length unc-34 cDNA. 35S labeledUNC-34 bound specifically to GST-SUT-1 fusion proteincoated beads but not GST coated beads (Fig. 5A), demonstrat-ing SUT-1 and UNC-34 proteins can interact in two indepen-dent assays. UNC-34 encodes a homolog of DrosophilaEnabled (ENA) and is a member of the ENA/vasodilator-stimulated phosphoprotein (ENA/VASP) protein family. Themammalian homolog of unc-34 and ena is encoded bymena. The C. elegans UNC-34 protein shares �37% aminoacid similarity with Mena.
UNC-34, Mena and other ENA/VASP proteins character-istically contain three distinct functional domains. These area proline-rich domain (PRD), Enabled/vasodilator-stimulatedphosphoprotein homology domain 1 (EVH1) and EVHdomain 2 (EVH2) (Fig. 5B) (27). EVH1 domains areprotein–protein interaction sequences that bind viaproline-rich motifs (e.g. FPPPP) to a variety of proteinsincluding cytoskeletal-associated proteins. The EVH2domain promotes multimerization and F-actin binding (28).The PRD domain binds profilin, an actin binding protein, pro-teins with Src homology 3 and some proteins with WW motifs(e.g. FE65) (27,29). To identify the UNC-34 domain respon-sible for SUT-1 binding, we generated three UNC-34 deletionconstructs each encoding an individual UNC-34 domain. Welabeled these truncated proteins with 35S and tested forbinding to GST-SUT-1. Only the EVH1 domain bound toSUT-1 (Fig. 5C). Thus, SUT-1 binds to the same domain ofUNC-34 that potentially binds to cytoskeletal associatedproteins.
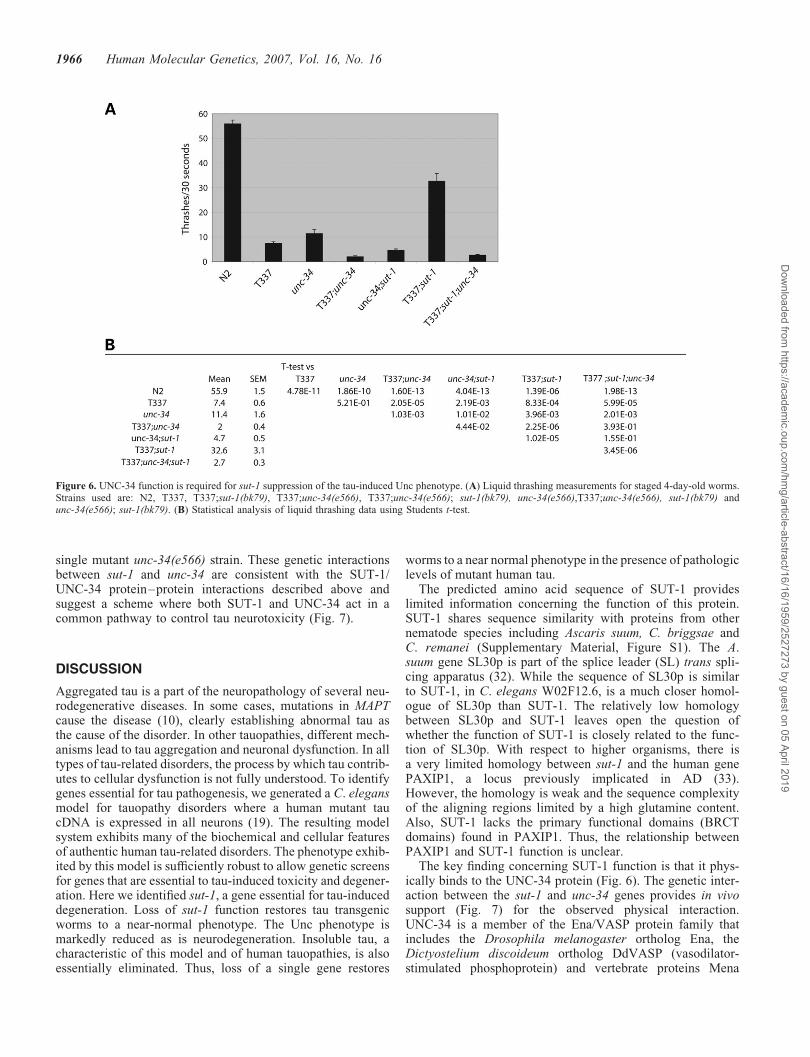
Since UNC-34 binds SUT-1, we investigated whetherunc-34 activity is required for suppression of the tau pheno-type by sut-1(bk79). We constructed double mutant animalsusing the loss of function mutation sut-1(bk79) and thepreviously characterized loss-of-function unc-34 allelee566. unc-34(e566) causes defects in axon pathfinding andneuronal migration leading to an Unc phenotype (30,31).We used the liquid thrashing assay to measure the effectsof loss-of-function sut-1 and unc-34 alleles, either singularlyor as double mutants, in the absence or presence of thetau transgene (Fig. 6). As described above, the sut-1 (bk79)allele suppresses the tau-induced Unc phenotype with theT337;sut-1(bk79) strain having higher thrash rates thanthe T337 strain alone (Fig. 6). However, when the unc-34loss-of-function allele is added, the resulting double mutantT337;sut-1(bk79);unc-34(e566) has a much lower thrash ratecompared to T337;sut-1(bk79) or T337 alone. Thus, unc-34activity is required for sut-1(bk79) to suppress the tau-inducedUnc phenotype. In addition, unc-34 influences the tau-induced phenotype in the presence of wild type sut-1. The
unc-34(e566) allele is an enhancer of the T337 phenotypewith T337;unc-34(e566) having lower thrash rates than T337alone. This result indicates that wild type unc-34 gene par-tially protects worms from the tau transgene-induced Unc phe-notype. Finally, the sut-1 and unc-34 genes also interact in theabsence of the tau transgene. While sut-1(bk79) has wild typethrash rates (Fig. 2E), sut-1 acts as an enhancer of the unc-34mutant as shown by the fact that the double mutantsut-1(bk79);unc-34 (e566) has lower thrash rates than the
Figure 5. SUT-1 protein binds to UNC-34. (A) In vitro translated full-lengthUNC-34 binds to recombinant GST-SUT-1, but not recombinant GST alone ina GST-pulldown assay. Multiple bands are likely due to internal initiation atnon-start methionines during translation in reticulocyte lysates. (B) UNC-34domains and single domain constructs. (C) Truncated SUT-1 proteins werelabeled with 35S by in vitro translation and assayed for GST-SUT-1 binding.The EVH1 variant bound to SUT-1 but not EVH2 or PRD constructs.
Human Molecular Genetics, 2007, Vol. 16, No. 16 1965
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019
single mutant unc-34(e566) strain. These genetic interactionsbetween sut-1 and unc-34 are consistent with the SUT-1/UNC-34 protein–protein interactions described above andsuggest a scheme where both SUT-1 and UNC-34 act in acommon pathway to control tau neurotoxicity (Fig. 7).
DISCUSSION
Aggregated tau is a part of the neuropathology of several neu-rodegenerative diseases. In some cases, mutations in MAPTcause the disease (10), clearly establishing abnormal tau asthe cause of the disorder. In other tauopathies, different mech-anisms lead to tau aggregation and neuronal dysfunction. In alltypes of tau-related disorders, the process by which tau contrib-utes to cellular dysfunction is not fully understood. To identifygenes essential for tau pathogenesis, we generated a C. elegansmodel for tauopathy disorders where a human mutant taucDNA is expressed in all neurons (19). The resulting modelsystem exhibits many of the biochemical and cellular featuresof authentic human tau-related disorders. The phenotype exhib-ited by this model is sufficiently robust to allow genetic screensfor genes that are essential to tau-induced toxicity and degener-ation. Here we identified sut-1, a gene essential for tau-induceddegeneration. Loss of sut-1 function restores tau transgenicworms to a near-normal phenotype. The Unc phenotype ismarkedly reduced as is neurodegeneration. Insoluble tau, acharacteristic of this model and of human tauopathies, is alsoessentially eliminated. Thus, loss of a single gene restores
worms to a near normal phenotype in the presence of pathologiclevels of mutant human tau.
The predicted amino acid sequence of SUT-1 provideslimited information concerning the function of this protein.SUT-1 shares sequence similarity with proteins from othernematode species including Ascaris suum, C. briggsae andC. remanei (Supplementary Material, Figure S1). The A.suum gene SL30p is part of the splice leader (SL) trans spli-cing apparatus (32). While the sequence of SL30p is similarto SUT-1, in C. elegans W02F12.6, is a much closer homol-ogue of SL30p than SUT-1. The relatively low homologybetween SL30p and SUT-1 leaves open the question ofwhether the function of SUT-1 is closely related to the func-tion of SL30p. With respect to higher organisms, there isa very limited homology between sut-1 and the human genePAXIP1, a locus previously implicated in AD (33).However, the homology is weak and the sequence complexityof the aligning regions limited by a high glutamine content.Also, SUT-1 lacks the primary functional domains (BRCTdomains) found in PAXIP1. Thus, the relationship betweenPAXIP1 and SUT-1 function is unclear.
The key finding concerning SUT-1 function is that it phys-ically binds to the UNC-34 protein (Fig. 6). The genetic inter-action between the sut-1 and unc-34 genes provides in vivosupport (Fig. 7) for the observed physical interaction.UNC-34 is a member of the Ena/VASP protein family thatincludes the Drosophila melanogaster ortholog Ena, theDictyostelium discoideum ortholog DdVASP (vasodilator-stimulated phosphoprotein) and vertebrate proteins Mena
Figure 6. UNC-34 function is required for sut-1 suppression of the tau-induced Unc phenotype. (A) Liquid thrashing measurements for staged 4-day-old worms.Strains used are: N2, T337, T337;sut-1(bk79), T337;unc-34(e566), T337;unc-34(e566); sut-1(bk79), unc-34(e566),T337;unc-34(e566), sut-1(bk79) andunc-34(e566); sut-1(bk79). (B) Statistical analysis of liquid thrashing data using Students t-test.
1966 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

and VASP in mammals (34). All members of this family haveEVH1, PRD and EVH2 domains. The EVH1 and PRDdomains bind other proteins and the EVH2 domain may beinvolved in oligermerization (27,28). In addition to SUT-1,other EVH1 and PRD binding proteins include profilin,FE65, dynamin binding protein (DMNBP), diaphanous, vincu-lin and zyxin (35).
Functionally, Ena/VASP proteins link signaling pathways toactin dynamics, and are involved in cell migration, adhesion,polarity, endocytosis and other processes. On a molecularlevel, Ena/VASP antagonizes actin fibril capping thereby pro-moting actin fiber extension and also inhibiting branching(34,36). On the cellular level, in worms, UNC-34 participatesin axonal pathfinding and in neuronal migration duringdevelopment (31). Both of these processes exhibit dynamic,actin-based plasma membrane protrusions (lamellipodia andfilopodia) and UNC-34 is required for filopodia formation(37). In higher organisms, Ena/VASP proteins localize to the
leading edge of lamellipodia and modulate the protrusionstep of motility. Env/VASP proteins, in conjunction withother leading edge protein binding partners such as profilinand lamellipodin (38), control the dynamic actin structuresinvolved in motility.
One UNC-34 interacting protein of particular interest in ourtauopathy model is the DNMBP, also known as TUBA (39).We previously demonstrated RNAi knockdown of the wormDNMBP ortholog exacerbates the tau-induced phenotype inour C. elegans model (40). Dynamin mediates vesicle fissionin endocytosis, and DNMBP which binds both dynamin andMena, connects this process to regulation of the actin cytoskele-ton (41,42). In rat brain, DNMBP is located in synapses where itis presumed to participate with dynamin in synaptic vesicleendocytosis (39). In addition, DNMBP was implicated in ADwhere a SNP allele has been associated with an increased riskin AD (43). The putative interaction between Mena andDNMBP ties a previously identified genetic enhancer of
Figure 7. Model for the relationship between SUT-1 and tau pathology. (A) Genetic pathway relating SUT-1 to pathological tau. (B) Potential SUT-1 Interactionnetwork. Dashed lines are genetic interactions or indirect interactions and solid lines are physical interactions. Blue arrows represent data from mammaliansystems. Black arrows represent data from this worm model. References for human protein–protein interactions: APP-FE6544;68; FE65-Mena29;44; Mena-Profilin69; Diaphanous-Profilin70; Diaphanous-Rho70; DNMBP-Mena39.
Human Molecular Genetics, 2007, Vol. 16, No. 16 1967
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

tauopathy (DMNBP) with the genetic suppressor found in thisstudy (sut-1(bk79)) suggesting they have opposing roles, andlikely both act through unc-34. This demonstrates a convergenceof the complementary screening approaches described pre-viously (40) and in this study (Fig. 7).
The site of Ena/VASP protein interactions with dynamic actinprocesses is at discrete cytoplasmic locations often associatedwith membranes. On the other hand, SUT-1 has primarily anuclear localization though some cytoplasmic staining is alsoin evidence (Fig. 2E–J). Another protein that has both anuclear function and binds to an Ena/VASP protein in the cyto-plasm is FE65. FE65 binds Mena (29) and can form a tripartitecomplex with the amyloid precursor protein (APP) (44). APP isimportant in AD because endolytic cleavage of this protein byb- and g-secretase produces Ab, a toxic peptide and primarycomponent of the amyloid plaques found in this disease. APPand FE65 co-localize with Mena in lamellipodia (44), linkingAPP/FE65 with the regulation of actin dynamics during cellmovement. FE65 and APP are also implicated in actin-basedneuronal growth cone regulation (44). Mena appears to phys-ically link FE65 and APP to processes involving the regulationof actin dynamics. The FE65–APP interaction also results inregulation of nuclear transcription. FE65, in an APP-dependentprocess, binds to TIP60, a histone acetyl-transferase (45) and thetranscription factor CP2/LSF/LBP1 (46), and the TIP60-FE65complex can then move to the nucleus and regulate geneexpression (47). Thus, FE65-APP complexes not only partici-pate in dynamic actin-mediated processes in the cytoplasm,but also act in the nucleus to regulate expression of genesrelated to these processes. Likewise, SUT-1 may also haveboth a cytoplasmic function when bound to UNC-34 and an asyet unknown nuclear function (Fig. 7).
The mechanism by which sut-1 influences the tau-inducedpathology in this model system is unclear though the inter-action of sut-1 with UNC-34 provides information concerningthe molecular pathways affected. Several lines of evidencesuggest sut-1 and unc-34 regulate the same process: first,genetic loss of sut-1 suppresses, while loss of unc-34 enhancestau toxicity; second, loss of sut-1 prevents the tau-inducedphenotype only when an unc-34 is present; and third, theloss of sut-1 exacerbates the Unc phenotype of an unc-34loss-of-function mutant (Fig. 7). The relationship betweenSUT-1 and UNC-34 may be antagonistic and presumablyresults from the direct interaction between these two proteins.The observation that most of SUT-1 is located in the nucleussuggests SUT-1 could shuttle between cytoplasmic UNC-34and the nucleus to regulate transcription in response toUNC-34-dependent processes. Precedent for such a signaltransduction mechanism exists for the FE65-Mena-APP inter-actions described above where FE65 complexes with Menaand APP to regulate actin-mediated processes, yet FE65 alsocan also shuttle to the nucleus to affect transcription. Otherproteins that, like SUT-1, bind to EVH1 domains of Ena/VASP proteins and shuttle between the cytoplasm andnucleus include zyxin (48), Homer (49), Profilin (50,51) andDiaphanous1 (52); the latter three proteins affect geneexpression changes (53,54).
Actin and related proteins have recently been implicated intau-related neurodegenerative disease. In a Drosophila tauopa-thy model, expression of FTDP-17 mutant human tau causes
neurodegeneration as well as inducing excess F-actin for-mation (55). In this fly model, over-expression of actin exacer-bates tau toxicity, while destabilizing actin by over-expressingcofilin suppresses tau toxicity. In the same model, rod-likedeposits of actin filaments, profilin and tau are observed.Similar deposits are also seen in a tau transgenic mousemodel of FTDP-17 (55). In humans, actin-containing proteindeposits called Hirano bodies were initially observed in tauo-pathies such as Guam ALS/PDC, AD, Pick’s disease and PSP(56) providing evidence for alternation of actin-mediated pro-cesses in neurodegenerative disease (57). Hirano bodiescontain actin filaments (58), cofilin and actin depolymerizationfactor (ADF) (two actin binding proteins) (59). However,Hirano deposits are also seen in normal aging and the actualrelationship between these deposits and disease processes isunclear, though it is not unusual for the proteins depositedin neurodegenerative diseases to be intimately connected tothe pathogenetic mechanisms involved.
It is tempting to speculate that the process affected in ourSUT-1 mutant animals is some sort of presynaptic actin-dependent process such as DMNBP-dependent vesicle fission.This idea is supported by the results of a genome-wide RNAiscreen that identified as enhancers not only DMNBP (seeabove) but also several other neurotransmission genes includ-ing aex-1 (human ortholog is UNC13D) which is involved insynaptic vesicle release and acr-14, a nicotinic acetyl cholinereceptor (40). Also, previous work showed tau can cause a pre-synaptic deficit in neurotransmission in this model (19). Thus,we propose UNC-34 may be required for generation or main-tenance of normal synapses, and SUT-1, either directly, orthrough nuclear gene regulation, may negatively modulatethis function. Under the stress of tau toxicity, SUT-1 suppres-sion of this UNC-34-dependent process is detrimental.
Here we show loss of a single gene can nearly eliminate thetoxic effects of tau in tau transgenic C. elegans. This suggestsin human tauopathies, functional inhibition of a single proteincould be a treatment for neurodegeneration. Thus, identifi-cation of SUT-1 reveals a pathway target for drug develop-ment. While there is no human ortholog of SUT-1, wedemonstrate via the interaction with UNC-34, that SUT-1 ispart of a pathway conserved in higher organisms includingman. The possibility exists that in higher organisms, there isa protein that is functionally equivalent to SUT-1 but doesnot have a similar sequence. This has been demonstrated fora number of worm genes. One recent example is for sys-1, afunctional beta-catenin, bearing no sequence homology tomammalian beta-catenins. Based on functional criteria, sys-1can substitute for the bar-1 beta-catenin and the encodedprotein shares similar biochemical and transcriptional regulat-ory properties (60). The situation with SUT-1 may be compar-able, except the relevant mammalian functional counterpartremains unknown. Identification of a mammalian analog ofSUT-1 that negatively modulates a Mena-dependent pathwaycould lead to novel therapeutic targets.
METHODS
Strains. Bristol strain N2 is the wild type C. elegans strainused (61). Tau transgenic line T337-1 (CK10) was used for
1968 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

the suppressor screen. Line CK10–bkIs10[Paex-3::Tau–337M), Pmyo-2::GFP] carries a chromasomally integratedtransgene encoding the 1N4R isoforms of human tau carryingthe 337M FTDP-17 mutation with expression driven by thepan-neuronal promoter aex-3, and has a pronounced Unc phe-notype (19). CK15 is the CK10 strain backcrossed to theHawaiian C. elegans isolate CB4586 12 times. CZ1200carries an integrated unc-25::GFP transgene expressed inGABAergic neurons. The original source of unc-34(e566)mutation is strain CB566. Double mutants were constructedby standard methods. Genotypes were confirmed by PCRand sequencing or restriction digests for all strains with non-obvious phenotypes.
Mutagenesis. Tau transgenic worms were mutagenized usingethyl nitrosurea (ENU) as described (62). Briefly, tau transgenicworms were exposed to 1 mM ENU in M9 for 4 h with gentlerocking. Approximately 50 000 mutagenized haploid genomeswere screened. Mutant F2 progeny that had restored motilityrelative to the parental tau transgenic progeny were selected.This motility selection consisted of washing F2 mutagenizedworms of all bacteria using M9. Washed worms were placedon a 245 � 245 mm square agar plate with food at one end.Worms are placed at the opposite end from the food, andworms with normal locomotion rapidly move to the food,while those that have impaired locomotion do not arrive at thefood until much later. The animals that reached food first werepicked as candidate mutants. Mutants that bred true were sub-jected to a secondary screen which consisted of testing mutantline for tau expression levels. Only lines with tau proteinlevels similar to the parental line were retained for mappingand further analysis. The line containing a sut-1 mutation wastested against other isolated alleles by complementationtesting. However, all other alleles isolated complementedsut-1, thus only a single sut-1 allele was isolated.
Positional Cloning of sut-1. Single nucleotide polymorphismmapping was conducted essentially as described (21) exceptthat CK15 was used as the Hawaiian strain. The simplifiedmethod of Davis et al. (63) was adopted for some mapping.Using these methods sut-1(bk79) was mapped to the intervalbetween markers uce2-577 and uce2-725. To rescue the sut-1phenotype, pools of cosmids were injected at 30 ng/ml eachwith Pmyo-2::dsRED at 10 ng/ml as a co-injection marker.The rescuing pool contained cosmids W09G6, M01A4,K02E7 and T13B5. These cosmids were subsequently injectedindividually at 30 ng/ml with 90 ng/ml pBluescript II KS(þ) ascarrier and 10 ng/ml of Pmyo-2::dsRED.
Mutation screen. The exons and flanking sequences forgenes on T13B5 were amplified by PCR and sequenced. Thesequences for sut-1 exons were compared with the sequencefor N2 and CK10. A single point mutation was identified inexon 3 of T13B5.8 in sut-1(bk79).
Behavioral assays. Liquid thrashing assays were performedin 20 ml of M9 media (42 mM Na2HPO4, 22 mM KH2PO4,86 mM NaCl, 1 mM MgSO4) on Teflon-printed slides as pre-viously described (19). Worms were allowed to settle andthrashes counted for 30 s.
Protein extraction. Tau fractions were obtained as describedpreviously (19). To determine if aggregated tau accumulatesor is absent from sut-1 animals strains were sequentiallyextracted using buffers of increasing solublizing strengths
and compared with the parental T337 strain (19,24). First,worms were homogenized in high salt reassembly buffer[RAB-High Salt (0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4,0.75 M NaCl, 0.02 M NaF, 0.5 mM PMSF, 0.1% proteaseinhibitor cocktail, pH 7.0)] and ultra-centrifuged at 50 000�gravity yielding the soluble fraction (supernatant) and an inso-luble pellet. Next, the RAB insoluble material wasre-extracted with an ionic and non-ionic detergent containingRIPA buffer (50 mM Tris,150 mM NaCl,1% NP40,5 mM
EDTA,0.5% DOC,0.1% SDS, 0.5 mM PMSF, 0.1% proteaseinhibitor cocktail, pH 8.0) and centrifuged as above yieldingabnormal tau in the supernatant. Finally, the detergent insolu-ble pellet was re-extracted with 70% FA to solublize detergentinsoluble tau. The three fractions were analyzed using quanti-tative western blotting with tau specific primary and125I-labeled secondary antibodies.
Immunoblotting. Protein samples were boiled 5 min andloaded onto 10% pre-cast SDS–PAGE gels (Biorad). Forquantitative immunoblotting, we detected human tau usingantibody 17026 at a dilution of 1:3000 (A gift of VirginiaLee) as described previously (19). We used anti-tubulinantibody at a dilution of 1:1000 (Developmental StudiesHybridoma Bank). SUT-1 antibody was prepared as describedbelow and used at a dilution of 1:1000. 125I-labled goat anti-mouse or goat-anti-rabbit IgG were the secondary antibodyreagents used at a dilution of 1:1000 (New EnglandNuclear). Signals were quantitated using a Packard Cyclonephosphorimager.
Immunocytochemistry. Worms were fixed in paraformalde-hyde and permeablized by freeze cracking as described (64).Fixed whole animals were stained with SUT-1 specific affinitypurified rabbit antibodies at a dilution of 1:500. AlexA 568conjugated anti-rabbit antibody (Molecular Probes) was usedas the secondary antibody at a dilution of 1:500.
Measurement of neuronal degeneration. The unc-25::GFPtransgene was crossed into the background of the tau trans-genic strains assayed. Fifty worms of each genotype weredevelopmentally staged and analyzed for neuronal structuredefects as previously described (19).
Yeast two hybrid screening. Yeast two hybrid screening wascarried out as described (65), except that yeast strain L40ura-carrying pLexA-SUT-1 plasmid was transformed with theLamdaACT RB-2 library and plated on SD-trp-leu-his,100 mM 1,2,4 3-amino triazole. Colonies were picked after 5days and cDNA-containing plasmids were rescued into E.coli. Recovered cDNA plasmids were reintroduced into L40containing either pLexA–SUT1 or pLexA–MS2 coatprotein. Those cDNAs that activated expression with SUT-1,but not MS2 coat protein, were analyzed further.
Recombinant protein purification. The SUT-1 proteinexpression construct was prepared by inserting the sut-1cDNA into the pGEX 6P-1 expression vector (Pharmacia) togenerate a construct encoding a GST-SUT-1 fusion protein.The GST moiety allows one-step affinity purification ofrecombinant protein on Glutathione coupled sepharosebeads. A log-phase culture of BL21(DE3) cells carrying thepGEX-SUT-1 vector was induced for 3 h at 37ºC withshaking. Glutathione sepharose (Pharmacia) was used as theaffinity resin; cells were harvested, lysed and recombinantprotein was purified as previously described (66).
Human Molecular Genetics, 2007, Vol. 16, No. 16 1969
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

SUT-1 antibody preparation. SUT-1 antibodies were pre-pared commercially using the Invitrogen Zymed antibodyservice. Purified recombinant GST-SUT-1 protein was usedas the immunogen. Antisera were affinity purified using pureSUT-1 protein cleaved from the GST moiety using theZymed antibody affinity purification service.
GST pulldown assays. In vitro protein-binding assays wereperformed essentially as described (65) except the bindingbuffer contained 0.5% Bovine Serum Albumin (BSA), 0.1%Tween-20, 100 mM NaCl, 5 mM DTT, 20 mM HEPES, pH7.4. GST or GST-SUT-1 fusion protein was bound toglutathione-sepharose as described (65). The amount of GSTor GST-SUT-1 bound was determined by eluting the boundprotein and analyzing them by SDS–PAGE followed by coo-masie blue staining (67). Radiolabeled (35S) UNC-34 was pro-duced using the TNT reticulocyte lysate (Promega) accordingto the manufacture’s methods. Labeled protein was then addedto equivalent amounts of glutathione beads to which eitherGST alone or GST-SUT-1 fusion protein was bound and incu-bated with beads at 48C with gentle rotation for 60 min. Beadswere pelleted, washed five times in binding buffer and elutedby boiling in SDS–PAGE sample buffer. Eluted, labeled pro-teins were analyzed by SDS–PAGE. In the figures, theamount of material shown in lanes marked ‘input’ is 10% ofthe amount used in the GST-pulldown experiments.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
This work was supported by a Department of Veterans Affairsincluding a Merit Review Entry Program Grant (BCK). Thework was also supported by NIA grant PO1 AG17586(GDS, V.-M. Lee, PI). We thank Dr. James H. Thomas foradvice regarding C. elegans genetics and strains. We thankthe C. elegans Genetics Center for providing strains. Wethank Leo Anderson, Harmony Danner, Elaine Loomis andLindsey Foley for outstanding technical assistance. Wethank Yuji Kohara for the sut-1 cDNAs yk459d2, yk636d5,yk657g8, Andrew Fire for providing C. elegans expressionvectors, Yishi Jin for strain CZ1200, Virginia Lee for antibody17026, Peter Davies for antibody PHF1, Peter Seubert for anti-body12E8 and the b-tubulin antibody E7 is from the Develop-mental Studies Hybridoma Bank (NICHD). We also thankVirginia Lee, Joeseph Buxbaum, Tom Blumenthal and PegMacMorris for critical reading of the manuscript.
Conflict of Interest statement. The authors state that they haveno conflict of interest.
REFERENCES
1. Feinstein, S.C. and Wilson, L. (2005) Inability of tau to properly regulateneuronal microtubule dynamics: a loss-of-function mechanism by whichtau might mediate neuronal cell death. Biochim. Biophys. Acta, 1739,268–279.
2. Lee, V.M.Y., Goedert, M. and Trojanowski, J.Q. (2001)Neurodegenerative tauopathies. Annu. Rev. Neurosci., 24, 1121–1159.
3. Gordon-Weeks, P.R. (2004) Microtubules and growth cone function.J. Neurobiol., 58, 70–83.
4. Yu, J.Z. and Rasenick, M.M. (2006) Tau associates with actin indifferentiating PC12 cells. FASEB J., 20, 1452–1461.
5. Sharma, V.M., Litersky, J.M., Bhaskar, K. and Lee, G. (2007) Tauimpacts on growth-factor-stimulated actin remodeling. J. Cell Sci., doi:10.1242/jcs.03378.
6. Mcdermott, J.B., Aamodt, S. and Aamodt, E. (1996) ptl-1, aCaenorhabditis elegans gene whose products are homologous to the taumicrotubule-associated proteins. Biochemistry, 35, 9415–9423.
7. Goedert, M., Baur, C.P., Ahringer, J., Jakes, R., Hasegawa, M.,Spillantini, M.G., Smith, M.J. and Hill, F. (1996) PTL-1, amicrotubule-associated protein with tau-like repeats from the nematodeCaenorhabditis elegans. J. Cell Sci., 109, 2661–2672.
8. Ingram, E.M. and Spillantini, M.G. (2002) Tau gene mutations: dissectingthe pathogenesis of FTDP-17. Trends Mol. Med., 8, 555–562.
9. Spillantini, M.G., Bird, T.D. and Ghetti, B. (1998) FrontotemporalDementia and Parkinsonism linked to chromosome 17: a new group oftauopathies. Brain Pathol., 8, 387–402.
10. Hutton, M., Lendon, C.L., Rizzu, P., Baker, M., Froelich, S., Houlden, H.,Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A. et al. (1998)Association of missense and 50-splice-site mutations in tau with theinherited dementia FTDP-17. Nature, 393, 702–705.
11. Poorkaj, P., Bird, T.D., Wijsman, E., Nemens, E., Garruto, R.M.,Anderson, L., Andreadis, A., Wiederholt, W.C., Raskind, M. andSchellenberg, G.D. (1998) Tau is a candidate gene for chromosome 17frontotemporal dementia. Ann. Neurol., 43, 815–825.
12. Spillantini, M.G., Murrell, J.R., Goedert, M., Farlow, M.R., Klug, A. andGhetti, B. (1998) Mutation in the tau gene in familial multiple systemtauopathy with presenile dementia. Proc. Natl Acad. Sci. USA, 95,7737–7741.
13. Roy, S., Zhang, B., Lee, V.M.Y. and Trojanowski, J.Q. (2005) Axonaltransport defects: a common theme in neurodegenerative diseases. Acta
Neuropathol., 109, 5–13.
14. Stamer, K., Vogel, R., Thies, E., Mandelkow, E. and Mandelkow, E.M.(2002) Tau blocks traffic of organelles, neurofilaments, and APP vesiclesin neurons and enhances oxidative stress. J. Cell Biol., 156, 1051–1063.
15. Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed,L., Miller, B.I., Geschwind, D.H., Bird, T.D., McKeel, D., Goate, A. et al.
(1998) Mutation-specific functional impairments in distinct Tau isoformsof hereditary FTDP-17. Science, 282, 1914–1917.
16. Goedert, M., Jakes, R. and Crowther, R.A. (1999) Effects offrontotemporal dementia FTDP-17 mutations on heparin-inducedassembly of tau filaments. FEBS Lett., 450, 306–311.
17. Nacharaju, P., Lewis, J., Easson, C., Yen, S., Hackett, J., Hutton, M. andYen, S.H. (1999) Accelerated filament formation from tau protein withspecific FTDP-17 missense mutations. FEBS Lett., 447, 195–199.
18. Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A. and Hof, P.R.(2000) Tau protein isoforms, phosphorylation and role inneurodegenerative disorders. Brain Res. Rev., 33, 95–130.
19. Kraemer, B.C., Zhang, B., Leverenz, J.B., Thomas, J.H., Trojanowski,J.Q. and Schellenberg, G.D. (2003) Neurodegeneration and defectiveneurotransmission in a Caenorhabditis elegans model of tauopathy. Proc.
Natl Acad. Sci. USA, 100, 9980–9985.
20. Iwasaki, K., Staunton, J., Saifee, O., Nonet, M. and Thomas, J.H. (1997)aex-3 encodes a novel regulator of presynaptic activity in C. elegans.Neuron, 18, 613–622.
21. Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H. and Plasterk, R.H.(2001) Rapid gene mapping in Caenorhabditis elegans using a highdensity polymorphism map. Nat. Genet., 28, 160–164.
22. Behm-Ansmant, I. and Izaurralde, E. (2006) Quality control of geneexpression: a stepwise assembly pathway for the surveillance complexthat triggers nonsense-mediated mRNA decay. Genes Dev., 20, 391–398.
23. Khajavi, M., Inoue, K. and Lupski, J.R. (2006) Nonsense-mediatedmRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum.
Genet, 14, 1074–1081.
24. Ishihara, T., Hong, M., Zhang, B., Nakagawa, Y., Lee, M.K.,Trojanowski, J.Q. and Lee, V.M.Y. (1999) Age-dependent emergence andprogression of a tauopathy in transgenic mice overexpressing the shortesthuman tau isoform. Neuron, 24, 751–762.
25. Jin, Y., Jorgensen, E., Hartwieg, E. and Horvitz, H.R. (1999) TheCaenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylaseand is required for synaptic transmission but not synaptic development.J. Neurosci., 19, 539–548.
1970 Human Molecular Genetics, 2007, Vol. 16, No. 16
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019

26. Fields, S. and Song, O. (1989) A novel genetic system to detect protein–protein interactions. Nature, 340, 245–246.
27. Krause, M., Dent, E.W., Bear, J.E., Loureiro, J.J. and Gertler, F.B. (2003)Ena/VASP proteins: regulators of the actin cytoskeleton and cellmigration. Annu. Rev. Cell Dev. Biol., 19, 541–564.
28. Bachmann, C., Fischer, L., Walter, U. and Reinhard, M. (1999) TheEVH2 domain of the vasodilator-stimulated phosphoprotein mediatestetramerization, F-actin binding, and actin bundle formation. J. Biol.
Chem., 274, 23549–23557.29. Ermekova, K.S., Zambrano, N., Linn, H., Minopoli, G., Gertler, F., Russo,
T. and Sudol, M. (1997) The WW domain of neural protein FE65 interactswith proline-rich motifs in Mena, the mammalian homolog of Drosophilaenabled. J. Biol. Chem., 272, 32869–32877.
30. Colavita, A. and Culotti, J.G. (1998) Suppressors of ectopic UNC-5growth cone steering identify eight genes involved in axon guidance inCaenorhabditis elegans. Dev. Biol., 194, 72–85.
31. Withee, J., Galligan, B., Hawkins, N. and Garriga, G. (2004)Caenorhabditis elegans WASP and Ena/VASP proteins playcompensatory roles in morphogenesis and neuronal cell migration.Genetics, 167, 1165–1176.
32. Denker, J.A., Zuckerman, D.M., Maroney, P.A. and Nilsen, T.W. (2002)New components of the spliced leader RNP required for nematodetrans-splicing. Nature, 417, 667–670.
33. Rademakers, R., Cruts, M., Sleegers, K., Dermaut, B., Theuns, J.,Aulchenko, Y., Weckx, S., DePooter, T., VandenBroeck, M., Corsmit, E.et al. (2005) Linkage and association studies identify a novel locus forAlzheimer disease at 7q36 in a Dutch population-based sample.Am. J. Hum. Gen., 77, 643–652.
34. Kwiatkowski, A.V., Gertler, F.B. and Loureiro, J.J. (2003) Function andregulation of Ena/VASP proteins. Trends Cell Biol., 13, 386–392.
35. Bundschu, K., Walter, U. and Schuh, K. (2006) The VASP-Spred-Sproutydomain puzzle. J. Biol. Chem., 281, 36477–36481.
36. Bear, J.E., Svitkina, T.M., Krause, M., Schafer, D.A., Loureiro, J.J.,Strasser, G.A., Maly, I.V., Chaga, O.Y., Cooper, J.A., Borisy, G.G. andGertler, F.B. (2002) Antagonism between Ena/VASP proteins and actinfilament capping regulates fibroblast motility. Cell, 109, 509–521.
37. Chang, C., Adler, C.E., Krause, M., Clark, S.G., Gertler, F.B.,Tessier-Lavigne, M. and Bargmann, C.I. (2006) MIG-10/lamellipodin andAGE-1/PI3K promote axon guidance and outgrowth in response to slit andnetrin. Curr. Biol., 16, 854–862.
38. Krause, M., Leslie, J.D., Stewart, M., Lafuente, E.M., Valderrama, F.,Jagannathan, R., Strasser, G.A., Rubinson, D.A., Liu, H., Way, M. et al.
(2004) Lamellipodin, an Ena/VASP ligand, is implicated in the regulationof lamellipodial dynamics. Dev. Cell, 7, 571–583.
39. Salazar, M.A., Kwiatkowski, A.V., Pellegrini, L., Cestra, G., Butler,M.H., Rossman, K.L., Serna, D.M., Sondek, J., Gertler, F.B. and DeCamilli, P. (2003) Tuba, a novel protein containing bin/amphiphysin/Rvsand Dbl homology domains, links dynamin to regulation of the actincytoskeleton. J. Biol. Chem., 278, 49031–49043.
40. Kraemer, B.C., Burgess, J.K., Chen, J.H., Thomas, J.H. and Schellenberg,G.D. (2006) Molecular pathways that influence human tau-inducedpathology in Caenorhabditis elegans. Hum. Mol. Genet., 15, 1483–1496.
41. Dawson, J.C., Legg, J.A. and Machesky, L.M. (2006) Bar domainproteins: a role in tubulation, scission and actin assembly inclathrin-mediated endocytosis. Trends Cell Biol., 16, 493–498.
42. Praefcke, G.J. and McMahon, H.T. (2004) The dynamin superfamily:universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell
Biol., 5, 133–147.43. Kuwano, R., Miyashita, A., Arai, H., Asada, T., Imagawa, M., Shoji, M.,
Higuchi, S., Urakami, K., Kakita, A., Takahashi, H. et al. (2006)Dynamin-binding protein gene on chromosome 10q is associated withlate-onset Alzheimer’s disease. Hum. Mol. Genet., 15, 170–2182.
44. Sabo, S.L., Ikin, A.F., Buxbaum, J.D. and Greengard, P. (2001) TheAlzheimer amyloid precursor protein (APP) and FE65, an APP-bindingprotein, regulate cell movement. J. Cell Biol., 153, 1403–1414.
45. Cao, X.W. and Sudhof, T.C. (2001) A transcriptively active complex ofAPP with Fe65 and histone acetyltransferase Tip60. Science, 293,115–120.
46. Zambrano, N., Minopoli, G., Decandia, P. and Russo, T. (1998) The Fe65adaptor protein interacts through its PID1 domain with the transcriptionfactor CP2/LSF/LBP1. J. Biol. Chem., 273, 20128–20133.
47. Muller, T., Concannon, C.G., Ward, M.W., Walsh, C.M., Tirniceriu, A.L.,Tribl, F., Kogel, D., Prehn, J.H. and Egensperger, R. (2007) Modulation ofgene expression and cytoskeletal dynamics by the amyloid precursorprotein intracellular domain (AICD). Mol. Biol. Cell, 18, 201–210.
48. Nix, D.A., Fradelizi, J., Bockholt, S., Menichi, B., Louvard, D.,Friederich, E. and Beckerle, M.C. (2001) Targeting of zyxin to sites ofactin membrane interaction and to the nucleus. J. Biol. Chem., 276,34759–34767.
49. Ishiguro, K. and Xavier, R. (2004) Homer-3 regulates activation of serumresponse element in T cells via its EVH1 domain. Blood, 103, 2248–2256.
50. Skare, P., Kreivi, J.P., Bergstrom, A. and Karlsson, R. (2003) Profilin Icolocalizes with speckles and Cajal bodies: a possible role in pre-mRNAsplicing. Exp. Cell Res., 286, 2–21.
51. Birbach, A., Verkuyl, J.M. and Matus, A. (2006) Reversible,activity-dependent targeting of profilin to neuronal nuclei. Exp. Cell Res.,312, 2279–2287.
52. Wallar, B.J. and Alberts, A.S. (2003) The formins: active scaffolds thatremodel the cytoskeleton. Trends Cell Biol., 13, 435–446.
53. Lederer, M., Jockusch, B.M. and Rothkegel, M. (2005) Profilin regulatesthe activity of p42POP, a novel Myb-related transcription factor. J. CellSci., 118, 331–341.
54. Staus, D.P., Blaker, A.L., Taylor, J.M. and Mack, C.P. (2007) Diaphanous1 and 2 regulate smooth muscle cell differentiation by activating themyocardin-related transcription factors. Arterioscler. Thromb. Vasc. Biol.,27, 478–486.
55. Fulga, T.A., Elson-Schwab, I., Khurana, V., Steinhilb, M.L., Spires, T.L.,Hyman, B.T. and Feany, M.B. (2007) Abnormal bundling andaccumulation of F-actin mediates tau-induced neuronal degeneration invivo. Nat. Cell Biol., 9, 139–148.
56. Hirano, A. (1994) Hirano bodies and related neuronal inclusions.Neuropathol. Appl. Neurobiol., 20, 3–11.
57. Hirano, A., Dembitzer, H.M., Kurland, L.T. and Zimmerman, H.M.(1968) The fine structure of some intraganglionic alterations.J. Neuropathol. Exp. Neurol., 27, 167–182.
58. Galloway, P.G., Perry, G. and Gambetti, P. (1987) Hirano body filamentscontain actin and actin-associated proteins. J. Neuropathol. Exp. Neurol.,46, 185–199.
59. Maciver, S.K. and Harrington, C.R. (1995) Two actin binding proteins,actin depolymerizing factor and cofilin, are associated with Hirano bodies.Neuroreport, 6, 1985–1988.
60. Kidd, A.R., Miskowski, J.A., Siegfried, K.R., Sawa, H. and Kimble,J. (2005) A beta-catenin identified by functional rather than sequencecriteria and its role in Wnt/MAPK signaling. Cell, 121, 761–772.
61. Brenner, S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77,71–94.
62. De Stasio, E.A. and Dorman, S. (2001) Optimization of ENU mutagenesisof Caenorhabditis elegans. Mutat. Res., 495, 81–88.
63. Davis, M.W., Hammarlund, M., Harrach, T., Hullett, P., Olsen, S. andJorgensen, E.M. (2005) Rapid single nucleotide polymorphism mappingin C. elegans. BMC Genomics, 6, 118.
64. Crittenden, S.L. and Kimble, J. (1999) Confocal methods forCaenorhabditis elegans. Meth. Mol. Biol., 122, 141–151.
65. Kraemer, B., Crittenden, S., Gallegos, M., Moulder, G., Barstead, R.,Kimble, J. and Wickens, M. (1999) NANOS-3 and FBF proteinsphysically interact to control the sperm-oocyte switch in Caenorhabditiselegans. Curr. Biol., 9, 1009–1018.
66. Frangioni, J.V. and Neel, B.G. (1993) Solubilization and purification ofenzymatically active glutathione S-transferase (pGEX) fusion proteins.Anal. Biochem., 210, 179–187.
67. Harlowe, E. and Lane, D. (1999) Using Antibodies: A laboratory Manual.Cold Spring Harbor Press, Cold Spring Harbor, NY, pp. 153–219.
68. Tanahashi, H. and Tabira, T. (1999) Molecular cloning of human Fe65L2and its interaction with the Alzheimer’s beta-amyloid precursor protein.Neurosci. Lett., 261, 143–146.
69. Gertler, F.B., Niebuhr, K., Reinhard, M., Wehland, J. and Soriano, P.(1996) Mena, a relative of VASP and Drosophila Enabled, is implicated inthe control of microfilament dynamics. Cell, 87, 227–239.
70. Watanabe, N., Madaule, P., Reid, T., Ishizaki, T., Watanabe, G.,Kakizuka, A., Saito, Y., Nakao, K., Jockusch, B.M. and Narumiya,S. (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, isa target protein for Rho small GTPase and is a ligand for profilin. EMBOJ., 16, 3044–3056.
Human Molecular Genetics, 2007, Vol. 16, No. 16 1971
Dow
nloaded from https://academ
ic.oup.com/hm
g/article-abstract/16/16/1959/2527273 by guest on 05 April 2019