Embed Size (px)
Citation preview

1
SUPPLEMENTARY INFORMATION
Secondary structure reshuffling modulates glycosyltransferase function
at the membrane
David Giganti1,2,3, David Albesa-Jové2,3, Saioa Urresti2,3, Ane Rodrigo-Unzueta2,3, Mariano
A. Martínez1, Natalia Comino2,3, Nathalie Barilone1, Marco Bellinzoni1, Alexandre Chenal4,
Marcelo E. Guerin2,3,5,* and Pedro M. Alzari1,*
1Institut Pasteur, Unité de Microbiologie Structurale & CNRS UMR 3528, 25 rue du Dr.
Roux, 75724, Paris Cedex 15, France, 2Unidad de Biofisica, Centro Mixto Consejo Superior de Investigaciones Cientificas -
Universidad del País Vasco/Euskal Herriko Unibertsitatea (CSIC,UPV/EHU), Barrio
Sarriena s/n, Leioa, Bizkaia, 48940, Spain, 3Departamento de Bioquímica, Universidad del País Vasco, Spain, 4Institut Pasteur, Unité de Biochimie des Interactions Macromoléculaires & CNRS UMR
3528, 28 rue du Dr. Roux, 75724, Paris Cedex 15, France, 5IKERBASQUE, Basque Foundation for Science, 48011, Bilbao, Spain.
6To whom correspondence should be addressed: Marcelo E. Guerin, Unidad de Biofisica,
Centro Mixto Consejo Superior de Investigaciones Cientificas - Universidad del País
Vasco/Euskal Herriko Unibertsitatea (CSIC,UPV/EHU), Barrio Sarriena s/n, Leioa, Bizkaia,
48940, Spain, Tel: +34 94 601 8052; Fax : +34 94 601 3360; E-mail:
[email protected]; and Pedro M. Alzari, Institut Pasteur, Unité de Microbiologie
Structurale & CNRS UMR 3528, 25 rue du Dr. Roux, 75724, Paris Cedex 15, France; Tel:
+33 145 688 607; Fax : +33 145 688 604; E-mail: [email protected]
Nature Chemical Biology: doi:10.1038/nchembio.1694

2
SUPPLEMENTARY RESULTS
SUPPLEMENTARY FIGURES
Supplementary Figure 1. Amino acid sequence of GSGA-PimA construct used for
heterologous production in E. coli. (a) The recombinant PimA contruct (430 residues)
contains a peptide extension of 44 residues (highlighted in orange) at the N-terminus of full-
length PimA from M. smegmatis (highlighted in yellow), including a poly-histidine tag and
the optimal Tobacco Etch Virus protease (TEV) cleavage site (ENLYFQG, underlined). The
introduction of a three-residues spacer (SerGlyAla, highlighted in red) between the TEV
cleavage site and the N-terminus of PimA was crucial to achieve efficient removal of the N-
terminal histidine tag. (b) SDS-PAGE showing purified PimA before and after TEV
treatment.
Nature Chemical Biology: doi:10.1038/nchembio.1694
3
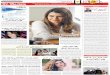
Supplementary Figure 2. Four crystallographically independent apo PimA monomers
reveal two different conformational states. (a). The asymmetric unit of the P3221 crystal
form contains four apo PimA molecules (shown here in yellow, cyan, orange and red). The
two independent molecules colored in yellow and cyan adopt a ‘compact’ conformational
state, in which the helical hairpin formed by helices α4 (residues 134 to 145) and α5
(residues 149 to 157) folds back against the N-terminal domain. The other two molecules in
the asymmetric unit (colored in orange and red) adopt an ‘extended’ state, in which the same
Nature Chemical Biology: doi:10.1038/nchembio.1694

4
helical hairpin is partially disordered and protrudes away from the N-terminal domain.
Panels (b) and (c) show the structural superposition of the two molecules in the compact and
extended conformations, respectively. Each chain is color-coded as in (a).
Nature Chemical Biology: doi:10.1038/nchembio.1694
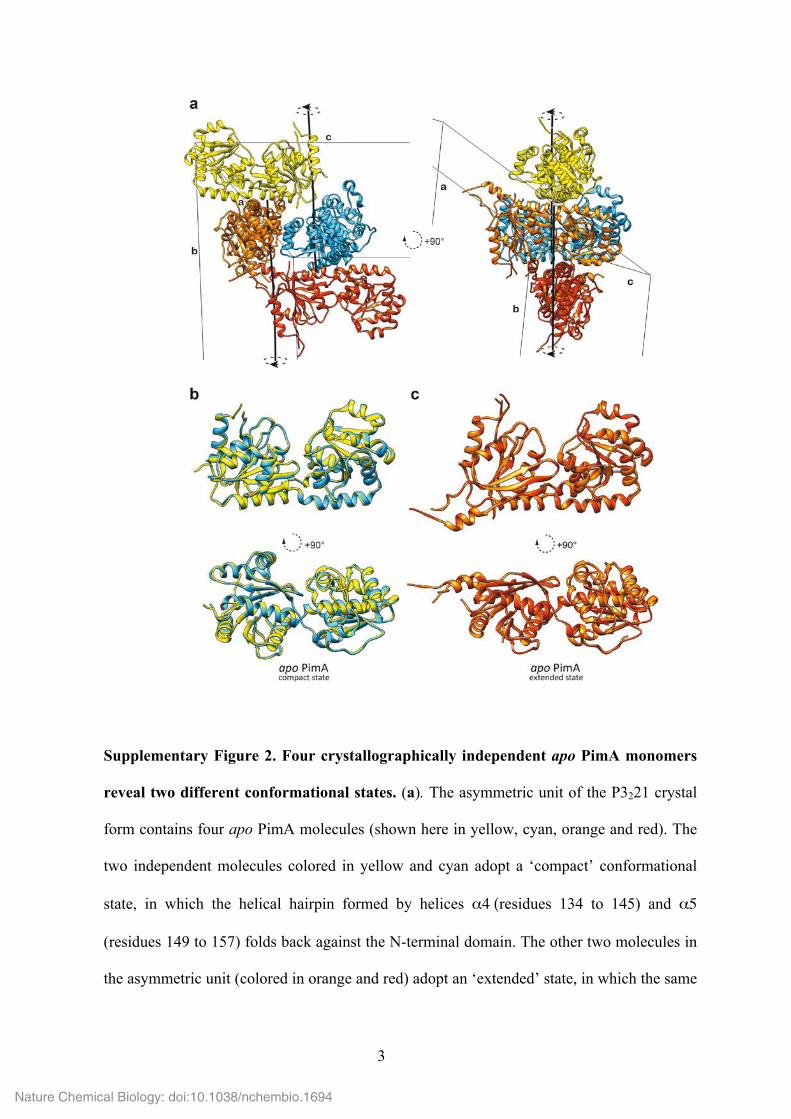
5
Supplementary Figure 3. Final 2Fo-Fc electron density maps of the compact and
extended conformational states of apo PimA. (a, b) Overall and close views of the N-
terminal domain of a PimA molecule B (compact state) of apo PimA contoured at 1σ, which
exhibits significant structural variability. (c, d) Similar views of apo PimA molecule A
(extended state).
Nature Chemical Biology: doi:10.1038/nchembio.1694

6
Supplementary Figure 4. The two apo forms of PimA display an ‘open’ conformation
compared to the sugar-nucleotide-bound enzyme. The figure shows the structural
superposition of GDP-bound PimA (shown in orange) with the apo enzyme in its compact
(a) and extended (b) conformations (shown in grey). Both PimA apo forms crystallized in
an ‘open’ conformation with a deep and wide cleft separating the N- and C-terminal
domains. These domains are covalently connected through two linker regions, the
connecting loop between β7 and α6 (residues 167 to 172) and a short kink (residues 347 to
349) between the last two α-helices of the protein, α13 (C-terminal domain) and α14 (N-
terminal domain). Both linkers ensure flexibility of the N- and C-terminal domains. Binding
of GDP or GDP-Man induces a 14° rotational reorientation of the N-terminal domain
relative to the C-terminal domain, stabilizing the enzyme in a ‘closed’ state.
Nature Chemical Biology: doi:10.1038/nchembio.1694
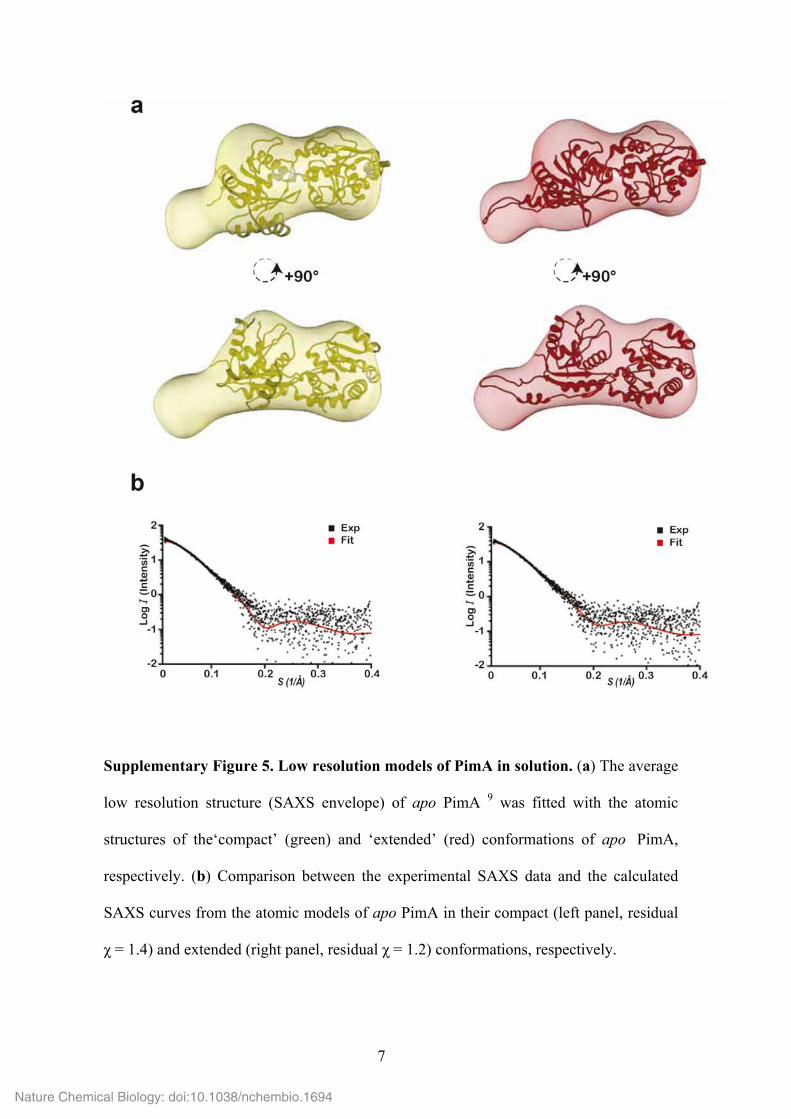
7
Supplementary Figure 5. Low resolution models of PimA in solution. (a) The average
low resolution structure (SAXS envelope) of apo PimA 9 was fitted with the atomic
structures of the‘compact’ (green) and ‘extended’ (red) conformations of apo PimA,
respectively. (b) Comparison between the experimental SAXS data and the calculated
SAXS curves from the atomic models of apo PimA in their compact (left panel, residual
χ = 1.4) and extended (right panel, residual χ = 1.2) conformations, respectively.
Nature Chemical Biology: doi:10.1038/nchembio.1694

8
Supplementary Figure 6. Hydrophobic surface patch exposed to the solvent in apo
PimA. A patch of hydrophobic residues within the N-terminal domain is largely exposed to
the solvent in the extended conformation of apo PimA. This patch is close to the amphipatic
helix α2 (shown in blue), which includes Trp82 as well as an exposed cluster of basic
residues likely involved in the interactions with phospholipids 7.
Nature Chemical Biology: doi:10.1038/nchembio.1694

9
Supplementary Figure 7. Multiple sequence alignment of the highly mobile region of
PimA (residues 110 to 170) in mycobacterial orthologs. Orthologs shown are from
Mycobacterium smegmatis (Msme, MSMEG2946), M. tuberculosis (Mtub, O06204), M.
leprae (Mlep, O07147), M. bovis (Mbov, Q7TY88), M. marinum (Mmar, B2HN44), M.
ulcerans (Mulc, A0PSZ0), M. avium (Mavi, A0QID0), M. phlei (Mphl, I0RIP4), M.
Nature Chemical Biology: doi:10.1038/nchembio.1694

10
abscessus (Mabs, B1MCK2), M. gilvum (Mgil, A4TD14), M. massiliense (Mmas, I8H153),
M. tusciae (Mtus, H1K855), M. vanbaalenii (Mvan, A1T872), M. rhodesiae (Mrho,
G8RJM2), M. xenopi (Mxen, I0RXA8), M. canettii (Mcan, L0QWY2), M. orygis (Mory,
M8CC24), M. parascrofulaceum (Mpar. D5PI67), M. intracellulare (Mint, H8JIX8), M.
vaccae (Mvac, K0UP21), M. colombiense (Mcol, J4TE97), M. indicus (Mind, J9WGS6), M.
chubuense (Mchu, I4BIF7), M. fortuitum (Mfor, K0UZF8), and M. liflandii (Mlif,
L7VBH2). Strictly conserved positions are shown in blue background. The secondary
structural elements corresponding to the four distinct 3D structures of PimA discussed in the
manuscript (the compact and extended apo PimA forms and the complexes of PimA with
GDP-Man and with GDP + diC8PI) are shown below the alignment. The linear regions
involved in the α-helix-to-β-strand and β-strand-to-α-helix transitions are indicated.
Nature Chemical Biology: doi:10.1038/nchembio.1694

11
Supplementary Figure 8. The canonical GT-B conformation (GDP-bound form) of
PimA, but not the atypical apo form, is functionally competent for nucleotide-sugar
binding and catalysis. The figure shows a detailed view of the active site of PimA as
observed for the apo form (a) and the PimA-GDP-Man complex (b). In apo PimA, the tip of
the β-hairpin β5-β6 (residues 117 to 123, N-terminal domain) interacts with the catalytic
loop β11-α10 (residues 268 to 278, C-terminal domain), stabilizing an ‘open’ state of the
enzyme. This structural arrangement of the catalytic loop would not only interfere with
GDP-Man binding, but also bring the lateral chain of the crucial Glu274 away from the
catalytic pocket, strongly suggesting that the observed ‘open’ state corresponds to an
inactive conformation. Binding of the donor sugar substrate (GDP-Man) is required to
achieve a ‘closed’ state 30 that brings together critical conserved residues making up a
functionally competent active site.
Nature Chemical Biology: doi:10.1038/nchembio.1694
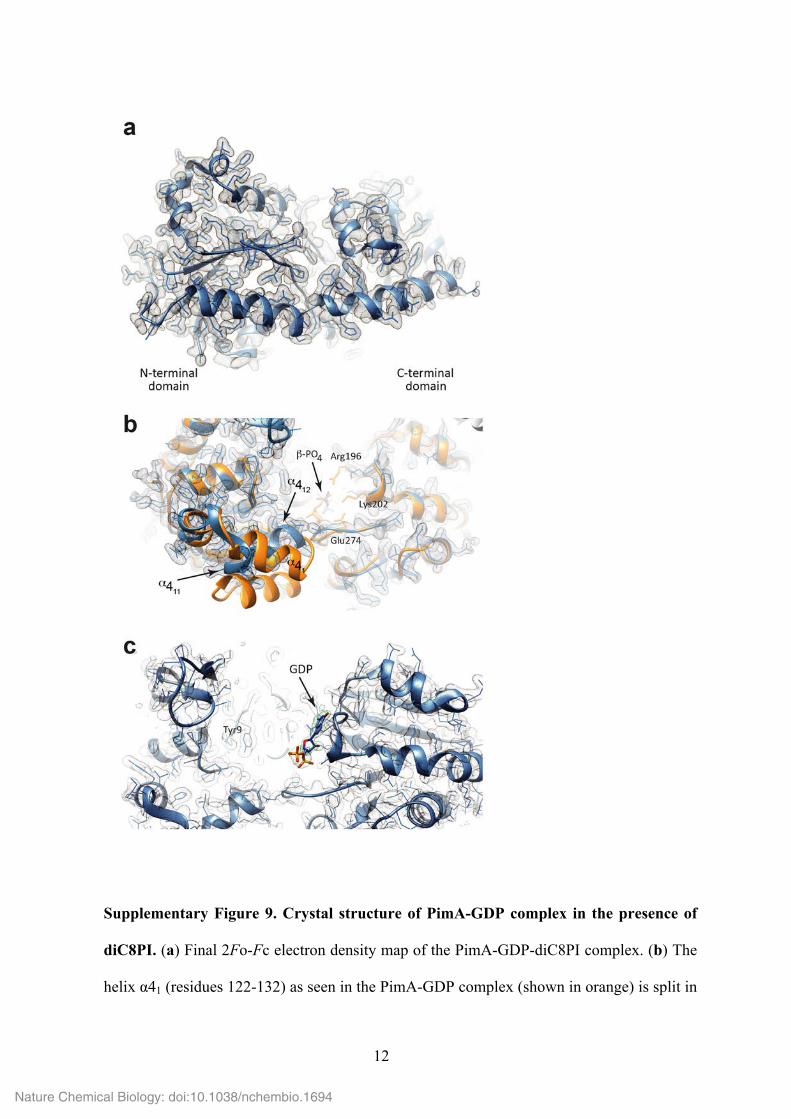
12
Supplementary Figure 9. Crystal structure of PimA-GDP complex in the presence of
diC8PI. (a) Final 2Fo-Fc electron density map of the PimA-GDP-diC8PI complex. (b) The
helix α41 (residues 122-132) as seen in the PimA-GDP complex (shown in orange) is split in
Nature Chemical Biology: doi:10.1038/nchembio.1694

13
two distinctly oriented α helices in the PimA-GDP-diC8PI complex (blue). (c) View of the
GDP binding site in the crystal structure of PimA GDP-diC8PI. Shown are the 2Fo-Fc map
contoured at 1 sigma (grey mesh) and the Fo-Fc omit map (after removal of GDP)
contoured at 4 sigma (green mesh).
Nature Chemical Biology: doi:10.1038/nchembio.1694
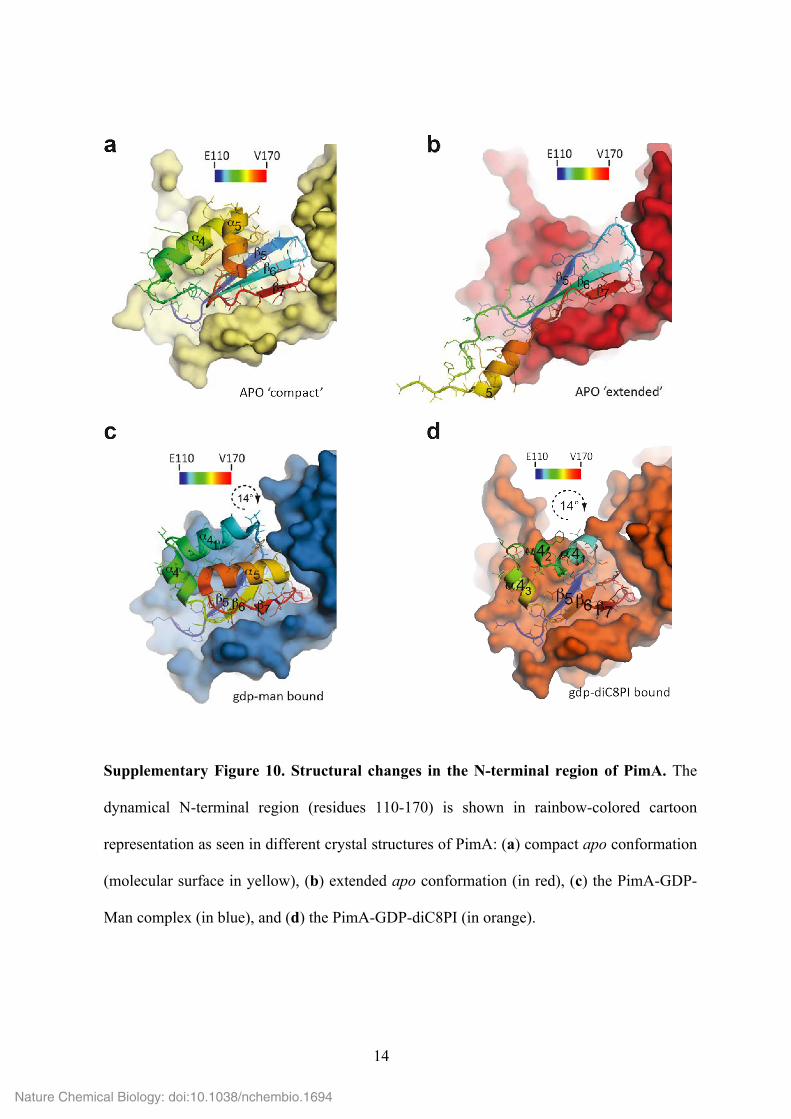
14
Supplementary Figure 10. Structural changes in the N-terminal region of PimA. The
dynamical N-terminal region (residues 110-170) is shown in rainbow-colored cartoon
representation as seen in different crystal structures of PimA: (a) compact apo conformation
(molecular surface in yellow), (b) extended apo conformation (in red), (c) the PimA-GDP-
Man complex (in blue), and (d) the PimA-GDP-diC8PI (in orange).
Nature Chemical Biology: doi:10.1038/nchembio.1694
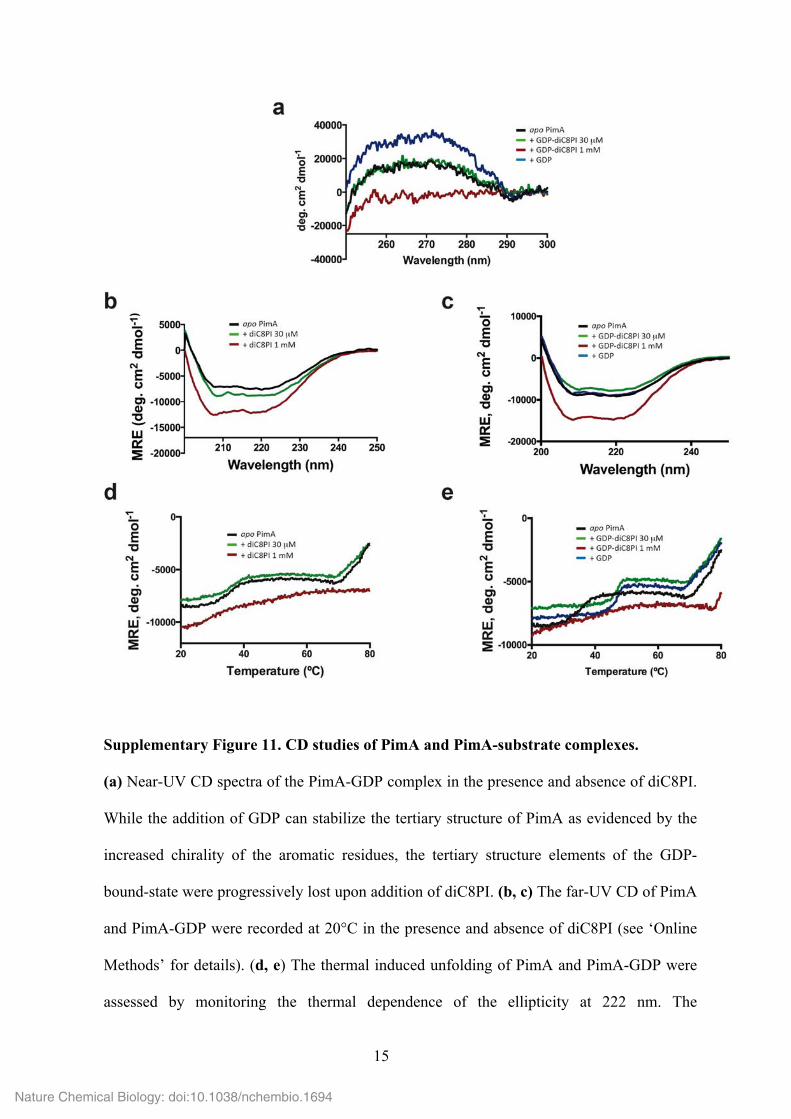
15
Supplementary Figure 11. CD studies of PimA and PimA-substrate complexes.
(a) Near-UV CD spectra of the PimA-GDP complex in the presence and absence of diC8PI.
While the addition of GDP can stabilize the tertiary structure of PimA as evidenced by the
increased chirality of the aromatic residues, the tertiary structure elements of the GDP-
bound-state were progressively lost upon addition of diC8PI. (b, c) The far-UV CD of PimA
and PimA-GDP were recorded at 20°C in the presence and absence of diC8PI (see ‘Online
Methods’ for details). (d, e) The thermal induced unfolding of PimA and PimA-GDP were
assessed by monitoring the thermal dependence of the ellipticity at 222 nm. The
Nature Chemical Biology: doi:10.1038/nchembio.1694

16
cooperativity of PimA unfolding in the absence and in the presence of sub-micellar
concentration of diC8PI revealed a three-state reaction with an intermediate state populated
from 40 to 70°C. The slopes of the transitions between the states reflect the complex set of
interactions involved in the stabilization of both secondary and tertiary structures. This
cooperativity is lost in the presence of 1 mM diC8PI, suggesting that PimA retains
secondary structures upon diC8PI interactions, while protein side-chains exhibit a high
degree of flexibility.
Nature Chemical Biology: doi:10.1038/nchembio.1694

17
Supplementary Figure 12. Tryptophan fluorescence spectroscopy of wild-type PimA in
the presence of small unilamellar vesicles (SUVs). (a) In addition to Trp154, wild-type
PimA has two other tryptophan residues (Trp82, Trp349) that occupy the same buried
positions within the N-terminal domain of the apo (yellow and red) and GDP-bound (blue)
structures of PimA. (b) The ratio of fluorescence intensities at 320 nm and 360 nm of 1 µM
wild-type PimA in the presence and absence of 1 mM SUVs are shown. The observation of
a decrease of the FIR 320/360 upon incubation with negatively charged SUVs (in contrast
with the increase observed for PimAW82F/W349F, see Fig. 3c) suggest that, in addition to the
changes observed for Trp154, the environment of at least one of the other two tryptophans is
also modified in the presence of membrane (i.e. Trp82 and/or Trp349 would become
exposed to a more hydrophilic environment).
Nature Chemical Biology: doi:10.1038/nchembio.1694
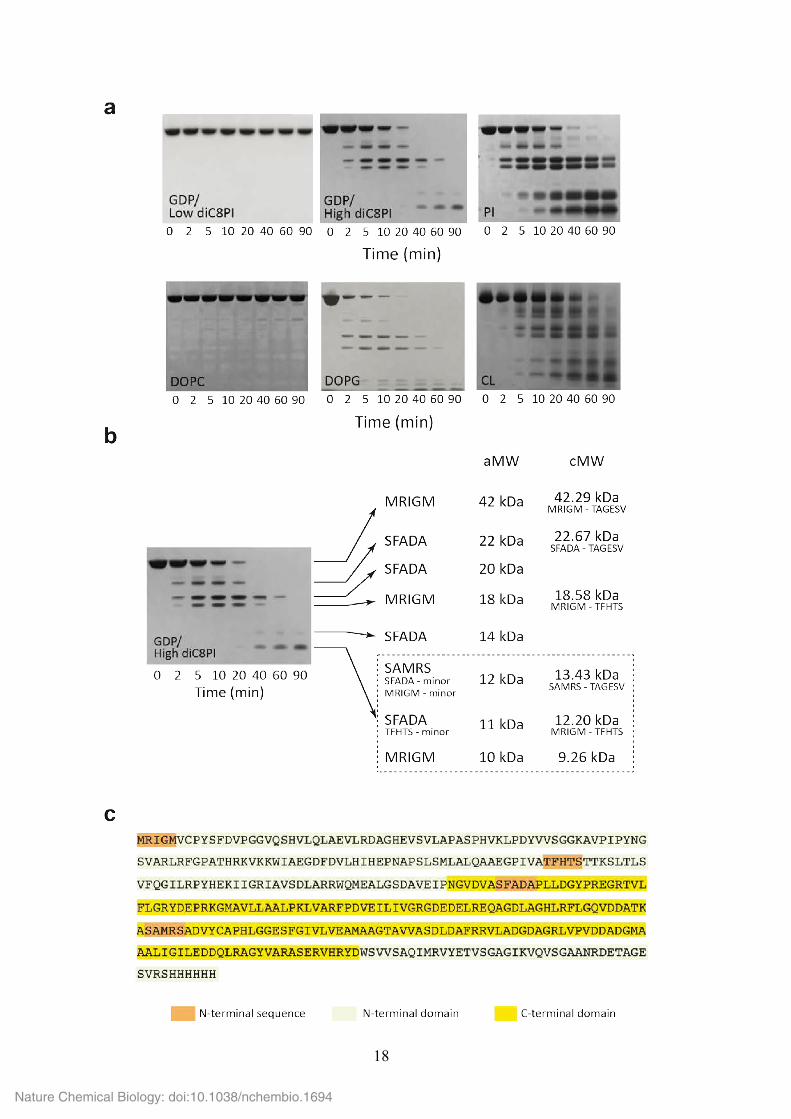
18
Nature Chemical Biology: doi:10.1038/nchembio.1694

19
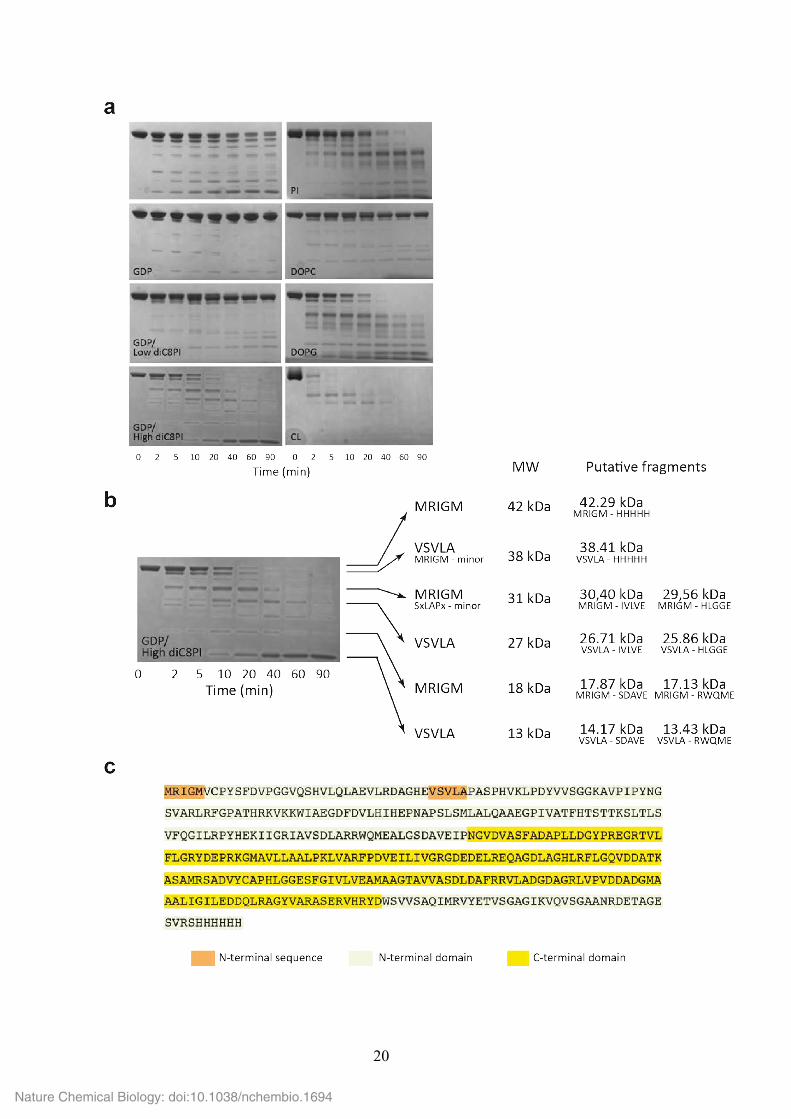
Supplementary Figure 13. Effect of substrate and non-substrate phospholipids on the
conformation of PimA. (a) When PimA was incubated with GDP and 30 µM diC8PI (a
concentration value below the CMC), the enzyme was protected from degradation by
elastase. In contrast, PimA became highly sensitive to proteolysis in the presence of GDP
and 1 mM diC8PI (~20 times the CMC). When the enzyme was incubated with elastase in
the presence of small unilamellar vesicles (SUVs) prepared from different lipids, PimA was
protected from proteolysis after the addition of 1,2-dioleoyl-sn-glycero-3-phosphocholine
(DOPC, neutral/zwitterionic phospholipid), whereas the presence of anionic phospholipids
such as PI, DOPG or cardiolipin (CL) promoted a conformational change in the protein that
increased its susceptibility to proteolysis. (b) PimA was incubated with elastase in the
presence of GDP and 1 mM diC8PI (~20 times the CMC) for 0–90 min at 37 °C (see
‘Online Methods’ for further details). Aliquots were removed at the indicated time intervals,
and subsequently analysed by SDS-PAGE. The N-terminal sequence obtained for each
proteolytic fragment and the corresponding molecular weight are indicated. It is worth
noting that elastase is a serine protease that preferentially cleaves at the C-terminus of
alanine, valine, serine, glycine, leucine or isoleucine residues. PimA displays 94 putative
cleavage sites 31. (c) Amino acid sequence of PimA showing the location of the proteolytic
sites identified by N-terminal protein sequencing (orange background).
Nature Chemical Biology: doi:10.1038/nchembio.1694
20
Nature Chemical Biology: doi:10.1038/nchembio.1694

21
Supplementary Figure 14. Limited proteolysis studies of PimA with Endoproteinase
Glu-C (V8 protease). Endoproteinase Glu-C is a serine proteinase which selectively cleaves
peptide bonds C-terminal to glutamic acid residues. PimA has 21 putative cleavage sites 31,
different from those predicted for elastase. (a) When PimA was incubated with GDP and 30
µM diC8PI (a concentration value below the CMC), the enzyme was protected from
degradation by endoproteinase Glu-C. In contrast, PimA became highly sensitive to
proteolysis in the presence of GDP and 1 mM diC8PI (~20 times the CMC). The other
panels show the endoproteinase Glu-C cleavage of PimA preincubated with GDP and 30 uM
diC8PI in the presence of different neutral (DOPC) and anionic (DOPG, CL, PI)
phospholipids. (b) PimA was incubated with elastase in the presence of GDP and 1 mM
diC8PI (~20 times the CMC) for 0–90 min at 37 °C (see ‘Online Methods’ for further
details). Aliquots were removed at the indicated time intervals, and subsequently analysed
by SDS-PAGE. The N-terminal sequence obtained for each proteolytic fragment, its
estimated molecular weight and the putative associated fragments are indicated. (c) Amino
acid sequence of PimA showing the location of the proteolytic sites identified by N-terminal
protein sequencing (orange background).
Nature Chemical Biology: doi:10.1038/nchembio.1694
22
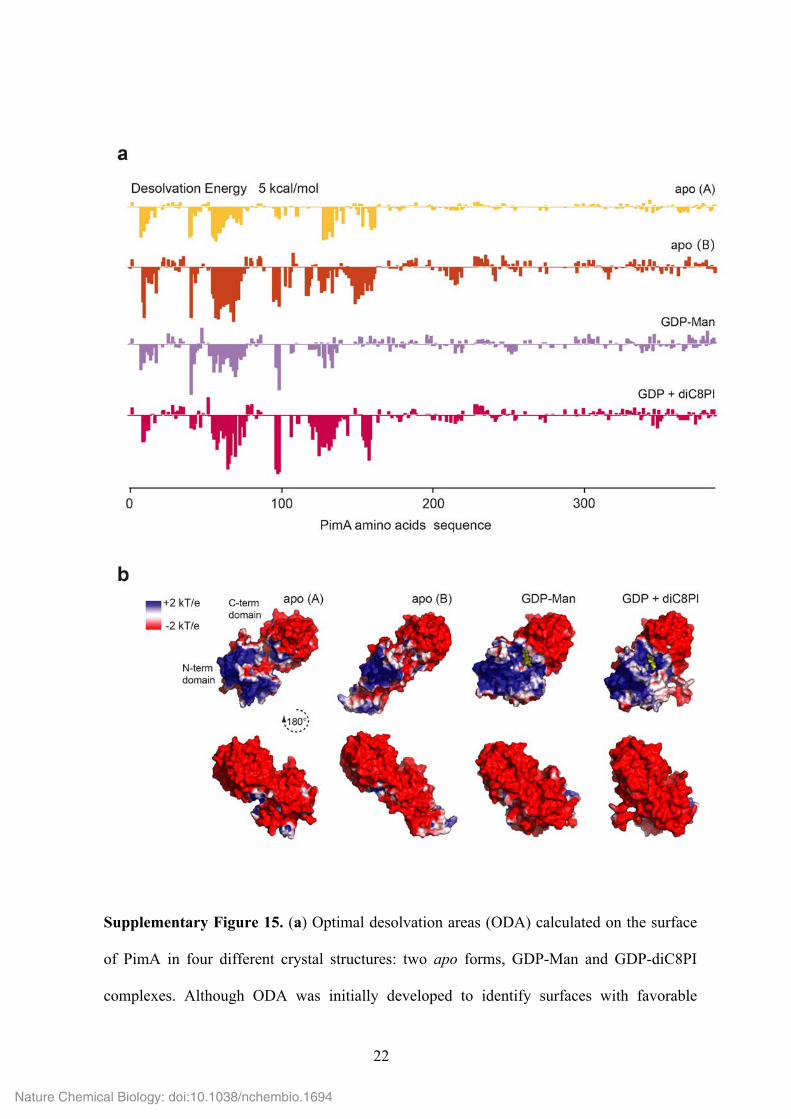
Supplementary Figure 15. (a) Optimal desolvation areas (ODA) calculated on the surface
of PimA in four different crystal structures: two apo forms, GDP-Man and GDP-diC8PI
complexes. Although ODA was initially developed to identify surfaces with favorable
Nature Chemical Biology: doi:10.1038/nchembio.1694

23
change of energy when buried upon protein-protein association 32, previous studies show
that ODA hot spots match well with membrane-associated regions of different GT-B
glycosyltransferases 6,7. (b) Electrostatic charge distribution for the same four PimA
structures. Electronegative regions are shown in red, electropositive regions are in blue, and
neutral regions are in white. Continuum electrostatics were calculated with the Poisson-
Boltzmann solver of APBS 1.3 33.
Nature Chemical Biology: doi:10.1038/nchembio.1694
24
SUPPLEMENTARY TABLE
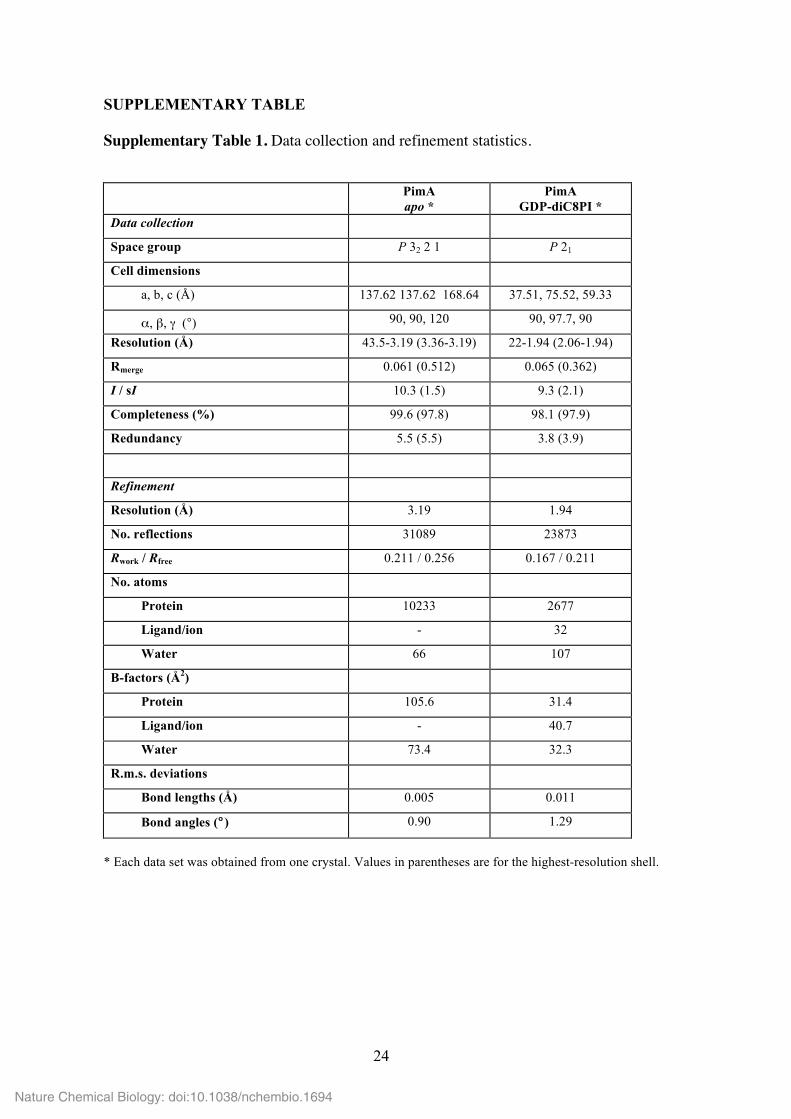
Supplementary Table 1. Data collection and refinement statistics.
PimA apo *
PimA GDP-diC8PI *
Data collection
Space group P 32 2 1 P 21
Cell dimensions
a, b, c (Å) 137.62 137.62 168.64 37.51, 75.52, 59.33
α, β, γ (°) 90, 90, 120 90, 97.7, 90
Resolution (Å) 43.5-3.19 (3.36-3.19) 22-1.94 (2.06-1.94)
Rmerge 0.061 (0.512) 0.065 (0.362)
I / sI 10.3 (1.5) 9.3 (2.1)
Completeness (%) 99.6 (97.8) 98.1 (97.9)
Redundancy 5.5 (5.5) 3.8 (3.9)
Refinement
Resolution (Å) 3.19 1.94
No. reflections 31089 23873
Rwork / Rfree 0.211 / 0.256 0.167 / 0.211
No. atoms
Protein 10233 2677
Ligand/ion - 32
Water 66 107
B-factors (Å2)
Protein 105.6 31.4
Ligand/ion - 40.7
Water 73.4 32.3
R.m.s. deviations
Bond lengths (Å) 0.005 0.011
Bond angles (°) 0.90 1.29
* Each data set was obtained from one crystal. Values in parentheses are for the highest-resolution shell.
Nature Chemical Biology: doi:10.1038/nchembio.1694

25
SUPPLEMENTARY VIDEOS
Supplementary Video 1. Structural transition between the extended and compact
conformational states of apo PimA. This movement corresponds to a swinging back-and-
forth movement of the helical hairpin α4-α5. To calculate the morph trajectory, we first used
Modeller (http://salilab.org/modeller/) to construct ab initio all missing residues and
polypeptide segments in the three crystallographic structures of PimA: the compact and
extended apo conformations, and the GDP-bound form. After superposition of their C-
terminal domains, we used Chimera (UCSF) to generate coarse conformational
intermediates by interpolating the atomic positions. The interpolation was based on the
Corkscrew method, as implemented in Chimera. We show 20 intermediates calculated
between the two apo PimA forms. All generated structures were minimized with a MMTK
routine and Amber parameters. PimA is colored in a rainbow gradient with the N-terminal
and C-terminal ends in blue and red, respectively; the unstructured C-terminal end Val372-
Val386 is not shown.
Supplementary Video 2. Structural transition between apo PimA and GDP-bound
PimA involving the reshuffling of secondary structural elements from the N-terminal
domain. Calculations were performed as described for Supplementary Video 1, but the
morph trajectory now includes 60 intermediate structures. To get a more clear illustration of
the secondary structure reshuffling, we imposed a route visiting one intermediate picked
from the previous compact-extended morph trajectory. The depicted trajectories are not
representative of the complex dynamics of PimA, but are intended as a visual support to
appreciate the large conformational changes.
Nature Chemical Biology: doi:10.1038/nchembio.1694

26
REFERENCES
6. Albesa-Jové, D., Giganti, D., Jackson, M., Alzari, P.M. & Guerin, M.E. Glycobiology
24, 108-124 (2014).
7. Guerin, M.E. et al. J. Biol. Chem. 282, 20705– 20714 (2007).
9. Giganti, D. et al. J. Biol. Chem. 288, 29797-29808 (2013).
30. Hayward, S. & Lee, R.A. J. Mol. Graph. Model. 21, 181-3 (2002).
31. Gasteiger, G.E. et al. In The Proteomics Protocols Handbook, Walker, J.M. (ed),
(Humana Press, 2005).
32. Fernandez-Recio, J., Trotov, M., Skorodumov, C. & Abagyan, R. Proteins 58, 134-43
(2005).
33. Honig, B. & Nicholls, A. Science 268, 1144-1149 (1995).
Nature Chemical Biology: doi:10.1038/nchembio.1694








![LISTINO - elettronicamannucci.it · Nastro Led 182 Led/m 3528 - 24Vdc - 14,4W/m - Bianco caldo [PolyBag 1 pz.] € 60,20 JO35014243WW Nastro Led 182 Led/m 3528 - 24Vdc - 14,4W/m -](https://img.dokumen.tips/doc/110x75/5c61b39809d3f266528b45a6/listino-nastro-led-182-ledm-3528-24vdc-144wm-bianco-caldo-polybag.jpg)