Embed Size (px)
Citation preview

Termodynamik
Studiet af omdannelse af energi
Arbejde, varme og energi2.5-2.10

Tilstandsfunktion
Værdien er kun afhængig af systemets øjeblikkeligetilstand og afhænger ikke af vejen. Entydigt givet ved en beskrivelse af systemets tilstand
∫=∆f
i
dhh Eksakt differential, uafhængigt af integrationsvejen
Vejfunktion
∫=f
i
dlL Ikke eksakt integralAfhænger af vejen, L er ikke en tilstandsfunktion
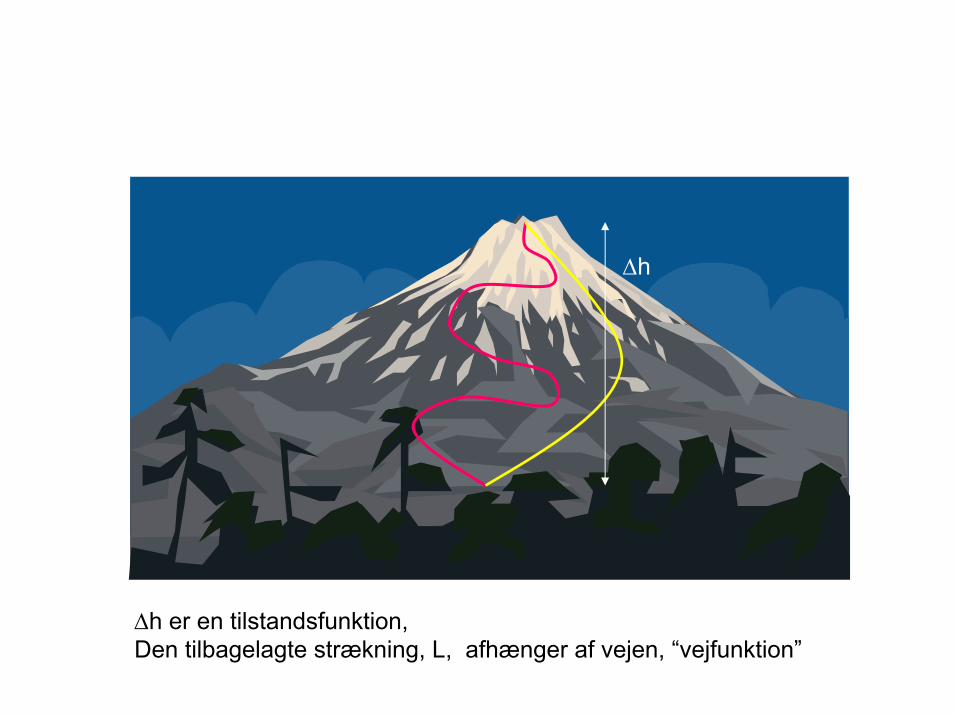
∆h
∆h er en tilstandsfunktion, Den tilbagelagte strækning, L, afhænger af vejen, “vejfunktion”

Tilstandsligning:Funktion, der udtrykker sammenhæng
mellem tre tilstandsstørrelserp=f(T,V,n) p=nRT/V
U(p,V,T) kan pga tilstandsligning skrivessom funktion af to uafhængige variable
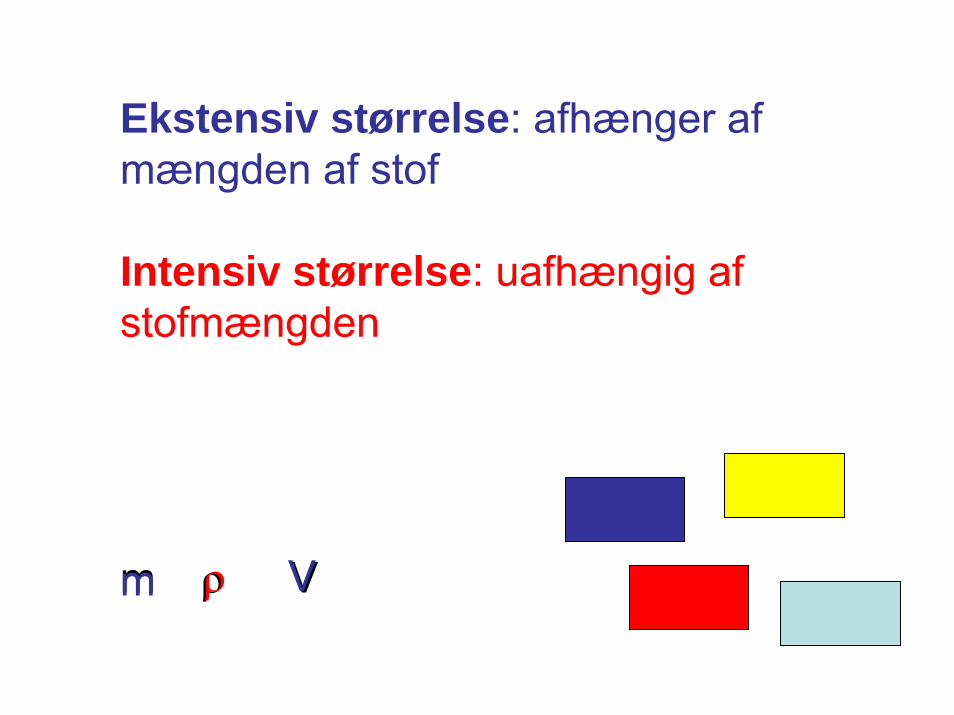
Ekstensiv størrelse: afhænger afmængden af stof
Intensiv størrelse: uafhængig afstofmængden
m ρ Vm ρ V

Varmekapacitet: Ekstensiv Cv
Molar varmekapacitet: Intentiv Cv,m=Cv/n

Enthalpi
H ≡ U + pV [J]Tilstandsfunktion∆H=∆U+∆(pV)
Hvorfor en ny energi-funktion?∆U=qV ∆H=qp Τ1
Konstant tryk, ikke noget “additional work”
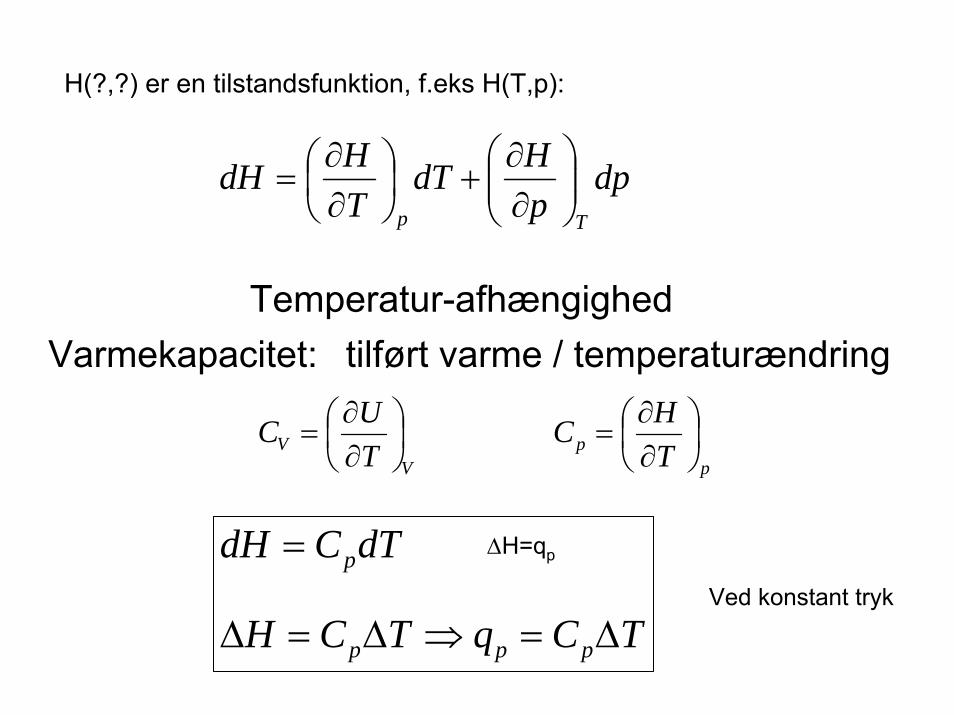
H(?,?) er en tilstandsfunktion, f.eks H(T,p):
dppHdT
THdH
Tp⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=
Temperatur-afhængighedVarmekapacitet: tilført varme / temperaturændring
pp T
HC ⎟⎠⎞
⎜⎝⎛
∂∂
=V
V TUC ⎟
⎠⎞
⎜⎝⎛
∂∂
=
TCqTCH
dTCdH
ppp
p
∆=⇒∆=∆
=Ved konstant tryk
∆H=qp

Hvordan bestemmer man enthalpienstemperaturafhængighed?
pkonstant ved),()()(
...
),(
2
1,12
2,
∫+=
+++=
⎟⎠⎞
⎜⎝⎛
∂∂
=
⎟⎠⎞
⎜⎝⎛
∂∂
=
T
Tmpmm
mp
pp
pp
dTpTCTHTH
TcbTaC
TqC
pTHandTHC
Mål Cp,m
T5

Enthalpi ændring for en reaktion som forbruger eller producererideal gas
H=U+pV = U + nRT∆H= ∆U + ∆(nRT) = ∆U + ∆n(RT)
Hvor stor er forskellen mellem ∆H og ∆U? T4
Cp>Cv som oftest.
Cp-Cv=nR for ideal gas
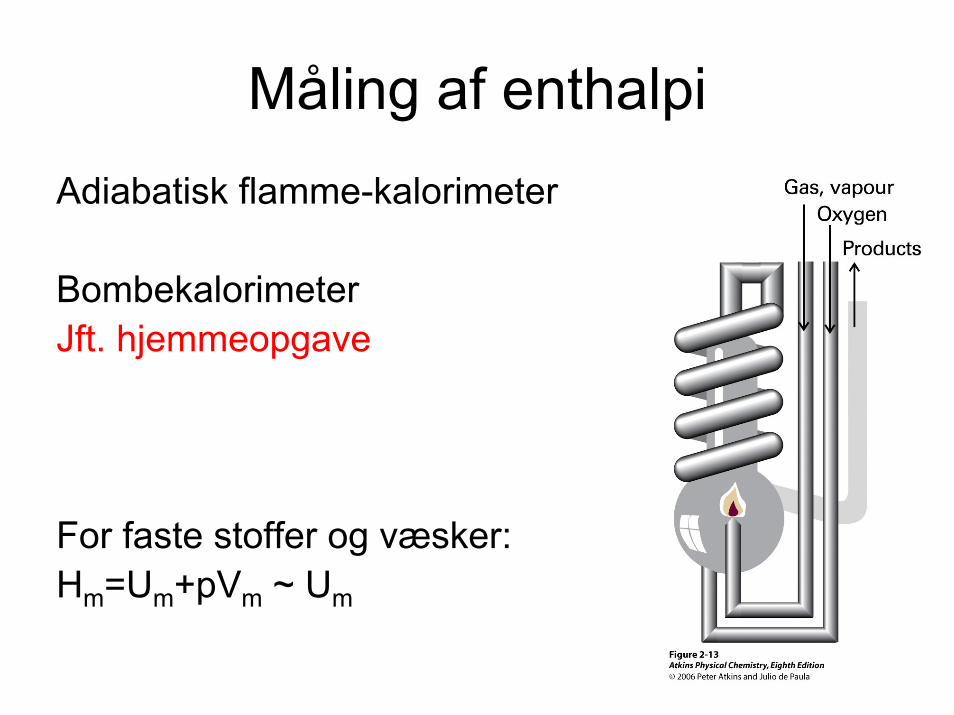
Måling af enthalpiAdiabatisk flamme-kalorimeter
BombekalorimeterJft. hjemmeopgave
For faste stoffer og væsker:Hm=Um+pVm ~ Um
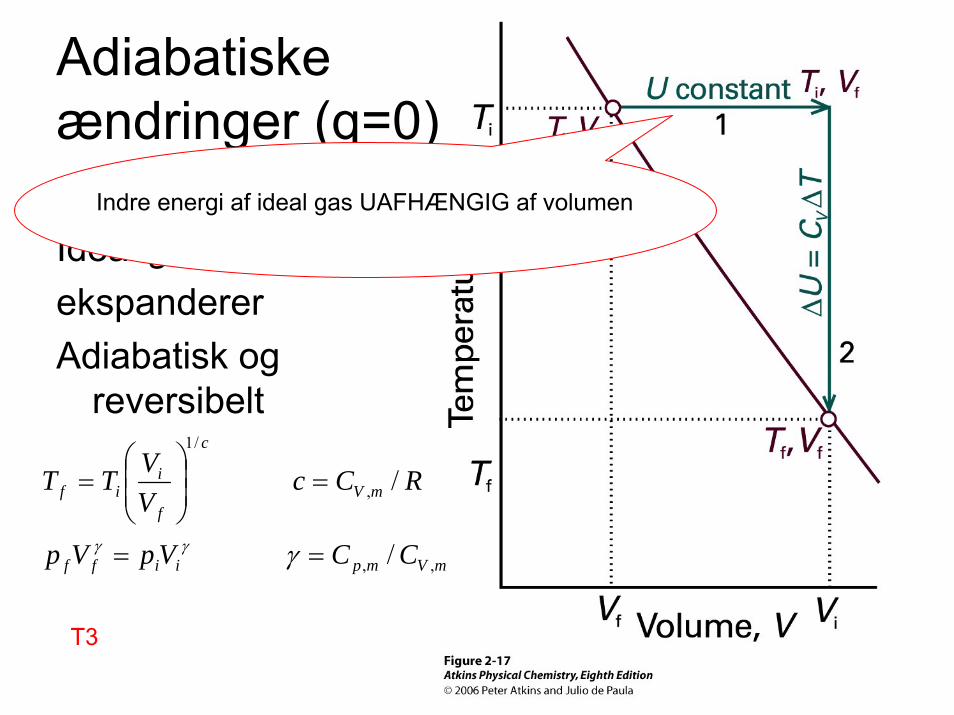
Adiabatiskeændringer (q=0)
Ideal gas ekspandererAdiabatisk og
reversibelt
mVmpiiff
mV
c
f
iif
CCVpVp
RCcVVTT
,,
,
/1
/
/
==
=⎟⎟⎠
⎞⎜⎜⎝
⎛=
γγγ
T3
Indre energi af ideal gas UAFHÆNGIG af volumen
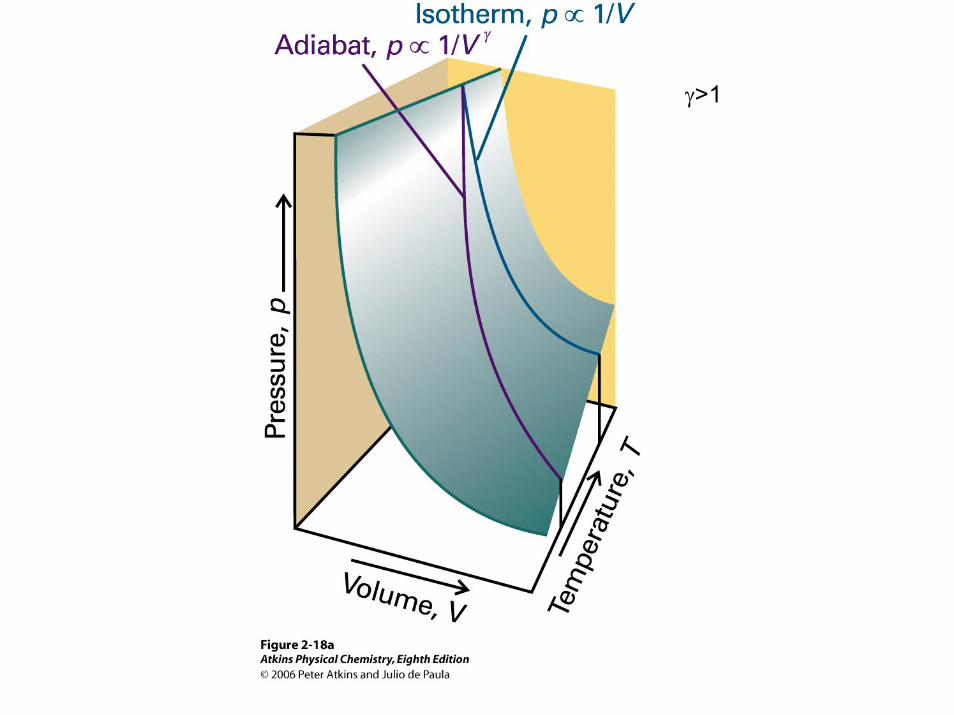
γ>1

Thermo-kemi
Standard enthalpi ændring: ændring i enthalpi for process hvor både udgansstof og “slut-stof” er i standard-tilstanden
Standard tilstanden ved given T: ren form ved trykket 1 bar
EksempelStandard fordampningsenthalpi ∆vapHӨ: 1 mol ren væske fordamper til gas ved trykket 1 barH2O(l) -> H2O (g) ∆vapHӨ(373K) = 40.66 kJ mol-1
Standard smelteenthalpi ∆fusHӨ: 1 mol is smelter til flydende vand ved trykket 1 barH2O(s) -> H2O (l) ∆fusHӨ(273K) = 6.01 kJ mol-1
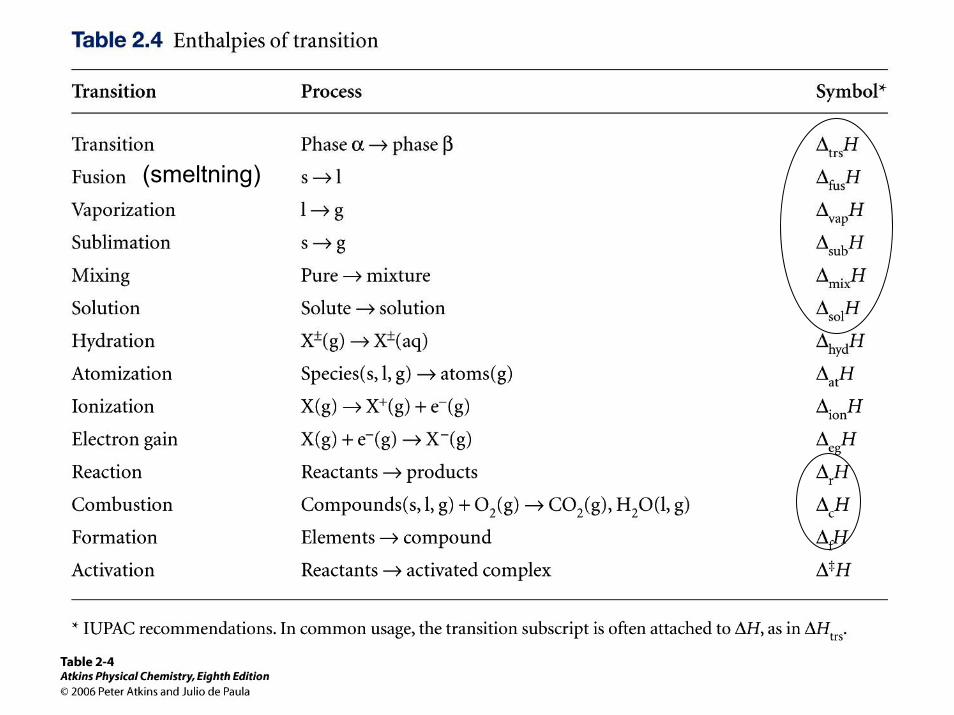
(smeltning)
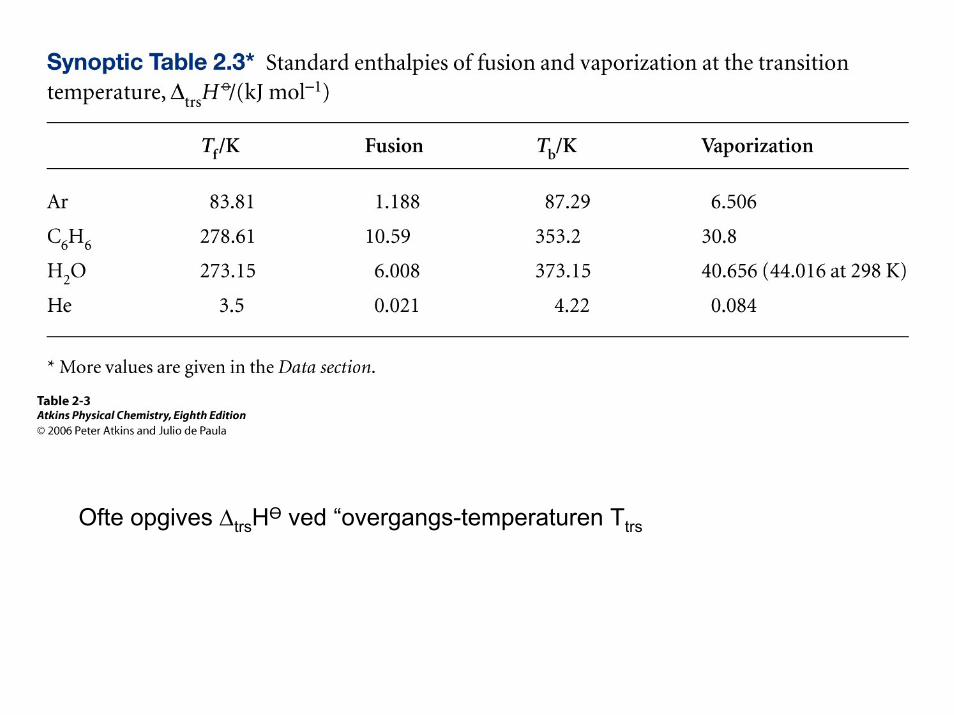
Ofte opgives ∆trsHӨ ved “overgangs-temperaturen Ttrs
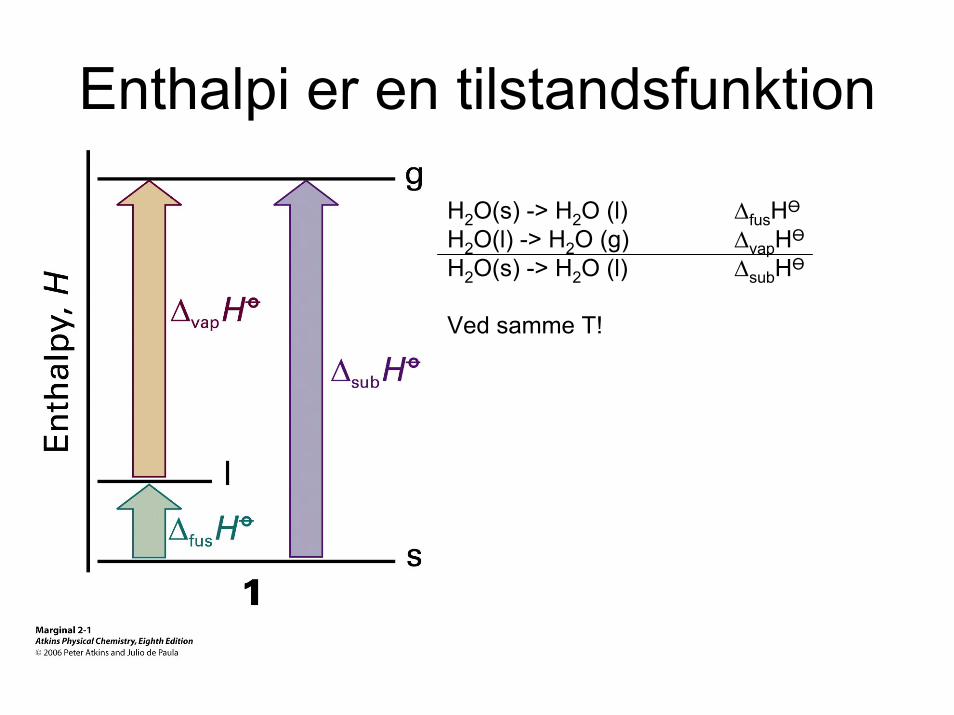
Enthalpi er en tilstandsfunktion
H2O(s) -> H2O (l) ∆fusHӨ
H2O(l) -> H2O (g) ∆vapHӨ
H2O(s) -> H2O (l) ∆subHӨ
Ved samme T!
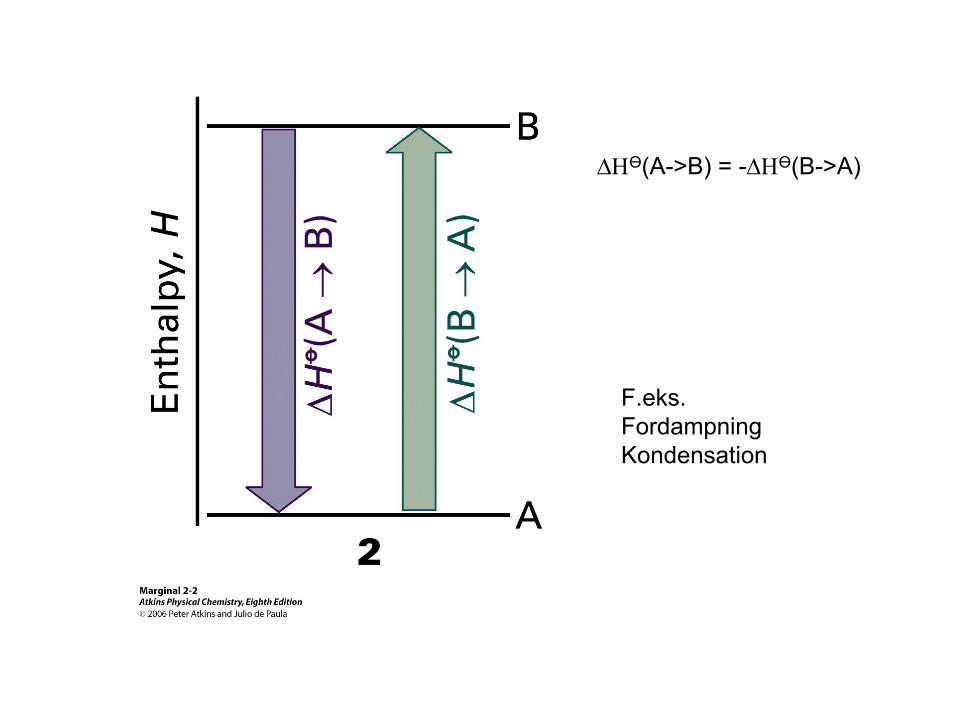
∆ΗӨ(A->B) = -∆ΗӨ(B->A)
F.eks.FordampningKondensation

Standard dannelses enthalpi
dU=dq+dw og dH=dU+d(pV)ikke noget nulpunkt, vi måler ændringer
Vi vælger et nulpunkt for energifunktionerne:
Enthalpien af et rent grundstof i detsnaturlige forekomst ved temperaturen T ogtrykket 1 bar sættes til 0
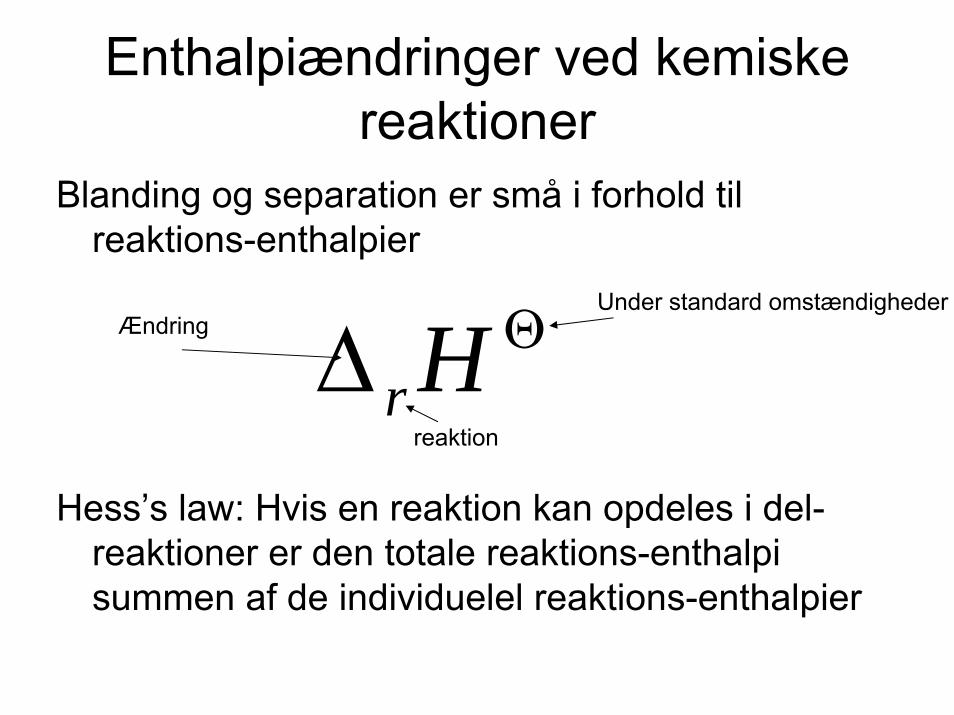
Enthalpiændringer ved kemiskereaktioner
Blanding og separation er små i forhold tilreaktions-enthalpier
Hess’s law: Hvis en reaktion kan opdeles i del-reaktioner er den totale reaktions-enthalpisummen af de individuelel reaktions-enthalpier
Θ∆ Hrreaktion
Under standard omstændighederÆndring

∆fHӨ, ∆rHӨ
aA+bB+… cC+dD+…
Eks: H2(g) + 0.5 O2(g) -> H2O(l)∆rHӨ=qp= ∆fHӨ(H2O(l)) = -285.83 kJ/molTABEL 2.5
...(B)Hb∆(A)Ha∆...(D)Hd∆(C)Hc∆H∆ θf
θf
θf
θf
θr −−−++=
==−=−= dcba dcba νννν
∑= θifi
θr H∆νH∆
FORTEGN!





![Intelligent udnyttelse af kulstof og energi på renseanlægmængder energi. Omkring 1,5 % af verdens udledning af drivhusgasser stammer fra spildevand [Andersen, 2008]. Udviklingsprojektet](https://img.dokumen.tips/doc/110x75/60a0dc6737e4d000767f57e5/intelligent-udnyttelse-af-kulstof-og-energi-p-renseanlg-mngder-energi-omkring.jpg)













![Lov om ændring af lov om fremme af vedvarende energi ~[…] …new.folketingsbilag.dk/files/1802459.pdf · 2017-10-13 · rende energi Dansk Energi takker for muligheden for at komme](https://img.dokumen.tips/doc/110x75/5e3ed718a0965d6ac92a27d2/lov-om-ndring-af-lov-om-fremme-af-vedvarende-energi-new-2017-10-13-rende.jpg)









