Embed Size (px)
Citation preview

Structure–Activity Relationship Studies of Sphingosine Kinase Inhibitors and Mitochondrial Uncouplers
Elizabeth S. Childress
Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
In Chemistry
Webster L. Santos, Committee Chair Paul R. Carlier
Richard D. Gandour David G. I. Kingston
June 20, 2017 Blacksburg, VA
Keywords: Structure–Activity Relationship, Sphingosine Kinase, Sphingosine 1-Phosphate, Mitochondrial Uncoupler, Protonophore, Oxygen Consumption Rate
Copyright 2017, Elizabeth S. Childress

Structure–Activity Relationship Studies of Sphingosine Kinase Inhibitors and Mitochondrial Uncouplers
Elizabeth S. Childress
Abstract
Sphingosine 1-phosphate (S1P) is a cellular signaling molecule that has been implicated
in a variety of diseases including cancer, fibrosis, Alzheimer’s, and sickle cell disease. It is
formed from the phosphorylation of sphingosine (Sph) by sphingosine kinase (SphK) and SphK
exists as two isoforms—SphK1 and SphK2, which differ with respect to their cellular activity
and localization. As the key mediators in the synthesis of S1P, SphKs have attracted attention as
viable targets for pharmaceutical inhibition. To validate their potential as therapeutic targets, we
aimed to develop potent, selective, and in vivo active inhibitors of SphK.
Herein, we describe the design, synthesis and biological evaluation of SphK2 inhibitors.
We first describe the development of six SphK2 inhibitors that assess the utility of replacing
lipophilic tail groups with heterocyclic rings. These six compounds demonstrate that the lipid
binding pocket for SphK2 cannot accommodate compounds with tail groups that are
conformationally restricted or positively charged. We then describe the development of
aminothiazole-based analogues of an SphK1-selective inhibitor. A library of 37 aryl-substituted
aminothiazole tail groups were synthesized, revealing a structure–activity relationship study that
examines electronic effects on the aryl-substituted aminothiazoles and the effect of modifying
the amino portion of the aminothiazole. These molecules show surprisingly good potency and
selectivity for SphK2. In particular, we highlight 3.20dd (SLC4101431), a biphenyl
aminothiazole that is the post potent and selective SphK2 inhibitor to date, with an SphK2 Ki
of 90 nM and 100-fold selectivity for SphK2. This molecule’s in vivo activity will also be
discussed.

iii
Mitochondrial uncouplers are small molecules that shuttle protons from the inter
membrane space to the mitochondrial matrix independent of ATP synthase, which disrupts
oxidative phosphorylation and promotes increased nutrient metabolism for homeostasis to be
maintained. Consequently, small molecule mitochondrial uncouplers have been pursued as
probes for mitochondrial function and as potential therapeutics for the treatment of obesity and
type 2 diabetes.
Herein, we describe the design, synthesis, and biological evaluation of small molecule
mitochondrial uncouplers. We report a library of 52 compounds that have good mitochondrial
uncoupling activity over a wide therapeutic range, including 5.16t (SHC4111522) and 5.17i
(SHC4091665), which have EC50 values of 0.63 µM and 1.53 µM, respectively, and achieve at
least 2-fold increase in oxygen consumption rates relative to basal levels. With these molecules,
we demonstrate that pKa and cLogP significantly contribute to uncoupling activity and must be
accounted for when developing new generation small molecule mitochondrial uncouplers.

iv
Structure–Activity Relationship Studies of Sphingosine Kinase Inhibitors and Mitochondrial Uncouplers
Elizabeth S. Childress
General Audience Abstract
Sphingosine kinase 1 and 2 (SphK1 and SphK2) are enzymes that facilitate the
production of the biomolecule sphingosine 1-phosphate (S1P), which plays an essential role in
cell growth and survival. However, overproduction of S1P has been linked to a number of
diseases including cancer, Alzheimer’s, and sickle cell disease. Therefore, because S1P is
involved in these diseases, the amount of available S1P must be controlled. This work describes
the design, development, and biological study of over 40 compounds that could be used as
potential inhibitors of SphK2 to help control S1P levels and, therefore, hopefully alleviate the
effects of disease. In particular, this work describes molecules that probe the SphK2 binding
pocket and demonstrates that the molecules cannot be rigid or positively charged when binding
to the hydrophobic portion of the SphK2 binding pocket. Additionally, this work describes the
most potent and selective reported SphK2 inhibitor to date, 3.20dd (SLC4101431).
Mitochondrial uncouplers are compounds that target our body's mitochondria and aim to
make ATP production challenging, causing the mitochondria to burn extra energy in the form of
glucose and fatty acids to allow normal levels of ATP to be produced. By making the
mitochondria burn extra energy, mitochondrial uncouplers have the potential to be treatments for
diseases such as obesity and diabetes. This works describes the design, development, and
biological study of over 50 mitochondrial uncouplers that are capable of increasing
mitochondrial activity over a wide concentration range, including 5.16t (SHC4111522) and 5.17i
(SHC4091665), which are very potent and effective uncouplers.

v
Dedication
For my grandma, Barbara Blanton, the truest Hokie I have ever known.
Acknowledgements
I would first like to thank my advisor Dr. Webster Santos, for all of his support, advice,
patience, and guidance. He has been such a wonderful role model and mentor throughout the
four-and-a-half years that I have worked for him. I clearly remember him coming to visit my
hood one day to discuss the progress of my research. I was having a difficult time purifying one
of my compounds. He chuckled and said, “You’ll figure it out.” He then walked away. His
response frustrated me at the time but that moment helped me develop my problemsolving skills
and become an independent scientist.
I would next like to thank my committee members Dr. Paul Carlier, Dr. Richard
Gandour, and Dr. David Kingston for their insights and constructive criticisms. You have helped
improve my critical thinking and communication skills and cultivated my scientific curiosity.
In addition, I would like to thank my collaborators Dr. Kevin Lynch and Yugesh Kharel
for conducting all of the biological studies for my sphingosine kinase inhibitors. I would also like
to thank my collaboratores Dr. Kyle Hoehn and Stefan Hargett for conducting all of the
biological studies for my mitochondrial uncouplers. I would also like to thank the Santos
group—Cheryl Peck, Russell Snead, Hao Li, Ashley Peralta, Yumin Dai, Jacob Murray, Eric
Medici, Chris Sibley, Russell Fritzemeier, Ashley Gates, Jose Santiago-Rivera, and Laura
Wonilowicz—for all of their support and for providing me with such a fun and wonderful
research environment. It has made the tedious and frustrating days bearable. I would like to
thank Karen Iannaccone for her patience and help, especially with scheduling meetings and
dealing with travel documentation. I would also like to thank Dr. Molly Congdon and Dr. Jessica

vi
Wynn for their guidance and friendship over the years. I am also grateful for Ashley Gates for
being the editor-in-chief for several of my dissertation’s chapters and for all of the walks to
Starbucks.
I would like to thank Christopher Presley for his unwavering love and support. I am so
thankful that graduate school brought you into my life. You are my number one. I would also
like to thank my family for their encouragement, faith, and unconditional love. They have always
believed in me, especially when I didn’t believe in myself. To my brother Michael, thank you for
pushing me to be the best version of myself and for always knowing how to make me laugh. To
my parents Floyd and Mary, thank you for your constant love and support and for all of the
sacrifices that you have made over the years to provide me with the opporutinites that I have
now. Everything that I am and have accomplished is because of you. Finally, I would like to
thank my grandparents Bob and Barbara Blanton for showing me how to live a life full of
kindness, grace, and generosity.

vii
Table of Contents
Dedication ....................................................................................................................................... v
Acknowledgements ......................................................................................................................... v
List of Figures…………………………………………………………………………………....xii
List of Tables……………………………………………………………………………………xiii
List of Schemes………………………………………………………………………………….xiv
1 The Chemical Biology of Sphingosine Kinase 2 ...................................................................... 1
1.1 Introduction ........................................................................................................................ 1
1.2 Sphingolipids ..................................................................................................................... 1
1.3 Sphingosine 1-Phosphate ................................................................................................... 2
1.4 Sphingosine Kinase ............................................................................................................ 5
1.5 Sphingosine Kinase 1 ......................................................................................................... 6
1.5.1 Localization and Regulation ....................................................................................... 6
1.5.2 Crystal Structure ......................................................................................................... 6
1.6 Sphingosine Kinase 2 ......................................................................................................... 7
1.6.1 Structure–Function Relationship ................................................................................ 7
1.6.2 Isoforms ...................................................................................................................... 8
1.6.3 Localization ................................................................................................................. 9
1.6.4 Localization–Function Relationship ......................................................................... 10
1.6.5 Regulation ................................................................................................................. 12
1.6.6 Activity in Disease .................................................................................................... 13
1.7 Sphingosine Kinase 2 Inhibitors ...................................................................................... 16
1.7.1 (R)-FTY720-OMe ..................................................................................................... 16

viii
1.7.2 SKI-II ........................................................................................................................ 17
1.7.3 ABC294640 .............................................................................................................. 17
1.7.4 VT-ME6 .................................................................................................................... 18
1.7.5 Amgen 82 .................................................................................................................. 18
1.7.6 K145 .......................................................................................................................... 19
1.7.7 SLR080811 ............................................................................................................... 19
1.7.8 SLP120701 ................................................................................................................ 20
1.7.9 SLM6031434 ............................................................................................................ 20
1.8 Conclusions ...................................................................................................................... 20
1.9 Dissertation Overview For Developing Inhibitors of Sphingosine Kinase 2 .................. 21
1.10 References ........................................................................................................................ 22
2 Structure-Activity Relationship Studies of the Lipophilic Tail Region of Sphingosine Kinase
2 Inhibitors .................................................................................................................................... 39
2.1 Contributions .................................................................................................................... 39
2.2 Abstract ............................................................................................................................ 39
2.3 Introduction ...................................................................................................................... 40
2.4 Results and Discussion .................................................................................................... 42
2.4.1 Inhibitor Development .............................................................................................. 42
2.4.2 Structure–Activity Relationship Studies and Biological Evaluation of Derivatives 45
2.5 Conclusions ...................................................................................................................... 48
2.6 Funding Sources ............................................................................................................... 49
2.7 References ........................................................................................................................ 50

ix
3 Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2 Selective
Inhibitors: Design, Synthesis, and in Vivo Activity ..................................................................... 53
3.1 Contributions .................................................................................................................... 53
3.2 Abstract ............................................................................................................................ 53
3.3 Introduction ...................................................................................................................... 54
3.4 Results and Discussion .................................................................................................... 57
3.4.1 Inhibitor Design and Development ........................................................................... 57
3.4.2 Inhibitor Synthesis .................................................................................................... 59
3.4.3 Structure–Activity Relationship Studies and Biological Evaluation of Derivatives 63
3.5 Conclusions ...................................................................................................................... 72
3.6 Funding Sources ............................................................................................................... 73
3.7 References ........................................................................................................................ 74
4 Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential ........................ 84
4.1 Contributions .................................................................................................................... 84
4.2 Introduction ...................................................................................................................... 84
4.3 Mitochondrial Uncoupling ............................................................................................... 85
4.4 Pharmacological Mimicry of Uncoupling Proteins ......................................................... 86
4.5 Uncouplers of Oxidative Phosphorylation ....................................................................... 87
4.5.1 DNP (2,4-Dinitrophenol) .......................................................................................... 90
4.5.2 FCCP (Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone) ........................... 92
4.5.3 Bupivacaine ............................................................................................................... 92
4.5.4 TTFB (4,5,6,7-Tetrachloro-2-trifluoromethylbenzimidazole) .................................. 93
4.5.5 C4R1 ......................................................................................................................... 94

x
4.5.6 SR4 ............................................................................................................................ 94
4.5.7 Ppc-1 ......................................................................................................................... 95
4.5.8 ES9 (Endosin 9) ........................................................................................................ 96
4.5.9 Niclosamide Ethanolamine ....................................................................................... 96
4.5.10 Ellipticine ................................................................................................................ 97
4.5.11 C12TPP .................................................................................................................... 97
4.5.12 S-13 ......................................................................................................................... 98
4.5.13 SF-6847/Tyrphostin A9/AG17 ............................................................................... 98
4.5.14 CZ5 ......................................................................................................................... 99
4.5.15 Cationic Triphenylphosphoniums ........................................................................... 99
4.5.16 BAM15 ................................................................................................................. 100
4.6 Conclusions .................................................................................................................... 101
4.7 Dissertation Overview for Developing Mitochondrial Uncouplers ............................... 101
4.8 References ...................................................................................................................... 103
5 Structure-Activity Relationship Studies of Uncouplers of Oxidative Phosphorylation ....... 127
5.1 Contributions .................................................................................................................. 127
5.2 Abstract .......................................................................................................................... 127
5.3 Introduction .................................................................................................................... 128
5.4 Results and Discussion .................................................................................................. 131
5.4.1 Inhibitor Development ............................................................................................ 131
5.4.2 Chemical Synthesis ................................................................................................. 131
5.4.3 Structure–Activity Relationship Studies and Biological Characterization of 5.13
Derivatives .......................................................................................................................... 133

xi
5.5 Current Efforts and Future Directions ........................................................................... 144
5.6 Conclusions .................................................................................................................... 144
5.7 References ...................................................................................................................... 145
6 Experimental Section ............................................................................................................ 154
6.1 Structure–Activity Relationship Studies of the Lipophilic Tail Region of Sphingosine
Kinase 2 Inhibitors .................................................................................................................. 154
6.1.1 Sphingosine Kinase Assays .................................................................................... 154
6.1.2 General Material and Synthetic Procedures ............................................................ 154
6.2 Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2
Selective Inhibitors: Design, Synthesis, and in Vivo Activity ................................................ 165
6.2.1 Sphingosine kinase assays ...................................................................................... 165
6.2.2 Sample Preparation and LC–MS–MS Analysis ...................................................... 165
6.2.3 Pharmacokinetic Analysis ....................................................................................... 166
6.2.4 Molecular Docking ................................................................................................. 167
6.2.5 General Material and Synthetic Procedures ............................................................ 167
6.3 Structure–Activity Relationship Studies of Uncouplers of Oxidative Phosphorylation 228
6.3.1 Oxygen Consumption Rate Seahorse Assay ........................................................... 228
6.3.2 pKa Determination .................................................................................................. 229
6.3.3 General Material and Synthetic Procedures ............................................................ 236
6.3.4 Oxygen Consumption Rate Data ............................................................................ 255
6.3.5 NMR Spectra .......................................................................................................... 259
6.4 References ...................................................................................................................... 327

xii
List of Figures
Figure 1.1. Cer–S1P rheostat .......................................................................................................... 2
Figure 1.2. S1P metabolism ............................................................................................................ 3
Figure 1.3. Intracellular and extracellular S1P signaling pathways. ............................................... 4
Figure 1.4. SphK1 and SphK2 sequence composition. ................................................................... 5
Figure 1.5. Crystal structure of SphK1 with Sph docked in the J-shaped binding pocket.
Reprinted with permission from reference 54. Copyright 2017 Elsevier. .............................. 7
Figure 1.6. SphK1-selective inhibitor 1.1 ....................................................................................... 7
Figure 1.7. Location of SphK2 localization sequences and binding domains. ............................... 8
Figure 1.8. SphK2 isoform distribution in human tissue. Reused with permission from reference
64. Copyright 2017 American Society for Biochemistry and Molecular Biology. ................ 9
Figure 1.9. SphK2 localization and activity. Reused (adapted) with permission from reference
46. Copyright 1999-2017 John Wiley & Sons. ..................................................................... 12
Figure 1.10. Structures of Select SphK2 inhibitors ...................................................................... 16
Figure 2.1. Structure of SphK2 Inhibitors .................................................................................... 41
Figure 2.2. Pharmacophore of guanidine-based inhibitors………………………………………42 Figure 3.1. Select SphK inhibitors ................................................................................................ 56
Figure 3.2. Tail group modifications toward the development of new "J-shaped" SphK1
inhibitors ............................................................................................................................... 57
Figure 3.3 Superimposition of the lowest energy docked poses of 3.2 and an aminothiazole
analogue of 3.3 in a homology model of SphK2………………………………………………...58
Figure 3.4. Effect of 3.20l and 3.20dd on sphingolipids in U937 cells………………………….70
Figure 3.5. Detected S1P blood levels in mice following injection with 3.20dd………………..71

xiii
Figure 4.1. Oxidative Phosphorylation and Mitochondrial Uncoupling ....................................... 86
Figure 5.1. Select Mitochondrial Uncouplers ............................................................................. 129
Figure 5.2. Pharmacophore of 5.13 ............................................................................................. 131
Figure 5.3. Changes in oxygen consumption rate relative to DMSO control for 5.5, 5.13, 5.16h,
5.16t, 5.17h, and 5.17i ........................................................................................................ 143
List of Tables
Table 1.1. Regulators of SphK2 .................................................................................................... 13
Table 2.1. Inhibitory Effects of Derivatives of 2.4 and 2.5 on SphK1 and SphK2 ...................... 47
Table 3.1. SphK1 and SphK2 Inhibitory Activity for Monosubstituted Aminothiazole Derivatives
............................................................................................................................................... 64
Table 3.2. SphK1 and SphK2 Inhibitory Activity for Disubstituted and Bulky Aminothiazole
Derivatives……………………………………………………………………………………….66
Table 3.3. Ki and cLogP Values of Selected Inhibitors of SphK1 and SphK2…………………..69
Table 4.1. Reported Mitochondrial Uncouplers ........................................................................... 88
Table 5.1. Activity of Symmetrical Derivatives of 5.13 ............................................................ 134
Table 5.2. Activity of Unsymmetrical Derivatives of 5.13 ......................................................... 137
Table 5.3. Activity of Amide Derivatives of 5.13 ...................................................................... 139
Table 5.4. pKa and cLogP Values of Select Uncouplers ............................................................. 141
Table 5.5. EC50 Values of Select Uncouplers with Activity at 40 µM ...................................... 143

xiv
List of Schemes
Scheme 2.1. Synthesis of derivatives of 2.4……………………………………………………..43
Scheme 2.2. Synthesis of derivatives of 2.5……………………………………………………..43
Scheme 2.3. Synthesis of amidopiperazine tail derivatives 2.16a–d…………………………….44
Scheme 2.4. Synthesis of 2.20…………………………………………………………………...44
Scheme 2.5. Synthesis of 2.23…………………………………………………………………...45
Scheme 2.6. Synthesis of derivatives of 2.4 lacking an internal phenyl ring……………………45
Scheme 3.1. Synthesis of substituted aminothiazole derivatives 3.20a–ee………………...…....60
Scheme 3.2. Synthesis of biphenyl aminothiazole derivatives 3.23a–d…………………………60
Scheme 3.3. Synthesis of the oxathiazole analogue of inhibitor 3.20k………………………….62
Scheme 3.4. Synthesis of the methylated analogue of 3.20dd…………………………………..62
Scheme 5.1. Synthesis of Symmetrical and Unsymmetrical Derivatives of 5.13 ....................... 132
Scheme 5.2. Synthesis of Amide Derivatives of 5.13 ................................................................. 132

1
1 The Chemical Biology of Sphingosine Kinase 2
1.1 Introduction
A key regulator in the production of the sphingolipid sphingosine 1-phosphate (S1P) is
sphingosine kinase (SphK). This enzyme exists as two isoforms in humans: sphingosine kinase 1
(SphK1) and sphingosine kinase 2 (SphK2). A multitude of studies have demonstrated SphK1’s
role in cell proliferation, growth, and survival pathways.1, 2 Less is known about SphK2’s role in
cellular processes, while much of the literature indicates that SphK2 is involved in pro-apoptotic
pathways.1, 2 Inhibition or even knockdown of SphK2 in disease-state models has demonstrated
that SphK2 may also be proliferative.1, 2 These contradictory roles make SphK2 an attractive
enzyme for study. This review will focus on the known chemical biology of SphK2, its role in
disease, as well as selective inhibitors for this enzyme.
1.2 Sphingolipids
Sphingolipids are biomolecules associated with the cell membrane that participate in a
variety of cellular processes including lipid bilayer regulation, cellular growth and migration, as
well as angiogenesis and programmed cell death (apoptosis).3-5 Common examples of
sphingolipids include ceramide (Cer), sphingosine (Sph), and S1P. These three sphingolipids
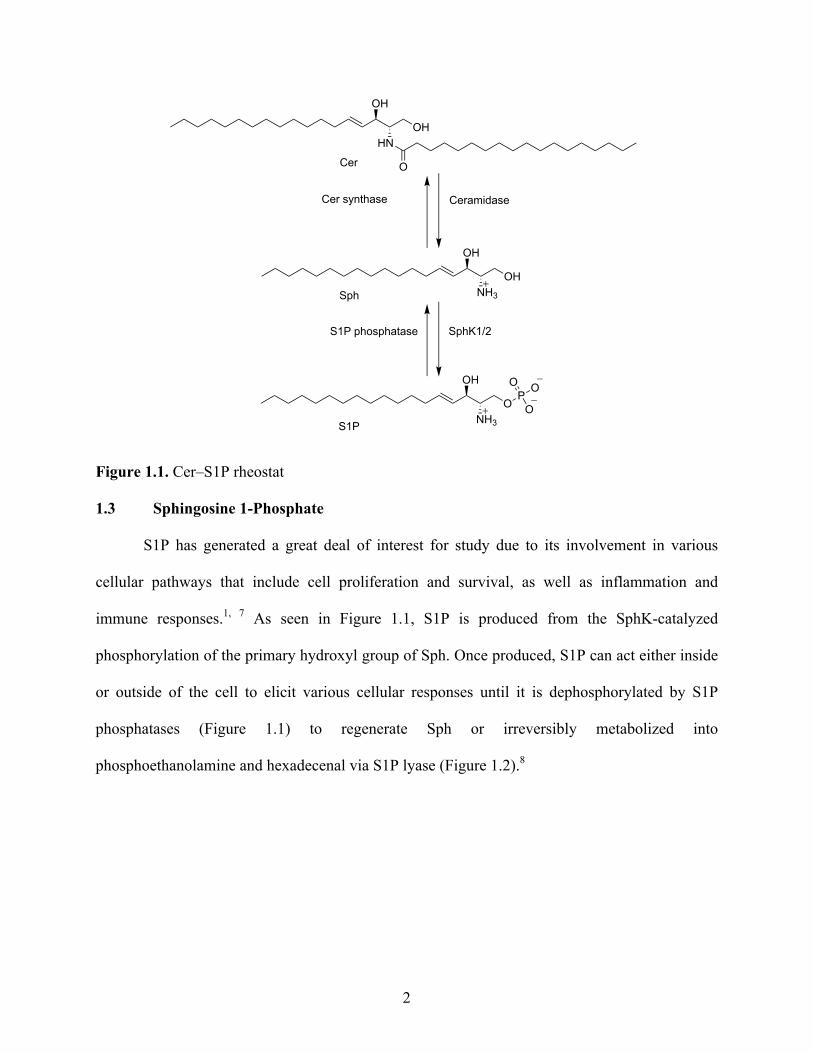
belong to a proposed reversible rheostat (Figure 1.1) that is finely controlled by a set of enzymes
that determine cell proliferation or apoptosis.6 Synthesis of Cer and Sph has been implicated in
pro-apoptotic pathways, while the production of S1P leads to pro-survival and growth pathways.
2
Figure 1.1. Cer–S1P rheostat
1.3 Sphingosine 1-Phosphate
S1P has generated a great deal of interest for study due to its involvement in various
cellular pathways that include cell proliferation and survival, as well as inflammation and
immune responses.1, 7 As seen in Figure 1.1, S1P is produced from the SphK-catalyzed
phosphorylation of the primary hydroxyl group of Sph. Once produced, S1P can act either inside
or outside of the cell to elicit various cellular responses until it is dephosphorylated by S1P
phosphatases (Figure 1.1) to regenerate Sph or irreversibly metabolized into
phosphoethanolamine and hexadecenal via S1P lyase (Figure 1.2).8
OH
OH
HN
Ceramidase
OH
OH
NH3
O
OH
NH3
PO
OO
Cer
Sph
O
S1P phosphatase SphK1/2
S1P
Cer synthase

3
Figure 1.2. S1P metabolism
S1P acts as an extracellular ligand for its G-protein coupled receptors, S1PR1–5, once
transported outside of the cell by the spinster 2 transporter.9-12 The binding of S1P to any one of
its receptors elicits a series of second messenger signals that eventually activate a variety of
targets involved in pro-survival processes, including the proteins Rho, Rac, Ras, and JNK along
with the MAPK/ERK pathway (Figure 1.3).3, 8 More recently, S1P receptor signaling has been
implicated in insulin signaling,13-15 energy homeostasis,16, 17 and as promoter of chromosome
segregation and spindle checkpoint relaxation during mitosis.18
Less is known about the intracellular signaling pathways of S1P (Figure 1.3), though S1P
is known to bind to and inhibit histone deacetylases 1 and 2 (HDAC 1/2), promoting epigenetic
modifications.19, 20 S1P also stimulates Ca2+ release from the endoplasmic reticulum (ER),
triggering pro-apoptotic pathways.2, 21 In addition, S1P can bind to the mitochondrial protein
prohibitin 2 to regulate cellular respiration, and it can also bind to the tumor necrosis factor
TRAF2, which stimulates E3 ubiquitin ligase activity, promoting proliferative and pro-
inflammatory pathways.22, 23 More recently, intracellular S1P was shown to increase erythrocyte
glycolysis and O2 release under hypoxic conditions.24
O
OH
NH3
PO
OO
S1P lyase
O
OP
O
OOH3N
HHexadecenal Phosphoethanolamine
S1P

4
Figure 1.3. Intracellular and extracellular S1P signaling pathways.
Due to its role in a variety of inflammatory and proliferative processes, S1P plays a key
role in a variety of diseases, most notably cancer, fibrosis, Alzheimer’s disease, and sickle cell
disease.8, 25-38 High levels of S1P have been reported in cancer cell lines, mouse models, and
cancer patient samples, implicating S1P as a potential mediator for cancer.1, 8, 27, 29 The role of
S1P in fibrosis is more complex than its role in cancer. Studies have indicated that S1P is a
“Janus-faced” participant in fibrosis, having contradictory roles depending on whether it is acting
as an intracellular or extracellular ligand.3, 37, 38 Intracellular S1P has been implicated as a
promoter of anti-fibrotic processes whereas extracellular S1P has been associated with pro-
fibrotic pathways.3, 37-39 S1P has also been shown to regulate the transmembrane protein BACE-
1, which is involved in the production of amyloid-ß peptide, a senile plaque commonly
associated with Alzheimer’s disease,25 and decreased S1P production has been implicated in the
progression of Alzheimer’s disease.35, 36 Furthermore, elevated S1P levels have been linked to
sickle cell disease.32, 34 Interestingly, these disease states were commonly reliant upon the
expression levels and activity of the S1P-producing enzymes SphK1 and SphK2.1, 3, 8, 27, 32, 34, 35
Therefore, controlling S1P synthesis via SphK inhibition may be crucial for the treatment of
these respective diseases.
S1P
S1P
S1PR1-5
Sph
HDAC 1/2,Prohibitin 2,
TRAF2
extracellular space
Rho, Rac, MAPK/ERK,
JNKSphK1/2
transporter
intracellular space

5
1.4 Sphingosine Kinase
The levels of pro-apoptotic Sph and pro-survival S1P are tightly regulated in the
proposed Cer-S1P rheostat. Two key regulators of this rheostat are the highly conserved enzymes
SphK1 and SphK2. These two isoforms have an approximate 47% amino acid sequence identity
and 83% amino acid sequence similarity.40 Additionally, both contain five conserved domains
(C1–C5) in their sequences, including a catalytic domain in the C1–C3 regions that contains the
ATP binding site (Figure 1.4).41 Although SphK1 and SphK2 share amino-acid-sequence identity
and similarity, they differ in their sequence makeup. SphK1 is shorter in sequence, comprising
384 amino acid residues compared to 618 for SphK2.41 A proline-rich domain and four
transmembrane domains contribute to the longer sequence length in SphK2.41
Figure 1.4. SphK1 and SphK2 sequence composition.
Enzymatic activity studies found that SphK1 is substrate specific for D-erythro-sphingosine,
the native ligand form of Sph.40 On the other hand, these same studies found that SphK2 is less
substrate-specific than SphK1. D-erythro-sphingosine is not the only substrate for SphK2; D-
erythro-dihydrosphingosine, D,L-threo-dihydrosphingosine, and phytosphingosine can also be
phosphorylated by SphK2.40 Not only do SphK1 and SphK2 differ in their substrate affinities,
they also differ in their binding affinity for Sph (Km). SphK1 has a Km of 10 µM for Sph, while
SphK2 has a Km of 5 µM for Sph.40
Knockout experiments in mice have illustrated the relevance of both SphK isoforms despite
both sharing the same substrate and function. Double knockout of SphK1 and SphK2 in mice

6
was found to be embryonically lethal, as this caused vascular and neuronal developmental issues;
however, single knockout of either SphK1 or SphK2 in mice was not physiologically
detrimental.42, 43 These results demonstrate that normal cellular function is maintained as long as
one isoform is functional.
1.5 Sphingosine Kinase 1
1.5.1 Localization and Regulation
SphK1 is a cytosolic enzyme that promotes cell proliferation and survival.1, 2, 44, 45 Post-
translational modification studies of SphK1 have shown that phosphorylation by the kinases
ERK 1/2 causes SphK1 to transfer from the cytosol to the plasma membrane.46, 47 This relocation
allows SphK1 to synthesize S1P that can be transported out of the cell and bind S1PRs.48 In
addition, phosphorylation of SphK1 by ERK 1/2 causes its activity to increase to 10 times higher
than normal.46, 47 The kinases Lyn and Fyn, epidermal growth factor (EGF), and eukaryotic
elongation factor 1A (eEF1A) are also known to elevate SphK1 activity.46, 49-53 Upregulation of
SphK1 activity, though, has been linked to numerous diseases such as cancer, fibrosis, and sickle
cell disease.1-3, 29, 32, 34
1.5.2 Crystal Structure
In 2013, Wang et al. published the first crystal structure of human SphK1 as an apoprotein
and as a haloprotein (PDB ID 3VZB and 3VZC, respectively).54 The crystal structure of SphK1
bound to Sph revealed that the binding pocket is J-shaped (Figure 1.5) and that SphK1 substrates
likely enter the binding pocket tail-first through a “tunneling mechanism.”54 The crystal structure
also revealed how ATP binds to SphK1, allowing molecular modeling of SphK1 catalytic
activity. The proposed model suggests that SphK1’s aspartic acid residue 81 activates Sph by
deprotonating the Sph primary hydroxyl group to allow nucleophilic attack on the γ-phosphate

7
group of ATP, forming S1P.54 Scientists at the pharmaceutical company Pfizer corroborated this
report by publishing a crystal structure (PDB ID 4V24) of SphK1 co-crystallized with the
SphK1-selective inhibitor 1.1 (PF-543 (Figure 1.6)).55 Information gathered from these crystal
structures has helped guide the development of SphK inhibitors.56-58
Figure 1.5. Crystal structure of SphK1 with Sph docked in the J-shaped binding pocket. Reprinted with permission from reference 54. Copyright 2017 Elsevier.
Figure 1.6. SphK1-selective inhibitor 1.1
1.6 Sphingosine Kinase 2
1.6.1 Structure–Function Relationship
Despite sharing five conserved domains, SphK2 deviates from SphK1 with respect to its
structural makeup. Several domains and amino acid sequences (Figure 1.7) contribute to the
SphK2 sequence length, which helps dictate its cellular function. The proline-rich region of the
SphK2 sequence binds the cytokine receptor IL-12Rβ1, activating IFN-γ to promote immune
responses.59 Another component of the SphK2 sequence is a Bcl-2 homology 3 (BH3)-like
domain, which is found in the N-terminus of the enzyme.60 Within this domain, SphK2 interacts
Overall StructureThe overall structure of SphK1 (residues 9–364) adopts a two-domain architecture that comprises nine a helices, 17 b strands,and a 310-helix (Figure 1). The N-terminal domain (NTD) (residues9–150 and 357–364, containing the C1–C3 domains) adopts ana/b fold and comprises six a helices (a1-a6) and six b strands(b1–b5 and b17), resembling the dinucleotide binding motif of aRossmann fold of a three-layer a/b/a sandwich. The core ofthe NTD is a twisted parallel four-stranded b sheet (strand order,b2, b1, b3, b4), which is extended by a substructure of antipar-allel strands b17 and b5, with strand b17 running in part parallelto strand b4. The central b sheet is flanked by three a helices oneach side: a2, a3, and a4 on one side and a1, a5, and a6 on theother side. Note that the C-terminal strand b17 forms the 6th bstrand of the NTD, a feature of the swapping of secondary-struc-
Figure 2. Structural Comparison of SphK1 with DGKand NAD Kinase(A) Superposition of human SphK1 (cyan) and DGK from
Staphylococcus aureus (DgkB) (gray, Protein data Bank
[PDB] code 2QVL).
(B) Superposition of human SphK1 and an NAD kinase from
Archaeoglobus fulgidus (magenta, PDB code 1ZOZ).
Figure 3. Lipid Substrate Binding in SphK1(A) Binding mode of sphingosine in the CTD of SphK1. The bound sphingosine lipid is shown in stick representation with an atomic color scheme of red, blue, and
purple for oxygen, nitrogen, and carbon atoms, respectively. SphK1 is colored as in Figure 1. A 2Fo-Fc electron density map for the lipid molecule is shown in teal
mesh and contoured at 1s.
(B) Detailed lipid-protein interactions between sphingosine and SphK1. Side chains of SphK1 within a distance of 5 A from the lipid are shown in thin stick
representation. Hydrogen-bond interactions are denoted by magenta dashed lines. A water molecule is shown as a red sphere.
ture elements between the two domains. TheC-terminal domain (CTD) (residues 151–356, con-taining the C4 and C5 domains) comprises 11 bstrands and four helices. The 11 b strands arearranged into a two-layer antiparallel b sandwich,with the ‘‘back’’ sheet comprising the antiparallelb strands b7, b6, b16, b13, and b9 and the ‘‘front’’sheet containing b15, b14, b8, b10, b11, and b12.Three a helices, a7–a9, and a 310 helix followinghelix a9, pack onto the front sheet of the sand-
wich, whereas a long coiled loop (loop b9–b10, residues 214–256) covers the back sheet.Protein sequence motif analysis and structure classification
suggested that SphKs belong to the phosphofructokinase(PFK)-like superfamily (Cheek et al., 2005; Labesse et al.,2002), sharing the same protein fold with NAD kinases, DGKs,and ceramide kinases, as well as PFKs that are more distantlyrelated to it. Indeed, the SphK1 structure bears no similarity toprotein kinases or other lipid kinases, such as PI3K. The overallfold of SphK1 manifests a two-domain architecture similar tothose of NAD kinases (Garavaglia et al., 2004; Liu et al., 2005;Mori et al., 2005; Poncet-Montange et al., 2007) and DGKs(Bakali et al., 2007; Miller et al., 2008; Nichols et al., 2007),even though SphK1 shares with them a very low overallsequence identity of 10%–20% over !250 residues (Figure 2).
Structure
Molecular Basis of Sphingosine Kinase 1 Regulation
Structure 21, 798–809, May 7, 2013 ª2013 Elsevier Ltd All rights reserved 801S O N
HO
O O
1.1 (PF-543)SphK1 Ki = 3.6 nM

8
with the pro-survival mitochondrial transmembrane molecule Bcl-xL; this interaction promotes
the release of pro-apoptotic factors BAK and BAX.60, 61
In addition to a BH3 domain, SphK2 also contains a nuclear localization sequence (NLS)
in the N-terminus of the enzyme. Studies demonstrated that SphK2 could trigger cell cycle arrest
by preventing the cell from entering the S1 phase of interphase in the cell cycle.20 The NLS in
SphK2 was found to be directly linked to this event as SphK2s lacking an NLS failed to prevent
cells from entering the S1 phase.20
While SphK2 contains an NLS to allow its localization to the nucleus, it also contains at
least one nuclear export sequence (NES) to allow its transport out of the nucleus.62 The identified
NES is located between residues 416 and 425 in the proline-rich domain.62 Phosphorylation of
the serine residues 419 and 421 in the NES causes SphK2 to enter the cytoplasm,62 afterwhich
SphK2 can catalyze the formation of S1P for receptor signaling.
The SphK2 sequence also contains a lipid-binding domain. Although its exact location
and amino acid makeup is unknown, studies determined that the lipid-binding domain is located
within the first 172 amino acid residues of the SphK2 sequence.63 Studies have implicated this
domain in SphK2 localization, as SphK2 was reported to bind sulfatide, a component of cell
membranes, and SphK2 lacking the first 172 residues failed to translocate from the cytosol.63
Figure 1.7. Location of SphK2 localization sequences and binding domains.
1.6.2 Isoforms
In humans, SphK2 exists as two isoforms—SphK2-S,40 consisting of 618 residues, and
SphK2-L, consisting of 654 residues.40, 64 The amino acid sequencing of SphK2-L is the same as
9
Other Procedures—Bromodeoxyuridine (BrdUrd) incorporation wasmeasured as described previously (12).
RESULTS
mRNA for SPHK2-L Is the Predominant Form in Various HumanTissues and Cells—BLAST searches of the human genome sequencedata base revealed the existence of SPHK2-L. The SPHK2-L gene is!10kb long and consists of 6 exons separated by 5 introns (Fig. 1A). Thededuced amino acid sequence of this cDNA contained 654 residues anda calculated molecular weight of 69,213. SPHK2-S, originally reportedby Liu et al. (9), lacks the exon 1, and the first initiation codon fortranslation was within the exon 2 (Fig. 1B). It is reasonable to assumethat SPHK2-S is generated by means of alternative splicing from the
same gene that encodes SPHK2-L. It should be noted that prior to theinitiating codon in mouse exon 2 there is a stop codon (Fig. 1B, doubleunderlined TGA), strongly suggesting that no SPHK2-L exists inmouse.
We quantified the amount of each splice variant in various humancell lines by real-time quantitative PCR using two pairs of primer sets(Fig. 2A). In primer set 1 the forward primer was located within exon 1and the reverse primer on exon 2 so that the specific mRNA encodingSPHK2-L could be detected, whereas in primer set 2 the forward primerwas located within exon 2 and the reverse primer on exon 3, whichenabled us to recognize mRNA encoding both SPHK2-L and SPHK2-S.Assuming that the molar ratio of SPHK2-L to SPHK2-S mRNA is 1:1,the ratio of the amount of PCR products obtained with these primerpairs is anticipated to be 1:2. Surprisingly, in various human tissues and
FIGURE 2. SPHK2-L and SPHK2-S mRNA expression levels determined by real-time quantitative reverse transcription-PCR in various human tissues and cell lines. A, forspecific detection of SPHK2-L, primers (Pair 1) overlapping the exon1/exon2 boundary that encodes the N-terminal-extended part of SPHK2-L were used. For detection of bothSPHK2-L and SPHK2-S, primers (Pair 2) overlapping the exon2/exon3 boundary were used. B and C, reverse transcription was performed on total RNA isolated from various human celllines (B) and tissues (C). The cDNAs were used for real-time PCR with primers specific for SPHK2-L (Pair 1), SPHK2-L plus SPHK2-S (Pair 2), and for the housekeeping gene, GAPDH. Valuesof mRNA amounts were normalized to GAPDH expression and expressed relative to SPHK2-S expression and are the mean " S.E. from three independent experiments done six times.
N-terminal-extended Form of SPHK2
36320 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 43 • OCTOBER 28, 2005 at VIVA, Virginia Tech on August 28, 2013http://www.jbc.org/Downloaded from
that for SphK2-S, which includes the BH3 domain, lipid-binding domain, NLS sequence, and
NES sequence; the exact role of the extra 36 N-terminal amino acid residues in SphK2-L
remains to be determined.
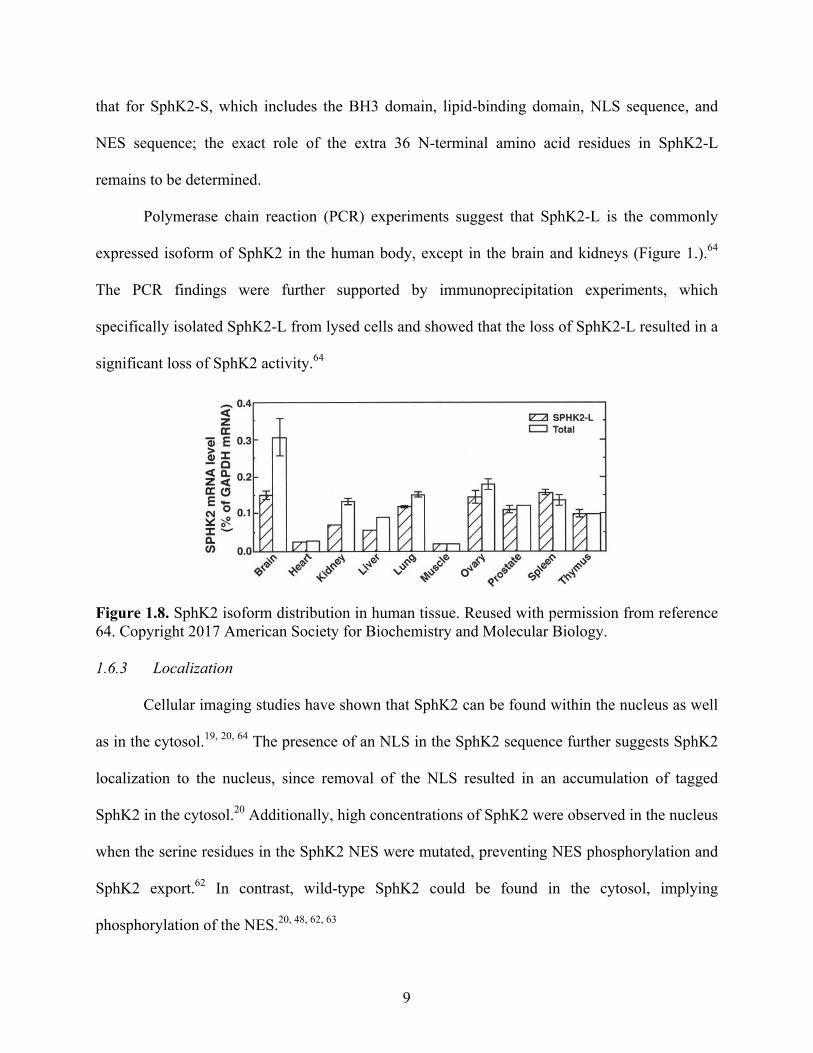
Polymerase chain reaction (PCR) experiments suggest that SphK2-L is the commonly
expressed isoform of SphK2 in the human body, except in the brain and kidneys (Figure 1.).64
The PCR findings were further supported by immunoprecipitation experiments, which
specifically isolated SphK2-L from lysed cells and showed that the loss of SphK2-L resulted in a
significant loss of SphK2 activity.64
Figure 1.8. SphK2 isoform distribution in human tissue. Reused with permission from reference 64. Copyright 2017 American Society for Biochemistry and Molecular Biology. 1.6.3 Localization
Cellular imaging studies have shown that SphK2 can be found within the nucleus as well
as in the cytosol.19, 20, 64 The presence of an NLS in the SphK2 sequence further suggests SphK2
localization to the nucleus, since removal of the NLS resulted in an accumulation of tagged
SphK2 in the cytosol.20 Additionally, high concentrations of SphK2 were observed in the nucleus
when the serine residues in the SphK2 NES were mutated, preventing NES phosphorylation and
SphK2 export.62 In contrast, wild-type SphK2 could be found in the cytosol, implying
phosphorylation of the NES.20, 48, 62, 63

10
Besides acting as a nucleic and cytosolic enzyme, SphK2 has also been associated with
the plasma membrane, ER, mitochondria, and extracellular space. Expression of SphK2 at the
plasma membrane has been demonstrated at basal conditions, synthesizing S1P that can be
exported out of the cell to engage S1PRs.65 Yet under cellular stress, SphK2 levels at the plasma
membrane were much lower, preventing activation of S1PR-mediated processes.65
SphK2 activity in the mitochondria, specifically the inner mitrochondrial membrane, has
been documented.22, 66 Direct interaction between the pro-apoptotic mitochondrial protein Bcl-xL
and the SphK2 BH3 domain further supports this localization.60 How SphK2 localizes to the
mitochondria from the nucleus or cytoplasm remains unclear, though cellular stress is known to
not be a trigger.65 However, cellular stress does promote ER localization.65
SphK2 localization to the extracellular space has been observed.67 Studies found that
during apoptosis, SphK2 localized to the extracellular space following the caspase-1-promoted
removal of the SphK2 N-terminus.67
1.6.4 Localization–Function Relationship
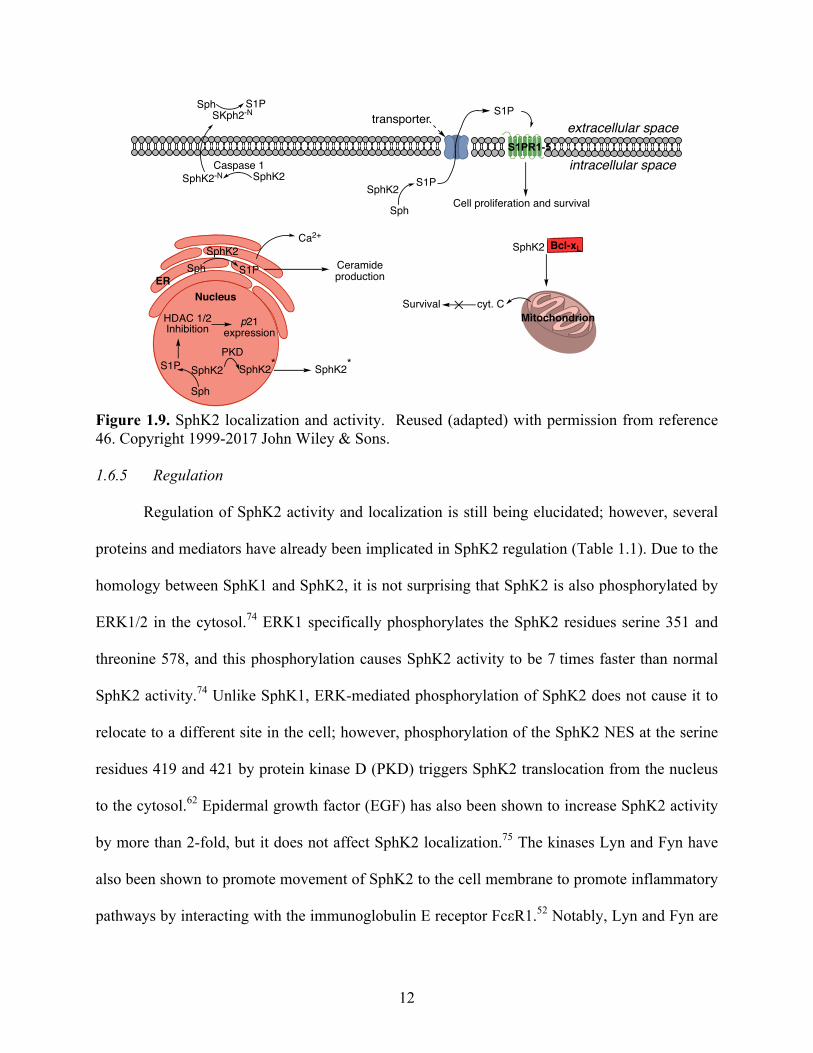
SphK2’s role within the cell is influenced by its localization (Figure 1.9). As stated
earlier, nuclear SphK2 can prevent the cell from entering the S1 phase of the cell cycle, which
then prevents DNA synthesis.20 Furthermore, S1P synthesized by nuclear SphK2 can prevent
HDAC 1/2 activity, promoting gene transcription and expression of p21, a protein associated
with cell cycle arrest.19, 68 A pair of studies, though, have reported that nuclear SphK2 can be
proliferative. S1P synthesized by overexpressed nuclear SphK2 prevented the anti-proliferative
activities of retinoic acid receptor beta (RARβ).69 S1P synthesized by nuclear SphK2 has been
shown to stabilize the catalytic portion of telomerase, thereby promoting telomerase stability and
cell proliferation.70

11
Mitochondrial SphK2 binds Bcl-xL, inducing the apoptotic BAK/BAX-mediated
emission of cytochrome c from mitochondria.60, 61 SphK2 in the ER can promote Ca2+ release
during cellular stress, and the Ca2+ activates pro-apoptotic mediators such as cytochrome C.21, 65
In addition, SphK2 in the ER can also increase intracellular levels of pro-apoptotic Cer.65
Extracellular SphK2 is proposed to have anti-inflammatory functions because S1P synthesized
extracellularly promoted lymphocyte localization to damaged cells.67
Multiple studies have reported elevated blood S1P levels in SphK2-null mice or mice
treated with SphK2-selective inhibitors, a phenomenon not observed with SphK1-null mice or
SphK1-selective inhibitors.42, 65, 71-73 Recent studies have elucidated these observations by
implicating SphK2 in the clearance of blood S1P. In particular, mass-labeled exogenous S1P was
observed to have slower clearance rates in SphK2-null mice and mice treated with SpK2-
selective inhibitors, suggesting SphK2 functions not only as a generator of S1P but also as aids
in the removal of blood S1P.72 The exact mechanism for how SphK2 contributes to the turnover
of blood S1P is unclear, and the localization of SphK2 during this process is currently unknown.
12
Figure 1.9. SphK2 localization and activity. Reused (adapted) with permission from reference 46. Copyright 1999-2017 John Wiley & Sons. 1.6.5 Regulation
Regulation of SphK2 activity and localization is still being elucidated; however, several
proteins and mediators have already been implicated in SphK2 regulation (Table 1.1). Due to the
homology between SphK1 and SphK2, it is not surprising that SphK2 is also phosphorylated by
ERK1/2 in the cytosol.74 ERK1 specifically phosphorylates the SphK2 residues serine 351 and
threonine 578, and this phosphorylation causes SphK2 activity to be 7 times faster than normal
SphK2 activity.74 Unlike SphK1, ERK-mediated phosphorylation of SphK2 does not cause it to
relocate to a different site in the cell; however, phosphorylation of the SphK2 NES at the serine
residues 419 and 421 by protein kinase D (PKD) triggers SphK2 translocation from the nucleus
to the cytosol.62 Epidermal growth factor (EGF) has also been shown to increase SphK2 activity
by more than 2-fold, but it does not affect SphK2 localization.75 The kinases Lyn and Fyn have
also been shown to promote movement of SphK2 to the cell membrane to promote inflammatory
pathways by interacting with the immunoglobulin E receptor FcεR1.52 Notably, Lyn and Fyn are
SphSphK2
S1P Ceramideproduction
Ca2+
Sph
SphK2S1P
HDAC 1/2Inhibition p21
expression
SphK2* SphK2*PKD
cyt. CSurvival
SphK2 Bcl-xL
Mitochondrion
NucleusER
Sph
S1P
S1P
S1PR1-5
SphK2-N SphK2Caspase 1
SKph2-NSph S1P
Cell proliferation and survival
transporter extracellular space
SphK2
intracellular space

13
unable to increase SphK2 activity.52 SphK2 activity, though, increases by almost 3-fold when
interacting with eEF1A.51 Although it is still unknown how eEF1A promotes SphK2 activity,
eEF1A is hypothesized to make the Sph/ATP-SphK2 binding interactions more efficient.51
Besides proteins and mediator molecules, cellular stress can also regulate SphK2 activity, as
hypoxia was found to increase SphK2 activity by almost 2-fold during hypoxia, resulting in an
increase in extracellular S1P.76
Regulator Activity
ERK1/2 Phosphorylates SphK2; increases SphK2 activity by 7-
fold. PKD Phosphorylates the SphK2
NES EFG Increases SphK2 activity by >
2-fold Lyn and Fyn Promotes SphK2 localization
to the cell membrane; promotes SphK2 interaction
with FcεR1 eEF1A Increases SphK2 activity by
3-fold Hypoxia Increases SphK2 activity by
2-fold Table 1.1. Regulators of SphK2
1.6.6 Activity in Disease
Reports on SphK2 activity in disease have been conflicting. In some cases SphK2 seems
to lessen or even prevent disease while in other cases SphK2 has shown to promote the
progression of disease. One such disease is cancer. Due to reports implicating SphK2 in pro-
apoptotic activity, it would seem that expression and even up-regulation of SphK2 would help
alleviate the progression of cancer. However, several studies2, 77-84 have reported that this may
not be the case. Overexpression of SphK2 has been observed in numerous cancer cell lines,

14
including non-small cell lung carcinoma, multiple myeloma, breast cancer, melanoma, and
leukemia.82, 85-87 Interestingly, in cases of SphK2 overexpression in cancer cell lines, SphK2 was
found to be highly localized to the cytosol and plasma membrane, a localization associated with
cell proliferation.82 Complete knockdown or down-regulation of SphK2 in kidney, breast, and
cerebral cell lines suppressed cancer cell proliferation and migration.80 The extent to which this
suppression occurred was greater than that observed for SphK1.2, 80 Breast cancer xenograft
mouse models have also demonstrated that knockdown of SphK2 can suppress the progression of
cancer by eliciting pro-inflammatory and anti-tumor activity.81 Inhibition of SphK2 in mice
models and cancer cell lines by SphK2-specific inhibitors have also shown anti-tumor activity.2,
78, 79 Additionally, breast cancer cell lines became susceptible to chemotherapy treatment with
the synergistic administration of chemotherapy and an SphK2-selective inhibitor.77 Inhibition of
SphK1 occurred concomitantly with SphK2 in this study, suggesting tumor cell chemoresistance
might not be solely SphK2-derived. Despite a growing body of evidence implicating SphK2 (as
well as SphK1) involvement in cancer progression, one study found that inhibition of SphK2
(and SphK1) activity does not cause tumor cell death, suggesting that pharmacologically
targeting SphK2 may not be the most feasible route for cancer treatment.88 However, more
studies are needed to corroborate this report.
SphK2 activity in inflammatory diseases and responses has also been documented.1, 79, 89-
92 Inhibition of SphK2 was shown to reduce the extent of colitis and arthritis in mice models.79,
89, 93 Inhibition of SphK2 has also been found to lessen inflammation in rat liver cell grafts
following liver transplants, demonstrating SphK2 facilitation of inflammation.91 However,
SphK2 activity can also be anti-inflammatory as demonstrated by either knockdown or inhibition
of SphK2 in murine arthritis.1, 90, 94 These conflicting reports appear to be disease model-specific,

15
suggesting more information is needed to truly understand SphK2 activity in inflammatory
diseases and responses.
SphK2’s role in fibrosis, specifically kidney fibrosis, is emerging. One recent study
showed that SphK2-null mice were less susceptible to kidney fibrosis upon folic acid treatment
as wild-type and SphK1-null mice.95 The SphK2 knockout mice also showed reduced fibrotic
and inflammatory markers. These observations were recapitulated when wild-type mice treated
with folic acid to induce kidney fibrosis showed reduced kidney fibrosis and fibrotic markers
upon administration of an SphK2-selective inhibitor.95 These results suggest that SphK2
inhibition may be a viable route for controlling kidney fibrosis.
SphK2 has also been implicated in ischemic reperfusion (IR) injury. Reports on its role in
IR are also conflicting. Inhibition of SphK2 in mice liver models protected against IR, whereas
knockout of SphK2 in mouse myocardia promoted IR, leading to tissue injury.8, 66, 96 These
results suggest that the method of preventing SphK2 activity and the tissue type influence the
outcome in IR studies.48
SphK2 may also be involved in neurodegenerative diseases such as Alzheimer’s disease
and Parkinson’s disease. High concentrations of SphK2 were observed in autopsy cerebral
samples of Alzheimer patients.25 Additionally, inhibition of SphK2 in mice resulted in a decrease
in the BACE-1-mediated production of amyloid-β (Aβ), a protein often found in Alzheimer’s
patient brains.25 In comparison, SphK2 was discovered to be down-regulated in cerebral samples
of Parkinson’s patients.97 Knockdown studies of SphK2 showed decreased S1P levels in
dopaminergic neurons, which was accompanied by a reduction in ATP levels and an increase in
radical oxygen species.98 SphK2 Cellular localization studies of dopaminergic neuronal cells
indicate that SphK2 is primarily localized to the mitochondria, suggesting down-regulation of
16
SphK2 in the Parkinson’s disease cerebral samples is associated with mitochondrial
dysfunction.98
1.7 Sphingosine Kinase 2 Inhibitors
As a mediator in the formation of S1P and implicated role in disease, SphK2 is emerging as
a potential therapeutic target for pharmaceutical inhibition. In comparison to SphK1-selective
inhibitors, there is a paucity of reported potent and selective SphK2 inhibitors. Nevertheless, a
number of SphK2 inhibitors have been developed, with some showing promising results in
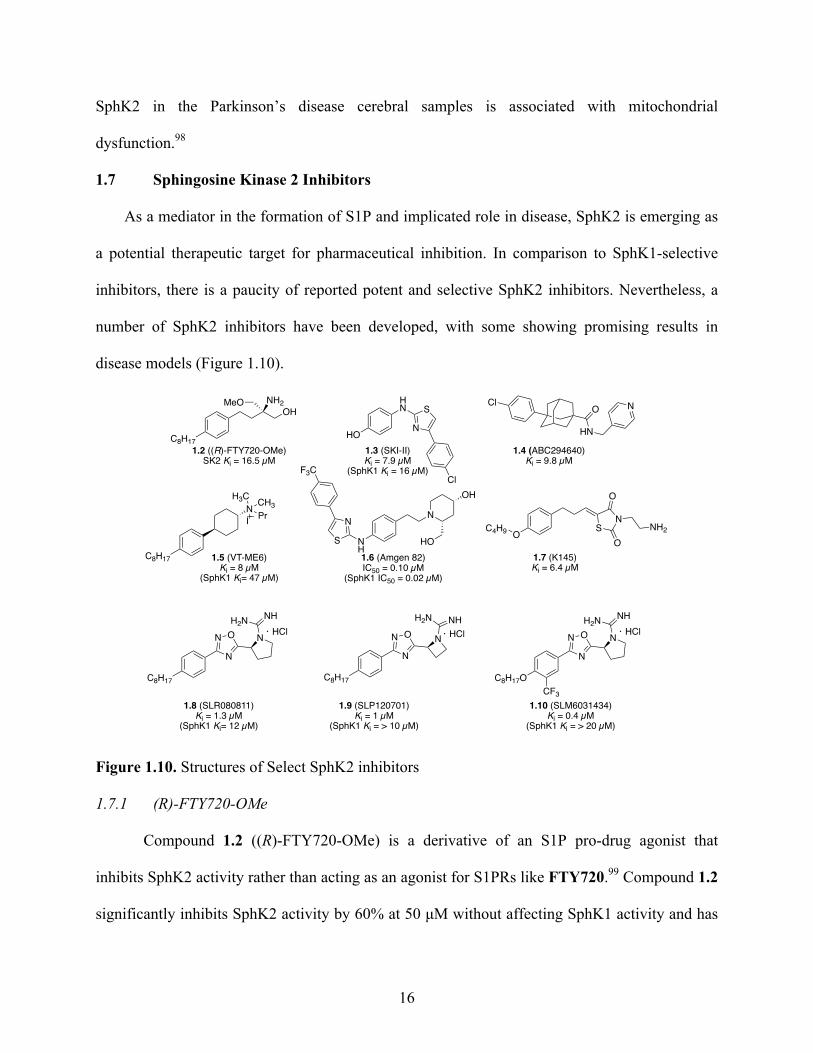
disease models (Figure 1.10).
Figure 1.10. Structures of Select SphK2 inhibitors
1.7.1 (R)-FTY720-OMe
Compound 1.2 ((R)-FTY720-OMe) is a derivative of an S1P pro-drug agonist that
inhibits SphK2 activity rather than acting as an agonist for S1PRs like FTY720.99 Compound 1.2
significantly inhibits SphK2 activity by 60% at 50 µM without affecting SphK1 activity and has
HN
N
S
Cl
HO1.3 (SKI-II) Ki = 7.9 µM
(SphK1 Ki = 16 µM)
C8H17
OHNH2MeO
1.2 ((R)-FTY720-OMe)SK2 Ki = 16.5 µM
O
HN
NCl
1.4 (ABC294640)Ki = 9.8 µM
N
C8H17
H3C
PrCH3
I
1.5 (VT-ME6)Ki = 8 µM
(SphK1 Ki= 47 µM)
C8H17
N
ON N
1.8 (SLR080811)Ki = 1.3 µM
(SphK1 Ki= 12 µM)
O SN
NH2
O
OC4H9
1.7 (K145) Ki = 6.4 µM
C8H17
N
ON N
N
OH
HONH
N
S
F3C
1.6 (Amgen 82)IC50 = 0.10 µM
(SphK1 IC50 = 0.02 µM)
1.9 (SLP120701)Ki = 1 µM
(SphK1 Ki = > 10 µM)
NHH2N H2N NHHClHCl
C8H17O
N
ON N
1.10 (SLM6031434)Ki = 0.4 µM
(SphK1 Ki = > 20 µM)
NHH2NHCl
CF3

17
an inhibition constant (Ki) of 16.5 µM for SphK2 (Figure 1.10).99 Studies have demonstrated that
1.2 promotes apoptosis in HEK 293 cells and reduces DNA synthesis in MCF-7 breast cancer
cells.8, 99 Recently, 1.2 was shown to enhance pulmonary vascular barrier function to reduce and
prevent vascular leak in acute respiratory distress syndrome models.100
1.7.2 SKI-II
Compound 1.3 (SKI-II) is an aminothiazole-based SphK inhibitor that was discovered
from a library screen.101 It is a non-selective SphK inhibitor, having Ki’s of 16 µM and 7.9 µM
for SphK1 and SphK2, respectively (Figure 1.10).102 Notably, it was utilized in the creation and
isolation of the first reported SphK1 crystal structure, as mentioned earlier in this review.54
Information derived from this work has led to the development of more potent SphK2 inhibitors,
including aminothiazole-based compounds reported by Vogt et al., Gustin et al., and Aurelio et
al.56, 103, 104 Multiple studies have demonstrated its efficacy in promoting apoptosis in cancer cell
lines such as T24 cells.8, 101, 102 However, 1.3 was recently shown to also inhibit dihydroceramide
desaturase (Des1), which facilitates the production of ceramide.105 Inhibition of Des1 has been
shown to promote cell cycle arrest. Thus, the apoptosis observed in cancer cell lines from 1.3
treatment result from a concomitant inhibition of SphK1/2 and Des1.
1.7.3 ABC294640
As a compound discovered from a high-throughput screening of a series of compounds,
1.4 (ABC294640) is an SphK2-selective inhibitor with a Ki of 9.8 µM (Figure 1.10).106 Studies
indicate its good oral bioavailability and low toxicity.106 Its anticancer potential as a suppressor
of tumor proliferation has been documented in numerous reports.78, 89, 91, 106-108 Additionally, it
has shown promise in alleviating the effects of inflammatory diseases such as arthritis and
colitis78, 89, 91, 106 Interestingly, 1.4 can also serve as an estrogen receptor antagonist, preventing

18
breast cancer proliferation and survival in MCF-7 breast cancer cell lines and preventing tumor
cell growth in mice models.109 Additionally, it has also been implicated as an inhibitor of
Des1.110 Regardless, it was recently tested in phase I clinical trials (clinicaltrials.gov registry
identifier NCT01488515). Twenty-two patients with various cancer types were dosed twice daily
for 3 years with an average dose of 500 mg 1.4, which resulted in reduced detected plasma S1P
levels within the first 12 h of treatment.111 Of the enrolled patients, one showed partial response
to treatment and 6 showed a stable response by the end of the trial, suggesting a therapeutic
potential for this molecule.111
1.7.4 VT-ME6
Developed from structure-activity relationship (SAR) studies conducted on an S1P pro-
drug agonist, 1.5 (VT-ME6) is an SphK2-selective inhibitor with a Ki of 8 µM (Figure 1.10).6
Tail length modification studies112 demonstrated that the tail length of 1.5 influences its
selectivity for SphK2 over SphK1. Tail groups containing alkyl chain lengths of 8 and 14 carbon
atoms imparted a 3-fold selectivity for SphK2 over SphK1, while all other tested tail lengths (6,
9, 10, 11, 12, 16) showed little to no selectivity for SphK2 over SphK1.112 Studies have shown
that 1.5 affects S1P signaling by preventing the phosphorylation of ERK; however, treatment of
leukemia U937 cells with this inhibitor did not cause a drop in S1P levels.6
1.7.5 Amgen 82
As a product of structure-based analysis of the SphK1 crystal structure, 1.6 (Amgen 82)
is a non-selective SphK inhibitor that targets the Sph binding pocket rather than the ATP binding
site.56 Assays found that this inhibitor has an average half maximal inhibitory concentration
(IC50) of 0.02 µM and 0.10 µM for SphK1 and SphK2, respectively.56 The SphK Ki’s for 1.6
were not determined. Compound 1.6 can inhibit SphK activity in multiple cancer cell lines,

19
including breast cancer and skin cancer cell lines.56, 88 Surprisingly, administration of the
inhibitor at low concentrations (0.001–1 µM) inhibited SphK activity without affecting tumor
cell viability.88 However, higher concentrations of 1.6 ( > 1 µM) were found to concomitantly
inhibit SphK activity and reduce tumor cell viability.88 This decreased tumor cell viability was
not linked to SphK inhibition; rather, it was attributed to a concentration-promoted, surfactant-
like behavior of 1.6, causing cell lysis.88 Nevertheless, 1.6 was shown to be orally bioavailable
with the ability to lower blood S1P by 72% at 100 mg/kg compared to controls.88 In a tumor
xenograph mouse model, 1.6 was unable to reduce tumor cell growth despite reducing blood S1P
levels,88 suggesting that reducing tumor cell growth in vivo requires an approach more complex
than solely reducing blood S1P levels.
1.7.6 K145
Another SphK2-selective inhibitor is 1.7 (K145), which has a Ki of 6.4 µM (Figure
1.10).113 In vitro studies found that 1.7 decreased S1P levels and reduced cell proliferation in
U937 cells.113 In addition, decreased ERK activity was also observed, indicating that SphK2
many not be the only target of 1.7.113 In vivo studies found that 1.7 is orally bioavailable and can
prevent leukemia and breast cancer cell growth.113
1.7.7 SLR080811
Derived from the SphK2-selective inhibitor 1.5, 1.8 (SLR080811) is a 10-fold SphK2-
selective inhibitor with a Ki of 1.3 µM (Figure 1.8).73 Various cell studies demonstrated that 1.8
can decrease the levels of intracellular S1P.73 Treatment of SphK1-null mice with 1.8 caused a
decrease in blood S1P levels 2 to 4 hours after injection, demonstrating its effect on SphK2;
however, treatment of wild-type mice with 1.8 resulted in elevated levels of blood S1P following
injection,73 a phenotype commonly observed in SphK2-null mice.42, 65, 71-73 Interestingly,

20
insertion of a methylene unit between 1.8’s guanylated pyrrolidine ring and oxadiazole ring
generates a potent SphK1-selective inhibitor that is capable of lowering S1P levels in vitro and in
vivo.114
1.7.8 SLP120701
As the azetidine analogue of 1.8, 1.9 (SLP120701) is a > 10-fold SphK2-selective
inhibitor with a Ki of 1.2 µM.114 Like 1.8, 1.9 decreases S1P levels in cultured cells and elevates
blood S1P in mice. Pharmacokinetic studies of 1.9 found that it has an improved half-life (t1/2) to
1.8, with a t1/2 of 8 h in comparison to 4h.114 Compound 1.9 was recently tested in a kidney
fibrosis mouse model, showing promise as an antifibrotic treatment, as treatment of mice with
induced kidney injury with 10 mg/kg of 1.9 showed reduced fibrotic markers and fibrosis of the
kidneys.95
1.7.9 SLM6031434
Developed from an SAR study of 1.8, 1.10 (SLM6031434) is a potent SphK2-selective
inhibitor with a Ki of 0.4 µM and 50-fold selectivity for SphK2. It has been shown to decrease
S1P levels in cultured cells in a dose dependent manor, and as observed with 1.8 and 1.9, causes
an elevation in blood S1P levels when given to wild-type mice. It has recently been utilized as a
chemical biology tool to probe SphK2 function, specifically to elucidate the elevated blood S1P
phenomenon observed when wild-type mice are treated with SphK2-selective inhibitors. 1.10
helped implicate SphK2 as a mediator in blood S1P clearance because wild-type mice treated
with 1.10 showed a remarkably decreased clearance rate of mass-labeled S1P from the blood.
1.8 Conclusions
The current understanding of the roles and regulation of SphK2 in normal cellular
function and in disease states will continue to evolve as more studies are conducted using

21
different types of cell lines, animal models, and SphK2 inhibitors. Although the crystal structure
of SphK2 has yet to be solved, current efforts to develop new and potent SphK2-selective
inhibitors have benefited from multiple publications of the SphK1 crystal structure and
information derived from the SphK2 inhibitors discussed herein.
1.9 Dissertation Overview For Developing Inhibitors of Sphingosine Kinase 2
Chapter 1 discussed the chemical biology of SphK2, disease states associated with its
activity, and select SphK2-selective inhibitors. Chapter 2 will disclose an SAR of the tail group
region of 1.8, revealing that the lipid binding pocket of SphK2 cannot accommodate charged tail
groups and is much larger than SphK1. Chapter 3 will disclose an SAR of aminothiazole
derivatives of an SphK1-selective inhibitor derived from 1.8. These derivatives show
surprisingly good potency and selectivity for SphK2. Chapter 6 provides the supplemental
information for the compounds synthesized and characterized by the author of this dissertation.
NMR spectra for the synthesized compounds can be found online (http://www.sciencedirect.com
/science/article/pii/S0960894X15002474 for compounds synthesized in Chapter 2 and http://pubs
.acs.org/doi/suppl/10.1021/acs.jmedchem.7b00233 for compounds synthesized in Chapter 3).

22
1.10 References
1. Kunkel, G. T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the Sphingosine-1-Phosphate
Axis in Cancer, Inflammation and Beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702.
2. Maceyka, M.; Harikumar, K. B.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate Signaling
and Its Role in Disease. Trends Cell Biol. 2012, 22, 50–60.
3. Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-Phosphate: A Janus-Faced Mediator
of Fibrotic Diseases. Biochim. Biophys. Acta 2013, 1831, 239–250.
4. Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingolipids in Cancer. Cancer Metastasis Rev.
2011, 30, 567–576.
5. Takabe, K.; Paugh, S. W.; Milstien, S.; Spiegel, S. "Inside-out" Signaling of Sphingosine-1-
Phosphate: Therapeutic Targets. Pharmacol. Rev. 2008, 60, 181-195.
6. Raje, M. R.; Knott, K.; Kharel, Y.; Bissel, P.; Lynch, K. R.; Santos, W. L. Design, Synthesis
and Biological Activity of Sphingosine Kinase 2 Selective Inhibitors. Bioorg. Med. Chem.
2012, 20, 183-194.
7. Olivera, A.; Spiegel, S. Sphingosine-1-Phosphate as Second Messenger in Cell Proliferation
Induced by PDGF and FCS Mitogens. Nature 1993, 365, 557–560.
8. Orr Gandy, K. A.; Obeid, L. M. Targeting the Sphingosine Kinase/Sphingosine 1-Phosphate
Pathway in Disease: Review of Sphingosine Kinase Inhibitors. Biochim. Biophys. Acta 2013,
1831, 157–166.
9. Blaho, V. A.; Hla, T. An Update on the Biology of Sphingosine 1-Phosphate Receptors. J.
Lipid Res. 2014, 55, 1596-1608.

23
10. Dyckman, A. J. Modulators of Sphingosine-1-Phosphate Pathway Biology: Recent
Advances of Sphingosine-1-Phosphate Receptor 1 (S1P1) Agonists and Future Perspectives.
J. Med. Chem. 2017, DOI: 10.1021/acs.jmedchem.6b01575.
11. Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-Phosphate Signalling. Development 2014,
141, 5–9.
12. Nagahashi, M.; Kim, E. Y.; Yamada, A.; Ramachandran, S.; Allegood, J. C.; Hait, N. C.;
Maceyka, M.; Milstien, S.; Takabe, K.; Spiegel, S. Spns2, a Transporter of Phosphorylated
Sphingoid Bases, Regulates Their Blood and Lymph Levels, and the Lymphatic Network.
FASEB J. 2013, 27, 1001–1011.
13. Fayyaz, S.; Henkel, J.; Japtok, L.; Krämer, S.; Damm, G.; Seehofer, D.; Püschel, G. P.;
Kleuser, B. Involvement of Sphingosine 1-Phosphate in Palmitate-Induced Insulin Resistance
of Hepatocytes via the S1P2 Receptor Subtype. Diabetologia 2014, 57, 373–382.
14. Japtok, L.; Schmitz, E. I.; Fayyaz, S.; Kramer, S.; Hsu, L. J.; Kleuser, B. Sphingosine 1-
Phosphate Counteracts Insulin Signaling in Pancreatic Beta-Cells via the Sphingosine 1-
Phosphate Receptor Subtype 2. FASEB J. 2015, 29, 3357–3369.
15. Kitada, Y.; Kajita, K.; Taguchi, K.; Mori, I.; Yamauchi, M.; Ikeda, T.; Kawashima, M.;
Asano, M.; Kajita, T.; Ishizuka, T.; Banno, Y.; Kojima, I.; Chun, J.; Kamata, S.; Ishii, I.;
Morita, H. Blockade of Sphingosine 1-Phosphate Receptor 2 Signaling Attenuates High-Fat
Diet-Induced Adipocyte Hypertrophy and Systemic Glucose Intolerance in Mice.
Endocrinology 2016, 157, 1839–1851.
16. Silva, V. R.; Micheletti, T. O.; Pimentel, G. D.; Katashima, C. K.; Lenhare, L.; Morari, J.;
Mendes, M. C.; Razolli, D. S.; Rocha, G. Z.; de Souza, C. T.; Ryu, D.; Prada, P. O.; Velloso,

24
L. A.; Carvalheira, J. B.; Pauli, J. R.; Cintra, D. E.; Ropelle, E. R. Hypothalamic S1P/S1PR1
Axis Controls Energy Homeostasis. Nat. Commun. 2014, 5, 1–15.
17. Silva, V. R. R.; Katashima, C. K.; Bueno Silva, C. G.; Lenhare, L.; Micheletti, T. O.;
Camargo, R. L.; Ghezzi, A. C.; Camargo, J. A.; Assis, A. M.; Tobar, N.; Morari, J.; Razolli,
D. S.; Moura, L. P.; Pauli, J. R.; Cintra, D. E.; Velloso, L. A.; Saad, M. J. A.; Ropelle, E. R.
Hypothalamic S1P/S1PR1 Axis Controls Energy Homeostasis in Middle-Aged Rodents: The
Reversal Effects of Physical Exercise. Aging (Albany NY) 2017, 9, 142–154.
18. Andrieu, G.; Ledoux, A.; Branka, S.; Bocquet, M.; Gilhodes, J.; Walzer, T.; Kasahara, K.;
Inagaki, M.; Sabbadini, R. A.; Cuvillier, O.; Hatzoglou, A. Sphingosine 1-Phosphate
Signaling through Its Receptor S1P5 Promotes Chromosome Segregation and Mitotic
Progression. Science Signal. 2017, 10, 1–11.
19. Hait, N. C.; Allegood, J.; Maceyka, M.; Strub, G. M.; Harikumar, K. B.; Singh, S. K.; Luo,
C.; Marmorstein, R.; Kordula, T.; Milstien, S.; Spiegel, S. Regulation of Histone Acetylation
in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325, 1254–1257.
20. Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine
Kinase 2 Is a Nuclear Protein and Inhibits DNA Synthesis. J. Biol. Chem. 2003, 278, 46832–
46839.
21. Ghosh, T. K.; Bian, J.; Gill, D. L. Sphingosine 1-Phosphate Generated in the Endoplasmic
Reticulum Membrane Activates Release of Stored Calcium. J. Biol. Chem. 1994, 269, 22628–
22635.
22. Strub, G. M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J. C.; Hait, N. C.; Maceyka, M.;
Price, M. M.; Chen, Q.; Simpson, D. C.; Kordula, T.; Milstien, S.; Lesnefsky, E. J.; Spiegel,
S. Sphingosine-1-Phosphate Produced by Sphingosine Kinase 2 in Mitochondria Interacts

25
with Prohibitin 2 to Regulate Complex IV Assembly and Respiration. FASEB J. 2011, 25,
600–612.
23. Alvarez, S. E.; Harikumar, K. B.; Hait, N. C.; Allegood, J.; Strub, G. M.; Kim, E. Y.;
Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; Milstien, S.; Spiegel, S. Sphingosine-1-
Phosphate Is a Missing Cofactor for the E3 Ubiquitin Ligase TRAF2. Nature 2010, 465,
1084–1088.
24. Sun, K.; Zhang, Y.; D'Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.;
Huang, A.; Wen, Y. E.; Bogdanov, M. V.; Vila, A.; O'Brien, J.; Kellems, R. E.; Dowhan, W.;
Subudhi, A. W.; Jameson-Van Houten, S.; Julian, C. G.; Lovering, A. T.; Safo, M.; Hansen,
K. C.; Roach, R. C.; Xia, Y. Sphingosine-1-Phosphate Promotes Erythrocyte Glycolysis and
Oxygen Release for Adaptation to High-Altitude Hypoxia. Nat. Commun. 2016, 7, 1–13.
25. Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.;
Yokoshima, S.; Fukuyama, T.; Lee, V. M.; Trojanowski, J. Q.; Tomita, T.; Iwatsubo, T.
BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci.
2011, 31, 6850–6857.
26. Prager, B.; Spampinato, S. F.; Ransohoff, R. M. Sphingosine 1-Phosphate Signaling at the
Blood–Brain Barrier. Trends Mol. Med. 2015, 21, 354–363.
27. Pyne, N. J.; Tonelli, F.; Lim, K. G.; Long, J. S.; Edwards, J.; Pyne, S. Sphingosine 1-
Phosphate Signalling in Cancer. Biochem. Soc. Trans. 2012, 40, 94–100.
28. Plano, D.; Amin, S.; Sharma, A. K. Importance of Sphingosine Kinase (Sphk) as a Target in
Developing Cancer Therapeutics and Recent Developments in the Synthesis of Novel SphK
Inhibitors. J. Med. Chem. 2014, 57, 5509–5524.

26
29. Pyne, N. J.; Pyne, S. Sphingosine 1-Phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–
503.
30. Yang, L.; Yue, S.; Yang, L.; Liu, X.; Han, Z.; Zhang, Y.; Li, L. Sphingosine
Kinase/Sphingosine 1-Phosphate (S1P)/S1P Receptor Axis Is Involved in Liver Fibrosis-
Associated Angiogenesis. J. Hepatol. 2013, 59, 114–123.
31. Gellings Lowe, N.; Swaney, J. S.; Moreno, K. M.; Sabbadini, R. A. Sphingosine-1-
Phosphate and Sphingosine Kinase Are Critical for Transforming Growth Factor-Beta-
Stimulated Collagen Production by Cardiac Fibroblasts. Cardiovasc. Res. 2009, 82, 303–312.
32. Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.;
Bogdanov, M.; Alexander, D. C.; Milburn, M. V.; Ahmed, M. H.; Lin, H.; Idowu, M.; Zhang,
J.; Kato, G. J.; Abdulmalik, O. Y.; Zhang, W.; Dowhan, W.; Kellems, R. E.; Zhang, P.; Jin, J.;
Safo, M.; Tsai, A. L.; Juneja, H. S.; Xia, Y. Elevated Sphingosine-1-Phosphate Promotes
Sickling and Sickle Cell Disease Progression. J. Clin. Invest. 2014, 124, 2750–2761.
33. Proia, R. L.; Hla, T. Emerging Biology of Sphingosine-1-Phosphate: Its Role in
Pathogenesis and Therapy. J. Clin. Invest. 2015, 125, 1379–1387.
34. Sun, K.; Zhang, Y.; Bogdanov, M. V.; Wu, H.; Song, A.; Li, J.; Dowhan, W.; Idowu, M.;
Juneja, H. S.; Molina, J. G.; Blackburn, M. R.; Kellems, R. E.; Xia, Y. Elevated Adenosine
Signaling Via Adenosine A2b Receptor Induces Normal and Sickle Erythrocyte Sphingosine
Kinase 1 Activity. Blood 2015, 125, 1643–1652.
35. Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste,
E.; Pitson, S.; Maurage, C. A.; Duyckaerts, C.; Cuvillier, O.; Delisle, M.-B. Reduced
Sphingosine Kinase-1 and Enhanced Sphingosine 1-Phosphate Lyase Expression Demonstrate

27
Deregulated Sphingosine 1-Phosphate Signaling in Alzheimer’s Disease. Acta Neuropath.
Comm. 2014, 2, 1–12.
36. Couttas, T. A.; Kain, N.; Daniels, B.; Lim, X. Y.; Shepherd, C.; Kril, J.; Pickford, R.; Li, H.;
Garner, B.; Don, A. S. Loss of the Neuroprotective Factor Sphingosine 1-Phosphate Early in
Alzheimer’s Disease Pathogenesis. Acta Neuropath. Comm. 2014, 2, 1–9.
37. Takuwa, Y.; Ikeda, H.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Sphingosine-1-Phosphate as
a Mediator Involved in Development of Fibrotic Diseases. Biochim. Biophys. Acta 2013,
1831, 185–192.
38. Pyne, N. J.; Dubois, G.; Pyne, S. Role of Sphingosine 1-Phosphate and Lysophosphatidic
Acid in Fibrosis. Biochim. Biophys. Acta 2013, 1831, 228–238.
39. Ikeda, H.; Watanabe, N.; Ishii, I.; Shimosawa, T.; Kume, Y.; Tomiya, T.; Inoue, Y.;
Nishikawa, T.; Ohtomo, N.; Tanoue, Y.; Iitsuka, S.; Fujita, R.; Omata, M.; Chun, J.; Yatomi,
Y. Sphingosine 1-Phosphate Regulates Regeneration and Fibrosis after Liver Injury via
Sphingosine 1-Phosphate Receptor 2. J. of Lipid Res. 2009, 50, 556–564.
40. Liu, H.; Sugiura, M.; Nava, V. E.; Edsall, L. C.; Kono, K.; Poulton, S.; Milstien, S.;
Kohama, T.; Spiegel, S. Molecular Cloning and Functional Characterization of a Novel
Mammalian Sphingosine Kinase Type 2 Isoform. J. Biol. Chem. 2000, 275, 19513–19520.
41. Spiegel, S.; Milstien, S. Sphingosine 1-Phosphate, a Key Cell Signaling Molecule. J. Biol.
Chem. 2002, 277, 25851–25854.
42. Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G. F.; Spiegel, S.; Proia, R. L. Essential
Role for Sphingosine Kinases in Neural and Vascular Development. Mol. Cell Biol. 2005, 25,
11113–11121.

28
43. Hait, N. C.; Oskeritzian, C. A.; Paugh, S. W.; Milstien, S.; Spiegel, S. Sphingosine Kinases,
Sphingosine 1-Phosphate, Apoptosis and Diseases. Biochim. Biophys. Acta 2006, 1758, 2016–
2026.
44. Sarkar, S.; Maceyka, M.; Hait, N. C.; Paugh, S. W.; Sankala, H.; Milstien, S.; Spiegel, S.
Sphingosine Kinase 1 Is Required for Migration, Proliferation and Survival of MCF-7 Human
Breast Cancer Cells. FEBS Letters 2005, 579, 5313–5317.
45. Yan, G.; Chen, S.; You, B.; Sun, J. Activation of Sphingosine Kinase-1 Mediates Induction
of Endothelial Cell Proliferation and Angiogenesis by Epoxyeicosatrienoic Acids. Cardiovasc
Res 2008, 78, 308–314.
46. Chan, H.; Pitson, S. M. Post-Translational Regulation of Sphingosine Kinases. Biochim.
Biophys. Acta 2013, 1831, 147–156.
47. Pitson, S. M.; Moretti, P. A. B.; Zebol, J. R.; Lynn, H. E.; Xia, P.; Vadas, M. A.;
Wattenberg, B. W. Activation of Sphingosine Kinase 1 by ERK1/2‐Mediated
Phosphorylation. EMBO J. 2003, 22, 5491–5500.
48. Neubauer, H. A.; Pitson, S. M. Roles, Regulation and Inhibitors of Sphingosine Kinase 2.
FEBS J. 2013, 280, 5317–5336.
49. Döll, F.; Pfeilschifter, J.; Huwiler, A. The Epidermal Growth Factor Stimulates Sphingosine
Kinase-1 Expression and Activity in the Human Mammary Carcinoma Cell Line MCF7.
Biochim. Biophys. Acta 2005, 1738, 72–81.
50. Leclercq, T. M.; Moretti, P. A.; Pitson, S. M. Guanine Nucleotides Regulate Sphingosine
Kinase 1 Activation by Eukaryotic Elongation Factor 1Aand Provide a Mechanism for
EEF1A-Associated Oncogenesis. Oncogene 2011, 30, 372–378.

29
51. Leclercq, T. M.; Moretti, P. A.; Vadas, M. A.; Pitson, S. M. Eukaryotic Elongation Factor
1A Interacts with Sphingosine Kinase and Directly Enhances Its Catalytic Activity. J. Biol.
Chem. 2008, 283, 9606–9614.
52. Olivera, A.; Urtz, N.; Mizugishi, K.; Yamashita, Y.; Gilfillan, A. M.; Furumoto, Y.; Gu, H.;
Proia, R. L.; Baumruker, T.; Rivera, J. Ige-Dependent Activation of Sphingosine Kinases 1
and 2 and Secretion of Sphingosine 1-Phosphate Requires Fyn Kinase and Contributes to
Mast Cell Responses. J. Biol. Chem. 2006, 281, 2515–2525.
53. Urtz, N.; Olivera, A.; Bofill-Cardona, E.; Csonga, R.; Billich, A.; Mechtcheriakova, D.;
Bornancin, F.; Woisetschlager, M.; Rivera, J.; Baumruker, T. Early Activation of Sphingosine
Kinase in Mast Cells and Recruitment to Fcepsilonri Are Mediated by Its Interaction with Lyn
Kinase. Mol. Cell. Biol. 2004, 24, 8765–8777.
54. Wang, Z.; Min, X.; Xiao, S. H.; Johnstone, S.; Romanow, W.; Meininger, D.; Xu, H.; Liu,
J.; Dai, J.; An, S.; Thibault, S.; Walker, N. Molecular Basis of Sphingosine Kinase 1 Substrate
Recognition and Catalysis. Structure 2013, 21, 798–809.
55. Wang, J.; Knapp, S.; Pyne, N. J.; Pyne, S.; Elkins, J. M. Crystal Structure of Sphingosine
Kinase 1 with Pf-543. ACS Med. Chem. Lett. 2014, 5, 1329–1333.
56. Gustin, D. J.; Li, Y.; Brown, M. L.; Min, X.; Schmitt, M. J.; Wanska, M.; Wang, X.;
Connors, R.; Johnstone, S.; Cardozo, M.; Cheng, A. C.; Jeffries, S.; Franks, B.; Li, S.; Shen,
S.; Wong, M.; Wesche, H.; Xu, G.; Carlson, T. J.; Plant, M.; Morgenstern, K.; Rex, K.;
Schmitt, J.; Coxon, A.; Walker, N.; Kayser, F.; Wang, Z. Structure Guided Design of a Series
of Sphingosine Kinase (SphK) Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4608–4616.
57. Schnute, M. E.; McReynolds, M. D.; Carroll, J.; Chrencik, J.; Highkin, M. K.; Iyanar, K.;
Jerome, G.; Rains, J. W.; Saabye, M.; Scholten, J. A.; Yates, M.; Nagiec, M. M. Discovery of

30
a Potent and Selective Sphingosine Kinase 1 Inhibitor through the Molecular Combination of
Chemotype-Distinct Screening Hits. J. Med. Chem. 2017, 60, 2562–2572.
58. Baek, D. J.; MacRitchie, N.; Anthony, N. G.; Mackay, S. P.; Pyne, S.; Pyne, N. J.; Bittman,
R. Structure–Activity Relationships and Molecular Modeling of Sphingosine Kinase
Inhibitors. J. Med. Chem. 2013, 56, 9310–9327.
59. Yoshimoto, T.; Furuhata, M.; Kamiya, S.; Hisada, M.; Miyaji, H.; Magami, Y.; Yamamoto,
K.; Fujiwara, H.; Mizuguchi, J. Positive Modulation of IL-12 Signaling by Sphingosine
Kinase 2 Associating with the Il-12 Receptor Β1 Cytoplasmic Region J. Immunol. 2003, 171,
1352–1359.
60. Liu, H.; Toman, R. E.; Goparaju, S. K.; Maceyka, M.; Nava, V. E.; Sankala, H.; Payne, S.
G.; Bektas, M.; Ishii, I.; Chun, J.; Milstien, S.; Spiegel, S. Sphingosine Kinase Type 2 Is a
Putative BH3-Only Protein That Induces Apoptosis. J. Biol. Chem. 2003, 278, 40330-40336.
61. Chipuk, J. E.; McStay, G. P.; Bharti, A.; Kuwana, T.; Clarke, C. J.; Siskind, L. J.; Obeid, L.
M.; Green, D. R. Sphingolipid Metabolism Cooperates with Bak and Bax to Promote the
Mitochondrial Pathway of Apoptosis. Cell 2012, 148, 988–1000.
62. Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S. K.; Jahangeer, S.; Okada, T.;
Nakamura, S. Protein Kinase D-Mediated Phosphorylation and Nuclear Export of
Sphingosine Kinase 2. J. Biol. Chem. 2007, 282, 27493–27502.
63. Don, A. S.; Rosen, H. A Lipid Binding Domain in Sphingosine Kinase 2. Biochem. Biophys.
Res. Commun. 2009, 380, 87–92.
64. Okada, T.; Ding, G.; Sonoda, H.; Kajimoto, T.; Haga, Y.; Khosrowbeygi, A.; Gao, S.; Miwa,
N.; Jahangeer, S.; Nakamura, S. Involvement of N-Terminal-Extended Form of Sphingosine

31
Kinase 2 in Serum-Dependent Regulation of Cell Proliferation and Apoptosis. J. Biol. Chem.
2005, 280, 36318–36325.
65. Maceyka, M.; Sankala, H.; Hait, N. C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.;
Zhang, M.; Satin, L. S.; Merrill, A. H., Jr.; Milstien, S.; Spiegel, S. SphK1 and SphK2,
Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J.
Biol. Chem. 2005, 280, 37118–37129.
66. Shi, Y.; Rehman, H.; Ramshesh, V. K.; Schwartz, J.; Liu, Q.; Krishnasamy, Y.; Zhang, X.;
Lemasters, J. J.; Smith, C. D.; Zhong, Z. Sphingosine Kinase-2 Inhibition Improves
Mitochondrial Function and Survival after Hepatic Ischemia-Reperfusion. J. Hepatol. 2012,
56, 137–145.
67. Weigert, A.; Cremer, S.; Schmidt, M. V.; von Knethen, A.; Angioni, C.; Geisslinger, G.;
Brune, B. Cleavage of Sphingosine Kinase 2 by Caspase-1 Provokes Its Release from
Apoptotic Cells. Blood 2010, 115, 3531–3540.
68. Sankala, H. M.; Hait, N. C.; Paugh, S. W.; Shida, D.; Lepine, S.; Elmore, L. W.; Dent, P.;
Milstien, S.; Spiegel, S. Involvement of Sphingosine Kinase 2 in p53-Independent Induction
of p21 by the Chemotherapeutic Drug Doxorubicin. Cancer Res 2007, 67, 10466–10474.
69. Sun, D. F.; Gao, Z. H.; Liu, H. P.; Yuan, Y.; Qu, X. J. Sphingosine 1-Phosphate Antagonizes
the Effect of All-Trans Retinoic Acid (ATRA) in a Human Colon Cancer Cell Line by
Modulation of Rarbeta Expression. Cancer Lett. 2012, 319, 182–189.
70. Panneer Selvam, S.; De Palma, R. M.; Oaks, J. J.; Oleinik, N.; Peterson, Y. K.; Stahelin, R.
V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C. D.; Ogretmen, B. Binding
of the Sphingolipid S1P to HTERT Stabilizes Telomerase at the Nuclear Periphery by
Allosterically Mimicking Protein Phosphorylation. Science Signal. 2015, 8, 1–13.

32
71. Allende, M. L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu,
R.; Rosenbach, M.; Keohane, C. A.; Mandala, S.; Spiegel, S.; Proia, R. L. Mice Deficient in
Sphingosine Kinase 1 Are Rendered Lymphopenic by FTY720. J. Biol. Chem. 2004, 279,
52487–52492.
72. Kharel, Y.; Morris, E. A.; Congdon, M. D.; Thorpe, S. B.; Tomsig, J. L.; Santos, W. L.;
Lynch, K. R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J.
Pharmacol. Exp. Ther. 2015, 355, 23–31.
73. Kharel, Y.; Raje, M.; Gao, M.; Gellett, A. M.; Tomsig, J. L.; Lynch, K. R.; Santos, W. L.
Sphingosine Kinase Type 2 Inhibition Elevates Circulating Sphingosine 1-Phosphate.
Biochem. J. 2012, 447, 149–157.
74. Hait, N. C.; Bellamy, A.; Milstien, S.; Kordula, T.; Spiegel, S. Sphingosine Kinase Type 2
Activation by ERK-Mediated Phosphorylation. J. Biol. Chem. 2007, 282, 12058–12065.
75. Hait, N. C.; Sarkar, S.; Le Stunff, H.; Mikami, A.; Maceyka, M.; Milstien, S.; Spiegel, S.
Role of Sphingosine Kinase 2 in Cell Migration toward Epidermal Growth Factor. J. Biol.
Chem. 2005, 280, 29462–29469.
76. Schnitzer, S. E.; Weigert, A.; Zhou, J.; Brune, B. Hypoxia Enhances Sphingosine Kinase 2
Activity and Provokes Sphingosine-1-Phosphate-Mediated Chemoresistance in A549 Lung
Cancer Cells. Mol. Cancer Res. 2009, 7, 393–401.
77. Antoon, J. W.; White, M. D.; Slaughter, E. M.; Driver, J. L.; Khalili, H. S.; Elliott, S.; Smith,
C. D.; Burow, M. E.; Beckman, B. S. Targeting NFĸB Mediated Breast Cancer
Chemoresistance through Selective Inhibition of Sphingosine Kinase-2. Cancer Biol. Ther.
2011, 11, 678–689.

33
78. Beljanski, V.; Lewis, C. S.; Smith, C. D. Antitumor Activity of Sphingosine Kinase 2
Inhibitor ABC294640 and Sorafenib in Hepatocellular Carcinoma Xenografts. Cancer Biol.
Ther. 2011, 11, 524–534.
79. Chumanevich, A. A.; Poudyal, D.; Cui, X.; Davis, T.; Wood, P. A.; Smith, C. D.; Hofseth,
L. J. Suppression of Colitis-Driven Colon Cancer in Mice by a Novel Small Molecule
Inhibitor of Sphingosine Kinase. Carcinogenesis 2010, 31, 1787–1793.
80. Gao, P.; Smith, C. D. Ablation of Sphingosine Kinase-2 Inhibits Tumor Cell Proliferation
and Migration. Mol. Cancer Res. 2011, 9, 1509-1519.
81. Weigert, A.; Schiffmann, S.; Sekar, D.; Ley, S.; Menrad, H.; Werno, C.; Grosch, S.;
Geisslinger, G.; Brune, B. Sphingosine Kinase 2 Deficient Tumor Xenografts Show Impaired
Growth and Fail to Polarize Macrophages Towards an Anti-Inflammatory Phenotype. Int. J.
Cancer 2009, 125, 2114–2121.
82. Neubauer, H. A.; Pham, D. H.; Zebol, J. R.; Moretti, P. A. B.; Leclercq, T. M.; Chan, H.;
Powell, J. A.; Pitman, M. R.; Samuel, M. S.; Bonder, C. S.; Gliddon, B. L.; Pitson, S. M.;
Neubauer, H. A.; Pham, D. H.; Chan, H.; Bonder, C. S.; Pitson, S. M.; Peterson, A. L.; Creek,
D. J.; Powell, J. A.; Samuel, M. S.; Bonder, C. S.; Pitson, S. M. An Oncogenic Role for
Sphingosine Kinase 2. Oncotarget 2016, 7, 64886–64899.
83. Wallington-Beddoe, C. T.; Powell, J. A.; Tong, D.; Pitson, S. M.; Bradstock, K. F.; Bendall,
L. J. Sphingosine Kinase 2 Promotes Acute Lymphoblastic Leukemia by Enhancing MYC
Expression. Cancer Res. 2014, 74, 2803–2815.
84. Xiao, M.; Liu, Y.; Zou, F. Sensitization of Human Colon Cancer Cells to Sodium Butyrate-
Induced Apoptosis by Modulation of Sphingosine Kinase 2 and Protein Kinase D. Exp. Cell
Res. 2012, 318, 43–52.

34
85. Wang, Q.; Li, J.; Li, G.; Li, Y.; Xu, C.; Li, M.; Xu, G.; Fu, S. Prognostic Significance of
Sphingosine Kinase 2 Expression in Non-Small Cell Lung Cancer. Tumor Biol. 2014, 35,
363–368.
86. Venkata, J. K.; An, N.; Stuart, R.; Costa, L. J.; Cai, H.; Coker, W.; Song, J. H.; Gibbs, K.;
Matson, T.; Garrett-Mayer, E.; Wan, Z.; Ogretmen, B.; Smith, C.; Kang, Y. Inhibition of
Sphingosine Kinase 2 Downregulates the Expression of C-MYC and MCL-1 and Induces
Apoptosis in Multiple Myeloma. Blood 2014, 124, 1915–1925.
87. Rhodes, D. R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.;
Pandey, A.; Chinnaiyan, A. M. Oncomine: A Cancer Microarray Database and Integrated
Data-Mining Platform. Neoplasia (Ann Arbor, MI, U. S.) 2004, 6, 1–6.
88. Rex, K.; Jeffries, S.; Brown, M. L.; Carlson, T.; Coxon, A.; Fajardo, F.; Frank, B.; Gustin,
D.; Kamb, A.; Kassner, P. D.; Li, S.; Li, Y.; Morgenstern, K.; Plant, M.; Quon, K.; Ruefli-
Brasse, A.; Schmidt, J.; Swearingen, E.; Walker, N.; Wang, Z.; Vivienne Watson, J. E.;
Wickramasinghe, D.; Wong, M.; Xu, G.; Holger, W. Sphingosine Kinase Activity is Not
Required for Tumor Cell Viability. PLoS One 2013, 8, 1–12.
89. Fitzpatrick, L. R.; Green, C.; Frauenhoffer, E. E.; French, K. J.; Zhuang, Y.; Maines, L. W.;
Upson, J. J.; Paul, E.; Donahue, H.; Mosher, T. J.; Smith, C. D. Attenuation of Arthritis in
Rodents by a Novel Orally-Available Inhibitor of Sphingosine Kinase.
Inflammopharmacology 2011, 19, 75–87.
90. Lai, W. Q.; Irwan, A. W.; Goh, H. H.; Melendez, A. J.; McInnes, I. B.; Leung, B. P. Distinct
Roles of Sphingosine Kinase 1 and 2 in Murine Collagen-Induced Arthritis. J. Immunol.
2009, 183, 2097–2103.

35
91. Liu, Q.; Rehman, H.; Shi, Y.; Krishnasamy, Y.; Lemasters, J. J.; Smith, C. D.; Zhong, Z.
Inhibition of Sphingosine Kinase-2 Suppresses Inflammation and Attenuates Graft Injury
after Liver Transplantation in Rats. PLoS One 2012, 7, e41834.
92. Imeri, F.; Schwalm, S.; Lyck, R.; Zivkovic, A.; Stark, H.; Engelhardt, B.; Pfeilschifter, J.;
Huwiler, A. Sphingosine Kinase 2 Deficient Mice Exhibit Reduced Experimental
Autoimmune Encephalomyelitis: Resistance to FTY720 but Not St-968 Treatments.
Neuropharmacology 2016, 105, 341–350.
93. Xi, M.; Ge, J.; Wang, X.; Sun, C.; Liu, T.; Fang, L.; Xiao, Q.; Yin, D. Development of
Hydroxy-Based Sphingosine Kinase Inhibitors and Anti-Inflammation in Dextran Sodium
Sulfate Induced Colitis in Mice. Bioorg. Med. Chem. 2016, 24, 3218–3230.
94. Baker, D. A.; Eudaly, J.; Smith, C. D.; Obeid, L. M.; Gilkeson, G. S. Impact of Sphingosine
Kinase 2 Deficiency on the Development of TNF-Alpha-Induced Inflammatory Arthritis.
Rheumatol. Int. 2013, 33, 2677–2681.
95. Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K. R.; Chroscicki, P.; Foley, L. S.;
Balogun, Z. A.; Alexander, K. J.; Park, H.; Okusa, M. D.; Bajwa, A.; Huang, L.; Kurmaeva,
E.; Ye, H.; Dondeti, K. R.; Chroscicki, P.; Foley, L. S.; Balogun, Z. A.; Alexander, K. J.;
Park, H.; Rosin, D. L.; Okusa, M. D.; Lynch, K. R.; Rosin, D. L. Sphingosine Kinase 2
Deficiency Attenuates Kidney Fibrosis Via IFN-γ. J. Am. Soc. Nephrol. 2017, 28, 1145–1161.
96. Vessey, D. A.; Li, L.; Jin, Z. Q.; Kelley, M.; Honbo, N.; Zhang, J.; Karliner, J. S. A
Sphingosine Kinase Form 2 Knockout Sensitizes Mouse Myocardium to
Ischemia/Reoxygenation Injury and Diminishes Responsiveness to Ischemic Preconditioning.
Oxid. Med. Cell. Longev. 2011, 2011, 961059.

36
97. Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L. T.; Krichevsky, A. M.; Andersen, S.
L.; Stephens, R. M.; Benes, F. M.; Sonntag, K. C. Gene Expression Profiling of Substantia
Nigra Dopamine Neurons: Further Insights into Parkinson's Disease Pathology. Brain 2009,
132, 1795–1809.
98. Sivasubramanian, M.; Kanagaraj, N.; Dheen, S. T.; Tay, S. S. W. Sphingosine Kinase 2 and
Sphingosine-1-Phosphate Promotes Mitochondrial Function in Dopaminergic Neurons of
Mouse Model of Parkinson's Disease and in Mpp+-Treated Mn9d Cells in Vitro.
Neuroscience (Amsterdam, Neth.) 2015, 290, 636–648.
99. Lim, K. G.; Sun, C.; Bittman, R.; Pyne, N. J.; Pyne, S. (R)-Fty720 Methyl Ether Is a Specific
Sphingosine Kinase 2 Inhibitor: Effect on Sphingosine Kinase 2 Expression in HEK 293 Cells
and Actin Rearrangement and Survival of MCF-7 Breast Cancer Cells. Cell Signal. 2011, 23,
1590–1595.
100. Camp, S. M.; Chiang, E. T.; Sun, C.; Usatyuk, P. V.; Bittman, R.; Natarajan, V.; Garcia, J.
G. N.; Dudek, S. M. Pulmonary Endothelial Cell Barrier Enhancement by Novel FTY720
Analogs: Methoxy-FTY720, Fluoro-FTY720, and Β-Glucuronide-FTY720. Chem. Phys.
Lipids 2015, 191, 16–24.
101. French, K. J.; Schrecengost, R. S.; Lee, B. D.; Zhuang, Y.; Smith, S. N.; Eberly, J. L.; Yun,
J. K.; Smith, C. D. Discovery and Evaluation of Inhibitors of Human Sphingosine Kinase.
Cancer Res. 2003, 63, 5962–5969.
102. Gao, G.; Peterson, U. K.; Smith, R. A.; Smith, C. D. Characterization of Isoenzyme-
Selective Inhibitors of Human Sphingosine Kinases. PLoS One 2012, 7, 1–11.
103. Aurelio, L.; Scullino, C. V.; Pitman, M. R.; Sexton, A.; Oliver, V.; Davies, L.; Rebello, R.
J.; Furic, L.; Creek, D. J.; Pitson, S. M.; Flynn, B. L. From Sphingosine Kinase to

37
Dihydroceramide Desaturase: A Structure–Activity Relationship (SAR) Study of the Enzyme
Inhibitory and Anticancer Activity of 4-((4-(4-chlorophenyl)thiazol-2-Yl)amino)phenol (SkI-
II). J. Med. Chem. 2016, 59, 965–984.
104. Vogt, D.; Weber, J.; Ihlefeld, K.; Bruggerhoff, A.; Proschak, E.; Stark, H. Design,
Synthesis and Evaluation of 2-Aminothiazole Derivatives as Sphingosine Kinase Inhibitors.
Bioorg. Med. Chem. 2014, 22, 5354–5367.
105. Cingolani, F.; Casasampere, M.; Sanllehi, P.; Casas, J.; Bujons, J.; Fabrias, G. Inhibition of
Dihydroceramide Desaturase Activity by the Sphingosine Kinase Inhibitor SkI-II. J. Lipid.
Res. 2014, 55, 1711–1720.
106. French, K. J.; Zhuang, Y.; Maines, L. W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J. J.;
Green, C. L.; Keller, S. N.; Smith, C. D. Pharmacology and Antitumor Activity of
ABC294640, a Selective Inhibitor of Sphingosine Kinase-2. J. Pharmacol. Exp. Ther. 2010,
333, 129–139.
107. Lewis, C. S.; Voelkel-Johnson, C.; Smith, C. D. Suppression of C-MYC and RRM2
Expression in Pancreatic Cancer Cells by the Sphingosine Kinase-2 Inhibitor ABC294640.
Oncotarget 2016, 7, 60181–60192.
108. Xun, C.; Chen, M.-B.; Qi, L.; Zhang, T.-N.; Peng, X.; Ning, L.; Chen, Z.-X.; Wang, L.-W.
Targeting Sphingosine Kinase 2 (SphK2) by ABC294640 Inhibits Colorectal Cancer Cell
Growth in Vitro and in Vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94/91–94/99.
109. Antoon, J. W.; White, M. D.; Meacham, W. D.; Slaughter, E. M.; Muir, S. E.; Elliott, S.;
Rhodes, L. V.; Ashe, H. B.; Wiese, T. E.; Smith, C. D.; Burow, M. E.; Beckman, B. S.
Antiestrogenic Effects of the Novel Sphingosine Kinase-2 Inhibitor ABC294640.
Endocrinology 2010, 151, 5124–5135.

38
110. Venant, H.; Rahmaniyan, M.; Jones, E. E.; Lu, P.; Lilly, M. B.; Garrett-Mayer, E.; Drake,
R. R.; Kraveka, J. M.; Smith, C. D.; Voelkel-Johnson, C. The Sphingosine Kinase 2 Inhibitor
ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of
Dihydroceramides in Vitro and in Vivo. Mol. Cancer Ther. 2015, 14, 2744–2752.
111. Britten, C. D.; Thomas, M. B.; Garrett-Mayer, E.; Chin, S. H.; Shirai, K.; Ogretmen, B.;
Bentz, T. A.; Brisendine, A.; Anderton, K.; Cusack, S. L.; Maines, L. W.; Zhuang, Y.; Smith,
C. D. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in
Patients with Advanced Solid Tumors. Clin. Cancer. Res. 2017, DOI: 10.1158/1078-
0432.CCR-16-2363.
112. Knott, K.; Kharel, Y.; Raje, M. R.; Lynch, K. R.; Santos, W. L. Effect of Alkyl Chain
Length on Sphingosine Kinase 2 Selectivity. Bioorg. Med. Chem. Lett. 2012, 22, 6817–6820.
113. Liu, K.; Guo, T. L.; Hait, N. C.; Allegood, J.; Parikh, H.; Xu, W.; Kellogg, G. E.; Grant, S.;
Spiegel, S.; Zhang, S. Biological Characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-
phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a Selective Sphingosine Kinase-2
Inhibitor and Anticancer Agent. PLoS One 2013, 8, 1–13.
114. Patwardhan, N. N.; Morris, E. A.; Kharel, Y.; Raje, M. R.; Gao, M.; Tomsig, J. L.; Lynch,
K. R.; Santos, W. L. Structure–Activity Relationship Studies and in Vivo Activity of
Guanidine-Based Sphingosine Kinase Inhibitors: Discovery of SphK1- and SphK2-Selective
Inhibitors. J. Med. Chem. 2015, 58, 1879–1899.

39
2 Structure-Activity Relationship Studies of the Lipophilic Tail Region of
Sphingosine Kinase 2 Inhibitors
2.1 Contributions
Multiple members of the Santos group contributed to this work. The reported compounds
were synthesized by the author, Dr. Molly Congdon, Dr. Neeraj Patwardhan, James
Grumkowski, and Emily Morris. Biological analyses of the reported compounds were conducted
by Dr. Yugesh Kharel of the University of Virginia Department of Pharmacology. The final
manuscript was written by Dr. Molly Congdon and Dr. Webster Santos. The author synthesized
and characterized final compounds 2.13d, 2.13e, and 2.16a-d and their corresponding
intermediates and contributed to the experimental write-up and revision of the manuscript. This
chapter is an adapted reprint with permission from Elsevier [Congdon, M.D.; Childress, E.S.;
Patwardhan, N.N.; Gumkowski, J.; Morris, E.A.; Kharel, Y.; Lynch, K.R. and Santos, W.L.,
Structure-Activity Relationship Studies of the Lipophilic Tail Region of Sphingosine Kinase 2
Inhibitors, Bioorg. Med. Chem. Lett. 2015, 25, 4956–4960, © 2015 Elsevier].
2.2 Abstract
Sphingosine 1-phosphate (S1P) is a ubiquitous, endogenous signaling molecule that is
synthesized by the isozymes sphingosine kinase 1 and 2. Alteration of S1P levels is linked to
several disease states including cancer, fibrosis, Alzheimer’s disease, and sickle cell disease,
making the S1P signaling pathway an attractive target for therapeutic intervention. While much
attention has been placed on developing SphK1-selective inhibitors, there is a dearth of reported
SphK2-selective agents. Herein, we report our investigations on the structure-activity
relationship studies on the lipophilic tail region of 2.4 (SLR080811), a SphK2-selective inhibitor.
Our studies demonstrate that the internal phenyl ring is a key structural feature in the scaffold of

40
2.4. Our studies also indicate that the SphK2 lipophilic binding pocket is not amenable for
charged heteroatom-bearing tailgroups. Further, we show the dependence of SphK2 activity and
selectivity on alkyl tail length, suggesting a larger lipid binding pocket in SphK2 compared to
SphK1.
2.3 Introduction
Sphingosine 1-phosphate (S1P) is a pleotropic sphingolipid that is synthesized from the
phosphorylation of sphingosine (Sph) by the two isoforms of sphingosine kinase: SphK1 and
SphK2. S1P functions as both an intracellular and extracellular signaling molecule, with S1P’s
primary function being an extracellular ligand for its G-protein coupled receptors S1P1-5. S1P
signaling has been associated with a variety of diseases including cancer, fibrosis, multiple
sclerosis, Alzheimer’s disease, and sickle cell disease.1-6 As a result of their key role in the
synthesis of S1P, regulation of SphKs has attracted an increasing amount of attention as a
therapeutic target. The ability to control SphKs function would also aid in the understanding of
their in vivo function, as well as their effects in the sphingolipid signaling pathway.
Many differences exist between SphK1 and SphK2 including size, cellular localization,
and function.7 While double knockout studies in mice suggest that SphKs are the sole source of
S1P, some functional redundancy exists as SphK1 or SphK2 null mice are viable and fertile.
Although inhibitor development towards SphK1 has been a focus of intense studies,8, 9 inhibitors
of SphK2 are emerging (Figure 2.1). For example, 2.1 (ABC294640 (Ki = 10 µM)) was the first
inhibitor with SphK2 activity that has been deployed in a variety of disease models including
lupus nephritis, diabetic nephropathy, Crohn’s disease, ulcerative colitis, and osteoarthritis.9, 10
However, it was recently shown to be a tamoxifen-like partial agonist of estrogen receptors in
breast cancer cells.11 Another inhibitor, thiazolidine-2,4-dione 2.2 (K145 (Ki = 6.4 µM)), which

41
is an analog of sphingosine was recently reported as a selective SphK2 inhibitor.12 2.2 was
shown to inhibit leukemia cell growth in vitro as well as in a xenograph mouse model.
Due to our interest in understanding the in vivo function of SphK2 and a paucity of
highly potent and selective inhibitors,12 we focused our studies in developing unique scaffolds to
achieve our goals. Our first generation inhibitor, 2.3 (VT-ME6), contained a quaternary
ammonium head group as a warhead and established that a positively charged moiety is
necessary for engaging key amino acid residues in the enzyme binding pocket.13, 14 This
compound is moderately potent (Ki = 8 µM) and displays three-fold selectivity for SphK2 over
SphK1. Subsequent improvements produced 2.4 (SLR080811), which features a 1,2,4-
oxadiazole linker and a guanylated pyrrolidine head group and possesses a Ki of 13.3 µM and 1.3
µM for SphK1 and SphK2, respectively.15 A significant discovery from these studies is that
pharmacological inhibition of SphK2 elevates blood S1P levels in mice. Further structure–
activity relationship studies on the guanidine head group revealed that the azetidine-containing
derivative 2.5 (SLP1201701) has an improved half-life of 8 h in mice (in comparison to 4 h).16 In
this report, we detail our investigations on the tail region of the guanidine-based scaffold (Figure
2.2). Our studies demonstrate that the internal phenyl ring is essential to maintain inhibitory
ClO
HN
N
2.1 (ABC294640)
O
MeS
N
O
O
NH2
Me NMe
MeMe
2.3 (VT-ME6)
2.2 (K145)
Me N
ON N
NHH2N ◊HCl
I
2.4 (SLR080811)
Me N
ON N
NHH2N◊HCl
2.5 (SLP120701)
Figure 2.1. Structure of SphK2 Inhibitors

42
activity for SphK2, the alkyl tail length has a significant effect on the potency and selectivity
towards SphK2, and that the SphK2 binding pocket cannot accommodate charged heteroatom-
bearing tail groups.
2.4 Results and Discussion
2.4.1 Inhibitor Development
The synthesis of derivatives of 2.4 with varying alkyl length as well as heterocycles
attached to the phenyl ring is shown in Schemes 2.1 and 2.2. In Scheme 2.1, 4-iodobenzonitrile
(2.6a) was cross-coupled to a series of alkynes or hydroborated intermediates under standard
Sonogashira or Suzuki–Miyaura conditions. Subsequent reaction with hydroxylamine afforded
amidoximes 2.7a–e, which were cyclized to 1,2,4-oxadiazoles 2.8a–f in the presence of HCTU
and Boc-L-proline. Deprotection with HCl and reduction of alkynyl groups with tosylhydrazine
at refluxing conditions yielded amines 2.9a–h. To install the guanidine moiety, the amines were
treated with DIEA and N,N’-Di-Boc-1H-pyrazole-1-carboxamidine for several days at room
temperature and deprotected with HCl to produce the desired derivatives 2.10a,d,f–h. A similar
synthetic strategy was employed to access the remaining phenyl/alkyl derivatives (2.13c and
2.13f–g); however, heterocycles 2.13d–e were obtained via Buchwald–Hartwig coupling
conditions as shown in Scheme 2.2. Similarly, Scheme 2.3 illustrates the synthesis of various
amidopiperazine tail surrogates 2.16a–d via Buchwald–Hartwig and acetylation conditions.
Me N
ON HN
NH
NH2
Tail
Linker
Head
Figure 2.2. Pharmacophore of guanidine-based inhibitors.
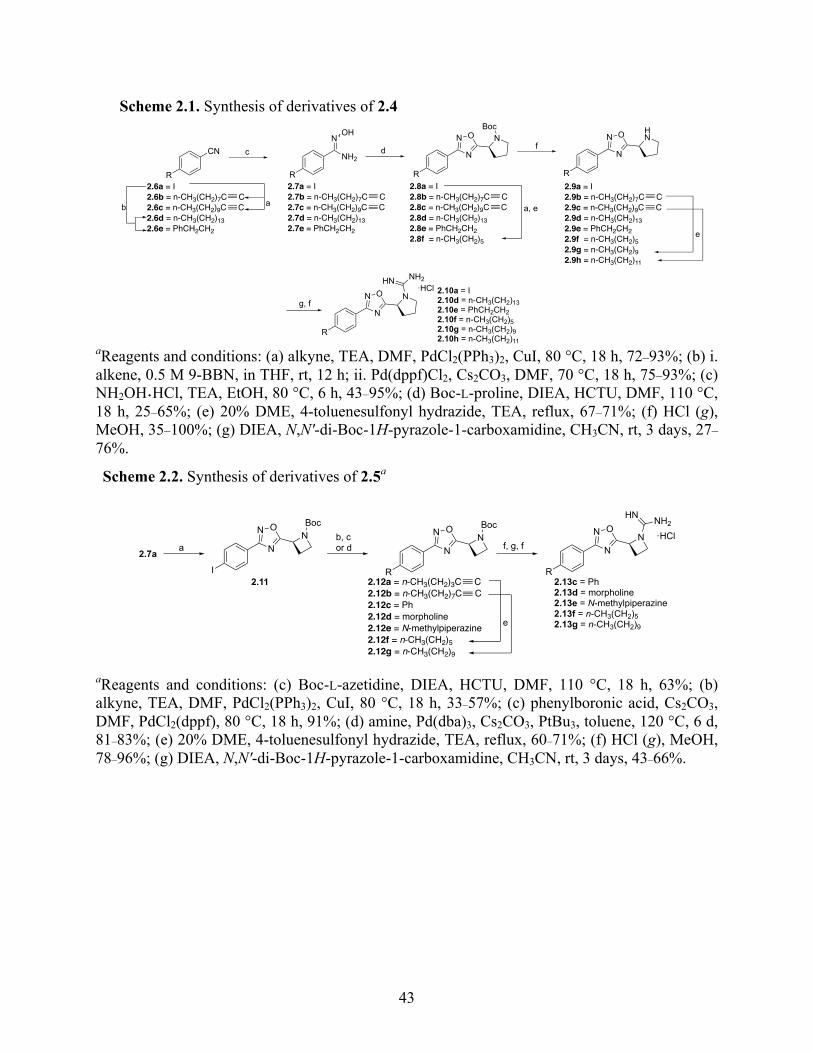
43
aReagents and conditions: (a) alkyne, TEA, DMF, PdCl2(PPh3)2, CuI, 80 °C, 18 h, 72–93%; (b) i. alkene, 0.5 M 9-BBN, in THF, rt, 12 h; ii. Pd(dppf)Cl2, Cs2CO3, DMF, 70 °C, 18 h, 75–93%; (c) NH2OH˖HCl, TEA, EtOH, 80 °C, 6 h, 43–95%; (d) Boc-L-proline, DIEA, HCTU, DMF, 110 °C, 18 h, 25–65%; (e) 20% DME, 4-toluenesulfonyl hydrazide, TEA, reflux, 67–71%; (f) HCl (g), MeOH, 35–100%; (g) DIEA, N,N'-di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 27–76%.
aReagents and conditions: (c) Boc-L-azetidine, DIEA, HCTU, DMF, 110 °C, 18 h, 63%; (b) alkyne, TEA, DMF, PdCl2(PPh3)2, CuI, 80 °C, 18 h, 33–57%; (c) phenylboronic acid, Cs2CO3, DMF, PdCl2(dppf), 80 °C, 18 h, 91%; (d) amine, Pd(dba)3, Cs2CO3, PtBu3, toluene, 120 °C, 6 d, 81–83%; (e) 20% DME, 4-toluenesulfonyl hydrazide, TEA, reflux, 60–71%; (f) HCl (g), MeOH, 78–96%; (g) DIEA, N,N'-di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 43–66%.
N
ON
R
N
HN NH2
R
N
ON NBoc
R
N
NH2
OH
R
CN
ab
R
N
ONHN
c d
a, e
f
e
g, f2.10a = I2.10d = n-CH3(CH2)132.10e = PhCH2CH22.10f = n-CH3(CH2)52.10g = n-CH3(CH2)92.10h = n-CH3(CH2)11
·HCl
2.8a = I2.8b = n-CH3(CH2)7C C2.8c = n-CH3(CH2)9C C2.8d = n-CH3(CH2)132.8e = PhCH2CH22.8f = n-CH3(CH2)5
2.7a = I2.7b = n-CH3(CH2)7C C2.7c = n-CH3(CH2)9C C2.7d = n-CH3(CH2)132.7e = PhCH2CH2
2.6a = I2.6b = n-CH3(CH2)7C C2.6c = n-CH3(CH2)9C C2.6d = n-CH3(CH2)132.6e = PhCH2CH2
2.9a = I2.9b = n-CH3(CH2)7C C2.9c = n-CH3(CH2)9C C2.9d = n-CH3(CH2)132.9e = PhCH2CH22.9f = n-CH3(CH2)52.9g = n-CH3(CH2)92.9h = n-CH3(CH2)11
Scheme 2.1. Synthesis of derivatives of 2.4
Scheme 2.2. Synthesis of derivatives of 2.5a
b, c or d N
ON
R
NBoc
e
N
ON
R
N
NH2HN
2.13c = Ph2.13d = morpholine2.13e = N-methylpiperazine2.13f = n-CH3(CH2)52.13g = n-CH3(CH2)9
f, g, fN
ON
I
NBoc
a2.7a
2.11
·HCl
2.12a = n-CH3(CH2)3C C2.12b = n-CH3(CH2)7C C2.12c = Ph2.12d = morpholine2.12e = N-methylpiperazine2.12f = n-CH3(CH2)52.12g = n-CH3(CH2)9

44
aReagents and conditions: (a) piperazine, Pd2(dba)3, PtBu3, Cs2CO3, toluene, 120 °C, 3 days, 52%; (b) acid chloride, CH2Cl2, 0 °C to rt, 2 h, 66–88%; (c) HCl (g), MeOH, 76–95%; (d) DIEA, N,N'-di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 23–74%.
Compounds 2.20 and 2.23 were synthesized as shown in Schemes 2.4 and 2.5,
respectively. 4-(3-ethoxymethyl)-5-methylbenzyl)benzonitrile 2.19 was formed in two steps via
mono-substitution of 1,3-bis(bromomethyl)-5-methylbenzene 2.17 and subsequent palladium-
catalyzed cross coupling reaction with 4-cyanophenylboronic acid to afford 2.19. Alternatively,
benzonitrile 2.22 was achieved using sodium benzenesulfonate and 2.21. Standard oxadiazole
formation, guanidylation, and deprotection afforded 2.20 and 2.23, respectively. Finally, a series
of alkyl tails directly linked to the oxadiazole ring were synthesized (Scheme 2.5). Treatment of
alkylbromides with potassium cyanide gave alkylnitriles 2.25a–c, which were converted to
amidoximes 2.26a–c. Transformation to oxadiazoles 2.27a–c was effected either by HCTU-
mediated cyclization at 110 °C or by two-step coupling/TBAF-catalyzed cyclization, which
eventually led to 2.28a–c.
aReagents and conditions: (a) NaH, EtOH, 0 °C to rt, 46%; (b) 4-cyanophenylboronic acid, Pd(PPh3)4, Na2CO3, 1:1 THF:H2O, 94%; (c) NH2OH·HCl, TEA, EtOH, 80 °C, 6 h, 71–93%; (d) Boc-L-proline, DIEA, HCTU, DMF, 110 °C, 18 h, 46–82%; (e) HCl (g), MeOH, 33–91%; (f) DIEA, N,N'-di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 53–83%.
2.8a N
ON N
NHN
Boc
2.14 2.16a = isovaleryl2.16b = 1-adamantanecarbonyl2.16c = phenylacetyl2.16d = benzyl
a bN
ON N
NN
R
Boc
c,d,c N
ON N
NN
R
HN NH2
·HCl
2.15a = isovaleryl2.15b = 1-adamantanecarbonyl2.15c = phenylacetyl2.15d = benzyl
CH3
BrBr EtO
CH3
Br EtO
CN
2.17 2.18
ba
CH3
2.19
c-f, e
CH3
EtON
ON N
NH2HN
2.20
·HCl
Scheme 2.3. Synthesis of amidopiperazine tail dervatives 2.16a–da
Scheme 2.4. Synthesis of 2.20a

45
aReagents and conditions: (a) sodium benzenesulfonate, DMF, 60 °C, 2 h, 89%; (b) NH2OH·HCl, TEA, EtOH, 80 °C, 6 h, 71–93%; (d) Boc-L-proline, DIEA, HCTU, DMF, 110 °C, 18 h, 46–82%; (e) HCl (g), MeOH, 33–91%; (f) DIEA, N,N'-Di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 53–83%.
aReagents and conditions: (a) KCN, 9:1 EtOH:H2O, 80 °C, 18 h, 20–93%; (b) NH2OH·HCl, TEA, EtOH, 80 °C, 12 h, 53–69%; (c) Boc-L-proline, DIEA, HCTU, DMF, 110 °C, 18 h, 50%; (d) Boc-L-proline, DIEA, HCTU, CH2Cl2, rt, 4 h, 57–80%; (e) 1 M TBAF, THF, rt, 1 h, 93–95%; (f) HCl (g), MeOH, 66–100%; (g) DIEA, N,N'-di-Boc-1H-pyrazole-1-carboxamidine, CH3CN, rt, 3 days, 51–71%.
2.4.2 Structure–Activity Relationship Studies and Biological Evaluation of Derivatives
With the library of putative inhibitors synthesized, the inhibitory effects of the
compounds were determined for hSphK1 and mSphK2 using a previously published protocol
(Table 1).16 Briefly, Sph and cell lysate containing recombinant SphK1 or SphK2 were incubated
with or without inhibitor in the presence of γ-[32P]ATP. After 20 minutes, the reaction mixtures
were extracted, separated using thin layer chromatography, and quantified using liquid
scintillation counting. The kinase inhibition was determined as the amount of 32P-S1P produced
as a function of inhibitor concentration. Compounds were screened at 10 µM inhibitor
concentrations.
Br
CN
2.21
aS
CN
Ph
O ON
ON
N
NHHN
Sb-e, d
2.22 2.23OO
·HCl
a bR
Br NR R NH2
NOH
2.24a = C8H172.24b = C12H252.24c = C16H33
2.25a = C8H172.25b = C12H252.25c = C16H33
2.26a = C8H172.26b = C12H252.26c = C16H33
2.28a = C8H172.28b = C12H252.28c = C16H33
N
N
O
R
NBoc
2.27a = C8H172.27b = C12H252.27c = C16H33
c or d, e f,g,fN
N
O
R
N
NHH2N ·HCl
Scheme 2.5. Synthesis of 2.23a
Scheme 2.6. Synthesis of derivatives of 2.4 lacking an internal phenyl ringa

46
As shown in Table 2.1, replacement of the octyl chain of 2.4 with iodide, phenyl or
phenethyl groups did not improve inhibitory activity (entries 1-3). Decreasing or increasing the
lipophilic alkyl tail length from hexyl to tetradecyl in two-carbon increments resulted in
compounds with similar inhibitory activity as 2.4 (entries 4-10), although the hexyl chain was
slightly less active. In cases where the kinase activity was similar to 2.4 at 10 µM, rescreening at
a more stringent inhibitor concentration (1 µM) was performed: the results indicated that none of
these analogs had improved activity compared to 2.4. We also note that the pyrrolidine and
azetidine rings have been shown to have similar potency, but with the advantage of improved in
vivo half-life for the azetidine derivatives. Interestingly, as the alkyl tail increased to a decyl
group, SphK2 selectivity decreased as SphK1 inhibition increased. However, as the chain length
increased further to a dodecyl and tetradecyl, inhibition of SphK1 decreased while maintaining
SphK2 activity. These results suggest that the lipid binding pocket in SphK2 is much larger than
that of SphK1 and is consistent with the prediction based on a crystal structure of SphK1 bound
to a SphK1-selective inhibitor.17 We next investigated the effect of the phenyl substituent next to
the 1,2,4-oxadiazole ring. Removal of this ring while maintaining the overall length of the
molecule resulted not only in diminished SphK2 selectivity but also inhibitory activity (entries
11-13). Our data indicate that the phenyl ring is necessary for selectivity and potency using this
scaffold.
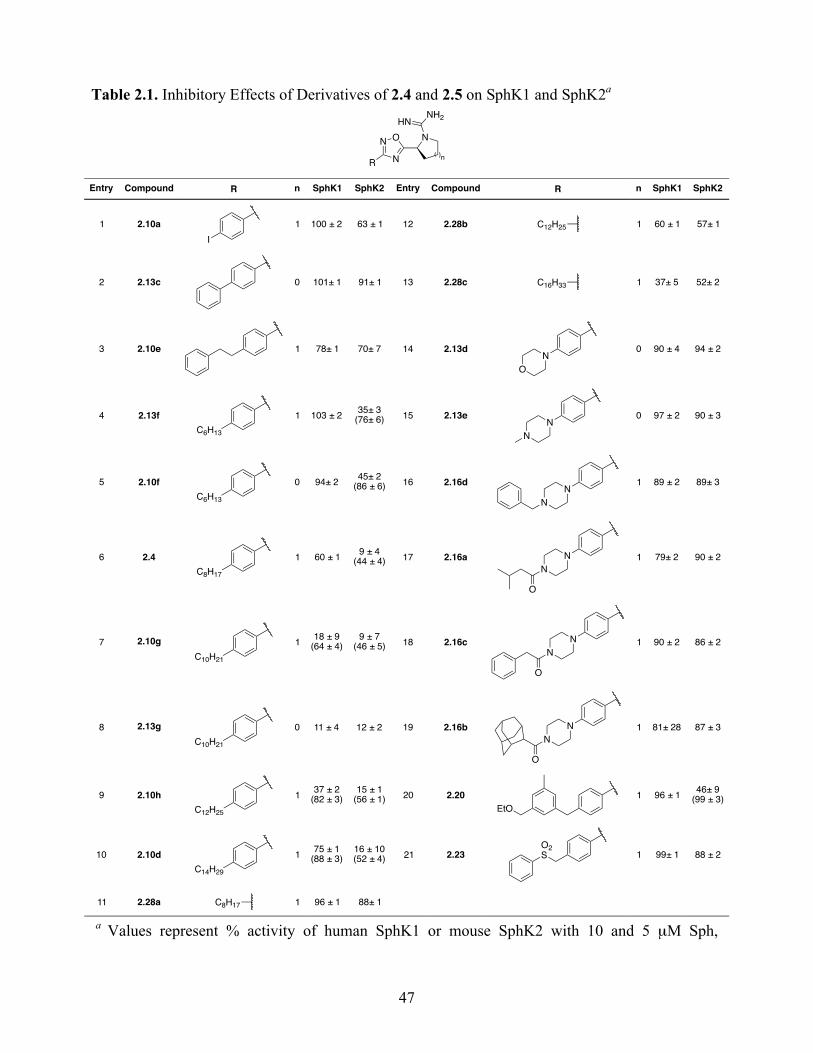
47
a Values represent % activity of human SphK1 or mouse SphK2 with 10 and 5 µM Sph,
Entry Compound R n SphK1 SphK2 Entry Compound R n SphK1 SphK2
1 2.10aI
1 100 ± 2 63 ± 1 12 2.28b C12H25 1 60 ± 1 57± 1
2 2.13c 0 101± 1 91± 1 13 2.28c C16H33 1 37± 5 52± 2
3 2.10e 1 78± 1 70± 7 14 2.13dN
O
0 90 ± 4 94 ± 2
4 2.13fC6H13
1 103 ± 2 35± 3(76± 6) 15 2.13e
NN
0 97 ± 2 90 ± 3
5 2.10fC6H13
0 94± 2 45± 2(86 ± 6) 16 2.16d
NN
1 89 ± 2 89± 3
6 2.4C8H17
1 60 ± 1 9 ± 4(44 ± 4) 17 2.16a N
N
O
1 79± 2 90 ± 2
7 2.10gC10H21
1 18 ± 9(64 ± 4)
9 ± 7(46 ± 5) 18 2.16c N
N
O
1 90 ± 2 86 ± 2
8 2.13gC10H21
0 11 ± 4 12 ± 2 19 2.16b NN
O
1 81± 28 87 ± 3
9 2.10hC12H25
1 37 ± 2(82 ± 3)
15 ± 1(56 ± 1) 20 2.20
EtO1 96 ± 1 46± 9
(99 ± 3)
10 2.10dC14H29
1 75 ± 1(88 ± 3)
16 ± 10(52 ± 4) 21 2.23
O2S 1 99± 1 88 ± 2
11 2.28a C8H17 1 96 ± 1 88± 1
N
N
O N
NH2HN
R n
Table 2.1. Inhibitory Effects of Derivatives of 2.4 and 2.5 on SphK1 and SphK2a

48
respectively, in the presence of 10 µM inhibitor. Each value is an average of two experiments. Values in parenthesis indicate compounds also assayed at 1 µM.
As there is no published crystal structure of SphK2, further modifications were necessary
for elucidation of the lipid-binding pocket. Morpholine and a series of heterocyclic ring
derivatives were synthesized to probe for potential non-covalent interactions that were not van
der Waals interactions (entries 14-19). A piperazine ring was an attractive selection because of
increased conformational rigidity and ability to function as an anchor point in which various
groups can be appended. The morpholine, N-methyl, and N-benzyl piperazine derivatives were
all found to be inactive toward SphK1 and SphK2, despite an increase in respective tail length.
As the N-methyl and N-benzyl piperazine are positively charged, their lack of inhibitory activity
can be attributed to the high likelihood that the lipid binding pocket is lined with hydrophobic
groups, as observed in published SphK1 crystal structures,17, 18 which would prevent effective
inhibitor binding. In an effort to avoid the creation of positively charged substituents, neutral
amide versions with increasing steric bulk were tested. Isovaleryl-, phenacetyl-, and
adamantylcarbonyl-substituted analogues were also inactive. As the SphK1 binding pocket is “J-
shaped”18 and the SphK2 binding pocket is also likely this conformation, the tested amides were
probably too rigid to adopt the necessary conformation needed for enzyme binding. Finally, the
trisubstituted aryl (2.20) and sulfonate (2.23) bearing groups featured in a SphK1-selective
inhibitor developed by Pfizer (PF-543) were tested but were also found to be poor inhibitors
(entries 20-21).19
2.5 Conclusions
In summary, a focused library of SphK2-selective inhibitor derivatives of 2.4 that
interrogated the lipophilic tail region of the pharmacophore were synthesized. Our studies
demonstrate the dependence of SphK2 inhibitory activity on alkyl chain length; the most optimal

49
length includes octyl and decyl substituents, which suggests an ideal ‘head-to-tail’ (positive
charge to terminal methyl group) length of approximately 18-21 atoms. Furthermore, our studies
provide evidence for the much larger lipophilic binding cavity in SphK2 over SphK1, and neither
enzyme’s binding cavity can accommodate positively charged or conformationally restricted tail
groups. In the scaffold of 2.4, the internal phenyl ring appears to be essential for activity and is
likely interacting with residues in the kinase binding pocket. These predictions can be aided by a
SphK2 crystal structure, which is currently unavailable.
2.6 Funding Sources
We acknowledge financial support by NIH (Grants R01 GM104366 and R01
GM067958).

50
2.7 References
1. Bigaud, M.; Guerini, D.; Billich, A.; Bassilana, F.; Brinkmann, V. Second Generation S1P
Pathway Modulators: Research Strategies and Clinical Developments. Biochim Biophys Acta
2014, 1841, 745–758.
2. Takuwa, N.; Du, W.; Kaneko, E.; Okamoto, Y.; Yoshioka, K.; Takuwa, Y. Tumor-
Suppressive Sphingosine-1-Phosphate Receptor-2 Counteracting Tumor-Promoting
Sphingosine-1-Phosphate Receptor-1 and Sphingosine Kinase 1. Am. J. Cancer Res. 2011, 1,
460–481.
3. Kunkel GT; Maceyka M; Milstien S; S., S. Targeting the Sphingosine-1-Phosphate Axis in
Cancer, Inflammation and Beyond. Nat Rev Drug Discov 2013, 12, 688–702.
4. Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.;
Bogdanov, M.; Alexander, D. C.; Milburn, M. V.; Ahmed, M. H.; Lin, H.; Idowu, M.; Zhang,
J.; Kato, G. J.; Abdulmalik, O. Y.; Zhang, W.; Dowhan, W.; Kellems, R. E.; Zhang, P.; Jin, J.;
Safo, M.; Tsai, A. L.; Juneja, H. S.; Xia, Y. Elevated Sphingosine-1-Phosphate Promotes
Sickling and Sickle Cell Disease Progression. J Clin Invest 2014, 124, 2750–2761.
5. Prager, B.; Spampinato, S. F.; Ransohoff, R. M. Sphingosine 1-Phosphate Signaling at the
Blood–Brain Barrier. Trends Mol. Med. 2015, 21, 354–363.
6. Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.;
Yokoshima, S.; Fukuyama, T.; Lee, V. M.; Trojanowski, J. Q.; Tomita, T.; Iwatsubo, T.
BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci.
2011, 31, 6850–6857.
7. Maceyka, M.; Sankala, H.; Hait, N. C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang,
M.; Satin, L. S.; Merrill, A. H., Jr.; Milstien, S.; Spiegel, S. SphK1 and SphK2, Sphingosine

51
Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J. Biol. Chem.
2005, 280, 37118–37129.
8. Plano, D.; Amin, S.; Sharma, A. K. Importance of Sphingosine Kinase (SphK) as a Target in
Developing Cancer Therapeutics and Recent Developments in the Synthesis of Novel SphK
Inhibitors. J Med Chem 2014, 57, 5509–5524.
9. Neubauer, H. A.; Pitson, S. M. Roles, Regulation and Inhibitors of Sphingosine Kinase 2.
FEBS J. 2013, 280, 5317–5336.
10. French, K. J.; Zhuang, Y.; Maines, L. W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J. J.;
Green, C. L.; Keller, S. N.; Smith, C. D. Pharmacology and Antitumor Activity of
ABC294640, a Selective Inhibitor of Sphingosine Kinase-2. J. Pharmacol. Exp. Ther. 2010,
333, 129–139.
11. Antoon, J. W.; White, M. D.; Meacham, W. D.; Slaughter, E. M.; Muir, S. E.; Elliott, S.;
Rhodes, L. V.; Ashe, H. B.; Wiese, T. E.; Smith, C. D.; Burow, M. E.; Beckman, B. S.
Antiestrogenic Effects of the Novel Sphingosine Kinase-2 Inhibitor ABC294640.
Endocrinology 2010, 151, 5124–5135.
12. Liu, K.; Guo, T. L.; Hait, N. C.; Allegood, J.; Parikh, H. I.; Xu, W.; Kellogg, G. E.; Grant,
S.; Spiegel, S.; Zhang, S. Biological Characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-
phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a Selective Sphingosine Kinase-2
Inhibitor and Anticancer Agent. PloS one 2013, 8, e56471.
13. Raje, M. R.; Knott, K.; Kharel, Y.; Bissel, P.; Lynch, K. R.; Santos, W. L. Design, Synthesis
and Biological Activity of Sphingosine Kinase 2 Selective Inhibitors. Bioorg. Med. Chem.
2012, 20, 183–194.

52
14. Knott, K.; Kharel, Y.; Raje, M. R.; Lynch, K. R.; Santos, W. L. Effect of Alkyl Chain
Length on Sphingosine Kinase 2 Selectivity. Bioorg. Med. Chem. Lett. 2012, 22, 6817–6820.
15. Kharel, Y.; Raje, M.; Gao, M.; Gellett, A. M.; Tomsig, J. L.; Lynch, K. R.; Santos, W. L.
Sphingosine Kinase Type 2 Inhibition Elevates Circulating Sphingosine 1-Phosphate
Biochem. J. 2012, 447, 149–157.
16. Patwardhan, N. N.; Morris, E. A.; Kharel, Y.; Raje, M. R.; Gao, M.; Tomsig, J. L.; Lynch,
K. R.; Santos, W. L. Structure-Activity Relationship Studies and in Vivo Activity of
Guanidine-Based Sphingosine Kinase Inhibitors: Discovery of Sphk1- and Sphk2-Selective
Inhibitors. J. Med. Chem. 2015, 58, 1879–1899.
17. Wang, J.; Knapp, S.; Pyne, N. J.; Pyne, S.; Elkins, J. M. Crystal Structure of Sphingosine
Kinase 1 with PF-543. ACS Med. Chem. Lett. 2014, 5, 1329–1333.
18. Wang, Z.; Min, X.; Xiao, S. H.; Johnstone, S.; Romanow, W.; Meininger, D.; Xu, H.; Liu,
J.; Dai, J.; An, S.; Thibault, S.; Walker, N. Molecular Basis of Sphingosine Kinase 1 Substrate
Recognition and Catalysis. Structure 2013, 21, 798–809.
19. Schnute, M. E.; McReynolds, M. D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J. W.; Hall,
T.; Chrencik, J.; Kraus, M.; Cronin, C. N.; Saabye, M.; Highkin, M. K.; Broadus, R.; Ogawa,
S.; Cukyne, K.; Zawadzke, L. E.; Peterkin, V.; Iyanar, K.; Scholten, J. A.; Wendling, J.;
Fujiwara, H.; Nemirovskiy, O.; Wittwer, A. J.; Nagiec, M. M. Modulation of Cellular S1P
Levels with a Novel, Potent and Specific Inhibitor of Sphingosine Kinase-1. Biochem. J.
2012, 444, 79–88.

53
3 Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2
Selective Inhibitors: Design, Synthesis, and in Vivo Activity
3.1 Contributions
The work in this chapter was done primarily by the author. The author synthesized and
characterized all final compounds and their corresponding intermediates. Dr. Yugesh Kharel of
the University of Virginia Department of Pharmacology conducted biological analyses of the
reported compounds. Dr. Anne M. Brown conducted the molecular modeling experiments. The
final manuscript was written by the author and Dr. Webster Santos. This chapter is a reprint with
permission from the Journal of Medicinal Chemistry [Childress, E. S.; Kharel, Y.; Brown, A. M.;
Bevan, D. R.; Lynch, K. R.; Santos, W. L. Transforming Sphingosine Kinase 1 Inhibitors into
Dual and Sphingosine Kinase 2 Selective Inhibitors: Design, Synthesis, and in Vivo Activity. J.
Med. Chem. 2017, 60, 33933–3957, © 2015 American Chemical Society].
3.2 Abstract
Sphingosine 1-phosphate (S1P) is a pleiotropic signaling molecule that interacts with its
five G-protein coupled receptors S1PR1–5 to regulate cell growth and survival and has been
implicated in a variety of diseases including cancer and sickle cell disease. As the key mediators
in the synthesis of S1P, sphingosine kinase (SphK) isoforms 1 and 2 have attracted attention as
viable targets for pharmaceutical inhibition. In this article, we describe the design, synthesis, and
biological evaluation of aminothiazole-based guanidine inhibitors of SphK. Surprisingly,
combining features of reported SphK1 inhibitors generated SphK1/2 dual inhibitor 3.20l
(SLC4011540) (hSphK1 Ki = 120 nM, hSphK2 Ki = 90 nM) and SphK2 inhibitor 3.20dd
(SLC4101431) (Ki = 90 nM, 100-fold SphK2 selectivity). These compounds effectively decrease

54
S1P levels in vitro. In vivo administration of 3.20dd validated that inhibition of SphK2 increases
blood S1P levels.
3.3 Introduction
Sphingosine 1-phosphate (S1P) is a ubiquitous cellular signaling molecule that has been
implicated in a variety of diseases including cancer,1-4 fibrosis,5-7 Alzheimer’s disease,8, 9 sickle
cell disease,10, 11 and viral infections such as Chikungunya virus.12 S1P interacts with proteins
both within and outside of the cell. As an extracellular ligand, S1P promotes cell migration and
survival by binding to five G-protein coupled receptors, S1PR1-5.13-16 Although its function as
an intracellular ligand is less well defined, S1P is reported to control epigenetic modifications
through regulation of HDAC1/2 activity via sphingosine kinase isotype 2 (SphK2) and alter
erythrocyte glycolysis to increase O2 release under hypoxic conditions.17-19 S1P is synthesized by
the phosphorylation of sphingosine (Sph) by SphK, which exists as two isotypes: SphK1 and
SphK2. SphK1 and SphK2 share approximately 50% sequence identity20 but differ with respect
to their function, in part due to their differing localization within the cell. SphK1 is a cytosolic
enzyme that promotes cell survival and proliferation,21, 22 whereas some SphK2 is found in the
nucleus17, 18 but can relocate to the cytosol on phosphorylation.23, 24 Depending on this
localization, SphK2 can promote either apoptosis12, 13 or cell proliferation.17, 18, 25, 26 Although
isotype-specific SphK null mice are viable, fertile, and phenotypically unremarkable,27 SphK1-
null mice have about a 2-fold reduction in blood S1P levels, whereas SphK2-null mice have 2-4
fold increased blood S1P levels.28-32 However, ablation of both SphK isotype genes in the mouse
is embryonically lethal in mid gestation as a consequence of complications during vascular and
neurological development.27, 32

55
The therapeutic potential of drugging the S1P pathway was realized by the approval of
fingolimod by the U. S. Food and Drug Administration for the treatment of multiple sclerosis. 33,
34 Fingolimod is a pro-drug that is phosphorylated by SphK2 to act as a functional antagonist of
S1P receptors, S1PR1/3. Other approaches have also been employed to manipulate S1P activity
including targeting SphKs to control S1P synthesis.3, 35-37 There are currently multiple reports of
potent SphK1-selective inhibitors and SphK1/SphK2-dual inhibitors (Figure 3.1). Inhibitors such
as SphK1-selective inhibitor 3.1 (PF-543)38, 39 and SphK1/SphK2-dual inhibitor 3.5 (SKI-II)40-42
have been co-crystallized with SphK1 and have been useful in developing next generation
inhibitors.43, 44 Indeed, medicinal chemists at Amgen improved 3.5 to realize a selective SphK1
inhibitor, 3.2 (Amgen 23),45 while preserving the aminothiazole region. While 3.5 is recently
reported to have off-target activity against dihydroceramide desaturase,46 3.1 has shown promise
for disease states such as sickle cell disease, where inflammation, cell sickling, and hemolysis
were reduced in treated mouse models.11
In contrast with SphK1 inhibitors, highly potent SphK2-selective inhibitors are lacking.
Early SphK2 inhibitors (3.8 (SLR080811),29, 47 3.9 (ABC294640),48 and 3.10 (K145)49, 50), which
have low micromolar potency and are modestly selective vs SphK1, are active in cultured cells,
as measured by their ability to lower cellular S1P levels. Among these, 3.9 has been employed in
a variety of animal disease models, including ulcerative colitis,51 Crohn’s disease,52
ischemia/reperfusion injury,53 osteoarthritis,54 and colon cancer.55 However, 3.9 has recently
been reported to have an off-target effect of acting as a tamoxifen-like molecule with the
estrogen receptor.56 The development of improved inhibitors that are SphK2 specific will help in
understanding the physiological function of SphK2 in vivo. Recently, SphK2 was shown to play
a role in endothelial cell barrier integrity as well as attenuation of kidney fibrosis through
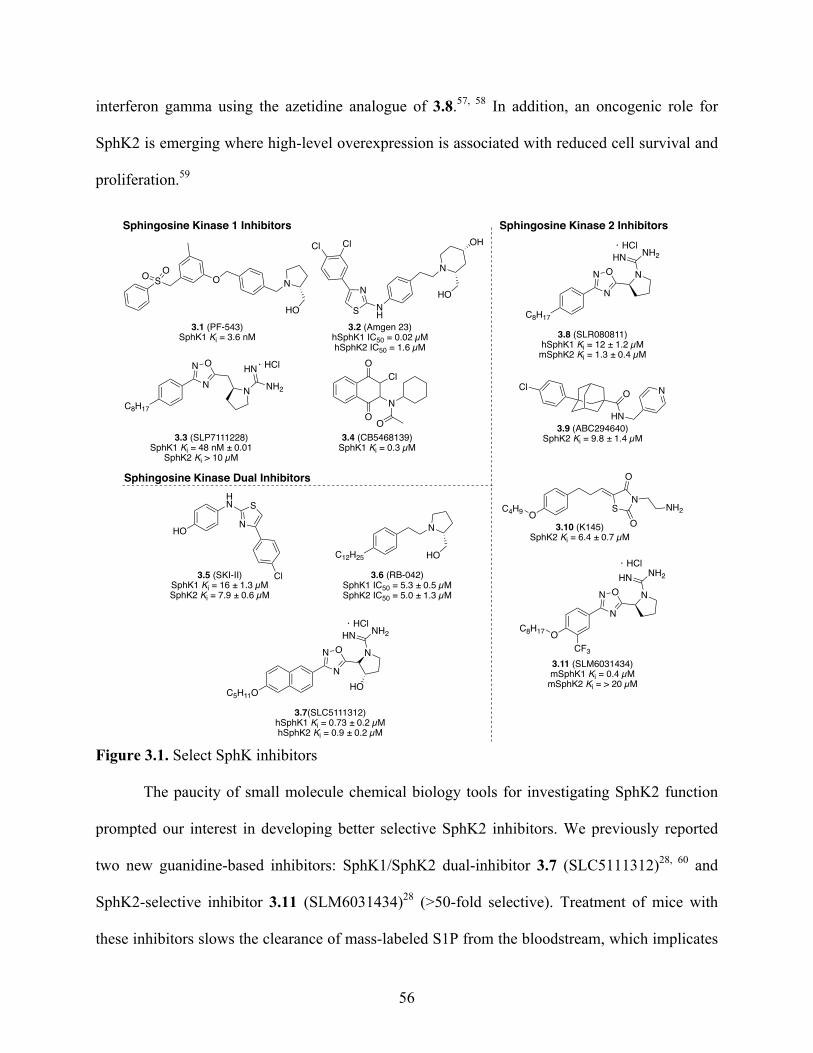
56
interferon gamma using the azetidine analogue of 3.8.57, 58 In addition, an oncogenic role for
SphK2 is emerging where high-level overexpression is associated with reduced cell survival and
proliferation.59
Figure 3.1. Select SphK inhibitors
The paucity of small molecule chemical biology tools for investigating SphK2 function
prompted our interest in developing better selective SphK2 inhibitors. We previously reported
two new guanidine-based inhibitors: SphK1/SphK2 dual-inhibitor 3.7 (SLC5111312)28, 60 and
SphK2-selective inhibitor 3.11 (SLM6031434)28 (>50-fold selective). Treatment of mice with
these inhibitors slows the clearance of mass-labeled S1P from the bloodstream, which implicates
HN
N
S
Cl
HO
C12H25
3.6 (RB-042)SphK1 IC50 = 5.3 ± 0.5 µMSphK2 IC50 = 5.0 ± 1.3 µM
N
HO
N
OH
HONH
N
S
Cl
3.2 (Amgen 23)hSphK1 IC50 = 0.02 µMhSphK2 IC50 = 1.6 µM
O
HN
NCl
3.9 (ABC294640)SphK2 Ki = 9.8 ± 1.4 µM
O SN
NH2
O
OC4H9
3.10 (K145) SphK2 Ki = 6.4 ± 0.7 µM
Sphingosine Kinase Dual Inhibitors
Sphingosine Kinase 1 Inhibitors Sphingosine Kinase 2 Inhibitors
C8H17
N
ON
3.3 (SLP7111228)SphK1 Ki = 48 nM ± 0.01
SphK2 Ki > 10 µM
N
HN
NH2
HCl
OCF3
N
ON N
3.11 (SLM6031434)mSphK1 Ki = 0.4 µM
mSphK2 Ki = > 20 µM
HN NH2HCl
C8H17
Cl
N
ON N
3.7(SLC5111312)hSphK1 Ki = 0.73 ± 0.2 µMhSphK2 Ki = 0.9 ± 0.2 µM
HN NH2HCl
C5H11O HO
3.5 (SKI-II) SphK1 Ki = 16 ± 1.3 µM SphK2 Ki = 7.9 ± 0.6 µM
O
O
Cl
N
O3.4 (CB5468139)
SphK1 Ki = 0.3 µM
S O N
HO
O O
3.1 (PF-543)SphK1 Ki = 3.6 nM
C8H17
N
ON N
3.8 (SLR080811)hSphK1 Ki = 12 ± 1.2 µMmSphK2 Ki = 1.3 ± 0.4 µM
HN NH2HCl

57
SphK2 in the disposal of circulating S1P. Although the mechanism whereby SphK2 influences
S1P clearance from blood is currently unclear, these results highlight the need for more potent
and selective SphK2 and SphK1/SphK2 dual inhibitors to explore this phenomenon further.
Herein, we report the synthesis, structure-activity relationship study, and biological evaluation of
guanidine-based aminothiazoles with in vivo activity as selective SphK2 inhibitors.
3.4 Results and Discussion
3.4.1 Inhibitor Design and Development
Our group reported the synthesis of 3.3 (SLP7111228),47 an SphK1-selective inhibitor
with a Ki of 48 nM and >100-fold selectivity for SphK1 over SphK2 for both the rat and human
enzymes. This compound was found to reduce S1P levels in both cultured human U937 leukemia
cells and in the bloodstream of rats. The latter result recapitulates the phenotype of SphK1 null
mice.32, 47 In developing this molecule, the addition of a methylene unit between the 1,2,4-
oxadiazole and pyrrolidine rings was key in converting a 10-fold SphK2-selective inhibitor (3.8)
into a >100-fold SphK1-selective inhibitor. In this work, we sought to improve 3.3’s potency and
selectivity further by replacing the octyl chain with an aminothiazole group, as present in SphK1
inhibitors such as 3.2 (Figure 3.2). Docking of 3.2 into the binding pocket of a homology model
of
C13H27 O PO
O OOH
NH3
S1P
C8H17
N
ON
N
HN
NH2
・HCl
3.3
NH
N
ON
N
HN
NH2
・HCl
S
NR
This Work
tail group
head group
key selectivity switch to SphK1
C8H17
N
ON N
3.8
HN NH2・HCl
Figure 3.2. Tail group modifications toward the development of new “J-shaped” SphK1 inhibitors.

58
SphK2 and superimposition with 3.2’s “aminothiazole tail” linked to an oxadiazole phenyl ring
suggested binding interactions mimicking 3.2 (Figure 3). We hypothesized that these altered
“tail” groups, represented by the 14-18 carbon aliphatic groups of sphingoid bases, would create
inhibitors that can adopt the necessary “J-shape” for strong binding interactions with the
sphingosine binding pocket, as established by published co-crystal structures of SphK1 with
aminothiazole-based inhibitors 3.2 and 3.5.44, 45 Gustin et al. recently developed potent
aminothiazole-based SphK1/SphK2 dual inhibitors derived from 3.5, including 3.2.45 Although
most compounds reported were dual inhibitors, some compounds in their series show a bias
toward SphK1 inhibition with attractive pharmacokinetic properties. Vogt and co-workers
subsequently reported the synthesis of aminothiazole-based derivatives of 3.5 that are
SphK1/SphK2-dual inhibitors with several inhibitors showing a slight bias for SphK2
selectivity.61 However, these molecules show only micromolar potency, and extensive biological
Figure 3.3. Superimposition of the lowest energy docked poses of 3.2 (purple sticks, colored by atom) and an aminothiazole analogue of 3.3 (teal sticks, color by atom) in a homology model of SphK2. Key residues in the binding cavity are shown as gray sticks, ATP is shown in green, and the overall protein structure is shown as a gray cartoon for perspective.

59
studies have yet to be reported. Aurelio et al. also recently reported the synthesis of derivatives
of 3.5 that show limited success as SphK1/SphK2 dual inhibitors and SphK1- and SphK2-
selective inhibitors.62 Because of the prominence of aminothiazole moieties in SphK inhibitor
scaffolds, we incorporated aminothiazole moieties into our guanidine-based scaffold, specifically
to achieve SphK1-selective inhibitors.
3.4.2 Inhibitor Synthesis
In prior studies,47, 63 we established that the guanidine, 1,2,4-oxadiazole, and internal
phenyl ring moieties were key features of the sphingosine kinase inhibitor scaffold. Therefore,
we focused our attention on the “tail” region by appending aminothiazoles decorated with
diverse aryl structures. The synthesis of aryl-substituted aminothiazoles is shown in Scheme 3.1.
4-Trifluoromethylacetamide benzonitrile 3.13 was synthesized by the acetylation of 4-
aminobenzonitrile 3.12 with trifluoroacetic anhydride. Benzonitrile 3.13 was then reacted with
hydroxylamine hydrochloride and triethylamine in ethanol in a microwave reactor to yield
amidoxime 3.14, which was reacted with homoproline using HCTU and Hunig’s base at 100 ºC
to afford 1,2,4-oxadiazole 3.15. The trifluoroacetate group was removed using lithium hydroxide
to afford amine 3.16. Treatment of 3.16 with thiocarbodiimidazole followed by ammonia
provided the key intermediate thiourea 3.17. Aminothiazoles 3.18a–ee were produced by
reacting the required α-bromoketone with thiourea 3.17 and Hunig’s base in ethanol in a
microwave reactor. The Boc group was removed with trifluoroacetic acid and immediately
reacted with N,N’-di-Boc-1H-pyrazole-1-carboxamidine in a microwave reactor to afford bis-
Boc-protected guanidines 3.19a–ee. Bubbling HCl gas subsequently provided the desired
guanidine derivatives 3.20a–ee (see Tables 3.1 and 3.2 for structures).

60
a Reagents and conditions: (a) trifluoroacetic anhydride, DCM, 0 ºC to rt, 19 h, 88%; (b) NH2OH.HCl, TEA, ACN, 150 ºC microwave, 6 min, 57%; (c) Boc-L-homoproline, HCTU, DIEA, DMF, rt to 100 ºC, 4 h, 67%; (d) 1:1 1 M LiOH:MeOH, 100 ºC, 3 h, 82%; (e) Thio-CDI, THF, rt, 4 h; (f) NH3(g), 1 min, 86%; (g) α-bromoketone, DIEA, EtOH, 100 ºC, microwave, 5 min, 30–90%; (h) 1:1 TFA:DCM; (i) N, N'-di-Boc-1H-pyrazole-1-carboxamidine, DIEA, ACN, 50 ºC, microwave, 2 h, 26–87%; (j) HCl (g), MeOH, 90–100%.
Biphenyl derivatives were synthesized as outlined in Scheme 3.2. Using either compound
3.18f or 3.18i, Suzuki-Miyaura cross-coupling with various aryl boronic acids yielded biaryl
aminothiazoles 3.21a–d. The standard synthetic sequence of deprotection, guanylation, and
deprotection was then followed to yield the desired biaryl aminothiazole derivatives 3.23 a–d.
aReagents and conditions: (a) aryl boronic acid, PdCl2(dppf), Cs2CO3, DMF, 150 ºC microwave, 90 min; (b) 1:1 TFA:DCM; (c) N, N'-di-Boc-1H-pyrazole-1-carboxamidine, DIEA, ACN, 50 ºC, microwave, 2 h; (d) HCl (g), MeOH.
Scheme 3.1. Synthesis of substituted aminothiazole derivatives 3.20a–eea
Scheme 3.2. Synthesis of biphenyl aminothiazole derivatives 3.23a–da
CN
NH
F3C
O
NH
N
NH2
OH
F3C
OCN
H2N NH
F3C
O N
ON
H2N
N
ONe, f
NH
N
ON
NBocH2N
S
NH
N
ON
NBoc
S
NR
j
3.12 3.13 3.14 3.15
3.163.17 3.18a–ee
3.19a–ee 3.20a–ee
NH
N
ON
N
S
NR NHBoc
BocN
NH
N
ON
N
S
NR NH2
HN
NBoc
NBoc
a b c
d
h, i
g
HCl
3.18f, R1 = Br, R2 = H3.18i, R1 = H, R2 = Br
3.22 a–d, 37–75%
3.23 a: R3 = H, R4 = Ph, 93% 3.23 b: R3 = 4-CF3-Ph, R4 = H, 100%3.23 c: R3 = 3-CF3-Ph, R4 = H, 100%3.23 d: R3 = 4-F-Ph, R4 = H, 90%
3.21 a–d, 53–88%
NH
N
ON
NBoc
S
N
HCl
NH
N
ON
N
S
N NH2
HN
NH
N
ON
N
S
N NHBoc
BocN
R3
R4
a b, c
R3
R4
d R1
R2
NH
N
ON
NBoc
S
NR1
R2

61
To assess the significance of the hydrogen bond effect in this series of aminothiazole, a
comparison of activity between a representative aminothiazole example of the series (3.20k,
Table 3.1) and its analogous oxathiazole was done. The corresponding oxathiazole derivative
was synthesized as presented in Scheme 3.3. 4-tert-Butoxy benzonitrile 3.25 was synthesized via
nucleophilic aromatic substitution of 4-fluorobenzonitrile 3.24 with 1 M potassium tert-butoxide.
Benzonitrile 3.25 was then reacted with hydroxylamine hydrochloride and triethylamine in
ethanol using a microwave reactor to yield its amidoxime, which was then reacted with
homoproline using HCTU and Hunig’s base at 100 ºC to afford 1,2,4-oxadiazole 3.26. Phenol
derivative 3.27 was achieved via treatment of 1,2,4-oxadiazole 3.26 with trifluoroacetic acid
followed by reprotection with Boc-anhydride. Nucleophilic aromatic substitution with phenol
derivative 3.27 on 2,4-dibromothiazole afforded oxathiazole 3.28. Suzuki–Miyaura cross-
coupling with 4-trifluoromethylphenyl boronic acid yielded the desired oxathiazole 3.29. The
standard synthetic sequence of deprotection, guanylation, and deprotection was followed for
compound 3.29 to yield the desired oxathiazole derivative 3.31.

62
aReagents and conditions: (a) 1 M KOtBu, THF, reflux, 15 h; (b) NH2OH.HCl, TEA, EtOH, 150 ºC microwave, 6 min; (c) Boc-L-homoproline, HCTU, DIEA, DMF, rt to 100 ºC, 4 h; (d) 1:1 TFA:DCM; (e) di-tert-butyl dicarbonate, TEA, dioxane, 0 ºC, 1 h; (f) 2,5-dibromothiazole, K2CO3, DMF, 135 ºC, 1 h; (g) 4-trifluoromethylphenyl boronic acid, PdCl2(dppf), Cs2CO3, DMF, 150 ºC, microwave, 90 min; (h) N, N'-di-Boc-1H-pyrazole-1-carboxamidine, DIEA, ACN, 50 ºC, microwave, 2 h; (i) HCl (g), MeOH.
To further probe the significance of the hydrogen bond effect of the amino moiety in
3.20dd, its methylated derivative was synthesized as illustrated in Scheme 3.4. Biaryl
aminothiazole 3.18dd was methylated with methyl iodine in refluxing acetone to yield 32. The
standard synthetic sequence of deprotection, guanylation, and deprotection was then followed to
yield the desired methylated aminothiazole derivative 3.34.
aReagents and conditions: (a) MeI, K2CO3, acetone, reflux, 17 h; (b) 1:1 TFA:DCM; (c) N, N'-di-Boc-1H-pyrazole-1-carboxamidine, DIEA, ACN, 50 ºC, microwave, 2 h; (d) HCl (g), MeOH.
O
N
ON
NBoc
3.26, 70%F
CN
O
CNa
3.25, 66%
N
ON
NBocHO
O
N
ON
NBoc
S
NBr
3.28, 39% 3.29, 53%
3.30, 83% 3.31, 100%O
N
ON
N
S
N NHBoc
BocN
O
N
ON
N
S
N NH2
HN
3.24 3.27, 40%
N
ONNBoc
O
N
S
F3C
b, c d, e
f g
i F3CF3Ch HCl
NH
N
ON
NBoc
S
N
3.18dd
3.33, 73% 3.34, 100%
HCl
N
N
ON
N
S
N NHBoc
BocN
N
N
ON
N
S
N NH2
HN
N
N
ON
NBoc
S
N
3.32, 73%H3C
CH3CH3
a
b, c d
Scheme 3.3. Synthesis of the oxathiazole analogue of inhibitor 3.20ka
Scheme 3.4. Synthesis of the methylated analogue of 3.20dda

63
3.4.3 Structure–Activity Relationship Studies and Biological Evaluation of Derivatives
With the goal of defining the structure-activity profile of SphKs, a focused library of
aminothiazoles bearing a guanidine group was synthesized. These analogues were assayed using
a previously described protocol.47, 64 In particular, synthesized inhibitors were tested at 0.3 µM
with human recombinant enzymes (Tables 1 & 2). All aminothiazoles vary with respect to the
number and type of substituents on the appended aryl ring as well as the substitution pattern on
this ring.
As shown in Table 3.1, substituting the thiazole ring with an unsubstituted phenyl ring
(3.20a) resulted in poor inhibition of either SphK isotype in our assay. Replacement of the
phenyl ring with either a 4’ (3.20b) or 3’ (3.20c) pyridyl ring also produced compounds that
were inactive with both SphK isotypes. Halogen substituents at the para position of the phenyl
ring inhibited both enzymes but with slight selectivity for SphK2. The selectivity and potency for
SphK2 was unanticipated because these compounds were expected to inhibit SphK1, as previous
work from our laboratories demonstrated that the homologated guanidine-pyrrolidine
headgroup47 generates an SphK1 inhibitor when the “tail” is an unsubstituted octyl group.
Furthermore, aminothiazoles in the Amgen series are also selective for SphK1,45 although their
scaffold differed in other aspects also. The identity of the halogen atom—fluorine, chlorine, or
bromine—did not greatly affect the compounds’ (3.20d–f) SphK2 potency reducing the enzyme
activity to ~ 50%. Moving the halogen group to the meta or ortho position (3.20g–j) resulted in a
retention of SphK2 potency, but with a loss in SphK2 selectivity, producing only moderately
SphK2-selective inhibitors.
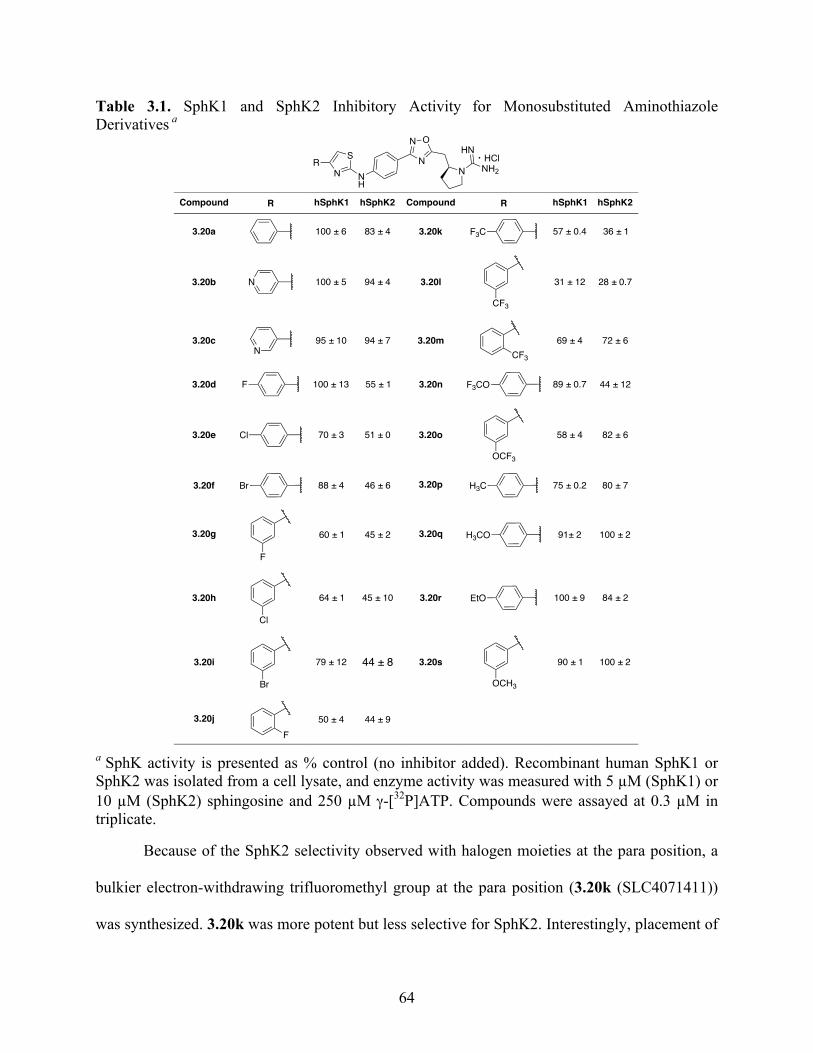
64
Table 3.1. SphK1 and SphK2 Inhibitory Activity for Monosubstituted Aminothiazole Derivatives a
a SphK activity is presented as % control (no inhibitor added). Recombinant human SphK1 or SphK2 was isolated from a cell lysate, and enzyme activity was measured with 5 µM (SphK1) or 10 µM (SphK2) sphingosine and 250 µM γ-[32P]ATP. Compounds were assayed at 0.3 µM in triplicate.
Because of the SphK2 selectivity observed with halogen moieties at the para position, a
bulkier electron-withdrawing trifluoromethyl group at the para position (3.20k (SLC4071411))
was synthesized. 3.20k was more potent but less selective for SphK2. Interestingly, placement of
Compound R hSphK1 hSphK2 Compound R hSphK1 hSphK2
3.20a 100 ± 6 83 ± 4 3.20k F3C 57 ± 0.4 36 ± 1
3.20b N 100 ± 5 94 ± 4 3.20l
CF3
31 ± 12 28 ± 0.7
3.20cN
95 ± 10 94 ± 7 3.20mCF3
69 ± 4 72 ± 6
3.20d F 100 ± 13 55 ± 1 3.20n F3CO 89 ± 0.7 44 ± 12
3.20e Cl 70 ± 3 51 ± 0 3.20o
OCF3
58 ± 4 82 ± 6
3.20f Br 88 ± 4 46 ± 6 3.20p H3C 75 ± 0.2 80 ± 7
3.20g
F
60 ± 1 45 ± 2 3.20q H3CO 91± 2 100 ± 2
3.20h
Cl
64 ± 1 45 ± 10 3.20r EtO 100 ± 9 84 ± 2
3.20i
Br
79 ± 12 44 ± 8 3.20s
OCH3
90 ± 1 100 ± 2
3.20jF
50 ± 4 44 ± 9
・HClNH
N
ON
N
S
NR NH2
HN

65
the trifluoromethyl group at the meta position (3.20l) produced a more potent SphK inhibitor
(28% inhibition). However, this molecule showed no selectivity for SphK1 vs SphK2. The ortho
trifluoromethyl analogue (3.20m) did not improve potency or selectivity. These results suggest
that the key binding interactions were lost with the ortho-substituted derivative likely due to a
loss of CF3 interactions with residues Cys533, His556, and Tyr566 that are found at the end of
the hydrophobic tunnel of the SphK2 binding pocket, which was shown60 by our group to be
important for strong inhibitor binding.
Replacement of the electron-withdrawing groups with electron-donating groups—methyl
(3.20n), methyl ether (3.20o), and ethyl ether (3.20p)—at the para position markedly diminished
SphK2 inhibition (<30%) (Table 3.1). Likewise, positioning the methyl ether to the meta
position (3.20q) did not improve inhibitory activity at either enzyme isotype. A trifluoromethyl
ether group was then tested at the para position for increased lipophilicity and to reestablish
deactivating electronics65, 66 to the aryl ring. The resulting molecule (3.20r (SLC4081418))
decreased SphK2 activity to ~ 40%, which is likely due to a reestablishment of the fluorine
bonding interactions mentioned (vide supra). Good selectivity was also observed with this
molecule, as SphK1 activity was diminished only by ~10%. However, shifting the
trifluoromethyl ether moiety from the para position to the meta position (3.20s) led to a switch in
SphK potency and selectivity. Collectively, these results indicate that placement of electron-
donating groups on the aryl ring is unfavorable for SphK2 inhibition.
The effects of disubstitution on the aryl ring of the aminothiazole were also explored
(Table 3.2). Although poor inhibition activities were observed with monosubstituted aryl rings
containing traditional electron-donating groups, we were curious about the effects of having

66
a SphK activity is presented as % control (no inhibitor added). Recombinant human SphK1 or SphK2 was isolated from a cell lysate, and enzyme activity was measured with 5 µM (SphK1) or 10 µM (SphK2) sphingosine and 250 µM γ-[32P]ATP. Compounds were assayed at 0.3 µM in triplicate.
disubstituted aryl rings. Compounds with either two methyl (3.20t) or two methyl ether moieties
(3.20u) showed minimal activity. Placement of a methyl ether at the para position and the
chlorine moiety at the meta position (3.20v) led to modest SphK2 activity (~70%) and no SphK1
inhibition. In contrast, disubstitution with electron-withdrawing groups reestablished SphK2
Table 3.2. SphK1 and SphK2 Inhibitory Activity for Disubstituted and Bulky Aminothiazole Derivatives a
Compound R X hSphK1 hSphK2 Compound R X hSphK1 hSphK2
3.20tH3C
H3CNH 91 ± 4 67 ± 12 3.20cc
F3C
F
NH 65 ± 2 48 ± 1
3.20uH3CO
H3CONH 100 ± 7 100 ± 7 3.20dd NH 100 ± 2 27 ± 12
3.20vH3CO
ClNH 100 ± 0.2 71 ± 11 3.23a NH 92 ± 8 73 ± 7
3.20wF
FNH 51 ± 5 51 ± 5 3.23b F NH 82 ± 9 31 ± 7
3.20xCl
ClNH 51 ± 4 46 ± 11 3.23c F3C NH 100 ± 2 96 ± 1
3.20yF
F3CNH 58 ± 5 33 ± 5 3.23d
F3CNH 99 ± 6 85 ± 3
3.20z
F
F
NH 68 ± 7 43 ± 8 3.20eeO
NH 68 ± 7 67 ± 5
3.20aa
F
F3C
NH 51 ± 4 30 ± 15 3.31 F3C O 62 ± 4 55 ± 2
3.20bb
F3C
F3C
NH 100 ± 4 63 ± 4 3.34 NCH3 100 ± 5 100 ± 5
HCl
X
N
ON
N
S
N NH2
HNR

67
potency, particularly fluorines (3.20w, 3.20z), chlorine (3.20x), or a combination of fluorine with
a trifluoromethyl group (3.20y (SLC4091423), 3.20aa (SLC4091424), 3.20cc). The substitution
pattern for these molecules did not significantly affect their SphK2 potency; however, they did
show SphK1/SphK2 dual inhibitor activity, affording good SphK2 inhibition (50–70%) and
modest to good SphK1 inhibition (30–50%). Disubstitution of the aryl ring with two
trifluoromethyl moieties (3.20bb) led to a loss in compound potency, but this loss in potency was
met with a gain in selectivity, as it was inactive toward SphK1 at concentrations up to 1 µM.
Together, these results reinforce the preference for an electron-deficient aryl ring.
Our previous studies63, 67 suggest that the SphK2 lipid-binding pocket is larger than that
of SphK1. To explore the effects of bulky moieties on the aminothiazole ring, biphenyl
derivatives were synthesized and tested (Table 3.2). We discovered that the para-substituted
biphenyl derivative (3.20dd) was not only potent toward SphK2 but also selective for SphK2.
The meta-substituted biphenyl derivative (3.23a) essentially lost activity. However, a fluorine on
the 4-position (3.23b) maintained the potency and selectivity observed with 3.20dd. Attempts to
substitute 3.20dd with a trifluoromethyl moiety at either the 4- (3.23c) or 3-position (3.23d) on
the biphenyl ring led to compounds inactive in both SphKs, suggesting that the molecules may
either be too large for the binding pocket or have poor solubility (cLogP = 6.78). In an effort to
introduce additional bulk onto the aminothiazole, a benzofuran moiety (3.20ee) was synthesized
but was found to be ineffective as an inhibitor. Collectively, these results suggest that the
biphenyl moiety has the optimal binding interaction as well as bend to fit in the J-shaped
hydrophobic tunnel.
In addition to investigating the effects of various aryl groups on the aminothiazoles, we
also explored the effects of modifying the exocyclic nitrogen of the aminothiazoles (Table 3.2).

68
The co-crystal structure of SphK1 with 3.5 reported by Wang et al. notes a key hydrogen
bonding interaction between the aminothiazole NH and SphK1 threonine-196.44 Although our
scaffold would most likely fit slightly deeper into the SphK1 binding pocket, we probed the
significance of this hydrogen bonding capability by either replacing the NH with an ether linkage
(3.31) or methylating (3.34) the amino group. N-Me derivative 3.34 was found to be completely
inactive in both SphKs up to 1 µM, whereas the ether derivative 3.31 showed moderate activity
towards both SphKs, decreasing SphK1 and SphK2 activity by ~ 45% at 0.3 µM. These results
suggest that the amino moiety plays a key role in enzyme binding likely through hydrogen
bonding (compare 3.34 with 3.20dd) although we cannot rule out the possibility that the loss in
SphK2 potency may also be due to steric effects created from methylation. Comparison of 3.31
with amino moiety 3.20k shows a loss in SphK2 potency with equipotent SphK1 inhibition,
indicating that the molecules’ hydrogen bonding role (donor vs. acceptor) at this position is more
significant in SphK2 than SphK1.
To quantify the aminothiazoles’ activity toward SphK1 and SphK2, we determined the
inhibitory constant (Ki) of the most potent compounds in the library (Table 3.3). Consistent with
the results of the initial screen (Tables 3.1 and 3.2), fluoro and trifluoromethyl positional isomers
of disubstituted aryls 3.20y and 3.20aa had good activity with both enzymes and partial
selectivity toward SphK2 (~5 fold) (entries 1 and 2). Fortunately, monosubstitution with a meta-

69
acLogP was calculated for the protonated inhibitor with Chemdraw Professional 13.0.
trifluoromethyl group (3.20l) by removal of the fluorine atom from 3.20y or 3.20aa resulted in
improved binding inhibition (entry 3). 3.20l is a dual inhibitor with Ki values of 120 nM and 90
nM for SphK1 and SphK2, respectively. Moving the trifluoromethyl group to the para position
had a negative inhibitory effect as expected (entry 4). However, switching to a trifluoromethyl
ether on the para position (3.20r) improved the Ki towards SphK2 (250 nM) while decreasing the
inhibitory activity against SphK1 (Ki 5 µM), affording 20-fold selectivity toward SphK2 (entry
5). The introduction of a more lipophilic and larger substituent such as a phenyl ring on the para
position (3.20dd) resulted in a potent and selective SphK2 inhibitor (Ki 90 nM). To the best of
our knowledge, 3.20dd is the most potent SphK2 reported to date; this compound is 100-fold
selective toward SphK2 vs SphK1. In comparing compounds 3.20k, 3.20r, and 3.20dd, it is
Table 3.3. Ki and cLogPa Values of Selected Inhibitors of SphK1 and SphK2 Entry Compound Structure hSphK1
Ki (µM)hSphK2 Ki (µM)
hSphK2 Selectivity cLogP
1 3.20yHCl
NH
N
ON
N
S
N NH2
HNF
F3C
0.7 ± 0.05 0.17 ± 0.15 4 5.33
2 3.20aaHCl
NH
N
ON
N
S
N NH2
HN
F3C
F
0.8 ± 0.07 0.17 ± 0.17 5 5.33
3 3.20lHCl
NH
N
ON
N
S
N NH2
HN
F3C
0.12 ± 0.04 0.09 ± 0.01 1 5.18
4 3.20kHCl
NH
N
ON
N
S
N NH2
HNF3C 0.82 ± 0.25 0.39 ± 0.09 2 5.18
5 3.20rHCl
NH
N
ON
N
S
N NH2
HNF3CO 5 ± 0.22 0.25 ± 0.10 20 5.42
6 3.20dd HCl
NH
N
ON
N
S
N NH2
HN9 ± 2 0.09 ± 0.02 100 6.18
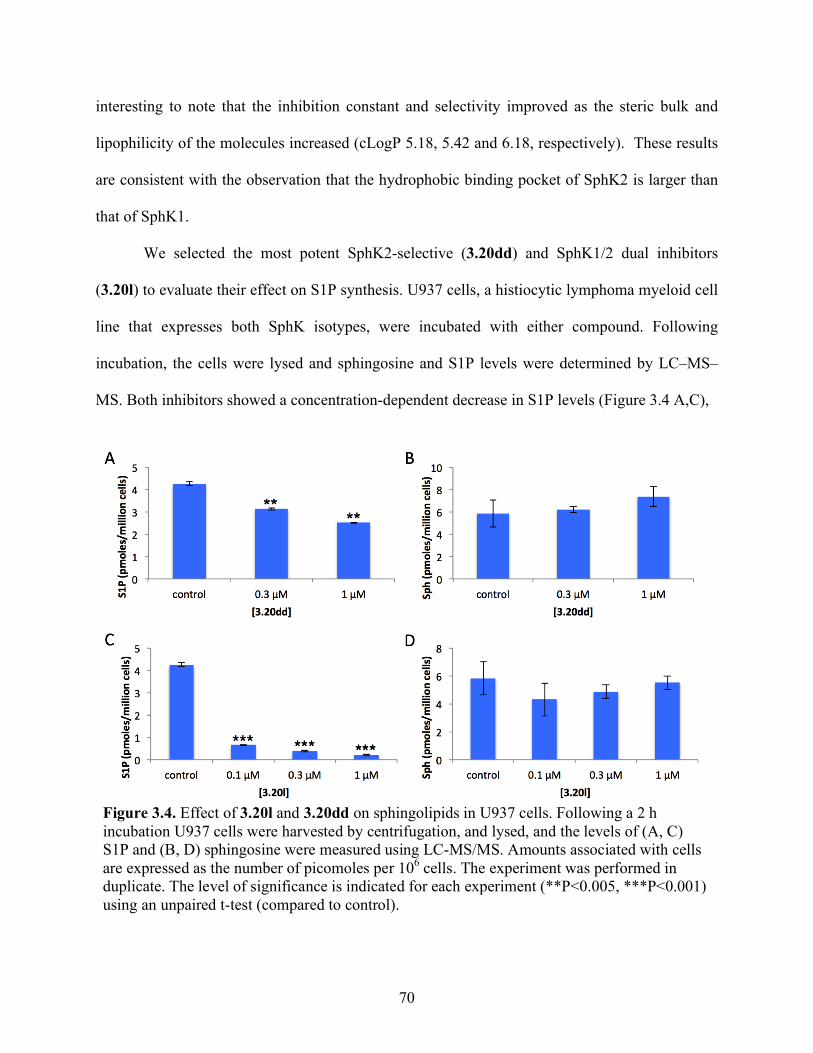
70
interesting to note that the inhibition constant and selectivity improved as the steric bulk and
lipophilicity of the molecules increased (cLogP 5.18, 5.42 and 6.18, respectively). These results
are consistent with the observation that the hydrophobic binding pocket of SphK2 is larger than
that of SphK1.
We selected the most potent SphK2-selective (3.20dd) and SphK1/2 dual inhibitors
(3.20l) to evaluate their effect on S1P synthesis. U937 cells, a histiocytic lymphoma myeloid cell
line that expresses both SphK isotypes, were incubated with either compound. Following
incubation, the cells were lysed and sphingosine and S1P levels were determined by LC–MS–
MS. Both inhibitors showed a concentration-dependent decrease in S1P levels (Figure 3.4 A,C),
Figure 3.4. Effect of 3.20l and 3.20dd on sphingolipids in U937 cells. Following a 2 h incubation U937 cells were harvested by centrifugation, and lysed, and the levels of (A, C) S1P and (B, D) sphingosine were measured using LC-MS/MS. Amounts associated with cells are expressed as the number of picomoles per 106 cells. The experiment was performed in duplicate. The level of significance is indicated for each experiment (**P<0.005, ***P<0.001) using an unpaired t-test (compared to control).
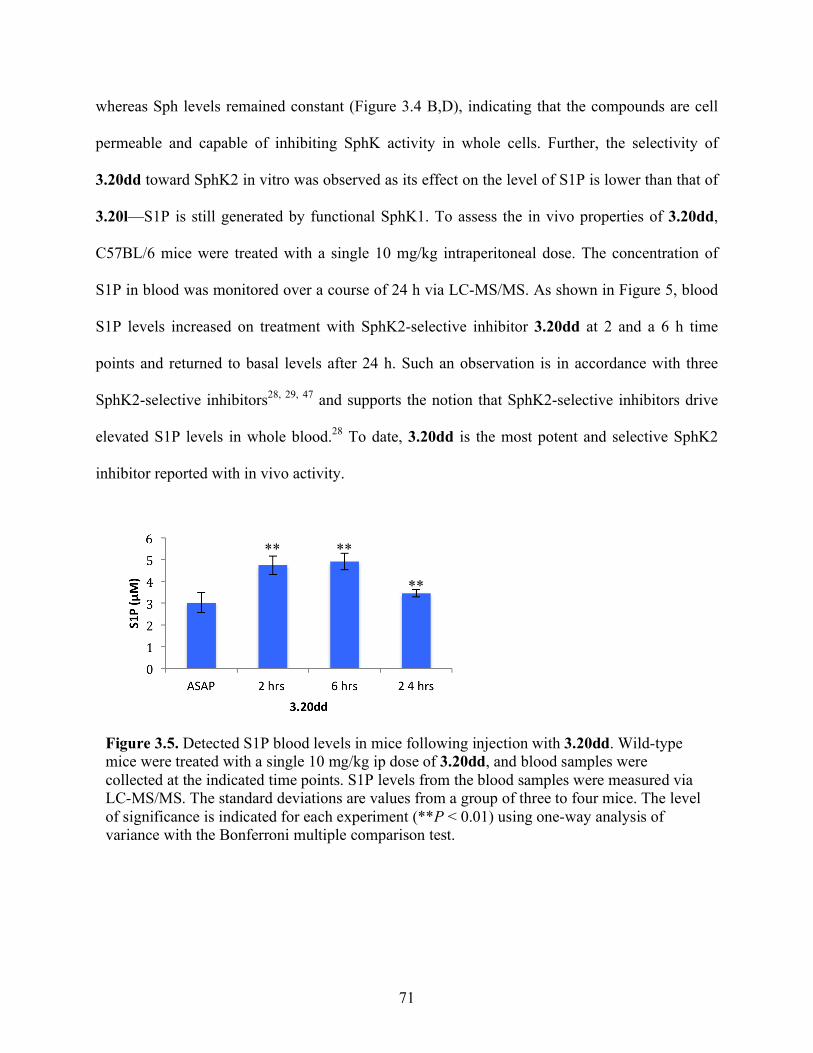
71
whereas Sph levels remained constant (Figure 3.4 B,D), indicating that the compounds are cell
permeable and capable of inhibiting SphK activity in whole cells. Further, the selectivity of
3.20dd toward SphK2 in vitro was observed as its effect on the level of S1P is lower than that of
3.20l—S1P is still generated by functional SphK1. To assess the in vivo properties of 3.20dd,
C57BL/6 mice were treated with a single 10 mg/kg intraperitoneal dose. The concentration of
S1P in blood was monitored over a course of 24 h via LC-MS/MS. As shown in Figure 5, blood
S1P levels increased on treatment with SphK2-selective inhibitor 3.20dd at 2 and a 6 h time
points and returned to basal levels after 24 h. Such an observation is in accordance with three
SphK2-selective inhibitors28, 29, 47 and supports the notion that SphK2-selective inhibitors drive
elevated S1P levels in whole blood.28 To date, 3.20dd is the most potent and selective SphK2
inhibitor reported with in vivo activity.
Figure 3.5. Detected S1P blood levels in mice following injection with 3.20dd. Wild-type mice were treated with a single 10 mg/kg ip dose of 3.20dd, and blood samples were collected at the indicated time points. S1P levels from the blood samples were measured via LC-MS/MS. The standard deviations are values from a group of three to four mice. The level of significance is indicated for each experiment (**P < 0.01) using one-way analysis of variance with the Bonferroni multiple comparison test.
** ** **

72
3.5 Conclusions
Herein, we disclose the discovery and development of the most potent and selective
SphK2 inhibitor reported to date. The scaffold contains guanidine head and aminothiazole tail
groups. A surprising result of our studies is that principles obtained from earlier SphK1
inhibitors—insertion of a methylene unit between the oxadiazole and pyrrolidine ring of 3.3 and
aminothiazole from 3.2—generated a potent and selective SphK2 inhibitor. Unfortunately, the X-
ray crystal structure of SphK2 is not yet available. Thus, docking of inhibitors in the binding site
using a validated homology model of SphK2 has been utilized to facilitate the development of
inhibitors. Structure-activity relationship studies of these inhibitors indicate that potent inhibition
of both SphK1 and SphK2 necessitates an electron-deficient phenyl ring, but these substituents
likely benefit from interacting with residues Cys533, His556 and Tyr566 at the end of the
binding pocket. Our investigations also support the importance of hydrogen bonding with the
exocyclic NH of the aminothiazole ring; removal of hydrogen bonding capacity resulted in a
significant loss in activity.
Biological analysis of aminothiazole inhibitors 3.20l and 3.20dd revealed that they
effectively lower S1P levels in U937 cells. In vivo study demonstrated that SphK2-selective
3.20dd caused elevated blood S1P levels in wild type mice, recapitulating our previous
findings28, 29, 47, 60 with SphK2 selective inhibitors. Collectively, our work provides a novel
chemical biology approach toward selective SphK2 and dual SphK inhibition. We expect that
these studies will aid in elucidating the in vivo function of SphK2 as well as the development of
improved SphK inhibitors.

73
3.6 Funding Sources
This work was supported by NIH (grants R01 GM104366 and R01 GM121075) and
Alzheimer’s & Related Dieases Research Award Fund.

74
3.7 References
1. Heffernan-Stroud, L. A.; Obeid, L. M. Sphingosine Kinase 1 in Cancer. Adv. Cancer Res.
2013, 117, 201–235.
2. Pyne, N. J.; Tonelli, F.; Lim, K. G.; Long, J. S.; Edwards, J.; Pyne, S. Sphingosine 1-
Phosphate Signalling in Cancer. Biochem. Soc. Trans. 2012, 40, 94–100.
3. Plano, D.; Amin, S.; Sharma, A. K. Importance of Sphingosine Kinase (Sphk) as a Target in
Developing Cancer Therapeutics and Recent Developments in the Synthesis of Novel
SphK Inhibitors. J Med Chem 2014, 57, 5509–5524.
4. Pyne, N. J.; Pyne, S. Sphingosine 1-Phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–
503.
5. Schwalm, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine-1-Phosphate: A Janus-Faced Mediator
of Fibrotic Diseases. Biochim. Biophys. Acta 2013, 1831, 239–250.
6. Yang, L.; Yue, S.; Yang, L.; Liu, X.; Han, Z.; Zhang, Y.; Li, L. Sphingosine
Kinase/Sphingosine 1-Phosphate (S1p)/S1p Receptor Axis Is Involved in Liver Fibrosis-
Associated Angiogenesis. J. Hepatol. 2013, 59, 114–123.
7. Gellings Lowe, N.; Swaney, J. S.; Moreno, K. M.; Sabbadini, R. A. Sphingosine-1-Phosphate
and Sphingosine Kinase Are Critical for Transforming Growth Factor-Beta-Stimulated
Collagen Production by Cardiac Fibroblasts. Cardiovasc. Res. 2009, 82, 303–312.
8. Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.;
Yokoshima, S.; Fukuyama, T.; Lee, V. M.; Trojanowski, J. Q.; Tomita, T.; Iwatsubo, T.
BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci.
2011, 31, 6850–6857.

75
9. Prager, B.; Spampinato, S. F.; Ransohoff, R. M. Sphingosine 1-Phosphate Signaling at the
Blood–Brain Barrier. Trends Mol. Med. 2015, 21, 354–363.
10. Proia, R. L.; Hla, T. Emerging Biology of Sphingosine-1-Phosphate: Its Role in
Pathogenesis and Therapy. J. Clin. Invest. 2015, 125, 1379–1387.
11. Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.;
Bogdanov, M.; Alexander, D. C.; Milburn, M. V.; Ahmed, M. H.; Lin, H.; Idowu, M.;
Zhang, J.; Kato, G. J.; Abdulmalik, O. Y.; Zhang, W.; Dowhan, W.; Kellems, R. E.;
Zhang, P.; Jin, J.; Safo, M.; Tsai, A. L.; Juneja, H. S.; Xia, Y. Elevated Sphingosine-1-
Phosphate Promotes Sickling and Sickle Cell Disease Progression. J. Clin. Invest. 2014,
124, 2750–2761.
12. Reid, S. P.; Tritsch, S. R.; Kota, K.; Chiang, C. Y.; Dong, L.; Kenny, T.; Brueggemann, E.
E.; Ward, M. D.; Cazares, L. H.; Bavari, S. Sphingosine Kinase 2 Is a Chikungunya Virus
Host Factor Co-Localized with the Viral Replication Complex. Emerg. Microbes Infect.
2015, 4, e61.
13. Rosen, H.; Gonzalez-Cabrera, P. J.; Sanna, M. G.; Brown, S. Sphingosine 1-Phosphate
Receptor Signaling. Annu. Rev. Biochem. 2009, 78, 743–768.
14. Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-Phosphate Signalling. Development 2014,
141, 5–9.
15. Blaho, V. A.; Hla, T. An Update on the Biology of Sphingosine 1-Phosphate Receptors. J.
Lipid Res. 2014, 55, 1596–1608.
16. Dyckman, A. J. Modulators of Sphingosine-1-Phosphate Pathway Biology: Recent
Advances of Sphingosine-1-Phosphate Receptor 1 (S1P1) Agonists and Future
Perspectives. J. Med. Chem. 2017, DOI: 10.1021/acs.jmedchem.6b01575.

76
17. Hait, N. C.; Allegood, J.; Maceyka, M.; Strub, G. M.; Harikumar, K. B.; Singh, S. K.; Luo,
C.; Marmorstein, R.; Kordula, T.; Milstien, S.; Spiegel, S. Regulation of Histone
Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325, 1254–1257.
18. Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine
Kinase 2 Is a Nuclear Protein and Inhibits DNA Synthesis. J. Biol. Chem. 2003, 278,
46832–46839.
19. Sun, K.; Zhang, Y.; D'Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.;
Huang, A.; Wen, Y. E.; Bogdanov, M. V.; Vila, A.; O'Brien, J.; Kellems, R. E.; Dowhan,
W.; Subudhi, A. W.; Jameson-Van Houten, S.; Julian, C. G.; Lovering, A. T.; Safo, M.;
Hansen, K. C.; Roach, R. C.; Xia, Y. Sphingosine-1-Phosphate Promotes Erythrocyte
Glycolysis and Oxygen Release for Adaptation to High-Altitude Hypoxia. Nat. Commun.
2016, 7, 12086.
20. Liu, H.; Sugiura, M.; Nava, V. E.; Edsall, L. C.; Kono, K.; Poulton, S.; Milstien, S.;
Kohama, T.; Spiegel, S. Molecular Cloning and Functional Characterization of a Novel
Mammalian Sphingosine Kinase Type 2 Isoform. J. Biol. Chem. 2000, 275, 19513–
19520.
21. Kunkel, G. T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the Sphingosine-1-
Phosphate Axis in Cancer, Inflammation and Beyond. Nat. Rev. Drug Discov. 2013, 12,
688–702.
22. Maceyka, M.; Harikumar, K. B.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate
Signaling and Its Role in Disease. Trends Cell Biol. 2012, 22, 50–60.

77
23. Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S. K.; Jahangeer, S.; Okada, T.;
Nakamura, S. Protein Kinase D-Mediated Phosphorylation and Nuclear Export of
Sphingosine Kinase 2. J. Biol. Chem. 2007, 282, 27493–27502.
24. Okada, T.; Ding, G.; Sonoda, H.; Kajimoto, T.; Haga, Y.; Khosrowbeygi, A.; Gao, S.; Miwa,
N.; Jahangeer, S.; Nakamura, S. Involvement of N-Terminal-Extended Form of
Sphingosine Kinase 2 in Serum-Dependent Regulation of Cell Proliferation and
Apoptosis. J. Biol. Chem. 2005, 280, 36318–36325.
25. Neubauer, H. A.; Pitson, S. M. Roles, Regulation and Inhibitors of Sphingosine Kinase 2.
FEBS J. 2013, 280, 5317–5336.
26. Sankala, H. M.; Hait, N. C.; Paugh, S. W.; Shida, D.; Lepine, S.; Elmore, L. W.; Dent, P.;
Milstien, S.; Spiegel, S. Involvement of Sphingosine Kinase 2 in p53-Independent
Induction of p21 by the Chemotherapeutic Drug Doxorubicin. Cancer Res. 2007, 67,
10466–10474.
27. Hait, N. C.; Oskeritzian, C. A.; Paugh, S. W.; Milstien, S.; Spiegel, S. Sphingosine Kinases,
Sphingosine 1-Phosphate, Apoptosis and Diseases. Biochim. Biophys. Acta 2006, 1758,
2016–2026.
28. Kharel, Y.; Morris, E. A.; Congdon, M. D.; Thorpe, S. B.; Tomsig, J. L.; Santos, W. L.;
Lynch, K. R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate
Levels. J. Pharmacol. Exp. Ther. 2015, 355, 23–31.
29. Kharel, Y.; Raje, M.; Gao, M.; Gellett, A. M.; Tomsig, J. L.; Lynch, K. R.; Santos, W. L.
Sphingosine Kinase Type 2 Inhibition Elevates Circulating Sphingosine 1-Phosphate.
Biochem. J. 2012, 447, 149–157.

78
30. Maceyka, M.; Sankala, H.; Hait, N. C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.;
Zhang, M.; Satin, L. S.; Merrill, A. H., Jr.; Milstien, S.; Spiegel, S. SphK1 and SphK2,
Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J.
Biol. Chem. 2005, 280, 37118–37129.
31. Allende, M. L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu,
R.; Rosenbach, M.; Keohane, C. A.; Mandala, S.; Spiegel, S.; Proia, R. L. Mice Deficient
in Sphingosine Kinase 1 Are Rendered Lymphopenic by FTY720. J. Biol. Chem. 2004,
279, 52487–52492.
32. Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G. F.; Spiegel, S.; Proia, R. L. Essential
Role for Sphingosine Kinases in Neural and Vascular Development. Mol. Cell Biol. 2005,
25, 11113–11121.
33. Cohen, J. A.; Barkhof, F.; Comi, G.; Hartung, H. P.; Khatri, B. O.; Montalban, X.; Pelletier,
J.; Capra, R.; Gallo, P.; Izquierdo, G.; Tiel-Wilck, K.; de Vera, A.; Jin, J.; Stites, T.; Wu,
S.; Aradhye, S.; Kappos, L. Oral Fingolimod or Intramuscular Interferon for Relapsing
Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 402–415.
34. Kappos, L.; Radue, E. W.; O'Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.;
Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; Burtin, P. A Placebo-Controlled Trial
of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–
401.
35. Pitman, M. R.; Costabile, M.; Pitson, S. M. Recent Advances in the Development of
Sphingosine Kinase Inhibitors. Cell Signal 2016, 28, 1349-1363.
36. Lynch, K. R.; Thorpe, S. B.; Santos, W. L. Sphingosine Kinase Inhibitors: A Review of
Patent Literature (2006-2015). Exp. Op. Ther. Pat. 2016, 26, 1409–1416.

79
37. Vogt, D.; Stark, H. Therapeutic Strategies and Pharmacological Tools Influencing S1P
Signaling and Metabolism. Med. Res. Rev. 2017, 37, 3–51.
38. Schnute, M. E.; McReynolds, M. D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J. W.; Hall,
T.; Chrencik, J.; Kraus, M.; Cronin, C. N.; Saabye, M.; Highkin, M. K.; Broadus, R.;
Ogawa, S.; Cukyne, K.; Zawadzke, L. E.; Peterkin, V.; Iyanar, K.; Scholten, J. A.;
Wendling, J.; Fujiwara, H.; Nemirovskiy, O.; Wittwer, A. J.; Nagiec, M. M. Modulation
of Cellular S1P Levels with a Novel, Potent and Specific Inhibitor of Sphingosine
Kinase-1. Biochem. J. 2012, 444, 79–88.
39. Schnute, M. E.; McReynolds, M. D.; Carroll, J.; Chrencik, J.; Highkin, M. K.; Iyanar, K.;
Jerome, G.; Rains, J. W.; Saabye, M.; Scholten, J. A.; Yates, M.; Nagiec, M. M.
Discovery of a Potent and Selective Sphingosine Kinase 1 Inhibitor through the
Molecular Combination of Chemotype-Distinct Screening Hits. J. Med. Chem. 2017, 60,
2562–2572.
40. French, K. J.; Schrecengost, R. S.; Lee, B. D.; Zhuang, Y.; Smith, S. N.; Eberly, J. L.; Yun,
J. K.; Smith, C. D. Discovery and Evaluation of Inhibitors of Human Sphingosine
Kinase. Cancer Res. 2003, 63, 5962–5969.
41. Gao, G.; Peterson, U. K.; Smith, R. A.; Smith, C. D. Characterization of Isoenzyme-
Selective Inhibitors of Human Sphingosine Kinases. PLoS One 2012, 7, 1–11.
42. Orr Gandy, K. A.; Obeid, L. M. Targeting the Sphingosine Kinase/Sphingosine 1-Phosphate
Pathway in Disease: Review of Sphingosine Kinase Inhibitors. Biochim. Biophys. Acta
2013, 1831, 157–166.
43. Wang, J.; Knapp, S.; Pyne, N. J.; Pyne, S.; Elkins, J. M. Crystal Structure of Sphingosine
Kinase 1 with Pf-543. ACS Med. Chem. Lett. 2014, 5, 1329–1333.

80
44. Wang, Z.; Min, X.; Xiao, S. H.; Johnstone, S.; Romanow, W.; Meininger, D.; Xu, H.; Liu,
J.; Dai, J.; An, S.; Thibault, S.; Walker, N. Molecular Basis of Sphingosine Kinase 1
Substrate Recognition and Catalysis. Structure 2013, 21, 798–809.
45. Gustin, D. J.; Li, Y.; Brown, M. L.; Min, X.; Schmitt, M. J.; Wanska, M.; Wang, X.;
Connors, R.; Johnstone, S.; Cardozo, M.; Cheng, A. C.; Jeffries, S.; Franks, B.; Li, S.;
Shen, S.; Wong, M.; Wesche, H.; Xu, G.; Carlson, T. J.; Plant, M.; Morgenstern, K.; Rex,
K.; Schmitt, J.; Coxon, A.; Walker, N.; Kayser, F.; Wang, Z. Structure Guided Design of
a Series of Sphingosine Kinase (SphK) Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23,
4608–4616.
46. Cingolani, F.; Casasampere, M.; Sanllehí, P.; Casas, J.; Bujons, J.; Fabrias, G. Inhibition of
Dihydroceramide Desaturase Activity by the Sphingosine Kinase Inhibitor SkI II. J. Lipid
Res. 2014, 55, 1711–1720.
47. Patwardhan, N. N.; Morris, E. A.; Kharel, Y.; Raje, M. R.; Gao, M.; Tomsig, J. L.; Lynch,
K. R.; Santos, W. L. Structure-Activity Relationship Studies and in Vivo Activity of
Guanidine-Based Sphingosine Kinase Inhibitors: Discovery of SphK1- and SphK2-
Selective Inhibitors. J. Med. Chem. 2015, 58, 1879–1899.
48. French, K. J.; Zhuang, Y.; Maines, L. W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J. J.;
Green, C. L.; Keller, S. N.; Smith, C. D. Pharmacology and Antitumor Activity of
Abc294640, a Selective Inhibitor of Sphingosine Kinase-2. J. Pharmacol. Exp. Ther.
2010, 333, 129-139.
49. Liu, K.; Guo, T. L.; Hait, N. C.; Allegood, J.; Parikh, H.; Xu, W.; Kellogg, G. E.; Grant, S.;
Spiegel, S.; Zhang, S. Biological Characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-

81
phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a Selective Sphingosine Kinase-2
Inhibitor and Anticancer Agent. PLoS One 2013, 8, 1–13.
50. Santos, W. L.; Lynch, K. R. Drugging Sphingosine Kinases. ACS Chem. Biol. 2015, 10,
225–233.
51. Maines, L. W.; Fitzpatrick, L. R.; French, K. J.; Zhuang, Y.; Xia, Z.; Keller, S. N.; Upson, J.
J.; Smith, C. D. Suppression of Ulcerative Colitis in Mice by Orally Available Inhibitors
of Sphingosine Kinase. Dig. Dis. Sci. 2008, 53, 997–1012.
52. Maines, L. W.; Fitzpatrick, L. R.; Green, C. L.; Zhuang, Y.; Smith, C. D. Efficacy of a
Novel Sphingosine Kinase Inhibitor in Experimental Crohn's Disease.
Inflammopharmacology 2010, 18, 73–85.
53. Shi, Y.; Rehman, H.; Ramshesh, V. K.; Schwartz, J.; Liu, Q.; Krishnasamy, Y.; Zhang, X.;
Lemasters, J. J.; Smith, C. D.; Zhong, Z. Sphingosine Kinase-2 Inhibition Improves
Mitochondrial Function and Survival after Hepatic Ischemia-Reperfusion. J. Hepatol.
2012, 56, 137–145.
54. Maines, L.; Fitzpatrick, L.; French, K.; Zhuang, Y.; Xia, Z.; Keller, S.; Upson, J.; Smith, C.
Suppression of Ulcerative Colitis in Mice by Orally Available Inhibitors of Sphingosine
Kinase. Dig. Dis. Sci. 2008, 53, 997–1012.
55. Chumanevich, A. A.; Poudyal, D.; Cui, X.; Davis, T.; Wood, P. A.; Smith, C. D.; Hofseth,
L. J. Suppression of Colitis-Driven Colon Cancer in Mice by a Novel Small Molecule
Inhibitor of Sphingosine Kinase. Carcinogenesis 2010, 31, 1787–1793.
56. Antoon, J. W.; White, M. D.; Meacham, W. D.; Slaughter, E. M.; Muir, S. E.; Elliott, S.;
Rhodes, L. V.; Ashe, H. B.; Wiese, T. E.; Smith, C. D.; Burow, M. E.; Beckman, B. S.

82
Antiestrogenic Effects of the Novel Sphingosine Kinase-2 Inhibitor ABC294640.
Endocrinology 2010, 151, 5124–5135.
57. Dimasi, D. P.; Pitson, S. M.; Bonder, C. S. Examining the Role of Sphingosine Kinase-2 in
the Regulation of Endothelial Cell Barrier Integrity. Microcirculation 2016, 23, 248–265.
58. Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K. R.; Chroscicki, P.; Foley, L. S.;
Balogun, Z. A.; Alexander, K. J.; Park, H.; Lynch, K. R.; Rosin, D. L.; Okusa, M. D.
Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis Via Ifn-Γ. J. Am. Soc.
Nephrol. 2017, 28, 1145–1161.
59. Neubauer, H. A.; Pham, D. H.; Zebol, J. R.; Moretti, P. A. B.; Peterson, A. L.; Leclercq, T.
M.; Chan, H.; Powell, J. A.; Pitman, M. R.; Samuel, M. S.; Bonder, C. S.; Creek, D. J.;
Gliddon, B. L.; Pitson, S. M. An Oncogenic Role for Sphingosine Kinase 2. Oncotarget
2016, 7, 64886–64899.
60. Congdon, M. D.; Kharel, Y.; Brown, A. M.; Lewis, S. N.; Bevan, D. R.; Lynch, K. R.;
Santos, W. L. Structure–Activity Relationship Studies and Molecular Modeling of
Naphthalene-Based Sphingosine Kinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7,
229–234.
61. Vogt, D.; Weber, J.; Ihlefeld, K.; Bruggerhoff, A.; Proschak, E.; Stark, H. Design, Synthesis
and Evaluation of 2-Aminothiazole Derivatives as Sphingosine Kinase Inhibitors. Bioorg.
Med. Chem. 2014, 22, 5354–5367.
62. Aurelio, L.; Scullino, C. V.; Pitman, M. R.; Sexton, A.; Oliver, V.; Davies, L.; Rebello, R.
J.; Furic, L.; Creek, D. J.; Pitson, S. M.; Flynn, B. L. From Sphingosine Kinase to
Dihydroceramide Desaturase: A Structure–Activity Relationship (SAR) Study of the

83
Enzyme Inhibitory and Anticancer Activity of 4-((4-(4-chlorophenyl)thiazol-2-
yl)amino)phenol (SkI-II). J. Med. Chem. 2016, 59, 965–984.
63. Congdon, M. D.; Childress, E. S.; Patwardhan, N. N.; Gumkowski, J.; Morris, E. A.; Kharel,
Y.; Lynch, K. R.; Santos, W. L. Structure–Activity Relationship Studies of the Lipophilic
Tail Region of Sphingosine Kinase 2 Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25,
4956–4960.
64. Kharel, Y.; Mathews, T. P.; Kennedy, A. J.; Houck, J. D.; Macdonald, T. L.; Lynch, K. R. A
Rapid Assay for Assessment of Sphingosine Kinase Inhibitors and Substrates. Anal.
Biochem. 2011, 411, 230–235.
65. Leroux, F. R.; Manteau, B.; Vors, J. P.; Pazenok, S. Trifluoromethyl Ethers-Synthesis and
Properties of an Unusual Substituent. Beilstein J. Org. Chem. 2008, 4, 1–13.
66. Sheppard, W. A. The Effect of Fluorine Substitution on the Electronic Properties of Alkoxy,
Alkylthio and Alkylsulfonyl Groups. J. Am. Chem. Soc. 1963, 85, 1314–1318.
67. Knott, K.; Kharel, Y.; Raje, M. R.; Lynch, K. R.; Santos, W. L. Effect of Alkyl Chain
Length on Sphingosine Kinase 2 Selectivity. Bioorg. Med. Chem. Lett. 2012, 22, 6817–
6820.

84
4 Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential
4.1 Contributions
This chapter is an adaptation of a manuscript currently being written by the author,
Stephanie J. Alexopoulos of the University of New South Wales, Dr. Kyle Hoehn of the
University of New South Wales, and Dr. Webster Santos for publication in a peer-reviewed
journal.
4.2 Introduction
Cellular respiration is a physiological process with a fundamental goal of producing
energy in the form of ATP. It is comprised of four main steps: glycolysis, pyruvate oxidation, the
citric acid cycle, and oxidative phosphorylation. In glycolysis, glucose derived from
carbohydrate catabolism and glycogenolysis is metabolized in the cytosol into two molecules of
pyruvate, which are translocated into the mitochondrial matrix (MM) to be converted into acetyl
coenzyme A (acetyl CoA) via pyruvate dehydrogenase. Acetyl CoA generated from pyruvate
oxidation and fatty acid oxidation is carried forward into the citric acid cycle, which proceeds as
a series of redox reactions to produce the reduced electron carriers NADH and FADH2.
In the final step of cellular respiration, oxidative phosphorylation, NADH and FADH2,
which are primarily produced during the citric acid cycle, transfer their electrons to complexes I
and II in the electron transport chain, respectively.1 This converts the cofactors into their
oxidized forms NAD+ and FAD, which are recycled in other steps of cellular respiration to
regenerate NADH and FADH2. Complexes I and II then pass their electrons along the electron
transport chain (ETC) to further complexes, ultimately resulting in the reduction of molecular
oxygen into water at complex IV.1 The movement of electrons along the ETC from complex I to
complex IV is an exergonic process that releases energy by pumping protons from the MM into

85
the mitochondrial inner membrane (MIM) space, generating an electrochemical gradient known
as a proton motive force (pmf).2, 3 ATP synthase harnesses the pmf to generate ATP by
transporting protons in the MIM space back into the MM.4
4.3 Mitochondrial Uncoupling
Transport of protons from the MIM space into the MM independent of ATP synthase is
known as mitochondrial uncoupling (Figure 4.1).5, 6 It is hypothesized that this physiological
process is a protective adaptation to prevent the production of reactive oxygen species (ROS)
during oxidative phosphorylation.7 Mitochondrial uncoupling can occur in both plants and
animals in either a passive or active fashion. Passive uncoupling is associated with proton leak
via the adenine nucleotide translocase (ANT) protein complexes, while active uncoupling is
known to occur via uncoupling proteins (UCPs) that are found along the mitochondrial inner
membrane.8 There are five known UCPs in mammals, UCP1–5,9 and of the five UCPs, the
literature is the richest on UCP1–3.8-11 UCP1 has been established as a promoter of non-
shivering thermogenesis12 while UCP2 and UCP3 are implicated7 in a protective role by
minimizing oxidative damage from ROS.
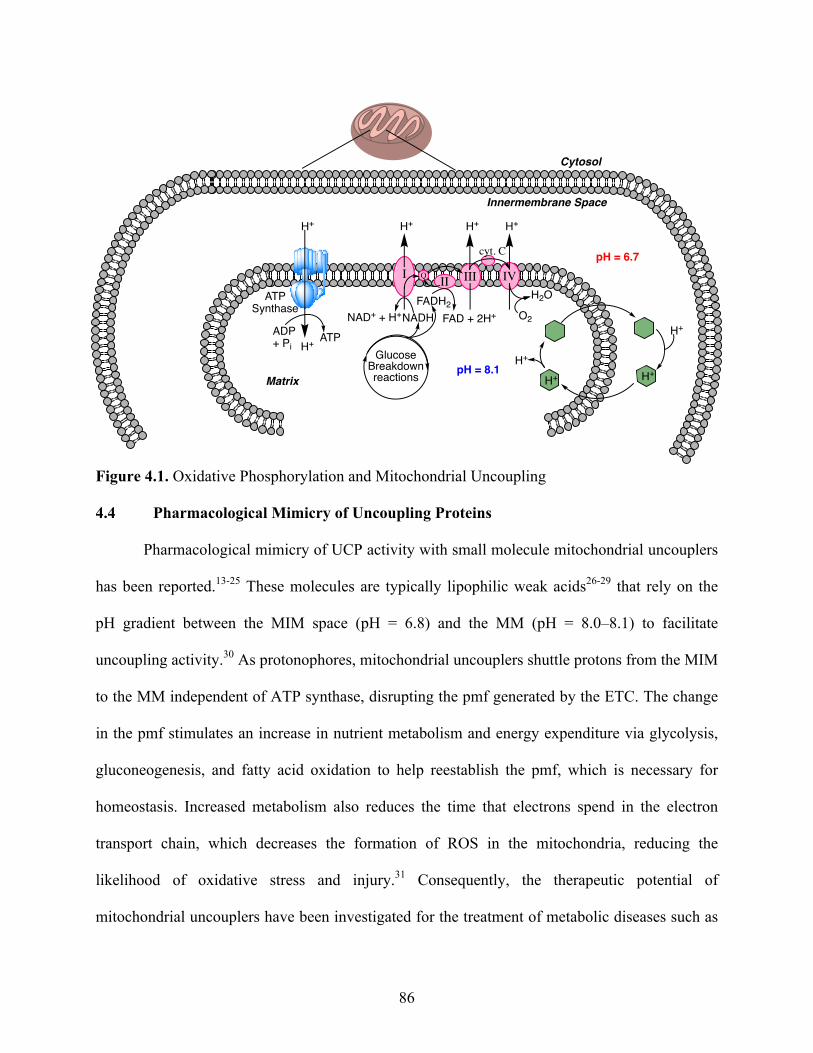
86
Figure 4.1. Oxidative Phosphorylation and Mitochondrial Uncoupling
4.4 Pharmacological Mimicry of Uncoupling Proteins
Pharmacological mimicry of UCP activity with small molecule mitochondrial uncouplers
has been reported.13-25 These molecules are typically lipophilic weak acids26-29 that rely on the
pH gradient between the MIM space (pH = 6.8) and the MM (pH = 8.0–8.1) to facilitate
uncoupling activity.30 As protonophores, mitochondrial uncouplers shuttle protons from the MIM
to the MM independent of ATP synthase, disrupting the pmf generated by the ETC. The change
in the pmf stimulates an increase in nutrient metabolism and energy expenditure via glycolysis,
gluconeogenesis, and fatty acid oxidation to help reestablish the pmf, which is necessary for
homeostasis. Increased metabolism also reduces the time that electrons spend in the electron
transport chain, which decreases the formation of ROS in the mitochondria, reducing the
likelihood of oxidative stress and injury.31 Consequently, the therapeutic potential of
mitochondrial uncouplers have been investigated for the treatment of metabolic diseases such as
ATP Synthase
I Q II III
cyt. C
IVH2O
H+ H+ H+
NADHNAD+ + H+FADH2
FAD + 2H+ O2ADP + Pi H+ ATP
Cytosol
Innermembrane Space
Matrix
Glucose Breakdown reactions
H+
H+
H+H+
H+pH = 8.1
pH = 6.7

87
obesity17, 19, 23, 32, 33 and type 2 diabetes (T2D),24, 32-35 as well as for neurodegenerative diseases
including Alzheimer’s disease36, 37 and Parkinson’s disease.37, 38 This perspective will review the
mitochondrial uncouplers reported to date and explore their potential as therapeutics.
4.5 Uncouplers of Oxidative Phosphorylation
Mitochondrial uncouplers with the ability to promote proton leakage from the MIM space
to the MM are listed in Table 4.1. For some, their mechanism of action is unclear but most are
hypothesized to have protonophoric activity. Early development of mitochondrial uncouplers
appears to emphasize compound acidity (pKa), as many of the early reported uncouplers such as
4.1 and 4.5 have pKa values below 6.8. These compounds are notorious for off-target effects and
toxicity, which has fostered general reluctance to use and develop mitochondrial uncouplers as
therapeutics. Whereas more recent reported mitochondrial uncouplers have pKas within the range
of 6.8–8.1 including 4.8 and 4.19, a shift towards the development of more weakly acidic
uncouplers to mitigate off-target effects and minimize toxicity is emerging. Indeed, greater
emphasis has been placed towards the development of uncouplers that are selective for the
mitochondria, as highlighted with the synthesis of 4.3, 4.14, and 4.19. However, the need for
potent, selective, and nontoxic mitochondrial uncouplers is evident, as there is a dearth of
reported compounds within the past 10 years. Reexamining approved clinical drugs for
mitochondrial uncoupling activity has partially met this need with the discovery that 4.12 is an
active mammalian mitochondrial uncoupler. However, phenotypic screens for mitochondrial
selectivity will also need to be generated to ensure that library hits are viable leads for structure–
activity relationship (SAR) studies in the discovery of next generation mitochondrial uncouplers.
Only once new mitochondrial uncouplers are shown to be selective for the mitochondria and to
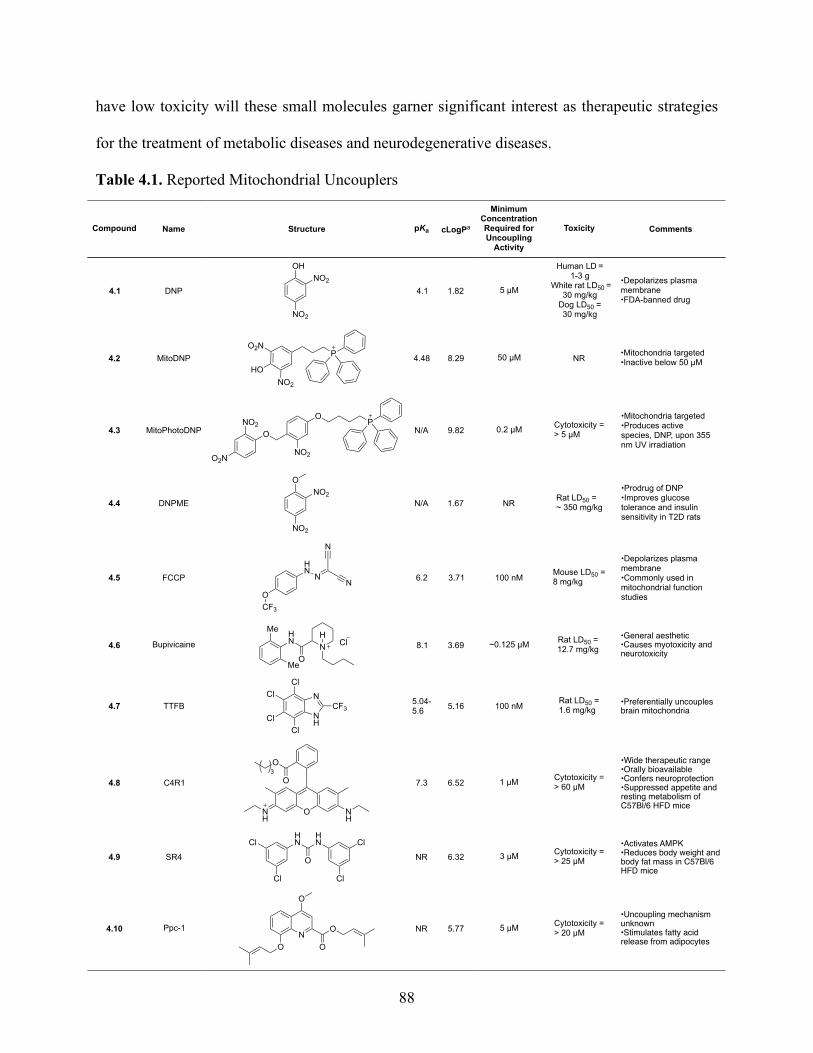
88
have low toxicity will these small molecules garner significant interest as therapeutic strategies
for the treatment of metabolic diseases and neurodegenerative diseases.
Table 4.1. Reported Mitochondrial Uncouplers
Compound Name Structure pKa cLogPa
Minimum Concentration Required for Uncoupling
Activity
Toxicity Comments
4.1 DNP
OHNO2
NO2
4.1 1.82 5 µM
Human LD = 1-3 g
White rat LD50 = 30 mg/kg
Dog LD50 = 30 mg/kg
•Depolarizes plasma membrane•FDA-banned drug
4.2 MitoDNPHO
O2N
NO2
P 4.48 8.29 50 µM NR•Mitochondria targeted•Inactive below 50 µM
4.3 MitoPhotoDNP O
O2N
NO2
NO2
OP
N/A 9.82 0.2 µM Cytotoxicity => 5 µM
•Mitochondria targeted•Produces active species, DNP, upon 355 nm UV irradiation
4.4 DNPME
ONO2
NO2
N/A 1.67 NRRat LD50 =~ 350 mg/kg
•Prodrug of DNP•Improves glucose tolerance and insulin sensitivity in T2D rats
4.5 FCCP
OCF3
HN
N
N
N 6.2 3.71 100 nMMouse LD50 = 8 mg/kg
•Depolarizes plasma membrane•Commonly used in mitochondrial function studies
4.6 Bupivicaine
Me
Me
HN
ONH
Cl 8.1 3.69 ~0.125 µM Rat LD50 = 12.7 mg/kg
•General aesthetic•Causes myotoxicity and neurotoxicity
4.7 TTFBNH
NCF3
ClCl
ClCl
5.04-5.6 5.16 100 nM
Rat LD50 = 1.6 mg/kg
•Preferentially uncouples brain mitochondria
4.8 C4R1
O NH
NH
O
O3
7.3 6.52 1 µM Cytotoxicity = > 60 µM
•Wide therapeutic range•Orally bioavailable•Confers neuroprotection•Suppressed appetite and resting metabolism of C57Bl/6 HFD mice
4.9 SR4
HN
HN
O
Cl
Cl
Cl
Cl
NR 6.32 3 µM Cytotoxicity = > 25 µM
•Activates AMPK•Reduces body weight and body fat mass in C57Bl/6 HFD mice
4.10 Ppc-1N
O
O
O
O
NR 5.77 5 µM Cytotoxicity => 20 µM
•Uncoupling mechanism unknown•Stimulates fatty acid release from adipocytes
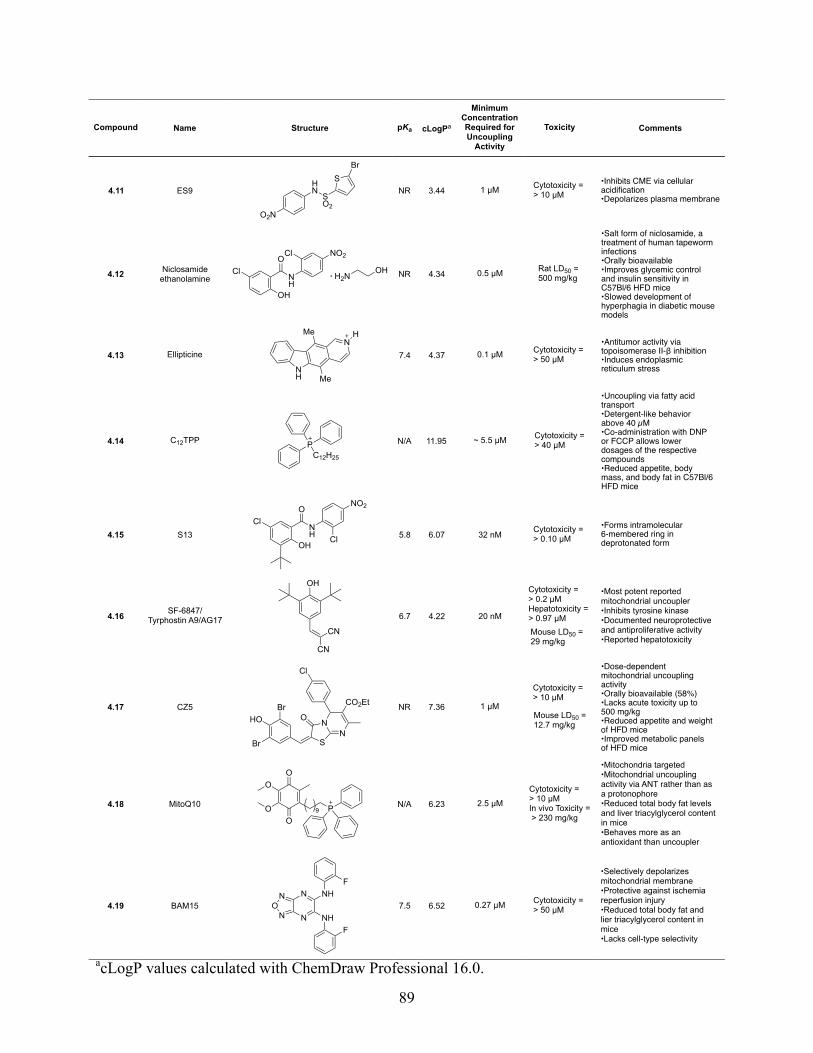
89
acLogP values calculated with ChemDraw Professional 16.0.
Compound Name Structure pKa cLogPa
Minimum Concentration Required for Uncoupling
Activity
Toxicity Comments
4.11 ES9HN
SO2
SBr
O2N
NR 3.44 1 µM Cytotoxicity = > 10 µM
•Inhibits CME via cellular acidification•Depolarizes plasma membrane
4.12 Niclosamide ethanolamine
OH
ClNH
OCl NO2
H2NOH NR 4.34 0.5 µM Rat LD50 =
500 mg/kg
•Salt form of niclosamide, a treatment of human tapeworm infections•Orally bioavailable•Improves glycemic control and insulin sensitivity in C57Bl/6 HFD mice•Slowed development of hyperphagia in diabetic mouse models
4.13 Ellipticine
NH
N
Me
Me H
7.4 4.37 0.1 µM Cytotoxicity = > 50 µM
•Antitumor activity via topoisomerase II-β inhibition•Induces endoplasmic reticulum stress
4.14 C12TPP PC12H25
N/A 11.95 ~ 5.5 µM Cytotoxicity => 40 µM
•Uncoupling via fatty acid transport•Detergent-like behavior above 40 µM•Co-administration with DNP or FCCP allows lower dosages of the respective compounds•Reduced appetite, body mass, and body fat in C57Bl/6 HFD mice
4.15 S13NH
Cl
OH
ONO2
Cl 5.8 6.07 32 nM Cytotoxicity = > 0.10 µM
•Forms intramolecular 6-membered ring in deprotonated form
4.16SF-6847/
Tyrphostin A9/AG17
OH
CN
CN
6.7 4.22 20 nM
Cytotoxicity => 0.2 µMHepatotoxicity = > 0.97 µMMouse LD50 = 29 mg/kg
•Most potent reported mitochondrial uncoupler•Inhibits tyrosine kinase•Documented neuroprotective and antiproliferative activity•Reported hepatotoxicity
4.17 CZ5HO
Br
Br S
NN
Cl
CO2Et
ONR 7.36 1 µM
Cytotoxicity = > 10 µM
Mouse LD50 = 12.7 mg/kg
•Dose-dependent mitochondrial uncoupling activity•Orally bioavailable (58%)•Lacks acute toxicity up to 500 mg/kg•Reduced appetite and weight of HFD mice•Improved metabolic panels of HFD mice
4.18 MitoQ10
O
O
O
O P9N/A 6.23 2.5 µM
Cytotoxicity = > 10 µMIn vivo Toxicity = > 230 mg/kg
•Mitochondria targeted•Mitochondrial uncoupling activity via ANT rather than as a protonophore•Reduced total body fat levels and liver triacylglycerol content in mice•Behaves more as an antioxidant than uncoupler
4.19 BAM15 ON
N
N
N
NH
NHF
F
7.5 6.52 0.27 µM Cytotoxicity = > 50 µM
•Selectively depolarizes mitochondrial membrane•Protective against ischemia reperfusion injury•Reduced total body fat and lier triacylglycerol content in mice•Lacks cell-type selectivity

90
4.5.1 DNP (2,4-Dinitrophenol)
Of the published mitochondrial uncouplers, 4.1 ((2,4-Dinitrophenol, DNP) is the most
well known. It was originally used during World War I in the preparation of munitions, but many
workers exposed to the chemical experienced weight loss, fever, nausea, and death.39 Due to its
weight loss inducing effects, DNP was explored as a treatment for obesity. Early studies found
that regulated doses of 4.1 successfully promoted weight loss in obese patients.40, 41 However,
cases of cataracts, blindness42-44 or death from drug-induced hyperthermia were reported.45-47
Consequently, the Food and Drug Administration (FDA) removed the drug from the market in
1938.48 Later studies found that the toxicity issues associated with 4.1 treatment result from a
narrow drug therapeutic window and a concomitant depolarization of the plasma membrane and
the MIM.49-52 Today, it is considered an illicit drug, but it is still used as an uncontrolled weight
loss supplement, particularly by body builders.53
Although 4.1’s therapeutic potential for the treatment of obesity and T2D is emerging, a
major obstacle as a pharmaceutical is its narrow therapeutic window. Caldeira da Silva et al. and
Goldgof et al. both found that under controlled dosing regimens, mice fed on a high fat diet
(HFD) showed improved blood glucose, insulin, and triglyceride levels and reduced body
mass.17, 54 Interestingly, mice receiving a controlled dosage of 4.1 lived longer,54 a common
observation in calorie-restricted diet studies.55, 56
A controlled release version of 4.1, CRMP, was more recently developed.34 This orally
bioavailable formulation facilitates a wide therapeutic window for 4.1, eliciting an effect up to
100 mg/kg without causing toxicity (4.1 began to show toxicity issues at 1 mg/kg). This
formulation also achieves a minimum effective dose of 0.5 mg/kg to lower blood, liver, and

91
muscle triacylglycerol content in rats, a dose 10-fold lower than that required for 4.1 (5 mg/kg).
Additionally, CRMP improved glucose tolerance and insulin sensitivity in T2D rats.
In an alternative approach to reduce toxicity due to off-target effects, Blaikie et al.
tethered 4.1 to triphenylphosphonium (TPP) (4.2, MitoDNP) to selectively target 4.1 to the
mitochondria.57 The dispersed positive charge in TPP affords selective targeting to mitochondria.
Unfortunately, this maneuver lacks uncoupling activity, failing to increase mitochondrial
respiration rates at concentrations below 50 µM (4.1 increases respiration rates at 5 µM).57 To
circumvent this failure, Chalmers and coworkers modified 4.2 by inserting a photocleavable
linker between 4.1 and the TPP moiety, creating 4.3 (MitoPhotoDNP).58 The TPP moiety
delivers 4.1 to the mitochondria and releases 4.1 upon 355 nm UV irradiation.58 4.3 is active at
concentrations as low as 0.2 µM and lacks cytotoxicity up to 5 µM.58 However, it has yet to be
tested for its efficacy in vivo.
Tissue targeted delivery of 4.1 also offers a promising strategy. For example, Perry et al.
developed a prodrug of 4.1 to produce 4.4 (DNPME).33 This methyl ether analogue of 4.1 has no
uncoupling activity, as it reduced circulating plasma levels of 4.1, with the majority of 4.1
accumulating in white adipose tissue. Bioactivation of 4.4 in the liver via P450-mediated
demethylation reveals 4.1 locally. Studies have shown that 4.4 improved glucose tolerance and
insulin sensitivity in T2D rats. This prodrug was also shown to have reduced toxicity, having a
~10-fold higher single dose LD50 (~350 mg/kg) than 4.1 (~45 mg/kg).33
The therapeutic potential of 4.1 is expanding, for example, toward treatment for spinal
cord injury and neurodegenerative diseases. Excitotoxic forces and physical trauma have both
been shown36, 37, 59 to lead to mitochondrial dysfunction, which includes increased ROS
production and extensive Ca2+ mitochondrial sequestration. In particular, over-activation of the

92
N-methyl-D-aspartate (NMDA)-sensitive glutamate receptor by the neurotransmitter glutamate
has been linked59 to neurodegenerative diseases like Alzheimer’s,36, 60 Parkinson’s,38, 61 and
Huntington’s diseases.62 4.1’s ability to reduce mitochondrial Ca2+ levels and ROS production
has been well documented,37, 63-67 which highlights the therapeutic utility of mitochondrial
uncouplers as a treatment for mitochondrial dysfunction.
4.5.2 FCCP (Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone)
The other mitochondrial uncoupler that has been extensively studied is the hydrazone 4.5
(Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone, FCCP). Its development and discovery
was first reported by Heytler et al. in 1962, along with its analogues such as carbonyl cyanide m-
chlorophenylhydrazone (CCCP).68 Compound 4.5’s and CCCP’s uncoupling activity in
mitochondria68, 69 and chloroplasts68-71 were noted early on, with 4.5 proving to be the more
potent uncoupler (4.5 IC50 = 0.04 µM, CCCP IC50 = 0.1 µM).72 Unfortunately, 4.5 also has a
narrow therapeutic window and causes off-target effects,51, 52, 73-75 such as depolarization of the
mitochondria and plasma membrane. Nevertheless, 4.5 serves as a valuable as a chemical probe
for mitochondrial and cellular function, as well as for studying neurodegenerative diseases. 37, 52,
74, 76-87
4.5.3 Bupivacaine
The effect of general aesthetics on oxidative phosphorylation have long been studied for
their ability to disrupt ATP synthesis, affect electron and Ca2+ transport, or act as mitochondrial
uncouplers has been reported.88 Compound 4.6 (Bupivacaine) has emerged89-93 as a general
aesthetic that uncouples oxidative phosphorylation. Dabadie et al. showed that it can increase
mitochondrial respiration by acting as a protonophore,89 which was confirmed by mitochondrial
swelling experiments.94, 95 Compound 4.6’s mitochondrial uncoupling activity is hypothesized to

93
arise from due to its liposolubility and pKa (8.1), which allows it to passively transfer between
the MIM space and MM. However, Terada et al. and van Dam et al. discovered,90, 96 the
protonophoric mitochondrial uncoupling occurs only in the presence of hydrophobic anions such
as picrate; otherwise, it acts as a decoupler by inducing a slip in the proton pumps during
oxidative phosphorylation. Sztark et al. later showed93 that 4.6 can be either a mitochondrial
uncoupler or decoupler, depending upon the respiration state of the mitochondria and the
membrane potential.
Unfortunately, 4.6 has been shown to have myotoxicity and neurotoxicity.97-101 Weinberg
and coworkers found102 that 4.6’s myotoxicity is in part due to the inhibition of carnitine
metabolism in cardiac mitochondria. Lirk et al. established100 that 4.6’s neurotoxicity is linked to
ROS activation103 of mitogen activated protein kinase (MAPK) pathway. Further, 4.6 has been
shown to promote ROS generation,91 which is thought to contribute to its toxicity. Although the
exact mechanism of the enhanced ROS generation is unclear, disruption of the electron transport
via reversible binding to complexes I and III plays a role. 4.6-induced cardiotoxicity in rats could
be reversed with lipid infusion using Intralipid.104 More recently, Chen and coworkers showed
that a lipid emulsion version of 4.6 helped to minimize myocardial toxicity.105
4.5.4 TTFB (4,5,6,7-Tetrachloro-2-trifluoromethylbenzimidazole)
Compound 4.7 (TTFB) was as a constituent of a benzimidazole library intended for
herbicidal use.106 Jones and Watson107 and Büchel et al.108 concurrently reported that 4.7 is a
mitochondrial uncoupler, showing that 0.03–0.08 µM was necessary to elicit the uncoupling. The
observed uncoupling activity is attributed to the compound’s pKa (NH) of 5.04–5.6,106-108
suggesting that the molecule’s active form is its anionic form, which is stabilized by the aromatic
imidazole ring.18, 109 However, due to reported mammalian toxicity106 and an uncoupling

94
preference for brain mitochondria over liver mitochondria,110 4.7 has not been pursued as a
therapy for metabolic or neurodegenerative diseases.
4.5.5 C4R1
Developed from the fluorescent dye rhodamine 19,14 4.8 (C4R1) is a mitochondrial
uncoupler (IC50 of ~ 1 µM) that can promote mitochondrial respiration up to ~ 60 µM,
highlighting the molecule’s wide therapeutic range.111 This mild uncoupling activity is attributed
to its measured pKa of 7.3.14 Studies found that 4.8 is a cationic protonophore that is selective for
the mitochondria with activity that is dependent upon membrane potential.14, 111
Compound 4.8 is orally bioavailable and offers neuroprotection by reducing brain
swelling and cognitive decline in a stroke model.19, 111 It also showed promise as a lead molecule
for the treatment of obesity by suppressing appetite and increasing resting metabolism in
C57Bl/6 HFD mice over a 30-day period.19 With a daily dosage of 30 µmol/kg/dayof 4.8,
overall body mass and body fat decreased by 19% and 20%, respectively, without observed
toxicity effects.19 Interestingly, 27 µmol/kg/day of 4.1 did not alter the metabolism of C57Bl/6
HFD mice.17
4.5.6 SR4
A library screen for anti-cancer activity led to the discovery of the dichlorophenylurea 4.9
(SR4).112 Treatment of HL-60 leukemia cells with 4.9 resulted in a mitochondria-induced
apoptosis, which was associated with a depolarization of the mitochondrial membrane.112 Further
investigation in melanoma cell lines and mouse models led to the discovery that it activates
AMP-activated kinase (AMPK) by promoting phosphorylation and induction of anti-tumor
mechanisms.113 Further study revealed that this process occurs in adipocytes and inhibits
adipogenesis, suggesting 4.9 has anti-obesity potential.114 C57Bl/6 HFD mice dosed with 5

95
mg/kg three times a week experienced a 16% reduction in body weight and ~ 35% body fat mass
compared to controls.32 Treatment with 4.9 also improved insulin sensitivity and glucose
metabolism and reduced liver lipid content as a direct consequence of AMPK activation32 and
mitochondrial uncoupling activity.16 Compound 4.9 increases mitochondrial respiration at
concentrations as low as 3 µM and up to 25 µM independent of the ANT and UCPs, indicating
protonophoric uncoupling activity. 16 However, selectivity for the mitochondria has not been
established, and 4.9 exhibits similar liver toxicity to 4.5.16
4.5.7 Ppc-1
Discovered from a natural product library screen, 4.10 (Ppc-1) is a secondary metabolite
of the slime mold Polysphondylium pseudo-candidum.115 Kikuchi and coworkers discovered116
that this molecule has mitochondrial uncoupling activity, increasing oxygen consumption at
concentrations as high 20 µM without causing toxicity or inhibiting mitochondrial respiration.117
It is currently unclear how 4.10 functions as a mitochondrial uncoupler, but studies have
established that its mitochondrial uncoupling activity is not from the formation of a permeability
transition pore (PTP), which improves ion permeability into the mitochondrial matrix.23, 118
Compound 4.10’s potential as a weight loss therapy has been explored. ICR mice fed on
a normal diet and dosed with 0.8 mg/kg/week maintained a normal appetite and constant weight
over an 8-week period versus control.23 Analysis of tissue and blood serum samples revealed
high concentrations of 4.10 in adipocytes and higher than normal blood fatty acid levels, which
is believed to be a consequence of 4.10-stimulated fatty acid release from adipocytes.23
Surprisingly, higher (4 mg/kg/week and 10 mg/kg/week) and lower doses (0.16 mg/kg/week) of
4.10 were ineffective in preventing weight gain.23

96
4.5.8 ES9 (Endosin 9)
Discovered from a small molecule library screen for inhibitors of tobacco pollen
germination and growth, 4.11 (ES9)119 was found to also inhibit clathrin-mediated endocytosis
(CME) with an IC50 of 5 µM.15 Because CME is dependent on ATP stores, 4.11’s impact on
ATP production was assessed, revealing mitochondrial uncoupling activity.15 Compound 4.11
increased mitochondrial respiration at concentrations as low as 1 µM15 but depleted ATP stores
above 10 µM, which was attributed to depolarization of the plasma membrane.15 Dejonghe et al.
discovered that 4.11 inhibits CME via cellular acidification through mitochondrial uncoupling
and depolarization of the plasma membrane.15 Compound 4.11 has yet to be explored as a
chemical probe mitochondrial function.
4.5.9 Niclosamide Ethanolamine
Compound 4.12 (niclosamide ethanolamine, NEN) is the salt form of niclosamide, an
FDA approved anthelmintic drug whose mode of action is mitochondrial uncoupling of the
parasite’s mitochondria.120 Compound 4.12 is orally bioavailable and has an excellent safety
profile,121, 122 Because of its mitochondrial activity, Tao et al. assessed 4.12 as a potential
treatment for T2D.24 Compound 4.12 uncouples mammalian mitochondria at concentrations as
low as 0.5 µM, and dosing mice fed on a HFD with 40 mg/kg improved glycemic control,
increased insulin sensitivity, reduced liver fat content, and limited weight gain versus control.24
In a mouse model for diabetes, treatment of db/db mice with 4.12 improved glycemic control
and slowed the development of hyperphagia.24 It aslo improved glucose metabolism without
increasing insulin sensitivity, which was later found to be a consequence of 4.12 functioning as a
blocker of the glucagon PKA signaling pathway.123

97
4.5.10 Ellipticine
Compound 4.13 (Ellipticine) is an alkaloid isolated from the plant Ochrosia elliptica with
antitumor activity and noted toxicity. Its toxicity results from mitochondrial uncoupling activity,
which increases respiration and highly depolarizes the mitochondrial membrane in both plant and
mammalian cells at concentrations as low as 0.1 µM.124-126 However, concentrations above 50
µM were found to inhibit mitochondrial respiration.125, 127 Compound 4.13’s cationic (pKa = 7.4)
and neutral forms are implicated in its mitochondrial uncoupling activity, and its membrane
permeability was demonstrated to be pH independent.126, 128, 129 Unfortunately, 4.13 is cytotoxic,
inducing endoplasmic reticulum stress130 and inhibiting of topoisomerase II-β.131 There are no
current reports of 4.13 as a chemical probe for mitochondrial function.
4.5.11 C12TPP
Because cationic TPPs accumulate in the mitochondria,132-138 they are commonly used as
a handle57 for mitochondrial drug delivery. Severin et al. and Sukhanova et al. discovered139 that
4.14 (C12TPP) in the presence of the fatty acid palmitate can induce mild mitochondrial
uncoupling in rat-heart mitochondria and decrease the amount of mitochondrial H2O2.139, 140 In
the absence of fatty acid additives, 4.14 can uncouple yeast mitochondria at concentrations as
low as ~5.5 µM, but this likely occurs due to the presence of endogenous fatty acids in the yeast
cells.140, 141 Although 4.14 lacks pro-oxidant activity at high concentrations,139, 140 it does inhibit
mitochondrial respiration above 40 µM, a consequence of mitochondrial swelling induced by
detergent behavior.140
Compound 4.14’s therapeutic potential has been explored. Synergistic administration of
4.14 with either 4.5 or 4.1 reduced the concentration of uncoupler needed to increase
mitochondrial respiration (0.4 µM vs. 0.6 µM and 10 µM vs. 55 µM, respectively).13 It is

98
hypothesized13 that 4.14 interacts with the anionic forms of 4.5 and 4.1 to facilitate
mitochondrial localization, minimizing off-target depolarization of the plasma membrane. This
system has yet to be tested in animals. Interestingly, dosing mice fed a HFD with 50
µmol/kg/day reduced food intake, body mass, and body fat without noticeable toxicity.142 These
results are a consequence of fatty acid-associated mitochondrial uncoupling and an UCP-1
independent up-regulation of brown adipose tissue.142
4.5.12 S-13
The salicylanilide 4.15 (S-13)143 increases mitochondrial respiration at concentrations as
low as 0.032 µM.144 Its anionic form (pKa = 5.8) is suggested to be the active form during
mitochondrial uncoupling activity.145 It is proposed that in its anionic state, 4.15 forms an
internal 6-membered ring, which retains the neutral state’s lipophilicity, permitting passive
diffusion through the mitochondrial membrane.144 However, 4.15 inhibits mitochondrial
respiration above 0.10 µM.146
4.5.13 SF-6847/Tyrphostin A9/AG17
Originally synthesized for agricultural fungicidal and acaricidal usage,147 4.16 (SF-
6847/Tyrphostin A9/AG17) increases mitochondrial respiration at concentrations as low as 0.020
µM.148 Its anionic state (pKa = 6.70) is hypothesized to be the active structure during the
uncoupling process.149 NMR studies and energy barrier calculations indicate150, 151 that a
restricted rotational barrier localizes the negative charge, which is also shielded by the two tert-
butyl moieties on the benzene ring, allowing 4.16 to retain its lipophilicity when deprotonated.152
Compound 4.16’s therapeutic potential has only been explored to a limited extent.
Concentrations as low as ~ 0.5 µM 4.16 have been shown to be protective against glutamate-
induced oxidative damage in neuronal cells.153 Additionally, 4.16 was shown to reduce the

99
impact of insulin on the white fat tissue cells; however, it is unclear if this observation is
associated with 4.16’s mitochondrial uncoupling activity.154 Unfortunately, 4.16 has reported
cytotoxicity above 0.20 µM,152 hepatotoxicity above 0.97 µM152 and a mouse LD50 of 29
mg/kg147. In addition to uncoupling the mitochondria, 4.16 inhibits tyrosine kinase155, 156 and
Ca2+ release.157
4.5.14 CZ5
Discovered from a high-throughput screen, 4.17 (CZ5) is a protonophoric mitochondrial
uncoupler158 that increases mitochondrial respiration dose-dependently and preferentially
uncouples myocyte and adipocyte mitochondria at concentrations as low as 1 µM.158 Higher
concentrations were required to increase mitochondrial respiration in hepatocytes.158
Additionally, 4.17 has minimal cytotoxicity up to 10 µM.
Compound 4.17 has good pharmacokinetics, is orally bioavailable (58%), and can be
administered up to 500 mg/kg in a single dose without causing acute toxicity.158 HFD fed mice
dosed with 30 mg/kg/day over a 5-week period consumed less food and had reduced body
weight and body fat content in comparison to controls.158 Improved metabolic panels were also
observed, including reduced cholesterol, triacylglycerol, and fasting blood glucose levels, and
improved insulin levels.158
4.5.15 Cationic Triphenylphosphoniums
In addition to 4.14, numerous TPP mitochondrial uncouplers have been reported, some of
which have shown success in vitro and in vivo. These compounds include MitoQ,159 SKQ1,13, 160
MitoFluo,161, 162 FF16-TPP,163 and MitoBHT.21 Of the TPPs reported, the literature is the richest
on 4.18 (MitoQ10).164 Initially designed as a method for delivering ubiquinone to the
mitochondria for antioxidant purposes,159 4.18 was found to have mitochondrial uncoupling

100
properties, stimulating mitochondrial respiration at concentrations as low as 2.5 µM via the
ANT;21 concentrations above 10 µM led to decreased membrane potential and cell death.159
However, later studies discovered that 4.18 has nanomolar uncoupling activity, although exact
concentrations were not specified. 4.18 displays antioxidant properties in addition to
mitochondrial uncoupling activity, as it was able to prevent H2O2 oxidation of the fatty acid cis-
parinaric acid.159
Compound 4.18 is orally bioavailable and lacks toxicity up to 230 mg/kg/day.165 Long-
term (28 weeks) dosing of 3.2 µmol/day/mouse decreased total mouse body fat and liver
triacylglycerol content without affecting mouse body mass.166 Other mouse studies with 4.18
have explored its effects on models of obesity,167 cardiac ischemia reperfusion,168 hypertension
and stroke,169 and sepsis.170 Two human phase II studies have also been conducted with 4.18.
One study explored its ability to slow the progression of Parkinson’s disease in newly diagnosed
patients;171 however, it produced no improvements in comparison to the placebo.171 The other
study explored its ability to reduce blood serum hepatitis C virus (HCV) RNA levels in patients
with chronic HCV, but instead of reducing HCV RNA levels, 4.18 reduced liver damage
associated with HCV.172 For all studies mentioned above, 4.18’s therapeutic use was as an
antioxidant rather than as a mitochondrial uncoupler.
4.5.16 BAM15
Discovered from a screen of a known library of compounds,20 4.19 (BAM15) was found
to have mitochondrial uncoupling activity with an EC50 of 0.27 µM.173 Unlike 4.1 and 4.5, 4.19 is
selective for the mitochondria over the plasma membrane, which was confirmed by
electrophysiology studies.20 Kenwood et al. hypothesized20 that the selectivity for the
mitochondria is due to the molecule’s pKa, (7.5). SAR studies established173 that the furazan and

101
pyrazine rings were essential for uncoupling.173 However, 4.19 lacks cell-type selectivity, as it
was capable of uncoupling, muscle, liver, heart, and connective tissue cells.20 Nonetheless, 4.19
showed protective effects in kidney ischemia reperfusion injury.20 More recent studies have
established174-176 its utility for the study of basic mitochondrial function, particularly due to its
limited off-target effects and wide therapeutic window.20
4.6 Conclusions
Mitochondrial uncoupling is a key physiological mechanism in mammals that has
evolved to provide endogenous heat and to counteract ROS prooduction during oxidative
phosphorylation, as indicated by the presence of multiple UCP isoforms. Pharmacological
mitochondrial uncoupling has evolved as a therapeutic for various metabolic and
neurodegenerative diseases. Cell toxicity from off-target effects such as depolarization of the
plasma membrane has generated reluctance towards the use of mitochondrial uncouplers in
medicine. However, fine-tuning of molecular electronics, pro-drug strategies, and mitochondrial
selective handles have produced mitochondrial uncouplers with excellent therapeutic potential.
Future mitochondrial uncouplers will need to be developed with pKa and lipophilicity in
consideration to promote mitochondrial selectivity and to maintain drug-likeness. Further SAR
studies will aid in the elucidation of mitochondrial function in disease states and will help bring
to fruition the development of mitochondrial uncoupler clinical candidates.
4.7 Dissertation Overview for Developing Mitochondrial Uncouplers
Chapter 4 discussed the process of mitochondrial uncoupling, small molecule
mitochondrial uncouplers, and the therapeutic potential of small molecule mitochondrial
uncouplers for the treatment of metabolic diseases and mitochondrial dysfunction. Chapter 5 will
disclose an SAR on the aniline portion of 4.19. These derivatives show good mitochondrial

102
uncoupling activity over a wide concentration range and reveal that pKa and cLogP contribute
equally to mitochondrial uncoupling activity. Chapter 6 provides the supplemental information
for the compounds synthesized and characterized by the author of this dissertation.

103
4.8 References
1. Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System.
Annu. Rev. Biochem. 1985, 54, 1015–1069.
2. Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation.
Biochim. Biophys. Acta 2011, 1807, 1507–1538.
3. Schultz, B. E.; Chan, S. I. Structures and Proton-Pumping Strategies of Mitochondrial
Respiratory Enzymes. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23–65.
4. Dimroth, P.; Kaim, G.; Matthey, U. Crucial Role of the Membrane Potential for ATP
Synthesis by F(1)F(O) ATP Synthases. J. Exp. Biol. 2000, 203, 51–59.
5. Loomis, W. F.; Lipmann, F. Reversible Inhibition of Coupling between Phosphorylation and
Oxidation. J. Biol. Chem. 1948, 173, 807–808.
6. Weinbach, E. C.; Garbus, J. Mechanism of Action of Reagents That Uncouple Oxidative
Phosphorylation. Nature 1969, 221, 1016–1018.
7. Divakaruni, A. S.; Brand, M. D. The Regulation and Physiology of Mitochondrial Proton
Leak. Physiology (Bethesda) 2011, 26, 192–205.
8. Ricquier, D.; Bouillaud, F. Mitochondrial Uncoupling Proteins: From Mitochondria to the
Regulation of Energy Balance. J. Physiol. 2000, 529, 3–10.
9. Erlanson-Albertsson, C. The Role of Uncoupling Proteins in the Regulation of Metabolism.
Acta Physiol. Scand. 2003, 178, 405–412.
10. Busiello, R. A.; Savarese, S.; Lombardi, A. Mitochondrial Uncoupling Proteins and Energy
Metabolism. Front. Physiol. 2015, 6, 1–36.

104
11. Rousset, S.; Alves-Guerra, M.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.; Bouillaud, F.;
Ricquier, D. The Biology of Mitochondrial Uncoupling Proteins. Diabetes Metab. Syndr.
Obes. 2004, 53, 130–135.
12. Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. Ucp1:
The Only Protein Able to Mediate Adaptive Non-Shivering Thermogenesis and Metabolic
Inefficiency. Biochem. Biophys. Acta 2001, 1504, 82–106.
13. Antonenko, Y. N.; Khailova, L. S.; Knorre, D. A.; Markova, O. V.; Rokitskaya, T. I.;
Il'yasova, T. M.; Severina, I.; Kotova, E. A.; Karavaeva, Y. E.; Prikhodko, A. S.; Severin, F.
F.; Skulachev, V. P. Penetrating Cations Enhance Uncoupling Activity of Anionic
Protonophores in Mitochondria. PLoS One 2013, 8, 1–13.
14. Antonenko, Y. N.; Avetisyan, A. V.; Cherepanov, D. A.; Knorre, D. A.; Korshunova, G. A.;
Markova, O. V.; Ojovan, S. M.; Perevoshchikova, I. V.; Pustovidko, A. V.; Rokitskaya, T. I.;
Severina, II; Simonyan, R. A.; Smirnova, E. A.; Sobko, A. A.; Sumbatyan, N. V.; Severin, F.
F.; Skulachev, V. P. Derivatives of Rhodamine 19 as Mild Mitochondria-Targeted Cationic
Uncouplers. J. Biol. Chem. 2011, 286, 17831–17840.
15. Dejonghe, W.; Kuenen, S.; Mylle, E.; Vasileva, M.; Keech, O.; Viotti, C.; Swerts, J.;
Fendrych, M.; Ortiz-Morea, F. A.; Mishev, K.; Delang, S.; Scholl, S.; Zarza, X.; Heilmann,
M.; Kourelis, J.; Kasprowicz, J.; Nguyen le, S. L.; Drozdzecki, A.; Van Houtte, I.; Szatmari,
A. M.; Majda, M.; Baisa, G.; Bednarek, S. Y.; Robert, S.; Audenaert, D.; Testerink, C.;
Munnik, T.; Van Damme, D.; Heilmann, I.; Schumacher, K.; Winne, J.; Friml, J.; Verstreken,
P.; Russinova, E. Mitochondrial Uncouplers Inhibit Clathrin-Mediated Endocytosis Largely
through Cytoplasmic Acidification. Nat. Commun. 2016, 7, 11710.

105
16. Figarola, J. L.; Singhal, J.; Tompkins, J. D.; Rogers, G. W.; Warden, C.; Horne, D.; Riggs,
A. D.; Awasthi, S.; Singhal, S. S. Sr4 Uncouples Mitochondrial Oxidative Phosphorylation,
Modulates AMP-Dependent Kinase (AMPK)-Mammalian Target of Rapamycin (MTOR)
Signaling, and Inhibits Proliferation of HepG2 Hepatocarcinoma Cells. J. Biol. Chem. 2015,
290, 30321–30341.
17. Goldgof, M.; Xiao, C.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Reitman, M. L. The
Chemical Uncoupler 2,4-Dinitrophenol (DNP) Protects against Diet-Induced Obesity and
Improves Energy Homeostasis in Mice at Thermoneutrality. J. Biol. Chem. 2014, 289, 19341–
19350.
18. Jones, O. T. G.; Watson, W. A. Properties of Substituted 2-Trifluoromethylbenzimidazoles
as Uncouplers of Oxidative Phosphorylation. Biochem. J. 1967, 102, 564–573.
19. Kalinovich, A. V.; Shabalina, I. G. Novel Mitochondrial Cationic Uncoupler C4R1 Is an
Effective Treatment for Combating Obesity in Mice. Biochemistry (Mosc) 2015, 80, 620–628.
20. Kenwood, B. M.; Weaver, J. L.; Bajwa, A.; Poon, I. K.; Byrne, F. L.; Murrow, B. A.;
Calderone, J. A.; Huang, L.; Divakaruni, A. S.; Tomsig, J. L.; Okabe, K.; Lo, R. H.; Coleman,
G. C.; Columbus, L.; Yan, Z.; Saucerman, J. J.; Smith, J. S.; Holmes, J. W.; Lynch, K. R.;
Ravichandran, K. S.; Uchiyama, S.; Santos, W. L.; Rogers, G. W.; Okusa, M. D.; Bayliss, D.
A.; Hoehn, K. L. Identification of a Novel Mitochondrial Uncoupler That Does Not
Depolarize the Plasma Membrane. Mol. Metab. 2014, 3, 114–123.
21. Lou, P. H.; Hansen, B. S.; Olsen, P. H.; Tullin, S.; Murphy, M. P.; Brand, M. D.
Mitochondrial Uncouplers with an Extraordinary Dynamic Range. Biochem. J. 2007, 407,
129–140.

106
22. McCracken, R. O.; Carr, A. W.; Stillwell, W. H.; Lipkowitz, K. B.; Boisvenue, R.;
O'Doherty, G. O. P.; Wickiser, D. I. Trifluoromethanesulfonamide Anthelmintics
Protonophoric Uncouplers of Oxidative Phosphorylation. Biochem. Pharmacol. 1993, 45,
1873–1880.
23. Suzuki, T.; Kikuchi, H.; Ogura, M.; Homma, M. K.; Oshima, Y.; Homma, Y. Weight Loss
by Ppc-1, a Novel Small Molecule Mitochondrial Uncoupler Derived from Slime Mold. PLoS
One 2015, 10, e0117088.
24. Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G. I.; Jin, S. Niclosamide Ethanolamine-Induced
Mild Mitochondrial Uncoupling Improves Diabetic Symptoms in Mice. Nat. Med. 2014, 20,
1263–1269.
25. Tollenaere, J. P. Structure-Activity Relationships of Three Groups of Uncouplers of
Oxidative Phosphorylation: Salicylanilides, 2-Trifluoromethylbenzimidazoles, and Phenols. J.
Med. Chem. 1973, 16, 791–796.
26. Matsuno-Yagi, A.; Hatefi, Y. Uncoupling of Oxidative Phosphorylation: Different Effects of
Lipophilic Weak Acids and Electrogenic Ionophores on the Kinetics of ATP Synthesis.
Biochemistry 1989, 28, 4367–4374.
27. Parker, V. H. Effect of Nitrophenols and Halogenophenols on the Enzymic Activity of Rat-
Liver Mitochondria. Biochem. J. 1958, 69, 306–311.
28. Hemker, H. C. Lipid Solubility as a Factor Influencing the Activity of Uncoupling Phenols.
Biochem. Biophys. Acta 1962, 63, 46–54.
29. Chopineaux-Courtois, V.; Reymond, F.; Bouchard, G.; Carrupt, P.; Testa, B.; Girault, H. H.
Effects of Charge and Intramolecular Structure on the Lipophilicity of Nitrophenols. J. Am.
Chem. Soc. 1999, 121, 1743–17467.

107
30. Casey, J. R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev.
Mol. Cell Biol. 2010, 11, 50–61.
31. Korshunov, S. S.; Skulachev, V. P.; Starkov, A. A. High Protonic Potential Actuates a
Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997,
416, 15–18.
32. Figarola, J. L.; Singhal, P.; Rahbar, S.; Gugiu, B. G.; Awasthi, S.; Singhal, S. S. Coh-Sr4
Reduces Body Weight, Improves Glycemic Control and Prevents Hepatic Steatosis in High
Fat Diet-Induced Obese Mice. PLoS One 2013, 8, e83801.
33. Perry, R. J.; Kim, T.; Zhang, X. M.; Lee, H. Y.; Pesta, D.; Popov, V. B.; Zhang, D.; Rahimi,
Y.; Jurczak, M. J.; Cline, G. W.; Spiegel, D. A.; Shulman, G. I. Reversal of
Hypertriglyceridemia, Fatty Liver Disease, and Insulin Resistance by a Liver-Targeted
Mitochondrial Uncoupler. Cell Metab. 2013, 18, 740–748.
34. Perry, R. J.; Zhang, D.; Zhang, X.; Boyer, J. L.; Shulman, G. I. Controlled-Release
Protonophore Reverses Diabetes and Steatohepatitis in Rats. Science 2015, 347, 1253–1256.
35. Anderson, E. J.; Lustig, M. E.; Boyle, K. E.; Woodlief, T. L.; Kane, D. A.; Lin, C.-T.; Price,
J. W., III; Kang, L.; Rabinovitch, P. S.; Szeto, H. H.; Houmard, J. A.; Cortright, R. N.;
Wasserman, D. H.; Neufer, P. D. Mitochondrial H2O2 Emission and Cellular Redox State
Link Excess Fat Intake to Insulin Resistance in Both Rodents and Humans. J. Clin. Invest.
2009, 119, 573–581.
36. Geisler, J. G.; Marosi, K.; Halpern, J.; Mattson, M. P. DNP, Mitochondrial Uncoupling, and
Neuroprotection: A Little Dab'll Do Ya. Alzheimers Dement. 2016, DOI:
10.1016/j.jalz.2016.08.001.

108
37. Lin, M. T.; Beal, M. F. Mitochondrial Dysfunction and Oxidative Stress in
Neurodegenerative Diseases. Nature 2006, 443, 787–795.
38. Wu, Y. N.; Munhall, A. C.; Johnson, S. W. Mitochondrial Uncoupling Agents Antagonize
Rotenone Actions in Rat Substantia Nigra Dopamine Neurons. Brain Res. 2011, 1395, 86–93.
39. Colman, E. Dinitrophenol and Obesity: An Early Twentieth-Century Regulatory Dilemma.
Regul. Toxicol. Pharmacol. 2007, 48, 115–117.
40. Tainter, M. L.; Cutting, W. C.; Hines, E. Effects of Moderate Doses of Dinitrophenol on the
Energy Exchange and Nitrogen Metabolism of Patients under Conditions of Restricted
Dietary. J. Pharmacol. Exp. Ther. 1935, 55, 326–353.
41. Tainter, M. L.; Stockton, A. B.; Cutting, W. C. Use of Dinitrophenol in Obesity and Related
Conditions. JAMA 1933, 101, 1472–1475.
42. Horner, W. D. Cataract Following Dinitrophenol Treatment for Obesity. Arch. Ophthalmol.
1936, 16, 447–461.
43. Rodin, F. H. Cataracts Following the Use of Dinitrophenol: A Summary of Thirty-Two
Cases. Cal. West Med. 1936, 44, 276–279.
44. Bettman, J. W. Experimental Dinitrophenol Cataract. Am. J. Ophthalmol. 1946, 29, 1388–
1395.
45. Bianchetti, A.; Pugliatti, C.; Jori, A. On the Hyperthermia Induced by 2,4-Dinitrophenol.
Med. Pharmacol. Exp. 1967, 17, 401–408.
46. Poole, F. E.; Haining, R. B. Sudden Death from Dinitrophenol Poisoning. JAMA 1934, 102,
1141–1147.
47. McFee, R. B.; Caraccio, T. R.; McGuigan, M. A.; Reynolds, S. A.; Bellanger, P. Dying to
Be Thin: A Dinitrophenol Related Fatality. Vet. Hum. Toxicol. 2004, 46, 251–254.

109
48. Toxicological Profile of Dinitrophenols. In USDHHS, Ed. 1995;
http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=729&tid=132. (accessed March 14, 2017)
49. Jackson, P. C.; St. John, J. B. Effects of 2,4-Dinitrophenol on Membrane Lipids of Roots.
Plant Physiol. 1982, 70, 858–862.
50. Ravesloot, J. H.; Rombouts, E. 2,4-Dinitrophenol Acutely Inhibits Rabbit Atrial Ca2+-
Sensitive Cl- Current (Ito2). Can. J. Physiol. Pharmacol. 2000, 78, 766–773.
51. Brismar, T.; Collins, V. P. Effect of External Cation Concentration and Metabolic Inhibitors
on Membrane Potential of Human Glial Cells. J. Physiol. 1993, 460, 365-383.
52. Buckler, K. J.; Vaughan-Jones, R. D. Effects of Mitochondrial Uncouplers on Intracellular
Calcium, pH, and Membrane Potential in Rat Carotid Body Type I Cells. J. Physiol. 1998,
513, 819–833.
53. Petróczi, A.; Ocampo, J. A. V.; Shah, I.; Jenkinson, C.; New, R.; James, R. A.; Taylor, G.;
Naughton, D. P. Russian Roulette with Unlicensed Fat-Burner Drug 2,4-Dinitrophenol
(DNP): Evidence from a Multidisciplinary Study of the Internet, Bodybuilding Supplements
and DNP Users. Sub. Abuse Treat. Prevent. Policy 2015, 10, 39.
54. Caldeira da Silva, C. C.; Cerqueira, F. M.; Barbosa, L. F.; Medeiros, M. H. G.;
Kowaltowski, A. J. Mild Mitochondrial Uncoupling in Mice Affects Energy Metabolism,
Redox Balance, and Longevity. Aging Cell 2008, 7, 552–560.
55. Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio,
A.; Cantoni, O.; Clementi, E.; Moncada, S.; Carruba, M. O. Calorie Restriction Promotes
Mitochondrial Biogenesis by Inducing the Expression of Enos. Science 2005, 310, 314–317.

110
56. Lambert, A. J.; Merry, B. J. Effect of Caloric Restriction on Mitochondrial Reactive Oxygen
Species Production and Bioenergetics: Reversal by Insulin. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 2003, 286, 71–79.
57. Blaikie, F. H.; Brown, S. E.; Samuelsson, L. M.; Brand, M. D.; Smith, R. A.; Murphy, M. P.
Targeting Dinitrophenol to Mitochondria: Limitations to the Development of a Self-Limiting
Mitochondrial Protonophore. Biosci. Rep. 2006, 26, 231–243.
58. Chalmers, S.; Caldwell, S. T.; Quin, C.; Prime, T. A.; James, A. M.; Cairns, A. G.; Murphy,
M. P.; McCarron, J. G.; Hartley, R. C. Selective Uncoupling of Individual Mitochondria
within a Cell Using a Mitochondria-Targeted Photoactivated Protonophore. J. Am. Chem. Soc.
2012, 134, 758–761.
59. Lipton, S. A. Paradigm Shift in Neuroprotection by NMDA Receptor Blockade: Memantine
and Beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170.
60. De Felice, F. G.; Ferreira, S. T. Novel Neuroprotective, Neuritogenic and Anti-
Amyloidogenic Properties of 2,4-Dinitrophenol: The Gentle Face of Janus. IUBMB Life 2006,
58, 185–191.
61. Abdel-Salam, O. M. E.; Youness, E. R.; Mohammed, N. A.; Shaffie, N.; Abouelfadi, D. M.;
A., S. A. The Effect of 2,4-Dinitrophenol on Oxidative Stree and Neuronal Damage in Rat
Brain Induced by Systemic Rotenone Injection. React. Oxy. Spec. 2017, 3, 135–147.
62. Maragos, W. F.; Rockich, K. T.; Dean, J. J.; Young, K. L. Pre- or Post-Treatment with the
Mitochondrial Uncoupler 2,4-Dinitrophenol Attenuates Striatal Quinolinate Lesions. Brain
Res. 2003, 966, 312–316.

111
63. Korde, A. S.; Sullivan, P. G.; Maragos, W. F. The Uncoupling Agent 2,4-Dinitrophenol
Improves Mitochondrial Homeostasis Following Striatal Quinolinic Acid Injections. J.
Neurotrauma 2005, 22, 1142–1149.
64. Madeiro da Costa, R. F.; Martinez, A. M. B.; Ferreira, S. T. 2,4-Dinitrophenol Blocks
Neurodeneration and Preserves Sciatic Nerve Function after Trauma. J. Neurotrauma 2010,
27, 829–841.
65. Y., J.; McEwen, M. L.; Nottingham, S. A.; Maragos, W. F.; Dragicevic, N. B.; Sullivan, P.
G.; Springer, J. E. The Mitochondrial Uncoupling Agent 2,4-Dinitrophenol Improves
Mitochondrial Function, Attenuates Oxidative Damage, and Increases White Matter Sparing
in the Contused Spinal Cord. J. Neurotrauma 2004, 21, 1396–1404.
66. Maragos, W. F.; Korde, A. S. Mitochondrial Uncoupling as a Potential Therapeutic Target in
Acute Central Nervous System Injury. J. Neurochem. 2004, 91, 257–262.
67. Liu, D.; Zhang, Y.; Gharavi, R.; Park, H. R.; Lee, J.; Siddiqui, S.; Telljohann, R.; Nassar, M.
R.; Cutler, R. G.; Becker, K. G.; Mattson, M. P. The Mitochondrial Uncoupler Dnp Triggers
Brain Cell Mtor Signaling Network Reprogramming and Creb Pathway up-Regulation. J.
Neurochem. 2015, 134, 677–692.
68. Heytler, P. G.; Prichard, W. W. A New Class of Uncoupling Agents - Carbonyl Cyanide
Phenylhydrazones. Biochem. Biophys. Res. Commun. 1962, 7, 272–275.
69. Parker, V. H. Uncouplers of Rat-Liver Mitochondrial Oxidative Phosphorylation. Biochem.
J. 1965, 97, 658–662.
70. Goldsby, R. A.; Heytler, P. G. Uncoupling of Oxydative Phosphorylation by Carbonyl
Cyanide Phenylhydrazones. Biochemistry 1963, 2, 1142–1147.

112
71. Heytler, P. G. Uncoupling of Oxidative Phosphorylation by Carbonyl Cyanide
Phenylhydrazones. . Biochemistry 1963, 2, 357–361.
72. Heytler, P. G. Uncouplers of Oxidative Phosphorylation. Pharmacol. Ther. 1981, 10, 461–
472.
73. Park, K. S.; Jo, I.; Pak, K.; Bae, S. W.; Rhim, H.; Suh, S. H.; Park, J.; Zhu, H.; So, I.; Kim,
K. W. Fccp Depolarizes Plasma Membrane Potential by Activating Proton and Na+ Currents
in Bovine Aortic Endothelial Cells. Pflugers Arch. 2002, 443, 344–352.
74. To, M. S.; Aromataris, E. C.; Castro, J.; Roberts, M. L.; Barritt, G. J.; Rychkov, G. Y.
Mitochondrial Uncoupler FCCP Activates Proton Conductance but Does Not Block Store-
Operated Ca(2+) Current in Liver Cells. Arch. Biochem. Biophys. 2010, 495, 152–158.
75. Juthberg, S. K. A.; Brismar, T. Effect of Metabolic Inhibitors on Membrane Potential and
Ion Conductance of Rat Astrocytes. Cell. Mol. Neurobio. 1997, 17, 367–377.
76. Caputo, C.; Bolaños, P. Effect of Mitochondria Poisoning by Fccp on Ca2+ Signaling in
Mouse Skeletal Muscle Fibers. Pflugers Arch. Eur. J. Physiol. 2008, 455, 733–743.
77. Chen, F.; De Diego, C.; Xie, L.; Yang, J.; Klitzner, T. S.; Weiss, J. N. Effects of Metabolic
Inhibition on Conduction, Ca Transients, and Arrhythmia Vulnerability in Embryonic Mouse
Hearts. J. Physiol. Heart Circ. Physiol. 2007, 293, 2472–2478.
78. Doebler, J. A. Effects of Protonophores on Membrane Electrical Characteristics in NG108-
15 Cells. Neurochem. Res. 2000, 25, 263–168.
79. Fukumoto, G. H.; Lamp, S. T.; Motter, C.; Bridge, J. H.; Garfinkel, A.; Goldhaber, J. I.
Metabolic Inhibition Alters Subcellular Calcium Release Patterns in Rat Ventricular
Myocytes: Implications for Defective Excitation-Contraction Coupling During Cardiac
Ischemia and Failure. Circ. Res. 2005, 96, 551–557.

113
80. Kim, G. H.; Kim, J. E.; Rhie, S. J.; Yoon, S. The Role of Oxidative Stress in
Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340.
81. Johri, A.; Beal, M. F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J.
Pharmacol. Exp. Ther. 2012, 342, 619-630.
82. Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.;
McLean, J. R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; Hargus, G.; Deleidi, M.; Lawson, T.;
Bogetofte, H.; Perez-Torres, E.; Clark, L.; Moskowitz, C.; Mazzulli, J.; Chen, L.; Volpicelli-
Daley, L.; Romero, N.; Jiang, H.; Uitti, R. J.; Huang, Z.; Opala, G.; Scarffe, L. A.; Dawson,
V. L.; Klein, C.; Feng, J.; Ross, O. A.; Trojanowski, J. Q.; Lee, V. M. Y.; Marder, K.;
Surmeier, D. J.; Wszolek, Z. K.; Przedborski, S.; Krainc, D.; Dawson, T. M.; Isacson, O.
Pharmacological Rescue of Mitochondrial Deficits in IPSC-Derived Neural Cells from
Patients with Familial Parkinson's Disease. Sci. Transl. Med. 2012, 4, 1–14.
83. Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A. A.; McKhann, G. M.; Yan, Y.; Wang, C.;
Zhang, H.; Molkentin, J. D.; Gunn-Moore, F. J.; Vonsattel, J. P.; Arancio, O.; Chen, J. X.;
Yan, S. D. Cyclophilin D Deficiency Attenuates Mitochondrial and Neuronal Perturbation
and Ameliorates Learning and Memory in Alzheimer's Disease. Nat. Med. 2008, 14, 1097–
1105.
84. Krebiehl, G.; Ruckerbauer, S.; Burbulla, L. F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg,
H.; Gizatullina, Z.; Gellerich, F. N.; Woitalla, D.; Riess, O.; Kahle, P. J.; Proikas-Cezanne, T.;
Kruger, R. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of
Parkinson's Disease-Associated Protein DJ-1. PLoS One 2010, 5, e9367.

114
85. Yao, J.; Irwin, R. W.; Zhao, L.; Nilsen, J.; Hamilton, R. T.; Brinton, R. D. Mitochondrial
Bioenergetic Deficit Precedes Alzheimer's Pathology in Female Mouse Model of Alzheimer's
Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675.
86. Jarmakani, J. M.; Nakazawa, M.; Gagatomo, T.; Langer, G. A. Effect of Hypoxia on
Mechanical Function in the Neonatal Mammalian Heart. Am. J. Physiol. 1978, 235, 469-474.
87. Regula, C. S.; Pfeiffer, J. R.; Berlin, R. D. Microtubule Assembly and Disassembly at
Alkaline Ph. J. Cell Bio. 1981, 89, 45–53.
88. Rottenberg, H. Uncoupling of Oxidative Phosphorylation in Rat Liver Mitochondria by
General Anesthetics. Proc. Natl. Acad. Sci. USA 1983, 80, 3313–3317.
89. Dabadie, P.; Bendriss, P.; Erny, P.; Mazat, J.-P. Uncoupling Effects of Local Anesthetics on
Rat Liver Mitochondria. FEBS Lett. 1987, 226, 77–82.
90. Terada, H.; Shima, O.; Yoshida, K.; Shinohara, Y. Effects of the Local Anesthetic
Bupivacaine on Oxidative Phosphorylation in Mitochondria. J. Biol. Chem. 1990, 265, 7837–
7842.
91. Cela, O.; Piccoli, C.; Scrima, R.; Quarato, G.; Marolla, A.; Cinnella, G.; Dambrosio, M.;
Capitanio, N. Bupivacaine Uncouples the Mitochondrial Oxidative Phosphorylation, Inhibits
Respiratory Chain Complexes I and III and Enhances Ros Production: Results of a Study on
Cell Cultures. Mitochondrion 2010, 10, 487–496.
92. Starkov, A. A.; Wallace, K. B. Structural Determinants of Fluorochemical-Induced
Mitochondrial Dysfunction. Tox. Sci. 2002, 66, 244–252.
93. Sztark, F.; Ouhabi, R.; Dabadie, P.; Mazat, J.-P. Effects of the Local Anesthetic Bupivacaine
on Mitochondrial Energy Metabolism: Change from Uncoupling to Decoupling Depending on
the Respiration State. Biochem. Mol. Biol. Int. 1997, 43, 997–1003.

115
94. Schönfeld, P.; Sztark, F.; Slimani, M.; Dabadie, P.; Mazat, J.-P. Is Bupivacaine a Decoupler,
a Protonophore or a Proton-Leak Inducer? FEBS Lett. 1992, 304, 273–276.
95. Sun, X.; Garlid, K. D. On the Mechanism by Which Bupivacaine Conducts Protons across
the Membranes of Mitochondria and Liposomes. J. Biol. Chem. 1992, 267, 19147–19154.
96. van Dam, K.; Shinohara, Y.; Unami, A.; Yoshida, J.; Terada, H. Slipping Pumps or Proton
Leaks in Oxidative Phosphorylation. FEBS Lett. 1990, 277, 131–133.
97. Irwin, W.; Fontaine, E.; Agnolucci, L.; Penzo, D.; Betto, R.; Bortolotto, S.; Reggiani, C.;
Salviati, G.; Bernardi, P. Bupivacaine Myotoxicity Is Mediated by Mitochondria. J. Biol.
Chem. 2002, 277, 12221–12227.
98. Perez-Castro, R.; Patel, S.; Garavito-Aguilar, Z. V.; Rosenberg, A.; Recio-Pinto, E.; Zhang,
J.; Blanck, T. J.; Xu, F. Cytotoxicity of Local Anesthetics in Human Neuronal Cells. Anesth.
Analg. 2009, 108, 997–1007.
99. Zink, W.; Graf, B. M. Local Anesthetic Myotoxicity. Reg. Anesth. Pain Med. 2004, 29, 333–
340.
100. Lirk, P.; Haller, I.; Colvin, H. P.; Lang, L.; Tomaselli, B.; Klimaschewski, L.; Gerner, P. In
Vitro, Inhibition of Mitogen-Activated Protein Kinase Pathways Protects against
Bupivacaine- and Ropivacaine-Induced Neurotoxicity. Anesth. Analg. 2008, 106, 1456–1464.
101. Onyuksel, H.; Sethi, V.; Weinberg, G. L.; Dudeja, P. K.; Rubinstein, I. Bupivacaine, but
Not Lidocaine, Disrupts Cardiolipin-Containing Small Biomimetic Unilamellar Liposomes.
Chem. Biol. Interact. 2007, 169, 154–159.
102. Weinberg, G. L.; Palmer, J. W.; VadeBoncouer, T.; Zuechner, M. B.; Edelman, G.; Hoppel,
C. L. Bupivacain Inhibits Acylcarnitine Exchange in Cardiac Mitochondria. Anesthesiology
2000, 92, 523–528.

116
103. Deng, Y. T.; Huang, H. C.; Lin, J. K. Rotenone Induces Apoptosis in MCF-7 Human
Breast Cancer Cell-Mediated Ros through Jnk and P38 Signaling. Mol. Carcinog. 2010, 49,
141–151.
104. Weinberg, G. L.; VadeBoncouer, T.; Ramaraju, G. A.; Garcia-Amaro, M. F.; Cwik, M. J.
Pretreatment or Resuscitation with a Lipid Infusion Shifts the Dose-Response to Bupivacaine-
Induced Asystole in Rats. Anesthesiology 1998, 88, 1071–1075.
105. Chen, Z.; Jin, Z.; Xia, Y.; Zhao, S.; Xu, X.; Papadimos, T. J.; Wang, Q. The Protective
Effect of Lipid Emulsion in Preventing Bupivacaine-Induced Mitochondrial Injury and
Apoptosis of H9C2 Cardiomyocytes. Drug Deliv. 2017, 24, 430–436.
106. Burton, D. E.; Lambie, A. J.; Ludgate, J. C. L.; Newbold, G. T.; Percival, A.; Saggers, T. 2-
Trifluorobenzimidazoles: A New Class of Herbicidal Compounds. Nature 1965, 208, 1166–
1169.
107. Jones, O. T. G.; Watson, W. A. Activity of 2-Trifluoromethylbenzimidazoles as
Uncouplers of Oxidative Phosphorylation. Nature 1965, 208, 1169–1170.
108. Büchel, K. H.; Korte, F.; Beechey, R. B. Uncoupling of the Oxidative Phosphorylation in
Mitochondria by NH-Acidic Benzimidazoles. Angew. Chem. Int. Ed. 1965, 4, 788–789.
109. Beechey, R. B. The Uncoupling of Respiratory-Chain Phosphorylation by 4,5,6,7-
Tetrachloro-2-Trifluoromethylbenzimidazole. Biochem. J. 1965, 98, 284–289.
110. Ilivicky, J.; Casida, J. E. Uncoupling Action of 2,4-Dinitrophenols, 2-
Trifluoromethylbenzimidazoles, and Certain Other Pesticide Chemicals Upon Mitochondria
from Different Sources and Its Relation to Toxicity. Biochem. Pharmacol. 1969, 18, 1389–
1401.

117
111. Khailova, L. S.; Silachev, D. N.; Rokitskaya, T. I.; Avetisyan, A. V.; Lyamsaev, K. G.;
Severina, II; Il'yasova, T. M.; Gulyaev, M. V.; Dedukhova, V. I.; Trendeleva, T. A.;
Plotnikov, E. Y.; Zvyagilskaya, R. A.; Chernyak, B. V.; Zorov, D. B.; Antonenko, Y. N.;
Skulachev, V. P. A Short-Chain Alkyl Derivative of Rhodamine 19 Acts as a Mild Uncoupler
of Mitochondria and a Neuroprotector. Biochim. Biophys .Acta 2014, 1837, 1739–1747.
112. Figarola, J. L.; Weng, Y.; Lincoln, C.; Horne, D.; Rahbar, S. Novel Dichlorophenyl Urea
Compounds Inhibit Proliferation of Human Leukemia Hl-60 Cells by Inducing Cell Cycle
Arrest, Differentiation and Apoptosis. Invest. New Drugs 2012, 30, 1413–1425.
113. Singhal, S. S.; Figarola, J.; Singhal, J.; Leake, K.; Nagaprashantha, L.; Lincoln, C.; Gabriel
Gugiu, B.; Horne, D.; Jove, R.; Awasthi, S.; Rahbar, S. 1,3-bis(3,5-dichlorophenyl) urea
Compound 'COH-SR4' Inhibits Proliferation and Activates Apoptosis in Melanoma. Biochem.
Pharmacol. 2012, 84, 1419–1427.
114. Figarola, J. L.; Rahbar, S. Smallmolecule COH-SR4 Inhibits Adipocyte Differentiation Via
AMPK Activation. Int. J. Mol. Med. 2013, 31, 1166–1176.
115. Kikuchi, H.; Ishiko, S.; Nakamura, K.; Kubohara, Y.; Oshima, Y. Novel Prenylated and
Geranylated Aromatic Compounds Isolated from Polysphondylium Cellular Slime Molds.
Tetrahedron 2010, 66, 6000–6007.
116. Homma, Y.; Suzuki, T.; Ogura, M.; Oshima, Y.; Kikuchi, H. Prenyloxyquinoline
Carboxylic-Acid Derivative WO2014061647 A1, 2014.
117. Kikuchi, H.; Suzuki, T.; Ogura, M.; Homma, M. K.; Homma, Y.; Oshima, Y. Synthesis of
Prenylated Quinolinecarboxylic Acid Derivatives and Their Anti-Obesity Activities. Bioorg.
Med. Chem. 2015, 23, 66–72.

118
118. Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and Regulation of the Permeability
Transition Pore. J. Bioenerg. Biomembr. 2017, 49, 27–47.
119. Drakakaki, G.; Robert, S.; Szatmari, A.-M.; Brown, M. Q.; Nagawa, S.; Damme, D. V.;
Leonard, M.; Yang, Z.; Girke, h.; Schmid, S. L.; Russinovab, E.; Friml, J.; Raikhel, N. V.;
Hicks, G. R. Clusters of Bioactive Compounds Target Dynamic Endomembrane Networks in
Vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17850–17855.
120. Frayha, G. J.; Smyth, J. D.; Gobert, J. G.; Savel, J. The Mechanisms of Action of
Antiprotozoal and Anthelmintic Drugs in Man. Gen. Pharmacol. 1997, 28, 273–299.
121. Andrews, P.; Thyssen, J.; Lorke, D. The Biology and Toxicology of Molluscicides,
Bayluscide. Pharmacol. Therapeut. 1982, 19, 245–295.
122. Hecht, G.; Gloxhuber, C. Studies on the Tolerance of 5,2'-Dichloro-4'-Nitrosalicylanilide
Ethanolamine Salt. Z. Tropenmed. Parasitol. 1962, 13, 1–8.
123. Chowdhury, M. K.; Turner, N.; Bentley, N. L.; Das, A.; Wu, L. E.; Richani, D.;
Bustamante, S.; Gilchrist, R. B.; Morris, M. J.; Shepherd, P. R.; Smith, G. C. Niclosamide
Reduces Glucagon Sensitivity via Hepatic PKA Inhibition in Obese Mice: Implications for
Glucose Metabolism Improvements in Type 2 Diabetes. Sci. Rep. 2017, 7, 40159.
124. Gosalvez, M.; Blanco, M.; Hunter, J.; Miko, M.; Chance, B. Effects of Anticancer Agents
on the Respiration of Isolated Mitochondria and Tumor Cells. Eur. J. Cancer 1974, 10, 567–
574.
125. Dupont, J.; Dodin, G.; Schwaller, M. A. Interactions between Ellipticine Derivatives and
the Mitochondrial Respiratory Chain. Plant Science 1988, 54, 109–115.

119
126. Schwaller, M.-A.; Allard, B.; Lescot, E.; Moreau, F. o. Protonophoric Activity of
Ellipticine and Isomers across the Energy-Transducing Membrane of Mitochondria. J. Biol.
Chem. 1995, 270, 22709–22713.
127. Dupont, J.; Schwaller, M. A.; Dodin, G. Ditercalinium, a Nucleic Acid Binder, Inhibits the
Respiratory Chain of Isolated Mammalian Mitochondria. Cancer Res. 1990, 50, 7966–7972.
128. Terce, F.; Tocanne, J. F.; Laneelle, G. Interactions of Ellipticine with Model or Natural
Membranes. A Spectrophotometric Study. Eur. J. Biochem. 1982, 125, 203–207.
129. Terce, F.; Tocanne, J. F.; Laneelle, G. Ellipticine-Induced Alteration of Model and Natural
Membranes. Biochem. Pharmacol. 1983, 32, 2189–2194.
130. Hägg, M.; Berndtsson, M.; Mandic, A.; Zhou, R.; Shoshan, M. C.; Linder, S. Induction of
Endoplasmic Reticulum Stress by Ellipticine Plant Alkaloids. Mole. Cancer Ther. 2004, 3,
489.
131. Tewey, K. M.; Chen, G. L.; Nelson, E. M.; Liu, L. F. Intercalative Antitumor Drugs
Interfere with the Breakage-Reunion Reaction of Mammalian DNA Topoisomerase II. J. Biol.
Chem. 1984, 259, 9182–9187.
132. Burns, R. J.; Murphy, M. P. Labeling of Mitochondrial Proteins in Living Cells by the
Thiol Probe Thiobutyltriphenylphosphonium Bromide. Arch. Biochem. Biophys. 1997, 339,
33–39.
133. Burns, R. J.; Smith, R. A. J.; Murphy, M. P. Synthesis and Characterization of
Thiobutyltriphenylphosphonium Bromide, a Novel Thiol Reagent Targeted to the
Mitochondrial Matrix. Arch. Biochem. Biophys. 1995, 322, 60–68.
134. Hoek, J. B.; Nicholls, D. G.; Williamson, J. R. Determination of the Mitochondrial
Protonmotive Force in Isolated Hepatocytes. J. Biol. Chem. 1980, 255, 1458–1464.

120
135. Liberman, E. A.; Topaly, V. P.; Tsofina, L. M.; Jasaitis, A. A.; Skulachev, V. P.
Mechanism of Coupling of Oxidative Phosphorylation and the Membrane Potential of
Mitochondria. Nature 1969, 222, 1076-1078.
136. Murphy, M. P. Selective Targeting of Bioactive Compounds to Mitochondria. Trends
Biotech. 1997, 15, 326–330.
137. Ross, M. F.; Kelso, G. F.; Blaikie, F. H.; James, A. M.; Cocheme, H. M.; Filipovska, A.;
Ros, T.; Hurd, T. R.; Smith, R. A. J.; Murphy, M. P. Lipophilic Triphenylphosphonium
Cations as Tools in Mitochondrial Bioenergetics and Free Radical Biology. Biochemistry
(Moscow) 2005, 70, 222–230.
138. Smith, R. A. J.; Porteous, C. M.; Coulter, C. V.; Murphy, M. P. Selective Targeting of an
Antioxidant to Mitochondria. Eur. J. Biochem. 1999, 263, 709–716.
139. Severin, F. F.; Severina, II; Antonenko, Y. N.; Rokitskaya, T. I.; Cherepanov, D. A.;
Mokhova, E. N.; Vyssokikh, M. Y.; Pustovidko, A. V.; Markova, O. V.; Yaguzhinsky, L. S.;
Korshunova, G. A.; Sumbatyan, N. V.; Skulachev, M. V.; Skulachev, V. P. Penetrating
Cation/Fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad.
Sci. USA 2010, 107, 663–668.
140. Sukhanova, E. I.; Trendeleva, T. A.; Zvyagilskaya, R. A. Interaction of Yeast Mitochondria
with Fatty Acids and Mitochondria-Targeted Lipophilic Caions. Biochemistry (Mosc) 2010,
75, 139–144.
141. Trendeleva, T. A.; Sukhanova, E. I.; Rogov, A. G.; Zvyagilskaya, R. A.; Seveina, II;
Ilyasova, T. M.; Cherepanov, D. A.; Skulachev, V. P. Role of Charge Screening and
Delocalization for Lipophilic Cation Permeability of Model and Mitochondrial Membranes.
Mitochondrion 2013, 13, 500–506.

121
142. Kalinovich, A. V.; Mattsson, C. L.; Youssef, M. R.; Petrovic, N.; Ost, M.; Skulachev, V.
P.; Shabalina, I. G. Mitochondria-Targeted Dodecyltriphenylphosphonium (C12TPP)
Combats High-Fat-Diet-Induced Obesity in Mice. Int. J. Obes. (Lond) 2016, 40, 1864–1874.
143. Williamson, R. L.; Metcalf, R. L. Salicylanilides: A New Group of Active Uncouplers of
Oxidative Phosphorylation. Science 1967, 158, 1694–1695.
144. Terada, H.; Goto, S.; Yamamoto, K.; Takeuchi, I.; Hamada, Y.; Miyake, K. Structural
Requirements of Salicylanilides for Uncoupling Activity in Mitochondria: Quantitative
Analysis of Structure–Uncoupling Relationships. Biochem. Biophys. Acta 1988, 936, 504–
512.
145. Kasianowicz, J.; Benz, R.; McLaughlin, S. How Do Protons Cross the Membrane-Solution
Interface? Kinetic Studie on Bilayer Membranes Exposed to the Protonophore S-13 (5-chloro-
3-tert-butyl-2’-chloro-4’-nitrosalicylanilide. J. Membrane Biol. 1987, 95, 73–89.
146. Kaplay, M.; Kurup, C. K. R.; Lam, K. W.; Sanadi, D. R. Oxidative Phosphorylation. XX.
Stoichiometric Aspects of Uncoupling of Oxidative Phosphorylation by a Salicylanilide
Derivative. Biochemistry 1970, 9, 3599–3604.
147. Horiuchi, F.; Fujimoto, K.; Ozaki, T.; Nishizawa, Y. 3, 5-dialkyl-4-
hydroxybenzylidenemalononitrile; a New Group of Fungicidal and Acaricidal Agents. Agr.
and Biol. Chem. 1971, 35, 2003–2007.
148. Muraoka, S.; Terada, H. 3,5-di-tert-butyl-4-hydroxybenzylidenemalononitrile; a New
Powerful Uncoupler of Respiratory-Chain Phosphorylation. Biochim. Biophys. Acta 1972,
275, 271–275.

122
149. Terada, H. Some Biochemical and Physicochemical Properties of the Potent Uncoupler SF
6847 (3,5-di-tert-butyl-4-hydroxybenzylidenemalononitrile). Biochim. Biophys. Acta 1975,
387, 519–532.
150. Yoshikawa, K.; Kumazawa, N.; Terada, H.; Akagi, K. Physicochemical Properties of SF
6847, a Potent Uncoupler of Oxidative Phosphorylation in Mitochondria in Relation to its
Activity. Int. J. Quantum Chem. 1980, 18, 539–544.
151. Terada, H.; Kumazawa, N.; Ju-Ichi, M.; Yoshikawa, K. Molecular Basis of the
Protonophoric and Uncoupling Activities of the Potent Uncoupler SF-6847 ((3,5-di-tert-butyl-
4-hydroxybenzylidene)malononitrile) and Derivatives. Regulation of Their Electronic
Structures by Restricted Intramolecular Rotation. Biochim. Biophys. Acta 1984, 767, 192–
199.
152. Hori, H.; Nagasawa, H.; Ishibashi, M.; Uto, Y.; Hirata, A.; Saijo, K.; Ohkura, K.; Kirk, K.
L.; Uehara, Y. TX-1123: An Antitumor 2-hydroxyarylidene-4-cyclopentene-1,3-dione as a
Protein Tyrosine Kinase Inhibitor Having Low Mitochondrial Toxicity. Bioorg. Med. Chem.
2002, 10, 3257–3265.
153. Sagara, Y.; Ishige, K.; Tsai, C.; Maher, P. Tyrphostins Protect Neuronal Cells from
Oxidative Stress. J. Biol. Chem. 2002, 277, 36204–36215.
154. Ohkura, K.; Hori, H. Modification of Cell Response to Insulin by Membrane-Acting
Agents in Rat White Adipocytes: Analysis of Structural Features by Computational
Simulation. Bioorg. Med. Chem. 2001, 9, 3023–3033.
155. Fuortes, M.; Melchior, M.; Han, H.; Lyon, G. J.; Nathan, C. Role of the Tyrosine Kinase
PYK2 in the Integrin-Dependent Activation of Human Neutrophils by Tnf. J. Clin. Invest.
1999, 104, 327–335.

123
156. Levitzki, A.; Mishani, E. Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu. Rev.
Biochem. 2006, 75, 93–109.
157. Marhaba, R.; Mary, F.; Pelassy, C.; Stanescu, A. T.; Aussel, C.; Breittmayer, J. P.
Tyrphostin A9 Inhibits Calcium Release-Dependent Phosphorylations and Calcium Entry via
Calcium Release-Activated Channel in Jurkat T Cells. J. of Immunol. 1996, 157, 1468–1473.
158. Fu, Y. Y.; Zhang, M.; Turner, N.; Zhang, L. N.; Dong, T. C.; Gu, M.; Leslie, S. J.; Li, J. Y.;
Nan, F. J.; Li, J. A Novel Chemical Uncoupler Ameliorates Obesity and Related Phenotypes
in Mice with Diet-Induced Obesity by Modulating Energy Expenditure and Food Intake.
Diabetologia 2013, 56, 2297–2307.
159. Kelso, G. F.; Porteous, C. M.; Coulter, C. V.; Hughes, G.; Porteous, W. K.; Ledgerwood,
E. C.; Smith, R. A.; Murphy, M. P. Selective Targeting of a Redox-Active Ubiquinone to
Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001,
276, 4588–4596.
160. Chernyak, B. V.; Antonenko, Y. N.; Domnina, L. V.; Ivanova, O. Y.; Lyamzaev, K. G.;
Pustovidko, A. V.; Rokitskaya, T. I.; Severina, I. I.; Simonyan, R. A.; Trendeleva, T. A.;
Zvyagilskaya, R. A. Novel Penetrating Cations for Targeting Mitochondria. Curr. Pharm.
Des. 2013, 19, 2795–2806.
161. Denisov, S. S.; Kotova, E. A.; Plotnikov, E. Y.; Tikhonov, A. A.; Zorov, D. B.;
Korshunova, G. A.; Antonenko, Y. N. A Mitochondria-Targeted Protonophoric Uncoupler
Derived from Fluorescein. Chem. Commun. 2014, 50, 15366–15369.
162. Antonenko, Y. N.; Denisov, S. S.; Silachev, D. N.; Khailova, L. S.; Jankauskas, S. S.;
Rokitskaya, T. I.; Danilina, T. I.; Kotova, E. A.; Korshunova, G. A.; Plotnikov, E. Y.; Zorov,
D. B. A Long-Linker Conjugate of Fluorescein and Triphenylphosphonium as Mitochondria-

124
Targeted Uncoupler and Fluorescent Neuro- and Nephroprotector. Biochim. Biophys. Acta
2016, 1860, 2463–2473.
163. Wu, S.; Cao, Q.; Wang, X.; Cheng, K.; Cheng, Z. Design, Synthesis and Biological
Evaluation of Mitochondria Targeting Theranostic Agents. Chem. Commun. 2014, 50, 8919–
8922.
164. Asin-Cayuela, J.; Manas, A. R.; James, A. M.; Smith, R. A.; Murphy, M. P. Fine-Tuning
the Hydrophobicity of a Mitochondria-Targeted Antioxidant. FEBS Lett. 2004, 571, 9–16.
165. Smith, R. A. J.; Porteous, C. M.; Gane, A. M.; Murphy, M. P. Delivery of Bioactive
Molecules to Mitochondria in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412.
166. Rodriguez-Cuenca, S.; Cocheme, H. M.; Logan, A.; Abakumova, I.; Prime, T. A.; Rose, C.;
Vidal-Puig, A.; Smith, A. C.; Rubinsztein, D. C.; Fearnley, I. M.; Jones, B. A.; Pope, S.;
Heales, S. J.; Lam, B. Y.; Neogi, S. G.; McFarlane, I.; James, A. M.; Smith, R. A.; Murphy,
M. P. Consequences of Long-Term Oral Administration of the Mitochondria-Targeted
Antioxidant Mitoq to Wild-Type Mice. Free Radic. Biol. Med. 2010, 48, 161–172.
167. Fink, B. D.; Guo, D. F.; Kulkarni, C. A.; Rahmouni, K.; Kerns, R. J.; Sivitz, W. I.
Metabolic Effects of a Mitochondrial-Targeted Coenzyme Q Analog in High Fat Fed Obese
Mice. Pharmacol. Res. Perspect. 2017, 5, 1–13.
168. Adlam, V. J.; Harrison, J. C.; Porteous, C. M.; James, A. M.; Smith, R. A. J.; Murphy, M.
P.; Sammut, I. A. Targeting an Antioxidant to Mitochondria Decreases Cardiac Ischemia-
Reperfusion Injury. FASEB J. 2005, 19, 1088–1095.
169. Graham, D.; Huynh, N. N.; Hamilton, C. A.; Beattie, E.; Smith, R. A. J.; Cocheme, H. M.;
Murphy, M. P.; Dominiczak, A. F. Mitochondria-Targeted Antioxidant MitoQ10 Improves
Endothelial Function and Attenuates Cardiac Hypertrophy. Hypertension 2009, 54, 322–328.

125
170. Lowes, D. A.; Thottakam, B. M.; Webster, N. R.; Murphy, M. P.; Galley, H. F. The
Mitochondria-Targeted Antioxidant MitoQ Protects against Organ Damage in a
Lipopolysaccharide-Peptidoglycan Model of Sepsis. Free Radic. Biol. Med. 2008, 45, 1559–
1565.
171. Snow, B. J.; Rolfe, F. L.; Lockhart, M. M.; Frampton, C. M.; O'Sullivan, J. D.; Fung, V.;
Smith, R. A. J.; Murphy, M. P.; Taylor, K. M. A Double-Blind, Placebo–Controlled Study to
Assess the Mitochondria-Targeted Antioxidant Mitoq as a Disease-Modifying Therapy in
Parkinson's Disease. Move. Disord. 2010, 25, 1670–1674.
172. Gane, E. J.; Weilert, F.; Orr, D. W.; Keogh, G. F.; Gibson, M.; Lockhart, M. M.; Frampton,
C. M.; Taylor, K. M.; Smith, R. A. J.; Murphy, M. P. The Mitochondria-Targeted Anti-
Oxidant Mitoquinone Decreases Liver Damage in a Phase Ii Study of Hepatitis C Patients.
Liver Int. 2010, 30, 1019–1026.
173. Kenwood, B. M.; Calderone, J. A.; Taddeo, E. P.; Hoehn, K. L.; Santos, W. L. Structure–
Activity Relationships of Furazano[3,4-B]Pyrazines as Mitochondrial Uncouplers. Bioorg.
Med. Chem. 2015, 25, 4858–4861.
174. Aw, W. C.; Bajracharya, R.; Towarnicki, S. G.; Ballard, J. W. Assessing Bioenergetic
Functions from Isolated Mitochondria in Drosphila Melanogaster. J. Biol. Methods 2016, 3,
1–6.
175. Kooragayala, K.; Gotoh, N.; Cogliati, T.; Nellissery, J.; Kaden, T. R.; French, S.; Balaban,
R. S.; Covian, R.; Swaroop, A. Quantification of Oxygen Consumption in Retina ex Vivo
Demonstrates Limited Reserve Capacity of Photoreceptor Mitochondria. Biochem. Mol. Biol.
2015, 56, 8428–8438.

126
176. Laker, R. C.; Xu, P.; Ryall, K. A.; Sujkowski, A.; Kenwood, B. M.; Chain, K. H.; Zhang,
M.; Royal, M. A.; Hoehn, K. L.; Driscoll, M.; Adler, P. N.; Wessells, R. J.; Saucerman, J. J.;
Yan, Z. A Novel Mitotimer Reporter Gene for Mitochondrial Content, Structure, Stress, and
Damage in Vivo. J. Biol. Chem. 2014, 289, 12005–12015.

127
5 Structure-Activity Relationship Studies of Uncouplers of Oxidative
Phosphorylation
5.1 Contributions
The work in this chapter was done primarily by the author. The author synthesized and
characterized all final compounds and their corresponding intermediates. Stefan Hargett of the
University of Virginia Department of Pharmacology conducted all biological analyses of the
reported compounds. Dr. Yumin Dai conducted all pKa determinations of the reported
compounds. This chapter is an adaptation of a manuscript currently being written by the author
and Dr. Webster Santos for publication in a peer-reviewed journal.
5.2 Abstract
Mitochondrial uncouplers are small molecules that leak protons from the mitochondrial
membrane space into the mitochondrial matrix independent of ATP synthase, causing a
disruption in the proton motive force generated during oxidative phosphorylation. In response,
nutrient metabolism increases to reestablish the proton gradient necessary for normal cellular
function. Consequently, mitochondrial uncouplers have generated interest over the years for their
therapeutic potential for the treatment of obesity and type 2 diabetes. In this article, we describe
the synthesis and biological evaluation of small molecule mitochondrial uncouplers. Altering the
electronics on the aniline ring of the mitochondrial uncoupler 5.13 (BAM15) generated
analogues with good mitochondrial uncoupling activity across a wide therapeutic range.
Evaluation of pKa and cLogP of analogues of 5.13 suggest the best uncoupling activity is
observed from molecules that have a pKa between 6.8 and 8.0 and a cLogP > 6.

128
5.3 Introduction
Oxidative phosphorylation is an essential aerobic process that utilizes the potential
energy in the form of a proton motive force (pmf) generated from the electron transport chain to
produce the high-energy biomolecule adenosine triphosphate (ATP) via ATP synthase.2 Once
formed, ATP can be used to fuel various biological reactions to maintain cellular function, or it
can be used as a signaling molecule.177 Leakage of protons from the mitochondrial inner
membrane space into the mitochondrial matrix independent of ATP synthase to disrupt the pmf
used to generate ATP is known as mitochondrial uncoupling.5, 6 The process of mitochondrial
uncoupling occurs naturally in mammals, particularly in brown adipose tissue, which use
uncoupling proteins (UCP) that are situated throughout the mitochondrial inner membrane.8-11, 178
Mammals have five known UCPs, UCP1–5. Of the five UCPs, UCP1–3 are the most studied and
well understood.8-11 UCP1 has long been established as a promoter of non-shivering
thermogenesis,12, 179 while UCP2–3 are thought7, 180 to play a protective role by minimizing
oxidative damage from radical oxygen species.
Small molecule mitochondrial uncouplers have been developed to pharmacologically
mimic UCP activity (Figure 5.1).6, 181, 182 These uncouplers are typically lipophilic weak acids
that can transport protons from the mitochondrial inner membrane space into the mitochondrial
matrix independent of ATP synthase. Their therapeutic potential for the treatment of various
disease states have been explored, particularly for the treatment of obesity19, 23, 183, 184 and type 2
diabetes (T2D)24, 33, 34 due to their ability to promote increased nutrient metabolism and energy
expenditure. Multiple reports have shown19, 24, 32-34, 142, 158 that mice fed a high fat diet
supplemented with small molecule mitochondrial uncouplers gained less weight than controls
and also had reduced blood glucose and lipid levels, in addition to improved insulin sensitivity,18,

129
20-25 highlighting the therapeutic benefits of mitochondrial uncouplers in the treatment of obesity
and T2D.
Figure 5.1. Select Mitochondrial Uncouplers
Over the past 80 years, numerous mitochondrial uncouplers have been developed and
reported. Of the published mitochondrial uncouplers, 5.1 (DNP) is the most well-known.
Originally used as an explosive,185 5.1 was found to have weight-loss-inducing effects,
promoting upwards of 50% increase in metabolism,40 which led this drug to be marketed as a
weight-loss supplement.186 However, 5.1 caused adverse side effects including drastic increases
in body temperature,41, 45-47 cataracts,42-44, 187 and blindness,187 which led the U. S. Food and Drug
Administration to remove this drug from the market in 1938.48 Studies now indicate that the
toxicity issues associated with 5.1 are due to a narrow therapeutic index and off-target effects
resulting from dual depolarization of the mitochondrial inner membrane and plasma
membrane.49-52, 75, 188 Unfortunately, a mitochondria-targeted analogue of 5.1 tethered to a
triphenylphosphonium cation (5.2, MitoDNP)57 severely lost uncoupling activity below 50 µM.
OHNO2
NO2
OCF3
HN N
N
N
5.5 (FCCP)
N
O
O
O
O
5.7 (Ppc-1)
HN
HN
O
Cl
Cl
Cl
Cl
5.8 (SR4)
ONO2
NO2
5.4 (DNPME)
OH
Cl NH
OCl NO2
H2N OH
5.6 (Niclosamide Ethanolamine)
O NH
NH
O
O3
5.9 (C4R1)
PC12H25
5.10 (C12TPP)
HN S
O2
SBr
O2N
5.11 (ES9)
5.1 (DNP)
HOBr
Br S
NN
Cl
CO2EtO
5.12 (CZ5)
O
O2N
NO2
NO2
O P
5.3 (MitoPhotoDNP)
HO
O2N
NO2
P
5.2 (MitoDNP)

130
A modified version of 5.2 caged with a photocleavable linker (5.3, MitoPhotoDNP)58
successfully released 5.1 in the mitochondria upon 355 nm UV irradiation. In vivo application of
this molecule has yet to be reported. More recently, Perry et al. reported a controlled-release
derivative of 5.1 that is orally bioavailable with an increased therapeutic range, reduced toxicity,
promoted insulin sensitivity, and improved glucose tolerance in T2D rats.34 Further, a prodrug
version of 5.1, 5.4 (DNPME,) was found to be non-toxic and liver specific while improving
glucose tolerance in T2D rats and increasing insulin sensitivity.33 Another notable potent
mitochondrial uncoupler is 5.5 (FCCP).68, 189, 190 Unfortunately, its very narrow therapeutic
window and off-target effects at the plasma membrane severely limit therapeutic applications in
vivo.51, 74, 75, 191 Nonetheless, the continued search for mitochondrial uncouplers has generated
5.6 (niclosamide ethanolamine),24 5.7 (Ppc-1),23 5.8 (SR4),16, 32 5.9 (C4R1),14, 19, 111 5.10 (ES9),15
5.11 (CZ5),158 and 5.12 (C12TPP).142 Despite this recent progress, the field has suffered from a
paucity of mitochondrial selective agents with a wide therapeutic window.
Recently, we reported the discovery and synthesis of a novel mitochondrial uncoupler,
5.13 (BAM15), which is selective for the mitochondria and has a wide therapeutic window.20, 173
Preliminary studies revealed that the furazan, pyrazine, and aniline moieties on 5.13 are
necessary for mitochondrial uncoupling activity (Figure 5.2). Although the aniline moiety cannot
be replaced with a phenol moiety, our studies highlighted the importance of the aniline rings in
mitochondrial respiration studies, as measured by oxygen consumption rate (OCR). Herein, we
report the synthesis, structure–activity relationship study (SAR), and biological evaluation of
derivatives of 5.13 that explore electronic effects of substituents on the aniline ring. These
analogues show good mitochondrial uncoupling activity with a wide therapeutic window.

131
Figure 5.2. Pharmacophore of 5.13
5.4 Results and Discussion
5.4.1 Inhibitor Development
Studies have established that mitochondrial uncouplers are typlically lipophilic, weak
acids.6, 27-29, 192 An issue most commonly associated with reported mitochondrial uncouplers is
selectivity for the mitochondria. Uncouplers such as 5.1 and 5.5 are notoriously nonselective for
the mitochondria with off target effects of depolarizing the plasma membrane. Our recently
reported mitochondrial uncoupler 5.13 was demonstrated to be selective for the mitochondria
over the plasma membrane. We hypothesize that its selectivity is due to its measured pKa, which
was found to be 7.56.173 This pKa value allows 5.13 to easily shuttle a proton between the
mitochondrial inner membrane space (pH = 6.8)30 and mitochondrial matrix (pH = 8.0–8.1).30 In
an effort to explore the effect of uncoupler pKa on uncoupling activity, we made modifications to
the aniline moiety of 5.13 by varying the electronics on the ring by having either electron-
donating or electron-withdrawing groups at different positions on the ring.
5.4.2 Chemical Synthesis
The synthesis of symmetrical and unsymmetrical derivatives of 5.13 is shown in Scheme
5.1. Dichlorofurazan 5.15 was synthesized from the reaction of diaminofurazan (5.14) with
oxalic acid in a 10% HCl solution, followed by treatment PCl5/POCl3.193-195 Compound 5.15 was
then reacted with various alkyl or aryl amines in THF to afford the desired symmetrical
ON
N
N
N NH
NHfurazan
pyrazine
aniline
F
F
5.13 (BAM15)

132
derivatives 5.16a–w. To synthesize the unsymmetrical derivatives, compound 5.15 was reacted
with 2-fluoroaniline and triethylamine in THF followed by the addition of the desired aryl amine
and triethylamine to yield the unsymmetrical derivatives 5.17a–x.
Scheme 5.1. Synthesis of Symmetrical and Unsymmetrical Derivatives of 5.13a
aReagents and conditions: (a) oxalic acid, 10% HCl, reflux, 3 h, quantitative; (b) PCl5, POCl3, reflux, 2 h, 30%; (c) amine, THF, reflux, 19 h, 19–95%; (d) 2-fluoroaniline, TEA, THF, 0 ºC, 0.25 to 1 h, then aryl amine, TEA, THF, 0 ºC to rt, 19 h, 13–59%.
The synthesis of amide derivatives of 5.13 is shown in Scheme 5.2. The diamine 5.18
was synthesized from the reaction between 5.15 and ammonium hydroxide. Compound 5.18 was
then reacted with either an anhydride or sulfonyl chloride and triethylamine in DCM to afford
compounds 5.19a–e.
Scheme 5.2. Synthesis of Amide Derivatives of 5.13a
aReagents and conditions: (a) NH4OH, ACN, 0 ºC to rt, 3 h, quantitative; (b) anhydride, TEA, DCM, 0 ºC to rt, 19 h, 40%; (c) sulfonyl chloride, TEA, DCM, 0 ºC to rt, 19 h, 8–29%.
a ON
N
N
N NH2
NH2
5.15
5.18
b or c ON
N
N
N NHR
NHR5.19a: R = CF3CO5.19b: R = (4-CH3)PhSO25.19c: R = (4-F)PhSO25.19d: R = (3-CF3)PhSO25.19e: R = (2-NO2)PhSO2
ON
N
NH2
NH2 ON
N
N
N
Cl
Cl
5.14 5.15
ON
N
N
N R
R
5.16a: R = (CH3)3CNH5.16b: R = (CH2)2CHNH5.16c: R = (CH2)4CHNH5.16d: R = (CH2)5CHNH5.16e: R = O(CH2CH2)2N5.16f: R = (2-Me)PhNH5.16g: R = (3-Me)PhNH5.16h: R = (4-Me)PhNH5.16i: R = (2-OMe)PhNH5.16j: R = (3-OMe)PhNH5.16k: R = (4-OMe)PhNH5.16l: R = (2-OEt)PhNH
ON
N
N
N
N
N H
Ar
H
5.17 a-x
a, b c
d
5.16m: R = (3-OEt)PhNH5.16n: R = (4-OEt)PhNH5.16o: R = (3-OH)PhNH5.16p: R = (4-NHCOCF3)PhNH5.16q: R = (2-OCF3)PhNH5.16r: R = (3-OCF3)PhNH5.16s: R = (4-OCF3)PhNH5.16t: R = (2-CF3)PhNH5.16u: R = (3-CF3)PhNH5.16v: R = (4-CF3)PhNH5.16w: R = (4-CN)PhNH
5.17a: Ar = Ph5.17b: Ar = 2-Pyr5.17c: Ar = (3-F)Ph5.17d: Ar =( 4-F)Ph5.17e: Ar = (2,3-F)Ph5.17f: Ar = (2,4-F)Ph5.17g: Ar = (2-Me)Ph5.17h: Ar = (3-Me)Ph5.17i: Ar = (4-Me)Ph5.17j: Ar = (2-OMe)Ph5.17k: Ar = (3-OMe)Ph5.17l: Ar = (4-OMe)Ph
5.17m: Ar = (2-OEt)Ph5.17n: Ar = (3-OEt)Ph5.17o: Ar = (4-OEt)Ph5.17p: Ar = (3-OH)Ph5.17q: Ar = (4-NHCOCF3)Ph5.17r: Ar = (2-OCF3)Ph5.17s: Ar = (3-OCF3)Ph5.17t: Ar = (4-OCF3)Ph5.17u: Ar = (2-CF3)Ph5.17v: Ar = (3-CF3)Ph5.17w: Ar = (4-CF3)Ph
F

133
5.4.3 Structure–Activity Relationship Studies and Biological Characterization of 5.13
Derivatives
We synthesized a series of derivatives of 5.13 that focused on modifications to the aniline
rings. Symmetrical derivatives maintain the aniline ring with either electron rich or electron
deficient substituents at varying positions. For unsymmetrical derivatives, the 2-fluoroaniline
was maintained while the electronics and sterics on the neighboring aniline ring was
investigated. Further, the 2-fluoroaniline moiety of 5.13 was replaced with amides or
sulfonamides to determine their effect on mitochondrial uncoupling. All compounds were tested
at concentrations of 0.1, 0.25, 0.5, 1, 5, 10, 20, and 40 µM for mitochondrial uncoupling activity
as a function of oxygen consumption rate (OCR). OCR serves as a gauge for mitochondrial
respiration because molecular oxygen is required for the production of ATP via oxidative
phosphorylation. The maximum percentage of basal (100%) respiration that can be stimulated at
40 µM is reported in Tables 5.1–5.3. We focused our attention to developing compounds that
have good efficacy over a wide therapeutic range. Therefore, to effectively identify compounds
that maintain uncoupling activity at high concentrations, we used 40 µM as a filter to prevent
championing compounds that may have good potency but lack efficacy and to highlight
compounds with poor potency but good efficacy.
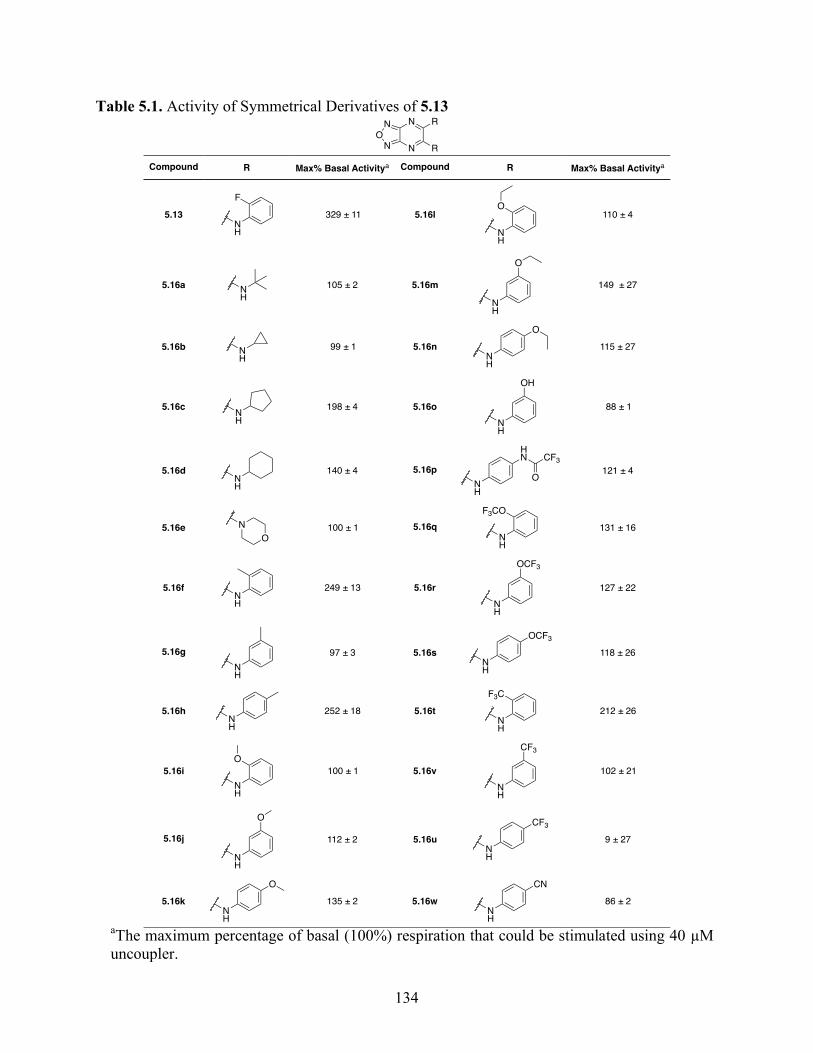
134
Table 5.1. Activity of Symmetrical Derivatives of 5.13
aThe maximum percentage of basal (100%) respiration that could be stimulated using 40 µM uncoupler.
Compound R Max% Basal Activitya Compound R Max% Basal Activitya
5.13NH
F329 ± 11 5.16l
NH
O110 ± 4
5.16a NH
105 ± 2 5.16m
NH
O
149 ± 27
5.16b NH
99 ± 1 5.16nNH
O115 ± 27
5.16c NH
198 ± 4 5.16oNH
OH
88 ± 1
5.16dNH
140 ± 4 5.16pNH
HN CF3
O121 ± 4
5.16e NO
100 ± 1 5.16qNH
F3CO131 ± 16
5.16fNH
249 ± 13 5.16rNH
OCF3
127 ± 22
5.16gNH
97 ± 3 5.16sNH
OCF3
118 ± 26
5.16hNH
252 ± 18 5.16tNH
F3C212 ± 26
5.16iNH
O100 ± 1 5.16v
NH
CF3
102 ± 21
5.16j
NH
O
112 ± 2 5.16uNH
CF3
9 ± 27
5.16kNH
O135 ± 2 5.16w
NH
CN86 ± 2
N
NNO
N
R
R

135
Using 5.13 as the standard, the effect of replacing the 2-fluoroaniline aniline moiety with
alkylamines was initially explored (Table 5.1). Substitution with the bulky tert-butylamine
(5.16a) or a cyclopropylamine (5.16b) did not increase OCR. Increasing the cyclic amine ring
size to five- (5.16c) and six- (5.16d, SHC4041644) membered resulted in a modest (> 2-fold)
increase in OCR. Replacement of the cyclic amines with a heterocyclic morpholine analogue
(5.16e) produced an inactive molecule (no increase in respiration).
We next switched our focus to altering the electronics of the aniline ring portion of 5.13
(Table 5.1). Installation of methyl substituents on the anline rings (5.16f–h) resulted in increased
OCR values relative to basal levels. While the meta-substituted 5.16g (SHC4051652) had no
effect on respiration, the ortho- (5.16f) and para-substituted (5.16h, SHC4081662) derivatives
increased OCR to 250%. Encouraged by these results, electron rich methyl (5.16i–k) and ethyl
(5.16l–n) ether derivatives were synthesized and tested. Unfortunately, all had minimal effect in
increasing OCR. To further confirm that strongly electron-donating moieties have deleterious
effects on respiration, phenol 16o and trifluoroacetamide 5.16p were tested and results indicate
that both compounds lack uncoupling activity. These studies suggest that increasing electron
density on the aniline ring of the 5.13 scaffold has a negligible effect on uncoupling activity.
To investigate the effect of increased lipophilicity and decreased ring electron density,
trifluoromethyl ether (5.16q–s) and trifluoromethyl (5.16t–v) derivatives at the ortho, meta, and
para positions were then synthesized.196, 197 5.16q–s equally had minimal effects on OCR,
increasing OCR up to 130%. Interestingly, the position of the trifluoromethyl group had
significant influence on respiration rates (ortho > meta > para) with 5.16t (SHC4111522)
stimulating 2-fold basal activity. Replacing the trifluoromethyl group with a nitrile (5.16w) also
produced an inactive uncoupler.

136
Among symmetrical derivatives tested, 5.13 remained as the best in class. Therefore,
unsymmetrical analogues were synthesized wherein the 2-fluoroaniline moiety was maintained.
As shown in Table 5.2, an unsymmetrical derivative bearing an aniline ring (5.17a) modestly
stimulated respiration (176%) up to 40 µM, which was similarly observed with an isoelectronic
replacement with a 2-aminopyridine ring (5.17b). Transposition of the fluorine atom in 5.13 to
either the meta (5.17c) or para position (5.17d) slightly diminished uncoupling activity.
Subsequent addition of an extra fluorine atom to the 2-fluoroaniline at either the 3- (5.17e) or 4-
position of the ring (5.17f) decreased the activity further.
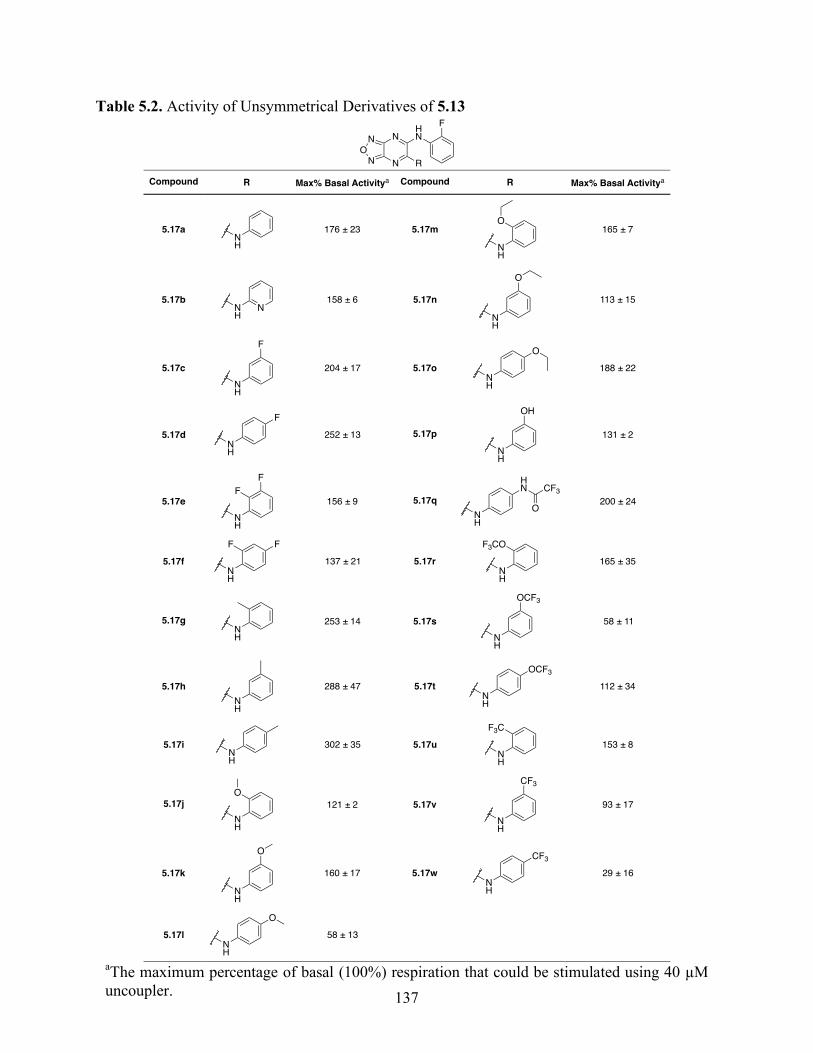
137
Table 5.2. Activity of Unsymmetrical Derivatives of 5.13
aThe maximum percentage of basal (100%) respiration that could be stimulated using 40 µM uncoupler.
Compound R Max% Basal Activitya Compound R Max% Basal Activitya
5.17aNH
176 ± 23 5.17m
NH
O165 ± 7
5.17bNH
N158 ± 6 5.17n
NH
O
113 ± 15
5.17cNH
F
204 ± 17 5.17oNH
O188 ± 22
5.17dNH
F252 ± 13 5.17p
NH
OH
131 ± 2
5.17eNH
FF
156 ± 9 5.17qNH
HN CF3
O200 ± 24
5.17fNH
FF137 ± 21 5.17r
NH
F3CO165 ± 35
5.17gNH
253 ± 14 5.17sNH
OCF3
58 ± 11
5.17hNH
288 ± 47 5.17tNH
OCF3
112 ± 34
5.17iNH
302 ± 35 5.17uNH
F3C153 ± 8
5.17jNH
O121 ± 2 5.17v
NH
CF3
93 ± 17
5.17k
NH
O
160 ± 17 5.17wNH
CF3
29 ± 16
5.17lNH
O58 ± 13
ON
N
N
NHN
R
F

138
In contrast, unsymmetrical derivatives with a methyl group at the ortho (5.17g), meta
(5.17h, SHC4081663), or para (5.17i, SHC4091665) position were found to have good efficacy
and produced OCRs comparable to 5.13. Replacement with the stronger electron-donating
methyl (5.17j-l) and ethyl (5.17m-o) ethers afforded moderate uncouplers, with the meta-
substituted methyl ether (5.17k) and the ortho- (5.17m) and para-substituted ethyl (5.17n) ethers
being the most efficacious and potent of their respective series. Phenol 5.17p and
trifluoroacetamide 5.17q also achieved moderate uncoupling activity. The position of the
electron-withdrawing trifluoromethyl ether and trifluoromethyl substituents significantly
influenced respiration rates with the ortho-substituted analogues (5.17r and 5.17u, SHC4111523)
producing the highest respiration rates of their respective series. The meta and para positions
(5.17s-t and 5.17v-w) were found to be relatively inactive.
To expand structural features on the current scaffold, the aniline moiety was replaced
with amide moieties (Table 5.3). Our previous work established that 5.15 lacked mitochondrial
uncoupling activity.173 Conversion of 5.15 into the diamine 5.18 did not afford an active
uncoupler. Acetylation of 5.18 with a trifluoromethylacetyl group (5.19a) also resulted in an
inactive uncoupler. Because sulfonamides have been shown15, 22, 92, 198 to have mitochondrial
uncoupling activity, sulfonamides 5.19b-e were synthesized. The tosyl (19c, SHC4031633) and
4-fluorobenzenesulfonamide (5.19d) were unable to significantly stimulate an increase in
respiration levels. Increasing the lipophilicity of the sulfonamide library with a 3-
trifluoromethylbenzenesulfonamide (5.19e) achieved moderate uncoupling activity (195%);
however, replacement of the trifluoromethyl moiety with a stronger deactivating nitro group
(5.19f) eliminated uncoupling activity. The general lack of mitochondrial uncoupling activity

139
with the amide and sulfonamide derivatives is most likely due to the compounds’ decreased pKa
(< 6.8), making protonophoric uncoupling challenging.
Table 5.3. Activity of Amide Derivatives of 5.13
aThe maximum percentage of basal (100%) respiration that could be stimulated using 40 µM uncoupler.
Because mitochondrial uncouplers are typically lipophilic weak acids, we decided to
determine the pKa and cLogP of select compounds (Table 5.4). We selected compounds across
the spectrum of activity: active (> 2-fold increase in OCR at 40 µM vs basal), moderately active
(1.3 ≤ 2-fold increase in OCR at 40 µM vs basal), and inactive (≤ 1.2-fold increase in OCR at 40
µM vs basal). To understand the link between uncoupling activity and physiochemical properties
of compounds, we measured the pKa of select compounds and related them to calculated cLogP
values. Evaluation of the calculated cLogP values revealed that moderately active (5.16d, 5.16r,
and 5.17u) and active (5.16h, 5.16t, 5.17h, 5.17i) compounds in the set have cLogP values > 6,
which is not surprising because these compounds must be membrane permeable to achieve any
uncoupling activity. Interestingly, moderately active uncouplers were found to have pKas around
6.8 and between 8.0 and 10 while a majority of active uncouplers were found to have pKas within
the ideal range of 6.8–8.0 with the exception of 5.16h, which was found to have a pKa of 10.75.
Compound # R Max% Basal
ActivityaCompound
# R Max% Basal Activitya
5.18 H 99 ± 2 5.19cSO2
F
111 ± 3
5.19aCF3
O99 ± 2 5.19d
SO2
CF3
195 ± 7
5.19b SO2
120 ± 6 5.19eSO2
O2N
102 ± 2
N
NNO
N
NHR
NHR

140
While there are examples of compounds in our library with cLogP > 6 that are inactive
(e.g. 5.16g, 5.16n, and 5.17s), 5.16g and 5.16n have high pKas far from the ideal range (> 10)
and 5.17s appears to be cytotoxic. Analogues with cLogP < 5.5 (e.g. 5.19c) were found to be
inactive as uncouplers; their measured pKas were also found to be below 6.8 (e.g. 5.19c), which
would collectively make uncoupling challenging. Therefore, these results suggest that compound
lipophilicity (cLogP values) and pKa equally contribute to uncoupling activity, as corroborated
by the OCR data provided in Tables 5.1–5.3, and must be taken into consideration when
evaluating the development of future uncouplers.
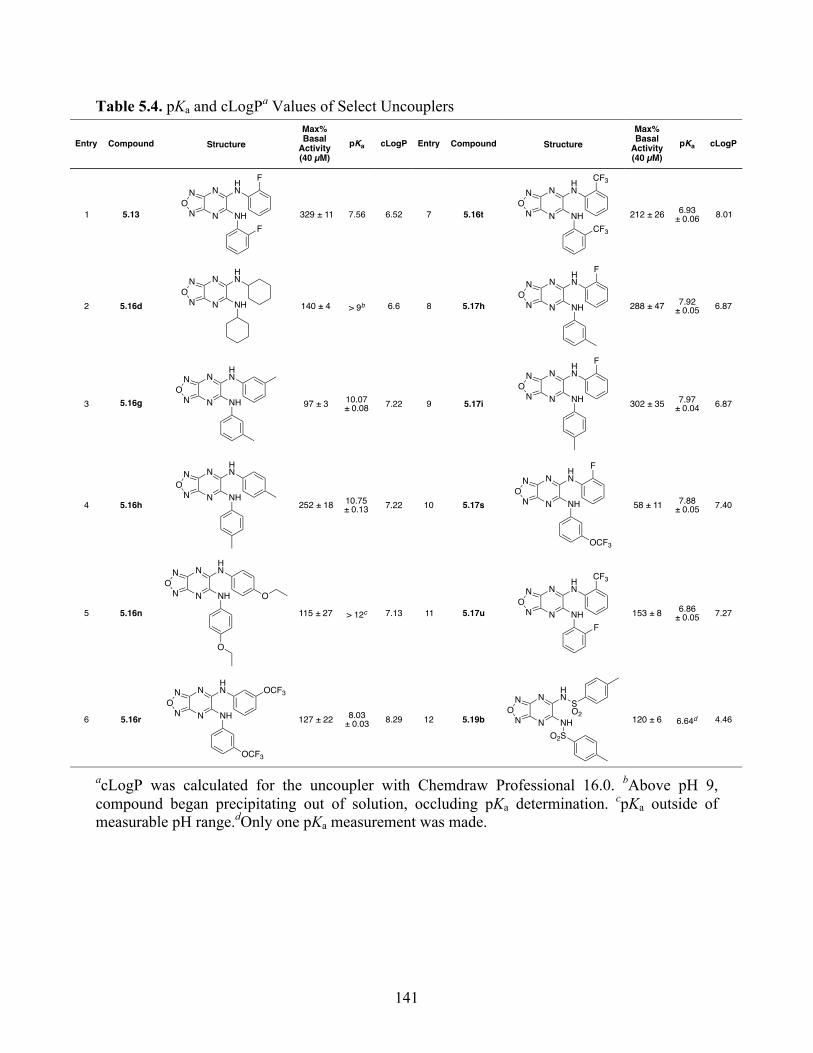
141
Table 5.4. pKa and cLogPa Values of Select Uncouplers
acLogP was calculated for the uncoupler with Chemdraw Professional 16.0. bAbove pH 9, compound began precipitating out of solution, occluding pKa determination. cpKa outside of measurable pH range.dOnly one pKa measurement was made.
Entry Compound StructureMax% Basal
Activity (40 µM)
pKa cLogP Entry Compound StructureMax% Basal
Activity (40 µM)
pKa cLogP
1 5.13O
N
N
N
NHN
NH
F
F329 ± 11 7.56 6.52 7 5.16t
ON
N
N
NHN
NHCF3
CF3
212 ± 26 6.93 ± 0.06 8.01
2 5.16dO
N
N
N
NHN
NH 140 ± 4 > 9b 6.6 8 5.17hO
N
N
N
NHN
NH
F
288 ± 47 7.92 ± 0.05 6.87
3 5.16gO
N
N
N
NHN
NH 97 ± 3 10.07 ± 0.08 7.22 9 5.17i
ON
N
N
NHN
NH
F
302 ± 35 7.97 ± 0.04 6.87
4 5.16h
ON
N
N
NHN
NH252 ± 18 10.75
± 0.13 7.22 10 5.17sO
N
N
N
NHN
NH
F
OCF3
58 ± 11 7.88 ± 0.05 7.40
5 5.16n
ON
N
N
NHN
NH O
O
115 ± 27 > 12c 7.13 11 5.17uO
N
N
N
NHN
NHF
CF3
153 ± 8 6.86 ± 0.05 7.27
6 5.16r
ON
N
N
NHN
NH
OCF3
OCF3
127 ± 22 8.03 ± 0.03 8.29 12 5.19b
ON
N
N
NHN
NHO2S
SO2 120 ± 6 6.64d 4.46

142
To quantify the uncoupling activity of derivatives of 5.13, we determined the half
maximal effective concentration (EC50) values of select compounds classified as active
uncouplers (Table 5.5) because of their ability to increase OCR > 200% up to 40 µM (Figure
5.3). The para methyl (5.16h) symmetrical derivative was found to be the least potent of the
selected active compounds, which is consistent with OCR data presented in Figure 5.3, as it
stimulated ~ 2-fold increase in OCR up to 40 µM. Interestingly, its has the highest pKa of the set
(Table 5.4). Conversely, the ortho trifluoromethyl (5.16t, SHC4111522) symmetrical derivative,
an isoelectronic analogue of 5.13, was found to be the most potent uncoupler of the set, having
an EC50 of 0.63, which is consistent with its measured pKa and cLogP (Table 5.4). All active
uncouplers with ~3-fold or higher OCR at 40 µM were found to be unsymmetrical derivatives
(5.17h and 5.17i). Consequently, their EC50 values were found to be low micromolar, and they
show similar efficacy to 5.13 (Figure 5.3).
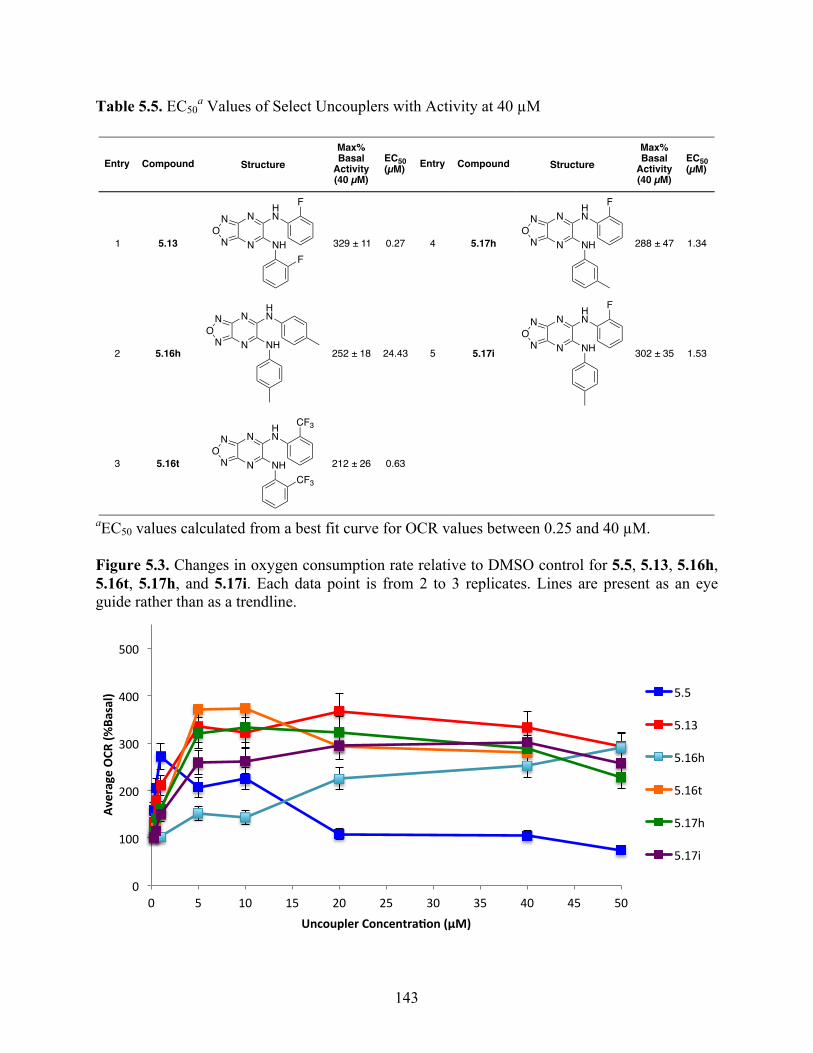
143
Table 5.5. EC50a Values of Select Uncouplers with Activity at 40 µM
aEC50 values calculated from a best fit curve for OCR values between 0.25 and 40 µM.
Figure 5.3. Changes in oxygen consumption rate relative to DMSO control for 5.5, 5.13, 5.16h, 5.16t, 5.17h, and 5.17i. Each data point is from 2 to 3 replicates. Lines are present as an eye guide rather than as a trendline.
Entry Compound StructureMax% Basal
Activity (40 µM)
EC50(µM) Entry Compound Structure
Max% Basal
Activity (40 µM)
EC50(µM)
1 5.13O
N
N
N
NHN
NH
F
F329 ± 11 0.27 4 5.17h
ON
N
N
NHN
NH
F
288 ± 47 1.34
2 5.16h
ON
N
N
NHN
NH252 ± 18 24.43 5 5.17i
ON
N
N
NHN
NH
F
302 ± 35 1.53
3 5.16tO
N
N
N
NHN
NHCF3
CF3
212 ± 26 0.63
0
100
200
300
400
500
0 5 10 15 20 25 30 35 40 45 50
AverageOCR
(%Ba
sal)
UncouplerConcentra8on(µM)
5.5
5.13
5.16h
5.16t
5.17h
5.17i

144
5.5 Current Efforts and Future Directions
From the data presented in Table 5.4, the most active uncouplers in our library have
cLogP values > 6 and have pKa values between 6.8 and 8.0. Because 5.13 has a pKa near the
median of this ideal range, we decided to select compounds that have similar potency to 5.13 but
with pKas abutting the two extremes of the ideal pKa range to test in vivo. Current efforts involve
dosing mice with 5.16t (EC50 = 0.63 µM, pKa = 6.93) and 5.17h (EC50 = 1.34 µM, pKa = 7.92) to
determine if these analogues have good oral bioavailability and improved pharmacokinetics to
5.13. These studies will also determine if 5.16t and 5.17h have similar or improved efficacy to
5.13 by assessing their ability to increase OCR in mice. Positive feedback from these studies will
lead to future studies to evaluate 5.16t and 5.17h’s ability to promote leanness in mouse models
of obesity.
5.6 Conclusions
Herein, we report the design and synthesis of potent and efficacious derivatives of 5.13, a
mitochondrial uncoupler that does not depolarize the plasma membrane. Modifications were
made to the aniline moiety of 5.13. Symmetrical, unsymmetrical, and amide derivatives were
synthesized. Symmetrical and unsymmetrical derivatives that increased respiration at least 2-fold
higher than normal basal levels were found to have cLogP values > 6 and pKa values between 6.8
and 8.0. Collectively, our work provides greater insight into the chemical property requirements
for developing new mitochondrial uncouplers and expands upon a novel chemical scaffold that
selectively uncouples the mitochondria. This work should help elucidate the mechanism of
pharmaceutical mitochondrial uncoupling and pave the way for the use of this therapeutic
strategy for the treatment of metabolic-related diseases.

145
5.7 References
1. Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. Biochim.
Biophys. Acta 2011, 1807, 1507-1538.
2. Khakh, B. S.; Burnstock, G. The Double Life of Atp. Sci. Am. 2009, 301, 84-92.
3. Loomis, W. F.; Lipmann, F. Reversible Inhibition of Coupling between Phosphorylation and
Oxidation. J. Biol. Chem. 1948, 173, 807-808.
4. Weinbach, E. C.; Garbus, J. Mechanism of Action of Reagents That Uncouple Oxidative
Phosphorylation. Nature 1969, 221, 1016-1018.
5. Busiello, R. A.; Savarese, S.; Lombardi, A. Mitochondrial Uncoupling Proteins and Energy
Metabolism. Front. Physiol. 2015, 6, 36.
6. Erlanson-Albertsson, C. The Role of Uncoupling Proteins in the Regulation of Metabolism. Acta
Physiol. Scand. 2003, 178, 405-412.
7. Ricquier, D.; Bouillaud, F. Mitochondrial Uncoupling Proteins: From Mitochondria to the
Regulation of Energy Balance. J. Physiol. 2000, 529, 3-10.
8. Rousset, S.; Alves-Guerra, M.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.; Bouillaud, F.;
Ricquier, D. The Biology of Mitochondrial Uncoupling Proteins. Diabetes Metab. Syndr. Obes.
2004, 53, 130-135.
9. Krauss, S.; Zhang, C.-Y.; Lowell, B. B. The Mitochondrial Uncoupling-Protein Homologues.
Nat. Rev. Mol. Cell Biol. 2005, 6, 248-261.
10. Gimeno, R. E.; Dembski, M.; Weng, X.; Deng, N.; Shyjan, A. W.; Gimeno, C. J.; Iris, F.; Ellis,
S. J.; Woolf, E. A.; Tartaglia, L. A. Cloning and Characterization of an Uncoupling Protein
Homolog: A Potential Molecular Mediator of Human Thermogenesis. Diabetes 1997, 46, 900-
906.

146
11. Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. Ucp1:
The Only Protein Able to Mediate Adaptive Non-Shivering Thermogenesis and Metabolic
Inefficiency. Biochem. Biophys. Acta 2001, 1504, 82-106.
12. Echtay, K. S.; Roussel, D.; St-Pierre, J.; Jekabsons, M. B.; Cadenas, S.; Stuart, J. A.; Harper, J.
A.; Roebuck, S. J.; Morrison, A.; Pickering, S.; Clapham, J. C.; Brand, M. D. Superoxide
Activates Mitochondrial Uncoupling Proteins. Nature 2002, 415, 96-99.
13. Divakaruni, A. S.; Brand, M. D. The Regulation and Physiology of Mitochondrial Proton Leak.
Physiology (Bethesda) 2011, 26, 192-205.
14. Kovacic, P.; Pozos, R. S.; Somanathan, R.; Shangari, N.; O'Brien, P. J. Mechanism of
Mitochondrial Uncouplers, Inhibitors, and Toxins: Focus on Electron Transfer, Free Radicals,
and Structure-Activity Relationships. Curr. Med. Chem. 2005, 12, 2601-2623.
15. Terada, H. Uncouplers of Oxidative Phosphorylation. Environ. Health Perspect. 1990, 87, 213-
218.
16. Harper, J. A.; Dickinson, K.; Brand, M. D. Mitochondrial Uncoupling as a Target for Drug
Development for the Treatment of Obesity. Obes. Rev. 2001, 2, 255-265.
17. Tseng, Y. H.; Cypess, A. M.; Kahn, C. R. Cellular Bioenergetics as a Target for Obesity
Therapy. Nat. Rev. Drug Discov. 2010, 9, 465-482.
18. Kalinovich, A. V.; Shabalina, I. G. Novel Mitochondrial Cationic Uncoupler C4r1 Is an
Effective Treatment for Combating Obesity in Mice. Biochemistry (Mosc) 2015, 80, 620-628.
19. Suzuki, T.; Kikuchi, H.; Ogura, M.; Homma, M. K.; Oshima, Y.; Homma, Y. Weight Loss by
Ppc-1, a Novel Small Molecule Mitochondrial Uncoupler Derived from Slime Mold. PLoS One
2015, 10, e0117088.

147
20. Perry, R. J.; Kim, T.; Zhang, X. M.; Lee, H. Y.; Pesta, D.; Popov, V. B.; Zhang, D.; Rahimi, Y.;
Jurczak, M. J.; Cline, G. W.; Spiegel, D. A.; Shulman, G. I. Reversal of Hypertriglyceridemia,
Fatty Liver Disease, and Insulin Resistance by a Liver-Targeted Mitochondrial Uncoupler. Cell
Metab. 2013, 18, 740-748.
21. Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G. I.; Jin, S. Niclosamide Ethanolamine-Induced Mild
Mitochondrial Uncoupling Improves Diabetic Symptoms in Mice. Nat. Med. 2014, 20, 1263-
1269.
22. Perry, R. J.; Zhang, D.; Zhang, X.; Boyer, J. L.; Shulman, G. I. Controlled-Release
Protonophore Reverses Diabetes and Steatohepatitis in Rats. Science 2015, 347, 1253-1256.
23. Figarola, J. L.; Singhal, P.; Rahbar, S.; Gugiu, B. G.; Awasthi, S.; Singhal, S. S. Coh-Sr4
Reduces Body Weight, Improves Glycemic Control and Prevents Hepatic Steatosis in High Fat
Diet-Induced Obese Mice. PLoS One 2013, 8, e83801.
24. Fu, Y. Y.; Zhang, M.; Turner, N.; Zhang, L. N.; Dong, T. C.; Gu, M.; Leslie, S. J.; Li, J. Y.;
Nan, F. J.; Li, J. A Novel Chemical Uncoupler Ameliorates Obesity and Related Phenotypes in
Mice with Diet-Induced Obesity by Modulating Energy Expenditure and Food Intake.
Diabetologia 2013, 56, 2297-2307.
25. Kalinovich, A. V.; Mattsson, C. L.; Youssef, M. R.; Petrovic, N.; Ost, M.; Skulachev, V. P.;
Shabalina, I. G. Mitochondria-Targeted Dodecyltriphenylphosphonium (C12tpp) Combats High-
Fat-Diet-Induced Obesity in Mice. Int. J. Obes. (Lond) 2016, 40, 1864-1874.
26. Parascandola, P. Dinitrophenol and Bioenergetics: A Historical Perspective. Mol. Cell.
Biochem. 1974, 5, 69-77.

148
27. Tainter, M. L.; Cutting, W. C.; Hines, E. Effects of Moderate Doses of Dinitrophenol on the
Energy Exchange and Nitrogen Metabolism of Patients under Conditions of Restricted Dietary.
J. Pharmacol. Exp. Ther. 1935, 55, 326-353.
28. Grundlingh, J.; Dargan, P. I.; El-Zanfaly, M.; Wood, D. M. 2,4-Dinitrophenol (Dnp): A Weight
Loss Agent with Significant Acute Toxicity and Risk of Death. J. Med. Toxicol. 2011, 7, 205-
212.
29. Tainter, M. L.; Stockton, A. B.; Cutting, W. C. Use of Dinitrophenol in Obesity and Related
Conditions. J. Amer. Med. Assoc. 1933, 101, 1472-1475.
30. Bianchetti, A.; Pugliatti, C.; Jori, A. On the Hyperthermia Induced by 2,4-Dinitrophenol. Med.
Pharmacol. Exp. 1967, 17, 401-408.
31. McFee, R. B.; Caraccio, T. R.; McGuigan, M. A.; Reynolds, S. A.; Bellanger, P. Dying to Be
Thin: A Dinitrophenol Related Fatality. Vet. Hum. Toxicol. 2004, 46, 251-254.
32. Poole, F. E.; Haining, R. B. Sudden Death from Dinitrophenol Poisoning. J. Am. Med. Assoc.
1934, 102, 1141-1147.
33. Horner, W. D. Cataract Following Dinitrophenol Treatment for Obesity. Arch. Ophthalmol.
1936, 16, 447-461.
34. Horner, W. D. A Study of Dinitrophenol and Its Relation to Cataract Formation. Trans Am.
Ophthalmol. Soc. 1941, 39, 405-437.
35. Bettman, J. W. Experimental Dinitrophenol Cataract. Am. J. Ophthalmol. 1946, 29, 1388-1395.
36. Rodin, F. H. Cataracts Following the Use of Dinitrophenol: A Summary of Thirty-Two Cases.
Cal. West Med. 1936, 44, 276-279.
37. Toxicological Profile of Dinitrophenols. In USDHHS, Ed. 1995;
http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=729&tid=132.

149
38. Jackson, P. C.; St. John, J. B. Effects of 2,4-Dinitrophenol on Membrane Lipids of Roots. Plant
Physiol. 1982, 70, 858-862.
39. Ravesloot, J. H.; Rombouts, E. 2,4-Dinitrophenol Acutely Inhibits Rabbit Atrial Ca2+-Sensitive
Cl- Current (Ito2). Can. J. Physiol. Pharmacol. 2000, 78, 766-773.
40. Juthberg, S. K. A.; Brismar, T. Effect of Metabolic Inhibitors on Membrane Potential and Ion
Conductance of Rat Astrocytes. Cell. Mol. Neurobio. 1997, 17, 367-377.
41. Wu, S. N.; Li, H. F.; Chiang, H. T. Characterization of Atp-Sensitive Potassium Channels
Functionally Expressed in Pituitary Gh 3 Cells. J. Membr. Biol. 2000, 178, 205-214.
42. Brismar, T.; Collins, V. P. Effect of External Cation Concentration and Metabolic Inhibitors on
Membrane Potential of Human Glial Cells. J. Physiol. 1993, 460, 365-383.
43. Buckler, K. J.; Vaughan-Jones, R. D. Effects of Mitochondrial Uncouplers on Intracellular
Calcium, Ph, and Membrane Potential in Rat Carotid Body Type I Cells. J. Physiol. 1998, 513,
819-833.
44. Blaikie, F. H.; Brown, S. E.; Samuelsson, L. M.; Brand, M. D.; Smith, R. A.; Murphy, M. P.
Targeting Dinitrophenol to Mitochondria: Limitations to the Development of a Self-Limiting
Mitochondrial Protonophore. Biosci. Rep. 2006, 26, 231-243.
45. Chalmers, S.; Caldwell, S. T.; Quin, C.; Prime, T. A.; James, A. M.; Cairns, A. G.; Murphy, M.
P.; McCarron, J. G.; Hartley, R. C. Selective Uncoupling of Individual Mitochondria within a
Cell Using a Mitochondria-Targeted Photoactivated Protonophore. J. Am. Chem. Soc. 2012, 134,
758-761.
46. Draber, W.; Büchel, K. H.; Schäfer, G. Quantitative Structure-Activity Studies of Hydrazones,
Uncouplers of Oxidative Phosphorylation. Z. Naturforsch., B: Chem. Sci. 1972, 27, 159-171.

150
47. Heytler, P. G.; Prichard, W. W. A New Class of Uncoupling Agents - Carbonyl Cyanide
Phenylhydrazones. Biochem. Biophys. Res. Commun. 1962, 7, 272-275.
48. Antalík, M.; Šturdík, E.; Sulo, P.; Propperová, A.; Mihalovová, E.; Podhradský, D.; Dzurila, M.
Uncoupling Effect of Protonophoric and Nonprotonophoric Analogs of Carbonyl Cyanide
Phenylhydrazone on Mitochondrial Oxidative Phosphorylation. Gen. Physiol. Biophys. 1988, 7,
517-528.
49. Park, K. S.; Jo, I.; Pak, K.; Bae, S. W.; Rhim, H.; Suh, S. H.; Park, J.; Zhu, H.; So, I.; Kim, K.
W. Fccp Depolarizes Plasma Membrane Potential by Activating Proton and Na+ Currents in
Bovine Aortic Endothelial Cells. Eur. J. Physiol. 2002, 443, 344-352.
50. To, M. S.; Aromataris, E. C.; Castro, J.; Roberts, M. L.; Barritt, G. J.; Rychkov, G. Y.
Mitochondrial Uncoupler Fccp Activates Proton Conductance but Does Not Block Store-
Operated Ca(2+) Current in Liver Cells. Arch. Biochem. Biophys. 2010, 495, 152-158.
51. Figarola, J. L.; Singhal, J.; Tompkins, J. D.; Rogers, G. W.; Warden, C.; Horne, D.; Riggs, A.
D.; Awasthi, S.; Singhal, S. S. Sr4 Uncouples Mitochondrial Oxidative Phosphorylation,
Modulates Amp-Dependent Kinase (Ampk)-Mammalian Target of Rapamycin (Mtor) Signaling,
and Inhibits Proliferation of Hepg2 Hepatocarcinoma Cells. J. Biol. Chem. 2015, 290, 30321-
30341.
52. Khailova, L. S.; Silachev, D. N.; Rokitskaya, T. I.; Avetisyan, A. V.; Lyamsaev, K. G.;
Severina, II; Il'yasova, T. M.; Gulyaev, M. V.; Dedukhova, V. I.; Trendeleva, T. A.; Plotnikov,
E. Y.; Zvyagilskaya, R. A.; Chernyak, B. V.; Zorov, D. B.; Antonenko, Y. N.; Skulachev, V. P.
A Short-Chain Alkyl Derivative of Rhodamine 19 Acts as a Mild Uncoupler of Mitochondria
and a Neuroprotector. Biochim. Biophys .Acta 2014, 1837, 1739-1747.

151
53. Antonenko, Y. N.; Avetisyan, A. V.; Cherepanov, D. A.; Knorre, D. A.; Korshunova, G. A.;
Markova, O. V.; Ojovan, S. M.; Perevoshchikova, I. V.; Pustovidko, A. V.; Rokitskaya, T. I.;
Severina, II; Simonyan, R. A.; Smirnova, E. A.; Sobko, A. A.; Sumbatyan, N. V.; Severin, F. F.;
Skulachev, V. P. Derivatives of Rhodamine 19 as Mild Mitochondria-Targeted Cationic
Uncouplers. J. Biol. Chem. 2011, 286, 17831-17840.
54. Dejonghe, W.; Kuenen, S.; Mylle, E.; Vasileva, M.; Keech, O.; Viotti, C.; Swerts, J.; Fendrych,
M.; Ortiz-Morea, F. A.; Mishev, K.; Delang, S.; Scholl, S.; Zarza, X.; Heilmann, M.; Kourelis,
J.; Kasprowicz, J.; Nguyen le, S. L.; Drozdzecki, A.; Van Houtte, I.; Szatmari, A. M.; Majda, M.;
Baisa, G.; Bednarek, S. Y.; Robert, S.; Audenaert, D.; Testerink, C.; Munnik, T.; Van Damme,
D.; Heilmann, I.; Schumacher, K.; Winne, J.; Friml, J.; Verstreken, P.; Russinova, E.
Mitochondrial Uncouplers Inhibit Clathrin-Mediated Endocytosis Largely through Cytoplasmic
Acidification. Nat. Commun. 2016, 7, 11710.
55. Kenwood, B. M.; Weaver, J. L.; Bajwa, A.; Poon, I. K.; Byrne, F. L.; Murrow, B. A.;
Calderone, J. A.; Huang, L.; Divakaruni, A. S.; Tomsig, J. L.; Okabe, K.; Lo, R. H.; Coleman, G.
C.; Columbus, L.; Yan, Z.; Saucerman, J. J.; Smith, J. S.; Holmes, J. W.; Lynch, K. R.;
Ravichandran, K. S.; Uchiyama, S.; Santos, W. L.; Rogers, G. W.; Okusa, M. D.; Bayliss, D. A.;
Hoehn, K. L. Identification of a Novel Mitochondrial Uncoupler That Does Not Depolarize the
Plasma Membrane. Mol. Metab. 2014, 3, 114-123.
56. Kenwood, B. M.; Calderone, J. A.; Taddeo, E. P.; Hoehn, K. L.; Santos, W. L. Structure-
Activity Relationships of Furazano[3,4-B]Pyrazines as Mitochondrial Uncouplers. Bioorg. Med.
Chem. 2015, 25, 4858-4861.
57. De Deken, R. H. Relations Entre La Structure Moléculaire Et Le Pouvoir Inhibiteur Des Nitro-
Et Halophénols. Biochem. Biophys. Acta 1955, 17, 494-502.

152
58. Parker, V. H. Effect of Nitrophenols and Halogenophenols on the Enzymic Activity of Rat-
Liver Mitochondria. Biochem. J. 1958, 69, 306-311.
59. Hemker, H. C. Lipid Solubility as a Factor Influencing the Activity of Uncoupling Phenols.
Biochem. Biophys. Acta 1962, 63, 46-54.
60. Chopineaux-Courtois, V.; Reymond, F.; Bouchard, G.; Carrupt, P.; Testa, B.; Girault, H. H.
Effects of Charge and Intramolecular Structure on the Lipophilicity of Nitrophenols. J. Am.
Chem. Soc. 1999, 121, 1743-17467.
61. Casey, J. R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular Ph. Nat. Rev.
Mol. Cell Biol. 2010, 11, 50-61.
62. Thottempudi, V.; Yin, P.; Zhang, J.; Parrish, D. A.; Shreeve, J. M. 1,2,3-Triazolo[4,5,-
E]Furazano[3,4,-B]Pyrazine 6-Oxide--a Fused Heterocycle with a Roving Hydrogen Forms a
New Class of Insensitive Energetic Materials. Chemistry 2014, 20, 542-548.
63. Fernandez, E.; Garcia-Ochoa, S.; Huss, S.; Mallo, A.; Bueno, J. M.; Micheli, F.; Paio, A.; Piga,
E.; Zarantonello, P. Solid-Phase Versus Solution Synthesis of Asymmetrically Disubstituted
Furazano[3,4-B]Pyrazines. Tetrahedron Lett. 2002, 43, 4741-4745.
64. Starchenkov, I. B.; Andrianov, V. G. Chemistry of Furazano[3,4-B]Pyrazines. 3. Method for
the Synthesis of 5,6-Disubstituted Furazano[3,4-B]Pyrazines. Chem. Heterocycl. Compd. (N. Y.)
1998, 33, 1219-1233.
65. Leroux, F. R.; Manteau, B.; Vors, J. P.; Pazenok, S. Trifluoromethyl Ethers--Synthesis and
Properties of an Unusual Substituent. Beilstein J. Org. Chem. 2008, 4, 13.
66. Sheppard, W. A. The Effect of Fluorine Substitution on the Electronic Properties of Alkoxy,
Alkylthio and Alkylsulfonyl Groups. J. Am. Chem. Soc. 1963, 85, 1314-1318.

153
67. Starkov, A. A.; Wallace, K. B. Structural Determinants of Fluorochemical-Induced
Mitochondrial Dysfunction. Tox. Sci. 2002, 66, 244-252.
68. McCracken, R. O.; Carr, A. W.; Stillwell, W. H.; Lipkowitz, K. B.; Boisvenue, R.; O'Doherty,
G. O. P.; Wickiser, D. I. Trifluoromethanesulfonamide Anthelmintics Protonophoric Uncouplers
of Oxidative Phosphorylation. Biochem. Pharmacol. 1993, 45, 1873-1880.
69. Schnellmann, R. G.; Manning, R. O. Perfluorooctane Sulfonamide: A Structurally Novel
Uncoupler of Oxidative Phosphorylation. Biochem. Biophys. Acta 1990, 1016, 344-348.

154
6 Experimental Section
6.1 Structure–Activity Relationship Studies of the Lipophilic Tail Region of
Sphingosine Kinase 2 Inhibitors
6.1.1 Sphingosine Kinase Assays
The inhibitory activity of the synthesized compounds on human SphK1 and SphK2 was
determined using a previously published method.199, 200 Recombinant human SphK1 or mouse
SphK2 isolated from a cell lysate was briefly incubated with (10 µM) or without compound,
sphingosine, and γ-[32P]ATP. The radiolabeled sphingosine 1-phosphate was isolated via
extraction and thin-layer chromatography and then quantified via scintillation counting.
6.1.2 General Material and Synthetic Procedures
All chemical reagents were purchased from commercial sources and used without further
purification. Thin layer chromatography (TLC) was performed on aluminum-backed silica gel,
200 µm, F254, and column chromatography was performed on flash grade silica gel, 40-63 µm,
using a Combiflash Rf purification system. 1H NMR spectra were recorded at 500 or 400 MHz;
the corresponding 13C NMR resonant frequencies were 126 and 101 MHz, respectively. 1H NMR
chemical shifts are reported in ppm with the solvent resonance as an internal standard (CDCl3:
7.26 ppm; CD3OD: 4.87 ppm). 13C NMR, chemical shifts are reported in ppm with the solvent
resonance as the internal standard (CDCl3: 77.16 ppm; CD3OD: 49.00 ppm). Data are reported as
follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad,
m= multiplet), coupling constants (Hz), and integration. Rotamers are denoted by an asterisk (*).
High resolution mass spectroscopy (HRMS) was performed on an LC/MS time–of–flight mass
spectrometer using electrospray ionization (ESI). HPLC analyses were performed with a Thermo
Electron TSQ triple quadrupole mass spectrometer equipped with an ESI source. All compounds

155
tested in biological assays are >95% pure by 1H NMR and HPLC analyses unless noted
otherwise. NMR spectra for the synthesized compounds can be found online
(http://www.sciencedirect.com/science/article/pii/S0960894X15002474).
General Procedure 2A: Synthesis of amide-oxime derivatives. TEA (2.7 equiv) was added to a
solution of benzonitrile 2.6a (1 equiv) with hydroxylamine hydrochloride (2.6 equiv) in ethanol
(0.47 M solution). The mixture was stirred at 80 ºC for 6 h. The organic solvent was removed
under reduced pressure, and the residue was purified by silica gel column chromatography to
yield the desired product.
General Procedure 2B: Coupling of amide-oxime derivatives with ((S)-1-(tert-
butoxycarbonyl)azetidine-2-carboxylic acid or (S)-1-(tert-butoxycarbonyl)pyrrolidine-2-
carboxylic acid. DIEA (1.80 equiv) was added to a solution of amide-oxime 2.7a (1 equiv) and
((S)-1-(tert-butoxycarbonyl)azetidine-2-carboxylic acid or (S)-1-(tert-
butoxycarbonyl)pyrrolidine-2-carboxylic acid in DMF (0.2 M solution). HCTU (1.5 equiv) was
then added to the resulting mixture at rt and stirred at 100 ºC for 4–8 h. The reaction progress
was followed by TLC. The solution was partitioned between EtOAc and LiBr aqueous solution.
The aqueous solution was washed with EtOAc three times, and the combined organic layers
were washed with brine, dried over Na2SO4, filtered, and concentrated via vacuum. The resulting
residue was purified by silica gel column chromatography.
General Procedure 2C: Buchwald–Hartwig coupling of aryl iodides with amines. The desired
amine (1.2 equiv) was added to a round bottom containing 2.6a (1 equiv) in toluene (0.120 M).
The reaction mixture was degassed for 10 min by bubbling N2 through the solution. Cs2CO3 (1.2
equiv), tri-tert-butylphosphine (0.4 equiv), and Pd2(dba)3 (0.1 equiv) were added together. The
resulting reaction mixture was then stirred at 100 oC for 24 h, after which it was poured into

156
water and extracted three times with EtOAc. The combined organic extracts were washed with
brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting brown residue
was purified by flash chromatography over silica gel.
General Procedure 2D: Deprotection of t-Boc protecting groups with TFA and guanylation of
amines. To a solution of t-Boc protected intermediate in DCM, a 1N TFA solution in DCM was
added. The reaction mixture was then stirred at rt for 4 h. At this time, TLC showed complete
conversion of starting material. The organic solvent was removed under reduced pressure. The
residue was then dissolved in ACN (0.02 M solution). Diisopropylethylamine (10 equiv) and
(Z)-tert-butyl (((tert-butoxycarbonyl)imino)(1H-pyrazol-1-yl)methyl)carbamate (1.05 equiv)
were added to the solution, and the resulting reaction mixture was left to mix at rt for 3 days after
which the organic solvent was removed under reduced pressure and the residue was purified by
silica gel column chromatography.
General Procedure 2E: Deprotection of t-Boc protecting groups with HCl (g). Hydrochloric acid
gas was bubbled through a solution of the N-Boc protected compound in methanol for 2-5
minutes, until complete consumption of starting material was observed by TLC. The reaction
mixture was concentrated under reduced pressure and triturated with diethyl ether to yield the
corresponding free amine hydrochloride salt.
General Procedure 2F: Acylation of amines. The desired acyl chloride (1.8 equiv) was added to
a round bottom containing tert-butyl (S)-2-(3-(4-(piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-
yl)pyrrolidine-1-carboxylate (1 equiv) and TEA (1.6 equiv) in DCM (0.045 M) cooled to 0 ºC.
The reaction mixture was then warmed to room temperature and stirred for 17 h. The organic
solvent was removed under reduced pressure and the residue was purified by silica gel column
chromatography.

157
(Z)-N'-hydroxy-4-iodobenzimidamide (2.7a). Synthesized by General Procedure 2A. 88%, white
solid. 1H NMR (400 MHz, CD3OD) δ 7.78 (dt, J=7.8, 3.6 Hz, 2H), 7.44 (dt, J=7.4, 0.1 Hz, 2H);
13C NMR (101 MHz, CD3OD) δ 154.5, 138.7, 133.8, 129.0, 96.2. HRMS (ESI+): calcd for
C7H8N2OI [M+H]+: 262.9681, found: 262.9695.
(S)-tert-butyl-2-(3-(4-iodophenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate (2.8a).
Synthesized by General Procedure 2B. 53%, yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.85-
7.76 (m, 4H), 5.22-4.99 (m, 1H), 3.75-3.43 (m, 2H), 2.46-2.30 (m, 1H), 2.20-1.94 (m, 3H), 1.45
(s, 3H), 1.28 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 181.0, 167.9, 153.6, 138.2, 129.0, 126.3,
98.1, 80.6, 53.9, 46.5, 32.5, 28.3, 23.8; HRMS (ESI+): Calcd for C17H20N3O3INa [M+Na]:
464.0447, Found: 464.0405.
(S)-tert-butyl 2-(3-(4-iodophenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-carboxylate (2.11).
Synthesized by General Procedure 2B. 11%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.83 (s,
4H), 5.41 (dd, J = 8, 4 Hz, 1H), 4.23-4.17 (m, 1H), 4.08-4.02 (m, 1H) 2.76-2.67 (m, 1H), 2.58-
2.52 (m, 1H), 1.35 (bs, 9H); 13C NMR (101 MHz, CDCl3) δ 178.76, 168.12, 138.26, 129.12,
126.30, 98.13, 80.81, 55.29, 47.66, 28.31, 22.00; HRMS (ESI+): calcd for C16H18IN3NaO3
[M+Na]+: 450.0291, found: 450.0314.
(S)-tert-butyl 2-(3-(4-morpholinophenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-carboxylate (2.12d).
Synthesized by General Procedure 2C. 81%, white-yellow solid. 1H NMR (500 MHz, CDCl3) δ
7.98 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 5.40 (dd, J = 8.8, 5.6 Hz, 1H), 4.20 (td, J =
8.7, 5.8 Hz, 1H), 4.04 (td, J = 8.6, 6.2 Hz, 4H), 3.89 – 3.85 (m, 4H), 3.29 – 3.24 (m, 4H), 2.75 –
2.65 (m, 1H), 2.58 – 2.48 (m, 1H), 1.34 (bs, 9H); 13C NMR (101 MHz, CDCl3) δ 178.02, 168.44,
153.26, 128.86, 117.41, 114.77, 80.69, 66.84, 48.31, 28.31, 22.02; HRMS (ESI+): calcd for
C20H27N4O4 [M+H]+: 387.2032, found: 387.2022.

158
(S)-tert-butyl 2-(3-(4-(4-methylpiperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-
carboxylate (2.12e). Synthesized by General Procedure 2C. 47%, white-yellow solid. 1H NMR
(400 MHz, CDCl3) δ 7.94 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 5.37 (dd, J = 8.6, 5.5 Hz,
1H), 4.17 (dd, J = 8.6, 5.5 Hz, 1H), 4.05 – 3.96 (m, 1H), 3.37 – 3.24 (m, 4H), 2.66 (m, 1H), 2.60
– 2.44 (m, 5H), 2.33 (s, 3H), 1.34 (br s, 9 H); 13C NMR (101 MHz, CDCl3) δ 177.86, 168.38,
153.06, 128.72, 116.74, 114.88, 80.56, 77.48, 77.16, 76.84, 54.89, 47.87, 46.16, 29.13, 28.22,
21.92; HRMS (ESI+): calcd for C21H30N5O3 [M+H]+: 400.2349, found: 400.2376.
(S)-tert-butyl (((tert-butoxycarbonyl)imino)(2-(3-(4-morpholinophenyl)-1,2,4-oxadiazol-5-
yl)azetidin-1-yl)methyl)carbamate. Synthesized by General Procedure 2D. 66%, clear oil. 1H
NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 6.08 – 5.94 (m,
1H), 4.63 (dd, J = 8.6 Hz, 1H), 4.19 (td, J = 9.4, 5.5 Hz, 1H), 3.92 – 3.83 (m, 4H), 3.33 – 3.22
(m, 4H), 2.90 – 2.77 (m, 1H), 2.63 – 2.50 (m, 1H), 1.47 (s, 18H); 13C NMR (101 MHz, CDCl3) δ
176.86, 168.43, 159.88, 153.30, 128.94, 117.24, 114.74, 80.10, 66.85, 48.30, 28.22, 22.53;
HRMS (ESI+): calcd for C26H37N6O6 [M+H]+: 529.2775, found: 529.2795.
(S)-tert-butyl (((tert-butoxycarbonyl)imino)(2-(3-(4-(4-methylpiperazin-1-yl)phenyl)-1,2,4-
oxadiazol-5-yl)azetidin-1-yl)methyl)carbamate. Synthesized by General Procedure 2D. 43%,
yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.84 (s, 1H), 7.99 – 7.92 (m, 2H), 6.99 – 6.93 (m,
2H), 6.02 (s, 1H), 4.67 – 4.56 (m, 1H), 4.18 (td, J = 9.4, 5.6 Hz, 1H), 3.36 – 3.30 (m, 4H), 2.90 –
2.77 (m, 1H), 2.61 – 2.51 (m, 5H), 2.36 (s, 3H), 1.54 – 1.37 (m, 18H); 13C NMR (101 MHz,
CDCl3) δ 191.21, 176.61, 162.69, 153.40, 128.91, 128.80, 125.54, 114.96, 114.92, 81.77, 78.93,
58.17, 54.97, 47.97, 46.24, 41.05, 31.39, 29.85, 29.22, 28.15, 22.53, 20.86, 20.52; HRMS
(ESI+): calcd for C27H40N7O5 [M+H]+: 542.3091 , found: 542.3108.

159
(S)-amino(2-(3-(4-morpholinophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-yl)methaniminium
chloride (2.13d). Synthesized by General Procedure 2E. 78%, white solid. 1H NMR (400 MHz,
CD3OD) δ 7.95 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 5.80 (dd, J = 9.4, 5.2 Hz, 1H), 4.37
(m, 1H), 4.26 (m, 1H), 3.86 – 3.82 (m, 4H), 3.30 – 3.26 (m, 4H), 3.10 – 3.00 (m, 1H), 2.62 (m,
1H).; 13C NMR (101 MHz, CD3OD) δ 177.78, 169.72, 158.56, 155.09, 129.63, 117.55, 115.78,
67.79, 58.43, 50.82, 22.94; HRMS (ESI+): calcd for C16H21N6O2 [M+H]+: 329.1729, found:
329.1719.
(S)-4-(4-(5-(1-(amino(iminio)methyl)azetidin-2-yl)-1,2,4-oxadiazol-3-yl)phenyl)-1-
methylpiperazin-1-ium chloride (2.13e). Synthesized by General Procedure 2E. 83%, white
solid. 1H NMR (400 MHz, CD3OD) δ 7.94 (d, J = 8.9 Hz, 2H), 7.07 (d, J = 8.9 Hz, 2H), 5.80
(m, 1H), 4.37 (m, 1H), 4.26 (m, 1H), 3.38 – 3.34 (m, 9H), 3.05 (m, 1H), 2.67 – 2.61 (m, 5H),
2.38 (s, 3H); 13C NMR (126 MHz, CD3OD) δ 177.31, 167.46, 157.18, 146.52, 128.99, 125.00,
120.04, 64.64, 57.08, 53.10, 49.60, 21.64; HRMS (ESI+): calcd for C17H25N7O [M+H]+:
343.2121, found: 343.2027.
tert-butyl (S)-2-(3-(4-(piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-carboxylate.
Synthesized by General Procedure 2C. 40%, beige solid. 1H NMR (400 MHz, CDCl3) δ 7.97 (d,
J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 5.40 (dd, J = 8.8, 5.7 Hz, 1H), 4.25 – 4.15 (m, 1H),
4.08 – 3.99 (m, 1H), 3.29 – 3.25 (m, 4H), 3.06 – 3.01 (m, 4H), 2.75 – 2.65 (m, 1H), 2.59 – 2.47
(m, 1H), 1.35 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.83, 168.28, 153.19, 128.69, 117.12,
115.07, 80.54, 55.25, 48.53, 45.45, 29.68, 28.15, 21.85; HRMS (ESI+): calcd for C20H28N5O3
[M+H]+: 386.2192, found: 386.2202.
tert-butyl (S)-2-(3-(4-(piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate
(2.14). Synthesized by General Procedure 2C. 50%, off-white solid. 1H NMR (400 MHz, CDCl3)

160
δ 7.93 (d, 2H), 6.94 (d, J = 8.5 Hz, 2H), 5.19 – 4.96 (m, 1H), 4.59 (s, 1H), 3.79 – 3.59 (m, 1H),
3.56 – 3.40 (m, 1H), 3.37 – 3.28 (m, 4H), 3.14 – 3.06 (m, 4H), 2.43 – 2.26 (m, 1H), 2.20 – 2.05
(m, 2H), 2.02 – 1.90 (m, 1H), 1.43* (s, 3H), 1.27* (s, 6H); 13C NMR (101 MHz, CDCl3) δ 180.19,
168.13, 153.11, 128.73, 117.39, 115.24, 80.45, 53.88, 48.33, 46.41, 45.32, 32.44, 29.76, 28.46,
28.22, 23.77; HRMS (ESI+): calcd for C21H30N5O3 [M+H]+: 400.2349, found: 400.2352.
tert-butyl (S)-2-(3-(4-(4-(3-methylbutanoyl)piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-
yl)pyrrolidine-1-carboxylate (2.15a). Synthesized by General Procedure 2F. 86%, beige
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.5 Hz, 2H), 6.95 (d, J = 8.3 Hz,
2H), 5.21 – 5.01 (m, 1H), 3.85 – 3.75 (m, 2H), 3.77 – 3.61 (m, 3H), 3.60 – 3.43 (m, 1H), 3.34 –
3.24 (m, 4H), 2.47 – 2.30 (m, 1H), 2.26 (d, J = 7.0 Hz, 2H), 2.22 – 2.06 (m, 4H), 2.05 – 1.92 (m,
1H), 1.46 (s, 3H), 1.29 (s, 6H), 0.99 (d, J = 6.5 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 180.34,
171.26, 168.19, 152.81, 128.88, 117.81, 115.45, 80.54, 53.97, 48.63, 48.37, 46.49, 45.60, 42.18,
41.32, 32.52, 28.55, 28.31, 25.97, 24.49, 23.85, 22.91; HRMS (ESI+): calcd for C26H38N5O4
[M+H]+: 484.2924, found: 484.2887.
tert-butyl (S)-2-(3-(4-(4-((3r,5r,7r)-adamantane-1-carbonyl)piperazin-1-yl)phenyl)-1,2,4-
oxadiazol-5-yl)pyrrolidine-1-carboxylate (2.15b). Synthesized by General Procedure 2F. 68%,
yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, 2H), 6.95 (d, J = 8.2 Hz, 2H),
5.21 – 5.00 (m, 1H), 3.92 – 3.81 (m, 4H), 3.75 – 3.61 (m, 1H), 3.60 – 3.42 (m, 1H), 3.33 – 3.23
(m, 4H), 2.45 – 2.28 (m, 1H), 2.21 – 2.11 (m, 2H), 2.07 – 1.95 (m, 9H), 1.80 – 1.68 (m, 7H),
1.46 (s, 3H), 1.29 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 191.60, 180.30, 175.99, 168.20,
152.91, 128.83, 117.64, 115.23, 80.53, 53.96, 48.55, 46.47, 45.08, 41.87, 39.27, 36.78, 36.66,
32.51, 28.61, 28.29, 28.10, 23.83; HRMS (ESI+): calcd for C32H44N5O4 [M+H]+: 562.3393,
found: 562.3384.

161
tert-butyl (S)-2-(3-(4-(4-(2-phenylacetyl)piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-
1-carboxylate (2.15c). Synthesized by General Procedure 2F. 67 %, beige amorphous solid. 1H
NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.4 Hz, 2H), 7.34 – 7.27 (m, 2H), 7.27 – 7.20 (m, 3H),
6.88 (d, J = 8.3 Hz, 2H), 5.22 – 4.97 (m, 1H), 3.85 – 3.75 (m, 3H), 3.73 – 3.40 (m, 4H), 3.29 –
3.19 (m, 2H), 3.12 – 3.02 (m, 2H), 2.44 – 2.24 (m, 1H), 2.20 – 2.04 (m, 2H), 2.02 – 1.91 (m,
1H), 1.44 (s, 3H), 1.27 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 180.32, 169.80, 168.14, 153.73,
152.66, 134.91, 129.48, 128.98, 128.83, 128.69, 128.65, 128.51, 127.27, 127.09, 117.76, 115.40,
80.55, 53.95, 48.27, 48.07, 46.47, 45.85, 41.59, 41.21, 32.49, 28.53, 28.28, 23.82; HRMS
(ESI+): calcd for C29H36N5O4 [M+H]+: 518.2767, found: 518.2812.
tert-butyl (S)-2-(3-(4-(4-benzylpiperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-
carboxylate (2.15d). Benzyl bromide (0.01 mL, 0.057 mmol) was added to a round bottom
containing tert-butyl (S)-2-(3-(4-(piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-
carboxylate (33 mg, 0.0.086 mmol) in DCM (0.18 mL). The reaction mixture was stirred for 17
h, after which it was poured into water and extracted three times with DCM. The combined
organic extracts were washed with brine, dried over Na2SO4 and concentrated under reduced
pressure. The resulting brown residue was purified by flash chromatography over silica gel (50 –
70% EtOAc in hexanes) to give the title compound (20 mg, 49%) as an off-white solid. 1H NMR
(400 MHz, CDCl3) δ 7.96 (d, J = 8.4 Hz, 2H), 7.38 – 7.31 (m, 4H), 7.30 – 7.27 (m, 1H), 6.94 (d,
J = 8.5 Hz, 2H), 5.39 (dd, J = 8.8, 5.6 Hz, 1H), 4.25 – 4.15 (m, 1H), 4.08 – 3.98 (m, 1H), 3.58 (s,
2H), 3.36 – 3.29 (m, 4H), 2.75 – 2.47 (m, 6H), 1.35 (s, 9H); 13C NMR (101 MHz, CDCl3) δ
177.93, 168.53, 153.29, 129.32, 128.82, 128.47, 127.37, 114.93, 80.70, 63.17, 52.99, 48.08,
28.33, 22.03; HRMS (ESI+): calcd for C27H34N5O3 [M+H]+: 476.2662, found: 476.2644.

162
tert-butyl (S,E)-(((tert-butoxycarbonyl)imino)(2-(3-(4-(4-(3-methylbutanoyl)piperazin-1-
yl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methyl)carbamate. Synthesized by General
Procedure 2D. 43%, off-white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.90 (d, J = 8.4
Hz, 2H), 6.87 (d, 2H), 5.50 (dd, J = 7.6, 4.5 Hz, 1H), 3.84 – 3.77 (m, 1H), 3.76 – 3.68 (m, 3H),
3.62 – 3.57 (m, 2H), 3.26 – 3.19 (m, 4H), 2.41 – 2.32 (m, 1H), 2.20 (d, J = 7.0 Hz, 2H), 2.14 –
2.04 (m, 2H), 2.01 – 1.92 (m, 1H), 1.39 (s, 18H), 0.93 (d, J = 6.6 Hz, 6H); 13C NMR (101 MHz,
CDCl3) δ 178.37, 171.38, 168.20, 153.49, 152.83, 131.98, 128.98, 128.76, 128.41, 125.42,
117.64, 115.41, 81.36, 55.61, 49.77, 48.63, 48.34, 45.62, 42.17, 41.35, 35.14, 31.46, 28.25,
28.11, 26.01, 24.10, 22.91; HRMS (ESI+): calcd for C32H48N7O6 [M+H]+: 626.3666, found:
626.3685.
tert-butyl ((E)-((S)-2-(3-(4-(4-((3r,5r,7r)-adamantane-1-carbonyl)piperazin-1-yl)phenyl)-1,2,4-
oxadiazol-5-yl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate. Synthesized by
General Procedure 2D. 26%, yellow amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J =
8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 5.58 (dd, J = 7.5, 4.4 Hz, 1H), 3.87 (dd, 5H), 3.81 (q, J =
6.4, 5.7 Hz, 1H), 3.29 (dd, J = 5.0 Hz, 4H), 2.47 – 2.38 (m, 1H), 2.29 – 2.22 (m, 1H), 2.21 – 2.12
(m, 1H), 2.09 – 2.00 (m, 10H), 1.78 – 1.68 (m, 6H), 1.46 (s, 18H); 13C NMR (101 MHz, CDCl3)
δ 178.59, 176.03, 175.69, 168.52, 168.20, 152.93, 128.94, 117.58, 115.20, 114.83, 103.86, 80.94,
55.40, 49.57, 48.53, 45.09, 44.77, 41.87, 39.27, 38.83, 36.78, 36.55, 31.56, 31.47, 31.21, 29.82,
29.41, 29.08, 28.61, 28.25, 27.80, 27.23, 24.05; HRMS (ESI+): calcd for C38H54N7O6 [M+H]+:
704.4136, found: 704.4191.
tert-butyl (S,E)-(((tert-butoxycarbonyl)imino)(2-(3-(4-(4-(2-phenylacetyl)piperazin-1-yl)phenyl)-
1,2,4-oxadiazol-5-yl)pyrrolidin-1-yl)methyl)carbamate. Synthesized by General Procedure 2D.
74%, off-white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 10.10 (s, 1H), 7.93 (d, J = 8.9

163
Hz, 2H), 7.36 – 7.31 (m, 2H), 7.28 – 7.27 (m, 3H), 6.89 (d, J = 8.8 Hz, 2H), 5.56 (dd, J = 6.1 Hz,
1H), 3.91 – 3.73 (m, 6H), 3.61 (dd, J = 5.2 Hz, 2H), 3.27 (dd, J = 5.3 Hz, 2H), 3.10 (dd, J = 5.1
Hz, 2H), 2.47 – 2.38 (m, 1H), 2.29 – 2.11 (m, 2H), 2.07 – 1.98 (m, 1H), 1.46 (s, 18H); 13C NMR
(126 MHz, CDCl3) δ 178.60, 169.70, 168.15, 153.81, 152.69, 134.96, 128.97, 128.67, 128.49,
127.78, 127.11, 117.73, 115.65, 115.36, 115.03, 114.82, 100.42, 100.12, 82.28, 79.81, 63.22,
55.41, 49.56, 48.29, 48.08, 45.86, 41.91, 41.57, 41.26, 31.70, 31.48, 28.26, 27.95; HRMS
(ESI+): calcd for C35H46N7O6 [M+H]+: 660.3510, found: 660.3520.
tert-butyl (S,Z)-((2-(3-(4-(4-benzylpiperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-
yl)((tert-butoxycarbonyl)imino)methyl)carbamate. Synthesized by General Procedure 2D. 44%,
yellow amorphous solid.1H NMR (400 MHz, CDCl3) δ 10.82 (s, 1H), 7.94 (d, J = 8.4 Hz, 2H),
7.39 – 7.31 (m, 4H), 7.30 – 7.27 (m, 1H), 6.94 (d, J = 8.5 Hz, 2H), 6.07 – 5.92 (m, 1H), 4.68 –
4.56 (m, 1H), 4.23 – 4.10 (m, 1H), 3.58 (s, 2H), 3.32 (dd, J = 5.0 Hz, 4H), 2.89 – 2.74 (m, 1H),
2.66 – 2.49 (m, 5H), 1.45 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 176.70, 168.50, 162.79,
153.32, 138.02, 129.31, 129.17, 128.87, 128.72, 128.45, 127.35, 116.56, 114.88, 82.51, 79.89,
63.15, 52.97, 48.07, 28.22, 28.14, 22.52; HRMS (ESI+): calcd for C33H44N7O5 [M+H]+:
618.3404, found: 618.3431.
(S)-amino(2-(3-(4-(4-(3-methylbutanoyl)piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-
1-yl)methaniminium chloride (2.16a). Synthesized by General Procedure 2E. 88%, white solid.
1H NMR (400 MHz, CD3OD) δ 8.03 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 5.47 (d, J =
7.4 Hz, 1H), 3.91 – 3.77 (m, 6H), 3.65 (q, J = 9.3 Hz, 1H), 3.48 (dt, J = 18.2, 4.2 Hz, 4H), 3.34
(dt, J = 3.3, 1.7 Hz, 4H), 2.66 – 2.45 (m, 2H), 2.39 (d, J = 7.0 Hz, 2H), 2.26 (q, J = 6.3 Hz, 1H),
2.19 – 2.06 (m, 1H), 1.03 (d, J = 6.5 Hz, 6H); 13C NMR (101 MHz, CD3OD) δ 175.78, 173.68,

164
169.36, 152.15, 130.02, 120.47, 118.26, 55.63, 51.20, 50.79, 47.44, 46.13, 42.71, 41.92, 30.22,
27.04, 24.53, 22.94; HRMS (ESI+): calcd for C22H32N7O2+
[M+H]+: 426.2617, found: 426.2617.
((S)-2-(3-(4-(4-((3r,5r,7r)-adamantane-1-carbonyl)piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-
yl)pyrrolidin-1-yl)(amino)methaniminium chloride (2.16b). Synthesized by General Procedure
2E. 91%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 7.94 – 7.89 (m, 2H), 7.09 – 7.04 (m,
2H), 5.40 (dd, J = 8.0, 1.9 Hz, 1H), 3.88 (t, J = 5.1 Hz, 4H), 3.80 – 3.73 (m, 1H), 3.60 (td, J =
9.7, 7.2 Hz, 1H), 3.34 – 3.31 (m, 4H), 2.59 – 2.43 (m, 2H), 2.22 (dddt, J = 9.6, 7.1, 5.0, 2.6 Hz,
1H), 2.05 (s, 10H), 1.80 (s, 6H); 13C NMR (126 MHz, CD3OD) δ 178.44, 177.95, 169.55,
157.08, 154.64, 129.63, 117.65, 116.13, 56.44, 46.33, 43.01, 40.13, 37.60, 32.69, 29.98, 24.33;
HRMS (ESI+): calcd for C28H38N7O2+
[M+H]+: 504.3087, found: 504.3059.
(S)-amino(2-(3-(4-(4-(2-phenylacetyl)piperazin-1-yl)phenyl)-1,2,4-oxadiazol-5-yl)pyrrolidin-1-
yl)methaniminium chloride (2.16c). Synthesized by General Procedure 2E. 92%, white solid. 1H
NMR (500 MHz, CD3OD) δ 7.94 (d, J = 8.6 Hz, 2H), 7.36 – 7.21 (m, 5H), 7.12 (d, J = 8.6 Hz,
2H), 5.40 (d, J = 7.4 Hz, 1H), 3.84 (s, 2H), 3.81 (t, J = 5.2 Hz, 2H), 3.75 (d, J = 6.7 Hz, 3H),
3.58 (ddd, J = 16.4, 9.8, 6.6 Hz, 2H), 3.35 (q, J = 6.0, 5.2 Hz, 3H), 3.22 (d, J = 4.9 Hz, 2H), 2.53
(dd, J = 12.8, 6.6 Hz, 1H), 2.44 (dd, J = 13.2, 6.4 Hz, 1H), 2.25 – 2.16 (m, 1H), 2.11 – 2.02 (m,
1H); 13C NMR (126 MHz, CD3OD) δ 178.61, 172.29, 169.36, 157.06, 153.74, 153.25, 136.30,
129.85, 129.83, 129.76, 127.99, 119.26, 119.05, 117.21, 117.03, 56.46, 49.97, 49.63, 46.68,
46.55, 44.62, 42.49, 41.30, 32.71, 24.34; HRMS (ESI+): calcd for C25H30N7O2+
[M+H]+:
460.2461, found: 460.2477.
(S)-4-(4-(5-(1-(amino(iminio)methyl)azetidin-2-yl)-1,2,4-oxadiazol-3-yl)phenyl)-1-
benzylpiperazin-1-ium chloride (2.16d). Synthesized by General Procedure 2E. 97 %, off-white
solid. 1H NMR (400 MHz, CD3OD) δ 8.05 – 7.99 (m, 2H), 7.65 (dd, J = 6.5, 3.0 Hz, 2H), 7.58 –

165
7.52 (m, 3H), 7.18 (d, J = 8.7 Hz, 2H), 5.85 (dd, J = 9.4, 5.2 Hz, 1H), 4.48 (s, 2H), 4.40 (td, J =
8.9, 6.1 Hz, 1H), 4.30 (ddd, J = 9.4, 8.2, 5.8 Hz, 1H), 4.08 (d, J = 13.2 Hz, 2H), 3.68 – 3.58 (m,
2H), 3.31 – 3.22 (m, 2H), 3.18 – 3.04 (m, 1H), 2.76 – 2.60 (m, 1H); 13C NMR (101 MHz,
CD3OD) δ 177.97, 169.51, 158.55, 153.28, 132.59, 131.42, 130.44, 129.99, 129.82, 119.06,
117.04, 61.58, 58.48, 52.57, 50.94, 46.56, 23.00; HRMS (ESI+): calcd for C23H29N7O2+
[M+2H]2+: 209.62115, Found: 209.6207.
6.2 Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2
Selective Inhibitors: Design, Synthesis, and in Vivo Activity
6.2.1 Sphingosine kinase assays
The inhibitory activity of the synthesized compounds on human SphK1 and SphK2 was
determined using a previously published method.199, 200 Recombinant human SphK1 or SphK2
isolated from a cell lysate was briefly incubated with (0.3 µM) or without compound,
sphingosine, and γ-[32P]ATP. The radiolabeled sphingosine 1-phosphate was isolated via
extraction and thin-layer chromatography and then quantified via scintillation counting.
6.2.2 Sample Preparation and LC–MS–MS Analysis
Sample preparation protocols were from our previous publication201 with minor modifications.
Cell pellets (2–3 × 106 cells), whole blood (10 µL) or plasma (10 µL) was mixed with 2 mL of a
methanol:chloroform solution (3:1) and transferred to a capped glass vial. Suspensions were
supplemented with 10 µL of internal standard solution containing 10 pmoles of deuterated (D7)
S1P and deuterated (D7) sphingosine. The mixture was placed in a bath sonicator for 10 min and
incubated at 48 °C for 16 h. The mixture was then cooled to ambient temperature and mixed with
200 µL of 1M KOH in methanol. The samples were again sonicated and incubated a further 2 h
at 37 °C. Samples were then neutralized by the addition of 20 µL of glacial acetic acid and

166
transferred to 2 mL microcentrifuge tubes. Samples were then centrifuged at 12,000 x g for 12
min at 4 °C. The supernatant fluid was collected in a separate glass vial and evaporated under a
stream of nitrogen gas. Immediately prior to LC–MS analysis, the dried material was dissolved
in 0.3 ml of methanol and centrifuged at 12,000 x g for 12 min at 4 °C. Fifty µL of the resulting
supernatant fluid were analyzed by Liquid Chromatography-ESI Mass Spectrometry (LC–MS)
using a triple quadrupole mass spectrometer (AB-Sciex 4000 Q-Trap) coupled to a Shimadzu
LC-20AD LC system. A binary solvent gradient with a flow rate of 1 mL/min was used to
separate sphingolipids and drugs by reverse phase chromatography using a Supelco Discovery
C18 column (50 mm × 2.1 mm, 5 µm bead size). Mobile phase A consisted of
water:methanol:formic acid (79:20:1) while mobile phase B was methanol : formic acid (99:1).
The run started with 100% A for 0.5 minutes. Solvent B was then increased linearly to 100% B
in 5.1 minutes and held at 100% for 4.3 min. The column was finally re-equilibrated to 100% A
for 1 min. Natural sphingolipids were detected using multiple reaction monitoring (MRM) as
follows: S1P (380.4 à 264.4); deuterated (D7)C18S1P (387.4 à 271.3); sphingosine (300.5 à
264.4); deuterated (D7) sphingosine (307.5 à 271.3).
6.2.3 Pharmacokinetic Analysis
Mouse studies were conducted using a previously reported method.199 3.20dd (10 mg/kg) was
administered intraperitoneally to groups of 3 to 4 mice (strain: C57BL6/j) or an equal volume of
vehicle (2% solution of hydroxypropyl-β-cyclodextrin (Cargill Cavitron 82004)). Blood samples
were then collected at the specified time points (ASAP time points were collected 1-2 min
following drug addition). The blood samples were analyzed via LC–MS, as described (vide
supra). Animal protocols were approved prior to experimentation by the University of Virginia’s
School of Medicine Animal Care and Use Committee.

167
6.2.4 Molecular Docking
Molecular docking was performed using compounds (3.2, 3.20x) to assess potential difference in
position in the binding pocket of SphK2. The SphK2 model, with ATP and Mg2+ bound, was
generated with Molecular Operating Environment (MOE) and energy minimized as previously
described.202 Marvin was used for drawing, displaying and characterizing chemical structures,
substructures and reactions for preparation in docking programs, Marvin 17.3.13, 2017,
ChemAxon (http://www.chemaxon.com). AutoDock Tools203 was used to prepare the protein
and ligand files, while AutoDock Vina204 was used to perform the docking for pose prediction.
The grid box was set to 20 x 20 x 28 Angstrom, with a 1.000 Å grid spacing was used. The
center of the box was placed at the approximate center of the ligand-binding cavity, with a part
of the ATP binding cavity included as previously performed202 and to ensure coverage and
interaction with key Asp residues. Up to ten docked poses were predicted for each compound.
The number of predicted poses is dependent on the fitness of the sampled compound
orientations. The lowest energy pose for each docked ligand in SphK2 was then used for analysis
of interactions with key residues in the SphK2 binding pocket. Free energy of binding scores
were cataloged for each docked compound and used as one level of comparison between
compounds.
6.2.5 General Material and Synthetic Procedures
All reactions conducted in a microwave were conducted in a Discover SP microwave synthesizer
(CEM Corporation). All solvents were dried using the PureSolv solvent purification system prior
to use. All chemical reagents were purchased from commercial sources and used without further
purification. Thin layer chromatography (TLC) was performed on aluminum-backed silica gel,
200 µm, F254, and column chromatography was performed on flash grade silica gel, 40-63 µm,

168
using a Combiflash Rf purification system. 1H NMR spectra were recorded at 500 or 400 MHz;
the corresponding 13C NMR resonant frequencies were 126 and 101 MHz, respectively; the
corresponding 19F NMR resonant frequencies were 471 and 376 MHz, respectively. 1H NMR
chemical shifts are reported in ppm with the solvent resonance as an internal standard (CDCl3:
7.26 ppm; CD3OD: 4.87 ppm; acetone-d6: 2.05 ppm). 13C NMR, chemical shifts are reported in
ppm with the solvent resonance as the internal standard (CDCl3: 77.16 ppm; CD3OD: 49.00 ppm;
acetone-d6: 206.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d
= doublet, t = triplet, q = quartet, br = broad, m= multiplet), coupling constants (Hz), and
integration. Rotamers are denoted by an asterisk (*). High resolution mass spectroscopy (HRMS)
was performed on an LC–MS time–of–flight mass spectrometer using electrospray ionization
(ESI). HPLC analyses were performed with a Thermo Electron TSQ triple quadrupole mass
spectrometer equipped with an ESI source. All compounds tested in biological assays are >95%
pure by 1H NMR and HPLC analyses unless noted otherwise. NMR spectra for the synthesized
compounds can be found online (http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.7b00233).
General Procedure 3A: Synthesis of amide-oxime derivatives. TEA (2.7 equiv) was added to a
solution of the appropriate benzonitrile (1 equiv) with hydroxylamine hydrochloride (2.6 equiv)
in ethanol (0.47 M solution). The mixture was reacted in a microwave for 6 min at 150 ºC. The
organic solvent was removed under reduced pressure, and the residue was purified by silica
gel column chromatography to yield the desired product.
General Procedure 3B: Coupling of amide-oxime derivatives with (S)-2-(1-(tert-
butoxycarbonyl)pyrrolidin-2-yl)acetic acid. DIEA (1.80 equiv) was added to a solution of the
appropriate amidoxime (1 equiv) and (S)-2-(1-(tert-butoxycarbonyl)pyrrolidin-2-yl)acetic acid in
DMF (0.2 M solution). HCTU (1.5 equiv) was then added to the resulting mixture at rt and

169
stirred at 100 ºC for 4 to 8 h. The reaction progress was followed by TLC. The solution was
partitioned between EtOAc and LiBr aqueous solution. The aqueous solution was washed with
EtOAc three times, and the combined organic layers were washed with brine, dried over Na2SO4,
filtered, and concentrated via vacuum. The resulting residue was purified by silica gel column
chromatography.
General Procedure 3C: Coupling of 3.17 with alpha-bromoketones. DIEA (2 equiv) was added
to a solution of tert-butyl (S)-2-((3-(4-thioureidophenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate 3.17 (1 equiv) and alpha-bromoketone (1 equiv) in ethanol
(0.2 M solution). The resulting reaction mixture was then reacted in a microwave at 100 °C for 5
min. The organic solvent was removed under reduced pressure, and the resulting residue was
purified by silica gel column chromatography.
General Procedure 3D: Suzuki coupling of aryl bromides with phenyl boronic acids. Cs2CO3 (2
equiv) was added to a solution of of t-Boc protected aryl bromide intermediate (1 equiv) and the
appropriate phenyl boronic acid derivative (3 equiv) in DMF (0.045 M solution). The resulting
mixture was degassed for 10 min by bubbling N2 through the solution. PdCl2(dppf) (0.03 equiv)
was then added to the mixture, and heated in a microwave reactor at 150 ºC for 90 min. The
solution was partitioned between EtOAc and LiBr aqueous solution. The aqueous solution was
washed with EtOAc three times, and the combined organic layers were washed with brine, dried
over Na2SO4, filtered, and concentrated via vacuum. The resulting residue was purified by silica
gel column chromatography.
General Procedure 3E: Deprotection of t-Boc protecting groups with TFA and guanylation of
amines. To a solution of t-Boc protected intermediate in DCM, a 1N TFA solution in DCM was
added. The reaction mixture was then stirred at rt for 4 h. At this time, TLC showed complete

170
conversion of starting material. The organic solvent was removed under reduced pressure. The
residue was then dissolved in ACN (0.02 M solution). Diisopropylethylamine (10 equiv) and
(Z)-tert-butyl (((tert-butoxycarbonyl)imino)(1H-pyrazol-1-yl)methyl)carbamate (1.05 equiv)
were added to the solution, and the resulting reaction mixture was reacted in a microwave reactor
at 50 °C for 2 h. The organic solvent was removed under reduced pressure and the residue was
purified by silica gel column chromatography.
General Procedure 3F: Deprotection of t-Boc protecting groups with HCl (g). Hydrochloric acid
gas was bubbled through a solution of the N-Boc protected compound in methanol for 2 to 5
minutes, until complete consumption of starting material was observed by TLC. The reaction
mixture was concentrated under reduced pressure and triturated with diethyl ether to yield the
corresponding free amine hydrochloride salt.
N-(4-cyanophenyl)-2,2,2-trifluoroacetamide (3.13). 4-aminobenzonitrile 3.12 (1.0 g, 8.46 mmol)
was dissolved in DCM (8.5 mL) and cooled to 0 °C. Triethylamine (1.3 mL, 9.31 mmol) was
added dropwise and allowed to stir at 0 °C for 10 min. Trifluoroacetic anhydride (1.8 mL, 9.31
mmol) was then added dropwise. The reaction mixture was allowed to warm up to rt and stirred
for 19 h. The resulting reaction mixture was quenched with sat. aq. NH4Cl, and extracted three
times with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4,
and concentrated via vacuum. The residue was purified by silica gel column chromatography
(30% EtOAc/ hexanes) to yield 3.13 (1.0 g, 88%) as white solid. 1H NMR (400 MHz, CD3OD) δ
7.89 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H); 13C NMR (101 MHz, CD3OD) δ 157.14 (q,
2JCF = 38.4 Hz), 156.76 (q, 2JCF = 38.4 Hz), 142.17, 122.19, 121.26 (q, 1JCF = 281.3 Hz), 119.35,
118.57 (q, 1JCF = 281.3 Hz), 115.70 (q, 1JCF = 281.3 Hz), 112.85 (q, 1JCF = 281.3 Hz), 109.84; 19F

171
NMR (376 MHz, CD3OD) δ -75.64 (s, 3F); HRMS (ESI-): calcd for C9H4F3N2O [M-H]-:
213.0275, found: 213.0273.
(Z)-2,2,2-trifluoro-N-(4-(N'-hydroxycarbamimidoyl)phenyl)acetamide (3.14). Synthesized by
General Procedure 3A. 57%, white solid. 1H NMR (400 MHz, CD3OD) δ 7.39 (d, J = 8.9 Hz,
2H), 6.70 (d, J = 8.9 Hz, 2H); 13C NMR (101 MHz, acetone-d6) δ 156.22 (q, 2JCF = 38.4 Hz),
155.85 (q, 2JCF = 38.4 Hz), 155.48 (q, 2JCF = 38.4 Hz), 155.11 (q, 2JCF = 38.4 Hz), 152.17,
138.14, 131.54, 127.11, 121.23, 120.91 (q, 1JCF = 280.2 Hz), 118.29 (q, 1JCF = 280.2 Hz), 115.42
(q, 1JCF = 280.2 Hz), 112.56 (q, 1JCF = 280.2 Hz); 19F NMR (471 MHz, acetone-d6) δ -76.14 (s,
3F); HRMS (ESI+): calcd for C9H8F3N3O2 [M+H]+: 248.0659, found: 248.1822.
tert-butyl (S)-2-((3-(4-(2,2,2-trifluoroacetamido)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.15). Synthesized by General Procedure B. 67%, yellow
oil. 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 8.12 – 7.93 (m, 2H), 7.75 (dd, J = 14.9, 8.2 Hz,
2H), 4.35 – 4.19 (m, 1H), 3.46 – 3.21 (m, 3H), 3.07 (dt, J = 15.3, 7.3 Hz, 1H), 2.06 (q, J = 8.0,
6.9 Hz, 1H), 1.94 – 1.73 (m, 3H), 1.43 (s, 9H); 13C NMR (101 MHz, cdcl3) δ 177.39, 167.67,
155.56, 155.19 (q, 2JCF = 53.5 Hz), 154.79 (q, 2JCF = 53.5 Hz), 154.47 (q, 2JCF = 53.5 Hz), 153.94
(q, 2JCF = 53.5 Hz) 138.54, 128.45, 124.30, 120.89, 120.11 (q, 1JCF = 289.9 Hz), 117.24 (q, 1JCF =
289.9 Hz), 114.37 (q, 1JCF = 289.9 Hz), 111.51 (q, 1JCF = 289.9 Hz), 80.54*, 80.00*, 55.32*,
55.18*, 46.82*, 46.37*, 31.68*, 31.05*, 30.27, 28.47, 23.53*, 22.81*; 19F NMR (470 MHz, CDCl3)
δ -74.47 (s, 3F); HRMS (ESI+): calcd for C20H23F3N4NaO4 [M+Na]+: 463.1569, found:
463.1546.
tert-butyl (S)-2-((3-(4-aminophenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate
(3.16). (S)-tert-butyl-2-(3-(4-iodophenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboxylate 3.15
(54 mg, 0.123 mmol) was dissolved in MeOH (5 mL) and then 1 M LiOH (5 mL) was added.

172
The reaction mixture was refluxed for 3 h. At this time, TLC showed complete conversion of
starting material. The resulting product was extracted with EtOAc, and the combined organic
layers were washed with brine, dried over Na2SO4, filtered, and concentrated via vacuum to
provide 3.16 (45 mg, 82%) as a clear solid without further purification. 1H NMR (400 MHz,
CDCl3) δ 7.83 (d, J = 8.5 Hz, 2H), 6.69 (d, J = 8.1 Hz, 2H), 4.34 – 4.17 (m, 1H), 4.04 (s, 2H),
3.49 – 3.19 (m, 3H), 3.13 – 2.90 (m, 1H), 2.02 (s, 1H), 1.93 – 1.69 (m, 3H), 1.45 (s, 9H); 13C
NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 177.14, 177.01, 168.62, 154.81,
154.59, 149.68, 129.26, 116.93, 116.78, 115.03, 114.71, 80.34*, 79.92*, 55.55, 47.11*, 46.70*,
32.16*, 31.35*, 31.14*, 30.38*, 28.85, 23.91*, 23.13*; HRMS (ESI+): calcd for C18H24N4NaO3
[M+Na]+: 367.1746, found: 367.1743.
tert-butyl (S)-2-((3-(4-thioureidophenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-
carboxylate (3.17). tert-butyl (S)-2-((3-(4-aminophenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate 3.16 (100 mg, 0.290 mmol) was dissolved in THF (4 mL).
Di(1H-imidazol-1-yl)methanethione (70 mg, 0.392 mmol) was added to the reaction mixture and
allowed to stir at rt until TLC showed complete conversion of starting material. Ammonia gas
was then passed through the solution for 1 min. The organic solvent was removed under reduced
pressure, and the residue was purified by silica gel column chromatography (35%–80%
EtOAc/hexane) to yield 3.17 (101 mg, 86%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ
8.48 (d, J = 13.6 Hz, 1H), 8.12 (d, J = 8.1 Hz, 2H), 7.42 – 7.32 (m, 2H), 6.33 (s, 2H), 4.29 (d, J =
30.3 Hz, 1H), 3.52 – 3.25 (m, 3H), 3.08 (t, J = 7.6 Hz, 1H), 2.07 (s, 0H), 1.93 – 1.76 (m, 3H),
1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 181.73, 177.91, 167.46, 154.36, 151.61, 139.13,
129.40, 125.85, 124.54, 80.28*, 79.89*, 55.28, 46.88*, 46.53*, 31.90*, 31.15*, 30.34, 28.62,
23.69*, 22.92.*; HRMS (ESI+): calcd for C19H25N5O3S [M+H]+ : 404.1756, found: 404.1751.

173
tert-butyl (S)-2-((3-(4-((4-phenylthiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18a). Synthesized by General Procedure 3C. 56%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.10 – 8.04 (m, 2H), 7.91 – 7.84 (m, 2H), 7.71 –
7.61 (m, 1H), 7.59 – 7.51 (m, 2H), 7.42 (dd, J = 8.4, 6.9 Hz, 2H), 7.37 – 7.30 (m, 1H), 6.91 (s,
1H), 4.30 (m, 1H), 3.40 (m, 3H), 3.07 (m, 1H), 2.08 (m, 1H), 1.93 – 1.81 (m, 3H), 1.48 (s, 9H);
13C NMR (101 MHz, CDCl3) δ 177.22, 168.00, 162.90, 154.37, 151.71, 142.84, 134.53, 128.96,
128.83, 128.19, 126.28, 120.75, 117.33, 102.84, 80.21, 55.28, 46.90*, 46.51*, 31.95*, 31.17*,
30.24*, 29.85*, 28.64, 23.72*, 22.93*; HRMS (ESI+): calcd for C27H29N5O3S [M+H]+: 504.2069,
found: 504.2024.
tert-butyl (S)-2-((3-(4-((4-(pyridin-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18b). Synthesized by General Procedure 3C. 39%, off-
white solid. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 2H), 8.09 (d, J = 8.3 Hz, 2H), 7.85 – 7.69 (m,
2H), 7.60 (t, J = 7.8 Hz, 2H), 7.15 (s, 1H), 4.41 – 4.21 (m, 1H), 3.51 – 3.27 (m, 3H), 3.15 – 2.99
(m, 1H), 2.15 – 2.01 (m, 1H), 1.96 – 1.78 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ
177.33, 169.03, 167.92, 163.43, 150.29, 149.18, 142.56, 141.56, 131.98, 129.01, 121.16, 120.56,
117.59, 106.73, 80.22, 55.32, 46.90*, 46.53*, 31.94*, 31.18*, 30.28*, 29.85*, 28.65, 23.72*,
22.95*; HRMS (ESI+): calcd for C26H29N6O3S [M+H]+: 505.2022, found: 505.2016.
tert-butyl (S)-2-((3-(4-((4-(pyridin-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18c). Synthesized by General Procedure 3C. 75%, off-
white solid. 1H NMR (400 MHz, CDCl3) δ 9.20 (d, J = 1.9 Hz, 1H), 8.57 (dd, J = 4.8, 1.7 Hz,
1H), 8.23 – 8.13 (m, 2H), 8.08 (d, J = 8.2 Hz, 2H), 7.65 – 7.54 (m, 2H), 7.36 (dd, J = 7.9, 4.7 Hz,
1H), 7.00 (s, 1H), 4.40 – 4.21 (m, 1H), 3.52 – 3.26 (m, 3H), 3.15 – 2.98 (m, 1H), 2.13 – 2.01 (m,
1H), 1.95 – 1.75 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.24, 167.95, 163.59,

174
154.40, 148.85, 148.62, 147.73, 142.84, 133.54, 130.48, 128.98, 123.73, 120.88, 117.47, 104.04,
80.23*, 79.81*, 55.29, 46.91*, 46.50*, 31.92*, 31.15*, 30.23*, 29.85*, 28.63, 23.71*, 22.93*;
HRMS (ESI+): calcd for C26H29N6O3S [M+H]+: 505.2022, found: 505.2012.
tert-butyl (S)-2-((3-(4-((4-(4-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18d). Synthesized by General Procedure C. 66%, light
yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.05 (ap t, J = 10, 2H), 7.95 (br s, 1H), 7.86 – 7.81
(m, 2H), 7.58 – 7.50 ( m, 2H), 7.09 (ap t, J = 10, 2H), 6.82 (s, 1H), 4.38 – 4.23 (m, 1H), 3.50 –
3.26 (m, 3H), 3.14 - 3.00 (m, 1H), 2.12 – 2.00 (m, 1H), 1.94 – 1.77 (m, 3H), 1.48 (br s, 9H); 3C
NMR (101 MHz, CDCl3) δ 177.23, 167.97, 164.00 (d,f 1JCF = 248.5 Hz), 163.13, 161.54 (d, 1JCF
= 248.5 Hz), 158.61, 154.41, 151.31, 150.70, 142.80, 130.86 (d, 4JCF = 3.0), 130.83 (d, 4JCF =
3.0 Hz), 129.23, 128.95, 128.03 (d, 3JCF = 8.1 Hz), 127.95 (d, 3JCF = 8.1 Hz), 125.54, 120.95,
117.37, 115.83 (d, 2JCF = 22.2), 115.6 (d, 2JCF = 22.2 Hz), 102.34, 80.24*, 79.82*, 55.34, 46.90*,
46.52*, 31.94*, 31.17*, 31.01*, 30.26*, 28.64, 23.71*, 22.94*; 19F NMR (376 MHz, CDCl3) δ -
109.71 to -116.29 (m, 1F); HRMS (ESI+): calcd for C27H28FN5O3S [M+H]+: 522.1975, found:
522.1966.
tert-butyl (S)-2-((3-(4-((4-(4-chlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18e). Synthesized by General Procedure 3C. 77%,
yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.05 (ap t, J = 10 Hz, 2H), 7.98 (br s, 1H), 7.79 (dt,
J = 10, 2.5 Hz, 2H), 7.59 – 7.50 ( m, 2H), 7.37 (dt, J = 10, 2.5 Hz, 2H), 6.87 (s, 1H), 4.40 – 4.23
(m, 1H), 3.50 – 3.26 (m, 3H), 3.14 - 3.00 (m, 1H), 2.12 – 2.03 (m, 1H), 1.94 – 1.77 (m, 3H),
1.48 (br s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.05, 167.80, 162.99, 154.50, 150.34, 142.63,
133.70, 132.87, 128.80, 128.77, 127.34, 120.62, 117.23, 102.96, 80.10*, 79.67*, 55.14, 46.74*,

175
46.36*, 31.75*, 31.00*, 30.10*, 29.68*, 28.48, 23.54*, 22.76*; HRMS (ESI+): calcd for
C27H28ClN5NaO3S [M+Na]+: 560.1499, found 560.1502.
tert-butyl (S)-2-((3-(4-((4-(4-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18f). Synthesized by General Procedure 3C. 84%, off-
white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.2 Hz, 2H), 7.77 – 7.70 (m,
2H), 7.53 (dd, J = 9.2, 2.7 Hz, 3H), 6.90 (s, 1H), 4.40 – 4.22 (m, 1H), 3.50-3.27 (m, 3H), 3.15 –
2.99 (m, 1H), 2.13 – 2.03 (m, 1H), 1.95-178 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3)
δ 177.12, 168.01, 163.27, 154.44, 150.39, 142.98, 142.86, 133.45, 131.84, 128.82, 127.76,
121.95, 120.50, 117.35, 103.14, 80.33*, 79.87*, 55.34*, 55.22*, 46.86*, 46.51*, 31.84*, 31.13*,
31.05*, 30.99*, 30.25*, 29.80*, 28.61, 23.64*, 22.88*; HRMS (ESI+): calcd for C27H28BrN5NaO3S
[M+Na]+: 604.0993, found: 604.0967.
tert-butyl (S)-2-((3-(4-((4-(3-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18g). Synthesized by General Procedure 3C. 90%, off-
white solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.2 Hz, 2H), 7.74 (br. s, 1H), 7.63 (d, J =
7.9 Hz, 1H), 7.60 – 7.51 (m, 3H), 7.37 (td, J = 8.0, 5.9 Hz, 1H), 7.01 (q, J = 8.5, 1H), 6.92 (s,
1H), 4.40 – 4.40 – 4.21 (m, 1H), 3.3.57 – 3.26 (m, 3H), 3.16 – 2.96 (m, 1H), 2.15 – 2.01 (m, 1H),
1.94 – 1.78 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.28, 167.98, 164.52(d, 1JCF
= 246.4 Hz), 163.06, 162.08 (d, 1JCF = 246.4 Hz), 154.42, 150.44, 142.71, 136.72 (d, 3JCF = 8.1
Hz), 136.64 (d, 3JCF = 8.1 Hz), 130.35 (d, 3JCF = 9.1 Hz), 130.26 (d, 3JCF = 9.1 Hz), 128.97,
121.80 (d, 4JCF = 2.0 Hz), 121.78 (d, 4JCF = 2.0 Hz), 121.11, 120.88 117.44, 115.05 (d, 2JCF =
21.2 Hz), 114.84 (d, 2JCF = 21.2 Hz), 113.36 (d, 2JCF = 23.2 Hz), 113.13 (d, 2JCF = 21.2 Hz),
103.77, 80.26*, 79.84*, 55.32, 46.91*, 46.52*, 31.94*, 31.16*, 30.27*, 29.85*, 28.64, 23.72*,

176
22.93*; 19F NMR (376 MHz, CDCl3) δ -112.92 to -113.24 (m, 1F); HRMS (ESI+): calcd for
C27H28FN5NaO3S [M+Na]+: 544.1795, found: 544.1784.
tert-butyl (S)-2-((3-(4-((4-(3-chlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18h). Synthesized by General Procedure 3C. 63%, white
solid. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.7 Hz, 3H), 7.90 – 7.79 (m, 1H), 7.76 – 7.68
(m, 1H), 7.56 (t, J = 9.6 Hz, 2H), 7.40 – 7.21 (m, 2H), 6.90 (s, 1H), 4.45 – 4.19 (m, 1H), 3.54 –
3.24 (m, 3H), 3.07 (dd, J = 14.7, 8.6 Hz, 1H), 2.16 – 1.99 (m, 1H), 1.87 (dd, J = 14.3, 7.1 Hz,
3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.19, 168.01, 163.18, 154.70, 154.44,
150.21, 142.87, 136.28, 134.76, 130.02, 128.90, 128.03, 126.36, 124.29, 120.69, 117.41, 103.78,
80.30*, 79.86*, 55.35*, 55.24*, 50.92*, 46.89*, 46.52*, 31.89*, 31.15*, 30.27*, 28.63*, 23.68*,
22.91*; HRMS (ESI+): calcd for C27H28ClN5O3S [M+H]+: 538.1680, found: 538.1679.
tert-butyl (S)-2-((3-(4-((4-(3-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18i). Synthesized by General Procedure 3C. 78%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.11 – 8.04 (m, 2H), 8.01 (d, J = 1.9 Hz, 1H),
7.78 (d, J = 8.0 Hz, 1H), 7.55 (t, J = 8.9 Hz, 1H), 7.47 – 7.41 (m, 1H), 7.27 (d, J = 5.5 Hz, 1H),
6.91 (s, 1H), 4.30 – 4.22 (m, 1H), 3.40 – 3.16 – 2.98 (m, 3H), 3.07 (m, 1H), 2.20 – 1.97 (m, 1H),
1.97 – 1.77 (m, 2H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.22, 167.99, 163.11,
154.39, 150.10, 142.70, 136.48, 131.01, 130.33, 129.28, 128.96, 128.63, 124.75, 122.99, 120.83,
117.40, 103.84, 80.25*, 79.80*, 55.34, 46.90*, 46.51*, 31.92*, 31.16*, 30.24*, 28.64*, 23.71*,
22.92*; HRMS (ESI+): calcd for C27H28BrN5NaO3S [M+Na]+: 604.0993, found: 604.0988.
tert-butyl (S)-2-((3-(4-((4-(2-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18j). Synthesized by General Procedure 3C. 25 mg, 56%,
off-white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.16 (td, J = 7.7, 2.0 Hz, 1H), 8.06 (d,

177
J = 7.2 Hz, 2H), 7.75 (s, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.32 – 7.18 (m, 3H), 7.13 (ddd, J = 12.0,
7.9, 1.5 Hz, 1H), 4.40 – 4.22 (m, 1H), 3.54 – 3.24 (m, 3H), 3.16 – 2.99 (m, 1H), 2.14 – 2.01 (m,
1H), 1.96 – 1.77 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 191.63, 177.19, 168.00,
161.95, 161.71 (d, 1JCF = 251.5 Hz), 159.22 (d, 1JCF = 251.5 Hz), 154.41, 145.30, 142.90, 130.07
(d, 4JCF = 3.0 Hz), 130.04 (d, 4JCF = 3.0 Hz), 129.17 (d, 3JCF = 9.1 Hz), 129.08 (d, 3JCF = 9.1
Hz), 128.92, 124.54, 124.50, 122.39 (d, 3JCF = 11.1 Hz), 122.28 (d, 3JCF = 11.1 Hz), 120.69,
117.34, 117.24, 116.15 (d, 2JCF = 22.2 Hz), 115.93 (d, 2JCF = 22.2 Hz), 108.05 (d, 3JCF = 16.2
Hz), 107.89 (d, 3JCF = 16.2 Hz), 80.25*, 79.80*, 55.35, 46.91*, 46.52*, 31.93*, 31.16*, 30.26,
28.65, 23.71*, 22.93*; 19F NMR (376 MHz, CDCl3) δ -114.11 (s, 1F); HRMS (ESI+): calcd for
C27H28FN5NaO3S [M+Na]+: 544.1795, found: 544.1782.
tert-butyl (S)-2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18k). Synthesized by General Procedure 3C.
70%, off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.17 (br s, 1H), 8.10-8.00 (m, 2H), 7.96 (ap
d, J = 8, 2H), 7.64 (ap d, J = 8, 2H), 7.62 – 7.53 (m, 2H), 6.98 (br s, 1H), 4.41 – 4.23 (m, 1H),
3.51 – 3.25 (m, 3H), 3.14 -3.01 (m, 1H), 2.14 – 2.02 (m, 1H), 1.95 – 1.78 (m, 3H), 1.48 (br s,
9H); 13C NMR (101 MHz, CDCl3) δ 177.19, 167.99, 163.35, 154.44, 150.17, 142.83, 137.74,
130.26 (q, 2JCF = 32.3 Hz), 129.94 (q, 2JCF = 32.3 Hz), 129.62 (q, 2JCF = 32.3 Hz), 129.30
(q, 2JCF = 32.3 Hz), 128.89, 126.38, 125.82 (q, 3JCF = 4.0 Hz), 125.78 (q, 3JCF = 4.0 Hz), 125.74
(q, 3JCF = 4.0 Hz), 125.70 (q, 3JCF = 4.0 Hz), 122.97 (q, 1JCF = 224.2 Hz), 120.90, 120.75 (q, 1JCF
= 224.2 Hz), 120.26, 117.44, 104.67, 104.65, 80.34*, 79.89*, 55.25, 46.89*, 46.53*, 31.86*,
31.13*, 30.27*, 29.84*, 28.62*, 23.67*, 22.90*; 19F NMR (376 MHz, CDCl3) δ -62.49 (S, 3F);
HRMS (ESI+): calcd for C28H29F3N5O3S [M+H]+: 572.1943, found: 572.1958.

178
tert-butyl (S)-2-((3-(4-((4-(3-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18l). Synthesized by General Procedure 3C.
79%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 12.3 Hz, 1H), 8.10 (s,
1H), 8.03 (dt, J = 14.0, 7.3 Hz, 3H), 7.54 (ddt, J = 23.4, 15.4, 8.1 Hz, 4H), 6.95 (s, 1H), 4.42 –
4.23 (m, 1H), 3.54 – 3.25 (m, 3H), 3.13 – 3.02 (m, 1H), 2.13 – 2.02 (m, 1H), 1.96 – 1.77 (m,
3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.12, 168.02, 163.39, 154.49, 150.03,
142.94, 135.24, 131.58 (q, 2JCF = 32.3 Hz), 131.25 (q, 2JCF = 32.3 Hz), 130.93 (q, 2JCF = 32.3
Hz), 130.61 (q, 2JCF = 32.3 Hz), 129.32, 129.22, 128.82, 125.58 (q, 1JCF = 165.2 Hz), 124.56
(q, 3JCF = 4.0 Hz), 124.52(q, 3JCF = 4.0 Hz), 124.49 (q, 3JCF = 4.0 Hz; q, 1JCF = 165.2 Hz)),
124.45 (q, 3JCF = 4.0 Hz), 122.95, 122.91 (q, 1JCF = 165.2 Hz), 122.87, 120.65 (q, 1JCF = 165.2
Hz), 120.50, 117.37, 103.95, 80.40*, 79.90*, 55.35*, 55.20*, 46.86*, 46.518, 31.81*, 31.10*,
30.99*, 30.25*, 28.59, 23.62*, 22.86*; 19F NMR (376 MHz, CDCl3) δ -62.71 (s, 3F). HRMS
(ESI+): calcd for C28H28F3N5NaO3S [M+Na]+: 594.1763, found: 594.1767.
tert-butyl (S)-2-((3-(4-((4-(2-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18m). Synthesized by General Procedure 3C.
79%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.30 (br s, 1H), 7.98 (d, J = 8.5 Hz,
2H), 7.78 – 7.73 (m, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.56 (td, J = 7.7, 1.5 Hz, 1H), 7.46 (t, J = 7.7
Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 6.76 (s, 1H), 4.38 – 4.22 (m, 1H), 3.49 – 3.27 (m, 3H), 3.16 –
2.97 (m, 1H), 2.13 – 2.00 (m, 1H), 1.95 – 1.77 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz,
CDCl3) δ 191.63, 177.20, 167.97, 162.95, 154.38, 148.62, 142.83, 134.47, 132.09, 131.72,
129.01 (q, 2JCF = 30.3 Hz), 128.86, 128.71 (q, 2JCF = 30.3 Hz), 128.40, 128.38, 128.32 (q, 2JCF =
30.3 Hz), 128.10 (q, 1JCF = 275.7 Hz), 126.66 (q, 3JCF = 6.1 Hz), 126.60 (q, 3JCF = 6.1 Hz),
126.55 (q, 3JCF = 6.1 Hz), 126.49 (q, 3JCF = 6.1 Hz), 125.59 (q, 1JCF = 275.7 Hz), 122.87 (q, 1JCF

179
= 275.7 Hz), 120.93, 120.73, 120.14 (q, 1JCF = 275.7 Hz), 117.48, 106.95, 106.91, 80.22*, 79.77*,
55.34, 46.90*, 46.51*, 31.95*, 31.16*, 30.98*, 30.23*, 29.84, 28.63*, 28.52*, 23.70*, 22.92*; 19F
NMR (376 MHz, CDCl3) δ -57.78 (s, 3F); HRMS (ESI+): calcd for C28H28F3N5NaO3S [M+H]+:
572.1943, found: 572.1942.
tert-butyl (S)-2-((3-(4-((4-(p-tolyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18n). Synthesized by General Procedure 3C. 47%, off-
white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.2 Hz, 2H), 7.75 (d, J = 7.8
Hz, 2H), 7.58 – 7.47 (m, 2H), 7.22 (d, J = 7.9 Hz, 2H), 6.84 (s, 1H), 4.41 – 4.21 (m, 1H), 3.42 (t,
J = 20.8 Hz, 3H), 3.18 – 2.97 (m, 1H), 2.38 (s, 3H), 2.16 – 2.01 (m, 1H), 1.87 (s, 3H), 1.48 (s,
9H); 13C NMR (101 MHz, CDCl3) δ 177.30, 167.99, 162.98, 151.64, 142.84, 138.08, 131.75,
129.52, 128.96, 126.20, 120.69, 117.32, 102.00, 80.22*, 79.80*, 55.35, 46.91*, 46.51*, 31.97*,
31.17*, 30.24*, 29.85*, 28.64, 23.72*, 22.93*, 21.43; HRMS (ESI+): calcd for C28H32N5O3S
[M+H]+: 518.2226, found: 518.2225.
tert-butyl (S)-2-((3-(4-((4-(4-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18o). Synthesized by General Procedure 3C. 75%, clear
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.15 – 7.96 (m, 3H), 7.80 (d, 2H), 7.61 – 7.44
(m, 2H), 6.93 (d, J = 8.7 Hz, 2H), 6.75 (s, 1H), 4.40 – 4.20 (m, 1H), 3.83 (s, 3H), 3.50 – 3.24 (m,
3H), 3.21 – 2.97 (m, 1H), 2.13 – 1.99 (m, 1H), 1.96 – 1.76 (m, 3H), 1.48 (s, 9H); 13C NMR (101
MHz, CDCl3) δ 177.14, 168.03, 162.96, 159.63, 154.64, 154.39, 151.36, 143.01, 128.85, 127.65,
127.55, 127.53, 120.60, 120.41, 117.23, 114.16, 100.95, 80.24*, 79.78*, 55.45*, 55.34, 55.21*,
46.88*, 46.50*, 31.89*, 31.13*, 30.96*, 30.21*, 28.62, 23.68*, 22.90*; HRMS (ESI+): calcd for
C28H32N5O4S [M+H]+: 534.2175, found: 534.2160.

180
tert-butyl (S)-2-((3-(4-((4-(4-ethoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18p). Synthesized by General Procedure 3C. 92%, light
yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.3 Hz, 2H), 7.82 (s, 1H),
7.79 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 7.7 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.75 (s, 1H), 4.39 –
4.21 (m, 1H), 4.07 (q, J = 7.0 Hz, 2H), 3.51 – 3.26 (m, 3H), 3.07 (m, 1H), 2.13 – 2.02 (m, 1H),
1.95 – 1.78 (m, 4H), 1.48 (s, 9H), 1.43 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3)
δ 177.09, 167.84, 162.73, 158.91, 154.25, 151.25, 142.74, 128.75, 127.38, 127.16, 120.41,
117.11, 114.58, 100.72, 80.06*, 79.63*, 63.49, 55.14, 46.72*, 46.34*, 31.77*, 30.99*, 30.05, 28.47,
23.54*, 22.75*, 14.80; HRMS (ESI+): calcd for C29H34N5O4S [M+H]+: 548.2332, found:
548.2308.
tert-butyl (S)-2-((3-(4-((4-(3-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18q). Synthesized by General Procedure 3C. 70%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.09 – 7.97 (m, 3H), 7.58 – 7.51 (m, 2H), 7.49 –
7.40 (m, 2H), 7.32 (t, J = 8.1 Hz, 1H), 6.96 – 6.81 (m, 2H), 4.43 – 4.18 (m, 1H), 3.86 (s, 3H),
3.54 – 3.24 (m, 4H), 3.19 – 2.97 (m, 1H), 2.17 – 2.00 (m, 1H), 1.97 – 1.74 (m, 3H), 1.48 (s, 9H);
13C NMR (101 MHz, CDCl3) δ 177.18, 167.97, 162.95, 160.02, 154.64, 154.40, 151.45, 142.89,
135.90, 129.82, 129.16, 128.88, 120.55, 118.71, 117.30, 113.91, 111.80, 103.11, 80.24*, 79.79*,
55.44*, 55.35*, 55.22*, 46.89*, 46.50*, 31.91*, 31.14*, 30.99*, 30.22*, 28.62, 23.69*, 22.91*;
HRMS (ESI+): calcd for C28H32N5O4S [M+H]+: 534.2175, found: 534.2182.
tert-butyl (S)-2-((3-(4-((4-(4-(trifluoromethoxy)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18r). Synthesized by General Procedure 3C.
30%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (ap. t, J = 7.5 Hz, 2H), 7.90 (s, 1H), 7.88
(d, J = 8.5 Hz, 2H), 7.59 – 7.51 (m, 2H), 7.28 – 7.23 (m, 2H), 6.88 (s, 1H), 4.39 – 4.22 (m, 1H),

181
3.54 – 3.27 (m, 2H), 3.14 – 2.99 (m, 1H), 2.13 – 1.99 (m, 1H), 1.95 – 1.77 (m, 3H), 1.48 (s, 9H);
13C NMR (101 MHz, CDCl3) δ 177.04, 167.73, 163.03, 154.22, 150.12, 148.80, 142.54, 133.08,
128.76, 127.46, 121.72 (q, 1JCF = 258.6 Hz), 121.09, 119.16 (q, 1JCF = 258.6 Hz), 117.22, 103.12,
80.09*, 79.64*, 55.18*, 55.06*, 46.73*, 46.34*, 31.73*, 30.98*, 30.83*, 30.06*, 29.68, 28.46*,
23.53*, 22.74*; 19F NMR(376 MHZ, CDCl3) δ -57.82 (s, 3F); HRMS (ESI+): calcd for
C28H28F3N5O4S [M+H]+: 588.1892, found: 588.1916.
tert-butyl (S)-2-((3-(4-((4-(4-(trifluoromethoxy)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18s). Synthesized by General Procedure 3C.
30%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.3 Hz, 2H), 7.78 (dt, J = 7.9, 1.3
Hz, 1H), 7.73 (dt, J = 2.9, 1.4 Hz, 1H), 7.64 (s, 1H), 7.57 (d, J = 7.7 Hz, 2H), 7.43 (t, J = 8.0 Hz,
1H), 7.17 (ddt, J = 8.1, 2.3, 1.1 Hz, 1H), 6.95 (s, 1H), 4.40 – 4.20 (m, 1H), 3.51 – 3.25(m, 3H),
3.16 – 2.98 (m, 1H), 2.16 – 2.00 (m, 1H), 1.96 – 1.76 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz,
CDCl3) δ 177.24, 167.96, 163.09, 154.40, 150.19, 149.84, 142.65, 136.56, 130.14, 128.98,
124.42, 121.97 (q, 1JCF = 156.6 Hz), 120.96, 120.32 (q, 1JCF = 156.6 Hz), 119.41, 118.87 (q, 1JCF
= 156.6 Hz), 117.44, 104.04, 80.23*, 79.82*, 55.33, 46.89*, 46.52*, 31.94,* 31.17*, 30.26, 28.64,
23.71*, 22.93*; 19F NMR(376 MHZ, CDCl3) δ -57.82 (s, 3F); HRMS (ESI+): calcd for
C28H28F3N5NaO4S [M+Na]+: 610.1712, found: 610.1721.
tert-butyl (S)-2-((3-(4-((4-(3,4-dimethylphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18t). Synthesized by General Procedure 3C. 75%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.30 – 8.08 (m, 1H), 8.03 (d, J = 8.1 Hz, 2H),
7.63 (s, 1H), 7.57 (d, J = 7.7, 2.1 Hz, 1H), 7.54 – 7.46 (m, 2H), 7.15 (d, J = 7.9 Hz, 1H), 6.82 (s,
1H), 4.30 (d, J = 29.1 Hz, 1H), 3.55 – 3.26 (m, 4H), 3.20 – 2.93 (m, 1H), 2.29 (s, 3H), 2.27 (s,
3H), 2.13 – 2.00 (m, 1H), 1.94 – 1.77 (m, 4H), 1.54 – 1.40 (m, 9H); 13C NMR (101 MHz, CDCl3)

182
δ 177.15, 168.03, 163.03, 154.64, 154.39, 151.76, 143.01, 136.92, 136.69, 132.19, 130.04,
128.84, 128.29, 127.48, 123.71, 120.40, 117.27, 101.87, 80.25*, 79.80*, 55.34*, 55.21*, 46.88*,
46.49*, 31.89*, 31.12*, 30.96*, 30.20*, 28.61, 23.67*, 22.89*, 20.07, 19.99, 19.72; HRMS (ESI+):
calcd for C29H34N5O3S [M+H]+: 532.2382, found: 532.2398.
tert-butyl (S)-2-((3-(4-((4-(3,4-dimethoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18u). Synthesized by General Procedure 3C. 63%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.23 – 7.98 (m, 3H), 7.58 – 7.48 (m, 2H), 7.46 –
7.37 (m, 2H), 6.90 (d, J = 8.6 Hz, 1H), 6.78 (s, 1H), 4.30 (d, J = 30.3 Hz, 1H), 3.94 (s, 3H), 3.91
(s, 3H), 3.53 – 3.25 (m, 3H), 3.19 – 2.95 (m, 1H), 2.15 – 1.99 (m, 1H), 1.97 – 1.75 (m, 3H), 1.47
(s, 9H); 13C NMR (101 MHz, cdcl3) δ 177.18, 167.99, 162.95, 154.37, 151.37, 149.16, 149.11,
142.96, 128.87, 128.32, 127.78, 120.50, 118.79, 117.22, 111.36, 109.63, 101.28, 80.23*, 79.78*,
56.18, 56.08*, 56.06*, 56.02*, 55.31*, 46.87*, 46.48*, 31.91*, 31.13*, 30.98*, 30.20*, 28.61,
23.68*, 22.89*; HRMS (ESI+): calcd for C29H34N5O5S [M+H]+: 564.2281, found: 564.2271.
tert-butyl (S)-2-((3-(4-((4-(3-chloro-4-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18v). Synthesized by General Procedure 3C.
91%, light yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.3 Hz, 2H), 7.85
(d, J = 2.2 Hz, 1H), 7.73 (dd, J = 8.6, 2.2 Hz, 1H), 7.59 – 7.50 (m, 2H), 6.95 (d, J = 8.6 Hz, 1H),
6.76 (s, 1H), 4.40 – 4.22 (m, 1H), 3.93 (s, 3H), 3.47 – 3.29 (m, 3H), 3.07 (ddd, J = 30.3, 14.3, 8.2
Hz, 1H), 2.14 – 2.02 (m, 1H), 1.94 – 1.78 (m, 4H), 1.47 (s, 9H); 13C NMR (101 MHz, CDCl3) δ
177.24, 167.90, 163.26, 155.01, 149.42, 142.47, 128.97, 128.11, 127.80, 125.73, 122.84, 117.56,
112.20, 101.65, 80.23*, 79.79*, 56.38, 55.34*, 55.23*, 46.90*, 46.50*, 31.93*, 31.15*, 30.99*,
30.23*, 28.63, 23.72*, 22.92*; HRMS (ESI+): calcd for C28H31ClN5O4S [M+H]+: 568.1785,
found: 568.1780.

183
tert-butyl (S)-2-((3-(4-((4-(3,4-difluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18w). Synthesized by General Procedure 3C. 58%, off-
white solid. 1H NMR (400 MHz, CDCl3) δ 8.11 – 8.03 (m, 2H), 7.72 – 7.62 (m, 2H), 7.61 – 7.50
(m, 3H), 7.19 (dd, J = 10.1, 8.3 Hz, 1H), 6.84 (s, 1H), 4.29 (t, J = 18.7 Hz, 1H), 3.50 – 3.26 (m,
3H), 3.15 – 2.99 (m, 1H), 2.16 – 2.01 (m, 1H), 1.96 – 1.77 (m, 3H), 1.48 (s, 9H); 13C NMR (101
MHz, CDCl3) δ 177.25, 167.94, 163.13, 154.42, 151.93 (dd, 2JCF = 36.4 Hz, 3JCF = 13.1 Hz),
151.80 (dd, 2JCF = 36.4 Hz, 3JCF = 13.1 Hz), 151.57 (dd, 2JCF = 36.4 Hz, 3JCF = 13.1 Hz), 151.45
(dd, 2JCF = 36.4 Hz, 3JCF = 13.1 Hz), 149.65, 149.47 (dd, 2JCF = 37.4 Hz, 3JCF = 13.1 Hz), 149.34
(dd, 2JCF = 37.4 Hz, 3JCF = 13.1 Hz), 149.10 (dd, 2JCF = 37.4 Hz, 3JCF = 13.1 Hz), 148.97 (dd,
2JCF = 37.4 Hz, 3JCF = 13.1 Hz), 142.64, 131.80 (d, 3JCF = 4.0 Hz), 131.76 (d, 3JCF = 4.0 Hz),
131.74 (d, 3JCF = 4.0 Hz), 131.70 (d, 3JCF = 4.0 Hz), 128.97, 122.21 (d, 3JCF = 4.0 Hz), 122.17 (d,
3JCF = 4.0 Hz), 122.15 (d, 3JCF = 4.0 Hz), 122.11 (d, 3JCF = 4.0 Hz), 120.97, 117.68 80 (dd, 1JCF =
227.8 Hz, 2JCF = 18.2 Hz), 117.50 (dd, 1JCF = 227.8 Hz, 2JCF = 18.2 Hz), 117.45, 115.43 (dd,
1JCF = 227.8 Hz, 2JCF = 19.2 Hz), 115.24 (dd, 1JCF = 227.8 Hz, 2JCF = 19.2 Hz), 103.26, 80.26*,
79.85*, 55.31, 46.91*, 46.53*, 31.93*, 31.17*, 30.28, 28.64, 23.71*, 22.94*; 19F NMR (376 MHz,
CDCl3) δ -137.57 (s, 1F), -138.63 (s, 1F); HRMS (ESI+): calcd for C27H27F2N5NaO3S [M+Na]+:
562.1695, found: 562.1666.
tert-butyl (S)-2-((3-(4-((4-(3,4-dichlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18x). Synthesized by General Procedure 3C. 59%, yellow
solid. 1H NMR (400 MHz, CDCl3) δ 8.09 – 7.98 (m, 2H), 7.91 (d, J = 8.5 Hz, 1H), 7.56 – 7.44
(m, 2H), 7.33 – 7.23 (m, 2H), 4.41 – 4.23 (m, 1H), 3.53 – 3.29 (m, 3H), 3.16 – 2.99 (m, 1H),
2.14 – 2.02 (m, 1H), 1.95 – 1.74 (m, 3H), 1.49 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.25,
167.95, 162.20, 154.67, 146.89, 142.77, 133.94, 132.53, 132.30, 131.74, 130.36, 128.90, 127.38,

184
117.45, 108.45, 80.27*, 79.84*, 55.34, 46.90*, 46.52*, 31.91*, 31.14*, 30.26, 28.64, 23.69*,
22.92*; HRMS (ESI+): calcd for C27H28Cl2N5O3S [M+H]+: 572.1290, found: 572.1244.
tert-butyl (S)-2-((3-(4-((4-(4-fluoro-3-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18y). Synthesized by General Procedure 3C.
24%, clear yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.15 – 7.98 (m, 4H), 7.82 (s, 1H), 7.60 –
7.51 (m, 2H), 7.31 – 7.17 (m, 1H), 6.90 (s, 1H), 4.40 – 4.23 (m, 1H), 3.52 – 3.27 (m, 3H), 3.15 –
3.01 (m, 1H), 2.14 – 2.03 (m, 1H), 1.94 – 1.81 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz,
CDCl3) δ 177.27, 167.92, 163.40, 160.64 (d, 1JCF = 257.6 Hz), 158.09 (d, 1JCF = 257.6 Hz),
154.42, 149.25, 142.64, 131.46 (d, 3JCF = 8.1 Hz), 131.38 (d, 3JCF = 8.1 Hz), 131.14 (d, 4JCF = 4.0
Hz), 131.10 (d, 4JCF = 4.0 Hz), 128.96, 126.76 (q, 1JCF = 272.7 Hz), 125.04 (qd, 3JCF = 5.1 Hz,
4JCF = 1.0 Hz), 125.03 (qd, 3JCF = 5.1 Hz, 4JCF = 1.0 Hz), 125.00 (qd, 3JCF = 5.1 Hz, 4JCF = 1.0
Hz), 124.98 (qd, 3JCF = 5.1 Hz, 4JCF = 1.0 Hz), 124.95, 124.94, 124.05 (q, 1JCF = 272.7 Hz),
121.35 (q, 1JCF = 272.7 Hz), 120.99, 118.96 (d, 3JCF = 13.1 Hz), 118.83 (d, 3JCF = 13.1 Hz),
118.63 (q, 1JCF = 272.7 Hz), 118.50, 117.48, 117.43, 117.38, 117.22, 103.56, 80.30*, 79.88*,
55.33, 46.90*, 46.54*, 31.91*, 31.17*, 30.29, 28.63, 23.69*, 22.93*; 19F NMR (376 MHz, CDCl3)
δ -61.47 (d, J = 12.6 Hz 3F), -115.74 (br. s, 1F); HRMS (ESI+): calcd for C28H27F4N5NaO3S
[M+Na]+: 612.1668, found: 612.1663.
tert-butyl (S)-2-((3-(4-((4-(3,5-difluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18z). Synthesized by General Procedure 3C. 76%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.06 (t, J = 10.4 Hz, 2H), 7.94 (s, 1H), 7.62 –
7.52 (m, 2H), 7.40 – 7.32 (m, 1H), 6.92 (s, 1H), 6.75 (tt, J = 8.9, 2.5 Hz, 1H), 4.44 – 4.21 (m,
1H), 3.53 – 3.24 (m, 3H), 3.15 – 3.00 (m, 1H), 2.15 – 2.01 (m, 1H), 1.95 – 1.75 (m, 3H), 1.48 (s,
9H); 13C NMR (101 MHz, CDCl3) δ 177.21, 168.01, 164.70 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1

185
Hz), 164.57 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1 Hz), 163.17, 162.24 (dd, 1JCF = 248.5 Hz, 3JCF =
13.1 Hz), 162.11 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1 Hz), 154.47, 149.46, 149.42, 142.70, 137.75
(d, 2JCF = 20.2 Hz), 137.65, 137.55 (d, 2JCF = 20.2 Hz), 128.93, 120.87, 117.48, 109.16 (d, 2JCF =
27.3 Hz), 109.08 (d, 3JCF = 11.1 Hz), 108.97 (d, 3JCF = 11.1 Hz), 108.89 (d, 2JCF = 27.3 Hz),
104.66, 103.48 (d, 2JCF = 51.5 Hz), 103.23, 102.97 (d, 2JCF = 51.5 Hz), 80.34*, 79.89*, 55.34,
46.90*, 46.54*, 31.88*, 31.16*, 30.30, 28.74*, 28.63*, 23.68*, 22.92*; 19F NMR (376 MHz,
CDCl3) δ -109.78 (br. S, 2F); HRMS (ESI+): calcd for C27H28F2N5O3S [M+H]+: 540.1881,
found: 540.1845.
tert-butyl (S)-2-((3-(4-((4-(3-fluoro-5-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18aa). Synthesized by General Procedure
3C. 44%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.12 – 8.02 (m, 2H), 7.91 (d, J
= 12.9 Hz, 2H), 7.75 (d, J = 9.6 Hz, 1H), 7.64 – 7.51 (m, 2H), 7.28 – 7.23 (m, 1H), 7.00 (s, 1H),
4.41 – 4.23 (m, 1H), 3.52 – 3.27 (m, 3H), 3.14 – 3.01 (m, 1H), 2.14 – 2.02 (m, 1H), 1.97 – 1.75
(m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.24, 167.98, 164.16(d, 1JCF = 248.5
Hz), 163.39, 161.70 (d, 1JCF = 248.5 Hz), 154.47, 149.04, 142.61, 137.75 (d, 3JCF = 8.1 Hz),
137.67 (d, 3JCF = 8.1 Hz), 133.44 (qd, 2JCF = 33.3 Hz, 3JCF = 8.1 Hz), 133.36 (qd, 2JCF = 33.3 Hz,
3JCF = 8.1 Hz), 133.11(qd, 2JCF = 33.3 Hz, 3JCF = 8.1 Hz), 133.03, 132.78 (qd, 2JCF = 33.3 Hz,
3JCF = 8.1 Hz), 132.70 (qd, 2JCF = 33.3 Hz, 3JCF = 8.1 Hz), 132.45 (qd, 2JCF = 33.3 Hz, 3JCF = 8.1
Hz), 132.37 (qd, 2JCF = 33.3 Hz, 3JCF = 8.1 Hz), 128.96, 127.50 (q, 1JCF = 272.7 Hz), 124.81 (q,
1JCF = 272.7 Hz), 122.10 (q, 1JCF = 272.7 Hz), 121.00, 119.36 (q, 1JCF = 272.7 Hz),118.70,
118.66, 118.63, 117.53, 116.59 (d, 2JCF = 23.2 Hz), 116.36 (d, 2JCF = 23.2 Hz),, 112.11 (dq, 2JCF
= 23.2 Hz, 3JCF = 4.0 Hz), 112.08 (dq, 2JCF = 23.2 Hz, 3JCF = 4.0 Hz), 111.87 (dq, 2JCF = 23.2 Hz,
3JCF = 4.0 Hz), 111.83 (dq, 2JCF = 23.2 Hz, 3JCF = 4.0 Hz), 105.06, 80.35*, 79.91*, 55.33, 46.90*,

186
46.54*, 31.89*, 31.16*, 30.31, 28.63, 23.69*, 22.93*; 19F NMR (376 MHz, CDCl3) δ 19F NMR
(376 MHz, CDCl3) δ -62.87 (s, 3F), -110.68 (q, J = 8.2, 7.6 Hz, 1F); HRMS (ESI+): calcd for
C28H28F4N5O3S [M+H]+: 590.1849, found: 590.1843.
tert-butyl (S)-2-((3-(4-((4-(3,5-bis(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18bb). Synthesized by General Procedure
3C. 49%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 2H), 8.08 (dd, J =
13.4, 8.2 Hz, 2H), 7.89 (s, 1H), 7.80 (s, 1H), 7.57 (t, J = 9.0 Hz, 2H), 7.08 (s, 1H), 4.41 – 4.23
(m, 1H), 3.52 – 3.26 (m, 3H), 3.16 – 3.00 (m, 1H), 2.15 – 2.03 (m, 1H), 1.97 – 1.80 (m, 3H),
1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.28, 167.93, 163.64, 154.46, 148.68, 142.53,
136.44, 132.65 (d, 2JCF = 33.3 Hz), 132.32 (q, 2JCF = 33.3 Hz), 131.99 (q, 2JCF = 33.3 Hz), 131.66
(d, 2JCF = 33.3 Hz), 129.00, 127.56 (q, 1JCF = 273.7 Hz), 126.12, 124.85 (q, 1JCF = 273.7 Hz),
122.14 (q, 1JCF = 273.7 Hz), 121.35, 119.42 (q, 1JCF = 273.7 Hz), 117.59, 105.45, 80.36*, 79.91*,
55.31, 46.90*, 46.55*, 31.91*, 31.18*, 30.32, 28.64, 23.69*, 22.94*; 19F NMR (376 MHz, CDCl3)
δ -62.96 (s, 6F); HRMS (ESI+): calcd for C29H27F6N5NaO3S [M+Na]+: 662.1636, found:
662.1627.
tert-butyl (S)-2-((3-(4-((4-(2-fluoro-5-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.18cc). Synthesized by General Procedure
3C. 78%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.47 (dd, J = 7.1, 2.5 Hz, 1H),
8.07 (ap. t, J = 9.6 Hz, 2H), 7.91 (s, 1H), 7.63 – 7.48 (m, 3H), 7.32 – 7.15 (m, 2H), 4.42 – 4.21
(m, 1H), 3.52 – 3.27 (m, 3H), 3.16 – 3.02 (m, 1H), 2.15 – 2.02 (m, 1H), 1.97 – 1.77 (m, 3H),
1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.24, 167.94, 163.19 (d, 1JCF = 256.5 Hz), 162.29,
160.65 (d, 1JCF = 256.6 Hz), 154.46, 143.80, 142.71, 128.95, 128.00 (q, 1JCF = 272.7 Hz), 127.72,
127.67, 127.61, 127.38 (q, 2JCF = 33.3 Hz), 127.05 (q, 2JCF = 33.3 Hz), 126.72 (q, 2JCF = 33.3

187
Hz), 126.00, 125.30 (q, 1JCF = 272.7 Hz), 123.11 (d, 3JCF = 12.1 Hz), 122.99 (d, 3JCF = 12.1 Hz),
122.59 (q, 1JCF = 272.7 Hz), 120.89, 119.89 (q, 1JCF = 272.7 Hz), 117.43, 117.08, 116.81 (d, 2JCF
= 24.2 Hz), 116.57 (d, 2JCF = 24.2 Hz), 109.27 (d, 2JCF = 16.2 Hz), 109.11 (d, 2JCF = 16.2 Hz),
80.34*, 79.88*, 55.31, 46.89*, 46.54*, 31.91*, 31.17*, 31.04*, 30.29*, 28.63, 23.69*, 22.92*; 19F
NMR (376 MHz, CDCl3) δ -62.08 (s, 3F), -109.18 (br. s, 1F); HRMS (ESI+): calcd for
C28H27F4N5NaO3S [M+Na]+: 612.1668, found: 612.1621.
tert-butyl (S)-2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18dd). Synthesized by General Procedure 3C. 19%, light
yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.3 Hz, 2H), 7.95 (d, J = 8.0
Hz, 2H), 7.74 – 7.53 (m, 3H), 7.46 (t, J = 7.6 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 6.95 (s, 1H), 4.40
– 4.22 (m, 1H), 3.52 – 3.27 (m, 3H), 3.17 – 2.98 (m, 1H), 2.14 – 2.02 (m, 1H), 1.95 – 1.78 (m,
3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.22, 167.98, 162.92, 154.64, 151.40,
142.85, 140.86, 140.81, 133.55, 129.00, 128.97, 128.95, 127.51, 127.31, 127.13, 126.67, 117.35,
102.90, 80.21*, 79.79*, 55.32, 46.52, 31.95*, 31.17*, 30.23, 28.65, 23.71*, 22.93*; HRMS (ESI+):
calcd for C33H34N5O3S [M+H]+: 580.2382, found: 580.2377.
tert-butyl (S)-2-((3-(4-((4-(benzofuran-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.18ee). Synthesized by General Procedure 3C. 71%, tan
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 8.07 (d, J = 8.3 Hz, 2H), 7.99 –
7.93 (m, 1H), 7.77 (s, 1H), 7.61 – 7.51 (m, 3H), 7.40 – 7.31 (m, 2H), 6.94 (s, 1H), 4.40 – 4.22
(m, 1H), 3.53 – 3.27 (m, 3H), 3.17 – 2.98 (m, 1H), 2.15 – 2.01 (m, 1H), 1.97 – 1.77 (m, 3H),
1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.25, 167.98, 163.02, 155.87, 154.39, 143.94,
143.76, 142.75, 128.96, 125.51, 124.82, 123.32, 120.99, 120.90, 117.43, 117.29, 117.13, 111.95,

188
103.23, 80.25*, 79.81*, 55.33, 46.90, 46.52, 31.94*, 31.18*, 31.02*, 30.26*, 28.65, 23.71*, 22.94*;
HRMS (ESI+): calcd for C29H29N5NaO4S [M+Na]+: 566.1838, found: 566.1813.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-phenylthiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19a). Synthesized by General
Procedure 3E. 82%, off-white oily solid. 1H NMR (500 MHz, CDCl3) δ 8.07 (d, J = 8.3 Hz, 2H),
7.87 (d, J = 7.6 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.34 (dd, J = 7.4 Hz,
1H), 6.90 (s, 1H), 4.79 (dd, J = 8.3, 4.9 Hz, 1H), 3.72 – 3.61 (m, 2H), 3.50 (s, 1H), 3.14 (dd, J =
15.2, 8.5 Hz, 1H), 2.32 – 2.25 (m, 1H), 1.93 (dt, J = 10.2, 5.1 Hz, 1H), 1.82 (dd, J = 14.0, 7.5 Hz,
2H), 1.48 (s, 18H); 13C NMR (126 MHz, CDCl3) δ 177.01, 167.91, 163.01, 154.20, 151.38,
142.61, 134.29, 129.06, 128.87, 128.28, 126.27, 117.38, 102.66, 77.73, 56.66, 50.25, 30.81,
30.45, 28.31; HRMS (ESI+): calcd for C33H40N7O5S [M+H]+: 646.2812, found: 646.2805.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(pyridin-4-yl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19b).
Synthesized by General Procedure 3E. 11%, off-white oily solid. 1H NMR (500 MHz, CDCl3) δ
10.41 (s, 1H), 8.65 (ap. s, 2H), 8.09 (s, 1H), 8.01 (d, J = 7.8 Hz, 2H), 7.74 (ap. s, 2H), 7.60 (d, J
= 7.9 Hz, 2H), 7.13 (s, 1H), 4.81 – 4.73 (s, 1H), 3.81 – 3.62 (m, 2H), 3.59 – 3.40 (m, 1H), 3.08
(dd, J = 15.2, 8.8 Hz, 1H), 2.36 – 2.26 (m, 1H), 1.98 – 1.89 (m, 1H), 1.84 – 1.75 (m, 2H), 1.48
(s, 18H); 13C NMR (126 MHz, CDCl3) δ 176.85, 167.83, 163.35, 162.48, 159.25, 156.79, 154.30,
150.41, 150.01, 149.24, 142.56, 141.49, 128.97, 127.21, 122.29, 121.09, 120.50, 117.42, 106.53,
82.01, 79.68, 56.60, 50.32, 30.94, 30.46, 28.32, 24.52; HRMS (ESI+): calcd for C32H39N8O5S
[M+H]+: 647.2764, found: 647.2772.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(pyridin-3-yl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19c).

189
Synthesized by General Procedure 3E. 78%, yellow oily solid. 1H NMR (400 MHz, CDCl3) δ
10.42 (s, 1H), 9.18 (s, 1H), 8.90 (s, 1H), 8.56 (d, J = 4.8 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.95
(d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.35 (dd, J = 8.0, 4.8 Hz, 1H), 6.96 (s, 1H), 4.81 –
4.71 (m, 1H), 3.85 – 3.62 (m, 2H), 3.59 – 3.42 (m, 1H), 3.04 (dd, J = 15.7, 9.0 Hz, 1H), 2.37 –
2.26 (m, 1H), 1.96 – 1.87 (m, 1H), 1.82 – 1.71 (m, 2H), 1.57 – 1.36 (m, 18H); 13C NMR (101
MHz, CDCl3) δ 176.57, 167.78, 163.53, 162.51, 154.41, 148.70, 148.51, 147.62, 142.92, 133.59,
130.59, 128.80, 123.71, 120.40, 117.17, 103.81, 82.25, 79.85, 56.54, 50.41, 31.02, 30.43, 28.29,
24.64; HRMS (ESI+): calcd for C32H38N8NaO5S [M+Na]+: 669.2584, found: 669.2589.
tert-butyl (S,E)-(((tert-butoxycarbonyl)amino)(2-((3-(4-((4-(3-fluorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methylene)carbamate (3.19d).
Synthesized by General Procedure 3E. 43%, light yellow oily solid. 1H NMR (400 MHz, CDCl3)
δ 10.40 (s, 1H), 8.01 (d, J = 8.7 Hz, 2H), 7.88 – 7.81 (m, 2H), 7.56 (d, J = 8.7 Hz, 2H), 7.14 –
7.06 (m, 2H), 6.81 (s, 1H), 4.82 – 4.73 (m, 1H), 3.79 – 3.62 (m, 2H), 3.57 – 3.46 (m, 1H), 3.09
(dd, J = 15.5, 8.8 Hz, 1H), 2.35 – 2.25 (m, 1H), 1.96 – 1.88 (m, 1H), 1.85 – 1.74 (m, 2H), 1.48
(s, 18H); 13C NMR (101 MHz, CDCl3) δ 176.79, 167.82, 163.97 (d, 1JCF = 247.5 Hz), 163.03,
161.52 (d, 1JCF = 247.5 Hz), 154.28, 150.72, 150.53, 142.67, 130.91, 130.76, 128.93, 128.00 (d,
3JCF = 8.1 Hz), 127.92 (d, 3JCF = 8.1 Hz), 120.72, 117.21, 115.83 (d, 2JCF = 21.2 Hz), 115.62 (d,
2JCF = 21.2 Hz), 102.16, 81.89, 79.74, 56.61, 50.34, 30.94, 30.44, 28.30; 19F NMR (376 MHz,
CDCl3) δ -113.86 (p, J = 6.7 Hz, 1F); HRMS (ESI+): calcd for C33H39FN7O5S [M+H]+:
664.2717, found: 664.2710.
tert-butyl (S,E)-(((tert-butoxycarbonyl)amino)(2-((3-(4-((4-(4-chlorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methylene)carbamate (3.19e).
Synthesized by General Procedure 3E. 57%, light yellow oily solid. 1H NMR (400 MHz, CDCl3)

190
δ 10.36 (s, 1H), 8.02 (d, J = 8.3 Hz, 2H), 7.85 (s, 1H), 7.81 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.3
Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 6.88 (s, 1H), 4.77 (qd, J = 7.2, 3.9 Hz, 1H), 3.79 – 3.62 (m,
2H), 3.57 – 3.45 (m, 1H), 3.09 (dd, J = 15.5, 8.8 Hz, 1H), 2.31 (dt, J = 12.3, 6.5 Hz, 1H), 1.96 –
1.88 (m, 1H), 1.86 – 1.74 (m, 1H), 1.72 – 1.59 (m, 1H), 1.48 (s, 18H); 13C NMR (101 MHz,
CDCl3) δ 176.84, 167.84, 163.03, 154.28, 153.95, 151.48, 150.56, 142.69, 133.84, 133.34,
133.10, 128.96, 127.52, 120.83, 117.25, 103.04, 102.80, 82.17, 81.67, 56.61, 50.28, 30.93, 30.46,
28.32; HRMS (ESI+): calcd for C33H38ClN7O5S [M+H]+: 680.2422, found: 680.2425.
tert-butyl (S,Z)-((2-((3-(4-((4-(4-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.19f). Synthesized by
General Procedure 3E. 83%, off-white oily solid. 1H NMR (400 MHz, CDCl3) δ 10.56 – 10.37
(m, 1H), 8.65 – 8.42 (m, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.2
Hz, 2H), 6.86 (s, 1H), 4.75 (dq, J = 11.1, 6.7, 6.0 Hz, 1H), 3.90 – 3.72 (m, 1H), 3.72 – 3.61 (m,
1H), 3.57 – 3.43 (m, 1H), 3.00 (dd, J = 15.9, 9.2 Hz, 1H), 2.39 – 2.27 (m, 1H), 1.97 – 1.85 (m,
1H), 1.82 – 1.70 (m, 1H), 1.48 (d, J = 5.7 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ 176.42,
167.73, 162.96, 154.49, 150.48, 142.82, 133.57, 131.86, 128.73, 127.78, 121.90, 120.23, 116.99,
102.96, 82.26, 79.99, 56.52, 50.47, 31.13, 30.44, 28.63, 28.29, 28.10, 24.60; HRMS (ESI+):
calcd for C33H39BrN7O5S [M+H]+: 724.1917, found: 724.1912.
tert-butyl (S,E)-(((tert-butoxycarbonyl)amino)(2-((3-(4-((4-(3-fluorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methylene)carbamate (3.19g).
Synthesized by General Procedure 3E. 57%, yellow oily solid. 1H NMR (400 MHz, CDCl3) δ
10.43 (s, 1H), 8.21 – 8.04 (m, 0H), 7.98 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.0 Hz, 1H), 7.58 (d, J =
8.6 Hz, 3H), 7.37 (ap q, J = 8.0, 5.8 Hz, 1H), 7.01 (td, J = 8.5, 2.7 Hz, 1H), 6.91 (s, 1H), 4.81 –
4.72 (m, 1H), 3.82 – 3.63 (m, 2H), 3.57 – 3.44 (m, 1H), 3.06 (dd, J = 15.6, 8.9 Hz, 1H), 2.38 –

191
2.27 (m, 1H), 1.96 – 1.87 (m, 1H), 1.84 – 1.72 (m, 2H), 1.49 (s, 18H); 13C NMR (101 MHz,
CDCl3) δ 176.61, 167.80, 164.50 (d, 1JCF = 245.4 Hz), 162.93, 162.55, 162.07 (d, 1JCF = 245.4
Hz), 154.39, 150.44, 142.75, 136.84 (d, 3JCF = 8.1 Hz), 136.76 (d, 3JCF = 8.1Hz), 130.30 (d, 3JCF
= 8.1 Hz), 130.22 (d, 3JCF = 8.1 Hz), 128.85, 121.76, 120.52, 117.11, 114.93 (d, 2JCF = 21.2 Hz),
114.72 (d, 2JCF = 21.2 Hz), 113.31 (d, 2JCF = 23.2 Hz), 113.08 (d, 2JCF = 23.2 Hz), 105.33,
103.58, 82.25, 79.80, 56.57, 50.45, 31.04, 30.43, 28.35, 28.25, 28.13; 19F NMR (376 MHz,
CDCl3) δ -113.07 to -113.17 (m, 1F); HRMS (ESI+): calcd for C33H39FN7O5S [M+H]+:
664.2717, found: 664.2727.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-chlorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19h).
Synthesized by General Procedure 3E. 77%, light yellow solid. 1H NMR (400 MHz, CDCl3) δ
10.43 (s, 1H), 8.35 (s, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.86 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.58
(d, J = 8.3 Hz, 2H), 7.37 – 7.24 (m, 2H), 6.89 (s, 1H), 4.80 – 4.71 (m, 1H), 3.86 – 3.73 (m, 1H),
3.72 – 3.62 (m, 1H), 3.57 – 3.45 (m, 1H), 3.02 (dd, J = 15.8, 9.1 Hz, 1H), 2.38 – 2.30 (m, 1H),
1.96 – 1.87 (m, 1H), 1.84 – 1.71 (m, 2H), 1.48 (ap d. rot, 18H); 13C NMR (101 MHz, CDCl3) δ
176.51, 167.78, 162.95, 162.52, 154.47, 150.43, 150.27, 142.77, 136.40, 134.73, 130.02, 128.80,
127.97, 126.32, 124.34, 120.42, 117.09, 103.61, 82.28, 79.89, 56.55, 50.43, 31.10, 30.46, 28.36,
28.25; HRMS (ESI+): calcd for C33H38ClN7O5S [M+H]+: 680.2422, found: 680.2435.
tert-butyl (S,Z)-((2-((3-(4-((4-(3-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.19i). Synthesized by
General Procedure 3E. 57%, off-white oily solid. 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 1H),
8.03 – 8.00 (m, 2H), 7.98 (s, 1H), 7.79 (dt, J = 7.8, 1.3 Hz, 1H), 7.66 (s, 1H), 7.58 – 7.54 (m,
1H), 7.46 – 7.42 (m, 1H), 7.29 (d, J = 7.9 Hz, 1H), 6.90 (s, 1H), 4.82 – 4.73 (m, 1H), 3.79 – 3.62

192
(m, 2H), 3.58 – 3.46 (m, 1H), 3.07 (dd, J = 15.6, 8.8 Hz, 1H), 2.37 – 2.26 (m, 1H), 1.97 – 1.87
(m, 1H), 1.86 – 1.73 (m, 2H), 1.48 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 176.60, 167.81,
163.23, 154.59, 154.32, 150.03, 142.82, 136.65, 130.91, 130.32, 129.23, 128.84, 124.80, 122.96,
120.53, 117.15, 103.57, 83.05, 56.54, 50.33, 31.02, 30.45, 28.30, 24.45; HRMS (ESI+): calcd for
C33H39BrN7O5S [M+H]+: 724.1917, found: 724.1909.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(2-fluorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19j).
Synthesized by General Procedure 3E. 86%, light yellow oily solid. 1H NMR (400 MHz, CDCl3)
δ 10.42 (s, 1H), 8.18 (td, J = 7.8, 2.0 Hz, 1H), 8.09 (s, 1H), 7.95 (d, J = 8.3 Hz, 2H), 7.63 – 7.56
(m, 2H), 7.25 – 7.18 (m, 2H), 7.17 – 7.09 (m, 1.5 Hz, 1H), 4.82 – 4.72 (m, 1H), 3.86 – 3.62 (m,
2H), 3.57 – 3.47 (d, J = 11.3 Hz, 1H), 3.04 (dd, J = 15.7, 9.0 Hz, 1H), 2.38 – 2.29 (m, 1H), 1.96
– 1.87 (m, 1H), 1.84 – 1.74 (m, 2H), 1.48 (ap. d., rot., 18H); 13C NMR (101 MHz, CDCl3) δ
176.57, 167.83, 162.58, 161.80, 161.73 (d, 1JCF = 250.5 Hz), 159.25 (d, 1JCF = 250.5 Hz), 154.46,
150.45, 145.33 (d, 4JCF = 3.0 Hz), 145.30 (d, 4JCF = 3.0 Hz), 142.87, 130.15 (d, 4JCF = 4.0 Hz),
130.11 (d, 4JCF = 4.0 Hz), 129.05 (d, 3JCF = 8.1 Hz), 128.97 (d, 3JCF = 8.1 Hz), 128.83, 124.55,
124.52, 122.48 (d, 3JCF = 11.1 Hz), 122.37 (d, 3JCF = 11.1Hz), 120.46, 117.06, 116.11 (d, 2JCF =
23.2 Hz), 115.88 (d, 2JCF = 23.2 Hz), 107.92(d, 3JCF = 16.2 Hz), 107.76 (d, 3JCF = 16.2 Hz),
82.24, 79.79, 77.36, 56.56, 50.42, 31.07, 30.46, 28.38, 28.25, 28.13; 19F NMR (376 MHz,
CDCl3) δ - 114.04 to -114.17 (m, 1F); HRMS (ESI+): calcd for C33H38FN7NaO5S [M+Na]+:
686.2537, found: 686.2550.
tert-butyl (S,E)-(((tert-butoxycarbonyl)amino)(2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methylene)carbamate (3.19k).
Synthesized by General Procedure 3E. 45%, off-white solid. 1H NMR (400 MHz, CDCl3) δ

193
10.45 (br s, 1H), 8.54 (br s, 1H), 7.98 (d, J= 8 Hz, 2H), 7.93 (d, J = 8 Hz, 2H), 7.65 (d, J = 8 Hz,
2H), 7.61 (dt. J = 8, 4 Hz, 2H), 6.98 (br s. 1H), 4.81 – 4.72 (m, 1H), 3.85 – 3.62 (m, 2H), 3.58 –
3.43 (m, 1H). 3.02 (dd, J = 16, 8 Hz, 1H), 2.39 - 2.27 (m, 2H), 1.97 – 1.87 (m, 1H), 1.82 – 1.70
(m, 2H), 1.52-1.44 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 176.49, 167.75, 163.18, 154.41,
150.11, 149.03, 142.73, 137.81, 130.19 (q, 2JCF = 32.8 Hz), 129.86 (q, 2JCF = 32.8 Hz), 129.54
(q, 2JCF = 32.8 Hz), 129.22 (q, 2JCF = 32.8 Hz), 128.77, 128.39, 126.38, 125.80 (q, 4JCF = 4.0 Hz),
125.75 (q, 4JCF = 4.0 Hz), 125.71 (q, 4JCF = 4.0 Hz), 125.68 (q, 4JCF = 4.0 Hz), 122.98, 120.45,
117.13, 104.45, 83.52, 66.00, 56.54, 50.45, 31.08, 30.42, 28.28, 28.10; 19F NMR(376 MHZ,
CDCl3) δ -62.53 (s, 3F); HRMS (ESI+): calcd for C34H39F3N7O5S [M+H]+: 714.2685, found:
714.2703.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-(trifluoromethyl)phenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19l).
Synthesized by General Procedure 3E. 6%, off-white oily solid. 1H NMR (400 MHz, CDCl3) δ
10.46 (s, 1H), 8.60 (s, 1H), 8.11 (s, 1H), 8.07 (d, J = 7.5 Hz, 1H), 7.89 (d, J = 8.3 Hz, 2H), 7.60
(d, J = 8.6 Hz, 2H), 7.57 – 7.48 (m, 2H), 6.95 (s, 1H), 4.81 – 4.71 (m, 1H), 3.87 – 3.74 (m, 1H),
3.73 – 3.62 (m, 1H), 3.57 – 3.46 (m, 1H), 2.99 (dd, J = 16.0, 9.3 Hz, 1H), 2.41 – 2.30 (m, 1H),
1.96 – 1.86 (m, 1H), 1.82 – 1.71 (m, 2H), 1.48 (d, rot., J = 22.4 Hz, 18H); 13C NMR (101 MHz,
CDCl3) δ 176.38, 167.74, 163.12, 162.49, 154.55, 150.41, 150.18, 142.79, 135.41, 131.61 (q,
2JCF = 32.3 Hz), 131.29 (q, 2JCF = 32.3 Hz), 130.97 (q, 2JCF = 32.3 Hz), 130.65 (q, 2JCF = 32.3
Hz), 129.45, 129.24, 128.88, 128.73, 125.66, 124.53 (q, 4JCF = 3.0 Hz), 124.48 (q, 4JCF = 3.0 Hz),
124.45 (q, 4JCF = 3.0 Hz), 124.41 (q, 4JCF = 3.0 Hz), 122.96 (q, 4JCF = 3.0 Hz), 122.92 (q, 4JCF =
3.0 Hz), 122.89 (q, 4JCF = 3.0 Hz), 122.85 (q, 4JCF = 3.0 Hz), 120.34, 117.04, 103.79, 82.32,

194
79.98, 56.50, 50.48, 31.17, 30.47, 28.35, 28.22, 28.09; 19F NMR (376 MHz, CDCl3) δ -62.66 (s,
3F); HRMS (ESI+): calcd for C34H39F3N7O5S [M+H]+: 714.2685, found: 714.2682.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(2-(trifluoromethyl)phenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19m).
Synthesized by General Procedure 3E. 37%, yellow oily solid. 1H NMR (400 MHz, CDCl3) δ
10.33 (s, 1H), 8.01 (d, J = 8.2 Hz, 2H), 7.84 – 7.74 (m, 2H), 7.70 (d, J = 7.7 Hz, 1H), 7.58 (t, J =
7.5 Hz, 1H), 7.52 – 7.44 (m, 3H), 6.77 (s, 1H), 4.78 (qd, J = 7.3, 4.1 Hz, 1H), 3.74 – 3.61 (m,
2H), 3.56 – 3.44 (m, 1H), 3.12 (dd, J = 15.4, 8.5 Hz, 1H), 2.33 – 2.23 (m, 1H), 1.92 (dq, J =
10.4, 4.8 Hz, 1H), 1.87 – 1.75 (m, 2H), 1.48 (d, J = 9.0 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ
191.61, 176.91, 167.90, 162.66 48 (q, 3JCF = 19.2 Hz), 162.47 48 (q, 2JCF = 319.2 Hz), 154.20,
150.48, 148.79, 142.69, 134.55 48 (q, 1JCF = 285.8 Hz), 132.10, 131.72 (q, 1JCF = 285.8 Hz),
128.97 48 (q, 1JCF = 285.8 Hz), 128.65, 128.32, 126.58 48 (q, 1JCF = 285.8 Hz), 120.91, 117.31,
107.12, 82.14, 79.54, 56.61, 50.27, 30.83, 30.46, 28.39*, 28.24*, 24.56; 19F NMR (376 MHz,
CDCl3) δ -57.78 (s, 3F); HRMS (ESI+): calcd for C34H38F3N7NaO5S [M+Na]+: 736.2505, found:
714.2500.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(p-tolyl)thiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19n). Synthesized by General
Procedure 3E. 82%, off-white oily solid. 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 1H), 8.00 (d, J
= 8.3 Hz, 2H), 7.76 (d, J = 7.8 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 6.83
(s, 1H), 4.81 – 4.73 (m, 1H), 3.80 – 3.60 (m, 2H), 3.56 – 3.45 (m, 1H), 3.08 (dd, J = 15.4, 8.8
Hz, 1H), 2.38 (s, 3H), 2.35 – 2.25 (m, 1H), 1.98 – 1.87 (m, 1H), 1.85 – 1.74 (m, 2H), 1.48 (s,
18H); 13C NMR (101 MHz, CDCl3) δ 176.75, 167.87, 162.80, 154.28, 151.70, 142.87, 137.94,
131.88, 129.49, 129.22, 128.90, 126.16, 120.47, 117.09, 101.88, 81.88, 79.77, 56.61, 50.32,

195
30.93, 30.44, 29.81, 28.30, 24.53, 21.44; HRMS (ESI+): calcd for C34H42N7O5S [M+H]+:
660.2968, found: 660.2991.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4-methoxyphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19o).
Synthesized by General Procedure 3E. 61%, clear oily solid. 1H NMR (400 MHz, CDCl3) δ
10.40 (s, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.84 – 7.75 (m, 2H), 7.62 (d, J = 2.1 Hz, 1H), 7.59 – 7.55
(m, 1H), 6.96 – 6.91 (m, 2H), 6.72 (s, 1H), 4.82 – 4.69 (m, 1H), 3.83 (s, 3H), 3.74 – 3.58 (m,
2H), 3.55 – 3.42 (m, 1H), 3.03 (dd, J = 15.7, 8.9 Hz, 1H), 2.36 – 2.23 (m, 1H), 1.96 – 1.83 (m,
1H), 1.81 – 1.71 (m, 1H), 1.58 – 1.35 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 176.75, 167.87,
162.80, 154.28, 151.70, 142.87, 137.94, 131.88, 129.49, 129.22, 128.90, 126.16, 120.47, 117.09,
101.88, 81.88, 79.77, 56.61, 50.32, 30.93, 30.44, 29.81, 28.30, 24.53, 21.44; HRMS (ESI+):
calcd for C34H42N7O6S [M+H]+: 676.2917, found: 676.2903.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4-ethoxyphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19p).
Synthesized by General Procedure 3E. 68%, light yellow oil. 1H NMR (400 MHz, CDCl3) δ
10.35 (s, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 6.94
(d, J = 8.4 Hz, 2H), 6.74 (s, 1H), 4.82 – 4.73 (m, 1H), 4.08 (q, J = 7.0 Hz, 2H), 3.77 – 3.62 (m,
2H), 3.56 – 3.45 (m, 1H), 3.11 (dd, J = 15.4, 8.6 Hz, 1H), 2.34 – 2.24 (m, 1H), 1.97 – 1.88 (m,
2H), 1.86 – 1.75 (m, 2H), 1.52 – 1.40 (m, 21H); 13C NMR (126 MHz, CDCl3) δ 176.96, 167.94,
162.78, 159.07, 153.98, 152.88, 151.53, 142.85, 134.03, 129.00, 127.54, 127.43, 117.19, 114.75,
105.35, 100.89, 83.18, 77.73, 63.67, 56.64, 50.25, 30.79, 30.45, 29.85, 28.31, 28.19, 15.00;
HRMS (ESI+): calcd for C35H44N7O6S [M+H]+: 690.3074, found: 690.3084.

196
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-methoxyphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19q).
Synthesized by General Procedure 3E. 51%, clear amorphous solid. 1H NMR (400 MHz, CDCl3)
δ 10.41 (s, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.63 (s, 1H), 7.57 (d, J = 8.3 Hz, 2H), 7.48 – 7.41 (m,
2H), 7.32 (t, J = 8.0 Hz, 1H), 6.92 – 6.83 (m, 2H), 6.36 (s, 1H), 4.82 – 4.71 (m, 1H), 3.86 (s,
3H), 3.80 – 3.61 (m, 2H), 3.58 – 3.44 (m, 1H), 3.05 (dd, J = 15.6, 8.9 Hz, 1H), 2.39 – 2.22 (m,
0H), 1.97 – 1.87 (m, 1H), 1.84 – 1.70 (m, 2H), 1.55 – 1.39 (m, 18H); 13C NMR (101 MHz,
CDCl3) δ 176.65, 167.84, 163.00, 162.58, 160.02, 154.31, 153.02, 151.37, 150.40, 142.95,
136.02, 133.88, 129.80, 128.83, 120.35, 118.71, 117.07, 113.79, 111.79, 105.30, 102.87, 82.19,
79.75, 56.56, 55.45, 50.33, 30.96, 30.43, 29.83, 28.29, 28.10, 24.50; HRMS (ESI+): calcd for
C34H42N7O6S [M+H]+: 676.2917, found: 676.2900.
tert-butyl (S,E)-(((tert-butoxycarbonyl)amino)(2-((3-(4-((4-(4-(trifluoromethoxy)phenyl)thiazol-
2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methylene)carbamate (3.19r).
Synthesized by General Procedure 3E. 70%, yellow amorphous solid. 1H NMR (500 MHz,
CDCl3) δ 10.41 (s, 1H), 8.02 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.85 (s, 1H), 7.57 (d, J
= 8.1 Hz, 2H), 6.88 (s, 1H), 4.81 – 4.71 (m, 1H), 3.79 – 3.61 (m, 2H), 3.56 – 3.44 (m, 1H), 3.14
– 3.04 (dd, J = 15.5, 8.6 Hz, 1H), 2.34 – 2.25 (m, 1H), 1.95 – 1.88 (m, 1H), 1.85 – 1.74 (m, 2H),
1.48 (s, 18H); 13C NMR (126 MHz, CDCl3) δ 176.70, 168.89, 167.68, 162.92, 154.13, 150.83,
150.19, 148.84, 142.49, 133.15, 128.82, 127.48, 121.10, 120.75, 119.45, 117.11, 103.06, 81.60,
79.46, 56.46, 50.14, 30.75, 30.29, 29.67, 28.16, 24.33; 19F NMR (470 MHz, CDCl3) δ -57.82 (s,
3F); HRMS (ESI+): calcd for C34H38F3N7O6S [M+H]+: 730.2634, found: 730.2672.

197
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-(trifluoromethoxy)phenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19s).
Synthesized by General Procedure 3E. 83%, yellow amorphous solid. 1H NMR (400 MHz,
CDCl3) δ 10.38 (s, 1H), 8.04 – 8.00 (m, 2H), 7.80 (ddd, J = 7.8, 1.6, 1.0 Hz, 1H), 7.73 (dt, J =
2.5, 1.3 Hz, 1H), 7.60 – 7.54 (m, 2H), 7.43 (t, J = 8.0 Hz, 1H), 7.19 – 7.15 (m, 1H), 6.94 (s, 1H),
4.82 – 4.73 (m, 1H), 3.82 – 3.67 (m, 2H), 3.57 – 3.45 (m, 1H), 3.09 (dd, J = 15.5, 8.8 Hz, 1H),
2.32 (d, J = 11.6 Hz, 1H), 1.97 – 1.88 (m, 1H), 1.86 – 1.75 (m, 1H), 1.67 – 1.56 (m, 1H), 1.50 (s
rot., 9H), 1.46 (s rot., 9H); 13C NMR (101 MHz, CDCl3) δ 176.81, 163.00, 150.47 (q, 3JCF = 32.8
Hz), 150.20 (q, 3JCF = 32.8 Hz), 149.82 (q, 3JCF = 32.8 Hz), 142.60, 136.61, 130.13, 128.96,
124.46, 120.90, 120.28, 118.85, 117.26, 103.93, 82.20, 79.66, 56.59, 50.34, 30.95, 30.47, 28.38*,
28.24*, 28.14; 19F NMR (376 MHz, CDCl3) δ -57.66 (s, 3F); HRMS (ESI+): calcd for
C34H38F3N7O6S [M+H]+: 730.2634, found: 730.2635.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3,4-dimethylphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19t).
Synthesized by General Procedure 3E. 92%, clear amorphous solid. 1H NMR (400 MHz, CDCl3)
δ 10.38 (s, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.67 – 7.51 (m, 6H), 7.17 (d, J = 7.8 Hz, 1H), 6.81 (s,
1H), 6.36 (d, J = 2.4 Hz, 1H), 4.82 – 4.72 (m, 1H), 3.81 – 3.60 (m, 2H), 3.50 (s, 1H), 3.07 (dd, J
= 15.5, 8.7 Hz, 1H), 2.30 (d, J = 11.3 Hz, 6H), 1.98 – 1.85 (m, 1H), 1.85 – 1.72 (m, 2H), 1.47 (s,
18H); 13C NMR (101 MHz, CDCl3) δ 176.70, 167.88, 163.10, 162.57, 154.72, 154.24, 153.05,
151.67, 150.44, 143.05, 136.92, 136.63, 133.77, 132.34, 130.07, 128.85, 127.44, 123.69, 120.31,
117.05, 105.24, 101.66, 82.97, 82.15, 79.69, 56.57, 50.32, 30.90, 30.42, 28.29, 28.11, 24.55,
20.04, 19.74; HRMS (ESI+): calcd for C35H44N7O5S [M+H]+: 674.3125, found: 674.3134.

198
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3,4-dimethoxyphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19u).
Synthesized by General Procedure 3E. 78%, clear amorphous solid. 1H NMR (400 MHz, CDCl3)
δ 10.41 (s, 1H), 8.56 – 8.24 (m, 1H), 7.95 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.47 –
7.39 (m, 2H), 7.22 (s, 1H), 6.76 (s, 1H), 4.76 (dq, J = 11.7, 7.1, 5.1 Hz, 1H), 3.93 (d, J = 18.3
Hz, 6H), 3.83 – 3.60 (m, 3H), 3.56 – 3.43 (m, 1H), 3.04 (dd, J = 15.6, 9.0 Hz, 1H), 2.38 – 2.23
(m, 1H), 1.97 – 1.84 (m, 1H), 1.82 – 1.70 (m, 2H), 1.54 – 1.38 (m, 18H); 13C NMR (101 MHz,
CDCl3) δ 176.63, 167.81, 162.82, 162.56, 154.36, 151.37, 150.41, 149.11, 142.93, 128.79,
127.90, 120.32, 118.80, 117.01, 111.35, 109.61, 101.06, 83.08, 82.20, 79.74, 56.54, 56.08, 56.06,
50.36, 30.98, 30.46, 28.26, 28.09; HRMS (ESI+): calcd for C35H44N7O7S [M+H]+: 706.3023,
found: 706.3028.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-chloro-4-methoxyphenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19v).
Synthesized by General Procedure 3E. 34%, light yellow oil. 1H NMR (500 MHz, CDCl3) δ
10.38 (s, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.87 (d, J = 2.2 Hz, 1H), 7.75 (dd, J = 8.5, 2.2 Hz, 1H),
7.59 – 7.51 (m, 2H), 6.97 (d, J = 8.6 Hz, 1H), 6.78 (s, 1H), 4.82 – 4.72 (m, 1H), 3.94 (s, 3H),
3.84 – 3.62 (m, 2H), 3.58 – 3.42 (m, 1H), 3.10 (dd, J = 15.4, 8.7 Hz, 1H), 2.36 – 2.23 (m, 1H),
1.97 – 1.87 (m, 1H), 1.88 – 1.74 (m, 1H), 1.74 – 1.56 (m, 1H), 1.54 – 1.40 (m, 18H); 13C NMR
(126 MHz, CDCl3) δ 176.89, 167.87, 162.97, 154.91, 150.17, 142.71, 128.98, 128.46, 128.11,
125.70, 122.84, 117.23, 112.21, 101.84, 100.14, 83.18, 77.73, 56.62, 56.40, 30.88, 30.46, 29.85,
28.31, 28.18; HRMS (ESI+): calcd for C34H41ClN7O6S [M+H]+: 710.2528, found: 710.2542.

199
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3,4-difluorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19w).
Synthesized by General Procedure 3E. 43%, off-white solid. 1H NMR (400 MHz, CDCl3) δ
10.42 (s, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.96 – 7.87 (m, 1H), 7.74 – 7.65 (m, 1H), 7.62 – 7.52 (m,
3H), 7.23 – 7.15 (m, 1H), 6.84 (s, 1H), 4.81 – 4.72 (m, 1H), 3.86 – 3.61 (m, 2H), 3.57 – 3.43 (m,
1H), 3.07 (dd, J = 15.6, 8.8 Hz, 1H), 2.38 – 2.26 (m, 1H), 1.97 – 1.88 (m, 1H), 1.84 – 1.72 (m,
2H), 1.48 (d, J = 17.1 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ 176.74, 167.79, 162.99, 162.59,
150.44, 149.63, 142.58, 128.91, 122.12, 120.79, (dd, 1JCF = 228.3 Hz, 3JCF = 17.2 Hz), 117.49
(dd, 1JCF = 228.3 Hz, 3JCF = 17.2 Hz), 117.19, 115.40 (dd, 1JCF = 228.3 Hz, 3JCF = 17.2 Hz),
115.22 (dd, 1JCF = 228.3 Hz, 3JCF = 17.2 Hz), 103.12, 82.23, 79.74, 56.57, 50.43, 31.01, 30.44,
28.36, 28.24; 19F NMR (376 MHz, CDCl3) δ -137.54 to -137.74 (m, 1F), -138.64 – -138.84 (m,
1F); HRMS (ESI+): calcd for C33H38F2N7O5S [M+H]+: 682.2623, found: 682.2590.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3,5-dichlorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19x).
Synthesized by General Procedure 3E. 45%, clear yellow solid. 1H NMR (400 MHz, CDCl3) δ
10.36 (s, 1H), 7.94 (d, J = 8.5 Hz, 3H), 7.55 – 7.49 (m, 2H), 7.45 (d, J = 2.1 Hz, 1H), 7.29 (dd, J
= 8.5, 2.2 Hz, 1H), 7.24 (s, 1H), 4.79 – 4.70 (m, 1H), 3.82 – 3.60 (m, 2H), 3.55 – 3.41 (m, 1H),
3.04 (dd, J = 15.6, 8.9 Hz, 1H), 2.33 – 2.23 (m, 1H), 1.95 – 1.85 (m, 1H), 1.82 – 1.68 (m, 2H),
1.48 (s, 12H), 1.44 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 176.68, 167.81, 162.57, 161.89,
150.45, 146.89, 142.75, 133.80, 132.44, 132.39, 131.76, 130.38, 128.87, 127.40, 120.65, 117.18,
108.44, 82.23, 79.76, 56.58, 50.34, 30.99, 30.45, 28.38, 28.24, 28.13; HRMS (ESI+): calcd for
C33H38Cl2N7O5S [M+H]+: 714.2032, found: 714.2042.

200
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4-fluoro-3-
(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-
yl)methyl)carbamate (3.19y). Synthesized by General Procedure 3E. 48%, off-white solid. 1H
NMR (400 MHz, CDCl3) δ 10.38 (s, 1H), 8.13 – 7.91 (m, 5H), 7.56 (d, J = 8.4 Hz, 2H), 7.23 (d,
J = 9.4 Hz, 1H), 6.90 (s, 1H), 4.84 – 4.71 (m, 1H), 3.83 – 3.63 (m, 2H), 3.57 – 3.46 (m, 1H),
3.07 (dd, J = 15.6, 8.9 Hz, 1H), 2.37 – 2.27 (m, 1H), 1.98 – 1.88 (m, 1H), 1.85 – 1.72 (m, 2H),
1.55 – 1.42 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 176.79, 167.79, 163.28, 162.59, 160.66,
158.10, 154.35, 150.46, 149.30, 142.55, 140.47, 137.78, 131.52 (d, 3JCF = 9.1 Hz), 131.43 (d,
3JCF = 9.1 Hz), 131.19, 128.94, 126.81 (q, 1JCF = 273.7 Hz), 124.94, 124.09 (q, 1JCF = 273.7 Hz),
121.38 (q, 1JCF = 273.7 Hz), 120.93, 119.32, 118.64 (q, 1JCF = 273.7 Hz), 118.51, 117.43, 117.29,
117.22, 103.45, 82.24, 79.76, 56.58, 50.34, 30.99, 30.49, 28.36*, 28.26*; 19F NMR (376 MHz,
CDCl3) δ -61.44 (d, J = 12.7 Hz, 3F), -115.54 to -116.19 (m, 1F); HRMS (ESI+): calcd for
C34H38F4N7O5S [M+H]+: 732.2591, found: 732.2542.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3,5-difluorophenyl)thiazol-2-
yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.19z).
Synthesized by General Procedure 3E. 79%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.83 (s,
1H), 8.92 (s, 1H), 8.26 (d, J = 8.3 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 7.5 Hz, 2H),
7.62 (s, 1H), 7.15 – 7.07 (m, 1H), 5.16 – 5.05 (m, 1H), 4.24 – 4.09 (m, 1H), 4.09 – 3.98 (m, 1H),
3.93 – 3.80 (m, 1H), 3.36 (dd, J = 15.9, 9.2 Hz, 1H), 2.78 – 2.65 (m, 1H), 2.34 – 2.23 (m, 1H),
2.18 – 2.05 (m, 2H), 1.92 – 1.74 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 176.41, 167.72,
164.69 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1 Hz), 164.56 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1 Hz),
162.95, 162.49, 162.23 (dd, 1JCF = 248.5 Hz, 3JCF = 13.1 Hz), 162.10 (dd, 1JCF = 248.5 Hz, 3JCF =
13.1 Hz), 154.55, 150.38, 149.46, 142.68, 137.86 (dd, 3JCF = 10.1 Hz), 137.76 (dd, 3JCF = 10.1

201
Hz), 137.66 (dd, 3JCF = 10.1 Hz), 128.77, 120.40, 117.07, 109.14 (dd, 3JCF = 19.2 Hz, 4JCF = 7.1
Hz), 109.07 (dd, 3JCF = 19.2 Hz, 4JCF = 7.1 Hz), 108.95 (dd, 3JCF = 19.2 Hz, 4JCF = 7.1 Hz),
108.88 (dd, 3JCF = 19.2 Hz, 4JCF = 7.1 Hz), 104.44, 103.40 (dd, 2JCF = 25.8 Hz), 103.14 (dd, 2JCF
= 25.8 Hz), 102.89 (dd, 2JCF = 25.8 Hz), 82.33, 79.97, 56.53, 50.56, 31.17, 30.44, 28.34*, 28.24*,
24.68; 19F NMR (376 MHz, CDCl3) δ -109.87 (t, J = 8.2 Hz, 2F); HRMS (ESI+): calcd for
C33H38F2N7O5S [M+H]+: 682.2623, found: 682.2615.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3-fluoro-5-
(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-
yl)methyl)carbamate (3.19aa). Synthesized by General Procedure 3E. 51%, clear yellow solid.
1H NMR (400 MHz, CDCl3) δ 10.43 (s, 1H), 8.34 (s, 1H), 7.94 (d, J = 8.4 Hz, 2H), 7.89 (s, 1H),
7.78 (dt, J = 9.6, 1.9 Hz, 1H), 7.61 – 7.55 (m, 2H), 7.25 (d, J = 6.2 Hz, 2H), 6.99 (s, 1H), 4.81 –
4.72 (m, 1H), 3.86 – 3.62 (m, 2H), 3.59 – 3.44 (m, 1H), 3.02 (dd, J = 15.7, 9.0 Hz, 1H), 2.41 –
2.29 (m, 1H), 1.98 – 1.87 (m, 1H), 1.83 – 1.71 (m, 2H), 1.51 (s, 8H), 1.45 (s, 10H); 13C NMR
(101 MHz, CDCl3) δ 176.54, 167.74, 164.19 (d, 1JCF = 248.5 Hz), 163.20, 162.52, 161.73 (d,
1JCF = 248.5 Hz), 154.49, 150.43, 149.10, 142.57, 137.89 (q, 3JCF = 9.1 Hz), 137.80 (d, 3JCF = 9.1
Hz), 133.40 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 133.32 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz),
133.08 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 132.99 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 132.74
(qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 132.66 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 132.41 (qd, 2JCF =
34.3 Hz, 3JCF = 9.1 Hz), 132.33 (qd, 2JCF = 34.3 Hz, 3JCF = 9.1 Hz), 128.84, 127.56 (q, 1JCF =
274.7 Hz), 124.85 (q, 1JCF = 274.7 Hz), 122.13 (q, 1JCF = 274.7 Hz), 120.72, 119.41 (q, 1JCF =
274.7 Hz), 118.61, 117.20, 116.68 (d, 2JCF = 23.2 Hz), 116.45 (d, 2JCF = 23.2 Hz), 112.02 (d, 2JCF
= 25.3 Hz), 111.77 (d, 2JCF = 25.3 Hz), 104.90, 82.32, 79.92, 56.54, 50.46, 31.13, 30.49, 28.37*,

202
28.24*, 24.62; 19F NMR (376 MHz, CDCl3) δ -62.84 (s, 3F) , -110.73 (t, J = 9.0 Hz, 1F); HRMS
(ESI+): calcd for C34H38F4N7O5S [M+H]+: 732.2591, found: 732.2602.
tert-butyl (S,Z)-((2-((3-(4-((4-(3,5-bis(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.19bb).
Synthesized by General Procedure 3E. 84%, off-white solid. 1H NMR (400 MHz, CDCl3) δ
10.40 (s, 1H), 8.30 (s, 2H), 8.11 (d, J = 19.0 Hz, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.80 (s, 1H), 7.56
(d, J = 8.6 Hz, 2H), 7.07 (s, 1H), 4.82 – 4.73 (m, 1H), 3.84 – 3.63 (m, 2H), 3.58 – 3.47 (m, 1H),
3.05 (dd, J = 15.6, 9.0 Hz, 1H), 2.39 – 2.28 (m, 1H), 1.99 – 1.88 (m, 1H), 1.85 – 1.74 (m, 2H),
1.48 (d, J = 18.2 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ 176.75, 167.74, 163.51, 162.55,
150.45, 148.74, 142.42, 136.53, 132.64 (q, 2JCF = 33.3 Hz), 132.31 (q, 2JCF = 33.3 Hz), 131.98
(q, 2JCF = 33.3 Hz), 131.66 (q, 2JCF = 33.3 Hz), 128.94, 127.60(q, 1JCF = 274.7 Hz), 126.15,
124.88 (q, 1JCF = 274.7 Hz), 122.16 (q, 1JCF = 274.7 Hz), 121.35, 121.05, 119.45 (q, 1JCF = 274.7
Hz), 117.35, 105.34, 82.27, 79.83, 56.56, 50.40, 31.04, 30.53, 28.37*, 28.24*; 19F NMR (376
MHz, CDCl3) δ -62.94 (s, 6F); HRMS (ESI+): calcd for C35H38F6N7O5S [M+H]+: 782.2559,
found: 782.2541.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(2-fluoro-5-
(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-
yl)methyl)carbamate (3.19cc). Synthesized by General Procedure 3E. 51%, clear yellow solid.
1H NMR (400 MHz, CDCl3) δ 10.34 (s, 1H), 8.48 (dd, J = 7.1, 2.5 Hz, 1H), 8.04 (d, J = 8.3 Hz,
2H), 7.83 (s, 1H), 7.55 (d, J = 8.4 Hz, 3H), 7.25 – 7.18 (m, 1H), 4.83 – 4.73 (m, 1H), 3.80 – 3.62
(m, 2H), 3.58 – 3.45 (m, 1H), 3.09 (dd, J = 16.9, 8.4 Hz, 1H), 2.36 – 2.24 (m, 1H), 1.99 – 1.89
(m, 1H), 1.86 – 1.72 (m, 2H), 1.49 (d, J = 9.5 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ 176.91,
167.83, 163.22 (d, 1JCF = 256.5 Hz), 162.25, 160.68 (d, 1JCF = 256.5 Hz), 158.33, 154.26, 148.80,

203
143.85, 142.54, 129.01, 127.72, 126.04, 123.36, 123.26 (q, 3JCF = 12.1 Hz), 123.15 (q, 3JCF =
12.1 Hz), 123.03 (q, 3JCF = 12.1 Hz), 122.90 (q, 3JCF = 12.1 Hz), 122.61, 121.09, 117.33, 116.82,
116.58, 109.22 (d, 3JCF = 16.2 Hz), 109.06 (d, 3JCF = 16.2 Hz), 83.56, 56.61, 50.26, 30.92, 30.50,
29.85, 28.31, 28.14; 19F NMR (376 MHz, CDCl3) δ -62.05 (s, 3F), -109.15 to -109.28 (m, 1F);
HRMS (ESI+): calcd for C34H38F4N7O5S [M+H]+: 732.2591, found: 732.2605.
tert-butyl (S,Z)-((2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-
5-yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.19dd).
Synthesized by General Procedure 3E. 83%, light yellow solid. 1H NMR (400 MHz, CDCl3) δ
10.43 – 10.31 (m, 1H), 8.05 (d, J = 8.3 Hz, 2H), 7.99 – 7.92 (m, 2H), 7.79 – 7.69 (m, 1H), 7.65
(dd, J = 8.5, 6.9 Hz, 4H), 7.61 – 7.56 (m, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H),
6.94 (s, 1H), 4.84 – 4.74 (m, 1H), 3.78 – 3.62 (m, 2H), 3.59 – 3.44 (m, 1H), 3.11 (dd, J = 15.5,
8.7 Hz, 1H), 2.36 – 2.25 (m, 1H), 1.97 – 1.88 (m, 1H), 1.87 – 1.75 (m, 2H), 1.53 – 1.44 (m,
18H); 13C NMR (101 MHz, CDCl3) δ 176.82, 167.87, 162.83, 162.58, 154.31, 151.36, 150.33,
142.77, 140.80, 133.58, 128.95, 128.67, 127.49, 127.27, 127.12, 126.93, 126.65, 120.68, 117.18,
113.25, 102.79, 82.17, 79.67, 56.60, 50.33, 30.91, 30.45, 29.86, 28.31, 24.60; HRMS (ESI+):
calcd for C39H44N7O5S [M+H]+: 722.3125, found: 722.3107.
tert-butyl (S,Z)-((2-((3-(4-((4-(benzofuran-2-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.19ee). Synthesized
by General Procedure 3E. 69%, off-white solid. 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 1H),
8.13 (br. s, 2H), 8.05 – 7.92 (m, 3H), 7.62 – 7.50 (m, 3H), 7.40 – 7.31 (m, 2H), 6.92 (s, 1H), 4.83
– 4.73 (m, 1H), 3.83 – 3.62 (m, 1H), 3.57 – 3.45 (m, 1H), 3.08 (dd, J = 15.3, 8.6 Hz, 1H), 2.36 –
2.27 (m, 1H), 1.97 – 1.87 (m, 1H), 1.85 – 1.71 (m, 2H), 1.49* (ap. d, J = 5.9 Hz, 18H); 13C NMR
(101 MHz, CDCl3) δ 176.75, 167.84, 163.01, 155.88, 154.34, 148.93, 143.88, 143.82, 142.76,

204
128.91, 125.53, 124.78, 123.31, 120.92, 120.74, 117.26, 117.16, 111.95, 103.02, 83.55, 82.15,
79.75, 56.58, 50.33, 36.83, 30.97, 30.48, 29.85, 28.32, 28.13, 24.84, 24.61; HRMS (ESI+): calcd
for C35H40N7O6S [M+H]+: 686.2761, found: 686.2760.
(S)-amino(2-((3-(4-((4-phenylthiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-
1-yl)methaniminium chloride (3.20a). Synthesized by General Procedure 3E. 85%, yellow solid.
1H NMR (500 MHz, CD3OD) δ 8.10 (d, J = 8.3 Hz, 2H), 7.85 (dd, J = 16.1, 8.0 Hz, 4H), 7.46 (t,
J = 7.6 Hz, 2H), 7.38 (t, J = 7.4 Hz, 1H), 7.19 (s, 1H), 4.56 – 4.50 (m, 1H), 3.56 (ddd, J = 12.1,
10.3, 5.8 Hz, 1H), 3.51 – 3.42 (m, 1H), 3.33 (s, 2H), 2.33 – 2.24 (m, 1H), 2.17 – 1.97 (m, 3H);
13C NMR (126 MHz, CD3OD) δ 177.97, 169.20, 166.40, 156.47, 144.36, 129.88, 129.72, 129.63,
127.27, 122.36, 119.87, 57.22, 31.61, 30.08, 23.59; HRMS (ESI+): calcd for C23H24N7OS
[M+H]+: 446.1763, found: 446.1755.
(S)-amino(2-((3-(4-((4-(pyridin-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium trifluoroacetate (3.20b). Synthesized by General
Procedure 3E. 100%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.75 (d, J = 6.0 Hz, 2H), 8.47
– 8.41 (m, 2H), 8.10 – 8.02 (m, 3H), 7.95 – 7.89 (m, 2H), 4.58 – 4.49 (m, 1H), 3.59 – 3.52 (m,
1H), 3.51 – 3.42 (m, 1H), 3.34 – 3.31 (m, 2H), 2.36 – 2.23 (m, 1H), 2.19 – 1.96 (m, 3H); 13C
NMR (101 MHz, CD3OD) δ 176.41, 167.86, 163.98, 155.01, 148.91, 146.25, 143.48, 142.84,
128.02, 122.18, 119.62, 117.04, 114.31, 55.77, 30.14, 28.63, 22.15; HRMS (ESI+): calcd for
C22H23N8OS [M+H]+: 447.1716, found: 447.1704. HPLC purity: 87%.
(S)-amino(2-((3-(4-((4-(pyridin-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20c). Synthesized by General Procedure
3E. 85%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 9.31 (s, 1H), 8.96 (dt, J = 8.4, 1.7 Hz,
1H), 8.69 (d, J = 5.6 Hz, 1H), 8.05 – 7.97 (m, 3H), 7.91 – 7.82 (m, 2H), 7.66 (s, 1H), 4.55 – 4.48

205
(m, 1H), 3.57 – 3.50 (m, 1H), 3.48 – 3.40 (m, 1H), 3.31 – 3.29 (m, 1H), 2.33 – 2.21 (m, 1H),
2.17 – 1.95 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 177.82, 169.29, 165.60, 156.45, 145.97,
145.00, 142.24, 142.04, 141.36, 135.43, 129.44, 128.13, 120.86, 118.37, 109.98, 57.18, 31.57,
30.06, 23.58; HRMS (ESI+): calcd for C22H23N8OS [M+H]+: 447.1716, found: 447.1698.
(S)-amino(2-((3-(4-((4-(4-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20d). Synthesized by General Procedure
3E. 100%, yellow solid. 1H NMR (500 MHz, CD3OD) δ 8.09 (d, J = 8.3 Hz, 2H), 7.89 (dd, J =
8.4, 5.3 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.19 (t, J = 8.6 Hz, 2H), 7.15 (s, 1H), 4.57 – 4.51 (m,
1H), 3.60 – 3.51 (m, 1H), 3.51 – 3.41 (m, 1H), 3.36 – 3.33 (m, 2H), 2.29 (ddd, J = 18.0, 10.1, 5.4
Hz, 1H), 3.39 – 3.36 (m, 0H), 2.20 – 1.96 (m, 3H); 13C NMR (126 MHz, CD3OD) δ 176.54,
167.77, 165.04, 163.86 (d, 1JCF = 199.0 Hz), 161.89 (d, 1JCF = 199.0 Hz), 155.04, 146.83, 142.85,
129.13, 128.29, 127.95 (d, 4JCF = 6.1 Hz), 127.89 (d, 4JCF = 6.1 Hz), 120.97, 118.47, 115.30 (d,
3JCF = 18.2 Hz), 115.12 (d, 3JCF = 18.2 Hz), 55.80, 36.84, 30.19, 28.67, 22.18; 19F NMR (376
MHz, CD3OD) δ -116.45 to -116.59 (m, 1F); HRMS (ESI+): calcd for C23H23FN7OS [M+H]+:
464.1669, found: 464.1681.
(S)-amino(2-((3-(4-((4-(4-chlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20e). Synthesized by General Procedure
3E. 100%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.12 – 8.05 (m, 2H), 7.92 – 7.80 (m,
4H), 7.50 – 7.42 (m, 2H), 7.22 (d, J = 4.4 Hz, 1H), 4.55 (q, J = 6.6 Hz, 1H), 3.60 – 3.52 (m, 1H),
3.52 – 3.43 (m, 1H), 2.35 – 2.24 (m, 1H), 2.19 – 1.98 (m, 3H); 13C NMR (151 MHz, CD3OD) δ
177.90, 169.24, 156.48, 148.86, 144.56, 135.11, 133.30, 129.92, 129.63, 128.72, 121.91, 119.47,
64.45, 57.24, 31.62, 30.10, 23.60; HRMS (ESI+): calcd for C23H23ClN7OS [M+H]+: 481.1452,
found: 481.1456.

206
(S)-amino(2-((3-(4-((4-(4-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20f). Synthesized by General Procedure
3E. 100% light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.06 (d, J = 8.7 Hz, 2H), 7.84 (dd, J
= 10.2, 8.6 Hz, 4H), 7.59 (d, J = 8.5 Hz, 2H), 7.22 (s, 1H), 4.59 – 4.50 (m, 1H), 3.59 – 3.51 (m,
1H), 3.51 – 3.43 (m, 1H), 3.30 – 3.28 (m, 2H), 2.35 – 2.23 (m, 1H), 2.19 – 1.98 (m, 3H); 13C
NMR (101 MHz, CD3OD) δ 177.71, 169.35, 164.59, 164.52, 156.43, 151.28, 151.16, 145.44,
135.19, 134.57, 132.73, 129.38, 128.78, 123.98, 122.39, 120.31, 118.08, 104.75, 57.20, 31.60,
30.06, 23.58; HRMS (ESI+): calcd for C23H23BrN7OS [M+H]+: 524.0868, found: 524.0873.
(S)-amino(2-((3-(4-((4-(3-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.9g). Synthesized by General Procedure 3E.
100%, yellow solid. 1H NMR (500 MHz, CD3OD) δ 8.08 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 8.7
Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 10.1 Hz, 1H), 7.49 – 7.41 (m, 1H), 7.26 (s, 1H),
7.09 (td, J = 8.4, 2.1 Hz, 1H), 4.56 – 4.48 (m, 1H), 3.56 – 3.50 (m, 1H), 3.48 – 3.41 (m, 1H),
3.32 – 3.30 (m, 2H), 2.33 – 2.22 (m, 1H), 2.17 – 1.97 (m, 3H); 13C NMR (126 MHz, CD3OD) δ
177.96, 169.19, 166.35, 165.52 (d, 1JCF = 195.9 Hz), 163.58 (d, 1JCF = 195.9 Hz), 156.46, 148.11,
144.28, 136.47 (d, 4JCF = 6.1 Hz), 136.41 (d, 4JCF = 6.1 Hz), 131.74 (d, 4JCF = 7.1 Hz), 131.67 (d,
4JCF = 7.1 Hz), 129.71, 123.04, 123.02, 122.35, 119.82, 116.20 (d, 3JCF = 17.2 Hz), 116.03 (d,
3JCF = 17.2 Hz), 114.06 (d, 3JCF = 18.2 Hz), 113.88 (d, 3JCF = 18.2 Hz), 105.55, 57.22, 31.62,
30.08, 23.60; 19F NMR (376 MHz, CD3OD) δ -77.20 (s, 1F); HRMS (ESI+): calcd for
C23H23FN7OS [M+H]+: 465.1747, found: 465.1724.
(S)-amino(2-((3-(4-((4-(3-chlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20h). Synthesized by General Procedure
3E. 100%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.13 (d, J = 8.2 Hz, 2H), 7.89 (s,

207
1H), 7.80 (t, J = 8.2 Hz, 3H), 7.51 – 7.39 (m, 2H), 7.31 (s, 1H), 4.61 – 4.52 (m, 1H), 3.63 – 3.53
(m, 1H), 3.53 – 3.44 (m, 1H), 2.37 – 2.25 (m, 1H), 2.20 – 2.00 (m, 3H); 13C NMR (101 MHz,
CD3OD) δ 178.03, 169.05, 167.18, 156.42, 143.65, 135.88, 135.09, 131.50, 129.81, 129.73,
127.24, 125.65, 123.22, 120.58, 57.19, 31.60, 30.09, 23.59; HRMS (ESI+): calcd for
C23H23ClN7OS [M+H]+: 480.1373, found: 480.1370.
(S)-amino(2-((3-(4-((4-(3-bromophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20i). Synthesized by General Procedure 3E.
100%, off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.09 – 8.05 (m, 1H), 8.04 – 7.98 (m, 2H),
7.91 – 7.82 (m, 3H), 7.47 – 7.42 (m, 1H), 7.31 (t, J = 7.9 Hz, 1H), 7.21 (s, 1H), 4.56 – 4.48 (m,
1H), 3.58 – 3.50 (m, 1H), 3.48 – 3.40 (m, 1H), 3.29 – 3.27 (m, 2H), 2.33 – 2.21 (m, 1H), 2.16 –
1.97 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 180.23, 171.87, 167.12, 158.95, 153.37, 147.95,
140.81, 134.06, 133.94, 132.42, 131.90, 128.20, 126.20, 122.87, 120.59, 107.96, 59.72, 34.12,
32.58, 26.10; HRMS (ESI+): calcd for C23H23BrN7OS [M+H]+: 524.0868, found: 524.0869.
(S)-amino(2-((3-(4-((4-(2-fluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20j). Synthesized by General Procedure
3E. 100%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.17 – 8.06 (m, 3H), 7.87 (d, J =
8.3 Hz, 2H), 7.47 – 7.39 (m, 1H), 7.38 – 7.21 (m, 3H), 4.62 – 4.54 (m, 1H), 3.66 – 3.56 (m, 1H),
3.55 – 3.44 (m, 1H), 3.39 – 3.35 (m, 2H), 2.40 – 2.27 (m, 1H), 2.22 – 2.02 (m, 3H); 13C NMR
(101 MHz, CD3OD) δ 177.92, 169.20, 165.30, 162.76 (d, 1JCF = 250.5 Hz), 160.28 (d, 1JCF =
250.5 Hz), 156.45, 156.01, 152.88, 148.68, 144.41, 143.41, 131.07, 130.97, 130.93, 129.66,
125.73 (d, 4JCF = 3.0 Hz), 125.70 (d, 4JCF = 3.0 Hz), 122.10, 121.98, 119.61, 117.13 (d, 2JCF =
23.2 Hz), 116.90 (d, 2JCF = 23.2 Hz), 57.22, 31.62, 30.09, 23.60; 19F NMR (471 MHz, CD3OD) δ

208
-116.05 to -116.22 (m, 1F); HRMS (ESI+): calcd for C23H23FN7OS [M+H]+: 464.1669, found:
464.1658.
(S)-amino(2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20k). Synthesized by General Procedure
3E. 100%, yellow solid. 1H NMR (500 MHz, CD3OD) δ 8.10 (d, J = 8.1 Hz, 2H), 8.06 (d, J = 8.6
Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.37 (s, 1H), 4.57 – 4.50 (m, 1H),
3.60 – 3.51 (m, 1H), 3.51 – 3.42 (m, 1H), 3.32 (s, 1H), 2.34 – 2.24 (m, 1H), 2.19 – 1.98 (m, 3H);
13C NMR (126 MHz, CD3OD) δ 177.81, 169.32, 165.27, 156.46, 149.94, 145.07, 139.07, 130.70
(q, 2JCF = 25.3 Hz), 130.45 (q, 2JCF = 25.3 Hz), 129.51, 127.53, 126.84, 126.70 (q, 4JCF = 3.0 Hz),
126.67 (q, 4JCF = 3.0 Hz), 126.64 (q, 4JCF = 3.0 Hz), 126.60 (q, 4JCF = 3.0 Hz), 124.69, 121.06,
118.72, 106.64, 57.24, 31.63, 30.09, 23.60; 19F NMR (471 MHz, CD3OD) δ -64.02 (s, 3F);
HRMS (ESI+): calcd for C24H23F3N7OS [M+H]+: 515.1715, found: 515.1718.
(S)-amino(2-((3-(4-((4-(3-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20l). Synthesized by General Procedure 3E.
100%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.21 (s, 1H), 8.19 – 8.15 (m, 1H), 8.06
– 7.98 (m, 2H), 7.89 – 7.84 (m, 2H), 7.63 – 7.56 (m, 2H), 7.32 (s, 1H), 4.56 – 4.49 (m, 1H), 3.63
– 3.59 (m, 1H), 3.53 – 3.45 (m, 1H), 2.34 – 2.22 (m, 1H), 2.18 – 1.96 (m, 3H); 13C NMR (101
MHz, CD3OD) δ 177.72, 169.34, 164.73, 156.46, 150.89, 145.42, 137.08, 134.58, 132.49 (q, 2JCF
= 31.3 Hz), 132.17 (q, 2JCF = 31.3 Hz), 131.86 (q, 2JCF = 31.3 Hz), 131.54 (q, 2JCF = 31.3 Hz),
130.49, 130.43, 129.95, 129.80 (q, 1JCF = 272.7 Hz), 129.37, 127.10, 126.52 (q, 1JCF = 272.7 Hz),
125.14 (q, 4JCF = 4.0 Hz), 125.11 (q, 4JCF = 4.0 Hz), 125.06 (q, 4JCF = 4.0 Hz), 125.03 (q, 4JCF =
4.0 Hz), 124.40 (q, 1JCF = 272.7 Hz), 123.98, 123.59 (q, 4JCF = 4.0 Hz), 123.56 (q, 4JCF = 4.0 Hz),
123.51 (q, 4JCF = 4.0 Hz), 123.48 (q, 4JCF = 4.0 Hz), 121.70 (q, 1JCF = 272.7 Hz), 120.38, 118.08,

209
105.77, 57.22; 19F NMR (376 MHz, CD3OD) δ -64.27 (s, 3F); HRMS (ESI+): calcd for
C24H23F3N7OS [M+H]+: 514.1637, found: 514.1625.
(S)-amino(2-((3-(4-((4-(2-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20m). Synthesized by General Procedure
3E. 93%, off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.15 – 8.09 (m, 2H), 7.89 (d, J = 7.8
Hz, 1H), 7.83 – 7.65 (m, 5H), 7.04 (s, 1H), 4.61 – 4.53 (m, 1H), 3.62 – 3.55 (m, 1H), 3.54 – 3.46
(m, 1H), 3.40 – 3.35 (m, 2H), 2.37 – 2.26 (m, 1H), 2.21 – 2.00 (m, 3H); 13C NMR (101 MHz,
CD3OD) δ 178.06, 169.06, 166.70, 156.45, 148.60, 144.53, 143.47, 133.49, 133.34, 132.47,
130.78, 129.82, 129.34 (q, 2JCF = 29.3 Hz), 129.05 (q, 2JCF = 29.3 Hz), 127.68 (q, 3JCF = 5.1 Hz),
127.63 (q, 4JCF = 5.1 Hz), 127.57 (q, 4JCF = 5.1 Hz), 127.52 (q, 4JCF = 5.1 Hz), 123.47, 120.86,
108.89, 57.20, 31.60, 30.08, 23.59; 19F NMR (376 MHz, CD3OD) δ -59.48 (s, 3F); HRMS
(ESI+): calcd for C24H23F3N7OS [M+H]+: 515.1715, found: 515.1663.
(S)-amino(2-((3-(4-((4-(p-tolyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20n). Synthesized by General Procedure
3E. 85%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.02 (d, J = 8.7 Hz, 2H), 7.88 (d, J =
8.8 Hz, 2H), 7.81 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 7.7 Hz, 2H), 7.06 (s, 1H), 4.57 – 4.49 (m, 1H),
3.58 – 3.55 (m, 1H), 3.50 – 3.46 (m, 1H), 3.30 – 3.24 (m, 2H), 2.37 (s, 3H), 2.34 – 2.22 (m, 1H),
2.19 – 1.98 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 177.71, 169.39, 164.38, 156.44, 152.69,
145.64, 138.70, 133.46, 130.22, 129.38, 126.99, 120.16, 117.99, 103.13, 57.22, 31.61, 30.06,
23.59, 21.28; HRMS (ESI+): calcd for C24H26N7OS [M+H]+: 460.1920, found: 460.1920.
(S)-amino(2-((3-(4-((4-(4-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20o). Synthesized by General Procedure
3E. 100%, off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.05 – 7.97 (m, 2H), 7.90 – 7.79 (m,

210
4H), 6.99 – 6.93 (m, 3H), 4.57 – 4.46 (m, 1H), 3.83 (s, 3H), 3.58 – 3.50 (m, 1H), 3.50 – 3.40 (m,
1H), 3.29 (d, J = 3.7 Hz, 1H), 2.34 – 2.22 (m, 1H), 2.17 – 1.95 (m, 4H); 13C NMR (101 MHz,
CD3OD) δ 178.23, 168.80, 168.62, 162.30, 156.38, 144.37, 142.34, 135.53, 131.81, 130.58,
130.11, 129.43, 128.98, 125.05, 123.38, 122.10, 118.64, 115.55, 114.71, 57.17, 56.02, 31.65,
30.14, 23.66; HRMS (ESI+): calcd for C24H26N7O2S+ [M+H]+: 476.1869, found: 476.1858.
HPLC purity: 85%.
(S)-amino(2-((3-(4-((4-(4-ethoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20p). Synthesized by General Procedure
3E. 76%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.15 (d, J = 8.2 Hz, 2H), 7.81 – 7.65 (m,
4H), 7.04 – 6.96 (m, 2H), 4.58 – 4.49 (m, 1H), 4.09 (q, J = 7.0 Hz, 2H), 3.62 – 3.51 (m, 1H),
3.49 – 3.42 (m, 1H), 3.35 – 3.30 (m, 2H), 2.35 – 2.22 (m, 1H), 2.18 – 1.97 (m, 3H), 1.41 (t, J =
7.0 Hz, 3H); 13C NMR (101 MHz, CD3OD) δ 178.18, 174.32, 168.95, 161.43, 156.42, 130.58,
130.00, 129.43, 128.88, 121.55, 118.52, 115.95, 115.22, 81.41, 64.74, 57.18, 31.59, 30.06, 23.59,
15.09; HRMS (ESI+): calcd for C25H28N7O2S [M+H]+: 490.2025, found: 490.2024.
(S)-amino(2-((3-(4-((4-(3-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20q). Synthesized by General Procedure
3E. 86%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.29 (s, 1H), 8.12 (d, J = 6.5 Hz, 2H),
7.78 – 7.66 (m, 2H), 7.40 – 7.27 (m, 3H), 6.97 (d, J = 7.1 Hz, 1H), 6.82 (s, 1H), 4.58 – 4.45 (m,
1H), 3.84 (s, 3H), 3.59 – 3.49 (m, 1H), 3.50 – 3.37 (m, 1H), 3.34 – 3.28 (m, 2H), 2.35 – 2.20 (m,
1H), 2.15 – 1.93 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 178.16, 168.87, 168.24, 161.59,
156.38, 145.44, 142.77, 132.82, 131.27, 130.05, 124.52, 121.75, 119.71, 116.02, 113.06, 57.21,
56.09, 31.69, 30.19, 23.69; HRMS (ESI+): calcd for C24H26N7O2S+ [M+H]+: 476.1869, found:
476.1847.

211
(S)-amino(2-((3-(4-((4-(4-(trifluoromethoxy)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-
5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20r). Synthesized by General Procedure
3E. 90%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.06 (d, J = 8.3 Hz, 4H), 7.92 (d, J =
8.3 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.24 (s, 1H), 4.61 – 4.50 (m, 1H), 3.63 – 3.55 (m, 1H),
3.54 – 3.45 (m, 2H), 3.33 – 3.21 (m, 1H), 2.20 – 2.14 (m, 1H), 2.14 – 2.02 (m, 3H); 13C NMR
(126 MHz, CD3OD) δ 177.73, 169.41, 164.67, 156.47, 151.16, 149.87, 145.53, 135.35, 129.40,
128.63, 122.19, 120.33, 118.09, 104.99, 57.25, 31.62, 30.08, 23.59; 19F NMR (470 MHz,
CD3OD) δ -59.47 (s, 3F); HRMS (ESI+): calcd for C24H23F3N7O2S [M+H]+: 531.1664, found:
531.1672. HPLC purity: 81%.
(S)-amino(2-((3-(4-((4-(3-(trifluoromethoxy)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-
5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20s). Synthesized by General Procedure
3E. 100%, light yellow solid. 1H NMR (400 MHz, v) δ 8.04 (d, J = 8.5 Hz, 2H), 7.96 – 7.83 (m,
4H), 7.51 (t, J = 8.0 Hz, 1H), 7.29 (s, 1H), 7.25 – 7.19 (m, 1H), 4.58 – 4.50 (m, 1H), 3.60 – 3.52
(m, 1H), 3.51 – 3.43 (m, 1H), 2.34 – 2.23 (m, 1H), 2.19 – 1.99 (m, 3H); 13C NMR (101 MHz,
CD3OD) δ 177.75, 169.36, 164.61, 156.44, 150.98, 150.87, 145.47, 138.41, 131.34, 129.38,
125.52, 120.85, 120.37, 119.46, 118.07, 105.73, 57.23, 31.60, 30.07, 23.58; 19F NMR (376 MHz,
CD3OD) δ -59.32 (s, 3F); HRMS (ESI+): calcd for C24H23F3N7O2S [M+H]+: 531.1664, found:
531.1712.
(S)-amino(2-((3-(4-((4-(3,4-dimethylphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20t). Synthesized by General Procedure
3E. 89%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.28 (s, 1H), 8.14 (d, J = 7.9 Hz, 2H),
7.73 (d, J = 8.2 Hz, 2H), 7.55 (s, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 6.82 (s,
1H), 4.60 – 4.48 (m, 1H), 3.61 – 3.40 (m, 2H), 3.37 – 3.30 (m, 1H), 2.33 (s, 3H), 2.30 (s, 3H),

212
2.19 – 1.94 (m, 3H), 1.40 – 1.12 (m, 1H); 13C NMR (101 MHz, CD3OD) δ 178.21, 168.91,
168.29, 156.42, 145.71, 142.81, 139.57, 138.58, 135.47, 131.26, 130.04, 129.15, 128.37, 124.92,
124.60, 121.76, 57.18, 31.62, 30.10, 23.62, 19.89, 19.69; HRMS (ESI+): calcd for C25H28N7O3S+
[M+H]+: 474.2076, found: 474.2077.
(S)-amino(2-((3-(4-((4-(3,4-dimethoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20u). Synthesized by General Procedure
3E. 100%, orange solid. 1H NMR (400 MHz, CD3OD) δ 8.17 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.6
Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.60 – 7.54 (m, 1H), 7.33 (d, J = 6.9
Hz, 2H), 7.06 (d, J = 8.5 Hz, 1H), 4.58 – 4.49 (m, 1H), 3.91 (s, 3H), 3.87 (s, 3H), 3.60 – 3.51 (m,
1H), 3.49 – 3.42 (m, 1H), 3.33 (d, J = 6.0 Hz, 2H), 2.34 – 2.23 (m, 1H), 2.17 – 2.01 (m, 3H); 13C
NMR (101 MHz, CD3OD) δ 178.31, 168.99, 168.80, 156.42, 152.07, 150.90, 143.92, 142.18,
130.17, 129.46, 125.53, 123.36, 122.47, 120.66, 118.81, 113.03, 112.57, 111.17, 57.17, 56.76,
56.53, 31.60, 30.09, 23.60; HRMS (ESI+): calcd for C25H28N7OS+ [M+H]+: 506.1969, found:
506.1948.
(S)-amino(2-((3-(4-((4-(3-chloro-4-methoxyphenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-
5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20v). Synthesized by General Procedure
3E. 100%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.07 (d, J = 8.5 Hz, 2H), 7.90 (s, 1H),
7.83 (d, J = 8.5 Hz, 2H), 7.80 (d, J = 8.6 Hz, 1H), 7.15 (d, J = 8.6 Hz, 1H), 7.10 (s, 1H), 4.57 –
4.52 (m, 1H), 3.94 (s, 3H), 3.60 – 3.52 (m, 1H), 3.50 – 3.42 (m, 2H), 2.33 – 2.25 (m, 1H), 2.19 –
1.99 (m, 3H); 13C NMR (126 MHz, CD3OD) δ 177.87, 169.26, 165.70, 156.49, 144.75, 129.59,
128.86, 128.54, 126.89, 123.76, 121.61, 119.20, 113.56, 57.24, 56.77, 31.63, 30.10, 23.60;
HRMS (ESI+): calcd for C24H25ClN7O2S [M+H]+: 510.1479, found: 510.1495.

213
(S)-amino(2-((3-(4-((4-(3,4-difluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20w). Synthesized by General Procedure
3E. 100%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.09 (d, J = 8.2 Hz, 2H), 7.91 – 7.79
(m, 3H), 7.77 – 7.70 (m, 1H), 7.35 (q, J = 8.9 Hz, 1H), 7.24 (s, 1H), 4.63 – 4.53 (m, 1H), 3.64 –
3.56 (m, 1H), 3.56 – 3.46 (m, 1H), 2.40 – 2.24 (m, 1H), 2.23 – 2.03 (m, 3H13C NMR (101 MHz,
CD3OD) δ 177.84, 169.26, 165.51, 156.44, 148.70, 144.82, 132.67, 129.56, 123.68 (dd, 4JCF =
4.0 Hz), 123.64 (dd, 4JCF = 4.0 Hz), 123.62 (dd, 4JCF = 4.0 Hz), 123.58 (dd, 4JCF = 4.0 Hz),
121.39, 118.99, 118.70 (d, 2JCF = 18.2 Hz), 118.52 (d, 2JCF = 18.2 Hz), 116.18 (d, 2JCF = 19.2
Hz), 115.99 (d, 2JCF = 19.2 Hz), 57.25, 31.66, 30.13, 23.63; 19F NMR (376 MHz, CD3OD) δ -
140.17 to -140.55 (m, 1F), -141.28 to -141.63 (m, 1F); HRMS (ESI+): calcd for C23H22F2N7OS
[M+H]+: 482.1575, found: 482.1571.
(S)-amino(2-((3-(4-((4-(3,4-dichlorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20x). Synthesized by General Procedure
3E. 100%, white solid. 1H NMR (500 MHz, CD3OD) δ 8.07 (d, J = 8.5 Hz, 2H), 7.90 (s, 1H),
7.83 (d, J = 8.5 Hz, 2H), 7.80 (d, J = 8.6 Hz, 1H), 7.15 (d, J = 8.6 Hz, 1H), 7.10 (s, 1H), 4.57 –
4.52 (m, 1H), 3.56 – 3.49 (m, 1H), 3.48 – 3.39 (m, 1H), 2.33 – 2.25 (m, 1H), 2.19 – 1.99 (m,
3H); 13C NMR (101 MHz, CD3OD) δ 177.85, 169.26, 156.45, 146.18, 144.76, 135.36, 133.91,
133.59, 132.58, 131.14, 129.55, 128.49, 121.49, 119.08, 57.23, 54.79, 31.62, 30.08, 23.59;
HRMS (ESI+): calcd for C23H22Cl2N7OS [M+H]+: 514.0984, found: 514.0973.
(S)-amino(2-((3-(4-((4-(4-fluoro-3-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20y). Synthesized by General
Procedure 3E. 100%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.23 – 8.10 (m, 2H), 8.04
(d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.2 Hz, 2H), 7.39 (t, J = 9.4 Hz, 1H), 7.29 (s, 1H), 4.55 – 4.47

214
(m, 1H), 3.57 – 3.48 (m, 1H), 3.48 – 3.38 (m, 1H), 2.31 – 2.21 (m, 1H), 2.14 – 1.95 (m, 3H); 13C
NMR (126 MHz, CD3OD) δ 177.94, 169.17, 166.32, 161.64 (d, 1JCF = 205.0 Hz), 159.61 (d, 1JCF
= 205.0 Hz), 156.44, 147.17, 144.34, 133.33 (d, 3JCF = 7.1 Hz), 133.26 (d, 3JCF = 7.1 Hz), 131.44
(d, 4JCF = 2.0 Hz), 131.42 (d, 4JCF = 2.0 Hz), 129.64, 127.31 (q, 1JCF = 218.2 Hz), 125.97 (q, 4JCF
= 4.0 Hz), 125.93 (q, 4JCF = 4.0 Hz), 125.90 (q, 4JCF = 4.0 Hz), 125.86 (q, 4JCF = 4.0 Hz), 125.14
(q, 1JCF = 218.2 Hz), 122.98 (q, 1JCF = 218.2 Hz), 122.16, 120.82 (q, 1JCF = 218.2 Hz), 119.63,
119.36 (q, 3JCF = 11.1 Hz), 119.25 (q, 3JCF = 11.1 Hz), 118.67 (d, 3JCF = 17.2 Hz), 118.50 (d, 3JCF
= 17.2 Hz), 57.22, 31.61, 30.10, 23.60; 19F NMR (376 MHz, CD3OD) δ -62.93 (d, J = 12.8 Hz,
3F), -117.55 to -117.83 (m, 1F); HRMS (ESI+): calcd for C24H22F4N7OS [M+H]+: 532.1543,
found: 532.1538.
(S)-amino(2-((3-(4-((4-(3,5-difluorophenyl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20z). Synthesized by General Procedure
3E. 100%, off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.03 (d, J = 8.4 Hz, 2H), 7.86 (d, J =
8.4 Hz, 2H), 7.56 – 7.48 (m, 2H), 7.31 (s, 1H), 6.91 – 6.83 (m, 1H), 4.59 – 4.48 (m, 1H), 3.71 (d,
J = 2.8 Hz, 1H), 3.60 – 3.51 (m, 1H), 3.49 – 3.41 (m, 1H), 3.16 – 3.10 (m, 1H), 2.34 – 2.22 (m,
1H), 2.18 – 1.97 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 177.73, 169.35, 166.08 (dd, 1JCF =
246.5 Hz), 165.95 (dd, 1JCF = 246.5 Hz), 164.57, 163.63 (dd, 1JCF = 246.5 Hz), 163.50 (dd, 1JCF =
246.5 Hz), 156.44, 150.23 (d, 4JCF = 4.0 Hz), 150.19 (d, 4JCF = 4.0 Hz), 145.35, 139.76 (dd, 3JCF
= 9.1 Hz), 139.66(dd, 3JCF = 9.1 Hz), 139.56 (dd, 3JCF = 9.1 Hz), 129.40, 120.43, 118.09, 109.79
(d, 2JCF = 27.3 Hz), 109.71 (d, 3JCF = 11.1 Hz), 109.59 (d, 3JCF = 11.1 Hz), 109.52 (d, 2JCF = 27.3
Hz), 106.65, 103.70 (dd, 2JCF = 26.3 Hz), 103.44 (dd, 3JCF = 26.3 Hz), 103.18 (dd, 2JCF = 26.3
Hz), 71.34*, 67.91*, 57.20, 40.56, 31.61, 30.05, 23.59; 19F NMR (376 MHz, CD3OD) δ -111.61

215
to -111.72 (m, 2F); HRMS (ESI+): calcd for C23H22F2N7OS [M+H]+: 482.1575, found:
482.1534.
(S)-amino(2-((3-(4-((4-(3-fluoro-5-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20aa). Synthesized by General
Procedure 3E. 100%, yellow solid. 1H NMR (500 MHz, CD3OD) δ 8.23 – 8.10 (m, 2H), 8.04 (d,
J = 8.2 Hz, 2H), 7.79 (d, J = 8.2 Hz, 2H), 7.39 (t, J = 9.4 Hz, 1H), 7.29 (s, 1H), 4.55 – 4.47 (m,
1H), 3.57 – 3.48 (m, 1H), 3.48 – 3.38 (m, 1H), 2.31 – 2.21 (m, 1H), 2.14 – 1.95 (m, 3H); 13C
NMR (126 MHz, CD3OD) δ 177.77, 169.32, 165.57, 164.91, 163.12, 156.44, 149.32, 145.21,
139.62, (d, 3JCF = 9.1 Hz) 139.53 (d, 3JCF = 9.1 Hz), 134.07 (d, 3JCF = 8.1 Hz), 133.99 (d, 3JCF =
8.1 Hz), 133.74 (d, 3JCF = 9.1 Hz), 133.65 (d, 3JCF = 9.1 Hz), 129.42, 126.24, 123.51, 120.70,
119.55 (q, 4JCF = 3.0 Hz), 119.52 (q, 4JCF = 3.0 Hz), 119.51 (q, 4JCF = 3.0 Hz), 119.48 (q, 4JCF =
3.0 Hz), 118.30, 117.37 (d, 3JCF = 17.2 Hz), 117.13 (d, 3JCF = 17.2 Hz), 112.44, 112.17, 107.28,
57.25, 31.64, 30.12, 23.61; 19F NMR (376 MHz, CD3OD) δ -64.34 (s, 1F) , -112.36 (t, J = 9.1
Hz, 1F); HRMS (ESI+): calcd for C24H22F4N7OS [M+H]+: 532.1543, found: 532.1520.
(S)-amino(2-((3-(4-((4-(3,5-bis(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20bb). Synthesized by General
Procedure 3E. 100%, white solid. 1H NMR (400 MHz, CD3OD) δ 8.51 (s, 2H), 8.07 (d, J = 8.4
Hz, 2H), 7.93 – 7.86 (m, 3H), 7.60 (s, 1H), 4.62 – 4.54 (m, 1H), 3.64 – 3.55 (m, 1H), 3.55 – 3.46
(m, 1H), 2.39 – 2.27 (m, 1H), 2.21 – 2.03 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 178.12,
168.96, 167.94, 156.43, 145.08, 143.15, 133.46, 133.42, 132.82 (q, 2JCF = 32.3 Hz), 132.50 (q,
2JCF = 32.3 Hz), 132.18 (q, 2JCF = 32.3 Hz), 131.85 (q, 2JCF = 32.3 Hz), 131.02, 130.99, 129.91,
126.79, 126.64 (q, 4JCF = 3.0 Hz), 126.61 (q, 4JCF = 3.0 Hz), 126.57 (q, 4JCF = 3.0 Hz), 126.53 (q,
4JCF = 3.0 Hz), 124.09, 124.06, 124.01, 123.98, 121.24, 57.18, 31.60, 30.09, 23.59; 19F NMR

216
(376 MHz, CD3OD) δ -64.49 (s, 3F); HRMS (ESI+): calcd for C25H22F6N7OS [M+H]+:
582.1511, found: 582.1516.
(S)-amino(2-((3-(4-((4-(2-fluoro-5-(trifluoromethyl)phenyl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20cc). Synthesized by General
Procedure 3E. 100%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.57 – 8.52 (m, 1H), 8.11 –
8.03 (m, 2H), 7.90 (d, J = 8.6 Hz, 2H), 7.73 – 7.66 (m, 1H), 7.50 – 7.40 (m, 2H), 4.62 – 4.54 (m,
1H), 3.65 – 3.56 (m, 1H), 3.56 – 3.46 (m, 1H), 2.40 – 2.27 (m, 1H), 2.22 – 2.04 (m, 3H); 13C
NMR (101 MHz, CD3OD) δ 183.96, 177.78, 175.04, 169.33, 164.57 (d, 1JCF = 256.5 Hz),
164.00, 162.03 (d, 1JCF = 256.5 Hz), 156.45, 145.34, 144.51, 129.37, 128.20 (q, 1JCF = 112.1 Hz),
127.09 (q, 1JCF = 112.1 Hz), 124.59 (d, 3JCF = 12.1 Hz), 124.47 (d, 3JCF = 12.1 Hz), 124.32,
123.78, 120.62, 118.54, 118.20, 117.98, 110.58, 106.88, 103.88, 57.26, 31.61, 30.11, 23.58; 19F
NMR (376 MHz, CD3OD) δ -63.69 (s, 3F), -110.28 to -110.99 (m, 1F); HRMS (ESI+): calcd for
C24H22F4N7OS [M+H]+: 532.1543, found: 532.1520. HPLC purity: 85% pure.
(S)-(2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)(amino)methaniminium chloride (3.20dd). Synthesized by General
Procedure 3E. 100%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.11 (d, J = 8.5 Hz,
2H), 8.03 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.73 (dd, J = 14.9, 7.9 Hz, 4H), 7.50 (t, J
= 7.6 Hz, 2H), 7.39 (t, J = 7.3 Hz, 1H), 7.26 (s, 1H), 4.65 – 4.51 (m, 1H), 3.65 – 3.56 (m, 1H),
3.55 – 3.47 (m, 1H), 2.41 – 2.27 (m, 1H), 2.24 – 2.02 (m, 4H); 13C NMR (101 MHz, CD3OD) δ
176.60, 167.69, 165.59, 155.03, 146.50, 142.48, 141.43, 140.13, 130.85, 128.55, 128.38, 127.30,
126.99, 126.46, 126.36, 121.61, 119.03, 55.78, 30.18, 28.66, 22.17; HRMS (ESI+): calcd for
C29H28N7OS [M+H]+: 522.2076, found: 522.2046.

217
(S)-amino(2-((3-(4-((4-(benzofuran-2-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.20ee). Synthesized by General Procedure
3E. 100%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.20 (d, J = 6.5 Hz, 1H), 7.98 (t, J = 7.7
Hz, 3H), 7.79 (d, J = 7.9 Hz, 2H), 7.49 (d, J = 7.5 Hz, 2H), 7.37 – 7.25 (m, 2H), 4.52 – 4.42 (m,
1H), 3.52 – 3.44 (m, 1H), 3.43 – 3.35 (m, 2H), 2.30 – 2.13 (m, 1H), 2.13 – 1.90 (m, 3H); 13C
NMR (101 MHz, CD3OD) δ 177.93, 168.95, 166.98, 156.94, 156.73, 156.32, 145.31, 143.60,
139.64, 129.74, 126.42, 126.20, 126.02, 124.57, 123.13, 121.58, 120.62, 115.61, 112.61, 57.14,
31.56, 30.08, 23.56; HRMS (ESI+): calcd for C25H24N7O2S [M+H]+: 486.1712, found: 486.1683.
tert-butyl (S)-2-((3-(4-((4-([1,1'-biphenyl]-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.21a). Synthesized by General Procedure 3D. 30%, yellow
amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.12 – 8.02 (m, 3H), 7.85 (dt, J = 7.7, 1.5 Hz,
1H), 7.77 (s, 1H), 7.69 – 7.62 (m, 2H), 7.59 – 7.43 (m, 6H), 7.40 – 7.33 (m, 1H), 6.97 (s, 1H),
4.40 – 4.22 (m, 1H), 3.52 – 3.27 (m, 3H), 3.17 – 2.98 (m, 1H), 2.14 – 2.02 (m, 1H), 1.95 – 1.78
(m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.20, 167.90, 162.96, 154.37, 151.61,
142.82, 141.84, 141.22, 135.00, 129.26, 128.96, 128.92, 127.54, 127.37, 127.04, 125.21, 125.15,
120.66, 117.40, 117.29, 103.13, 80.23, 79.78*, 55.35, 55.22*, 46.90*, 46.50*, 31.93, 31.15*,
30.98*, 30.21*, 29.85*, 28.63, 23.71*, 22.91*; HRMS (ESI+): calcd for C33H34N5O3S [M+H]+:
580.2382, found: 580.2384.
tert-butyl (S)-2-((3-(4-((4-(4'-fluoro-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.21b). Synthesized by General Procedure 3D.
34%, off-white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.3 Hz, 2H), 7.93 (d,
J = 8.0 Hz, 2H), 7.70 – 7.65 (br s, 1H), 7.63 – 7.50 (m, 6H), 7.14 (t, J = 8.7 Hz, 2H), 6.95 (s,
1H), 4.39 – 4.22 (m, 1H), 3.50 – 3.27 (m, 3H), 3.15 – 2.97 (m, 1H), 2.13 – 2.01 (m, 1H), 1.95 –

218
1.79 (m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.22, 168.00, 163.88 (d, 1JCF =
247.5 Hz), 162.96, 161.43 (d, 1JCF = 247.5 Hz), 154.37, 151.25, 150.57, 142.75, 139.84, 136.93
(d, 4JCF = 3.0 Hz), 136.90 (d, 4JCF = 3.0 Hz), 133.53, 133.45, 131.92, 128.95, 128.84, 128.72 (d,
3JCF = 8.1 Hz), 128.64 (d, 3JCF = 8.1 Hz), 127.80, 127.35, 126.71, 122.07, 120.73, 117.39,
117.35, 115.92 (d, 2JCF = 21.2 Hz), 115.71 (d, 2JCF = 21.2 Hz), 103.27, 102.96, 80.23, 55.34,
46.90*, 46.51*, 31.93*, 31.15*, 31.00*, 30.23*, 29.85*, 28.64*, 23.71*, 22.92*; 19F NMR (376
MHz, CDCl3) δ -114.71 to -116.15 (m, 1F); HRMS (ESI+): calcd for C33H33FN5O3S [M+H]+:
598.2288, found: 598.2234.
tert-butyl (S)-2-((3-(4-((4-(4'-(trifluoromethyl)-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.21c). Synthesized by General
Procedure 3D. 37%, yellow amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.2 Hz,
2H), 7.97 (d, J = 8.0 Hz, 1H), 7.78 – 7.54 (m, 8H), 6.98 (s, 1H), 4.40 – 4.21 (m, 1H), 3.52 – 3.27
(m, 3H), 3.17 – 2.97 (m, 1H), 2.13 – 2.00 (m, 1H), 1.96 – 1.78 (m, 3H), 1.48 (s, 9H); 13C NMR
(101 MHz, CDCl3) δ 177.41, 167.99, 163.01, 154.38, 151.06, 144.29, 142.73, 139.28, 134.41,
129.69 (q, 2JCF = 32.3 Hz), 129.37 (q, 2JCF = 32.3 Hz), 128.98, 128.83 (q, 2JCF = 32.3 Hz),
127.67, 127.54, 127.37, 127.23, 126.84, 125.91 (q, 4JCF = 4.0 Hz), 125.87 (q, 4JCF = 4.0 Hz),
125.79 (q, 4JCF = 4.0 Hz), 117.39, 103.40, 80.23*, 79.81*, 55.30, 46.90*, 46.50*, 31.93*, 31.15*,
30.23*, 29.83*, 28.63, 23.71*, 22.92*; 19F NMR (376 MHz, CDCl3) δ -62.43 (s, 3F); HRMS
(ESI+): calcd for C34H32F3N5NaO3S [M+H]+: 670.2075, found: 670.2068.
tert-butyl (S)-2-((3-(4-((4-(3'-(trifluoromethyl)-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.21d). Synthesized by General
Procedure 3D. 35%, tan amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.12 – 8.03 (m, 2H),
7.98 (d, J = 7.9 Hz, 2H), 7.88 (s, 1H), 7.81 (d, J = 7.5 Hz, 1H), 7.70 – 7.51 (m, 6H), 6.98 (s, 1H),

219
4.39 – 4.23 (m, 1H), 3.52 – 3.27 (m, 3H), 3.15 – 2.96 (m, 1H), 2.13 – 2.01 (m, 1H), 1.95 – 1.79
(m, 3H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.08, 167.78, 162.84, 154.24, 150.90,
142.60, 141.42, 139.12, 134.10, 131.77, 131.65, 131.34 (q, 2JCF = 30.3 Hz), 131.02 (q, 2JCF =
30.3 Hz), 130.22, 129.27, 128.81, 128.68, 127.64 (q, 1JCF = 155.5 Hz), 127.41, 126.69, 126.10
(q, 1JCF = 155.5 Hz), 125.51, 123.99 (q, 2JCF = 30.3 Hz), 123.69 (q, 2JCF = 30.3 Hz), 122.81,
120.63, 117.20, 103.18, 80.10, 55.15, 46.74*, 46.35*, 31.75*, 30.98*, 30.07*, 28.47, 23.54*,
22.76*; 19F NMR (376 MHz, CDCl3) δ -62.60 (s, 3F); HRMS (ESI+): calcd for C34H33F3N5O3S
[M+H]+: 648.2256, found: 648.2209.
tert-butyl (S,Z)-((2-((3-(4-((4-([1,1'-biphenyl]-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-
5-yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.22a). Synthesized
by General Procedure 3E. 68%, off-white oily solid. 1H NMR (500 MHz, CDCl3) δ 10.39 (s,
1H), 8.09 (s, 1H), 8.02 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 7.5 Hz, 1H), 7.66 (d, J = 7.4 Hz, 2H),
7.59 – 7.53 (m, 4H), 7.52 – 7.44 (m, 3H), 7.37 (t, J = 7.3 Hz, 1H), 6.96 (s, 1H), 4.82 – 4.72 (m,
1H), 3.81 – 3.62 (m, 3H), 3.57 – 3.43 (m, 1H), 3.09 (dd, 1H), 2.36 – 2.26 (m, 1H), 1.95 – 1.87
(m, 1H), 1.85 – 1.73 (m, 2H), 1.48 (d, J = 14.1 Hz, 18H); 13C NMR (126 MHz, CDCl3) δ 176.87,
167.90, 162.91, 162.64, 154.28, 151.66, 150.48, 142.80, 141.84, 141.27, 135.10, 129.26, 128.95,
127.54, 127.38, 127.00, 125.26, 125.12, 120.74, 117.20, 103.03, 82.17, 79.61, 56.62, 50.29,
30.88, 30.47, 28.38, 28.25, 24.51; HRMS (ESI+): calcd for C39H44N7O5S [M+H]+: 722.3125,
found: 722.3107.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4'-fluoro-[1,1'-biphenyl]-4-yl)thiazol-
2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.22b).
Synthesized by General Procedure 3E. 50%, off-white oily solid. 1H NMR (400 MHz, CDCl3) δ
10.39 (s, 1H), 8.03 (d, J = 8.1 Hz, 2H), 7.94 (d, J = 8.0 Hz, 2H), 7.89 – 7.78 (m, 1H), 7.65 – 7.55

220
(m, 5H), 7.13 (t, J = 8.5 Hz, 2H), 6.94 (s, 1H), 4.84 – 4.71 (m, 1H), 3.81 – 3.61 (m, 2H), 3.58 –
3.43 (m, 1H), 3.09 (dd, J = 15.4, 8.7 Hz, 1H), 2.37 – 2.24 (m, 1H), 1.96 – 1.87 (m, 1H), 1.86 –
1.72 (m, 2H), 1.54 – 1.41 (m, 18H); 13C NMR (126 MHz, CDCl3) δ 176.85, 167.88, 163.65 (d,
1JCF = 100 Hz), 162.89, 162.65 (d, 1JCF = 100 Hz), 154.31, 151.29, 150.51, 142.78, 139.81,
136.97, 133.61, 131.92, 128.97, 128.71 (d, 4JCF = 7.1 Hz), 128.64 (d, 4JCF = 7.1 Hz), 127.82,
127.35 (d, 2JCF = 63.6 Hz), 126.72 (d, 2JCF = 63.6 Hz), 120.77, 117.23, 115.91 (d, 3JCF = 17.2
Hz), 115.74 (d, 3JCF = 17.2 Hz), 103.17, 102.86, 82.17, 79.64, 56.61, 50.31, 30.92, 30.46, 29.85,
28.32, 28.17, 24.56; 19F NMR (376 MHz, CDCl3) δ -115.56 to -115.66 (m, 1F); HRMS (ESI+):
calcd for C39H43FN7O5S [M+H]+: 740.3030, found: 740.2979.
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4'-(trifluoromethyl)-[1,1'-biphenyl]-
4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate
(3.22c). Synthesized by General Procedure 3E. 75%, off-white oily solid. 1H NMR (400 MHz,
CDCl3) δ 10.32 (s, 1H), 8.09 (d, J = 8.3 Hz, 2H), 7.98 (d, J = 7.9 Hz, 2H), 7.81 – 7.63 (m, 6H),
7.58 (d, J = 8.3 Hz, 2H), 7.46 (s, 1H), 6.98 (s, 1H), 4.79 (dt, J = 11.9, 5.8 Hz, 1H), 3.79 – 3.62
(m, 2H), 3.57 – 3.44 (m, 1H), 3.15 (dd, J = 15.4, 8.3 Hz, 1H), 2.33 – 2.22 (m, 1H), 1.98 – 1.91
(m, 1H), 1.89 – 1.74 (m, 2H), 1.53 – 1.44 (m, 18H); 13C NMR (126 MHz, CDCl3) δ 176.80,
167.74, 162.82, 162.54, 154.05, 150.95, 148.99, 144.17, 142.55, 139.09, 134.34, 129.52, 128.86,
127.50, 127.21, 126.69, 125.78 (q, 4JCF = 2.0 Hz), 125.75 (q, 4JCF = 2.0 Hz), 125.73 (q, 4JCF = 2.0
Hz), 125.70 (q, 4JCF = 2.0 Hz), 121.05 (q, 4JCF = 2.0 Hz), 120.79, 117.15, 103.17, 101.06, 83.41,
81.99, 56.47, 50.10, 30.69, 30.30, 29.69, 28.15, 27.98; 19F NMR (376 MHz, CDCl3) δ -62.44 (s,
3F); HRMS (ESI+): calcd for C40H43F3N7O5S [M+H]+: 790.2998, found: 790.2989.

221
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(3'-(trifluoromethyl)-[1,1'-biphenyl]-
4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate
(3.22d). Synthesized by General Procedure 3E. 37%, off-white oily solid. 1H NMR (400 MHz,
CDCl3) δ 10.39 (s, 1H), 8.06 – 7.94 (m, 4H), 7.87 (s, 1H), 7.80 (d, J = 7.6 Hz, 1H), 7.68 – 7.52
(m, 6H), 6.96 (s, 1H), 4.82 – 4.73 (m, 1H), 3.82 – 3.62 (m, 2H), 3.57 – 3.45 (m, 1H), 3.09 (dd, J
= 15.5, 8.8 Hz, 1H), 2.31 (dt, J = 11.5, 6.7 Hz, 1H), 1.97 – 1.88 (m, 1H), 1.84 – 1.75 (m, 1H),
1.71 – 1.62 (m, 2H), 1.55 – 1.42 (m, 18H); 13C NMR (126 MHz, CDCl3) δ 176.83, 167.87,
163.01, 151.07, 150.47, 142.78, 141.61, 139.21, 134.37, 131.91, 131.75 (q, 2JCF = 25.3 Hz),
131.49 (q, 2JCF = 25.3 Hz), 131.24 (q, 2JCF = 25.3 Hz), 130.98 (q, 2JCF = 25.3 Hz), 130.36,
129.42, 128.96, 128.81 (q, 1JCF = 100.0 Hz), 127.81 (q, 1JCF = 100.0 Hz), 127.55, 126.86, 126.26,
125.41, 124.17 (q, 4JCF = 3.0 Hz), 124.14 (q, 4JCF = 3.0 Hz), 124.11 (q, 4JCF = 3.0 Hz), 124.09 (q,
4JCF = 3.0 Hz), 123.90 (q, 4JCF = 3.0 Hz), 123.87 (q, 4JCF = 3.0 Hz), 123.84 (q, 4JCF = 3.0 Hz),
123.81 (q, 4JCF = 3.0 Hz), 123.25, 120.75, 117.24, 117.16, 105.35, 103.20, 82.19, 79.67, 56.61,
50.32, 30.92, 30.45, 28.36, 28.28, 28.16; 19F NMR (376 MHz, CDCl3) δ -62.63 (s, 3F); HRMS
(ESI+): calcd for C40H43F3N7O5S [M+H]+: 790.2998, found: 790.2997.
(S)-(2-((3-(4-((4-([1,1'-biphenyl]-3-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)(amino)methaniminium chloride (3.23a). Synthesized by General
Procedure 3E. 93%, light yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.19 – 8.14 (m, 1H), 8.02
(d, J = 8.7 Hz, 2H), 7.95 – 7.86 (m, 3H), 7.73 – 7.66 (m, 2H), 7.60 – 7.55 (m, 1H), 7.53 – 7.44
(m, 3H), 7.40 – 7.33 (m, 1H), 7.25 (s, 1H), 7.22 (s, 1H), 4.57 – 4.50 (m, 1H), 3.55 (ddd, J = 9.8,
8.0, 4.2 Hz, 1H), 3.51 – 3.41 (m, 1H), 2.36 – 2.19 (m, 1H), 2.19 – 1.97 (m, 3H); 13C NMR (101
MHz, CD3OD) δ 177.73, 169.37, 164.53, 156.44, 152.44, 145.59, 142.93, 142.47, 136.66,
130.19, 129.92, 129.40, 128.48, 128.08, 127.47, 126.06, 125.64, 120.26, 118.04, 104.46, 57.23,

222
31.60, 30.07, 23.58; HRMS (ESI+): calcd for C29H28N7OS [M+H]+: 522.2076, found: 522.2072.
HPLC purity: 88%.
(S)-amino(2-((3-(4-((4-(4'-fluoro-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.23b). Synthesized by General
Procedure 3E. 90%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.09 (d, J = 8.5 Hz, 2H),
7.94 (d, J = 7.8 Hz, 2H), 7.84 (d, J = 8.5 Hz, 3H), 7.72 – 7.63 (m, 3H), 7.23 (s, 1H), 7.22 – 7.15
(m, 2H), 4.57 – 4.49 (m, 1H), 3.58 – 3.50 (m, 1H), 3.49 – 3.42 (m, 1H), 3.33 – 3.31 (m, 2H),
2.35 – 2.22 (m, 1H), 2.18 – 1.97 (m, 3H); 13C NMR (126 MHz, CD3OD) δ 177.96, 169.20,
167.04, 166.33 (d, 1JCF = 128.3 Hz), 165.06 (d, 1JCF = 128.3 Hz), 156.46, 149.01, 144.38, 141.46,
138.03, 132.86, 129.76, 129.70, 129.54, 128.90, 128.26 (d, 2JCF = 49.5 Hz), 127.77 (d, 2JCF =
49.5 Hz), 119.81, 118.95, 116.73 (d, 3JCF = 17.3 Hz), 116.56 (d, 3JCF = 17.3 Hz), 57.22, 31.62,
30.08, 28.13, 23.59; 19F NMR (376 MHz, CD3OD) δ -117.18 to -117.54 (m, 1F); HRMS (ESI+):
calcd for C29H27FN7OS [M+H]+: 540.1982, found: 540.1978.
(S)-amino(2-((3-(4-((4-(4'-(trifluoromethyl)-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.23c). Synthesized by
General Procedure 3E. 100%, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.11 – 7.99 (m,
3H), 7.96 – 7.83 (m, 4H), 7.80 – 7.73 (m, 2H), 7.58 (d, J = 8.4 Hz, 1H), 7.28 – 7.18 (m, 2H),
4.58 – 4.51 (m, 1H), 3.59 – 3.51 (m, 1H), 3.51 – 3.45 (m, 1H), 2.36 – 2.25 (m, 1H), 2.19 – 1.99
(m, 3H); 13C NMR (126 MHz, CD3OD) δ 177.72, 169.41, 156.48, 152.04, 151.44, 145.62,
145.54, 139.94, 136.20, 135.32, 132.74, 131.64, 129.40, 128.80, 128.45, 128.38, 127.72, 126.84
(q, 4JCF = 3.0 Hz), 126.81 (q, 4JCF = 3.0 Hz), 126.78 (q, 4JCF = 3.0 Hz), 126.75 (q, 4JCF = 3.0 Hz),
122.39, 120.29, 118.08, 104.78, 57.26, 31.63, 30.09, 23.60; 19F NMR (376 MHz, CD3OD) δ -
63.98 (s, 3F): calcd for C30H27F3N7OS [M+H]+: 590.1950, found: 590.1954.

223
(S)-amino(2-((3-(4-((4-(3'-(trifluoromethyl)-[1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-
1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.23d). Synthesized by
General Procedure 3E. 100%, off-white solid. 1H NMR (400 MHz, CD3OD) δ 8.16 – 8.03 (m,
2H), 8.02 – 7.97 (m, 1H), 7.94 (d, J = 4.1 Hz, 2H), 7.88 – 7.82 (m, 2H), 7.79 – 7.75 (m, 2H),
7.68 – 7.65 (m, 2H), 4.57 – 4.50 (m, 1H), 3.60 – 3.50 (m, 1H), 3.50 – 3.41 (m, 1H), 3.34 – 3.31
(m, 1H), 2.34 – 2.22 (m, 1H), 2.17 – 2.00 (m, 4H); 13C NMR (101 MHz, CD3OD) δ 177.96,
169.17, 166.36, 156.43, 148.71, 144.30, 142.73, 140.75, 133.87, 132.47, 131.64, 130.89, 129.70,
128.53, 127.96, 127.34, 127.06, 125.26 (q, 4JCF = 4.0 Hz), 125.22 (q, JCF = 4.0 Hz), 125.18 (q,
JCF = 4.0 Hz), 125.14 (q, JCF = 4.0 Hz), 124.41, 124.37, 122.32, 119.83, 57.20, 31.60, 30.07,
23.59; 19F NMR (376 MHz, CD3OD) δ -64.14 (s, 3F); HRMS (ESI+): calcd for C30H27F3N7OS
[M+H]+: 590.1950, found: 590.1949.
N-(4-cyanophenyl)-2,2,2-trifluoroacetamide (3.25). 4-Fluorobenzonitrile (0.5 g, 4.13 mmol) was
dissolved in THF (20.6 mL) and 1 M potassium tert-butoxide in THF (10.3 mL, 10.32 mmol)
was then added. The reaction mixture was refluxed for 4 h, after which the organic solvent was
removed under reduced pressure, and the resulting residue was purified by silica gel column
chromatography (5% EtOAc/ hexanes) to yield 2 (207 mg, 29%) as a clear liquid. 1H NMR (400
MHz, CDCl3) δ 7.52 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 1.38 (s, 9H); 13C NMR (101
MHz, CDCl3) δ 159.90, 133.36, 122.94, 119.11, 105.58, 80.17, 28.80; HRMS (ESI+): calcd for
C11H14NO [M+H]+: 176.1075, found: 176.1083.
(Z)-4-(tert-butoxy)-N'-hydroxybenzimidamide. Synthesized by General Procedure 3A. 87% as
white solid. 1H NMR (400 MHz, acetone-d6) δ 9.11 (s, 1H), 7.64 (d, J = 8.6 Hz, 2H), 6.99 (d, J =
8.6 Hz, 1H), 5.48 (s, 2H), 1.34 (s, 9H); 13C NMR (101 MHz, acetone-d6) δ 157.36, 151.95,

224
128.89, 126.94, 123.92, 78.87, 28.95; HRMS (ESI+): calcd for C11H17N2O2 [M+H]+: 209.1290,
found: 209.1290.
tert-butyl (S)-2-((3-(4-(tert-butoxy)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-
carboxylate (3.26). Synthesized by General Procedure 3B. 52%, yellow oil. 1H NMR (400 MHz,
CDCl3) δ 7.98 – 7.93 (m, 2H), 7.05 (d, J = 8.3 Hz, 2H), 4.35 – 4.18 (m, 1H), 3.46 – 3.23 (m,
3H), 3.04 (ddd, J = 37.3, 14.5, 8.7 Hz, 1H), 2.04 (q, J = 6.4, 4.4 Hz, 1H), 1.91 – 1.74 (m, 3H),
1.44 (s, 9H), 1.37 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 177.30, 168.09, 158.39, 154.24,
128.43, 123.86, 121.52, 80.05, 79.37, 55.26, 46.82, 46.41, 31.83, 31.03, 30.86, 30.10, 28.97,
28.54, 23.63, 22.84; HRMS (ESI+): calcd for C22H32N3O4 [M+H]+: 402.2393, found: 402.2407.
tert-butyl (S)-2-((3-(4-hydroxyphenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate
(3.27). 3.26 (212 mg, 0.529 mmol) was dissolved in DCM (2 mL) and then 1 N TFA (2 mL) was
added. The reaction mixture was stirred for 4 h. At this time, TLC showed complete conversion
of starting material. Organic solvent was removed under reduced pressure. The resulting product
was then dissolved in dioxane (1 mL), and a mixture of di-tert-butyl dicarbonate (0.146 mL,
0.635 mmol) and TEA (0.192 mL, 1.38 mmol) was added dropwise to the solution. The reaction
mixture was stirred for 1 h, after which the organic solvent was removed under reduced pressure,
and the residue was purified by silica gel column chromatography (30% EtOAc/hexane) to
provide 27(73 mg, 40%) as a white solid. 1H NMR (400 MHz, acetone-d6) δ 7.92 (d, J = 8.5 Hz,
2H), 6.98 (d, J = 8.5 Hz, 2H), 4.29 – 4.20 (m, 1H), 3.41 – 3.25 (m, 2H), 3.13 (dd, J = 14.6, 8.2
Hz, 1H), 2.95 (s, 1H), 2.15 – 2.06 (m, 1H), 1.95 – 1.80 (m, 3H), 1.47 – 1.39 (m, 9H); 13C NMR
(101 MHz, CD3OD) δ 178.74, 169.35, 161.71, 156.09, 130.01, 118.99, 116.75, 81.52, 81.01,
56.89*, 56.53*, 47.81*, 47.31*, 32.38*, 32.10*, 31.53*, 31.16*, 28.73*, 28.59*, 24.33*, 23.51*;
HRMS (ESI+): calcd for C18H22N3O4 [M-H]-: 344.1610, found: 344.1620.

225
tert-butyl (S)-2-((3-(4-((4-bromothiazol-2-yl)oxy)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.28). 2, 4-dibromothiazole (0.025 g, 0.101 mmol) was
added to a mixture of 27 (0.035 g, 0.101 mmol) and K2CO3 (0.017 g, 0.122 mmol) in DMF (1
mL). The reaction mixture was refluxed for 17 h, after which the solution was partitioned
between EtOAc and LiBr aqueous solution. The aqueous solution was washed with EtOAc three
times, and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and
concentrated via vacuum. The resulting residue was purified by silica gel column
chromatography (30% EtOAc/ hexane) to yield 28 (20 mg, 39%) as an off-white solid. 1H NMR
(400 MHz, CDCl3) δ 8.17 – 8.10 (m, 2H), 7.40 (d, J = 8.4 Hz, 2H), 6.78 (s, 1H), 4.38 – 4.21 (m,
1H), 3.49 – 3.28 (m, 3H), 3.27 – 2.98 (m, 1H), 2.13 – 2.01 (m, 1H), 1.93 – 1.77 (m, 3H), 1.47 (s,
9H); 13C NMR (101 MHz, CDCl3) δ 177.64, 172.10, 167.55, 156.88, 154.30, 129.48, 129.19,
124.79, 120.39, 120.10, 119.76, 110.97, 80.18*, 79.77*, 55.27*, 55.20*, 46.88*, 46.49*, 31.95*,
31.16*, 31.01*, 30.24*, 28.62, 23.71*, 22.92*; HRMS (ESI+): calcd for C21H23BrN4NaO4S
[M+Na]+: 529.0466, found: 529.0493.
tert-butyl (S)-2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)oxy)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidine-1-carboxylate (3.29). Synthesized by General Procedure 3D. 45%, off-
white solid. 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.1 Hz, 2H),
7.64 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 8.3 Hz, 2H), 7.17 (s, 1H), 4.39 – 4.21 (m, 1H), 3.51 – 3.30
(m, 3H), 3.17 – 3.00 (m, 1H), 2.14 – 2.03 (m, 1H), 1.94 – 1.80 (m, 3H), 1.48 (s, 9H); 13C NMR
(101 MHz, CDCl3) δ 177.78, 172.08, 167.71, 157.42, 148.58, 137.36, 129.50, 129.41, 126.27,
125.83, 124.50, 120.36, 110.97, 108.87, 80.20*, 79.80*, 55.29, 46.89*, 46.52*, 31.96*, 31.17*,
30.27*, 28.64, 23.73*, 22.94*; 19F NMR (376 MHz, CDCl3) δ -62.61 (s, 3F); HRMS (ESI+):
calcd for C21H23BrN4NaO4S [M+Na]+: 529.0466, found: 529.0493.

226
tert-butyl (S,Z)-(((tert-butoxycarbonyl)imino)(2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-
yl)oxy)phenyl)-1,2,4-oxadiazol-5-yl)methyl)pyrrolidin-1-yl)methyl)carbamate (3.30).
Synthesized by General Procedure 3E. 83%, white solid. 1H NMR (400 MHz, CDCl3) δ 10.33 (s,
1H), 8.21 – 8.13 (m, 2H), 7.91 (d, J = 8.2 Hz, 2H), 7.64 (d, J = 8.2 Hz, 2H), 7.50 – 7.45 (m, 2H),
7.42 – 7.37 (m, 1H), 7.17 (s, 1H), 4.85 – 4.75 (m, 1H), 3.74 – 3.61 (m, 2H), 3.56 – 3.43 (m, 1H),
3.20 (dd, J = 15.2, 8.2 Hz, 1H), 2.32 – 2.23 (m, 1H), 1.97 – 1.89 (m, 1H), 1.88 – 1.76 (m, 2H),
1.47 (d, J = 7.6 Hz, 18H); 13C NMR (101 MHz, CDCl3) δ 177.49, 172.13, 167.61, 162.67,
157.36, 156.85, 154.23, 150.45, 148.55, 137.35, 130.41, 130.24, 129.92, 129.58, 129.49, 126.26,
125.89 (q, 4JCF = 3.5 Hz), 125.85 (q, 4JCF = 3.5 Hz), 125.81 (q, 4JCF = 3.5 Hz), 125.77 (q, 4JCF =
3.5 Hz), 124.69, 121.91, 120.41, 120.35, 119.78, 110.93, 108.84, 82.12, 79.45, 56.61, 50.28,
30.76, 30.45, 28.36, 28.23, 24.54; 19F NMR (376 MHz, CDCl3) δ -62.62 (s, 3F); HRMS (ESI+):
calcd for C34H38F3N6O6S [M+H]+: 715.2526, found: 715.2499.
(S)-amino(2-((3-(4-((4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)oxy)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)methaniminium chloride (3.31). Synthesized by General Procedure 3E.
100%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.24 – 8.15 (m, 2H), 8.02 (d, J = 8.1 Hz,
2H), 7.70 (d, J = 8.2 Hz, 2H), 7.63 (s, 1H), 7.61 – 7.53 (m, 2H), 4.59 – 4.51 (m, 1H), 3.61 – 3.52
(m, 1H), 3.53 – 3.43 (m, 1H), 3.38 – 3.34 (m, 2H), 2.38 – 2.25 (m, 1H), 2.21 – 2.01 (m, 3H); 13C
NMR (101 MHz, cd3od) δ 178.31, 173.59, 168.87, 159.02, 156.45, 149.34, 138.97, 130.41,
130.32, 127.36, 126.75 (q, 4JCF = 4.0 Hz), 126.71 (q, 4JCF = 4.0 Hz), 126.67 (q, 4JCF = 4.0 Hz),
126.64 (q, 4JCF = 4.0 Hz), 125.48, 121.77, 121.60, 113.12, 111.34, 57.17, 31.60, 30.05, 23.58; 19F
NMR (376 MHz, CD3OD) δ -64.16 (s, 3F); HRMS (ESI+): calcd for C24H22F3N6O2S [M+H]+:
515.1477, found: 515.1445.

227
tert-butyl (S)-2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)(methyl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidine-1-carboxylate (3.32). To a solution of tert-butyl (S)-2-((3-(4-
3.18dd (0.025 g, 0.043 mmol) in acetone (1 mL) was added K2CO3 (0.024 g, 0.172 mmol) and
methyl iodide (0.061 g, 0.431 mmol). The reaction mixture was refluxed for 17 h. The organic
solvent was removed under reduced pressure, and the residue was purified by silica gel column
chromatography (30% EtOAc/ hexane) to yield 3.32 (15 mg, 73%) as an off-white solid. 1H
NMR (400 MHz, CDCl3) δ 8.13 (d, J = 8.3 Hz, 2H), 7.94 (d, J = 7.9 Hz, 2H), 7.63 (t, J = 7.2 Hz,
5H), 7.45 (t, J = 7.5 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 6.85 (s, 1H), 4.40 – 4.22 (m, 1H), 3.68 (s,
3H), 3.52 – 3.28 (, 3H), 3.20 – 2.99 (m, 1H), 2.16 – 2.02 (m, 1H), 1.97 – 1.78 (m, 3H), 1.49 (s,
9H); 13C NMR (101 MHz, CDCl3) δ 177.62, 168.41, 154.64, 151.48, 148.51, 140.96, 140.54,
134.00, 128.93, 128.79, 127.43, 127.40, 127.33, 127.12, 126.59, 123.52, 102.76, 80.17, 55.29,
46.92*, 46.53*, 40.39, 31.98*, 31.13*, 30.25*, 29.85*, 28.65, 23.74*, 22.94*; HRMS (ESI+): calcd
for C34H36N5O3S [M+H]+: 594.2538, found: 594.2553
tert-butyl (S,Z)-((2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)(methyl)amino)phenyl)-1,2,4-
oxadiazol-5-yl)methyl)pyrrolidin-1-yl)((tert-butoxycarbonyl)imino)methyl)carbamate (3.33).
Synthesized by General Procedure 3E. 73%, off-white solid. 1H NMR (400 MHz, CDCl3) δ
10.33 (s, 1H), 8.15 (d, J = 8.2 Hz, 2H), 7.94 (d, J = 8.0 Hz, 2H), 7.62 (dd, J = 10.9, 8.2 Hz, 6H),
7.45 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 6.84 (s, 1H), 4.84 – 4.76 (m, 1H), 3.73 – 3.61
(m, 5H), 3.55 – 3.48 (m, 1H), 3.19 (dd, J = 15.2, 8.2 Hz, 1H), 2.32 – 2.24 (m, 1H), 1.98 – 1.90
(m, 1H), 1.89 – 1.76 (m, 2H), 1.48 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 177.30, 168.44,
167.82, 158.56, 155.56, 154.18, 151.41, 148.46, 140.94, 140.50, 133.98, 128.91, 128.85, 128.71,
127.39, 127.11, 126.57, 123.65, 123.55, 102.70, 81.99, 79.53, 56.63, 50.23, 40.37, 30.76, 30.48,
28.30, 28.12, 24.54; HRMS (ESI+): calcd for C40H46N7O5S [M+H]+: 736.3281, found: 736.3274.

228
(S)-(2-((3-(4-((4-([1,1'-biphenyl]-4-yl)thiazol-2-yl)amino)phenyl)-1,2,4-oxadiazol-5-
yl)methyl)pyrrolidin-1-yl)(amino)methaniminium chloride (3.34). Synthesized by General
Procedure 3F. 100% yield, off-white solid. 1H NMR (500 MHz, CD3OD) δ 8.13 (d, J = 8.6 Hz,
2H), 7.93 (d, J = 8.1 Hz, 2H), 7.71 (d, J = 8.6 Hz, 2H), 7.66 – 7.59 (m, 5H), 7.43 (t, J = 7.6 Hz,
2H), 7.33 (t, J = 7.3 Hz, 1H), 7.10 (s, 1H), 4.58 – 4.50 (m, 1H), 3.66 (s, 3H), 3.55 (td, J = 9.2,
8.5, 3.9 Hz, 1H), 3.49 – 3.42 (m, 1H), 3.33 – 3.31 (m, 2H), 2.34 – 2.22 (m, 1H), 2.17 – 2.12 (m,
1H), 2.11 – 1.97 (m, 2H); 13C NMR (126 MHz, CD3OD) δ 178.12, 169.97, 169.08, 156.47,
152.27, 150.03, 141.96, 141.78, 135.08, 129.89, 129.60, 128.41, 128.07, 127.81, 127.58, 124.81,
124.41, 57.20, 40.80, 31.61, 30.09, 23.59; HRMS (ESI+): calcd for C30H30N7OS [M*]+:
536.2233, found: 536.2230.
6.3 Structure–Activity Relationship Studies of Uncouplers of Oxidative
Phosphorylation
6.3.1 Oxygen Consumption Rate Seahorse Assay
The oxygen consumption rates of the synthesized compounds was determined using a previously
published method20 with a Seahorse XF-24 Flux Analyzer (Seahorse Biosciences, North
Billerica, MA). L6 myoblast cells were seeded in a Seahorse 24-well tissue culture plate at a
density of 3.5 x 104 cells/well. The cells were then allowed to adhere for 24 h. Prior to the assay,
the media was changed to unbuffered DMEM containing pyruvate and glutamine (Gibco
#12800-017, pH =7.4 at 37 ºC) and the cells were equilibrated for 30 mins at 37 ºC. Compounds
were injected during the assay and OCR was measured using 2 min measurement periods. The
mitochondrial uncoupling agents 5.5 and 5.13 were used as a positive controls for increasing
oxygen consumption.
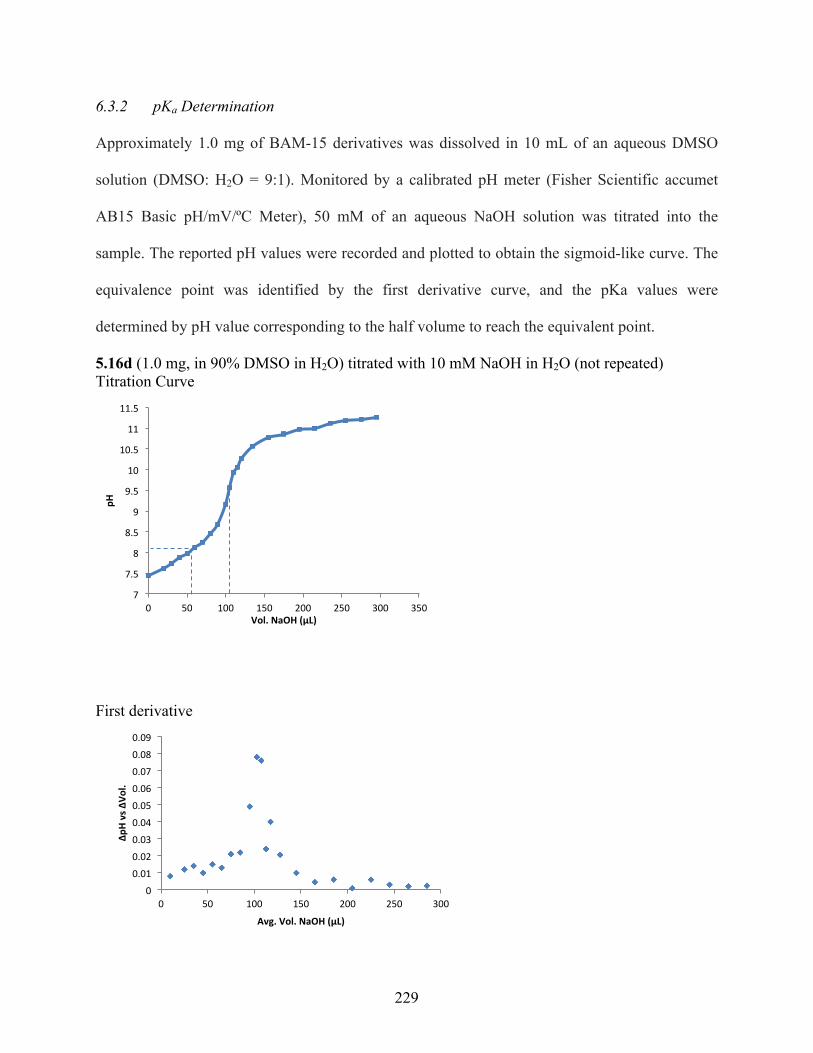
229
6.3.2 pKa Determination
Approximately 1.0 mg of BAM-15 derivatives was dissolved in 10 mL of an aqueous DMSO
solution (DMSO: H2O = 9:1). Monitored by a calibrated pH meter (Fisher Scientific accumet
AB15 Basic pH/mV/ºC Meter), 50 mM of an aqueous NaOH solution was titrated into the
sample. The reported pH values were recorded and plotted to obtain the sigmoid-like curve. The
equivalence point was identified by the first derivative curve, and the pKa values were
determined by pH value corresponding to the half volume to reach the equivalent point.
5.16d (1.0 mg, in 90% DMSO in H2O) titrated with 10 mM NaOH in H2O (not repeated) Titration Curve
First derivative
7
7.5
8
8.5
9
9.5
10
10.5
11
11.5
0 50 100 150 200 250 300 350
pH
Vol.NaOH(µL)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0 50 100 150 200 250 300
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
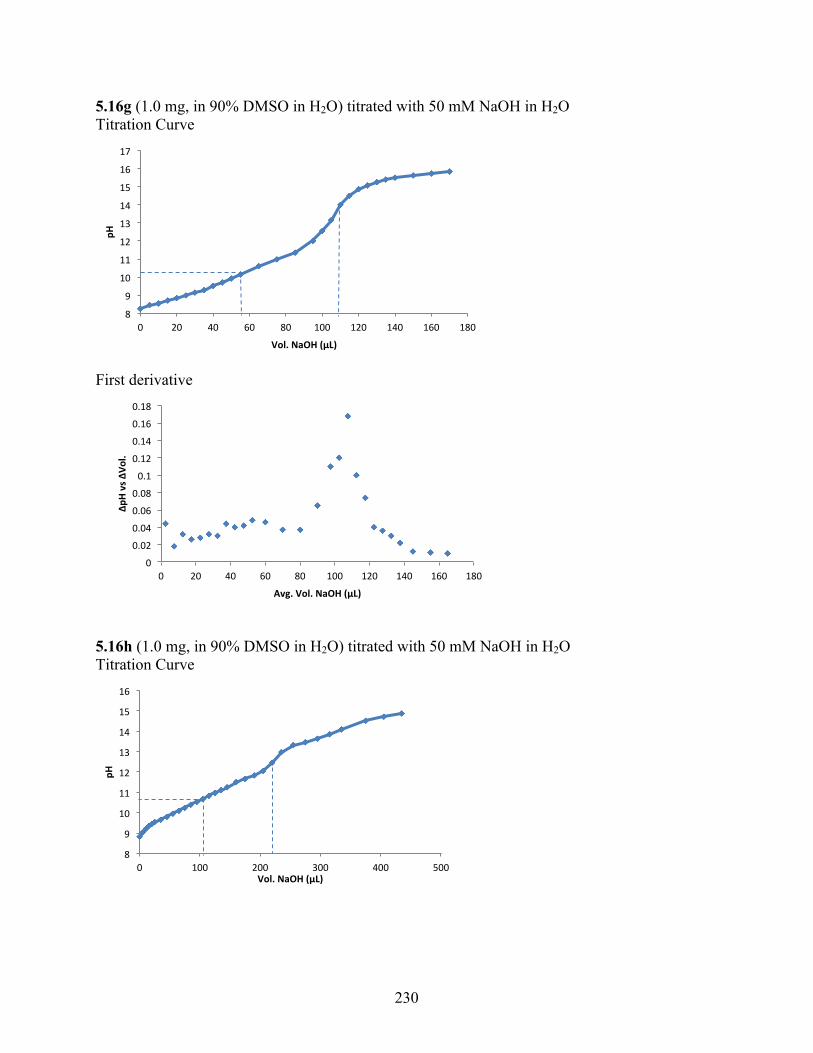
230
5.16g (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
5.16h (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
8
9
10
11
12
13
14
15
16
17
0 20 40 60 80 100 120 140 160 180
pH
Vol.NaOH(µL)
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0.18
0 20 40 60 80 100 120 140 160 180
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
8
9
10
11
12
13
14
15
16
0 100 200 300 400 500
pH
Vol.NaOH(µL)
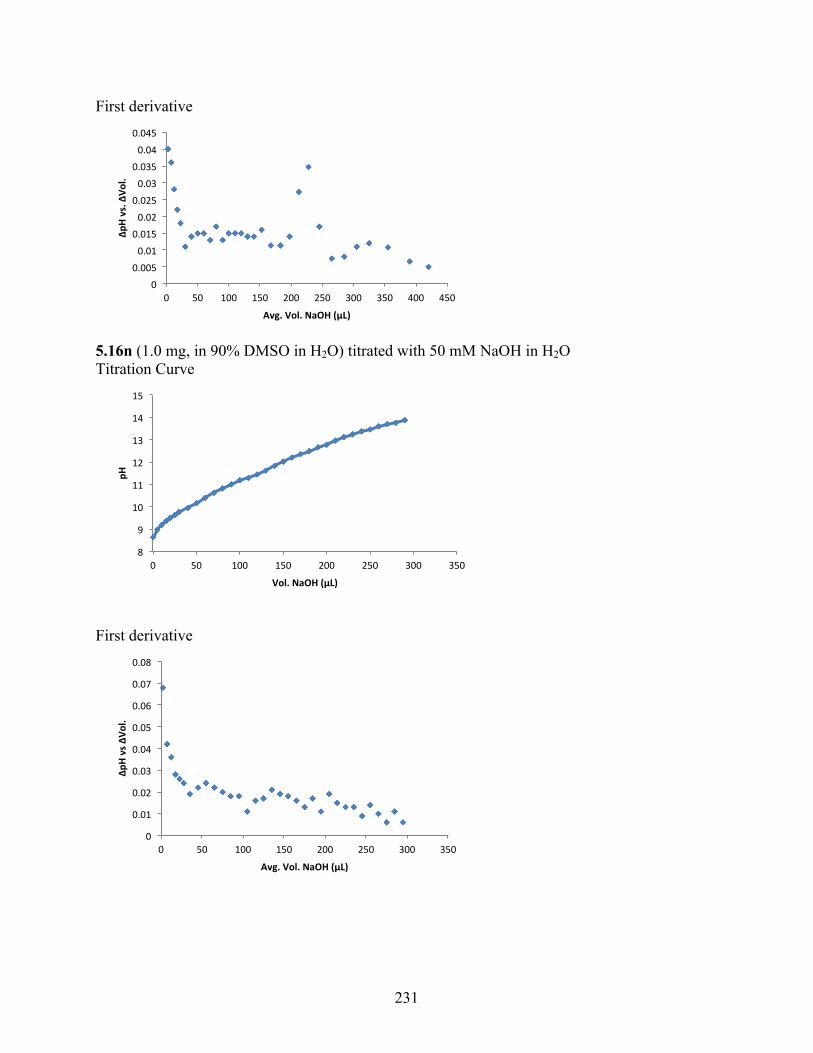
231
First derivative
5.16n (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0.045
0 50 100 150 200 250 300 350 400 450
ΔpHvs.Δ
Vol.
Avg.Vol.NaOH(µL)
8
9
10
11
12
13
14
15
0 50 100 150 200 250 300 350
pH
Vol.NaOH(µL)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 50 100 150 200 250 300 350
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
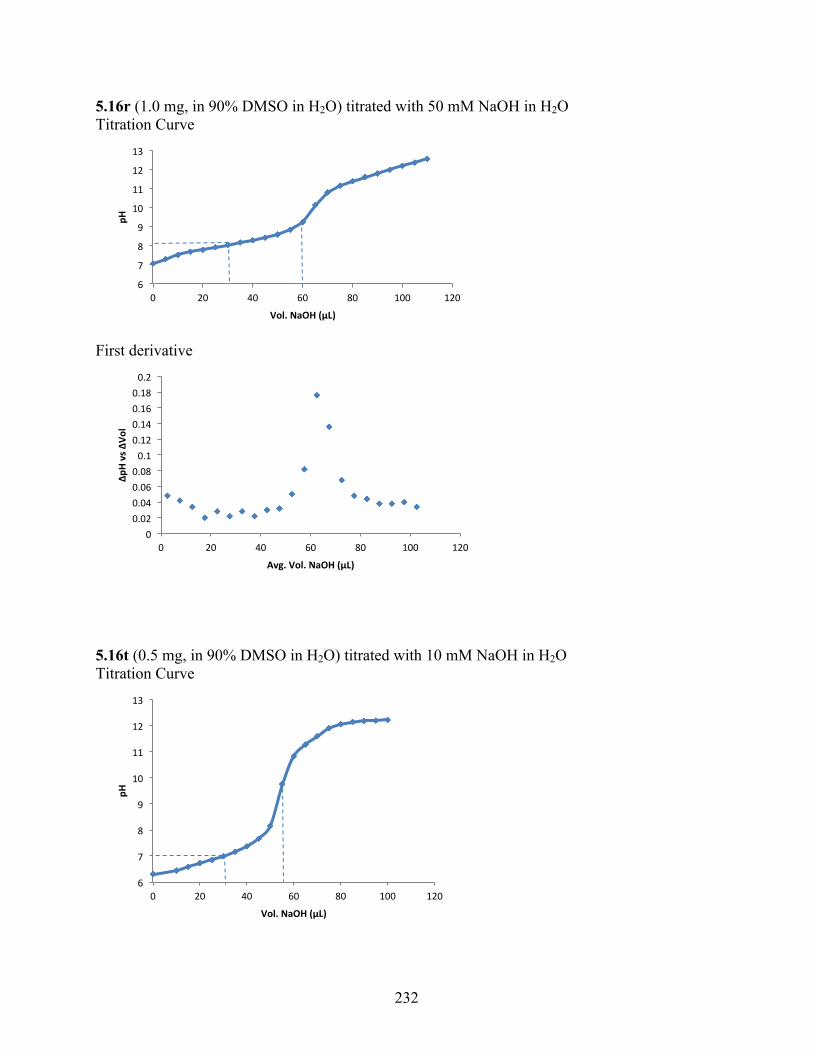
232
5.16r (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
5.16t (0.5 mg, in 90% DMSO in H2O) titrated with 10 mM NaOH in H2O Titration Curve
6
7
8
9
10
11
12
13
0 20 40 60 80 100 120
pH
Vol.NaOH(µL)
00.020.040.060.080.1
0.120.140.160.180.2
0 20 40 60 80 100 120
ΔpHvsΔVo
l
Avg.Vol.NaOH(µL)
6
7
8
9
10
11
12
13
0 20 40 60 80 100 120
pH
Vol.NaOH(µL)
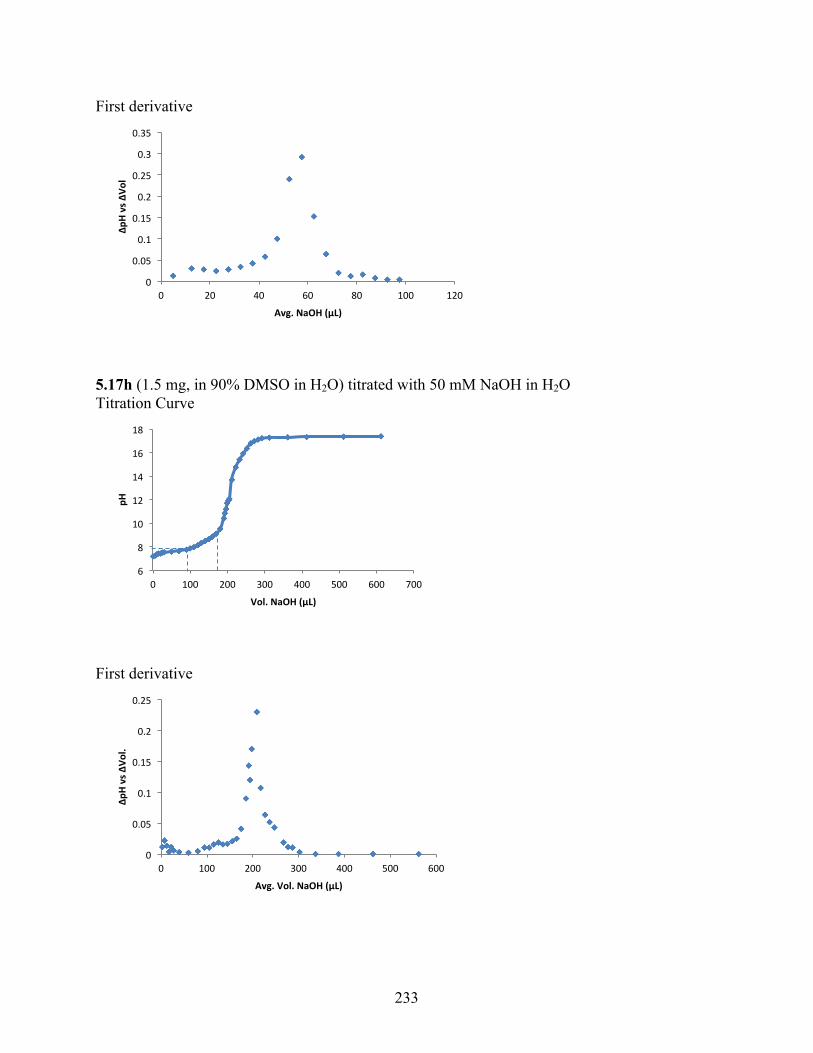
233
First derivative
5.17h (1.5 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0 20 40 60 80 100 120
ΔpHvsΔVo
l
Avg.NaOH(µL)
6
8
10
12
14
16
18
0 100 200 300 400 500 600 700
pH
Vol.NaOH(µL)
0
0.05
0.1
0.15
0.2
0.25
0 100 200 300 400 500 600
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
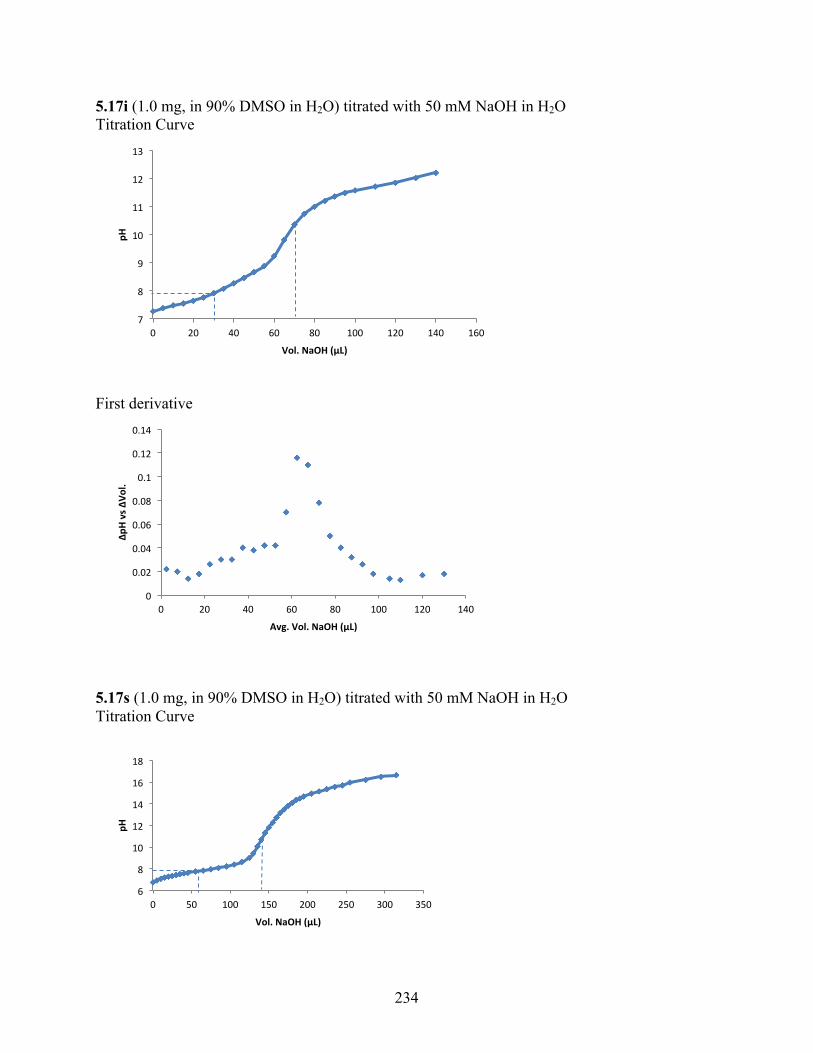
234
5.17i (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
5.17s (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
7
8
9
10
11
12
13
0 20 40 60 80 100 120 140 160
pH
Vol.NaOH(µL)
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0 20 40 60 80 100 120 140
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
6
8
10
12
14
16
18
0 50 100 150 200 250 300 350
pH
Vol.NaOH(µL)
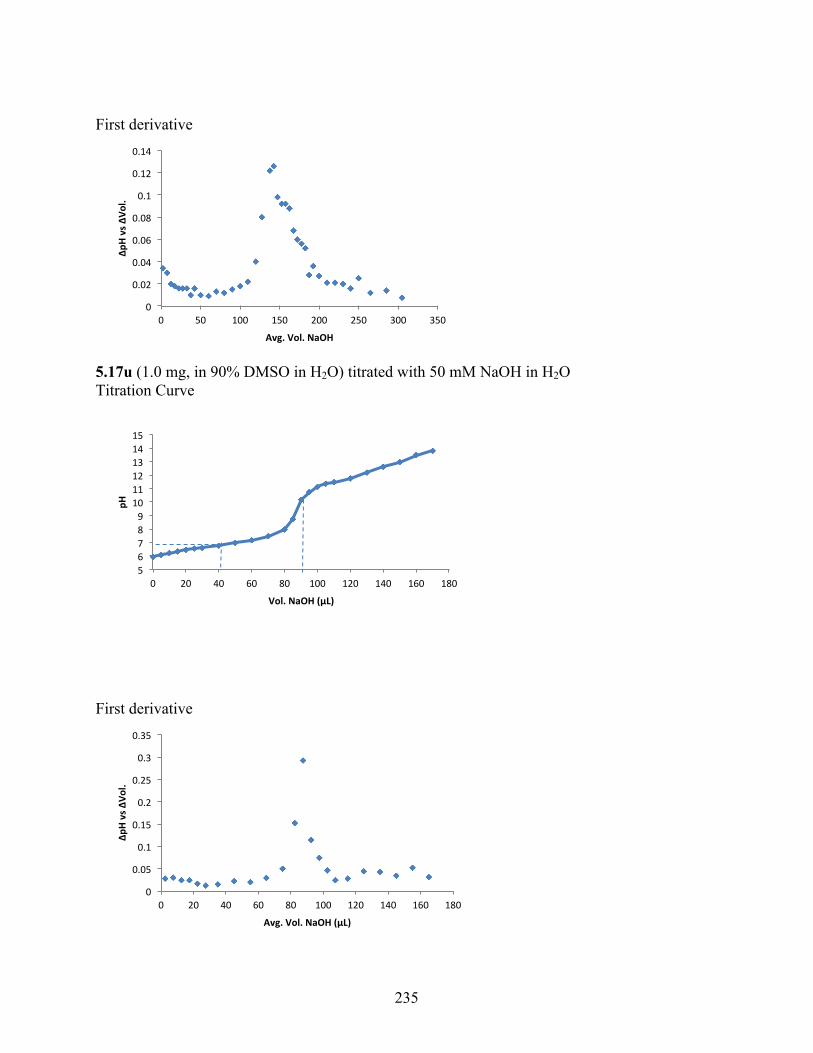
235
First derivative
5.17u (1.0 mg, in 90% DMSO in H2O) titrated with 50 mM NaOH in H2O Titration Curve
First derivative
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0 50 100 150 200 250 300 350
ΔpHvsΔVo
l.
Avg.Vol.NaOH
56789
101112131415
0 20 40 60 80 100 120 140 160 180
pH
Vol.NaOH(µL)
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0 20 40 60 80 100 120 140 160 180
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)
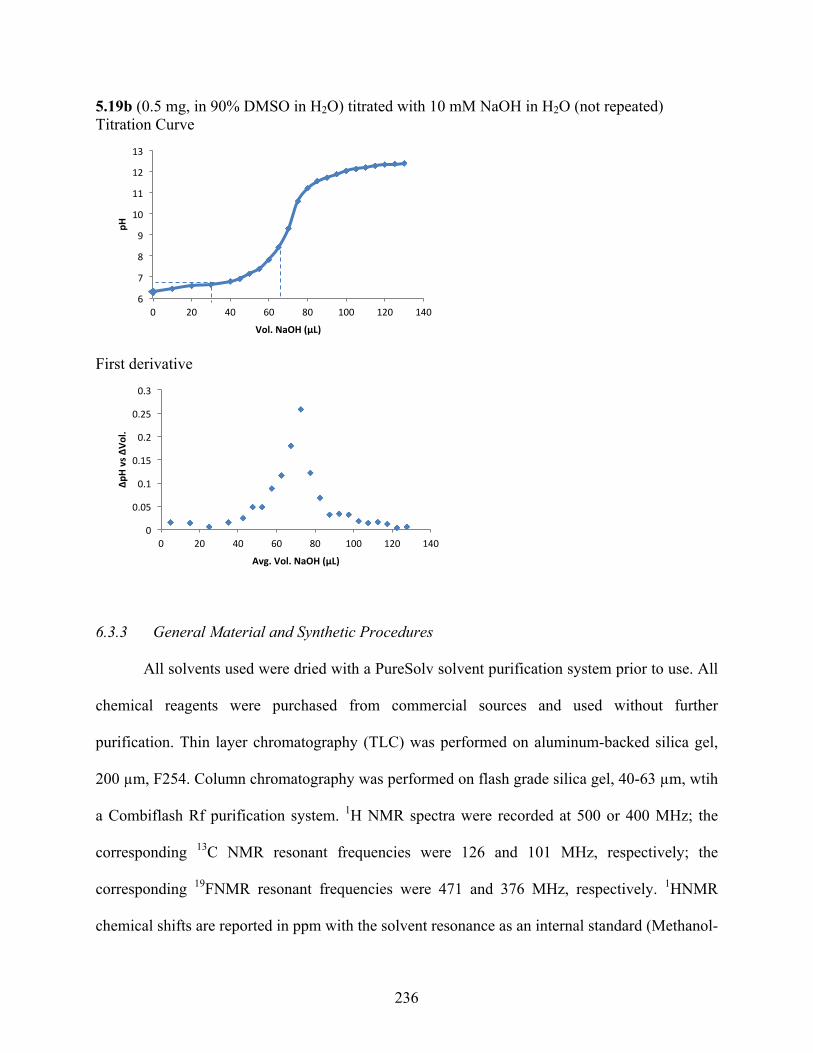
236
5.19b (0.5 mg, in 90% DMSO in H2O) titrated with 10 mM NaOH in H2O (not repeated) Titration Curve
First derivative
6.3.3 General Material and Synthetic Procedures
All solvents used were dried with a PureSolv solvent purification system prior to use. All
chemical reagents were purchased from commercial sources and used without further
purification. Thin layer chromatography (TLC) was performed on aluminum-backed silica gel,
200 µm, F254. Column chromatography was performed on flash grade silica gel, 40-63 µm, wtih
a Combiflash Rf purification system. 1H NMR spectra were recorded at 500 or 400 MHz; the
corresponding 13C NMR resonant frequencies were 126 and 101 MHz, respectively; the
corresponding 19FNMR resonant frequencies were 471 and 376 MHz, respectively. 1HNMR
chemical shifts are reported in ppm with the solvent resonance as an internal standard (Methanol-
6
7
8
9
10
11
12
13
0 20 40 60 80 100 120 140
pH
Vol.NaOH(µL)
0
0.05
0.1
0.15
0.2
0.25
0.3
0 20 40 60 80 100 120 140
ΔpHvsΔVo
l.
Avg.Vol.NaOH(µL)

237
d4: 4.87 ppm; acetone-d6: 2.05 ppm; acetonitrile-d3: 1.94 ppm). 13C NMR, chemical shifts are
reported in ppm with the solvent resonance as the internal standard (Methanol-d4: 49.00 ppm;
acetone-d6: 206.26 ppm; acetonitrile-d3: 118.26 ppm). Data are reported as follows: chemical
shift, multiplicity (s = singlet, d = doublet, t= triplet, q = quartet, br = broad, m= multiplet),
coupling constants (Hz), and integration. Rotomers and tautomers are denoted by an asterisk (*).
High resolution mass spectroscopy (HRMS) was performed on an LC/MS time-of-flight mass
spectrometer using electrospray ionization (ESI). HPLC analyses were performed with a Thermo
Electron TSQ triple quadrupole mass spectrometer equipped with an ESI source. All compounds
tested in biological assays are >95% pure by 1H NMR and HPLC analyses unless noted
otherwise.
General Procedure 5A: Symmetric derivative synthesis
5,6-dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (1 equiv) was dissolved in THF (0.6 M solution).
The desired substituted arylamines or alkylamines (4 equiv) was then added. The reaction
mixture was allowed reflux for 13 h. After which, the solution was concentrated via vacuum, and
the residue was purified by silica gel column to yield the title compound.
General Procedur 5B: Unsymmetric derivative synthesis
5,6-dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (1 equiv) was dissolved in THF (0.52 M solution)
and cooled to 0 ºC. The desired substituted arylamines (0.9 equiv) was then added dropwise and
allowed to stir for 10 min. Triethylamine (1 equiv) was then added dropwise and allowed to stir
at 0 °C for 1 h. The second desired substituted arylamine was then added dropwise and allowed
to stir for 10 min. Triethylamine (1 equiv) was then added dropwise, and the reaction was then
allowed to warm to rt and allowed to stir for 19 h. After which, the solution was concentrated via
vacuum, and the residue was purified by silica gel column to yield the title compound.

238
General Procedure 5C: Amide Derivative Synthesis
[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (1 equiv) 20 mg, 0.131 mmol) was dissolved in
DCM (0.44 M solution) with TEA (2 equiv) and cooled to 0 °C. The select sulfonyl chloride or
anhydride (3 equiv) was then added dropwise. The reaction mixture was allowed to warm up to
rt and stirred for 17 h. The resulting reaction mixture was then concentrated via vacuum, and the
residue was purified by silica gel column chromatography to yield the title compound.
5,6-dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (5.15). 1,2,5-oxadiazole-3,4-diamine (1 equiv.)
was dissolved in 10% HCl (6 M solution). Oxalic acid (1.5 equiv) was added to the solution, and
the mixture was heated to reflux for 3 h. After which, the mixture was cooled to rt, filtered, and
dried. The resulting dihydroxy white solid was then carried forward crude (1 equiv) by adding
phosphorous pentachloride (3 equiv) and phosphorous oxychloride (2 M solution). The resulting
mixture was heated to 95 ºC for 2 h, and excess phosphorous oxychloride was removed via
vacuum distillation. The mixture was cooled to 0 ºC, and cold water was added (15–20 mL),
causing the title compound to precipitate out of solution. The precipitate was purified via
recrystallization using an acetone/water mixture, producing the title compound as a white solid,
23%. Analytical data matches with the literature.205
N5,N6-di-tert-butyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16a). Synthesized by
General Procedure 5A. 79%, white solid. 1H NMR (400 MHz, Acetone-d6) δ 7.96 (s, 2H), 2.68
(s, 24H); 13C NMR (101 MHz, Acetone-d6) δ 150.35, 149.17, 54.65, 28.47; HRMS (ESI+): calcd
for C12H21N6O [M+H]+: 265.1777, found: 265.1776.
N5,N6-dicyclopropyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16b). Synthesized by
General Procedure 5A. 33%, white solid. 1H NMR (400 MHz, Acetone-d6) δ 7.32 (s, 2H), 3.01
(tt, J = 7.3, 3.9 Hz, 2H), 0.89 – 0.82 (m, 4H), 0.64 – 0.57 (m, 4H); 13C NMR (101 MHz,

239
Acetone-d6) δ 151.67, 150.97, 25.51, 6.94; HRMS (ESI+): calcd for C10H13N6O [M+H]+:
233.1151, found: 233.1137.
N5,N6-dicyclopentyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.15c). Synthesized by
General Procedure 5A. 40%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ 7.15 (d, J = 6.6
Hz, 2H), 4.44 (s, 2H), 2.14 – 1.96 (m, 4H), 1.74 – 1.42 (m, 12H); 13C NMR (101 MHz, Acetone-
d6) δ 151.44, 149.33, 54.54, 32.86, 24.62; HRMS (ESI+): calcd for C14H21N6O [M+H]+:
289.1777, found: 289.1774.
N5,N6-dicyclohexyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16d). Synthesized by
General Procedure 5A. 72%, clear solid. 1H NMR (400 MHz, Acetone-d6) δ 8.26 (d, 2H), 5.38 –
5.23 (m, 2H), 3.34 – 3.19 (m, 4H), 2.95 (dt, J = 13.5, 3.6 Hz, 4H), 2.84 (dt, J = 12.8, 3.6 Hz,
2H), 2.59 (qt, J = 12.9, 3.4 Hz, 4H), 2.51 – 2.28 (m, 6H); 13C NMR (101 MHz, Acetone-d6) δ
151.38, 148.70, 51.79, 32.78, 26.33, 25.85; HRMS (ESI+): calcd for C16H25N6O [M+H]+:
317.2090, found: 317.2089.
5,6-dimorpholino-[1,2,5]oxadiazolo[3,4-b]pyrazine (5.16e). Synthesized by General Procedure
5A. 95%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ 3.81 (s, 1H); 13C NMR (101 MHz,
Acetone-d6) δ 154.04, 151.75, 66.80, 49.45; HRMS (ESI+): calcd for C12H17N6O3 [M+H]+:
293.1362, found: 293.1361. HPLC Purity: 86% pure.
N5,N6-di-o-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16f). Synthesized by General
Procedure 5A. 35%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 9.36 (s, 2H), 7.55 (dd, J =
6.9, 2.3 Hz, 2H), 7.40 – 7.25 (m, 6H), 2.34 (s, 6H); 13C NMR (101 MHz, Acetone-d6) δ 152.83*,
152.45*, 151.25*, 151.05*, 136.42, 135.48, 131.69, 128.54, 127.69, 127.45, 18.18; HRMS
(ESI+): calcd for C18H17N6O [M+H]+: 333.1464, found: 333.1470.

240
N5,N6-di-m-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16g). Synthesized by General
Procedure 5A. 60%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 8.82 (s, 2H), 7.61 – 7.27
(m, 6H), 7.06 (d, J = 7.5 Hz, 2H), 2.38 (s, 6H); 13C NMR (101 MHz, Acetonitrile-d3) δ 140.16,
130.01, 126.88, 123.25, 119.96, 116.00, 112.50, 21.52; HRMS (ESI+): calcd for C18H17N6O
[M+H]+: 333.1464, found: 333.1464.
N5,N6-di-p-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16h). Synthesized by General
Procedure 5A. 69%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ 9.56 (s, 2H), 7.62 (s,
4H), 7.25 (d, J = 8.1 Hz, 4H), 2.34 (s, 6H); 13C NMR (101 MHz, Acetonitrile-d3) δ 151.17,
148.92, 136.01, 130.41, 123.54, 115.47, 20.97; HRMS (ESI+): calcd for C18H17N6O [M+H]+:
333.1464, found: 333.1441.
N5,N6-bis(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16i). Synthesized
by General Procedure 5A. 16%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 9.03 – 8.64 (m,
2H), 7.28 – 6.97 (m, 8H), 3.92 (s, 6H); 13C NMR (126 MHz, Acetone-d6) δ 151.28, 150.77,
128.30, 126.82, 121.81, 115.25, 112.88, 111.67, 56.40; HRMS (ESI+): calcd for C18H17N6O3
[M+H]+: 365.1362, found: 365.1345.
N5,N6-bis(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16j). Synthesized
by General Procedure 5A. 32%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ 9.64 (s, 2H),
7.53 (s, 4H), 7.34 (d, J = 8.1 Hz, 2H), 6.77 (dd, 2H), 3.82 (s, 6H); 13C NMR (101 MHz, Acetone-
d6) δ 161.40, 150.72, 140.11, 130.83, 114.61, 111.20, 108.21, 55.85, 55.72, 55.60; HRMS
(ESI+): calcd for C18H17N6O3 [M+H]+: 365.1362, found: 365.1370.
N5,N6-bis(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16k). Synthesized
by General Procedure 5A. 47%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 9.26 (d, J =
70.1 Hz, 2H), 7.70 (s, 4H), 7.04 – 6.95 (m, 4H), 3.83 (s, 6H); 13C NMR (101 MHz, Acetone-d6)

241
δ 158.27, 151.21, 125.17, 123.20, 115.85, 114.89, 49.87; HRMS (ESI+): calcd for C18H17N6O3
[M+H]+: 365.1362, found: 365.1367.
N5,N6-bis(2-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16l). Synthesized
by General Procedure 5A. 54%, light yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 10.75 –
10.04 (m, 2H), 9.04 – 8.05 (m, 2H), 7.11 (dq, J = 28.7, 9.3, 8.3 Hz, 6H), 4.37 – 3.97 (m, 4H),
1.56 – 1.18 (m, 6H); 13C NMR (101 MHz, Acetone-d6) δ 150.31, 126.29, 121.87, 113.51, 65.18,
15.27; HRMS (ESI+): calcd for C20H21N6O3 [M+H]+: 393.1675, found: 393.1674.
N5,N6-bis(3-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16m). Synthesized
by General Procedure 5A. 27%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 9.61 (s, 2H),
7.79 – 7.37 (m, 2H), 7.32 (t, J = 8.1 Hz, 4H), 6.81 – 6.65 (m, 2H), 4.08 (q, J = 7.0 Hz, 4H), 1.38
(t, J = 7.0 Hz, 6H); 13C NMR (101 MHz, Acetone-d6) δ 160.74, 130.83, 111.78, 111.01, 108.80,
64.22, 15.22; HRMS (ESI+): calcd for C20H21N6O3 [M+H]+: 393.1675, found: 393.1656.
N5,N6-bis(4-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16n). Synthesized
by General Procedure 5A. 73%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ 9.15 (s, 2H),
7.74 (s, 4H), 7.04 – 6.94 (m, 4H), 4.07 (q, J = 7.0 Hz, 4H), 1.38 (t, J = 6.9 Hz, 6H); 13C NMR
(101 MHz, Acetone-d6) δ 157.38, 151.05, 148.33, 131.76, 124.81, 115.46, 64.24, 15.17; HRMS
(ESI+): calcd for C20H21N6O3 [M+H]+: 393.1675, found: 393.1673.
3,3'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))diphenol (5.16o). Synthesized by
General Procedure 5A. 43%, light yellow solid. H NMR (500 MHz, Acetone-d6) δ 7.91 – 7.65
(m, 0H), 7.48 (d, J = 91.0 Hz, 2H), 7.09 – 6.87 (m, 1H), 8.71 (s, 2H), 7.23 (t, J = 7.9 Hz, 4H),
6.74 – 6.57 (m, 2H); 13C NMR (126 MHz, Acetone-d6) δ 158.94, 150.93, 150.64, 140.40, 130.72,
113.53, 112.80, 109.46; HRMS (ESI+): calcd for C16H13N6O3 [M+H]+: 337.1049, found:
337.1044.

242
N,N'-(([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))bis(4,1-phenylene))bis(2,2,2-
trifluoroacetamide) (5.16p). Synthesized by General Procedure 5A. 22%, yellow solid. 1H NMR
(400 MHz, Acetone-d6) δ 10.38 (s, 2H), 9.61 (s, 2H), 8.00 – 7.94 (m, 4H), 7.86 – 7.81 (m, 4H);
13C NMR (101 MHz, Acetone-d6) δ 156.27 (q, 2JCF = 37.4 Hz), 155.90 (q, 2JCF = 37.4 Hz),
155.53 (q, 2JCF = 37.4 Hz), 155.16 (q, 2JCF = 37.4 Hz), 152.34, 150.84, 150.01, 135.59*, 135.15*,
124.51, 122.03, 121.28 (q, 1JCF = 288.9 Hz), 118.42 (q, 1JCF = 288.9 Hz), 115.56 (q, 1JCF = 288.9
Hz), 112.69 (q, 1JCF = 288.9 Hz); 19F NMR (376 MHz, acetone) δ -76.58 (s, 6F); HRMS (ESI+):
calcd for C20H13F6N8O3 [M+H]+: 527.1015, found: 527.1009.
N5,N6-bis(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16q).
Synthesized by General Procedure 5A. 20%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ
8.02 (s, 2H), 7.54-7.46 (m, 6H), 7.38-7.29 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 147.75,
145.73, 141.01, 135.57, 129.33, 126.71, 125.52 (q, 1JCF = 258.5 Hz), 123.87, 123.07, 122.95,
120.40 (q, 1JCF = 258.5 Hz), 117.84 (q, 1JCF = 258.5 Hz); 19F NMR (376 MHz, Acetone-d6) δ -
58.69 (s, 6F); HRMS (ESI-): calcd for C18H9F6N6O3 [M-H]: 471.0646, found: 471.0657.
N5,N6-bis(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16r).
Synthesized by General Procedure 5A. 69%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ
10.00 (s, 2H), 8.01 – 7.64 (m, 4H), 7.58 (t, J = 8.1 Hz, 2H), 7.17 (d, 2H); 13C NMR (101 MHz,
Acetone-d6) δ 150.42, 131.64, 125.33 (q, 1JCF = 256.5 Hz), 122.78 (q, 1JCF = 258.5 Hz), 121.11,
120.24 (q, 1JCF = 258.5 Hz), 117.74, 117.69 (q, 1JCF = 258.5 Hz), 115.00; 19F NMR (376 MHz,
Acetone-d6) δ -58.47 (s, 6F); HRMS (ESI+): calcd for C18H11F6N6O3 [M+H]+: 473.0797, found:
473.0773.
N5,N6-bis(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16s).
Synthesized by General Procedure 5A. 40%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ

243
9.39 – 8.65 (m, 2H), 7.80 (s, 4H), 7.49 (d, J = 8.5 Hz, 4H); 13C NMR (101 MHz, Acetone-d6) δ
149.92, 146.42, 138.74, 125.30 (q, 1JCF = 256.5 Hz), 123.98, 122.76, 120.22 (q, 1JCF = 256.5 Hz),
117.68 (q, 1JCF = 256.5 Hz); 19F NMR (376 MHz, Acetone-d6) δ -58.78 (s, 6F); HRMS (ESI+):
calcd for C18H11F6N6O3 [M+H]+: 473.0797, found: 473.0792.
N5,N6-bis(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16t).
Synthesized by General Procedure 5A. 52%, light yellow solid. 1H NMR (400 MHz, Acetone-d6)
δ 9.40 (s, 2H), 8.03 – 7.76 (m, 6H), 7.64 (dd, J = 7.7 Hz, 2H); 13C NMR (101 MHz, Acetone-d6)
δ 152.64*, 152.08*, 152.05*, 151.32*, 135.93, 134.66, 134.66, 134.64, 130.82, 130.68, 129.37,
129.06 (q, 1JCF = 274.7 Hz), 128.12 (q, 4JCF = 5.1 Hz), 128.07 (q, 4JCF = 5.1 Hz), 128.02 (q, 4JCF
= 5.1 Hz), 127.97 (q, 4JCF = 5.1 Hz), 127.31 (q, 2JCF = 30.3 Hz), 127.01 (q, 2JCF = 30.3 Hz),
126.34 (q, 1JCF = 274.7 Hz), 123.63 (q, 1JCF = 274.7 Hz), 120.92 (q, 1JCF = 274.7 Hz); 19F NMR
(376 MHz, Acetone-d6) δ -61.18 (s, 6F); HRMS (ESI+): calcd for C18H11F6N6O [M+H]+:
441.0899, found: 441.0892.
N5,N6-bis(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16u).
Synthesized by General Procedure 5A. 50%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ
10.11 (s, 2H), 8.07 (s, 4H), 7.70 (t, J = 7.9 Hz, 2H), 7.55 (d, J = 7.8 Hz, 2H); 13C NMR (101
MHz, Acetone-d6) δ 149.32, 141.84, 132.29 (q, 2JCF = 32.3 Hz), 131.97 (q, 2JCF = 32.3 Hz),
131.65 (q, 2JCF = 32.3 Hz), 131.19, 129.27 (q, 1JCF = 272.7 Hz), 126.56 (q, 1JCF = 272.7 Hz),
126.08, 123.86 (q, 1JCF = 272.7 Hz), 122.09, 121.16 (q, 1JCF = 272.7 Hz), 119.00; 19F NMR (376
MHz, Acetone-d6) δ -63.20 (s, 6F); HRMS (ESI+): calcd for C18H11F6N6O [M+H]+: 441.0899,
found: 441.0887.
N5,N6-bis(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.16v).
Synthesized by General Procedure 5A. 63%, light yellow solid. 1H NMR (400 MHz, Acetone-d6)

244
δ 10.17 (s, 2H), 7.93 (s, 4H), 7.80 (d, J = 8.3 Hz, 4H); 13C NMR (101 MHz, Acetone-d6) δ
148.83, 145.22, 129.53 (q, 1JCF = 273.7 Hz), 127.38 (q, 4JCF = 4.0 Hz), 127.34 (q, 4JCF = 4.0 Hz),
127.30 (q, 4JCF = 4.0 Hz), 127.27 (q, 4JCF = 4.0 Hz), 127.03 (q, 2JCF = 26.3 Hz), 126.90 (q, 1JCF =
273.7 Hz), 126.84 (q, 2JCF = 26.3 Hz), 126.58 (q, 2JCF = 26.3 Hz), 126.25 (q, 2JCF = 26.3 Hz),
124.15 (q, 1JCF = 273.7 Hz), 122.60, 121.45 (q, 1JCF = 273.7 Hz); 19F NMR (376 MHz, Acetone-
d6) δ -62.53 (s, 6F); HRMS (ESI+): calcd for C18H11F6N6O [M+H]+: 441.0899, found: 441.0875.
4,4'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))dibenzonitrile (5.16w).
Synthesized by General Procedure 5A. 19%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ
9.78 (s, 2H), 8.25 – 8.17 (m, 4H), 7.92 – 7.85 (m, 4H); 13C NMR (101 MHz, Acetone-d6) δ
152.28, 152.07, 150.92, 150.02, 142.45, 134.08, 133.99, 123.89, 123.78, 119.25, 109.63; HRMS
(ESI+): calcd for C18H11N8O [M+H]+: 355.1056, found: 355.1040.
N5-(2-fluorophenyl)-N6-phenyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17a).
Synthesized by General Procedure 5B. 44%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ
9.63 (s, 2H), 8.14 – 7.58 (m, 3H), 7.45 (t, J = 7.8 Hz, 3H), 7.35 – 7.14 (m, 6H); 13C NMR (101
MHz, Acetonitrile-d3) δ 156.74, 156.54, 154.96, 154.29, 149.81, 148.02, 140.27 (d, 1JCF = 204.2
Hz), 138.25 (d, 1JCF = 204.2 Hz), 130.15, 127.83, 127.75, 127.65, 126.16 (d, 2JCF = 18.2 Hz),
125.98 (d, 2JCF = 18.2 Hz), 125.54, 122.65, 118.26, 117.22 (d, 2JCF = 19.2 Hz), 117.03 (d, 2JCF =
19.2 Hz); 19F NMR (376 MHz, Acetonitrile-d3) δ -122.05 to -130.05 (m, 1F); HRMS (ESI+):
calcd for C16H12FN6O [M+H]+: 323.1057, found: 323.1051. HPLC Purity: 90% pure.
N5-(2-fluorophenyl)- N6-(pyridin-2-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17b).
Synthesized by General Procedure 5B. 12%, off-white solid. 1H NMR (400 MHz, Acetonitrile-
d3) δ 10.16 (s, 1H), 8.65 (dd, J = 8.0 Hz, 1H), 8.22 – 8.14 (m, 1H), 8.02 (ddd, J = 8.9, 7.1, 1.9
Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.37 – 7.11 (m, 5H); 13C NMR (126 MHz, Acetone-d6) δ

245
156.95, 155.85, 153.89, 152.67 (d, 1JCF = 157.6 Hz), 152.22, 151.11 (d, 1JCF = 157.6 Hz), 150.35,
143.49, 137.61, 127.27 (d, 3JCF = 9.1 Hz), 127.18 (d, 3JCF = 9.1 Hz), 126.24 (d, 3JCF = 6.1 Hz),
126.18 (d, 3JCF = 6.1f Hz), 125.63 (d, 4JCF = 3.0 Hz), 125.60 (d, 4JCF = 3.0 Hz), 125.09, 123.50,
117.87, 117.40, 116.11 (d, 2JCF = 15.2 Hz), 115.96 (d, 2JCF = 15.2 Hz); 19F NMR (376 MHz,
Acetonitrile-d3) δ -129.95 to -130.15 (m, 1F); HRMS (ESI+): calcd for C15H11FN7O [M+H]+:
324.1009, found: 324.0997.
N5-(2-fluorophenyl)- N6-(3-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17c).
Synthesized by General Procedure 5B. 42%, off-white solid. 1H NMR (400 MHz, Acetone-d6) δ
11.15 (s, 1H), 9.82 (s, 1H), 7.60 (s, 5H), 7.48 (ddd, J = 8.2, 6.6 Hz, 1H), 7.35 – 7.21 (m, 4H),
6.98 (td, J = 8.8, 2.5 Hz, 1H); 13C NMR (101 MHz, a Acetone-d6) δ 165.21, 164.63, 162.75,
162.41, 131.64, 127.35, 125.92, 118.20, 116.93, 112.31 (d, 2JCF = 20.2 Hz), 112.10 (d, 2JCF =
20.2 Hz), 109.50 (d, 2JCF = 28.3 Hz), 109.22 (d, 2JCF = 28.3 Hz); 19F NMR (376 MHz, Acetone-
d6) δ -113.18 to -113.24 (m, 1F), -113.24 to -113.29 (m, 1F); HRMS (ESI+): calcd for
C16H11F2N6O [M+H]+: 341.0962, found: 341.0956.
N5-(2-fluorophenyl)- N6-(4-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17d).
Synthesized by General Procedure 5B. 28%, off-white solid. 1H NMR (500 MHz, Acetone-d6) δ
10.82 (s, 1H), 9.72 (s, 1H), 8.28 – 7.56 (m, 4H), 7.36 – 7.17 (m, 4H); 13C NMR (126 MHz,
Acetone-d6) δ 160.70, 158.78, 148.45, 146.74, 126.32, 126.15, 124.87, 123.46, 116.17, 116.01,
115.85, 115.69 (d, 2JCF = 18.2 Hz), 115.51 (d, 2JCF = 18.2 Hz); 19F NMR (376 MHz, Acetone-d6)
δ -118.35 to -120.14 (m, 1F), -129.69 (dt, J = 8.8, 4.3 Hz, 1F); HRMS (ESI+): calcd for
C16H11F2N6O [M+H]+: 341.0962, found: 341.0967.
N5-(2,3-difluorophenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17e). Synthesized by General Procedure 5B. 32%, light yellow solid. 1H NMR (400 MHz,

246
Acetone-d6) δ 10.91 (s, 1H), 9.75 (s, 1H), 8.64 – 8.03 (m, 1H), 7.86 – 7.42 (m, 1H), 7.35 – 7.22
(m, 4H), 7.22 – 7.13 (m, 1H); 13C NMR (101 MHz, Acetone-d6) δ 152.14, 152.08, 149.71,
149.62, 144.02 (d, 1JCF = 246.4 Hz), 143.89 (d, 1JCF = 247.5 Hz), 141.58 (d, 1JCF = 246.4 Hz),
141.44 (d, 1JCF = 247.5 Hz), 124.79 (dd, 3JCF = 6.1 Hz), 124.72 (dd, 3JCF = 6.1 Hz), 124.66 (dd,
3JCF = 6.1 Hz), 119.03, 113.39 (d, 2JCF = 18.2 Hz), 113.21 (d, 2JCF = 18.2 Hz); 19F NMR (376
MHz, Acetone-d6) δ -139.18 to -139.86 (m, 1F), -148.96 to -154.99 (m, 2F); HRMS (ESI+):
calcd for C16H10F3N6O [M+H]+: 359.0868, found: 359.0858.
N5-(2,4-difluorophenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17f). Synthesized by General Procedure 5B. 16%, light yellow solid. 1H NMR (400 MHz,
Acetone-d6) δ 10.90 (s, 1H), 9.76 (s, 1H), 8.29 – 7.55 (m, 2H), 7.34 – 7.25 (m, 2H), 7.24 – 7.16
(m, 2H), 7.17 – 7.08 (m, 2H); 13C NMR (101 MHz, Acetone-d6) δ 161.14, 161.02, 158.70,
158.59, 155.67 (d, 1JCF = 247.5 Hz), 155.38 (d, 1JCF = 249.5 Hz), 153.22 (d, 1JCF = 247.5 Hz),
152.91 (d, 1JCF = 249.5 Hz), 147.46, 126.17, 125.16, 124.86, 124.82, 123.68, 115.81 (d, 2JCF =
21.2 Hz), 115.60 (d, 2JCF = 21.2 Hz), 111.70 (d, 2JCF = 22.2 Hz), 111.48 (d, 2JCF = 22.2 Hz),
104.60 (dd, 2JCF = 25.3 Hz), 104.35 (dd, 2JCF = 25.3 Hz), 104.09; 19F NMR (376 MHz, Acetone-
d6) δ -114.32 to -116.24 (m, 2F), -119.15 to -121.61 (m, 1F); HRMS (ESI+): calcd for
C16H10F3N6O [M+H]+: 359.0868, found: 359.0862.
N5-(2-fluorophenyl)-N6-(o-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17g).
Synthesized by General Procedure 5B. 17%, off-white solid. 1H NMR (500 MHz, Acetone-d6) δ
9.60 (s, 1H), 8.63 (s, 1H), 7.47 – 7.41 (m, 1H), 7.40 – 7.36 (m, 2H), 7.35 – 7.25 (m, 5H), 2.28 (s,
3H); 13C NMR (126 MHz, Acetone-d6) δ 152.69, 152.61 (d, 1JCF = 131.3 Hz), 151.31 (d, 1JCF =
131.3 Hz), 150.93, 135.83 (d, 3JCF = 7.1 Hz), 135.76 (d, 3JCF = 7.1 Hz), 131.78, 129.76, 128.88,
127.72 (d, 3JCF = 10.1 Hz), 127.62 (d, 3JCF = 10.1 Hz), 125.72, 125.61, 124.36, 17.98; 19F NMR

247
(376 MHz, Acetonitrile-d3) δ -122.81 to -123.46 (m, 1F); HRMS (ESI+): calcd for C17H14FN6O
[M+H]+: 337.1213, found: 337.1200
N5-(2-fluorophenyl)-N6-(m-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17h).
Synthesized by General Procedure 5B. 43%, yellow solid. 1H NMR (400 MHz, Acetonitrile-d3) δ
9.10 (s, 1H), 7.62 (s, 1H), 7.47 (s, 2H), 7.39 – 7.21 (m, 5H), 7.07 (d, J = 7.5 Hz, 1H), 2.39 (s,
3H); 13C NMR (101 MHz, Acetonitrile-d3) δ 156.74, 154.29, 140.21, 130.05, 127.79 (d, 3JCF =
7.1 Hz), 127.72 (d, 3JCF = 7.1 Hz), 126.92, 126.01, 125.97, 125.50, 123.07, 119.75, 117.20 (d,
2JCF = 19.2 Hz), 117.01 (d, 2JCF = 19.2 Hz), 21.52; 19F NMR (376 MHz, Acetone-d6) δ -118.84 to
-127.22 (m, 1F); HRMS (ESI+): calcd for C17H14FN6O [M+H]+: 337.1213, found: 337.1203.
N5-(2-fluorophenyl)-N6-(p-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17i).
Synthesized by General Procedure 5B. 40%, light yellow solid. 1H NMR (400 MHz, Acetone-d6)
δ 9.68 (s, 2H), 8.01 – 7.52 (m, 3H), 7.37 – 7.18 (m, 5H), 2.35 (s, 3H); 13C NMR (101 MHz,
Acetone-d6) δ 156.52, 154.28, 140.77, 135.42, 130.49, 127.36, 125.88, 124.79, 122.46, 117.08
(d, 3JCF = 19.6 Hz), 116.90 (d, 3JCF = 19.6 Hz), 21.07; 19F NMR (376 MHz, Acetone-d6) δ -
117.35 to -129.39 (m, 1F); HRMS (ESI+): calcd for C17H14FN6O [M+H]+: 337.1213, found:
337.1201.
N5-(2-fluorophenyl)-N6-(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17j). Synthesized by General Procedure 5B. 55%, yellow solid. 1H NMR (400 MHz, Acetone-
d6) δ 10.05 (s, 1H), 8.78 (s, 1H), 7.35 – 7.03 (m, 8H), 3.97 (s, 3H); 13C NMR (126 MHz,
Acetone-d6) δ 157.15, 155.87, 149.72, 128.48, 126.31, 126.19, 126.09, 125.57, 125.04, 124.85,
123.88 (d, 2JCF = 34.3 Hz), 123.54 (d, 2JCF = 34.3 Hz), 120.72, 116.43 (d, 3JCF = 6.1 Hz), 116.27
(d, 3JCF = 6.1 Hz), 110.69, 55.60; 19F NMR (376 MHz, Acetone-d6) δ -123.87 to -126.02 (m, 1F);
HRMS (ESI-): calcd for C17H12FN6O2 [M-H]-: 351.1011, found: 351.1015.

248
N5-(2-fluorophenyl)-N6-(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17k). Synthesized by General Procedure 5B. 50%, yellow solid. 1H NMR (400 MHz,
Acetone-d6) δ 10.05 (s, 1H), 8.78 (s, 1H), 7.35 – 7.03 (m, 8H), 3.97 (s, 3H); 13C NMR (126 MHz,
Acetone-d6) δ 161.39, 156.28, 154.31, 130.84, 127.27, 125.85, 125.82, 116.97 (d, 2JCF = 19.1
Hz), 116.81 (d, 2JCF = 19.1 Hz), 114.50, 111.29, 108.09, 55.71; 19F NMR (376 MHz, Acetone-d6)
δ -124.26 to -126.08 (m, 1F); HRMS (ESI-): calcd for C17H12FN6O2 [M-H]-: 351.1011, found:
351.1003.
N5-(2-fluorophenyl)-N6-(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17l). Synthesized by General Procedure 5B. 38%, yellow solid. 1H NMR (400 MHz, Acetone-
d6) δ 10.65 (s, 1H), 9.65 (s, 1H), 8.20 – 7.46 (m, 3H), 7.27 (dt, J = 5.4, 2.7 Hz, 3H), 7.16 – 6.90
(m, 2H), 3.83 (s, 3H); 13C NMR (101 MHz, Acetone-d6) δ 166.60, 165.69, 158.31, 154.18,
127.55, 126.05, 124.30, 117.27 (d, 2JCF = 19.2 Hz), 117.08 (d, 2JCF = 19.2 Hz), 115.31, 56.05; 19F
NMR (376 MHz, Acetone-d6) δ -124.80 (s, 1F); HRMS (ESI+): calcd for C17H14FN6O2 [M+H]+:
353.1162, found: 353.1177.
N5-(2-ethoxyphenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine
(5.17m). Synthesized by General Procedure 5B. 39%, yellow solid. 1H NMR (400 MHz,
Acetone-d6) δ 10.81 (s, 1H), 10.20 (s, 1H), 8.76 (s, 1H), 7.36 (s, 1H), 7.31 – 7.20 (m, 3H), 7.20 –
7.04 (m, 3H), 4.22 (q, J = 7.0 Hz, 2H), 1.42 (t, J = 6.9 Hz, 3H); 13C NMR (126 MHz, Acetone-
d6) δ 156.44, 154.37, 151.11, 150.02, 127.34 (d, 4JCF = 3.0 Hz), 127.31 (d, 4JCF = 3.0 Hz),
126.21, 126.20, 126.04, 124.99, 121.93, 121.59, 117.53 (d, 3JCF = 14.1 Hz), 117.39 (d, 3JCF =
14.1 Hz), 113.04; 19F NMR (376 MHz, Acetone-d6) δ -123.40 to -126.20 (m, 1F); HRMS (ESI+):
calcd for C18H16FN6O2 [M+H]+: 367.1319, found: 367.1322. HPLC Purity: 85% pure.

249
N5-(3-ethoxyphenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17n).
Synthesized by General Procedure 5B. 22%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ
10.76 (s, 1H), 9.77 (s, 1H), 7.95 – 7.41 (m, 2H), 7.39 – 7.20 (m, 5H), 6.82 – 6.71 (m, 1H), 4.09
(q, J = 7.0 Hz, 2H), 1.39 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, Acetonitrile-d3) δ 160.63,
156.32, 154.39, 131.03, 130.76, 127.66, 125.98, 125.10, 123.94, 118.48, 118.26, 118.03, 117.03
(d, 3JCF = 16.2 Hz), 116.87 (d, 3JCF = 16.2 Hz), 114.53, 112.01, 108.78, 108.01, 104.20 (d, 1JCF =
275.7 Hz), 101.47 (d, 1JCF = 275.7 Hz), 64.44, 15.02; 19F NMR (376 MHz, Acetonitrile-d3) δ -
121.14 to -130.75 (m, 1F); HRMS (ESI+): calcd for C18H16FN6O2 [M+H]+: 367.1319, found:
367.1324.
N5-(4-ethoxyphenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.17o).
Synthesized by General Procedure 5B. 55%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ
9.64 (s, 2H), 7.80 (s, 3H), 7.27 (dd, J = 7.4, 4.0 Hz, 3H), 7.03 – 6.96 (m, 2H), 4.08 (q, J = 7.0
Hz, 2H), 1.38 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, acetone) δ 157.24, 156.79 (d, 1JCF =
247.5 Hz), 154.34 (d, 1JCF = 247.5 Hz), 150.29, 148.35 (d, 3JCF = 11.1 Hz), 148.24 (d, 3JCF = 11.1
Hz), 145.51, 133.19, 130.96 (d, 3JCF = 12.2 Hz), 130.84 (d, 3JCF = 12.2 Hz), 127.05 (d, 3JCF = 8.1
Hz), 126.97 (d, 3JCF = 8.1 Hz), 125.66, 125.63, 123.95, 116.92 (d, 2JCF = 19.2 Hz), 116.73 (d,
2JCF = 19.2 Hz), 115.52, 64.26, 15.20; 19F NMR (376 MHz, Acetone-d6) δ -125.05 to -127.41
(m); HRMS (ESI+): calcd for C18H16FN6O2 [M+H]+: 367.1319, found: 367.1313.
3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol (5.17p).
Synthesized by General Procedure 5B. 33%, yellow solid. 1H NMR (400 MHz, Acetone-d6) δ
9.42 (s, 1H), 8.27 – 8.17 (m, 1H), 7.42 – 7.27 (m, 3H), 7.19 (t, J = 8.1 Hz, 1H), 6.73 (t, J = 2.2
Hz, 1H), 6.68 (ddd, J = 8.1, 2.2, 0.9 Hz, 1H), 6.63 (ddd, J = 8.1, 2.3, 0.9 Hz, 1H), 5.00 (s, 2H);
13C NMR (101 MHz, Acetone-d6) δ 157.61 (d, 1JCF = 248.5 Hz), 156.02 (d, 1JCF = 243.4 Hz),

250
155.15 (d, 1JCF = 248.5 Hz), 153.61 (d, 1JCF = 243.4 Hz), 152.07, 151.25, 150.74, 148.59, 130.93,
128.35 (d, 3JCF = 7.1 Hz), 128.28 (d, 3JCF = 7.1 Hz), 126.68, 126.18 (d, 3JCF = 11.1 Hz), 126.07
(d, 3JCF = 11.1 Hz), 125.56, 125.52, 116.73 (d, 2JCF = 19.2 Hz), 116.54 (d, 2JCF = 19.2 Hz),
113.58, 110.08, 108.03; 19F NMR (376 MHz, Acetone-d6) δ -124.79 to -124.95 (m, 1F); HRMS
(ESI+): calcd for C16H12FN6O2 [M+H]+: 339.1006, found: 339.1004.
2,2,2-trifluoro-N-(4-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-
yl)amino)phenyl)acetamide (5.17q). Synthesized by General Procedure 5B. 18%, yellow solid.
1H NMR (400 MHz, Acetonitrile-d3) δ 9.23 (s, 2H), 7.87 – 7.46 (m, 6H), 7.36 – 7.21 (m, 3H);
13C NMR (126 MHz, CD3CN) δ 156.39, 156.24 (q, 2JCF = 29.3 Hz), 155.95 (q, 2JCF = 29.3 Hz),
155.65 (q, 2JCF = 29.3 Hz), 155.35 (q, 2JCF = 29.3 Hz), 154.59, 149.83, 133.91, 127.79, 125.99,
125.49, 123.19, 122.75, 120.35 (q, 1JCF = 230.3 Hz), 117.21 (d, 3JCF = 16.2 Hz), 117.05 (d, 3JCF =
16.2 Hz), 115.78 (q, 1JCF = 230.3 Hz), 113.49 (q, 1JCF = 230.3 Hz); 19F NMR (376 MHz,
Acetonitrile-d3) δ -76.46 to -76.75 (m, 3F), -125.89 (s, 1F); HRMS (ESI+): calcd for
C18H12F4N7O2 [M+H]+: 434.0989, found: 434.0988.
N5-(2-fluorophenyl)-N6-(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17r). Synthesized by General Procedure 5B. 14%, off-white solid. 1H NMR (400
MHz, Acetone-d6) δ 11.10 (s, 1H), 9.88 (s, 1H), 7.56 – 7.40 (m, 2H), 7.39 – 7.23 (m, 6H); 13C
NMR (101 MHz, Acetone-d6) δ 155.32, 155.11, 152.93, 152.69, 140.24, 128.33, 126.13, 125.96,
125.86, 124.88, 124.50 (q, 1JCF = 258.6 Hz), 123.69, 123.15, 122.12, 121.95 (q, 1JCF = 258.6 Hz),
119.39 (q, 1JCF = 258.6 Hz), 116.83, 115.88, 115.74, 115.57; 19F NMR (376 MHz, Acetone-d6) δ
-58.48 (s, 3F), -123.69 to -131.84 (m, 1F); HRMS (ESI+): calcd for C17H11F4N6O2 [M+H]+:
407.0880, found: 407.0897.

251
N5-(2-fluorophenyl)-N6-(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17s). Synthesized by General Procedure 5B. 59%, yellow solid. 1H NMR (400 MHz,
Acetone-d6) δ 10.83 – 9.59 (m, 2H), 7.87 (d, J = 49.5 Hz, 3H), 7.58 (t, J = 8.2 Hz, 1H), 7.33 –
7.22 (m, 3H), 7.20 – 7.14 (m, 1H); 13C NMR (101 MHz, Acetone-d6) δ 156.45, 154.01, 150.43,
131.64, 127.36, 125.91 (d, 4JCF = 3.0 Hz), 125.88 (d, 4JCF = 3.0 Hz), 125.38, 125.10, 122.84 (q,
1JCF = 257.6 Hz), 121.09, 120.29 (q, 1JCF = 257.6 Hz), 117.76, 117.19 (q, 3JCF = 15.2 Hz), 117.04
(q, 3JCF = 15.2 Hz), 116.86 (q, 3JCF = 15.2 Hz), 116.71 (q, 3JCF = 15.2 Hz), 114.97; 19F NMR
(376 MHz, Acetone-d6) δ -58.45 (s, 3F), -120.56 to -130.21 (m, 1F); HRMS (ESI+): calcd for
C17H11F4N6O2 [M+H]+: 407.0880, found: 407.0887.
N5-(2-fluorophenyl)-N6-(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17t). Synthesized by General Procedure 5B. 28%, light yellow solid. 1H NMR (400
MHz, Acetone-d6) δ 10.96 (s, 1H), 9.80 (s, 1H), 8.36 – 7.53 (m, 3H), 7.47 – 7.37 (m, 2H), 7.34 –
7.11 (m, 3H); 13C NMR (101 MHz, acetone) δ 150.62, 131.83, 127.47, 126.06, 126.02, 125.54
(q, 1JCF = 257.6 Hz), 122.99 (q, 1JCF = 257.6 Hz), 121.27, 120.45 (q, 1JCF = 257.6 Hz), 117.93,
117.19 (d, 2JCF = 17.2 Hz), 117.02 (d, 2JCF = 17.2 Hz), 115.15; 13C NMR (126 MHz, Acetone-d6)
δ 155.35, 153.39, 145.48, 126.36, 124.89 (d, 4JCF = 3.0 Hz), 124.86 (d, 4JCF = 3.0 Hz), 123.64 (q,
1JCF = 205.0 Hz), 123.00, 121.87, 121.61 (q, 1JCF = 205.0 Hz), 119.58 (q, 1JCF = 205.0 Hz),
117.55 (q, 1JCF = 205.0 Hz), 116.01 (d, 3JCF = 15.2 Hz), 115.86 (d, 3JCF = 15.2 Hz); 19F NMR
(376 MHz, Acetone-d6) δ -59.45 (s, 3F), -122.88 to -126.12 (m, 1F); HRMS (ESI+): calcd for
C17H11F4N6O2 [M+H]+: 407.0880, found: 407.0872.
N5-(2-fluorophenyl)-N6-(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17u). Synthesized by General Procedure 5B. 27%, off-white solid. 1H NMR (400
MHz, Acetone-d6) δ 11.13 (s, 1H), 9.82 (s, 1H), 8.67 (s, 1H), 7.85 – 7.67 (m, 2H), 7.51 – 7.19

252
(m, 5H); 13C NMR (101 MHz, Acetone-d6) δ 155.85, 153.41, 151.47, 150.72, 144.88, 139.26,
134.76, 129.15 (q, 1JCF = 273.7 Hz), 127.85 (q, 4JCF = 4.0 Hz), 127.81 (q, 4JCF = 4.0 Hz), 127.76
(q, 4JCF = 4.0 Hz), 127.72 (q, 4JCF = 4.0 Hz), 126.63 (d, 4JCF = 7.1 Hz), 126.56 (d, 4JCF = 7.1 Hz),
126.44 (q, 1JCF = 273.7 Hz), 125.96, 125.83 (d, 4JCF = 3.0 Hz), 125.80 (d, 4JCF = 3.0 Hz), 123.73
(q, 1JCF = 273.7 Hz), 123.40, 121.02 (q, 1JCF = 273.7 Hz), 116.28 (d, 3JCF = 18.2 Hz), 116.10 d,
3JCF = 18.2 Hz); 19F NMR (376 MHz, Acetone-d6) δ -62.71 (s, 3F), -131.01 to -131.82 (m, 1F);
HRMS (ESI+): calcd for C17H11F4N6O [M+H]+: 391.0930, found: 391.0932.
N5-(2-fluorophenyl)-N6-(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17v). Synthesized by General Procedure 5B. 49%, off-white solid. 1H NMR (400
MHz, Acetone-d6) δ 10.82 (s, 1H), 9.90 (s, 1H), 8.49 – 7.92 (m, 3H), 7.70 (dd, J = 8.0 Hz, 1H),
7.55 (d, J = 7.8 Hz, 1H), 7.36 – 7.19 (m, 3H); 13C NMR (101 MHz, Acetone-d6) δ 157.30,
154.11, 132.04 (d, 2JCF = 37.4 Hz), 131.67 (d, 2JCF = 37.4 Hz), 131.20, 129.34 (q, 1JCF = 272.7
Hz), 127.35, 127.19, 126.64 (q, 1JCF = 272.7 Hz), 126.11, 125.93 (d, 4JCF = 3.0 Hz), 125.90 (d,
4JCF = 3.0 Hz), 125.25, 123.94 (q, 1JCF = 272.7 Hz), 122.12, 119.03, 117.23 (q, 2JCF = 20.2 Hz),
117.03 23 (q, 2JCF = 20.2 Hz), 116.84 23 (q, 2JCF = 20.2 Hz), 111.02; 19F NMR (376 MHz,
Acetone-d6) δ -63.20 , -123.46 to -132.24 (m); HRMS (ESI+): calcd for C17H11F4N6O [M+H]+:
391.0930, found: 391.0917. HPLC Purity: 92% pure.
N5-(2-fluorophenyl)-N6-(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-
diamine (5.17w). Synthesized by General Procedure 5B. 44%, off-white solid. 1H NMR (400
MHz, Acetone-d6) δ 10.97 (s, 1H), 9.87 (s, 1H), 8.60 – 7.85 (m, 3H), 7.80 (d, J = 8.4 Hz, 2H),
7.28 (dt, J = 10.7, 4.1 Hz, 3H); 13C NMR (101 MHz, Acetone-d6) δ 161.37, 158.62, 152.49,
131.70, 127.16, 126.70, 126.40, 125.88, 125.75 (d, 4JCF = 1.0 Hz), 125.74 (d, 4JCF = 1.0 Hz),
124.84, 124.01, 122.38, 121.78, 117.05 (q, 3JCF = 15.2 Hz), 116.90 (q, 3JCF = 15.2 Hz), 116.71

253
(q, 3JCF = 15.2 Hz), 116.57 (q, 3JCF = 15.2 Hz); 19F NMR (376 MHz, Acetone-d6) δ -62.51 (s,
3F), -124.05 to -131.33 (m, 1F); HRMS (ESI+): calcd for C17H10F4N6NaO [M+Na]+: 413.0750,
found: 413.0740. HPLC Purity: 86% pure.
[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (5.18). 5.15 (1 equiv.) was dissolved in ACN (6M
solution), and 30% aqueous ammonium (2M solution) was added to the solution at 0 ºC. The
reaction mixture was allowed to warm to rt and stirred for 3 h, resulting in precipitate formation.
The precipitated was filtered, washed with water, and dried, giving the title compound as a
yellow solid in quantitative yield that was carried forward as is. Analytical data matches with the
literature.205
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2,2,2-trifluoroacetamide) (5.19a).
Synthesized by General Procedure 5C. 40%, clear solid. 13C NMR (126 MHz, CD3OD) δ 152.18,
151.10, 124.52 (q, 1JCF = 229.3 Hz), 122.25 (q, 1JCF = 229.3 Hz), 119.98 (q, 1JCF = 229.3 Hz),
117.71 (q, 1JCF = 229.27 Hz; 19F NMR (376 MHz, CD3OD) δ -86.90 (s, 6F); GCMS (EI): calcd
for C8H2F6N6O3 [M*-2F]: 306.0, found: 306.1.
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-methylbenzenesulfonamide) (5.19b).
Synthesized by General Procedure 5C. 13%, off-white solid. 1H NMR (400 MHz, CD3OD) δ
7.60 (d, J = 7.9 Hz, 4H), 7.05 (d, J = 7.9 Hz, 4H), 2.34 (s, 6H); 13C NMR (126 MHz, CD3OD) δ
152.40*, 152.04*, 142.81*, 142.10*, 129.68*, 128.78*, 127.74*, 115.52*, 115.04*, 21.35;
HRMS (ESI-): calcd for C18H20N7O5S2 [M+NH4+]+: 478.0962, found: 478.0997.
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-fluorobenzenesulfonamide) (5.19c).
Synthesized by General Procedure 5C. 13%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.77 –
7.70 (m, 4H), 7.09 – 7.02 (m, 4H); 13C NMR (101 MHz, CD3OD) δ 166.77 (d, 1JCF = 251.5 Hz),
164.28 (d, 1JCF = 251.5 Hz), 152.41, 152.03, 141.82, 130.52 (d, 3JCF = 8.1 Hz), 130.43 (d, 3JCF =

254
8.1 Hz), 116.13 (d, 2JCF = 23.2 Hz), 115.90 (d, 2JCF = 23.2 Hz); 19F NMR (376 MHz, CD3OD) δ -
111.07 to -111.22 (m, 2F); HRMS (ESI-): calcd for C16H13F2N7O5S2 [M+NH4+]+: 485.0388,
found: 485.0124.
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(3-(trifluoromethyl)benzenesulfonamide)
(5.19d). Synthesized by General Procedure 5C. 13%, golden yellow solid. . 1H NMR (400 MHz,
CD3OD) δ 7.95 (dt, J = 7.9, 1.4 Hz, 2H), 7.90 (t, J = 1.9 Hz, 2H), 7.70 (dt, J = 7.9, 1.9, 1.0 Hz,
2H), 7.57 – 7.51 (m, 2H); 13C NMR (126 MHz, Methanol-d4) δ 152.41, 152.04, 146.39, 131.97
(q, 2JCF = 26.3 Hz), 131.71 (q, 2JCF = 26.3 Hz), 131.45 (q, 2JCF = 26.3 Hz), 131.39, 131.19 (q,
2JCF = 26.3 Hz), 130.47, 128.84, 128.81, 128.78, 128.23 (q, 1JCF = 218.2 Hz), 126.07 (q, 1JCF =
218.2 Hz), 124.55, 124.52, 124.49, 124.47, 123.91 (q, 1JCF = 218.2 Hz), 121.75 (q, 1JCF = 218.2
Hz); 19F NMR (376 MHz, Methanol-d4) δ -64.27 (s, 3F); HRMS (ESI-): calcd for
C18H14F6N7O5S2 [M+NH4+]+: 586.0402, found: 586.0390.
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2-nitrobenzenesulfonamide (5.19e).
Synthesized by General Procedure 5C. 29%, yellow solid. 1H NMR (400 MHz, CD3OD) δ 8.07
(s, 1H), 8.05 (s, 1H), 7.65 – 7.54 (m, 8H); 13C NMR (101 MHz, CD3OD) δ 152.40, 152.04,
149.47, 137.53, 133.40, 132.11, 131.52, 124.49; HRMS (ESI+): calcd for C16H14N9O9S2
[M+NH4+]+: 540.0356, found: 540.0319.
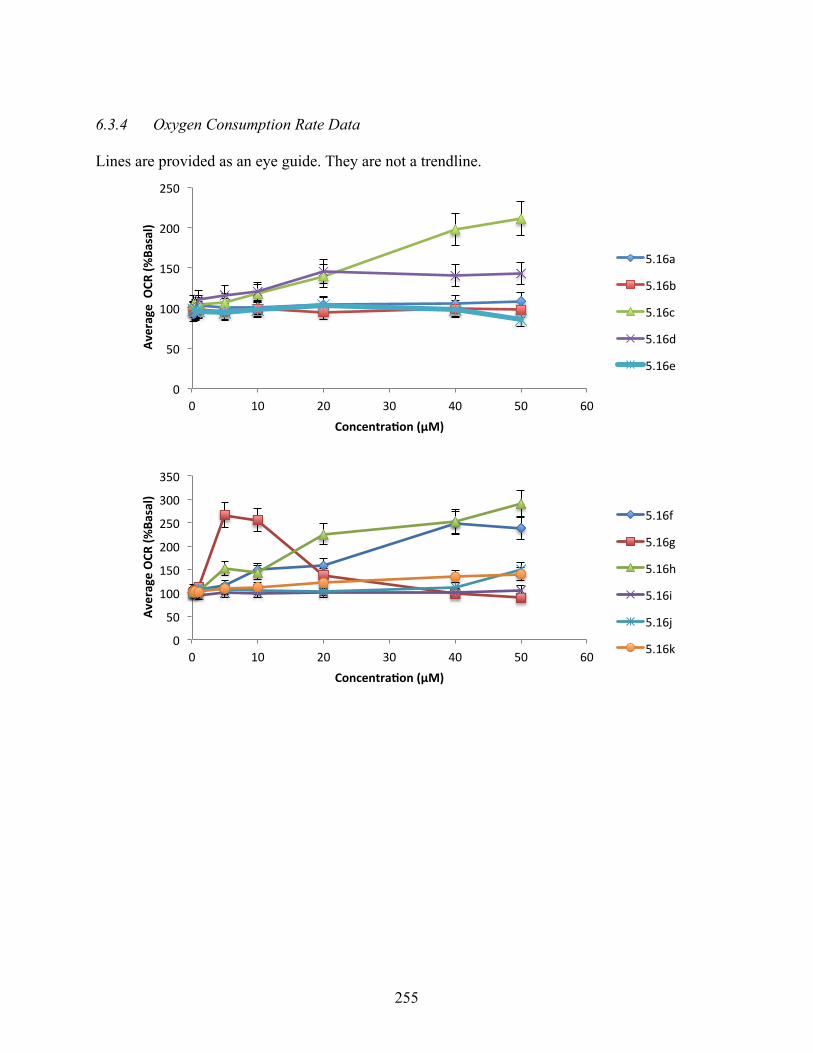
255
6.3.4 Oxygen Consumption Rate Data
Lines are provided as an eye guide. They are not a trendline.
0
50
100
150
200
250
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.16a
5.16b
5.16c
5.16d
5.16e
0
50
100
150
200
250
300
350
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.16f
5.16g
5.16h
5.16i
5.16j
5.16k
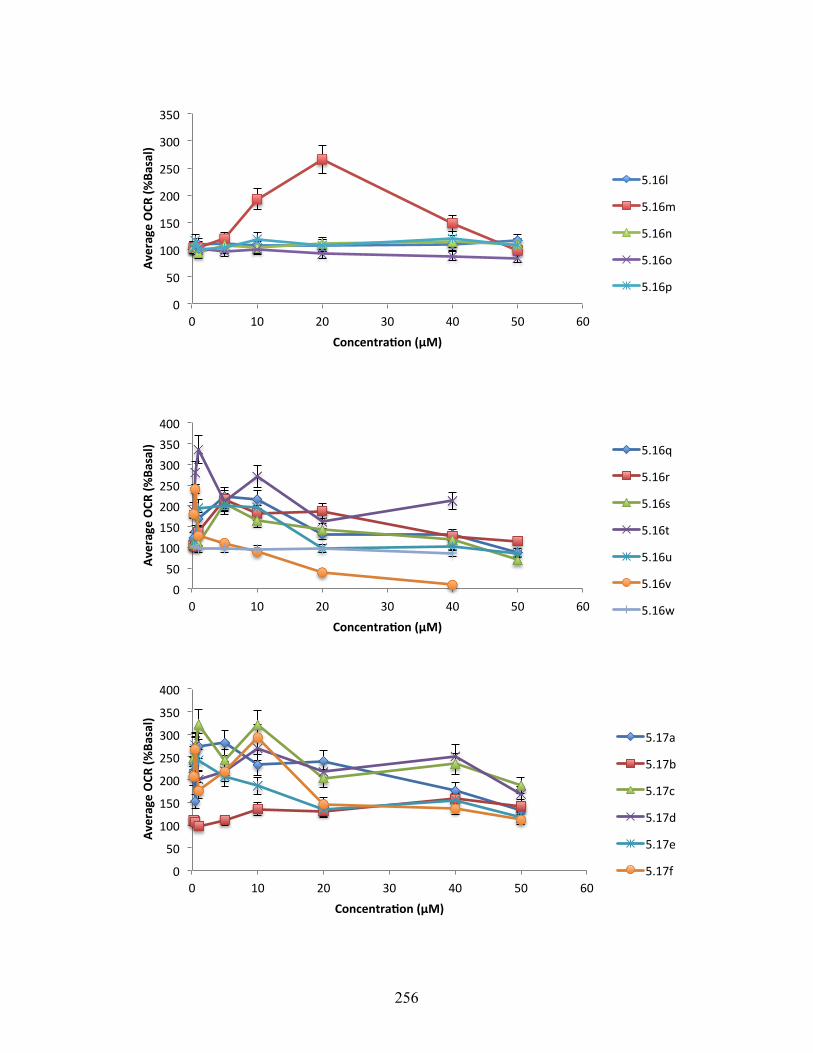
256
0
50
100
150
200
250
300
350
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.16l
5.16m
5.16n
5.16o
5.16p
050100150200250300350400
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.16q
5.16r
5.16s
5.16t
5.16u
5.16v
5.16w
0
50
100
150
200
250
300
350
400
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.17a
5.17b
5.17c
5.17d
5.17e
5.17f
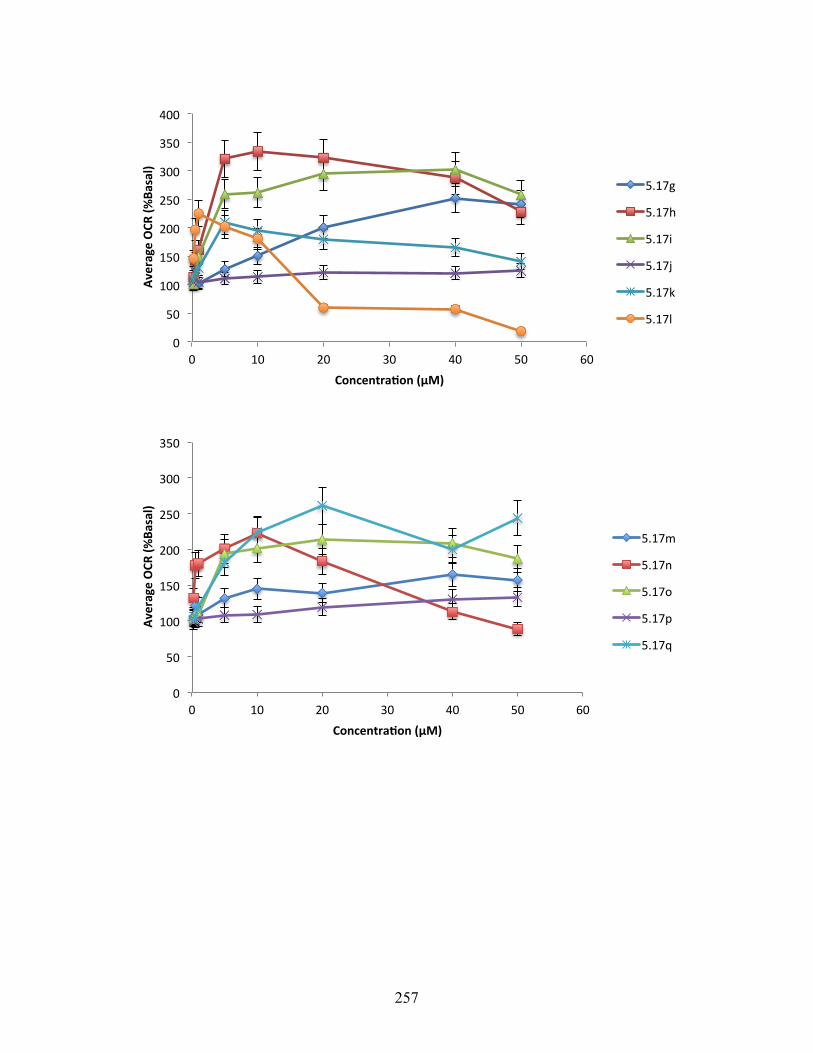
257
0
50
100
150
200
250
300
350
400
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.17g
5.17h
5.17i
5.17j
5.17k
5.17l
0
50
100
150
200
250
300
350
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.17m
5.17n
5.17o
5.17p
5.17q
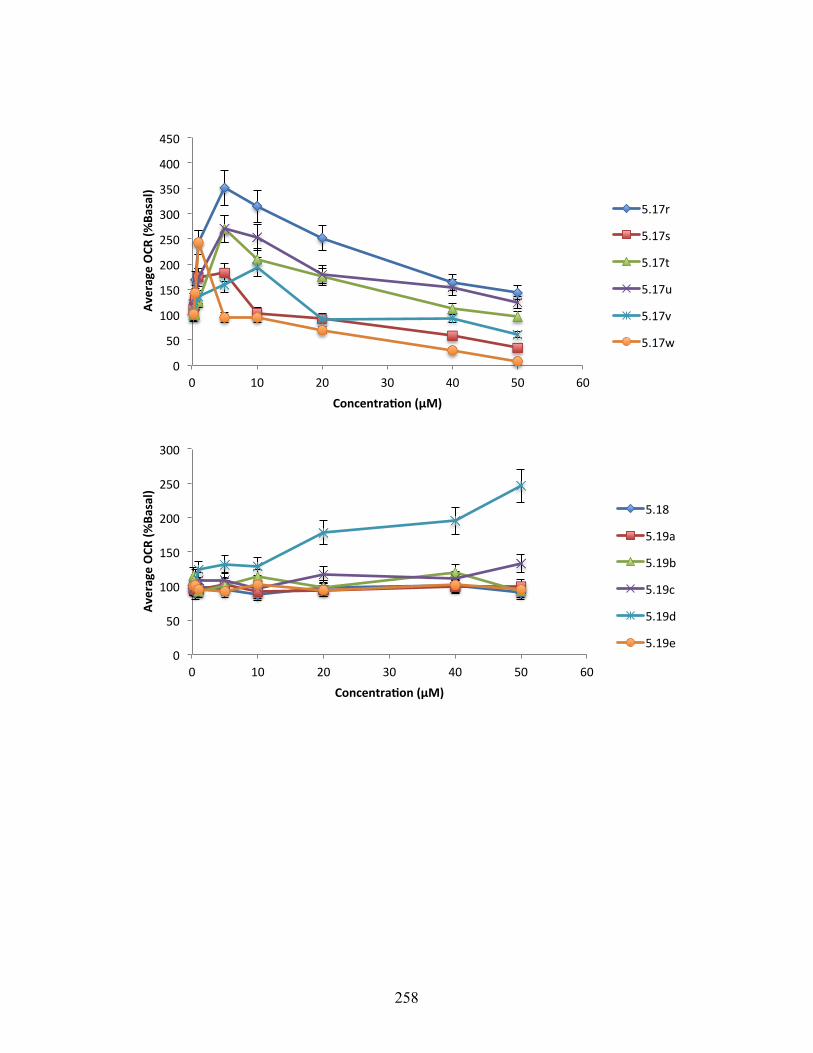
258
0
50
100
150
200
250
300
350
400
450
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.17r
5.17s
5.17t
5.17u
5.17v
5.17w
0
50
100
150
200
250
300
0 10 20 30 40 50 60
AverageOCR
(%Ba
sal)
Concentra8on(µM)
5.18
5.19a
5.19b
5.19c
5.19d
5.19e
259
6.3.5 NMR Spectra
5,6-dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine 13CNMR (5.13)
N5,N6-di-tert-butyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16a)
0102030405060708090100110120130140150160170f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190MU-dCl_CARBON_01-3
151.85
156.22
O1
N2
3
4
N5
N6
7
8
N9
Cl10
Cl11
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700MU-50_PROTON_01-2
18.00
1.53
2.68
7.96
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
CH314
CH315
CH316
CH317
CH318
CH319
X
260
N5,N6-di-tert-butyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16a)
N5,N6-dicyclopropyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16b)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
MU-50_CARBON_01
28.47
54.65
149.17
150.35
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
CH314
CH315
CH316
CH317
CH318
CH319
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
MU-63_re_PROTON_01-2
4.10
4.16
2.00
1.79
0.59
0.60
0.60
0.61
0.61
0.62
0.63
0.83
0.84
0.84
0.85
0.86
0.86
0.87
0.88
2.99
2.99
3.00
3.01
3.02
3.03
3.04
3.07
7.32
7.32
X
261
N5,N6-dicyclopropyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16b)
N5,N6-dicyclopentyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR(5.16c)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600MU-63_re_C_CARBON_01
6.94
25.51
150.97
151.67
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
MU-65_re_PROTON_01
12.00
3.95
2.00
2.09
1.51
1.51
1.52
1.52
1.53
1.54
1.54
1.55
1.56
1.57
1.57
1.57
1.58
1.58
1.59
1.59
1.60
1.61
1.62
1.62
1.62
1.63
1.63
1.64
1.64
1.65
1.65
1.66
1.66
1.67
1.68
1.68
1.69
1.70
1.71
1.71
1.71
1.72
1.72
2.06
2.07
2.07
2.08
2.09
2.09
2.10
2.10
2.11
2.12
2.13
2.13
2.13
2.14
2.14
2.15
4.46
4.48
4.49
4.51
7.16
7.16
7.17
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
1415
16
17
18
19
20
21
X
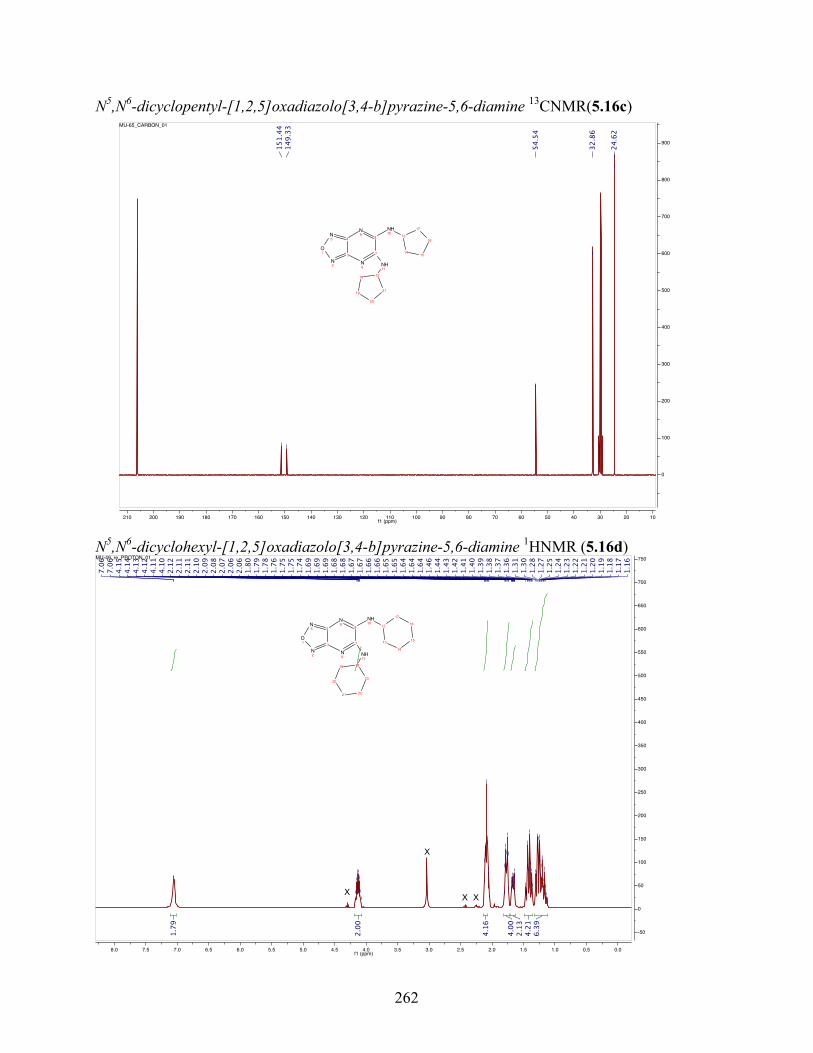
262
N5,N6-dicyclopentyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR(5.16c)
N5,N6-dicyclohexyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16d)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750MU-49_re_PROTON_01
6.39
4.21
2.13
4.00
4.16
2.00
1.79
1.16
1.17
1.18
1.19
1.20
1.21
1.22
1.23
1.24
1.25
1.27
1.28
1.30
1.31
1.36
1.37
1.38
1.39
1.40
1.41
1.42
1.43
1.44
1.46
1.64
1.64
1.64
1.65
1.65
1.66
1.66
1.67
1.67
1.68
1.68
1.69
1.69
1.69
1.74
1.75
1.75
1.76
1.78
1.79
1.80
2.06
2.06
2.07
2.08
2.09
2.10
2.11
2.11
2.12
4.10
4.11
4.12
4.13
4.14
4.15
7.06
7.06
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
X
X
X X
102030405060708090100110120130140150160170180190200210f1 (ppm)
0
100
200
300
400
500
600
700
800
900
MU-65_CARBON_01
24.62
32.86
54.54
149.33
151.44
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
1415
16
17
18
19
20
21
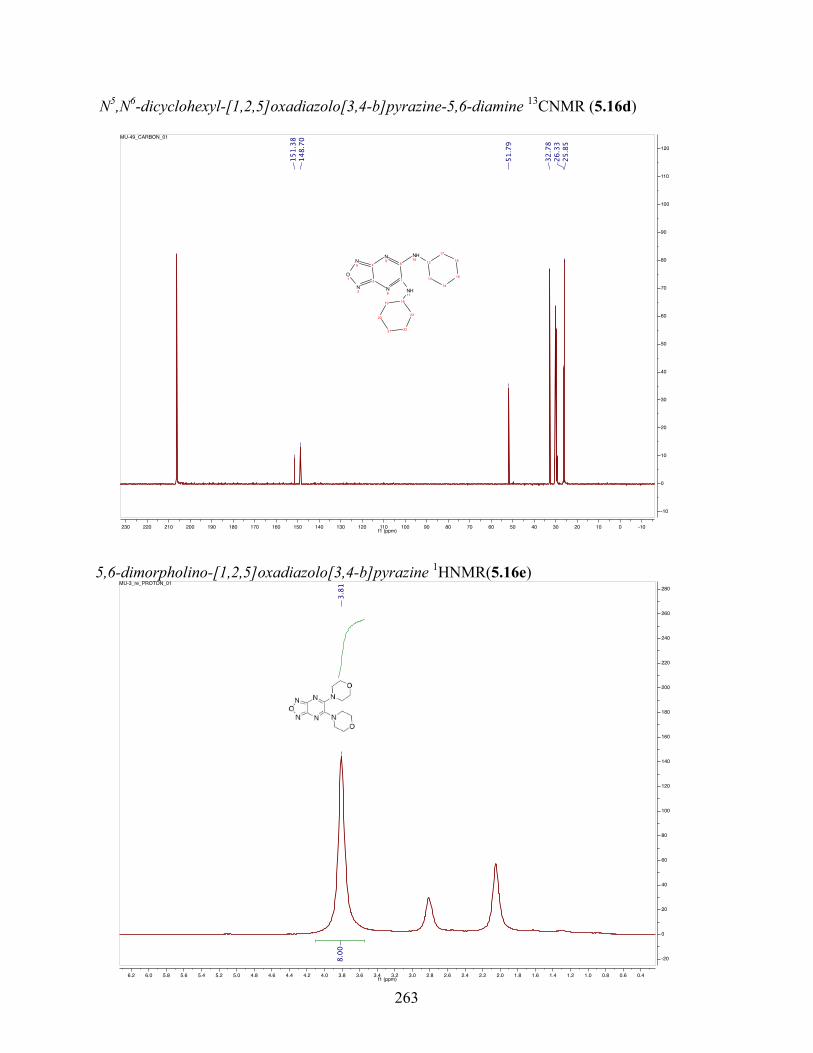
263
N5,N6-dicyclohexyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16d)
5,6-dimorpholino-[1,2,5]oxadiazolo[3,4-b]pyrazine 1HNMR(5.16e)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
MU-49_CARBON_01
25.85
26.33
32.78
51.79
148.70
151.38
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
0.40.60.81.01.21.41.61.82.02.22.42.62.83.03.23.43.63.84.04.24.44.64.85.05.25.45.65.86.06.2f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280MU-3_re_PROTON_01
8.00
3.81
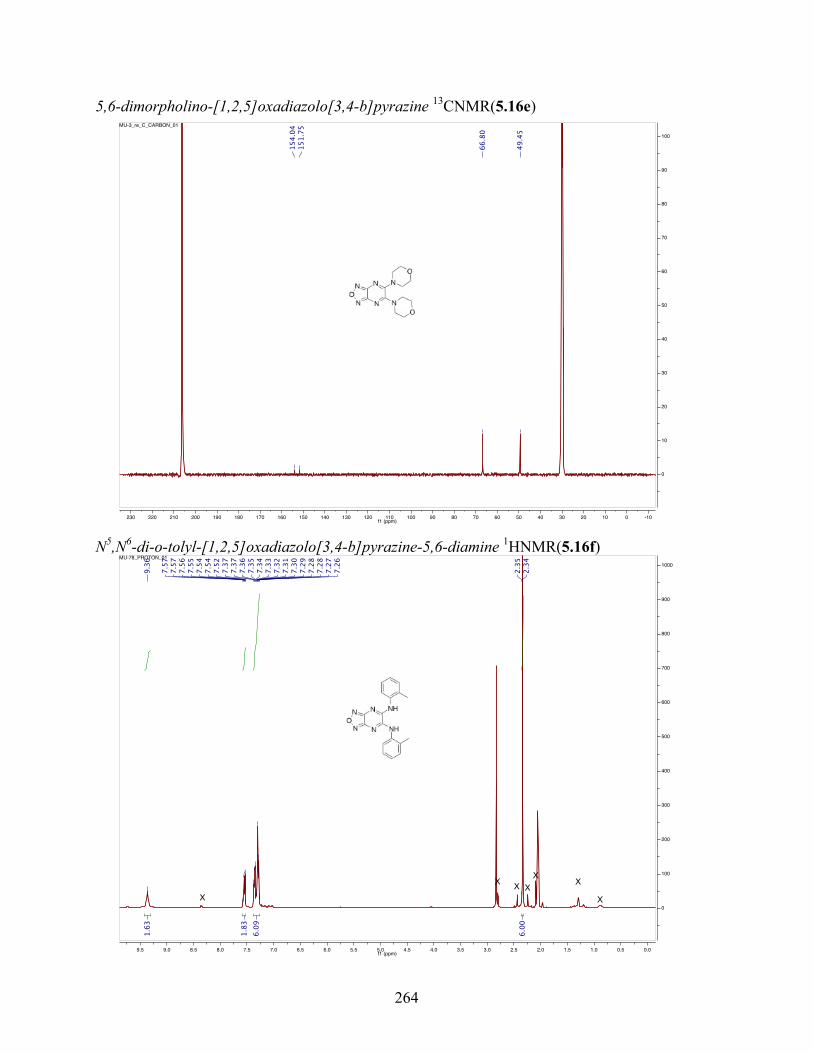
264
5,6-dimorpholino-[1,2,5]oxadiazolo[3,4-b]pyrazine 13CNMR(5.16e)
N5,N6-di-o-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR(5.16f)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
0
10
20
30
40
50
60
70
80
90
100
MU-3_re_C_CARBON_01
49.45
66.80
151.75
154.04
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.5f1 (ppm)
0
100
200
300
400
500
600
700
800
900
1000
MU-78_PROTON_01
6.00
6.09
1.83
1.63
2.34
2.35
7.26
7.27
7.28
7.28
7.29
7.30
7.31
7.32
7.33
7.34
7.35
7.36
7.37
7.37
7.52
7.54
7.54
7.55
7.56
7.57
7.57
9.36
X
X X
XX
X
X
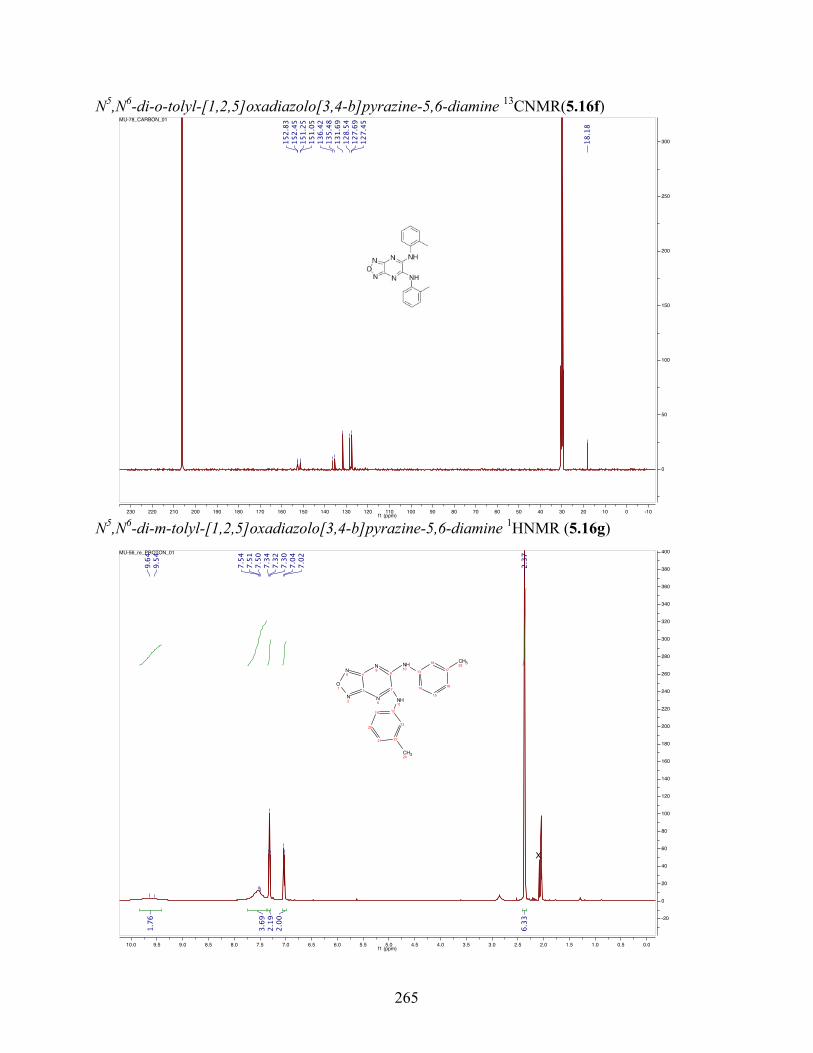
265
N5,N6-di-o-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR(5.16f)
N5,N6-di-m-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16g)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
0
50
100
150
200
250
300
MU-78_CARBON_01
18.18
127.45
127.69
128.54
131.69
135.48
136.42
151.05
151.25
152.45
152.83
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
380
400MU-56_re_PROTON_01
6.33
2.00
2.19
3.69
1.76
2.37
7.02
7.04
7.30
7.32
7.34
7.50
7.51
7.54
9.54
9.64
X
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
2122
23
CH324
CH325
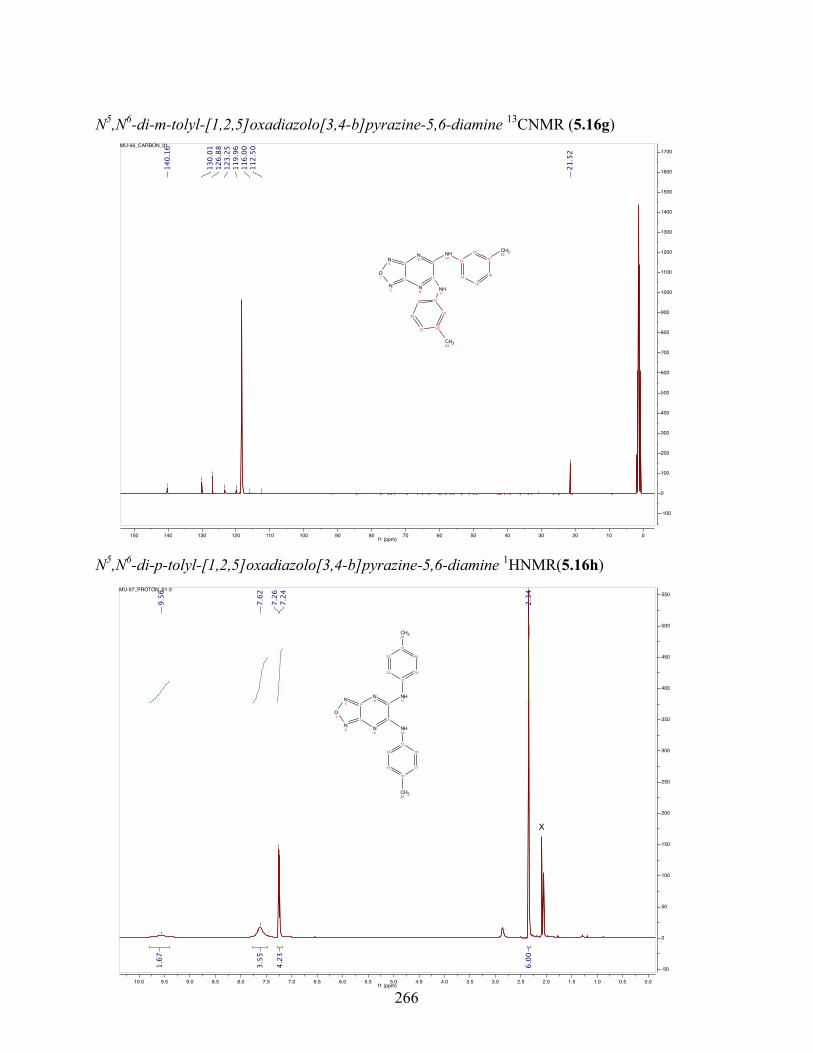
266
N5,N6-di-m-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16g)
N5,N6-di-p-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR(5.16h)
0102030405060708090100110120130140150f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
MU-56_CARBON_01
21.52
112.50
116.00
119.96
123.25
126.88
130.01
140.16
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
2122
23
CH324
CH325
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550MU-57_PROTON_01-3
6.00
4.23
3.55
1.67
2.34
7.24
7.26
7.62
9.56
X
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
CH324
CH325
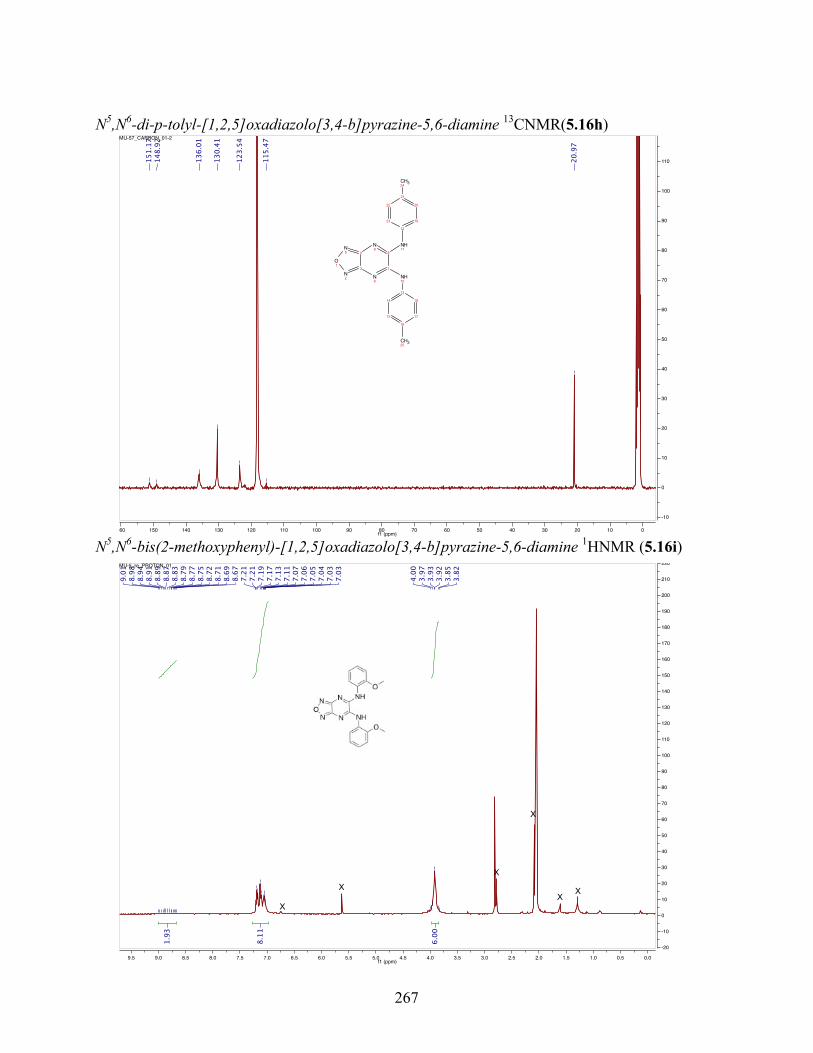
267
N5,N6-di-p-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR(5.16h)
N5,N6-bis(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16i)
0102030405060708090100110120130140150160f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
MU-57_CARBON_01-2
20.97
115.47
123.54
130.41
136.01
148.92
151.17
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
CH324
CH325
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.5f1 (ppm)
-20
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220MU-5_re_PROTON_01
6.00
8.11
1.93
3.82
3.85
3.92
3.93
3.97
4.00
7.03
7.03
7.04
7.05
7.06
7.07
7.11
7.13
7.17
7.19
7.21
7.21
8.67
8.69
8.71
8.72
8.75
8.77
8.79
8.83
8.87
8.89
8.91
8.94
8.98
9.01
X
X
X
X
XX
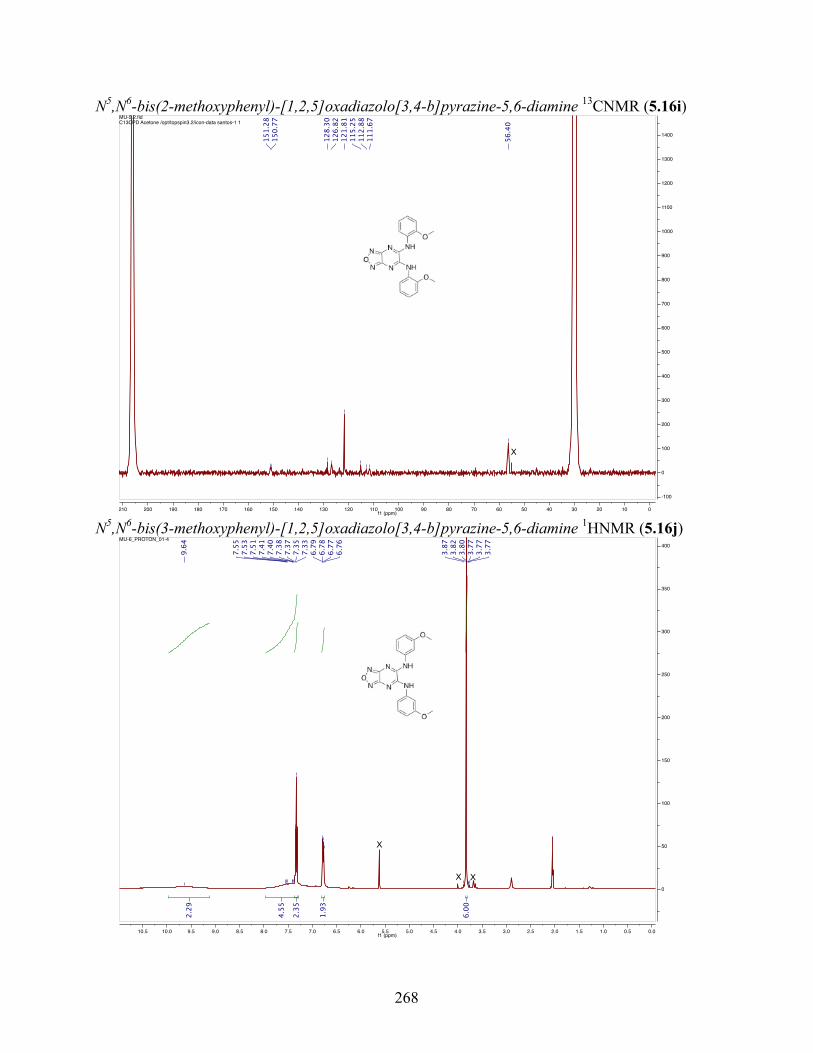
268
N5,N6-bis(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16i)
N5,N6-bis(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16j)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
MU-5.2.fidC13CPD Acetone /opt/topspin3.2/icon-data santos-1 1
56.40
111.67
112.88
115.25
121.81
126.82
128.30
150.77
151.28
X
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
0
50
100
150
200
250
300
350
400
MU-6_PROTON_01-4
6.00
1.93
2.35
4.55
2.29
3.77
3.77
3.77
3.80
3.82
3.87
6.76
6.77
6.78
6.79
7.33
7.35
7.37
7.38
7.40
7.41
7.51
7.53
7.55
9.64
X
X X
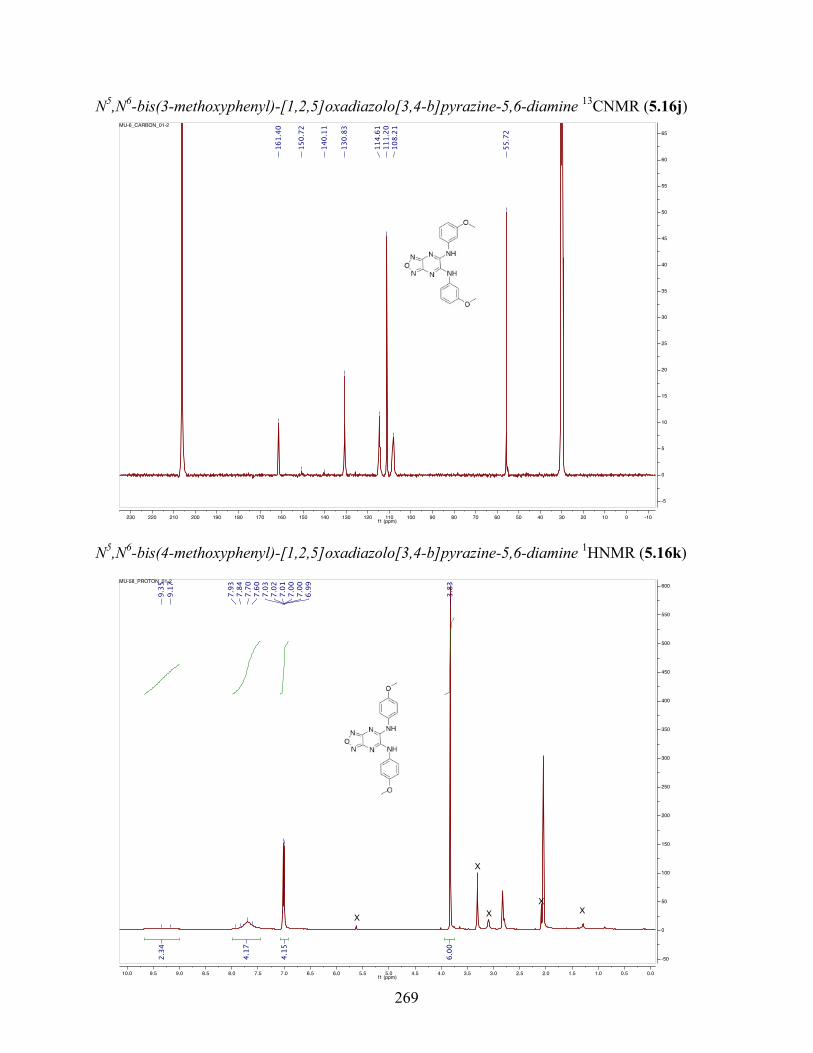
269
N5,N6-bis(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16j)
N5,N6-bis(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16k)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
MU-6_CARBON_01-2
55.72
108.21
111.20
114.61
130.83
140.11
150.72
161.40
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600MU-58_PROTON_01-2
6.00
4.15
4.17
2.34
3.83
6.99
7.00
7.00
7.01
7.02
7.03
7.60
7.70
7.84
7.93
9.17
9.35
X
XX
X
X
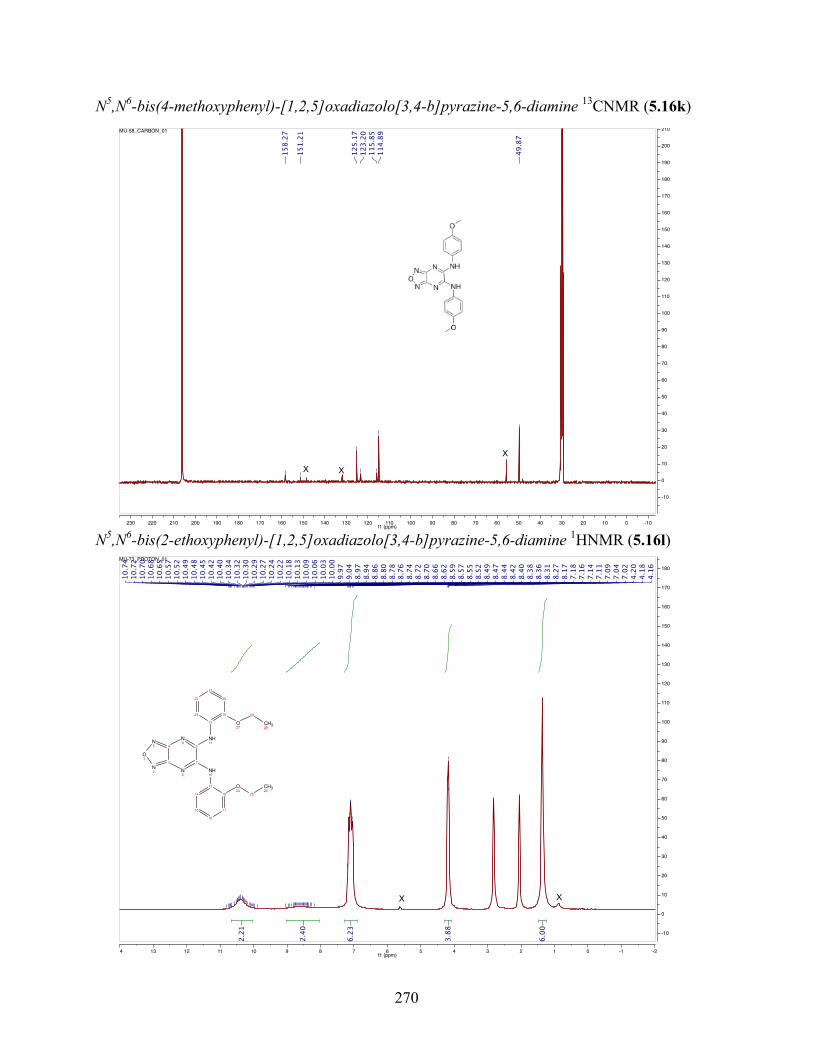
270
N5,N6-bis(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16k)
N5,N6-bis(2-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16l)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210MU-58_CARBON_01
49.87
114.89
115.85
123.20
125.17
151.21
158.27
X
X X
-2-101234567891011121314f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
MU-73_PROTON_01
6.00
3.88
6.23
2.40
2.21
4.16
4.18
4.20
7.02
7.04
7.09
7.11
7.14
7.16
7.18
8.17
8.27
8.31
8.36
8.38
8.40
8.42
8.44
8.47
8.49
8.52
8.55
8.57
8.59
8.62
8.66
8.70
8.72
8.74
8.76
8.78
8.80
8.86
8.94
8.97
9.04
9.97
10.00
10.03
10.06
10.09
10.13
10.18
10.22
10.24
10.27
10.29
10.30
10.32
10.34
10.40
10.42
10.45
10.48
10.49
10.52
10.57
10.65
10.68
10.70
10.72
10.74
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
25
CH326
O27
28
CH329
XX
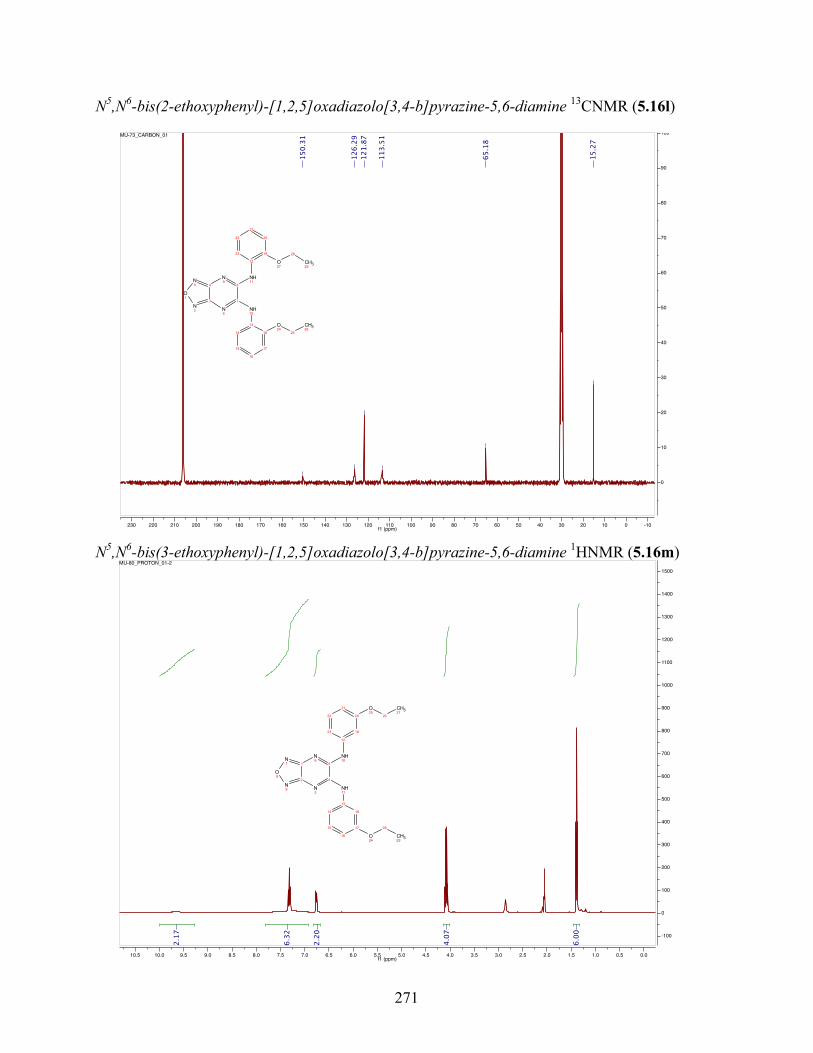
271
N5,N6-bis(2-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16l)
N5,N6-bis(3-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16m)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
0
10
20
30
40
50
60
70
80
90
100MU-73_CARBON_01
15.27
65.18
113.51
121.87
126.29
150.31
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
25
CH326
O27
28
CH329
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
MU-80_PROTON_01-2
6.00
4.07
2.20
6.32
2.17
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
O25
26
CH327
28
CH329
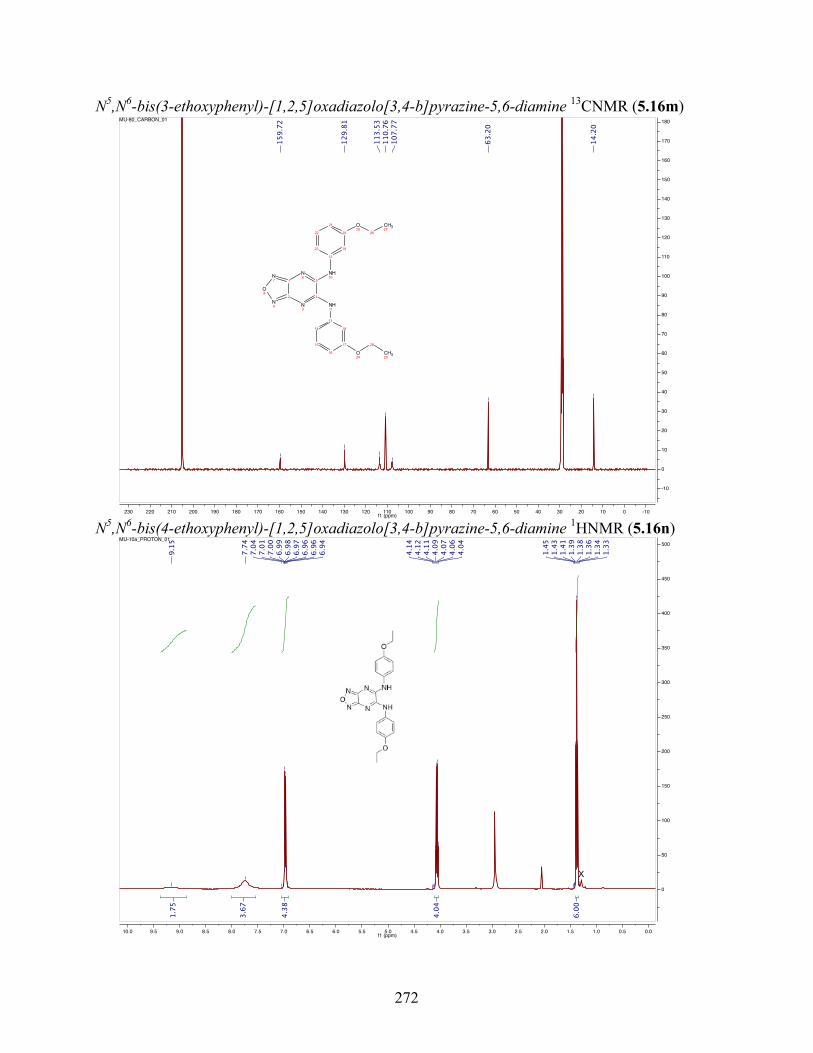
272
N5,N6-bis(3-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16m)
N5,N6-bis(4-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16n) 0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0
f1 (ppm)
0
50
100
150
200
250
300
350
400
450
500MU-10a_PROTON_01
6.00
4.04
4.38
3.67
1.75
1.33
1.34
1.36
1.38
1.39
1.41
1.43
1.45
4.04
4.06
4.07
4.09
4.11
4.12
4.14
6.94
6.96
6.96
6.97
6.98
6.99
7.00
7.01
7.04
7.74
9.15
X
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180MU-80_CARBON_01
14.20
63.20
107.77
110.76
113.53
129.81
159.72
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
O25
26
CH327
28
CH329
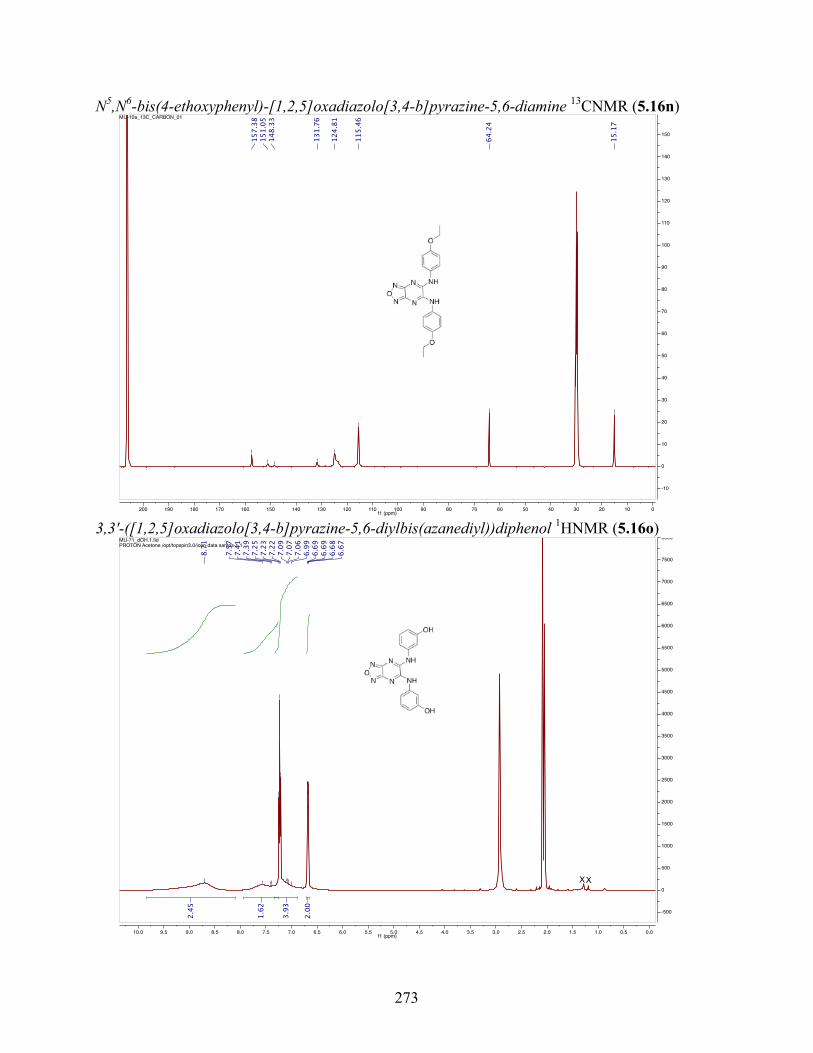
273
N5,N6-bis(4-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16n)
3,3'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))diphenol 1HNMR (5.16o)
0102030405060708090100110120130140150160170180190200f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
MU-10a_13C_CARBON_01
15.17
64.24
115.46
124.81
131.76
148.33
151.05
157.38
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000MU-71_dOH.1.fidPROTON Acetone /opt/topspin3.0/icon-data santos-1 1
2.00
3.93
1.62
2.45
6.67
6.68
6.69
6.69
6.99
7.06
7.07
7.09
7.22
7.23
7.25
7.39
7.41
7.57
8.71
XX
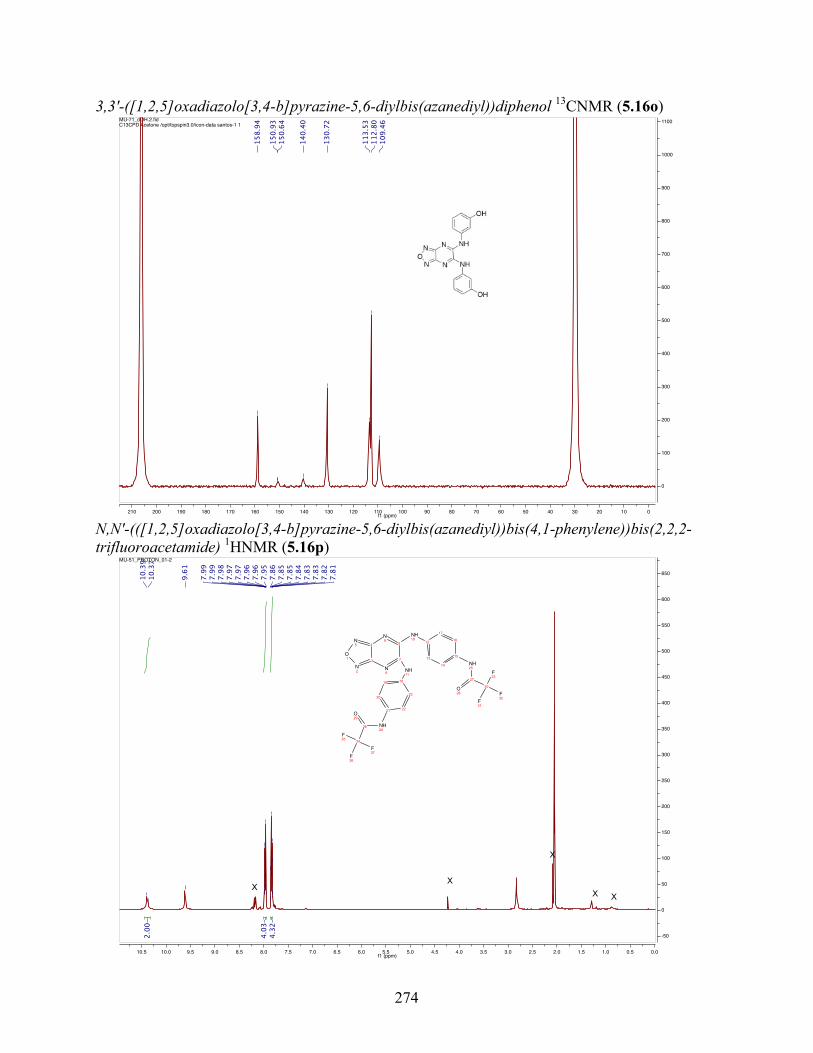
274
3,3'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))diphenol 13CNMR (5.16o)
N,N'-(([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))bis(4,1-phenylene))bis(2,2,2-trifluoroacetamide) 1HNMR (5.16p)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
0
100
200
300
400
500
600
700
800
900
1000
1100MU-71_dOH.2.fidC13CPD Acetone /opt/topspin3.0/icon-data santos-1 1
109.46
112.80
113.53
130.72
140.40
150.64
150.93
158.94
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
MU-51_PROTON_01-2
4.32
4.03
2.00
7.81
7.82
7.83
7.83
7.84
7.85
7.85
7.86
7.95
7.96
7.96
7.97
7.97
7.98
7.99
7.99
9.61
10.37
10.39
X
X
XX
X
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
NH24
NH25
26
27
O28
O29
30
F31
F32
F33
34
F35
F36
F37
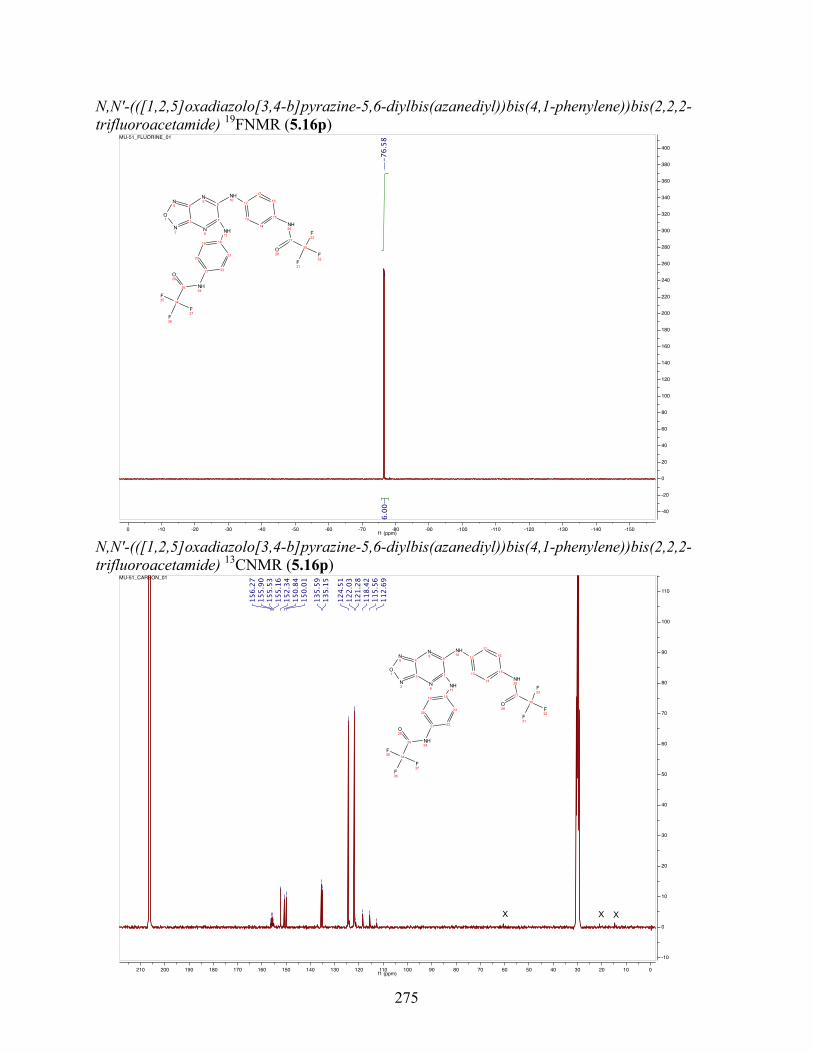
275
N,N'-(([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))bis(4,1-phenylene))bis(2,2,2-trifluoroacetamide) 19FNMR (5.16p)
N,N'-(([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))bis(4,1-phenylene))bis(2,2,2-trifluoroacetamide) 13CNMR (5.16p)
-150-140-130-120-110-100-90-80-70-60-50-40-30-20-100f1 (ppm)
-40
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
380
400
MU-51_FLUORINE_01
6.00
-76.58
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
NH24
NH25
26
27
O28
O29
30
F31
F32
F33
34
F35
F36
F37
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
MU-51_CARBON_01
112.69
115.56
118.42
121.28
122.03
124.51
135.15
135.59
150.01
150.84
152.34
155.16
155.53
155.90
156.27
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
NH24
NH25
26
27
O28
O29
30
F31
F32
F33
34
F35
F36
F37
X X X
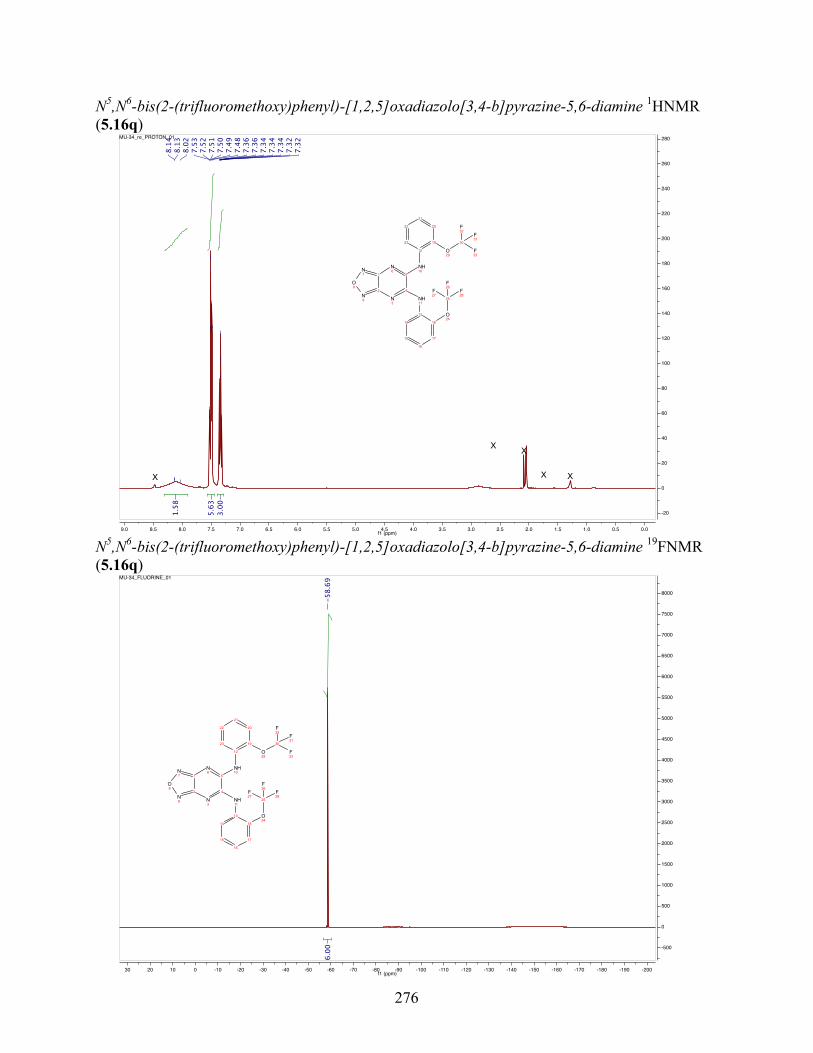
276
N5,N6-bis(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16q)
N5,N6-bis(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16q)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280MU-34_re_PROTON_01
3.00
5.63
1.58
7.32
7.32
7.34
7.34
7.34
7.36
7.36
7.48
7.49
7.50
7.51
7.52
7.53
8.02
8.13
8.14
X
X X
X
X
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
25
F26
F27
F28
O29
30
F31
F32
F33
-200-190-180-170-160-150-140-130-120-110-100-90-80-70-60-50-40-30-20-100102030f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
MU-34_FLUORINE_01
6.00
-58.69
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
25
F26
F27
F28
O29
30
F31
F32
F33
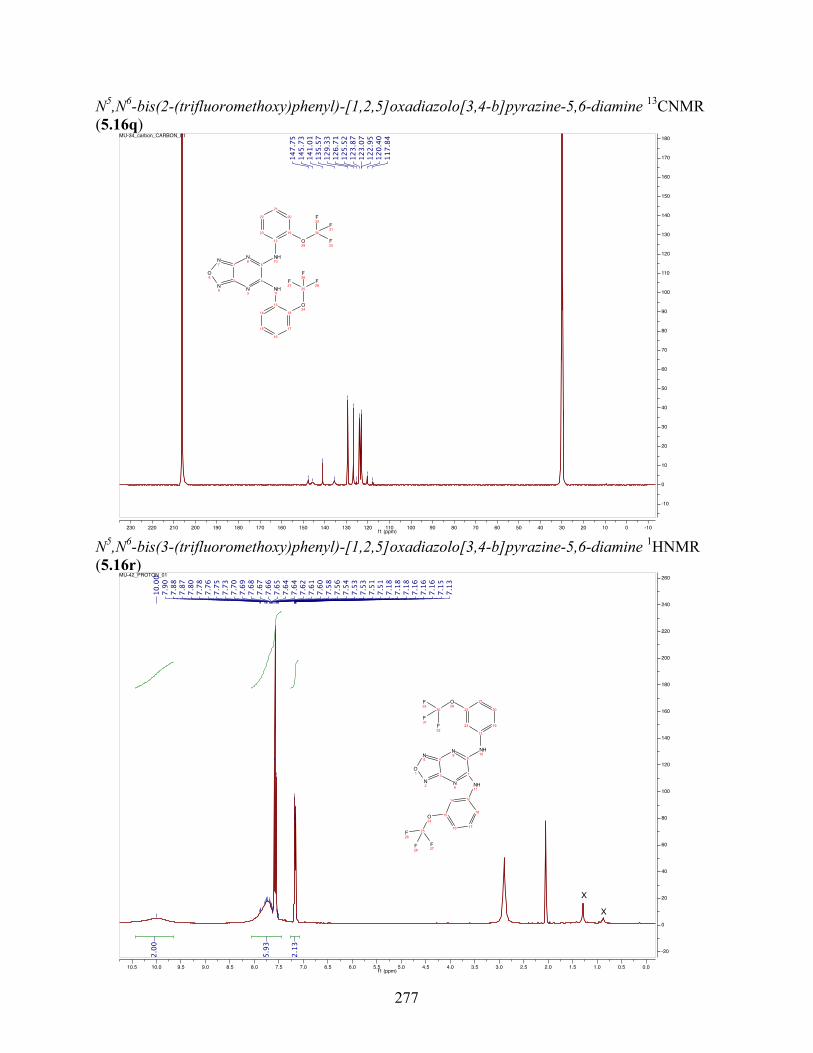
277
N5,N6-bis(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16q)
N5,N6-bis(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16r)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260MU-42_PROTON_01
2.13
5.93
2.00
7.13
7.15
7.16
7.16
7.16
7.18
7.18
7.18
7.51
7.51
7.53
7.53
7.54
7.56
7.58
7.60
7.61
7.62
7.64
7.64
7.65
7.66
7.67
7.68
7.69
7.70
7.73
7.75
7.76
7.78
7.80
7.87
7.88
7.90
10.00
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
1617
18
19
20
21
22
23
O24
25
F26
F27
F28
O29
30
F31
F32
F33
X
X
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180MU-34_carbon_CARBON_01
117.84
120.40
122.95
123.07
123.87
125.52
126.71
129.33
135.57
141.01
145.73
147.75
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
O24
25
F26
F27
F28
O29
30
F31
F32
F33
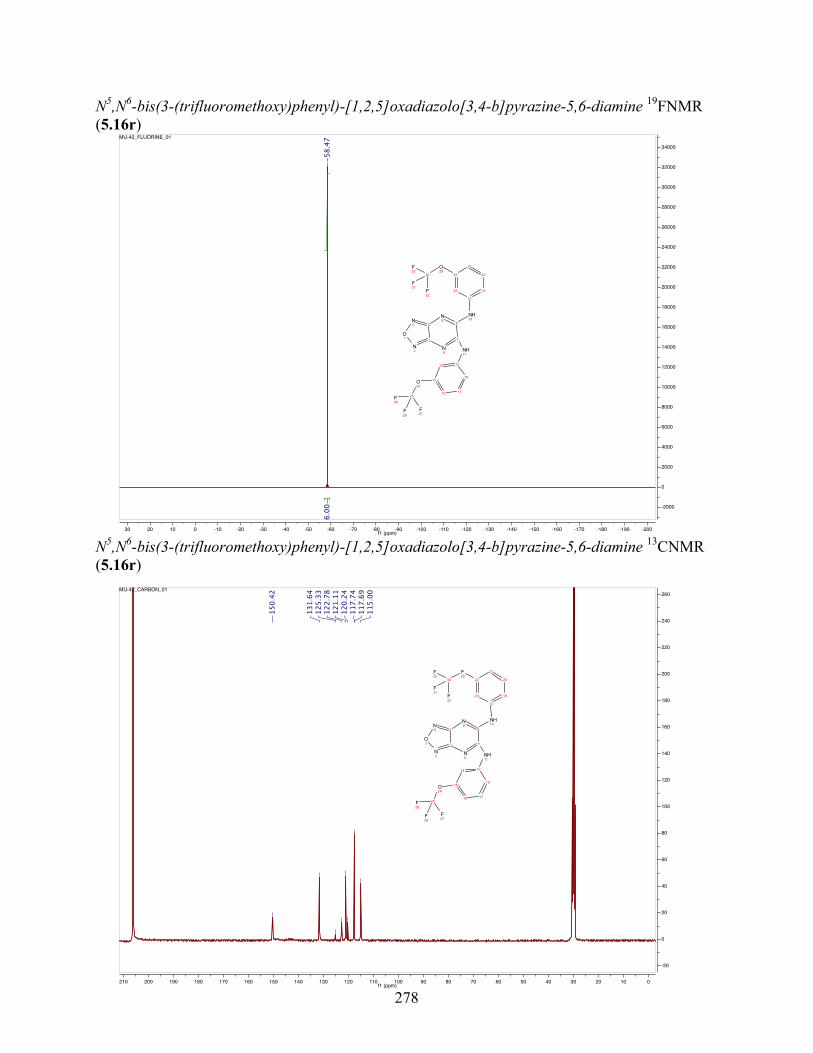
278
N5,N6-bis(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16r)
N5,N6-bis(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16r)
-200-190-180-170-160-150-140-130-120-110-100-90-80-70-60-50-40-30-20-100102030f1 (ppm)
-2000
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
30000
32000
34000
MU-42_FLUORINE_01
6.00
-58.47
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
1617
18
19
20
21
22
23
O24
25
F26
F27
F28
O29
30
F31
F32
F33
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260MU-42_CARBON_01
115.00
117.69
117.74
120.24
121.11
122.78
125.33
131.64
150.42
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
1617
18
19
20
21
22
23
O24
25
F26
F27
F28
F29
30
F31
F32
F33
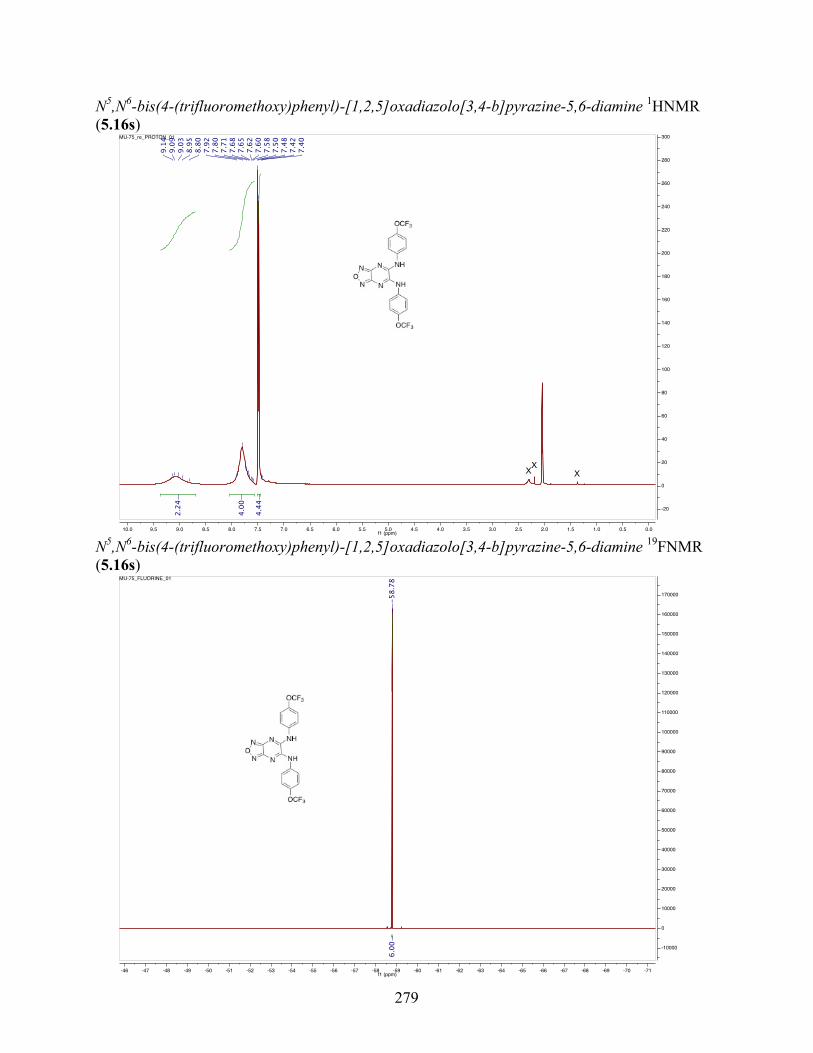
279
N5,N6-bis(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16s)
N5,N6-bis(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16s)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300MU-75_re_PROTON_01
4.44
4.00
2.24
7.40
7.42
7.48
7.50
7.58
7.60
7.62
7.65
7.68
7.71
7.80
7.92
8.80
8.95
9.03
9.09
9.14
X
XX
-71-70-69-68-67-66-65-64-63-62-61-60-59-58-57-56-55-54-53-52-51-50-49-48-47-46f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
110000
120000
130000
140000
150000
160000
170000
MU-75_FLUORINE_01
6.00
-58.78
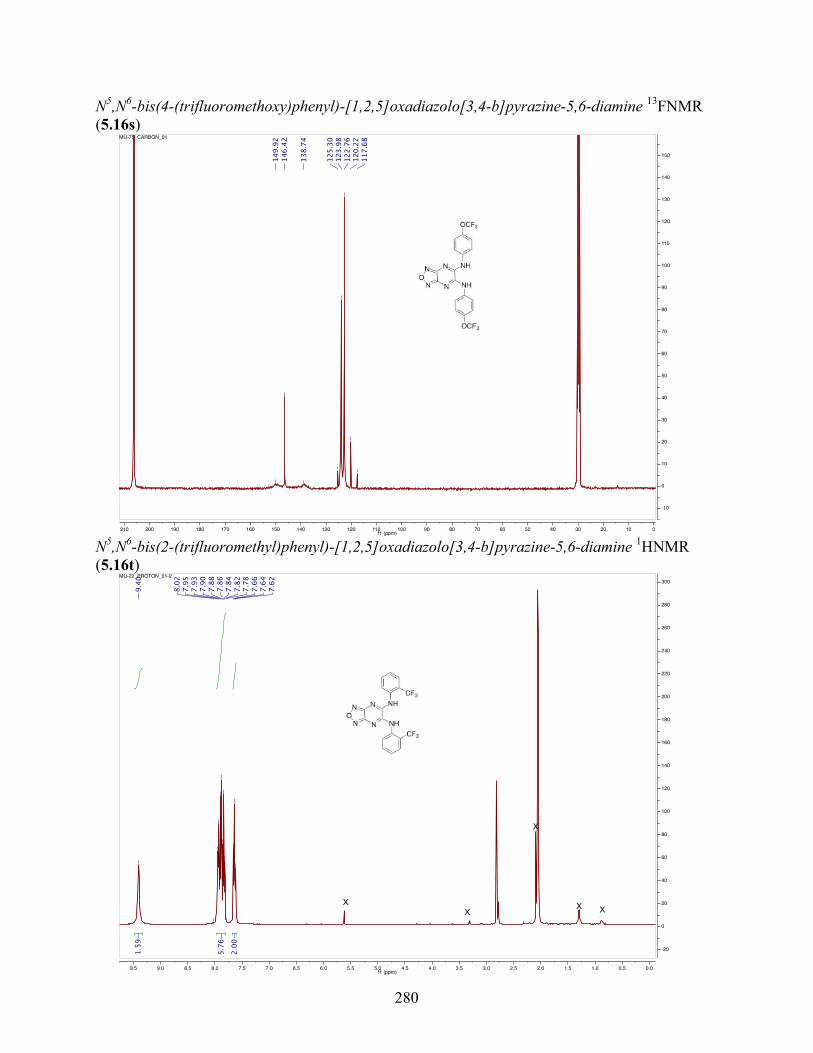
280
N5,N6-bis(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13FNMR (5.16s)
N5,N6-bis(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16t)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
MU-75_CARBON_01
117.68
120.22
122.76
123.98
125.30
138.74
146.42
149.92
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300MU-22_PROTON_01-2
2.00
5.76
1.59
7.62
7.64
7.66
7.78
7.82
7.84
7.86
7.88
7.90
7.93
7.95
8.02
9.40
X
XX
X
X
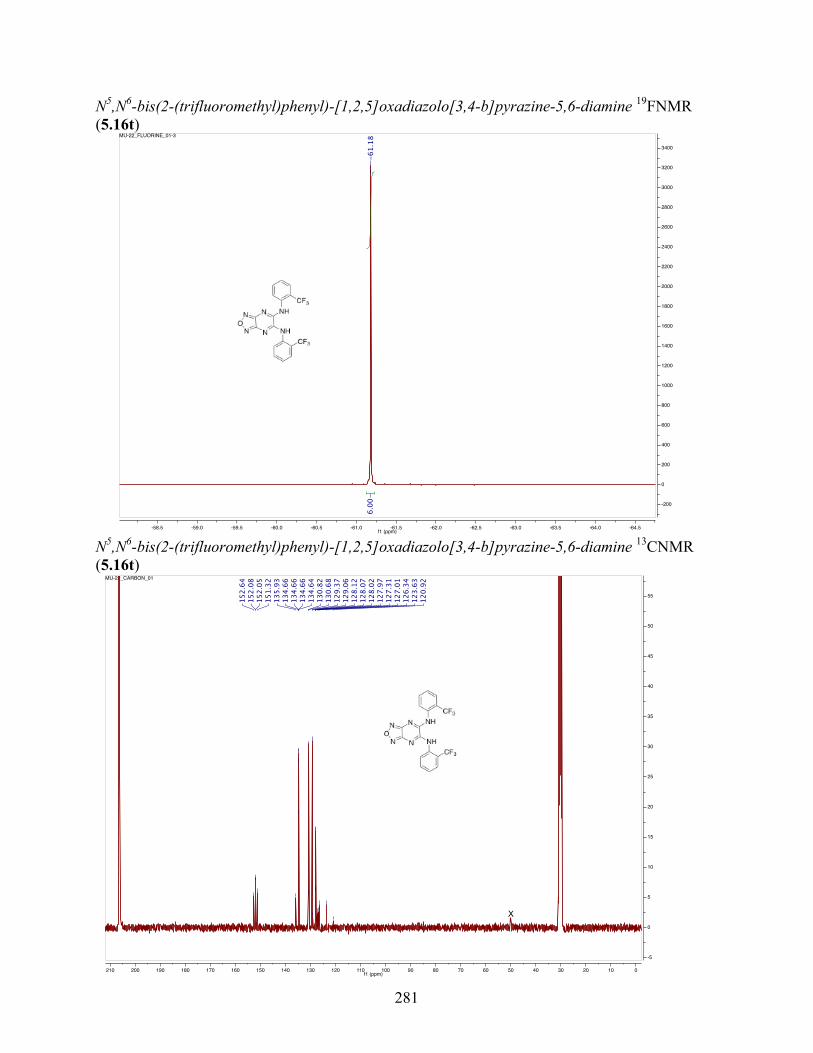
281
N5,N6-bis(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16t)
N5,N6-bis(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16t)
-64.5-64.0-63.5-63.0-62.5-62.0-61.5-61.0-60.5-60.0-59.5-59.0-58.5f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
MU-22_FLUORINE_01-3
6.00
-61.18
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
MU-22_CARBON_01
120.92
123.63
126.34
127.01
127.31
127.97
128.02
128.07
128.12
129.06
129.37
130.68
130.82
134.64
134.66
134.66
134.66
135.93
151.32
152.05
152.08
152.64
X
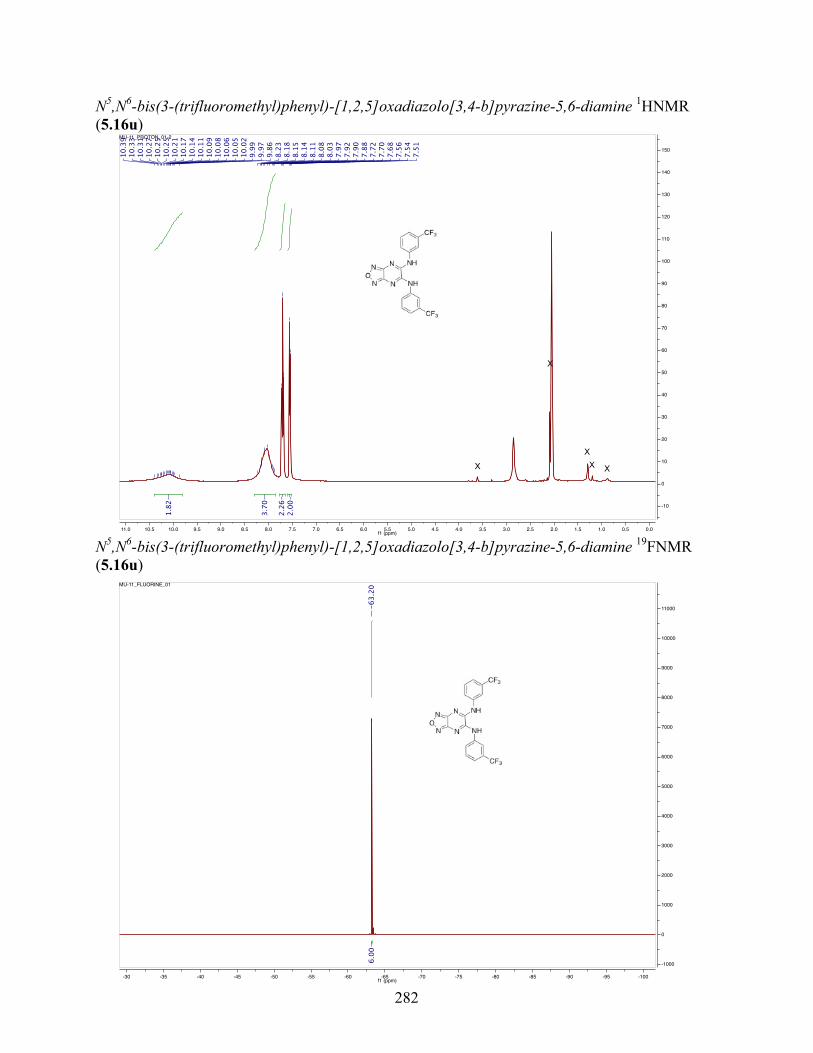
282
N5,N6-bis(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16u)
N5,N6-bis(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16u)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.0f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
MU-11_PROTON_01-2
2.00
2.26
3.70
1.82
7.51
7.54
7.56
7.68
7.70
7.72
7.88
7.90
7.92
7.97
8.03
8.08
8.11
8.14
8.15
8.18
8.23
9.86
9.97
9.99
10.02
10.05
10.06
10.08
10.09
10.11
10.14
10.17
10.21
10.25
10.25
10.27
10.31
10.33
10.39
X
X
X
XX
-100-95-90-85-80-75-70-65-60-55-50-45-40-35-30f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
MU-11_FLUORINE_01
6.00
-63.20
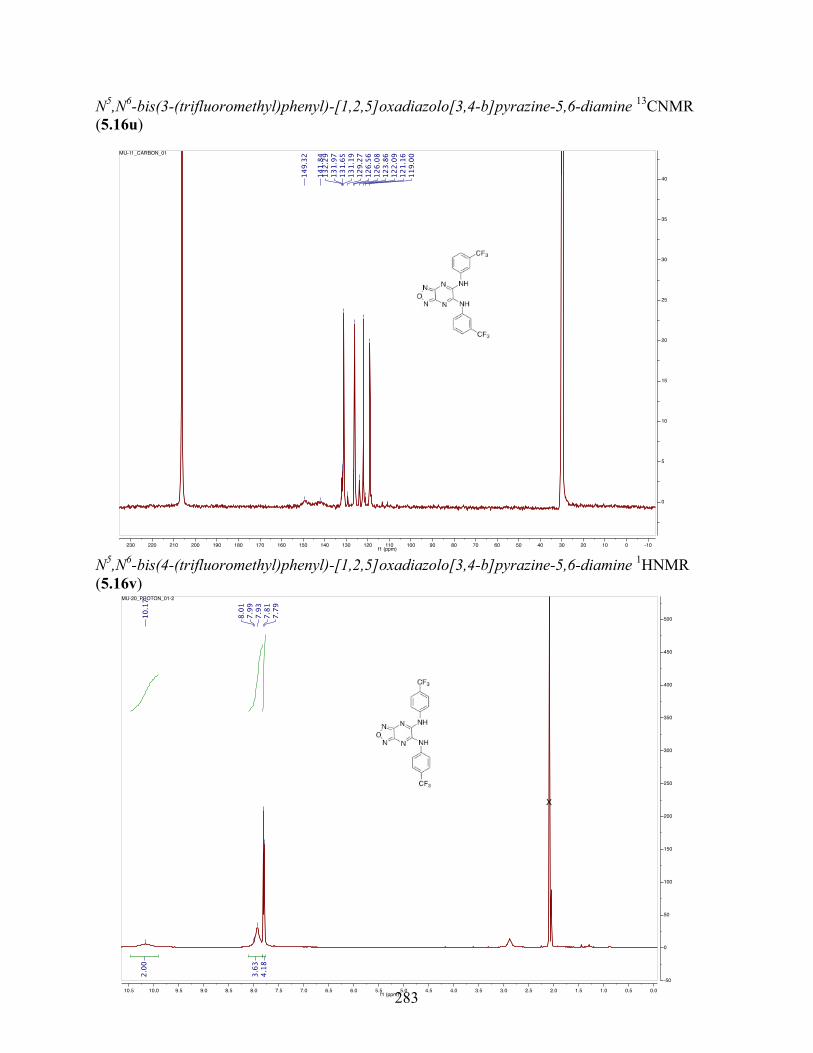
283
N5,N6-bis(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16u)
N5,N6-bis(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.16v)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
0
5
10
15
20
25
30
35
40
MU-11_CARBON_01
119.00
121.16
122.09
123.86
126.08
126.56
129.27
131.19
131.65
131.97
132.29
141.84
149.32
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
MU-20_PROTON_01-2
4.18
3.63
2.00
7.79
7.81
7.93
7.99
8.01
10.17
X

284
N5,N6-bis(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.16v)
N5,N6-bis(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.16v)
-69.0-68.5-68.0-67.5-67.0-66.5-66.0-65.5-65.0-64.5-64.0-63.5-63.0-62.5-62.0-61.5-61.0-60.5-60.0-59.5-59.0-58.5-58.0-57.5-57.0-56.5f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
MU-20_FLUORINE_01-2
6.00
-62.53
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110MU-20_CARBON_01
121.45
122.60
124.14
126.25
126.57
126.84
126.89
127.03
127.26
127.30
127.34
127.38
129.53
145.22
148.83
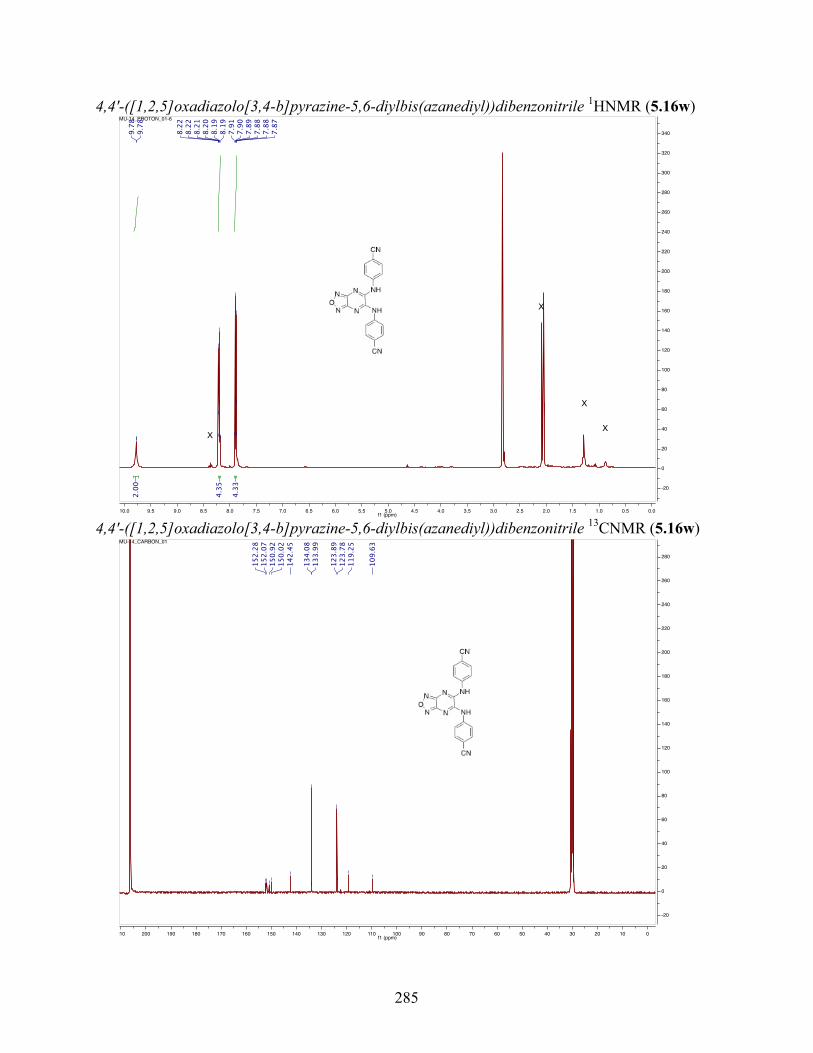
285
4,4'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))dibenzonitrile 1HNMR (5.16w)
4,4'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))dibenzonitrile 13CNMR (5.16w)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
MU-14_PROTON_01-6
4.33
4.35
2.00
7.87
7.88
7.88
7.89
7.90
7.91
8.19
8.19
8.20
8.21
8.22
8.22
9.78
9.78
X
X
XX
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
MU-14_CARBON_01
109.63
119.25
123.78
123.89
133.99
134.08
142.45
150.02
150.92
152.07
152.28
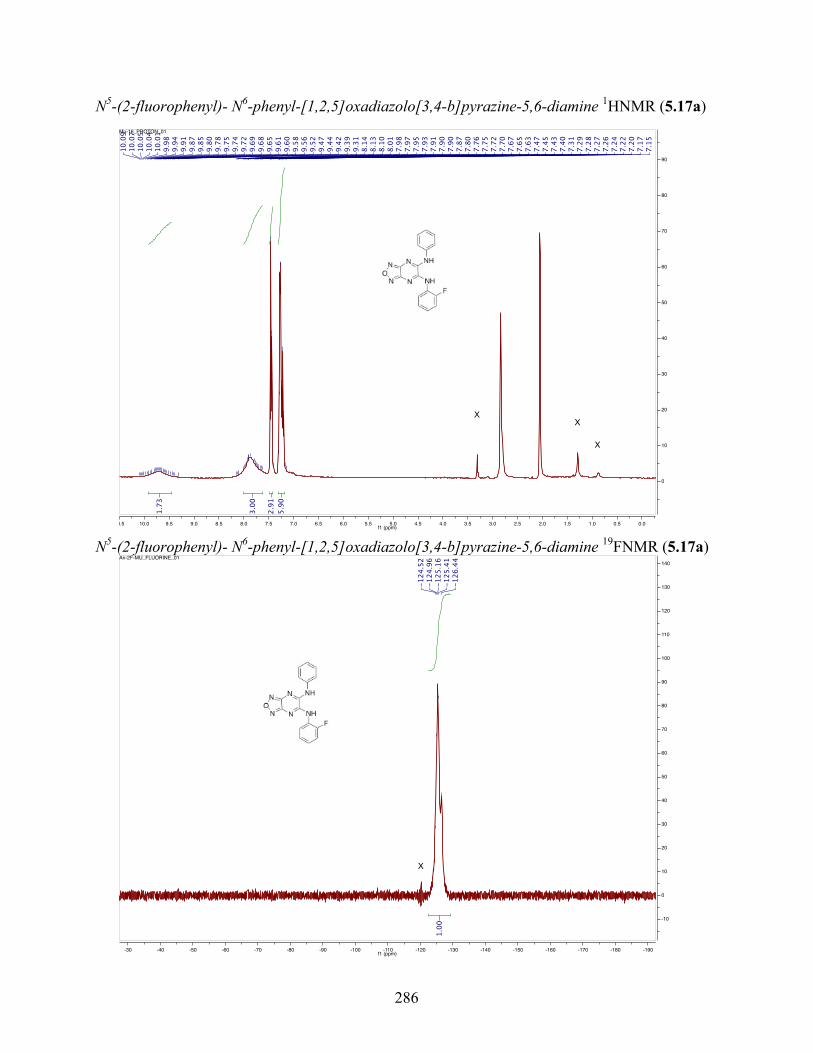
286
N5-(2-fluorophenyl)- N6-phenyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17a)
N5-(2-fluorophenyl)- N6-phenyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17a)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
0
10
20
30
40
50
60
70
80
90
Mu-16_PROTON_01
5.90
2.91
3.00
1.73
7.15
7.17
7.20
7.22
7.24
7.26
7.27
7.28
7.29
7.31
7.40
7.43
7.45
7.47
7.63
7.65
7.67
7.70
7.72
7.75
7.76
7.80
7.87
7.90
7.90
7.91
7.93
7.95
7.97
7.98
8.01
8.10
8.13
8.14
9.31
9.39
9.42
9.44
9.47
9.52
9.56
9.58
9.60
9.61
9.65
9.68
9.69
9.72
9.74
9.75
9.78
9.80
9.85
9.87
9.91
9.94
9.98
10.01
10.04
10.05
10.07
10.09
X
X
X
-190-180-170-160-150-140-130-120-110-100-90-80-70-60-50-40-30f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140An-2F-MU_FLUORINE_01
1.00
-126.44
-125.41
-125.16
-124.96
-124.52
X
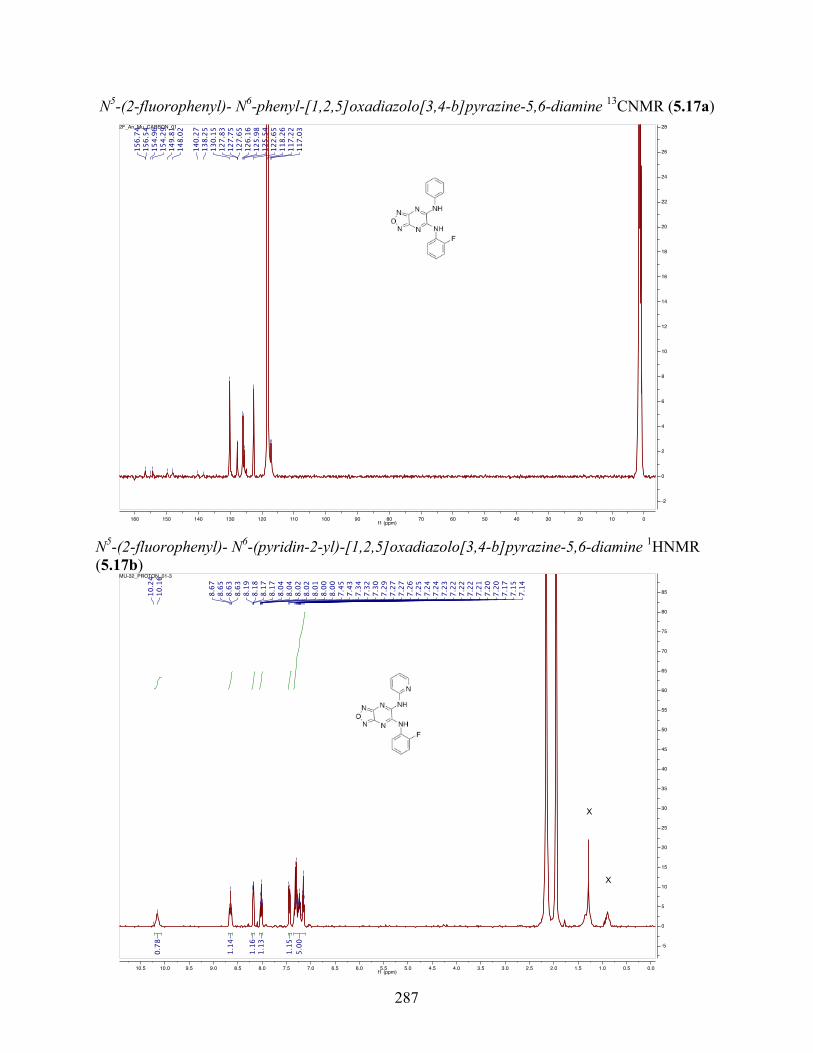
287
N5-(2-fluorophenyl)- N6-phenyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17a)
N5-(2-fluorophenyl)- N6-(pyridin-2-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17b)
0102030405060708090100110120130140150160f1 (ppm)
-2
0
2
4
6
8
10
12
14
16
18
20
22
24
26
282F_An_Mu_CARBON_01
117.03
117.22
118.26
122.65
125.54
125.98
126.16
127.65
127.75
127.83
130.15
138.25
140.27
148.02
149.81
154.29
154.96
156.54
156.74
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
MU-32_PROTON_01-3
5.00
1.15
1.13
1.16
1.14
0.78
7.14
7.15
7.17
7.20
7.20
7.21
7.22
7.22
7.22
7.23
7.24
7.24
7.25
7.26
7.27
7.27
7.29
7.30
7.32
7.34
7.43
7.45
8.00
8.00
8.01
8.02
8.02
8.04
8.04
8.17
8.17
8.18
8.19
8.63
8.63
8.65
8.67
10.16
10.24
X
X
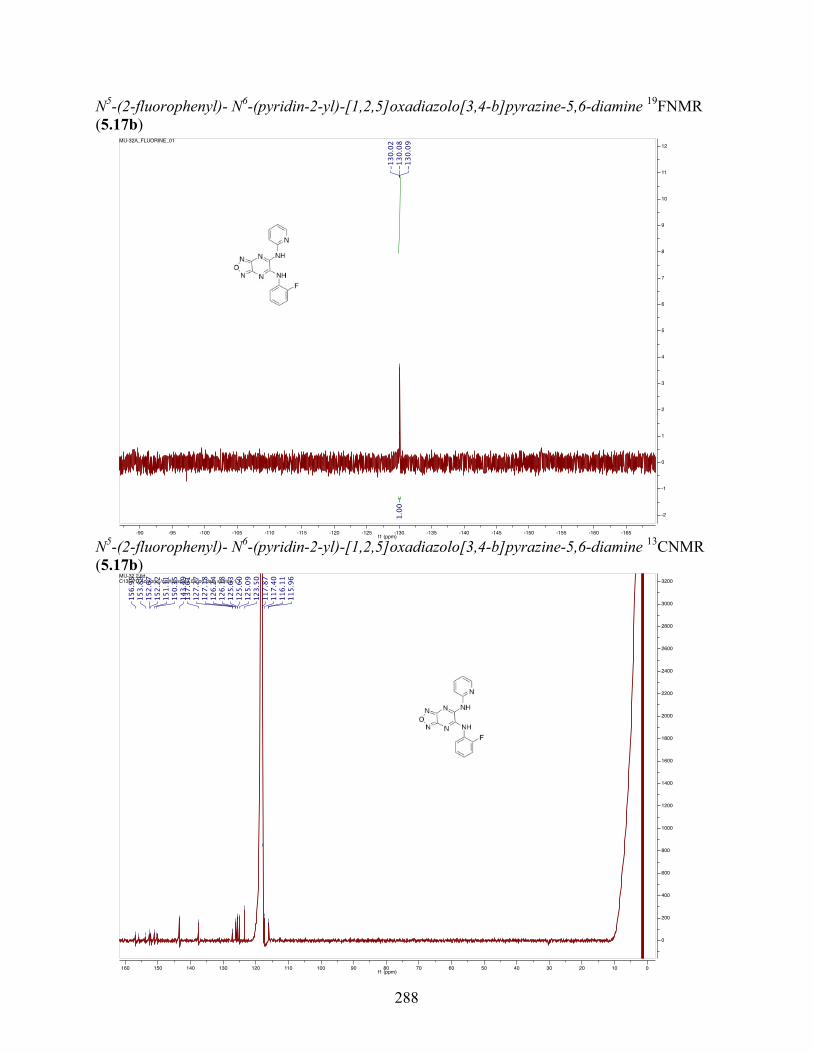
288
N5-(2-fluorophenyl)- N6-(pyridin-2-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17b)
N5-(2-fluorophenyl)- N6-(pyridin-2-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17b)
-165-160-155-150-145-140-135-130-125-120-115-110-105-100-95-90f1 (ppm)
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12MU-32A_FLUORINE_01
1.00
-130.09
-130.08
-130.02
0102030405060708090100110120130140150160f1 (ppm)
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200MU-32.2.fidC13CPD Acetone /opt/topspin3.0/icon-data santos-1 1
115.96
116.11
117.40
117.87
123.50
125.09
125.60
125.63
126.18
126.24
127.18
127.27
137.61
143.49
150.35
151.11
152.22
152.67
153.89
156.95
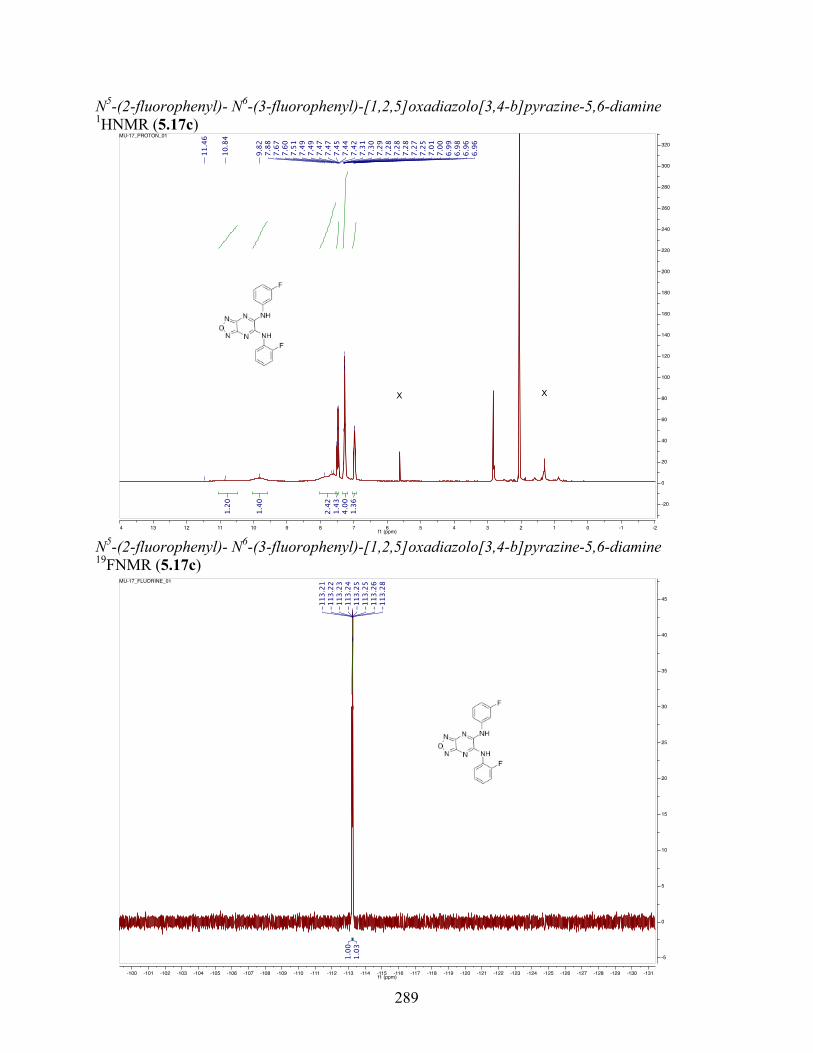
289
N5-(2-fluorophenyl)- N6-(3-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17c)
N5-(2-fluorophenyl)- N6-(3-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17c)
-2-101234567891011121314f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
MU-17_PROTON_01
1.36
4.00
1.43
2.42
1.40
1.20
6.96
6.96
6.98
6.99
7.00
7.01
7.25
7.27
7.28
7.28
7.28
7.29
7.30
7.31
7.42
7.44
7.45
7.47
7.47
7.49
7.49
7.51
7.60
7.67
7.88
9.82
10.84
11.46
XX
-131-130-129-128-127-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111-110-109-108-107-106-105-104-103-102-101-100f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
MU-17_FLUORINE_01
1.03
1.00
-113.28
-113.26
-113.25
-113.25
-113.24
-113.23
-113.22
-113.21
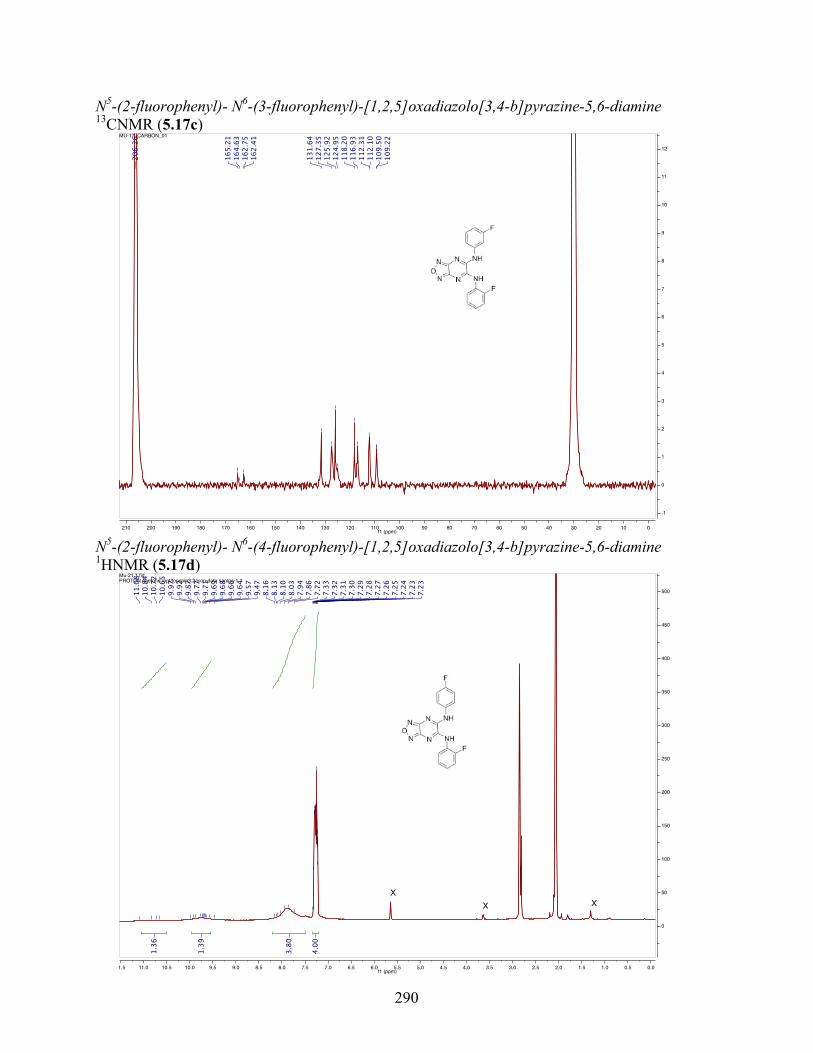
290
N5-(2-fluorophenyl)- N6-(3-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17c)
N5-(2-fluorophenyl)- N6-(4-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17d)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
MU-17_CARBON_01
109.22
109.50
112.10
112.31
116.93
118.20
124.95
125.92
127.35
131.64
162.41
162.75
164.63
165.21
206.26
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
0
50
100
150
200
250
300
350
400
450
500
Mu-21.1.fidPROTON Acetone /opt/topspin3.2/icon-data santos-1 1
4.00
3.80
1.39
1.36
7.23
7.23
7.24
7.25
7.26
7.27
7.28
7.29
7.30
7.31
7.32
7.33
7.72
7.86
7.94
8.03
8.10
8.13
8.16
9.47
9.57
9.64
9.66
9.68
9.69
9.71
9.77
9.87
9.92
9.97
10.65
10.72
10.84
11.08
X
X X
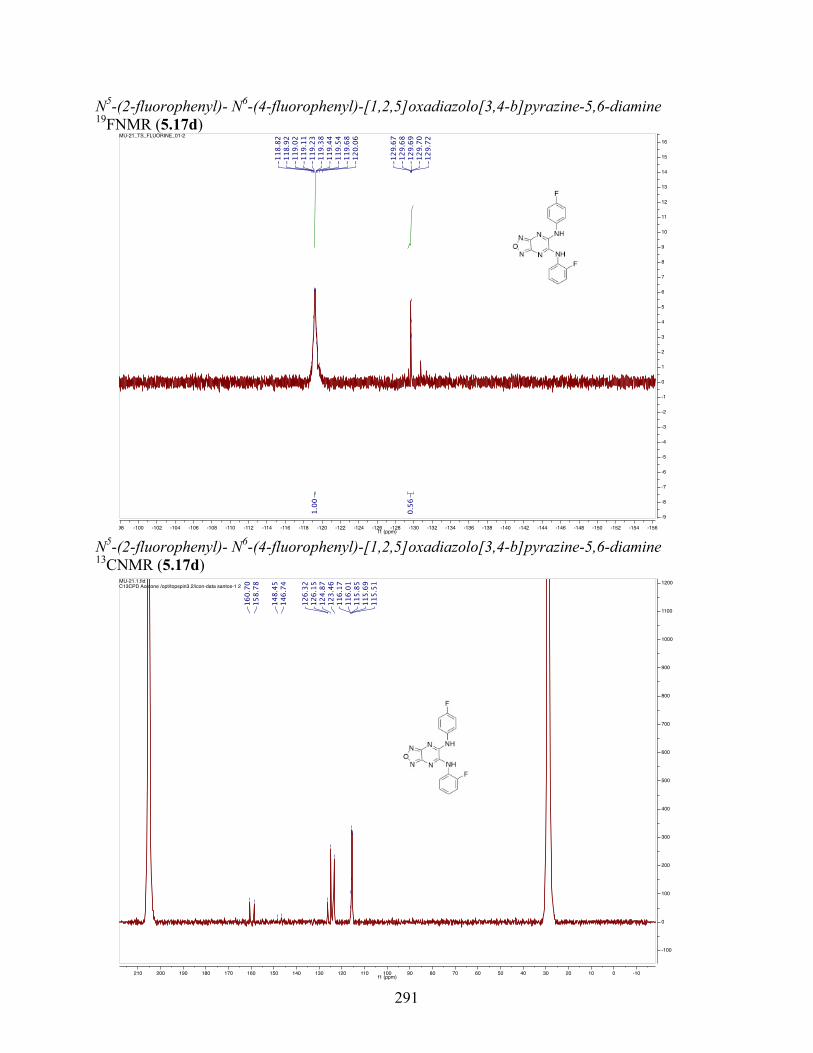
291
N5-(2-fluorophenyl)- N6-(4-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17d)
N5-(2-fluorophenyl)- N6-(4-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17d)
-156-154-152-150-148-146-144-142-140-138-136-134-132-130-128-126-124-122-120-118-116-114-112-110-108-106-104-102-100-98f1 (ppm)
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
MU-21_TS_FLUORINE_01-2
0.56
1.00
-129.72
-129.70
-129.69
-129.68
-129.67
-120.06
-119.68
-119.54
-119.44
-119.38
-119.23
-119.11
-119.02
-118.92
-118.82
-100102030405060708090100110120130140150160170180190200210f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200MU-21.1.fidC13CPD Acetone /opt/topspin3.2/icon-data santos-1 2
115.51
115.69
115.85
116.01
116.17
123.46
124.87
126.15
126.32
146.74
148.45
158.78
160.70
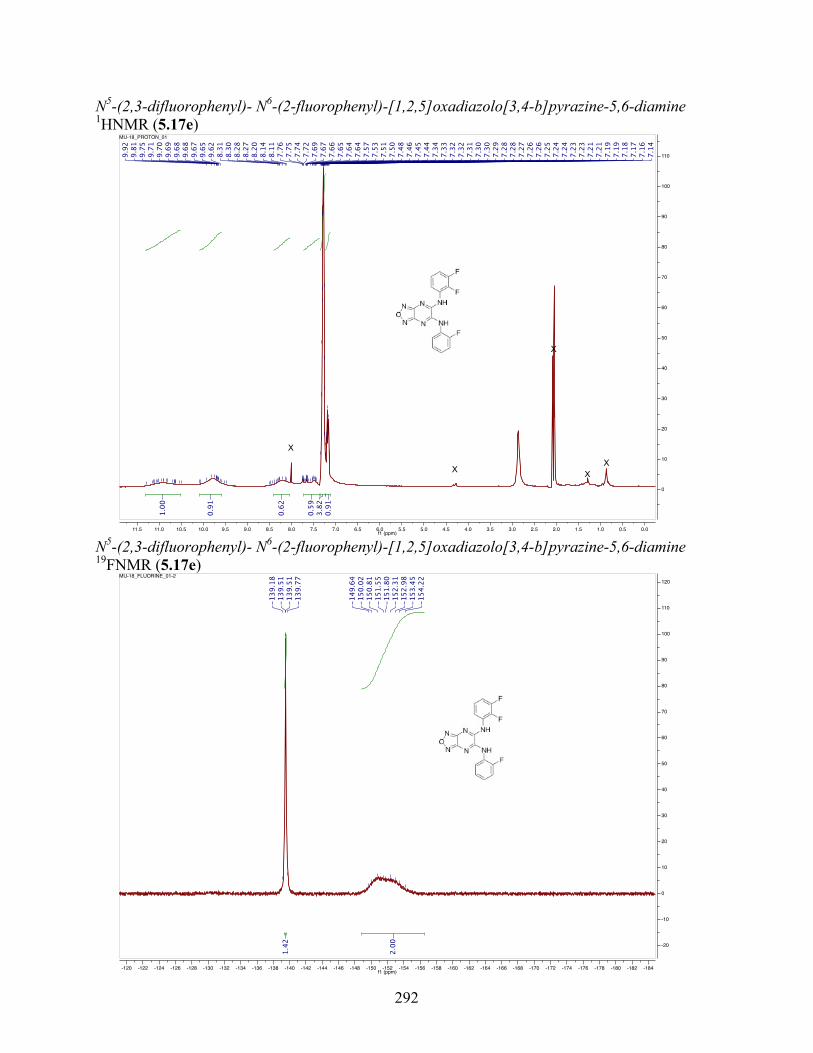
292
N5-(2,3-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17e)
N5-(2,3-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17e)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
0
10
20
30
40
50
60
70
80
90
100
110
MU-18_PROTON_01
0.91
3.82
0.59
0.62
0.91
1.00
7.14
7.16
7.17
7.18
7.19
7.19
7.21
7.21
7.23
7.23
7.24
7.24
7.25
7.26
7.26
7.27
7.28
7.28
7.29
7.30
7.30
7.31
7.32
7.32
7.33
7.34
7.44
7.45
7.46
7.48
7.50
7.51
7.53
7.57
7.64
7.64
7.65
7.66
7.67
7.69
7.72
7.74
7.75
7.76
8.11
8.14
8.20
8.27
8.28
8.30
8.31
9.62
9.65
9.67
9.68
9.68
9.69
9.70
9.71
9.75
9.81
9.92
X
X
X
X
X
-184-182-180-178-176-174-172-170-168-166-164-162-160-158-156-154-152-150-148-146-144-142-140-138-136-134-132-130-128-126-124-122-120f1 (ppm)
-20
-10
0
10
20
30
40
50
60
70
80
90
100
110
120MU-18_FLUORINE_01-2
2.00
1.42
-154.22
-153.45
-152.98
-152.31
-151.80
-151.55
-150.81
-150.02
-149.64
-139.77
-139.51
-139.51
-139.18
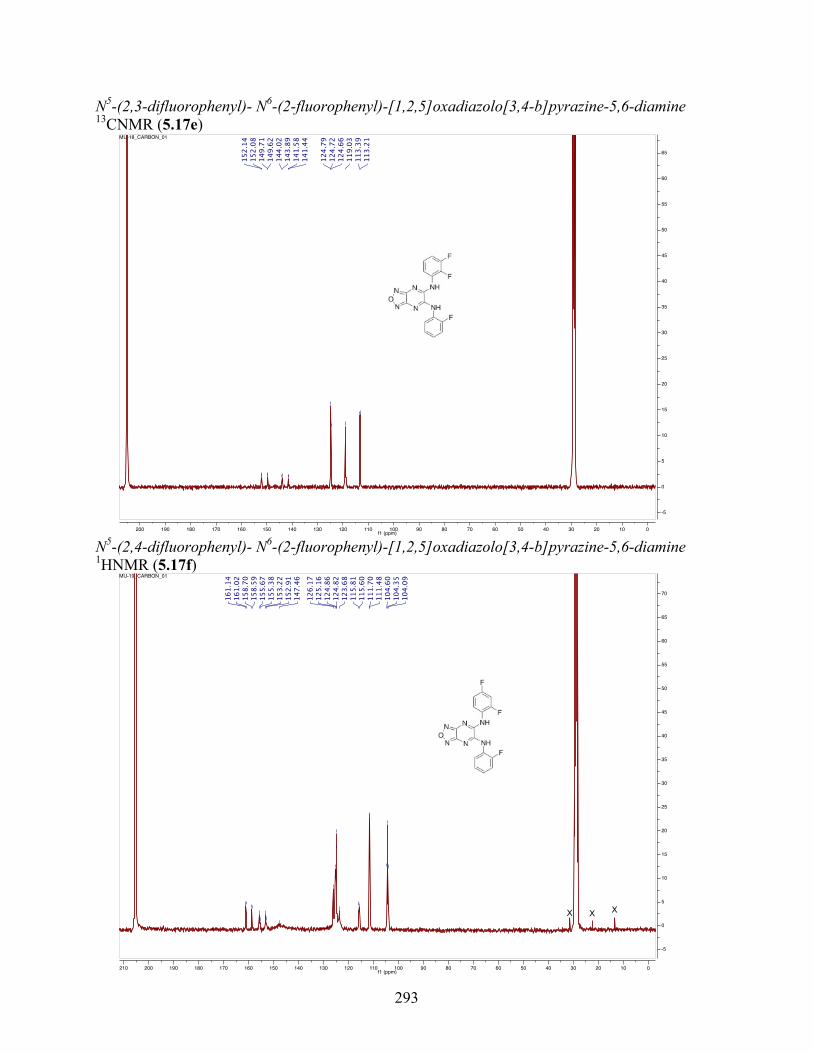
293
N5-(2,3-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17e)
N5-(2,4-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17f)
0102030405060708090100110120130140150160170180190200f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
MU-18_CARBON_01
113.21
113.39
119.03
124.66
124.72
124.79
141.44
141.58
143.89
144.02
149.62
149.71
152.08
152.14
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
MU-19_CARBON_01
104.09
104.35
104.60
111.48
111.70
115.60
115.81
123.68
124.82
124.86
125.16
126.17
147.46
152.91
153.22
155.38
155.67
158.59
158.70
161.02
161.14
X XX
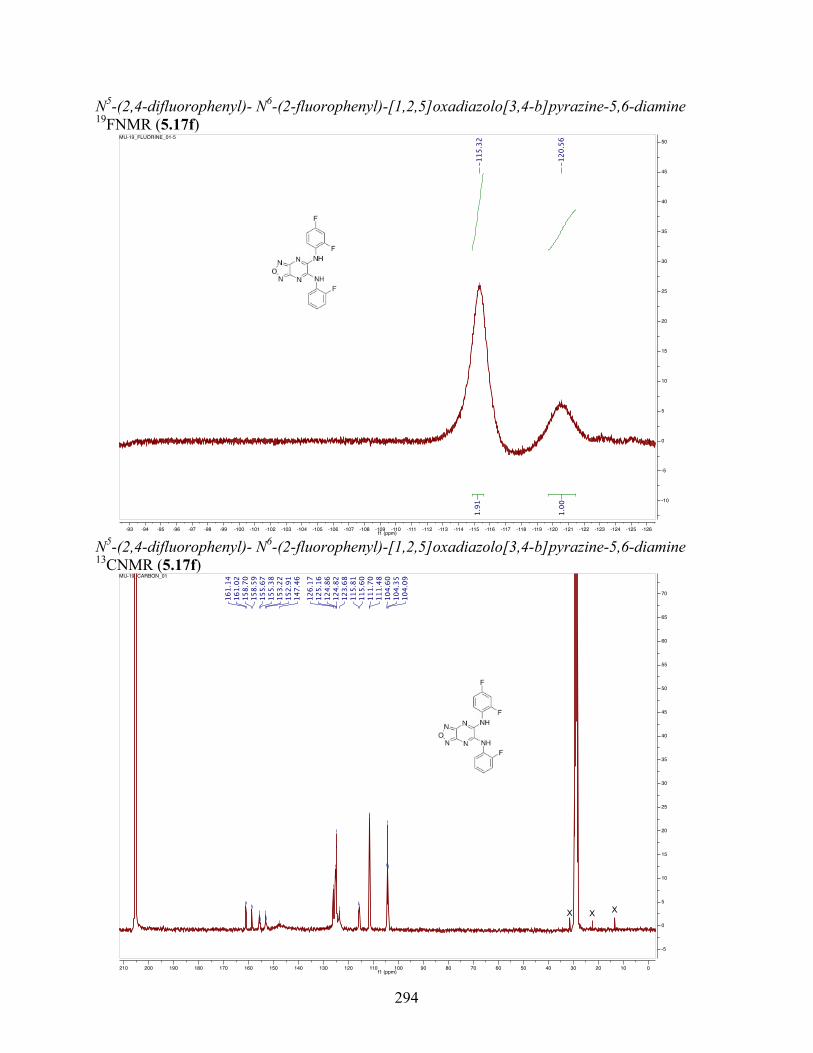
294
N5-(2,4-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17f)
N5-(2,4-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17f)
-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111-110-109-108-107-106-105-104-103-102-101-100-99-98-97-96-95-94-93f1 (ppm)
-10
-5
0
5
10
15
20
25
30
35
40
45
50MU-19_FLUORINE_01-5
1.00
1.91
-120.56
-115.32
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
MU-19_CARBON_01
104.09
104.35
104.60
111.48
111.70
115.60
115.81
123.68
124.82
124.86
125.16
126.17
147.46
152.91
153.22
155.38
155.67
158.59
158.70
161.02
161.14
X XX
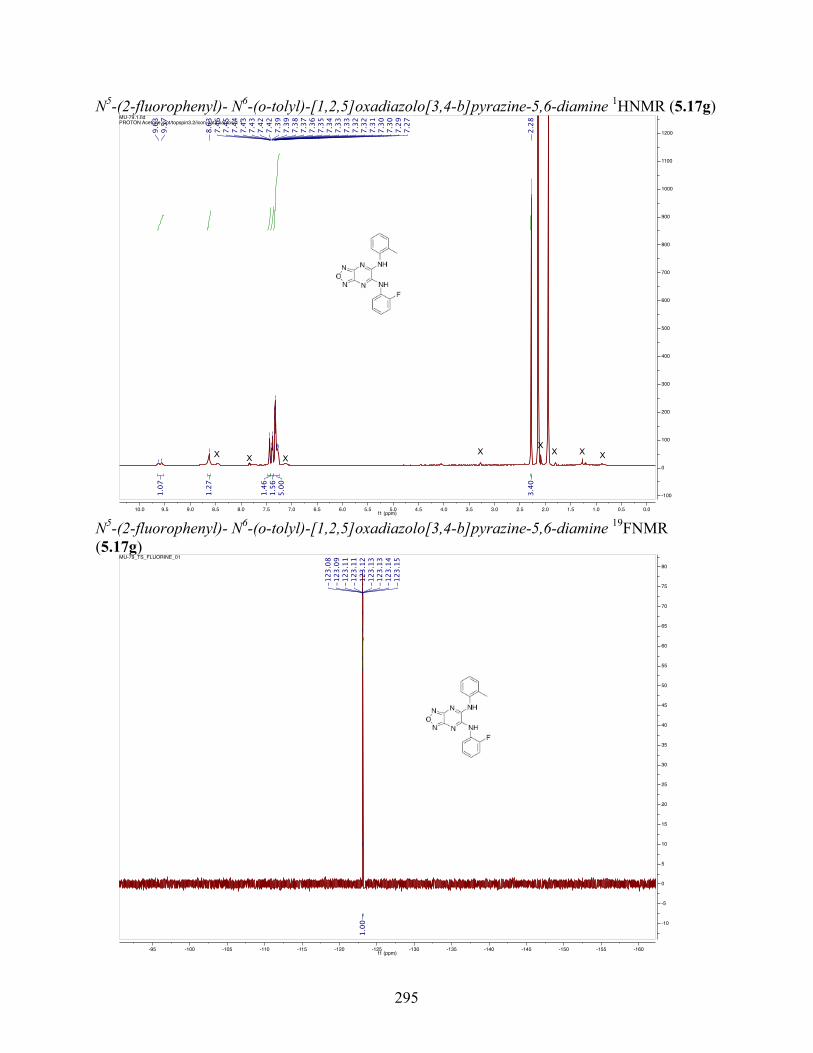
295
N5-(2-fluorophenyl)- N6-(o-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17g)
N5-(2-fluorophenyl)- N6-(o-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17g)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
MU-79.1.fidPROTON Acetone /opt/topspin3.2/icon-data santos-1 1
3.40
5.00
1.56
1.46
1.27
1.07
2.28
7.27
7.29
7.30
7.30
7.31
7.32
7.32
7.33
7.33
7.34
7.35
7.36
7.37
7.38
7.39
7.39
7.42
7.42
7.43
7.43
7.44
7.45
7.46
8.63
9.57
9.63
XX X
XX
X XX
-160-155-150-145-140-135-130-125-120-115-110-105-100-95f1 (ppm)
-10
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
MU-79_TS_FLUORINE_01
1.00
-123.15
-123.14
-123.13
-123.13
-123.12
-123.11
-123.11
-123.09
-123.08
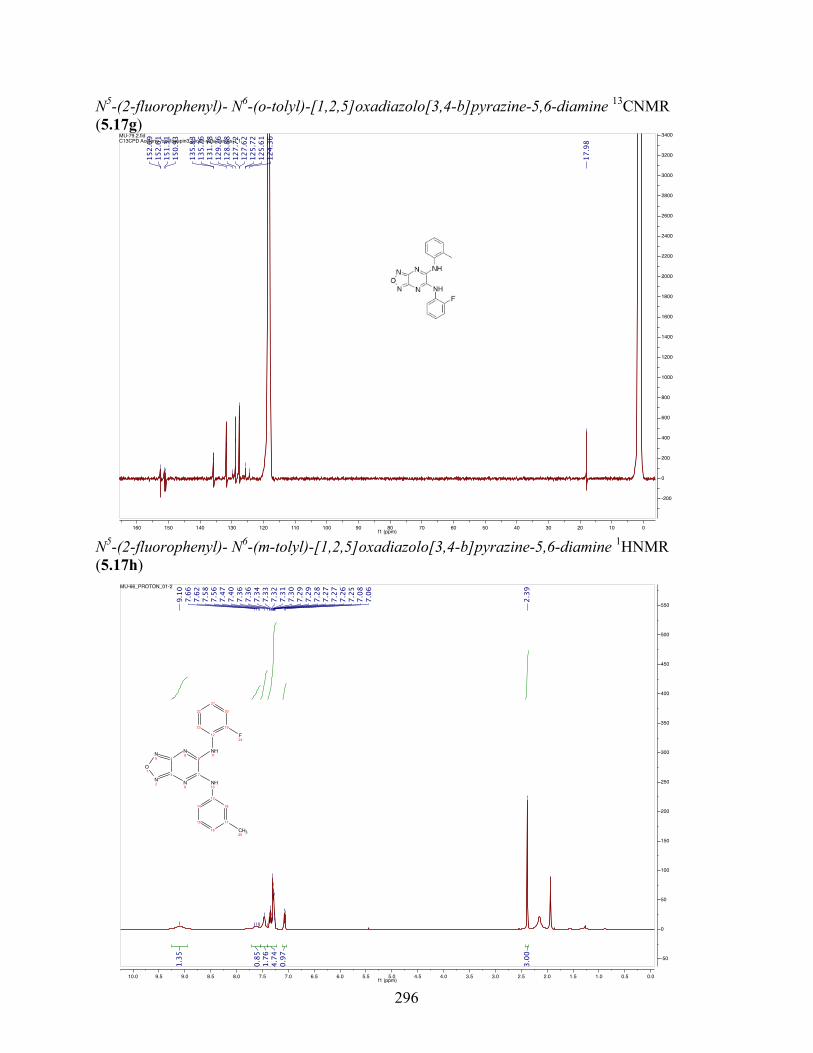
296
N5-(2-fluorophenyl)- N6-(o-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17g)
N5-(2-fluorophenyl)- N6-(m-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17h)
0102030405060708090100110120130140150160f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400MU-79.2.fidC13CPD Acetone /opt/topspin3.2/icon-data santos-1 1
17.98
124.36
125.61
125.72
127.62
127.72
128.88
129.76
131.78
135.76
135.83
150.93
151.31
152.61
152.69
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
MU-66_PROTON_01-2
3.00
0.97
4.74
1.76
0.85
1.35
2.39
7.06
7.08
7.25
7.26
7.27
7.27
7.28
7.29
7.29
7.30
7.31
7.32
7.33
7.34
7.36
7.36
7.40
7.47
7.56
7.58
7.62
7.66
9.10
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
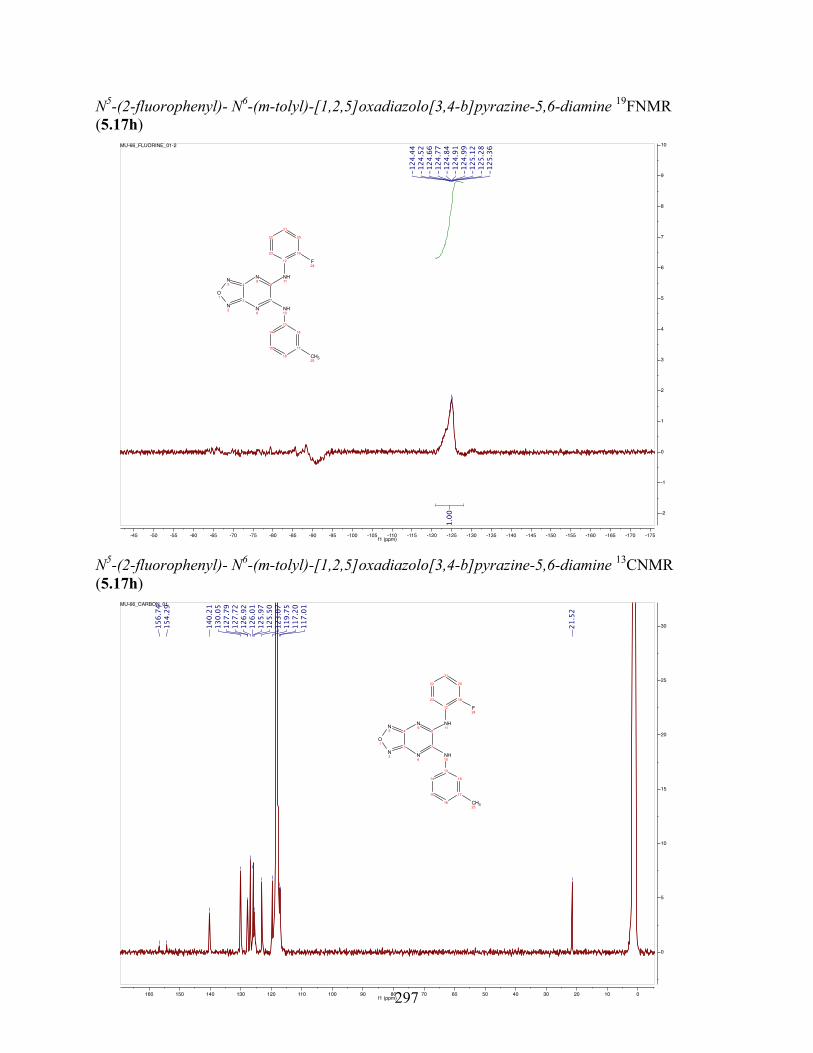
297
N5-(2-fluorophenyl)- N6-(m-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17h)
N5-(2-fluorophenyl)- N6-(m-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17h)
-175-170-165-160-155-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50-45f1 (ppm)
-2
-1
0
1
2
3
4
5
6
7
8
9
10MU-66_FLUORINE_01-2
1.00
-125.36
-125.28
-125.12
-124.99
-124.91
-124.84
-124.77
-124.66
-124.52
-124.44
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
0102030405060708090100110120130140150160f1 (ppm)
0
5
10
15
20
25
30
MU-66_CARBON_01
21.52
117.01
117.20
119.75
123.07
125.50
125.97
126.01
126.92
127.72
127.79
130.05
140.21
154.29
156.74
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
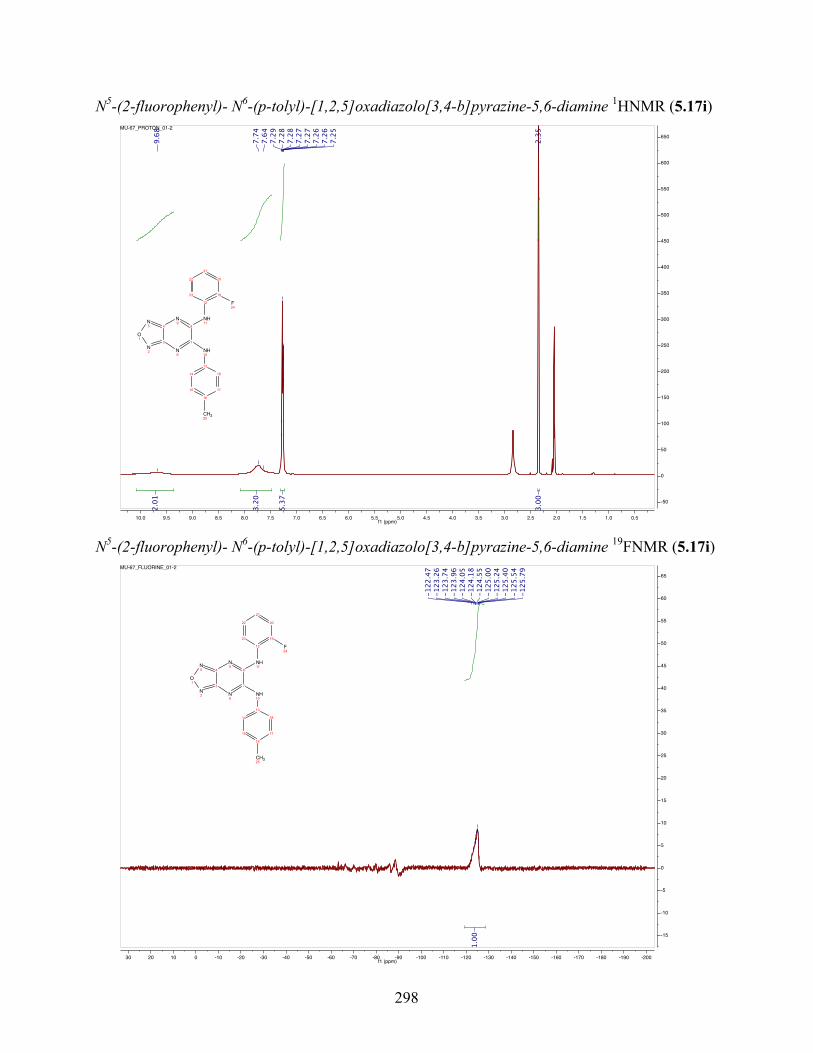
298
N5-(2-fluorophenyl)- N6-(p-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17i)
N5-(2-fluorophenyl)- N6-(p-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17i)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
MU-67_PROTON_01-2
3.00
5.37
3.20
2.01
2.35
7.25
7.26
7.26
7.27
7.27
7.28
7.28
7.29
7.64
7.74
9.68
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
-200-190-180-170-160-150-140-130-120-110-100-90-80-70-60-50-40-30-20-100102030f1 (ppm)
-15
-10
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
MU-67_FLUORINE_01-2
1.00
-125.79
-125.54
-125.40
-125.24
-125.00
-124.55
-124.18
-124.05
-123.96
-123.74
-123.26
-122.47
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
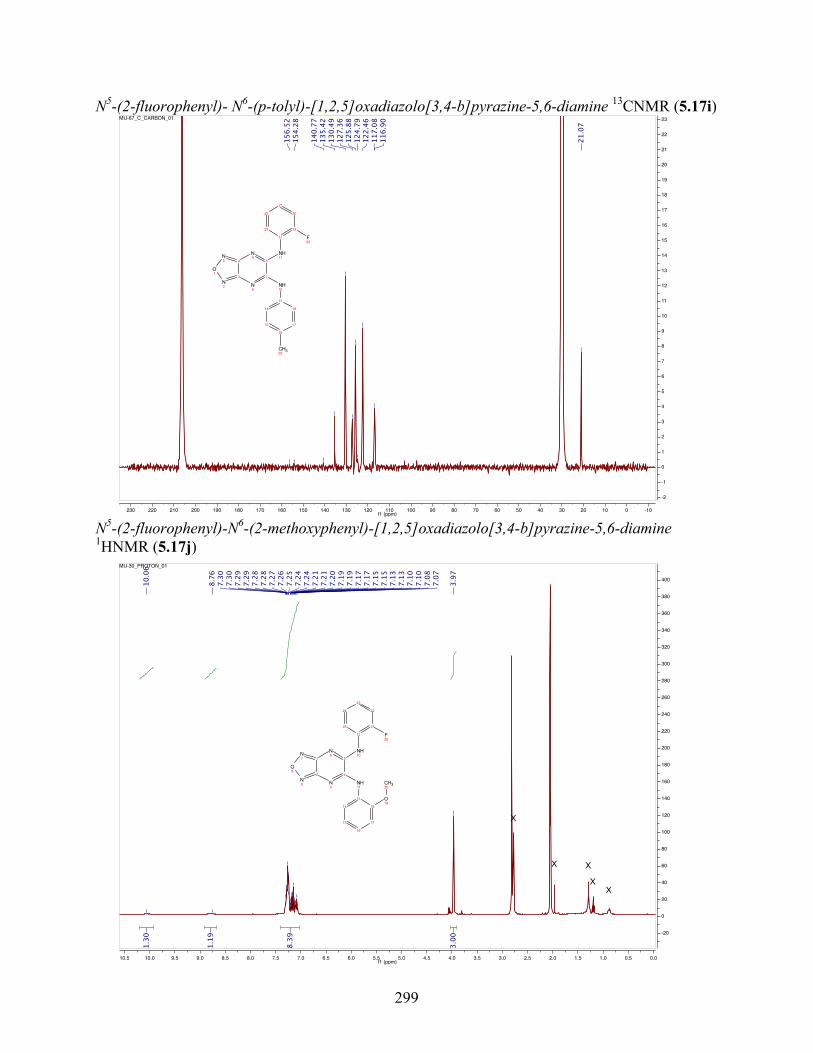
299
N5-(2-fluorophenyl)- N6-(p-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17i)
N5-(2-fluorophenyl)-N6-(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17j)
-100102030405060708090100110120130140150160170180190200210220230f1 (ppm)
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23MU-67_C_CARBON_01
21.07
116.90
117.08
122.46
124.79
125.88
127.36
130.49
135.42
140.77
154.28
156.52
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
CH325
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
380
400
MU-30_PROTON_01
3.00
8.39
1.19
1.30
3.97
7.07
7.08
7.10
7.10
7.13
7.13
7.15
7.15
7.17
7.17
7.19
7.19
7.20
7.21
7.21
7.24
7.24
7.25
7.26
7.27
7.28
7.28
7.29
7.29
7.30
7.30
8.76
10.06
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
O19
CH320
21
22
23
24
25
F26
X X
X
X
X
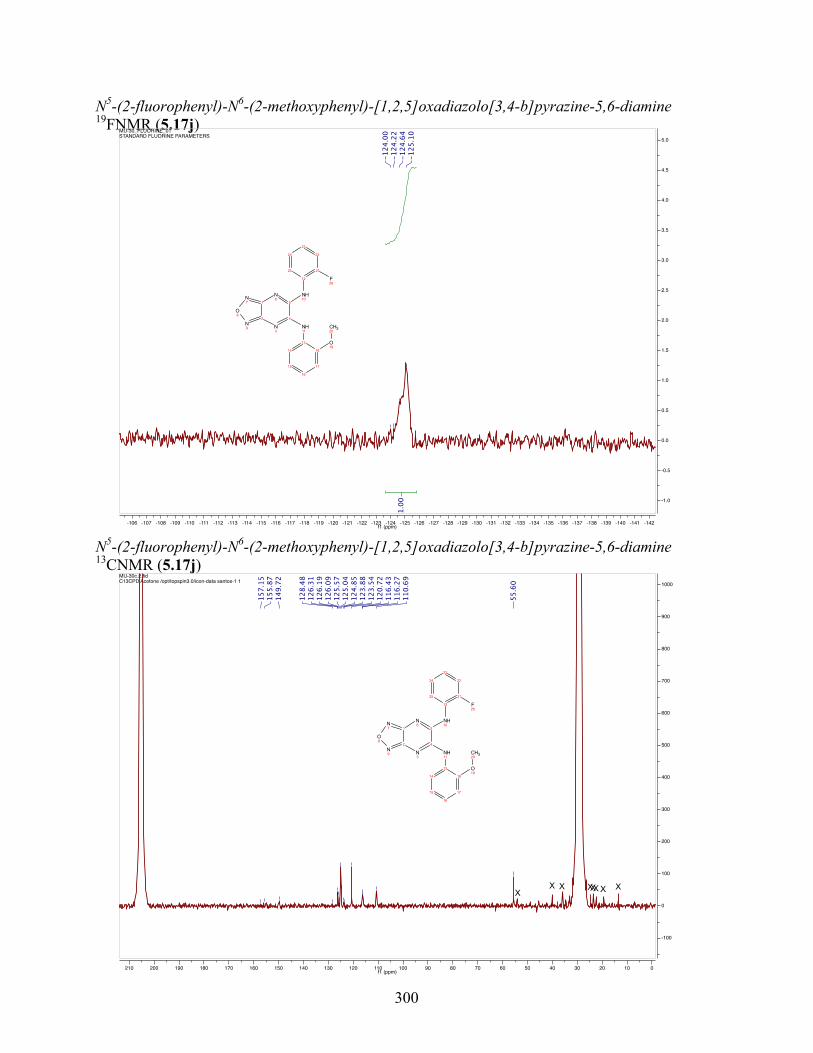
300
N5-(2-fluorophenyl)-N6-(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17j)
N5-(2-fluorophenyl)-N6-(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17j)
-142-141-140-139-138-137-136-135-134-133-132-131-130-129-128-127-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111-110-109-108-107-106f1 (ppm)
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
MU-30_FLUORINE_01STANDARD FLUORINE PARAMETERS
1.00
-125.10
-124.64
-124.22
-124.00
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
O19
CH320
21
22
23
24
25
F26
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
MU-30c.2.fidC13CPD Acetone /opt/topspin3.0/icon-data santos-1 1
55.60
110.69
116.27
116.43
120.72
123.54
123.88
124.85
125.04
125.57
126.09
126.19
126.31
128.48
149.72
155.87
157.15
X X XXXX XX
1
2
N3
4
5
N6N
7
O8
N9
NH10
NH11
12
13
14
15
16
17
18
O19
CH320
21
22
23
24
25
F26
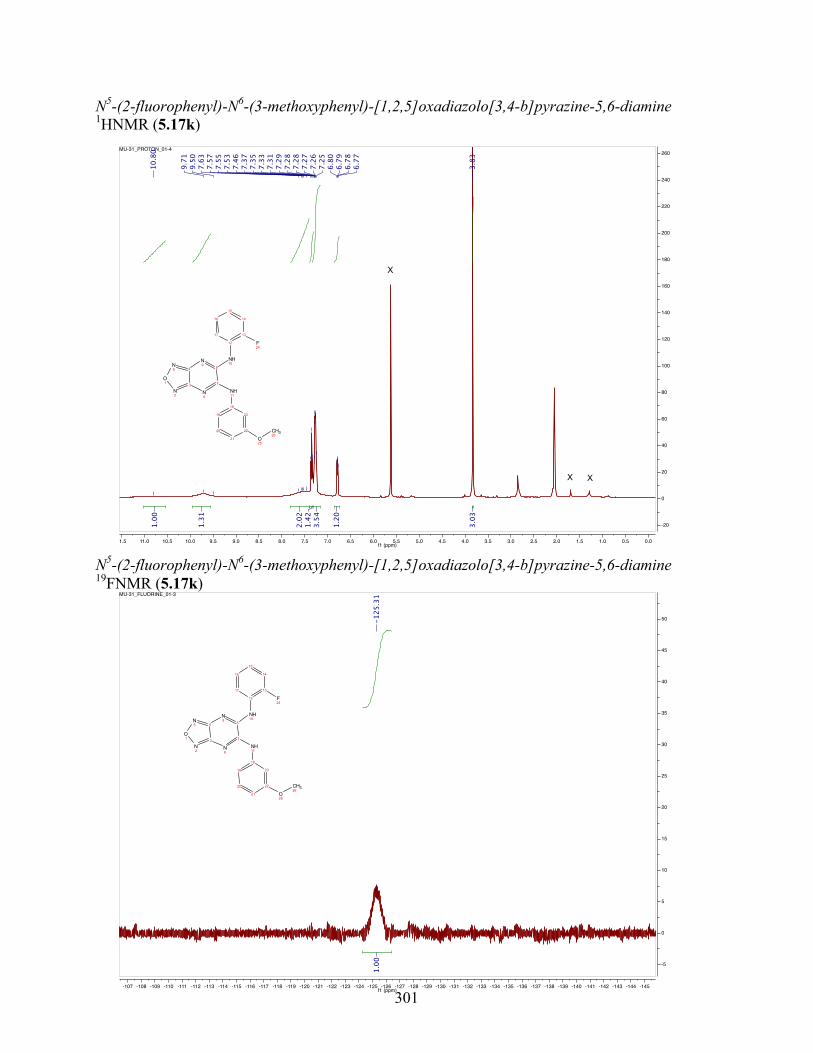
301
N5-(2-fluorophenyl)-N6-(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17k)
N5-(2-fluorophenyl)-N6-(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17k)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260MU-31_PROTON_01-4
3.03
1.20
3.54
1.42
2.02
1.31
1.00
3.83
6.77
6.78
6.79
6.80
7.25
7.26
7.27
7.28
7.28
7.29
7.31
7.33
7.35
7.37
7.46
7.53
7.55
7.57
7.63
9.50
9.71
10.80
X
X X
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
O25
CH326
-145-144-143-142-141-140-139-138-137-136-135-134-133-132-131-130-129-128-127-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111-110-109-108-107f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
MU-31_FLUORINE_01-3
1.00
-125.31
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
O25
CH326
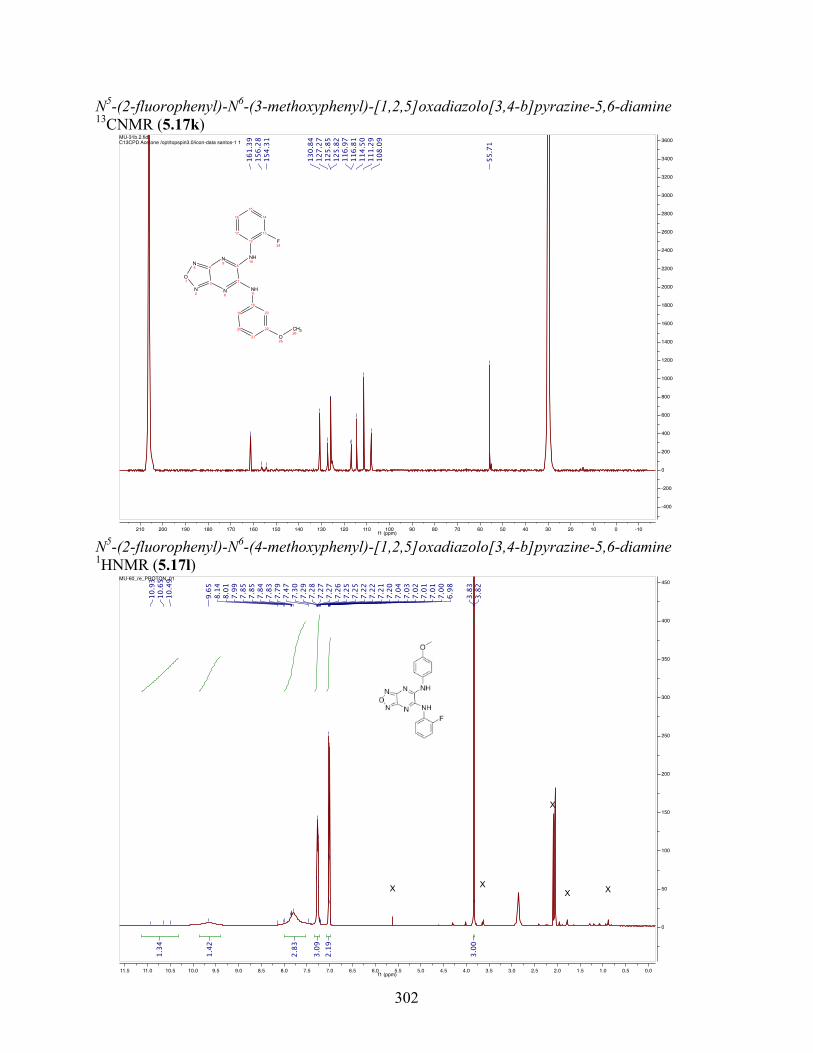
302
N5-(2-fluorophenyl)-N6-(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17k)
N5-(2-fluorophenyl)-N6-(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17l)
-100102030405060708090100110120130140150160170180190200210f1 (ppm)
-400
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600MU-31b.2.fidC13CPD Acetone /opt/topspin3.0/icon-data santos-1 1
55.71
108.09
111.29
114.50
116.81
116.97
125.82
125.85
127.27
130.84
154.31
156.28
161.39
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
O25
CH326
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
0
50
100
150
200
250
300
350
400
450MU-60_re_PROTON_01
3.00
2.19
3.09
2.83
1.42
1.34
3.82
3.83
6.98
7.00
7.01
7.01
7.02
7.03
7.04
7.20
7.21
7.22
7.22
7.25
7.25
7.26
7.27
7.27
7.28
7.29
7.30
7.47
7.79
7.83
7.84
7.85
7.85
7.99
8.01
8.14
9.65
10.49
10.65
10.93
XX
X
XX
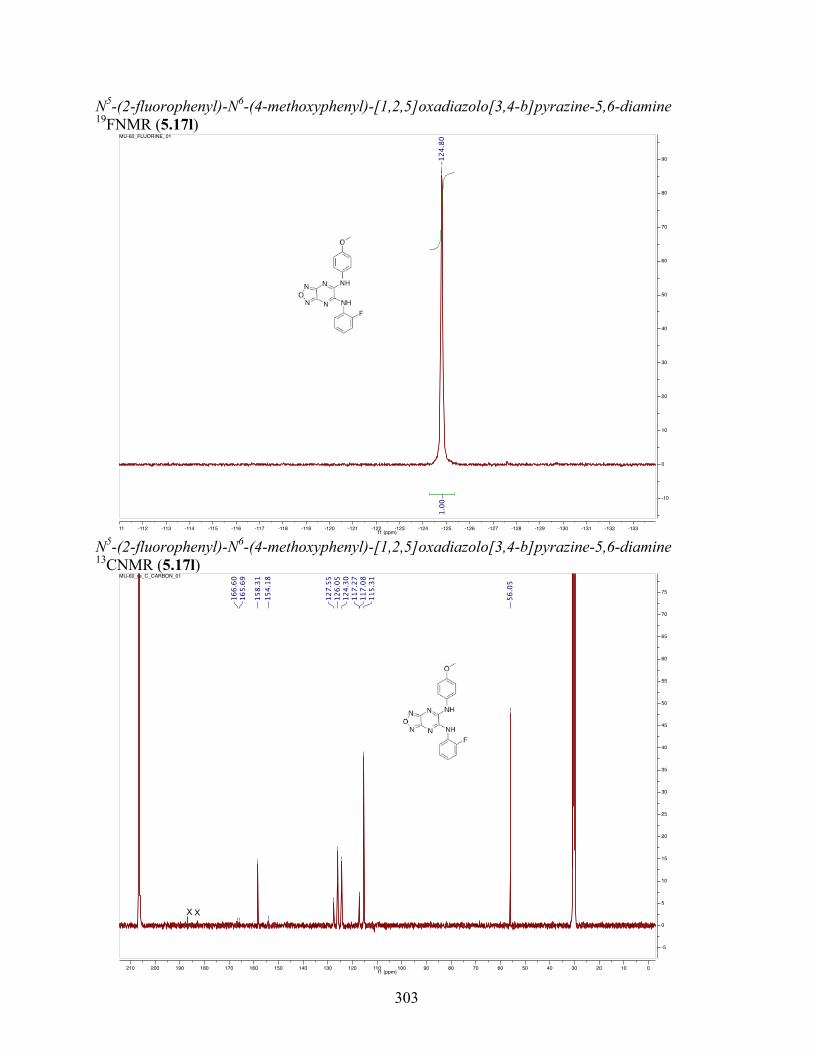
303
N5-(2-fluorophenyl)-N6-(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17l)
N5-(2-fluorophenyl)-N6-(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17l)
-133-132-131-130-129-128-127-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
MU-60_FLUORINE_01
1.00
-124.80
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
MU-60_re_C_CARBON_01
56.05
115.31
117.08
117.27
124.30
126.05
127.55
154.18
158.31
165.69
166.60
X X
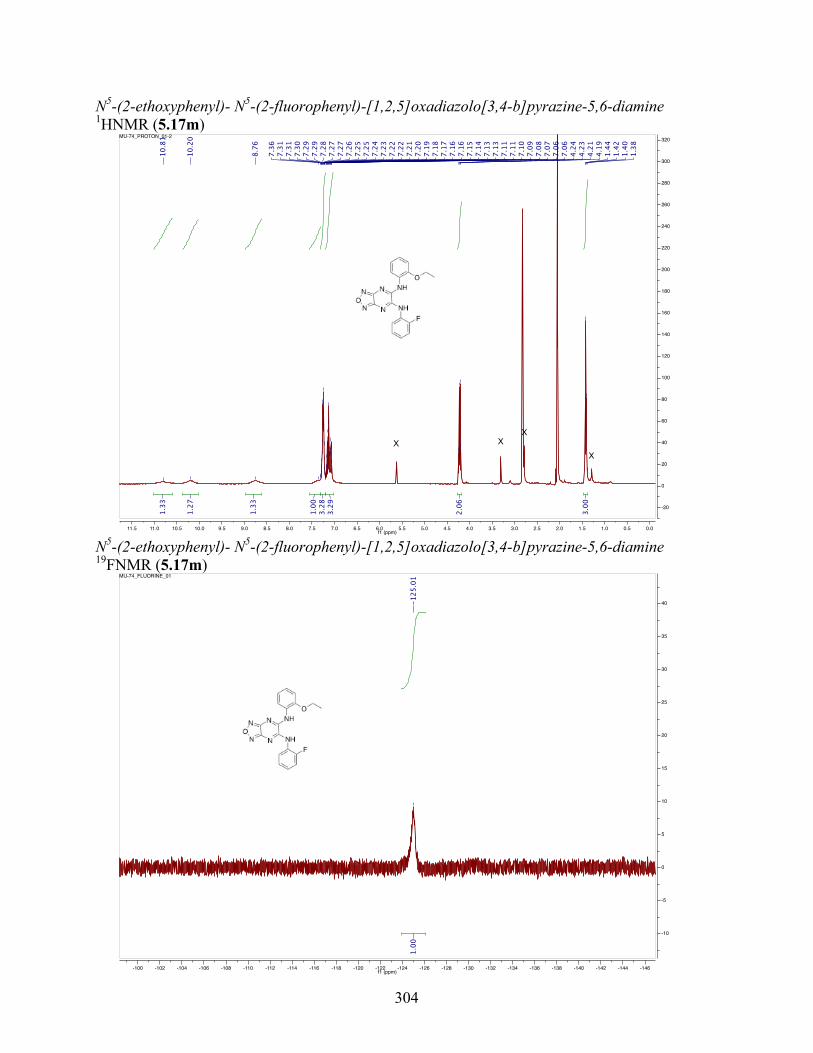
304
N5-(2-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17m)
N5-(2-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17m)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320MU-74_PROTON_01-2
3.00
2.06
3.29
3.28
1.00
1.33
1.27
1.33
1.38
1.40
1.42
1.44
4.19
4.21
4.23
4.24
7.06
7.06
7.07
7.08
7.09
7.10
7.11
7.11
7.13
7.13
7.14
7.15
7.16
7.16
7.17
7.18
7.19
7.20
7.21
7.22
7.22
7.23
7.24
7.25
7.25
7.26
7.27
7.27
7.28
7.29
7.29
7.30
7.31
7.31
7.36
8.76
10.20
10.81
X
X
XX
-146-144-142-140-138-136-134-132-130-128-126-124-122-120-118-116-114-112-110-108-106-104-102-100f1 (ppm)
-10
-5
0
5
10
15
20
25
30
35
40
MU-74_FLUORINE_01
1.00
-125.01
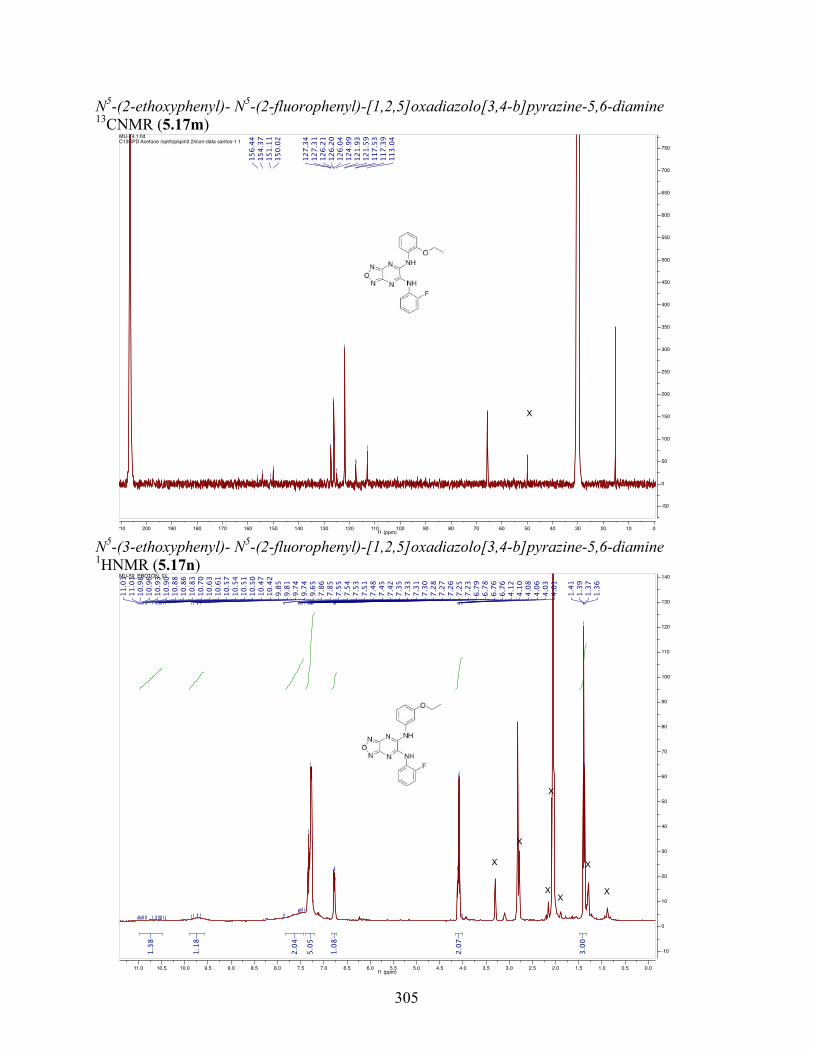
305
N5-(2-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17m)
N5-(3-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17n)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
MU-74.1.fidC13CPD Acetone /opt/topspin3.2/icon-data santos-1 1
113.04
117.39
117.53
121.59
121.93
124.99
126.04
126.20
126.21
127.31
127.34
150.02
151.11
154.37
156.44
X
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.0f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140MU-59_PROTON_01
3.00
2.07
1.08
5.05
2.04
1.18
1.38
1.36
1.37
1.39
1.41
4.01
4.03
4.06
4.08
4.10
4.12
6.76
6.76
6.78
6.79
7.23
7.25
7.26
7.27
7.28
7.30
7.31
7.33
7.35
7.42
7.45
7.48
7.51
7.53
7.54
7.55
7.85
7.86
9.65
9.74
9.74
9.81
9.85
10.42
10.47
10.50
10.51
10.54
10.57
10.61
10.63
10.70
10.83
10.86
10.88
10.90
10.93
10.96
10.98
11.01
11.03
X
XX
X
X
X
X
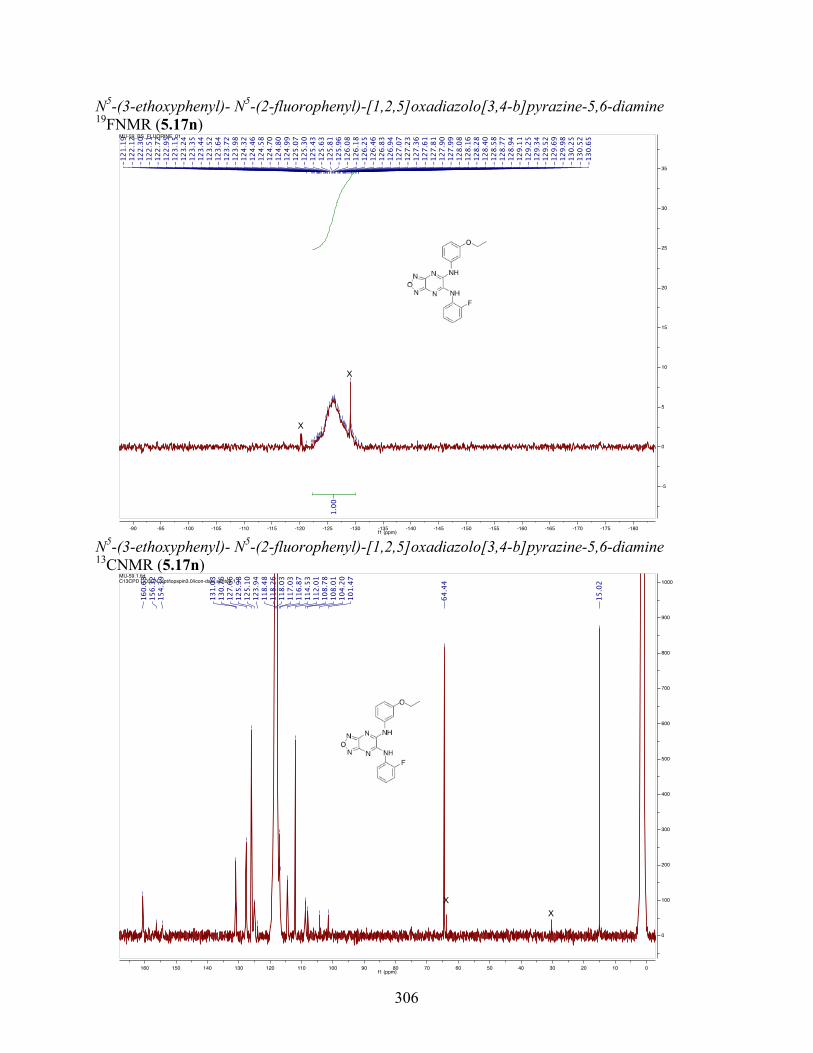
306
N5-(3-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17n)
N5-(3-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17n)
-180-175-170-165-160-155-150-145-140-135-130-125-120-115-110-105-100-95-90f1 (ppm)
-5
0
5
10
15
20
25
30
35
MU-59_BS_FLUORINE_01
1.00
-130.65
-130.52
-130.25
-129.98
-129.69
-129.52
-129.34
-129.25
-129.11
-128.94
-128.77
-128.58
-128.40
-128.28
-128.16
-128.08
-127.99
-127.90
-127.81
-127.61
-127.36
-127.23
-127.07
-126.94
-126.83
-126.46
-126.25
-126.18
-126.08
-125.96
-125.81
-125.63
-125.43
-125.30
-125.07
-124.99
-124.80
-124.70
-124.58
-124.46
-124.32
-123.98
-123.72
-123.64
-123.52
-123.44
-123.35
-123.24
-123.15
-122.95
-122.72
-122.51
-122.30
-122.12
-121.19
X
X
0102030405060708090100110120130140150160f1 (ppm)
0
100
200
300
400
500
600
700
800
900
1000
MU-59.1.fidC13CPD CD3CN /opt/topspin3.0/icon-data santos-1 1
15.02
64.44
101.47
104.20
108.01
108.78
112.01
114.53
116.87
117.03
118.03
118.26
118.48
123.94
125.10
125.98
127.66
130.76
131.03
154.39
156.32
160.63
X
X
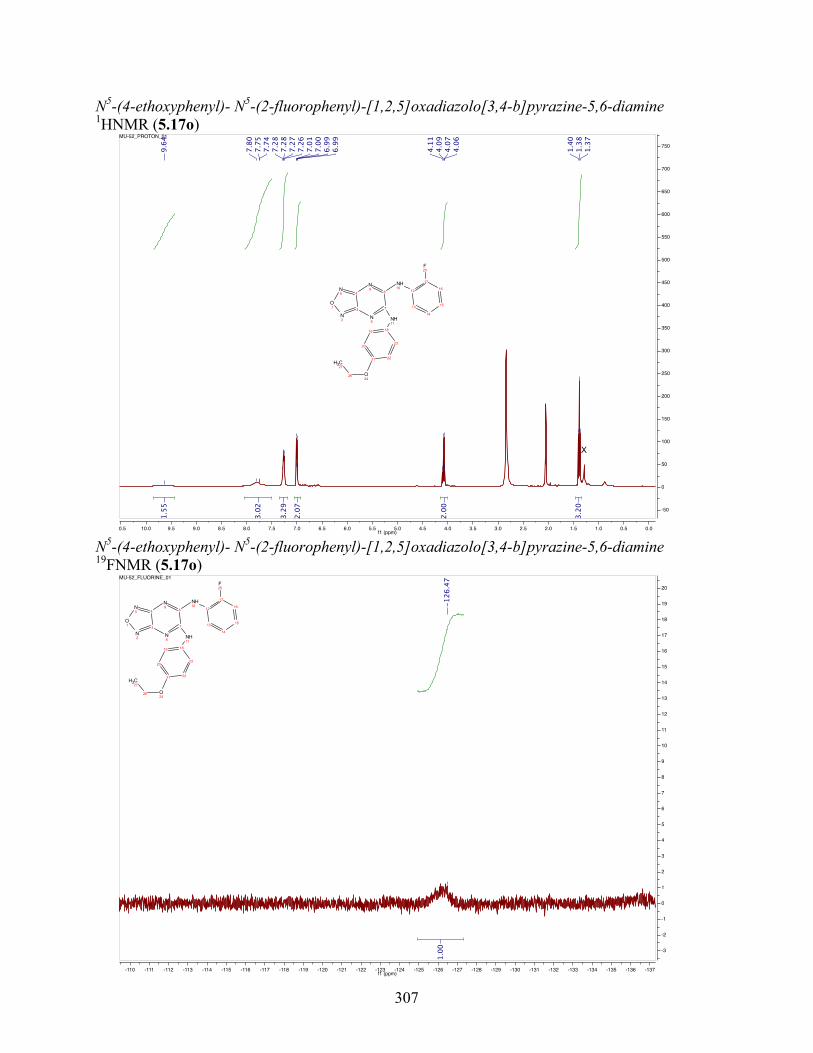
307
N5-(4-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17o)
N5-(4-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17o)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
MU-52_PROTON_01
3.20
2.00
2.07
3.29
3.02
1.55
1.37
1.38
1.40
4.06
4.07
4.09
4.11
6.99
6.99
7.00
7.01
7.26
7.27
7.28
7.28
7.74
7.75
7.80
9.64
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
O24
F25
26
CH327
X
-137-136-135-134-133-132-131-130-129-128-127-126-125-124-123-122-121-120-119-118-117-116-115-114-113-112-111-110f1 (ppm)
-3
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
MU-52_FLUORINE_01
1.00
-126.47
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
O24
F25
26
CH327
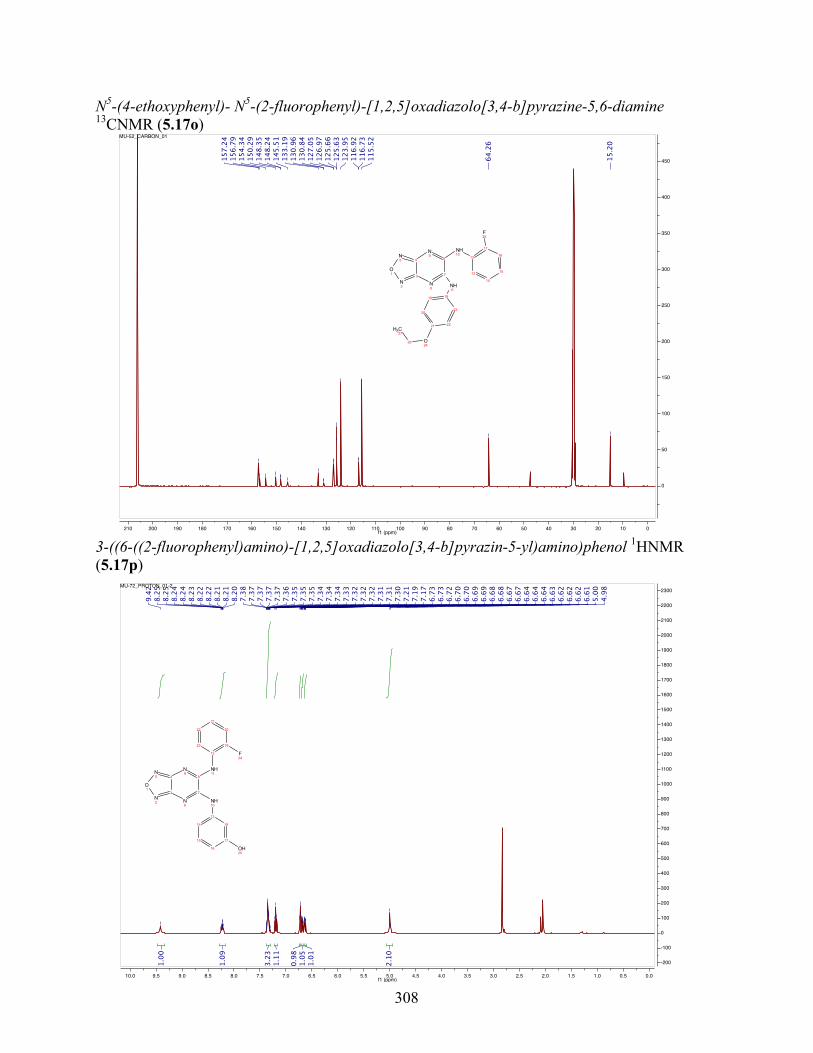
308
N5-(4-ethoxyphenyl)- N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17o)
3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol 1HNMR (5.17p)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
0
50
100
150
200
250
300
350
400
450
MU-52_CARBON_01
15.20
64.26
115.52
116.73
116.92
123.95
125.63
125.66
126.97
127.05
130.84
130.96
133.19
145.51
148.24
148.35
150.29
154.34
156.79
157.24
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
1819
20
2122
23
O24
F25
26
CH327
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0f1 (ppm)
-200
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
2100
2200
2300MU-72_PROTON_01-2
2.10
1.01
1.05
0.98
1.11
3.23
1.09
1.00
4.98
5.00
6.61
6.62
6.62
6.62
6.63
6.64
6.64
6.64
6.67
6.67
6.68
6.68
6.69
6.69
6.70
6.70
6.72
6.73
6.73
7.17
7.19
7.21
7.30
7.31
7.31
7.32
7.32
7.32
7.33
7.34
7.34
7.34
7.35
7.35
7.35
7.36
7.37
7.37
7.37
7.37
7.38
8.20
8.21
8.21
8.22
8.22
8.23
8.24
8.24
8.25
8.25
9.42
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
OH25
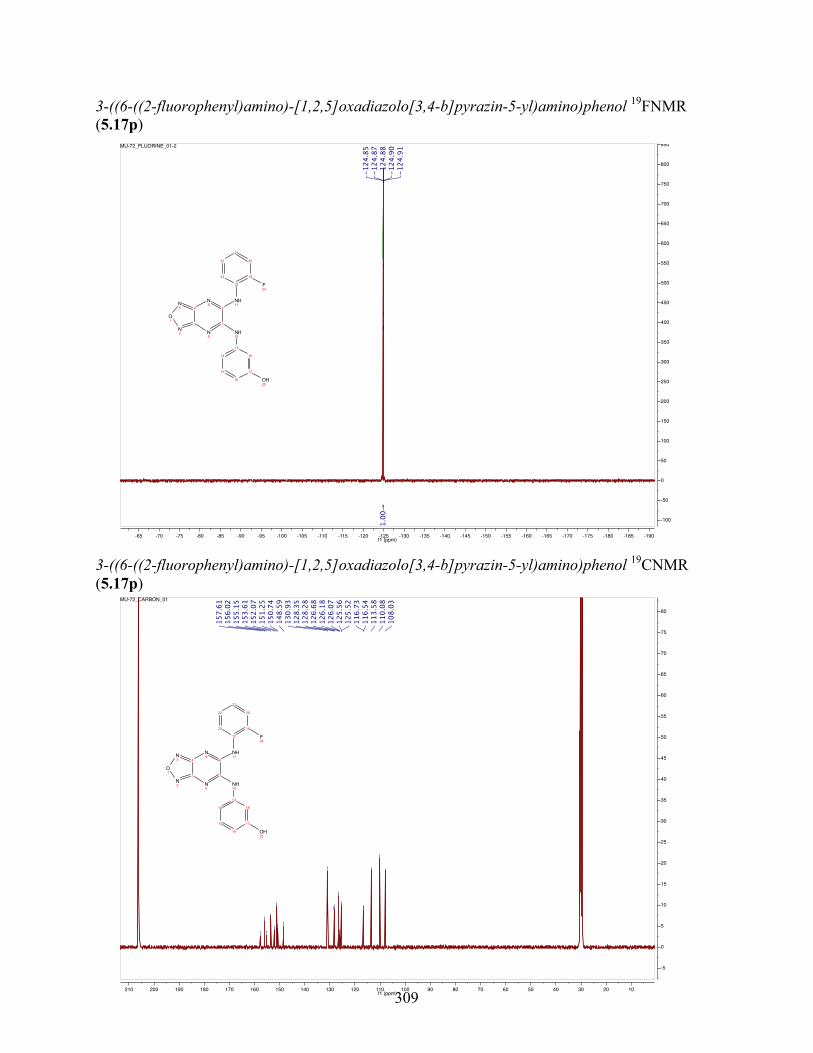
309
3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol 19FNMR (5.17p)
3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol 19CNMR (5.17p)
-190-185-180-175-170-165-160-155-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65f1 (ppm)
-100
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
850MU-72_FLUORINE_01-2
1.00
-124.91
-124.90
-124.88
-124.87
-124.85
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
OH25
102030405060708090100110120130140150160170180190200210f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
MU-72_CARBON_01
108.03
110.08
113.58
116.54
116.73
125.52
125.56
126.07
126.18
126.68
128.28
128.35
130.93
148.59
150.74
151.25
152.07
153.61
155.15
156.02
157.61
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
21
22
23
F24
OH25
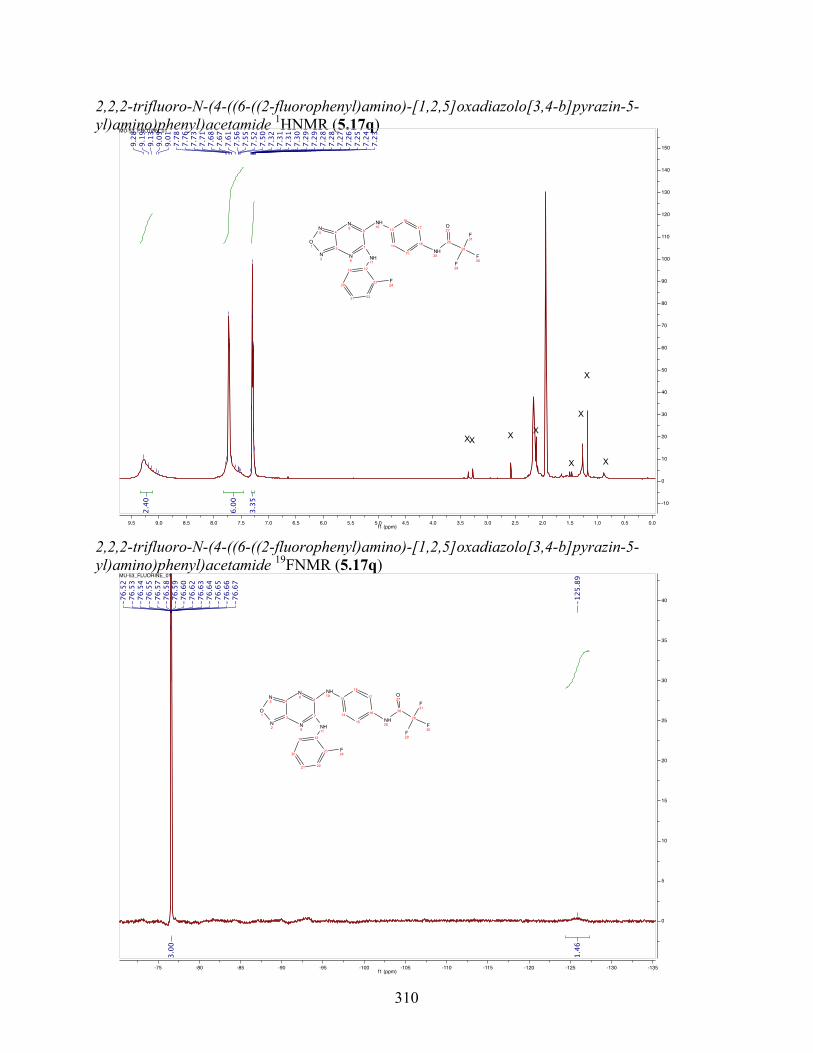
310
2,2,2-trifluoro-N-(4-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenyl)acetamide 1HNMR (5.17q)
2,2,2-trifluoro-N-(4-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenyl)acetamide 19FNMR (5.17q)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.5f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
MU-53_PROTON_01
3.35
6.00
2.40
7.23
7.24
7.25
7.26
7.27
7.28
7.28
7.29
7.29
7.30
7.31
7.31
7.32
7.50
7.52
7.55
7.56
7.61
7.67
7.68
7.71
7.73
7.76
7.78
9.01
9.05
9.13
9.19
9.28
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
2122
23 F24
NH25
26
O27
28
F29
F30
F31
XXX
X
X
X
XX
-135-130-125-120-115-110-105-100-95-90-85-80-75f1 (ppm)
0
5
10
15
20
25
30
35
40
MU-53_FLUORINE_011.46
3.00
-125.89
-76.67
-76.66
-76.65
-76.64
-76.63
-76.62
-76.60
-76.59
-76.58
-76.57
-76.55
-76.54
-76.53
-76.52
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
2122
23 F24
NH25
26
O27
28
F29
F30
F31
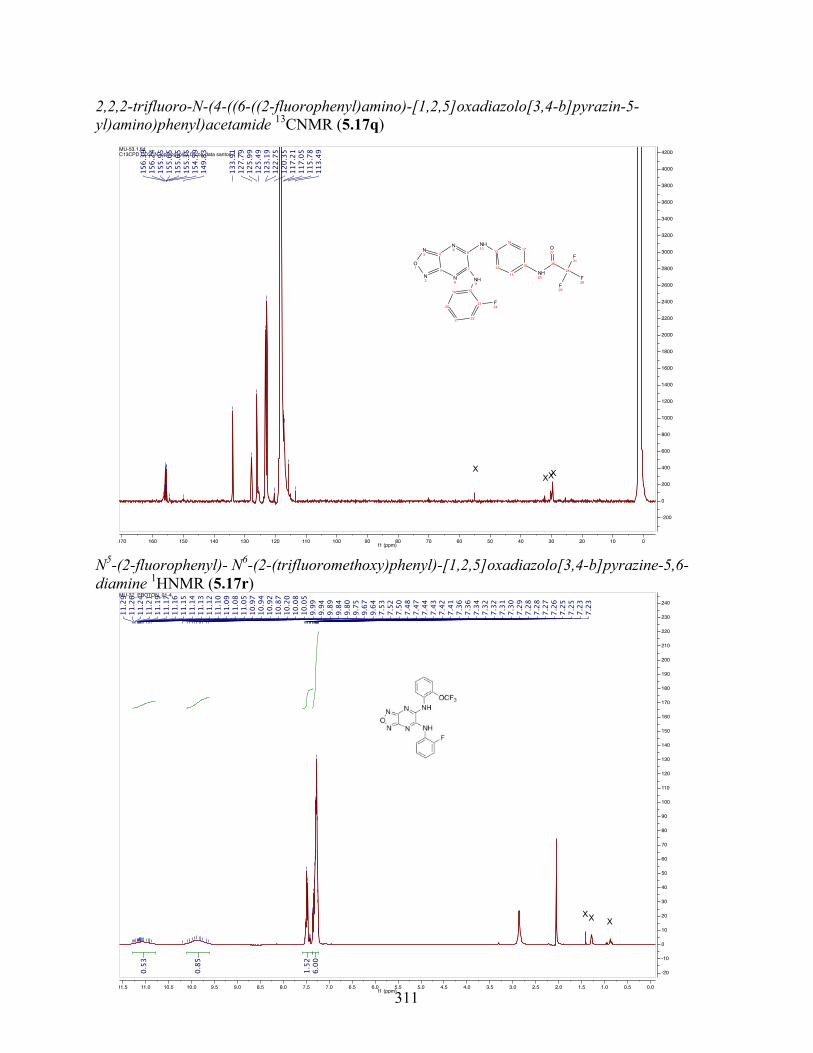
311
2,2,2-trifluoro-N-(4-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenyl)acetamide 13CNMR (5.17q)
N5-(2-fluorophenyl)- N6-(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17r)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-20
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
230
240
MU-37_PROTON_01-4
6.00
1.52
0.85
0.53
7.23
7.23
7.25
7.25
7.26
7.27
7.28
7.28
7.29
7.30
7.31
7.32
7.32
7.34
7.36
7.36
7.41
7.42
7.43
7.44
7.47
7.48
7.50
7.52
7.53
9.64
9.67
9.75
9.80
9.84
9.89
9.94
9.99
10.05
10.08
10.20
10.87
10.92
10.94
10.97
11.05
11.08
11.09
11.10
11.12
11.13
11.14
11.15
11.16
11.17
11.19
11.21
11.24
11.26
11.29
XX
X
0102030405060708090100110120130140150160170f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600
3800
4000
4200MU-53.1.fidC13CPD CD3CN /opt/topspin3.0/icon-data santos-1 1
113.49
115.78
117.05
117.21
120.35
122.75
123.19
125.49
125.99
127.79
133.91
149.83
154.59
155.35
155.65
155.65
155.95
156.24
156.39
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
18
19
20
2122
23 F24
NH25
26
O27
28
F29
F30
F31
X
XXX
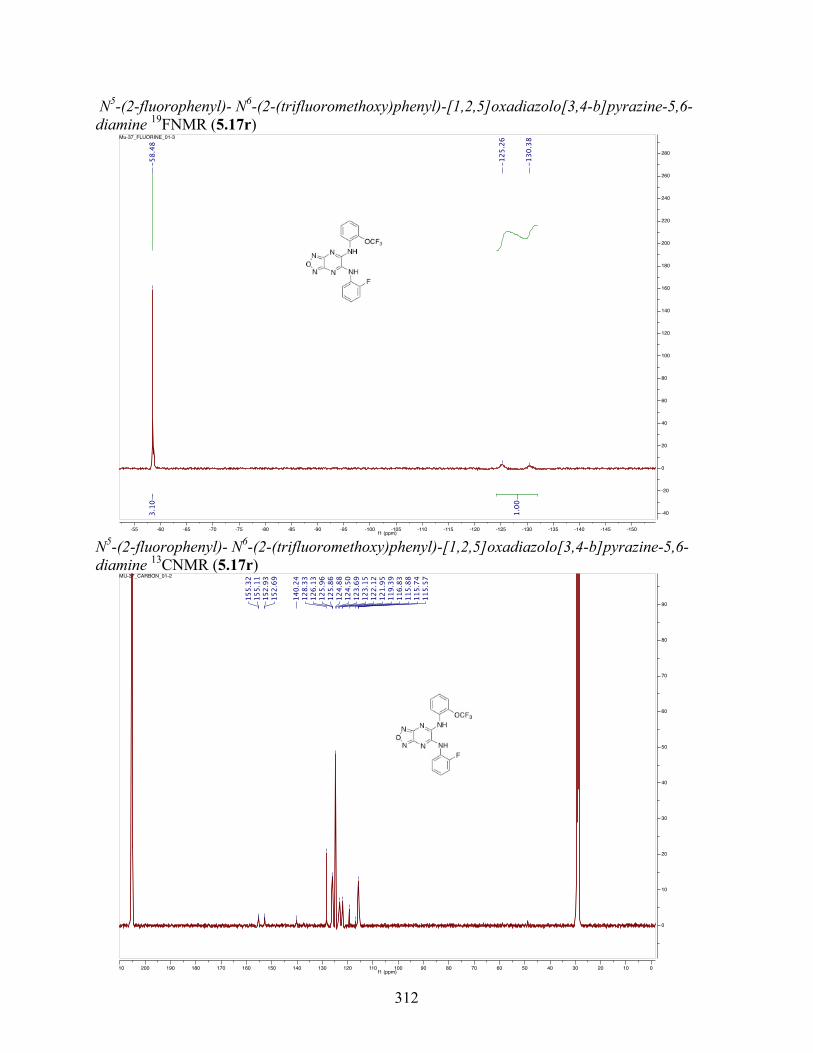
312
N5-(2-fluorophenyl)- N6-(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17r)
N5-(2-fluorophenyl)- N6-(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17r)
-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55f1 (ppm)
-40
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
Mu-37_FLUORINE_01-3
1.00
3.10
-130.38
-125.26
-58.48
0102030405060708090100110120130140150160170180190200210f1 (ppm)
0
10
20
30
40
50
60
70
80
90
MU-37_CARBON_01-2
115.57
115.74
115.88
116.83
119.39
121.95
122.12
123.15
123.69
124.50
124.88
125.86
125.96
126.13
128.33
140.24
152.69
152.93
155.11
155.32
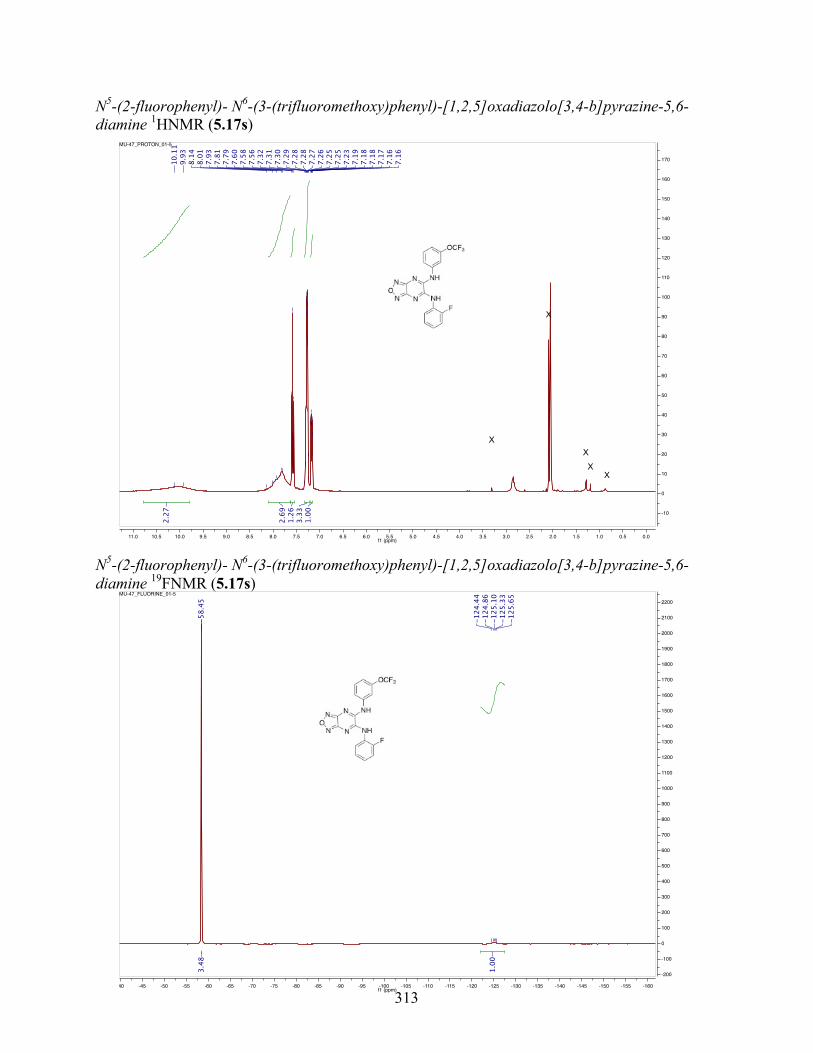
313
N5-(2-fluorophenyl)- N6-(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17s)
N5-(2-fluorophenyl)- N6-(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17s)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.0f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
MU-47_PROTON_01-5
1.00
3.33
1.26
2.69
2.27
7.16
7.16
7.17
7.18
7.18
7.19
7.23
7.25
7.25
7.26
7.27
7.28
7.28
7.29
7.30
7.31
7.32
7.56
7.58
7.60
7.79
7.81
7.93
8.01
8.14
9.93
10.11
X
X
X
X
X
-160-155-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50-45-40f1 (ppm)
-200
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
2100
2200
MU-47_FLUORINE_01-5
1.00
3.48
-125.65
-125.33
-125.10
-124.86
-124.44
-58.45
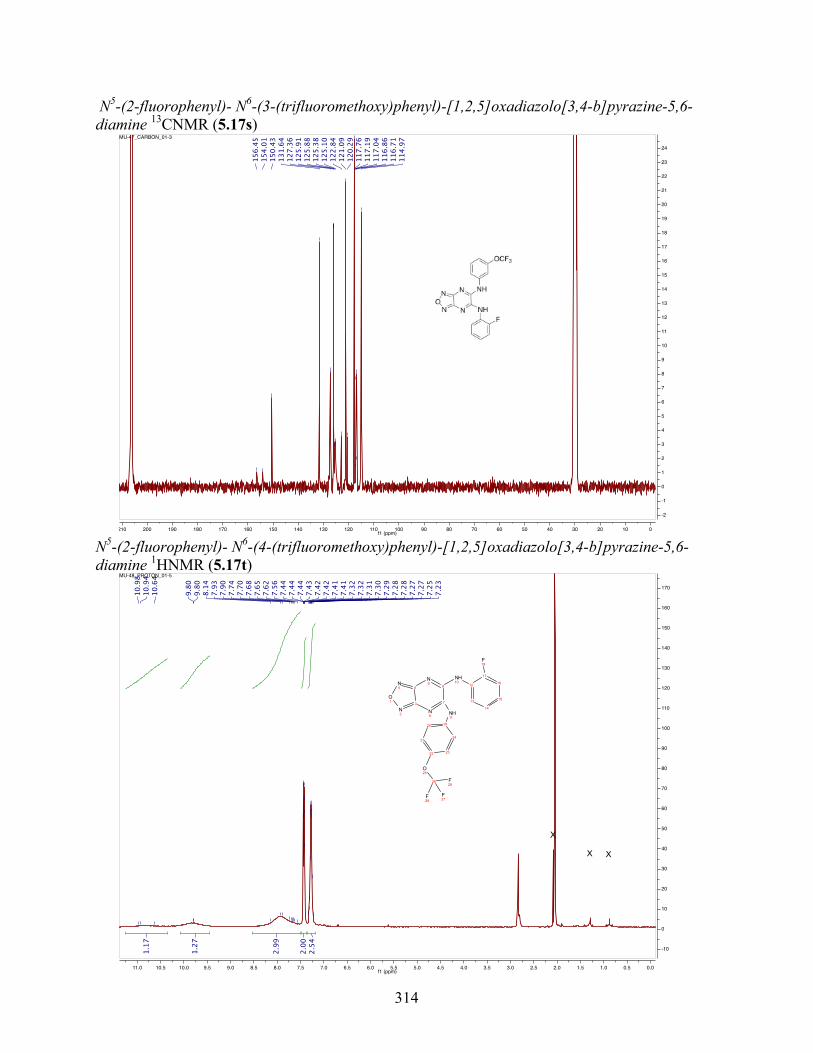
314
N5-(2-fluorophenyl)- N6-(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17s)
N5-(2-fluorophenyl)- N6-(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17t)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
MU-47_CARBON_01-3
114.97
116.71
116.86
117.04
117.19
117.76
120.29
121.09
122.84
125.10
125.38
125.88
125.91
127.36
131.64
150.43
154.01
156.45
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.0f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
MU-48_PROTON_01-5
2.54
2.00
2.99
1.27
1.17
7.23
7.25
7.27
7.27
7.28
7.28
7.29
7.30
7.31
7.32
7.32
7.41
7.41
7.42
7.42
7.43
7.44
7.44
7.44
7.56
7.62
7.65
7.68
7.70
7.74
7.90
7.93
8.14
9.80
9.80
10.64
10.94
10.98
X
X X
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
F18
1920
21
2223
24
O25
26
F27
F28
F29
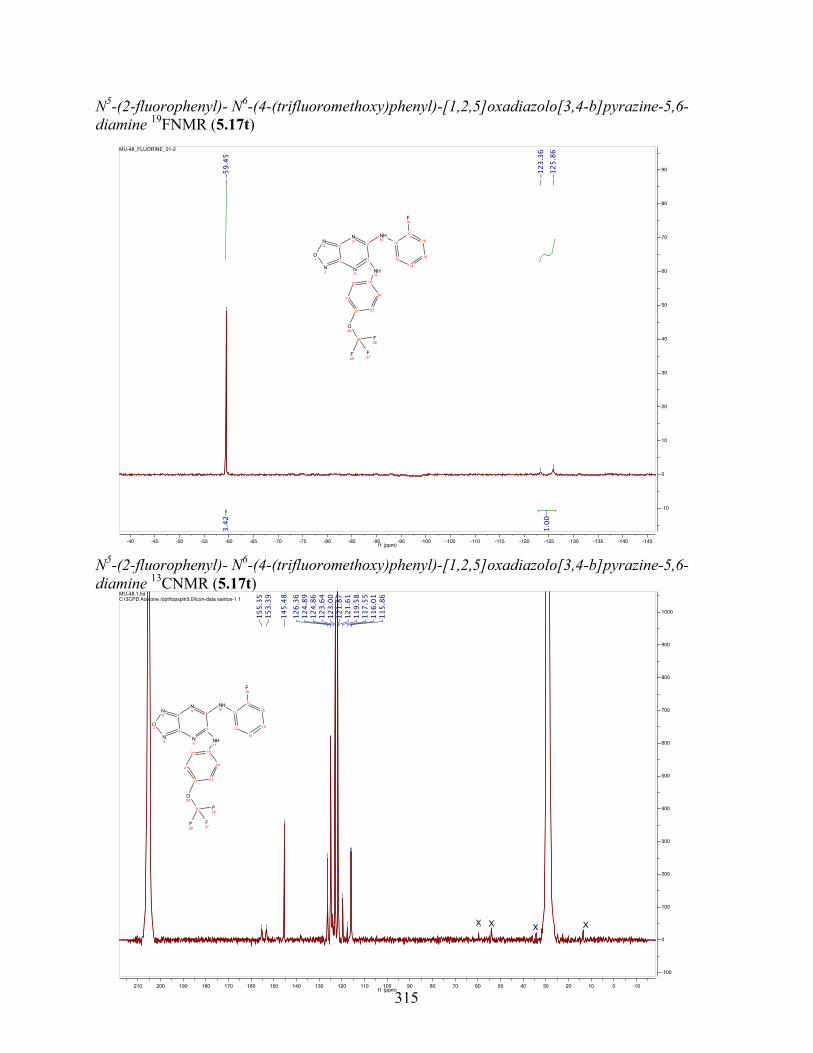
315
N5-(2-fluorophenyl)- N6-(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17t)
N5-(2-fluorophenyl)- N6-(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17t)
-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50-45-40f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
MU-48_FLUORINE_01-2
1.00
3.42
-125.86
-123.36
-59.45
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
F18
1920
21
2223
24
O25
26
F27
F28
F29
-100102030405060708090100110120130140150160170180190200210f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
MU-48.1.fidC13CPD Acetone /opt/topspin3.0/icon-data santos-1 1
115.86
116.01
117.55
119.58
121.61
121.87
123.00
123.64
124.86
124.89
126.36
145.48
153.39
155.35
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15
16
17
F18
1920
21
2223
24
O25
26
F27
F28
F29
X XX X
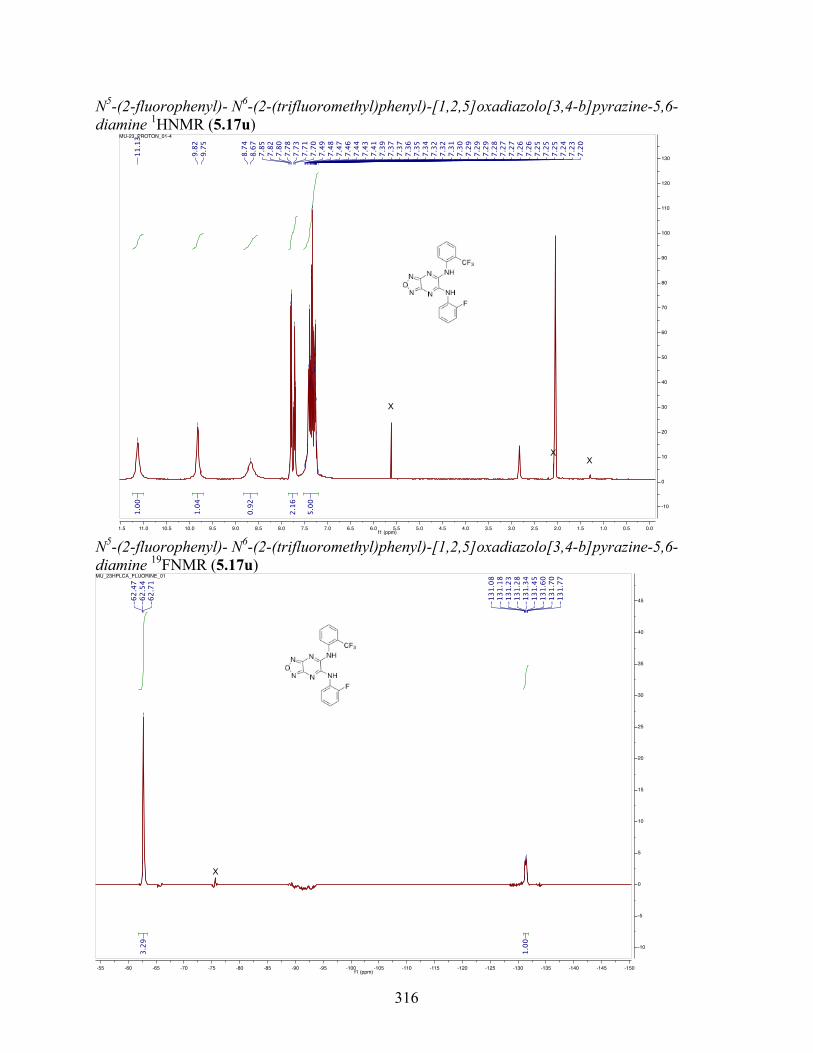
316
N5-(2-fluorophenyl)- N6-(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17u)
N5-(2-fluorophenyl)- N6-(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17u)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
MU-23_PROTON_01-4
5.00
2.16
0.92
1.04
1.00
7.20
7.23
7.24
7.25
7.25
7.25
7.26
7.26
7.27
7.27
7.28
7.29
7.29
7.29
7.30
7.31
7.32
7.32
7.34
7.35
7.36
7.37
7.37
7.39
7.41
7.43
7.44
7.46
7.47
7.48
7.49
7.70
7.71
7.73
7.78
7.80
7.82
7.85
8.67
8.74
9.75
9.82
11.13
X
X
X
-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55f1 (ppm)
-10
-5
0
5
10
15
20
25
30
35
40
45
MU_23HPLCA_FLUORINE_01
1.00
3.29
-131.77
-131.70
-131.60
-131.45
-131.34
-131.28
-131.23
-131.18
-131.08
-62.71
-62.54
-62.47
X
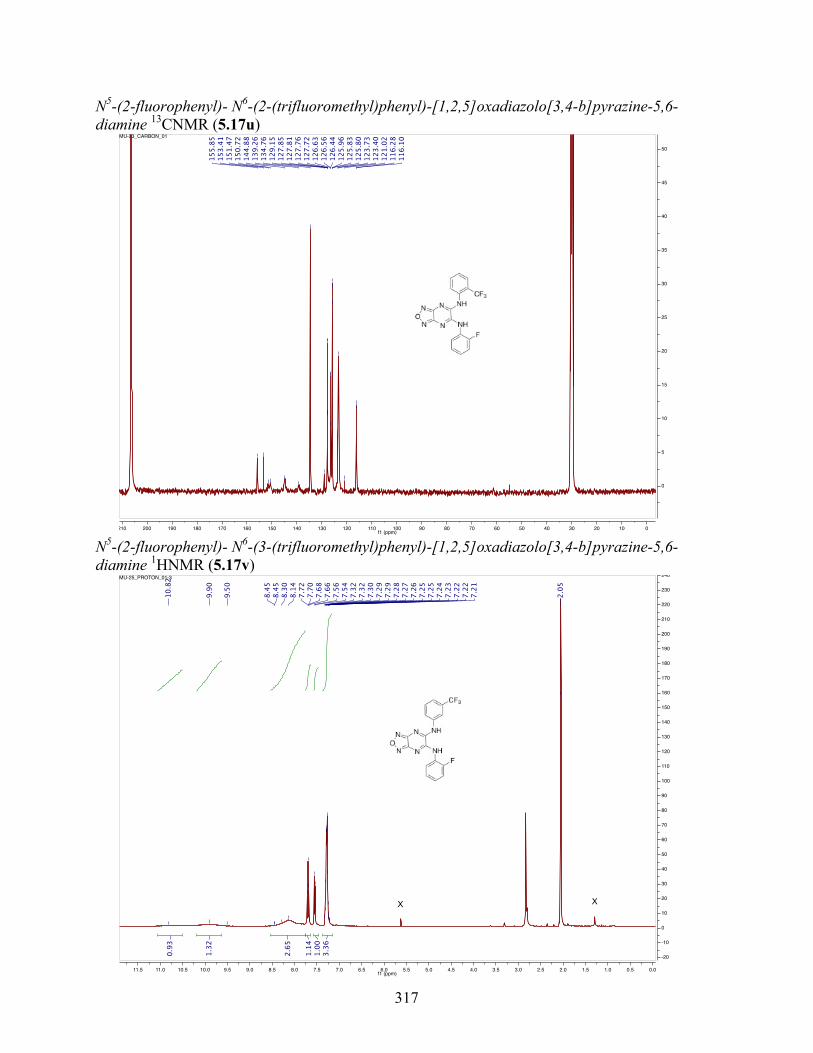
317
N5-(2-fluorophenyl)- N6-(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17u)
N5-(2-fluorophenyl)- N6-(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17v)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
0
5
10
15
20
25
30
35
40
45
50
MU-23_CARBON_01
116.10
116.28
121.02
123.40
123.73
125.80
125.83
125.96
126.44
126.56
126.63
127.72
127.76
127.81
127.85
129.15
134.76
139.26
144.88
150.72
151.47
153.41
155.85
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-20
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
230
240MU-25_PROTON_01-3
3.36
1.00
1.14
2.65
1.32
0.93
2.05
7.21
7.22
7.22
7.23
7.24
7.25
7.25
7.26
7.27
7.28
7.29
7.29
7.30
7.32
7.32
7.54
7.56
7.66
7.68
7.70
7.72
8.14
8.30
8.45
8.45
9.50
9.90
10.82
X X
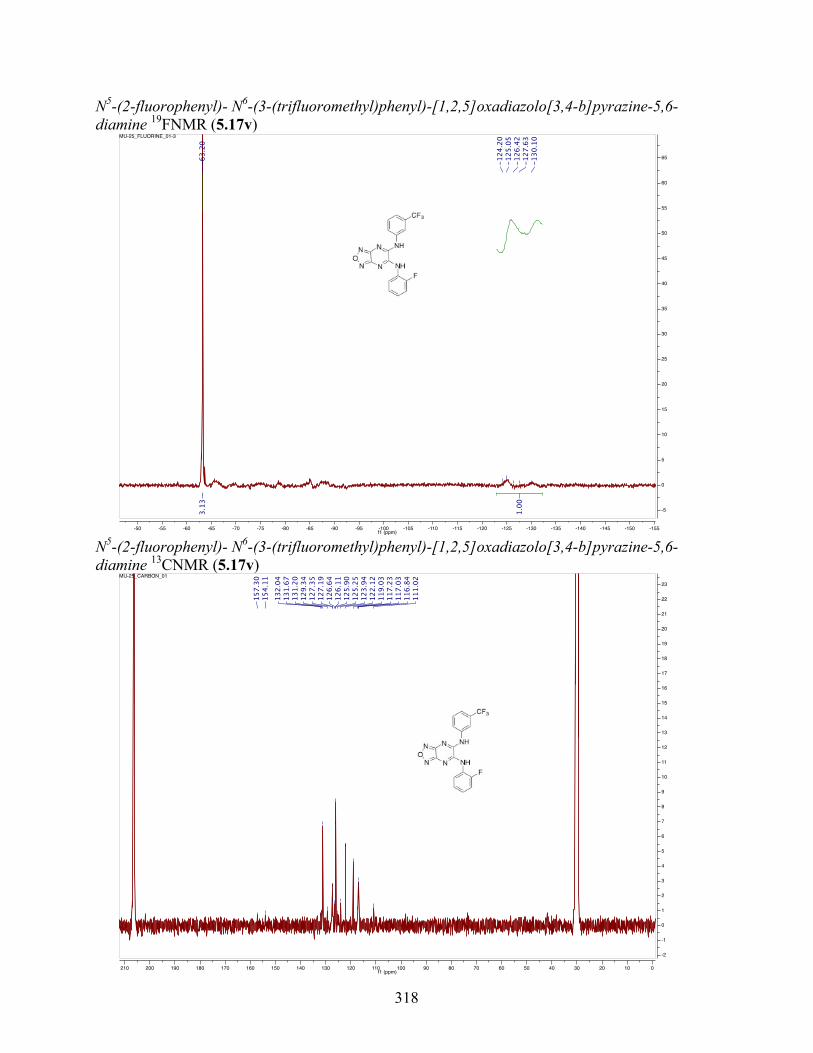
318
N5-(2-fluorophenyl)- N6-(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17v)
N5-(2-fluorophenyl)- N6-(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17v)
-155-150-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
MU-25_FLUORINE_01-3
1.00
3.13
-130.10
-127.63
-126.42
-125.05
-124.20
-63.20
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-2
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
MU-25_CARBON_01
111.02
116.84
117.03
117.23
119.03
122.12
123.94
125.25
125.90
126.11
126.64
127.19
127.35
129.34
131.20
131.67
132.04
154.11
157.30
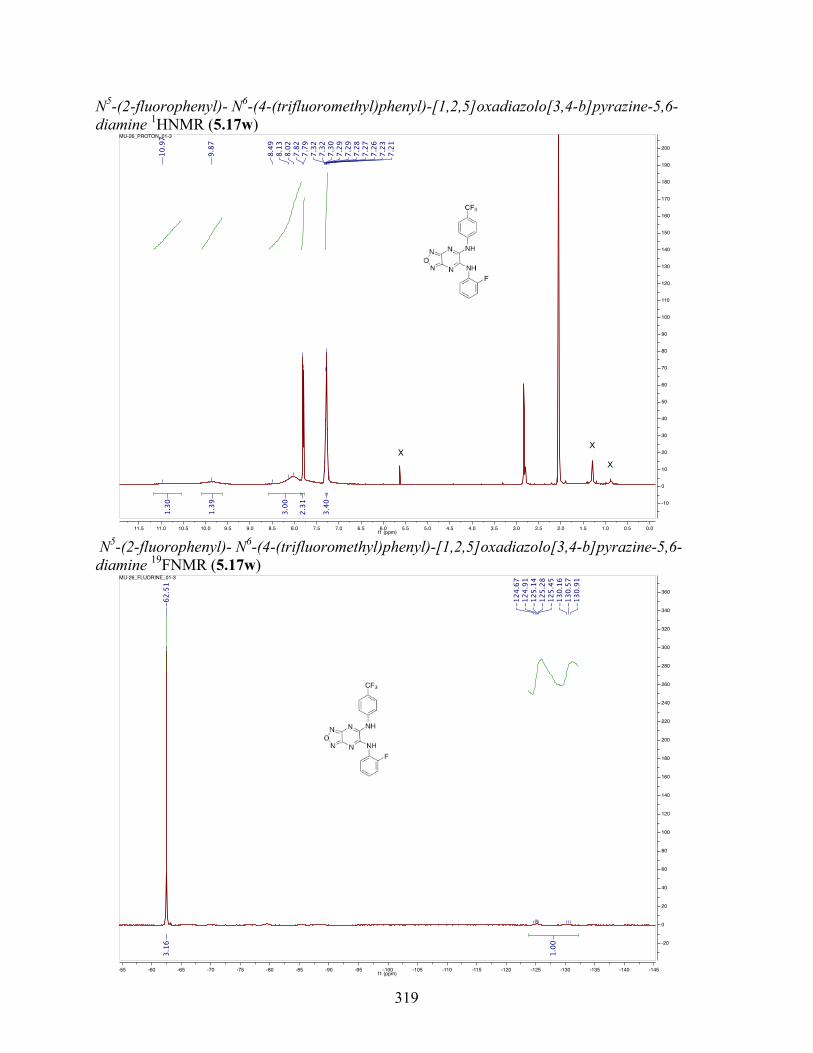
319
N5-(2-fluorophenyl)- N6-(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 1HNMR (5.17w)
N5-(2-fluorophenyl)- N6-(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 19FNMR (5.17w)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.511.011.5f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
170
180
190
200
MU-26_PROTON_01-3
3.40
2.31
3.00
1.39
1.30
7.21
7.23
7.26
7.27
7.28
7.29
7.29
7.30
7.32
7.32
7.79
7.82
8.02
8.13
8.49
9.87
10.97
X
X
X
-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
MU-26_FLUORINE_01-3
1.00
3.16
-130.91
-130.57
-130.16
-125.45
-125.28
-125.14
-124.91
-124.67
-62.51
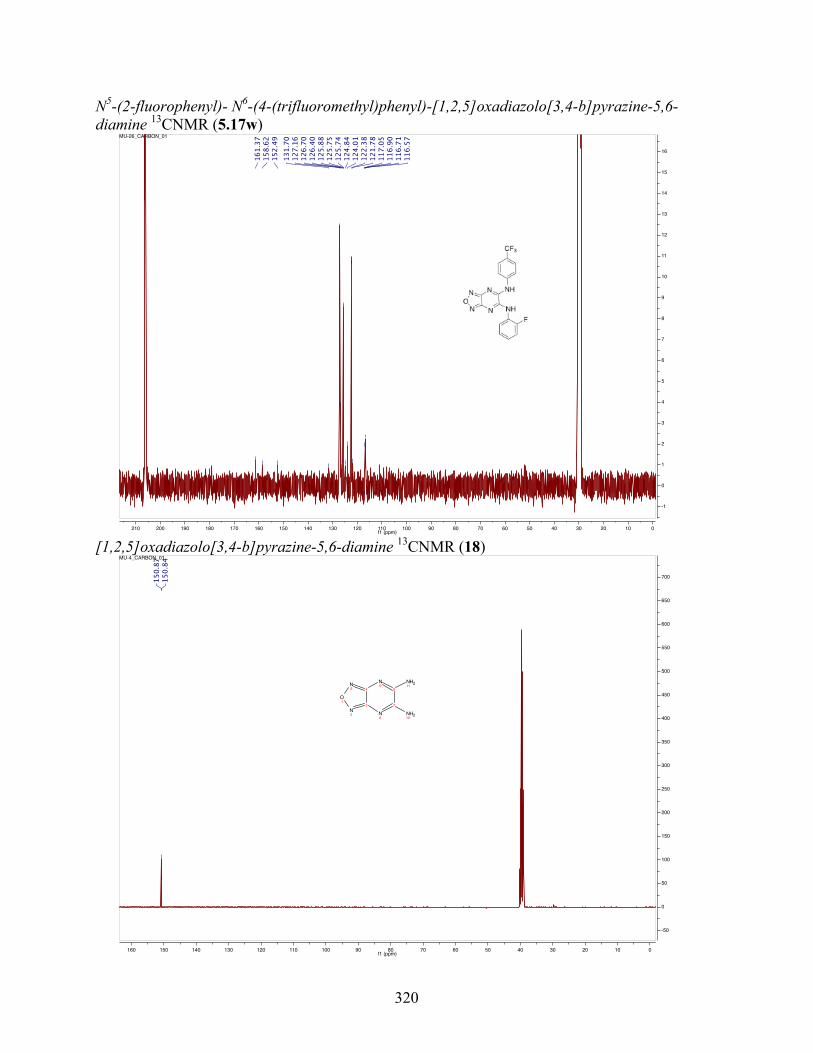
320
N5-(2-fluorophenyl)- N6-(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (5.17w)
[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine 13CNMR (18)
0102030405060708090100110120130140150160170180190200210f1 (ppm)
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
MU-26_CARBON_01
116.57
116.71
116.90
117.05
121.78
122.38
124.01
124.84
125.74
125.75
125.88
126.40
126.70
127.16
131.70
152.49
158.62
161.37
0102030405060708090100110120130140150160f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
MU-4_CARBON_01
150.84
150.87
O1
N2
3
4
N5
N6
7
8
N9
NH210
NH211
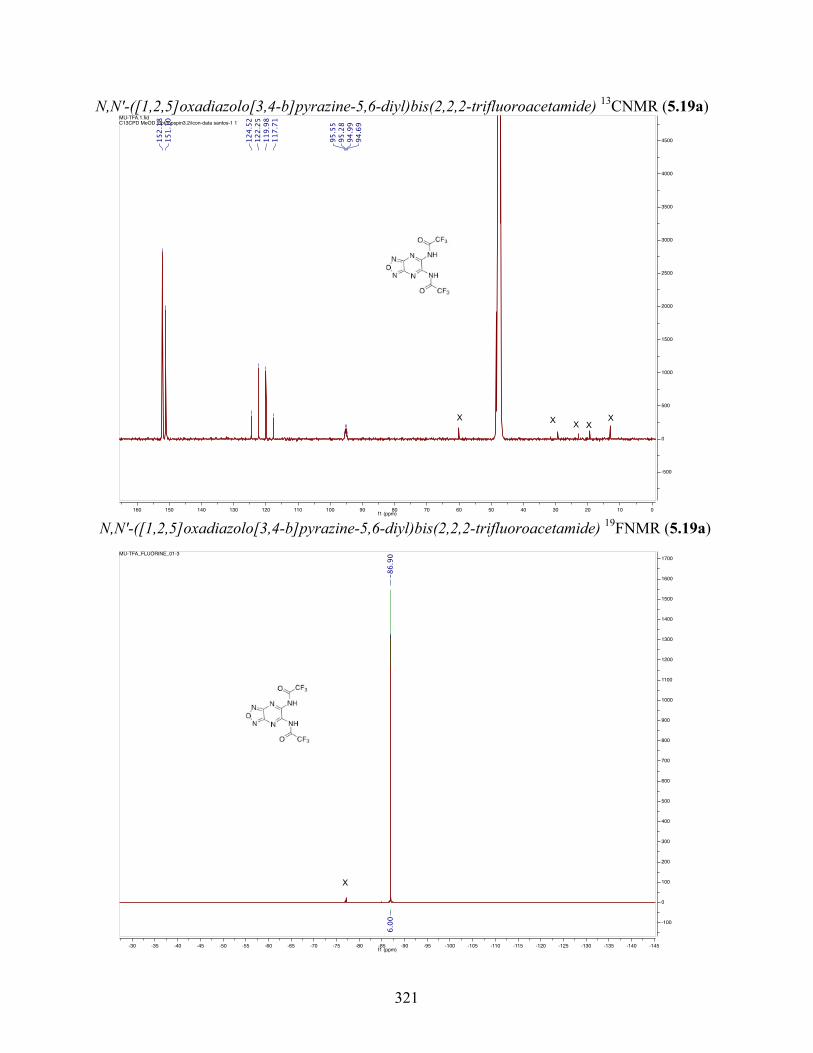
321
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2,2,2-trifluoroacetamide) 13CNMR (5.19a)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2,2,2-trifluoroacetamide) 19FNMR (5.19a)
0102030405060708090100110120130140150160f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
MU-TFA.1.fidC13CPD MeOD /opt/topspin3.2/icon-data santos-1 1
94.69
94.99
95.28
95.55
117.71
119.98
122.25
124.52
151.10
152.18
X XX X
X
-145-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50-45-40-35-30f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700MU-TFA_FLUORINE_01-3
6.00
-86.90
X
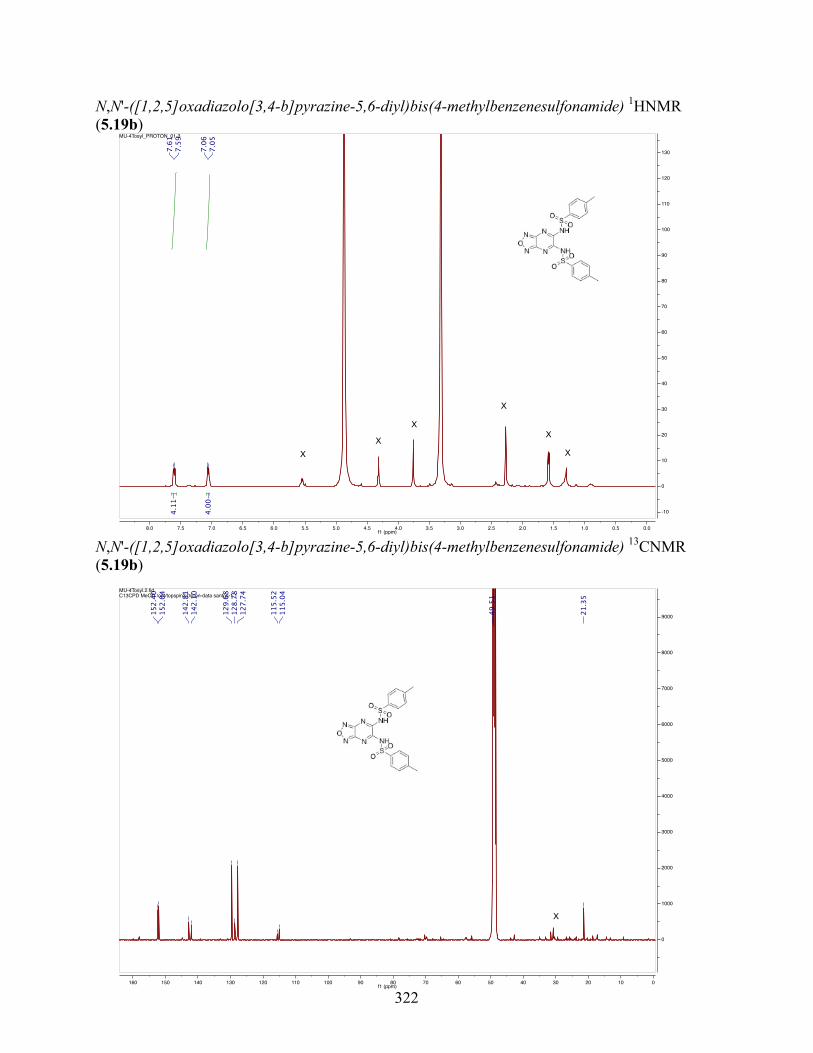
322
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-methylbenzenesulfonamide) 1HNMR (5.19b)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-methylbenzenesulfonamide) 13CNMR (5.19b)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
MU-4Tosyl_PROTON_01-3
4.00
4.11
7.05
7.06
7.59
7.61
X
X
X
X
X
X
0102030405060708090100110120130140150160f1 (ppm)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
MU-4Tosyl.2.fidC13CPD MeOD /opt/topspin3.0/icon-data santos-1 1
21.35
49.51
115.04
115.52
127.74
128.78
129.68
142.10
142.81
152.04
152.40
X
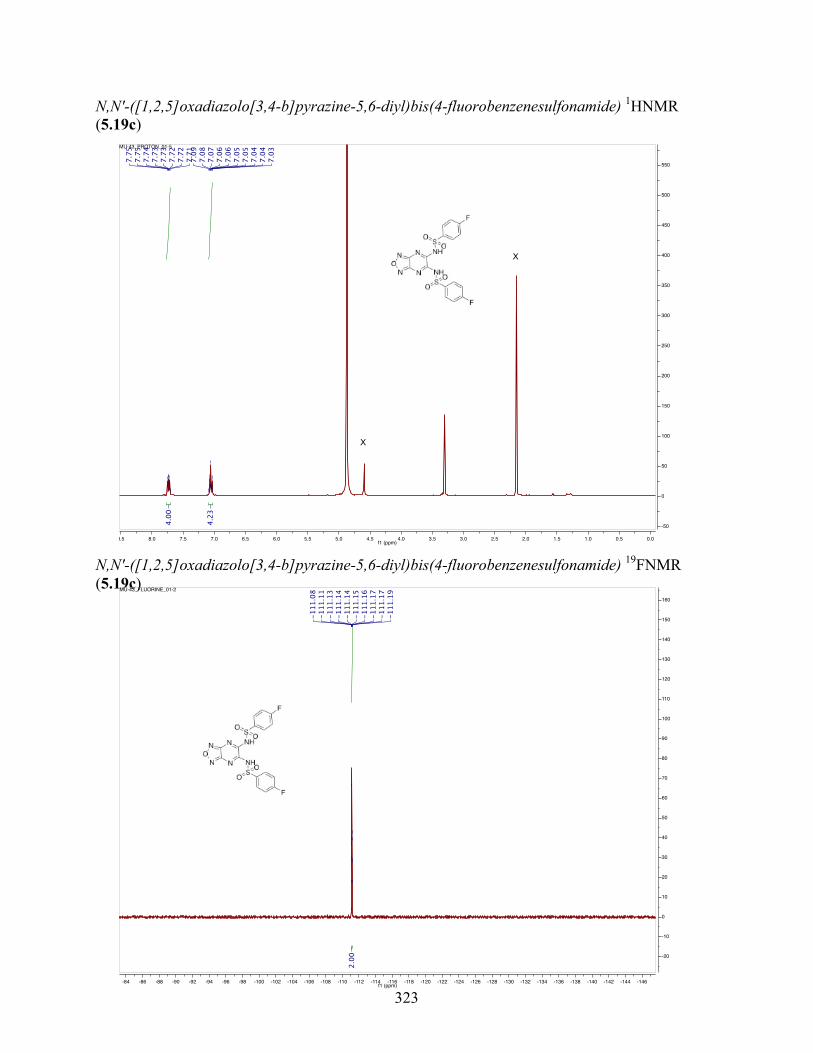
323
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-fluorobenzenesulfonamide) 1HNMR (5.19c)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-fluorobenzenesulfonamide) 19FNMR (5.19c)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
MU-43_PROTON_01-5
4.23
4.00
7.03
7.04
7.04
7.05
7.05
7.06
7.06
7.07
7.08
7.09
7.71
7.72
7.72
7.73
7.73
7.74
7.75
7.75
X
X
-146-144-142-140-138-136-134-132-130-128-126-124-122-120-118-116-114-112-110-108-106-104-102-100-98-96-94-92-90-88-86-84f1 (ppm)
-20
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
MU-43_FLUORINE_01-2
2.00
-111.19
-111.17
-111.17
-111.16
-111.15
-111.14
-111.14
-111.13
-111.11
-111.08
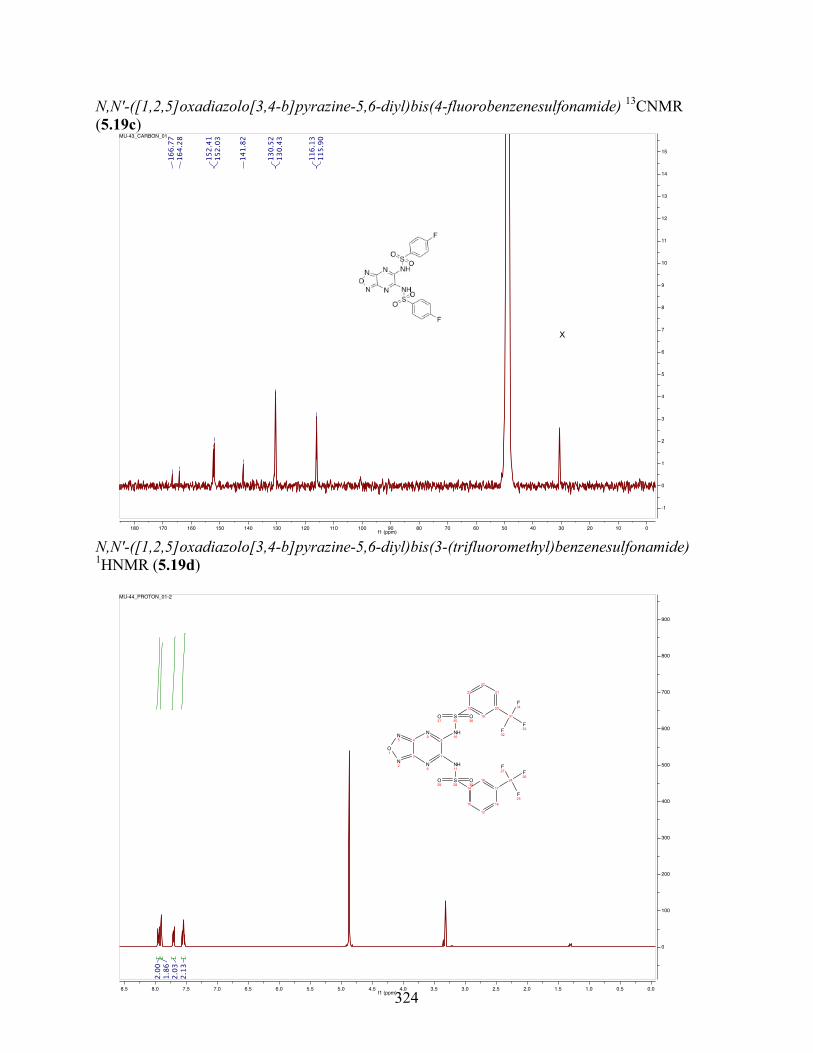
324
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(4-fluorobenzenesulfonamide) 13CNMR (5.19c)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(3-(trifluoromethyl)benzenesulfonamide) 1HNMR (5.19d)
0102030405060708090100110120130140150160170180f1 (ppm)
-1
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
MU-43_CARBON_01
115.90
116.13
130.43
130.52
141.82
152.03
152.41
164.28
166.77
X
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
0
100
200
300
400
500
600
700
800
900
MU-44_PROTON_01-2
2.13
2.03
1.86
2.00
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15 16
17
18
19
20
21
22
23
24
F25
F26
F27
S28
O29
O30
31
F32
F33
F34
S35
O36
O37
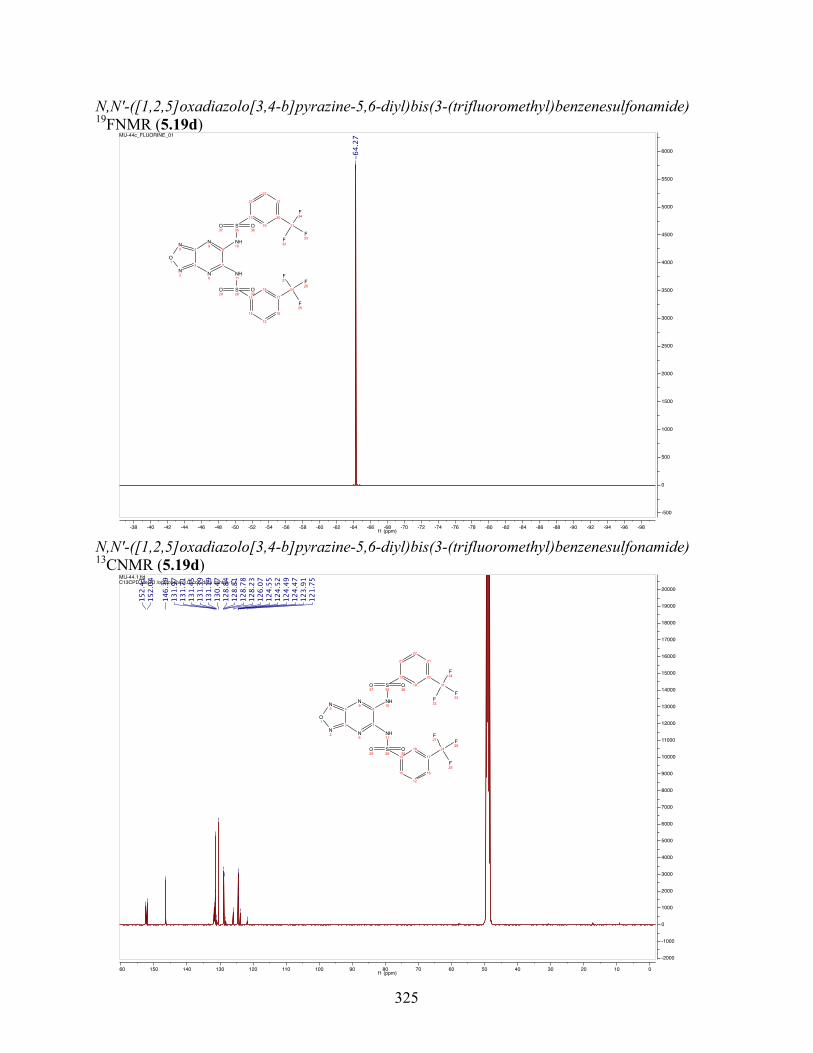
325
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(3-(trifluoromethyl)benzenesulfonamide) 19FNMR (5.19d)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(3-(trifluoromethyl)benzenesulfonamide) 13CNMR (5.19d)
0102030405060708090100110120130140150160f1 (ppm)
-2000
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
20000
MU-44.1.fidC13CPD MeOD /opt/topspin3.0/icon-data santos-1 1
121.75
123.91
124.47
124.49
124.52
124.55
126.07
128.23
128.78
128.81
128.84
130.47
131.19
131.39
131.45
131.71
131.97
146.39
152.04
152.41
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15 16
17
18
19
20
21
22
23
24
F25
F26
F27
S28
O29
O30
31
F32
F33
F34
S35
O36
O37
-98-96-94-92-90-88-86-84-82-80-78-76-74-72-70-68-66-64-62-60-58-56-54-52-50-48-46-44-42-40-38f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
MU-44c_FLUORINE_01
-64.27
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15 16
17
18
19
20
21
22
23
24
F25
F26
F27
S28
O29
O30
31
F32
F33
F34
S35
O36
O37
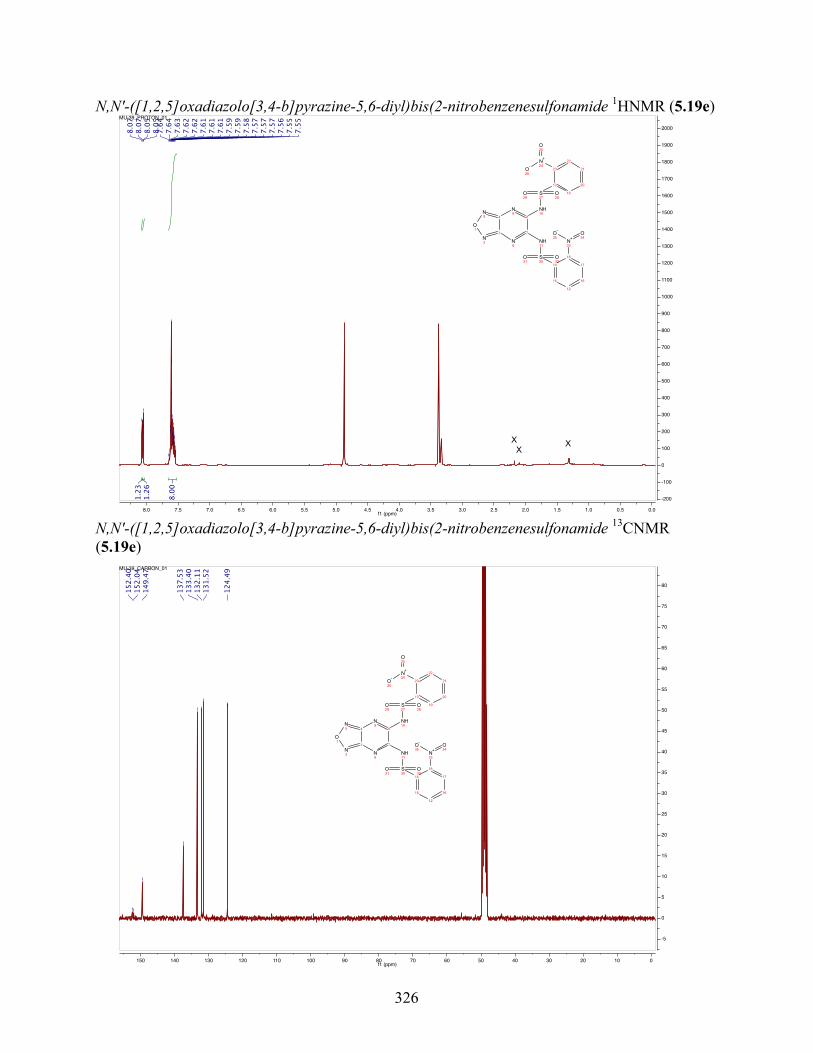
326
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2-nitrobenzenesulfonamide 1HNMR (5.19e)
N,N'-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diyl)bis(2-nitrobenzenesulfonamide 13CNMR (5.19e)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-200
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
1900
2000
MU-39_PROTON_01
8.00
1.26
1.23
7.55
7.55
7.56
7.57
7.57
7.57
7.58
7.59
7.59
7.61
7.61
7.61
7.62
7.62
7.63
7.64
7.64
8.05
8.05
8.07
8.07
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15 16
17
18
19
20
21
22
23
N+
24
O25
O-
26
S27
O28
O29
S30
O31
O32
N+
33
O34
O-
35
X
XX
0102030405060708090100110120130140150f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
MU-39_CARBON_01
124.49
131.52
132.11
133.40
137.53
149.47
152.04
152.40
O1
N2
3
4
N5
N6
7
8
N9
NH10
NH11
12
13
14
15 16
17
18
19
20
21
22
23
N+
24
O25
O-
26
S27
O28
O29
S30
O31
O32
N+
33
O34
O-
35

327
6.4 References
1. Patwardhan, N. N.; Morris, E. A.; Kharel, Y.; Raje, M. R.; Gao, M.; Tomsig, J. L.; Lynch, K.
R.; Santos, W. L. Structure–Activity Relationship Studies and in Vivo Activity of Guanidine-
Based Sphingosine Kinase Inhibitors: Discovery of SphK1- and SphK2-Selective Inhibitors. J.
Med. Chem. 2015, 58, 1879–1899.
2. Kharel, Y.; Morris, E. A.; Congdon, M. D.; Thorpe, S. B.; Tomsig, J. L.; Santos, W. L.;
Lynch, K. R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J.
Pharmacol. Exp. Ther. 2015, 355, 23–31.
3. Kharel, Y.; Raje, M.; Gao, M.; Gellett, A. M.; Tomsig, J. L.; Lynch, K. R.; Santos, W. L.
Sphingosine Kinase Type 2 Inhibition Elevates Circulating Sphingosine 1-Phosphate. Biochem.
J. 2012, 447, 149–157.
4. Congdon, M. D.; Kharel, Y.; Brown, A. M.; Lewis, S. N.; Bevan, D. R.; Lynch, K. R.; Santos,
W. L. Structure–Activity Relationship Studies and Molecular Modeling of Naphthalene-Based
Sphingosine Kinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 229–234.
5. Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson,
A. J. Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J.
Comput. Chem. 2009, 30, 2785–2791.
6. Trott, O.; Olson, A. J. Autodock Vina: Improving the Speed and Accuracy of Docking with a
New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31,
455–461.
7. Kenwood, B. M.; Weaver, J. L.; Bajwa, A.; Poon, I. K.; Byrne, F. L.; Murrow, B. A.;
Calderone, J. A.; Huang, L.; Divakaruni, A. S.; Tomsig, J. L.; Okabe, K.; Lo, R. H.; Coleman, G.
C.; Columbus, L.; Yan, Z.; Saucerman, J. J.; Smith, J. S.; Holmes, J. W.; Lynch, K. R.;

328
Ravichandran, K. S.; Uchiyama, S.; Santos, W. L.; Rogers, G. W.; Okusa, M. D.; Bayliss, D. A.;
Hoehn, K. L. Identification of a Novel Mitochondrial Uncoupler That Does Not Depolarize the
Plasma Membrane. Mol. Metab. 2014, 3, 114–123.
8. Thottempudi, V.; Yin, P.; Zhang, J.; Parrish, D. A.; Shreeve, J. M. 1,2,3-Triazolo[4,5,-
E]Furazano[3,4,-B]Pyrazine 6-Oxide-a Fused Heterocycle with a Roving Hydrogen Forms a
New Class of Insensitive Energetic Materials. Chem. Eur. J. 2014, 20, 542–548.





























