Embed Size (px)
Citation preview

Classe de première
SPÉCIALITÉ PHYSIQUE CHIMIE
Wulfran Fortin - 2019

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
ii

Table des matières
1 Détermination de la composition d’un système chimique à l’état initial 11.1 Masse molaire M et quantité de matière n . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.2 Spectroscopie d’absorbance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2 Suivi et modélisation de l’évolution d’un système chimique 52.1 Réaction d’oxydo-réduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52.2 Tableau d’avancement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
3 Détermination d’une quantité de matière grâce à une transformation chimique 93.1 Titrage par suivi colorimétrique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93.2 Dosage des ions Fe2+ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
4 De la structure à la polarité d’une entité 114.1 Schéma de Lewis d’une entité . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114.2 Géométrie d’une entité . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134.3 Électronégativité des atomes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144.4 Polarisation d’une liaison chimique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144.5 Polarité d’une molécule . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5 De la structure des entités à la cohésion et à la solubilté/miscibilité d’espèces chimiques 155.1 Cohésion des solides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155.2 Cohésion des solides ioniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155.3 Cohésion des solides moléculaires . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155.4 Dissolution des solides ioniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165.5 Solubilité d’une espèce dans un solvant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185.6 Extraction solvant solvant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185.7 Hydrophilie, lipophilie et amphiphilie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
6 Structure des entités organiques 216.1 Molécules organiques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216.2 Formule brutes et semi-développées . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216.3 Squelette carboné saturé . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216.4 Groupes caractéristiques et familles fonctionnelles . . . . . . . . . . . . . . . . . . . . . . . . . . 226.5 Spectroscopie infrarouge . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
7 Synthèse d’espèces chimiques organiques 277.1 Étapes d’un protocole . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 277.2 Rendement de synthèses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
8 Conversion de l’énergie stockée dans la matière organique 318.1 Les combustibles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 318.2 Problématiques liées aux combustibles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 318.3 Combustion en chimie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 328.4 Réaction exothermique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
iii

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
9 Interactions fondamentales et notion de champ 359.1 Interaction électrostatique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 359.2 Interaction gravitationnelle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 379.3 Analogies entre interactions de gravitation et électrostatique . . . . . . . . . . . . . . . . . . . 38
10 Description d’un fluide au repos 3910.1 Les fluides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3910.2 Description qualitative d’un fluide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3910.3 Force de pression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4010.4 Loi de Boyle et Mariotte - XVIIesiècle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4010.5 Loi fondamentale de la statique des fluides - XVIIesiècle . . . . . . . . . . . . . . . . . . . . . . 41
11 Mouvement d’un système 4311.1 Vecteur vitesse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4311.2 Vecteur variation de vitesse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4411.3 Variation de la vitesse et résultante des forces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
12 Aspects énergétiques des phénomènes électriques 4912.1 Courant électrique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4912.2 Source réelle de tension continue . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5012.3 Puissance et énergie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5012.4 Effet Joule . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5112.5 Rendement d’un convertisseur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
13 Aspects énergétiques des phénomènes mécaniques 5313.1 Énergie cinétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5313.2 Travail d’une force . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5313.3 Forces conservatives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5513.4 Forces non conservatives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5513.5 Théorème de l’énergie cinétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5513.6 Énergie potentielle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5613.7 Énergie mécanique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5713.8 Conservation et non conservation de l’énergie mécanique . . . . . . . . . . . . . . . . . . . . . 57
14 Ondes mécaniques 5914.1 Onde mécanique progressive . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.2 Célérité et retard . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.3 Onde mécanique périodique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.4 Onde mécanique sinusoïdale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.5 Période, longueur d’onde et célérité . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.6 Exercices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5914.7 Correction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
15 Images et couleurs 6115.1 Optique géométrique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6115.2 Colorimétrie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6115.3 Exercices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6115.4 Correction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
16 Optique ondulatoire 6316.1 Ondes électromagnétiques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.2 Longueur d’onde, célérité et fréquence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.3 Le photon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.4 Interaction onde-matière . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.5 Quantification des niveaux d’énergie des atomes . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.6 Exercices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6316.7 Correction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
iv

Chapitre 1
Détermination de la composition d’un sys-tème chimique à l’état initial
IntroductionLa chimie a pour objectif de modifier desmolécules lors de réactions.Il faut être capable de compter rapidementles molécules mises en présences dans le mi-lieu réactionnel. Ce chapitre présente deuxoutils pour faire cela : le concept de massemolaire qui permet de compter en pesant,et une technique expérimentale de mesurede concentration molaire : la spectrophoto-métrie d’absorption.
1.1 Masse molaire M et quantité de matièren
1.1.1 Masse molaire atomique
Définition La masse molaire atomique M est lamasse d’une mole d’atomes identiques de masse in-dividuelle matome en g. Elle est exprimée en g.mol−1.
M = NA×matome
Exemple Les masses molaires atomiques sont ins-crites dans chaque case du tableau périodique. AinsiM(C) = 12.0 g.mol−1, M(H) = 1.0 g.mol−1,M(O) = 16.0 g.mol−1, etc...
On peut ainsi calculer la masse d’un atome à partirde la valeur de la masse molaire et de la constanted’Avogadro : comme M = NA×matome alors matome =MNA
exprimée en g. Un atome de carbone a donc une
masse matome carbone =12
6.022×1023 = 2.00× 10−23 g.
1.1.2 Masse molaire d’une espèce chimique
Définition La masse molaire M d’une espèce chi-mique est la masse d’une mole de cette entité chi-mique.
Pour calculer cette masse molaire, il fautconnaître la formule brute de l’espèce chimiquepour faire l’inventaire des atomes présents.
Ensuite, connaissant les masses molaires ato-miques, on peut calculer la masse molaire de l’es-pèce chimique.
Exemple Le saccharose est le sucre alimentaireque l’on trouve dans nos cuisines. Il a pour formulebrute C12H22O11, donc une molécule de saccharosecontient 12 atomes de carbone, 22 atomes d’hydro-gène et 11 atomes d’oxygène, donc pour une mole desaccharose, on aura 12 moles d’atomes de carbone,22 moles atomes d’hydrogène et 11 moles atomesd’oxygène. On peut alors calculer la masse molairemoléculaire du saccharose
M(C12H22O11) =12×M(C) + 22×M(H) + 11×M(O)= 12× 12.0+ 22× 1.0+ 11× 16.0
= 342 g.mol−1
1.1.3 Quantité de matière n et masse molaire M
Définition Si on a un échantillon de masse m en gd’une espèce chimique de masse molaire moléculaireM en g.mol−1 alors la masse totale m de notre es-pèce chimique sera le produit entre la masse d’unemole de l’espèce par la quantité de matière n de cetteespèce
m= n×M
On pourra alors utiliser cette formule pour détermi-ner la quantité de matière n présente dans un échan-tillon de masse m d’une espèce chimique de massemolaire moléculaire M en isolant le paramètre ndans l’équation précédente
n=mM
Exemple On a 1 kg de saccharose, calculons laquantité de matière n présente dans cet échantillon :m = 1 kg = 1 × 103 g et M = 342 g.mol−1 doncn= 1×103 g
342 g.mol−1 = 2.92 mol.
1

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
1.1.4 Volume molaire d’un gaz
Définition Un gaz constitué d’une quantité de ma-tière n d’atomes ou molécules occupe un volume Ven L qui dépend seulement de la pression et la tem-pérature du gaz. La nature chimique du gaz n’a pasd’influence sur le volume occupé.
Le volume occupé par une mole de gaz s’appelle levolume molaire Vm.
On a la relation suivante entre le volume Vgaz oc-cupé par une quantité de matière n de gaz et le vo-lume molaire Vm
Vgaz = n× Vm
Le volume molaire Vm dépend de la température etde la pression. En général, on prendra
— Vm = 22.4 L.mol−1 à pression atmosphériquenormale et 0oC (CNTP : Conditions Normalesde Température et de Pression )
— Vm = 24 L.mol−1 à pression atmosphériquenormale et à 20oC
Exemple Calculons la quantité de matière de di-oxygène présente dans 1 m3 de ce gaz dans lesCNTP. V = 1 m3 = 1000 L, dans les CNTP Vm =22.4 L.mol−1, et comme Vgaz = n×Vm on peut isolerl’inconnue n
n=Vgaz
Vm=
100022.4
= 44.6 mol
1.1.5 Concentration en quantité de matière
Définition Si on dissout une quantité de matière n(en mol) dans un solvant pour former un volume V(en L) de solution, alors on définit la concentrationen quantité de matière C comme étant la quantité
C =nV
et elle se mesure en mol.L−1.
Définition La quantité de matière n (en mol) d’unsoluté contenue dans un volume V (en L) de solutionde concentration massique t (en g.L−1) se calcule dela façon suivante :
— la quantité de matière n (en mol) peut se cal-culer par la formule suivante, si on connaît samasse m (en g) et sa masse molaire M (eng.mol−1) :
n=mM
— on peut calculer la masse m à partir de la défi-nition de la concentration massique t = m
V enisolant m et on obtient
m= t × V
On utilise cette dernière équation et la précé-dente pour exprimer la quantité de matière n
n=t × V
M
Définition La quantité de matière n d’un solutédans un volume V de solution de concentration enquantité de matière C s’obtient en isolant n dans ladéfinition d’une concentration en quantité de ma-tière
C =nV
doncn= C × V
Définition En comparant les deux méthodes pré-cédentes pour calculer la quantité de matière n, onpeut démontrer la relation suivante :
Comme n= t×VM et n= C × V alors
t × VM
= C × V
et en simplifiant par V on a C = tM ou encore
t = C ×M
1.2 Spectroscopie d’absorbance
1.2.1 Spectre d’absorption
Définition Le spectre d’absorption d’une espècechimique décrit la manière dont la lumière incidenteest absorbée par l’espèce en fonction de la longueurd’onde de la lumière. On trace un graphe de l’inten-sité lumineuse absorbée (ou transmise) en fonctionde la longueur d’onde de la radiation lumineuse.Voir figure 1.2.
1.2.2 Couleur d’une espèce en solution
Définition La couleur d’une espèce chimiqueéclairée par une lumière blanche est la couleur com-plémentaire de la couleur absorbée. Elle est le ré-sultat du mélange des couleurs des radiations trans-mises.
1.2.3 Absorbance
Définition On éclaire une cuve de longueur l avecune lumière colorée (autour de la longueur d’ondeλmax). On mesure l’intensité I0 du faisceau traver-sant une cuve remplie de solvant, et l’intensité I dufaisceau tranversant la cuve remplie d’une solutioncolorée de concentration C en mol.L−1 (figure 1.1).
2

CHAPITRE 1. DÉTERMINATION DE LA COMPOSITION D’UN SYSTÈME CHIMIQUE À L’ÉTAT INITIAL
On appelle transmitance T le rapport T = II0
. Ce rap-port varie de 0 % à 100 % .On appelle absorbance A la quantité
A= log1T= log
I0
I
Les ordres de grandeurs pour A vont de 0 à 4 pourde bons appareils de mesure.
Figure 1.1 – Mesure de l’absorbance d’une solution
Figure 1.2 – Spectre d’absorption UV-Visible d’une so-lution de permanganate de potassium. L’absorbancemaximale dans le visible se situe vers 528 nm (réf. TheRoyal Society of Chemistry)
1.2.4 Loi de Beer-Lamber
Définition Une solution colorée de concentrationC en mol.L−1, traversée par un faisceau lumineuxde longueur d’onde λmax sur une longueur l, en cm(figure 1.1), et de coefficient d’extinction molaire ε,en mol−1.L.cm−1 vérifie la loi de Beer Lambert quirelie l’absorbance A de la solution à sa concentrationC par la formule
A= ε× l × C
Cette loi permet de mesurer la concentration d’unesolution colorée à partir de la mesure de son absor-bance optique dans le visible.
1.2.5 Dosage par spectrophotométrie
Spectrophotomètre Le spectrophotomètre est unappareil de laboratoire qui permet de mesurer l’ab-sorbance A d’une solution colorée, à une certainelongueur d’onde, en lumière visible, et parfois dansle proche ultra violet.Il contient une source de lumière colorée dont onpeut régler la longueur d’onde, un support de cuvequi sera traversé par la lumière, un capteur de lu-mière pour mesurer l’intensité transmise et un sys-tème de calcul de l’absorbance.Les spectrophotomètres peuvent afficher l’absor-bance pour les plus simples et tracer le spectre d’ab-sorbance complet pour les plus évolués. C’est un ap-pareil standard présent dans les laboratoires de chi-mie.
Figure 1.3 – Schéma du spectrophotomètre visible-ultraviolet CARY 60 de Agilent Technologies
Dosage par colorimétrie Un dosage consiste à me-surer la concentration d’une espèce chimique en so-lution. Pour doser une espèce colorée, on va réaliserune courbe d’étalonnage en traçant l’absorbance Aen fonction de la concentration C . Ensuite, on me-sure au spectrophotomètre l’absorbance de la solu-tion inconnue et on en déduit graphiquement la va-leur de la concentration (voir figure 1.4).Pour mesurer une absorbance au spectrophoto-mètre, on procède en plusieurs étapes :
— déterminer la longueur d’onde λmax où l’ab-sorbance est maximale (figure 1.2).
3

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
— choisir cette longueur d’onde pour la couleurde la source lumineuse du spectrophotomètre
— "faire le blanc", c’est-à-dire mesurer l’absor-bance du solvant pur, qui servira de référence
— mesurer l’absorbance de la solutionLors des mesures d’absorbances, il faut impérative-ment garder les faces des cuves propres et bien rin-cer une cuve ayant contenu une solution de forteconcentration.
Figure 1.4 – Absorbance en fonction de la concentra-tion pour une solution de I2
4

Chapitre 2
Suivi et modélisation de l’évolution d’un sys-tème chimique
IntroductionLes réactions d’oxydo réduction consistentau transfert d’électrons lors de la réactionchimique entre divers réactifs. Cette famillede réaction est de grande importance dansle domaine du vivant ainsi que des appli-cations technologiques.Pour pouvoir prévoir l’évolution les quanti-tés de réactifs et de produits lors d’une réac-tion chimique dont on connaît la l’équationde réaction, on utilise un outil mathéma-tique appelé tableau d’avancement.
2.1 Réaction d’oxydo-réduction
2.1.1 Couple Oxydant Réducteur
Définition Un réducteur est une espèce chimiquecapable de libérer un ou plusieurs électrons.
Définition Un oxydant est une espèce chimique ca-pable de capturer un ou plusieurs électrons.
Définition Un couple oxydant/réducteur est formépar deux espèces chimiques qui sont reliées par unedemi équation électronique (figure 2.1).
Figure 2.1 – Couple oxydoréducteur et demi réactionassociée
Exemples de couples oxydoréducteurs On in-dique le couple Ox/Red et la demi équation asso-ciée. On utilise le signe = à la place du signe −→pour écrire la demi équation (table 2.1).
2.1.2 Réaction d’oxydo-réduction
Définition Une réaction d’oxydoréduction meten présence deux couples Ox/Red et un transfertd’électrons du réducteur d’un couple vers l’oxydantde l’autre couple.Pour équilibrer une telle réaction d’oxydoréduction,il est essentiel de bien retenir qu’il s’agit d’untransfert d’électrons.Ce transfert d’électron peut avoir lieu dans lemélange réactionnel, ou via un circuit électrique(cas des piles et les accumulateurs).
Définition Pour établir l’équation bilan d’une ré-action d’oxydoréduction, les étapes à suivre sont lessuivantes
— écrire les demi équations électroniques dechaque couple Ox/Red et équilibrer ces demiéquations
— écrire ces demi équations de manière à avoirtous les réactifs à gauche
— si nécessaire, multiplier les demi équations pardes coefficients de manière à ce que tous lesélectrons d’une demi équation puissent êtretransférés à l’autre demi équation
— sommer ces deux demi équations pour obtenirl’équation bilan de la réaction
Exemple On fait réagir deux couples oxydoréduc-teurs Cu2+/Cu et Al3+/Al. Les réactifs sont Al etCu2+.
— On équilibre des demi équations Ox/RedCu2+ + 2e− = CuAl3+ + 3e− = Al
5

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Couple Ox/Red Demi équation Redox
Fe2+/Fe Fe2+ + 2e− = FeCu2+/Cu Cu2+ + 2e− = CuAg+/Ag Ag+ + e− = Ag
H3O+/H2 H3O+ + e− = 12 H2 +H2O
MnO−4 /Mn2+ MnO−4 + 8H+ + 5e− = Mn2+ + 4H2OCH3CHO/CH3CH2OH CH3CHO+ 2H+ + 2e− = CH3CH2OH
etc ... etc ...
Table 2.1 – Quelques couples Ox/red
— On place les réactifs à gaucheCu2+ + 2e− = CuAl = Al3+ + 3e−
— On multiplie de manière à échanger le mêmenombre d’électrons (ici 6 électrons)
Cu2+ + 2e− = Cu
× 3
Al = Al3+ + 3e−
× 2— On fait le bilan
3Cu2+ + 2Al −→ 3Cu+ 2Al3+
Remarque Pour équilibrer certaines demi équa-tion redox, il faut faire intervenir des ions H+, c’està dire qu’il faudra être en milieu acide. Exemple del’ion permanganate dans le couple MnO−4 /Mn2+
— On écrit simplement le début de la demi équa-tion électroniqueMnO−4 + e− = Mn2+
— Comme il y a 4 atomes d’oxygène, on ajoute àdroite 4 molécules d’eau H2OMnO−4 + e− = Mn2+ + 4H2O
— On ajoute ensuite le nombre suffisant d’ionsH+ pour conserver l’élément hydrogène (ici 8)MnO−4 + 8H+ + e− = Mn2+ + 4H2O
— Enfin, on ajoute le nombre nécessaire d’élec-trons pour avoir la conservation de la chargeélectriqueMnO−4 + 8H+ + 5e− = Mn2+ + 4H2O
2.2 Tableau d’avancement
2.2.1 Quantité de matière
Mesurer la quantité de matière n (en mol) d’uneespèce chimique, c’est compter le nombre d’entité(atome, molécule, ion) de cette espèce chimique enles regroupant en paquet contenant NA = 6.022 ×1023 objets, appelé une mole.Si on connaît la masse molaire M (en g.mol−1) del’espèce et la masse m de l’échantillon de l’espècechimique alors
n=mM
Pour un liquide pur, dont on connaît la masse volu-mique ρ (en g.mL−1) et le volume de l’échantillon
(en mL) alors la quantité de matière est
n=ρ × V
M
Pour une espèce chimique en solution de concentra-tion molaire C (en mol.L−1) dont on prélève un vo-lume V (en L), la quantité de matière sera
n= C × V
Pour un gaz dont on connaît le volume V expriméen L, à partir du volume molaire Vm en L.mol−1
pris dans les mêmes conditions de température etde pression, la quantité de matière n présente dansle gaz sera
n=VVm
2.2.2 Réaction chimique
Une réaction chimique met en présence des réac-tifs qui vont former progressivement des produits.Quand un des réactifs disparaît, alors la réactions’arrête. Ce réactif disparu en premier est le réactiflimitant.On peut faire le bilan de matière de ce systèmechimique, c’est à dire faire l’inventaire des espècesen présence et donner les quantités de matière dechaque espèce.Une réaction chimique est décrite par une équationbilan qui donne les proportions dans les quelles lesréactifs disparaissent pour former les produits, demanière à conserver la masse et la charge électriquede notre système chimique.
Exemples
CH3CH2OH + 3 O2 −→ 2 CO2 + 3 H2O
Cu2+ + 2 HO− −→ Cu(OH)2
2.2.3 Tableau d’avancement
Avancement de réaction x L’avancement x d’uneréaction est un compteur exprimé en mol qui per-met de savoir combien de fois la réaction chimique
6

CHAPITRE 2. SUIVI ET MODÉLISATION DE L’ÉVOLUTION D’UN SYSTÈME CHIMIQUE
s’est réalisée au niveau microscopique. Il vaut zéroau départ et augmente progressivement. On a donctoujours x ≥ 0 mol.
Tableau d’avancement Le tableau d’avancementdécrit l’évolution d’un système chimique, depuisl’état initial, le moment où la réaction démarre,jusqu’à l’état final, quand la réaction se termine etque le système n’évolue plus.
Le tableau donne également l’état intermédiairedu système lors de son évolution.Dans ce tableau les réactifs sont consommés, ilsdisparaissent, les produits sont fabriqués, ils appa-raissent.
Quant le réactif limitant disparaît en premier,l’avancement à cet instant est l’avancement maximalxmax . Pour rechercher sa valeur, on calcule la valeurde x permettant de faire disparaître chaque réactifet on gardera la valeur la plus petite, car elle seraatteinte en premier, x partant de zéro. Le réactifcorrespondant sera le réactif limitant.
La dernière ligne du tableau donne le bilan de ma-tière du système, c’est à dire les quantités de matièresdes réactifs et des produits à la fin de la réaction.
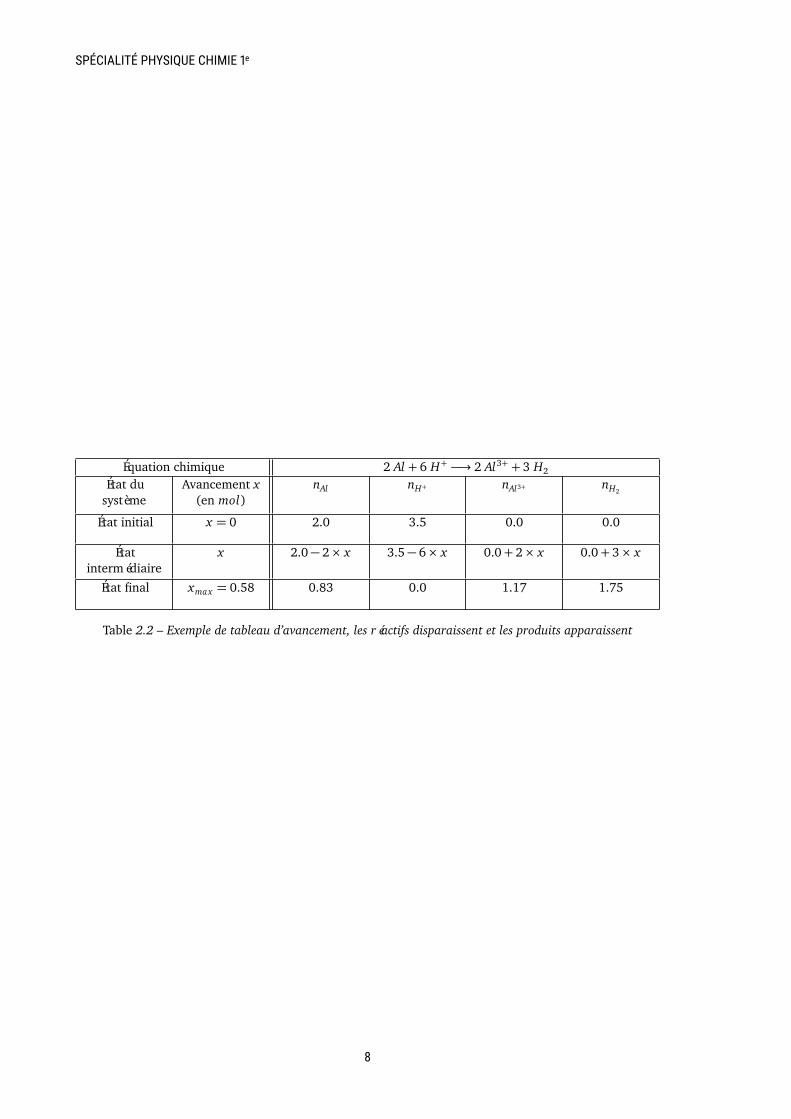
Exemples Voir le tableau 2.2. Pour trouver l’avan-cement maximum, on cherche les valeurs de x per-mettant d’annuler les quantités de matière nAl etnH+ . On a donc deux équations à résoudre
2.0− 2× x1 = 0
et3.5− 6× x2 = 0
qui ont pour solution x1 = 1.0 et x2 = 0.58. Ongarde la plus petite valeur et donc xmax = 0.58 molet H+ est le réactif limitant.
2.2.4 Avancement final et maximal
L’avancement final correspond à l’avancement ef-fectivement observé quand la réaction s’arrête etqu’elle n’évolue plus.L’avancement maximal est l’avancement qui théori-quement pourrait être atteint quand un des réactifsdisparaît.Il existe des cas où l’avancement final est plus faibleque l’avancement maximal, la réaction semble s’ar-rêter avant l’épuisement de l’un des réactifs.
2.2.5 Transformations totale et non totale
Pour une transformation totale
xfinal = xmaximal
Pour une transformation non totale
xfinal ≤ xmaximal
2.2.6 Mélanges stœchiométriques
Dans un mélange stœchiométrique, les quantitésde réactifs sont dans les proportions données par lescoefficients stœchiométriques de l’équation bilan dela réaction. Quand la réaction s’arrête, les réactifsdisparaissent en même temps.
7
SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Équation chimique 2 Al + 6 H+ −→ 2 Al3+ + 3 H2
État dusystème
Avancement x(en mol)
nAl nH+ nAl3+ nH2
État initial x = 0 2.0 3.5 0.0 0.0
Étatintermédiaire
x 2.0− 2× x 3.5− 6× x 0.0+ 2× x 0.0+ 3× x
État final xmax = 0.58 0.83 0.0 1.17 1.75
Table 2.2 – Exemple de tableau d’avancement, les réactifs disparaissent et les produits apparaissent
8

Chapitre 3
Détermination d’une quantité de matièregrâce à une transformation chimique
IntroductionL’objectif de ce chapitre est de découvrir uneméthode permettant de mesurer une quan-tité de matière présente dans un milieu enréalisant une réaction avec un autre réac-tif, dont un paramètre physique mesurablechangera lors de la disparition complèted’un des réactifs. Le paramètre observablepeut être la couleur, un potentiel électriqueou la conductivité électrique du milieu ré-actionnel.
3.1 Titrage par suivi colorimétrique
3.1.1 Principe général d’un dosage
Définition Le dosage consiste à déterminer laconcentration d’une espèce en solution. Pour cela ilexiste plusieurs méthodes
— le dosage par colorimétrie— le dosage par étalonnage— le titrage par mesure d’une grandeur physique
3.1.2 Dosage par colorimétrie
Définition Pour un dosage par colorimétrie, onpeut utiliser un erlenmeyer vu qu’aucun instrumentde mesure n’est nécessaire (voir figure 3.1. L’équiva-lence est atteinte lorsqu’on observe un changementd’aspect du milieu réactionnel (changement de cou-leur). Les dosages se font à la goutte près. Il estdonc recommandé d’effectuer un premier dosage ra-pide de manière à situer approximativement le vo-lume de solution titrante versé à l’équivalence. Ledeuxième dosage se fera alors lentement et précisé-ment (goutte à goutte) lorsqu’on sera proche de cevolume à l’équivalence.
3.1.3 Dosage redox
Équation bilan de l’équation de dosage On a unoxydant noté Ox1 qui se réduira selon la demi équa-
Figure 3.1 – Montage expérimental pour réaliser letitrage avec suivi colorimétrique
tion électronique
Ox1 + a× e− = Red1
On a un réducteur noté Red2 qui se réduira selon lademi équation électronique
Red2 = Ox2 + b× e−
L’équation bilan de la réaction s’obtient en multi-pliant les coefficients de la première équation parb et ceux de la deuxième par a puis on ajoute lesdeux équations électroniques membres à membres
b× (Ox1 + a× e− = Red1)a× (Red2 = Ox2 + b× e−)
b×Ox1 + a× Red2 −→ a×Ox2 + b×Ox2
Relation fondamentale des dosages redoxD’après l’équation de réaction b moles d’Ox1
9

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
réagissent avec a moles de Red2 quand on est àla stœchiométrie. Lors du dosage, au moment oùl’équivalence est faite, les proportions de réactifssont stœchiométriques. Si n1 est la quantité dematière de Ox1 et n2 est la quantité de matière deRed2 alors on aura à l’équivalence
n1
b=
n2
a
3.2 Dosage des ions Fe2+
3.2.1 Objectif
Une solution contient des ions Fer II Fe2+ donton veut mesurer la concentration C . On réalise undosage en utilisant une réaction d’oxydoréductionentre les ions Fer II et l’ion permanganate MnO−4 .On prélève un volume V de solution de Fer II à ti-trer, de concentration C inconnue, et on utilise unesolution titrante de permanganate de potatiun deconcentration Co dont on aura versé un volume Véq.à l’équivalence.
3.2.2 Réaction du dosage
Les couples oxydants réducteurs utilisés sont
MnO−4 /Mn2+
Fe3+/Fe2+
Les demi équations rédox de ces couples sont
MnO−4 + 8H+ + 5e− = Mn2+ + 4H2O
Fe3+ + e− = Fe2+
On place les réactifs à gauche et les produits àdroite, on multiplie les demie équations de manièreà avoir un transfert de 5 électrons puis on les ajoutemembres à membres
1× (MnO−4 + 8H+ + 5e− = Mn2+ + 4H2O)5× (Fe2+ = Fe3+ + e−)
MnO−4 + 8H+ + 5Fe2+ −→Mn2+ + 4H2O+ 5Fe3+
3.2.3 Quantités de matière à l’équivalence
On exprime les quantités de matière à partir desvolumes et des concentrations des solutions ayantréagis dans le bécher.Pour les ions fer II
n(Fe2+) = C × V
Pour les ions permanganates
n(MnO−4 ) = Co × Véquiv.
D’après l’équation de réaction 1 ions MnO−4 réagitavec 5 ions Fe2+ donc on doit garder la proportionet
n(MnO−4 )
1=
n(Fe2+)5
On peut alors écrire qu’à l’équivalence
n(Fe2+) = 5× n(MnO−4 )
et en utilisant les concentrations et les volumes
C × V = 5× Co × Véquiv.
on peut alors calculer la concentration inconnue
C =5× Co × Véquiv.
V
10

Chapitre 4
De la structure à la polarité d’une entité
IntroductionLes atomes cherchent à acquérir une struc-ture électronique proche de celle d’un gaznoble. Ils pourront alors engager des liai-sons avec d’autres atomes, en partageantdes électrons. On explique ainsi la for-mation des molécules et d’ions polyato-miques. La présence de ces électrons sur ladernière couche, qui vont formé des dou-blets, expliquera aussi la forme des mo-lécules dans l’espace. Enfin, comme cer-tains éléments attirent plus facilement lesélectrons que d’autres, une molécule, bienque neutre électriquement, pourra inter-agir électro statiquement avec ses voisines,ce qui va induire certaines propriétés phy-sico chimiques à notre échelle.
4.1 Schéma de Lewis d’une entité
4.1.1 Stabilité d’un élément
En 1916, Lewis propose que la liaison entre deuxatomes dans une molécule (liaison covalente) ré-sulte de la mise en communs d’électrons entre lesdeux atomes qui permet à chaque élément d’aug-menter sa stabilité en complétant sa couche électro-nique externe de manière à être similaire à celle dugaz noble le plus proche. Voir figure 4.1.
atome A A B atome B
liaison covalente
Figure 4.1 – Mutualisation des électrons d’une liaisoncovalente entre deux atomes
4.1.2 Représentation de Lewis
Principe Autour d’un élément, les électrons vontformer des doublets (des paires), ceux engagés dansune liaison chimique seront des doublets liants, lesautres seront des doublets non liants. Un électronnon engagé dans un doublet sera qualifié d’électroncélibataire.
Limite du modèle Ce modèle a ses limites à partirde la troisième période (ligne) du tableau des élé-ments, par exemple le phosphore P peut engager 5doublets et donc avoir 10 électrons sur sa dernièrecouche.
Atomes On dessine uniquement les électrons de ladernière couche par un point. Les électrons céliba-taires seront engagés dans des doublets liants de ma-nière à compléter la couche de façon similaire à celled’un gaz noble.Par exemple, le carbone ayant 4 électrons sur sa der-nière couche, il doit engager 4 liaisons pour parta-ger 4 électrons et arriver ainsi à la structure de ladernière couche du néon. Par contre, l’azote ayant 5électrons sur sa dernière couche, il n’a besoin que de3 liaisons pour apporter les 3 électrons manquants,il aura donc sur sa dernière couche un doublet nonliant et trois électrons célibataires. Voir figure 4.2.
N O ClH C
N O ClH C
Figure 4.2 – Modèle de Lewis de quelques atomes etliaisons covalentes possibles (en rouge)
Molécules Pour dessiner le schéma de Lewis d’unemolécule, on procède aux étapes suivantes
11

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
1. compter le nombre total d’électrons de va-lences à partir de la configuration électroniquede chaque atome de la molécule.
2. en déduire le nombre total de doublets dispo-nible en divisant ce nombre par deux.
3. on place au centre de la molécule l’atome de-vant gagner le plus d’électrons, puis on établitdes liaisons avec les autres atomes et on ajoutedes doublets non liants
4. vérifier que tous les éléments dans la moléculeont acquis une struture électronique stable,identique à celle du gaz noble le plus proche.
Exemple L’acide cyanhydrique a pour formulebrute HCN . On va dessiner sa structure de Lewis.Voir figure 4.3.
1. l’hydrogène possède 1 électron sur sa coucheexterne, le carbone 4 et l’azote 5, il y a en toutN = 1+ 4+ 5= 10 électrons de valence.
2. les 10 électrons de valence vont former 102 = 5
doublets d’électrons.
3. le carbone sera au centre car il doit former 4liaisons. L’azote doit former 3 liaisons et l’hy-drogène une seule. Il y aura donc 4 doubletsliants. Le 5e doublet sera non liant et situé surl’azote pour le stabiliser.
4. l’hydrogène a sa couche ressemblant à cellede l’hélium, l’azote et le carbone ressemblentmaintenant au néon avec 8 électrons sur ladernière couche.
N
C
H H
C
N
H
C
N
NCH
NCH
Figure 4.3 – Formation des liaisons covalente dansl’acide cyanhydrique
Ions Pour un ion polyatomique on procède commepour une molécule, mais on corrige le nombre to-tal d’électrons de valence disponibles en fonction dela charge électrique de l’ion. Un anion a des élec-trons en plus, un cation a des électrons en moins.On ajoute ensuite la charge globale de l’ion.
Exemple L’ion oxonium a pour formule bruteH3O+. On va dessiner son schéma de Lewis. Voir fi-gure 4.4.
1. les trois atomes d’hydrogènes ont 1 électronde valence, l’oxygène 6, il y a donc théorique-ment 9 électrons, cependant, comme l’ion aune charge positive, il faut retirer 1 électron.Donc on aura finalement 8 électrons dispo-nibles.
2. à partir des 8 électrons disponibles, nous pour-rons former 4 doublets.
3. l’oxygène est au centre de l’ion moléculaire,les trois hydrogènes l’entourent, il y a troisdoublets liants. Le quatrième doublet sera nonliant, et placé sur l’oxygène.
4. les trois hydrogènes ont la structure électro-nique de l’hélium, et l’oxygène a la structureélectronique du néon.
5. on ajoute enfin le signe + pour préciser lacharge globale de l’ion moléculaire.
O
H H
O
charge positive
H HH H
H HH
+
OH
+
H
H
Figure 4.4 – Schéma de Lewis de l’ion oxonium H3O+
4.1.3 Lacune électronique
Un atome à qui il manque un doublet d’électronspour obtenir la même configuration électroniquestable que celle du gaz noble le plus proche possèdeune lacune électronique , qu’on dessine par un pe-tit rectangle vide à la place d’un doublet. Voir figure4.5.
H+
ion H+ Molécule AlCl3
Al
Cl
ClCl
Figure 4.5 – Schéma de Lewis d’espèces avec lacunesélectroniques
12

CHAPITRE 4. DE LA STRUCTURE À LA POLARITÉ D’UNE ENTITÉ
4.2 Géométrie d’une entité
4.2.1 Principe
Les doublets électroniques autour d’un atome serepoussent entre eux à cause des forces électrosta-tiques répulsives. Cela va donner une forme géomé-trique en trois dimensions à la molécule.
4.2.2 Géométrie de molécules ou d’ions molécu-laires
Tétraèdre C’est le cas d’un atome central entouréde quatre doublets liants à des atomes identiques,comme CH4 ou NH+4 . Voir figure 4.6.
H C
H
H
H
H N
H
H
H
+
H
H
H
H
Figure 4.6 – Structure tétraédrique du méthane et del’ion ammonium.
Pyramide à base triangulaire L’atome central estrelié à trois atomes identique et possède un doubletnon liant, comme NH3 ou H3O+. Voir figure 4.7.
H N
H
H
H O
H
H
+
H
H
H
doubletnon liant
Figure 4.7 – Structure pyramidale de l’ammoniac etde l’ion hydronium.
Coudée L’atome central possède deux doubletsnon liants et engage deux liaisons avec deux atomesidentiques, comme H2O. Voir figure 4.8.
OHH
HH
doubletnon liant
doubletnon liant
Figure 4.8 – Structure coudée de l’eau.
Triangulaire plane L’atome central engage deuxliaisons simples et une double liaison, toutes ces liai-sons sont dans le même plan, comme pour les car-bones de CH2CH2. Voir figure 4.9.
CH
HC
H
H
H
H
C
Figure 4.9 – Structure triangulaire plane de l’éthylène.
Linéaire L’atome centrale engage deux doublesliaisons avec deux autres atomes, la molécule est li-néaire, comme pour CO2. Voir figure 4.10.
O C O
C
Figure 4.10 – Structure linéaire du dioxyde de car-bone.
13

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
4.3 Électronégativité des atomes
L’électronégativité χ d’un atome traduit sa capa-cité à attirer vers lui les électrons d’un doublet liantquand il est engagé dans une liaison chimique. Plusson électronégativité est grande, plus il attire verslui les électrons.Voir figure 4.11.
élément électronégativité χH 2.2He non définiLi 1.1Be 1.6B 2.0C 2.5N 3.0O 3.5F 4.0
Ne non définiNa 0.9Mg 1.3Al 1.6Si 1.9P 2.2S 2.6Cl 3.2Ar non défini
Table 4.1 – Valeur de l’électronégativité de Pauling destrois premières périodes.
1
0
3
2
4
Figure 4.11 – Échelle d’électronégativité de Paulingdes trois premières périodes.
4.4 Polarisation d’une liaison chimique
Quand deux atomes différents partagent unemême liaison chimique, cette liaison sera polarisési la différence d’électronégativité entre les deuxatomes est supérieure ou égale à 0.4.
L’atome le plus électronégatif portera une chargeélectrique partielle −δ et l’autre atome une chargeélectrique partielle +δ. Voir figure 4.12.
C H
O H
C Opolarisées
non polarisée
Figure 4.12 – Une liaison engageant deux atomes dontla différence d’électronégativité est supérieure ou égaleà 0.4 est polarisée.
4.5 Polarité d’une molécule
Une molécule est dite «polaire» si elle contientune ou plusieurs liaisons polarisées qui ne se com-pensent pas. On a alors le centre géométrique descharges positives différent du centre géométriquedes charges négatives. La molécule se comportecomme un dipôle électrique, globalement neutre,mais ayant une extrémité positive, l’autre négative.Cela va influencer des propriétés macroscopiquescomme les températures de changement de phase,la solubilité et la réactivité chimique de ces espèces,car les interactions entre molécules sont plus impor-tantes. Voir figure 4.13.
O HC H
H
H
CH
H
H
C
H
H
CH
H
H
C OO OH
H
NH
HH
a) b)
c)
d)
e)
Figure 4.13 – a) n’est pas polaire, aucune liaison n’estpolarisée. c) n’est pas polaire, ses deux liaisons polari-sées se compensent car la molécule est linéaires. Dansles trois autres cas, b), d) et e), des liaisons sont po-larisées et elles ne se compensent pas du fait de la géo-métrie dans l’espace de la molécule.
14

Chapitre 5
De la structure des entités à la cohésion et àla solubilté/miscibilité d’espèces chimiques
IntroductionLes interactions au niveau microscopiqueentre les entités constituants une espècesous forme solide vont permettre d’expli-quer les propriétés physico-chimiques del’espèce à notre échelle, comme la solubi-lité dans différents solvants, ou les tempé-ratures de changement de phase.
5.1 Cohésion des solides
Selon l’espèce chimique, la cohésion d’un solideest assurée par des liaisons covalentes (diamant),un partage d’électrons par tous les atomes du solide(métaux). Il existe également des solides où il n’ya pas d’ordre au niveau microscopique, on les ap-pelle des solides amorphes, c’est le cas du verre parexemple.Nous allons étudier dans ce chapitre le cas où la co-hésion se fait par
— interaction entre ions— interaction entre molécules polarisées— interaction par pont hydrogène
Selon la nature de l’interaction au niveau microsco-pique, l’énergie de cohésion peut changer de façonimportante. Voir table 5.1
5.2 Cohésion des solides ioniques
5.2.1 Solide ionique
Un solide ionique est un arrangement très régulierd’anions et de cations, il est électriquement neutre.Sa formule statistique indique la nature et la propor-tion d’anions et de cations présents dans le cristal.Voir figure 5.1.
Cl-Cl- Na+
Cl-Na+ Na+
Cl-Cl- Na+
Cl-Na+
Cl- Na+
Cl-Na+
Cl-Na+
Cl- Na+
Cl-Na+
Cl-Na+ Na+
Cl-Cl- Na+
Cl- Na+
Cl-Na+
Cl- Na+
Cl-Na+
formulestatistique
NaCl
anion
cation
Figure 5.1 – Le sel de cuisine (chlorure de sodium) estun cristal ionique constitué par un empilement régu-lier de cations Na+ et d’anions Cl−.
5.2.2 Cohésion
La cohésion d’un cristal ionique est assurée pardes forces d’attractions électrostatiques entre les ca-tions et les anions. Il y a également une compétitionavec les forces de répulsions entre les ions de mêmecharge électrique. Enfin, les électrons des couchesinternes des ions forment comme une sphère dureinfranchissable, et la structure du cristal ionique dé-pendra aussi de la taille des différents ions.Ces forces de cohésions sont intenses, et l’éner-gie nécessaire pour les vaincre est importante. Parexemple, pour faire fondre le chlorure de sodium, ilfaut le chauffer à plus de 800 oC .
5.3 Cohésion des solides moléculaires
5.3.1 Solide moléculaire
Un solide moléculaire est une espèce chimiquepure dont les entités qui le forme sont des molécules,régulièrement empilées pour former une structurecristalline.On peut citer par exemple la glace, cristal formé parles molécules d’eau, les cristaux de sucres que l’onutilise en cuisine sont formés par des molécules desaccharose régulièrement empilée.
15

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Force Type Énergie (kJ .mol−1) Exempleionique cation-anion 400 à 4000 NaCldipôle-dipôle Keesom 5 à 25 HCl · · ·HCldipôle-dipôle induit Debye 2 à 10 HCl · · ·Cl2dipôle instantané-dipôle induit
London 0.05 à 40 F2 · · · F2
dipôle - dipôle liaison hydrogène 2 à 40 H2O · · ·H −O−H
Table 5.1 – Ordre de grandeur des énergies des forces intermoléculaires
5.3.2 Cohésion par interaction de van der Waals
Dans un cristal moléculaire, les forces qui per-mettent aux molécules d’interagir entre elles sontbasées sur le fait qu’une molécule peut être pola-risée. Cette polarisation peut être permanente, in-duite ou instantanée. Voir le tableau 5.2.Entre les molécules polarisées, il existe donc desinteractions électrostatiques attractives, faibles et àcourte portée, qui vont permettre au cristal de se for-mer. Ces interactions sont appelées interactions devan der Waals. Voir figure 5.2.
Figure 5.2 – Dans un cristal moléculaire, des molé-cules polarisées (spontanément, par induction ou ins-tantanément) interagissent entre elles par des forcesde van der Waals.
5.3.3 Cohésion par pont hydrogène
Une liaison hydrogène se forme entre un atome btrès électronégatif et porteur d’un doublet non liantavec un atome d’hydrogène formant une liaison co-valente avec un atome a très électronégatif (figure5.3).
Un exemple de molécule formant des ponts hy-drogènes est l’eau : ces ponts qu’il faut rompre pourfaire changer de phase l’eau, sont responsables desvaleurs inhabituellement hautes des températuresde fusion et de vaporisation de l’eau, comparées àcelles de molécules de structures similaires. Voir fi-gure 5.4.
Les atomes a et b peut être F, O, N ou Cl.
a H b
liaison covalente
doublet non liant
liaison hydrogène
Figure 5.3 – Formation d’une liaison hydrogène entredeux molécules contenant l’atome a et l’atome b
5.4 Dissolution des solides ioniques
5.4.1 L’eau
L’eau est un solvant polaire, la molécule H2O estplutôt négative coté oxygène et positive coté hydro-gène, car les liaisons oxygène-hydrogène sont for-tement polarisées. La molécule d’eau va donc êtrecapable d’attirer des anions ou des cations selon sonorientation dans l’espace. Voir figure 5.5.
5.4.2 La dissolution
Quand un solide ionique est plongé dans l’eau, onobserve sa dissolution, le solide se désagrège et lesions se dispersent en solution dans l’eau. Ce proces-sus se réalise en trois étapes. Voir figure 5.6.
1. dissociation L’eau étant polaire, elle attire àelle les ions qui sont extraient de leur réseaucristallin au niveau de la surface du cristal.
2. solvatation Les ions libérés du cristal sont ra-pidement entourés par des molécules d’eauqui masquent ainsi la charge électrique del’ion qui ne pourra pas être attiré par les autresions du cristal.
3. dispersion dans l’eau liquide, les moléculesont un mouvement d’agitation permanent, dûà la température. Progressivement, les ionsvont se disperser dans le liquide
16

CHAPITRE 5. DE LA STRUCTURE DES ENTITÉS À LA COHÉSION ET À LA SOLUBILTÉ/MISCIBILITÉ D’ESPÈCESCHIMIQUES
Dipôle permanent Il est dû à une différence d’électronégativité des deux atomes de la liaison cova-lente, un des deux atomes attire plus fortement les électrons.
Dipôle induit La liaison entre les deux atomes se polarise sous l’effet d’une charge électriqueexterne (un autre dipôle par exemple), les électrons de la liaison sont repoussésou attirés et il apparaît un dipôle induit : il disparaîtra dès que la source extérieures’éloignera.
Dipôle instantané Pendant un court intervalle de temps, le moment dipolaire d’un atome ou d’unemolécule n’est pas nul : il a une certaine valeur dans une certaine direction, mêmesi en moyenne, sur une durée plus longue ce moment dipolaire est nul. Pendantde brefs instants, on a donc une interaction entre atomes ou molécules avec desdipôles inducteurs et induits.
Table 5.2 – Forces d’interaction entre les molécules
-100
0
100
-80
-60
-40
-20
20
40
60
80
H2Te H2Se H2S H2O
tem
péra
ture
(°C
)
Figure 5.4 – Températures de fusion et de vaporisationpour les éléments de la colonne de l’oxygène, formantsdes molécules du type H2X . On observe que l’eau estune anomalie, à cause de la présence de fortes interac-tions entre molécules, dues aux ponts hydrogène.
5.4.3 Équation de réaction de dissolution
Une dissolution peut être décrite par une équa-tion de dissolution qui doit conserver la masse et lacharge électrique. On part d’un solide (le sel) pourobtenir des ions en solution. La solution sera neutreélectriquement.
Exemples
NaCl(s) −→ Na+(aq) + Cl−(aq)
CoCl2(s) −→ Co2+(aq) + 2Cl−(aq)
FeCl3(s) −→ Fe3+(aq) + 3Cl−(aq)
Anion(négatif)
Cation(positif)
Figure 5.5 – La molécule d’eau très polaire peut attirerdes cations ou des anions selon son orientation relativeà l’ion.
dissociation solvatation dispersion
+ - + - +- + - + - ++ - + - +
--
+
- + - +- + - + - ++ - + - + -
-+ - + - +
+ - + - ++ - + - + -
-+
Figure 5.6 – La dissolution d’un solide ionique dansl’eau se fait en trois étapes : la dissociation, la solva-tation et la dispersion.
5.4.4 Concentration en ion
On place un certain nombre de moles nS de so-luté dans un volume V de solvant. On a donc uneconcentration C en soluté apporté qui sera
C =nS
V
Cependant, selon le type de sel, une mole de sel peutdonner plusieurs moles d’ions en solution. On préci-sera alors la concentration en ion
[cat ion] =ncat ion
V
17

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
et
[anion] =nanion
V
Ces concentrations seront supérieures ou égales à C .
Exemple Dissolution du chlorure de Fer III dansl’eau. On peut écrire le tableau d’avancement sui-vant 5.3. On peut ensuite calculer la concentrationmolaire en soluté apporté
C =n (FeCl3)
Vsol=
n(S)Vsol
et les concentrations molaires effectives des ions
Fe3+
=n(Fe3+)
Vsol=
n(S)Vsol
Cl−
=n(Cl−)
Vsol=
3× n(S)Vsol
5.5 Solubilité d’une espèce dans un solvant
5.5.1 Définition
La solubilité s d’une espèce chimique est la quan-tité de matière maximale nmax qu’il est possible dedissoudre dans un volume V de solvant à une tem-pérature donnée
s =nmax
V
nmax est en mol, V en L, et s en mol.L−1.
5.5.2 Type de solvant
La solubilité d’une espèce chimique dépend dusolvant dans lequel on essaie de la dissoudre.
espèces ioniques Les espèces ioniques sont so-lubles dans les solvants polaires, car les ions serontentourés par les molécules polaires du solvant, graceà des interactions électrostatiques.
espèces moléculaires Pour les espèces molécu-laires, on distingue le cas où l’espèce est polaire etcelui où l’espèce est apolaire.
— un soluté polaire sera miscible dans un sol-vant polaire grâce à des interaction de van derWaals, et parfois il est aussi possible de réali-ser des ponts hydrogènes.
— un soluté apolaire sera miscible dans un sol-vant apolaire grâce à des interactions de vander Waals.
5.6 Extraction solvant solvant
5.6.1 Principe
L’extraction solvant solvant permet de faire passerune espèce chimique d’un solvant où elle est moinssoluble à un autre solvant où elle est plus soluble.Les deux solvants ne doivent pas être miscibles pourpouvoir facilement les séparer par décantation.On utilise cette technique en fin de synthèse parexemple, pour extraire l’espèce synthétisée du mé-lange réactionnel. Elle passe dans un solvant quisera ensuite évaporé pour isoler l’espèce synthétiséesous forme liquide ou cristalline.
5.6.2 Protocole
Pour une extraction liquide liquide, on utilise uneampoule à décanter. Voir figure 5.7. Les étapes duprotocole sont
1. vérifier que le robinet de l’ampoule est ferméet placer un bécher sous l’ampoule
2. introduire le premier solvant avec l’espèce àisoler
3. introduire le solvant d’extraction, fermer l’am-poule
4. agiter vigoureusement l’ampoule pour bras-ser les solvants, dégazer régulièrement en ou-vrant le robinet, pour libérer la pression quipeut s’accumuler
5. reposer l’ampoule sur son support, ôter le bou-chon
6. laisser décanter les deux phases
7. récupérer la phase avec le solvant d’extraction
5.7 Hydrophilie, lipophilie et amphiphilie
5.7.1 Définition
Une molécule amphiphile est une molécule quipossède une partie soluble dans l’eau (hydrophile)et une partie soluble dans les graisses (lipophile).Voir figure 5.8.
5.7.2 Exemples
Les molécules amphiphiles ont un rôle importantdans notre quotidien (les savons), pour la vie (mem-branes de cellules), des applications industrielles etscientifiques (tensioactifs pour stabiliser les émul-sions) et l’industrie agroalimentaire (stabilisationdes émulsions et des mousses).
18

CHAPITRE 5. DE LA STRUCTURE DES ENTITÉS À LA COHÉSION ET À LA SOLUBILTÉ/MISCIBILITÉ D’ESPÈCESCHIMIQUES
Équation chimique FeCl3(s) −→ Fe3+(aq) + 3Cl−(aq)
État du système Avancement x(en mol)
nFeCl3 nFe3+ nCl−
État initial x = 0 n(S) 0.0 0.0
Étatintermédiaire
x n(S)− x 0.0+ x 0.0+ 3× x
État final xmax = n(S) n(S)− xmax = 0 0.0+ n(S) 0.0+ 3× n(S)
Table 5.3 – Exemple de tableau d’avancement d’une réaction de dissolution
5.7.3 Le savon
Un savon contient des molécules d’oléate de so-dium qui sont amphiphiles. Voir figure 5.8.
Elles permettent de former une fine couche entreles corps gras et l’eau et de former des micelles, quiseront entraînées par l’eau lors du lavage.La partie lipophile du savon se fixe sur la partiegrasse et la partie hydrophile permet l’entraînementpar l’eau.Une micelle est une «bulle de gras» enveloppée parune fine couche de savon, sa taille est inférieure aumicromètre.
solvant d'extractionsolvant initial
espèce à extraire
ampoule à décanter
bouchon
bécher
dégazage !
Figure 5.7 – Pour l’extraction liquide liquide, on utiliseune ampoule à décanter. L’espèce à extraire est dans unpremier solvant. On ajoute le solvant d’extraction, nonmiscible avec le premier solvant, on agite vigoureuse-ment l’ampoule en prenant soin de dégazer régulière-ment, puis on laisse décanter les deux solvants, l’espèceà isoler a migré dans le solvant d’extraction.
19

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
O
O-Na+
lipophilehydrophile
Formule topologique
schéma simplifié
Figure 5.8 – Molécule d’oléate de sodium dans le sa-von, avec la partie hydrophile et la partie lipophile.
eau
corps gras hydrophobe
fine couche de moléculesamphophiles
Figure 5.9 – Les molécules du savon forment une finemembrane autour des corps gras permettant qu’ilssoient entraînés par l’eau.
20

Chapitre 6
Structure des entités organiques
IntroductionIl existe plus de 12 millions de composésorganiques connus, ayants leurs proprespropriétés physiques et chimique. Cepen-dant, les chimistes ont observés qu’il étaitpossible de classer ces composés dans unnombre réduit de familles en fonction deleurs caractéristiques structurales, et qu’ausein d’une même famille, la réactivité chi-mique des composées est très similaire etprévisible.
6.1 Molécules organiques
Définition Les entités organiques sont principale-ment composées d’atomes de carbone, d’hydrogène,d’oxygène et d’azote. Souvent, elles ont pour ori-gines le monde du vivant, soit en étant directementextraites du vivant végétal ou animal, soit en prove-nant de roches fossiles comme le pétrole et le char-bon. Elles peuvent être également synthétiques, c’està dire fabriquées par la chimie de synthèse, ou mêmeartificielles car étant une création de l’Être Humain.
6.2 Formule brutes et semi-développées
Définition Une formule brute d’une espèce chi-mique fait l’inventaire des atomes présents dans l’es-pèce, en indiquant en indice le nombre de fois quel’atome apparaît dans l’espèce. Toute informationsur la manière donc les atomes sont reliés entre euxest perdue.
Définition Une formule semi-développée d’une es-pèce chimique indique en deux dimension la ma-nière dont sont reliés entre eux les atomes, et notam-ment la présence de double ou triple liaisons, à l’ex-ception des atomes d’hydrogènes qu’on représentesimplement accolé à l’atome avec lequel ils engagentune liaison, et en indice, on précise le nombre d’hy-drogènes ayant une liaison avec l’atome.
Exemple La formule brute C2H5O peut corres-pondre à deux formules semi-développées
CH3 O CH3CH3 CH2 OH
6.3 Squelette carboné saturé
6.3.1 Carbone saturé
On dit qu’un atome de carbone est saturé quandil engage quatre liaisons covalentes avec quatreatomes différents, il n’y a pas de double liaison oude triple liaison.
6.3.2 Squelette carboné
Dans une molécule organique, le squelette car-boné est la structure que forment les atomes de car-bones reliés entre eux par des liaisons covalents.Sur cette chaîne carbonée s’ajoutent ensuite d’autresatomes, notamment l’hydrogène, de manière à quetous les carbones engagent quatre liaisons cova-lentes.
6.3.3 Squelette carboné saturé
Dans une telle molécule, les atomes de carbonesne partagent que des liaisons simples avec leurs voi-sins, il n’y a pas de liaisons double ou triple entredeux carbones.
6.3.4 Les alcanes
Structure
Les alcanes sont des hydrocarbures linéaires ouramifiés, saturés, c’est à dire des molécules necontenant que des atomes C et H, les atomes Cengagent systématiquement quatre liaisons diffé-rentes, et forment des chaînes et des ramifications,mais sans boucles (cycles). La formule brute géné-rale d’un alcane est
CnH2n+2
21

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
avec n= 1,2, 3,4.... Pour une formule brute donnée,il existe plusieurs isomères
— un alcane linéaire— parfois plusieurs alcanes ramifiés
Exemple L’alcane C5H12 peut correspondre auxformules semi-développées suivantes
CH3 CH2 CH2 CH2 CH3
CH3 C
CH3
CH3
CH3
Nomenclature
chaîne carbonée linéaireLe nom d’un alcane linéaire est constitué d’un pré-fixe qui indique le nombre des atomes de carbonesuivi de la terminaison "ane" (table 6.1).
n formule nom1 CH4 méthane2 C2H6 éthane3 C3H8 propane4 C4H10 butane5 C5H12 pentane6 C6H14 hexane7 C7H16 heptane8 C8H18 octane9 C9H20 nonane
Table 6.1 – Nom des premiers alcanes linéaires
groupe nom?− CH3 méthyle
?− CH2 − CH3 éthyle
Table 6.2 – Groupes alkyles
chaîne carbonée ramifiée Pour nommer un al-cane ramifié, on procède selon les étapes suivantes
— chercher la chaîne carbonée la plus longue.— compter le nombre de carbones de cette
chaîne qui donnera le nom de l’alcane (table6.1).
— numéroter les carbones de la chaîne de ma-nière à ce que le numéro de la première rami-fication soit le plus petit possible.
— le nom de l’alcane ramifié se construit en pla-çant les groupes alkyles dans l’ordre alphabé-tique, précédés de leur indice de position, suividu nom de l’alcane linéaire de la chaîne prin-cipale (table 6.2).
— le "e" final des groupes alkyles est supprimé,ils sont précédés du préfixe di, tri, tétra si legroupe est présent plusieurs fois.
Exemple Voir figure 6.1
CH3 CH2 CH2 CH CH3
CH3
14 3 25
CH3 CH CH2 CH2 CH3
CH3
CH
CH2 CH3
1 2 3 4 5 6
2-méthyl pentane
3-éthyl-2-méthyl hexane
Figure 6.1 – Exemple d’usage de la nomenclature desalcanes.
6.4 Groupes caractéristiques et famillesfonctionnelles
6.4.1 Définition
Un groupe caractéristique, ou groupe fonctionnel,est une partie d’une molécule qui est composée d’unatome ou d’un groupe d’atomes ayant un compor-tement chimique caractéristique. Chimiquement, ungroupe fonctionnel donné se comporte généralementde la même façon, quelle que soit la molécule danslaquelle il se trouve.
6.4.2 Famille alcool
Définition Le groupe caractéristique des composésde type alcool est constitué d’un atome d’oxygène liéd’une part à un hydrogène et d’autre part à un car-bone ayant quatre liaisons différentes. On appelle cegroupe un groupe hydroxyle.
C O H
Nomenclature Le nom d’un alcool dérive de celuide l’alcane de même chaîne carbonée en remplaçantle "e" final par la terminaison "ol", précédée si né-cessaire de l’indice de position du groupe hydroxyledans la chaîne carbonée principale. Voir figure 6.2.
22

CHAPITRE 6. STRUCTURE DES ENTITÉS ORGANIQUES
CH3 CH2 CH CH2 CH3
CH3
CH
OH
6 5 34 2 1
5-méthylhexan-3-ol
4
CH3 CH CH2 CH3
OH
31
butan-2-ol
2
Figure 6.2 – Exemple d’usage de la nomenclature desalcools
Exemple
CH3 CH2 CH2 OH
Cette molécule est le propan-1-ol
CH3 CH1
OH
CH3
Cette molécule est le propan-2-ol
6.4.3 Famille aldéhyde
Définition Le groupe caractéristique des composésde type aldéhyde est constitué d’un atome de car-bone lié d’une part à un hydrogène et à un oxy-gène, et d’autre part à un carbone ayant quatre liai-sons différentes. L’ensemble d’atomes C O s’ap-pelle groupe carbonyle.
C CO
H
Nomenclature Le nom d’un aldéhyde est consti-tué de celui de l’alcane de la chaîne carbonée la pluslongue portant le groupe carbonyle, son préfixe estsuivi du suffixe -al.
Exemple
CH3 CH2 CH2 C
H
O
Molécule de butanal.
6.4.4 Famille cétone
Définition Le groupe caractéristique des composésde type cétone est constitué d’un atome de carbonelié d’une part à un oxygène, et d’autre part à deuxcarbones ayant quatre liaisons différentes.
C C
O
C
Nomenclature Son nom est constitué de celui del’alcane de la chaîne carbonée la plus longue portantle groupe carbonyle. Son préfixe est suivi du numérodu carbone qui porte l’atome d’oxygène et de -one.
Exemple
CH3 CH2 CH2 CH2
O
CH3
Molécule de pentan-2-one.
6.4.5 Famille acide carboxylique
Définition Le groupe caractéristique des composésde type acide carboxylique est constitué d’un atomede carbone lié d’une part à un oxygène et à ungroupe oxygène hydrogène, et d’autre part à un car-bone ayant quatre liaisons différentes. Ce grouped’atomes s’appelle groupe carboxyle.
C CO
O H
Nomenclature Son nom est constitué de celui del’alcane de la chaîne carbonée la plus longue portantle groupe carboxyle. Son préfixe est précédé du monacide et suivi du suffixe -oïque.
Exemple
CH3 CH2 COH
OCette molécule est l’acide propanoïque.
6.4.6 Remarque
Il existe de nombreuses autres familles fonc-tionnelles, qui impliquent d’autres atomes, commel’azote, le soufre et les halogènes.
23
SPÉCIALITÉ PHYSIQUE CHIMIE 1e
6.5 Spectroscopie infrarouge
6.5.1 Principe
Une molécule est constituée d’atomes reliés entreeux par des liaisons covalentes, résultat de la mu-tualisation d’électrons. Une molécule n’est pas unobjet parfaitement rigide, on peut le modéliser enphysique par un ensemble de billes (les atomes) re-liées par des ressorts (les liaisons).Les atomes ont des mouvements d’oscillations lelong de la liaison (élongation) ou de part et d’autrede cette liaison (flexion). La fréquence de ces vibra-tion dépend de la masse des atomes de la liaison etde la «raideur» du ressort, et donc du type de liaison.Ce mouvement va absorber une certaine énergie dela lumière incidente, qui correspond à celle trans-portée par des photons dans l’infra rouge de 2.5 µmà 25 µm. En mesurant le spectre d’absorption infrarouge d’une substance, on va avoir des informationssur le type de liaisons présentes, et donc la présenceou l’absence de certains groupes fonctionnels.
6.5.2 Spectre d’absorption infra rouge
Un spectre d’absorption infrarouge représente lepourcentage d’intensité lumineuse transmise à tra-vers l’échantillon en fonction du nombre d’onde σen cm−1
σ =1λ
avec λ la longueur d’onde en cm. σ représente unefréquence à laquelle vibre la liaison. On gradue legraphique de droite à gauche.
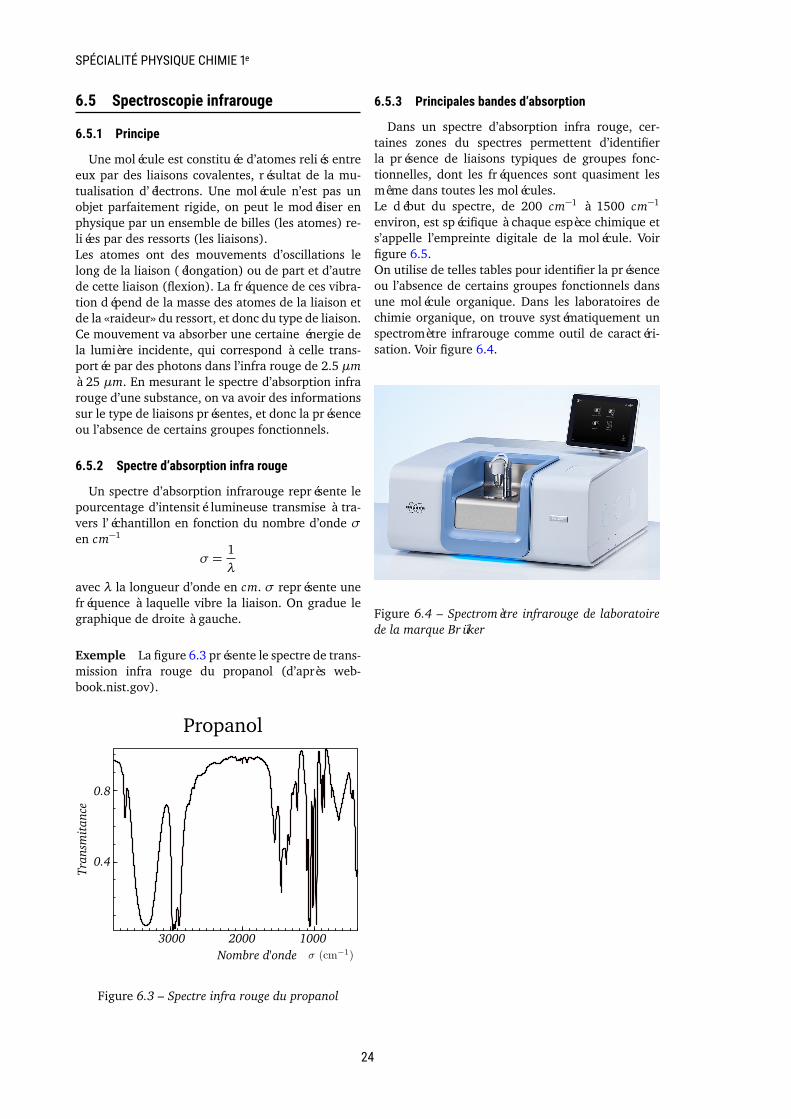
Exemple La figure 6.3 présente le spectre de trans-mission infra rouge du propanol (d’après web-book.nist.gov).
100020003000
0.4
0.8
Tran
smit
ance
Nombre d'onde
Propanol
Figure 6.3 – Spectre infra rouge du propanol
6.5.3 Principales bandes d’absorption
Dans un spectre d’absorption infra rouge, cer-taines zones du spectres permettent d’identifierla présence de liaisons typiques de groupes fonc-tionnelles, dont les fréquences sont quasiment lesmême dans toutes les molécules.Le début du spectre, de 200 cm−1 à 1500 cm−1
environ, est spécifique à chaque espèce chimique ets’appelle l’empreinte digitale de la molécule. Voirfigure 6.5.On utilise de telles tables pour identifier la présenceou l’absence de certains groupes fonctionnels dansune molécule organique. Dans les laboratoires dechimie organique, on trouve systématiquement unspectromètre infrarouge comme outil de caractéri-sation. Voir figure 6.4.
Figure 6.4 – Spectromètre infrarouge de laboratoirede la marque Brüker
24

CHAPITRE 6. STRUCTURE DES ENTITÉS ORGANIQUES
1000
Nombre d'onde
200030004000
C-H
O-H
C=O
C=O
C=OO-H
C-H
C-H
Zone de l'empreinte digitalespécifique à chaque molécule
Alcane
Alcool
Aldéhyde
Cétone
Acide carboxylique
Figure 6.5 – Carte simplifiée des bandes d’absorptions infra rouge de quelques groupes fonctionnels
25

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
26

Chapitre 7
Synthèse d’espèces chimiques organiques
IntroductionLa synthèse d’une espèce chimique en chi-mie organique est une suite d’étape où onsynthétise, isole, purifie et caractérise unproduit d’une réaction.
7.1 Étapes d’un protocole
7.1.1 Principe
Les étapes successives de la synthèse d’une espècechimique en chimie organique sont
— la transformation des réactifs (ou synthèse)— isolement de l’espèce chimique— purification du produit synthétisé— analyse du produit (identification et pureté)
Pour chaque étape, il existe différentes techniquesexpérimentales à connaître.
7.1.2 Transformation des réactif
Chauffage à reflux Le montage de chauffage àreflux permet d’accélérer la réaction chimique enchauffant les réactifs, et à condenser leur vapeurpour les garder dans le mélange réactionnel. Voir fi-gures 7.1 et 7.2.
Chauffage avec réfrigérant à air Pour les espècespeu volatiles, il est facile de condenser les vapeursdes espèces à l’aide d’un long tube en verre refroidipar l’air ambiant. C’est un montage plus simple etmoins cher à mettre en œuvre.
7.1.3 Isolement d’une espèce
Filtration simple Si l’espèce synthétisée est unsolide cristallisé, il suffit de réaliser une filtrationsimple. Voir figure 7.4.
Filtration Büchner Si l’espèce synthétisée est unsolide cristallisé très fin qui donne un aspect pâteux,ou que le filtrat est trop visqueux pour utiliser une
réfrigérant à boule
ballon monocol
chauffe ballon
support élévateur
eau
mélange réactionnelpierre ponce
eau
ballon bicol
ampoule de coulée
Figure 7.1 – Montage d’un chauffage à reflux. Le ré-frigérant à boule permet de condenser les vapeurs etne pas perdre les réactifs et produits par évapora-tion. Les grains de pierre ponce permettent d’avoir uneébullition stable et progressive. Le support élévateurpermet d’arrêter rapidement le chauffage et le mon-tage/démontage du ballon. On peut utiliser une am-poule de coulée si la réaction dégage beaucoup d’éner-gie et si il faut introduire lentement un réactif.
27

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
réfrigérant à air(tube en verre)
plaque chauffante
mélange réactionnel
erlenmeyer
Figure 7.2 – Chauffage à reflux avec réfrigérant à air,dans le cas d’espèces peu volatiles.
bécher
mélangeréactionnel
filtrat
entonnoir
papier filtre
cristaux
Figure 7.3 – Montage de filtration simple, il fonc-tionne bien si les cristaux sont gros et si le filtratest fluide, pour passer rapidement à travers le papierfiltre.
fiole à vide
aspiration
papier filtrefiltre Büchner
mélangeréactionnel
Figure 7.4 – Filtre Büchner pour filtrer des mélangessolides liquides avec un liquide visqueux ou un solidepâteux.
filtration simple, on utilise un filtre Büchner, où l’as-piration d’air permet de forcer le passage du filtratà travers le disque de papier filtre. Voir figure ??.
Séparation liquide-liquide Si l’espèce synthétiséeest liquide, pour la séparer du mélange réaction-nel, on utilise un autre solvant, non miscible avecle mélange réactionnel, mais où l’espèce synthétiséeest beaucoup plus soluble. Pour faire cela, on utiliseune ampoule à décanter pour mettre en contact lesolvant avec le mélange réactionnel. On procède engénéral à deux ou trois extractions successives pourisoler le produit, puis on évapore le solvant d’extrac-tion. Voir figure 7.5
7.1.4 Purification d’une espèce
Recristallisation Pour purifier un produit qui estsous forme solide cristallisé, il faut le dissoudre dansun solvant, puis évaporer lentement ce solvant pourrecristalliser le solide. Le processus de recristallisa-tion permet d’évacuer les impuretés incluses acci-dentellement dans la première cristallisation lors dela synthèse.
Distillation Pour purifier un liquide, on peut ledistiller, car les autres liquides correspondants auximpuretés n’ont pas les même température de vapo-risation. Voir figure 7.7.
7.1.5 Analyse d’une espèce
Chromatographie sur couche mince La chroma-tographie sur couche mince permet de vérifier la pu-reté du produit synthétisé. Elle permet également devérifier qu’il correspond à l’espèce que l’on voulait
28

CHAPITRE 7. SYNTHÈSE D’ESPÈCES CHIMIQUES ORGANIQUES
mélangeréactionnel
solvant du milieuréactionnel
solvantd'extraction
espèce Aespèce B
Figure 7.5 – Extraction liquide-liquide à l’aide d’uneampoule à décanter et d’un solvant d’extraction.
colonne de distillation
réfrigérant droit
chauffe ballon
support élévateur
thermomètre
mélange de liquides
liquide le plus volatile
eau
eau
Figure 7.6 – La distillation fractionnée permet de pu-rifier une espèce synthétisée qui est liquide.
ligne de dépôt
plaque recouvertede silice
espèces puresespèce à analyser
éluant
front du solvant
séchage révélation
h
H
Figure 7.7 – Montage de chromatographie sur couchemince pour vérifier la pureté de l’espèce synthétisée.
synthétiser. Il faut cependant déjà disposer d’une es-pèce pure de référence. Voir figure 7.7.
Banc Köfler Le banc Köfler permet de mesurer latempérature de fusion d’une espèce solide, et de vé-rifier qu’elle est conforme à celle de l’espèce pureque l’on vise à synthétiser. Voir figure 7.8.
Autres méthodes Il existe d’autres méthodes decaractérisation des espèces synthétisées
— mesure de température d’ébullition— mesure d’indice optique— spectroscopie de flamme— spectroscopie ultraviolette et visible— spectroscopie infra rouge— spectroscopie par résonance magnétique— spectrométrie de masse— chromatographie sous phase gazeuse— diffraction par rayon X
7.2 Rendement de synthèses
Lors d’une synthèse, la quantité de matière ou lamasse de l’espèce synthétisée nobtenue peut être in-férieure à celle attendue si la réaction était totale
29

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
70oC260oC
poudres d'étalonnage des températures
zone de test
index de lecturede la température
Figure 7.8 – Banc Köfler pour mesurer la températurede fusion d’une espèce solide sous forme de poudre.
nattendue.On définit alors le rendement de la réaction R
R=nobtenue
nattendue× 100
La valeur du rendement dépend de plusieurs fac-teurs
— il existe des réaction qui ne sont pas totales(estérification par exemple)
— on peut perdre de la matière aux différentesétapes de la réaction (évaporation, à traversles filtres, dans les solvants, ...)
— réactions parasites lors de la synthèse quiconsomment les réactifs ou les produits.
30

Chapitre 8
Conversion de l’énergie stockée dans la ma-tière organique
IntroductionLa domestication du feu a permis aux êtreshumains de se procurer un moyen de chauf-fage, d’éclairage, de cuisson de leurs ali-ments et de défense contre les prédateurs.Ce fût aussi un moyen pour fabriquer denouveaux matériaux et objets grâce à lamétallurgie et à la poterie.Enfin, lors de la révolution industrielle auXIXe siècle, la combustion du charbon, puisplus tard du pétrole, a permis de produiredu mouvement, utilisé pour faire fonction-ner des machines, pour transporter despassagers et des objets et pour fabriquer del’électricité.Cela est encore de toute première impor-tance de nos jours et de nombreux conflitsactuels s’expliquent par la lutte pour accé-der aux sources d’énergies fossiles combus-tibles, telles le pétrole et le gaz.
8.1 Les combustibles
8.1.1 Combustibles fossiles
Les combustibles fossiles ont pour origine des ma-tériaux organiques (plantes ou planctons) qui furentrapidement enfouis dans des sédiments au fond delacs ou d’océans, il y a environ 300 millions d’an-nées, puis qui furent dégradés lentement par la pres-sion et de la chaleur souterraine pour augmenter laquantité relative de carbone et d’hydrogène.Les combustibles fossiles contiennent essentielle-ment du carbone, de l’hydrogène, de l’oxygène, del’azote, du souffre.On peut citer
— le charbon dont le nom dépend de sa concen-tration en carbone (houille, lignite , anthra-cite, ...)
— le pétrole (dont l’aspect dépend aussi de sacomposition, brun très fluide à noir très vis-
queux)— le gaz naturel (méthane, propane, butane)
8.1.2 Combustibles issus de la biomasse
Ces combustibles sont directement pris dans lemonde du vivant.
— le bois est le plus ancien combustible utilisé— le charbon de bois est du bois passé dans un
four spécial pour l’enrichir en carbone— du méthane issu de la fermentation de maté-
riaux organiques— le méthanol issu de matériaux organiques— des corps gras, animaux ou végétaux (pour
l’éclairage)
8.2 Problématiques liées aux combustibles
8.2.1 Sécurité des personnes
Une combustion présente des dangers pour les in-dividus
— incendies et brûlures— intoxication par du monoxyde de carbone qui
bloque le transport de l’oxygène par l’hémo-globine
— intoxication par des résidus de combustion po-tentiellement cancérigènes (poussières fines,goudrons, composés organiques, ...)
8.2.2 Impact environnemental
L’environnement est fortement impacté par la pré-sence des combustibles fossiles
— impact climatique dû à l’enrichissement en di-oxyde de carbone de l’atmosphère (réchauffe-ment climatique)
— exploitation polluante (gaz de schistes, oléo-ducs qu fuient, marées noires)
— pluies acides (à cause du souffre)— poussières
31

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
8.2.3 Tensions géopolitiques
Le contrôle de l’approvisionnement en pétrole estabsolument stratégique et vital pour toutes les so-ciétés modernes, et cela se traduit par une lutte trèsféroce pour contrôler l’exploitation du pétrole et letransport de l’énergie fossile.La plus part des conflits actuels s’expliquent par laprésence de sources d’énergies fossiles à contrôler
— les différentes guerres dans les pays du golfpersique (Irak, Iran, Syrie, Arabie Saoudite)pour le contrôle des plus grandes réservesmondiales de pétrole et de gaz
— Amérique du Sud, avec le Venezuela, grandproducteur de pétrole
— Ukraine, pays traversé par des gazoducs four-nissant presque la moitié du gaz pour l’Europede l’Est
8.3 Combustion en chimie
8.3.1 Oxydoréduction
Définition Une combustion est une réaction d’oxy-doréduction impliquant un combustible (le réduc-teur) et le dioxygène (l’oxydant).
8.3.2 Alcanes
Définition La combustion dans le dioxygène d’unalcane de formule brute CnH2n+2 (avec n un en-tier) produit du dioxyde de carbone et de l’eau selonl’équation bilan
CnH2n+2 +3n+ 1
2O2 −→ nCO2 + (n+ 1)H2O
Exemple La combustion du méthane CH4 estune réaction d’oxydoréduction entre deux couplesOx/Red
CO2/CH4
O2/H2O
Les deux demi équations électroniques sont
CO2 + 8H+ + 8e− = CH4 + 2H2O
O2 + 4H+ + 4e− = 2H2O
L’équation bilan de la réaction d’oxydoréductionsera alors
CH4 + 2H2O = CO2 + 8H+ + 8e−
2× (O2 + 4H+ + 4e− = 2H2O)CH4 + 2H2O+ 2O2 + 8H+ −→
CO2 + 8H+ + 4H2Oet en simplifiant
CH4 + 2O2 −→ CO2 + 2H2O
8.3.3 Alcools
Définition La combustion dans le dioxygène d’unalcool de formule brute CnH2n+1OH avec n entier,produit du dioxyde de carbone et de l’eau selonl’équation bilan
CnH2n+1OH +3n2
O2 −→ nCO2 + (n+ 1)H2O
Exemple La combustion de l’éthanol C2H5OH estune réaction d’oxydoréduction entre deux couplesOx/Red
CO2/C2H6O
O2/H2O
Les deux demi équations électroniques sont
2CO2 + 12H+ + 12e− = C2H6O+ 3H2O
O2 + 4H+ + 4e− = 2H2O
L’équation bilan de la réaction d’oxydoréductionsera alors
C2H6O+ 3H2O = 2CO2 + 12H+ + 12e−
3× (O2 + 4H+ + 4e− = 2H2O)C2H6O+ 3H2O+ 3O2 + 12H+ −→
2CO2 + 12H+ + 6H2Oet en simplifiant
C2H6O+ 3O2 −→ 2CO2 + 3H2O
8.4 Réaction exothermique
8.4.1 Réaction de combustion
Une combustion est une réaction chimique exo-thermique, elle libère de l’énergie qui était stockéedans les molécules des réactifs.
8.4.2 Énergie molaire de liaisons
Définition L’énergie molaire de liaison Ei d’une mo-lécule diatomique A B est l’énergie qu’il fautfournir pour rompre les liaisons d’une mole de molé-cule AB(gaz) à l’état gazeux en ses deux atomes A(gaz)et B(gaz) à l’état gazeux. Cette énergie est toujourspositive, et s’exprime en joules par mole (J .mol−1).
Définition L’énergie molaire de liaison d’une molé-cule polyatomique est la somme des énergies de liai-son de toutes les liaisons présentes dans la molécule.On dessine le schéma de Lewis de la molécule, onfait l’inventaire de toutes les liaisons présentes et onsomme les énergies molaires de liaisons.
32

CHAPITRE 8. CONVERSION DE L’ÉNERGIE STOCKÉE DANS LA MATIÈRE ORGANIQUE
Exemple La formule de Lewis de l’éthanol est
C
H
H
H
C
H
H
O H
On fait l’inventaire des types de liaisons présentes— Il y a 5 liaisons C H d’énergie de liaison
415 kJ .mol−1
— Il y a 1 liaison O H d’énergie de liaison463 kJ .mol−1
— Il y a 1 liaison C O d’énergie de liaison350 kJ .mol−1
— Il y a 1 liaison C C d’énergie de liaison348 kJ .mol−1
L’énergie moléculaire de liaison de l’éthanol est donc
El(C2H6O) = 5× 415+ 1× 463+ 1× 350+ 1× 348(8.1)
= 3236 kJ .mol−1 (8.2)
(8.3)
8.4.3 Énergie molaire de réaction
Définition L’énergie molaire de réaction Er estl’énergie libérée lors de la réaction de combustionde 1 mole de carburant. Si on calcule la somme desénergies moléculaire de liaisons des réactifs (figure8.1) et la somme des énergies moléculaire de liaisonsdes produits (figure 8.2), on observe une différencenégative (figure 8.3) qui est perdue par le systèmechimique car libérée vers l’extérieur.
Er = Eliaison(réactif 1) + Eliaison(réactif 2) (8.4)
− (Eliaison(produit 1) + Eliaison(produit 2)) (8.5)
Exemple On étudie la combustion du méthane
CH4 + 2O2 −→ CO2 + 2H2O
On calcule les énergies de liaisons des différentesmolécules.
— Le méthane
C H
H
H
Ha pour énergie de liaison
El(méthane) = 4× 415= 1660 kJ .mol−1
— Le dioxygèneO O
a pour énergie de liaison
El(dioxygène) = 498 kJ .mol−1
énergie dusystème
CH4 O2 O2
réactifs
C H H H H O O O O
atomes libres
énergie à fournir pour briser les liaisons chimique
Figure 8.1 – Pour briser toutes les liaisons chimiquesdes réactifs, il faut fournir une certaines énergie.
énergie dusystème
CO2 H2O H2O
produits
C H H H H O O O O
atomes libres
énergie à fournir pour briser les liaisons chimique
Figure 8.2 – Pour briser toutes les liaisons chimiquesdes produits, il faut fournir une certaines énergie.
33

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Le système perd de l'énergie
réactifs
atomes libres
produits
atomes libres
Figure 8.3 – Si on compare les énergies de liaisonsdes réactifs et des produits, on observe une différencenégative.
— Le dioxyde de carboneO C O
a pour énergie de liaison
El(dioxyde carbone) = 2×804= 1608 kJ .mol−1
— L’eauH O H
a pour énergie de liaison
El(eau) = 2× 463= 926 kJ .mol−1
— Finalement, l’énergie de réaction sera donc
Er = El(méthane) + 2× El(dioxygène) (8.6)
− (2× El(eau) + El(dioxyde carbone))(8.7)
= 1660+ 2× 498− 2× 926− 1608 (8.8)
= −804 kJ .mol−1 (8.9)
La combustion complète de 1 mole d’éthanol valibérer une énergie de 804 kJ .mol−1.
8.4.4 Pouvoir calorifique
Définition Le pouvoir calorifique d’un combustiblePC est l’énergie E (en J) qu’une masse m (en kg)de combustible peut libérer lors d’une combustion.Il s’exprime en joules par kilogramme (J .kg−1).
PC =Em
Exemple Pour le butane PC = 47.6 MJ .kg−1. Lacombustion d’une masse de 850 g de butane va li-
bérer une énergie
E = 850 g × 47.6 MJ .kg−1 (8.10)
= 0.850 kg × 47.6× 106 J .kg−1 (8.11)
= 4.05× 107 J (8.12)
= 40.5 MJ (8.13)
34

Chapitre 9
Interactions fondamentales et notion dechamp
IntroductionDeux interactions fondamentales agissentà notre échelle entre les objets si ils pos-sèdent une masse et une charge électrique.Les objets massifs vont interagir entre euxdu fait de leur masse grâce à l’interactiongravitationnelle.Les objets possédant une charge électriquevont interagir entre eux via une interactionélectrostatique.La compréhension de ces deux interactionsest essentielle pour décrire les mouvementsdes planètes, les chutes des corps, l’électri-cité, les propriétés des molécules et atomes.Cette compréhension de ces interactionsa également permis le développement detrès nombreuses applications modernes ensciences, en médecine, pour les télécommu-nication, en ingénierie, etc. ...
9.1 Interaction électrostatique
9.1.1 Charges électriques
Définition Un objet peut posséder une charge élec-trique q dont le signe est positif ou négatif. La charges’exprime en Coulomb, dont le symbole est C . La pluspetite charge qu’il est possible d’avoir est la chargeélémentaire e qui a pour valeur
e = 1.6× 10−19 C
Production de charges Un des moyens les plusancien pour produire des charges électriques est defrotter un tissus, une fourrure ou de la laine contreun autre matériau (voir figure 9.1).
— pour produire des charges positives, on utiliseun barreau en verre auquel les électrons sontarrachés
— pour produire des charges négatives, on utiliseun barreau en matière organique (ambre, ébo-
nite, plastique) qui se couvre d’électrons arra-chés au tissu
laine
verre
plastique
électronsarrachés
électronsarrachés
Figure 9.1 – Production de charges électriques posi-tives et négatives par frottement de laine contre unetige en verre ou en matière plastique
9.1.2 Interactions électrostatiques
Définition Les charges électriques de signes iden-tiques se repoussent. Les charges électriques designes opposés s’attirent (voir figure 9.2).
répulsion attraction
Figure 9.2 – Des charges de même signe se repoussent,des charges de signe contraire s’attirent.
Loi de Coulomb La loi de Coulomb décrit quantita-tivement l’interaction entre deux charges électriquesqA et qB (voir figure 9.3).Si le système étudié est la charge qA placée en un pointA, et si en un point B on place une charge électriqueqB alors la charge qA va subir une force ~FB/A telle que
— sa direction est la droite AB
35

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
— son point d’application est la charge qA— son sens est vers B si elle est attractive, oppo-
sée à B si elle est répulsive— sa norme est donnée par la formule
FB/A =k× qA× qB
AB2
avec k = 9× 109 u.s.i..— qA et qB sont en Coulomb (C), AB en mètre
(m), FA/B en newton (N).On peut aussi donner cette loi sous une forme vec-torielle
~FB/A =k× qA× qB
AB2~uBA
avec ~uBA un vecteur unitaire orienté de B vers A.On observe également que cette interaction se fait àdistance, les charges ne sont pas en contact.
A
B
qA
qB
uBA
FB/A
qB et qA sont de signes opposés
sur ce schéma
Le système étudié est qA
A
B
qA
qB
uBA
FB/A
qB et qA sont de même signesur ce schéma
Figure 9.3 – La loi de Coulomb permet de calculer levecteur force qui s’exerce sur une charge qA à cause dela présence d’une autre charge qB.
9.1.3 Champ électrostatique ~E
Définition Si on place une charge électrique qdans une zone de l’espace où règne en chaque pointun champ électrostatique ~E alors la charge élec-trique q va subir une force ~F telle que
~F = q~E
F s’exprime en newton (N), le champ E s’exprimeen volt par mètre (V.m−1) et la charge q en coulomb(C). Voir figure 9.4.
q > 0
E
F
q < 0E
F
Figure 9.4 – Une charge q placée dans une région oùrègne un champ électrostatique ~E subit une force ~F.
Définition Un champ électrostatique ~E est créépar une ou plusieurs charges électriques placéesdans l’espace.Pour une charge unique q, on peut trouver facile-ment l’expression du champ créé. Voir figure 9.5. Sion place à proximité une autre charge Q, cette der-nière subit une force électrostatique
~F =k×Q× q
d2~uq vers Q
On peut écrire cette relation sous la forme
~F =Q~E
si on pose
~E =k× q
d2~uq vers Q
Q < 0
q > 0
EF
E'
E''
Figure 9.5 – Une charge ponctuelle q crée un champélectrique radial, qui pointe vers l’extérieur si la chargeest positive ou vers le centre si la charge est négative.Le champ est plus intense à proximité de la charge élec-trique.
Définition Le long d’une ligne de champ, le vec-teur champ électrostatique mesuré en chaque pointde la ligne est tangent à la ligne de champ. Voir fi-gure 9.6.
36

CHAPITRE 9. INTERACTIONS FONDAMENTALES ET NOTION DE CHAMP
E
ligne de champ
Figure 9.6 – Une ligne de champ est une courbe tan-gente en chaque point au champ vectoriel ~E.
Définition On peut produire un champ électrosta-tique uniforme entre deux plaques d’un condensa-teur plan. Les vecteurs champs auront même sens,même direction et même intensité dans le volumecompris entre les deux armatures. Les lignes dechamp seront des droites parallèles en elles, per-pendiculaires aux armatures du condensateur. Voirfigure 9.7.
E
armature
V+
V-
potentiel le plus haut
potentiel le plus bas
armature
zone de champ électrostatique uniforme
E
EE
E
Figure 9.7 – Un condensateur plan produit entre sesdeux armatures métalliques un champ électrique uni-forme orienté de l’électrode à haut potentiel vers l’élec-trode à bas potentiel.
Applications On utilise un tel système pour modi-fier la vitesse de particules chargées ou les dévier.Cela est utilisé notamment dans un appareil appeléspectromètre de masse (à quadrupôle ou à mesurede temps de vol) pour faite des analyses chimiquesd’échantillons de très petites tailles.
9.2 Interaction gravitationnelle
9.2.1 La masse
Définition La masse d’un objet se mesure en kilo-gramme (kg). Elle peut se mettre en évidence par
— l’inertie que va posséder l’objet, c’est à dire ladifficulté à lui faire changer son mouvement
— l’attraction gravitationnelle qui pourra s’exer-cer entre l’objet et un autre objet massif
9.2.2 Loi d’interaction de gravitation
Définition On considère que le système étudié estun objet placé au point A possédant une masse mA.Il va subit de la part d’un second objet placé au pointB et ayant une masse mB une force d’attraction ~FB/Aselon la loi de la gravitation universelle
~FB/A = −G ×mA×mB
(AB)2~uBA
avec mA et mB en kg, la distance AB en m, G =6.67× 10−11 N .m2.kg−2 la constante de gravitationuniverselles, et ~uBA un vecteur unitaire orienté de Bvers A. Voir figure 9.8.
A
B
mA
mB
uBA
FB/A
Le système étudié est A
Figure 9.8 – La loi de la gravitation universelle de cal-culer le vecteur force qui s’exerce sur une masse mA àcause de la présence d’une autre masse mB.
9.2.3 Champ de gravitation
Définition Un champ gravitationnel ~G est créé parun ou plusieurs objets massifs dans leur voisinage.Si un objet de masse m est placé dans ce champ, ilva subit une force gravitationnelle ~F telle que
~F = m ~G
Définition Le champ de gravitation ~G créé par unemasse ponctuelle M est radial, orienté vers la masseponctuelle, et dépend de la distance d entre la masseet le point où s’exerce le champ
~G = −G ×M
d2~u
avec G constante de gravitation universelle, Mmasse en kg , d en m et le vecteur ~u est orienté versl’extérieur. Voir figure 9.9.
37

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
M
G
G '
G ''
u
u
u
Figure 9.9 – Une masse ponctuelle M crée un champgravitationnel radial. Le champ est plus intense àproximité de la masse M.
9.3 Analogies entre interactions de gravita-tion et électrostatique
On observe différentes analogies entre les lois deCoulomb et de gravitation universelle
— ce sont des actions à distance— l’intensité des forces décroît inversement pro-
portionnellement à la distance au carré— la direction des forces d’interaction passe par
le centre de chaque objet massif ou chargé.— les deux interaction peuvent être attractives,
mais seule la force électrostatique peut être ré-pulsive.
38

Chapitre 10
Description d’un fluide au repos
IntroductionL’étude des fluides, c’est à dire les liquideset les gaz, fut importante pour dévelop-per de nouvelles techniques de transportset d’irrigation (canaux), ainsi que de nou-velles machines pour remplacer la tractionanimale (moulins, turbines à eau, vérinshydrauliques, machines à vapeur, moteursthermiques). Cette nouvelle discipline, lamécanique des fluides et la thermodyna-mique, se développa à partir du XVIIesièclepuis durant le XIXesiècle.
10.1 Les fluides
Définition Par opposition à un solide, un fluide n’apas de forme propre, on peut le déformer très facile-ment, contrairement à un solide. On distingue deuxtypes de fluides
— les gaz occupent tout le volume qui est dispo-nible, on peut aussi les comprimer, faire chan-ger leur volume
— les liquides ont un volume constant, ils sontincompressible, il n’est pas possible de fairechanger leur volume
Exemples Les gaz et les liquides sont des fluides.On remarquera que la notion de fluide dépend del’échelle de temps considérée : si le déplacementest très rapide, un impact sur l’eau aura les mêmesconséquences funestes qu’un impact sur un sol dur.À l’opposé, une roche soumise à une forte pression etforte température dans le sous sol terrestre aura uncomportement d’un fluide sur une échelle de tempsgéologique.
10.2 Description qualitative d’un fluide
10.2.1 Fluide à l’échelle microscopique
On décrit le fluide comme étant un ensembledésordonné de molécules ou atomes. Voir figure9.1. Pour un liquide ces entités chimiques seront en
contacts, très proches les unes des autres. Dans ungaz, elles seront éloignées les unes des autres, avecde grands espaces vides entre elles. Dans un fluide,les entités qui le compose ont un mouvement d’agi-tation permanent ce qui explique qu’un fluide puissese déformer facilement et qu’un gaz occupe tout levolume mis à sa disposition.
solide liquide gaz
fluides
Figure 10.1 – Dans les fluides (gaz et liquide), les enti-tés chimiques (atomes ou molécules) sont dans un étatdésordonné, ce qui explique les propriétés physiquesdes fluides.
10.2.2 Masse du fluide m
La masse du fluide mesurée à notre échelle serala somme des masses des molécules ou atomes com-posants ce fluide. Elle sera mesurée en kilogramme.
10.2.3 Volume du fluide V
Le volume occupé par un fluide sera mesuré enmètre au cube.
10.2.4 Masse volumique ρ
La masse volumiqueρ d’un fluide est le coefficientde proportionnalité entre son volume V et sa massem
m= ρ × V
39

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Les unités sont— la masse m est en kg— le volume V est en m3
— la masse volumique ρ est en kg.m−3
La masse volumique dépend beaucoup de la pres-sion et de la température pour un gaz.
Exemples La masse volumique de l’eau est de1000 kg.m−3, la masse volumique de l’air est de1.20 kg.m−3 à pression atmosphérique et à 20oC .
10.2.5 Pression p - XVIIIeet XIXesiècles
Un fluide exerce une pression sur les parois durécipient où il est enfermé, car au niveau microsco-pique, les entités qui le compose ont un mouvementd’agitation permanent et viennent frapper les paroisqui subissent en retour une force. Voir figure 10.2.
entité chimique(atome, molécule)
force due à l'impact surla paroi
Figure 10.2 – La pression dans un fluide est due auxchocs des entités chimiques contre les parois du réser-voir.
10.2.6 Température T ou θ
La température d’un gaz ou d’un liquide que nousmesurons représente l’énergie cinétique moyenneque possèdent les entités composant ce fluide et quiont un mouvement d’agitation permanent. Si la tem-pérature augmente, cela signifie qu’au niveau mi-croscopie, les entités ont acquis plus d’énergie ciné-tique.
Exemples À température ambiante, les moléculesde gaz de l’atmosphère N2 et O2 ont une vitesse d’en-viron 500 m.s−1. Si la température augmente, cettevitesse augmente aussi.
10.3 Force de pression
Définition Un fluide placé dans une enceinte (voirfigure 10.3) et ayant une pression p exprimée en
Pascal (Pa) exerce sur une surface S de cette en-ceinte, exprimée en m2, une force de pression P ennewton (N) dont la direction est perpendiculaire àla surface, vers l’extérieur selon la loi
F = p× S
fluide à lapression ppiston de rayon r
et de surface S = π.r²
force pressante F = p.S
Figure 10.3 – Le vérin utilise un fluide à la pression ppour exercer sur un piston de surface S une force F =p×S. Ce dispositif est notamment utilisé sur des enginsde chantier, la pression de l’huile hydraulique étant del’ordre de quelques centaines de bar, soit 10 M Pa .
Exemples Un vérin agricole utilisé dans une fen-deuse à bûche a un piston de 80 mm de diamètreet il est alimenté avec une huile hydraulique à unepression de 180 bar. On va calculer la force dispo-nible à l’extrémité du piston mobile.On calcule en mètre carré la surface du pistonS = π× ( D
2 )2 = π× ( 80×10−3 m
2 )2 = 5.0× 10−3 m2
On calcule la pression du fluide en hecto pascal sa-chant que 1 bar = 105 Pa.p = 180 bar = 1.80× 107 PaOn calcule la force pressante F = p × S = 9,0 ×104 N , le vérin est capable de soulever un poids de9.0× 104 N qui correspond à une masse de 9 t.
10.4 Loi de Boyle et Mariotte - XVIIesiècle
Définition Un gaz parfait à une température don-née constante obéit à une relation simple entre sapression p et son volume occupé V , la relation deBoyle - Mariotte (voir figure 10.4)
p× V = Constante
Exemple Soit un gaz occupant un volume V1 avecune pression p1. On modifie le volume de manièreà avoir un nouveau volume V2 trois fois plus faible.On va calculer la nouvelle pression du gaz.Le nouveau volume sera V2 =
V13 . D’après la loi de
Boyle Mariotte, le produit p1 × V1 est constant etdonc
p1 × V1 = p2 × V2
40

CHAPITRE 10. DESCRIPTION D’UN FLUIDE AU REPOS
p1 , V1
p2 , V2
Figure 10.4 – Un gaz parfait enfermé dans une en-ceinte hermétique et qui reste à température constantevérifie la relation suivante liant sa pression p et le vo-lume occupé V p1 × V1 = p2 × V2 = constant
On remplace la valeur de V2
p1 × V1 = p2 ×V1
3
puis on isole p2 pour obtenir
p2 = p1 × 3
La pression a été multipliée par 3.
10.5 Loi fondamentale de la statique desfluides - XVIIesiècle
Définition Dans un fluide incompressible et au re-pos, la différence de pression entre deux points Aet B du fluide est donné par la relation (voir figure10.5)
pB − pA = ρ × g × (zA− zB)
avec— la pression pA au point A en pascal (Pa)— la pression pB au point B en pascal (Pa)— l’altitude zA du point A en mètre (m)— l’altitude zB du point B en mètre (m)— la masse volumique ρ du fluide (kg.m−3)— l’intensité de la pesanteur (N .kg−1)
Exemple L’eau a pour masse volumique ρ =1000 kg.m−3. À une profondeur de 10 m, la pres-sion sera
pB − pA = 1000× 9.81× (10 m− 0 m) = 980 hPa
soit presque une fois de plus que la pression atmo-sphérique.
zAA
B zB
altitude
pression pA
pression pB
g
masse volumique ρ
Figure 10.5 – La loi fondamentale de la statique desfluides donne une relation entre eux pressions pA et pBdans le fluide de masse volumique ρ dans le champ depesanteur ~g à des altitudes zA et zB.
41

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
42

Chapitre 11
Mouvement d’un système
IntroductionUn système en mécanique est un objetayant une certaine masse et qui subit lesactions d’objets du reste de l’Univers. Cesactions vont modifier le mouvement du sys-tème uniquement si elles ne se compensentpas. Le système change son mouvement sisa vitesse change, ou si sa trajectoire n’estplus rectiligne. Ce changement se fait d’au-tant plus facilement que l’objet a une massefaible.
11.1 Vecteur vitesse
11.1.1 Définition
Rappel La vitesse v d’un système («speed» en an-glais) est le rapport entre la distance parcourue d etla durée du parcours ∆t, c’est à dire
v =d∆t
Dans cette définition, on ne sait pas dans quel sensni dans quelle direction l’objet se déplace.
Définition Le vecteur vitesse−→Vi («velocity» en an-
glais) est le vecteur construit à partir du vecteur dé-placement d’un objet
−−−−−→Mi Mi+1 et la durée du dépla-
cement ∆t entre les deux points Mi et Mi+1 de latrajectoire
−→Vi =
1∆t−−−−−→Mi Mi+1
Le vecteur−→Vi sera tangent à la trajectoire au point
Mi . Il donne toutes les informations suivantes
— la vitesse du déplacement v = ||−→V ||
— le sens et la direction du déplacement, il esttangent à la trajectoire au point Mi
11.1.2 Mesure expérimentale
Méthode graphique Sur une trajectoire obtenuepar une méthode chrono photographique, on peut
tracer le vecteur vitesse entre deux points consécu-tifs de la manière suivantes (voir figure 11.1)
1. mesurer la distance réelle d entre les deuxpoints consécutifs, en ligne droite. On utiliseraéventuellement une échelle.
2. déterminer la durée ∆t du trajet entre cesdeux points
3. calculer la valeur de la vitesse v = ||−→Vi ||=
d∆t
4. choisir une échelle des vitesses pour tracer levecteur vitesse
−→Vi
5. tracer à l’échelle le vecteur−→Vi au point Mi de
façon qu’il soit tangent à la trajectoire. Il estdans le sens du déplacement.
trajectoire
M1
M2
t2
t1
en m
distance d durée Δt = t2 - t1
a) mesure de d et Δt
trajectoire
M1
M2
t2
t1
en m en m.s-1
V1b) tracé de V1
le vecteur V1 est tangent à la trajectoire !
Figure 11.1 – Détermination graphique du vecteur vi-tesse ~V1 au point M1. On mesure dans un premiertemps la distance parcourue et la durée du parcours(a) puis on trace à l’échelle le vecteur vitesse (b).
Méthode par coordonnées À partir d’une trajec-toire obtenue par une méthode chrono photogra-phique, on définit un repère orthonormé et on me-sure la position des points sur la trajectoire en don-nant leur coordonnées dans ce repère ainsi que la
43

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
date à laquelle l’objet est passé à ce point (voir figure11.2). Pour calculer le vecteur vitesse
−→Vi entre deux
points Mi et Mi+1 on utilise la méthode suivantes
1. Récupérer les coordonnées du point Mi et ladate de passage à ce point t i
Mi(x i , yi); t i
2. Récupérer les coordonnées du point Mi+1 et ladate de passage à ce point t i+1
Mi+1(x i+1, yi+1); t i+1
3. Calculer les coordonnées du vecteur déplace-ment
−−−−−→Mi Mi+1
−−−−−→Mi Mi+1 = (x i+1 − x i)~i + (yi+1 − yi)~j
4. Calculer la durée du déplacement entre cesdeux points
∆t = t i+1 − t i
5. Calculer le vecteur vitesse
−→Vi =
x i+1 − x i
∆t~i +
yi+1 − yi
∆t~j
Remarque très importante La méthode décrite cidessus avec les coordonnées de deux positions suc-cessives est imprécise si la trajectoire est très courbe,car le vecteur vitesse ainsi calculé ne sera pas tan-gent. Cette méthode est satisfaisante à condition quela courbure soit très faible
— c’est le cas des mouvements rectilignes— c’est le cas de points très proches sur une
trajectoire courbe, on approxime l’arc de lacourbe à un segment.
Des méthodes plus précises existent mais il faut uti-liser plus de points sur la trajectoire. Certaines deces méthodes seront vues en terminale.
11.2 Vecteur variation de vitesse
11.2.1 Définition
On mesure sur une trajectoire deux vecteurs vi-tesses consécutifs
−−→Vi−1 et
−→Vi . Si on appelle vecteur
variation de vitesse−−→∆Vi , alors on a la relation sui-
vante−→Vi =
−−→Vi−1 +
−−→∆Vi
On peut alors mettre cette égalité sous une formequi sera utilisée pour mesurer la variation du vecteurvitesse
−−→∆Vi =
−→Vi −
−−→Vi−1
trajectoire
M1
M2
t2
t1
x1 x2 (x en m)
(y en m)
y1
y2
t1 x1 y1
t2 y2x2
date t (s) x (m) y (m) Vx (m.s-1) Vy (m.s-1)
V1x =x1x2
t2 t1V1y =
y1y2t2 t1
etc. ... etc. ... etc. ...
etc. ... etc. ...
Figure 11.2 – Détermination par calcul des coor-donnes du vecteur vitesse ~V1 au point M1. On mesureles coordonnées des points de la trajectoire dans unrepère à différentes dates, puis on calcule à partir deces valeurs les coordonnes du vecteur vitesse à chaquedate.
11.2.2 Mesure expérimentale
Méthode graphique
1. On trace en deux point consécutifs les vecteursvitesse
−−→Vi−1 et
−→Vi (voir paragraphes précédents
et figure 11.3)
2. on fait la construction géométrique de lasomme vectorielle
−→Vi =
−−→Vi−1 +
−−−→∆Vi−1
3. le vecteur−−→∆Vi est tracé sur le schéma, au ni-
veau du point Mi .
trajectoire
M1
M2
en men m.s-1
V1
M3V2
V2 = +V1 ΔV2
ΔV2
ΔV2V1
V2
Figure 11.3 – Le vecteur variation de vitesse seconstruit à partir de deux vecteurs vitesses mesurés endeux points consécutifs.
44

CHAPITRE 11. MOUVEMENT D’UN SYSTÈME
Méthode par coordonnées1. on calcule les coordonnées des vecteurs vi-
tesses−→Vi et
−−→Vi−1 (voir paragraphes précé-
dents)2. on calcule les coordonnes du vecteur variation
de vitesse−−→∆Vi = (V x i − V x i−1)~i + (V yi − V yi−1)~j
11.3 Variation de la vitesse et résultantedes forces
11.3.1 Deuxième loi de Newton
Définition Un objet de masse m qui subit pendantune durée breve ∆t une force
−→Fi au point Mi de sa
trajectoire va avoir son mouvement modifié. La force−→Fi va provoquer une variation de vitesse
−−→∆Vi en une
durée ∆t de façon à ce que
−→Fi = m
−−→∆Vi
∆tVoir figure 11.4.
M1
V1
M2
M3
V2
ΔV2 = 0
F2 = 0
trajectoire
M1
M2
en m
en m.s-1
V1
M3V2
ΔV2
F2ΔV2=mΔt en N
F2
trajectoire
a) pas de force appliquée, trajectoire rectiligne uniforme
b) force appliquée pendant un bref instant au point M2, le vecteur vitesse change au point M2
Figure 11.4 – Une force qui s’exerce sur l’objet au pointM2 pendant une durée ∆t très brève va provoquer unchangement du vecteur vitesse ~V2 qui se calcule à l’aidede la deuxième loi de Newton.
11.3.2 Cas d’une force nulle
Si la force est nulle, on a
m−−→∆Vi
∆t=−→0
donc −−→∆Vi =
−→0
Il n’y a pas de variation du vecteur vitesse, le mou-vement se fait en ligne droite et à vitesse constante.Voir figure 11.5.
trajectoire
M1
t1
en men m.s-1
V1
M2
t2
M3
t3
M4
t4
V2
V3
V4
Figure 11.5 – En l’absence de force exercée sur le sys-tème, le vecteur vitesse est constant et le mouvementest rectiligne et uniforme.
11.3.3 Influence de la masse m
Comme−→Fi = m
−−→∆Vi
∆talors
−−→∆Vi =
∆tm−−→∆Vi
donc si la masse augmente, la variation du vecteurvitesse est plus petite, le mouvement est moins mo-difié. Quand un objet est très massif, il a beaucoupd’inertie, il est difficile de faire changer son mouve-ment.
11.3.4 Cas d’une force constante ~Fi
C’est par exemple le cas du poids d’un objet, quiest constant et toujours orienté vers le bas. Comme
−→Fi = m
−−→∆Vi
∆t
alors−−→∆Vi =
∆tm−→Fi
est constant, on a toujours le même vecteur variationde vitesse. On peut ainsi dessiner une trajectoire pa-rabolique. Voir figure 11.6.
45

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
ΔV constant
ΔV
M1
V1
V2
M2
M3
V2*
V2
V1
V2 V1 ΔV = +
Une force s'applique en M2 pendantun bref instant ce qui modifie le vecteur vitesse
Figure 11.6 – En présence d’une force constante exer-cée sur le système, le vecteur variation de vitesse estconstant et le mouvement est parabolique.
46

CHAPITRE 11. MOUVEMENT D’UN SYSTÈME
M1 M2 M3 M4 M5M5 M6 M7
1 m
Figure 11.7 – Calculer et représenter les vecteurs vitesses aux points M2 et M6, préciser l’échelle utilisée pourles vitesses.
M1 M2 M3 M4 M5M5 M6 M7
1 m
Figure 11.8 – Calculer et représenter les vecteurs variation de vitesse aux points M2 et M6, préciser l’échelleutilisée pour les variations de vitesses.
1 m
M1
M2
M3
M4
M5M5 M6
M7
M8
M9
M1
M2
M3
M4
M5M5 M6
M7
M8
M9
Figure 11.9 – Calculer et représenter les vecteurs variation de vitesse aux points M3 et M6, préciser l’échelleutilisée pour les variations de vitesses.
47

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
1 m
M1
M2
M3
M6M7 M8
M12
M13
M14
O
Figure 11.10 – Calculer et représenter les vecteurs variation de vitesse aux points M2, M7 et M13, préciserl’échelle utilisée pour les variations de vitesses.
48

Chapitre 12
Aspects énergétiques des phénomènes élec-triques
IntroductionLes énergies fossiles ou renouvelables sontconverties en énergie électrique qui est plusfacile à transporter et à distribuer dansles différents lieux d’habitat ou industriels,puis cette énergie est convertie en énergiecinétique grâce à des moteurs, en énergielumineuse à l’aide de différents types delampes, ou en énergie thermique via l’effetJoule.
12.1 Courant électrique
12.1.1 Porteurs de charge électrique
Métaux Dans les métaux, les porteurs de chargeélectrique sont les électrons sur les dernièrescouches des atomes du métal qui ne sont pas liésà leur atomes, ils se déplacent sous l’effet d’une ten-sion électrique appliquée sur le métal. Voir figure12.1.
atome (noyau positif et nuage électronique)
électron libre
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
haut potentiel
V+
baspotentiel
V-
I
Figure 12.1 – Dans les métaux, les porteurs de chargeélectrique qui sont mobiles sont des électrons descouches externes des atomes du métal, qui peuvent cir-culer librement d’atomes en atomes. Sous l’effet d’unetension électrique, ils circulent du bas potentiel vers lehaut potentiel.
Solutions ioniques Les porteurs de charge élec-triques sont les cations et les anions qui migrenten sens opposé, les cations de l’électrode positivevers l’électrode négative, les anions prennent le senscontraire. 12.2.
électrode
haut potentiel
V+
baspotentiel
V- I
-
-
-
-
++
+
+
+
-
cation
anion
Figure 12.2 – Dans les solutions, les porteurs de chargeélectrique qui sont mobiles sont des cations et desanions.
Plasma Lors d’une étincelle ou d’un éclair dans ungaz, on a un mécanisme de circulation de chargesélectriques, impliquant des électrons arrachés à desmolécules et des cations issus de cette ionisation.12.3.
12.1.2 Courant électrique
Définition Le courant électrique est le débit decharges électriques . L’intensité I d’un courant élec-trique est égal à la valeur absolue de la charge élec-trique Q qui traverse à chaque seconde une sectionquelconque du conducteur.
I =|Q|∆t
avec— l’intensité I en A (Ampère)
49

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
électrode
hautpotentiel
V+
baspotentiel
V-
I
molécule gazcationélectron libre
déplacement électrons
déplacements cations
Figure 12.3 – Dans un plasma, des électrons sont ar-rachés à des atomes d’un gaz.
— la quantité de charges |Q| en C (Coulomb)— la durée du fonctionnement∆t en s (seconde)
Exemple Un courant électrique de 300 mA circuledans un conducteur pendant 5 min. Calculons lacharge électrique s’étant écoulée :
Q = I ×∆t = 300 mA× 5 min
donc en utilisant les bonnes unités
Q = I ×∆t = 0, 300 A× 300 s = 90 C
On peut calculer le nombre N d’électrons qui se sontdéplacés, sachant que chaque électron porte unecharge élémentaire q = −1.6× 10−19 C
N =|q|Q=
901.6× 10−19
= 5.6× 1020
Cela correspond à 0.9 mmol d’électrons, c’est à direque peu d’électrons du métal participent au courant.
12.2 Source réelle de tension continue
12.2.1 Source parfaite de tension
Définition Une source de tension parfaite de forceélectromotrice E (en volts) est capable de fournir unetension E constante quelque soit le courant I qui tra-verse ce générateur parfait. 12.4.
12.2.2 Source réelle de tension
Définition Une source de tension réelle de forceélectromotrice E et de résistance interne r fournitune tension U quand un courant I traverse le géné-rateur tel que
U = E − r × I
avec— E et U en V (volts)— r en ω (ohms)— I en A (Ampère)12.5.
a )
I
UE
U(volt)
I (ampère)
E
b )
Figure 12.4 – Une source de tension parfaite de tensioninterne E a une tension U = E à ses bornes quelquesoit le courant I qu’elle fournit. Le sens du courant estimposé par le générateur de tension.
b )
U(volt)
I (ampère)
E
c )
I
U
E
I
U
a )
u r=
r.I
U = E - ur = E - r x I
coefficient directeur: -r
Imax
destruction du générateur
Figure 12.5 – Une source de tension réelle de tensioninterne E et de résistance interne r a une tension U =E − r × I à ses bornes qui décroît avec le courant.
Remarque importante La présence d’une résis-tance interne r dans un générateur réel de tensiona des conséquences importantes dès que le courantI devient important
— La tension U chute de la quantité −r × I— le générateur réel possède une résistance in-
terne r responsable de pertes d’énergies pareffet Joule, le générateur va chauffer.
L’échauffement du générateur peut provoquer sadestruction (explosion d’une batterie, coupure ducircuit par fusion d’un câble électrique métallique,incendie, etc... ).
12.3 Puissance et énergie
12.3.1 Puissance P
Définition Si un dispositif peut fournir ou absor-ber une quantité d’énergie E pendant une durée ∆t,alors il fournit ou absorbe une puissance P telle que
P =E∆t
avec— la puissance P est en Watt (W )— l’énergie est en Joules (J)— la durée de fonctionnement ∆t est en se-
condes (s)
50

CHAPITRE 12. ASPECTS ÉNERGÉTIQUES DES PHÉNOMÈNES ÉLECTRIQUES
12.3.2 Puissance électrique
Définition La puissance P fournie ou consomméepar un appareil électrique traversé par un courant Iet ayant une tension U à ses bornes est
P = U × I
avec— la puissance P en Watt (W )— la tension U en Volts (V )— le courant I en Ampères (A)
12.3.3 Ordres de grandeur de puissance absorbée ouproduite par certains dispositifs
— montre électronique 1 µW— éclairage domestique à DEL 10 W— moteur électrique d’outillage 500 W— fer à repasser 1000 W— four électrique domestique 1 kW— voiture électrique 100 kW— locomotive électrique 2000 à 6000 kW— barrage de Kembs 160 MW— centrale de Fessenheim 1800 MW— centrale de Cattenom 5200 MW— barrage des Trois Gorges (Chine) 22500 MW
12.4 Effet Joule
Définition L’effet Joules est la conversion del’énergie électrique en énergie thermique dans undipôle résistif de résistance R traversé par un cou-rant I et soumis à une tension U . On a les relationssuivantes
— loi d’Ohm U = R× I— puissance électrique P = U × I
On peut alors exprimer la puissance thermique dis-sipée par effet Joule
PJoules = R× I2
ou encore
PJoules =U2
Ravec
— U en Volts (V )— I en Ampères (I)— R en Ohms (Ω)— P en Watts (W )
12.5 Rendement d’un convertisseur
12.5.1 Bilan de puissance
Définition Faire le bilan de puissance électrique,c’est faire l’inventaire de l’énergie électrique fournieau circuit, et celle que le circuit va transformer en
une autre forme utilisable et celle qu’il va perdre,c’est à dire sous forme d’énergie que l’on ne peutpas utiliser.
énergie fournie= énergie perdue+ énergie utile
12.5.2 Convertisseur
Définition Un convertisseur d’énergie transformeune forme d’énergie en une autre forme d’énergie.Voir figure 12.6.
énergiefournie
énergieutilisable
énergie perdue
Convertisseur d'énergie
Figure 12.6 – Un convertisseur d’énergie transformeune forme d’énergie en une autre forme avec une perted’énergie.
Exemples— une lampe électrique convertit l’énergie élec-
trique en énergie lumineuse— une batterie transforme l’énergie chimique en
énergie électrique— un moteur électrique transforme l’énergie
électrique en énergie cinétique (voir figure12.7)
— un chauffage électrique transforme l’énergieélectrique en énergie thermique
énergieélectrique
énergiecinétique
énergie thermique
Moteur industriel électrique type SIMOTICS (Siemens)
énergiecinétique
énergieélectrique
énergie thermique
énergiecinétique
Figure 12.7 – Un moteur électrique transforme l’éner-gie électrique en énergie cinétique mais chauffe durantson fonctionnement.
51

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
12.5.3 Rendement
Définition Un convertisseur d’énergie transformel’énergie entrante en une autre forme pour l’éner-gie sortante utilisable, mais cette conversion peut sefaire avec des pertes d’énergies qu’on ne peut pasutiliser. Le rendement du convertisseur est alors
rendement=énergie utilisableénergie entrante
< 100%
Exemple Un moteur électrique reçoit de l’énergieélectrique, mais à cause de l’effet Joules et des frotte-ments, il chauffe, et cette énergie thermique est uneperte, on ne peut pas l’utiliser. L’énergie cinétiqueutilisable sera plus faible que l’énergie électriquefournie, le rendement est inférieur à 1. Un moteurélectrique a un rendement de l’ordre de 90%.
12.5.4 Puissance ou énergie ?
Remarque Pour calculer le rendement, on peutraisonner en terme de puissances si on considèrel’énergie consommée en une seconde. Il faut simple-ment ne pas mélanger les deux grandeurs physiques(puissance ou énergie) dans les formules !Si l’énergie fournie pendant une durée de fonction-nement ∆t est
Efournie = Pfournie ×∆t
et l’énergie utilisable est
Eutile = Putile ×∆t
alors le rendement est
r =Eutile
Efournie
=Putile ×∆t
Pfournie ×∆t
=Putile
Pfournie
Exemple Un moteur électrique consomme unepuissance de 3 W pendant 10 minutes et fournit unepuissance mécanique de 2.5 W . Calculez son rende-ment de conversion.Si on raisonne en terme d’énergie :
— énergie consommée Ec = 3 W × 10 × 60 s =1800 J
— énergie utilisable Ed = 2.5 W × 10 × 60 s =1500 J
— rendement r = EdEc= 1500 J
1800 J = 0.83Si on raisonne en terme d’énergie consommée ouproduite en 1 seconde, on utilise les puissances :
— puissance consommée Pc = 3 W— puissance utilisable Pd = 2.5 W— rendement r = Pd
Pc= 2.5 W
3.0 W = 0.83
52

Chapitre 13
Aspects énergétiques des phénomènes mé-caniques
IntroductionL’énergie est une grandeur caractéristiqued’un corps qui peut décrire son état demouvement, sa température, sa déforma-tion, sa charge électrique. L’énergie de cecorps va pouvoir être modifiée par le tra-vail qu’il effectue ou qu’on lui fournit. LaPhysique peut être ainsi décrite commel’étude d’échanges d’énergies sous diffé-rentes formes entre les objets de l’Univers.
13.1 Énergie cinétique
13.1.1 Système étudié
On se restreint au cas où le mouvement du sys-tème étudié se fait en ligne droite, c’est à dire unmouvement de translation.Tout se passe alors comme si on pouvait résuméle système étudié à un simple point possédant unemasse m égale à celle du système.C’est le modèle du point matériel qui n’est valide quedans le cas où on n’étudie pas la rotation sur luimême du système étudié. Voir figure 13.1.
camion
route
camion
poids
réaction de la route
Figure 13.1 – Le modèle du point matériel est une sim-plification de la réalité qui résume un système com-plexe à un point ayant la même masse et sur lequels’exercent les actions extérieures.
13.1.2 L’énergie cinétique en translation
Définition Si un système de masse m a un mou-vement de translation à la vitesse ~v dans un référen-tiel dans lequel on étudie son mouvement, alors ilpossède une énergie due à son mouvement appeléeénergie cinétique Ec que l’on calcule par la relation
Ec =12×m× v2
avec les unités suivantes— l’énergie cinétique Ec est en Joule (J)— la masse m est en kilogramme (kg)— la vitesse v est en mètre par seconde (m.s−1)
Exemple Un objet de masse m = 50 g qui se dé-place à une vitesse v = 1200 km.h−1 a une énergiecinétique Ec égale à
Ec =12×m× v2
=12× 50 g ×
1200 km.h−12
=12× 50× 10−3 kg ×
1200× 103 m3600 s
2
= 2780 J
= 2, 78 kJ
13.2 Travail d’une force
13.2.1 Introduction
Intuitivement, on peut décrire que le travailconsiste à exercer une force sur un objet qui se dé-place pour échanger de l’énergie, en lui fournissantl’énergie pour accélérer son mouvement ou en dimi-nuant son énergie pour freiner son mouvement. Voirfigure 13.2.
53

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
Figure 13.2 – Quand les bœufs tirent la charrue, ilsexercent une force qui se déplace et il y a une trans-mission d’énergie vers le sol retourné.
13.2.2 Principe
Un système est en mouvement, il se déplace d’unpoint A à un point B en ligne droite. On peut alorsdéfinir le vecteur déplacement de A à B en deux di-mensions par
−→AB = (xB − xA)~i + (yB − yA)~j
Le système étudié est soumis à une force extérieure−→F qui s’applique à lui tout le long de son déplace-ment.Le vecteur déplacement
−→AB et le vecteur force
−→F
font un angle α entre eux, α étant compris dans l’in-tervalle [0o, 180o].
A
B
Figure 13.3 – Travail WAB
~F
d’une force constante ~Fsur un déplacement de A à B de son point d’applica-tion.
13.2.3 Définition
Le travail WAB(−→F ) de la force
−→F s’exerçant sur le
système durant son déplacement de A à B est égalau produit scalaire du vecteur déplacement
−→AB et de
la force−→F . Voir 13.3.
WAB(−→F ) =
−→AB ·−→F
= AB × F × cos(α)
avec les unités suivantes— le travail WAB(
−→F ) est en Joule (J)
— le déplacement AB est en mètre (m)— la force F est en Newton (N)
La valeur du travail peut être— positive pour une force moteur— nulle quand la force est perpendiculaire au dé-
placement— négative pour une force résistante ou de frei-
nageVoir figure 13.4.
A
B
A
B
A
B
90o
travail moteur
travail nul
travail résistant
Figure 13.4 – Selon l’angle entre le vecteur force et levecteur déplacement, le travail peut être moteur, nulou résistant.
13.2.4 Exemples
Un objet soumis à son poids P = 100 N se dé-place du point A au point B selon trois trajectoirespossibles. Voir figure 13.5.Calculer le travail du poids de l’objet pour chaquetrajectoire.
— la trajectoire 1 se décompose en deux seg-ments de droite, un premier segment horizon-tal de 2 m où le poids est perpendiculaire audéplacement et le travail est donc nul. Puis unsegment vertical de 5.5 m où le poids est co-linéaire au déplacement , le travail est W =5.5×100= 550 J . Donc le travail total pour lapremière trajectoire est W = 0+550= 550 J .
— la trajectoire 2 est une ligne droite faisant unangle constant avec le poids qui est vertical.On calcule les coordonnées du vecteur
−→AB = 2~i − 5.5~j
le poids a pour coordonnées
−→P = −100 ~j
54

CHAPITRE 13. ASPECTS ÉNERGÉTIQUES DES PHÉNOMÈNES MÉCANIQUES
1 mtrajectoire 1
trajectoire 2
trajectoire 3
A
B
Figure 13.5 – Calcul du travail du poids ~P ayant pournorme P = 100 N pour trois trajectoires allant dupoint A au point B.
et donc le produit scalaire correspondant autravail W est
W = 2× 0+ (−5.5)× (−100) = 550 J
— la trajectoire 3 consiste en un segment ver-tical de 3.5 m puis d’un segment incliné à45o. Pour le premier segment, le travail estW = 100 × 3.5 = 350 J . Pour le deuxièmesegment, en appliquant Pythagore à un tri-angle de 2 m de coté, on a que la longueurdu segment vaut
p22 + 22 = 2.83 m et donc
le travail du poids sur ce segment vaut W =2.83×100×cos 45o = 200 J . Finalement, pourla totalité de la trajectoire le travail du poidsvaut
W = 350+ 200= 550 J
On remarque que le travail du poids ne dépend quedes altitudes de la position de départ et de la po-sition d’arrivée mais pas du chemin emprunté. Deplus, comme l’objet perd de l’altitude, le travail dupoids est ici positif, le travail du poids est moteur.
13.3 Forces conservatives
13.3.1 Définition
Une force−→F est dite conservative si son travail
pour aller d’un point A à un point B est indépendantdu chemin suivi pour aller de A à B.
13.3.2 Exemple
Le poids d’un objet est une force conservative, seulcompte la position du point de départ et celle dupoint d’arrivée.
13.4 Forces non conservatives
13.4.1 Définition
Une force−→F est dite non conservative si son travail
pour aller d’un point A à un point B est dépendant duchemin suivi pour aller de A à B.
13.4.2 Exemple
Les forces de frottement (contre un solide ou dansdes fluides) sont non conservatives, car leur travailest toujours résistant et il dépend de la distance par-courue.
13.5 Théorème de l’énergie cinétique
13.5.1 Définition
Le théorème de l’énergie cinétique dit que la varia-tion de l’énergie cinétique ∆Ec d’un objet au coursd’un déplacement d’un point A à un point B est égalà la somme des travaux de toutes les forces appliquéesà l’objet au cours de son déplacement de A à B
∆Ec = Ecfinale− Ecinitiale
=∑
WAB
−−−→Fapplic.
13.5.2 Exemple
Chute libre sans vitesse initiale À l’aide du théo-rème de l’énergie cinétique, on peut calculer la vi-tesse atteinte par un objet soumis uniquement à sonpoids lors d’une chute libre où la vitesse initiale estnulle. Voir figure 13.6.Comme la vitesse initiale est nulle, Ecinitiale
= 0 J .
A position initiale, vitesse nulle
B position finale, vitesse vfinale
hauteur de chute h
Figure 13.6 – Chute libre d’un objet soumis à sonpoids.
55

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
La seule force exercée sur l’objet étant son poids−→P ,
vertical vers le bas, le travail du poids au bout d’unehauteur de chute h sera
W−→
P
= h× P
Ce travail est positif, le poids est moteur.Au bout d’une hauteur de chute h, l’objet a une éner-gie cinétique Ecfinale
et d’après le théorème de l’éner-gie cinétique
Ecfinale− Ecinitiale
=W−→
P
Ecfinale− 0= h× P
12×m× v2
finale = h×m× g
12× v2
finale = h× g
vfinale =p
2× h× g
On remarque que la vitesse de chute libre d’un objetdans le vide ne dépend pas de sa masse. Cela peutse confirmer expérimentalement à l’aide d’un tubede Newton.
Chute libre sans vitesse initiale On lance verti-calement vers le haut un projectile avec une vitesseinitiale de 5.0 m.s−1 dans le champ de pesanteur ter-restre (g = 9.81 m.s−2). Voir figure 13.7.Calculer l’altitude maximale hmax atteinte par le pro-
B position finale, vitesse nulle
B position initiale, vitesse vinitiale
hauteur de chute h
Figure 13.7 – Mouvement d’un d’un objet soumis à sonpoids et jeté verticalement avec une vitesse initiale.
jectile.L’altitude maximale sera atteinte quand la vitesse del’objet devient nulle (ensuite, l’objet retombe). Onaura donc une énergie cinétique finale nulle.Le poids
−→P de l’objet est colinéaire durant le mouve-
ment mais vers le bas, son travail est donc résistantet négatif
W−→
P
= −hmax × P
On applique ensuite le théorème de l’énergie ciné-tique pour isoler la valeur de hmax
Ecfinale− Ecinitiale
=W−→
P
0−12×m× v2
initiale = −hmax × P
−12×m× v2
initiale = −hmax ×m× g
−12× v2
initiale = −hmax × g
12× v2
initiale = hmax × g
hmax =v2
initiale
2× g
hmax =5.02
2× 9.81hmax = 1.27 m
13.6 Énergie potentielle
13.6.1 Définition
En général, une énergie qui ne dépend que de laposition d’un objet dans l’espace et qui ne dépendpas de sa vitesse est appelée énergie potentielle.La variation de l’énergie potentielle est égale au tra-vail fourni à l’objet pour le déplacer d’un point initialA à un point final B.
13.6.2 Définition
L’énergie potentielle de pesanteur Ep d’un objet estl’énergie que possède cet objet du fait de sa positionpar rapport à la Terre.Pour définir Ep, on mesure l’altitude z de l’objet surun axe vertical dirigé vers le haut, l’origine est choi-sie de façon arbitraire. Voir figure 13.8
objet de masse m
0
z
altitude (m)
le choix de l'origine est arbitraire
champ de pesanteur
Figure 13.8 – Définition de l’énergie potentielle de pe-santeur d’un objet de masse m.
Ep (z) = m× g × z + constante
56

CHAPITRE 13. ASPECTS ÉNERGÉTIQUES DES PHÉNOMÈNES MÉCANIQUES
On choisit d’avoir la constante nulle pour l’énergiepotentielle à l’altitude nulle
Ep (z) = m× g × z
avec— m en kilogramme (kg)— g = 9.81 m.s−2 sur la Terre— z l’altitude en mètre (m)— Ep est en joule (J)
13.6.3 Remarque
On cherche en général à déterminer la variationde l’énergie potentielle∆Ep = Ep(zfinale)−Ep(zinitiale)entre deux position différentes.
13.7 Énergie mécanique
13.7.1 Définition
Si un objet passe en un point avec une certaine vi-tesse et en étant à une certaine distance de la Terre,alors il possède une énergie mécanique Em qui est lasomme de son énergie cinétique Ec et de son énergiepotentielle Ep
Em = Ec + Ep
Toutes ces énergies s’expriment en joule (J).
13.8 Conservation et non conservation del’énergie mécanique
13.8.1 Cas de conservation de l’énergie mécanique
Forces conservatives
Dans le cas de forces conservatives commele poids, l’énergie mécanique d’un système resteconstante pendant son mouvement, et l’énergie setransfert entre énergie cinétique et énergie potentielde manière à ce que la somme reste constante.
La chute libre sans frottement
Un objet en chute libre sans frottement a uneénergie mécanique Em égale à la somme de son éner-gie cinétique Ec et énergie potentielle Ep. Voir figure13.9.Cette énergie mécanique est constante. On peutavoir certaines situations particulières. L’énergie ci-nétique sera nulle pour l’altitude maximale (le débutde la chute).
Em = Ec + Ep = 0+m× g × zmax
Puis au fur et à mesure de la chute, l’énergie poten-tielle devient de plus en plus petite et même négative
car z décroît. Comme la somme de l’énergie poten-tielle et de l’énergie cinétique reste constante, c’estl’énergie cinétique qui va croître, et donc la vitesseaugmente. Tant que rien n’arrête la chute, la vitesseaugmente.
objet de masse m en chute libre
0
z
altitude (m)
champ de pesanteur
vitesse
Figure 13.9 – Énergie mécanique d’un objet en chutelibre sans frottement.
Oscillation d’un pendule
En physique, un pendule est un petit objet demasse m accroché à un fil de longueur l. Si on écartelégèrement de sa position d’équilibre l’objet et qu’onle lâche, on observe une oscillation régulière dontla période dépend uniquement de la longueur l dufil et de l’accélération de pesanteur g. Les oscilla-tions peuvent durer presque une heure si le fil esttrès raide et très fin. Voir figure 13.10.L’énergie mécanique est constante, il y a deux casextrêmes.
— si l’énergie potentielle est nulle (minimumd’altitude, point le plus bas), alors l’énergiemécanique est égale à l’énergie cinétique, lavitesse est maximale au passage du point leplus bas.
— si l’énergie cinétique est nulle alors l’énergiemécanique est égale à l’énergie potentielle quisera maximale, on est au point le plus haut(maximum d’altitude zmax).
L’oscillation consiste à un transfert permanent entrel’énergie cinétique et l’énergie potentielle de ma-nière à ce que la somme reste constante.
13.8.2 Cas de non conservation de l’énergie méca-nique
Forces non conservatives
Dans le cas de forces non conservatives commeles forces de frottement, l’énergie mécanique du sys-tème décroît d’une quantité égale au travail de cesforces non conservatives durant le mouvement.
57

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
objet de masse m accroché à un fil de longueur l
0
z
altitude (m)
champ de pesanteur
vitesse
zmax
S
Figure 13.10 – Énergie mécanique d’un pendule pe-sant.
Frottement durant un mouvement
Quand une voiture freine, des mâchoires viennentplaquer fortement des patins sur un disque de frein,provoquant d’importants frottements qui échauffentle disque de frein, il peut même devenir incandes-cent sous l’effet de la chaleur. On transforme l’éner-gie cinétique en énergie thermique qui est dissipéegrâce à la ventilation du disque et à l’air, l’énergiemécanique de la voiture diminue et elle perd de lavitesse. Voir figure 13.11. Au point A, l’énergie mé-canique de la voiture est
Em(A) =12×m× v2
A + Ep
Au point B, son énergie potentielle est identiquemais l’énergie cinétique est moindre du fait de la
force de freinage−→f exercée entre les positions A et
B, l’énergie mécanique est alors
Em(B) =12×m× v2
B + Ep
La variation d’énergie mécanique sera
Em(B)− Em(A) = Ec(B)− Ec(A)
et donc d’après le théorème de l’énergie cinétique
Em(B)− Em(A) =−→AB ·−→f
Comme le travail de la force de freinage est néga-tif, la différence d’énergie mécanique est négative,l’énergie mécanique finale est plus petite, car l’éner-gie cinétique a diminuée.
Rebond d’une balle avec pertes
Quand une balle de tennis ou de ping-pong re-bondit sur une surface dure, elle se déforme au mo-ment de l’impact. Cette déformation provoque une
disque de freinplaquettesde frein
altitude (m)
0A B0
Figure 13.11 – Énergie mécanique d’un pendule pe-sant.
perte d’énergie à cause de frottements dans la ma-tière composant l’enveloppe de la balle, l’énergiemécanique est diminuée à chaque rebond, et la ballemontera alors moins haut. Elle finit par s’immobili-ser quand toute l’énergie mécanique sera dissipée etque l’énergie cinétique et potentielles seront nulles.
58

Chapitre 14
Ondes mécaniques
IntroductionÀ écrire.
14.1 Onde mécanique progressive
14.2 Célérité et retard
14.3 Onde mécanique périodique
14.4 Onde mécanique sinusoïdale
14.5 Période, longueur d’onde et célérité
14.6 Exercices
Corps purs et composition des mélanges
1 a. Donnez la définition d’une espèce chimique,d’un corps pur, d’un mélange homogène et d’un mé-lange hétérogène.b. Donnez dans chaque cas précédent un exemple.
14.7 Correction
1 Correction ... 2 Correction ... 3 Cor-rection ...
59

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
60

Chapitre 15
Images et couleurs
IntroductionÀ écrire.
15.1 Optique géométrique
15.2 Colorimétrie
15.3 Exercices
Corps purs et composition des mélanges
1 a. Donnez la définition d’une espèce chimique,d’un corps pur, d’un mélange homogène et d’un mé-lange hétérogène.b. Donnez dans chaque cas précédent un exemple.
15.4 Correction
1 Correction ... 2 Correction ... 3 Cor-rection ...
61

SPÉCIALITÉ PHYSIQUE CHIMIE 1e
62

Chapitre 16
Optique ondulatoire
IntroductionÀ écrire.
16.1 Ondes électromagnétiques
16.2 Longueur d’onde, célérité et fréquence
16.3 Le photon
16.4 Interaction onde-matière
16.5 Quantification des niveaux d’énergiedes atomes
16.6 Exercices
Corps purs et composition des mélanges
1 a. Donnez la définition d’une espèce chimique,d’un corps pur, d’un mélange homogène et d’un mé-lange hétérogène.b. Donnez dans chaque cas précédent un exemple.
16.7 Correction
1 Correction ... 2 Correction ... 3 Cor-rection ...
63





























