Embed Size (px)
Citation preview

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
1 od 23
SAŽETAK KARAKTERISTIKA LEKA
Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 2000i.j./0.5 mLPakovanje: ukupno 6 kom, napunjen injekcioni špric, 6 x 0.5 mL
Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 10000i.j./1 mL Pakovanje: ukupno 6 kom, napunjen injekcioni špric, 6 x 1 mL
Proizvođač: Cilag AG
Adresa: Hochstrasse 201, Schaffhausen, Švajcarska
Podnosilac zahteva: Predstavništvo Janssen-Cilag KFT Beograd
Adresa: Omladinskih brigada 88b, Novi Beograd

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
2 od 23
1. IME LEKA, INTERNACIONALNO NEZAŠTIĆENO IME LEKA (INN)
Eprex ®
INN: epoetin alfa2. KVALITATIVNI I KVANTITATIVNI SASTAV
Epoetin alfa*......................................................................10000 I.J/mL (84,0 mikrograma po mililitru)
Epoetin alfa* .......................................................................2000 I.J./0.5mL (16.8 mikrograma po mililitru)
Napunjeni injekcioni špric od 1.0 mL sadrži 10000 I.J. (84,0 mikrograma) epoetina alfa
Napunjeni injekcioni špric od 0.5 mL sadrži 2000 I.J. (16,8 mikrograma) epoetina alfa
* Proizveden rekombinantnom DNK tehnologijom u kulturi jajnih ćelija kineskog hrčka (Chinese Hamster Ovary cells – CHO).
Kompletan spisak pomoćnih supstanci dat je u delu 6.1.
3. FARMACEUTSKI OBLIK
Rastvor za injekciju u napunjenom injekcionom špricu. Bistar, bezbojan rastvor, bez mehaničkih onečišćenja..
4. KLINIČKI PODACI
4.1. Terapijske indikacije
Simptomatska anemija koja je posledica hronične renalne insuficijencije (CRF) kod dece i odraslih:
Anemija koja je posledica hronične renalne insuficijencije kod dece i odraslih pacijenata na hemodijalizi i odraslih pacijenata na peritonealnoj dijalizi.
Teška anemija bubrežnog porekla praćena kliničkim simptomima kod odraslih pacijenata sa renalnom insuficijencijom u predijaliznom stadijumu.
Terapija anemije i smanjenje potrebe za transfuzijom kod odraslih osoba sa malignim oboljenjima koji primaju hemioterapiju (solidni tumori, maligni limfomi, multipli mijelom) i kod pacijenata kod kojih je rizično primeniti transfuziju, na osnovu procene opšteg stanja pacijenta ( npr. kardiovaskularni status, postojanje anemije na početku hemioterapije).

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
3 od 23
Eprex se može primeniti za povećanje količine autologne krvi kod pacijenata uključenih u program predonacije. Upotrebu epoetina u ovoj indikaciji treba proceniti u odnosu na rizik od tromboembolije. Terapiju bi trebalo primeniti samo kod pacijenata sa umerenom anemijom (Hb 10-13 g/dL [6,2-8,1 mmoL/L], bez deficita gvožđa), ukoliko sakupljanje krvi nije moguće ili je nedovoljno za predviđenu veliku elektivnu operaciju koja zahteva veliki volumen krvi (4 ili više jedinica krvi za žene ili 5 ili više jedinica krvi za muškarce).
Eprex se može primeniti u cilju smanjenja transfuzije alogene krvi kod odraslih pacijenata, bez deficita gvožđa, pre velikih elektivnih ortopedskih operacija, a kod kojih postoji visoki rizik od komplikacija usled transfuzije.Ovu primenu treba ograničiti na pacijente sa umerenom anemijom (npr. Hb 10-13g/dL) kod kojih nije bilo moguće primeniti program kolekcije autologne krvi i kod kojih se očekuje umereni gubitak krvi (900-1800mL).
U perioperativnom okruženju treba uvek primenjivati Dobru praksu u rukovanju krvlju.
4.2. Doziranje i način primene
Način primene
Kao i kod ostalih lekova za parenteralnu primenu, pre ubrizgavanja proverite da li postoje čestice u rastvoru ili promena boje.
a) intravenska injekcija: daje se tokom najmanje jednog do pet minuta, u zavisnosti od ukupne doze. Kod pacijenata na hemodijalizi, može se dati bolusna injekcija tokom hemodijalize u venski put dijalizne linije. Alternativno, injekcija se može dati na kraju hemodijalize kroz kanilu arteriovenske fistule, koju zatim treba isprati sa 10 mL izotoničnog rastvora natrijum hlorida kako bi se obezbedilo potpuno ubrizgavanje leka u cirkulaciju.
Sporije ubrizgavanje je poželjno kod pacijenata koji reaguju na lek simptomima sličnim gripu.
Eprex ne treba primenjivati u obliku intravenske infuzije, niti mešati sa drugim lekovima.
b) supkutana injekcija: ne treba premašiti maksimalnu zapreminu od 1mL po mestu ubrizgavanja injekcije.U slučaju većih zapremina, lek treba ubrizgati na veći broj mesta.
Injekcije se daju u ekstremitete ili prednji zid abdomena.
U situacijama kada doktor odluči da pacijent ili negovatelj mogu samostalno bezbedno i efikasno da primene lek Eprex supkutano, potrebno je obezbediti odgovarajuću obuku za adekvatno doziranje i primenu.
Pogledajte deo 3. Kako se upotrebljava lek Eprex (Uputstvo za upotrebu leka Eprex ) Uputstva za pacijenta
Simptomatska anemija koja je posledica hronične renalne insuficijencije kod dece i odraslih:

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
4 od 23
Kod pacijenata sa hroničnom bubrežnom insuficijencijom kod kojih je venski put rutinski dostupan (pacijenti na hemodijalizi) prednost treba dati intravenskom načinu primene. Ukoliko intravenski put nije lako dostupan
(pacijenti u predijaliznom stadijumu i pacijenti na peritonealnoj dijalizi) Eprex se može primeniti supkutano.
Simptomi anemije i posledice mogu biti različiti u odnosu na starost, pol, kao i na već postojeće bolesti ili poremećaje; zbog toga je neophodno da lekar individualno proceni stanje i lečenje svakog pacijenta.
Eprex je potrebno davati da bi se koncentracija hemoglobina povećala na ne više od 12 g/dL ( 7.5 mmoL/L). Treba izbegavati povećanje hemoglobina više od 2 g/dL (1.25 mmoL/L) u toku 4 nedelje. Ukoliko se to dogodi, potrebno je prilagoditi dozu.
Usled intra-individualnih razlika, kod jednog pacijenta se mogu povremeno javiti koncentracije hemoglobina koje su veće ili manje od onih koje se žele postići. Variranje hemoglobina treba regulisati prilagođavanjem doze sa ciljnom koncentracijom hemoglobina izmedju 10 i 12 g/dL (6,2-7,5 mmoL/L), osim kod dece, kod kojih koncentracija hemoglobina treba da bude izmedju 9,5 i 11 g/dL (5,9-6,8 mmoL/L).
Treba izbegavati održavanje nivoa hemoglobina u koncentraciji većoj od 12g/L (7,5 mmoL/L). Ukoliko je povećanje hemoglobina veće od 2 g/l (1.25 mmol/l) mesečno, ili održavana koncentracija hemoglobina pređe nivo od 12 g/L (7.5mmoL/L), smanjiti dozu epoetina alfa za 25%. Ukoliko koncentracija hemoglobina pređe nivo od 13 g/L ( 8.1 mmoL/L) potrebno je obustaviti terapiju dok koncentracija hemoglobina ne padne ispod 12 g/dL (7.5mmoL/L). Tada se opet uvodi terapija epoetinom alfa u redukovanoj dozi za 25%.
Potrebno je pažljivo pratiti pacijente da bi se obezbedilo da najniža dozvoljena doza leka Eprex koristi za adekvatnu kontrolu anemije i simptoma anemije
Nivo gvoždja treba kontrolisati pre i tokom lečenja, a preparate za nadoknadu gvoždja treba primeniti ukoliko je neophodno. Pored toga, pre započinjanja terapije epoetinom alfa, treba isključiti i druge uzroke anemije, kao što je na primer deficit vitamina B12 ili folata. Ukoliko odgovor na terapiju epoetinom alfa izostane, treba tragati za drugim uzrocima anemije (deficit gvoždja, folata ili vitamina B12; trovanje aluminijumom; interkurentne infekcije; inflamatorne ili traumatske epizode; okultni gubitak krvi; hemoliza i fibroza koštane srži bilo kog porekla).
Odrasli pacijenti na hemodijalizi:
Kod pacijenata na hemodijalizi, kod kojih je intravenski put lako dostupan, prednost treba dati intravenskom načinu primene.
Lečenje je podeljeno u dve faze:
Korektivna faza:
50 IJ/kg, tri puta nedeljno, Ukoliko je neophodno prilagođavanje doze, doza se postepeno povećava ili smanjuje za po 25 IJ/kg tri puta nedeljno, u intervalima od bar 4 nedelje.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
5 od 23
Faza održavanja:
Podešavanje doziranja da bi se vrednosti hemoglobina (Hb) održale na željenom nivou: Hb između 10 i 12 g/dL(6,2 – 7,5 mmoL/L).
Preporučena ukupna nedeljna doza iznosi izmedju 75 i 300 IJ/kg.
Raspoloživi klinički podaci ukazuju da pacijentima sa veoma niskom početnom vrednošću hemoglobina (< 6 g/dL ili < 3,75 mmoL/L) može da bude potrebna veća doza održavanja nego onima čija je početna anemija manje teška (Hb > 8 g/dL ili > 5 mmoL/L).
Pedijatrijski pacijenti na hemodijalizi:
Lečenje je podeljeno u dve faze:
Korektivna faza:
50 IJ/kg tri puta nedeljno, intravenski. Ukoliko je neophodno, podešavanje doze treba vršiti za po 25 IJ/kg tri puta nedeljno u intervalima od najmanje 4 nedelje, sve dok se ne postigne željena vrednost.
Faza održavanja:
Podešavanje doziranja da bi se vrednosti hemoglobina (Hb) održale na željenom nivou: Hb između 9,5 i 11 g/dL (5,9 – 6,8 mmoL/L).
Deci sa telesnom masom ispod 30 kg obično je potrebna veća doza održavanja nego deci sa telesnom masomiznad 30 kg i odraslima. Na primer, u kliničkom ispitivanju nakon 6 meseci lečenja utvrđene su sledeće doze održavanja:
Doza (IJ/kg 3x nedeljno)Težina (kg) Srednja vrednost Uobičajena doza održavanja
< 10 100 75-15010-30 75 60-150> 30 33 30-100
Raspoloživi klinički podaci ukazuju da pacijentima, čija je početna vrednost hemoglobina veoma niska (< 6,8 g/dL ili < 4,25 mmoL/L), može biti potrebna veća doza održavanja nego onima čija je početna vrednost hemoglobina viša (> 6,8 g/dL ili > 4,25 mmoL/L).
Odrasli pacijenti sa insuficijencijom bubrega koji još uvek nisu podvrgnuti dijalizi:
Ukoliko venski put nije lako dostupan, Eprex se može primeniti supkutano.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
6 od 23
Lečenje je podeljeno u dve faze:
Korektivna faza:
Početna doza od 50 IJ/kg tri puta nedeljno, a zatim, ukoliko je neophodno, sledi povećanje doze za po 25 IJ/kg tri puta nedeljno, sve dok se ne postignu željene vrednosti, u intervalima od bar četiri nedelje.Alterativno, ako se Eprex primenjuje subkutano, može se primeniti inicijalna doza od 120 IJ/kg (do maksimalno 10000IJ) kao pojedinačna injekcija jednom nedeljno.
Ako povećanje hemoglobina nije adekvatno (manje od 1g/dL (0,6mmoL/L) za četiri nedelje), povećati dozu za oko 25%. Povećanje doze se ne sme vršiti više od jednom u toku 4 nedelje.
Faza održavanja:
Tokom faze održavanja, Eprex se može primenjivati ili 3 puta nedeljno, odnosno, jednom nedeljno ili jednom na svake dve nedelje ukoliko se primenjuje subkutano.
Potrebno je da se adekvatno podesi doziranje ili interval doziranja da bi se vrednosti hemoglobina (Hb) održale na željenom nivou: Hb između 10 i 12 g/dL (6,2-7,5 mmoL/L). Produženje intervala doziranja može zahtevati povećanje doze.
Maksimalna doza ne sme biti veća od 150 IJ/kg, 3 puta nedeljno, 240IJ/kg (do maksimalne vrednosti od 20000IJ) jednom nedeljno, ili 480Ij/kg (do maksimalne vrednosti od 40000IJ) jednom na svake dve nedelje.
Odrasli pacijenti na peritonealnoj dijalizi:
Ukoliko venski put nije lako dostupan, Eprex se može primeniti supkutano.
Lečenje je podeljeno u dve faze:
Korektivna faza:
Početna doza od 50 IJ/kg, dva puta nedeljno.
Faza održavanja:
Podešavanje doziranja da bi se vrednosti hemoglobina (Hb) održale na željenom nivou: Hb između 10 i 12 g/dL(6,2-7,5 mmoL/L) (doza održavanja između 25 i 50 IJ/kg, dva puta nedeljno u dve jednake injekcije).
Odrasli pacijenti sa malignim oboljenjem koji primaju hemioterapiju:

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
7 od 23
Eprex treba primeniti supkutano pacijentima sa anemijom (npr. Hb ≤ 10 g/dL [6,2 mmoL/L]). Simptomi anemije i posledice mogu biti različiti u odnosu na starost, pol, kao i na već postojeće bolesti ili poremećaje; zbog toga je neophodno da lekar individualno proceni stanje i lečenje svakog pacijenta.
Usled intra-individualnih razlika, kod jednog pacijenta se mogu povremeno javiti koncentracije hemoglobina koje su veće ili manje od onih koje se žele postići. Variranje hemoglobina treba regulisati prilagođavanjem doze sa ciljnom koncentracijom hemoglobina izmedju 10 i 12 g/dL (6,2-7,5 mmoL/L). Treba izbegavati održavanje nivoa hemoglobina u koncentraciji većoj od 12g/L (7,5 mmoL/L); prilagođavanje doze leka kada koncentracijahemoglobina pređe 12 g/L (7.5 mmoL/L), prethodno je opisano.
Terapiju epoetinom alfa treba nastaviti mesec dana po završetku hemioterapije.
Početna doza je 150 IJ/kg, primenjena supkutano, tri puta nedeljno. Alternativo, Eprex se može dati u početnoj dozi od 450 IJ/kg supkutano, jednom nedeljno Ukoliko je vrednost hemoglobina porasla bar za 1 g/dL (0,62 mmoL/L) ili je broj retikulocita porastao za ≥ 40.000 ćelija/µL iznad bazalne vrednosti posle 4 nedelje lečenja, doza treba da ostane na nivou od 150 IJ/kg tri puta nedeljno i 450 IJ/kg, jednom nedeljno. Ukoliko porast vrednosti hemoglobina iznosi < 1 g/dL (< 0,62 mmoL/L), a broj retikulocita poraste za < 40.000 ćelija/µL iznad bazalne vrednosti, dozu treba povećati na 300 IJ/kg tri puta nedeljno. Ukoliko posle dodatnog perioda od 4 nedelje terapije sa 300 IJ/kg,vrednost hemoglobina poraste za ≥ 1 g/dL (0,62 mmoL/L) ili poraste broj retikulocita za ≥ 40.000 ćelija/µL, doza treba da ostane na nivou od 300 IJ/kg tri puta nedeljno. Medjutim, ukoliko je vrednost hemoglobina porasla za < 1 g/dL (< 0,62 mmoL/L), a broj retikulocita poraste za < 40.000 ćelija/µL iznad bazalne vrednosti, verovatno nema odgovora i lečenje treba prekinuti.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
8 od 23
Preporučeni režim doziranja prikazan je na sledećem dijagramu:
150IJ/kg 3 x nedeljno ili 450IJ/kg1 nedeljno
4 nedelje
4 nedelje
4 nedelje
Potrebno je pažljivo pratiti pacijente kako bi se obezbedilo da se adekvatna kontrola simptoma anemije postigne najnižim dozvoljenim dozama agensa stimulacije eritropoeze (ESA).
Podešavanje doze da bi se održala koncentracija hemoglobina između 10g/dL i 12g/dL:
Ukoliko je porast vrednosti hemoglobina veći od 2 g/dL (1,25 mmoL/L) mesečno, ili su vrednosti hemoglobina veće od 12 g/dL (>7,5mmoL/L), smanjiti dozu epoetina alfa za 25-50%. Ukoliko vrednosti hemoglobina pređu 13 g/dL (7,5 mmoL/L), terapiju treba obustaviti sve dok njegova vrednost ne padne ispod 12 g/dL (7,5 mmoL/L), nakon čega treba ponovo započeti primenu epoetina alfa u dozi 25% nižoj od prethodne.
Odrasli hirurški pacijenti u programu pripreme za autolognu donaciju:
Treba koristiti intravenski način primene. U toku donacije krvi epoetin alfa treba primeniti nakon završetka procedure donacije.
Blago anemični pacijenti (hematokrit od 33-39%) kojima je potreban predepozit od ≥ 4 jedinice krvi, treba da dobijaju epoetin alfa u dozi od 600 IJ/kg, dva puta nedeljno tokom 3 nedelje pre operacije. Primenom ovog režima, bilo je moguće povući ≥ 4 jedinice krvi od 81% pacijenata lečenih epoetinom alfa u poredjenju sa 37%
150IJ/kg 3 x nedeljnoili450IJ/kg1 nedeljno
Porast br. Retikulocita >40000/ml ili porast Hb > 1g/dl
Porast br.retikulocita < 40000/ml Iliporast Hb
< 1g/dl
300IJ/kg 3 x nedeljno
Porast br. Retikulocita >40000/ml ili porast Hb> 1g/dl
Porast br.retikulocita < 40000/ml Iliporast Hb
< 1g/dl
Prekid terapije
Ciljna koncentracija Hb 12g/dl

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
9 od 23
pacijenata tretiranih placebom. Terapija epoetinom alfa smanjuje rizik od izlaganja homolognoj krvi za 50% u poređenju sa pacijentima koji nisu primali epoetin alfa.
Svi pacijenti koji se leče epoetinom alfa treba da prime adekvatnu nadoknadu gvožđa (npr. 200 mg elementalnog gvožđa dnevno, oralno) tokom celokupne terapije epoetinom alfa. Sa nadoknadom gvoždja treba započeti što je pre moguće, čak nekoliko nedelja pre nego što se započne sa stvaranjem autolognog predepozita, kako bi se napravile velike zalihe gvoždja pre početka terapije epoetinom alfa.
Odrasli pacijenti koji se pripremaju za veliku elektivnu ortopedsku operaciju:
Treba koristiti supkutani način primene.
Preporučen režim doziranja iznosi 600 IJ/kg epoetina alfa, jednom nedeljno tokom tri nedelje (21, 14. i 7. dana) pre operacije i na dan operacije. U slučaju da postoji medicinska potreba da se skrati period pre operacija na manje od tri nedelje, treba davati 300 IJ/kg epoetina alfa dnevno tokom 10 uzastopnih dana pre operacije, na dan operacije i tokom četiri dana neposredno posle operacije. Ukoliko se hematološkom analizom u preoperativnom periodu ustanovi da je nivo hemoglobina dostigao vrednost od 15 g/dL ili više, treba prestati sa primenom epoetina alfa i sledeću dozu ne treba primeniti.
Treba voditi računa da na početku lečenja pacijenti nemaju deficit gvoždja. Svi pacijenti koji se leče epoetinom alfa treba da prime adekvatnu nadoknadu gvoždja (npr. 200 mg elementalnog gvoždja dnevno, oralno) tokom terapije epoetinom alfa. Ukoliko je moguće, sa nadoknadom gvoždja treba započeti pre početka lečenja epoetinom alfa, da bi se napravile adekvatne zalihe gvoždja.
4.3. Kontraindikacije
Pacijenti kod kojih se javi aplazija crvenih krvnih zrnaca (Pure Red Cell Aplasia - PRCA) posle lečenja nekim od eritropoetina, ne smeju primiti Eprex ili neki drugi eritropoetin (videti odeljak 4.4 – Aplazija crvenih krvnih zrnaca).
Nekontrolisana hipertenzija.
Sve kontraindikacije povezane sa programima predoniranja autologne krvi treba poštovati kod pacijenata koji se leče epoetinom alfa.
Poznata preosetljivost na aktivnu supstancu ili bilo koju pomoćnu supstancu ovog leka.
Korišćenje epoetina alfa kod pacijenata koji se pripremaju za veliku elektivnu ortopedsku operaciju, a koji ne učestvuju u programu predoniranja autologne krvi, kontraindikovano je kod pacijenata sa teškim koronarnim, perifernim arterijskim, karotidnim ili cerebrovaskularnim oboljenjem, uključujući pacijente sa nedavno preležanim infarktom miokarda ili cerebrovaskularnim insultom.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
10 od 23
Hirurški pacijenti koji iz bilo kog razloga ne mogu da primaju adekvatnu antitrombotičku profilaksu.
4.4. Posebna upozorenja i mere opreza pri upotrebi leka
Opšta upozorenja i mere opreza
Kod svih pacijenata koji primaju epoetin alfa, krvni pritisak treba brižljivo pratiti i regulisati prema potrebi. Epoetin alfa treba primenjivati oprezno u slučaju postojanja nelečene, neadekvatno lečene ili loše regulisane hipertenzije. Može da bude potrebno korigovanje antihipertenzivne terapije. Ukoliko krvni pritisak ne može da se kontroliše, terapiju epoetinom alfa treba prekinuti.
Epoetin alfa takodje treba primenjivati oprezno u slučaju postojanja epilepsije i hronične insuficijencije jetre.
Koncentracije hemoglobina kod pacijenata sa hroničnom bubrežnom insuficijencijom i pacijenata sa malignitetom treba redovno pratiti sve do postizanja stabilnih vrednosti, nakon čega treba nastaviti sa periodičnim praćenjem vrednosti hemoglobina.
Vrednosti hemoglobina treba pratiti kod svih pacijenata, zbog mogućeg povećanog rizika od tromboembolijskih poremećaja i fatalnog ishoda, ukoliko se terapija primenjuje kod pacijenata sa vrednostima hemoglobina višim od ciljnih za određenu terapijsku indikaciju.
Tokom lečenja epoetinom alfa, može doći do umerenog dozno-zavisnog porasta broja trombocita u okviru normalnog opsega. Broj trombocita se vraća na prethodni nivo tokom nastavka terapije. Takođe, primećena je i trombocitemija iznad normalnih vrednosti. Preporučuje se da se broj trombocita redovno prati tokom prvih 8 nedelja terapije.
Svi drugi uzroci anemije (manjak gvoždja, hemoliza, gubitak krvi, manjak vitamina B12 ili folata) treba da se razmotre i leče pre započinjanja terapije epoetinom alfa. U većini slučajeva, vrednosti feritina u serumu padaju simultano sa porastom volumena krvnih ćelija bez tečne plazme. Kako bi se obezbedio optimalan odgovor na epoetin alfa, treba obezbediti adekvatne zalihe gvoždja:
nadoknada gvoždja, npr. 200-300 mg/dan oralno (100-200 mg/dan kod dece) preporučuje se kod pacijenata sa hroničnom insuficijencijom bubrega koji imaju nivoe feritina u serumu ispod 100 ng/mL.
oralna supstitucija gvoždja od 200-300 mg/dan preporučuje se kod svih pacijenata sa malignim oboljenjima kod kojih je saturacija transferinom ispod 20%.
Sve navedene, dodatne faktore anemije treba takođe pažljivo razmotriti prilikom donošenja odluke o povećanju doze epoetina alfa kod pacijenata sa malignitetom.
Nastanak ili pogoršanje porfirije su se veoma retko ispoljavali kod pacijenata koji su primali epoetin alfa. Epoetin alfa treba primenjivati s oprezom kod pacijenata obolelih od porfirije.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
11 od 23
U cilju boljeg praćenja agenasa stimulacije eritropoeze (Erythropoiesis-stimulating agents - ESAs), zaštićeni naziv propisanog ESA treba da bude jasno naznačen u kartonu pacijenta.
Promena terapije pacijenta sa jednog, na drugi ESA, može se izvršiti samo uz odgovarajući nadzor lekara specijaliste.
Aplazija crvenih krvnih zrnaca
Aplazija crvenih krvnih zrnaca (Pure Red Cell Aplasia - PRCA) prouzrokovana stvaranjem antitela je veoma retko prijavljivana posle nekoliko meseci do nekoliko godina supkutane primene epoetina. Takođe su prijavljeni slučajevi kod pacijenata sa hepatitisom C koji su lečeni interferonom, ribavirinom i ESAs. Lek EPREX nije odobren za lečenje anemije udružene sa hepatitisom C.
Kod pacijenata kod kojih se pojavi iznenadan nedostatak efikasnosti koji se definiše padom nivoa hemoglobina (1 do 2 g/dL mesečno) uz povećanu potrebu za transfuzijama, treba proveriti broj retikulocita i treba bi istražiti tipične uzroke odsustva odgovora (npr. deficit gvoždja, folata ili vitamina B12, intoksikacija aluminijumom, infekcija ili zapaljenje, gubitak krvi i hemoliza).
Paradoksalni pad hemoglobina i razvoj teške anemije udružene sa niskim brojem retikulocita zahteva brz prekid terapije lekom Eprex i ispitivanje na anti-eritropoetinska antitela.Treba, takođe, razmotriti biopsiju koštane srži radi postavljanja dijagnoze PRCA.
Nikakvu drugu eritropoetinsku terapiju ne treba započinjati zbog rizika od ukrštene reakcije.
Pacijenti sa hroničnom insuficijencijom bubrega
Kod pacijenata sa hroničnom insuficijencijom bubrega, porast hemoglobina treba da iznosi približno 1 g/dL(0,62 mmoL/L) mesečno, i ne treba da predje 2 g/dL (1,25 mmoL/L) mesečno, kako bi se na minimum sveli rizici od povećanja hipertenzije.
Kod pacijenata sa hroničnom bubrežnom insuficijencijom koncentracija hemoglobina ne sme preći preporučenu gornju granicu ciljne vrednosti hemoglobina, datu u delu 4.2 – Doziranje i način primene. U kliničkim ispitivanjima zapažen je povećani rizik od smrti i ozbiljnih kardiovaskularnih događaja prilikom primene ESA u cilju dostizanja vrednosti hemoglobina većih od 12 g/dL (7,5mmoL/L).
Kontrolisanim kliničkim ispitivanjima nije pokazana značajnija prednost koja bi se mogla dovesti u vezu sa primenom epoetina u cilju postizanja vrednosti koncentracije hemoglobina veće od one koja je neophodna za kontrolu simptoma anemije i izbegavanje transfuzije krvi.
Pacijente sa hroničnom insuficijencijom bubrega na terapiji lekom Eprex primenjenim supkutano, treba redovno pratiti u pogledu gubitka efikasnosti, koji se definiše kao odsustvo ili smanjenje odgovora na terapiju lekom Eprex kod pacijenata koji su prethodno reagovali na takvu terapiju. Ovo karakteriše stalan pad nivoa
hemoglobina uprkos povećanju doze leka Eprex .

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
12 od 23
Neki pacijenti sa produženim intervalima doziranja epoetinom alfa (više od jednom nedeljno) možda neće dostići adekvatne vrednosti hemoglobina (videti deo 5.1) i mogu zahtevati povećanje doze epoetina alfa. Vrednosti hemoglobina treba redovno pratiti.
Tromboze šanta su se javile kod pacijenata koji su na hemodijalizi, naročito kod onih koji imaju tendenciju za hipotenziju ili kod kojih su se javljale komplikacije arteriovenske fistule ( npr stenoze, aneurizme i sl.). Ovim pacijentima se preporučuje rana revizija šanta i tromboembolijska profilaksa davanjem acetilsalicilne kiseline.
Hiperkalijemija se javila u izolovanim slučajevima čija uzročnost još nije utvrđena. Potrebno je kontrolisati serumske nivoe elektrolita kod pacijenata sa hroničnom renalnom insuficijencijom. Ukoliko je otkriven povišen nivo koncentracije kalijuma u serumu, pored adekvatne terapije hiperkalijemije potrebno je razmotriti dalju primenu epoetina alfa dok se ne koriguje nivo serumskog kalijuma.
Povećanje doze heparina tokom hemodijalize je često potrebno tokom trajanja terapije epoetinom alfa, kao rezultat povećanog volumena krvnih ćelija bez plazme. Može doći do okluzije sistema za dijalizu ako heparinizacija nije optimalna.
Na osnovu raspoloživih podataka, korekcija anemije primenom epoetina alfa kod odraslih pacijenata sa hroničnom insuficijencijom bubrega koji se još uvek ne podvrgavaju dijalizi, ne ubrzava stopu napredovanja insuficijencije bubrega.
Pacijenti sa anemijom indukovanom hemoterapijom
Epoetini su faktori rasta koji prvenstveno stimulišu proizvodnju crvenih krvnih zrnaca. Eritropoetinski receptori mogu da se pojave na površini različitih ćelija tumora. Kao i kod svih faktora rasta, postoji bojazan da bi eritropoetini mogli da stimulišu rast bilo koje vrste tumora. U nekoliko kontrolisanih kliničkih ispitivanja, epoetini nisu pokazali produženje preživljavanja ili smanjenje rizika od progresije tumora kod pacijenata sa anemijom koja je posledica malignog oboljenja.
U kontrolisanim kliničkom ispitivanjima sa lekom Eprex i drugim ESA pokazana je: smanjena lokalna kontrola pokretljivosti kod pacijenata sa uznapredovalim malignim tumorom glave i
vrata koji primaju radioterapiju, kada je ciljna vrednost hemoglobina veća od 14 g/dL (8.7 mmoL/L),
skraćeno ukupno preživljavanje i povećana smrtnost, povezani sa progresijom bolesti u četvrtom mesecu kod pacijenata sa metastaskim tumorom dojke koji primaju hemioterapiju, kada je ciljna vrednost hemoglobina 12 – 14 g/dL (7,5-8,7mmoL/L),
povećan rizik od smrti kada je ciljna vrednost hemoglobina 12g/dL (7,5mmoL/L) kod pacijenata sa aktivnim malignim oboljenjem koji ne primaju hemioterapiju niti radioterapiju. Primena ESA nije indikovana kod ove grupe pacijenata.
U odnosu na gore navedeno, u određenim kliničkim situacijama transfuzija krvi bi bila prihvatljivija terapija za lečenje anemije kod pacijenata sa malignim oboljenjem. Odluku o davanju rekombinantnog eritropoetina treba

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
13 od 23
zasnovati na osnovu odnosa potencijalne koristi od potencijalnog rizika za svakog pacijenta individualno. Faktori koji bi trebalo da utiču na tu odluku uključuju tip tumora i stadijum; stepen anemije, očekivano preživljavanje; sredinu u kojoj je pacijent lečen i karakteristike pacijenta, (videti deo 5.1).
Kod pacijenata sa malignitetom koji primaju hemioterapiju, treba uzeti u obzir period kašnjenja od 2-3 nedeljeizmedju primene ESAi pojave eritropoetinom-indukovanih crvenih krvnih zrnaca, prilikom procene toga da li je terapija epoetinom alfa odgovarajuća (pacijenti izloženi riziku od transfuzije).
Budući da je zapažena povećana incidenca trombotičkih vaskularnih epizoda (TVE), kod pacijenata sa malignitetom koji su na terapiji agensima stimulacije eritropoeze (videti deo 4.8 - Neželjena dejstva), ovaj rizik treba pažljivo odmeriti u odnosu na korist koja će se dobiti lečenjem epoetinom alfa, posebno kod pacijenata sa malignitetom, koji imaju povećan rizik od trombotičkih vaskularnih epizoda, na primer gojaznost, i kod pacijenata sa prethodnom istorijom TVE (npr. duboka venska tromboza ili plućna embolija). Kliničko ispitivanje (BEST) dizajnirano je sa ciljem da se ispita da li terapija epoetinom alfa, koja prevazilazi terapijske limite korekcije anemije, kod žena sa metastatskim tumorima dojke, može poboljšati odgovor na terapiju. U ovom kliničkom ispitivanju incidenca fatalnih tromboembolijskih poremećaja je bila veća kod pacijenata koji su primali epoetin alfa od pacijenata koji su primali placebo.
Hirurški pacijenti u programu pripreme za autolognu predonaciju
Sva upozorenja i posebne mere opreza povezane sa programom pripreme za autolognu predonaciju, naročito rutinsku zamenu volumena, treba poštovati.
Pacijenti koji se pripremaju za velike elektivne ortopedske operacije
Kod pacijenata koji se pripremaju za velike elektivne ortopedske operacije, uzrok anemije bi trebalo da se ustanovi i leči, ukoliko je moguće, pre početka lečenja epoetinom alfa. Trombotičke epizode mogu da predstavljaju rizik u ovoj populaciji i ovaj rizk bi trebalo prižljivo odmeriti u odnosu na korist koja će se dobiti lečenjem ove grupe pacijenata.
Pacijenti koji se pripremaju za velike elektivne ortopedske operacije treba da prime odgovarajuću antitrombotičku profilaksu, jer trombotičke i vaskularne epizode mogu da se pojave kod hirurških pacijenata, naročito kod onih sa osnovnim kardiovaskularnim oboljenjem. Pored toga, posebne mere opreza treba primeniti kod pacijenata sa predispozicijom za razvoj duboke venske tromboze. Pored toga, kod pacijenata sa bazalnim hemoglobinom > 13 g/dL, mogućnost da lečenje epoetinom alfa bude povezano sa povećanim rizikom od postoperativnih trombotičkih/vaskularnih epizoda ne može da se isključi. Stoga, ova terapija ne treba da se primenjuje kod pacijenata sa bazalnim hemoglobinom > 13 g/dL.
Lek Eprex sadži manje od 1mmoL natrijuma (23mg) po dozi i smatra se da je ''bez natrijuma''.
4.5. Interakcije sa drugim lekovima i druge vrste interakcija
Ne postoje dokazi koji ukazuju da terapija epoetinom alfa menja metabolizam drugih lekova. Medjutim, s obzirom da se ciklosporin vezuje za eritrocite postoji potencijal interakcije lekova. Ukoliko se epoetin alfa daje

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
14 od 23
istovremeno sa ciklosporinom, treba pratiti nivoe ciklosporina u krvi i podesiti dozu ciklosporina prema porastu hematokrita.
Ne postoje dokazi koji ukazuju na interakciju izmedju epoetina alfa i G-CSF ili GM-CSF u pogledu hematološke diferencijacije ili proliferacije uzoraka biopsije tumora in vitro.
4.6. Primena u periodu trudnoće i dojenja
Ne postoje adekvatne i dobro kontrolisane studije kod trudnica. U ispitivanjima na životinjama utvrđena je reproduktivna toksičnost (Videti odeljak 5.3), tako da:
Kod pacijentkinja sa hroničnom insuficijencijom bubrega, epoetin alfa treba upotrebljavati tokom trudnoće samo ako je potencijalna korist za pacijentkinju veća od potencijalnog rizika za fetus.
Pacijentkinje koje treba hirurški zbrinuti koje su trudne ili koje doje, a koje učestvuju u programu kolekcije autologne krvi, upotreba epoetina alfa se ne preporučuje.
Nije poznato da li se egzogeni epoetin alfa izlučuje u humano mleko. Epoetin alfa treba sa oprezom primenjivati kod dojilja. Za odluku o nastavku/prekidu dojenja ili nastavku/prekidu terapije sa epoetinom alfa treba uzeti u obzir korist od dojenja za dete u odnosu na korist epoetina alfa na lečenje majke.
4.7. Uticaj na psihofizičke sposobnosti prilikom upravljanja motornim vozilom i rukovanja mašinama
Nema uticaja.
4.8. Neželjena dejstva
Opšta
Kod pacijenata sa malignim oboljenjem ili pacijenata sa hroničnom insuficijencijom bubrega najčešće neželjene reakcije tokom lečenja epoetinom alfa su dozno-zavisan porast krvnog pritiska ili pogoršanje postojeće hipertenzije. Preporučuje se praćenje krvnog pritiska i to naročito na početku terapije (videti deo 4.4).Ostale česte neželjene reakcije koje su prijavljene u kliničkim ispitivanjima epoetina alfa su duboka venska tromboza, plućna embolija, epileptički napadi, dijareja, nauzeja, glavobollja, simptomi ''slični gripu'', pireksija, osip, i povraćanje. Simptomi slični gripu, koji uključuju glavobolju, artralgiju, mijalgiju, i pireksiju mogu se javiti naročito na početku lečenja.Učestalost varira u zavisnosti od indikacije (videti tabelu).
Kongestija respiratornog trakta koja uključuje slučajeve kongestije gornjeg respiratornog trakta, nazalna kongestija i nazofaringitis, prijavljeni su u ispitivanjima produženog intervala doziranja kod odraslih pacijenata sa renalnom insuficijencijom koji još uvek nisu na dijalizi.
Ozbiljne neželjene reakcije uključuju vensku i arterijsku trombozu i embolizam (uključujući i smrtne ishode) kao što su duboka venska tromboza, plućna embolija, arterijska tromboza (uključujući infarkt miokarda i

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
15 od 23
ishemiju miokarda), tromboza retine i tromboza šanta (uključujući dijalizni sistem). Dodatno, u kliničkim ispitivanjima epoetina alfa, prijavljeni su i cerebrovaskularni poremećaji (uključujući i cerebralni infarkt i cerebralnu hemoragiju) i prolazni ishemijski napadi .
Prijavljene su aneurizme.
Prijavljene su hipersenzitivne reakcije, uključujući slučajeve osipa, urtikarije, anafilaktičke reakcije i angioneurotskog edema.
Tokom lečenja epoetinom alfa kod pacijenata sa prethodno normalnim ili nižim krvnim pritiskom javile su se hipertenzivne krize sa encefalopatijom i epileptičkim napadima koje su zahtevale neodložnu pažnju lekara i intezivnu medicinsku negu. Posebno treba obratiti pažnju na iznenadne glavobolje koje liče na migrene kao mogući signal upozorenja.
Aplazija crvenih krvnih zrnaca izazvana stvaranjem antitela je veoma retko prijavljena kod <1/10000 slučajeva
po pacijentu godišnje, nakon nekoliko meseci ili godina lečenja lekom Eprex (videti odeljak 4.4).
U dvostruko, slepo placebo kontrolisanom kliničkom ispitivanju ispitivana je opšta bezbednost leka Eprex kod 142 ispitanika sa hroničnom insuficijencijom bubrega i 765 ispitanika sa malignim oboljenjem. Neželjene reakcije su bile prijavljene kod >0,2 ispitanika ovog kliničkog ispitivanja, dodatnih kliničkih ispitivanja i postmarketinškog iskustva koji su lečeni lekom Eprex upisane su u sledeću tabelu.
Učestalost neželjenih reakcija se definiše kao: veoma česte (>1/10); česte (>1/100, <1/10); povremene(>1/1000, <1/100); retke (>1/10000, <1/1000 ); veoma retke (<1/10000).Učestalost je definisana kao nepoznata ukoliko neželjeno dejstvo nije prijavljeno u placebo kontrolisanim dvostruko slepim registracionim kliničkim ispitivanjima ili kada se učestalost ne može dobiti iz dostupnih podataka.
U okviru svake grupe učestalosti, neželjene reakcije su prikazane u opadajućem redosledu sa aspekta ozbiljnosti.
Sistem organa Učestalost Neželjeno dejstvo leka
Poremećaji na nivou krvi i limfnog sistema
Povremeno Trombocitemija (pacijenti sa malignim oboljenjem)
Nepoznata učestalost Aplazija crvenih krvnih zrnaca (PRCA) prouzrokovana stvaranjem antitela na eritropoetin1
Trombocitemija (pacijenti sa hroničnom insuficijencijom bubrega )
Infekcije i infestacije Nepoznata učestalost Anafilaktička reakcijaHipersenzitivnost
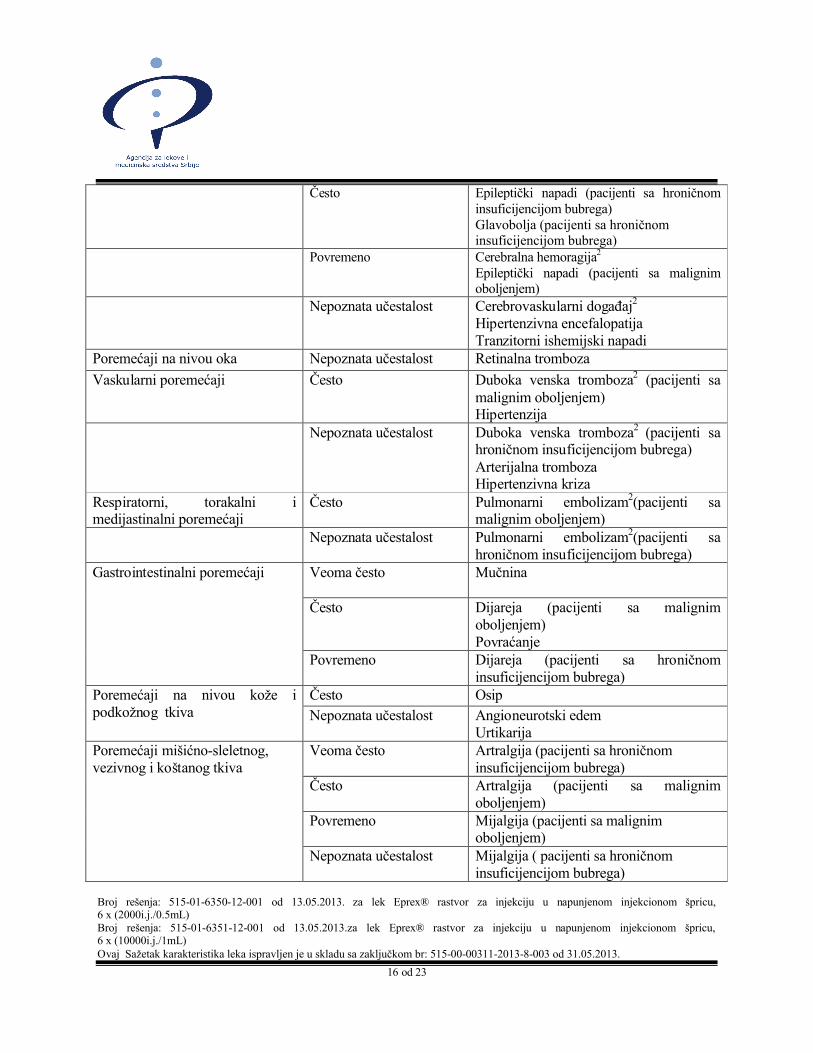
Poremećaj nervnog sistema Veoma često Glavobolja (pacijenti sa malignim oboljenjem)
Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
16 od 23
Često Epileptički napadi (pacijenti sa hroničnom insuficijencijom bubrega)Glavobolja (pacijenti sa hroničnom insuficijencijom bubrega)
Povremeno Cerebralna hemoragija2
Epileptički napadi (pacijenti sa malignim oboljenjem)
Nepoznata učestalost Cerebrovaskularni događaj2
Hipertenzivna encefalopatijaTranzitorni ishemijski napadi
Poremećaji na nivou oka Nepoznata učestalost Retinalna tromboza
Vaskularni poremećaji Često Duboka venska tromboza2 (pacijenti sa malignim oboljenjem)Hipertenzija
Nepoznata učestalost Duboka venska tromboza2 (pacijenti sa hroničnom insuficijencijom bubrega)Arterijalna trombozaHipertenzivna kriza
Respiratorni, torakalni i medijastinalni poremećaji
Često Pulmonarni embolizam2(pacijenti sa malignim oboljenjem)
Nepoznata učestalost Pulmonarni embolizam2(pacijenti sa hroničnom insuficijencijom bubrega)
Gastrointestinalni poremećaji Veoma često Mučnina
Često Dijareja (pacijenti sa malignim oboljenjem) Povraćanje
Povremeno Dijareja (pacijenti sa hroničnom insuficijencijom bubrega)
Poremećaji na nivou kože i podkožnog tkiva
Često Osip
Nepoznata učestalost Angioneurotski edemUrtikarija
Poremećaji mišićno-sleletnog, vezivnog i koštanog tkiva
Veoma često Artralgija (pacijenti sa hroničnom insuficijencijom bubrega)
Često Artralgija (pacijenti sa malignim oboljenjem)
Povremeno Mijalgija (pacijenti sa malignim oboljenjem)
Nepoznata učestalost Mijalgija ( pacijenti sa hroničnom insuficijencijom bubrega)

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
17 od 23
Kongenitalni i nasledni/genetskiporemećaji
Nepoznata učestalost Porfirija
Opšti poremećaji i reakcije na mestu primene
Veoma često Pireksija (pacijenti sa malignim oboljenjem)Simptomi slični gripu (pacijenti sa hroničnom insuficijencijom bubrega)
Često Simptomi slični gripu (pacijenti sa malignim oboljenjem)
Nepoznata učestalost Neefikasnost lekaPeriferni edem Pireksija (pacijenti sa hroničnom insuficijencijom bubrega)Reakcija na mestu davanja injekcije
Laboratorijska ispitivanja Nepoznata učestalost Pozitivan nalaz antieritropoetin antitela1
Hirurške i medicinske procedure Često Tromboza šanta uključujući i opremu za dijalizu (pacijenti sa hroničnom insuficijencijom bubrega)
1 Učestalost nije moguće proceniti na osnovu kliničkih ispitivanja2.Uključujući slučajeve smrtnog ishoda
Pacijenti sa hroničnom insuficijencijom bubrega
Kod pacijenata koji imaju hroničnu renalnu insuficijenciju, nivo hemoglobina veći od 12 g/dL može biti udružen sa većim rizikom od kardiovaskularnih događaja, uključujući i smrt (videti deo 4.4).
Tromboza šanta se javila kod pacijenata koji su na hemodijalizi, naročito kod onih koji imaju tendenciju ka hipotenziji ili komplikacije arteriovenske fisule. (videti deo 4.4).
Pacijenti sa malignim oboljenjem
Povećana incidenca od tromboembolijskih poremećaja je prijavljena kod pacijenata sa malignim oboljenjem koji su primali agens stimulacije eritropoeze (ESAs), uključujući i epoetin alfa. (videti deo 4.4).
Pacijenti koji se pripremaju za velike elektivne ortopedske operacije
Kod pacijenata koji se pripremaju za veliku elektivnu ortopedsku operaciju, sa bazalnim hemoglobinom od 10 do 13 g/dL, incidenca trombotičkih/vaskularnih komplikacija (od kojih je većina bila duboka venska tromboza), u celokupnoj populaciji pacijenata u kliničkim ispitivanjima, bila je slična u različitim grupama pacijenata tretiranih različitim dozama epoetina alfa i u grupi koja je primala placebo, mada je kliničko iskustvo ograničeno.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
18 od 23
Osim toga, kod pacijenata sa bazalnim koncentracijama hemoglobinom > 13 g/dL, ne može se isključiti mogućnost da se lečenje epoetinom alfa može povezati sa povećanim rizikom od postoperativnih trombotičkih/vaskularnih poremećaja.
4.9. Predoziranje
Terapijska granica epoetina alfa je veoma široka. Prekomerno doziranje epoetina alfa može da proizvede efekte koji predstavljaju produžetak farmakoloških efekata ovog hormona. Ukoliko dođe do preterano visokih nivoa hemoglobina može se izvršiti flebotomija. Ukoliko je neophodno, treba pružiti i dodatnu suportivnu negu.
5. FARMAKOLOŠKI PODACI
5.1. Farmakodinamski podaci
Farmakoterapijska grupa: ANTIANEMICI, STIMULATORI ERITROPOEZE
ATC kod: B03XA01
Eritropoetin je glikoprotein koji stimuliše stvaranje eritrocita iz prekursorskih ćelija, kao faktor koji stimuliše mitozu i diferencirajući hormon.
Molekulska težina eritropoetina iznosi 32.000 do 40.000 daltona. Na proteinsku frakciju molekula otpada oko 58% i ona se sastoji od 165 amino kiselina. Četiri ugljeno-hidratna lanca su vezana za protein preko tri N-glikozidne veze i jedne O-glikozidne veze. Epoetin alfa dobijen genskom tehnologijom je identičan po svom sastavu amino-kiselina i ugljenih-hidrata sa endogenim humanim eritropoetinom koji je izolovan iz urina anemičnih pacijenata.
Epoetin alfa ima najveći mogući stepen čistoće prema sadašnjim standardima. Pri koncentracijama aktivnog sastojka koje se primenjuju kod ljudi, ne mogu se naći rezidue ćelijske linije koje se koristi za njegovu proizvodnju.
Biološka efikasnost epoetina alfa je pokazana kod različitih životinjskih modela in vivo (normalni i anemični pacovi, policitemični miševi). Posle primene epoetina alfa, broj eritrocita, vrednosti Hb i broj retikulocita rastu, kao i povećano vezivanje 59Fe.
Otkriveno je povećano vezivanje 3H-timidina u eritroidnim ćelijama slezine in vitro (ćelijska kultura slezine miša) posle inkubacije sa epoetinom alfa.
Primenom kulture ćelija humane koštane srži može se pokazati da epoetin alfa specifično stimuliše eritropoezu i da ne utiče na stvaranje leukocita (leukopoezu). Citotoksična dejstva epoetina alfa na ćelije koštane srži nisu otkrivena.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
19 od 23
Odrasli pacijenti sa renalnom insuficijencijom koji jos uvek nisu na dijalizi
U dva ispitivanja produženog intervala doziranja lekom Eprex ( 3 puta nedeljno, jednom nedeljno, jednom u toku 2 nedelje i jednom u toku 4 nedelje) neki pacijenti sa dužim intervalima doziranja nisu održavali adekvatne vrednosti hemoglobina i dostigli su protokolom definisan kriterijum povlačenja (0% u grupi koja je primala Eprex jednom nedeljno, 3,7% u grupi koja je primala Eprex jednom u dve nedelje i 3,3% u grupi koja je primala Eprex jednom u četiri nedelje)
Terapija kod pacijenata sa anemijom koja je izazvana hemioterapijom
Grupa od 721 pacijenta sa karcinomom koji su primali ne-platinsku hemioterapiju bila je uključena u tri placebom-kontrolisana klinička ispitivanja, 389 pacijenata sa hematološkim malignitetima (221 multipli mijelom, 144 non-Hočkinov limfom, i 24 ostali hematološki maligniteti) i 332 sa solidnim tumorima (172 dojke, 64 ginekološki, 23 pluća, 22 prostate, 21 gastro-intestinalni i 30 ostale vrste tumora). U dva velika, otvorena klinička ispitivanja, bilo je uključeno 2697 pacijenata sa karcinomom koji su primali ne-platinsku hemioterapiju, 1895 sa solidnim tumorima (683 dojke, 260 pluća, 174 ginekološki, 300 gastro-intestinalni, i 478 ostale vrste tumora) i 802 sa hematološkim malignitetima.
U jednom prospektivnom, randomizovanom, dvostruko-slepom, placebo-kontrolisanom ispitivanju sprovedenom kod 375 anemičnih pacijenata sa različitim ne-mijeloidnim malignitetima koji su primali ne-platinsku hemioterapiju, zabeleženo je značajno smanjenje posledica vezanih za anemiju (npr. zamor, smanjena energija i smanjenje aktivnosti), mereno sledećim instrumentima i skalama: FACT-An opšta skala, FACT-An skala zamora i CLAS. U druga dva manja, randomizovana, placebo-kontrolisana ispitivanja nije pokazano značajno poboljšanje u pogledu parametara kvaliteta života na skali EORTC-QLQ-C30, odnosno skali CLAS.
Eritropoetin je faktor rasta koji prvenstveno stimuliše proizvodnju crvenih krvnih zrnaca. Eritropoetinski receptori mogu da se pojave na površini čitavog niza ćelija tumora.
Preživljavanje i napredovanje tumora ispitivano je u pet velikih kontrolisanih kliničkih ispitivanja koja su ukupno obuhvatala 2833 pacijenta, od kojih su četiri klinička ispitivanja bila dvostruko slepa, placebo kontrolisana i jedno otvoreno kliničko ispitivanje. U klinička ispitivanja uključivani su ili pacijenti koji su bili na hemoterapiji (dva ispitivanja) ili pacijenti kod kojih ESA nisu indikovani: anemija kod pacijenata sa tumorom koji su na hemoterapiji, i pacijenti sa kancerom glave i vrata koji nisu na radioterapiji. Ciljna koncentracija hemoglobina u dva ispitivanja bila je >13g/dL; a u tri preostala ispitivanja 12-14g/dL. U otvorenom kliničkom ispitivanju nije bilo razlike u ukupnom preživljavanju između pacijenata na terapiji rekombinantnim humanim eritropoetinima i u kontrolnoj grupi. U četiri placebo kontrolisana ispitivanja granične vrednosti ukupnog preživljavanja kretale su se između 1,25 i 2,47 u korist kontrolne grupe. Ova ispitivanja su pokazala konzistentno, neobjašnjivo statistički značajno povećanje smrtnosti, kod pacijenata sa anemijom uzrokovanom različitim čestim oblicima kancera koji su na terapiji rekombinantnim humanim eritropoetinima u odnosu na kontrolnu grupu. Ukupno preživljavanje u kliničkim ispitivanjima ne može se objasniti, na zadovoljavajući način, razlikama u incidenci od tromboza i sličnih komplikacija između grupa koje su primale rekombinantni humani eritropoetin i kontrolne grupe.

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
20 od 23
Izvršena je analiza podataka istraživanja koje je obuhvatilo više od 13 900 pacijenata obolelih od kancera (koji su podvrgnuti hemoterapiji, radioterapiji, hemoradioterapiji ili koji nisu bili na terapiji). Pacijenti su bili uključeni u 53 kontrolisane kliničke studije sa nekoliko epoetina. Meta analizom podataka o ukupnom preživljavanju procenjen je odnos rizika od 1,06 u korist kontrolne grupe (95% Cl:1,00; 1,12; 53 studije i 13 933 pacijenta) i za pacijente obolele od kancera koji su podvrgnuti hemoterapiji, odnos rizika od 1,04 (95% Cl:0,97; 1,11; 38 studija i 10 441 pacijent). Meta analize ukazuju na značajno povećanje relativnog rizika od tromboembolijskih događaja kod pacijenata obolelih od kancera koji su na terapiji rekombinantnim humanim eritropoetinom (Videti odeljak 4.4).
5.2. Farmakokinetički podaci
Intravenski način primene (i.v.)
Posle multiplih intravenskih doza poluvreme eliminacije epoetina alfa je približno 4 sata kod zdravih dobrovoljaca, a nešto duže kod pacijenata sa insuficijencijom bubrega, približno 5 sati. Kod dece poluvreme eliminacije je približno 6 sati.
Supkutani način primene (s.c.)
Koncentracije epoetina alfa u serumu posle supkutane injekcije su mnogo niže nego posle intravenske injekcije. Nivoi u serumu se polako povećavaju i dostižu maksimum izmedju 12 i 18 sati posle primene. Maksimalna vrednost je mnogo niža nego posle intravenske primene (približno 1/20 vrednosti).
Akumulacije nema i koncentracija je ista, kada se izmeri 24 sata posle prve injekcije ili 24 sata posle poslednje injekcije.
Poluvreme eliminacije se teško utvrdjuje kod supkutanog načina primene, a procenjuje se da iznosi oko 24 sata.
Biološka raspoloživost supkutano primenjenog epoetina alfa je mnogo niža nego posle intravenske primene, približno 20%.
5.3. Predklinički podaci o bezbednosti leka
U nekim pretkliničkim toksikološkim ispitivanjima na psima i pacovima, ali ne i na majmunima, terapija epoetinom alfa je bila povezana sa subkliničkom fibrozom koštane srži. (Fibroza koštane srži je poznata komplikacija hronične insuficijencije bubrega kod ljudi i može da bude povezana sa sekundarnim hiperparatireoidizmom ili nepoznatim faktorima. Incidenca fibroze koštane srži nije bila povećana u jednoj studiji kod pacijenata na hemodijalizi koji su lečeni epoetinom alfa tokom 3 godine u poredjenju sa odgovarajućom kontrolnom grupom pacijenata na dijalizi koji nisu lečeni epoetinom alfa.)
U studijama na životinjama, epoetin alfa dovodio do smanjenja fetalne mase, odložene osifikacije i povećanja fetalnog mortaliteta kada je primenjivan u nedeljnim dozama približno 20 puta većim od preporučenih nedeljnih

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
21 od 23
doza u humanoj populaciji. Ove promene su interpretirane kao sekundarna posledica smanjenja telesne mase majke.
Epoetin alfa nije doveo do promene u bakterijskoj i kulturi ćelija sisara prilikom testova mutagenosti i in vivo u toku mikronukleus testa na miševima.
Dugotrajne studije karcinogenosti nisu sprovođene. Postoje oprečni izveštaji u literature po pitanju uloge eritropoetina u tumorskoj proliferaciji. Ovi izveštaji su bazirani na in vitro nalazima na uzorcima humanih tumora, ali nije siguran klinički značaj ovog nalaza.
6. FARMACEUTSKI PODACI
6.1. Lista pomoćnih supstanci
Polisorbat 80GlicinVoda za injekcije
Pomoćne supstance sa poznatom aktivnošću ili efektom (prisutni u leku u koncentraciji <1mmoL):
Dinatrijum-fosfat dihidratNatrijum-dihidrogenfosfat, dihidratNatrijum-hlorid
6.2. Inkompatibilnost
Zbog nedostatka studija o kompatibilnosti, ovaj lek ne treba mešati sa drugim lekovima.
6.3. Rok upotrebe
18 meseci.
6.4. Posebne mere upozorenja pri čuvanju
Čuvati u frižideru (2° - 8°C). Lek treba čuvati u ovom temperaturnom opsegu sve do primene kod pacijenta.
Čuvati u originalnom pakovanju, zaštićeno od svetlosti.
Ne zamrzavati i ne mućkati.
Za ambulantnu primenu, lek Eprex se može izvaditi iz frižidera i čuvati na temperaturi do 25°C u periodnu od najviše 3 dana, jednokratno.
6.5. Priroda i sadržaj kontaktne ambalaže

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
22 od 23
0,5 ml (2000 IJ) ) rastvora za injekciju u napunjenom injekcionom špricu (staklo tipa I) sa klipom (teflonom obložena guma) i iglom sa zaštitnim poklopcem (guma sa polipropilenskom zaštitom) i zaštitnikom za iglu (polikarbonat) dodatim uz špric– pakovanje sa 6 napunjenih špriceva.
1 mL (10000 IJ) ) rastvora za injekciju u napunjenom injekcionom špricu (staklo tipa I) sa klipom (teflonom obložena guma) i iglom sa zaštitnim poklopcem (guma sa polipropilenskom zaštitom) i zaštitnikom za iglu (polikarbonat) dodatim uz špric– pakovanje sa 6 napunjenih špriceva.
6.6. Uputstvo za upotrebu i rukovanje i mere opreza pri odlaganju materijala koji treba odbaciti nakon primene leka
Ne primenjivati u intravenskoj infuziji i ne mešati sa drugim rastvorima lekova.
Pre upotrebe, ostaviti napunjeni špric leka Eprex da dostigne sobnu temperaturu. Za ovo je obično potrebno između 15 i 30 minuta.
Lek Eprex možete izvaditi iz frižidera i držati na sobnoj temperaturi (do 25 oC) u naredna tri dana. Ukoliko se napunjeni injekcioni špric izvadi iz frižidera i ostavi da dostigne sobnu temperaturu (do 25 oC) mora da se iskoristi ili uništi.
Lek ne treba koristiti:
- ako je sigurnosni žig oštećen;- ako je rastvor promenio boju ili ako se vide čestice u rastvoru- ako znate ili mislite da je lek slučajno bio zamrzavan.- ako je došlo do kvara na frižideru.
Iz jednog šprica uzeti samo jednu dozu leka Eprex. Neželjenu količinu tečnosti iz šprica odstranite pre uzimanja injekcije. Pogledajte deo 3. Kako se upotrebljava lek Eprex (Uputstvo za upotrebu leka EPREX) u Uputstvu za pacijenta.
Rastvor za injekciju u napunjenom injekcionom špricu (PROTECS) da bi se izbegle povrede od uboda iglom, nakon upotrebe. Uputstvo za lek uključuje kompletno uputstvo i rukovanje napunjenih injekcionih špriceva.
Neiskorišćeni lek ili otpad treba uništiti u skladu sa lokalnom regulativom.
7. NOSILAC DOZVOLE I PROIZVOĐAČ
Naosilac dozvole:Predstavništvo Janssen-Cilag KFT Beograd, Novi Beograd, Omladinskih brigada 88b.
Proizvođač:

Broj rešenja: 515-01-6350-12-001 od 13.05.2013. za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) Broj rešenja: 515-01-6351-12-001 od 13.05.2013.za lek Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL)Ovaj Sažetak karakteristika leka ispravljen je u skladu sa zaključkom br: 515-00-00311-2013-8-003 od 31.05.2013.
23 od 23
Cilag AG, Hochstrasse 201, Schaffhausen, Švajcarska
8. BROJ PRVE DOZVOLE I OBNOVE DOZVOLE
EprexBroj prve dozvole Broj obnove dozvole
Rastvor za injekciju u napunjenom injekcionom špricu:
6 x 2000 I.J./0,5 mL 03-6795/96
03-6792/96
437/2008/12
438/2008/126 x 10000 I.J/mL
Broj poslednje obnove dozvole za stavljanje u promet leka Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL) : 515-01-6350-12-001
Broj poslednje obnove dozvole za stavljanje u promet leka Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL): 515-01-6351-12-001 .
9. DATUM PRVE DOZVOLE I DATUM OBNOVE DOZVOLE
EprexDatum prve dozvole Datum obnove dozvole
Rastvor za injekciju u napunjenom injekcionom špricu:
6 x 2000 I.J./0,5 mL 27. 12.1996.
12.02.2008.
12.02.2008.6 x 10000 IJ/mL
Datum poslednje obnove dozvole za stavljanje u promet leka Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (2000i.j./0.5mL): 13.05.2013.
Datum poslednje obnove dozvole za stavljanje u promet leka Eprex® rastvor za injekciju u napunjenom injekcionom špricu, 6 x (10000i.j./1mL): 13.05.2013.
10. DATUM REVIZIJE TEKSTA
Mart 2013.





























