Embed Size (px)
Citation preview

CASO CLÍNICO
Natalia Solís – Francisco RodríguezRotación Segunda Infancia
2012

• A.C.V• Adolescente 13 años• Sexo femenino.• Antecedente de cuadro reciente (3 días
previos a cuadro actual) de diarrea y vómitos, que cedió espontáneamente.

• Acude a SUI el miércoles 24.10.2012 por un cuadro de 3 días de evolución, caracterizado por paresia de extremidades superiores (predominio a derecha), progresiva, y que luego compromete a EI izquierda, con imposibilidad de mantenerse de pie sin apoyo, de carácter intermitente.
• Inicialmente consultó en Hospital de Villarrica, el 23.10, donde fue evaluada por Neurólogo. En el examen físico destacó:– Hiporreflexia de EEII– Paresia de Extremidades.
• Fue derivada con Obs. Sd Guillain Barre

• Al Examen físico el 24.11, destaca:– Ex neurológico:
• Glasgow 15• Signos meníngeos (-)• Pares Craneanos Normales• Sensibilidad Conservada, simétrica.• ROT (+) • Babinski (-)• Disminucion de la fuerza muscular en cuádriceps de
ambas EEII. • Tono y Fuerza del resto del cuerpo normal.

• Exámenes de Laboratorio:– Hemograma y PCR (N)– Glucosa (N)– Urea, BUN (N)– Creatinina (N)– Transaminasas (N)– Bilirrubina Total y Directa (N)– ELP (N)– CK total y Mb (N)

Enfermedades Neuromusculares en
Pediatría:Aproximación Diagnóstica

Introducción
• Son causa frecuente de morbilidad• Son motivo de consulta a diferentes especialistas
infantiles• Tienen una amplia variedad de motivos de consulta y
síntomas iniciales inespecíficos, lo que a veces dificulta la aproximación diagnóstica
• Muchas de ellas son causa de discapacidad progresiva en el niño, generando un alto impacto a nivel individual, familiar y social
• Es fundamental, por ende, sospecharlas precozmente e iniciar acciones que lleven a un diagnóstico y manejo oportunos

Definición
• Grupo de enfermedades que afectan cualquiera de los componentes de la Unidad Motora, es decir, la unidad funcional constituida por:
1.- El cuerpo de la Motoneurona del Asta Anterior de la Médula Espinal 2.- Su axón (Nervio Periférico) 3.- Todas las fibras musculares inervadas por esta motoneurona

Clasificación
• 1.-Atrofias Neurogénicas: incluyen las enfermedades de la motoneurona y del nervio (neuropatías)
• 2.-Miopatías: patologías primarias del músculo sin alteraciones estructurales en nervio periférico
• 3.-Trastornos de la Unión Neuromuscular

Clasificación
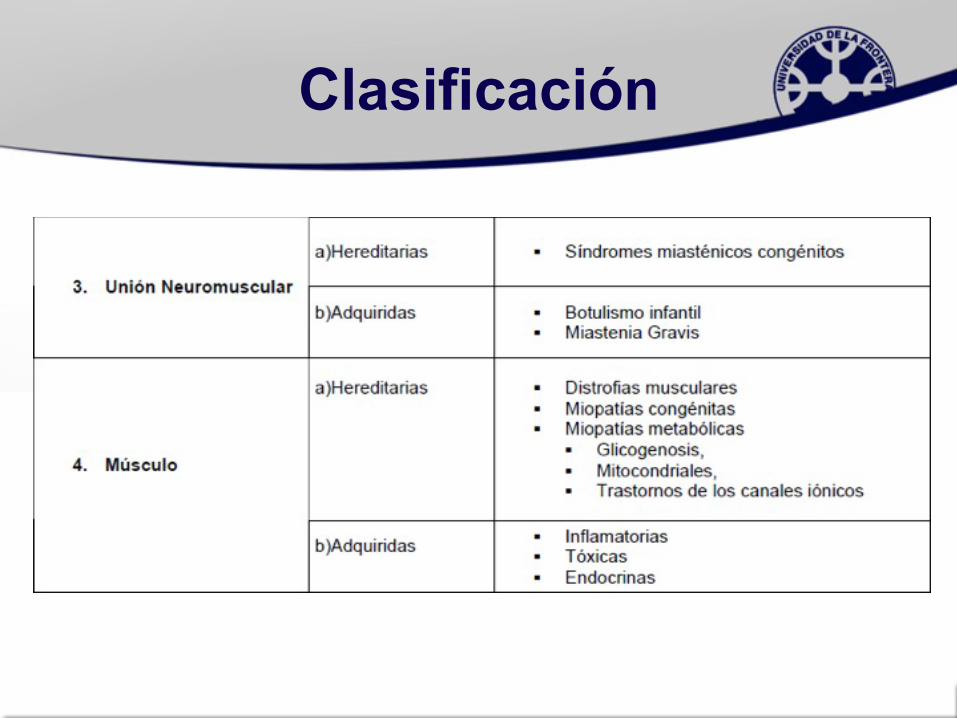
Clasificación

Etiología
• 1.- Hereditariasa) Autosómicas Dominantes (ej: Distrofia
Miotónica)b) Autosómicas Recesivas (ej: Distrofias de
Cinturas, Atrofias Espinales)c) Recesivas Ligadas al Sexo (ej: Distrofia
muscular de Duchenne)• 2.- Adquiridasa) Autoinmunes (ej: Miastenia Gravis,
Polimiositisb) Infecciosasc) Toxico-Medicamentosasd) Endocrino-Metabólicas

Presentación Clínica
• Depende principalmente de las características de la enfermedad específica y la edad de inicio
• Según el curso de la misma puede ser aguda o crónica
• Hay una amplia variedad de formas, la mayoría inespecíficas

Aproximación Diagnóstica
• 1.- Motivo de consulta, síntomas y signos al examen físico Diagnóstico Sindromático
• 2.- Establecer Diagnóstico Topográfico• 3.- Establecer Diagnóstico Etiológico• 4.- Considerar Diagnósticos Diferenciales

Anamnesis• 1.- Identificar el Motivo de Consulta (ej: debilidad, retraso
en el desarrollo psicomotor)• 2.- Identificar síntomas asociados (ej: Duchenne: retraso
del lenguaje, dificultades de aprendizaje)• 3.- Definir la edad de inicio de los síntomas y
correlacionar con la adquisición de hitos del DSM. Ej. 1: En DM de Duchenne 50% de los pacientes logran
caminar después de los 18 meses. Ej. 2: a) Debilidad de inicio desde el nacimiento: Distrofia
Miotónica Congénita, miopatías congénitas, distrofias musculares congénitas y menos frecuentemente a una neuropatía congénita por hipomielinización o algunas formas de enfermedades de motoneurona.
b) Debilidad de inicio en el preescolar orienta a algunas distrofias musculares, atrofias musculares espinales tipo 2 o 3 o algunos cuadros adquiridos, como las miopatías o polineuropatías inflamatorias.
c) Debilidad de inicio en el escolar: polineuropatías.

Anamnesis
• 4.- Establecer el Perfil Temporal del Cuadro: a) Agudo: de días a semanas b) Subagudo: de semanas a pocos meses c) Crónico: años a) y b) orientan a causas adquiridas• 5.- Definir Evolución de los Síntomas: a) Estacionaria (síntomas estables) b) Progresiva: distrofias musculares progresivas c) Fluctuante: unión NM, miopatías metabólicas
o canalopatías • 6.- Indagar Antecedentes del Embarazo:
patología materna, disfunción de la UFP, distocia, trauma, etc

Anamnesis• 7.- Indagar Antecedentes Neonatales: hipotonía, dificultades
respiratorias o deglutorias• 8.- Investigar dirigidamente exposición a tóxicos (alcohol,
plomo, arsénico, N-hexano) o fármacos que pueden producir una polineuropatía sensitivo motora adquirida. Esto en cuadros de debilidad de inicio agudo o subagudo en el escolar o adolescente.
• 9.- Evaluar Enfermedades Asociadas:• a)Diabetes: causa de neuropatía en cuadros de algunos años
de evolución y se asocia a cuadros neuromusculares con compromiso multisistémico como la Distrofia Miotónica de Steinert y las citopatías mitocondriales
• b)Hipoacusia: neuropatías hereditarias, miopatías mitocondriales, enfermedad de Refsum, etc.
• c)Cardiopatía: de relativa frecuencia en distrofinopatías, distrofias facio-escápulo-humeral, distrofia de Emery Dreifuss, laminopatías, miopatías metabólicas, mitocondriales y otras.
• d)Compromiso ocular: de cámaras (distrofias musculares congénitas con compromiso ocular), cataratas (asociadas a neuropatías específicas y distrofia miotónica entre otras), etc.

Anamnesis
• 10.- Indagar Historia Familiar: Recabar información detallada y realizar árbol genealógico completo,
consignando individuos afectados y no afectados, patología central y otras aparentemente no relacionadas, poniendo especial énfasis en sexo de los afectados, severidad de los síntomas en generaciones sucesivas y consanguinidad. Con varios individuos afectados, es posible definir el tipo de herencia: autosómica dominante o recesiva, ligada al X, o mitocondrial. Los casos aislados pueden corresponder a trastornos recesivos o ligados al X con bajo números de hijos en distintas generaciones (familia no informativa).

Examen Físico• Inspección:
– Aspecto general y facial:• Debilidad Facial (Distrofia
Miotónica y Facio-escápulo-humeral, Miopatía Nemalínica)
• Aspecto de facies alargada, escasa mímica facial, boca entreabierta o en V invertida.
• Indemnidad facial con severa debilidad de extremidades (Motoneurona: Atrofia Muscular Espinal tipo 1)

• Trofismo:– Observar constitución y masa
muscular general: Hipo-hipertrofias.– Precisar distribución y asimetrías:
atrofias, de una o más extremidades, localizada en segmentos (proximal, distal) o más específicas: lengua, compartimiento posterior del muslo, piernas.
– Hipertrofias (aspecto “musculoso”)• Generalizada: Miotonia congénita de
Thomsen• Localizada: Distrofia Duchenne
(Gemelos, vasto lateral cuádriceps, lengua, deltoides, glúteos)
Examen Físico
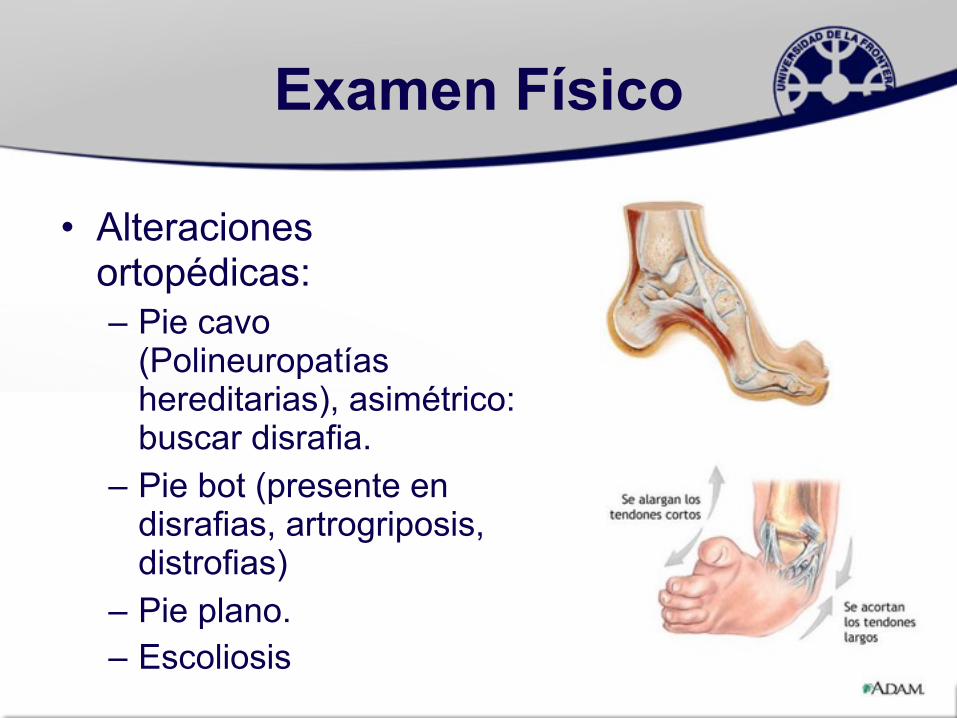
• Alteraciones ortopédicas:– Pie cavo
(Polineuropatías hereditarias), asimétrico: buscar disrafia.
– Pie bot (presente en disrafias, artrogriposis, distrofias)
– Pie plano.– Escoliosis
Examen Físico

• Fasciculaciones: Signo de denervación– Lengua: nervios hipoglosos– Temblor postural y/o acción:
polineuropatías hereditarias– Temblor fino distal de los dedos:
poliminimioclonus: atrofias espinales.
• Postura y marcha: – Marcha anadina (pato) o bamboleo
pelviano: debilidad músculos proximales miopatías
– Marcha estepada: Polineuropatías y miopatías distales.
Examen Físico

• Incorporación del niño del suelo:– Maniobra de
Gowers: Debilidad proximal.
Rev. Ped. Elec. [en línea] 2005, Vol 2, N° 1. ISSN 0718-0918
Examen Físico

Fuerza Muscular
• Evaluar musculatura proximal y distal de extremidades.
• Grupos musculares: – EESS: deltoides, bíceps, tríceps, flexores y
extensores muñeca.– EEII: Glúteos mayores, medios, cuádriceps,
tibiales anteriores, gemelos y peroneos. • Miopatías: Debilidad proximal• Polineuropatías: Debilidad distal
Examen Físico

Rev. Ped. Elec. [en línea] 2005, Vol 2, N° 1. ISSN 0718-0918
Examen Físico

Palpación Muscular
• Definir: consistencia, sensibilidad, calcificaciones, contracturas.
• Consistencia gomosa en zonas de hipertrofia muscular: Distrofias musculares.
• Sensibilidad dolorosa: Miopatías metabólicas o inflamatorias
Examen Físico

Movilidad Articular
• Identificar contracturas: – Distrofias musculares– Tardías en enfermedades
de motoneurona.
• Duchenne: flexores de cadera y tobillos
Examen Físico

• Percusión Muscular Miotonía, dificultad del
músculo para relajarse– Extremidades– Lengua
• Examen de Tono– Hipotonía: mayoría de
cuadros neuromusculares.
Examen Físico

Reflejos osteotendíneos
• Definir topografía de afección.• Enf. Motoneurona: Hipo o
arreflexia: global o predominio proximal.
• Neuropatías: Hipo o arreflexia de predominio distal.
• Miopatías: Reflejos distales más vivos que proximales.
• Radiculopatías o plexopatías: hipo o arreflexia dependerá de segmento comprometido
Examen Físico
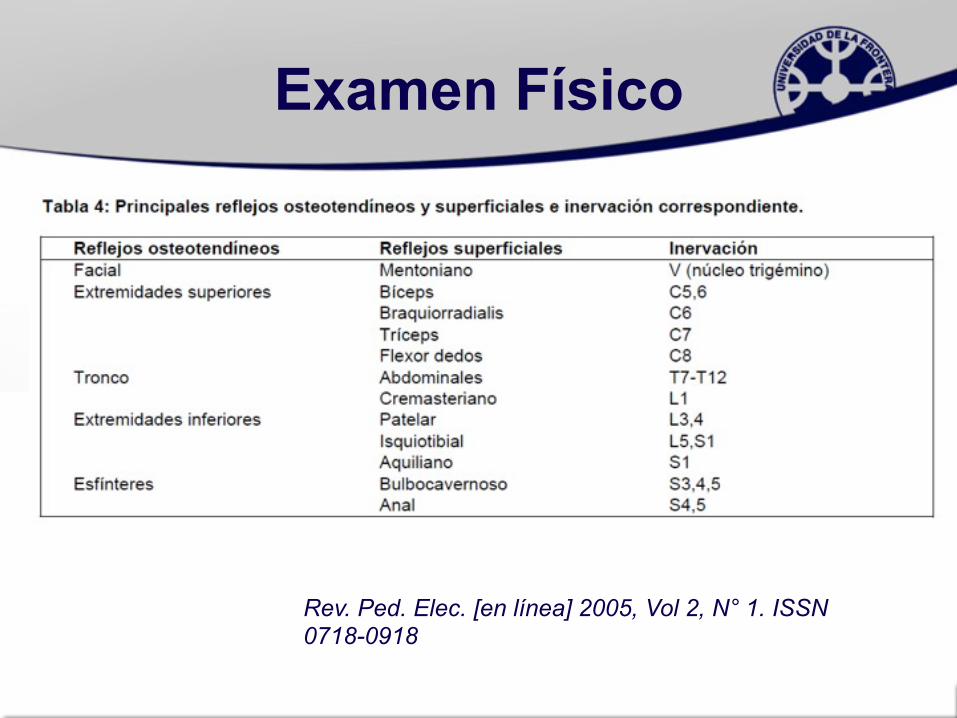
Rev. Ped. Elec. [en línea] 2005, Vol 2, N° 1. ISSN 0718-0918
Examen Físico
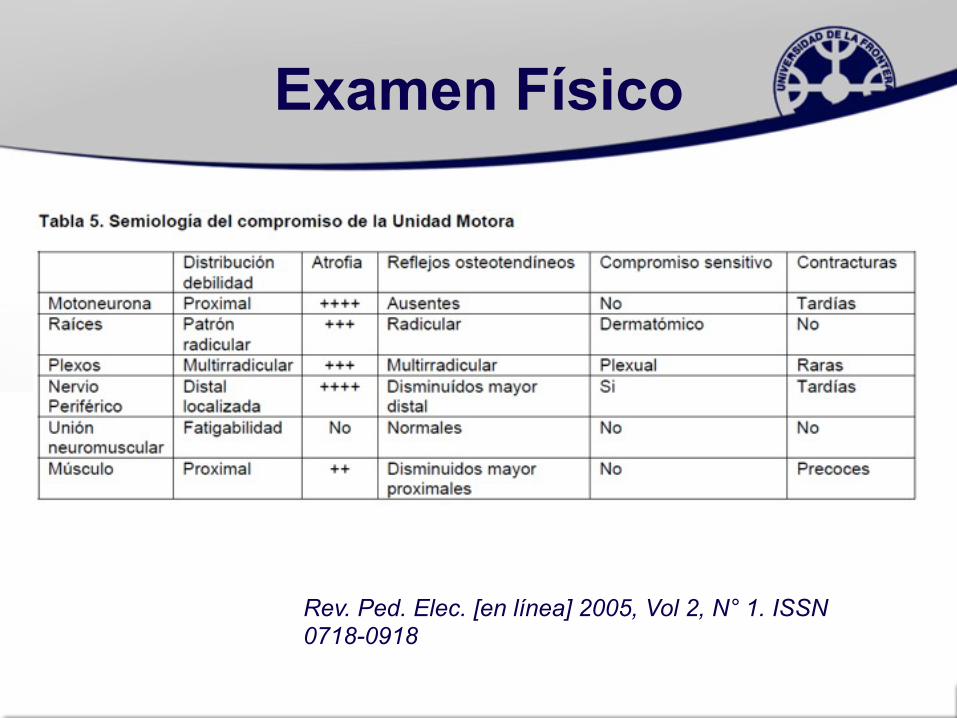
Rev. Ped. Elec. [en línea] 2005, Vol 2, N° 1. ISSN 0718-0918
Examen Físico

Laboratorio• Enzimas Musculares: Creatinkinasa o creatin-fosfoquinasa
(CPK) • Estudios electro-fisiológicos
– Velocidad de conducción nerviosa – Electromiografía – Estudios (electromiografía) de fibra única – Test estimulación repetitiva
• Estudios de respuesta a anticolinesterásicos: – Test de Tensilón MR (cloruro de edrofonio) o neostigmina
• Estudios histopatológicos – Biopsia muscular – Biopsia de nervio
• Estudios imagenológicos – Ecografía, tomografía axial computada (TAC) y resonancia magnética
de músculos • Estudios bioquímicos y genéticos específicos.

Enzimas Musculares• Principalmente creatinkinasa (CK)• 3 isoformas:
– BB (encéfalo)– MM (Tejido muscular esquelético y cardíaco)– MB (Varios tejidos, M. esquelético 5%, hasta
25% RN)– Elevada ante necrosis de fibras musculares
(también puede ser elevada por trauma muscular, parto)

Imágenes• Ecografía, TAC y RMN de músculo:
– Diagnóstico miopatías inflamatorias– Dirigir Biopsia

Estudio Electrofisiológico
• Electromiografía:– Analiza:
• Actividad de inserción de la aguja (daño mecánico): aumentada en procesos inflamatorios.
• Presencia de actividad espontánea en reposo (anormal)
• Potenciales de acción de unidad motora: voluntarios, analizados según duración, amplitud y fases.
– Breves, pequeños y polifásicos: procesos miopáticos– Mayor amplitud, anchos y polifásicos: neuropáticos
crónicos.

Estudio Electrofisiológico
• Estudio de velocidad de conducción nerviosa– Polineuropatías:
• Desmielinizantes: prolongación de latencias y disminución de VCN
• Axonales: Latencias y velocidades normales, con amplitudes reducidas.

• Histopatología:– Biopsia: Diagnóstico.
• Genética Molecular– Analisis ADN: Diagnóstico– No permite distinguir fenotipos leves y severos
causadas por misma alteración genética.– Correlacionar con clínica y laboratorio

Otros estudios• Pesquisar complicaciones:
– Respiratorias– Nutricionales– Endocrinas– Gastrointestinales– Cardiacas– Ortopédicas

Bibliografía
• Rev. Ped. Elec. [en línea] 2005, Vol 2, N° 1. ISSN 0718-0918: “Enfermedades Neuromusculares en Pediatría”, Dra. Karin Kleinsteuber S., Dra. María de los Ángeles Avaria B., Departamento de Pediatría y Cirugía Infantil, Facultad de Medicina Norte, Universidad de Chile, Unidad de Neurología, Hospital de Niños Roberto del Río.





























