Embed Size (px)
Citation preview

PRENATAL DIAGNOSIS, VOL. 12,637-64 1 (1 992)
RADIOGRAPHIC, HAEMATOLOGICAL, AND BIOCHEMICAL FINDINGS IN A FETUS WITH
CAFFEY DISEASE
B. LECOLIER*, G. BERCAUt, M. GONZALlbt, R. AFRIATt, D. RAMBAUD? N. MULLIEZS
AND s. DE KERMADEC~ * Laboratoire de Biologie Foetale. Hhpital Notre-Dame de Bon-Secours. 75014 Paris, France; tService de GynPcologie-Obsritrique et Centre de Diagnostic Prinatal. Hhpital Notre-Dame de Bon-Secours, ?SO14
Paris, France; $Service de Foetopathologie. Hbpital Saint-Antoine. 75012, Paris, France
SUMMARY An early case of prenatal Caffey disease is reported. Ultrasound examination performed at 20 weeks showed major angulations oflong bones, but both ultrasound scan and X-rays failed to make the differential diagnosis between Caffey disease and lethal osteogenesis imperfecta. A cordocentesis allowed us to find important biological abnormalities. The pregnancy was terminated after the rapid development of hydrops fetalis. The definitive diagnosis of Caffey disease was obtained by special X-ray and pathological study.
KEY WORDS Caffey disease Prenatal diagnosis Cordocentesis Biological syndrome
INTRODUCTION Caffey disease or infantile cortical hyperostosis is a multifocal bone disease which generally occurs before the fifth month of life (Caffey and Silverman, 1945). The prenatal diagnosis of Caffey disease is rare. The cases recognized in utero are frequently associated with polyhydramnios and have a very poor prognosis.
CASE REPORT
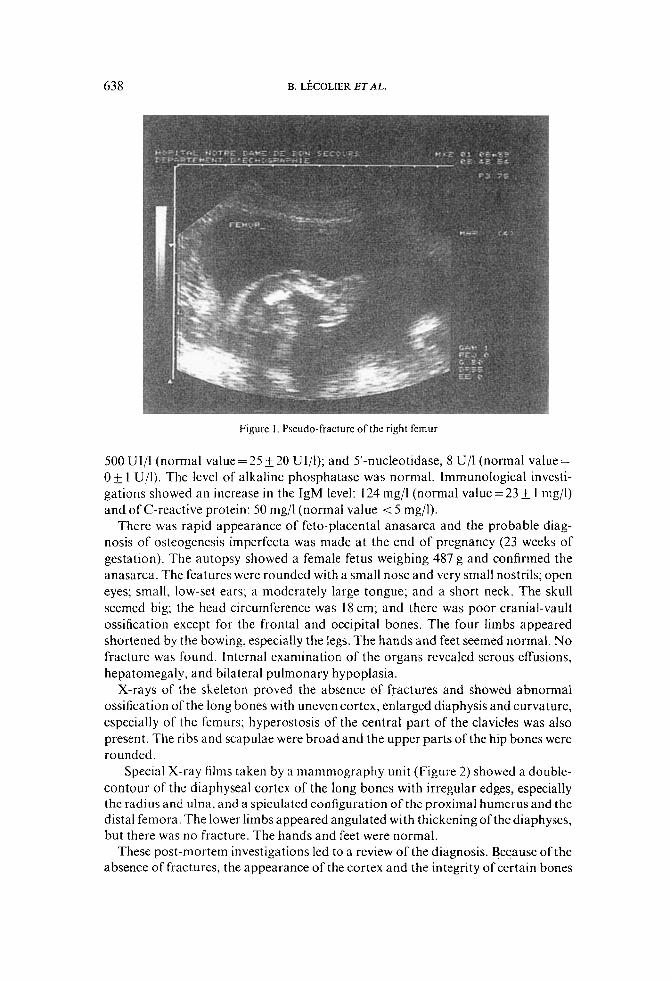
At 20 weeks of gestation, Mrs A, a 30-year-old G,P, was referred for routine ultra- sound scanning, which revealed major angulations of the following long bones: femurs (Figure I) , tibias, fibula, humerus, and right ulna. These angulations were so pronounced that fractures were strongly suspected. The thorax appeared short and not closed in the front. The last right rib seemed to be fractured. At this time, polyhydramnios was also noticed.
X-rays of the uterus, indicated by the ultrasound findings, confirmed the evidence of angulations of long bones but failed to prove clearly any fracture or osteopenia. However, the irregularities of the ribs suggested multiple callus formation, as in lethal osteogenesis imperfecta (type I1 A of Sillence).
Cordocentesis was performed and allowed us to determine that the karyotype was normal (46,XX) and to carry out various tests. Haematological examination showed normal red cell and platelet counts, a normal haemoglobin level (1 19 g/l) and haematocrit (36 per cent) but a marked leukocytosis: 25 x lo9 per litre (normal value = 4.3 f 1.2 x lo9 per litre) mainly due to neutrophil polynuclears (58 per cent), band cells (10 per cent), and metamyelocytes (3 per cent). Biochemical tests revealed increased serum levels of hepatic enzymes: gamma-glutamyl transpeptidase,
0197-3851/92/08063745$07.50 0 1992 by John Wiley & Sons, Ltd.
Received 15 July 1991 Accepted 6 October 1991
638 B. LBCOLIER ET AL.
Figure I . Pseudo-fracture of the right femur
500 Ul/l (normal value = 25 20 UI/l); and 5’-nucleotidase, 8 U/1 (normal value = 0 f. 1 Ujl). The level of alkaline phosphatase was normal. Immunological investi- gations showed an increase in the IgM level: 124 mg/l (normal value = 23 f. 1 mg/l) and of C-reactive protein: 50 mg/l (normal value < 5 mg/l).
There was rapid appearance of feto-placental anasarca and the probable diag- nosis of osteogenesis imperfecta was made at the end of pregnancy (23 weeks of gestation). The autopsy showed a female fetus weighing 487 g and confirmed the anasarca. The features were rounded with a small nose and very small nostrils; open eyes; small, low-set ears; a moderately large tongue; and a short neck. The skull seemed big; the head circumference was 18 cm; and there was poor cranial-vault ossification except for the frontal and occipital bones. The four limbs appeared shortened by the bowing, especially the legs. The hands and feet seemed normal. No fracture was found. Internal examination of the organs revealed serous effusions, hepatomegaly, and bilateral pulmonary hypoplasia.
X-rays of the skeleton proved the absence of fractures and showed abnormal ossification of the long bones with uneven cortex, enlarged diaphysis and curvature, especially of the femurs; hyperostosis of the central part of the clavicles was also present. The ribs and scapulae were broad and the upper parts of the hip bones were rounded.
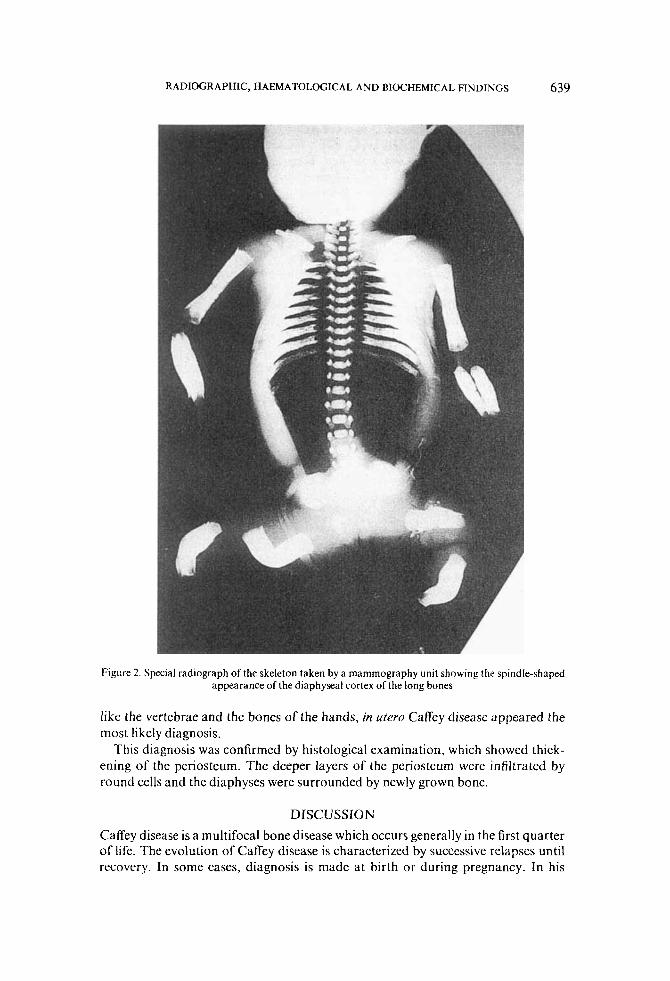
Special X-ray films taken by a mammography unit (Figure 2) showed a double- contour of the diaphyseal cortex of the long bones with irregular edges, especially the radius and ulna, and a spiculated configuration of the proximal humerus and the distal femora. The lower limbs appeared angulated with thickening of the diaphyses, but there was no fracture. The hands and feet were normal.
These post-mortem investigations led to a review of the diagnosis. Because ofthe absence of fractures, the appearance of the cortex and the integrity of certain bones
639 RADIOGRAPHIC, HAEMATOLOGICAL AND BIOCHEMICAL FINDINGS
Figure 2. Special radiograph of the skeleton taken by a mammography unit showing the spindle-shaped appearance of the diaphyseal cortex of the long bones
like the vertebrae and the bones of the hands, in utero Caffey disease appeared the most likely diagnosis.
This diagnosis was confirmed by histological examination, which showed thick- ening of the periosteum. The deeper layers of the periosteum were infiltrated by round cells and the diaphyses were surrounded by newly grown bone.
DISCUSSION Caffey disease is a multifocal bone disease which occurs generally in the first quarter of life. The evolution of Caffey disease is characterized by successive relapses until recovery. In some cases, diagnosis is made at birth or during pregnancy. In his

640 B. LECOLIER E T A L.
literature review, Labrune found 21 cases of antenatal Caffey disease, 10 of which were discovered in utero (Labrune et al., 1983); his one case diagnosed in utero was associated with hydramnios, anasarca, and bilateral pulmonary hypoplasia. The fetus died during delivery at 27 weeks of gestation. Since then, three other cases diagnosed in utero have been reported (Bellufi et al., 1984; Langer and Kaufmann, 1985; Kozlowski and Tsuruta, 1989), all with polyhydramnios. The first child sur- vived 5; months, the second 16h and the third was stillborn. In the first case, pulmonary hypoplasia and hepatomegaly were reported. The two children who survived suffered from severe cardiac and pulmonary failure. In our case, pulmon- ary hypoplasia and hepatomegaly were also present. These three recent cases are very similar to the one reported by Bennett and Nelson (1953). Feto-placental anasarca and polyhydramnios seem to have a very poor prognosis. It is important to note that the major angulations of long bones are present only in the antenatally occurring forms of Caffey disease. These angulations can lead to three different diagnoses: hypophosphatasia, camptomelic dysplasia, and osteogenesis imperfecta. Hypophosphatasia shows very poor mineralization and camptomelic dysplasia is associated with hypoplastic fibulae, relative sparing of the upper limbs, and frequent talipes equinovarus. The following features distinguish Caffey disease from osteo- genesis imperfecta: integrity of certain bones (spine, hands, feet) and relative sparing of the metaphyses, well seen in the forearms, are characteristic of Caffey disease; transparency of the skeleton, thinness of cortices, and fractures are the signs of osteogenesis imperfecta.
However, this case demonstrates the limits of ultrasound examination and standard X-rays. A definitive diagnosis was obtained only post-mortem, by a special X-ray film, and confirmed by histological examination.
The cause of Caffey disease is still unknown. The hypothesis of a genetic trans- mission has been supported, since familial cases of Caffey disease are not infrequent (Newberg and Tampas, 1981). An infectious aetiology has never been proven but an inflammatory syndrome is commonly found. Our findings lead us to support the hypothesis of an infectious disease acquired in utero (Caffey, 1957; Silverman, 1976; Langer and Kaufmann, 1985; Marshall et al., 1987; Tabardel et al., 1988). Indeed, elevated IgM and C-reactive protein levels, which have been previously reported (Temperley et al., 1972; Rachmander and Ramkissoon, 1978; Tabardel et al., 1988), are useful signs of neonatal infection, and high levels of IgM and hepatic enzymes are observed in utero in cases of congenital rubella or toxoplasmosis (Daffos and Forestier, 1988). We did not attempt to isolate any micro-organism because of the initial diagnosis of lethal osteogenesis imperfecta, but it appears important, in the future, to try to show evidence of an infectious cause of Caffey disease.
In conclusion, the prenatal diagnosis of CafTey disease should be considered when angulations of long bones are found by ultrasound examination. Because of the very poor prognosis when presenting in fetal life, the name of lethal prenatal cortical hyperostosis is proposed to distinguish it from the generally benign postnatal disease, until the cause of this disease is discovered.
ACKNOWLEDGEMENT
We are grateful to Dr Pierre Maroteaux for his advice and helpful suggestions.

RADIOGRAPHIC, HAEMATOLOGICAL AND BIOCHEMICAL FINDINGS 64 1
REFERENCES Bellufi, G., Chirico, G., Colombo, A., Ceciliani, L., Dell’Orbo, C., Fiori, P., Pazzaglia, U.,
Quacci, D. (1984). Report of a new case of neonatal cortical hyperostosis, Ann. Radiol. (Paris), 27,7948.
Bennett, H.S., Nelson, T.R. (1953). Prenatal cortical hyperostosis, Br. J. Radiol., 26,47-49. Caffey, J. (1957). Infantile cortical hyperostosis; a review of the clinical and radiographic
Caffey, J., Silverman, W.A. ( 1 945). Infantile cortical hyperostoses, preliminary report of a
Daffos, F., Forestier, F. (1988). Mkdecine et Biobgie du Foetus Humain, Paris: Maloine. Kozlowski, K., Tsuruta, T. (1989). Dysplastic cortical hyperostosis: a new form of lethal
neonatal dwarfism, Br. J . Radiol., 62,376-378. Labrune, M., Guedj, G., Vial, M., Bessis, R., Roset, M., Kerbrat, V. (1983). Maladie de
Caffey a ditbut anttnatal, Arch. Fr. Pkdiatr., 40,39-43. Langer, R., Kaufmann, H.J. (1985). Pranatale Diagnosestellung bei Caffey’scher Erkrankung
(infantile korticale Hyperostose), Klin. Padiat., 197,473-476. Marshall, G.S., Edwards, K.M., William, B.W. (1987). Sporadic congenital Caffey’s disease,
Clin. Pediafr. (Phila), 26, 177-180. Newberg, A.H., Tampas, J.P. (I98 I) . Familial infantile cortical hyperostosis: an update, Am.
J . Roentgenol., 137,93-96. Rachmander, V., Ramkissoon, R. (1978). Infantile cortical hyperostosis with raised
immunoglobulins, Arch. Dis. Child., 53,426-428. Silverman, F.N. (1976). Virus diseases of bone. Do they exist? The Neuhauser lecture, Am. J .
Roentgenol., 126,677-703. Tarbardel, Y., Seghaye, M.C., Senterre, J. (1988). Maladie de Caffey-Sylverman ntonatale
avec thrombocytose, augmentation de la C-rtactive prottine et des immunoglobulines, Arch. Fr. Pkdiatr., 45,263-265.
Temperley, I.J., Douglas, S.J., Rees, J.P.R. (1972). Raised immunoglobulin levels and thrombocytosis in infantile cortical hyperostosis, Arch. Dis. Child., 47,982-983.
features, Proc. R. SOC. Med., 50,347-354.
new syndrome, Am. J. Roentgenol., 54,l-16.





























