Embed Size (px)
Citation preview

RADICAL POLYMERIZATION OF VINYL CHLORIDE:KINETICS AND MECHANISM OF BULK AND
EMULSION POLYMERIZATION
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI and E. SUND
The Norwegian Institute of Technology,7034 Trondheim NTH, Norway
ABSTRACTBulk and emulsion polymerization have as a common featufe that there arealready formed, from the start, particles which are one-phase systems consistingof polymers swollen with monomer. The composition of the particles is thesame in the two cases and is constant up to about 70 per cent conversion at50°C. Bulk and emulsion polymerization differ in that in the former case theouter phase is practically pure monomer while in the latter it consists of adilute aqueous solution of vinyl chloride (—0.t mole/I. at 50°C).
Mechanisms of bulk and emulsion polymerization are discussed. A mechanisminvolving desorption and reabsorption of radicals into the particles is treatedin more detail. The result of the calculations seems to indicate that the bulkand emulsion polymerization are similar in that already at low conversionthe main reactions, both polymerization and termination, take place in theparticles. Even for bulk polymerization the number of radicals in the outerphase is, under ordinary conditions, practically independent of the rate ofradical termination in that phase. The present treatment does not involve anyassumption of an established thermodynamically determined equilibriumdistribution of radicals in the two phases. In both these respects the presenttheory differs from previously advanced theories for bulk polymerization.The subdivision of the reaction zone in discrete particles has a similar effect onthe bulk polymerization as on emulsion polymerization. It increases the rate ofpolymerization compared to that which would be obtained if the particlesformed a continuous phase. The present theory may readily explain theexperimentally observed drop in rate at precipitation, as well as the autocatalyticcourse of the reaction. The increase in the initial rate of bulk polymerization byaddition of chain transfer agents results from an increase in the effective rateconstant for desorption. Bulk and emulsion polymerization will responddifferently to a change in the rate of desorption and absorption of radicals in theparticles. This is due to the difference in the concentration of monomer in theouter phase in the two cases. In the case of bulk polymerization an increase inthe rate of desorption and a decrease in the rate of absorption of radicals intothe particles will tend to increase the rate of reaction whereas the opposite isexpected for emulsion polymerization.
New experimental evidence concerning emulsion polymerization with mixedemulsifiers consisting of sodium dodecylsulphate and different h-fatty alcoholsis presented. A marked decrease in the rate is obtained by use of n-hexadecanol.The mixed emulsifier is compared with the pure anionic emulsifier at seedpolymerization with a given amount of seed and number of particles. With thepure anionic emulsifier the rate of polymerization is independent of the degree
121
P.A.c.—26/28

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
of coverage with emulsifier. A complete coverage of the particles with a mixtureof sodium dodecylsulphate and n-hexadecanol in a certain ratio leads to amarked decrease in rate. It is suggested that these results may be due to themixed emulsifier forming a condensed layer on the surface of the particleswhich reduces the effective absorption constant.
INTRODUCTION
The following significant features of kinetics of radical polymerizationof vinyl chloride are common for bulk-, suspension- and emulsionpolymerization.
1. The reaction is autocatalytic from the onset of reaction.2. The reaction order, with respect to the initiator, is close to 0.5.3. Molecular weight does not depend upon the conversion or the initiator
concentration. Molecular weight and molecular weight distribution aresimilar for bulk and emulsion polymerization.
This similarity in the kinetics of bulk and emulsion polymerization, whichwill be outlined in more detail, is typical for vinyl chloride.
Although several theories, which may account for some of the experimentalresults have been put forward, there still seems to exist great uncertaintyas to the mechanism of the polymerization both in bulk and emulsion.Talamini and Peggion1 have recently given an extensive review of thepolymerization of vinyl chloride covering the literature up to 1965. Thereforein the present paper special attention has been paid to new experimentalevidence and theoretical approaches to the problem of kinetics and mecha-nisms of the radical polymerization processes.
A. BULK POLYMERIZATION
Vinyl chloride is a very poor solvent of its own polymer. Therefore thepolymerization system, from the start, separates into two phases; a dilutephase with practically no polymer and a concentrated phase consisting ofpolymer particles swollen with monomer. The activity of the monomer inthe dilute phase is approximately equal to unity and the Flory—Hugginsequation for the partial free energy of the monomer in the particles atsaturation pressure is given by:
=RTn
i + (1 - + = o (1)
where V1 is the molar volume of the monomer, a the interfacial tension, r theparticle radius, q1 and c52 the volume fractions of monomer and polymerin the particles respectively, and A the interaction constant. The value of thelatter is 0.88 at 50°C2 which corresponds to a concentration of monomer inthe particles of 6 mole/i. Therefore in the bulk polymerization of vinylchloride the system consists of two different phases each of them having aconstant composition up to about 70 per cent conversion. The influence of
122
RADICAL POLYMERIZATION OF VINYL CHLORIDE
the interfacial energy term in limiting the dilution of the particles withmonomer is negligible compared to the effect of the interaction energy term.
The concentrated phase in bulk polymerization and generally under
C0. 4N
0)E>'00
Table 1. Particle size and number as a function ofconversion C in the bulk polymerization of vinyl
chloride at 50C3
CDiameter
d x io (cm) N < 10- '
0.0017 < 1.0 > 23.20.009 2.90 5.100.045 5.10 4.650.096 5.90 6.410.148 7.20 5.400.300 9.6 4.70
N5 number of particles perg of initial amount of vinyl chloride.
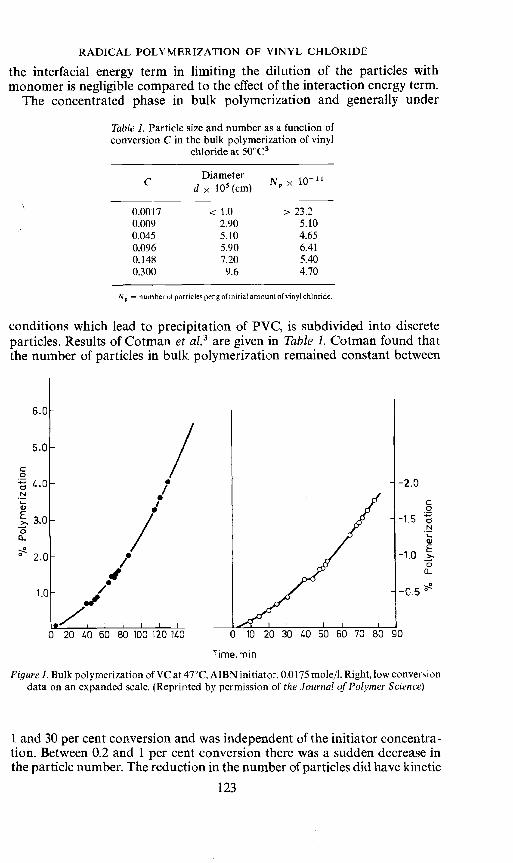
conditions which lead to precipitation of PVC, is subdivided into discreteparticles. Results of Cotman et al.3 are given in Table 1. Cotman found thatthe number of particles in bulk polymerization remained constant between
3.0
2.0
—2.0
C0-1,5
N
0)E-1.0 ?'00
0 20 40 60 80 100 120 140 10 20 30 40 50 60 70 80 90
Time,min
Figure 1. Bulk polymerization of VC at 47©C, AIBN initiator, 0.0175 mole/l. Right, low conversiondata on an expanded scale. (Reprinted by permission of the Journal of Polymer Science)
1 and 30 per cent conversion and was independent of the initiator concentra-tion. Between 0.2 and 1 per cent conversion there was a sudden decrease inthe particle number. The reduction in the number of particles did have kinetic
123

J. UGELSTAD, I-I. LERVIK, B. GARDINOVACKI AND E. SUND
consequences also. As indicated by the results given in Figure 1, a decreasein rate coincides with the sharp decrease in the particle number. Cotmanconcluded that the absence of secondary nucleation of particles indicated apredominant absorption of radicals in the particles in contact with monomer.
Bort et al.4' reported that at relatively high rates of radical productionsingle particles with a smooth surface were formed. The number of particleswas about the same as that found by Cotman and corresponded to 4.5 x 1014particles per I of the monomer at start. At low rates of radical productionaggregates are formed consisting of a number of globular particles. Animportant observation made by Bort was that both number and morphologyof the particles (single particles or aggregates) were determined by the initialconditions of polymerization, below 1 per cent conversion. A change inrate of polymerization after 1—2 per cent conversion did not bring about anychange in either number or morphology of the particles. Bort, as well asCotman, concludes that the polymerization mainly takes place in theparticles already at a very low conversion.
Previous authors dealing with the kinetics of the bulk polymerization ofvinyl chloride ascribed the autocatalytic behaviour of the polymerizationto a trapping of chain radicals in the polymer particles. The high viscosityin the particles was not expected to influence the value of the propagationconstant but was suggested to lead to a decrease or complete stop of thetermination reaction. Schindler and Breitenbach6 assumed the steady statecondition for the radicals. Moreover they assumed that the value of thetermination constant in the particles was lower than that in the monomerphase. In the calculation of an expression for the reaction rate they made useof an overall termination constant which was suggested to decrease withincreasing conversion according to a purely empirical equation. BothBengough and Norrisb7, and Mickley, Michaels and Moore8 assumed intheir mechanisms that the termination in the particles was effectively stopped.The steady state condition for the radicals in the particles was maintained bychain transfer of radical activity to the monomer and desorption of monomerradicals from the particles. The termination took place in the monomer phaseonly. The complete stop of termination in the particles may seem unreasonablein view of the relatively high content of monomer in the particles. Monomerhas been found to be a very effective plasticizer for PVC
An interesting new contribution to the theory of the bulk polymerizationhas recently been given by Talamini and coworkers. In a number of papers9they have underlined the fact that in bulk polymerization of vinyl chloridethe reaction takes place in two phases from a very low conversion onwards.With increasing conversion the amount of the dilute phase decreases whilethat of the concentrated phase increases. The two phases have constantcomposition in the whole range of their coexistence. Therefore, although thevalues of the termination constants may be different in the two phases, theindividual values will not be expected to vary up to about 70 per cent con-version.
The total rate expressed in moles of monomer reacted per unit time isgiven by
rp kP[R]L[M]LVL + k{R][MIv (2)
124

RADICAL POLYMERIZATION OF VINYL CHLORIDE
where [R]L and EMIL are the radical and monomer concentration in thedilute phase respectively, and VL is the volume of the dilute phase. ER],[M] and V refer to the concentrated phase. Introducing the degree ofconversion C into equation 2 gives:
dCkR ML kR Mp[ [ ()
where M0 is the number of moles of monomer at the start. Talamini introducesthe specific rates rL and rp in the two phases, defined as degree of conversionper unit time:
dC ML M--— = rj7 + rp - (4)ut ivi0 ivi0
if C0 is the degree of conversion at which phase separation occurs and A themonomer to polymer weight ratio in the particles, one obtains:
= r[1 — C T A(C — C0)J + rA(C — C0) (5)
The conversion at which separation occurs is very low, hence with a goodapproximation equation 5 can be rewritten:
dC = rL(l — C — AC) + ACrE (6)
Talamini states that because of the constant composition of the two phasesthe ratio of the corresponding specific rates will stay constant:
rP=QxrL (7)
From equations 3 and 4 this is equivalent to a constant ratio of radicalconcentrations in the two phases
= Q{R]L (8)
Talamini proceeds as follows: from equations 6 and 7:
dC- = TL — (1 + A — AQ)CrL (9)
Substitution of —(1 + A — AQ) = q gives:
dC = rL + qrC (10)
Upon integration:C = — [exp(qrt) —1] (11)
By expanding the exponential term in series it results in:
(12)qL.. n!n= 1
125

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
Talamini furthermore assumes that rL may be expressed as:
IL = k[I] = k1,(k1.) [I] (13)
where [I] apparently stands for the overall concentration of initiator in thesystem and ktL is the termination constant in the dilute phase. Inserting forTL = k[I]+ into equation 12 gives:
C=_>—_-,_[I]'t (14)
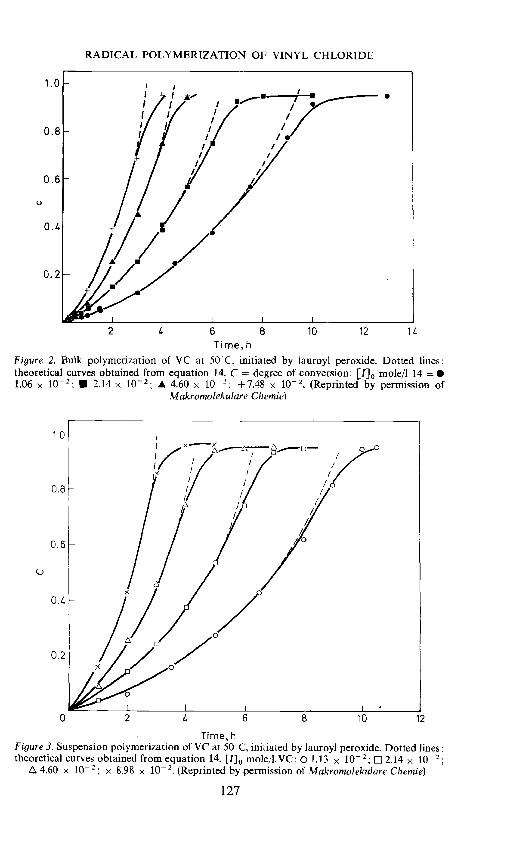
This equation is found to agree well with experimental results both frombulk and suspension polymerization over a wide range of conversion,initiator type and concentration. Examples are shown in Figures 2 and 3h1•By limiting to the second term in the series one obtains from equation 14an equation of the form:
C = c1[I]t + X2{I] x (15)
where and 2 are constants.This should, according to Talamini, lead to a reaction order, with respect
to the initiator, which varied with conversion. The order would be 0.5 attime zero and may increase with time as the second term becomes moreimportant. However, if the differential rates with different initiator con-centration are compared at the same conversion, which is obviously correct,equation 15 predicts a constant order of 0.5, independent of conversion.Solving for t from equation 15 and inserting in the rate expression
= o[I] + 2c2[I]t (16)
gives:
= [I](4cx2C + (17)
It seems to the present authors that the main assumptions made by Talaminiin the evaluation of equation 14 may be open to some doubt. These are theassumptions of a constant ratio of polymerization rates in the two phases(equation 7) and moreover the assumption that the concentration of radicalsin the dilute phase at a constant rate of radical production is determinedsolely by the rate of termination in the dilute phase. The argument againstthis is as follows: one may, in accordance with many authors, assume a quasisteady state for the number of moles of radicals in the two phases:
dRL = 2k[I1LVL — ka[R]L + kdc[R]p —2ktL[R]VL = 0 (18)
= 2kj[I]pvp + ka[R]L — kdc[R]p — 2k[R]v 0 (19)
126
RADICAL POLYMERIZATION OF VINYL CHLORIDE
Figure 2. Bulk polymerization of VC at 50CC, initiated by lauroyl peroxide.theoretical curves obtained from equation 14. C = degree of conversion: [']1.06 x 10 2. U 2.14 x 102; A 4.60 x 10 : + 7.48 x 10-2. (Reprinted by
Makrotnolekulare Cheinie)
Time, hFigure 3. Suspension polymerization of VC at 50°C, initiated by lauroyl peroxide. Dotted lines:theoretical curves obtained from equation 14. ['] mole/l.VC: 01.13 x 102; 2.14 x 102;
4.60 x 10—2; x 8.98 x 2• (Reprinted by permission of Makroinolekulare Che?nie)
127
1.0- I
/2".
I
/'—U-—
'I/70.8 - II/0.6 -
U
0.4 -
0.2—
2 4 6 8 10 12 14
Time, h
Dotted lines:mole/i 14=•permission of
U
to
0.8
0.6
0./.
0.2
0

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
Here are included the terms describing transport of radicals from one phaseto the other in addition to those giving the formation and destruction ofradicals by termination in the two phases. It is assumed that the rate ofabsorption of radicals from the dilute phase into the particles is proportionalto the concentration of radicals in the dilute phase and vice versa for thedesorption of radicals from the concentrated phase. The values of ka andkdc are of course expected to be constant at a given conversion and particlenumber only. Addition of equations 18 and 19 gives:
1 r — 1 ri,.l2rz L I ID.12T7— tLL JL L "tPL JP Pwhich is a completely general equation. It gives the connection between thetotal rate of radical production kI, which may be considered to be constant,and the total rate of radical destruction. For sake of simplicity the efficiencyfactor is throughout this discussion assumed to be equal to 1. Generally, theconcentration of radicals in one of the phases will also depend upon the rateof radical destruction in the other phase. It is furthermore obvious that insuch cases, where the radical concentrations are determined by quasisteady state conditions, the ratio of the concentration of radicals in the twophases will not generally be governed by an established, thermodynamicallydetermined equilibrium. For this to hold, the value of ka and kdc must be sohigh that the terms describing the absorption and desorption of radicals arethe dominating ones in one or both phases. Under these conditions one has:
[R]p/[R]L = ka/kdC Q (21)
Inserting from equation 21 into equation 20 gives:
[R] L = [kI/(ktLVL + Q2kV)3 (22)
Even in this case the radical concentration in the liquid phase will generallyalso depend upon the termination in the concentrated phase and vice versa.Two limiting cases may be considered:
A: Q2kPVP ktV (23)
(kI/kV (24)
Q(kII/kLVL) (25)B: Q2kPVP (26)
[R•]L = (kI/kV)4 (27)
[R] = (k1I/kV) (28)
In case A the termination in the particles is negligible. The rate is:
dM 7k.i— — = ( —---) kP(ML + QMP) (29)dt \'tLVL/
or expressed in degree of conversion:
dC 7k.i4k(1—C—AC+ACQ) (30)tLL
128
RADICAL POLYMERIZATION OF VINYL CHLORIDE
These rate expressions are in principle similar to that obtained by Mickleyet al. Also at low conversion where kI/VL is approximately constant, equation30 is identical to that obtained by Talamini (equation 9). As discussed belowboth the assumption of a thermodynamically determined equilibriumdistribution of radicals in the two phases and the assumption of a negligibletermination in the particles may be questioned from a theoretical point ofview. The latter also seems to be contradicted by experimental evidence.According to equation 24 the formation of the particles should not influencethe number of moles of radicals in the dilute phase at low conversion whereVL is approximately constant, and therefore not even the rate of polymeriza-tion in the dilute phase. As discussed below the experimental fact is that theformation of polymer particles leads to a marked decrease in rate. A moredetailed treatment of the kinetics of bulk polymerization is given below,after presentation of new experimental evidence on the effect of the precipita-tion and the effect of addition of chain transfer agents.
Effect of precipitationMost interesting contributions to the problem of the mechanism of the
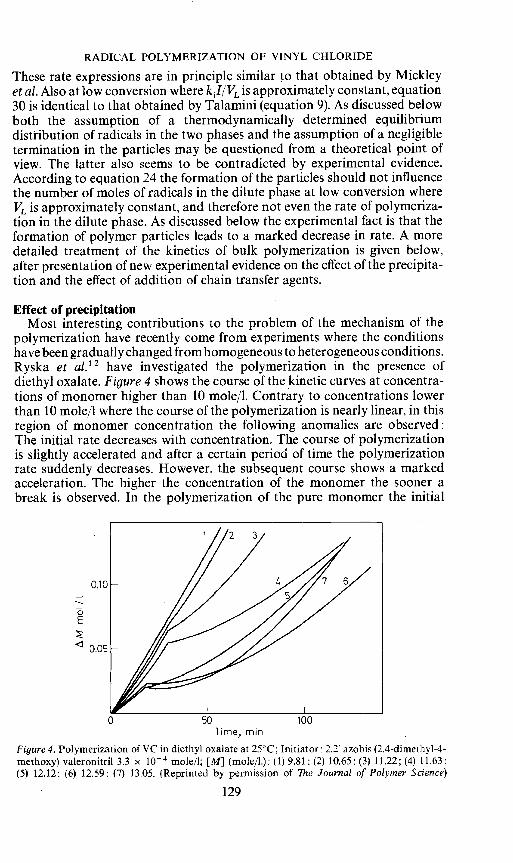
polymerization have recently come from experiments where the conditionshave been gradually changed from homogeneous to heterogeneous conditions.Ryska et al.12 have investigated the polymerization in the presence ofdiethyl oxalate. Figure 4 shows the course of the kinetic curves at concentra-tions of monomer higher than 10 mole/i. Contrary to concentrations lowerthan 10 mole/I where the course of the polymerization is nearly linear, in thisregion of monomer concentration the following anomalies are observed:The initial rate decreases with concentration. The course of polymerizationis slightly accelerated and after a certain period of time the polymerizationrate suddenly decreases. However, the subsequent course shows a markedacceleration. The higher the concentration of the monomer the sooner abreak is observed. In the polymerization of the pure monomer the initial
0E
Figure 4. Polymerization of VC in diethyl oxalate at 25CC; Initiator: 2.2 azobis(2.4-dimethyl-4-methoxy) valeronitril 3.3 x iO mole/I; [M] (mole/i.): (1) 9.81: (2) 10.65; (3) 11.22; (4) 11.63:(5) 12.12: (6) 12.59: (7) 13.05. (Reprinted by permission of The Journal of Polymer Science)
129
Time, mm
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
almost linear course was not observed. As mentioned earlier, however,Cotman observed a similar break in the curve at very low conversion, alsowith pure monomer, at the point where the number of particles suddenlydecreased, although the effect was less pronounced than that shown inFigure 4.
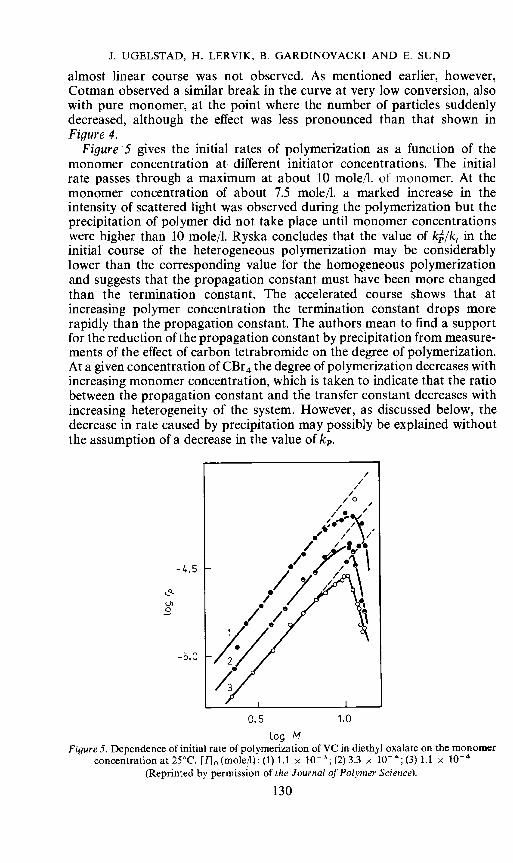
Figure 5 gives the initial rates of polymerization as a function of themonomer concentration at different initiator concentrations. The initialrate passes through a maximum at about 10 mole/i. of monomer. At themonomer concentration of about 7.5 moleil. a marked increase in theintensity of scattered light was observed during the polymerization but theprecipitation of polymer did not take place until monomer concentrationswere higher than 10 moie/l. Ryska concludes that the value of k2/k in theinitial course of the heterogeneous polymerization may be considerablylower than the corresponding value for the homogeneous polymerizationand suggests that the propagation constant must have been more changedthan the termination constant. The accelerated course shows that atincreasing polymer concentration the termination constant drops morerapidly than the propagation constant. The authors mean to find a supportfor the reduction of the propagation constant by precipitation from measure-ments of the effect of carbon tetrabromide on the degree of polymerization.At a given concentration of CBr4 the degree of polymerization decreases withincreasing monomer concentration, which is taken to indicate that the ratiobetween the propagation constant and the transfer constant decreases withincreasing heterogeneity of the system. However, as discussed below, thedecrease in rate caused by precipitation may possibly be explained withoutthe assumption of a decrease in the value of k.
-4.5
0)0
-5.0
Log MFigure 5. Dependence of initial rate of polymerization of VC in diethyl oxalate on the monomer
concentration at 25°C. [I] (mole/I): (1) 1.1 x 10; (2) 3.3 x 1O; (3) 1.1 x iO(Reprinted by permission of the Journal of Polymer Science).
130
0.5 1.0
1.4
1.2
1.0
0.6
0.40.2
Figure 6. Rate of polymerization of VC in 1,2-dichlorethane at 50°C asconcentration. —0-—- initial polymerization rates; —+ +—indicated degrees of conversion. Initiator = lauroyl peroxide, 4.64 x
permission of Makrosnolekulare Cheinie)
cD
a function of the monomerpolymerization rates at
10-2 mole/i. (Reprinted by
Figure 7. Molecular weight as a function of the monomer concentration for the polymerizationof VC in 1,2-dichlorethane at 50°C. [I] = 4.64 x o 2 mole/I. (Reprinted by permission of
Makrornolekulare Che,nie)
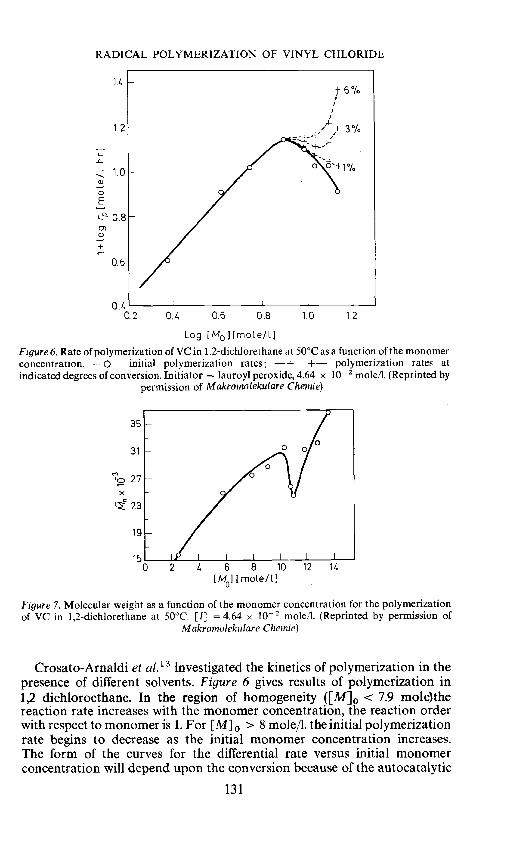
Crosato-Arnaldi et al.'3 investigated the kinetics of polymerization in thepresence of different solvents. Figure 6 gives results of polymerization in1,2 dichloroethane. In the region of homogeneity ([M]0 <7.9 mole)thereaction rate increases with the monomer concentration, the reaction orderwith respect to monomer is 1. For [M]0 > 8 mole/I. the initial polymerizationrate begins to decrease as the initial monomer concentration increases.The form of the curves for the differential rate versus initial monomerconcentration will depend upon the conversion because of the autocatalytic
131
RADICAL POLYMERIZATION OF VINYL CHLORIDE
0E
L 0.80)0+
0.4 0.6 0.8 1.0 1.2
Log [M0][molelLJ
0 2 4 6 8 10 12 14
EM0] [mole/L]

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
effect in the heterogeneous region. Molecular weight of polymer formed up to6 per cent conversion shows a similar behaviour (Figure 7). Obviously, thedecrease in rate caused by precipitation is accompanied by a decrease in thedegree of polymerization. The decrease in rate caused by precipitation isexplained by Crosato—Arnaldi et a!. in the following way: the polymerizationstarts and proceeds in solution (homogeneous conditions) until the polymerconcentration reaches the value at which the polymerization system separatesinto two phases, one concentrated and the other very dilute in polymer.If V is the volume of the system and p the number of moles of radicalsproduced in unit time, the steady state number of moles of radicals underhomogeneous conditions is:
=(p.v)4 (31)
When the polymerization system separates into two phases it is assumed thatthe volume does not vary and the radical production rate is unaffected.Indicating with VL the volume of the dilute phase and with L the number ofmoles of radicals, produced per unit of time, which remain in this phase, thenumber of radicals at steady state, RL is given by:
RL = ('Y (32)
Analogously, if V is the volume of the concentrated phase and cx the numberof radicals produced per unit of time in this phase, the number of radicals atsteady state condition R is:
=(y (33)
The overall steady state radical concentration [RU] in the polymerizationsystem is given by:
[RU].RL+RP
(34)
Denoting by cbL and çb the volume fraction of the dilute and concentratedphase respectively, the relation 34 can be rewritten in the form:
/ A'\4 7 A\4I L'f'L I P'!'P\—
k\2kLV)+
k\2kpV)With the assumption that krL k and indicating with fL = cjp andf, = the fractions of the radicals going in the dilute and concentratedphase respectively, the ratio y = ([R]/R/V) of the overall radical steadystate concentration in the polymerization system constituted of the twophases, to that in the system before phase separation, is given by the followingequation: ky =f4 + () (1 -fL)(1 — L) (36)
132

RADICAL POLYMERIZATION OF VINYL CHLORIDE
If it is assumed that the growing radicals, are more soluble in the concentratedphase, fL is always lower than çb. Numerical solution of equation 36 yieldsthat if = kL phase separation will lead to a sharp decrease in rate andsuccessively to an autocatalytic effect. If kL/kp > 1, the decrease in rate dueto phase separation is reduced and, the successive autocatalytic effectincreased.
In the view of the present authors some of the basic assumptions of thistreatment may be open to doubt. As in• the pre'viously cited paper byTalamini et al.'°, it is assumed that the steady state concentration of theradicals in one phase is governed solely by the rate of radical production andthe rate of radical destruction by termination in that phase. By comparisonwith equations 18 and 19 it is seen that equations 32 and 33 involve that
PL — ka 1L + = 2kfL (37)
= + k — = 2k (38)
where PL and p are the rates of radical production from the initiator inthe dilute and concentrated phase respectively. The relationship between thenumber of radicals in the two phases is obviously more complex thanassumed by Crosato-Arnaldi. The number of radicals in one phase maydepend upon the rate of termination in the other phase. Moreover, thenumber of radicals in each phase is not generally proportional to the squareroot of the radical production in this phase.
Effect of chain transfer agentsV
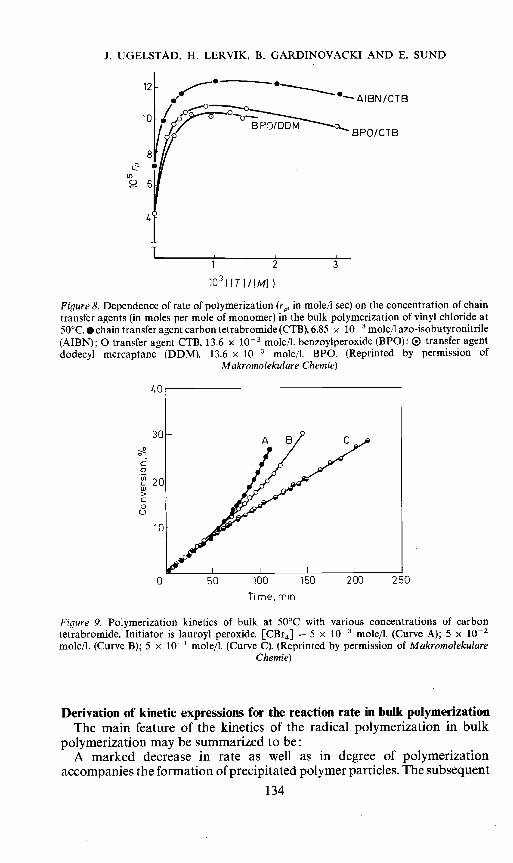
Another type of experiment which has been used in the discussion ofmechanisms is measurement of the effect of chain transfer agents. Breitenbachand Schindler14 found that addition of small amounts of chain transferagents such as CBr4 and dodecyl mercaptane lead to a very marked increasein the initial rate of reaction (Figure 8) and at the same time caused a reductionor complete removal of the autocatalytic effect. These results have recentlybeen confirmed by several authors'2' 15 Figure 9 gives results obtained byVidotto et al.'5 which illustrate the reduction of the autocatalytic effectcaused by CBr4.
This most interesting effect of an increase in rate with the addition ofchain transfer agents has been given different explanations in the literature.As discussed above, Talamini assumes an established, thermodynamicallydetermined equilibrium distribution of radicals between the two phases.In accordance with wellknown principles the decrease in polymer weight,caused by the chain transfer agent, will be expected to increase the preferencefor polymeric species and thus for the growing radicals in the liquid phase andlead to an increase in the rate. Cotman3 suggests that the lower averagedegree of polymerization leads to an increasing fraction of soluble radicalsand, moreover, that the insoluble fraction, by virtue of the lower molecularweight, may coalesce more slowly. Finally Breitenbach suggests that theeffect may be ascribed to a transfer of rather unreactive monomer radicalsto active radicals by the chain transfer agents.
133
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
U,0
10
4
T
••____________,1• —AIBNfCTB
103([T]f EM]
BPO/CTB
Figure 8. Dependence of rate of polymerization (re, in mole/i sec) on the concentration of chaintransfer agents (in moles per mole of monomer) in the bulk polymerization of vinyl chloride at50°C.• chain transfer agent carbon tetrabromide (CTB), 6.85 x iO mole7l azo-isobutyronitrile(AIBN): 0 transfer agent CTB, 13.6 x io- mole/I. benzoylperoxide (BPO): ® transfer agentdodecyl mercaptane (DDM). t3.6 x t0 mole/I. BPO. (Reprinted by permission of
Makrornolekul are Chernie)
C0U,
'U>C00
250
Figure 9. Polymerization kinetics of bulk at 50°C with various concentrations of carbontetrabromide. Initiator is lauroyl peroxide. [CBr4] = 5 x io mole/i. (Curve A); 5 x 10_2mole/I. (Curve B); 5 x 10_i mole/l. (Curve C). (Reprinted by permission of Makroinolekulare
Chemie)
Derivation of kinetic expressions for the reaction rate in bulk polymerizationThe main feature of the kinetics of the radical polymerization in bulk
polymerization may be summarized to be:A marked decrease in rate as well as in degree of polymerization
accompanies the formation of precipitated polymer particles. The subsequent134
12
8
2 3
lirnemin

RADICAL POLYMERIZATION OF VINYL CHLORIDE
course is characterized by a marked autocatalytic effect. There is a significantinfluence of particle number on the reaction rate at low conversion.
Addition of certain chain transfer agents leads to a marked increase of theinitial rate of bulk polymerization and at the same time the autocatalyticeffect disappears.
In order to derive an expression for the rate of reaction, equation 2 isconveniently rewritten in the form
— kp[M]R• + k[M]R (39)
or in degree of conversion:
=—(kP[M]LR•L + k[M]R.) (40)
The concentrations of monomer in the dilute phase, {M] I. and concentratedphase [M] are constant up to -.70 per cent conversion and equal to 13.6and 6 mole/I respectively, at 50°C. The number of moles of radicals in thedilute phase, RL, and in the particles, R, are calculated from equations 18and 19 without applying any a priori assumption on the distribution ofradicals between the two phases. In accordance with Mickley et al.8 it isassumed that the rates of radical absorption and desorption are determinedand may be expressed as diffusion processes.
ka = 41tDLNr (41)kdc = 4itDNr (42)
where N is the number of particles, r is the particle radius and DL and Dare the diffusion coefficients in the dilute phase and in the particles,respectively.
The values 3.6 x 108 1/mole sec and 2 x 10h1 dm2/sec are used forand D respectively. These values and the form of kdc, being determined
by a diffusion process within the particles, are based upon experimentalresults with emulsion polymerization, as described later. The experimentallydetermined quantities are in fact k/k and The absolute valuesgiven for and DL are based upon Burnett and Wright's value ofk = 1.1 x iO I/mole sec, at 50°C16. The value of absorption of radicals isnot known. If it is determined by the diffusion of the radicals in the dilutephase, the value of DL is probably of the order of magnitude of 108 to 10dm2/sec. The comparatively low value of D is possibly due to the fact that thedesorption of a radical from a particle has to be preceded by a chain transferto monomer33. The distribution of the initiator between the two phases isalso unknown. If this distribution is solely determined by entropy factors, avalue of [I]p/[I]L = 0.5 is estimated. The value of kL is uncertain. Burnettand Wright give a value of kIL = 2 x i09 1/mole sec, at 50°C which givesa value of k/k = 0.24. Several authors have reported that this value may betoo high'2' 13, 1719 Because of the uncertainty as to the value of kL, thecalculations have been carried out for ktL/kp = 5 (as obtained by use of theBurnett and Wright values), 20 and 40. The last value is obtained by use of the
135
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
value of k as given by Burnett and Wright = 1.1 x 10 1./mole sec and thevalue of k/k = 0.089 (1+ mole sec) given by Tkachenko et al.18. Thedefinition of the kL applied in the calculation of k/l4 is uncertain. In mostpapers it is apparently, by definition, twice the value applied in this paper.In this case a value kL/ktp = 20 would be more correct It should be pointedout that the value of ktL will affect the value for the number of moles of radicalsin the particles and in the dilute phase only in conditions where the termina-tion in the dilute phase has a significant influence on the total rate of termina-tionj.
In Figure 10 calculated curves with ktL/kp = 20 and DL = iO and 10 8dm2/sec are compared with an experimental curve from Figure 2 at a givenrate of radical production. In this calculation a value of k1 = 1.15 x 106sec' for the decomposition of LPO at 50°C and an efficiency factor1' ofI has been assumed. It. appears from Figure 10 that while the shape of thetheoretical curves at conversions above 2 per cent is similar to theexperimental curve, the calculated curves predict a considerably lower rateat low conversion. It does not seem possible by this simple treatment toexplain the high rate at low conversion without accepting a rather un-reasonable low rate of radical absorption into the particles. Calculationscarried out with ktL/ktp = 5 and 40 showed that the value of kL hardlyhad any effect on the number of radicals in the particles and in the dilutephase even at very low conversion under the given conditions.
0.5
0.4
u Q3
0.2
0.1
100 200 300 400
Time, mm
Figure 10. Bulk polymerization at 50CC of VC. Solid curve: experimental curve taken fromFigure 2 with [I] = 1.06 x 10—2 mole/I.: dotted curves calculated by help of equations 10,
18 and l9:——DL = 108;———DL = 107dm2/sec
Crosato-Arnaldi's1 1, as well as Mickley's8 expressions seems to fit betterthe experimental results at low conversion, as their treatment assumes
136
It is of course possible that the value of k given by Burnett et a!. is too high and their k,Lis correct. Calculations show that this leads to the same general conclusions as presented below.

RADICAL POLYMERIZATION OF VINYL CHLORIDE
contribution to the rate from a practically constant rate of polymerizationin the dilute phase at low conversion. As discussed earlier, this assumptionmay be open to doubt. The question is whether it is possible to explain therelatively high rate at low conversion as found experimentally even if it isaccepted that the absorption of radicals into the particles and terminationof radicals in the particles, already at very low conversion, plays a dominantrole in determining the total number of radicals in the system. This is whatthe experiments with precipitation polymerization would suggest, as wellas the observations of Cotman3 and Bort5 on particle formation. The sub-division of the reaction zone in discrete particles may possibly account for therelatively high rate at low conversions. For the calculation of this effectone may use the Stockmayer2° and O'Toole21 treatment, as modified byUgeistad et a!.22 to take into account the reabsorption of desorbed radicalsinto the particles. The effect of the subdivision may only be expected to be ofimportance at low conversion. In that case the formation of radicals frominitiator decomposition inside the particles may be neglected and one maymake use of the recursion formula, which applies for the case when theradicals are liberated in the outer phase:
(PA/N)Pfl = [kd + nkp/v] (n + 1)P 1 + (kp/v) (n + 1) (n + 2)P2 (43)PA is the rate of radical absorption into the particles, N is the number ofparticles per I of initial volume and kd is the rate constant for desorption ofradicals from a particle. (The rate of desorption from a particle containingn radicals is now expressed as r = n x kd x N. Therefore kd =k4/Vp).Also P is the probability of an n fold occupancy in a particle and v is thevolume of one particle. The solution of this equation as given by O'Toolefor the average number of radicals per particle is:
fl = I(1)/I,_ 1(a) (44)
where I, and 'rn-i are modified Bessel functions.
a = (8c) = (8pAv/Nktp) (45)
m = kdv/kp (46)
The rate of absorption of radicals into the particles may be expressed as:
PA L + knN — 2ktLJ (47)
where L is the rate of formation of radicals in the dilute phase, L the numberof radicals in the dilute phase. The rate of absorption is assumed to beproportional to the concentration of radicals in the liquid phase, i.e.PA = ka(flL/VL). By insertion of (nL/VL) = PA/ka into equation 47 andmultiplying each term by v/kpN one obtains:
(48)where
= PLV/N2k (49)137

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
Y = 2N2ktpkLVL/kVp (50)
By means of equations 44 and 48 n may be calculated as a function of ',m and Yas described in a previous paper22. The total number of radicals inthe particles, n, is obtained as N x n and the number of radicals in thedilute phase is given by:
flL—YXc(X (51)
The values of PA, L ktL, and k1 in equations 43—5 1 are expressed in units ofmolecules. m is a measure of the relative chance of radical escape from theparticle, Yis a measure of the chance of reabsorption. The higher the value ofYat a given particle number, the lower is the chance for reabsorption. Highvalues of m and Ywill at a given particle number tend to decrease the overallnumber of radicals in the particles and to increase the number of radicalsin the liquid phase.
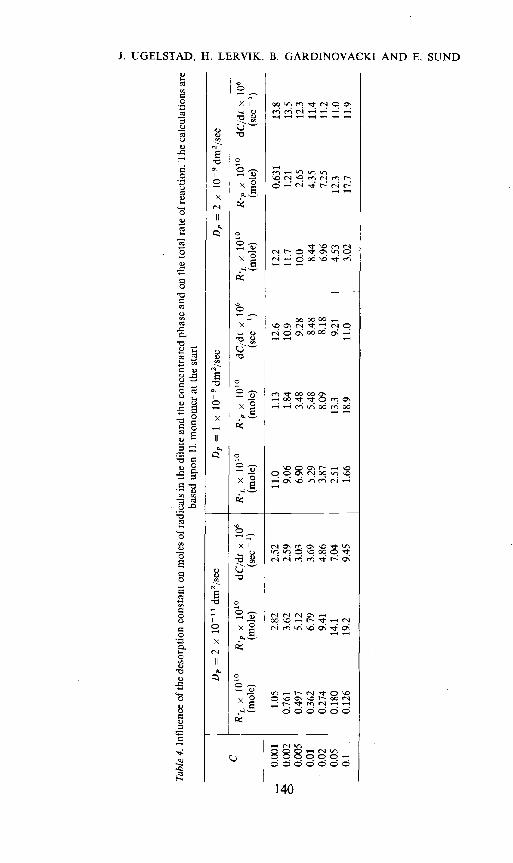
Results of calculations based upon the above treatment, which takes intoaccount the subdivision of the concentrated phase are given in Tables 2Table 2. Effect of subdivision of the concentrated phase on the number of moles of radicals in thedilute, RL, and the concentrated phase, R•, and the total rate of polymerization. The calculationsare based upon 11. monomer at the start. Rate of radical production = p = 2.4 x 10—8mole/sec; k1 = 3.6 x 108 I/mole sec; D = 2 x 10h1 dm2/sec; N = 4.5 x 1014 particles;
kL/ktP = 20 and 4QX
c RL x 1010(mole)
R• x 1010(mole)
dC/dt x 106(sec 1)
0.001 1.05 (0.871) 2.82 (1.87) 2.52 (1.87)0.002 0.761 (0.660) 3.62 (2.65) 2.59 (2.01)0.005 0.497 (0.458) 5.12 (4.19) 3.03 (2.54)0.01 0.362 (0.346) 6.7 (5.93) 3.69 (3.26)0.02 0.274 (0.260) 9.4 (8.39) 4.86 (4.36)0.05 0.180 (0.173) 14.1 (13.3) 7.04 (6.63)0.1 0.126 (0.120) 19.2 (18.8) 9.45 (9.24)
= 20 and 40 give the same values. Initial rate without precipitation: k,L,k,p = 20: (dC/dc) = 1.4 x 10 sec.kL/IcP 40: (dC/dc) = I x 10' sec
and 3. The rate of radical production is the same as in Figure 10 and thevalue of DL is iO-. The values given in the brackets are those obtained whenthe effect of subdivision on the termination reaction in the particles is nottaken into account. The effect of the subdivision of th concentrated phase isto decrease the chance of termination in the particles at low conversion.As seen from Table 2 the result is a marked increase in the number of moles ofradicals in the particles, and also a small increase in the dilute phase, at lowconversion. The effect decreases with increasing conversion. Therefore theeffect of the subdivision will be to increase the rate of polymerization at lowconversion, and thus bring the theory more into accordance with theexperimental result. If one uses the values given in Table 2 to compare therate of termination in the two phases, it is easily calculated that the terminationin the particles is already quite dominating at low conversion. Therefore notonly the number of moles of radicals in the particles, but also in the dilutephase is practically completely determined by the rate of termination in the
138

RADICAL POLYMERIZATION OF VINYL CHLORIDE
particles. The value of kL will not be expected to influence the value of thenumber of radicals in either of the two phases over quite a wide range ofkL. Thus calculation with kL values corresponding to ktL/ktp = 5 andktL/ktp 40 have only negligible influence on the radical number in thetwo phases. In this respect the present theory is completely different from theone of Mickley et al. and Crosato-Arnaldi et a!. where the number of radicalsin the dilute phase is considered to be determined by the termination inthat phase.
The rate which would be obtained in the absence of precipitation is givenat the bottom of Table 2, and therefore this treatment may also directlyexplain the decrease in rate by precipitation. The decrease in the molecularweight accompanying the decrease in rate due to precipitation followsnaturally from the high concentration of radicals in the precipitated phaseat low conversion.
It is noteworthy that even at ktL/klp values of 40 the precipitation will beexpected to lead to a marked decrease in rate. Absorption of radicals into theparticles leads to a decrease in the effective volume and thereby to an increasein the total rate of the second order termination reaction. The effect is partlycounteracted by the effect of subdivision of the reaction zone and bydesorption of radicals from the particles. Furthermore the values given inTable 2 predict that the particles are the dominating reaction zone for thepolymerization reaction already at very low conversion.
Table 3. Influence of particle number on the total rate of polymerization. The calculations arebased upon 11. monomer at the start
C
dC/dt x 106 (sec)
N = 4.5 x 1014 N = 4.5 x 1015 N = 4.5 x 1016(particles) (particles) (particles)
0.0010.0020.0050.01
2.52 3.59 4.912.59 3.90 5.313.03 4.77 6.533.69 5.79 8.00
Table 3 illustrates the calculated effect of an increase in the number ofparticles on the rate of reaction. The increase in subdivision caused by ahigher particle number is expected to decrease further the overall rate oftermination in the particles. As seen from Table 3 this leads to a furtherincrease in the rate of polymerization. The experimental evidence is that thenumber of particles at low conversion, below 1 per cent, is considerablyhigher than 4.5 x 1014. It seems that when this is taken into account, theagreement between the theoretical and experimental results is improved.It should be pointed out that the calculations in Tables 2 and 3 are basedupon a rather high value of DL. A lower value of DL will lead to an increasein the rate at low conversion.
The presence of a chain transfer agent is, by the present theory, expectedto increase the value of m by increasing the effective diffusion coefficient.If the experimental value of the diffusion coefficient is given by Dk TM/kP33
139
rn
r:d,
Thb
le 4
. Inf
luen
ce o
f the
deso
rptio
n con
stan
t on
mol
es of
radi
cals
in th
e dilu
te a
nd t
he c
once
ntra
ted p
hase
and
on th
e to
tal r
ate o
f rea
ctio
n. T
he c
alcu
latio
ns a
re
base
d upo
n 11
. m
onom
er a
t the
sta
rt
—
0.00
1 0.
002
0.00
5 0.
01
0.02
0.
05
0.1 C
D =
2 x
10—
11 d
m2/
sec
D =
1 >
< l0
dm2/s
ec
= 2
x 1
09dm
2/se
c
R x
1010
R
x 10
10
dC/d
t x 1
06
(mol
e)
(mol
e)
(sec
1)
R
L x
101
0 x
lOb
dC/d
t >
<
106
(mol
e)
(mol
e)
(sec
')
RL
x 1
010
R•
x 10
10
dC/d
t x 1
06
(mol
e)
(mol
e)
(sec
) 1.
05
2.82
2.
52
0.76
1 3.
62
2.59
0.
497
5.12
3.
03
0.36
2 6.
79
3.69
0.
274
9.41
4.
86
0.18
0 14
.1
7.04
0.
126
19.2
9.
45
11.0
1.
13
12.6
9.
06
1.84
10
.9
6.90
3.
48
9.28
5.
29
5.48
8.
48
3.87
8.
09
8.18
2.
51
13.3
9.
21
1.66
18
.9
11.0
122
11.7
10
.0
8.44
6.
96
4.53
3.
02
0.63
1 1.
21
2.65
4.
35
7.25
12
.3
17.7
13,8
13
.5
12.3
11.4
11
.2
11.0
0 z

RADICAL POLYMERIZATION OF VINYL CHLORIDE
(where D is the diffusion coefficient for monomer and transfer agent radicalsand kfM is the transfer constant to monomer) the ratio of the values of inwith and without chain transfer agents, 7, is given by: kfT[T]/kfM[M] wherekfT is the chain transfer constant to Tand [M], and [T] refers to the concen-tration in the particles.
Table 4 shows the results of calculations of an increase in in by factors of50 and 100. An increase in m leads to a higher number of moles of radicalsin the liquid phase and a lower number of radicals in the particles. Theoverall rate at low conversion is increased, the autocatalytic effect is absent.At sufficiently high values of m the theory predicts that the rate at lowconversion should be independent of the value of m. This seems to be inaccordance with the experimental results (Figure 9). It should be pointed outagain that this treatment does not involve any a priori assumption of a thermo-dynamically established equilibrium distribution of radicals between the twophases.
B. EMULSION POLYMERIZATION
All authors who have studied the emulsion polymerization of vinylchloride agree that the reaction does not follow case 2 of the Smith—Ewarttheory The main deviations from this theory are:
1. The number of latex particles varies strongly with the emulsifierconcentration while the polymerization rate changes relatively very little.
2. The number of particles is independent of the initiator concentration.3. The rate of reaction increases with increasing initiator concentration
at a constant number of particles. (The reaction order found by the differentauthors varies between 0.52 and 0.82.)
4. The conversion versus time curves show an autocatalytic behaviourup to high conversion.
In order to explain the small effect of particle number on the rate ofreaction, Giskehaug24 suggested a considerable contribution from polymeri-zation in the water phase. He then had to assume a value of k/k of about5 (l mo1es sect), which may seem unreasonably high compared to thevalue obtained by other authors. Gerrens et al.2 found that the experimentalvalues of rate and particle number when inserted in the rate expression
kPCM -rp= T xNxn'A
leads to a value of considerably below 0.5, which indicated that at a givenmoment only a small fraction of particles contained a radical. In order toexplain the low value of n, Gerrens suggested a degradative chain transferreaction to polymer. He found a reaction order of 0.8 with respect to initiator.This was taken in support of the assumption of a degradative chain transferreaction. This high order with respect to initiator has not been confirmedby other authors23' 24 Peggion et al.25 suggest that the deviation from theSmith—Ewart theory may be due both to the relatively high solubility ofmonomer in water and to the low solubility of polymer in monomer. Theconcentration of monomer in the particles at equilibrium is given by equation
141
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
1. In cases when the monomer is a good solvent for its polymer, the restrictedswelling of the particles, which is obtained in emulsion polymerization, isdetermined by a balance between the swelling force and the interfacialenergy26. In the case of vinyl chloride, however, the interfacial energy termwill even in emulsion polymerization be of minor importance, compared tothe term containing the interaction energy, in limiting the swelling of theparticles with monomer27. It is not easy to see why this difference should leadto any deviation from the Smith—Ewart theory. Also in the case of vinylchloride the reaction zones, the particles, are one-phase systems of a constantcomposition. The composition of the particles is the same as in bulkpolymerization, i.e. they contain a considerable amount of monomer.
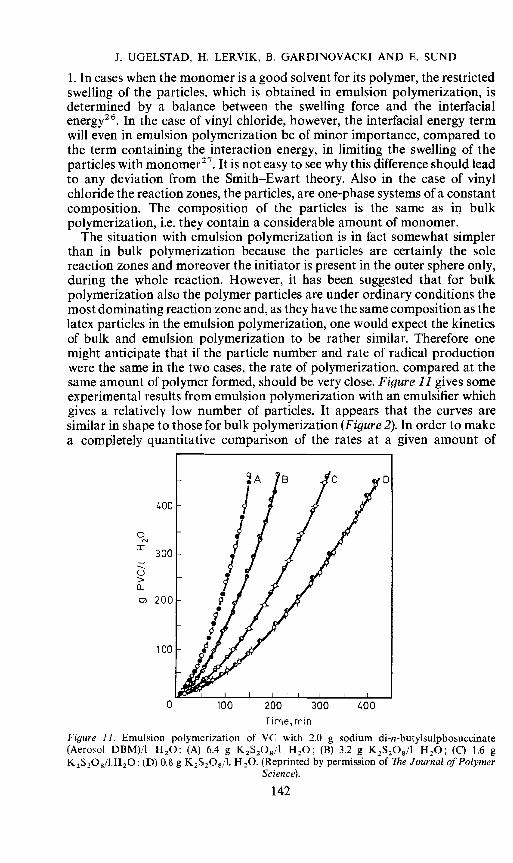
The situation with emulsion polymerization is in fact somewhat simplerthan in bulk polymerization because the particles are certainly the solereaction zones and moreover the initiator is present in the outer sphere only,during the whole reaction. However, it has been suggested that for bulkpolymerization also the polymer particles are under ordinary conditions themost dominating reaction zone and, as they have the same composition as thelatex particles in the emulsion polymerization, one would expect the kineticsof bulk and emulsion polymerization to be rather similar. Therefore onemight anticipate that if the particle number and rate of radical productionwere the same in the two cases, the rate of polymerization, compared at thesame amount of polymer formed, should be very close. Figure 11 gives someexperimental results from emulsion polymerization with an emulsifier whichgives a relatively low number of particles. It appears that the curves aresimilar in shape to those for bulk polymerization (Figure 2). In order to makea completely quantitative comparison of the rates at a given amount of
Figure 11. Emulsion polymerization of VC with 2.0 g sodium di-n-butylsulphosuccinate(Aerosol DBM)/l H20: (A) 6.4 g K2S203/1 H20; (B) 3.2 g K2S208/l H20; (C) 1.6 gK2S,08/l.H20: (D) 0.8 g K2S208/l. H20. (Reprinted by permission of The Journal of Polymer
Science).
142
0:i:
0>ci0)
400
300
200
100
0 100 200 300 400Tme, mm
RADICAL POLYMERIZATION OF VINYL CHLORIDE
Log (particle number)
Figure 12. Log differential rate (g PVC1 I. H20 h) versus log (number of particles per 1 1120)at a conversion of 100gPVC/LH2O. Concentration of K2S208 = 1.6 g/l.H20.• No emulsifier;e sodium octylsuiphate; D sodium decylsulphate; 0 sodium dodecylsuiphate (Empicol-8043);esodium tetradecylsulphate; sodium hexadecylsuiphate; sodium di-n-butylsulphosuccinate;EIIK-30, C12 K-30, C15; K-30 is a mixture of various paraffinsulphonates with average chain
length of 12 and 15 C-atoms respectively
0
C-)>0cy)
Time, mm
Figure 13. Comparison of g PVC formed as a function of time with different initiator systems.Solid curves: [K2S208] = 6 x 1O3mole/1. H20; 0 [K2S208] =2 x 10 mole/I. H20;[HSOfl = 1.2 x iO mole/i. H20, [CuSO4] = 7.9 x i0 mole/i. H20; e x,'-azobis
(methylbutyronirile-y-N4-sulphonate (AMBN-S) = 5.4 >< 10 mole/i. 1120.
143
ci)
ci
C)0
2.1 "02.0 - ,A
1'5
0 100 200 300 400

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
polymer formed, one would have to compare experiments where both therate of radical production and particle number were the same. Such data arenot available. However, one may from the experimental effect of the particlenumber on the rate (Figure 12) extrapolate the result of emulsion polymeri-zation to the corresponding conditions for bulk polymerization. Such anextrapolation does indicate that the rates are nearly the same.
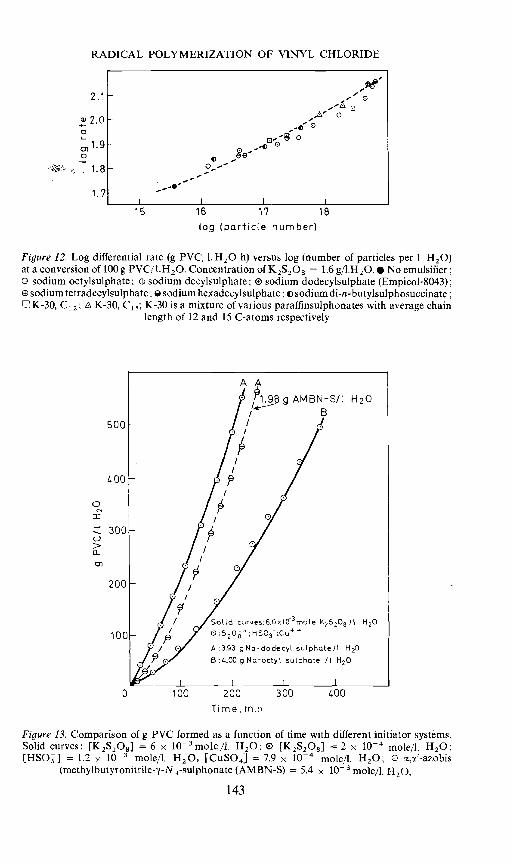
Figure 12 gives log differential rate at 100g PVC formed as a function oflog particle number with different types and amounts of emulsifiers. Thisfigure includes results with a larger number of emulsifiers than in a previouspaper23. It appears that the rate of emulsion polymerization is determined,not by the type of emulsifier, but by the number of particles formed. Thereaction order with respect to the number of particles is very low. It increaseswith increasing number of particles up to the value of about 0.15.
In view of the fact that the rate of decomposition of persulphate is verysensitive to the presence of any foreign agents in the water28, it might havebeen suggested that the effect shown in Figure 12 was due to an effect on therate of radical production from persuiphate. To clarify this question, therates obtained with different emulsifiers and persulphate as initiator werecompared with the same emulsifier systems using a redox system composedof persulphate, bisulphite and copper ions29 as the initiator system. Anyeffect on the rate of radical production from persulphate, caused by theparticles, emulsifiers or other agents present, may be expected to becompletely outbalanced by the effect of the bisulphite—copper system. Theresults of this investigation are shown in Figure 13. The redox system wasadjusted to give the same rate as 6 x 10 moles of pure persuiphate perI H20 at a given amount of sodium lauryl sulphate. Exactly the same initiatorsystems were then applied with a given amount of sodium octylsuiphate.The ratio of particle number obtained with lauryl sulphate to that obtainedwith octylsulphate is about 50. Octylsulphate gives a considerably lowerrate and again the kinetic curves with the two initiator systems practicallyoverlap. This result confirms the previously assumed effect of particle numberon the polymerization rate, at a constant rate of radical production. Figure 13includes the result of a run with a water soluble azo compound also. Thedecomposition rate constant for this compound is 3.35 x 106 sec' at 50°Cand is reported to be quite unaffected by the presence of any foreignsubstances30. It may be anticipated from a comparison of the rates in the twocases that the rate of radical production from persuiphate in the presence ofvinyl chloride is 3.4 times higher than that which would be expected from therate constant for decomposition in pure water31. Breitenbach et al.32 foundthat the rate of radical production from persulphate was increased by afactor of 10 when the solution was saturated with vinyl acetate.
The results of emulsion colymerization of vinyl chloride have led tothe conclusion that the mean number of radicals per particle is verylow, iO — 10-1, dependent upon the particle number2' 23, Ugelstad.et al.22'23'33 have shown that the low value of n may be explained by inter-particle termination brought about by a rapid desorption and reabsorption ofradicals. Such a mechanism may be especially favourable in the case ofvinyl chloride because of the chain transfer to monomer. Approximateexpressions for the rate of polymerization, valid at low values of n (4 0.5),
144

RADICAL POLYMERIZATION OF VINYL CHLORIDE
were evaluated. This was done by assuming that at low values of ii onlyparticles with 0,1, and 2 radicals need to be considered, which led to the follow-ing simplified expression for the rate of reaction23:
rp = kPCMp%4 (i + : ) (52)
pW and are expressed in units of molecules, rp is the rate of reaction inmoles of monomer, pW is the rate of radical production in the water phase,V is the total volume of the particles and Nv is the number of particles, allvalues defined per I H 20. CM is the concentration of monomer in theparticles, kd is the rate 'constant' of desorption of radicals.
If the desorption is determined by a diffusion process within the particles(kd = 4rrDr/v), equation 52 takes the form:
= kPCMpW+ (_ + N'*v)4 (53)
Equation 53 was found to describe the experimental results of emulsionpolymerization over a wide range of particle number and conversion.The value of and D applied in the treatment of bulk polymerization hasbeen obtained from the experimental data for emulsion polymerization(Figure 12 and equation 53). The values given for and D in the presentcase are higher than those previously reported23. This is due to the highervalue of the radical production from persuiphate, which the results givenin Figure 13 indicated, that has been applied in the present calculation.
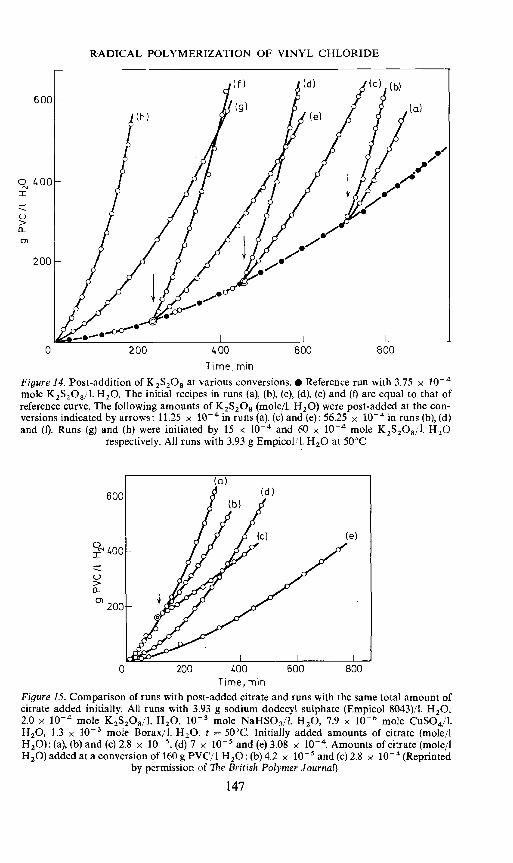
The evidence for the low values of n was based upon calculations from thefamiliar rate equation for emulsion polymerization, rp =kCMn x N/NA, byapplication of experimental values for CM and N, and with Burnett andWright's value of k. As discussed above the value of k is somewhat uncertain.Independent evidence for the low value of n in emulsion polymerization ofvinyl chloride has recently been obtained by experiments where the rate ofradical production was changed during the run33. Figure 14 gives resultsobtained with post addition of initiator. With the type and amount ofemulsifier applied, the number of particles was 8 x 1018 per I H20. In theexperiments with post addition of initiator the reaction was initiated withthe same amount of initiator as in the reference run. After three differentconversions the concentration of initiator was increased 4 and 16 timesrespectively. It appears that the differential rate of reaction at a givenconversion is independent of the time at which post addition took place.A fourfold increase in the initiator concentration led to a doubling of therate in accordance with a half order with respect to initiator. The importantfact concerning the question of the value of n is that the 'new' rate after postaddition of initiator was established very rapidly, in less than one minute.After addition of initiator, and establishment of a rate twice that of thereference run, the total number of radicals in the system must accordinglybe twice that before addition, i.e. 16 x 1018 x ñ. The establishment of the
145

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
new rate therefore requires the production of at least 8 x 1018 x i radicals.The time t required to produce this number of radicals is:
t = 8 x 10'8n/p (54)
where p is the rate of radical production from persuiphate after a fourfoldincrease in the persuiphate concentration. Inserting into equation 54,t = 1 mm, and the experimental value for p, gives a value of n <0.05.Figure 15 gives results where the rate of radical production has been reducedafter a certain conversion. In these experiments the redox system ofpersuiphate, bisuiphite and copper ions has been applied and the rate ofradical production has been reduced by an increase in the amount of citratein the system. Again the 'new' rates corresponding to the new rate of radicalproduction are established very rapidly.
Although these effects of change in rate of radical production refer toemulsion polymerization, the similarity of emulsion and bulk polymerizationof vinyl chloride may possibly allow general conclusions to be drawn. In theauthors' opinion the rapid establishment of the new' rate corresponding to agiven conversion and rate of radical production argues strongly for thejustification of the application of the quasi steady state treatment and speaksagainst the non-steady state treatment suggested by Magat34.
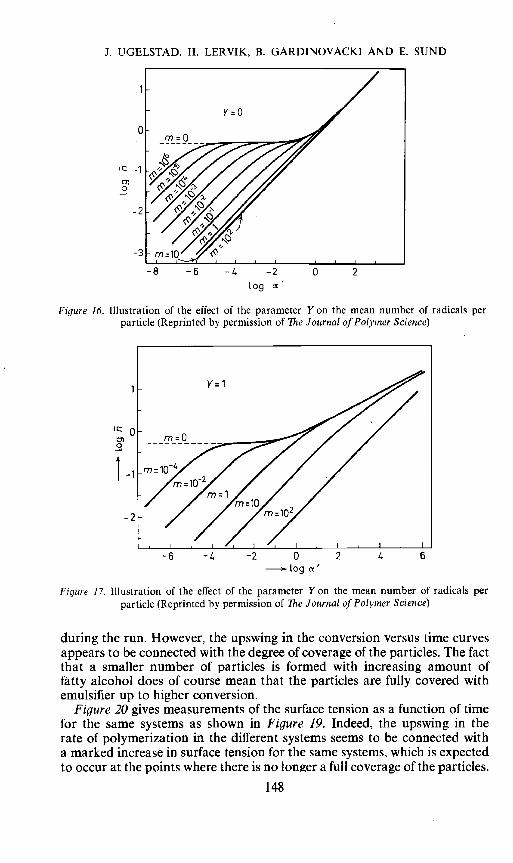
In comparison of emulsion polymerization with bulk polymerization itshould be brought to mind that in bulk polymerization an increase in therate of desorption of radicals from the particles leads to an increase in rateat low conversions. In the same way a decrease in the rate of absorption wasshown theoretically to be expected to increase the rate. In the case of emulsionpolymerization the opposite is true. The concentration of monomer in thewater phase is so low that an increase in desorption or a decrease inabsorption is expected to lead to a decrease in rate, due to a decrease in thenumber of radicals in the particles. The effect of a decrease in ka is to increasethe parameter Y Figures 16 and 17 illustrate the calculated effect of anincrease in Y Comparing at the same values of 'and m, an increase in Y leadsto a decrease in the number of radicals in the particles, which in the case ofemulsion polymerization leads to a decrease in rate. Possibly, such a decreasein rate of radical absorption is responsible for the effects with mixedemulsifiers3 .
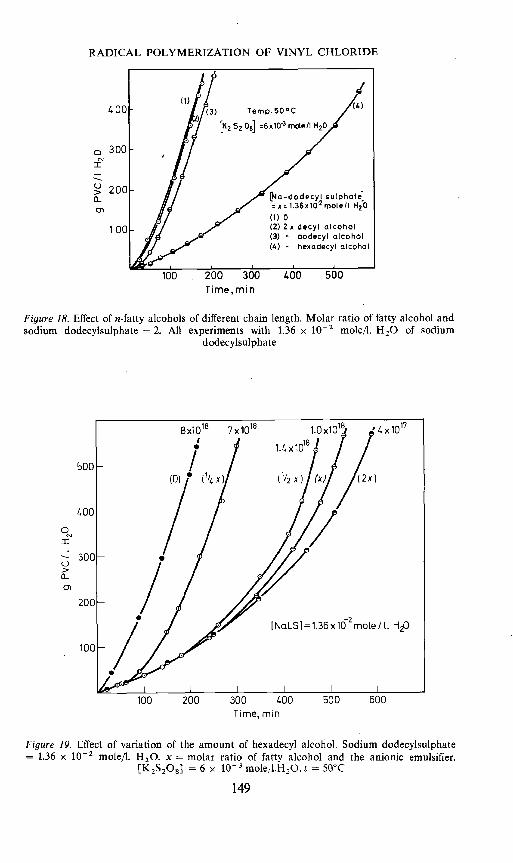
Figure 18 gives results with Na-laurylsulphate and mixtures of Na-laurylsuiphate with fatty alcohols of different chain length as emulsifiers.It appears that hexadecyl alcohol leads to a strong decrease in the rate ofreaction and number of particles, while the lower alcohols have hardly anyeffect in either respect.
Figure 19 gives the result of experiments where the amount of hexadecylalcohol has been varied. An increase in the amount of the fatty alcoholleads to a steady reduction in the particle number. The initial rate is stronglyreduced and rather surprisingly is the same in all cases, independent of theamount of fatty alcohol. An increase in the amount of fatty alcohol does,however, lead to a delay in the upswing of the curve. One might, at first sight,think that this would mean that in all cases where fatty alcohol was applied,the particle number at low conversion was the same and very low. This did notappear to be the case. There is only a small change in the particle number
146
RADICAL POLYMERIZATION OF VINYL CHLORIDE
Time, mm
Figure 14. Post-addition of K2S2O8 at various conversions.• Reference run with 3.75 x l0mole K2S2O8/l. H2O. The initial recipes in runs (a), (b), (c), (d), (e) and (I) are equal to that ofreference curve. The following amounts of K2S208 (mole/l. 1120) were post-added at the con-versions indicated by arrows: 11.25 x 1O' in runs (a), (c) and (e); 56.25 x 10 in runs (b), (d)and (I). Runs (g) and (h) were initiated by 15 x io- and 60 x iO- mole K2S208/l. 1120
respectively. All runs with 3.93 g Empicol/l. H20 at 50°C
Time, mm
Figure 15. Comparison of runs with post-added citrate and runs with the same total amount ofcitrate added initially. All runs with 3.93 g sodium dodecyl sulphate (Empicol 8043)/I. 1120,2.0 x 10 mole K2S2O8/l. H2O, io mole NaHSO3/I. H2O, 7.9 x 10-6 mole CuSO4/LH20, 1.3 x l0 mole Borax/I. 1120. t = 50°C. Initially added amounts of citrate (mole/IH20): (a), (b) and (c) 2.8 x ltV5, (d) 7 x l0 and (e) 3.08 x 10g. Amounts of citrate (mole/lH20) added at a conversion of 160 g PVC/l H2O: (b) 4.2 x 10 and (c) 2.8 x io (Reprinted
by permission of The British Polymer Journal)
147
0 200 400 600 800
401
C)>0-0)
0 200 400 600 800
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
0)0
Figure 16. Illustration of the effect of the parameter Y on the mean number of radicals perparticle (Reprinted by permission of The Journal of Polymer Science)
Figure 17. Illustration of the effect of the parameter Y on the mean number of radicals perparticle (Reprinted by permission of The Journal of Polymer Science)
during the run. However, the upswing in the conversion versus time curvesappears to be connected with the degree of coverage of the particles. The factthat a smaller number of particles is formed with increasing amount offatty alcohol does of course mean that the particles are fully covered withemulsifier up to higher conversion.
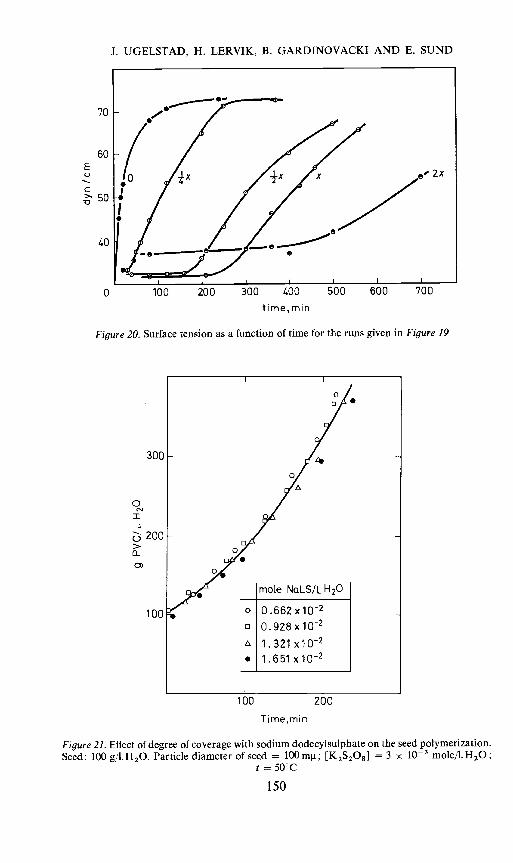
Figure 20 gives measurements of the surface tension as a function of timefor the same systems as shown in Figure 19. Indeed, the upswing in therate of polymerization in the different systems seems to be connected witha marked increase in surface tension for the same systems, which is expectedto occur at the points where there is no longer a full coverage of the particles.
148
log x
-2
-6 —1. —2 0 2 4 6
0I0>ILa,
RADICAL POLYMERIZATION OF VINYL CHLORIDE
200 300
Time, mm
Figure 18. Effect of n-fatty alcohols of different chain length. Molar ratio of fatty alcohol andsodium dodecylsuiphate = 2. All experiments with 1.36 x 10-2 mole/I. H20 of sodium
dodecylsulphate
501
401
0I3000>0
0,200
100
100 200 300 400 500 600Time, mm
Figure 19. Effect of variation of the amount of hexadecyl alcohol. Sodium dodecylsulphate= 1.36 x 10_2 mole/I. H2O. x = molar ratio of fatty alcohol and the anionic emulsifier.
[K2S203] = 6 x 10 mole7l.H20. = 50CC.
149
70
60E0
40
0
J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
Figure 20. Surface tension as a function of time for the runs given in Figure 19
0I3 200>00)
100
Time,min
200
Figure 21. Effect of degree of coverage with sodium dodecylsuiphate on the seed polymerization.Seed: 100 g/l.H20. Particle diameter of seed = 100 mJL; [K2S208] = 3 x iO mole/LH2O;
= 50°C
150
time, mm
300
mole NQLS/LH2O
00
0.662x102
0.928x102
1.321 x1021.651 x102
0
0>0
RADICAL POLYMERIZATION OF VINYL CHLORIDE
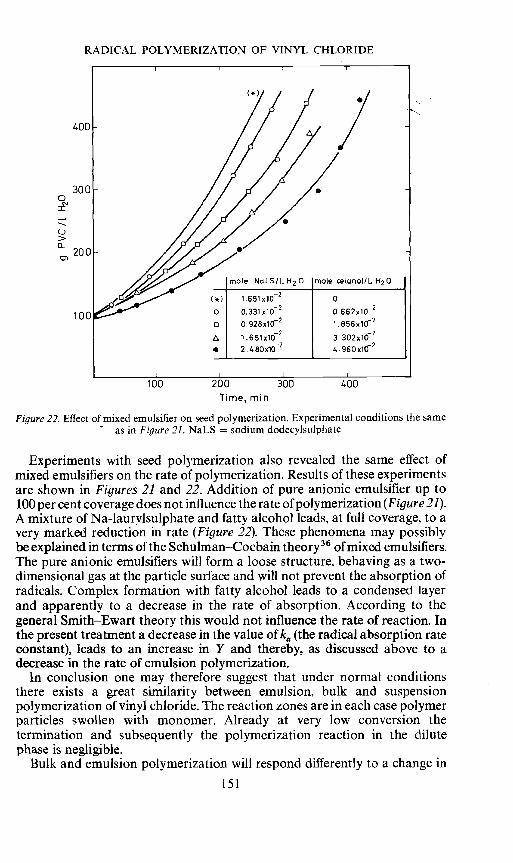
Figure 22. Effect of mixed emulsifier on seed polymerization. Experimental conditions the sameas in Figure 21. NaLS = sodium dodecylsuiphate
Experiments with seed polymerization also revealed the same effect ofmixed emulsifiers on the rate of polymerization. Results of these experimentsare shown in Figures 21 and 22. Addition of pure anionic emulsifier up to100 per cent coverage does not influence the rate of polymerization (Figure 21).A mixture of Na-laurlsulphate and fatty alcohol leads, at full coverage, to avery marked reduction in rate (Figure 22). These phenomena may possiblybe explained in terms of the Schulman—Cocbain theory36 of mixed emulsifiers.The pure anionic emulsifiers will form a loose structure, behaving as a two-dimensional gas at the particle surface and will not prevent the absorption ofradicals. Complex formation with fatty alcohol leads to a condensed layerand apparently to a decrease in the rate of absorption. According to thegeneral Smith—Ewart theory this would not influence the rate of reaction. Inthe present treatment a decrease in the value of ka (the radical absorption rateconstant), leads to an increase in Y and thereby, as discussed above to adecrease in the rate of emulsion polymerization.
In conclusion one may therefore suggest that under normal conditionsthere exists a great similarity between emulsion, bulk and suspensionpolymerization of vinyl chloride. The reaction zones are in each case polymerparticles swollen with monomer. Already at very low conversion thetermination and subsequently the polymerization reaction in the dilutephase is negligible.
Bulk and emulsion polymerization will respond differently to a change in
151
41
Time, mm

J. UGELSTAD, H. LERVIK, B. GARDINOVACKI AND E. SUND
the rate of radical desorption or absorption due to the difference in themonomer concentration in• the liquid phase, in the two cases. A generaltheory based upon desorption and reabsorption of radicals in the particlesis able to explain the experimental results both for bulk and emulsionpolymerization.
REFERENCES1 0. Talamini and F. Peggion. Chap. 5 of Vinyl Polymerization, edited by G. Ham, Vol. I,
Part I, Marcel Dekker Inc., New York 1967.2 H. Gerrens, W. Fink and F. Kohnlein. Preprints I.U.P.A.C. International Symposium on
Macromolecular Chemistry, Prague, p. 331, 1965.J. D. Cotman Jr., M. F. Gonzales and G. C. Claver. J. Polymer Sci. A-i, 5, 1137 (1967):D. N. Bort, V. G. Marinin, A. J. Kalinin and V. A. Kargin, Vysokomolekul. Soedin. AlO, 2574(1968).D. N. Bort, V. 0. Marinin, A. J. Kaiinin and V. A. Kargin. Dokl. Akad. Nauk SSSR, 183, 1080.(1968).
6 A. Schindler and J. W. Breitenbach. Ric. Xci. Suppl. 25, 34 (1955).W. J. Bengough and R. 0. W. Norrish. Proc. Roy. Soc. (London) A 200, 301 (1950).H. S. Mickley, A. S. Michaels and A. L. Moore. J. Polymer Sci. 60, 121 (1962).G. Talamini. J. Polymer Sci. A-2, 4, 535 (1966).
10 G. Talamini and G. Vidotto. Chim. delle Macromol. 118 (1968).A. Crosato-Arnaldi, P. Gasparini and G. Talamini. Makromol. Chem. 117, 140 (1968).
12 M. Ryska, M. Kolinsky and D. Lim. J. Polymer Sd. C. 16, 621 (1967).13 A. Crosato-Arnaldi, 0. Talamini and 0. Vidotto. Makromol. Chem. 111, 123 (1968).14 J w Breitenbach and A. Schindler. Mh. Chein. 86, 437 (1955).
J. W. Breitenbach, 0. F. Olaj, H. Reiff and A. Schindler. Makromoi. Chem. 122.51 (1969).15 G. Vidotto, S. Brugnaro and 0. Talamini. Makromol. Chem. in press.16 G. M. Burnett and W. Wright. Proc. Roy. Soc. (London), A221, 28 (1954).17 G. V. Tkachenko, P. M. Khomokovskii and S. S. Medvedev. Zh. Phys. Khim. 25, 823 (1951).18 0. V. Tkachenko, G. G. Petlukov and V. A. Dodonov. Vysokomolekul. Soedin. 3, 1549 (1961).19 W. H. Atkinson, C. H. Bamford and G. C. Eastmond. Trans. Faraday Soc. 66, 1446 (1970).20 W. H. Stockmayer. J. Polymer Sd. 24, 314 (1957).21 J. T. O'Toole. J. Polymer Sd. 9, 1291 (1965).22 Ugelstad, P. C. Mork and J. Aasen. J. AppL Polymer Sci. A-i, 5, 2281 (1967).23 J Ugelstad, P. C. Mørk, P. Dahl and P. Rangnes. J. Polymer SeE. C 27,49 (1969).24 K. Giskehaug. Soc. Chem. Intl. (London) Monograph, 20, 235 (1966). The symposium, The
Chemistry of Polymerization Processes, held April 1965.25 E. Peggion, F. Testa and 0. Talamini. Makromol. Cheat. 71, 173. (1964).26 M. Morton, S. Kaizerman and M. W. Altier. J. Colloid Sci. 9,300 (1954).27 H. Gerrens. Dechema Monograph. 49, 53 (1963).28 C. F. M. Morris and A. 0. Parts. Makromol. Chem. 119, 212 (1968).29 P. C. Merk and J. Ugelstad. Makromol. Cheat. 128, 83 (1969).30 J W. Breitenbach and K. Kuchner. Mh. Chem. 97, 662 (1966).31 j M. Kolthoff and!. K. Miller. J. Chem. Soc. 73,3055(1951).32 w Breitenbach, K. Kuchner, M. Fritze and H. Tarnowescki. British Polymer J. 2,13(1970).
J. Ugelstad and P. C. Mark. British Polymer J. 2, 31(1970).M. Magat. J. Polymer Sci. 16, 491 (1955).J. Ugelstad, H. Lervik and B. Gardinovacki. To be published.
36 J. H. Schulman and E. 0. Cockbain. Trans. Faraday Soc. 36, 651 (1940).F. Collins. Personal communication.
152





















![Vinyl chloride power point[1]](https://img.dokumen.tips/doc/110x75/5565724cd8b42a7b518b4ebd/vinyl-chloride-power-point1-558497ad57a9c.jpg)







