Embed Size (px)
Citation preview

PYRIDOXAL PHOSPHATE ENZYMES:Mechanistic, Structural, and EvolutionaryConsiderations
Andrew C. Eliot1 and Jack F. Kirsch2
1Department of Chemistry and 2Departments of Chemistry and Molecular and CellBiology, University of California, Berkeley, California 94720-3206; email:[email protected], [email protected]
Key Words substrate specificity, reaction type specificity, enzyme inhibition,enzyme mechanism
f Abstract Pyridoxal phosphate (PLP)-dependent enzymes are unrivaled in thediversity of reactions that they catalyze. New structural data have paved the way fortargeted mutagenesis and mechanistic studies and have provided a framework forinterpretation of those results. Together, these complementary approaches yield newinsight into function, particularly in understanding the origins of substrate andreaction type specificity. The combination of new sequences and structures enablesbetter reconstruction of their evolutionary heritage and illuminates unrecognizedsimilarities within this diverse group of enzymes. The important metabolic roles ofmany PLP-dependent enzymes drive efforts to design specific inhibitors, which arenow guided by the availability of comprehensive structural and functional databases.Better understanding of the function of this important group of enzymes is crucial notonly for inhibitor design, but also for the design of improved protein-based catalysts.
CONTENTS
INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384MECHANISTIC VERSATILITY OF PLP . . . . . . . . . . . . . . . . . . . . . . . . . 385STRUCTURAL DIVERSITY . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 387DETERMINANTS OF REACTION TYPE . . . . . . . . . . . . . . . . . . . . . . . . . 394
Dunathan Stereoelectronic Hypothesis . . . . . . . . . . . . . . . . . . . . . . . . . . 394Transamination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 395Racemization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398Role of the Closed Conformation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 399
DETERMINANTS OF SUBSTRATE SPECIFICITY. . . . . . . . . . . . . . . . . . . 399Role of the Closed Conformation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 399Dual Specificity of Aminotransferases . . . . . . . . . . . . . . . . . . . . . . . . . . 399
EVOLUTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 403MECHANISMS OF INHIBITION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 406
Annu. Rev. Biochem. 2004. 73:383–415doi: 10.1146/annurev.biochem.73.011303.074021
Copyright © 2004 by Annual Reviews. All rights reservedFirst published online as a Review in Advance on April 2, 2004
3830066-4154/04/0707-0383$14.00
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Noncovalent Inactivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 406Activated Nucleophiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 408Activated Electrophiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 409Reversible Competitive Inhibition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 410The Challenge of Inhibitor Specificity . . . . . . . . . . . . . . . . . . . . . . . . . . 411
CONCLUSIONS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 411
INTRODUCTION
Following its identification in 1951 as one of the active vitamers of vitamin B6
(1), pyridoxal 5�-phosphate (PLP) has been the subject of extensive researchdirected toward understanding its unequaled catalytic versatility.1 As a result, thebasic mechanisms of PLP-assisted reactions, both in solution and enzyme-associated, have been well characterized and are now a staple of biochemistrytextbooks (for example, see 2, 3). A number of reviews have addressed thesubject in greater detail, and readers are directed particularly to the recent articlesby Hayashi (4) and John (5). Transaminases, edited by Christen & Metzler (6),remains an excellent and thorough source of information about these enzymes.More detailed reviews concentrate on the function of a number of specificenzymes, including tryptophan synthase (7), O-acetylserine sulfhydrylase (8),�-aminolevulinate synthase (9), serine hydroxymethyltransferase (SHMT) (10),and branched chain amino acid aminotransferase (BCAT) (11).
In addition to their versatility as catalysts, PLP-dependent enzymes haveattracted attention because of their widespread involvement in cellular processes.These enzymes are principally involved in the biosynthesis of amino acids andamino acid-derived metabolites, but they are also found in the biosyntheticpathways of amino sugars (12) and other amine-containing compounds. Theirimportance is further underscored by the number identified as drug targets. Forexample, inhibitors of �-aminobutyric acid aminotransferase (GABA ATase) areused in the treatment of epilepsy (13), SHMT has been identified as a target forcancer therapy (14), and inhibitors of ornithine decarboxylase (ODC) areemployed in the treatment of African sleeping sickness (15). Functional defectsin PLP enzymes have furthermore been implicated in a number of diseasepathologies, including homocystinuria, which is most frequently caused bymutations in cystathionine �-synthase (16, 17).
1Abbreviations: AATase, aspartate aminotransferase; ACC, 1-aminocyclopropane-1-carboxylate; AlaP, 1-aminoethylphosphonate; ALR, alanine racemase; AONS, 8-amino-7-oxononanoate synthase; ATase, aminotransferase; BCAT, branched chain amino acidaminotransferase; DAAT, D-amino acid aminotransferase; DGD, dialkylglycine decar-boxylase; GABA, �-aminobutyric acid; OAT, ornithine aminotransferase; ODC, ornithinedecarboxylase; PLP, pyridoxal 5�-phosphate; PMP, pyridoxamine 5�-phosphate; SAM,S-adenosyl-L-methionine; SHMT, serine hydroxymethyl transfease; and TATase, aro-matic amino acid aminotransferase.
384 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Despite the long history of research in the field, we are only now beginningto answer some of the most exciting questions. In particular, technologicaladvances that have allowed ever more rapid determination of enzyme structuresand the accumulation of large sequence databases have also enhanced ourunderstanding of enzymatic catalysis. Analyses of sequences and structures yieldsignificant insight regarding the evolution of this diverse class of enzymes.Additionally, crystal structures elegantly demonstrate how PLP enzymes harnessthe potential of the cofactor to accelerate the rates only of specific reactions, andthey provide the basis for targeted mutagenesis.
This review describes many of the new insights that have come from recentstructural and mechanistic studies, with a primary focus on determinants ofsubstrate specificity and reaction type, as well as the design of inhibitors for thisclass of enzyme. The mechanisms and structures are briefly discussed to providebackground.
MECHANISTIC VERSATILITY OF PLP
PLP-catalyzed reaction types can be divided according to the position at whichthe net reaction occurs. Reactions at the � position include transamination,decarboxylation, racemization, and elimination and replacement of an electro-philic R group. Those at the � or � position include elimination or replacement.Examples of each of these reactions are shown in Scheme 1, and the basicmechanisms are shown in Schemes 2 (�) and 3 (� and �). Exceptions to thesecommon types include the formation of a cyclopropane ring from S-adenosyl-L-methionine (SAM), catalyzed by 1-aminocyclopropane-1-carboxylate (ACC)synthase (18), and the cleavage of ACC to �-ketobutyrate and ammonia,catalyzed by ACC deaminase (19); these are not discussed here. Because manyof the reaction pathways share common intermediates, a number of enzymes alsocatalyze reactions that are combinations of the basic types, such as the decar-boxylation-dependent transamination (aminoisobutyrate � pyruvate 3 acetone� CO2 � L-Ala) catalyzed by dialkylglycine decarboxylase (DGD; 20) or the�-elimination and �-replacement (O-phospho-L-homoserine � H2O 3 L-Thr �Pi) catalyzed by threonine synthase (21).
Although the scope of PLP-catalyzed reactions initially appears to be bewil-deringly diverse, there is a simple unifying principle. The cofactor in all casesfunctions to stabilize negative charge development at C� in the transition statethat is formed after condensation of the amino acid substrate with PLP to forma Schiff base (referred to as the external aldimine).2 The fully formed carbanion
2There are two exceptions to this basic principle. The glycogen phosphorylase family ofenzymes utilizes the phosphate group of PLP for catalysis [reviewed in (106)], and theaminomutase family catalyzes a radical-initiated reaction on PLP-bound amino acidsubstrates [reviewed in (107)]. Neither of these families will be discussed in this review.
385PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.
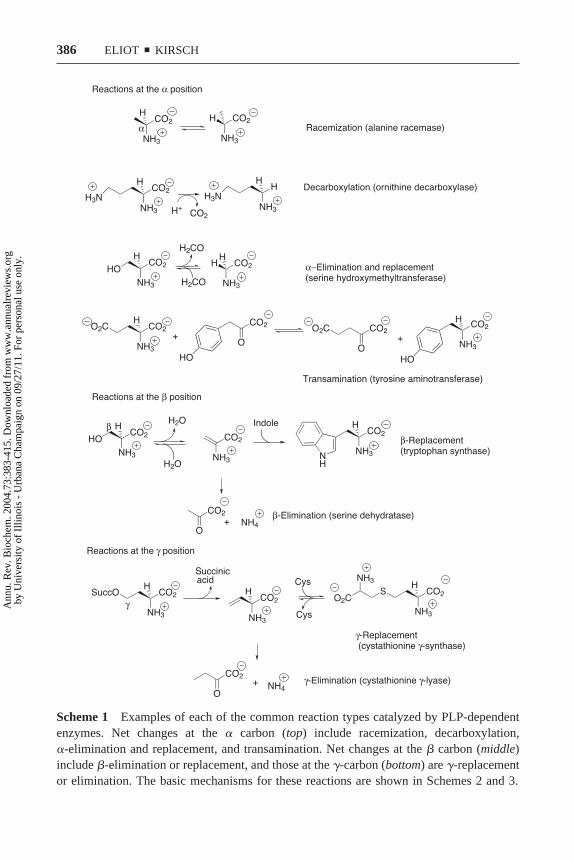
Scheme 1 Examples of each of the common reaction types catalyzed by PLP-dependentenzymes. Net changes at the � carbon (top) include racemization, decarboxylation,�-elimination and replacement, and transamination. Net changes at the � carbon (middle)include �-elimination or replacement, and those at the �-carbon (bottom) are �-replacementor elimination. The basic mechanisms for these reactions are shown in Schemes 2 and 3.
386 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

is referred to as the quinonoid intermediate (Scheme 4). The pKa for loss of theC�-proton from an amino acid in the absence of PLP is � 30 (22); therefore, thatanion, formation of which is required in order to enable all of the describedchemistry, is ordinarily inaccessible under physiological conditions.3 The stabi-lization of the C�-anion is facilitated by delocalization of the negative chargethrough the pi system of the cofactor, and for this reason PLP is often describedas an electron sink. This factor allows PLP in the absence of enzyme to catalyzemany of the possible reactions slowly (reviewed in 6). The function of the proteinapoenzyme, therefore, is to enhance this innate catalytic potential and to enforceselectivity of substrate binding and reaction type. In most cases, this selectivityis exquisite—potential side reactions are limited to barely detectable levels.
STRUCTURAL DIVERSITY
Although PLP enzymes were for a time underrepresented in protein structuredatabases (5), the situation has been rectified in recent years, as an abundance ofnew structures have been solved. It was initially postulated that the structures ofPLP enzymes would correlate with the reaction type (24), but it has since beenfound that each of the major structural classes contains representatives ofmultiple reaction types, the evolutionary implications of which are discussedbelow. All PLP enzymes whose structures have been solved to date belong to oneof five fold types, which have been described in detail in two recent reviews (25,26), and therefore are only briefly summarized here. Figure 1 shows singlerepresentatives of Fold Types I-IV, whose mechanisms are discussed in thisreview.
The majority of known structures are of Fold Type I (aspartate aminotrans-ferase family) enzymes, a group that includes many of the best-characterized PLPenzymes. They invariably function as homodimers or higher-order oligomers,with two active sites per dimer. The active sites lie on the dimer interface, andeach monomer contributes essential residues to both active sites. In general, thetwo active sites are independent, but asymmetry has been observed in a fewcases. Negative cooperativity, for example, has been reported in GABA ATase,where a dimer with two functional active sites exhibits the same specific activityas a dimer with only one functional active site (27). The two active sites in eachdimer of glutamate-1-semialdehyde aminomutase also exhibit different reactiv-ities, as evidenced by the biphasic kinetics of reduction of the enzyme-PLPaldimine by sodium borohydride (28). Each monomer of Fold Type I enzymeshas a large and a small domain. In a number of cases [e.g., aspartate amino-transferase (AATase)], these domains move significantly upon association with
3Certain non-PLP-dependent amino acid racemases, such as glutamate racemase (23),remain puzzling exceptions. If these reactions proceed through a transition state that hassubstantial anion character, it is not apparent how that state is stabilized.
387PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

388 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

substrate, creating a closed conformation that may contribute to the specificity forboth the substrate and the reaction type (see below).
The structures of Fold Type II (tryptophan synthase family) enzymes aresimilar to those of Fold Type I, but the proteins are evolutionarily distinct (29).One significant difference is that the active sites of Fold Type II enzymes arecomposed entirely of residues from one monomer [first observed in tryptophansynthase (30)]. Nevertheless, the functional form remains a homodimer orhigher-order oligomer. These enzymes also differ from those of Fold Type I inthat they often contain additional regulatory domains. Examples include threo-nine synthase (31) and cystathionine �-synthase (32), which are allostericallyregulated by SAM, and threonine deaminase, which is regulated by isoleucineand valine (reviewed in 33).
The Fold Type IV (D-amino acid aminotransferase family) enzymes aresuperficially similar to Fold Types I and II, in that they are also functionalhomodimers, and the catalytic portion of each monomer is composed of a smalland a large domain. The cofactor is bound in a site that is a near mirror imageof the Fold Types I and II binding sites, so that the re rather than si face is solventexposed (34).
Fold Types III (alanine racemase family) and V (glycogen phosphorylasefamily) are strikingly different from the other PLP enzymes. The Fold Type Venzymes are mechanistically distinct in utilizing the phosphate group of thecofactor for catalysis and are not considered further. The Fold Type III enzymesconsist of a classical �/� barrel and a second �-strand domain. Interestingly, themode of binding of PLP is similar to that of other fold types, with the phosphate
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Scheme 2 Examples of mechanisms of reactions involving net change at the � position.All reactions are shown starting with the substrate aldimine, which is formed by transaldi-mination of the lysine-bound PLP. Racemization: the bacterial alanine racemase utilizes atyrosine residue (51, 104) to deprotonate L-alanine, forming the quinonoid intermediate,which is reprotonated by a lysine residue on the opposite face of the cofactor to produceD-alanine. Decarboxylation: the reaction begins with loss of CO2 from the substratealdimine, producing the quinonoid intermediate. Protonation by an unidentified active siteresidue in ornithine decarboxylase generates the product aldimine. �-Replacement: thewell-studied serine hydroxymethyltransferase (10) initiates the retro-aldol cleavage ofserine by deprotonation of the hydroxyl group. Formaldehyde is released to generate thequinonoid intermediate. Protonation of the quinonoid at C� by the lysine produces theproduct aldimine of glycine. Transamination: the first half-reaction catalyzed by tyrosineaminotransferase involves initial proton abstraction from the glutamate aldimine at C� bythe active site lysine, yielding the quinonoid intermediate. Reprotonation at C4� of thecofactor by that lysine generates the ketimine intermediate, which is subsequently hydro-lyzed to release �-ketoglutarate, leaving the enzyme in the PMP form. A complete catalyticcycle involves subsequent reaction with hydroxyphenylpyruvate to give tyrosine and toregenerate the PLP form of the enzyme.
389PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

390 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

group anchored at the N terminus of an ��helix, H-bond interactions made to the3�-OH, and the presence of the ubiquitous lysine Schiff base. Furthermore, theseenzymes are also obligate dimers, as each monomer contributes residues to bothactive sites (35).
Multiple scaffolds have clearly evolved to bind PLP and to assist in catalysisby this cofactor. In no case does the fold type dictate the reaction type, as eachfold type contains multiple reaction types, and all common reaction types arefound in at least two fold types.4
4Although only one racemase structure has been solved (alanine racemase Fold Type III),serine racemase is predicted to be in Fold Type II based on sequence (36), and the fungalalanine racemase in Fold Type I (37).
Scheme 4 The primary function of PLP is to stabilize anions generated at C�. Thenegative charge is delocalized by resonance in the pi system of the cofactor in the quinonoidintermediate after loss of a proton from the external aldimine.
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Scheme 3 Examples of mechanisms of reactions at the � and � positions. �-Replacement:tryptophan synthase (7) catalyzes a net �-replacement by first deprotonating the serinealdimine at C�, producing the quinonoid intermediate. Protonation of the hydroxyl group bythe active site lysine promotes its elimination, generating the aminoacrylate aldimine.Indole adds to C� to form a second quinonoid that is subsequently protonated at C� togenerate the product aldimine. �-Replacement: in the cystathionine �-synthase-catalyzed�-replacement reaction (105), O-succinyl homoserine is deprotonated at C� to produce thequinonoid intermediate that is subsequently protonated at C4� of the cofactor to give theketimine intermediate. Proton abstraction at C� by an unknown active site base results inelimination of the succinyl group, which may occur in either a step-wise or concerted(shown) manner. Michael addition of cysteine to the �,�-unsaturated ketimine and subse-quent proton transfers yield a second quinonoid intermediate that is protonated at C� to formthe product aldimine.
391PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

392 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Fig
ure
1R
ibbo
ndi
agra
ms
ofre
pres
enta
tive
enzy
mes
ofFo
ldT
ypes
I-
IV.E
ach
stru
ctur
ede
pict
sa
hom
odim
erw
ithth
ein
divi
dual
mon
omer
sdi
stin
guis
hed
byco
lor.
The
PLP
cofa
ctor
issh
own
inre
d(t
ople
ft).
Fold
Typ
eI
(E.
coli
aspa
rtat
eam
inot
rans
fera
se;
pdb
file
1asn
)(t
opri
ght)
.Fo
ldT
ype
II(S
alm
onel
laty
phim
uriu
mO
-ace
tyls
erin
esu
lfhy
-dr
ylas
e;1o
as)
(bot
tom
left
).Fo
ldT
ype
III
(Bac
illu
sst
earo
ther
mop
hilu
sal
anin
era
cem
ase;
1sft
)(b
otto
mri
ght)
.Fo
ldT
ype
IV(t
herm
ophi
licB
acil
lus
sp.
D-a
min
oac
idam
inot
rans
fera
se;
1daa
).T
hefig
ure
was
prep
ared
with
Ras
Mol
.
393PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

DETERMINANTS OF REACTION TYPE
Dunathan Stereoelectronic Hypothesis
Well before the lack of correlation between fold type and reaction type wasrecognized, much research was directed toward understanding the determinantsof reaction type specificity. Dunathan postulated in 1966 (38) that the topologyof the amino acid aldimine determined the bond to C� that would be broken. Hesuggested that that bond must be situated so that it will align perpendicularly withthe pyridine ring of the cofactor in the transition state of the reaction (Scheme 5).The ensuing carbanion is stabilized by conjugation with the extended pi system.This hypothesis was later confirmed when the structure of the aspartate amino-transferase/phosphopyridoxyl aspartate complex was solved (39). All subse-quently determined structures are consistent with this idea. Of particular interestin this respect are enzymes that catalyze C�-deprotonation and decarboxylationat different points during their catalytic cycle.
8-AMINO-7-OXONONANOATE SYNTHASE (AONS) AONS, the second of fourenzymes in the biotin biosynthetic pathway, catalyzes the decarboxylation andaddition of a pimeloyl group to alanine (Scheme 6). Interestingly, the reactionmechanism involves initial deprotonation rather than decarboxylation (40). Thequinonoid thus formed attacks pimeloyl-CoA, forming a �-keto acid intermedi-ate, which is decarboxylated and reprotonated to form the product aldimine(Scheme 7). Decarboxylation of a �-keto acid is a facile reaction that may notrequire the participation of the cofactor, but the observation of the expectedquinonoid intermediate indicates that it is likely involved (41). Although nostructure is available of the intermediate aldimine formed prior to decarboxyl-ation, the structure of the product aldimine shows that pimeloyl addition occurs
Scheme 5 The Dunathan stereoelectronic hypothesis. Substrates are bound to PLPsuch that the bond to C� that is to be broken is aligned with the pi orbitals of thecofactor. Control of the substrate orientation thus enables the enzyme to distinguishbetween, for example, decarboxylation and deprotonation.
394 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

on the face opposite from that of deprotonation, resulting in the carboxylategroup occupying nearly the same position as that previously held by the proton(Scheme 7).
DIALKYLGLYCINE DECARBOXYLASE (DGD) DGD is both an aminotransferase anda decarboxylase. A complete catalytic cycle involves decarboxylation andtransamination of dialkylglycine to generate a ketone product and the pyridox-amine phosphate (PMP) form of the enzyme followed by reaction with pyruvatein a typical transamination to generate alanine and to restore the PLP form of theenzyme (Scheme 8). Structural and mechanistic studies demonstrated that thedecarboxylation of the dialkyl amino acids is forced by large side chains, whichare not accommodated in the same site as the methyl side chain of alanine. Theyinstead occupy the same position as the alanine carboxylate. This reorientationresults in the scissile bond being that between C� and the carboxylate rather thanthat between C� and the proton (42) (Scheme 9). Thus decarboxylation isprefered over deprotonation.
These examples demonstrate how PLP enzymes can strictly control the initialbond-breakage step and limit other potential reactions by restricting the boundsubstrate to a specific orientation. Control of the steps subsequent to the initialbond breakage is less well understood. C�-deprotonation, for example, can leadto �- or �-elimination/replacement, racemization, or transamination (Schemes 2,3). To promote the desired reaction, an enzyme must rigorously govern theelectron flow and proton transfers. The structures of PLP enzymes have shownhow active site residues are positioned to promote particular reaction types andto restrict possible side reactions. These principles are readily appreciatedthrough a comparison of the structures of enzymes catalyzing transamination tothat of a racemase.
Transamination
The first structures of a PLP-dependent enzyme to be determined were of themitochondrial and cytosolic aspartate aminotransferases (AATase) (39, 43, 44).They explained much of the available mechanistic data and inspired fruitful
Scheme 6 The reaction catalyzed by 8-amino-7-oxononanoate synthase. The carboxylategroup of alanine is replaced by a pimeloyl moiety. The mechanism of this reaction is shownin Scheme 7.
395PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

targeted mutagenesis investigations (45–49). The electron sink nature of thecofactor is enhanced by a close interaction of the pyridinium nitrogen with anaspartate residue (D222), which acts to maintain the cofactor in the protonatedform (Scheme 10). The displaced Schiff base lysine residue (K258) is positionedto transfer a proton to and from C� and C4�. Since the substrates of AATase,aspartate and glutamate, cannot undergo �- or �- elimination, the primary sidereaction that must be minimized is racemization. This objective is achieved byclosure of the enzyme around the substrate, so that solvent water molecules donot access the quinonoid intermediate to protonate the re face (50). Furthermore,there are no active site acids in position to donate a proton to the re face of thatintermediate. Subsequently solved structures of other aminotransferases con-
Scheme 7 The mechanism of the 8-amino-7-oxononanoate synthase-catalyzedreaction. The alanine aldimine is initially deprotonated, and the resulting quinonoidattacks the pimeloyl CoA thioester. Addition of the pimeloyl group to the faceopposite from the deprotonation event causes the carboxylate to reorient perpendic-ular to the pyridine ring, resulting in subsequent decarboxylation. Protonation of thissecond quinonoid produces the product aldimine. Adapted from Reference 41.
396 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

firmed that these are general properties of the entire class of enzymes. TheD-amino acid aminotransferase (Fold Type IV) is particularly interesting becauseits active site is in part a mirror image of the L-amino acid aminotransferases (34),in that the active site lysine is positioned on the re face and the si face is solventexposed, neatly accounting for its opposite stereospecificity.
Scheme 8 The reaction catalyzed by dialkylglycine decarboxylase. In the firsthalf-reaction, a dialkyl substrate is decarboxylated and transaminated, producingCO2, a ketone, and the PMP form of the enzyme. The latter form subsequently reactswith pyruvate in a transamination half-reaction to give alanine and to restore the PLPform of the enzyme.
Scheme 9 Steric control of reaction type by dialkylglycine decarboxylase. Thethree substituents on C� occupy distinct binding sites, labeled A, B, and C. Whereasthe A and B sites are tolerant of carboxylates and hydrogen or large alkyl groups,respectively, the C site only accommodates small alkyl groups. Amino acid substrateswith small side chains, such as alanine, bind preferably with their carboxylatemoieties in the B site, placing the C�-proton in the reactive A site. Substrates with alarge side chain (such as the phenylglycine shown), however, bind with that group inthe B site, forcing the carboxylate into the reactive A site. For this reason, thedistinction between decarboxylation or deprotonation is a consequence of the sub-strate structure. Adapted from Reference 42.
397PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Racemization
Although the structure of alanine racemase (ALR) from Bacillus stearother-mophilus (51) is the only one presently available, the catalytic mechanism isapparent from the unique architecture of its active site. An active site Brønstedacid is expected to be on the opposite re face to complement the lysine on the siface, and a tyrosine provides this function here (Scheme 2). Like AATase, ALRacts on substrates incapable of undergoing elimination; therefore, the primaryside reaction that must be limited is transamination. A major factor in promotingthat restriction is likely the arginine that replaces the aspartate near the pyri-dinium nitrogen (Scheme 10). The positively charged arginine prevents proto-nation of the cofactor (51), thereby negating its electron sink properties.Although it might be expected that this interaction would also impair the normalfunction of the enzyme, the racemization reaction may require less delocalizationof electron density into the cofactor. In this regard, it is notable that some knownamino acid racemases do not require a cofactor (see Footnote 1). Althoughracemization is generally presumed to proceed through a fully formed carbanionat C� (52), the instability of this species elicits consideration of an SE2mechanism where the incoming proton develops some bonding with C� in aconcerted transition state. Jencks pointed out (53) that stepwise mechanisms aregenerally preferred because of entropy considerations, but that mechanismsbecome concerted when the intermediate that would develop in the stepwiseprocess is too unstable to exist.
Interestingly, some of the insights gained from this structure cannot begenerally applied to all PLP-dependent racemases. Paiardini et al. (54) reported
Scheme 10 Control of the electron sink properties of PLP by the amino acid sidechain positioned nearest to the pyridinium nitrogen atom. An aspartate residueoccupies this locus in aspartate aminotransferase and all other known Fold Type Ienzymes, thereby maintaining ring protonation. The corresponding residue is anarginine in the bacterial alanine racemase, which is expected to maintain the cofactorin the unprotonated form. Lack of a proton at this position would greatly diminish theelectron withdrawing properties of PLP.
398 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

an alanine racemase that is predicted to be a member of Fold Type I and to havean aspartate residue that interacts with the pyridinium nitrogen, as do all otherFold Type I enzymes.
Role of the Closed Conformation
Although it is not clear if the observed conformational changes of PLP enzymescontribute to substrate specificity (see below), the existence of the closedconformation undoubtedly contributes to reaction type specificity. Wolfendenpointed out the advantages of a closed conformation that makes more contactswith the bound substrate than are possible in a conformation from which thesubstrate must be able to dissociate (55). The closed conformation can favorspecific reactions by providing greater control over proton transfers and solventaccessibility. A well-studied example is serine hydroxymethyltransferase, whichcatalyzes a variety of side reactions (transamination, decarboxylation, racemiza-tion, etc.) when presented with substrates other than serine. The open form of theenzyme is unable to discriminate between the various reaction types in the waythat the closed form can (37, 56). Therefore, these reactions occur, at least in part,because the substrates in question do not induce the closed conformation.
DETERMINANTS OF SUBSTRATE SPECIFICITY
Role of the Closed Conformation
The potential contribution of induced fit to enzymatic substrate specificity hasbeen much debated (for a recent overview, see 57), but it is now accepted that aconformational change induced only by certain substrates does not necessarilyresult in increased specificity for those substrates. Thus, it has been argued thatthe conformational change observed in AATase and many other PLP enzymesdoes not contribute to their substrate specificities (4). Exceptions to the generalrule, however, include cases where substrate association or product dissociationis rate-determining for good substrates, whereas chemistry is rate-determining forpoor substrates (58). Since it has been shown that release of the productoxalacetate is partially rate determining for the reaction of AATase with aspartateand �-ketoglutarate (�KG) (59), AATase is an example of an induced fit enzymein which the conformational change may contribute to substrate specificity. It isnot yet clear whether this conclusion extends to other PLP enzymes.
Dual Specificity of Aminotransferases
The basis for the dual specificity of aminotransferases has been elucidated byrecent structural information. Because the aminotransferase reaction requires twodifferent substrates to bind in succession to the same cofactor in the active site,these enzymes must be able to accommodate both structures while discriminating
399PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

against all others. One possible solution would be for the PLP itself to movebetween two different substrate binding sites, but such movement has never beenobserved. An alternative is to take advantage of flexible side chains to positionthe functional groups into exclusive binding sites. This is the case for histidinolphosphate aminotransferase, which reacts with both histidinol phosphate andglutamate. The recently solved structures of substrate complexes of this enzyme(60) show that, although the phosphate and carboxylate groups interact primarilywith the same arginine residue, the side chains of the two substrates have separatebinding sites (Scheme 11).
Most known aminotransferases, however, adopt strategies for binding bothsubstrates in the same site. Because many use the common substrate glutamate(thereby linking other amino acids to the cellular nitrogen pool), the problem ofdual specificity is generally that of accommodating the negatively charged�-carboxylate of glutamate in a site that must also accept a neutral or positivelycharged side chain. Two common solutions to this problem have been found: theuse of an arginine switch and an extended hydrogen bond network.
ARGININE SWITCHES The first example of an arginine switch was observed in anengineered enzyme that was constructed by introducing six mutations intoAATase. These changes resulted in a substantial increase in activity towardaromatic substrates (61). The subsequent determination of the structure of the
Scheme 11 Dual specificity of histidinol phosphate aminotransferase. The phosphate andcarboxylate moieties of glutamate and histidinol phosphate, respectively, bind in the samesite, although an additional arginine does interact with the phosphate group. In contrast, theoppositely charged imidazole and carboxylate side chains occupy spatially distinct sites andinteract with different active site residues. The enzyme thus recognizes each substratespecifically. Adapted from Reference 60.
400 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

mutant enzyme (62) showed that the large aromatic substrates are accommodatedby movement of Arg292 out of the active site. Dicarboxylate substrates bind their�- or �-carboxylate via direct interaction with this arginine through a bidentatehydrogen bond/ion pair in the canonical AATase conformation (Scheme 12). Theposition of Arg292 is locked in AATases; therefore, substrates lacking a carbox-ylate side chain are effectively excluded.
More recently, directed evolution techniques have been utilized to broaden thesubstrate specificity of AATase to include branched chain (63) or aromatic aminoacids (64). Crystal structures of mutants with increased activity toward branchedchain amino acids (65, 66) indicated that they also have acquired the ability to switchArg292 out of the active site when uncharged substrates are bound, indicating thatthis trait is easily induced in AATase and is crucial for dual specificity.
Arginine switches are not unique to engineered enzymes. Tyrosine (aromatic)aminotransferase (TATase) is a well-characterized Fold Type I enzyme that hasnatural specificity for the aromatic amino acids tyrosine, phenylalanine, andtryptophan, as well as for the dicarboxylic amino acids aspartate and glutamate.A number of structures are now available of the Paracoccus denitrificansTATase that clearly demonstrate the arginine switch (67, 68).
Crystallographic and modeling studies illuminated a similar strategy employedby the GABA (69, 70) and ornithine (71) aminotransferases, which react with both
Scheme 12 Schematic of the arginine switch in aminotransferases that react withboth dicarboxylic and aromatic amino acids. The �-carboxylate of glutamate (left)interacts closely with Arg292. This residue reorients to point out of the active sitewhen aromatic substrates bind (right). This movement allows the enzyme to acceptboth types of substrates. Adapted from Reference 62.
401PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

�-amino acid substrates and the common substrate glutamate. GABA ATase, likeAATase and TATase, binds the dicarboxylic acid substrate via two conservedarginines. The carboxylate of the �-amino acid, GABA, occupies the same positionas the �-carboxylate of glutamate, thereby taking advantage of the similar distancebetween the amino and carboxylate groups of GABA and the amino and �-carbox-ylate groups of glutamate. The second arginine (equivalent to Arg386 of AATase)moves to interact with a conserved glutamate near the active site (Scheme 13).
HYDROGEN BOND NETWORKS Arginine switches, however, are not ubiquitous,even among TATases. Early sequence alignments indicated that a subgroup ofaminotransferases (designated I�) lack an Arg292 equivalent (72). The onlystructure available for a TATase of this group is that of the unligandedPyrococcus horikoshii enzyme (73). Modeling of the substrates in the active sitesuggests that this enzyme binds glutamate via an extended hydrogen-bondingnetwork, as has been observed in the AATase from this same organism (74)(Scheme 14). The absence of a positively charged residue in this TATase makesit much easier to accommodate the uncharged substrates by simple rearrange-ment of the hydrogen bond network. Moreover, absence of the flexible arginineside chain allows this enzyme to distinguish between glutamate and aspartate.The specificity ratio (kcat/Km
Glu)/(kcat/KmAsp) for the P. horikoshii TATase is
3400 (73), compared to 0.27 for the Escherichia coli TATase (75), a typicalmember of the I� family.
Scheme 13 Schematic of the arginine switch in GABA aminotransferase. As in the caseof AATase, GABA ATase binds the dicarboxylic acid substrate glutamate via twoconserved arginines. In order to accommodate GABA, Arg445 moves away from thecofactor to engage in a salt bridge with a nearby glutamate residue. Adapted fromReferences 69 and 70.
402 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.
The recently solved structures of the natural E. coli branched chain amino acidaminotransferase (BCAT) (76) show that this enzyme also takes advantage ofdifferent hydrogen bond interactions. BCAT reacts with the branched chainaliphatic amino acids isoleucine, leucine, and valine, as well as with glutamate.The hydrophobic substrates and glutamate all bind in the same site, which is ahydrophobic pocket consisting largely of aromatic residues. In contrast to theTATases, there is no large-scale rearrangement of the H-bond network toaccommodate the charged substrate. Instead, the glutamate side chain, which isslightly longer than that of the other substrates, extends into a pocket where fourresidues (Arg97, Tyr31, Tyr129, and the backbone amide of Val109) hydrogenbond to the carboxylate. The arginine in this case forms only a monodentateinteraction rather than the bidentate H-bond network seen in other enzymes, andit is extensively H-bonded to neighboring residues, allowing it to remain inposition when the small hydrophobic substrates are bound (Scheme 15).
EVOLUTION
Evolutionary relationships among PLP-dependent enzymes have been exten-sively examined (25, 29, 72). The following discussion is therefore limited torecent insights emanating from the increased number of available structures.
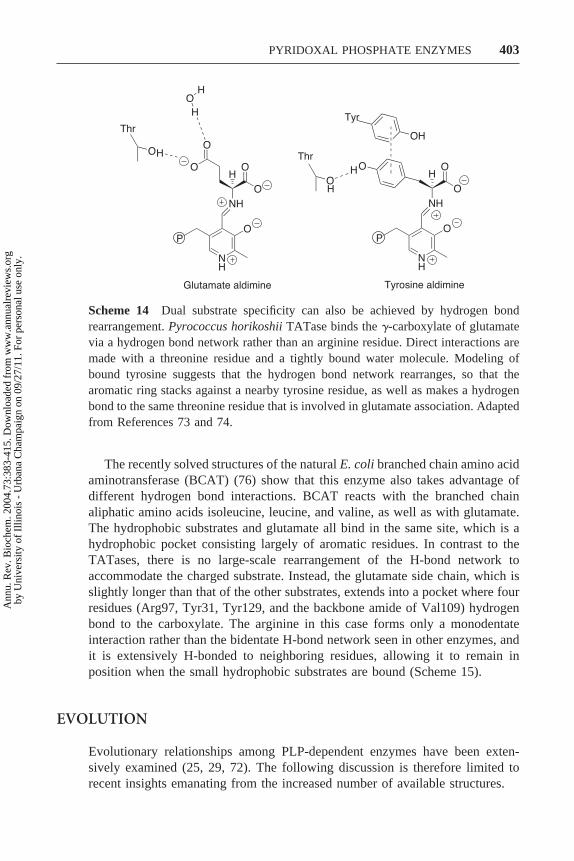
Scheme 14 Dual substrate specificity can also be achieved by hydrogen bondrearrangement. Pyrococcus horikoshii TATase binds the �-carboxylate of glutamatevia a hydrogen bond network rather than an arginine residue. Direct interactions aremade with a threonine residue and a tightly bound water molecule. Modeling ofbound tyrosine suggests that the hydrogen bond network rearranges, so that thearomatic ring stacks against a nearby tyrosine residue, as well as makes a hydrogenbond to the same threonine residue that is involved in glutamate association. Adaptedfrom References 73 and 74.
403PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

The analyses of sequences and structures that led to the categorization of PLPenzymes into the five recognized fold types also indicated that the fold types areevolutionarily distinct (29). The similarities of the cofactor binding sites thusprovide an excellent example of convergent evolution. It is believed that reactiontype generally evolved first within each fold type, followed by narrowingsubstrate specificity (29). A number of enzymes, however, group most closelywith those that catalyze reactions of a different type, suggesting that theirreaction type-specificity arose later in evolution. In these cases, the change inreaction type can often be explained as a consequence of altered substratespecificity. For example, enzymes catalyzing �-elimination are found in manyevolutionary subgroups, often among enzymes that catalyze transamination or�-elimination. Since �-elimination of a good leaving group is a very facilereaction, it is easy to imagine that the acquisition of improved binding of asubstrate with a � leaving group could readily lead to a change in the reactionspecificity to favor elimination. Another example of an enzyme where a substratespecificity change effects a change in reaction type is dialkylglycine decarbox-ylase (see above). DGD is fundamentally an aminotransferase like most of itsclosest evolutionary relatives, but it also catalyzes decarboxylation of dialkylsubstrates that bind in a unique orientation.
Scheme 15 Dual substrate recognition by branched chain amino acid aminotransferase.The substrate binding pocket is composed primarily of aromatic residues, and the hydro-phobic substrate isoleucine is surrounded by five of them, only three of which are shown(Phe36, Tyr164, and Tyr31). The longer glutamate substrate extends far enough to formhydrogen bonds with the hydroxyl groups of two tyrosines and the guanidino group of anarginine residue. Note that the orientation of the substrate C�-H bond is opposite that foundin other fold types. Adapted from Reference 76.
404 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

L-Threonine-O-3-phosphate decarboxylase, an enzyme that is also mostclosely related to aminotransferases, is unusual because the change in reactiontype from transamination to decarboxylation requires a different substrate ori-entation with respect to the cofactor, and therefore mandates substantial rear-rangement of the substrate binding mode. How this change was achievedevolutionarily is evident in the fact that the closest evolutionary relative of theenzyme is histidinol-phosphate aminotransferase, which has evolved to bind aphosphate group in the position usually occupied by the substrate carboxylate.One can imagine that from this starting point (or something similar), it is easy toimprove binding of threonine-phosphate in the same site. With the phosphate asan anchor point in the usual carboxylate site, the carboxylate is positioned forbond breakage (Scheme 16). It is not clear, however, how decarboxylation-dependent transamination is averted, but presumably the mechanism is similar tothose of other decarboxylases and is related to the lack of a proton on the activesite lysine after decarboxylation. Solvent exclusion can also prevent hydrolysis ofthe ketimine intermediate formed by protonation at C4� (78).
Possibly the most interesting group of PLP enzymes from an evolutionarystandpoint is that of Fold Type IV (D-amino acid aminotransferase (DAAT)family). This family is unique in containing both a D-amino acid and an L-aminoacid aminotransferase (the previously described BCAT). Since the active sitelysine can only catalyze proton transfer on one face (79), DAAT and BCAT mustbind their substrates in opposite orientations (Scheme 17), as evidenced in thecrystal structures of these enzymes (76, 80). Either the binding mode of the
Scheme 16 Possible evolutionary route to threonine phosphate decarboxylase. Histidinolphosphate aminotransferase is quite similar to Fold Type I amino acid aminotransferasesand must share a common ancestral aminotransferase. However, it acquired additionalaffinity for a phosphate group. Relatively minor changes are required for that enzymeto accommodate threonine phosphate. With the large phosphate group as an anchor,the carboxylate is forced into the position occupied by the C� proton in the relatedaminotransferases, thereby effecting a change in the reaction type from transamination todecarboxylation.
405PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

substrate was reversed at some point during evolution, or the enzymes share acommon ancestor with broad specificity.
MECHANISMS OF INHIBITION
The prominent role of PLP enzymes in metabolism has generated a great deal ofinterest in the mechanisms of their inhibition. Although they, like nearly allenzymes, are susceptible to simple competitive inhibition, the catalytic versatilityof the cofactor enhances their potential susceptibility to natural or designedmechanism-based inhibitors. Because such inhibition is often irreversible, it is ofmuch greater practical utility than competitive inhibition (81). The inhibitedcomplexes are also particularly useful for crystallographic studies, as they oftenmimic substrate or reaction intermediate complexes. A large number of inhibitorsof PLP enzymes have now been identified (for more detailed reviews, see 6, 82),and they have been generally grouped into three categories according to theirmode of inactivation.
Noncovalent Inactivation
The simplest mechanism-based inhibitors of PLP enzymes are those that formexceptionally stable complexes that often resemble normal reaction intermedi-ates. Although the inhibitor is covalently bound to the cofactor, the affinity forthe protein is through noncovalent interactions. The combined affinity may bevery high. A recently reported example is the stable ketimine intermediateformed in the reaction of ACC synthase with L-aminoethoxyvinylglycine (83)(Scheme 18). The ketimine is an intermediate in the reaction catalyzed by
Scheme 17 Reverse orientation of substrate binding by D-amino acid aminotrans-ferase (DAAT) and branched chain amino acid aminotransferase (BCAT). Theseclosely related enzymes are both in Fold Type IV, where the active site lysine ispositioned on the re face of the cofactor, opposite from its position in Fold Type I andII enzymes. As a result, D-amino acids (shown bound to DAAT) are bound with thecarboxylate pointing away from, and L-amino acids (shown bound to BCAT) arebound with the carboxylate proximal to, the cofactor phosphate group.
406 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

aminotransferases but not in the ACC synthase-catalyzed elimination reaction;thus the enzyme is unable to catalyze its hydrolysis. Of particular interest is thesubset of these inhibitors whose stability is the result of enzyme-catalyzedaromatization. An example is the inhibition of GABA ATase by (S)-4-amino-4,5-dihydro-2-thiophenecarboxylic acid (84) (Scheme 19). Since the products ofthese reactions are not covalently attached to the enzyme, the inhibited enzymescan often be reactivated by dialysis in the presence of PLP, so that thePLP-inhibitor species is replaced with fresh PLP (for example, see 83), but thismode of reactivation does not generally occur on a physiologically relevanttimescale.
Scheme 18 Inhibition of 1-aminocyclopropane-1-carboxylate synthase by amino-ethoxyvinylglycine. The inhibitor reacts to form a stable ketimine that is nothydrolyzed and remains tightly bound to the enzyme. Dissociation by dialysisremoves the cofactor together with the covalently bound inhibitor (83).
Scheme 19 Inhibition of �-aminobutyrate aminotransferase by (S)-4-amino-4,5-dihydro-2-thiophenecarboxylic acid. The reaction proceeds identically to the aminotransferasereaction up to the formation of the ketimine intermediate. At this point, deprotonation of a� carbon yields a very stable aromatic product that does not react further and remains in theactive site (84).
407PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Activated Nucleophiles
A number of amino acids with good �-leaving groups, such as �-chloroalanine,inhibit several types of PLP enzymes (6). The general mechanism for thisreaction is by initial formation and release of an enamine intermediate, which isa potent nucleophile and can attack C4� of the cofactor (85) (Scheme 20).Inactivation is irreversible, because the cofactor remains covalently bound to theenzyme. An alternative proposed inactivation pathway (86) is by direct Michaeladdition of an active site nucleophile to the aminoacrylate aldimine. Freeaminoacrylate can also diffuse out of the enzyme (as it does in natural �-elimination reactions), where it spontaneously decomposes to pyruvate andammonia, leaving the enzyme in an active form. Because of this possibility ofturnover as well as inhibition, the effectiveness of these inhibitors is oftenquantitated by the inactivation/turnover ratio.
Scheme 20 Mechanism of inactivation of a PLP-dependent enzyme by �-chloroala-nine. The reaction follows the normal pathway to �-elimination up to the formationof the aminoacrylate aldimine. From there, the aminoacrylate may be released bytransaldimination and may subsequently attack the cofactor nucleophilically. Subse-quent hydrolysis of the imine yields the final inactivated enzyme. A number of othermechanisms are possible, as noted in the text.
408 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Activated Electrophiles
Inhibition can also result from rearrangement of the inhibitor to generate anelectrophile that subsequently reacts irreversibly with an active site nucleophile(often the active site lysine). Two common types of inhibitors of this group areacetylenic compounds such as propargylglycine, which reacts to form a highlyreactive allene intermediate (87) (Scheme 21), and vinylic compounds such asvinylglycine, which reacts to form an �,�-unsaturated imine Michael acceptor(88) (Scheme 22). The reactivity of the potential intermediates formed from theseinhibitors allows for alternative reaction fates in addition to those shown. In thecase of vinylglycine, for instance, an aminocrotonate aldimine, a potentialMichael acceptor, can be formed from the quinonoid (89). The aminocrotonatemay also be released by transaldimination, at which point it can nucleophilicallyattack the cofactor in the manner described above for aminoacrylate (90). A thirdpossible fate is diffusion off the enzyme, where it decomposes spontaneously to�-ketobutyrate and ammonia (91), leaving the enzyme in the active PLP form.
Scheme 21 Mechanism of inactivation by propargylglycine. The reaction parallelsa �-elimination mechanism through the formation of the enamine intermediate. Atthis point, the acetylene moiety is rearranged to form a highly reactive allene.Nucleophilic attack by an active site residue results in covalent attachment to theenzyme. The mechanism shown is slightly altered from that originally proposed byAbeles & Walsh (87) to include a ketimine intermediate in accord with the mostrecent proposal for the mechanism of �-elimination (105).
409PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

The vinylglycine ketimine can also be hydrolyzed to release the potentially toxicMichael acceptor 2-ketobut-3-enoic acid, leaving the enzyme in the PMP form(92). Effective inhibitor design requires limiting these possible alternativepathways.
Reversible Competitive Inhibition
Competitive inhibitors have proven to be exceptionally useful in studies ofenzyme function and as unreactive substrate mimics in crystallographic studies.There are two general classes of competitive inhibitors—those that bind nonco-valently and those that react reversibly with the enzyme to form an aldimine thatdoes not react further. A classical example of the former is maleate, whichinhibits AATase by binding in the aspartate site (6). This association inducesclosure of the enzyme into the active form; thus this inhibitor has proven usefulfor crystallographic studies of this form of the enzyme.
The most common of the inhibitors that form unreactive aldimines are�-methyl substrate analogs and amino-oxy or hydrazine analogs. The closesimilarity of the �-methyl compounds to the substrates makes them particularlyuseful for crystallography, and structures of complexes of AATase and phos-phoserine aminotransferase with �-methylaspartate and �-methylglutamate,respectively, have been reported (93–95). Recent examples of the use of amino-oxy compounds for structure determination are those of ACC synthase with theamino-oxy analogue of SAM (96) (Scheme 23) and of ornithine aminotransferasewith L-canaline, an analogue of ornithine (97) (Scheme 23). The amino-oxyadducts with PLP are sufficiently stable so that these compounds often need notbe substrate analogs. Hydroxylamine itself binds to AATase to form a PLP-oxime whose Ki � 700 nM, a figure less than that for any of the dicarboxylicinhibitor complexes (6). A final example of an inhibitor that forms an unreactivealdimine is that of 1-aminoethyl phosphonate (AlaP), which binds tightly to alanine
Scheme 22 Mechanism of inactivation by vinylglycine. The reaction parallels an amino-transferase mechanism through the formation of the ketimine intermediate. Michael addi-tion by an active site nucleophile to the vinylglycine ketimine results in a covalent adduct.
410 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

racemase (98). Although this molecule has a C� proton, it does not undergodeprotonation. The stability of this complex enabled determination of the enzymestructure with the inhibitor in the active site, providing a mimic of the complex withthe natural substrate (99).
The Challenge of Inhibitor Specificity
Although the variety of possible mechanisms of inhibition makes it easy todesign effective inhibitors, specificity remains as a major challenge for use invivo. Gabaculine (Scheme 23), for example, is a potent inhibitor of GABAATase and also inhibits the closely related ornithine aminotransferase (100),making it unsuitable for pharmaceutical applications. One promising approach isto incorporate the reactive functional groups described above in structures thatare very close substrate analogs. An example is �-vinylGABA (vigabatrin;Scheme 23), a specific inhibitor of GABA ATase, which is used in the treatmentof epilepsy (reviewed in 101). In this case, the vinylic group is appended to thenatural substrate GABA to form a potential electrophile analogous to vinylgly-cine. Another example is the ornithine aminotransferase (OAT) inhibitor 5-fluoromethylornithine (102) (Scheme 23). The fluoride ion is susceptible to�-elimination, which generates an enamine capable of nucleophilic attack on thecofactor in the manner described above (71, 103), while the ornithine scaffoldprovides OAT specificity.
Understanding mechanisms of inhibition is crucial not only to enable the designof better inhibitors, but also to understand the control of reaction mechanisms by thisimportant class of enzymes. It is also of interest to ask how enzymes whose naturalreaction pathways include reactive intermediates, such as the aminoacrylate aldimineand vinylglycine ketimine, manage to avoid inactivation.
CONCLUSIONS
The recent effusion of X-ray structures for PLP-dependent enzymes—more thantwice as many were deposited in the protein data bank between 1997 and 2001as in the preceding five years—has done much to provide a visual framework in
Scheme 23 The structures of the inhibitors gabaculine, vigabatrin, 5-fluoromethylorni-thine, canaline, and amino-oxy SAM.
411PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

which the mechanistic concepts secured over the past half-century can beinterpreted. The findings served to focus more penetrating targeted mutagenesisexperiments and, taken together with those from newer studies capitalizing upondirected evolution methods, helped to elucidate some of the fundamental prin-ciples of protein design.
We now have a good understanding of the mechanisms of dual substraterecognition by aminotransferases and of how the fates of the common C� anionare directed. Many questions remain unanswered, particularly with regard to thecontrol of reaction pathways of both natural substrates and mechanism-basedinhibitors. Of the four fold types, only Fold Type I is well represented in thestructural database, and many of the reaction types are only represented by oneor two structures. We need additional structures of underrepresented fold typesin order to direct further functional studies. The understanding of the substrateand reaction type-specificity of PLP enzymes is crucial for the design not only ofspecific and medicinally useful inhibitors, but also of improved protein-basedcatalysts.
ACKNOWLEDGMENT
The authors thank Susan Aitken and Kathryn McElroy for critically reviewingthe manuscript.
The Annual Review of Biochemistry is online at http://biochem.annualreviews.org
LITERATURE CITED
1. Heyl D, Luz E, Harris SA, Folkers K.1951. J. Am. Chem. Soc. 73:3430–33
2. Stryer L. 1995. Biochemistry. New York:Freeman
3. Voet D, Voet J. 1995. Biochemistry.New York: Wiley
4. Hayashi H. 1995. J. Biochem. 118:463–73
5. John RA. 1995. Biochim. Biophys. Acta1248:81–96
6. Christen P, Metzler DE, eds. 1985.Transaminases. New York: Wiley
7. Miles EW. 2001. Chem. Rec. 1:140–518. Tai CH, Cook PF. 2001. Acc. Chem. Res.
34:49–599. Ferreira GC, Gong J. 1995. J. Bioenerg.
Biomembr. 27:151–5910. Rao NA, Talwar R, Savithri HS. 2000.
Int. J. Biochem. Cell Biol. 32:405–16
11. Hutson S. 2001. Prog. Nucleic Acid Res.Mol. Biol. 70:175–206
12. He XM, Liu HW. 2002. Annu. Rev. Bio-chem. 71:701–54
13. Kleppner SR, Tobin AJ. 2001. EmergingTher. Targets 5:219–39
14. Snell K, Riches D. 1989. Cancer Lett.44:217–20
15. Wang CC. 1995. Annu. Rev. Pharmacol.Toxicol. 35:93–127
16. Mudd SH, Laster L, Finkelstein JD,Irreverre F. 1964. Science 143:1443–45
17. Kraus JP, Janosik M, Kozich V, MandellR, Shih V, et al. 1999. Hum. Mutat.13:362–75
18. Adams DO, Yang SF. 1979. Proc. Natl.Acad. Sci. USA 76:170–74
19. Honma M, Shimomura T. 1978. Agric.Biol. Chem. 42:1825–31
412 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

20. Keller JW, Baurick KB, Rutt GC,O’Malley MV, Sonafrank NL, et al.1990. J. Biol. Chem. 265:5531–39
21. Watanabe Y, Shimura K. 1956. J. Bio-chem. 43:283–94
22. Rios A, Amyes TL, Richard JP. 2000.J. Am. Chem. Soc. 122:9373–85
23. Gallo KA, Knowles JR. 1993. Biochem-istry 32:3981–90
24. Alexander FW, Sandmeier E, Mehta PK,Christen P. 1994. Eur. J. Biochem. 219:953–60
25. Jansonius JN. 1998. Curr. Opin. Struct.Biol. 8:759–69
26. Schneider G, Kack H, Lindqvist Y.2000. Struct. Fold. Des. 8:R1–6
27. Churchich JE, Moses U. 1981. J. Biol.Chem. 256:1101–4
28. Hennig M, Grimm B, Contestabile R,John RA, Jansonius JN. 1997. Proc.Natl. Acad. Sci. USA 94:4866–71
29. Mehta PK, Christen P. 2000. Adv. Enzy-mol. Relat. Areas Mol. Biol. 74:129–84
30. Hyde CC, Ahmed SA, Padlan EA, MilesEW. 1988. J. Biol. Chem. 263:17857–71
31. Madison JT, Thompson JF. 1976. Bio-phys. Biochem. Res. Commun. 71:684–91
32. Finkelstein JD, Kyle WE, Martin JJ, PickAM. 1975. Biophys. Biochem. Res. Com-mun. 66:81–87
33. Gallagher DT, Gilliland GL, Xiao G,Zondlo J, Fisher KE, et al. 1998. Struc-ture 6:465–75
34. Sugio S, Petsko GA, Manning JM, SodaK, Ringe D. 1995. Biochemistry 34:9661–69
35. Kern AD, Oliveira MA, Coffino P,Hackert ML. 1999. Struct. Fold. Des.7:567–81
36. Wolosker H, Blackshaw S, Snyder SH.1999. Proc. Natl. Acad. Sci. USA96:13409–14
37. Contestabile R, Paiardini A, PascarellaS, di Salvo ML, D’Aguanno S, Bossa F.2001. Eur. J. Biochem. 268:6508–25
38. Dunathan HC. 1966. Proc. Natl. Acad.Sci. USA 55:712–16
39. Kirsch JF, Eichele G, Ford GC, VincentMG, Jansonius JN, et al. 1984. J. Mol.Biol. 174:497–525
40. Ploux O, Marquet A. 1996. Eur. J. Bio-chem. 236:301–8
41. Webster SP, Alexeev D, CampopianoDJ, Watt RM, Alexeeva M, et al. 2000.Biochemistry 39:516–28
42. Sun S, Zabinski RF, Toney MD. 1998.Biochemistry 37:3865–75
43. Ford GC, Eichele G, Jansonius JN. 1980.Proc. Natl. Acad. Sci. USA 77:2559–63
44. Borisov VV, Borisova SN, Sosfenov NI,Vainshtein BK. 1980. Nature 284:189–90
45. Kuramitsu S, Inoue Y, Tanase S, MorinoY, Kagamiyama H. 1987. Biochem. Bio-phys. Res. Commun. 146:416–21
46. Kochhar S, Finlayson WL, Kirsch JF,Christen P. 1987. J. Biol. Chem. 262:11446–48
47. Toney MD, Kirsch JF. 1987. J. Biol.Chem. 262:12403–5
48. Cronin CN, Kirsch JF. 1988. Biochemis-try 27:4572–79
49. Hayashi H, Kuramitsu S, Inoue Y,Morino Y, Kagamiyama H. 1989. Bio-chem. Biophys. Res. Commun. 159:337–42
50. Kochhar S, Christen P. 1992. Eur. J. Bio-chem. 203:563–69
51. Shaw JP, Petsko GA, Ringe D. 1997.Biochemistry 36:1329–42
52. Albery WJ, Knowles JR. 1986. Biochem-istry 25:2572–77
53. Jencks WP. 1985. Chem. Rev. 85:511–2754. Paiardini A, Contestabile R, D’Aguanno
S, Pascarella S, Bossa F. 2003. Biochim.Biophys. Acta 1647:214–19
55. Wolfenden R. 1974. Mol. Cell. Biochem.3:207–11
56. Schirch V, Shostak K, Zamora M,Guatam-Basak M. 1991. J. Biol. Chem.266:759–64
57. Pasternak A, White A, Jeffery CJ,Medina N, Cahoon M, et al. 2001. Pro-tein Sci. 10:1331–42
413PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

58. Herschlag D. 1988. Bioorg. Chem.16:62–96
59. Goldberg JM, Kirsch JF. 1996. Biochem-istry 35:5280–91
60. Haruyama K, Nakai T, Miyahara I,Hirotsu K, Mizuguchi H, et al. 2001.Biochemistry 40:4633–44
61. Onuffer JJ, Kirsch JF. 1995. Protein Sci.4:1750–57
62. Malashkevich VN, Onuffer JJ, KirschJF, Jansonius JN. 1995. Nat. Struct. Biol.2:548–53
63. Yano T, Oue S, Kagamiyama H. 1998.Proc. Natl. Acad. Sci. USA 95:5511–15
64. Rothman SC, Kirsch JF. 2003. J. Mol.Biol. 327:593–608
65. Oue S, Okamoto A, Yano T, Kaga-miyama H. 1999. J. Biol. Chem. 274:2344–49
66. Oue S, Okamoto A, Yano T, Kaga-miyama H. 2000. J. Biochem. 127:337–43
67. Okamoto A, Nakai Y, Hayashi H,Hirotsu K, Kagamiyama H. 1998. J. Mol.Biol. 280:443–61
68. Okamoto A, Ishii S, Hirotsu K,Kagamiyama H. 1999. Biochemistry38:1176–84
69. Toney MD, Pascarella S, De Biase D.1995. Protein Sci. 4:2366–74
70. Storici P, Capitani G, De Biase D, MoserM, John RA, et al. 1999. Biochemistry38:8628–34
71. Storici P, Capitani G, Muller R,Schirmer T, Jansonius JN. 1999. J. Mol.Biol. 285:297–309
72. Jensen RA, Gu W. 1996. J. Bacteriol.178:2161–71
73. Matsui I, Matsui E, Sakai Y, Kikuchi H,Kawarabayasi Y, et al. 2000. J. Biol.Chem. 275:4871–79
74. Ura H, Harata K, Matsui I, Kuramitsu S.2001. J. Biochem. 129:173–78
75. Hayashi H, Inoue K, Nagata T,Kuramitsu S, Kagamiyama H. 1993. Bio-chemistry 32:12229–39
76. Goto M, Miyahara I, Hayashi H,
Kagamiyama H, Hirotsu K. 2003. Bio-chemistry 42:3725–33
77. Deleted in proof78. Eliot AC, Kirsch JF. 2003. Acc. Chem.
Res. 36:757–6579. Soda K, Yoshimura T, Esaki N. 2001.
Chem. Rec. 1:373–8480. Peisach D, Chipman DM, Van Ophem
PW, Manning JM, Ringe D. 1998. Bio-chemistry 37:4958–67
81. Silverman RB. 1988. J. Enzyme Inhib.2:73–90
82. Nanavati SM, Silverman RB. 1989.J. Med. Chem. 32:2413–21
83. Capitani G, McCarthy DL, Gut H, Grut-ter MG, Kirsch JF. 2002. J. Biol. Chem.277:49735–42
84. Fu M, Nikolic D, Van Breemen RB, Sil-verman RB. 1999. J. Am. Chem. Soc.121:7751–59
85. Likos JJ, Ueno H, Feldhaus RW, MetzlerDE. 1982. Biochemistry 21:4377–86
86. Kishore GM. 1984. J. Biol. Chem. 259:10669–74
87. Abeles RH, Walsh CT. 1973. J. Am.Chem. Soc. 95:6124–25
88. Rando RR. 1974. Biochemistry 13:3859–63
89. Soper TS, Manning JM, Marcotte PA,Walsh CT. 1977. J. Biol. Chem. 252:1571–75
90. Nanavati SM, Silverman RB. 1991.J. Am. Chem. Soc. 113:9341–49
91. Miles EW. 1975. Biochem. Biophys. Res.Commun. 66:94–102
92. Choi S, Storici P, Schirmer M, Silver-man RB. 2002. J. Am. Chem. Soc. 124:1620–24
93. McPhalen CA, Vincent MG, Picot D,Jansonius JN, Lesk AM, Chothia C.1992. J. Mol. Biol. 227:197–213
94. Okamoto A, Higuchi T, Hirotsu K,Kuramitsu S, Kagamiyama H. 1994.J. Biochem. 116:95–107
95. Hester G, Stark W, Moser M, Kallen J,Markovic-Housley Z, Jansonius JN.1999. J. Mol. Biol. 286:829–50
96. Capitani G, Eliot AC, Gut H, Khomutov
414 ELIOT y KIRSCH
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

RM, Kirsch JF, Grutter MG. 2003. Bio-chim. Biophys. Acta 1647:55–60
97. Shah SA, Shen BW, Brunger AT. 1997.Structure 5:1067–75
98. Badet B, Walsh C. 1985. Biochemistry24:1333–41
99. Stamper GF, Morollo AA, Ringe D,Stamper CG. 1998. Biochemistry37:10438–45
100. Jung MJ, Seiler N. 1978. J. Biol. Chem.253:7431–39
101. Mumford JP, Cannon DJ. 1994. Epilep-sia 35:S25–28
102. Daune G, Gerhart F, Seiler N. 1988. Bio-chem. J. 253:481–88
103. Bolkenius FN, Knodgen B, Seiler N.1990. Biochem. J. 268:409–14
104. Sun S, Toney MD. 1999. Biochemistry38:4058–65
105. Brzovic P, Holbrook EL, Greene RC,Dunn MF. 1990. Biochemistry 29:442–51
106. Livanova NB, Chebotareva NA, EroninaTB, Kurganov BI. 2002. Biochemistry67:1089–98
107. Frey PA. 2001. Annu. Rev. Biochem.70:121–48
415PYRIDOXAL PHOSPHATE ENZYMES
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

Annual Review of Biochemistry Volume 73, 2004
CONTENTS
THE EXCITEMENT OF DISCOVERY, Alexander Rich 1
MOLECULAR MECHANISMS OF MAMMALIAN DNA REPAIR AND THE DNA DAMAGE CHECKPOINTS, Aziz Sancar, Laura A. Lindsey-Boltz, Keziban Ünsal-Kaçmaz, Stuart Linn 39
CYTOCHROME C -MEDIATED APOPTOSIS, Xuejun Jiang, Xiaodong Wang 87NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY OF HIGH-MOLECULAR-WEIGHT PROTEINS, Vitali Tugarinov, Peter M. Hwang, Lewis E. Kay 107
INCORPORATION OF NONNATURAL AMINO ACIDS INTO PROTEINS, Tamara L. Hendrickson, Valérie de Crécy-Lagard, Paul Schimmel 147
REGULATION OF TELOMERASE BY TELOMERIC PROTEINS, Agata Smogorzewska, Titia de Lange 177CRAWLING TOWARD A UNIFIED MODEL OF CELL MOBILITY: Spatial and Temporal Regulation of Actin Dynamics, Susanne M. Rafelski, Julie A. Theriot 209
ATP-BINDING CASSETTE TRANSPORTERS IN BACTERIA, Amy L. Davidson, Jue Chen 241
STRUCTURAL BASIS OF ION PUMPING BY CA-ATPASE OF THE SARCOPLASMIC RETICULUM, Chikashi Toyoshima, Giuseppe Inesi 269DNA POLYMERASE , THE MITOCHONDRIAL REPLICASE, Laurie S. Kaguni 293
LYSOPHOSPHOLIPID RECEPTORS: Signaling and Biology, Isao Ishii, Nobuyuki Fukushima, Xiaoqin Ye, Jerold Chun 321
PROTEIN MODIFICATION BY SUMO, Erica S. Johnson 355
PYRIDOXAL PHOSPHATE ENZYMES: Mechanistic, Structural, and Evolutionary Considerations, Andrew C. Eliot, Jack F. Kirsch 383
THE SIR2 FAMILY OF PROTEIN DEACETYLASES, Gil Blander, Leonard Guarente 417
INOSITOL 1,4,5-TRISPHOSPHATE RECEPTORS AS SIGNAL INTEGRATORS, Randen L. Patterson, Darren Boehning, Solomon H. Snyder 437
STRUCTURE AND FUNCTION OF TOLC: The Bacterial Exit Duct for Proteins and Drugs, Vassilis Koronakis, Jeyanthy Eswaran, Colin Hughes 467
ROLE OF GLYCOSYLATION IN DEVELOPMENT, Robert S. Haltiwanger, John B. Lowe 491
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.

STRUCTURAL INSIGHTS INTO THE SIGNAL RECOGNITION PARTICLE, Jennifer A. Doudna, Robert T. Batey 539
PALMITOYLATION OF INTRACELLULAR SIGNALING PROTEINS: Regulation and Function, Jessica E. Smotrys, Maurine E. Linder 559
FLAP ENDONUCLEASE 1: A Central Component of DNA Metabolism, Yuan Liu, Hui-I Kao, Robert A. Bambara 589
EMERGING PRINCIPLES OF CONFORMATION-BASED PRION INHERITANCE, Peter Chien, Jonathan S. Weissman, Angela H. DePace 617
THE MOLECULAR MECHANICS OF EUKARYOTIC TRANSLATION, Lee D. Kapp, Jon R. Lorsch 657
MECHANICAL PROCESSES IN BIOCHEMISTRY, Carlos Bustamante, Yann R. Chemla, Nancy R. Forde, David Izhaky 705
INTERMEDIATE FILAMENTS: Molecular Structure, Assembly Mechanism, and Integration Into Functionally Distinct Intracellular Scaffolds, Harald Herrmann, Ueli Aebi 749
DIRECTED EVOLUTION OF NUCLEIC ACID ENZYMES, Gerald F. Joyce 791
USING PROTEIN FOLDING RATES TO TEST PROTEIN FOLDING THEORIES, Blake Gillespie, Kevin W. Plaxco 837
EUKARYOTIC mRNA DECAPPING, Jeff Coller, Roy Parker 861
NOVEL LIPID MODIFICATIONS OF SECRETED PROTEIN SIGNALS, Randall K. Mann, Philip A. Beachy 891
RETURN OF THE GDI: The GoLoco Motif in Cell Division, Francis S. Willard, Randall J. Kimple, David P. Siderovski 925
OPIOID RECEPTORS, Maria Waldhoer, Selena E. Bartlett, Jennifer L. Whistler 953
STRUCTURAL ASPECTS OF LIGAND BINDING TO AND ELECTRON TRANSFER IN BACTERIAL AND FUNGAL P450S, Olena Pylypenko, Ilme Schlichting 991
ROLES OF N-LINKED GLYCANS IN THE ENDOPLASMIC RETICULUM, Ari Helenius, Markus Aebi 1019
ANALYZING CELLULAR BIOCHEMISTRY IN TERMS OF MOLECULAR NETWORKS, Yu Xia, Haiyuan Yu, Ronald Jansen, Michael Seringhaus, Sarah Baxter, Dov Greenbaum, Hongyu Zhao, Mark Gerstein 1051
Ann
u. R
ev. B
ioch
em. 2
004.
73:3
83-4
15. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by U
nive
rsity
of
Illin
ois
- U
rban
a C
ham
paig
n on
09/
27/1
1. F
or p
erso
nal u
se o
nly.





























