Embed Size (px)
DESCRIPTION
Process Validation Presentation Final Version
Citation preview

PROCESS VALIDATIONPROCESS VALIDATIONin a Regulated Industryin a Regulated Industry
Doug FrankSr. Director of Quality

Program OverviewProgram Overview Terms & DefinitionsTerms & Definitions Systems for Implementing a Validation Program Systems for Implementing a Validation Program Essential RequirementsEssential Requirements Software QualificationsSoftware Qualifications Specific TestsSpecific Tests

Why Validate?Why Validate?
Required by:Required by:– FDA’s QSRFDA’s QSR– ISO 13485: 2003ISO 13485: 2003

ADDITIONAL REASONS FOR ADDITIONAL REASONS FOR VALIDATIONVALIDATION
Customer satisfaction: Non-conforming product can lead to lost customers.
Customer Mandated: Provision for securing new business
Product liability: Conformance to product specifications must be maintained.
Reduced production costs: PV leads to reduced inspections, testing, scrap and rework. Shifts costs from production to prevention.
Supports improvements: Testing data can be used to support improvements in the process or the development of the next generation of the process.
Regulatory requirement (CFR Title 21, Part 820.75)

Basic PrinciplesBasic Principles Establish that the process equipment has the capability of operating
within required parameters.
Demonstrate that controlling, monitoring, and/or measuring equipment and instrumentation are capable of operating within the parameters prescribed for the process equipment.
Perform replicate cycles (runs) representing the required operational range of the equipment to demonstrate that the processes have been operated within the prescribed parameters for the process and that the output or product consistently meets predetermined specifications for quality and function.
Monitor the validated process during routine operation. As needed, requalify and recertify the equipment.

Terms & DefinitionsTerms & Definitions
Validation: Confirmation by examination and provision of objective evidence
that the particular requirements for a specific intended use can be consistently fulfilled (820.3(z)).
Process validation: Establishing documented evidence which provides a high degree of
assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes (CMI).

Terms & DefinitionsTerms & Definitions
Installation qualification (IQ):
Establishing by objective evidence that all key aspects of the process equipment and ancillary system installation adhere to the manufacturer’s approved specification and that the recommendations of the supplier of the equipment are suitably considered (GHTF/SG3/N99-10:2004).

Terms & DefinitionsTerms & DefinitionsOperational qualification (OQ):
Studies which are designed to challenge the process and process equipment, and establish objective evidence that the process meets predetermined requirements throughout all anticipated operating ranges (CMI).

Performance qualification (PQ): Studies conducted to demonstrate the process will consistently
produce acceptable product under normal operating conditions. Challenges to the process should simulate conditions that will be encountered during actual manufacturing (CMI).
Terms & DefinitionsTerms & Definitions

Validation protocol: A written plan stating how validation will be conducted,
including test parameters, product characteristics, production equipment, and decision points on what constitutes acceptable test results (FDA Guidance).
Verification: Confirmation by examination and provision of objective evidence
that specified requirements have been fulfilled (820.3(aa)).
Terms & DefinitionsTerms & Definitions

WHEN SHOULD PROCESSES BE WHEN SHOULD PROCESSES BE VALIDATED?VALIDATED?
Where process results cannot be fully verified during routine production by inspection and Where process results cannot be fully verified during routine production by inspection and test, the process must be validated according to established procedures [820.75(a)]. test, the process must be validated according to established procedures [820.75(a)].
AIs Process
OutputVerifiable
BIs VerificationSufficient &
Cost Effective
CVerify &Control
the Process
DValidate
ERedesign Product
and/orProcess
YES YES
NO NO
The following model may be useful in determining whether or not a process should validated:
(GHTF/SG3/N99-10:2004)

WHEN SHOULD PROCESSES BE VALIDATED?WHEN SHOULD PROCESSES BE VALIDATED?
Routine end-product tests have insufficient sensitivity to verify the Routine end-product tests have insufficient sensitivity to verify the desired safety and efficacy of the finished devicesdesired safety and efficacy of the finished devices..
Clinical or destructive testing would be required to show that the Clinical or destructive testing would be required to show that the manufacturing process has produced the desired result or product.manufacturing process has produced the desired result or product.
Routine end-product tests do not reveal all variations in safety and Routine end-product tests do not reveal all variations in safety and efficacy that may occur in the finished devices.efficacy that may occur in the finished devices.
The process capability is unknown, or it is suspected that the process The process capability is unknown, or it is suspected that the process is barely capable of meeting the device specifications. is barely capable of meeting the device specifications.

Relation of Risk AnalysisRelation of Risk Analysis
For complex or critical processes, a risk analysis is normally conducted prior to establishing validation requirements. The risk analysis will reveal which output parameters should be concentrated upon, and assist in the development of appropriate tests.

Getting StartedGetting Started
Develop a Validation Program that Defines:Develop a Validation Program that Defines: ResponsibilitiesResponsibilities Process Type DefinitionProcess Type Definition Flow Chart With Decision PointsFlow Chart With Decision Points MethodsMethods FormsForms DocumentationDocumentation OversightOversight

CarboMedics SystemCarboMedics System Policies Implemented for :Policies Implemented for :
Process ValidationProcess Validation Design Controls…Design ChangeDesign Controls…Design Change Test Plan & Protocol PoliciesTest Plan & Protocol Policies Risk AnalysisRisk Analysis Corrective & Preventative ActionCorrective & Preventative Action Events & ComplaintsEvents & Complaints Documentation & RecordsDocumentation & Records Management ReviewManagement Review

CarboMedics SystemCarboMedics SystemProcess Validation Policy provides Requirements Process Validation Policy provides Requirements for:for:
Validation Review Board/Design Change (VRB/DCN Validation Review Board/Design Change (VRB/DCN Committee)Committee)
Validation Master Plan (PVMP)Validation Master Plan (PVMP) Request for Validation Status (RFVS)Request for Validation Status (RFVS) Process Validation Summary Report (PVSR)Process Validation Summary Report (PVSR)

CarboMedics SystemCarboMedics SystemValidation Review Board Validation Review Board
Oversees and ensures complianceOversees and ensures compliance Reviews and Approves PVMP’sReviews and Approves PVMP’s Review RFVS’s & Selected Test Plans/Protocol’sReview RFVS’s & Selected Test Plans/Protocol’s Provides consultation & Guidance on validation Provides consultation & Guidance on validation
approachapproach

CarboMedics SystemCarboMedics System
PROCESS VALIDATION MASTER PLAN Provides the roadmap for conducting validations.
Establishes criticality of the process.
Defines the validation approach.
Documents rationale for decisions to “not validate”.
Provides documented evidence of the validation
Provides an easy to follow trail to locate relevant validation documents and test data.
Establishes requirements for process changes.


Request for Validation StatusRequest for Validation Status
A CarboMedics form used to communicate planned validations to the VRB that have not been documented on a PVMP.
Alternatively, the form is also used to present formal questions or issues that may or may not be covered in the policy to the VRB for disposition.


Process Validation Summary ReportProcess Validation Summary Report PVSR is a controlled document which lists all current validation
documentation to demonstrate that processes are validated and specifies any revalidation requirements.
Each PVSR document number is referenced on the Process Validation Master Plan, for ease of document retrieval.
The PVSR contains:• Process title• Applicable uncontrolled documentation (attached)• List of all validation documentation (IQ, OQ and PQ)• Revalidation requirements• Recurring validation requirements


Process TypeProcess Type
CarboMedics bases process validation requirements on the Process Type.
Four process types have been established to clarify what qualifications are required, and when these qualifications must be completed.
Verified - A process which is fully verified by subsequent inspections or tests, and does not have any equipment that would require an Installation Qualification. No validation is required.
Type I - A process which is fully verified by subsequent inspections or tests, but relies on relevant equipment. An Installation Qualification and a Software Qualification (if applicable) are required prior to pilot or production use.
Type II - A process the results of which cannot be fully verified by subsequent inspection and tests. Process validation must be completed prior to production use.

Type III - A Type II process that if not satisfactorily performed could result in serious injury to the patient. Process validation must be completed prior to shipment of product for human surgical use.
Examples are:o Sterilizationo Aseptic Transfero Performance testso Injection Molding of critical partso Blending / Mixing of critical solutions
Note: Serious injury is defined as an injury that (1) is life threatening, (2) results in permanent impairment of a body function or permanent damage to body structure,
or (3) necessitates medical or surgical intervention by a health care professional.
Process TypeProcess Type

Test Plans and Test ReportsTest Plans and Test Reports
Qualifications are planned, conducted and reported using controlled plans and reports or Test Protocols.
Test Plans are required to include:o Scope statement that includes the objectives of the testo Hypothesis/Expected Outcomeo Sample requirements and description including a rationale for
the number of samples to be testedo Test methods or procedureso Requirements for test reporto Reference Documents (if required)

Test Reports are required to include:
• Reference Documents (if not previously listed in the Test Plan)• Any deviations from the original Test Plan, along with a
rationale for acceptability of the deviation • Test results as required by the Test Plan• Discussion and interpretation of the test results as required by
the Test Plan• Conclusions drawn directly from and supported by the test data• Traceability documentation of test parts or specimens (as
required)• Raw data attached or reference to a laboratory notebook
Validation test parts are required to be retained with the Test Report.
All original Test Plans and Test Reports are retained in a central Documentation Center.

Protocol Plans and ReportsProtocol Plans and Reports
In some cases, it is necessary to evaluate a change using production parts. An example would be a minor change in a polishing process, where a large sample is desired to ensure accurate results.
In these cases, CarboMedics uses a Protocol Plan and Report system, which requires approval from Manufacturing, Quality Control, Quality Assurance, Engineering, and Regulatory Affairs management.
Unless specifically stated in the protocol plan, the product must be
held until approval and disposition is obtained for the protocol report.
The protocol plan must contain:
• Title - the process or item to be evaluated• Purpose - the proposed change and the reason for making it.• Scope - the range of products, processes, or equipment to which the protocol
applies.• Hypothesis / Expected Outcome - the criteria by which success will be judged.• Reference Documents - Any procedures or test methods to be used, or other
forms, diagrams, or relevant documents needed to conduct the protocol.• Sample Requirements - A rationale for the number and type of parts to be tested.• Test Description - The required test parameters, quantity, and frequency that will
result in sufficient data for evaluation.• Procedure - A step-by-step description of the work to be done.• Disposition Of Parts – both during and after the protocol• Attachments - Any forms or relevant documents needed to conduct the protocol

The Protocol Report will include:
• Title - the process or item that was evaluated
• Purpose & Scope - as described above
• Reference Documents - only if not included in the Protocol Plan
• Results - The results of the testing, and identification of all tested product
• Discussion - The meaning of the test results as compared to the purpose
• Deviations & Additions - Identify all deviations and additions from the plan, with justification for the changes.
• Conclusion - A summary of the results and a statement as to the “success” or “failure” of the protocol.

Laboratory NotebooksLaboratory Notebooks Laboratory notebooks are frequently used to document collected test data.
Raw data should be physically attached to the laboratory notebook.
Notebooks are numbered sequentially and assigned to specific individuals.
Each page is signed by the owner and supervisor.
After the laboratory notebook has been filled, it is stored in a central library.

Raw DataRaw Data
Raw data should be filed with the test report.
Forms provided to manufacturing for documenting results should be signed and dated by the responsible individual.
Any errors on the raw data should be lined out, initialed and dated.

RevalidationRevalidation All or a portion of a validation that is required to be repeated
when changes that affect the original validation are made. Examples of changes requiring revalidation
• Changes to product specifications• Process parameters• Equipment (type, function, location, control system,
major repairs)• Raw materials• Manufacturing materials

Recurring ValidationRecurring Validation
Recurring validation is all or a portion of a validation that is required to be repeated periodically, and demonstrates continuing compliance of a validated process or operation.
Examples include sterilization and aseptic systems.
CarboMedics uses a Calibration Tracking System to track due dates for recurring validations and shelf life studies.

Essential RequirementsEssential Requirements
Industry generally satisfies by conducting:
Installation Qualification
Operational Qualification
Performance Qualification

Examples of IQ Elements (as appropriate)
Installation conditions (utilities, wiring, etc.) Installation check (ball bars, laser alignment, vibration analysis,
etc.)Verification of installation drawings or instructions
Equipment documentation (drawings, schematics, spare parts list, manuals, etc.)
Purchased software documentation Critical equipment features (i. e. materials of construction,
cleanability, etc.) Environmental conditions (such as clean room requirements,
temperature, humidity) Preventative maintenance schedule Calibration requirements Identification of repair operations that require requalification

Operational QualificationOperational Qualification
Studies which are designed to challenge the process and process equipment, and establish objective evidence that the process meets predetermined requirements throughout all anticipated operating ranges.
NOTE: Process and built-in monitoring equipment (instruments) should be calibrated at the beginning of the OQ, and the calibration should be checked at the end of the study to establish confidence in the validation of the process. Where required, independent measurement instruments used as part of a validation shall have an acceptable and current calibration status. In situations where these independent instruments might be affected by the actual testing (such as thermocouples exposed to high temperatures), the instruments should also be calibrated upon completion of the study.

Examples of OQ Elements (as appropriate)
Verification of all systems and subsystem functions
Confirmation of all safety devices and systems
Software qualification
Evaluate impact of key parameters on the process (DOE, worst-case testing, etc.)
Measurement system suitability
Bias and/or Repeatibility & Reproducibility of measurement systems
Operator training and qualification

Performance QualificationPerformance Qualification
Studies conducted to demonstrate the process will consistently produce acceptable product under normal operating conditions. Challenges to the process should simulate conditions that will be encountered during actual manufacturing.

Examples of PQ Elements (as appropriate)
Verification of released process documentation (manufacturing procedures, inspection procedures, product specifications, engineering drawings, tool drawings, material specifications, related forms, etc.)
Dimensional verification (first article, layout inspection, etc.)
Process stability (x-bar & R charts)
Process capability (Cp, Cpk)
Fault seeding
Product performance evaluation (impact of the process on follow-on operations, product functionality, material and physical properties, etc.)
Cleanliness tests (extraction tests, ESCA, etc.)
Biological tests (bioburden, cytotoxicity, hemolysis, etc.)
Product sterility
Manned and unmanned testing of Controlled Environments, etc.

Software QualificationSoftware Qualification
Required For Critical Software Systems
Critical software systems are those systems, which are used to support regulatory requirements and/or systems, which affect product fit, form, function, or processes.
Required for both purchased and proprietary software.

Level Of Software Concern
Major • Could result in death or serious injury to the patient, or
complications that require extended medical care or surgical intervention.
• Documentation and validation will be most rigorous and extensive
Moderate • Could result in moderate inconvenience to the patient, or minor
patient complications not expected to require extended medical care or surgical intervention to correct.
• Documentation and validation will be rigorous and extensive, with special emphasis on the most critical modules.
Minor• Would not be expected to affect the product’s form, fit, or function
or result in latent complications.• Documentation and validation requirements will be reasonable to
verify proper functioning of the various modules.

Software Development/Qualification Requirements Document will be developed by the requestor.
Design Document will be prepared by the software developer.
The software developer translates the design into code.
Operating instructions are developed to assure that operators have sufficient documentation for successful execution of the software.
Software Qualification conducted by Test Plan and Test Report.
Training for all end users of the software (documented where applicable).
Release of controlled documentation for software. Carbomedics uses a Software Specification (SS) which contains:
• Cover Sheet / Index (optional)• Requirements Document• Design Document• Software
Release of all applicable operating procedures.

Test Plan SpecificsTest Plan Specifics
Test Plan - Describe the methods of qualification (inspection, analysis, demonstration, or test) to be used to verify the following:
• Specified requirements in the Design and Requirements Documents have been met.
• The software consistently performs to it’s predetermined specifications and quality attributes.
• The software acts appropriately when error conditions are encountered.
• The software operates under normal operating conditions, anticipated production variability, and under worst case / maximum loads.
Software Qualification
Test Cases are used to validate software, should be designed to show that the software meets its essential requirements. Each test case will list:
• the system requirement• the method for testing the requirement• the expected result• the actual result (recorded during validation
Code Review - The source code is reviewed by a second individual, and results are documented in the test report.
Problem Reports – Problems encountered during software validation will be documented and filed as part of the test report. All problems must be resolved before the validation process can be completed.
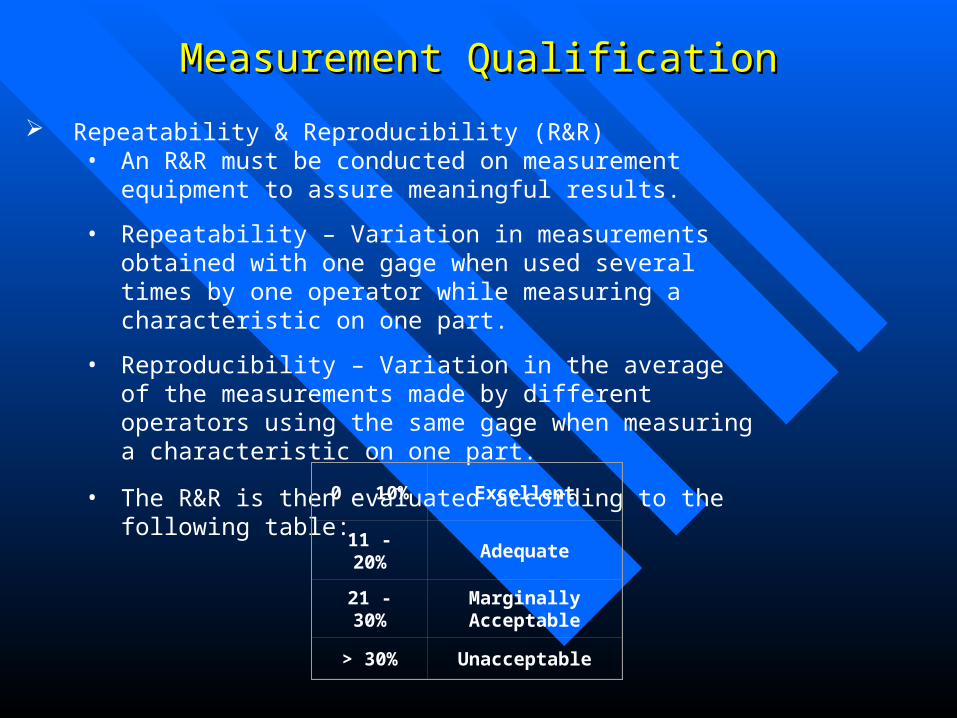
Measurement QualificationMeasurement Qualification
Repeatability & Reproducibility (R&R) • An R&R must be conducted on measurement equipment to assure
meaningful results.
• Repeatability – Variation in measurements obtained with one gage when used several times by one operator while measuring a characteristic on one part.
• Reproducibility – Variation in the average of the measurements made by different operators using the same gage when measuring a characteristic on one part.
• The R&R is then evaluated according to the following table:
0 - 10% Excellent
11 - 20% Adequate
21 - 30%Marginally Acceptable
> 30% Unacceptable

Calibration
• Measurement equipment should be calibrated prior to use in production, and placed on a calibration schedule for periodic recalibration.
Bias
• When multiple measurement equipment are used, the bias between measurement equipment is sometimes used to qualify measurement of complex geometries.
• The maximum bias generally allowed is 10% for critical dimensions and 20% for non-critical dimensions.
Specific TestsSpecific Tests

Specific TestsSpecific Tests
X BAR & R CHARTS
Prior to evaluation of process capability, the process must first be demonstrated to be in a state of control.
Xbar charts plot the mean of successive groups, and compares these values to established control limits.
R charts plot the range of successive groups, and compares these values to established control limits.
Points which lie outside of the control limits should be evaluated for potential causes. If the cause is identified and corrected, then the point may be excluded from the graph or repeat the reading.

PROCESS CAPABILITY
Process capability (Cp) provides a comparison of the measured variance to the specified tolerance.
Cp = (USL – LSL) 6
where: Cp = process capabilityUSL = upper specification limitLSL = lower specification limit = measured standard deviation
Specific TestsSpecific Tests

However, Cp as defined only provides the potential capability. If the process mean is not centered within the process, discrepant product may still result. A more revealing measure is Cpk, which is sensitive to where the mean lies within the
tolerance band.
Cpk = Min {(USL – Xbar) or (Xbar – LSL)}
3 CarboMedics has typically accepted Cp values of 1.0 or greater for processes where the operator can adjust the mean, and Cpk values of 1.33 for processes that cannot be adjusted by the operator.
Specific TestsSpecific Tests

F& t Testing, Tukey AnalysisF& t Testing, Tukey Analysis
F&t TESTING, TUKEY ANALYSIS
Often, the effects of a process change will be determined by comparing product from the new process to that of the old process.
For two individual groups, F & t testing is appropriate. The F test first compares the variance from each group, and then the t test compares the means based on equal or unequal variance.
The Tukey analysis is used when more than two groups are being compared.

Design of Experiments (DOE) TestingDesign of Experiments (DOE) Testing
During the Operational Qualification, the process should be characterized to ensure that the specified requirements are met under all conditions.
A screening experiment is sometimes employed first to determine which process parameters have significant effect on the process output.
The significant process parameters are then tested using a DOE. This ensures acceptable product under all potential conditions, as long as the parameters are within the defined limits.
A number of DOE models are available, including Taguchi, factorial, response surface, D-optimal, etc…
Results of the DOE should also be analyzed for optimal process conditions.

Worst Case TestingWorst Case Testing
For certain processes, DOE testing may not be appropriate. Worst case testing should be employed in these situations.
Worst case testing is the set of conditions encompassing upper and lower processing limits and circumstances, which pose the greatest chance of process or product failure when compared to ideal conditions. Such conditions do not necessarily induce product or process failure.

Worst Case TestingWorst Case Testing
FAULT SEED TESTING
For inspection processes, documented evidence must be provided that the process is capable of rejecting product that does not meet specified requirements.
Inspection processes that only yield a pass/fail result cannot be qualified using standard R&R techniques, and fault seed testing should be used.
Fault seed testing is conducted by randomly testing unacceptable product, and verifying the inspection process rejects this product.
The inspector should not know which product is acceptable, and ideally should be unaware that the process is being tested.
Acceptable results are then based on the criticality of the attribute being inspected. For automated processes, generally all fault seeded product must be rejected.

The sample size for each test should be established in the test plan, and should be based on the criticality of the process.
Sample size is normally based on the level of confidence desired to ensure a certain portion of the population is within the sample range. This may be calculated by the Wilks equation:
npn-1 – (n-1)pn = 1 -
where:n = sample sizep = proportion of population contained within the sample range = confidence level
Typically, a sample size of 30 is adequate for most testing, which covers 90% of the population with 80% confidence. Occasionally, a sample size as low as 3 may be used in development activities, where 50% of the population is covered with 50% confidence.
Sample Size RationaleSample Size Rationale

• Quality System Regulation, Title 21 Part 820 of the Code of Federal Regulations(www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm? CFRPart=820, Apr 2003)
• Medical Device Quality Systems Manual: A Small Entity Compliance Guide (www.fda.gov/cdrh/qsr/intro.html, Dec 1996)
• General Principles of Software Validation (www.fda.gov/cdrh/comp/guidance/938.html, Jan 2002)
• Guidance for Industry Part 11, Electronic Records; Electronic Signatures — Scope and Application (http://www.fda.gov/cder/guidance/5667fnl.doc, Aug 2003)
• Guideline on General Principles of Process Validation (www.fda.gov/cder/guidance/pv.htm, May 1987)
• Guide to Inspections of Medical Device Manufacturers (www.fda.gov/ora/inspect_ref/igs/med_dev_mnfct/cover.html, Dec 1997)
• Replacement Heart Valve Guidance - Draft Document, Division of Cardiovascular, Respiratory and Neurological Devices (www.fda.gov/cdrh/ode/3751.html, Oct 1994)
FDA ReferencesFDA References

Non-FDA ReferencesNon-FDA References
• ISO 9001 - Quality Systems - Model for quality assurance in design, development, production, installation and servicing
• ISO 13485 - Quality Systems - Medical Devices - Particular requirements for the application of ISO 9001
• EN 46001 - Application of EN ISO 9001 to the manufacture of medical devices
• Medical Device Directive, Council Directive 93/42/EEC of 14 June 1993 concerning medical devices
• GHTF/SG3/N99-10:2004 (Edition 2), “Process Validation Guidance” (http://www.ghtf.org/sg3/inventorysg3/sg3_fd_n99-10_edition2.pdf, Jan 2004)

COMMITMENT !!!COMMITMENT !!!

QUESTIONS ?QUESTIONS ?





























