Embed Size (px)
Citation preview

University of Tennessee at Chattanooga University of Tennessee at Chattanooga
UTC Scholar UTC Scholar
Honors Theses Student Research, Creative Works, and Publications
5-2019
Preparation of organometallic cobalt(III) complexes containing Preparation of organometallic cobalt(III) complexes containing
bidentate chiral amine ligands as potential transfer hydrogenation bidentate chiral amine ligands as potential transfer hydrogenation
catalysts catalysts
Luccas C. Do Carmo University of Tennessee at Chattanooga, [email protected]
Follow this and additional works at: https://scholar.utc.edu/honors-theses
Part of the Chemistry Commons
Recommended Citation Recommended Citation Do Carmo, Luccas C., "Preparation of organometallic cobalt(III) complexes containing bidentate chiral amine ligands as potential transfer hydrogenation catalysts" (2019). Honors Theses.
This Theses is brought to you for free and open access by the Student Research, Creative Works, and Publications at UTC Scholar. It has been accepted for inclusion in Honors Theses by an authorized administrator of UTC Scholar. For more information, please contact [email protected].

1
Preparation of Organometallic Cobalt(III) Complexes Containing Bidentate Chiral Amine
Ligands as Potential Transfer Hydrogenation Catalysts
Luccas C. Do Carmo
Departmental Honors Thesis
University of Tennessee at Chattanooga
Department of Chemistry and Physics
Project Director: Dr. John Lee
Examination Date:
March 29, 2019
Committee Members:
Dr. John Lee
Dr. Jisook Kim
Dr. Gretchen Potts
_____________________________________
Dr. John Lee
Project Director
_____________________________________
Dr. Jisook Kim
Departmental Examiner
_____________________________________
Dr. Gretchen Potts
Departmental Examiner

2
Abstract
Mixed coordination sphere cobalt(III) complexes containing a chiral bidentate amine ligand and
an organic carbocyclic ligand have been prepared and characterized. The integration of this
mixed ligand environment involving an organometallic and Werner-type ligand along with the
earth abundant metal in a high oxidation state has the potential for interesting reactivity.
Complexes of the type [Co(CpR)(X-DPEN)(I)](I), where CpR = cyclopentadienyl (Cp, R = H) or
pentamethylcyclopentadienyl (Cp*, R = Me) and X = meso-1,2-, 1R,2R-, and 1S,2S-, have been
prepared by ligand substitution from [Co(CpR)(I)2(CO)] with 1,2-diphenyletheylenediamine
(DPEN). The outer sphere iodide is readily exchangeable with sodium tetrakis[3,5-
bis(trifluoromethyl)phenyl]borate (NaBArF) or cesium acetate (CsOAc), and both iodides can be
exchanged with silver(I) acetate (AgOAc). The latter reactions with AgOAc results in either
[Co(CpR)(X-DPEN)(OH2)](OAc)2 or [Co(CpR)(X-DPEN)(OAc)](OAc) depending on the solvent
profile, and partial iodide removal to produce [Co(Cp*)(1S,2S-DPEN)(OAc)](I) in the presence
of CsOAc. The complexes are isolated as diamagnetic black, dark purple, or, in the case of
acetate complexes, pink solids and characterized by 1H-NMR, 13C-NMR and UV-vis
spectroscopy as well as either elemental analysis or mass spectrometry. Four complexes,
[Co(Cp)(1R,2R-DPEN)(I)](I) (1), [Co(Cp)(1S,2S-DPEN)(I)](BArF) (2a), [Co(Cp*)(1S,2S-
DPEN)(I)](I) (4), and [Co(Cp*)(1S,2S-DPEN)(OAc)](I) (13) have been characterized by single-
crystal X-ray diffraction. The preparation, characterization, and initial reactivity of these
complexes as potential catalysts for aqueous hydrogen transfer as well as the extension to the
substituted chiral amine (R,R)-N-(2-amino-1,2-diphenylethyl)-p-toluenesulfonamide (Ts-DPEN)
will be discussed.

3
Table of Contents
Cover Sheet………………………………………………………………………………………. 1
Abstract…………………………………………………………………………………………... 2
Table of Contents ………………………………………………………………………………...3
Glossary ………………………………………………………….……………………………….4
Chapter 1: History and use of Cobalt and the DPEN ligand in
catalysis……………………………………………………………………………………………5
1. Use of Transition Metals in Asymmetric Transfer Hydrogenation ………………..6
2. 1st Row Transition Metals for Catalysis …………………………………………..9
3. Cobalt in Catalysis ………………………………………………………………..11
4. Use of Cobalt with Achiral and Chiral Amine Ligands …………………………..12
Chapter 2: Synthesis and reactivity of Co(III) complexes with bidentate amine ligands for
catalytic asymmetric transfer hydrogenation ………………………………………………...15
1. Introduction ……………………………………………………………………….16
2. Experimental Section ……………………………………………………………..19
3. Results and Discussion – Part 1 Synthesis and Characterization …………………26
4. Results and Discussion – Part 2 Reactivity ……………………………………….41
5. Conclusions and Future Work …………………………………………………….44
Chapter 3: Exploration of aqueous Co(III) acetate complexes for catalytic activity ………46
1. Introduction ……………………………………………………………………….47
2. Experimental Section ..……………………………………………………………49
3. Results and Discussion – Part 1 Synthesis and Characterization …………………57
4. Results and Discussion – Part 2 Reactivity ……………………………………….69
5. Conclusions and Future Work …………………………………………………….71
Chapter 4: Synthesis and reactivity of Co(III) complexes bearing a Ts-DPEN ligand …….74
1. Introduction ……………………………………………………………………….75
2. Experimental Section ……………………………………………………………..76
3. Results and Discussion – Part 1 Synthesis and Characterization …………………78
4. Results and Discussion – Part 2 Reactivity ……………………………………….80
5. Conclusions and Future Work …………………………………………………….80
Appendix A: Crystallography Data ……………………………………………………………...83
Acknowledgments………………………………………………………………………………..85

4
Glossary
Bpy………………………………………………………………………………… 2,2'-Bipyridine
CO…………………………………………………………………....Carbonyl (carbon monoxide)
Cp………………………………………………………………………………...Cyclopentadienyl
Cp*…………………………………………………………………..Pentamethylcyclopentadienyl
CpR………………………………………………………………………………………Cp or Cp*
DPEN…………………………………………………………..……1,2-diphenylethylenediamine
I………………………………………………………………………………………………Iodide
OAc…………………………………………………………………………………………Acetate
OH2…………………………………………………………………………………………...Aqua
Ts………………………………………………………………………………….Toluenesulfonyl

5
Chapter 1: History and Use of Cobalt and DPEN Ligand in
Catalysis

6
1. Use of Transition Metals in Asymmetric Transfer Hydrogenation
The discovery of homogeneous transition metal catalysis made an impact in industrial
chemical transfomations such as hydrogenation, hydroformylation, and polymerization
reactions.1 This has led to many new chemical processes that have technological, industrial, and
medical significance. A key discovery among these transition metal catalysts was the synthesis
of the so called Noyori Catalysts. In 1996, Noyori and Ikariya synthesized
[RuCl(η6-arene)(N-arylsulfonyl-DPEN)]; DPEN = 1,2-diphenylethylene-1,2-diamine
(Figure 1), which won the Nobel Prize 2001.2 These complexes were reported to catalyze
aromatic ketones and imines with great stereoselectivity and activity using either 2-PrOH or
formic acid as a hydrogen source.2,3,4
These metal catalysts drive the kinetics and enantioselectivity of the reaction products. In
catalytic transfer hydrogenation, double bonds are reduced to single bonds with the addition of
H2 from non-gaseous H2 sources. Transfer hydrogenation dates back to 1903 where Knoevenagel
successfully used palladium to promote the disproportionation of dimethyl 1,4-
dihydroterephthalate to dimethyl terephthalate and cis-hexahydroterephthalate.5,6 Types of
transfer hydrogenation techniques include Meerwein−Ponndorf−Verley (MPV) reductions
(Scheme 1), late transition metal-catalyzed reactions, organocatalytic, enzyme-catalyzed,
Figure 1. Noyori Catalyst
7
thermal, and base-catalyzed, processes.6 My research will focus on late transition metal catalyzed
reactions.
A significant milestone in the study of transfer hydrogenation was the discovery of late
transition metal catalysts which include second- and third-row transition metal groups.6 For
example, in the 1980s, Ru based catalysts were beginning to be synthesized and were
successfully facilitating transfer hydrogenation reactions.7,8 With the emergence of these novel
catalysts, further discoveries led to the development of asymmetric transfer hydrogenation. This
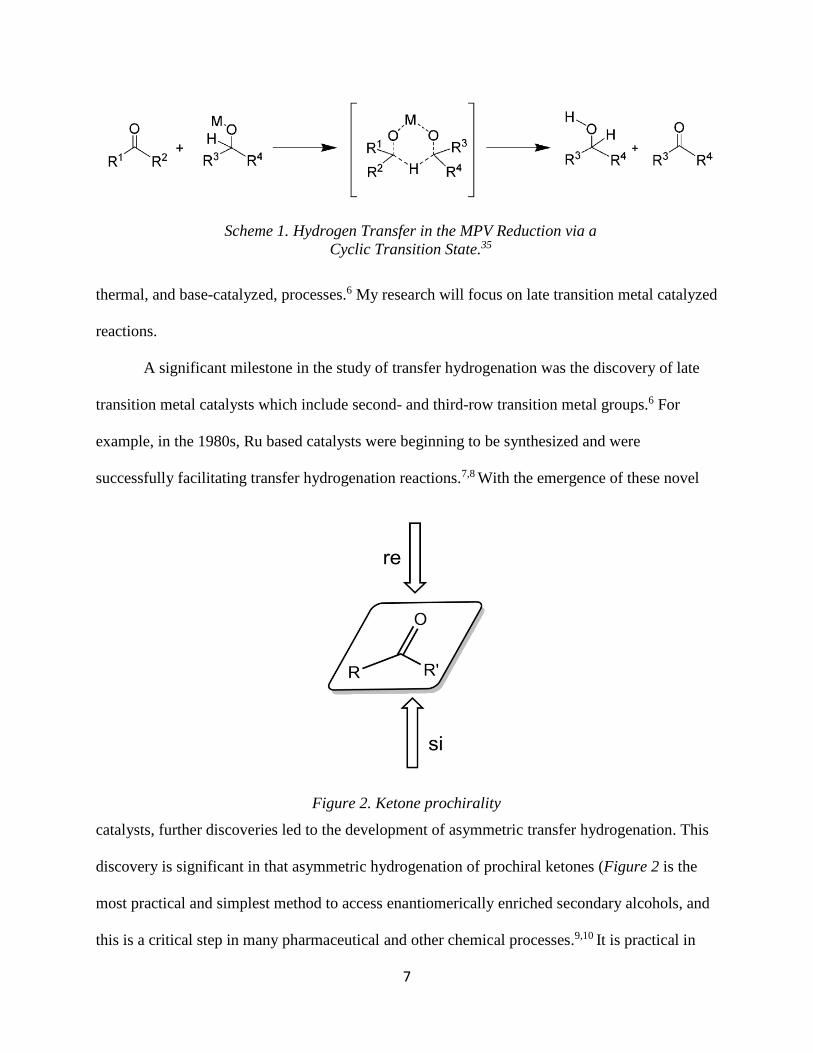
discovery is significant in that asymmetric hydrogenation of prochiral ketones (Figure 2 is the
most practical and simplest method to access enantiomerically enriched secondary alcohols, and
this is a critical step in many pharmaceutical and other chemical processes.9,10 It is practical in
Scheme 1. Hydrogen Transfer in the MPV Reduction via a
Cyclic Transition State.35
Figure 2. Ketone prochirality
8
the sense that hydrogenation requires working with flammable H2 gas, while transfer
hydrogenation requires a hydrogen source such as an alcohol. The widely used catalysts for
transfer hydrogenation involve Ir, Rh, and Ru complexes in a variety of forms which include
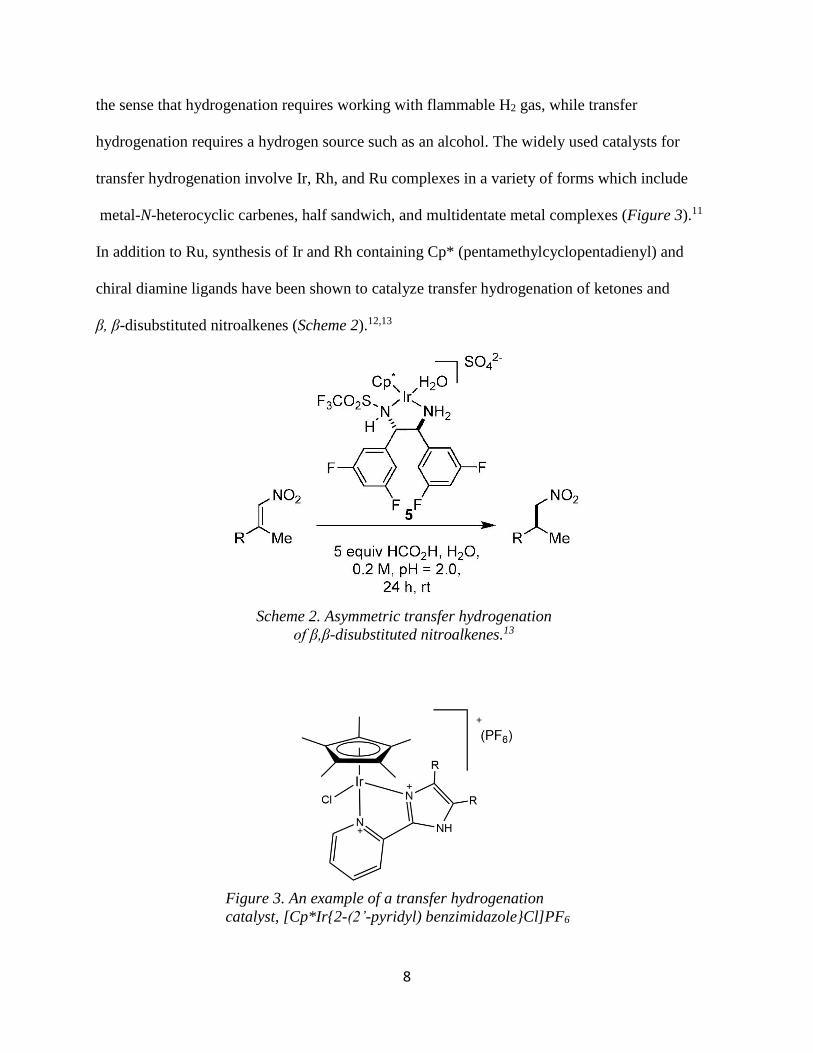
metal-N-heterocyclic carbenes, half sandwich, and multidentate metal complexes (Figure 3).11
In addition to Ru, synthesis of Ir and Rh containing Cp* (pentamethylcyclopentadienyl) and
chiral diamine ligands have been shown to catalyze transfer hydrogenation of ketones and
β, β-disubstituted nitroalkenes (Scheme 2).12,13
Figure 3. An example of a transfer hydrogenation
catalyst, [Cp*Ir{2-(2’-pyridyl) benzimidazole}Cl]PF6
Scheme 2. Asymmetric transfer hydrogenation
of β,β-disubstituted nitroalkenes.13

9
2. 1st Row Transition Metals for Catalysis
Metal Cost per gram
Ru $8.55
Rh $103.20
Ir $46.94
Co $0.03
Along with the development of Ir, Rh, and Ru based catalysts, other late transition metals
have been explored and utilized for transfer hydrogenation catalysis, which includes: Au, Co, Ni,
Os, Pd, Pt, and Re.14-20 Due to variety of active metals for transfer hydrogenation, there has been
a growing interest in exploring reactivities of 1st row transition metals. As mentioned before, the
reduction of carbonyls, such as aldehydes, ketones, esters, carboxylic acids, and acid anhydrides,
are key processes in organic chemistry. However, well-designed catalysts are mainly synthesized
from precious metals such as Ir, Rh, and Ru. Because precious metals are rare, and the demand
for catalysts is growing, exploring reactivities of Earth abundant metals is sensible for economic
reasons as well as to reduce environmental toxicity (Table 1).8,21 However, because these
transition metals have access to multiple 1-electron oxidation states and weak metal to carbon
bonding, it is challenging to design catalytic systems with these metals.22
Table 1. Metal Prices. http://www.infomine.com/investment/precious-metals/ (as of 3/20/19)

10
Despite the design challenges of 1st row metal systems, several complexes have been
synthesized and show catalytic activity. For example, Fe catalyzed transfer hydrogenation has
been studied profusely in the last decade with great progress.23 In 2011, the first Fe mediated
transfer hydrogenation of imines was reported using the Fe precursor [Et3NH][HFe3(CO)11] in
the presences of a N,N′-bis[o-(bis(4-trifluoromethylphenyl)phosphino)benzylidene]-(1S,2S)-
diaminocyclohexane (PNNP) ligand.24 Another example is the Fe catalyzed homogeneous
hydrogenation of alkenes using (PNHPiPr)Fe(H)2(CO) (PNHPiPr = NH(CH2CH2PiPr2)2), which
was done under mild conditions.25 Ni based catalysts have also been synthesized and shown
activity in the reduction of unsaturated compounds via transfer hydrogenation (Scheme 3).26
Scheme 3. Nickel‐Catalyzed Asymmetric Transfer
Hydrogenation of Hydrazones

11
3. Cobalt in Catalysis
For the project described in subsequent chapters, we are interested in the design of Earth
abundant metals for catalysis to
possibly improve the current method of
hydrogenation. Current literature
reveals that there are cobalt based
systems that facilitate hydrogenation
reactions. One design includes the
preparation of C1-symmetric bis-
(imino) pyridines in which one imine is
secured by a large 2,6-diisopropylaryl ring and the other is arranged from a single enantiomer of
a chiral alkyl amine (Figure 4).27 The complex is activated in the presence of H2 to release
methane and make a Co(I) hydride.28 The oxidation state of the metal remains the same during
the catalytic reaction.
Moreover, carbon dioxide can be thermally reduced to methanol using a homogeneous
cobalt-based catalyst. Jacob Schneidewind and coworkers have developed the first homogenous
non-noble metal catalyst for the hydrogenation of CO2 to methanol. Their catalyst design is
developed from [Co(acac)3], where acac = acetylacetone, Triphos
(bis(diphenylphosphinoethyl)phenylphosphine), and HNTf2 (triflimide).29 The use of this
complex allows the hydrogenation reaction to run at 100 °C without decreasing any catalytic
activity. All of the chemical systems mentioned indicate that cobalt has unique properties that
drives its catalytic potential. The oxidation states of the metal do not change since the ligands are
Figure 4. enantiopure, C1-symmetric
bis(imino)pyridine Co(I) methyl complexes for
asymmetric olefin hydrogenation.27

12
redox active and facilitate the catalytic reactions. Our proposed system will be different as we
will be working with Co(III) and and will utilize the DPEN ligand for an H+ and the Co-H as H-
as reported for the Noyori catalyst.2
4. Use of Cobalt with Achiral and Chiral Amine Ligands
Co(III)-DPEN (DPEN = 1,2-diphenylethylenediamine) compounds have been
synthesized and been utilized for catalysis. These compounds have been reported to facilitate
asymmetric C-C bond forming reactions.30 These compounds are a derivative of the Werner
compounds, which were the first inorganic compounds that were resolved enantiomerically about
a century ago.31 The [Co((S,S)-DPEN)3]3+ 3X– ((S,S)-33+ 3X–; X = Cl, ClO4) compounds were
able to catalyze Michael addition of malonate esters to nitroalkenes in high enantioselectivity
without the need of an inert atmosphere environment.
Co(III) mixed sandwich complexes of the type [Co(CpR)([9]aneX3)](PF6)2, where
[9]aneX3 = thioether ([9]aneS3) or azaether ([9]aneN3), have been prepared from
[Co(CpR)(CO)(I)2] which includes the organometallic cyclopentadienyl (Cp) or
pentamethylcyclopentadienyl (Cp*) ligands.32 These complexes show the lability of CO and
iodide ligands are labile and therefore can serve as synthetic starting point for a mixed
coordination sphere. Our proposed complex includes an organometallic ligand, Cp and/or Cp*
and the Werner DPEN ligand. The addition of a hydride to the complex [Co(CpR)(X,X-
DPEN)(I)](I) will also explored as it plays a key role in transfer hydrogenation. However,
isolation and characterization of Co(III)-H complexes are rare, which makes these complexes
interesting from a structural viewpoint.33,34 These examples give rationale to further explore
Co(III) as a potential catalyst for transfer hydrogenation reactions and the proposed complex will
be discussed in the next chapter.

13
References
1. Pasrshall, G. W.; Putscher, R. E. J. Chem. Educ., 1986, 63, 189.
2. Uematsu, N.; Fuji, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc.
1996, 118, 4916-4917.
3. Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. J. Am. Chem.Soc. 1996,
118, 2521−2522.
4. Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Sandoval, C.; Noyori, R. J. Am. Chem.
Soc. 2006, 128, 8724−8725.
5. Knoevenagel, E.; Bergdolt, B. Chem. Ber. 1903, 36, 2857−2860.
6. Wang, D.; Astruc, D. Chemical Reviews 2015, 115, 6621-6686.
7. Bianchi, M.; Matteol, U.; Menchi, G.; Frediani, P.; Pratesi, U.; Piacenti, F.; Botteghi, C. J.
Organomet. Chem. 1980, 198, 73−80.
8. Matteoli, U.; Frediani, P.; Bianchi, M.; Botteghi, C.; Gladiali, S. J. Mol. Catal. 1981, 12,
265−319.
9. Noyori, R., Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). J. Angew.
Chem., Int. Ed. 2002, 41, 2008-2022.
10. Stefane, B.; Pozgan, F., Asymmetric Hydrogenation and Transfer Hydrogenation of
Ketones. In Hydrogenation, Karamé, I., Ed. InTech: Rijeka, 2012; p Ch. 02.
11. Pachhunga, K.; Therrien, B.; Kreisel, K. A.; Yap, G. P.; Kollipara, M. R. Polyhedron,
2007, 26, 3638-3644.
12. Murata, K.; Ikariya, T.; Noyori, R. J. Org. Chem. 1999, 64, 2186−2187.
13. Soltani, O.; Ariger, M. A.; Carreira, E. M. Org. Lett. 2009, 11, 4196−4198.
14. He, L.; Ni, J.; Wang, L.-C.; Yu, F.-J.; Cao, Y.; He, H.-Y.; Fan, K.-N. Chem-Eur. J. 2009,
15, 11833−11836.
15. Bullock, R. M. Science 2013, 342, 1054−1055.
16. Polshettiwar, V.; Baruwati, B.; Varma, R. S. Green Chem. 2009, 11, 127−131.
17. Chelucci, G.; Baldino, S.; Baratta, W. Acc. Chem. Res. 2015, 48, 363−379.
18. Hauwert, P.; Maestri, G.; Sprengers, J. W.; Catellani, M.; Elsevier, C. J. Angew. Chem.,
Int. Ed. 2008, 47, 3223−3226.
19. Alonso, F.; Riente, P.; Rodriguez-Reinoso, F.; Ruiz-Martínez, J.; Sepulveda-
Escribano, A.; Yus, M. J. Catal. 2008, 260, 113−118.
20. Jiang, Y.; Blacque, O.; Fox, T.; Frech, C. M.; Berke, H. Organometallics. 2009, 28, 5493
−5504.
21. Chirik, P. J.; Morris, R. Acc. Chem. Res. 2015, 48, 2495−2495.
22. Chakraborty, S.; Guan, H. Dalton Trans. 2010, 39, 7427-7436.
23. Bata, P.; Notheisz, F.; Kluson, P,; Zsignmond, Á. Appl. Organemet. Chem. 2015, 29,
45-49.
24. Zhou, S.; Fleischer, S.; Junge, K.; Das, S.; Addis, D.; Beller, M. Angew. Chem Int. Ed.
2010, 49, 8121-8125.
25. Xu, R.; Chakraborty, S.; Bellows, S. M.; Yuan, H.; Cundari, T. R.; Jones, W. D. ACS
Cat. 2016, 6, 2127-2135.
26. Xu, , H.; Yang, P.; Chuanprasit, P.; Hirao, H.; Zhou, J. Angew. Chem., Int. Ed. 2015, 54,
5112-5116.

14
27. Monfette, S.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. J. A. Chem. Soc. 2012, 134,
4561-4564.
28. Luca, O. R.; Crabtree, R. H. Chem. Soc. Rev. 2013, 42, 1440-1459. (29)
29. Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Angew. Chem., Int.
Ed. 2017, 56, 1890-1893.
30. Lewis, K. G.; Ghosh, S. K.; Bhuvanesh, N.; Gladysz, J. A. ACS Cent. Sci.,
2015, 1, 50-56.
31. Werner, A. Chem. Ber. 1912, 45, 121-130.
32. Lee, J. P.; Latendrese, T. P.; Henson, K. A.; Mehne, L. F. Inorg. Chim. Acta. 2019, 485,
200-208.
33. Wiedner, E. S.; Roberts, J. A. S.; Dougherty, W. G.; Kassel, W. S.; DuBois, D. L.;
Bullock, R. M., Inorg. Chem. 2013, 52, 9975-9988.
34. Lacy, D. C.; Roberts, G. M.; Peters, J. C. J. Am. Chem. Soc., 2015, 137, 4860–4864.
35. Wang, D.; Astruc, D. Chem. Rev., 2015, 115, 6621–6686.

15
Chapter 2: Synthesis and reactivity of Co(III) complexes
with bidentate amine ligands for catalytic asymmetric
transfer hydrogenation

16
1. Introduction
The purpose of this research is to develop a cobalt(III) catalyst to facilitate homogenous
asymmetric hydrogenation (Scheme 1). Currently, the majority of the catalysts being used are
precious metals such as ruthenium, rhodium, and iridium.1 These reactions are responsible for
synthesizing biochemical compounds that can be useful for making pharmaceutical drugs
(Scheme 2).2 For example, Noyori et. al. developed a ruthenium system featuring Ts-DPEN and
para-cymene ligands, and others have used this as a starting point to make changes to the chiral
ligand that is able to catalyze asymmetric hydrogenation reactions (Figure 1).3 My goal is to
replace expensive metals with first-row transition metals by attempting to extend this chemistry
to Earth-abundant Co(III) homogeneous complexes. This is proposed to be accomplished by
taken advantage of the affinity of Co(III) for sp3-hybridized-amine ligands, which are present in
the Noyori catalyst, and the subsequent inertness of the Co-N bond.4,5
Scheme 1. Selective preparation of chiral alkanes, alcohols or amines from
prochiral ketones and imines
Figure 1. Noyori Catalyst

17
It would be beneficial to switch to first–row transition metals as they are more abundant and
thus more cost effective. Indeed, finding ways to replace precious metals with Earth abundant
metal-based catalysts is a growing field among synthetic chemists.1 The synthesis of enantiopure
compounds using abundant Earth metals is challenging due to the redox flexibility of the metal
(i.e., many1-electron oxidation states are typically available). However, literature suggests that
cobalt complexes containing chiral ligands have been synthesized and has been used for
asymmetric alkene hydrogenation reactions.1,6
A mixed coordination sphere cobalt complex was successfully synthesized by our group and
has been fully characterized excluding 13C NMR (Figure 2). The complex features the addition
of a stabilizing anionic cyclopentadienyl (Cp) or pentamethylcyclopentadienyl (Cp*) to occupy
three coordination sites of the octahedral complex, which will direct reactivity, similar to the
Scheme 2. Synthesis of Levofloxacin
Figure 2. [Co(CpR)(X,X-DPEN)(I)](I)

18
aromatic p-cymene ligand on the Noyori catalyst. Also, the complex contains a 1,2-
diphenylethane-1,2-diamine (DPEN) chiral ligand which will provide an asymmetric
environment and a proton source. Lastly, the complex contains an iodide in both the inner sphere
bound to the Co(III) and outer sphere with the former likely to be labile and the latter to
potentially provide unique solubility opportunities via anion exchange.
Transfer hydrogenation, as mentioned already, involves the addition of H2 across a C-C
double bond. Although H2 in of itself is a cleaner gas than hydrocarbons, there are environmental
issues during modern production of H2 gas. Currently, 90% of H2 production is done by steam
reforming of natural gas or high temperature oil fraction.7 H2 production from natural gas,
however, is always associated with greenhouse gas emissions.8 For this reason, alternative
sources H2 have been of most interest among chemists and environmental researchers. The two
main alternatives include H2O splitting as well as electrocatalytic CO2 reduction. However, these
have their challenges as well due to high activation energies.9
A key step in the preparation of a transfer hydrogenation catalyst from [Co(CpR)(X,X-
DPEN)(I)](I) will be the synthesis of a cobalt hydride (Co-H). The isolation and synthesis of
substituting the inner sphere iodide with a hydride to the complex will be challenging as Co(III)
hydride as isolated examples are rare.10

19
2. Experimental Section
2.1. Materials
All solvents and reagents were used as received. The ligands R,R-DPEN, S,S-DPEN, as well
as both silver acetate (AgOAc) and sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate (NaBArF)
were purchased from either Aldrich Chemical Company or Acros Organics and used as received.
The metal reagents [Co(Cp)(CO)(I)2] and [Co(Cp*)(CO)(I)2] were prepared as previously
reported.11,12
2.2. Measurements
1H and 13C NMR spectra were obtained on a JEOL ECX 400 MHz spectrometer (operating
frequency for 13C NMR was 100 MHz) and referenced against tetramethylsilane using residual
proton signals (1H NMR) or the 13C resonances of the deuterated solvent (13C NMR). UV-vis
spectra were obtained on a Varian Cary UV-vis spectrophotometer in dichloromethane using 1
cm quartz cuvettes. Elemental analyses were performed by Atlantic Microlab, Inc in Norcross,
GA. Mass spectra were obtained via electrospray ionization on a Waters Quattro Micro
quadrupole mass spectrometer operated in the positive mode with a 2.0 kV capillary and 35 V
cone voltage. Samples dissolved in acetonitrile were introduced at a flow rate of 22 µL/min
using the direct infusion syringe pump. Mass spectra were obtained over the range m/z 200 –
1400 with a scan speed of 1200 amu/sec.
2.3.Syntheses
2.3.1. Preparation of [Co(Cp)(R,R-DPEN)(I)](I) (1).
A 50 mL round-bottom flask was charged with [Co(Cp)(CO)(I)2] (0.1027 g, 0.2531
mmol) and toluene (20 mL). To the dark purple solution was added (1R,2R)
diphenylethylenediamine (R,R-DPEN) (0.0658 g, 0.310 mmol), and the mixture was allowed to

20
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a dark purple residue, and hexanes were used to precipitate a solid
product. The dark purple solid was collected by vacuum filtration and dried in vacuo. The final
product weighed 0.141 g, 95% yield. 1H NMR (3a-1H-LD), 13C NMR (3a-13C-LD).
1H NMR (CDCl3, δ): 5.85 (5H, s, Cp), 4.33 (1H, td, CH), 3.38 (1H, td, CH), 3.30 (1H, t, NH),
4.60 (1H, s, NH), 7.21-7.32 (8H, m, DPEN aromatic and NH ). 13C {1H} NMR (CDCl3, δ):
129.3, 129.1, 129.0, 128.9, 128.7, 128.7, 128.6, 128.5, 128.2, 128.1, 128.0, 128.0 (12C, DPEN
aromatic C), 83.8 (5C, Cp), 64.9 (1C, CH), 63.7 (1C, CH). Anal. Calcd for C19H21N2I2Co: H,
3.59; N, 4.75. Found: H, 3.84; N, 5.10. UV-Vis (CH2Cl2, nm (L•M-1cm-1)): 555 (1130), 279
(22800).
2.3.2 Preparation of [Co(Cp)(S,S-DPEN)(I)](I) (2).
A 50 mL round-bottom flask was charged with [Co(Cp)(CO)(I)2] (0.109 g, 0.268 mmol)
and dichloromethane (20 mL).To the dark purple solution was added (1S,2S)-
diphenylethylenediamine (S,S-DPEN) (0.0691 g, 0.326 mmol), and the mixture was allowed to
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a dark purple residue, and hexanes were used to precipitate a solid
product. The dark purple solid was collected by vacuum filtration and dried in vacuo. The final
product weighed 0.135 g, 85% yield. 1H NMR (7b-1H-LD) 13C NMR (33b-13C-JM). 1H NMR
(CDCl3, δ): 6.06 (5H, s, Cp), 4.71 (1H, m, CH), 3.53 (1H, m, CH), 5.25 (1H, m, NH), 7.17-7.30
(9H, m, DPEN aromatic and NH). 13C {1H} NMR (CD3CN, δ): 128.4-130.0 (12C, DPEN
aromatic C), 84.6 (5C, Cp), 65.7 (1C, CH), 64.6 (1C, CH). Anal. Calcd for C19H21N2I2Co: C,
31.74; H, 4.46. Found: C, 31.15; H, 4.51. UV-Vis (CH2Cl2, nm (L•M-1cm-1)): 576 (1290), 295
(24800).

21
2.3.3 Preparation of [Co(Cp*)(R,R-DPEN)(I)](I) (3).
A 50 mL round-bottom flask was charged with [Co(Cp*)(CO)(I)2] (0.110 g, 0.230 mmol)
and dichloromethane (20 mL). To the dark purple solution was added (1R,2R)-
diphenylethylenediamine (R,R-DPEN) (0.0612 g, 0.288 mmol), and the mixture was allowed to
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a dark purple residue, and hexanes were used to precipitate a solid
product. The dark purple solid was collected by vacuum filtration and dried in vacuo. The final
product weighed 0.119 g, 86% yield. 1H NMR (1f-1H-LD), 13C NMR (1a-13C-LD). 1H NMR
(acetone-d6, δ): 1.97 (15H, s, Cp*), 3.70 (1H, td, CH), 4.57 (1H, td, CH), 3.88 (1H, t, NH), 4.71
(1H, t, NH), 5.41 (1H, t, NH), 6.08 (1H, t, NH), 7.18-7.30 (12C, m, DPEN aromatic CH).
13C{1H} NMR (CD3CN, δ): 11.1 (5C, Cp* aromatic CH3), 63.7 (1C, CH), 65.0 (1C, CH), 93.6
(5C, Cp* aromatic C), 138.5, 137.6, 128.7, 128.7, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1,
128.0, 127.9 (12C, DPEN aromatic C). Anal. Calcd for C19H21N2I2Co: H, 4.73. Found: H, 5.02.
UV-Vis (CH2Cl2, nm (L•M-1cm-1)): 565 (1170), 358 (4540), 289 (25900).
2.3.4 Preparation of [Co(Cp*)(S,S-DPEN)(I)](I) (4).
A 50 mL round-bottom flask was charged with [Co(Cp)(CO)(I)2] (0.107 g,
0.225 mmol) and dichloromethane (20 mL). To the dark purple solution was added (1S,2S)-
diphenylethylenediamine (S,S-DPEN) (0.0594 g, 0.280 mmol), and the mixture was allowed to
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a dark purple residue, and hexanes were used to precipitate a solid
product. The dark purple solid was collected by vacuum filtration and dried in vacuo. The final
product weighed 0.0666g, 45% yield. 1H NMR (2b-1H-LD), 13C (2a-13C-LD). 1H NMR
(CD3CN, δ): 1.75 (15H, s, Cp*), 4.11 (1H, td, CH), 3.51 (1H, td, CH), 3.91 (1H, t, NH), 4.66

22
(1H, d, NH), 5.06 (1H, d, NH), 7.12-7.25 (11H, m , DPEN aromatic and NH). 13C {1H} NMR
(CD3CN, δ): 137.9, 137.1, 129.6, 129.2, 129.1, 129.0, 128.9, 128.8, 128.7, 128.6, 128.2, 127.7
(12C, DPEN arom. C), 93.6 (5C, Cp* arom. C), 64.6 (1C, CH), 64.2 (1C, CH), 11.1 (5C, Cp*
arom. CH3). Anal. Calcd for C19H21N2I2Co: C, 43.65; H, 4.73; N, 4.24. Found: C, 43.36; H, 4.91;
N, 4.44. UV-Vis (CH2Cl2, nm (L•M-1cm-1)): 565 (1230), 357 (4150), 288 (24600).
2.3.5. Preparation of [Co(Cp)(meso-DPEN)(I)](I) (5).
A 50 mL round-bottom flask was charged with [Co(Cp)(CO)(I)2] (0.103 g, 0.253 mmol)
and dichloromethane (15 mL). To the dark purple solution was added meso-1,2-
diphenylethylenediamine (meso-DPEN) (0.0690 g, 0.325 mmol), and the mixture was allowed to
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a black residue, and hexanes were used to precipitate a solid product.
The black solid was collected by vacuum filtration and dried in vacuo. 1H NMR (32a-1H-JM),
13C (32b-13C-JM). 1H NMR (CDCl3, δ): 5.86 (5H, s, Cp), 6.22 (2H, m, CH or NH), 4.59 (2H, m,
CH or NH), 4.06 (1H, m, CH or NH), 7.05-7.36 (15H, m, DPEN aromatic ). 13C {1H} NMR
(CD3CN, δ): 135.7, 128.6, 128.4, 128.2, 128.1, 128.0, 127.9, 127.9, 127.0, 127.7, 127.6, 127.5
(12C, DPEN aromatic C), 84.6 (5C, Cp), 65.7 (1C, CH), 64.6 (1C, CH). Data suggest two
diastereomers in a 10:1 ratio.
2.3.6 Attempted Preparation of [Co(Cp*)(meso-DPEN)(I)](I) (6).
A 50 mL round-bottom flask was charged with [Co(Cp*)(CO)(I)2] (0.102 g, 0.214 mmol)
and dichloromethane (15 mL). To the dark purple solution was added meso-1,2-
diphenylethylenediamine (meso-DPEN) (0.0545 g, 0.257 mmol), and the mixture was allowed to
stir at room temperature for approximately 24 h under a nitrogen atmosphere. The volatiles were
reduced in vacuo, leaving a purple residue, and hexanes were used to precipitate a solid product.

23
The purple solid was collected by vacuum filtration and dried in vacuo 13C {1H} NMR (CD3CN,
δ) 93.0 (5C, s, Cp), 92.7 (5C, s, Cp), 63.3 (1C, s, DPEN), 59.3 (1C, s, DPEN),
12.0 (5C, s, Cp* CH3). Data suggest two diastereomers in a 1:2 ratio.
2.3.7. Preparation of [Co(Cp)(S,S-DPEN)(I)](BArF) (2a)
A 10 mL round-bottom flask was charged with [Co(Cp)(S,S-DPEN)(I)](I) (0.0104 g,
0.0176 mmol), dichloromethane (3mL), NaBArF (0.0241 g, 0.0272 mmol), and H2O (3 mL). The
dark purple mixture was vigorously stirred for 2 h at 35°C. The organic phase was separated and
was evaporated. 1H NMR (32a-1H-LD), 13C NMR (32a-13C-LD) (0.0181 g). 1H NMR (acetone-
d6, δ): 7.77 (14H, m, ortho-H BArF), 7.66 (7H, s, para-H BArF), 7.15-7.32 (12H, m, DPEN
aromatic and NH), 6.78 (1H, t, NH), 5.95 (5H, s, Cp), 5.42 (1H, t, NH), 4.56 (1H, td, CH), 3.70
(1H, t, NH), 3.46 (1H, td, CH).13C {1H} NMR (acetone-d6, δ): 161.8 (4C, quartet, C-B), 134.7,
129.7, 129.3, 129.1, 129.3, 128.9, 128.9, 128.7, 128.6, 127.9, 127.4 (11C, m, ortho-C BArF and
DPEN aromatic) 124.5 (4C, quartet, CF3), 85.0 (5C, Cp), 66.3 (1C, CH), 64.5 (1C, CH). Anal.
Calcd for C51H33N2BCoF24I2: C, 46.18; H, 4.73; N, 2.11. Found: C, 46.28; H, 2.64; N, 2.06.
2.3.8. Preparation of [Co(Cp*)(R,R-DPEN)(I)](BArF) (3a)
50 mL round-bottom flask was charged with [Co(Cp*)(R,R-DPEN)(I)](I) (0.1758 g,
0.2663 mmol), dichloromethane (15 mL), NaBArF (0.3539 g, 0.3993 mmol), and H2O (15 mL).
The dark purple mixture was vigorously stirred for 2 h at 35°C. The organic phase was separated
and was evaporated. 1H NMR (37d-1H-LD). 1H NMR(acetone-d6, δ): 7.79 (18H, m, ortho-H
BArF), 7.67 (9H, s, para-H BArF), 7.12-7.48 (16H, m, DPEN aromatic), 5.95 (1H, t, NH), 5.42
(1H, s, NH), 4.77 (1H, t, NH), 4.37 (1H, td, CH), 3.97 (1H, t, NH) 3.66 (1H, td, CH), 1.94 (15H,
s, Cp*). Anal. Calcd for C56H43N2BCoF24I2: C, 47.38; H, 2.77; N, 1.97. Found: C, 48.01; H, 2.79
; N, 1.63.

24
2.3.9 Preparation of [Co(Cp*)(S,S-DPEN)(I)](BArF) (4a).
50 mL round-bottom flask was charged with [Co(Cp*)(S,S-DPEN)(I)](I) (0.0706 g,
0.1069 mmol), Dichloromethane (15 mL), NaBArF (0.1427 g, 0.1610 mmol), and H2O (15 mL).
The dark purple mixture was vigorously stirred for 1.5 hr at 35°C. The organic phase was
separated and was reduced in vacuo. A dark purple solid was isolated. The final product weighed
0.092 g. 1H NMR (41a-1H-LD). 1H NMR(acetone-d6, δ): 7.79 (21H, m, ortho-H BArF), 7.67
(10H, s, para-H BArF), 7.12-7.48 (18H, m, DPEN aromatic), 5.95 (1H, t, NH) 5.41 (1H, t, NH),
4.77 (1H, t, NH), 4.38 (1H, td, CH), 3.96 (1H, t, NH) 3.68 (1H, td, CH), 1.94 (15H, s, Cp*).
Anal. Calcd for C56H43N2BCoF24I2: C, 47.38; H, 2.77; N, 1.97. Found: C, 48.18; H, 3.12; N,
1.67.
2.4.10. Attempted Preparation of [Co(Cp*)(R,R-DPEN)(CH3)]I
From the glovebox, a 100 mL round-bottom flask was charged with 3 (0.0603g, 0.0913
mmol), dichloromethane (20 mL), and granulated Zn (0.0315 g, 0.481 mmol). The dark purple
solution was stirred at either room temperature or reflux for ~22 h. A solid, presumably ZnI, was
removed from solution through filtration. The filtrate was collected. CH3I (0.15 mL, 5 mol
equivalent) was then added to the solution. There were no observable color changes. The
solution stirred for 3 h and formed a cloudy precipitate. Solution was dried under vacuo until a
solid was isolated. The final product weighed 0.053g. 1H NMR (9a-1H-LD). The NMR spectra
revealed that the complex was paramagnetic, and therefore there were no significant peaks to be
observed.

25
2.4.11. Attempted Catalytic Preparation of dimethyl 2-(2-nitro-1-phenylethyl) malonate
A 1H NMR of dimethyl malonate (a) and trans--nitrostyrene (b) were obtained
separately. Added (a) (0.004 mL, 0.0352 mmol) and (b) (0.0053g, 0.0355 mmol) into a vial and
dissolved them in 0.75 mL acetone-d6. Added 4a (0.005g, 0.0035 mmol) into the solution. The
solution was then transferred to a screw cap NMR tube. Added triethylamine (0.005 mL, 0.0356
mmol) to the solution and was left at room temperature for ~3.5 h. A slight color change
occurred from dark to light purple. After 72 h, the solution color changed to red-orange. 1H
NMR (84a-1H-LD). There were no observable changes in the organic substrates per NMR
spectra.
2.5. X-Ray Crystallography Data Collection and Processing
Appendix A contains crystal data, collection parameters, and refinement criteria for the
crystal structures of 1, 2a, and 4. All crystals were grown by diffusion of diethyl ether into a
dichloromethane solution of the complex, or hexane layering onto a dichloromethane solution of
the complex. Crystals were mounted on the tip of a Bruker SPINE-pin mount and X-ray intensity
data were measured at low temperature using an Oxford Cryosystems Desktop Cooler (200(2) K)
for all structures with a graphite monochromated Mo kα radiation (λ = 0.71073 Å) on a Bruker
SMART X2S Benchtop diffractometer. Integration, data reduction and scaling were carried out
with the programs SAINT and SADABS in the Bruker APEX2 suite of software. Each structure
was solved (XS) using direct methods and refined using full matrix least squares refinement
(SHELXL2017) within Olex2. A direct-methods solution was calculated that provided the non-
hydrogen atoms from the E-map. All non-hydrogen atoms were refined with anisotropic
displacement parameters. All of the hydrogen atoms in each structure were placed in ideal
positions and refined as riding atoms.

26
3. Results and Discussion – Part 1 Synthesis and Characterization
Synthesis of [Co(CpR)(X,X-DPEN)(I)](Y)
The classic coordination chemistry of studies by Werner with Co(III) and amine ligands
showed the affinity of Co(III) for amine ligands,4,5 and with the lability of the CO and I (with
bidentate ligands) on complex [Co(CpR)(CO)(I)2] , our starting point was meso-DPEN in order to
optimize synthetic procedure due to the low cost of the ligand. Complexes of the type
[Co(Cp*)(P-P)I]I had been successfully prepared where P-P is a bidentate phosphine.13
Optimization reactions were performed for both Cp (5) and Cp* (6) starting [Co(CpR)(CO)(I)2]
complexes. The products are isolated as black or purple solids for [Co(Cp)(meso-DPEN)(I)]I (5)
and [Co(Cp*)(meso-DPEN)(I)]I (6), respectively, with the Cp* shown in Scheme 3. As shown by
NMR spectroscopy, the reaction of the meso ligand with [Co(CpR)(CO)(I)2] results in the
isolation of a mixture of two diastereomers (Scheme 3) where the ratio is sterically driven and the
Cp complex is a 1:10 ratio of products while the Cp* complex is a 1:2 ratio as observed by 13C
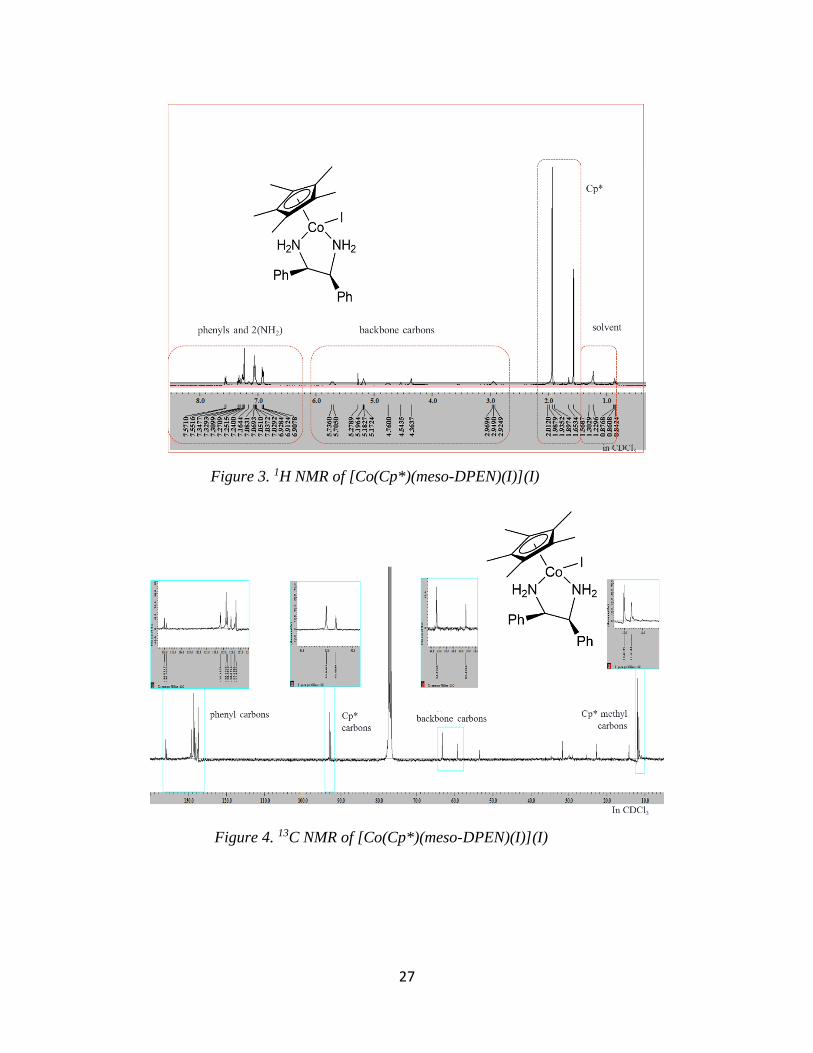
NMR spectroscopy. Seven resonances are expected in the 13C NMR for both complexes.
However, fourteen resonances are observed. Furthermore, based on relative peak heights, this is
representative of a 2:1 ratio of diastereomers where all 1H and 13C NMR resonances have been
resolved for both complexes (Figures 3 and 4).
Scheme 3. Synthesis of [Co(Cp*)(meso-DPEN)(I)](I)
27
Figure 3. 1H NMR of [Co(Cp*)(meso-DPEN)(I)](I)
Figure 4. 13C NMR of [Co(Cp*)(meso-DPEN)(I)](I)
28
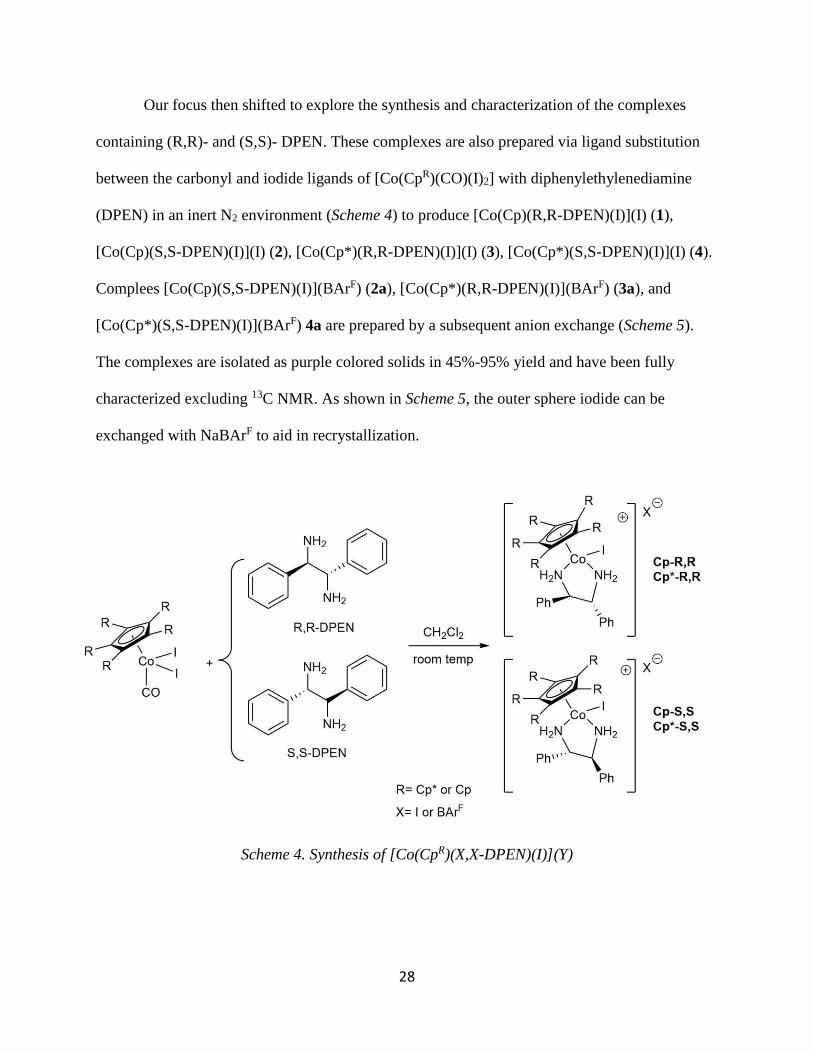
Our focus then shifted to explore the synthesis and characterization of the complexes
containing (R,R)- and (S,S)- DPEN. These complexes are also prepared via ligand substitution
between the carbonyl and iodide ligands of [Co(CpR)(CO)(I)2] with diphenylethylenediamine
(DPEN) in an inert N2 environment (Scheme 4) to produce [Co(Cp)(R,R-DPEN)(I)](I) (1),
[Co(Cp)(S,S-DPEN)(I)](I) (2), [Co(Cp*)(R,R-DPEN)(I)](I) (3), [Co(Cp*)(S,S-DPEN)(I)](I) (4).
Complees [Co(Cp)(S,S-DPEN)(I)](BArF) (2a), [Co(Cp*)(R,R-DPEN)(I)](BArF) (3a), and
[Co(Cp*)(S,S-DPEN)(I)](BArF) 4a are prepared by a subsequent anion exchange (Scheme 5).
The complexes are isolated as purple colored solids in 45%-95% yield and have been fully
characterized excluding 13C NMR. As shown in Scheme 5, the outer sphere iodide can be
exchanged with NaBArF to aid in recrystallization.
Scheme 4. Synthesis of [Co(CpR)(X,X-DPEN)(I)](Y)
)
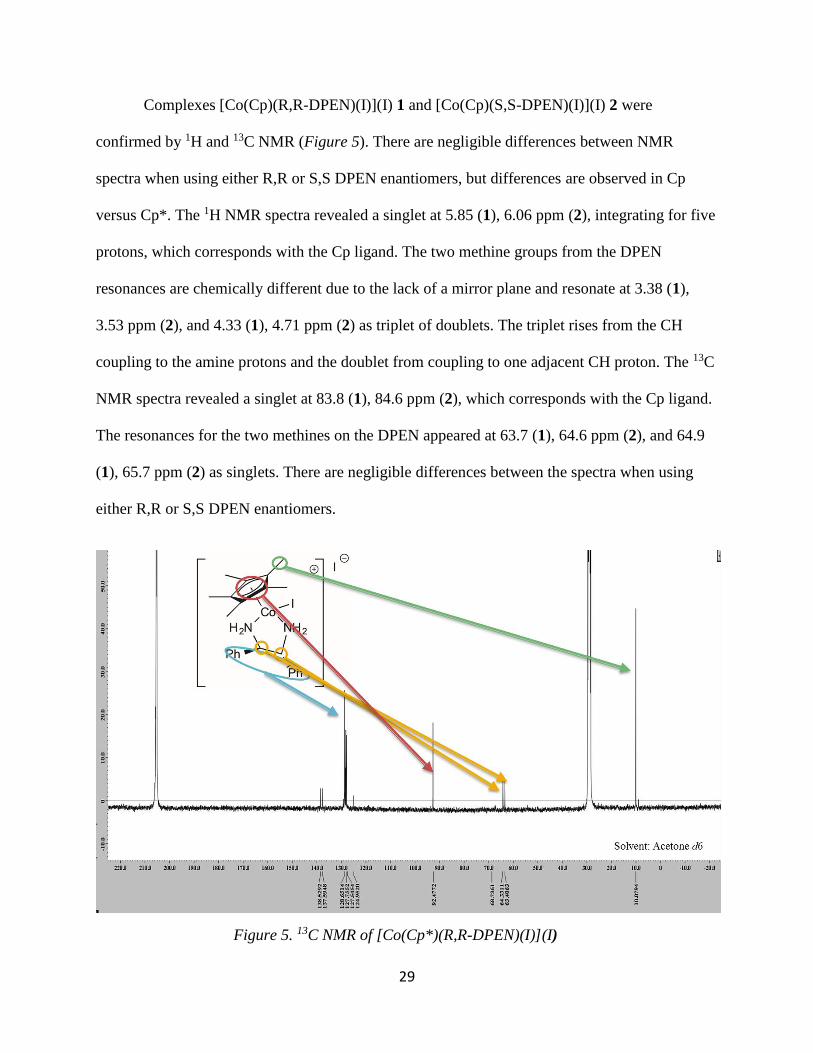
29
Complexes [Co(Cp)(R,R-DPEN)(I)](I) 1 and [Co(Cp)(S,S-DPEN)(I)](I) 2 were
confirmed by 1H and 13C NMR (Figure 5). There are negligible differences between NMR
spectra when using either R,R or S,S DPEN enantiomers, but differences are observed in Cp
versus Cp*. The 1H NMR spectra revealed a singlet at 5.85 (1), 6.06 ppm (2), integrating for five
protons, which corresponds with the Cp ligand. The two methine groups from the DPEN
resonances are chemically different due to the lack of a mirror plane and resonate at 3.38 (1),
3.53 ppm (2), and 4.33 (1), 4.71 ppm (2) as triplet of doublets. The triplet rises from the CH
coupling to the amine protons and the doublet from coupling to one adjacent CH proton. The 13C
NMR spectra revealed a singlet at 83.8 (1), 84.6 ppm (2), which corresponds with the Cp ligand.
The resonances for the two methines on the DPEN appeared at 63.7 (1), 64.6 ppm (2), and 64.9
(1), 65.7 ppm (2) as singlets. There are negligible differences between the spectra when using
either R,R or S,S DPEN enantiomers.
Figure 5. 13C NMR of [Co(Cp*)(R,R-DPEN)(I)](I)

30
Complexes [Co(Cp*)(R,R-DPEN)(I)](I) 3 and [Co(Cp*)(S,S-DPEN)(I)](I) 4 were
confirmed by 1H and 13C NMR. The 1H NMR spectra revealed a singlet at 1.97 (3), 1.75 ppm
(4), integrating for fifteen protons, which corresponds with the Cp* analogue. The two methine
groups from the DPEN resonances were revealed at 3.70 (3), 3.51 ppm (4), and 4.57 (3), 4.11
ppm (4). The 13C NMR spectra revealed a singlet at 93.6 (3), 93.6 ppm (4), which corresponds
with the Cp* aromatic C analogue. The resonances for the methyl groups on the Cp* appeared at
11.1 (3), 11.1 ppm (4). The two methine carbon resonances appeared at 63.7 (3), 64.1 ppm (4),
and 65.0 (3), 64.6 ppm (4). A 1H-13C HMQC NMR spectrum confirmed the assignments for the
DPEN methine backbone in 1H and 13C NMR spectra for 3 (Figure 6).
Figure 6. 1H-13C HMQC of [Co(Cp*)(R,R-DPEN)(I)](I)
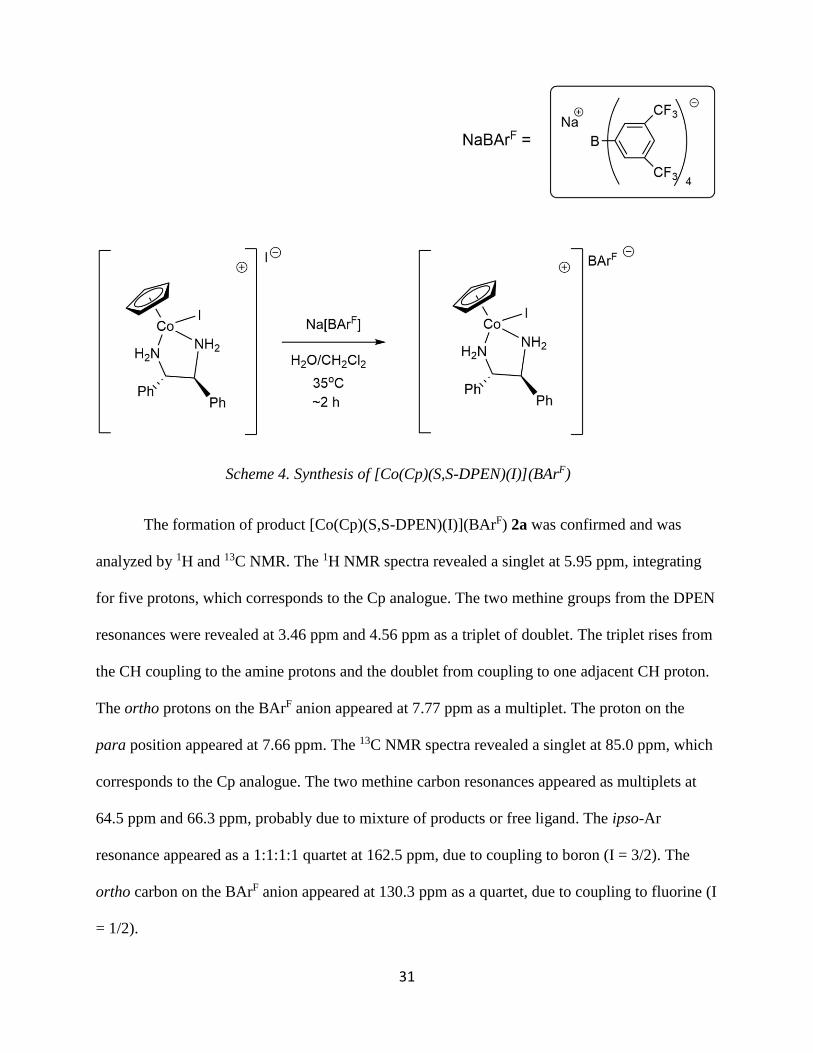
31
The formation of product [Co(Cp)(S,S-DPEN)(I)](BArF) 2a was confirmed and was
analyzed by 1H and 13C NMR. The 1H NMR spectra revealed a singlet at 5.95 ppm, integrating
for five protons, which corresponds to the Cp analogue. The two methine groups from the DPEN
resonances were revealed at 3.46 ppm and 4.56 ppm as a triplet of doublet. The triplet rises from
the CH coupling to the amine protons and the doublet from coupling to one adjacent CH proton.
The ortho protons on the BArF anion appeared at 7.77 ppm as a multiplet. The proton on the
para position appeared at 7.66 ppm. The 13C NMR spectra revealed a singlet at 85.0 ppm, which
corresponds to the Cp analogue. The two methine carbon resonances appeared as multiplets at
64.5 ppm and 66.3 ppm, probably due to mixture of products or free ligand. The ipso-Ar
resonance appeared as a 1:1:1:1 quartet at 162.5 ppm, due to coupling to boron (I = 3/2). The
ortho carbon on the BArF anion appeared at 130.3 ppm as a quartet, due to coupling to fluorine (I
= 1/2).
Scheme 4. Synthesis of [Co(Cp)(S,S-DPEN)(I)](BArF)

32
The formation of product [Co(Cp*)(R,R-DPEN)(I)](BArF) 3a was confirmed by 1H. The
1H NMR spectra revealed a singlet at 1.94 ppm, integrating for fifteen protons, which
corresponds to the Cp* analogue. The two methine groups from the DPEN resonances were
revealed at 3.66 ppm and 4.37 ppm as triplet of doublets. The ortho protons on the BArF
appeared at 7.79 ppm as a multiplet and the para protons on the BArF appeared at 7.67 ppm as a
singlet. The BArF proton integrations were relatively higher than expected. This is potentially
due to excess ligand in the product. The proton on the para position appeared at 7.67 ppm. 13C
NMR analysis will be explored in the future.
Scheme 5. Synthesis of [Co(Cp*)(R,R-DPEN)(I)](BArF)

33
The formation of product [Co(Cp*)(S,S-DPEN)(I)](BArF) 4a was confirmed by 1H NMR.
The 1H NMR spectra revealed a singlet at 1.94 ppm, integrating for fifteen protons, which
corresponds with the Cp* analogue. The two methine groups from the DPEN resonances were
revealed at 3.68 ppm and 4.38 ppm as triplet of doublets. The ortho protons on the BArF
appeared at 7.79 ppm as a multiplet and the para protons on the BArF appeared at 7.67 ppm as a
singlet. The BArF proton integrations were relatively higher than expected. This is potentially
due to excess ligand in the product. The proton on the para position appeared at 7.67 ppm. 13C
NMR analysis will be explored in the future.
Scheme 6. Synthesis of [Co(Cp*)(S,S-DPEN)(I)](BArF)

34
UV-vis Analysis of [Co(CpR)(X,X-DPEN)(I)](I)
UV-Vis Spectroscopy of transition metal complexes experiments are done to quantify
electronic transitions of the metal center and the coordinating ligands. UV-Vis experiments were
done for products 1, 2, 3, and 4. Stock solutions of each product of roughly 1.20-1.40 mM in
dichloromethane. A summary of the results is listed in Table 2. An example spectrum is shown
in Figure 7.
Complex Concentration (µM) λmax (nm) Abs ε (L/cm mol) Transition type
1
680
57.0
555
279
0.766
1.30
1130
22800
d-d
CT or ligand
2
690
55.2
558
280
0.888
1.37
1290
24800
d-d
CT or ligand
3
248
248
49.6
565
358
289
0.289
1.13
1.29
1170
4540
25900
d-d
Cp*
CT or ligand
4
610
122
48.8
565
357
288
0.748
0.506
1.20
1230
4150
24600
d-d
Cp*
CT or ligand
For products 1 and 2, the samples revealed transitions at 279 (1), 280 nm (2), and 555 (1),
558 nm (2), with λmax values of 1.30 (1), 1.37 (2), and 0.766 (1), 0.888 (2), respectively. The
molar absorptivities were calculated to be 22833 (1), 24750 L mol-1 cm-1 (2), and 1125 (1), 1287
L mol-1 cm-1 (2), respectively. For the 1:2 dilution, there was an unresolved shoulder near 400
Table 2. UV-vis analysis of products 1, 2, 3, and 4

35
nm at 1.40 Abs (1 and 2). The transition at 555 (1), 558 nm (2) is a d-d electronic distribution
since the transition occurred in the visible region of the spectrum and has a ε value consistent
with a d-d transition lacking a center of inversion. The transition at 279 (1), 280 nm (2) indicated
a charge transfer between the metal and ligands, or completely ligand based due to the absorption
in the UV region of the spectrum and the large magnitude of the ε.
For products 3 and 4, the samples revealed transitions at 289 (3), 288 (4), 358 (3), 357
(4), and 565 (3), 565 nm (4), with λmax values of 1.29 (3), 1.20 (4), 1.13 (3), 0.506 (4), and 0.289
(3), 0.748 (4) Abs, respectively. The molar absorptivities were calculated to be 25990 (3), 24578
(4), 4537 (3), 4151 (4), and 1166 (3), 1227 L mol-1 cm-1 (4), respectively. For the 1:5 dilution,
there was an unresolved shoulder near 440 nm at 0.300 (3), 0.700 Abs (4). The transition at 565
(3), 565 nm (4), is a d-d electronic distribution since the transition occurred in the visible region
of the spectrum and has a ε value consistent with a d-d transition lacking a center of inversion.
The transition at 358 (3), 357 nm (4), could be Cp* based as it is absorbed in the UV region. The
transition at 289 (3), 288 nm (4) indicated a charge transfer between the metal and ligands, or
completely ligand based due to the absorption in the UV region of the spectrum and the large
magnitude of the ε.

36
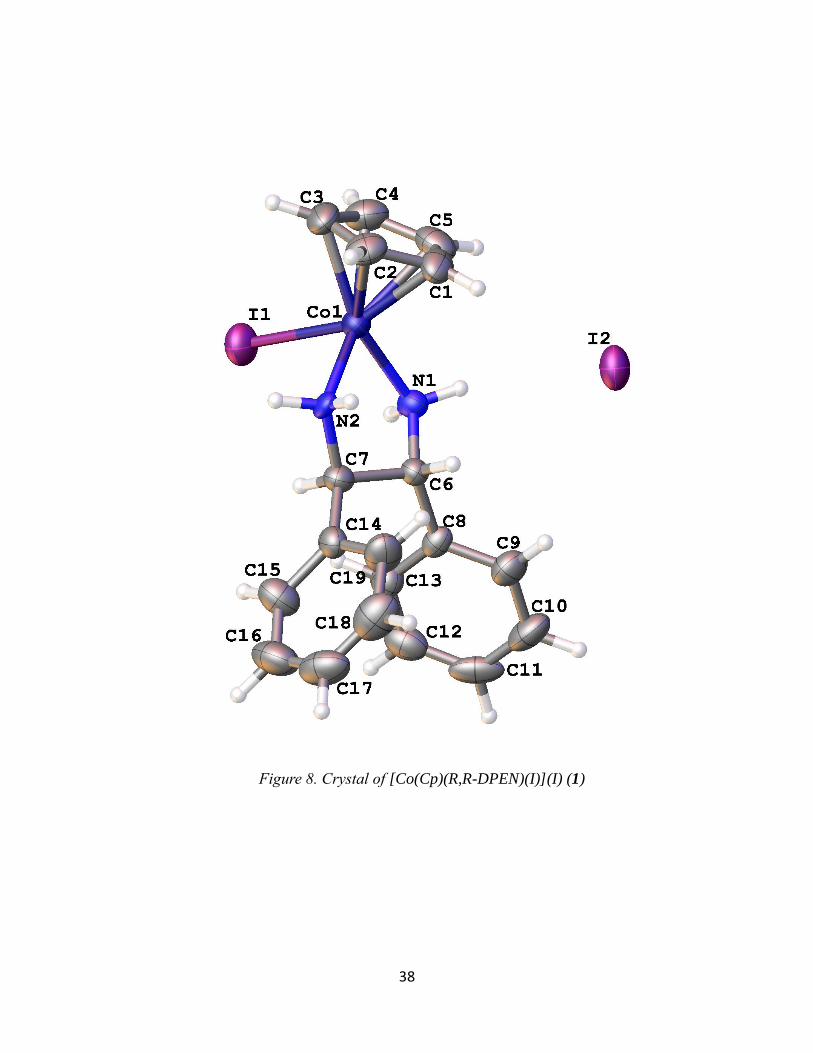
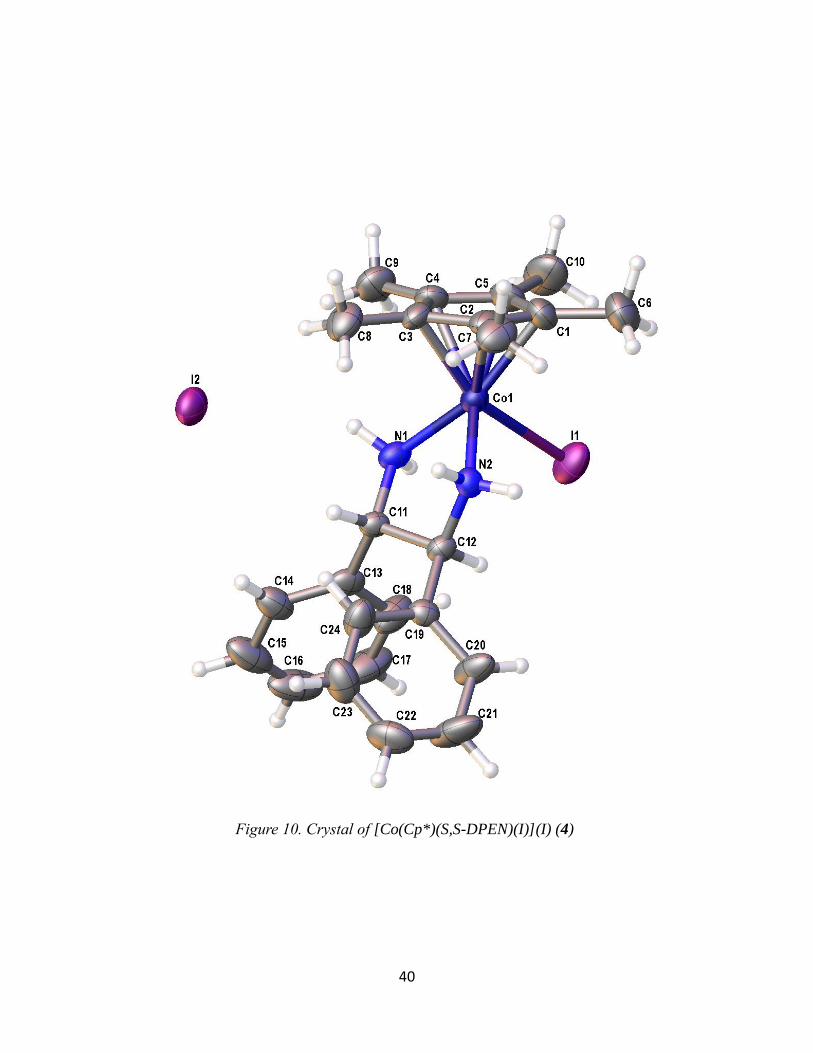
X-Ray Crystallography of [Co(CpR)(X,X-DPEN)(I)](Y)
Crystal structures were obtained for products 1, 2a, and 4 and a summary of the bond
lengths and angles are listed in Table 3. These are the first single-crystal X-ray structures of
Co(III) compounds with both Cp or Cp* ligands and a bidentate amine ligand in its coordination
sphere. Among the products obtained, all of them show similar bond lengths and angles. The
bond lengths of the Co-N range from 1.965-2.012 Å and the Co-I range from 2.588-2.639 Å.
The bond angles for N1-Co-N2 of all three products show a bite angle between 82-84°
while the I-Co-N is 90-95°. These data suggest that the complex is best described as octahedral.
It is slightly distorted due to the chelate of the bidentate, making it sterically locked in place at
the reduced chelate angle. This steric phenomenon is likely due to the size of the N atom. Indeed,
Figure 7. An example UV-vis Spectra of [Co(Cp*)(R,R-DPEN)(I)](I)
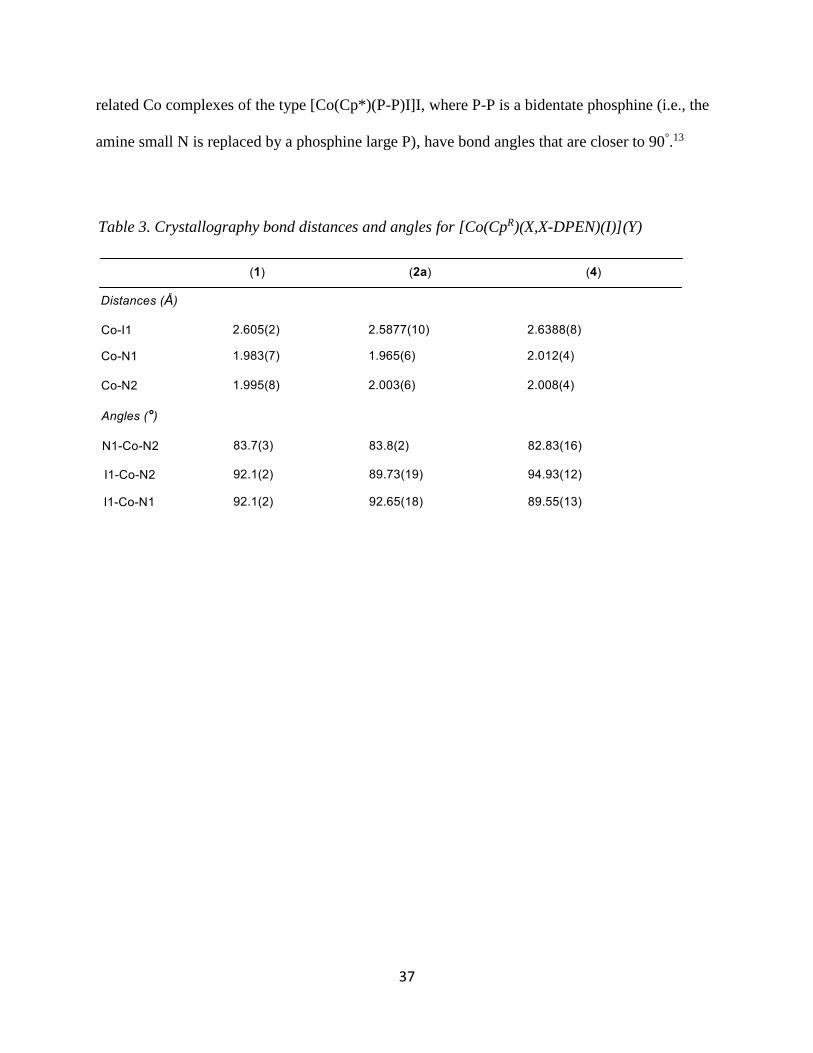
37
related Co complexes of the type [Co(Cp*)(P-P)I]I, where P-P is a bidentate phosphine (i.e., the
amine small N is replaced by a phosphine large P), have bond angles that are closer to 90°.13
Table 3. Crystallography bond distances and angles for [Co(CpR)(X,X-DPEN)(I)](Y)
38
Figure 8. Crystal of [Co(Cp)(R,R-DPEN)(I)](I) (1)
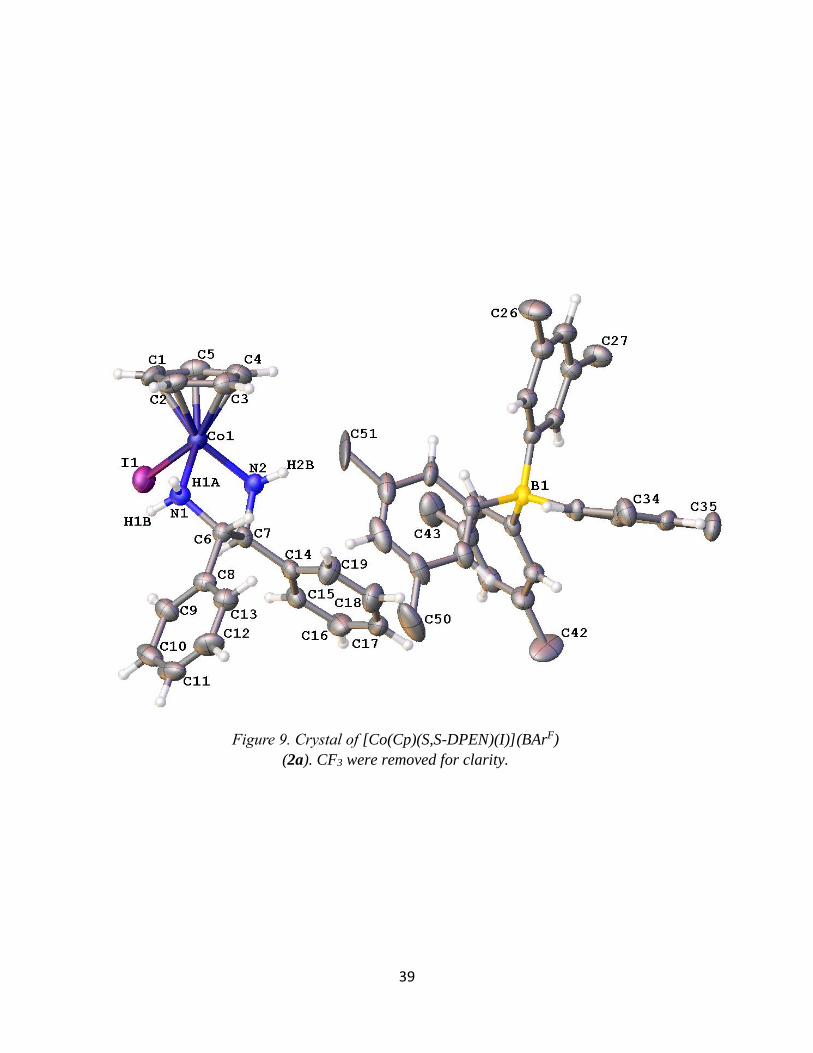
39
Figure 9. Crystal of [Co(Cp)(S,S-DPEN)(I)](BArF)
(2a). CF3 were removed for clarity.
40
Figure 10. Crystal of [Co(Cp*)(S,S-DPEN)(I)](I) (4)

41
4. Results and Discussion – Part 2 Reactivity
It is important to consider that transition metal hydrides are critical intermediates in
homogeneous organometallic catalysis and are involved in multiple catalytic cycles.14 Rhodium
hydride complexes have been synthesized and have been shown to be reactive, making them
effective catalysts.15 Since Rh is in the same periodic group as Co, it was in our interest to
explore the possible synthesis and isolation of a Co(III) hydride complex for asymmetric
hydrogen transfer reactions. Our initial approach was to use LiAlH4, KBH4, and LiBHEt3 as
hydride donating reagents to add the hydride to the cobalt (Scheme 7). However, these strong
reducing agents reduced the metal to Co(II) as indicated by a paramagnetic 1H-NMR spectrum.
To bypass that route, it was proposed to reduce the complex by 2 e- and then add a source
of electrophilic H+ or CH3+. The source of H+ to be used was [p-bromoanilium]BF4, which was
successfully isolated and has been used before to prepare a Co(III)-H in the complex
[HCo(L2)(CH3CN)]2+, where L2 = PnC-PPh22N
Ph2 (1,5-diphenyl-3,7-
bis((diphenylphosphino)alkyl)-1,5-diaza-3,7-diphosphacyclooctane; alkyl = (CH2)2, n = 2 (L2).10
In initial attempts, CH3I was used to add an electrophilic methyl group. The reason is due to
precedence for oxidative addition to the metal complexes, and the fact that isolating Co(III) with
hydrides are rare.10,16 The reaction proposed is illustrated in Scheme 8, where the Co(III)
Scheme 7. Attempted synthesis of [Co(CpR)(X,X-DPEN)(H)](I)

42
complex is reduced by Zn to Co(I) and then the addition of CH3I would oxidize the metal back to
a +3 oxidation state. 1H-NMR data showed the Cp* ligand to be absent, and the presence of free
DPEN ligand. If the zinc reduced Co(III) to Co(I), the resulting complex likely decomposed (see
below).
The Co(I) complex [Co(Cp*)(CO)2], which is a precursor to the starting point for all
previous reactions, could react with DPEN to replace the two carbonyls. However, even in
refluxing toluene for 17 h, no reaction was observed as shown by IR spectrum (1931 and 1995
cm-1) which suggests both CO ligands of [Co(Cp*)(CO)2] were still present after the reaction
time (Scheme 9). Furthermore, this result suggests that if Zn did reduce [Co(Cp*)(R,R-
DPEN)(CH3)](I), as described above, the complex likely decomposed at the elevated
temperatures.
Scheme 8. Attempted synthesis of [Co(Cp*)(R,R-DPEN)(CH3)](I)
Scheme 9. Attempted synthesis of [Co(Cp*)(R,R-DPEN)]

43
Attempts to do a Michael addition
With the successful synthesis of the BArF coordination complexes 2a, 3a, and 4a, we
decided to explore its reactivity by attempting the Michael addition reaction. This is a known
reaction that is catalyzed by the related [Δ/Λ-Co(S,S-DPEN)3](BArF)(Cl)2, a cobalt(III) based
catalyst.5 The reaction between dimethyl malonate and trans--nitrostyrene is catalyzed by [Δ/Λ-
Co(S,S-DPEN)3](BArF)(Cl)2, and due to the similarities to our systems I attempted this reaction
with complex 4a. Although there was a slight color change when the solution was let to sit
overnight, the 1H NMR spectrum revealed no observable changes in the chemical shifts. Heating
the solution could possibly kinetically initiate the reaction, however, the catalyst was found to
decompose at high temperatures.
Anion exchange reactions
A series of outer sphere anion exchange reactions were tested on [Co(CpR)(X,X-DPEN)(I)](I) for
recrystallization purposes. The anions tested include: sodium tetrafluoroborate (NaBF4),
ammonium hexafluorophosphate (NH4PF6), sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate
(NaBArF), sodium perchlorate (NaClO4), and silver acetate (AgOAc). Out of the five anions
tested, the reaction with NaBArF gave the best recrystallization and elemental analysis results, as
discussed above. Interestingly, the AgOAc reaction produced an unexpected solubility property.
Scheme 10. Attempted Michael Addition of dimethyl malonate and trans--nitrostyrene

44
The complexes became water soluble upon exchanging iodide for acetate, which is significant as
this can open up several reactivity pathways in the organic as well as biochemical fields. More
details on the acetate complexes will be discussed in Chapter 3.
5. Conclusions and Future Work
Four complexes of the type [Co(CpR)(X,X-DPEN)(I)](I) have been synthesized. The
related meso complexes show a mixture of two diastereomers that appears to be dependent on
sterics. All four of the enantiomerically pure complexes have been fully characterized by 1H/13C
NMR, Low-Res MS, UV-Vis, elemental analysis, and, for three complexes, single-crystal X-ray
diffraction. Attempts were made to prepare a Co(III)-H complex via direct hydride/iodide ligand
exchange as well as 2-e- reduction followed by an oxidative addition. However, the reduction
was unsuccessful, suggesting that the DPEN ligand addition solely favors Co(III) complexes.
Conditions were found for outer-sphere anion exchange; where exchange for the BArF anion
results in crystalline material that produces publishable elemental analysis results and with the
OAc anion results in water soluble complexes. The next step will be exploring the reactivity of
the complex with cis-1,2-diaminocyclohexane (DIAC). This ligand will serve as an electron
donating system to the complex, which can help mediate the net positive charge to result in a
more covalent system that can potentially provide better access to a Co(III)-H complex.

45
References
1. Monfette, S.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. J. A. Chem. Soc. 2012, 134,
4561-4564.
2. Noyori, R. Angew. Chem. Int. Ed. 2002, 41, 2008-2022.
3. Uematsu, N.; Fuji, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc.
1996, 118, 4916-4917.
4. Friend, J. A.; Nunn, E.K., J. Chem Soc. 1958, 0, 1567-1571.
5. Lewis, K. G.; Ghosh, S. K.; Bhuvanesh, N.; Gladysz, J. A. ACS Cent. Sci. 2015, 1, 50-56.
6. Wang, D.; Astruc, D. Chemical Reviews 2015, 115, 6621-6686
7. Das, D.; Veziroglu, T. N. Int. J. Hydrogen Energy. 2001, 26, 13-28.
8. Harynto, A.; Fernando, S.; Murali, N.; Adhikari, S. Energy Fuels. 2005, 19, 2098-2106.
9. Dempsey, J.; Brunschwig, B. S.; Winkler, J. R.; Gray, H. B. Acc. Chem. Res. 2009, 42,
1995-2004.
10. Wiedner, E. S.; Roberts, J. A. S.; Dougherty, W. G.; Kassel, W. S.; DuBois, D. L.;
Bullock, R. M., Inorg. Chem. 2013, 52, 9975-9988.
11. King, R. B. Inorg. Chem. 1966, 5, 82-87.
12. King, R. B.; Efraty, A.; Douglas, W. M. Organomet. Chem. 1973, 56, 345.
13. Bao, Q. B.; Landon, S. J.; Rheingold, A. L.; Haller, T. M.; Brill, T. B. Inorg. Chem.
1985, 24, 900-908.
14. Cheng, T.; Bullock, R. M. Organometallics. 2002, 21, 2325-2331.
15. Price, A. J.; Ciancanelli, R.; Noll, B. C.; Curtis, C. J.; DuBois, D. L.; DuBois, M. R.
Organometallics. 2002, 21, 4833-4839.
16. Hartwig, J. F. Inorg. Chem. 2013, 52, 9975-9968.

46
Chapter 3: Exploration of aqueous Co(III) acetate
complexes for catalytic activity

47
1. Introduction
There is a growing interest in green synthetic methodology among researchers in
academic and industrial fields focusing on eliminating chlorinated solvents.1 As a result,
alternative solvents have been developed to combat environmental concerns.2 Of these, water is
an ideal solvent for synthesis due to its availability, cost, nonflammability, environmental
congruity, and nontoxicity. Recently, there has been a growing interest in aqueous
organometallic catalysis, a field that takes advantage of this green solvent3
Specific examples of organometallic reactions that take advantage of water as a solvent,
specifically utilizing rare earth metals, include the addition of H2 to trans-Ru(P2)2Cl2 (P2 = a
bidentate phosphine) to form trans-[Ru(P2)2(H2)H]+ dihydrogen complexes. Another example is
the molybdocene-catalyzed hydration of nitriles.4,5 Generally, metal chlorides, perchlorates, and
other species that contain first row transition metals, Ni, Fe, Cu, and Zn, exhibit high catalytic
activity due to optimal Lewis acid characteristics in aqueous media.2 This Lewis acid feature is
dependent on the hydrolysis constants (pKh) as well as water exchange rate constants (WERCs).6
The WERC is a significant factor of Lewis acid strength in aqueous catalysis.2 As the value of
WERC increases, the catalytic activity increases. An example of aqueous catalytic reactions
include an Fe(III) tosylate-catalyzed (tosylate = 4-toluenesulfonate) deprotection of aromatic
Scheme 1. Fe(III) Tosylate-Catalyzed Deprotection of Aromatic acetals in water

48
acetals in water, where the acetals were deprotected in the presence of Fe(III) tosylate under mild
conditions (Scheme 1).7
There have been several reports of transfer hydrogenation in aqueous conditions using
transition metals. A surfactant type Rh complex successfully catalyzed a transfer hydrogenation
of ketones in water with 94% yield and with 84% ee (Scheme 2).2,8 This surfactant system
showed high activity on a variety of aliphatic ketones with functional groups that might be
formed into intermediates of anticoagulants.
Another example of a successful catalyzed transfer hydrogenation of ketones using Rh
was demonstrated by Himeda et al.3 Their group utilized achiral [Cp*Rh(bpy)Cl]Cl,
Figure 1. Acetate complex
soluble in aqueous phase
Scheme 2. Asymmetric Transfer Hydrogenation by a
Surfactant-Type Rh Catalyst in Water.2

49
[Cp*Ir(bpy)Cl]Cl, and [(C6Me6)Ru(bpy)Cl]Cl (C6Me6 = hexamethylbenzene) complexes with
aqueous formic acid and sodium formate as a hydrogen source, resulting in a 89-99% yield of 1-
phenylethanol. Also, Thorpe et al. prepared Rh and Ir complexes using aminosulfonamide
ligands [Ts-DPEN and N-(p-toluenesulfonyl)-cyclohexanediamine] and [Cp*MCl2]2 (M = Rh,
Ir), in 51% water to catalyze transfer hydrogenation of ketones (Scheme 3).9
As mentioned in Chapter 2, our group conducted a series of outer anion exchange
reactions on [Co(CpR)(X,X-DPEN)(I)](I) for recrystallization purposes. Out of the five anions
experimented, the reaction using silver acetate (AgOAc) produced an unanticipated water-
soluble product [Co(Cp)(S,S-DPEN)(OH2)](OAc)2 ∙ nH2O] (8) (Figure 1). This is interesting as
our complexes are not typically water soluble. Nonetheless, since aqueous catalysis has been
known and experimented on with organometallic transition metal complexes, it was in our
interest to explore the synthesis and reactivity of this new product. In this chapter, the synthesis
and characterization of seven new acetate complexes will be discussed. Also, the reactivity of
these complexes will be analyzed, specifically if they were able to catalyze transfer
hydrogenation of ketones using water and sodium formate and/or a mixture of formic acid and
triethylamine.
2. Experimental Section
2.1. Materials
All solvents and reagents were used as received. The ligands R,R-DPEN, S,S-DPEN, as well as
both silver acetate (AgOAc) and cesium acetate (CsOAc) were purchased from either Aldrich
Chemical Company or Alfa Aesar and used as received. The metal reagents [Co(Cp)(CO)(I)2]
and [Co(Cp*)(CO)(I)2] were prepared as previously reported.10,11

50
2.2. Measurements
1H and 13C NMR spectra were obtained on a JEOL ECX 400 MHz spectrometer
(operating frequency for 13C NMR was 100 MHz) and referenced against tetramethylsilane using
residual proton signals (1H NMR) or the 13C resonances of the deuterated solvent (13C NMR).
Elemental analyses were performed by Atlantic Microlab, Inc in Norcross, GA.
2.3.Syntheses
2.3.1. Preparation of [Co(Cp)(R,R-DPEN)(OH2)](OAc)2 ∙ nH2O (7).
A 100 mL beaker was charged with 1 (0.0806 g, 0.0137 mmol) and dichloromethane (10
mL), silver acetate (0.0692 g, 0.415 mmol), and H2O (10 mL). Immediately, the aqueous layer
began to turn bright purple/pink. The solution was vigorously stirred for 10 min at room
temperature. The aqueous layer was collected and filtered. The filter paper was washed with
water. The resultant filtrate was dried in the fume hood, leaving a light purple solid. The final
product weighed 0.0195 g. 1H NMR (39a-1H-LD-5), 13C NMR (39a-13C-LD).
1H NMR (CDCl3, δ): 6.92-7.24 (12H, m, DPEN aromatic and NH), 5.84 (5H, s, Cp), 5.01 (1H,
td, CH), 4.63 (1H, d, NH), 3.98 (1H, td, CH), 2.22-2.28 (2H, m, OH2), 2.20 (6H, s, OAc). 13C
{1H} NMR (CDCl3, δ): 182 (1C, s, C=O), 138.2 (1C, s, ipso C arene), 136.7 (1C, s, ipso C arene)
129.3, 129.2, 129.1, 128.8, 128.8, 128.6, 128.4, 128.0, 127.9, 127.2 (10C, m, Aromatic C
DPEN), 84.4 (5C, s, Cp), 64.1 (1C, s, DPEN CH), 62.4 (1C, s, DPEN CH), 24.7 (1C, s, OAc
CH3). Anal. Calcd for C21H26O3N2CoI: C, 46.7; N, 5.19; H, 5.93. Found: C, 46.4; N, 5.53; H,
4.73, I, 20.8.
2.3.2. Preparation of [Co(Cp)(S,S-DPEN)(OH2)](OAc)2 ∙ nH2O (8).
A 50 mL round-bottom flask was charged with 2 (0.100 g, 0.170 mmol) and dichloromethane
(10 mL), silver acetate (0.0568 g, 0.340 mmol), and H2O (10 mL). Immediately, the aqueous

51
layer began to turn bright purple/pink. The solution was vigorously stirred for 10 min at room
temperature. The aqueous layer was collected and filtered. The filter paper was washed with
water. The resultant filtrate was transferred into a round-bottom flask and was dried in vacuo,
leaving a light purple solid. Note: Extensive foaming and bumping occurred during solvent
removal. The final product weighed 0.0626 g. 1H NMR (50b-1H-LD), 13C NMR (50a-13C-LD).
1H NMR (CDCl3, δ): 6.88-7.35 (12H, m, DPEN aromatic and NH), 6.02 (5H, s, Cp), 5.04 (1H,
td, CH), 4.65 (1H, d, NH), 4.04 (1H, td, CH), 2.22-2.33 (3H, m, OH2), 2.21 (6H, s, OAc). 13C
{1H} NMR (CDCl3, δ): 182 (1C, s, C=O), 137.9 (1C, s, ipso C arene), 136.4 (1C, s, ipso C arene)
129.5, 129.3, 129.2, 128.9, 128.8, 128.7, 128.3, 128.2, 127.6, 127.2 (10C, m, Aromatic C
DPEN), 84.5 (5C, s, Cp), 64.0 (1C, s, DPEN CH), 62.7 (1C, s, DPEN CH), 24.6 (1C, s, OAc
CH3). Anal. Calcd for C27H39N2Co: N, 6.14; H, 5.93. Found: N, 6.14; H, 5.93. Anal. Calcd for
C21H26O3N2CoI: C, 58.47; N, 5.93; H, 6.14. Found: C, 57.94, N, 5.93; H, 6.14.
2.3.3. Preparation of [Co(Cp*)(R,R-DPEN)(OH2)](OAc)2 ∙ nH2O (9).
A 50 mL round-bottom flask was charged with 3 (0.1001 g, 0.1516 mmol) and dichloromethane
(10 mL), silver acetate (0.0506 g, 0.3032 mmol), and H2O (10 mL). Immediately, the aqueous
layer began to turn bright purple/pink. The solution was vigorously stirred for 10 min at room
temperature. The aqueous layer was collected and filtered. The filter paper was washed with
water. The resultant filtrate was transferred into a round-bottom flask and was dried in vacuo,
leaving a light purple solid. Note: Extensive foaming and bumping occurred during solvent
removal. The final product weighed 0.0611 g. 1H NMR (51b-1H-LD), 13C NMR (51a-13C-LD).
1H NMR (CDCl3, δ): 7.02-7.30 (12H, m, DPEN aromatic and NH), 6.34 (1H, t, NH), 5.58 (1H,
td, CH), 5.00 (1H, t, NH), 4.90 (1H, t, NH), 4.58 (1H, td, CH), 2.23 (5H, s, OAc), 1.99 (4H, m,
OH2), 1.56 (15H, s, Cp*). 13C {1H} NMR (CDCl3, δ): 181.87 (1C, s, C=O), 138.5 (1C, s, ipso C

52
arene), 137.0 (1C, s, ipso C arene) 128.9, 128.8, 128.7, 128.5, 128.4, 128.4, 128.3, 127.5, 127.3,
127.2 (10C, m, Aromatic C DPEN), 92.3 (5C, s, Cp), 63.2 (1C, s, DPEN CH), 61.2 (1C, s,
DPEN CH), 24.5 (1C, s, OAc CH3), 9.43 (5C, s, Cp* CH3). Anal. Calcd for C21H26O3N2CoI: C,
46.7; N, 5.19; H, 5.93. Found: C, 46.4; N, 5.53; H, 4.73, I, 20.8.
2.3.4. Preparation of [Co(Cp*)(S,S-DPEN)(OH2)](OAc)2 ∙ nH2O (10).
A 50 mL round-bottom flask was charged with 4 (0.0300 g, 0.0454 mmol) and
dichloromethane (10 mL), silver acetate (0.0506 g, 0.102 mmol), and H2O (10 mL).
Immediately, the aqueous layer began to turn bright purple/pink. The solution was vigorously
stirred for 30 min at room temperature. The aqueous layer was collected and filtered. The filter
paper was washed with water. The resultant filtrate was evaporated in the fume hood, leaving a
light purple solid. The final product weighed 0.0200 g. 1H NMR (68a-1H-LD-5), 13C NMR (68a-
13C-LD).
1H NMR (CDCl3, δ): 7.00-7.38 (12H, m, DPEN aromatic and NH), 6.27 (1H, t, NH), 4.88 (1H,
td, CH), 4.77 (1H, d, NH), 4.53 (1H, td, CH), 3.07 (6H, s, OAc), 2.22 (6H, s, OAc), 1.91 (3H, m,
OH2), 1.51 (15H, s, Cp*). 13C {1H} NMR (CDCl3, δ): 182 (1C, s, C=O), 138.7 (1C, s, ipso C
arene), 137.2 (1C, s, ipso C arene), 129.1, 128.9, 128.8, 128.6, 128.5, 128.3, 128.3, 128.1, 127.5,
127.1 (10C, m, Aromatic C DPEN), 92.3 (5C, s, Cp), 63.2 (1C, s, DPEN CH), 60.9 (1C, s,
DPEN CH), 26.7 (1C, s, OAc CH3), 9.38 (5C, s, Cp* CH3).
2.3.5. Preparation of [Co(Cp*)(R,R-DPEN)(OAc)](OAc) ∙ nH2O (11).
A 50 mL round-bottom flask was charged with [Co(Cp*)(CO)(I)2]
(0.2500 g, 0.525 mmol), dichloromethane (25 mL), followed by silver acetate (0.175 g, 1.05
mmol). The solution was vigorously stirred for 30 min at room temperature. After the 30 min,
the solution appeared bright blue. The round-bottom was then charged with R,R-DPEN (0.1675

53
g, 0.789 mmol). The solution immediately turned bright purple upon stirring. The solution
remained stirring for 30 min. The solution was then transferred to an evaporating dish. The dried
product was then washed with hexanes. The final product weighed 0.284 g. 1H NMR (74a-1H-
LD), 13C NMR (74b-13C-LD).
1H NMR (CDCl3, δ): 6.96-7.36 (16H, m, DPEN aromatic and NH), 6.43 (1H, t, NH), 5.68 (1H, t,
NH), 4.94 (1H, m, CH), 4.58 (1H, td, CH), 2.79 (7H, s, OAc/H2O) 2.23 (5H, s, OAc/H2O), 1.92
(4H, s, OAc/H2O), 1.55 (15H, s, Cp*) ppm. 13C {1H} NMR (CDCl3, δ): 182 (1C, s, C=O), 138.8
(1C, s, ipso C arene), 137.2 (1C, s, ipso C arene), 129.3, 128.9, 128.8, 128.6, 128.4, 128.3, 127.5,
127.2, 127.0, 126.60 (10C, m, Aromatic C DPEN), 92.3 (5C, s, Cp), 63.2 (1C, s, DPEN CH),
60.9 (1C, s, DPEN CH), 26.7 (1C, s, OAc CH3), 9.42 (15C, s, Cp*) ppm.
2.3.6. Preparation of [Co(Cp*)(R,R-DPEN)(OAc)](I) ∙ nH2O (12).
A 50 mL round-bottom flask was charged with [Co(Cp*)(CO)(I)2]
(0.2503 g, 0.526 mmol), dichloromethane (15 mL), followed by cesium acetate (0.094 g, 0.490
mmol). The solution was vigorously stirred for 30 min at room temperature. After the 30 min,
there were no observable color changes. The cesium acetate was still visible as yellow clumps.
The round-bottom was then charged with R,R-DPEN (0.1674 g, 0.789 mmol). The solution
began to gradually turn a brighter purple upon stirring. The yellow CsOAc clumps turned white,
indicating a possible change into CsI. The solution remained stirring for 30 min. The solution
was then filtered through celite and then transferred to an evaporating dish. The dried product
was then washed with hexanes. The final product weighed 0.358 g. 1H NMR (80a-1H-LD),
1H NMR (CDCl3, δ): 6.99-7.52 (14H, m, DPEN aromatic and NH), 5.57 (2H, m, NH), 4.89 (2H,
m, NH;CH), 4.53 (1H, td, CH), 2.26 (1H, m, OH2), 2.23 (6H, s, OAc), 2.17 (3H, m, OH2), 1.96
(6H, s, OAc), 1.53 (15H, s, Cp*).

54
2.3.7. Preparation of [Co(Cp*)(S,S-DPEN)(OAc)](I) ∙ 3H2O (13).
A 50 mL round-bottom flask was charged with 4 (0.0400 g, 0.0606 mmol) and
dichloromethane (10 mL), cesium acetate (0.0463 g, 0.241 mmol), and H2O (2 mL).
Immediately, the aqueous layer began to turn bright purple/pink. The solution was vigorously
stirred for 1 h 30 min at room temperature. The aqueous layer was collected and filtered. The
filter paper was washed with water. The resultant filtrate was evaporated in the fume hood,
leaving a light purple solid. No recorded yield. 1H NMR (63a-1H-LD-5), 13C NMR (63a-13C-
LD).
1H NMR (CDCl3, δ): 7.08-7.37 (13H, m, DPEN aromatic and NH), 5.57 (2H, m, NH), 4.98 (1H,
td, CH), 4.40 (2H, m, NH;CH), 2.33 (1H, m, OH2), 2.24 (3H, s, OAc), 2.22 (1H, s, OH2), 1.81
(9H, m, OH2 and OAc), 1.66 (2H, m, OH2), 1.62 (15H, s, Cp*). 13C {1H} NMR (CDCl3, δ):
181.87 (1C, s, C=O), 137.7 (1C, s, ipso C arene), 133.7 (1C, s, ipso C arene) 129.4, 129.3, 129.2,
129.1, 128.8, 128.2, 128.1, 128.0, 127.5, 127.2 (10C, m, Aromatic C DPEN), 93.0 (5C, s, Cp),
63.5 (1C, s, DPEN CH), 62.4 (1C, s, DPEN CH), 26.5 (1C, s, OAc CH3), 10.7 (5C, s, Cp* CH3).
NMR data revealed a mixture of products.
2.3.8. VT 1H NMR of [Co(Cp*)(S,S-DPEN)(OH2)](OAc)2 (10)
A sample of 10 was dissolved CDCl3 and was first heated from room temperature in 10°
increments to 55 °C in 10 °C increments. Peaks at 3.05, 2.20, and 1.92 ppm coalesced at 55 °C.
Afterwards, the sample was cooled incrementally 55 °C to -40 °C. No significant changes
occurred during cooling. (68a-h-1H-LD).

55
2.3.9. VT 1H NMR of [Co(Cp*)(R,R-DPEN)(OAc)](OAc) (11).
A sample of 11 was dissolved CDCl3 and was first cooled from room temperature in
increments leading down to -20 °C in 10 °C increments. No significant changes occurred during
cooling. Afterwards, the sample was heated incrementally from -20 °C to 45 °C in 10 °C
increments. Peaks at 2.20 and 1.92 coalesced at 45 °C. (76a-f-1H-LD).
2.3.10. Attempt 1 of Transfer Hydrogenation of Acetophenone
A 5 mL round-bottom flask was charged with 7 (0.0105 g, 0.0201 mmol), H2O (3 mL),
sodium formate (0.695 g, 10.2 mmol), and acetophenone (0.24 mL, 2.06 mmol). The dark purple
solution was mixed at 75 °C for 21 h. After stirring, two layers were formed. The lower aqueous
phase appeared a hazy-beige color, while the upper organic phase appeared dark brown. It
appeared that the sodium formate precipitated in between the phases. The organic layer was
extracted with diethyl ether. It was filtered via gravity filtration and was then transferred into an
evaporating dish. After the volatiles were evaporated, the dried product was analyzed via 1H
NMR. There is no indication that 1-phenylethanol was present in the product.
2.3.11. Attempt 2 of Transfer Hydrogenation of Acetophenone
A 5 mL round-bottom flask was charged with 7 (0.0100 g, 0.0191 mmol), D2O (2 mL),
sodium formate (0.6517 g, 9.58 mmol), and acetophenone (0.24 mL, 2.06 mmol). The reactants
were mixed starting at 45 °C and then constant at 50 °C for about 2 h. The initial solution color
was dark purple. After 1 h, a color changed occurred. The bottom aqueous layer turned light pink
and the top organic layer turned brown. A 5 mL sample was taken from the aqueous layer for 1H
NMR analysis. The data revealed a large acetophenone peak. The remaining solution continued
to react at 80 °C for another 1 h. A 1H NMR sample was taken in DMSO-d6 to check for
evidence of 1-phenylethanol. There was no indication of expected product.

56
2.3.12. Attempt 3 of Transfer Hydrogenation of Acetophenone
A 5 mL round-bottom flask was charged with formic acid (0.82 mL) and triethylamine (1.2
mL). The flask was then charged with acetophenone (0.25 mL, 2.14 mmol), and 9 (0.0139 g,
0.0235 mmol). The bright purple solution was stirred for about 3 h at 40 °C. The product was
isolated by solvent removal via diethyl ether extraction followed by diethyl ether removal in
vacuo. A viscous liquid (acetophenone and other organic product) was left over. A 1H NMR
sample was taken and revealed no indication of 1-phenylethanol.
2.6. X-Ray Crystallography Data Collection and Processing
Appendix A contains crystal data, collection parameters, and refinement criteria for the crystal
structure of 13. The crystal was grown by slow evaporation from a dichloromethane solution.
The crystal was mounted on the tip of a Bruker SPINE-pin mount and X-ray intensity data were
measured at low temperature using an Oxford Cryosystems Desktop Cooler (200(2) K) for the
structure with a graphite monochromated Mo kα radiation (λ = 0.71073 Å) on a Bruker SMART
X2S Benchtop diffractometer. Integration, data reduction and scaling were carried out with the
programs SAINT and SADABS in the Bruker APEX2 suite of software. The structure was
solved (XS) using direct methods and refined using full matrix least squares refinement
(SHELXL2017) within Olex2. A direct-methods solution was calculated that provided the non-
hydrogen atoms from the E-map. All non-hydrogen atoms were refined with anisotropic
displacement parameters. All of the hydrogen atoms in the structure was placed in ideal positions
and refined as riding atoms.
57
3. Results and Discussion – Part 1 Synthesis and Characterization
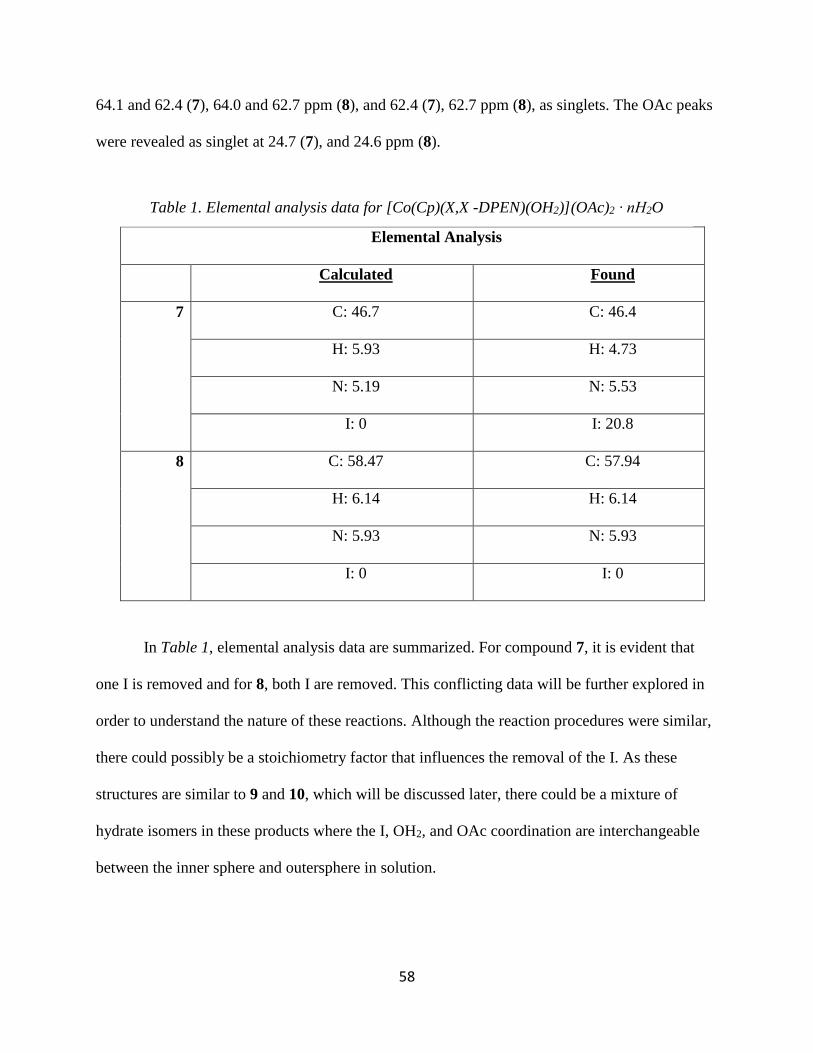
Synthesis of [Co(Cp)(X,X -DPEN)(OH2)](OAc)2 ∙ nH2O (7,8)
During the anion exchange experiments mentioned Chapter 2, a water soluble complex of
the type [Co(Cp)(X,X-DPEN)(OH2)](OAc)2 ∙ nH2O was synthesized. These products can be
obtained by reacting [Co(CpR)(X,X-DPEN)(I)](I) with AgOAc in a water/dichloromethane
solvent mixture. Complexes [Co(Cp)(R,R-DPEN)(OH2)](OAc)2 ∙ nH2O 7 and [Co(Cp)(S,S-
DPEN)(OH2)](OAc)2 ∙ nH2O 8 were confirmed by 1H and 13C NMR. The 1H NMR spectra
revealed a singlet at 5.84 (7), 6.02 ppm (8), respectively, corresponding to the five protons on the
the Cp ligand. The two methine protons from the DPEN resonances are chemically different due
to the lack of a mirror plane and resonate at 5.01 (7), 5.04 ppm (8) and 3.98 (7), 4.04 ppm (8) as
triplet of doublets. The triplet rises from the CH coupling to the amine protons and the doublet
from coupling to one adjacent CH proton. The two OAc resonances appeared as singlets at 2.20
(7), 2.22 ppm (8). The 13C NMR spectra revealed a singlet at 84.4 (7), 84.5 ppm (8), which
corresponds with the Cp ligand. The resonances for the two methines on the DPEN appeared at
Scheme 4. Synthesis of [Co(Cp)(X,X-DPEN)(OH2)](OAc)2 ∙ nH2O
58
64.1 and 62.4 (7), 64.0 and 62.7 ppm (8), and 62.4 (7), 62.7 ppm (8), as singlets. The OAc peaks
were revealed as singlet at 24.7 (7), and 24.6 ppm (8).
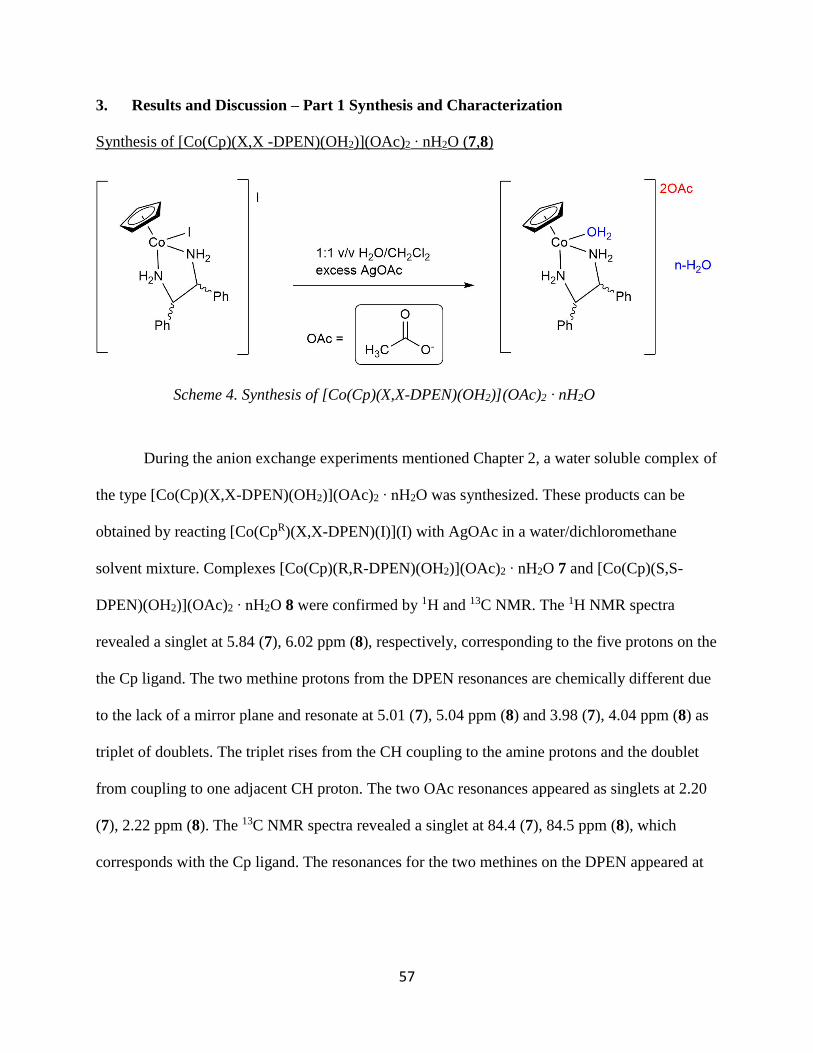
Elemental Analysis
Calculated Found
7 C: 46.7 C: 46.4
H: 5.93 H: 4.73
N: 5.19 N: 5.53
I: 0 I: 20.8
8 C: 58.47 C: 57.94
H: 6.14 H: 6.14
N: 5.93 N: 5.93
I: 0 I: 0
In Table 1, elemental analysis data are summarized. For compound 7, it is evident that
one I is removed and for 8, both I are removed. This conflicting data will be further explored in
order to understand the nature of these reactions. Although the reaction procedures were similar,
there could possibly be a stoichiometry factor that influences the removal of the I. As these
structures are similar to 9 and 10, which will be discussed later, there could be a mixture of
hydrate isomers in these products where the I, OH2, and OAc coordination are interchangeable
between the inner sphere and outersphere in solution.
Table 1. Elemental analysis data for [Co(Cp)(X,X -DPEN)(OH2)](OAc)2 ∙ nH2O
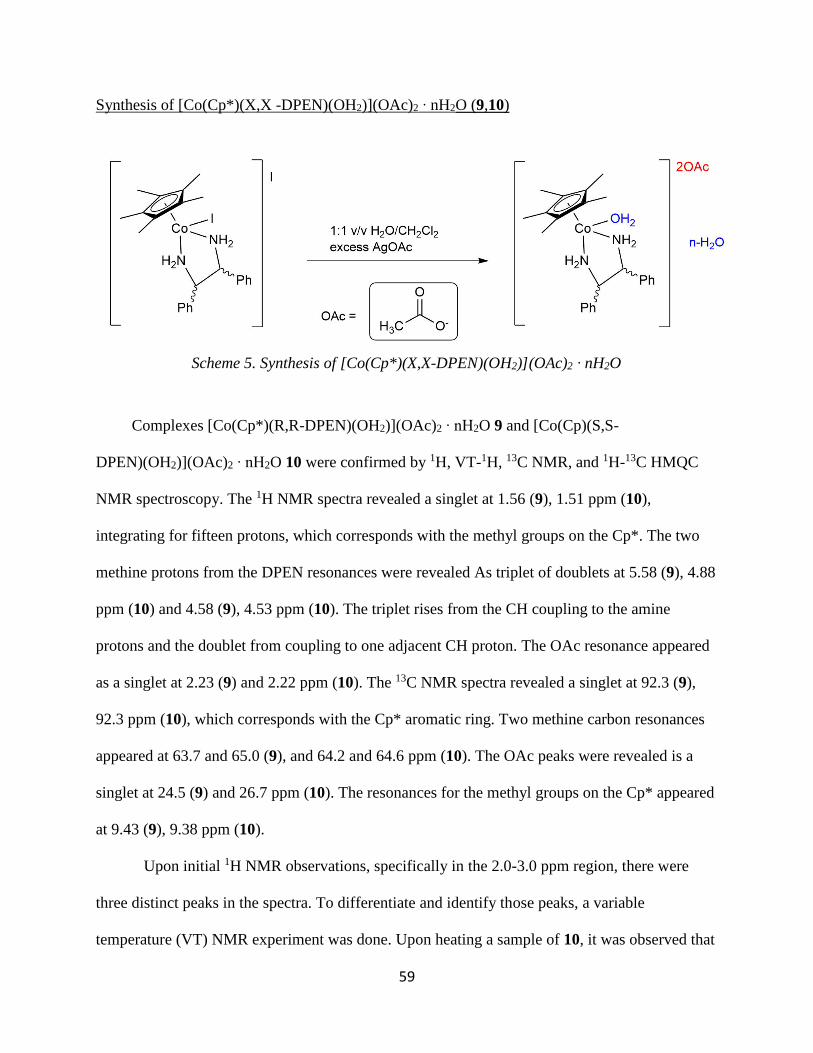
59
Synthesis of [Co(Cp*)(X,X -DPEN)(OH2)](OAc)2 ∙ nH2O (9,10)
Complexes [Co(Cp*)(R,R-DPEN)(OH2)](OAc)2 ∙ nH2O 9 and [Co(Cp)(S,S-
DPEN)(OH2)](OAc)2 ∙ nH2O 10 were confirmed by 1H, VT-1H, 13C NMR, and 1H-13C HMQC
NMR spectroscopy. The 1H NMR spectra revealed a singlet at 1.56 (9), 1.51 ppm (10),
integrating for fifteen protons, which corresponds with the methyl groups on the Cp*. The two
methine protons from the DPEN resonances were revealed As triplet of doublets at 5.58 (9), 4.88
ppm (10) and 4.58 (9), 4.53 ppm (10). The triplet rises from the CH coupling to the amine
protons and the doublet from coupling to one adjacent CH proton. The OAc resonance appeared
as a singlet at 2.23 (9) and 2.22 ppm (10). The 13C NMR spectra revealed a singlet at 92.3 (9),
92.3 ppm (10), which corresponds with the Cp* aromatic ring. Two methine carbon resonances
appeared at 63.7 and 65.0 (9), and 64.2 and 64.6 ppm (10). The OAc peaks were revealed is a
singlet at 24.5 (9) and 26.7 ppm (10). The resonances for the methyl groups on the Cp* appeared
at 9.43 (9), 9.38 ppm (10).
Upon initial 1H NMR observations, specifically in the 2.0-3.0 ppm region, there were
three distinct peaks in the spectra. To differentiate and identify those peaks, a variable
temperature (VT) NMR experiment was done. Upon heating a sample of 10, it was observed that
Scheme 5. Synthesis of [Co(Cp*)(X,X-DPEN)(OH2)](OAc)2 ∙ nH2O

60
three peaks coalesced into one peak that integrated for six hydrogens, suggesting one acetate
environment at ~2.0 ppm (Figure 2). When heated, the energy barrier allows ligand exchange to
occur. This is manifested when the three peaks coalesced into one, which is a time average of the
three signals.
Furthermore, a 1H-13C HMQC NMR analysis confirmed the consistency of the observed
data from 1H (VT) and 13C NMR spectra of 10. This analysis affirmed that the peak at 2.20 ppm
was the acetate peak (Figure 3). However, based on the elemental analysis data from 7 and 8, it
is possible that there is also innersphere-outersphere ligand coordination exchange among the I,
OH2, and OAc in the solution, resulting in hydrate isomers. This will be further explored in the
future.
Figure 2. VT of [Co(Cp*)(S,S-DPEN)(OH2)](OAc)2 ∙ nH2O

61
Figure 3. 1H-13C HMQC of [Co(Cp*)(S,S-DPEN)(OH2)](OAc)2 ∙ H2O

62
Synthesis of [Co(Cp*)(R,R -DPEN)(OAc)](OAc) ∙ nH2O (11)
To determine if the water/dichloromethane solvent mixture was necessary for the
synthesis of the acetate complexes, our group explored a different synthetic pathway. Reacting
[Co(CpR)(CO)(I)2] with AgOAc followed by DPEN addition, with only dichloromethane as the
solvent, produced the complex [Co(Cp*)(R,R-DPEN)(OAc)](OAc) ∙ nH2O. Complex 11 was
confirmed by 1H, VT-1H, 13C NMR, and 1H-13C HMQC. The 1H NMR spectra revealed a singlet
at 1.55 ppm, integrating for fifteen protons, which corresponds with the Cp* analogue. Two
methine protons from the DPEN resonances were revealed at 4.94 and 4.58 ppm as triplet of
doublets. The triplet rises from the CH coupling to the amine protons and the doublet from
coupling to one adjacent CH proton. The OAc resonance appeared as two sharp singlets at 2.23
and 1.92 ppm as well as abroad singlet at 2.79 ppm, vide infra. The 13C NMR spectra revealed a
singlet at 92.3 ppm, which corresponds with the Cp* aromatic ring. Two methine carbon
resonances appeared at 63.2 and 60.9 ppm. The OAc peak resonates as a singlet at 26.7 ppm. The
resonances for the methyl groups on the Cp* appeared at 9.42 ppm.
Upon initial 1H NMR observations, there were three peaks downfield to the Cp* analogue
that had ambiguous identities. To differentiate and identify those peaks, a variable temperature
Scheme 6. Synthesis of [Co(Cp*)(R,R-DPEN)(OAc)](OAc) ∙ nH2O
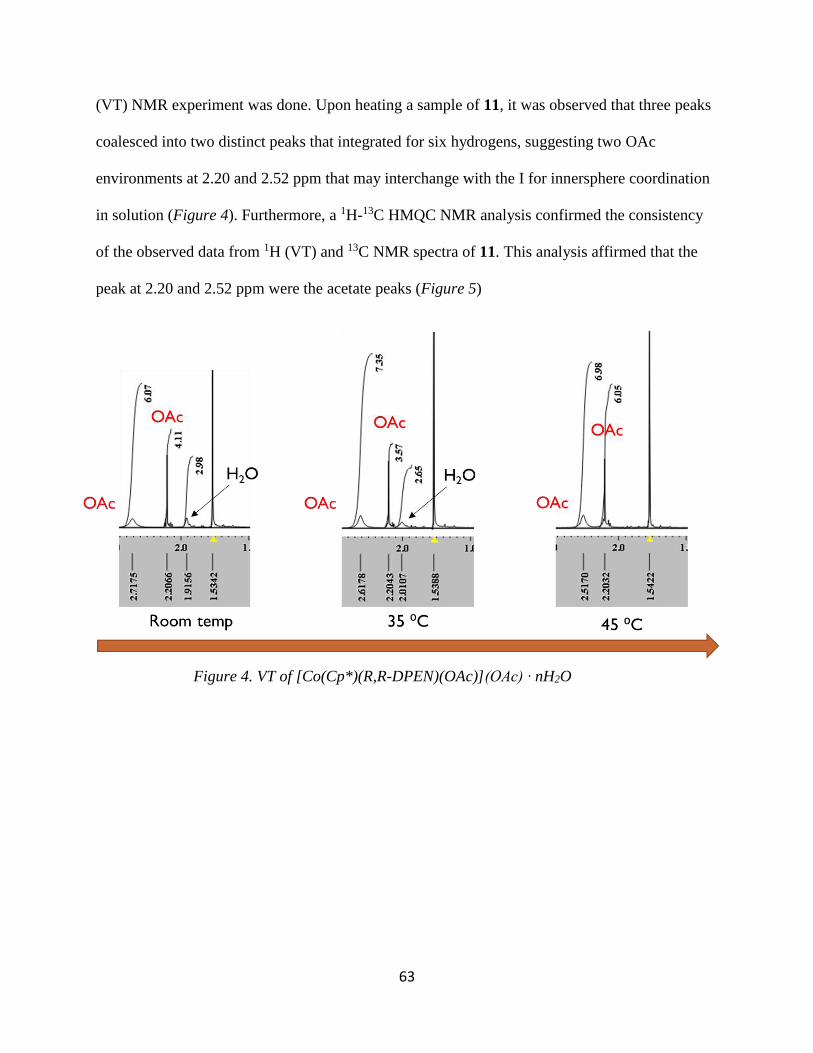
63
(VT) NMR experiment was done. Upon heating a sample of 11, it was observed that three peaks
coalesced into two distinct peaks that integrated for six hydrogens, suggesting two OAc
environments at 2.20 and 2.52 ppm that may interchange with the I for innersphere coordination
in solution (Figure 4). Furthermore, a 1H-13C HMQC NMR analysis confirmed the consistency
of the observed data from 1H (VT) and 13C NMR spectra of 11. This analysis affirmed that the
peak at 2.20 and 2.52 ppm were the acetate peaks (Figure 5)
Figure 4. VT of [Co(Cp*)(R,R-DPEN)(OAc)](OAc) ∙ nH2O
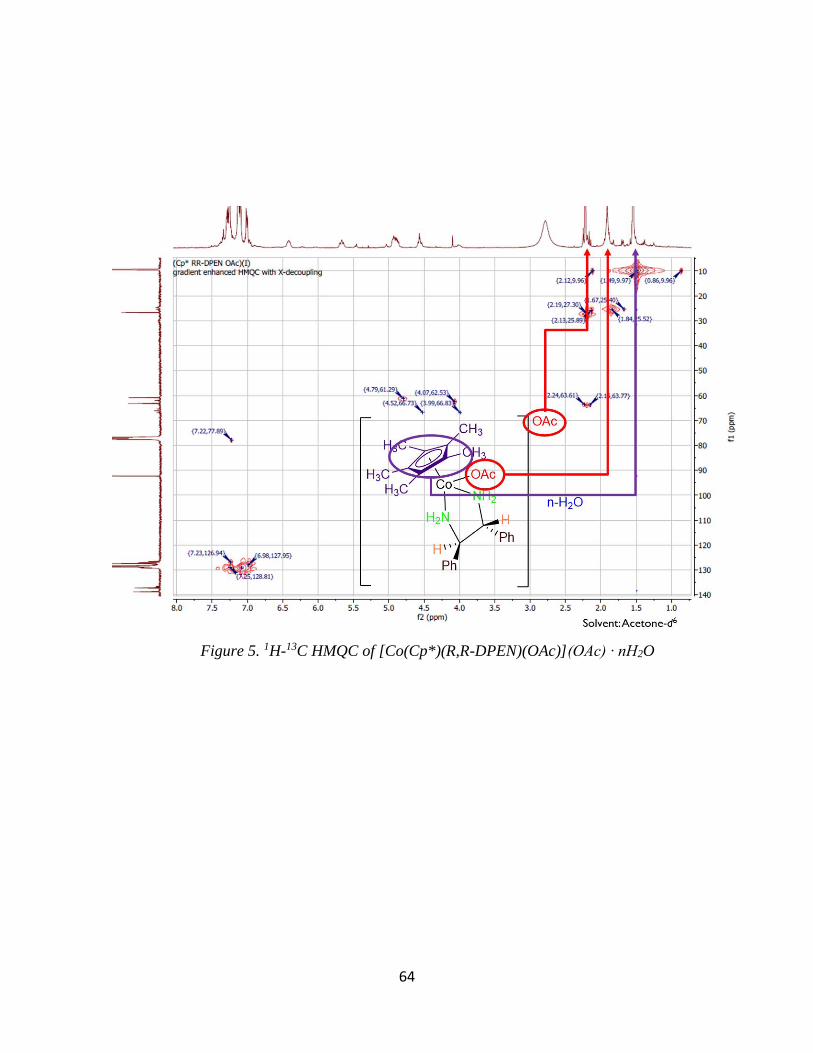
64
Figure 5. 1H-13C HMQC of [Co(Cp*)(R,R-DPEN)(OAc)](OAc) ∙ nH2O

65
Synthesis of [Co(Cp*)(R,R-DPEN)(OAc)](I) ∙ nH2O (12)
Our group then explored the addition of CsOAc to the complex [Co(CpR)(CO)(I)2] in
dichloromethane. This reaction displaced the iodide from the innersphere and an OAc molecule
coordinated to the metal. The iodide relocated to the outersphere. Complex 12 was confirmed by
1H and 13C NMR. The 1H NMR spectra revealed a singlet at 1.53 ppm, integrating for fifteen
protons, which corresponds to the methyl resonances on the Cp*. Two methine protons from the
DPEN resonances were revealed at 4.89 and 4.53 ppm. Also, at 4.89, there appears to be overlap
of the amine protons, which is why 1H reveals two protons in that region instead of one. This
behavior is typical for these complexes. The OAc resonance appeared as sharp singlet at 2.23 as
well as abroad singlet at 1.96 ppm. 13C NMR analysis for this complex will be explored in the
future.
Scheme 7. Synthesis of [Co(Cp*)(R,R-DPEN)(OAc)](I) ∙ nH2O
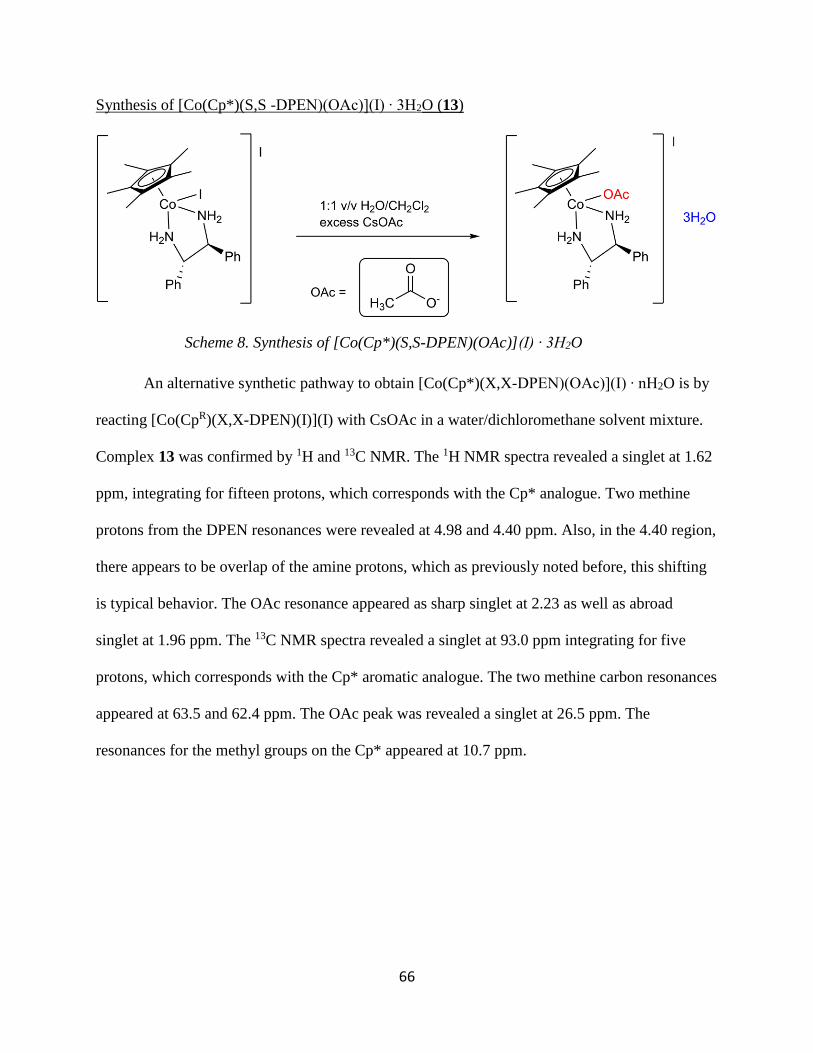
66
Synthesis of [Co(Cp*)(S,S -DPEN)(OAc)](I) ∙ 3H2O (13)
An alternative synthetic pathway to obtain [Co(Cp*)(X,X-DPEN)(OAc)](I) ∙ nH2O is by
reacting [Co(CpR)(X,X-DPEN)(I)](I) with CsOAc in a water/dichloromethane solvent mixture.
Complex 13 was confirmed by 1H and 13C NMR. The 1H NMR spectra revealed a singlet at 1.62
ppm, integrating for fifteen protons, which corresponds with the Cp* analogue. Two methine
protons from the DPEN resonances were revealed at 4.98 and 4.40 ppm. Also, in the 4.40 region,
there appears to be overlap of the amine protons, which as previously noted before, this shifting
is typical behavior. The OAc resonance appeared as sharp singlet at 2.23 as well as abroad
singlet at 1.96 ppm. The 13C NMR spectra revealed a singlet at 93.0 ppm integrating for five
protons, which corresponds with the Cp* aromatic analogue. The two methine carbon resonances
appeared at 63.5 and 62.4 ppm. The OAc peak was revealed a singlet at 26.5 ppm. The
resonances for the methyl groups on the Cp* appeared at 10.7 ppm.
Scheme 8. Synthesis of [Co(Cp*)(S,S-DPEN)(OAc)](I) ∙ 3H2O
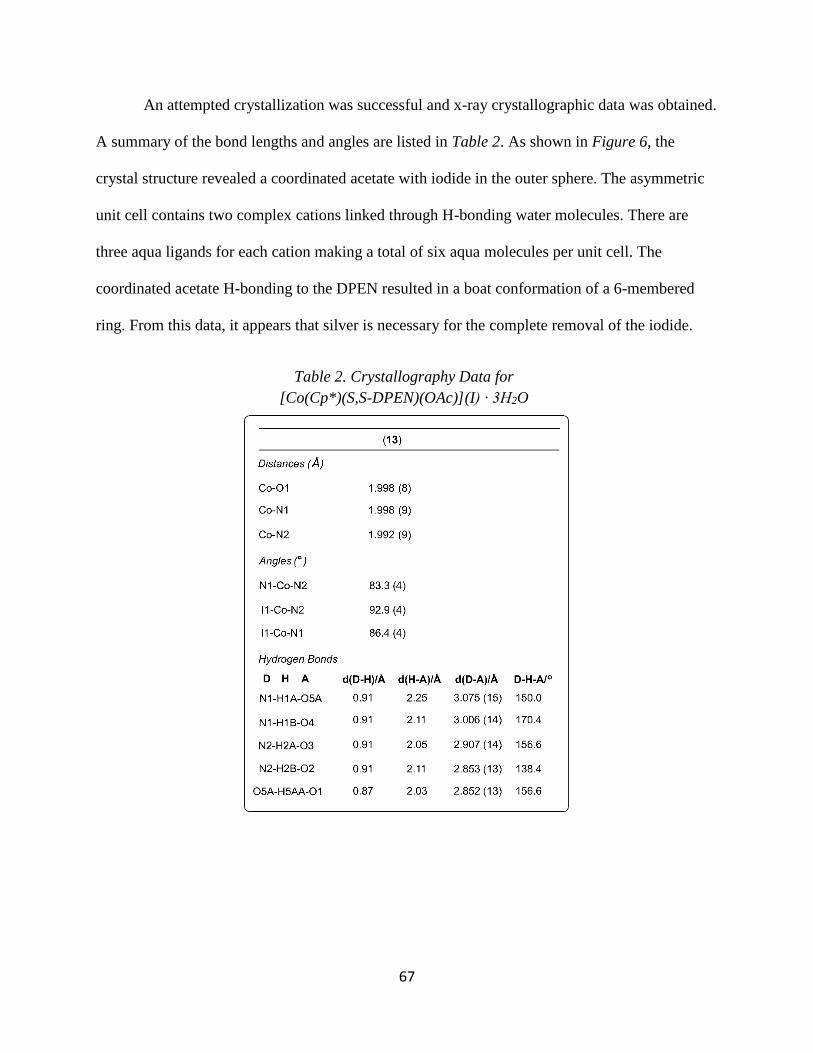
67
An attempted crystallization was successful and x-ray crystallographic data was obtained.
A summary of the bond lengths and angles are listed in Table 2. As shown in Figure 6, the
crystal structure revealed a coordinated acetate with iodide in the outer sphere. The asymmetric
unit cell contains two complex cations linked through H-bonding water molecules. There are
three aqua ligands for each cation making a total of six aqua molecules per unit cell. The
coordinated acetate H-bonding to the DPEN resulted in a boat conformation of a 6-membered
ring. From this data, it appears that silver is necessary for the complete removal of the iodide.
Table 2. Crystallography Data for
[Co(Cp*)(S,S-DPEN)(OAc)](I) ∙ 3H2O
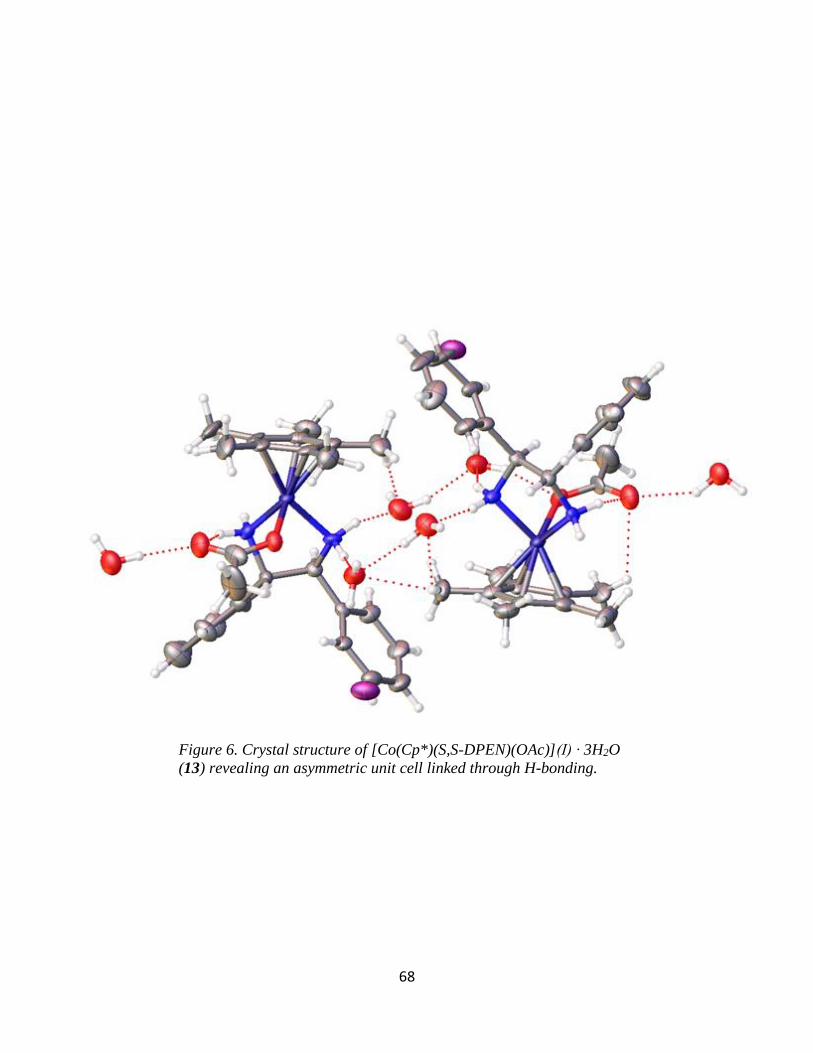
68
Figure 6. Crystal structure of [Co(Cp*)(S,S-DPEN)(OAc)](I) ∙ 3H2O
(13) revealing an asymmetric unit cell linked through H-bonding.
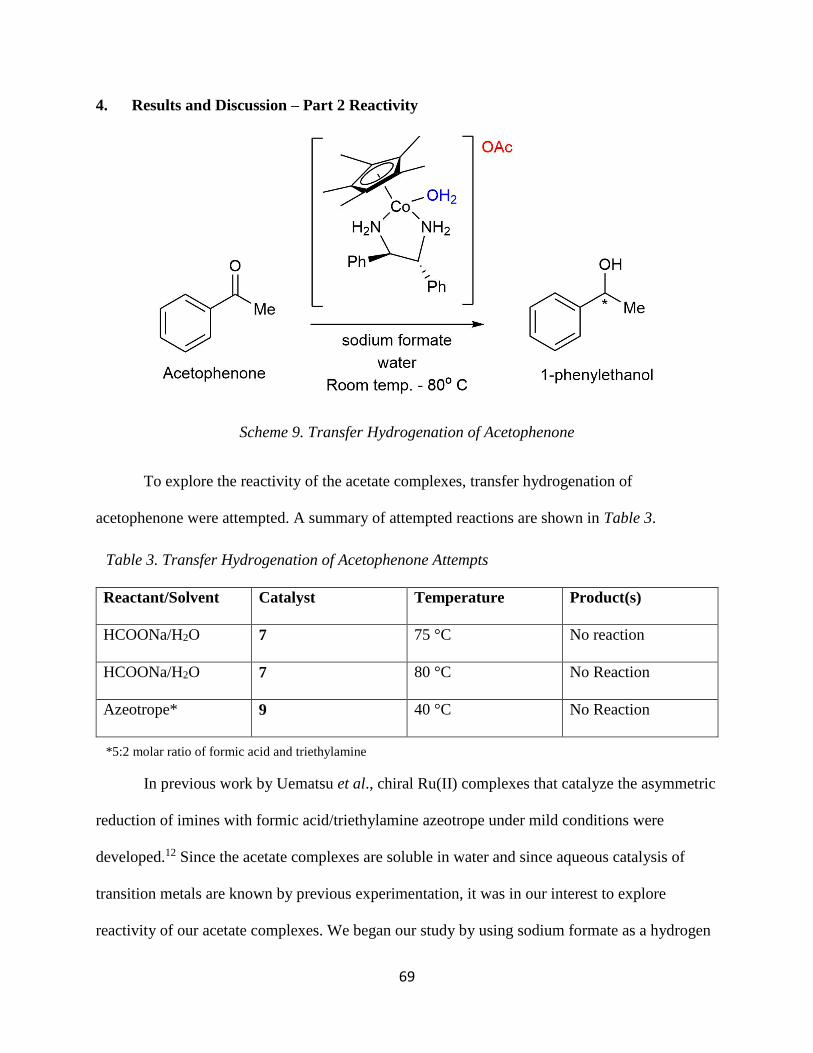
69
4. Results and Discussion – Part 2 Reactivity
To explore the reactivity of the acetate complexes, transfer hydrogenation of
acetophenone were attempted. A summary of attempted reactions are shown in Table 3.
Reactant/Solvent Catalyst Temperature Product(s)
HCOONa/H2O 7 75 °C No reaction
HCOONa/H2O 7 80 °C No Reaction
Azeotrope* 9 40 °C No Reaction
In previous work by Uematsu et al., chiral Ru(II) complexes that catalyze the asymmetric
reduction of imines with formic acid/triethylamine azeotrope under mild conditions were
developed.12 Since the acetate complexes are soluble in water and since aqueous catalysis of
transition metals are known by previous experimentation, it was in our interest to explore
reactivity of our acetate complexes. We began our study by using sodium formate as a hydrogen
Scheme 9. Transfer Hydrogenation of Acetophenone
*5:2 molar ratio of formic acid and triethylamine
Table 3. Transfer Hydrogenation of Acetophenone Attempts

70
source in aqueous solvent to assist in catalyzing the substrate, acetophenone. The expected
product was 1-phenylethanol. The first attempt, with a reaction temperature of 75 °C, resulted in
no reaction per 1H NMR. In another attempt, when heated to 80 °C, there was also no reaction. A
third attempt involved the use of an azeotropic mixture of formic acid and triethylamine as the
reactant and solvent, and heating the solution to 40 °C. There was also no desired product as
indicated by 1H NMR. A probable reason for the complex impotency can be explained by the VT
NMR studies that were previously done on the complexes. When heating the complex above 45
°C, it becomes unstable and decomposes. These reactions might require some heat to overcome
an activation energy, which results in catalyst decomposition. Alternatively the complex is not
reducing enough to remove a hydride from the formate and produce CO2. For the first
explanation it can be concluded that the acetate complexes were unstable and could not facilitate
transfer hydrogenation reactions. For the second explanation, future work will involve exploring
catalytic hydrogenation of CO2, which would be the reverse reaction of transfer hydrogenation
from formate, to see if the catalyst is competent for that reaction.

71
5. Conclusions and Future Work
Seven complexes of the types [Co(CpR)(X,X-DPEN)(OH2)](OAc)2 ∙ nH2O,
[Co(Cp*)(R,RDPEN)(OAc)](OAc) ∙ nH2O, [Co(Cp*)(R,R-DPEN)(OAc)](I) ∙ nH2O, and
[Co(Cp*)(S,SDPEN)(OAc)](I) ∙ 3H2O have been synthesized successfully. These products can
be obtained either by reacting [Co(CpR)(X,X-DPEN)(I)](I) with AgOAc, in pot via reaction of
[Co(CpR)(CO)(I)2] with AgOAc followed by DPEN addition, or [Co(CpR)(CO)(I)2] with CsOAc
followed by DPEN addition. It appears that depending on solvent choice and/or reagent choice,
produces variations of complexes. These include complexes with an iodide likely coordinated to
the metal and two OAc in the outer-sphere; a complex with an aqua molecule coordinated to the
metal and one OAc in the outer-sphere; and a complex with an OAc coordinated to the metal and
an iodide in the outer-sphere.
1H-VT, 13C, HMQC NMR spectroscopy as well as Single Crystal X-Ray Crystallography
confirm the presence of acetate and aqua molecules in the complex. Elemental analysis confirms
the presence of iodide in the majority of the complexes, although synthetic details should be
explored further to understand the nature of these acetate complexes. The cesium acetate
Scheme 6. Nitroaldol Reaction of Benzaldehyde

72
complexes still need to be characterized further. VT-NMR and attempted transfer hydrogenation
reactions reveal OAc complex instability or the complex is not reducing enough to remove a
hydride from the formate and produce CO2. Future work will be determining the reactivity limits
of the complexes via catalytic reduction of CO2 or attempting the nitroaldol reaction of
benzaldehyde in aqueous solution (Scheme 6).13

73
References
1. Welton, T. Proc. R. Soc. London, Ser, A. 2015, 471, 20150502
2. Kitanosono, T.; Masida, K.; Xu, P.; Kobayashi, S. Chem Rev. 2018, 118, 679-746
3. Himeda, Y.; Onozawa-Komatsuzaki, N.; Sugihara, H.; Arakawa, H.; Kasuga, K. J. Mol.
Catal. A: Chem. 2003, 195, 95-100.
4. Szymczak, N. K.; Braden, D. A.; Crossland, J. L.; Turov, Y.; Zakharov, L. N.; Tyler, D.
Inorg. Chem. 2009, 48, 2676-2984.
5. Ahmed, T. J.; Fox, B. R.; Knapp, S.; Yelle, R. B.; Juliette, J. J.; Tyler, D. Inorg. Chem.
2009, 48, 7828-7837.
6. Kobayashi, S.; Nagayama, S.; Busujima, T. J. Am. Chem. Soc. 1998, 120, 8287-8288
7. Olaon, M. E.; Carolan, J. P.; Chiodo, M. V.; Lazzara, P. R.; Mohan, R.S. Tetrahedron
Lett. 2010, 51, 3969-3971.
8. Li, J.; Tang, Y.; Wang, Q.; Li, X.; Cun, L.; Zhang, X.; Zhu, J.; Li, L.; Deng, J. J. Am.
Chem. Soc. 2012, 134, 18522−18525.
9. Thorpe, T.; Blacker, J.; Brown, S. M.; Bubert, C.; Crosby, J.; Fitzjohn, S.; Muxworhty, J.
P.; Williams, J. M. J. Tetrahedron Lett. 2001, 42, 4041-4043.
10. King, R. B.; Efraty, A.; Douglas, W. M. Organomet. Chem. 1973, 56, 345.
11. Uematsu, N.; Fuji, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc.
1996, 118, 4916-4917.
12. Uematsu, N.; Fujii, Ak.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996,
118, 4916-4917.
13. Ma, Z.; Gurbanov, A. V.; Maharramov, A. M.; Guseinov, F. I.; Kopylovich, M.; Zubkov,
F. I.; Mahmudov, K. T.; Pombeiro, A. J. L. J. Mol. Catal. A: Chem. 2017, 426, 526-533.

74
Chapter 4: Synthesis and reactivity of Co(III) complexes
bearing a Ts-DPEN ligand

75
1. Introduction
Though catalysis was not observed with the acetate complexes to facilitate transfer
hydrogenation reactions, their unique solubility gives room for more research projects.
Nonetheless, it was in our interest to explore the possibility of utilizing the Ts-DPEN [N-(p-
toluenesulfonyl)-1,2-diphenylethylenediamine] ligand, which has been shown to catalyze
transfer hydrogenation reactions of ketones.1 Early experiments with this ligand involved the use
of [RuCl2(benzene)]2 mixed with a base in 2-propanol. Later experiments revealed the proposed
mechanism of the catalytic reduction of ketones and imines using [Ru(p-cymene)(Ts-DPEN)(H)]
in 2-propanol that involved a concerted cycle. A variety of chiral alcohols were produced in high
yields and ee (Scheme 1).2,3 The proton transfer from the hydride and the NH2 to the substrate
gives a cyclic six-membered transition state to produce an alcohol. The 2-propanol then supplies
a hydride to the metal and a proton to the ligand, leaving acetone as a byproduct. There is no
metal-substrate coordination in this proposed mechanism. The chiral ligand provides a chiral
pocket for either the re or si face of the prochiral molecules, whether it is ketones being reduced
to alcohols or imines to amines.4 In contrast to ketone reduction, which requires 2-propanol,
Scheme 1. Ru Catalyzed Transfer Hydrogenation of Ketones.2

76
reduction of imines are performed in an azeotropic mixture (5:2 molar ratio) of formic acid-
triethylamine and has been shown to be catalyzed by Ru complexes with Ts-DPEN derivatives.3
Although this reaction is historically catalyzed by precious metals in combination with the Ts-
DPEN ligand, it is hypothesized that an extension to octahedral Co(III) would be feasible.
Therefore, our group set out to prepare and study the reactivity of the Ts-DPEN with Co(III).
The syntheses and reactivities will be discussed in this chapter.
2. Experimental Section
2.1. Materials
All solvents and reagents were used as received. The ligand (R,R)-TsDPEN was purchased from
Alfa Aesar and used as received. The metal reagents [Co(Cp)(CO)(I)2] and [Co(Cp*)(CO)(I)2]
were prepared as previously reported.5,6 GC-MS spectra were obtained from Trace GC Ultra Gas
Chromatograph. A Thermo Polaris Q Mass Spectrometer was used to determine molecular
weights.
2.2. Measurements
1H and 13C NMR spectra were obtained on a JEOL ECX 400 MHz spectrometer (operating
frequency for 13C NMR was 100 MHz) and referenced against tetramethylsilane using residual
proton signals (1H NMR) or the 13C resonances of the deuterated solvent (13C NMR). Elemental
analyses were performed by Atlantic Microlab, Inc in Norcross, GA.

77
2.3 Syntheses
5.3.1. Preparation of [Co(Cp*)(R,R-Ts(H)DPEN)(I)](I) (14).
In a nitrogen filled glovebox, a 50 mL round-bottom flask was charged with
[Co(Cp*)(CO)(I)2] (0.0507 g, 0.107 mmol), dichloromethane (25 mL), and (R,R)-Ts(H)DPEN
(0.0469 g, 0.128 mmol) There was a slight color change from dark purple to a lighter purple. The
mixture was stirred at room temperature for about 16 h. The solvent was removed in vacuo and
the resulting product was washed with hexanes. The recovered green/gray product had a final
product weight of 0.0741 g. (85% yield). 1H NMR (87a-1H-LD), 13C NMR (87a-13C-LD).
1H NMR (acetone-d6, δ): 7.05-7.49 (20H, m, Ts-DPEN aromatic and NH), 4.70 (2H, m, CH),
2.23 (15H, s, Cp*), 1.26 (3H, s, Ts-CH3). 13C {1H} NMR (acetone-d6, δ): 142.4 (1C, s, ipso C
arene), 129.1, 128.3, 127.9, 127.8, 127.8, 127.7, 127.6, 127.4, 127.3, 127.1, 126.8 (11C, m,
Aromatic C DPEN), 102 (5C, s, Cp), 71.9 (1C, s, DPEN CH), 69.1 (1C, s, DPEN CH), 20.3 (1C,
s, Ts-CH3), 10.6 (5C, s, Cp* CH3).
5.3.2. Attempted Preparation of [Co(Cp*)(R,R-TsDPEN)(I)] (15).
A 50 mL round-bottom flask was charged with 14 (0.0250 g, 0.0307 mmol),
dichloromethane (5 mL), and KOH (0.0260 g, 0.463 mmol) Upon vigorous stirring, after 5 min,
H2O was added (5 mL). The aqueous layer changed to purple color. After 10 min, the organic
layer began to turn yellow/green. The layers were separated into two separate evaporating dishes.
The product from the organics that remained was a green/gray color. The final product weighed
0.0107 g. 1H NMR (94a-1H-LD). The NMR spectra revealed that the complex was
paramagnetic, and therefore there were no significant peaks to be observed.

78
5.3.3. Attempted Transfer Hydrogenation of Acetophenone
In a fume hood, a 3 mL conical flask was charged with formic acid/triethylamine (5:2
mol). The mixture was let to react for 5 min and then cool to room temperature. 14 (0.00820 g,
10 µmol) was then added. There was a color change from brown to dark blue. The flask was then
charged with acetophenone (0.12 mL, 1 mmol) and the solution began to stir at room
temperature. After 1 h, the solution color changed to light pink. The solution stirred for 48 h and
was then transferred into a 10 mL round-bottom flask. The solution was dissolved in 2 mL ethyl
acetate (EtOAc) and washed with 2 mL NaHCO3 (10%). The organic layer was separated and
placed in an evaporating dish. There was no recorded yield. A 1H NMR and GC-MS sample was
taken and revealed no indication of 1-phenylethanol.
3. Results and Discussion – Part 1 Synthesis and Characterization
Synthesis of [Co(Cp*)(R,R-Ts(H)DPEN)(I)](I) (14)
It was initially hypothesized that the Co(III) complex bearing the Ts-DPEN ligand would
be synthesized in an alcohol mixture with triethylamine under refluxing conditions.4 This would
deprotonate the amine, making the overall complex neutral. However, 1H NMR data suggested
Scheme 2. Synthesis of [Co(Cp*)(R,R-Ts(H)DPEN)(I)](I)

79
the complex was likely reduced to paramagnetic Co(II). We decided to synthesize the complex
similar to our original iodide complexes. Therefore, [Co(Cp*)(CO)(I)2] was reacted with excess
Ts-DPEN in dichloromethane. This synthetic route allowed for the formation the stable cationic
complex 14. The results were confirmed by 1H and 13C NMR. The 1H NMR spectra revealed a
singlet at 2.23 ppm, integrating for fifteen protons, which corresponds with the methyl groups on
the Cp* ligand. The Ts-CH3 resonance appeared as a singlet at 1.26 ppm. The two methine
protons from the DPEN ligand appeared at 4.70 ppm and integrated for two protons. The amine
protons appear to be buried in the aromatics region. The 13C NMR spectra revealed a singlet at
102 ppm which corresponds with the Cp* aromatic ring. The two methine carbon resonances
were confirmed and appeared at 71.9 and 69.1 ppm. The Ts-CH3 peak was revealed as a singlet
at 20.3 ppm. The resonances for the methyl groups on the Cp* appeared at 10.6 ppm.
Synthesis of [Co(Cp*)(R,R-TsDPEN)(I)] (15)
With the successful product isolation of cationic [Co(Cp*)(R,R-Ts(H)DPEN)(I)](I), it
was in our interest to continue the attempts at deprotonating the complex to synthesize the
desired neutral variant of the complex. The synthesis of complex 15 was attempted by reacting
14 under basic conditions. The product was analyzed by 1H NMR. The spectra did not reveal any
Scheme 3. Synthesis of [Co(Cp*)(R,R-TsDPEN)](I)

80
significant peaks. The Cp* resonance did not appear at all. This suggests the complex was likely
reduced to Co(II) and thus paramagnetic. Therefore, the addition of a strong base under the
conditions experimented was shown to be an ineffective way to deprotonate the secondary amine
on the Ts(H) DPEN ligand.
4. Results and Discussion – Part 2 Reactivity
Catalytic asymmetric transfer hydrogenation of acetophenone was attempted using 14 as
the catalyst. Per 1H NMR, a quintuplet was revealed at 4.6 ppm which corresponds to the CH3.
However, the acetophenone peak was ambiguous. A sample of the product was analyzed on the
GC-MS. A peak on the chromatogram at 5.40 min revealed a m/z that accounted for
acetophenone. There was no evidence of 1-phenylethanol, the desired product.
5. Conclusions and Future Work
The synthesis of complexes of the type [Co(Cp*)(Ts-DPEN)(I)](I) was attempted. There
is evidence in the successful synthesis and stability of the cationic variant, [Co(Cp*)(R,R-
Ts(H)DPEN)(I)](I), as revealed by both 1H and 13C NMR. Attempts to deprotonate the complex,
Scheme 4. Transfer Hydrogenation of Acetophenone

81
however, were inconclusive. Furthermore, attempts to use the cationic complex 14 for the
catalytic reduction of acetophenone revealed no evidence of the desired product, 1-
phenylethanol. The next step will be to continue attempts to deprotonate the cationic variant
under the most favorable conditions.
The reactivity of these complexes to be used as a catalyst is also another step in the
continuation of this project. In our attempts, an azeotropic mixture of formic acid and
triethylamine was used as the solvent and hydrogen source for the catalytic cycle. As this is the
most commonly used solvent for the hydrogen transfer of ketones and imines using the Ts-
DPEN, literature studies reveal the reaction rates using this methodology are slow.7 In fact, when
the ratio of formic acid and triethylamine is lowered, the reaction rates increase.8,9,10 Also, it was
also reported that asymmetric hydrogen transfer using the Ts-DPEN ligand with water as the
solvent increases the reaction rate and is pH dependent.11 It is evident then, that higher pH favors
both higher reaction rates and enantioselectivities.12 Since the catalytic reduction of
acetophenone in aqueous conditions using Ts-DPEN ligand was successful using Ir (Scheme
5),7,13 it will therefore be beneficial to utilize reaction conditions to an aqueous environment for
the future work of our complexes.
Scheme 5. Ir Catalyzed Transfer Hydrogenation of Acetophenone.13

82
References
1. Haack. K.-J.; Hashiguchi, S.; Fuji, A.; Ikariya, T.; Noyori, R.. Angew. Chem. Int. Ed.
1997, 36, 285-288.
2. Samec, J. S. M.; Backvall, J-E.; Andersson, P.G.; Brandt, P. Chem. Soc. Rev. 2006, 35,
237–248.
3. D. G. I. Petra, J. N. H. Reek, J.-W. Handgraaf, E. J. Meijer, P. Dierkes, P. C. Kamer, J.
Brusse, H. E. Schoemaker and P. W. N. M. van Leeuwen, Chem.–Eur. J., 2000, 6, 2818.
4. Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996,
118, 4916-4917.
5. King, R. B. Inorg. Chem. 1966, 5, 82-87.
6. King, R. B.; Efraty, A.; Douglas, W. M. Organomet. Chem. 1973, 56, 345.
7. Wu, X.; Li, X.; Zanotti-Gerosa, A.; Pettman, A.; Liu, J.; Mills, A. J.; Xiao, J. Chem. Eur.
J. 2008, 14, 2209-222.
8. Blacker, J.; Martin J. In Asymmetric Catalysis on Industrial Scale: Challenges,
Approaches and Solutions. Blaser, H. U., Schmidt, E., Eds.; VCH: Weinheim, 2004; pp 201-216.
9. Blackmond, D.G.; Ropic, M.; Stefinovic, M. Org. Process Res. Dev. 2006, 10, 457.
10. Miyagi, M.; Takehara, J.; Collet, S.; Okano, K. Org. Process Res. Dev. 2000, 4, 346.
11. X. F. Wu, X. G. Li, W. Hems, F. King, J. L. Xiao. Org. Biomol. Chem. 2004, 2, 1818.
12. X. F. Wu, X. G.Li, F. King, J. L. Xiao, Angew. Chem. 2005, 117, 3473.
13. Kitanosono, T.; Masida, K.; Xu, P.; Kobayashi, S. Chem Rev. 2018, 118, 679-746.

83
Appendix A: Crystallography Data
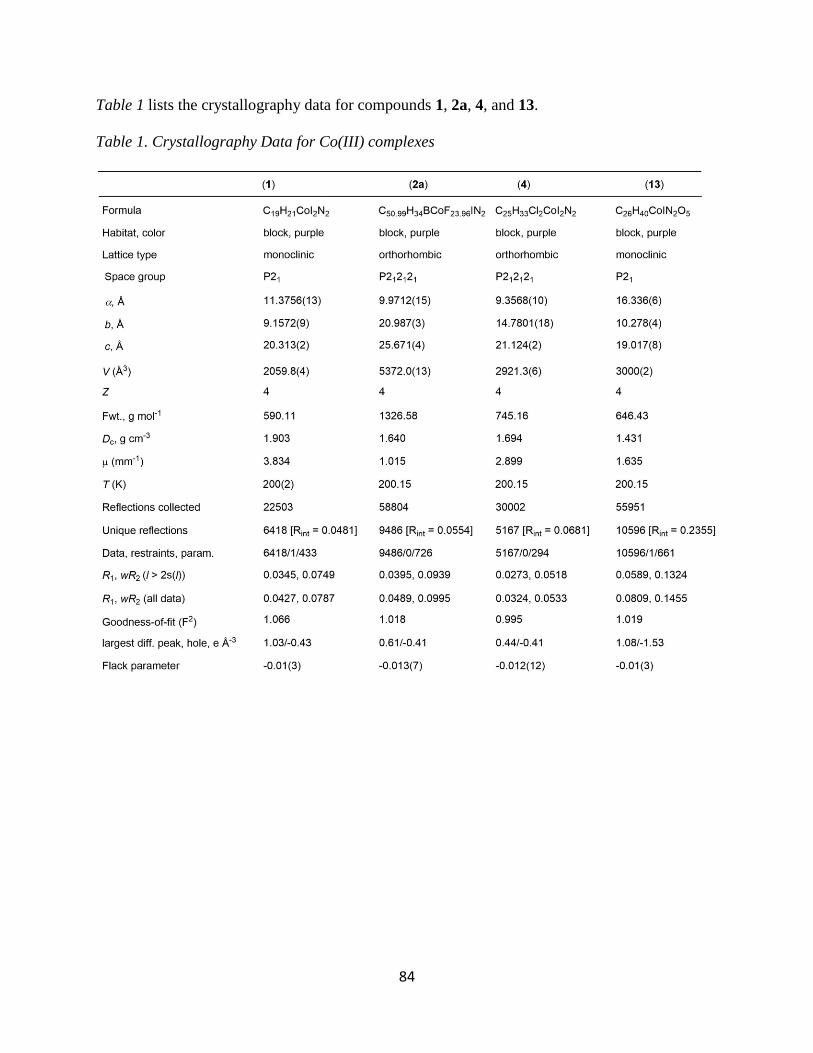
84
Table 1 lists the crystallography data for compounds 1, 2a, 4, and 13.
Table 1. Crystallography Data for Co(III) complexes

85
Acknowledgements
Departments:
UTC Chemistry & Physics Department
I would like to thank the department for giving all students opportunity to actively
participate in research. This experience has definitely taught me to embrace responsibilities,
improve time management, and has challenged and improved my critical thinking skills. The
faculty-student interaction is phenomenal and I hope that this department will maintain this
powerful educational feature.
UTC Honors College
I would like to thank the Honors College for providing support for students to participate
in interdisciplinary research. These opportunities are crucial and essential for higher learning and
future success.
Individuals:
Advisor
I would like to thank my research advisor, Dr. John Lee for giving me the opportunity to
participate in his research group. I have learned so much chemistry and gained many research
and instrumental skills as his apprentice. I have grown to love the research process even when
there are stressful times associated with it. Thanks for pushing me to get things done and settings
those very beneficial deadlines. Thanks to you, I will be able to take this knowledge and these
skills to be a blessing to society in some way. You’re the best!
Committee Members
I would like to thank Dr. Kim and Dr. Potts for being willing to be part of my committee.
You were my first chemistry professors when I transferred to UTC and I enjoyed your lectures.
Thanks for being supportive and kind throughout my journey in classes and throughout the
writing of this thesis. Keep up the great work as professors!
Faculty
I would like to thank Dr. Pienkos for being super supportive and helping with my thesis
revisions.

86
Students
Connor Frye, Trevor Paratore, Chelsey Mertens, Cintly Guzman, Kevin Lee, Olivia Gregerson,
and Mary Nie:
I would like to personally thank the listed students for being real and immensely supportive
throughout the duration I have been actively participating in research in Grote Hall. You have no
idea how much you have been helpful to me!
Funding:
Grote Chemistry Fund
SEARCH Grant (2018-2019)
Westbrook Scholarship





























