Embed Size (px)
Citation preview

PRACOWNIA CHEMII NIEORGANICZNEJ II
REGULAMIN PRACOWNI CHEMII NIEORGANICZNEJ II
Pracownia składa się z 8 ćwiczeń, każde oceniane w skali 0-10 punktów.
Przystąpienie do wykonania ćwiczenia jest możliwe po zdaniu kolokwium
dopuszczającego z akresu materiału objętego wymaganiami..
Zaliczenie ćwiczenia polega na jego wykonaniu, zdaniu kolokwium i zaliczeniu opisu.
Opis ćwiczenia powinien być oddany na następnej pracowni. Opóźnienie oddania opisu
spowoduje obniżenie oceny o 1 punkt za każdy tydzień opóźnienia.
Warunkiem zaliczenia pracowni jest zaliczenie wszystkich ćwiczeń oraz uzyskanie
minimum 41 punków.
Ocena końcowa z Pracowni jest określana na podstawie sumy punktów uzyskanych z
poszczególnych ćwiczeń według następującego schematu:
0-40 pkt. niedostatecznie
41-54pkt. dostatecznie
55-58pkt. dostatecznie plus
59-68pkt. dobrze
69-72pkt. dobrze plus
73-80pkt. bardzo dobrze.

SPIS ĆWICZEŃ
Ćwiczenie 1: Kropki kwantowe. Synteza i właściwości optyczne nanokryształów
półprzewodnikowych
dr Marcin Karbarz, . e-mail: [email protected], mgr Joanna Małecka sala 149-9
Ćwiczenie 2 Elektrochemiczne badanie reakcji elektrokatalitycznej redukcja tlenu.
dr Adam Lewera [email protected], mgr Marta Gierwatowska, sala 349
Ćwiczenie 3: Własności i zastosowanie amalgamatów
dr Cezary Gumiński, e-mail: [email protected], sala 349
Ćwiczenie 4: Elektroosadzanie stopowych powłok ochronnych
dr hab. Mikołaj Donten, e-mail: [email protected], sala 149-2
Ćwiczenie 5: Badanie liczby niesparowanych elektronow w kompleksach metali
przejsciowych metoda NMR
dr Włodzimierz Makulski, e-mail:, [email protected], sala 149-10
Ćwiczenie 6 : Badanie szybkości reakcji podstawienia w kompleksach
oktaedrycznych,
dr Jadwiga Stroka, e-mail: [email protected], mgr Małgorzata Głowienka sala 349
Ćwiczenie 7: Chemiczne reakcje oscylacyjne
dr Rafał Jurczakowski, e-mail [email protected] dr Anna Nowicka sala 149-3
Ćwiczenie 8: Samorzutna organizacja cząsteczek alkanotioli na powirzchni złota,
dr Agnieszka Więckowska, e-mail [email protected] mgr Joanna Juchaniewicz
sala 149-8.

Ćwiczenie 1.
Kropki kwantowe. Synteza i właściwości optyczne nanokryształów
półprzewodnikowych.
Dr Marcin Karbarz, e-mail: [email protected], mgr Tomasz Rapecki
sala 149-9
Wymagania
Cząstka w pudle potencjału i jej poziomy energetyczne. Kwantowy model atomu wodoru.
Podstawy teorii pasmowej ciała stałego: metale, półprzewodniki, izolatory. Pojęcie masy
efektywnej elektronu i dziury. Otrzymywanie, struktura i właściwości siarczków i selenków
cynkowców. Podstawy absorpcyjnej i emisyjnej spektroskopii UV/Vis. Właściwości koloidów.
Cel ćwiczenia
Otrzymanie nanokryształów półprzewodników (kropek kwantowych) na przykładzie syntezy
CdS i CdSe. Określenie wpływu warunków syntezy na rozmiar nanokryształów oraz wpływu
rozmiarów kryształów na ich widma UV/Vis, porównanie właściwości nanokryształów i
kryształów „klasycznych”.
Wprowadzenie
Pod pojęciem kropek kwantowych rozumiemy struktury / kryształy o rozmiarach nie
większych niż kilka nm. Typowymi przykładami są nanokryształy takich półprzewodników jak
CdS lub CdSe.
Półprzewodniki mogą absorbować lub emitować (po uprzednim wzbudzeniu)
promieniowanie elektromagnetyczne, o długości fali zależnej od szerokości przerwy
energetyczne (Eg). Zmniejszając kryształy półprzewodnika do odpowiednio niewielkich
rozmiarów okazuje się że wielkość przerwy energetycznej zwiększa się (Eg + ∆E).
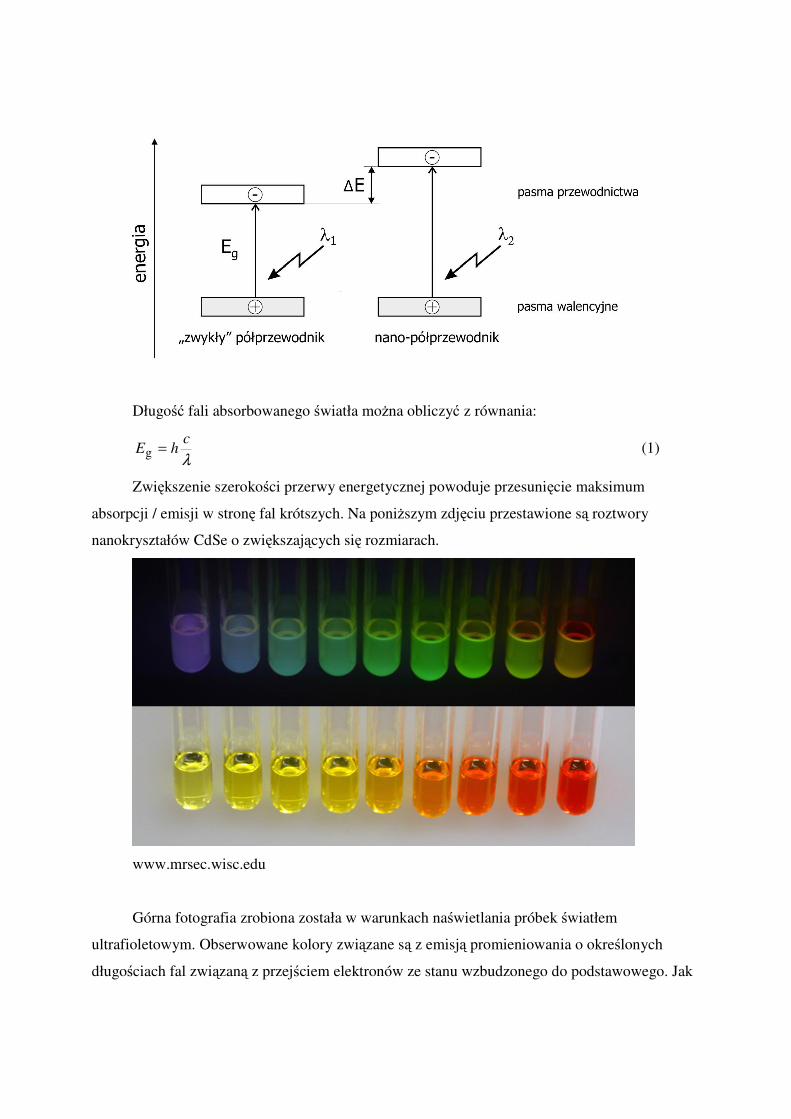
Długość fali absorbowanego światła można obliczyć z równania:
λ
chE =g (1)
Zwiększenie szerokości przerwy energetycznej powoduje przesunięcie maksimum
absorpcji / emisji w stronę fal krótszych. Na poniższym zdjęciu przestawione są roztwory
nanokryształów CdSe o zwiększających się rozmiarach.
www.mrsec.wisc.edu
Górna fotografia zrobiona została w warunkach naświetlania próbek światłem
ultrafioletowym. Obserwowane kolory związane są z emisją promieniowania o określonych
długościach fal związaną z przejściem elektronów ze stanu wzbudzonego do podstawowego. Jak

widać zwiększaniu rozmiarów nanokryształów towarzyszy zwiększanie się długości fal
promieniowania emitowanego (do światła fioletowego do żółto zielonego). Natomiast barwy
próbek obserwowanych w świetle widzialnym zmieniają się od żółtej do czerwonej co
odpowiada (po uwzględnieniu zjawiska przesunięcia Stokesa) barwom dopełniającym. Za te
zmiany odpowiedzialne jest zjawisko, znane w chemii i fizyce kwantowej jako efekt „cząstki w
pudle potencjału”, czyli cząstki poruszającej się w ograniczonej przestrzeni. Rozpatrując
przypadek jednowymiarowy, po rozwiązaniu równania Schrödingera uzyskuje się równanie
opisujące energię cząstki na poszczególnych poziomach energetycznych, En (n = 1, 2, 3..) w
pudle o szerokości L:
2
22
8mL
hnEn = (2)
gdzie h jest stałą Plancka, m – masą cząstki.
Z równania tego wynika, że im mniejszy rozmiar L, tym większa różnica energii
poszczególnych poziomów.
Różnicę w energii absorpcji lub emisji nanokryształów i klasycznych kryształów
półprzewodników można opisać równaniem podobnym do równania 2:
+=∆
**2
2 11
8 he mmR
hE (3)
gdzie R jest promieniem nanokryształu, me* i mh
* są efektywnymi masami elektronu i
dziury w półprzewodniku.
Półprzewodnikowe kropki kwantowe wykazują silny efekt fluorescencji, a barwa
emitowanego światła zależy od ich rozmiarów. Dzięki temu kropki kwantowe znajdują
zastosowanie m.in. w biologii i medycynie do znakowania cząsteczek oraz w technice do
konstruowania wydajnych źródeł światła.
Wykonanie ćwiczenia
W trakcie ćwiczenia prowadzone są dwie syntezy:
I. Nanokryształów CdS przez zmieszanie roztworów Cd(NO3)2 i Na2S, w obecności
polimeru poli(etylenoiminy). Polimer ten adsorbuje się na powstających kryształkach
półprzewodnika i dzięki temu zapobiega ich agregacji i wytrąceniu osadu CdS.

II. Nanokryształów CdSe przez zmieszanie roztworu Se i CdO w podwyższonej
temperaturze w obecności trioktylofosfiny, 1-oktadekenu i kwasu oleinowego.
Dla tak otrzymanych kryształów zostaną przeprowadzone badania fluorescencji oraz
zarejestrowane widma absorpcyjne.
I. Synteza kryształów siarczku kadmu
Studenci wykonują trzy warianty syntezy kryształów CdS. Różnice dotyczą użycia
polimeru zapobiegającego wytrącaniu siarczku kadmu. Przed przystąpieniem do syntezy
konieczne jest przygotowanie roztworów:
1) 50 ml roztworu azotanu (V) kadmu o stężeniu 2*10-2 M w metanolu,
2) 50 ml roztworu siarczku sodu o stężeniu 2*10-2 M w metanolu.
UWAGA: konieczne jest zaopatrzenie się w rękawice i okulary ochronne.
Wariant 1 (synteza nanokryształów CdS)
W celu przeprowadzenia syntezy należy rozpuścić niewielką ilość poli(etylenoiminy) o
wysokiej masie cząsteczkowej (Mn = ~10000 lub Mn = ~60000) w 10 ml metanolu (stężenie
polimeru powinno wynosić około 4.5*10-4 M). Rozpuszczanie należy przeprowadzić w
płaskodennej kolbie stożkowej. Po dokładnym rozpuszczeniu się polimeru w metanolu kolbę
przenosimy do lodówki lub łaźni chłodzącej w celu osiągnięcia temperatury około 10oC.
Następnie kolbę ze schłodzonym roztworem stawiamy na mieszadle magnetycznym,
włączamy umiarkowanie szybkie mieszanie i pipetą automatyczną wprowadzamy uprzednio
przygotowane roztwory azotanu (V) kadmu i siarczku sodu naprzemiennie w objętości po 0.5 ml
każdy. W celu utrzymania stałego tempa dodawania roztworów po każdej porcji należy liczyć
powoli do 5. Po dodaniu całkowitej objętości 5 ml każdego z roztworów kolbę zostawiamy na
mieszadle na kolejne 5 do 10 min. Po tym czasie odstawiamy roztwór do dalszych badań.
Wariant 2 (synteza koloidalnego CdS)
Procedura postępowania jest podobna do powyższej. Należy użyć wcześniej
przygotowanych roztworów 1) i 2). Natomiast w tym przypadku inhibitorem będzie
poli(etylenoimina) o niskiej masie cząsteczkowej (Mn = ~423).
Wariant 3 (otrzymywanie zawiesiny CdS)
W wariancie tym postępuje się analogicznie jak wcześniej, jednakże nie używa się
czynnika inhibitującego przyrost kryształów (polimeru).

Otrzymane roztwory wykorzystujemy do obserwowania fluorescencji w świetle
nadfioletowym oraz do rejestracji widm absorpcyjnych.
II. Synteza nanokryształów selenku kadmu
Syntezę nanokryształów CdSe przeprowadza się z użyciem roztworów CdO i Se. Na tym
przykładzie badany będzie wpływ czasu reakcji na rozmiar powstających nanokryształów.
Krok 1. Przygotowanie prekursora Se.
Odważyć 6 mg Se i dodać go do 1 ml 1-oktadekenu C18H36 (tech., 90%) odmierzonego
wcześniej do kolby o objętości 10 ml. Do tego roztworu dodać 0.2 ml trioktylofosfiny
[CH3(CH2)7]3P. Następnie umieścić w kolbie element mieszający oraz ustawić kolbę na
mieszadle magnetycznym (można podgrzać aby ułatwić rozpuszczanie).
Krok 2. Przygotowanie prekursora Cd.
W kolbie o objętości 25 ml umieścić 7.5 mg CdO. Następnie dodać 0.3 ml kwasu
oleinowego (kwas cis-9-oktadekenowy, CH3[CH2]7CH=CH[CH2]7COOH) oraz 5 ml
1-oktadekenu C18H36.
Krok 3.Wykonanie eksperymentu.
Kolbę z przygotowanym powyżej roztworem CdO (patrz Krok 1.) ogrzewać. Po
osiągnięciu temperatury 225oC wprowadzić 1 ml roztworu Se (przygotowanego w Kroku 2.).
Roztwór Se jest w temperaturze pokojowej.
Charakterystyka produktów silnie zależy od czasu reakcji, należy więc rozpocząć pomiar
czasu natychmiast po dodaniu porcji prekursora Se. Próbki należ pobierać w sekwencji
co 3 sekundy, jednak pierwszą próbkę można pobrać tak szybko jak to tylko możliwe.
Próbki pobierać pipetami Pasteura i umieszczać je w probówkach do schłodzenia uważnie
obserwując zachodzące zmiany.
Po otrzymaniu pełnego zestawu próbek wstawić je pod lampę UV notując obserwowane
barwy dla każdego z roztworów, a następnie zmierzyć ich widma absorpcyjne w celu znalezienia
maksimów absorbancji.
Sprawozdanie z wykonania ćwiczenia
1. Opisz krótko otrzymane rezultaty, w tym wynik obserwacji fluorescencji. Opisz wpływ czasu
reakcji na widma i rozmiar nanokryształów CdSe. Postaraj się skorelować obserwowane barwy z

długościami fal odpowiadającymi maksimum w widmach absorpcyjnych CdSe.
2. Wykreśl zależność zmiany przerwy energetycznej (∆E) od promienia kryształu
półprzewodnika (równanie 3). Dla jakich rozmiarów kryształów można mówić, że ma się do
czynienia z kropkami kwantowymi? W tym celu pomocne będzie wykreślenie procentowego
udziału ∆E w całkowitej wartości energii przerwy energetycznej (∆E+Eg). (Eg=4×10-9 J).
3. Na podstawie równań (1) i (3) wykreśl teoretyczną zależność ∆λ (nm) od promienia
nanokryształu CdS, R. ∆λ to różnica długości fal odpowiadających maksimum absorpcji
nanokryształów (λnano) i kryształów klasycznych (λklas). Przyjmij, że me* i mh
* to, odpowiednio,
0.19 i 0.80 masy swobodnego elektronu.
4. Oblicz wartość ∆λ wykorzystując wyniki eksperymentów przeprowadzonych dla
poszczególnych próbek (jako λklas przyjmij wartość uzyskaną dla koloidalnego CdS lub wartość
literaturową λklas =363nm). Na podstawie wykresu sporządzonego w punkcie 3 wyznacz promień
nanokryształów.
5. Wykorzystując wartości promieni jonowych Cd2+ i S2-, rCd i rS, i przyjmując, że para Cd i S
tworzy kulę o promieniu rCd + rS, oszacuj liczbę par Cd-S w nanokrysztale, dzieląc objętość
nanokryształu przez objętość zajmowaną przez parę Cd-S.
6. Zakładając 100% wydajność reakcji syntezy CdS i uwzględniając wyznaczoną średnią liczbę
atomów Cd i S w nanokrysztale, oblicz stężenie (mol/dm3) nanokryształów
w roztworze. Wykorzystując prawo Lamberta-Beera, na podstawie obliczonego stężenia
i wyznaczonej wartości absorbancji oblicz wartość molowego współczynnika absorpcji kropek
kwantowych CdS, ε. Porównaj otrzymany wynik z typową wartością ε wynoszącą około 104
dm3mol-1cm-1. Wyniki krótko skomentuj.
7. Krótko opisać jedną możliwość praktycznego zastosowania kropek kwantowych.
Literatura
1. „Kropki kwantowe w biotechnologii i medycynie” D. Frąckowiak, E. Staśkowiak, J.
Łukasiewicz, Postępy Fizyki, 56, 1 (2005) 12-19.
2. A. Bielański, Podstawy chemii nieorganicznej, Wydawnictwo Naukowe PWN, Warszawa
2002.
3. K. Pigoń, Z. Ruziewicz, Chemia fizyczna, Wydawnictwo Naukowe PWN, Warszawa 2005
lub W. Kołos, Chemia kwantowa, PWN, Warszawa 1978.

4. T. Lipiec, Z. S. Szmal, Chemia analityczna z elementami analizy instrumentalnej, PZWL,
Warszawa 1980.
5. A.F. Wells, Strukturalna chemia nieorganiczna, WNT, Warszawa 1993.
6. N. N. Greenwood, A. Ernshaw, Chemistry of the elements, Pergamon Press, 1985.
7. “A Safer, Easier, Faster Synthesis for CdSe Quantum Dot Nanocrystals” E. M. Boatman, G.
C. Lisensky, Journal of Chemical Education, 82, 11 (2005) 1697-1699. (dostępne u
prowadzącego)
8. “Semiconductor Nanocrystals: A Powerful Visual Aid for Introducing the Particle in a Box”
T. Kippeny, et al. Journal of Chemical Education, 79, 9 (2002) 1094-1100. (dostępne u
prowadzącego)
9. L. Kolditz, Chemia nieorganiczna, Wydawnictwo Naukowe PWN, Warszawa 1994.
10. C. Kittel, Wstęp do fizyki ciała stałego, PWN, Warszawa 1976.
Wymagania
Cząstka w pudle potencjału i jej poziomy energetyczne. Kwantowy model atomu wodoru.
Podstawy teorii pasmowej ciała stałego: metale, półprzewodniki, izolatory, pojęcie masy
efektywnej elektronu i dziury. Otrzymywanie, struktura i właściwości siarczków i selenków
cynkowców. Podstawy absorpcyjnej i emisyjnej spektroskopii UV/Vis. Właściwości optyczne
koloidów.

Ćwiczenie 2
Elektrochemiczne badanie reakcji elektrokatalitycznej redukcja tlenu.
Dr Krzysztof Miecznikowski e-mail [email protected] dr Adam Lewera e-mail
[email protected], prof. dr hab. P. Kulesza; e-mail [email protected] , pokój
360A
1. Wymagania
Zagadnienia opisane w materiałach dodatkowych, w szczególności pojęcie katalizy,
elektrokatalizy i katalizatora. Kataliza homo i heterogeniczna. Selektywność katalizatora.
Nanostruktury węglowe (w tym nanorurki i nanocząstki). Mechanizm reakcji katalitycznej
redukcji tlenu (uproszczony). Metody elektrochemiczne (woltamperometria cykliczna,
chronoamperometria). Kataliza na powierzchni metali szlachetnych. Prąd elektryczny: potencjał,
natężenie, praca, moc. Pojęcie i zasada działania ogniwa paliwowego. Podstawowe pojęcia
termodynamiczne: entalpia, entalpia swobodna, entropia, ze szczególnym uwzględnieniem
termodynamiki procesów elektrochemicznych. Termodynamika ogniwa paliwowego.
Zaznajomienie się z instrukcją do ćwiczenia.
2. Cel ćwiczenia
Celem ćwiczenia jest zapoznanie się z koncepcją elektrokatalizy na przykładzie reakcji
redukcji tlenu. Wprowadzone zostaną, na przykładach konkretnych katalizatorów, pojęcia
aktywacji powierzchniowej i molekularnej. Przedmiotem ćwiczenia będzie określenie aktywności
katalitycznej kilku typowych materiałów z wykorzystaniem klasycznych metod
elektrochemicznych (woltamperometria cykliczna, chronoamperometria) oraz dodatkowo
określenie aktywności tych katalizatorów w modelowym ogniwie paliwowym, metodami
galwanostatycznymi i galwanodynamicznymi. Badania elektrokatalityczne będą przeprowadzone
na przykładzie redukcji tlenu na katalizatorach typu czerni platynowej (z podkładem węglowym i
bez niego), nanocząstek innych metali i układów wieloskładnikowych, układów organicznych
typu porfiryn, ftalocyjanin czy Co-polipirolu lub innych układów, zasugerowanych przez
prowadzącego ćwiczenie.

3. Wstęp
Znaczenie poznania mechanizmu reakcji elektrokatalitycznej redukcji tlenu, jak również wytypowanie
taniego i efektywnego katalizatora tej reakcji jest trudne do przecenienia, ze względu chociażby na jej praktyczne
wykorzystanie w ogniwach paliwowych. Przebiega ona według poniższego (uproszczonego) schematu:
Sumaryczne równanie reakcji redukcji tlenu jest następujące:
O2 + 4H+ + 4e- ↔ 2H2O (1)
Z mechanistycznego punktu widzenia proces może zachodzić na dwa sposoby: jako bezpośrednia 4-elektronowa
redukcja (jak w równaniu sumarycznym 1) lub w postaci dwóch 2-elektronowych procesów z nadtlenkiem wodoru
(H2O2) jako substancją pośrednią (według mechanizmu przedstawionego poniżej na równaniach 2-3). Bezpośrednia
redukcja tlenu (równanie 1) zachodzi przy standardowym potencjale ok. 1.23 V względem normalnej elektrody
wodorowej (NHE). Natomiast w przypadku redukcji tlenu w środowisku kwaśnym z udziałem etapu pośredniego
prowadzącego do wytworzenia nadtlenku wodoru:
O2 + 2H+ + 2e- → H2O2 (2)
proces ten przebiega przy standardowym potencjale 0.67 V względem NHE, a następnie obserwowana jest dalsza
redukcja formy przejściowej (H2O2) do wody:
H2O2 + 2H+ + 2e- → 2H2O (3)
Ten ostatni proces zachodzi teoretycznie przy znacznie wyższym potencjale standardowym 1.76 V (względem
NHE). W rzeczywistości wszystkie wymienione procesy zachodzą ze znacznymi nadnapięciami, to znaczy przy
potencjałach bardziej ujemnych. W ogniwie paliwowym 4-elektronowa reakcja redukcji tlenu jest energetycznie
znacznie bardziej wydajna w porównaniu do procesu przebiegającego poprzez formę pośrednią (H2O2).
Jako katalizatory reakcji redukcji tlenu przedmiotem zainteresowania są głównie cztery klasy materiałów:
głównie wykorzystuje się metale szlachetne, a szczególne miejsce zajmuje tutaj platyna, stosowana w różnych
postaciach (np. nanocząstek, czy stopów z innymi metalami). Alternatywnymi układami dla platyny w procesie
redukcji tlenu są makrocykliczne związki metali przejściowych (np. porfiryny lub ftalocyjaniny kobaltu i żelaza)
oraz niektóre tlenki metali (np. spinele, czy perowskity). Ostatnią grupę układów o potencjalnym znaczeniu do

konstrukcji katody w metanolowym ogniwie paliwowym stanowią nanocząstki wytwarzane na bazie rutenu i
modyfikowane różnymi pierwiastkami, np. Se, S, czy Mo i S. Ostatnia wymieniona grupa elektrokatalizatorów
wydaje się być ciekawą alternatywą dla układów na bazie platyny, ponieważ wykazuje się znaczną aktywnością i
selektywnością względem redukcji tlenu. Czynnik selektywności tych katalizatorów wydaje się być istotny w
kontekście zastosowania w ogniwie metanolowym, ponieważ w trakcie pracy tego ogniwa znajdujące się w obszarze
anodowym paliwo (metanol) może przedostać się poprzez membranę do katody (ang. methanol crossover) i
spowodować obniżenie jej aktywności katalitycznej. To niepożądane zjawisko zwykle pogłębia się w trakcie pracy
ogniwa. Z tego powodu prowadzi się bardzo intensywne badania nad opracowaniem nowego typu katalizatorów
redukcji tlenu, które byłyby nie tylko reaktywne, ale również i selektywne względem metanolu (ang. metanol
tolerance).
4. Wykonanie ćwiczenia
(1) Przygotowanie odczynników i aparatury
Elektrody płytkowe z węgla szklistego (wszystkie) czyścić z użyciem tlenku glinu o średnicy ziaren 0.05 µm,
dokładnie opłukać i przetrzeć bibułą.
Przygotować dwa naczyńka pomiarowe (zlewki 50ml), wlać 20 cm3 roztworu elektrolitu podstawowego (0.5 mol
dm-3 H2SO4), zamknąć wieczkiem, umieścić w statywie i wprowadzić szklane rurki doprowadzające gazy z butli (argon i
tlen). Rurki doprowadzające gazy powinny być zanurzone praktycznie do samego dna zlewek. Odkręcić butle i
rozpocząć natlenianie w jednej zlewce i argonowanie w drugiej. Zanotować czas. Argonowanie i natlenianie powinno
trwać min. 15 minut.
Ważne: Należy pamiętać iż do badania katalizatorów zawierających platynę wykorzystuje się specjalnie
oznaczoną płytkę! Przygotować zawiesiny badanych katalizatorów w nafionie w sposób następujący: 20 mg każdego
katalizatora odważyć w osobnej fiolce. Zanotować wagę i nazwę katalizatora. Do każdej fiolki dodać 230ul 5% roztworu
nafionu i 770 ul etanolu. Otrzymana zawiesina zawiera około 2mg katalizatora na 100ul (pomijając objętość dodanego
katalizatora). Zawiesiny mieszać na mieszadle magnetycznym w ciągu minimum 15 minut. Pobrać po 100ul każdej
zawiesiny i kolejno osadzać je na błyszczącej stronie płytek z węgla szklistego (pamiętając, iż katalizator zawierający
platynę osadza się na specjalnie oznaczonej płytce), pokrywając równomiernie powierzchnię całej płytki (4cm2).
Obliczyć masę katalizatora przypadającą na jednostkę powierzchni płytki.
(2) Charakterystyka elektrochemiczna katalizatorów w tlenie z wykorzystaniem metod elektrochemicznych
Każdy katalizator badać wg poniższego schematu:
Gdy osadzony na płytce z węgla szklistego katalizator wyschnie, płytkę umieścić w uchwycie i wprowadzić go do
naczyńka pomiarowego zawierającego roztwór H2SO4 nasycony argonem, jako elektrodę pracującą. Czystą płytkę z
węgla szklistego, umieszczoną w uchwycie użyć jako elektrodę pomocniczą, elektrodę Ag/AgCl użyć jako elektrody
odniesienia. Po 15 minutach od rozpoczęcia argonowania przesunąć rurkę doprowadzającą Ar pod powierzchnię
roztworu, umieszczając ją nad powierzchnią roztworu i zarejestrować 10 cykli woltamperometrycznych (20 segmentów)
przy prędkości 50mV s-1 . Zakres potencjałów zależy od użytego katalizatora- dla nanocząstek Ru/Se należy zastosować
+0.7 do –0.3V vs. Ag/AgCl, a dla pozostałych katalizatorów należy zastosować +1.0 do –0.3V vs. Ag/AgCl,
rozpoczynając od potencjału najwyższego (tj. 0.7 lub 1.0 V). Proces ten ma na celu aktywację katalizatora (oczyszczenie,
nawodnienie membrany nafionowej). Należy prowadzić go do uzyskania powtarzalnych woltamogramów (maksymalnie
10 cykli woltamperometrycznych).
Po zakończonym procesie aktywacji zarejestrować w argonie jedną krzywą woltamperometryczną w takim samym
zakresie potencjałów jak aktywacja, przy szybkości polaryzacji 10mV s-1, w Ar, następnie zanurzyć rurkę
doprowadzającą Ar pod powierzchnię roztworu i przełożyć komplet elektrod do zlewki z natlenionym roztworem H2SO4.
Przesunąć rurkę doprowadzającą tlen ponad powierzchnię roztworu i zarejestrować kolejny woltamogram w O2 nie
zmieniając parametrów. Nałożyć oba woltamogramy i wydrukować. Wskazać proces redukcji tlenu.
(3) Charakterystyka katalizatorów w ogniwie paliwowym
Nanieść zawiesinę katalizatorów na podkład przewodzący, poczekać aż wyschnie, zanotować powierzchnię i ilość
użytego katalizatora (10mg/cm2) i zestawić kompletne ogniwo paliwowe, pamiętając o rozseparowaniu anody i katody
membraną z nafionu. Podłączyć mierniki (woltomierz do biegunów ogniwa, amperomierz wraz z opornicą dekadową w
szeregu jako obciążenie ogniwa). Tymczasowo odłączyć jeden z przewodów od opornicy dekadowej (co jest
równoznaczne z nieskończonym oporem elektrycznym – tzw. warunki bezprądowe, ponieważ prąd elektryczny nie
będzie płynął w takim układzie). Rozpocząć zasilanie gazami z elektrolizera. Rejestrować napięcie na biegunach ogniwa
w sytuacji bezprądowej do momentu jego stabilizacji. Zanotować napięcie na biegunach ogniwa paliwowego w
warunkach bezprądowych:
Napięcie między biegunami ogniwa paliwowego w warunkach bezprądowych:................................................
Ustawić 1kΩ na opornicy dekadowej i podłączyć ją do biegunów ogniwa (przyłączając uprzednio odłączony
przewód z opornicy dekadowej). Poprzez zmianę oporu zmieniać płynący prąd i rejestrować zmiany napięcia na
biegunach ogniwa. Zarejestrować około 20 punktów w zakresie od 900 do 200 mV (najlepiej rozseparowane o ok. 20
mV) i wyniki zamieścić w poniższej tabeli:
Opór, R /Ω Napięcie, U/V Prąd I /A (I=U/R) Moc P /W (P=I*U)

Rozpocząć pobór prądu, tak dobierając opór by napięcie wynosiło ok. 0.6V. Zanotować stosowany opór:
Opór na opornicy: .........................................
Nie zmieniać oporu, gdy napięcie ulegnie zmianie w trakcie eksperymentu! Notować zmianę napięcia w funkcji
czasu, wypełniając poniższą tabelę:
Czas Napięcie /V
Czas Napięcie /V


Ćwiczenie 2
Elektrochemiczna charakterystyka katalizatorów procesu redukcji tlenu i
przykład praktycznego wykorzystania tej reakcji w ogniwach paliwowych.
Prowadzący: dr Adam Lewera ([email protected], pokój 360A, tel. 524), mgr
Marta Gierwatowska
2. Wymagania
Zagadnienia opisane w materiałach dodatkowych, w szczególności pojęcie katalizy,
elektrokatalizy i katalizatora. Kataliza homo i heterogeniczna. Selektywność katalizatora.
Nanostruktury węglowe (w tym nanorurki i nanocząstki). Mechanizm reakcji katalitycznej
redukcji tlenu (uproszczony). Metody elektrochemiczne (woltamperometria cykliczna,
chronoamperometria). Kataliza na powierzchni metali szlachetnych. Równanie stanu gazu
doskonałego. Prąd elektryczny: potencjał, natężenie, praca, moc. Pojęcie i zasada działania
ogniwa paliwowego. Podstawowe pojęcia termodynamiczne: entalpia, entalpia swobodna,
entropia, ze szczególnym uwzględnieniem termodynamiki procesów elektrochemicznych.
Termodynamika ogniwa paliwowego. Zaznajomienie się z instrukcją do ćwiczenia.
2. Cel ćwiczenia
Celem ćwiczenia jest zapoznanie się z koncepcją elektrokatalizy na przykładzie
reakcji redukcji tlenu. Wprowadzone zostaną, na przykładach konkretnych katalizatorów,
pojęcia aktywacji powierzchniowej i molekularnej. Przedmiotem ćwiczenia będzie określenie
aktywności katalitycznej kilku typowych materiałów z wykorzystaniem klasycznych metod
elektrochemicznych (woltamperometria cykliczna, chronoamperometria) oraz dodatkowo
określenie aktywności tych katalizatorów w modelowym ogniwie paliwowym, metodami
galwanostatycznymi i galwanodynamicznymi. Badania elektrokatalityczne będą
przeprowadzone na przykładzie redukcji tlenu na katalizatorach typu czerni platynowej (z
podkładem węglowym i bez niego), nanocząstek innych metali i układów
wieloskładnikowych, układów organicznych typu porfiryn, ftalocyjanin czy Co-polipirolu lub
innych układów, zasugerowanych przez prowadzącego ćwiczenie.
3. Wstęp
Znaczenie poznania mechanizmu reakcji elektrokatalitycznej redukcji tlenu, jak również wytypowanie taniego i
efektywnego katalizatora tej reakcji jest trudne do przecenienia, ze względu chociażby na jej praktyczne

wykorzystanie w ogniwach paliwowych. Mechanizm tego procesu jest bardzo złożony, w mocno uproszczonej
wersji można go znaleźć w materiałach dodatkowych do tego ćwiczenia.
Sumaryczne równanie reakcji redukcji tlenu jest następujące:
O2 + 4H+ + 4e- ↔ 2H2O (1)
Z mechanistycznego punktu widzenia proces może zachodzić na dwa sposoby: jako bezpośrednia 4-elektronowa
redukcja (jak w równaniu sumarycznym 1) lub w postaci dwóch 2-elektronowych procesów z nadtlenkiem
wodoru (H2O2) jako substancją pośrednią (według mechanizmu przedstawionego poniżej na równaniach 2-3).
Bezpośrednia redukcja tlenu (równanie 1) zachodzi przy standardowym potencjale ok. 1.23 V względem
normalnej elektrody wodorowej (NEW). Natomiast w przypadku redukcji tlenu w środowisku kwaśnym z
udziałem etapu pośredniego prowadzącego do wytworzenia nadtlenku wodoru:
O2 + 2H+ + 2e- → H2O2 (2)
proces ten przebiega przy standardowym potencjale 0.67 V względem NEW, a następnie obserwowana jest
dalsza redukcja formy przejściowej (H2O2) do wody:
H2O2 + 2H+ + 2e- → 2H2O (3)
Ten ostatni proces zachodzi teoretycznie przy znacznie wyższym potencjale standardowym (1.76 V względem
NEW). W rzeczywistości wszystkie wymienione procesy zachodzą ze znacznymi nadnapięciami, to znaczy przy
potencjałach bardziej ujemnych. W ogniwie paliwowym 4-elektronowa reakcja redukcji tlenu jest energetycznie
znacznie bardziej wydajna w porównaniu do procesu przebiegającego do H2O2.
Jako katalizatory reakcji redukcji tlenu przedmiotem zainteresowania są głównie cztery klasy
materiałów: zwykle wykorzystuje się metale szlachetne, a szczególne miejsce zajmuje tutaj platyna, stosowana
w różnych postaciach (np. nanocząstek, czy stopów z innymi metalami). Alternatywnymi układami dla platyny
w procesie redukcji tlenu są makrocykliczne związki metali przejściowych (np. porfiryny lub ftalocyjaniny
kobaltu i żelaza) oraz niektóre tlenki metali (np. spinele, czy perowskity). Ostatnią grupę układów o
potencjalnym znaczeniu do konstrukcji katody w metanolowym ogniwie paliwowym stanowią nanocząstki
wytwarzane na bazie rutenu i modyfikowane różnymi pierwiastkami, np. Se, S, czy Mo i S. Ostatnia
wymieniona grupa elektrokatalizatorów wydaje się być ciekawą alternatywą dla układów na bazie platyny,
ponieważ wykazuje się znaczną aktywnością i selektywnością względem redukcji tlenu. Czynnik selektywności
tych katalizatorów wydaje się być istotny w kontekście zastosowania w ogniwie metanolowym, ponieważ w

trakcie pracy tego ogniwa znajdujące się w obszarze anodowym paliwo (metanol) może przedostać się poprzez
membranę do katody (ang. methanol crossover) i spowodować obniżenie jej aktywności katalitycznej. To
niepożądane zjawisko zwykle pogłębia się w trakcie pracy ogniwa. Z tego powodu prowadzi się bardzo
intensywne badania nad opracowaniem nowego typu katalizatorów redukcji tlenu, które byłyby nie tylko
reaktywne, ale również i selektywne względem metanolu (ang. metanol tolerance).
4. Wykonanie ćwiczenia
(2) Przygotowanie odczynników i aparatury
Elektrody płytkowe z węgla szklistego (wszystkie) czyścić na tkaninie pokrytej wodną zawiesiną tlenku glinu o
średnicy ziaren 0.05 µm, dokładnie opłukać i przetrzeć bibułą.
Przygotować dwa naczynka pomiarowe (zlewki 50ml), do każdej wlać około 20 cm3 roztworu elektrolitu
podstawowego (0.5 mol dm-3 H2SO4), zamknąć wieczkiem, umieścić w statywie i wprowadzić szklane rurki
doprowadzające gazy z butli (argon i tlen). Rurki doprowadzające gazy powinny być zanurzone praktycznie do
samego dna zlewek. Odkręcić butle i rozpocząć natlenianie w jednej zlewce i argonowanie w drugiej. Zanotować
czas. Argonowanie i natlenianie powinno trwać min. 15 minut.
Przygotować zawiesiny badanych katalizatorów w nafionie w sposób następujący: 20 mg każdego katalizatora
odważyć w osobnej fiolce. Zanotować wagę i nazwę katalizatora. Do każdej fiolki dodać 230µl 5% roztworu
nafionu i 770 µl etanolu. Otrzymana zawiesina zawiera około 2mg katalizatora na 100µl (pomijając objętość
dodanego katalizatora). Zawiesiny mieszać na mieszadle magnetycznym w ciągu minimum 15 minut. Pobrać po
100µl każdej zawiesiny i kolejno osadzać je na błyszczącej stronie płytek z węgla szklistego, pokrywając
równomiernie powierzchnię całej płytki (4cm2). Obliczyć masę katalizatora przypadającą na jednostkę powierzchni
płytki.
(2) Charakterystyka elektrochemiczna katalizatorów w tlenie z wykorzystaniem metod elektrochemicznych
Każdy katalizator badać wg poniższego schematu:
Gdy osadzony na płytce z węgla szklistego katalizator wyschnie, płytkę umieścić w uchwycie i wprowadzić go
do naczynka pomiarowego zawierającego roztwór H2SO4 nasycony argonem, jako elektrodę pracującą. Czystą
płytkę z węgla szklistego, umieszczoną w uchwycie użyć jako elektrodę pomocniczą, elektrodę Ag/AgCl użyć jako
elektrodę odniesienia. Po 15 minutach od rozpoczęcia argonowania przesunąć rurkę doprowadzającą Ar
umieszczając ją nad powierzchnią roztworu i zarejestrować 10 cykli woltamperometrycznych (20 segmentów) przy
prędkości 100mV s-1 (zobacz rysunek poniżej). Zakres potencjałów zależy od użytego katalizatora: dla nanocząstek
Ru/Se należy zastosować +0.75 do –0.3V vs. Ag/AgCl, a dla pozostałych katalizatorów należy zastosować +1.0 do –
0.3V vs. Ag/AgCl, rozpoczynając od potencjału najwyższego (tj. 0.75 lub 1.0 V). Proces ten ma na celu aktywację

katalizatora (oczyszczenie, nawodnienie membrany nafionowej). Należy prowadzić go do uzyskania powtarzalnych
woltamogramów (maksymalnie 10 cykli woltamperometrycznych).
Ustawienia przyrządu dla woltamperometrii cyklicznej. Wartość „Init” określa potencjał początkowy (1 lub 0.75V, w
zależności od katalizatora). Wartość „High” określa najwyższy potencjał w czasie przemiatania (wartość powinna
być identyczna jak „Init”), wartość „Low” określa najniższy potencjał w czasie przemiatania (-0.3V). Wartość
„Scan rate” to szybkość przemiatania potencjałem (0.1 V/s w procesie aktywacji, 0.01 V/s podczas badania
charakterystyk w Ar i O2). Wartość „Sweep segments” określa liczbę segmentów – każdy cykl woltamperometryczny
to dwa segmenty, więc w procesie aktywacji należy podać wartość 20, podczas charakterystyki – wartość 2.
„Sensitivity” to wartość wzmocnienia prądowego („czułość”), zwykle używa się wartości 1e-003, w przypadku gdy
przyrząd zakomunikuje „overflow” należy zmienić tę wartość na 1e-002. Pozostałe parametry należy zostawić bez
zmian.
Po zakończonym procesie aktywacji zarejestrować w argonie jedną krzywą woltamperometryczną w takim samym
zakresie potencjałów jak aktywacja, przy szybkości polaryzacji 10mV s-1, następnie zanurzyć rurkę doprowadzającą
Ar pod powierzchnię roztworu i przełożyć komplet elektrod do zlewki z natlenionym roztworem H2SO4. Przesunąć
rurkę doprowadzającą tlen ponad powierzchnię roztworu i zarejestrować kolejny woltamogram w O2 nie zmieniając
parametrów. Nałożyć oba woltamogramy i wydrukować. Wskazać proces redukcji tlenu.
(3) Charakterystyka katalizatorów w ogniwie paliwowym
Nanieść zawiesinę katalizatorów na podkład przewodzący, poczekać aż wyschnie, zanotować powierzchnię i
ilość użytego katalizatora (10mg/cm2) i zestawić kompletne ogniwo paliwowe, pamiętając o rozseparowaniu anody i
katody membraną z nafionu. Zmierzyć panującą temperaturę i ciśnienie.
Temperatura otoczenia:............................. Ciśnienie..........................................
Podłączyć mierniki (woltomierz do biegunów ogniwa, amperomierz wraz z opornicą dekadową w szeregu jako
obciążenie ogniwa). Tymczasowo odłączyć jeden z przewodów od opornicy dekadowej (co jest równoznaczne z
nieskończonym oporem elektrycznym – tzw. warunki bezprądowe, ponieważ prąd elektryczny nie będzie płynął w
takim układzie). Rozpocząć zasilanie gazami z elektrolizera. Rejestrować napięcie na biegunach ogniwa w sytuacji
bezprądowej do momentu jego stabilizacji. Zanotować napięcie na biegunach ogniwa paliwowego w warunkach
bezprądowych:
Napięcie między biegunami ogniwa paliwowego w warunkach bezprądowych:................................................
Ustawić 1kΩ na opornicy dekadowej i podłączyć ją do biegunów ogniwa (przyłączając uprzednio odłączony
przewód z opornicy dekadowej). Poprzez zmianę oporu zmieniać płynący prąd i rejestrować zmiany napięcia na
biegunach ogniwa. Zarejestrować około 20 punktów w zakresie od 900 do 200 mV (najlepiej rozseparowane o ok.
20 mV) i wyniki zamieścić w poniższej tabeli:
Opór, R /Ω Napięcie, U/V Prąd I /mA Moc P /mW (P=I*U)

Rozpocząć pobór prądu, tak dobierając opór by napięcie wynosiło ok. 0.6V. Zanotować stosowany opór:
Opór na opornicy: .........................................
Nie zmieniać oporu, gdy napięcie ulegnie zmianie w trakcie eksperymentu! Notować zmianę napięcia w funkcji
czasu, wypełniając poniższą tabelę:

Czas Napięcie /V
Czas Napięcie /V

23
Określanie sprawności konwersji w wodorowo-tlenowym ogniwie paliwowym
Ta część ćwiczenia ma na celu określenie, jak procent substratów reakcji
zachodzących w ogniwie paliwowym jest wykorzystywany („sprawność konwersji”). W
tym celu należy zmierzyć szybkość przepływu gazów przez ogniwo paliwowe
(zakładając, że jest równe wydajności elektrolizera), tj. dopełnić zbiornik z wodą w
elektrolizerze do poziomu „0”, zamknąć odprowadzenie gazów z elektrolizera i
jednocześnie uruchomić elektrolizer i rozpocząć pomiar czasu. Zanotować czas, po
którym poziom gazów osiągnie następujące wartości:
Czas od
rozpoczęcia
pomiaru /s
Objętość
H2 /cm3
Czas od
rozpoczęcia
pomiaru /s
Objętość
O2 /cm3
20 10
40 20
60 30
80
40
By określić wielkość przepływu można użyć metody regresji liniowej (najlepiej),
lub wyliczyć wartość średnią z otrzymanych wyników.
Przepływ = .................................. cm3 O2/s lub .............................. cm
3 H2/s
Dla punktów charakterystyki prądowo-napięciowej ogniwa, znając natężenie
płynącego prądu, obliczyć sprawność konwersji jako procent zużycia substratów w
funkcji napięcia na biegunach ogniwa. Jako iż przepływ prądu jest skutkiem
zachodzących reakcji redoks, to procent zużycia substratów można obliczyć jako iloraz
wartości natężenia prądu czerpanego z ogniwa (z charakterystyki prądowo – napięciowej)
do maksymalnej możliwej wartości natężenia prądu, możliwej do uzyskania z danej ilości
substratów (gazów, wyznaczonej z pomiarów przepływu powyżej). Wskazówka: użyć
stałej Faradaya by obliczyć maksymalny prąd możliwy do uzyskania z wyznaczonej

24
szybkości przepływu gazów i porównać tę wielkość z wartością zmierzoną. Użyć
równania stanu gazu doskonałego ze zmierzonymi wartościami temperatury i ciśnienia. Z
porównania tego wyprowadzić wzór na wydajność substratową w funkcji mierzonego
prądu DLA WŁASNEGO OGNIWA, tj. doprowadzić do najprostszej postaci,
podstawiając wielkości zmierzone, i użyć go do wypełnienia odpowiedniej tabelki w
części „opracowanie wyników”.
Określanie sprawności termodynamicznej ogniwa.
Korzystając z zależności termodynamicznych obliczyć sprawność
termodynamiczną ogniwa paliwowego dla każdego z punktów zależności prądowo –
napięciowej. Wskazówka: sprawność termodynamiczna ogniwa paliwowego, wodorowo-
tlenowego, w warunkach standardowych równa się:
η = 0,675 x U
Wyprowadzenie powyższego wzoru znajduje się w materiałach dodatkowych do
ćwiczenia.
5. Opracowanie wyników
(1) Z otrzymanych woltamogramów odczytać potencjał, przy którym rozpoczyna się proces redukcji tlenu
oraz maksymalny zarejestrowany prąd związany z tym procesem i zamieścić w poniższej tabelce:
Katalizator Potencjał
redukcji tlenu
/mV
Maksymalny
prąd redukcji tlenu
/mA
Powierzchnia
elektrody z
katalizatorem /cm2
Masa
katalizatora
/g
Gęstość
prądu reakcji
redukcji tlenu
/mA cm-2
reakcji
redukcji
tlenu na
jednostk
masy
katalizatora
/mA g
25
(2) Opisać otrzymane wyniki w kategoriach aktywności danego materiału w
elektrokatalitycznej reakcji redukcji tlenu. Wytypować katalizator najbardziej aktywny.
(3) Wyniki charakterystyki prądowo-napięciowej, oraz wielkości z nich wyliczone
zamieścić w poniższej tabelce:
Opór, R /Ω Napięcie, U/V Prąd I /mA Moc P /mW
(P=I*U)
Wydajność
substratowa /%
Wydajność
termodynamiczna
/%
(4) Wykreślić i skomentować zależność napięcia na biegunach ogniwa od pobieranego prądu. Z otrzymanej
zależności prądowo-napięciowej dla ogniwa paliwowego obliczyć oddawaną moc w funkcji prądu i wykreślić
na tym samym rysunku (patrz rysunek poniżej). Porównać maksymalną moc z mocą niezbędną do zasilania
typowych urządzeń elektrycznych. Określić optymalne warunki pracy przebadanego ogniwa paliwowego
(napięcie przy którym oddawana moc jest maksymalna).

26
0 100 200 300 4000.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
0
20
40
60
80
100
120
E / V
j / mA cm-2
b
a
b'
a'
Mo
c (m
W/c
m2)
(Krzywe a i b to wykresy gęstości mocy dla pewnego katalizatora A i B w
funkcji gęstości prądu, krzywe a’ oraz b’ to charakterystyka napięciowo-
prądowa odpowiednio dla tych samych katalizatorów (tj. a odpowiada a’ i b
odpowiada b’).
(5) Wykreślić i skomentować zarejestrowane zależności napięcia na biegunach
ogniwa paliwowego w funkcji czasu (trwałość katalizatora) dla wszystkich badanych
katalizatorów (na jednym wykresie).
(6) Na jednym wykresie wykreślić i skomentować procent zużycia substratów
(wydajność substratową) i wydajność termodynamiczną ogniwa w funkcji napięcia na
jego biegunach. Jakie wnioski praktyczne płyną z tego wyniku? Wskazówka:
wyprowadzić lub przeanalizować wyprowadzenie wzoru na sprawność termodynamiczną
ogniwa paliwowego w funkcji napięcia z części praktycznej.
(7) Podsumowanie i komentarz do wyników: Jak przebiega proces elektrokatalitycznej redukcji tlenu, i
jakie cechy elektrokatalizatora tej redukcji mogą być określone elektrochemicznie przy pomocy
woltamperometrii cyklicznej? Jak eksperymenty woltamperometryczne przekładają się na wyniki w
ogniwie paliwowym? Jakie są zalety ogniw paliwowych? Jakie zastosowanie praktyczne mogło by mieć

27
ogniwo przebadane na ćwiczeniu? Co należało by zrobić, by poprawić praktyczną sprawność badanego
ogniwa?
6. Zalecana literatura
1. Skrypt w wersji elektronicznej 2. A. Cygański, „Podstawy Metod Elektroanalitycznych”, WNT, Warszawa, 1999 3. Andrzej Czerwiński, „Akumulatory, baterie, ogniwa”, WKŁ, Warszawa, 2005 4. Andrzej Czerwiński , « Współczesne źródła energii », Wydawnictwa Wydziału
Chemii UW, Warszawa 2001. 5. S. Litster, G. McLean, PEM fuel cell electrodes, J. Power Sources 130 (2004) 61 6. R. Bashyam, P. Zelenay, A class of non-precious metal composite catalysts for fuel
cells, Nature 443 (2007) 63 7. E. Antolini, Recent developments in polymer electrolyte fuel cell electrodes, J. Appl.
Electrochem, 34 (2004) 563 8. J. W. Lee, B. N. Popov, Ruthenium-based electrocatalysts for oxygen reduction
reaction – a review, J. Solid State Electrochem., 11 (2007) 1355 9. M. Winter, R. J. Brodd, What Are Batteries, Fuel Cells and Supercapacitors?, Chem.
Rev. 104 (2004) 4245

28
Ćwiczenie 3.
Własności i zastosowanie amalgamatów
dr Cezary Gumiński, e-mail: [email protected], + doktorantka sala 349
1 Wymagania
Charakterystyka stopów (związki i roztwory stałe, typy sieci krystalograficznych,
bertolidy i daltonidy, wiązania metaliczne). Definicja i podział amalgamatów.
Otrzymywanie amalgamatów. Zastosowanie amalgamatów. Fizykochemiczne badania
własności i struktury amalgamatów; badania elektrochemiczne. Podstawy potencjometrii
i kulometrii. Wyznaczanie rozpuszczalności w układach metal stały – metal ciekły.
Własności chemiczne rtęci i jej związków, ze szczególnym uwzględnieniem specyfiki
Hg(I).
2 Cel ćwiczenia
Celem ćwiczenia jest:
wyznaczenie rozpuszczalności miedzi w rtęci posługując się kulometrycznym
preparowaniem i potencjometrycznym badaniem amalgamatu o różnych stężeniach.
badanie procesu wiązania rtęci przez stop dentystyczny wykorzystując pomiar
jego twardości.
3 Wykonanie ćwiczenia
(i) Przygotować klucze elektrolityczne. Napełnić naczyńko roztworem siarczanu
miedzi, i po zamontowaniu kluczy oraz elektrody wiszącej, przedmuchiwać roztwór
argonem przez 45 min. Połączyć i uaktywnić układ elektryczny (generator prądu, miernik
prądu, potencjometr). Wykręcić tłoczkiem kroplę Hg na ½ obrotu. Dokonywać elektrolizy
stałym prądem 2000 µA przez 60,0 s. Po przerwaniu elektrolizy odczytywać potencjał
elektrody z wytworzonym amalgamatem miedzi po 3, 30, 60, 90, 120, 150 i 180 s.
Eksperyment powtarzać na nowych kroplach Hg stosując za każdym razem inny prąd:
generowania: 1000, 500, 200, 100, 50, 20, 10, 5, 2, 1 i 0.5 µA.

29
(ii) Odważyć 3.0-3.5 g Hg i sproszkowany stop dentystyczny w proporcji ok.
1:1. Szybko utrzeć proszek wraz z Hg w naczynku. Po napełnieniu i ubiciu próbek w
stalowej formie mierzyć ich twardość przy pomocy twardościomierza w pewnych
odstępach czasu pomiędzy 4 a 30 min od momentu zakończenia ucierania.
4 Opis wykonanego ćwiczenia
(i) Sporządzić wykresy zależności potencjał elektrody amalgamatowej – czas
dla różnych stężeń miedzi (na jednym wykresie). Wyznaczone w ten sposób potencjały
równowagowe odłożyć względem logarytmu prądu wytwarzania amalgamatu.
Przeprowadzić interpolację liniową dla niskich stężeń miedzi (małych prądów
generowania) i ekstrapolację plateau dla najwyższych stężeń w kierunku niskich stężeń.
Wybrzuszenie w okolicach punktu nasycenia jest związane z brakiem kompletnego
wykrystalizowania fazy stałej w ciągu 150 s obserwacji i nie należy go brać pod uwagę.
Na przecięciu prostoliniowych zależności odczytać prąd generowania dla amalgamatu
nasyconego i potencjał równowagowy dla tegoż amalgamatu. Z uzyskanej wartości prądu
obliczyć stężenie nasyconego amalgamatu miedzi w mol/dm3, wiedząc że proces
elektrodowy jest 2 elektronowy, wydajność prądu jest 100 %, a kropelka Hg jest kulą o
promieniu 0.43 mm. Znając stężenie elektrolitu i nasyconego amalgamatu oraz
odpowiadający nasyceniu potencjał równowagowy ( Er) obliczyć EoCu2+/Cu(Hg) z równania:
Er = EoCu2+/Cu(Hg) + (RT/nF) ln [CuSO4]/[Cu(Hg)]
Przyjmujemy, że współczynniki aktywności Cu w amalgamacie i jonów Cu w
roztworze wynoszą jeden. Obliczamy energię swobodną ∆G ex procesu amalgamowania
miedzi ze wzoru:
∆Gex = -nF(EoCu2+/Cu – Eo
Cu2+/Cu(Hg)) w kJ/mol Cu
jeżeli EoCu2+/Cu wynosi 0.103 V względem nasyconej elektrody kalomelowej.
Skomentować wartość energii swobodnej (duża czy mała jak na reakcje chemiczną).
(ii) Odłożyć zmierzone wartości twardości amalgamatu dentystycznego w
czasie. Skomentować otrzymaną krzywa kinetyczną. Podać inne przykłady wiązania
rozpuszczalnika przez ciało stałe z chemii nieorganicznej.

30
5 Podstawy teoretyczne (skrót)
Charakterystyka wiązań międzymetalicznych
Stopy metali są często pomijane w podręcznikach chemii nieorganicznej, bowiem
traktuje się je przeważnie jako roztwory lub mieszaniny, zaliczając badania w tej
dziedzinie raczej do chemii fizycznej, fizyki, a przede wszystkim metaloznawstwa. Stop
metaliczny dwu i więcej składnikowy może być w fazie stałej roztworem lub mieszaniną
roztworów ,ale także związkiem chemicznym lub mieszaniną związków chemicznych. W
fazie ciekłej mamy do czynienia najczęściej z roztworami właściwymi lecz niekiedy dwie
fazy ciekłe nie mieszają się ze sobą (np. Ga-Hg) lub faza ciekła zawiera silnie
rozdrobniony metal jakby w stanie koloidalnym, choć jego rozpuszczalność jest
przekroczona wiele razy (np. Fe w Hg).
Każdy pierwiastek w stanie stałym posiada ściśle określoną sieć krystalograficzną.
Większość metali krystalizuje w jednej z trzech struktur najgęstszego ułożenia atomów:
regularnej przestrzennie centrowanej, regularnej zewnętrznie centrowanej oraz
heksagonalnej. Jeżeli do jednego metalu będziemy wprowadzać atomy drugiego metalu,
atomy te mogą albo zastępować atomy pierwszego metalu w jego sieci, albo
wbudowywać się w przestrzenie międzywęzłowe, przy czym typ struktury
krystalograficznej pierwszego metalu powinien zostać zachowany w tym procesie.
Wprowadzanie atomów drugiego metalu może jednak spowodować powstanie nowej
fazy o strukturze krystalograficznej odmiennej od tej jaką posiadały pierwszy i drugi
metal przed zmieszaniem; tworzy się wtedy związek między składnikami. Stanowi to
podstawę definicji roztworów i związków chemicznych (czy międzymetalicznych) w
sensie krystalograficznym.
Roztworem stałym nazwiemy określoną fazę charakteryzującą się ułożeniem
atomów w sieci identycznym z ułożeniem atomów jednego ze składników, a parametry
sieci, odległości atomowe i kąty między wiązaniami są ciągłą funkcją składu. Najczęściej
takie rozpuszczalności są niewielkie, ale w przypadku metali z tej samej grupy
rozpuszczalność wzajemna jest możliwa w każdej proporcji (np. Hf-Zr, K-Rb, Bi-Sb),
gdyż posiadają one analogiczne struktury krystalograficzne a związki międzymetaliczne
nie tworzą się.

31
Związkiem międzymetalicznym nazwiemy fazę różniącą się typem sieci od swych
składników lub wykazującą skokową zmianę parametrów sieciowych przy zmianie
składu (np. AuIn, Hg4Na, Mg3Pr, Ca2Zn3). Taka klasyfikacja pozwala na łatwe
odróżnienie związków od roztworów stałych za pomocą dyfraktogramów rentgenowskich
czy badań metalograficznych. Pojawienie się nowego rodzaju widma rentgenowskiego
czy widzialnych pod mikroskopem nowych ugrupowań krystalicznych, gdy zmieniamy
wzajemne proporcje składników, świadczy o tworzeniu się nowej fazy, którą może być
związek lub nowy roztwór stały.
Wydaje się, że poznane połączenia są w większości daltonidami, a więc związkami
o ściśle określonym składzie. Jest to słuszne tylko dla części związków chemicznych (np.
dla NaCl, BaCO3, H2O, PtSn, Pb3Tb). Olbrzymią grupę stanowią bertolidy, a więc
związki o składzie zmiennym w pewnych granicach. Mogą to być stopy: jak B0.8-1.0Li czy
AgHg1.26-1.31, tlenki jak TiO0.8-1.3, siarczki jak FeS1.00-1.15, selenki jak VSe0.98-1.20, wodorki
CrH1-2 a nawet wieloskładnikowe związki jak Na0.3-0.9WO3. W przypadku połączeń
międzymetalicznych olbrzymia ich liczba ma charakter właśnie bertolidów. Oprócz
terminu „związek międzymetaliczny”, często używa się terminu „fazy metaliczne
pośrednie”. Pośrednie w sensie składu, bo struktura w całym zakresie istnienia bertolidu
powinna być taka sama.
Aby zrozumieć dlaczego w przypadku metali granica między roztworami,
mieszaninami a związkami jest trudniej uchwytna należy zapoznać się z istotą wiązania
metalicznego. Wiązania metaliczne występujące zarówno w czystych metalach jak i
stopach są podobne do wiązań atomowych. Zasadnicza różnica polega na
bezkierunkowości wiązania metalicznego, a ścisłej kierunkowości atomowego. Objawia
się to szczególnie, gdy porównamy plastyczność Al., czy mosiądzu, z kruchością
kryształów S czy CSi. Wiązania atomowe i metaliczne są zasadniczo homogenne, a więc
siły wiążące atomy są związane z wytworzeniem wspólnej chmury elektronowej.
Różnica tkwi w ich strukturze. W przypadku wiązań atomowych chmury elektronowe są
zlokalizowane najczęściej na określonych orbitalach wypełnianych dwoma elektronami o
przeciwnych spinach, a poszczególne orbitale oddziela znaczna bariera energetyczna. W
metalach chmury elektronowe nakrywają się częściowo i nie muszą wypełniać
całkowicie poszczególnych orbitali. Elektron z jednego atomu metalu może łatwo się

32
znaleźć na nie zajętym orbitalu w atomie drugiego metalu, gdyż przejście takie nie
oddziela znaczniejsza bariera energetyczna i nie mają one trwałego charakteru jak w
przypadku tworzenia się jonów. Elektrony w metalach i stopach tworzą więc coś w
rodzaju „gazu elektronowego” znajdującego się w periodycznym polu kationów metali
obsadzających węzły sieci krystalograficznej. Kwantowy opis takich wiązań jest bardzo
złożony.
Oprócz wiązań o charakterze typowo metalicznym (np. czyste metale, Ni-Zn, Al.-
Mg, In-U, w których elektroujemność jest zbliżona) mogą występować wiązania o
charakterze metaliczno-atomowym i metaliczno-jonowym. Wiązania o charakterze
metaliczno-atomowym są w dużym stopniu ukierunkowane i tworzą się między
pierwiastkami o podobnej elektroujemności z tendencją do tworzenia oktetu z elektronów
walencyjnych (np.AsGa, Be2Sn, HgTe, InSb), a stechiometria połączenia często wynika z
typowych wartościowości chemicznych. Wiązanie o charakterze metaliczno-jonowym
tworzy się między atomami o znacznie różnej elektroujemności (np. Rb4Sn, KTl, Na3Tl,
AuCs, Bi2Ca3) a stechiometria może, lecz nie musi, wynikać z typowych wartościowości
komponentów.
Stechiometrie wielu połączeń międzymetalicznych nie wynikają ze stosunku
wartościowości (np. Bi2Ce, CoSi3, Cr3Ti2), a np. w układzie In-Li powstają połączenia:
InLi, In4Li5, In2Li3, InLi2, InLi3, In3Li13, InLi6, z których tylko dwa związki posiadają
przewidywalną stechiometrię z punktu widzenia chemii.
O typie struktury i składzie tworzącego się połączenia międzymetalicznego
decydują:
Stosunki promieni atomowych składników, które rzutują na liczby koordynacyjne,
bowiem metale mają tendencję do maksymalnego upakowania atomów w przestrzeni.
Stężenie elektronów, czyli stosunek ogólnej ilości elektronów walencyjnych do
ogólnej ilości atomów.
Różnica elektroujemności składników, a więc ich położenie w układzie
okresowym i własności polaryzacyjne.
Czynniki te w poszczególnych przypadkach wywierają wpływ w różnym stopniu i
ich ścisłe zróżnicowanie stanowi trudne zagadnienie.

33
Podział amalgamatów
Amalgamatami nazywamy stopy metali z rtęcią. Nazwa ta obejmuje zarówno
układy jednofazowe (homogenne) stałe albo ciekłe, jak dwu- i wielo-fazowe
(heterogenne), które mogą zawierać kryształy będące w równowadze z roztworem
nasyconym, ciecze nie mieszające się, lub mieszaninę kryształów. Amalgamat prosty
zawiera oprócz Hg jeden metal, amalgamat złożony zawiera co najmniej dwa metale i
Hg. Pomimo, że rtęć i amalgamaty były znane i wykorzystywane już w starożytności (np.
złocenie), potem przez setki lat frapowały alchemików i farmaceutów,
systematyzowaniem się ich własności i strukturą zajęto się dopiero na przełomie XIX/XX
wieku. Najczęściej ciekłe amalgamaty proste posiadają wiele cech podobnych do stopów
stałych. Mogą więc być homogenne jak i heterogenne, oraz zawierać, lub nie, połączenia
międzymetaliczne z Hg. Z tego względu możemy podzielić amalgamaty proste na cztery
grupy:
Amalgamaty metali alkalicznych (w tym także rodniki amonu i alkiloamoniowe),
ziem alkalicznych (lecz bez Be), lantanowców i aktynowców. Wszystkie te metale
reagują z Hg tworząc połączenia międzymetaliczne o rozpuszczalności większej od 10-3
% mol ( w temperaturze 298 K). Powstawaniu tych amalgamatów towarzyszy
wydzielanie znacznego ciepła i wyraźnie ujemna energia swobodna procesu
amalgamowania (kilkadziesiąt kJ/mol). Tworzące się połączenia międzymetaliczne
zachowują trwałość powyżej temperatury wrzenia Hg. Silne oddziaływanie M-Hg
prowadzi do bardzo niskich współczynników aktywności tych metali w amalgamacie
(poniżej 10-3) co objawia się również silnym ujemnym odchyleniem od praw roztworów
idealnych (prawa Raoulta i Henry’ego). Potencjały normalne tych amalgamatów są do
około 1 V bardziej dodatnie niż odpowiadające im potencjały czystych metali.
Amalgamaty z niskotopliwymi pierwiastkami bloku p i d (Al., Ga, In, Tl, Sn, Pb,
Bi, Mn, Cu, Ag, Au, Zn, Cd). Metale te po rozpuszczeniu tworzą w Hg roztwory
właściwe o stężeniach wyższych od 10-3 % mol. Energia swobodna oddziaływania M-Hg
jest zbliżona do energii wiązania w czystych metalach M-M stąd ewentualnie tworzące
się połączenia z Hg nie są zbyt trwałe (rozpadają się zwykle poniżej temperatury wrzenia
Hg) lub nie powstają w ogóle. Procesy rozpuszczania metali charakteryzują się
niewielkim efektem cieplnym dodatnim lub ujemnym. Większość amalgamatów jest

34
bliska spełnienia w stanie ciekłym prawa roztworów idealnych. Potencjały normalne
amalgamatów metali mniej szlachetnych od Hg są zbliżone do potencjałów czystych
metali (dla metali bardziej szlachetnych od Hg potencjały amalgamatów nie mają sensu,
bo w ogniwie zachodzi reakcja chemiczna, np. Au3+ z Hg).
Amalgamaty pierwiastków wysokotopliwych bloku p (często amfoterycznych)
oraz część bloku d (B, C, Si, Ge, As, Sb, V, Nb, Ta, Cr, Mo, W, Re, Fe, Ru, Os, Co, Ir).
Ponieważ wiązania M-M w tych pierwiastkach są silne a wiązania M-Hg bardzo słabe
proces rozpuszczania kończy się tworzeniem mocno rozcieńczonych roztworów
nasyconych o stężeniach niższych niż 10-3 % mol, a towarzyszy temu procesowi znaczny
efekt endotermiczny. Wprowadzone do Hg większe ilości tych metali (np. na drodze
elektrolizy) odkładają się w postaci prawie czystych faz krystalicznych. Ze względu na
bardzo niskie stężenia metali w Hg i hetrogeniczny charakter powierzchni takich
amalgamatów znaczną rolę mogą odgrywać zanieczyszczenia i reakcje uboczne
powodując, że pomiar potencjałów (zwanych wtedy mieszanymi) bywa niepewny, choć
potencjały normalne takich amalgamatów powinny być bardziej ujemne niż czystych
metali. Współczynniki aktywności metali w takich amalgamatach są znacznie wyższe od
jedności, co powinno rzutować na dodatnie odchylenia od praw Raoulta i Henry’ego
(efekt praktycznie niemierzalny ze względu na niskie stężenia).
Amalgamaty pozostałych pierwiastków bloku d (Ti, Zr, Hf, Rh, Ni, Pd, Pt) oraz
prawdopodobnie Be. Oddziaływania międzyatomowe w tych metalach są wprawdzie
silne, lecz tworzą one połączenia z Hg o znacznej trwałości. Powstałe związki słabo
rozpuszczają się w Hg (poniżej 10-3 % mol). Całkowity proces rozpuszczania jest reakcją
o niewielkim efekcie cieplnym dodatnim lub ujemnym. Reakcje te przebiegają wolno,
gdyż metal i Hg oddziela po jakimś czasie stały związek M-Hg tworzący się na styku faz.
Ze względu na niskie stężenia rozpuszczonego metalu w fazie ciekłej występują, podobne
jak poprzednio, problemy z pomiarami potencjometrycznymi, a zmierzone potencjały
równowagowe nie odpowiadają potencjałom czystych metali. Współczynniki aktywności
metali w takich amalgamatach są mniejsze od jedności.
Pozostałe pierwiastki niemetaliczne, albo wykazują względem Hg obojętność (jak
gazy szlachetne, N, P) albo (jak O, S, Se, Te, F, Cl, Br, I) tworzą trwałe związki o
charakterze jonowym. Ponieważ rozpuszczalności ostatnich połączeń są bardzo niskie,

35
należałoby sklasyfikować te układy jako zbliżone do czwartej grupy podziału
amalgamatów.
Można też podzielić amalgamaty z punktu widzenia przynależności pierwiastków
do odpowiednich bloków elektronów s, p, d, f, lecz jak widać z powyżej przedstawionej
charakterystyki, podział taki przebiegałby niekiedy sztucznie, gdyż np. amalgamat Fe
wykazuje więcej podobieństw do amalgamatu Si niż Mn.
Wiązania o charakterze najbardziej metalicznym powinny występować dla Hg z
metalami bloku p. Ze względu na znaczne różnice w elektroujemności pierwiastki bloku
s i f tworzą wiązania o charakterze metaliczno-jonowym, a dla bloku d należałoby
oczekiwać charakteru metliczno-atomowego.
Otrzymywanie amalgamatów
W zależności od przynależności metalu do odpowiedniej grupy podziału możemy
zastosować jeden z czterech sposobów otrzymywania amalgamatów.
Bezpośrednie rozpuszczanie metalu w Hg. Proces taki przebiega szybko, gdy
powierzchnia metalu jest wolna od tlenku a rozpuszczalność wysoka. W ten sposób
można otrzymać amalgamaty pierwszej i drugiej grupy podziału, a czwartej jedynie w
podwyższonej temperaturze.
Elektroredukcja jonów metali na katodzie Hg w środowisku wodnym, a w
przypadku aktywnych metali w środowisku bezwodnym lub stopionych solach. Metodą
tą można uzyskać również amalgamaty metali źle rozpuszczalnych (trzecia i czwarta
grupa podziału) z dużą wydajnością. Nadmiar metalu lub połączenia międzymetalicznego
odkłada się w postaci drobno krystalicznej w głębi lub na powierzchni Hg. Część
amalgamatów można otrzymać przez jednoczesną redukcję na elektrodzie obojętnej
(grafit, metal szlachetny, trudnotopliwy) jonów Hg i wybranego metalu. Amalgamaty
metali szlachetnych (Ag, Au, Pd, Pt) można uzyskać redukując jony Hg bezpośrednio na
tych metalach. Amalgamaty rodnika amonowego lub alkiloamoniowego można
najprościej otrzymać właśnie na drodze elektrolizy.
Cementacja przy użyciu amalgamatów metali bardziej aktywnych z roztworu
metali bardziej szlachetnych, np.:
Ce(III) + 3Na(Hg) Ce(Hg) + 3Na(I)

36
2Au(III) + 5Hg 2Au(Hg) + 3Hg(II)
Hg(II) + 2Cd Cd(Hg) + Cd(II)
NH4(I) + K(Hg) NH4.(Hg) + K(I)
Ostatnią reakcję wykorzystał po raz pierwszy Davy do otrzymania amalgamatu
rodnika amonowego NH4.(Hg) z soli amonowej w warunkach nieutleniających.
Reakcje chemiczne wykorzystując karbonylki, wodorki i związki
metaloorganiczne, np.:
Ni(CO)4 + Hg Ni(Hg) + 4CO
2CuH + Hg2Cl2 2Cu(Hg) + 2HCl
2NaC2H5 + 2Hg 2Na(Hg) + C2H4 + C2H6
Zastosowanie amalgamatów
Proces amalgamowania metali szlachetnych (Au, Ag, Pt, Pd, Rh, Ir) wykorzystuje
się do odzysku tych metali z rud. Z amalgamatu odseparowanego od resztki rudy
otrzymuje się te metale w stanie czystym po oddestylowaniu Hg. Proces powinien być
prowadzony w hermetycznie zamkniętej aparaturze. Niestety wykorzystują go na własną
rękę ludzie prymitywni (np. Indianie w dorzeczu Amazonki) poważnie zatruwając siebie
i środowisko.
Hg można wykorzystać do oddzielania metali. Np. ze stopu Ce-Fe-Si rozpuści się
w Hg tylko Ce; po odfiltrowaniu Fe i Si, oraz termicznym oddzieleniu Hg z amalgamatu
otrzymamy czysty Ce. Rafinacji metali dokonuje się w warunkach elektrolitycznych
polaryzując ogniwa tak, aby po lewej stronie wydzielał się czysty metal lub prosty
amalgamat
M1-M2-M3(Hg)\M1z+\M1(Hg) lub M1-M2-M3(Hg)\M1
z+\M1
gdzie M1 jest metalem rafinowanym, a M2 i M3 zanieczyszczeniami. W trakcie
procesu metale bardziej szlachetne pozostają w amalgamacie (po lewej stronie), a mniej
szlachetne przechodzą do roztworu w postaci jonów. Można prowadzić odzysk
niektórych metali ze ścieków przemysłowych metodą elektrolizy stosując Hg jako
katodę.

37
Jednym ze sposobów otrzymywania metali alkalicznych i ich związków (np. KOH,
Na2O2) jest redukcja jonów tych metali na Hg i poddawanie amalgamatu dalszym
reakcjom, np.
2SO2 + 2Na(Hg) Na2S2O4 + 2Hg
NO2 + Na(Hg) NaNO2 + Hg
2CO2 + 2Na(Hg) Na2C2O4 + 2Hg
TiCl4 + 4Na(Hg) Ti + 4NaCl + 4Hg
W chemii organicznej używa się amalgamatów do redukcji, rozkładu wiązań,
generowania wolnych rodników i otrzymywania połączeń rtęcio-organicznych.
Hg bywa składnikiem stopów stałych i ciekłych mających określone zastosowanie:
np. TeHg i Cd-Te-Hg stosuje się jako materiały półprzewodnikowe, Zn-Hg w technice
laserowej, a szereg amalgamatów pierwszej i drugiej grupy w lampach
luminescencyjnych (zastąpienie Hg amalgamatami pozwala obniżyć toksyczne działanie
par Hg).
Sproszkowane Au lub stop Ag-Sn z niewielkim dodatkiem Cu służy w dentystyce
po zmieszaniu z Hg jako materiał do bardzo trwałych plomb. Stop taki ulega nieznacznej
korozji, posiada podobną twardość, ścieralność i rozszerzalność termiczną jak kość, a
najprawdopodobniej jeszcze przeciwdziała powstawaniu próchnicy. Jak wykazały
badania toksyczność takich plomb jest mniejsza lub porównywalna z toksycznością
przeciętnego środowiska miejskiego. Twardnienie takiego stopu polega na reakcji z Hg z
wytworzeniem heterogennej, stałej mieszaniny połączeń międzymetalicznych:
Ag-Cu-Sn + Hg Ag2Hg3 + Cu6Sn5 + HgSn7 (brak współczynników
stechiometrii)
Z amalgamatów prostych przez oddestylowanie Hg otrzymuje się całkiem czyste
proszki metali (np. najczystszy U lub Pu). Analogicznie z amalgamatów złożonych
można uzyskać proszki połączeń zawierających Hg (np. Fe6Zn2Hg, Hg3Te4Tl2, HgInSe2)
lub jej nie zawierających (np. AuSn, InSb, Al3Ni, Co-Fe, La5Ni).
Amalgamaty mają zastosowanie przy rozdziale izotopów, np. według reakcji:
7Li(Hg) + 6Li+ 6Li(Hg) + 7Li+

38
Fizykochemiczne metody badania składu i struktury amalgamatów
Metody elektrochemiczne są najczęściej stosowane do badań, gdyż zarówno Hg
jak i amalgamaty są często używane jako materiał elektrodowy, a elektrochemia w
dużym stopniu zawdzięcza swój rozwój właśnie Hg. Metody elektrochemiczne
charakteryzuje duża precyzja i bardzo szeroki zakres badanych stężeń 10-8-100 % mol.
Można wyodrębnić dwie grupy technik: bezprądowych (potencjometria i
konduktometria) i z przepływem prądu (elektroliza, polarografia amalgamatowa,
kulometria, chronowoltamperometria – z włączeniem technik pulsowych,
chronopotencjometria, chronoamperometria, chronokulometria).
Metoda pomiaru siły elektromotorycznej (sem) amalgamatowych ogniw
stężeniowych opiera się na wykorzystaniu zależności Nernsta:
E = EoMz+/M(Hg) + RT/zF ln [Mz+]/[M(Hg)]
Równanie to jest w zasadzie słuszne dla aktywności i tylko wtedy, gdy metal M
jest mniej szlachetny od Hg, o stężeniu większym od 10-6 mol/dm3, a układ jest
homogenny i termodynamicznie odwracalny. W przypadku, gdy oba półogniwa
zawierają amalgamat o różnych stężeniach stosuje się równanie Turina:
∆E = RT/zF ln [M(Hg)1]/[M(Hg)2]
Gdy [M(Hg)1] = [M(Hg)2], to ∆E = 0. Gdy potencjały zmierzony i wyliczony nie
spełniają równania Turina należy wnioskować o występowaniu reakcji ubocznych w
półogniwie (tworzenie połączeń międzymetalicznych typu M-M’ lub M-M’-Hg w
przypadku układów trójskładnikowych), lub wydzielaniu się fazy stałej z przesyconego
amalgamatu ciekłego. Ten drugi przypadek wykorzystuje się do wyznaczania
rozpuszczalności metalu w Hg. Mierząc sem ogniwa:
M(Hg)\ Mz+\ M gdy [M(Hg)] = 1 mol/dm3
można określić potencjał termodynamiczny ∆G tworzenia 1 mol/dm3 amalgamatu z
czystego metalu i Hg zgodnie z zależnością:
∆G = -zF(EoM(Hg)/Mz+ - Eo
M/Mz+)

39
Jeżeli nie można otrzymać odpowiednio stężonego amalgamatu, można oszacować
EoM(Hg)/Mz+ używając bardziej rozcieńczonego amalgamatu i posłużyć się równaniem
Turina. Oczywiście ∆G można określić, gdy znamy potencjały normalne metalu i
amalgamatu względem tej samej elektrody odniesienia.
Elektrochemiczne utlenianie metali jest od dawna wykorzystywane w badaniach
stopów Hg. Gdy w klasycznej polarografii i wszystkich metodach pokrewnych zastąpimy
Hg amalgamatem i zaczniemy polaryzować go od potencjałów ujemnych do dodatnich,
możemy rejestrować krzywe utlenienia. W warunkach polarograficznych (używając
elektrody kapiącej) prąd fali opisuje wzór:
id = k z [M(Hg)] DM1/2 m2/3 t1/6
gdzie [M(Hg)] – stężenie metalu w amalgamacie, DM – współczynnik dyfuzji metalu w
Hg, m – wydajność kapilary, t – czas trwania kropli. Podobnie jak to było w przypadku
potencjometrii, gdy w badanym amalgamacie mamy reakcje uboczne lub jest on
przesycony, powyższe równanie nie jest spełnione. Znając stężenie homogennego
amalgamatu można wyznaczyć ta metodą DM. W przypadku utleniania amalgamatu w
warunkach chronowoltamperometrycznych z płaskiej elektrody o stałej powierzchni prąd
piku opisuje wzór:
ip = k z2/3 [M(Hg)] A DM1/2 v1/2
gdzie A – powierzchnia elektrody i v – szybkość zmian potencjału. Gdy elektrodę
stanowi wisząca kropla lub cienka warstwa odpowiednie równanie jest bardziej
skomplikowane. W przypadku stosowania technik zmiennoprądowych czy pulsowych
obowiązują inne wzory, lecz zawsze rejestrowany sygnał prądowy jest wprost
proporcjonalny do stężenia M(Hg), powierzchni elektrody i pierwiastka ze
współczynnika dyfuzji.
Metoda kulometryczna polega na pomiarze ładunku jaki przepływa podczas
procesu elektrodowego (utlenienia frakcji, czy całości metalu w amalgamacie). Aby
poprawnie przeprowadzić takie eksperymenty, należy uprzednio zapoznać się z
krzywymi woltamperometrycznymi badanego układu. Elektrolizę należy prowadzić albo
przy stałym potencjale integrując prąd w czasie, albo stałym prądem przez określony czas
wiedząc, że potencjał elektrody zmienia się w trakcie elektrolizy. Metoda kulometryczna

40
prowadzi do sensownych wyników, gdy wydajność prądowa wynosi 100 % (brak
procesów ubocznych) lub jest ściśle określona.
Do badań własności i struktury amalgamatów używa się szeregu innych technik
fizykochemicznych. Wykorzystanie analizy rentgenowskiej (lub dyfrakcji wiązek
elektronów lub neutronów) jest możliwe tylko dla stężonych amalgamatów ze względu
na znaczne pochłanianie promieni X przez Hg. Analiza otrzymanych widm umożliwia
projekcję struktury takich ciekłych stopów.
Parametry termodynamiczne procesu tworzenia amalgamatu mogą być
wyznaczone z pomiarów potencjometrycznych lub prężności par przeprowadzonych w
różnych temperaturach. Tak można precyzyjnie wyznaczyć aktywności i ∆G, ale mniej
precyzyjnie ∆S (entropia) i ∆H (entalpia). Jedynie precyzyjną metodę pomiaru entalpii
stanowi kalorymetria. Niewielkie zmiany entalpii rozpuszczania metalu w Hg sugerują
tworzenie się niezbyt trwałych wiązań między M i Hg. Znaczne ujemne wartości entalpii
świadczą o tworzeniu się w fazie amalgamatu cząsteczek MHgx. Przy tworzeniu się
takich cząsteczek spada aktywność metalu, natomiast rośnie ona powyżej jedności, gdy
M i Hg wykazują tendencję do wzajemnej niemieszalności.
Pomiary podatności magnetycznej ujawniają tworzenie się niektórych połączeń w
stopach. Metal wprowadzany stopniowo do Hg zmienia łagodnie podatność magnetyczną
wraz ze składem, tylko wtedy gdy nie powstają w tym zakresie połączenia
międzymetaliczne. Nagłe zmiany podatności sygnalizują powstawanie związku. W
analogiczny sposób przeprowadza się i interpretuje pomiary przewodnictwa
elektrycznego.
Pewne informacje o strukturze ciekłych amalgamatów można uzyskać z pomiarów
lepkości, gęstości właściwej, czy napięcia powierzchniowego. Nagłe zmiany tych
parametrów wraz ze zmieniającym się składem sugerują zmiany dokonujące się
wewnątrz fazy lub powstawanie nowej. Amalgamaty stałe możemy badać mierząc
dodatkowo ich twardość.
Bardzo istotną rolę w badaniu stopów (amalgamatów) odgrywa analiza termiczna,
która polega na pomiarze temperatur przemian zachodzących w stopie przy zmiennym
składzie. Technika ta nadal jest rozwijana i często stosowana, gdyż uzyskane podczas
rejestracji krzywe ogrzewania lub chłodzenia pozwalają na konstrukcje diagramów
41
fazowych, które są rodzajem mapy dla wybranego układu dwóch lub więcej metali.
Wyniki z analizy termicznej bywają często uzupełniane informacjami zdobytymi przy
wykorzystaniu wymienionych wcześniej innych technik fizykochemicznych.
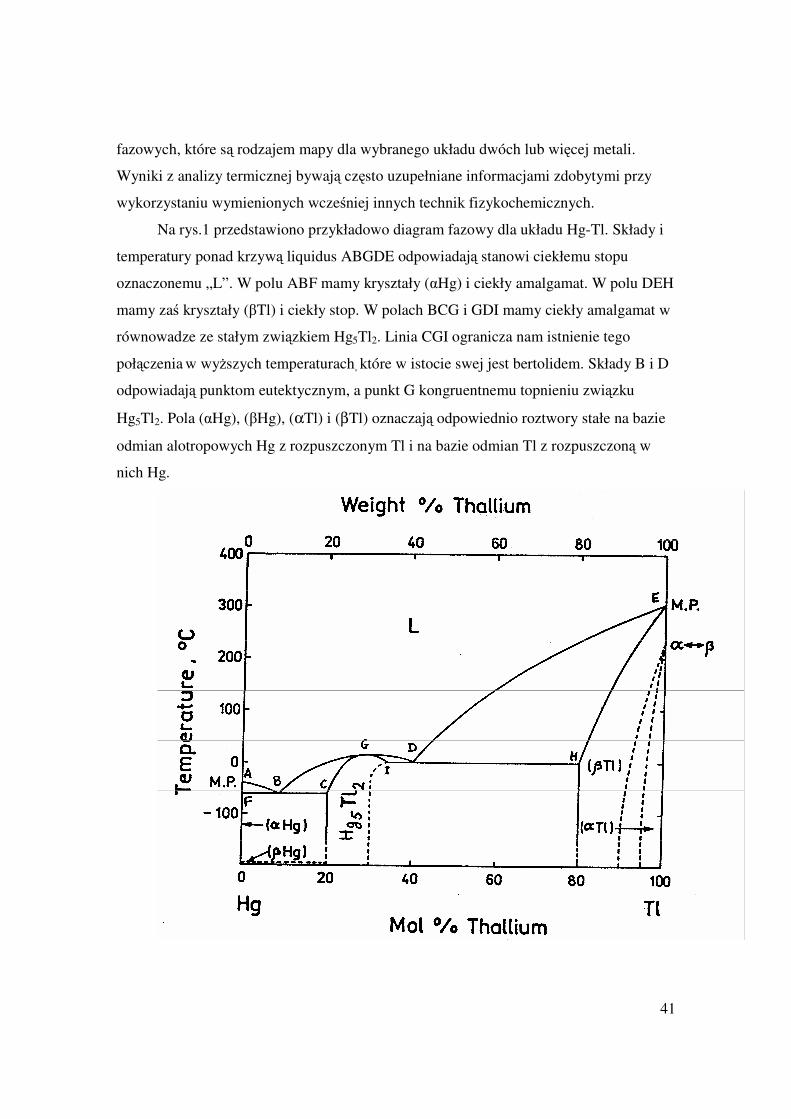
Na rys.1 przedstawiono przykładowo diagram fazowy dla układu Hg-Tl. Składy i
temperatury ponad krzywą liquidus ABGDE odpowiadają stanowi ciekłemu stopu
oznaczonemu „L”. W polu ABF mamy kryształy (αHg) i ciekły amalgamat. W polu DEH
mamy zaś kryształy (βTl) i ciekły stop. W polach BCG i GDI mamy ciekły amalgamat w
równowadze ze stałym związkiem Hg5Tl2. Linia CGI ogranicza nam istnienie tego
połączenia w wyższych temperaturach, które w istocie swej jest bertolidem. Składy B i D
odpowiadają punktom eutektycznym, a punkt G kongruentnemu topnieniu związku
Hg5Tl2. Pola (αHg), (βHg), (αTl) i (βTl) oznaczają odpowiednio roztwory stałe na bazie
odmian alotropowych Hg z rozpuszczonym Tl i na bazie odmian Tl z rozpuszczoną w
nich Hg.

42
Rys. 1 Diagram fazowy układu Hg-Tl.
Pewne informacje o strukturze prostych amalgamatów można uzyskać analizując
wielkość współczynnika dyfuzji metalu w Hg. Są to wielkości rzędu 10-5 cm2/s. Jeśli w
prostym, ciekłym, homogennym amalgamacie atomy rozpuszczonego metalu nie są
specyficznie solwatowane przez atomy Hg, współczynnik dyfuzji można przewidzieć w
oparciu o równanie Suhterlanda-Einsteina:
DM = k T / 4 π ηHg rM
gdzie k – stała Boltzmana, T – temperatura bezwzględna, ηHg – lepkość Hg
(rozcieńczonego amalgamatu), rM - promień atomu.
Jeśli atom metalu jest solwatowany przez atomy Hg, a zdarza się to w
przypadkach, gdy między M i Hg tworzy się trwałe połączenie międzymetaliczne (grupy
podziału amalgamatów pierwsza i czwarta), wtedy współczynnik DM jest wyraźnie niższy
niż przewidzianego powyższym równaniem. Oszacować go wtedy trudno i trzeba
wyznaczyć eksperymentalnie różnymi metodami fizykochemicznymi z użyciem
izotopów promieniotwórczych lub pomiaru sem. Można ocywiście zastosować techniki z
przepływem prądu, gdy w opisujących je równaniach występuje DM.
Hg może dyfundować w głąb stałego metalu. Proces taki jest znacznie wolniejszy.
DHg wynosi 10-8-10-14 cm2/s i silnie zależy od struktury krystalograficznej metalu,
rozpuszczalności w nim Hg i tendencji do tworzenia połączeń międzymetalicznych
danego metalu z Hg.
Wyznaczanie rozpuszczalności metali w Hg
Do wyznaczania rozpuszczalności metali w Hg można wykorzystywać niemal
wszystkie wymienione metody badań fizykochemicznych amalgamatów. W każdym
przypadku nagłe zmiany mierzonego parametru wraz ze zmianą składu mogą świadczyć
o odkładaniu się fazy stałej z roztworu przesyconego. Można tu wykorzystać: analizę
termiczną, pomiary podatności magnetycznej, przewodnictwa, gęstości właściwej,
prężności par, lepkości, napięcia powierzchniowego, kinetyki rozkładu metodą

43
chemiczną lub elektrochemiczną, współczynnika dyfuzji, transmisji wiązki neutronów,
miareczkowania kalorymetrycznego. Największe znaczenie praktyczne mają pomiary
sem elektrod amalgamatowych ze względu na to, że metody oparte na przepływie prądu
(jak chronowoltamperometria amalgamatowa) nie dostarczają wartości rónowagowych
rozpuszczalności, a o takie nam chodzi, lecz lekko zaburzone czy to kinetyką
rozpuszczania, czy krystalizacji.
Aby wyznaczyć rozpuszczalnośc danego metalu w rtęci, należy sporządzić szereg
amalgamatów badanego metalu o stężeniach zmieniających się od 10-5 mol/dm3 do
wartości znacznie przekraczajacej poziom rozpuszczalności. Po odczycie potencjału
równowagowego amalgamatu należy sporządzić wykres w układzie współrzędnych:
potencjał – logarytm stężenia metalu i z punktu przecięcia odcinków liniowych
ekstrapolowanych od najniższych i największych stężeń określić punkt przełomu
zależności odpowiadający stężeniu nasyconego amalgamatu.
Rys. 2 Zależność potencjału elektrody Mn(II)/Mn(Hg) (względem nasyconej elektrody
kalomelowej) od logarytmu stężenia Mn w amalgamacie. Roztwór 1.00x10-2 mol/dm3
MnCl2 w 0.1 mol/dm3 KCl.

44
Na rys. 2 pokazano przykładowo zależność potencjału elektrody od logarytmu
stężenia manganu w amalgamacie. W zakresie stężeń manganu 1x10-4 – 8.7x10-3
mol/dm3 potencjał elektrody zmienia się wraz ze stężeniem zgodnie z równaniem
Nernsta. Powyżej 8.7x10-3 mol/dm3 potencjał elektrody praktycznie nie zależy od
zawartości metalu wprowadzonego do Hg. Wtedy bowiem przekroczyliśmy granicę
rozpuszczalności, stężęnie manganu pozostaje stałe, a jego nadmiar wytrąca się w postaci
kryształów równowagowej fazy stałej Hg5Mn2.
Jak to zostało wcześniej wspomniane, metoda ta ma zastosowanie do badań metali
bardziej elektroaktywnych niż Hg, tworzących odwracalne układy elektrochemiczne i
gdy stężenie metalu (rozpuszczalnośc) jest wyższe od 10-5 mol/dm3. Poniżej tego stężenia
(w specjalnie nieoczyszczanych roztworach) znajdują się inne potencjałotwórcze
pierwiastki i ich jony na porównywalnym poziomie, a potencjał określa wtedy nie tylko
układ Mz+/M(Hg), ale i te zanieczyszczenia. Atomy i jony bardziej aktywne ulegają
wymianie na mniej aktywne na drodze cementacji i potencjał układu praktycznie płynie.
Amalgamaty mogą także ulegać korozji pod wpływem tlenu i jonów wodorowych, toteż
należy dbać aby stężenie tych form było jak najniższe. Równowaga w układzie powinna
ustalać się szybko zarówno ze względu na proces elektrodowy jak i kinetykę
krystalizacji, która zależy od stopnia przesycenia amalgamatu. Szereg amalgamatów
krystalizuje wolno i stan wielokrotnego przesycenia potrafi się utrzymywać bardzo długo
(np. Fe, Co).
6. Literatura
A. Bielański, Chemia Nieorganiczna, PWN, Warszawa, 1998, rozdz. „Cynkowce”.
J.D. Lee, Zwięzła Chemia Nieorganiczna, PWN, Warszawa, 1994, rozdz.
„Cynkowce”.
F.A. Cotton, G. Wilkinson, P.L. Gaus, Chemia Nieorganiczna, PWN, Warszawa,
1995, rozdz. „Zn, Cd, Hg”.
A. Cygański, Podstawy Metod Elektroanalitycznych, WNT, Warszawa, 1999,
rozdz. „Potencjometria i Kulometria”.
C. Gumiński, Intermetallic Compounds, J.H. Westbrook & R.L. Fleischer, Eds.,
Wiley, Chichester, 2002, rozdz. „Amalgams” (po angielsku).

45
C. Gumiński, „Podstawy teoretyczne”, w tym skrypcie poniżej.

46
Ćwiczenie 4
Elektroosadzanie stopowych powłok ochronnych.
dr hab. Mikołaj Donten, e-mail: [email protected] mgr Tomasz Rapecki,
sala 149-2
2 Wymagania:
Zagadnienia dotyczące elektrolizy i podstaw kinetyki reakcji elektrodowych:
- potencjał standardowy i formalny układu red-ox
- pojęcie odwracalności i nieodwracalności reakcji elektrodowych
- zjawisko elektrolizy w roztworach wodnych
- I i II prawo elektrolizy Faraday'a
Podstawowe zagadnienia dotyczące procesów elektroosadzania powłok metali i
stopów:
- skład kąpieli galwanicznych do prądowego wytwarzania powłok (skład
podstawowy i dodatki)
Skaningowy mikroskop elektronowy:
- zasada działania (pobieżnie)
Podstawowe zagadnienia dotyczące rentgenowskiej analizy jakościowej i
ilościowej:
- powstawanie promieniowania rentgenowskiego (lampa rentgenowska), widmo ciągle
(niechrakterystyczne) i widmo charakterystyczne
- Podstawowe zagadnienia dotyczace rentgenowskiej analizy strukturalnej:
- dyfrakcja promieniowania rentgenowskiego
- rownanie Bragga
2 Cel ćwiczenia:
Celem ćwiczenia jest zapoznanie studentów z procesami wytwarzania i metodami
badania ochronnych powłok galwanicznych.

47
3 Wstęp:
Stopowe powłoki amorficzne zawierające wolfram i metale z grupy żelazowców
mogą być z powodzeniem elektroosadzane z cytrynianowych kąpieli galwanicznych.
Uzyskiwane ta metoda pokrycia charakteryzują się dużą odpornością mechaniczną i
korozyjna. Typowa kąpielą z której łatwo jest otrzymać powłokę galwaniczną dobrej
jakości jest kąpiel zawierająca w swoim składzie wolframian sodu (80g/L), Ni, Co lub Fe
(2.5g/L), cytrynian dwusodowy lub dwuamonowy (60g/L) borany i fosforany w ilości 5-
10g/L amoniak dodany do uzyskania pH 8.5 i dodatki. Dodatkami najczęściej są środki
zwilżające jak FSK lub rokafenol i wybłyszczacze: butyn 2 diol 1,4, czasami także
siarczan dodecylu. Elektroosadzanie stopów odbywa się w podwyższonej temperaturze
65oC.
Powłoki stopowe uzyskiwane z kąpieli galwanicznych o składzie przedstawionym
powyżej wyróżniają się specyficzną, pseudoamorficzną strukturą, są twarde i zawierają
duże ilości wolframu, 20 do 35 %at. w zależności od rodzaju żelazowca tworzącego stop.
Metodami stałoprądowymi nie udaje się uzyskać stopów o strukturze szklistej
zawierającej większe ilości wolframu.
4 Wykonanie ćwiczenia
Grupa studentów dzieli się na dwuosobowe zespoły a zadaniem kazdego zesołu
jest osadzenie powłoki stopowej. Zespoły osadzać bedą ten sam stop ale procesy
prowadzone bedą w róznych warunkach. Ostatecznym celem będzie ustalenie
parametrów osadzanej powłoki w zależności od zastosowanych warunków osadzania.
Przed przystąpieniem do wykonania cwiczenia należy sprawdzić stan roztworu
kąpieli galwanicznej:
- Skontrolować i ewentualnie uzupełnić poziom wody destylowanej w łaźni
wodnej. Ustawić termostat na 65 oC i włączyć łaźnię wodna.
- Roztwór kąpieli galwanicznej przeznaczonej do osadzania stopu niklowo-
wolframowego należy przesączyć i doprowadzić do pH ok. 8.5 dodając, w zależności od
potrzeby, NH3 aq lub roztworu kwasu siarkowego.
- Po konsultacji z prowadzącym ćwiczenie asystentem uzupełnić zawartość
dodatków i niklu w roztworze.

48
- Roztwór kąpieli galwanicznej przelać do naczynia elektrolitycznego, przestrzenie
anodowe napełnić roztworem siarczanu sodu. Naczynie umieścić w łaźni wodnej.
Dwuosobowe zespoły przystępują do wykonania niezależnych eksperymentów:
Katodę z mosiądzowanej folii stalowej o szerokości ok 5mm i całkowitej
powierzchni ok 4cm2 umyć w wodzie z detergentem, odtłuścić w acetonie, zanurzyć na
kilkanaście sekund w roztworze kwasu siarkowego lub solnego, opłukać woda, wysuszyć
i zważyć.
Po zważeniu katodę umieścić w uchwycie, aktywować przez 15 sekundowe
zanurzenie w 10% roztworze kwasu siarkowego, opłukać w wodzie i umieścić w
naczyniu.
Przez ok. 20 minut prowadzić elektrolizę przy gęstości prądu ustaloną z
prowadzącym ćwiczenie asystentem.
Po zakończeniu procesu osadzania odłączyć źródło prądu (galwanostat) wyjąc z
naczynia elektrody, pokrytą warstwa stopu Ni-W katodę dokładnie opłukać wodą
destylowaną, wysuszyć i zważyć.
Powierzchnię wysuszonej katody obejrzeć pod mikroskopem optycznym
zwracając uwagę na strukturę i jednorodność pokrycia. Sczególną uwagę należy zwrócic
na wygląd krawędzi pokrywanej blachy.
Przy użyciu skaningowego mikroskopu elektronowego wykonać zdjęcia
powierzchni powłoki galwanicznej, oszacować jej grubość i wykonać analizę składu
osadzonej powłoki
6 Opis wyników ćwiczenia
Opisać przebieg wykonanych czynności z zaznaczeniem celowości działania (co
zostało zrobione i po co zostało zrobione).
Na podstawie zdjęć powierzchni i wyników analizy elementarnej ustosunkować się
do zwartości, gładkości i jednorodności uzyskanej powłoki.
Przedstawić wyniki pomiarów (przyrost masy katody, wyniki analizy elementarnej
powłoki i wyniki pomiaru grubośći warstwy)
Określić szybkość narastania powłoki (miligramy/cm2/godz.)

49
Obliczyć średnia grubość powłoki w mikrometrach. Do obliczeń przyjąć gęstość
stopu jako średnia ważoną gęstości składników (niklu i wolframu). Wagami są ułamki
molowe składników.
Wyznaczyć szybkość narastania powłoki w mikrometrach/godz.
Na podstawie pomiaru masy i składu osadzonego stopu oraz ładunku elektrolizy
obliczyć wydajność prądową procesu osadzania stopu. Do obliczeń przyjąć, że redukcja
niklu przebiega dwuelektronowo, a redukcja wolframu szescioelektronowo.
Określić wpływ kontrolowanego w eksperymencie parametru osadzania stopu na
skład i właściwosci powłoki.
Gotowy opis dostarczyć prowadzącemu ćwiczenie w trakcie następnej pracowni z
chemii nieorganicznej.
7 Zalecana literatura
L.Jones P.Atkins „Chemia ogólna”( rozdział 18 Elektrochemia), WNT, Warszawa
2004
P.Atkins „Chemia fizyczna” (Procesy elektrochemiczne- Elektroliza,
Charakterystyka ogniwa pracującego) PWN, Warszwa 2003
„Poradnik galwanotechnika” praca zbiorowa WNT, Warszawa
Część 1 rozdział 1 „podstawowe pojęcia galwanotechniczne
B.D. Cutley "Podstawy dyfrakcji promieni rentgenowskich" PWN, Warszawa
1964
M.L.Donten, M.Donten Skaningowy mikroskop elektronowy Analityka 2002(1)
M.L.Donten, M.Donten Mikrosonda elektronowa EDS Analityka 2001(4)

50
Ćwiczenie 5
Badanie liczby niesparowanych elektronów w kompleksach metali
przejściowych metodą NMR
Dr Włodzimierz Makulski, sala 149-10, e-mail: [email protected]
1 Cel:
wyznaczanie parametrów magnetycznych substancji (podatność magnetyczna)
oraz wnioskowanie na tej podstawie o strukturze substancji
Wstęp
Jedną z podstawowych cech decydujących o strukturze kompleksu metalu
przejściowego jest rozmieszczenie elektronów na poziomach energetycznych w atomie
centralnym. Wywiera ono bezpośredni wpływ na własności kompleksu. Spośród różnych
metod stosowanych do wyznaczenia struktury elektronowej szczególnie przydatne są
metody magnetyczne, umożliwiające wyznaczenie momentu magnetycznego cząsteczki,
a na tej podstawie – liczby niesparowanych elektronów.
Zachowanie substancji w polu magnetycznym
Jeżeli dowolną substancję umieścimy w polu magnetycznym, to natężenie pola
magnetycznego wewnątrz tej substancji (indukcja magnetyczna B) różnić się będzie od
natężenia pola magnetycznego w próżni (H). Różnica pomiędzy B a H związana jest z
namagnesowaniem I, czyli momentem magnetycznym przypadającym na jednostkę
objętości substancji
B = µ0(H + I) (1)
Jeśli wielkość I jest ujemna, indukcja magnetyczna jest mniejsza od natężenia pola
magnetycznego i substancję taką nazywamy diamagnetykiem. Jeśli B > H to substancja
nazywana jest paramagnetykiem.
Równanie (1) można też przedstawić w równoważnej postaci
B = µ0(1+χ)H (2)

51
gdzie µ0 jest przenikalnością magnetyczną próżni, zaś χ - tzw. objętościową podatnością
magnetyczną substancji (wielkość bezwymiarowa).
W praktyce chemicznej wygodnie jest posługiwać się molową podatnością
magnetyczną χM określoną równaniem
H
IMMM
ρχ
ρχ == (m3/mol) (3)
czyli wielkością określającą namagnesowanie 1 mola substancji znajdującego się w
jednostkowej objętości, wywołanego polem o jednostkowym natężeniu.
Wielkość podatności magnetycznej zależy od dwóch czynników: polaryzowalności
magnetycznej αM i momenu magnetycznego µM. Teoretyczna zależność pomiędzy tymi
wielkościami przedstawiona jest równaniem
µ
+α=χkT
N MMAM 3
2
(4)
gdzie NA jest liczbą Avogadro, zaś k – stałą Boltzmanna. Polaryzowalność wiąże się
blisko z powstawaniem indukowanego momentu magnetycznego, skierowanego
przeciwnie do zewnętrznego pola magnetycznego (αM < 0) i odpowiedzialnego za
osłabienie pola magnetycznego wewnątrz substancji. Indukowany moment magnetyczny,
występujący w każdej substancji, jest odpowiedzialny za efekty diamagnetyczne. W
przypadku gdy cząsteczki substancji nie posiadają trwałego momentu magnetycznego µM
(np. większość związków organicznych), molowa przenikalnośc magnetyczna jest
zdeterminowana przez polaryzowalność magnetyczną αM i substancja jest
diamagnetykiem.
W odróżnieniu od indukowanego momentu magnetycznego, wyjaśnianego na
gruncie fizyki klasycznej jako efektu orbitalnego ruchu elektronów, trwałe momenty
magnetyczne cząsteczek lub jonów wywołane są głównie spinami elektronów. Spinowy
moment magnetyczny opisany jest równaniem
)1( +µ=µ SSg BeM (5)
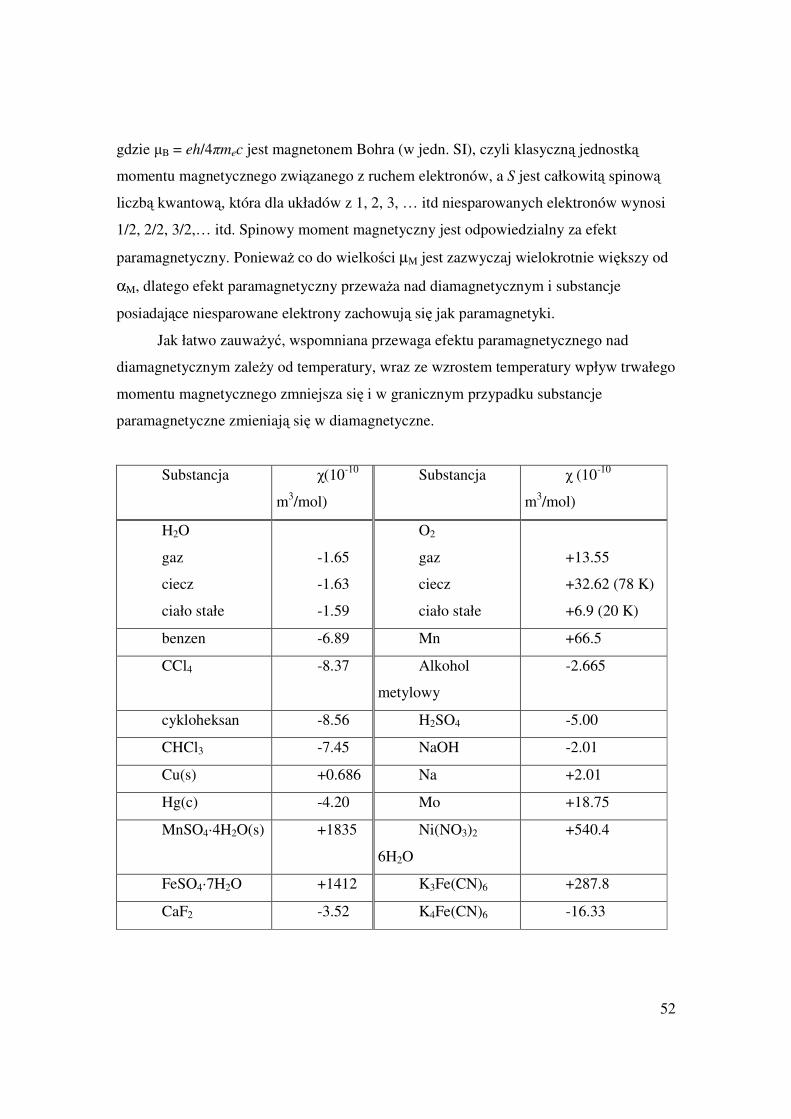
52
gdzie µB = eh/4πmec jest magnetonem Bohra (w jedn. SI), czyli klasyczną jednostką
momentu magnetycznego związanego z ruchem elektronów, a S jest całkowitą spinową
liczbą kwantową, która dla układów z 1, 2, 3, … itd niesparowanych elektronów wynosi
1/2, 2/2, 3/2,… itd. Spinowy moment magnetyczny jest odpowiedzialny za efekt
paramagnetyczny. Ponieważ co do wielkości µM jest zazwyczaj wielokrotnie większy od
αM, dlatego efekt paramagnetyczny przeważa nad diamagnetycznym i substancje
posiadające niesparowane elektrony zachowują się jak paramagnetyki.
Jak łatwo zauważyć, wspomniana przewaga efektu paramagnetycznego nad
diamagnetycznym zależy od temperatury, wraz ze wzrostem temperatury wpływ trwałego
momentu magnetycznego zmniejsza się i w granicznym przypadku substancje
paramagnetyczne zmieniają się w diamagnetyczne.
Substancja χ(10-10
m3/mol)
Substancja χ (10-10
m3/mol)
H2O
gaz
ciecz
ciało stałe
-1.65
-1.63
-1.59
O2
gaz
ciecz
ciało stałe
+13.55
+32.62 (78 K)
+6.9 (20 K)
benzen -6.89 Mn +66.5
CCl4 -8.37 Alkohol
metylowy
-2.665
cykloheksan -8.56 H2SO4 -5.00
CHCl3 -7.45 NaOH -2.01
Cu(s) +0.686 Na +2.01
Hg(c) -4.20 Mo +18.75
MnSO4·4H2O(s) +1835 Ni(NO3)2
6H2O
+540.4
FeSO4·7H2O +1412 K3Fe(CN)6 +287.8
CaF2 -3.52 K4Fe(CN)6 -16.33

53
Zakładając, że udział polaryzowalności magnetycznej jest pomijalny, molowa
podatność magnetyczna substancji paramagnetycznej może być powiązana z całkowitym
spinem elektronowym cząsteczki za pomocą wyrażenia
)1(3
4)1(
3
222
+µ
≈+µ
=χ SSkT
NSS
kT
Ng ABABeM (6)
Teoretyczne wartości podatności magnetycznej wynikające z liczby
niesparowanych elektronów, dla temperatury 25°C, podane są w poniższej tabeli.
Liczba
niesparowanych
elektronów
S
µM
(J/T)
χM (10-
10m3/mol)
w temp. 25oC
1 1/2 1.73µB 158
2 2/2 2.83µB 422
3 3/2 3.87µB 790
4 4/2 4.90µB 1269
5 5/2 5.92µB 1847
6 6/2 6.93µB 2162
7 7/2 7.94µB 2476
Wyznaczanie podatności magnetycznej substancji
Klasyczną metodą wyznaczania podatności magnetycznej jest waga Gouy'a, gdzie
próbka zawieszona na szalce wagi umieszczona jest w silnym polu magnetycznym. W
przypadku diamagnetyków próbka jest wypychana z pola i pozornie traci na wadze.
Substancje paramagnetyczne wciągane są wgłąb pola, przez co pozornie przybierają na
wadze. Wielkość tego efektu zależy od natężenia pola magnetycznego H.
Wadą powyższej metody jest konieczność stosowania bardzo silnych magnesów
oraz pracochłonność wykonania. W niniejszym ćwiczeniu zastosujemy inną metodę,
opartą na pomiarze NMR. Ponieważ pole magnetyczne działające na jądro
"magnetyczne" zależy od przenikalności magnetycznej próbki, obserwowana częstość

54
rezonansowa takiego jądra – a więc położenie sygnału w widmie NMR – zmieniać się
będzie ze zmianami w podatności magnetycznej otoczenia. Porównując częstości
rezonansowe jądra zmierzone w próbkach o różnej podatności magnetycznej można
wyznaczyć stosunek tych podatności. Metoda ta została zaproponowana w 1959 r. przez
D.F. Evansa i odtąd wiąże się z jego nazwiskiem.
Różnica częstości rezonansowych obserwowana dla tego samego jądra w próbkach
o różnej podatności magnetycznej wynosi
( )00
0
31)(
χχν
νν−−=
−i
i (7)
gdzie χi i χ0 są objętościowymi podatnościami magnetycznymi, zaś współczynnik 1/3
ma charakter geometryczny (1/3 gdy kierunek pola równoległy do kierunku wirowania,
1/6 – gdy prostopadły; w spektrometrach a magnesem nadprzewodzącym kierunek jest
równoległy).
Wyznaczenie podatności magnetycznej i liczby niesparowanych elektronów
W naszym przypadku eksperyment polega na zmierzeniu i porównaniu częstości
rezonansowych protonów substancji wskaźnikowej – t-butanolu – w wodzie i w wodnym
roztworze związku kompleksowego. Z różnicy w podatności magnetycznej można
wyznaczyć molową podatnośćmagnetyczną związku kompleksowego i w konsekwencji
liczbę niesparowanych elektronów przypadającą na cząsteczkę tego związku.
Wykonanie ćwiczenia
Ćwiczenie wykonujemy w grupach dwuosobowych. Każda grupa realizuje przepis
analityczny oraz pomiar NMR. Następuje podział na trzy grupy ćwiczeniowe: a), b) i c).
W kolbach miarowych o poj. 5 ml rozpuścić w wodzie po ok. 0.25 mmola uwodnionego
siarczanu żelaza (II)(grupa a), heksacyjanożelazianu(II) potasu (grupa b) oraz
żelazicyjanku potasowego(II) (grupa c). Następnie odpipetować do kolbek o poj. 1 ml po
0.5 ml przygotowanych roztworów soli, dodać 30 µl t-butanolu i dopełnić do kreski H2O.
Do czwartej kolbki o poj. 1 ml dodać pipetą 0.5 ml wody destylowanej, 30 µl t-
butanolu i dopełnić do kreski przy pomocy D2O. Napenić tak przygotowanym roztworem
kapilarę i zatopić jej koniec.

55
Do trzech probówek NMR dodać po 0.5 ml przygotowanych roztworów soli żelaza
w H2O. Roztwór w kapilarze służy jako wzorzec do pomiaru dla wszystkich trzech
podgrup a), b) i c).
Ustawić temperaturę sondy na 25oC, wybrać standardowe parametry pomiarowe
dla jądra 1H, wyregulować jednorodność pola magnetycznego i zarejestrować widmo
wzorcowe z kapilary oraz 3 widma: roztworów a), b) i c) w probówkach z umieszczoną
wewnątrz kapilarą.
Zidentyfikować wszystkie sygnały widoczne w widmach NMR. Zmierzyć różnicę
częstotliwości pomiędzy sygnałami t-butanolu w probówce i w kapilarze. Na podstawie
tej różnicy wyznaczyć molową podatność magnetyczną FeSO4⋅7H2O, K4[Fe(CN)6] oraz
K3[Fe(CN)6a następnie liczbę niesparowanych elektronów w każdym z tych
kompleksów.
W trakcie ćwiczenia poszczególne grupy przygotowują informacje w oparciu o
literaturę przedmiotu:
a) pochodzenie barwy zielono-niebieskiej roztworu wodnego soli FeSO4⋅7H2O
oraz charakterystyka chemiczna witriolu żelaza.
b) konfiguracja elektronowa jonu Fe(CN)6-4.
c) pochodzenie zabarwienia żelazicyjanku, konfiguracja elektronowa jonu
Fe(CN)6-3.
W opracowywaniu ćwiczenia korzystamy z Internetu w zdobyciu informacji
spektroskopowych o badanych solach kompleksowych.
W trakcie drugiej pracowni następuje: poprawa kolokwiów wejściowych oraz
wspólne omówienie sposobu wykonania sprawozdania przez poszczególnych studentów.
Sprawozdanie
W sprawozdaniu zamieścić: wstęp teoretyczny tyczący się spektroskopii związków
kompleksowych ze szczególnym uwzględnieniem metody NMR, krótki opis
przygotowania próbki, oraz kompletne obliczenia molowej podatności magnetycznej oraz

56
liczby niesparowanych elektronów w atomie centralnym kompleksów. W opisie należy
uwzględnić dane literaturowe dotyczące własności spektroskopowych badanych
związków chemicznych.
Literatura:
P.B.Dorain, ”Symetria w chemii nieorganicznej”, PWN Warszawa, 1968.
A.Bartecki, ”Spektroskopia elektronowa związków nieorganicznych i
kompleksowych”, PWN, Warszawa, 1971.
P.W. Atkins, "Chemia Fizyczna", PWN Warszawa, 2001
G.M. Barrow, "Chemia Fizyczna", PWN, Warszawa, 1973
S. Braun, H.-O. Kalinowski, S. Berger, "100 and More basic NMR Experiments",
Wiley-VCH, New York, 1998, rozdz. 8.8
D.F. Evans, J. Chem. Soc. 1959, 2003-2005
J.L. Deutsch, S.M. Poling, J. Chem. Educ. 46 (1969) 167-168
Uwaga: pozycje 4-7 używają systemu jednostek CGS!

57
Ćwiczenie 6.
Badanie szybkości reakcji podstawienia w kompleksach oktaedrycznych.
dr Jadwiga Stroka, e-mail [email protected], sala 349
1. Wymagania
Budowa elektronowa atomów i jonów metali przejściowych. Właściwości metali
przejściowych i właściwości ligandów tłumaczące ich zdolność do tworzenia związków
kompleksowych. Struktury związków koordynacyjnych. Typy ligandów. Izomeria
związków koordynacyjnych.
Teoria pola krystalicznego i teoria orbitali molekularnych. Właściwości
magnetyczne kompleksów. Strukturalne i termodynamiczne konsekwencje
rozszczepienia orbitali d.
Praktyczna interpretacja widm. Termy spektroskopowe. Przejścia d-d.
Kinetyka reakcji podstawienia ligandów w kompleksach. Kompleksy inertne i
labilne. Mechanizmy reakcji podstawienia, kryteria ich występowania. Rodzaj
mechanizmu, a symetria kompleksu. Reakcje podstawienia w kompleksach
oktaedrycznych i kompleksach kwadratowych. Metody stosowane do badania reakcji
związków kompleksowych.
2. Cel ćwiczenia
Celem ćwiczenia jest badanie reakcji uwodnienia kompleksów: trans-[Co(NH3)4Cl2]+,
[Co(NH3)5Cl]2+ i trans-[Co(en)2Cl2]+
oraz synteza wybranego kompleksu.
3. Wstęp
Dużą rolę w klasycznej chemii koordynacyjne odgrywają kompleksy jonów metali
z nieorganicznymi ligandami jednokleszczowymi takimi jak: Cl−, Br−, I−, NH3 , H2O,
OH−, SO32−, pirydyna, SCN−,CN− i NO2
− oraz dwukleszczowymi takimi jak
etylenodiamina, anion kwasu szczawiowego lub węglanowego, bipirydyl i 1,10-
fenatrolina.
Często stosuje się dogodny podział kompleksów na dwie klasy: kompleksy labilne
i kompleksy inertne. Pierwsza klasa obejmuje większość kompleksów metali grup

58
głównych i wiele kompleksów metali przejściowych. W kompleksach drugiej klasy np. w
kompleksach Co(III) i Cr(III) wymiana ligandów przebiega bardzo powoli. Kompleksy
[Co(NH3)6]3+ i [Cr(NH3)6]
3+ są bardzo stabilne w wodnych kwaśnych roztworach w
przeciwieństwie do kompleksu [Ni(NH3)6]2+, który w tych samych warunkach ulega
szybkiej reakcji podstawienia ligandu cząsteczkami wody. W wyniku tej reakcji powstaje
kompleks [Ni(H2O)6]2+. Badania szybkości wymiany ligandów stworzyły podstawy
chemii metali przejściowych. Szybkość wymiany ligandów można wytłumaczyć na
gruncie teorii pola krystalicznego lub teorii orbitali molekularnych.
Rozważmy rekcje uwodnienia kompleksów oktaedrycznych Co(III).
[MeL5X]n+ + H2O → [MeL5H2O]m+ + X (1)
Kompleksy występujące w roztworze wodnym ulegają reakcjom hydrolizy, w których
wymieniany ligand zostaje zastąpiony w pierwszej sferze koordynacyjnej cząsteczką
wody. Ponieważ stężenie [H2O] w roztworze wodnym jest praktycznie stałe, hydrolizę
kwasową można opisać równaniem:
v=kobs*[kompleksu] (2)
Znajomość wyrażenia (2) nie pozwala na wyciagnięcie żadnych wniosków
odnośnie wpływu stężenia wody na kinetykę reakcji hydrolizy.
W wyniku przebadania reakcji hydrolizy szeregu kompleksów oktaedrycznych
stwierdzono, że reakcja ta przebiega według mechanizmu SN1. Potwierdzają to badania
wpływu grupy wymienianej, efektów przestrzennych i efektów związanych z ładunkiem
na badania kinetyczne. W Tabeli 1 przedstawiono przykładowe dane stałych szybkości
hydrolizy kompleksów Co(III) w środowisku kwaśnym:
[Co(AA)2Cl2]+ + H2O → [Co(AA)2H2OCl]2+ + Cl- (3)
[Co(AA)2H2OCl]2+ + H2O →[Co(AA)2(H2O)2]3+ + Cl- (4)
Reakcja (4) jest około 100 razy wolniejsza od reakcji (3), w związku z tym w warunkach
eksperymentu będziemy wyznaczać szybkość tej reakcji (3)
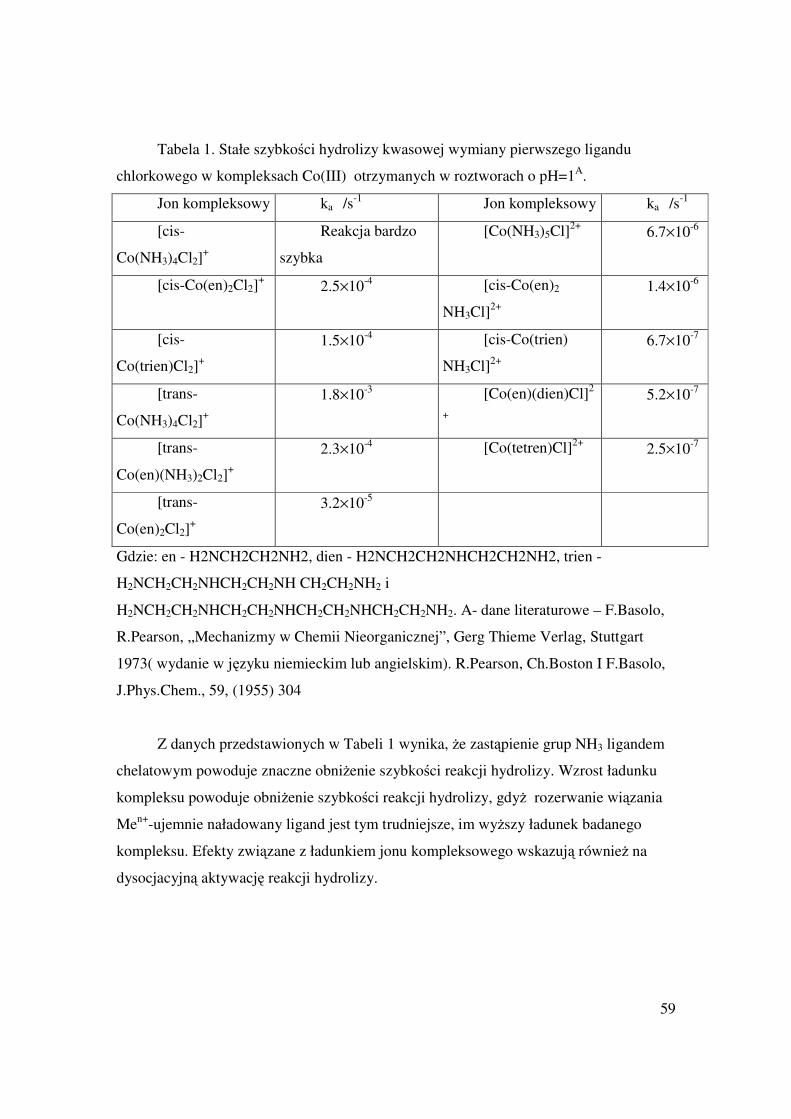
59
Tabela 1. Stałe szybkości hydrolizy kwasowej wymiany pierwszego ligandu
chlorkowego w kompleksach Co(III) otrzymanych w roztworach o pH=1A.
Jon kompleksowy ka /s-1 Jon kompleksowy ka /s
-1
[cis-
Co(NH3)4Cl2]+
Reakcja bardzo
szybka
[Co(NH3)5Cl]2+ 6.7×10-6
[cis-Co(en)2Cl2]+ 2.5×10-4 [cis-Co(en)2
NH3Cl]2+
1.4×10-6
[cis-
Co(trien)Cl2]+
1.5×10-4 [cis-Co(trien)
NH3Cl]2+
6.7×10-7
[trans-
Co(NH3)4Cl2]+
1.8×10-3 [Co(en)(dien)Cl]2
+
5.2×10-7
[trans-
Co(en)(NH3)2Cl2]+
2.3×10-4 [Co(tetren)Cl]2+ 2.5×10-7
[trans-
Co(en)2Cl2]+
3.2×10-5
Gdzie: en - H2NCH2CH2NH2, dien - H2NCH2CH2NHCH2CH2NH2, trien -
H2NCH2CH2NHCH2CH2NH CH2CH2NH2 i
H2NCH2CH2NHCH2CH2NHCH2CH2NHCH2CH2NH2. A- dane literaturowe – F.Basolo,
R.Pearson, „Mechanizmy w Chemii Nieorganicznej”, Gerg Thieme Verlag, Stuttgart
1973( wydanie w języku niemieckim lub angielskim). R.Pearson, Ch.Boston I F.Basolo,
J.Phys.Chem., 59, (1955) 304
Z danych przedstawionych w Tabeli 1 wynika, że zastąpienie grup NH3 ligandem
chelatowym powoduje znaczne obniżenie szybkości reakcji hydrolizy. Wzrost ładunku
kompleksu powoduje obniżenie szybkości reakcji hydrolizy, gdyż rozerwanie wiązania
Men+-ujemnie naładowany ligand jest tym trudniejsze, im wyższy ładunek badanego
kompleksu. Efekty związane z ładunkiem jonu kompleksowego wskazują również na
dysocjacyjną aktywację reakcji hydrolizy.

60
4. Wykonanie ćwiczenia
Jako przykładowe reakcje uwodnienia kompleksu wybrano reakcję podstawienia
jonu chlorkowego cząsteczką wodą w kompleksach: trans-[Co(NH3)4Cl2]Cl, trans-
[Co(en)2Cl2]Cl lub [Co(NH3)5Cl]Cl2, reakcje te będziemy badać metodą
spektrofotometryczną. W ramach ćwiczenia należy zbadać dwa z zaproponowanych
kompleksów.
4.a Wyznaczenie stałej szybkości uwodnienia kompleksu trans-[Co(NH3)4Cl2]Cl.
Dla kompleksu trans-[Co(NH3)4Cl2]Cl zachodzi następująca reakcja:
trans-[Co(NH3)4Cl2]+ + H2O → trans-[Co(NH3)4ClH2O]+2 + Cl- (5)
W trzech zlewkach o obj. 25ml umieszczamy po około 50 mg kompleksu trans-
[Co(NH3)4Cl2]Cl, każdą próbkę rozpuszczamy w wodnym roztworze 0.1M HNO3
dopiero przed samym pomiarem.
Otrzymanym roztworem napełniamy kuwetę.
I. Rejestrujemy widma w zakresie widzialnym od 400 do 800 nm w funkcji czasu.
Stosując odpowiedni program aparaturowy rejestrujemy 15 widm co 150 sekund.
Następnie wyznaczmy długość fali odpowiadającą maksimum adsorpcji produktu i
substratu, a otrzymane dane drukujemy.
II. Próbkę z drugiej zlewki analizujemy przy długości fali odpowiadającej
maksimum absorpcji badanego substratu trans-[Co(NH3)4Cl2]Cl mierząc absorpcję co 6
sekund przez 30 minut. W tym celu stosujemy program pomiarowy KINETYKA.
Otrzymane dane zapisujemy na dyskietce oraz drukujemy w postaci numerycznej i
wykresu.
III. Próbkę z trzeciej zlewki analizujemy (jeżeli dysponujemy wystarczającą
ilością czasu) przy długości fali odpowiadającej maksimum absorpcji badanego produktu.
[Co(NH3)4ClH2O]Cl2 mierząc absorpcję co 6 sekund przez 30-40 minut. W tym celu
stosujemy program pomiarowy KINETYKA. Otrzymane dane zapisujemy na dyskietce
oraz drukujemy w postaci numerycznej i wykresu.
Ponieważ w warunkach eksperymentu stężenie wody nie zmienia się, to szybkość
tej reakcji można opisać równaniem (2)

61
Stałą szybkości reakcji pseudo-pierwszego rzędu można otrzymać z nachylenia
prostej ln(A-A∞)=f(t), gdzie A i A∞ jest odpowiednio absorpcją odpowiadającą
maksimum długości fali substratu lub produktu w danym czasie i po całkowitym zajściu
reakcji. A∞ można wyznaczyć z zależności A=(1 / t), obliczenia A∞ należy wykonać z
kilku ostatnich punktów pomiarowych.
4.b Wyznaczenie stałej szybkości uwodnienia kompleksu trans-[Co(en)2Cl2]+.
W tym przypadku zachodząca reakcja jest opisana równaniem:
trans-[Co(en)2Cl2]+ + H2O → trans-[Co(en)2ClH2O]+2 + Cl- (6)
Ze względu na niższą szybkość reakcji procedura pomiarowa będzie następująca.
W zlewce o obj. 25ml umieszczamy po około 50mg kompleksu [Co(en)2Cl2]Cl, próbkę
rozpuszczamy w wodnym roztworze 0.1M HNO3. Otrzymanym roztworem napełniamy
kuwetę.
I. Stosując odpowiedni program rejestrujemy widma w zakresie widzialnym od
400 do 800nm. Należy zarejestrować około 6-8 widm co 15-20 minut, dla których
wyznaczmy absorpcję dla długości fali odpowiadającej maksimum absorpcji substratu.
Otrzymane widma zapisujemy na dysku. Po zakończenie rejestracji należy nanieść
wszystkie widma na jeden wykres, a otrzymane dane wydrukować.
Dodatkowo z widm można odczytywać absorpcję odpowiadającą Λ=530nm
W tym przypadku można również wyznaczyć szybkość reakcji z równania (2).
Podobnie jak uprzednio stałą szybkości reakcji pseudo-pierwszego rzędu można
otrzymać z nachylenia prostej ln(A-A∞)=f(t). Ponieważ dla tego kompleksu reakcja
akwatacji jest wolna w związku z tym przyjmujemy dla substartu A=0 lub po upływie 24
godzin mierzymy widmo badanego kompleksu i wyznaczmy wartość A∞ dla długości fali
odpowiadającej maksimum absorpcji substratu.
4.c Wyznaczenie stałej szybkości uwodnienia kompleksu [Co(NH3)5Cl]Cl
Zachodząca reakcja akwatacji jest opisana równaniem:
[Co(NH3)5Cl]+2 + H2O → [Co(NH3)5H2O]+3+ Cl- (7)

62
Ze względu na małą szybkość reakcji (7) w temperaturze pokojowej, badania będą
prowadzone w temperaturze 60 oC.
W tym celu należy umieścić na 20 minut cztery kolby o pojemności 100ml w
termostacie nastawionym na 60 oC. Kolby te zawierają odpowiednio 0.05M, 0.1M, 0.3M
oraz 0.6M HNO3
(stężony kwas azotowy jest około 15.9M). Następnie do każdej kolby dodać tyle
związku [Co(NH3)5Cl]Cl, aby jego stężenie wynosiło 1.2x10-2M. Po rozpuszczeniu soli
należy umieścić kolby na 15 minut w termostacie, tak aby roztwór osiągnął temperaturę
60 oC.
Następnie z każdej kolby należy pobrać roztwór i zarejestrować widmo w zakresie
od 350 do 700nm. Należy wykonać 8 pomiarów dla każdej próbki co 15 minut.
W tym przypadku szybkość reakcji jest opisana równaniem:
v=k*[kompleksu]*[H+]n (8)
Jeżeli reakcja jest katalizowana przez jony wodorowe, które nie są zużywane w
trakcie reakcji, jej szybkość opisuje równanie:
v=kobs*[kompleksu]
Stałą szybkości reakcji pseudo-pierwszego rzędu można otrzymać z nachylenia
prostej ln(A-A∞)=f(t), gdzie A i A∞ jest odpowiednio absorpcją roztworu w danym czasie
i po całkowitym zajściu reakcji. A∞ można zmierzyć po dwu dniach lub obliczyć z
równania Lamberta-Berra wiedząc, że współczynnik absorpcji dla długości fali 550 nm
wynosi 21 cm-1 mol-1.
5.Opis ćwiczenia.
Opisz wykonaną syntezę, napisz zachodzące reakcje oraz oblicz wydajność.
Wyznacz stałe szybkości uwodnienia dwóch wybranych kompleksów. Podaj mechanizm
reakcji podstawienia. Wyjaśnij jakie czynniki wpływają na szybkość badanych reakcji
podstawienia. Zaproponuj inne metody badania takich reakcji.

63
6. Synteza kompleksu trans-[Co(NH3)4Cl2]Cl
Metoda I
Do 4 g [Co(NH3)4CO3]Cl dodać 25 cm3 lodowato zimnego stężonego kwasu
solnego. Utrzymywać przy tym temperaturę 0oC. Gdy ustanie wydzielanie się CO2 do
fioletowej zawiesiny dodać kroplami, ostrożnie, pod wyciągiem, 25 cm3 kwasu
siarkowego. Cały czas należy chłodzić mieszaninę w łaźni z lodem. Następnie
pozostawić mieszaninę na 2 - 3 dni w temperaturze pokojowej, aż osad przybierze czysto
zieloną barwę. Wtedy zdekantować ciecz znad osadu trans-[Co(NH3)4Cl2]HSO4, osad
przenieść na sączek za pomocą 20 % H2SO4 i przesączyć. Następnie przemyć otrzymany
produkt niewielką ilością tego samego kwasu a później alkoholem. Wydajność 5 g
Odpowiedni chlorek tworzy się, gdy zimny, nasycony roztwór wodorosiarczanu
wlać do nadmiaru zimnego, stężonego HCl lub do nasyconego NH4Cl.
Źródło: Schlessinger G.G., "Inorganic laboratory preparation" Chemical
Publishing Co., Inc.; New York 1962
Metoda II
Inną metodę syntezy kompleksu trans- [Co(NH3)4Cl2]Cl zaproponowano ostatnio
w literaturze, gdzie związek te syntetyzowano z kompleksu [Co(NH3)4CO3]NO3
(kompleks ten był otrzymany w trakcie Pracowni Chemii Nieorganicznej I ). Łaźnię
wodna podgrzać do temperatury 80 oC .
6 g [Co(NH3)4CO3]NO3 umieścić w zlewce na 250 ml i rozpuścić w 25ml wody.
Roztwór silnie mieszając podgrzewać przez 3 minuty w 50-60 oC na łaźni. Dodać 16.5
ml stężonego roztworu HCl, należy uważać, aby roztwór nie wykipiał. Tak
przygotowany roztwór, silnie mieszając, ogrzewać do 80 oC na łaźni przez 5 minut
(kontrola temperatury jest bardzo ważna). W trakcie reakcji powstaje ciemnozielony osad
kompleksu trans- [Co(NH3)4Cl2]Cl. Roztwór należy szybko ochłodzić do temperatury
pokojowej umieszczając zlewkę w naczyniu z lodem. Następnie odsączyć otrzymany
osad. Osad należy rozpuścić w małej ilości lodowatej wody, w celu rozpuszczenia form
cis i po odsączeniu otrzymamy czysty izomer trans. Kryształy trans- [Co(NH3)4Cl2]Cl

64
przemyć na sączku 10 ml lodowatego etanolu i pozostawić do wysuszenia w
temperaturze pokojowej. Wydajność od 45-55%
Źródło: L. L.Borer, H.W.Erdman , J.Chem.Educ., 71, 332, 1994
7a. Synteza kompleksu cis- i trans- [Co(en)2Cl2]Cl
4 CoCl2 + 8 en (=etylenodiamina) + 6 H2O + O2 --> 4 trans-
[Co(en)2(OH)H2O]Cl2
trans-[Co(en)2(OH)H2O]Cl2 + 2 HCl --> trans-[Co(en)2Cl2]Cl*HCl + 2 H2O
trans-[Co(en)2Cl2]Cl --> (ogrzewanie) --> cis-[Co(en)2Cl2]Cl
I. izomer trans-[Co(en)2Cl2]Cl
Do mieszaniny 540 cm3 wody i 60 g bezwodnej etylenodiaminy (en) dodać
roztwór 106 g CoCl2*6H2O w 500 cm3 wody. Czerwonawy roztwór przenieść do 2-
litrowej kolby i przez 6 godzin przepuszczać przez niego gwałtowny strumień powietrza.
Kolor powinien zmienić się na ciemny, rubinowoczerwony. Utleniony roztwór umieścić
w dużym krystalizatorze lub parownicy i dodać 350 cm3 stężonego HCl. Mieszaninę
zatężyć na łaźni wodnej do około 550 cm3 i ochłodzić w lodzie. Wytrąconą w formie
intensywnie zielonych płytek sól [Co(en)2Cl2]Cl*HCl*2H2O odsączyć i dokładnie
wysuszyć na sączku. Wydajność 93 - 103 g
Jeśli przesącz zatęży się ponownie do ok. 300 cm3 to otrzyma się dodatkową
porcję produktu. Będzie ona jednak zanieczyszczona [Co(en)3]Cl3 i en*2HCl. Z tej
frakcji otrzymamy, po wysuszeniu w 100oC, ok. 18g soli.
Główny produkt utrzeć w dużym moździerzu z metanolem. Kolor soli zmieni się
na jasnozielony gdy odłączy się cząsteczka kwasu. Po wysuszeniu w 105-120oC
przemiana w obojętną trans- sól jest całkowita. Wydajność 83 - 93 g
II. izomer cis-
Obojętną trans-sól rozpuścić w minimalnej ilości wody o temperaturze 90oC i
odparować do sucha na łaźni wodnej (lepiej na łaźni powietrznej o temp. 103-105oC).
Pozostałość powtórnie potraktować taką samą ilością gorącej wody i odparowywanie

65
powtórzyć. Otrzymuje się cis-sól w postaci czysto fioletowego proszku, gorzej
rozpuszczalnego w wodzie niż odpowiednia trans- sól. Niewielkie ilości zanieczyszczeń
mogą zostać usunięte z produktu przez przemywanie go bardzo małymi porcjami
lodowatej wody aż przesącz przybierze barwę czysto fioletową.
Wydajność 70 g
Źródło: Schlessinger G.G., "Inorganic laboratory preparation" Chemical
Publishing Co., Inc.; New York 1962
6. Zalecana literatura
Skrypt-Ćwiczenia z chemii nieorganicznej , Wyd. Uniwersytetu Warszawskiego.
F.A.Cotton, G.Wilkinson, P.L.Gaus, „Chemia nieorganiczna”, PWN Warszawa
1995.
J.D.Lee, Zwięzła Chemia Nieorganiczna, PWN 1993
K.Schwetlik, „Kinetyczne metody badania mechanizmów reakcji”, PWN 1975.
A.Bartecki, „Spektroskopia elektronowa związków nieorganicznych i
kompleksowych”,
PWN 1971.
Z.Kęcki, „Podstawy spektroskopii molekularnej”.
W.Kołos, „Chemia kwantowa”, PWN 1975
S. F. A. Kettle, „Fizyczna chemia nieorganiczna”, PWN 1999.
L. Kolditz „Chemia nieorganiczna”, PWN 1994,
P.W.Atkins, “Chemia fizyczna”, PWN 2001.

66
Ćwiczenie 7.
Chemiczne reakcje oscylacyjne.
Dr Rafał Jurczakowski, e-mail dr hab. Marek Orlik, prof. UW,e-mail
[email protected], sala 149-3, e-mail:
Wymagania:
Właściwości manganu, żelaza, rtęci i ceru na różnych stopniach utlenienia,
równania reakcji redoks
Struktury dyssypatywne – pojęcie, tworzenie w procesach procesów
nieodwracalnych. Przykłady fizyczne i chemiczne. Pojęcia: atraktora, bifurkacji i
bistabilności – przykłady w fizyce, chemii i układach modelowych.
Chemiczne reakcje oscylacyjne jako przykłady struktur dyssypatywnych. Ogólne
warunki występowania oscylacji. Najprostsze modele reakcji oscylacyjnych: Lotki,
Lotki-Volterry i Brukselator.
Reakcja Biełousowa-Żabotyńskiego (BŻ) w reaktorach różnych typów –
przykładowe sposoby realizacji. Formy chemiczne ulegające oscylacyjnym zmianom w
czasie. Zarys mechanizmu reakcji BŻ.
Podstawowe informacje o reakcjach Braya-Liebhafsky’ego i Briggsa-Rauschera.
Zjawiska Lieseganga i typowe sposoby ich realizacji
Oscylacje elektrochemiczne. Pulsujące rtęciowe serce. Oscylacje w procesach
polarograficznych: rola ujemnego oporu i opis wybranego przykładu oscylacji.
Pojęcie chaosu deterministycznego i jego eksperymentalne przejawy. Dziwny
atraktor jako obraz chaosu.
Cel projektu
Opanowanie podstaw teoretycznych i przeprowadzenie wybranych reakcji
oscylacyjnych w układach homogenicznych i heterogenicznych; zapoznanie się z
właściwościami pierwiastków i związków nieorganicznych biorących udział w tych
procesach.

67
Wstęp:
W oddaleniu od stanu równowagi niektóre procesy fizykochemiczne przebiegają w
sposób wykazujący spontaniczną organizację (samoorganizację). Mimo utrzymywania
niezmiennych w czasie warunków prowadzenia takich procesów w układzie może dojść
do spontanicznych oscylacji stężeń niektórych reagentów, np. przemiennej dominacji
form Ox i Red katalizatora. Jeśli formy Ox i Red mają różną barwę, zjawiska te
ujawniają się jako periodyczne lub chaotyczne (z możliwym ukrytym porządkiem !)
zmiany barwy roztworu. Jeśli roztwór nie jest mieszany, zmiany te mogą nie następować
jednocześnie w całej jego objętości i wtedy powstają wędrujące „fale chemiczne” lub
stacjonarne struktury przestrzenne. Uporządkowania tego typu, ponieważ zachodzą w
oddaleniu od stanu równowagi, gdy ma miejsce dyssypacja (rozpraszanie) energii
nazywane są strukturami dyssypatywnymi – czasowymi, czasowo-przestrzennymi lub
przestrzennymi, w zależności od tego, czy dynamiczne zmiany stanu układu następują
tylko w funkcji współrzędnej czasu, czy także lub wyłącznie w funkcji współrzędnej
przestrzennej.
Tego typu zjawiska występują w różnych układach fizykochemicznych, o ile
spełnione są odpowiednie warunki. W przypadku procesów chemicznych obserwujemy
ich powstawanie w układach homogenicznych, w reakcjach z wytrącającymi się osadami,
w reakcjach przebiegających na powierzchniach katalizatorów stałych, w różnego typu
procesach elektrochemicznych itp. Teoria i szczegółowy opis mechanizmu tych zjawisk
są często dość skomplikowane, ale eksperymentalna realizacja niektórych procesów
oscylacyjnych jest prosta. Najsłynniejsze ich przykłady to reakcja Biełousowa-
Żabotyńskiego i tzw. „pulsujące rtęciowe serce”.
Przebieg ćwiczenia:
Po zdanym kolokwium z materiału zawartego w wymaganiach wykonanie
ćwiczenia polega na samodzielnym przygotowaniu przez studentów odczynników i
przeprowadzeniu wybranych, efektownych reakcji oscylacyjnych w różnych układach
homogenicznych i heterogenicznych. Badana są następujące procesy: 1) reakcja
Biełousowa-Żabotyńskiego katalizowana jonami ceru (z pomiarem czasu indukcji i
okresu oscylacji, 2) reakcja Biełousowa-Żabotyńskiego katalizowana ferroiną (z

68
pomiarem okresu oscylacji), w warunkach układu mieszanego (oscylacje synchroniczne)
i niemieszanego (gdy tworzą się fale chemiczne), 3) trójbarwny układ oscylacyjny
Briggsa-Rauschera oraz 4) pulsujące rtęciowe serce jako przykład oscylatora
heterogenicznego
Szczegółowe opisy ćwiczeń
Ćwiczenie 6.1 Reakcja Biełousowa-Żabotyńskiego katalizowana cerem
Do zlewki o poj. 150 - 200 cm3 należy wlać (odmierzone cylindrem miarowym) 58
cm3 wody destylowanej i rozpuścić w niej 5,2 g krystalicznego kwasu malonowego. Do
tego roztworu dodać 16 cm3 nasyconego roztworu KBrO3, 24 cm3 6 mol·dm-3 H2SO4 i
wrzucić mieszadełko magnetyczne. Postawić zlewkę na mieszadle i włączyć
umiarkowanie szybkie mieszanie.
Inicjowanie oscylacji. Przygotować stoper. Za pomocą pipety automatycznej
wstrzyknąć do mieszanego roztworu 1 cm3 0,10 mol·dm-3 roztworu Ce(NO3)3, włączając
jednocześnie stoper. Zwykle natychmiast pojawia się żółte zabarwienie, które szybko
zanika. Po kilkuminutowym okresie indukcji (który należy zmierzyć) powinny zacząć się
oscylacje, widoczne jako przejścia między bezbarwnym i żółtym roztworem, czemu
towarzyszy zauważalne wydzielanie się pęcherzyków CO2. Mierzyć stoperem okres co
najmniej kilkunastu kolejnych oscylacji, zapisując wyniki.
Doświadczenie można powtórzyć dla innej ilości katalizatora (np. 0,5 lub 2 cm3),
obserwując wpływ tej zmiany na charakterystykę oscylacji.
Ćwiczenie 6.2. Reakcja Biełousowa-Żabotyńskiego, katalizowana ferroiną
I. Przygotowanie odczynników
Roztwór A: 0,025 mol·dm-3 ferroina. W kolbce miarowej o poj. 100 cm3 rozpuścić
0,70 g FeSO4·7H2O lub 0,99 g Fe(NH4)2(SO4)2 (soli Mohra), dodać 1,49 g
1,10-fenantroliny (ale nie w postaci chlorowodorku !), przy czym natychmiast powstaje
intensywnie czerwono zabarwiony kompleks [Fe(phen)3]2+. Całość wytrząsa się przez
kilkanaście minut, dopełnia wodą destylowaną do kreski i miesza starannie zawartość
kolbki.

69
Roztwór B: kwas bromomalonowy (przygotować ex tempore !). W kolbce
Erlenmeyera z korkiem szlifowym umieścić 15 cm3 wody destylowanej i rozpuścić w
niej 1,6 g stałego KBr i 2,1 g stałego kwasu malonowego. Do tego roztworu dodać
następnie 13 cm3 6 mol·dm-3 H2SO4 i tak sporządzoną mieszaninę miareczkować małymi
porcjami (pod wyciągiem !!!) nasyconym roztworem KBrO3 z biurety. Uwaga: przy
napełnianiu biurety zwrócić uwagę, aby nie dostały się do niej kryształki KBrO3, które
mogą zatkać wylot biurety! W tym celu nie należy mieszać zawartości butelki z
roztworem KBrO3 tuż przed napełnieniem biurety.
W czasie miareczkowania zawartość kolbki musi być starannie mieszana po
dodaniu każdej niewielkiej (max. 1 cm3) porcji titranta, aż do zaniku brązowej barwy
wydzielanego bromu (silnie toksyczna substancja !!!). Powstający kwas
monobromomalonowy jest nietrwały i ulega powolnej dekarboksylacji, więc ewentualne
jego przechowywanie wymaga umieszczenia zamkniętej kolbki Erlenmeyera w lodówce.
Objętość zużytego roztworu KBrO3 powinna wynosić ok. 15 cm3. Uwaga: niewielkie
przemiareczkowanie próbki, jak też dodanie niewielkiego nadmiaru H2SO4, nie jest
szkodliwe, może nawet czasem okazać się pożyteczne, natomiast pozostawienie w
układzie nadmiaru jonów Br- zwykle uniemożliwia oscylacje.
II. Eksperymenty:
A. Oscylacje synchroniczne w roztworze mieszanym
W zlewce o poj. 100 - 150 cm3 umieścić 50 - 60 cm3 nasyconego roztworu KBrO3,
20 cm3 świeżo przyrządzonego roztworu kwasu bromomalonowego i uruchomić
mieszanie magnetyczne. Włączając stoper dodać do mieszanego roztworu 1 cm3
roztworu ferroiny za pomocą pipety automatycznej. Obserwować powstające oscylacyjne
zmiany barwy roztworu (jakie substancja jest za to odpowiedzialna ?) i mierzyć
(stoperem) okres co najmniej kilkunastu kolejnych oscylacji. Zapisać wyniki.
II. Oscylacje czasowo-przestrzenne w cienkiej warstwie nieruchomego roztworu
(koncentryczne fale chemiczne)
Do czystej suchej szalki Petriego o średnicy min. 10 cm, umieszczonej na
poziomym podłożu, odmierzyć kolejno następujące roztwory: 1) nasyconego
bromianu(V) potasu KBrO3, 2) świeżo przyrządzonego roztworu kwasu

70
bromomalonowego, 3) ferroiny - w proporcji objętościowej 10 : 4 : 1,5 (dla szalki
Petriego o typowych rozmiarach liczby te odpowiadają objętościom w cm3), tak aby
grubość warstwy roztworu w szalce wynosiła 1 - 2 mm. Zawartość szalki należy
niezwłocznie zamieszać (kolistymi ruchami) dla uzyskania jednorodnego roztworu do
chwili pojawienia się jego niebieskiej barwy i wtedy zostawić szalkę w spokoju. Powinna
szybko powrócić czerwona, choć o nieco innym odcieniu, barwa. W ciągu kilku minut
powinny pojawić się pojedyncze niebieskie centra wiodące, z których rozchodzą się
koncentryczne fale chemiczne. Gdy obraz staje się już mało interesujący, roztwór należy
zamieszać dla przywrócenia jednolitej barwy i poczekać na utworzenie nowego układu
fal. Jeśli zjawiska te nie występują, można dodać nieco 6 mol·dm-3 roztworu H2SO4.
Oszacować wielkość przestrzennego okresu oscylacji w różnych miejscach
roztworu.
Ćwiczenie 6.3. Reakcja Briggsa - Rauschera
I. Przygotowanie odczynników:
(A) 0,2 mol·dm-3 KIO3 + 0,16 mol·dm-3 HClO4 (kwas dobrze jest dodać ex
tempore, z powodu wytrącającego się powoli osadu KClO4, co jednak nie przeszkadza w
powstaniu oscylacji);
(B) H2O2, sporządzony przez zmieszanie 33 % perhydrolu cz. d. a z wodą
destylowaną w stosunku objętościowym 2 : 3,
(C) 0,15 mol·dm-3 kwas malonowy z dodatkiem 0,03 % skrobi (uwaga: odważkę
skrobi rozpuszczalnej ogrzewa się w probówce z niewielką ilością wody do całkowitego
rozpuszczenia, a otrzymany zol wlewa do sporządzonego wcześniej roztworu kwasu
malonowego i dodaje wody do żądanej objętości);
(D) 1,0 mol·dm-3 MnSO4.
II. Eksperyment. W zlewce o poj. 100 cm3 należy umieścić po 30 cm3 roztworów
A, B i C i włączyć dość szybkie mieszanie magnetyczne. Następnie za pomocą pipety
automatycznej dodać 1 cm3 roztworu katalizatora (D). Niemal natychmiast rozpoczynają
się oscylacje, na które składają się trzy barwy - roztwór staje się kolejno bezbarwny,

71
złocisty, niebieski, znów bezbarwny itd. Wstępne schłodzenie roztworów (w lodówce)
wydłuża czas obserwacji oscylacji.
Po zaobserwowaniu pierwszych dwóch-trzech cykli oscylacyjnych można
wyłączyć mieszanie, dla zaobserwowania przebiegu reakcji oscylacyjnej w tych
warunkach. Kiedy przebieg zjawiska staje się nieefektowny, należy na powrót włączyć
mieszanie.
Ćwiczenie 6.4. Pulsujące rtęciowe serce
Dużą kroplę rtęci ( o średnicy ok. 1 - 1,5 cm) umieścić na szkiełku zegarkowym,
ustawionym na poziomym podłożu. Dodać tyle wody destylowanej, aby niemal przykryć
jej warstwą kroplę rtęci oraz tyle 6 mol dm-3 kwasu siarkowego, aby roztwór przykrył
rtęć z pewnym nadmiarem (niekiedy dobre wyniki osiąga się dla nierozcieńczonego 6
mol dm-3 H2SO4). Dodać kilka kropli wodnego roztworu KMnO4 tak aby zawartość
szkiełka przybrała ciemnoróżową barwę (kropla rtęci powinna być widoczna !).
Odpowiednio wygiętym stalowym drutem, umieszczonym w drewnianym uchwycie,
wymieszać roztwór a następnie dotknąć kropli rtęci. Jeśli kropla kurczy się, a po
odsunięciu drutu - ponownie rozpłaszcza, drut ten mocuje się na stałe, tak aby kropla Hg,
kurcząc się, traciła z nim kontakt, a rozpłaszczając się ów kontakt odzyskiwała.
Uzyskaniu efektownych oscylacji o charakterze dwóch przechodzących w siebie
trójkątów sprzyja duży rozmiar kropli rtęci oraz zdecydowanie boczne (w miejscach o
największej średnicy), a nie przesunięte ku górze ustawienie stalowego drutu. Przy
odrobinie wprawy można uzyskać jeszcze bardziej złożone pulsacje, np. takie, w trakcie
których kropla rtęci przyjmuje kształt pięciokątny. Zamiast KMnO4 można użyć
K2Cr2O7.
Uwagi:
jeśli kropla rtęci słabo reaguje na dotknięcie stalowym drutem, można dodać kilka
kropli więcej roztworu KMnO4 lub kilka cm3 roztworu H2SO4;
jeśli kropla rtęci nie zmienia w ogóle kształtu pod wpływem kontaktu ze stalowym
drutem, należy przygotować nowy układ, używając starannie wymytego szkiełka
zegarkowego i drutu stalowego o oczyszczonej końcówce.

72
Sposób przygotowania opisu ćwiczenia:
1. Reakcje Biełousowa-Żabotyńskiego i Briggsa-Rauschera
Zwięzły opis wyników wykonanych eksperymentów, w tym:
- podanie składów poszczególnych roztworów i sposobów ich przygotowania
- opis zjawisk i ich charakterystyka ilościowa (okres indukcji i oscylacji dla BŻ)
- opis najważniejszych elementów mechanizmu FKN reakcji Biełousowa-
Żabotyńskiego
2. Pulsujące rtęciowe serce:
- opis eksperymentu
- dokładny opis mechanizmu działania pulsującego rtęciowego serca, ze
wskazaniem na rolę poszczególnych reagentów.
Literatura:
M. Orlik: „Reakcje oscylacyjne – porządek i chaos”, WNT Warszawa 1996,
rozdziały: 1, 2.1, 2.2.1, 2.3, 3.1 – 3.3, 3.4.1, 3.4.2 (bez teoretycznej analizy stabilności
stanu stacjonarnego w rozdziale 3), 4.1.1 – 4.1.3, 4.3.1., 4.3.2, 5.2.1 – 5.2.4, 6.1, 6.2, 6.6,
7.1 7.3.
A. L. Kawczyński: „Reakcje chemiczne – od równowagi przez struktury
dyssypatywne do chaosu”, WNT, Warszawa 1990
A. Bielański, „Podstawy chemii nieorganicznej”, PWN, Warszawa 2002

73
Ćwiczenie 8.
Samorzutna organizacja cząsteczek alkanotioli na powirzchni złota.
dr Agnieszka Więckowskae-mail [email protected] dr Anna Nowicka sala
149-8.
Wymagania:

74
Przed przystąpieniem do ćwiczenia student powinien zapoznać się z
następującymi zagadnieniami:
• Budowa podwójnej warstwy elektrycznej – model Helmholtza;
• Potencjał formalny układu redoks;
• Podstawy chronowoltamperometrii cyklicznej – układ pomiarowy, przebieg
woltamogramu (w szczególności wymagana jest znajomość takich pojęć jak elektrolit
podstawowy, depolaryzator, dyfuzja, prąd pojemnościowy, prąd faradajowski,
nadpotencjał, wykres Tafela);
• Adsorpcja chemiczna i fizyczna, izoterma adsorpcji Langmuira;
• Efekt tunelowy;
• Zasada działania Skaningowego Mikroskopu Tunelowego
Powyższe zagadnienia są omówione m. in. w następujących podręcznikach:
1. P.W. Atkins, Chemia Fizyczna, PWN, Warszawa 2001.
2. K. Pigoń, Z. Ruziewicz, Chemia Fizyczna cz. 1, PWN, Warszawa 1993.
3. W. Szczepaniak, Metody instrumentalne w analizie chemicznej, PWN, Warszawa
2004.
Ponadto zalecane jest zapoznanie się z załączonym wstępem teoretycznym do
ćwiczenia 8.


76
WSTĘP
Alkanotiole oraz dwusiarczki alkilowe o ogólnych wzorach RSH oraz RSSR, gdzie R
jest łańcuchem n-alkilowym, posiadają zdolność do tworzenia dobrze zorganizowanych
monowarstw na powierzchniach metali takich jak złoto, srebro, miedź, platyna i rtęć. Jednak
najczęściej wykorzystywanym podłożem jest złoto, ze względu na brak przeszkadzających
tlenków na powierzchni. Monowarstwy alkanotiolowe uzyskuje się w procesie
samoorganizacji cząsteczek na powierzchni metalu. Proces ten jest spontaniczny dzięki
silnemu oddziaływaniu siarki z metalicznym podłożem (chemisorpcja), kohezji łańcuchów
węglowodorowych oraz oddziaływaniu pomiędzy grupami końcowymi. W praktyce
przygotowanie monowarstwy polega na zanurzeniu substratu w rozcieńczonym roztworze
odpowiedniego związku siarki – tiolu lub dwusiarczku. Jakość otrzymywanej monowarstwy
w dużym stopniu zależy od:
• stężenia tiolu w roztworze,
• czasu przez jaki substrat pozostaje zanurzony w roztworze,
• rodzaju tiolu lub dwusiarczku,
• temperatury w której prowadzimy proces.
Niskie stężenie stosowanych roztworów (zazwyczaj 1mM) służy spowolnieniu
adsorpcji cząsteczek na powierzchni, co z kolei sprzyja tworzeniu dobrze zorganizowanych
monowarstw. Czas na jaki substrat pozostaje zanurzony w roztworze zależy natomiast od
rodzaju adsorbowanego związku, dla alkanotioli wynosi on zazwyczaj od kilku minut do
kilku godzin, natomiast dla dwusiarczków oraz siarczków, kilka dni. Jakość monowarstwy
zależy również w dużym stopniu od długości łańcuchów tworzących ją cząsteczek, dla n-
alkanotioli uporządkowanie monowarstwy jest tym lepsze, im dłuższy jest łańcuch alkilowy.
Wiąże się to z faktem, iż energia oddziaływań van der Waalsa jest zależna od ilości grup
metylenowych w łańcuchu. Kolejnym istotnym elementem przy otrzymywaniu dobrze
uporządkowanych monowarstw jest temperatura. Zazwyczaj proces samoorganizacji
prowadzi się w temperaturze pokojowej, jednak zwiększenie jej do około 500C może
prowadzić do znacznego obniżenia ilości defektów w monowarstwie. Dalszy wzrost
temperatury powoduje zazwyczaj pogorszenie uporządkowania, czego przyczyną może być
częściowa desorpcja tiolu lub zmiana konformacji łańcuchów alkilowych (defekty gauche).
Odpowiedni dobór omówionych powyżej parametrów pozwala na modyfikację powierzchni
złota monowarstwą alkanotiolową o wysokim stopniu uporządkowania.

77
Cząsteczki alkanotioli lub dwusiarczków alkilowych wiążą się z powierzchnią złota dzięki
oddziaływaniu siarki z atomami złota na powierzchni, przy czym oddziaływanie to ma
charakter wiązania kowalencyjnego. Zachodzące w trakcie procesu adsorpcji tiolu lub
dwusiarczku reakcje prowadzą do utworzenia tiolanów, co umownie można zapisać w
następujący sposób:
RSSRAuRSAuAuRSH nn
H
2
1Au 1
2/1n
2 +←− →+ −+−
−
Równowagowa odległość atomów siarki od powierzchni metalu wynosi około 2 Å,
przy czym atomy siarki zlokalizowane są pomiędzy trzema atomami złota (patrz rysunek
poniżej).
Rys.1.
Cząsteczki alkanotioli zaadsorbowane na powierzchni złota wykazują upakowanie
heksagonalne, a odległość pomiędzy sąsiadującymi łańcuchami wynosi około 5 Å. Daje to
średnią powierzchnię przypadającą na pojedynczą cząsteczkę równą około 21 Å2. Łańcuchy
alkilowe w monowarstwach alkanotiolowych na złocie są nachylone pod kątem około 300
względem normalnej do powierzchni substratu (rys. 1), co powoduje, że realna grubość
monowarstwy jest mniejsza niż długość tworzących ją cząsteczek. Nie zmienia to jednak
faktu, iż zachowana jest wprost proporcjonalna zależność pomiędzy grubością warstwy i
długością łańcuchów alkilowych.
Au

78
Pomiar pojemności
Stosunkowo często stosowaną metodą weryfikacji jakości monowarstw jest
elektrochemiczny pomiar pojemności. Na granicy faz metal-roztwór elektrolitu występuje
tzw. podwójna warstwa elektryczna, którą w uproszczeniu można przedstawić jako
kondensator o przeciwnie naładowanych okładkach, przy czym jedną okładkę stanowi
powierzchnia metalu, drugą natomiast nieruchoma warstwa jonów z roztworu przylegająca do
metalu (model Helmholtza). Stosując metody elektrochemiczne możliwe jest wyznaczenie
pojemności tego umownego kondensatora, czyli podwójnej warstwy elektrycznej. Rozważmy
przypadek chronowoltamperometrii cyklicznej, gdzie mamy sytuację, w której do metalu
(elektrody) zanurzonego w roztworze elektrolitu podstawowego przykładamy potencjał
zmieniający się w czasie. Potencjał zmienia się liniowo od początkowej wartości E1 do
wartości E2 po czym ponownie wraca do E1, co ilustruje rysunek poniżej:
Rys.2.
W efekcie zmiany potencjału elektrody pracującej następuje ładowanie warstwy
podwójnej. Na woltamogramie (krzywa prąd-potencjał) obserwujemy tzw. prąd
pojemnościowy. W związku z tym, że potencjał elektrody zmienia się według schematu
E1→E2→E1 rejestrujemy prąd pojemnościowy zarówno anodowy (ia) jak i katodowy (ic).
Przykładowy woltamogram przedstawiono na rysunku poniżej:
-3.0E-07
-2.0E-07
-1.0E-07
0.0E+00
1.0E-07
2.0E-07
3.0E-07
4.0E-07
-0.2 -0.1 0 0.1 0.2 0.3 0.4 0.5 0.6
E [V]
i[A
]
c
pot

79
Rys.3.
Na postawie mierzonego prądu możemy obliczyć pojemność (F/cm2) zgodnie z
zależnością:
A
v
|i||i|
C
ca
total
+
=2
1
(1)
gdzie ia oraz ic są odpowiednio prądami pojemnościowymi anodowym i katodowym
(A), A jest powierzchnią elektrody pracującej (cm2), natomiast ν jest szybkością zmiany
potencjału elektrody (V/s), czyli szybkością polaryzacji elektrody.
Cząsteczki alkanotiolu zaadsorbowane na powierzchni złota powodują znaczny spadek
prądów pojemnościowych. Efekt ten jest związany z niską wartością stałej dielektrycznej
warstwy oddzielającej metal elektrody od roztworu. W rezultacie mierzona pojemność jest o
rząd wielkości niższa od pojemności uzyskiwanych dla niezmodyfikowanych elektrod
złotych. W oparciu o model Helmholtza, całkowitą mierzoną pojemność możemy opisać
poprzez układ szeregowo połączonych kondensatorów odpowiadających pojemności
monowarstwy Cm oraz pojemności warstwy podwójnej Cdl. Przy wysokich stężeniach
elektrolitu podstawowego (powyżej 0,1 mol/l) pojemność warstwy podwójnej jest zazwyczaj
wysoka. W rezultacie mierzona całkowita pojemność jest zdominowana przez pojemność
monowarstwy. Ta ostatnia może być wyrażona następującą zależnością:
dC m
m
0εεεεεεεε= (2)
gdzie εm jest względną stałą dielektryczną monowarstwy (zwykle około 2.3); ε0 jest
przenikalnością elektryczną próżni równą 8.854×10-12 F/m; d jest grubością warstwy
dielektryka. Powyższy wzór wiąże mierzoną pojemność z grubością warstwy i jej
własnościami dielektrycznymi. Pojemność stanowi jedną z podstawowych wielkości
charakteryzujących monowarstwy pod względem ich jakości. Na podstawie pomiarów
pojemnościowych możemy określić w jakim stopniu monowarstwa jest penetrowana przez
wodę lub jony. Przykładowo, pojemność dla niezmodyfikowanej elektrody złotej zwykle
wynosi około 20 µF/cm2, podczas gdy dla elektrod pokrytych dobrze zorganizowaną
monowarstwą alkanotiolową uzyskuje się wartości w zakresie 1-2 µF/cm2. Pośrednie wartości
pomiędzy 2-20 µF/cm2 świadczą o obecności defektów w monowarstwie. Jako defekt należy
rozumieć taki obszar na powierzchni zmodyfikowanej elektrody, gdzie monowarstwa może
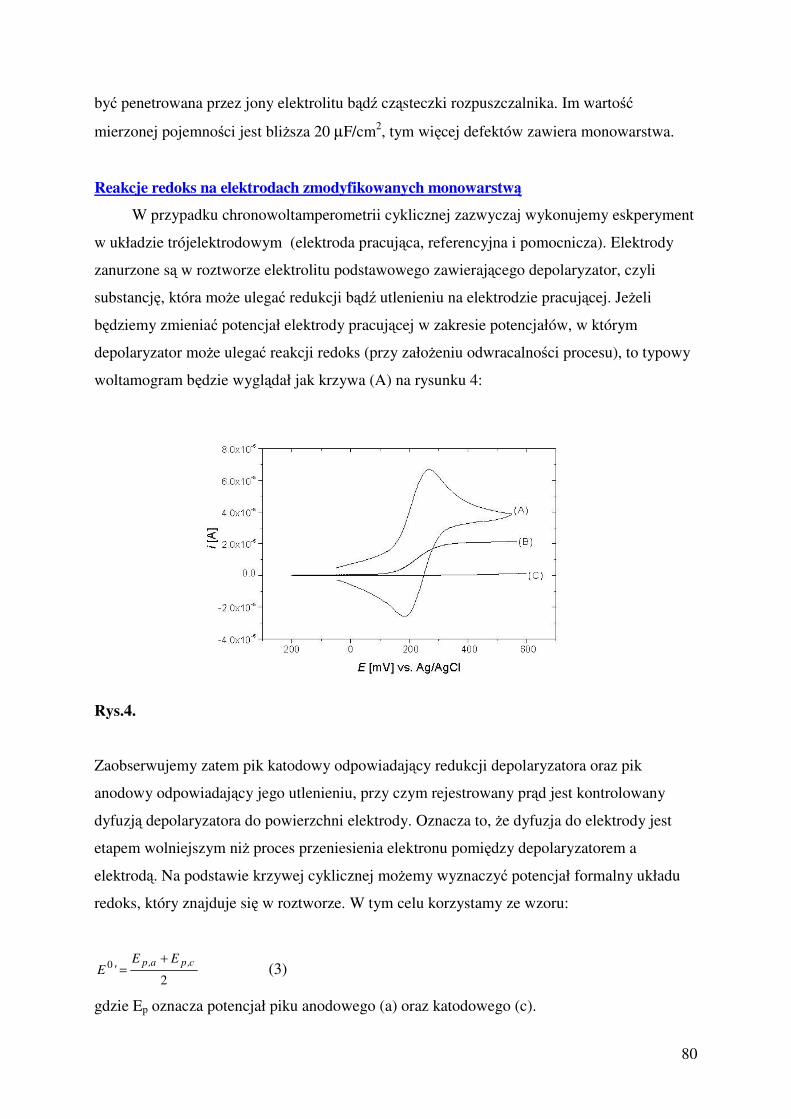
80
być penetrowana przez jony elektrolitu bądź cząsteczki rozpuszczalnika. Im wartość
mierzonej pojemności jest bliższa 20 µF/cm2, tym więcej defektów zawiera monowarstwa.
Reakcje redoks na elektrodach zmodyfikowanych monowarstwą
W przypadku chronowoltamperometrii cyklicznej zazwyczaj wykonujemy eskperyment
w układzie trójelektrodowym (elektroda pracująca, referencyjna i pomocnicza). Elektrody
zanurzone są w roztworze elektrolitu podstawowego zawierającego depolaryzator, czyli
substancję, która może ulegać redukcji bądź utlenieniu na elektrodzie pracującej. Jeżeli
będziemy zmieniać potencjał elektrody pracującej w zakresie potencjałów, w którym
depolaryzator może ulegać reakcji redoks (przy założeniu odwracalności procesu), to typowy
woltamogram będzie wyglądał jak krzywa (A) na rysunku 4:
Rys.4.
Zaobserwujemy zatem pik katodowy odpowiadający redukcji depolaryzatora oraz pik
anodowy odpowiadający jego utlenieniu, przy czym rejestrowany prąd jest kontrolowany
dyfuzją depolaryzatora do powierzchni elektrody. Oznacza to, że dyfuzja do elektrody jest
etapem wolniejszym niż proces przeniesienia elektronu pomiędzy depolaryzatorem a
elektrodą. Na podstawie krzywej cyklicznej możemy wyznaczyć potencjał formalny układu
redoks, który znajduje się w roztworze. W tym celu korzystamy ze wzoru:
20 c,pa,p EE'E
+= (3)
gdzie Ep oznacza potencjał piku anodowego (a) oraz katodowego (c).

81
Rozważmy teraz co dzieje się w układzie, gdy depolaryzator znajduje się w
bezpośrednim sąsiedztwie elektrody. Weźmy pod uwagę tylko redukcję depolaryzatora. W
naszym eksperymencie kontrolujemy potencjał elektrody pracującej, co jest równoważne z
kontrolowaniem energii elektronów wewnątrz elektrody. Jeśli zaczniemy zmieniać potencjał
elektrody do coraz bardziej ujemnych wartości to wówczas energia elektronów wzrośnie i
mogą one osiągnąć wystarczającą energię, aby przejść na nieobsadzone poziomy elektronowe
(LUMO) depolaryzatora, który znajduje się w roztworze. Podobnie dla procesu utleniania,
zmieniając potencjał elektrody na coraz bardziej dodatni, energia elektronów może być
obniżona, przy czym przy pewnej energii (a zatem przy określonym potencjale) elektrony z
zajętych poziomów elektronowych (HOMO) depolaryzatora mogą być przeniesione do
elektrody. Potencjały przy których te procesy zaczynają zachodzić zależą od wartości
potencjału formalnego E0’ przypisanego do danego układu redoks.
Powyższe rozważania dotyczą sytuacji, gdy elektroda pracująca jest niezmodifikowana,
w przeciwnym wypadku proces przenoszenia elektronów pomiędzy depolaryzatorem, a
elektrodą jest nieco bardziej złożony. W przypadku, gdy elektroda zmodyfikowana jest
monowarstwą alkanotiolową, transport elektronów zachodzi za pośrednictwem cząsteczek
zaadsorbowanych na powierzchni elektrody, oczywiście pod warunkiem, że monowarstwa
rownomiernie pokrywa całą powierzchnię elektrody i nie posiada defektów. Zrozumiałe jest,
że modyfikacja monowarstwą alkanotiolową powoduje oddzielenie powierzchni elektrody od
depolaryzatora znajdującego się w roztworze, na dystans określony grubością monowarstwy.
Rozdzielenie donora i akceptora elektronów powoduje, że przeniesienie elektronów powinno
zachodzić na zasadzie tunelowania za pośrednictwem cząsteczek tworzących monowarstwę.
Elektron musi zatem pokonać pewną barierę energetyczną, którą stanowi monowarstwa. W
takiej sytuacji elektrony nie są przenoszone bezpośrednio pomiędzy poziomami
elektronowymi elektrody i depolaryzatora, ale dzieje się to za pośrednictwem orbitali HOMO
i LUMO cząsteczek alkanotioli.
W przypadku modyfikacji elektrody monowarstwą o wysokim stopniu uporządkowania,
proces elektrodowy jest kontrolowany kinetyką przeniesienia elektronu, zatem przeniesienie
elektronu jest wolniejsze niż transport (dyfuzja) depolaryzatora do powierzchni elektrody. W
takim przypadku krzywe woltamperometryczne charakteryzują się eksponencjalnym
wzrostem prądu wraz z rosnącym nadpotencjałem. Jako nadpotencjał należy rozumieć różnicę
pomiędzy potencjałem przyłożonym do elektrody pracującej a potencjałem formalnym układu
redoks znajdującego się w roztworze. Przykładowa krzywa cykliczna wygląda wówczas jak
krzywa (C) na rusunku 4.

82
Jak już wcześniej wspomniano powyższe rozważania są prawdziwe dla sytuacji gdy
monowarstwa alkanotiolowa jest dobrze uporządkowana i nie zawiera defektów. Obecność
defektów powoduje, że przeniesienie elektronu pomiędzy elektrodą a znajdującym się w
roztworze depolaryzatorem zachodzi nie tylko za pośrednictwem monowarstwy, ale również
w wyniku bezpośredniego kontaktu depolaryzatora z powierzchnią elektrody. Zatem
mechanizm wymiany elektronów jest mieszany – zachodzi zarówno na zasadzie tunelowania
przez cząsteczki alkanotioli, jak również bezpośrednio pomiędzy elektrodą i depolaryzatorem.
Wówczas przebieg woltamogramu zależy od ilości defektów oraz ich rozmiarów. Gdy
defektami są niewielkie i odpowiednio oddzielone od siebie odsłonięte fragmenty
powierzchni elektrody, przebieg krzywej cyklicznej może być analogiczny jak dla zespołu
submikroelektrod – uzyskujemy wówczas tzw. prąd stacjonarny (patrz krzywa (B) rysunek 4).
Sytuacja, gdy uzyskujemy prąd stacjonarny ma miejsce jednak tylko wtedy, gdy obszary
dyfuzyjne poszczególnych submikroelektrod są niezależne, czyli nie nakładają się. W
przypadku przenikania się obszarów dyfuzyjnych na woltamogramie zaobserwujemy piki
(krzywa (A) na rysunku 4), co wynika ze zwiększonego udziału dyfuzji liniowej do
powierzchni elektrody. Należy jednak dodać, że w takim przypadku szybkość procesu
elektrodowego jest niższa niż w przypadku niezmodyfikowanej elektrody, dlatego też często
obserwuje się zwiększenie różnicy potencjałów pików anodowego i katodowego.
W celu określenia jakości monowarstwy, można wprowadzić tzw. współczynnik
blokowania procesu elektrochemicznego, który określa w jakim stopniu dostęp
depolaryzatora do powierzchni elektrody jest “zablokowany”. Współczynnik blokowania
procesu elektrochemicznego (EBE) oblicza się za pomocą wzoru:
−=
)(2/1
)(2/11n
m
i
iEBE (4)
gdzie i1/2 to prąd faradajowski mierzony przy potencjale formalnym, odpowiednio w
obecności monowarstwy (m) oraz na niezmodyfikowanej elektrodzie (n). Im wartość
współczynnika jest bliższa jedności tym lepiej „zablokowany” jest dostęp depolaryzatora do
powierzchni elektrody, zatem tym lepsze jest uporządkowanie monowarstwy.
Niezależnie od tego, czy monowarstwa jest dobrze uporządkowana, czy też zawiera
dużą ilość defektów, możliwe jest wyznaczenie średniej stałej szybkości dla procesu
przeniesienia elektronu. Aby wyznaczyć wspomnianą stałą, eksperyment
woltamperometryczny powinien być wykonany tak, aby uzyskać kinetyczną kontrolę procesu
elektrodowego. W praktyce rejestruje się woltamogramy dla kolejno zwiększających się

83
szybkości polaryzacji elektrody, tak aby uzyskać różnicę potencjałów pików katodowego i
anodowego większą niż 300 mV. Wówczas należy sporządzić zależność ln(ip) od Ep-E0’, przy
czym z przecięcia tej zależności z osią y będzie możliwe wyznaczenie wartości standardowej
stałej szybkości k0, zgodnie ze wzorem:
−−= )'EE(
RT
nFexpknFAC.i p
*p
0002270
α (5)
gdzie n jest liczbą wymienianych elektronów, F jest stałą Faradaya, A jest
powierzchnią elektrody, C0* jest stężeniem depolaryzatora, Ep oznacza potencjał piku, E0’ jest
potencjałem formalnym, R stałą gazową, T oznacza temperaturę, α jest współczynnikiem
przejścia. Uzyskana wartość standardowej stałej szybkości zależy od stopnia pokrycia
elektrody monowarstwą i jest tym niższa im większa powierzchnia elektrody jest pokryta
monowarstwą. Zależność tą wyraża wzór:
0
0
1n
m
k
k=−θ (6)
gdzie θ jest stopniem pokrycia elektrody monowarstwą, natomiast k0 jest standardową
stałą szybkości przeniesienia elektronu zmierzoną w obecności monowarstwy (m) oraz na
niezmodyfikowanej elektrodzie (n).
Elektrochemiczna desorpcja monowarstw tiolowych
Zarówno adsorpcja jak i desorpcja cząsteczek tioli z powierzchni złota może być
kontrolowana elektrochemicznie. Przykładowo, jeśli elektroda złota zmodyfikowana
monowarstwą tiolową zanurzona jest w roztworze 0.5M KOH i przyłożymy do niej
wystarczająco ujemny potencjał (zwykle przynajmniej –0.7 V vs. Ag/AgCl), następuje desorpcja
cząsteczek tioli z powierzchni złota. Proces ten można opisać jako reakcję redukcji tiolanów
zgodnie z równaniem:
( ) ( )+−+−
−+− −+ →++− MSRAuMeRSAuAu 0
ndesorpcja
1n
gdzie M+ jest kationem obecnym w roztworze elektrolitu, np. K+. Proces ten nie jest jednak
nieodwracalny, albowiem przyłożenie bardziej dodatniego potencjału niż potencjał desorpcji
spowoduje ponowną adsorpcję cząsteczek tiolu na powierzchni metalu. W warunkach
eksperymentu woltamperometrycznego, gdy polaryzujemy elektrodę ujemnie, na krzywej
cyklicznej możemy zaobserwować pik redukcji związany z desorpcją tiolu natomiast po
zawróceniu potencjału pojawi się pik utleniania związany z ponowną adsorpcją tiolu. Pik
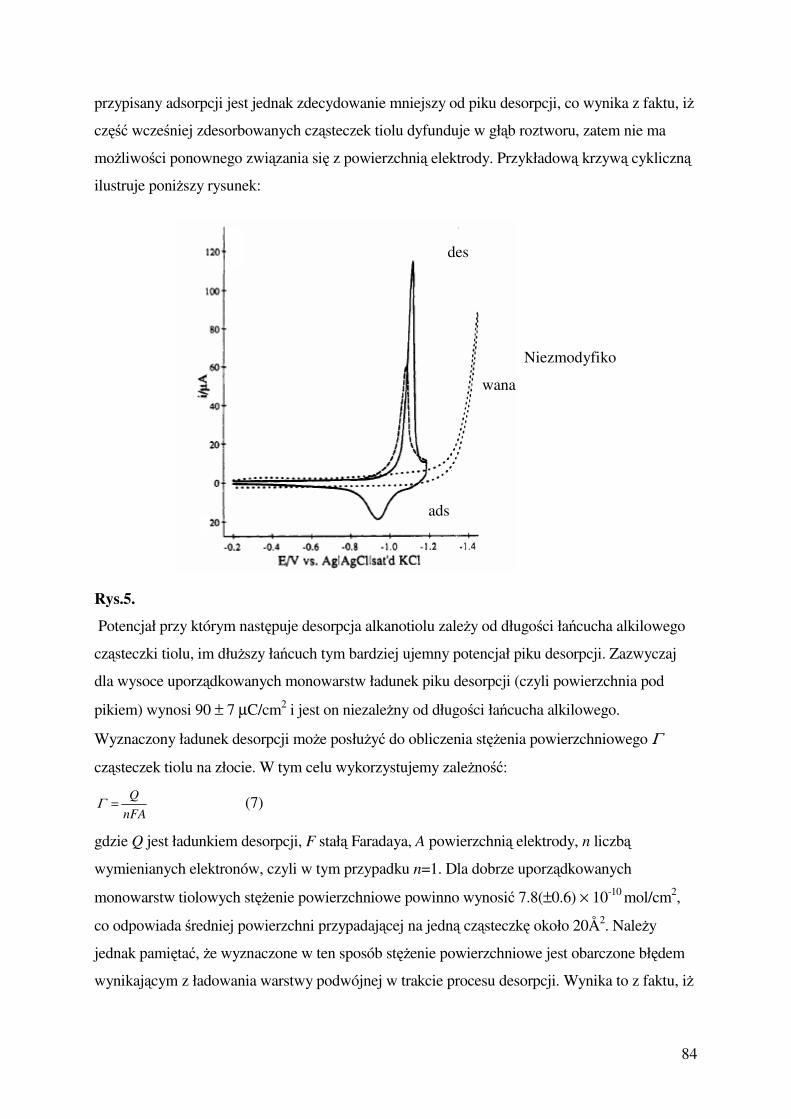
84
przypisany adsorpcji jest jednak zdecydowanie mniejszy od piku desorpcji, co wynika z faktu, iż
część wcześniej zdesorbowanych cząsteczek tiolu dyfunduje w głąb roztworu, zatem nie ma
możliwości ponownego związania się z powierzchnią elektrody. Przykładową krzywą cykliczną
ilustruje poniższy rysunek:
Rys.5.
Potencjał przy którym następuje desorpcja alkanotiolu zależy od długości łańcucha alkilowego
cząsteczki tiolu, im dłuższy łańcuch tym bardziej ujemny potencjał piku desorpcji. Zazwyczaj
dla wysoce uporządkowanych monowarstw ładunek piku desorpcji (czyli powierzchnia pod
pikiem) wynosi 90 ± 7 µC/cm2 i jest on niezależny od długości łańcucha alkilowego.
Wyznaczony ładunek desorpcji może posłużyć do obliczenia stężenia powierzchniowego Γ
cząsteczek tiolu na złocie. W tym celu wykorzystujemy zależność:
nFA
Q=Γ (7)
gdzie Q jest ładunkiem desorpcji, F stałą Faradaya, A powierzchnią elektrody, n liczbą
wymienianych elektronów, czyli w tym przypadku n=1. Dla dobrze uporządkowanych
monowarstw tiolowych stężenie powierzchniowe powinno wynosić 7.8(±0.6) × 10-10 mol/cm2,
co odpowiada średniej powierzchni przypadającej na jedną cząsteczkę około 20Å2. Należy
jednak pamiętać, że wyznaczone w ten sposób stężenie powierzchniowe jest obarczone błędem
wynikającym z ładowania warstwy podwójnej w trakcie procesu desorpcji. Wynika to z faktu, iż
des
ads
Niezmodyfiko
wana
elektroda

85
w trakcie procesu desorpcji pojemność zmienia się od 1-2 µF/cm2 do około 20 µF/cm2. Zatem
zwykle oszacowane w powyższy sposób stężenie powierzchniowe jest zawyżone.
Zasada działania Skaningowego Mikroskopu Tunelowego
Skaningowy mikroskop tunelowy (STM) został skonstruowany w 1982 roku przez
Bininga i Rohrera, za który to wynalazek twórcy zostali nagrodzeni Nagrodą Nobla w
dziedzinie fizyki w 1986 roku. STM należy do grupy mikroskopów skaningowych, w których
do uzyskania obrazów nie stosuje się soczewek. Ich miejsce zajęło ostrze przymocowane do
skanera piezoelektrycznego, który zmienia w niewielkim stopniu swe wymiary pod wpływem
napięcia elektrycznego. Tym samym zmienia położenie igły umożliwiając jej przesuwanie się
nad próbką we wszystkich trzech płaszczyznach. Dzięki temu możliwe jest uzyskanie obrazu
powierzchni próbki z rozdzielczością rzędu pojedynczego atomu. Jest to jednak możliwe
tylko w przypadku, gdy badana próbka jest wykonana z materiału przewodzącego prąd
elektryczny. Schematycznie zasadę działania skaningowego mikroskopu tunelowego
przedstawia poniższy rysunek.
Rys. 6 Zasada działania skaningowego mikroskopu tunelowego
Skaningowy mikroskop tunelowy działa o oparciu o zjawisko kwantowego efektu
tunelowego, czyli możliwość przejścia, z pewnym prawdopodobieństwem, cząstki przez
barierę potencjału o energii potencjalnej większej niż energia potencjalna cząstki.
Prawdopodobieństwo przejścia przez tę barierę określa równanie falowe Schroedingera.

86
Igła, którą może być drut wolframowy lub platynowo – irydowy o średnicy 0,2 – 0,5
mm, zawiera na końcu kryształ ustawiony wierzchołkiem w stronę ostrza. Dzięki temu
zakończeniem sondy jest dokładnie jeden atom. W czasie pomiarów STM igła mikroskopu
znajduje się w odległości rzędu kilku angstremów od powierzchni próbki. Ten niewielki
dystans umożliwia przeniesienie elektronu od ostrza igły przez barierę potencjału do badanej
próbki właśnie dzięki zjawisku tunelowemu. Elektrony mogą tunelować z ostrza przez
powietrze (lub próżnię) do próbki lub odwrotnie, w zależności od kierunku przyłożonego
napięcia. Wartość prądu tunelowego zależy eksponencjalnie od szerokości bariery potencjału,
zgodnie z równaniem:
I ∝ BVe-Cd
gdzie B i C są wartościami stałymi, I oznacza wartość prądu tunelowania, V jest
napięciem przyłożonym między ostrze a próbkę, zaś d odpowiada odległości igły od próbki.
Igła idealna
Igła rzeczywista
Rys.7 Skanowanie powierzchni próbki atomowo płaskiej przez sondę STM.
Komputer analizuje i zapamiętuje mapę prądów tunelowych dla każdego
punktu próbki i na tej podstawie tworzony jest później obraz próbki. W
przypadku badania substancji zbudowanych z różnych atomów (nie
pierwiastków) wartość prądu zależy od siły wiązania elektronu przez atom.
Pomiar tego prądu pozwala obrazować strukturę atomową powierzchni próbki,
tzn. że zdolność rozdzielcza mikroskopu tunelowego pozwala rozróżniać
pojedyncze atomy.

87
Technika skaningowej mikroskopii tunelowej umożliwia odwzorowanie z
nanometrową rozdzielczością topografii powierzchni materiałów przewodzących.
Mikroskop STM może działać w kilku trybach:
trybie stałoprądowym
trybie stałej wysokości
trybie skaningowej spektroskopii tunelowej
W trybie stałoprądowym igła może oddalać się i przybliżać do próbki skanując jej
powierzchnię. W czasie pomiaru prąd tunelowania utrzymywany jest blisko stałej wartości, za
co odpowiada zmiana położenia igły względem pionowej osi z. Zatem, w tym trybie ruch igły
odwzorowuje obrazowaną powierzchnię próbki. Technika stałoprądowa jest historycznie
pierwszą metodą pomiarową w skaningowej mikroskopii tunelowej, a jej niewątpliwą zaletą
jest możliwość stosowania jej do różnego rodzaju, niekoniecznie płaskich, powierzchni.
W trybie stałej wysokości igła porusza się na stałej wysokości nad próbką, a rejestrowane
są jedynie zmiany prądu tunelowego. Zatem w tym przypadku za powstanie obrazu
odpowiadają zmiany prądu tunelowania. Jednakże pomiary tą techniką można stosować tylko
dla próbek o równej powierzchni. Wgłębienia na powierzchni próbki mogą powodować zanik
obrazu, zaś wypukłości grożą kolizją igły z metalem. Zastosowanie tej techniki pomiarowej
umożliwia znacznie szybsze zobrazowanie atomowo płaskiej powierzchni niż w przypadku
metody stałoprądowej, co minimalizuje ryzyko zniekształcenia obrazu odwzorowującego
topografię powierzchni.
Skaningowa mikroskopia tunelowa umożliwia wizualizację nanoświata oraz zrozumienie
procesów zachodzących na poziomie molekularnym. Jednakże mierzona wartość prądu
tunelowania dostarcza więcej informacji niż tylko o geometrii powierzchni. Bariera
potencjału, przez którą przechodzi elektron, składa się z niejednorodnych stanów
elektronowych. Zatem prąd tunelowania może również odzwierciedlać różnice energetyczne
w obszarze bariery potencjału. Różnice w wartości prądu przy danej różnicy potencjału i
stałej odległości próbka – igła umożliwiają uzyskanie spektroskopowych informacji o
powierzchni elektrody bądź o cząsteczce znajdującej się między substratem a igłą
mikroskopu. W skaningowej spektroskopii tunelowej (STS), dla danego położenia igły,
wyznacza się zależność natężenia prądu tunelowania od przyłożonego napięcia. Aby to
umożliwić należy rozważyć zależność między prądem tunelowania a elektronową strukturą
próbki.

88
Szczególnym przypadkiem jest tunelowanie przez molekułę, która unieruchomiona
między substratem a igłą mikroskopu tworzy złącze tunelowe. W takim układzie cząsteczka
pełni rolę bariery, przez którą przechodzi elektron.
Rys.8 Atomy żelaza na miedzi (111) Rys.9 Powierzchnia
krzemu
Zastosowanie
Poza obrazowaniem struktury atomowej i profilu powierzchni
skanowanej próbki, skaningowy mikroskop tunelowy znajduje inne
zastosowania. Eksperymenty z mikroskopem STM doprowadziły do
niesamowitego odkrycia. Jeżeli do igły przyłoży się większe napięcie niż przy
skanowaniu, to może ona oderwać pojedynczy atom z powierzchni próbki i
przełożyć go w inne miejsce. W ten sposób możliwa jest obróbka materiału na
poziomie atomowym. Mikroskop STM jest pierwszym prawdziwym narzędziem
nanotechnologii.
Rys. 8 pochodzi ze strony : http://www.almaden.ibm.com/vis/stm/atomo.html
Rys.9 pochodzi ze strony:
http://www-03.ibm.com/ibm/history/exhibits/vintage/images/4506VV3181.jpg
Zastosowanie monowarstw tiolowych
Obecnie monowarstwy oparte na związkach tiolowych lub dwusiarczkowych znajdują coraz
liczniejsze zastosowania. Wykorzystuje się je m. in. do:
• uzyskiwania powierzchni o charakterze katalitycznym, w szczególności zaś
biokatalitycznym (przykładowo monowarstwa pozwala na wiązanie enzymów do
powierzchni elektrody bez utraty ich aktywności);

89
• uzyskiwania powierzchni o zróżnicowanej reaktywności i zwilżalności;
• zabezpieczania przed korozją (monowarstwy spowalniają proces utleniania powierzchni
metalu);
• konstruowania elektrochemicznych sensorów;
• badań nad transportem elektronowym przez cząsteczki organiczne (monowarstwy
zbudowane z odpowiednio zmodyfikowanych tioli mogą służyć jako uproszczone modele
biologicznych układów redoks).
Zastosowania monowarstw obejmują zatem m. in. chemię materiałową, chemię analityczną oraz
chemię bionieorganiczną.

90
WYKONANIE ĆWICZENIA
Przy każdej zmianie roztworu należy naczynko elektrochemiczne, rurkę do odtleniania oraz
wszystkie używane elektrody dokładnie opłukać wodą destylowaną!!!
I. Przygotowanie złotych elektrod do pomiarów elektrochemicznych
1. Do wykonania pomiarów należy przygotować trzy dyskowe elektrody złote. Przed
przystąpieniem do pomiarów elektrody wypolerować za pomocą tlenku glinu, następnie
przemyć obficie wodą destylowaną.
2. W celu wyznaczenia powierzchni rzeczywistej elektrody pracującej przygotowujemy 50
ml roztworu kwasu siarkowego (VI) o stężeniu 0.1 M. Następnie rejestrujemy krzywe
woltamperometryczne w zakresie potencjałów –0.3 ÷ 1.5 V z szybkością polaryzacji 50
mV/s do momentu uzyskania stabilnego woltamperogramu. Z wartości ładunku piku
utleniania obliczamy właściwą powierzchnię elektrody (UWAGA! – wyznaczając
powierzchnię rzeczywistą elektrody należy pamiętać, że ładunek dla elektrody o
powierzchni 1 cm2 wynosi 390 µC), którą porównujemy z powierzchnią geometryczną
elektrody.
II. Przygotowanie monowarstw tiolowych oraz pomiar ich pojemności.
1. Przygotowujemy 100ml wodnego roztworu NaClO4xH2O (M=140.46 g/mol) o stężeniu 1
mol/l. Roztwór umieszczamy w naczynku elektrochemicznym, podłączamy elektrody -
referencyjną oraz pomocniczą po czym roztwór odtleniamy za pomocą azotu lub argonu.
Czas odtleniania wynosi około 10 minut.
2. Przygotowujemy 10 ml roztworu dodekanotiolu (M=202.35 g/mol; gęstość wynosi 0.844
g/cm3) w etanolu. Stężenie dodekanotiolu powinno wynosić około 1 mmol/l.
3. Przygotowujemy 5 ml roztworu disiarczku bis-2-((ferrocenylokarbonylo)
amino)etylowego (M=576.08 g/mol) w etanolu. Stężenie powinno wynosić również około
2 mmol/l.
4. Jedną z elektrod dyskowych umieścić w przygotowanym roztworze dodekanotiolu (30
minut), natomiast drugą w roztworze disiarczku bis-2-((ferrocenylokarbonylo)-
amino)etylowy na około 60 minut.
5. Trzecią (czystą) elektrodę dyskową umieścić w naczynku elektrochemicznym i podłączyć
jako elektrodę pracującą.

91
6. Zarejestrować 5 wotltamogramów w zakresie od –0.1 V do 0.5 V oraz od 0.1 V do 0.8 V
dla szybkości polaryzacji 1V/s.
7. Na podstawie każdej z zarejestrowanych krzywych obliczyć pojemność warstwy
podwójnej zgodnie ze wzorem (1), wyznaczyć średnią wartość pojemności.
8. Punkty 5-7 powtórzyć dla elektrody zmodyfikowanej dodekanotiolem dla zakresu
potencjału: -0.1÷ 0.5 V. Należy pamiętać aby po wyjęciu elektrody złotej z roztworu
dekanotiolu przepłukać ją etanolem oraz wodą destylowaną. Dopiero tak przygotowaną
elektrodę umieszczamy w naczynku elektrochemicznym.
9. Po wykonaniu pomiaru elektrodę z dodekanotiolem należy opłukać etanolem i ponownie
umieścić w roztworze tiolu.
10. Po wyznaczeniu pojemności dla elektrod zmodyfikowanych monowarstwami obliczamy
grubość monowarstw korzystając ze wzoru (2).
11. Ponadto korzystając ze wzoru (2) należy obliczyć oczekiwaną/teoretyczną wartość
pojemności dla elektrody złotej zmodyfikowanej dodekanotiolem, przyjmując, że długość
cząsteczki dodekanotiolu wynosi 1.65 nm. (UWAGA! – szacując grubość monowarstwy
należy pamiętać, że cząsteczki alkanotioli zaadsorbowane na złocie są nachylone pod
kątem 300 względem normalnej do powierzchni w przypadku alkanotiolu, dla disiarczku
bis-2-((ferrocenylokarbonylo)amino)etylowego kąt nachylenia wynosi 180). Uzyskane
wartości eksperymentalną i teoretyczną należy porównać i na tej podstawie ocenić jakość
monowarstwy. Czy uzyskana monowarstwa zawiera dużą czy raczej niewielką ilość
defektów?
III. Badanie przepuszczalności mowarstwy dla jonów
1. W naczynku elektrochemicznym umieszczamy roztwór 0.2mM Fc_CH2COONa
(M=266.04 g/mol) w 1 M NaClO4 oraz podłączamy elektrody – referencyjną, pomocniczą
oraz pracującą. Jako elektrodę pracującą podłączamy czystą elektrodę złotą. Roztwór
odtleniamy argonem lub azotem przez około 10-15 minut.
2. Rejestrujemy wotlamogram w zakresie -0.5V do 0.5V przy szybkości polaryzacji 0.05
V/s.
3. Na podstawie zarejestrowanej krzywej wyznaczamy potencjał formalny układu redoks. W
tym celu wykorzystujemy równanie (3).

92
4. Następnie w naczynku umieszczamy elektrodę złotą zmodyfikowaną dodekanotiolem i
podłączamy ją jako elektrodę pracującą.
5. Rejestrujemy wotlamogramy w zakresie -0.5V do 0.5V zwiększając stopniowo szybkość
polaryzacji od 0.05 V/s do coraz wyższych wartości, tak aby uzyskać różnicę pomiędzy
potencjałami pików większą niż 300 mV.
6. Wykorzystując krzywe cykliczne zarejestrowane (przy tej samej szybkości polaryzacji tj.
0.05 V/s) dla elektrody złotej i elektrody pokrytej monowarstwą tiolu obliczamy
współczynnik EBE zgodnie ze wzorem (4). Czy uzyskana wartość EBE świadczy o
wysokim czy raczej niskim stopniu uporządkowania monowarstwy?
7. Obliczamy stałą szybkości przeniesienia elektronu zgodnie z opisem we wstępie.
8. Korzystając z równania (6) obliczamy stopień pokrycia elektrody tiolem (jako wartość
stałej szybkości przeniesienia elektronu dla Fc_CH2COONa na niezmodyfikowanej
elektrodzie przyjmujemy 6×10-2 cm/s). Czy stopień pokrycia elektrody odzwierciedla
rezultaty uzyskane w badaniach pojemności?
IV. Elektrochemiczna charakterystyka monowarstwy zmodyfikowanej
próbnikiem redoks.
1. W naczynku elektrochemicznym z 1 M roztworem NaClO4 umieszczamy elektrody –
referencyjną, pomocniczą oraz pracującą zmodyfikowaną disiarczkiem bis-2-
((ferrocenylokarbonylo)amino)etylowym. Roztwór przed pomiarem odtleniamy argonem
przez około 10-15 minut.
2. Rejestrujemy wotlamogramy w zakresie 0.1V do 0.8V przy różnych szybkościach
polaryzacji: 0.01; 0.02; 0.05; 0.1 V/s. Dla każdej szybkości polaryzacji pomiar
wykonujemy 2-krotnie.
3. Z zarejestrowanych krzywych woltamperometrycznych wyznaczamy wysokości piku
anodowego oraz wykreślić zależność: wartość prądu piku anodowego względem
szybkości polaryzacji elektrody.
4. Korzystając z równania (7) obliczamy stopień pokrycia elektrody tiolem
zmodyfikowanym próbnikiem redoks. (UWAGA! – obliczając stopień pokrycia elektrody
tiolem elektroaktywnym należy pamiętać, że Q jest to ładunek obliczony z sygnału
anodowego próbnika redoks. Obliczenia należy wykonać dla każdej stosowanej szybkości
polaryzacji.). Następnie na podstawie otrzymanego średniego ładunku obliczamy stężenie

93
powierzchniowe elektroaktywnego tiolu na elektrodzie Γ Czy stopień pokrycia elektrody
odzwierciedla rezultaty uzyskane w badaniach pojemności?
5. Punkty 5-7 z rozdziału II powtórzyć dla elektrody zmodyfikowanej oraz disiarczkiem bis-
2-((ferrocenylokarbonylo)-amino)etylowym dla zakresu potencjału: 0.1÷ 0.8 V . Należy
pamiętać aby po wyjęciu elektrody złotej z roztworu tiolu przepłukać ją etanolem oraz
wodą destylowaną. Dopiero tak przygotowaną elektrodę umieszczamy w naczynku
elektrochemicznym.
6. Po wykonaniu pomiaru elektrodę z monowarstwą należy opłukać etanolem i ponownie
umieścić w roztworze tiolu.
7. Po wyznaczeniu pojemności dla elektrod zmodyfikowanych monowarstwami obliczamy
grubość monowarstw korzystając ze wzoru (2).
8. Ponadto korzystając ze wzoru (2) należy obliczyć oczekiwaną/teoretyczną wartość
pojemności dla elektrody złotej zmodyfikowanej disiarczkiem bis-2-
((ferrocenylokarbonylo)amino)etylowym wynosi 1.33 nm (UWAGA! – szacując grubość
monowarstwy należy pamiętać, że cząsteczki alkanotioli zaadsorbowane na złocie są
nachylone pod kątem 300 względem normalnej do powierzchni w przypadku alkanotiolu,
dla disiarczku bis-2-((ferrocenylokarbonylo)amino)etylowego kąt nachylenia wynosi 180).
Uzyskane wartości eksperymentalną i teoretyczną należy porównać i na tej podstawie
ocenić jakość monowarstwy. Czy uzyskana monowarstwa zawiera dużą czy raczej
niewielką ilość defektów?
V. Elektrochemiczna desorpcja cząsteczek alkanotioli z powierzchni złota
1. Przygotować 100 ml wodnego roztworu KOH (M=56.11 g/mol) o stężeniu 0.5 mol/l.
Następnie roztwór umieszczamy w naczynku elektrochemicznym oraz podłączamy
elektrody – referencyjną, pomocniczą oraz pracującą. Jako elektrodę pracującą
podłączamy czystą elektrodę złotą. Roztwór odtleniamy przez około 10-15 minut.
2. Rejestrujemy woltamogram w zakresie od –0.2V do –1.1V, przy szybkości polaryzacji
0.05V/s. (Czułość prądu ustawiamy na 1e-5
A).
3. Powtarzamy punkty 1 i 2 stosując zmodyfikowane dodekanotiolem, a następnie
disiarczkiem bis-2-((ferrocenylokarbonylo)amino)etylowym elektrody jako elektrody
pracujące. Porównujemy otrzymane woltamogramy.

94
4. Z woltamogramu zarejestrowanego dla elektrod pokrytych monowarstwami obu tioli
wyznaczamy ładunek związany z redukcją zaadsorbowanego tiolanu (pik desorpcji).
Następnie na podstawie otrzymanego ładunku obliczamy stężenie powierzchniowe tiolu
na elektrodzie Γ oraz stopień pokrycia elektrody θ (zakładając, że maksymalne pokrycie
alkanotiolem wynosi Γmax=7.8×10-10 mol/cm2, natomiast disiarczkiem bis-2-
((ferrocenylokarbonylo)amino)etylowym Γmax=4.5×10-10 mol/cm2 oraz przyjmując, że θ =
Γ/Γmax). Porównujemy uzyskany wynik z tym, który otrzymano w poprzedniej częsci
ćwiczenia. Czy uzyskane rezultaty są takie same czy różnią się od siebie? Jeśli się różnią,
jaka jest przyczyna? W przypadku monowarstwy z ferrocenową pochodną porównać
otrzymaną wartość Γ w podpunkcie IV.4 z wartością Γ z desorpcji.
VI. Charakterystyka topografii powierzchnii zmodyfikowanej elektrody za pomocą
mikroskopu STM
1. Do badań za pomocą Skaningowego Mikroskopu Tunelowego należy przygotować złotą
elektrodę płytkową. Przed pomiarem, elektrodę należy wyżarzyć w płomieniu palnika, a
następnie schłodzić do temperatury pokojowej i powtórzyć wyżarzanie. Procedura ta ma na
celu rekonstrukcje powierzchni zlota (111), i tym samym zminimalizowanie defektów
monowarstwy. W powyższy sposób przygotowaną elektrodę umieszczamy w roztworze
dodekanotiolu na około 1h. Następnie elektrodę płuczemy obficie etanolem i wodą
destylowaną. Topografię powierzchni monowarstwy badamy wykonując zdjęcia przy
małej wartości napięcia i dużym prądzie tunelowym, oraz przy dużej wartości napięcia i
małej wartości prądu tunelowania. Porównujemy uzyskane obrazy i warunki pomiarów.
Na zakończenie, dysponując zebranymi danymi, studenci powinni ocenić (według
własnego zdania), do jakiego typu z wymienionych we wstępie zastosowań nadawałaby się
przygotowana przez nich monowarstwa (każda rozsądnie uzasadniona odpowiedź będzie
akceptowana).