Embed Size (px)
Citation preview
www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
POSTER SESSION 3 P3. GENETICS, GENOMICS, AND PROTEOMICS
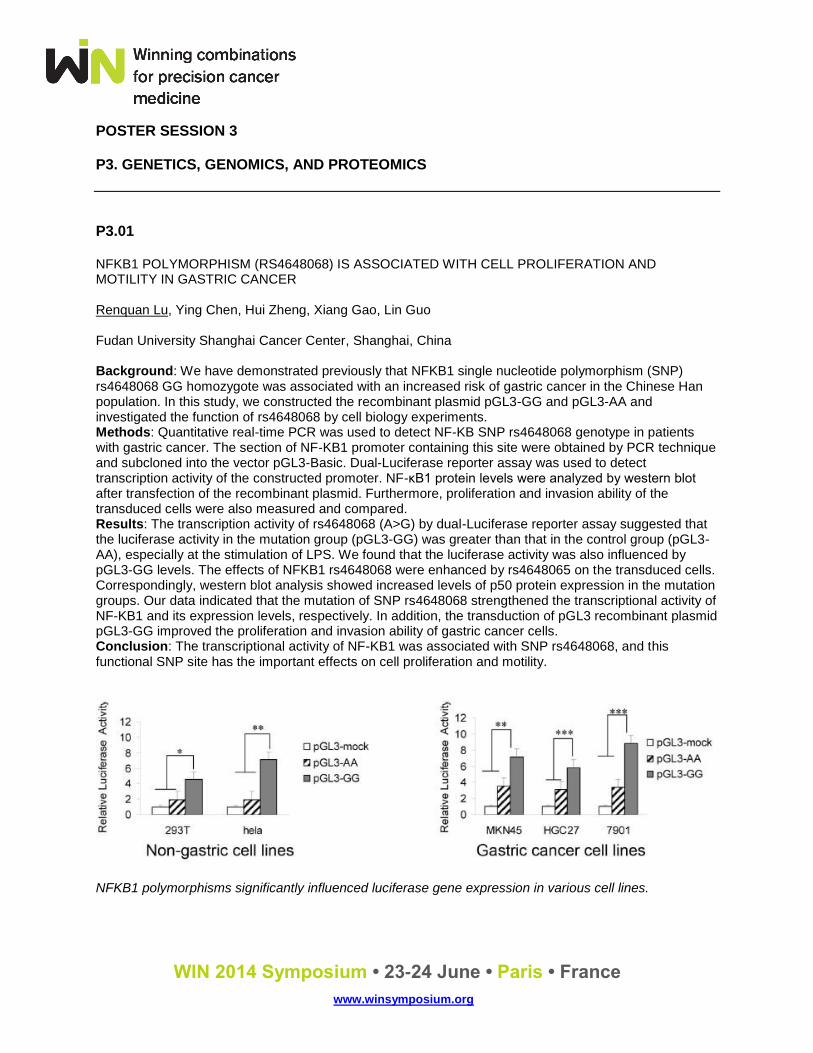
P3.01 NFKB1 POLYMORPHISM (RS4648068) IS ASSOCIATED WITH CELL PROLIFERATION AND MOTILITY IN GASTRIC CANCER Renquan Lu, Ying Chen, Hui Zheng, Xiang Gao, Lin Guo Fudan University Shanghai Cancer Center, Shanghai, China Background: We have demonstrated previously that NFKB1 single nucleotide polymorphism (SNP) rs4648068 GG homozygote was associated with an increased risk of gastric cancer in the Chinese Han population. In this study, we constructed the recombinant plasmid pGL3-GG and pGL3-AA and investigated the function of rs4648068 by cell biology experiments. Methods: Quantitative real-time PCR was used to detect NF-KB SNP rs4648068 genotype in patients with gastric cancer. The section of NF-KB1 promoter containing this site were obtained by PCR technique and subcloned into the vector pGL3-Basic. Dual-Luciferase reporter assay was used to detect transcription activity of the constructed promoter. NF-κB1 protein levels were analyzed by western blot after transfection of the recombinant plasmid. Furthermore, proliferation and invasion ability of the transduced cells were also measured and compared. Results: The transcription activity of rs4648068 (A>G) by dual-Luciferase reporter assay suggested that the luciferase activity in the mutation group (pGL3-GG) was greater than that in the control group (pGL3-AA), especially at the stimulation of LPS. We found that the luciferase activity was also influenced by pGL3-GG levels. The effects of NFKB1 rs4648068 were enhanced by rs4648065 on the transduced cells. Correspondingly, western blot analysis showed increased levels of p50 protein expression in the mutation groups. Our data indicated that the mutation of SNP rs4648068 strengthened the transcriptional activity of NF-KB1 and its expression levels, respectively. In addition, the transduction of pGL3 recombinant plasmid pGL3-GG improved the proliferation and invasion ability of gastric cancer cells. Conclusion: The transcriptional activity of NF-KB1 was associated with SNP rs4648068, and this functional SNP site has the important effects on cell proliferation and motility.
NFKB1 polymorphisms significantly influenced luciferase gene expression in various cell lines.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.02 SOMATIC MUTATIONAL PROFILING AND ITS ASSOCIATION WITH SURVIVAL IN LUNG SQUAMOUS CELL CARCINOMA Wanling Zeng
1, Wan-Teck Lim
2, Daniel Shao-Weng Tan
2, Sakktee Sai Krishna
2, Eng-Huat Tan
2, Tong
Kiat-Hon Lim3, Wan-Teck Lim
2
1Duke-NUS Graduate Medical School, Singapore, Singapore
2National Cancer Centre, Singapore, Singapore
3Singapore General Hospital
Background:Lung cancer is the leading cause of cancer death globally. Lung squamous cell carcinoma (SCC) is the second most common lung cancer subtype and molecular alterations of lung SCC are not well-characterized. Currently, there is no approved targeted agent for the treatment of lung SCC. We aim to indentify genetic alterations in samples of patients with lung SCC and to investigate the impact of genetic alterations on survival. Methods: We extracted DNA from formalin-fixed, paraffin-embedded tissue samples from 79 patients diagnosed with lung SCC at the National Cancer Centre Singapore (NCCS) from 1998 to 2010. Targeted mutational profiling of the tissue samples were performed using the Sequenom MassArray platform with LungCarta panel (SEQUENOM Inc., San Diego, CA). Overall survival was estimated using the Kaplan Meier method and its association with mutational status was assessed in the context of Cox proportional hazards models Results: In total, 79 patients with pathologically confirmed lung SCC were included in the study. At least one somatic mutation was detected in 53.2% of the tissue samples. Genetic mutations that were more commonly detected include STK11 (12.7%), TP53 (11.4%), EGFR (10.1%), EPHA2/3 (10.1%), PIK3CA (10.1%), NTRK1/2 (8.9%) and NFE2L2 (8.9%). Mutations in KRAS, DDR2, BRAF, MET, NOTCH1, JAK2, PTEN, FGFR4 and AKT were also found. There were 3 never-smokers and among the never-smokers, genetic alterations were identified in 2 out of 3 never-smokers. In survival analyses, there was no statistically significant association between mutational status of the patients and overall survival after adjusting for confounding variables. Conclusion: The identification of somatic mutations will contribute towards characterizing the molecular landscape of lung SCC and ultimately the development of targetable therapeutics for the treatment of lung SCC.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.03 AMPLIFICATION OF SINGLE NUCLEOTIDE POLYMORPHISMS OF THE EGFR GENE PROMOTER SEQUENCE IN NSCLC PATIENTS Vladimir Jurisic, Jasmina Obradovic University of Kragujevac, Kragujevac, Serbia Epidermal growth factor receptor (EGFR) is an important regulator of tumor growth and metastasis to be overexpressed in many tumours including NSCLC also. Based on variation in EGFR gene, not all NSCLC patients respond equally to this therapy. Therefore, personalisation of drug therapy in NSCLC patients requires genotyping of EGFR gene. The aim of this study was to test the effects several additives in various concentrations for amplification single nucleotide polymorphisms in promoter sequence of the EGFR since the promoter sequence containing the multiple GC regions which are difficult for the amplification. PureLink™ Genomic DNA Kits (Invitrogen/ Life Technologies, Carlsbad, CA) were used for extracion of DNA from formalin-fixed paraffin-embedded lung cancer tissue. EGFR polymorphisms -216G>T/ and -191C>A were genotyped using the PCR-RFLP method. Sequencing was conducted using ABI PRISM® BigDyeTM Terminator v 3.1 Cycle Sequencing Kit in both forward and reverse direction. Resultes showed that between several tested additives including; glycerol, DMSO, formamide, Tween 20, Triton X-100, PEG and BSA, only a two have effectiveness including glycerol and DMSO, and with best results at concentrations of 15% and 5% respectively. Comparison of the obtained sequence with the reference sequence of EGFR promoter region (http://www.ncbi.nlm.nih.gov; GenBank reference: M11234.1) revealed that the PCR amplification was highly specific. We have shown that using appropriate co-solvent it is possible amplify promoter region of EGFR for single nucleotide polymorphisms -216G>T or -191C>A, that contained multiple GC regions.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.04 WHOLE GENOME SEQUENCING INFORMS THERAPEUTIC SELECTION FOR PANCREATIC CANCER David Chang
1, Amber Johns
2, Anne-Marie Patch
1, Andrew Biankin, David Miller
1, John Pearson
1,
Elizabeth Musgrove1, Marina Pajic
1, Nicola Waddell
2, Peter Bailey
3, Sean Grimmond
1
1University of Glasgow, Glasgow, United Kingdom
2Garvan Institute of Medical Research, Sydney, Australia
3University of Queensland, Brisbane, Australia
Background: Pancreatic cancer remains one of the most lethal malignancies with profound molecular heterogeneity. Faced with this diversity, it is not surprising that therapeutic development using an unselected approach to patient recruitment has been challenging. Method and Material: We performed whole genome sequencing and CNV analysis of 100 pancreatic ductal adenocarcinomas recruited through the Australian Pancreatic Cancer Genome Initiative as part of the International Cancer Genome Consortium. Results: Chromosomal rearrangements leading to gene disruption was common, including those known to be important in pancreatic cancer (TP53, SMAD4, CDKN2A, ARID1A, ROBO2) and novel candidates (KDM6A, RNF43 and PREX2). Patterns of structural variation classified PDAC into 4 subtypes with potential clinical utility: stable, locally rearranged, scattered and unstable. A third of locally rearranged tumours harboured focal amplifications, many of which contained druggable oncogenes (ERBB2, MET, FGFR1, CDK6, PIK3R3, and PIK3CA), but at low individual frequency. Genomic instability co-segregated with inactivation of DNA maintenance genes (BRCA1, BRCA2, PALB2 or RPA1), and a mutational signature of DNA damage repair deficiency. This subgroup was associated with response to platinum-based therapy and defines candidate biomarkers of therapeutic responsiveness to platinum and potentially PARP inhibitors. Conclusions: This study provides the most comprehensive description of the genomic events that characterise pancreatic cancer to date and demonstrates that structural variation is a prominent mechanism of genomic damage in this disease. This approach may provide new opportunities in the development of biomarkers of therapeutic responsiveness to platinum-based therapies and PARP inhibitors for a number of cancer types including pancreas, breast, ovary and prostate.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.05 THE MYCN-HMGA2-CDKN2A PATHWAY IN NON-SMALL CELL LUNG CARCINOMA: IMPACT ON PROGNOSIS. Hanne Astrid Eide
1, Rita Halvorsen
1, Åslaug Helland
1, Cecilie Kiserud
2, Hossein Piri
3, Lars Jørgensen
4,
Maria Moksnes Bjaanæs1, Odd Terje Brustugun
1, Steinar Solberg
4,
1Oslo University Hospital-Radiumhospital, Institute for Cancer Research, Oslo, Norway
2Oslo University Hospital, Department of Oncology, Oslo, Norway
3Qazvin University of Medical Sciences, Quazvin, Iran
4Oslo University Hospital-Rikshospitalet, Oslo, Norway
Background:Extensive research in lung cancer has improved our understanding of the molecular, genetic and epigenetic alterations needed for tumor development in this deadly disease. Investigation on the functional importance of these alterations might lead to new paradigms in lung cancer treatment and eventually to improved survival. Activated MYCN-HMGA2-CDKN2A pathway has previously been shown to be associated with poor patient outcome in ovarian cancer amongst others. Could this also hold true for non-small cell lung cancers? Materials and methods:Tumor samples, collected from curatively intended non-small cell lung cancer surgery at Oslo-University Hospital, Oslo, Norway, were analyzed with RT-qPCR in regards of expression of 4 selected genes, MYCN, CDKN2A, DICER and HMGA2. Clinical data and histopathological features of the patients were collected from hospital medical journals. Micro-RNA profiling was also done in adenocarcinoma samples by using Agilent microarrays. Lastly, HMGA2 protein expression levels were determined by immunohistochemistry on tissue micro arrays (TMAs). Results:175 patients were included in the study with a median follow up time of 57 months. During follow up, 33.2% of the participants presented relapse of disease, either with local recurrence or metastasis. Of the four selected genes, MYCN expression levels were significantly associated with progression free survival. 55.1% of the samples had a high expression level of the HMGA2 protein. In histological subsets, 89.5% of the squamous cell carcinomas expressed high levels of while 46.6% of the adenocarcinomas showed overexpression of the HGMA2 protein. In survival analysis we saw no correlation of HMGA2 protein to progression free survival. Conclusions:High levels of MYCN in non-small cell lung cancer are associated with reduced progression free survival.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.07 CLINICAL NEXT GENERATION SEQUENCING (NGS) OF A LARGE SERIES OF ADVANCED LUNG ADENOCARCINOMA REVEALS A HIGH FREQUENCY OF TARGETABLE GENOMIC ALTERATIONS Gary Palmer, Norma Palma, Doron Lipson, Geoff Otto, Geneva Young, Jie He, Jeffrey Ross, Phil Stephens, Roman Yelensky, Siraj Ali, Vincent Miller Foundation Medicine, Cambridge, MA, USA Background: Targeted therapies and molecular diagnostics play major roles in the contemporary management of metastatic or relapsed lung adenocarcinoma (LA). Although LA genomics has been studied in various cohorts, accurate estimates of targetable alteration prevalence in advanced disease are not yet established. Here we describe our experience with a clinical next generation sequencing (NGS)-based assay, optimized for routine FFPE specimens, on a large series of LA samples. Methods: Hybridization capture of all exons of 236 cancer-related genes and 47 introns of 19 commonly-
rearranged genes was applied to ≥50ng of DNA extracted from 724 recent consecutive FFPE LA
specimens received in our CLIA laboratory and sequenced to a median depth of >500x. Specialized algorithms were applied to identify driver alterations including base substitutions, short indels, copy-number alterations and genomic rearrangements at low mutant allele frequencies. Results: NGS was performed on patients with relapsed/refractory disease in the majority of cases. Alterations were identified in over 150 genes, including the expected common alterations of KRAS (28%), EGFR (23%), CDKN2A (20%), and STK11 (14%). Less common targetable alterations occurred in ERBB2 (8%), MDM2 (7%), NF1 (6%), PIK3CA (5%), BRAF (5%), and MET (3%). Most genes were altered by multiple mechanisms, exemplified by ERBB2, which harbored both amplifications (5%) and activating indels (3%). Known and novel fusion events were identified in 10% of cases, including in ALK (5%), RET (3%), and ROS1 (2%). Follow-up of patients with novel ALK and RET rearrangements has demonstrated sensitivity to crizotinib and cabozantinib, respectively. Conclusions: NGS-based genomic profiling successfully performed on clinical FFPE lung adenocarcinoma samples uncovered a high frequency of alterations which can inform targeted treatment decisions. This real-world cohort represents the largest survey of these targetable alterations to-date. The ability of NGS to identify a wide range of known and novel alterations of all classes can result in immediate benefit for patients with metastatic disease.

www.winsymposium.org
WIN 2014 Symposium • 23-24 June • Paris • France
P3.08 CHARACTERIZATION OF A COLORECTAL CANCER PREDISPOSITION POLYMORPHISM AT THE POLD3 LOCUS Tessa Sandberg, Ian Tomlinson University of Oxford, Oxford, United Kingdom Many germline mutations have been identified that are associated with colorectal cancer (CRC) predisposition. However, CRC also develops in genetically susceptible individuals that are not carriers of any known mutations. A whole-genome sequencing study identified germline mutations in the polymerase genes POLE and POLD1, which were associated with a multi-generational increased risk of CRC and had high penetrance and dominant inheritance. A meta-analysis of five genome-wide association studies identified a polymorphism rs3824999 at the POLD3 locus associated with an increased risk in CRC. POLD3 is a regulatory subunit of the polymerase δ that plays a major role in ensuring polymerase processivity. In the following research, we aim to characterize the functional variation at/near rs3824999 and to investigate the functional mechanism resulting in CRC. Furthermore, we will relate this mechanism to the germline mutations in POLE and POLD1. To achieve these aims, we have refined the original signal by genetic fine-mapping followed by assessment of functional information in the ENCODE database. Secondly, analysis of chromatin in the region by chromatin immunoprecipitation and formaldehyde-assisted isolation of regulatory elements has been performed to find the functional-relevant SNP. Using this data we are currently testing the effect of the functional variation on gene expression an in vitro luciferase report assay and measuring allele-specific POLD3 expression in cell lines heterozygous for our top SNPs. Finally, in order to investigate the mechanism resulting in CRC, the processivity of polymerase δ will be measured by sequencing and compared between isogenic cell lines of high- and low-risk genotypes induced by CRISPR. The results will be presented on the day of the symposium.






























