Embed Size (px)
Citation preview

Universität Bremen
Porphyrin– und Porphyrin–Fulleren–Derivate –
Synthese und Immobilisierung auf Goldoberflächen
Dissertation
zur Erlangung des Grades eines
Doktors der Naturwissenschaften
(Dr. rer. nat.)
dem Fachbereich 2 (Biologie/Chemie)
im September 2008 vorgelegt
von
Christoph S. Eberle
Bremen 2008


Schriftliche Erklärung nach §6 Abb. 5 der Promotionsordnung vom 23. Mai 1984:
Ich, Christoph Stefan Eberle, habe die vorliegende Arbeit ohne unerlaubte fremde Hilfe
angefertigt, keine anderen als die von mir angegebenen Quellen und Hilfsmittel benutzt und
inhaltlich oder wörtlich entnommene Stellen aus benutzten Werken als solche kenntlich
gemacht.
1. Gutachter: Prof. Dr. Franz–Peter Montforts
2. Gutachter: Prof. Dr. Wolf–Dieter Stohrer
Tag des Rigorosums: 18. November 2008

Zeit
Aller Dinge Gehalt, er wird durch dich nur entschieden,
Leise Gottheit, auch mich richtest du, richte gelind.
Johann Wolfgang Goethe / Friedrich Schiller

D a n k s a g u n g
Experimentelle Arbeiten für diese Dissertation wurden durchgeführt von Juni 2005 bis
August 2008 im Institut für Organische Chemie an der Universität Bremen. Meinem
Doktorvater, Herrn Prof. Dr. Franz–Peter Montforts, gehört mein erster Dank: für seine
ausgezeichnete Betreuung, für hilfreiche Diskussionen, für konstruktive Kritik wie für
Freiheiten, die er mir bei der Bearbeitung dieses vielschichtigen Themas gewährt hat. Jede der
folgenden Seiten drückt diesen Dank am sichtbarsten aus. Für die Anfertigung des
Zweitgutachtens danke ich Herrn Prof. Dr. Wolf–Dieter Stohrer, der Universität Bremen
für die Gewährung eines Promotionsabschluss–Stipendiums. Ich schulde ferner allen, Dank
zu sagen, allen, die während der vergangenen Jahre diese Arbeit unterstützt, zu ihrem
Gelingen beigetragen haben:
Herrn Dr. Thomas Dülcks und Frau Dipl.-Ing. Dorit Kemken aus der instrumental–
analytischen Abteilung von Prof. Dr. Dieter Leibfritz für die Aufnahme zahlreicher Massen–
spektren, Herrn Dipl.-Ing. Johannes Stelten für seine Hilfe bei der Strukturaufklärung mittels
ein– und zweidimensionaler NMR–Experimente; Herrn PD Dr. Andreas Hartwig, Herrn
Dr. Klaus Rischka, Frau Dipl.-Chem. Katharina Richter und Herrn Dr. Michael Nöske vom
Fraunhofer Institut für Materialforschung und Angewandte Fertigungstechnik (IFAM) in
Bremen für ihre Hilfe bei der Untersuchung modifizierter Goldelektroden mittels Röntgen–
photoelektronenspektroskopie (XPS); Herrnge Rechnungen von Metalloporphyrin–Fulleren–
Dyaden, die in dieser Arbeit vorgestellt werden; den Mitarbeitern aus der Zentralen
Serviceeinrichtung Ver– und Entsorgung, Strahlenschutz (ZVES) des Fachbereichs 2, vor
allem Frau Corinna Knorr, Herrn Peter Ude und Herrn Walter Ohse, für angenehme
Zusammenarbeit auf allen Dienstwegen und für ihre „Ekstase aus Schweineleber“; den für
mich zuständigen Mitarbeitern aus der Universitäts– und Fachbereichsverwaltung Bremen:
über drei Jahre ließ ich mich gerne von ihnen verwalten.
Frau Prof. Dr. Luisa Maria Abrantes für ihre Einladungen nach Lissabon, um innerhalb des
DAAD–Projekts Nr. 40200248 elektrochemische und physikalische Daten an modifizierten
Goldoberflächen erheben zu können. Darüber hat sie steten Anteil am Fortgang dieser Arbeit
und meiner Portugiesisch–Kenntnisse genommen (diese selbst haben mir manche Entdeckung
v

bereitet und einen Rekord erhebender Begegnungen); Frau Dr. Ana Viana danke ich
für rastertunnelmikroskopische Aufnahmen, Herrn Dr. Jorge Correia für die ellipsometrische
Vermessung modifizierter Goldelektroden und beiden dafür, dass sie mich ebenso geduldig
wie kenntnisreich mit gängiger Oberflächenanalytik vertraut gemacht haben, darunter
Rastertunnelmikroskopie, Ellipsometrie und Cyclovoltammetrie. Auch danke ich Ana und
ihrem Mann Luis für viele Kleinigkeiten, die sie getan haben, damit ich zurückkehre und
mich willkommen fühle in der Stadt, „die die halbe Welt entdeckte“. Dies schließt meine
Lissabonner ein, jeden auf seine Weise: Frau Prof. Dr. Luisa Maria Abrantes, Herrn Dr.
Rodrigo da Alameda, Frau Dr. Ana Tenheiro, Herrn Dr. Jorge Correia, Frau Dr. Ana
Mourato, Frau Dr. Ana Melato, Frau Dr. Elisabete Valerio, Frau Virginia Ferreia MSc., Frau
Joana Cabrita MSc., Frau Dr. Alda Fundo, Herrn Luis Santos MSc., Herrn Dr. Antonio
Cascalheira (Lumisense) und Herrn Dr. Miguel Freitas (Lumisense) – hier rufe ich nochmals:
Muito obrigado a todos de primeiro cavalheiro lisboeta da alemanha! Und dafür, mich in
portugiesische Kultur eingeführt zu haben: von Bacalhau über Fado bis Fernando Pessoa.
Meinen ehemaligen und jetzigen Kollegen wie auch allen akademischen Gästen unseres
Arbeitskreises: Frau Idania Adams MSc., Herrn Lic. Rudy Martin, Herrn Dr. Nguyen Van
Dau, Frau Prof. Dr. Nguyen Thi Hui, Herrn Prof. Dr. Nguyen Dinh Thanh, Frau Prof. Dr. Ana
Margarita Esteva Guas, Herrn Prof. Dr. Luis Montero Cabrera, Herrn Prof. Dr. Lechosław
Latos–Gra�y�ski, Frau Dr. Genevieve Adukpo, Frau Dr. Ana Ruiz, Frau Dr. Rosa Saez, Frau
Dr. Agnieszka Kozielec, Herrn Dr. Mauricio Santos, Herrn Dr. Touraj Shokati, Herrn
Dr. Thorsten Könekamp, Herrn Dr. Doan Duy Tien, Frau Dr. Barbara Panek–Bryła, Herrn
Dipl.-Chem. Jan–Erik Damke, Herrn Dipl.-Chem. Martin Erbacher, Frau Dipl.-Chem.
Yvonne Neumann, Frau Dipl.-Chem. Daniela Bauer, Herrn Dipl.-Chem. Sebastian Bischoff,
Herrn Dipl.-Chem. Torben König, Herrn Hauptmann d. R. Dr. Stephan Leupold, Frau Ngyuen
Thi Viet Thanh MSc., Herrn Dr. Vladimir Azov, Frau Barbara Szyma�ska MSc., Frau
Izabella Baraniec MSc., Herrn Andrzej J�drzejczuk MSc., Herrn Maciej Skibi�ski MSc.,
Herrn Dennis Leupold, Herrn Mathias Dücker, Frau Dr. Martina Osmers, Frau Ursula
Lücking, Frau Anngret Lincke und Frau Jessica Schmal. Mit Bremen bleiben Zeiten
verknüpft, die wir miteinander teilen. An beides erinnere ich mich gern, doch bleibt
wesentlicher neben dem Erinnern das, was wir verinnerlichen. Gemeinsame Zeiten, drei Jahre
dauerten sie, sogar ein wenig länger, und wenn der Vorhang fällt, zählen wir Verweil–Dochs,
vi

die Augenblicke, die groß sind und sein mögen. Für alle Zeiten sollten wir –wie Rilke uns–
nur eines wünschen: „Tiefbesiegt von immer Größerem zu sein.“
Meinen Forschungspraktikanten Katrin Lummer, Andre Wichmann, Andreas Schnieber und
Jan Würriehausen danke ich für tatkräftige Mitarbeit und ihr über das Synthetische hinaus
bekundete Interesse an den Projekten. Beides sei hier noch einmal gewürdigt.
Folgenden Autoren, Urheberrechteinhabern und lizenzgebenden Verlagen danke ich für
die Genehmigung des Nachdrucks von Abbildungen und Zitaten in dieser Arbeit: Verlag
Wiley–VCH (Weinheim), Teubner–Verlag (Wiesbaden), Gesellschaft Deutscher Chemiker
(Frankfurt a. M.), Amerikanische Chemische Gesellschaft (Washington, DC), Herrn Prof. Dr.
Franz–Peter Montforts (Universität Bremen), Frau Prof. Dr. Anke Krüger (Universität Kiel),
Frau Dr. Ana Viana (Universität Lissabon, Portugal), Herrn John Robert Marlow („Nano“),
Herrn Mike Treder und Herrn Chris Phoenix (Center for Responsible Nanotechnolgy, USA),
Herrn Dr. Mihail Roco (National Science Foundation, USA), Herrn Dr. Miroslaw Cygler
(Biotechnology Research Institute, Canada), Herrn Prof. Dr. Stephen Mann FRS (University
of Bristol, Großbritannien), Herrn Prof. Dr. Robert Langer (Massachusetts Institute of
Technology, USA) sowie Herrn Prof. Dr. Devens Gust (Arizona State University, USA).
Denen, deren kritische Durchsicht geholfen hat, das Manuskript in seine endgültige Form zu
bringen, an sie richtet sich besonderer Dank: Herrn Dr. Vladimir Azov, Frau Dipl.-Chem.
Daniela Bauer und Frau Dipl.-Chem. Yvonne Neumann. Kathy (Tacoma, Washington), Jean
und Edward (Silver City, New Mexico) bin ich immer wieder zu Dank verpflicht wie sie sich
Korrekturen meiner englischsprachigen Texte annehmen, einschließlich Kapitel 6 dieser
Arbeit.
Ausgesprochen und von ganzem Herzen dankte ich früher schon Kathy, Rudy, Rayser, Maité,
Krzysztof, Barbara und Szymon, die selbst der Richtung folgen, in die sie weisen, ebenso der
gesamten Gemeinschaft „Ja Pan“ – und für das, was uns einander unsichtbar verbindet. Noch
einmal: allen Freunden, Bekannten und Weggefährten, die diese Arbeit aus der Nähe oder
Ferne verfolgt haben, neugierig, unausgesetzt, korrespondierend, danke ich wie meiner
Familie, die es nie bereute, mich gewähren zu lassen. Wie ich auch.– Ohne ihn zu sehen,
vielleicht war immer schon mein Weg dort angelegt, von wo ich ausging. Die mit mir
Strecken gehen und gegangen sind, ihnen ist diese Arbeit gewidmet.
vii

In h a l t s v e r z e i c h n i s
Danksagung v
1. EINLEITUNG 12
1.1 Metalloporphyrinoide als Biokatalysatoren und künstliche
Photosynthese–Systeme 16
1.2 Das Prinzip Selbstorganisation von Molekülen und dessen Anwendung
in der Nanotechnologie 22
1.3 Selbstorganisierte Monoschichten 28
2. AUFGABENSTELLUNG 35
2.1 Synthese von immobilisierbaren Porphyrinoiden, Metalloporphyrinoiden
und Fullerenen 35
2.2 Aufbau von Donor–Akzeptor–Systemen, organischen Sensoren und
Katalysatoren 35
3. DURCHFÜHRUNG DER SYNTHESEN 37
3.1 Gewinnung enantiomerenangereicherter Liponsäure mittels kinetischer
Racematspaltung mit Lipase aus C. rugosa 37
3.2 Synthese einfacher Liponsäureester und –amide 42
3.3 Synthese eines Fulleren–Liponsäure–Derivates 44
3.4 Aufbau einer Porphyrin–Fulleren–Dyade auf Goldoberflächen 44
3.5 Synthese von Porphyrinbisestern 48
4. UNTERSUCHUNG VON MODIFIZIERTEN ELEKTRODEN 50
4.1 Immobilisierung auf Goldoberflächen 50
4.2 Cyclovoltammetrie 51
4.2.1 Elektrochemische Charakterisierung von Fulleren–Monoschichten auf
Goldoberflächen 51
viii

4.2.2 Elektrochemische Charakterisierung von Metalloporphyrin–Fulleren–
Monoschichten auf Goldoberflächen 55
4.2.3 Elektrochemische Charakterisierung von Metalloporphyrin–Monoschichten
auf Goldoberflächen 65
4.3 Physikalische Charakterisierung 70
4.3.1 Röntgenphotoelektronenspektroskopie (XPS) 70
4.3.2 Rastertunnelmikroskopie 73
4.3.3 Ellipsometrie 77
4.4 Semiempirische Rechnungen 79
5. ZUSAMMENFASSUNG UND AUSBLICK 81
5.1 Synthese von immobilisierbaren Porphyrinoiden, Metalloporphyrinoiden
und Fulleren–Derivaten 81
5.2 Ausblick 83
6. SUMMARY AND OUTLOOK 84
6.1 Synthesis and immobilisation of porphyrinoids, metalloporphyrinoids and
fullerene derivatives 84
6.2 Outlook 86
7. EXPERIMENTELLER TEIL 87
7.1 Abkürzungen und Symbole 87
7.2 Allgemeine experimentelle Bedingungen 90
7.2.1 Analytische Methoden 90
7.2.2 Chromatographie 93
7.2.3 Qualität verwendeter Lösungsmittel 93
7.2.4 Software und Datenbank 93
7.3 Darstellung von Octyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 1a und
5-[(3R)-1,2-dithiolan-3-yl]pentansäure 1 95
7.3.1 Darstellung von Hexyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 1b und
5-[(3R)-1,2-dithiolan-3-yl]pentansäure 1 98
ix

7.4 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]-N-[3-(1H-imidazol-1-l)propyl]
pentanamid 3a 100
7.4.1 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]-N-[2-(1H-imidazol-5-yl)ethyl]
pentanamid 3b 102
7.5 Darstellung von 3-(1H-pyrrol-1-yl)propyl 5-[(3R)-1,2-dithiolan-3-yl]
pentanoat 4a 104
7.5.1 Darstellung von 2-(1H-pyrrol-1-yl)-1-(1H-pyrrol-1-ylmethyl)ethyl 5-[(3R)-1,2-
dithiolan-3-yl]pentanoat 4b 106
7.6 Darstellung von Phytyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 5 108
7.7 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]pentan-1-ol 6 110
7.8 Darstellung von (3’’R)-(1,1-Dioxido-2,5-dihydrothien-3,4-diyl) bis(methylen)
di-5’-(1“,2“-dithiolan-3“-yl) pentanoat 8 112
7.8.1 Darstellung von (3”-R)–Cyclohex-4’-en [1’.2’:1.9] (C60-Ih) [5,6]Fulleren-4’,5’-
diyl bis(methylen)di-5-(1”.2”-dithiolan-3”-yl) pentanoat 9 114
7.9 Darstellung von 3,3’-[2,7,12,18-Tetramethyl-porphyrin-13,17-diyl]-
dipropionsäure-dimethylester 13 116
7.9.1 Darstellung von [13,17]-Bis(3-hydroxypropyl)-2,7,12,18-tetramethyl-
porphyrinato]-cobalt(II) 15c 118
7.9.2 Darstellung von [13,17]-Bis(3-hydroxypropyl)-2,7,12,18-tetramethyl-
porphyrinato]-mangan(III)-chlorid 15d 120
7.9.3 Darstellung von [13,17]-Bis(3-hydroxypropyl)-2,7,12,18-tetramethyl-
porphyrinato]-cobalt(III)-chlorid 15e 122
8. Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl}-porphyrin 16a 124
8.1 Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl-porphyrinato}-zink(II) 16b 126
8.1.2 Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl-porphyrinato}-nickel(II) 16c 128
8.1.3 Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl-porphyrinato}-cobalt(II) 16d 130
8.1.4 Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl-porphyrinato}-mangan(III)-chlorid 16e 132
x

8.1.5 Darstellung von {13,17-Bis[2-(4-[(3R)-(1,2-dithiolan-3-yl]-pentyl)]-carbonyl-
ethoxy]-2,7,12,18-tetramethyl-porphyrinato}-cobalt(III)-chlorid 16f 134
9. LITERATURVERZEICHNIS 136
10. LEBENSLAUF 143
xi

Dissertation -12- Christoph S. Eberle
1. EINLEITUNG
“For nature is not in a hurry and mankind is.” Giacomo Ciamician
Dem Treibhauseffekt entgegenzuwirken, ist Ziel des Kyoto–Protokolls. Staaten, die das
Vertragswerk ratifizieren, nimmt es in die Selbstverpflichtung. Seine Vorgaben
verlangen, den Kohlendioxid–Ausstoß über Jahrzehnte erheblich zu senken. Neben der
Rahmenkonvention der Vereinten Nationen über Klimaänderungen bekundet das 2005 in
Kraft getretene Kyoto–Protokoll erstmals eine politische Absicht der Völkergemeinschaft,
gemeinsam Auswirkungen der Erderwärmung im 21. Jahrhundert begegnen zu wollen. Da die
heutige Erderwärmung vom Menschen verursacht wird, wird sie zum Politikum, denn ihre
Folgen betreffen die Beziehungen zwischen den Menschen. Bliebe nämlich der langfristige
Klimawandel unbeachtet, drohten der Weltwirtschaft Folgeschäden von umgerechnet bis zu
5.5 Billionen Euro bis zum Jahr 2050. So lautet die Schätzung, die der „Stern Report on the
Economices of Climate Change“ nennt, eine Studie, die von der britischen Regierung in
Auftrag gegeben und Ende 2006 veröffentlicht worden war.[1]
Daneben hat das Kyoto–Protokoll entscheidende Fragen aufgeworfen: die Frage, welche
Rohstoffquellen künftig den Energiebedarf der Menschheit decken sollen, ohne die Umwelt
zu schädigen; die Frage nach effizienterer Nutzung von Primärenergie und nach umfassenden
Maßnahmen, Energie nachhaltig einzusparen. Gleichzeitig wächst mit der Weltbevölkerung
der Druck, dieser Problematik Herr zu werden. Denn sie geht einher mit politischen
Interessen und dem Kampf um Wettbewerbsfähigkeit von Unternehmen. Beides wird
verschärft durch den kraftvollen Aufschwung, den die Volkswirtschaften einstiger
Entwicklungsländer genommen haben, vor allem die Chinas und Indiens. Immer höherer
Energieverbrauch ist und wird der Preis dafür sein, dass Bedürfnisse von immer mehr
Menschen befriedigt werden können. Doch erst die Ressourcen von vier Planeten ähnlich der
Erde könnten ausreichen, um die Lebensverhältnisse in allen Ländern auf OECD–Standard zu
bringen.[2] Aus diesen Gründen ist die Chemie in all ihren Disziplinen besonders gefordert, da
sie Antworten geben kann, die zur Lösung dieser Fragen besonders beitragen. Verwertbare
Lösungen setzen auf beiden Seiten der Energiebilanz an, um aus erneuerbaren Ressourcen mit
höchstmöglichem Wirkungsgrad Energie zu erzeugen, um ihren Verbrauch rentabel zu halten
und Überschüsse zu speichern. Anstrengungen, die darauf gerichtet sind, lohnen nur, wenn
sich Grenzen der technischen Machbarkeit erweitern lassen. In ihnen verläuft der Weg, auf

Dissertation -13- Christoph S. Eberle
dem Fortschritte erzielt werden können; außerhalb dieser Grenzen beginnt das Utopische.
Nanotechnologie, wie sie nun erdacht wird, hat mit beidem zu tun; sie gewinnt erst Gestalt
über vielerlei mögliche und möglich gemachte Anwendungen. Sie wiederum wecken große
Erwartungen.
„Die viel diskutierte Photovoltaik beispielsweise erfordert völlig neue Materialen, um
wirtschaftlich zu werden. Hier sind die Anorganiker, die Organiker, die Polymerchemiker, die
Analytiker und Wissenschaftler anderer Fachrichtungen gefordert. Ähnliches gilt für neue
Materialien in leistungsfähigen Batteriesystemen oder Brennstoffzellen“, schreibt Dieter Jahn,
Präsident der Gesellschaft Deutscher Chemiker, im Vorwort zu den Nachrichten aus der
GDCh–Energieinitiative vom April 2007. Daher betont er, dass das Forum, um verschiedene
Ansätze zu diskutieren geschaffen werden muss, innerhalb von Fachkreisen und außerhalb.
Außer mit der breiten Öffentlichkeit, gilt es, so Jahn weiter, das Gespräch zu suchen mit
Verantwortlichen in Politik und Verwaltung, damit die nötigen Fördergelder für
wissenschaftliche Projekte bereitgestellt werden. „SusChem, die europäische Technologie–
plattform für nachhaltige Chemie, hat Energieforschung zu einem Schwerpunkt gemacht, und
auch auf nationaler Ebene, in der Hightech–Strategie der Bundesregierung steht Energie–
forschung ganz oben auf der Prioritätenliste.“
Dies zwingt dazu, auch das bisherige System des Wirtschaftens zu überdenken, wie es sich
seit Beginn der Industrialisierung entwickelt hat. Es ist wesentlich auf die Verbrennung
von Kohle, Gas und Öl angewiesen. Immer noch bestreiten diese Brennstoffe über Dreiviertel
des Energiebedarfs weltweit. Seit zwanzig Jahren aber übersteigt die Abbaurate fossiler
Bodenschätze die Zahl von neu erschließbaren Lagerstätten. Je mehr diese Quellen versiegen,
je unmöglicher Neufunde werden, desto dringender bedarf es Rohstoffe, die fossile
wirtschaftlich ersetzen können. Dies hat dazu geführt, dass am Weltmarkt das Angebot
beständig hinter der Nachfrage bleibt. Schon heute werden Rohölsorten weit teurer gehandelt
als zu Zeiten des ersten Preisschocks in den 1970er Jahren. Es steht zu vermuten, dass das
weltweite Fördermaximum bald erreicht werden könnte (so genannter Ölpunkt, engl. „Oil
Peak“ oder „Depletion Peak“), je weniger die OPEC–Staaten in der Lage scheinen,
Kapazitäten auszuweiten, insbesondere in Saudi–Arabien. Nimmt man an, dass sich die
Gesamtmenge förderbaren Erdöls als Glockenkurve beschreiben lässt, dann ist an genanntem
Punkt die Hälfte gefördert worden. Solch natürliche Verknappung verteuert die zweite Hälfte
danach dauerhaft. So hat der von frühen Fürsprechern beschworene Paradigmenwechsel[3]
begonnen, sich in der Weltpolitik bemerkbar zu machen: statt Sonnenenergie nur in
erdgeschichtlich gespeicherten Formen zu nutzen, gilt es, sich aus der solaren Quelle primär

Dissertation -14- Christoph S. Eberle
zu versorgen, um das Energieproblem zu lösen mit all seinen Aspekten. Bisher stellten fossile
Brennstoffe die Energie der Sonne am konzentriertesten bereit. Auf ihrem gigantischen
Konsum hat die moderne Zivilisation Wohlstand und Fortschritt errichtet, der ungleich
verteilt ist. Damit hat sie sich in eine Abhängigkeit begeben, die paradox anmutet, da mehr
Sonnenlicht auf die Erde einstrahlt als dort genutzt wird und da ihre fossilen Speicher für die
Fertigung anderer Industrieprodukte zu wertvoll sind, als dass diese weiter verbrannt werden
dürften. Das Verdienst, auf dieses Paradoxon zuerst hingewiesen zu haben, gebührt dem
italienischen Forscher Giacomo Ciamician. 1912 fragte er in seiner weitblickenden Vorlesung
„The Photochemistry of the Future“[4], wie die Menschheit von der Natur lernen könne, die
Strahlungsenergie der Sonne besser einzusetzen und sinnvoller als mit fossilen Stoffen. Lernte
sie mit Hilfe der Photochemie eine solche Lektion, würde dies in Zukunft sowohl die
Grundlagen als auch den Fortschritt moderner Zivilisation sichern. Ciamicians Prophetie ist
uns heute näher denn je. Mittlerweile zeichnet sich ab, dass die praktikabelste und
vernünftigste Alternative darin besteht, auf eine Kombination aus erneuerbaren
Energieträgern zurückzugreifen. Unter ihnen werden denjenigen wesentliche Bedeutung
zuwachsen, die Sonnenlicht nutzen, mittelbar oder unmittelbar.[5]
Die gegenwärtige Energieversorgung
gesamte Primärenergieversorgung weltweit 478.7 Exajoule gesamte Primärenergieversorgung Deutschland 14.4 Exajoule Stromverbrauch weltweit 16695 TWh Stromverbrauch Deutschland 586 TWh Kraftwerkskapazität Deutschland (Elektrizität) 119 GW Anteil regenerativer Energien am Stromverbrauch Deutschland 14.3 % (2007)
Tabelle 1: Daten zur gegenwärtigen Energieversorgung. Falls nicht anders vermerkt, beziehen sich alle Zahlen auf das Jahr 2005 (nach Zweibel, Mason, Fthenakis, 2008).[7]
Ca. 1.2 x 1017 W[6] strahlt die Sonne täglich zur Erde. Ungleichmäßig und diffus verteilt sich
diese elektromagnetische Strahlung auf der Planetenoberfläche (ca. 170 Wm-2). Doch genügte
ein Bruchteil davon, den gegenwärtigen Energieverbrauch weltweit zu decken. Dieser beträgt
in etwa 2.5% der solaren Leistung oder –anders gesagt– vierzig Minuten ihrer täglichen
Einstrahlzeit.[7] Diese Kraftquelle treibt geologische Kreisläufe an und biochemische
Prozesse, deren bedeutendster die Photosynthese ist. Algen, Pflanzen und Bakterien, die dazu
in der Lage sind, sichern letzten Endes die stofflichen Grundlagen für das natürliche
Gleichgewicht der Biosphäre und das Überleben in ihr; sie machten immer höher entwickeltes
aerobes Leben erst möglich. Aus all diesen Gründen ist der heutigen Forschung zweierlei

Dissertation -15- Christoph S. Eberle
aufgegeben: zu verstehen, wie Licht– und Dunkelreaktion bei der Photosynthese verlaufen
und Mittel und Möglichkeiten zu finden, sie sich technisch anzueignen.

Dissertation -16- Christoph S. Eberle
1.1 Metalloporphyrinode als Biokatalysatoren und künstliche
Photosynthese–Systeme
Die Bruttogleichung der Photosynthese beschreibt vereinfacht folgende endergonische
Reaktion (�G° = 2880 kJ/mol):
6 CO2 + 6 H2O C6H12O6 + 6 O2
Ihr Ergebnis ist die Sauerstofferzeugung aus Wasser, während Lichtenergie der Sonne
in chemische Bindungsenergie eingespeist wird. Diese Energieumwandlung setzt mehrere
Redoxreaktionen voraus, bei denen Elektronen und Protonen wandern. Dies ermöglicht z.B.
Pflanzen, an der Thylakoidmembran ihrer Chloroplasten ein Potentialgefälle zu erzeugen,
dessen sie sich bedient, um ATP und NADPH zu synthetisieren. Diese Reduktionsäquivalente
werden in den Calvin–Zyklus eingeschleust, um Kohlendioxid, durch Rubisco fixiert, für
den Glukoseaufbau zu verbrauchen. Elektronentransfers gekoppelt mit der gleichzeitigen
Verschiebung von Protonen kommen in der Photosynthese ebenso vor wie in vielen
metabolischen Stoffwechselwegen: im mitochrondrialen Elektronentransport über die
Enzyme der Atmungskette, während des Fettsäureabbaus, der O2–Bindung an Hemerythrin
oder der DNA–Replikation und Reparatur.[8] Ohne den mechanistischen Beitrag von
Metalloporphyrinoiden liefen all diese Prozesse nicht so ab, wie sie uns bekannt sind.
Eingebunden als Kofaktoren, werden sie von ihrer jeweiligen Proteinumgebung in den dafür
günstigen Konformationen gehalten, so dass insgesamt die Reorganisationsenergie gemäß der
Marcus–Theorie gering bleibt. Allgemein spielen gekoppelte Transfers von Elektronen und
Protonen überall dort eine Rolle, wo Energie gewonnen, gespeichert oder ineinander
überführt werden soll.
Den natürlichen photoinduzierten Elektronentransfer im Detail zu verstehen, war und
ist Gegenstand vielfältiger Forschungsvorhaben.[9] Die theoretischen Grundlagen dieses
Gebiets stammen von R. A. Marcus, der dafür 1992 mit dem Chemienobelpreis geehrt wurde.
Seit Jahren widmet sich die Fachliteratur ausgiebig unter verschiedenartigen Aspekten der
Konstruktion von leistungsfähigen Modellsystemen (Abb. 1). Sie sollen die lichtinduzierte
Ladungstrennung nachahmen, die den initialen Schritt des photosynthetischen Prozesses
darstellt. Denn mit Absorption von Licht im spektralen Bereich der Chlorophylle gehen
diese in einen energetisch angeregten Zustand über (S0 –> S1 Übergang). Dadurch kommt
ein Redoxpotential zustande, das ausreicht, alle darauf folgenden Transferreaktionen in Gang

Dissertation -17- Christoph S. Eberle
zu setzen. Genau diesen ladungsseparierten Zustand zu erzeugen und ihn so lange zu erhalten,
bis er in ein technisch nutzbares Potential überführt werden kann, auf diesem Prinzip beruhen
Abbildung 1: Ausgewählte Beispiele photosynthetischer Modellsysteme.
künstliche Photosynthese–Systeme (Abb. 2). Noch erreichen heutige technische
Photosynthese–Systeme bei Weitem nicht den Wirkungsgrad der natürlichen Photosynthese
von etwa 30%. Um Sonnenlicht mit Hilfe solch künstlicher Systeme effizienter zu nutzen,
genügt es nicht allein, nach Ladungstrennung die verschiedenen hochenergetischen Zustände
zu durchlaufen und möglichst lange zu erhalten. Zuvor muss Sonnenlicht gebündelt und am
Ende in eine leicht speicher– und transportierbare Energieform umgewandelt werden, wie
dies in Pflanzen–, Bakterien– oder Algenpigmenten gelingt. Modellsysteme zum Studium
des lichtinduzierten Elektronentransfers bestehen aus einem Donor und Akzeptor. Als
Redoxpartner werden diese zu einer molekularen Dyade vereint. Entweder hält eine starre
Brücke beide Partner zusammen oder sie sind flexibel kovalent miteinander verknüpft.
Diskutiert wird neuerdings, die durch lichtinduzierten Elektronentransfer induzierten
Radikalionenpaare in wohl abgestimmten Netzwerken zu organisieren.[10] Als kooperierende
Dyaden sollen sie ferner für binärlogische Funktionen taugen, und von ihnen erwartet man
schnelle Ladungstrennung, aber eine verlangsamte Relaxation aus ihrem angeregten in den
Grundzustand. Der angeregte Zustand wird zunächst so erreicht, dass nach Absorption
N N
N N
H3C
CH3
CH3H3C
O O
Zn
O O
N N
N N
H3C
CH3
CH3
CH3
Zn
H3C
NNH
NHN
NH
O
O
ONH
O

Dissertation -18- Christoph S. Eberle
eines Lichtquants ein Elektron aus dem HOMO des Donors in dessen LUMO gelangt.
Von dort ist die Übertragung ins entsprechende LUMO des Akzeptors möglich, ehe
Ladungsrekombination das System in den Grundzustand zurückführt. Insgesamt konkurrieren
folgende Reaktionen zur Ladungstrennung (vgl. Jablonski–Schema): die strahlungslose
Rückkehr in den Grundzustand, indem Wärme an die Umgebung abgegeben wird („internal
conversion“), Relaxation durch Fluoresenz in den Grundzustand oder der Übergang in den
Triplettzustand unter Spinumkehr („intersystem crossing“), dessen Deaktivierung länger
dauert. Geschwindigkeitskonstanten all dieser Reaktionen fließen in die Berechnung der
Quantenausbeute ein, die die Effizienz des Elektronentransfers (5) angibt.
1 Anregung 2 Interne Konversion (“Internal Conversion”) 3 Fluoreszenz 4 Intersystemwechsel (“Intersystem Crossing”) 5 Elektronentransfer 6 Ladungsrekombination
Abbildung 2: Jablonski–Schema für die möglichen elektronischen Zustände einer Porphyrin–Chinon–Dyade nach Lichtabsorption.
Es liegt nahe, auch bei der Konstruktion künstlicher Systeme sich die an den natürlichen
photosynthetischen Reaktionen beteiligten Biomoleküle zum Vorbild zu nehmen. Dies
sind Chlorophyll–Chromophore, die in leicht unterschiedlicher struktureller Abwandlung
in allen photosynthetischen Organismen vorkommen. Abgeleitet sind die Chlorophylle von
einem gemeinsamen Biosynthesevorläufer, dem Uroporphyrinogen III. In Anlehnung an
diese natürlichen Vorbilder dienen in künstlichen Systemen meist porphyrinoide Grundkörper
NH N
N NH
O OO
O
O
O
CH3 CH3
CH3
Energie (eV)
1.0
2.0
0
1D*-A
1 2 3
3D*-A
D.+-A.-
0.1 ns
D-A
4
5
6

Dissertation -19- Christoph S. Eberle
als Donoren, vor allem Porphyrin– und Chlorinderivate, die sowohl partial– als auch
totalsynthetisch zugänglich sind. Beide Konzepte wurden in vorangehenden Arbeiten in
unserer Gruppe entwickelt.[11] Als Elektronenakzeptoren eignen sich neben Chinonen ganz
besonders Buckminster–Fullerene, weil sie über gut dokumentierte photophysikalische
Eigenschaften verfügen.[12] Mit diesen zu Dyaden kombinierten Bausteinen konnten
strukturelle Aspekte der photosynthetischen Reaktionszentren aufgeklärt und Faktoren
verstanden werden, die den lichtgetriebenen Elektronentransfer beeinflussen. Er hängt ab vom
Abstand des Donor–Akzeptorpaares, deren Orientierung zueinander und von deren
Redoxpotentialen. Aber auch Orbitalsymmetrien üben beträchtlichen Einfluss aus, da sie für
die Ladungstrennung und –rekombination bestimmte Übergänge jeweils erlauben oder
verbieten.[11c, 13] Um Einsichten in den Mechanismus zu gewinnen, wie Elektronen in Dyaden
genau übertragen werden, muss letztlich eine spezifische Brücke gewählt werden. Entweder
vermittelt sie eine elektronische Kopplung über Bindungen oder durch den Raum zwischen
Donor und Akzeptor. Im ersten Fall bedeutet dies Übertragung von Elektronen entlang von
Bindungen („through bond“), im zweiten Fall über das Lösungsmittel („through space“).
Abbildung 3: Prinzip einer molekularen Batterie. Sie beruht auf lichtgetriebenem Elektronentransfer in künstlichen Donor–Akzeptor–Systemen (nach Gust, 1997).[20] Wie schon angeklungen, nehmen porphyrinoide Naturstoffe vielfältige Aufgaben wahr;
darunter fallen die wichtigsten in den Bereich der Bioenergetik und Biokatalyse. All dies ist
möglich aufgrund des Ladungswechsels ihres jeweiligen Metallzentralatoms. Als Kofaktor
von Enzymen fügen sie sich in die eigens für sie geschaffene molekulare Umgebung, wobei
die Variation der Substituenten und der jeweilige Sättigungsgrad ihres chromophoren Systems
auffällt.[14] In der Natur gehören der rote Blutfarbstoff Häm, ein Eisenkomplex mit

Dissertation -20- Christoph S. Eberle
Porphyringrundgerüst, das grüne Pflanzenpigment Chlorophyll a, ein mit Magnesium
komplexierter, teilgesättigter Tetrapyrrolzyklus, der prototypisch ist für die Strukturmerkmale
von natürlichen Chlorinen, daneben Bakteriochlorophylle, denen entsprechend eine
Bakteriochlorinstruktur zugrundeliegt, ferner der Faktor F 430 in methanogenen Bakterien,
ein Nickelporphyrinoid, sowie das für Menschen essentielle Vitamin B12, ein Cobaltkomplex
mit corrinoidem Grundmuster zu den wichtigsten Porphyrinoiden.
Schema 1: Elektrokatalytische Epoxidierung von Ethylen mit Hilfe eines durch Elektropolymerisation von Pyrroleinheiten immobilisierten MnCl–Porphyrins.[16]
In unserem Laboratorium wurden partialsynthetische Routen erarbeitet, um Porphyrinoide
und Metalloporphyrinoide herzustellen.[11a,b,d,f], [15] Als Derivate natürlicher Vertreter sind
sie von vornherein als Modellsysteme maßgeschneidert, um biologische Funktionen
nachzuahmen, aber auch andere Reaktionen zu induzieren. Anders als bei totalsynthetischen
Konzepten werden die Porphyrinoide aus natürlichen Quellen gewonnen und danach
abgewandelt, um sie auf leitfähigen Oberflächen immobilisieren. So ließ sich z.B. ein mit
Pyrrolringen verknüpftes MnCl–Porphyrin auf Graphitelektroden elektropolymerisieren. An
einer solchen durch Elektropolymerisation gewonnenen Elektrodenschicht gelingt es, Ethylen
mit Luftsauerstoff zu epoxidieren.[16] Welche Reaktionsphasen bei diesem katalytischen
Zyklus durchlaufen werden, veranschaulicht Schema 1. Zunächst wird elektrochemisch
Mn(III) zu Mn(II) reduziert, wonach unter Aufnahme eines weiteren Elektrons Luftsauerstoff
Mn (V)
O
Mn (III)
O
O-
Mn (II)
Mn (III)
Ph O
O O
Ph
Ph O-
O
2
CH2 CH2
O
e-
e-O2 /

Dissertation -21- Christoph S. Eberle
als Peroxid an das Metallzentrum bindet, das nun in der Oxidationsstufe III vorliegt. In
Gegenwart von Benzoesäureanhydrid bildet sich eine reaktive Oxo–Mn(V)–Zwischenstufe,
nachdem das Anhydrid den terminalen Sauerstoff des Peroxidanions für eine Eliminierung
aktiviert hat. Indem der mangangebundene Sauerstoff auf Ethylen weiter übertragen wird,
regeneriert sich aus dieser Oxomangan–Zwischenstufe der ursprüngliche Mn(III)–Komplex.
Weitere Metalloporphyrinoide wurden schon für andere biomimetische Reaktionen
eingesetzt, z.B. Fe–haltige Porphyrine zum Nachweis von Cyanid[17], Co–haltige zur
Nitritdetektion[18], Co– und FeCl–haltige Derivate zur elektrokatalytischen Reduktion von
Luftsauerstoff[19] oder entsprechende Ru–Verbindungen zur NO–Fixierung[19c]. In künftigen
Modellsystemen sollten auch solche Transferreaktionen, wie sie z.B. von Vitamin B12 und
dessen Derivaten bekannt sind, untersucht werden (Abb. 3). Insgesamt scheinen die
katalytischen Möglichkeiten enorm, die mit biomimetischen porphyrinoiden Systemen
erschlossen werden können; sie reichen von der Umweltentgiftung durch Beseitigung
organischer Halogenide über die Oxidation organischer Verbindungen vermittelt durch
Metalloporphyrine bis zur photoelektrochemischen Reduktion von Protonen. Umgekehrt
ergeben sich für die Sensorik Anwendungen: zum Nachweis von Gasen wie CO, O2 oder
NH3, von Umweltgiften, von Biomolekülen wie etwa Glucose oder ganz allgemein zur
theoretischen Exploration von Ligandenaustausch–Reaktionen.
CoI CoIIIRX
CoII CoII+ R
R-X
+R
+ X -
X+
+ X -
+ R. .
Abbildung 3: Denkbarer Katalysezyklus mit Hilfe von Co–Corrinoid– und Co–Porphyrinoid–Monoschichten auf Elektrodenoberflächen zur Erzeugung von Kohlenstoffradikalen.
+ e-
+X

Dissertation -22- Christoph S. Eberle
1.2 Das Prinzip Selbstorganisation von Molekülen und dessen Anwendung in
der Nanotechnologie
„Technology is not magic: it must obey the laws of physics. But it can seem like magic.“
Chris Phoenix
Selbstorganisation kennzeichnet biologische Systeme. Sie ist das Prinzip, nach dem frühste
Stufen der Evolution abliefen. So brachte sie Gebilde hervor, die eine Voraussetzung für
Leben sind: bis zu den mannigfachen Eigenschaften der heute an ihre Umgebung angepassten
Spezies. Die Zelle selbst, mit der Leben im kleinsten beginnt, hat dieses Prinzip am
sichtbarsten verwirklicht. Dort bewirkt es, dass mit dem wunderbaren Zusammenspiel der
Organelle in Kompartimenten komplexe Funktionen möglich werden, wie die enormen
Stoffwechselleistungen, RNA– und DNA–Replikation, Proteinbiosynthese, Signaltransduk–
tion oder Photosynthese. Dies erfordert, dass sich Strukturen organisieren, damit sie sich
selbst vervielfältigen, auf Außenreize reagieren und die verschiedenen chemischen
Reaktionen stattfinden können. So ordnen sie sich in der Nanometerdimension zu
Molekülarchitekturen, so wachsen sie zu einem zellulären Ganzen, das sich selbst erschafft,
dessen Teile zum Geschaffenen gehören, aber nicht mit ihm identisch sind (Abb. 4). Wo diese
übergehen, ein lebendes System zu bilden, diese Schwelle versuchten Manturana und Varela
erkenntnistheoretisch zu finden.[21] Ihre Theorie der Autopoiese fasst die Essenz des Lebens
zusammen, erweitert dessen Definition über die zellulären Formen hinaus, wie sie
von den biologischen Wissenschaften beschrieben werden. Um demnach als lebend zu
gelten, muss nicht nur ein nach außen begrenztes Ensemble vorhanden sein aus
irgendwie gearteten molekularen Akteuren. Wichtiger ist vielmehr deren Aktivität im Inneren.
Diese Aktivität knüpft ein Netz von Beziehungen, das Merkmale annimmt, die biologischem
Leben zugeschrieben werden. Dieses ist auf seiner untersten Stufe kognitiv, und erst mit der
minimalsten Stufe von Kognition beginnt Autopoiese. Daraus ist geschlossen worden, dass
sich Leben zwar autopoietisch entwickelt, doch diese Bedingung reicht allein nicht aus
für seine Entstehung.[22] Als autopoietisches System erhält es sich in der Umgebung,
aus der seine Bestandteile stammen. Hören dagegen seine Bestandteile auf im System
zusammenzuarbeiten, zerfällt auch das System.
Ohne Selbstorganisation einzelner Atome bis zu den hierarchischen Anordnungen der
Moleküle fehlte dem Leben buchstäblich die stoffliche Basis. Dessen Strukturen selbst sind
es, die auf jeder Stufe wiederum Bedingungen schaffen, sich höher zu organisieren. Auf der

Dissertation -23- Christoph S. Eberle
Vielfalt ihrer Aufgaben, die diese Nanostrukturen übernehmen, beruht die biologische Vielfalt
selbst. Sie entspringt dem genetischen Informationsfluss von der ‚DNA–Software’ zur
‚Protein–Hardware’, durch den sich zelluläres Leben auszeichnet.[23] Was es ausmacht, findet
im Verborgenen statt, wirkt sich aber in der makroskopischen Welt aus. Ist der Nanomaßstab
wesentlich, damit die molekularen Träger diese Information speichern, verarbeiten, vernetzen
und übertragen können, wie sie es können? Wie müssen die molekularen Träger beschaffen
sein, ihren Aufgaben gerecht werden? Warum sind sie als Maschinen so effizient und genau,
wie sie es sind: dies sind Fragen, die die Nanowissenschaften uns vorlegen, je näher sie an
die Biologie heranrücken. Sie beinhalten neben den wissenschaftlichen Aspekten auch
philosophische. Sie zu beantworten, wird sich produktiv auswirken auf jeden weiteren
Versuch, künstliche Systeme zu entwerfen, die zelluläres Leben nachahmen sollen. Technisch
verwertbare Lösungen auf diese Fragen zu finden, dies sind die Motive, die die
Nanowissenschaften vorantreiben.
Abbildung 4: Das Leben als autopoietische Einheit (Mann, 2008).[23] Umwelt und Zellinneres sind je ein Ganzes, wirken aber über ihre nanoskaligen Bestandteile aufeinander ein. An der Schnittstelle zwischen beiden findet beständiger Austausch statt.
Dass Selbstorganisation ein in der Natur allgültiges Prinzip ist, wird gerade dort offenbar, wo
Materie sich entwickelt und organisiert. Abseits dieser Beobachtungen dient sie als ein
Ansporn, mit dem der Mensch seine eigene kulturelle Evolution vorantreiben kann. Stets
beruhte sie auf der Herstellung neuer Materialien. Dazu mussten die nötigen technischen
Neuerungen eingeführt werden. Gewaltige Erwartungen hat nun ein technologisches Konzept
geweckt, das einzigartig als Anwendung dieses Prinzips wahrgenommen wird.

Dissertation -24- Christoph S. Eberle
Die Geburtsstunde dessen, was heute Nanotechnologie genannt wird, liegt ein halbes
Jahrhundert zurück. 1959 hielt Richard Feynman, der sechs Jahre später den Nobelpreis für
Physik entgegennehmen durfte, am California Institute of Technology seinen inspirierenden
Vortrag „There is plenty of room at the bottom“. Die Vorstellung, Materie auf der Ebene
einzelner Atome beherrschen und neu schaffen zu können, verband sich ihm mit grandiosen
Möglichkeiten:
„…I would like to describe a field, in which little has been done, but in which enormous
amount can be done…What I want to talk about is the problem of manipulating and
controlling things on a small scale…What I have demonstrated is that there is room –
that you can decrease the size of things in a practical way…I will not discuss how we
are going to do it, but only what is possible in principle…What could we do with layered
structures with just the right layers? What would the properties of the materials be if we
would really arrange the atoms the way we want them?... At the atomic level, we have
new kinds of forces and new kinds of possibilities, new kinds of effects…”[24]
Feynman erklärte, dass Raum für einen solchen Eingriff im atomaren Aufbau der Materie
existiere. Denn dort, wo er stattfindet, sollten die Prinzipien der Physik ihn erlauben. Dieser
Raum vergrößert sich, je kleiner die Dinge werden, und jenseits menschlichen Augenscheins
lässt sich viel und vielerlei in ihm bewerkstelligen. Ohne diesen Begriff geprägt zu haben
oder ihn gar in dem Sinne zu verwenden, der ihm heute unterlegt wird, standen
Feynmans Denkanstöße seitdem der nanotechnologischen Forschung Pate, die politische
und wirtschaftliche Bedeutung errungen hat. Ebenfalls am Ort von Feynmans Vortrag
verkündete Präsident Clinton im Jahr 2000 die Auflage eines mehrere Milliarden US–Dollar
umfassenden Regierungsprogramms zur koordinierten Förderung dieser Technologie in den
Vereinigten Staaten. Auch darum wurde dieser nationale Vorstoß (U.S. National
Nanotechnology Initiative, NNI) eingeleitet, weil Amerika militärisches Potential sieht und
fürchtet, dass es der künftigen Entwicklung von Waffen auf Nanobasis zugrunde liegen
kann.[25], [26] Seine führende Rolle in einem sehr interdisziplinären Bereich der Forschung
sollen vorsorglich eben jene Maßnahmen sichern, um von Staats wegen ausgesuchte Projekte
langfristig zu finanzieren, vornehmlich solche, die entweder ganz neue großtechnische
Anwendungen begründen könnten oder solche, die den Fertigungsprozess bestehender
Industrieprodukte vereinfachen. Mehr noch geht es darum, den Begriff Nanotechnologie
genauer und also enger zu fassen, um wissenschaftliche Fakten von Fiktion zu scheiden. Da
miteinander konkurrierende Definitionen herangezogen werden, erschwert dies die politische
Debatte, besonders, wenn die Risken neuer Produkte bewertet werden sollen, die aus dieser

Dissertation -25- Christoph S. Eberle
Technologie entstehen können, etwa wenn von möglichen Gesundheitsschäden durch
Nanopartikel die Rede ist. Übereinstimmend und allgemein gilt Nanotechnologie als die
Beschäftigung mit Objekten, die kleiner sind als 100 nm und neue Eigenschaften aufweisen.
Dies beinhaltet neben der Sichtbarmachung derartiger Gebilde und Oberflächen, sie zu
modifizieren und kontrolliert von ihren Atomen aufwärts zu restrukturieren. Hiermit versucht
der so genannte ‚bottom-up’–Ansatz[27], Feynmans Vision von miniaturisierten Maschinen zu
verwirklichen. Dennoch nicht allein die Verkleinerung auf wenige Nanometer bestimmt
darüber, wieweit sich die Eigenschaften von Objekten ändern bis zu neuen Quanteneffekten.
Allgemeiner hängt dies ab vom Verhältnis Oberfläche zu Volumen, denn nanodimensionierte
Partikel bestehen größtenteils aus Atomen, die an einer Oberfläche liegen. Verringert sich
die räumliche Ausdehnung eines Objektes, nimmt dessen Volumen stärker ab als dessen
Oberfläche. Dies bringt Gebilde hervor, die fast nur noch Oberfläche sind mit ganz
anderen optischen, elektronischen und physikalischen Eigenschaften oder Reaktivitäten und
Beweglichkeiten als die, die ein makroskopischer Stoff aufweist. Graduell lässt sich jede
diese Eigenschaften variieren, so dass auf der Nanoskala Grenz– und Oberflächen als vierter
Aggregatzustand der Materie angesehen werden.[28]
Abbildung 5: Die vier Stadien der nanotechnologischen Entwicklung nach Roco.[29]

Dissertation -26- Christoph S. Eberle
Derweil sehen Vertreter der U.S.–Nanotechnologie–Initative vier Stadien, die diese
technologische Entwicklung durchläuft und die sich zum Teil überschneiden (Abb. 5).[29] Bei
den Materialien der ersten Generation handelt es sich noch um passive Nanostrukturen,
konstruiert, um einen bestimmten Zweck zu erfüllen. Diesen folgen aktive Strukturen, die in
der Lage sind, mehrere Aufgaben bei elektronischen, photonischen, biologischen oder
mechanochemischen Anwendungen auszuüben. Indem Nanostrukturen wie diese zu einem
System zusammengefügt werden, dessen Bestandteile hierarchisch durchgliedert und
funktionell aufeinander abgestimmt sind, könnte man Schritt für Schritt zu künstlichen Zellen
gelangen. Die biologische Maschinerie zu kopieren und in selbst entworfenen Nanofabriken
nach Wunsch produzieren zu können, in diesem Sinne würde Nanotechnologie möglich, wie
sie Eric Drexler ursprünglich gedacht und mit seinem Konzept des ‚molecular manufacturing’
popularisiert hatte.[30] Sein futuristisches Buch „Engines of Creation“ steht am Anfang
einer seit den 1980er Jahren dauernden Kontroverse, die neben Hoffnungen auch öffentliche
Ängste und Mißverständisse nährt, je mehr die Wirklichkeit Feynmans und Drexlers
Visionen einholt. Da sich weiterhin ihre wissenschaftliche Grundlage verbreitet, nimmt
man ihre Implikationen ernster. Sie betreffen alle Wirtschaftszweige, die Güter des
täglichen Gebrauchs herstellen. Dieser Weg, geebnet von geschäftlichen und wissenschaft–
lichen Interessen, verläuft ähnlich dem, den die Informations– und Biotechnologie in der
zweiten Hälfte des zwanzigsten Jahrhunderts gegangen sind. Vielfach regte auch in ihrem Fall
Forschung die eigene Anwendung an und umgekehrt. So zeigt sich heute schon, wovon
erwartet wird, dass es sich in Zukunft noch mehr fortsetzt: Die Vermarktung kommender
Generationen von Nanoprodukten übernehmen neue Gründerfirmen nicht allein. Dies fällt
ebenso etablierten Unternehmen und Konsortien zu, da die Nanotechnologie traditionelle
Technologien ergänzen wird, mit ihnen verschmilzt und sich in bestehende
Produktionsverfahren einfügt. Entsprechend nehmen die weltweiten Investitionen zu, die
in Programme zur ihrer Erforschung und Entwicklung fließen. Von staatlichen Stellen
verfünffachte sich die Summe auf ca. 4.1 Mrd. USD weltweit. Insgesamt schätzt man, dass
2015 international Waren im Wert von bis zu 1 Billion USD gehandelt werden, die in
irgendeiner Form Nanotechnologie enthalten (Roco, 2005). Doch bleibt umstritten, wann und
inwieweit deren weitere Entwicklung jene Allmöglichkeiten überhaupt erschließen kann, die
man heute in ihr sieht. Darum wird sie begleitet werden von ethischen Fragen, weil nicht alles
Mögliche auch wünschenswert ist.
Folgt man den Argumenten eines der Pioniere in diesem Forschungszweig, nämlich
George Whitesides, dann gab es Technologien im Nanomaßstab, noch ehe dieses Schlagwort

Dissertation -27- Christoph S. Eberle
Mitte der 1970er Jahre erstmals durch den Japaner Norio Taniguchi aufkam.[31] Seither
sind sie schleichend ins allgemeine Bewusstsein gedrungen. Ihre Anfänge liegen in
der Miniaturisierung elektronischer Bauteile in Computern und auch in Pionierleistungen der
Biochemie seit Schrödingers (1887 – 1961) Frage „Was ist Leben?“[32]. Vom Verständnis,
was biologische Phänomene ausmacht, lag der Schritt nahe, sie zu beherrschen, da sie von
Molekülen angetrieben sind. Whitesides[33] sieht die Nanowissenschaften, wie sie sich ihm
darstellen, als Dömane der Chemie, da sich hinter ihnen –pointiert formuliert– Tätigkeiten
und Methoden verbergen, die Chemiker seit je ausüben und die zu deren ureigener Routine
gehören, nämlich die chemische Synthese: Atome durch chemische Bindungen zu immer
neuen Molekülen zu verknüpfen, die tägliches Leben wenn nicht verändern, so doch
zumindest verbessern sollen. Aus diesen Gründen wird deutlich, so Whitesides weiter, dass
Chemiker die führende Rolle, die ihnen in diesem Bereich der Forschung zukommt, bewusst
übernehmen müssen. Denn zunächst sind Durchbrüche am ehesten dort zu erwarten,
wo es gilt, neue Materialen für Anwendungen zu ersinnen, aus denen kommerzialisierte
Technologien werden. Dieses revolutionäre Potential in den Grenzen der Material–
wissenschaften lässt sich nur mit Hilfe der Chemie freisetzen, da sie deren Grundlage ist.
„…materials are usually the products of chemical processes…Nanoscience is now a
thread woven into many fields of science. Nanotechnology–certainly evolutionary, and
perhaps revolutionary–will emerge from it…”[33]
Ein inzwischen gängiger Weg zu neuen Materialen ist der über selbstorganisierte
Monoschichten.

Dissertation -28- Christoph S. Eberle
1.3 Selbstorganisierte Monoschichten
Seit den Pionierarbeiten von Nuzzo und Allara[34] erweisen sich selbstorganisierte
Monoschichten (engl. self assembled monolayers, SAMs) als vielseitiges Werkzeug
in der Nanotechnologie.[28, 35-38] Schwefelorganische Verbindungen (Thiole, Disulfide,
Sulfide) scheiden sich spontan und sehr schnell auf Metalloberflächen ab, entweder aus in der
Regel ethanolischen Lösungen (~ 1–10 mM) bei Raumtemperatur oder aus der Gasphase im
Hochvakuum. Organische Moleküle sind so auf einer Reihe von Oberflächen immobilisierbar.
Neben Gold dienen andere Übergangsmetalle wie Silber, Kupfer, Platin, Palladium oder gar
Quecksilber als Substrate, aber auch Indium– und Galliumverbindungen wurden bereits
verwendet. Adsorbate auf all diesen Materialen lassen sich als aus drei Teilen zusammen–
gesetzt beschreiben (Abb. 6): einer Kopfgruppe, einem Linker und einer terminalen Gruppe,
deren chemische Funktionalität die Oberflächeneigenschaften der gesamten Monoschicht
bestimmt. Hydrophobe Alkylketten des Linkers wechselwirken vorwiegend durch Van–der–
Waals Kräfte miteinander. Durch Einführung entsprechender Gruppen ergeben sich weitere
sterische und elektrostatische Effekte. Auch kommen chirale Detailstrukturen hier zum
Tragen.[38] Entscheidend bleibt die Kopfgruppe, mit der das organische Molekül an
der Metalloberfläche verankert wird, da letztlich von der Stärke der Metall–Schwefel–
Bindung die Stabilität der einzelnen Moleküle in der Monoschicht abhängt. Eine Stabilität
durch Oberflächenkoordination wie die von schwefelorganischen Verbindungen gewährleistet
auch die Affinität von Phosphonsäuren für Titan(IV)oxid. Technisch von Bedeutung sind
ferner die Elektropolymerisation von Pyrrolen und die Elektrooxidation von Aminen an
Kohlenstoffmaterialen. Mit all diesen Verfahren können Elektrodenoberflächen chemisch
modifiziert werden. Neben kovalenten Verknüpfungen gibt es Möglichkeiten zur nicht–
kovalenten Bindung. Darunter fallen schwache Van–der–Waals Kräfte, mit denen
Oberflächenhaftung erzielt werden kann, so z.B. bei Monoschichten auf HOPG.
In der einschlägigen Fachliteratur findet man selbstorganisierte Monoschichten aus Thiolen
und Disulfiden als die am häufigsten beschriebenen und daher am besten charakterisierten
Systeme. Dies hängt zusammen mit der Präferenz für Gold als Standard für derartige
Anwendungen, weil es ein chemisch inertes Metall ist. Seine Leitfähigkeit erlaubt,
modifizierte Goldoberflächen mit elektrochemischen Standardverfahren wie der Cyclo–
voltammetrie zu charakterisieren. Unterhalb seines Schmelzpunkts oxidiert Gold nicht und
kann in verschiedenen Formen bearbeitet werden. Mit bestimmten Chemikalien lässt sich
seine Oberfläche ätzen, was die Lithographie gezielt ausnutzt. Darüber hinaus fand man, dass

Dissertation -29- Christoph S. Eberle
es für Schwefelverbindungen besonders affin ist und deren Adsorbate längere Zeit unversehrt
bleiben. Insgesamt ist der Adsorptionsprozess gut reproduzierbar und verläuft ohne
unerwünschte Nebenreaktionen. Auch wirken Monoschichten auf Goldoberflächen (anders
als Silber) nicht toxisch, wenn sie mit Zellen oder anderem biologischem Material in Kontakt
gebracht werden. Diese Biokompatibilität ist eine wesentliche Voraussetzung, um solche
integrierten Systeme für Sensorik, als Protein–, RNA–, DNA–Chips oder Microarrays
einzusetzen.[39] Praktisch gibt es einige Parameter, die diesen Prozess steuern und die sich
über die experimentellen Bedingungen einstellen lassen, um organische Monoschichten mit
größtmöglicher Ordnung zu erzeugen. Dazu gehören vor allem Lösungsmittel, Temperatur,
Adsorptionszeit und die Qualität des Metallsubstrats, daneben die Konzentration, Reinheit
und Struktur der zu immobilisierenden Moleküle oder allgemein die Zusammensetzung der
Lösung ebenso wie deren Sauerstoffgehalt.
Abbildung 6: Idealtypischer Aufbau einer selbstorganisierten Monoschicht aus n-Alkylthiolen auf einer Goldoberfläche mit (111)–Textur (nach Viana, 2002).[86]
Thermodynamisch ist die Bildung der Au–S Bindung begünstigt, da bei der Reaktion die
Gesamtenthalpie abnimmt und dadurch die Chemisorption spontan vonstatten geht. Bei
diesem Prozess handelt es sich um einen insgesamt exothermen, wenn die Bildung von
molekularem Wasserstoff unterstellt wird.[40, 86] Es bilden sich sowohl bei Thiolen als auch
mit Disulfiden die gleichen Au(I)–Thiolatspezies, die eine kovalente Bindung mit leicht
polarem Charakter eingegangen sind. Die Energie dieser Bindung beträgt 168–189 kJ/mol,
und bei einem Winkel von etwa 120° zwischen Alkankette und Goldoberfläche liegt das
chemisch gebundene Schwefelatom sp2–hybridisiert vor. Gleichzeitig schwächt die Bildung
der Au–S Bindung die oberflächennahen Au–Au Bindungen, und die in die Chemisorption
Endgruppe: Redoxaktivität ~ 30º
Linker (Alkylketten): Van–der–Waals Wechselwirkungen
Kopfgruppe (SH oder RSSR): Chemisorption auf dem Substrat als Thiolat [Au(I)–S]
Au (111)

Dissertation -30- Christoph S. Eberle
einbezogenen Goldatome werden aus der monokristallinen Textur herausgehoben und an der
Oberfläche beweglich.[41] Wenn Thiole adsorbieren, entsteht formal Wasserstoff. Dessen
Nachweis und möglicher Verbleib konnte in Experimenten noch nicht vollends geklärt
werden. Im Vakuum in der Gasphase könnte die Freisetzung von Wasserstoff gemäß
Gleichung (1) stattfinden. Dies setzt homolytische Spaltung der S–H Bindung voraus, wobei
dem Wasserstoffradikal die Weiterreaktion mit mehreren denkbaren Partnern offen steht: mit
Lösemittelmolekülen, mit einem zweiten Wasserstoffradikal oder mit der Goldoberfläche, um
dort eliminiert zu werden. Letzteres ist jedoch ein nur gering aktivierter Prozess.[28] Bei
Chemisorption in Lösung liegen die Verhältnisse anders. Befindet sich Sauerstoff im
Medium, kann die S–H Bindung heterolytisch gespalten werden wie in Gleichung (2). Damit
ist oxidative Umwandlung in Wasser möglich.
R-SH + Au0
R-SH + Au0
RSSR + Au0
R-S- - Au+ + 1/2 H2 (1)
R-S- - Au+ + H+ + e- (Au) (2)
2 R-S- - Au+ (3)
Nanometerdicke organische Filme vor allem auf Goldoberflächen sind vielfach mit etablierten
analytischen und spektroskopischen Verfahren vermessen worden. Darunter fallen
elektrochemische wie die Cyclovoltammetrie oder physikalische wie die Ellipsometrie,
XPS, STM, AFM, IRRAS und QCM. Als gängiges Untersuchungsobjekt behaupten
selbstorganisierte Monoschichten ihren Platz in den Nanowissenschaften, weshalb sie für
vielerlei potentielle Anwendungen geeignet sind.
Abbildung 7: a) Schematische Darstellung der Kettengeometrie von adsorbierten Alkylthiolen auf Goldoberflächen. b) „Odd-Even“–Effekt bei adsorbierten Alkylthiolen (nach Love et al., 2005).[28]
Au
S
H
H
H
S
HH
H H
H
b)
Au
a)
S
H
H
H
+ α- α
S
β
x
z
y

Dissertation -31- Christoph S. Eberle
Die Adsorbate formen dicht gepackte Monoschichten mit all–trans Konformation der
Moleküle (Abb. 7a). Allgemein werden zwei Parameter herangezogen, um die Geometrie von
langkettigen Resten zu beschreiben. Der Winkel � markiert die Neigung zur Metalloberfläche,
während � die Rotation um die Längsachse des Moleküls angibt. Man beobachtet ferner einen
sogenannten „Odd–Even“–Effekt bei Monoschichten aus n–Alkylthiolen mit verdrehbaren
Methylenketten (Abb. 7b). Experimentelle Daten bekräftigen den konservierten Wert für
� � 30°, der mit den unterschiedlichen Oberflächenprojektionen der Methylengruppen
zusammenhängt, je nachdem ob ihre Anzahl gerade oder ungerade ist. Enthalten die Linker
eine ungerade Anzahl von Methylengruppen, ist die freie Energie der Monoschichten etwas
höherer als im umgekehrten Fall.
Abbildung 8: Schematische Darstellung der (3 × 3)R30° Elementaranordnung von n-Alkylthiolen (dunkle Kreise) in selbstorganisierten Monoschichten auf einer monokristallinen Goldoberfläche (helle Keise). Beide Strukturen sind hexagonal, wobei die Moleküle in der Monoschicht um 30° geneigt sind gegenüber der unterliegenden Goldtextur. Darin stehen zwei benachbarte Goldatome (Abstand 2.88 Å) um den Faktor 3 näher zusammen als zwei benachbarte Schwefelatome (Viana, 2002).[86]
Wie sich herausstellte, beeinflusst die kristalline Struktur des Metallsubstrats die Architektur
der organischen Monoschichten. Dies führt bei Adsorption auf Au(100) zu einer c(2 × 2)
Anordnung anders als bei Goldoberflächen mit (111)–Orientierung, bei der die Adsorbate
eine hexagonale (3 × 3)R30° Grundform bilden, die mit einer c(4 × 2) Sekundärstruktur
einhergeht. Sie ist in Abbildung 8 schematisch wiedergegeben. Eine solche Struktur wurde als
Folge der Aktivierung der S–H Bindung an der Goldoberfläche erklärt.[42] In der
Vergangenheit wurden modifizierte Oberflächen eingehender untersucht, u.a. mittels
Röntgendiffraktion, STM– und AFM–Techniken. Das Bild, dass sie lieferten, ließ jedoch
schlussfolgern, dass zum Teil Adsorbate paarweise über S–S Bindungen miteinander
verbrückt sein müssten. Dieses Disulfidmodell widerspricht der ursprünglich vorgeschlagenen

Dissertation -32- Christoph S. Eberle
Struktur. Alternative Deutungen der experimentellen Befunde stimmen wiederum besser mit
der Theorie überein, so dass in der Literatur dem Thiolatmodell mit seiner hexagonalen
Einheitszelle wieder der Vorzug gegeben wird. Fischer et al.[43] simulierten mittels PBE–DFT
Algorithmen die Struktur von selbstorganisierten Alkanthiolmonoschichten auf Au(111). Die
Rechnungen ergaben, dass für Adsorbate die Thiolatform thermodynamisch bevorzugt ist
gegenüber disulfidverbrückten. Letztere destabilisiert die Au–S Bindung, in der der Abstand
beider Atome um ca. 0.3 Å größer ist. Beim S–S Dimer handelt es sich um eine kovalente
Brücke, wobei jedes Schwefelatom nur schwach an das Metallsubstrat bindet. Dennoch sind
die genannten strukturellen Aspekte weiterhin Gegenstand von Überlegungen. Denn zu klären
bleibt weiterhin die Frage, wie die Oberflächenstellen des Substrats die Bindung der Thiolate
unterstützt. Es scheint, dass Schwefelatome sowohl an Hohlstellen zwischen drei Goldatomen
als auch an Brückenstellen gebunden werden (vgl. Abb. 8).[44] Alle Modelle, die die
Struktur dieser Monoschichten beschreiben, belegen jedoch, dass zwei Faktoren den
Selbstorganisationsprozess grundsätzlich beeinflussen: die Ausbildung der Metall–Schwefel
Bindung und laterale Wechselwirkungen zwischen den organischen Resten, die das Adsorbat
stabilisieren.
Abbildung 9: Selbstorganisierte Monoschicht aus 16–Mercaptohexadecansäure (MHA), die sich auf einer monokristallinen Goldelektrode abgeschieden hat (nach Langer et al., 2003; Choi et al., 2006).[45, 46] Nach Immobilisierung der Vorläuferverbindung wurde die freie Säure durch Hydrolyse gewonnen. Durch Aufnahme oder Abgabe von Elektronen lässt sich, mit der Redoxreaktion gekoppelt, zwischen gerader und gekrümmter Molekülkonformation hin– und herschalten.
Da es sich bei den durch Selbstorganisation entstehenden Monoschichten um
dreidimensionale, zum Teil hochgeordnete Gebilde handelt, können sie verschiedene
Konformationen einnehmen. Ein Konformationswechsel lässt sich auch elektrochemisch

Dissertation -33- Christoph S. Eberle
steuern (Abb. 9)[45, 46]. All diese Beschaffenheiten können Materialien hervorbringen mit
neuen Eigenschaften. Doch setzt ihre Anwendung ein tieferes Verständnis aller kinetischer
und thermodynamischer Faktoren voraus, die den Selbstorganisationsprozess an Oberflächen
steuern, einschließlich des noch wenig verstandenen Einflusses, den Temperatur und die
Wahl des Lösungsmittels haben.
Bestimmte Anwendungen erfordern nach Selbstorganisation der Monoschicht diese selbst
zu verändern. Solche Postmodifikationen zielen ab auf grundlegende Materialeigenschaften
oder die molekulare Zusammensetzung der Oberflächenfilme. Solches lässt sich entweder
über chemische Reaktionen oder physikalische Mittel erreichen. Daneben gibt es die
Koadsorption von synthetisierten schwefelorganischen Verbindungen, um sofort gemischte
Monoschichten zu erhalten. Schritt für Schritt können damit bestimmte molekulare
Umgebungen eingerichtet oder –allgemeiner formuliert– neue Grenzflächen in diesen
Systemen geschaffen werden. Abbildung 10 fasst schematisch die Möglichkeiten zur
Postmodifikation selbstorganisierter Monoschichten zusammen.
Abbildung 10: Häufige Verfahren zur kovalenten und nicht–kovalenten Modifikation von selbst–organisierten Monoschichten (Love et al., 2005).[28] (a) Einführung anderer Adsorbate in Fehlstellen. (b) Chemische Reaktionen oder Adsorption biologischen oder chemischen Materials an funktionellen Kopfgruppen der Monoschicht (helle Kreise).
Techniken, bei denen selbstorganisierte Monoschichten hergestellt oder benutzt werden,
haben sich seit Jahrzehnten verfeinert; sie konnten vielfach die Langmuir–Blodgett

Dissertation -34- Christoph S. Eberle
Methode[47] ersetzen, die bis weit in die 1980er Jahre angewandt wurde, um ultradünne,
geordnete organische Filme auf Substrate zu bringen. Ihr gegenüber bietet die alternative
Strategie der Selbstorganisation von Adsorbatmolekülen einige Vorteile. Sie bestehen darin,
dass Monoschichten leicht und reproduzierbar herzustellen sind und höhere Stabilität
aufgrund der Immobilisierung von Molekülen durch chemische Bindungen besitzen.

Dissertation -35- Christoph S. Eberle
2. AUFGABENSTELLUNG
2.1 Synthese von immobilisierbaren Porphyrinoiden, Metalloporphyrinoiden
und Fullerenen
Gestützt auf in unserem Laboratorium vorangegangenen Dissertationen[11a,b,d] sollten
porphyrinoide Vorläufer aus Hämin hergestellt werden. Die vorliegende Arbeit verfolgte
partialsynthetische Ansätze. Sie zielten darauf ab, in die porphyrinoiden Vorläufer Metalle
einzubauen und mit geeigneten Linkern zu versehen, die Disulfidbrücken tragen. Mit diesen
Linkern sollte auch Fulleren–C60 derivatisiert werden. Daneben waren Disulfidlinker mit
Pyrrol–, Imidazol– und aliphatischen Einheiten unterschiedlicher Kettenlänge herzustellen.
Da Porphyrine aus natürlichen Quellen hiermit zugänglich sind, können Synthesen auch in
größerem Maßstab durchgeführt und der Weg zu Derivaten verkürzt werden.
2.2 Aufbau von Donor–Akzeptor–Systemen, organischen Sensoren und
Katalysatoren
Die so synthetisierte Substanzbibliothek aus Porphyrin– und Fulleren–Derivaten diente
dazu, ein schon entwickeltes Baukastenkonzept zu ergänzen. Dessen Anwendbarkeit konnten
Vorgängerarbeiten in unserem Laboratorium beispielhaft demonstrieren[11a,b,d], die aus dem
COST–Projekt1 „Redox chemistry and catalysis in the microenvironment of electrode
surfaces“ entstanden waren. Daran anknüpfend sollten nach Bedarf die hier dargestellten
Porphyrinoide, Metalloporphyrinoide, Fullerene oder Linkermoleküle zusammengestellt
werden für Immobilisierungen an monokristallinen Goldoberflächen. Dies sollte auch mit
Porphyrin–Fulleren–Dyaden[11d] geschehen, die gezielt durch Diels–Alder Reaktion an vorab
immobilisiertem Fulleren C60 aufgebaut werden sollten. Dabei liegt die Idee zugrunde, mittels
Selbstorganisation Systeme zu schaffen, denen bestimmte Aufgaben zugedacht werden
können: als Katalysatoren, Sensoren oder organische Photovoltaikzellen, je nach Reaktivität
des Metallatoms im Makrotetrazyklus. Daher wurden ZnII, NiII, CoII, CoClIII und und MnClIII
gewählt, die sich mittels standardisierter präparativer Verfahren einbauen lassen.[48] Innerhalb
einer vom DAAD2 finanzierten wissenschaftlichen Kooperation mit der Arbeitsgruppe
von Prof. Dr. Luisa Maria Abrantes an der Universität Lissabon (Portugal) sollten
elektrochemische und physikalische Untersuchungen an modifizierten Goldelektroden

Dissertation -36- Christoph S. Eberle
vorgenommen werden, um zunächst die jeweilige Belegungsdichte zu ermitteln sowie
Dicke und Aufbau der organischen Monoschichten zu bestimmen. Desweiteren sollten
deren Eigenschaften im Sinne der oben genannten Anwendungen getestet werden. All diese
Untersuchungen liefen unter dem Projekttitel: “Immobilisierung von Metalloporphyrinoiden,
Fullerenen, Porphyrin–Fulleren–Dyaden an Metalloberflächen und deren Charakterisierung
sowie elektrochemische Aktivität”. Die Untersuchung von modifizierten Au(111)–Elektroden
erlaubt unmittelbaren Vergleich mit veröffentlichten Daten, da Chemisorbate auf solchen
Metalloberflächen gut dokumentierte Untersuchungsobjekte darstellen.
___________________________ 1 European cooperation in the field of scientific and technical research 2 Deutscher Akademischer Austauschdienst

Dissertation -37- Christoph S. Eberle
3. DURCHFÜHRUNG DER SYNTHESEN
3.1 Gewinnung enantiomerenangereicherter Liponsäure mittels kinetischer
Racematspaltung mit Lipase aus C. rugosa
C. rugosa, ehemals C. cylindracea genannt, ist ein nicht–pathogener Hefestamm, der sich
asporisch vermehrt; sein extrazellulares Sekret enthält verschiedene Isoformen von Lipasen,
deren Tertiärstruktur in den 1990er Jahren von Grochulski et al. röntgenkristallographisch
aufgeklärt wurde.[49] Bekannt ist mittlerweile, dass fünf Gene (LIP1 – LIP5) die C. rugosa
Lipasen kodieren, die zusammengefasst CRL genannt werden.[50] Jede ihre Polypeptidketten
ist 543 Aminosäuren lang mit einem apparenten Molekulargewicht von etwa 60 kDa. Diese
Produkte der LIP Genfamilie stimmen untereinander in ihren Sequenzen stark überein (84%
Ähnlichkeit), doch auch im Vergleich zu Enzymen aus verwandten Organismen wie
Geotrichum candidum fällt große Sequenzhomologie auf. Da alle Lipasen hier von
Pseudogenen produziert werden, verwunderte somit die Entdeckung nicht, dass es dabei zu
Umprogrammierungen im genetischen Code gekommen war: bei der Translation in C. rugosa
steht –evolutionär bedingt– das universelle CUG–Codon für Serin statt für Leucin.[51] Die
optimale Anpassung an unterschiedliche Wachstumsbedingungen scheint ausschlaggebend
dafür zu sein, dass solche Isoenzyme exprimiert werden.[52] Dies belegt die Bandbreite
an Spezifitäten, mit der sie als Biokatalysatoren verschiedenartige Substrate umsetzen.
Bei großtechnischen Prozessen ermöglichen sie neben der Produktkontrolle milde,
umweltverträgliche Reaktionsbedingungen und die betriebswirtschaftlich relevante
Reduzierung von Kosten. Die enzymatische Maschinerie knüpft oder spaltet chemische
Bindungen an bestimmter Stelle. Nach den Gesetzen der Kinetik kann sie dies gezielter und
allgemein schneller bewerkstelligen im Vergleich zum Verlauf einer gängigen chemischen
Reaktion.[53] Daher haben enzymkatalysierte Methoden mehr und mehr Eingang in die
organische Synthese gefunden.
C. rugosa gehört zu den am häufigsten untersuchten Mikroorganismen, weil sich ihre
Exolipasen (CRL) für die verschiedensten biotechnologischen Umsetzungen eignen: in der
Nahrungs–, Kosmetik– und Kunstlederindustrie genauso wie bei der Herstellung von
Pharmazeutika, Zuckerestern, Pestiziden oder Detergenzien.[52, 54] Für analytische Zwecke
arbeitet man üblicherweise mit immobilisierten Formen dieser Lipasen, die entweder in eine
Matrix eingebettet oder an ihr verankert werden. Zahlreiche Autoren haben eine Reihe
von katalysierten Reaktionen beschrieben in ungewöhnlichen organischen Lösungsmitteln[55],

Dissertation -38- Christoph S. Eberle
in Mehrphasensystemen[56] und w/o Mikroemulsionen[57]. Dabei zeichnet sich CRL durch
hohe Stabilität und Stereoselektivität gleichermaßen aus. Sie wird darum zur Racemat–
trennung eingesetzt, die bei Ibuprofen, Naproxen oder Menthol beispielsweise mit
vollständigem Enantiomerenüberschuss gelingt. Während klassische Methoden mit chiralen
Hilfreagenzien arbeiten, beruht die Trennung der racemischen Verbindungen durch CRL
entweder auf Veresterung oder Hydrolyse einer der beiden enantiomeren Vorläufer. In
wässriger Umgebung überwiegen die hydrolytischen, in organischen Solventien die
Veresterungsreaktionen. Man beobachtet, dass Lipasen in beiden Fällen an der Grenze
zwischen wässriger und organischer Phase ihre Substrate binden und dort katalytisch am
aktivsten sind. Diese so genannte Grenzschichtaktivierung erklärt die Stabilität in polaren und
unpolaren Lösemitteln. Sie ist zurückzuführen auf besondere konformationelle Änderungen,
die reversibel sind und sich auf der Proteinoberfläche auswirken. Sie erstrecken sich bis zum
aktiven Zentrum, das unter einer Helixstruktur verborgen liegt. In unpolarer Umgebung wird
dieser Helix-Deckel geöffnet, um das aktive Zentrum freizugeben. Dieser thermodynamisch
begünstigte Zustand, den man bei CRL ebenso wie bei vielen anderen Lipasen findet, ist
stabilisiert durch Wechselwirkungen zwischen den beteiligten Strukturteilen. Abb. 11 stellt
die geöffnete Form des Enzyms der geschlossenen gegenüber.[49c]
Abbildung 11: GRASP–Modellierung der molekularen Oberfläche der Candida rugosa Lipase (CRL) in ihren beiden konformationellen Zuständen (Grochulski et al., 1994).[49c] Stickstoffatome, Carbonyl–kohlenstoffatome und Sauerstoffatome sind im Original verschiedenfarbig dargestellt. Der Pfeil (links im Bild) deutet auf das aktive Zentrum in der Substratbindungstasche. Dort bilden die Aminosäure–reste Ser 209, His 449 und Glu 341 die katalytische Triade. Zwischen den beiden anderen Pfeilen liegt die so genannte Deckel–Helix (engl. Flap), ein amphiphiler Oberflächenloop, dessen Sekundärstruktur flexibel ist. Eine Neuausrichtung dieser Sekundärstruktur bringt das Enzym von seiner offenen in die geschlossene Konformation und umgekehrt. Im offenen Zustand ist die vom Lösemittel umgebene Oberfläche hydrophober und für Substrate der Einlass zum aktiven Zentrum erleichtert.

Dissertation -39- Christoph S. Eberle
Kommerziell erhältliche Lipase Typ VII aus C. rugosa (E.C.3.1.1.3) katalysiert
enantioselektiv die Veresterung von racemischer Liponsäure rac–1.[58] �–Liponsäure[59]
selbst übernimmt wesentliche Aufgaben im Metabolismus: als Wachstumsfaktor
verschiedener Bakterienstämme und als Kofaktor für Multienzymkomplexe, die oxidative
Decarboxylierungen katalysieren, wie etwa die Pyruvat– oder �–Ketoglutarat–Dehydrogenase.
Calvin et al. beschrieben erstmals die Rolle dieser natürlich vorkommenden Säure bei
der Lichtreaktion der Photosynthese[60], was systematische Studien über die Chemie dieser
und ähnlicher Schwefelverbindungen anregte.[61,62] Ihre heterocyclische Disulfideinheit
prädestiniert Liponsäure für die Immobilisierung auf Goldoberflächen. Ihr Racemat kann
gespalten werden durch enzymatische Veresterung, die zu enantiomerenreinen Estern 1a,b
mit (S)–Konfiguration führt und zu angereicherter (R)–Liponsäure 1 als Nebenprodukt
(Schema 2). Fadnavis und Koteshwar entdeckten, dass die Enantioselektivität von der
Kettenlänge des aliphatischen Alkohols abhängt, der umgesetzt wird. Für die vorliegende
Arbeit wurde das enzymatisch erzeugte Diastereomerengemisch mittels fraktionierter
Kristallisation getrennt. Das nicht umgesetzte Liponsäure–Enantiomer fällt in der Kälte als
Feststoff aus, während das veresterte (S)–Enantiomer in Lösung bleibt. Dadurch lässt sich der
Schema 2: Enzymkatalysierte, enantioselektive Veresterung von racemischer �–Liponsäure nach Fadnavis und Koteshwar.[58] Enantiomerenüberschüsse wurden in der Originalpublikation jeweils chromatographisch bestimmt mit Hilfe chiraler Säulen (Chiralcel O.D. und Chiracel OJ, beide 25×0.46 cm, Diacel, Japan). Den Enantiomerenüberschuss von (R)–Liponsäure bei Veresterung mit n–Hexanol geben die Autoren mit 20.1% an. Im Falle der Veresterung mit n–Octanol beträgt der ee–Wert entsprechend 23.8%. Enantiomerenüberschüsse des (S)–Octylesters 1a wurden mit 58%, des (S)–Hexylesters 1b mit 72% angegeben.
S S
O
OHH
+
S S
O
ORH
ROH
R = n-Hexyl, n-Octyl
S S
O
OHH
rac-1
(S)
+
(R)
1
CRL
n-Hexan,30°C
1a,b

Dissertation -40- Christoph S. Eberle
Umsatz nur bis maximal 50% steigern anders als in Reaktionen, bei denen prochirale
Verbindungen eingesetzt werden, aus denen das gewünschte Produkt entsteht. Bei einem
Minimum an Substanzverlust, Kosten und experimentellem Aufwand liefert dieses Verfahren
angereicherte �–Liponsäure 1 mit (R)–Konfiguration. Sie dient als Edukt für die
Partialsynthese der angestrebten Fulleren– und Porphyrinderivate und Porphyrin–Fulleren–
Dyaden. Die Ester 1a,b wiederum sind nützliche Additive bei der Koimmobilisierung mit
porphyrinoiden Metallkomplexen, die ihrerseits Disulfidgruppen tragen. Angereicherte
(R)–Liponsäure 1 bietet sich ferner an als ideale Spacereinheit, um u.a. Fullerene, Imidazole
oder Pyrrole auf Metalloberflächen zu immobilisieren, wo sie mit vorab adsorbierten
Porphyrinoiden wechselwirken können. Die optische Reinheit der gewonnenen (R)–
Liponsäurefraktionen betrug 23% bzw. 40%. Auf die Bestimmung von ee–Werten nach der
Racematspaltung mittels NMR–Spektroskope wurde verzichtet, da bekannte homochirale
Verschiebungsreagenzien nicht in der Umgebung des Chiralitätszentrums wechselwirken.
Zusammen mit früheren Beobachtungen zur Substratspezifität[63] postulierten Fadnavis et al.
zwei hydrophobe Bindungstaschen in Nachbarschaft zum aktiven Zentrum (Abb. 12). Die
Lipase aus C. rugosa bindet darin Kohlenwasserstoffketten der Säure und des aliphatischen
Alkohols. Dieser Strukturvorschlag ähnelt dem von Toone et al. für die Schweineleber–
Esterase[64]. Dazu wird der Vereinfachung wegen auf biochemische und räumliche Details
verzichtet. Dennoch gestattet diese Sichtweise Einblicke in Struktur–Funktions–Beziehungen
bei Lipasen wie der aus C. rugosa, um auch für neue Substrate eine mögliche Spezifität
vorhersagen zu können. Über die vier Methylengruppen, die in die kleinere der beiden
Taschen passen, erkennt das Enzym den Dithiolanring der Liponsäure, während die
Carboxylatgruppe ins aktive Zentrum ragt und der aliphatische Alkohol von der großen
Bindungstasche aufgenommen wird.
Den ’induced fit’ dieser Reaktion bestimmt die Struktur des Alkohols, der nukleophil das
Acyl–Enzym–Intermediat angreift. Je genauer von den beiden Substraten dieses in seine
Tasche passt, desto höher ist die Enantioselektivität der Veresterung. Als optimal für die
Umsetzung erwies sich eine Kettenlänge von sechs Kohlenstoffatomen bei unverzweigten
Alkoholen.[58] Es scheint, dass (S)–Liponsäure spezifisch gebunden wird, wobei das andere
Enantiomer als kompetitiver Inhibitor wirkt. Dies erklärt, warum chirale Ester entstehen,
obwohl vier Kohlenstoffatome zwischen dem Stereozentrum und der katalytischen Triade im
aktiven Zentrum liegen, in dem die enzymatische Reaktion stattfindet.

Dissertation -41- Christoph S. Eberle
Abbildung 12: Schematische Darstellung der molekularen Erkennung von Substraten durch C. rugosa Lipase (nach Fadnavis und Koteshwar, 1998).[58]
S
S
H(CH2)4
OH
O
CO R
aktives Zentrum
molekulareErkennungsseite
hydrophobe Bindungstasche 1
hydrophobeBindungstasche 2
R = n-Octyl oder n-Hexyl

Dissertation -42- Christoph S. Eberle
3.2 Synthese einfacher Liponsäureester und –amide
Nach Vaidyanathan et al.[65] wurde in der vorliegenden Arbeit ein ähnlicher experimenteller
Ansatz gewählt. Dieser beruht auf einem leicht modifizier– und durchführbarem
Eintopfverfahren mit kommerziell vertriebenem N,N’–Carbonyldiimidazol 2 (CDI). Dabei
handelt es sich um ein gängiges Reagenz, das Carbonsäuregruppen aktiviert unter Bildung
eines intermediären Imidazolids. Gleichzeitig wird Kohlendioxid freigesetzt, der die
Kupplung mit primären und sekundären Aminen begünstigt. Dieser katalytische Effekt macht
sich gerade dort bemerkbar, wo sterische oder elektronische Gründe die Reaktivität mindern.
Tatsächlich kann Kohlendioxid die Umsatzraten aber nur bis zu einer bestimmten kritischen
Menge steigern. In Gegenwart überschüssigen Allylbromids gelang es Kamijo et al.[66],
Veresterungen auch mit sterisch gehinderten Reaktionspartnern durchzuführen.
Ingesamt erlaubt das hier angewandte Eintopfverfahren, die Menge an entstehendem Imidazol
gering zu halten, da CDI nur in leichtem Überschuss vorliegt. Bei milden Temperaturen wird
die in trockenem THF gelöste, enantiomerenangereicherte (R)–Liponsäure 1 vollständig
acyliert. Dies lässt sich mittels Dünnschichtchromatographie nach etwa 45 Minuten
feststellen. Für die anschließenden Veresterungen oder Amidierungen wurde das in situ
erzeugte CO2 nicht entfernt. So konnten verschiedene, optisch aktive �–Liponsäureamide und
–ester hergestellt werden (Schema 3). In der Regel lagen Ausbeuten reproduzierbar zwischen
64 – 74%. Amide oder Alkohole, die mit aktivierter Liponsäure umgesetzt wurden, waren
entweder kommerziell erhältlich oder nach Literaturangaben[66, 67] darstellbar.
Derivate wie die hier beschriebenen können als Additive dienen bei der Koimmobilisierung
mit Metalloporhyrinoiden auf Goldoberflächen. Dort koordinieren sie das Zentralatom des
tetrapyrrolischen Ringsystems, oder sie formen eigene Monoschichten mit polaren Enden, an
denen u.a. Elektropolymerisationen durchgeführt werden können. Auf diesem Weg
kontrolliert leitfähige Polymere herzustellen, wird zurzeit mit Hilfe von immobilisierten
Liponsäurepyrrolylestern (4a und 4b)[68] weiter verfolgt. Entsprechende Untersuchungen
finden in der eingangs genannten Kooperation mit der Arbeitsgruppe Abrantes an der
Universität Lissabon statt.

Dissertation -43- Christoph S. Eberle
Schema 3: Darstellung der optisch aktiven Liponsäureester und –amide 3 a,b und 4 a,b nach Vaidyanathan et al.[65] Auf die gleiche Weise wurde das in dieser Arbeit beschriebenen Phytolderivat 5 erhalten. Reagenzien und Bedingungen: (a) THF, CDI 2 (1.2 Äq.), 45°C, 64 – 74%.
N
N
H
S
S
O
OH
H
N N
S
S
O
NH
R
H
1 3
N
S
S
OOH
H
S
S
O
OR
H
1 4
4 a R =
3 a R =
3 b R =
a
2
a
24 b R =
N
N

Dissertation -44- Christoph S. Eberle
3.3 Synthese eines Fulleren–Liponsäure–Derivats
Durch Aktivierung mit N,N’–dicyclohexylcarbodiimid (DCC) und DMAP–Kokatalyse bei
Raumtemperatur wurde der Sulfolenbisester 8 aus dem literaturbekannten Bisakohol 7[69] und
einem Überschuss enantiomerenangereicherter (R)–Liponsäure 1 hergestellt. Bei 190°C
entsteht in situ durch Schwefeldioxid–Extrusion aus dem Bisester 8 das eigentliche Dien, das
mit dienophilem Fulleren–C60 eine Diels–Alder Reaktion eingeht (Schema 4)[70]. So gelingt in
Trichlorbenzol über eine [4+2]–Cycloaddition die Derivatisierung von Fulleren–C60 zu 9.
Dieses neuartige Addukt erwiese sich als ausgezeichneter Akzeptor in kooperierenden
Dyaden[10]. Mit Einführung zweier Liponsäureketten können vier Au–S Bindungen
ausgebildet werden. (R)–konfigurierte Liponsäure wurde gewählt, um stereochemisch
wohldefinierte Fulleren–Liponsäure–Derivate zu gewinnen. Deren Immobilisierung auf
Goldoberflächen soll Monoschichten höheren Ordnungsgrades aufbauen.
Schema 4: Synthese eines Fulleren–Liponsäure–Derivats 9 zur Immobilisierung auf Goldoberflächen. Reagenzien und Bedingungen: (a) DCC, DMAP, THF, RT, 82 %. (b) 1,2,4–Trichlorbenzol, 190°C, 61 %.[70]
3.4 Aufbau einer Porphyrin–Fulleren–Dyade
An chemisorbiertes Fulleren–C60 9[70] kann Zn–Sulfolenoporphyrin 10[71] kovalent gebunden
werden (Schema 5). Dies gelingt über eine zweite Diels–Alder Reaktion unter den gleichen
Bedingungen, wie in Kapitel 3.3 geschildert. In 1,2,4–Trichlorbenzol bei 190°C bildet sich in
S S S
O O
S
OO
H H
9
b
87
S
OO
OHOH
a
S S
O O
H
SS
OO
H
S
O O
1
S
S
H
CO2H
2+
12
1'
5'
3''
1''
1''
3''
5
1
1'2'
3'
1''

Dissertation -45- Christoph S. Eberle
situ aus dem Sulfolen 10 erneut das Dien, das auf die Oberfläche der Fullerenmonoschicht
trifft, welche als Dienophil reagiert. Diese Variante, um Porphyrine an Fullerenakzeptoren
zu knüpfen, ist ebenso auf Biline mit annelierten Sulfolenoringen übertragbar. Aus ihnen
entstanden nach Cyclisierung und [4+2]–Cycloaddition Chlorin–Fulleren–Dyaden.[72,78]
Dieser synthetische Zugang zu künstlichen Photosynthese–Systemen bewährte sich bei
zahlreichen Arbeiten in unserem Laboratorium. Gleichzeitig bedienten sich Kräutler[73],
Gunter[74], Cavaleiro[75] und Smith[76] dieser Variante, um Porphyrin–Fulleren–Derivate
herzustellen.
Schema 5: Partialsynthetische Herstellung der Zn–Porphyrin–Fulleren–Dyade 11. Das Fulleren–Liponsäure–Derivat 9 schied sich zuvor als organische Monoschicht auf einer Goldelektrode mit (111)–Textur ab. Die in situ erzeugte Dienkomponente 10 geht mit der dienophilen C60–Monoschicht eine [4+2]–Cycloaddition ein.
Die Dyade 11 in Lösung zu synthetisieren, ergab bei früheren Versuchen kaum trennbare
Gemische aus Konstitutionsisomeren. Nach thermischer Schwefeldioxid–Extrusion kann
nämlich das Zn–Porphyrin 10 mit seinem exocyclischem olefinischem Rest in
unterschiedlichen Positionen mehrfach an Fulleren–C60 addieren. Dadurch entstehen
Au
S
S
S
O
O
S
O
O
H
H
Δ 1,2,4-Trichlorbenzol
Au
N
N
N
N
CH3
CH3
CH3
CH3O
O
O
O
ZnSO
O
+
9 10
N
N
N
N
CH3
CH3
CH3
CH3O
O
O
O
Zn
S
S
S
O
O
S
O
O
H
H
Pt
e- e-
e- e- hν
Elektrolytlösunge-
e-
- SO2
11

Dissertation -46- Christoph S. Eberle
Konstitutionsisomere oder Mehrfachaddukte. Bemerkenswert ist die Ausbeute von 41%[71]
bzw. 59%[11d] für den Cycloadditionsschritt, obwohl drastische Bedingungen erforderlich sind.
Wenn solche isomeren Addukte auf Goldoberflächen immobilisiert werden, haben die
unterschiedlichen molekularen Anordnungen Einfluss auf Packung und Organisation.
Sie bedingen Unregelmäßigkeiten in der Monoschicht, weil sich dasselbe Donor– und
Akzeptorzentrum in unterschiedlichen molekularen Anordnungen befindet. Aus diesen
Gründen schien die alternative Strategie zielführender, nämlich, die neuartige
Metalloporphyrin–Fulleren–Dyade 11 durch Oberflächenmodifikation herzustellen nachdem
ihr Akzeptorteil bereits immobilisiert ist. Dabei erwies sich die Gold–Schwefel Bindung
stabiler als bislang angenommen, denn trotz harscher Bedingungen, die für Diels–Alder
Reaktionen nötig sind, wurde diese nicht gespalten. Kapitel 4.2. geht im zweiten Teil auf die
elektrochemischen Ergebnisse ein, die den Befund bestätigen. Die Literatur enthält Beispiele,
bei denen Monoschichten auf Metalloberflächen in der Regel bei Raumtemperatur weiter
modifiziert worden sind.[76]
Abbildung 13: (a) Schematischer Überblick über mögliche Konstitutionsisomere und deren Nomenklatur bei Zweitaddition von exocyclischen Dienkomponenten an Fulleren–C60. Bezogen auf den Ort des Erstangriffs unterscheidet man Substitutionen in derselben Hemisphäre (cis), in der gegenüberliegenden (trans) und am Äquator (e). Dies setzt voraus, dass die jeweiligen Reste von gleicher Struktur sind. (b) Hexakis–Addukte an Fulleren–C60 (A. Krüger, 2007).[77c]
Dienkomponenten wie die aus 10 erzeugten können in verschiedenen Positionen und
mehrfach mit C60 reagieren, da die Regioselektivität von Additionen sehr gering ist.[77–78]
Aufgrund sterischer Hinderung vermittels einer dicht gepackten Monoschicht sind hier cis–
Produkte ausgeschlossen. Die in situ gebildete Dienkomponente 10 kann, da ihr der laterale

Dissertation -47- Christoph S. Eberle
Zugang verwehrt ist, nur in der gegenüberliegenden Hemisphäre angreifen, so dass
theoretisch vier Bereiche in Frage kommen (Abb. 13). Hingegen wären an Fehlstellen in der
Monoschicht und an deren Rändern auch andere Positionen substituierbar. In Lösung können
bei der oben durchgeführten Diels–Alder Reaktion insgesamt acht Produkte gebildet werden,
wenn alle dafür möglichen (6,6)–Bindungen betroffen sind und die Substituenten gleiche
Symmetrie aufweisen. Experimentell lassen sich jedoch meist weniger Addukte beobachten,
da die Regiochemie an Fullerenen nachgewiesenen Gesetzmäßigketen folgt.
Um eine Photovoltaikzelle zu verwirklichen, in der lichtinduziert vom Porphyrin–Donor zum
Fulleren–Akzeptor Elektronen fließen, wird häufig auf Derivate solcher Dyadenbausteine
zurückgegriffen, wie sie hier verwendet werden. Sie gelten als vielversprechend zum
Aufbau elektronischer Systeme zu verschiedenen Zwecken.[80–83] Je kontrollierbarer
molekulare Strukturen sich selbst organisieren sollen, umso wichtiger ist hierzu das
Verständnis, wie diese konstruiert sein müssen. Zugenommen haben in den letzten Jahren die
Anstrengungen, künstliche Systeme nach zellulärem Vorbild zu schaffen. Fortschritte in der
Genomforschung ermöglichen weiter, dass immer mehr biologische Phänomene mit Begriffen
der Chemie verstanden werden können. Dies wirkt inspirierend bei Problemen, bei denen
versucht wird, sich Struktur–Funktions–Beziehungen zu Nutze zu machen, die die Natur
evolutionär entwickelt hat.[84] Neu synthetisierte Moleküle müssen ebenso in der Lage
sein, übergreifende, stabile Ordnungen zu bilden. Je besser sie diese Fähigkeit erfüllen, desto
besser schreitet die Entwicklung von Materialen voran, die zugleich funktionell und
nanostrukturiert sind.

Dissertation -48- Christoph S. Eberle
3.5 Synthese von Porphyrinbisestern
Auch Porphyrinoide lassen sich ähnlich wie Fulleren C60 soweit derivatisieren, dass
sie zur Immobilisierung auf Goldoberflächen geeignet sind (Schema 6). Kommerziell
erhältliches Hämin 12, dessen Reinheitsgrad etwa 75% beträgt, wird einer Resorcinschmelze
unterzogen, wodurch zunächst beide Vinylgruppen abgebaut werden (bekannt als
Schummsche Protiodevinylierungsreaktion).[79] Nach Entfernung des Zentralatoms mit
Eisen(II)–sulfat unter Einleitung von HCl–Gas und Veresterung der Propionsäureseitenketten
in methanolischer Lösung entsteht Deuteroporphyrin–IX–dimethylester 13. Dieser dient nach
Aufreinigung durch Chromatographie und Kristallisation als Edukt für alle weiteren
Syntheseschritte.
Reduktion der Methylestergruppen mit überschüssigem Lithiumaluminiumhyrid in siedendem
Tetrahydrofuran macht den entsprechenden Deuteroporphyrin–IX–bisalkohol 14 zugänglich.
Um den Makrotetracyclus zu komplexieren, wurde auf literaturbekannte Verfahren[48]
zurückgegriffen. Bei den Zink–, Nickel– und Kobalt–Komplexen gelang der Metalleinbau
aus den jeweiligen Acetylacetonatsalzen. In siedendem Dimethylformamid wurden die
Kobaltchlorid– und Manganchlorid–Verbindungen dargestellt durch Einbau entsprechender
Metallchloride. In beiden Fällen beobachtet man fast quantitative Umsätze (ausgenommen bei
der Verwendung von Kobalt(II)–acetylacetonat). Diese Komplexierungen schützen das Innere
des Porphyrinringes, und sie erlauben zugleich verschiedenste biomimetische Anwendungen
nach Immobilisierung auf Goldoberflächen: als Katalysatoren, Sensoren oder photovoltaische
Bauteile. Die Porphyrinbisakohole 14, 15a–e reagierten mit enantiomerenangereicherter
�–(R)–Liponsäure 1 unter PPA–Aktivierung mit DMAP als Kokatalysator zu den neuen
Porphyrinbisestern 16a–f. Diese ließen sich chromatographisch von nicht umgesetzten
Edukten trennen. Mit 58 – 70.6 % liegen die Ausbeuten für diese Veresterung klar über denen
früherer Arbeiten aus unserem Laboratorium.[11a,b] Ergebnisse von Veresterungsversuchen mit
anderen Aktivierungsreagenzien sind in Tabelle 2 zusammengefasst.
Tabelle 2: Veresterungsversuche mit gängigen Carbonsäureaktivatoren.
Reagenz Ansatzgröße Ergebnis N,N’–Dicyclohexylcarbodiimid 2.6 Äq. kein Produkt isolierbar
N,N’–Carbonyldiimidazol 1.5 Äq. Monoester
N,N’–Carbonyldiimidazol 3.0 Äq. Monoester

Dissertation -49- Christoph S. Eberle
�–Liponsäure aus der kinetischen Racematspaltung (vgl. Kapitel 3.1) wurde nach einem
modifizierten Literaturverfahren[85] in getrocknetem THF in Gegenwart überschüssigen
LiAlH4 zum entsprechenden Alkohol 6 (Lipol) reduziert. Dabei entsteht zunächst das Dithiol,
das sich mit KI/I2–Lösung zum Disulfid reoxidieren lässt. Mit diesem Linker können auch
Porphyrin– und Chlorinderivate mit freien Carbonsäureresten für die Immobilisierung auf
Goldoberflächen verestert werden. Solche Derivate wurden in unserem Laboratorium
erarbeiteten partialsynthetischen Routen benutzt.
Schema 6: Partialsynthese funktionalisierter Metalloporphyrinoide zur Immobilisierung an Gold–oberflächen. Reagenzien und Bedingungen. (a) 1. THF, LiAlH4, Rückfluß, 4 h, 2. H2O, Rückfluß, 10 min, EtOH, Rückfluß, 5 d, 89%. (b) THF, M(II)–acetylacetonat oder M(II)Cl, DMF, Rückfluß, 24 h, 64.5 – 88.7%. (c) THF, NEt3, DMAP, PPA, �–(R)–Liponsäure, 24 h, 58 – 70.6%.
12 13
N N
N N
CH3
CH3
CH3CH3
CO2H CO2H
CH2
CH2
Fe
ClNH N
N NH
CH3
CH3
CH3CH3
CO2CH3 CO2CH3
NH N
N NH
CH3
CH3
CH3CH3
HO OH
16
14
M = 2H
M = Zn 15a
M = Ni 15b
M = Co 15c
M = MnCl 15d
M = CoCl 15e
b
N N
N N
CH3
CH3
CH3CH3
M
HO OH
15
a
cN N
N N
CH3
CH3
CH3CH3
M
O O
O
SS
H
O
S S
H
M = 2H 16a
M = Zn 16b
M = Ni 16c
M = Co 16d
M = MnCl 16e
M = CoCl 16f

Dissertation -50- Christoph S. Eberle
4. UNTERSUCHUNG MODIFIZIERTER ELEKTRODEN
4.1 Immobilisierung auf Goldoberflächen
Alle in den Experimenten zur Immobilisierung benutzten Wafer wurden von der Firma
Arrandee bezogen. Glass wurde hierfür mit einer haftvermittelnden Zwischenschicht (2 –
4 nm) aus Chrom überzogen und darüber Gold in 200 nm Dicke aufgetragen. Diese
Elektroden wurden vor Gebrauch in „Piranha“–Lösung [30 % H2SO4 : 10 % H2O2, (v:v)]
kurz gereinigt, gründlich mit Ethanol und Ultra–Q–Wasser gespült und unter Stickstoffstrom
getrocknet. Es folgte eine Behandlung in einer Bunsenbrennerflamme, um einheitlich
flache Oberflächen mit vorherrschend (111)–kristalliner Orientierung zu erzeugen
(„flame annealing“). Mehrmaliges Wiederholen dieser Prozedur lieferte die ausgedehnte
Terrassenstruktur, typisch für Au(111) (vgl. Abb. 31a). Solche Substrate für die
voltammetrischen und physikalischen Untersuchungen (vgl. Kapitel 4.2 – 4.4) zu wählen,
stellt sicher, dass Ergebnisse mit denen früherer Studien vergleichbar und konsistent sind.
Ferner lässt die einheitliche Goldoberfläche bessere Wechselwirkung mit immobilisierbaren
Molekülen erwarten.
Modifiziert wurden die vorbehandelten Elektroden im Nassverfahren, das sich sehr einfach
handhaben lässt. Bei diesem so genannten „dip coating“ lagern die Elektroden, eingetaucht in
eine Lösung der aufzutragenden Substanz bei Temperaturen von ca. 4°C, die Möleküle an.
Die Substanzen mit ihren disulfidischen Kopfgruppen, dadurch zur Chemisorption an
Goldoberflächen befähigt, wurden dazu in 0.1 bis 1 mM konzentrierten ethanolischen
Stammlösungen vorbereitet. Die kovalente, leicht polare Au–S Bindung formt sich spontan
durch S–S Bindungsbruch, wodurch sich organische Moleküle aus homogener Lösung als
selbstorganisierte Monoschichten an Metalloberflächen abscheiden. Anschließend wurden die
modifizierten Elektroden gründlich mit Ethanol und Ultra–Q–Wasser gespült und im
Stickstoffstrom getrocknet.
Seit der Entdeckung von Nuzzo und Allara[34], dass schwefelorganische Verbindungen an
Goldoberflächen spontan und dabei sehr geordnet chemisorbiert werden, haben sich
Forschungsgruppen in zahlreichen Arbeiten mit solchen Systemen beschäftigt. Inzwischen
gibt es unterschiedliche Ansätze, die die Fachliteratur beschreibt. So konnten auch
komplexere Moleküle mit Schwefelgruppen derivatisiert werden, darunter Fullerene,
Porphyrinoide, Metalloporphyrinoide, leitfähige Polymere oder Kohlenstoffröhren, um
selbstorganisierte Monoschichten aufzubauen (vgl. Kapitel 1.3). Deren Merkmale und

Dissertation -51- Christoph S. Eberle
Beschaffenheit im Detail zu beschreiben, gelingt heute mit Standardtechniken, von denen
einige auch in dieser Arbeit angewandt wurden.
4.2 Cyclovoltammetrie
4.2.1 Elektrochemische Charakterisierung von Fulleren–Monoschichten auf
Goldoberflächen
Zur elektrochemischen Charakterisierung von modifizierten Goldoberflächen diente eine
standardisierte Meßzelle (Abb. 14). Darin wurden Wafer mechanisch gegen einen
teflonbeschichteten Gummiring gepresst, der eine Fläche von 0.57 cm2 definiert.
Abbildung 14: Für die Cyclovoltammetrie verwendete Teflonzelle (Viana, 2002).[86] Eine gesättigte Kalomelelektrode (SCE) diente als Referenz bei allen elektrochemischen Messungen.
Immobilisierte Moleküle geben eine andere elektrochemische Antwort als solche, die sich
in Lösung befinden.[86, 107] Voltammogramme sind verschieden, weil von den Reaktanden eine
bestimmte Menge an die Metalloberfläche adsorbiert. Da diese Menge in der Regel
gleichbleibend ist, kann der diffusionsbedingte Stofftransport zur Elektrode vernachlässigt
werden. Verläuft der Elektronentransfer reversibel, resultieren Kurven, bei denen idealerweise
das Peakpotential der reduzierten und oxidierten Form genau gleich ist (Ep = 0). Dies gilt für
ein Adsorptionsverhalten, das durch die Langmuir–Isotherme ausgedrückt wird, und lässt auf
Goldwafer
Gummiring
Referenzelektrode
Sekundärelektrode (Pt) Stickstoffzufuhr
Leitung für elektrischen Kontakt

Dissertation -52- Christoph S. Eberle
keinerlei Interaktion zwischen den Redoxzentren schließen. Selbstorganisierte Monoschichten
stellen nur selten ein solch ideales System dar. Häufig ergeben sich über Fehlstellen
im Substrat eine schlechtere Selbstorganisation der Adsorbate neben Mehrschichtenbildung
oder stark intermolekularen Wechselwirkungen zwischen den Redoxpaaren. All dieses
verursacht Abweichungen vom idealen Verhalten. Abhängig von Konzentration und
chemischer Struktur, beeinflussen Elektrolyte den Redoxprozess wesentlich, da sie auch
in Monoschichten eindringen können. Nachfolgend wird detaillierter und vergleichend
eingegangen auf elektrochemische Eigenschaften von auf Au(111) immobilisierten
Molekülen, die in dieser Arbeit synthetisiert wurden. Ihre verschiedenen funktionellen
Endgruppen ermöglichen es, Ladungstransfermechanismen an Grenzflächen zu studieren.
Dabei lassen sich Redox– und elektrokatalytische Aktivitäten der organischen
Oberflächenfilme festzustellen.
Abbildung 15: Cyclovoltammetrie einer Goldelektrode, beschichtet mit dem Fulleren–Liponsäure–Derivat 9, in 0.1 mol dm-3 TEAP in Propylencarbonat. [70] Das Voltammogramm wurde mit 200 mVs-1
aufgezeichnet. Die eingefügte Grafik zeigt die lineare Beziehung zwischen Stromdichte und Vorschubgeschwindigkeit (gezeigt von 20 bis 400 mVs-1).
-1.2 -0.9 -0.6 -0.3 0.0
-30
-15
0
15
30
i / μ
Acm
-2
E / V vs. SCE
0.0 0.1 0.2 0.3 0.4
0
10
20
30
40
i pox/ μ
Acm
-2
ν / mVs-1
R2 = 0.9933
– 0.61 V

Dissertation -53- Christoph S. Eberle
Goldelektroden, an denen das Fulleren–Liponsäure–Derivat 9[70] abgeschieden wurde, ließen
sich in 0.1 mol dm-3 TEAP in Propylencarbonat vermessen (Abb. 15). Andere Elektrolyte
gleicher Konzentration, z.B. 0.1 mol dm-3 LiClO4 in Acetonitril, lieferten weniger
reproduzierbare Ergebnisse. Bei ca. –0.65 V und –1.05 V zeigt das Voltammogramm zwei
Redoxpeaks, die zum ersten und zweiten Ein–Elektronentransfer im Fullerenredoxprozess
gehören. Beide Potentiale stimmen überein mit Literaturwerten für auf Gold ab–
geschiedene Monoschichten aus chemisch modifizierten Fullerenen.[80, 87] Es ist bekannt,
dass solche Derivatisierungen Einfluss üben auf das Redoxverhalten, da Doppelbindungen
gesättigt werden und so das über das Kohlenstoffgerüst ausgedehnte �–System eingeschränkt
wird. Dies verschiebt Redoxpotentiale hin zu negativen Werten. Unmodifiziert kann
Fulleren–C60 reversibel bis zu sechs Elektronen aufnehmen: entweder in organischen
Lösemittelgemischen[88] oder in flüssigem Ammoniak bei niedrigen Temperaturen.[89]
Auf monokristallinen Goldoberflächen bleiben die immobilisierten Fulleren–Liponsäure–
Derivate 9 zwar elektroaktiv, doch spalten sich beide Redoxpeaks weiter auf als bei
immobilisierten Systemen dieser Art erwartet werden darf. Daneben veranschaulicht Abb. 15,
dass die Stromdichte linear ansteigt mit der Vorschubgeschwindigkeit, ein Zusammenhang,
der beispielhaft für den Oxidationsprozess bei –0.61 V gezeigt ist.
Abbildung 16: Reduktive Desorption einer Monoschicht, abgeschieden aus Derivat 9 auf einer monokristallinen Goldoberfläche, in 0.1 mol dm-3 NaOH. Die Voltammogramme wurden bei 20 mV s-1 aufgezeichnet nach einstündiger Entgasung der Elektrolytlösung.[70]
In stark alkalischer Lösung lösen sich Monoschichten bei negativen Potentialen wieder
von der Goldoberfläche ab. Daraus lässt sich sowohl auf die Stärke der Au–S Bindung
-1.4 -1.2 -1.0 -0.8 -0.6 -0.4 -0.2 0.0
-45
-30
-15
0
1st cycle 2nd cycle
i / μ
A c
m-2
E / V vs. SCE
1. Zyklus
2. Zyklus
– 1.225 V

Dissertation -54- Christoph S. Eberle
schließen als auch auf Menge der adsobierten Moleküle. Diese reduktive Desorption stellt
somit das Gegenexperiment dar zur vorangehenden Immobilisierung (vgl. Gleichung 4).
Der cyclovoltammetrisch verfolgte Prozess für eine abgelöste Monoschicht des Fulleren–
Liponsäure–Derivats 9 ist in Abb. 16 gezeigt. Man beobachtet ein Peakpotential bei
–1.225 V, das leicht negativer liegt als bei vergleichbarer Desorption einer Thiolmonoschicht
mit bloßen Kohlenwasserstoffketten (–1.115 V)[90]. Seiner Reproduzierbarkeit wegen erklärt
der zu negativen Potentialen verschobene Peak die größere Stabilität der C60–Monoschicht
über ihren Bisdisulfidanker. Dass Moleküle mit disulfidischen Kopfgruppen Thiolate bilden,
die stabile Monoschichten aufbauen, konnten frühere Arbeiten elektrochemisch und mittels
XPS bestätigen.[12, 91] In Abb. 2 wie auch bei reduktiver Desorption anderer, in dieser Arbeit
beschriebener Liponsäurederivate finden sich keine Hinweise auf S–oxidierte Spezies. Solche
Disulfinate und Disulfonate können in der Adsorptionslösung vorhanden sein. Sie wurden
beobachtet von Dong et al., die sie mit einem zweiten Peak identifizierten, der ebenfalls bei
negativerem Potential auftauchte als der Reduktionspeak (– 0.94 V).[92]
Unterhalb des Reduktionspeaks gibt im obigen Voltammogramm die Integration der
eingeschlossenen Fläche die Ladung aller immobilisierten Fullerenmoleküle an. Da das
kathodische Peakpotential nahe an dem für die Wasserstoffbildung liegt, fällt eine exakte
Integration schwer. Zugrunde gelegt wird für die weiteren Berechnungen, dass vier
Elektronen übertragen wurden, um den Gold–Schwefel–Bindungsbruch herbeizuführen.
Berechnet aus der reduktiven Desorption des Fulleren–Liponsäure–Derivats 4 von einer
monokristallinen Goldoberfläche gemäß Gl. 5, ergibt sich der Bedeckungsgrad �:
� = Q / nFA (5)
Der um den Faktor 1.2 korrigierte Endwert betrug durchschnittlich 1.4×10-10 mol cm-2 oder
Z = 8 × 1013 Moleküle cm-2. Dies ergibt für das Fulleren–Liponsäure–Derivat 4 einen
Platzbedarf von 1.19 nm2 auf der Goldoberfläche, der dem für ein C60–Molekül zu
erwartenden räumlichen Ausdehnung entspricht (~ 1nm2 / Molekül).
S(CH2)nX + e-Au Au0 + X(CH2)nS- (4)

Dissertation -55- Christoph S. Eberle
4.2.2 Elektrochemische Charakterisierung von Metalloporphyrin–Fulleren–
Monoschichten auf Goldoberflächen
Um die Stärke der Au–S Bindung zu testen, wurden aus 1 mM konzentrierten ethanolischen
Stammlösungen der Liponsäurederivate 1a und 3b Monoschichten an monokristallinen
Goldoberflächen abgeschieden. Nachdem die so modifizierten Elektroden 40 Minuten lang
siedendem Toluol ausgesetzt waren, wurden sie cyclovoltammetrisch untersucht. Abbildung
17 und 18 zeigen die jeweiligen Reduktionspeaks bei ca. 1.0 V, die auf die Anwesenheit
intakt gebliebener Monoschichten hinweisen. Bei nachfolgenden Zyklen verschieben sich die
Peaks zu positiveren Potentialen. Mit jedem weiteren kathodischen Durchlauf nähert sich das
elektrochemische Verhalten dem unmodifizierter Goldelektroden an. Sobald das Potential
umschlägt und wieder positivere Werte annimmt, beobachtet man teilweise Resorption der in
Lösung gegangenen Liponsäurederivate. Deren erneute Ablösung gelingt anschließend
früher, was auf abnehmende Stabilität der in kürzerer Zeit gebildeten Monoschichten deutet.
Nach reduktiver Desorption und Integration der Fläche unterhalb des ersten Reduktionspeaks
ließ sich anhand von Gl. 5 der Bedeckungsgrad der betreffenden Goldoberfläche ermitteln.
Der Wert von 6.5 × 10-10 mol cm-2 spricht für eine dicht gepackte Monoschicht, wobei der
nötige Korrekturfaktor von 1.2 für diese Art Oberflächen einbezogen wurde (Abb. 17).
Abbildung 17: Reduktive Desorption einer mit 3b modifizierten Goldelektrode mit vorherrschend (111)–kristalliner Orientierung. Die Elektrode wurde zuvor für 40 min siedendem Toluol ausgesetzt. 100 mM NaOH–Lösung diente als Elektrolyt, und das Voltammogramm wurde mit 20 mVs-1 aufgezeichnet.
-1,6 -1,2 -0,8 -0,4 0,0-60
-40
-20
0
i / μ
Α c
m−
2
E / V vs. SCE
1. Zyklus 2. Zyklus 3. Zyklus 4. Zyklus
Ep
red, I = -1.01 V
Ep
red, II = -0.95 V
N
N
S
S
O
NH
H
3b
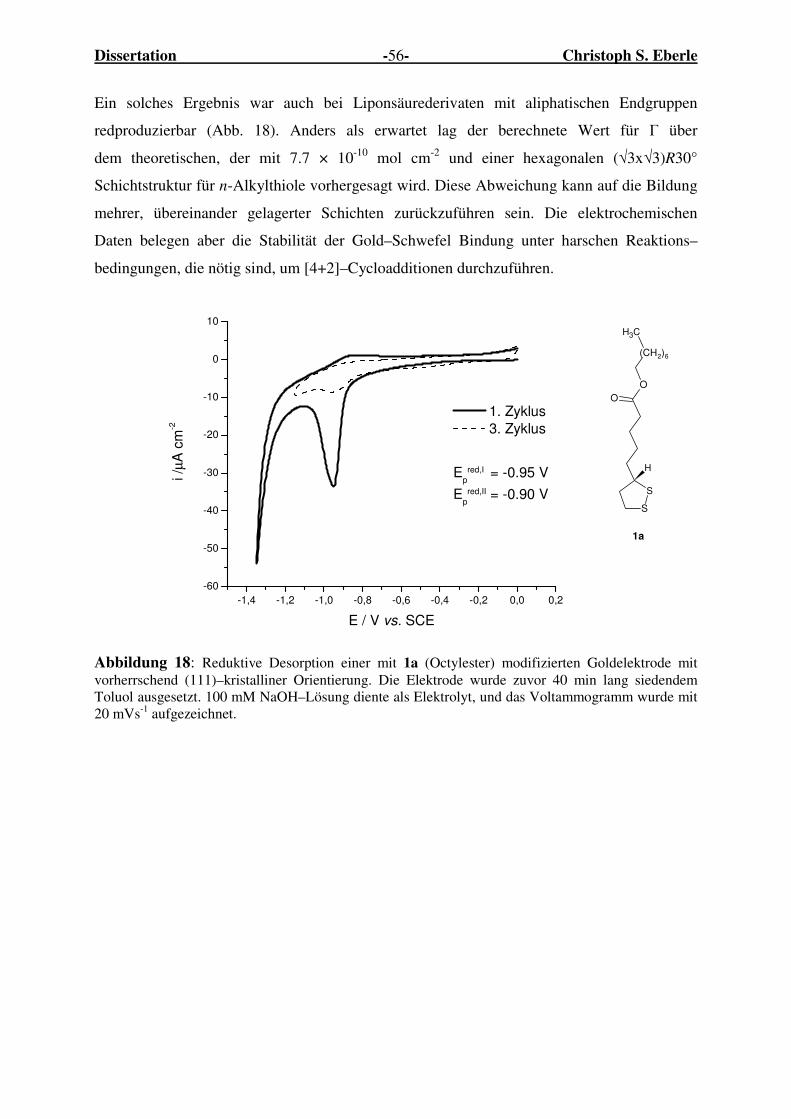
Dissertation -56- Christoph S. Eberle
Ein solches Ergebnis war auch bei Liponsäurederivaten mit aliphatischen Endgruppen
redproduzierbar (Abb. 18). Anders als erwartet lag der berechnete Wert für über
dem theoretischen, der mit 7.7 × 10-10 mol cm-2 und einer hexagonalen (3x3)R30°
Schichtstruktur für n-Alkylthiole vorhergesagt wird. Diese Abweichung kann auf die Bildung
mehrer, übereinander gelagerter Schichten zurückzuführen sein. Die elektrochemischen
Daten belegen aber die Stabilität der Gold–Schwefel Bindung unter harschen Reaktions–
bedingungen, die nötig sind, um [4+2]–Cycloadditionen durchzuführen.
Abbildung 18: Reduktive Desorption einer mit 1a (Octylester) modifizierten Goldelektrode mit vorherrschend (111)–kristalliner Orientierung. Die Elektrode wurde zuvor 40 min lang siedendem Toluol ausgesetzt. 100 mM NaOH–Lösung diente als Elektrolyt, und das Voltammogramm wurde mit 20 mVs-1 aufgezeichnet.
-1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0 0,2-60
-50
-40
-30
-20
-10
0
10
i /μA
cm
-2
E / V vs. SCE
1. Zyklus 3. Zyklus
Epred,I = -0.95 V
Epred,II = -0.90 V
S
S
OO
(CH2)6
H
CH3
1a

Dissertation -57- Christoph S. Eberle
Um selbstorganisierte Monoschichten aus der Metalloporphyrin–Fulleren–Dyade auf Au(111)
aufzubauen, wurden zwei Ansätze in dieser Arbeit verfolgt. Im ersten wurde die
Zielverbindung partialsynthetisch in Lösung hergestellt.[11d] Dabei entsteht ein Gemisch aus
Konstitutionsisomeren, die zur Immobilisierung auf Goldoberflächen herangezogen wurden.
Alternativ erfolgte der letzte Syntheseschritt an bereits immobilisierten dienophilen
Fullerenmonoschichten, deren elektrochemische Charakterisierung das vorangehende Kapitel
beschreibt. Diese wurden Diels–Alder Reaktionen mit dem Zn–Porphyrin 10 unterzogen (vgl.
Kapitel 3.4).
Nach dieser Zweitfunktionalisierung der Monoschicht wurden entsprechende Goldelektroden
elektrochemisch untersucht. Das Voltammogramm 19a veranschaulicht den reversiblen
Oxidationsverlauf der Zn–Porphyrineinheit der Dyade 11 verglichen mit einer 0.3 mM
Lösung des Sulfolenoporphyrins 10. In beiden Fällen wird durch zwei Peaks bei 0.61 V (I)
und 0.81 V (II) die schrittweise Ein– und Zwei–Elektronen–Oxidation des Metalloporphyrins
angezeigt. Ferner kann der Reduktionprozess der Fullereneinheit sowohl für das
Konstituionsisomerengemisch der Dyade nachvollzogen werden als für die immobilisierte
Dyade nach Diels–Alder–Reaktion mit dem Porphyrinteil (Abb 19b). Bei –0.54 V und
–0.96 V zeigt das Voltammogramm 19b für die Dyade 11 (Diels–Alder–Addukt) zwei
reversible Redoxpeaks, die dem ersten und zweiten Ein–Elektronentransfer im Fulleren–
redoxprozess entsprechen. Diese Belege zusammen mit der Desorbierbarkeit der Dyade nach
der Cycloaddition zeigen, dass eine über Dithiolanlinker verankerte Monoschicht mit einem
Fulleren–Akzeptor und einem Zn–Porphyrin–Donor auf der Goldoberfläche vorhanden ist.
Noch bleibt zu klären, warum das elektrochemische Verhalten des Porphyrinredoxzentrums
verschieden ist von dem in Lösung beobachteten, in der die Dyade 11 als Isomerengemisch
vorlag.

Dissertation -58- Christoph S. Eberle
(a)
(b) Abbildung 19: Elektrochemische Charakterisierung einer Monoschicht aus der Dyade 11. Die Voltammogramme wurden in 0.1 TMAP/PC. Gezeigt sind die Ausschnitte für den Porphyrinbereich (a) und für den Fullerenbereich (b). Die Vorschubgeschwindigkeit betrug jeweils (a) 50 mV s-1 und (b) 200 mVs-1 oder 50 mVs-1.
Ellipsometrische Messungen ergänzen diese Beobachtungen, denn sie sind konsistent mit
der Bildung einer C60–Monoschicht, die anschließend weiter chemisch modifiziert wurde und
die immobilisierte Zn–Porphyrin–Fulleren–Dyade 11 nachweist (vgl. Kapitel 4.3.3).
Kontrollexperimente mit der Dienkomponente 10 ergaben, dass ohne vorhergehende
immobilisierbare Fullerenmonoschicht die verwendeten Elektroden unter Diels–Alder
Reaktionsbedingungen ebenfalls modifizierbar sind. Allerdings konnte ein Redoxprozess
erwartungsgemäß nicht detektiert werden. Voltamogramme 20a und 20b bilden die
0,0 0,2 0,4 0,6 0,8 1,0
0
10
20
30
I
II
Au/Zn-Porphyrin-C60
SAM Zn-Sulfolenoporphyrin 10 (0.3 mM Lösung)
i / μ
A c
m-2
E / V vs. SCE
-1,2 -1,0 -0,8 -0,6 -0,4 -0,2
-30
-20
-10
0
10 Au/Zn-Porphyrin-C60
SAM Dyade 11 (3.5 mM Lösung)
E / V vs. SCE
i / μ
A c
m-2
200 mV s-1
50 mV s-1
-40
-20
0
i / μA cm
-2

Dissertation -59- Christoph S. Eberle
entsprechenden Desorptionsversuche ab. Sie belegen schwache Chemisorption des Zn–
Sulfolenoporphyrins 10 an der monokristallinen Goldoberfläche. Dies trat sowohl bei
Raumtemperatur als auch bei Erhitzen bis ca. 200°C auf.
Damit zeigen die experimentellen Daten, dass das Sulfolenoporphyrin 10 chemisch teilweise
am Substrat bindet, ehe die Reaktionstemperatur erreicht wird und SO2 extruiert. Zwar
widmet die Literatur sich ausführlich der Chemie schwefelorganischer Verbindungen zur
Modifikation von Metallsubstraten, doch ist die Zahl der Beispiele gering, bei denen
Chemisorption und die damit verbundenen Prozesse der Selbstorganisation auf molekularer
Ebene genauer agbebildet werden, insbesondere für komplexere Adsorbate. Fernandes et al.
führten computergestützte Studien mit 1–Dekanthiol auf Au(111) durch, um zu simulieren,
wie die Metalloberfläche mit Thiolaten wechselwirkt.[93] Mittels BOC–MP Methoden
analysierten Shustorovich und Sellers die Reaktivität von Schwefeloxiden vorwiegend
an monokristallinen Übergangsmetallen.[94] Wie verschiedene schwefelhaltige Verbindungen
an Goldoberflächen binden, untersuchten dagegen Rodriguez et al. mit modernen
spektroskopischen Verfahren systematisch.[95] Bisher wurden derartige theoretische oder
experimentelle Untersuchungen noch nicht mit Sulfolenen unternommen. Entsprechend lässt
sich anhand der zuvor beschriebenen elektrochemischen Indizien keine Aussage treffen über
die Natur einer möglicherweise kovalenten Gold–Sulfolen–Bindung. Eine solche ist aber
anzunehmen, da SO2 allein auf Au(111) bei tiefen Temperaturen zunächst nicht zerfällt,
sondern schwach chemisorbiert. Unklar bleibt dabei, welche SO2–Atome die Bindung
vermitteln.[96] Diese wird geschwächt bei Metallen, die Elektronen zum LUMO von SO2
übertragen können, wie etwa Kupfer. Sowohl Gold als auch Silber sind dazu weniger in der
Lage aufgrund ihrer energetisch tiefer liegenden d–Orbitale. Deshalb kann das unbesetzte
LUMO des Schwefeldioxids nicht S–O antibindend wirken, so dass Chemisorption statt
Zersetzung resultiert.[95, 96]

Dissertation -60- Christoph S. Eberle
(a)
(b)
Abbildung 20: Reduktive Desorption des Zn–Porphyrinsulfolens 10. Eine 1 mM Lösung des Sulfolens in 1,2,4–Trichlorbenzol wurde jeweils 60 min. bei Raumtemperatur (a) und bei 190°C (b) mit einer monokristallinen Goldelektrode inkubiert Die Voltammogramme wurden mit 20 mV s-1 in 100 mM NaOH–Lösung aufgezeichnet.
Bei Temperaturen von 190°C, die für die [4+2]–Cycloaddition nötig sind, sollten schwache
Wechselwirkungen des Sulofenoporphyrins 10 mit Au(111) aufgehoben werden. Doch sollte
auch die in situ erzeugte Dieneinheit an Gold chemisorbieren. Ähnliches ist bekannt
von metallorganischen Komplexen, an denen Olefine kontrolliert gebunden und freigesetzt
werden können. Darauf beruhen elektrochemische Verfahren zur Trennung von Olefin–
gemischen, die in den letzten Jahren entwickelt wurden. Sie nutzen die steuerbare Affinität
-1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0
-15
-10
-5
0
5
1. Zyklus 2. Zyklus 3. Zyklus
i / μ
A c
m-2
E / V vs. SCE
-1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0
-20
-15
-10
-5
0
5
1. Zyklus 2. Zyklus 3. Zyklus 4. Zyklusi /
μA
cm
-2
E / V vs. SCE

Dissertation -61- Christoph S. Eberle
einzelner Komponenten in diesen Gemischen für Übergangsmetallkomplexe.[97] Solche lassen
sich auch katalytisch einsetzen. Vor Kurzem stellten Li et al. Beispiele für organische
Reaktionen zusammen, bei denen die Bindung von Olefinen an Gold eine Rolle spielen.[98]
Dass in beiden Fällen –wie die Voltammogramme 20a und 20b zeigen– eine Immobilisierung
durch chemische Bindung stattgefunden hat, bestätigen ellipsometrische Daten. Zurück–
zuführen ist dieser Befund auf die Reaktion der Sulfolengruppe (oder des in situ generierten
Schwefeldioxids) an der monokristallinen Goldoberfläche. Ein solche Wechselwirkung
erklärt z.T. die höhere Belegungsdichte, die nach Desorption der Monoschicht, bestehend aus
dem Diels–Alder Addukt 11, berechnet werden konnte (Abb. 22b). Mit 3.9 × 10-10 mol cm-2
verdoppelte sie sich in etwa gegenüber der Belegungsdichte der als Konstitutionsisomeren–
gemisch immobilisierten Dyade (� = 2.3 × 10-10 mol cm-2). Daraus folgt, dass im Falle des
Gemischs pro Molekül ca. 7.19 nm2 beansprucht werden, während sich für das Diels–Alder
Addukt der Platzbedarf auf durchschnittlich 4.26 nm2 verringert.
Zum einen kommen sich die Redoxzentren in der nach der Diels–Alder Reaktion
vorhandenen Monoschicht sehr nahe, ausgedrückt durch einen scharfen Reduktionspeak.
Desorptionsverhalten wie die hier gezeigten konnten in allen Voltammogrammen beobachtet
werden. Die harschen Bedingungen der [4+2]–Cycloaddition destabilisieren die Au–S
Bindung, ohne sie zu spalten, ausgedrückt durch das um durchschnittlich 200 mV positivere
Reduktionspotential (vgl. Abb. 22b). Eine solche Verschiebung war bei Desorption des
Isomerengemischs nicht zu beobachten. Aus den voltammetrischen wie auch den
ellipsometrische Daten lässt sich schließen, dass die Dyade 11 auf der Goldelektrode gebildet
wurde. Gleichwohl gibt es experimentelle Anzeichen für einen teilweisen Abbau der
Fullerenmonoschicht. Dies geschieht entweder unter den hohen Temperaturen oder
durch Austausch mit SO2 und dem Sulfolenoporphyrin 10 während der Reaktion. Zwar
besteht auch bei der Variante, das Sulfolenoporphyrin 10 durch Schwefeldioxid–Extrusion
kovalent an eine C60–Monoschicht zu knüpfen, die Gefahr, dass Mehrfachadditionen der
Dienkomponente stattfinden. Jedoch sind hierbei die konstitutionsisomeren Möglichkeiten
weitestgehend eingeschränkt. Sowohl die Unterseiten–Abschirmung durch die Fulleren–
monoschicht als auch die Größe des Addenden (vgl. Kapitel 3.4) schließt die Bildung von
cis–Produkten aus. Außerdem wurde schon früher beobachtet, dass thermische Behandlung
von organischen Monoschichten nicht nur die laterale Adsorbatemobilität auf der Gold–
oberfläche erhöht, sondern auch Defekte im Metallsubstrat ausbessern kann. Gleichzeitig
vermindert dies Defekte in der Monoschicht selbst, deren Qualität sichtbar verbessert wird.[99]
Somit lässt sich vermuten, dass auch nach der Diels–Alder Reaktion die oben beschriebenen,

Dissertation -62- Christoph S. Eberle
termisch induzierten Effekte am Substrat zu einer dichter gepackten Monoschicht geführt
haben. Zum anderen müssen die konstitutionsisomeren Diels–Alder Produkte die
Belegungsdichte signifikant verringern, sofern sie als Gemisch auf Goldoberflächen
immobilisiert werden. Dies folgt aus dem größeren Platzbedarf der verschiedenartig
angeordneten, sterisch anspruchsvollen Bisaddukte (Abb. 21). Gemäß früherer Befunde[79]
darf dabei unterstellt werden, dass in Lösung die e– und trans–substitutierten Produkte die
Hauptfraktion stellen.

Dissertation -63- Christoph S. Eberle
Abbildung 21: Selbstorganisierte Monoschichten der Metalloporphyrin–Fulleren–Dyade 11 auf Goldelektroden. (a) nach Diels–Alder Reaktion an einer vorangehend immobilisierten Fulleren–oberfläche. (b) Immobilisierung der in Lösung synthetisierten Dyade. Scheidet diese sich als Gemisch ab, so resultiert eine weniger dichte Packung der Konstitutionsisomere in der Monoschicht.
N N
N N
CH3
CH3
CH3CH3
O OO O
Zn
S S S
O O
S
OO
HH
Au
N N
N N
CH3
CH3
CH3CH3
O OO O
Zn
S S S
O O
S
OO
HH
N N
N N
CH3
CH3
CH3CH3
O OO O
Zn
S S S
O O
S
OO
HH
N N
N N
CH3
CH3
CH3CH3
O OO O
Zn
S S S
O O
S
OO
HH
Au
N N
N N
CH3
CH3
CH3CH3
O OO O
Zn
S S S
O O
S
OO
HH
NN
NN
CH3
CH3
CH3
CH3
O
O
O
O
Zn
S S S
O O
S
OO
HH
N
NN
N
CH3
CH3
CH3
CH3
O
O
O
O
Zn
S S S
O O
S
OO
HH
a)
b)

Dissertation -64- Christoph S. Eberle
(a)
(b)
Abbildung 22: Reduktive Desorption der Zn–Porphyrin–Fulleren C60–Dyade 11 von einer Au(111)–Oberfläche. (a) Desorption des Konstitutionsisomerengemischs der Dyade und (b) nach Diels–Alder–Reaktion an einer vorab immobilisierten C60–Monoschicht. Die Voltammogramme wurden in 100 mM NaOH–Lösung mit 20 mV s-1 aufgezeichnet.
-1,6 -1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0 0,2
-50
-40
-30
-20
-10
0
10
Ered
= -1.225 V
i / μ
A c
m-2
E / V vs. SCE
1. Zyklus
-1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0
-100
-80
-60
-40
-20
0
20
Ered
= -1.007 V
1. Zyklus
i / μ
A c
m-2
E / V vs. SCE

Dissertation -65- Christoph S. Eberle
4.2.3 Charakterisierung von Metalloporphyrin–Monoschichten auf
Goldoberflächen
Monoschichten aus schwefelorganischen Derivaten behaupten ihren Platz als einfaches
Studienobjekt, wenn Oberflächen chemisch modifiziert werden sollen. Seit Jahren kommen
sie nun in vielerlei Bereichen zum Einsatz: als chemische und biologische Sensoren,
elektronische Bauteile u.a. für Quantencomputer und in der Katalysatorforschung (vgl.
Kapitel 1.3). Um Modellsysteme zu schaffen, die z.B. als Elektrokatalysatoren wirken
können, ist es nötig, Moleküle zuvor in Lösung auf die gewünschte Anwendung zu testen.
Deren individuelle Eigenschaften müssen erhalten bleiben in einem Film auf festen,
leitfähigen Oberflächen, die im atomaren Bereich spezifisch ausgerichtet sind
(Nanostrukturierung). Solche Monoschichten entstehen durch Selbstorganisation ihrer
Komponenten auf dem Substrat. Um sie dort reproduzierbar wachsen zu lassen, müssen die
Zielverbindungen mit molekularen Ankern versehen werden, die robuste Haftung vermitteln.
Gemäß dieser Strategie sind im Folgenden die elektrokatalytischen Eigenschaften des
neu synthetisierten Co–Porphyrin–Liponsäurebisesters 16d vorgestellt (vgl. Kapitel 3.5). Zur
Immobilisierung auf Au(111) dienten zwei Dithiolangruppen pro Porphyrinring. Beide
Gruppen beteiligen sich an der Chemisorption, weshalb der Kobaltkomplex in hoher Dichte
auf die Oberfläche gelangt. Belegungsraten wurden –wie beschrieben– anhand der reduktiven
Desorption der organischen Monoschicht ermittelt (Abb. 24a). Der scharfe Reduktionspeak
im ersten Cyclovoltammogramm trat auf bei –1.10 V. Integration der Potentialfläche ergab
einen Wert von 2.3 × 10-10 mol cm-2, wobei die Annahme zugrunde lag, dass vier Elektronen
(je eines pro Schwefelatom) übertragen wurden. Dies stimmt überein mit früheren
elektrochemischen und ellipsometrischen Untersuchungen[19], die die Anwesenheit gut
gepackter Monoschichten belegen. In ihnen sind die Porphyrinringe zur Goldoberfläche
geneigt (vornehmlich perpendiculare Orientierung). Bei weiteren kathodischen Durchläufen
resultieren kleinere und positiv verschobene Reduktionspeaks. Sie gehören zu Thiolatspezies,
die nach Ablösung von der Goldelektrode erneut readsorbieren. Dieser Prozess kann nur
in sehr kurzen Zeiträumen ablaufen, so dass ihre Zahl auf der Oberfläche fortlaufend
abnimmt. Zwischen den nun immobilisierten Metalloporphyrinderivaten bestehen keine
starken Wechselwirkungen mehr, weshalb sich die ungeordnetere Monoschicht leichter
desorbieren lässt. Abbildung 23 veranschaulicht das reversible Oxidationsverhalten der
Vorläuferverbindung 15c. Jeweils ein Oxidationspeak bei 0.98 V mit dem korrespondierenden

Dissertation -66- Christoph S. Eberle
Reduktions–potential bei 0.917 V war identifizierbar, die der zweifachen Oxidation des
Porphyrinrings zum Dikation entsprechen. Dagegen sind die beiden zum Co(II)/Co(III)–
Wechsel gehörenden Redoxpeaks im Voltammogramm zu schlecht aufgelöst, um zugeordnet
werden zu können.
Abbildung 23: Cyclovoltammetrie des Co–Bisalkohols 15c. 10 mg des Metalloporphyrins wurden in 0.1 M TBAP/PC auf einer unmodifizierten monokristallinen Goldelektrode mit 50 mV s-1 vermessen.
Nach Immobilisierung büßt das katalytische Metallzentrum seine Aktivität nicht ein (Abb.
24b). Im ersten Cyclovoltammogramm bildet der klar definierte große kathodische Peak bei
–120 mV die elektrokatalytische Reduktion gelösten Sauerstoffs ab. Die Reaktion trat auf
in 500 mM wässriger, sauerstoffhaltiger H2SO4. Somit kann unterstellt werden, dass die Co–
Porphyrinmonoschicht in oxidierter Form vorliegt. Co(II), das für die Sauerstoffbindung
verantwortlich ist, kann elektrochemisch zu Co(III) regeneriert werden. Für eine ähnlich
organisierte Co–Porphyrinmonoschicht auf Au(111)[19b] wurden entsprechende Versuche
in 100 mM sauerstoffhaltiger H2SO4–Lösung durchgeführt und ein weit positiveres
Reduktionspotential festgestellt (0.085 V). Verglichen mit der hier untersuchten Monoschicht,
fällt auf, dass bei fast gleicher Belegungsdichte (2.3 × 10-10 mol cm-2) die an der Reaktion
beteiligte Stromdichte in etwa um das 2.5–fache geringer ist (~60 μA cm-2). Das heißt, ein
immobilisierter Co–Porphyrin–liponsäurebisesters 16d beansprucht durchschnittlich eine
Fläche von 7.22 nm2, was dem Platzbedarf für ein cystaminverbücktes Co–Porphyrinderivat
(6.64 nm2)[19b] ähnelt. Die vorliegenden Ergebnisse deuten ferner auf Entstehung
verschiedener Produkte während des elektrochemischen Stoffumsatzes. Wahrscheinlich setzt
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4
-10
0
10
20
30
40
50
i / μ
Α c
m-2
E / V vs. SCE

Dissertation -67- Christoph S. Eberle
sich zunächst gebildetes Wasserstoffperoxid in der stark sauren Umgebung sofort zu Wasser
um.
(a)
(b)
Abbildung 24: (a) Reduktive Desorption einer mit 16d beschichteten Au(111)–Elektrode. 100 mM NaOH–Lösung diente als Elektrolyt. Alle Voltammogramme wurden mit 20 mV s-1 aufgezeichnet. (b) Elektrokatalytische Aktivität des immobilisierten Co–Porphyrin–Liponsäurederivats 16d. Beide Voltammogramme wurden in einer 500 mM konzentrierten, sauerstoffhaltigen H2SO4–Lösung aufgezeichnet. Die Vorschubgeschwindigkeit betrug 20 mV s-1.
-0,4 -0,2 0,0 0,2 0,4 0,6
-60
-50
-40
-30
-20
-10
0
10
1. Zyklus 2. Zyklus
i / μ
Α c
m-2
E / V vs. SCE
-1,6 -1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0 0,2-30
-25
-20
-15
-10
-5
0
5
i / μ
A c
m-2
E / V vs. SCE
1. Zyklus 2. Zyklus 3. Zyklus

Dissertation -68- Christoph S. Eberle
Auch gemischte Monoschichten aus Metalloporphyrin– und Liponsäurederivaten wurden
voltammetrisch untersucht (Abb. 25). STM–Aufnahmen derart modifizierter Goldelektroden
müssen letzten Endes zeigen, wie beide Komponenten in der Monoschicht verteilt sind:
entweder in jeweils eigenen Domänen oder indem die Liponsäurederivate die Lücken
zwischen immobilisierten Metalloporphyrinen auffüllen.
Abbildung 25: Mögliche Anordnung einer gemischten Monoschicht aus Ni–Porphyrin–Liponsäurebisester 16c und (S)–Liponsäurealkylester 1a (Octylester) auf Au(111)–Elektroden.
Abbildung 26: Reduktive Desorption einer gemischten Monoschicht aus Ni–Porphyrin–Liponsäurebiseter 16c und (S)–Liponsäurealkylester 1a (Octylester). Die beiden Komponenten wurden aus einer 1:1 (v:v) Mischung in CH2Cl2 auf der Goldelektrode abgeschieden. Die Konzentration der Stamm–lösungen betrug 1 mM. Voltammogramme der in 0.1 M NaOH-Lösung desorbierten Spezies wurden mit 20 mV s-1 aufgezeichnet.
-1,6 -1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0 0,2
-25
-20
-15
-10
-5
0
5
i / μ
A c
m-2
E / V vs. SCE
1. Zyklus 2. Zyklus
Ep
II= -1.069 V
Ep
I = ~ -0.980 V
Au
H
O
O
S S
CH3
H
O
O
S S
CH3
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
Ni
H
O
O
S S
CH3
H
O
O
S S
CH3

Dissertation -69- Christoph S. Eberle
Elektrochemische Desorption gemischter Monoschichten ergab zwei Peaks bei -1.069 V
und -0.980 V. Übereinstimmend mit früheren Ergebnissen kann derjenige bei negativerem
Potential den von der Goldelektrode abgelösten Ni–Porphyrinen zugeordnet werden, während
das zweite Reduktionspotential der Desorption der (S)–Liponsäureester entspricht. Da
letzteres als Schulter des Hauptpeaks erscheint, erschwert dies, die Belegungsdichte für beide
Spezies voneinander getrennt und eindeutig zu bestimmen. Auffällig ist in diesem
Zusammenhang, dass beim zweiten kathodischen Durchlauf der zweite Peak stark positiv
verschoben ist. Offenbar resorbieren aus einer (1:1)–Mischung der beiden Komponenten
die (S)–Liponsäureester schlechter an der Goldoberfläche als die Ni–Porphyrin–
Liponsäurebisester. Eine unimolekulare Monoschicht kann dagegen bei vergleichbaren
Potentialen desorbiert und mit 2.78 × 10-10 mol cm-2 des Ni–Porphyrinderivats 16c bedeckt
werden (vgl. Abb. 27).
Abbildung 27: Reduktive Desorption einer mit Ni–Porphyrin–Liponsäurebisester 16c beschichteten Au(111)–Elektrode. 100 mM NaOH–Lösung diente als Elektrolyt, und die Vorschubgeschwindigkeit betrug 20 mV s-1.
-1,6 -1,4 -1,2 -1,0 -0,8 -0,6 -0,4 -0,2 0,0 0,2
-30
-25
-20
-15
-10
-5
0
5
Ep
I = -1.11 V
i / μ
A c
m-2
E / V vs. SCE
1. Zyklus 2. Zyklus

Dissertation -70- Christoph S. Eberle
4.3 Physikalische Charakterisierung
4.3.1 Röntgenphotoelektronenspektroskopie (XPS)
Außer mit den zuvor beschriebenen elektrochemischen Verfahren diente XPS* zur weiteren
Untersuchung immobilisierter Spezies. Für die Analyse von Monoschichten und gemischten
Monoschichten aus Porphyrin– und Liponsäurederivaten wird nachfolgend ausschließlich die
S 2p Region der Spektren herangezogen. In keinem der hier durchgeführten Experimente ließ
sich Schwefeloxidation feststellen. Bekanntermaßen reagieren nämlich Thiole und Disulfide
unter Licht und Luftsauerstoff zu Sulfonaten.[100] Dies liegt an ihren schwachen S–H oder
S–S Bindungen, wodurch Monoschichten von der Oberfläche abgelöst werden können. Die
Oxidation geht bis Stufe +4, die den Sulfonsäuren entspricht. Angenommen wird, dass UV–
Licht Sauerstoff spaltet und Ozon gebildet wird, das die Adsorbate angreift. Wahrscheinlich
ist auch Singuletsauerstoff daran beteiligt, der durch Ozonabbau an Metalloberflächen
entsteht. Bisher gelang es nicht diese Schritte, die die Photooxidation von Monoschichten
einleiten, detailliert aufzuklären.[28]
Abbildung 28: XPS–Spektrum einer gemischten Monoschicht aus Ni–Deuteroporphyrin–Liponsäurebiseter 16c und (S)–Liponsäurealkylester 1a (Octylester). Beide Komponenten wurden in einer 1:1 (v:v) Mischung in Dichlormethan 24 h auf einer polykristallinen Goldelektrode adsorbiert. Das Substrat wurde zuvor einer Flammbehandlung unterzogen. Gezeigt ist der Bereich der S 2p Signale von 158 bis 164 eV.
Abbildung 28 zeigt die Peaks aus der S 2p Region für eine gemischte Monoschicht
aus Ni–Deuterporphyrin– und (S)–Liponsäureesterderivaten, die sich auf polykristallinen

Dissertation -71- Christoph S. Eberle
Goldoberflächen abschied. Das im Porphyrinmakrozyklus komplexierte Metallatom wurde
nachgewiesen durch ein Signal bei 852 eV, das Ni2p3/2 entspricht. Peakbreiten von 160 bis
164 eV deuten auf mehrere Molekülstrukturen in der Monoschicht. Diese gehören sowohl zu
gebundenen als auch ungebundenen Schwefelspezies, deren Verhältnis 3:1 beträgt. Dabei
lassen sich die Signale bei ~160 eV und ~161 eV chemisorbierten Schwefelatomen (S2p3/2)
zuordnen. Dagegen rührt das Signal bei ~162.5 eV von Schwefelatomen (S2p3/2) her, die
nicht kovalent auf der Goldoberfläche binden. Beide Komponenten lösen sich sehr gut
in Dichlormethan, worin die Monoschichten auf das Goldsubstrat aufgetragen wurden.
Möglicherweise lassen sich somit Ablagerungen einer von beiden Derivaten auf dem
Oberflächenfilm ausschließen, ebenso aufgrund des Prozederes der Probenvorbereitung (vgl.
Kapitel 4.1). Stattdessen könnten vereinzelt (S)–Liponsäureoctylester darin eingelagert
sein wegen starker, lateraler Wechselwirkungen mit den Methylenketten von bereits
immobilisierten Ni–Porphyrinen. Trotz S–S Bindungsbruch ist nicht auszuschließen, dass die
Liponsäureanker mit ihren beiden heterocyclischen Schwefelatomen an der Goldoberfläche
gebunden werden. Wahrscheinlich verläuft der Adsorptionsprozess der Metalloporphyrine
unter den gegebenen Bedingungen nicht einheitlich. Übereinstimmend mit Annahmen von
Shirai et al. ließen sich so unterschiedliche Selbstanordnungen der Kopf– und terminalen
Gruppe unterstellen.[101] Beide strukturelle Alternativen veranschaulicht Abb. 30.
Abbildung 29: XPS–Spektrum einer selbstorganisierten Monoschicht aus Zn–Porphyrin–Liponsäurebisester 16b. Das Metalloporphyrin wurde in Dichlormethan 24 h auf einer polykristallinen Goldelektrode adsorbiert. Das Substrat wurde zuvor einer Flammbehandlung unterzogen. Gezeigt ist der Bereich der S 2p Signale von 158 bis 164 eV.

Dissertation -72- Christoph S. Eberle
Zum Vergleich wurden unimolekulare Monoschichten aus dem Zn–Porphyrinderivat 16b auf
denselben Goldsubstraten vermessen. Auch diesmal enthält der S 2p Ausschnitt des XPS–
Spektrums enthält einen breiten Peak zwischen 158 und 163 eV (Abb. 29). Hierbei kann
ein genaues Verhältnis von gebundenem zu ungebundenem Schwefel nicht angegeben
werden. Das Signal bei 160.7 eV entspricht chemisorbierten Schwefelatomen (S2p3/2) auf der
Goldoberfläche. Ein Signal bei 1018 eV korrespondiert mit Zn2p und deutet auf die
Anwesenheit komplexierten Zinks im Porphyrinderivat.
Abbildung 30: Selbstorganisierte Monoschicht aus Metalloporphyrinen mit jeweils entgegengesetzter Anordnung von Kopf– und terminaler Gruppe (nach Shirai et al., 2006).[101] (a) Monoschicht aus rein kovalent gebundenen Metalloporphyrinen und (b) mit physisorbierten Metalloporphyrinen.
______________________________ *Röntgenphotoelektronenspektroskopie (XPS) wurde durchgeführt von Dr. Michael Nöske am
Bremer Fraunhofer Institut für Materialforschung und Angewandte Fertigungstechnik (IFAM).
Au
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
M
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
M
Au
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
M
NN
NN
CH3
CH3
CH3 CH3
OO
O
H
O
H
S SSS
M
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
M
N N
N N
CH3
CH3
CH3CH3
O O
O
H
O
H
SS S S
M
a)
b)

Dissertation -73- Christoph S. Eberle
4.3.2 Rastertunnelmikroskopie
Um einzeln immobilisierte Moleküle sichtbar zu machen, wurden modifizierte
Goldelektroden mittels Rastertunnelmikroskopie* (engl. Scanning Tunneling Microscopy,
STM) untersucht. Abb. 31b–e zeigt die Morphologie einer auf Au(111) abgeschiedenen
Monoschicht des Fulleren–Liponsäure–Derivats 9.[70] Durch Vergleich mit der unbehandelten
Goldelektrode, deren charakteristische (111)-Textur erkennbar ist (Abb. 31a), verdeutlicht
sich die Oberflächenmodifikation anhand der zugenommenen Unebenheiten (Abb. 31b, c).
Nach 24 Stunden in toluolischer Lösung treten diese über den gesamten mikroskopierten
Bereich auf. Sie gehen zurück auf hohe Fullerenbedeckung und deuten auf eine
wohlgeordnete Monoschicht, in der C60–Moleküle maximal 1.5 bis 1.7 nm voneinander
entfernt stehen. Ferner ergab die cross section–Analyse einen Durchmesser von ca. 1 nm für
einzelne absorbierte Fullerenmoleküle (Abb. 31e), die sich als aufgehellte, runde Flecke vom
Goldsubstrat abheben. Damit folgt der ermittelte Durchmesser der in der Literatur postulierten
Größe eines Fullerens[12b]. Diese um die elektrochemischen Daten vervollständigten Befunde
bestätigen die Anwesenheit einer zwar dicht gepackten Monoschicht, die jedoch
kein übergreifendes strukturelles Muster aufweist, das sie charakterisierte (Abb. 31d).
Dennoch findet man die zu erwartende hexagonale Elementaranordnung, wie für
Monoschichten aus n-Alkylthiolen beobachtet (Abb. 31e). Zuvor wird sich sterische
Hinderung sicher auf die Fullerenverteilung über die Goldoberfläche auswirken, während der
Selbstorganisationsprozess abläuft. Nach Chemisorption, an der pro Derivat vier
Schwefelatome beteiligt sind, erhöht sich die Stabilität der C60–Adsorbate, doch bleiben deren
Redoxeigenschaften hinter idealen Erwartungen zurück. Dies zeigen die beiden weit
auseinander liegenden Redoxpeaks im Cyclovoltammogramm (vgl. Abb. 15). Wohl lässt
sich das elektrochemische Verhalten der Monoschicht durch Koimmobilisierung verbessern,
wenn Additive zusätzlich verschiedene Endgruppen tragen. Dazu müssen optimale
Bedingungen gefunden werden, damit sich Additive wie Fullerenderivate mengenmäßig
in einer gemischten Monoschicht gleich verteilten (vgl. Abb. 32). Da dieses Derivat in einem
Schritt über beide Disulfidenden in hoher Konzentration auf Goldoberflächen verankert
wird, bietet es sich an als Alternative gegenüber entsprechenden Thiolaten: etwa bei
der Konstruktion elektronischer Bauteile[102] oder künstlicher Photosynthese–Systeme, wenn
in gemischten Monoschichten das Akzeptorzentrum koimmobilisiert wird mit einem
Elektronendonor, wie es Porphyrine[19, 20, 103] oder Chlorine[104] sind.

Dissertation -74- Christoph S. Eberle
Abbildung 31: STM–Charakterisierung[70] einer unmodifizierten Goldelektrode (a) und einer abgeschiedenen Monoschicht des Fulleren–Liponsäure–Derivats 9 (b) 120×120 nm2; (c) 50×50 nm2; (d) 10×10 nm2 und (e) „cross section“ zur diskreten Markierung eines C60–Moleküls; der Tunnelstrom lag bei 800 pA bis 1 nA für die Aufnahmen (a) und (b) und 500 pA bis 700 pA für die Aufnahmen (c) bis (e). Die elektrische Spannung betrug 0.1 V bis 0.2 V. Scanraten lagen bei allen Aufnahmen zwischen 5 Hz bis 8 Hz. Es wurden erste Versuche unternommen, das Adsorptionsverhalten aus Mischungen zu
studieren.[70] Dieses ist im Allgemeinen noch immer sehr wenig verstanden. Die Morphologie
einer gemischten Monoschicht nach Selbstorganisation zeigt Abb. 32. Wie diese vorläufigen
Daten belegen, sinkt in Gegenwart von n–Hexanthiol die Belegungsdichte für das Fulleren–
Liponsäure–Derivat 9 signifikant. Dennoch sind keine voneinander gesonderten Domänen auf
der Goldoberfläche auszumachen, die von jeweils einem der beiden Komponenten gebildet
a b
c d
e 1.09 nm

Dissertation -75- Christoph S. Eberle
werden. In einzelnen Regionen finden sich typische Merkmale von Alkanthiol–
Einzelschichten (Abb. 32c). Trotz dieser relativen Kontrollierbarkeit ließ sich mit dem 1:1
Ansatz noch keine gleichmäßige Verteilung beider Komponenten erreichen. Sowohl
die molekulare Packung als auch die elektrochemische Antwort von chemisorbierten
Fullerenderivaten zu verbessern, soll in fortgesetzten Forschungskooperationen erreicht
werden. Versuche dieser Art wurden in der Arbeitsgruppe Abrantes an der Universität
Lissabon auch die zuvor beschriebenen Metalloporphyrine ausgeweitet (Abb. 33).
Abbildung 32: STM–Aufnahme einer gemischten Monoschicht. Diese schied sich ab an einer monokristallinen Goldelektrode aus einer 1:1 Mischung des C60–Derivats 9 und CH3(CH2)5SH. Die Konzentration der Stammlösungen betrugen 1 mM (C60–Derivat) und 0.5 mM (n–Hexanthiol). (a) 55×55 nm2; (b) 20×20 nm2; (c) 20×20 nm2. Der Pfeil deutet auf die Alkanthiolatregion innerhalb der Monoschicht.[70] Morphologien von porphyrinhaltigen Funktionsschichten sind schwerer im Detail abzubilden.
Dies hängt zusammen mit der Planarität solcher Strukturen im Gegensatz zu Fullerenen.
Daher gelangt es nicht, im katalytisch aktiven Monofilm (vgl. Kap. 4.2.3) einzelne,
immobilisierte Metalloporphyrine aufzulösen. Als Indiz gilt auch hier: Je höher die Co-
Porphyrine als Monolage organisiert sind, desto aufgerauter erscheint die Substratoberfläche.
Chemische Vorreinigung und Tempern der Goldelektroden sorgen für ausgedehnt ebene
Oberflächen; sie weisen (111)–kristalline Textur auf mit vertikalen Abständen von 0.24 nm
(Abb. 33a). Erwartungsgemäß beobachtet man kraterartige Vertiefungen („pits“), die typisch
sind für Schwefeladsorptionen auf Gold (Abb. 33b,c). Da vier Gold–Schwefel Bindungen pro
Porphyrin entstehen, erhöht dies ebenso die Stabilität der Monoschicht wie es die
Beweglichkeit der Schwefeladsorbate mindern könnte. Aus der elektrochemisch ermittelten
Belegungsdichte ergibt sich ein durchschnittlicher Platzbedarf von 61Å2 pro Molekül. Diese
Abschätzung bestätigt die Anwesenheit einer dicht gepackten Monoschicht, in der Co–
Porphyrine sich eher perpendicular (30Å2) als parallel (170 Å2) zur Goldoberfläche anordnen.
a b c b

Dissertation -76- Christoph S. Eberle
Bekanntermaßen beeinflusst dies die katalytische Aktivität solcher Adsorbate, abhängig
davon auf welchem Substrat sie absorbiert sind (z.B. Gold oder Graphit). Der Schätzwert lässt
zunächst keine einheintlich ausgerichtete, monomolekulare Funktiosschicht vermuten.
Abbildung 33: STM–Aufnahme einer Co–Porphyrin–Monoschicht. Diese schied sich ab an einer monokristallinen Goldelektrode aus einer 1 mM konzentrierten, ethanolischen Lösung des Co–Porphyrinbisesters 16d. (a) 150×150 nm2; (b) 150×150 nm2; (c) 50×50 nm2.
______________________________ *Rastertunnelmikroskopische Aufnahmen stammen von Dr. Ana Viana aus der Arbeitsgruppe von
Prof. Dr. Luisa Maria Abrantes an der Universität Lissabon (Portugal).
a b c
Z = 1.5 nm
Flame–annealing

Dissertation -77- Christoph S. Eberle
4.3.3 Ellipsometrie
Um laterale Schichtdicken modifizierter Goldelektroden zu bestimmen, wurde Ellipsometrie*
angewandt. Bei ihr handelt es sich um eine optische Methode, die noch bis zu 1 Å
dünne organische Filme nachweisen kann. Dadurch dass sich der Polarisationszustand
monochromatischen Lichts vor und nach der Reflektion (oder Transmission) an einer
Oberfläche ändert, liefert die ellipsometrische Messung Information über die optischen
Eigenschaften der Probe. Anhand der Änderung der Messparameter � und zwischen der
unbehandelten und der modifizierten Goldelektrode wurden die jeweiligen Schichtdicken
ermittelt. Allgemein ist dafür der –Wert signifikanter, über den auf die Anwesenheit von
organischen Filmen geschlossen werden kann. �–Werte dagegen schwanken weniger, da
Monoschichten vor allem aus n–Alkylthiolen kein Licht absorbieren sollten. Dennoch legt
man eben diese Annahme zugrunde, um die ellipsometrischen Daten für die Analyse zu
benutzen. Obwohl Extinktionskoeffizienten für ultradünne Oberflächenfilme sehr niedrig
sind, verhalten sie sich keinesfalls transparent (k � 0). Wird ein solcher Film bei diskreter
Wellenlänge unter einem bestimmten Winkel bestrahlt, gilt allgemein ein
Dreiphasenmodell[104]. Dieses betrachtet den Messgegenstand genauer zusammengesetzt aus
Elektrode, Monoschicht und Luft. Anderseits kompliziert sich dadurch die Beziehung der
Fresnelschen Koeffizienten zueinander wie folgt:
�, = �, >−
n 1 (�), >−
n 2 (�), >−
n 3 (�), �1, �, d (6)
Absorbierende Schichten auf Oberflächen werden bei jeder Wellenlänge � durch drei Größen
beschrieben: durch den Real- und Imaginärteil von n2 sowie der Schichtdicke d, während nur
die Änderung der Parameter �, verfolgt werden kann. Unterstellt wurde, dass der Realteil
des komplexen Brechungsindex 1.45 – 1.50 für Oberflächenfilme beträgt, die in dieser Arbeit
vermessen wurden. Dieser Wert findet sich häufig bei der Ellipsometrie charakterisierter
Einzelschichten aus n–Alkylthiolen und Porphyrinen.[105, 106] Mit diesem Wert wurde die
Schichtdicke einer mit dem Fulleren-Liponsäure-Derivat 9 überzogenen Goldelektrode auf
durchschnittlich 2.2 nm bestimmt bei einem beobachteten Extinktionskoeffizienten von ca.
0.07. Nach Diels-Alder Reaktion an dieser dienophilen Fullerenoberfläche (vgl. Kapitel 3.4)
konnte eine ungefähre Verdopplung der Schichtdicke festgestellt werden. Aus der �– und
–Änderung ergab sich ein durchschnittlicher Wert von 4.4 nm. Dies spricht für das

Dissertation -78- Christoph S. Eberle
Vorhandensein einer relativ geordneten und einheitlichen Monoschicht, die sich aus einzelnen
Zn-Porphyrin-Fulleren-Dyaden 11 zusammensetzt, wie in Abb. 33 dargestellt.
Abbildung 34: Ellipsometrisch gemessene Schichtdicke der Zn–Porphyrin–Fulleren–Dyade 11. Der ermittelte Wert von 2.4 nm entspricht einer Fullerenmonoschicht vor [4+2]–Cycloaddition mit dem Porphyrinteil. ______________________________
*Ellipsometrische Messungen wurden durchgeführt von Dr. Jorge Correia aus der Arbeitsgruppe von Prof. Dr. Luisa Maria Abrantes an der Universität Lissabon (Portugal).
N N
N N
CH3
CH3
CH3CH3
O OO O
S S S
O O
S
OO
HH
Zn
11
Au
4.4 nm
2.4 nm

Dissertation -79- Christoph S. Eberle
4.4 Semiempirische Rechnungen
Semiemiprische PM3–Methoden halfen, die bevorzugte Konformation von Porphyrin–
Fulleren–Dyaden zu berechnen.[71] Ergebnisse ihrer Immobilisierung auf monokristallinen
Goldoberflächen wurde in vorangehenden Kapiteln behandelt. Wie die Berechnung ergab,
liegt die energetisch günstigere Konformation 10.6 kcal mol-1 niedriger als die gestreckte
(Abb. 35a). Demnach nimmt die Dyade eine bevorzugt gekrümmte Konformation ein.
Diese wird vorgegeben durch die molekulare Geometrie der Brücke zwischen Porphyrin und
Fulleren (Abb. 35b). Deren Rückgrat hat Zickzack–Form, in der energetisch am wenigsten
ungünstige gauche–Wechselwirkungen vorkommen. Als Folge dieser Krümmung ist das
Zinkatom im Porphyrinring etwa 10 Å entfernt von der Mitte des C60–Ikosaeders und etwa
6.3 Å von der Peripherie des Fullerengerüsts. Über eine Distanz dieser Länge zeigen sich
keinerlei attraktive Wechselwirkungen zwischen Donor und Akzeptor. Können sich diese
weiter einander annähern, bildeten sich solche Wechselwirkungen aus, wie im Falle etlicher
Porphyrin–Fulleren–Dyaden, die literaturbekannt sind.[107]
Abbildung 35: Optimierte Konformation/Konfiguration der Porphyrin–Fullerene–Dyade 11 ohne Liponsäureanker. Rechnungen erfolgten mittels semiemprischer PM3–Verfahren.[71]
Gleiche Rechnungen mit dem PM3–Formalismus wurden auch auf die Dyade 11 angewandt.
Die optimierte Struktur zeigt, dass die Liponsäureanker unsymmetrisch orientiert sind
aufgrund freier Drehbarkeit um die C–C Einfachbindungen des Methylenrückgrats (Abb. 36).
Ferner schließt die große Entfernung Interaktionen zwischen den Liponsäureeinheiten
aus. Von den Schwefelatomen im Ditholanring liegt das Methinbrückenproton im Zn–
Porphyrinteil ca. 3.6 Å entfernt. Diese Moleküllänge stimmt überein mit dem ellipsometrisch
ermittelten Durchmesser der immobilisierten Dyade nach der Diels–Alder Reaktion (vgl.
Kapitel 4.3.3).
a) b)

Dissertation -80- Christoph S. Eberle
Abbildung 36: Optimierte Konformation/Konfiguration der Metalloporphyrin–Fulleren–Dyade 11. Rechnungen erfolgten mittels semiemprischer PM3–Verfahren.

Dissertation -81- Christoph S. Eberle
5. ZUSAMMENFASSUNG UND AUSBLICK 5.1 Synthese von immobilisierbaren Porphyrinen, Metalloporphyrinen und
Fullerenen
Um so genannte kooperierende Dyaden zu entwerfen[10], zielte unser Konzept auf die
Herstellung geeigneter Donoren, Akzeptoren und Brückenmoleküle. Gemäß des partial–
synthetischen Konzepts, das dieser Arbeit zugrunde liegt, wird jeder Baustein in eigenen
Ansätzen zugänglich gemacht. Disulfidgruppen, die dadurch jeder Baustein trägt, befähigen
ihn zur Adsorption an Goldoberflächen. Als natürlicher Linker diente �-Liponsäure, deren
Racemat enzymatisch nach Fadnavis et al. gespalten werden kann.[58] Neben (S)–
konfigurierten Liponsäureestern blieb enantiomerenangereicherte (R)–Liponsäure mit 40%
optischer Reinheit zurück; sie war Edukt in allen beschriebenen Partialsynthesen, während
die (S)–Ester als Additive für die Koimmobilisierung mit derivatisierten Fullerenen und
Porphyrinen zur Verfügung standen. Trotz Racematspaltung kann sämtlliches Startmaterial
weiter verwertet werden. Um Additive mit unterschiedlichen funktionellen Gruppen zu
erhalten, wurden außerdem einfache Liponsäureester und –amide dargestellt. Von
kommerziell erhältlichem Hämin führt der Syntheseweg in fünf Schritten zu
dithiolanverbrückten Metalloporphyrinen (Abb. 37).
Die kovalente, leicht polare Gold–Schwefel Bindung formt sich spontan durch S–S
Bindungsbruch, weshalb organische Moleküle aus homogener Lösung als selbstorganisierte
Monoschicht an Goldoberflächen abgeschieden werden.[28] Eigenschaften von auf diese
Weise mit Metalloporphyrinen und Fullerenen[70] modifizierten Elektroden wurden mittels
Cyclovoltammetrie, STM, XPS und Ellipsometrie untersucht. Elektrochemische Daten
weisen darauf hin, dass alle vier Schwefelatome der Liponsäurereste an der Chemisorption
beteiligt sind. Dadurch formen sich Monoschichten sehr hoher Stabilität. Auch kann chirale
Information aus enantiomerenangereicherter Liponsäure in den Selbstorganisationsprozess
getragen werden, um Monoschichten höheren Ordnungsgrads aufzubauen. Elektrochemische
Befunde zeigen ferner, dass die Au–S Bindung in 1,2,4–Trichlorbenzol unter den harschen
Bedingungen einer Diels–Alder–Reaktion nicht gespalten wird. Nach Bedarf lässt sich
so in situ eine Metalloporphyrin–Fulleren–Dyade 11 aufbauen, die gleichzeitig auf der
Goldoberfläche immobilisiert bleibt. Die Dicke der Monoschicht vor und nach [4+2]–
Cycloaddition wurde ellipsometrisch ermittelt. Man findet reproduzierbar eine Verdopplung
auf in etwa 4.5 nm, die für das Vorhandensein der Metalloporphyrin–Fulleren–Dyade 11
spricht. Dies wird bestätigt sich durch Vergleich der immobilisierten Dyade (Diels–Alder

Dissertation -82- Christoph S. Eberle
Addukt) mit dem direkt immobilisierten Gemisch aus Konstitutionsisomeren der Dyade 11,
wo ähnliche Schichtdicken festgestellt wurden (5.1 nm). Mit dieser Anordnung solten sich
organische Photovoltaikzellen verwirklichen lassen (vgl. Schema 5).
Abbildung 37: Dithiolanverbrückte Metalloporphyrine und Fullerene zur Immobilisierung auf Metalloberflächen.
5.2 Ausblick
Der Vergleich einer unimolekularen Monoschicht der Dyade 11 mit einer gemischten
Monoschicht, bestehend aus separaten Donor–, Akzeptor– und Brückenmolekülen, soll
Aufschluss geben über die Möglichkeit zum Aufbau kooperierender Dyaden.[10] Wenn
verschiedene molekulare Bausteine in die durch Selbstorganisation aufgebauten
Monoschichten integriert werden, erlaubt dies, Oberflächen in der Nanodimension
noch vielfältiger und feiner zu strukturieren. Nach Bedarf können diese Oberflächen
weiter modifiziert werden, chemisch, elektrochemisch oder physikalisch. Solche Eingriffe
dienen dazu, gezielt Sensoren, Katalysatoren oder biomimetische Photovoltaikzellen
M = 2H
M = Zn
M = Ni
M = Co
M = MnCl
M = CoCl
N N
N N
CH3
CH3
CH3CH3
M
O O
O
SS
H
O
S S
H
N N
N N
CH3
CH3
CH3CH3
O OO O
S S S
O O
S
OO
HH
Zn
S S S
O O
S
OO
HH

Dissertation -83- Christoph S. Eberle
herzustellen (Abb. 38). Hierzu ließe sich das in unserem Laboratorium ausgearbeitete
Baukastenprinzip auch auf die Modifizierung von Chlorinstrukturen ausweiten, um sie mit
den für die Immobilisierung nötigen molekularen Ankern zu versehen.
Abbildung 38: Gemischte Monoschichten auf Goldelektroden aus derivatisierten Fullerenen, Thioalkanen und Metalloporphyrinoiden.
Die vorliegende Arbeit hat Ansätze in dieser Richtung fortgeführt und neue aufgezeigt.
Das Hauptaugenmerk richtet sich dabei auf die elektrochemische und physikalische
Vermessung von Modellsystemen. Sie soll verstehen helfen, welche Faktoren den
Selbstorganisationsprozess beeinflussen, wenn er an Metalloberflächen aus Mischungen
abläuft. Sie soll weitere Einsichten bringen in die mechanistischen Details des
Elektronentransfers und den mit diesem Transfer gekoppelten Reaktionen, wie er in
organischen Monoschichten beobachtet werden kann oder –allgemeiner formuliert– an
Grenzschichten in derartigen Systemen.[108] Neben den gängigen leitfähigen Substraten bieten
sich für diese Studien auch Kohlenstoffmaterialien an. Mit Kohlenstoffnanoröhren
beschichtete Elektroden beispielsweise versprechen größere Oberflächen zur Immobilisierung
elektroaktiver Spezies, ebenso Goldnanopartikel.
Au
S S S
O O
S
OO
HH
SS
N N
N N
CH3
CH3
CH3CH3
O ONH NH
Fe
Cl
H
O
O
S S
CH3
H
O
O
S S
CH3
CH3O
O
SS
N N
N N
CH3
CH3CH3
O ONH NH
Co
CO2CH3CO2CH3

Dissertation -84- Christoph S. Eberle
6. SUMMARY AND OUTLOOK 6.1 Synthesis and immobilisation of porphyrinoid, metalloporphyrinoid and
fullerene derivatives
For the design of cooperating dyads[10] this work was aimed at the synthesis of suitable
donors, acceptors and spacers according a tool box concept. Thus, all components carry
disulfide groups for robust binding on gold. �-Lipoic acid served as natural spacer unit. The
enzymatic resolution of its racemic mixture according to a modified procedure as described
by Fadnavis et al.[58] yielded (S)–configurated lipoic acid alkyl esters and (R)–lipoic acid of
40% optical purity. The acid was used as starting material in all partial synthetic routes,
whereas the (S)–esters served as additives for coimmobilizations with functional fullerenes
and porphyrins. Notwithstanding of the enzymatic resolution all of the starting material could
employed for different synthetic purposes. For obtaining additives bearing different terminal
functional groups, lipoic acid esters and amides were prepared. Starting from commercially
available hemine a reaction sequence was developed leading to dithiolane bridged
metalloporphyrins within five steps (Figure 37).
The covalent S–Au bond is formed spontanously via S–S bond fission of lipoic acid
derivatives, thus immobilizing organic molecules as self-assembled monolayers (SAMs)
on gold surfaces.[28] Properties of metalloporphyrin and fullerene SAMs were investigated by
STM, XPS, ellipsometry and cyclic voltammetry in scientific cooperations in Bremen and
Lisbon. Electrochemical data indicated a concomitant four electron reduction. Therefore
monolayers with very high stability were formed by these disulphides. Due to the application
of optical active lipoic acids chiral information could be transferred to the self assembly
process on gold substrates resulting in monolayers of higher order. Voltammetric data
demonstrated that the S–Au bond is not cleaved in 1,2,4 trichlorobenzene under extreme
reaction conditions applied for Diels-Alder reactions. An immobilised fullerene was formed
on demand on gold surfaces. At 190°C the immobilized fullerene was functionalised via [4+2]
cycloaddition by a porphyrin to form adduct 11. By in situ extrusion of SO2 the
sulfolenoporphyrin 10 reacts as diene with the dienophilic fullerene C60 at the surface[70]. The
dyad represents a model system for future electron transfer studies. Ellipsometric data gives
further evidence for the presence of monolayers of the metalloporphyrin fullerene derivative.
Average thickness of 5.1 nm and 4.4 nm has been estimated for direct self-assembly of the
dyad and after Diels–Alder reaction on fullerene modified gold, respectively. These data are

Dissertation -85- Christoph S. Eberle
consistent with the formation of a C60 SAM on the bare gold surface, which is then modified
with a layer of zinc porphyrin after Diels–Alder reaction, supporting the desired formation of
an immobilised dyad through covalent bonds. Surface coverage determined from
electrochemical desorption of dyad 11 after immobilisation as mixture of constitutional
isomers by self-assembly was calculated as 2.3 × 10-10 mol cm-2, taking into account a
concomitant four electron reduction. Compared to desorption experiments of the Diels–Alder
adduct (3.9 × 10-10 mol cm-2), the lower value can be explained by sterical demands of the
different C60 isomers, formed during Diels–Alder reaction in solution. Since it is well known
that control of regioselectivity on fullerenes cannot easily exercised during addition
reactions.[77, 78] The alternative approach as described here increases the regioselectivity of the
second [4+2] cycloaddition by excluding the possibility of forming cis– and e–products on the
preadsorbed fullerene SAM. Light induced electron transfer from the porphyrin donor to the
fullerene acceptor (Scheme 5) should allow designing a photovoltaic cell.
Figure 37. Dithiolane functionalised metalloporphyrins and fullerenes for immobilisation on metal surfaces.
M = 2H
M = Zn
M = Ni
M = Co
M = MnCl
M = CoCl
N N
N N
CH3
CH3
CH3CH3
M
O O
O
SS
H
O
S S
H
N N
N N
CH3
CH3
CH3CH3
O OO O
S S S
O O
S
OO
HH
Zn
S S S
O O
S
OO
HH

Dissertation -86- Christoph S. Eberle
6.2 Outlook Comparsion of a unimolecular monolayer of dyad 11 with a mixed monolayer of single
donor, acceptor and spacer moieties, should give cooperating dyads. Integration of different
components into the self assembled monolayers should enable engineering of tailor made
sensors, catalysts and artifical photosynthetic systems (Figure 38). For that purpose
monolayer surfaces should be further modified chemically, electrochemically or physically.
The tool box concept as developed in our laboratory is also to be extended to the modification
of chlorin structures by attaching suitable linkers.
Scientific cooperations should lead to the construction of similar systems to be tested for the
mentioned applications. In this regards the main focus is on electrochemical and physical
investigation of model systems. The goals are to understand the factors influencing self
assembly process on metal surfaces from a heterogenous solution, and to get insights in
mechanistic details of electron transfer and reactions associated in organic monolayers or at
inferfaces in surface films in general.[108] Beside common metal substrates such studies should
be performed with carbon materials. For instance, electrodes covered with carbon nanotubes
as well as gold nanoparticles may promise a higher surface area for immobilisation of
electroactive species.
Figure 38. Mixed self–assembled monolayers (SAMs) on gold electrodes derived from functional metalloporphyrinoids, fullerenes and thioalkanes.
Au
S S S
O O
S
OO
HH
SS
N N
N N
CH3
CH3
CH3CH3
O ONH NH
Fe
Cl
H
O
O
S S
CH3
H
O
O
S S
CH3
CH3O
O
SS
N N
N N
CH3
CH3CH3
O ONH NH
Co
CO2CH3CO2CH3

Dissertation -87- Christoph S. Eberle
7. EXPERIMENTELLER TEIL
7.1 Abkürzungen und Symbole
Die Beziehung zwischen absoluter Konfiguration einer Verbindung und ihrem Kon–
figurationsformelbild ist eindeutig. Handelt es sich um chirale Verbindungen, kennzeichnet
eine fettgedruckte arabische Kennziffer exakt die im zugehörigen Formelbild angegebene
Konfiguration. Bei Racematen wird das Präfix „rac“ vorangestellt. Abkürzungen richten sich
nach den allgemeinen Vorgaben der Gesellschaft Deutscher Chemiker (GDCh) für die
Zeitschrift Angewandte Chemie.[109] Weiterhin kommen im Text folgende Abkürzungen und
Symbole vor:
� chemische Verschiebung [ppm]
� Wellenlänge [nm]
� Wellenzahl [cm-1]
Äq. Äquivalente
AFM Atomic Force Microscopy
ATP Adenosin–5’–triphosphat
BRN Beilstein Registry Number
BOP–MP Bond Order Conservation–Morse Potential
CAS Chemical Abstracts Registry Number
CDI Carbonyldimidazol
C. rugosa Candida rugosa
DC Dünnschichtchromatographie
DCC Dicyclohexylcarbodiimid
DMAP N,N–dimethylaminopyridin
DMF Dimethylformamid
DNA Desoxy Ribonucleic Acid
d. Th. der Theorie
ee Enantiomerenüberschuß
EI Elektroionization
ESI Elektrospray–Ionization
Ep Readoxpeakabstand |Eox – Ered| [V]
F Faraday–Konstante, 9.648523415(39) × 104 C mol-1

Dissertation -88- Christoph S. Eberle
g Gramm
h Plank–Konstante, 6.62606876(52) × 10-34 J s
HOMO Highest Occupied Molecular Orbital
HOPG Highly Oriented Polycrystalline Graphite
HPLC High Pressure Liquid Chromatography
HR-MS hochauflösende Massenspektrometrie
Hz Hertz
NMR Nuclear Magnetic Resonance
NOE Nuclear Overhauser Effect
i Stromdichte [A cm-2]
IR Infrarotspektroskopie
IPR Isolated Pentagon Rule
IRRAS Infrared Reflection Absorption Spectroscopy
J Kopplungskonstante
k Extinktionskoeffozient
l Liter
LUMO Lowest Unoccupied Molecular Orbital
M Molare Masse
MHz Megahertz
mg Miligramm
ml Mililiter
MS Massenspektrometrie
n Brechungsindex oder Elektronenanzahl
N Normal
NA Avogadro–Konstante, 6.02214199(47) × 1023
NADPH Nicotinamid–Adenin–Dinucleotid–Phosphat (reduzierte Form)
NEt3 Triethylamin
PC Propylencarbonat
PM3 Parameterized Model number 3
PPA n–Propylphosphorsäureanhydrid
ppm parts per million
PBE–DFT Perdew–Burke–Ernzerhopf Density Functional Theory
QCM Quartz Crystal Microbalance
Rf Retentionsfaktor

Dissertation -89- Christoph S. Eberle
Rfl. Rückfluss
RT Raumtemperatur
RNA Ribonucleic Acid
SAM Self–Assembled Monolayer
SCE Saturated Calomel Electrode
Sdp. Siedepunkt
Smp. Schmelzpunkt
STM Scanning Tunnelling Microscopy
TBAP Tetrabutylammoniumperchlorat
TEAP Tetraethylammoniumperchlorat
THF Tetrahydrofuran
TMS Tetramethylsilan
UV Ultraviolett
VIS Visible
w/o wässrig/organisch
XPS X–Ray Photoelectron Spectroscopy
� Belegungsdichte [mol cm-1]
ellipsometrischer Phasenparameter
� ellipsometrischer Amplitudenparameter
� Neigungswinkel bei ellipsometrischer Messung
� molarer Extinktionskoeffizient [cm2�mmol-1]

Dissertation -90- Christoph S. Eberle
7.2 Allgemeine Experimentelle Bedingungen 7.2.1 Analytische Methoden Schmelzpunktbestimmung (Schmelzpunkt): Schmelzpunkte wurden bestimmt an einem
Reichert–Thermovar Heiztischmikroskop nach Kofler der Firma Reichert und an einem
Schmelzpunktgerät der Firma Gallenkamp. Die Bestimmungen sind jeweils unkorrigiert.
Polarimetrie: Die Messungen wurden an einem Polarimeter 243 der Firma Perkin–Elmer
durchgeführt. Der spezifische Drehwinkel [ ]Tλα ergab sich nach folgender Formel:
[ ][ ] dc
T
⋅=
αα λ
[ ]Tλα = spezifischer Drehwinkel bei einer Wellenlänge � und einer Temperatur T in deg
[deg cm2 / 10g]
� = gemessener Drehwert in deg
c = Konzentration in g/ml
d = Schichtdicke in cm
Infrarotspektroskopie (IR): Alle Spektren wurden an einem Paragon 500 FT–IR
Spektrometer der Firma Perkin–Elmer aufgenommen. Die Auflösung betrug 4.0 cm-1.
Relative Intensitäten der Banden wurden mit s (Bande starker Intensität), m (Bande mittlerer
Intensität), w (Bande schwacher Intensität) und br (breite Bande) bezeichnet.
Ultraviolett- und VIS-Spektroskopie (UV/Vis): Alle Spektren wurden an einem Cary 50
Spektrometer der Firma Varian aufgenommen. Für quantitative Messungen wurden 10-5 bis
10-7 molare Lösungen eingesetzt. Absorptionsmaxima bei einer Wellenlänge � wurden mit
dem molaren Extinktionskoeffizienten � angegeben gemäß folgender Beziehung:
dcI
IA ⋅⋅== ε0log
A = gemessen Absorption bei einer Wellenlänge �
� = molarer Extinktionskoeffizient in cm2�mmol-1 oder mol-1�dm3�cm-1
c = Konzentration mol�L-1
d = Schichtdicke in cm

Dissertation -91- Christoph S. Eberle
Kernresonanzspektroskopie (1H–NMR, 13C–NMR, DEPT, H,H–COSY, HSQC, HMBC,
NOESY): Alle Experimente wurden durchgeführt an einem DPX–200 Advance (�: 1H = 200
MHz, 13C = 50 MHz) oder an einem AM–360 NMR-Gerät (�: 1H = 360 MHz, 13C = 90 MHz)
der Firma Bruker–Daltonic und mit 5 mm durchmessenden NMR–Röhrchen bei Raum–
temperatur aufgenommen. Die Spektrometersoftware der Herstellerfirma gab den Standard
vor. Das „Lock-in“ erfolgte auf das jeweils angegebene Lösungsmittel. Die chemische
Verschiebung � (in ppm) wurde mittels undeuterierter Lösungsmittelreste bestimmt. Die
Feinstruktur der Protonensignale wurde mit s (Singulett), bs (breites Singulett), d (Dublett), t
(Triplett), q (Quartett), dt (Dublett von Tripletts), m (Multiplett) beschrieben. Kopplungs–
konstanten xJ beziehen sich auf 1H,1H–Kopplungen, wobei x für die Anzahl der Bindungen
zwischen den koppelnden Kernen steht. 13C–Spektren wurden mit 1H–Breitband–
Entkopplung aufgenommen, sodass nur Singulett–Signale erschienen. In den experimentellen
Daten zu jeder Substanz sind nur die jeweiligen 1H–NMR–Daten aufgelistet. Zur exakten
Zuordnung von Signalen dienten zweidimensionale NMR–Methoden und NOE–Experimente.
Massenspektrometrie (MS): Alle Spektren wurden aufgenommen an einem
doppelfokussierenden Massenspektrometer MAT 8200, MAT 95 und an einem MAT 95 XL
Trap mit ESI Quelle der Firma Finnigen MAT sowie an einem Elektrospray
Massenspektrometer Esquire LC der Firma Bruker Daltronic. Die Elektronenstoßionisation
(EI) wurde mit einer Ionisierungsenergie von 70 eV bei einer Temperatur von 200°C
durchgeführt. Verdampfungstemperaturen wurden jeweils angegeben. Die Elektrospray–
ionisierung (ESI) erfolgte mit angegebenem Lösungsmittel und einer Probenzugabe von
2 �L/min mittels Direkteinlass. Spektren wurden kontinuierlich aufgenommen und zur
Auswertung dasjenige Spektrum herangezogen, welches die prozentual größte Molekülpeak–
gruppe zeigte. Soweit für die Strukturaufklärung nicht entscheidend, wurden nur Peaks mit
einer relativen Intensität von mehr als 10% zur Analyse herangezogen. Es wurden keine
Matrixsignale mit angegeben.
Hochauflösende Massenspektrometrie (HR–MS): Hochauflösungen wurden an einem
doppelfokussierenden Massenspektrometer MAT 8200 der Firma Finnigan MAT nach der
peak-matching Methode und an einem APEX Qe 9,4T (superconducting magnet) mit Apollo II
Elektrospray Quelle und Qh unit (Quadrupol–Filter) aufgenommen. Als Referenzsubstanz
diente Perfluorkerosin (PFK).

Dissertation -92- Christoph S. Eberle
Röntgenphotoelektronenspektroskopie (XPS): Die Untersuchungen erfolgten mit einem
Kratos-Ultra-System der Firma Kratos. Folgende Geräteparameter wurden dabei eingestellt:
Hybridmode, Abnahmewinkel der Photoelektronen 0°, monochromatisierte AlK�–Anregung,
CAE-Mode mit 160 eV Passenergie in Übersichtsspektren und 20 eV in hochaufgelösten
Linienspektren. Die Analysenfläche betrug ca. 0.3*0.7 mm und die Neutralisation der Proben
erfolgte mit niederenergetischen Elektronen (2.6 eV).
Cyclovoltammetrie: Für elektrochemische Studien wurden alle Glasgeräte zuvor mit
Chromsäurelösung gereinigt und danach ausgiebig mit ddH2O gespült. Cyclovoltammetrische
Untersuchungen wurden durchgeführt an einem PARSTAT 2263 Potentiostaten der Firma
Perkin-Elmer. Vor jeder Messung wurden die jeweiligen Elektrolytlösungen eine Stunde lang
mit Stickstoff (99.9999 %) entgast. Die Elektrolytlösungen wurden in eine Einkompartiment–
Teflonzelle gegeben mit einer Pt-Gegenelektrode und einer gesättigten Kalomelektrode
(SCE), welche als Referenz diente. Zuvor wurden die modifizierten Goldwafer gegen einen
Gummiring geklemmt, der eine Fläche von 0.57 cm2 beschrieb.
Ellipsometrie: Ellipsometrische Untersuchungen wurden bei einem Neigungswinkel von 70°
durchgeführt an einem rotierenden analyser type Ellipsometer (SE 400 SENTECH
Instruments GmbH, Berlin) mit einem He-Ne Laser (632.8 nm).
Rastertunnelmikroskopie: Alle Messungen wurden mit Hilfe eines Nanoscope IIIa
multimode Mikroskop (Digital Instruments, Veeco) durchgeführt bei Raumtemperatur und an
Luft. STM–Probenköpfe wurden hergestellt aus Pt/Ir und ebenfalls von der Firma Veeco
bezogen.

Dissertation -93- Christoph S. Eberle
7.2.2 Chromatographie
Dünnschichtchromatographie (DC): Es wurden DC–Alufolien (20*20 cm) mit einer
Kieselgel 60 F254 und einer Aluminiumoxid ALOX/UV254 Beschichtung von 0.2 mm der Firma
Fluka verwendet. Die Detektion der Banden erfolgte mit einer Fluoreszenz–Lampe bei
Wellenlängen von 254 bzw. 366 nm. Die verwendeten Laufmittelsysteme sind jeweils
angegeben.
Säulenchromatographie: Es wurde Aluminiumoxid (ALOXN II–III, neutral, Aktivität II–III)
der nach Rockmann der Firma ICN Biomedicals und Kieselgel 32–63 �m 60 Å der Firma
ICN Biomedicals verwendet. Die Säulen wurden im Falle von Aluminiumoxid nach der
Sedimentationsmethode (Vorlegen des Eluenten) gepackt oder im Falle von Kieselgel
nach der Slurry–Methode (Aufschlämmen des Adsorbens) mit leichtem Überdruck. Alle
Trennungen erfolgten bei Normaldruck.
7.2.3 Qualität verwendeter Chemikalien und Lösungsmittel
Für sämtliche Reaktionen und Aufreinigungsschritte wurden einfach destillierte
Lösungsmittel eingesetzt. Die Chemikalien wurden in „zur Synthese“–Qualität von den
Firmen Sigma Aldrich, Acros, Fluka, VMR (Merck), Riedel–de–Häen, Lancaster und
TCI bezogen. Als Schutzgas wurde technisches Argon 4.8 der Firma Air Liquide und Argon
4.6 der Firma Linde eingesetzt. Die für die HPLC– und UV–Analysen verwendeten
Lösungsmittel wurden in „HPLC“–Qualität eingesetzt. Die mit * markierten Reagenzien und
Lösungsmittel wurden wie folgt destilliert:
N,N–Dimethylformamid (DMF) über CaH2 abdestilliert,
Tetrahydrofuran (THF) über Na/Benzophenon abdestilliert,
1,2,4–Trichlorbenzol (C6H3Cl3) über CaH2 abdestilliert,
Triethylamin (Et3N) über CaH2 abdestilliert.
7.2.4 Software und Datenbanken Formelbilder wurden mittels ISIS DRAW 2.5 der Firma MDL Information Systems, Inc.
oder mit ChemSketch Freeware 8.17 der Firma ACD, Inc. erstellt. Die Auswertung der
NMR–Spektren erfolgte mit Win1D-NMR der Firma Bruker Daltronik oder mit MestReC

Dissertation -94- Christoph S. Eberle
4.8.1.1 Test Version der Firma Mestrelab Research. Literaturrecherchen wurden durchgeführt
mit dem Beilstein Crossfire Commander 7.0 (Beilstein PlusReactions 2006/02) und mit dem
SciFinder Scholar 2006 (CAPLUS, MEDLINE, REGISTRY, CASREACT). Falls zum
Zeitpunkt der Literaturrecherche Einträge in den entsprechenden Datenbanken vorhanden
waren, stehen die Registriernummern (BRN oder CAS) im Anschluss an die analytischen
Daten der Experimente.

Dissertation -95- Christoph S. Eberle
7.3 Darstellung von Octyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 1a und 5-[(3R)-1,2-dithiolan-3-yl]pentansäure 1[58]
S
S
OOH
H
(S)
C8H14O2S2
206.33
C16H30O2S2
318.53
S
S
OOH
H
(R)
+
C8H14O2S2
206.33
1a 1rac-1
S
S
OO
(CH2)6
H
CH3
206 mg racemische Liponsäure rac–1 (1 mmol), 650 mg Octanol (5 mmol, 0.79 ml) und
50 mg Lipase Typ VII aus C. rugosa (E.C.3.1.1.3) wurden in 60 ml n–Hexan gelöst.
Das Gemisch wurde 72 Stunden bei 30°C gerührt und der Fortgang der Reaktion mittels
Dünnschichtchromatographie verfolgt. Danach wurde das pulverförmige Enzym durch
Filtration entfernt. Das Reaktionsgemisch wurde über Nacht bei –20°C aufbewahrt.
Der auskristallisierte Feststoff (gelbe Kristalle) wurde mittels einer Glasfritte abgesaugt,
gründlich mit n–Hexan gespült und trocken gesaugt. Die organische Phase wurde am
Rotationsverdampfer eingeengt und anschließend über Kieselgel mit Dichlormethan als
Laufmittel chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im Feinvakuum
erhielt man (S)-Liponsäure–n–octylester 1a als gelbes Öl.
Ausbeute*: 149.3 mg (0.468 mmol, 46.8 % d. Th.).-

Dissertation -96- Christoph S. Eberle
DC [Kieselgel, Dichlormethan (100 %)]: Rf = 0.86.-
IR (NaCl, Film): � = 2927 cm-1 (s, CH), 2855 (s, CH), 1736 (s, CO, Ester), 1460 (s),
1358 (m), 1242 (m), 1175 (s), 1072 (w), 724 (w).-
1H–NMR (200 MHz, CDCl3): � = 0.89 (t, 3H, 3J = 6.74 Hz, CH2CH3), 1.3 – 2.5 (m, 22H),
3.14 (t, 2H, 3J = 6.75 Hz, SCH2CH2), 3.57 (t, 1H, 3J = 6.95 Hz, SCH), 4.06 (t, 2H, 3J =
6.76 Hz, OCH2CH2) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (% rel. Intensität): 318 (100) [M+], 206 (95) [M+ – C8H16],
189 (34), 173 (44), 141 (47).-
Optische Aktivität: [�]D 20 = –32.9 (c 0.4 in CHCl3) (Lit. [58]: [�]D = –33.3).-
Präzisionsmasse: C16H30O2S2 berechnet 318.16872
gefunden 318.16979
�–(R)–Liponsäure 1
Ausbeute*: 95.3 mg (0.462 mmol, 46.2 % d. Th.).-
Schmelzpunkt: 55 – 56°C (Lit. [61a] 46 – 48°C)
DC: [Kieselgel, Dichlormethan (100%)]: Rf = 0.01.-
IR (KBr): � = 2938 cm-1 (s, CH), 1691 (s, CO, Säure), 1466 (s), 1428 (s), 1407 (s), 1306 (s),
1250 (s), 1202 (s), 1078 (m), 931 (s), 733 (m), 671 (m), 520 (m).-
1H–NMR (360 MHz, CDCl3): � = 1.39 – 1.61 (m, 6H, 3xCH2), 1.82 und 2.37 (m, 2H,
SCH2CH2), 2.28 – 2.30 (m, 1H, COCH2), 3.02 – 3.07 (m, 2H, SCH2), 3.48 (m, 1H, SCH),
10.45 (s, br, 1H, COOH) ppm.-

Dissertation -97- Christoph S. Eberle
MS (EI, 70 eV, Diekteinlass): m/z (% rel. Intensität): 206 (100) [M+], 173 (10), 155 (12), 123
(59), 105 (16), 95 (41), 81 (75).-
Optische Aktivität: [�]D 20 = + 40.2 (c = 0.16 in Benzol) (Lit. [61a]: [�]D 20 = + 96.7).-
Präzisionsmasse: C8H14O2S2 berechnet 206.04352
gefunden 206.04410
CAS: 1077-27-6.- BRN: 81851.-
______________________________ *Ausbeuten beziehen sich auf die angegebene Stoffmenge racemischer Liponsäure rac–1.

Dissertation -98- Christoph S. Eberle
7.3.1 Darstellung von Hexyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 1b
und 5-[(3R)-1,2-dithiolan-3-yl]pentansäure 1[58]
S
S
OOH
H
(S)
C16H30O2S2
290.48
S
S
OOH
H
(R)
+
C8H14O2S2
206.33
1b 1rac-1
S
S
OO
(CH2)4
H
CH3
C8H14O2S2
206.33
206 mg racemische Liponsäure rac–1 (1 mmol), 510 mg Hexanol (5 mmol, 0.63 ml) und
50 mg Lipase Typ VII aus C. rugosa (E.C.3.1.1.3) wurden in 60 ml n–Hexan gelöst.
Das Gemisch wurde 72 Stunden bei 30°C gerührt und der Fortgang der Reaktion mittels
Dünnschichtchromatographie verfolgt. Danach wurde das pulverförmige Enzym durch
Filtration entfernt. Das Reaktionsgemisch wurde über Nacht bei –20°C aufbewahrt.
Der auskristallisierte Feststoff (gelbe Kristalle) wurde über eine Glasfritte abgesaugt,
gründlich mit n–Hexan gespült und trocken gesaugt. Die organische Phase wurde am
Rotationsverdampfer eingeengt und anschließend über Kieselgel mit Dichlormethan als
Laufmittel chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im Feinvakuum
erhielt man (S)–Liponsäure–n–hexylester 1b als gelbes Öl.
Ausbeute*: 136.6 mg (0.470 mmol, 47 % d. Th.).-

Dissertation -99- Christoph S. Eberle
DC [Kieselgel, Dichlormethan (100 %)]: Rf = 0.80.-
IR (NaCl, Film): � = 2929 cm-1 (s, CH), 2858 (s, CH), 1732 (s, CO, Ester), 1456 (m),
1177 (s), 1072 (m).-
1H–NMR (200 MHz, CDCl3): � = 0.91 (t, 3H, 3J = 6.60 Hz, CH2CH3), 1.4 – 2.6 (m, 18H),
3.15 (t, 2H, 3J = 6.60 Hz, SCH2CH2), 3.59 (t, 1H, 3J = 7.09 Hz, SCH), 4.08 (t, 2H, 3J =
6.84 Hz, OCH2CH2) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (% rel. Intensität): 290 (100) [M+], 206 (70) [M+ – C6H13],
189 (40), 173 (40), 141 (38).-
Optische Aktivität: [�]D 20 = –29.1 (c = 0.4 in CHCl3) (Lit. [58]: [�]D 20 = –29.6).-
Präzisionsmasse: C14H26O2S2 berechnet 290.13742
gefunden 290.13730
BRN: 7688459.-
�-(R)-Liponsäure 1
Ausbeute: 90.8 mg (0.44 mmol, 44 % d. Th.).-
Schmelzpunkt: 56 – 57°C (Lit. [61a] 46 – 48°C)
DC: [Kieselgel, Dichlormethan (100%)]: Rf = 0.01.-
Optische Aktivität: [�]D 20 = + 23.3 (c = 0.3 in Benzol) (Lit. [61a]: [�]D 20 = + 96.7).-
CAS: 1077-27-6.- BRN: 81851.- ______________________________
*Ausbeuten beziehen sich auf die angegebene Stoffmenge racemischer Liponsäure rac–1.

Dissertation _ -100- _ _ _ Christoph S. Eberle
7.4 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]-N-[2-(1H-imidazol-5-yl)ethyl]pentanamid 3a
S
S
OOH
H
S
S
O
NH
H
NN H
C8H14O2S2
206.33
3a1
C13H21N3OS2
299.45
206 mg �–(R)–Liponsäure 1 (1 mmol) und 197 mg N,N’–Carbonyldiimidazol (1.2 mmol)
wurden in 20 ml THF* gelöst. Nach einstündigem Rühren bei 45°C unter Argonatmosphäre
wurden zur Reaktionsmischung 333 mg Histamin (3 mmol) in 10 ml THF* getropft. Das
Gemisch rührte weiter bei 45°C über Nacht unter Argon. Das Lösungsmittel wurde
anschließend am Rotationsverdampfer entfernt. Über Aluminiumoxid (Akt. II–III, neutral)
wurde das Rohprodukt mit Dichlormethan/Methanol (19:1) als Laufmittelgemisch gereinigt
(Säulendurchmesser: 2 cm). Nach Trocknen im Feinvakuum erhielt man hellgelbe Kristalle.
Ausbeute: 222 mg (0.74 mmol, 74 % d. Th.).-
Schmelzpunkt: 71 – 72°C.-
DC [Alox, Dichlormethan/Methanol (19:1)]: Rf = 0.79.-

Dissertation _ -101- _ _ _ Christoph S. Eberle
IR (KBr): � = 3235 cm-1 (s, NH), 2938 (s, CH), 1638 (s, CO, Amid), 1560 (s), 1534 (s),
1483 (m), 1438 (m), 1328 (m), 1258 (m), 1192 (w), 1139 (w), 1095 (w), 1065 (s), 1027 (m),
930 (m), 914 (m), 828 (m), 752 (m), 663 (m), 618 (m).-
1H–NMR (360 MHz, CDCl3/DMSO)*: � = 1.33 – 1.59 (m, 2H, 3xCH2), 1.81 und 2.36 (m,
2H, SCH2CH2), 2.08 (t, 2H, 3J = 7.2 MHz, COCH2CH2), 2.69 (m, 2H, CH2), 3.02 und 3.07
(m, SCH2, 2H), 3.41 (m, 2H, NCH2), 3.47 (m, 1H, SCH), 6.70 (s, 1H, CH im Imidazolring),
6.80 (s, br, 1H, CONH), 7.45 (s, 1H, CH im Imidazolring) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (%): 299 (59) [M+], 266 (100), 234 (14).-
Optische Aktivität: [�]D 20 = + 11.3 (c = 0.004 in CH2Cl2).-
Präzisionsmasse: C13H21ON3S2 berechnet 299.11261
gefunden 299.11143
______________________________ *Das N–H Signal im Imidazolring konnte nicht zugeordnet werden.

Dissertation _ -102- _ _ _ Christoph S. Eberle
7.4.1 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]-N-[3-(1H-imidazol-1-yl)propyl]pentanamid 3b
S
S
OOH
H
N
N
S
S
O
NH
H
C8H14O2S2
206.33
C14H23N3S2
313.48
1 3b
206 mg �–(R)–Liponsäure 1 (1 mmol) und 197 mg N,N’–Carbonyldiimidazol (1.2 mmol)
wurden in 20 ml THF* gelöst. Nach einstündigem Rühren bei 45°C unter Argonatmosphäre
wurden zur Reaktionslösung 380 mg 1–(3–Aminopropyl)–imidazol (3 mmol, 0.36 ml) in
10 ml THF* getropft. Das Gemisch rührte weiter bei 45°C über Nacht unter Argon. Das
Lösungsmittel wurde anschließend am Rotationsverdampfer eingeengt. Über Kieselgel wurde
das Rohprodukt mit Dichlormethan/Methanol/Cyclohexan (7:2:1) als Laufmittelgemisch
gereinigt. (Säulendurchmesser: 2 cm). Nach Trocknen im Feinvakuum erhielt man das
Produkt als gelbes Öl.
Ausbeute: 223 mg (0.735 mmol, 73.5 % d. Th.).-
DC [Kieselgel, Dichlormethan/Methanol/Cyclohexan (7:2:1)]: Rf = 0.53.-

Dissertation _ -103- _ _ _ Christoph S. Eberle
DC [Kieselgel, Dichlormethan/MeOH (19:1)] Rf = 0.79.-
IR (NaCl, Film): � = 3280 cm-1 (s, NH), 2931 (s, CH), 1651 (s, CONH), 1557 (s), 1511 (m),
1436 (s), 1373 (w), 1281 (w), 1232 (m), 1109 (m), 1081 (m), 917 (m), 818 (m), 736 (s),
667 (m), 625 (w).-
1H–NMR (200 MHz, CDCl3): � = 1.3 – 2.6 (m, 14H, 7xCH2), 3.1 – 3.3 (m, 2H, SCH2), 3.59
(t, 1H, 3J = 7.09 Hz, SCH), 4.09 (t, 2H, 3J = 6.84 Hz, NHCH2CH2), 6.12 (s, 1H, NH), 7.02 (s,
1H, CH im Imidazolring), 7.14 (s, 1H, CH im Imidazolring), 8.04 (s, 1H, CH im
Imidazolring) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (%): 313 (52) [M+], 280 (100), 266 (10), 249 (15), 212
(48).-
Optische Aktivität: [�]D 20 = + 9.3 (c = 0.004 in CH2Cl2).-
Präzisionsmasse: C14H23ON3S2 berechnet 313.12826
gefunden 313.12823
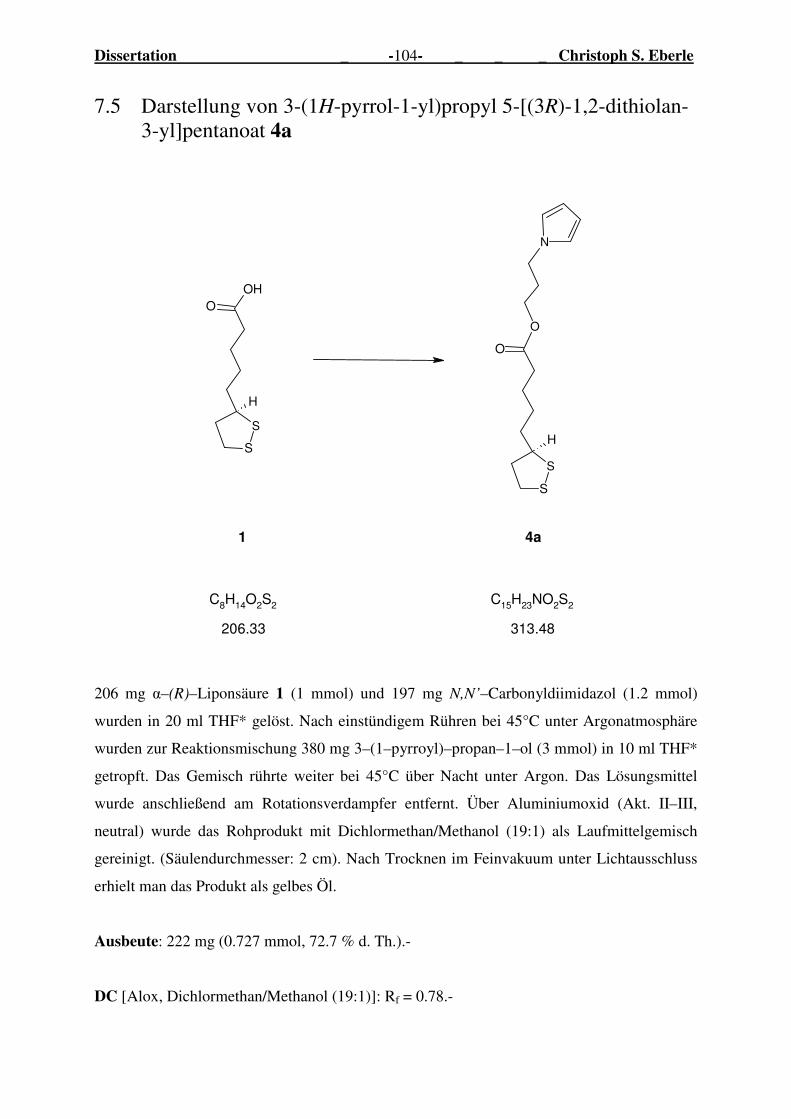
Dissertation _ -104- _ _ _ Christoph S. Eberle
7.5 Darstellung von 3-(1H-pyrrol-1-yl)propyl 5-[(3R)-1,2-dithiolan-3-yl]pentanoat 4a
S
S
OOH
H
S
S
O
O
H
N
C8H14O2S2
206.33
1 4a
C15H23NO2S2
313.48
206 mg �–(R)–Liponsäure 1 (1 mmol) und 197 mg N,N’–Carbonyldiimidazol (1.2 mmol)
wurden in 20 ml THF* gelöst. Nach einstündigem Rühren bei 45°C unter Argonatmosphäre
wurden zur Reaktionsmischung 380 mg 3–(1–pyrroyl)–propan–1–ol (3 mmol) in 10 ml THF*
getropft. Das Gemisch rührte weiter bei 45°C über Nacht unter Argon. Das Lösungsmittel
wurde anschließend am Rotationsverdampfer entfernt. Über Aluminiumoxid (Akt. II–III,
neutral) wurde das Rohprodukt mit Dichlormethan/Methanol (19:1) als Laufmittelgemisch
gereinigt. (Säulendurchmesser: 2 cm). Nach Trocknen im Feinvakuum unter Lichtausschluss
erhielt man das Produkt als gelbes Öl.
Ausbeute: 222 mg (0.727 mmol, 72.7 % d. Th.).-
DC [Alox, Dichlormethan/Methanol (19:1)]: Rf = 0.78.-

Dissertation _ -105- _ _ _ Christoph S. Eberle
IR (NaCl, Film): � = 3099 cm-1 (w), 2932 (s, CH), 1733 (s, CO, Ester), 1500 (s), 1456 (m),
1393 (m), 1360 (m), 1282 (s), 1176 (m), 1090 (m), 1054 (m), 968 (w), 822 (w), 727 (s), 660
(w), 618 (w).-
1H–NMR (360 MHz, CDCl3): � = 1.31 – 1.47 (m, 2H, CH2), 1.51 – 1.65 (m, 4H, 2xCH2),
1.77 – 1.86 (m, 2H, SCH2CH2), 1.95 – 2.02 (m, 2H, OCH2CH2), 2.24 (t, 2H, 3J = 7.2 Hz,
COCH2CH2), 2.98 – 3.11 (m, 2H, SCH2), 3.44 – 3.54 (m, 1H, SCH), 3.84 – 3.91 (m, 2H,
NCH2), 3.95 – 3.99 (m, 2H, OCH2), 6.04 (t, 2H, 3J = 2.16 Hz, 2xNCHCH), 6.64 (t, 2H, 3J =
2.16 Hz, 2xNCH) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (%): 313 (100) [M+], 280 (15), 249 (10).-
Optische Aktivität: [�]D 20 = + 9.5 (c = 0.004 in CH2Cl2).-
Präzisionsmasse: C15H23O2NS2 berechnet 313.11702
gefunden 313.11739

Dissertation _ -106- _ _ _ Christoph S. Eberle
7.5.1 Darstellung von 2-(1H-pyrrol-1-yl)-1-(1H-pyrrol-1-ylmethyl)ethyl 5-[(3R)-1,2-dithiolan-3-yl]pentanoat 4b
S
S
OOH
H
4b
C8H14O2S2
206.33
C19H26NO2S2
378.55
S
S
O
O
H
N
N
1
206 mg �–(R)–Liponsäure 1 (1 mmol) und 197 mg N,N’–Carbonyldiimidazol (1.2 mmol)
wurden in 20 ml THF* gelöst. Nach einstündigem Rühren bei 45°C unter Argonatmosphäre
wurden zur Reaktionsmischung 380 mg 1,3–di–1H–pyrrol–1–ylpropan–2–ol (3 mmol) in
10 ml THF* getropft. Das Gemisch rührte weiter bei 45°C über Nacht unter Argon. Das
Lösungsmittel wurde anschließend am Rotationsverdampfer entfernt. Über Aluminiumoxid
(Akt. II–III, neutral) wurde das Rohprodukt mit Dichlormethan/Methanol (19:1) als
Laufmittelgemisch gereinigt. (Säulendurchmesser: 2 cm). Nach Trocknen im Feinvakuum
unter Lichtausschluss erhielt man das Produkt als gelb–braunes Öl.*
Ausbeute: 243 mg (0.64 mmol, 64 % d. Th.).-
DC [Alox, Dichlormethan/Methanol (19:1)]: Rf = 0.84.-

Dissertation _ -107- _ _ _ Christoph S. Eberle
IR (NaCl, Film): � = 3101 cm-1 (w), 2931 (s, CH), 1733 (s, CO, Ester), 1532 (m), 1498 (m),
1436 (s), 1376 (m), 1286 (s), 1173 (s), 1075 (s), 1047 (s), 1014 (s), 867 (w), 730 (s), 666 (s).-
1H–NMR (200 MHz, CDCl3): � = 1.18 – 1.73 (m, 6H, 3xCH2), 2.32 – 3.71 (m, 7H), 3.78 –
3.94 (m, 4H, 2xOCHCH2), 5.0 – 5.18 (m, 1H, OCH), 6.19 (t, 2H, 3J = 2.1 Hz, 2xNCHCH),
6.69 (t, 2H, 3J = 2.1 Hz, 2xNCH) ppm.-
Optische Aktivität: [�]D 20 = + 6.0 (c = 0.004 in CH2Cl2).-
MS (EI, 70 eV, Direkteinlass, T = 146°C): m/z (%): 378 (100) [M+], 189 (5) [M+ –
C11H13ON2], 173 (15) [M+ – C11H13O2N2], 106 (100) [M+ – C16H20O2N2].-
______________________________ *Produkt zersetzt sich leicht.

Dissertation _ -108- _ _ _ Christoph S. Eberle
7.6 Darstellung von Phytyl 5-[(3S)-1,2-dithiolan-3-yl]pentanoat 5
S
S
O
OR
H
S
S
OOH
H
1
C8H14O2S2
206.33
C29H49O2S2
484.84
H
CH3
CH3
CH3
CH3
CH3
R =
5
206 mg �–(R)–Liponsäure 1 (1 mmol) und 197 mg N,N’–Carbonyldiimidazol (1.2 mmol)
wurden in 20 ml THF* gelöst. Nach einstündigem Rühren bei 45°C unter Argonatmosphäre
wurden zur Reaktionsmischung 889 mg Phytol (3 mmol, 1 ml) in 10 ml THF* zugetropft.
Das Gemisch rührte weiter bei 45°C über Nacht unter Argon. Das Lösungsmittel wurde
anschließend am Rotationsverdampfer entfernt. Über Kieselgel wurde das Rohprodukt
mit Dichlormethan chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als gelbes Öl.
Ausbeute: 378 mg (0.78 mmol, 78 % d. Th.).-
DC [Kieselgel, Dichlormethan (100%)]: Rf = 0.71.-

Dissertation _ -109- _ _ _ Christoph S. Eberle
IR (NaCl, Film): � = 2927 cm-1 (s, CH), 2867 (s, CH), 1736 (s, CO, Ester), 1669 (w), 1462
(m), 1378 (m), 1237 (m), 1170 (m), 963 (w), 736 (w).-
1H–NMR (360 MHz, CDCl3): � = 0.71 – 0.81 (m, 12H, 4xCH3), 0.9 – 1.5 (m, 30H), 1.51 –
1.65 (m, 3H, C=CCH3), 1.76 – 2.01 und 2.3 – 2.4 (m, 2H, SCH2CH2), 2.21 (dt, 2H, 3J =
7.38 Hz, COCH2CH2), 2.97 – 3.11 (m, 2H, SCH2), 3.43 – 3.51 (m, 1H, SCH), 4.48 (t, 2H, 3J = 7.92 Hz, OCH2CH2), 5.2 – 5.3 (m, 1H, CH2CH=CCH3) ppm.-
MS (EI, 70 eV, Direkteinlass): m/z (%): 484 (60) [M+], 206 (100) [M+ – C20H38], 187 (30).-
Optische Aktivität: [�]D 20 = + 14.4 (c = 0.008 in CH2Cl2).-
Präzisionsmasse: C28H52O2S2 berechnet 484.34087
gefunden 484.34032

Dissertation _ -110- _ _ _ Christoph S. Eberle
7.7 Darstellung von 5-[(3R)-1,2-dithiolan-3-yl]pentan-1-ol 6[85]
S
S
O
H
H
S
S
OOH
H
C8H14O2S2
206.33
C8H16OS2
192.34
1 6
500 mg �–(R)–Liponsäure 1 (2.42 mmol) wurden unter Argonatmosphäre in 50 ml THF*
gelöst und im Eisbad auf 0°C abgekühlt. Unter Rühren wurden vorsichtig 220 mg LiAlH4
(5.8 mmol, 2.4 Äq.) zugegeben. Das Gemisch rührte drei Stunden bei Raumtemperatur.
Danach wurden 50 ml H2O zugesetzt und mit 1N HCl auf pH 3 angesäuert. Anschließend
wurde fünfmal mit je 50 ml Chloroform ausgeschüttelt. Die organische Phase wurde über
Watte filtriert und am Rotationsverdampfer eingeengt.
Zu dem in Chloroform gelösten Dithiol wurde unter Rühren langsam eine 10% (g/g) KI–I2–
Lösung in THF gegeben, bis leichter Iodüberschuss vorhanden war. Das Gemisch rührte zwei
Stunden bei Raumtemperatur. Danach wurde überschüssiges Iod mit 10%iger (v/v) NaHSO3–
Lösung reduziert. Die organische Phase wurde viermal mit 25 ml halbgesättigter NaHCO3–
Lösung und viermal mit 25 ml H2O gewaschen und anschließend über Watte filtriert. Das
Lösungsmittel wurde am Rotationsverdampfer entfernt. Über Aluminiumoxid (Akt. II–III)
wurde das Rohprodukt mit Ethylacetat chromatographiert (Säulendurchmesser: 2 cm). Nach
Trocknen im Feinvakuum unter Lichtausschluss erhielt man das Produkt als gelbes Öl, das
sich leicht zersetzt.
Ausbeute: 130.84 mg (0.68 mmol, 28.1 % d. Th.).-
DC [Alox, Ethylacetat (100%)]: Rf = 0.68.-

Dissertation _ -111- _ _ _ Christoph S. Eberle
IR (NaCl, Film): � = 3369 (s, OH), 2929 (s, CH), 2856 (s, CH), 1658 (m), 1565 (w),
1461 (m), 1455 (m), 1440 (m), 1377 (m), 1348 (m), 1314 (m), 1255 (m), 1216 (m), 1053 (m),
873 (w), 755 (s), 665 (s) cm-1.-
1H–NMR (200 MHz, CDCl3): � = 1.15 – 2.18 (m, 10H, 5xCH2), 2.40 – 2.49 (m, 2H,
SCH2CH2), 3.1 – 3.3 (m, 1H, SCH2), 3.52 – 3.79 (m, 1H, SCH), 5.1 (s, br, 1H, OH) ppm.-
MS (ESI, CH2Cl2/Methanol): positiv: 215 [M + Na+], 231 [M + K+].-
MS (EI, 70 eV, Direkteinlass, T = 64°C): m/z (%): 192 (46) [M+], 159 (26) [M+ – SH], 141
(30), 109 (41), 81 (25), 67 (100).-
Optische Aktivität: [�]D 20 = + 10.0 (c = 0.006 in CH2Cl2).-
Präzisionsmasse: C8H16OS2 berechnet 192.06426
gefunden 192.06430

Dissertation _ -112- _ _ _ Christoph S. Eberle
7.8 Darstellung von (3’’R)-(1,1-Dioxido-2,5-dihydrothien-3,4-diyl) bis(methylen)di-5’-(1“,2“-dithiolan-3“-yl) pentanoat 8[70]
13.8 mg (0.077 mmol) Sulfolenbisalkohol 7, 5.4 mg (0.044 mmol) 4–N,N–Dimethyl–
aminopyridin und 43.5 mg (0.21 mmol) N,N’–Dicyclohexylcarbodiimid wurden in
15 ml THF* gelöst. Dazu wurden 49.9 mg �–(R)–Liponsäure 1 (0.242 mmol, 2.7 Äq.) in 5 ml
THF* gegeben. Das Gemisch wurde fünf Tage lang unter Argonatmosphäre bei Raum–
temperatur gerührt. Danach wurde die Reaktionslösung am Rotationsverdampfer eingeengt.
Über Kieselgel chromatographierte man das Rohprodukt mit Dichlormethan/Ethylacetat
(10:1) als Laufmittelgemisch (Säulendurchmesser: 2 cm). Nach Trocknen im Feinvakuum
erhielt man das Produkt als gelbliches Öl.
Ausbeute: 35 mg (0.063 mmol, 82 %).- DC [(Kieselgel, CH2Cl2/Ethylacetat (10:1)]: Rf = 0.59.- IR (NaCl, Film): � = 2931 cm-1 (s, CH), 2855 (s, CH), 1738 (s, CO, Ester), 1704 (m), 1699
(m), 1661 (w), 1652 (m), 1645 (m), 1520 (m), 1452 (w), 1386 (m), 1318 (m, SO2), 1229 (s,
CO), 1165 (m, SO2), 1121 (w, SO2) 1082 (w), 893 (m).-
1H–NMR (200 MHz, CDCl3): � = 1.1 – 1.9 (m, 20H), 3.1 – 3.3 (m, 4H, SCH2), 3.59 (t, 2H, 3J = 6.85, SCH), 3.92 (s, 4H, SO2CH2), 4.81 (s, 4H, OCH2) ppm.-
7
SS
OH
O
H
SS OH
O
H
SO
O
OH
OH
1
+
S S
O O
H
SS
OO
H
S
O O
C8H14O2S2
206.33
C6H10O4S
178.21
C22H34O6S5
554.83
8

Dissertation _ -113- _ _ _ Christoph S. Eberle
UV/Vis (CH2Cl2): λmax = 321 nm.-
MS (ESI, CH2Cl2/Methanol (1:100): positiv: 577 [M+Na+], negativ: 589 [M+Cl-].-
MS (EI, 70 eV, Direkteinlass, T = 240°C): m/z (%) = 554 (14) [M+], 490 (78) [M+ – SO2],
412 (27), 287 (100), 254 (51), 223 (66), 189 (100).-
Präzisionsmasse: C22H34O6S5-SO2 berechnet 490.13400
gefunden 490.13539

Dissertation _ -114- _ _ _ Christoph S. Eberle
7.8.1 Darstellung von (3”-R)–Cyclohex-4’-en [1’.2’:1.9] (C60-Ih) [5,6]Fulleren-4’,5’-diyl bis(methylen)di-5-(1”.2”-dithiolan-3”-yl) pentanoat 9[70]
15 mg Fulleren C60 (0.02 mmol) wurden in 10 ml 1,2,4–Trichlorbenzol* im Ultraschallbad
bei 25°C gelöst und 8 mg Liponsäurebisester 8 (0.014 mmol) in 5 ml 1,2,4–Trichlorbenzol*
zugegeben. Zuvor wurde die Reaktionsapparatur gründlich mit Argon begast und das
Gemisch unter Argonatmosphäre 120 min auf 190°C erhitzt. Nach dem Abkühlen wurde das
Lösungsmittel am Kugelrohr bei 50–60°C destilliert. Über Kieselgel wurde unreagiertes
Fulleren C60 zunächst mit Toluol eluiert, danach das Produkt mit Dichlormethan/Ethylacetat
(19:1) als Laufmittelgemisch chromatographiert (Säulendurchmesser: 2 cm). Nach Trocknen
im Feinvakuum erhielt man das Produkt als braunen Feststoff.
Ausbeute: 10.7 mg (8.8 �mol, 61 %).- DC: [Kieselgel, CH2Cl2/Ethylacetat (19:1)]: Rf = 0.87.-
S S S
O O
S
OO
H H
8
S S
O O
H
SS
OO
H
S
O O
C22H34O6S5
554.83
C82H34O2S2
1211.40
9

Dissertation _ -115- _ _ _ Christoph S. Eberle
IR (KBr): � = 2924 cm-1 (s, CH), 2854 (m), 1728 (s, CO, Ester), 1458 (m), 1377 (w), 1301
(m), 1126 (s), 572 (w), 526 (m).-
1H–NMR (360 MHz, CDCl3): � = 1.38 – 1.47 (m, 4H, 2xCH2) 1.60 – 1.74 (m, 4H, 2xCH2),
1.81 – 1.91 (m, 4H, 2xCH2), 2.58 – 2.70 (m, 2H, 2xCH), 2.19 – 2.37 (m, 6H, 2xCH, 2xCH2),
3.49 – 3.64 (m, 4H, 2xCH, 2xCH), 3.38 – 3.49 (m, 2H, 2xCH), 4.14 (s, 4H, 2xCH2), 5.27 und
5.30 (2s, 4H, 2xCH2) ppm.-
UV/Vis (CH2Cl2): λmax = 257 nm.-
MS [ESI, CH2Cl2/MeOH (1:100)]: positiv: 1233 [M+Na+]; negativ: 1211 [M-], 1243
[M+CH3O-].-
Präzisionsmasse: C82H34O4S4 berechnet 1210.1340
gefunden 1210.1695

Dissertation _ -116- _ _ _ Christoph S. Eberle
12 13
NH N
N NH
CH3
CH3
CH3CH3
CO2CH3CO2CH3
C34H32N4O4FeCl
651.94
C32H34N4O4
538.64
N N
N N
CH3
CH3
CH3CH3
CO2H
Fe
Cl
CO2H
CH2
CH2
7.9 Darstellung von 3,3’-[2,7,12,18-Tetramethyl-porphyrin-13,17-diyl]-dipropionsäure-dimethylester 13[11d, 48]
10 g ca. 75%iges Hämin 12 (11.5 mol) wurden mit 40 g Resorcin (363.2 mol, 64 Äq.) versetzt
und in einem Dreihalskolben mit KPG–Rührer und Rückflußkühler unter Argonatmosphäre
auf einem Metallbad 45 Minuten bei 165°C erhitzt. Danach wurden über den Rückflußkühler
zügig 300 ml Ether zur Schmelze gegossen. Es entstand eine braune Suspension, die über eine
Glasfritte filtriert wurde. Der zähe Rückstand wurde solange mit Ether gewaschen, bis ein
braun-schwarzes Pulver zurückblieb, das von Etherresten im Feinvakuum befreit wurde.
Dieses rohe Deuterohämin wurde in einer aus Mischung aus 60 ml Pyridin und 200 ml gelöst.
Zur tiefroten Lösung gab man 50 g Eisen(II)–sulfat–Heptahydrat, kühlte auf 0°C ab und
leitete bei dieser Temperatur vier Stunden lang trockenes HCl–Gas ein. Danach wurde die
Lösung auf Eis gegossen und mit Dichlormethan erschöpfend extrahiert. Die gesammelten
organischen Phasen wurden über Watte filtriert und die Lösung am Rotationsverdampfer
eingeengt. Über Aluminiumoxid (Akt. II–III, neutral, 5 cm Säule) wurde das Rohprodukt mit
Dichlormethan filtriert. Nach Trocknen im Feinvakuum erhielt man Deuteroporphyrin–IX–
dimethylester 13 als violetten Feststoff.
Ausbeute: 4.71 g (8.74 mmol, 76 % d. Th.).-

Dissertation _ -117- _ _ _ Christoph S. Eberle
Schmelzpunkt: 223°C
DC: [Alox, CH2Cl2]: Rf = 0.57.-
IR (KBr): � = 3313 cm-1 (w, NH), 2940 (w, CH), 2905 (w, CH), 1728 (s, C=O), 1438 (m,
�(CH2)), 1365 (m), 1299 (m), 1196 (m), 1170 (s, C-O), 1106 (m), 980 (m), 845 (m), 737 (s),
676 (m).-
1H–NMR (200 MHz, CDCl3): � = -3.88 (s, 2H, NH), 3.29 (t, 4H, 3J =7.72 Hz, 2xCH2 � zu
Carbonyl), 3.63 (s, 3H, 12-CH3), 3.65, 3.66, 3.67 (s, je 3H, 18-CH3 und OCH3), 3.73 (s, 3H,
7-CH3), 3.75 (s, 3H, 2-CH3), 4.43 (q, 4H, 3J = 7.71 Hz, 2xCH2 � zu Carbonyl), 9.08 (s, 1H,
8-CH), 9.10 (s, 1H, 3-CH), 10.03 (s, 1H, 10-CH), 10.07 (s, 1H, 5-CH), 10.09 (s, 1H, 15-CH),
10.13 (s, 1H, 20-CH) ppm.-
UV/Vis (CH2Cl2): λmax (�) = 400 nm (181006), 496 (14449), 530 (8393), 566 (6473), 620
(4344).-
MS [EI, 70eV, Direkteinlass, T = 371°C] m/z (% relative Intensität) = 538 (100) [M+], 479
(5), 465 (35) [M+ – CH2CO2CH3], 392 (15) [M+ – 2 CH2CO2CH3].-
Präzisionsmasse: C34H34N4O4 berechnet 538.25801
gefunden 538.25758
CAS: 10589-94-3.- BRN: 635089.-

Dissertation _ -118- _ _ _ Christoph S. Eberle
13
N N
N N
CH3
CH3
CH3CH3
OH OH
Co
C32H34N4O4
538.64
C30H32N4O2Co
539.53
NH N
N NH
CH3
CH3
CH3CH3
CO2CH3CO2CH3
15c
7.9.1 Darstellung von [13,17-Bis(3-hydroxypropyl)-2,7,12,18-tetramethyl-21H,23H-porphyrinato]-cobalt(II) 15c*
1.1 g (2.04 mmol) Deuteroporphyrin–IX–dimethylester 13 wurden mit 309.67 mg LiAlH4
(8.16 mmol, 4 Äq.) in THF* unter Argonatmosphäre gelöst und vier Stunden zum Rückfluß
erhitzt. Danach wurden 2 ml Wasser zugegeben und erneut 10 min zum Rückfluß erhitzt.
Das Lösungsmittel wurde am Rotationsverdampfer entfernt und der bräunlich–violette
Rückstand im Feinvakuum getrocknet. Anschließend überführte man das Rohprodukt in eine
Extraktionshülse und erhitzte fünf Tage lang in einer Soxhlettapparatur mit Ethanol zum
Rückfluß. Nach Entfernen der ethanolischen Lösung am Rotationsverdampfer und Trocknen
im Feinvakuum blieb der Bisalkohol 14 als violetter Feststoff zurück, der weiter umgesetzt
wurde.
300 mg des Deuteroporphyrin–bis–alkohols 14 (0.62 mmol) und 956.56 mg (3.72 mmol,
6 Äq.) Co(II)–acetylacetonat erhitzte man in 80 ml THF* unter Argonatmosphäre und
Lichtausschluss über Nacht zum Rückfluss. Danach überführte man die dunkelrote Lösung
mit Dichlormethan in einen Scheidetrichter und wusch fünfmal mit gesättigter NaCl–Lösung.
Die gesammelten organischen Phasen wurden über Watte filtriert und über Aluminiumoxid
(Akt. II–III) mit Methanol abgenutscht. Das Lösungsmittel wurde am Rotationsverdampfer
entfernt. Über Aluminiumoxid (Akt. II–III) wurde das Rohprodukt mit Methanol filtriert

Dissertation _ -119- _ _ _ Christoph S. Eberle
(Säulendurchmesser: 5 cm). Nach Trocknen im Feinvakuum erhielt man das Produkt als
rotbraunen Feststoff.
Ausbeute: 216.33 mg (0.40 mmol, 64.5 % d. Th.).-
Schmelzpunkt: >300°C
DC: [Kieselgel, CH2Cl2/THF (3:2)]: Rf = 0.57.-
DC: [Alox, Methanol]: Rf = 0.93.-
IR (KBr): � = 3422 (s, OH), 2918 (m, CH), 2841 (m, CH), 1629 (m, C=C), 1575 (m, C=C),
1557 (m), 1507 (w), 1457 (w), 1435 (w), 1383 (w), 1248 (m), 1122 (w), 1036 (m), 986 (w),
844 (m), 795 (w) cm-1.-
1H–NMR (360 MHz, CDCl3): � = 2.30 (t, 4H, 3J = 6.60 Hz, Propyl), 3.42, 3.46, 3.55, 3.58 (s,
je 3H, CH3), 3.74 (t, 4H, 3J = 5.80, Propyl), 4.08 (dt, 6H, 3J = 5.79, Propyl), 9.12, 9.15 (2s, je
1H, 3-CH, 8-CH) 9.91, 9.96 (2s, je 1H, 5-CH, 10-CH), 10.01 (s, 1H, 20-CH), 10.48 (s, 1H,
15-CH) ppm.-
UV/Vis (CH2Cl2): λmax (�) = 392 (145192), 416 (189230), 527 (24423), 554 (22692) nm.-
MS [EI, 70eV, Direkteinlass, T = 323°C] m/z (% relative Intensität) = 539 (25) [M+], 494
(15), 449 (17).-
Präzisionsmasse: C30H32N4O2Co berechnet 539.18572
gefunden 539.18576
______________________________ *Der metallfreie Komplex ebenso wie die jeweiligen Zink- und Nickel-Komplexe wurden nach
Vorschriften in der Dissertation von A. Walter[11a] und M. Wedel[11b] hergestellt und charakterisiert.

Dissertation _ -120- _ _ _ Christoph S. Eberle
14 15d
N N
N N
CH3
CH3
CH3CH3
OH OH
Mn
Cl
C30H34N4O2
482.63
C30H32N4O2Mn
570.99
NH N
N NH
CH3
CH3
CH3CH3
OH OH
7.9.2 Darstellung von [13,17-Bis(3-hydroxypropyl)-2,7,12,18-tetramethyl-21H,23H-porphyrinato]-mangan(III)-chlorid 15d
300 mg Deuteroporphyrin–bis–alkohol 14 (0.62 mmol) und 390.1 mg (3.1 mmol, 5 Äq.)
MnCl2 wurden in 100 ml DMF* unter Argonatmosphäre und Lichtausschluss über Nacht
zum Rückfluss erhitzt. Danach überführte man die dunkelbraune Lösung mit Dichlormethan
in einen Scheidetrichter und wusch mehrmals mit gesättigter NaCl–Lösung.
Gesammelte organische Phasen wurden über Watte filtriert und das Lösungsmittel am
Rotationsverdampfer entfernt. Über Aluminiumoxid wurde das Rohprodukt mit Dichlor–
methan/Methanol (4:1) chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als dunkelbraunen Feststoff.
Ausbeute: 311.9 mg (0.546 mmol, 88.1 % d. Th.).-
Schmelzpunkt: >330°C
DC: [Alox, CH2Cl2/Methanol (4:1)]: Rf = 0.79.-

Dissertation _ -121- _ _ _ Christoph S. Eberle
IR (KBr): � = 3401 (s, OH), 2916 (s, CH), 2856 (s, CH), 1630 (m, C=C), 1476 (m), 1438 (m),
1376 (m), 1326 (m), 1239 (m), 1126 (m), 1037 (s), 1025 (s), 986 (m), 905 (m), 844 (m), 728
(w), 695 (w), 672 (w), 601 (w), 523 (w) cm-1.-
1H–NMR: paramagnetisch.-
UV/Vis (CH2Cl2): λmax (�) = 357 nm (22588), 395 (14380), 470 (13766), 560 (2973).-
MS [EI, 70eV, Direkteinlass, T = 370°C] m/z (% relative Intensität) = 535 (70) [M+ – Cl],
490 (19), 445 (24).-

Dissertation _ -122- _ _ _ Christoph S. Eberle
14 16e
N N
N N
CH3
CH3
CH3CH3
OH OH
Co
Cl
C30H34N4O2
482.63
C30H32N4O2Co
574.97
NH N
N NH
CH3
CH3
CH3CH3
OH OH
7.9.3 Darstellung von [13,17-Bis(3-hydroxypropyl)-2,7,12,18- tetramethyl-21H,23H-porphyrinato]-cobalt(III)-chlorid 15e
300 mg Deuteroporphyrin–bis–alkohol 14 (0.62 mmol) und 402.5 mg (3.1 mmol, 5 Äq.)
CoCl2 wurden in 100 ml DMF* unter Argonatmosphäre und Lichtausschluss über
Nacht zum Rückfluss erhitzt. Danach überführte man die rotbraune Lösung mit Dichlor–
methan in einen Scheidetrichter und wusch mehrmals mit gesättigter NaCl–Lösung.
Gesammelte organische Phasen wurden über Watte filtriert und das Lösungsmittel am
Rotationsverdampfer entfernt. Über Aluminiumoxid wurde das Rohprodukt mit Dichlor–
methan/Methanol (4:1) chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als rotbraunen Feststoff.
Ausbeute: 313 mg (0.54 mmol, 87 % d. Th.).-
Schmelzpunkt: >330°C
DC: [Alox, CH2Cl2/Methanol (4:1)]: Rf = 0.87.-
IR (KBr): � = 3413 (s, OH), 2917 (m, CH), 2856 (m, CH), 1630 (m, C=C), 1440 (m), 1376
(m), 1326 (w), 1247 (m), 1127 (w), 1038 (m), 904 (w), 845 (m), 730 (w), 671 (w) cm-1.-

Dissertation _ -123- _ _ _ Christoph S. Eberle
1H–NMR: paramagnetisch.-
UV/Vis (CH2Cl2): λmax (�) = 391 nm (43698), 548 (3220).-
MS [EI, 70eV, Direkteinlass, T = 323°C] m/z (% relative Intensität) = 539 (38) [M+ – Cl],
494 (25), 449 (15).-

Dissertation _ -124- _ _ _ Christoph S. Eberle
NH N
N NH
CH3
CH3
CH3CH3
HO OH
14 16a
C46H58N4O4S4
859.24
C30H34N4O2
482.61
NH N
N NH
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
8. Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrin 16a
25.0 mg Deuteroporphyrin–bis–alkohol 14 (45.75 �mmol), 125.5 mg �–(R)–Liponsäure 1
(0.608 mmol) und 13.0 mg DMAP (0.11 mmol) wurden in 30 ml THF* gelöst. Nach
zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen Lösung von PPA in
Ethylacetat zu und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die
Mischung über Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml
Dichlormethan zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und
halbgesättigter NaHCO3–Lösung ausgeschüttelt. Gesammelte organische Phasen wurden zur
Trocknung über Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt. Über
Kieselgel wurde das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als

Dissertation _ -125- _ _ _ Christoph S. Eberle
Laufmittelgemisch chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als dunkelroten Feststoff.
Ausbeute: 25.5 mg (29.7 �mol, 64.9 % d. Th.).-
Schmelzpunkt: 116°C
DC: [Kieselgel, CH2Cl2/Methanol/n-Hexan (19:1:1)]: Rf = 0.88.-
IR (KBr): � = 3429 (s), 2926 (m, CH), 2852 (m, CH), 1728 (s, C=O, Ester), 1638 (m), 1555
(w), 1437 (m), 1304 (m), 1228 (m), 1173 (s), 1129 (m), 975 (w), 841 (m), 731 (m), 708 (w)
671 (w) cm-1.-
1H–NMR (360 MHz, CDCl3/Pyridin-d5): � = -3.82 (s, 2H, NH), 1.22 – 1.47 (m, 12H,
6xCH2), 1.67 und 2.23 (m, 4H, 2xSCHCH2), 2.20 (m, 4H, 2xCOCH2), 2.62 (m, 4H, 2xCH2 �
zum Porphyrinring), 3.01 (m, 4H, 2xSCH2), 3.33 (m, 2H, SCH), 3.58 (s, 3H, 12-CH3), 3.62
(s, 3H, 18-CH3), 3.70 (s, 3H, 7-CH3), 3.72 (s, 3H, 2-CH3), 4.14 (m, 4H, 2xCH2 � zum
Porphyrinring), 4.38 (m, 4H, 2xOCH2), 9.06 (s, 1H, 8-CH), 9.08 (s, 1H, 3-CH), 10.01 (s, 1H,
15-H), 10.02 (s, 1H, 10-H), 10.06 (s, 1H, 5-H), 10.13 (s, 1H, 20-H) ppm.-
UV/Vis (CH2Cl2): λmax (�) = 398 nm (100862), 496 (12241), 528 (9655), 567 (8448), 618
(7413) nm.-
Optische Aktivität: [�]D 20 = + 4.0 (c = 0.002 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:100)]: positiv: 859 [M + H+], 881 [M + Na+], 897 [M + K+];
negativ: 857 [M-], 889 [M + CH3O-].-

Dissertation _ -126- _ _ _ Christoph S. Eberle
N N
N N
CH3
CH3
CH3CH3
HO OH
Zn
15a 16b
C46H56N4O2S4 Zn
920.63
N N
N N
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
Zn
C30H32N4O2Zn
545.99
8.1 Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrinato}-zink(II) 16b
25.0 mg des Zink(II)–Deuteroporphyrin–bis–alkohol 15a (45.75 �mmol), 125.5 mg �–(R)–
Liponsäure 1 (0.608 mmol) und 13.0 m DMAP (0.11 mmol) wurden in 30 ml THF* gelöst.
Nach zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen Lösung von PPA in
Ethylacetat zu und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die
Mischung über Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml
Dichlormethan zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und
halbgesättigter NaHCO3–Lösung ausgeschüttelt. Gesammelte organische Phasen wurden zur
Trocknung über Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt. Über
Kieselgel wurde das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als

Dissertation _ -127- _ _ _ Christoph S. Eberle
Laufmittelgemisch chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als violetten Feststoff.
Ausbeute: 24.8 mg (32.3 �mol, 70.6 % d. Th.).-
Schmelzpunkt: 161.3°C
DC: [Kieselgel, CH2Cl2/Methanol/n-Hexan (19:1:1)]: Rf = 0.89.-
IR (KBr): � = 2914 (m, CH), 2853 (m, CH), 1729 (s, C=O, Ester), 1629 (m), 1452 (m), 1373
(w), 1305 (m), 1237 (m), 1173 (s), 1128 (s), 1083 (w), 1032 (w), 1017 (w), 978 (w), 922 (w),
892 (w), 844 (m), 754 (w) cm-1.-
1H–NMR (360 MHz, CDCl3/Pyridin-d5): � = 1.19 – 1.44 (m, 12H, 6xCH2), 1.65 und 2.20 (m,
4H, 2xSCHCH2), 2.17 (m, 4H, 2xCOCH2), 2.58 (m, 4H, 2xCH2 � zum Porphyrinring), 2.96
(m, 4H, 2xSCH2), 3.30 (m, 2H, 2xSCH), 3.70 (s, 3H, 2-CH3), 3.54 (s, 3H, 12-CH3), 3.58 (s,
3H, 18-CH3), 3.67 (s, 3H, 7-CH3), 4.13 (m, 4H, 2xCH2 � zum Porphyrinring), 4.33 (m, 4H,
2xOCH2), 9.02 (s, 1H, 8-CH), 9.05 (s, 1H, 3-CH), 9.92 (s, 1H, 10-CH), 9.95 (s, 1H, 15-CH),
9.98 (s, 1H, 5-CH), 10.06 (s, 1H, 20-CH) ppm.-
UV/Vis (CH2Cl2): λmax (�) = 285 nm (302663), 323 nm (210654), 399 (1929782), 530
(174334), 566 (193705).-
Optische Aktivität: [�]D 20 = + 4.3 (c = 0.0014 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:10)]: positiv: 921 [M + H+], 943 [M + Na+], 959 [M + K+];
negativ: 956 [M + Cl-].-

Dissertation _ -128- _ _ _ Christoph S. Eberle
N N
N N
CH3
CH3
CH3CH3
HO OH
Ni
15b 16c
C46H56N4O2S4Ni
915.91
N N
N N
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
Ni
C30H32N4O2Ni
538.19
8.1.2 Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrinato}-nickel(II) 16c
25.0 mg Nickel(II)–Deuteroporphyrin–bis–alkohol 15b (45.75 �mmol), 125.5 mg �–(R)–
Liponsäure 1 (0.608 mmol) und 13.0 mg DMAP (0.11 mmol) wurden in 30 ml THF* gelöst.
Nach zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen PPA-Lösung in Ethylacetat zu
und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die Mischung über
Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml Dichlormethan
zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und halbgesättigter
NaHCO3–Lösung ausgeschüttelt. Gesammelte organische Phasen wurden zur Trocknung über
Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt. Über Kieselgel wurde
das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als Laufmittelgemisch
chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im Feinvakuum erhielt man

Dissertation _ -129- _ _ _ Christoph S. Eberle
das Produkt als rotes Öl. Es gelang nicht, das Produkt aus CH2Cl2/n–Pentan isotherm
umzukristallisieren.
Ausbeute: 29.16 mg (31.84 �mol, 69.6 % d. Th.).-
DC: [Kieselgel, CH2Cl2/Methanol/n-Hexan (19:1:1)]: Rf = 0.87.-
IR (NaCl, Film): � = 2924 (s, CH), 2854 (s, CH), 1713 (s, C=O, Ester), 1456 (s), 1374 (m),
1259 (m), 1171 (m), 742 (w) cm-1.-
1H–NMR (360 MHz, CDCl3/Pyridin-d5): � = 1.1 – 3.2 (m, 30H), 3.36 (s, 3H, 12-CH3), 3.40
(s, 3H, 18-CH3), 3.45 (s, 3H, 7-CH3), 3.50 (s, 3H, 2-CH3), 3.69 – 3.84 (m, 4H, 2xCH2 � zum
Porphyrinring), 4.3 – 4.4 (m, 4H, 2xOCH2), 9.09 (s, 1H, 8-CH), 9.11 (s, 1H, 3-CH), 9.50 (s,
1H, 15-CH), 9.51 (s, 1H, 10-CH), 9.54 (s, 1H, 5-CH), 9.62 (s, 1H, 20-CH) ppm.-
UV/Vis (CH2Cl2): λmax = 290, 390, 483, 513, 549 nm.-
Optische Aktivität: [�]D 20 = + 4.1 (c = 0.002 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:10)]: negativ: 913 [M- – H].-
MS [EI, 70 eV, Direkteinlass, T = 364°C] m/z (%) = 914 (6) [M+], 726 (5) [M+ – C8H13OS2],
493 (5) [M+ – C10H17O2S2].-

Dissertation _ -130- _ _ _ Christoph S. Eberle
N N
N N
CH3
CH3
CH3CH3
HO OH
Co
15c 16d
C46H56N4O2S4
916.15
C30H32N4O2Co
539.53
N N
N N
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
Co
8.1.3 Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrinato}-cobalt(II) 16d
25.0 mg Cobalt(II)–Deuteroporphyrin–bis–alkohol 15c (45.75 �mmol), 125.5 mg �–(R)–
Liponsäure 1 (0.608 mmol) und 13.0 mg DMAP (0.11 mmol) wurden in 30 ml THF* gelöst.
Nach zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen Lösung von PPA in
Ethylacetat zu und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die
Mischung über Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml
Dichlormethan zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und
halbgesättigter NaHCO3–Lösung ausgeschüttelt. Gesammelte organische Phasen wurden zur
Trocknung über Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt. Über
Kieselgel wurde das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als
Laufmittelgemisch chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im

Dissertation _ -131- _ _ _ Christoph S. Eberle
Feinvakuum erhielt man das Produkt als dunkelrotes Öl. Isotherme Umkristallisation aus
CH2Cl2/n–Pentan lieferte das Produkt als rotbraunen Feststoff*.
Ausbeute: 21.78 mg (23.77 �mol, 52 % d. Th.).-
Schmelzpunkt: 164.2°C
DC: [Kieselgel, CH2Cl2/Methanol/n-Hexan (19:1:1)]: Rf = 0.63.-
IR (KBr): � = 2922 (m, CH), 2856 (m, CH), 1777 (m), 1634 (s, CO, Ester), 1505 (m), 1436
(m), 1383 (s), 1228 (m), 1126 (m), 1096 (m), 1062 (m), 912 (m), 801 (w), 737 (m) cm-1.-
1H–NMR (360 MHz, CDCl3/Pyridin-d5): � = 1.0 – 2.3 und 2.8 – 3.0 (m, 30H), 2.50 (s, 4H,
2 x CH2 � zum Porphyrinring), 3.51 (s, 3H, 2-CH3), 3.52 (s, 3H, 18-CH3), 3.54 (s, 3H, 7-
CH3), 3.60 (s, 3H, 2-CH3), 4.13 (s, 4H, 2 x CH2 � zum Porphyrinring), 9.24 (s, 1H, 8-CH),
9.27 (s, 1H, 3-CH), 9.99 (s, 1H, 15-CH), 10.05 (s, 1H, 10-CH), 10.09 (s, 1H, 5-CH), 10.12 (s,
1H, 20-CH) ppm.-
UV/Vis (CH2Cl2): λmax (�) = 396 nm (211337), 404 (305320), 470 (25857), 532 (36300), 570
(32322) nm.-
Optische Aktivität: [�]D 20 = + 3.9 (c = 0.002 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:10)]: positiv: 915 [M+].-
______________________________ *Das Produkt enthielt weiterhin Spuren von Verunreinigungen, die trotz mehrtägiger isothermer
Umkristallisation aus CH2Cl2/n–Pentan nicht vollständig entfernt werden konnten.

Dissertation _ -132- _ _ _ Christoph S. Eberle
N N
N N
CH3
CH3
CH3CH3
HO OH
Mn
Cl
15d 16e
C46H56N4O2S4
947.61
C30H32N4O2
570.99
N N
N N
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
Mn
Cl
8.1.4 Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrinato}-mangan(III)-chlorid 16e
25.0 mg des MnCl(III)–Deuteroporphyrin–bis–alkohols 15d (45.75 �mmol), 125.5 mg �–(R)–
Liponsäure 1 (0.608 mmol) und 13.0 mg DMAP (0.11 mmol) wurden in 30 ml THF* gelöst.
Nach zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen Lösung von PPA in
Ethylacetat zu und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die
Mischung über Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml
Dichlormethan zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und
halbgesättigter NaHCO3–Lösung ausgeschüttelt. Gesammelte organischen Phasen wurden
zur Trocknung über Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt.
Über Kieselgel wurde das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als

Dissertation _ -133- _ _ _ Christoph S. Eberle
Laufmittelgemisch chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als dunkelbraunen Feststoff.
Ausbeute: 25.22 mg (26.61 �mol, 58.2 % d. Th.).-
Schmelzpunkt: 92.5°C.-
DC: [Alox, CH2Cl2/Methanol (9:1)]: Rf = 0.51.-
IR (KBr): � = 2928 (m, CH), 2824 (m, CH), 1733 (w), 1634 (s, CO, Ester), 1602 (s), 1535
(m), 1520 (m), 1444 (m), 1376 (m), 1223 (s), 1124 (w), 1102 (w), 1067 (w), 987 (m), 942
(w), 806 (s), 748 (w) cm-1.-
1H–NMR: paramagnetisch.-
UV/Vis (CH2Cl2): λmax (�) = 411 nm (520984), 457 (73806), 542 (36179), 578 (21708).-
Optische Aktivität: [�]D 20 = + 7.4 (c = 0.004 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:10)]: positiv: 911 [M+ – Cl].-

Dissertation _ -134- _ _ _ Christoph S. Eberle
N N
N N
CH3
CH3
CH3CH3
HO OH
Co
Cl
15e 16f
C46H56N4O2S4CoCl
951.61
C30H32N4O2CoCl
574.99
N N
N N
CH3
CH3
CH3CH3
O
O
S
S
H
O
O
S
S
H
Co
Cl
8.1.5 Darstellung von 13,17-Bis{3-(5’-[(3’’R)-(1,2-dithiolan-3’’-yl]-pentanoyl)-oxy-propanyl}-2,7,12,18-tetramethyl-21H,23H-porphyrinato}-cobalt(III)-chlorid 16f
25.0 mg des CoCl(III)–Deuteroporphyrin–bis–alkohols 15e (45.75 �mmol), 125.5 mg (R)–
Liponsäure 1 (0.608 mmol) und 13.0 mg DMAP (0.11 mmol) wurden in 30 ml THF* gelöst.
Nach zehnminütigem Rühren bei Raumtemperatur unter Argonatmosphäre wurden 3 ml
Triethylamin* zugegeben und das Gemisch im Eisbad auf 0°C abgekühlt. Danach spritzte
man über ein Septum langsam 1 ml (1.7 mmol) einer 50%igen Lösung von PPA in
Ethylacetat zu und ließ die Mischung weitere 40 min bei 0°C rühren. Danach ließ man die
Mischung über Nacht bei Raumtemperatur rühren. Zur Aufarbeitung wurden 50 ml
Dichlormethan zugegeben und im Scheidetrichter nacheinander mehrmals mit H2O und
halbgesättigter NaHCO3–Lösung ausgeschüttelt. Gesammelte organische Phasen wurden
zur Trocknung über Watte filtriert und das Lösungsmittel am Rotationsverdampfer entfernt.
Über Kieselgel wurde das Rohprodukt mit Dichlormethan/Methanol/n–Hexan (19:1:1) als

Dissertation _ -135- _ _ _ Christoph S. Eberle
Laufmittelgemisch chromatographiert (Säulendurchmesser: 5 cm). Nach Trocknen im
Feinvakuum erhielt man das Produkt als rotbraunen Feststoff.
Ausbeute: 27.31 mg (28.70 �mol, 62.7 % d. Th.).-
Schmelzpunkt: 104.5°C.-
DC: [Kieselgel, CH2Cl2/Methanol/n–Hexan (19:1.1)]: Rf = 0.65.-
IR (KBr): � = 3091 cm-1 (m), 3043 (m), 2932 (s, CH), 2825 (m), 1737 (w, CO, Ester), 1598
(s), 1539 (s), 1522 (s), 1445 (s), 1378 (s), 1346 (m), 1265 (s), 1228 (s), 1181 (m), 1106 (m),
1065 (m), 988 (s), 950 (m), 896 (w), 805 (s), 736 (s), 702 (s), 665 (m).-
1H–NMR: paramagnetisch.-
UV/Vis (CH2Cl2): λmax (�) = 412 nm (1531148), 531 (140984), 557 (108197).-
Optische Aktivität: [�]D 20 = + 7 (c = 0.004 in CH2Cl2).-
MS [ESI, CH2Cl2/MeOH (1:10)]: positiv: 915 [M+ – Cl].-

Dissertation _ -136- _ _ _ Christoph S. Eberle
9. LITERATURVERZEICHNIS
___________________
[1] http://www.hm-treasury.gov.uk/independent_reviews
/stern_review_economics_climate_change/stern_review_report.cfm
[2] http://www.wind-energie.de/de/themen/mensch-umwelt/klimaschutz/
[3] F. Capra, „Wendezeit. Bausteine für ein neues Weltbild“, dt. 20. Auflage, Scherz, Bern,
München, Wien, 1991.
[4] G. Ciamican, Science, 1912, 36, 385-394.
[5] J. L. Dempsey, A. J. Esswein, D. R. Manke, J. Rosenthal, J. D. Soper, D. G. Nocera,
Inorg. Chem., 2005, 44, 6879-6892.
[6] V. Balzani, A. Credi, M. Venturi, ChemSusChem, 2008, 1, 2-35.
[7] K. Zweibel, J. Mason, V. Fthenakis, Spektrum der Wissenschaft, 2008, 3, 60-70.
[8] M. H. V. Huynh, T. J. Meyer, Chem. Rev., 2007, 107, 5004-5064.
[9] a) H. Kurreck, M. Huber, Angew. Chem. Int. Ed. Engl., 1995, 34, 849-866. b) K.
Ohkubo, P. J. Sintic, N. V. Tkachenko, H. Lemmetyinen, W. E., Z. Ou, J. Shao, K. M.
Kadish, M. J. Crossley, S. Fukuzumi, Chem. Phys., 2006, 326, 3-14. c) M. R.
Wasielewski, J. Org. Chem., 2006, 71, 5051-5066. d) F. D’Souza, R. Chitta, S. Gadde,
L. M. Rogers, P. A. Karr, M. E. Zandler, A. S. D. Sandanayaka, Y. Araki, O. Ito,
Chem. Eur. J., 2007, 13, 916-922. e) T. Könekamp, A. Ruiz, J. Duwenhorst, W.
Schmidt, T. Borrmann, W. D. Stohrer, F.-P. Montforts, Chem. Eur. J., 2007, 13, 6595-
6604. f) A. C. Benniston, Phys. Chem. Chem. Phys., 2007, 9, 5739-5747.
[10] A. Harriman, Angew. Chem., 2004, 116, 5093-5095.
[11] a) A. Walter, Dissertation, 1997, Bremen. b) M. Wedel, Dissertation, 2000, Bremen.
c) J. Ceron, Dissertation, 2002, Bremen. d) S. Leupold, Dissertation, 2004, Bremen.
e) T. Könekamp, Dissertation, 2007, Bremen. f) D. D. Tien, Dissertation, 2007,
Bremen. g) B. Panek-Bryła, Dissertation, 2008, Bremen.
[12] a) D. Bonifazi, Dissertation ETH No. 15599, 2004, Zürich. b) D. Bonifazi, O. Enger,
F. Diederich, Chem. Soc. Rev., 2007, 36, 390-414.
[13] a) J. S. Lindsay, J. K. Delany, D. C. Mauzerall, H. Linschitz, J. Am. Chem. Soc., 1988,
110, 3610-3621. b) Y. Abel, F.-P. Montforts, Tetrahedron Lett., 1997, 38, 1745-1748.
c) Y. Abel, Dissertation, 1998, Bremen.
[14] a) F.-P. Montforts, M. Glasenapp-Breiling, in: “Houben-Weyl-Methods of Organic
Chemistry, Vol. E9d”, E. Schaumann (Ed.), Thieme, Stuttgart, New-York, 1998. b)
D. Pippin, F. Lower, S. H. Leung, I. Ghosh, S. Lanz, P. A. Jacobi, Org. Lett., 2001, 3,

Dissertation _ -137- _ _ _ Christoph S. Eberle
3061. c) F.-P. Montforts, M. Glasenapp-Breiling, in: “Progress in the Chemistry of
Organic Natural Products”, Vol. 84, W. Herz, H. Falk, G. W. Kirby (Eds.), Springer,
Wien, New York, 2002.
[15] M. Wedel, A. Walter, F.-P. Montforts, Eur. J. Org. Chem., 2001, 9, 1681-1687.
[16] S. Cosnier, C. Gondran, K. Gorgy, R. Wessel, F.-P. Montforts, M. Wedel,
Electrochem. Commun., 2002, 4, 426-430.
[17] J. A. Legako, B. J. White, H. J. Harmon, Sens. Actuators B, 2003, 1, 128-132.
[18] S. Cosnier, C. Gondran, R. Wessel, M. Wedel, F.-P. Montforts, Sens. Actuators B,
2003, 3, 213-222.
[19] a) C. M. Cordas, A. S. Viana, S. Leupold, F.-P. Montforts, L. M. Abrantes,
Electrochem. Commun., 2003, 5, 36-41. b) A. S. Viana, S. Leupold, F.-P. Montforts,
L. M. Abrantes, Electrochim. Acta, 2005, 50, 2807-2813. c) R. Martin, R. Cao, A. M.
Esteva, F.-P. Montforts, J. Porphyrins Phtalocyanines, in press.
[20] http://photoscience.la.asu.edu/photosyn/faculty/gust/index.htm
[21] F. G. Varela, H. R. Manturana, R. Uribe, Biosystems, 1974, 5, 187-196.
[22] M. Bitbol, P. L. Luisi, J. R. Interface, 2004, 1, 99-107.
[23] S. Mann, Angew. Chem., 2008, 120, 2-18.
[24] http://www.its.caltech.edu/~feynman/plenty.html
[25] J. R. Marlow, „Nano“, 1st Ed., Tom Doherty Associates, LLC, New York, 2004.
[26] J. Altmann, “Military Nanotechnology: Potential Applications and Preventive Arms
Control”, 1st Ed., Routledge, Kentucky, 2006.
[27] a) E. Katz, I. Willner, Angew. Chem., 2004, 116, 6166-625. b.) M. Kanehara, E.
Kodzuka, T. Teranishi, J. Am. Chem. Soc., 2006, 128, 13084-13094. c) J. A. A. W.
Elemans, R. v. Hameren, R. J. M. Nolte, A. E. Rowan, Adv. Mater., 2006, 18, 1251-
1266. d) W. Auwärter, A. Weber-Bargioini, S. Brink, A. Riemann, A. Schriffrin, M.
Ruben, J. V. Barth, ChemPhysChem, 2007, 2, 250-254.
[28] J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo, G. M. Whitesides, Chem. Rev.,
2005, 105, 1103-1169.
[29] a) M. C. Roco, W. S. Bainbridge, J. Nanoparticle Res., 2005, 7, 1-13. b) M. C. Roco,
“National Nanotechnology Initiative – Past, present, Future”, in: “Science,
Engineering and Technology”, 2nd Ed., Taylor and Francis, 2007, p. 3.1-3.26.
[30] K. E. Drexler, Proc. Natl. Acad. Sci. USA, 1981, 78, 5275-5278.
[31] N. Taniguchi, “On the Basic Concept of 'Nano-Technology'”, Proc. Intl. Conf. Prod.
Eng. Tokyo, Part II, Japan Society of Precision Engineering, 1974.

Dissertation _ -138- _ _ _ Christoph S. Eberle
[32] E. Schrödinger, “Was ist Leben? Die lebende Zelle mit den Augen des Physikers
betrachtet“, 2. Aufl., Leo Lehnen (Sammlung Dalp), München,1951.
[33] G. M. Whitesides, Small, 2005, 2, 172-179.
[34] R. G. Nuzzo, D. L. Allara, J. Am. Chem. Soc., 1983, 105, 4481-4483.
[35] A. Ulman, “An Introduction to Ultrathin Films from Langmuir-Blodgett to Self-
Assembly”, Academic Press, San Diego, 1991.
[36] H. O. Finklea, “Elektrochemistry of Organised Monolayer of Thiols and Related
Molecules on Electrodes”, in: “Electroanalytical Chemistry, A series of Advances”, A.
J. Bard, I. Rubinstein (Eds.), Vol. 19, p. 109, New York, 1996.
[37] a) Y.-T. Kim, R. L. McCarley, A. J. Bard, Langmuir, 1993, 9, 1941-1944. b) J. J.
Gooding, F. Mearns, W. Yang, J. Liu, Electroanalysis, 2003, 15, 81-96. c) T. Kondo,
K. Uosaki, J. Photochem. Photobiol. C: Photochem. Rev., 2006, 8, 1-17. d) A. H.
Ell, G. Csjernyik, V. F. Slagt, J.-E. Bäckvall, S. Berner, C. Puglia, G. Ledung, S.
Oscarsson, Eur. J. Org. Chem., 2006, 5, 1193-1999. e) S. M. Flores, A. Shaporenko,
C. Vavilala, H.-J. Butt, M. Schmittel, M. Zharnikov, R. Berger, Surf. Sci., 2006, 600,
2847-2856. f) C. Vericat, G. A. Benitez, M. E. Vela, R. C. Salvarezza, N. G. Tognalli,
A. Fainstein, Langmuir, 2007, 23, 1152-1159. g) G. Pace, A. Petitjean, M.-N. Lalloz-
Vogel, J. Harrowfield, J.-M. Lehn, P. Samori, Angew. Chem., 2008, 120, 2518-2522.
h) L. Ban, M. Mrksich, Angew. Chem., 2008, 120, 3444-3447. i) V. Ferri, M. Elbing,
G. Pace, M. D. Dickey, M. Zharnikov, P. Samori, M. Mayor, M. A. Rampi, Angew.
Chem., 2008, 120, 3455-3457.
[38] F. Tao, S. L. Bernasek, Chem. Rev., 2007, 107, 1408-1453.
[39] a) M. Mrksich, Cell. Mol. Life Sci., 1998, 54, 653-662. b) L. Liu, S. Chen, C. M.
Giachelli, B. D. Ratner, S. Jiang, J. Biomed. Mater. Res., 2005, 74A, 23-31. c) Y.
Song, Z. Li, Z. Liu, G. Wie, L. Sun, Microscopy Research Technique, 2005, 68, 59-64.
d) J. Kunze, J. Leitch, A. L. Schwan, R. J. Faragher, R. Naumann, S. Schiller, W.
Knoll, J. R. Dutcher, J. Lipkowski, Langmuir, 2006, 22, 5509-5519. e) F. Zhang, S. S.
Cho, S. H. Yang, S. S. Seo, G. S. Cha, H. Nam, Electroanalysis, 2006, 3, 217-222.
f) S. K. Dondapati, J. M. Montornes, P. L. Sanchez, J. L. A. Sanchez, C. O’Sullivan,
I. Katakis, Electronanalysis, 2006, 19-20, 1879-1884. g) C. D. Han, C. Leitner,
T. Weinbrenner, R. Schlapak, A. Tinazli, R. Tampe, B. Lacker, C. Steindl, P.
Hinterdorfer, H. J. Gruber, M. Hölzl, Bioconjugate Chem. 2007, 18, 247-253. h) H.-C.
Kim, S.-K. Lee, Y.-S. Sohn, H.-K. Ryu, S. W. Jeong, Macromol. Symp., 2007, 249-
250, 71-75.

Dissertation _ -139- _ _ _ Christoph S. Eberle
[40] J. B. Schenoff, M. Li, H. Ly, J. Am. Chem. Soc., 1995, 117, 12528-12536.
[41] M. M. Biener, J. Biener, C. M. Friend, Surf. Sci., 2007, 601, 1659-1667.
[42] L. H. Dubois, R. G. Nuzzo, Ann. Rev. Phys. Chem., 1992, 43, 437-463.
[43] D. Fischer, A. Curioni, W. Andreoni, Langmuir, 2003, 19, 3567-3571.
[44] L. Sun, R. M. Crooks, Langmuir, 1993, 9, 1951-1954. b) V. De Renzi, R. Di Felice, D.
Marchetto, R. Biagi, U. del Pennino, A. Selloni, J. Phys. Chem. B, 2004, 108, 16-20.
[45] J. Lahann, S. Mitragotri, T.-N. Tran, H. Kaido, J. Sundaram, I. S. Choi, S. Hoffer, G.
A. Somorjai, R. Langer, Science, 2003, 299, 371-374.
[46] I. S. Choi, Y. S. Chi, Angew. Chem., 2006, 118, 5014-5018.
[47] K. B. Blodgett, I. Langmuir, Phys. Rev., 1937, 51, 964-982.
[48] K. M. Kadish, K. M. Smith, R. Guilard, “The Porphyrin Handbook: Synthesis and
Organic Chemistry”, Vol. I., Academic press: New York, 2000, Chapter 1.
[49] a) P. Grochulski, Y. Li, J. D. Schrag, F. Bouthillier, P. Smith, D. Harrison, B. Rubin,
M. Cygler, J. Biol. Chem., 1993, 268, 12843-12847. b) M. Cygler, P. Grochulski, R. J.
Kazlauskas, J. D. Schrag, F. Bouthiller, B. Rubin, A. N. Serreqi, A. K. Gupta, J. Am.
Chem. Soc., 1994, 116, 3180-3186. c) P. Grochulski, Y. Li, J. D. Schrag, M. Cygler,
Prot. Sci., 1994, 3, 82-91. d) J. Zuegg, H. Hönig, J. D. Schrag, M. Cygler, J. Mol. Cat.
B: Enz., 1997, 3, 83-98.
[50] M. Lotti, A. Tramontano, S. Longhi, Protein Eng., 1994, 7, 531-535.
[51] L. Alberghina, M. Lotti, J. Mol. Cat. B: Enz., 1997, 3, 37-41.
[52] S. Benjamin, A. Pandey, Yeast, 1998, 14, 1069-1087.
[53] L. H. Posorske, J. Am. Oil Chem. Soc., 1984, 61, 1758-1760.
[54] a) M. Goto, S. Noda, N. Kamiya, F. Nakashio, Biotechnol. Lett., 1996, 18, 839-844.
b) S.-W. Tsai, C.-C. Lu, C.-S. Chang, Biotechnol. Bioeng., 1996, 51, 148-156. c) F.
Bellezza, A. Cipiciani, G. Cruciani, F. Fringuelli, J. Chem. Soc. Perkin Trans. 1, 2000,
4439-4444.
[55] a) H. Michor, R. Marr, T. Gamse, T. Schilling, E. Klingsbichel, H. Schwab,
Biotechnol. Letters, 1996, 18, 79-84. b) W.-H. Wu, C. C. Akoh, R. S. Phillips, Enz.
Microbial Technol., 1996, 18, 536-539.
[56] B. Cambou, A. M. Klibanov, Biotechnol. Bioeng., 1984, 16,1449-1454.
[57] B. Orlich, Dissertation, 2000, Berlin.
[58] N. W. Fadnavis, K. Koteshwar, Tetrahedron: Asymmetry, 1997, 8, 337-339.
[59] L. J. Reed, I. C. Counsalus, B. G. Debusk, C. S. Homberger, Jr., Science, 1951, 114,
93-94.

Dissertation _ -140- _ _ _ Christoph S. Eberle
[60] a) M. Calvin, J. A. Barltrop, J. Am. Chem. Soc., 1952, 74, 6153-6154. b) J. A.
Barltrop, P. M. Hayes, M. Calvin, J. Am. Chem. Soc., 1954, 76, 4348-4367.
[61] a) C. S. Hornberger, R. F. Heitmiller, I. C. Gunsalus, G. H. F. Schnakenberg, J. Am.
Chem. Soc., 1952, 74, 2382. b) E. Walter, A. F. Wagner, L. H. Peterson, F. W. Holly,
K. Folkers, J. Am. Chem. Soc., 1954, 76, 4748. c) A. F. Wagner, E. Walton, G. E.
Boxer, M. P. Pruss, F. W. Holly, K. Folkers, J. Am. Chem. Soc., 1956, 78, 5079-5081.
[62] K. Mislow, W. C. Meluch, J. Am. Chem. Soc., 1956, 78, 5920-5921.
[63] a) M. H. Brookes, B. T. Golding, D. A. Howes, A. T. Hudson, J. Chem. Soc., Chem.
Commun., 1983, 1051-1052. b) L. Dasaradhi, N. W. Fadnavis, U. T. Bhalerao, J.
Chem. Soc., Chem. Commun., 1990, 729-730. c) P. C. B. Page, C. M. Rayner, I. O.
Sutherland, J. Chem. Soc. Perkin Trans. 1, 1990, 1615-1618. d) A. S. Gopalan, H. K.
Jacobs, J. Chem. Soc. Perkin Trans 1, 1990, 1897-1900. e) T. T. Upadhya, M. D.
Nikalje, A. Sudalai, Tetrahedron Lett., 2001, 42, 4891-4893.
[64] a) E. J. Toone, M. J. Werth, J. B. Jones, J. Am. Chem. Soc., 1990, 112, 4946-4952. b)
U. T. Bhalera, L. Dasaradhi, P. Neelakantan, N. W. Fadnavis, J. Chem. Soc. Chem.
Commun., 1991, 1197-1198.
[65] R. Vaidyanathan, V. G. Kalthod, D. P. Ngo, J. M. Manley, S. P. Lapekas, J. Org.
Chem., 2004, 69, 2565-2568.
[66] T. Kamijo, H. Harada, K. Iizuka, Chem. Pharm. Bull., 1984, 32, 5044-5047.
[66] A. Treibs, A. Dietl, Liebigs Ann. Chem., 1958, 619, 80-95.
[67] G. Bidan, Tetrahedron Lett., 1985, 26, 735-736.
[68] a) J. Cabrita, C. Eberle, F.-P. Montforts, L. M. Abrantes, unpublished results. b) J.
Cabrita, Master Thesis, University of Lisbon, 2008.
[69] H. Takayama, N. Akadegawa, K. Ando, J. Chem. Soc., Perkin Trans. 1, 1993, 2263-
2268.
[70] A. S. Viana, S. Leupold, C. Eberle, T. Shokati, F.-P. Montforts, L. M. Abrantes, Surf.
Sci., 2007, 601, 5062-5068.
[71] S. Leupold, T. Shokati, C. Eberle, T. Borrmann, F.-P. Montforts, Eur. J. Org. Chem.,
2008, 15, 2621-2627.
[72] F.-P. Montforts, O. Kutzki, Angew. Chem. Int. Ed. Engl., 2000, 39, 599-601.
[73] a) A. Rieder, C. S. Sheehan, B. Kräutler, Helv. Chim. Acta, 2000, 83, 583-591. b) A.
Rieder, B. Kräutler, J. Am. Chem. Soc., 2000, 122, 9050-9051.
[74] a) R. N. Warrener, H. Tang, M. J. Gunter, Chem. Commun., 1999, 803-804. b) R. N.
Warrener, H. Tang, M. J. Gunter, J. Porphyrins Phtalocyanines, 2002, 6, 673-684.

Dissertation _ -141- _ _ _ Christoph S. Eberle
[75] M. G. H. Vicente, A. C. Tome, A. Walter, J. A. S. Cavaleiro, Tetrahedron Lett., 1997,
38, 3639-3642.
[76] S. H. Lee, K. M. Smith, Tetrahedron Lett., 2005, 46, 2009-2013.
[77] a) A. Hirsch, “The Chemistry of the Fullerenes”, Thieme, Stuttgart. New-York, 1994.
b) A. Hirsch, M. Brettreich, ”Fullerenes: Chemistry and Reactions”, Wiley-VCH,
Weinheim, 2004. c) A. Krüger, “Neue Kohlenstoffmaterialien. Eine Einführung“, 1.
Aufl., Teubner, Wiesbaden, 2007.
[78] O. Kutzki, A. Walter, F.-P. Montforts, Helv. Chim. Acta, 2000, 83, 2231-2245.
[79] a) O. Schumm, Hoppe Seylers Z. Phys. Chem., 1928, 176, 122. b) O. Schumm, Hoppe
Seylers Z. Phys. Chem., 1929, 181, 141. c) R. Bonnet, I. H. Campion-Smith, A. J.
Page, J. Chem. Soc., Perkin Trans. 1, 1977, 68-71. d) K. M. Smith, J. A. S. Cavaleiro,
Heteocycles, 1987, 26, 1947-1950.
[80] a) S. Lee, Y.-S. Shon, T. R. Lee, S. S. Perry, Thin Solid Films, 2000, 358, 152-158. b)
R. R. Sahoo, A. Patnaik, J. Colloid Interface Sci., 2003, 268, 43-49. c) T. Gu, J. K.
Whitsell, M. A. Fox, J. Org. Chem., 2004, 69, 4075-4080.
[81] a) A. Ruxandra-Tenea, Dissertation, 2004, Bonn. b) S. Zhang, L. Echegoyen, J. Org.
Chem., 2005, 70, 9874-9881.
[82] T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T.
Hirakawa, S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216-1228.
[83] a) T. Inomata, K. Konishi, Chem. Commun., 2003, 1282-1283. b) A. Satake, Y.
Kobuke, Tetrahedron, 2005, 61, 13-41.
[84] T. Norin, U. Pandit, Chem. Int., 2008, 30, 4-7.
[85] A. F. Wagner, E. Walton, G. E. Boxer, M. P. Pruss, F. W. Holly, K. Folkers, J. Am.
Chem. Soc., 1956, 78, 5079-5081.
[86] A. S. Viana, PhD Thesis, University of Lisbon, 2002.
[87] a) N. Martin, L. Sanchez, B. Illescas, I. Perez, Chem. Rev., 1998, 98, 2527-2547. b) S.
Saha, E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink, J. F. Stoddart, Chem. Eur. J.,
2005, 11, 6846-6858.
[88] Q. Xie, E. Perez-Cordero, L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978-3980.
[89] F. Zhou, C. Jehoulet, A. J. Bard, J. Am. Chem. Soc., 1992, 114, 11004-11006.
[90] A. S. Viana, M. Kalaji, L. M. Abrantes, Electrochim. Acta, 2002, 47, 1587-1594.
[91] H. A. Biebuyck, C. D. Brain, G. M. Whitesides, Langmuir, 1994, 10, 1825-1831.
[92] Y. Dong, S. Abaci, C. Shannon, Langmuir, 2003, 19, 8922-8926.

Dissertation -142- Christoph S. Eberle
[93] R. P. S. Fartaria, F. F. M. Freitas, F. M. S. S. Fernandes, J. Electroanal. Chem., 2005,
574, 321-331.
[94] H. Sellers, E. Shustorovich, J. Mol. Cat. A: Chem., 1997, 119, 367-375.
[95] G. Liu, J. A. Rodriguez, J. Dvorak, J. Hrbek, T. Jirsak, Surf. Sci., 2002, 505, 295-307.
[96] J. A. Rodriguez, J. M. Ricart, A. Clotet, F. Illas, J. Phys. Chem., 2001, 115, 454-465.
[97] a) T. Suzuki, R. D. Noble, C. A. Koval, Inorg. Chem., 1997, 36, 136-140. b) K. Wang,
E. I. Stiefel, Science, 2001, 291, 106-109.
[98] Z. Li, C. Brouwer, C. He, Chem. Rev., 2008, 108, 3239-3265.
[99] J.-P. Bucher, L. Santesson, K. Kern, Langmuir, 1994, 10, 979-983.
[100] J. Huang, J. C. Hemminger, J. Am. Chem. Soc., 1993, 115, 3342-3343.
[101] Y. Shirai, L. Cheng, B. Chen, J. M. Tour, J. Am. Chem. Soc., 2006, 128, 13479-13489.
[102] a) A. Credi, Angew. Chem., 2007, 119, 5568-5572. b) Z. Yao, J. Bhaumik, S.
Dhanalekshmi, M. Ptaszek, P. A. Rodriguez, J. S. Lindsey, Tetrahedron, 2007, 10657-
10670.
[103] X. Lu, L. Zhang, M. Li, X. Wang, Y. Zhang, X. Liu, G. Zuo, ChemPhysChem, 2006,
7, 854-862.
[104] a) Y. Saga, H. Tamiaki, J. Photochem. Photobiol. B: Biol., 2004, 73, 29-34. b) M.
Kondo, Y. Nakamura, K. Fujii, M. Nagata, Y. Suemori, T. Dewa, K. Iida, A. T.
Gardiner, R. J. Cogdell, M. Nango, Biomacromolecules, 2007, 8, 2457-2463.
[105] S. Gottesfeld, Y. Kim, A. Redondo, in: I. Rubinstein (Ed.), “Physical
Electrochemistry: Principles, Methodes and Applications”, Marcel Dekker, New
York, 1995, p. 393.
[106] D. A. Offord, S. B. Sachs, M. S. Ennis, T. A. Eberspacher, J. H. Griffin, C. E. D.
Chidsey, J. P. Collman, J. Am. Chem. Soc., 1998, 120, 4478-4487.
[107] a) T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T.
Hirakawa, S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216 -1228. b) H. Spillmann,
A. Kiebele, M. Stöhr, T. Jung, D. Bonifazi, F. Cheng, F. Diederich, Adv. Mater., 2006,
18, 275 -279.
[108] B. Ulgut, H. D. Abruna, Chem. Rev., 2008, 108, 2721-2736.
[109] Instructions for Autors, Angew. Chem., 2000, 112, 19-23.

Dissertation -143- Christoph S. Eberle
10. LEBENSLAUF
Persönliches: Christoph Stefan Eberle, geboren am 21. Mai 1979
in Neunkirchen (Saar)
___________________________________________________________________________
Schulbildung:
1985 – 1989 Grundschule Hirzweiler/Welschbach
1990 – 1995 Gesamtschule Schiffweiler
1995 – 1998 gymnasiale Oberstufe der Gesamtschulen Schiffweiler
und Neunkirchen am Krebsberggymnasium in
Neunkirchen (Saar)
22. Juni 1998 Abitur
26. Juni 1998 Abiturpreis des Landkreises Neunkirchen (Saar)
___________________________________________________________________________
Zivildienst:
August 1998 – Zivildienstleistender im Palotti–Haus und im St. Josef
August 1999 Krankenhaus in Neunkirchen (Saar)
zuletzt eingesetzt als Pflegehelfer
___________________________________________________________________________
Hochschulbildung:
Oktober 1999 – Studium der Biochemie an der Universität Bayreuth
Oktober 2004
24. Oktober 2001 Vordiplom in Biochemie
Januar – Juli 2004 Diplomarbeit mit dem Titel „Chirale Carbocyclen aus (2R,3R) –
Weinsäure“ unter Anleitung von Prof. Dr. Rainer Schobert
am Lehrstuhl Organische Chemie I in der Fakultät Biologie,
Chemie und Geowissenschaften
28. Juli 2004 Diplom in Biochemie

Dissertation _ -144- _ _ _ Christoph S. Eberle
Mai 2005 Annahme als Doktorand im Fachbereich 2 (Biologie/Chemie)
der Universität Bremen
seit Juni 2005 Anfertigung vorliegender Dissertation im Institut für Organische
Chemie unter Anleitung von Prof. Dr. Franz–Peter Montforts
Juni 2007 – Mai 2008 Wissenschaftlicher Mitarbeiter für Forschung und Lehre im
Fachbereich 2 (Biologie/Chemie)
Juni – November 2008 Promotionsabschluss–Stipendium der Universität Bremen
___________________________________________________________________________
Auslandsaufenthalte:
1. – 12. Oktober 2006
3. – 17. September 2007
25. März – 24. April 2008
o Wissenschaftleraustausch innerhalb des DAAD–Projekts Nr. 40200248 mit der
Arbeitsgruppe von Prof. Dr. Luisa Maria Abrantes an der Universität Lissabon
(Portugal)
___________________________________________________________________________
Sonstige und ehrenamtliche Tätigkeiten:
April 2001 Studentische Hilfskraft am Lehrstuhl Didaktik der Biologie
(Prof. Dr. Siegfried Klautke) in der Fakultät Biologie, Chemie
und Geowissenschaften der Universität Bayreuth
August – Oktober 2001 Studentische Hilfskraft am Lehrstuhl Biochemie (Prof. Dr.
Dr. h. c. Mathias Sprinzl) in der Fakultät Biologie, Chemie und
Geowissenschaften der Universität Bayreuth
Januar – März 2002 Studentische Hilfskraft am Lehrstuhl Anorganische Chemie I
(Prof. Dr. Harald Hillebrecht) in der Fakultät Biologie, Chemie
und Geowissenschaften der Universität Bayreuth
Januar – Juli 2004 Studentische Hilfskraft am Lehrstuhl Organische Chemie I
(Prof. Dr. Rainer Schobert) in der Fakultät Biologie, Chemie
und Geowissenschaften der Universität Bayreuth
November 2005 – Stellvertretender Regionalsprecher des JungChemikerForums
November 2007 (JCF) Bremen in der Gesellschaft Deutscher Chemiker (GDCh)

Dissertation _ -145- _ _ _ Christoph S. Eberle
___________________________________________________________________________
Mitgliedschaften:
Gesellschaft Deutscher Chemiker (GDCh)
Verband angestellter Akademiker und leitender Angestellter der
chemischen Industrie (VAA)
International Union of Pure and Applied Chemistry (IUPAC)
Bundesverband junger Autoren und Autorinnen (BVjA)
___________________________________________________________________________
Konferenzbeiträge und Veröffentlichungen:
Poster: A. S. Viana, C. Eberle, F.-P. Montforts, L. M. Abrantes, 59th
Annual Meeting of the International Society of
Electrochemistry, 2008, Seville, Book of Abstracts, s04-P-076.
Poster: C. Eberle, A. S. Viana, F.-P. Montforts, L. M. Abrantes, XV.
Meeting of the Portuguese Electrochemical Society, 2008,
Lisbon, Book of Abstracts, p. 48.
Poster: C. S. Eberle, A. S. Viana, L. M. Abrantes, F.-P. Montforts,
9th GDCh–JCF Spring Symposium, 2007, Chemnitz,
Book of Abstracts, p. 84.
Poster: C. S. Eberle, S. Leupold, F.-P. Montforts, J. Porphyrins
Phtalocyanines 2006, 10, 618.
Artikel: S. Leupold, T. Shokati, C. Eberle, T. Borrmann, F.-P. Montforts,
Eur. J. Org. Chem., 2008, 15, 2621-2627.
Artikel: A. S. Viana, S. Leupold, C. Eberle, T. Shokati, F.-P. Montforts,
L. M. Abrantes, Surf. Sci., 2007, 601, 5062-5068.





























