Embed Size (px)
Citation preview

1
Platform Manufacturing of Biopharmaceuticals: Putting Accumulated Data and Experience to Work Table of Contents
1. Objective and Scope 2. Definitions 3. Introduction 4. Platform Manufacturing Approaches and their Application: Phases of
Product Development and Commercial Product Manufacture 5. Leveraging Platform Process and Platform Manufacturing Data Throughout
Drug Development Programs and Manufacturing Operations 5.1 Viral clearance / inactivation steps are usually ‘platformed’ 5.2 A platform process generates data that are relevant to multiple
products 5.3. How much data are required to specify a platform?
6. Presentation of Platform Process Data for Regulatory Review 7. Conclusions
Contributors:
Contributor / Coordinator: Enda Moran, Pfizer Contributor: Wolfgang Kuhne, F. Hoffmann - La Roche Ltd Contributor: Kris Barnthouse, Janssen Biologics Contributor: David Beattie, Merck Contributor: Ranga Godavarti, Pfizer Contributor: Piers Allin, EBE

2
Platform Manufacturing of Biopharmaceuticals: Putting Accumulated Data and Experience to Work
1. Objective and Scope
Technology and process platforms are now used extensively in the biopharmaceutical
industry, both throughout the various phases of product development and in the facilities
used to manufacture clinical and commercial biopharmaceutical products. This document
provides background information on the practical application of technology &
manufacturing platforms within the biopharmaceutical industry. While examples and
references are primarily made to proteins, the principles and approaches may equally be
applied to other biopharmaceutical product classes such as conjugate molecular vaccines,
live viral vaccines and so on. The value of data and knowledge gathered from platform
manufacturing or platform-based process development is explored, and the advantages of
the application and use of platforms, both to drug sponsors and the drug regulatory
agencies, are considered.
2. Definitions
Platform technology / platform process: a common or standard method, equipment,
procedure or work practice that may be applied to the research, development or
manufacture of different products
Platform manufacturing: implementation of standard technologies, systems and
work practices within manufacturing facilities, and their use for the manufacture of
different products
OR
the approach of developing a production strategy for a new drug starting from
manufacturing processes similar to those used by the same manufacturer to
manufacture other drugs of the same type
QbD: Quality by Design

3
IND: Investigational New Drug
IMPD: Investigational Medicinal Product Dossier
EBE: European Biopharmaceutical Enterprises
Mab or mAb: Monoclonal antibody
HCP: Host cell protein
CHO: Chinese hamster ovary
GS-NS0: Glutamine synthetase-NS0
3. Introduction
A platform technology or process may be generally defined as a common or standard
method, equipment, procedure or work practice that may be applied across multiple
products under development or manufacture. For example, ‘platform’ may be used to
refer to an expression system such as Chinese Hamster Ovary (CHO) cells, a high
throughput screening system based on robotics, an analytical method such as capillary
electrophoresis, a drug product formulation, a mode of cell culture such as perfusion
culture, a process unit operation such as affinity chromatography or even a complete
process comprising multiple unit operations, Figure 1.
Figure 1 Examples of different types of platform-based tools or methods
in biotechnology product development and manufacture. The
term ‘platform’ can be applied broadly.
A cell line (expression
system) Robotics
Affinity
Capture
(ProA)
Ion
exchange
(AEX)
NanofiltrationCentrifuge UF/DF
A process
Low pH
Inactivation

4
The use of platform technologies emerged in biopharmaceutical product
development programs during the 1990s and primarily focused on monoclonal antibody
production processes, Figure 2. As companies’ biologics portfolios expanded to more
than just a few molecules, the benefits of consistency and similarity of tools and methods
across product development programs was becoming apparent. There are multiple
benefits of using platform approaches for development and manufacturing.
Standardisation of approaches and tools across multiple products leads to improved
quality and consistency, substantial cost-savings primarily as a result of more efficient
resource utilisation (equipment/people), and faster process and product development.
Platform approaches provide deep understanding of the impact of the process design and
control stategy on process performance and product quality. This would have benefits in
the implementation of QbD, and in understanding the likely impacts of manufacturing
deviations or process changes. The use of platform technologies represents considerable
opportunities to reduce risk to patient safety, providing access to platform technologies
with proven performance and known safety profiles. Platform technologies applied to a
particular product type, for example monoclonal antibodies (Mabs), introduces a degree
of consistency to the products under development thereby benefitting their process and
clinical development. Modern commercial biotech manufacturing facilities are now
beginning to use platform technologies and standardised work practices to create
efficiencies and flexibility, and to be capable of the manufacture of multiple products
using a consistent set of well-established technologies.
In the past few years, platform approaches have evolved to a mature state within
manufacturing operations of the biopharmaceutical industry. For many reasons,
principally economic ones, commercial manufacturing facilities of the future must be
capable of the manufacture of multiple products. Some newer facilities constructed in
the past few years will have this flexibility built-in, and will have the capability of
manufacturing multiple products usually in a particular product family e.g., Mabs. The
evolution of multi-product facilities will be enabled through the implementation of
standard technologies and work practices within the facilities (platform manufacturing),

5
1990s
2000+
Mature Platforms within process and product development programs
Many manufacturing facilities using platforms and standardised practices and capable of multi-product manufacture
‘New/different’products (to a standard Mab) can still pose implementation issues for manufacture in established facilities
Companies often developing just one lead product
Innovative and research-driven environment
Little focus on ‘manufacturability’. Any of a multitude of technologies selected to make product
No concept of “Platform”
Maturation of Platform Technologies/Processes
Biotechnology products approved
New manufacturing facilities established
Broad array of technologies used e.g., batch, fed-batch, perfusion (multiple systems), roller-bottle culture, different cell lines etc. – ‘whatever worked!’
‘Inflexible’ facilities
First Platform approaches introduced within company development programs
1980s 1990s
1990s
2000+
Mature Platforms within process and product development programs
Many manufacturing facilities using platforms and standardised practices and capable of multi-product manufacture
‘New/different’products (to a standard Mab) can still pose implementation issues for manufacture in established facilities
Companies often developing just one lead product
Innovative and research-driven environment
Little focus on ‘manufacturability’. Any of a multitude of technologies selected to make product
No concept of “Platform”
Maturation of Platform Technologies/Processes
Biotechnology products approved
New manufacturing facilities established
Broad array of technologies used e.g., batch, fed-batch, perfusion (multiple systems), roller-bottle culture, different cell lines etc. – ‘whatever worked!’
‘Inflexible’ facilities
First Platform approaches introduced within company development programs
1980s 1990s
and the development of similar processes from product to product using standardised
technology and unit operations (platform processes) for transfer into these facilities.
Figure 2 The evolution of platform-based approaches in biotechnology product
development and manufacture. In the 1980s, platform approaches were not
employed. Currently, platforms are widespread within product development
organisations and many production facilities.
4. Platform Manufacturing Approaches and their Application: Phases of Product Development and Commercial Product Manufacture
The pharmaceutical industry is continuously striving to increase the efficiency of
process development in order to move promising products faster and more cost-
effectively into clinical studies. One preferred approach for rapid implementation of
manufacturing processes for new biologics is to develop and implement early-stage
platform processes that are designed to be suitable for the greater part of a company’s
product pipeline. A major intent of the approach is to prevent process development and
clinical material manufacture becoming bottlenecks in the overall clinical development of
new products. For example, production cell lines are created using the same parental cell

6
line, a similar vector and transfection method, and selected using a standardised
approach (usually a high-throughput technology). The final choice of the production cell
line would be based on its performance in the ‘platform process’ using standardized
culture media and culture conditions and the product would be purified using the same
series of downstream process steps: this is different to past approaches to process
development whereby a new process would be developed to accommodate the
recombinant cell line. The product would be formulated in a standard formulation (fixed
buffer composition) and presented in a consistent format, generally lyophilised or liquid.
Process platforms should, by definition, meet a number of a company’s pre-determined
requirements regarding yield, process operability, product quality and so on for early
stage development programs.
A platform manufacturing process used in the clinical development phases would
be expected to deliver the product requirements for supply of phase I clinical studies and
would usually be satisfactory for phase II study supply. In some cases, a need for changes
or improvements might emerge, e.g., to improve the robustness of the phase I process or
to adjust or modify product quality attributes for phase II supply, but these changes would
generally be slight modifications of the platform. Pivotal (phase III) clinical studies are
usually supplied by the final version of the manufacturing process, and often at the
intended manufacturing scale for commercial product supply. Late stage process
development for phase III clinical material manufacture is usually based on the initial
phase I/II platform process. Depending on the company’s development strategy, the need
for optimization, and the availability of alternative innovative technologies, single or
multiple steps of the phase I/II process might be re-developed or optimised in order to
specify the final, commercial manufacturing process.
The platform process concept allows the reconciliation of what might, at first pass,
appear to be incompatible requirements and outcomes of process and product
development. That is, reduced costs and duration of development programs (a concern

7
for the drug sponsors, and the patients waiting for drugs) can be matched with increased
process understanding, increased consistency of quality and heightened assurance of
safety (a concern for patients, the drug sponsors and the Health Authorities). The quality
of a new product can be assessed much earlier in the development process owing to the
availability of standard platform analytical methods which would be evaluated, adapted
and optimized on previous projects. In addition, the knowledge accumulated on previous
projects may be leveraged in order to predict process capabilities under a given set of
operating conditions. The increased process knowledge accumulated from several years
of experience with a platform process therefore creates opportunities to employ QbD-
based approaches to process development, enabling the definition of unit operation and
process design spaces and at the same time, decreasing the development workload for
any given project. These benefits are linked to, and dependent on, the implementation
and use of effective data and knowledge management systems.
Another opportunity is the concept of setting ‘platform specifications’ for new
molecules for those product quality attributes that are common across most, if not all,
mAb programs. The broad experience the industry has with multiple mAb programs in
terms of patient exposure means that the specification-setting exercise for a new program
can be simplified for a number of “standard” attributes by defaulting to a template list of
limits. This would primarily encompass those attributes that are related to “safety” of the
product, for example purity, percent aggregate, percent residual DNA, percent residual
HCP, and endotoxin (if normalized based on highest target dose). In keeping with
regulatory expectations, these specifications could be relatively broad for phase I clinical
programs, so that clinical trial data are acquired with product that has some range of
attribute variability, and then systematically tightened as more manufacturing experience
with the particular molecule is gained and process capability is understood. An extension
of this concept would be to apply “platform specifications” even to those structural
characteristics, such as oxidation, disulphide content, N-terminal and C-terminal

8
modification, glycosylation variants, etc., that have been shown through structure-activity
studies to be not significant for the activity of a particular new product.
Platform processes are currently most established in the biopharmaceutical industry
for the manufacture of monoclonal antibodies. Monoclonal antibodies are regarded as
well-characterized molecules and are known to exhibit very similar physico-chemical
properties. In these cases, development and application of a platform process is regarded
as cost-reducing, efficiency-increasing and in general, accelerative of programs. Most of
the process platforms developed for monoclonal antibodies are based on a standard
process structure: fed-batch cell culture usually in chemically-defined media, clarification
by centrifugation and/or depth filtration, affinity chromatography capture, low pH viral
inactivation step, followed by a combination of polishing steps including an anion
exchange chromatography step (bind/elute or flow-through mode), a cation exchange
chromatography step, a virus filtration step, and the final UF/DF and filtration steps. The
main benefits of an established platform process such as this include;
o Reduction of process development effort, time and costs.
o Prior platform data informs risk assessments on new process weak-points, and
focuses development effort where most needed.
o Consistency in process performance and product quality (especially important
when developing a particular class of products, e.g., monoclonal antibodies
o Simplification of technology transfer activities to production facilities
o Improved asset utilisation: one facility / same equipment for multiple products
o Documentation preparation can be simplified; e.g., only minor adjustments to
production batch records may be required from one process (product) to the next
o Ability to translate learnings from one product to another as process database
grows. Greater significance of a ‘multi-product’ dataset as compared to a ‘one-off’
study on a single product.
o Raw materials and consumables are standardized allowing the use of materials
with safety profiles proven to be acceptable, cost reductions through volume

9
purchasing and waste reduction as inventory stocks may be used across several
different products
o Reduction in time and resources leading up to and including pre-clinical and
clinical studies
o Reduction in failure rates during manufacturing due to accumulated process
experience
o Reduced personnel training burden owing to similar processes and equipment
o Improved speed through repetition
o Routine procedures for in-process and batch release testing lead to reduction of
errors
o ‘Platform specifications’ for early-phase clinical programs
o Broad database and experience to speed troubleshooting of manufacturing
processes
o Preparation of INDs/IMPDs/marketing authorisation applications may be
facilitated more readily: pre-populated templates may be created which reduce
the time necessary for manufacturers to author and prepare the submissions
A platform process should not be regarded as a static, non-changing procedure. Its
performance should be reviewed on a periodic basis. The manufacturer should ensure
that the platform process is delivering a minimum acceptable level of process robustness,
product yield, product quality etc. during clinical development of different products:
these criteria would be developed based on a company’s own specific needs. Changes to
the platform should be introduced through controlled implementation of improvements
and uptake of innovations. Such controls are important to understand the impact of
improvements on the platform.

10
5. Leveraging Platform Process and Platform Manufacturing Data Throughout Drug Development Programs and Manufacturing Operations
The biotechnology industry has matured to the point that many established drug
developers and manufacturers can now claim considerable experience and knowledge
gained through the development and manufacturing of multiple drug candidates.
Extensive and valuable knowledge databases may be accumulated when platform
processes and methods are applied to the development and manufacture of these
biopharmaceutical drugs. Under these circumstances, the question is posed: can we
leverage process and product data gathered across a portfolio of molecules, and apply
these data to the development of similar molecules in the development pipeline? The
benefits of such an approach have been discussed above and include speedier
submissions if certain studies or validations do not need to be executed for each new
product, faster file reviews at the regulatory agency owing to reduced data requirements
in submissions, and greater significance and reliability can be attached to a ‘multi-product’
dataset compared to a one-off study on a single product. Specific examples of platforms,
the data derived from them, and the value of these data are discussed below.
5.1 Viral clearance / inactivation steps are usually ‘platformed’
The viral clearance steps in a mammalian cell-derived biopharmaceutical
manufacturing process are almost always considered when the use of platform process
data/knowledge are discussed. There are good reasons for this. The same virus
clearance studies are executed multiple times under the same protocol on different
protein molecules. The different proteins, especially those from the same ‘family’ of
molecules, e.g., monoclonal antibodies, generally show similar behaviour at the different
clearance steps (filtration/low pH or chemical inactivation/chromatography) which are
operated the same way (platform process) for each product. These repeat studies tend
to yield consistent and similar results from product to product.

11
Numerous published studies have demonstrated the capability of established
downstream process steps for viral clearance. For example, the clearance of the simian
virus type 40 (SV40) at an ion exchange step using Q-Sepharose Fast Flow (QSFF) resin
and under different operating conditions was consistently > 5 log10 for a number of
monoclonal antibody preparations, Table 1.
Table 1 Clearance of the SV40 virus at an ion exchange step using QSFF resin, under different operating conditions and for a number of monoclonal antibody preparations (mAb 1, mAb 2, mAb 3). After Curtis, S. et al. (2003) Generic/Matrix Evaluation of SV40 Clearance by Anion Exchange Chromatography in Flow-Through Mode. Biotechnol. Bioeng., 84, 2, p179
Product Bed Height (cm)
Linear flow rate
(cm/h)
Load capacity (g mAb/L
QSFF resin)
Log10 SV40 clearance ±
95% CIa
Resin type
mAb 1 19 76 50 ≥5.8 ± 0.4 Naïve resin
mAb 1 19 76 50 ≥5.9 ± 0.4 Naïve resin
mAb 1 19 76 50 ≥5.5 ± 0.4 Reused resin b
mAb 2 19 76 50 ≥5.1 ± 0.0 Naïve resin
mAb 2 19 76 50 ≥5.5 ± 0.4 Naïve resin
mAb 2 19 76 50 ≥5.1 ± 0.4 Reused resin b
mAb 3 20 100 100 ≥5.7 ± 0.2 Naïve resin
mAb 3 20 100 100 ≥5.8 ± 0.1 Naïve resin
mAb 3 20 100 100 ≥7.0 ± 0.1 Reused resin b
aConfidence interval. bThe resin was reused more than 50 times under commercial operation conditions prior to
being evaluated for SV40 clearance.
The data in Table 1 are useful reference material to support the principle of
generic or modular viral clearance steps, but a drug manufacturer must develop its own
in-house data to support its case that a step that can be applied to multiple products
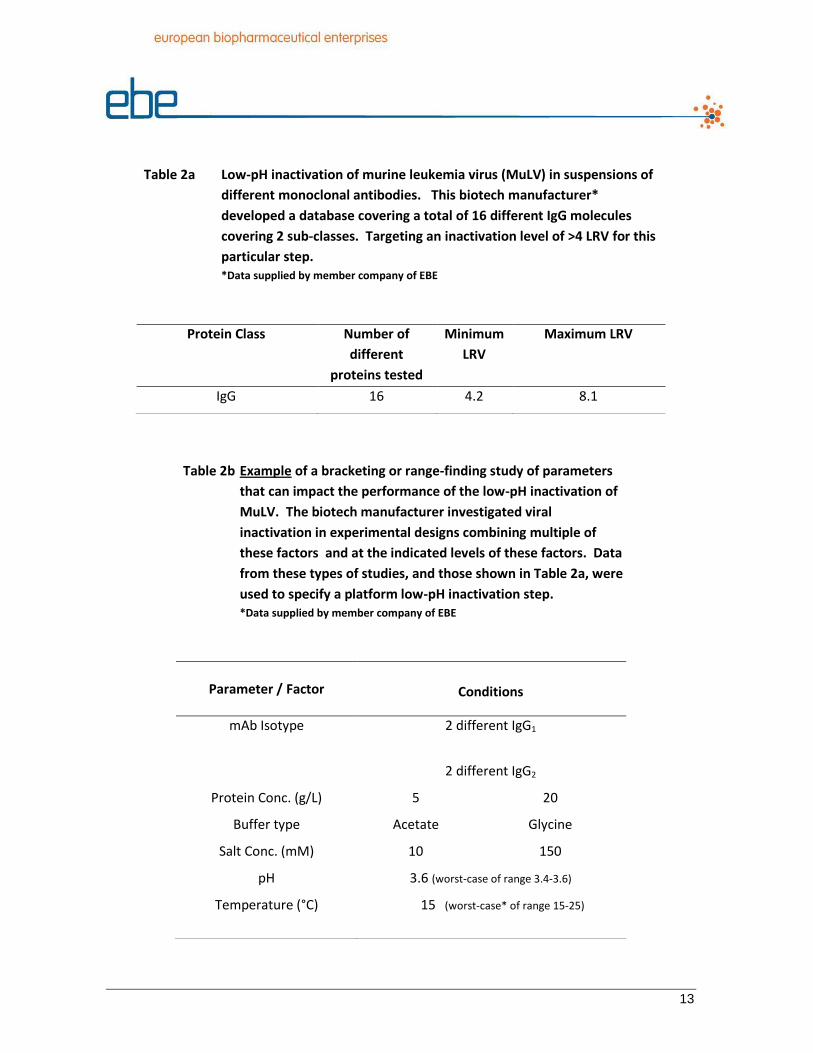
without additional testing. The data in Table 2a are from a large drug manufacturer’s
development and marketed product portfolio, and show the inactivation of murine
leukemia virus (MuLV) due to low-pH treatment of suspensions of sixteen different
monoclonal antibodies. After experimentally executing low-pH inactivations for these

12
sixteen different IgG molecules, the step has been proven capable of achieving the
minimum acceptable log reduction values (LRV) under a variety of conditions. These
different conditions including buffer type, monoclonal antibody sub-type, protein
concentration etc. were further examined in range-finding studies to assess the degree of
robustness of the step, Table 2b. Taken together, the body of data is used to specify the
conditions of operation of the platform step.
This determination of the robustness in itself is a useful benefit to the
manufacturer, as no further development or optimisation work needs to be executed on
this platform step. However, there are opportunities to further exploit this rich database.
For example, the data indicate there is little or no incremental value in executing a further
inactivation study on another IgG protein in the pipeline: further data will only verify what
is already proven. Therefore, there is an opportunity to eliminate testing and concentrate
the saved time and expense on other aspects of the development process. Indeed, the
recent CHMP Guideline on Virus Safety Evaluation of Biotechnological Investigational
Medicinal Products (2008) acknowledges that prior experience and data may be used to
support the reduction in virus clearance testing for investigational products under clinical
development. For now, there are clear expectations for virus clearance testing identified
in ICH Q5A (Virus Safety Evaluation of Biotechnology Products from Cell Lines of Human
or Animal Origin), but this opportunity for streamlining and reducing unnecessary burden
in the drug development and regulatory submissions process is an important one to
consider for the near future.
13
Table 2a Low-pH inactivation of murine leukemia virus (MuLV) in suspensions of
different monoclonal antibodies. This biotech manufacturer*
developed a database covering a total of 16 different IgG molecules
covering 2 sub-classes. Targeting an inactivation level of >4 LRV for this
particular step. *Data supplied by member company of EBE
Protein Class Number of
different
proteins tested
Minimum
LRV
Maximum LRV
IgG 16 4.2 8.1
Table 2b Example of a bracketing or range-finding study of parameters
that can impact the performance of the low-pH inactivation of
MuLV. The biotech manufacturer investigated viral
inactivation in experimental designs combining multiple of
these factors and at the indicated levels of these factors. Data
from these types of studies, and those shown in Table 2a, were
used to specify a platform low-pH inactivation step. *Data supplied by member company of EBE
Parameter / Factor
Conditions
mAb Isotype 2 different IgG1
2 different IgG2
Protein Conc. (g/L) 5 20
Buffer type Acetate Glycine
Salt Conc. (mM) 10 150
pH 3.6 (worst-case of range 3.4-3.6)
Temperature (°C) 15 (worst-case* of range 15-25)

14
5.2 A platform process generates data that are relevant to multiple products
The data in Table 3 show the successful application of a platform method for
cleaning of an ultrafiltration system post-use for the processing of two different
monoclonal antibodies. In situations such as this where the protein concentration and
buffer constituents are similar, the platform cleaning method negates the need to
perform cleaning cycle development for each product, with concomitant savings in time,
materials, documentation preparation, and automation system modifications.
Table 3 Application of a platform method for the cleaning of an ultrafiltration system. Only two measures used to assess cleaning performance are shown. Measures are executed post-processing. Examples presented for two different monoclonal antibodies (Mabs). *Data supplied by member company of EBE
Mab 1 Mab 2
* ‘Worst-case’ from perspective of inactivating an enveloped virus
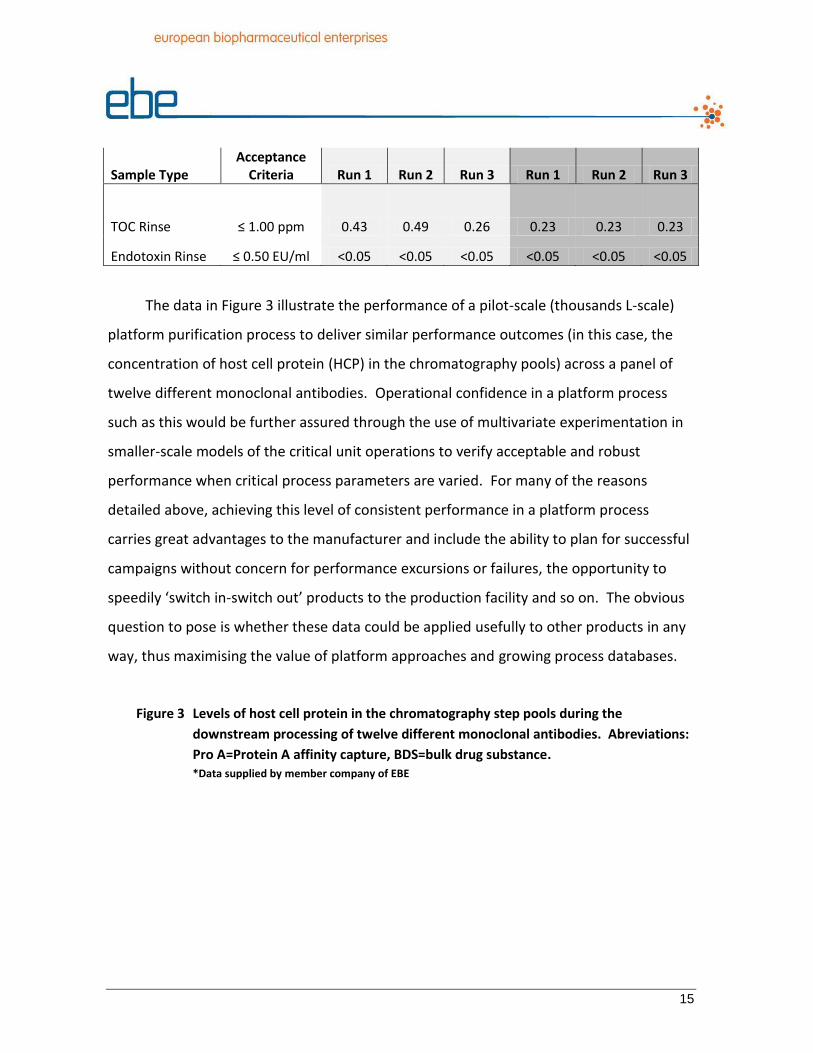
15
Sample Type Acceptance
Criteria Run 1 Run 2 Run 3 Run 1 Run 2 Run 3
TOC Rinse ≤ 1.00 ppm 0.43 0.49 0.26 0.23 0.23 0.23
Endotoxin Rinse ≤ 0.50 EU/ml <0.05 <0.05 <0.05 <0.05 <0.05 <0.05
The data in Figure 3 illustrate the performance of a pilot-scale (thousands L-scale)
platform purification process to deliver similar performance outcomes (in this case, the
concentration of host cell protein (HCP) in the chromatography pools) across a panel of
twelve different monoclonal antibodies. Operational confidence in a platform process
such as this would be further assured through the use of multivariate experimentation in
smaller-scale models of the critical unit operations to verify acceptable and robust
performance when critical process parameters are varied. For many of the reasons
detailed above, achieving this level of consistent performance in a platform process
carries great advantages to the manufacturer and include the ability to plan for successful
campaigns without concern for performance excursions or failures, the opportunity to
speedily ‘switch in-switch out’ products to the production facility and so on. The obvious
question to pose is whether these data could be applied usefully to other products in any
way, thus maximising the value of platform approaches and growing process databases.
Figure 3 Levels of host cell protein in the chromatography step pools during the
downstream processing of twelve different monoclonal antibodies. Abreviations:
Pro A=Protein A affinity capture, BDS=bulk drug substance. *Data supplied by member company of EBE

16
For example (and referring to Figure 3), if a subsequent product (mAb #13) is under
manufacture, the opportunity presents to remove in-process sampling and testing at the
ProA pool for the HCP attribute. Taken further, the process database may expand (more
products) such that confidence of always meeting specification (or target) develops to the
point that all sampling and testing for HCP could be removed. Indeed, and looking at it
from a scientific perspective, a case could be built using platform and product-specific
verification studies to remove the attribute from the final specification list of the
substance or product. Note that HCP is used here as an example but the principle applies
to any process or product-related attribute. Using platform process data in this way
would represent a powerful application of data and risk management to the manufacture
and testing of biopharmaceuticals.
5.3 How much data are required to specify a platform?
There are a number of important and recurring questions to address when
considering the robustness and generic applicability of data derived from platform
operations or platform processes. Firstly, the drug manufacturer will at some point
transition from ‘developing the platform’ to considering the platform specified and

17
established: the question is when this transition occurs? The reality is that this ‘transition’
will depend on the quality and amount of data the manufacturer has gathered on the
platform step or process, and the type of process step or process under consideration.
When we speak of “quality” of data, it is implied that certain types of data such as those
from multi-factorial experimentation will provide greater insights into the degree of
robustness and stability of a process or process step than those data from single factor
studies. Regarding the “amount of data” question, and considering extreme scenarios just
for illustration purposes, it would be difficult to claim a ‘platform’ based on studies or
process runs of just one or two molecules, but when the number of molecules studied
expands to ten or greater as in the example shown in Figure 3 above, the platform could
well be considered established. The amount of data required to verify a platform will also
depend on the characteristics of a process step or unit operation. For example, an
aggressive cleaning regime on an ultrafiltration system dirtied or challenged by a protein
suspended in a simple buffer is unlikely to show substantially varied performance from
one protein molecule to another. In this case, a performance evaluation across two or
three molecules would provide sufficient evidence to the manufacturer that the platform
cleaning step is robust and generically applicable to the processing of similar protein
molecules. In contrast, much more process experience and data would be required if a
platform fed-batch culture step was being developed for a particular class of molecule,
owing to the biological (different cell lines, impact of certain raw materials etc.) and
process variability inherent in this type of unit operation. In summary, there can be no
rigid guiding rules on the amount of data and experience required to specify a platform
process or process step. The drug manufacturer will always be in the best position to
determine for its own particular cases what is appropriate and justifiable based on many
considerations including prior process experience, degree of understanding of the factors
(chemistry, biology etc.) influencing the performance of the process or process step, the
ability to control the process or process step and so on.
The second question concerns platform evolution, and asks whether a change to a
platform step or platform process would constitute a minor modification to the existing

18
platform, or instead, signals the initiation of a ‘new’ platform. Since there is a continuum
of potential changes from minor to major, there can be no hard and fast rules as to what
constitutes a transition to a new platform. For example, and at opposite ends of the
spectrum, a few-degrees centigrade (°C) change in the temperature of the cleaning
solution of an ultrafiltration system would very likely be considered a minor modification
to a platform step, whereas a transition to a new mammalian cell expression system e.g.,
from CHO to GS-NS0, would very likely be considered a transition to a new platform
process. Drug developers and manufacturers that have invested significant years and
resource into developing process platforms tend to carefully modify and adjust those
platforms in stages rather than make frequent, dramatic leaps into new platforms, since in
the latter case, the lengthy phase of data gathering to assess process performance and
robustness would have to start again. Finally, it is worth emphasising the potential value
of the drug sponsor considering early communication with the regulatory agencies to
discuss the justification and rationale for leveraging platform data, if there is to be
significant reliance on platform process data in a forthcoming license variation or new
submission.
6. Presentation of Platform Process Data for Regulatory Review
Regulatory submissions efforts become more efficient through the use of platform
process-based data and knowledge. Internal to companies, templates may be created for
sections of IND/IMPD and commercial licensure submissions which reduce the time
necessary for manufacturers to author the submissions. Regulatory agency reviewers will
also appreciate submissions that make use of platform-based knowledge to demonstrate
more complete understanding of the manufacturing process and the product. Site
inspections, especially for multi-product plants, can also be simplified when platform
approaches have been used, owing to the reduced number of unique manufacturing

19
processes and the concomitant reduction in complexity of the associated quality systems
necessary to manage them.
Platform data should also prove useful to streamline the filing process for post-
approval changes to a manufacturing process or analytical method. For example, in
instances where there is sufficient platform and product-specific evidence that changes to
manufacturing steps and/or equipment or changes to analytical methods etc. have no
impact on the quality, safety or efficacy of the product, then it may be possible to
downgrade the classification of the post-approval submission.
The application of platform-based experience and data to newer products under
development without necessarily having to re-do every study for every product signals a
maturation of the science, experience and knowledge underpinning biotechnological drug
development. While there will always be some product-specific nuances to deal with in
every development program, we are entering the phase of evolution of the industry
where platform data are available to be presented for review to support clinical trial and
marketing authorisation applications. These platform data packages might include those
describing viral clearance capabilities of process steps, clearance of process related
impurities, cleaning regimes for process equipment etc. How might these data be made
available for review? Some possible means are considered here using a viral clearance
step as an example (Figure 4), but as stated previously, the principles are not exclusive to
viral clearance/inactivation unit operations. Current filing structures and mechanisms in
place in many global regions at present do not work as described in Figure 4: this
discussion below is intended to explore possibilities.
Platform-process data could be compiled for multiple products to demonstrate
generic applicability of the viral clearance step. Additional development data could also
be presented, demonstrating the robustness of the step across the intended ranges of
critical process parameters e.g., pH, temperature, time etc. and other process conditions
providing assurance that the step performs acceptably within the operating ranges

20
intended for manufacture. This data package as a whole could be submitted for each
product in each clinical trial application or alternatively, might be lodged in a reference
document similar to a Drug Master File at the agency and would be available for
consultation by reviewers at each new clinical trial application submission by the drug
sponsor. The reference document or ‘Master File’ could be updated by the drug sponsor
if a significant process change was introduced to the viral clearance step, or if a new
replacement clearance step was introduced. If available data were insufficient to support
the generic applicability of the changed or replacement clearance step across multiple
products, product-specific virus clearance data would be submitted under normal practice
until data accumulate to support the step as a platform or ‘generic’ step. The ‘Master File’
would then be updated with new data, and the package applied to subsequent products
under clinical trial or marketing authorisation application.

21
Figure 4 Possibilities for presentation of platform-derived data for regulatory review. A ‘generic’ virus clearance data package is shown as an example, but this illustration could apply to any package of platform process data. Data could be presented for review by submitting the same data package with each new clinical trials application (Path A) or presented once to the agency to be maintained as a type of ‘Master File’ (Path B). The principles shown could equally be applied to Marketing Authorisation Applications.
7. Conclusions
The evolution and benefits of platform technology and processes throughout
biopharmaceutical drug development and manufacture have been discussed. The
benefits of using a platform approach are apparent: standardisation of approaches and
tools across multiple products leads to improved quality and consistency, substantial
cost-savings, efficient resource utilisation (equipment/people) and faster process and

22
product development. Modern commercial manufacturing facilities use
standard/platform technologies and work practices to improve efficiencies and flexibility,
and to be more capable of the manufacture of multiple products. Platform process-
derived data at many drug developers/manufacturers have accumulated to extensive
databases. The biopharmaceutical industry, including the regulatory bodies/health
authorities, is now in a position to exploit these data to improve biopharmaceutical drug
development, manufacture and regulation.





























